User login
New Onset of in Hospitalized Patients
A 78‐year‐old otherwise healthy man with longstanding hypertension is admitted to the hospital with pneumonia. During the second hospital day, he develops atrial fibrillation (AF) with rapid ventricular response, but his hemodynamics remain stable. He is given oral metoprolol for rate control. A transthoracic echocardiogram (TTE) shows mild concentric left ventricular hypertrophy, normal left ventricular size and function, and mild left atrial enlargement. The AF spontaneously terminates after 24 hours and does not recur during the hospitalization. What treatment and monitoring are recommended at the time of discharge for this patient's AF?
BACKGROUND
AF is a common dysrhythmia that clinicians often encounter while caring for hospitalized patients. Although many patients will have carried a diagnosis of AF prior to hospital admission, this review will pertain to patients for whom a first documented episode of AF occurs during hospitalization. These patients can be conceptually separated into 2 categories: those who have had undiagnosed AF for some time (and are now diagnosed due to continuous inpatient telemetry monitoring) and those whose AF is secondary to their acute medical illness. Although practically speaking, this distinction is not easy to make, markers of chronic AF may include left atrial enlargement and a clinical history of longstanding palpitations.
INCIDENCE
The prevalence of AF in the general population is estimated at 0.4% to 1.0%.[1, 2] Prevalence increases with advancing age. Compared to the general population, the population of hospitalized patients is inherently older and enriched for comorbidities that are known risk factors for the development of AF (such as congestive heart failure, hypertension, and obstructive sleep apnea) or are associated with paroxysmal AF (such as stroke or transient ischemic attack [TIA]). As a result, the prevalence of occult AF is necessarily higher in this population than that of a general cohort. The risk of developing AF is further increased in hospitalized patients simply by the acute illness (or postoperative state), whose associated catecholamine surge and systemic proinflammatory state are well‐known precipitants for AF.[3] AF is common after cardiac surgery (25%30%)[4, 5] and occurs in about 3% of patients undergoing major noncardiac surgery.[6]
In the setting of severe medical illness such as sepsis, the incidence of new onset AF has recently been estimated at around 6%.[7] Among patients hospitalized with stroke, 2% to 5% will have a new diagnosis of AF made by the admission electrocardiogram (ECG).[8, 9, 10] Subsequent cardiac monitoring with inpatient telemetry or Holter monitoring will detect previously undiagnosed AF in another 5% to 8% of patients admitted with stroke.[11, 12]
PATHOPHYSIOLOGY
AF is a supraventricular tachyarrhythmia characterized by uncoordinated atrial activation; this chaotic atrial activation translates into atrial mechanical dysfunction. [13] Patients who develop AF may have atrial substrate, such as patchy atrial myocyte fibrosis, that increases their propensity to develop atrial dysrhythmias.[14] Other factors contributing to the likelihood of developing AF are anisotropic conduction, atrial chamber dilation, systemic inflammation, hyperadrenergic state, and atrial ischemia.[3, 15, 16, 17] Atrial flutter, on the other hand, is an organized macro‐reentrant supraventricular arrhythmia that typically rotates around the tricuspid annulus.
RISK FACTORS
Risk factors for the development of AF are well defined. The risk factors in the chronic setting remain the same as those for the development of AF in the setting of medical illness or in the postoperative state: advancing age, male gender, prior diagnosis of AF, congestive heart failure, hypertension, and obstructive sleep apnea.[1, 5, 18] Surgical procedures, due to the sympathetic surge and proinflammatory state that occur in the postoperative period, increase the risk of AF. Cardiac and thoracic procedures, which involve direct manipulation of the heart and adjacent structures, proffer the highest risk of AF.[19, 20] Although not necessarily a risk factor for the development of AF, patients with recent ischemic stroke are at high risk of harboring AF; up to 7% of patients are diagnosed with AF in the 3 months following ischemic stroke.[21]
DIAGNOSIS
In the inpatient setting, the diagnosis of AF is typically made through telemetry monitoring, which reveals irregularly spaced QRS complexes and an absence of organized atrial activity (ie, no discernible P waves or flutter waves). For patients not on a continuous cardiac monitor, the diagnosis of AF is made by 12‐lead ECG, which is triggered by patient complaint (palpitations, lightheadedness, dyspnea, or chest pain), physical exam findings, or review of vital sign measurements (ie, sudden changes in heart rate). The dysrhythmia should sustain for at least 30 seconds for a diagnosis of AF to be made.
INITIAL WORKUP
When AF is suspected (or has been diagnosed by telemetry), a 12‐lead ECG should be immediately obtained (Table 1). This will help to confirm the diagnosis of AF (as distinct from atrial flutter) and begin the investigation for underlying causes (ie, analysis of ST‐segment shifts for evidence of myocardial ischemia or pericarditis). A focused history, physical exam, and review of vital signs can quickly determine if there are any urgent indications for cardioversion, such as the development of pulmonary edema, the presence of angina pectoris, or rhythm‐related hypotension. A TTE should be obtained to assess for structural heart disease (left atrial enlargement, valvular disease, cardiac tumor) that may serve as a substrate for AF. The echocardiogram will also provide an assessment of left ventricular function, which will inform the treating physician regarding the safety of using atrioventricular (AV) nodal blocking agents, such as ‐blockers and nondihydropyridine calcium channel blockers, which may also act as negative inotropes. Although occult hyperthyroidism is a rare cause of AF,[22] a serum thyroid‐stimulating hormone test should be obtained to rule out this reversible cause. Electrolytes should be monitored and serum potassium and magnesium levels should be maintained at >4.0 mmol/L and >2.0 mEq mmol/L, respectively. Measurement of serum B‐type natriuretic peptide can be helpful in determining prognosis and likelihood of left ventricular dysfunction in patients with AF.[23, 24]
|
| Confirmatory study |
| 12‐lead electrocardiogram |
| Assessment of clinical stability |
| History (chest pain, shortness of breath, syncope/presyncope) |
| Physical exam (blood pressure, heart rate, pulmonary rales, jugular venous distension) |
| Evaluation for structural heart disease |
| Physical exam (pathologic murmurs, third heart sound, abnormal PMI, friction rub) |
| Transthoracic echocardiogram |
| Metabolic triggers |
| Serum potassium and magnesium |
| Serum thyroid stimulating hormone |
| Prognostic indicators |
| Serum brain natriuretic peptide |
| Other investigations (as guided by clinical suspicion) |
| Chest CT angiogram |
| Serum troponin |
| Blood cultures |
Other investigations should be guided by the clinical suspicion for other secondary causes. Examples include assessment for infection in the postoperative patient, ruling out myocardial infarction in patients with chest pain and risk factors for coronary artery disease, evaluating for pericarditis following cardiac surgery, and having a high suspicion for pulmonary embolism in patients with prolonged immobilization, hypercoagulable state, or recent knee/hip replacement surgery.
STRATEGIES FOR PREVENTION/SCREENING
AF prevention and screening strategies are not practical for patients admitted for medical illnesses. When used for perioperative prophylaxis, however, amiodarone has been shown to clearly reduce postoperative AF (and shorten hospitalizations) after coronary artery bypass graft surgery.[4, 25] Statin use has been associated with a decrease in postoperative AF following major noncardiac surgery.[26] Patients hospitalized with acute ischemic stroke or TIA should undergo cardiac monitoring throughout their hospitalization if feasible, or for at least 24 hours.[27] Recent data indicate that either Holter monitoring or continuous cardiac telemetry are acceptable methods of screening stroke patients for underlying AF.[11]
THERAPIES
In all cases of AF, underlying causes of the dysrhythmia (such as heart failure, infection, electrolyte disturbances, and pain) should be sought and treated.
AF associated with unstable symptoms (heart failure, angina, hypotension) calls for urgent rhythm control. In this setting, cardioversion should be performed immediately; anticoagulation should be initiated concomitantly unless a contraindication to anticoagulation exists. Stable patients should be assessed for indications for elective cardioversion and acute anticoagulation. Generally speaking, it is desirable to perform transesophageal echocardiography (TEE) and cardioversion prior to discharge from the hospital in patients whose new‐onset AF has persisted, assuming that they are candidates for therapeutic anticoagulation. This is particularly true for patients who are at all symptomatic from their AF. Allowing patients to remain in AF for weeks to months will increase their risk of developing long‐standing persistent AF.
AF is a well‐recognized risk factor for the development of atrial thrombi and resultant thromboembolic events. Thrombus formation is thought to be a result of stasis of blood in the atria during AF as well as a localized hypercoagulable state in the left atrium in patients with AF.[28] Left atrial thrombus can develop in patients with AF of duration <3 days.[29] Echocardiographic evidence suggests that left atrial appendage function can be transiently depressed following cardioversion, which may help to explain the finding of increased risk of thromboembolism immediately after cardioversion.[30, 31] In fact, 98% of thromboembolic events after cardioversion occur within 10 days.[31] Studies using serial TEE show that atrial thrombi typically resolve after 3 to 4 weeks of anticoagulation.[28] These data are the basis for the recommendation that patients with AF that has lasted 48 hours or more should receive 4 weeks of therapeutic anticoagulation prior to cardioversion that is not TEE guided. Importantly, administration of antiarrhythmic agents, such as amiodarone, should be considered an attempt at rhythm control, and therefore anticoagulation should be used in the same way during antiarrhythmic drug initiation as with direct‐current cardioversion. Medications most commonly used to acutely terminate AF are ibutilide, propafenone, and flecainide.
In the inpatient setting, nonemergent cardioversion in patients who have had AF for more than 48 hours should be TEE guided, unless the onset of the arrhythmia was clearly documented and therapeutic anticoagulation was initiated within 48 hours of the onset. Patients should be receiving therapeutic anticoagulation at the time of the TEE. Contrast‐enhanced magnetic resonance imaging is a promising noninvasive option for assessing for intracardiac thrombus, but this modality has not yet been widely adopted as an acceptable alternative to TEE.[32]
Anticoagulation in the short term can be rapidly achieved using heparins (intravenous unfractionated heparin, subcutaneous enoxaparin) or the newer oral anticoagulants such as dabigatran (a thrombin inhibitor) or rivaroxaban and apixaban (factor Xa inhibitors). Importantly, should significant bleeding occur, options for reversal of these new oral anticoagulant agents are limited.[33] Vitamin K antagonists such as warfarin remain a viable option for long‐term anticoagulation, but usually require 4 to 5 days to reach peak effect; the goal international normalized ratio (INR) is 2.0 to 3.0. In patients with chronic kidney disease, the newer oral anticoagulants (dabigatran, rivaroxiban, and apixaban), as well as low molecular weight heparins, should be dose adjusted in patients with moderate renal dysfunction and avoided altogether in patients with severe renal dysfunction.
Ventricular response rate control, rather than rhythm control, is a reasonable initial strategy for patients who do not have significant symptoms from AF. Rate control can be achieved using traditional AV nodal blocking agents (‐blockers and nondihydropyridine calcium channel blockers). Initially, the use of intravenous (IV) agents is reasonable. IV metoprolol and IV diltiazem are useful because they both have a rapid onset of action, which allows for repeated bolus dosing at closely spaced intervals. Both IV agents have a 2‐ to 4‐hour half‐life. Once rate control has been achieved, the amount of IV drug required to achieve heart rate control can be tallied and converted into oral dosing. Cardiac glycosides can also be used to rate‐control AF; digitalis works by exerting a vagotonic effect via alterations in calcium handling in the AV node. Digoxin is most effective in the rate control of patients with persistent AF rather than those with recent onset AF.[34] Even in patients with persistent AF, digoxin only lowers average heart rate during rest and not during exertion/stress.[35] In patients with marginal blood pressure, digoxin can be safely used because it does not have any negative inotropic effects. In patients receiving a rate control strategy, the decision of whether to anticoagulate should be based on the risk of thromboembolic stroke as determined by clinical risk factors. In general, patients with a CHADS2 score[36] of 0 can be treated with aspirin (325 mg daily)[37] for thromboembolism prevention, and those with a score of 2 or more should receive therapeutic anticoagulation. Patients with a CHADS2 score of 1 can reasonably be treated with either regimen, and a more nuanced assessment of bleeding and stroke risk is required. The more recently described CHA2DS2‐VASc score allows for better stroke risk discrimination among patients with low CHADS2 scores (Table 2).[38]
| CHADS2 Elements | CHADS2 Score | Annual Stroke Risk |
|---|---|---|
| ||
| CHF | 0 | 1.2% |
| Hypertension | 1 | 2.8% |
| Age 75 years | 2 | 3.6% |
| Diabetes | 3 | 6.4% |
| Stroke/TIA (2 points) | 4 | 8.0% |
| 56 | 11.4% | |
| CHA2DS2‐VASc Elements | CHA2DS2‐VASc Score | Annual Stroke Risk a |
| CHF | 0 | 0.0% |
| Hypertension | 1 | 0.7% |
| Age 75 years (2 points) | 2 | 1.9% |
| Age 6574 years | 3 | 4.7% |
| Diabetes | 4 | 2.3% |
| Stroke/TIA (2) | 5 | 3.9% |
| Vascular disease | 6 | 4.5% |
| Female gender | 7 | 10.1% |
| 89 | 20% | |
Additionally, the HAS‐BLED scoring system (which incorporates hypertension, abnormal renal/liver function, stroke, bleeding history, labile INR, and drugs/alcohol) provides a convenient method for estimating a patient's risk of major bleeding with therapeutic anticoagulation.[39]
Patients who are hospitalized with acute stroke and are found to have new onset AF require special consideration in regard to the timing of anticoagulation and rate‐control strategies. Although these patients are at risk for recurrent cardioembolism during their hospitalization, they are also at increased risk of hemorrhagic conversion of their cerebral infarct. Randomized studies comparing lowmolecular‐weight heparins versus antiplatelet agents for acute cardioembolic stroke indicate no net benefit of anticoagulation in thefirst 2 weeks after stroke.[40, 41] However, anticoagulation is probably safe within 14 days for patients with minor stroke because they are at less risk of hemorrhagic conversion.[27] Therefore, a reasonable approach is to start anticoagulation immediately after TIA, 5 to 7 days after a minor stroke, and 10 to 14 days after a major stroke. Furthermore, patients with acute ischemic stroke are particularly susceptible to infarct extension from even minor degrees of blood pressure reduction,[42] and therefore their AF must be managed with this hemodynamic consideration in mind.
SHORT‐TERM SEQUELAE
Increased hospital stay length, hospital cost, and morbidity have been well described to be increased in patients with postoperative AF following cardiac surgery[5] and noncardiac surgery.[43] In a recent study of patients with severe sepsis, those who developed new onset AF had a significantly increased risk of stroke and in‐hospital mortality.[7]
LONG‐TERM THERAPIES/MONITORING
Among patients with newly diagnosed AF during a hospitalization, those with multiple major risk factors for stroke (CHADS2 score >1 or CHA2DS2VASc score >2) should receive long‐term anticoagulation, unless monitoring is performed (Holter monitor, event monitor, implantable loop recorder) and shows an absence of AF. In patients with hypertension or coronary artery disease, prescription of a ‐blocker should be considered. Outpatient clinic follow‐up with a general cardiologist or electrophysiologist is important to help guide these decisions regarding rhythm monitoring, continuation of anticoagulation, and continuation of any antiarrhythmic drugs that were prescribed.
LONG‐TERM SEQUELAE
AF has recently been shown to have adverse long‐term consequences, even in a relatively healthy cohort of patients.[44] Postoperative AF has been associated with poor neurocognitive outcomes following CABG surgery.[45] Although data are lacking with regard to the prognostic significance of AF in the setting of hospitalization, it is reasonable to presume that it is a predictor for future episodes of AF. We know that 15% to 20% of all strokes occur in patients with AF,[2] and the group of patients with a new diagnosis of AF during hospital admission is almost certainly enriched for stroke risk. This underscores the importance of either starting long‐term anticoagulation upon discharge in patients at medium‐high risk of stroke, or ensuring timely communication of a new AF diagnosis to patients' outpatient physicians so that appropriate antithrombotic drugs can be started soon after discharge.
CONCLUSIONS
AF is a common problem among patients hospitalized for medical illness or in the postoperative state. Diagnosis of the dysrhythmia and identification of any reversible causes are the key first steps in management. Oftentimes, rate and rhythm control strategies are both reasonable courses of action, although it is important to include appropriate anticoagulation as part of both approaches. Cardiology consultation can be helpful in the decision‐making process.
In the vignette described at the beginning, we have a patient with a CHADS2 score of 2 (age, hypertension) and newly diagnosed paroxysmal AF during hospitalization. The dysrhythmia was likely triggered by his medical illness, but we have no way of knowing whether he has had asymptomatic paroxysms of AF in the past. Oral anticoagulation along with a ‐blocker should be prescribed at discharge. Clinic follow‐up with a cardiologist should be arranged prior to discharge, and consideration of withdrawing anticoagulation in the future should be guided by outpatient rhythm monitoring.
Disclosure
Nothing to report.
- , , , et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–2375.
- , , , , . Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–473.
- , , , et al. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–3010.
- , , , et al. Prophylactic Oral Amiodarone for the Prevention of Arrhythmias that Begin Early After Revascularization, Valve Replacement, or Repair: PAPABEAR: a randomized controlled trial. JAMA. 2005;294:3093–3100.
- , , , et al. Atrial fibrillation following coronary artery bypass graft surgery: predictors, outcomes, and resource utilization. MultiCenter Study of Perioperative Ischemia Research Group. JAMA. 1996;276:300–306.
- , , , , . Incidence, predictors, and outcomes associated with postoperative atrial fibrillation after major noncardiac surgery. Am Heart J. 2012;164: 918–924.
- , , , , . Incident stroke and mortality associated with new‐onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA. 2011;306:2248–2254.
- , , . Usefulness of cardiovascular investigations in stroke management: clinical relevance and economic implications. Stroke. 2007;38:1956–1958.
- , , , . Value of cardiac monitoring and echocardiography in TIA and stroke patients. Stroke. 1985;16:950–956.
- , , . Ambulatory electrocardiographic monitoring in patients with transient focal cerebral ischaemia. J Neurol Neurosurg Psychiatry. 1984;47:256–259.
- , , , et al. Continuous stroke unit electrocardiographic monitoring versus 24‐hour holter electrocardiography for detection of paroxysmal atrial fibrillation after stroke. Stroke. 2012;43:2689–2694.
- , , , , . Noninvasive cardiac monitoring for detecting paroxysmal atrial fibrillation or flutter after acute ischemic stroke: a systematic review. Stroke. 2007;38:2935–2940.
- , , , et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:651–745.
- , , , , , . Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184.
- , , . Role of inflammation in initiation and perpetuation of atrial fibrillation: a systematic review of the published data. J Am Coll Cardiol. 2007;50:2021–2028.
- , , , et al. Atrial fibrillation after coronary artery bypass grafting is associated with sympathetic activation. Ann Thorac Surg. 1995;60:1709–1715.
- , , , et al. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation. 2005;111:2881–2888.
- , , , et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–571.
- , , , . Mechanisms, prevention, and treatment of atrial fibrillation after cardiac surgery. J Am Coll Cardiol. 2008;51:793–801.
- , , , . Incidence of arrhythmias after thoracic surgery: thoracotomy versus video‐assisted thoracoscopy. J Cardiothorac Vasc Anesth. 1998;12:659–661.
- , , , et al. Delayed detection of atrial fibrillation after ischemic stroke. J Stroke Cerebrovasc Dis. 2009;18:453–457.
- , , , et al. How useful is thyroid function testing in patients with recent‐onset atrial fibrillation? The Canadian Registry of Atrial Fibrillation Investigators. Arch Intern Med. 1996;156:2221–2224.
- , , , , , . Natriuretic peptide levels in atrial fibrillation: a prospective hormonal and Doppler‐echocardiographic study. J Am Coll Cardiol. 2000;35:1256–1262.
- , , . Relationship between brain natriuretic peptide and recurrence of atrial fibrillation after successful electrical cardioversion: a meta‐analysis. J Int Med Res. 2011;39:1618–1624.
- , , , et al. Amiodarone prophylaxis for atrial fibrillation of high‐risk patients after coronary bypass grafting: a prospective, double‐blinded, placebo‐controlled, randomized study. Eur Heart J. 2006;27:1584–1591.
- , , , , . Statin use and postoperative atrial fibrillation after major noncardiac surgery. Heart Rhythm. 2012;9:163–169.
- , , , et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227–276.
- , , , . Cardioversion of nonrheumatic atrial fibrillation. Reduced thromboembolic complications with 4 weeks of precardioversion anticoagulation are related to atrial thrombus resolution. Circulation. 1995;92:160–163.
- , , , . Left atrial appendage thrombus is not uncommon in patients with acute atrial fibrillation and a recent embolic event: a transesophageal echocardiographic study. J Am Coll Cardiol. 1995;25:452–459.
- , , , et al. Impact of electrical cardioversion for atrial fibrillation on left atrial appendage function and spontaneous echo contrast: characterization by simultaneous transesophageal echocardiography. J Am Coll Cardiol. 1993;22:1359–1366.
- , , , . Pulsed Doppler evaluation of atrial mechanical function after electrical cardioversion of atrial fibrillation. J Am Coll Cardiol. 1989;13:617–623.
- , , , et al. Detection and characterization of intracardiac thrombi on MR imaging. AJR Am J Roentgenol. 2002;179:1539–1544.
- , , , , , . Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo‐controlled, crossover study in healthy subjects. Circulation. 2011;124:1573–1579.
- , , , et al. Conversion of atrial fibrillation to sinus rhythm and rate control by digoxin in comparison to placebo. Eur Heart J. 1997;18:643–648.
- , , , et al. The evidence regarding the drugs used for ventricular rate control. J Fam Pract. 2000;49:47–59.
- , , , , , . Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864–2870.
- Stroke prevention in atrial fibrillation study. Final results. Circulation. 1991;84:527–539.
- , , , , . Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor‐based approach: the Euro Heart Survey on atrial fibrillation. Chest. 2010;137:263–272.
- , , , , , . A novel user‐friendly score (HAS‐BLED) to assess 1‐year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093–1100.
- , , , . Low molecular‐weight heparin versus aspirin in patients with acute ischaemic stroke and atrial fibrillation: a double‐blind randomised study. HAEST Study Group. Heparin in Acute Embolic Stroke Trial. Lancet. 2000;355:1205–1210.
- The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19435 patients with acute ischaemic stroke. International Stroke Trial Collaborative Group. Lancet. 1997;349:1569–1581.
- , , , et al. The angiotensin‐receptor blocker candesartan for treatment of acute stroke (SCAST): a randomised, placebo‐controlled, double‐blind trial. Lancet. 2011;377:741–750.
- , , , , . Supraventricular arrhythmia in patients having noncardiac surgery: clinical correlates and effect on length of stay. Ann Intern Med. 1998;129:279–285.
- , , , et al. Risk of death and cardiovascular events in initially healthy women with new‐onset atrial fibrillation. JAMA. 2011;305:2080–2087.
- , , , et al. The impact of postoperative atrial fibrillation on neurocognitive outcome after coronary artery bypass graft surgery. Anesth Analg. 2002;94:290–295, table of contents.
A 78‐year‐old otherwise healthy man with longstanding hypertension is admitted to the hospital with pneumonia. During the second hospital day, he develops atrial fibrillation (AF) with rapid ventricular response, but his hemodynamics remain stable. He is given oral metoprolol for rate control. A transthoracic echocardiogram (TTE) shows mild concentric left ventricular hypertrophy, normal left ventricular size and function, and mild left atrial enlargement. The AF spontaneously terminates after 24 hours and does not recur during the hospitalization. What treatment and monitoring are recommended at the time of discharge for this patient's AF?
BACKGROUND
AF is a common dysrhythmia that clinicians often encounter while caring for hospitalized patients. Although many patients will have carried a diagnosis of AF prior to hospital admission, this review will pertain to patients for whom a first documented episode of AF occurs during hospitalization. These patients can be conceptually separated into 2 categories: those who have had undiagnosed AF for some time (and are now diagnosed due to continuous inpatient telemetry monitoring) and those whose AF is secondary to their acute medical illness. Although practically speaking, this distinction is not easy to make, markers of chronic AF may include left atrial enlargement and a clinical history of longstanding palpitations.
INCIDENCE
The prevalence of AF in the general population is estimated at 0.4% to 1.0%.[1, 2] Prevalence increases with advancing age. Compared to the general population, the population of hospitalized patients is inherently older and enriched for comorbidities that are known risk factors for the development of AF (such as congestive heart failure, hypertension, and obstructive sleep apnea) or are associated with paroxysmal AF (such as stroke or transient ischemic attack [TIA]). As a result, the prevalence of occult AF is necessarily higher in this population than that of a general cohort. The risk of developing AF is further increased in hospitalized patients simply by the acute illness (or postoperative state), whose associated catecholamine surge and systemic proinflammatory state are well‐known precipitants for AF.[3] AF is common after cardiac surgery (25%30%)[4, 5] and occurs in about 3% of patients undergoing major noncardiac surgery.[6]
In the setting of severe medical illness such as sepsis, the incidence of new onset AF has recently been estimated at around 6%.[7] Among patients hospitalized with stroke, 2% to 5% will have a new diagnosis of AF made by the admission electrocardiogram (ECG).[8, 9, 10] Subsequent cardiac monitoring with inpatient telemetry or Holter monitoring will detect previously undiagnosed AF in another 5% to 8% of patients admitted with stroke.[11, 12]
PATHOPHYSIOLOGY
AF is a supraventricular tachyarrhythmia characterized by uncoordinated atrial activation; this chaotic atrial activation translates into atrial mechanical dysfunction. [13] Patients who develop AF may have atrial substrate, such as patchy atrial myocyte fibrosis, that increases their propensity to develop atrial dysrhythmias.[14] Other factors contributing to the likelihood of developing AF are anisotropic conduction, atrial chamber dilation, systemic inflammation, hyperadrenergic state, and atrial ischemia.[3, 15, 16, 17] Atrial flutter, on the other hand, is an organized macro‐reentrant supraventricular arrhythmia that typically rotates around the tricuspid annulus.
RISK FACTORS
Risk factors for the development of AF are well defined. The risk factors in the chronic setting remain the same as those for the development of AF in the setting of medical illness or in the postoperative state: advancing age, male gender, prior diagnosis of AF, congestive heart failure, hypertension, and obstructive sleep apnea.[1, 5, 18] Surgical procedures, due to the sympathetic surge and proinflammatory state that occur in the postoperative period, increase the risk of AF. Cardiac and thoracic procedures, which involve direct manipulation of the heart and adjacent structures, proffer the highest risk of AF.[19, 20] Although not necessarily a risk factor for the development of AF, patients with recent ischemic stroke are at high risk of harboring AF; up to 7% of patients are diagnosed with AF in the 3 months following ischemic stroke.[21]
DIAGNOSIS
In the inpatient setting, the diagnosis of AF is typically made through telemetry monitoring, which reveals irregularly spaced QRS complexes and an absence of organized atrial activity (ie, no discernible P waves or flutter waves). For patients not on a continuous cardiac monitor, the diagnosis of AF is made by 12‐lead ECG, which is triggered by patient complaint (palpitations, lightheadedness, dyspnea, or chest pain), physical exam findings, or review of vital sign measurements (ie, sudden changes in heart rate). The dysrhythmia should sustain for at least 30 seconds for a diagnosis of AF to be made.
INITIAL WORKUP
When AF is suspected (or has been diagnosed by telemetry), a 12‐lead ECG should be immediately obtained (Table 1). This will help to confirm the diagnosis of AF (as distinct from atrial flutter) and begin the investigation for underlying causes (ie, analysis of ST‐segment shifts for evidence of myocardial ischemia or pericarditis). A focused history, physical exam, and review of vital signs can quickly determine if there are any urgent indications for cardioversion, such as the development of pulmonary edema, the presence of angina pectoris, or rhythm‐related hypotension. A TTE should be obtained to assess for structural heart disease (left atrial enlargement, valvular disease, cardiac tumor) that may serve as a substrate for AF. The echocardiogram will also provide an assessment of left ventricular function, which will inform the treating physician regarding the safety of using atrioventricular (AV) nodal blocking agents, such as ‐blockers and nondihydropyridine calcium channel blockers, which may also act as negative inotropes. Although occult hyperthyroidism is a rare cause of AF,[22] a serum thyroid‐stimulating hormone test should be obtained to rule out this reversible cause. Electrolytes should be monitored and serum potassium and magnesium levels should be maintained at >4.0 mmol/L and >2.0 mEq mmol/L, respectively. Measurement of serum B‐type natriuretic peptide can be helpful in determining prognosis and likelihood of left ventricular dysfunction in patients with AF.[23, 24]
|
| Confirmatory study |
| 12‐lead electrocardiogram |
| Assessment of clinical stability |
| History (chest pain, shortness of breath, syncope/presyncope) |
| Physical exam (blood pressure, heart rate, pulmonary rales, jugular venous distension) |
| Evaluation for structural heart disease |
| Physical exam (pathologic murmurs, third heart sound, abnormal PMI, friction rub) |
| Transthoracic echocardiogram |
| Metabolic triggers |
| Serum potassium and magnesium |
| Serum thyroid stimulating hormone |
| Prognostic indicators |
| Serum brain natriuretic peptide |
| Other investigations (as guided by clinical suspicion) |
| Chest CT angiogram |
| Serum troponin |
| Blood cultures |
Other investigations should be guided by the clinical suspicion for other secondary causes. Examples include assessment for infection in the postoperative patient, ruling out myocardial infarction in patients with chest pain and risk factors for coronary artery disease, evaluating for pericarditis following cardiac surgery, and having a high suspicion for pulmonary embolism in patients with prolonged immobilization, hypercoagulable state, or recent knee/hip replacement surgery.
STRATEGIES FOR PREVENTION/SCREENING
AF prevention and screening strategies are not practical for patients admitted for medical illnesses. When used for perioperative prophylaxis, however, amiodarone has been shown to clearly reduce postoperative AF (and shorten hospitalizations) after coronary artery bypass graft surgery.[4, 25] Statin use has been associated with a decrease in postoperative AF following major noncardiac surgery.[26] Patients hospitalized with acute ischemic stroke or TIA should undergo cardiac monitoring throughout their hospitalization if feasible, or for at least 24 hours.[27] Recent data indicate that either Holter monitoring or continuous cardiac telemetry are acceptable methods of screening stroke patients for underlying AF.[11]
THERAPIES
In all cases of AF, underlying causes of the dysrhythmia (such as heart failure, infection, electrolyte disturbances, and pain) should be sought and treated.
AF associated with unstable symptoms (heart failure, angina, hypotension) calls for urgent rhythm control. In this setting, cardioversion should be performed immediately; anticoagulation should be initiated concomitantly unless a contraindication to anticoagulation exists. Stable patients should be assessed for indications for elective cardioversion and acute anticoagulation. Generally speaking, it is desirable to perform transesophageal echocardiography (TEE) and cardioversion prior to discharge from the hospital in patients whose new‐onset AF has persisted, assuming that they are candidates for therapeutic anticoagulation. This is particularly true for patients who are at all symptomatic from their AF. Allowing patients to remain in AF for weeks to months will increase their risk of developing long‐standing persistent AF.
AF is a well‐recognized risk factor for the development of atrial thrombi and resultant thromboembolic events. Thrombus formation is thought to be a result of stasis of blood in the atria during AF as well as a localized hypercoagulable state in the left atrium in patients with AF.[28] Left atrial thrombus can develop in patients with AF of duration <3 days.[29] Echocardiographic evidence suggests that left atrial appendage function can be transiently depressed following cardioversion, which may help to explain the finding of increased risk of thromboembolism immediately after cardioversion.[30, 31] In fact, 98% of thromboembolic events after cardioversion occur within 10 days.[31] Studies using serial TEE show that atrial thrombi typically resolve after 3 to 4 weeks of anticoagulation.[28] These data are the basis for the recommendation that patients with AF that has lasted 48 hours or more should receive 4 weeks of therapeutic anticoagulation prior to cardioversion that is not TEE guided. Importantly, administration of antiarrhythmic agents, such as amiodarone, should be considered an attempt at rhythm control, and therefore anticoagulation should be used in the same way during antiarrhythmic drug initiation as with direct‐current cardioversion. Medications most commonly used to acutely terminate AF are ibutilide, propafenone, and flecainide.
In the inpatient setting, nonemergent cardioversion in patients who have had AF for more than 48 hours should be TEE guided, unless the onset of the arrhythmia was clearly documented and therapeutic anticoagulation was initiated within 48 hours of the onset. Patients should be receiving therapeutic anticoagulation at the time of the TEE. Contrast‐enhanced magnetic resonance imaging is a promising noninvasive option for assessing for intracardiac thrombus, but this modality has not yet been widely adopted as an acceptable alternative to TEE.[32]
Anticoagulation in the short term can be rapidly achieved using heparins (intravenous unfractionated heparin, subcutaneous enoxaparin) or the newer oral anticoagulants such as dabigatran (a thrombin inhibitor) or rivaroxaban and apixaban (factor Xa inhibitors). Importantly, should significant bleeding occur, options for reversal of these new oral anticoagulant agents are limited.[33] Vitamin K antagonists such as warfarin remain a viable option for long‐term anticoagulation, but usually require 4 to 5 days to reach peak effect; the goal international normalized ratio (INR) is 2.0 to 3.0. In patients with chronic kidney disease, the newer oral anticoagulants (dabigatran, rivaroxiban, and apixaban), as well as low molecular weight heparins, should be dose adjusted in patients with moderate renal dysfunction and avoided altogether in patients with severe renal dysfunction.
Ventricular response rate control, rather than rhythm control, is a reasonable initial strategy for patients who do not have significant symptoms from AF. Rate control can be achieved using traditional AV nodal blocking agents (‐blockers and nondihydropyridine calcium channel blockers). Initially, the use of intravenous (IV) agents is reasonable. IV metoprolol and IV diltiazem are useful because they both have a rapid onset of action, which allows for repeated bolus dosing at closely spaced intervals. Both IV agents have a 2‐ to 4‐hour half‐life. Once rate control has been achieved, the amount of IV drug required to achieve heart rate control can be tallied and converted into oral dosing. Cardiac glycosides can also be used to rate‐control AF; digitalis works by exerting a vagotonic effect via alterations in calcium handling in the AV node. Digoxin is most effective in the rate control of patients with persistent AF rather than those with recent onset AF.[34] Even in patients with persistent AF, digoxin only lowers average heart rate during rest and not during exertion/stress.[35] In patients with marginal blood pressure, digoxin can be safely used because it does not have any negative inotropic effects. In patients receiving a rate control strategy, the decision of whether to anticoagulate should be based on the risk of thromboembolic stroke as determined by clinical risk factors. In general, patients with a CHADS2 score[36] of 0 can be treated with aspirin (325 mg daily)[37] for thromboembolism prevention, and those with a score of 2 or more should receive therapeutic anticoagulation. Patients with a CHADS2 score of 1 can reasonably be treated with either regimen, and a more nuanced assessment of bleeding and stroke risk is required. The more recently described CHA2DS2‐VASc score allows for better stroke risk discrimination among patients with low CHADS2 scores (Table 2).[38]
| CHADS2 Elements | CHADS2 Score | Annual Stroke Risk |
|---|---|---|
| ||
| CHF | 0 | 1.2% |
| Hypertension | 1 | 2.8% |
| Age 75 years | 2 | 3.6% |
| Diabetes | 3 | 6.4% |
| Stroke/TIA (2 points) | 4 | 8.0% |
| 56 | 11.4% | |
| CHA2DS2‐VASc Elements | CHA2DS2‐VASc Score | Annual Stroke Risk a |
| CHF | 0 | 0.0% |
| Hypertension | 1 | 0.7% |
| Age 75 years (2 points) | 2 | 1.9% |
| Age 6574 years | 3 | 4.7% |
| Diabetes | 4 | 2.3% |
| Stroke/TIA (2) | 5 | 3.9% |
| Vascular disease | 6 | 4.5% |
| Female gender | 7 | 10.1% |
| 89 | 20% | |
Additionally, the HAS‐BLED scoring system (which incorporates hypertension, abnormal renal/liver function, stroke, bleeding history, labile INR, and drugs/alcohol) provides a convenient method for estimating a patient's risk of major bleeding with therapeutic anticoagulation.[39]
Patients who are hospitalized with acute stroke and are found to have new onset AF require special consideration in regard to the timing of anticoagulation and rate‐control strategies. Although these patients are at risk for recurrent cardioembolism during their hospitalization, they are also at increased risk of hemorrhagic conversion of their cerebral infarct. Randomized studies comparing lowmolecular‐weight heparins versus antiplatelet agents for acute cardioembolic stroke indicate no net benefit of anticoagulation in thefirst 2 weeks after stroke.[40, 41] However, anticoagulation is probably safe within 14 days for patients with minor stroke because they are at less risk of hemorrhagic conversion.[27] Therefore, a reasonable approach is to start anticoagulation immediately after TIA, 5 to 7 days after a minor stroke, and 10 to 14 days after a major stroke. Furthermore, patients with acute ischemic stroke are particularly susceptible to infarct extension from even minor degrees of blood pressure reduction,[42] and therefore their AF must be managed with this hemodynamic consideration in mind.
SHORT‐TERM SEQUELAE
Increased hospital stay length, hospital cost, and morbidity have been well described to be increased in patients with postoperative AF following cardiac surgery[5] and noncardiac surgery.[43] In a recent study of patients with severe sepsis, those who developed new onset AF had a significantly increased risk of stroke and in‐hospital mortality.[7]
LONG‐TERM THERAPIES/MONITORING
Among patients with newly diagnosed AF during a hospitalization, those with multiple major risk factors for stroke (CHADS2 score >1 or CHA2DS2VASc score >2) should receive long‐term anticoagulation, unless monitoring is performed (Holter monitor, event monitor, implantable loop recorder) and shows an absence of AF. In patients with hypertension or coronary artery disease, prescription of a ‐blocker should be considered. Outpatient clinic follow‐up with a general cardiologist or electrophysiologist is important to help guide these decisions regarding rhythm monitoring, continuation of anticoagulation, and continuation of any antiarrhythmic drugs that were prescribed.
LONG‐TERM SEQUELAE
AF has recently been shown to have adverse long‐term consequences, even in a relatively healthy cohort of patients.[44] Postoperative AF has been associated with poor neurocognitive outcomes following CABG surgery.[45] Although data are lacking with regard to the prognostic significance of AF in the setting of hospitalization, it is reasonable to presume that it is a predictor for future episodes of AF. We know that 15% to 20% of all strokes occur in patients with AF,[2] and the group of patients with a new diagnosis of AF during hospital admission is almost certainly enriched for stroke risk. This underscores the importance of either starting long‐term anticoagulation upon discharge in patients at medium‐high risk of stroke, or ensuring timely communication of a new AF diagnosis to patients' outpatient physicians so that appropriate antithrombotic drugs can be started soon after discharge.
CONCLUSIONS
AF is a common problem among patients hospitalized for medical illness or in the postoperative state. Diagnosis of the dysrhythmia and identification of any reversible causes are the key first steps in management. Oftentimes, rate and rhythm control strategies are both reasonable courses of action, although it is important to include appropriate anticoagulation as part of both approaches. Cardiology consultation can be helpful in the decision‐making process.
In the vignette described at the beginning, we have a patient with a CHADS2 score of 2 (age, hypertension) and newly diagnosed paroxysmal AF during hospitalization. The dysrhythmia was likely triggered by his medical illness, but we have no way of knowing whether he has had asymptomatic paroxysms of AF in the past. Oral anticoagulation along with a ‐blocker should be prescribed at discharge. Clinic follow‐up with a cardiologist should be arranged prior to discharge, and consideration of withdrawing anticoagulation in the future should be guided by outpatient rhythm monitoring.
Disclosure
Nothing to report.
A 78‐year‐old otherwise healthy man with longstanding hypertension is admitted to the hospital with pneumonia. During the second hospital day, he develops atrial fibrillation (AF) with rapid ventricular response, but his hemodynamics remain stable. He is given oral metoprolol for rate control. A transthoracic echocardiogram (TTE) shows mild concentric left ventricular hypertrophy, normal left ventricular size and function, and mild left atrial enlargement. The AF spontaneously terminates after 24 hours and does not recur during the hospitalization. What treatment and monitoring are recommended at the time of discharge for this patient's AF?
BACKGROUND
AF is a common dysrhythmia that clinicians often encounter while caring for hospitalized patients. Although many patients will have carried a diagnosis of AF prior to hospital admission, this review will pertain to patients for whom a first documented episode of AF occurs during hospitalization. These patients can be conceptually separated into 2 categories: those who have had undiagnosed AF for some time (and are now diagnosed due to continuous inpatient telemetry monitoring) and those whose AF is secondary to their acute medical illness. Although practically speaking, this distinction is not easy to make, markers of chronic AF may include left atrial enlargement and a clinical history of longstanding palpitations.
INCIDENCE
The prevalence of AF in the general population is estimated at 0.4% to 1.0%.[1, 2] Prevalence increases with advancing age. Compared to the general population, the population of hospitalized patients is inherently older and enriched for comorbidities that are known risk factors for the development of AF (such as congestive heart failure, hypertension, and obstructive sleep apnea) or are associated with paroxysmal AF (such as stroke or transient ischemic attack [TIA]). As a result, the prevalence of occult AF is necessarily higher in this population than that of a general cohort. The risk of developing AF is further increased in hospitalized patients simply by the acute illness (or postoperative state), whose associated catecholamine surge and systemic proinflammatory state are well‐known precipitants for AF.[3] AF is common after cardiac surgery (25%30%)[4, 5] and occurs in about 3% of patients undergoing major noncardiac surgery.[6]
In the setting of severe medical illness such as sepsis, the incidence of new onset AF has recently been estimated at around 6%.[7] Among patients hospitalized with stroke, 2% to 5% will have a new diagnosis of AF made by the admission electrocardiogram (ECG).[8, 9, 10] Subsequent cardiac monitoring with inpatient telemetry or Holter monitoring will detect previously undiagnosed AF in another 5% to 8% of patients admitted with stroke.[11, 12]
PATHOPHYSIOLOGY
AF is a supraventricular tachyarrhythmia characterized by uncoordinated atrial activation; this chaotic atrial activation translates into atrial mechanical dysfunction. [13] Patients who develop AF may have atrial substrate, such as patchy atrial myocyte fibrosis, that increases their propensity to develop atrial dysrhythmias.[14] Other factors contributing to the likelihood of developing AF are anisotropic conduction, atrial chamber dilation, systemic inflammation, hyperadrenergic state, and atrial ischemia.[3, 15, 16, 17] Atrial flutter, on the other hand, is an organized macro‐reentrant supraventricular arrhythmia that typically rotates around the tricuspid annulus.
RISK FACTORS
Risk factors for the development of AF are well defined. The risk factors in the chronic setting remain the same as those for the development of AF in the setting of medical illness or in the postoperative state: advancing age, male gender, prior diagnosis of AF, congestive heart failure, hypertension, and obstructive sleep apnea.[1, 5, 18] Surgical procedures, due to the sympathetic surge and proinflammatory state that occur in the postoperative period, increase the risk of AF. Cardiac and thoracic procedures, which involve direct manipulation of the heart and adjacent structures, proffer the highest risk of AF.[19, 20] Although not necessarily a risk factor for the development of AF, patients with recent ischemic stroke are at high risk of harboring AF; up to 7% of patients are diagnosed with AF in the 3 months following ischemic stroke.[21]
DIAGNOSIS
In the inpatient setting, the diagnosis of AF is typically made through telemetry monitoring, which reveals irregularly spaced QRS complexes and an absence of organized atrial activity (ie, no discernible P waves or flutter waves). For patients not on a continuous cardiac monitor, the diagnosis of AF is made by 12‐lead ECG, which is triggered by patient complaint (palpitations, lightheadedness, dyspnea, or chest pain), physical exam findings, or review of vital sign measurements (ie, sudden changes in heart rate). The dysrhythmia should sustain for at least 30 seconds for a diagnosis of AF to be made.
INITIAL WORKUP
When AF is suspected (or has been diagnosed by telemetry), a 12‐lead ECG should be immediately obtained (Table 1). This will help to confirm the diagnosis of AF (as distinct from atrial flutter) and begin the investigation for underlying causes (ie, analysis of ST‐segment shifts for evidence of myocardial ischemia or pericarditis). A focused history, physical exam, and review of vital signs can quickly determine if there are any urgent indications for cardioversion, such as the development of pulmonary edema, the presence of angina pectoris, or rhythm‐related hypotension. A TTE should be obtained to assess for structural heart disease (left atrial enlargement, valvular disease, cardiac tumor) that may serve as a substrate for AF. The echocardiogram will also provide an assessment of left ventricular function, which will inform the treating physician regarding the safety of using atrioventricular (AV) nodal blocking agents, such as ‐blockers and nondihydropyridine calcium channel blockers, which may also act as negative inotropes. Although occult hyperthyroidism is a rare cause of AF,[22] a serum thyroid‐stimulating hormone test should be obtained to rule out this reversible cause. Electrolytes should be monitored and serum potassium and magnesium levels should be maintained at >4.0 mmol/L and >2.0 mEq mmol/L, respectively. Measurement of serum B‐type natriuretic peptide can be helpful in determining prognosis and likelihood of left ventricular dysfunction in patients with AF.[23, 24]
|
| Confirmatory study |
| 12‐lead electrocardiogram |
| Assessment of clinical stability |
| History (chest pain, shortness of breath, syncope/presyncope) |
| Physical exam (blood pressure, heart rate, pulmonary rales, jugular venous distension) |
| Evaluation for structural heart disease |
| Physical exam (pathologic murmurs, third heart sound, abnormal PMI, friction rub) |
| Transthoracic echocardiogram |
| Metabolic triggers |
| Serum potassium and magnesium |
| Serum thyroid stimulating hormone |
| Prognostic indicators |
| Serum brain natriuretic peptide |
| Other investigations (as guided by clinical suspicion) |
| Chest CT angiogram |
| Serum troponin |
| Blood cultures |
Other investigations should be guided by the clinical suspicion for other secondary causes. Examples include assessment for infection in the postoperative patient, ruling out myocardial infarction in patients with chest pain and risk factors for coronary artery disease, evaluating for pericarditis following cardiac surgery, and having a high suspicion for pulmonary embolism in patients with prolonged immobilization, hypercoagulable state, or recent knee/hip replacement surgery.
STRATEGIES FOR PREVENTION/SCREENING
AF prevention and screening strategies are not practical for patients admitted for medical illnesses. When used for perioperative prophylaxis, however, amiodarone has been shown to clearly reduce postoperative AF (and shorten hospitalizations) after coronary artery bypass graft surgery.[4, 25] Statin use has been associated with a decrease in postoperative AF following major noncardiac surgery.[26] Patients hospitalized with acute ischemic stroke or TIA should undergo cardiac monitoring throughout their hospitalization if feasible, or for at least 24 hours.[27] Recent data indicate that either Holter monitoring or continuous cardiac telemetry are acceptable methods of screening stroke patients for underlying AF.[11]
THERAPIES
In all cases of AF, underlying causes of the dysrhythmia (such as heart failure, infection, electrolyte disturbances, and pain) should be sought and treated.
AF associated with unstable symptoms (heart failure, angina, hypotension) calls for urgent rhythm control. In this setting, cardioversion should be performed immediately; anticoagulation should be initiated concomitantly unless a contraindication to anticoagulation exists. Stable patients should be assessed for indications for elective cardioversion and acute anticoagulation. Generally speaking, it is desirable to perform transesophageal echocardiography (TEE) and cardioversion prior to discharge from the hospital in patients whose new‐onset AF has persisted, assuming that they are candidates for therapeutic anticoagulation. This is particularly true for patients who are at all symptomatic from their AF. Allowing patients to remain in AF for weeks to months will increase their risk of developing long‐standing persistent AF.
AF is a well‐recognized risk factor for the development of atrial thrombi and resultant thromboembolic events. Thrombus formation is thought to be a result of stasis of blood in the atria during AF as well as a localized hypercoagulable state in the left atrium in patients with AF.[28] Left atrial thrombus can develop in patients with AF of duration <3 days.[29] Echocardiographic evidence suggests that left atrial appendage function can be transiently depressed following cardioversion, which may help to explain the finding of increased risk of thromboembolism immediately after cardioversion.[30, 31] In fact, 98% of thromboembolic events after cardioversion occur within 10 days.[31] Studies using serial TEE show that atrial thrombi typically resolve after 3 to 4 weeks of anticoagulation.[28] These data are the basis for the recommendation that patients with AF that has lasted 48 hours or more should receive 4 weeks of therapeutic anticoagulation prior to cardioversion that is not TEE guided. Importantly, administration of antiarrhythmic agents, such as amiodarone, should be considered an attempt at rhythm control, and therefore anticoagulation should be used in the same way during antiarrhythmic drug initiation as with direct‐current cardioversion. Medications most commonly used to acutely terminate AF are ibutilide, propafenone, and flecainide.
In the inpatient setting, nonemergent cardioversion in patients who have had AF for more than 48 hours should be TEE guided, unless the onset of the arrhythmia was clearly documented and therapeutic anticoagulation was initiated within 48 hours of the onset. Patients should be receiving therapeutic anticoagulation at the time of the TEE. Contrast‐enhanced magnetic resonance imaging is a promising noninvasive option for assessing for intracardiac thrombus, but this modality has not yet been widely adopted as an acceptable alternative to TEE.[32]
Anticoagulation in the short term can be rapidly achieved using heparins (intravenous unfractionated heparin, subcutaneous enoxaparin) or the newer oral anticoagulants such as dabigatran (a thrombin inhibitor) or rivaroxaban and apixaban (factor Xa inhibitors). Importantly, should significant bleeding occur, options for reversal of these new oral anticoagulant agents are limited.[33] Vitamin K antagonists such as warfarin remain a viable option for long‐term anticoagulation, but usually require 4 to 5 days to reach peak effect; the goal international normalized ratio (INR) is 2.0 to 3.0. In patients with chronic kidney disease, the newer oral anticoagulants (dabigatran, rivaroxiban, and apixaban), as well as low molecular weight heparins, should be dose adjusted in patients with moderate renal dysfunction and avoided altogether in patients with severe renal dysfunction.
Ventricular response rate control, rather than rhythm control, is a reasonable initial strategy for patients who do not have significant symptoms from AF. Rate control can be achieved using traditional AV nodal blocking agents (‐blockers and nondihydropyridine calcium channel blockers). Initially, the use of intravenous (IV) agents is reasonable. IV metoprolol and IV diltiazem are useful because they both have a rapid onset of action, which allows for repeated bolus dosing at closely spaced intervals. Both IV agents have a 2‐ to 4‐hour half‐life. Once rate control has been achieved, the amount of IV drug required to achieve heart rate control can be tallied and converted into oral dosing. Cardiac glycosides can also be used to rate‐control AF; digitalis works by exerting a vagotonic effect via alterations in calcium handling in the AV node. Digoxin is most effective in the rate control of patients with persistent AF rather than those with recent onset AF.[34] Even in patients with persistent AF, digoxin only lowers average heart rate during rest and not during exertion/stress.[35] In patients with marginal blood pressure, digoxin can be safely used because it does not have any negative inotropic effects. In patients receiving a rate control strategy, the decision of whether to anticoagulate should be based on the risk of thromboembolic stroke as determined by clinical risk factors. In general, patients with a CHADS2 score[36] of 0 can be treated with aspirin (325 mg daily)[37] for thromboembolism prevention, and those with a score of 2 or more should receive therapeutic anticoagulation. Patients with a CHADS2 score of 1 can reasonably be treated with either regimen, and a more nuanced assessment of bleeding and stroke risk is required. The more recently described CHA2DS2‐VASc score allows for better stroke risk discrimination among patients with low CHADS2 scores (Table 2).[38]
| CHADS2 Elements | CHADS2 Score | Annual Stroke Risk |
|---|---|---|
| ||
| CHF | 0 | 1.2% |
| Hypertension | 1 | 2.8% |
| Age 75 years | 2 | 3.6% |
| Diabetes | 3 | 6.4% |
| Stroke/TIA (2 points) | 4 | 8.0% |
| 56 | 11.4% | |
| CHA2DS2‐VASc Elements | CHA2DS2‐VASc Score | Annual Stroke Risk a |
| CHF | 0 | 0.0% |
| Hypertension | 1 | 0.7% |
| Age 75 years (2 points) | 2 | 1.9% |
| Age 6574 years | 3 | 4.7% |
| Diabetes | 4 | 2.3% |
| Stroke/TIA (2) | 5 | 3.9% |
| Vascular disease | 6 | 4.5% |
| Female gender | 7 | 10.1% |
| 89 | 20% | |
Additionally, the HAS‐BLED scoring system (which incorporates hypertension, abnormal renal/liver function, stroke, bleeding history, labile INR, and drugs/alcohol) provides a convenient method for estimating a patient's risk of major bleeding with therapeutic anticoagulation.[39]
Patients who are hospitalized with acute stroke and are found to have new onset AF require special consideration in regard to the timing of anticoagulation and rate‐control strategies. Although these patients are at risk for recurrent cardioembolism during their hospitalization, they are also at increased risk of hemorrhagic conversion of their cerebral infarct. Randomized studies comparing lowmolecular‐weight heparins versus antiplatelet agents for acute cardioembolic stroke indicate no net benefit of anticoagulation in thefirst 2 weeks after stroke.[40, 41] However, anticoagulation is probably safe within 14 days for patients with minor stroke because they are at less risk of hemorrhagic conversion.[27] Therefore, a reasonable approach is to start anticoagulation immediately after TIA, 5 to 7 days after a minor stroke, and 10 to 14 days after a major stroke. Furthermore, patients with acute ischemic stroke are particularly susceptible to infarct extension from even minor degrees of blood pressure reduction,[42] and therefore their AF must be managed with this hemodynamic consideration in mind.
SHORT‐TERM SEQUELAE
Increased hospital stay length, hospital cost, and morbidity have been well described to be increased in patients with postoperative AF following cardiac surgery[5] and noncardiac surgery.[43] In a recent study of patients with severe sepsis, those who developed new onset AF had a significantly increased risk of stroke and in‐hospital mortality.[7]
LONG‐TERM THERAPIES/MONITORING
Among patients with newly diagnosed AF during a hospitalization, those with multiple major risk factors for stroke (CHADS2 score >1 or CHA2DS2VASc score >2) should receive long‐term anticoagulation, unless monitoring is performed (Holter monitor, event monitor, implantable loop recorder) and shows an absence of AF. In patients with hypertension or coronary artery disease, prescription of a ‐blocker should be considered. Outpatient clinic follow‐up with a general cardiologist or electrophysiologist is important to help guide these decisions regarding rhythm monitoring, continuation of anticoagulation, and continuation of any antiarrhythmic drugs that were prescribed.
LONG‐TERM SEQUELAE
AF has recently been shown to have adverse long‐term consequences, even in a relatively healthy cohort of patients.[44] Postoperative AF has been associated with poor neurocognitive outcomes following CABG surgery.[45] Although data are lacking with regard to the prognostic significance of AF in the setting of hospitalization, it is reasonable to presume that it is a predictor for future episodes of AF. We know that 15% to 20% of all strokes occur in patients with AF,[2] and the group of patients with a new diagnosis of AF during hospital admission is almost certainly enriched for stroke risk. This underscores the importance of either starting long‐term anticoagulation upon discharge in patients at medium‐high risk of stroke, or ensuring timely communication of a new AF diagnosis to patients' outpatient physicians so that appropriate antithrombotic drugs can be started soon after discharge.
CONCLUSIONS
AF is a common problem among patients hospitalized for medical illness or in the postoperative state. Diagnosis of the dysrhythmia and identification of any reversible causes are the key first steps in management. Oftentimes, rate and rhythm control strategies are both reasonable courses of action, although it is important to include appropriate anticoagulation as part of both approaches. Cardiology consultation can be helpful in the decision‐making process.
In the vignette described at the beginning, we have a patient with a CHADS2 score of 2 (age, hypertension) and newly diagnosed paroxysmal AF during hospitalization. The dysrhythmia was likely triggered by his medical illness, but we have no way of knowing whether he has had asymptomatic paroxysms of AF in the past. Oral anticoagulation along with a ‐blocker should be prescribed at discharge. Clinic follow‐up with a cardiologist should be arranged prior to discharge, and consideration of withdrawing anticoagulation in the future should be guided by outpatient rhythm monitoring.
Disclosure
Nothing to report.
- , , , et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–2375.
- , , , , . Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–473.
- , , , et al. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–3010.
- , , , et al. Prophylactic Oral Amiodarone for the Prevention of Arrhythmias that Begin Early After Revascularization, Valve Replacement, or Repair: PAPABEAR: a randomized controlled trial. JAMA. 2005;294:3093–3100.
- , , , et al. Atrial fibrillation following coronary artery bypass graft surgery: predictors, outcomes, and resource utilization. MultiCenter Study of Perioperative Ischemia Research Group. JAMA. 1996;276:300–306.
- , , , , . Incidence, predictors, and outcomes associated with postoperative atrial fibrillation after major noncardiac surgery. Am Heart J. 2012;164: 918–924.
- , , , , . Incident stroke and mortality associated with new‐onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA. 2011;306:2248–2254.
- , , . Usefulness of cardiovascular investigations in stroke management: clinical relevance and economic implications. Stroke. 2007;38:1956–1958.
- , , , . Value of cardiac monitoring and echocardiography in TIA and stroke patients. Stroke. 1985;16:950–956.
- , , . Ambulatory electrocardiographic monitoring in patients with transient focal cerebral ischaemia. J Neurol Neurosurg Psychiatry. 1984;47:256–259.
- , , , et al. Continuous stroke unit electrocardiographic monitoring versus 24‐hour holter electrocardiography for detection of paroxysmal atrial fibrillation after stroke. Stroke. 2012;43:2689–2694.
- , , , , . Noninvasive cardiac monitoring for detecting paroxysmal atrial fibrillation or flutter after acute ischemic stroke: a systematic review. Stroke. 2007;38:2935–2940.
- , , , et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:651–745.
- , , , , , . Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184.
- , , . Role of inflammation in initiation and perpetuation of atrial fibrillation: a systematic review of the published data. J Am Coll Cardiol. 2007;50:2021–2028.
- , , , et al. Atrial fibrillation after coronary artery bypass grafting is associated with sympathetic activation. Ann Thorac Surg. 1995;60:1709–1715.
- , , , et al. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation. 2005;111:2881–2888.
- , , , et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–571.
- , , , . Mechanisms, prevention, and treatment of atrial fibrillation after cardiac surgery. J Am Coll Cardiol. 2008;51:793–801.
- , , , . Incidence of arrhythmias after thoracic surgery: thoracotomy versus video‐assisted thoracoscopy. J Cardiothorac Vasc Anesth. 1998;12:659–661.
- , , , et al. Delayed detection of atrial fibrillation after ischemic stroke. J Stroke Cerebrovasc Dis. 2009;18:453–457.
- , , , et al. How useful is thyroid function testing in patients with recent‐onset atrial fibrillation? The Canadian Registry of Atrial Fibrillation Investigators. Arch Intern Med. 1996;156:2221–2224.
- , , , , , . Natriuretic peptide levels in atrial fibrillation: a prospective hormonal and Doppler‐echocardiographic study. J Am Coll Cardiol. 2000;35:1256–1262.
- , , . Relationship between brain natriuretic peptide and recurrence of atrial fibrillation after successful electrical cardioversion: a meta‐analysis. J Int Med Res. 2011;39:1618–1624.
- , , , et al. Amiodarone prophylaxis for atrial fibrillation of high‐risk patients after coronary bypass grafting: a prospective, double‐blinded, placebo‐controlled, randomized study. Eur Heart J. 2006;27:1584–1591.
- , , , , . Statin use and postoperative atrial fibrillation after major noncardiac surgery. Heart Rhythm. 2012;9:163–169.
- , , , et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227–276.
- , , , . Cardioversion of nonrheumatic atrial fibrillation. Reduced thromboembolic complications with 4 weeks of precardioversion anticoagulation are related to atrial thrombus resolution. Circulation. 1995;92:160–163.
- , , , . Left atrial appendage thrombus is not uncommon in patients with acute atrial fibrillation and a recent embolic event: a transesophageal echocardiographic study. J Am Coll Cardiol. 1995;25:452–459.
- , , , et al. Impact of electrical cardioversion for atrial fibrillation on left atrial appendage function and spontaneous echo contrast: characterization by simultaneous transesophageal echocardiography. J Am Coll Cardiol. 1993;22:1359–1366.
- , , , . Pulsed Doppler evaluation of atrial mechanical function after electrical cardioversion of atrial fibrillation. J Am Coll Cardiol. 1989;13:617–623.
- , , , et al. Detection and characterization of intracardiac thrombi on MR imaging. AJR Am J Roentgenol. 2002;179:1539–1544.
- , , , , , . Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo‐controlled, crossover study in healthy subjects. Circulation. 2011;124:1573–1579.
- , , , et al. Conversion of atrial fibrillation to sinus rhythm and rate control by digoxin in comparison to placebo. Eur Heart J. 1997;18:643–648.
- , , , et al. The evidence regarding the drugs used for ventricular rate control. J Fam Pract. 2000;49:47–59.
- , , , , , . Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864–2870.
- Stroke prevention in atrial fibrillation study. Final results. Circulation. 1991;84:527–539.
- , , , , . Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor‐based approach: the Euro Heart Survey on atrial fibrillation. Chest. 2010;137:263–272.
- , , , , , . A novel user‐friendly score (HAS‐BLED) to assess 1‐year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093–1100.
- , , , . Low molecular‐weight heparin versus aspirin in patients with acute ischaemic stroke and atrial fibrillation: a double‐blind randomised study. HAEST Study Group. Heparin in Acute Embolic Stroke Trial. Lancet. 2000;355:1205–1210.
- The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19435 patients with acute ischaemic stroke. International Stroke Trial Collaborative Group. Lancet. 1997;349:1569–1581.
- , , , et al. The angiotensin‐receptor blocker candesartan for treatment of acute stroke (SCAST): a randomised, placebo‐controlled, double‐blind trial. Lancet. 2011;377:741–750.
- , , , , . Supraventricular arrhythmia in patients having noncardiac surgery: clinical correlates and effect on length of stay. Ann Intern Med. 1998;129:279–285.
- , , , et al. Risk of death and cardiovascular events in initially healthy women with new‐onset atrial fibrillation. JAMA. 2011;305:2080–2087.
- , , , et al. The impact of postoperative atrial fibrillation on neurocognitive outcome after coronary artery bypass graft surgery. Anesth Analg. 2002;94:290–295, table of contents.
- , , , et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–2375.
- , , , , . Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–473.
- , , , et al. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–3010.
- , , , et al. Prophylactic Oral Amiodarone for the Prevention of Arrhythmias that Begin Early After Revascularization, Valve Replacement, or Repair: PAPABEAR: a randomized controlled trial. JAMA. 2005;294:3093–3100.
- , , , et al. Atrial fibrillation following coronary artery bypass graft surgery: predictors, outcomes, and resource utilization. MultiCenter Study of Perioperative Ischemia Research Group. JAMA. 1996;276:300–306.
- , , , , . Incidence, predictors, and outcomes associated with postoperative atrial fibrillation after major noncardiac surgery. Am Heart J. 2012;164: 918–924.
- , , , , . Incident stroke and mortality associated with new‐onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA. 2011;306:2248–2254.
- , , . Usefulness of cardiovascular investigations in stroke management: clinical relevance and economic implications. Stroke. 2007;38:1956–1958.
- , , , . Value of cardiac monitoring and echocardiography in TIA and stroke patients. Stroke. 1985;16:950–956.
- , , . Ambulatory electrocardiographic monitoring in patients with transient focal cerebral ischaemia. J Neurol Neurosurg Psychiatry. 1984;47:256–259.
- , , , et al. Continuous stroke unit electrocardiographic monitoring versus 24‐hour holter electrocardiography for detection of paroxysmal atrial fibrillation after stroke. Stroke. 2012;43:2689–2694.
- , , , , . Noninvasive cardiac monitoring for detecting paroxysmal atrial fibrillation or flutter after acute ischemic stroke: a systematic review. Stroke. 2007;38:2935–2940.
- , , , et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:651–745.
- , , , , , . Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184.
- , , . Role of inflammation in initiation and perpetuation of atrial fibrillation: a systematic review of the published data. J Am Coll Cardiol. 2007;50:2021–2028.
- , , , et al. Atrial fibrillation after coronary artery bypass grafting is associated with sympathetic activation. Ann Thorac Surg. 1995;60:1709–1715.
- , , , et al. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation. 2005;111:2881–2888.
- , , , et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–571.
- , , , . Mechanisms, prevention, and treatment of atrial fibrillation after cardiac surgery. J Am Coll Cardiol. 2008;51:793–801.
- , , , . Incidence of arrhythmias after thoracic surgery: thoracotomy versus video‐assisted thoracoscopy. J Cardiothorac Vasc Anesth. 1998;12:659–661.
- , , , et al. Delayed detection of atrial fibrillation after ischemic stroke. J Stroke Cerebrovasc Dis. 2009;18:453–457.
- , , , et al. How useful is thyroid function testing in patients with recent‐onset atrial fibrillation? The Canadian Registry of Atrial Fibrillation Investigators. Arch Intern Med. 1996;156:2221–2224.
- , , , , , . Natriuretic peptide levels in atrial fibrillation: a prospective hormonal and Doppler‐echocardiographic study. J Am Coll Cardiol. 2000;35:1256–1262.
- , , . Relationship between brain natriuretic peptide and recurrence of atrial fibrillation after successful electrical cardioversion: a meta‐analysis. J Int Med Res. 2011;39:1618–1624.
- , , , et al. Amiodarone prophylaxis for atrial fibrillation of high‐risk patients after coronary bypass grafting: a prospective, double‐blinded, placebo‐controlled, randomized study. Eur Heart J. 2006;27:1584–1591.
- , , , , . Statin use and postoperative atrial fibrillation after major noncardiac surgery. Heart Rhythm. 2012;9:163–169.
- , , , et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227–276.
- , , , . Cardioversion of nonrheumatic atrial fibrillation. Reduced thromboembolic complications with 4 weeks of precardioversion anticoagulation are related to atrial thrombus resolution. Circulation. 1995;92:160–163.
- , , , . Left atrial appendage thrombus is not uncommon in patients with acute atrial fibrillation and a recent embolic event: a transesophageal echocardiographic study. J Am Coll Cardiol. 1995;25:452–459.
- , , , et al. Impact of electrical cardioversion for atrial fibrillation on left atrial appendage function and spontaneous echo contrast: characterization by simultaneous transesophageal echocardiography. J Am Coll Cardiol. 1993;22:1359–1366.
- , , , . Pulsed Doppler evaluation of atrial mechanical function after electrical cardioversion of atrial fibrillation. J Am Coll Cardiol. 1989;13:617–623.
- , , , et al. Detection and characterization of intracardiac thrombi on MR imaging. AJR Am J Roentgenol. 2002;179:1539–1544.
- , , , , , . Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo‐controlled, crossover study in healthy subjects. Circulation. 2011;124:1573–1579.
- , , , et al. Conversion of atrial fibrillation to sinus rhythm and rate control by digoxin in comparison to placebo. Eur Heart J. 1997;18:643–648.
- , , , et al. The evidence regarding the drugs used for ventricular rate control. J Fam Pract. 2000;49:47–59.
- , , , , , . Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864–2870.
- Stroke prevention in atrial fibrillation study. Final results. Circulation. 1991;84:527–539.
- , , , , . Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor‐based approach: the Euro Heart Survey on atrial fibrillation. Chest. 2010;137:263–272.
- , , , , , . A novel user‐friendly score (HAS‐BLED) to assess 1‐year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093–1100.
- , , , . Low molecular‐weight heparin versus aspirin in patients with acute ischaemic stroke and atrial fibrillation: a double‐blind randomised study. HAEST Study Group. Heparin in Acute Embolic Stroke Trial. Lancet. 2000;355:1205–1210.
- The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19435 patients with acute ischaemic stroke. International Stroke Trial Collaborative Group. Lancet. 1997;349:1569–1581.
- , , , et al. The angiotensin‐receptor blocker candesartan for treatment of acute stroke (SCAST): a randomised, placebo‐controlled, double‐blind trial. Lancet. 2011;377:741–750.
- , , , , . Supraventricular arrhythmia in patients having noncardiac surgery: clinical correlates and effect on length of stay. Ann Intern Med. 1998;129:279–285.
- , , , et al. Risk of death and cardiovascular events in initially healthy women with new‐onset atrial fibrillation. JAMA. 2011;305:2080–2087.
- , , , et al. The impact of postoperative atrial fibrillation on neurocognitive outcome after coronary artery bypass graft surgery. Anesth Analg. 2002;94:290–295, table of contents.
Human papillomavirus vaccine: Safe, effective, underused
The vaccines against human papillomavirus (HPV) are the only ones designed to prevent cancer caused by a virus1,2—surely a good goal. But because HPV is sexually transmitted, HPV vaccination has met with public controversy.3 To counter the objections and better protect their patients’ health, primary care providers and other clinicians need a clear understanding of the benefits and the low risk of HPV vaccination—and the reasons so many people object to it.3
In this article, we will review:
- The impact of HPV-related diseases
- The basic biologic features of HPV vaccines
- The host immune response to natural HPV infection vs the response to HPV vaccines
- The clinical efficacy and safety of HPV vaccines
- The latest guidelines for HPV vaccination
- The challenges to vaccination implementation
- Frequently asked practical questions about HPV vaccination.
HPV-RELATED DISEASES: FROM BOTHERSOME TO DEADLY
Clinical sequelae of HPV infection include genital warts; cancers of the cervix, vulva, vagina, anus, penis, and oropharynx; and recurrent respiratory papillomatosis.4–6
Genital warts
HPV types 6 and 11 are responsible for more than 90% of the 1 million new cases of genital warts diagnosed annually in the United States.7–10
Bothersome and embarrassing, HPV-related genital warts can cause itching, burning, erythema, and pain, as well as epithelial erosions, ulcerations, depigmentation, and urethral and vaginal bleeding and discharge.11,12 Although they are benign in the oncologic sense, they can cause a good deal of emotional and financial stress. Patients may feel anxiety, embarrassment,13 and vulnerability. Adolescents and adults who have or have had genital warts need to inform their current and future partners or else risk infecting them—and facing the consequences.
Direct health care costs of genital warts in the United States have been estimated to be at least $200 million per year.14
Cervical cancer
Cervical cancer cannot develop unless the cervical epithelium is infected with one of the oncogenic HPV types. Indeed, oncogenic HPV is present in as many as 99.8% of cervical cancer specimens.15 HPV 16 and 18 are the most oncogenic HPV genotypes and account for 75% of all cases of cervical cancer. Ten other HPV genotypes account for the remaining 25%.16
In 2012, there were an estimated 12,170 new cases of invasive cervical cancer in the United States and 4,220 related deaths.17 The cost associated with cervical cancer screening, managing abnormal findings, and treating invasive cervical cancer in the United States is estimated to be $3.3 billion per year.18
Although the incidence and the mortality rates of cervical cancer have decreased more than 50% in the United States over the past 3 decades thanks to screening,19 cervical cancer remains the second leading cause of death from cancer in women worldwide. Each year, an estimated 500,000 women contract the disease and 240,000 die of it.20
Anal cancer
A recent study indicated that oncogenic HPV can also cause anal cancer, and the proportion of such cancers associated with HPV 16 or HPV 18 infection is as high as or higher than for cervical cancers, and estimated at 80%.21
The incidence of anal cancer is increasing by approximately 2% per year in both men and women in the general population,22 and rates are even higher in men who have sex with men and people infected with the human immunodeficiency virus.23
Hu and Goldie24 estimated that the lifetime costs of caring for all the people in the United States who in just 1 year (2003) acquired anal cancer attributable to HPV would total $92 million.
Oropharyngeal cancer
HPV types 16, 18, 31, 33, and 35 also cause oropharyngeal cancer. HPV 16 accounts for more than 90% of cases of HPV-related oropharyngeal cancer.25
Chaturvedi et al6 tested tissue samples from three national cancer registries and found that the number of oropharyngeal cancers that were HPV-positive increased from 16.3% in 1984–1989 to 71.7% in 2000–2004, while the number of HPV-negative oropharyngeal cancers fell by 50%, paralleling the drop in cigarette smoking in the United States.
Hu and Goldie24 estimated that the total lifetime cost for all new HPV-related oropharyngeal cancers that arose in 2003 would come to $38.1 million.24
Vulvar and vaginal cancers
HPV 16 and 18 are also responsible for approximately 50% of vulvar cancers and 50% to 75% of vaginal cancers.4,5
Recurrent respiratory papillomatosis
HPV 6 and 11 cause almost all cases of juvenile- and adult-onset recurrent respiratory papillomatosis.26 The annual cost for surgical procedures for this condition in the United States has been estimated at $151 million.27
HPV VACCINES ARE NONINFECTIOUS AND NONCARCINOGENIC
Currently, two HPV vaccines are available: a quadrivalent vaccine against types 6, 11, 16, and 18 (Gardasil; Merck) and a bivalent vaccine against types 16 and 18 (Cervarix; Glaxo-SmithKline). The quadrivalent vaccine was approved by the US Food and Drug Administration (FDA) in 2006, and the bivalent vaccine was approved in 2009.28,29
Both vaccines contain virus-like particles, ie, viral capsids that contain no DNA. HPV has a circular DNA genome of 8,000 nucleotides divided into two regions: the early region, for viral replication, and the late region, for viral capsid production. The host produces neutralizing antibodies in response to the L1 capsid protein, which is different in different HPV types.
In manufacturing the vaccines, the viral L1 gene is incorporated into a yeast genome or an insect virus genome using recombinant DNA technology (Figure 1). Grown in culture, the yeast or the insect cells produce the HPV L1 major capsid protein, which has the intrinsic capacity to self-assemble into virus-like particles.30–33 These particles are subsequently purified for use in the vaccines.34
Recombinant virus-like particles are morphologically indistinguishable from authentic HPV virions and contain the same typespecific antigens present in authentic virions. Therefore, they are highly effective in inducing a host humoral immune response. And because they do not contain HPV DNA, the recombinant HPV vaccines are noninfectious and noncarcinogenic.35
VACCINATION INDUCES A STRONGER IMMUNE RESPONSE THAN INFECTION
HPV infections trigger both a humoral and a cellular response in the host immune system.
The humoral immune response to HPV infection involves producing neutralizing antibody against the specific HPV type, specifically the specific L1 major capsid protein. This process is typically somewhat slow and weak, and only about 60% of women with a new HPV infection develop antibodies to it.36,37
HPV has several ways to evade the host immune system. It does not infect or replicate within the antigen-presenting cells in the epithelium. In addition, HPV-infected keratinocytes are less susceptible to cytotoxic lymphocytic-mediated lysis. Moreover, HPV infection cause very little tissue destruction. And finally, natural cervical HPV infection does not result in viremia. As a result, antigen-presenting cells have no chance to engulf the virions and present virion-derived antigen to the host immune system. The immune system outside the epithelium has limited opportunity to detect the virus because HPV infection does not have a blood-borne phase.38,39
The cell-mediated immune response to early HPV oncoproteins may help eliminate established HPV infection.40 In contrast to antibodies, the T-cell response to HPV has not been shown to be specific to HPV type.41 Clinically, cervical HPV infection is common, but most lesions go into remission or resolve as a result of the cell-mediated immune response.40,41
In contrast to the weak, somewhat ineffective immune response to natural HPV infection, the antibody response to HPV vaccines is rather robust. In randomized controlled trials, almost all vaccinated people have seroconverted. The peak antibody concentrations are 50 to 10,000 times greater than in natural infection. Furthermore, the neutralizing antibodies induced by HPV vaccines persist for as long as 7 to 9 years after immunization.42 However, the protection provided by HPV vaccines against HPV-related cervical intraepithelial neoplasia does not necessarily correlate with the antibody concentration.43–47
Why does the vaccine work so well?
Why are vaccine-induced antibody responses so much stronger than those induced by natural HPV infection?
The first reason is that the vaccine, delivered intramuscularly, rapidly enters into blood vessels and the lymphatic system. In contrast, in natural intraepithelial infection, the virus is shed from mucosal surfaces and does not result in viremia.48
In addition, the strong immunogenic nature of the virus-like particles induces a robust host antibody response even in the absence of adjuvant because of concentrated neutralizing epitopes and excellent induction of the T-helper cell response.35,49,50
The neutralizing antibody to L1 prevents HPV infection by blocking HPV from binding to the basement membrane as well as to the epithelial cell receptor during epithelial microabrasion and viral entry. The subsequent micro-wound healing leads to serous exudation and rapid access of serum immunoglobulin G (IgG) to HPV virus particles and encounters with circulatory B memory cells.
Furthermore, emerging evidence suggests that even very low antibody concentrations are sufficient to prevent viral entry into cervical epithelial cells.46–48,51–53
THE HPV VACCINES ARE HIGHLY EFFECTIVE AND SAFE
The efficacy and safety of the quadrivalent and the bivalent HPV vaccines have been evaluated in large randomized clinical trials.23,28,29,54,55 Table 1 summarizes the key findings.
The Females United to Unilaterally Reduce Endo/ectocervical Disease (FUTURE I)54 and FUTURE II28 trials showed conclusively that the quadrivalent HPV vaccine is 98% to 100% efficacious in preventing HPV 16- and 18-related cervical intraepithelial neoplasia, carcinoma in situ, and invasive cervical cancer in women who had not been infected with HPV before. Similarly, the Papilloma Trial against Cancer in Young Adults (PATRICIA) concluded that the bivalent HPV vaccine is 93% efficacious.29
Giuliano et al55 and Palefsky et al23 conducted randomized clinical trials of the quadrivalent HPV vaccine for preventing genital disease and anal intraepithelial neoplasia in boys and men; the efficacy rates were 90.4%55 and 77.5%.23
A recent Finnish trial in boys age 10 to 18 found 100% seroconversion rates for HPV 16 and HPV 18 antibodies after they received bivalent HPV vaccine.56 Similar efficacy has been demonstrated for the quadrivalent HPV vaccine in boys.57
Adverse events after vaccination
After the FDA approved the quadrivalent HPV vaccine for girls in 2006, the US Centers for Disease Control and Prevention (CDC) conducted a thorough survey of adverse events after immunization from June 1, 2006 through December 31, 2008.58 There were about 54 reports of adverse events per 100,000 distributed vaccine doses, similar to rates for other vaccines. However, the incidence rates of syncope and venous thrombosis were disproportionately higher, according to data from the US Vaccine Adverse Event Reporting System. The rate of syncope was 8.2 per 100,000 vaccine doses, and the rate of venous thrombotic events was 0.2 per 100,000 doses.58
There were 32 reports of deaths after HPV vaccination, but these were without clear causation. Hence, this information must be interpreted with caution and should not be used to infer causal associations between HPV vaccines and adverse outcomes. The causes of death included diabetic ketoacidosis, pulmonary embolism, prescription drug abuse, amyotrophic lateral sclerosis, meningoencephalitis, influenza B viral sepsis, arrhythmia, myocarditis, and idiopathic seizure disorder.58
Furthermore, it is important to note that vasovagal syncope and venous thromboembolic events are more common in young females in general.59 For example, the background rates of venous thromboembolism in females age 14 to 29 using oral contraceptives is 21 to 31 per 100,000 woman-years.60
Overall, the quadrivalent HPV vaccine is well tolerated and clinically safe. Postlicensure evaluation found that the quadrivalent and bivalent HPV vaccines had similar safety profiles.61
Vaccination is contraindicated in people with known hypersensitivity or prior severe allergic reactions to vaccine or yeast or who have bleeding disorders.
HPV VACCINATION DOES MORE THAN PREVENT CERVICAL CANCER IN FEMALES
The quadrivalent HPV vaccine was licensed by the FDA in 2006 for use in females age 9 to 26 to prevent cervical cancer, cervical cancer precursors, vaginal and vulval cancer precursors, and anogenital warts caused by HPV types 6, 11, 16, and 18. The CDC’s Advisory Committee on Immunization Practices (ACIP) issued its recommendation for initiating HPV vaccination for females age 11 to 12 in March 2007. The ACIP stated that the vaccine could be given to girls as early as age 9 and recommended catch-up vaccinations for those age 13 to 26.62,63
The quadrivalent HPV vaccine was licensed by the FDA in 2009 for use in boys and men for the prevention of genital warts. In December 2010, the quadrivalent HPV vaccine received extended licensure from the FDA for use in males and females for the prevention of anal cancer. In October 2011, the ACIP voted to recommend routine use of the quadrivalent HPV vaccine for boys age 11 to 12; catch-up vaccination should occur for those age 13 to 22, with an option to vaccinate men age 23 to 26.
These recommendations replace the “permissive use” recommendations from the ACIP in October 2009 that said the quadrivalent HPV vaccine may be given to males age 9 to 26.64 This shift from a permissive to an active recommendation connotes a positive change reflecting recognition of rising oropharyngeal cancer rates attributable to oncogenic, preventable HPV, rising HPV-related anal cancer incidence, and the burden of the disease in female partners of infected men, with associated rising health care costs.

The bivalent HPV vaccine received FDA licensure in October 2009 for use in females age 10 to 25 to prevent cervical cancer and precursor lesions. The ACIP included the bivalent HPV vaccine in its updated recommendations in May 2010 for use in girls age 11 to 12. Numerous national and international organizations have endorsed HPV vaccination.65–71
Table 2 outlines the recommendations from these organizations.
HPV VACCINATION RATES ARE STILL LOW
HPV vaccine offers us the hope of eventually eradicating cervical cancer. However, the immunization program still faces many challenges, since HPV vaccination touches on issues related to adolescent sexuality, parental autonomy, and cost. As a result, HPV immunization rates remain relatively low in the United States according to several national surveys. Only 40% to 49% of girls eligible for the vaccine received even one dose, and of those who received even one dose, only 32% to 53.3% came back for all three doses.72–75 Furthermore, indigent and minority teens were less likely to finish the three-dose HPV vaccine series.
Why are the vaccination rates so low?
Parental barriers. In one survey,73 reasons that parents gave for not having their daughters vaccinated included:
- Lack of knowledge of the vaccine (19.4%)
- Lack of perceived need for the vaccine (18.8%)
- Belief that their daughter was not sexually active (18.3%)
- Clinician not recommending vaccination (13.1%).
In an effort to improve HPV vaccination rates,41 several states proposed legislation for mandatory HPV vaccination of schoolgirls shortly after licensure of the quadrivalent HPV vaccine.3 Since then, we have seen a wave of public opposition rooted in concerns and misinformation about safety, teenage sexuality, governmental coercion, and cost. Widespread media coverage has also highlighted unsubstantiated claims about side effects attributable to the vaccine that can raise parents’ mistrust of vaccines.76 Concerns have also been raised about a threat to parental autonomy in how and when to educate their children about sex.77
Moreover, the vaccine has raised ethical concerns in some parents and politicians that mandatory vaccination could undermine abstinence messages in sexual education and may alter sexual activity by condoning risky behavior.78 However, a recent study indicated that there is no significant change in sexual behavior related to HPV vaccination in young girls.79
In 2012, Mullins et al80 also found that an urban population of adolescent girls (76.4% black, 57.5% sexually experienced) did not feel they could forgo safer sexual practices after first HPV vaccination, although the girls did perceive less risk from HPV than from other sexually transmitted infections after HPV vaccination (P < .001).80 Inadequate knowledge about HPV-related disease and HPV vaccine correlated with less perceived risk from HPV after vaccination among the girls, and a lack of knowledge about HPV and less communication with their daughters about HPV correlated with less perceived risk from HPV in the mothers of the study population.81
Health-care-provider barriers. Physician endorsement of vaccines represents a key predictor of vaccine acceptance by patients, families, and other clinicians.82–84 In 2008, a cross-sectional, Internet-based survey of 1,122 Texas pediatricians, family practice physicians, obstetricians, gynecologists, and internal medicine physicians providing direct patient care found that only 48.5% always recommended HPV vaccination to girls.74 Of all respondents, 68.4% were likely to recommend the vaccine to boys, and 41.7% agreed with mandated vaccination. Thus, more than half of the physicians were not following the current recommendations for universal HPV vaccination for 11- to -12-year-olds.
In a survey of 1,013 physicians during the spring and summer of 2009, only 34.6% said they always recommend HPV vaccination to early adolescents, 52.7% to middle adolescents, and 50.2% to late adolescents and young adults.85 Pediatricians were more likely than family physicians and obstetrician-gynecologists to always recommend HPV vaccine across all age groups (P < .001). Educational interventions targeting various specialties may help overcome physician-related barriers to immunization.85
Financial barriers. HPV vaccine, which must be given in three doses, is more expensive than other vaccines, and this expense is yet another barrier, especially for the uninsured.86 Australia launched a government-funded program of HPV vaccination (with the quadrivalent vaccine) in schools in 2007, and it has been very successful. Garland et al87 reported that new cases of genital warts have decreased by 73% since the program began, and the rate of high-grade abnormalities on Papanicolaou testing has declined by a small but significant amount.
For HPV vaccination to have an impact on public health, vaccination rates in the general population need to be high. In order to achieve these rates, we need to educate our patients on vaccine safety and efficacy and counsel vaccine recipients about the prevention of sexually transmitted infections and the importance of regular cervical cancer screening after age 21. Clinicians can actively “myth-bust” with patients, who may not realize that the vaccine should be given despite a history of HPV infection or abnormal Pap smear.
FREQUENTLY ASKED QUESTIONS
What if the patient is late for a shot?
The current recommended vaccination schedule for the bivalent and quadrivalent HPV vaccines is a three-dose series administered at 0, 2, and 6 months, given as an intramuscular injection, preferably in the deltoid muscle. The minimal dosing interval is 4 weeks between the first and second doses and 12 weeks between the second and third doses.
The vaccines use different adjuncts with different specific mechanisms for immunogenicity; therefore, it is recommended that the same vaccine be used for the entire three-dose series. However, if circumstances preclude the completion of a series with the same vaccine, the other HPV vaccine may be used.63 Starting the series over is not recommended.
Long-term studies demonstrated clinical efficacy 8.5 years after vaccination.47 Amnestic response by virtue of activation of pools of memory B cells has been demonstrated, suggesting the vaccine may afford lifelong immunity.88
Is a pregnancy test needed before HPV vaccination?
The ACIP states that pregnancy testing is not required before receiving either of the available HPV vaccines.
A recent retrospective review of phase III efficacy trials and pregnancy registry surveillance data for both vaccines revealed no increase in spontaneous abortions, fetal malformations, or adverse pregnancy outcomes.89 Data are limited on bivalent and quadrivalent HPV vaccine given within 30 days of pregnancy and subsequent pregnancy and fetal outcomes. Both vaccines have been assigned a pregnancy rating of category B; however, the ACIP recommends that neither vaccine be given if the recipient is known to be pregnant. If pregnancy occurs, it is recommended that the remainder of the series be deferred until after delivery.62
It is not known whether the vaccine is excreted in breast milk. The manufacturers of both the bivalent and quadrivalent HPV vaccines recommend caution when vaccinating lactating women.30,31
Can HPV vaccine be given with other vaccines?
In randomized trials, giving the bivalent HPV vaccine with the combined hepatitis A, hepatitis B, meningococcal conjugate and the combined tetanus, diphtheria, and acellular pertussis vaccines did not interfere with the immunogenic response, was safe, and was well tolerated.90,91 Coadministration of the quadrivalent HPV vaccine has been studied only with hepatitis B vaccine, with similar safety and efficacy noted.
The ACIP recommends giving HPV vaccine at the same visit with other age-appropriate immunizations to increase the likelihood of adherence to recommended vaccination schedules.62
Is HPV vaccination cost-effective?
Kim and Goldie86 performed a cost-effectiveness analysis of HPV vaccination of girls at age 12 and catch-up vaccination up to the ages of 18, 21, and 26. For their analysis, they considered prevention of cancers associated with HPV types 16 and 18, of genital warts associated with types 6 and 11, and of recurrent respiratory papillomatosis. They also assumed that immunity would be lifelong, and current screening practices would continue.
They calculated that routine vaccination of 12-year-old girls resulted in an incremental cost-effective ratio of $34,900 per quality-adjusted life-year (QALY) gained. A threshold of less than $50,000 per QALY gained is considered reasonably cost-effective, with an upper limit of $100,000 considered acceptable.92
In the same analysis by Kim and Goldie,86 catch-up vaccination of girls through age 18 resulted in a cost of $50,000 to $100,000 per QALY gained, and catch-up vaccination of females through age 26 was significantly less cost-effective at more then $130,000 per QALY gained. The vaccine was also significantly less cost-effective if 5% of the population was neither screened nor vaccinated, if a 10-year booster was required, and if frequent cervical cancer screening intervals were adopted.
This analysis did not include costs related to the evaluation and treatment of abnormal Pap smears and cross-protection against other HPV-related cancers.
The cost-effectiveness of HPV vaccination depends on reaching more girls at younger ages (ideally before sexual debut) and completing the three-dose schedule to optimize duration of immunity.92 Appropriate modification of the current recommendations for the intervals of cervical cancer screening for vaccinated individuals will further improve the cost-effectiveness of vaccination. The inclusion of male vaccination generally has more favorable cost per QALY in scenarios in which female coverage rates are less than 50%93 and among men who have sex with men.94
TO ERADICATE CERVICAL CANCER
Given the remarkable efficacy and expected long-term immunogenicity of HPV vaccines, we anticipate a decline in HPV-related cervical cancer and other related diseases in the years to come. However, modeling studies predicting the impact of HPV vaccination suggest that although substantial reductions in diseases can be expected, the benefit, assuming high vaccination rates, will not be apparent for at least another decade.95 Furthermore, the current HPV vaccines contain only HPV 16 and 18 L1 protein for cancer protection and, therefore, do not provide optimal protection against all oncogenic HPV-related cancers.
The real hope of eradicating cervical cancer and all HPV-related disease relies on a successful global implementation of multivalent HPV vaccination, effective screening strategies, and successful treatment.
- Baden LR, Curfman GD, Morrissey S, Drazen JM. Human papillomavirus vaccine—opportunity and challenge. N Engl J Med 2007; 356:1990–1991.
- Schiffman M, Wacholder S. From India to the world—a better way to prevent cervical cancer. N Engl J Med 2009; 360:1453–1455.
- Colgrove J, Abiola S, Mello MM. HPV vaccination mandates—law-making amid political and scientific controversy. N Engl J Med 2010; 363:785–791.
- World Health Organization. WHO/ICO Information Centre on Human Papilloma Virus (HPV) and Cervical Cancer. 2010Summary Report. www.who.int/hpvcentre. Accessed November 12, 2012.
- Muñoz N, Bosch FX, de Sanjosé S, et al; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348:518–527.
- Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011; 29:4294–4301.
- Jansen KU, Shaw AR. Human papillomavirus vaccines and prevention of cervical cancer. Annu Rev Med 2004; 55:319–331.
- Fleischer AB, Parrish CA, Glenn R, Feldman SR. Condylomata acuminata (genital warts): patient demographics and treating physicians. Sex Transm Dis 2001; 28:643–647.
- Clifford GM, Rana RK, Franceschi S, Smith JS, Gough G, Pimenta JM. Human papillomavirus genotype distribution in low-grade cervical lesions: comparison by geographic region and with cervical cancer. Cancer Epidemiol Biomarkers Prev 2005; 14:1157–1164.
- Schiffman M, Solomon D. Findings to date from the ASCUS-LSIL Triage Study (ALTS). Arch Pathol Lab Med 2003; 127:946–949.
- Insinga RP, Dasbach EJ, Myers ER. The health and economic burden of genital warts in a set of private health plans in the United States. Clin Infect Dis 2003; 36:1397–1403.
- Kodner CM, Nasraty S. Management of genital warts. Am Fam Physician 2004; 70:2335–2342.
- Maw RD, Reitano M, Roy M. An international survey of patients with genital warts: perceptions regarding treatment and impact on lifestyle. Int J STD AIDS 1998; 9:571–578.
- Insinga RP, Dasbach EJ, Elbasha EH. Assessing the annual economic burden of preventing and treating anogenital human papillomavirus-related disease in the US: analytic framework and review of the literature. Pharmacoeconomics 2005; 23:1107–1122.
- Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
- de Sanjose S, Quint WG, Alemany L, et al; Retrospective International Survey and HPV Time Trends Study Group. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol 2010; 11:1048–1056.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Insinga RP, Glass AG, Rush BB. The health care costs of cervical human papillomavirus—related disease. Am J Obstet Gynecol 2004; 191:114–120.
- Horner MJ, Ries LAG, Krapcho M, et al. SEER Cancer Statistics Review,1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER Web site, 2009. Accessed November 12, 2012.
- Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine 2006; 24(suppl 3):S3/11–S3/25.
- Hoots BE, Palefsky JM, Pimenta JM, Smith JS. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int J Cancer 2009; 124:2375–2383.
- Johnson LG, Madeleine MM, Newcomer LM, Schwartz SM, Daling JR. Anal cancer incidence and survival: the surveillance, epidemiology, and end results experience, 1973–2000. Cancer 2004; 101:281–288.
- Palefsky JM, Giuliano AR, Goldstone S, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med 2011; 365:1576–1585.
- Hu D, Goldie S. The economic burden of noncervical human papillomavirus disease in the United States. Am J Obstet Gynecol 2008; 198:500.e1–500.e7.
- Herrero R, Castellsagué X, Pawlita M, et al; IARC Multicenter Oral Cancer Study Group. Human papillomavirus and oral cancer: the International Agency for Research on Cancer multicenter study. J Natl Cancer Inst 2003; 95:1772–1783.
- Lacey CJ, Lowndes CM, Shah KV. Chapter 4: Burden and management of non-cancerous HPV-related conditions: HPV-6/11 disease. Vaccine 2006; 24(suppl 3):S3/35–S3/41.
- Derkay CS. Task force on recurrent respiratory papillomas. A preliminary report. Arch Otolaryngol Head Neck Surg 1995; 121:1386–1391.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
- Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Patient information about Gardasil (human papillomavirus quadrivalent type 6,11,16 and 18 vaccine, recombinant. http://www.gardasil.com/. Accessed November 12, 2012.
- GlaxoSmithKline Biologicals, Rixensart, Belgium. Highlights of prescribing information. Cervarix (human papillomavirus bivalent type 16 and 18 vaccine, recombinant. http://us.gsk.com/products/assets/us_cervarix.pdf. Accessed November 12, 2012.
- Zhou J, Sun XY, Stenzel DJ, Frazer IH. Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology 1991; 185:251–257.
- Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci USA 1992; 89:12180–12184.
- Schiller JT, Lowy DR. Papillomavirus-like particles and HPV vaccine development. Semin Cancer Biol 1996; 7:373–382.
- Harro CD, Pang YY, Roden RB, et al. Safety and immunogenicity trial in adult volunteers of a human papillomavirus 16 L1 virus-like particle vaccine. J Natl Cancer Inst 2001; 93:284–292.
- af Geijersstam V, Kibur M, Wang Z, et al. Stability over time of serum antibody levels to human papillomavirus type 16. J Infect Dis 1998; 177:1710–1714.
- Safaeian M, Porras C, Schiffman M, et al; Costa Rican Vaccine Trial Group. Epidemiological study of anti-HPV16/18 seropositivity and subsequent risk of HPV16 and -18 infections. J Natl Cancer Inst 2010; 102:1653–1662.
- Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer 2002; 2:59–65.
- Scott M, Nakagawa M, Moscicki AB. Cell-mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol 2001; 8:209–220.
- Roden R, Wu TC. Preventative and therapeutic vaccines for cervical cancer. Expert Rev Vaccines 2003; 2:495–516.
- Wang SS, Hildesheim A. Chapter 5: Viral and host factors in human papillomavirus persistence and progression. J Natl Cancer Inst Monogr 2003; 31:35–40.
- De Carvalho N, Teixeira J, Roteli-Martins CM, et al. Sustained efficacy and immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine up to 7.3 years in young adult women. Vaccine 2010; 28:6247–6255.
- Harper DM, Franco EL, Wheeler CM, et al; HPV Vaccine Study group. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet 2006; 367:1247–1255.
- Villa LL, Costa RL, Petta CA, et al. High sustained efficacy of a prophylactic quadrivalent human papillomavirus types 6/11/16/18 L1 virus-like particle vaccine through 5 years of follow-up. Br J Cancer 2006; 95:1459–1466.
- Villa LL, Ault KA, Giuliano AR, et al. Immunologic responses following administration of a vaccine targeting human papillomavirus Types 6, 11, 16, and 18. Vaccine 2006; 24:5571–5583.
- Smith JF, Brownlow M, Brown M, et al. Antibodies from women immunized with Gardasil cross-neutralize HPV 45 pseudovirions. Hum Vaccin 2007; 3:109–115.
- Rowhani-Rahbar A, Mao C, Hughes JP, et al. Longer term efficacy of a prophylactic monovalent human papillomavirus type 16 vaccine. Vaccine 2009; 27:5612–5619.
- Stanley M. HPV - immune response to infection and vaccination. Infect Agent Cancer 2010; 5:19.
- Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol 2010; 117(suppl 2):S5–S10.
- Yan M, Peng J, Jabbar IA, et al. Activation of dendritic cells by human papillomavirus-like particles through TLR4 and NF-kappaB-mediated signalling, moderated by TGF-beta. Immunol Cell Biol 2005; 83:83–91.
- Roberts JN, Buck CB, Thompson CD, et al. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 2007; 13:857–861.
- Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci USA 2009; 106:20458–20463.
- Day PM, Kines RC, Thompson CD, et al. In vivo mechanisms of vaccine-induced protection against HPV infection. Cell Host Microbe 2010; 8:260–270.
- Garland SM, Hernandez-Avila M, Wheeler CM, et al; Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007; 356:1928–1943.
- Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV Infection and disease in males. N Engl J Med 2011; 364:401–411.
- Petäjä T, Keränen H, Karppa T, et al. Immunogenicity and safety of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine in healthy boys aged 10–18 years. J Adolesc Health 2009; 44:33–40.
- Reisinger KS, Block SL, Lazcano-Ponce E, et al. Safety and persistent immunogenicity of a quadrivalent human papillomavirus types 6, 11, 16, 18 L1 virus-like particle vaccine in preadolescents and adolescents: a randomized controlled trial. Pediatr Infect Dis J 2007; 26:201–209.
- Slade BA, Leidel L, Vellozzi C, et al. Postlicensure safety surveillance for quadrivalent human papillomavirus recombinant vaccine. JAMA 2009; 302:750–757.
- Block SL, Brown DR, Chatterjee A, et al. Clinical trial and post-licensure safety profile of a prophylactic human papillomavirus (types 6, 11, 16, and 18) l1 virus-like particle vaccine. Pediatr Infect Dis J 2010; 29:95–101.
- Farmer RD, Lawrenson RA, Thompson CR, Kennedy JG, Hambleton IR. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet 1997; 349:83–88.
- Labadie J. Postlicensure safety evaluation of human papilloma virus vaccines. Int J Risk Saf Med 2011; 23:103–112.
- Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER; Centers for Disease Control and Prevention (CDC). Quadrivalent human papillomavirus vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2007; 56:1–24.
- Centers for Disease Control and Prevention (CDC). FDA licensure of bivalent human papillomavirus vaccine (HPV2, Cervarix) for use in females and updated HPV vaccination recommendations from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2010; 59:626–629.
- Centers for Disease Control and Prevention (CDC). FDA licensure of quadrivalent human papillomavirus vaccine (HPV4, Gardasil) for use in males and guidance from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2010; 59:630–632.
- World Health Organization (WHO). Weekly Epidemiological Record (WER). January 2009; 84:1–16. http://www.who.int/wer/2009/wer8401_02/en/index.html. Accessed November 12, 2012.
- Saslow D, Castle PE, Cox JT, et al. American Cancer Society guideline for human papillomavirus (HPV) vaccine use to prevent cervical cancer and its precursors. CA Cancer J Clin 2007; 57:7–28.
- Committee opinion no. 467: human papillomavirus vaccination. Obstet Gynecol 2010; 116:800–803.
- American College of Physicians. ACP Guide to Adult Immunization. 4th ed. 2011:58–60. http://immunization.acponline.org/. Accessed November 12, 2012.
- Vaughn JA, Miller RA. Update on immunizations in adults. Am Fam Physician 2011; 84:1015–1020.
- American Academy of Pediatrics Committee on Infectious Diseases. Prevention of human papillomavirus infection: provisional recommendations for immunization of girls and women with quadrivalent human papillomavirus vaccine. Pediatrics 2007; 120:666–668.
- Friedman L, Bell DL, Kahn JA, et al. Human papillomavirus vaccine: an updated position statement of the Society for Adolescent Health and Medicine. J Adolesc Health 2011; 48:215–216.
- Centers for Disease Control and Prevention (CDC). National and state vaccination coverage among adolescents aged 13 through 17 years--United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:1117–1123.
- Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
- Kahn JA, Cooper HP, Vadaparampil ST, et al. Human papillomavirus vaccine recommendations and agreement with mandated human papillomavirus vaccination for 11-to-12-year-old girls: a statewide survey of Texas physicians. Cancer Epidemiol Biomarkers Prev 2009; 18:2325–2332.
- Schwartz JL, Caplan AL, Faden RR, Sugarman J. Lessons from the failure of human papillomavirus vaccine state requirements. Clin Pharmacol Ther 2007; 82:760–763.
- Cooper LZ, Larson HJ, Katz SL. Protecting public trust in immunization. Pediatrics 2008; 122:149–153.
- Olshen E, Woods ER, Austin SB, Luskin M, Bauchner H. Parental acceptance of the human papillomavirus vaccine. J Adolesc Health 2005; 37:248–251.
- Zimmerman RK. Ethical analysis of HPV vaccine policy options. Vaccine 2006; 24:4812–4820.
- Al Romaih WRR, Srinivas A, Shahtahmasebi S, Omar HA. No significant change in sexual behavior in association with human papillomavirus vaccination in young girls. Int J Child Adolesc Health 2011; 4:1–5.
- Mullins TL, Zimet GD, Rosenthal SL, et al. Adolescent perceptions of risk and need for safer sexual behaviors after first human papillomavirus vaccination. Arch Pediatr Adolesc Med 2012; 166:82–88.
- Middleman AB, Tung JS. School-located immunization programs: do parental p predict behavior? Vaccine 2011; 29:3513–3516.
- Samoff E, Dunn A, VanDevanter N, Blank S, Weisfuse IB. Predictors of acceptance of hepatitis B vaccination in an urban sexually transmitted diseases clinic. Sex Transm Dis 2004; 31:415–420.
- Gnanasekaran SK, Finkelstein JA, Hohman K, O’Brien M, Kruskal B, Lieu T. Parental perspectives on influenza vaccination among children with asthma. Public Health Rep 2006; 121:181–188.
- Daley MF, Crane LA, Chandramouli V, et al. Influenza among healthy young children: changes in parental attitudes and predictors of immunization during the 2003 to 2004 influenza season. Pediatrics 2006; 117:e268–e277.
- Vadaparampil ST, Kahn JA, Salmon D, et al. Missed clinical opportunities: provider recommendations for HPV vaccination for 11–12 year old girls are limited. Vaccine 2011; 29:8634–8641.
- Kim JJ, Goldie SJ. Health and economic implications of HPV vaccination in the United States. N Engl J Med 2008; 359:821–832.
- Garland SM, Skinner SR, Brotherton JM. Adolescent and young adult HPV vaccination in Australia: achievements and challenges. Prev Med 2011; 53(suppl 1):S29–S35.
- Rowhani-Rahbar A, Alvarez FB, Bryan JT, et al. Evidence of immune memory 8.5 years following administration of a prophylactic human papillomavirus type 16 vaccine. J Clin Virol 2012; 53:239–243.
- Forinash AB, Yancey AM, Pitlick JM, Myles TD. Safety of the HPV bivalent and quadrivalent vaccines during pregnancy (February) Ann Pharmacother 2011; [epub ahead of print]
- Wheeler CM, Harvey BM, Pichichero ME, et al. Immunogenicity and safety of human papillomavirus-16/18 AS04-adjuvanted vaccine coadministered with tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine and/or meningococcal conjugate vaccine to healthy girls 11 to 18 years of age: results from a randomized open trial. Pediatr Infect Dis J 2011; 30:e225–e234.
- Pedersen C, Breindahl M, Aggarwal N, et al. Randomized trial: immunogenicity and safety of coadministered human papillomavirus-16/18 AS04-adjuvanted vaccine and combined hepatitis A and B vaccine in girls. J Adolesc Health 2012; 50:38–46.
- Eichler HG, Kong SX, Gerth WC, Mavros P, Jönsson B. Use of costeffectiveness analysis in health-care resource allocation decisionmaking: how are cost-effectiveness thresholds expected to emerge? Value Health 2004; 7:518–528.
- Chesson HW. HPV vaccine cost-effectiveness: updates and review. Presentation before the Advisory Committee on Immunization Practices (ACIP), June 22, 2011. Atlanta, GA: US Department of Health and Human Services, CDC; 2011. http://www.cdc.gov/vaccines/recs/acip/downloads/mtg-slides-jun11/07-5-hpv-cost-effect.pdf. Accessed August 31, 2012.
- Kim JJ. Targeted human papillomavirus vaccination of men who have sex with men in the USA: a cost-effectiveness modelling analysis. Lancet Infect Dis 2010; 10:845–852.
- Cuzick J, Castañón A, Sasieni P. Predicted impact of vaccination against human papillomavirus 16/18 on cancer incidence and cervical abnormalities in women aged 20–29 in the UK. Br J Cancer 2010; 102:933–939.
The vaccines against human papillomavirus (HPV) are the only ones designed to prevent cancer caused by a virus1,2—surely a good goal. But because HPV is sexually transmitted, HPV vaccination has met with public controversy.3 To counter the objections and better protect their patients’ health, primary care providers and other clinicians need a clear understanding of the benefits and the low risk of HPV vaccination—and the reasons so many people object to it.3
In this article, we will review:
- The impact of HPV-related diseases
- The basic biologic features of HPV vaccines
- The host immune response to natural HPV infection vs the response to HPV vaccines
- The clinical efficacy and safety of HPV vaccines
- The latest guidelines for HPV vaccination
- The challenges to vaccination implementation
- Frequently asked practical questions about HPV vaccination.
HPV-RELATED DISEASES: FROM BOTHERSOME TO DEADLY
Clinical sequelae of HPV infection include genital warts; cancers of the cervix, vulva, vagina, anus, penis, and oropharynx; and recurrent respiratory papillomatosis.4–6
Genital warts
HPV types 6 and 11 are responsible for more than 90% of the 1 million new cases of genital warts diagnosed annually in the United States.7–10
Bothersome and embarrassing, HPV-related genital warts can cause itching, burning, erythema, and pain, as well as epithelial erosions, ulcerations, depigmentation, and urethral and vaginal bleeding and discharge.11,12 Although they are benign in the oncologic sense, they can cause a good deal of emotional and financial stress. Patients may feel anxiety, embarrassment,13 and vulnerability. Adolescents and adults who have or have had genital warts need to inform their current and future partners or else risk infecting them—and facing the consequences.
Direct health care costs of genital warts in the United States have been estimated to be at least $200 million per year.14
Cervical cancer
Cervical cancer cannot develop unless the cervical epithelium is infected with one of the oncogenic HPV types. Indeed, oncogenic HPV is present in as many as 99.8% of cervical cancer specimens.15 HPV 16 and 18 are the most oncogenic HPV genotypes and account for 75% of all cases of cervical cancer. Ten other HPV genotypes account for the remaining 25%.16
In 2012, there were an estimated 12,170 new cases of invasive cervical cancer in the United States and 4,220 related deaths.17 The cost associated with cervical cancer screening, managing abnormal findings, and treating invasive cervical cancer in the United States is estimated to be $3.3 billion per year.18
Although the incidence and the mortality rates of cervical cancer have decreased more than 50% in the United States over the past 3 decades thanks to screening,19 cervical cancer remains the second leading cause of death from cancer in women worldwide. Each year, an estimated 500,000 women contract the disease and 240,000 die of it.20
Anal cancer
A recent study indicated that oncogenic HPV can also cause anal cancer, and the proportion of such cancers associated with HPV 16 or HPV 18 infection is as high as or higher than for cervical cancers, and estimated at 80%.21
The incidence of anal cancer is increasing by approximately 2% per year in both men and women in the general population,22 and rates are even higher in men who have sex with men and people infected with the human immunodeficiency virus.23
Hu and Goldie24 estimated that the lifetime costs of caring for all the people in the United States who in just 1 year (2003) acquired anal cancer attributable to HPV would total $92 million.
Oropharyngeal cancer
HPV types 16, 18, 31, 33, and 35 also cause oropharyngeal cancer. HPV 16 accounts for more than 90% of cases of HPV-related oropharyngeal cancer.25
Chaturvedi et al6 tested tissue samples from three national cancer registries and found that the number of oropharyngeal cancers that were HPV-positive increased from 16.3% in 1984–1989 to 71.7% in 2000–2004, while the number of HPV-negative oropharyngeal cancers fell by 50%, paralleling the drop in cigarette smoking in the United States.
Hu and Goldie24 estimated that the total lifetime cost for all new HPV-related oropharyngeal cancers that arose in 2003 would come to $38.1 million.24
Vulvar and vaginal cancers
HPV 16 and 18 are also responsible for approximately 50% of vulvar cancers and 50% to 75% of vaginal cancers.4,5
Recurrent respiratory papillomatosis
HPV 6 and 11 cause almost all cases of juvenile- and adult-onset recurrent respiratory papillomatosis.26 The annual cost for surgical procedures for this condition in the United States has been estimated at $151 million.27
HPV VACCINES ARE NONINFECTIOUS AND NONCARCINOGENIC
Currently, two HPV vaccines are available: a quadrivalent vaccine against types 6, 11, 16, and 18 (Gardasil; Merck) and a bivalent vaccine against types 16 and 18 (Cervarix; Glaxo-SmithKline). The quadrivalent vaccine was approved by the US Food and Drug Administration (FDA) in 2006, and the bivalent vaccine was approved in 2009.28,29
Both vaccines contain virus-like particles, ie, viral capsids that contain no DNA. HPV has a circular DNA genome of 8,000 nucleotides divided into two regions: the early region, for viral replication, and the late region, for viral capsid production. The host produces neutralizing antibodies in response to the L1 capsid protein, which is different in different HPV types.
In manufacturing the vaccines, the viral L1 gene is incorporated into a yeast genome or an insect virus genome using recombinant DNA technology (Figure 1). Grown in culture, the yeast or the insect cells produce the HPV L1 major capsid protein, which has the intrinsic capacity to self-assemble into virus-like particles.30–33 These particles are subsequently purified for use in the vaccines.34
Recombinant virus-like particles are morphologically indistinguishable from authentic HPV virions and contain the same typespecific antigens present in authentic virions. Therefore, they are highly effective in inducing a host humoral immune response. And because they do not contain HPV DNA, the recombinant HPV vaccines are noninfectious and noncarcinogenic.35
VACCINATION INDUCES A STRONGER IMMUNE RESPONSE THAN INFECTION
HPV infections trigger both a humoral and a cellular response in the host immune system.
The humoral immune response to HPV infection involves producing neutralizing antibody against the specific HPV type, specifically the specific L1 major capsid protein. This process is typically somewhat slow and weak, and only about 60% of women with a new HPV infection develop antibodies to it.36,37
HPV has several ways to evade the host immune system. It does not infect or replicate within the antigen-presenting cells in the epithelium. In addition, HPV-infected keratinocytes are less susceptible to cytotoxic lymphocytic-mediated lysis. Moreover, HPV infection cause very little tissue destruction. And finally, natural cervical HPV infection does not result in viremia. As a result, antigen-presenting cells have no chance to engulf the virions and present virion-derived antigen to the host immune system. The immune system outside the epithelium has limited opportunity to detect the virus because HPV infection does not have a blood-borne phase.38,39
The cell-mediated immune response to early HPV oncoproteins may help eliminate established HPV infection.40 In contrast to antibodies, the T-cell response to HPV has not been shown to be specific to HPV type.41 Clinically, cervical HPV infection is common, but most lesions go into remission or resolve as a result of the cell-mediated immune response.40,41
In contrast to the weak, somewhat ineffective immune response to natural HPV infection, the antibody response to HPV vaccines is rather robust. In randomized controlled trials, almost all vaccinated people have seroconverted. The peak antibody concentrations are 50 to 10,000 times greater than in natural infection. Furthermore, the neutralizing antibodies induced by HPV vaccines persist for as long as 7 to 9 years after immunization.42 However, the protection provided by HPV vaccines against HPV-related cervical intraepithelial neoplasia does not necessarily correlate with the antibody concentration.43–47
Why does the vaccine work so well?
Why are vaccine-induced antibody responses so much stronger than those induced by natural HPV infection?
The first reason is that the vaccine, delivered intramuscularly, rapidly enters into blood vessels and the lymphatic system. In contrast, in natural intraepithelial infection, the virus is shed from mucosal surfaces and does not result in viremia.48
In addition, the strong immunogenic nature of the virus-like particles induces a robust host antibody response even in the absence of adjuvant because of concentrated neutralizing epitopes and excellent induction of the T-helper cell response.35,49,50
The neutralizing antibody to L1 prevents HPV infection by blocking HPV from binding to the basement membrane as well as to the epithelial cell receptor during epithelial microabrasion and viral entry. The subsequent micro-wound healing leads to serous exudation and rapid access of serum immunoglobulin G (IgG) to HPV virus particles and encounters with circulatory B memory cells.
Furthermore, emerging evidence suggests that even very low antibody concentrations are sufficient to prevent viral entry into cervical epithelial cells.46–48,51–53
THE HPV VACCINES ARE HIGHLY EFFECTIVE AND SAFE
The efficacy and safety of the quadrivalent and the bivalent HPV vaccines have been evaluated in large randomized clinical trials.23,28,29,54,55 Table 1 summarizes the key findings.
The Females United to Unilaterally Reduce Endo/ectocervical Disease (FUTURE I)54 and FUTURE II28 trials showed conclusively that the quadrivalent HPV vaccine is 98% to 100% efficacious in preventing HPV 16- and 18-related cervical intraepithelial neoplasia, carcinoma in situ, and invasive cervical cancer in women who had not been infected with HPV before. Similarly, the Papilloma Trial against Cancer in Young Adults (PATRICIA) concluded that the bivalent HPV vaccine is 93% efficacious.29
Giuliano et al55 and Palefsky et al23 conducted randomized clinical trials of the quadrivalent HPV vaccine for preventing genital disease and anal intraepithelial neoplasia in boys and men; the efficacy rates were 90.4%55 and 77.5%.23
A recent Finnish trial in boys age 10 to 18 found 100% seroconversion rates for HPV 16 and HPV 18 antibodies after they received bivalent HPV vaccine.56 Similar efficacy has been demonstrated for the quadrivalent HPV vaccine in boys.57
Adverse events after vaccination
After the FDA approved the quadrivalent HPV vaccine for girls in 2006, the US Centers for Disease Control and Prevention (CDC) conducted a thorough survey of adverse events after immunization from June 1, 2006 through December 31, 2008.58 There were about 54 reports of adverse events per 100,000 distributed vaccine doses, similar to rates for other vaccines. However, the incidence rates of syncope and venous thrombosis were disproportionately higher, according to data from the US Vaccine Adverse Event Reporting System. The rate of syncope was 8.2 per 100,000 vaccine doses, and the rate of venous thrombotic events was 0.2 per 100,000 doses.58
There were 32 reports of deaths after HPV vaccination, but these were without clear causation. Hence, this information must be interpreted with caution and should not be used to infer causal associations between HPV vaccines and adverse outcomes. The causes of death included diabetic ketoacidosis, pulmonary embolism, prescription drug abuse, amyotrophic lateral sclerosis, meningoencephalitis, influenza B viral sepsis, arrhythmia, myocarditis, and idiopathic seizure disorder.58
Furthermore, it is important to note that vasovagal syncope and venous thromboembolic events are more common in young females in general.59 For example, the background rates of venous thromboembolism in females age 14 to 29 using oral contraceptives is 21 to 31 per 100,000 woman-years.60
Overall, the quadrivalent HPV vaccine is well tolerated and clinically safe. Postlicensure evaluation found that the quadrivalent and bivalent HPV vaccines had similar safety profiles.61
Vaccination is contraindicated in people with known hypersensitivity or prior severe allergic reactions to vaccine or yeast or who have bleeding disorders.
HPV VACCINATION DOES MORE THAN PREVENT CERVICAL CANCER IN FEMALES
The quadrivalent HPV vaccine was licensed by the FDA in 2006 for use in females age 9 to 26 to prevent cervical cancer, cervical cancer precursors, vaginal and vulval cancer precursors, and anogenital warts caused by HPV types 6, 11, 16, and 18. The CDC’s Advisory Committee on Immunization Practices (ACIP) issued its recommendation for initiating HPV vaccination for females age 11 to 12 in March 2007. The ACIP stated that the vaccine could be given to girls as early as age 9 and recommended catch-up vaccinations for those age 13 to 26.62,63
The quadrivalent HPV vaccine was licensed by the FDA in 2009 for use in boys and men for the prevention of genital warts. In December 2010, the quadrivalent HPV vaccine received extended licensure from the FDA for use in males and females for the prevention of anal cancer. In October 2011, the ACIP voted to recommend routine use of the quadrivalent HPV vaccine for boys age 11 to 12; catch-up vaccination should occur for those age 13 to 22, with an option to vaccinate men age 23 to 26.
These recommendations replace the “permissive use” recommendations from the ACIP in October 2009 that said the quadrivalent HPV vaccine may be given to males age 9 to 26.64 This shift from a permissive to an active recommendation connotes a positive change reflecting recognition of rising oropharyngeal cancer rates attributable to oncogenic, preventable HPV, rising HPV-related anal cancer incidence, and the burden of the disease in female partners of infected men, with associated rising health care costs.
The bivalent HPV vaccine received FDA licensure in October 2009 for use in females age 10 to 25 to prevent cervical cancer and precursor lesions. The ACIP included the bivalent HPV vaccine in its updated recommendations in May 2010 for use in girls age 11 to 12. Numerous national and international organizations have endorsed HPV vaccination.65–71
Table 2 outlines the recommendations from these organizations.
HPV VACCINATION RATES ARE STILL LOW
HPV vaccine offers us the hope of eventually eradicating cervical cancer. However, the immunization program still faces many challenges, since HPV vaccination touches on issues related to adolescent sexuality, parental autonomy, and cost. As a result, HPV immunization rates remain relatively low in the United States according to several national surveys. Only 40% to 49% of girls eligible for the vaccine received even one dose, and of those who received even one dose, only 32% to 53.3% came back for all three doses.72–75 Furthermore, indigent and minority teens were less likely to finish the three-dose HPV vaccine series.
Why are the vaccination rates so low?
Parental barriers. In one survey,73 reasons that parents gave for not having their daughters vaccinated included:
- Lack of knowledge of the vaccine (19.4%)
- Lack of perceived need for the vaccine (18.8%)
- Belief that their daughter was not sexually active (18.3%)
- Clinician not recommending vaccination (13.1%).
In an effort to improve HPV vaccination rates,41 several states proposed legislation for mandatory HPV vaccination of schoolgirls shortly after licensure of the quadrivalent HPV vaccine.3 Since then, we have seen a wave of public opposition rooted in concerns and misinformation about safety, teenage sexuality, governmental coercion, and cost. Widespread media coverage has also highlighted unsubstantiated claims about side effects attributable to the vaccine that can raise parents’ mistrust of vaccines.76 Concerns have also been raised about a threat to parental autonomy in how and when to educate their children about sex.77
Moreover, the vaccine has raised ethical concerns in some parents and politicians that mandatory vaccination could undermine abstinence messages in sexual education and may alter sexual activity by condoning risky behavior.78 However, a recent study indicated that there is no significant change in sexual behavior related to HPV vaccination in young girls.79
In 2012, Mullins et al80 also found that an urban population of adolescent girls (76.4% black, 57.5% sexually experienced) did not feel they could forgo safer sexual practices after first HPV vaccination, although the girls did perceive less risk from HPV than from other sexually transmitted infections after HPV vaccination (P < .001).80 Inadequate knowledge about HPV-related disease and HPV vaccine correlated with less perceived risk from HPV after vaccination among the girls, and a lack of knowledge about HPV and less communication with their daughters about HPV correlated with less perceived risk from HPV in the mothers of the study population.81
Health-care-provider barriers. Physician endorsement of vaccines represents a key predictor of vaccine acceptance by patients, families, and other clinicians.82–84 In 2008, a cross-sectional, Internet-based survey of 1,122 Texas pediatricians, family practice physicians, obstetricians, gynecologists, and internal medicine physicians providing direct patient care found that only 48.5% always recommended HPV vaccination to girls.74 Of all respondents, 68.4% were likely to recommend the vaccine to boys, and 41.7% agreed with mandated vaccination. Thus, more than half of the physicians were not following the current recommendations for universal HPV vaccination for 11- to -12-year-olds.
In a survey of 1,013 physicians during the spring and summer of 2009, only 34.6% said they always recommend HPV vaccination to early adolescents, 52.7% to middle adolescents, and 50.2% to late adolescents and young adults.85 Pediatricians were more likely than family physicians and obstetrician-gynecologists to always recommend HPV vaccine across all age groups (P < .001). Educational interventions targeting various specialties may help overcome physician-related barriers to immunization.85
Financial barriers. HPV vaccine, which must be given in three doses, is more expensive than other vaccines, and this expense is yet another barrier, especially for the uninsured.86 Australia launched a government-funded program of HPV vaccination (with the quadrivalent vaccine) in schools in 2007, and it has been very successful. Garland et al87 reported that new cases of genital warts have decreased by 73% since the program began, and the rate of high-grade abnormalities on Papanicolaou testing has declined by a small but significant amount.
For HPV vaccination to have an impact on public health, vaccination rates in the general population need to be high. In order to achieve these rates, we need to educate our patients on vaccine safety and efficacy and counsel vaccine recipients about the prevention of sexually transmitted infections and the importance of regular cervical cancer screening after age 21. Clinicians can actively “myth-bust” with patients, who may not realize that the vaccine should be given despite a history of HPV infection or abnormal Pap smear.
FREQUENTLY ASKED QUESTIONS
What if the patient is late for a shot?
The current recommended vaccination schedule for the bivalent and quadrivalent HPV vaccines is a three-dose series administered at 0, 2, and 6 months, given as an intramuscular injection, preferably in the deltoid muscle. The minimal dosing interval is 4 weeks between the first and second doses and 12 weeks between the second and third doses.
The vaccines use different adjuncts with different specific mechanisms for immunogenicity; therefore, it is recommended that the same vaccine be used for the entire three-dose series. However, if circumstances preclude the completion of a series with the same vaccine, the other HPV vaccine may be used.63 Starting the series over is not recommended.
Long-term studies demonstrated clinical efficacy 8.5 years after vaccination.47 Amnestic response by virtue of activation of pools of memory B cells has been demonstrated, suggesting the vaccine may afford lifelong immunity.88
Is a pregnancy test needed before HPV vaccination?
The ACIP states that pregnancy testing is not required before receiving either of the available HPV vaccines.
A recent retrospective review of phase III efficacy trials and pregnancy registry surveillance data for both vaccines revealed no increase in spontaneous abortions, fetal malformations, or adverse pregnancy outcomes.89 Data are limited on bivalent and quadrivalent HPV vaccine given within 30 days of pregnancy and subsequent pregnancy and fetal outcomes. Both vaccines have been assigned a pregnancy rating of category B; however, the ACIP recommends that neither vaccine be given if the recipient is known to be pregnant. If pregnancy occurs, it is recommended that the remainder of the series be deferred until after delivery.62
It is not known whether the vaccine is excreted in breast milk. The manufacturers of both the bivalent and quadrivalent HPV vaccines recommend caution when vaccinating lactating women.30,31
Can HPV vaccine be given with other vaccines?
In randomized trials, giving the bivalent HPV vaccine with the combined hepatitis A, hepatitis B, meningococcal conjugate and the combined tetanus, diphtheria, and acellular pertussis vaccines did not interfere with the immunogenic response, was safe, and was well tolerated.90,91 Coadministration of the quadrivalent HPV vaccine has been studied only with hepatitis B vaccine, with similar safety and efficacy noted.
The ACIP recommends giving HPV vaccine at the same visit with other age-appropriate immunizations to increase the likelihood of adherence to recommended vaccination schedules.62
Is HPV vaccination cost-effective?
Kim and Goldie86 performed a cost-effectiveness analysis of HPV vaccination of girls at age 12 and catch-up vaccination up to the ages of 18, 21, and 26. For their analysis, they considered prevention of cancers associated with HPV types 16 and 18, of genital warts associated with types 6 and 11, and of recurrent respiratory papillomatosis. They also assumed that immunity would be lifelong, and current screening practices would continue.
They calculated that routine vaccination of 12-year-old girls resulted in an incremental cost-effective ratio of $34,900 per quality-adjusted life-year (QALY) gained. A threshold of less than $50,000 per QALY gained is considered reasonably cost-effective, with an upper limit of $100,000 considered acceptable.92
In the same analysis by Kim and Goldie,86 catch-up vaccination of girls through age 18 resulted in a cost of $50,000 to $100,000 per QALY gained, and catch-up vaccination of females through age 26 was significantly less cost-effective at more then $130,000 per QALY gained. The vaccine was also significantly less cost-effective if 5% of the population was neither screened nor vaccinated, if a 10-year booster was required, and if frequent cervical cancer screening intervals were adopted.
This analysis did not include costs related to the evaluation and treatment of abnormal Pap smears and cross-protection against other HPV-related cancers.
The cost-effectiveness of HPV vaccination depends on reaching more girls at younger ages (ideally before sexual debut) and completing the three-dose schedule to optimize duration of immunity.92 Appropriate modification of the current recommendations for the intervals of cervical cancer screening for vaccinated individuals will further improve the cost-effectiveness of vaccination. The inclusion of male vaccination generally has more favorable cost per QALY in scenarios in which female coverage rates are less than 50%93 and among men who have sex with men.94
TO ERADICATE CERVICAL CANCER
Given the remarkable efficacy and expected long-term immunogenicity of HPV vaccines, we anticipate a decline in HPV-related cervical cancer and other related diseases in the years to come. However, modeling studies predicting the impact of HPV vaccination suggest that although substantial reductions in diseases can be expected, the benefit, assuming high vaccination rates, will not be apparent for at least another decade.95 Furthermore, the current HPV vaccines contain only HPV 16 and 18 L1 protein for cancer protection and, therefore, do not provide optimal protection against all oncogenic HPV-related cancers.
The real hope of eradicating cervical cancer and all HPV-related disease relies on a successful global implementation of multivalent HPV vaccination, effective screening strategies, and successful treatment.
The vaccines against human papillomavirus (HPV) are the only ones designed to prevent cancer caused by a virus1,2—surely a good goal. But because HPV is sexually transmitted, HPV vaccination has met with public controversy.3 To counter the objections and better protect their patients’ health, primary care providers and other clinicians need a clear understanding of the benefits and the low risk of HPV vaccination—and the reasons so many people object to it.3
In this article, we will review:
- The impact of HPV-related diseases
- The basic biologic features of HPV vaccines
- The host immune response to natural HPV infection vs the response to HPV vaccines
- The clinical efficacy and safety of HPV vaccines
- The latest guidelines for HPV vaccination
- The challenges to vaccination implementation
- Frequently asked practical questions about HPV vaccination.
HPV-RELATED DISEASES: FROM BOTHERSOME TO DEADLY
Clinical sequelae of HPV infection include genital warts; cancers of the cervix, vulva, vagina, anus, penis, and oropharynx; and recurrent respiratory papillomatosis.4–6
Genital warts
HPV types 6 and 11 are responsible for more than 90% of the 1 million new cases of genital warts diagnosed annually in the United States.7–10
Bothersome and embarrassing, HPV-related genital warts can cause itching, burning, erythema, and pain, as well as epithelial erosions, ulcerations, depigmentation, and urethral and vaginal bleeding and discharge.11,12 Although they are benign in the oncologic sense, they can cause a good deal of emotional and financial stress. Patients may feel anxiety, embarrassment,13 and vulnerability. Adolescents and adults who have or have had genital warts need to inform their current and future partners or else risk infecting them—and facing the consequences.
Direct health care costs of genital warts in the United States have been estimated to be at least $200 million per year.14
Cervical cancer
Cervical cancer cannot develop unless the cervical epithelium is infected with one of the oncogenic HPV types. Indeed, oncogenic HPV is present in as many as 99.8% of cervical cancer specimens.15 HPV 16 and 18 are the most oncogenic HPV genotypes and account for 75% of all cases of cervical cancer. Ten other HPV genotypes account for the remaining 25%.16
In 2012, there were an estimated 12,170 new cases of invasive cervical cancer in the United States and 4,220 related deaths.17 The cost associated with cervical cancer screening, managing abnormal findings, and treating invasive cervical cancer in the United States is estimated to be $3.3 billion per year.18
Although the incidence and the mortality rates of cervical cancer have decreased more than 50% in the United States over the past 3 decades thanks to screening,19 cervical cancer remains the second leading cause of death from cancer in women worldwide. Each year, an estimated 500,000 women contract the disease and 240,000 die of it.20
Anal cancer
A recent study indicated that oncogenic HPV can also cause anal cancer, and the proportion of such cancers associated with HPV 16 or HPV 18 infection is as high as or higher than for cervical cancers, and estimated at 80%.21
The incidence of anal cancer is increasing by approximately 2% per year in both men and women in the general population,22 and rates are even higher in men who have sex with men and people infected with the human immunodeficiency virus.23
Hu and Goldie24 estimated that the lifetime costs of caring for all the people in the United States who in just 1 year (2003) acquired anal cancer attributable to HPV would total $92 million.
Oropharyngeal cancer
HPV types 16, 18, 31, 33, and 35 also cause oropharyngeal cancer. HPV 16 accounts for more than 90% of cases of HPV-related oropharyngeal cancer.25
Chaturvedi et al6 tested tissue samples from three national cancer registries and found that the number of oropharyngeal cancers that were HPV-positive increased from 16.3% in 1984–1989 to 71.7% in 2000–2004, while the number of HPV-negative oropharyngeal cancers fell by 50%, paralleling the drop in cigarette smoking in the United States.
Hu and Goldie24 estimated that the total lifetime cost for all new HPV-related oropharyngeal cancers that arose in 2003 would come to $38.1 million.24
Vulvar and vaginal cancers
HPV 16 and 18 are also responsible for approximately 50% of vulvar cancers and 50% to 75% of vaginal cancers.4,5
Recurrent respiratory papillomatosis
HPV 6 and 11 cause almost all cases of juvenile- and adult-onset recurrent respiratory papillomatosis.26 The annual cost for surgical procedures for this condition in the United States has been estimated at $151 million.27
HPV VACCINES ARE NONINFECTIOUS AND NONCARCINOGENIC
Currently, two HPV vaccines are available: a quadrivalent vaccine against types 6, 11, 16, and 18 (Gardasil; Merck) and a bivalent vaccine against types 16 and 18 (Cervarix; Glaxo-SmithKline). The quadrivalent vaccine was approved by the US Food and Drug Administration (FDA) in 2006, and the bivalent vaccine was approved in 2009.28,29
Both vaccines contain virus-like particles, ie, viral capsids that contain no DNA. HPV has a circular DNA genome of 8,000 nucleotides divided into two regions: the early region, for viral replication, and the late region, for viral capsid production. The host produces neutralizing antibodies in response to the L1 capsid protein, which is different in different HPV types.
In manufacturing the vaccines, the viral L1 gene is incorporated into a yeast genome or an insect virus genome using recombinant DNA technology (Figure 1). Grown in culture, the yeast or the insect cells produce the HPV L1 major capsid protein, which has the intrinsic capacity to self-assemble into virus-like particles.30–33 These particles are subsequently purified for use in the vaccines.34
Recombinant virus-like particles are morphologically indistinguishable from authentic HPV virions and contain the same typespecific antigens present in authentic virions. Therefore, they are highly effective in inducing a host humoral immune response. And because they do not contain HPV DNA, the recombinant HPV vaccines are noninfectious and noncarcinogenic.35
VACCINATION INDUCES A STRONGER IMMUNE RESPONSE THAN INFECTION
HPV infections trigger both a humoral and a cellular response in the host immune system.
The humoral immune response to HPV infection involves producing neutralizing antibody against the specific HPV type, specifically the specific L1 major capsid protein. This process is typically somewhat slow and weak, and only about 60% of women with a new HPV infection develop antibodies to it.36,37
HPV has several ways to evade the host immune system. It does not infect or replicate within the antigen-presenting cells in the epithelium. In addition, HPV-infected keratinocytes are less susceptible to cytotoxic lymphocytic-mediated lysis. Moreover, HPV infection cause very little tissue destruction. And finally, natural cervical HPV infection does not result in viremia. As a result, antigen-presenting cells have no chance to engulf the virions and present virion-derived antigen to the host immune system. The immune system outside the epithelium has limited opportunity to detect the virus because HPV infection does not have a blood-borne phase.38,39
The cell-mediated immune response to early HPV oncoproteins may help eliminate established HPV infection.40 In contrast to antibodies, the T-cell response to HPV has not been shown to be specific to HPV type.41 Clinically, cervical HPV infection is common, but most lesions go into remission or resolve as a result of the cell-mediated immune response.40,41
In contrast to the weak, somewhat ineffective immune response to natural HPV infection, the antibody response to HPV vaccines is rather robust. In randomized controlled trials, almost all vaccinated people have seroconverted. The peak antibody concentrations are 50 to 10,000 times greater than in natural infection. Furthermore, the neutralizing antibodies induced by HPV vaccines persist for as long as 7 to 9 years after immunization.42 However, the protection provided by HPV vaccines against HPV-related cervical intraepithelial neoplasia does not necessarily correlate with the antibody concentration.43–47
Why does the vaccine work so well?
Why are vaccine-induced antibody responses so much stronger than those induced by natural HPV infection?
The first reason is that the vaccine, delivered intramuscularly, rapidly enters into blood vessels and the lymphatic system. In contrast, in natural intraepithelial infection, the virus is shed from mucosal surfaces and does not result in viremia.48
In addition, the strong immunogenic nature of the virus-like particles induces a robust host antibody response even in the absence of adjuvant because of concentrated neutralizing epitopes and excellent induction of the T-helper cell response.35,49,50
The neutralizing antibody to L1 prevents HPV infection by blocking HPV from binding to the basement membrane as well as to the epithelial cell receptor during epithelial microabrasion and viral entry. The subsequent micro-wound healing leads to serous exudation and rapid access of serum immunoglobulin G (IgG) to HPV virus particles and encounters with circulatory B memory cells.
Furthermore, emerging evidence suggests that even very low antibody concentrations are sufficient to prevent viral entry into cervical epithelial cells.46–48,51–53
THE HPV VACCINES ARE HIGHLY EFFECTIVE AND SAFE
The efficacy and safety of the quadrivalent and the bivalent HPV vaccines have been evaluated in large randomized clinical trials.23,28,29,54,55 Table 1 summarizes the key findings.
The Females United to Unilaterally Reduce Endo/ectocervical Disease (FUTURE I)54 and FUTURE II28 trials showed conclusively that the quadrivalent HPV vaccine is 98% to 100% efficacious in preventing HPV 16- and 18-related cervical intraepithelial neoplasia, carcinoma in situ, and invasive cervical cancer in women who had not been infected with HPV before. Similarly, the Papilloma Trial against Cancer in Young Adults (PATRICIA) concluded that the bivalent HPV vaccine is 93% efficacious.29
Giuliano et al55 and Palefsky et al23 conducted randomized clinical trials of the quadrivalent HPV vaccine for preventing genital disease and anal intraepithelial neoplasia in boys and men; the efficacy rates were 90.4%55 and 77.5%.23
A recent Finnish trial in boys age 10 to 18 found 100% seroconversion rates for HPV 16 and HPV 18 antibodies after they received bivalent HPV vaccine.56 Similar efficacy has been demonstrated for the quadrivalent HPV vaccine in boys.57
Adverse events after vaccination
After the FDA approved the quadrivalent HPV vaccine for girls in 2006, the US Centers for Disease Control and Prevention (CDC) conducted a thorough survey of adverse events after immunization from June 1, 2006 through December 31, 2008.58 There were about 54 reports of adverse events per 100,000 distributed vaccine doses, similar to rates for other vaccines. However, the incidence rates of syncope and venous thrombosis were disproportionately higher, according to data from the US Vaccine Adverse Event Reporting System. The rate of syncope was 8.2 per 100,000 vaccine doses, and the rate of venous thrombotic events was 0.2 per 100,000 doses.58
There were 32 reports of deaths after HPV vaccination, but these were without clear causation. Hence, this information must be interpreted with caution and should not be used to infer causal associations between HPV vaccines and adverse outcomes. The causes of death included diabetic ketoacidosis, pulmonary embolism, prescription drug abuse, amyotrophic lateral sclerosis, meningoencephalitis, influenza B viral sepsis, arrhythmia, myocarditis, and idiopathic seizure disorder.58
Furthermore, it is important to note that vasovagal syncope and venous thromboembolic events are more common in young females in general.59 For example, the background rates of venous thromboembolism in females age 14 to 29 using oral contraceptives is 21 to 31 per 100,000 woman-years.60
Overall, the quadrivalent HPV vaccine is well tolerated and clinically safe. Postlicensure evaluation found that the quadrivalent and bivalent HPV vaccines had similar safety profiles.61
Vaccination is contraindicated in people with known hypersensitivity or prior severe allergic reactions to vaccine or yeast or who have bleeding disorders.
HPV VACCINATION DOES MORE THAN PREVENT CERVICAL CANCER IN FEMALES
The quadrivalent HPV vaccine was licensed by the FDA in 2006 for use in females age 9 to 26 to prevent cervical cancer, cervical cancer precursors, vaginal and vulval cancer precursors, and anogenital warts caused by HPV types 6, 11, 16, and 18. The CDC’s Advisory Committee on Immunization Practices (ACIP) issued its recommendation for initiating HPV vaccination for females age 11 to 12 in March 2007. The ACIP stated that the vaccine could be given to girls as early as age 9 and recommended catch-up vaccinations for those age 13 to 26.62,63
The quadrivalent HPV vaccine was licensed by the FDA in 2009 for use in boys and men for the prevention of genital warts. In December 2010, the quadrivalent HPV vaccine received extended licensure from the FDA for use in males and females for the prevention of anal cancer. In October 2011, the ACIP voted to recommend routine use of the quadrivalent HPV vaccine for boys age 11 to 12; catch-up vaccination should occur for those age 13 to 22, with an option to vaccinate men age 23 to 26.
These recommendations replace the “permissive use” recommendations from the ACIP in October 2009 that said the quadrivalent HPV vaccine may be given to males age 9 to 26.64 This shift from a permissive to an active recommendation connotes a positive change reflecting recognition of rising oropharyngeal cancer rates attributable to oncogenic, preventable HPV, rising HPV-related anal cancer incidence, and the burden of the disease in female partners of infected men, with associated rising health care costs.
The bivalent HPV vaccine received FDA licensure in October 2009 for use in females age 10 to 25 to prevent cervical cancer and precursor lesions. The ACIP included the bivalent HPV vaccine in its updated recommendations in May 2010 for use in girls age 11 to 12. Numerous national and international organizations have endorsed HPV vaccination.65–71
Table 2 outlines the recommendations from these organizations.
HPV VACCINATION RATES ARE STILL LOW
HPV vaccine offers us the hope of eventually eradicating cervical cancer. However, the immunization program still faces many challenges, since HPV vaccination touches on issues related to adolescent sexuality, parental autonomy, and cost. As a result, HPV immunization rates remain relatively low in the United States according to several national surveys. Only 40% to 49% of girls eligible for the vaccine received even one dose, and of those who received even one dose, only 32% to 53.3% came back for all three doses.72–75 Furthermore, indigent and minority teens were less likely to finish the three-dose HPV vaccine series.
Why are the vaccination rates so low?
Parental barriers. In one survey,73 reasons that parents gave for not having their daughters vaccinated included:
- Lack of knowledge of the vaccine (19.4%)
- Lack of perceived need for the vaccine (18.8%)
- Belief that their daughter was not sexually active (18.3%)
- Clinician not recommending vaccination (13.1%).
In an effort to improve HPV vaccination rates,41 several states proposed legislation for mandatory HPV vaccination of schoolgirls shortly after licensure of the quadrivalent HPV vaccine.3 Since then, we have seen a wave of public opposition rooted in concerns and misinformation about safety, teenage sexuality, governmental coercion, and cost. Widespread media coverage has also highlighted unsubstantiated claims about side effects attributable to the vaccine that can raise parents’ mistrust of vaccines.76 Concerns have also been raised about a threat to parental autonomy in how and when to educate their children about sex.77
Moreover, the vaccine has raised ethical concerns in some parents and politicians that mandatory vaccination could undermine abstinence messages in sexual education and may alter sexual activity by condoning risky behavior.78 However, a recent study indicated that there is no significant change in sexual behavior related to HPV vaccination in young girls.79
In 2012, Mullins et al80 also found that an urban population of adolescent girls (76.4% black, 57.5% sexually experienced) did not feel they could forgo safer sexual practices after first HPV vaccination, although the girls did perceive less risk from HPV than from other sexually transmitted infections after HPV vaccination (P < .001).80 Inadequate knowledge about HPV-related disease and HPV vaccine correlated with less perceived risk from HPV after vaccination among the girls, and a lack of knowledge about HPV and less communication with their daughters about HPV correlated with less perceived risk from HPV in the mothers of the study population.81
Health-care-provider barriers. Physician endorsement of vaccines represents a key predictor of vaccine acceptance by patients, families, and other clinicians.82–84 In 2008, a cross-sectional, Internet-based survey of 1,122 Texas pediatricians, family practice physicians, obstetricians, gynecologists, and internal medicine physicians providing direct patient care found that only 48.5% always recommended HPV vaccination to girls.74 Of all respondents, 68.4% were likely to recommend the vaccine to boys, and 41.7% agreed with mandated vaccination. Thus, more than half of the physicians were not following the current recommendations for universal HPV vaccination for 11- to -12-year-olds.
In a survey of 1,013 physicians during the spring and summer of 2009, only 34.6% said they always recommend HPV vaccination to early adolescents, 52.7% to middle adolescents, and 50.2% to late adolescents and young adults.85 Pediatricians were more likely than family physicians and obstetrician-gynecologists to always recommend HPV vaccine across all age groups (P < .001). Educational interventions targeting various specialties may help overcome physician-related barriers to immunization.85
Financial barriers. HPV vaccine, which must be given in three doses, is more expensive than other vaccines, and this expense is yet another barrier, especially for the uninsured.86 Australia launched a government-funded program of HPV vaccination (with the quadrivalent vaccine) in schools in 2007, and it has been very successful. Garland et al87 reported that new cases of genital warts have decreased by 73% since the program began, and the rate of high-grade abnormalities on Papanicolaou testing has declined by a small but significant amount.
For HPV vaccination to have an impact on public health, vaccination rates in the general population need to be high. In order to achieve these rates, we need to educate our patients on vaccine safety and efficacy and counsel vaccine recipients about the prevention of sexually transmitted infections and the importance of regular cervical cancer screening after age 21. Clinicians can actively “myth-bust” with patients, who may not realize that the vaccine should be given despite a history of HPV infection or abnormal Pap smear.
FREQUENTLY ASKED QUESTIONS
What if the patient is late for a shot?
The current recommended vaccination schedule for the bivalent and quadrivalent HPV vaccines is a three-dose series administered at 0, 2, and 6 months, given as an intramuscular injection, preferably in the deltoid muscle. The minimal dosing interval is 4 weeks between the first and second doses and 12 weeks between the second and third doses.
The vaccines use different adjuncts with different specific mechanisms for immunogenicity; therefore, it is recommended that the same vaccine be used for the entire three-dose series. However, if circumstances preclude the completion of a series with the same vaccine, the other HPV vaccine may be used.63 Starting the series over is not recommended.
Long-term studies demonstrated clinical efficacy 8.5 years after vaccination.47 Amnestic response by virtue of activation of pools of memory B cells has been demonstrated, suggesting the vaccine may afford lifelong immunity.88
Is a pregnancy test needed before HPV vaccination?
The ACIP states that pregnancy testing is not required before receiving either of the available HPV vaccines.
A recent retrospective review of phase III efficacy trials and pregnancy registry surveillance data for both vaccines revealed no increase in spontaneous abortions, fetal malformations, or adverse pregnancy outcomes.89 Data are limited on bivalent and quadrivalent HPV vaccine given within 30 days of pregnancy and subsequent pregnancy and fetal outcomes. Both vaccines have been assigned a pregnancy rating of category B; however, the ACIP recommends that neither vaccine be given if the recipient is known to be pregnant. If pregnancy occurs, it is recommended that the remainder of the series be deferred until after delivery.62
It is not known whether the vaccine is excreted in breast milk. The manufacturers of both the bivalent and quadrivalent HPV vaccines recommend caution when vaccinating lactating women.30,31
Can HPV vaccine be given with other vaccines?
In randomized trials, giving the bivalent HPV vaccine with the combined hepatitis A, hepatitis B, meningococcal conjugate and the combined tetanus, diphtheria, and acellular pertussis vaccines did not interfere with the immunogenic response, was safe, and was well tolerated.90,91 Coadministration of the quadrivalent HPV vaccine has been studied only with hepatitis B vaccine, with similar safety and efficacy noted.
The ACIP recommends giving HPV vaccine at the same visit with other age-appropriate immunizations to increase the likelihood of adherence to recommended vaccination schedules.62
Is HPV vaccination cost-effective?
Kim and Goldie86 performed a cost-effectiveness analysis of HPV vaccination of girls at age 12 and catch-up vaccination up to the ages of 18, 21, and 26. For their analysis, they considered prevention of cancers associated with HPV types 16 and 18, of genital warts associated with types 6 and 11, and of recurrent respiratory papillomatosis. They also assumed that immunity would be lifelong, and current screening practices would continue.
They calculated that routine vaccination of 12-year-old girls resulted in an incremental cost-effective ratio of $34,900 per quality-adjusted life-year (QALY) gained. A threshold of less than $50,000 per QALY gained is considered reasonably cost-effective, with an upper limit of $100,000 considered acceptable.92
In the same analysis by Kim and Goldie,86 catch-up vaccination of girls through age 18 resulted in a cost of $50,000 to $100,000 per QALY gained, and catch-up vaccination of females through age 26 was significantly less cost-effective at more then $130,000 per QALY gained. The vaccine was also significantly less cost-effective if 5% of the population was neither screened nor vaccinated, if a 10-year booster was required, and if frequent cervical cancer screening intervals were adopted.
This analysis did not include costs related to the evaluation and treatment of abnormal Pap smears and cross-protection against other HPV-related cancers.
The cost-effectiveness of HPV vaccination depends on reaching more girls at younger ages (ideally before sexual debut) and completing the three-dose schedule to optimize duration of immunity.92 Appropriate modification of the current recommendations for the intervals of cervical cancer screening for vaccinated individuals will further improve the cost-effectiveness of vaccination. The inclusion of male vaccination generally has more favorable cost per QALY in scenarios in which female coverage rates are less than 50%93 and among men who have sex with men.94
TO ERADICATE CERVICAL CANCER
Given the remarkable efficacy and expected long-term immunogenicity of HPV vaccines, we anticipate a decline in HPV-related cervical cancer and other related diseases in the years to come. However, modeling studies predicting the impact of HPV vaccination suggest that although substantial reductions in diseases can be expected, the benefit, assuming high vaccination rates, will not be apparent for at least another decade.95 Furthermore, the current HPV vaccines contain only HPV 16 and 18 L1 protein for cancer protection and, therefore, do not provide optimal protection against all oncogenic HPV-related cancers.
The real hope of eradicating cervical cancer and all HPV-related disease relies on a successful global implementation of multivalent HPV vaccination, effective screening strategies, and successful treatment.
- Baden LR, Curfman GD, Morrissey S, Drazen JM. Human papillomavirus vaccine—opportunity and challenge. N Engl J Med 2007; 356:1990–1991.
- Schiffman M, Wacholder S. From India to the world—a better way to prevent cervical cancer. N Engl J Med 2009; 360:1453–1455.
- Colgrove J, Abiola S, Mello MM. HPV vaccination mandates—law-making amid political and scientific controversy. N Engl J Med 2010; 363:785–791.
- World Health Organization. WHO/ICO Information Centre on Human Papilloma Virus (HPV) and Cervical Cancer. 2010Summary Report. www.who.int/hpvcentre. Accessed November 12, 2012.
- Muñoz N, Bosch FX, de Sanjosé S, et al; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348:518–527.
- Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011; 29:4294–4301.
- Jansen KU, Shaw AR. Human papillomavirus vaccines and prevention of cervical cancer. Annu Rev Med 2004; 55:319–331.
- Fleischer AB, Parrish CA, Glenn R, Feldman SR. Condylomata acuminata (genital warts): patient demographics and treating physicians. Sex Transm Dis 2001; 28:643–647.
- Clifford GM, Rana RK, Franceschi S, Smith JS, Gough G, Pimenta JM. Human papillomavirus genotype distribution in low-grade cervical lesions: comparison by geographic region and with cervical cancer. Cancer Epidemiol Biomarkers Prev 2005; 14:1157–1164.
- Schiffman M, Solomon D. Findings to date from the ASCUS-LSIL Triage Study (ALTS). Arch Pathol Lab Med 2003; 127:946–949.
- Insinga RP, Dasbach EJ, Myers ER. The health and economic burden of genital warts in a set of private health plans in the United States. Clin Infect Dis 2003; 36:1397–1403.
- Kodner CM, Nasraty S. Management of genital warts. Am Fam Physician 2004; 70:2335–2342.
- Maw RD, Reitano M, Roy M. An international survey of patients with genital warts: perceptions regarding treatment and impact on lifestyle. Int J STD AIDS 1998; 9:571–578.
- Insinga RP, Dasbach EJ, Elbasha EH. Assessing the annual economic burden of preventing and treating anogenital human papillomavirus-related disease in the US: analytic framework and review of the literature. Pharmacoeconomics 2005; 23:1107–1122.
- Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
- de Sanjose S, Quint WG, Alemany L, et al; Retrospective International Survey and HPV Time Trends Study Group. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol 2010; 11:1048–1056.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Insinga RP, Glass AG, Rush BB. The health care costs of cervical human papillomavirus—related disease. Am J Obstet Gynecol 2004; 191:114–120.
- Horner MJ, Ries LAG, Krapcho M, et al. SEER Cancer Statistics Review,1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER Web site, 2009. Accessed November 12, 2012.
- Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine 2006; 24(suppl 3):S3/11–S3/25.
- Hoots BE, Palefsky JM, Pimenta JM, Smith JS. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int J Cancer 2009; 124:2375–2383.
- Johnson LG, Madeleine MM, Newcomer LM, Schwartz SM, Daling JR. Anal cancer incidence and survival: the surveillance, epidemiology, and end results experience, 1973–2000. Cancer 2004; 101:281–288.
- Palefsky JM, Giuliano AR, Goldstone S, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med 2011; 365:1576–1585.
- Hu D, Goldie S. The economic burden of noncervical human papillomavirus disease in the United States. Am J Obstet Gynecol 2008; 198:500.e1–500.e7.
- Herrero R, Castellsagué X, Pawlita M, et al; IARC Multicenter Oral Cancer Study Group. Human papillomavirus and oral cancer: the International Agency for Research on Cancer multicenter study. J Natl Cancer Inst 2003; 95:1772–1783.
- Lacey CJ, Lowndes CM, Shah KV. Chapter 4: Burden and management of non-cancerous HPV-related conditions: HPV-6/11 disease. Vaccine 2006; 24(suppl 3):S3/35–S3/41.
- Derkay CS. Task force on recurrent respiratory papillomas. A preliminary report. Arch Otolaryngol Head Neck Surg 1995; 121:1386–1391.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
- Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Patient information about Gardasil (human papillomavirus quadrivalent type 6,11,16 and 18 vaccine, recombinant. http://www.gardasil.com/. Accessed November 12, 2012.
- GlaxoSmithKline Biologicals, Rixensart, Belgium. Highlights of prescribing information. Cervarix (human papillomavirus bivalent type 16 and 18 vaccine, recombinant. http://us.gsk.com/products/assets/us_cervarix.pdf. Accessed November 12, 2012.
- Zhou J, Sun XY, Stenzel DJ, Frazer IH. Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology 1991; 185:251–257.
- Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci USA 1992; 89:12180–12184.
- Schiller JT, Lowy DR. Papillomavirus-like particles and HPV vaccine development. Semin Cancer Biol 1996; 7:373–382.
- Harro CD, Pang YY, Roden RB, et al. Safety and immunogenicity trial in adult volunteers of a human papillomavirus 16 L1 virus-like particle vaccine. J Natl Cancer Inst 2001; 93:284–292.
- af Geijersstam V, Kibur M, Wang Z, et al. Stability over time of serum antibody levels to human papillomavirus type 16. J Infect Dis 1998; 177:1710–1714.
- Safaeian M, Porras C, Schiffman M, et al; Costa Rican Vaccine Trial Group. Epidemiological study of anti-HPV16/18 seropositivity and subsequent risk of HPV16 and -18 infections. J Natl Cancer Inst 2010; 102:1653–1662.
- Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer 2002; 2:59–65.
- Scott M, Nakagawa M, Moscicki AB. Cell-mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol 2001; 8:209–220.
- Roden R, Wu TC. Preventative and therapeutic vaccines for cervical cancer. Expert Rev Vaccines 2003; 2:495–516.
- Wang SS, Hildesheim A. Chapter 5: Viral and host factors in human papillomavirus persistence and progression. J Natl Cancer Inst Monogr 2003; 31:35–40.
- De Carvalho N, Teixeira J, Roteli-Martins CM, et al. Sustained efficacy and immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine up to 7.3 years in young adult women. Vaccine 2010; 28:6247–6255.
- Harper DM, Franco EL, Wheeler CM, et al; HPV Vaccine Study group. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet 2006; 367:1247–1255.
- Villa LL, Costa RL, Petta CA, et al. High sustained efficacy of a prophylactic quadrivalent human papillomavirus types 6/11/16/18 L1 virus-like particle vaccine through 5 years of follow-up. Br J Cancer 2006; 95:1459–1466.
- Villa LL, Ault KA, Giuliano AR, et al. Immunologic responses following administration of a vaccine targeting human papillomavirus Types 6, 11, 16, and 18. Vaccine 2006; 24:5571–5583.
- Smith JF, Brownlow M, Brown M, et al. Antibodies from women immunized with Gardasil cross-neutralize HPV 45 pseudovirions. Hum Vaccin 2007; 3:109–115.
- Rowhani-Rahbar A, Mao C, Hughes JP, et al. Longer term efficacy of a prophylactic monovalent human papillomavirus type 16 vaccine. Vaccine 2009; 27:5612–5619.
- Stanley M. HPV - immune response to infection and vaccination. Infect Agent Cancer 2010; 5:19.
- Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol 2010; 117(suppl 2):S5–S10.
- Yan M, Peng J, Jabbar IA, et al. Activation of dendritic cells by human papillomavirus-like particles through TLR4 and NF-kappaB-mediated signalling, moderated by TGF-beta. Immunol Cell Biol 2005; 83:83–91.
- Roberts JN, Buck CB, Thompson CD, et al. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 2007; 13:857–861.
- Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci USA 2009; 106:20458–20463.
- Day PM, Kines RC, Thompson CD, et al. In vivo mechanisms of vaccine-induced protection against HPV infection. Cell Host Microbe 2010; 8:260–270.
- Garland SM, Hernandez-Avila M, Wheeler CM, et al; Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007; 356:1928–1943.
- Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV Infection and disease in males. N Engl J Med 2011; 364:401–411.
- Petäjä T, Keränen H, Karppa T, et al. Immunogenicity and safety of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine in healthy boys aged 10–18 years. J Adolesc Health 2009; 44:33–40.
- Reisinger KS, Block SL, Lazcano-Ponce E, et al. Safety and persistent immunogenicity of a quadrivalent human papillomavirus types 6, 11, 16, 18 L1 virus-like particle vaccine in preadolescents and adolescents: a randomized controlled trial. Pediatr Infect Dis J 2007; 26:201–209.
- Slade BA, Leidel L, Vellozzi C, et al. Postlicensure safety surveillance for quadrivalent human papillomavirus recombinant vaccine. JAMA 2009; 302:750–757.
- Block SL, Brown DR, Chatterjee A, et al. Clinical trial and post-licensure safety profile of a prophylactic human papillomavirus (types 6, 11, 16, and 18) l1 virus-like particle vaccine. Pediatr Infect Dis J 2010; 29:95–101.
- Farmer RD, Lawrenson RA, Thompson CR, Kennedy JG, Hambleton IR. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet 1997; 349:83–88.
- Labadie J. Postlicensure safety evaluation of human papilloma virus vaccines. Int J Risk Saf Med 2011; 23:103–112.
- Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER; Centers for Disease Control and Prevention (CDC). Quadrivalent human papillomavirus vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2007; 56:1–24.
- Centers for Disease Control and Prevention (CDC). FDA licensure of bivalent human papillomavirus vaccine (HPV2, Cervarix) for use in females and updated HPV vaccination recommendations from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2010; 59:626–629.
- Centers for Disease Control and Prevention (CDC). FDA licensure of quadrivalent human papillomavirus vaccine (HPV4, Gardasil) for use in males and guidance from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2010; 59:630–632.
- World Health Organization (WHO). Weekly Epidemiological Record (WER). January 2009; 84:1–16. http://www.who.int/wer/2009/wer8401_02/en/index.html. Accessed November 12, 2012.
- Saslow D, Castle PE, Cox JT, et al. American Cancer Society guideline for human papillomavirus (HPV) vaccine use to prevent cervical cancer and its precursors. CA Cancer J Clin 2007; 57:7–28.
- Committee opinion no. 467: human papillomavirus vaccination. Obstet Gynecol 2010; 116:800–803.
- American College of Physicians. ACP Guide to Adult Immunization. 4th ed. 2011:58–60. http://immunization.acponline.org/. Accessed November 12, 2012.
- Vaughn JA, Miller RA. Update on immunizations in adults. Am Fam Physician 2011; 84:1015–1020.
- American Academy of Pediatrics Committee on Infectious Diseases. Prevention of human papillomavirus infection: provisional recommendations for immunization of girls and women with quadrivalent human papillomavirus vaccine. Pediatrics 2007; 120:666–668.
- Friedman L, Bell DL, Kahn JA, et al. Human papillomavirus vaccine: an updated position statement of the Society for Adolescent Health and Medicine. J Adolesc Health 2011; 48:215–216.
- Centers for Disease Control and Prevention (CDC). National and state vaccination coverage among adolescents aged 13 through 17 years--United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:1117–1123.
- Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
- Kahn JA, Cooper HP, Vadaparampil ST, et al. Human papillomavirus vaccine recommendations and agreement with mandated human papillomavirus vaccination for 11-to-12-year-old girls: a statewide survey of Texas physicians. Cancer Epidemiol Biomarkers Prev 2009; 18:2325–2332.
- Schwartz JL, Caplan AL, Faden RR, Sugarman J. Lessons from the failure of human papillomavirus vaccine state requirements. Clin Pharmacol Ther 2007; 82:760–763.
- Cooper LZ, Larson HJ, Katz SL. Protecting public trust in immunization. Pediatrics 2008; 122:149–153.
- Olshen E, Woods ER, Austin SB, Luskin M, Bauchner H. Parental acceptance of the human papillomavirus vaccine. J Adolesc Health 2005; 37:248–251.
- Zimmerman RK. Ethical analysis of HPV vaccine policy options. Vaccine 2006; 24:4812–4820.
- Al Romaih WRR, Srinivas A, Shahtahmasebi S, Omar HA. No significant change in sexual behavior in association with human papillomavirus vaccination in young girls. Int J Child Adolesc Health 2011; 4:1–5.
- Mullins TL, Zimet GD, Rosenthal SL, et al. Adolescent perceptions of risk and need for safer sexual behaviors after first human papillomavirus vaccination. Arch Pediatr Adolesc Med 2012; 166:82–88.
- Middleman AB, Tung JS. School-located immunization programs: do parental p predict behavior? Vaccine 2011; 29:3513–3516.
- Samoff E, Dunn A, VanDevanter N, Blank S, Weisfuse IB. Predictors of acceptance of hepatitis B vaccination in an urban sexually transmitted diseases clinic. Sex Transm Dis 2004; 31:415–420.
- Gnanasekaran SK, Finkelstein JA, Hohman K, O’Brien M, Kruskal B, Lieu T. Parental perspectives on influenza vaccination among children with asthma. Public Health Rep 2006; 121:181–188.
- Daley MF, Crane LA, Chandramouli V, et al. Influenza among healthy young children: changes in parental attitudes and predictors of immunization during the 2003 to 2004 influenza season. Pediatrics 2006; 117:e268–e277.
- Vadaparampil ST, Kahn JA, Salmon D, et al. Missed clinical opportunities: provider recommendations for HPV vaccination for 11–12 year old girls are limited. Vaccine 2011; 29:8634–8641.
- Kim JJ, Goldie SJ. Health and economic implications of HPV vaccination in the United States. N Engl J Med 2008; 359:821–832.
- Garland SM, Skinner SR, Brotherton JM. Adolescent and young adult HPV vaccination in Australia: achievements and challenges. Prev Med 2011; 53(suppl 1):S29–S35.
- Rowhani-Rahbar A, Alvarez FB, Bryan JT, et al. Evidence of immune memory 8.5 years following administration of a prophylactic human papillomavirus type 16 vaccine. J Clin Virol 2012; 53:239–243.
- Forinash AB, Yancey AM, Pitlick JM, Myles TD. Safety of the HPV bivalent and quadrivalent vaccines during pregnancy (February) Ann Pharmacother 2011; [epub ahead of print]
- Wheeler CM, Harvey BM, Pichichero ME, et al. Immunogenicity and safety of human papillomavirus-16/18 AS04-adjuvanted vaccine coadministered with tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine and/or meningococcal conjugate vaccine to healthy girls 11 to 18 years of age: results from a randomized open trial. Pediatr Infect Dis J 2011; 30:e225–e234.
- Pedersen C, Breindahl M, Aggarwal N, et al. Randomized trial: immunogenicity and safety of coadministered human papillomavirus-16/18 AS04-adjuvanted vaccine and combined hepatitis A and B vaccine in girls. J Adolesc Health 2012; 50:38–46.
- Eichler HG, Kong SX, Gerth WC, Mavros P, Jönsson B. Use of costeffectiveness analysis in health-care resource allocation decisionmaking: how are cost-effectiveness thresholds expected to emerge? Value Health 2004; 7:518–528.
- Chesson HW. HPV vaccine cost-effectiveness: updates and review. Presentation before the Advisory Committee on Immunization Practices (ACIP), June 22, 2011. Atlanta, GA: US Department of Health and Human Services, CDC; 2011. http://www.cdc.gov/vaccines/recs/acip/downloads/mtg-slides-jun11/07-5-hpv-cost-effect.pdf. Accessed August 31, 2012.
- Kim JJ. Targeted human papillomavirus vaccination of men who have sex with men in the USA: a cost-effectiveness modelling analysis. Lancet Infect Dis 2010; 10:845–852.
- Cuzick J, Castañón A, Sasieni P. Predicted impact of vaccination against human papillomavirus 16/18 on cancer incidence and cervical abnormalities in women aged 20–29 in the UK. Br J Cancer 2010; 102:933–939.
- Baden LR, Curfman GD, Morrissey S, Drazen JM. Human papillomavirus vaccine—opportunity and challenge. N Engl J Med 2007; 356:1990–1991.
- Schiffman M, Wacholder S. From India to the world—a better way to prevent cervical cancer. N Engl J Med 2009; 360:1453–1455.
- Colgrove J, Abiola S, Mello MM. HPV vaccination mandates—law-making amid political and scientific controversy. N Engl J Med 2010; 363:785–791.
- World Health Organization. WHO/ICO Information Centre on Human Papilloma Virus (HPV) and Cervical Cancer. 2010Summary Report. www.who.int/hpvcentre. Accessed November 12, 2012.
- Muñoz N, Bosch FX, de Sanjosé S, et al; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348:518–527.
- Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011; 29:4294–4301.
- Jansen KU, Shaw AR. Human papillomavirus vaccines and prevention of cervical cancer. Annu Rev Med 2004; 55:319–331.
- Fleischer AB, Parrish CA, Glenn R, Feldman SR. Condylomata acuminata (genital warts): patient demographics and treating physicians. Sex Transm Dis 2001; 28:643–647.
- Clifford GM, Rana RK, Franceschi S, Smith JS, Gough G, Pimenta JM. Human papillomavirus genotype distribution in low-grade cervical lesions: comparison by geographic region and with cervical cancer. Cancer Epidemiol Biomarkers Prev 2005; 14:1157–1164.
- Schiffman M, Solomon D. Findings to date from the ASCUS-LSIL Triage Study (ALTS). Arch Pathol Lab Med 2003; 127:946–949.
- Insinga RP, Dasbach EJ, Myers ER. The health and economic burden of genital warts in a set of private health plans in the United States. Clin Infect Dis 2003; 36:1397–1403.
- Kodner CM, Nasraty S. Management of genital warts. Am Fam Physician 2004; 70:2335–2342.
- Maw RD, Reitano M, Roy M. An international survey of patients with genital warts: perceptions regarding treatment and impact on lifestyle. Int J STD AIDS 1998; 9:571–578.
- Insinga RP, Dasbach EJ, Elbasha EH. Assessing the annual economic burden of preventing and treating anogenital human papillomavirus-related disease in the US: analytic framework and review of the literature. Pharmacoeconomics 2005; 23:1107–1122.
- Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
- de Sanjose S, Quint WG, Alemany L, et al; Retrospective International Survey and HPV Time Trends Study Group. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol 2010; 11:1048–1056.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Insinga RP, Glass AG, Rush BB. The health care costs of cervical human papillomavirus—related disease. Am J Obstet Gynecol 2004; 191:114–120.
- Horner MJ, Ries LAG, Krapcho M, et al. SEER Cancer Statistics Review,1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER Web site, 2009. Accessed November 12, 2012.
- Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine 2006; 24(suppl 3):S3/11–S3/25.
- Hoots BE, Palefsky JM, Pimenta JM, Smith JS. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int J Cancer 2009; 124:2375–2383.
- Johnson LG, Madeleine MM, Newcomer LM, Schwartz SM, Daling JR. Anal cancer incidence and survival: the surveillance, epidemiology, and end results experience, 1973–2000. Cancer 2004; 101:281–288.
- Palefsky JM, Giuliano AR, Goldstone S, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med 2011; 365:1576–1585.
- Hu D, Goldie S. The economic burden of noncervical human papillomavirus disease in the United States. Am J Obstet Gynecol 2008; 198:500.e1–500.e7.
- Herrero R, Castellsagué X, Pawlita M, et al; IARC Multicenter Oral Cancer Study Group. Human papillomavirus and oral cancer: the International Agency for Research on Cancer multicenter study. J Natl Cancer Inst 2003; 95:1772–1783.
- Lacey CJ, Lowndes CM, Shah KV. Chapter 4: Burden and management of non-cancerous HPV-related conditions: HPV-6/11 disease. Vaccine 2006; 24(suppl 3):S3/35–S3/41.
- Derkay CS. Task force on recurrent respiratory papillomas. A preliminary report. Arch Otolaryngol Head Neck Surg 1995; 121:1386–1391.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
- Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Patient information about Gardasil (human papillomavirus quadrivalent type 6,11,16 and 18 vaccine, recombinant. http://www.gardasil.com/. Accessed November 12, 2012.
- GlaxoSmithKline Biologicals, Rixensart, Belgium. Highlights of prescribing information. Cervarix (human papillomavirus bivalent type 16 and 18 vaccine, recombinant. http://us.gsk.com/products/assets/us_cervarix.pdf. Accessed November 12, 2012.
- Zhou J, Sun XY, Stenzel DJ, Frazer IH. Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology 1991; 185:251–257.
- Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci USA 1992; 89:12180–12184.
- Schiller JT, Lowy DR. Papillomavirus-like particles and HPV vaccine development. Semin Cancer Biol 1996; 7:373–382.
- Harro CD, Pang YY, Roden RB, et al. Safety and immunogenicity trial in adult volunteers of a human papillomavirus 16 L1 virus-like particle vaccine. J Natl Cancer Inst 2001; 93:284–292.
- af Geijersstam V, Kibur M, Wang Z, et al. Stability over time of serum antibody levels to human papillomavirus type 16. J Infect Dis 1998; 177:1710–1714.
- Safaeian M, Porras C, Schiffman M, et al; Costa Rican Vaccine Trial Group. Epidemiological study of anti-HPV16/18 seropositivity and subsequent risk of HPV16 and -18 infections. J Natl Cancer Inst 2010; 102:1653–1662.
- Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer 2002; 2:59–65.
- Scott M, Nakagawa M, Moscicki AB. Cell-mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol 2001; 8:209–220.
- Roden R, Wu TC. Preventative and therapeutic vaccines for cervical cancer. Expert Rev Vaccines 2003; 2:495–516.
- Wang SS, Hildesheim A. Chapter 5: Viral and host factors in human papillomavirus persistence and progression. J Natl Cancer Inst Monogr 2003; 31:35–40.
- De Carvalho N, Teixeira J, Roteli-Martins CM, et al. Sustained efficacy and immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine up to 7.3 years in young adult women. Vaccine 2010; 28:6247–6255.
- Harper DM, Franco EL, Wheeler CM, et al; HPV Vaccine Study group. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet 2006; 367:1247–1255.
- Villa LL, Costa RL, Petta CA, et al. High sustained efficacy of a prophylactic quadrivalent human papillomavirus types 6/11/16/18 L1 virus-like particle vaccine through 5 years of follow-up. Br J Cancer 2006; 95:1459–1466.
- Villa LL, Ault KA, Giuliano AR, et al. Immunologic responses following administration of a vaccine targeting human papillomavirus Types 6, 11, 16, and 18. Vaccine 2006; 24:5571–5583.
- Smith JF, Brownlow M, Brown M, et al. Antibodies from women immunized with Gardasil cross-neutralize HPV 45 pseudovirions. Hum Vaccin 2007; 3:109–115.
- Rowhani-Rahbar A, Mao C, Hughes JP, et al. Longer term efficacy of a prophylactic monovalent human papillomavirus type 16 vaccine. Vaccine 2009; 27:5612–5619.
- Stanley M. HPV - immune response to infection and vaccination. Infect Agent Cancer 2010; 5:19.
- Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol 2010; 117(suppl 2):S5–S10.
- Yan M, Peng J, Jabbar IA, et al. Activation of dendritic cells by human papillomavirus-like particles through TLR4 and NF-kappaB-mediated signalling, moderated by TGF-beta. Immunol Cell Biol 2005; 83:83–91.
- Roberts JN, Buck CB, Thompson CD, et al. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 2007; 13:857–861.
- Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci USA 2009; 106:20458–20463.
- Day PM, Kines RC, Thompson CD, et al. In vivo mechanisms of vaccine-induced protection against HPV infection. Cell Host Microbe 2010; 8:260–270.
- Garland SM, Hernandez-Avila M, Wheeler CM, et al; Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007; 356:1928–1943.
- Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV Infection and disease in males. N Engl J Med 2011; 364:401–411.
- Petäjä T, Keränen H, Karppa T, et al. Immunogenicity and safety of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine in healthy boys aged 10–18 years. J Adolesc Health 2009; 44:33–40.
- Reisinger KS, Block SL, Lazcano-Ponce E, et al. Safety and persistent immunogenicity of a quadrivalent human papillomavirus types 6, 11, 16, 18 L1 virus-like particle vaccine in preadolescents and adolescents: a randomized controlled trial. Pediatr Infect Dis J 2007; 26:201–209.
- Slade BA, Leidel L, Vellozzi C, et al. Postlicensure safety surveillance for quadrivalent human papillomavirus recombinant vaccine. JAMA 2009; 302:750–757.
- Block SL, Brown DR, Chatterjee A, et al. Clinical trial and post-licensure safety profile of a prophylactic human papillomavirus (types 6, 11, 16, and 18) l1 virus-like particle vaccine. Pediatr Infect Dis J 2010; 29:95–101.
- Farmer RD, Lawrenson RA, Thompson CR, Kennedy JG, Hambleton IR. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet 1997; 349:83–88.
- Labadie J. Postlicensure safety evaluation of human papilloma virus vaccines. Int J Risk Saf Med 2011; 23:103–112.
- Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER; Centers for Disease Control and Prevention (CDC). Quadrivalent human papillomavirus vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2007; 56:1–24.
- Centers for Disease Control and Prevention (CDC). FDA licensure of bivalent human papillomavirus vaccine (HPV2, Cervarix) for use in females and updated HPV vaccination recommendations from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2010; 59:626–629.
- Centers for Disease Control and Prevention (CDC). FDA licensure of quadrivalent human papillomavirus vaccine (HPV4, Gardasil) for use in males and guidance from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2010; 59:630–632.
- World Health Organization (WHO). Weekly Epidemiological Record (WER). January 2009; 84:1–16. http://www.who.int/wer/2009/wer8401_02/en/index.html. Accessed November 12, 2012.
- Saslow D, Castle PE, Cox JT, et al. American Cancer Society guideline for human papillomavirus (HPV) vaccine use to prevent cervical cancer and its precursors. CA Cancer J Clin 2007; 57:7–28.
- Committee opinion no. 467: human papillomavirus vaccination. Obstet Gynecol 2010; 116:800–803.
- American College of Physicians. ACP Guide to Adult Immunization. 4th ed. 2011:58–60. http://immunization.acponline.org/. Accessed November 12, 2012.
- Vaughn JA, Miller RA. Update on immunizations in adults. Am Fam Physician 2011; 84:1015–1020.
- American Academy of Pediatrics Committee on Infectious Diseases. Prevention of human papillomavirus infection: provisional recommendations for immunization of girls and women with quadrivalent human papillomavirus vaccine. Pediatrics 2007; 120:666–668.
- Friedman L, Bell DL, Kahn JA, et al. Human papillomavirus vaccine: an updated position statement of the Society for Adolescent Health and Medicine. J Adolesc Health 2011; 48:215–216.
- Centers for Disease Control and Prevention (CDC). National and state vaccination coverage among adolescents aged 13 through 17 years--United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:1117–1123.
- Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
- Kahn JA, Cooper HP, Vadaparampil ST, et al. Human papillomavirus vaccine recommendations and agreement with mandated human papillomavirus vaccination for 11-to-12-year-old girls: a statewide survey of Texas physicians. Cancer Epidemiol Biomarkers Prev 2009; 18:2325–2332.
- Schwartz JL, Caplan AL, Faden RR, Sugarman J. Lessons from the failure of human papillomavirus vaccine state requirements. Clin Pharmacol Ther 2007; 82:760–763.
- Cooper LZ, Larson HJ, Katz SL. Protecting public trust in immunization. Pediatrics 2008; 122:149–153.
- Olshen E, Woods ER, Austin SB, Luskin M, Bauchner H. Parental acceptance of the human papillomavirus vaccine. J Adolesc Health 2005; 37:248–251.
- Zimmerman RK. Ethical analysis of HPV vaccine policy options. Vaccine 2006; 24:4812–4820.
- Al Romaih WRR, Srinivas A, Shahtahmasebi S, Omar HA. No significant change in sexual behavior in association with human papillomavirus vaccination in young girls. Int J Child Adolesc Health 2011; 4:1–5.
- Mullins TL, Zimet GD, Rosenthal SL, et al. Adolescent perceptions of risk and need for safer sexual behaviors after first human papillomavirus vaccination. Arch Pediatr Adolesc Med 2012; 166:82–88.
- Middleman AB, Tung JS. School-located immunization programs: do parental p predict behavior? Vaccine 2011; 29:3513–3516.
- Samoff E, Dunn A, VanDevanter N, Blank S, Weisfuse IB. Predictors of acceptance of hepatitis B vaccination in an urban sexually transmitted diseases clinic. Sex Transm Dis 2004; 31:415–420.
- Gnanasekaran SK, Finkelstein JA, Hohman K, O’Brien M, Kruskal B, Lieu T. Parental perspectives on influenza vaccination among children with asthma. Public Health Rep 2006; 121:181–188.
- Daley MF, Crane LA, Chandramouli V, et al. Influenza among healthy young children: changes in parental attitudes and predictors of immunization during the 2003 to 2004 influenza season. Pediatrics 2006; 117:e268–e277.
- Vadaparampil ST, Kahn JA, Salmon D, et al. Missed clinical opportunities: provider recommendations for HPV vaccination for 11–12 year old girls are limited. Vaccine 2011; 29:8634–8641.
- Kim JJ, Goldie SJ. Health and economic implications of HPV vaccination in the United States. N Engl J Med 2008; 359:821–832.
- Garland SM, Skinner SR, Brotherton JM. Adolescent and young adult HPV vaccination in Australia: achievements and challenges. Prev Med 2011; 53(suppl 1):S29–S35.
- Rowhani-Rahbar A, Alvarez FB, Bryan JT, et al. Evidence of immune memory 8.5 years following administration of a prophylactic human papillomavirus type 16 vaccine. J Clin Virol 2012; 53:239–243.
- Forinash AB, Yancey AM, Pitlick JM, Myles TD. Safety of the HPV bivalent and quadrivalent vaccines during pregnancy (February) Ann Pharmacother 2011; [epub ahead of print]
- Wheeler CM, Harvey BM, Pichichero ME, et al. Immunogenicity and safety of human papillomavirus-16/18 AS04-adjuvanted vaccine coadministered with tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine and/or meningococcal conjugate vaccine to healthy girls 11 to 18 years of age: results from a randomized open trial. Pediatr Infect Dis J 2011; 30:e225–e234.
- Pedersen C, Breindahl M, Aggarwal N, et al. Randomized trial: immunogenicity and safety of coadministered human papillomavirus-16/18 AS04-adjuvanted vaccine and combined hepatitis A and B vaccine in girls. J Adolesc Health 2012; 50:38–46.
- Eichler HG, Kong SX, Gerth WC, Mavros P, Jönsson B. Use of costeffectiveness analysis in health-care resource allocation decisionmaking: how are cost-effectiveness thresholds expected to emerge? Value Health 2004; 7:518–528.
- Chesson HW. HPV vaccine cost-effectiveness: updates and review. Presentation before the Advisory Committee on Immunization Practices (ACIP), June 22, 2011. Atlanta, GA: US Department of Health and Human Services, CDC; 2011. http://www.cdc.gov/vaccines/recs/acip/downloads/mtg-slides-jun11/07-5-hpv-cost-effect.pdf. Accessed August 31, 2012.
- Kim JJ. Targeted human papillomavirus vaccination of men who have sex with men in the USA: a cost-effectiveness modelling analysis. Lancet Infect Dis 2010; 10:845–852.
- Cuzick J, Castañón A, Sasieni P. Predicted impact of vaccination against human papillomavirus 16/18 on cancer incidence and cervical abnormalities in women aged 20–29 in the UK. Br J Cancer 2010; 102:933–939.
KEY POINTS
- Two HPV vaccines are available: a quadrivalent vaccine against HVP types 6, 11, 16, and 18, and a bivalent vaccine against types 16 and 18.
- HPV causes cervical cancer, genital warts, oropharyngeal cancer, anal cancer, and recurrent respiratory papillomatosis, creating a considerable economic and health burden.
- The host immune response to natural HPV infection is slow and weak. In contrast, HPV vaccine induces a strong and long-lasting immune response.
- The HPV vaccines have greater than 90% efficacy in preventing cervical dysplasia and genital warts that are caused by the HPV types the vaccine contains. They are as safe as other common prophylactic vaccines.
- HPV vaccination has been challenged by public controversy over the vaccine’s safety, teenage sexuality, mandatory legislation, and the cost of the vaccine.
A short story of the short QT syndrome
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
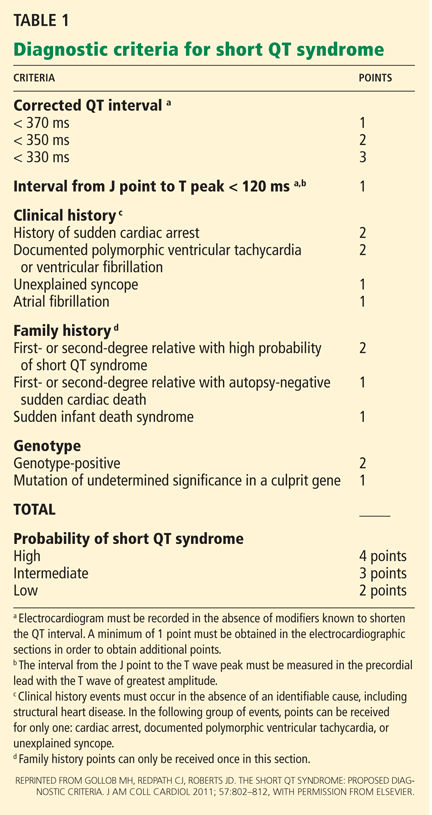
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
KEY POINTS
- Short QT syndrome is a genetic disease described initially in young patients who had atrial fibrillation or who died suddenly with no apparent structural heart disease.
- The diagnosis is established by the finding of a short QT interval. However, other factors including personal and family history are also important in establishing the diagnosis.
- The current recommendations for managing patients with short QT syndrome are not evidence-based. We encourage consultation with centers that have special interest in QT-interval-related disorders.
- Placement of an implantable cardioverter-defibrillator is considered the standard of care, especially in survivors of sudden cardiac death, ventricular fibrillation, or ventricular tachycardia. Unfortunately, a higher incidence of inappropriate shocks adds to the challenges of managing this potentially deadly disease.
VISUALS: Determining the cause of hallucinations in children and adolescents
Discuss this article at www.facebook.com/CurrentPsychiatry
Visual hallucinations in children and adolescents can be caused by many conditions other than psychosis. To prevent misdiagnosis and unnecessary antipsychotic use, it is important to rule out other causes of visual hallucinations. The mnemonic VISUALS reminds us of common causes.
Visions that are culturally sanctioned occur in non-Western societies—eg, images of fairy-like spirits are accepted and reinforced as part of the Filipino culture—and in several Christian denominations in the United States. Positive cultural connotations may increase the frequency of visual hallucinations as well as produce varied attitudes and emotional reactions to them.1
Imaginary friends often fulfill a child’s need for a relationship, although even social children may have these “friends.” Children often refer to imaginary friends in conversations and play with them. Usually they are also children. They may be extensions of people the child admires or be named after characters from stories, movies, or television. Children rarely are able to explain the imaginary friend’s appearance and more than half the time there is no trigger for the appearance of such friends.2,3
Stress and anxiety in preschool children may precipitate the onset of visual or tactile hallucinations. They often happen at night but also can occur when the child is awake. Typical visual hallucinations may include monsters, bugs, pets, or toys.2
Urine drug screens should be conducted for all adolescents and children. Cocaine, methamphetamine, and amphetamines—including high doses of prescribed stimulants—can cause visual hallucinations. Lysergic acid diethylamide (“LSD”), phencyclidine (“PCP”), 3,4-methylenedioxymethamphetamine (“ecstasy”), marijuana, nitrous oxide, and mescaline often cause visual hallucinations, although these substances may not be identified in a routine urine toxicology. Other considerations are withdrawal from benzodiazepines, sedative-hypnotics, or alcohol, and rare adverse reactions to antidepressants, antibiotics, or anticonvulsants.4,5
Age and developmental immaturity may make it difficult for children to distinguish between reality and non-reality, including dreams and shadows in the dark. Underdeveloped communication may make it difficult to interpret what the child is trying to communicate.2
Look into other medical explanations, such as migraines, seizures, tumors, ophthalmologic disease, delirium, or metabolic disorders (Table).4,5
Table
Medical causes of visual hallucinations in children and adolescents
| Medical condition | Symptoms |
|---|---|
| Neurologic | Migraine with aura; migraine coma; familial hemiplegic migraines; temporal or occipital lobe seizures; ictal, postictal, or interictal psychosis; tumors in occipital or temporal lobes |
| Ophthalmologic | Cataracts, retinal diseases, glaucoma |
| Inborn errors of metabolism | Homocysteine remethylation defects; urea cycle disorders; GM2 gangliosidoses; Niemann-Pick disease, type C; alpha-mannosidosis |
| Delirium | Metabolic disturbance, infection, intracranial process |
| Metabolic encephalopathy | Cardiopulmonary insufficiency, uremia, hepatic disease, vitamin deficiencies, inflammatory disease |
| Source:References 4,5 | |
Sleep-onset visual hallucinations (hypnagogic) and hallucinations upon awakening (hypnopompic) often are bizarre and dream-like. They may consist of geometric patterns, landscapes, faces, or figures. They mainly occur with narcolepsy but can be seen in insomnia or excessive daytime sleepiness.4,5
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. al-Issa I. The illusion of reality or the reality of illusion. Hallucinations and culture. Br J Psychiatry. 1995;166(3):368-373.
2. Tsai LY, Champine DJ. Schizophrenia and other psychotic disorders. In: Wiener JM Dulcan MK, eds. Textbook of child and adolescent psychiatry. 3rd ed. Washington, DC: American Psychiatric Publishing, Inc.; 2004:379–409.
3. Gleason TR, Sebanc AM, Hartup WW. Imaginary companions of preschool children. Dev Psychol. 2000;36(4):419-428.
4. Cummings JL, Miller BL. Visual hallucinations. Clinical occurrence and use in differential diagnosis. West J Med. 1987;146(1):46-51.
5. Teeple RC, Caplan JP, Stern TA. Visual hallucinations: differential diagnosis and treatment. Prim Care Companion J Clin Psychiatry. 2009;11(1):26-32.
Discuss this article at www.facebook.com/CurrentPsychiatry
Visual hallucinations in children and adolescents can be caused by many conditions other than psychosis. To prevent misdiagnosis and unnecessary antipsychotic use, it is important to rule out other causes of visual hallucinations. The mnemonic VISUALS reminds us of common causes.
Visions that are culturally sanctioned occur in non-Western societies—eg, images of fairy-like spirits are accepted and reinforced as part of the Filipino culture—and in several Christian denominations in the United States. Positive cultural connotations may increase the frequency of visual hallucinations as well as produce varied attitudes and emotional reactions to them.1
Imaginary friends often fulfill a child’s need for a relationship, although even social children may have these “friends.” Children often refer to imaginary friends in conversations and play with them. Usually they are also children. They may be extensions of people the child admires or be named after characters from stories, movies, or television. Children rarely are able to explain the imaginary friend’s appearance and more than half the time there is no trigger for the appearance of such friends.2,3
Stress and anxiety in preschool children may precipitate the onset of visual or tactile hallucinations. They often happen at night but also can occur when the child is awake. Typical visual hallucinations may include monsters, bugs, pets, or toys.2
Urine drug screens should be conducted for all adolescents and children. Cocaine, methamphetamine, and amphetamines—including high doses of prescribed stimulants—can cause visual hallucinations. Lysergic acid diethylamide (“LSD”), phencyclidine (“PCP”), 3,4-methylenedioxymethamphetamine (“ecstasy”), marijuana, nitrous oxide, and mescaline often cause visual hallucinations, although these substances may not be identified in a routine urine toxicology. Other considerations are withdrawal from benzodiazepines, sedative-hypnotics, or alcohol, and rare adverse reactions to antidepressants, antibiotics, or anticonvulsants.4,5
Age and developmental immaturity may make it difficult for children to distinguish between reality and non-reality, including dreams and shadows in the dark. Underdeveloped communication may make it difficult to interpret what the child is trying to communicate.2
Look into other medical explanations, such as migraines, seizures, tumors, ophthalmologic disease, delirium, or metabolic disorders (Table).4,5
Table
Medical causes of visual hallucinations in children and adolescents
| Medical condition | Symptoms |
|---|---|
| Neurologic | Migraine with aura; migraine coma; familial hemiplegic migraines; temporal or occipital lobe seizures; ictal, postictal, or interictal psychosis; tumors in occipital or temporal lobes |
| Ophthalmologic | Cataracts, retinal diseases, glaucoma |
| Inborn errors of metabolism | Homocysteine remethylation defects; urea cycle disorders; GM2 gangliosidoses; Niemann-Pick disease, type C; alpha-mannosidosis |
| Delirium | Metabolic disturbance, infection, intracranial process |
| Metabolic encephalopathy | Cardiopulmonary insufficiency, uremia, hepatic disease, vitamin deficiencies, inflammatory disease |
| Source:References 4,5 | |
Sleep-onset visual hallucinations (hypnagogic) and hallucinations upon awakening (hypnopompic) often are bizarre and dream-like. They may consist of geometric patterns, landscapes, faces, or figures. They mainly occur with narcolepsy but can be seen in insomnia or excessive daytime sleepiness.4,5
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Visual hallucinations in children and adolescents can be caused by many conditions other than psychosis. To prevent misdiagnosis and unnecessary antipsychotic use, it is important to rule out other causes of visual hallucinations. The mnemonic VISUALS reminds us of common causes.
Visions that are culturally sanctioned occur in non-Western societies—eg, images of fairy-like spirits are accepted and reinforced as part of the Filipino culture—and in several Christian denominations in the United States. Positive cultural connotations may increase the frequency of visual hallucinations as well as produce varied attitudes and emotional reactions to them.1
Imaginary friends often fulfill a child’s need for a relationship, although even social children may have these “friends.” Children often refer to imaginary friends in conversations and play with them. Usually they are also children. They may be extensions of people the child admires or be named after characters from stories, movies, or television. Children rarely are able to explain the imaginary friend’s appearance and more than half the time there is no trigger for the appearance of such friends.2,3
Stress and anxiety in preschool children may precipitate the onset of visual or tactile hallucinations. They often happen at night but also can occur when the child is awake. Typical visual hallucinations may include monsters, bugs, pets, or toys.2
Urine drug screens should be conducted for all adolescents and children. Cocaine, methamphetamine, and amphetamines—including high doses of prescribed stimulants—can cause visual hallucinations. Lysergic acid diethylamide (“LSD”), phencyclidine (“PCP”), 3,4-methylenedioxymethamphetamine (“ecstasy”), marijuana, nitrous oxide, and mescaline often cause visual hallucinations, although these substances may not be identified in a routine urine toxicology. Other considerations are withdrawal from benzodiazepines, sedative-hypnotics, or alcohol, and rare adverse reactions to antidepressants, antibiotics, or anticonvulsants.4,5
Age and developmental immaturity may make it difficult for children to distinguish between reality and non-reality, including dreams and shadows in the dark. Underdeveloped communication may make it difficult to interpret what the child is trying to communicate.2
Look into other medical explanations, such as migraines, seizures, tumors, ophthalmologic disease, delirium, or metabolic disorders (Table).4,5
Table
Medical causes of visual hallucinations in children and adolescents
| Medical condition | Symptoms |
|---|---|
| Neurologic | Migraine with aura; migraine coma; familial hemiplegic migraines; temporal or occipital lobe seizures; ictal, postictal, or interictal psychosis; tumors in occipital or temporal lobes |
| Ophthalmologic | Cataracts, retinal diseases, glaucoma |
| Inborn errors of metabolism | Homocysteine remethylation defects; urea cycle disorders; GM2 gangliosidoses; Niemann-Pick disease, type C; alpha-mannosidosis |
| Delirium | Metabolic disturbance, infection, intracranial process |
| Metabolic encephalopathy | Cardiopulmonary insufficiency, uremia, hepatic disease, vitamin deficiencies, inflammatory disease |
| Source:References 4,5 | |
Sleep-onset visual hallucinations (hypnagogic) and hallucinations upon awakening (hypnopompic) often are bizarre and dream-like. They may consist of geometric patterns, landscapes, faces, or figures. They mainly occur with narcolepsy but can be seen in insomnia or excessive daytime sleepiness.4,5
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. al-Issa I. The illusion of reality or the reality of illusion. Hallucinations and culture. Br J Psychiatry. 1995;166(3):368-373.
2. Tsai LY, Champine DJ. Schizophrenia and other psychotic disorders. In: Wiener JM Dulcan MK, eds. Textbook of child and adolescent psychiatry. 3rd ed. Washington, DC: American Psychiatric Publishing, Inc.; 2004:379–409.
3. Gleason TR, Sebanc AM, Hartup WW. Imaginary companions of preschool children. Dev Psychol. 2000;36(4):419-428.
4. Cummings JL, Miller BL. Visual hallucinations. Clinical occurrence and use in differential diagnosis. West J Med. 1987;146(1):46-51.
5. Teeple RC, Caplan JP, Stern TA. Visual hallucinations: differential diagnosis and treatment. Prim Care Companion J Clin Psychiatry. 2009;11(1):26-32.
1. al-Issa I. The illusion of reality or the reality of illusion. Hallucinations and culture. Br J Psychiatry. 1995;166(3):368-373.
2. Tsai LY, Champine DJ. Schizophrenia and other psychotic disorders. In: Wiener JM Dulcan MK, eds. Textbook of child and adolescent psychiatry. 3rd ed. Washington, DC: American Psychiatric Publishing, Inc.; 2004:379–409.
3. Gleason TR, Sebanc AM, Hartup WW. Imaginary companions of preschool children. Dev Psychol. 2000;36(4):419-428.
4. Cummings JL, Miller BL. Visual hallucinations. Clinical occurrence and use in differential diagnosis. West J Med. 1987;146(1):46-51.
5. Teeple RC, Caplan JP, Stern TA. Visual hallucinations: differential diagnosis and treatment. Prim Care Companion J Clin Psychiatry. 2009;11(1):26-32.
Pharmacotherapy for comorbid depression and alcohol dependence
Discuss this article at www.facebook.com/CurrentPsychiatry
With a lifetime prevalence of 30%, alcohol use disorders (AUDs)—which in DSM-IV-TR include alcohol abuse and alcohol dependence—are among the most common psychiatric disorders.1 Depressive disorders, including major depressive disorder (MDD) and dysthymia, frequently co-occur with AUDs.2-4 This pattern of comorbidity adversely affects the prognosis, course, and treatment of both MDD and AUDs.5 High severity in 1 of these disorders is associated with high severity in the other.2,6 Alcohol dependence appears to prolong the course of depression7,8 and increases the risk of suicidal symptoms and behaviors.9,10 Patients with depression and AUDs are at increased risk of relapse to heavy drinking.7,11
Whether the high comorbidity rate of depressive disorders and AUDs is a result of 1 disorder causing the other (ie, AUDs leading to depression or vice versa) or can be attributed to shared etiology is unknown. Clinicians need to consider this question when treating patients with this pattern of comorbidity because distinguishing primary depression from secondary depression influences treatment decisions.12
There is a great need for pharmacologic interventions that can concurrently treat both depression and AUDs. This article reviews the evidence for current treatments for dually diagnosed patients and highlights novel agents that are worthy of further study for this complex patient population.
Current treatment options
Pharmacotherapy for MDD and alcohol dependence is common when these conditions occur alone. FDA-approved medications for treating depression include monoamine oxidase inhibitors, tricyclic antidepressants (TCAs), tetracyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors.
SSRIs are the most widely used class of antidepressants. They gained FDA approval based on studies conducted in non-comorbid patients because patients with comorbid conditions usually are excluded from research studies.13 Few trials have evaluated patients with depression and AUDs; TCAs and SSRIs are best studied in these patients.
Serotonergic antidepressants
SSRIs are first-line medications for MDD because of their low abuse potential, favorable side effect profile, and relative safety in overdose.
Table 1
Serotonergic antidepressants for patients with AUDs and depression
| Study | Sample | Results |
|---|---|---|
| Cornelius et al, 199714 | Outpatients with severe major depression and AD 1. Fluoxetine (20 to 40 mg/d; n = 25) 2. Placebo (n = 26) | Greater reductions in depressive symptoms and drinking in patients treated with fluoxetine compared with placebo |
| Roy, 199815 | Inpatients with current major depression and AD who were abstinent for ≥2 weeks 1. Sertraline (100 mg/d; n = 18) 2. Placebo (n = 18) | Greater reductions in depressive symptoms in patients treated with sertraline compared with placebo. Drinking outcomes were not emphasized because 35 of 36 patients reported continuous abstinence throughout the trial |
| Kranzler et al, 199516 | Outpatients with AD. Fourteen percent had current major depression. All received weekly individual or group CBT focused on relapse prevention and skills building 1. Fluoxetine (mean daily dose: 48 mg; n = 51) 2. Placebo (n = 50) | Significant decrease in alcohol consumption for both groups during the trial. No significant differences in alcohol consumption between groups. Among those with current depression, patients treated with fluoxetine experienced greater reduction in depressive symptoms vs placebo |
| Moak et al, 200317 | Currently depressed, actively drinking, alcohol-dependent outpatients. All received individual CBT 1. Sertraline (mean daily dose: 186 mg; n = 38) 2. Placebo (n = 44) | Sertraline had an advantage over placebo in reducing depressive symptoms in women but not in men. Sertraline reduced drinks per drinking day but not other drinking-related outcomes |
| Cornelius et al, 200918 | Adolescents (age 15 to 20) with AA or AD and MDD. All received intensive manual-based therapy (CBT for MDD and AUD, MET for AUD) almost weekly 1. Fluoxetine (20 mg/d; n = 24) 2. Placebo (n = 26) | All improved during the course of trial. No significant differences between fluoxetine and placebo groups in depression- or drinking-related outcomes |
| Roy-Byrne et al, 200019 | Actively drinking alcohol-dependent outpatients with history of ≥1 depressive episode. All received weekly group therapy for alcoholism 1. Nefazodone (mean daily dose: 460 mg; n = 32) 2. Placebo (n = 32) | Greater reduction in depressive symptoms but not in drinking-related outcomes in patients treated with nefazodone |
| Hernandez-Avila et al, 200420 | Outpatients with AD and current major depression. All received supportive psychotherapy for 10 weeks 1. Nefazodone (mean daily dose: 413 mg; n = 21) 2. Placebo (n = 20) | Depressive and anxiety symptoms declined significantly over time, but no statistically significant differences in depressive or anxiety symptoms between nefazodone and placebo. Patients treated with nefazodone had significantly greater reductions in heavy drinking days and in total drinks compared with placebo-treated patients |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; CBT: cognitive-behavioral therapy; MDD: major depressive disorder; MET: motivational enhancement therapy | ||
In a study of adolescents, Cornelius et al18 failed to find any differences between fluoxetine and placebo in any depression or drinking-related outcomes. This study compared the efficacy of fluoxetine, 20 mg/d, with placebo in 50 adolescents with MDD and AUDs who also received intensive, manual-based cognitive-behavioral therapy and motivational enhancement therapy. All patients improved during the trial, but there were no significant differences between fluoxetine- and placebo-treated adolescents.
Other serotonergic medications. Two studies have evaluated nefazodone, a serotonin (5-HT2) antagonist, in dually diagnosed patients. In a 12-week trial, Roy-Byrne et al19 evaluated the efficacy of nefazodone (mean daily dose: 460 mg) vs placebo in 64 actively drinking alcohol-dependent patients who had ≥1 prior episode of depression; all participated in a weekly psychoeducation group on alcoholism. Nefazodone was associated with significantly greater reduction in depressive symptoms but no reductions in drinking compared with placebo. However, a 10-week study of nefazodone20 (mean daily dose: 413 mg) vs placebo in 41 alcohol-dependent patients with current major depression found that those who received nefazodone significantly reduced heavy drinking days compared with the placebo group. There were no significant differences in depressive symptoms between groups.
Conflicting evidence on TCAs
Although several studies suggest TCAs may help reduce depressive symptoms in patients with AUDs, results on their ability to reduce drinking are conflicting (Table 2).24-26 In 1 study, 6 months of desipramine (mean daily dose: 200 mg) reduced drinking in 28 alcohol-dependent individuals with secondary depression24; in another, 12 weeks of imipramine plus weekly relapse prevention psychotherapy did not affect drinking-related outcomes in 69 actively drinking alcoholic outpatients with current depressive disorders.25
Table 2
Limited evidence supports TCAs for comorbid depression and AUDs
| Study | Sample | Results |
|---|---|---|
| Mason et al, 199624 | Outpatients with AD and secondary depression. Part of larger study including non-depressed patients with AD (N = 71) 1. Desipramine (mean daily dose 200 mg; n = 15) 2. Placebo (n = 13) | Greater reduction in depressive symptoms and drinking in desipramine-treated patients compared with placebo-treated patients |
| McGrath et al, 199625 | Outpatients with AD or AA and major depression, dysthymia, or depressive disorder NOS 1. Imipramine (mean daily dose 260 mg; n = 36) 2. Placebo (n = 33) | Greater reduction in depressive symptoms for imipramine-treated patients compared with placebo-treated patients. Drinking-related outcomes were not directly affected by medication except improvements in mood led to reduced alcohol use |
| Altintoprak et al, 200826 | Inpatients with AD and MDD 1. Mirtazapine (30 mg/d; n = 24) 2. Amitriptyline (100 mg/d; n = 20) | Drinking-related outcomes were not emphasized because all patients were required to abstain from drinking during the study. Both treatments reduced depressive symptoms; there were no significant differences between groups |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder; NOS: not otherwise specified; TCAs: tricyclic antidepressants | ||
Altintoprak et al26 compared the efficacy of the antidepressant mirtazapine, 30 mg/d, with the TCA amitriptyline, 100 mg/d, in 44 patients with comorbid alcohol dependence and MDD. All patients were required to abstain from drinking alcohol during the study. Both medications resulted in steady reductions in depressive symptoms and alcohol cravings; however, researchers found no significant differences between the 2 treatment groups.
Analyses of combined studies
Pettinati27 conducted a qualitative review of antidepressants for patients with depression and alcohol dependence that included 8 controlled clinical trials (2 on TCAs and 6 on serotonergic medications) conducted between 1994 and 2004. In this review, both TCAs and serotonergic medications were similarly effective in reducing depressive symptoms but not consistently effective in reducing drinking.
27 this review suggested that antidepressants can reduce depressive symptoms but not drinking. The authors also found evidence that the more the antidepressant reduced depressive symptoms, the more it reduced alcohol use. Studies published after these reviews have not substantially altered these findings.
Alcohol abuse medications
Four medications are FDA-approved for treating alcohol dependence:
- disulfiram
- naltrexone (in 2 formulations: oral and long-acting injectable)
- acamprosate.
Table 3
Can medications that target alcohol use also improve depression?
| Study | Sample | Results |
|---|---|---|
| Petrakis et al, 200729 | Outpatients with AD and an axis I disorder, including depression (secondary analysis of Petrakis et al, 200531) 1. Naltrexone (50 mg/d; n = 34) 2. Disulfiram (250 mg/d; n = 43) 3. Naltrexone + disulfiram (n = 28) 4. Placebo (n = 34) | Generally, medication was more effective than no medication. No advantage of 1 medication over the other. There was no relationship between depression diagnosis and medication condition, which suggests that for patients with depression, there was no advantage to medication |
| Pettinati et al, 201030 | Outpatients with AD and MDD 1. Sertraline (200 mg/d; n = 40) 2. Naltrexone (100 mg/d; n = 49) 3. Sertraline + naltrexone (n = 42) 4. Placebo (n = 39) | Greater proportion of patients in combined medication group abstained from alcohol and refrained from heavy alcohol use during the trial compared with those in sertraline-only or naltrexone-only groups. No significant differences among groups on depression-related outcomes |
| AD: alcohol dependence; MDD: major depressive disorder | ||
Nevertheless, these studies suggest that medications for treating depression or AUDs have, at best, only a modest effect in patients with both disorders.
Novel agents
Several novel medications have been evaluated as possible treatments for comorbid depression and AUDs because they target the underlying neurobiology of both disorders:
- agents that target the neurotransmitter glutamate, including the N-methyl-d-aspartate glutamate receptor antagonists memantine and ketamine
- dopaminergic agents such as quetiapine
- corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists.
In a case study, a 55-year-old man with treatment-resistant major depression and co-occurring alcohol and benzodiazepine dependence who received a single dose of IV ketamine, 0.5 mg/kg over 50 minutes, experienced “significant improvements” in depressive symptoms that lasted throughout the 7-day follow-up.33 This study did not report on ketamine’s effects on his alcohol use.
The atypical antipsychotic quetiapine acts as a serotonin (5-HT1A and 5-HT2) and dopamine (D1 and D2) antagonist, and reports suggest it reduces alcohol craving and affective symptoms in patients with AUDs.34,35 In a 16-week, open-label study, quetiapine decreased alcohol consumption, alcohol craving, and intensity of some psychiatric symptoms in 28 alcohol-dependent patients with bipolar disorder, schizoaffective disorder, or borderline personality disorder.36
See the Box for a description of the role CRF1 antagonists may play in treating patients with concurrent MDD and AUDs. See Table 4 for studies of memantine and quetiapine in treating depression with AUDs.
Corticotropin-releasing factor (CRF) has a well-established role in stress and has been implicated for treating anxiety and depressive disorders. Evidence also suggests that CRF receptor 1 (CRF1) may be a treatment target for alcohol use disorders (AUDs). Acute alcohol withdrawal and prolonged alcohol use are associated with elevated levels of extrahypothalamic CRF and correlated anxiety. CRF antagonists can reduce the anxiogenic effects of alcohol withdrawal and reduce some symptoms of alcohol dependence, including excessive alcohol self-administration and stress-induced relapse to alcohol use in rats with alcohol dependence, but not in those without dependence. Therefore, CRF1 receptor antagonists may be especially helpful for individuals who use alcohol to reduce negative emotional states such as anxiety or dysphoria, including those with concurrent major depressive disorder and AUDs.
Bibliography
Funk CK, Zorrilla EP, Lee MJ, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61(1):78-86.
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27(10):2718-2726.
Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32(9):1535-1542.
Other agents may play a role in treating depression with AUDs
| Study | Sample | Results |
|---|---|---|
| Muhonen et al, 200832 | Outpatients with MDD and AD 1. Memantine (20 mg/d; n = 40) | Both treatments reduced depressive and anxiety symptoms. No significant differences between groups. Study did not examine alcohol-related outcomes |
| Martinotti et al, 200833 | Outpatients with comorbid AD and either bipolar disorder, schizoaffective disorder or borderline personality disorder. Open-label study 1. Quetiapine (300 to 800 mg/d; n = 28) | Quetiapine was associated with reduced alcohol consumption, alcohol craving, and intensity of psychiatric symptoms |
| AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder | ||
Interpreting the evidence
These findings underscore the importance of thorough evaluations. SSRIs are a first-line treatment for depression and as such probably should be the first choice for patients with comorbid AUDs. Drinking should be monitored closely and abstinence encouraged. Using medications that target AUDs is safe and modestly effective in patients with comorbid depression. Evidence suggests that treating both disorders simultaneously is more effective than treating either alone. Medications should be prescribed as part of a comprehensive treatment plan that also includes psychotherapy.
Related Resources
- Pettinati H. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
- Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
- Acamprosate • Campral
- Amitriptyline • Elavil
- Desipramine • Norpramin
- Disulfiram • Antabuse
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Ketamine • Ketalar
- Memantine • Namenda
- Mirtazapine • Remeron
- Naltrexone • Revia, Vivitrol
- Nefazodone • Serzone
- Quetiapine • Seroquel
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Petrakis receives research or grant support from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, the U.S. Department of Defense, and the U.S. Department of Veterans Affairs.
This work has been supported by a grant from the Veterans Affairs New England Mental Illness Research, Education, and Clinical Center and by the National Institute of Mental Health (T32MH062994-07).
Acknowledgements
The authors thank Elizabeth Guidone for her helpful comments and Diana Limoncelli for her assistance in manuscript preparation.
1. Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: results of a national survey. Drug Alcohol Depend. 1995;39(3):197-206.
3. Kessler RC, Crum RM, Warner LA, et al. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
4. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
5. Burns L, Teesson M, O’Neill K. The impact of comorbid anxiety and depression on alcohol treatment outcomes. Addiction. 2005;100(6):787-796.
6. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
7. Hasin DS, Tsai WY, Endicott J, et al. Five-year course of major depression: effects of comorbid alcoholism. J Affect Disord. 1996;41(1):63-70.
8. Mueller TI, Lavori PW, Keller MB, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
9. Cornelius JR, Salloum IM, Mezzich J, et al. Disproportionate suicidality in patients with comorbid major depression and alcoholism. Am J Psychiatry. 1995;152(3):358-364.
10. Sher L, Oquendo MA, Galfalvy HC, et al. The relationship of aggression to suicidal behavior in depressed patients with a history of alcoholism. Addict Behav. 2005;30(6):1144-1153.
11. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
12. Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9(5):374-380.
13. Oslin DW. Treatment of late-life depression complicated by alcohol dependence. Am J Geriatr Psychiatry. 2005;13(6):491-500.
14. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
15. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
16. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
17. Moak DH, Anton RF, Latham PK, et al. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
18. Cornelius JR, Bukstein OG, Wood DS, et al. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addict Behav. 2009;34(10):905-909.
19. Roy-Byrne PP, Pages KP, Russo JE, et al. Nefazodone treatment of major depression in alcohol-dependent patients: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2000;20(2):129-136.
20. Hernandez-Avila CA, Modesto-Lowe V, Feinn R, et al. Nefazodone treatment of comorbid alcohol dependence and major depression. Alcohol Clin Exp Res. 2004;28(3):433-440.
21. Kranzler HR, Burleson JA, Brown J, et al. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20(9):1534-1541.
22. Naranjo CA, Bremner KE, Lanctôt KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake dependence and problems. Addiction. 1995;90(1):87-99.
23. Pettinati HM, Volpicelli JR, Kranzler HR, et al. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24(7):1041-1049.
24. Mason BJ, Kocsis JH, Ritvo EC, et al. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275(10):761-767.
25. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
26. Altintoprak AE, Zorlu N, Coskunol H, et al. Effectiveness and tolerability of mirtazapine and amitriptyline in alcoholic patients with co-morbid depressive disorder: a randomized, double-blind study. Hum Psychopharmacol. 2008;23(4):313-319.
27. Pettinati HM. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
28. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
29. Petrakis I, Ralevski E, Nich C, et al. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
31. Petrakis IL, Poling J, Levinson C, et al. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
32. Muhonen LH, Lönnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392-399.
33. Liebrenz M, Borgeat A, Leisinger R, et al. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137(15-16):234-236.
34. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled pilot trial of quetiapine for the treatment of Type A and Type B alcoholism. J Clin Psychopharmacol. 2007;27(4):344-351.
35. Croissant B, Klein O, Gehrlein L, et al. Quetiapine in relapse prevention in alcoholics suffering from craving and affective symptoms: a case series. Eur Psychiatry. 2006;21(8):570-573.
36. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
Discuss this article at www.facebook.com/CurrentPsychiatry
With a lifetime prevalence of 30%, alcohol use disorders (AUDs)—which in DSM-IV-TR include alcohol abuse and alcohol dependence—are among the most common psychiatric disorders.1 Depressive disorders, including major depressive disorder (MDD) and dysthymia, frequently co-occur with AUDs.2-4 This pattern of comorbidity adversely affects the prognosis, course, and treatment of both MDD and AUDs.5 High severity in 1 of these disorders is associated with high severity in the other.2,6 Alcohol dependence appears to prolong the course of depression7,8 and increases the risk of suicidal symptoms and behaviors.9,10 Patients with depression and AUDs are at increased risk of relapse to heavy drinking.7,11
Whether the high comorbidity rate of depressive disorders and AUDs is a result of 1 disorder causing the other (ie, AUDs leading to depression or vice versa) or can be attributed to shared etiology is unknown. Clinicians need to consider this question when treating patients with this pattern of comorbidity because distinguishing primary depression from secondary depression influences treatment decisions.12
There is a great need for pharmacologic interventions that can concurrently treat both depression and AUDs. This article reviews the evidence for current treatments for dually diagnosed patients and highlights novel agents that are worthy of further study for this complex patient population.
Current treatment options
Pharmacotherapy for MDD and alcohol dependence is common when these conditions occur alone. FDA-approved medications for treating depression include monoamine oxidase inhibitors, tricyclic antidepressants (TCAs), tetracyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors.
SSRIs are the most widely used class of antidepressants. They gained FDA approval based on studies conducted in non-comorbid patients because patients with comorbid conditions usually are excluded from research studies.13 Few trials have evaluated patients with depression and AUDs; TCAs and SSRIs are best studied in these patients.
Serotonergic antidepressants
SSRIs are first-line medications for MDD because of their low abuse potential, favorable side effect profile, and relative safety in overdose.
Table 1
Serotonergic antidepressants for patients with AUDs and depression
| Study | Sample | Results |
|---|---|---|
| Cornelius et al, 199714 | Outpatients with severe major depression and AD 1. Fluoxetine (20 to 40 mg/d; n = 25) 2. Placebo (n = 26) | Greater reductions in depressive symptoms and drinking in patients treated with fluoxetine compared with placebo |
| Roy, 199815 | Inpatients with current major depression and AD who were abstinent for ≥2 weeks 1. Sertraline (100 mg/d; n = 18) 2. Placebo (n = 18) | Greater reductions in depressive symptoms in patients treated with sertraline compared with placebo. Drinking outcomes were not emphasized because 35 of 36 patients reported continuous abstinence throughout the trial |
| Kranzler et al, 199516 | Outpatients with AD. Fourteen percent had current major depression. All received weekly individual or group CBT focused on relapse prevention and skills building 1. Fluoxetine (mean daily dose: 48 mg; n = 51) 2. Placebo (n = 50) | Significant decrease in alcohol consumption for both groups during the trial. No significant differences in alcohol consumption between groups. Among those with current depression, patients treated with fluoxetine experienced greater reduction in depressive symptoms vs placebo |
| Moak et al, 200317 | Currently depressed, actively drinking, alcohol-dependent outpatients. All received individual CBT 1. Sertraline (mean daily dose: 186 mg; n = 38) 2. Placebo (n = 44) | Sertraline had an advantage over placebo in reducing depressive symptoms in women but not in men. Sertraline reduced drinks per drinking day but not other drinking-related outcomes |
| Cornelius et al, 200918 | Adolescents (age 15 to 20) with AA or AD and MDD. All received intensive manual-based therapy (CBT for MDD and AUD, MET for AUD) almost weekly 1. Fluoxetine (20 mg/d; n = 24) 2. Placebo (n = 26) | All improved during the course of trial. No significant differences between fluoxetine and placebo groups in depression- or drinking-related outcomes |
| Roy-Byrne et al, 200019 | Actively drinking alcohol-dependent outpatients with history of ≥1 depressive episode. All received weekly group therapy for alcoholism 1. Nefazodone (mean daily dose: 460 mg; n = 32) 2. Placebo (n = 32) | Greater reduction in depressive symptoms but not in drinking-related outcomes in patients treated with nefazodone |
| Hernandez-Avila et al, 200420 | Outpatients with AD and current major depression. All received supportive psychotherapy for 10 weeks 1. Nefazodone (mean daily dose: 413 mg; n = 21) 2. Placebo (n = 20) | Depressive and anxiety symptoms declined significantly over time, but no statistically significant differences in depressive or anxiety symptoms between nefazodone and placebo. Patients treated with nefazodone had significantly greater reductions in heavy drinking days and in total drinks compared with placebo-treated patients |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; CBT: cognitive-behavioral therapy; MDD: major depressive disorder; MET: motivational enhancement therapy | ||
In a study of adolescents, Cornelius et al18 failed to find any differences between fluoxetine and placebo in any depression or drinking-related outcomes. This study compared the efficacy of fluoxetine, 20 mg/d, with placebo in 50 adolescents with MDD and AUDs who also received intensive, manual-based cognitive-behavioral therapy and motivational enhancement therapy. All patients improved during the trial, but there were no significant differences between fluoxetine- and placebo-treated adolescents.
Other serotonergic medications. Two studies have evaluated nefazodone, a serotonin (5-HT2) antagonist, in dually diagnosed patients. In a 12-week trial, Roy-Byrne et al19 evaluated the efficacy of nefazodone (mean daily dose: 460 mg) vs placebo in 64 actively drinking alcohol-dependent patients who had ≥1 prior episode of depression; all participated in a weekly psychoeducation group on alcoholism. Nefazodone was associated with significantly greater reduction in depressive symptoms but no reductions in drinking compared with placebo. However, a 10-week study of nefazodone20 (mean daily dose: 413 mg) vs placebo in 41 alcohol-dependent patients with current major depression found that those who received nefazodone significantly reduced heavy drinking days compared with the placebo group. There were no significant differences in depressive symptoms between groups.
Conflicting evidence on TCAs
Although several studies suggest TCAs may help reduce depressive symptoms in patients with AUDs, results on their ability to reduce drinking are conflicting (Table 2).24-26 In 1 study, 6 months of desipramine (mean daily dose: 200 mg) reduced drinking in 28 alcohol-dependent individuals with secondary depression24; in another, 12 weeks of imipramine plus weekly relapse prevention psychotherapy did not affect drinking-related outcomes in 69 actively drinking alcoholic outpatients with current depressive disorders.25
Table 2
Limited evidence supports TCAs for comorbid depression and AUDs
| Study | Sample | Results |
|---|---|---|
| Mason et al, 199624 | Outpatients with AD and secondary depression. Part of larger study including non-depressed patients with AD (N = 71) 1. Desipramine (mean daily dose 200 mg; n = 15) 2. Placebo (n = 13) | Greater reduction in depressive symptoms and drinking in desipramine-treated patients compared with placebo-treated patients |
| McGrath et al, 199625 | Outpatients with AD or AA and major depression, dysthymia, or depressive disorder NOS 1. Imipramine (mean daily dose 260 mg; n = 36) 2. Placebo (n = 33) | Greater reduction in depressive symptoms for imipramine-treated patients compared with placebo-treated patients. Drinking-related outcomes were not directly affected by medication except improvements in mood led to reduced alcohol use |
| Altintoprak et al, 200826 | Inpatients with AD and MDD 1. Mirtazapine (30 mg/d; n = 24) 2. Amitriptyline (100 mg/d; n = 20) | Drinking-related outcomes were not emphasized because all patients were required to abstain from drinking during the study. Both treatments reduced depressive symptoms; there were no significant differences between groups |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder; NOS: not otherwise specified; TCAs: tricyclic antidepressants | ||
Altintoprak et al26 compared the efficacy of the antidepressant mirtazapine, 30 mg/d, with the TCA amitriptyline, 100 mg/d, in 44 patients with comorbid alcohol dependence and MDD. All patients were required to abstain from drinking alcohol during the study. Both medications resulted in steady reductions in depressive symptoms and alcohol cravings; however, researchers found no significant differences between the 2 treatment groups.
Analyses of combined studies
Pettinati27 conducted a qualitative review of antidepressants for patients with depression and alcohol dependence that included 8 controlled clinical trials (2 on TCAs and 6 on serotonergic medications) conducted between 1994 and 2004. In this review, both TCAs and serotonergic medications were similarly effective in reducing depressive symptoms but not consistently effective in reducing drinking.
27 this review suggested that antidepressants can reduce depressive symptoms but not drinking. The authors also found evidence that the more the antidepressant reduced depressive symptoms, the more it reduced alcohol use. Studies published after these reviews have not substantially altered these findings.
Alcohol abuse medications
Four medications are FDA-approved for treating alcohol dependence:
- disulfiram
- naltrexone (in 2 formulations: oral and long-acting injectable)
- acamprosate.
Table 3
Can medications that target alcohol use also improve depression?
| Study | Sample | Results |
|---|---|---|
| Petrakis et al, 200729 | Outpatients with AD and an axis I disorder, including depression (secondary analysis of Petrakis et al, 200531) 1. Naltrexone (50 mg/d; n = 34) 2. Disulfiram (250 mg/d; n = 43) 3. Naltrexone + disulfiram (n = 28) 4. Placebo (n = 34) | Generally, medication was more effective than no medication. No advantage of 1 medication over the other. There was no relationship between depression diagnosis and medication condition, which suggests that for patients with depression, there was no advantage to medication |
| Pettinati et al, 201030 | Outpatients with AD and MDD 1. Sertraline (200 mg/d; n = 40) 2. Naltrexone (100 mg/d; n = 49) 3. Sertraline + naltrexone (n = 42) 4. Placebo (n = 39) | Greater proportion of patients in combined medication group abstained from alcohol and refrained from heavy alcohol use during the trial compared with those in sertraline-only or naltrexone-only groups. No significant differences among groups on depression-related outcomes |
| AD: alcohol dependence; MDD: major depressive disorder | ||
Nevertheless, these studies suggest that medications for treating depression or AUDs have, at best, only a modest effect in patients with both disorders.
Novel agents
Several novel medications have been evaluated as possible treatments for comorbid depression and AUDs because they target the underlying neurobiology of both disorders:
- agents that target the neurotransmitter glutamate, including the N-methyl-d-aspartate glutamate receptor antagonists memantine and ketamine
- dopaminergic agents such as quetiapine
- corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists.
In a case study, a 55-year-old man with treatment-resistant major depression and co-occurring alcohol and benzodiazepine dependence who received a single dose of IV ketamine, 0.5 mg/kg over 50 minutes, experienced “significant improvements” in depressive symptoms that lasted throughout the 7-day follow-up.33 This study did not report on ketamine’s effects on his alcohol use.
The atypical antipsychotic quetiapine acts as a serotonin (5-HT1A and 5-HT2) and dopamine (D1 and D2) antagonist, and reports suggest it reduces alcohol craving and affective symptoms in patients with AUDs.34,35 In a 16-week, open-label study, quetiapine decreased alcohol consumption, alcohol craving, and intensity of some psychiatric symptoms in 28 alcohol-dependent patients with bipolar disorder, schizoaffective disorder, or borderline personality disorder.36
See the Box for a description of the role CRF1 antagonists may play in treating patients with concurrent MDD and AUDs. See Table 4 for studies of memantine and quetiapine in treating depression with AUDs.
Corticotropin-releasing factor (CRF) has a well-established role in stress and has been implicated for treating anxiety and depressive disorders. Evidence also suggests that CRF receptor 1 (CRF1) may be a treatment target for alcohol use disorders (AUDs). Acute alcohol withdrawal and prolonged alcohol use are associated with elevated levels of extrahypothalamic CRF and correlated anxiety. CRF antagonists can reduce the anxiogenic effects of alcohol withdrawal and reduce some symptoms of alcohol dependence, including excessive alcohol self-administration and stress-induced relapse to alcohol use in rats with alcohol dependence, but not in those without dependence. Therefore, CRF1 receptor antagonists may be especially helpful for individuals who use alcohol to reduce negative emotional states such as anxiety or dysphoria, including those with concurrent major depressive disorder and AUDs.
Bibliography
Funk CK, Zorrilla EP, Lee MJ, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61(1):78-86.
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27(10):2718-2726.
Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32(9):1535-1542.
Other agents may play a role in treating depression with AUDs
| Study | Sample | Results |
|---|---|---|
| Muhonen et al, 200832 | Outpatients with MDD and AD 1. Memantine (20 mg/d; n = 40) | Both treatments reduced depressive and anxiety symptoms. No significant differences between groups. Study did not examine alcohol-related outcomes |
| Martinotti et al, 200833 | Outpatients with comorbid AD and either bipolar disorder, schizoaffective disorder or borderline personality disorder. Open-label study 1. Quetiapine (300 to 800 mg/d; n = 28) | Quetiapine was associated with reduced alcohol consumption, alcohol craving, and intensity of psychiatric symptoms |
| AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder | ||
Interpreting the evidence
These findings underscore the importance of thorough evaluations. SSRIs are a first-line treatment for depression and as such probably should be the first choice for patients with comorbid AUDs. Drinking should be monitored closely and abstinence encouraged. Using medications that target AUDs is safe and modestly effective in patients with comorbid depression. Evidence suggests that treating both disorders simultaneously is more effective than treating either alone. Medications should be prescribed as part of a comprehensive treatment plan that also includes psychotherapy.
Related Resources
- Pettinati H. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
- Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
- Acamprosate • Campral
- Amitriptyline • Elavil
- Desipramine • Norpramin
- Disulfiram • Antabuse
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Ketamine • Ketalar
- Memantine • Namenda
- Mirtazapine • Remeron
- Naltrexone • Revia, Vivitrol
- Nefazodone • Serzone
- Quetiapine • Seroquel
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Petrakis receives research or grant support from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, the U.S. Department of Defense, and the U.S. Department of Veterans Affairs.
This work has been supported by a grant from the Veterans Affairs New England Mental Illness Research, Education, and Clinical Center and by the National Institute of Mental Health (T32MH062994-07).
Acknowledgements
The authors thank Elizabeth Guidone for her helpful comments and Diana Limoncelli for her assistance in manuscript preparation.
Discuss this article at www.facebook.com/CurrentPsychiatry
With a lifetime prevalence of 30%, alcohol use disorders (AUDs)—which in DSM-IV-TR include alcohol abuse and alcohol dependence—are among the most common psychiatric disorders.1 Depressive disorders, including major depressive disorder (MDD) and dysthymia, frequently co-occur with AUDs.2-4 This pattern of comorbidity adversely affects the prognosis, course, and treatment of both MDD and AUDs.5 High severity in 1 of these disorders is associated with high severity in the other.2,6 Alcohol dependence appears to prolong the course of depression7,8 and increases the risk of suicidal symptoms and behaviors.9,10 Patients with depression and AUDs are at increased risk of relapse to heavy drinking.7,11
Whether the high comorbidity rate of depressive disorders and AUDs is a result of 1 disorder causing the other (ie, AUDs leading to depression or vice versa) or can be attributed to shared etiology is unknown. Clinicians need to consider this question when treating patients with this pattern of comorbidity because distinguishing primary depression from secondary depression influences treatment decisions.12
There is a great need for pharmacologic interventions that can concurrently treat both depression and AUDs. This article reviews the evidence for current treatments for dually diagnosed patients and highlights novel agents that are worthy of further study for this complex patient population.
Current treatment options
Pharmacotherapy for MDD and alcohol dependence is common when these conditions occur alone. FDA-approved medications for treating depression include monoamine oxidase inhibitors, tricyclic antidepressants (TCAs), tetracyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors.
SSRIs are the most widely used class of antidepressants. They gained FDA approval based on studies conducted in non-comorbid patients because patients with comorbid conditions usually are excluded from research studies.13 Few trials have evaluated patients with depression and AUDs; TCAs and SSRIs are best studied in these patients.
Serotonergic antidepressants
SSRIs are first-line medications for MDD because of their low abuse potential, favorable side effect profile, and relative safety in overdose.
Table 1
Serotonergic antidepressants for patients with AUDs and depression
| Study | Sample | Results |
|---|---|---|
| Cornelius et al, 199714 | Outpatients with severe major depression and AD 1. Fluoxetine (20 to 40 mg/d; n = 25) 2. Placebo (n = 26) | Greater reductions in depressive symptoms and drinking in patients treated with fluoxetine compared with placebo |
| Roy, 199815 | Inpatients with current major depression and AD who were abstinent for ≥2 weeks 1. Sertraline (100 mg/d; n = 18) 2. Placebo (n = 18) | Greater reductions in depressive symptoms in patients treated with sertraline compared with placebo. Drinking outcomes were not emphasized because 35 of 36 patients reported continuous abstinence throughout the trial |
| Kranzler et al, 199516 | Outpatients with AD. Fourteen percent had current major depression. All received weekly individual or group CBT focused on relapse prevention and skills building 1. Fluoxetine (mean daily dose: 48 mg; n = 51) 2. Placebo (n = 50) | Significant decrease in alcohol consumption for both groups during the trial. No significant differences in alcohol consumption between groups. Among those with current depression, patients treated with fluoxetine experienced greater reduction in depressive symptoms vs placebo |
| Moak et al, 200317 | Currently depressed, actively drinking, alcohol-dependent outpatients. All received individual CBT 1. Sertraline (mean daily dose: 186 mg; n = 38) 2. Placebo (n = 44) | Sertraline had an advantage over placebo in reducing depressive symptoms in women but not in men. Sertraline reduced drinks per drinking day but not other drinking-related outcomes |
| Cornelius et al, 200918 | Adolescents (age 15 to 20) with AA or AD and MDD. All received intensive manual-based therapy (CBT for MDD and AUD, MET for AUD) almost weekly 1. Fluoxetine (20 mg/d; n = 24) 2. Placebo (n = 26) | All improved during the course of trial. No significant differences between fluoxetine and placebo groups in depression- or drinking-related outcomes |
| Roy-Byrne et al, 200019 | Actively drinking alcohol-dependent outpatients with history of ≥1 depressive episode. All received weekly group therapy for alcoholism 1. Nefazodone (mean daily dose: 460 mg; n = 32) 2. Placebo (n = 32) | Greater reduction in depressive symptoms but not in drinking-related outcomes in patients treated with nefazodone |
| Hernandez-Avila et al, 200420 | Outpatients with AD and current major depression. All received supportive psychotherapy for 10 weeks 1. Nefazodone (mean daily dose: 413 mg; n = 21) 2. Placebo (n = 20) | Depressive and anxiety symptoms declined significantly over time, but no statistically significant differences in depressive or anxiety symptoms between nefazodone and placebo. Patients treated with nefazodone had significantly greater reductions in heavy drinking days and in total drinks compared with placebo-treated patients |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; CBT: cognitive-behavioral therapy; MDD: major depressive disorder; MET: motivational enhancement therapy | ||
In a study of adolescents, Cornelius et al18 failed to find any differences between fluoxetine and placebo in any depression or drinking-related outcomes. This study compared the efficacy of fluoxetine, 20 mg/d, with placebo in 50 adolescents with MDD and AUDs who also received intensive, manual-based cognitive-behavioral therapy and motivational enhancement therapy. All patients improved during the trial, but there were no significant differences between fluoxetine- and placebo-treated adolescents.
Other serotonergic medications. Two studies have evaluated nefazodone, a serotonin (5-HT2) antagonist, in dually diagnosed patients. In a 12-week trial, Roy-Byrne et al19 evaluated the efficacy of nefazodone (mean daily dose: 460 mg) vs placebo in 64 actively drinking alcohol-dependent patients who had ≥1 prior episode of depression; all participated in a weekly psychoeducation group on alcoholism. Nefazodone was associated with significantly greater reduction in depressive symptoms but no reductions in drinking compared with placebo. However, a 10-week study of nefazodone20 (mean daily dose: 413 mg) vs placebo in 41 alcohol-dependent patients with current major depression found that those who received nefazodone significantly reduced heavy drinking days compared with the placebo group. There were no significant differences in depressive symptoms between groups.
Conflicting evidence on TCAs
Although several studies suggest TCAs may help reduce depressive symptoms in patients with AUDs, results on their ability to reduce drinking are conflicting (Table 2).24-26 In 1 study, 6 months of desipramine (mean daily dose: 200 mg) reduced drinking in 28 alcohol-dependent individuals with secondary depression24; in another, 12 weeks of imipramine plus weekly relapse prevention psychotherapy did not affect drinking-related outcomes in 69 actively drinking alcoholic outpatients with current depressive disorders.25
Table 2
Limited evidence supports TCAs for comorbid depression and AUDs
| Study | Sample | Results |
|---|---|---|
| Mason et al, 199624 | Outpatients with AD and secondary depression. Part of larger study including non-depressed patients with AD (N = 71) 1. Desipramine (mean daily dose 200 mg; n = 15) 2. Placebo (n = 13) | Greater reduction in depressive symptoms and drinking in desipramine-treated patients compared with placebo-treated patients |
| McGrath et al, 199625 | Outpatients with AD or AA and major depression, dysthymia, or depressive disorder NOS 1. Imipramine (mean daily dose 260 mg; n = 36) 2. Placebo (n = 33) | Greater reduction in depressive symptoms for imipramine-treated patients compared with placebo-treated patients. Drinking-related outcomes were not directly affected by medication except improvements in mood led to reduced alcohol use |
| Altintoprak et al, 200826 | Inpatients with AD and MDD 1. Mirtazapine (30 mg/d; n = 24) 2. Amitriptyline (100 mg/d; n = 20) | Drinking-related outcomes were not emphasized because all patients were required to abstain from drinking during the study. Both treatments reduced depressive symptoms; there were no significant differences between groups |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder; NOS: not otherwise specified; TCAs: tricyclic antidepressants | ||
Altintoprak et al26 compared the efficacy of the antidepressant mirtazapine, 30 mg/d, with the TCA amitriptyline, 100 mg/d, in 44 patients with comorbid alcohol dependence and MDD. All patients were required to abstain from drinking alcohol during the study. Both medications resulted in steady reductions in depressive symptoms and alcohol cravings; however, researchers found no significant differences between the 2 treatment groups.
Analyses of combined studies
Pettinati27 conducted a qualitative review of antidepressants for patients with depression and alcohol dependence that included 8 controlled clinical trials (2 on TCAs and 6 on serotonergic medications) conducted between 1994 and 2004. In this review, both TCAs and serotonergic medications were similarly effective in reducing depressive symptoms but not consistently effective in reducing drinking.
27 this review suggested that antidepressants can reduce depressive symptoms but not drinking. The authors also found evidence that the more the antidepressant reduced depressive symptoms, the more it reduced alcohol use. Studies published after these reviews have not substantially altered these findings.
Alcohol abuse medications
Four medications are FDA-approved for treating alcohol dependence:
- disulfiram
- naltrexone (in 2 formulations: oral and long-acting injectable)
- acamprosate.
Table 3
Can medications that target alcohol use also improve depression?
| Study | Sample | Results |
|---|---|---|
| Petrakis et al, 200729 | Outpatients with AD and an axis I disorder, including depression (secondary analysis of Petrakis et al, 200531) 1. Naltrexone (50 mg/d; n = 34) 2. Disulfiram (250 mg/d; n = 43) 3. Naltrexone + disulfiram (n = 28) 4. Placebo (n = 34) | Generally, medication was more effective than no medication. No advantage of 1 medication over the other. There was no relationship between depression diagnosis and medication condition, which suggests that for patients with depression, there was no advantage to medication |
| Pettinati et al, 201030 | Outpatients with AD and MDD 1. Sertraline (200 mg/d; n = 40) 2. Naltrexone (100 mg/d; n = 49) 3. Sertraline + naltrexone (n = 42) 4. Placebo (n = 39) | Greater proportion of patients in combined medication group abstained from alcohol and refrained from heavy alcohol use during the trial compared with those in sertraline-only or naltrexone-only groups. No significant differences among groups on depression-related outcomes |
| AD: alcohol dependence; MDD: major depressive disorder | ||
Nevertheless, these studies suggest that medications for treating depression or AUDs have, at best, only a modest effect in patients with both disorders.
Novel agents
Several novel medications have been evaluated as possible treatments for comorbid depression and AUDs because they target the underlying neurobiology of both disorders:
- agents that target the neurotransmitter glutamate, including the N-methyl-d-aspartate glutamate receptor antagonists memantine and ketamine
- dopaminergic agents such as quetiapine
- corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists.
In a case study, a 55-year-old man with treatment-resistant major depression and co-occurring alcohol and benzodiazepine dependence who received a single dose of IV ketamine, 0.5 mg/kg over 50 minutes, experienced “significant improvements” in depressive symptoms that lasted throughout the 7-day follow-up.33 This study did not report on ketamine’s effects on his alcohol use.
The atypical antipsychotic quetiapine acts as a serotonin (5-HT1A and 5-HT2) and dopamine (D1 and D2) antagonist, and reports suggest it reduces alcohol craving and affective symptoms in patients with AUDs.34,35 In a 16-week, open-label study, quetiapine decreased alcohol consumption, alcohol craving, and intensity of some psychiatric symptoms in 28 alcohol-dependent patients with bipolar disorder, schizoaffective disorder, or borderline personality disorder.36
See the Box for a description of the role CRF1 antagonists may play in treating patients with concurrent MDD and AUDs. See Table 4 for studies of memantine and quetiapine in treating depression with AUDs.
Corticotropin-releasing factor (CRF) has a well-established role in stress and has been implicated for treating anxiety and depressive disorders. Evidence also suggests that CRF receptor 1 (CRF1) may be a treatment target for alcohol use disorders (AUDs). Acute alcohol withdrawal and prolonged alcohol use are associated with elevated levels of extrahypothalamic CRF and correlated anxiety. CRF antagonists can reduce the anxiogenic effects of alcohol withdrawal and reduce some symptoms of alcohol dependence, including excessive alcohol self-administration and stress-induced relapse to alcohol use in rats with alcohol dependence, but not in those without dependence. Therefore, CRF1 receptor antagonists may be especially helpful for individuals who use alcohol to reduce negative emotional states such as anxiety or dysphoria, including those with concurrent major depressive disorder and AUDs.
Bibliography
Funk CK, Zorrilla EP, Lee MJ, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61(1):78-86.
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27(10):2718-2726.
Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32(9):1535-1542.
Other agents may play a role in treating depression with AUDs
| Study | Sample | Results |
|---|---|---|
| Muhonen et al, 200832 | Outpatients with MDD and AD 1. Memantine (20 mg/d; n = 40) | Both treatments reduced depressive and anxiety symptoms. No significant differences between groups. Study did not examine alcohol-related outcomes |
| Martinotti et al, 200833 | Outpatients with comorbid AD and either bipolar disorder, schizoaffective disorder or borderline personality disorder. Open-label study 1. Quetiapine (300 to 800 mg/d; n = 28) | Quetiapine was associated with reduced alcohol consumption, alcohol craving, and intensity of psychiatric symptoms |
| AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder | ||
Interpreting the evidence
These findings underscore the importance of thorough evaluations. SSRIs are a first-line treatment for depression and as such probably should be the first choice for patients with comorbid AUDs. Drinking should be monitored closely and abstinence encouraged. Using medications that target AUDs is safe and modestly effective in patients with comorbid depression. Evidence suggests that treating both disorders simultaneously is more effective than treating either alone. Medications should be prescribed as part of a comprehensive treatment plan that also includes psychotherapy.
Related Resources
- Pettinati H. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
- Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
- Acamprosate • Campral
- Amitriptyline • Elavil
- Desipramine • Norpramin
- Disulfiram • Antabuse
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Ketamine • Ketalar
- Memantine • Namenda
- Mirtazapine • Remeron
- Naltrexone • Revia, Vivitrol
- Nefazodone • Serzone
- Quetiapine • Seroquel
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Petrakis receives research or grant support from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, the U.S. Department of Defense, and the U.S. Department of Veterans Affairs.
This work has been supported by a grant from the Veterans Affairs New England Mental Illness Research, Education, and Clinical Center and by the National Institute of Mental Health (T32MH062994-07).
Acknowledgements
The authors thank Elizabeth Guidone for her helpful comments and Diana Limoncelli for her assistance in manuscript preparation.
1. Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: results of a national survey. Drug Alcohol Depend. 1995;39(3):197-206.
3. Kessler RC, Crum RM, Warner LA, et al. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
4. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
5. Burns L, Teesson M, O’Neill K. The impact of comorbid anxiety and depression on alcohol treatment outcomes. Addiction. 2005;100(6):787-796.
6. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
7. Hasin DS, Tsai WY, Endicott J, et al. Five-year course of major depression: effects of comorbid alcoholism. J Affect Disord. 1996;41(1):63-70.
8. Mueller TI, Lavori PW, Keller MB, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
9. Cornelius JR, Salloum IM, Mezzich J, et al. Disproportionate suicidality in patients with comorbid major depression and alcoholism. Am J Psychiatry. 1995;152(3):358-364.
10. Sher L, Oquendo MA, Galfalvy HC, et al. The relationship of aggression to suicidal behavior in depressed patients with a history of alcoholism. Addict Behav. 2005;30(6):1144-1153.
11. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
12. Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9(5):374-380.
13. Oslin DW. Treatment of late-life depression complicated by alcohol dependence. Am J Geriatr Psychiatry. 2005;13(6):491-500.
14. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
15. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
16. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
17. Moak DH, Anton RF, Latham PK, et al. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
18. Cornelius JR, Bukstein OG, Wood DS, et al. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addict Behav. 2009;34(10):905-909.
19. Roy-Byrne PP, Pages KP, Russo JE, et al. Nefazodone treatment of major depression in alcohol-dependent patients: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2000;20(2):129-136.
20. Hernandez-Avila CA, Modesto-Lowe V, Feinn R, et al. Nefazodone treatment of comorbid alcohol dependence and major depression. Alcohol Clin Exp Res. 2004;28(3):433-440.
21. Kranzler HR, Burleson JA, Brown J, et al. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20(9):1534-1541.
22. Naranjo CA, Bremner KE, Lanctôt KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake dependence and problems. Addiction. 1995;90(1):87-99.
23. Pettinati HM, Volpicelli JR, Kranzler HR, et al. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24(7):1041-1049.
24. Mason BJ, Kocsis JH, Ritvo EC, et al. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275(10):761-767.
25. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
26. Altintoprak AE, Zorlu N, Coskunol H, et al. Effectiveness and tolerability of mirtazapine and amitriptyline in alcoholic patients with co-morbid depressive disorder: a randomized, double-blind study. Hum Psychopharmacol. 2008;23(4):313-319.
27. Pettinati HM. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
28. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
29. Petrakis I, Ralevski E, Nich C, et al. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
31. Petrakis IL, Poling J, Levinson C, et al. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
32. Muhonen LH, Lönnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392-399.
33. Liebrenz M, Borgeat A, Leisinger R, et al. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137(15-16):234-236.
34. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled pilot trial of quetiapine for the treatment of Type A and Type B alcoholism. J Clin Psychopharmacol. 2007;27(4):344-351.
35. Croissant B, Klein O, Gehrlein L, et al. Quetiapine in relapse prevention in alcoholics suffering from craving and affective symptoms: a case series. Eur Psychiatry. 2006;21(8):570-573.
36. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
1. Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: results of a national survey. Drug Alcohol Depend. 1995;39(3):197-206.
3. Kessler RC, Crum RM, Warner LA, et al. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
4. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
5. Burns L, Teesson M, O’Neill K. The impact of comorbid anxiety and depression on alcohol treatment outcomes. Addiction. 2005;100(6):787-796.
6. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
7. Hasin DS, Tsai WY, Endicott J, et al. Five-year course of major depression: effects of comorbid alcoholism. J Affect Disord. 1996;41(1):63-70.
8. Mueller TI, Lavori PW, Keller MB, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
9. Cornelius JR, Salloum IM, Mezzich J, et al. Disproportionate suicidality in patients with comorbid major depression and alcoholism. Am J Psychiatry. 1995;152(3):358-364.
10. Sher L, Oquendo MA, Galfalvy HC, et al. The relationship of aggression to suicidal behavior in depressed patients with a history of alcoholism. Addict Behav. 2005;30(6):1144-1153.
11. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
12. Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9(5):374-380.
13. Oslin DW. Treatment of late-life depression complicated by alcohol dependence. Am J Geriatr Psychiatry. 2005;13(6):491-500.
14. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
15. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
16. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
17. Moak DH, Anton RF, Latham PK, et al. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
18. Cornelius JR, Bukstein OG, Wood DS, et al. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addict Behav. 2009;34(10):905-909.
19. Roy-Byrne PP, Pages KP, Russo JE, et al. Nefazodone treatment of major depression in alcohol-dependent patients: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2000;20(2):129-136.
20. Hernandez-Avila CA, Modesto-Lowe V, Feinn R, et al. Nefazodone treatment of comorbid alcohol dependence and major depression. Alcohol Clin Exp Res. 2004;28(3):433-440.
21. Kranzler HR, Burleson JA, Brown J, et al. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20(9):1534-1541.
22. Naranjo CA, Bremner KE, Lanctôt KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake dependence and problems. Addiction. 1995;90(1):87-99.
23. Pettinati HM, Volpicelli JR, Kranzler HR, et al. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24(7):1041-1049.
24. Mason BJ, Kocsis JH, Ritvo EC, et al. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275(10):761-767.
25. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
26. Altintoprak AE, Zorlu N, Coskunol H, et al. Effectiveness and tolerability of mirtazapine and amitriptyline in alcoholic patients with co-morbid depressive disorder: a randomized, double-blind study. Hum Psychopharmacol. 2008;23(4):313-319.
27. Pettinati HM. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
28. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
29. Petrakis I, Ralevski E, Nich C, et al. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
31. Petrakis IL, Poling J, Levinson C, et al. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
32. Muhonen LH, Lönnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392-399.
33. Liebrenz M, Borgeat A, Leisinger R, et al. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137(15-16):234-236.
34. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled pilot trial of quetiapine for the treatment of Type A and Type B alcoholism. J Clin Psychopharmacol. 2007;27(4):344-351.
35. Croissant B, Klein O, Gehrlein L, et al. Quetiapine in relapse prevention in alcoholics suffering from craving and affective symptoms: a case series. Eur Psychiatry. 2006;21(8):570-573.
36. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
One twin has cerebral palsy; $103M verdict … and more
AFTER PREMATURE RUPTURE OF MEMBRANES at 25 weeks’ gestation, a woman went to the emergency department (ED) and was later released. Eight days later, she returned to the ED with abdominal pain; a soporific drug was administered. After several hours, it was determined that she was in labor. Twins were delivered vaginally. One child has cerebral palsy and requires assistance in daily activities, although her cognitive function is intact.
PARENTS’ CLAIM The mother should not have been released after premature rupture of her membranes. The nurses and ObGyns failed to timely recognize that the mother was in labor, and failed to prevent premature delivery. Proper recognition of contractions would have allowed for administration of a tocolytic to delay delivery. That drug had been effectively administered during the first two trimesters of the pregnancy. A cesarean delivery should have been performed.
DEFENDANTS’ DEFENSE There was no negligence. The hospital argued that fetal heart-rate monitors did not suggest contractions.
VERDICT A $103 million New York verdict was returned against the hospital; a defense verdict was returned for the physicians.
Perforated uterus and severed iliac artery after D&C
A GYNECOLOGIC SURGEON performed a dilation and curettage (D&C) on a 47-year-old woman. During surgery, the patient suffered a perforated uterus and a severed iliac artery, resulting in a myocardial infarction.
PATIENT’S CLAIM The surgeon failed to dilate the cervix appropriately to assess the cervical and endometrial cavity length, and then failed to use proper instrumentation in the uterus. He did not assess uterine shape before the D&C. The patient suffered cognitive and emotional injuries, and will require additional surgery.
PHYSICIAN’S DEFENSE The patient’s anatomy is abnormal. A perforation is a known complication of a D&C.
VERDICT A $350,000 Wisconsin settlement was reached.
Failure to monitor a high-risk patient
A WOMAN WITH A HEART CONDITION who routinely took a beta-blocker plus migraine medication also had lupus. Her pregnancy was therefore at high risk for developing intrauterine growth restriction. Her US Navy ObGyn was advised by a maternal-fetal medicine (MFM) specialist to monitor the pregnancy closely with frequent ultrasonography and other tests that were never performed.
The baby was born by emergency cesarean delivery at 36 weeks’ gestation. The child suffered severe hypoxia and a brain hemorrhage just before delivery, which caused serious, permanent physical and neurologic injuries. He needs 24-hour care, is confined to a wheelchair, and requires a feeding tube.
PATIENT’S CLAIM The ObGyn failed to monitor the mother for fetal growth restriction as recommended by the MFM specialist.
DEFENDANTS’ DEFENSE There was no negligence; the mother was treated properly.
VERDICT After a $28 million Virginia verdict was awarded, the parties continued to dispute whether the judgment would be paid under California law (where the child was born) or Virginia law (where the case was filed). Prior to a rehearing, a $25 million settlement was reached.
Uterine cancer went undiagnosed
A WOMAN IN HER 50s saw her gynecologist in March 2004 to report vaginal staining. She did not return to the physician’s office until January 2005, when she reported daily vaginal bleeding. Ultrasonography showed a 4-cm mass in the endometrial cavity, consistent with a large polyp. A hysteroscopy and biopsy revealed that the woman had uterine cancer. She underwent a hysterectomy and radiation therapy, but the cancer metastasized to her lungs and she died in October 2006.
ESTATE’S CLAIM The gynecologist failed to diagnose uterine cancer in a timely manner.
PHYSICIAN’S DEFENSE The patient’s cancer was aggressive; an earlier diagnosis would not have changed the outcome.
VERDICT A $820,000 Massachusetts settlement was reached.
WHEN A 51-YEAR-OLD WOMAN NOTICED A BULGE in her vagina, she consulted her gynecologist. He determined the cause to be a cystocele and rectocele, and recommended a tension-free vaginal tape–obturator (TVT-O) procedure with anterior and posterior colporrhaphy.
The patient awoke from surgery in severe pain and was told that she had lost a lot of blood. Two weeks later, the physician explained that the stitches, not yet absorbed, were causing an abrasion, and that more vaginal tissue had been removed than planned.
Two more weeks passed, and the patient used a mirror to look at her vagina but could not see the opening. The TVT-O tape had created a ridge of tissue in the anterior vagina, causing severe stenosis. Vaginal dilators were required to expand the vagina. Entrapment of the dorsal clitoral nerve by the TVT-O tape was also discovered. The patient continues to experience dyspareunia and groin pain.
PATIENT’S CLAIM The gynecologist failed to tell her that, 2 months before surgery, the FDA had issued a public health warning about complications associated with transvaginal placement of surgical mesh during prolapse and urinary incontinence repair. Nor was she informed that the defendant had just completed training in TVT-O surgery, was not fully credentialed, and was proctored during the procedure.
PHYSICIAN’S DEFENSE The case was settled before the trial concluded.
VERDICT A $390,000 Virginia settlement was reached.
Lumpectomy, though no mass palpated
A 52-YEAR-OLD WOMAN FOUND A LUMP in her left breast. Her internist ordered mammography, which identified a 2-cm oval, asymmetrical density in the upper inner quadrant of the left breast. The radiologist recommended ultrasonography (US).
The patient consulted a surgical oncologist, who performed fine-needle aspiration. Pathology identified “clusters of malignant cells consistent with carcinoma,” and suggested a confirmatory biopsy. The oncologist recommended lumpectomy and sentinel node biopsy.
On the day of surgery, the patient could not locate the mass. The oncologist testified that he had palpated it. During surgery, gross examination did not show a mass or tumor. Frozen sections of sentinel nodes did not reveal evidence of cancer.
The patient suffered postsurgical seromas and lymphedema. The lymphedema has partially resolved, but causes pain in her left arm and breast.
PATIENT’S CLAIM The surgical oncologist should have performed US before surgery. It was negligent to continue with surgery when there were negative intraoperative findings for cancer or a mass.
PHYSICIAN’S DEFENSE Proper care was provided.
VERDICT A $950,000 Illinois verdict was returned.
Genetic testing fails to identify cystic fibrosis in one twin
AFTER HAVING ONE CHILD with cystic fibrosis (CF), parents underwent genetic testing. Embryos were prepared for in vitro fertilization (IVF) and sent to a genetic-testing laboratory. The lab reported that the embryos were negative for CF. Two embryos were implanted, and the mother gave birth to twins, one of which has CF.
PARENTS’ CLAIM Multiple errors by the genetic-testing laboratory led to an incorrect report on the embryos. The parents claimed wrongful birth.
DEFENDANTS’ DEFENSE The testing laboratory and physician owner argued that amniocentesis should have been performed during the pregnancy to rule out CF.
VERDICT The trial judge denied the use of the amniocentesis defense because an abortion would have been the only option available, and abortion is against the public policy of Tennessee. The court entered summary judgment on liability for the parents.
A $13 million verdict was returned, including $7 million to the parents for emotional distress.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
AFTER PREMATURE RUPTURE OF MEMBRANES at 25 weeks’ gestation, a woman went to the emergency department (ED) and was later released. Eight days later, she returned to the ED with abdominal pain; a soporific drug was administered. After several hours, it was determined that she was in labor. Twins were delivered vaginally. One child has cerebral palsy and requires assistance in daily activities, although her cognitive function is intact.
PARENTS’ CLAIM The mother should not have been released after premature rupture of her membranes. The nurses and ObGyns failed to timely recognize that the mother was in labor, and failed to prevent premature delivery. Proper recognition of contractions would have allowed for administration of a tocolytic to delay delivery. That drug had been effectively administered during the first two trimesters of the pregnancy. A cesarean delivery should have been performed.
DEFENDANTS’ DEFENSE There was no negligence. The hospital argued that fetal heart-rate monitors did not suggest contractions.
VERDICT A $103 million New York verdict was returned against the hospital; a defense verdict was returned for the physicians.
Perforated uterus and severed iliac artery after D&C
A GYNECOLOGIC SURGEON performed a dilation and curettage (D&C) on a 47-year-old woman. During surgery, the patient suffered a perforated uterus and a severed iliac artery, resulting in a myocardial infarction.
PATIENT’S CLAIM The surgeon failed to dilate the cervix appropriately to assess the cervical and endometrial cavity length, and then failed to use proper instrumentation in the uterus. He did not assess uterine shape before the D&C. The patient suffered cognitive and emotional injuries, and will require additional surgery.
PHYSICIAN’S DEFENSE The patient’s anatomy is abnormal. A perforation is a known complication of a D&C.
VERDICT A $350,000 Wisconsin settlement was reached.
Failure to monitor a high-risk patient
A WOMAN WITH A HEART CONDITION who routinely took a beta-blocker plus migraine medication also had lupus. Her pregnancy was therefore at high risk for developing intrauterine growth restriction. Her US Navy ObGyn was advised by a maternal-fetal medicine (MFM) specialist to monitor the pregnancy closely with frequent ultrasonography and other tests that were never performed.
The baby was born by emergency cesarean delivery at 36 weeks’ gestation. The child suffered severe hypoxia and a brain hemorrhage just before delivery, which caused serious, permanent physical and neurologic injuries. He needs 24-hour care, is confined to a wheelchair, and requires a feeding tube.
PATIENT’S CLAIM The ObGyn failed to monitor the mother for fetal growth restriction as recommended by the MFM specialist.
DEFENDANTS’ DEFENSE There was no negligence; the mother was treated properly.
VERDICT After a $28 million Virginia verdict was awarded, the parties continued to dispute whether the judgment would be paid under California law (where the child was born) or Virginia law (where the case was filed). Prior to a rehearing, a $25 million settlement was reached.
Uterine cancer went undiagnosed
A WOMAN IN HER 50s saw her gynecologist in March 2004 to report vaginal staining. She did not return to the physician’s office until January 2005, when she reported daily vaginal bleeding. Ultrasonography showed a 4-cm mass in the endometrial cavity, consistent with a large polyp. A hysteroscopy and biopsy revealed that the woman had uterine cancer. She underwent a hysterectomy and radiation therapy, but the cancer metastasized to her lungs and she died in October 2006.
ESTATE’S CLAIM The gynecologist failed to diagnose uterine cancer in a timely manner.
PHYSICIAN’S DEFENSE The patient’s cancer was aggressive; an earlier diagnosis would not have changed the outcome.
VERDICT A $820,000 Massachusetts settlement was reached.
WHEN A 51-YEAR-OLD WOMAN NOTICED A BULGE in her vagina, she consulted her gynecologist. He determined the cause to be a cystocele and rectocele, and recommended a tension-free vaginal tape–obturator (TVT-O) procedure with anterior and posterior colporrhaphy.
The patient awoke from surgery in severe pain and was told that she had lost a lot of blood. Two weeks later, the physician explained that the stitches, not yet absorbed, were causing an abrasion, and that more vaginal tissue had been removed than planned.
Two more weeks passed, and the patient used a mirror to look at her vagina but could not see the opening. The TVT-O tape had created a ridge of tissue in the anterior vagina, causing severe stenosis. Vaginal dilators were required to expand the vagina. Entrapment of the dorsal clitoral nerve by the TVT-O tape was also discovered. The patient continues to experience dyspareunia and groin pain.
PATIENT’S CLAIM The gynecologist failed to tell her that, 2 months before surgery, the FDA had issued a public health warning about complications associated with transvaginal placement of surgical mesh during prolapse and urinary incontinence repair. Nor was she informed that the defendant had just completed training in TVT-O surgery, was not fully credentialed, and was proctored during the procedure.
PHYSICIAN’S DEFENSE The case was settled before the trial concluded.
VERDICT A $390,000 Virginia settlement was reached.
Lumpectomy, though no mass palpated
A 52-YEAR-OLD WOMAN FOUND A LUMP in her left breast. Her internist ordered mammography, which identified a 2-cm oval, asymmetrical density in the upper inner quadrant of the left breast. The radiologist recommended ultrasonography (US).
The patient consulted a surgical oncologist, who performed fine-needle aspiration. Pathology identified “clusters of malignant cells consistent with carcinoma,” and suggested a confirmatory biopsy. The oncologist recommended lumpectomy and sentinel node biopsy.
On the day of surgery, the patient could not locate the mass. The oncologist testified that he had palpated it. During surgery, gross examination did not show a mass or tumor. Frozen sections of sentinel nodes did not reveal evidence of cancer.
The patient suffered postsurgical seromas and lymphedema. The lymphedema has partially resolved, but causes pain in her left arm and breast.
PATIENT’S CLAIM The surgical oncologist should have performed US before surgery. It was negligent to continue with surgery when there were negative intraoperative findings for cancer or a mass.
PHYSICIAN’S DEFENSE Proper care was provided.
VERDICT A $950,000 Illinois verdict was returned.
Genetic testing fails to identify cystic fibrosis in one twin
AFTER HAVING ONE CHILD with cystic fibrosis (CF), parents underwent genetic testing. Embryos were prepared for in vitro fertilization (IVF) and sent to a genetic-testing laboratory. The lab reported that the embryos were negative for CF. Two embryos were implanted, and the mother gave birth to twins, one of which has CF.
PARENTS’ CLAIM Multiple errors by the genetic-testing laboratory led to an incorrect report on the embryos. The parents claimed wrongful birth.
DEFENDANTS’ DEFENSE The testing laboratory and physician owner argued that amniocentesis should have been performed during the pregnancy to rule out CF.
VERDICT The trial judge denied the use of the amniocentesis defense because an abortion would have been the only option available, and abortion is against the public policy of Tennessee. The court entered summary judgment on liability for the parents.
A $13 million verdict was returned, including $7 million to the parents for emotional distress.
AFTER PREMATURE RUPTURE OF MEMBRANES at 25 weeks’ gestation, a woman went to the emergency department (ED) and was later released. Eight days later, she returned to the ED with abdominal pain; a soporific drug was administered. After several hours, it was determined that she was in labor. Twins were delivered vaginally. One child has cerebral palsy and requires assistance in daily activities, although her cognitive function is intact.
PARENTS’ CLAIM The mother should not have been released after premature rupture of her membranes. The nurses and ObGyns failed to timely recognize that the mother was in labor, and failed to prevent premature delivery. Proper recognition of contractions would have allowed for administration of a tocolytic to delay delivery. That drug had been effectively administered during the first two trimesters of the pregnancy. A cesarean delivery should have been performed.
DEFENDANTS’ DEFENSE There was no negligence. The hospital argued that fetal heart-rate monitors did not suggest contractions.
VERDICT A $103 million New York verdict was returned against the hospital; a defense verdict was returned for the physicians.
Perforated uterus and severed iliac artery after D&C
A GYNECOLOGIC SURGEON performed a dilation and curettage (D&C) on a 47-year-old woman. During surgery, the patient suffered a perforated uterus and a severed iliac artery, resulting in a myocardial infarction.
PATIENT’S CLAIM The surgeon failed to dilate the cervix appropriately to assess the cervical and endometrial cavity length, and then failed to use proper instrumentation in the uterus. He did not assess uterine shape before the D&C. The patient suffered cognitive and emotional injuries, and will require additional surgery.
PHYSICIAN’S DEFENSE The patient’s anatomy is abnormal. A perforation is a known complication of a D&C.
VERDICT A $350,000 Wisconsin settlement was reached.
Failure to monitor a high-risk patient
A WOMAN WITH A HEART CONDITION who routinely took a beta-blocker plus migraine medication also had lupus. Her pregnancy was therefore at high risk for developing intrauterine growth restriction. Her US Navy ObGyn was advised by a maternal-fetal medicine (MFM) specialist to monitor the pregnancy closely with frequent ultrasonography and other tests that were never performed.
The baby was born by emergency cesarean delivery at 36 weeks’ gestation. The child suffered severe hypoxia and a brain hemorrhage just before delivery, which caused serious, permanent physical and neurologic injuries. He needs 24-hour care, is confined to a wheelchair, and requires a feeding tube.
PATIENT’S CLAIM The ObGyn failed to monitor the mother for fetal growth restriction as recommended by the MFM specialist.
DEFENDANTS’ DEFENSE There was no negligence; the mother was treated properly.
VERDICT After a $28 million Virginia verdict was awarded, the parties continued to dispute whether the judgment would be paid under California law (where the child was born) or Virginia law (where the case was filed). Prior to a rehearing, a $25 million settlement was reached.
Uterine cancer went undiagnosed
A WOMAN IN HER 50s saw her gynecologist in March 2004 to report vaginal staining. She did not return to the physician’s office until January 2005, when she reported daily vaginal bleeding. Ultrasonography showed a 4-cm mass in the endometrial cavity, consistent with a large polyp. A hysteroscopy and biopsy revealed that the woman had uterine cancer. She underwent a hysterectomy and radiation therapy, but the cancer metastasized to her lungs and she died in October 2006.
ESTATE’S CLAIM The gynecologist failed to diagnose uterine cancer in a timely manner.
PHYSICIAN’S DEFENSE The patient’s cancer was aggressive; an earlier diagnosis would not have changed the outcome.
VERDICT A $820,000 Massachusetts settlement was reached.
WHEN A 51-YEAR-OLD WOMAN NOTICED A BULGE in her vagina, she consulted her gynecologist. He determined the cause to be a cystocele and rectocele, and recommended a tension-free vaginal tape–obturator (TVT-O) procedure with anterior and posterior colporrhaphy.
The patient awoke from surgery in severe pain and was told that she had lost a lot of blood. Two weeks later, the physician explained that the stitches, not yet absorbed, were causing an abrasion, and that more vaginal tissue had been removed than planned.
Two more weeks passed, and the patient used a mirror to look at her vagina but could not see the opening. The TVT-O tape had created a ridge of tissue in the anterior vagina, causing severe stenosis. Vaginal dilators were required to expand the vagina. Entrapment of the dorsal clitoral nerve by the TVT-O tape was also discovered. The patient continues to experience dyspareunia and groin pain.
PATIENT’S CLAIM The gynecologist failed to tell her that, 2 months before surgery, the FDA had issued a public health warning about complications associated with transvaginal placement of surgical mesh during prolapse and urinary incontinence repair. Nor was she informed that the defendant had just completed training in TVT-O surgery, was not fully credentialed, and was proctored during the procedure.
PHYSICIAN’S DEFENSE The case was settled before the trial concluded.
VERDICT A $390,000 Virginia settlement was reached.
Lumpectomy, though no mass palpated
A 52-YEAR-OLD WOMAN FOUND A LUMP in her left breast. Her internist ordered mammography, which identified a 2-cm oval, asymmetrical density in the upper inner quadrant of the left breast. The radiologist recommended ultrasonography (US).
The patient consulted a surgical oncologist, who performed fine-needle aspiration. Pathology identified “clusters of malignant cells consistent with carcinoma,” and suggested a confirmatory biopsy. The oncologist recommended lumpectomy and sentinel node biopsy.
On the day of surgery, the patient could not locate the mass. The oncologist testified that he had palpated it. During surgery, gross examination did not show a mass or tumor. Frozen sections of sentinel nodes did not reveal evidence of cancer.
The patient suffered postsurgical seromas and lymphedema. The lymphedema has partially resolved, but causes pain in her left arm and breast.
PATIENT’S CLAIM The surgical oncologist should have performed US before surgery. It was negligent to continue with surgery when there were negative intraoperative findings for cancer or a mass.
PHYSICIAN’S DEFENSE Proper care was provided.
VERDICT A $950,000 Illinois verdict was returned.
Genetic testing fails to identify cystic fibrosis in one twin
AFTER HAVING ONE CHILD with cystic fibrosis (CF), parents underwent genetic testing. Embryos were prepared for in vitro fertilization (IVF) and sent to a genetic-testing laboratory. The lab reported that the embryos were negative for CF. Two embryos were implanted, and the mother gave birth to twins, one of which has CF.
PARENTS’ CLAIM Multiple errors by the genetic-testing laboratory led to an incorrect report on the embryos. The parents claimed wrongful birth.
DEFENDANTS’ DEFENSE The testing laboratory and physician owner argued that amniocentesis should have been performed during the pregnancy to rule out CF.
VERDICT The trial judge denied the use of the amniocentesis defense because an abortion would have been the only option available, and abortion is against the public policy of Tennessee. The court entered summary judgment on liability for the parents.
A $13 million verdict was returned, including $7 million to the parents for emotional distress.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Vitamin deficiencies and mental health: How are they linked?
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
Options for treating antidepressant-induced sweating
Excessive sweating—diaphoresis—affects up to 22% of patients who take antidepressants.1 Diaphoresis may interfere with social and occupational activities, which can lead to medication discontinuation and prevent effective treatment. Stopping, decreasing, or changing antidepressants are options, but patients may be reluctant if the current dose has relieved their depressive symptoms. Adding a medication to reduce diaphoresis may be appropriate.
Sympathetic division of the peripheral nervous system signals cholinergic neurons to stimulate sweat gland secretion. In the CNS, thermoregulation occurs in the hypothalamus through a balanced and complex interaction among serotonergic and dopaminergic neurons.1 Consequently, oral medications to decrease sweating target peripheral or CNS neurons. Although evidence is limited to case reports, consider cholinergic and serotonergic antagonists and dopamine partial agonists to relieve antidepressant-induced diaphoresis.
Pharmacologic options
Peripherally, the anticholinergic agent benztropine reduced or eliminated diaphoresis at doses ranging from 0.5 mg every other day to 1 mg/d.2,3 Dry mouth was the only reported side effect.
Centrally acting serotonin antagonists may decrease diaphoresis through the 5-HT2A receptor, which signals the hypothalamus to raise body temperature. Cyproheptadine is an antihistamine with serotonin receptor antagonism. In case reports, it reduced or eliminated sweating in doses of 4 mg once or twice daily.4 Mild sedation was the only noted adverse effect. The norepinephrine and serotonin antagonist mirtazapine reduced diaphoresis within 2 weeks of initiation at 15 mg/d with no adverse effects.5 Sweating resolved after mirtazapine was titrated to 60 mg/d.
In addition to excess serotonin activity, diaphoresis may result from decreased dopaminergic tone in the hypothalamus. Centrally acting dopamine agonists—even partial agonists—may restore homeostasis and decrease sweating. Aripiprazole, 10 to 20 mg/d, reduced sweating in 2 patients; no adverse effects were reported.6
Agents to avoid
Antiadrenergic medications such as clonidine have decreased or exacerbated diaphoresis in studies.1 Similarly, paroxetine may alleviate or cause sweating. It is difficult to attribute paroxetine’s occasional effectiveness in reducing sweating solely to its anticholinergic properties because improvement may be attributed to an initial anxiolytic effect or efficacy in treating the underlying anxiety disorder.1
Disclosure
Dr. Scarff reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Marcy TR, Britton ML. Antidepressant-induced sweating. Ann Pharmacother. 2005;39(4):748-752.
2. Pierre JM, Guze BH. Benztropine for venlafaxine-induced night sweats. J Clin Psychopharmacol. 2000;20(2):269.-
3. Garber A, Gregory RJ. Benztropine in the treatment of venlafaxine-induced sweating. J Clin Psychiatry. 1997;58(4):176-177.
4. Ashton AK, Weinstein WL. Cyproheptadine for drug-induced sweating. Am J Psychiatry. 2002;159(5):874-875.
5. Buecking A, Vandeleur CL, Khazaal Y, et al. Mirtazapine in drug-induced excessive sweating. Eur J Clin Pharmacol. 2005;61(7):543-544.
6. Lu BY, Cullen CE, Eide CE, et al. Antidepressant-induced sweating alleviated by aripiprazole. J Clin Psychopharmacol. 2008;28(6):710-711.
Excessive sweating—diaphoresis—affects up to 22% of patients who take antidepressants.1 Diaphoresis may interfere with social and occupational activities, which can lead to medication discontinuation and prevent effective treatment. Stopping, decreasing, or changing antidepressants are options, but patients may be reluctant if the current dose has relieved their depressive symptoms. Adding a medication to reduce diaphoresis may be appropriate.
Sympathetic division of the peripheral nervous system signals cholinergic neurons to stimulate sweat gland secretion. In the CNS, thermoregulation occurs in the hypothalamus through a balanced and complex interaction among serotonergic and dopaminergic neurons.1 Consequently, oral medications to decrease sweating target peripheral or CNS neurons. Although evidence is limited to case reports, consider cholinergic and serotonergic antagonists and dopamine partial agonists to relieve antidepressant-induced diaphoresis.
Pharmacologic options
Peripherally, the anticholinergic agent benztropine reduced or eliminated diaphoresis at doses ranging from 0.5 mg every other day to 1 mg/d.2,3 Dry mouth was the only reported side effect.
Centrally acting serotonin antagonists may decrease diaphoresis through the 5-HT2A receptor, which signals the hypothalamus to raise body temperature. Cyproheptadine is an antihistamine with serotonin receptor antagonism. In case reports, it reduced or eliminated sweating in doses of 4 mg once or twice daily.4 Mild sedation was the only noted adverse effect. The norepinephrine and serotonin antagonist mirtazapine reduced diaphoresis within 2 weeks of initiation at 15 mg/d with no adverse effects.5 Sweating resolved after mirtazapine was titrated to 60 mg/d.
In addition to excess serotonin activity, diaphoresis may result from decreased dopaminergic tone in the hypothalamus. Centrally acting dopamine agonists—even partial agonists—may restore homeostasis and decrease sweating. Aripiprazole, 10 to 20 mg/d, reduced sweating in 2 patients; no adverse effects were reported.6
Agents to avoid
Antiadrenergic medications such as clonidine have decreased or exacerbated diaphoresis in studies.1 Similarly, paroxetine may alleviate or cause sweating. It is difficult to attribute paroxetine’s occasional effectiveness in reducing sweating solely to its anticholinergic properties because improvement may be attributed to an initial anxiolytic effect or efficacy in treating the underlying anxiety disorder.1
Disclosure
Dr. Scarff reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Excessive sweating—diaphoresis—affects up to 22% of patients who take antidepressants.1 Diaphoresis may interfere with social and occupational activities, which can lead to medication discontinuation and prevent effective treatment. Stopping, decreasing, or changing antidepressants are options, but patients may be reluctant if the current dose has relieved their depressive symptoms. Adding a medication to reduce diaphoresis may be appropriate.
Sympathetic division of the peripheral nervous system signals cholinergic neurons to stimulate sweat gland secretion. In the CNS, thermoregulation occurs in the hypothalamus through a balanced and complex interaction among serotonergic and dopaminergic neurons.1 Consequently, oral medications to decrease sweating target peripheral or CNS neurons. Although evidence is limited to case reports, consider cholinergic and serotonergic antagonists and dopamine partial agonists to relieve antidepressant-induced diaphoresis.
Pharmacologic options
Peripherally, the anticholinergic agent benztropine reduced or eliminated diaphoresis at doses ranging from 0.5 mg every other day to 1 mg/d.2,3 Dry mouth was the only reported side effect.
Centrally acting serotonin antagonists may decrease diaphoresis through the 5-HT2A receptor, which signals the hypothalamus to raise body temperature. Cyproheptadine is an antihistamine with serotonin receptor antagonism. In case reports, it reduced or eliminated sweating in doses of 4 mg once or twice daily.4 Mild sedation was the only noted adverse effect. The norepinephrine and serotonin antagonist mirtazapine reduced diaphoresis within 2 weeks of initiation at 15 mg/d with no adverse effects.5 Sweating resolved after mirtazapine was titrated to 60 mg/d.
In addition to excess serotonin activity, diaphoresis may result from decreased dopaminergic tone in the hypothalamus. Centrally acting dopamine agonists—even partial agonists—may restore homeostasis and decrease sweating. Aripiprazole, 10 to 20 mg/d, reduced sweating in 2 patients; no adverse effects were reported.6
Agents to avoid
Antiadrenergic medications such as clonidine have decreased or exacerbated diaphoresis in studies.1 Similarly, paroxetine may alleviate or cause sweating. It is difficult to attribute paroxetine’s occasional effectiveness in reducing sweating solely to its anticholinergic properties because improvement may be attributed to an initial anxiolytic effect or efficacy in treating the underlying anxiety disorder.1
Disclosure
Dr. Scarff reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Marcy TR, Britton ML. Antidepressant-induced sweating. Ann Pharmacother. 2005;39(4):748-752.
2. Pierre JM, Guze BH. Benztropine for venlafaxine-induced night sweats. J Clin Psychopharmacol. 2000;20(2):269.-
3. Garber A, Gregory RJ. Benztropine in the treatment of venlafaxine-induced sweating. J Clin Psychiatry. 1997;58(4):176-177.
4. Ashton AK, Weinstein WL. Cyproheptadine for drug-induced sweating. Am J Psychiatry. 2002;159(5):874-875.
5. Buecking A, Vandeleur CL, Khazaal Y, et al. Mirtazapine in drug-induced excessive sweating. Eur J Clin Pharmacol. 2005;61(7):543-544.
6. Lu BY, Cullen CE, Eide CE, et al. Antidepressant-induced sweating alleviated by aripiprazole. J Clin Psychopharmacol. 2008;28(6):710-711.
1. Marcy TR, Britton ML. Antidepressant-induced sweating. Ann Pharmacother. 2005;39(4):748-752.
2. Pierre JM, Guze BH. Benztropine for venlafaxine-induced night sweats. J Clin Psychopharmacol. 2000;20(2):269.-
3. Garber A, Gregory RJ. Benztropine in the treatment of venlafaxine-induced sweating. J Clin Psychiatry. 1997;58(4):176-177.
4. Ashton AK, Weinstein WL. Cyproheptadine for drug-induced sweating. Am J Psychiatry. 2002;159(5):874-875.
5. Buecking A, Vandeleur CL, Khazaal Y, et al. Mirtazapine in drug-induced excessive sweating. Eur J Clin Pharmacol. 2005;61(7):543-544.
6. Lu BY, Cullen CE, Eide CE, et al. Antidepressant-induced sweating alleviated by aripiprazole. J Clin Psychopharmacol. 2008;28(6):710-711.
Stiff person syndrome: What psychiatrists need to know
Stiff person syndrome (SPS) is a rare autoimmune condition characterized by stiffness and rigidity in the lower limb muscles. Because SPS often is misdiagnosed as a psychiatric illness and psychiatric comorbidities are common in patients with this disorder,1 awareness and recognition of this unique condition is essential.
An insidious presentation
Patients with SPS present with:2
- axial muscle stiffness slowly progressing to proximal muscles
- unremarkable motor, sensory, and cranial nerve examinations with normal intellectual functioning
- normal muscle strength, although electromyography shows continuous motor activity
- spasms evoked by sudden movements, jarring noise, and emotional distress
- slow and cautious gait to avoid triggering spasms and falls.
Symptoms start slowly and insidiously. Axial muscle stiffness can result in spinal deformity. Involvement is asymmetrical, with a predilection for proximal lower limb and lumbar paraspinal muscles. Affected muscles reveal tight, hard, board-like rigidity. In later stages of SPS, mild atrophy and muscle weakness are likely.
Frequent misdiagnosis
Because facial muscle spasticity is prominent, SPS patients may be misdiagnosed with Parkinson’s disease, primary lateral sclerosis, or multiple sclerosis. Spasms affecting respiratory and thoracic paraspinal muscles (status spasticus) may be misdiagnosed as an anxiety-related condition. These spasms can be life-threatening and require IV diazepam and supportive measures.
More than 60% of SPS patients have a comorbid psychiatric disorder.3 Anxiety disorders—generalized anxiety disorder, agoraphobia, and panic disorder—major depression, and alcohol abuse are the most frequent psychiatric comorbidities seen in SPS patients.3
SPS patients who panic when in public may be misdiagnosed with agoraphobia.3 Emotional stimuli may cause muscle spasms leading to falls. Treating muscle spasticity with γ-aminobutyric acid (GABA) agonists and narcotics can lead to drug abuse and dependence. Muscle spasticity can fluctuate from hour to hour, abate with sleep, and get worse with emotional distress. These findings are why approximately 70% of SPS patients are initially misdiagnosed; conversion disorder is a frequent misdiagnosis.4 Mood disorder in SPS patients may be resistant to antidepressants until these patients are treated with immunotherapy.4
Treating SPS patients
Although early intervention can reduce long-term disability, approximately 50% of SPS patients eventually have to use a wheelchair as a result of pain and immobility.5
Antibodies to glutamic acid decarboxylase, which is the rate-limiting enzyme for GABA synthesis, are present in 85% of SPS patients.5 Therefore, treatment usually includes GABA-enhancing drugs, including sedative anxiolytics (clonazepam and diazepam), antiepileptics (gabapentin, levetiracetam, tiagabine, and vigabatrin), antispasticity drugs (baclofen, dantrolene, and tizanidine), and immunotherapy (corticosteroids, IV immunoglobulins, and rituximab).5 Antidepressants, biofeedback, and relaxation training also can offer relief. Psychotherapy and substance dependency interventions may be needed.
To achieve optimum outcomes in SPS patients, a close collaborative relationship among all treating clinicians—including primary care physicians, neurologists, anesthesiologists, and psychiatrists—is necessary.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Tinsley JA, Barth EM, Black JL, et al. Psychiatric consultations in stiff-man syndrome. J Clin Psychiatry. 1997;58(10):444-449.
2. Egwuonwu S, Chedebeau F. Stiff-person syndrome: a case report and review of the literature. J Natl Med Assoc. 2010;102(12):1261-1263.
3. Black JL, Barth EM, Williams DE, et al. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998;39(1):38-44.
4. Culav-Sumić J, Bosnjak I, Pastar Z, et al. Anxious depression and the stiff-person plus syndrome. Cogn Behav Neurol. 2008;21(4):242-245.
5. Hadavi S, Noyce AJ, Leslie RD, et al. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282.
Stiff person syndrome (SPS) is a rare autoimmune condition characterized by stiffness and rigidity in the lower limb muscles. Because SPS often is misdiagnosed as a psychiatric illness and psychiatric comorbidities are common in patients with this disorder,1 awareness and recognition of this unique condition is essential.
An insidious presentation
Patients with SPS present with:2
- axial muscle stiffness slowly progressing to proximal muscles
- unremarkable motor, sensory, and cranial nerve examinations with normal intellectual functioning
- normal muscle strength, although electromyography shows continuous motor activity
- spasms evoked by sudden movements, jarring noise, and emotional distress
- slow and cautious gait to avoid triggering spasms and falls.
Symptoms start slowly and insidiously. Axial muscle stiffness can result in spinal deformity. Involvement is asymmetrical, with a predilection for proximal lower limb and lumbar paraspinal muscles. Affected muscles reveal tight, hard, board-like rigidity. In later stages of SPS, mild atrophy and muscle weakness are likely.
Frequent misdiagnosis
Because facial muscle spasticity is prominent, SPS patients may be misdiagnosed with Parkinson’s disease, primary lateral sclerosis, or multiple sclerosis. Spasms affecting respiratory and thoracic paraspinal muscles (status spasticus) may be misdiagnosed as an anxiety-related condition. These spasms can be life-threatening and require IV diazepam and supportive measures.
More than 60% of SPS patients have a comorbid psychiatric disorder.3 Anxiety disorders—generalized anxiety disorder, agoraphobia, and panic disorder—major depression, and alcohol abuse are the most frequent psychiatric comorbidities seen in SPS patients.3
SPS patients who panic when in public may be misdiagnosed with agoraphobia.3 Emotional stimuli may cause muscle spasms leading to falls. Treating muscle spasticity with γ-aminobutyric acid (GABA) agonists and narcotics can lead to drug abuse and dependence. Muscle spasticity can fluctuate from hour to hour, abate with sleep, and get worse with emotional distress. These findings are why approximately 70% of SPS patients are initially misdiagnosed; conversion disorder is a frequent misdiagnosis.4 Mood disorder in SPS patients may be resistant to antidepressants until these patients are treated with immunotherapy.4
Treating SPS patients
Although early intervention can reduce long-term disability, approximately 50% of SPS patients eventually have to use a wheelchair as a result of pain and immobility.5
Antibodies to glutamic acid decarboxylase, which is the rate-limiting enzyme for GABA synthesis, are present in 85% of SPS patients.5 Therefore, treatment usually includes GABA-enhancing drugs, including sedative anxiolytics (clonazepam and diazepam), antiepileptics (gabapentin, levetiracetam, tiagabine, and vigabatrin), antispasticity drugs (baclofen, dantrolene, and tizanidine), and immunotherapy (corticosteroids, IV immunoglobulins, and rituximab).5 Antidepressants, biofeedback, and relaxation training also can offer relief. Psychotherapy and substance dependency interventions may be needed.
To achieve optimum outcomes in SPS patients, a close collaborative relationship among all treating clinicians—including primary care physicians, neurologists, anesthesiologists, and psychiatrists—is necessary.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Stiff person syndrome (SPS) is a rare autoimmune condition characterized by stiffness and rigidity in the lower limb muscles. Because SPS often is misdiagnosed as a psychiatric illness and psychiatric comorbidities are common in patients with this disorder,1 awareness and recognition of this unique condition is essential.
An insidious presentation
Patients with SPS present with:2
- axial muscle stiffness slowly progressing to proximal muscles
- unremarkable motor, sensory, and cranial nerve examinations with normal intellectual functioning
- normal muscle strength, although electromyography shows continuous motor activity
- spasms evoked by sudden movements, jarring noise, and emotional distress
- slow and cautious gait to avoid triggering spasms and falls.
Symptoms start slowly and insidiously. Axial muscle stiffness can result in spinal deformity. Involvement is asymmetrical, with a predilection for proximal lower limb and lumbar paraspinal muscles. Affected muscles reveal tight, hard, board-like rigidity. In later stages of SPS, mild atrophy and muscle weakness are likely.
Frequent misdiagnosis
Because facial muscle spasticity is prominent, SPS patients may be misdiagnosed with Parkinson’s disease, primary lateral sclerosis, or multiple sclerosis. Spasms affecting respiratory and thoracic paraspinal muscles (status spasticus) may be misdiagnosed as an anxiety-related condition. These spasms can be life-threatening and require IV diazepam and supportive measures.
More than 60% of SPS patients have a comorbid psychiatric disorder.3 Anxiety disorders—generalized anxiety disorder, agoraphobia, and panic disorder—major depression, and alcohol abuse are the most frequent psychiatric comorbidities seen in SPS patients.3
SPS patients who panic when in public may be misdiagnosed with agoraphobia.3 Emotional stimuli may cause muscle spasms leading to falls. Treating muscle spasticity with γ-aminobutyric acid (GABA) agonists and narcotics can lead to drug abuse and dependence. Muscle spasticity can fluctuate from hour to hour, abate with sleep, and get worse with emotional distress. These findings are why approximately 70% of SPS patients are initially misdiagnosed; conversion disorder is a frequent misdiagnosis.4 Mood disorder in SPS patients may be resistant to antidepressants until these patients are treated with immunotherapy.4
Treating SPS patients
Although early intervention can reduce long-term disability, approximately 50% of SPS patients eventually have to use a wheelchair as a result of pain and immobility.5
Antibodies to glutamic acid decarboxylase, which is the rate-limiting enzyme for GABA synthesis, are present in 85% of SPS patients.5 Therefore, treatment usually includes GABA-enhancing drugs, including sedative anxiolytics (clonazepam and diazepam), antiepileptics (gabapentin, levetiracetam, tiagabine, and vigabatrin), antispasticity drugs (baclofen, dantrolene, and tizanidine), and immunotherapy (corticosteroids, IV immunoglobulins, and rituximab).5 Antidepressants, biofeedback, and relaxation training also can offer relief. Psychotherapy and substance dependency interventions may be needed.
To achieve optimum outcomes in SPS patients, a close collaborative relationship among all treating clinicians—including primary care physicians, neurologists, anesthesiologists, and psychiatrists—is necessary.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Tinsley JA, Barth EM, Black JL, et al. Psychiatric consultations in stiff-man syndrome. J Clin Psychiatry. 1997;58(10):444-449.
2. Egwuonwu S, Chedebeau F. Stiff-person syndrome: a case report and review of the literature. J Natl Med Assoc. 2010;102(12):1261-1263.
3. Black JL, Barth EM, Williams DE, et al. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998;39(1):38-44.
4. Culav-Sumić J, Bosnjak I, Pastar Z, et al. Anxious depression and the stiff-person plus syndrome. Cogn Behav Neurol. 2008;21(4):242-245.
5. Hadavi S, Noyce AJ, Leslie RD, et al. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282.
1. Tinsley JA, Barth EM, Black JL, et al. Psychiatric consultations in stiff-man syndrome. J Clin Psychiatry. 1997;58(10):444-449.
2. Egwuonwu S, Chedebeau F. Stiff-person syndrome: a case report and review of the literature. J Natl Med Assoc. 2010;102(12):1261-1263.
3. Black JL, Barth EM, Williams DE, et al. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998;39(1):38-44.
4. Culav-Sumić J, Bosnjak I, Pastar Z, et al. Anxious depression and the stiff-person plus syndrome. Cogn Behav Neurol. 2008;21(4):242-245.
5. Hadavi S, Noyce AJ, Leslie RD, et al. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282.
Statins and diabetes risk: Fact, fiction, and clinical implications
On february 28, 2012, the US Food and Drug Administration (FDA) updated its labeling requirements for statins. In addition to revising its recommendations for monitoring liver function and its alerts about reports of memory loss, the FDA also warned of the possibility of new-onset diabetes mellitus and worse glycemic control in patients taking statin drugs.1
This change stoked an ongoing debate about the risk of diabetes with statin use and the implications of such an effect. To understand the clinical consequences of this alert and its effect on treatment decisions, we need to consider the degree to which statins lower the risk of cardiovascular disease in patients at high risk (including diabetic patients), the magnitude of the risk of developing new diabetes while on statin therapy, and the ratio of risk to benefit in treated populations.
This review will discuss the evidence for this possible adverse effect and the implications for clinical practice.
DO STATINS CAUSE DIABETES?
Individual controlled trials dating back more than a decade have had conflicting results about new diabetes and poorer diabetic control in patients taking statins.
The West of Scotland Coronary Prevention Study (WOSCOPS)2 suggested that the incidence of diabetes was 30% lower in patients taking pravastatin (Pravachol) 40 mg/day than with placebo. However, this was not observed with atorvastatin (Lipitor) 10 mg/day in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid-Lowering Arm (ASCOT-LLA)3 in hypertensive patients or in the Collaborative Atorvastatin Diabetes Study (CARDS)4 in diabetic patients,4 nor was it noted with simvastatin (Zocor) 40 mg/day in the Heart Protection Study (HPS).5
The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER),6 using the more potent agent rosuvastatin (Crestor) 20 mg/day in patients with elevated levels of C-reactive protein (CRP), was stopped early when an interim analysis found a 44% lower incidence of the primary end point. However, the trial also reported a 26% higher incidence of diabetes in follow-up of less than 2 years.
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER),7 with a mean age at entry of 75, there was a 32% higher incidence of diabetes with pravastatin therapy.7
Results of meta-analyses
Several meta-analyses have addressed these differences.
Rajpathak et al8 performed a meta-analysis, published in 2009, of six trials—WOSCOPS,2 ASCOT-LLA,3 JUPITER,6 HPS,5 the Long-term Intervention With Pravastatin in Ischaemic Disease (LIPID) study,9 and the Controlled Rosuvastatin Multinational Study in Heart Failure (CORONA),10 with a total of 57,593 patients. They calculated that the incidence of diabetes was 13% higher (an absolute difference of 0.5%) in statin recipients, which was statistically significant. In their initial analysis, the authors excluded WOSCOPS, describing it as hypothesis-generating. The relative increase in risk was less—6%—and was not statistically significant when WOSCOPS was included.
Sattar et al,11 in a larger meta-analysis published in 2010, included 91,140 participants in 13 major statin trials conducted between 1994 and 2009; each trial had more than 1,000 patients and more than 1 year of follow-up.2,3,5–7,9,10,12–17 New diabetes was defined as physician reporting of new diabetes, new diabetic medication use, or a fasting glucose greater than 7 mmol/L (126 mg/dL).
New diabetes occurred in 2,226 (4.89%) of the statin recipients and in 2,052 (4.5%) of the placebo recipients, an absolute difference of 0.39%, or 9% more (odds ratio [OR] 1.09; 95% confidence interval [CI] 1.02–1.17) (Figure 1).
The incidence of diabetes varied substantially among the 13 trials, with only JUPITER6 and PROSPER7 finding statistically significant increases in rates (26% and 32%, respectively). Of the other 11 trials, 4 had nonsignificant trends toward lower incidence,2,9,13,17 while the 7 others had nonsignificant trends toward higher incidence.
Does the specific statin make a difference?
Questions have been raised as to whether the type of statin used, the intensity of therapy, or the population studied contributed to these differences. Various studies suggest that factors such as using hydrophilic vs lipophilic statins (hydrophilic statins include pravastatin and rosuvastatin; lipophilic statins include atorvastatin, lovastatin, and simvastatin), the dose, the extent of lowering of low-density lipoprotein cholesterol (LDL-C), and the age or clinical characteristics of the population studied may influence this relationship.18–20
Yamakawa et al18 examined the effect of atorvastatin 10 mg/day, pravastatin 10 mg/day, and pitavastatin (Livalo) 2 mg/day on glycemic control over 3 months in a retrospective analysis. Random blood glucose and hemoglobin A1c levels were increased in the atorvastatin group but not in the other two.18
A prospective comparison of atorvastatin 20 mg vs pitavastatin 4 mg in patients with type 2 diabetes, presented at the American College of Cardiology’s 2011 annual meeting, reported a significant increase in fasting glucose levels with atorvastatin, particularly in women, but not with pitavastatin.19
In the Compare the Effect of Rosuvastatin With Atorvastatin on Apo B/Apo A-1 Ratio in Patients With Type 2 Diabetes Mellitus and Dyslipidaemia (CORALL) study,20 both high-dose rosuvastatin (40 mg) and high-dose atorvastatin (80 mg) were associated with significant increases in hemoglobin A1c, although the mean fasting glucose levels were not significantly different at 18 weeks of therapy.
A meta-analysis by Sattar et al11 did not find a clear difference between lipophilic statins (OR 1.10 vs placebo) and hydrophilic statins (OR 1.08). In analysis by statin type, the combined rosuvastatin trials were statistically significant in favor of a higher diabetes risk (OR 1.18, 95% CI 1.04–1.44). Nonsignificant trends were noted for atorvastatin trials (OR 1.14) and simvastatin trials (OR 1.11) and less so for pravastatin (OR 1.03); the OR for lovastatin was 0.98. This may suggest that there is a stronger effect with more potent statins or with greater lowering of LDL-C.
Meta-regression analysis in this study demonstrated that diabetes risk with statins was higher in older patients but was not influenced by body mass index or by the extent that LDL-C was lowered.
Statin dose as a risk factor
Intensive-dose statin therapy has been shown to reduce cardiovascular risk more than low-dose or moderate-dose therapy, thus supporting more aggressive treatment of LDL-C in higher-risk patients. However, some controlled studies comparing more-potent with less-potent statin regimens suggest that there may also be a higher risk of incident diabetes at higher doses.21–24
In a post hoc analysis of the Pravastatin or Atorvastatin Evaluation and Infection Therapy– Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial,21 patients who had experienced an acute coronary syndrome had a greater increase in hemoglobin A1c if treated with atorvastatin 80 mg/day than with pravastatin 40 mg/day.
Waters et al23 reported a higher risk of new diabetes with atorvastatin 80 mg than with placebo and a trend toward a higher risk with atorvastatin 80 mg than with atorvastatin 10 mg or simvastatin 20 mg.
In contrast, a review by Yousef et al24 of the data from the Enhanced Feedback for Effective Cardiac Treatment (EFFECT) study did not find a higher diabetes risk with more intensive statin therapy based on the magnitude of LDL-C reduction. A propensity-matched examination of deaths, recurrent acute ischemic events, or new diabetes in patients previously hospitalized with myocardial infarction found no differences in these end points each year out to 5 years. The risk of diabetes was in fact lower (but the difference was not statistically significant) in the high-dose groups out to 5 years. The risk of myocardial infarction or death was numerically different in the high-dose groups, but the difference was not statistically significant.
Preiss et al25 in 2011 performed a meta-analysis of the impact of intensity of statin therapy on diabetes risk. They examined data from 32,752 participants without diabetes at baseline in five randomized controlled trials with more than 1,000 participants and more than 1 year of follow-up, comparing high-dose therapy against moderate-dose statin therapy.21,22,26–28 New diabetes was considered present if there was an adverse event report of diabetes, if glucose-lowering drugs were started, or if two fasting plasma glucose measurements were higher than 7 mmol/L (126 mg/dL).
Diabetes developed in 1,449 (8.8%) of the intensive-therapy group and 1,300 (8.0%) of the moderate-therapy group (OR 1.12, 95% CI 1.04–1.22). In contrast, incident cardiovascular disease occurred in 3,134 (19.1%) of the intensive-therapy group and 3,550 (21.7%) of the moderate-therapy group (OR 0.84, 95% CI 0.75–0.94). Therefore, there was an 0.8% absolute increase in diabetes cases on high-dose statins and a 2.6% absolute reduction in adverse cardiovascular events.
CAUTION IN INTERPRETING THESE DATA
There are many reasons for caution in interpreting these studies.
The trials were not designed to look for diabetes
The data supporting the relationship between statin therapy and higher risk of diabetes are primarily from observational studies. These studies were not prospectively designed to address this question, and we therefore need to view this as association and not as causation.
The definition of diabetes varied between trials, and new-onset diabetes was often not rigorously screened for. In many trials the outcome of diabetes was at least partially based on nonstandardized, nonadjudicated physician reporting.
Consequently, if statins reduce the risk of diabetes, the results from WOSCOPS may overstate the reduction, since this study used a non-standard definition of incident diabetes (fasting plasma glucose > 126 mg/dL plus a > 36 mg/dL increase from baseline). When Sattar et al11 reanalyzed WOSCOPS data using a more standard definition, they found a smaller effect.
On the other hand, nonstandardized physician reporting may overstate an adverse effect. Sattar et al11 also found that when fasting plasma glucose levels alone were used as the definition for diabetes, the overall risk was attenuated and was no longer statistically significant (OR 1.07, 95% CI 0.97–1.17).
Perhaps statin therapy uncovers diabetes only in people at risk of diabetes
Perhaps statin therapy uncovers diabetes only in people at higher baseline risk of developing diabetes. Therefore, this adverse effect may be restricted to certain groups and not applicable to the general population.
In JUPITER, one of the two trials in which, on independent analysis, statin use was associated with new diabetes, 77% of patients in the rosuvastatin group who developed diabetes had impaired fasting glucose at entry and therefore were at higher risk of developing diabetes.6
Possibly, the relationship is driven by preexisting metabolic syndrome or other risk factors for diabetes. In the two studies that reported a statistically significantly higher incidence of new diabetes, more than 40% of patients in JUPITER met the criteria for metabolic syndrome, and metabolic syndrome, which increases in prevalence with age, was likely more prominent in the elderly population in PROSPER.
Waters et al23 grouped patients according to whether they had risk factors for diabetes (impaired fasting glucose, obesity, elevated triglycerides, and hypertension) and found that those who had none or one of these risk factors had no difference in the rate of new-onset diabetes with either moderate or intensive statin therapy, but the risk was pronounced in those who had three or four risk factors.
Ridker et al29 reanalyzed the JUPITER data from patients who did not have cardiovascular disease at baseline. Overall, for every 54 new cases of diabetes in follow-up, 134 cardiovascular events or deaths were prevented. In subgroup analysis, those who had one or more risk factors for diabetes at baseline (metabolic syndrome, impaired fasting glucose, obesity, or hemoglobin A1c > 6%) had a 39% reduction in the primary end point and a 28% increase in new diabetes. Those who had none of these risk factors had a 52% lower rate of cardiovascular events but no increase in diabetes.
Other confounding factors
Bias and confounding factors are difficult to control for in studies without prospectively defined, recognized, and analyzed outcomes.
Although it may be a bit of a stretch, residual confounding factors such as myalgia side effects while on statins may reduce exercise in the statin-treatment groups. Perhaps a change to a healthier lifestyle after cardiovascular events may be more common in placebo groups. Improved survival with statins may allow more people at risk of diabetes to live longer and present with the diagnosis.30
POSSIBLE EXPLANATIONS, BUT NO UNIFYING MECHANISM
If mechanisms could be identified to explain the association between statins and diabetes, this would strengthen the argument that it is a cause-and-effect relationship. Many explanations have been proposed as to how statins may influence glucose metabolism and insulin sensitivity.31–34 These are possible explanations based on other observations.
In theory, statins may improve insulin sensitivity via their anti-inflammatory effect, since inflammatory markers and proinflammatory cytokines have been linked with insulin resistance. However, other effects of statins may adversely affect glycemic control.
In vivo analysis has shown that some but not all statins increase insulin levels and decrease insulin sensitivity in a dose-dependent fashion. Some statins decrease adiponectin and may worsen glycemic control through loss of adiponectin’s proposed protective anti-proliferative and antiangiogenic properties. In vitro studies and animal studies have demonstrated a decrease in expression of insulin-responsive glucose transporter 4 (GLUT4) with atorvastatin, and an increase in GLUT1. It has been hypothesized that reduction in isoprenoid biosynthesis or decreased insulin signaling may explain these effects and that changes in glucose transport in adipocytes may cause insulin resistance. Other studies suggest that dysregulation of cellular cholesterol may attenuate beta-cell function. Impaired biosynthesis of ubiquinones may result in delayed production of adenosine triphosphate and consequently diminish insulin release.
But different effects have been reported for atorvastatin, simvastatin, and pravastatin, arguing against a unifying explanation or, alternatively, suggesting that differences in lipophilicity and potency among statins are important. Hydrophilic statins may be less likely to be taken up by extrahepatic cells such as pancreatic cells and adipocytes, possibly lessening these effects. However, the strong association between rosuvastatin (which is hydrophilic) and new diabetes would not support this hypothesis.
Despite these speculations, lack of conformity in response to different statins and discrepancies in the clinical outcomes noted in trials fail to clearly identify a common causative mechanism.
OTHER COMMON THERAPIES MAY INFLUENCE GLYCEMIC CONTROL
Statins are not the first drugs for reducing cardiovascular risk that have been shown to affect glucose levels during treatment.
Niacin
Niacin has been known to increase glucose levels but has long been used as a treatment for dyslipidemia despite this caution. Reduced glycemic control during niacin treatment in diabetic patients does not seem to alter the beneficial effects of treatment.35–37
In a post hoc analysis of the Coronary Drug Project (CDP), in patient subgroups defined by baseline fasting plasma glucose and compared with placebo, niacin reduced the 6-year risk of recurrent myocardial infarction and the combined end point of coronary heart disease death or nonfatal myocardial infarction similarly (interactive P value nonsignificant) across all levels of baseline fasting plasma glucose, including levels of 126 mg/dL or higher at study entry.36
In another post hoc analysis of CDP patient subgroups defined by the change in glycemic status from baseline to 1 year, niacin reduced the 6-year risk of the same end points similarly (interactive P value nonsignificant) across all levels of change in fasting plasma glucose from baseline to year 1, whether baseline fasting plasma glucose levels decreased, stayed the same, or increased to 10 mg/dL or higher on niacin therapy.36
Therefore, the beneficial effect of niacin of reducing the rate of recurrent nonfatal myocardial infarction and coronary heart disease events was not significantly diminished when impaired fasting glucose or diabetes was present when therapy was started or by on-therapy increases from baseline fasting plasma glucose.
In addition, on-therapy changes in glycemic control may be dose-related and minimized by surveillance and therapy adjustments. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT)38 found that changes in glycemic control were minimal as measured by fasting glucose and hemoglobin A1c; were associated with a higher niacin dose (1.5 g/day vs 1 g/day); and, when present, were successfully managed by adjusting the diabetes treatment regimen.
Antihypertensive drugs
Diuretics as well as beta-blockers have been reported to increase the incidence of diabetes in patients with hypertension.15,38–40
A retrospective longitudinal cohort study40 in 2009 examined the development of new-onset diabetes (defined as a new ICD-9 code for diabetes or initiation of diabetes treatment) in 24,688 treated hypertensive patients without diabetes at baseline; 4,385 (17.8%) of the patients developed diabetes. After adjusting for sex and age, the risk of new diabetes was significant in users of diuretics (OR 1.10), beta-blockers (OR 1.12), and calcium channel blockers (OR 1.10) compared with users of angiotensin-converting enzyme inhibitors, (OR 0.92), angiotensin receptor blockers (OR 0.90), or alpha-blockers (OR 0.88).
However, the increase in blood glucose does not seem to attenuate the beneficial effects of reducing cardiovascular events. In the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT),15 a long-term follow-up of those developing new-onset diabetes while taking chlorthalidone (Hygroton) found no difference in the risk of death from cardiovascular disease or from any cause (hazard ratio = 1.04).15
CLINICAL IMPLICATIONS
Balancing the benefits and risks of statins
It is important to examine how the 0.4% increase in absolute risk of new-onset diabetes as calculated in meta-analyses compares with the benefits of statin treatment in terms of cardiovascular risk reduction.
Using data from the Cholesterol Treatment Trialists (CTT) meta-analysis of statin trials in 71,370 participants, Sattar et al11 estimated that statin treatment is associated with 5.4 fewer deaths from coronary heart disease and cases of nonfatal myocardial infarction per 255 patients treated over 4 years for each 1-mmol/L (39 mg/dL) reduction in LDL-C compared with controls. The benefit would be even greater if stroke, revascularization, and hospitalization are included as end points. This benefit is contrasted with the risk of developing 1 additional case of diabetes for every 255 patients treated with statins over the same period.
Preiss et al25 calculated that there were 2 more cases of diabetes per 1,000 patient-years in patients receiving intensive doses than in those receiving moderate doses (18.9 vs 16.9), corresponding to 1 additional case of diabetes for every 498 patients treated per year. However, there were 6.5 fewer first major cardiovascular events per 1,000 patient-years (44.5 vs 51.0), corresponding to a number needed to treat per year to prevent 1 cardiovascular event of 155. Most of the benefit was due to fewer revascularizations, followed by nonfatal myocardial infarctions. The 12% increase in new diabetes with high-dose therapy contrasted with a 16% reduction in new cardiovascular disease combined events (OR 0.84, 95% CI 0.75–0.94).
As previously noted, in the JUPITER trial, the benefits of preventing cardiovascular events with statin therapy outweighed the risk of new diabetes in people both with and without baseline risk factors for diabetes.29 Similar to the observations with niacin and some antihypertensive drugs, the increase in blood glucose with statins does not appear to reduce the benefits of cardiovascular risk reduction in these patients at moderate to high risk, even when used at high doses.
People with diabetes need aggressive lipid-lowering—with statins
Diabetes is a coronary heart disease risk equivalent and is associated with high risk of cardiovascular events.41–46 Overall, the risk for these adverse events is two to four times greater in people with diabetes than without. Atherosclerosis-related events account for approximately 65% to 75% of all deaths in people with diabetes, and 75% of these events are coronary. Lipid abnormalities are strongly correlated with the risk of cardiovascular disease in people with diabetes, and aggressive treatment of risk factors, particularly lipid abnormalities, has been shown to reduce this risk.47–49 And data from multiple clinical trials support the use of statins to lower LDL-C as the first-line therapy for dyslipidemia in people with diabetes, just as it is in the general population.3–7,9,13,23,50–61
Analyses of diabetic subgroups encompassing 18,000 to 20,000 patients in the large statin trials have clearly demonstrated the benefits of statin therapy. A recent metaanalysis of 10 placebo-controlled trials that included approximately 16,000 patients with diabetes and 54,000 without diabetes demonstrated a 30% reduction in coronary heart disease, a 19% reduction in strokes, and a 12% reduction in mortality.54 Furthermore, in another meta-analysis of 14 trials, a similar 22% reduction in coronary heart disease was noted in people with diabetes whether or not they had a history of cardiovascular disease.55
Therefore, aggressive treatment of lipid abnormalities with statins as primary treatment has generally been adopted as a standard of care in diabetic patients, particularly those with clinical cardiovascular disease or one or more risk factors. The Adult Treatment Panel III guidelines recommend a minimum LDL-C goal of less than 100 mg/dL and a goal of less than 70 mg/dL as an option for patients with diabetes (Table 1).41,62 Similar recommendations have been issued by the American Diabetes Association together with the American College of Cardiology (Table 2),30 the American Diabetes Association by itself,63 and the American Academy of Pediatrics.6
Is new-onset diabetes as dangerous as established diabetes?
In studies to date, there did not appear to be more events in those who developed new-onset diabetes.
Waters et al,24 evaluating three trials of high-dose atorvastatin therapy, found that major cardiovascular events occurred in 11.3% of those with new-onset diabetes, 10.8% of those without new-onset diabetes (HR 1.02, 95% CI 0.77–1.35), and 17.5% of those who had diabetes at baseline.
Therefore, it may not be appropriate to extrapolate the glucose changes seen on statin therapy to an equivalent increase in adverse cardiovascular events as seen in other diabetic patients. The beneficial reduction in cardiovascular events does not appear to be diminished in those developing diabetes. It is not clear that the increase in glucose on statins has the same implications of a new diagnosis of diabetes. Does this elevation in glucose represent true diabetes or some downstream effect? For example, thiazide diuretics have been known to increase blood glucose levels, but the levels drop when these drugs are discontinued, even after many years of treatment.
On the other hand, it is possible that follow-up of 5 years or less in clinical trials has not allowed sufficient time to examine the influence of the increase in new-onset diabetes on future cardiovascular events. In addition, because of the widespread use of statins across a broad range of cardiovascular risk, even if the effect is small in absolute terms, the potential adverse effects are magnified, particularly in a low-risk population in which the cardiovascular benefits are smaller.
The association is real, but questions remain
In view of the evidence, it is difficult to refute that an association exits between statin use and new-onset diabetes, at least in some subgroups. The dose response noted in some studies further reinforces the conclusion that the association is real. However, many questions remain unanswered regarding mechanism of effect, whether there are differences depending on the particular statin or dose used, or differential effects in the populations treated (such as patients with metabolic syndrome or the elderly).
Until the contradictory observations can be resolved and plausible mechanisms of action elucidated, causality cannot be established. From a clinical standpoint there is no current evidence suggesting that the elevations in blood glucose seen while on lipid-lowering or blood-pressure-lowering therapy are associated with an increased risk of cardiovascular events or that they attenuate the beneficial effects of the therapy.
Statins should continue to be used in patients at high risk
Until further studies are done, statins should continue to be used, after assessing the risks and the benefits.
Primary prevention patients at moderate to high risk and secondary prevention patients stand to gain from statin therapy, and it should not be denied or doses reduced on the basis of concerns about the development of new-onset diabetes. The recognized modest risk of developing diabetes does not appear to blunt the cardioprotective effects of statin therapy in these moderate-to high-risk groups.
Rather than stop statins in patients at risk of diabetes such as the elderly or those with prediabetes, insulin resistance, or metabolic syndrome who are on therapy for appropriate reasons, it is reasonable to continue these drugs, monitoring glucose more closely and emphasizing the importance of weight reduction, diet, and aerobic exercise for preventing diabetes. The Diabetes Prevention Program Research Group, for example, reduced the incidence of diabetes by 58% over 2.8 years of follow-up with intensive lifestyle interventions (a low-calorie, low-fat diet plus moderate physical activity 150 minutes per week) vs usual care in at-risk populations.65
Should statins be used more cautiously in patients at lower risk?
The most recent Cholesterol Treatment Trialists meta-analysis of 27 randomized clinical trials (22 placebo-controlled, 134,537 people; 5 high-dose vs low-dose, 39,612 people) reported that reducing LDL-C with statins lowered cardiovascular risk even in low-risk patients.66 Overall, there were 21% fewer major cardiovascular events (coronary heart disease, stroke, or coronary revascularization) for every 1-mmol/L reduction in LDL-C.
The proportional reduction in events was at least as large in the two lowest-risk groups (estimated 5-year risk of < 5% and 5% to < 10%, 53,152 people) as in the higher-risk groups. This was reflected mainly in fewer nonfatal myocardial infarctions and coronary revascularizations. In these groups, the absolute reduction in risk for each 1-mmol/L reduction in LDL-C was 11 per 1,000 patients over 5 years. Even in this low-risk population, the reduction in cardiovascular risk seems to compare favorably with the small estimated increase risk of diabetes.
However, even in the lowest-risk group studied, the average baseline LDL-C level was greater than 130 mg/dL.
Therefore, in groups in which the benefits of statins on cardiovascular risk reduction are less robust (eg, low-risk primary prevention groups without significant elevations in LDLC, particularly the elderly), it would not be difficult to justify the case for more cautious use of statin therapy. If statins are used in these low-risk groups, restricting their use to those with at least moderate LDL-C elevation, using less aggressive LDL-C-lowering targets, and regular monitoring of fasting glucose seem reasonable until further information is available.
- US Food and Drug Administration. Statin drugs—drug safety communication: class labeling change. February 28, 2012. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm293670.htm.
- Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357–362.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R; for the Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009; 32:1924–1929.
- Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose—results from the LIPID trial. Diabetes Care 2003; 26:2713–2721.
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735–742.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Scandinavian Simvastatin Survival Study study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Barzilay JI, Davis BR, Pressel SL, et al; ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: the ALLHAT Diabetes Extension Study. Circ Cardiovasc Qual Outcomes 2012; 5:153–162.
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231–1239.
- GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? Ital Heart J 2000; 1:810–820.
- Yamakawa T, Takano T, Tanaka S, Kadonosono K, Terauchi Y. Influence of pitavastatin on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb 2008; 15:269–275.
- Kryzhanovski V, Gumprecht J, Zhu B, Yu CY, Hounslow N, Sponseller CA. Atorvastatin but not pitavastatin significantly increases fasting plasma glucose in patients with type 2 diabetes and combined dyslipidemia (abstract). J Am Coll Cardiol 2011; 57:E575.
- Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med 2012; 29:628–631.
- Sabatine MS, Morrow DA, Giugliano RP, et al. Implications of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Circulation 2004; 110(suppl III):834–880.
- Shepherd J, Barter P, Carmena R, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220–1226.
- Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535–1545.
- Yousef A, Tu JV, Wang J, Donovan L, Ko DT. The association of intensive statin therapy on long-term risks of cardiovascular events and diabetes following acute myocardial infarction (abstract). Circulation 2012; 125:e859.
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a metaanalysis. JAMA 2011; 305:2556–2564.
- de Lemos JA, Blazing MA, Wiviott SD, et al; A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Pedersen TR, Faegeman O, Kastelein JJ, et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Armitage J, Bowman L, Wallendszus K, et al; Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010; 37:1658–1669.
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012; 380:565–571.
- Brunzell JD, Davidson M, Furberg CD, et al; American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811–822.
- Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010; 55:1209–1216.
- Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204:483–490.
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006; 49:1881–1892.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 1999; 126:1205–1213.
- Guyton JR, Fazio S, Adewale AJ, et al. Effect of extended-release niacin on new-onset diabetes among hyperlipidemic patients treated with ezetimibe/simvastatin in a randomized controlled trial. Diabetes Care 2012; 35:857–860.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254–257.
- Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial. Arch Intern Med 2002; 162:1568–1576.
- Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NRAnglo-Scandinavian Cardiac Outcomes Trial Investigators. Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31:982–988.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201–207.
- Jong JP, Chang MH, Tien L, et al. Antihypertensive drugs and new-onset diabetes: a retrospective longitudinal cohort study. Cardiovasc Ther 2009; 27:159–163.
- Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study Lancet 2002; 359:2140–2144.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Sprafka JM, Burke GL, Folsom AR, McGovbern PG, Hahn LP. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991; 14:537–543.
- Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In:Harris MI, Cowie CC, Stern MP, et al, editors. Diabetes in America. 2nd ed. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 1995:233–257.
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–444.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348:383–393.
- Gaede P, Pederson O. Intensive integrated therapy of type 2 diabetes: implications for long-term prognosis. Diabetes 2004; 53:S39–S47.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the cholesterol and recurrent events (CARE) trial. The Care Investigators. Circulation 1998; 98:2513–2519.
- Pyðrälä K, Pedersen TR, Kjekshus J, Faergeman O, Olsson AG, Thorgeirsson G. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 1997; 20:614–620.
- Vijan S, Hayward RA; American College of Physicians. Pharmacologic lipid-lowering therapy in type 2 diabetes mellitus: background paper for the American College of Physicians. Ann Intern Med 2004; 140:650–658.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. Errata in Lancet 2008; 371:2084, Lancet 2005; 366:1358.
- Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009; 338:b2376.
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Baigent C; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Nissen SE, Nicholls SJ, Sipahi I, et al; ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes et al. N Engl J Med 2004; 350:1495–1504.
- LaRosa JC, Grundy SM, Waters DD, et al; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998; 339:1349–1357.
- Grundy SM, Cleeman JI, Bairey Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S5–S10.
- Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590.
On february 28, 2012, the US Food and Drug Administration (FDA) updated its labeling requirements for statins. In addition to revising its recommendations for monitoring liver function and its alerts about reports of memory loss, the FDA also warned of the possibility of new-onset diabetes mellitus and worse glycemic control in patients taking statin drugs.1
This change stoked an ongoing debate about the risk of diabetes with statin use and the implications of such an effect. To understand the clinical consequences of this alert and its effect on treatment decisions, we need to consider the degree to which statins lower the risk of cardiovascular disease in patients at high risk (including diabetic patients), the magnitude of the risk of developing new diabetes while on statin therapy, and the ratio of risk to benefit in treated populations.
This review will discuss the evidence for this possible adverse effect and the implications for clinical practice.
DO STATINS CAUSE DIABETES?
Individual controlled trials dating back more than a decade have had conflicting results about new diabetes and poorer diabetic control in patients taking statins.
The West of Scotland Coronary Prevention Study (WOSCOPS)2 suggested that the incidence of diabetes was 30% lower in patients taking pravastatin (Pravachol) 40 mg/day than with placebo. However, this was not observed with atorvastatin (Lipitor) 10 mg/day in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid-Lowering Arm (ASCOT-LLA)3 in hypertensive patients or in the Collaborative Atorvastatin Diabetes Study (CARDS)4 in diabetic patients,4 nor was it noted with simvastatin (Zocor) 40 mg/day in the Heart Protection Study (HPS).5
The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER),6 using the more potent agent rosuvastatin (Crestor) 20 mg/day in patients with elevated levels of C-reactive protein (CRP), was stopped early when an interim analysis found a 44% lower incidence of the primary end point. However, the trial also reported a 26% higher incidence of diabetes in follow-up of less than 2 years.
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER),7 with a mean age at entry of 75, there was a 32% higher incidence of diabetes with pravastatin therapy.7
Results of meta-analyses
Several meta-analyses have addressed these differences.
Rajpathak et al8 performed a meta-analysis, published in 2009, of six trials—WOSCOPS,2 ASCOT-LLA,3 JUPITER,6 HPS,5 the Long-term Intervention With Pravastatin in Ischaemic Disease (LIPID) study,9 and the Controlled Rosuvastatin Multinational Study in Heart Failure (CORONA),10 with a total of 57,593 patients. They calculated that the incidence of diabetes was 13% higher (an absolute difference of 0.5%) in statin recipients, which was statistically significant. In their initial analysis, the authors excluded WOSCOPS, describing it as hypothesis-generating. The relative increase in risk was less—6%—and was not statistically significant when WOSCOPS was included.
Sattar et al,11 in a larger meta-analysis published in 2010, included 91,140 participants in 13 major statin trials conducted between 1994 and 2009; each trial had more than 1,000 patients and more than 1 year of follow-up.2,3,5–7,9,10,12–17 New diabetes was defined as physician reporting of new diabetes, new diabetic medication use, or a fasting glucose greater than 7 mmol/L (126 mg/dL).
New diabetes occurred in 2,226 (4.89%) of the statin recipients and in 2,052 (4.5%) of the placebo recipients, an absolute difference of 0.39%, or 9% more (odds ratio [OR] 1.09; 95% confidence interval [CI] 1.02–1.17) (Figure 1).
The incidence of diabetes varied substantially among the 13 trials, with only JUPITER6 and PROSPER7 finding statistically significant increases in rates (26% and 32%, respectively). Of the other 11 trials, 4 had nonsignificant trends toward lower incidence,2,9,13,17 while the 7 others had nonsignificant trends toward higher incidence.
Does the specific statin make a difference?
Questions have been raised as to whether the type of statin used, the intensity of therapy, or the population studied contributed to these differences. Various studies suggest that factors such as using hydrophilic vs lipophilic statins (hydrophilic statins include pravastatin and rosuvastatin; lipophilic statins include atorvastatin, lovastatin, and simvastatin), the dose, the extent of lowering of low-density lipoprotein cholesterol (LDL-C), and the age or clinical characteristics of the population studied may influence this relationship.18–20
Yamakawa et al18 examined the effect of atorvastatin 10 mg/day, pravastatin 10 mg/day, and pitavastatin (Livalo) 2 mg/day on glycemic control over 3 months in a retrospective analysis. Random blood glucose and hemoglobin A1c levels were increased in the atorvastatin group but not in the other two.18
A prospective comparison of atorvastatin 20 mg vs pitavastatin 4 mg in patients with type 2 diabetes, presented at the American College of Cardiology’s 2011 annual meeting, reported a significant increase in fasting glucose levels with atorvastatin, particularly in women, but not with pitavastatin.19
In the Compare the Effect of Rosuvastatin With Atorvastatin on Apo B/Apo A-1 Ratio in Patients With Type 2 Diabetes Mellitus and Dyslipidaemia (CORALL) study,20 both high-dose rosuvastatin (40 mg) and high-dose atorvastatin (80 mg) were associated with significant increases in hemoglobin A1c, although the mean fasting glucose levels were not significantly different at 18 weeks of therapy.
A meta-analysis by Sattar et al11 did not find a clear difference between lipophilic statins (OR 1.10 vs placebo) and hydrophilic statins (OR 1.08). In analysis by statin type, the combined rosuvastatin trials were statistically significant in favor of a higher diabetes risk (OR 1.18, 95% CI 1.04–1.44). Nonsignificant trends were noted for atorvastatin trials (OR 1.14) and simvastatin trials (OR 1.11) and less so for pravastatin (OR 1.03); the OR for lovastatin was 0.98. This may suggest that there is a stronger effect with more potent statins or with greater lowering of LDL-C.
Meta-regression analysis in this study demonstrated that diabetes risk with statins was higher in older patients but was not influenced by body mass index or by the extent that LDL-C was lowered.
Statin dose as a risk factor
Intensive-dose statin therapy has been shown to reduce cardiovascular risk more than low-dose or moderate-dose therapy, thus supporting more aggressive treatment of LDL-C in higher-risk patients. However, some controlled studies comparing more-potent with less-potent statin regimens suggest that there may also be a higher risk of incident diabetes at higher doses.21–24
In a post hoc analysis of the Pravastatin or Atorvastatin Evaluation and Infection Therapy– Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial,21 patients who had experienced an acute coronary syndrome had a greater increase in hemoglobin A1c if treated with atorvastatin 80 mg/day than with pravastatin 40 mg/day.
Waters et al23 reported a higher risk of new diabetes with atorvastatin 80 mg than with placebo and a trend toward a higher risk with atorvastatin 80 mg than with atorvastatin 10 mg or simvastatin 20 mg.
In contrast, a review by Yousef et al24 of the data from the Enhanced Feedback for Effective Cardiac Treatment (EFFECT) study did not find a higher diabetes risk with more intensive statin therapy based on the magnitude of LDL-C reduction. A propensity-matched examination of deaths, recurrent acute ischemic events, or new diabetes in patients previously hospitalized with myocardial infarction found no differences in these end points each year out to 5 years. The risk of diabetes was in fact lower (but the difference was not statistically significant) in the high-dose groups out to 5 years. The risk of myocardial infarction or death was numerically different in the high-dose groups, but the difference was not statistically significant.
Preiss et al25 in 2011 performed a meta-analysis of the impact of intensity of statin therapy on diabetes risk. They examined data from 32,752 participants without diabetes at baseline in five randomized controlled trials with more than 1,000 participants and more than 1 year of follow-up, comparing high-dose therapy against moderate-dose statin therapy.21,22,26–28 New diabetes was considered present if there was an adverse event report of diabetes, if glucose-lowering drugs were started, or if two fasting plasma glucose measurements were higher than 7 mmol/L (126 mg/dL).
Diabetes developed in 1,449 (8.8%) of the intensive-therapy group and 1,300 (8.0%) of the moderate-therapy group (OR 1.12, 95% CI 1.04–1.22). In contrast, incident cardiovascular disease occurred in 3,134 (19.1%) of the intensive-therapy group and 3,550 (21.7%) of the moderate-therapy group (OR 0.84, 95% CI 0.75–0.94). Therefore, there was an 0.8% absolute increase in diabetes cases on high-dose statins and a 2.6% absolute reduction in adverse cardiovascular events.
CAUTION IN INTERPRETING THESE DATA
There are many reasons for caution in interpreting these studies.
The trials were not designed to look for diabetes
The data supporting the relationship between statin therapy and higher risk of diabetes are primarily from observational studies. These studies were not prospectively designed to address this question, and we therefore need to view this as association and not as causation.
The definition of diabetes varied between trials, and new-onset diabetes was often not rigorously screened for. In many trials the outcome of diabetes was at least partially based on nonstandardized, nonadjudicated physician reporting.
Consequently, if statins reduce the risk of diabetes, the results from WOSCOPS may overstate the reduction, since this study used a non-standard definition of incident diabetes (fasting plasma glucose > 126 mg/dL plus a > 36 mg/dL increase from baseline). When Sattar et al11 reanalyzed WOSCOPS data using a more standard definition, they found a smaller effect.
On the other hand, nonstandardized physician reporting may overstate an adverse effect. Sattar et al11 also found that when fasting plasma glucose levels alone were used as the definition for diabetes, the overall risk was attenuated and was no longer statistically significant (OR 1.07, 95% CI 0.97–1.17).
Perhaps statin therapy uncovers diabetes only in people at risk of diabetes
Perhaps statin therapy uncovers diabetes only in people at higher baseline risk of developing diabetes. Therefore, this adverse effect may be restricted to certain groups and not applicable to the general population.
In JUPITER, one of the two trials in which, on independent analysis, statin use was associated with new diabetes, 77% of patients in the rosuvastatin group who developed diabetes had impaired fasting glucose at entry and therefore were at higher risk of developing diabetes.6
Possibly, the relationship is driven by preexisting metabolic syndrome or other risk factors for diabetes. In the two studies that reported a statistically significantly higher incidence of new diabetes, more than 40% of patients in JUPITER met the criteria for metabolic syndrome, and metabolic syndrome, which increases in prevalence with age, was likely more prominent in the elderly population in PROSPER.
Waters et al23 grouped patients according to whether they had risk factors for diabetes (impaired fasting glucose, obesity, elevated triglycerides, and hypertension) and found that those who had none or one of these risk factors had no difference in the rate of new-onset diabetes with either moderate or intensive statin therapy, but the risk was pronounced in those who had three or four risk factors.
Ridker et al29 reanalyzed the JUPITER data from patients who did not have cardiovascular disease at baseline. Overall, for every 54 new cases of diabetes in follow-up, 134 cardiovascular events or deaths were prevented. In subgroup analysis, those who had one or more risk factors for diabetes at baseline (metabolic syndrome, impaired fasting glucose, obesity, or hemoglobin A1c > 6%) had a 39% reduction in the primary end point and a 28% increase in new diabetes. Those who had none of these risk factors had a 52% lower rate of cardiovascular events but no increase in diabetes.
Other confounding factors
Bias and confounding factors are difficult to control for in studies without prospectively defined, recognized, and analyzed outcomes.
Although it may be a bit of a stretch, residual confounding factors such as myalgia side effects while on statins may reduce exercise in the statin-treatment groups. Perhaps a change to a healthier lifestyle after cardiovascular events may be more common in placebo groups. Improved survival with statins may allow more people at risk of diabetes to live longer and present with the diagnosis.30
POSSIBLE EXPLANATIONS, BUT NO UNIFYING MECHANISM
If mechanisms could be identified to explain the association between statins and diabetes, this would strengthen the argument that it is a cause-and-effect relationship. Many explanations have been proposed as to how statins may influence glucose metabolism and insulin sensitivity.31–34 These are possible explanations based on other observations.
In theory, statins may improve insulin sensitivity via their anti-inflammatory effect, since inflammatory markers and proinflammatory cytokines have been linked with insulin resistance. However, other effects of statins may adversely affect glycemic control.
In vivo analysis has shown that some but not all statins increase insulin levels and decrease insulin sensitivity in a dose-dependent fashion. Some statins decrease adiponectin and may worsen glycemic control through loss of adiponectin’s proposed protective anti-proliferative and antiangiogenic properties. In vitro studies and animal studies have demonstrated a decrease in expression of insulin-responsive glucose transporter 4 (GLUT4) with atorvastatin, and an increase in GLUT1. It has been hypothesized that reduction in isoprenoid biosynthesis or decreased insulin signaling may explain these effects and that changes in glucose transport in adipocytes may cause insulin resistance. Other studies suggest that dysregulation of cellular cholesterol may attenuate beta-cell function. Impaired biosynthesis of ubiquinones may result in delayed production of adenosine triphosphate and consequently diminish insulin release.
But different effects have been reported for atorvastatin, simvastatin, and pravastatin, arguing against a unifying explanation or, alternatively, suggesting that differences in lipophilicity and potency among statins are important. Hydrophilic statins may be less likely to be taken up by extrahepatic cells such as pancreatic cells and adipocytes, possibly lessening these effects. However, the strong association between rosuvastatin (which is hydrophilic) and new diabetes would not support this hypothesis.
Despite these speculations, lack of conformity in response to different statins and discrepancies in the clinical outcomes noted in trials fail to clearly identify a common causative mechanism.
OTHER COMMON THERAPIES MAY INFLUENCE GLYCEMIC CONTROL
Statins are not the first drugs for reducing cardiovascular risk that have been shown to affect glucose levels during treatment.
Niacin
Niacin has been known to increase glucose levels but has long been used as a treatment for dyslipidemia despite this caution. Reduced glycemic control during niacin treatment in diabetic patients does not seem to alter the beneficial effects of treatment.35–37
In a post hoc analysis of the Coronary Drug Project (CDP), in patient subgroups defined by baseline fasting plasma glucose and compared with placebo, niacin reduced the 6-year risk of recurrent myocardial infarction and the combined end point of coronary heart disease death or nonfatal myocardial infarction similarly (interactive P value nonsignificant) across all levels of baseline fasting plasma glucose, including levels of 126 mg/dL or higher at study entry.36
In another post hoc analysis of CDP patient subgroups defined by the change in glycemic status from baseline to 1 year, niacin reduced the 6-year risk of the same end points similarly (interactive P value nonsignificant) across all levels of change in fasting plasma glucose from baseline to year 1, whether baseline fasting plasma glucose levels decreased, stayed the same, or increased to 10 mg/dL or higher on niacin therapy.36
Therefore, the beneficial effect of niacin of reducing the rate of recurrent nonfatal myocardial infarction and coronary heart disease events was not significantly diminished when impaired fasting glucose or diabetes was present when therapy was started or by on-therapy increases from baseline fasting plasma glucose.
In addition, on-therapy changes in glycemic control may be dose-related and minimized by surveillance and therapy adjustments. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT)38 found that changes in glycemic control were minimal as measured by fasting glucose and hemoglobin A1c; were associated with a higher niacin dose (1.5 g/day vs 1 g/day); and, when present, were successfully managed by adjusting the diabetes treatment regimen.
Antihypertensive drugs
Diuretics as well as beta-blockers have been reported to increase the incidence of diabetes in patients with hypertension.15,38–40
A retrospective longitudinal cohort study40 in 2009 examined the development of new-onset diabetes (defined as a new ICD-9 code for diabetes or initiation of diabetes treatment) in 24,688 treated hypertensive patients without diabetes at baseline; 4,385 (17.8%) of the patients developed diabetes. After adjusting for sex and age, the risk of new diabetes was significant in users of diuretics (OR 1.10), beta-blockers (OR 1.12), and calcium channel blockers (OR 1.10) compared with users of angiotensin-converting enzyme inhibitors, (OR 0.92), angiotensin receptor blockers (OR 0.90), or alpha-blockers (OR 0.88).
However, the increase in blood glucose does not seem to attenuate the beneficial effects of reducing cardiovascular events. In the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT),15 a long-term follow-up of those developing new-onset diabetes while taking chlorthalidone (Hygroton) found no difference in the risk of death from cardiovascular disease or from any cause (hazard ratio = 1.04).15
CLINICAL IMPLICATIONS
Balancing the benefits and risks of statins
It is important to examine how the 0.4% increase in absolute risk of new-onset diabetes as calculated in meta-analyses compares with the benefits of statin treatment in terms of cardiovascular risk reduction.
Using data from the Cholesterol Treatment Trialists (CTT) meta-analysis of statin trials in 71,370 participants, Sattar et al11 estimated that statin treatment is associated with 5.4 fewer deaths from coronary heart disease and cases of nonfatal myocardial infarction per 255 patients treated over 4 years for each 1-mmol/L (39 mg/dL) reduction in LDL-C compared with controls. The benefit would be even greater if stroke, revascularization, and hospitalization are included as end points. This benefit is contrasted with the risk of developing 1 additional case of diabetes for every 255 patients treated with statins over the same period.
Preiss et al25 calculated that there were 2 more cases of diabetes per 1,000 patient-years in patients receiving intensive doses than in those receiving moderate doses (18.9 vs 16.9), corresponding to 1 additional case of diabetes for every 498 patients treated per year. However, there were 6.5 fewer first major cardiovascular events per 1,000 patient-years (44.5 vs 51.0), corresponding to a number needed to treat per year to prevent 1 cardiovascular event of 155. Most of the benefit was due to fewer revascularizations, followed by nonfatal myocardial infarctions. The 12% increase in new diabetes with high-dose therapy contrasted with a 16% reduction in new cardiovascular disease combined events (OR 0.84, 95% CI 0.75–0.94).
As previously noted, in the JUPITER trial, the benefits of preventing cardiovascular events with statin therapy outweighed the risk of new diabetes in people both with and without baseline risk factors for diabetes.29 Similar to the observations with niacin and some antihypertensive drugs, the increase in blood glucose with statins does not appear to reduce the benefits of cardiovascular risk reduction in these patients at moderate to high risk, even when used at high doses.
People with diabetes need aggressive lipid-lowering—with statins
Diabetes is a coronary heart disease risk equivalent and is associated with high risk of cardiovascular events.41–46 Overall, the risk for these adverse events is two to four times greater in people with diabetes than without. Atherosclerosis-related events account for approximately 65% to 75% of all deaths in people with diabetes, and 75% of these events are coronary. Lipid abnormalities are strongly correlated with the risk of cardiovascular disease in people with diabetes, and aggressive treatment of risk factors, particularly lipid abnormalities, has been shown to reduce this risk.47–49 And data from multiple clinical trials support the use of statins to lower LDL-C as the first-line therapy for dyslipidemia in people with diabetes, just as it is in the general population.3–7,9,13,23,50–61
Analyses of diabetic subgroups encompassing 18,000 to 20,000 patients in the large statin trials have clearly demonstrated the benefits of statin therapy. A recent metaanalysis of 10 placebo-controlled trials that included approximately 16,000 patients with diabetes and 54,000 without diabetes demonstrated a 30% reduction in coronary heart disease, a 19% reduction in strokes, and a 12% reduction in mortality.54 Furthermore, in another meta-analysis of 14 trials, a similar 22% reduction in coronary heart disease was noted in people with diabetes whether or not they had a history of cardiovascular disease.55
Therefore, aggressive treatment of lipid abnormalities with statins as primary treatment has generally been adopted as a standard of care in diabetic patients, particularly those with clinical cardiovascular disease or one or more risk factors. The Adult Treatment Panel III guidelines recommend a minimum LDL-C goal of less than 100 mg/dL and a goal of less than 70 mg/dL as an option for patients with diabetes (Table 1).41,62 Similar recommendations have been issued by the American Diabetes Association together with the American College of Cardiology (Table 2),30 the American Diabetes Association by itself,63 and the American Academy of Pediatrics.6
Is new-onset diabetes as dangerous as established diabetes?
In studies to date, there did not appear to be more events in those who developed new-onset diabetes.
Waters et al,24 evaluating three trials of high-dose atorvastatin therapy, found that major cardiovascular events occurred in 11.3% of those with new-onset diabetes, 10.8% of those without new-onset diabetes (HR 1.02, 95% CI 0.77–1.35), and 17.5% of those who had diabetes at baseline.
Therefore, it may not be appropriate to extrapolate the glucose changes seen on statin therapy to an equivalent increase in adverse cardiovascular events as seen in other diabetic patients. The beneficial reduction in cardiovascular events does not appear to be diminished in those developing diabetes. It is not clear that the increase in glucose on statins has the same implications of a new diagnosis of diabetes. Does this elevation in glucose represent true diabetes or some downstream effect? For example, thiazide diuretics have been known to increase blood glucose levels, but the levels drop when these drugs are discontinued, even after many years of treatment.
On the other hand, it is possible that follow-up of 5 years or less in clinical trials has not allowed sufficient time to examine the influence of the increase in new-onset diabetes on future cardiovascular events. In addition, because of the widespread use of statins across a broad range of cardiovascular risk, even if the effect is small in absolute terms, the potential adverse effects are magnified, particularly in a low-risk population in which the cardiovascular benefits are smaller.
The association is real, but questions remain
In view of the evidence, it is difficult to refute that an association exits between statin use and new-onset diabetes, at least in some subgroups. The dose response noted in some studies further reinforces the conclusion that the association is real. However, many questions remain unanswered regarding mechanism of effect, whether there are differences depending on the particular statin or dose used, or differential effects in the populations treated (such as patients with metabolic syndrome or the elderly).
Until the contradictory observations can be resolved and plausible mechanisms of action elucidated, causality cannot be established. From a clinical standpoint there is no current evidence suggesting that the elevations in blood glucose seen while on lipid-lowering or blood-pressure-lowering therapy are associated with an increased risk of cardiovascular events or that they attenuate the beneficial effects of the therapy.
Statins should continue to be used in patients at high risk
Until further studies are done, statins should continue to be used, after assessing the risks and the benefits.
Primary prevention patients at moderate to high risk and secondary prevention patients stand to gain from statin therapy, and it should not be denied or doses reduced on the basis of concerns about the development of new-onset diabetes. The recognized modest risk of developing diabetes does not appear to blunt the cardioprotective effects of statin therapy in these moderate-to high-risk groups.
Rather than stop statins in patients at risk of diabetes such as the elderly or those with prediabetes, insulin resistance, or metabolic syndrome who are on therapy for appropriate reasons, it is reasonable to continue these drugs, monitoring glucose more closely and emphasizing the importance of weight reduction, diet, and aerobic exercise for preventing diabetes. The Diabetes Prevention Program Research Group, for example, reduced the incidence of diabetes by 58% over 2.8 years of follow-up with intensive lifestyle interventions (a low-calorie, low-fat diet plus moderate physical activity 150 minutes per week) vs usual care in at-risk populations.65
Should statins be used more cautiously in patients at lower risk?
The most recent Cholesterol Treatment Trialists meta-analysis of 27 randomized clinical trials (22 placebo-controlled, 134,537 people; 5 high-dose vs low-dose, 39,612 people) reported that reducing LDL-C with statins lowered cardiovascular risk even in low-risk patients.66 Overall, there were 21% fewer major cardiovascular events (coronary heart disease, stroke, or coronary revascularization) for every 1-mmol/L reduction in LDL-C.
The proportional reduction in events was at least as large in the two lowest-risk groups (estimated 5-year risk of < 5% and 5% to < 10%, 53,152 people) as in the higher-risk groups. This was reflected mainly in fewer nonfatal myocardial infarctions and coronary revascularizations. In these groups, the absolute reduction in risk for each 1-mmol/L reduction in LDL-C was 11 per 1,000 patients over 5 years. Even in this low-risk population, the reduction in cardiovascular risk seems to compare favorably with the small estimated increase risk of diabetes.
However, even in the lowest-risk group studied, the average baseline LDL-C level was greater than 130 mg/dL.
Therefore, in groups in which the benefits of statins on cardiovascular risk reduction are less robust (eg, low-risk primary prevention groups without significant elevations in LDLC, particularly the elderly), it would not be difficult to justify the case for more cautious use of statin therapy. If statins are used in these low-risk groups, restricting their use to those with at least moderate LDL-C elevation, using less aggressive LDL-C-lowering targets, and regular monitoring of fasting glucose seem reasonable until further information is available.
On february 28, 2012, the US Food and Drug Administration (FDA) updated its labeling requirements for statins. In addition to revising its recommendations for monitoring liver function and its alerts about reports of memory loss, the FDA also warned of the possibility of new-onset diabetes mellitus and worse glycemic control in patients taking statin drugs.1
This change stoked an ongoing debate about the risk of diabetes with statin use and the implications of such an effect. To understand the clinical consequences of this alert and its effect on treatment decisions, we need to consider the degree to which statins lower the risk of cardiovascular disease in patients at high risk (including diabetic patients), the magnitude of the risk of developing new diabetes while on statin therapy, and the ratio of risk to benefit in treated populations.
This review will discuss the evidence for this possible adverse effect and the implications for clinical practice.
DO STATINS CAUSE DIABETES?
Individual controlled trials dating back more than a decade have had conflicting results about new diabetes and poorer diabetic control in patients taking statins.
The West of Scotland Coronary Prevention Study (WOSCOPS)2 suggested that the incidence of diabetes was 30% lower in patients taking pravastatin (Pravachol) 40 mg/day than with placebo. However, this was not observed with atorvastatin (Lipitor) 10 mg/day in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid-Lowering Arm (ASCOT-LLA)3 in hypertensive patients or in the Collaborative Atorvastatin Diabetes Study (CARDS)4 in diabetic patients,4 nor was it noted with simvastatin (Zocor) 40 mg/day in the Heart Protection Study (HPS).5
The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER),6 using the more potent agent rosuvastatin (Crestor) 20 mg/day in patients with elevated levels of C-reactive protein (CRP), was stopped early when an interim analysis found a 44% lower incidence of the primary end point. However, the trial also reported a 26% higher incidence of diabetes in follow-up of less than 2 years.
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER),7 with a mean age at entry of 75, there was a 32% higher incidence of diabetes with pravastatin therapy.7
Results of meta-analyses
Several meta-analyses have addressed these differences.
Rajpathak et al8 performed a meta-analysis, published in 2009, of six trials—WOSCOPS,2 ASCOT-LLA,3 JUPITER,6 HPS,5 the Long-term Intervention With Pravastatin in Ischaemic Disease (LIPID) study,9 and the Controlled Rosuvastatin Multinational Study in Heart Failure (CORONA),10 with a total of 57,593 patients. They calculated that the incidence of diabetes was 13% higher (an absolute difference of 0.5%) in statin recipients, which was statistically significant. In their initial analysis, the authors excluded WOSCOPS, describing it as hypothesis-generating. The relative increase in risk was less—6%—and was not statistically significant when WOSCOPS was included.
Sattar et al,11 in a larger meta-analysis published in 2010, included 91,140 participants in 13 major statin trials conducted between 1994 and 2009; each trial had more than 1,000 patients and more than 1 year of follow-up.2,3,5–7,9,10,12–17 New diabetes was defined as physician reporting of new diabetes, new diabetic medication use, or a fasting glucose greater than 7 mmol/L (126 mg/dL).
New diabetes occurred in 2,226 (4.89%) of the statin recipients and in 2,052 (4.5%) of the placebo recipients, an absolute difference of 0.39%, or 9% more (odds ratio [OR] 1.09; 95% confidence interval [CI] 1.02–1.17) (Figure 1).
The incidence of diabetes varied substantially among the 13 trials, with only JUPITER6 and PROSPER7 finding statistically significant increases in rates (26% and 32%, respectively). Of the other 11 trials, 4 had nonsignificant trends toward lower incidence,2,9,13,17 while the 7 others had nonsignificant trends toward higher incidence.
Does the specific statin make a difference?
Questions have been raised as to whether the type of statin used, the intensity of therapy, or the population studied contributed to these differences. Various studies suggest that factors such as using hydrophilic vs lipophilic statins (hydrophilic statins include pravastatin and rosuvastatin; lipophilic statins include atorvastatin, lovastatin, and simvastatin), the dose, the extent of lowering of low-density lipoprotein cholesterol (LDL-C), and the age or clinical characteristics of the population studied may influence this relationship.18–20
Yamakawa et al18 examined the effect of atorvastatin 10 mg/day, pravastatin 10 mg/day, and pitavastatin (Livalo) 2 mg/day on glycemic control over 3 months in a retrospective analysis. Random blood glucose and hemoglobin A1c levels were increased in the atorvastatin group but not in the other two.18
A prospective comparison of atorvastatin 20 mg vs pitavastatin 4 mg in patients with type 2 diabetes, presented at the American College of Cardiology’s 2011 annual meeting, reported a significant increase in fasting glucose levels with atorvastatin, particularly in women, but not with pitavastatin.19
In the Compare the Effect of Rosuvastatin With Atorvastatin on Apo B/Apo A-1 Ratio in Patients With Type 2 Diabetes Mellitus and Dyslipidaemia (CORALL) study,20 both high-dose rosuvastatin (40 mg) and high-dose atorvastatin (80 mg) were associated with significant increases in hemoglobin A1c, although the mean fasting glucose levels were not significantly different at 18 weeks of therapy.
A meta-analysis by Sattar et al11 did not find a clear difference between lipophilic statins (OR 1.10 vs placebo) and hydrophilic statins (OR 1.08). In analysis by statin type, the combined rosuvastatin trials were statistically significant in favor of a higher diabetes risk (OR 1.18, 95% CI 1.04–1.44). Nonsignificant trends were noted for atorvastatin trials (OR 1.14) and simvastatin trials (OR 1.11) and less so for pravastatin (OR 1.03); the OR for lovastatin was 0.98. This may suggest that there is a stronger effect with more potent statins or with greater lowering of LDL-C.
Meta-regression analysis in this study demonstrated that diabetes risk with statins was higher in older patients but was not influenced by body mass index or by the extent that LDL-C was lowered.
Statin dose as a risk factor
Intensive-dose statin therapy has been shown to reduce cardiovascular risk more than low-dose or moderate-dose therapy, thus supporting more aggressive treatment of LDL-C in higher-risk patients. However, some controlled studies comparing more-potent with less-potent statin regimens suggest that there may also be a higher risk of incident diabetes at higher doses.21–24
In a post hoc analysis of the Pravastatin or Atorvastatin Evaluation and Infection Therapy– Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial,21 patients who had experienced an acute coronary syndrome had a greater increase in hemoglobin A1c if treated with atorvastatin 80 mg/day than with pravastatin 40 mg/day.
Waters et al23 reported a higher risk of new diabetes with atorvastatin 80 mg than with placebo and a trend toward a higher risk with atorvastatin 80 mg than with atorvastatin 10 mg or simvastatin 20 mg.
In contrast, a review by Yousef et al24 of the data from the Enhanced Feedback for Effective Cardiac Treatment (EFFECT) study did not find a higher diabetes risk with more intensive statin therapy based on the magnitude of LDL-C reduction. A propensity-matched examination of deaths, recurrent acute ischemic events, or new diabetes in patients previously hospitalized with myocardial infarction found no differences in these end points each year out to 5 years. The risk of diabetes was in fact lower (but the difference was not statistically significant) in the high-dose groups out to 5 years. The risk of myocardial infarction or death was numerically different in the high-dose groups, but the difference was not statistically significant.
Preiss et al25 in 2011 performed a meta-analysis of the impact of intensity of statin therapy on diabetes risk. They examined data from 32,752 participants without diabetes at baseline in five randomized controlled trials with more than 1,000 participants and more than 1 year of follow-up, comparing high-dose therapy against moderate-dose statin therapy.21,22,26–28 New diabetes was considered present if there was an adverse event report of diabetes, if glucose-lowering drugs were started, or if two fasting plasma glucose measurements were higher than 7 mmol/L (126 mg/dL).
Diabetes developed in 1,449 (8.8%) of the intensive-therapy group and 1,300 (8.0%) of the moderate-therapy group (OR 1.12, 95% CI 1.04–1.22). In contrast, incident cardiovascular disease occurred in 3,134 (19.1%) of the intensive-therapy group and 3,550 (21.7%) of the moderate-therapy group (OR 0.84, 95% CI 0.75–0.94). Therefore, there was an 0.8% absolute increase in diabetes cases on high-dose statins and a 2.6% absolute reduction in adverse cardiovascular events.
CAUTION IN INTERPRETING THESE DATA
There are many reasons for caution in interpreting these studies.
The trials were not designed to look for diabetes
The data supporting the relationship between statin therapy and higher risk of diabetes are primarily from observational studies. These studies were not prospectively designed to address this question, and we therefore need to view this as association and not as causation.
The definition of diabetes varied between trials, and new-onset diabetes was often not rigorously screened for. In many trials the outcome of diabetes was at least partially based on nonstandardized, nonadjudicated physician reporting.
Consequently, if statins reduce the risk of diabetes, the results from WOSCOPS may overstate the reduction, since this study used a non-standard definition of incident diabetes (fasting plasma glucose > 126 mg/dL plus a > 36 mg/dL increase from baseline). When Sattar et al11 reanalyzed WOSCOPS data using a more standard definition, they found a smaller effect.
On the other hand, nonstandardized physician reporting may overstate an adverse effect. Sattar et al11 also found that when fasting plasma glucose levels alone were used as the definition for diabetes, the overall risk was attenuated and was no longer statistically significant (OR 1.07, 95% CI 0.97–1.17).
Perhaps statin therapy uncovers diabetes only in people at risk of diabetes
Perhaps statin therapy uncovers diabetes only in people at higher baseline risk of developing diabetes. Therefore, this adverse effect may be restricted to certain groups and not applicable to the general population.
In JUPITER, one of the two trials in which, on independent analysis, statin use was associated with new diabetes, 77% of patients in the rosuvastatin group who developed diabetes had impaired fasting glucose at entry and therefore were at higher risk of developing diabetes.6
Possibly, the relationship is driven by preexisting metabolic syndrome or other risk factors for diabetes. In the two studies that reported a statistically significantly higher incidence of new diabetes, more than 40% of patients in JUPITER met the criteria for metabolic syndrome, and metabolic syndrome, which increases in prevalence with age, was likely more prominent in the elderly population in PROSPER.
Waters et al23 grouped patients according to whether they had risk factors for diabetes (impaired fasting glucose, obesity, elevated triglycerides, and hypertension) and found that those who had none or one of these risk factors had no difference in the rate of new-onset diabetes with either moderate or intensive statin therapy, but the risk was pronounced in those who had three or four risk factors.
Ridker et al29 reanalyzed the JUPITER data from patients who did not have cardiovascular disease at baseline. Overall, for every 54 new cases of diabetes in follow-up, 134 cardiovascular events or deaths were prevented. In subgroup analysis, those who had one or more risk factors for diabetes at baseline (metabolic syndrome, impaired fasting glucose, obesity, or hemoglobin A1c > 6%) had a 39% reduction in the primary end point and a 28% increase in new diabetes. Those who had none of these risk factors had a 52% lower rate of cardiovascular events but no increase in diabetes.
Other confounding factors
Bias and confounding factors are difficult to control for in studies without prospectively defined, recognized, and analyzed outcomes.
Although it may be a bit of a stretch, residual confounding factors such as myalgia side effects while on statins may reduce exercise in the statin-treatment groups. Perhaps a change to a healthier lifestyle after cardiovascular events may be more common in placebo groups. Improved survival with statins may allow more people at risk of diabetes to live longer and present with the diagnosis.30
POSSIBLE EXPLANATIONS, BUT NO UNIFYING MECHANISM
If mechanisms could be identified to explain the association between statins and diabetes, this would strengthen the argument that it is a cause-and-effect relationship. Many explanations have been proposed as to how statins may influence glucose metabolism and insulin sensitivity.31–34 These are possible explanations based on other observations.
In theory, statins may improve insulin sensitivity via their anti-inflammatory effect, since inflammatory markers and proinflammatory cytokines have been linked with insulin resistance. However, other effects of statins may adversely affect glycemic control.
In vivo analysis has shown that some but not all statins increase insulin levels and decrease insulin sensitivity in a dose-dependent fashion. Some statins decrease adiponectin and may worsen glycemic control through loss of adiponectin’s proposed protective anti-proliferative and antiangiogenic properties. In vitro studies and animal studies have demonstrated a decrease in expression of insulin-responsive glucose transporter 4 (GLUT4) with atorvastatin, and an increase in GLUT1. It has been hypothesized that reduction in isoprenoid biosynthesis or decreased insulin signaling may explain these effects and that changes in glucose transport in adipocytes may cause insulin resistance. Other studies suggest that dysregulation of cellular cholesterol may attenuate beta-cell function. Impaired biosynthesis of ubiquinones may result in delayed production of adenosine triphosphate and consequently diminish insulin release.
But different effects have been reported for atorvastatin, simvastatin, and pravastatin, arguing against a unifying explanation or, alternatively, suggesting that differences in lipophilicity and potency among statins are important. Hydrophilic statins may be less likely to be taken up by extrahepatic cells such as pancreatic cells and adipocytes, possibly lessening these effects. However, the strong association between rosuvastatin (which is hydrophilic) and new diabetes would not support this hypothesis.
Despite these speculations, lack of conformity in response to different statins and discrepancies in the clinical outcomes noted in trials fail to clearly identify a common causative mechanism.
OTHER COMMON THERAPIES MAY INFLUENCE GLYCEMIC CONTROL
Statins are not the first drugs for reducing cardiovascular risk that have been shown to affect glucose levels during treatment.
Niacin
Niacin has been known to increase glucose levels but has long been used as a treatment for dyslipidemia despite this caution. Reduced glycemic control during niacin treatment in diabetic patients does not seem to alter the beneficial effects of treatment.35–37
In a post hoc analysis of the Coronary Drug Project (CDP), in patient subgroups defined by baseline fasting plasma glucose and compared with placebo, niacin reduced the 6-year risk of recurrent myocardial infarction and the combined end point of coronary heart disease death or nonfatal myocardial infarction similarly (interactive P value nonsignificant) across all levels of baseline fasting plasma glucose, including levels of 126 mg/dL or higher at study entry.36
In another post hoc analysis of CDP patient subgroups defined by the change in glycemic status from baseline to 1 year, niacin reduced the 6-year risk of the same end points similarly (interactive P value nonsignificant) across all levels of change in fasting plasma glucose from baseline to year 1, whether baseline fasting plasma glucose levels decreased, stayed the same, or increased to 10 mg/dL or higher on niacin therapy.36
Therefore, the beneficial effect of niacin of reducing the rate of recurrent nonfatal myocardial infarction and coronary heart disease events was not significantly diminished when impaired fasting glucose or diabetes was present when therapy was started or by on-therapy increases from baseline fasting plasma glucose.
In addition, on-therapy changes in glycemic control may be dose-related and minimized by surveillance and therapy adjustments. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT)38 found that changes in glycemic control were minimal as measured by fasting glucose and hemoglobin A1c; were associated with a higher niacin dose (1.5 g/day vs 1 g/day); and, when present, were successfully managed by adjusting the diabetes treatment regimen.
Antihypertensive drugs
Diuretics as well as beta-blockers have been reported to increase the incidence of diabetes in patients with hypertension.15,38–40
A retrospective longitudinal cohort study40 in 2009 examined the development of new-onset diabetes (defined as a new ICD-9 code for diabetes or initiation of diabetes treatment) in 24,688 treated hypertensive patients without diabetes at baseline; 4,385 (17.8%) of the patients developed diabetes. After adjusting for sex and age, the risk of new diabetes was significant in users of diuretics (OR 1.10), beta-blockers (OR 1.12), and calcium channel blockers (OR 1.10) compared with users of angiotensin-converting enzyme inhibitors, (OR 0.92), angiotensin receptor blockers (OR 0.90), or alpha-blockers (OR 0.88).
However, the increase in blood glucose does not seem to attenuate the beneficial effects of reducing cardiovascular events. In the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT),15 a long-term follow-up of those developing new-onset diabetes while taking chlorthalidone (Hygroton) found no difference in the risk of death from cardiovascular disease or from any cause (hazard ratio = 1.04).15
CLINICAL IMPLICATIONS
Balancing the benefits and risks of statins
It is important to examine how the 0.4% increase in absolute risk of new-onset diabetes as calculated in meta-analyses compares with the benefits of statin treatment in terms of cardiovascular risk reduction.
Using data from the Cholesterol Treatment Trialists (CTT) meta-analysis of statin trials in 71,370 participants, Sattar et al11 estimated that statin treatment is associated with 5.4 fewer deaths from coronary heart disease and cases of nonfatal myocardial infarction per 255 patients treated over 4 years for each 1-mmol/L (39 mg/dL) reduction in LDL-C compared with controls. The benefit would be even greater if stroke, revascularization, and hospitalization are included as end points. This benefit is contrasted with the risk of developing 1 additional case of diabetes for every 255 patients treated with statins over the same period.
Preiss et al25 calculated that there were 2 more cases of diabetes per 1,000 patient-years in patients receiving intensive doses than in those receiving moderate doses (18.9 vs 16.9), corresponding to 1 additional case of diabetes for every 498 patients treated per year. However, there were 6.5 fewer first major cardiovascular events per 1,000 patient-years (44.5 vs 51.0), corresponding to a number needed to treat per year to prevent 1 cardiovascular event of 155. Most of the benefit was due to fewer revascularizations, followed by nonfatal myocardial infarctions. The 12% increase in new diabetes with high-dose therapy contrasted with a 16% reduction in new cardiovascular disease combined events (OR 0.84, 95% CI 0.75–0.94).
As previously noted, in the JUPITER trial, the benefits of preventing cardiovascular events with statin therapy outweighed the risk of new diabetes in people both with and without baseline risk factors for diabetes.29 Similar to the observations with niacin and some antihypertensive drugs, the increase in blood glucose with statins does not appear to reduce the benefits of cardiovascular risk reduction in these patients at moderate to high risk, even when used at high doses.
People with diabetes need aggressive lipid-lowering—with statins
Diabetes is a coronary heart disease risk equivalent and is associated with high risk of cardiovascular events.41–46 Overall, the risk for these adverse events is two to four times greater in people with diabetes than without. Atherosclerosis-related events account for approximately 65% to 75% of all deaths in people with diabetes, and 75% of these events are coronary. Lipid abnormalities are strongly correlated with the risk of cardiovascular disease in people with diabetes, and aggressive treatment of risk factors, particularly lipid abnormalities, has been shown to reduce this risk.47–49 And data from multiple clinical trials support the use of statins to lower LDL-C as the first-line therapy for dyslipidemia in people with diabetes, just as it is in the general population.3–7,9,13,23,50–61
Analyses of diabetic subgroups encompassing 18,000 to 20,000 patients in the large statin trials have clearly demonstrated the benefits of statin therapy. A recent metaanalysis of 10 placebo-controlled trials that included approximately 16,000 patients with diabetes and 54,000 without diabetes demonstrated a 30% reduction in coronary heart disease, a 19% reduction in strokes, and a 12% reduction in mortality.54 Furthermore, in another meta-analysis of 14 trials, a similar 22% reduction in coronary heart disease was noted in people with diabetes whether or not they had a history of cardiovascular disease.55
Therefore, aggressive treatment of lipid abnormalities with statins as primary treatment has generally been adopted as a standard of care in diabetic patients, particularly those with clinical cardiovascular disease or one or more risk factors. The Adult Treatment Panel III guidelines recommend a minimum LDL-C goal of less than 100 mg/dL and a goal of less than 70 mg/dL as an option for patients with diabetes (Table 1).41,62 Similar recommendations have been issued by the American Diabetes Association together with the American College of Cardiology (Table 2),30 the American Diabetes Association by itself,63 and the American Academy of Pediatrics.6
Is new-onset diabetes as dangerous as established diabetes?
In studies to date, there did not appear to be more events in those who developed new-onset diabetes.
Waters et al,24 evaluating three trials of high-dose atorvastatin therapy, found that major cardiovascular events occurred in 11.3% of those with new-onset diabetes, 10.8% of those without new-onset diabetes (HR 1.02, 95% CI 0.77–1.35), and 17.5% of those who had diabetes at baseline.
Therefore, it may not be appropriate to extrapolate the glucose changes seen on statin therapy to an equivalent increase in adverse cardiovascular events as seen in other diabetic patients. The beneficial reduction in cardiovascular events does not appear to be diminished in those developing diabetes. It is not clear that the increase in glucose on statins has the same implications of a new diagnosis of diabetes. Does this elevation in glucose represent true diabetes or some downstream effect? For example, thiazide diuretics have been known to increase blood glucose levels, but the levels drop when these drugs are discontinued, even after many years of treatment.
On the other hand, it is possible that follow-up of 5 years or less in clinical trials has not allowed sufficient time to examine the influence of the increase in new-onset diabetes on future cardiovascular events. In addition, because of the widespread use of statins across a broad range of cardiovascular risk, even if the effect is small in absolute terms, the potential adverse effects are magnified, particularly in a low-risk population in which the cardiovascular benefits are smaller.
The association is real, but questions remain
In view of the evidence, it is difficult to refute that an association exits between statin use and new-onset diabetes, at least in some subgroups. The dose response noted in some studies further reinforces the conclusion that the association is real. However, many questions remain unanswered regarding mechanism of effect, whether there are differences depending on the particular statin or dose used, or differential effects in the populations treated (such as patients with metabolic syndrome or the elderly).
Until the contradictory observations can be resolved and plausible mechanisms of action elucidated, causality cannot be established. From a clinical standpoint there is no current evidence suggesting that the elevations in blood glucose seen while on lipid-lowering or blood-pressure-lowering therapy are associated with an increased risk of cardiovascular events or that they attenuate the beneficial effects of the therapy.
Statins should continue to be used in patients at high risk
Until further studies are done, statins should continue to be used, after assessing the risks and the benefits.
Primary prevention patients at moderate to high risk and secondary prevention patients stand to gain from statin therapy, and it should not be denied or doses reduced on the basis of concerns about the development of new-onset diabetes. The recognized modest risk of developing diabetes does not appear to blunt the cardioprotective effects of statin therapy in these moderate-to high-risk groups.
Rather than stop statins in patients at risk of diabetes such as the elderly or those with prediabetes, insulin resistance, or metabolic syndrome who are on therapy for appropriate reasons, it is reasonable to continue these drugs, monitoring glucose more closely and emphasizing the importance of weight reduction, diet, and aerobic exercise for preventing diabetes. The Diabetes Prevention Program Research Group, for example, reduced the incidence of diabetes by 58% over 2.8 years of follow-up with intensive lifestyle interventions (a low-calorie, low-fat diet plus moderate physical activity 150 minutes per week) vs usual care in at-risk populations.65
Should statins be used more cautiously in patients at lower risk?
The most recent Cholesterol Treatment Trialists meta-analysis of 27 randomized clinical trials (22 placebo-controlled, 134,537 people; 5 high-dose vs low-dose, 39,612 people) reported that reducing LDL-C with statins lowered cardiovascular risk even in low-risk patients.66 Overall, there were 21% fewer major cardiovascular events (coronary heart disease, stroke, or coronary revascularization) for every 1-mmol/L reduction in LDL-C.
The proportional reduction in events was at least as large in the two lowest-risk groups (estimated 5-year risk of < 5% and 5% to < 10%, 53,152 people) as in the higher-risk groups. This was reflected mainly in fewer nonfatal myocardial infarctions and coronary revascularizations. In these groups, the absolute reduction in risk for each 1-mmol/L reduction in LDL-C was 11 per 1,000 patients over 5 years. Even in this low-risk population, the reduction in cardiovascular risk seems to compare favorably with the small estimated increase risk of diabetes.
However, even in the lowest-risk group studied, the average baseline LDL-C level was greater than 130 mg/dL.
Therefore, in groups in which the benefits of statins on cardiovascular risk reduction are less robust (eg, low-risk primary prevention groups without significant elevations in LDLC, particularly the elderly), it would not be difficult to justify the case for more cautious use of statin therapy. If statins are used in these low-risk groups, restricting their use to those with at least moderate LDL-C elevation, using less aggressive LDL-C-lowering targets, and regular monitoring of fasting glucose seem reasonable until further information is available.
- US Food and Drug Administration. Statin drugs—drug safety communication: class labeling change. February 28, 2012. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm293670.htm.
- Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357–362.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R; for the Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009; 32:1924–1929.
- Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose—results from the LIPID trial. Diabetes Care 2003; 26:2713–2721.
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735–742.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Scandinavian Simvastatin Survival Study study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Barzilay JI, Davis BR, Pressel SL, et al; ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: the ALLHAT Diabetes Extension Study. Circ Cardiovasc Qual Outcomes 2012; 5:153–162.
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231–1239.
- GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? Ital Heart J 2000; 1:810–820.
- Yamakawa T, Takano T, Tanaka S, Kadonosono K, Terauchi Y. Influence of pitavastatin on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb 2008; 15:269–275.
- Kryzhanovski V, Gumprecht J, Zhu B, Yu CY, Hounslow N, Sponseller CA. Atorvastatin but not pitavastatin significantly increases fasting plasma glucose in patients with type 2 diabetes and combined dyslipidemia (abstract). J Am Coll Cardiol 2011; 57:E575.
- Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med 2012; 29:628–631.
- Sabatine MS, Morrow DA, Giugliano RP, et al. Implications of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Circulation 2004; 110(suppl III):834–880.
- Shepherd J, Barter P, Carmena R, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220–1226.
- Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535–1545.
- Yousef A, Tu JV, Wang J, Donovan L, Ko DT. The association of intensive statin therapy on long-term risks of cardiovascular events and diabetes following acute myocardial infarction (abstract). Circulation 2012; 125:e859.
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a metaanalysis. JAMA 2011; 305:2556–2564.
- de Lemos JA, Blazing MA, Wiviott SD, et al; A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Pedersen TR, Faegeman O, Kastelein JJ, et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Armitage J, Bowman L, Wallendszus K, et al; Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010; 37:1658–1669.
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012; 380:565–571.
- Brunzell JD, Davidson M, Furberg CD, et al; American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811–822.
- Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010; 55:1209–1216.
- Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204:483–490.
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006; 49:1881–1892.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 1999; 126:1205–1213.
- Guyton JR, Fazio S, Adewale AJ, et al. Effect of extended-release niacin on new-onset diabetes among hyperlipidemic patients treated with ezetimibe/simvastatin in a randomized controlled trial. Diabetes Care 2012; 35:857–860.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254–257.
- Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial. Arch Intern Med 2002; 162:1568–1576.
- Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NRAnglo-Scandinavian Cardiac Outcomes Trial Investigators. Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31:982–988.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201–207.
- Jong JP, Chang MH, Tien L, et al. Antihypertensive drugs and new-onset diabetes: a retrospective longitudinal cohort study. Cardiovasc Ther 2009; 27:159–163.
- Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study Lancet 2002; 359:2140–2144.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Sprafka JM, Burke GL, Folsom AR, McGovbern PG, Hahn LP. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991; 14:537–543.
- Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In:Harris MI, Cowie CC, Stern MP, et al, editors. Diabetes in America. 2nd ed. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 1995:233–257.
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–444.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348:383–393.
- Gaede P, Pederson O. Intensive integrated therapy of type 2 diabetes: implications for long-term prognosis. Diabetes 2004; 53:S39–S47.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the cholesterol and recurrent events (CARE) trial. The Care Investigators. Circulation 1998; 98:2513–2519.
- Pyðrälä K, Pedersen TR, Kjekshus J, Faergeman O, Olsson AG, Thorgeirsson G. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 1997; 20:614–620.
- Vijan S, Hayward RA; American College of Physicians. Pharmacologic lipid-lowering therapy in type 2 diabetes mellitus: background paper for the American College of Physicians. Ann Intern Med 2004; 140:650–658.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. Errata in Lancet 2008; 371:2084, Lancet 2005; 366:1358.
- Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009; 338:b2376.
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Baigent C; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Nissen SE, Nicholls SJ, Sipahi I, et al; ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes et al. N Engl J Med 2004; 350:1495–1504.
- LaRosa JC, Grundy SM, Waters DD, et al; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998; 339:1349–1357.
- Grundy SM, Cleeman JI, Bairey Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S5–S10.
- Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590.
- US Food and Drug Administration. Statin drugs—drug safety communication: class labeling change. February 28, 2012. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm293670.htm.
- Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357–362.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R; for the Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009; 32:1924–1929.
- Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose—results from the LIPID trial. Diabetes Care 2003; 26:2713–2721.
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735–742.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Scandinavian Simvastatin Survival Study study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Barzilay JI, Davis BR, Pressel SL, et al; ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: the ALLHAT Diabetes Extension Study. Circ Cardiovasc Qual Outcomes 2012; 5:153–162.
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231–1239.
- GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? Ital Heart J 2000; 1:810–820.
- Yamakawa T, Takano T, Tanaka S, Kadonosono K, Terauchi Y. Influence of pitavastatin on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb 2008; 15:269–275.
- Kryzhanovski V, Gumprecht J, Zhu B, Yu CY, Hounslow N, Sponseller CA. Atorvastatin but not pitavastatin significantly increases fasting plasma glucose in patients with type 2 diabetes and combined dyslipidemia (abstract). J Am Coll Cardiol 2011; 57:E575.
- Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med 2012; 29:628–631.
- Sabatine MS, Morrow DA, Giugliano RP, et al. Implications of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Circulation 2004; 110(suppl III):834–880.
- Shepherd J, Barter P, Carmena R, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220–1226.
- Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535–1545.
- Yousef A, Tu JV, Wang J, Donovan L, Ko DT. The association of intensive statin therapy on long-term risks of cardiovascular events and diabetes following acute myocardial infarction (abstract). Circulation 2012; 125:e859.
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a metaanalysis. JAMA 2011; 305:2556–2564.
- de Lemos JA, Blazing MA, Wiviott SD, et al; A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Pedersen TR, Faegeman O, Kastelein JJ, et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Armitage J, Bowman L, Wallendszus K, et al; Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010; 37:1658–1669.
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012; 380:565–571.
- Brunzell JD, Davidson M, Furberg CD, et al; American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811–822.
- Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010; 55:1209–1216.
- Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204:483–490.
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006; 49:1881–1892.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 1999; 126:1205–1213.
- Guyton JR, Fazio S, Adewale AJ, et al. Effect of extended-release niacin on new-onset diabetes among hyperlipidemic patients treated with ezetimibe/simvastatin in a randomized controlled trial. Diabetes Care 2012; 35:857–860.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254–257.
- Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial. Arch Intern Med 2002; 162:1568–1576.
- Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NRAnglo-Scandinavian Cardiac Outcomes Trial Investigators. Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31:982–988.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201–207.
- Jong JP, Chang MH, Tien L, et al. Antihypertensive drugs and new-onset diabetes: a retrospective longitudinal cohort study. Cardiovasc Ther 2009; 27:159–163.
- Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study Lancet 2002; 359:2140–2144.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Sprafka JM, Burke GL, Folsom AR, McGovbern PG, Hahn LP. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991; 14:537–543.
- Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In:Harris MI, Cowie CC, Stern MP, et al, editors. Diabetes in America. 2nd ed. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 1995:233–257.
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–444.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348:383–393.
- Gaede P, Pederson O. Intensive integrated therapy of type 2 diabetes: implications for long-term prognosis. Diabetes 2004; 53:S39–S47.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the cholesterol and recurrent events (CARE) trial. The Care Investigators. Circulation 1998; 98:2513–2519.
- Pyðrälä K, Pedersen TR, Kjekshus J, Faergeman O, Olsson AG, Thorgeirsson G. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 1997; 20:614–620.
- Vijan S, Hayward RA; American College of Physicians. Pharmacologic lipid-lowering therapy in type 2 diabetes mellitus: background paper for the American College of Physicians. Ann Intern Med 2004; 140:650–658.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. Errata in Lancet 2008; 371:2084, Lancet 2005; 366:1358.
- Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009; 338:b2376.
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Baigent C; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Nissen SE, Nicholls SJ, Sipahi I, et al; ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes et al. N Engl J Med 2004; 350:1495–1504.
- LaRosa JC, Grundy SM, Waters DD, et al; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998; 339:1349–1357.
- Grundy SM, Cleeman JI, Bairey Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S5–S10.
- Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590.
KEY POINTS
- The evidence from individual clinical trials is mixed, but meta-analyses indicate that statin therapy is associated with approximately a 9% higher risk of diabetes (an absolute difference of about 0.4%).
- We need to interpret this information cautiously. Many potentially confounding factors are involved, and rigorous prospective trials are needed to examine this issue.
- The benefit of preventing serious cardiovascular events seems to outweigh the higher risks of diabetes and poorer glycemic control, and we should continue to give statins to patients at moderate to high risk, including those with diabetes, with vigilance for these side effects.