User login
Cellulitis pearls
A 38-year-old man is admitted to the hospital with a painful, swollen left leg. This was not the first instance of this kind for him. He had been admitted for the same problem 3 months earlier. During the earlier admission, he was diagnosed with cellulitis and treated with intravenous cefazolin for 4 days, then discharged on cephalexin with resolution of his swelling and pain. Today, his blood pressure is 120/70, pulse is 90, temperature is 38.2°C, his left leg is edematous from the mid-calf to the ankle, and he has erythema and warmth over the calf. His white blood cell count is 13,000, and a diagnosis of cellulitis is made. Which of the following treatments is most likely to shorten his hospital stay?
A. Vancomycin therapy instead of cefazolin.
B. Piperacillin/tazobactam therapy instead of cefazolin.
C. Prednisolone therapy in addition to antibiotics.
D. Furosemide therapy in addition to antibiotics.
The correct answer is C, prednisolone therapy in addition to antibiotics. Corticosteroids have been used as therapy for a number of infectious diseases, and steroid use has been shown to improve survival in patients with bacterial meningitis, tuberculous meningitis, tuberculous pericarditis, severe typhoid fever, tetanus, or pneumocystis pneumonia with moderate to severe hypoxemia.1 Corticosteroid use in many other infections has been studied, and for many infections, symptomatic benefit has been shown. Berkvist and Sjobeck studied 112 patients admitted to the hospital with lower-extremity erysipelas/cellulitis and randomized the patients to receive prednisolone or placebo in addition to antibiotic treatment.2 The prednisolone-treated patients had a shorter hospital stay (5 days vs. 6 days; P less than .01), and had a shorter length of intravenous antibiotic treatment ( 3 days vs. 4 days; P less than .05). The same researchers followed up the study cohort a year later to see if there was any difference in relapse between the steroid- and placebo-treated patients.3 There was no statistically significant difference in relapse (six patients treated with prednisolone relapsed, compared with 13 who received placebo). Solomon et al. did a retrospective study of patients admitted with erysipelas/cellulitis over a 7-year period.4 The control group was defined as patients who received antibiotics but did not receive prednisone, while the other patients in the study received both antibiotics and prednisone. The patients who received antibiotics and prednisone had more severe cellulitis (most had bullous cellulitis) than the patients in the control group. Long-term follow-up showed a higher incidence of erythema and recurrence of cellulitis in the control group. The return to full function was faster in the prednisone-treated patients than in the control group.
Back to the case. Which of the following is most important to do for this patient to help prevent future episodes of cellulitis?
A. Daily penicillin.
B. Treatment of tinea pedis.
C. Hydrochlorothiazide treatment for leg edema.
D. Topical triamcinolone treatment of dry skin on legs.
The correct answer here is treatment of concurrent tinea pedis infection. Antibiotic prophylaxis is considered in patients who have multiple recurrent episodes. This patient’s unilateral edema is most likely attributable to the cellulitis and should resolve with therapy, so diuretics would not be indicated. Risk factors for recurrent cellulitis are tinea pedis, obesity, venous insufficiency, and lymphedema.5
Concheiro and colleagues did a retrospective study of 122 cases of cellulitis and found tinea pedis in 33% of the cases.6 Muller et al. studied the importance of toe web microorganisms and erysipelas and found that the presence of interdigital tinea pedis was correlated with recurrent infection.7 Treatment of tinea pedis is an easily modifiable risk factor in patients with recurrent cellulitis.
Pearls: Consider adding a short course of steroids in patients with more severe erysipelas/cellulitis, as it can decrease hospital stay and IV antibiotics.
Look for tinea pedis and treat if present in patients who have erysipelas/cellulitis.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Arch Intern Med. 2008 May 26;168(10):1034-46.
2. Scand J Infect Dis 1997;29(4):377-82.
3. Scand J Infect Dis. 1998;30(2):206-7.
4. Isr Med Assoc J. 2018 Mar;20(3):137-40.
5. J Dtsch Dermatol Ges. 2004 Feb;2(2):89-95.
6. Actas Dermosifiliogr. 2009 Dec;100(10):888-94.
7. J Dtsch Dermatol Ges. 2014 Aug;12(8):691-5.
A 38-year-old man is admitted to the hospital with a painful, swollen left leg. This was not the first instance of this kind for him. He had been admitted for the same problem 3 months earlier. During the earlier admission, he was diagnosed with cellulitis and treated with intravenous cefazolin for 4 days, then discharged on cephalexin with resolution of his swelling and pain. Today, his blood pressure is 120/70, pulse is 90, temperature is 38.2°C, his left leg is edematous from the mid-calf to the ankle, and he has erythema and warmth over the calf. His white blood cell count is 13,000, and a diagnosis of cellulitis is made. Which of the following treatments is most likely to shorten his hospital stay?
A. Vancomycin therapy instead of cefazolin.
B. Piperacillin/tazobactam therapy instead of cefazolin.
C. Prednisolone therapy in addition to antibiotics.
D. Furosemide therapy in addition to antibiotics.
The correct answer is C, prednisolone therapy in addition to antibiotics. Corticosteroids have been used as therapy for a number of infectious diseases, and steroid use has been shown to improve survival in patients with bacterial meningitis, tuberculous meningitis, tuberculous pericarditis, severe typhoid fever, tetanus, or pneumocystis pneumonia with moderate to severe hypoxemia.1 Corticosteroid use in many other infections has been studied, and for many infections, symptomatic benefit has been shown. Berkvist and Sjobeck studied 112 patients admitted to the hospital with lower-extremity erysipelas/cellulitis and randomized the patients to receive prednisolone or placebo in addition to antibiotic treatment.2 The prednisolone-treated patients had a shorter hospital stay (5 days vs. 6 days; P less than .01), and had a shorter length of intravenous antibiotic treatment ( 3 days vs. 4 days; P less than .05). The same researchers followed up the study cohort a year later to see if there was any difference in relapse between the steroid- and placebo-treated patients.3 There was no statistically significant difference in relapse (six patients treated with prednisolone relapsed, compared with 13 who received placebo). Solomon et al. did a retrospective study of patients admitted with erysipelas/cellulitis over a 7-year period.4 The control group was defined as patients who received antibiotics but did not receive prednisone, while the other patients in the study received both antibiotics and prednisone. The patients who received antibiotics and prednisone had more severe cellulitis (most had bullous cellulitis) than the patients in the control group. Long-term follow-up showed a higher incidence of erythema and recurrence of cellulitis in the control group. The return to full function was faster in the prednisone-treated patients than in the control group.
Back to the case. Which of the following is most important to do for this patient to help prevent future episodes of cellulitis?
A. Daily penicillin.
B. Treatment of tinea pedis.
C. Hydrochlorothiazide treatment for leg edema.
D. Topical triamcinolone treatment of dry skin on legs.
The correct answer here is treatment of concurrent tinea pedis infection. Antibiotic prophylaxis is considered in patients who have multiple recurrent episodes. This patient’s unilateral edema is most likely attributable to the cellulitis and should resolve with therapy, so diuretics would not be indicated. Risk factors for recurrent cellulitis are tinea pedis, obesity, venous insufficiency, and lymphedema.5
Concheiro and colleagues did a retrospective study of 122 cases of cellulitis and found tinea pedis in 33% of the cases.6 Muller et al. studied the importance of toe web microorganisms and erysipelas and found that the presence of interdigital tinea pedis was correlated with recurrent infection.7 Treatment of tinea pedis is an easily modifiable risk factor in patients with recurrent cellulitis.
Pearls: Consider adding a short course of steroids in patients with more severe erysipelas/cellulitis, as it can decrease hospital stay and IV antibiotics.
Look for tinea pedis and treat if present in patients who have erysipelas/cellulitis.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Arch Intern Med. 2008 May 26;168(10):1034-46.
2. Scand J Infect Dis 1997;29(4):377-82.
3. Scand J Infect Dis. 1998;30(2):206-7.
4. Isr Med Assoc J. 2018 Mar;20(3):137-40.
5. J Dtsch Dermatol Ges. 2004 Feb;2(2):89-95.
6. Actas Dermosifiliogr. 2009 Dec;100(10):888-94.
7. J Dtsch Dermatol Ges. 2014 Aug;12(8):691-5.
A 38-year-old man is admitted to the hospital with a painful, swollen left leg. This was not the first instance of this kind for him. He had been admitted for the same problem 3 months earlier. During the earlier admission, he was diagnosed with cellulitis and treated with intravenous cefazolin for 4 days, then discharged on cephalexin with resolution of his swelling and pain. Today, his blood pressure is 120/70, pulse is 90, temperature is 38.2°C, his left leg is edematous from the mid-calf to the ankle, and he has erythema and warmth over the calf. His white blood cell count is 13,000, and a diagnosis of cellulitis is made. Which of the following treatments is most likely to shorten his hospital stay?
A. Vancomycin therapy instead of cefazolin.
B. Piperacillin/tazobactam therapy instead of cefazolin.
C. Prednisolone therapy in addition to antibiotics.
D. Furosemide therapy in addition to antibiotics.
The correct answer is C, prednisolone therapy in addition to antibiotics. Corticosteroids have been used as therapy for a number of infectious diseases, and steroid use has been shown to improve survival in patients with bacterial meningitis, tuberculous meningitis, tuberculous pericarditis, severe typhoid fever, tetanus, or pneumocystis pneumonia with moderate to severe hypoxemia.1 Corticosteroid use in many other infections has been studied, and for many infections, symptomatic benefit has been shown. Berkvist and Sjobeck studied 112 patients admitted to the hospital with lower-extremity erysipelas/cellulitis and randomized the patients to receive prednisolone or placebo in addition to antibiotic treatment.2 The prednisolone-treated patients had a shorter hospital stay (5 days vs. 6 days; P less than .01), and had a shorter length of intravenous antibiotic treatment ( 3 days vs. 4 days; P less than .05). The same researchers followed up the study cohort a year later to see if there was any difference in relapse between the steroid- and placebo-treated patients.3 There was no statistically significant difference in relapse (six patients treated with prednisolone relapsed, compared with 13 who received placebo). Solomon et al. did a retrospective study of patients admitted with erysipelas/cellulitis over a 7-year period.4 The control group was defined as patients who received antibiotics but did not receive prednisone, while the other patients in the study received both antibiotics and prednisone. The patients who received antibiotics and prednisone had more severe cellulitis (most had bullous cellulitis) than the patients in the control group. Long-term follow-up showed a higher incidence of erythema and recurrence of cellulitis in the control group. The return to full function was faster in the prednisone-treated patients than in the control group.
Back to the case. Which of the following is most important to do for this patient to help prevent future episodes of cellulitis?
A. Daily penicillin.
B. Treatment of tinea pedis.
C. Hydrochlorothiazide treatment for leg edema.
D. Topical triamcinolone treatment of dry skin on legs.
The correct answer here is treatment of concurrent tinea pedis infection. Antibiotic prophylaxis is considered in patients who have multiple recurrent episodes. This patient’s unilateral edema is most likely attributable to the cellulitis and should resolve with therapy, so diuretics would not be indicated. Risk factors for recurrent cellulitis are tinea pedis, obesity, venous insufficiency, and lymphedema.5
Concheiro and colleagues did a retrospective study of 122 cases of cellulitis and found tinea pedis in 33% of the cases.6 Muller et al. studied the importance of toe web microorganisms and erysipelas and found that the presence of interdigital tinea pedis was correlated with recurrent infection.7 Treatment of tinea pedis is an easily modifiable risk factor in patients with recurrent cellulitis.
Pearls: Consider adding a short course of steroids in patients with more severe erysipelas/cellulitis, as it can decrease hospital stay and IV antibiotics.
Look for tinea pedis and treat if present in patients who have erysipelas/cellulitis.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Arch Intern Med. 2008 May 26;168(10):1034-46.
2. Scand J Infect Dis 1997;29(4):377-82.
3. Scand J Infect Dis. 1998;30(2):206-7.
4. Isr Med Assoc J. 2018 Mar;20(3):137-40.
5. J Dtsch Dermatol Ges. 2004 Feb;2(2):89-95.
6. Actas Dermosifiliogr. 2009 Dec;100(10):888-94.
7. J Dtsch Dermatol Ges. 2014 Aug;12(8):691-5.

2018 FDA-approved new drugs
In 2018, the Food and Drug Administration approved a record 58 new drugs for humans. One of these agents, Annovera (segesterone acetate and ethinyl estradiol), is a vaginal ring to prevent pregnancy and is not relevant in this article. A second drug, Asparlas (calaspargase pegol-mknl), indicated to treat acute lymphoblastic leukemia, has not yet been released by its manufacturer. The agents with molecular weights (MW) less than 1,000 probably cross the placenta, but nearly all, regardless of MW, will cross in the second half of pregnancy.

There is no human pregnancy data for these agents, but there are five drugs included in pregnancy registries. However, it will take some time before the outcomes of these drugs are published. The routine absence of pregnancy data for most drugs was pointed out in a reference that I coauthored (“Should pregnant women be included in phase 4 clinical drug trials?” Am J Obstet Gynecol. 2015 Dec;213[6]:810-5). The article makes a strong argument for including some drugs in these trials.
Amyloidosis
Onpattro (patisiran) is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. The drug caused embryo-fetal death and reduced fetal body weight in rabbits at doses also associated with maternal toxicity. No developmental toxicity was observed in rats.
Anti-infectives
Aemcolo (rifamycin), which has a MW of 720, is indicated for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli. No adverse fetal effects were observed in rats and rabbits that received close to human doses.
Krintafel (tafenoquine) is an antimalarial agent that is used to prevent relapse in patients who are receiving appropriate antimalarial therapy for Plasmodium vivax infection. The drug may cause hemolytic anemia in a fetus deficient in glucose-6-phosphate dehydrogenase. In rabbits, the drug caused dose-related abortions and maternal toxicity was observed in rabbits and rats. Treatment with this drug in pregnancy is not recommended, according to the manufacturer.
Tpoxx (tecovirimat monohydrate), which has a MW of about 394, is indicated for the treatment of smallpox disease. The drug did not cause embryo-fetal toxicity in pregnant mice and rabbits, but the maximum exposure in rabbits was only 0.4 times the human exposure.
Xofluza (baloxavir marboxil), which has a MW of about 572, is a prodrug that is converted by hydrolysis to baloxavir. It is indicated for the treatment of acute uncomplicated influenza. No adverse developmental effects were observed in rats and rabbits.
Zemdri (plazomicin), which has a MW of about 593, is an aminoglycoside indicated for the treatment of complicated urinary tract infections including pyelonephritis. The drug did not cause fetal harm in rats and rabbits at doses that did not cause maternal toxicity; however, prolonged use of an aminoglycoside (such as streptomycin) has caused irreversible, bilateral congenital deafness in children exposed in utero to prolonged use and is a potential complication.
Three new drugs in 2018 are indicated for treating HIV-1:
Biktarvy is a three-drug combination that includes bictegravir, emtricitabine, and tenofovir. The latter two drugs are included in the 11th edition of my book (“Drugs in Pregnancy and Lactation,” 11th ed. [Riverwoods, Ill.: Wolters Kluwer, 2017) and are not included here. Both are classified as compatible in pregnancy. Bictegravir has a MW of about 471. No adverse embryo-fetal effects in rats and rabbits were observed with this agent.
Trogarzo (ibalizumab-uiyk), which has a MW of about 150,000, is a monoclonal antibody antiretroviral agent used in combination with other antiretrovirals. There are no animal data. Although the MW is very high, monoclonal antibodies are transported across the placenta as pregnancy progresses.
Pifeltro (doravirine), which has a MW of about 426, is a nonnucleoside reverse transcriptase inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1. The drug caused no significant toxicologic effects on embryo-fetal rats and rabbits.

If Biktarvy, Pifeltro, or Trogarzo are used in pregnancy, health care providers are encouraged to register the patient in the Antiretroviral Pregnancy Registry by calling 1-800-258-4263.
There are three new agents in the tetracycline class.
Nuzyra (omadacycline), which has a MW of about 729, is for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections.
Seysara (sarecycline), which has a MW of about 524, is for inflammatory lesions of nonnodular, moderate to severe acne vulgaris.
Xerava (eravacycline), which has a MW of about 632, is for complicated intra-abdominal infection.
The various dose-related toxicities observed with the three drugs in rats and rabbits included maternal deaths; increased postimplantation loss; reduced fetal body weights; delays in skeletal ossification; and fetal malformations of the skeleton, heart, and lung. Use of these drugs in the last half of pregnancy may cause permanent discoloration of the teeth and enamel hypoplasia, as well as inhibition of bone growth.
Antilipemic agents
Crysvita (burosumab-twza), which has a MW of about 147,000, is a fibroblast growth factor–blocking antibody indicated for the treatment of X-linked hypophosphatemia. In pregnant cynomolgus monkeys, doses slightly higher than the human dose were not teratogenic. The drug was detected in fetal serum indicating that it crossed the monkey placenta.
Tegsedi (inotersen), which has a MW of about 7,601, is an amyloidosis inhibitor used for polyneuropathy of hereditary transthyretin-mediated amyloidosis. It is available only through a restricted program. The drug was not teratogenic in mice and rabbits; however, it does decrease vitamin A levels, so supplementation with the vitamin is recommended.
Antineoplastics
The manufacturers recommend avoiding these drugs during pregnancy. Effective contraception should be used.
Daurismo (glasdegib), which has a MW of about 491, is a hedgehog pathway inhibitor indicated in combination with low-dose cytarabine for newly diagnosed acute myeloid leukemia (AML). The drug caused embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits at doses less than the human dose.
Erleada (apalutamide), which has a MW of about 477, is an androgen receptor inhibitor indicated for nonmetastatic, castration-resistant prostate cancer. Animal studies were not conducted because the drug should not be used in females.
Elzonris (tagraxofusp-erzs), which has a MW of 57,695, is a cytotoxin indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm. Animal studies have not been conducted.
Lumoxiti (moxetumomab pasudotox–tdfk) which as a MW of about 63,000, is indicated for relapsed or refractory hairy cell leukemia. Studies have not been conducted in pregnant animals. Two life-threatening outcomes have occurred with the drug: capillary leak syndrome and hemolytic uremic syndrome. The drug should be discontinued if either occurs.
Lutathera (lutetium Lu 177 dotatate), which has a MW of about 1,610, is a radiolabeled somatostatin analogue given as a single intravenous dose every 8 weeks for four doses for the treatment of gastroenteropancreatic neuroendocrine tumors. Reproductive studies in animals have not been conducted. However, all radiopharmaceuticals have the potential to cause embryo-fetal harm. They also can cause infertility in males and females.
Talzenna (talazoparib), which has a MW of about 553, is a poly (ADP-ribose) polymerase inhibitor indicated for the treatment of certain types of breast cancer. At doses much less then the human dose, the drug caused fetal malformations and embryo-fetal death in rats.
Tibsovo (ivosidenib), which has a MW of 583, is an isocitrate dehydrogenase 1 inhibitor used for patients with relapsed or refractory AML. The drug caused embryo-fetal toxicity in rats and rabbits at doses slighter higher than the human dose.
There are seven new kinase inhibitors.
Braftovi (encorafenib), which has a MW of 540, is indicated in combination with Mektovi for patients with a specific type of metastatic melanoma. The drug caused embryo-fetal toxicity in rats and rabbits.
Copiktra (duvelisib), which has a MW of about 435, is indicated for treatment of chronic lymphocytic leukemia and follicular lymphoma. In rats and rabbits, the drug caused embryo-fetal death, lower fetal weights, and malformations.
Lorbrena (lorlatinib), which has a MW of about 406, is given for the treatment of metastatic non–small cell lung cancer. In rats and rabbits, the drug caused abortions, decreased fetal body weight, and major malformations.
Mektovi (binimetinib), which has a MW of about 441, is used in combination with Braftovi for patients with a specific type of melanoma. The drug was embryotoxic and abortifacient in rabbits.
Vitrakvi (larotrectinib), which has a MW of about 527, is used for patients with solid tumors. Studies in rats revealed fetal anasarca (extreme generalized edema) and omphalocele in rabbits.
Vizimpro (dacomitinib), which has a MW of about 488, is indicated for metastatic non–small cell lung cancer. The drug caused embryo-fetal toxicity in rats and mice.
Xospata (gilteritinib), which has a MW of about 1,222, is indicated for relapsed or refractory AML. In rats, the drug caused embryo-fetal death, suppressed fetal growth, and caused multiple malformations.
Three drugs are classified as monoclonal antibodies.
Gamifant (emapalumab), which has a MW of about 148,000, is indicated for primary hemophagocytic lymphohistiocytosis. A murine surrogate antimouse antibody was given to pregnant mice throughout gestation and no fetal harm was observed.
Libtayo (cemiplimab-rwlc), which has a MW of 146,000, is indicated for patients with metastatic or locally advanced cutaneous squamous cell carcinoma. Animal reproduction studies have not been conducted; however, based on its mechanism, increased rates of abortion or stillbirth may occur if the drug is used in human pregnancy.
Poteligeo (mogamulizumab-kpkc), which has a MW of about 149,000, is given for relapsed/refractory mycosis fungoides or Sézary syndrome. In pregnant monkeys, there was no embryo-fetal lethality, teratogenicity, fetal growth restriction, spontaneous abortion, or increased fetal death.
Central nervous system
There are three antimigraine agents that are monoclonal antibodies given as a subcutaneous injection.
Aimovig (erenumab-aooe), which has a MW of about 150,000, caused no adverse effects in monkey offspring.
Ajovy (fremanezumab-vfrm), which has a MW of about 148,000, had no adverse effect in rat and rabbit offspring.
Emgality (galcanezumab-gnlm), which has a MW of about 147,000, produced no adverse effects in rat and rabbit offspring.
Diacomit (stiripentol), which has a MW of about 234, is an anticonvulsant used to treat seizures associated with Dravet syndrome. The drug caused severe embryo-fetal toxicity in mice, rabbits, and rats. The drug is included in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. Patients can enroll themselves by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Epidiolex (cannabidiol), which has a MW of about 314, is an anticonvulsant indicated for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. In pregnant rats, doses up to about 16 times the recommended human dose (RHD) caused no embryo-fetal adverse effects. The drug caused decreased fetal body weights, increased fetal structural variations, and maternal toxicity when the drug was given to pregnant rabbits throughout organogenesis. The no-effect dose for embryo-fetal toxicity was less than the human dose. Patients can enroll themselves in the NAAED Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Firdapse (amifampridine), a potassium channel blocker with a MW of about 201, is used for the treatment of Lambert-Eaton myasthenic syndrome. No adverse effects on embryo-fetal development were observed in rats and rabbits given the drug throughout organogenesis. However, in rats given the drug throughout pregnancy and lactation, there was an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups.
Lucemyra (lofexidine), which has a MW of about 296, is used to mitigate opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. The drug caused severe toxicity in the fetuses of rats and rabbits.
Olumiant (baricitinib), which has a MW of about 371, is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis. The drug was teratogenic in pregnant rats given doses about 20 times greater than the maximum RHD based on area under the curve. In rabbits, embryo death and rib anomalies were observed with doses 84 times greater than the maximum RHD, but no developmental toxicity was seen with doses 12 times greater than the maximum RHD.
Orilissa (elagolix), which has a MW of about 654, is a gonadotropin-releasing hormone receptor antagonist indicated for the management of pain associated with endometriosis. The drug caused abortions in rats and rabbits. Because the drug may increase the risk of early pregnancy loss, the manufacturer classifies it as contraindicated in pregnancy.
Dermatologic agents
Ilumya (tildrakizumab), which has a MW of about 147,000, is given by subcutaneous injection for the treatment of moderate to severe plaque psoriasis. When given during organogenesis in monkeys, no maternal or embryo-fetal toxicities were observed. However, when given throughout pregnancy a few neonatal deaths occurred, but the clinical significance of these nonclinical findings were unknown.
Fabry disease
Galafold (migalastat), which has a MW of about 200, is an alpha-galactosidase A pharmacologic chaperone indicated for the treatment of Fabry disease. Three pregnant women with Fabry disease were exposed to the drug in clinical studies but no information was provided on the pregnancy outcomes. No adverse developmental effects were observed in pregnant rats and rabbits.
Gastrointestinal agents
Akynzeo (netupitant or fosnetupitant palonosetron), which have MWs of about 579, 333, and 762, respectively, is available as an oral capsule (netupitant + palonosetron) and as an intravenous formulation (fosnetupitant + palonosetron). They are indicated, in combination with dexamethasone, for the prevention of nausea and vomiting related to cancer chemotherapy. Netupitant and fosnetupitant produced no embryo-fetal adverse effects in rats but were toxic to rabbit embryos. Palonosetron caused no embryo-fetal adverse effects in rats and rabbits.
Motegrity (prucalopride), which has a MW of about 486, is indicated for chronic idiopathic constipation. No adverse embryo-fetal developmental effects were observed in rats and rabbits.
Hematologic agents
Doptelet (avatrombopag), which has a MW of about 766, is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. No embryo-fetal effects were observed in rats, but in rabbits the drug was associated with spontaneous abortions.
Lokelma (sodium zirconium cyclosilicate) is a nonabsorbed zirconium silicate that exchanges potassium for hydrogen and sodium. Animal studies have not been conducted. Because it is not absorbed, it is not expected to result in fetal exposure to the drug.
Mulpleta (lusutrombopag), which has a MW of about 592, a thrombopoietin receptor agonist, is indicated for the treatment of thrombocytopenia in patients with chronic liver disease. High levels of the drug in pregnant rats were associated with adverse developmental outcomes. No adverse embryo-fetal effects were seen in pregnant rabbits.
Palynziq (pegvaliase-pqpz), which has a MW of about 1,000,000, is a phenylalanine-metabolizing enzyme indicated to reduce blood phenylalanine concentrations in patients with phenylketonuria. In pregnant rats, the drug caused an increase in skeletal variations. In rabbits, the drug caused a high incidence of multiple malformations.
Takhzyro (lanadelumab-flyo), which has a MW of about 49,000, is a monoclonal antibody indicated for prophylaxis to prevent attacks of hereditary angioedema. The drug caused no fetal harm in monkeys.
Tavalisse (fostamatinib disodium hexahydrate), which has a MW of about 733, is a kinase inhibitor used for the treatment of thrombocytopenia. In pregnant rats and rabbits, the drug caused adverse developmental outcomes including embryo-fetal mortality, lower fetal weights, and structural anomalies.
Ultomiris (ravulizumab), which has a MW of about 148,000, is a humanized monoclonal antibody indicated for adult patients with paroxysmal nocturnal hemoglobinuria. In mice, the drug was associated with increased rates of developmental abnormalities and an increased rate of dead and moribund offspring.
Immunologic agent
Revcovi (elapegademase-lvlr), which has a MW of about 113,000, is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency. Animal studies in pregnancy have not been conducted.
Nutrient/Nutritional supplement
Fish oil is indicated as a source of calories and fatty acids in pediatric patients with parenteral nutrition-associated cholestasis. Animal reproduction studies have not been conducted. It is doubtful if this product will be used in pregnancy.
Ophthalmic – nerve growth factor
Oxervate (cenegermin-bkbj), which has a MW of 13,266, is a solution that contains 118 amino acids. It is a recombinant human nerve growth factor indicated for neurotrophic keratitis. In rats and rabbits given the drug during organogenesis, there was a slight increase in postimplantation loss at doses greater than or equal to 267 times the human dose.
Respiratory drugs
Symdeko (tezacaftor + ivacaftor), which have MWs of about 521 and 392, is indicated for the treatment of patients with cystic fibrosis who are homozygous for the F508del mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator gene. There were no adverse developmental effects in pregnant rats and rabbits when the drugs were used separately or combined.
Yupelri (revefenacin), which has a MW of about 598, is an anticholinergic drug. It is an inhaled solution for the maintenance treatment of chronic obstructive pulmonary disease. In rats and rabbits, doses that were about 209 times the RHD produced no evidence of fetal harm.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. Mr. Briggs reported no relevant financial disclosures. Email him at [email protected].
In 2018, the Food and Drug Administration approved a record 58 new drugs for humans. One of these agents, Annovera (segesterone acetate and ethinyl estradiol), is a vaginal ring to prevent pregnancy and is not relevant in this article. A second drug, Asparlas (calaspargase pegol-mknl), indicated to treat acute lymphoblastic leukemia, has not yet been released by its manufacturer. The agents with molecular weights (MW) less than 1,000 probably cross the placenta, but nearly all, regardless of MW, will cross in the second half of pregnancy.
There is no human pregnancy data for these agents, but there are five drugs included in pregnancy registries. However, it will take some time before the outcomes of these drugs are published. The routine absence of pregnancy data for most drugs was pointed out in a reference that I coauthored (“Should pregnant women be included in phase 4 clinical drug trials?” Am J Obstet Gynecol. 2015 Dec;213[6]:810-5). The article makes a strong argument for including some drugs in these trials.
Amyloidosis
Onpattro (patisiran) is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. The drug caused embryo-fetal death and reduced fetal body weight in rabbits at doses also associated with maternal toxicity. No developmental toxicity was observed in rats.
Anti-infectives
Aemcolo (rifamycin), which has a MW of 720, is indicated for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli. No adverse fetal effects were observed in rats and rabbits that received close to human doses.
Krintafel (tafenoquine) is an antimalarial agent that is used to prevent relapse in patients who are receiving appropriate antimalarial therapy for Plasmodium vivax infection. The drug may cause hemolytic anemia in a fetus deficient in glucose-6-phosphate dehydrogenase. In rabbits, the drug caused dose-related abortions and maternal toxicity was observed in rabbits and rats. Treatment with this drug in pregnancy is not recommended, according to the manufacturer.
Tpoxx (tecovirimat monohydrate), which has a MW of about 394, is indicated for the treatment of smallpox disease. The drug did not cause embryo-fetal toxicity in pregnant mice and rabbits, but the maximum exposure in rabbits was only 0.4 times the human exposure.
Xofluza (baloxavir marboxil), which has a MW of about 572, is a prodrug that is converted by hydrolysis to baloxavir. It is indicated for the treatment of acute uncomplicated influenza. No adverse developmental effects were observed in rats and rabbits.
Zemdri (plazomicin), which has a MW of about 593, is an aminoglycoside indicated for the treatment of complicated urinary tract infections including pyelonephritis. The drug did not cause fetal harm in rats and rabbits at doses that did not cause maternal toxicity; however, prolonged use of an aminoglycoside (such as streptomycin) has caused irreversible, bilateral congenital deafness in children exposed in utero to prolonged use and is a potential complication.
Three new drugs in 2018 are indicated for treating HIV-1:
Biktarvy is a three-drug combination that includes bictegravir, emtricitabine, and tenofovir. The latter two drugs are included in the 11th edition of my book (“Drugs in Pregnancy and Lactation,” 11th ed. [Riverwoods, Ill.: Wolters Kluwer, 2017) and are not included here. Both are classified as compatible in pregnancy. Bictegravir has a MW of about 471. No adverse embryo-fetal effects in rats and rabbits were observed with this agent.
Trogarzo (ibalizumab-uiyk), which has a MW of about 150,000, is a monoclonal antibody antiretroviral agent used in combination with other antiretrovirals. There are no animal data. Although the MW is very high, monoclonal antibodies are transported across the placenta as pregnancy progresses.
Pifeltro (doravirine), which has a MW of about 426, is a nonnucleoside reverse transcriptase inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1. The drug caused no significant toxicologic effects on embryo-fetal rats and rabbits.
If Biktarvy, Pifeltro, or Trogarzo are used in pregnancy, health care providers are encouraged to register the patient in the Antiretroviral Pregnancy Registry by calling 1-800-258-4263.
There are three new agents in the tetracycline class.
Nuzyra (omadacycline), which has a MW of about 729, is for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections.
Seysara (sarecycline), which has a MW of about 524, is for inflammatory lesions of nonnodular, moderate to severe acne vulgaris.
Xerava (eravacycline), which has a MW of about 632, is for complicated intra-abdominal infection.
The various dose-related toxicities observed with the three drugs in rats and rabbits included maternal deaths; increased postimplantation loss; reduced fetal body weights; delays in skeletal ossification; and fetal malformations of the skeleton, heart, and lung. Use of these drugs in the last half of pregnancy may cause permanent discoloration of the teeth and enamel hypoplasia, as well as inhibition of bone growth.
Antilipemic agents
Crysvita (burosumab-twza), which has a MW of about 147,000, is a fibroblast growth factor–blocking antibody indicated for the treatment of X-linked hypophosphatemia. In pregnant cynomolgus monkeys, doses slightly higher than the human dose were not teratogenic. The drug was detected in fetal serum indicating that it crossed the monkey placenta.
Tegsedi (inotersen), which has a MW of about 7,601, is an amyloidosis inhibitor used for polyneuropathy of hereditary transthyretin-mediated amyloidosis. It is available only through a restricted program. The drug was not teratogenic in mice and rabbits; however, it does decrease vitamin A levels, so supplementation with the vitamin is recommended.
Antineoplastics
The manufacturers recommend avoiding these drugs during pregnancy. Effective contraception should be used.
Daurismo (glasdegib), which has a MW of about 491, is a hedgehog pathway inhibitor indicated in combination with low-dose cytarabine for newly diagnosed acute myeloid leukemia (AML). The drug caused embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits at doses less than the human dose.
Erleada (apalutamide), which has a MW of about 477, is an androgen receptor inhibitor indicated for nonmetastatic, castration-resistant prostate cancer. Animal studies were not conducted because the drug should not be used in females.
Elzonris (tagraxofusp-erzs), which has a MW of 57,695, is a cytotoxin indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm. Animal studies have not been conducted.
Lumoxiti (moxetumomab pasudotox–tdfk) which as a MW of about 63,000, is indicated for relapsed or refractory hairy cell leukemia. Studies have not been conducted in pregnant animals. Two life-threatening outcomes have occurred with the drug: capillary leak syndrome and hemolytic uremic syndrome. The drug should be discontinued if either occurs.
Lutathera (lutetium Lu 177 dotatate), which has a MW of about 1,610, is a radiolabeled somatostatin analogue given as a single intravenous dose every 8 weeks for four doses for the treatment of gastroenteropancreatic neuroendocrine tumors. Reproductive studies in animals have not been conducted. However, all radiopharmaceuticals have the potential to cause embryo-fetal harm. They also can cause infertility in males and females.
Talzenna (talazoparib), which has a MW of about 553, is a poly (ADP-ribose) polymerase inhibitor indicated for the treatment of certain types of breast cancer. At doses much less then the human dose, the drug caused fetal malformations and embryo-fetal death in rats.
Tibsovo (ivosidenib), which has a MW of 583, is an isocitrate dehydrogenase 1 inhibitor used for patients with relapsed or refractory AML. The drug caused embryo-fetal toxicity in rats and rabbits at doses slighter higher than the human dose.
There are seven new kinase inhibitors.
Braftovi (encorafenib), which has a MW of 540, is indicated in combination with Mektovi for patients with a specific type of metastatic melanoma. The drug caused embryo-fetal toxicity in rats and rabbits.
Copiktra (duvelisib), which has a MW of about 435, is indicated for treatment of chronic lymphocytic leukemia and follicular lymphoma. In rats and rabbits, the drug caused embryo-fetal death, lower fetal weights, and malformations.
Lorbrena (lorlatinib), which has a MW of about 406, is given for the treatment of metastatic non–small cell lung cancer. In rats and rabbits, the drug caused abortions, decreased fetal body weight, and major malformations.
Mektovi (binimetinib), which has a MW of about 441, is used in combination with Braftovi for patients with a specific type of melanoma. The drug was embryotoxic and abortifacient in rabbits.
Vitrakvi (larotrectinib), which has a MW of about 527, is used for patients with solid tumors. Studies in rats revealed fetal anasarca (extreme generalized edema) and omphalocele in rabbits.
Vizimpro (dacomitinib), which has a MW of about 488, is indicated for metastatic non–small cell lung cancer. The drug caused embryo-fetal toxicity in rats and mice.
Xospata (gilteritinib), which has a MW of about 1,222, is indicated for relapsed or refractory AML. In rats, the drug caused embryo-fetal death, suppressed fetal growth, and caused multiple malformations.
Three drugs are classified as monoclonal antibodies.
Gamifant (emapalumab), which has a MW of about 148,000, is indicated for primary hemophagocytic lymphohistiocytosis. A murine surrogate antimouse antibody was given to pregnant mice throughout gestation and no fetal harm was observed.
Libtayo (cemiplimab-rwlc), which has a MW of 146,000, is indicated for patients with metastatic or locally advanced cutaneous squamous cell carcinoma. Animal reproduction studies have not been conducted; however, based on its mechanism, increased rates of abortion or stillbirth may occur if the drug is used in human pregnancy.
Poteligeo (mogamulizumab-kpkc), which has a MW of about 149,000, is given for relapsed/refractory mycosis fungoides or Sézary syndrome. In pregnant monkeys, there was no embryo-fetal lethality, teratogenicity, fetal growth restriction, spontaneous abortion, or increased fetal death.
Central nervous system
There are three antimigraine agents that are monoclonal antibodies given as a subcutaneous injection.
Aimovig (erenumab-aooe), which has a MW of about 150,000, caused no adverse effects in monkey offspring.
Ajovy (fremanezumab-vfrm), which has a MW of about 148,000, had no adverse effect in rat and rabbit offspring.
Emgality (galcanezumab-gnlm), which has a MW of about 147,000, produced no adverse effects in rat and rabbit offspring.
Diacomit (stiripentol), which has a MW of about 234, is an anticonvulsant used to treat seizures associated with Dravet syndrome. The drug caused severe embryo-fetal toxicity in mice, rabbits, and rats. The drug is included in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. Patients can enroll themselves by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Epidiolex (cannabidiol), which has a MW of about 314, is an anticonvulsant indicated for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. In pregnant rats, doses up to about 16 times the recommended human dose (RHD) caused no embryo-fetal adverse effects. The drug caused decreased fetal body weights, increased fetal structural variations, and maternal toxicity when the drug was given to pregnant rabbits throughout organogenesis. The no-effect dose for embryo-fetal toxicity was less than the human dose. Patients can enroll themselves in the NAAED Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Firdapse (amifampridine), a potassium channel blocker with a MW of about 201, is used for the treatment of Lambert-Eaton myasthenic syndrome. No adverse effects on embryo-fetal development were observed in rats and rabbits given the drug throughout organogenesis. However, in rats given the drug throughout pregnancy and lactation, there was an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups.
Lucemyra (lofexidine), which has a MW of about 296, is used to mitigate opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. The drug caused severe toxicity in the fetuses of rats and rabbits.
Olumiant (baricitinib), which has a MW of about 371, is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis. The drug was teratogenic in pregnant rats given doses about 20 times greater than the maximum RHD based on area under the curve. In rabbits, embryo death and rib anomalies were observed with doses 84 times greater than the maximum RHD, but no developmental toxicity was seen with doses 12 times greater than the maximum RHD.
Orilissa (elagolix), which has a MW of about 654, is a gonadotropin-releasing hormone receptor antagonist indicated for the management of pain associated with endometriosis. The drug caused abortions in rats and rabbits. Because the drug may increase the risk of early pregnancy loss, the manufacturer classifies it as contraindicated in pregnancy.
Dermatologic agents
Ilumya (tildrakizumab), which has a MW of about 147,000, is given by subcutaneous injection for the treatment of moderate to severe plaque psoriasis. When given during organogenesis in monkeys, no maternal or embryo-fetal toxicities were observed. However, when given throughout pregnancy a few neonatal deaths occurred, but the clinical significance of these nonclinical findings were unknown.
Fabry disease
Galafold (migalastat), which has a MW of about 200, is an alpha-galactosidase A pharmacologic chaperone indicated for the treatment of Fabry disease. Three pregnant women with Fabry disease were exposed to the drug in clinical studies but no information was provided on the pregnancy outcomes. No adverse developmental effects were observed in pregnant rats and rabbits.
Gastrointestinal agents
Akynzeo (netupitant or fosnetupitant palonosetron), which have MWs of about 579, 333, and 762, respectively, is available as an oral capsule (netupitant + palonosetron) and as an intravenous formulation (fosnetupitant + palonosetron). They are indicated, in combination with dexamethasone, for the prevention of nausea and vomiting related to cancer chemotherapy. Netupitant and fosnetupitant produced no embryo-fetal adverse effects in rats but were toxic to rabbit embryos. Palonosetron caused no embryo-fetal adverse effects in rats and rabbits.
Motegrity (prucalopride), which has a MW of about 486, is indicated for chronic idiopathic constipation. No adverse embryo-fetal developmental effects were observed in rats and rabbits.
Hematologic agents
Doptelet (avatrombopag), which has a MW of about 766, is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. No embryo-fetal effects were observed in rats, but in rabbits the drug was associated with spontaneous abortions.
Lokelma (sodium zirconium cyclosilicate) is a nonabsorbed zirconium silicate that exchanges potassium for hydrogen and sodium. Animal studies have not been conducted. Because it is not absorbed, it is not expected to result in fetal exposure to the drug.
Mulpleta (lusutrombopag), which has a MW of about 592, a thrombopoietin receptor agonist, is indicated for the treatment of thrombocytopenia in patients with chronic liver disease. High levels of the drug in pregnant rats were associated with adverse developmental outcomes. No adverse embryo-fetal effects were seen in pregnant rabbits.
Palynziq (pegvaliase-pqpz), which has a MW of about 1,000,000, is a phenylalanine-metabolizing enzyme indicated to reduce blood phenylalanine concentrations in patients with phenylketonuria. In pregnant rats, the drug caused an increase in skeletal variations. In rabbits, the drug caused a high incidence of multiple malformations.
Takhzyro (lanadelumab-flyo), which has a MW of about 49,000, is a monoclonal antibody indicated for prophylaxis to prevent attacks of hereditary angioedema. The drug caused no fetal harm in monkeys.
Tavalisse (fostamatinib disodium hexahydrate), which has a MW of about 733, is a kinase inhibitor used for the treatment of thrombocytopenia. In pregnant rats and rabbits, the drug caused adverse developmental outcomes including embryo-fetal mortality, lower fetal weights, and structural anomalies.
Ultomiris (ravulizumab), which has a MW of about 148,000, is a humanized monoclonal antibody indicated for adult patients with paroxysmal nocturnal hemoglobinuria. In mice, the drug was associated with increased rates of developmental abnormalities and an increased rate of dead and moribund offspring.
Immunologic agent
Revcovi (elapegademase-lvlr), which has a MW of about 113,000, is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency. Animal studies in pregnancy have not been conducted.
Nutrient/Nutritional supplement
Fish oil is indicated as a source of calories and fatty acids in pediatric patients with parenteral nutrition-associated cholestasis. Animal reproduction studies have not been conducted. It is doubtful if this product will be used in pregnancy.
Ophthalmic – nerve growth factor
Oxervate (cenegermin-bkbj), which has a MW of 13,266, is a solution that contains 118 amino acids. It is a recombinant human nerve growth factor indicated for neurotrophic keratitis. In rats and rabbits given the drug during organogenesis, there was a slight increase in postimplantation loss at doses greater than or equal to 267 times the human dose.
Respiratory drugs
Symdeko (tezacaftor + ivacaftor), which have MWs of about 521 and 392, is indicated for the treatment of patients with cystic fibrosis who are homozygous for the F508del mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator gene. There were no adverse developmental effects in pregnant rats and rabbits when the drugs were used separately or combined.
Yupelri (revefenacin), which has a MW of about 598, is an anticholinergic drug. It is an inhaled solution for the maintenance treatment of chronic obstructive pulmonary disease. In rats and rabbits, doses that were about 209 times the RHD produced no evidence of fetal harm.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. Mr. Briggs reported no relevant financial disclosures. Email him at [email protected].
In 2018, the Food and Drug Administration approved a record 58 new drugs for humans. One of these agents, Annovera (segesterone acetate and ethinyl estradiol), is a vaginal ring to prevent pregnancy and is not relevant in this article. A second drug, Asparlas (calaspargase pegol-mknl), indicated to treat acute lymphoblastic leukemia, has not yet been released by its manufacturer. The agents with molecular weights (MW) less than 1,000 probably cross the placenta, but nearly all, regardless of MW, will cross in the second half of pregnancy.
There is no human pregnancy data for these agents, but there are five drugs included in pregnancy registries. However, it will take some time before the outcomes of these drugs are published. The routine absence of pregnancy data for most drugs was pointed out in a reference that I coauthored (“Should pregnant women be included in phase 4 clinical drug trials?” Am J Obstet Gynecol. 2015 Dec;213[6]:810-5). The article makes a strong argument for including some drugs in these trials.
Amyloidosis
Onpattro (patisiran) is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. The drug caused embryo-fetal death and reduced fetal body weight in rabbits at doses also associated with maternal toxicity. No developmental toxicity was observed in rats.
Anti-infectives
Aemcolo (rifamycin), which has a MW of 720, is indicated for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli. No adverse fetal effects were observed in rats and rabbits that received close to human doses.
Krintafel (tafenoquine) is an antimalarial agent that is used to prevent relapse in patients who are receiving appropriate antimalarial therapy for Plasmodium vivax infection. The drug may cause hemolytic anemia in a fetus deficient in glucose-6-phosphate dehydrogenase. In rabbits, the drug caused dose-related abortions and maternal toxicity was observed in rabbits and rats. Treatment with this drug in pregnancy is not recommended, according to the manufacturer.
Tpoxx (tecovirimat monohydrate), which has a MW of about 394, is indicated for the treatment of smallpox disease. The drug did not cause embryo-fetal toxicity in pregnant mice and rabbits, but the maximum exposure in rabbits was only 0.4 times the human exposure.
Xofluza (baloxavir marboxil), which has a MW of about 572, is a prodrug that is converted by hydrolysis to baloxavir. It is indicated for the treatment of acute uncomplicated influenza. No adverse developmental effects were observed in rats and rabbits.
Zemdri (plazomicin), which has a MW of about 593, is an aminoglycoside indicated for the treatment of complicated urinary tract infections including pyelonephritis. The drug did not cause fetal harm in rats and rabbits at doses that did not cause maternal toxicity; however, prolonged use of an aminoglycoside (such as streptomycin) has caused irreversible, bilateral congenital deafness in children exposed in utero to prolonged use and is a potential complication.
Three new drugs in 2018 are indicated for treating HIV-1:
Biktarvy is a three-drug combination that includes bictegravir, emtricitabine, and tenofovir. The latter two drugs are included in the 11th edition of my book (“Drugs in Pregnancy and Lactation,” 11th ed. [Riverwoods, Ill.: Wolters Kluwer, 2017) and are not included here. Both are classified as compatible in pregnancy. Bictegravir has a MW of about 471. No adverse embryo-fetal effects in rats and rabbits were observed with this agent.
Trogarzo (ibalizumab-uiyk), which has a MW of about 150,000, is a monoclonal antibody antiretroviral agent used in combination with other antiretrovirals. There are no animal data. Although the MW is very high, monoclonal antibodies are transported across the placenta as pregnancy progresses.
Pifeltro (doravirine), which has a MW of about 426, is a nonnucleoside reverse transcriptase inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1. The drug caused no significant toxicologic effects on embryo-fetal rats and rabbits.
If Biktarvy, Pifeltro, or Trogarzo are used in pregnancy, health care providers are encouraged to register the patient in the Antiretroviral Pregnancy Registry by calling 1-800-258-4263.
There are three new agents in the tetracycline class.
Nuzyra (omadacycline), which has a MW of about 729, is for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections.
Seysara (sarecycline), which has a MW of about 524, is for inflammatory lesions of nonnodular, moderate to severe acne vulgaris.
Xerava (eravacycline), which has a MW of about 632, is for complicated intra-abdominal infection.
The various dose-related toxicities observed with the three drugs in rats and rabbits included maternal deaths; increased postimplantation loss; reduced fetal body weights; delays in skeletal ossification; and fetal malformations of the skeleton, heart, and lung. Use of these drugs in the last half of pregnancy may cause permanent discoloration of the teeth and enamel hypoplasia, as well as inhibition of bone growth.
Antilipemic agents
Crysvita (burosumab-twza), which has a MW of about 147,000, is a fibroblast growth factor–blocking antibody indicated for the treatment of X-linked hypophosphatemia. In pregnant cynomolgus monkeys, doses slightly higher than the human dose were not teratogenic. The drug was detected in fetal serum indicating that it crossed the monkey placenta.
Tegsedi (inotersen), which has a MW of about 7,601, is an amyloidosis inhibitor used for polyneuropathy of hereditary transthyretin-mediated amyloidosis. It is available only through a restricted program. The drug was not teratogenic in mice and rabbits; however, it does decrease vitamin A levels, so supplementation with the vitamin is recommended.
Antineoplastics
The manufacturers recommend avoiding these drugs during pregnancy. Effective contraception should be used.
Daurismo (glasdegib), which has a MW of about 491, is a hedgehog pathway inhibitor indicated in combination with low-dose cytarabine for newly diagnosed acute myeloid leukemia (AML). The drug caused embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits at doses less than the human dose.
Erleada (apalutamide), which has a MW of about 477, is an androgen receptor inhibitor indicated for nonmetastatic, castration-resistant prostate cancer. Animal studies were not conducted because the drug should not be used in females.
Elzonris (tagraxofusp-erzs), which has a MW of 57,695, is a cytotoxin indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm. Animal studies have not been conducted.
Lumoxiti (moxetumomab pasudotox–tdfk) which as a MW of about 63,000, is indicated for relapsed or refractory hairy cell leukemia. Studies have not been conducted in pregnant animals. Two life-threatening outcomes have occurred with the drug: capillary leak syndrome and hemolytic uremic syndrome. The drug should be discontinued if either occurs.
Lutathera (lutetium Lu 177 dotatate), which has a MW of about 1,610, is a radiolabeled somatostatin analogue given as a single intravenous dose every 8 weeks for four doses for the treatment of gastroenteropancreatic neuroendocrine tumors. Reproductive studies in animals have not been conducted. However, all radiopharmaceuticals have the potential to cause embryo-fetal harm. They also can cause infertility in males and females.
Talzenna (talazoparib), which has a MW of about 553, is a poly (ADP-ribose) polymerase inhibitor indicated for the treatment of certain types of breast cancer. At doses much less then the human dose, the drug caused fetal malformations and embryo-fetal death in rats.
Tibsovo (ivosidenib), which has a MW of 583, is an isocitrate dehydrogenase 1 inhibitor used for patients with relapsed or refractory AML. The drug caused embryo-fetal toxicity in rats and rabbits at doses slighter higher than the human dose.
There are seven new kinase inhibitors.
Braftovi (encorafenib), which has a MW of 540, is indicated in combination with Mektovi for patients with a specific type of metastatic melanoma. The drug caused embryo-fetal toxicity in rats and rabbits.
Copiktra (duvelisib), which has a MW of about 435, is indicated for treatment of chronic lymphocytic leukemia and follicular lymphoma. In rats and rabbits, the drug caused embryo-fetal death, lower fetal weights, and malformations.
Lorbrena (lorlatinib), which has a MW of about 406, is given for the treatment of metastatic non–small cell lung cancer. In rats and rabbits, the drug caused abortions, decreased fetal body weight, and major malformations.
Mektovi (binimetinib), which has a MW of about 441, is used in combination with Braftovi for patients with a specific type of melanoma. The drug was embryotoxic and abortifacient in rabbits.
Vitrakvi (larotrectinib), which has a MW of about 527, is used for patients with solid tumors. Studies in rats revealed fetal anasarca (extreme generalized edema) and omphalocele in rabbits.
Vizimpro (dacomitinib), which has a MW of about 488, is indicated for metastatic non–small cell lung cancer. The drug caused embryo-fetal toxicity in rats and mice.
Xospata (gilteritinib), which has a MW of about 1,222, is indicated for relapsed or refractory AML. In rats, the drug caused embryo-fetal death, suppressed fetal growth, and caused multiple malformations.
Three drugs are classified as monoclonal antibodies.
Gamifant (emapalumab), which has a MW of about 148,000, is indicated for primary hemophagocytic lymphohistiocytosis. A murine surrogate antimouse antibody was given to pregnant mice throughout gestation and no fetal harm was observed.
Libtayo (cemiplimab-rwlc), which has a MW of 146,000, is indicated for patients with metastatic or locally advanced cutaneous squamous cell carcinoma. Animal reproduction studies have not been conducted; however, based on its mechanism, increased rates of abortion or stillbirth may occur if the drug is used in human pregnancy.
Poteligeo (mogamulizumab-kpkc), which has a MW of about 149,000, is given for relapsed/refractory mycosis fungoides or Sézary syndrome. In pregnant monkeys, there was no embryo-fetal lethality, teratogenicity, fetal growth restriction, spontaneous abortion, or increased fetal death.
Central nervous system
There are three antimigraine agents that are monoclonal antibodies given as a subcutaneous injection.
Aimovig (erenumab-aooe), which has a MW of about 150,000, caused no adverse effects in monkey offspring.
Ajovy (fremanezumab-vfrm), which has a MW of about 148,000, had no adverse effect in rat and rabbit offspring.
Emgality (galcanezumab-gnlm), which has a MW of about 147,000, produced no adverse effects in rat and rabbit offspring.
Diacomit (stiripentol), which has a MW of about 234, is an anticonvulsant used to treat seizures associated with Dravet syndrome. The drug caused severe embryo-fetal toxicity in mice, rabbits, and rats. The drug is included in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. Patients can enroll themselves by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Epidiolex (cannabidiol), which has a MW of about 314, is an anticonvulsant indicated for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. In pregnant rats, doses up to about 16 times the recommended human dose (RHD) caused no embryo-fetal adverse effects. The drug caused decreased fetal body weights, increased fetal structural variations, and maternal toxicity when the drug was given to pregnant rabbits throughout organogenesis. The no-effect dose for embryo-fetal toxicity was less than the human dose. Patients can enroll themselves in the NAAED Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Firdapse (amifampridine), a potassium channel blocker with a MW of about 201, is used for the treatment of Lambert-Eaton myasthenic syndrome. No adverse effects on embryo-fetal development were observed in rats and rabbits given the drug throughout organogenesis. However, in rats given the drug throughout pregnancy and lactation, there was an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups.
Lucemyra (lofexidine), which has a MW of about 296, is used to mitigate opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. The drug caused severe toxicity in the fetuses of rats and rabbits.
Olumiant (baricitinib), which has a MW of about 371, is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis. The drug was teratogenic in pregnant rats given doses about 20 times greater than the maximum RHD based on area under the curve. In rabbits, embryo death and rib anomalies were observed with doses 84 times greater than the maximum RHD, but no developmental toxicity was seen with doses 12 times greater than the maximum RHD.
Orilissa (elagolix), which has a MW of about 654, is a gonadotropin-releasing hormone receptor antagonist indicated for the management of pain associated with endometriosis. The drug caused abortions in rats and rabbits. Because the drug may increase the risk of early pregnancy loss, the manufacturer classifies it as contraindicated in pregnancy.
Dermatologic agents
Ilumya (tildrakizumab), which has a MW of about 147,000, is given by subcutaneous injection for the treatment of moderate to severe plaque psoriasis. When given during organogenesis in monkeys, no maternal or embryo-fetal toxicities were observed. However, when given throughout pregnancy a few neonatal deaths occurred, but the clinical significance of these nonclinical findings were unknown.
Fabry disease
Galafold (migalastat), which has a MW of about 200, is an alpha-galactosidase A pharmacologic chaperone indicated for the treatment of Fabry disease. Three pregnant women with Fabry disease were exposed to the drug in clinical studies but no information was provided on the pregnancy outcomes. No adverse developmental effects were observed in pregnant rats and rabbits.
Gastrointestinal agents
Akynzeo (netupitant or fosnetupitant palonosetron), which have MWs of about 579, 333, and 762, respectively, is available as an oral capsule (netupitant + palonosetron) and as an intravenous formulation (fosnetupitant + palonosetron). They are indicated, in combination with dexamethasone, for the prevention of nausea and vomiting related to cancer chemotherapy. Netupitant and fosnetupitant produced no embryo-fetal adverse effects in rats but were toxic to rabbit embryos. Palonosetron caused no embryo-fetal adverse effects in rats and rabbits.
Motegrity (prucalopride), which has a MW of about 486, is indicated for chronic idiopathic constipation. No adverse embryo-fetal developmental effects were observed in rats and rabbits.
Hematologic agents
Doptelet (avatrombopag), which has a MW of about 766, is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. No embryo-fetal effects were observed in rats, but in rabbits the drug was associated with spontaneous abortions.
Lokelma (sodium zirconium cyclosilicate) is a nonabsorbed zirconium silicate that exchanges potassium for hydrogen and sodium. Animal studies have not been conducted. Because it is not absorbed, it is not expected to result in fetal exposure to the drug.
Mulpleta (lusutrombopag), which has a MW of about 592, a thrombopoietin receptor agonist, is indicated for the treatment of thrombocytopenia in patients with chronic liver disease. High levels of the drug in pregnant rats were associated with adverse developmental outcomes. No adverse embryo-fetal effects were seen in pregnant rabbits.
Palynziq (pegvaliase-pqpz), which has a MW of about 1,000,000, is a phenylalanine-metabolizing enzyme indicated to reduce blood phenylalanine concentrations in patients with phenylketonuria. In pregnant rats, the drug caused an increase in skeletal variations. In rabbits, the drug caused a high incidence of multiple malformations.
Takhzyro (lanadelumab-flyo), which has a MW of about 49,000, is a monoclonal antibody indicated for prophylaxis to prevent attacks of hereditary angioedema. The drug caused no fetal harm in monkeys.
Tavalisse (fostamatinib disodium hexahydrate), which has a MW of about 733, is a kinase inhibitor used for the treatment of thrombocytopenia. In pregnant rats and rabbits, the drug caused adverse developmental outcomes including embryo-fetal mortality, lower fetal weights, and structural anomalies.
Ultomiris (ravulizumab), which has a MW of about 148,000, is a humanized monoclonal antibody indicated for adult patients with paroxysmal nocturnal hemoglobinuria. In mice, the drug was associated with increased rates of developmental abnormalities and an increased rate of dead and moribund offspring.
Immunologic agent
Revcovi (elapegademase-lvlr), which has a MW of about 113,000, is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency. Animal studies in pregnancy have not been conducted.
Nutrient/Nutritional supplement
Fish oil is indicated as a source of calories and fatty acids in pediatric patients with parenteral nutrition-associated cholestasis. Animal reproduction studies have not been conducted. It is doubtful if this product will be used in pregnancy.
Ophthalmic – nerve growth factor
Oxervate (cenegermin-bkbj), which has a MW of 13,266, is a solution that contains 118 amino acids. It is a recombinant human nerve growth factor indicated for neurotrophic keratitis. In rats and rabbits given the drug during organogenesis, there was a slight increase in postimplantation loss at doses greater than or equal to 267 times the human dose.
Respiratory drugs
Symdeko (tezacaftor + ivacaftor), which have MWs of about 521 and 392, is indicated for the treatment of patients with cystic fibrosis who are homozygous for the F508del mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator gene. There were no adverse developmental effects in pregnant rats and rabbits when the drugs were used separately or combined.
Yupelri (revefenacin), which has a MW of about 598, is an anticholinergic drug. It is an inhaled solution for the maintenance treatment of chronic obstructive pulmonary disease. In rats and rabbits, doses that were about 209 times the RHD produced no evidence of fetal harm.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. Mr. Briggs reported no relevant financial disclosures. Email him at [email protected].
Unintended consequences in the drive to simplify computerized test ordering
“X marks the spot!”
It’s one of the classic pirate tropes, bringing to mind images of Long John Silver, buried treasure, and a secret map with an “X” to show the hidden gold.
Today that “X” (or, in some cases, a check mark or radio button) seems to be indicating where the money is to be lost, rather than found.
Hospital computer systems are increasingly reliant on preprogrammed order lists that you check off rather than the actual test itself. We’ve gone from having to write out the tests we want, to typing them into a box, to checking them off with a mouse.
I’ve seen systems where you’re offered a menu such as:
A. Brain MRI (noncontrast)
B. Brain MRI (w/wo contrast)
C. Head MRA (noncontrast)
D. Head MRA (with contrast)
E. Neck MRA (noncontrast)
F. Neck MRA (with contrast)
G. Brain MRI and head/neck MRA (noncontrast)
H. Brain MRI and head/neck MRA (w/wo contrast)
And that’s just for the brain and its vascular supply. Expand that to the rest of the nervous system, then to the whole body, then to other tests (labs) ... and you get the idea.
I suppose the driving force here is to make the system easier to use. Doctors are busy. It saves time just have to check a box if you want three tests, rather than note all of them individually.
But it’s really not that hard to check off three. Probably less than 5 seconds (as of my last time on call). And this is where, to me, X marks the spot where the money isn’t.
Humans, like most animals, are pretty good at defaulting to a low-energy setting. So if you only have to check off one box instead of three, or five, or whatever, why bother?
If the patient is being admitted for a stroke/TIA, then it makes sense to do the brain MRI and head/neck MRA. But what if it’s just headaches, or a new seizure, or a concussion? I see plenty of times when more tests are done than necessary, simply because the ordering physician either didn’t know what was really needed or because it was easier to just check the box.
This is not, in my experience, rare. I’d say anywhere from one-third to half of patients I’ve consulted on had an overkill neurological work-up, in which tests with no medical indications had been ordered. They’ve generally already been put in the system, or even done, before I get to the bedside.

I suppose one could say they should wait for the specialist to get there before any of the costly tests are ordered, but that opens up another can of worms. What if a critical finding that needed to be acted upon isn’t found in time because of such a rule? Not only that, but waiting for me to show up and order tests means it will take longer to get them done, adding onto the hospital stay, and (again) running up costs.
So that’s not an answer, either. There really isn’t one, unfortunately.
But, in our haste to make things easier, or faster, or even just flashier, the trend seems to be at the cost of doing things reasonably. At the same time that we’re trying to save money, the single “X” may be marking the spot where we’re actually throwing it away.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
“X marks the spot!”
It’s one of the classic pirate tropes, bringing to mind images of Long John Silver, buried treasure, and a secret map with an “X” to show the hidden gold.
Today that “X” (or, in some cases, a check mark or radio button) seems to be indicating where the money is to be lost, rather than found.
Hospital computer systems are increasingly reliant on preprogrammed order lists that you check off rather than the actual test itself. We’ve gone from having to write out the tests we want, to typing them into a box, to checking them off with a mouse.
I’ve seen systems where you’re offered a menu such as:
A. Brain MRI (noncontrast)
B. Brain MRI (w/wo contrast)
C. Head MRA (noncontrast)
D. Head MRA (with contrast)
E. Neck MRA (noncontrast)
F. Neck MRA (with contrast)
G. Brain MRI and head/neck MRA (noncontrast)
H. Brain MRI and head/neck MRA (w/wo contrast)
And that’s just for the brain and its vascular supply. Expand that to the rest of the nervous system, then to the whole body, then to other tests (labs) ... and you get the idea.
I suppose the driving force here is to make the system easier to use. Doctors are busy. It saves time just have to check a box if you want three tests, rather than note all of them individually.
But it’s really not that hard to check off three. Probably less than 5 seconds (as of my last time on call). And this is where, to me, X marks the spot where the money isn’t.
Humans, like most animals, are pretty good at defaulting to a low-energy setting. So if you only have to check off one box instead of three, or five, or whatever, why bother?
If the patient is being admitted for a stroke/TIA, then it makes sense to do the brain MRI and head/neck MRA. But what if it’s just headaches, or a new seizure, or a concussion? I see plenty of times when more tests are done than necessary, simply because the ordering physician either didn’t know what was really needed or because it was easier to just check the box.
This is not, in my experience, rare. I’d say anywhere from one-third to half of patients I’ve consulted on had an overkill neurological work-up, in which tests with no medical indications had been ordered. They’ve generally already been put in the system, or even done, before I get to the bedside.
I suppose one could say they should wait for the specialist to get there before any of the costly tests are ordered, but that opens up another can of worms. What if a critical finding that needed to be acted upon isn’t found in time because of such a rule? Not only that, but waiting for me to show up and order tests means it will take longer to get them done, adding onto the hospital stay, and (again) running up costs.
So that’s not an answer, either. There really isn’t one, unfortunately.
But, in our haste to make things easier, or faster, or even just flashier, the trend seems to be at the cost of doing things reasonably. At the same time that we’re trying to save money, the single “X” may be marking the spot where we’re actually throwing it away.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
“X marks the spot!”
It’s one of the classic pirate tropes, bringing to mind images of Long John Silver, buried treasure, and a secret map with an “X” to show the hidden gold.
Today that “X” (or, in some cases, a check mark or radio button) seems to be indicating where the money is to be lost, rather than found.
Hospital computer systems are increasingly reliant on preprogrammed order lists that you check off rather than the actual test itself. We’ve gone from having to write out the tests we want, to typing them into a box, to checking them off with a mouse.
I’ve seen systems where you’re offered a menu such as:
A. Brain MRI (noncontrast)
B. Brain MRI (w/wo contrast)
C. Head MRA (noncontrast)
D. Head MRA (with contrast)
E. Neck MRA (noncontrast)
F. Neck MRA (with contrast)
G. Brain MRI and head/neck MRA (noncontrast)
H. Brain MRI and head/neck MRA (w/wo contrast)
And that’s just for the brain and its vascular supply. Expand that to the rest of the nervous system, then to the whole body, then to other tests (labs) ... and you get the idea.
I suppose the driving force here is to make the system easier to use. Doctors are busy. It saves time just have to check a box if you want three tests, rather than note all of them individually.
But it’s really not that hard to check off three. Probably less than 5 seconds (as of my last time on call). And this is where, to me, X marks the spot where the money isn’t.
Humans, like most animals, are pretty good at defaulting to a low-energy setting. So if you only have to check off one box instead of three, or five, or whatever, why bother?
If the patient is being admitted for a stroke/TIA, then it makes sense to do the brain MRI and head/neck MRA. But what if it’s just headaches, or a new seizure, or a concussion? I see plenty of times when more tests are done than necessary, simply because the ordering physician either didn’t know what was really needed or because it was easier to just check the box.
This is not, in my experience, rare. I’d say anywhere from one-third to half of patients I’ve consulted on had an overkill neurological work-up, in which tests with no medical indications had been ordered. They’ve generally already been put in the system, or even done, before I get to the bedside.
I suppose one could say they should wait for the specialist to get there before any of the costly tests are ordered, but that opens up another can of worms. What if a critical finding that needed to be acted upon isn’t found in time because of such a rule? Not only that, but waiting for me to show up and order tests means it will take longer to get them done, adding onto the hospital stay, and (again) running up costs.
So that’s not an answer, either. There really isn’t one, unfortunately.
But, in our haste to make things easier, or faster, or even just flashier, the trend seems to be at the cost of doing things reasonably. At the same time that we’re trying to save money, the single “X” may be marking the spot where we’re actually throwing it away.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Making an effective referral is surprisingly complex
Referrals actually are a complex procedure that can result in crucial health, developmental, and mental health benefits, yet patients attend referred services at wildly variable rates of 11%-81%, and for mental health and early intervention (EI) less than half the time.1 When surveyed, primary care providers (PCP) say that they want to share in the care of 75% of patients they refer, especially for mental health concerns. Yet after decades of practice, I can count on one hand the number of children I have referred to mental health or EI services for whom I received feedback from the specialist (here meaning agencies or providers outside the office). Lately, if the specialist is using the same EHR, I sometimes discover their note when reviewing the document list, but I was not cc’d. In fact, the most common outcome is that the patient never sees the specialist and we don’t find out until the next visit, often months later when precious time for intervention has passed. Less than 50% of children with a mental health issue that qualifies as a disorder are detected by PCPs, and less than half of those children complete a referral. But there are lots of reasons for that, you say, such as a lack of specialists. But less than half of referrals for toddlers with developmental delays are completed to EI services even when such services are available and free of cost.

What makes referrals so complicated? Lack of referral completion can come from structural factors and interpersonal factors. We and our patients both are frustrated by lack of specialty resources, specialists who do not accept our patient’s insurance (or any insurance), distance, transportation, hours of operation issues, overall life burdens or priorities of families, and of course, cost. We can help with a few of these, either with our own list or ideally with the help of a care coordinator or social worker keeping a list identifying local specialists, payment methods accepted, and perhaps reduced-cost care options or financial assistance. However, the interpersonal issues that can make or break a referral definitely are within our reach.
Some of the reasons patients report for not following through on a referral include not feeling that their PCP evaluated the situation adequately through history or that the PCP failed to perform tests, such as screens. Because 27% of referrals are made based on the first phone contact about an issue (a dump?), and most are made at the first visit an issue is considered (two-thirds for mental health referrals), this feeling is unsurprising and likely true.2 Families often do not know what kind of expertise we have to size up a need, especially if discussion about development or mental health have not been a regular part of care before a problem is detected. Parents of children with developmental delays who declined referral felt they were more expert on their child’s development than the PCP. Another reason given for not attending a referral is that the condition being referred for and what to expect from the referral, including logistics, was not clear to the parents of the patient. Low-literacy parents (30% of low-income samples) did not find written materials helpful. Parents referred to EI services, for example, sometimes thought they were being sent to Child Protective Services or feared notification of immigration. PCPs who have more time for visits and/or had a care navigator available to explain the process have more successful referrals (80%), especially if the manager makes the phone contact, which takes a parent on average seven calls to EI. In some cases, the parent does not agree that a consultation is needed. If this had been part of the referral discussion, a shared understanding might have been attained or an intermediate step chosen.

In many practices, language, literacy, and cultural differences are major barriers. Other barriers come from the parent or another family member denying there is an issue, not believing that the intervention being suggested is effective, concern over stigma for diagnoses such as mental illness or autism, not prioritizing therapies we recommend over other potential solutions such as home efforts or herbal medicine, or simple fear. The key here is for us to both give information and nonjudgmentally listen to the parent’s (or child’s) point of view and barriers, showing empathy by echoing their feelings, then using a motivational interviewing approach to weighing pros and cons of taking steps towards a referral. Requesting a “Talk Back” from the parent of what you tried to convey can assure understanding. The “warm hand off” to a smiling colocated professional that is so helpful at overcoming fear has recently been simulated by onsite tele-intake visits, resulting in 80% of patients keeping a visit for mental health care.3
For collaborative and cost-efficient care, we need to provide the specialist with data we have gathered, what questions we want answered, how best to communicate back, and what role we want in subsequent care. Referral completion is three times higher when PCPs schedule the appointment and communicate with the specialist. We need back a timely note or call about their impression, any tests or treatments initiated, and their ideas about sharing care going forward. A structured referral template makes for more satisfactory communication, but the key is actually sending and receiving it! Most PCPs surveyed count on the family to convey information back from a specialist. This respects their ownership of the issue, but what they tell us may be inaccurate, incomplete, and/or miss concerns the specialist may not have wanted to tell the patient, such as rare but serious possibilities being considered or delicate social issues uncovered.
Great discrepancies have been found between the frequency PCPs report providing information to specialists (69%) and what specialists report about frequency of receipt (38%). PCPs report hearing back about 21% of mental health referrals.4 Both may be true if referral information is lost in the system somewhere. Simply faxing the referral form to EI programs (that routinely contact families) rather than just giving families a phone number (33%) increased referral success to 58%! Text reminders also hold promise. Finally, with such low completion rates, tracking referrals made and information back is crucial, yet only 6 of 17 practices in one study did so.5 Apart from intra-EHR referral, newer software-as-a-service systems can transmit consent forms that include permission and information for the specialist to contact the patient and report on kept appointments (such as CHADIS) as well as exchanging results (such as Salesforce) that hold promise for closing the loop.
New interest by health care systems in better referrals is not just caused by care considerations, but for financial reasons. Specialty care costs more than primary care management, and missed specialist appointments are not only missed opportunities but also costly! And one-half of all outpatient visits are for referrals! This may become the best motivator for your practice or system to undertake a quality improvement project to improve this crucial primary care procedure.
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She reported no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to MDedge News. Email her at [email protected]
References
1. Acad Pediatr. 2014 May-Jun;14(3):315-23.
2. Hosp Community Psychiatry. 1992 May;43(5):489-93.
3. Pediatrics. 2019 Mar 1;143(3): e20182738.
4. Arch Pediatr Adolesc Med. 2000 May;154(5):499-506.
5. Pediatrics. 2010 Feb 1. doi: 10.1542/peds.2009-0388.
Referrals actually are a complex procedure that can result in crucial health, developmental, and mental health benefits, yet patients attend referred services at wildly variable rates of 11%-81%, and for mental health and early intervention (EI) less than half the time.1 When surveyed, primary care providers (PCP) say that they want to share in the care of 75% of patients they refer, especially for mental health concerns. Yet after decades of practice, I can count on one hand the number of children I have referred to mental health or EI services for whom I received feedback from the specialist (here meaning agencies or providers outside the office). Lately, if the specialist is using the same EHR, I sometimes discover their note when reviewing the document list, but I was not cc’d. In fact, the most common outcome is that the patient never sees the specialist and we don’t find out until the next visit, often months later when precious time for intervention has passed. Less than 50% of children with a mental health issue that qualifies as a disorder are detected by PCPs, and less than half of those children complete a referral. But there are lots of reasons for that, you say, such as a lack of specialists. But less than half of referrals for toddlers with developmental delays are completed to EI services even when such services are available and free of cost.
What makes referrals so complicated? Lack of referral completion can come from structural factors and interpersonal factors. We and our patients both are frustrated by lack of specialty resources, specialists who do not accept our patient’s insurance (or any insurance), distance, transportation, hours of operation issues, overall life burdens or priorities of families, and of course, cost. We can help with a few of these, either with our own list or ideally with the help of a care coordinator or social worker keeping a list identifying local specialists, payment methods accepted, and perhaps reduced-cost care options or financial assistance. However, the interpersonal issues that can make or break a referral definitely are within our reach.
Some of the reasons patients report for not following through on a referral include not feeling that their PCP evaluated the situation adequately through history or that the PCP failed to perform tests, such as screens. Because 27% of referrals are made based on the first phone contact about an issue (a dump?), and most are made at the first visit an issue is considered (two-thirds for mental health referrals), this feeling is unsurprising and likely true.2 Families often do not know what kind of expertise we have to size up a need, especially if discussion about development or mental health have not been a regular part of care before a problem is detected. Parents of children with developmental delays who declined referral felt they were more expert on their child’s development than the PCP. Another reason given for not attending a referral is that the condition being referred for and what to expect from the referral, including logistics, was not clear to the parents of the patient. Low-literacy parents (30% of low-income samples) did not find written materials helpful. Parents referred to EI services, for example, sometimes thought they were being sent to Child Protective Services or feared notification of immigration. PCPs who have more time for visits and/or had a care navigator available to explain the process have more successful referrals (80%), especially if the manager makes the phone contact, which takes a parent on average seven calls to EI. In some cases, the parent does not agree that a consultation is needed. If this had been part of the referral discussion, a shared understanding might have been attained or an intermediate step chosen.
In many practices, language, literacy, and cultural differences are major barriers. Other barriers come from the parent or another family member denying there is an issue, not believing that the intervention being suggested is effective, concern over stigma for diagnoses such as mental illness or autism, not prioritizing therapies we recommend over other potential solutions such as home efforts or herbal medicine, or simple fear. The key here is for us to both give information and nonjudgmentally listen to the parent’s (or child’s) point of view and barriers, showing empathy by echoing their feelings, then using a motivational interviewing approach to weighing pros and cons of taking steps towards a referral. Requesting a “Talk Back” from the parent of what you tried to convey can assure understanding. The “warm hand off” to a smiling colocated professional that is so helpful at overcoming fear has recently been simulated by onsite tele-intake visits, resulting in 80% of patients keeping a visit for mental health care.3
For collaborative and cost-efficient care, we need to provide the specialist with data we have gathered, what questions we want answered, how best to communicate back, and what role we want in subsequent care. Referral completion is three times higher when PCPs schedule the appointment and communicate with the specialist. We need back a timely note or call about their impression, any tests or treatments initiated, and their ideas about sharing care going forward. A structured referral template makes for more satisfactory communication, but the key is actually sending and receiving it! Most PCPs surveyed count on the family to convey information back from a specialist. This respects their ownership of the issue, but what they tell us may be inaccurate, incomplete, and/or miss concerns the specialist may not have wanted to tell the patient, such as rare but serious possibilities being considered or delicate social issues uncovered.
Great discrepancies have been found between the frequency PCPs report providing information to specialists (69%) and what specialists report about frequency of receipt (38%). PCPs report hearing back about 21% of mental health referrals.4 Both may be true if referral information is lost in the system somewhere. Simply faxing the referral form to EI programs (that routinely contact families) rather than just giving families a phone number (33%) increased referral success to 58%! Text reminders also hold promise. Finally, with such low completion rates, tracking referrals made and information back is crucial, yet only 6 of 17 practices in one study did so.5 Apart from intra-EHR referral, newer software-as-a-service systems can transmit consent forms that include permission and information for the specialist to contact the patient and report on kept appointments (such as CHADIS) as well as exchanging results (such as Salesforce) that hold promise for closing the loop.
New interest by health care systems in better referrals is not just caused by care considerations, but for financial reasons. Specialty care costs more than primary care management, and missed specialist appointments are not only missed opportunities but also costly! And one-half of all outpatient visits are for referrals! This may become the best motivator for your practice or system to undertake a quality improvement project to improve this crucial primary care procedure.
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She reported no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to MDedge News. Email her at [email protected]
References
1. Acad Pediatr. 2014 May-Jun;14(3):315-23.
2. Hosp Community Psychiatry. 1992 May;43(5):489-93.
3. Pediatrics. 2019 Mar 1;143(3): e20182738.
4. Arch Pediatr Adolesc Med. 2000 May;154(5):499-506.
5. Pediatrics. 2010 Feb 1. doi: 10.1542/peds.2009-0388.
Referrals actually are a complex procedure that can result in crucial health, developmental, and mental health benefits, yet patients attend referred services at wildly variable rates of 11%-81%, and for mental health and early intervention (EI) less than half the time.1 When surveyed, primary care providers (PCP) say that they want to share in the care of 75% of patients they refer, especially for mental health concerns. Yet after decades of practice, I can count on one hand the number of children I have referred to mental health or EI services for whom I received feedback from the specialist (here meaning agencies or providers outside the office). Lately, if the specialist is using the same EHR, I sometimes discover their note when reviewing the document list, but I was not cc’d. In fact, the most common outcome is that the patient never sees the specialist and we don’t find out until the next visit, often months later when precious time for intervention has passed. Less than 50% of children with a mental health issue that qualifies as a disorder are detected by PCPs, and less than half of those children complete a referral. But there are lots of reasons for that, you say, such as a lack of specialists. But less than half of referrals for toddlers with developmental delays are completed to EI services even when such services are available and free of cost.
What makes referrals so complicated? Lack of referral completion can come from structural factors and interpersonal factors. We and our patients both are frustrated by lack of specialty resources, specialists who do not accept our patient’s insurance (or any insurance), distance, transportation, hours of operation issues, overall life burdens or priorities of families, and of course, cost. We can help with a few of these, either with our own list or ideally with the help of a care coordinator or social worker keeping a list identifying local specialists, payment methods accepted, and perhaps reduced-cost care options or financial assistance. However, the interpersonal issues that can make or break a referral definitely are within our reach.
Some of the reasons patients report for not following through on a referral include not feeling that their PCP evaluated the situation adequately through history or that the PCP failed to perform tests, such as screens. Because 27% of referrals are made based on the first phone contact about an issue (a dump?), and most are made at the first visit an issue is considered (two-thirds for mental health referrals), this feeling is unsurprising and likely true.2 Families often do not know what kind of expertise we have to size up a need, especially if discussion about development or mental health have not been a regular part of care before a problem is detected. Parents of children with developmental delays who declined referral felt they were more expert on their child’s development than the PCP. Another reason given for not attending a referral is that the condition being referred for and what to expect from the referral, including logistics, was not clear to the parents of the patient. Low-literacy parents (30% of low-income samples) did not find written materials helpful. Parents referred to EI services, for example, sometimes thought they were being sent to Child Protective Services or feared notification of immigration. PCPs who have more time for visits and/or had a care navigator available to explain the process have more successful referrals (80%), especially if the manager makes the phone contact, which takes a parent on average seven calls to EI. In some cases, the parent does not agree that a consultation is needed. If this had been part of the referral discussion, a shared understanding might have been attained or an intermediate step chosen.
In many practices, language, literacy, and cultural differences are major barriers. Other barriers come from the parent or another family member denying there is an issue, not believing that the intervention being suggested is effective, concern over stigma for diagnoses such as mental illness or autism, not prioritizing therapies we recommend over other potential solutions such as home efforts or herbal medicine, or simple fear. The key here is for us to both give information and nonjudgmentally listen to the parent’s (or child’s) point of view and barriers, showing empathy by echoing their feelings, then using a motivational interviewing approach to weighing pros and cons of taking steps towards a referral. Requesting a “Talk Back” from the parent of what you tried to convey can assure understanding. The “warm hand off” to a smiling colocated professional that is so helpful at overcoming fear has recently been simulated by onsite tele-intake visits, resulting in 80% of patients keeping a visit for mental health care.3
For collaborative and cost-efficient care, we need to provide the specialist with data we have gathered, what questions we want answered, how best to communicate back, and what role we want in subsequent care. Referral completion is three times higher when PCPs schedule the appointment and communicate with the specialist. We need back a timely note or call about their impression, any tests or treatments initiated, and their ideas about sharing care going forward. A structured referral template makes for more satisfactory communication, but the key is actually sending and receiving it! Most PCPs surveyed count on the family to convey information back from a specialist. This respects their ownership of the issue, but what they tell us may be inaccurate, incomplete, and/or miss concerns the specialist may not have wanted to tell the patient, such as rare but serious possibilities being considered or delicate social issues uncovered.
Great discrepancies have been found between the frequency PCPs report providing information to specialists (69%) and what specialists report about frequency of receipt (38%). PCPs report hearing back about 21% of mental health referrals.4 Both may be true if referral information is lost in the system somewhere. Simply faxing the referral form to EI programs (that routinely contact families) rather than just giving families a phone number (33%) increased referral success to 58%! Text reminders also hold promise. Finally, with such low completion rates, tracking referrals made and information back is crucial, yet only 6 of 17 practices in one study did so.5 Apart from intra-EHR referral, newer software-as-a-service systems can transmit consent forms that include permission and information for the specialist to contact the patient and report on kept appointments (such as CHADIS) as well as exchanging results (such as Salesforce) that hold promise for closing the loop.
New interest by health care systems in better referrals is not just caused by care considerations, but for financial reasons. Specialty care costs more than primary care management, and missed specialist appointments are not only missed opportunities but also costly! And one-half of all outpatient visits are for referrals! This may become the best motivator for your practice or system to undertake a quality improvement project to improve this crucial primary care procedure.
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She reported no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to MDedge News. Email her at [email protected]
References
1. Acad Pediatr. 2014 May-Jun;14(3):315-23.
2. Hosp Community Psychiatry. 1992 May;43(5):489-93.
3. Pediatrics. 2019 Mar 1;143(3): e20182738.
4. Arch Pediatr Adolesc Med. 2000 May;154(5):499-506.
5. Pediatrics. 2010 Feb 1. doi: 10.1542/peds.2009-0388.
A new era of TTP treatment
Earlier this year, the Food and Drug Administration approved Cablivi (caplacizumab-yhdp) (Sanofi Genzyme, Cambridge, Mass.) for the treatment of acquired thrombotic thrombocytopenic purpura (TTP), making it the first medication specifically indicated for the treatment of TTP.

The approval of caplacizumab and the clinical trial results that approval is based on are the most promising developments in the treatment of TTP since the introduction of plasma exchange (PE) therapy. However, many questions remain about how to best administer caplacizumab, specifically, which patients should receive it? Should all TTP patients start on caplacizumab therapy or should it be limited to patients with histories of TTP or those slow to respond to standard therapy with PE and immunosuppression?
TTP is a rare thrombotic microangiopathy characterized by thrombocytopenia and microangiopathic hemolytic anemia caused by the inhibition of ADAMTS13, a metalloproteinase, which cleaves large-molecular-weight von Willebrand factor (vWF) multimers. Caplacizumab is a humanized bivalent, variable domain-only immunoglobulin fragment. The drug targets the A1 domain of vWF and inhibits the binding between vWF and the platelet glycoprotein Ib-IX-V receptor, preventing the formation of the microvascular thrombi and platelet loss associated with TTP.
FDA approval of caplacizumab came shortly after the publication of the results of the HERCULES trial in the New England Journal of Medicine by Marie Scully, MD, and her colleagues (N Engl J Med. 2019; 380[4]: 335-46).
HERCULES was an international phase 3, double blinded, placebo-controlled, randomized study designed to evaluate the efficacy and safety of caplacizumab. In total, 145 patients participated in the trial. Caplacizumab or placebo were given in addition to standard therapy of plasma exchange (PE) and immunosuppression. Caplacizumab or placebo were administered as an intravenous loading dose prior to the first PE after randomization and subcutaneously once daily until 30 days after the last PE. All patients received daily PE until 2 days after platelet count normalization.
The primary measure of the study was the time to platelet count response of greater than 150 x 109/L following the cessation of daily PE. Secondary measures included TTP-related death; TTP relapse; major thromboembolic events; proportion of subjects with refractory TTP; normalization of organ damage markers including lactate dehydrogenase, cardiac troponin I, and serum creatinine; and other adverse events.
The authors found that the median time of normalization of the platelet count was shorter in the caplacizumab group, compared with placebo, with the caplacizumab group being 1.55 times more likely to have a normalized platelet count at any given time point in the study. While statistically significant differences were identified in the rate of platelet normalization, the median number of days of PE until normalization was only 2 days less in the caplacizumab group (five treatments) than in the placebo group (seven treatments), which may not be clinically significant for the treating physician.
In fact, the secondary endpoints of the study seem much more clinically promising in the treatment of TTP. The composite rate of TTP-related death, TTP recurrence, or major thromboembolic events during the treatment period was significantly lower in the caplacizumab group (12%) versus placebo (49%). No TTP-related deaths occurred in the caplacizumab group. The caplacizumab group was also statistically less likely to have a TTP exacerbation, defined as disease recurrence within 30 days from the last PE, than the placebo group.
End organ damage serum markers also improved faster in the caplacizumab group, although there was no significant difference between groups. Overall hospitalization (median of 9 vs. 12 days) and ICU stays (median of 3 vs. 5 days) were shorter in the caplacizumab group, compared with the placebo group.
While several relapses, defined as disease recurrence after 30 days from the last PE, occurred in the caplacizumab group, the relapses were only found in patients with ADAMTS13 activity of less than 10% at the end of the treatment period. Mild side effects, such as mucocutaneous bleeding were more frequent in the caplacizumab group. No major bleeding complications were observed.
The HERCULES trial generates more questions about the role of ADAMTS13 activity testing to monitor treatment response and to make therapy decisions. Extremely low ADAMTS13 activity levels at the cessation of therapy may be a sign of treatment inadequacy and may warrant closer follow-up of at-risk patients on caplacizumab.
Sanofi Genzyme estimates that the U.S. list price will be approximately $270,000 for a standard treatment course, according to a news release from the company. Whether payers will add it to formularies remains uncertain, but the high drug cost may be countered by potential savings in the reduction of hospital and ICU days with caplacizumab therapy. Sanofi Genzyme will also have a patient support program for eligible patients.
Caplacizumab has been approved in Europe since August 2018, but is not readily available in the United States. Given the dearth of clinical experience with the drug outside of the TITAN and HERCULES trials, strong recommendations for when and how to initiate therapy remain elusive.
As caplacizumab is further introduced into clinical practice, more studies are needed to identify which patient groups will benefit most from therapy. The current data for caplacizumab shows that it will be used as an adjunct to standard PE therapy, rather than as a replacement. How the drug is used in combination with current TTP treatments – such as corticosteroids, rituximab, bortezomib, vincristine, N-acetylcysteine, and splenectomy – should be evaluated to identify which treatment combinations not only improve platelet counts, but also reduce mortality and morbidity while remaining cost effective.
Dr. Ricci is a staff physician and Apheresis Director at the Taussig Cancer Institute at the Cleveland Clinic. She reported having no conflicts of interest.
Earlier this year, the Food and Drug Administration approved Cablivi (caplacizumab-yhdp) (Sanofi Genzyme, Cambridge, Mass.) for the treatment of acquired thrombotic thrombocytopenic purpura (TTP), making it the first medication specifically indicated for the treatment of TTP.
The approval of caplacizumab and the clinical trial results that approval is based on are the most promising developments in the treatment of TTP since the introduction of plasma exchange (PE) therapy. However, many questions remain about how to best administer caplacizumab, specifically, which patients should receive it? Should all TTP patients start on caplacizumab therapy or should it be limited to patients with histories of TTP or those slow to respond to standard therapy with PE and immunosuppression?
TTP is a rare thrombotic microangiopathy characterized by thrombocytopenia and microangiopathic hemolytic anemia caused by the inhibition of ADAMTS13, a metalloproteinase, which cleaves large-molecular-weight von Willebrand factor (vWF) multimers. Caplacizumab is a humanized bivalent, variable domain-only immunoglobulin fragment. The drug targets the A1 domain of vWF and inhibits the binding between vWF and the platelet glycoprotein Ib-IX-V receptor, preventing the formation of the microvascular thrombi and platelet loss associated with TTP.
FDA approval of caplacizumab came shortly after the publication of the results of the HERCULES trial in the New England Journal of Medicine by Marie Scully, MD, and her colleagues (N Engl J Med. 2019; 380[4]: 335-46).
HERCULES was an international phase 3, double blinded, placebo-controlled, randomized study designed to evaluate the efficacy and safety of caplacizumab. In total, 145 patients participated in the trial. Caplacizumab or placebo were given in addition to standard therapy of plasma exchange (PE) and immunosuppression. Caplacizumab or placebo were administered as an intravenous loading dose prior to the first PE after randomization and subcutaneously once daily until 30 days after the last PE. All patients received daily PE until 2 days after platelet count normalization.
The primary measure of the study was the time to platelet count response of greater than 150 x 109/L following the cessation of daily PE. Secondary measures included TTP-related death; TTP relapse; major thromboembolic events; proportion of subjects with refractory TTP; normalization of organ damage markers including lactate dehydrogenase, cardiac troponin I, and serum creatinine; and other adverse events.
The authors found that the median time of normalization of the platelet count was shorter in the caplacizumab group, compared with placebo, with the caplacizumab group being 1.55 times more likely to have a normalized platelet count at any given time point in the study. While statistically significant differences were identified in the rate of platelet normalization, the median number of days of PE until normalization was only 2 days less in the caplacizumab group (five treatments) than in the placebo group (seven treatments), which may not be clinically significant for the treating physician.
In fact, the secondary endpoints of the study seem much more clinically promising in the treatment of TTP. The composite rate of TTP-related death, TTP recurrence, or major thromboembolic events during the treatment period was significantly lower in the caplacizumab group (12%) versus placebo (49%). No TTP-related deaths occurred in the caplacizumab group. The caplacizumab group was also statistically less likely to have a TTP exacerbation, defined as disease recurrence within 30 days from the last PE, than the placebo group.
End organ damage serum markers also improved faster in the caplacizumab group, although there was no significant difference between groups. Overall hospitalization (median of 9 vs. 12 days) and ICU stays (median of 3 vs. 5 days) were shorter in the caplacizumab group, compared with the placebo group.
While several relapses, defined as disease recurrence after 30 days from the last PE, occurred in the caplacizumab group, the relapses were only found in patients with ADAMTS13 activity of less than 10% at the end of the treatment period. Mild side effects, such as mucocutaneous bleeding were more frequent in the caplacizumab group. No major bleeding complications were observed.
The HERCULES trial generates more questions about the role of ADAMTS13 activity testing to monitor treatment response and to make therapy decisions. Extremely low ADAMTS13 activity levels at the cessation of therapy may be a sign of treatment inadequacy and may warrant closer follow-up of at-risk patients on caplacizumab.
Sanofi Genzyme estimates that the U.S. list price will be approximately $270,000 for a standard treatment course, according to a news release from the company. Whether payers will add it to formularies remains uncertain, but the high drug cost may be countered by potential savings in the reduction of hospital and ICU days with caplacizumab therapy. Sanofi Genzyme will also have a patient support program for eligible patients.
Caplacizumab has been approved in Europe since August 2018, but is not readily available in the United States. Given the dearth of clinical experience with the drug outside of the TITAN and HERCULES trials, strong recommendations for when and how to initiate therapy remain elusive.
As caplacizumab is further introduced into clinical practice, more studies are needed to identify which patient groups will benefit most from therapy. The current data for caplacizumab shows that it will be used as an adjunct to standard PE therapy, rather than as a replacement. How the drug is used in combination with current TTP treatments – such as corticosteroids, rituximab, bortezomib, vincristine, N-acetylcysteine, and splenectomy – should be evaluated to identify which treatment combinations not only improve platelet counts, but also reduce mortality and morbidity while remaining cost effective.
Dr. Ricci is a staff physician and Apheresis Director at the Taussig Cancer Institute at the Cleveland Clinic. She reported having no conflicts of interest.
Earlier this year, the Food and Drug Administration approved Cablivi (caplacizumab-yhdp) (Sanofi Genzyme, Cambridge, Mass.) for the treatment of acquired thrombotic thrombocytopenic purpura (TTP), making it the first medication specifically indicated for the treatment of TTP.
The approval of caplacizumab and the clinical trial results that approval is based on are the most promising developments in the treatment of TTP since the introduction of plasma exchange (PE) therapy. However, many questions remain about how to best administer caplacizumab, specifically, which patients should receive it? Should all TTP patients start on caplacizumab therapy or should it be limited to patients with histories of TTP or those slow to respond to standard therapy with PE and immunosuppression?
TTP is a rare thrombotic microangiopathy characterized by thrombocytopenia and microangiopathic hemolytic anemia caused by the inhibition of ADAMTS13, a metalloproteinase, which cleaves large-molecular-weight von Willebrand factor (vWF) multimers. Caplacizumab is a humanized bivalent, variable domain-only immunoglobulin fragment. The drug targets the A1 domain of vWF and inhibits the binding between vWF and the platelet glycoprotein Ib-IX-V receptor, preventing the formation of the microvascular thrombi and platelet loss associated with TTP.
FDA approval of caplacizumab came shortly after the publication of the results of the HERCULES trial in the New England Journal of Medicine by Marie Scully, MD, and her colleagues (N Engl J Med. 2019; 380[4]: 335-46).
HERCULES was an international phase 3, double blinded, placebo-controlled, randomized study designed to evaluate the efficacy and safety of caplacizumab. In total, 145 patients participated in the trial. Caplacizumab or placebo were given in addition to standard therapy of plasma exchange (PE) and immunosuppression. Caplacizumab or placebo were administered as an intravenous loading dose prior to the first PE after randomization and subcutaneously once daily until 30 days after the last PE. All patients received daily PE until 2 days after platelet count normalization.
The primary measure of the study was the time to platelet count response of greater than 150 x 109/L following the cessation of daily PE. Secondary measures included TTP-related death; TTP relapse; major thromboembolic events; proportion of subjects with refractory TTP; normalization of organ damage markers including lactate dehydrogenase, cardiac troponin I, and serum creatinine; and other adverse events.
The authors found that the median time of normalization of the platelet count was shorter in the caplacizumab group, compared with placebo, with the caplacizumab group being 1.55 times more likely to have a normalized platelet count at any given time point in the study. While statistically significant differences were identified in the rate of platelet normalization, the median number of days of PE until normalization was only 2 days less in the caplacizumab group (five treatments) than in the placebo group (seven treatments), which may not be clinically significant for the treating physician.
In fact, the secondary endpoints of the study seem much more clinically promising in the treatment of TTP. The composite rate of TTP-related death, TTP recurrence, or major thromboembolic events during the treatment period was significantly lower in the caplacizumab group (12%) versus placebo (49%). No TTP-related deaths occurred in the caplacizumab group. The caplacizumab group was also statistically less likely to have a TTP exacerbation, defined as disease recurrence within 30 days from the last PE, than the placebo group.
End organ damage serum markers also improved faster in the caplacizumab group, although there was no significant difference between groups. Overall hospitalization (median of 9 vs. 12 days) and ICU stays (median of 3 vs. 5 days) were shorter in the caplacizumab group, compared with the placebo group.
While several relapses, defined as disease recurrence after 30 days from the last PE, occurred in the caplacizumab group, the relapses were only found in patients with ADAMTS13 activity of less than 10% at the end of the treatment period. Mild side effects, such as mucocutaneous bleeding were more frequent in the caplacizumab group. No major bleeding complications were observed.
The HERCULES trial generates more questions about the role of ADAMTS13 activity testing to monitor treatment response and to make therapy decisions. Extremely low ADAMTS13 activity levels at the cessation of therapy may be a sign of treatment inadequacy and may warrant closer follow-up of at-risk patients on caplacizumab.
Sanofi Genzyme estimates that the U.S. list price will be approximately $270,000 for a standard treatment course, according to a news release from the company. Whether payers will add it to formularies remains uncertain, but the high drug cost may be countered by potential savings in the reduction of hospital and ICU days with caplacizumab therapy. Sanofi Genzyme will also have a patient support program for eligible patients.
Caplacizumab has been approved in Europe since August 2018, but is not readily available in the United States. Given the dearth of clinical experience with the drug outside of the TITAN and HERCULES trials, strong recommendations for when and how to initiate therapy remain elusive.
As caplacizumab is further introduced into clinical practice, more studies are needed to identify which patient groups will benefit most from therapy. The current data for caplacizumab shows that it will be used as an adjunct to standard PE therapy, rather than as a replacement. How the drug is used in combination with current TTP treatments – such as corticosteroids, rituximab, bortezomib, vincristine, N-acetylcysteine, and splenectomy – should be evaluated to identify which treatment combinations not only improve platelet counts, but also reduce mortality and morbidity while remaining cost effective.
Dr. Ricci is a staff physician and Apheresis Director at the Taussig Cancer Institute at the Cleveland Clinic. She reported having no conflicts of interest.
What is your diagnosis?
A skin biopsy of one of the lesions on the right toe showed dermal edema with an associated lymphohistiocytic infiltrate. There are scattered areas of perieccrine involvement and areas of vasculitis. Laboratory work up showed a normal complete blood count, a negative antinuclear antibodies (ANA) titer, a negative double-stranded DNA, normal levels of inflammatory markers, and negative cryoglobulins and cold agglutinins. The patient was diagnosed with pernio. The lesions improved within several weeks. She now wears thicker socks when she is ice skating.
Children, women, and the elderly are at a higher risk.1 This condition is frequently described in Northwestern Europe and the United Kingdom, especially in those living in houses without central heating.2
Clinically, the lesions appear a few hours or days after cold exposure on the toes, fingers, and in some unusual cases on the nose and the ears. The lesions present as erythematous to violaceous macules, papules, or nodules that in severe cases may blister and ulcerate. The lesions may be asymptomatic, pruritic, or tender. In children, pernio can be associated with the presence of cryoglobulins, cold agglutinins, anorexia nervosa, and genetic interferonopathy; it may precede the diagnosis of chronic myelomonocytic leukemia and may occur as a presenting sign of a blast crisis in acute lymphoblastic leukemia.3,4 The skin lesions usually resolve within days to a few weeks. Histopathologic analysis shows dermal edema with associated superficial and deep lymphohistiocytic infiltrate and perieccrine involvement.
The differential diagnosis of pernio includes other cold-induced syndromes such as Raynaud’s syndrome, cold panniculitis, cold urticaria, livedo reticularis, acrocyanosis, and chilblain lupus. In chilblain lupus (a form of chronic cutaneous lupus), the lesions may be very similar to pernio but the histopathology is consistent with changes of discoid lupus. Lesions of idiopathic palmoplantar hidradenitis present as erythematous tender nodules on the palms and the soles.5 The lesions can be triggered by vigorous physical activity, exposure to moisture, and excessive sweating. White, blue, and red discoloration of the fingers is seen in Raynaud’s phenomenon rather than the fixed erythematous to violaceous macules, papules, or nodules seen in pernio. Patients with erythromelalgia present with red painful palms and soles triggered by heat and, in contrast to pernio, relieved by cooling. Sweet syndrome, a febrile neutrophilic dermatoses, is characterized by tender erythematous papules and plaques with associated systemic symptoms. These patients may have an associated internal malignancy or infection, or the disorder may be triggered by medications or pregnancy.

Our patient had no systemic symptoms, and the pathology didn’t show any neutrophils. When the diagnosis is in doubt, a skin biopsy may help elucidate the diagnosis.
Once the diagnosis of pernio is made, it is recommended to order a complete blood count to rule out blood malignancies and cryoproteins.
Treatment of this condition consists of rewarming the extremity. If rewarming does not improve the patient’s symptoms, systemic treatment with nifedipine may be warranted.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Matiz said she had no relevant financial disclosures. Email her at [email protected].
References
1. Pediatrics. 2005 Sep;116(3):e472-5.
2. Mayo Clin Proc. 2014 Feb;89(2):207-15.
3. Pediatr Dermatol. 2018 Jan;35(1):e74-5.
4. Pediatr Dermatol. 2000 Mar-Apr;17(2):97-9.
5. Eur J Pediatr. 2001 Mar;160(3):189-91.
A skin biopsy of one of the lesions on the right toe showed dermal edema with an associated lymphohistiocytic infiltrate. There are scattered areas of perieccrine involvement and areas of vasculitis. Laboratory work up showed a normal complete blood count, a negative antinuclear antibodies (ANA) titer, a negative double-stranded DNA, normal levels of inflammatory markers, and negative cryoglobulins and cold agglutinins. The patient was diagnosed with pernio. The lesions improved within several weeks. She now wears thicker socks when she is ice skating.
Children, women, and the elderly are at a higher risk.1 This condition is frequently described in Northwestern Europe and the United Kingdom, especially in those living in houses without central heating.2
Clinically, the lesions appear a few hours or days after cold exposure on the toes, fingers, and in some unusual cases on the nose and the ears. The lesions present as erythematous to violaceous macules, papules, or nodules that in severe cases may blister and ulcerate. The lesions may be asymptomatic, pruritic, or tender. In children, pernio can be associated with the presence of cryoglobulins, cold agglutinins, anorexia nervosa, and genetic interferonopathy; it may precede the diagnosis of chronic myelomonocytic leukemia and may occur as a presenting sign of a blast crisis in acute lymphoblastic leukemia.3,4 The skin lesions usually resolve within days to a few weeks. Histopathologic analysis shows dermal edema with associated superficial and deep lymphohistiocytic infiltrate and perieccrine involvement.
The differential diagnosis of pernio includes other cold-induced syndromes such as Raynaud’s syndrome, cold panniculitis, cold urticaria, livedo reticularis, acrocyanosis, and chilblain lupus. In chilblain lupus (a form of chronic cutaneous lupus), the lesions may be very similar to pernio but the histopathology is consistent with changes of discoid lupus. Lesions of idiopathic palmoplantar hidradenitis present as erythematous tender nodules on the palms and the soles.5 The lesions can be triggered by vigorous physical activity, exposure to moisture, and excessive sweating. White, blue, and red discoloration of the fingers is seen in Raynaud’s phenomenon rather than the fixed erythematous to violaceous macules, papules, or nodules seen in pernio. Patients with erythromelalgia present with red painful palms and soles triggered by heat and, in contrast to pernio, relieved by cooling. Sweet syndrome, a febrile neutrophilic dermatoses, is characterized by tender erythematous papules and plaques with associated systemic symptoms. These patients may have an associated internal malignancy or infection, or the disorder may be triggered by medications or pregnancy.
Our patient had no systemic symptoms, and the pathology didn’t show any neutrophils. When the diagnosis is in doubt, a skin biopsy may help elucidate the diagnosis.
Once the diagnosis of pernio is made, it is recommended to order a complete blood count to rule out blood malignancies and cryoproteins.
Treatment of this condition consists of rewarming the extremity. If rewarming does not improve the patient’s symptoms, systemic treatment with nifedipine may be warranted.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Matiz said she had no relevant financial disclosures. Email her at [email protected].
References
1. Pediatrics. 2005 Sep;116(3):e472-5.
2. Mayo Clin Proc. 2014 Feb;89(2):207-15.
3. Pediatr Dermatol. 2018 Jan;35(1):e74-5.
4. Pediatr Dermatol. 2000 Mar-Apr;17(2):97-9.
5. Eur J Pediatr. 2001 Mar;160(3):189-91.
A skin biopsy of one of the lesions on the right toe showed dermal edema with an associated lymphohistiocytic infiltrate. There are scattered areas of perieccrine involvement and areas of vasculitis. Laboratory work up showed a normal complete blood count, a negative antinuclear antibodies (ANA) titer, a negative double-stranded DNA, normal levels of inflammatory markers, and negative cryoglobulins and cold agglutinins. The patient was diagnosed with pernio. The lesions improved within several weeks. She now wears thicker socks when she is ice skating.
Children, women, and the elderly are at a higher risk.1 This condition is frequently described in Northwestern Europe and the United Kingdom, especially in those living in houses without central heating.2
Clinically, the lesions appear a few hours or days after cold exposure on the toes, fingers, and in some unusual cases on the nose and the ears. The lesions present as erythematous to violaceous macules, papules, or nodules that in severe cases may blister and ulcerate. The lesions may be asymptomatic, pruritic, or tender. In children, pernio can be associated with the presence of cryoglobulins, cold agglutinins, anorexia nervosa, and genetic interferonopathy; it may precede the diagnosis of chronic myelomonocytic leukemia and may occur as a presenting sign of a blast crisis in acute lymphoblastic leukemia.3,4 The skin lesions usually resolve within days to a few weeks. Histopathologic analysis shows dermal edema with associated superficial and deep lymphohistiocytic infiltrate and perieccrine involvement.
The differential diagnosis of pernio includes other cold-induced syndromes such as Raynaud’s syndrome, cold panniculitis, cold urticaria, livedo reticularis, acrocyanosis, and chilblain lupus. In chilblain lupus (a form of chronic cutaneous lupus), the lesions may be very similar to pernio but the histopathology is consistent with changes of discoid lupus. Lesions of idiopathic palmoplantar hidradenitis present as erythematous tender nodules on the palms and the soles.5 The lesions can be triggered by vigorous physical activity, exposure to moisture, and excessive sweating. White, blue, and red discoloration of the fingers is seen in Raynaud’s phenomenon rather than the fixed erythematous to violaceous macules, papules, or nodules seen in pernio. Patients with erythromelalgia present with red painful palms and soles triggered by heat and, in contrast to pernio, relieved by cooling. Sweet syndrome, a febrile neutrophilic dermatoses, is characterized by tender erythematous papules and plaques with associated systemic symptoms. These patients may have an associated internal malignancy or infection, or the disorder may be triggered by medications or pregnancy.
Our patient had no systemic symptoms, and the pathology didn’t show any neutrophils. When the diagnosis is in doubt, a skin biopsy may help elucidate the diagnosis.
Once the diagnosis of pernio is made, it is recommended to order a complete blood count to rule out blood malignancies and cryoproteins.
Treatment of this condition consists of rewarming the extremity. If rewarming does not improve the patient’s symptoms, systemic treatment with nifedipine may be warranted.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Matiz said she had no relevant financial disclosures. Email her at [email protected].
References
1. Pediatrics. 2005 Sep;116(3):e472-5.
2. Mayo Clin Proc. 2014 Feb;89(2):207-15.
3. Pediatr Dermatol. 2018 Jan;35(1):e74-5.
4. Pediatr Dermatol. 2000 Mar-Apr;17(2):97-9.
5. Eur J Pediatr. 2001 Mar;160(3):189-91.
An 8-year-old girl comes to our pediatric dermatology clinic in the company of her mother for evaluation of painless purple spots on her toes. The lesions have been present for about 2 weeks. She has not been treated with any medications or creams. She denies any fevers, weight loss, mouth ulcers, sun sensitivity, joint pain, or any other symptoms. The patient has been a very healthy girl with occasional colds and no recent illnesses. The girl has never been admitted to the hospital. All her vaccinations are up to date. She takes no chronic medications. She lives in San Diego with her parents and two siblings. The girl recently started practicing ice-skating several times a week. There is no family history of any chronic medical conditions. She has no pets.
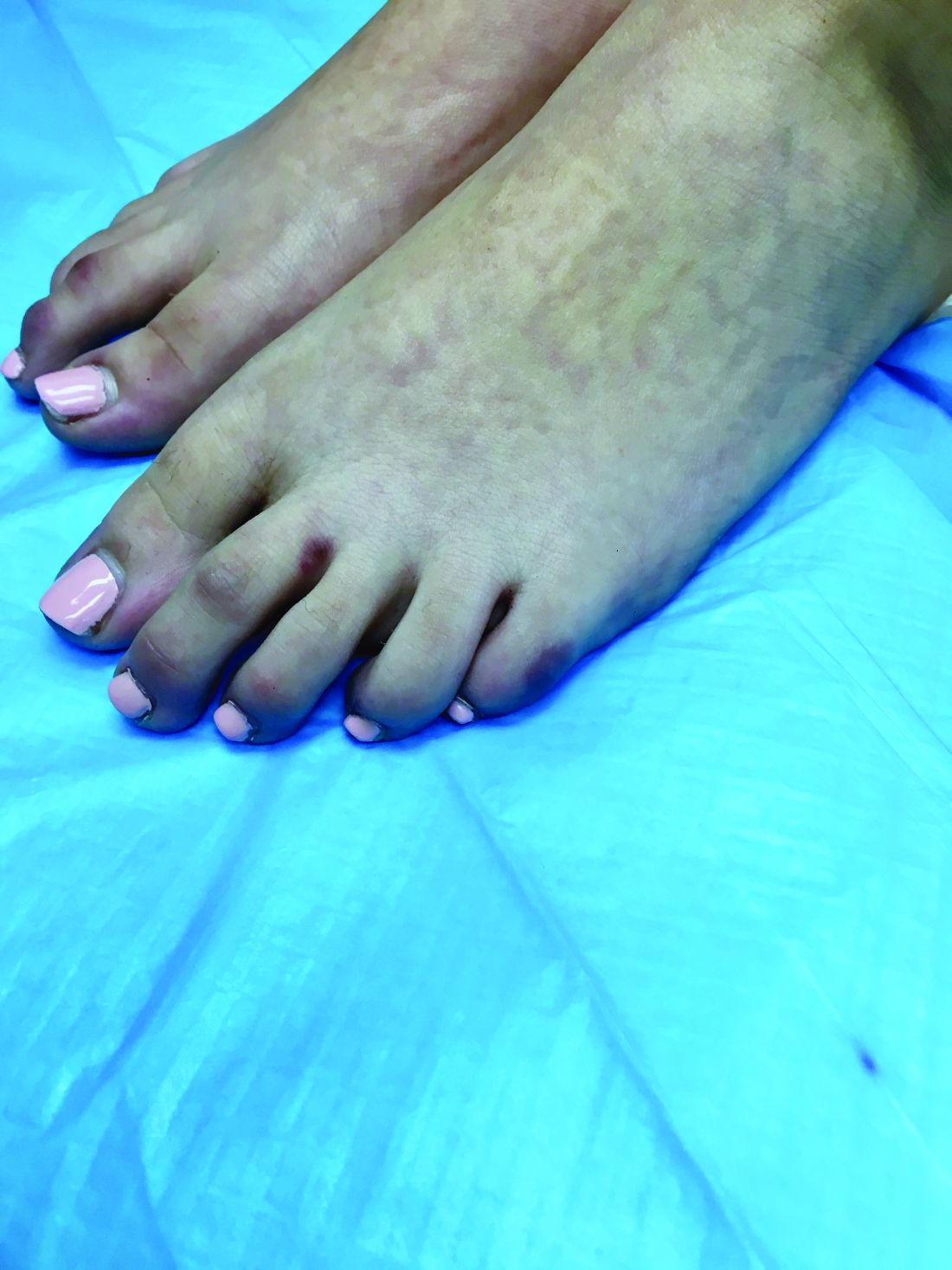
Firing patients
After last month’s

One might assume that, just as patients are free to choose or reject their doctors, physicians have an equal right to reject their patients; and to a certain extent, that’s true. There are no specific laws prohibiting a provider from terminating a patient relationship for any reason, other than a discriminatory one – race, nationality, religion, age, sex, sexual orientation, and so on. However, our ethical obligations to “do no harm” and to place our patients’ welfare above our own self-interests dictate that dismissing a patient should be the absolute last resort, after all other options have been exhausted.
First, to avoid charges of arbitrary termination, you should draw up a specific list of situations that could merit a dismissal from your office, and add it to your office manual. Every list will probably differ in some respects, but for the sake of example, here is mine:
- Threats or violence toward physicians or staff.
- Inappropriate sexual advances toward physicians or staff.
- Providing false or misleading medical history.
- Repeated rude or disruptive behavior.
- Demands for unapproved, unindicated, or inappropriate treatments or medications (particularly controlled substances).
- Refusal to adhere to agreed-upon treatment plans.
- Repeated failure to keep scheduled appointments.
- Repeated failure to pay medical bills.
As with pretty much everything in a private practice, accurate and written documentation of dismissible behavior is essential. Record all incidents and assemble as much material evidence as possible from all available sources.
In most cases (except the first two infractions on our list, for which we have zero tolerance), we make every effort to resolve the problem amicably. We communicate with the patients in question, explain our concerns, and discuss options for resolution. I also may send a letter, repeating my concerns and proposed solutions, as further documentation of our efforts to achieve an amicable resolution. All verbal and written warnings are, of course, documented as well. If the patient has a managed care policy, we review the managed care contract, which sometimes includes specific requirements for dismissal of its patients.
When such efforts fail, we send the patient two letters – one certified with return receipt, the other by conventional first class, in case the patient refuses the certified copy – explaining the reason for dismissal, and that care will be discontinued in 30 days from the letter’s date. (Most attorneys and medical associations agree that 30 days is sufficient reasonable notice.) We offer to provide care during the interim period, include a list of names and contact information for potential alternate providers, and offer to transfer records after receiving written permission.
Following these precautions will usually protect you from charges of “patient abandonment,” which is generally defined as the unilateral severance by the physician of the physician-patient relationship without giving the patient sufficient advance notice to obtain the services of another practitioner, and at a time when the patient still requires medical attention.
Some states have their own unique definitions of patient abandonment. You should check with your state’s health department, and your attorney, for any unusual requirements in your state, because violating these could lead to intervention by your state licensing board. There also is the risk of civil litigation, which typically is not covered by malpractice policies and may not be covered by your general liability policy either.
Patients who feel that termination was unjustified also may respond with negative reviews on social media, which I’ve discussed in recent columns, and will again, soon.
If something untrue is posted about you on a doctor-rating site, take action. Reputable sites have their own reputations to protect and can usually be persuaded to remove anything that is demonstrably false, although you may need a lawyer’s letter to get their attention. Try to get the error removed entirely or corrected within the original posting. An erratum on some distant page of the website is likely to be ignored, and will leave the false information online, intact.
Unfair comments are unlikely to be removed unless they are blatantly libelous; but many sites allow you to post a response, giving your side of the story. (More on that in the near future.) Also, there is nothing wrong with encouraging happy patients to write favorable reviews on those same sites. Sauce for the goose, and all that.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].
After last month’s
One might assume that, just as patients are free to choose or reject their doctors, physicians have an equal right to reject their patients; and to a certain extent, that’s true. There are no specific laws prohibiting a provider from terminating a patient relationship for any reason, other than a discriminatory one – race, nationality, religion, age, sex, sexual orientation, and so on. However, our ethical obligations to “do no harm” and to place our patients’ welfare above our own self-interests dictate that dismissing a patient should be the absolute last resort, after all other options have been exhausted.
First, to avoid charges of arbitrary termination, you should draw up a specific list of situations that could merit a dismissal from your office, and add it to your office manual. Every list will probably differ in some respects, but for the sake of example, here is mine:
- Threats or violence toward physicians or staff.
- Inappropriate sexual advances toward physicians or staff.
- Providing false or misleading medical history.
- Repeated rude or disruptive behavior.
- Demands for unapproved, unindicated, or inappropriate treatments or medications (particularly controlled substances).
- Refusal to adhere to agreed-upon treatment plans.
- Repeated failure to keep scheduled appointments.
- Repeated failure to pay medical bills.
As with pretty much everything in a private practice, accurate and written documentation of dismissible behavior is essential. Record all incidents and assemble as much material evidence as possible from all available sources.
In most cases (except the first two infractions on our list, for which we have zero tolerance), we make every effort to resolve the problem amicably. We communicate with the patients in question, explain our concerns, and discuss options for resolution. I also may send a letter, repeating my concerns and proposed solutions, as further documentation of our efforts to achieve an amicable resolution. All verbal and written warnings are, of course, documented as well. If the patient has a managed care policy, we review the managed care contract, which sometimes includes specific requirements for dismissal of its patients.
When such efforts fail, we send the patient two letters – one certified with return receipt, the other by conventional first class, in case the patient refuses the certified copy – explaining the reason for dismissal, and that care will be discontinued in 30 days from the letter’s date. (Most attorneys and medical associations agree that 30 days is sufficient reasonable notice.) We offer to provide care during the interim period, include a list of names and contact information for potential alternate providers, and offer to transfer records after receiving written permission.
Following these precautions will usually protect you from charges of “patient abandonment,” which is generally defined as the unilateral severance by the physician of the physician-patient relationship without giving the patient sufficient advance notice to obtain the services of another practitioner, and at a time when the patient still requires medical attention.
Some states have their own unique definitions of patient abandonment. You should check with your state’s health department, and your attorney, for any unusual requirements in your state, because violating these could lead to intervention by your state licensing board. There also is the risk of civil litigation, which typically is not covered by malpractice policies and may not be covered by your general liability policy either.
Patients who feel that termination was unjustified also may respond with negative reviews on social media, which I’ve discussed in recent columns, and will again, soon.
If something untrue is posted about you on a doctor-rating site, take action. Reputable sites have their own reputations to protect and can usually be persuaded to remove anything that is demonstrably false, although you may need a lawyer’s letter to get their attention. Try to get the error removed entirely or corrected within the original posting. An erratum on some distant page of the website is likely to be ignored, and will leave the false information online, intact.
Unfair comments are unlikely to be removed unless they are blatantly libelous; but many sites allow you to post a response, giving your side of the story. (More on that in the near future.) Also, there is nothing wrong with encouraging happy patients to write favorable reviews on those same sites. Sauce for the goose, and all that.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].
After last month’s
One might assume that, just as patients are free to choose or reject their doctors, physicians have an equal right to reject their patients; and to a certain extent, that’s true. There are no specific laws prohibiting a provider from terminating a patient relationship for any reason, other than a discriminatory one – race, nationality, religion, age, sex, sexual orientation, and so on. However, our ethical obligations to “do no harm” and to place our patients’ welfare above our own self-interests dictate that dismissing a patient should be the absolute last resort, after all other options have been exhausted.
First, to avoid charges of arbitrary termination, you should draw up a specific list of situations that could merit a dismissal from your office, and add it to your office manual. Every list will probably differ in some respects, but for the sake of example, here is mine:
- Threats or violence toward physicians or staff.
- Inappropriate sexual advances toward physicians or staff.
- Providing false or misleading medical history.
- Repeated rude or disruptive behavior.
- Demands for unapproved, unindicated, or inappropriate treatments or medications (particularly controlled substances).
- Refusal to adhere to agreed-upon treatment plans.
- Repeated failure to keep scheduled appointments.
- Repeated failure to pay medical bills.
As with pretty much everything in a private practice, accurate and written documentation of dismissible behavior is essential. Record all incidents and assemble as much material evidence as possible from all available sources.
In most cases (except the first two infractions on our list, for which we have zero tolerance), we make every effort to resolve the problem amicably. We communicate with the patients in question, explain our concerns, and discuss options for resolution. I also may send a letter, repeating my concerns and proposed solutions, as further documentation of our efforts to achieve an amicable resolution. All verbal and written warnings are, of course, documented as well. If the patient has a managed care policy, we review the managed care contract, which sometimes includes specific requirements for dismissal of its patients.
When such efforts fail, we send the patient two letters – one certified with return receipt, the other by conventional first class, in case the patient refuses the certified copy – explaining the reason for dismissal, and that care will be discontinued in 30 days from the letter’s date. (Most attorneys and medical associations agree that 30 days is sufficient reasonable notice.) We offer to provide care during the interim period, include a list of names and contact information for potential alternate providers, and offer to transfer records after receiving written permission.
Following these precautions will usually protect you from charges of “patient abandonment,” which is generally defined as the unilateral severance by the physician of the physician-patient relationship without giving the patient sufficient advance notice to obtain the services of another practitioner, and at a time when the patient still requires medical attention.
Some states have their own unique definitions of patient abandonment. You should check with your state’s health department, and your attorney, for any unusual requirements in your state, because violating these could lead to intervention by your state licensing board. There also is the risk of civil litigation, which typically is not covered by malpractice policies and may not be covered by your general liability policy either.
Patients who feel that termination was unjustified also may respond with negative reviews on social media, which I’ve discussed in recent columns, and will again, soon.
If something untrue is posted about you on a doctor-rating site, take action. Reputable sites have their own reputations to protect and can usually be persuaded to remove anything that is demonstrably false, although you may need a lawyer’s letter to get their attention. Try to get the error removed entirely or corrected within the original posting. An erratum on some distant page of the website is likely to be ignored, and will leave the false information online, intact.
Unfair comments are unlikely to be removed unless they are blatantly libelous; but many sites allow you to post a response, giving your side of the story. (More on that in the near future.) Also, there is nothing wrong with encouraging happy patients to write favorable reviews on those same sites. Sauce for the goose, and all that.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].

How much difference will Eli Lilly’s half-price insulin make?
When Erin Gilmer filled her insulin prescription at a Denver-area Walgreens in January, she paid $8.50. U.S. taxpayers paid another $280.51.
“It eats at me to know that taxpayer money is being wasted,” said Gilmer, who has Medicare and was diagnosed with type 1 diabetes while a sophomore at the University of Colorado in 2002.
The diagnosis meant that for the rest of her life she’d require daily insulin shots to stay alive. But the price of that insulin is skyrocketing.
Between 2009 and 2017 the wholesale price of a single vial of Humalog, the Eli Lilly and Co.–manufactured insulin Gilmer uses, nearly tripled – rising from $92.70 to $274.70, according to data from IBM Watson Health.
Six years ago, Gilmer qualified for Social Security Disability Insurance – and thus, Medicare – because of a range of health issues. At the time, the insulin she needed cost $167.70 per vial, according to IBM Watson Health.
“When it’s taxpayer money paying for medication for someone like me, it makes it a national issue, not just a diabetic issue,” Gilmer said.
Stories about people with type 1 diabetes dying when they couldn’t afford insulin have made headlines. Patient activists like Gilmer have protested high prices outside Lilly’s headquarters in Indianapolis.
Last October in Minnesota, State Attorney General Lori Swanson sued insulin manufacturers, alleging price gouging. Pharmaceutical executives were grilled about high drug prices by the Senate Finance Committee on Feb. 26.
This is the backdrop for Lilly’s announcement March 4 that it is rolling out a half-priced, generic version of Humalog called “insulin lispro.” The list price: $137.35 per vial.
“Patients, doctors and policymakers are demanding lower list prices for medicines and lower patient costs at the pharmacy counter,” Eli Lilly CEO David Ricks wrote in a blog post about the move. “You might be surprised to hear that we agree – it’s time for change in our system and for consumer prices to come down.”
No panacea
When Lilly’s Humalog, the first short-acting insulin, came to market in 1996, the list price was about $21 per vial. The price didn’t reach $275 overnight, but yearly price increases added up.
In February 2009, for example, the wholesale price was $92.70, according to IBM Watson Health. It rose to $99.65 in December 2009, then to $107.60 in September 2010, $115.70 in May 2011, and so on.
“There’s no justification for why prices should keep increasing at an average rate of 10% every year,” said Inmaculada Hernandez of the University of Pittsburgh School of Pharmacy, who was lead author of a January report in Health Affairs attributing the skyrocketing cost of prescription drugs to accumulated yearly price hikes.
“The public perception that we have in general is that drugs are so expensive because we need to pay for research and development, and that’s true,” Hernandez said. “However, usually research and development is paid for in the first years of life of a drug.”
At $137.35 per vial, Lilly’s generic insulin is priced at about the same level as Humalog was in 2012, 16 years after it came to market.
“We want to recognize that this is not a panacea,” said company spokesman Greg Kueterman. “This is an option that we hope can help people in the current system that we work with.”
It’s worth noting that Humalog is a rapid-acting insulin, but that’s only one of the two types of insulin most people with type 1 diabetes use every day. The second kind is long-lasting. Lilly makes one called Basaglar. The most popular long-lasting insulin is Lantus, produced by Sanofi. Neither has a lower-cost alternative.
Still, Lilly’s move on Humalog could put pressure on the other two big makers of insulin to act.
Novo Nordisk called Lilly’s lower-priced generic insulin “an important development,” in an emailed statement.
“Bringing affordable insulin to the market requires ideas from all stakeholders,” Novo Nordisk’s Ken Inchausti said in an email, which also listed steps the company has taken, such as a patient assistance program. The statement didn’t say whether Novo Nordisk is considering offering a lower-priced version of its popular insulin Novolog, a rival of Humalog.
A statement from Sanofi, the third major insulin maker, also didn’t say whether the company would offer lower-priced versions of its insulins.
“Sanofi supports any actions that increase access to insulins for patients living with diabetes at an affordable price,” spokeswoman Ashleigh Koss said in the email, which also touted the company’s patient assistance program.
A different kind of generic
One twist in this story is that Lilly’s new insulin is just a repackaged version of Humalog, minus the brand name. It’s called an “authorized generic.”
“Whoever came up with the term ‘authorized generic’?” Dr. Vincent Rajkumar said, laughing. Rajkumar is a hematologist at the Mayo Clinic in Rochester, Minn.
“It’s the same exact drug” as the brand name, he continued.
Typically, Rajkumar said, authorized generics are introduced by brand-name drugmakers to compete with generic versions of their drugs made by rival companies.
But in the case of Humalog and other insulins, there are no generics made by competitors, as there are for, say, the cholesterol medicine Lipitor or even other diabetes drugs, such as metformin.
So when Lilly’s authorized generic comes to market, the company will have both Humalog insulin and the authorized generic version of that medicine on the market.
Rajkumar said it’s a public relations move.
“There’s outrage over the price of insulin that is being discussed in Congress and elsewhere. And so the company basically says, ‘Hey, we will make the identical product available at half price.’ On the surface that sounds great,” Rajkumar said.
“But you look at the problems and you think, ‘OK, how crazy is this that someone is actually going to be buying the brand-name drug?’ ”
In fact, it’s possible that Lilly could break even or profit off its authorized generic compared to the name-brand Humalog, according to University of Pittsburgh’s Hernandez.
The profit margin would depend on the rebates paid by the company to insurers and pharmacy benefit managers. Rebates are getting a lot of attention these days as one factor that pushes drug prices higher. They’re usually not disclosed and increase as a drug’s price increases, providing an incentive to some companies to raise prices.
“Doing an authorized generic is nothing else than giving insurers two options,” Hernandez said: Pay the full list price for a brand-name drug and receive a higher rebate, or pay the lower price for the authorized generic and receive a presumably smaller rebate.
“What we really need to get insulin prices down is to get generics into the market, and we need more than one,” Hernandez said, adding that previous research has shown that prices begin to go down when two or three generics are competing in the marketplace.
Even so, Lillly’s Kueterman said the authorized generic insulin “is going to help hopefully move the system toward a more sustainable model.”
“I can guarantee you the reason that we’re doing this is to help people,” Kueterman said, noting the company’s Diabetes Solution Center has also helped “10,000 people each month pay significantly less for their insulin” since it opened in August.
For Erin Gilmer, the news about an authorized generic insulin from Lilly has left her mildly encouraged.
“It sounds really good, and it will help some people, which is great,” Gilmer said. “It’s Eli Lilly and pharma starting to understand that grassroots activism has to be taken seriously, and we are at a tipping point.”
This story is part of a partnership that includes NPR and Kaiser Health News. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
When Erin Gilmer filled her insulin prescription at a Denver-area Walgreens in January, she paid $8.50. U.S. taxpayers paid another $280.51.
“It eats at me to know that taxpayer money is being wasted,” said Gilmer, who has Medicare and was diagnosed with type 1 diabetes while a sophomore at the University of Colorado in 2002.
The diagnosis meant that for the rest of her life she’d require daily insulin shots to stay alive. But the price of that insulin is skyrocketing.
Between 2009 and 2017 the wholesale price of a single vial of Humalog, the Eli Lilly and Co.–manufactured insulin Gilmer uses, nearly tripled – rising from $92.70 to $274.70, according to data from IBM Watson Health.
Six years ago, Gilmer qualified for Social Security Disability Insurance – and thus, Medicare – because of a range of health issues. At the time, the insulin she needed cost $167.70 per vial, according to IBM Watson Health.
“When it’s taxpayer money paying for medication for someone like me, it makes it a national issue, not just a diabetic issue,” Gilmer said.
Stories about people with type 1 diabetes dying when they couldn’t afford insulin have made headlines. Patient activists like Gilmer have protested high prices outside Lilly’s headquarters in Indianapolis.
Last October in Minnesota, State Attorney General Lori Swanson sued insulin manufacturers, alleging price gouging. Pharmaceutical executives were grilled about high drug prices by the Senate Finance Committee on Feb. 26.
This is the backdrop for Lilly’s announcement March 4 that it is rolling out a half-priced, generic version of Humalog called “insulin lispro.” The list price: $137.35 per vial.
“Patients, doctors and policymakers are demanding lower list prices for medicines and lower patient costs at the pharmacy counter,” Eli Lilly CEO David Ricks wrote in a blog post about the move. “You might be surprised to hear that we agree – it’s time for change in our system and for consumer prices to come down.”
No panacea
When Lilly’s Humalog, the first short-acting insulin, came to market in 1996, the list price was about $21 per vial. The price didn’t reach $275 overnight, but yearly price increases added up.
In February 2009, for example, the wholesale price was $92.70, according to IBM Watson Health. It rose to $99.65 in December 2009, then to $107.60 in September 2010, $115.70 in May 2011, and so on.
“There’s no justification for why prices should keep increasing at an average rate of 10% every year,” said Inmaculada Hernandez of the University of Pittsburgh School of Pharmacy, who was lead author of a January report in Health Affairs attributing the skyrocketing cost of prescription drugs to accumulated yearly price hikes.
“The public perception that we have in general is that drugs are so expensive because we need to pay for research and development, and that’s true,” Hernandez said. “However, usually research and development is paid for in the first years of life of a drug.”
At $137.35 per vial, Lilly’s generic insulin is priced at about the same level as Humalog was in 2012, 16 years after it came to market.
“We want to recognize that this is not a panacea,” said company spokesman Greg Kueterman. “This is an option that we hope can help people in the current system that we work with.”
It’s worth noting that Humalog is a rapid-acting insulin, but that’s only one of the two types of insulin most people with type 1 diabetes use every day. The second kind is long-lasting. Lilly makes one called Basaglar. The most popular long-lasting insulin is Lantus, produced by Sanofi. Neither has a lower-cost alternative.
Still, Lilly’s move on Humalog could put pressure on the other two big makers of insulin to act.
Novo Nordisk called Lilly’s lower-priced generic insulin “an important development,” in an emailed statement.
“Bringing affordable insulin to the market requires ideas from all stakeholders,” Novo Nordisk’s Ken Inchausti said in an email, which also listed steps the company has taken, such as a patient assistance program. The statement didn’t say whether Novo Nordisk is considering offering a lower-priced version of its popular insulin Novolog, a rival of Humalog.
A statement from Sanofi, the third major insulin maker, also didn’t say whether the company would offer lower-priced versions of its insulins.
“Sanofi supports any actions that increase access to insulins for patients living with diabetes at an affordable price,” spokeswoman Ashleigh Koss said in the email, which also touted the company’s patient assistance program.
A different kind of generic
One twist in this story is that Lilly’s new insulin is just a repackaged version of Humalog, minus the brand name. It’s called an “authorized generic.”
“Whoever came up with the term ‘authorized generic’?” Dr. Vincent Rajkumar said, laughing. Rajkumar is a hematologist at the Mayo Clinic in Rochester, Minn.
“It’s the same exact drug” as the brand name, he continued.
Typically, Rajkumar said, authorized generics are introduced by brand-name drugmakers to compete with generic versions of their drugs made by rival companies.
But in the case of Humalog and other insulins, there are no generics made by competitors, as there are for, say, the cholesterol medicine Lipitor or even other diabetes drugs, such as metformin.
So when Lilly’s authorized generic comes to market, the company will have both Humalog insulin and the authorized generic version of that medicine on the market.
Rajkumar said it’s a public relations move.
“There’s outrage over the price of insulin that is being discussed in Congress and elsewhere. And so the company basically says, ‘Hey, we will make the identical product available at half price.’ On the surface that sounds great,” Rajkumar said.
“But you look at the problems and you think, ‘OK, how crazy is this that someone is actually going to be buying the brand-name drug?’ ”
In fact, it’s possible that Lilly could break even or profit off its authorized generic compared to the name-brand Humalog, according to University of Pittsburgh’s Hernandez.
The profit margin would depend on the rebates paid by the company to insurers and pharmacy benefit managers. Rebates are getting a lot of attention these days as one factor that pushes drug prices higher. They’re usually not disclosed and increase as a drug’s price increases, providing an incentive to some companies to raise prices.
“Doing an authorized generic is nothing else than giving insurers two options,” Hernandez said: Pay the full list price for a brand-name drug and receive a higher rebate, or pay the lower price for the authorized generic and receive a presumably smaller rebate.
“What we really need to get insulin prices down is to get generics into the market, and we need more than one,” Hernandez said, adding that previous research has shown that prices begin to go down when two or three generics are competing in the marketplace.
Even so, Lillly’s Kueterman said the authorized generic insulin “is going to help hopefully move the system toward a more sustainable model.”
“I can guarantee you the reason that we’re doing this is to help people,” Kueterman said, noting the company’s Diabetes Solution Center has also helped “10,000 people each month pay significantly less for their insulin” since it opened in August.
For Erin Gilmer, the news about an authorized generic insulin from Lilly has left her mildly encouraged.
“It sounds really good, and it will help some people, which is great,” Gilmer said. “It’s Eli Lilly and pharma starting to understand that grassroots activism has to be taken seriously, and we are at a tipping point.”
This story is part of a partnership that includes NPR and Kaiser Health News. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
When Erin Gilmer filled her insulin prescription at a Denver-area Walgreens in January, she paid $8.50. U.S. taxpayers paid another $280.51.
“It eats at me to know that taxpayer money is being wasted,” said Gilmer, who has Medicare and was diagnosed with type 1 diabetes while a sophomore at the University of Colorado in 2002.
The diagnosis meant that for the rest of her life she’d require daily insulin shots to stay alive. But the price of that insulin is skyrocketing.
Between 2009 and 2017 the wholesale price of a single vial of Humalog, the Eli Lilly and Co.–manufactured insulin Gilmer uses, nearly tripled – rising from $92.70 to $274.70, according to data from IBM Watson Health.
Six years ago, Gilmer qualified for Social Security Disability Insurance – and thus, Medicare – because of a range of health issues. At the time, the insulin she needed cost $167.70 per vial, according to IBM Watson Health.
“When it’s taxpayer money paying for medication for someone like me, it makes it a national issue, not just a diabetic issue,” Gilmer said.
Stories about people with type 1 diabetes dying when they couldn’t afford insulin have made headlines. Patient activists like Gilmer have protested high prices outside Lilly’s headquarters in Indianapolis.
Last October in Minnesota, State Attorney General Lori Swanson sued insulin manufacturers, alleging price gouging. Pharmaceutical executives were grilled about high drug prices by the Senate Finance Committee on Feb. 26.
This is the backdrop for Lilly’s announcement March 4 that it is rolling out a half-priced, generic version of Humalog called “insulin lispro.” The list price: $137.35 per vial.
“Patients, doctors and policymakers are demanding lower list prices for medicines and lower patient costs at the pharmacy counter,” Eli Lilly CEO David Ricks wrote in a blog post about the move. “You might be surprised to hear that we agree – it’s time for change in our system and for consumer prices to come down.”
No panacea
When Lilly’s Humalog, the first short-acting insulin, came to market in 1996, the list price was about $21 per vial. The price didn’t reach $275 overnight, but yearly price increases added up.
In February 2009, for example, the wholesale price was $92.70, according to IBM Watson Health. It rose to $99.65 in December 2009, then to $107.60 in September 2010, $115.70 in May 2011, and so on.
“There’s no justification for why prices should keep increasing at an average rate of 10% every year,” said Inmaculada Hernandez of the University of Pittsburgh School of Pharmacy, who was lead author of a January report in Health Affairs attributing the skyrocketing cost of prescription drugs to accumulated yearly price hikes.
“The public perception that we have in general is that drugs are so expensive because we need to pay for research and development, and that’s true,” Hernandez said. “However, usually research and development is paid for in the first years of life of a drug.”
At $137.35 per vial, Lilly’s generic insulin is priced at about the same level as Humalog was in 2012, 16 years after it came to market.
“We want to recognize that this is not a panacea,” said company spokesman Greg Kueterman. “This is an option that we hope can help people in the current system that we work with.”
It’s worth noting that Humalog is a rapid-acting insulin, but that’s only one of the two types of insulin most people with type 1 diabetes use every day. The second kind is long-lasting. Lilly makes one called Basaglar. The most popular long-lasting insulin is Lantus, produced by Sanofi. Neither has a lower-cost alternative.
Still, Lilly’s move on Humalog could put pressure on the other two big makers of insulin to act.
Novo Nordisk called Lilly’s lower-priced generic insulin “an important development,” in an emailed statement.
“Bringing affordable insulin to the market requires ideas from all stakeholders,” Novo Nordisk’s Ken Inchausti said in an email, which also listed steps the company has taken, such as a patient assistance program. The statement didn’t say whether Novo Nordisk is considering offering a lower-priced version of its popular insulin Novolog, a rival of Humalog.
A statement from Sanofi, the third major insulin maker, also didn’t say whether the company would offer lower-priced versions of its insulins.
“Sanofi supports any actions that increase access to insulins for patients living with diabetes at an affordable price,” spokeswoman Ashleigh Koss said in the email, which also touted the company’s patient assistance program.
A different kind of generic
One twist in this story is that Lilly’s new insulin is just a repackaged version of Humalog, minus the brand name. It’s called an “authorized generic.”
“Whoever came up with the term ‘authorized generic’?” Dr. Vincent Rajkumar said, laughing. Rajkumar is a hematologist at the Mayo Clinic in Rochester, Minn.
“It’s the same exact drug” as the brand name, he continued.
Typically, Rajkumar said, authorized generics are introduced by brand-name drugmakers to compete with generic versions of their drugs made by rival companies.
But in the case of Humalog and other insulins, there are no generics made by competitors, as there are for, say, the cholesterol medicine Lipitor or even other diabetes drugs, such as metformin.
So when Lilly’s authorized generic comes to market, the company will have both Humalog insulin and the authorized generic version of that medicine on the market.
Rajkumar said it’s a public relations move.
“There’s outrage over the price of insulin that is being discussed in Congress and elsewhere. And so the company basically says, ‘Hey, we will make the identical product available at half price.’ On the surface that sounds great,” Rajkumar said.
“But you look at the problems and you think, ‘OK, how crazy is this that someone is actually going to be buying the brand-name drug?’ ”
In fact, it’s possible that Lilly could break even or profit off its authorized generic compared to the name-brand Humalog, according to University of Pittsburgh’s Hernandez.
The profit margin would depend on the rebates paid by the company to insurers and pharmacy benefit managers. Rebates are getting a lot of attention these days as one factor that pushes drug prices higher. They’re usually not disclosed and increase as a drug’s price increases, providing an incentive to some companies to raise prices.
“Doing an authorized generic is nothing else than giving insurers two options,” Hernandez said: Pay the full list price for a brand-name drug and receive a higher rebate, or pay the lower price for the authorized generic and receive a presumably smaller rebate.
“What we really need to get insulin prices down is to get generics into the market, and we need more than one,” Hernandez said, adding that previous research has shown that prices begin to go down when two or three generics are competing in the marketplace.
Even so, Lillly’s Kueterman said the authorized generic insulin “is going to help hopefully move the system toward a more sustainable model.”
“I can guarantee you the reason that we’re doing this is to help people,” Kueterman said, noting the company’s Diabetes Solution Center has also helped “10,000 people each month pay significantly less for their insulin” since it opened in August.
For Erin Gilmer, the news about an authorized generic insulin from Lilly has left her mildly encouraged.
“It sounds really good, and it will help some people, which is great,” Gilmer said. “It’s Eli Lilly and pharma starting to understand that grassroots activism has to be taken seriously, and we are at a tipping point.”
This story is part of a partnership that includes NPR and Kaiser Health News. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Patient, heal thyself!
Octavio has prostate cancer. His prostate growth is large but localized.
“What do your doctors suggest?” I asked him.
“They sent me to two specialists at the medical center,” he said. “One does robotic surgery, the other does radiation. Each one told me why they recommend their technique.”
“How will you decide?”
“I’ll do some reading,” he said.
“What about the doctor who sent you to them?”
“He hasn’t discussed the choice with me, just sent me to get opinions. I have to make up own mind.”
Out of training for some time, I gather from students and family medical interactions that patient autonomy is now a reigning principle. Here is one definition:
. Patient autonomy does allow for health care providers to educate the patient but does not allow the health care provider to make the decision for the patient.
This sounds sensible, even admirable: no more paternalistic physicians talking down to patients and ordering them around. Yet a closer look shows a contradiction:
1. The second sentence says that patient autonomy “does not allow the health care provider to make the decision for the patient.”
2. But the first one says that patients should decide, “without their health care provider trying to influence the decision.”
Is “trying to influence” the same as making the decision for the patient?
Some would argue that it is: The power discrepancy between the parties makes a doctor’s attempt to influence amount to coercion.
Do you agree, esteemed colleagues, those of you who, like me, treat patients all day? If the choice is between freezing an actinic keratosis, burning it, or using topical chemotherapy, do you just lay all three options out there and ask the patient to pick one? What if your patient works in public and doesn’t have 2 weeks to wait while the reaction to topical 5-fluorouracil that makes his skin look like raw lobster subsides? Can you point that out? Or would that be “trying to influence” and thus not allowed?
You and I can think of many other examples, about medical choices large and small, where we could pose similar questions. This is not abstract philosophy; it is what we do all day.
Look up robot-assisted surgery and radiation for prostate cancer. You will find proponents of both, each making claims concerning survival, recurrence, discomfort, complications. Which is more important – a 15% greater chance of living 2 years longer or a 22% lower risk of incontinence? Will reading such statistics make your choice easier? What if other studies show different numbers?
Octavio chose surgery. I asked him how he decided.
“I talked with an internist I know socially,” he said. “He shared his experience with patients he’s referred for my problem and advised surgery as the better choice. I also saw a story online about a lawyer who chose one method, then 5 years later had to do the other.”
Octavio is sophisticated and well read. He lives near Boston, the self-described hub of medical expertise and academic excellence. Yet he makes up his mind the way everybody does: by asking a trusted adviser, by hearing an arresting anecdote. It’s not science. It’s how people think.
You don’t have to be a behavioral psychologist to know how hard it is for patients, especially frightened ones, to interpret statistical variances or compare disparate categories. Which is better – shorter life with less pain or longer life with more? How much less? How much more? There are ways to address such questions, but having an expert, trusted, and sympathetic adviser is a pretty good way to start. Only an abstract ethicist with no practical exposure to (or sympathy with) actual existing patients and their actual existing providers could possibly think otherwise.
“Let’s freeze those actinics off,” I suggest to a patient. “That won’t scar, you won’t need a dozen shots of lidocaine, and you won’t have to hide for 3 weeks.”

Did I influence her health care decision? Sure. Guilty as charged, with no apologies. When I am a patient, I want nothing less for myself: sympathetic, experienced guidance, shared by someone who knows me and appears to care one way or the other how I do.
Lord preserve us, doctors and patients both, from dogmatists who would demand otherwise.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
Octavio has prostate cancer. His prostate growth is large but localized.
“What do your doctors suggest?” I asked him.
“They sent me to two specialists at the medical center,” he said. “One does robotic surgery, the other does radiation. Each one told me why they recommend their technique.”
“How will you decide?”
“I’ll do some reading,” he said.
“What about the doctor who sent you to them?”
“He hasn’t discussed the choice with me, just sent me to get opinions. I have to make up own mind.”
Out of training for some time, I gather from students and family medical interactions that patient autonomy is now a reigning principle. Here is one definition:
. Patient autonomy does allow for health care providers to educate the patient but does not allow the health care provider to make the decision for the patient.
This sounds sensible, even admirable: no more paternalistic physicians talking down to patients and ordering them around. Yet a closer look shows a contradiction:
1. The second sentence says that patient autonomy “does not allow the health care provider to make the decision for the patient.”
2. But the first one says that patients should decide, “without their health care provider trying to influence the decision.”
Is “trying to influence” the same as making the decision for the patient?
Some would argue that it is: The power discrepancy between the parties makes a doctor’s attempt to influence amount to coercion.
Do you agree, esteemed colleagues, those of you who, like me, treat patients all day? If the choice is between freezing an actinic keratosis, burning it, or using topical chemotherapy, do you just lay all three options out there and ask the patient to pick one? What if your patient works in public and doesn’t have 2 weeks to wait while the reaction to topical 5-fluorouracil that makes his skin look like raw lobster subsides? Can you point that out? Or would that be “trying to influence” and thus not allowed?
You and I can think of many other examples, about medical choices large and small, where we could pose similar questions. This is not abstract philosophy; it is what we do all day.
Look up robot-assisted surgery and radiation for prostate cancer. You will find proponents of both, each making claims concerning survival, recurrence, discomfort, complications. Which is more important – a 15% greater chance of living 2 years longer or a 22% lower risk of incontinence? Will reading such statistics make your choice easier? What if other studies show different numbers?
Octavio chose surgery. I asked him how he decided.
“I talked with an internist I know socially,” he said. “He shared his experience with patients he’s referred for my problem and advised surgery as the better choice. I also saw a story online about a lawyer who chose one method, then 5 years later had to do the other.”
Octavio is sophisticated and well read. He lives near Boston, the self-described hub of medical expertise and academic excellence. Yet he makes up his mind the way everybody does: by asking a trusted adviser, by hearing an arresting anecdote. It’s not science. It’s how people think.
You don’t have to be a behavioral psychologist to know how hard it is for patients, especially frightened ones, to interpret statistical variances or compare disparate categories. Which is better – shorter life with less pain or longer life with more? How much less? How much more? There are ways to address such questions, but having an expert, trusted, and sympathetic adviser is a pretty good way to start. Only an abstract ethicist with no practical exposure to (or sympathy with) actual existing patients and their actual existing providers could possibly think otherwise.
“Let’s freeze those actinics off,” I suggest to a patient. “That won’t scar, you won’t need a dozen shots of lidocaine, and you won’t have to hide for 3 weeks.”
Did I influence her health care decision? Sure. Guilty as charged, with no apologies. When I am a patient, I want nothing less for myself: sympathetic, experienced guidance, shared by someone who knows me and appears to care one way or the other how I do.
Lord preserve us, doctors and patients both, from dogmatists who would demand otherwise.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
Octavio has prostate cancer. His prostate growth is large but localized.
“What do your doctors suggest?” I asked him.
“They sent me to two specialists at the medical center,” he said. “One does robotic surgery, the other does radiation. Each one told me why they recommend their technique.”
“How will you decide?”
“I’ll do some reading,” he said.
“What about the doctor who sent you to them?”
“He hasn’t discussed the choice with me, just sent me to get opinions. I have to make up own mind.”
Out of training for some time, I gather from students and family medical interactions that patient autonomy is now a reigning principle. Here is one definition:
. Patient autonomy does allow for health care providers to educate the patient but does not allow the health care provider to make the decision for the patient.
This sounds sensible, even admirable: no more paternalistic physicians talking down to patients and ordering them around. Yet a closer look shows a contradiction:
1. The second sentence says that patient autonomy “does not allow the health care provider to make the decision for the patient.”
2. But the first one says that patients should decide, “without their health care provider trying to influence the decision.”
Is “trying to influence” the same as making the decision for the patient?
Some would argue that it is: The power discrepancy between the parties makes a doctor’s attempt to influence amount to coercion.
Do you agree, esteemed colleagues, those of you who, like me, treat patients all day? If the choice is between freezing an actinic keratosis, burning it, or using topical chemotherapy, do you just lay all three options out there and ask the patient to pick one? What if your patient works in public and doesn’t have 2 weeks to wait while the reaction to topical 5-fluorouracil that makes his skin look like raw lobster subsides? Can you point that out? Or would that be “trying to influence” and thus not allowed?
You and I can think of many other examples, about medical choices large and small, where we could pose similar questions. This is not abstract philosophy; it is what we do all day.
Look up robot-assisted surgery and radiation for prostate cancer. You will find proponents of both, each making claims concerning survival, recurrence, discomfort, complications. Which is more important – a 15% greater chance of living 2 years longer or a 22% lower risk of incontinence? Will reading such statistics make your choice easier? What if other studies show different numbers?
Octavio chose surgery. I asked him how he decided.
“I talked with an internist I know socially,” he said. “He shared his experience with patients he’s referred for my problem and advised surgery as the better choice. I also saw a story online about a lawyer who chose one method, then 5 years later had to do the other.”
Octavio is sophisticated and well read. He lives near Boston, the self-described hub of medical expertise and academic excellence. Yet he makes up his mind the way everybody does: by asking a trusted adviser, by hearing an arresting anecdote. It’s not science. It’s how people think.
You don’t have to be a behavioral psychologist to know how hard it is for patients, especially frightened ones, to interpret statistical variances or compare disparate categories. Which is better – shorter life with less pain or longer life with more? How much less? How much more? There are ways to address such questions, but having an expert, trusted, and sympathetic adviser is a pretty good way to start. Only an abstract ethicist with no practical exposure to (or sympathy with) actual existing patients and their actual existing providers could possibly think otherwise.
“Let’s freeze those actinics off,” I suggest to a patient. “That won’t scar, you won’t need a dozen shots of lidocaine, and you won’t have to hide for 3 weeks.”
Did I influence her health care decision? Sure. Guilty as charged, with no apologies. When I am a patient, I want nothing less for myself: sympathetic, experienced guidance, shared by someone who knows me and appears to care one way or the other how I do.
Lord preserve us, doctors and patients both, from dogmatists who would demand otherwise.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].

Boosting Alzheimer’s trial participation via Medicare Advantage ‘memory fitness programs’
Clinical trials represent future hope for patients seeking better care, and there is no disease more in need of better care than Alzheimer’s disease. While death rates among most cancers, as well as heart disease, HIV-related illness, and other categories, have declined in the past decade, there has been no progress for Alzheimer’s disease. Better health and wellness overall may be having a beneficial effect that has produced a reduction in age-adjusted dementia rates, but with the aging of the population there are a greater absolute number of dementia cases than ever before, and that number is expected to continue rising. Finding a disease-modifying therapy seems to be the best hope for changing this dim outlook. Clinical trials intend to do just that but are hampered by patient enrollment rates that remain low. Far fewer eligible patients enroll than are needed, causing studies to take longer to complete, driving up their costs and essentially slowing progress. There is a need to increase patient enrollment, and there has been a variety of efforts intended to address this, not the least of which has been an explosion of media coverage of Alzheimer’s disease.

The Global Alzheimer’s Platform (GAP) Foundation, a nonprofit, self-described patient-centric entity dedicated to reducing the time and cost of Alzheimer’s disease clinical trials, recently announced an initiative to increase participation in Alzheimer’s clinical trials by supporting and collaborating with “memory fitness programs” through select Medicare Advantage plans. At worst, this seems a harmless way to increase attention and hopefully interest in clinical trial participation. At best, this may be a cost-effective way to increase enrollment and even improve dementia care. Dementia is notoriously underdiagnosed, especially by overworked, busy primary care providers who simply lack the time to perform the time-consuming testing that is typically required to diagnose and follow such patients.
There are some caveats to consider. First, memory fitness programs are of dubious benefit. They generally fit the description of being harmless, but there is little compelling evidence that they preserve or improve memory.
Second, enrollment in a clinical trial, for a patient, is not always a winning proposition. To date, there has been little success and in the absence of benefit, any downside – even if simply an inconvenience – is a net negative. Recently at the 2018 Clinical Trials on Alzheimer’s Disease meeting, Merck reported that patients with mild cognitive impairment receiving active treatment in the BACE1 inhibitor verubecestat trial actually declined at a more rapid rate than did those on placebo. While the absolute difference was small, and one could argue whether it was clinically significant or simply a random occurrence, it was a reminder that intervention with an experimental agent is not necessarily benign.
Third, Medicare Advantage plans, while popular in some circles, are not considered advantageous to providers so that the proliferation of inadequate reimbursement will potentially fuel the accelerating number of providers who opt out of insurance plans altogether. This is not necessarily an issue for the GAP Foundation specifically but is nonetheless an issue for anything that promotes MA plans).
Finally, it remains important to help patients and families maintain a positive outlook, especially when we have nothing better to offer. Alzheimer’s disease is not a death sentence for every patient affected. While many have difficult and heartbreaking courses, some have slowly progressive courses with relatively little impairment for an extended period of time. There are also the dementia-phobic, cognitively unimpaired individuals (or who simply have normal age-associated cognitive changes) in whom the continued drumbeat of dementia awareness and memory testing raises their paranoia ever higher. We treat deficits (or try to), but we have to live based on our preserved skills. The challenge clinicians must face with patients and families is how to maximize function while compensating for deficits and making sure that patients and families maintain their hope.
Dr. Caselli is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center.
Clinical trials represent future hope for patients seeking better care, and there is no disease more in need of better care than Alzheimer’s disease. While death rates among most cancers, as well as heart disease, HIV-related illness, and other categories, have declined in the past decade, there has been no progress for Alzheimer’s disease. Better health and wellness overall may be having a beneficial effect that has produced a reduction in age-adjusted dementia rates, but with the aging of the population there are a greater absolute number of dementia cases than ever before, and that number is expected to continue rising. Finding a disease-modifying therapy seems to be the best hope for changing this dim outlook. Clinical trials intend to do just that but are hampered by patient enrollment rates that remain low. Far fewer eligible patients enroll than are needed, causing studies to take longer to complete, driving up their costs and essentially slowing progress. There is a need to increase patient enrollment, and there has been a variety of efforts intended to address this, not the least of which has been an explosion of media coverage of Alzheimer’s disease.
The Global Alzheimer’s Platform (GAP) Foundation, a nonprofit, self-described patient-centric entity dedicated to reducing the time and cost of Alzheimer’s disease clinical trials, recently announced an initiative to increase participation in Alzheimer’s clinical trials by supporting and collaborating with “memory fitness programs” through select Medicare Advantage plans. At worst, this seems a harmless way to increase attention and hopefully interest in clinical trial participation. At best, this may be a cost-effective way to increase enrollment and even improve dementia care. Dementia is notoriously underdiagnosed, especially by overworked, busy primary care providers who simply lack the time to perform the time-consuming testing that is typically required to diagnose and follow such patients.
There are some caveats to consider. First, memory fitness programs are of dubious benefit. They generally fit the description of being harmless, but there is little compelling evidence that they preserve or improve memory.
Second, enrollment in a clinical trial, for a patient, is not always a winning proposition. To date, there has been little success and in the absence of benefit, any downside – even if simply an inconvenience – is a net negative. Recently at the 2018 Clinical Trials on Alzheimer’s Disease meeting, Merck reported that patients with mild cognitive impairment receiving active treatment in the BACE1 inhibitor verubecestat trial actually declined at a more rapid rate than did those on placebo. While the absolute difference was small, and one could argue whether it was clinically significant or simply a random occurrence, it was a reminder that intervention with an experimental agent is not necessarily benign.
Third, Medicare Advantage plans, while popular in some circles, are not considered advantageous to providers so that the proliferation of inadequate reimbursement will potentially fuel the accelerating number of providers who opt out of insurance plans altogether. This is not necessarily an issue for the GAP Foundation specifically but is nonetheless an issue for anything that promotes MA plans).
Finally, it remains important to help patients and families maintain a positive outlook, especially when we have nothing better to offer. Alzheimer’s disease is not a death sentence for every patient affected. While many have difficult and heartbreaking courses, some have slowly progressive courses with relatively little impairment for an extended period of time. There are also the dementia-phobic, cognitively unimpaired individuals (or who simply have normal age-associated cognitive changes) in whom the continued drumbeat of dementia awareness and memory testing raises their paranoia ever higher. We treat deficits (or try to), but we have to live based on our preserved skills. The challenge clinicians must face with patients and families is how to maximize function while compensating for deficits and making sure that patients and families maintain their hope.
Dr. Caselli is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center.
Clinical trials represent future hope for patients seeking better care, and there is no disease more in need of better care than Alzheimer’s disease. While death rates among most cancers, as well as heart disease, HIV-related illness, and other categories, have declined in the past decade, there has been no progress for Alzheimer’s disease. Better health and wellness overall may be having a beneficial effect that has produced a reduction in age-adjusted dementia rates, but with the aging of the population there are a greater absolute number of dementia cases than ever before, and that number is expected to continue rising. Finding a disease-modifying therapy seems to be the best hope for changing this dim outlook. Clinical trials intend to do just that but are hampered by patient enrollment rates that remain low. Far fewer eligible patients enroll than are needed, causing studies to take longer to complete, driving up their costs and essentially slowing progress. There is a need to increase patient enrollment, and there has been a variety of efforts intended to address this, not the least of which has been an explosion of media coverage of Alzheimer’s disease.
The Global Alzheimer’s Platform (GAP) Foundation, a nonprofit, self-described patient-centric entity dedicated to reducing the time and cost of Alzheimer’s disease clinical trials, recently announced an initiative to increase participation in Alzheimer’s clinical trials by supporting and collaborating with “memory fitness programs” through select Medicare Advantage plans. At worst, this seems a harmless way to increase attention and hopefully interest in clinical trial participation. At best, this may be a cost-effective way to increase enrollment and even improve dementia care. Dementia is notoriously underdiagnosed, especially by overworked, busy primary care providers who simply lack the time to perform the time-consuming testing that is typically required to diagnose and follow such patients.
There are some caveats to consider. First, memory fitness programs are of dubious benefit. They generally fit the description of being harmless, but there is little compelling evidence that they preserve or improve memory.
Second, enrollment in a clinical trial, for a patient, is not always a winning proposition. To date, there has been little success and in the absence of benefit, any downside – even if simply an inconvenience – is a net negative. Recently at the 2018 Clinical Trials on Alzheimer’s Disease meeting, Merck reported that patients with mild cognitive impairment receiving active treatment in the BACE1 inhibitor verubecestat trial actually declined at a more rapid rate than did those on placebo. While the absolute difference was small, and one could argue whether it was clinically significant or simply a random occurrence, it was a reminder that intervention with an experimental agent is not necessarily benign.
Third, Medicare Advantage plans, while popular in some circles, are not considered advantageous to providers so that the proliferation of inadequate reimbursement will potentially fuel the accelerating number of providers who opt out of insurance plans altogether. This is not necessarily an issue for the GAP Foundation specifically but is nonetheless an issue for anything that promotes MA plans).
Finally, it remains important to help patients and families maintain a positive outlook, especially when we have nothing better to offer. Alzheimer’s disease is not a death sentence for every patient affected. While many have difficult and heartbreaking courses, some have slowly progressive courses with relatively little impairment for an extended period of time. There are also the dementia-phobic, cognitively unimpaired individuals (or who simply have normal age-associated cognitive changes) in whom the continued drumbeat of dementia awareness and memory testing raises their paranoia ever higher. We treat deficits (or try to), but we have to live based on our preserved skills. The challenge clinicians must face with patients and families is how to maximize function while compensating for deficits and making sure that patients and families maintain their hope.
Dr. Caselli is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center.