User login
Importance of Recognizing Hypertrophic Cardiomyopathy in the Preoperative Clinic
Importance of Recognizing Hypertrophic Cardiomyopathy in the Preoperative Clinic
Hypertrophic cardiomyopathy (HCM) is a relatively common inherited condition characterized by abnormal asymmetric left ventricular (LV) thickening. This can lead to LV outflow tract (LVOT) obstruction, which has important implications for anesthesia management. This article describes a case of previously undiagnosed HCM discovered during a preoperative physical examination prior to a routine surveillance colonoscopy.
CASE PRESENTATION
A 55-year-old Army veteran with a history of a sessile serrated colon adenoma presented to the preadmission testing clinic prior to planned surveillance colonoscopy under monitored anesthesia care. His medical history included untreated severe obstructive sleep apnea (53 apnea-hypopnea index score), diet-controlled hypertension, prediabetes (6.3% hemoglobin A1c), hypogonadism, and obesity (41 body mass index). Medications included semaglutide 1.7 mg injected subcutaneously weekly and testosterone 200 mg injected intramuscularly every 2 weeks, as well as lisinopril-hydrochlorothiazide 10 to 12.5 mg daily, which had recently been discontinued because his blood pressure had improved with a low-sodium diet.
A review of systems was unremarkable except for progressive weight gain. The patient had no family history of sudden cardiac death. On physical examination, the patient’s blood pressure was 119/81 mm Hg, pulse was 86 beats/min, and respiratory rate was 18 breaths/min. The patient was clinically euvolemic, with no jugular venous distention or peripheral edema, and his lungs were clear to auscultation. There was, however, a soft, nonradiating grade 2/6 systolic murmur that had not been previously documented. The murmur decreased substantially with the Valsalva maneuver, with no change in hand grip.
Laboratory studies revealed hemoglobin and renal function were within the reference range. A routine 12-lead electrocardiogram (ECG) was unremarkable. A transthoracic echocardiogram revealed moderate pulmonary hypertension (59 mm Hg right ventricular systolic pressure), asymmetric LV hypertrophy (2.1 cm septal thickness), and severe LVOT obstruction (131.8 mm Hg gradient). Severe systolic anterior motion of the mitral valve was also present. The LV ejection fraction was 60% to 65%, with normal cavity size and systolic function. These findings were consistent with severe hypertrophic obstructive cardiomyopathy (HOCM). Upon more detailed questioning, the patient reported that over the previous 5 years he had experienced gradually decreasing exercise tolerance and mild dyspnea on exertion, particularly in hot weather, which he attributed to weight gain. He also reported a presyncopal episode the previous month while working in his garage in hot weather for a prolonged period of time.
The patient’s elective colonoscopy was canceled, and he was referred to cardiology. While awaiting cardiac consultation, he was instructed to maintain good hydration and avoid any heavy physical activity beyond walking. He was told not to resume his use of lisinopril-hydrochlorothiazide. A screening 7-day Holter monitor showed no ventricular or supraventricular ectopy. After cardiology consultation, the patient was referred to a HCM specialty clinic, where a cardiac magnetic resonance imaging confirmed severe asymmetric hypertrophy with resting obstruction (Figures 1-4). Treatment options were discussed with the patient, and he underwent a trial with the Β—blocker metoprolol 50 mg daily, which he could not tolerate. Verapamil extended-release 180 mg orally once daily was then initiated; however, his dyspnea persisted. He was amenable to surgical therapy and underwent septal myectomy, with 12 g of septal myocardium removed. He did well postoperatively, with a follow-up echocardiogram showing normal LV systolic function and no LVOT gradient detectable at rest or with Valsalva maneuver. His fatigue and exertional dyspnea significantly improved. Once the patient underwent septal myectomy and was determined to have no detectable LVOT gradient, he was approved for colonoscopy which has been scheduled but not completed.
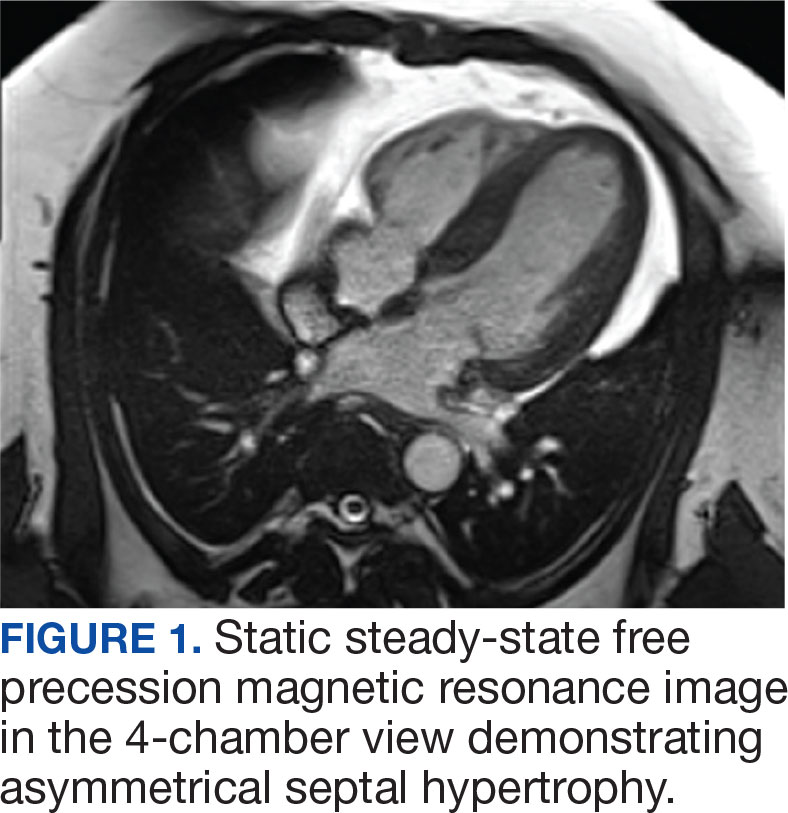
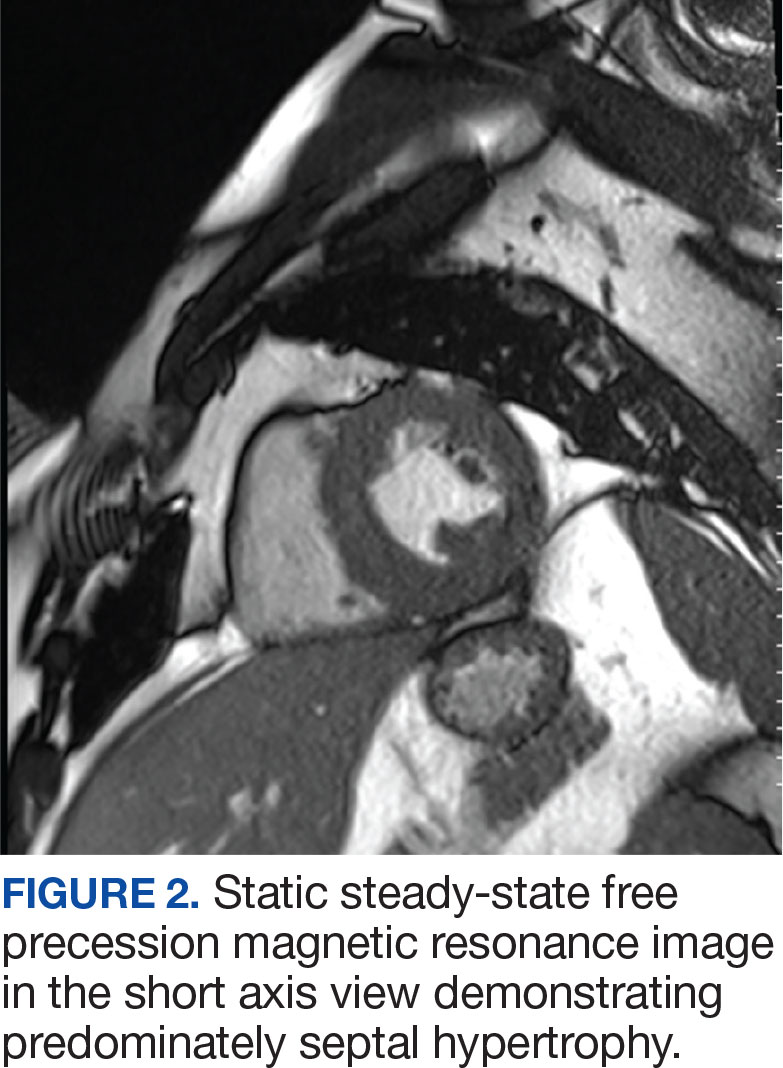
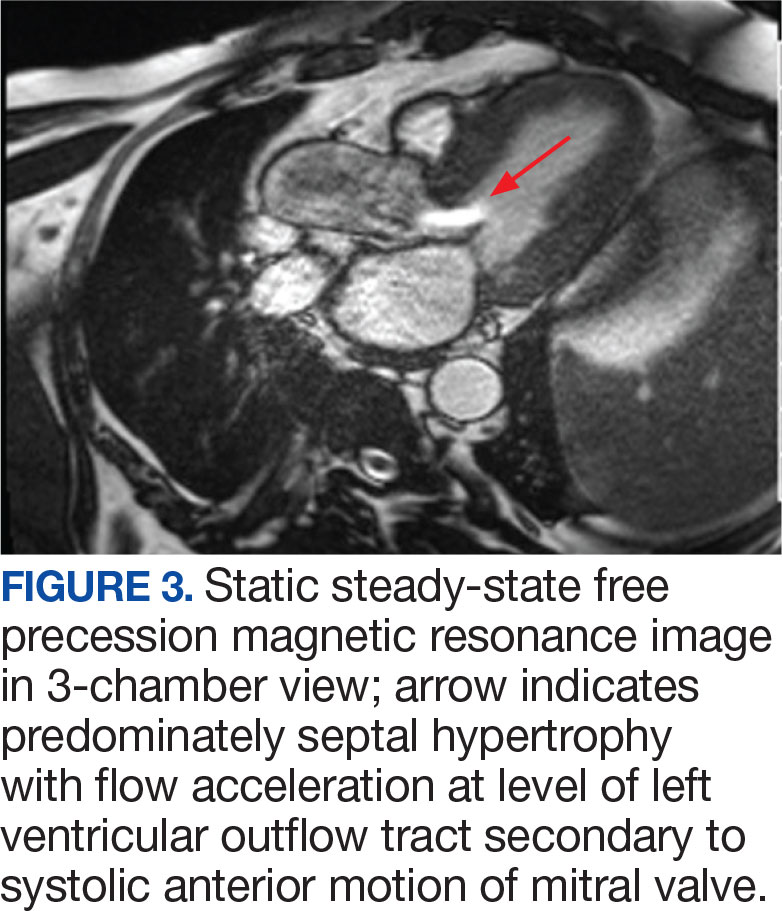
DISCUSSION
Once thought rare, HCM is now considered to be a relatively common inherited disorder, occurring in about 1 in 500 persons, with some suggesting that the actual prevalence is closer to 1 in 200 persons.1,2 Most often caused by mutations in ≥ 1 of 11 genes responsible for encoding cardiac sarcomere proteins, HCM is characterized by abnormal LV thickening without chamber enlargement in the absence of any identifiable cause, such as aortic valve stenosis or uncontrolled hypertension. The hypertrophy is often asymmetric, and in cases of asymmetric septal hypertrophy, dynamic LVOT obstruction can occur (known as HOCM). The condition is inherited in an autosomal dominant pattern with variable expression and is associated with myocardial fiber disarray, which can occur years before symptom onset.3 This myocardial disarray can lead to remodeling and an increased wall-to-lumen ratio of the coronary arteries, resulting in impaired coronary reserve.
Depending on the degree of LVOT obstruction, patients with HCM may be classified as nonobstructive, labile, or obstructive at rest. Patients without obstruction have an outflow gradient ≤ 30 mm Hg that is not provoked with Valsalva maneuver, administration of amyl nitrite, or exercise treadmill testing.3 Patients classified as labile do not have LVOT obstruction at rest, but obstruction may be induced by provocative measures. Finally, about one-third of patients with HCM will have LVOT gradients of > 30 mm Hg at rest. These patients are at increased risk for progression to symptomatic heart failure and may be candidates for surgical myectomy or catheter-based alcohol septal ablation.4 The patient in this case had a resting LVOT gradient of 131.8 mm Hg on echocardiography. The magnitude of this gradient placed the patient at a significantly higher risk of ventricular dysrhythmias and sudden cardiac death.5
Wall thickness also has prognostic implications. 6 Although any area of the myocardium can be affected, the septum is involved in about 90% cases. In their series of 48 patients followed over 6.5 years, Spirito et al found that the risk of sudden death in patients with HCM increased as wall thickness increased. For patients with a wall thickness of < 15 mm, the risk of death was 0 per 1000 person-years; however, this increased to 18.2 per 1000 person-years for patients with a wall thickness of > 30 mm.7
While many patients with HCM are asymptomatic, others may report dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, chest pain, palpitations, presyncope/ syncope, postural lightheadedness, fatigue, or edema. Symptomatology, however, is quite variable and does not necessarily correlate with the degree of outflow obstruction. Surprisingly, some patients with significant LVOT may have minimal symptoms, such as the patient in this case, while others with a lesser degree of LVOT obstruction may be very symptomatic.3,4
Physical examination of a patient with HCM may be normal or may reveal nonspecific findings such as a fourth heart sound or a systolic murmur. In general, physical examination abnormalities are related to LVOT obstruction. Those patients without significant outflow obstruction may have a normal cardiac examination. While patients with HCM may have a variety of systolic murmurs, the 2 most common are those related to outflow tract obstruction and mitral regurgitation caused by systolic anterior motion of the mitral valve.4 The systolic murmur associated with significant LVOT obstruction has been described as a harsh, crescendo-decrescendo type that begins just after S1 and is heard best at the apex and lower left sternal border.4 It may radiate to the axilla and base but not generally into the neck. The murmur usually increases with Valsalva maneuver and decreases with handgrip or going from a standing to a sitting/ squatting position. The initial examination of the patient in this case was not suggestive of HOCM, as confirmed by 2 practitioners (a cardiologist and an internist), each with > 30 years of clinical experience. This may have been related to the patient’s hydration status at the time, with Valsalva maneuver increasing obstruction to the point of reduced flow.
About 90% of patients with HCM will have abnormalities on ECG, most commonly LV hypertrophy with a strain pattern. Other ECG findings include: (1) prominent abnormal Q waves, particularly in the inferior (II, III, and aVF) and lateral leads (I, aVL, and V4-V6), reflecting depolarization of a hypertrophied septum; (2) left axis deviation; (3) deeply inverted T waves in leads V2 through V4; and (4) P wave abnormalities indicative of left atrial (LA) or biatrial enlargement. 8 It is notable that the patient in this case had a normal ECG, given that a minority of patients with HCM have been shown to have a normal ECG.9
Echocardiography plays an important role in diagnosing HCM. Diagnostic criteria include the presence of asymmetric hypertrophy (most commonly with anterior septal involvement), systolic anterior motion of the mitral valve, a nondilated LV cavity, septal immobility, and premature closure of the aortic valve. LV thickness is measured at both the septum and free wall; values ≥ 15 mm, with a septal-to-free wall thickness ratio of ≥ 1.3, are suggestive of HCM. Asymmetric LV hypertrophy can also be seen in other segments besides the septum, such as the apex.10
HCM/HOCM is the most common cause of sudden cardiac death in young people. The condition also contributes to significant functional morbidity due to heart failure and increases the risk of atrial fibrillation and subsequent stroke. Treatments tend to focus on symptom relief and slowing disease progression and include the use of medications such as Β—blockers, nondihydropyridine calcium channel blockers, and the myosin inhibitor mavacamten.11 Select patients, such as those with severe LVOT obstruction and symptoms despite treatment with Β—blockers or nondihydropyridine calcium channel blockers, may be offered septal myectomy or catheter-based alcohol septal ablation, coupled with insertion of an implantable cardiac defibrillator to prevent sudden cardiac death in patients at high arrhythmic risk.1,12
Patients with HCM, particularly those with LVOT obstruction, pose distinct challenges to the anesthesiologist because they are highly sensitive to decreases in preload and afterload. These patients frequently experience adverse perioperative events such as myocardial ischemia, systemic hypotension, and supraventricular or ventricular arrhythmias. Acute congestive heart failure may also occur, presumably due to concomitant diastolic dysfunction. Patients with previously unrecognized HCM are of particular concern, as they may manifest unexpected and sudden hypotension with the induction of anesthesia. There may then be a paradoxical response to vasoactive drugs and anesthetic agents, which accentuate LVOT obstruction. In these circumstances, undiagnosed HCM should be considered, and intraoperative rescue transesophageal echocardiography be performed.13 Once the diagnosis is confirmed, efforts should be made to reduce myocardial contractility and sympathetic discharge (eg, with Β—blockers), increase afterload (eg, with α1 agonists), and improve preload with adequate hydration. Proper resuscitation of hypotensive patients with HCM requires a thorough understanding of disease pathology, as effective interventions may seem to be counterintuitive. Inotropic agents such as epinephrine are contraindicated in HCM because increased inotropy and chronotropy worsen LVOT obstruction. Volume status is often tenuous; while adequate preload is important, overly aggressive fluid resuscitation may promote heart failure. It is important to keep in mind that even patients without resting LVOT obstruction may develop dynamic obstruction with anesthesia induction due to sudden reductions in preload and afterload. It is also important to note that the degree of LV hypertrophy is directly correlated with arrhythmic sudden death. Those patients with LV wall thickness ≥ 30 mm are at increased risk for potentially lethal tachyarrhythmias in the operating room.14
These considerations reinforce the need for proper preoperative identification of patients with HCM. Heightened awareness is key, given the fact that HCM is relatively common and tends to be underdiagnosed in the general population. These patients are generally young, otherwise healthy, and often undergo minor operative procedures in outpatient settings. It is incumbent upon the preoperative evaluator to take a thorough medical history and perform a careful physical examination. Clues to the diagnosis include exertional dyspnea, fatigue, angina, syncope/presyncope, or a family history of sudden cardiac death or HCM. A systolic ejection murmur, particularly one that increases with standing or Valsalva maneuver, and decreases with squatting or handgrip may also raise clinical suspicion. These patients should undergo a full cardiac evaluation, including echocardiography.
CONCLUSIONS
HCM is a common condition that is important to diagnose in the preoperative clinic. Failure to do so can lead to catastrophic complications during induction of anesthesia due to the sudden reduction in preload and afterload, which may cause a significant increase in LVOT obstruction. A high index of suspicion is essential, as clinical diagnosis can be challenging. The physical examination may be deceiving and symptoms are often subtle and nonspecific. It is imperative to alert the anesthesiologist before surgery so the complex hemodynamic management of patients with HOCM can be appropriately managed.
- Cheng Z, Fang T, Huang J, Guo Y, Alam M, Qian H. Hypertrophic cardiomyopathy: from phenotype and pathogenesis to treatment. Front Cardiovasc Med. 2021;8:722340. doi:10.3389/fcvm.2021.722340
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254. doi:10.1016/j.jacc.2015.01.019
- Hensley N, Dietrich J, Nyhan D, Mitter N, Yee MS, Brady M. Hypertrophic cardiomyopathy: a review. Anesth Analg. 2015;120(3):554-569. doi:10.1213/ ANE.0000000000000538
- Maron BJ, Desai MY, Nishimura RA, et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79(4):372–389. doi:10.1016/j.jacc.2021.12.002
- Jorda P, Garcia-Alvarez A. Hypertrophic cardiomyopathy: sudden cardiac death risk stratification in adults. Glob Cardiol Sci Pract. 2018;3(25). doi:10.21542/gcsp.2018.25
- Wigle ED, Sasson Z, Henderson MA, et al. Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis. 1985;28(1):1-83. doi:10.1016/0033-0620(85)90024-6
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342(24):1778–1785. doi:10.1056/ NEJM200006153422403
- Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy Lancet. 2017;389(10075):1253-1267. doi:10.1016/S0140-6736(16)31321-6
- Rowin EJ, Maron BJ, Appelbaum E, et al. Significance of false negative electrocardiograms in preparticipation screening of athletes for hypertrophic cardiomyopathy. Am J Cardiol. 2012;110(7):1027-1032. doi:10.1016/j. amjcard.2012.05.035
- Losi MA, Nistri S, Galderisi M et al. Echocardiography in patients with hypertrophic cardiomyopathy: usefulness of old and new techniques in the diagnosis and pathophysiological assessment. Cardiovasc Ultrasound. 2010;8(7). doi:10.1186/1476-7120-8-7
- Tian Z, Li L, Li X, et al. Effect of mavacamten on chinese patients with symptomatic obstructive hypertrophic cardiomyopathy: the EXPLORER-CN randomized clinical trial. JAMA Cardiol. 2023;8(10):957-965. doi:10.1001/ jamacardio.2023.3030
- Fang J, Liu Y, Zhu Y, et al. First-in-human transapical beating-heart septal myectomy in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol. 2023;82(7):575-586. doi:10.1016/j.jacc.2023.05.052
- Jain P, Patel PA, Fabbro M 2nd. Hypertrophic cardiomyopathy and left ventricular outflow tract obstruction: expecting the unexpected. J Cardiothorac Vasc Anesth. 2018;32(1):467-477. doi:10.1053/j.jvca.2017.04.054
- Poliac LC, Barron ME, Maron BJ. Hypertrophic cardiomyopathy. Anesthesiology. 2006;104(1):183-192. doi:10.1097/00000542-200601000-00025
Hypertrophic cardiomyopathy (HCM) is a relatively common inherited condition characterized by abnormal asymmetric left ventricular (LV) thickening. This can lead to LV outflow tract (LVOT) obstruction, which has important implications for anesthesia management. This article describes a case of previously undiagnosed HCM discovered during a preoperative physical examination prior to a routine surveillance colonoscopy.
CASE PRESENTATION
A 55-year-old Army veteran with a history of a sessile serrated colon adenoma presented to the preadmission testing clinic prior to planned surveillance colonoscopy under monitored anesthesia care. His medical history included untreated severe obstructive sleep apnea (53 apnea-hypopnea index score), diet-controlled hypertension, prediabetes (6.3% hemoglobin A1c), hypogonadism, and obesity (41 body mass index). Medications included semaglutide 1.7 mg injected subcutaneously weekly and testosterone 200 mg injected intramuscularly every 2 weeks, as well as lisinopril-hydrochlorothiazide 10 to 12.5 mg daily, which had recently been discontinued because his blood pressure had improved with a low-sodium diet.
A review of systems was unremarkable except for progressive weight gain. The patient had no family history of sudden cardiac death. On physical examination, the patient’s blood pressure was 119/81 mm Hg, pulse was 86 beats/min, and respiratory rate was 18 breaths/min. The patient was clinically euvolemic, with no jugular venous distention or peripheral edema, and his lungs were clear to auscultation. There was, however, a soft, nonradiating grade 2/6 systolic murmur that had not been previously documented. The murmur decreased substantially with the Valsalva maneuver, with no change in hand grip.
Laboratory studies revealed hemoglobin and renal function were within the reference range. A routine 12-lead electrocardiogram (ECG) was unremarkable. A transthoracic echocardiogram revealed moderate pulmonary hypertension (59 mm Hg right ventricular systolic pressure), asymmetric LV hypertrophy (2.1 cm septal thickness), and severe LVOT obstruction (131.8 mm Hg gradient). Severe systolic anterior motion of the mitral valve was also present. The LV ejection fraction was 60% to 65%, with normal cavity size and systolic function. These findings were consistent with severe hypertrophic obstructive cardiomyopathy (HOCM). Upon more detailed questioning, the patient reported that over the previous 5 years he had experienced gradually decreasing exercise tolerance and mild dyspnea on exertion, particularly in hot weather, which he attributed to weight gain. He also reported a presyncopal episode the previous month while working in his garage in hot weather for a prolonged period of time.
The patient’s elective colonoscopy was canceled, and he was referred to cardiology. While awaiting cardiac consultation, he was instructed to maintain good hydration and avoid any heavy physical activity beyond walking. He was told not to resume his use of lisinopril-hydrochlorothiazide. A screening 7-day Holter monitor showed no ventricular or supraventricular ectopy. After cardiology consultation, the patient was referred to a HCM specialty clinic, where a cardiac magnetic resonance imaging confirmed severe asymmetric hypertrophy with resting obstruction (Figures 1-4). Treatment options were discussed with the patient, and he underwent a trial with the Β—blocker metoprolol 50 mg daily, which he could not tolerate. Verapamil extended-release 180 mg orally once daily was then initiated; however, his dyspnea persisted. He was amenable to surgical therapy and underwent septal myectomy, with 12 g of septal myocardium removed. He did well postoperatively, with a follow-up echocardiogram showing normal LV systolic function and no LVOT gradient detectable at rest or with Valsalva maneuver. His fatigue and exertional dyspnea significantly improved. Once the patient underwent septal myectomy and was determined to have no detectable LVOT gradient, he was approved for colonoscopy which has been scheduled but not completed.
DISCUSSION
Once thought rare, HCM is now considered to be a relatively common inherited disorder, occurring in about 1 in 500 persons, with some suggesting that the actual prevalence is closer to 1 in 200 persons.1,2 Most often caused by mutations in ≥ 1 of 11 genes responsible for encoding cardiac sarcomere proteins, HCM is characterized by abnormal LV thickening without chamber enlargement in the absence of any identifiable cause, such as aortic valve stenosis or uncontrolled hypertension. The hypertrophy is often asymmetric, and in cases of asymmetric septal hypertrophy, dynamic LVOT obstruction can occur (known as HOCM). The condition is inherited in an autosomal dominant pattern with variable expression and is associated with myocardial fiber disarray, which can occur years before symptom onset.3 This myocardial disarray can lead to remodeling and an increased wall-to-lumen ratio of the coronary arteries, resulting in impaired coronary reserve.
Depending on the degree of LVOT obstruction, patients with HCM may be classified as nonobstructive, labile, or obstructive at rest. Patients without obstruction have an outflow gradient ≤ 30 mm Hg that is not provoked with Valsalva maneuver, administration of amyl nitrite, or exercise treadmill testing.3 Patients classified as labile do not have LVOT obstruction at rest, but obstruction may be induced by provocative measures. Finally, about one-third of patients with HCM will have LVOT gradients of > 30 mm Hg at rest. These patients are at increased risk for progression to symptomatic heart failure and may be candidates for surgical myectomy or catheter-based alcohol septal ablation.4 The patient in this case had a resting LVOT gradient of 131.8 mm Hg on echocardiography. The magnitude of this gradient placed the patient at a significantly higher risk of ventricular dysrhythmias and sudden cardiac death.5
Wall thickness also has prognostic implications. 6 Although any area of the myocardium can be affected, the septum is involved in about 90% cases. In their series of 48 patients followed over 6.5 years, Spirito et al found that the risk of sudden death in patients with HCM increased as wall thickness increased. For patients with a wall thickness of < 15 mm, the risk of death was 0 per 1000 person-years; however, this increased to 18.2 per 1000 person-years for patients with a wall thickness of > 30 mm.7
While many patients with HCM are asymptomatic, others may report dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, chest pain, palpitations, presyncope/ syncope, postural lightheadedness, fatigue, or edema. Symptomatology, however, is quite variable and does not necessarily correlate with the degree of outflow obstruction. Surprisingly, some patients with significant LVOT may have minimal symptoms, such as the patient in this case, while others with a lesser degree of LVOT obstruction may be very symptomatic.3,4
Physical examination of a patient with HCM may be normal or may reveal nonspecific findings such as a fourth heart sound or a systolic murmur. In general, physical examination abnormalities are related to LVOT obstruction. Those patients without significant outflow obstruction may have a normal cardiac examination. While patients with HCM may have a variety of systolic murmurs, the 2 most common are those related to outflow tract obstruction and mitral regurgitation caused by systolic anterior motion of the mitral valve.4 The systolic murmur associated with significant LVOT obstruction has been described as a harsh, crescendo-decrescendo type that begins just after S1 and is heard best at the apex and lower left sternal border.4 It may radiate to the axilla and base but not generally into the neck. The murmur usually increases with Valsalva maneuver and decreases with handgrip or going from a standing to a sitting/ squatting position. The initial examination of the patient in this case was not suggestive of HOCM, as confirmed by 2 practitioners (a cardiologist and an internist), each with > 30 years of clinical experience. This may have been related to the patient’s hydration status at the time, with Valsalva maneuver increasing obstruction to the point of reduced flow.
About 90% of patients with HCM will have abnormalities on ECG, most commonly LV hypertrophy with a strain pattern. Other ECG findings include: (1) prominent abnormal Q waves, particularly in the inferior (II, III, and aVF) and lateral leads (I, aVL, and V4-V6), reflecting depolarization of a hypertrophied septum; (2) left axis deviation; (3) deeply inverted T waves in leads V2 through V4; and (4) P wave abnormalities indicative of left atrial (LA) or biatrial enlargement. 8 It is notable that the patient in this case had a normal ECG, given that a minority of patients with HCM have been shown to have a normal ECG.9
Echocardiography plays an important role in diagnosing HCM. Diagnostic criteria include the presence of asymmetric hypertrophy (most commonly with anterior septal involvement), systolic anterior motion of the mitral valve, a nondilated LV cavity, septal immobility, and premature closure of the aortic valve. LV thickness is measured at both the septum and free wall; values ≥ 15 mm, with a septal-to-free wall thickness ratio of ≥ 1.3, are suggestive of HCM. Asymmetric LV hypertrophy can also be seen in other segments besides the septum, such as the apex.10
HCM/HOCM is the most common cause of sudden cardiac death in young people. The condition also contributes to significant functional morbidity due to heart failure and increases the risk of atrial fibrillation and subsequent stroke. Treatments tend to focus on symptom relief and slowing disease progression and include the use of medications such as Β—blockers, nondihydropyridine calcium channel blockers, and the myosin inhibitor mavacamten.11 Select patients, such as those with severe LVOT obstruction and symptoms despite treatment with Β—blockers or nondihydropyridine calcium channel blockers, may be offered septal myectomy or catheter-based alcohol septal ablation, coupled with insertion of an implantable cardiac defibrillator to prevent sudden cardiac death in patients at high arrhythmic risk.1,12
Patients with HCM, particularly those with LVOT obstruction, pose distinct challenges to the anesthesiologist because they are highly sensitive to decreases in preload and afterload. These patients frequently experience adverse perioperative events such as myocardial ischemia, systemic hypotension, and supraventricular or ventricular arrhythmias. Acute congestive heart failure may also occur, presumably due to concomitant diastolic dysfunction. Patients with previously unrecognized HCM are of particular concern, as they may manifest unexpected and sudden hypotension with the induction of anesthesia. There may then be a paradoxical response to vasoactive drugs and anesthetic agents, which accentuate LVOT obstruction. In these circumstances, undiagnosed HCM should be considered, and intraoperative rescue transesophageal echocardiography be performed.13 Once the diagnosis is confirmed, efforts should be made to reduce myocardial contractility and sympathetic discharge (eg, with Β—blockers), increase afterload (eg, with α1 agonists), and improve preload with adequate hydration. Proper resuscitation of hypotensive patients with HCM requires a thorough understanding of disease pathology, as effective interventions may seem to be counterintuitive. Inotropic agents such as epinephrine are contraindicated in HCM because increased inotropy and chronotropy worsen LVOT obstruction. Volume status is often tenuous; while adequate preload is important, overly aggressive fluid resuscitation may promote heart failure. It is important to keep in mind that even patients without resting LVOT obstruction may develop dynamic obstruction with anesthesia induction due to sudden reductions in preload and afterload. It is also important to note that the degree of LV hypertrophy is directly correlated with arrhythmic sudden death. Those patients with LV wall thickness ≥ 30 mm are at increased risk for potentially lethal tachyarrhythmias in the operating room.14
These considerations reinforce the need for proper preoperative identification of patients with HCM. Heightened awareness is key, given the fact that HCM is relatively common and tends to be underdiagnosed in the general population. These patients are generally young, otherwise healthy, and often undergo minor operative procedures in outpatient settings. It is incumbent upon the preoperative evaluator to take a thorough medical history and perform a careful physical examination. Clues to the diagnosis include exertional dyspnea, fatigue, angina, syncope/presyncope, or a family history of sudden cardiac death or HCM. A systolic ejection murmur, particularly one that increases with standing or Valsalva maneuver, and decreases with squatting or handgrip may also raise clinical suspicion. These patients should undergo a full cardiac evaluation, including echocardiography.
CONCLUSIONS
HCM is a common condition that is important to diagnose in the preoperative clinic. Failure to do so can lead to catastrophic complications during induction of anesthesia due to the sudden reduction in preload and afterload, which may cause a significant increase in LVOT obstruction. A high index of suspicion is essential, as clinical diagnosis can be challenging. The physical examination may be deceiving and symptoms are often subtle and nonspecific. It is imperative to alert the anesthesiologist before surgery so the complex hemodynamic management of patients with HOCM can be appropriately managed.
Hypertrophic cardiomyopathy (HCM) is a relatively common inherited condition characterized by abnormal asymmetric left ventricular (LV) thickening. This can lead to LV outflow tract (LVOT) obstruction, which has important implications for anesthesia management. This article describes a case of previously undiagnosed HCM discovered during a preoperative physical examination prior to a routine surveillance colonoscopy.
CASE PRESENTATION
A 55-year-old Army veteran with a history of a sessile serrated colon adenoma presented to the preadmission testing clinic prior to planned surveillance colonoscopy under monitored anesthesia care. His medical history included untreated severe obstructive sleep apnea (53 apnea-hypopnea index score), diet-controlled hypertension, prediabetes (6.3% hemoglobin A1c), hypogonadism, and obesity (41 body mass index). Medications included semaglutide 1.7 mg injected subcutaneously weekly and testosterone 200 mg injected intramuscularly every 2 weeks, as well as lisinopril-hydrochlorothiazide 10 to 12.5 mg daily, which had recently been discontinued because his blood pressure had improved with a low-sodium diet.
A review of systems was unremarkable except for progressive weight gain. The patient had no family history of sudden cardiac death. On physical examination, the patient’s blood pressure was 119/81 mm Hg, pulse was 86 beats/min, and respiratory rate was 18 breaths/min. The patient was clinically euvolemic, with no jugular venous distention or peripheral edema, and his lungs were clear to auscultation. There was, however, a soft, nonradiating grade 2/6 systolic murmur that had not been previously documented. The murmur decreased substantially with the Valsalva maneuver, with no change in hand grip.
Laboratory studies revealed hemoglobin and renal function were within the reference range. A routine 12-lead electrocardiogram (ECG) was unremarkable. A transthoracic echocardiogram revealed moderate pulmonary hypertension (59 mm Hg right ventricular systolic pressure), asymmetric LV hypertrophy (2.1 cm septal thickness), and severe LVOT obstruction (131.8 mm Hg gradient). Severe systolic anterior motion of the mitral valve was also present. The LV ejection fraction was 60% to 65%, with normal cavity size and systolic function. These findings were consistent with severe hypertrophic obstructive cardiomyopathy (HOCM). Upon more detailed questioning, the patient reported that over the previous 5 years he had experienced gradually decreasing exercise tolerance and mild dyspnea on exertion, particularly in hot weather, which he attributed to weight gain. He also reported a presyncopal episode the previous month while working in his garage in hot weather for a prolonged period of time.
The patient’s elective colonoscopy was canceled, and he was referred to cardiology. While awaiting cardiac consultation, he was instructed to maintain good hydration and avoid any heavy physical activity beyond walking. He was told not to resume his use of lisinopril-hydrochlorothiazide. A screening 7-day Holter monitor showed no ventricular or supraventricular ectopy. After cardiology consultation, the patient was referred to a HCM specialty clinic, where a cardiac magnetic resonance imaging confirmed severe asymmetric hypertrophy with resting obstruction (Figures 1-4). Treatment options were discussed with the patient, and he underwent a trial with the Β—blocker metoprolol 50 mg daily, which he could not tolerate. Verapamil extended-release 180 mg orally once daily was then initiated; however, his dyspnea persisted. He was amenable to surgical therapy and underwent septal myectomy, with 12 g of septal myocardium removed. He did well postoperatively, with a follow-up echocardiogram showing normal LV systolic function and no LVOT gradient detectable at rest or with Valsalva maneuver. His fatigue and exertional dyspnea significantly improved. Once the patient underwent septal myectomy and was determined to have no detectable LVOT gradient, he was approved for colonoscopy which has been scheduled but not completed.
DISCUSSION
Once thought rare, HCM is now considered to be a relatively common inherited disorder, occurring in about 1 in 500 persons, with some suggesting that the actual prevalence is closer to 1 in 200 persons.1,2 Most often caused by mutations in ≥ 1 of 11 genes responsible for encoding cardiac sarcomere proteins, HCM is characterized by abnormal LV thickening without chamber enlargement in the absence of any identifiable cause, such as aortic valve stenosis or uncontrolled hypertension. The hypertrophy is often asymmetric, and in cases of asymmetric septal hypertrophy, dynamic LVOT obstruction can occur (known as HOCM). The condition is inherited in an autosomal dominant pattern with variable expression and is associated with myocardial fiber disarray, which can occur years before symptom onset.3 This myocardial disarray can lead to remodeling and an increased wall-to-lumen ratio of the coronary arteries, resulting in impaired coronary reserve.
Depending on the degree of LVOT obstruction, patients with HCM may be classified as nonobstructive, labile, or obstructive at rest. Patients without obstruction have an outflow gradient ≤ 30 mm Hg that is not provoked with Valsalva maneuver, administration of amyl nitrite, or exercise treadmill testing.3 Patients classified as labile do not have LVOT obstruction at rest, but obstruction may be induced by provocative measures. Finally, about one-third of patients with HCM will have LVOT gradients of > 30 mm Hg at rest. These patients are at increased risk for progression to symptomatic heart failure and may be candidates for surgical myectomy or catheter-based alcohol septal ablation.4 The patient in this case had a resting LVOT gradient of 131.8 mm Hg on echocardiography. The magnitude of this gradient placed the patient at a significantly higher risk of ventricular dysrhythmias and sudden cardiac death.5
Wall thickness also has prognostic implications. 6 Although any area of the myocardium can be affected, the septum is involved in about 90% cases. In their series of 48 patients followed over 6.5 years, Spirito et al found that the risk of sudden death in patients with HCM increased as wall thickness increased. For patients with a wall thickness of < 15 mm, the risk of death was 0 per 1000 person-years; however, this increased to 18.2 per 1000 person-years for patients with a wall thickness of > 30 mm.7
While many patients with HCM are asymptomatic, others may report dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, chest pain, palpitations, presyncope/ syncope, postural lightheadedness, fatigue, or edema. Symptomatology, however, is quite variable and does not necessarily correlate with the degree of outflow obstruction. Surprisingly, some patients with significant LVOT may have minimal symptoms, such as the patient in this case, while others with a lesser degree of LVOT obstruction may be very symptomatic.3,4
Physical examination of a patient with HCM may be normal or may reveal nonspecific findings such as a fourth heart sound or a systolic murmur. In general, physical examination abnormalities are related to LVOT obstruction. Those patients without significant outflow obstruction may have a normal cardiac examination. While patients with HCM may have a variety of systolic murmurs, the 2 most common are those related to outflow tract obstruction and mitral regurgitation caused by systolic anterior motion of the mitral valve.4 The systolic murmur associated with significant LVOT obstruction has been described as a harsh, crescendo-decrescendo type that begins just after S1 and is heard best at the apex and lower left sternal border.4 It may radiate to the axilla and base but not generally into the neck. The murmur usually increases with Valsalva maneuver and decreases with handgrip or going from a standing to a sitting/ squatting position. The initial examination of the patient in this case was not suggestive of HOCM, as confirmed by 2 practitioners (a cardiologist and an internist), each with > 30 years of clinical experience. This may have been related to the patient’s hydration status at the time, with Valsalva maneuver increasing obstruction to the point of reduced flow.
About 90% of patients with HCM will have abnormalities on ECG, most commonly LV hypertrophy with a strain pattern. Other ECG findings include: (1) prominent abnormal Q waves, particularly in the inferior (II, III, and aVF) and lateral leads (I, aVL, and V4-V6), reflecting depolarization of a hypertrophied septum; (2) left axis deviation; (3) deeply inverted T waves in leads V2 through V4; and (4) P wave abnormalities indicative of left atrial (LA) or biatrial enlargement. 8 It is notable that the patient in this case had a normal ECG, given that a minority of patients with HCM have been shown to have a normal ECG.9
Echocardiography plays an important role in diagnosing HCM. Diagnostic criteria include the presence of asymmetric hypertrophy (most commonly with anterior septal involvement), systolic anterior motion of the mitral valve, a nondilated LV cavity, septal immobility, and premature closure of the aortic valve. LV thickness is measured at both the septum and free wall; values ≥ 15 mm, with a septal-to-free wall thickness ratio of ≥ 1.3, are suggestive of HCM. Asymmetric LV hypertrophy can also be seen in other segments besides the septum, such as the apex.10
HCM/HOCM is the most common cause of sudden cardiac death in young people. The condition also contributes to significant functional morbidity due to heart failure and increases the risk of atrial fibrillation and subsequent stroke. Treatments tend to focus on symptom relief and slowing disease progression and include the use of medications such as Β—blockers, nondihydropyridine calcium channel blockers, and the myosin inhibitor mavacamten.11 Select patients, such as those with severe LVOT obstruction and symptoms despite treatment with Β—blockers or nondihydropyridine calcium channel blockers, may be offered septal myectomy or catheter-based alcohol septal ablation, coupled with insertion of an implantable cardiac defibrillator to prevent sudden cardiac death in patients at high arrhythmic risk.1,12
Patients with HCM, particularly those with LVOT obstruction, pose distinct challenges to the anesthesiologist because they are highly sensitive to decreases in preload and afterload. These patients frequently experience adverse perioperative events such as myocardial ischemia, systemic hypotension, and supraventricular or ventricular arrhythmias. Acute congestive heart failure may also occur, presumably due to concomitant diastolic dysfunction. Patients with previously unrecognized HCM are of particular concern, as they may manifest unexpected and sudden hypotension with the induction of anesthesia. There may then be a paradoxical response to vasoactive drugs and anesthetic agents, which accentuate LVOT obstruction. In these circumstances, undiagnosed HCM should be considered, and intraoperative rescue transesophageal echocardiography be performed.13 Once the diagnosis is confirmed, efforts should be made to reduce myocardial contractility and sympathetic discharge (eg, with Β—blockers), increase afterload (eg, with α1 agonists), and improve preload with adequate hydration. Proper resuscitation of hypotensive patients with HCM requires a thorough understanding of disease pathology, as effective interventions may seem to be counterintuitive. Inotropic agents such as epinephrine are contraindicated in HCM because increased inotropy and chronotropy worsen LVOT obstruction. Volume status is often tenuous; while adequate preload is important, overly aggressive fluid resuscitation may promote heart failure. It is important to keep in mind that even patients without resting LVOT obstruction may develop dynamic obstruction with anesthesia induction due to sudden reductions in preload and afterload. It is also important to note that the degree of LV hypertrophy is directly correlated with arrhythmic sudden death. Those patients with LV wall thickness ≥ 30 mm are at increased risk for potentially lethal tachyarrhythmias in the operating room.14
These considerations reinforce the need for proper preoperative identification of patients with HCM. Heightened awareness is key, given the fact that HCM is relatively common and tends to be underdiagnosed in the general population. These patients are generally young, otherwise healthy, and often undergo minor operative procedures in outpatient settings. It is incumbent upon the preoperative evaluator to take a thorough medical history and perform a careful physical examination. Clues to the diagnosis include exertional dyspnea, fatigue, angina, syncope/presyncope, or a family history of sudden cardiac death or HCM. A systolic ejection murmur, particularly one that increases with standing or Valsalva maneuver, and decreases with squatting or handgrip may also raise clinical suspicion. These patients should undergo a full cardiac evaluation, including echocardiography.
CONCLUSIONS
HCM is a common condition that is important to diagnose in the preoperative clinic. Failure to do so can lead to catastrophic complications during induction of anesthesia due to the sudden reduction in preload and afterload, which may cause a significant increase in LVOT obstruction. A high index of suspicion is essential, as clinical diagnosis can be challenging. The physical examination may be deceiving and symptoms are often subtle and nonspecific. It is imperative to alert the anesthesiologist before surgery so the complex hemodynamic management of patients with HOCM can be appropriately managed.
- Cheng Z, Fang T, Huang J, Guo Y, Alam M, Qian H. Hypertrophic cardiomyopathy: from phenotype and pathogenesis to treatment. Front Cardiovasc Med. 2021;8:722340. doi:10.3389/fcvm.2021.722340
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254. doi:10.1016/j.jacc.2015.01.019
- Hensley N, Dietrich J, Nyhan D, Mitter N, Yee MS, Brady M. Hypertrophic cardiomyopathy: a review. Anesth Analg. 2015;120(3):554-569. doi:10.1213/ ANE.0000000000000538
- Maron BJ, Desai MY, Nishimura RA, et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79(4):372–389. doi:10.1016/j.jacc.2021.12.002
- Jorda P, Garcia-Alvarez A. Hypertrophic cardiomyopathy: sudden cardiac death risk stratification in adults. Glob Cardiol Sci Pract. 2018;3(25). doi:10.21542/gcsp.2018.25
- Wigle ED, Sasson Z, Henderson MA, et al. Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis. 1985;28(1):1-83. doi:10.1016/0033-0620(85)90024-6
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342(24):1778–1785. doi:10.1056/ NEJM200006153422403
- Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy Lancet. 2017;389(10075):1253-1267. doi:10.1016/S0140-6736(16)31321-6
- Rowin EJ, Maron BJ, Appelbaum E, et al. Significance of false negative electrocardiograms in preparticipation screening of athletes for hypertrophic cardiomyopathy. Am J Cardiol. 2012;110(7):1027-1032. doi:10.1016/j. amjcard.2012.05.035
- Losi MA, Nistri S, Galderisi M et al. Echocardiography in patients with hypertrophic cardiomyopathy: usefulness of old and new techniques in the diagnosis and pathophysiological assessment. Cardiovasc Ultrasound. 2010;8(7). doi:10.1186/1476-7120-8-7
- Tian Z, Li L, Li X, et al. Effect of mavacamten on chinese patients with symptomatic obstructive hypertrophic cardiomyopathy: the EXPLORER-CN randomized clinical trial. JAMA Cardiol. 2023;8(10):957-965. doi:10.1001/ jamacardio.2023.3030
- Fang J, Liu Y, Zhu Y, et al. First-in-human transapical beating-heart septal myectomy in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol. 2023;82(7):575-586. doi:10.1016/j.jacc.2023.05.052
- Jain P, Patel PA, Fabbro M 2nd. Hypertrophic cardiomyopathy and left ventricular outflow tract obstruction: expecting the unexpected. J Cardiothorac Vasc Anesth. 2018;32(1):467-477. doi:10.1053/j.jvca.2017.04.054
- Poliac LC, Barron ME, Maron BJ. Hypertrophic cardiomyopathy. Anesthesiology. 2006;104(1):183-192. doi:10.1097/00000542-200601000-00025
- Cheng Z, Fang T, Huang J, Guo Y, Alam M, Qian H. Hypertrophic cardiomyopathy: from phenotype and pathogenesis to treatment. Front Cardiovasc Med. 2021;8:722340. doi:10.3389/fcvm.2021.722340
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254. doi:10.1016/j.jacc.2015.01.019
- Hensley N, Dietrich J, Nyhan D, Mitter N, Yee MS, Brady M. Hypertrophic cardiomyopathy: a review. Anesth Analg. 2015;120(3):554-569. doi:10.1213/ ANE.0000000000000538
- Maron BJ, Desai MY, Nishimura RA, et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79(4):372–389. doi:10.1016/j.jacc.2021.12.002
- Jorda P, Garcia-Alvarez A. Hypertrophic cardiomyopathy: sudden cardiac death risk stratification in adults. Glob Cardiol Sci Pract. 2018;3(25). doi:10.21542/gcsp.2018.25
- Wigle ED, Sasson Z, Henderson MA, et al. Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis. 1985;28(1):1-83. doi:10.1016/0033-0620(85)90024-6
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342(24):1778–1785. doi:10.1056/ NEJM200006153422403
- Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy Lancet. 2017;389(10075):1253-1267. doi:10.1016/S0140-6736(16)31321-6
- Rowin EJ, Maron BJ, Appelbaum E, et al. Significance of false negative electrocardiograms in preparticipation screening of athletes for hypertrophic cardiomyopathy. Am J Cardiol. 2012;110(7):1027-1032. doi:10.1016/j. amjcard.2012.05.035
- Losi MA, Nistri S, Galderisi M et al. Echocardiography in patients with hypertrophic cardiomyopathy: usefulness of old and new techniques in the diagnosis and pathophysiological assessment. Cardiovasc Ultrasound. 2010;8(7). doi:10.1186/1476-7120-8-7
- Tian Z, Li L, Li X, et al. Effect of mavacamten on chinese patients with symptomatic obstructive hypertrophic cardiomyopathy: the EXPLORER-CN randomized clinical trial. JAMA Cardiol. 2023;8(10):957-965. doi:10.1001/ jamacardio.2023.3030
- Fang J, Liu Y, Zhu Y, et al. First-in-human transapical beating-heart septal myectomy in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol. 2023;82(7):575-586. doi:10.1016/j.jacc.2023.05.052
- Jain P, Patel PA, Fabbro M 2nd. Hypertrophic cardiomyopathy and left ventricular outflow tract obstruction: expecting the unexpected. J Cardiothorac Vasc Anesth. 2018;32(1):467-477. doi:10.1053/j.jvca.2017.04.054
- Poliac LC, Barron ME, Maron BJ. Hypertrophic cardiomyopathy. Anesthesiology. 2006;104(1):183-192. doi:10.1097/00000542-200601000-00025
Importance of Recognizing Hypertrophic Cardiomyopathy in the Preoperative Clinic
Importance of Recognizing Hypertrophic Cardiomyopathy in the Preoperative Clinic
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Muscle-related complaints occur in 7% to 25% of patients taking statin medications.1 In most instances, these adverse effects are quickly resolved when the medication is discontinued, but in rare occurrences, the statin can trigger an autoimmune response that progresses even after stopping use. This uncommon condition is typically accompanied by symmetrical proximal muscle weakness and an elevated CPK leading to a necrotizing myopathy requiring treatment with immunosuppressive therapy. Although less common, some patients may also present with dysphagia, myalgia, weight loss, and/or skin rash.1
Statin medications have been the cornerstone of lipid-lowering therapy due to their mechanism of inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-limiting step within the cholesterol synthesis pathway to produce mevalonic acid. There is a proven genetic association with human leukocyte antigen (HLA)-DRB1*11:01 in adults and anti-HMGCR–associated myopathy.1 The incidence of statin-induced necrotizing autoimmune myopathy (SINAM) in relation to each specific statin agent remains unknown; however, a systematic review of case reports found higher correlations for atorvastatin and simvastatin.2
There are 2 ways to confirm a SINAM diagnosis. The first and simplest includes checking for the presence of antibodies against HMGCR. The anti-HMGCR antibody test is typically used as a definitive diagnosis because it has a high specificity for SINAM.3 The second and more invasive diagnosis method involves a muscle biopsy, which is identified as positive if the biopsy shows the presence of necrotic muscle fibers.1,3
The anti-HMGCR antibody test can serve as a marker for disease activity because the antibodies are strongly correlated with CPK levels.1 CPK levels indicate the severity of muscle injury and is often used in addition to either of the confirmatory tests because it is faster and less expensive. Anti-HMGCR titers may remain positive while CPK returns to baseline when SINAM is dormant. In addition, clinicians may use an electromyography (EMG) test to measure the muscle response in association to nerve stimulation. 1 This test can show potential features of myopathic lesions such as positive sharp waves, spontaneous fibrillations, or myotonic repetitive potentials.
Typical treatment includes glucocorticoids as first-line agents, but SINAM can be difficult to treat due to its complicated pathophysiology processes.3 Escalation of therapy is sometimes required beyond a single agent; in these complex scenarios, methotrexate and/or intravenous (IV) immunoglobulin (IVIG) therapy are frequently added to the steroid therapy. There have been concerns with steroid use in specific patient populations due to the undesired adverse effect (AE) profile, and as a result IVIG has been used as monotherapy at a dose of 2 g/kg per month.3 Studies looking at IVIG monotherapy showed a reduction in CPK levels and improvement in strength after just 2 to 3 rounds of monthly treatment.3 Some patients receiving IVIG monotherapy even achieved baseline strength and no longer reported muscle-related symptoms, although the total treatment duration varied. A systematic review of 39 articles where glucocorticoids, IVIG, methotrexate and/or a combination were used to treat SINAM found an average time to remission of 8.6 months. Additionally, this systematic review observed more patients returned to baseline or experienced improvement in symptoms when being treated with a combination of glucocorticoid plus IVIG plus methotrexate.2 Suggested dosing recommendations are available in Table 1.

Patients diagnosed with HMGCR antibody myopathy are contraindicated for future statin therapy.1 Rechallenge of statins in this patient population has led to worsening of disease and therefore these patients should have a severe statin allergy listed in their medical documentation record.
CASE PRESENTATION
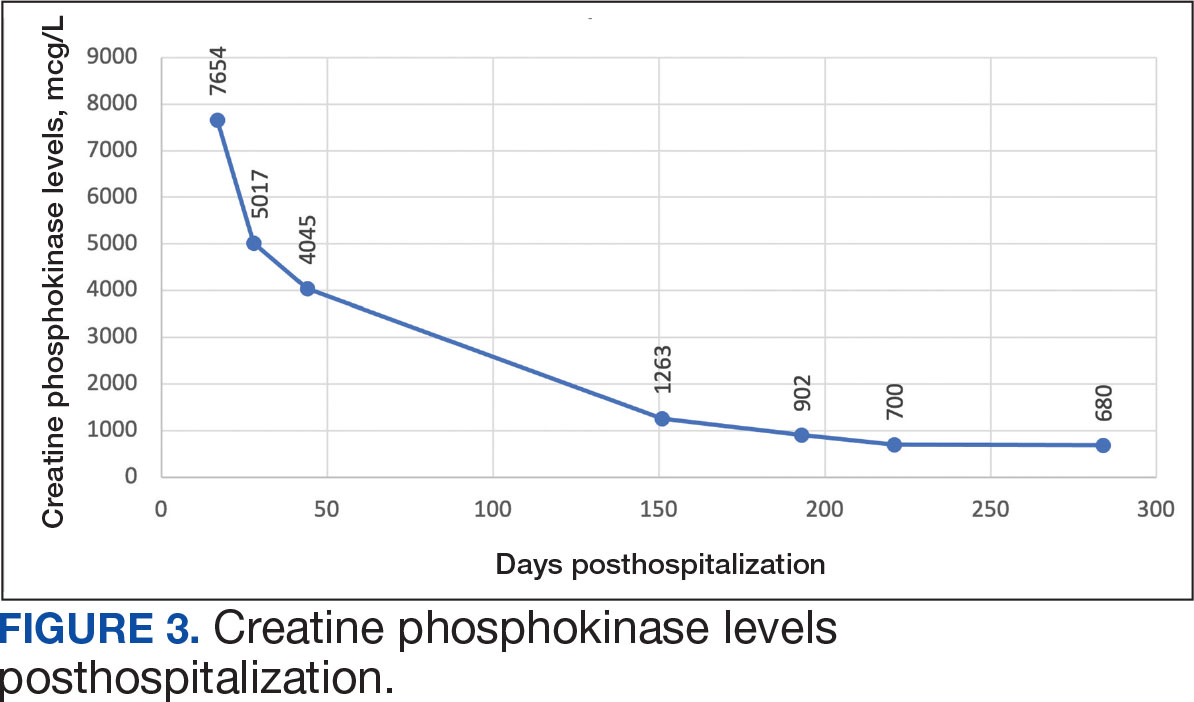
A 59-year-old male patient with a medical history including atrial fibrillation, peripheral vascular disease, type 2 diabetes mellitus (T2DM), hypertension, and peripheral neuropathy was referred by his primary care clinical pharmacist practitioner for an outpatient neurology consult. The patient reported a 4-month history of fatigue, lower extremity paresthesia, and progressive proximal muscle weakness which began in his legs, mostly noticeable when walking upstairs but quickly developed into bilateral arm weakness. The patient reported significant impact on his quality of life: he could no longer lift his arms above his head and had difficulty with daily activities such as brushing his hair or getting up from a chair. He reported multiple falls at home, and began to use a cane for assistance with ambulation. He confirmed adherence to atorvastatin over the past year. Laboratory testing on the day of the visit revealed an elevated CPK level at 9729 mcg/L (reference range for men, 30-300 mcg/L).
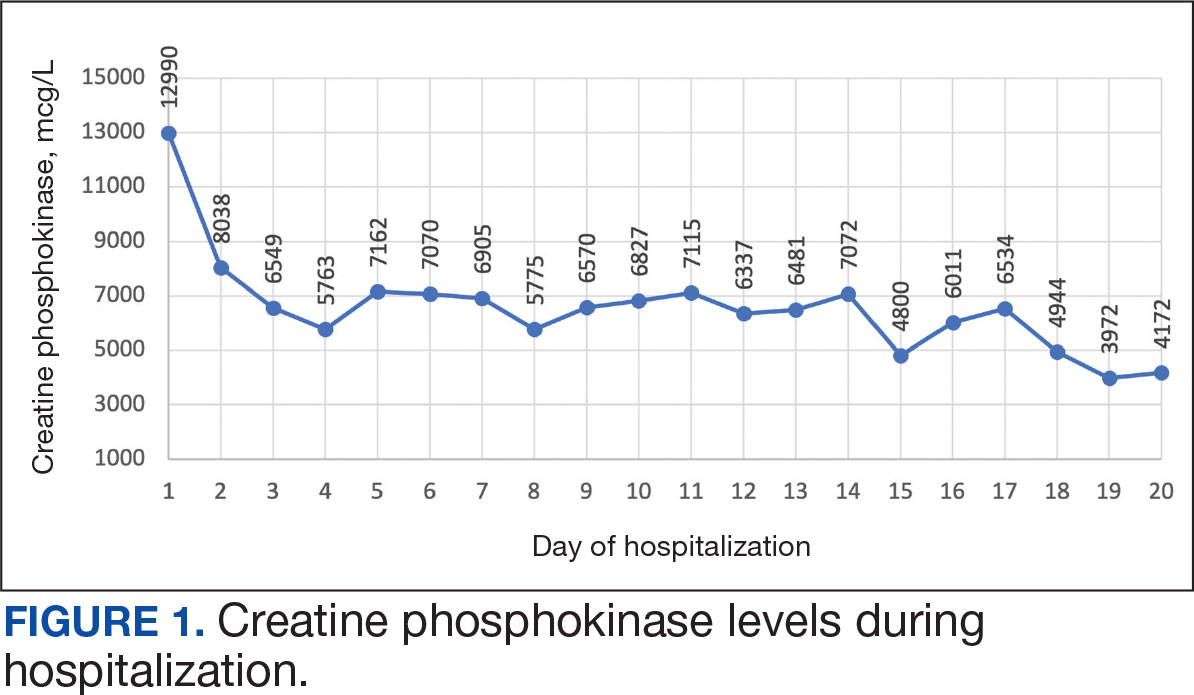
The patient was urged to go to the emergency department where his CPK level had increased to 12,990 mcg/L (Figure 1). The workup began to find the source of rhabdomyolysis and elevated liver enzymes differentiating autoimmune vs medication-induced myopathy. Upon admission atorvastatin was discontinued, anti-HMGCR antibody level was ordered, and IV fluids were started.
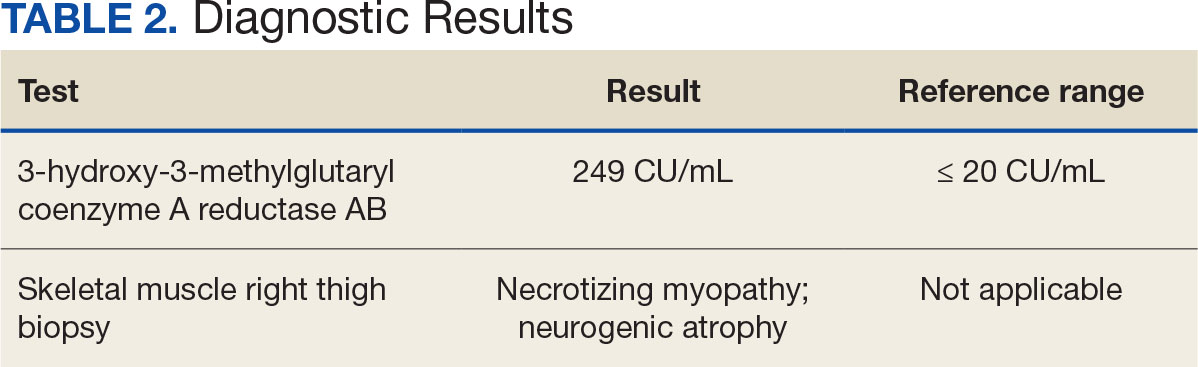
After 8 days of hospital admission with minimal improvement, Rheumatology and Neurology services were consulted in the setting of persistent CPK elevation and the potential neuropathic component of muscle weakness. Both consulting services agreed to consider muscle biopsy and EMG if the patient did not begin to show signs of improvement. The patient’s CPK levels remained elevated with minimal change in muscle weakness. The next step was a right quadricep muscle biopsy performed on Day 14 of admission. Sixteen days after admission, the anti-HMGCR antibody test (originally obtained upon admission) was positive and elevated at 249 CU/mL (reference range, < 20 CU/mL negative; reference range, ≥ 60 CU/mL strong positive), which confirmed the SINAM diagnosis (Table 2).
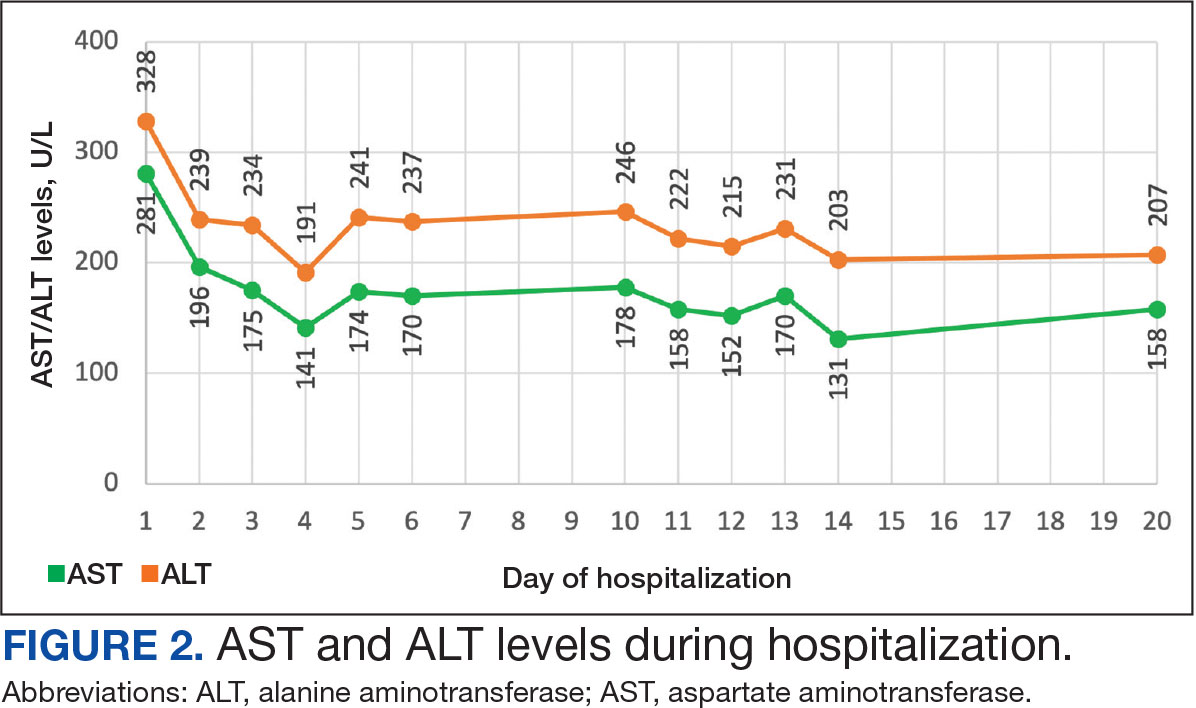
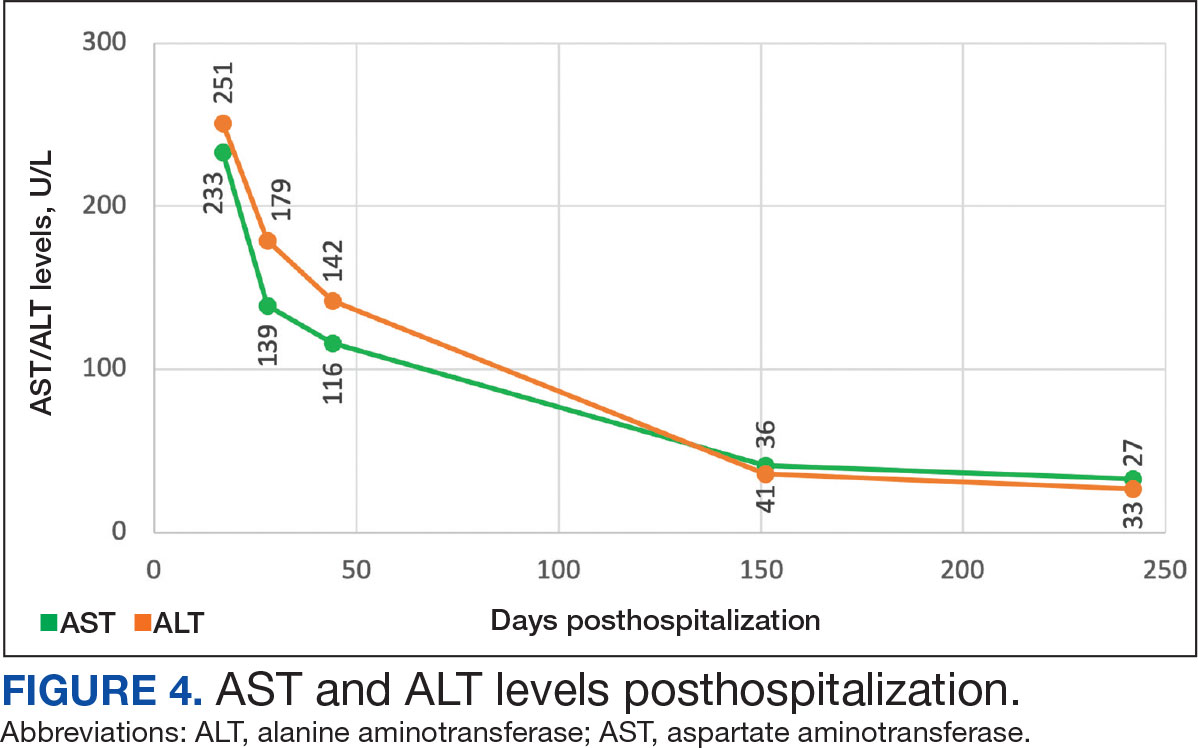
On Day 17 of hospitalization, the Neurology service initiated IVIG monotherapy to avoid the undesired glycemic AEs associated with glucocorticoids. The patient had a history of T2DM that was difficult to manage and his hemoglobin A1c level was the best it had ever been (6.2%) relative to a peak A1c of 11.0% 9 months prior. The patient was treated with a total IVIG dose of 2 g/kg divided into 3 daily doses while still obtaining CPK levels with daily laboratory tests to assist with trending the extent of disease severity improvement (Figures 2-4). After a 20-day hospital stay, the patient was discharged home with rehabilitation services and a scheduled outpatient EMG the following week.
The patient continued to report generalized body weakness, pain, and deconditioning upon discharge and was unable to attend the EMG neurology appointment. The patient did eventually attend a follow-up appointment about 6 weeks after hospital discharge and reported continued weakness. The Neurology service prescribed a 2-day IVIG regimen (total dose = 2 g/kg) monthly for the next 2 months. The patient returned to the neurology clinic 8 weeks later following 2 rounds of IVIG posthospitalization and reported that his muscle strength was returning, and he was able to slowly reintroduce exercise into his daily routine. During a follow-up appointment about 11 months after the initial hospitalization, the patient’s primary care clinical pharmacist provided education of effective management of cholesterol without statins, including use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors as recommended by the Neurology service. At this time, the patient’s calculated low-density lipoprotein (LDL) was 110 mg/dL (reference range, 0-99 mg/dL). The patient preferred to work on a healthy diet and positive lifestyle choices before trialing any lipid lowering therapies.
The patient appeared to tolerate this treatment regimen following 7 rounds of IVIG. He noted fatigue for about 24 hours after his infusion sessions but otherwise reported no additional AEs. He has continued to attend weekly physical therapy sessions and is able to walk without the assistance of a cane. He can now walk a mile before he begins to feel fatigued or experience bilateral lower leg pain. The pain appears neuropathic in nature, as the patient reports ongoing “pins and needles” sensation in his legs and feet. The patient has noticed a major improvement in his overall function, strength, and exercise tolerance since starting IVIG treatments and although he is not yet back to his baseline, he is motivated to continue his recovery. Neurology is considering ongoing treatment with IVIG monthly infusions given his continued clinical improvement.
DISCUSSION
There is limited evidence on the use of IVIG monotherapy for SINAM, although it may be a viable option for patients deemed poor candidates for glucocorticoid or methotrexate therapy. This particularly applies to patients with DM for which there may be concerns for managing blood glucose levels with steroid use. The Johns Hopkins Myositis Center evaluated 3 patients with SINAM who declined glucocorticoid therapy and had documented DM and weakness in the proximal arms and legs. Following 2 to 3 monthly rounds of IVIG 2 g/kg monotherapy, these patients had reduced CPK levels and had improvement in both arm and hip-flexion strength. Two patients reported no muscle-related symptoms after completing IVIG monotherapy treatment for 9 and 19 months.3
The optimal treatment duration for IVIG monotherapy for SINAM is still uncertain given the limited available data. The patient in this case report showed clinically significant muscle-related improvement following 7 monthly rounds of 2 g/kg IVIG treatments. The mechanism of action for IVIG in this setting is still unknown, although the medication may allow muscle regeneration to surpass muscle destruction, thus leading to resolution of the muscle-related symptoms.3
There are numerous concerns with IVIG use to consider prior to initiating treatment, including expense, AEs, patient response, and comorbidities. IVIG is considerably more expensive than glucocorticoid and methotrexate alternatives. Systemic reactions have been shown to occur in 5% to 15% of patients receiving IVIG infusion.4 The majority of these infusion reactions occur early during infusion or within a few hours after administration is complete.5 Early AEs to monitor for include injection site reactions, flu-like symptoms, dermatologic reactions, anaphylaxis, transfusion-related acute lung injury, and transfusion-associated circulatory overload. Additional AEs may be delayed, including thromboembolic events, acute kidney injury, aseptic meningitis, hemolysis, neutropenia, and blood-borne infection.6 IVIG has a boxed warning for thrombosis, renal dysfunction, and acute renal failure risk.7 There are multiple strategies documented to reduce the risk of IVIG reactions including slowing the infusion rate, ensuring adequate hydration, and/or giving analgesics, antihistamines, or steroids prior to infusion.6 The patient in this case had monthly IVIG infusions without the need of any pretreatment medications and only reported fatigue for about 24 hours following the infusion.
An essential question is how to provide safe cholesterol management for patients with SINAM. Some evidence has suggested that other lipid-lowering medications that avoid the mevalonate pathway, such as fenofibrate or ezetimibe, may be used cautiously initially at lower doses.1 Due to the severity of SINAM, it is crucial to closely monitor and ensure tolerability as new lipid-lowering agents are introduced. More evidence suggests that PCSK9 inhibitors are a safer option.8 PCSK9 inhibitors avoid the mevalonate pathway and block PCSK9 from binding to LDL receptors, allowing LDL to be removed from circulation.
Tiniakou et al followed 8 individuals for a mean 1.5 years who had anti-HMGCR immune-mediated myopathy at high cardiovascular risk. Muscle strength, CPK levels, and serum anti-HMGCR antibody titers were assessed at baseline and again after initiation of PCSK9 inhibitor. None of the patients experienced a decline in their muscle strength. CPK, anti-HMGCR antibody levels, and LDL trended down in all participants and 2 patients were able to reduce their immunosuppression treatment while still achieving clinical improvement. Tiniakou et al suggest that PCSK9 inhibitors are a safe and effective option to lower cholesterol in patients with SINAM.8
Alirocumab is the preferred PCSK9 inhibitor for patients at the US Department of Veterans Affairs (VA). The VA Pharmacy Benefits Management (PBM) Service guidance recommends alirocumab for patients with a history of atherosclerotic cardiovascular disease (ASCVD) or severe hypercholesterolemia.9 PBM guidance suggests alirocumab use for patients with a contraindication, intolerance, or insufficient LDL reduction with a maximally tolerated dose of statin and ezetimibe with a desire to reduce ASCVD risk by lowering LDL. Per the PBM Criteria for Use guidance, patients should follow the stepwise approach and trial ezetimibe prior to being considered for PCSK9 inhibitor therapy. Given the patient’s contraindication to future statin use and severity of myopathy, in this case the Neurology Service felt that the safest option to reach goal LDL reduction would be a PCSK9 inhibitor. Consideration can be made for alirocumab use when considering an alternative lipid lowering therapy.
CONCLUSIONS
This report demonstrates a case of SINAM caused by atorvastatin therapy. Patients presenting with proximal muscle weakness and elevated CPK even after statin discontinuation should be considered for a full workup to determine whether SINAM may be involved. This uncommon form of myopathy can be diagnosed based on the detection of anti-HMGCR antibodies and/or presence of necrosis on muscle biopsy. A combination of glucocorticoid, methotrexate, and IVIG is recommended for a patient’s best chance of muscle symptom improvement. IVIG monotherapy should be considered for patients with glycemic control concerns.
- Tiniakou E. Statin-associated autoimmune myopathy: current perspectives. Ther Clin Risk Manag. 2020;16:483-492. doi:10.2147/TCRM.S197941
- Somagutta MKR, Shama N, Pormento MKL, et al. Statin-induced necrotizing autoimmune myopathy: a systematic review. Reumatologia. 2022;60(1):63-69. doi:10.5114/reum.2022.114108
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. 2015;373(17):1680-1682. doi:10.1056/NEJMc1506163
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev. 2013;27(3):171-178. doi:10.1016/j.tmrv.2013.05.004
- Ameratunga R, Sinclair J, Kolbe J. Increased risk of adverse events when changing intravenous immunoglobulin preparations. Clin Exp Immunol. 2004;136(1):111-113. doi:10.1111/j.1365-2249.2004.02412.x
- Abbas A, Rajabally YA. Complications of immunoglobulin therapy and implications for treatment of inflammatory neuropathy: a review. Curr Drug Saf. 2019;14(1):3-13. doi:10.2174/1574886313666181017121139
- Privigen. Prescribing information. CSL Behring LLC; 2022. Accessed March 17, 2025. https://labeling.cslbehring.com/PI/US/Privigen/EN/Privigen-Prescribing-Information.pdf
- Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/Kexin Type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheumatol. 2019;71(10):1723-1726. doi:10.1002/art.40919
- US Department of Veterans Affairs, Pharmacy Benefits Management (PBM) Services. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9 Inhibitor) (Alirocumabpreferred, Evolocumab-non-preferred) Criteria for Use. June 2024. Accessed March 25, 2025. https://www.va.gov/formularyadvisor/DOC/128
- Jayatilaka S, Desai K, Rijal S, Zimmerman D. Statin-induced autoimmune necrotizing myopathy. J Prim Care Community Health. 2021;12:21501327211028714. doi:10.1177/21501327211028714
Muscle-related complaints occur in 7% to 25% of patients taking statin medications.1 In most instances, these adverse effects are quickly resolved when the medication is discontinued, but in rare occurrences, the statin can trigger an autoimmune response that progresses even after stopping use. This uncommon condition is typically accompanied by symmetrical proximal muscle weakness and an elevated CPK leading to a necrotizing myopathy requiring treatment with immunosuppressive therapy. Although less common, some patients may also present with dysphagia, myalgia, weight loss, and/or skin rash.1
Statin medications have been the cornerstone of lipid-lowering therapy due to their mechanism of inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-limiting step within the cholesterol synthesis pathway to produce mevalonic acid. There is a proven genetic association with human leukocyte antigen (HLA)-DRB1*11:01 in adults and anti-HMGCR–associated myopathy.1 The incidence of statin-induced necrotizing autoimmune myopathy (SINAM) in relation to each specific statin agent remains unknown; however, a systematic review of case reports found higher correlations for atorvastatin and simvastatin.2
There are 2 ways to confirm a SINAM diagnosis. The first and simplest includes checking for the presence of antibodies against HMGCR. The anti-HMGCR antibody test is typically used as a definitive diagnosis because it has a high specificity for SINAM.3 The second and more invasive diagnosis method involves a muscle biopsy, which is identified as positive if the biopsy shows the presence of necrotic muscle fibers.1,3
The anti-HMGCR antibody test can serve as a marker for disease activity because the antibodies are strongly correlated with CPK levels.1 CPK levels indicate the severity of muscle injury and is often used in addition to either of the confirmatory tests because it is faster and less expensive. Anti-HMGCR titers may remain positive while CPK returns to baseline when SINAM is dormant. In addition, clinicians may use an electromyography (EMG) test to measure the muscle response in association to nerve stimulation. 1 This test can show potential features of myopathic lesions such as positive sharp waves, spontaneous fibrillations, or myotonic repetitive potentials.
Typical treatment includes glucocorticoids as first-line agents, but SINAM can be difficult to treat due to its complicated pathophysiology processes.3 Escalation of therapy is sometimes required beyond a single agent; in these complex scenarios, methotrexate and/or intravenous (IV) immunoglobulin (IVIG) therapy are frequently added to the steroid therapy. There have been concerns with steroid use in specific patient populations due to the undesired adverse effect (AE) profile, and as a result IVIG has been used as monotherapy at a dose of 2 g/kg per month.3 Studies looking at IVIG monotherapy showed a reduction in CPK levels and improvement in strength after just 2 to 3 rounds of monthly treatment.3 Some patients receiving IVIG monotherapy even achieved baseline strength and no longer reported muscle-related symptoms, although the total treatment duration varied. A systematic review of 39 articles where glucocorticoids, IVIG, methotrexate and/or a combination were used to treat SINAM found an average time to remission of 8.6 months. Additionally, this systematic review observed more patients returned to baseline or experienced improvement in symptoms when being treated with a combination of glucocorticoid plus IVIG plus methotrexate.2 Suggested dosing recommendations are available in Table 1.
Patients diagnosed with HMGCR antibody myopathy are contraindicated for future statin therapy.1 Rechallenge of statins in this patient population has led to worsening of disease and therefore these patients should have a severe statin allergy listed in their medical documentation record.
CASE PRESENTATION
A 59-year-old male patient with a medical history including atrial fibrillation, peripheral vascular disease, type 2 diabetes mellitus (T2DM), hypertension, and peripheral neuropathy was referred by his primary care clinical pharmacist practitioner for an outpatient neurology consult. The patient reported a 4-month history of fatigue, lower extremity paresthesia, and progressive proximal muscle weakness which began in his legs, mostly noticeable when walking upstairs but quickly developed into bilateral arm weakness. The patient reported significant impact on his quality of life: he could no longer lift his arms above his head and had difficulty with daily activities such as brushing his hair or getting up from a chair. He reported multiple falls at home, and began to use a cane for assistance with ambulation. He confirmed adherence to atorvastatin over the past year. Laboratory testing on the day of the visit revealed an elevated CPK level at 9729 mcg/L (reference range for men, 30-300 mcg/L).
The patient was urged to go to the emergency department where his CPK level had increased to 12,990 mcg/L (Figure 1). The workup began to find the source of rhabdomyolysis and elevated liver enzymes differentiating autoimmune vs medication-induced myopathy. Upon admission atorvastatin was discontinued, anti-HMGCR antibody level was ordered, and IV fluids were started.
After 8 days of hospital admission with minimal improvement, Rheumatology and Neurology services were consulted in the setting of persistent CPK elevation and the potential neuropathic component of muscle weakness. Both consulting services agreed to consider muscle biopsy and EMG if the patient did not begin to show signs of improvement. The patient’s CPK levels remained elevated with minimal change in muscle weakness. The next step was a right quadricep muscle biopsy performed on Day 14 of admission. Sixteen days after admission, the anti-HMGCR antibody test (originally obtained upon admission) was positive and elevated at 249 CU/mL (reference range, < 20 CU/mL negative; reference range, ≥ 60 CU/mL strong positive), which confirmed the SINAM diagnosis (Table 2).
On Day 17 of hospitalization, the Neurology service initiated IVIG monotherapy to avoid the undesired glycemic AEs associated with glucocorticoids. The patient had a history of T2DM that was difficult to manage and his hemoglobin A1c level was the best it had ever been (6.2%) relative to a peak A1c of 11.0% 9 months prior. The patient was treated with a total IVIG dose of 2 g/kg divided into 3 daily doses while still obtaining CPK levels with daily laboratory tests to assist with trending the extent of disease severity improvement (Figures 2-4). After a 20-day hospital stay, the patient was discharged home with rehabilitation services and a scheduled outpatient EMG the following week.
The patient continued to report generalized body weakness, pain, and deconditioning upon discharge and was unable to attend the EMG neurology appointment. The patient did eventually attend a follow-up appointment about 6 weeks after hospital discharge and reported continued weakness. The Neurology service prescribed a 2-day IVIG regimen (total dose = 2 g/kg) monthly for the next 2 months. The patient returned to the neurology clinic 8 weeks later following 2 rounds of IVIG posthospitalization and reported that his muscle strength was returning, and he was able to slowly reintroduce exercise into his daily routine. During a follow-up appointment about 11 months after the initial hospitalization, the patient’s primary care clinical pharmacist provided education of effective management of cholesterol without statins, including use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors as recommended by the Neurology service. At this time, the patient’s calculated low-density lipoprotein (LDL) was 110 mg/dL (reference range, 0-99 mg/dL). The patient preferred to work on a healthy diet and positive lifestyle choices before trialing any lipid lowering therapies.
The patient appeared to tolerate this treatment regimen following 7 rounds of IVIG. He noted fatigue for about 24 hours after his infusion sessions but otherwise reported no additional AEs. He has continued to attend weekly physical therapy sessions and is able to walk without the assistance of a cane. He can now walk a mile before he begins to feel fatigued or experience bilateral lower leg pain. The pain appears neuropathic in nature, as the patient reports ongoing “pins and needles” sensation in his legs and feet. The patient has noticed a major improvement in his overall function, strength, and exercise tolerance since starting IVIG treatments and although he is not yet back to his baseline, he is motivated to continue his recovery. Neurology is considering ongoing treatment with IVIG monthly infusions given his continued clinical improvement.
DISCUSSION
There is limited evidence on the use of IVIG monotherapy for SINAM, although it may be a viable option for patients deemed poor candidates for glucocorticoid or methotrexate therapy. This particularly applies to patients with DM for which there may be concerns for managing blood glucose levels with steroid use. The Johns Hopkins Myositis Center evaluated 3 patients with SINAM who declined glucocorticoid therapy and had documented DM and weakness in the proximal arms and legs. Following 2 to 3 monthly rounds of IVIG 2 g/kg monotherapy, these patients had reduced CPK levels and had improvement in both arm and hip-flexion strength. Two patients reported no muscle-related symptoms after completing IVIG monotherapy treatment for 9 and 19 months.3
The optimal treatment duration for IVIG monotherapy for SINAM is still uncertain given the limited available data. The patient in this case report showed clinically significant muscle-related improvement following 7 monthly rounds of 2 g/kg IVIG treatments. The mechanism of action for IVIG in this setting is still unknown, although the medication may allow muscle regeneration to surpass muscle destruction, thus leading to resolution of the muscle-related symptoms.3
There are numerous concerns with IVIG use to consider prior to initiating treatment, including expense, AEs, patient response, and comorbidities. IVIG is considerably more expensive than glucocorticoid and methotrexate alternatives. Systemic reactions have been shown to occur in 5% to 15% of patients receiving IVIG infusion.4 The majority of these infusion reactions occur early during infusion or within a few hours after administration is complete.5 Early AEs to monitor for include injection site reactions, flu-like symptoms, dermatologic reactions, anaphylaxis, transfusion-related acute lung injury, and transfusion-associated circulatory overload. Additional AEs may be delayed, including thromboembolic events, acute kidney injury, aseptic meningitis, hemolysis, neutropenia, and blood-borne infection.6 IVIG has a boxed warning for thrombosis, renal dysfunction, and acute renal failure risk.7 There are multiple strategies documented to reduce the risk of IVIG reactions including slowing the infusion rate, ensuring adequate hydration, and/or giving analgesics, antihistamines, or steroids prior to infusion.6 The patient in this case had monthly IVIG infusions without the need of any pretreatment medications and only reported fatigue for about 24 hours following the infusion.
An essential question is how to provide safe cholesterol management for patients with SINAM. Some evidence has suggested that other lipid-lowering medications that avoid the mevalonate pathway, such as fenofibrate or ezetimibe, may be used cautiously initially at lower doses.1 Due to the severity of SINAM, it is crucial to closely monitor and ensure tolerability as new lipid-lowering agents are introduced. More evidence suggests that PCSK9 inhibitors are a safer option.8 PCSK9 inhibitors avoid the mevalonate pathway and block PCSK9 from binding to LDL receptors, allowing LDL to be removed from circulation.
Tiniakou et al followed 8 individuals for a mean 1.5 years who had anti-HMGCR immune-mediated myopathy at high cardiovascular risk. Muscle strength, CPK levels, and serum anti-HMGCR antibody titers were assessed at baseline and again after initiation of PCSK9 inhibitor. None of the patients experienced a decline in their muscle strength. CPK, anti-HMGCR antibody levels, and LDL trended down in all participants and 2 patients were able to reduce their immunosuppression treatment while still achieving clinical improvement. Tiniakou et al suggest that PCSK9 inhibitors are a safe and effective option to lower cholesterol in patients with SINAM.8
Alirocumab is the preferred PCSK9 inhibitor for patients at the US Department of Veterans Affairs (VA). The VA Pharmacy Benefits Management (PBM) Service guidance recommends alirocumab for patients with a history of atherosclerotic cardiovascular disease (ASCVD) or severe hypercholesterolemia.9 PBM guidance suggests alirocumab use for patients with a contraindication, intolerance, or insufficient LDL reduction with a maximally tolerated dose of statin and ezetimibe with a desire to reduce ASCVD risk by lowering LDL. Per the PBM Criteria for Use guidance, patients should follow the stepwise approach and trial ezetimibe prior to being considered for PCSK9 inhibitor therapy. Given the patient’s contraindication to future statin use and severity of myopathy, in this case the Neurology Service felt that the safest option to reach goal LDL reduction would be a PCSK9 inhibitor. Consideration can be made for alirocumab use when considering an alternative lipid lowering therapy.
CONCLUSIONS
This report demonstrates a case of SINAM caused by atorvastatin therapy. Patients presenting with proximal muscle weakness and elevated CPK even after statin discontinuation should be considered for a full workup to determine whether SINAM may be involved. This uncommon form of myopathy can be diagnosed based on the detection of anti-HMGCR antibodies and/or presence of necrosis on muscle biopsy. A combination of glucocorticoid, methotrexate, and IVIG is recommended for a patient’s best chance of muscle symptom improvement. IVIG monotherapy should be considered for patients with glycemic control concerns.
Muscle-related complaints occur in 7% to 25% of patients taking statin medications.1 In most instances, these adverse effects are quickly resolved when the medication is discontinued, but in rare occurrences, the statin can trigger an autoimmune response that progresses even after stopping use. This uncommon condition is typically accompanied by symmetrical proximal muscle weakness and an elevated CPK leading to a necrotizing myopathy requiring treatment with immunosuppressive therapy. Although less common, some patients may also present with dysphagia, myalgia, weight loss, and/or skin rash.1
Statin medications have been the cornerstone of lipid-lowering therapy due to their mechanism of inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-limiting step within the cholesterol synthesis pathway to produce mevalonic acid. There is a proven genetic association with human leukocyte antigen (HLA)-DRB1*11:01 in adults and anti-HMGCR–associated myopathy.1 The incidence of statin-induced necrotizing autoimmune myopathy (SINAM) in relation to each specific statin agent remains unknown; however, a systematic review of case reports found higher correlations for atorvastatin and simvastatin.2
There are 2 ways to confirm a SINAM diagnosis. The first and simplest includes checking for the presence of antibodies against HMGCR. The anti-HMGCR antibody test is typically used as a definitive diagnosis because it has a high specificity for SINAM.3 The second and more invasive diagnosis method involves a muscle biopsy, which is identified as positive if the biopsy shows the presence of necrotic muscle fibers.1,3
The anti-HMGCR antibody test can serve as a marker for disease activity because the antibodies are strongly correlated with CPK levels.1 CPK levels indicate the severity of muscle injury and is often used in addition to either of the confirmatory tests because it is faster and less expensive. Anti-HMGCR titers may remain positive while CPK returns to baseline when SINAM is dormant. In addition, clinicians may use an electromyography (EMG) test to measure the muscle response in association to nerve stimulation. 1 This test can show potential features of myopathic lesions such as positive sharp waves, spontaneous fibrillations, or myotonic repetitive potentials.
Typical treatment includes glucocorticoids as first-line agents, but SINAM can be difficult to treat due to its complicated pathophysiology processes.3 Escalation of therapy is sometimes required beyond a single agent; in these complex scenarios, methotrexate and/or intravenous (IV) immunoglobulin (IVIG) therapy are frequently added to the steroid therapy. There have been concerns with steroid use in specific patient populations due to the undesired adverse effect (AE) profile, and as a result IVIG has been used as monotherapy at a dose of 2 g/kg per month.3 Studies looking at IVIG monotherapy showed a reduction in CPK levels and improvement in strength after just 2 to 3 rounds of monthly treatment.3 Some patients receiving IVIG monotherapy even achieved baseline strength and no longer reported muscle-related symptoms, although the total treatment duration varied. A systematic review of 39 articles where glucocorticoids, IVIG, methotrexate and/or a combination were used to treat SINAM found an average time to remission of 8.6 months. Additionally, this systematic review observed more patients returned to baseline or experienced improvement in symptoms when being treated with a combination of glucocorticoid plus IVIG plus methotrexate.2 Suggested dosing recommendations are available in Table 1.
Patients diagnosed with HMGCR antibody myopathy are contraindicated for future statin therapy.1 Rechallenge of statins in this patient population has led to worsening of disease and therefore these patients should have a severe statin allergy listed in their medical documentation record.
CASE PRESENTATION
A 59-year-old male patient with a medical history including atrial fibrillation, peripheral vascular disease, type 2 diabetes mellitus (T2DM), hypertension, and peripheral neuropathy was referred by his primary care clinical pharmacist practitioner for an outpatient neurology consult. The patient reported a 4-month history of fatigue, lower extremity paresthesia, and progressive proximal muscle weakness which began in his legs, mostly noticeable when walking upstairs but quickly developed into bilateral arm weakness. The patient reported significant impact on his quality of life: he could no longer lift his arms above his head and had difficulty with daily activities such as brushing his hair or getting up from a chair. He reported multiple falls at home, and began to use a cane for assistance with ambulation. He confirmed adherence to atorvastatin over the past year. Laboratory testing on the day of the visit revealed an elevated CPK level at 9729 mcg/L (reference range for men, 30-300 mcg/L).
The patient was urged to go to the emergency department where his CPK level had increased to 12,990 mcg/L (Figure 1). The workup began to find the source of rhabdomyolysis and elevated liver enzymes differentiating autoimmune vs medication-induced myopathy. Upon admission atorvastatin was discontinued, anti-HMGCR antibody level was ordered, and IV fluids were started.
After 8 days of hospital admission with minimal improvement, Rheumatology and Neurology services were consulted in the setting of persistent CPK elevation and the potential neuropathic component of muscle weakness. Both consulting services agreed to consider muscle biopsy and EMG if the patient did not begin to show signs of improvement. The patient’s CPK levels remained elevated with minimal change in muscle weakness. The next step was a right quadricep muscle biopsy performed on Day 14 of admission. Sixteen days after admission, the anti-HMGCR antibody test (originally obtained upon admission) was positive and elevated at 249 CU/mL (reference range, < 20 CU/mL negative; reference range, ≥ 60 CU/mL strong positive), which confirmed the SINAM diagnosis (Table 2).
On Day 17 of hospitalization, the Neurology service initiated IVIG monotherapy to avoid the undesired glycemic AEs associated with glucocorticoids. The patient had a history of T2DM that was difficult to manage and his hemoglobin A1c level was the best it had ever been (6.2%) relative to a peak A1c of 11.0% 9 months prior. The patient was treated with a total IVIG dose of 2 g/kg divided into 3 daily doses while still obtaining CPK levels with daily laboratory tests to assist with trending the extent of disease severity improvement (Figures 2-4). After a 20-day hospital stay, the patient was discharged home with rehabilitation services and a scheduled outpatient EMG the following week.
The patient continued to report generalized body weakness, pain, and deconditioning upon discharge and was unable to attend the EMG neurology appointment. The patient did eventually attend a follow-up appointment about 6 weeks after hospital discharge and reported continued weakness. The Neurology service prescribed a 2-day IVIG regimen (total dose = 2 g/kg) monthly for the next 2 months. The patient returned to the neurology clinic 8 weeks later following 2 rounds of IVIG posthospitalization and reported that his muscle strength was returning, and he was able to slowly reintroduce exercise into his daily routine. During a follow-up appointment about 11 months after the initial hospitalization, the patient’s primary care clinical pharmacist provided education of effective management of cholesterol without statins, including use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors as recommended by the Neurology service. At this time, the patient’s calculated low-density lipoprotein (LDL) was 110 mg/dL (reference range, 0-99 mg/dL). The patient preferred to work on a healthy diet and positive lifestyle choices before trialing any lipid lowering therapies.
The patient appeared to tolerate this treatment regimen following 7 rounds of IVIG. He noted fatigue for about 24 hours after his infusion sessions but otherwise reported no additional AEs. He has continued to attend weekly physical therapy sessions and is able to walk without the assistance of a cane. He can now walk a mile before he begins to feel fatigued or experience bilateral lower leg pain. The pain appears neuropathic in nature, as the patient reports ongoing “pins and needles” sensation in his legs and feet. The patient has noticed a major improvement in his overall function, strength, and exercise tolerance since starting IVIG treatments and although he is not yet back to his baseline, he is motivated to continue his recovery. Neurology is considering ongoing treatment with IVIG monthly infusions given his continued clinical improvement.
DISCUSSION
There is limited evidence on the use of IVIG monotherapy for SINAM, although it may be a viable option for patients deemed poor candidates for glucocorticoid or methotrexate therapy. This particularly applies to patients with DM for which there may be concerns for managing blood glucose levels with steroid use. The Johns Hopkins Myositis Center evaluated 3 patients with SINAM who declined glucocorticoid therapy and had documented DM and weakness in the proximal arms and legs. Following 2 to 3 monthly rounds of IVIG 2 g/kg monotherapy, these patients had reduced CPK levels and had improvement in both arm and hip-flexion strength. Two patients reported no muscle-related symptoms after completing IVIG monotherapy treatment for 9 and 19 months.3
The optimal treatment duration for IVIG monotherapy for SINAM is still uncertain given the limited available data. The patient in this case report showed clinically significant muscle-related improvement following 7 monthly rounds of 2 g/kg IVIG treatments. The mechanism of action for IVIG in this setting is still unknown, although the medication may allow muscle regeneration to surpass muscle destruction, thus leading to resolution of the muscle-related symptoms.3
There are numerous concerns with IVIG use to consider prior to initiating treatment, including expense, AEs, patient response, and comorbidities. IVIG is considerably more expensive than glucocorticoid and methotrexate alternatives. Systemic reactions have been shown to occur in 5% to 15% of patients receiving IVIG infusion.4 The majority of these infusion reactions occur early during infusion or within a few hours after administration is complete.5 Early AEs to monitor for include injection site reactions, flu-like symptoms, dermatologic reactions, anaphylaxis, transfusion-related acute lung injury, and transfusion-associated circulatory overload. Additional AEs may be delayed, including thromboembolic events, acute kidney injury, aseptic meningitis, hemolysis, neutropenia, and blood-borne infection.6 IVIG has a boxed warning for thrombosis, renal dysfunction, and acute renal failure risk.7 There are multiple strategies documented to reduce the risk of IVIG reactions including slowing the infusion rate, ensuring adequate hydration, and/or giving analgesics, antihistamines, or steroids prior to infusion.6 The patient in this case had monthly IVIG infusions without the need of any pretreatment medications and only reported fatigue for about 24 hours following the infusion.
An essential question is how to provide safe cholesterol management for patients with SINAM. Some evidence has suggested that other lipid-lowering medications that avoid the mevalonate pathway, such as fenofibrate or ezetimibe, may be used cautiously initially at lower doses.1 Due to the severity of SINAM, it is crucial to closely monitor and ensure tolerability as new lipid-lowering agents are introduced. More evidence suggests that PCSK9 inhibitors are a safer option.8 PCSK9 inhibitors avoid the mevalonate pathway and block PCSK9 from binding to LDL receptors, allowing LDL to be removed from circulation.
Tiniakou et al followed 8 individuals for a mean 1.5 years who had anti-HMGCR immune-mediated myopathy at high cardiovascular risk. Muscle strength, CPK levels, and serum anti-HMGCR antibody titers were assessed at baseline and again after initiation of PCSK9 inhibitor. None of the patients experienced a decline in their muscle strength. CPK, anti-HMGCR antibody levels, and LDL trended down in all participants and 2 patients were able to reduce their immunosuppression treatment while still achieving clinical improvement. Tiniakou et al suggest that PCSK9 inhibitors are a safe and effective option to lower cholesterol in patients with SINAM.8
Alirocumab is the preferred PCSK9 inhibitor for patients at the US Department of Veterans Affairs (VA). The VA Pharmacy Benefits Management (PBM) Service guidance recommends alirocumab for patients with a history of atherosclerotic cardiovascular disease (ASCVD) or severe hypercholesterolemia.9 PBM guidance suggests alirocumab use for patients with a contraindication, intolerance, or insufficient LDL reduction with a maximally tolerated dose of statin and ezetimibe with a desire to reduce ASCVD risk by lowering LDL. Per the PBM Criteria for Use guidance, patients should follow the stepwise approach and trial ezetimibe prior to being considered for PCSK9 inhibitor therapy. Given the patient’s contraindication to future statin use and severity of myopathy, in this case the Neurology Service felt that the safest option to reach goal LDL reduction would be a PCSK9 inhibitor. Consideration can be made for alirocumab use when considering an alternative lipid lowering therapy.
CONCLUSIONS
This report demonstrates a case of SINAM caused by atorvastatin therapy. Patients presenting with proximal muscle weakness and elevated CPK even after statin discontinuation should be considered for a full workup to determine whether SINAM may be involved. This uncommon form of myopathy can be diagnosed based on the detection of anti-HMGCR antibodies and/or presence of necrosis on muscle biopsy. A combination of glucocorticoid, methotrexate, and IVIG is recommended for a patient’s best chance of muscle symptom improvement. IVIG monotherapy should be considered for patients with glycemic control concerns.
- Tiniakou E. Statin-associated autoimmune myopathy: current perspectives. Ther Clin Risk Manag. 2020;16:483-492. doi:10.2147/TCRM.S197941
- Somagutta MKR, Shama N, Pormento MKL, et al. Statin-induced necrotizing autoimmune myopathy: a systematic review. Reumatologia. 2022;60(1):63-69. doi:10.5114/reum.2022.114108
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. 2015;373(17):1680-1682. doi:10.1056/NEJMc1506163
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev. 2013;27(3):171-178. doi:10.1016/j.tmrv.2013.05.004
- Ameratunga R, Sinclair J, Kolbe J. Increased risk of adverse events when changing intravenous immunoglobulin preparations. Clin Exp Immunol. 2004;136(1):111-113. doi:10.1111/j.1365-2249.2004.02412.x
- Abbas A, Rajabally YA. Complications of immunoglobulin therapy and implications for treatment of inflammatory neuropathy: a review. Curr Drug Saf. 2019;14(1):3-13. doi:10.2174/1574886313666181017121139
- Privigen. Prescribing information. CSL Behring LLC; 2022. Accessed March 17, 2025. https://labeling.cslbehring.com/PI/US/Privigen/EN/Privigen-Prescribing-Information.pdf
- Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/Kexin Type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheumatol. 2019;71(10):1723-1726. doi:10.1002/art.40919
- US Department of Veterans Affairs, Pharmacy Benefits Management (PBM) Services. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9 Inhibitor) (Alirocumabpreferred, Evolocumab-non-preferred) Criteria for Use. June 2024. Accessed March 25, 2025. https://www.va.gov/formularyadvisor/DOC/128
- Jayatilaka S, Desai K, Rijal S, Zimmerman D. Statin-induced autoimmune necrotizing myopathy. J Prim Care Community Health. 2021;12:21501327211028714. doi:10.1177/21501327211028714
- Tiniakou E. Statin-associated autoimmune myopathy: current perspectives. Ther Clin Risk Manag. 2020;16:483-492. doi:10.2147/TCRM.S197941
- Somagutta MKR, Shama N, Pormento MKL, et al. Statin-induced necrotizing autoimmune myopathy: a systematic review. Reumatologia. 2022;60(1):63-69. doi:10.5114/reum.2022.114108
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med. 2015;373(17):1680-1682. doi:10.1056/NEJMc1506163
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev. 2013;27(3):171-178. doi:10.1016/j.tmrv.2013.05.004
- Ameratunga R, Sinclair J, Kolbe J. Increased risk of adverse events when changing intravenous immunoglobulin preparations. Clin Exp Immunol. 2004;136(1):111-113. doi:10.1111/j.1365-2249.2004.02412.x
- Abbas A, Rajabally YA. Complications of immunoglobulin therapy and implications for treatment of inflammatory neuropathy: a review. Curr Drug Saf. 2019;14(1):3-13. doi:10.2174/1574886313666181017121139
- Privigen. Prescribing information. CSL Behring LLC; 2022. Accessed March 17, 2025. https://labeling.cslbehring.com/PI/US/Privigen/EN/Privigen-Prescribing-Information.pdf
- Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/Kexin Type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheumatol. 2019;71(10):1723-1726. doi:10.1002/art.40919
- US Department of Veterans Affairs, Pharmacy Benefits Management (PBM) Services. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9 Inhibitor) (Alirocumabpreferred, Evolocumab-non-preferred) Criteria for Use. June 2024. Accessed March 25, 2025. https://www.va.gov/formularyadvisor/DOC/128
- Jayatilaka S, Desai K, Rijal S, Zimmerman D. Statin-induced autoimmune necrotizing myopathy. J Prim Care Community Health. 2021;12:21501327211028714. doi:10.1177/21501327211028714
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
Statin-Induced Necrotizing Autoimmune Myopathy in a Patient With Complex Diabetes Management
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
Prosthetic joint infection (PJI) occurs in about 1% to 2% of joint replacements. 1 Risk factors include immunosuppression, diabetes, chronic illnesses, and prolonged operative time.2 Bacterial infections constitute most of these infections, while fungal pathogens account for about 1%. Candida (C.) species, predominantly C. albicans, are responsible for most PJIs.1,3 In contrast, C. glabrata is a rare cause of fungal PJI, with only 18 PJI cases currently reported in the literature.4 C. glabrata PJI occurs more frequently among immunosuppressed patients and is associated with a higher treatment failure rate despite antifungal therapy.5 Treatment of fungal PJI is often complicated, involving multiple surgical debridements, prolonged antifungal therapy, and in some cases, prosthesis removal.6 However, given the rarity of C. glabrata as a PJI pathogen, no standardized treatment guidelines exist, leading to potential delays in diagnosis and tailored treatment.7,8
CASE PRESENTATION
A male Vietnam veteran aged 75 years presented to the emergency department in July 2023 with a fluid collection over his left hip surgical incision site. The patient had a complex medical history that included chronic kidney disease, well-controlled type 2 diabetes, hypertension, and osteoarthritis. His history was further complicated by nonalcoholic steatohepatitis with hepatocellular carcinoma that was treated with transarterial radioembolization and yttrium-90. The patient had undergone a left total hip arthroplasty in 1996 and subsequent open reduction and internal fixation about 9 months prior to his presentation. The patient reported the fluid had been present for about 6 weeks, while he received outpatient monitoring by the orthopedic surgery service. He sought emergency care after noting a moderate amount of purulent discharge on his clothing originating from his hip. In the week prior to admission, the patient observed progressive erythema, warmth, and tenderness over the incision site. Despite these symptoms, the patient remained ambulatory and able to walk long distances with the use of an assistive device.
Upon presentation, the patient was afebrile and normotensive. Laboratory testing revealed an elevated erythrocyte sedimentation rate of 77 mm/h (reference range, 0-20 mm/h) and a C-reactive protein of 9.8 mg/L (reference range, 0-2.5 mg/L), suggesting an underlying infectious process. A physical examination revealed a well-healed incision over the left hip with a poorly defined area of fluctuance and evidence of wound dehiscence. The left lower extremity was swollen with 2+ pitting edema, but tenderness was localized to the incision site. Magnetic resonance imaging of the left hip revealed a multiloculated fluid collection abutting the left greater trochanter with extension to the skin surface and inferior extension along the entire length of the surgical fixation hardware (Figure).
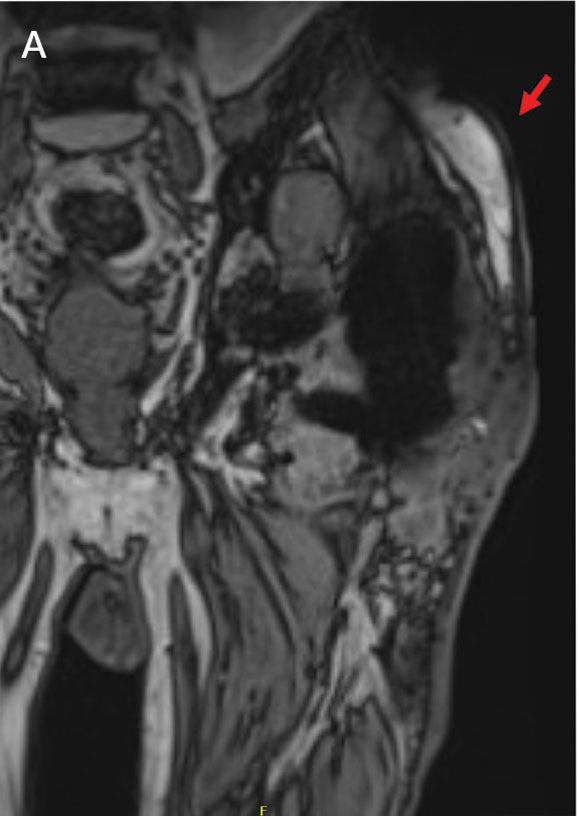
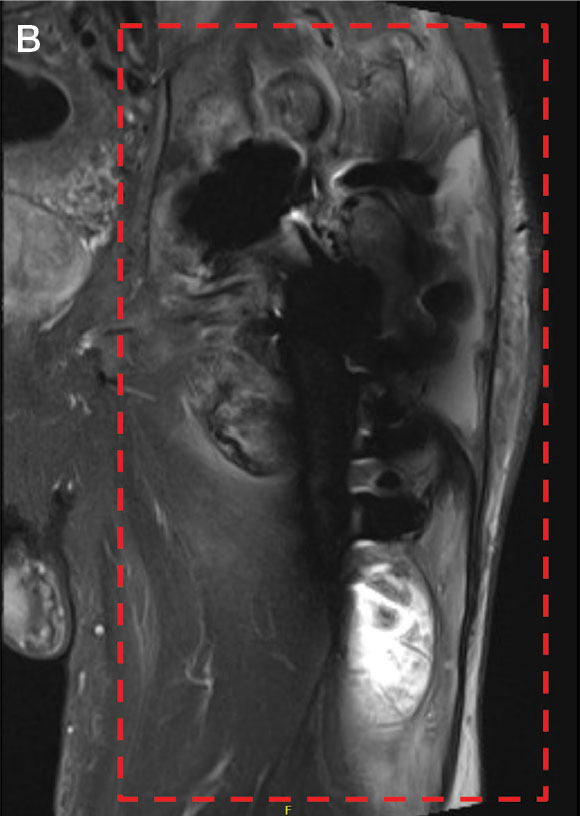
Upon admission, orthopedic surgery performed a bedside aspiration of the fluid collection. Samples were sent for analysis, including cell count and bacterial and fungal cultures. Initial blood cultures were sterile. Due to concerns for a bacterial infection, the patient was started on empiric intravenous (IV) ceftriaxone 2 g/day and IV vancomycin 1250 mg/day. Synovial fluid analysis revealed an elevated white blood cell count of 45,000/ìL, but bacterial cultures were negative. Five days after admission, the fungal culture from the left hip wound was notable for presence of C. glabrata, prompting an infectious diseases (ID) consultation. IV micafungin 100 mg/day was initiated as empiric antifungal therapy.
ID and orthopedic surgery teams determined that a combined medical and surgical approach would be best suited for infection control. They proposed 2 main approaches: complete hardware replacement with washout, which carried a higher morbidity risk but a better chance of infection resolution, or partial hardware replacement with washout, which was associated with a lower morbidity risk but a higher risk of infection persistence and recurrence. This decision was particularly challenging for the patient, who prioritized maintaining his functional status, including his ability to continue dancing for pleasure. The patient opted for a more conservative approach, electing to proceed with antifungal therapy and debridement while retaining the prosthetic joint.
After 11 days of hospitalization, the patient was discharged with a peripherally inserted central catheter for long-term antifungal infusions of micafungin 150 mg/day at home. Fungal sensitivity test results several days after discharge confirmed susceptibility to micafungin.
About 2 weeks after discharge, the patient underwent debridement and implant retention (DAIR). Wound cultures were positive for C. glabrata, Enterococcus faecalis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. Based on susceptibilities, he completed a 2-month course of IV micafungin 150 mg daily and daptomycin 750 mg daily, followed by an oral suppressive regimen consisting of doxycycline 100 mg twice daily, amoxicillin-clavulanate 2 g twice daily, and fluconazole initially 800 mg daily adjusted to 400 mg daily. The patient continued wound management with twice-daily dressing changes.
Nine months after DAIR, the patient remained on suppressive antifungal and antibacterial therapy. He continued to experience serous drainage from the wound, which greatly affected his quality of life. After discussion with his family and the orthopedic surgery team, he agreed to proceed with a 2-staged revision arthroplasty involving prosthetic explant and antibiotic spacer placement. However, the surgery was postponed due to findings of anemia (hemoglobin, 8.9 g/dL) and thrombocytopenia (platelet count, 73 x 103/λL). At the time of this report, the patient was being monitored closely with his multidisciplinary care team for the planned orthopedic procedure.
DISCUSSION
PJI is the most common cause of primary hip arthroplasty failure; however, fungal species only make up about 1% of PJIs.3,9-11 Patients are typically immunocompromised, undergoing antineoplastic therapies for malignancy, or have other comorbid conditions such as diabetes.12,13 C. glabrata presents a unique diagnostic and therapeutic challenge as it is not only rare but also notorious for its resistance to common antifungal agents. C. glabrata is known to develop multidrug resistance through the rapid accumulation of genomic mutations.14 Its propensity towards forming protective biofilm also arms it with intrinsic resistance to agents like fluconazole.15 Furthermore, based on a review of the available reports in the literature, C. glabrata PJIs are often insidious and present with symptoms closely mimicking those of bacterial PJIs, as it did in the patient in this case.16
Synovial fluid analysis, fungal cultures, and sensitivity testing are paramount for ensuring proper diagnosis for fungal PJI. The patient in this case was empirically treated with micafungin based on recommendations from the ID team. When the sensitivities results were reviewed, the same antifungal therapy was continued. Echinocandins have a favorable toxicity profile in long-term use, as well as efficacy against biofilm-producing organisms like C. glabrata.17,18
While there are a few cases citing DAIR as a feasible surgical strategy for treating fungal PJI, more recent studies have reported greater success with a 2-staged revision arthroplasty involving some combination of debridement, placement of antibiotic-loaded bone cement spacers, and partial or total exchange of the infected prosthetic joint.4,19-23 In this case, complete hardware replacement would have offered the patient the most favorable outlook for eliminating this fungal infection. However, given the patient’s advanced age, significant underlying comorbidities, and functional status, medical management with antifungal therapy and DAIR was favored.
Based on the discussion from the 6-month follow-up visit, the patient was experiencing progressive and persistent wound drainage and frequent dressing changes, highlighting the limitations of medical management for PJI in the setting of retained prosthesis. If the patient ultimately proceeds with a more invasive surgical intervention, another important consideration will be the likelihood of fungal PJI recurrence. At present, fungal PJI recurrence rates following antifungal and surgical treatment have been reported to range between 0% to 50%, which is too imprecise to be considered clinically useful.22-24
Given the ambiguity surrounding management guidelines and limited treatment options, it is crucial to emphasize the timeline of this patient’s clinical presentation and subsequent course of treatment. Upon presentation to the ED in late July, fungal PJI was considered less likely. Initial blood cultures from presentation were negative, which is common with PJIs. It was not until 5 days later that the left hip wound culture showed moderate growth of C. glabrata. Identifying a PJI is clinically challenging due to the lack of standardized diagnostic criteria. However, timely identification and diagnosis of fungal PJI with appropriate antifungal therapy, in patients with limited curative options due to comorbidities, can significantly improve quality of life and overall outcomes.25 Routine fungal and mycobacterial cultures are not currently recommended in PJI guidelines, but this case illustrates it is imperative in immunocompromised hosts.26
This case and the current paucity of similar cases in the literature stress the importance of clinicians publishing their experience in the management of fungal PJI. We strongly recommend that clinicians approach each suspected PJI with careful consideration of the patient’s unique risk factors, comorbidities, and goals of care, when deciding on a curative vs suppressive approach to therapy.
CONCLUSIONS
This case report highlights the importance of considering fungal pathogens for PJIs, especially in high-risk patients, the value of obtaining fungal cultures, the necessity of a multidisciplinary approach, the role of antifungal susceptibility testing, and consideration for the feasibility of a surgical intervention. It underscores the challenges in diagnosis and treatment of C. glabrata-associated PJI, emphasizing the importance of clinician experience-sharing in developing evidence-based management strategies. As the understanding of fungal PJI evolves, continued research and clinical data collection remain crucial for improving patient outcomes in the management of these complex cases.
- Osmon DR, Berbari EF, Berendt AR, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1-10. doi:10.1093/cid/cis966
- Eka A, Chen AF. Patient-related medical risk factors for periprosthetic joint infection of the hip and knee. Ann Transl Med. 2015;3(16):233. doi:10.3978/j.issn.2305-5839.2015.09.26
- Darouiche RO, Hamill RJ, Musher DM, Young EJ, Harris RL. Periprosthetic candidal infections following arthroplasty. Rev Infect Dis. 1989;11(1):89-96. doi:10.1093/clinids/11.1.89
- Koutserimpas C, Zervakis SG, Maraki S, et al. Non-albicans Candida prosthetic joint infections: a systematic review of treatment. World J Clin Cases. 2019;7(12):1430- 1443. doi:10.12998/wjcc.v7.i12.1430
- Fidel PL Jr, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80-96. doi:10.1128/CMR.12.1.80
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Lee YR, Kim HJ, Lee EJ, Sohn JW, Kim MJ, Yoon YK. Prosthetic joint infections caused by candida species: a systematic review and a case series. Mycopathologia. 2019;184(1):23-33. doi:10.1007/s11046-018-0286-1
- Herndon CL, Rowe TM, Metcalf RW, et al. Treatment outcomes of fungal periprosthetic joint infection. J Arthroplasty. 2023;38(11):2436-2440.e1. doi:10.1016/j.arth.2023.05.009
- Delaunay C, Hamadouche M, Girard J, Duhamel A; SoFCOT. What are the causes for failures of primary hip arthroplasties in France? Clin Orthop Relat Res. 2013;471(12): 3863-3869. doi:10.1007/s11999-013-2935-5
- Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91(1): 128-133. doi:10.2106/JBJS.H.00155
- Furnes O, Lie SA, Espehaug B, Vollset SE, Engesaeter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586. doi:10.1302/0301-620x.83b4.11223
- Gonzalez MR, Bedi ADS, Karczewski D, Lozano-Calderon SA. Treatment and outcomes of fungal prosthetic joint infections: a systematic review of 225 cases. J Arthroplasty. 2023;38(11):2464-2471.e1. doi:10.1016/j.arth.2023.05.003
- Gonzalez MR, Pretell-Mazzini J, Lozano-Calderon SA. Risk factors and management of prosthetic joint infections in megaprostheses-a review of the literature. Antibiotics (Basel). 2023;13(1):25. doi:10.3390/antibiotics13010025
- Biswas C, Chen SC, Halliday C, et al. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect. 2017;23(9):676.e7-676.e10. doi:10.1016/j.cmi.2017.03.014
- Hassan Y, Chew SY, Than LTL. Candida glabrata: pathogenicity and resistance mechanisms for adaptation and survival. J Fungi (Basel). 2021;7(8):667. doi:10.3390/jof7080667
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Pierce CG, Uppuluri P, Tristan AR, et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3(9):1494-1500. doi:10.1038/nport.2008.141
- Koutserimpas C, Samonis G, Velivassakis E, Iliopoulou- Kosmadaki S, Kontakis G, Kofteridis DP. Candida glabrata prosthetic joint infection, successfully treated with anidulafungin: a case report and review of the literature. Mycoses. 2018;61(4):266-269. doi:10.1111/myc.12736
- Brooks DH, Pupparo F. Successful salvage of a primary total knee arthroplasty infected with Candida parapsilosis. J Arthroplasty. 1998;13(6):707-712. doi:10.1016/s0883-5403(98)80017-x
- Merrer J, Dupont B, Nieszkowska A, De Jonghe B, Outin H. Candida albicans prosthetic arthritis treated with fluconazole alone. J Infect. 2001;42(3):208-209. doi:10.1053/jinf.2001.0819
- Koutserimpas C, Naoum S, Alpantaki K, et al. Fungal prosthetic joint infection in revised knee arthroplasty: an orthopaedic surgeon’s nightmare. Diagnostics (Basel). 2022;12(7):1606. doi:10.3390/diagnostics12071606
- Gao Z, Li X, Du Y, Peng Y, Wu W, Zhou Y. Success rate of fungal peri-prosthetic joint infection treated by 2-stage revision and potential risk factors of treatment failure: a retrospective study. Med Sci Monit. 2018;24:5549-5557. doi:10.12659/MSM.909168
- Hwang BH, Yoon JY, Nam CH, et al. Fungal periprosthetic joint infection after primary total knee replacement. J Bone Joint Surg Br. 2012;94(5):656-659. doi:10.1302/0301-620X.94B5.28125
- Ueng SW, Lee CY, Hu CC, Hsieh PH, Chang Y. What is the success of treatment of hip and knee candidal periprosthetic joint infection? Clin Orthop Relat Res. 2013;471(9):3002-3009. doi:10.1007/s11999-013-3007-6
- Nodzo, Scott R. MD; Bauer, Thomas MD, PhD; Pottinger, et al. Conventional diagnostic challenges in periprosthetic joint infection. J Am Acad Orthop Surg. 2015;23 Suppl:S18-S25. doi:10.5435/JAAOS-D-14-00385
- American Academy of Orthopaedic Surgeons. Diagnosis and prevention of periprosthetic joint infections. March 11, 2019. Accessed February 5, 2025. https://www.aaos.org/pjicpg
Prosthetic joint infection (PJI) occurs in about 1% to 2% of joint replacements. 1 Risk factors include immunosuppression, diabetes, chronic illnesses, and prolonged operative time.2 Bacterial infections constitute most of these infections, while fungal pathogens account for about 1%. Candida (C.) species, predominantly C. albicans, are responsible for most PJIs.1,3 In contrast, C. glabrata is a rare cause of fungal PJI, with only 18 PJI cases currently reported in the literature.4 C. glabrata PJI occurs more frequently among immunosuppressed patients and is associated with a higher treatment failure rate despite antifungal therapy.5 Treatment of fungal PJI is often complicated, involving multiple surgical debridements, prolonged antifungal therapy, and in some cases, prosthesis removal.6 However, given the rarity of C. glabrata as a PJI pathogen, no standardized treatment guidelines exist, leading to potential delays in diagnosis and tailored treatment.7,8
CASE PRESENTATION
A male Vietnam veteran aged 75 years presented to the emergency department in July 2023 with a fluid collection over his left hip surgical incision site. The patient had a complex medical history that included chronic kidney disease, well-controlled type 2 diabetes, hypertension, and osteoarthritis. His history was further complicated by nonalcoholic steatohepatitis with hepatocellular carcinoma that was treated with transarterial radioembolization and yttrium-90. The patient had undergone a left total hip arthroplasty in 1996 and subsequent open reduction and internal fixation about 9 months prior to his presentation. The patient reported the fluid had been present for about 6 weeks, while he received outpatient monitoring by the orthopedic surgery service. He sought emergency care after noting a moderate amount of purulent discharge on his clothing originating from his hip. In the week prior to admission, the patient observed progressive erythema, warmth, and tenderness over the incision site. Despite these symptoms, the patient remained ambulatory and able to walk long distances with the use of an assistive device.
Upon presentation, the patient was afebrile and normotensive. Laboratory testing revealed an elevated erythrocyte sedimentation rate of 77 mm/h (reference range, 0-20 mm/h) and a C-reactive protein of 9.8 mg/L (reference range, 0-2.5 mg/L), suggesting an underlying infectious process. A physical examination revealed a well-healed incision over the left hip with a poorly defined area of fluctuance and evidence of wound dehiscence. The left lower extremity was swollen with 2+ pitting edema, but tenderness was localized to the incision site. Magnetic resonance imaging of the left hip revealed a multiloculated fluid collection abutting the left greater trochanter with extension to the skin surface and inferior extension along the entire length of the surgical fixation hardware (Figure).
Upon admission, orthopedic surgery performed a bedside aspiration of the fluid collection. Samples were sent for analysis, including cell count and bacterial and fungal cultures. Initial blood cultures were sterile. Due to concerns for a bacterial infection, the patient was started on empiric intravenous (IV) ceftriaxone 2 g/day and IV vancomycin 1250 mg/day. Synovial fluid analysis revealed an elevated white blood cell count of 45,000/ìL, but bacterial cultures were negative. Five days after admission, the fungal culture from the left hip wound was notable for presence of C. glabrata, prompting an infectious diseases (ID) consultation. IV micafungin 100 mg/day was initiated as empiric antifungal therapy.
ID and orthopedic surgery teams determined that a combined medical and surgical approach would be best suited for infection control. They proposed 2 main approaches: complete hardware replacement with washout, which carried a higher morbidity risk but a better chance of infection resolution, or partial hardware replacement with washout, which was associated with a lower morbidity risk but a higher risk of infection persistence and recurrence. This decision was particularly challenging for the patient, who prioritized maintaining his functional status, including his ability to continue dancing for pleasure. The patient opted for a more conservative approach, electing to proceed with antifungal therapy and debridement while retaining the prosthetic joint.
After 11 days of hospitalization, the patient was discharged with a peripherally inserted central catheter for long-term antifungal infusions of micafungin 150 mg/day at home. Fungal sensitivity test results several days after discharge confirmed susceptibility to micafungin.
About 2 weeks after discharge, the patient underwent debridement and implant retention (DAIR). Wound cultures were positive for C. glabrata, Enterococcus faecalis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. Based on susceptibilities, he completed a 2-month course of IV micafungin 150 mg daily and daptomycin 750 mg daily, followed by an oral suppressive regimen consisting of doxycycline 100 mg twice daily, amoxicillin-clavulanate 2 g twice daily, and fluconazole initially 800 mg daily adjusted to 400 mg daily. The patient continued wound management with twice-daily dressing changes.
Nine months after DAIR, the patient remained on suppressive antifungal and antibacterial therapy. He continued to experience serous drainage from the wound, which greatly affected his quality of life. After discussion with his family and the orthopedic surgery team, he agreed to proceed with a 2-staged revision arthroplasty involving prosthetic explant and antibiotic spacer placement. However, the surgery was postponed due to findings of anemia (hemoglobin, 8.9 g/dL) and thrombocytopenia (platelet count, 73 x 103/λL). At the time of this report, the patient was being monitored closely with his multidisciplinary care team for the planned orthopedic procedure.
DISCUSSION
PJI is the most common cause of primary hip arthroplasty failure; however, fungal species only make up about 1% of PJIs.3,9-11 Patients are typically immunocompromised, undergoing antineoplastic therapies for malignancy, or have other comorbid conditions such as diabetes.12,13 C. glabrata presents a unique diagnostic and therapeutic challenge as it is not only rare but also notorious for its resistance to common antifungal agents. C. glabrata is known to develop multidrug resistance through the rapid accumulation of genomic mutations.14 Its propensity towards forming protective biofilm also arms it with intrinsic resistance to agents like fluconazole.15 Furthermore, based on a review of the available reports in the literature, C. glabrata PJIs are often insidious and present with symptoms closely mimicking those of bacterial PJIs, as it did in the patient in this case.16
Synovial fluid analysis, fungal cultures, and sensitivity testing are paramount for ensuring proper diagnosis for fungal PJI. The patient in this case was empirically treated with micafungin based on recommendations from the ID team. When the sensitivities results were reviewed, the same antifungal therapy was continued. Echinocandins have a favorable toxicity profile in long-term use, as well as efficacy against biofilm-producing organisms like C. glabrata.17,18
While there are a few cases citing DAIR as a feasible surgical strategy for treating fungal PJI, more recent studies have reported greater success with a 2-staged revision arthroplasty involving some combination of debridement, placement of antibiotic-loaded bone cement spacers, and partial or total exchange of the infected prosthetic joint.4,19-23 In this case, complete hardware replacement would have offered the patient the most favorable outlook for eliminating this fungal infection. However, given the patient’s advanced age, significant underlying comorbidities, and functional status, medical management with antifungal therapy and DAIR was favored.
Based on the discussion from the 6-month follow-up visit, the patient was experiencing progressive and persistent wound drainage and frequent dressing changes, highlighting the limitations of medical management for PJI in the setting of retained prosthesis. If the patient ultimately proceeds with a more invasive surgical intervention, another important consideration will be the likelihood of fungal PJI recurrence. At present, fungal PJI recurrence rates following antifungal and surgical treatment have been reported to range between 0% to 50%, which is too imprecise to be considered clinically useful.22-24
Given the ambiguity surrounding management guidelines and limited treatment options, it is crucial to emphasize the timeline of this patient’s clinical presentation and subsequent course of treatment. Upon presentation to the ED in late July, fungal PJI was considered less likely. Initial blood cultures from presentation were negative, which is common with PJIs. It was not until 5 days later that the left hip wound culture showed moderate growth of C. glabrata. Identifying a PJI is clinically challenging due to the lack of standardized diagnostic criteria. However, timely identification and diagnosis of fungal PJI with appropriate antifungal therapy, in patients with limited curative options due to comorbidities, can significantly improve quality of life and overall outcomes.25 Routine fungal and mycobacterial cultures are not currently recommended in PJI guidelines, but this case illustrates it is imperative in immunocompromised hosts.26
This case and the current paucity of similar cases in the literature stress the importance of clinicians publishing their experience in the management of fungal PJI. We strongly recommend that clinicians approach each suspected PJI with careful consideration of the patient’s unique risk factors, comorbidities, and goals of care, when deciding on a curative vs suppressive approach to therapy.
CONCLUSIONS
This case report highlights the importance of considering fungal pathogens for PJIs, especially in high-risk patients, the value of obtaining fungal cultures, the necessity of a multidisciplinary approach, the role of antifungal susceptibility testing, and consideration for the feasibility of a surgical intervention. It underscores the challenges in diagnosis and treatment of C. glabrata-associated PJI, emphasizing the importance of clinician experience-sharing in developing evidence-based management strategies. As the understanding of fungal PJI evolves, continued research and clinical data collection remain crucial for improving patient outcomes in the management of these complex cases.
Prosthetic joint infection (PJI) occurs in about 1% to 2% of joint replacements. 1 Risk factors include immunosuppression, diabetes, chronic illnesses, and prolonged operative time.2 Bacterial infections constitute most of these infections, while fungal pathogens account for about 1%. Candida (C.) species, predominantly C. albicans, are responsible for most PJIs.1,3 In contrast, C. glabrata is a rare cause of fungal PJI, with only 18 PJI cases currently reported in the literature.4 C. glabrata PJI occurs more frequently among immunosuppressed patients and is associated with a higher treatment failure rate despite antifungal therapy.5 Treatment of fungal PJI is often complicated, involving multiple surgical debridements, prolonged antifungal therapy, and in some cases, prosthesis removal.6 However, given the rarity of C. glabrata as a PJI pathogen, no standardized treatment guidelines exist, leading to potential delays in diagnosis and tailored treatment.7,8
CASE PRESENTATION
A male Vietnam veteran aged 75 years presented to the emergency department in July 2023 with a fluid collection over his left hip surgical incision site. The patient had a complex medical history that included chronic kidney disease, well-controlled type 2 diabetes, hypertension, and osteoarthritis. His history was further complicated by nonalcoholic steatohepatitis with hepatocellular carcinoma that was treated with transarterial radioembolization and yttrium-90. The patient had undergone a left total hip arthroplasty in 1996 and subsequent open reduction and internal fixation about 9 months prior to his presentation. The patient reported the fluid had been present for about 6 weeks, while he received outpatient monitoring by the orthopedic surgery service. He sought emergency care after noting a moderate amount of purulent discharge on his clothing originating from his hip. In the week prior to admission, the patient observed progressive erythema, warmth, and tenderness over the incision site. Despite these symptoms, the patient remained ambulatory and able to walk long distances with the use of an assistive device.
Upon presentation, the patient was afebrile and normotensive. Laboratory testing revealed an elevated erythrocyte sedimentation rate of 77 mm/h (reference range, 0-20 mm/h) and a C-reactive protein of 9.8 mg/L (reference range, 0-2.5 mg/L), suggesting an underlying infectious process. A physical examination revealed a well-healed incision over the left hip with a poorly defined area of fluctuance and evidence of wound dehiscence. The left lower extremity was swollen with 2+ pitting edema, but tenderness was localized to the incision site. Magnetic resonance imaging of the left hip revealed a multiloculated fluid collection abutting the left greater trochanter with extension to the skin surface and inferior extension along the entire length of the surgical fixation hardware (Figure).
Upon admission, orthopedic surgery performed a bedside aspiration of the fluid collection. Samples were sent for analysis, including cell count and bacterial and fungal cultures. Initial blood cultures were sterile. Due to concerns for a bacterial infection, the patient was started on empiric intravenous (IV) ceftriaxone 2 g/day and IV vancomycin 1250 mg/day. Synovial fluid analysis revealed an elevated white blood cell count of 45,000/ìL, but bacterial cultures were negative. Five days after admission, the fungal culture from the left hip wound was notable for presence of C. glabrata, prompting an infectious diseases (ID) consultation. IV micafungin 100 mg/day was initiated as empiric antifungal therapy.
ID and orthopedic surgery teams determined that a combined medical and surgical approach would be best suited for infection control. They proposed 2 main approaches: complete hardware replacement with washout, which carried a higher morbidity risk but a better chance of infection resolution, or partial hardware replacement with washout, which was associated with a lower morbidity risk but a higher risk of infection persistence and recurrence. This decision was particularly challenging for the patient, who prioritized maintaining his functional status, including his ability to continue dancing for pleasure. The patient opted for a more conservative approach, electing to proceed with antifungal therapy and debridement while retaining the prosthetic joint.
After 11 days of hospitalization, the patient was discharged with a peripherally inserted central catheter for long-term antifungal infusions of micafungin 150 mg/day at home. Fungal sensitivity test results several days after discharge confirmed susceptibility to micafungin.
About 2 weeks after discharge, the patient underwent debridement and implant retention (DAIR). Wound cultures were positive for C. glabrata, Enterococcus faecalis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. Based on susceptibilities, he completed a 2-month course of IV micafungin 150 mg daily and daptomycin 750 mg daily, followed by an oral suppressive regimen consisting of doxycycline 100 mg twice daily, amoxicillin-clavulanate 2 g twice daily, and fluconazole initially 800 mg daily adjusted to 400 mg daily. The patient continued wound management with twice-daily dressing changes.
Nine months after DAIR, the patient remained on suppressive antifungal and antibacterial therapy. He continued to experience serous drainage from the wound, which greatly affected his quality of life. After discussion with his family and the orthopedic surgery team, he agreed to proceed with a 2-staged revision arthroplasty involving prosthetic explant and antibiotic spacer placement. However, the surgery was postponed due to findings of anemia (hemoglobin, 8.9 g/dL) and thrombocytopenia (platelet count, 73 x 103/λL). At the time of this report, the patient was being monitored closely with his multidisciplinary care team for the planned orthopedic procedure.
DISCUSSION
PJI is the most common cause of primary hip arthroplasty failure; however, fungal species only make up about 1% of PJIs.3,9-11 Patients are typically immunocompromised, undergoing antineoplastic therapies for malignancy, or have other comorbid conditions such as diabetes.12,13 C. glabrata presents a unique diagnostic and therapeutic challenge as it is not only rare but also notorious for its resistance to common antifungal agents. C. glabrata is known to develop multidrug resistance through the rapid accumulation of genomic mutations.14 Its propensity towards forming protective biofilm also arms it with intrinsic resistance to agents like fluconazole.15 Furthermore, based on a review of the available reports in the literature, C. glabrata PJIs are often insidious and present with symptoms closely mimicking those of bacterial PJIs, as it did in the patient in this case.16
Synovial fluid analysis, fungal cultures, and sensitivity testing are paramount for ensuring proper diagnosis for fungal PJI. The patient in this case was empirically treated with micafungin based on recommendations from the ID team. When the sensitivities results were reviewed, the same antifungal therapy was continued. Echinocandins have a favorable toxicity profile in long-term use, as well as efficacy against biofilm-producing organisms like C. glabrata.17,18
While there are a few cases citing DAIR as a feasible surgical strategy for treating fungal PJI, more recent studies have reported greater success with a 2-staged revision arthroplasty involving some combination of debridement, placement of antibiotic-loaded bone cement spacers, and partial or total exchange of the infected prosthetic joint.4,19-23 In this case, complete hardware replacement would have offered the patient the most favorable outlook for eliminating this fungal infection. However, given the patient’s advanced age, significant underlying comorbidities, and functional status, medical management with antifungal therapy and DAIR was favored.
Based on the discussion from the 6-month follow-up visit, the patient was experiencing progressive and persistent wound drainage and frequent dressing changes, highlighting the limitations of medical management for PJI in the setting of retained prosthesis. If the patient ultimately proceeds with a more invasive surgical intervention, another important consideration will be the likelihood of fungal PJI recurrence. At present, fungal PJI recurrence rates following antifungal and surgical treatment have been reported to range between 0% to 50%, which is too imprecise to be considered clinically useful.22-24
Given the ambiguity surrounding management guidelines and limited treatment options, it is crucial to emphasize the timeline of this patient’s clinical presentation and subsequent course of treatment. Upon presentation to the ED in late July, fungal PJI was considered less likely. Initial blood cultures from presentation were negative, which is common with PJIs. It was not until 5 days later that the left hip wound culture showed moderate growth of C. glabrata. Identifying a PJI is clinically challenging due to the lack of standardized diagnostic criteria. However, timely identification and diagnosis of fungal PJI with appropriate antifungal therapy, in patients with limited curative options due to comorbidities, can significantly improve quality of life and overall outcomes.25 Routine fungal and mycobacterial cultures are not currently recommended in PJI guidelines, but this case illustrates it is imperative in immunocompromised hosts.26
This case and the current paucity of similar cases in the literature stress the importance of clinicians publishing their experience in the management of fungal PJI. We strongly recommend that clinicians approach each suspected PJI with careful consideration of the patient’s unique risk factors, comorbidities, and goals of care, when deciding on a curative vs suppressive approach to therapy.
CONCLUSIONS
This case report highlights the importance of considering fungal pathogens for PJIs, especially in high-risk patients, the value of obtaining fungal cultures, the necessity of a multidisciplinary approach, the role of antifungal susceptibility testing, and consideration for the feasibility of a surgical intervention. It underscores the challenges in diagnosis and treatment of C. glabrata-associated PJI, emphasizing the importance of clinician experience-sharing in developing evidence-based management strategies. As the understanding of fungal PJI evolves, continued research and clinical data collection remain crucial for improving patient outcomes in the management of these complex cases.
- Osmon DR, Berbari EF, Berendt AR, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1-10. doi:10.1093/cid/cis966
- Eka A, Chen AF. Patient-related medical risk factors for periprosthetic joint infection of the hip and knee. Ann Transl Med. 2015;3(16):233. doi:10.3978/j.issn.2305-5839.2015.09.26
- Darouiche RO, Hamill RJ, Musher DM, Young EJ, Harris RL. Periprosthetic candidal infections following arthroplasty. Rev Infect Dis. 1989;11(1):89-96. doi:10.1093/clinids/11.1.89
- Koutserimpas C, Zervakis SG, Maraki S, et al. Non-albicans Candida prosthetic joint infections: a systematic review of treatment. World J Clin Cases. 2019;7(12):1430- 1443. doi:10.12998/wjcc.v7.i12.1430
- Fidel PL Jr, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80-96. doi:10.1128/CMR.12.1.80
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Lee YR, Kim HJ, Lee EJ, Sohn JW, Kim MJ, Yoon YK. Prosthetic joint infections caused by candida species: a systematic review and a case series. Mycopathologia. 2019;184(1):23-33. doi:10.1007/s11046-018-0286-1
- Herndon CL, Rowe TM, Metcalf RW, et al. Treatment outcomes of fungal periprosthetic joint infection. J Arthroplasty. 2023;38(11):2436-2440.e1. doi:10.1016/j.arth.2023.05.009
- Delaunay C, Hamadouche M, Girard J, Duhamel A; SoFCOT. What are the causes for failures of primary hip arthroplasties in France? Clin Orthop Relat Res. 2013;471(12): 3863-3869. doi:10.1007/s11999-013-2935-5
- Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91(1): 128-133. doi:10.2106/JBJS.H.00155
- Furnes O, Lie SA, Espehaug B, Vollset SE, Engesaeter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586. doi:10.1302/0301-620x.83b4.11223
- Gonzalez MR, Bedi ADS, Karczewski D, Lozano-Calderon SA. Treatment and outcomes of fungal prosthetic joint infections: a systematic review of 225 cases. J Arthroplasty. 2023;38(11):2464-2471.e1. doi:10.1016/j.arth.2023.05.003
- Gonzalez MR, Pretell-Mazzini J, Lozano-Calderon SA. Risk factors and management of prosthetic joint infections in megaprostheses-a review of the literature. Antibiotics (Basel). 2023;13(1):25. doi:10.3390/antibiotics13010025
- Biswas C, Chen SC, Halliday C, et al. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect. 2017;23(9):676.e7-676.e10. doi:10.1016/j.cmi.2017.03.014
- Hassan Y, Chew SY, Than LTL. Candida glabrata: pathogenicity and resistance mechanisms for adaptation and survival. J Fungi (Basel). 2021;7(8):667. doi:10.3390/jof7080667
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Pierce CG, Uppuluri P, Tristan AR, et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3(9):1494-1500. doi:10.1038/nport.2008.141
- Koutserimpas C, Samonis G, Velivassakis E, Iliopoulou- Kosmadaki S, Kontakis G, Kofteridis DP. Candida glabrata prosthetic joint infection, successfully treated with anidulafungin: a case report and review of the literature. Mycoses. 2018;61(4):266-269. doi:10.1111/myc.12736
- Brooks DH, Pupparo F. Successful salvage of a primary total knee arthroplasty infected with Candida parapsilosis. J Arthroplasty. 1998;13(6):707-712. doi:10.1016/s0883-5403(98)80017-x
- Merrer J, Dupont B, Nieszkowska A, De Jonghe B, Outin H. Candida albicans prosthetic arthritis treated with fluconazole alone. J Infect. 2001;42(3):208-209. doi:10.1053/jinf.2001.0819
- Koutserimpas C, Naoum S, Alpantaki K, et al. Fungal prosthetic joint infection in revised knee arthroplasty: an orthopaedic surgeon’s nightmare. Diagnostics (Basel). 2022;12(7):1606. doi:10.3390/diagnostics12071606
- Gao Z, Li X, Du Y, Peng Y, Wu W, Zhou Y. Success rate of fungal peri-prosthetic joint infection treated by 2-stage revision and potential risk factors of treatment failure: a retrospective study. Med Sci Monit. 2018;24:5549-5557. doi:10.12659/MSM.909168
- Hwang BH, Yoon JY, Nam CH, et al. Fungal periprosthetic joint infection after primary total knee replacement. J Bone Joint Surg Br. 2012;94(5):656-659. doi:10.1302/0301-620X.94B5.28125
- Ueng SW, Lee CY, Hu CC, Hsieh PH, Chang Y. What is the success of treatment of hip and knee candidal periprosthetic joint infection? Clin Orthop Relat Res. 2013;471(9):3002-3009. doi:10.1007/s11999-013-3007-6
- Nodzo, Scott R. MD; Bauer, Thomas MD, PhD; Pottinger, et al. Conventional diagnostic challenges in periprosthetic joint infection. J Am Acad Orthop Surg. 2015;23 Suppl:S18-S25. doi:10.5435/JAAOS-D-14-00385
- American Academy of Orthopaedic Surgeons. Diagnosis and prevention of periprosthetic joint infections. March 11, 2019. Accessed February 5, 2025. https://www.aaos.org/pjicpg
- Osmon DR, Berbari EF, Berendt AR, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1-10. doi:10.1093/cid/cis966
- Eka A, Chen AF. Patient-related medical risk factors for periprosthetic joint infection of the hip and knee. Ann Transl Med. 2015;3(16):233. doi:10.3978/j.issn.2305-5839.2015.09.26
- Darouiche RO, Hamill RJ, Musher DM, Young EJ, Harris RL. Periprosthetic candidal infections following arthroplasty. Rev Infect Dis. 1989;11(1):89-96. doi:10.1093/clinids/11.1.89
- Koutserimpas C, Zervakis SG, Maraki S, et al. Non-albicans Candida prosthetic joint infections: a systematic review of treatment. World J Clin Cases. 2019;7(12):1430- 1443. doi:10.12998/wjcc.v7.i12.1430
- Fidel PL Jr, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80-96. doi:10.1128/CMR.12.1.80
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Lee YR, Kim HJ, Lee EJ, Sohn JW, Kim MJ, Yoon YK. Prosthetic joint infections caused by candida species: a systematic review and a case series. Mycopathologia. 2019;184(1):23-33. doi:10.1007/s11046-018-0286-1
- Herndon CL, Rowe TM, Metcalf RW, et al. Treatment outcomes of fungal periprosthetic joint infection. J Arthroplasty. 2023;38(11):2436-2440.e1. doi:10.1016/j.arth.2023.05.009
- Delaunay C, Hamadouche M, Girard J, Duhamel A; SoFCOT. What are the causes for failures of primary hip arthroplasties in France? Clin Orthop Relat Res. 2013;471(12): 3863-3869. doi:10.1007/s11999-013-2935-5
- Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91(1): 128-133. doi:10.2106/JBJS.H.00155
- Furnes O, Lie SA, Espehaug B, Vollset SE, Engesaeter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586. doi:10.1302/0301-620x.83b4.11223
- Gonzalez MR, Bedi ADS, Karczewski D, Lozano-Calderon SA. Treatment and outcomes of fungal prosthetic joint infections: a systematic review of 225 cases. J Arthroplasty. 2023;38(11):2464-2471.e1. doi:10.1016/j.arth.2023.05.003
- Gonzalez MR, Pretell-Mazzini J, Lozano-Calderon SA. Risk factors and management of prosthetic joint infections in megaprostheses-a review of the literature. Antibiotics (Basel). 2023;13(1):25. doi:10.3390/antibiotics13010025
- Biswas C, Chen SC, Halliday C, et al. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect. 2017;23(9):676.e7-676.e10. doi:10.1016/j.cmi.2017.03.014
- Hassan Y, Chew SY, Than LTL. Candida glabrata: pathogenicity and resistance mechanisms for adaptation and survival. J Fungi (Basel). 2021;7(8):667. doi:10.3390/jof7080667
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Pierce CG, Uppuluri P, Tristan AR, et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3(9):1494-1500. doi:10.1038/nport.2008.141
- Koutserimpas C, Samonis G, Velivassakis E, Iliopoulou- Kosmadaki S, Kontakis G, Kofteridis DP. Candida glabrata prosthetic joint infection, successfully treated with anidulafungin: a case report and review of the literature. Mycoses. 2018;61(4):266-269. doi:10.1111/myc.12736
- Brooks DH, Pupparo F. Successful salvage of a primary total knee arthroplasty infected with Candida parapsilosis. J Arthroplasty. 1998;13(6):707-712. doi:10.1016/s0883-5403(98)80017-x
- Merrer J, Dupont B, Nieszkowska A, De Jonghe B, Outin H. Candida albicans prosthetic arthritis treated with fluconazole alone. J Infect. 2001;42(3):208-209. doi:10.1053/jinf.2001.0819
- Koutserimpas C, Naoum S, Alpantaki K, et al. Fungal prosthetic joint infection in revised knee arthroplasty: an orthopaedic surgeon’s nightmare. Diagnostics (Basel). 2022;12(7):1606. doi:10.3390/diagnostics12071606
- Gao Z, Li X, Du Y, Peng Y, Wu W, Zhou Y. Success rate of fungal peri-prosthetic joint infection treated by 2-stage revision and potential risk factors of treatment failure: a retrospective study. Med Sci Monit. 2018;24:5549-5557. doi:10.12659/MSM.909168
- Hwang BH, Yoon JY, Nam CH, et al. Fungal periprosthetic joint infection after primary total knee replacement. J Bone Joint Surg Br. 2012;94(5):656-659. doi:10.1302/0301-620X.94B5.28125
- Ueng SW, Lee CY, Hu CC, Hsieh PH, Chang Y. What is the success of treatment of hip and knee candidal periprosthetic joint infection? Clin Orthop Relat Res. 2013;471(9):3002-3009. doi:10.1007/s11999-013-3007-6
- Nodzo, Scott R. MD; Bauer, Thomas MD, PhD; Pottinger, et al. Conventional diagnostic challenges in periprosthetic joint infection. J Am Acad Orthop Surg. 2015;23 Suppl:S18-S25. doi:10.5435/JAAOS-D-14-00385
- American Academy of Orthopaedic Surgeons. Diagnosis and prevention of periprosthetic joint infections. March 11, 2019. Accessed February 5, 2025. https://www.aaos.org/pjicpg
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
Violaceous Papules on Face
Violaceous Papules on Face
Discussion
The patient’s violaceous papule on the nose with an apple jelly appearance is consistent with lupus pernio—a cutaneous form of sarcoidosis associated with respiratory involvement. Lupus pernio disproportionately affects African Americans, which further supports this diagnosis.1 Lupus pernio is characterized by violaceous, indurated plaques predominantly on the face. It has a strong association with systemic sarcoidosis and often involves the lungs and other organs, as seen in this case. The laboratory results support this diagnosis. Hypercalcemia is a common systemic manifestation of sarcoidosis due to increased production of 1,25-dihydroxyvitamin D by activated macrophages with granulomas.2 Elevated chitotriosidase, an enzyme produced by macrophages, is another biomarker of sarcoidosis reflecting granuloma burden.3
The differential diagnoses included Langerhans cell histiocytosis (LCH), discoid lupus erythematosus, granulomatosis with polyangiitis, and granuloma annulare. However, these diagnoses did not fully align with the entirety of the patient’s clinical presentation and laboratory findings. LCH is a rare neoplastic disorder characterized by the abnormal proliferation and accumulation of Langerhans cells, a type of dendritic cell involved in immune response, in various tissues such as the skin and bone. Dermatologic findings in LCH include brown/purple papules and an erythematous papular rash rather than the violaceous plaques/papules in lupus pernio. LCH can have lung involvement; it typically presents with nodular or cystic changes in the upper lobes as opposed to the bibasilar opacities seen in this case.
Discoid lupus erythematosus presents with characteristic round, erythematous, scaly plaques on the cheeks, scalp, and ears. This is different from the apple jelly appearance seen in this case and does not present with systemic granulomatous involvement.
Typical manifestations of granulomatosis with polyangiitis, formerly known as Wegener’s granulomatosis, include renal disease, upper and lower respiratory tract involvement, or necrotizing vasculitis. Cutaneous manifestions of granulomatosis with polyangiitis typically include purpura or ulcers rather than the violaceous plaques seen in lupus pernio. Patients with granulomatosis with polyangiitis would also present with nonspecific systemic symptoms such as fever, weight loss, and malaise, which are not depicted in this case.4
Granuloma annulare is a benign condition that often presents with annular plaques that are skin-colored rather than violaceous. These plaques are often found on the hands and feet rather than the face. This condition also lacks the systemic manifestations seen in this case.
In primary care, encountering violaceous papule and plaques on the face, especially on the nasal alae or ear, should be concerning for possible lupus pernio, particularly in high-risk populations such as young African Americans. These lesions generally have a more indurated “deep” and “doughy” appearance and can result in scarring, distinguishing them from other types of cutaneous sarcoidosis. An apple jelly appearance seen on diascopy with a glass slide can further support the diagnosis. While the lesions are typically asymptomatic, patients may be concerned about potential cosmetic disfigurement. Given the potential for scarring and the association with systemic sarcoidosis, a dermatology referral is recommended for further evaluation and management.
A detailed patient history, physical examination, and laboratory exams are essential to accurately diagnose lupus pernio. Biopsy of a skin lesion, serum markers, and imaging studies were utilized to help assess systemic involvement and further confirm diagnosis in this patient. Following the diagnosis, the patient was started on his current regimen of prednisone, methotrexate, and hydroxychloroquine, which are standard therapies for managing both cutaneous and systemic sarcoidosis.
This case shows the importance of recognizing lupus pernio, a distinct form of cutaneous sarcoidosis, in patients presenting with characteristic skin lesions and systemic involvement. It is essential to differentiate it from other granulomatous and inflammatory skin conditions to ensure appropriate management and prevent complications.
Federal Practitioner thanks the Association of Military Dermatologists (militaryderm.org) for their assistance in developing the Image Challenge. Submissions based on photographs, radiography, or any other visual medium are welcomed.
- Lai J, Almazan E, Le T, Taylor MT, Alhariri J, Kwatra SG. Demographics, cutaneous manifestations, and comorbidities associated with progressive cutaneous sarcoidosis: a retrospective cohort study. Medicines (Basel). 2023;10(10):57. doi:10.3390/medicines10100057
- Burke RR, Rybicki BA, Rao DS. Calcium and vitamin D in sarcoidosis: how to assess and manage. Semin Respir Crit Care Med. 2010;31(4):474-484. doi:10.1055/s-0030-1262215
- Bargagli E, Maggiorelli C, Rottoli P. Human chitotriosidase: a potential new marker of sarcoidosis severity. Respiration. 2008;76(2):234-238. doi:10.1159/000134009
- Kubaisi B, Abu Samra K, Foster CS. Granulomatosis with polyangiitis (Wegener’s disease): An updated review of ocular disease manifestations. Intractable Rare Dis Res. 2016;5(2):61-69. doi:10.5582/irdr.2016.01014
Discussion
The patient’s violaceous papule on the nose with an apple jelly appearance is consistent with lupus pernio—a cutaneous form of sarcoidosis associated with respiratory involvement. Lupus pernio disproportionately affects African Americans, which further supports this diagnosis.1 Lupus pernio is characterized by violaceous, indurated plaques predominantly on the face. It has a strong association with systemic sarcoidosis and often involves the lungs and other organs, as seen in this case. The laboratory results support this diagnosis. Hypercalcemia is a common systemic manifestation of sarcoidosis due to increased production of 1,25-dihydroxyvitamin D by activated macrophages with granulomas.2 Elevated chitotriosidase, an enzyme produced by macrophages, is another biomarker of sarcoidosis reflecting granuloma burden.3
The differential diagnoses included Langerhans cell histiocytosis (LCH), discoid lupus erythematosus, granulomatosis with polyangiitis, and granuloma annulare. However, these diagnoses did not fully align with the entirety of the patient’s clinical presentation and laboratory findings. LCH is a rare neoplastic disorder characterized by the abnormal proliferation and accumulation of Langerhans cells, a type of dendritic cell involved in immune response, in various tissues such as the skin and bone. Dermatologic findings in LCH include brown/purple papules and an erythematous papular rash rather than the violaceous plaques/papules in lupus pernio. LCH can have lung involvement; it typically presents with nodular or cystic changes in the upper lobes as opposed to the bibasilar opacities seen in this case.
Discoid lupus erythematosus presents with characteristic round, erythematous, scaly plaques on the cheeks, scalp, and ears. This is different from the apple jelly appearance seen in this case and does not present with systemic granulomatous involvement.
Typical manifestations of granulomatosis with polyangiitis, formerly known as Wegener’s granulomatosis, include renal disease, upper and lower respiratory tract involvement, or necrotizing vasculitis. Cutaneous manifestions of granulomatosis with polyangiitis typically include purpura or ulcers rather than the violaceous plaques seen in lupus pernio. Patients with granulomatosis with polyangiitis would also present with nonspecific systemic symptoms such as fever, weight loss, and malaise, which are not depicted in this case.4
Granuloma annulare is a benign condition that often presents with annular plaques that are skin-colored rather than violaceous. These plaques are often found on the hands and feet rather than the face. This condition also lacks the systemic manifestations seen in this case.
In primary care, encountering violaceous papule and plaques on the face, especially on the nasal alae or ear, should be concerning for possible lupus pernio, particularly in high-risk populations such as young African Americans. These lesions generally have a more indurated “deep” and “doughy” appearance and can result in scarring, distinguishing them from other types of cutaneous sarcoidosis. An apple jelly appearance seen on diascopy with a glass slide can further support the diagnosis. While the lesions are typically asymptomatic, patients may be concerned about potential cosmetic disfigurement. Given the potential for scarring and the association with systemic sarcoidosis, a dermatology referral is recommended for further evaluation and management.
A detailed patient history, physical examination, and laboratory exams are essential to accurately diagnose lupus pernio. Biopsy of a skin lesion, serum markers, and imaging studies were utilized to help assess systemic involvement and further confirm diagnosis in this patient. Following the diagnosis, the patient was started on his current regimen of prednisone, methotrexate, and hydroxychloroquine, which are standard therapies for managing both cutaneous and systemic sarcoidosis.
This case shows the importance of recognizing lupus pernio, a distinct form of cutaneous sarcoidosis, in patients presenting with characteristic skin lesions and systemic involvement. It is essential to differentiate it from other granulomatous and inflammatory skin conditions to ensure appropriate management and prevent complications.
Federal Practitioner thanks the Association of Military Dermatologists (militaryderm.org) for their assistance in developing the Image Challenge. Submissions based on photographs, radiography, or any other visual medium are welcomed.
Discussion
The patient’s violaceous papule on the nose with an apple jelly appearance is consistent with lupus pernio—a cutaneous form of sarcoidosis associated with respiratory involvement. Lupus pernio disproportionately affects African Americans, which further supports this diagnosis.1 Lupus pernio is characterized by violaceous, indurated plaques predominantly on the face. It has a strong association with systemic sarcoidosis and often involves the lungs and other organs, as seen in this case. The laboratory results support this diagnosis. Hypercalcemia is a common systemic manifestation of sarcoidosis due to increased production of 1,25-dihydroxyvitamin D by activated macrophages with granulomas.2 Elevated chitotriosidase, an enzyme produced by macrophages, is another biomarker of sarcoidosis reflecting granuloma burden.3
The differential diagnoses included Langerhans cell histiocytosis (LCH), discoid lupus erythematosus, granulomatosis with polyangiitis, and granuloma annulare. However, these diagnoses did not fully align with the entirety of the patient’s clinical presentation and laboratory findings. LCH is a rare neoplastic disorder characterized by the abnormal proliferation and accumulation of Langerhans cells, a type of dendritic cell involved in immune response, in various tissues such as the skin and bone. Dermatologic findings in LCH include brown/purple papules and an erythematous papular rash rather than the violaceous plaques/papules in lupus pernio. LCH can have lung involvement; it typically presents with nodular or cystic changes in the upper lobes as opposed to the bibasilar opacities seen in this case.
Discoid lupus erythematosus presents with characteristic round, erythematous, scaly plaques on the cheeks, scalp, and ears. This is different from the apple jelly appearance seen in this case and does not present with systemic granulomatous involvement.
Typical manifestations of granulomatosis with polyangiitis, formerly known as Wegener’s granulomatosis, include renal disease, upper and lower respiratory tract involvement, or necrotizing vasculitis. Cutaneous manifestions of granulomatosis with polyangiitis typically include purpura or ulcers rather than the violaceous plaques seen in lupus pernio. Patients with granulomatosis with polyangiitis would also present with nonspecific systemic symptoms such as fever, weight loss, and malaise, which are not depicted in this case.4
Granuloma annulare is a benign condition that often presents with annular plaques that are skin-colored rather than violaceous. These plaques are often found on the hands and feet rather than the face. This condition also lacks the systemic manifestations seen in this case.
In primary care, encountering violaceous papule and plaques on the face, especially on the nasal alae or ear, should be concerning for possible lupus pernio, particularly in high-risk populations such as young African Americans. These lesions generally have a more indurated “deep” and “doughy” appearance and can result in scarring, distinguishing them from other types of cutaneous sarcoidosis. An apple jelly appearance seen on diascopy with a glass slide can further support the diagnosis. While the lesions are typically asymptomatic, patients may be concerned about potential cosmetic disfigurement. Given the potential for scarring and the association with systemic sarcoidosis, a dermatology referral is recommended for further evaluation and management.
A detailed patient history, physical examination, and laboratory exams are essential to accurately diagnose lupus pernio. Biopsy of a skin lesion, serum markers, and imaging studies were utilized to help assess systemic involvement and further confirm diagnosis in this patient. Following the diagnosis, the patient was started on his current regimen of prednisone, methotrexate, and hydroxychloroquine, which are standard therapies for managing both cutaneous and systemic sarcoidosis.
This case shows the importance of recognizing lupus pernio, a distinct form of cutaneous sarcoidosis, in patients presenting with characteristic skin lesions and systemic involvement. It is essential to differentiate it from other granulomatous and inflammatory skin conditions to ensure appropriate management and prevent complications.
Federal Practitioner thanks the Association of Military Dermatologists (militaryderm.org) for their assistance in developing the Image Challenge. Submissions based on photographs, radiography, or any other visual medium are welcomed.
- Lai J, Almazan E, Le T, Taylor MT, Alhariri J, Kwatra SG. Demographics, cutaneous manifestations, and comorbidities associated with progressive cutaneous sarcoidosis: a retrospective cohort study. Medicines (Basel). 2023;10(10):57. doi:10.3390/medicines10100057
- Burke RR, Rybicki BA, Rao DS. Calcium and vitamin D in sarcoidosis: how to assess and manage. Semin Respir Crit Care Med. 2010;31(4):474-484. doi:10.1055/s-0030-1262215
- Bargagli E, Maggiorelli C, Rottoli P. Human chitotriosidase: a potential new marker of sarcoidosis severity. Respiration. 2008;76(2):234-238. doi:10.1159/000134009
- Kubaisi B, Abu Samra K, Foster CS. Granulomatosis with polyangiitis (Wegener’s disease): An updated review of ocular disease manifestations. Intractable Rare Dis Res. 2016;5(2):61-69. doi:10.5582/irdr.2016.01014
- Lai J, Almazan E, Le T, Taylor MT, Alhariri J, Kwatra SG. Demographics, cutaneous manifestations, and comorbidities associated with progressive cutaneous sarcoidosis: a retrospective cohort study. Medicines (Basel). 2023;10(10):57. doi:10.3390/medicines10100057
- Burke RR, Rybicki BA, Rao DS. Calcium and vitamin D in sarcoidosis: how to assess and manage. Semin Respir Crit Care Med. 2010;31(4):474-484. doi:10.1055/s-0030-1262215
- Bargagli E, Maggiorelli C, Rottoli P. Human chitotriosidase: a potential new marker of sarcoidosis severity. Respiration. 2008;76(2):234-238. doi:10.1159/000134009
- Kubaisi B, Abu Samra K, Foster CS. Granulomatosis with polyangiitis (Wegener’s disease): An updated review of ocular disease manifestations. Intractable Rare Dis Res. 2016;5(2):61-69. doi:10.5582/irdr.2016.01014
Violaceous Papules on Face
Violaceous Papules on Face
A 40-year-old man with no significant medical history or comorbidities presented with a violaceous papule involving his nasal tip and scaly, violaceous plaques with associated alopecia involving his beard (Figure). Skin biopsy confirmed granulomatous dermatitis. Additional workup was notable for hypercalcemia (10.5 mg/dL; reference range, 8.4-10.2 mg/dL), elevated chitotriosidase (317 nmol/h/mL; reference range, < 150 nmol/h/mL), and bibasilar opacities with left perihilar consolidation on chest X-ray. The patient had a prolonged PR interval (207 ms; reference range, 120-200 ms) on electrocardiogram. A cardiac positron emission tomography revealed low level fluorodeoxyglucose uptake in the left ventricle. No ocular involvement was noted on evaluation by ophthalmology. The patient’s pharmacotherapy included prednisone 10 mg daily, methotrexate 7.5 mg weekly, and hydroxychloroquine 200 mg daily.
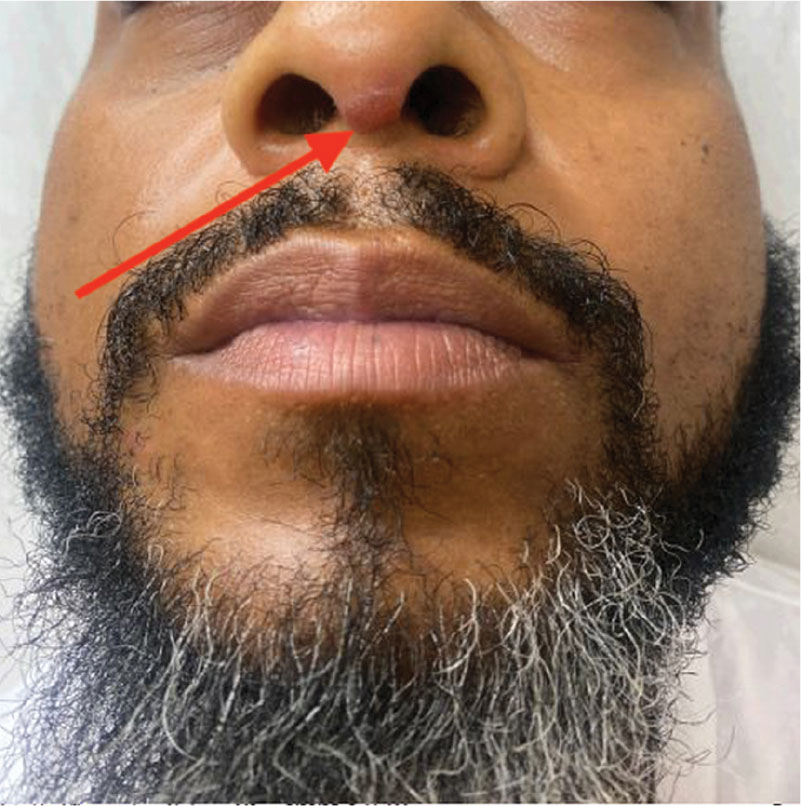
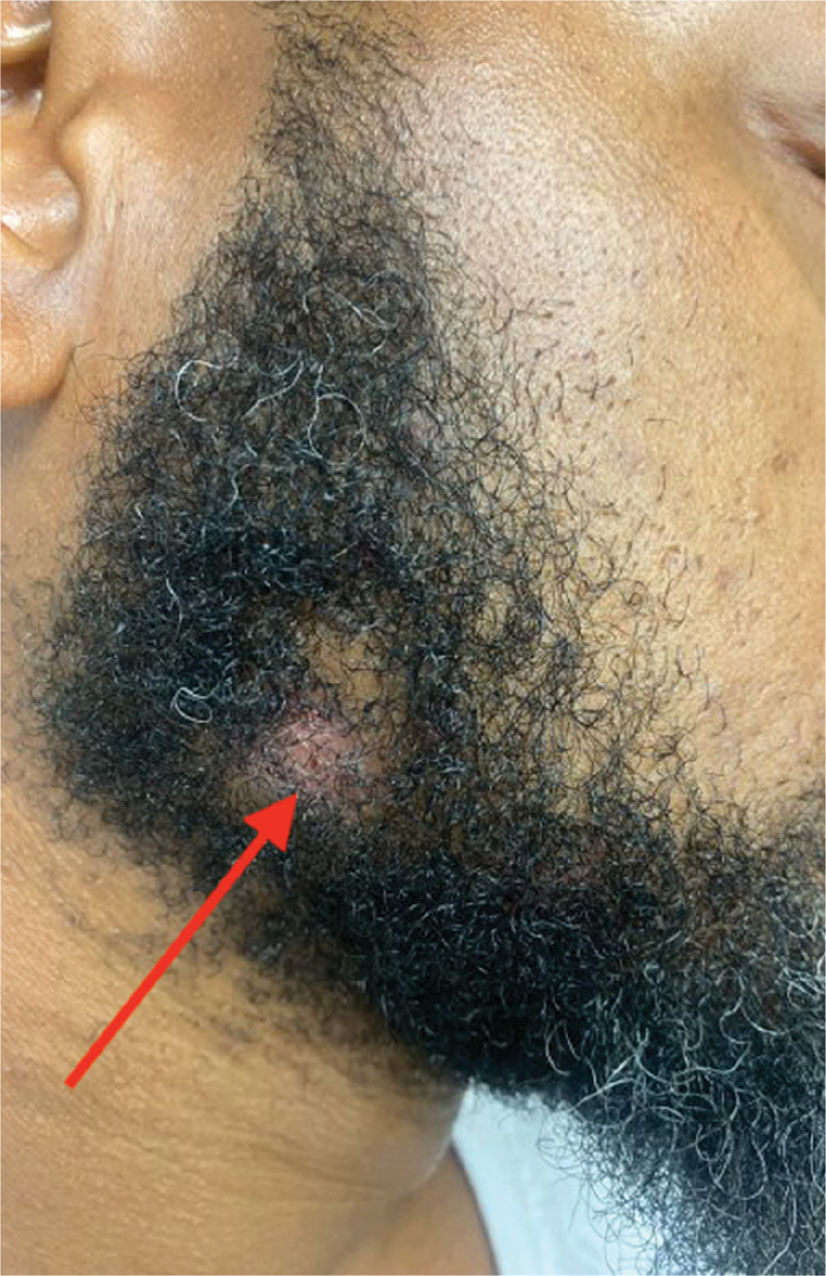

Dupilumab in the Treatment of Pemphigoid Gestationis
Dupilumab in the Treatment of Pemphigoid Gestationis
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
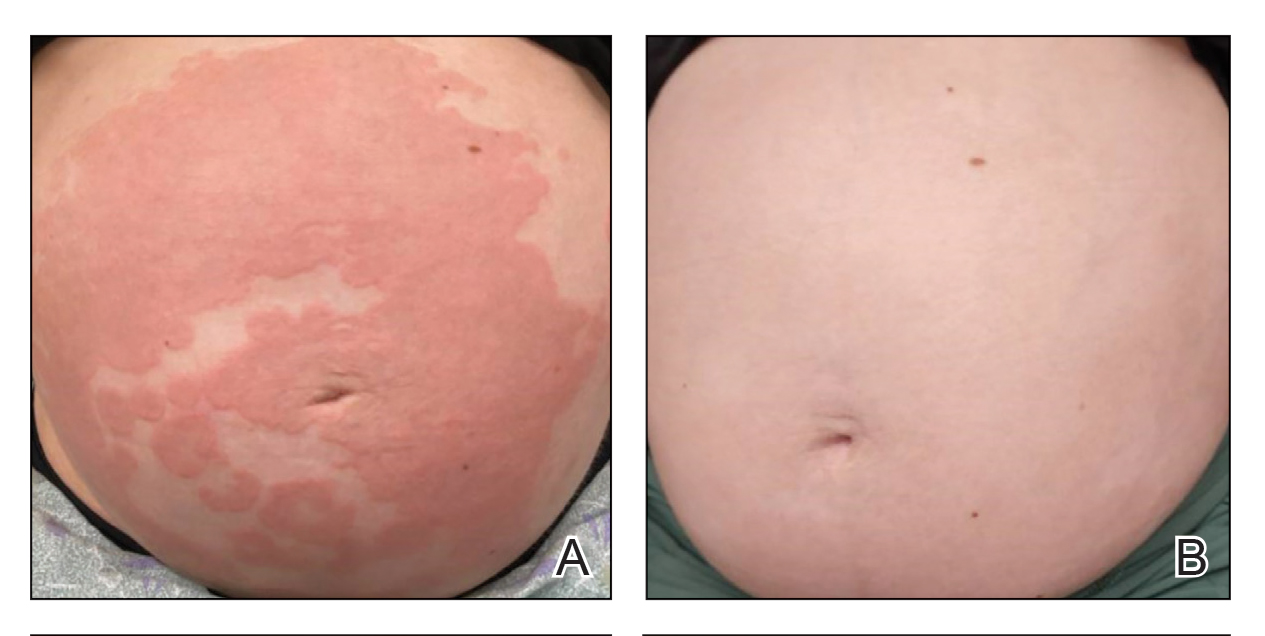
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
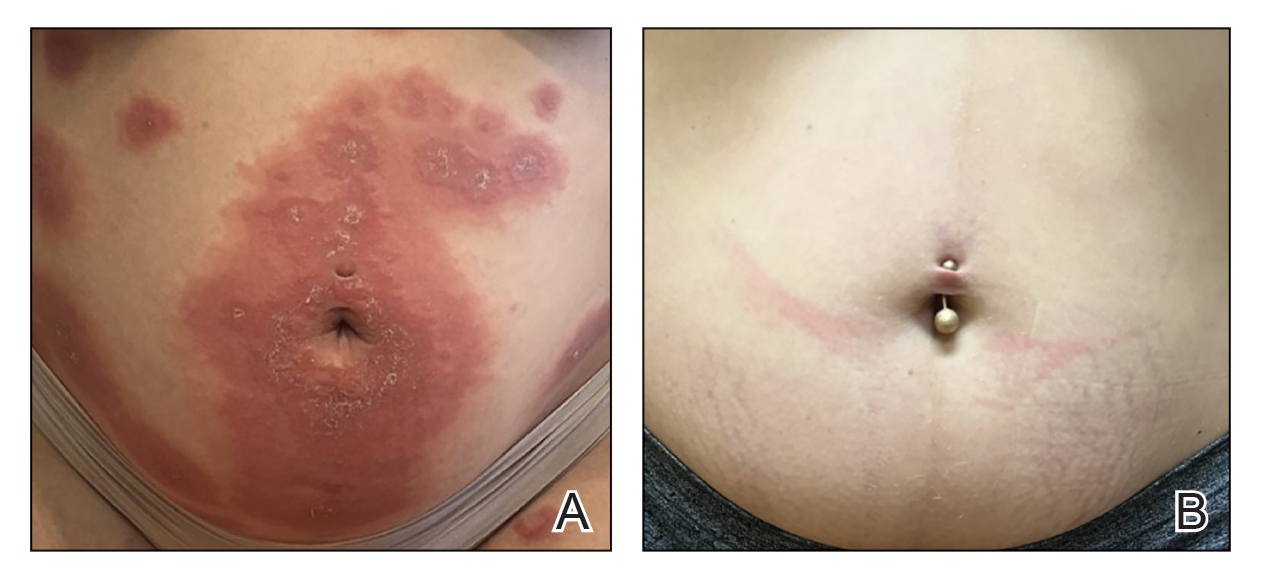
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
Dupilumab in the Treatment of Pemphigoid Gestationis
Dupilumab in the Treatment of Pemphigoid Gestationis
PRACTICE POINTS
- Dupilumab inhibits the IL-4Rα subunit, which is bound by IL‐4 and IL‐13, thereby reducing type 2 inflammation associated with pemphigoid gestationis (PG).
- Dupilumab may reduce the dose and duration of systemic corticosteroid therapy for PG, and its use in the second and third trimesters of pregnancy has been supported by emerging safety data.

A Veteran Presenting With Symptomatic Postprandial Episodes
A Veteran Presenting With Symptomatic Postprandial Episodes
Idiopathic postprandial syndrome (IPP), initially termed reactive hypoglycemia, presents with hypoglycemic-like symptoms in the absence of biochemical hypoglycemia and remains a diagnosis of exclusion. Its pathophysiology is poorly understood. The diagnosis requires thorough evaluation of cardiac, metabolic, neurologic, and gastrointestinal causes, as well as Whipple triad criteria. Dietary modifications, including reduced carbohydrate intake, increased protein and fiber, and frequent small meals, remain the cornerstone of IPP management. Continuous glucose monitoring (CGM) may be a useful adjunct in correlating symptoms with glucose trends, but its role is still evolving.
In the evaluation of patients with symptoms suggestive of hypoglycemia (Figure 1), patients should first be assessed for Whipple triad: symptoms consistent with hypoglycemia, blood glucose level < 55 mg/dL, and reversal of symptoms with glucose.1 Patients who meet Whipple triad criteria should be investigated to identify further etiologies of hypoglycemia. They may include insulinoma, medication-induced (insulin, sulfonylurea, meglitinide, or β blocker use), postbariatric surgery complications, noninsulinoma pancreatogenous hypoglycemia syndrome, ackee fruit consumption, or familial conditions.2 The presence of hypoglycemic symptoms in the postprandial or fasting state can provide valuable insights into underlying etiology.
Patients who do not meet Whipple triad criteria, but exhibit postprandial symptoms consistent with hypoglycemia, as in this case, present a diagnostic dilemma. IPP is defined as hypoglycemic symptoms occuring after carbohydrate ingestion without biochemical hypoglycemia. Initially termed reactive hypoglycemia, it was renamed in 1981to reflect the absence of low blood glucose levels.3
The understanding of this diagnosis has not significantly progressed since the 1980s. Its prevalence, incidence, risk factors, and societal burden remain unclear. IPP is a challenging diagnosis due to nonspecific symptoms that overlap with a myriad of conditions. These symptoms may include adrenergic symptoms such as diaphoresis, tremulousness, palpitations, anxiety, and hunger. Potentially severe neuroglycopenic symptoms, including weakness, dizziness, behavior changes, confusion, and coma, are not typically observed.4 Given that objective criteria are not well established, IPP remains a diagnosis of exclusion. It is imperative to rule out alternative etiologies, particularly cardiac, gastrointestinal, and neurologic causes.
CASE PRESENTATION
A male aged 41 years presented to primary care for evaluation of acute on chronic symptomatic postprandial episodes. He reported a history of symptomatic sinus bradycardia in the setting of sick sinus syndrome following dual-chamber pacemaker placement, posttraumatic stress disorder, and gastroesophageal reflux disease. He was a retired Navy sailor without any known occupational exposures who worked in the real estate industry. The patient reported feeling lightheaded, tremulous, and anxious most afternoons after lunch for several years. He also reported that meals heavy in carbohydrates exacerbated his symptoms, whereas skipping meals or lying down alleviated his symptoms. The patient also reported concomitant arm numbness, shortness of breath, palpitations, and nausea during these episodes. Review of systems was otherwise negative, including no weight changes, fever, chills, night sweats, chest pain, or syncope.
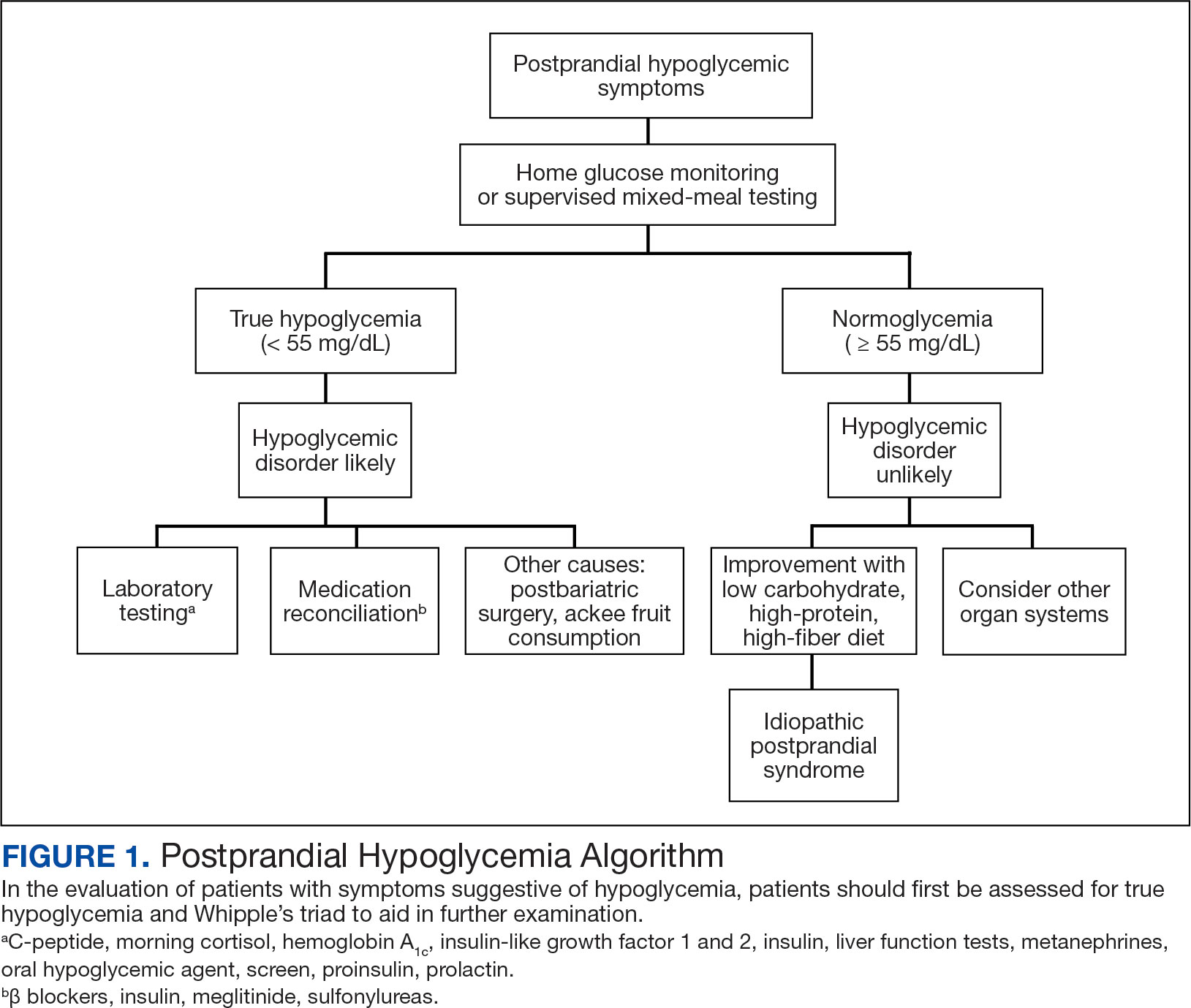
The patient’s medications included ferrous sulfate 325 mg once every other day, bupropion 200 mg once daily, metoprolol succinate 25 mg once daily, and as-needed lorazepam 1 mg once daily. The patient reported no current substance use but reported previous tobacco use 3 years prior (maximum 1 pack/week) and alcohol use 5 years prior (750 ml/day for 15 years). The patient did not exercise and typically ate oatmeal for breakfast, a sandwich or salad for lunch, and taquitos or salad for dinner, with snacks throughout the day. Notable family history included a maternal grandmother with colon cancer. The patient’s vital signs included a 36.8 °C temperature, heart rate 87 beats/min, 118/71 mm Hg blood pressure, oxygen saturation 98% on room air, 125.2 kg weight, and 38.5 body mass index. There were no orthostatic vital sign changes. A physical examination demonstrated obesity with an unremarkable cardiopulmonary and volume examination.
Additional testing included Gallium-68 dototate positron emission tomography/computed tomography, brain magnetic resonance imaging, echocardiogram, electromyogram, exercise tolerance test, Holter monitoring, invasive cardiopulmonary exercise testing, pacemaker interrogation, pulmonary function testing, stress echocardiogram, tilt table test, and venogram computed tomography of the chest, but the results were unremarkable (Appendix). His afternoon nonfasting glucose level was 138 mg/dL with a concurrent hemoglobin A1c of 5.2%. The patient had a fasting C-peptide level of 3.7 ng/mL (reference range 0.5-2.0 ng/mL), fasting insulin level 19.1 mIU/L (reference range < 25 mIU/L), and a fasting glucose level of 93 mg/dL (reference range 70-99 mg/dL). The patient’s urine 5-HIAA, plasma metanephrines, urine metanephrines, insulin-like growth factor 1, prolactin, corticotropin, fasting cortisol, and thyrotropin yielded results within reference ranges (Table). The veteran was prescribed a CGM, which demonstrated normal glucose levels (≥ 55 mg/dL) during symptomatic episodes (Figure 2).
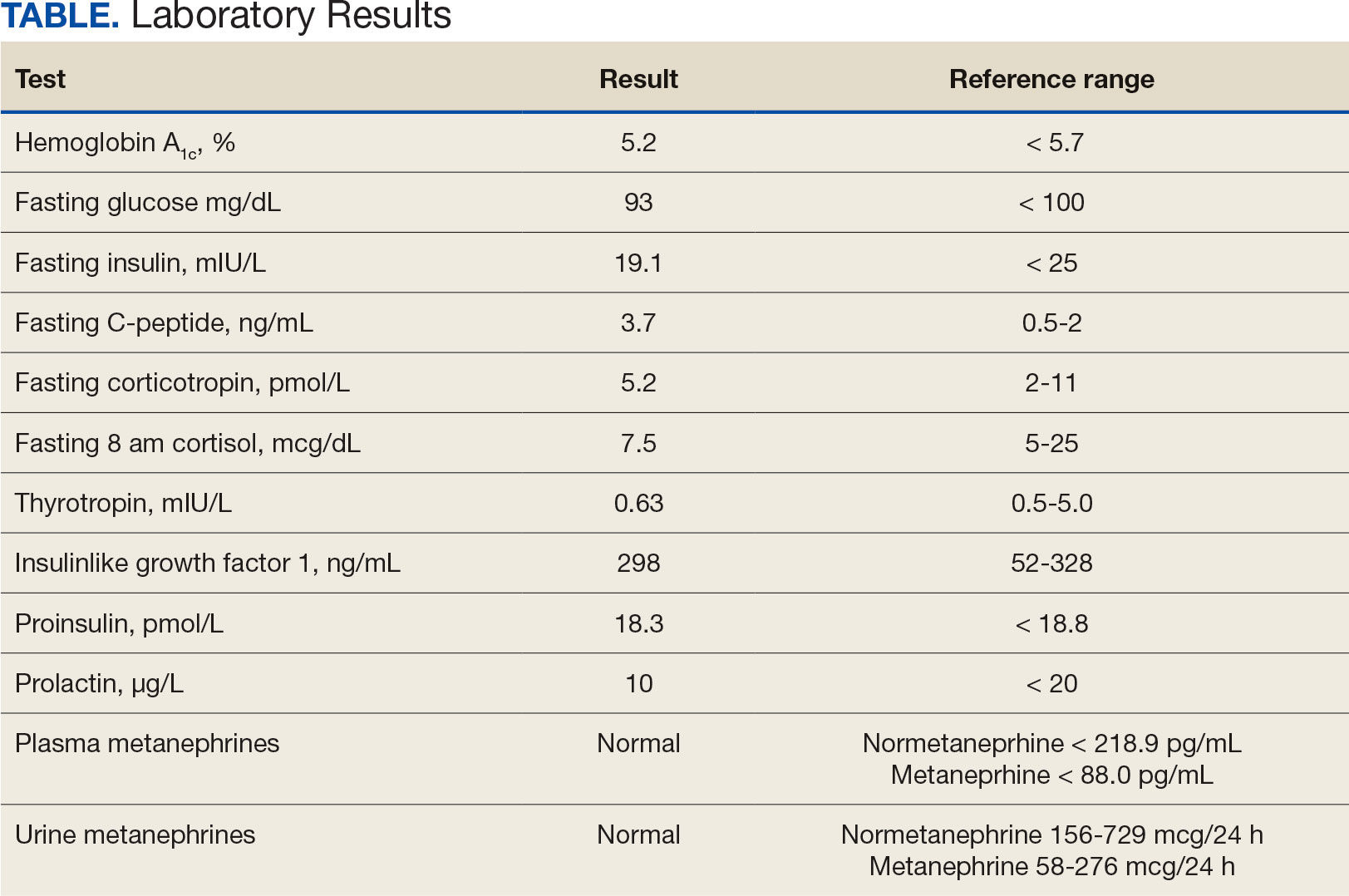
The patient was diagnosed with IPP given normoglycemia, exclusion of alternative diagnoses, and symptomatic improvement with dietary changes. He was referred to a nutritionist for a high-protein, high-fiber, and low-carbohydrate diet.
DISCUSSION
Seemingly simple diagnostic tools can lead to diagnostic pitfalls. Home glucose monitoring with the use of a standard glucometer during an episode is the typical first step in identifying hypoglycemia, as it is both pragmatic and accurate, with a mean absolute relative difference (MARD) of about 10% in hypoglycemic ranges.5 While the advent of CGM provides real-time data and can reveal clinically relevant fluctuations, it reveals mild hypoglycemia (54 to 70 mg/dL) of no clinical significance in a large proportion of individuals.
Additionally, CGM is less accurate than glucometers with a MARD of about 20% in hypoglycemia ranges.6 CGM technology, however, is rapidly evolving and undergoing further investigation for hypoglycemia detection. Therefore, CGM may be considered in select patients as prospective study results are established; the newest CGMs have MARDs very similar to fingerstick blood glucose data.7,8 In the patient described in this case, CGM helped corroborate the diagnosis, given that symptomatic episodes correlated with lower glucose levels. Provocative testing with oral glucose tolerance testing can frequently result in false positive hypoglycemic readings and is not recommended.9 Supervised mixed meal testing can also be used, which entails monitoring after consuming a mixed macronutrient meal. The test concludes after hypoglycemic symptoms develop or 5 hours elapse, whichever occurs first.1
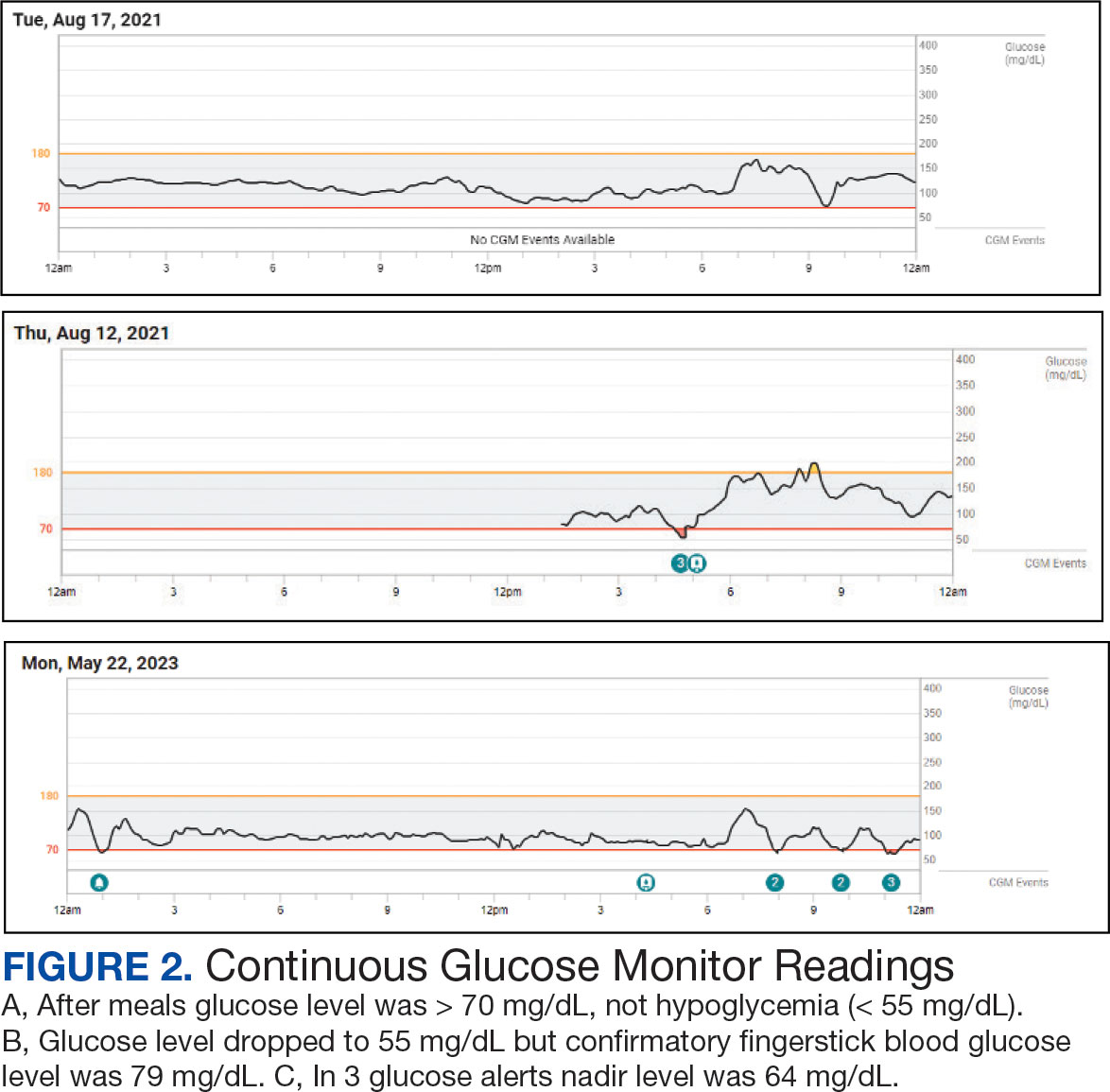
The pathophysiology of IPP is poorly understood. Proposed mechanisms include increased insulin sensitivity, increased adrenergic sensitivity, impaired glucagon regulation, emotional distress, insulin resistance, and increased glucagon-like peptide-1 production.10-13 Research suggests this may occur as pancreatic β cells fail in early type 2 diabetes mellitus, with diminished first-phase insulin release leading to an initial exuberant rise in blood glucose, an overshooting of the second phase of insulin secretion, and the feeling of the postprandial blood glucose falling, even though the final glucose level achieved is not truly low.13 There are contradictory studies in the literature demonstrating no association between insulin resistance and hypoglycemic symptoms.14 In 2022, Kosuda and colleagues looked at homeostatic model assessment for insulin resistance in patients with postprandial syndrome. They found that the patients were slightly insulin resistant but had normal or exaggerated insulin secretory capacity compared to an oral glucose load, whereas glucagon levels were robustly suppressed by a glucose load. The observed hormonal responses may result in the glycemic patterns and symptoms observed; further study is warranted to elucidate the mechanism.15
Dietary modification is the cornerstone treatment for postprandial syndrome, including reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals. There is also evidence that a Mediterranean diet may be beneficial for managing hypoglycemic symptoms.16 Furthermore, α-glucosidase inhibitors, whose mechanism of action delays the digestion of carbohydrates, have demonstrated promise. This medication class has demonstrated significance in raising postprandial glucose levels and alleviating hypoglycemic symptoms in patients with true postprandial hypoglycemia.17
CONCLUSIONS
IPP is a benign diagnosis encompassing hypoglycemic symptoms without biochemical hypoglycemia. It is not a true hypoglycemic disorder. IPP is challenging to diagnose, given that it is an interpretation of exclusion, supported by symptom improvement with dietary changes (ie, reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals). Supervised mixed meal testing or CGM can be used to assist with diagnosis. Even though CGM is undergoing further study in this patient population, it corroborated the diagnosis in the patient described in this case.
For hypoglycemic symptoms, physicians should first assess for evidence of Whipple triad to evaluate for true biochemical hypoglycemia. For true hypoglycemia (< 55 mg/dL), physicians may conduct an examination in conjunction with an endocrinologist. For normoglycemia (≥ 55 mg/dL), physicians should first exclude alternative etiologies (including cardiac and neurologic), and subsequently consider IPP.
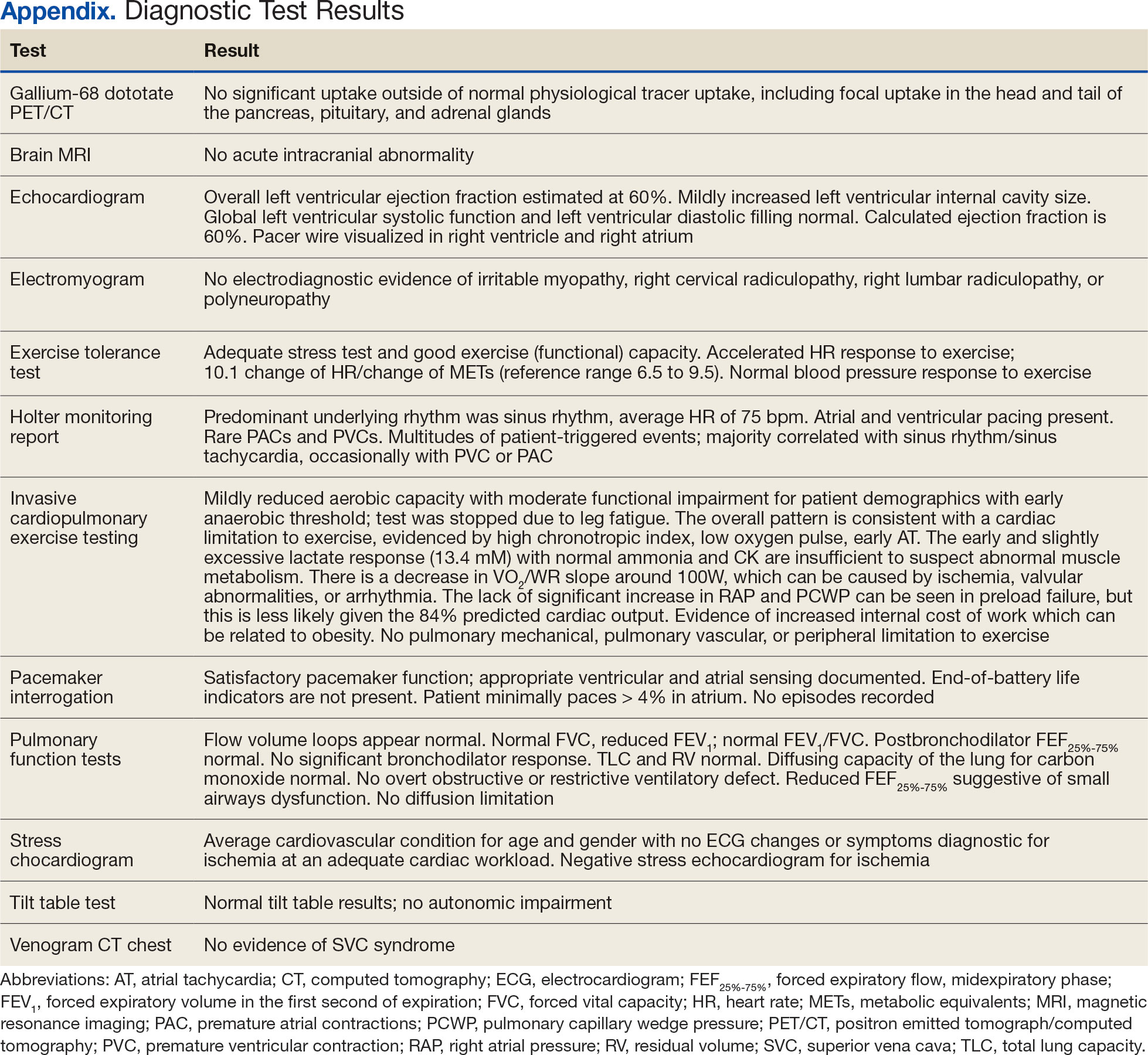
Bansal N, Weinstock RS. Non-Diabetic Hypoglycemia. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. MDText.com, Inc.; 2000.
Service FJ. Hypoglycemic disorders. New Engl J Med. 1995;332(17):1144-1152.doi:10.1056/NEJM199504273321707
Charles MA, Hofeldt F, Shackelford A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30(6):465-470.
Douillard C, Jannin A, Vantyghem MC. Rare causes of hypoglycemia in adults. Ann Endocrinol (Paris). 2020;81(2-3):110-117. doi:10.1016/j.ando.2020.04.003
Ekhlaspour L, Mondesir D, Lautsch N, et al. Comparative accuracy of 17 point-of-care glucose meters. J Diabetes Sci Technol. 2017;11(3):558-566. doi:10.1177/1932296816672237
Alitta Q, Grino M, Adjemout L, Langar A, Retornaz F, Oliver C. Overestimation of hypoglycemia diagnosis by FreeStyle Libre continuous glucose monitoring in long-term care home residents with diabetes. J Diabetes Sci Technol. 2018;12(3):727-728. doi:10.1177/1932296817747887
Mongraw-Chaffin M, Beavers DP, McClain DA. Hypoglycemic symptoms in the absence of diabetes: pilot evidence of clinical hypoglycemia in young women. J Clin Transl Endocrinol. 2019;18:100202. doi:10.1016/j.jcte.2019.100202
Shah VN, DuBose SN, Li Z, et al. Continuous glucose monitoring profiles in healthy nondiabetic participants: a multicenter prospective study. J Clin Endocrinol Metab. 2019;104(10):4356-4364. doi:10.1210/jc.2018-02763
Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2009;94(3):709-728. doi:10.1210/jc.2008-1410
Galati SJ, Rayfield EJ. Approach to the patient with postprandial hypoglycemia. Endocr Pract. 2014;20(4):331-340. doi:10.4158/EP13132.RA
Altuntas Y. Postprandial reactive hypoglycemia. Sisli Etfal Hastan Tip Bul. 2019;53(3):215-220.doi:10.14744/SEMB.2019.59455
HARRIS S. HYPERINSULINISM AND DYSINSULINISM. JAMA. 1924;83(10):729-733.doi:10.1001/jama.1924.02660100003002
Harris S. HYPERINSULINISM AND DYSINSULINISM (INSULOGENIC HYPOGLYCBMIA). Endocrinology. 1932;16(1):29-42. doi:10.1210/endo-16-1-29
Hall M, Walicka M, Panczyk M, Traczyk I. Metabolic parameters in patients with suspected reactive hypoglycemia. J Pers Med. 2021;11(4):276. doi:10.3390/jpm11040276
Kosuda M, Watanabe K, Koike M, et al. Glucagon response to glucose challenge in patients with idiopathic postprandial syndrome. J Nippon Med Sch. 2022;89(1):102-107. doi:10.1272/jnms.JNMS.2022_89-205
Hall M, Walicka M, Panczyk M, Traczyk I. Assessing long-term impact of dietary interventions on occurrence of symptoms consistent with hypoglycemia in patients without diabetes: a one-year follow-up study. Nutrients. 2022;14(3):497. doi:10.3390/nu14030497
Ozgen AG, Hamulu F, Bayraktar F, et al. Long-term treatment with acarbose for the treatment of reactive hypoglycemia. Eat Weight Disord. 1998;3(3):136-140. doi:10.1007/BF03340001
Idiopathic postprandial syndrome (IPP), initially termed reactive hypoglycemia, presents with hypoglycemic-like symptoms in the absence of biochemical hypoglycemia and remains a diagnosis of exclusion. Its pathophysiology is poorly understood. The diagnosis requires thorough evaluation of cardiac, metabolic, neurologic, and gastrointestinal causes, as well as Whipple triad criteria. Dietary modifications, including reduced carbohydrate intake, increased protein and fiber, and frequent small meals, remain the cornerstone of IPP management. Continuous glucose monitoring (CGM) may be a useful adjunct in correlating symptoms with glucose trends, but its role is still evolving.
In the evaluation of patients with symptoms suggestive of hypoglycemia (Figure 1), patients should first be assessed for Whipple triad: symptoms consistent with hypoglycemia, blood glucose level < 55 mg/dL, and reversal of symptoms with glucose.1 Patients who meet Whipple triad criteria should be investigated to identify further etiologies of hypoglycemia. They may include insulinoma, medication-induced (insulin, sulfonylurea, meglitinide, or β blocker use), postbariatric surgery complications, noninsulinoma pancreatogenous hypoglycemia syndrome, ackee fruit consumption, or familial conditions.2 The presence of hypoglycemic symptoms in the postprandial or fasting state can provide valuable insights into underlying etiology.
Patients who do not meet Whipple triad criteria, but exhibit postprandial symptoms consistent with hypoglycemia, as in this case, present a diagnostic dilemma. IPP is defined as hypoglycemic symptoms occuring after carbohydrate ingestion without biochemical hypoglycemia. Initially termed reactive hypoglycemia, it was renamed in 1981to reflect the absence of low blood glucose levels.3
The understanding of this diagnosis has not significantly progressed since the 1980s. Its prevalence, incidence, risk factors, and societal burden remain unclear. IPP is a challenging diagnosis due to nonspecific symptoms that overlap with a myriad of conditions. These symptoms may include adrenergic symptoms such as diaphoresis, tremulousness, palpitations, anxiety, and hunger. Potentially severe neuroglycopenic symptoms, including weakness, dizziness, behavior changes, confusion, and coma, are not typically observed.4 Given that objective criteria are not well established, IPP remains a diagnosis of exclusion. It is imperative to rule out alternative etiologies, particularly cardiac, gastrointestinal, and neurologic causes.
CASE PRESENTATION
A male aged 41 years presented to primary care for evaluation of acute on chronic symptomatic postprandial episodes. He reported a history of symptomatic sinus bradycardia in the setting of sick sinus syndrome following dual-chamber pacemaker placement, posttraumatic stress disorder, and gastroesophageal reflux disease. He was a retired Navy sailor without any known occupational exposures who worked in the real estate industry. The patient reported feeling lightheaded, tremulous, and anxious most afternoons after lunch for several years. He also reported that meals heavy in carbohydrates exacerbated his symptoms, whereas skipping meals or lying down alleviated his symptoms. The patient also reported concomitant arm numbness, shortness of breath, palpitations, and nausea during these episodes. Review of systems was otherwise negative, including no weight changes, fever, chills, night sweats, chest pain, or syncope.
The patient’s medications included ferrous sulfate 325 mg once every other day, bupropion 200 mg once daily, metoprolol succinate 25 mg once daily, and as-needed lorazepam 1 mg once daily. The patient reported no current substance use but reported previous tobacco use 3 years prior (maximum 1 pack/week) and alcohol use 5 years prior (750 ml/day for 15 years). The patient did not exercise and typically ate oatmeal for breakfast, a sandwich or salad for lunch, and taquitos or salad for dinner, with snacks throughout the day. Notable family history included a maternal grandmother with colon cancer. The patient’s vital signs included a 36.8 °C temperature, heart rate 87 beats/min, 118/71 mm Hg blood pressure, oxygen saturation 98% on room air, 125.2 kg weight, and 38.5 body mass index. There were no orthostatic vital sign changes. A physical examination demonstrated obesity with an unremarkable cardiopulmonary and volume examination.
Additional testing included Gallium-68 dototate positron emission tomography/computed tomography, brain magnetic resonance imaging, echocardiogram, electromyogram, exercise tolerance test, Holter monitoring, invasive cardiopulmonary exercise testing, pacemaker interrogation, pulmonary function testing, stress echocardiogram, tilt table test, and venogram computed tomography of the chest, but the results were unremarkable (Appendix). His afternoon nonfasting glucose level was 138 mg/dL with a concurrent hemoglobin A1c of 5.2%. The patient had a fasting C-peptide level of 3.7 ng/mL (reference range 0.5-2.0 ng/mL), fasting insulin level 19.1 mIU/L (reference range < 25 mIU/L), and a fasting glucose level of 93 mg/dL (reference range 70-99 mg/dL). The patient’s urine 5-HIAA, plasma metanephrines, urine metanephrines, insulin-like growth factor 1, prolactin, corticotropin, fasting cortisol, and thyrotropin yielded results within reference ranges (Table). The veteran was prescribed a CGM, which demonstrated normal glucose levels (≥ 55 mg/dL) during symptomatic episodes (Figure 2).
The patient was diagnosed with IPP given normoglycemia, exclusion of alternative diagnoses, and symptomatic improvement with dietary changes. He was referred to a nutritionist for a high-protein, high-fiber, and low-carbohydrate diet.
DISCUSSION
Seemingly simple diagnostic tools can lead to diagnostic pitfalls. Home glucose monitoring with the use of a standard glucometer during an episode is the typical first step in identifying hypoglycemia, as it is both pragmatic and accurate, with a mean absolute relative difference (MARD) of about 10% in hypoglycemic ranges.5 While the advent of CGM provides real-time data and can reveal clinically relevant fluctuations, it reveals mild hypoglycemia (54 to 70 mg/dL) of no clinical significance in a large proportion of individuals.
Additionally, CGM is less accurate than glucometers with a MARD of about 20% in hypoglycemia ranges.6 CGM technology, however, is rapidly evolving and undergoing further investigation for hypoglycemia detection. Therefore, CGM may be considered in select patients as prospective study results are established; the newest CGMs have MARDs very similar to fingerstick blood glucose data.7,8 In the patient described in this case, CGM helped corroborate the diagnosis, given that symptomatic episodes correlated with lower glucose levels. Provocative testing with oral glucose tolerance testing can frequently result in false positive hypoglycemic readings and is not recommended.9 Supervised mixed meal testing can also be used, which entails monitoring after consuming a mixed macronutrient meal. The test concludes after hypoglycemic symptoms develop or 5 hours elapse, whichever occurs first.1
The pathophysiology of IPP is poorly understood. Proposed mechanisms include increased insulin sensitivity, increased adrenergic sensitivity, impaired glucagon regulation, emotional distress, insulin resistance, and increased glucagon-like peptide-1 production.10-13 Research suggests this may occur as pancreatic β cells fail in early type 2 diabetes mellitus, with diminished first-phase insulin release leading to an initial exuberant rise in blood glucose, an overshooting of the second phase of insulin secretion, and the feeling of the postprandial blood glucose falling, even though the final glucose level achieved is not truly low.13 There are contradictory studies in the literature demonstrating no association between insulin resistance and hypoglycemic symptoms.14 In 2022, Kosuda and colleagues looked at homeostatic model assessment for insulin resistance in patients with postprandial syndrome. They found that the patients were slightly insulin resistant but had normal or exaggerated insulin secretory capacity compared to an oral glucose load, whereas glucagon levels were robustly suppressed by a glucose load. The observed hormonal responses may result in the glycemic patterns and symptoms observed; further study is warranted to elucidate the mechanism.15
Dietary modification is the cornerstone treatment for postprandial syndrome, including reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals. There is also evidence that a Mediterranean diet may be beneficial for managing hypoglycemic symptoms.16 Furthermore, α-glucosidase inhibitors, whose mechanism of action delays the digestion of carbohydrates, have demonstrated promise. This medication class has demonstrated significance in raising postprandial glucose levels and alleviating hypoglycemic symptoms in patients with true postprandial hypoglycemia.17
CONCLUSIONS
IPP is a benign diagnosis encompassing hypoglycemic symptoms without biochemical hypoglycemia. It is not a true hypoglycemic disorder. IPP is challenging to diagnose, given that it is an interpretation of exclusion, supported by symptom improvement with dietary changes (ie, reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals). Supervised mixed meal testing or CGM can be used to assist with diagnosis. Even though CGM is undergoing further study in this patient population, it corroborated the diagnosis in the patient described in this case.
For hypoglycemic symptoms, physicians should first assess for evidence of Whipple triad to evaluate for true biochemical hypoglycemia. For true hypoglycemia (< 55 mg/dL), physicians may conduct an examination in conjunction with an endocrinologist. For normoglycemia (≥ 55 mg/dL), physicians should first exclude alternative etiologies (including cardiac and neurologic), and subsequently consider IPP.
Idiopathic postprandial syndrome (IPP), initially termed reactive hypoglycemia, presents with hypoglycemic-like symptoms in the absence of biochemical hypoglycemia and remains a diagnosis of exclusion. Its pathophysiology is poorly understood. The diagnosis requires thorough evaluation of cardiac, metabolic, neurologic, and gastrointestinal causes, as well as Whipple triad criteria. Dietary modifications, including reduced carbohydrate intake, increased protein and fiber, and frequent small meals, remain the cornerstone of IPP management. Continuous glucose monitoring (CGM) may be a useful adjunct in correlating symptoms with glucose trends, but its role is still evolving.
In the evaluation of patients with symptoms suggestive of hypoglycemia (Figure 1), patients should first be assessed for Whipple triad: symptoms consistent with hypoglycemia, blood glucose level < 55 mg/dL, and reversal of symptoms with glucose.1 Patients who meet Whipple triad criteria should be investigated to identify further etiologies of hypoglycemia. They may include insulinoma, medication-induced (insulin, sulfonylurea, meglitinide, or β blocker use), postbariatric surgery complications, noninsulinoma pancreatogenous hypoglycemia syndrome, ackee fruit consumption, or familial conditions.2 The presence of hypoglycemic symptoms in the postprandial or fasting state can provide valuable insights into underlying etiology.
Patients who do not meet Whipple triad criteria, but exhibit postprandial symptoms consistent with hypoglycemia, as in this case, present a diagnostic dilemma. IPP is defined as hypoglycemic symptoms occuring after carbohydrate ingestion without biochemical hypoglycemia. Initially termed reactive hypoglycemia, it was renamed in 1981to reflect the absence of low blood glucose levels.3
The understanding of this diagnosis has not significantly progressed since the 1980s. Its prevalence, incidence, risk factors, and societal burden remain unclear. IPP is a challenging diagnosis due to nonspecific symptoms that overlap with a myriad of conditions. These symptoms may include adrenergic symptoms such as diaphoresis, tremulousness, palpitations, anxiety, and hunger. Potentially severe neuroglycopenic symptoms, including weakness, dizziness, behavior changes, confusion, and coma, are not typically observed.4 Given that objective criteria are not well established, IPP remains a diagnosis of exclusion. It is imperative to rule out alternative etiologies, particularly cardiac, gastrointestinal, and neurologic causes.
CASE PRESENTATION
A male aged 41 years presented to primary care for evaluation of acute on chronic symptomatic postprandial episodes. He reported a history of symptomatic sinus bradycardia in the setting of sick sinus syndrome following dual-chamber pacemaker placement, posttraumatic stress disorder, and gastroesophageal reflux disease. He was a retired Navy sailor without any known occupational exposures who worked in the real estate industry. The patient reported feeling lightheaded, tremulous, and anxious most afternoons after lunch for several years. He also reported that meals heavy in carbohydrates exacerbated his symptoms, whereas skipping meals or lying down alleviated his symptoms. The patient also reported concomitant arm numbness, shortness of breath, palpitations, and nausea during these episodes. Review of systems was otherwise negative, including no weight changes, fever, chills, night sweats, chest pain, or syncope.
The patient’s medications included ferrous sulfate 325 mg once every other day, bupropion 200 mg once daily, metoprolol succinate 25 mg once daily, and as-needed lorazepam 1 mg once daily. The patient reported no current substance use but reported previous tobacco use 3 years prior (maximum 1 pack/week) and alcohol use 5 years prior (750 ml/day for 15 years). The patient did not exercise and typically ate oatmeal for breakfast, a sandwich or salad for lunch, and taquitos or salad for dinner, with snacks throughout the day. Notable family history included a maternal grandmother with colon cancer. The patient’s vital signs included a 36.8 °C temperature, heart rate 87 beats/min, 118/71 mm Hg blood pressure, oxygen saturation 98% on room air, 125.2 kg weight, and 38.5 body mass index. There were no orthostatic vital sign changes. A physical examination demonstrated obesity with an unremarkable cardiopulmonary and volume examination.
Additional testing included Gallium-68 dototate positron emission tomography/computed tomography, brain magnetic resonance imaging, echocardiogram, electromyogram, exercise tolerance test, Holter monitoring, invasive cardiopulmonary exercise testing, pacemaker interrogation, pulmonary function testing, stress echocardiogram, tilt table test, and venogram computed tomography of the chest, but the results were unremarkable (Appendix). His afternoon nonfasting glucose level was 138 mg/dL with a concurrent hemoglobin A1c of 5.2%. The patient had a fasting C-peptide level of 3.7 ng/mL (reference range 0.5-2.0 ng/mL), fasting insulin level 19.1 mIU/L (reference range < 25 mIU/L), and a fasting glucose level of 93 mg/dL (reference range 70-99 mg/dL). The patient’s urine 5-HIAA, plasma metanephrines, urine metanephrines, insulin-like growth factor 1, prolactin, corticotropin, fasting cortisol, and thyrotropin yielded results within reference ranges (Table). The veteran was prescribed a CGM, which demonstrated normal glucose levels (≥ 55 mg/dL) during symptomatic episodes (Figure 2).
The patient was diagnosed with IPP given normoglycemia, exclusion of alternative diagnoses, and symptomatic improvement with dietary changes. He was referred to a nutritionist for a high-protein, high-fiber, and low-carbohydrate diet.
DISCUSSION
Seemingly simple diagnostic tools can lead to diagnostic pitfalls. Home glucose monitoring with the use of a standard glucometer during an episode is the typical first step in identifying hypoglycemia, as it is both pragmatic and accurate, with a mean absolute relative difference (MARD) of about 10% in hypoglycemic ranges.5 While the advent of CGM provides real-time data and can reveal clinically relevant fluctuations, it reveals mild hypoglycemia (54 to 70 mg/dL) of no clinical significance in a large proportion of individuals.
Additionally, CGM is less accurate than glucometers with a MARD of about 20% in hypoglycemia ranges.6 CGM technology, however, is rapidly evolving and undergoing further investigation for hypoglycemia detection. Therefore, CGM may be considered in select patients as prospective study results are established; the newest CGMs have MARDs very similar to fingerstick blood glucose data.7,8 In the patient described in this case, CGM helped corroborate the diagnosis, given that symptomatic episodes correlated with lower glucose levels. Provocative testing with oral glucose tolerance testing can frequently result in false positive hypoglycemic readings and is not recommended.9 Supervised mixed meal testing can also be used, which entails monitoring after consuming a mixed macronutrient meal. The test concludes after hypoglycemic symptoms develop or 5 hours elapse, whichever occurs first.1
The pathophysiology of IPP is poorly understood. Proposed mechanisms include increased insulin sensitivity, increased adrenergic sensitivity, impaired glucagon regulation, emotional distress, insulin resistance, and increased glucagon-like peptide-1 production.10-13 Research suggests this may occur as pancreatic β cells fail in early type 2 diabetes mellitus, with diminished first-phase insulin release leading to an initial exuberant rise in blood glucose, an overshooting of the second phase of insulin secretion, and the feeling of the postprandial blood glucose falling, even though the final glucose level achieved is not truly low.13 There are contradictory studies in the literature demonstrating no association between insulin resistance and hypoglycemic symptoms.14 In 2022, Kosuda and colleagues looked at homeostatic model assessment for insulin resistance in patients with postprandial syndrome. They found that the patients were slightly insulin resistant but had normal or exaggerated insulin secretory capacity compared to an oral glucose load, whereas glucagon levels were robustly suppressed by a glucose load. The observed hormonal responses may result in the glycemic patterns and symptoms observed; further study is warranted to elucidate the mechanism.15
Dietary modification is the cornerstone treatment for postprandial syndrome, including reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals. There is also evidence that a Mediterranean diet may be beneficial for managing hypoglycemic symptoms.16 Furthermore, α-glucosidase inhibitors, whose mechanism of action delays the digestion of carbohydrates, have demonstrated promise. This medication class has demonstrated significance in raising postprandial glucose levels and alleviating hypoglycemic symptoms in patients with true postprandial hypoglycemia.17
CONCLUSIONS
IPP is a benign diagnosis encompassing hypoglycemic symptoms without biochemical hypoglycemia. It is not a true hypoglycemic disorder. IPP is challenging to diagnose, given that it is an interpretation of exclusion, supported by symptom improvement with dietary changes (ie, reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals). Supervised mixed meal testing or CGM can be used to assist with diagnosis. Even though CGM is undergoing further study in this patient population, it corroborated the diagnosis in the patient described in this case.
For hypoglycemic symptoms, physicians should first assess for evidence of Whipple triad to evaluate for true biochemical hypoglycemia. For true hypoglycemia (< 55 mg/dL), physicians may conduct an examination in conjunction with an endocrinologist. For normoglycemia (≥ 55 mg/dL), physicians should first exclude alternative etiologies (including cardiac and neurologic), and subsequently consider IPP.
Bansal N, Weinstock RS. Non-Diabetic Hypoglycemia. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. MDText.com, Inc.; 2000.
Service FJ. Hypoglycemic disorders. New Engl J Med. 1995;332(17):1144-1152.doi:10.1056/NEJM199504273321707
Charles MA, Hofeldt F, Shackelford A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30(6):465-470.
Douillard C, Jannin A, Vantyghem MC. Rare causes of hypoglycemia in adults. Ann Endocrinol (Paris). 2020;81(2-3):110-117. doi:10.1016/j.ando.2020.04.003
Ekhlaspour L, Mondesir D, Lautsch N, et al. Comparative accuracy of 17 point-of-care glucose meters. J Diabetes Sci Technol. 2017;11(3):558-566. doi:10.1177/1932296816672237
Alitta Q, Grino M, Adjemout L, Langar A, Retornaz F, Oliver C. Overestimation of hypoglycemia diagnosis by FreeStyle Libre continuous glucose monitoring in long-term care home residents with diabetes. J Diabetes Sci Technol. 2018;12(3):727-728. doi:10.1177/1932296817747887
Mongraw-Chaffin M, Beavers DP, McClain DA. Hypoglycemic symptoms in the absence of diabetes: pilot evidence of clinical hypoglycemia in young women. J Clin Transl Endocrinol. 2019;18:100202. doi:10.1016/j.jcte.2019.100202
Shah VN, DuBose SN, Li Z, et al. Continuous glucose monitoring profiles in healthy nondiabetic participants: a multicenter prospective study. J Clin Endocrinol Metab. 2019;104(10):4356-4364. doi:10.1210/jc.2018-02763
Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2009;94(3):709-728. doi:10.1210/jc.2008-1410
Galati SJ, Rayfield EJ. Approach to the patient with postprandial hypoglycemia. Endocr Pract. 2014;20(4):331-340. doi:10.4158/EP13132.RA
Altuntas Y. Postprandial reactive hypoglycemia. Sisli Etfal Hastan Tip Bul. 2019;53(3):215-220.doi:10.14744/SEMB.2019.59455
HARRIS S. HYPERINSULINISM AND DYSINSULINISM. JAMA. 1924;83(10):729-733.doi:10.1001/jama.1924.02660100003002
Harris S. HYPERINSULINISM AND DYSINSULINISM (INSULOGENIC HYPOGLYCBMIA). Endocrinology. 1932;16(1):29-42. doi:10.1210/endo-16-1-29
Hall M, Walicka M, Panczyk M, Traczyk I. Metabolic parameters in patients with suspected reactive hypoglycemia. J Pers Med. 2021;11(4):276. doi:10.3390/jpm11040276
Kosuda M, Watanabe K, Koike M, et al. Glucagon response to glucose challenge in patients with idiopathic postprandial syndrome. J Nippon Med Sch. 2022;89(1):102-107. doi:10.1272/jnms.JNMS.2022_89-205
Hall M, Walicka M, Panczyk M, Traczyk I. Assessing long-term impact of dietary interventions on occurrence of symptoms consistent with hypoglycemia in patients without diabetes: a one-year follow-up study. Nutrients. 2022;14(3):497. doi:10.3390/nu14030497
Ozgen AG, Hamulu F, Bayraktar F, et al. Long-term treatment with acarbose for the treatment of reactive hypoglycemia. Eat Weight Disord. 1998;3(3):136-140. doi:10.1007/BF03340001
Bansal N, Weinstock RS. Non-Diabetic Hypoglycemia. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. MDText.com, Inc.; 2000.
Service FJ. Hypoglycemic disorders. New Engl J Med. 1995;332(17):1144-1152.doi:10.1056/NEJM199504273321707
Charles MA, Hofeldt F, Shackelford A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30(6):465-470.
Douillard C, Jannin A, Vantyghem MC. Rare causes of hypoglycemia in adults. Ann Endocrinol (Paris). 2020;81(2-3):110-117. doi:10.1016/j.ando.2020.04.003
Ekhlaspour L, Mondesir D, Lautsch N, et al. Comparative accuracy of 17 point-of-care glucose meters. J Diabetes Sci Technol. 2017;11(3):558-566. doi:10.1177/1932296816672237
Alitta Q, Grino M, Adjemout L, Langar A, Retornaz F, Oliver C. Overestimation of hypoglycemia diagnosis by FreeStyle Libre continuous glucose monitoring in long-term care home residents with diabetes. J Diabetes Sci Technol. 2018;12(3):727-728. doi:10.1177/1932296817747887
Mongraw-Chaffin M, Beavers DP, McClain DA. Hypoglycemic symptoms in the absence of diabetes: pilot evidence of clinical hypoglycemia in young women. J Clin Transl Endocrinol. 2019;18:100202. doi:10.1016/j.jcte.2019.100202
Shah VN, DuBose SN, Li Z, et al. Continuous glucose monitoring profiles in healthy nondiabetic participants: a multicenter prospective study. J Clin Endocrinol Metab. 2019;104(10):4356-4364. doi:10.1210/jc.2018-02763
Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2009;94(3):709-728. doi:10.1210/jc.2008-1410
Galati SJ, Rayfield EJ. Approach to the patient with postprandial hypoglycemia. Endocr Pract. 2014;20(4):331-340. doi:10.4158/EP13132.RA
Altuntas Y. Postprandial reactive hypoglycemia. Sisli Etfal Hastan Tip Bul. 2019;53(3):215-220.doi:10.14744/SEMB.2019.59455
HARRIS S. HYPERINSULINISM AND DYSINSULINISM. JAMA. 1924;83(10):729-733.doi:10.1001/jama.1924.02660100003002
Harris S. HYPERINSULINISM AND DYSINSULINISM (INSULOGENIC HYPOGLYCBMIA). Endocrinology. 1932;16(1):29-42. doi:10.1210/endo-16-1-29
Hall M, Walicka M, Panczyk M, Traczyk I. Metabolic parameters in patients with suspected reactive hypoglycemia. J Pers Med. 2021;11(4):276. doi:10.3390/jpm11040276
Kosuda M, Watanabe K, Koike M, et al. Glucagon response to glucose challenge in patients with idiopathic postprandial syndrome. J Nippon Med Sch. 2022;89(1):102-107. doi:10.1272/jnms.JNMS.2022_89-205
Hall M, Walicka M, Panczyk M, Traczyk I. Assessing long-term impact of dietary interventions on occurrence of symptoms consistent with hypoglycemia in patients without diabetes: a one-year follow-up study. Nutrients. 2022;14(3):497. doi:10.3390/nu14030497
Ozgen AG, Hamulu F, Bayraktar F, et al. Long-term treatment with acarbose for the treatment of reactive hypoglycemia. Eat Weight Disord. 1998;3(3):136-140. doi:10.1007/BF03340001
A Veteran Presenting With Symptomatic Postprandial Episodes
A Veteran Presenting With Symptomatic Postprandial Episodes
Whipple Disease With Central Nervous System Involvement
Whipple Disease With Central Nervous System Involvement
Whipple disease is a chronic, rare, infectious disease that manifests with systemic symptoms. This disease is caused by the gram-positive bacterium Tropheryma whipplei (T. whipplei). Common manifestations include gastrointestinal symptoms indicative of malabsorption, such as chronic diarrhea, unintentional weight loss (despite normal nutrient intake), and greasy, voluminous, foul-smelling stool. Other, less common manifestations include cardiovascular, endocrine, musculoskeletal, neurologic, and renal signs and symptoms. The prevalence of the disease is rare, affecting 3 in 1 million patients.1 This case highlights the importance of considering Whipple disease when treating patients with multiple symptoms and concurrent disease processes.
Case Presentation
A 53-year-old male with a medical history of hypertension, hyperlipidemia, hypothyroidism, and microcytic anemia presented with an 8-month history of persistent diarrhea associated with abdominal bloating, abdominal discomfort, and a 30-lb weight loss. He also reported fatigue, headaches, inability to concentrate, memory distortion, and visual disturbances involving flashes and floaters. The patient reported no fever, chills, nuchal rigidity, or prior neurologic symptoms. He reported intermittent bilateral hand and knee arthralgias. An autoimmune evaluation for arthralgia was negative, and a prior colonoscopy had been normal.
The patient’s hobbies included gardening, hiking, fishing, and deer hunting in Wyoming and Texas. He had spent time around cattle, dogs, and cats. He consumed alcohol twice weekly but reported no tobacco or illicit drug use or recent international travel. The patient’s family history was positive for rheumatoid arthritis, diabetes mellitus, and hypertension.
The patient’s vital signs were all within reference ranges, and lung auscultation revealed clear breathing sounds with no cardiac murmurs, gallops, or rubs. An abdominal examination revealed decreased bowel sounds, while the rest of the physical examination was otherwise normal.
Initial laboratory results showed that his sodium was 134 mEq/L (reference range, 136-145 mEq/L), hemoglobin was 9.3 g/dL (reference range for men, 14.0-18.0 g/dL), and hematocrit was 30.7% (reference range for men 42%-52%). His white blood cell (WBC) count and thyroid-stimulating hormone level were within normal limits. A cerebrospinal fluid (CSF) analysis revealed the following: WBCs 1.0/μL (0-5/μL), segmented neutrophils 10% (reference range, 7%), lymphocytes 80% (reference range, 40-80%), macrophages 10% (reference range, 2%), red blood cells 3 × 106 /μL (reference range, 4.3- 5.9 × 106 /µL), protein 23.5 mg/dL (reference range, 15-60 mg/dL), and glucose 44 mg/dL (reference range, 50-80 mg/dL).
Upper endoscopy with duodenal biopsy showed benign duodenal mucosa. Histopathologic evaluation revealed abundant foamy macrophages within lamina propria. Periodic acid–Schiff (PAS) stain was positive, diastase-resistant material was visualized within the macrophages (Figures 1 and 2). Polymerase chain reaction (PCR) testing of duodenal biopsy tissue was positive for T. whipplei. A lumbar puncture was performed, and PCR testing of CSF for T. whipplei was also positive. A stool PCR test was positive for Giardia. Transthoracic echocardiogram and brain magnetic resonance imaging were normal.
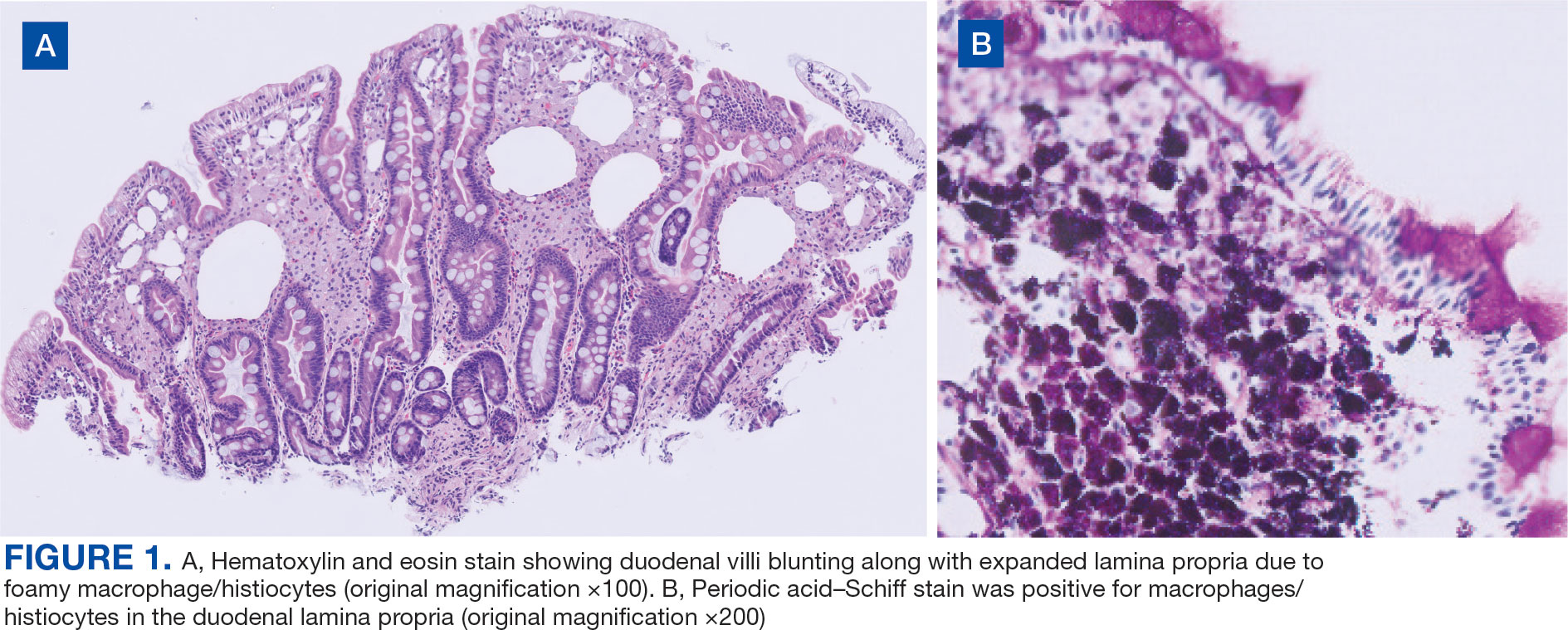
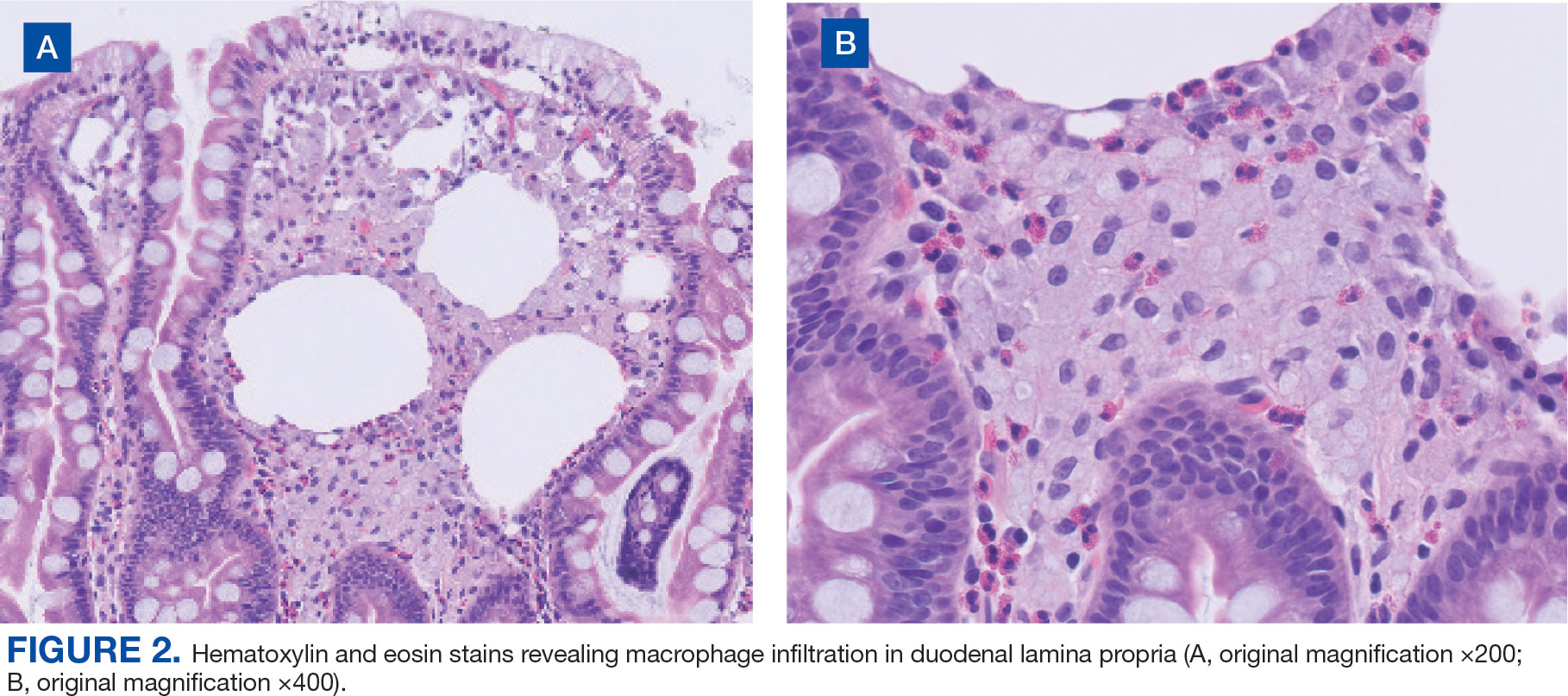
We treated the patient’s giardiasis with a single dose of oral tinidazole 2 g. To treat Whipple disease with central nervous system (CNS) involvement, we started the patient on ceftriaxone 2 g intravenous every 24 hours for 4 weeks, followed by oral trimethoprim and sulfamethoxazole (TMPSMX) 160/800 mg twice daily with an expected 1-year course.
Two months into TMP-SMX therapy, the patient developed an acute kidney injury with hyperkalemia (potassium, 5.5 mEq/L). We transitioned the therapy to doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily to complete 18 months of therapy. A lumbar puncture for CSF PCR and duodenal biopsy was planned for 6 months and 1 year after diagnosis.
Discussion
Whipple disease is often overlooked when making a diagnosis due to the nonspecific nature of its associated signs and symptoms. Classic Whipple disease has 2 stages: an initial prodromal stage marked by intermittent arthralgias, followed by a second gastrointestinal stage that involves chronic diarrhea, abdominal pain, and weight loss.1-3 Infection can sometimes be misdiagnosed as seronegative rheumatoid arthritis and a definite diagnosis can be missed for extended periods, with 1 case taking up to 8 years to diagnose after the first joint manifestations.2,4,5 Blood culture-negative endocarditis has also been well documented.1-5
The most common CNS clinical manifestations of Whipple disease include cognitive changes (eg, dementia), ocular movement disturbances (eg, oculomasticatory myorhythmia, which is pathognomonic for Whipple disease), involuntary movements, and hypothalamic dysfunction.1,6 Other neurologic symptoms include seizures, ataxia, meningitis, and myelopathy. Cerebrospinal fluid studies vary, with some results being normal and others revealing elevated protein counts.1
Disease Course
A retrospective study by Compain and colleagues reports that Whipple disease follows 3 patterns of clinical CNS involvement: classic Whipple disease with neurologic involvement, Whipple disease with isolated neurologic involvement, and neurologic relapse of previously treated Whipple disease.6 Isolated neurologic involvement is roughly 4% to 8%.6-8 Previous studies showed that the average delay from the presentation of neurologic symptoms to diagnosis is about 30 months.9
Diagnosis can be made with histologic evaluation of duodenal tissue using hematoxylin-eosin and PAS stains, which reveal foamy macrophages in expanded duodenal lamina propria, along with a positive tissue PCR.1,5 The slow replication rate of T. whipplei limits the effectiveness of bacterial cultures. After adequate treatment, relapses are still possible and regularly involve the CNS.1,4
Treatment typically involves blood-brain barrier-crossing agents, such as 2 weeks of meropenem 1 g every 24 hours or 2 to 4 weeks of ceftriaxone 2 g every 24 hours, followed by 1 year of TMP-SMX 160/800 mg twice daily. Doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily have also been shown to be effective, as seen in our patient.
Mortality rates vary for patients with Whipple disease and CNS involvement. One study reported poor overall prognosis in patients with CNS involvement, with mortality rates as high as 27%.10 However, rates of early detection and appropriate treatment may be improving, with 1 case series reporting 11% mortality in 18 patients with Whipple disease.6
Diagnosis
Because Whipple disease mimics many other diseases, misdiagnosis as infectious and noninfectious etiologies is common. PAS stain and tissue PCR helped uncover Whipple disease in a patient erroneously diagnosed with refractory Crohn disease.11
Weight loss, diarrhea, arthralgias, and cognitive impairment can also be seen in celiac disease. However, dermatologic manifestations, metabolic bone disease, and vitamin deficiencies are characteristics of celiac disease and can help distinguish it from T. whipplei infection.12
Whipple disease can also be mistaken for tropical sprue. Both can manifest with chronic diarrhea and duodenal villous atrophy; however, tropical sprue is more prevalent in specific geographic areas, and clinical manifestations are primarily gastrointestinal. Weight loss, diarrhea, steatorrhea, and folate deficiency are unique findings in tropical sprue that help differentiate it from Whipple disease.13 Likewise, other infectious diseases can be misdiagnosed as Whipple disease. Duodenal villi blunting and positive PAS staining have been reported in a Mycobacterium avium complex intestinal infection in a patient with AIDS, leading to a misdiagnosis of Whipple disease.14
Some parasitic infections have gastrointestinal symptoms similar to those of Whipple disease and others, such as giardiasis, are known to occur concurrently with Whipple disease.15-17 Giardiasis can also account for weight loss, malabsorptive symptoms, and greasy diarrhea. One case report hypothesized that 1 disease may predispose individuals to the other, as they both affect villous architecture.17 Additional research is needed to determine where the case reports have left off and to explore the connection between the 2 conditions.
Conclusions
The diagnosis of Whipple disease is challenging and frequently missed due to the rare and protean nature of the disease. This case highlights the importance of clinical suspicion for Whipple disease, especially in patients presenting with chronic seronegative arthritis, gastrointestinal abnormalities, and cognitive changes. Furthermore, this case points to the importance of additional testing for Whipple disease, even when a concurrent infection, such as giardiasis, has been identified.
- Biagi F, Balduzzi D, Delvino P, Schiepatti A, Klersy C, Corazza GR. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-1348. doi:10.1007/s10096-015-2357-2
- Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356(1):55-66. doi:10.1056/NEJMra062477
- El-Abassi R, Soliman MY, Williams F, England JD. Whipple’s disease. J Neurol Sci. 2017;377:197-206. doi:10.1016/j.jns.2017.01.048
- Melas N, Amin R, Gyllemark P, Younes AH, Almer S. Whipple’s disease: the great masquerader-a high level of suspicion is the key to diagnosis. BMC Gastroenterol. 2021;21(1):128. doi:10.1186/s12876-021-01664-1
- Boumaza A, Azzouz EB, Arrindell J, Lepidi H, Mezouar S, Desnues B. Whipple’s disease and Tropheryma whipplei infections: from bench to bedside. Lancet Infect Dis. 2022;22(10):e280-e291. doi:10.1016/S1473-3099(22)00128-1
- Compain C, Sacre K, Puéchal X, et al. Central nervous system involvement in Whipple disease: clinical study of 18 patients and long-term follow-up. Medicine (Baltimore). 2013;92(6):324-330. doi:10.1097/MD.0000000000000010
- Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2-5. doi:10.1136/jnnp.68.1.2
- Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457. doi:10.1097/00005792-200211000-00005
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore). 1997;76(3):170-184. doi:10.1097/00005792-199705000-00003
- Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8(9):899-903.
- Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of Whipple’s disease in a patient mistakenly on anti-TNF therapy. ACG Case Rep J. 2013;1(1):25-28. doi:10.14309/crj.2013.11
- . Therrien A, Kelly CP, Silvester JA. Celiac disease: extraintestinal manifestations and associated conditions. J Clin Gastroenterol. 2020;54(1):8-21. doi:10.1097/MCG.0000000000001267
- Murray JA, Rubio-Tapia A. Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol. 2012;26(5):581-600. doi:10.1016/j.bpg.2012.11.013
- Chirayath S, Bin Liaquat H, Bahirwani J, Labeeb A, Chaput K, Kaza C. Mycobacterium avium complex infection imitating Whipple disease in an immunocompromised patient with newly diagnosed acquired immunodeficiency syn - drome. ACG Case Rep J. 2021;8(5):e00588. doi:10.14309/crj.0000000000000588
- Fenollar F, Lepidi H, Gérolami R, Drancourt M, Raoult D. Whipple disease associated with giardiasis. J Infect Dis. 2003;188(6):828-834. doi:10.1086/378093
- Ruiz JAG, Simón PG, Aparicio Duque R, Mayor Jerez JL. Association between Whipple’s disease and Giardia lamblia infection. Rev Esp Enferm Dig. 2005;97(7)521-526. doi:10.4321/s1130-01082005000700007
- Gisbertz IA, Bergmans DC, van Marion-Kievit JA, Haak HR. Concurrent Whipple’s disease and Giardia lamblia infection in a patient presenting with weight loss. Eur J Intern Med. 2001;12(6):525-528. doi:10.1016/s0953-6205(01)00165-0
Whipple disease is a chronic, rare, infectious disease that manifests with systemic symptoms. This disease is caused by the gram-positive bacterium Tropheryma whipplei (T. whipplei). Common manifestations include gastrointestinal symptoms indicative of malabsorption, such as chronic diarrhea, unintentional weight loss (despite normal nutrient intake), and greasy, voluminous, foul-smelling stool. Other, less common manifestations include cardiovascular, endocrine, musculoskeletal, neurologic, and renal signs and symptoms. The prevalence of the disease is rare, affecting 3 in 1 million patients.1 This case highlights the importance of considering Whipple disease when treating patients with multiple symptoms and concurrent disease processes.
Case Presentation
A 53-year-old male with a medical history of hypertension, hyperlipidemia, hypothyroidism, and microcytic anemia presented with an 8-month history of persistent diarrhea associated with abdominal bloating, abdominal discomfort, and a 30-lb weight loss. He also reported fatigue, headaches, inability to concentrate, memory distortion, and visual disturbances involving flashes and floaters. The patient reported no fever, chills, nuchal rigidity, or prior neurologic symptoms. He reported intermittent bilateral hand and knee arthralgias. An autoimmune evaluation for arthralgia was negative, and a prior colonoscopy had been normal.
The patient’s hobbies included gardening, hiking, fishing, and deer hunting in Wyoming and Texas. He had spent time around cattle, dogs, and cats. He consumed alcohol twice weekly but reported no tobacco or illicit drug use or recent international travel. The patient’s family history was positive for rheumatoid arthritis, diabetes mellitus, and hypertension.
The patient’s vital signs were all within reference ranges, and lung auscultation revealed clear breathing sounds with no cardiac murmurs, gallops, or rubs. An abdominal examination revealed decreased bowel sounds, while the rest of the physical examination was otherwise normal.
Initial laboratory results showed that his sodium was 134 mEq/L (reference range, 136-145 mEq/L), hemoglobin was 9.3 g/dL (reference range for men, 14.0-18.0 g/dL), and hematocrit was 30.7% (reference range for men 42%-52%). His white blood cell (WBC) count and thyroid-stimulating hormone level were within normal limits. A cerebrospinal fluid (CSF) analysis revealed the following: WBCs 1.0/μL (0-5/μL), segmented neutrophils 10% (reference range, 7%), lymphocytes 80% (reference range, 40-80%), macrophages 10% (reference range, 2%), red blood cells 3 × 106 /μL (reference range, 4.3- 5.9 × 106 /µL), protein 23.5 mg/dL (reference range, 15-60 mg/dL), and glucose 44 mg/dL (reference range, 50-80 mg/dL).
Upper endoscopy with duodenal biopsy showed benign duodenal mucosa. Histopathologic evaluation revealed abundant foamy macrophages within lamina propria. Periodic acid–Schiff (PAS) stain was positive, diastase-resistant material was visualized within the macrophages (Figures 1 and 2). Polymerase chain reaction (PCR) testing of duodenal biopsy tissue was positive for T. whipplei. A lumbar puncture was performed, and PCR testing of CSF for T. whipplei was also positive. A stool PCR test was positive for Giardia. Transthoracic echocardiogram and brain magnetic resonance imaging were normal.
We treated the patient’s giardiasis with a single dose of oral tinidazole 2 g. To treat Whipple disease with central nervous system (CNS) involvement, we started the patient on ceftriaxone 2 g intravenous every 24 hours for 4 weeks, followed by oral trimethoprim and sulfamethoxazole (TMPSMX) 160/800 mg twice daily with an expected 1-year course.
Two months into TMP-SMX therapy, the patient developed an acute kidney injury with hyperkalemia (potassium, 5.5 mEq/L). We transitioned the therapy to doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily to complete 18 months of therapy. A lumbar puncture for CSF PCR and duodenal biopsy was planned for 6 months and 1 year after diagnosis.
Discussion
Whipple disease is often overlooked when making a diagnosis due to the nonspecific nature of its associated signs and symptoms. Classic Whipple disease has 2 stages: an initial prodromal stage marked by intermittent arthralgias, followed by a second gastrointestinal stage that involves chronic diarrhea, abdominal pain, and weight loss.1-3 Infection can sometimes be misdiagnosed as seronegative rheumatoid arthritis and a definite diagnosis can be missed for extended periods, with 1 case taking up to 8 years to diagnose after the first joint manifestations.2,4,5 Blood culture-negative endocarditis has also been well documented.1-5
The most common CNS clinical manifestations of Whipple disease include cognitive changes (eg, dementia), ocular movement disturbances (eg, oculomasticatory myorhythmia, which is pathognomonic for Whipple disease), involuntary movements, and hypothalamic dysfunction.1,6 Other neurologic symptoms include seizures, ataxia, meningitis, and myelopathy. Cerebrospinal fluid studies vary, with some results being normal and others revealing elevated protein counts.1
Disease Course
A retrospective study by Compain and colleagues reports that Whipple disease follows 3 patterns of clinical CNS involvement: classic Whipple disease with neurologic involvement, Whipple disease with isolated neurologic involvement, and neurologic relapse of previously treated Whipple disease.6 Isolated neurologic involvement is roughly 4% to 8%.6-8 Previous studies showed that the average delay from the presentation of neurologic symptoms to diagnosis is about 30 months.9
Diagnosis can be made with histologic evaluation of duodenal tissue using hematoxylin-eosin and PAS stains, which reveal foamy macrophages in expanded duodenal lamina propria, along with a positive tissue PCR.1,5 The slow replication rate of T. whipplei limits the effectiveness of bacterial cultures. After adequate treatment, relapses are still possible and regularly involve the CNS.1,4
Treatment typically involves blood-brain barrier-crossing agents, such as 2 weeks of meropenem 1 g every 24 hours or 2 to 4 weeks of ceftriaxone 2 g every 24 hours, followed by 1 year of TMP-SMX 160/800 mg twice daily. Doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily have also been shown to be effective, as seen in our patient.
Mortality rates vary for patients with Whipple disease and CNS involvement. One study reported poor overall prognosis in patients with CNS involvement, with mortality rates as high as 27%.10 However, rates of early detection and appropriate treatment may be improving, with 1 case series reporting 11% mortality in 18 patients with Whipple disease.6
Diagnosis
Because Whipple disease mimics many other diseases, misdiagnosis as infectious and noninfectious etiologies is common. PAS stain and tissue PCR helped uncover Whipple disease in a patient erroneously diagnosed with refractory Crohn disease.11
Weight loss, diarrhea, arthralgias, and cognitive impairment can also be seen in celiac disease. However, dermatologic manifestations, metabolic bone disease, and vitamin deficiencies are characteristics of celiac disease and can help distinguish it from T. whipplei infection.12
Whipple disease can also be mistaken for tropical sprue. Both can manifest with chronic diarrhea and duodenal villous atrophy; however, tropical sprue is more prevalent in specific geographic areas, and clinical manifestations are primarily gastrointestinal. Weight loss, diarrhea, steatorrhea, and folate deficiency are unique findings in tropical sprue that help differentiate it from Whipple disease.13 Likewise, other infectious diseases can be misdiagnosed as Whipple disease. Duodenal villi blunting and positive PAS staining have been reported in a Mycobacterium avium complex intestinal infection in a patient with AIDS, leading to a misdiagnosis of Whipple disease.14
Some parasitic infections have gastrointestinal symptoms similar to those of Whipple disease and others, such as giardiasis, are known to occur concurrently with Whipple disease.15-17 Giardiasis can also account for weight loss, malabsorptive symptoms, and greasy diarrhea. One case report hypothesized that 1 disease may predispose individuals to the other, as they both affect villous architecture.17 Additional research is needed to determine where the case reports have left off and to explore the connection between the 2 conditions.
Conclusions
The diagnosis of Whipple disease is challenging and frequently missed due to the rare and protean nature of the disease. This case highlights the importance of clinical suspicion for Whipple disease, especially in patients presenting with chronic seronegative arthritis, gastrointestinal abnormalities, and cognitive changes. Furthermore, this case points to the importance of additional testing for Whipple disease, even when a concurrent infection, such as giardiasis, has been identified.
Whipple disease is a chronic, rare, infectious disease that manifests with systemic symptoms. This disease is caused by the gram-positive bacterium Tropheryma whipplei (T. whipplei). Common manifestations include gastrointestinal symptoms indicative of malabsorption, such as chronic diarrhea, unintentional weight loss (despite normal nutrient intake), and greasy, voluminous, foul-smelling stool. Other, less common manifestations include cardiovascular, endocrine, musculoskeletal, neurologic, and renal signs and symptoms. The prevalence of the disease is rare, affecting 3 in 1 million patients.1 This case highlights the importance of considering Whipple disease when treating patients with multiple symptoms and concurrent disease processes.
Case Presentation
A 53-year-old male with a medical history of hypertension, hyperlipidemia, hypothyroidism, and microcytic anemia presented with an 8-month history of persistent diarrhea associated with abdominal bloating, abdominal discomfort, and a 30-lb weight loss. He also reported fatigue, headaches, inability to concentrate, memory distortion, and visual disturbances involving flashes and floaters. The patient reported no fever, chills, nuchal rigidity, or prior neurologic symptoms. He reported intermittent bilateral hand and knee arthralgias. An autoimmune evaluation for arthralgia was negative, and a prior colonoscopy had been normal.
The patient’s hobbies included gardening, hiking, fishing, and deer hunting in Wyoming and Texas. He had spent time around cattle, dogs, and cats. He consumed alcohol twice weekly but reported no tobacco or illicit drug use or recent international travel. The patient’s family history was positive for rheumatoid arthritis, diabetes mellitus, and hypertension.
The patient’s vital signs were all within reference ranges, and lung auscultation revealed clear breathing sounds with no cardiac murmurs, gallops, or rubs. An abdominal examination revealed decreased bowel sounds, while the rest of the physical examination was otherwise normal.
Initial laboratory results showed that his sodium was 134 mEq/L (reference range, 136-145 mEq/L), hemoglobin was 9.3 g/dL (reference range for men, 14.0-18.0 g/dL), and hematocrit was 30.7% (reference range for men 42%-52%). His white blood cell (WBC) count and thyroid-stimulating hormone level were within normal limits. A cerebrospinal fluid (CSF) analysis revealed the following: WBCs 1.0/μL (0-5/μL), segmented neutrophils 10% (reference range, 7%), lymphocytes 80% (reference range, 40-80%), macrophages 10% (reference range, 2%), red blood cells 3 × 106 /μL (reference range, 4.3- 5.9 × 106 /µL), protein 23.5 mg/dL (reference range, 15-60 mg/dL), and glucose 44 mg/dL (reference range, 50-80 mg/dL).
Upper endoscopy with duodenal biopsy showed benign duodenal mucosa. Histopathologic evaluation revealed abundant foamy macrophages within lamina propria. Periodic acid–Schiff (PAS) stain was positive, diastase-resistant material was visualized within the macrophages (Figures 1 and 2). Polymerase chain reaction (PCR) testing of duodenal biopsy tissue was positive for T. whipplei. A lumbar puncture was performed, and PCR testing of CSF for T. whipplei was also positive. A stool PCR test was positive for Giardia. Transthoracic echocardiogram and brain magnetic resonance imaging were normal.
We treated the patient’s giardiasis with a single dose of oral tinidazole 2 g. To treat Whipple disease with central nervous system (CNS) involvement, we started the patient on ceftriaxone 2 g intravenous every 24 hours for 4 weeks, followed by oral trimethoprim and sulfamethoxazole (TMPSMX) 160/800 mg twice daily with an expected 1-year course.
Two months into TMP-SMX therapy, the patient developed an acute kidney injury with hyperkalemia (potassium, 5.5 mEq/L). We transitioned the therapy to doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily to complete 18 months of therapy. A lumbar puncture for CSF PCR and duodenal biopsy was planned for 6 months and 1 year after diagnosis.
Discussion
Whipple disease is often overlooked when making a diagnosis due to the nonspecific nature of its associated signs and symptoms. Classic Whipple disease has 2 stages: an initial prodromal stage marked by intermittent arthralgias, followed by a second gastrointestinal stage that involves chronic diarrhea, abdominal pain, and weight loss.1-3 Infection can sometimes be misdiagnosed as seronegative rheumatoid arthritis and a definite diagnosis can be missed for extended periods, with 1 case taking up to 8 years to diagnose after the first joint manifestations.2,4,5 Blood culture-negative endocarditis has also been well documented.1-5
The most common CNS clinical manifestations of Whipple disease include cognitive changes (eg, dementia), ocular movement disturbances (eg, oculomasticatory myorhythmia, which is pathognomonic for Whipple disease), involuntary movements, and hypothalamic dysfunction.1,6 Other neurologic symptoms include seizures, ataxia, meningitis, and myelopathy. Cerebrospinal fluid studies vary, with some results being normal and others revealing elevated protein counts.1
Disease Course
A retrospective study by Compain and colleagues reports that Whipple disease follows 3 patterns of clinical CNS involvement: classic Whipple disease with neurologic involvement, Whipple disease with isolated neurologic involvement, and neurologic relapse of previously treated Whipple disease.6 Isolated neurologic involvement is roughly 4% to 8%.6-8 Previous studies showed that the average delay from the presentation of neurologic symptoms to diagnosis is about 30 months.9
Diagnosis can be made with histologic evaluation of duodenal tissue using hematoxylin-eosin and PAS stains, which reveal foamy macrophages in expanded duodenal lamina propria, along with a positive tissue PCR.1,5 The slow replication rate of T. whipplei limits the effectiveness of bacterial cultures. After adequate treatment, relapses are still possible and regularly involve the CNS.1,4
Treatment typically involves blood-brain barrier-crossing agents, such as 2 weeks of meropenem 1 g every 24 hours or 2 to 4 weeks of ceftriaxone 2 g every 24 hours, followed by 1 year of TMP-SMX 160/800 mg twice daily. Doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily have also been shown to be effective, as seen in our patient.
Mortality rates vary for patients with Whipple disease and CNS involvement. One study reported poor overall prognosis in patients with CNS involvement, with mortality rates as high as 27%.10 However, rates of early detection and appropriate treatment may be improving, with 1 case series reporting 11% mortality in 18 patients with Whipple disease.6
Diagnosis
Because Whipple disease mimics many other diseases, misdiagnosis as infectious and noninfectious etiologies is common. PAS stain and tissue PCR helped uncover Whipple disease in a patient erroneously diagnosed with refractory Crohn disease.11
Weight loss, diarrhea, arthralgias, and cognitive impairment can also be seen in celiac disease. However, dermatologic manifestations, metabolic bone disease, and vitamin deficiencies are characteristics of celiac disease and can help distinguish it from T. whipplei infection.12
Whipple disease can also be mistaken for tropical sprue. Both can manifest with chronic diarrhea and duodenal villous atrophy; however, tropical sprue is more prevalent in specific geographic areas, and clinical manifestations are primarily gastrointestinal. Weight loss, diarrhea, steatorrhea, and folate deficiency are unique findings in tropical sprue that help differentiate it from Whipple disease.13 Likewise, other infectious diseases can be misdiagnosed as Whipple disease. Duodenal villi blunting and positive PAS staining have been reported in a Mycobacterium avium complex intestinal infection in a patient with AIDS, leading to a misdiagnosis of Whipple disease.14
Some parasitic infections have gastrointestinal symptoms similar to those of Whipple disease and others, such as giardiasis, are known to occur concurrently with Whipple disease.15-17 Giardiasis can also account for weight loss, malabsorptive symptoms, and greasy diarrhea. One case report hypothesized that 1 disease may predispose individuals to the other, as they both affect villous architecture.17 Additional research is needed to determine where the case reports have left off and to explore the connection between the 2 conditions.
Conclusions
The diagnosis of Whipple disease is challenging and frequently missed due to the rare and protean nature of the disease. This case highlights the importance of clinical suspicion for Whipple disease, especially in patients presenting with chronic seronegative arthritis, gastrointestinal abnormalities, and cognitive changes. Furthermore, this case points to the importance of additional testing for Whipple disease, even when a concurrent infection, such as giardiasis, has been identified.
- Biagi F, Balduzzi D, Delvino P, Schiepatti A, Klersy C, Corazza GR. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-1348. doi:10.1007/s10096-015-2357-2
- Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356(1):55-66. doi:10.1056/NEJMra062477
- El-Abassi R, Soliman MY, Williams F, England JD. Whipple’s disease. J Neurol Sci. 2017;377:197-206. doi:10.1016/j.jns.2017.01.048
- Melas N, Amin R, Gyllemark P, Younes AH, Almer S. Whipple’s disease: the great masquerader-a high level of suspicion is the key to diagnosis. BMC Gastroenterol. 2021;21(1):128. doi:10.1186/s12876-021-01664-1
- Boumaza A, Azzouz EB, Arrindell J, Lepidi H, Mezouar S, Desnues B. Whipple’s disease and Tropheryma whipplei infections: from bench to bedside. Lancet Infect Dis. 2022;22(10):e280-e291. doi:10.1016/S1473-3099(22)00128-1
- Compain C, Sacre K, Puéchal X, et al. Central nervous system involvement in Whipple disease: clinical study of 18 patients and long-term follow-up. Medicine (Baltimore). 2013;92(6):324-330. doi:10.1097/MD.0000000000000010
- Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2-5. doi:10.1136/jnnp.68.1.2
- Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457. doi:10.1097/00005792-200211000-00005
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore). 1997;76(3):170-184. doi:10.1097/00005792-199705000-00003
- Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8(9):899-903.
- Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of Whipple’s disease in a patient mistakenly on anti-TNF therapy. ACG Case Rep J. 2013;1(1):25-28. doi:10.14309/crj.2013.11
- . Therrien A, Kelly CP, Silvester JA. Celiac disease: extraintestinal manifestations and associated conditions. J Clin Gastroenterol. 2020;54(1):8-21. doi:10.1097/MCG.0000000000001267
- Murray JA, Rubio-Tapia A. Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol. 2012;26(5):581-600. doi:10.1016/j.bpg.2012.11.013
- Chirayath S, Bin Liaquat H, Bahirwani J, Labeeb A, Chaput K, Kaza C. Mycobacterium avium complex infection imitating Whipple disease in an immunocompromised patient with newly diagnosed acquired immunodeficiency syn - drome. ACG Case Rep J. 2021;8(5):e00588. doi:10.14309/crj.0000000000000588
- Fenollar F, Lepidi H, Gérolami R, Drancourt M, Raoult D. Whipple disease associated with giardiasis. J Infect Dis. 2003;188(6):828-834. doi:10.1086/378093
- Ruiz JAG, Simón PG, Aparicio Duque R, Mayor Jerez JL. Association between Whipple’s disease and Giardia lamblia infection. Rev Esp Enferm Dig. 2005;97(7)521-526. doi:10.4321/s1130-01082005000700007
- Gisbertz IA, Bergmans DC, van Marion-Kievit JA, Haak HR. Concurrent Whipple’s disease and Giardia lamblia infection in a patient presenting with weight loss. Eur J Intern Med. 2001;12(6):525-528. doi:10.1016/s0953-6205(01)00165-0
- Biagi F, Balduzzi D, Delvino P, Schiepatti A, Klersy C, Corazza GR. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-1348. doi:10.1007/s10096-015-2357-2
- Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356(1):55-66. doi:10.1056/NEJMra062477
- El-Abassi R, Soliman MY, Williams F, England JD. Whipple’s disease. J Neurol Sci. 2017;377:197-206. doi:10.1016/j.jns.2017.01.048
- Melas N, Amin R, Gyllemark P, Younes AH, Almer S. Whipple’s disease: the great masquerader-a high level of suspicion is the key to diagnosis. BMC Gastroenterol. 2021;21(1):128. doi:10.1186/s12876-021-01664-1
- Boumaza A, Azzouz EB, Arrindell J, Lepidi H, Mezouar S, Desnues B. Whipple’s disease and Tropheryma whipplei infections: from bench to bedside. Lancet Infect Dis. 2022;22(10):e280-e291. doi:10.1016/S1473-3099(22)00128-1
- Compain C, Sacre K, Puéchal X, et al. Central nervous system involvement in Whipple disease: clinical study of 18 patients and long-term follow-up. Medicine (Baltimore). 2013;92(6):324-330. doi:10.1097/MD.0000000000000010
- Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2-5. doi:10.1136/jnnp.68.1.2
- Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457. doi:10.1097/00005792-200211000-00005
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore). 1997;76(3):170-184. doi:10.1097/00005792-199705000-00003
- Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8(9):899-903.
- Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of Whipple’s disease in a patient mistakenly on anti-TNF therapy. ACG Case Rep J. 2013;1(1):25-28. doi:10.14309/crj.2013.11
- . Therrien A, Kelly CP, Silvester JA. Celiac disease: extraintestinal manifestations and associated conditions. J Clin Gastroenterol. 2020;54(1):8-21. doi:10.1097/MCG.0000000000001267
- Murray JA, Rubio-Tapia A. Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol. 2012;26(5):581-600. doi:10.1016/j.bpg.2012.11.013
- Chirayath S, Bin Liaquat H, Bahirwani J, Labeeb A, Chaput K, Kaza C. Mycobacterium avium complex infection imitating Whipple disease in an immunocompromised patient with newly diagnosed acquired immunodeficiency syn - drome. ACG Case Rep J. 2021;8(5):e00588. doi:10.14309/crj.0000000000000588
- Fenollar F, Lepidi H, Gérolami R, Drancourt M, Raoult D. Whipple disease associated with giardiasis. J Infect Dis. 2003;188(6):828-834. doi:10.1086/378093
- Ruiz JAG, Simón PG, Aparicio Duque R, Mayor Jerez JL. Association between Whipple’s disease and Giardia lamblia infection. Rev Esp Enferm Dig. 2005;97(7)521-526. doi:10.4321/s1130-01082005000700007
- Gisbertz IA, Bergmans DC, van Marion-Kievit JA, Haak HR. Concurrent Whipple’s disease and Giardia lamblia infection in a patient presenting with weight loss. Eur J Intern Med. 2001;12(6):525-528. doi:10.1016/s0953-6205(01)00165-0
Whipple Disease With Central Nervous System Involvement
Whipple Disease With Central Nervous System Involvement

Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Acute agranulocytosis and aseptic meningitis are serious adverse effects (AEs) associated with sulfamethoxazole-trimethoprim. Acute agranulocytosis is a rare, potentially life-threatening blood dyscrasia characterized by a neutrophil count of < 500 cells per μL, with no relevant decrease in hemoglobin or platelet levels.1 Patients with agranulocytosis may be asymptomatic or experience severe sore throat, pharyngitis, or tonsillitis in combination with high fever, rigors, headaches, or malaise. These AEs are commonly classified as idiosyncratic and, in most cases, attributable to medications. If drug-induced agranulocytosis is suspected, the patient should discontinue the medication immediately.1
Meningitis is an inflammatory disease typically caused by viral or bacterial infections; however, it may also be attributed to medications or malignancy. Inflammation of the meninges with a negative bacterial cerebrospinal fluid culture is classified as aseptic meningitis. Distinguishing between aseptic and bacterial meningitis is crucial due to differences in illness severity, treatment options, and prognosis.2 Symptoms of meningitis may include fever, headache, nuchal rigidity, nausea, or vomiting.3 Several classes of medications can cause drug-induced aseptic meningitis (DIAM), but the most commonly reported antibiotic is sulfamethoxazole-trimethoprim.
DIAM is more prevalent in immunocompromised patients, such as those with a history of HIV/AIDS, organ transplant, collagen vascular disease, or malignancy, who may be prescribed sulfamethoxazoletrimethoprim for prophylaxis or treatment of infection.2 The case described in this article serves as a distinctive example of acute agranulocytosis complicated with aseptic meningitis caused by sulfamethoxazole-trimethoprim in an immunocompetent patient.
Case Presentation
A healthy male veteran aged 39 years presented to the Fargo Veterans Affairs Medical Center emergency department (ED) for worsening left testicular pain and increased urinary urgency and frequency for about 48 hours. The patient had no known medication allergies, was current on vaccinations, and his only relevant prescription was valacyclovir for herpes labialis. The evaluation included urinalysis, blood tests, and scrotal ultrasound. The urinalysis, blood tests, and vitals were unremarkable for any signs of systemic infection. The scrotal ultrasound was significant for left focal area of abnormal echogenicity with absent blood flow in the superior left testicle and a significant increase in blood flow around the left epididymis. Mild swelling in the left epididymis was present, with no significant testicular or scrotal swelling or skin changes observed. Urology was consulted and prescribed oral sulfamethoxazole-trimethoprim 800-160 mg every 12 hours for 30 days for the treatment of left epididymo-orchitis.
The patient returned to the ED 2 weeks later with fever, chills, headache, generalized body aches, urinary retention, loose stools, and nonspecific chest pressure. A serum blood test revealed significant neutropenia and leukopenia. The patient was admitted for observation, and sulfamethoxazole-trimethoprim was discontinued. The patient received sodium chloride intravenous (IV) fluid, oral potassium chloride supplementation, IV ondansetron, and analgesics, including oral acetaminophen, oral ibuprofen, and IV hydromorphone as needed. Repeated laboratory tests were completed with no specific findings; serum laboratory work, urinalysis, chest and abdominal X-rays, and echocardiogram were all unremarkable. The patient’s neutrophil count dropped from 5100 cells/µL at the initial ED presentation to 900 cells/µL (reference range, 1500-8000 cells/µL) (Table 1). Agranulocytosis quickly resolved after antibiotic discontinuation.
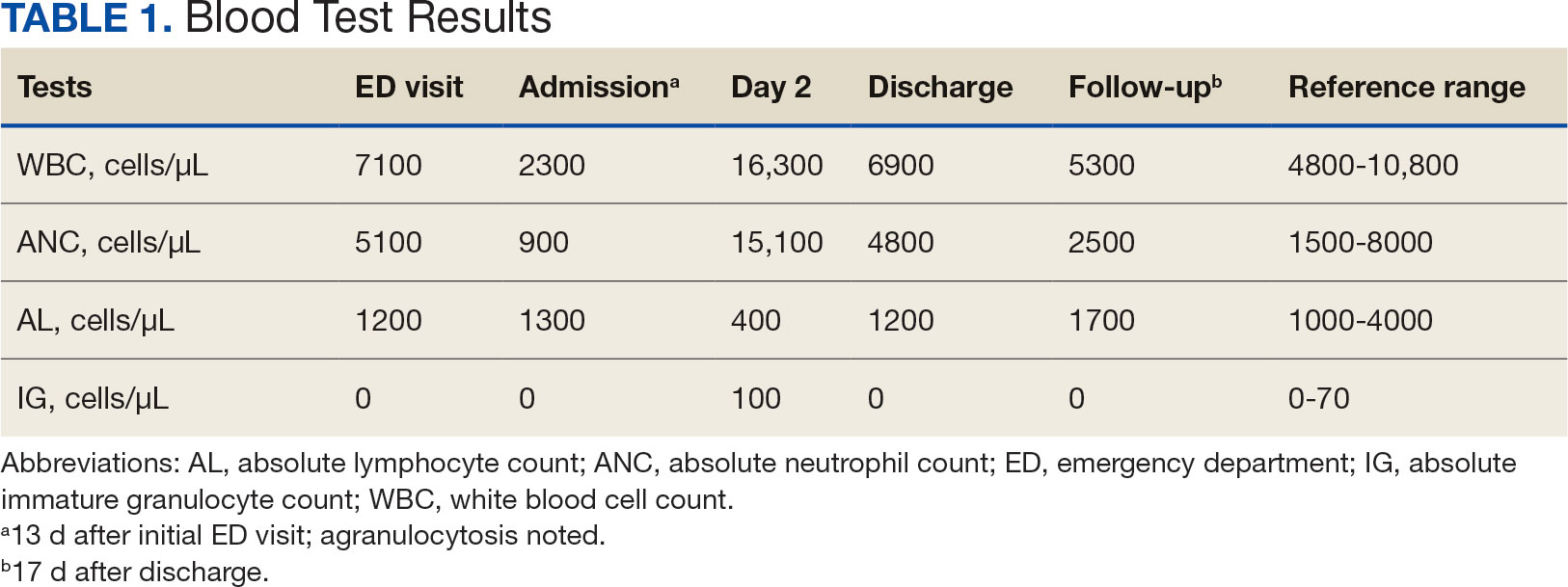
Upon further investigation, the patient took the prescribed sulfamethoxazole-trimethoprim for 10 days before stopping due to the resolution of testicular pain and epididymal swelling. The patient reported initial AEs of loose stools and generalized myalgias when first taking the medication. The patient restarted the antibiotic to complete the course of therapy after not taking it for 2 days. Within 20 minutes of restarting the medication, he experienced myalgias with pruritus, prompting him to return to the ED. Agranulocytosis and aseptic meningitis developed within 12 days after he was prescribed sulfamethoxazole-trimethoprim, though the exact timeframe is unknown.
The patient’s symptoms, except for a persistent headache, resolved during hospitalization. Infectious disease was consulted, and a lumbar puncture was performed due to prior fever and ongoing headaches to rule out a treatable cause of meningitis. The lumbar puncture showed clear spinal fluid, an elevated white blood cell count with neutrophil predominance, and normal protein and glucose levels. Cultures showed no aerobic, anaerobic, or fungal organisms. Herpes virus simplex and Lyme disease testing was not completed during hospitalization. Respiratory panel, legionella, and hepatitis A, B, and C tests were negative (Table 2). The negative laboratory test results strengthened the suspicion of aseptic meningitis caused by sulfamethoxazole-trimethoprim. The neurology consult recommended no additional treatments or tests.

The patient spontaneously recovered 2 days later, with a normalized complete blood count and resolution of headache. Repeat scrotal ultrasounds showed resolution of the left testicle findings. The patient was discharged and seen for a follow-up 14 days later. The final diagnosis requiring hospitalization was aseptic meningitis secondary to a sulfamethoxazole-trimethoprim.
Discussion
Sulfamethoxazole-trimethoprim is a commonly prescribed antibiotic for urinary tract infections, pneumocystis pneumonia, pneumocystis pneumonia prophylaxis, and methicillin-resistant Staphylococcus aureus skin and soft tissue infections. Empiric antibiotics for epididymo-orchitis caused by enteric organisms include levofloxacin or ofloxacin; however sulfamethoxazole-trimethoprim may be considered as alternative.5,6 Agranulocytosis induced by sulfamethoxazole-trimethoprim may occur due to the inhibition on folic acid metabolism, which makes the highly proliferating cells of the hematopoietic system more susceptible to neutropenia. Agranulocytosis typically occurs within 7 days of treatment initiation and generally resolves within a month after discontinuation of the offending agent.7 In this case, agranulocytosis resolved overnight, resulting in leukocytosis. One explanation for the rapid increase in white blood cell count may be the concurrent diagnosis of aseptic meningitis. Alternatively, the patient’s health and immunocompetence may have helped generate an adequate immune response. Medication-induced agranulocytosis is often a diagnosis of exclusion because it is typically difficult to diagnose.7 In more severe or complicated cases of agranulocytosis, filgrastim may be indicated.1
Sulfamethoxazole-trimethoprim is a lipophilic small-molecule medication that can cross the blood-brain barrier and penetrate the tissues of the bone, prostate, and central nervous system.8 Specifically, the medication can pass into the cerebrospinal fluid regardless of meningeal inflammation.9 The exact mechanism for aseptic meningitis caused by sulfamethoxazole-trimethoprim is unknown; however, it may accumulate in the choroid plexus, causing destructive inflammation of small blood vessels and resulting in aseptic meningitis.10 The onset of aseptic meningitis can vary from 10 minutes to 10 days after initiation of the medication. Pre-exposure to the medication typically results in earlier onset of symptoms, though patients do not need to have previously taken the medication to develop aseptic meningitis. Patients generally become afebrile with resolution of headache and mental status changes within 48 to 72 hours after stopping the medication or after about 5 to 7 half-lives of the medication are eliminated.11 Some patients may continue to experience worsening symptoms after discontinuation because the medication is already absorbed into the serum and tissues.
DIAM is an uncommon drug-induced hypersensitivity AE of the central nervous system. Diagnosing aseptic meningitis caused by sulfamethoxazole-trimethoprim can be challenging, as antibiotics are given to treat suspected infections, and the symptoms of aseptic meningitis can be hard to differentiate from those of infectious meningitis.11 Close monitoring between the initiation of the medication and the onset of clinical symptoms is necessary to assist in distinguishing between aseptic and infectious meningitis.3 If the causative agent is not discontinued, symptoms can quickly worsen, progressing from fever and headache to confusion, coma, and respiratory depression. A DIAM diagnosis can only be made with resolution of aseptic meningitis after stopping the contributory agent. If appropriate, this can be proven by rechallenging the medication in a controlled setting. The usual treatment for aseptic meningitis is supportive care, including hydration, antiemetics, electrolyte supplementation, and adequate analgesia.3
Differential diagnoses in this case included viral infection, meningitis, and allergic reaction to sulfamethoxazole-trimethoprim. The patient reported history of experiencing symptoms after restarting his antibiotic, raising strong suspicion for DIAM. Initiation of this antibiotic was the only recent medication change noted. Laboratory testing for infectious agents yielded negative results, including tests for aerobic and anaerobic bacteria as well as viral and fungal infections. A lumbar puncture and cerebrospinal fluid culture was clear, with no organisms shown on gram stain. Bacterial or viral meningitis was presumed less likely due to the duration of symptoms, correlation of symptoms coinciding with restarting the antibiotic, and negative cerebrospinal fluid culture results.
It was concluded that agranulocytosis and aseptic meningitis were likely induced by sulfamethoxazole-trimethoprim as supported by the improvement upon discontinuing the medication and subsequent worsening upon restarting it. Concurrent agranulocytosis and aseptic meningitis is rare, and there is typically no correlation between the 2 reactions. Since agranulocytosis may be asymptomatic, this case highlights the need to monitor blood cell counts in patients using sulfamethoxazole-trimethoprim. The possibility of DIAM should be considered in patients presenting with flu-like symptoms, and a lumbar puncture may be collected to rule out aseptic meningitis if the patient’s AEs are severe following the initiation of an antibiotic, particularly in immunosuppressed patients taking sulfamethoxazole-trimethoprim. This case is unusual because the patient was healthy and immunocompetent.
This case may not be generalizable and may be difficult to compare to other cases. Every case has patient-specific factors affecting subjective information, including the patient’s baseline, severity of symptoms, and treatment options. This report was based on a single patient case and corresponding results may be found in similar patient cases.
Conclusions
This case emphasizes the rare but serious AEs of acute agranulocytosis complicated with aseptic meningitis after prescribed sulfamethoxazole-trimethoprim. This is a unique case of an immunocompetent patient developing both agranulocytosis and aseptic meningitis after restarting the antibiotic to complete therapy. This case highlights the importance of monitoring blood cell counts and monitoring for signs and symptoms of aseptic meningitis, even during short courses of therapy. Further research is needed to recognize characteristics that may increase the risk for these AEs and to develop strategies for their prevention.
- Garbe E. Non-chemotherapy drug-induced agranulocytosis. Expert Opin Drug Saf. 2007;6(3):323-335. doi:10.1517/14740338.6.3.323
- Jha P, Stromich J, Cohen M, Wainaina JN. A rare complication of trimethoprim-sulfamethoxazole: drug induced aseptic meningitis. Case Rep Infect Dis. 2016;2016:3879406. doi:10.1155/2016/3879406
- Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4(2):285-297. doi:10.1517/14740338.4.2.285
- Jarrin I, Sellier P, Lopes A, et al. Etiologies and management of aseptic meningitis in patients admitted to an internal medicine department. Medicine (Baltimore). 2016;95(2):e2372. doi:10.1097/MD.0000000000002372
- Street EJ, Justice ED, Kopa Z, et al. The 2016 European guideline on the management of epididymo-orchitis. Int J STD AIDS. 2017;28(8):744-749. doi:10.1177/0956462417699356
- Kbirou A, Alafifi M, Sayah M, Dakir M, Debbagh A, Aboutaieb R. Acute orchiepididymitis: epidemiological and clinical aspects: an analysis of 152 cases. Ann Med Surg (Lond). 2022;75:103335. doi:10.1016/j.amsu.2022.103335
- Rattay B, Benndorf RA. Drug-induced idiosyncratic agranulocytosis - infrequent but dangerous. Front Pharmacol. 2021;12:727717. doi:10.3389/fphar.2021.727717
- Elmedani S, Albayati A, Udongwo N, Odak M, Khawaja S. Trimethoprim-sulfamethoxazole-induced aseptic meningitis: a new approach. Cureus. 2021;13(6):e15869. doi:10.7759/cureus.15869
- Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858-883. doi:10.1128/CMR.00007-10
- Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Intern Med. 1999;159(11):1185- 1194. doi:10.1001/archinte.159.11.1185
- Bruner KE, Coop CA, White KM. Trimethoprim-sulfamethoxazole-induced aseptic meningitis-not just another sulfa allergy. Ann Allergy Asthma Immunol. 2014;113(5):520-526. doi:10.1016/j.anai.2014.08.006
Acute agranulocytosis and aseptic meningitis are serious adverse effects (AEs) associated with sulfamethoxazole-trimethoprim. Acute agranulocytosis is a rare, potentially life-threatening blood dyscrasia characterized by a neutrophil count of < 500 cells per μL, with no relevant decrease in hemoglobin or platelet levels.1 Patients with agranulocytosis may be asymptomatic or experience severe sore throat, pharyngitis, or tonsillitis in combination with high fever, rigors, headaches, or malaise. These AEs are commonly classified as idiosyncratic and, in most cases, attributable to medications. If drug-induced agranulocytosis is suspected, the patient should discontinue the medication immediately.1
Meningitis is an inflammatory disease typically caused by viral or bacterial infections; however, it may also be attributed to medications or malignancy. Inflammation of the meninges with a negative bacterial cerebrospinal fluid culture is classified as aseptic meningitis. Distinguishing between aseptic and bacterial meningitis is crucial due to differences in illness severity, treatment options, and prognosis.2 Symptoms of meningitis may include fever, headache, nuchal rigidity, nausea, or vomiting.3 Several classes of medications can cause drug-induced aseptic meningitis (DIAM), but the most commonly reported antibiotic is sulfamethoxazole-trimethoprim.
DIAM is more prevalent in immunocompromised patients, such as those with a history of HIV/AIDS, organ transplant, collagen vascular disease, or malignancy, who may be prescribed sulfamethoxazoletrimethoprim for prophylaxis or treatment of infection.2 The case described in this article serves as a distinctive example of acute agranulocytosis complicated with aseptic meningitis caused by sulfamethoxazole-trimethoprim in an immunocompetent patient.
Case Presentation
A healthy male veteran aged 39 years presented to the Fargo Veterans Affairs Medical Center emergency department (ED) for worsening left testicular pain and increased urinary urgency and frequency for about 48 hours. The patient had no known medication allergies, was current on vaccinations, and his only relevant prescription was valacyclovir for herpes labialis. The evaluation included urinalysis, blood tests, and scrotal ultrasound. The urinalysis, blood tests, and vitals were unremarkable for any signs of systemic infection. The scrotal ultrasound was significant for left focal area of abnormal echogenicity with absent blood flow in the superior left testicle and a significant increase in blood flow around the left epididymis. Mild swelling in the left epididymis was present, with no significant testicular or scrotal swelling or skin changes observed. Urology was consulted and prescribed oral sulfamethoxazole-trimethoprim 800-160 mg every 12 hours for 30 days for the treatment of left epididymo-orchitis.
The patient returned to the ED 2 weeks later with fever, chills, headache, generalized body aches, urinary retention, loose stools, and nonspecific chest pressure. A serum blood test revealed significant neutropenia and leukopenia. The patient was admitted for observation, and sulfamethoxazole-trimethoprim was discontinued. The patient received sodium chloride intravenous (IV) fluid, oral potassium chloride supplementation, IV ondansetron, and analgesics, including oral acetaminophen, oral ibuprofen, and IV hydromorphone as needed. Repeated laboratory tests were completed with no specific findings; serum laboratory work, urinalysis, chest and abdominal X-rays, and echocardiogram were all unremarkable. The patient’s neutrophil count dropped from 5100 cells/µL at the initial ED presentation to 900 cells/µL (reference range, 1500-8000 cells/µL) (Table 1). Agranulocytosis quickly resolved after antibiotic discontinuation.
Upon further investigation, the patient took the prescribed sulfamethoxazole-trimethoprim for 10 days before stopping due to the resolution of testicular pain and epididymal swelling. The patient reported initial AEs of loose stools and generalized myalgias when first taking the medication. The patient restarted the antibiotic to complete the course of therapy after not taking it for 2 days. Within 20 minutes of restarting the medication, he experienced myalgias with pruritus, prompting him to return to the ED. Agranulocytosis and aseptic meningitis developed within 12 days after he was prescribed sulfamethoxazole-trimethoprim, though the exact timeframe is unknown.
The patient’s symptoms, except for a persistent headache, resolved during hospitalization. Infectious disease was consulted, and a lumbar puncture was performed due to prior fever and ongoing headaches to rule out a treatable cause of meningitis. The lumbar puncture showed clear spinal fluid, an elevated white blood cell count with neutrophil predominance, and normal protein and glucose levels. Cultures showed no aerobic, anaerobic, or fungal organisms. Herpes virus simplex and Lyme disease testing was not completed during hospitalization. Respiratory panel, legionella, and hepatitis A, B, and C tests were negative (Table 2). The negative laboratory test results strengthened the suspicion of aseptic meningitis caused by sulfamethoxazole-trimethoprim. The neurology consult recommended no additional treatments or tests.
The patient spontaneously recovered 2 days later, with a normalized complete blood count and resolution of headache. Repeat scrotal ultrasounds showed resolution of the left testicle findings. The patient was discharged and seen for a follow-up 14 days later. The final diagnosis requiring hospitalization was aseptic meningitis secondary to a sulfamethoxazole-trimethoprim.
Discussion
Sulfamethoxazole-trimethoprim is a commonly prescribed antibiotic for urinary tract infections, pneumocystis pneumonia, pneumocystis pneumonia prophylaxis, and methicillin-resistant Staphylococcus aureus skin and soft tissue infections. Empiric antibiotics for epididymo-orchitis caused by enteric organisms include levofloxacin or ofloxacin; however sulfamethoxazole-trimethoprim may be considered as alternative.5,6 Agranulocytosis induced by sulfamethoxazole-trimethoprim may occur due to the inhibition on folic acid metabolism, which makes the highly proliferating cells of the hematopoietic system more susceptible to neutropenia. Agranulocytosis typically occurs within 7 days of treatment initiation and generally resolves within a month after discontinuation of the offending agent.7 In this case, agranulocytosis resolved overnight, resulting in leukocytosis. One explanation for the rapid increase in white blood cell count may be the concurrent diagnosis of aseptic meningitis. Alternatively, the patient’s health and immunocompetence may have helped generate an adequate immune response. Medication-induced agranulocytosis is often a diagnosis of exclusion because it is typically difficult to diagnose.7 In more severe or complicated cases of agranulocytosis, filgrastim may be indicated.1
Sulfamethoxazole-trimethoprim is a lipophilic small-molecule medication that can cross the blood-brain barrier and penetrate the tissues of the bone, prostate, and central nervous system.8 Specifically, the medication can pass into the cerebrospinal fluid regardless of meningeal inflammation.9 The exact mechanism for aseptic meningitis caused by sulfamethoxazole-trimethoprim is unknown; however, it may accumulate in the choroid plexus, causing destructive inflammation of small blood vessels and resulting in aseptic meningitis.10 The onset of aseptic meningitis can vary from 10 minutes to 10 days after initiation of the medication. Pre-exposure to the medication typically results in earlier onset of symptoms, though patients do not need to have previously taken the medication to develop aseptic meningitis. Patients generally become afebrile with resolution of headache and mental status changes within 48 to 72 hours after stopping the medication or after about 5 to 7 half-lives of the medication are eliminated.11 Some patients may continue to experience worsening symptoms after discontinuation because the medication is already absorbed into the serum and tissues.
DIAM is an uncommon drug-induced hypersensitivity AE of the central nervous system. Diagnosing aseptic meningitis caused by sulfamethoxazole-trimethoprim can be challenging, as antibiotics are given to treat suspected infections, and the symptoms of aseptic meningitis can be hard to differentiate from those of infectious meningitis.11 Close monitoring between the initiation of the medication and the onset of clinical symptoms is necessary to assist in distinguishing between aseptic and infectious meningitis.3 If the causative agent is not discontinued, symptoms can quickly worsen, progressing from fever and headache to confusion, coma, and respiratory depression. A DIAM diagnosis can only be made with resolution of aseptic meningitis after stopping the contributory agent. If appropriate, this can be proven by rechallenging the medication in a controlled setting. The usual treatment for aseptic meningitis is supportive care, including hydration, antiemetics, electrolyte supplementation, and adequate analgesia.3
Differential diagnoses in this case included viral infection, meningitis, and allergic reaction to sulfamethoxazole-trimethoprim. The patient reported history of experiencing symptoms after restarting his antibiotic, raising strong suspicion for DIAM. Initiation of this antibiotic was the only recent medication change noted. Laboratory testing for infectious agents yielded negative results, including tests for aerobic and anaerobic bacteria as well as viral and fungal infections. A lumbar puncture and cerebrospinal fluid culture was clear, with no organisms shown on gram stain. Bacterial or viral meningitis was presumed less likely due to the duration of symptoms, correlation of symptoms coinciding with restarting the antibiotic, and negative cerebrospinal fluid culture results.
It was concluded that agranulocytosis and aseptic meningitis were likely induced by sulfamethoxazole-trimethoprim as supported by the improvement upon discontinuing the medication and subsequent worsening upon restarting it. Concurrent agranulocytosis and aseptic meningitis is rare, and there is typically no correlation between the 2 reactions. Since agranulocytosis may be asymptomatic, this case highlights the need to monitor blood cell counts in patients using sulfamethoxazole-trimethoprim. The possibility of DIAM should be considered in patients presenting with flu-like symptoms, and a lumbar puncture may be collected to rule out aseptic meningitis if the patient’s AEs are severe following the initiation of an antibiotic, particularly in immunosuppressed patients taking sulfamethoxazole-trimethoprim. This case is unusual because the patient was healthy and immunocompetent.
This case may not be generalizable and may be difficult to compare to other cases. Every case has patient-specific factors affecting subjective information, including the patient’s baseline, severity of symptoms, and treatment options. This report was based on a single patient case and corresponding results may be found in similar patient cases.
Conclusions
This case emphasizes the rare but serious AEs of acute agranulocytosis complicated with aseptic meningitis after prescribed sulfamethoxazole-trimethoprim. This is a unique case of an immunocompetent patient developing both agranulocytosis and aseptic meningitis after restarting the antibiotic to complete therapy. This case highlights the importance of monitoring blood cell counts and monitoring for signs and symptoms of aseptic meningitis, even during short courses of therapy. Further research is needed to recognize characteristics that may increase the risk for these AEs and to develop strategies for their prevention.
Acute agranulocytosis and aseptic meningitis are serious adverse effects (AEs) associated with sulfamethoxazole-trimethoprim. Acute agranulocytosis is a rare, potentially life-threatening blood dyscrasia characterized by a neutrophil count of < 500 cells per μL, with no relevant decrease in hemoglobin or platelet levels.1 Patients with agranulocytosis may be asymptomatic or experience severe sore throat, pharyngitis, or tonsillitis in combination with high fever, rigors, headaches, or malaise. These AEs are commonly classified as idiosyncratic and, in most cases, attributable to medications. If drug-induced agranulocytosis is suspected, the patient should discontinue the medication immediately.1
Meningitis is an inflammatory disease typically caused by viral or bacterial infections; however, it may also be attributed to medications or malignancy. Inflammation of the meninges with a negative bacterial cerebrospinal fluid culture is classified as aseptic meningitis. Distinguishing between aseptic and bacterial meningitis is crucial due to differences in illness severity, treatment options, and prognosis.2 Symptoms of meningitis may include fever, headache, nuchal rigidity, nausea, or vomiting.3 Several classes of medications can cause drug-induced aseptic meningitis (DIAM), but the most commonly reported antibiotic is sulfamethoxazole-trimethoprim.
DIAM is more prevalent in immunocompromised patients, such as those with a history of HIV/AIDS, organ transplant, collagen vascular disease, or malignancy, who may be prescribed sulfamethoxazoletrimethoprim for prophylaxis or treatment of infection.2 The case described in this article serves as a distinctive example of acute agranulocytosis complicated with aseptic meningitis caused by sulfamethoxazole-trimethoprim in an immunocompetent patient.
Case Presentation
A healthy male veteran aged 39 years presented to the Fargo Veterans Affairs Medical Center emergency department (ED) for worsening left testicular pain and increased urinary urgency and frequency for about 48 hours. The patient had no known medication allergies, was current on vaccinations, and his only relevant prescription was valacyclovir for herpes labialis. The evaluation included urinalysis, blood tests, and scrotal ultrasound. The urinalysis, blood tests, and vitals were unremarkable for any signs of systemic infection. The scrotal ultrasound was significant for left focal area of abnormal echogenicity with absent blood flow in the superior left testicle and a significant increase in blood flow around the left epididymis. Mild swelling in the left epididymis was present, with no significant testicular or scrotal swelling or skin changes observed. Urology was consulted and prescribed oral sulfamethoxazole-trimethoprim 800-160 mg every 12 hours for 30 days for the treatment of left epididymo-orchitis.
The patient returned to the ED 2 weeks later with fever, chills, headache, generalized body aches, urinary retention, loose stools, and nonspecific chest pressure. A serum blood test revealed significant neutropenia and leukopenia. The patient was admitted for observation, and sulfamethoxazole-trimethoprim was discontinued. The patient received sodium chloride intravenous (IV) fluid, oral potassium chloride supplementation, IV ondansetron, and analgesics, including oral acetaminophen, oral ibuprofen, and IV hydromorphone as needed. Repeated laboratory tests were completed with no specific findings; serum laboratory work, urinalysis, chest and abdominal X-rays, and echocardiogram were all unremarkable. The patient’s neutrophil count dropped from 5100 cells/µL at the initial ED presentation to 900 cells/µL (reference range, 1500-8000 cells/µL) (Table 1). Agranulocytosis quickly resolved after antibiotic discontinuation.
Upon further investigation, the patient took the prescribed sulfamethoxazole-trimethoprim for 10 days before stopping due to the resolution of testicular pain and epididymal swelling. The patient reported initial AEs of loose stools and generalized myalgias when first taking the medication. The patient restarted the antibiotic to complete the course of therapy after not taking it for 2 days. Within 20 minutes of restarting the medication, he experienced myalgias with pruritus, prompting him to return to the ED. Agranulocytosis and aseptic meningitis developed within 12 days after he was prescribed sulfamethoxazole-trimethoprim, though the exact timeframe is unknown.
The patient’s symptoms, except for a persistent headache, resolved during hospitalization. Infectious disease was consulted, and a lumbar puncture was performed due to prior fever and ongoing headaches to rule out a treatable cause of meningitis. The lumbar puncture showed clear spinal fluid, an elevated white blood cell count with neutrophil predominance, and normal protein and glucose levels. Cultures showed no aerobic, anaerobic, or fungal organisms. Herpes virus simplex and Lyme disease testing was not completed during hospitalization. Respiratory panel, legionella, and hepatitis A, B, and C tests were negative (Table 2). The negative laboratory test results strengthened the suspicion of aseptic meningitis caused by sulfamethoxazole-trimethoprim. The neurology consult recommended no additional treatments or tests.
The patient spontaneously recovered 2 days later, with a normalized complete blood count and resolution of headache. Repeat scrotal ultrasounds showed resolution of the left testicle findings. The patient was discharged and seen for a follow-up 14 days later. The final diagnosis requiring hospitalization was aseptic meningitis secondary to a sulfamethoxazole-trimethoprim.
Discussion
Sulfamethoxazole-trimethoprim is a commonly prescribed antibiotic for urinary tract infections, pneumocystis pneumonia, pneumocystis pneumonia prophylaxis, and methicillin-resistant Staphylococcus aureus skin and soft tissue infections. Empiric antibiotics for epididymo-orchitis caused by enteric organisms include levofloxacin or ofloxacin; however sulfamethoxazole-trimethoprim may be considered as alternative.5,6 Agranulocytosis induced by sulfamethoxazole-trimethoprim may occur due to the inhibition on folic acid metabolism, which makes the highly proliferating cells of the hematopoietic system more susceptible to neutropenia. Agranulocytosis typically occurs within 7 days of treatment initiation and generally resolves within a month after discontinuation of the offending agent.7 In this case, agranulocytosis resolved overnight, resulting in leukocytosis. One explanation for the rapid increase in white blood cell count may be the concurrent diagnosis of aseptic meningitis. Alternatively, the patient’s health and immunocompetence may have helped generate an adequate immune response. Medication-induced agranulocytosis is often a diagnosis of exclusion because it is typically difficult to diagnose.7 In more severe or complicated cases of agranulocytosis, filgrastim may be indicated.1
Sulfamethoxazole-trimethoprim is a lipophilic small-molecule medication that can cross the blood-brain barrier and penetrate the tissues of the bone, prostate, and central nervous system.8 Specifically, the medication can pass into the cerebrospinal fluid regardless of meningeal inflammation.9 The exact mechanism for aseptic meningitis caused by sulfamethoxazole-trimethoprim is unknown; however, it may accumulate in the choroid plexus, causing destructive inflammation of small blood vessels and resulting in aseptic meningitis.10 The onset of aseptic meningitis can vary from 10 minutes to 10 days after initiation of the medication. Pre-exposure to the medication typically results in earlier onset of symptoms, though patients do not need to have previously taken the medication to develop aseptic meningitis. Patients generally become afebrile with resolution of headache and mental status changes within 48 to 72 hours after stopping the medication or after about 5 to 7 half-lives of the medication are eliminated.11 Some patients may continue to experience worsening symptoms after discontinuation because the medication is already absorbed into the serum and tissues.
DIAM is an uncommon drug-induced hypersensitivity AE of the central nervous system. Diagnosing aseptic meningitis caused by sulfamethoxazole-trimethoprim can be challenging, as antibiotics are given to treat suspected infections, and the symptoms of aseptic meningitis can be hard to differentiate from those of infectious meningitis.11 Close monitoring between the initiation of the medication and the onset of clinical symptoms is necessary to assist in distinguishing between aseptic and infectious meningitis.3 If the causative agent is not discontinued, symptoms can quickly worsen, progressing from fever and headache to confusion, coma, and respiratory depression. A DIAM diagnosis can only be made with resolution of aseptic meningitis after stopping the contributory agent. If appropriate, this can be proven by rechallenging the medication in a controlled setting. The usual treatment for aseptic meningitis is supportive care, including hydration, antiemetics, electrolyte supplementation, and adequate analgesia.3
Differential diagnoses in this case included viral infection, meningitis, and allergic reaction to sulfamethoxazole-trimethoprim. The patient reported history of experiencing symptoms after restarting his antibiotic, raising strong suspicion for DIAM. Initiation of this antibiotic was the only recent medication change noted. Laboratory testing for infectious agents yielded negative results, including tests for aerobic and anaerobic bacteria as well as viral and fungal infections. A lumbar puncture and cerebrospinal fluid culture was clear, with no organisms shown on gram stain. Bacterial or viral meningitis was presumed less likely due to the duration of symptoms, correlation of symptoms coinciding with restarting the antibiotic, and negative cerebrospinal fluid culture results.
It was concluded that agranulocytosis and aseptic meningitis were likely induced by sulfamethoxazole-trimethoprim as supported by the improvement upon discontinuing the medication and subsequent worsening upon restarting it. Concurrent agranulocytosis and aseptic meningitis is rare, and there is typically no correlation between the 2 reactions. Since agranulocytosis may be asymptomatic, this case highlights the need to monitor blood cell counts in patients using sulfamethoxazole-trimethoprim. The possibility of DIAM should be considered in patients presenting with flu-like symptoms, and a lumbar puncture may be collected to rule out aseptic meningitis if the patient’s AEs are severe following the initiation of an antibiotic, particularly in immunosuppressed patients taking sulfamethoxazole-trimethoprim. This case is unusual because the patient was healthy and immunocompetent.
This case may not be generalizable and may be difficult to compare to other cases. Every case has patient-specific factors affecting subjective information, including the patient’s baseline, severity of symptoms, and treatment options. This report was based on a single patient case and corresponding results may be found in similar patient cases.
Conclusions
This case emphasizes the rare but serious AEs of acute agranulocytosis complicated with aseptic meningitis after prescribed sulfamethoxazole-trimethoprim. This is a unique case of an immunocompetent patient developing both agranulocytosis and aseptic meningitis after restarting the antibiotic to complete therapy. This case highlights the importance of monitoring blood cell counts and monitoring for signs and symptoms of aseptic meningitis, even during short courses of therapy. Further research is needed to recognize characteristics that may increase the risk for these AEs and to develop strategies for their prevention.
- Garbe E. Non-chemotherapy drug-induced agranulocytosis. Expert Opin Drug Saf. 2007;6(3):323-335. doi:10.1517/14740338.6.3.323
- Jha P, Stromich J, Cohen M, Wainaina JN. A rare complication of trimethoprim-sulfamethoxazole: drug induced aseptic meningitis. Case Rep Infect Dis. 2016;2016:3879406. doi:10.1155/2016/3879406
- Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4(2):285-297. doi:10.1517/14740338.4.2.285
- Jarrin I, Sellier P, Lopes A, et al. Etiologies and management of aseptic meningitis in patients admitted to an internal medicine department. Medicine (Baltimore). 2016;95(2):e2372. doi:10.1097/MD.0000000000002372
- Street EJ, Justice ED, Kopa Z, et al. The 2016 European guideline on the management of epididymo-orchitis. Int J STD AIDS. 2017;28(8):744-749. doi:10.1177/0956462417699356
- Kbirou A, Alafifi M, Sayah M, Dakir M, Debbagh A, Aboutaieb R. Acute orchiepididymitis: epidemiological and clinical aspects: an analysis of 152 cases. Ann Med Surg (Lond). 2022;75:103335. doi:10.1016/j.amsu.2022.103335
- Rattay B, Benndorf RA. Drug-induced idiosyncratic agranulocytosis - infrequent but dangerous. Front Pharmacol. 2021;12:727717. doi:10.3389/fphar.2021.727717
- Elmedani S, Albayati A, Udongwo N, Odak M, Khawaja S. Trimethoprim-sulfamethoxazole-induced aseptic meningitis: a new approach. Cureus. 2021;13(6):e15869. doi:10.7759/cureus.15869
- Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858-883. doi:10.1128/CMR.00007-10
- Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Intern Med. 1999;159(11):1185- 1194. doi:10.1001/archinte.159.11.1185
- Bruner KE, Coop CA, White KM. Trimethoprim-sulfamethoxazole-induced aseptic meningitis-not just another sulfa allergy. Ann Allergy Asthma Immunol. 2014;113(5):520-526. doi:10.1016/j.anai.2014.08.006
- Garbe E. Non-chemotherapy drug-induced agranulocytosis. Expert Opin Drug Saf. 2007;6(3):323-335. doi:10.1517/14740338.6.3.323
- Jha P, Stromich J, Cohen M, Wainaina JN. A rare complication of trimethoprim-sulfamethoxazole: drug induced aseptic meningitis. Case Rep Infect Dis. 2016;2016:3879406. doi:10.1155/2016/3879406
- Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4(2):285-297. doi:10.1517/14740338.4.2.285
- Jarrin I, Sellier P, Lopes A, et al. Etiologies and management of aseptic meningitis in patients admitted to an internal medicine department. Medicine (Baltimore). 2016;95(2):e2372. doi:10.1097/MD.0000000000002372
- Street EJ, Justice ED, Kopa Z, et al. The 2016 European guideline on the management of epididymo-orchitis. Int J STD AIDS. 2017;28(8):744-749. doi:10.1177/0956462417699356
- Kbirou A, Alafifi M, Sayah M, Dakir M, Debbagh A, Aboutaieb R. Acute orchiepididymitis: epidemiological and clinical aspects: an analysis of 152 cases. Ann Med Surg (Lond). 2022;75:103335. doi:10.1016/j.amsu.2022.103335
- Rattay B, Benndorf RA. Drug-induced idiosyncratic agranulocytosis - infrequent but dangerous. Front Pharmacol. 2021;12:727717. doi:10.3389/fphar.2021.727717
- Elmedani S, Albayati A, Udongwo N, Odak M, Khawaja S. Trimethoprim-sulfamethoxazole-induced aseptic meningitis: a new approach. Cureus. 2021;13(6):e15869. doi:10.7759/cureus.15869
- Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858-883. doi:10.1128/CMR.00007-10
- Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Intern Med. 1999;159(11):1185- 1194. doi:10.1001/archinte.159.11.1185
- Bruner KE, Coop CA, White KM. Trimethoprim-sulfamethoxazole-induced aseptic meningitis-not just another sulfa allergy. Ann Allergy Asthma Immunol. 2014;113(5):520-526. doi:10.1016/j.anai.2014.08.006
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Alopecia Induced by Poly-L-Lactic Acid Injection
Cosmetic procedures carry inherent risks of adverse events. Transient and permanent alopecia are rare complications of these procedures. Although they have not been fully elucidated, several pathologic mechanisms for hair loss following cosmetic procedures have been proposed, including extravascular compression (a phenomenon that has been well documented in bedridden patients) as well as intravascular occlusion leading to inflammation and necrosis, which has been associated with hyaluronic acid (HA) fillers.¹ Cases of alopecia also have been reported following mesotherapy and calcium hydroxyapatite, deoxycholic acid, and botulinum toxin injections.² We report a case of alopecia resulting from poly-L-lactic acid (PLLA) injection in a 35-year-old woman with the intent to raise awareness of this rare adverse event.
Case Report
A healthy 35-year-old woman received aesthetic PLLA injections on the face and frontal hairline performed by an outside dermatologist using the vector technique. During the procedure, the patient experienced intense itchiness at the right temporal artery vascular territory and reported a substantial headache the next day. She also presented with erythema and edema of the frontal and right parietal scalp with a well-delimited livedoid vascular area along the temporal artery territory on the right side of the head 1 day after the procedure (Figure 1). These signs were reported to the outside dermatologist who performed the procedure, but they were not assumed to be adverse events at that time.
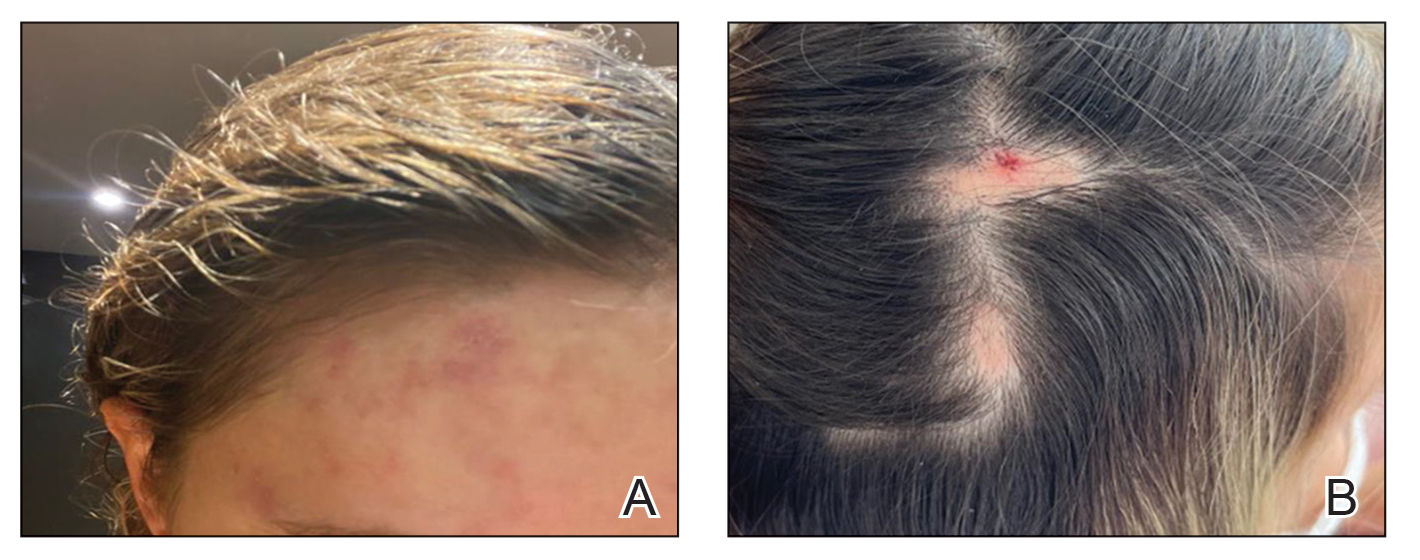
The condition persisted for 4 days followed by the development of an irregular 3×2-cm patch of alopecia on the right parietal scalp. A 3-day course of self-administered oral prednisolone 0.2 mg/kg/d was prescribed.
Twenty-seven days after the procedure, the patient presented to our trichology clinic for evaluation of a single patch of nonscarring alopecia on the right parietal scalp. Trichoscopy showed multiple yellow and black dots, broken hairs, pigment deposits, and an erythematous background mainly composed of linear telangiectatic vessels (Figure 2). Histopathologic analysis revealed a lymphocytic inflammatory infiltrate surrounding the follicular units that was compatible with an alopecia areata–like pattern as well as PLLA deposits in the subcutaneous tissue forming foreign body granulomas (Figure 3). The diagnosis of PLLA-induced alopecia was made based on the detection of PLLA at the biopsy site within the patchy alopecia.
Intralesional triamcinolone acetonide 5 mg/mL was administered at 1-cm intervals in the subdermal space (0.1 mL/puncture site). After 14 days, the patient developed an additional patch of alopecia in the same vascular territory as the right temporal artery, positioned just beneath the initial patch, with similar trichoscopy findings. The patches were treated with intralesional triamcinolone acetonide for 3 additional sessions, administered every 4 weeks. Long-term monitoring of the patient revealed regrowth with comparable hair count to the unaffected contralateral scalp, indicative of a nonscarring alopecia.
Comment
Poly-L-lactic acid is a biostimulator synthesized from the α-hydroxy acid family in 1954 that has been safely used in suture materials, resorbable plates, and orthopedic screws.4 Alopecia has been reported as a systemic allergic reaction to biodegradable screws following an orthopedic procedure.5 Prior reports of embolization and retinal ischemia with PLLA have raised concerns regarding its occlusive potential.6-9
Approved by the US Food and Drug Administration in 2004 for soft tissue restoration in HIV-related lipoatrophy, PLLA was expanded to cosmetic applications in 2009. As previously reported with HA fillers, we hypothesize that extravascular compression resulting from the placement of the filler material (due to the volume injected in the scalp area) contributes to the development of alopecia plus PLLA embolism–induced ischemic alopecia in the affected areas.10 In our case, the diagnosis of PLLA-induced alopecia was confirmed based on the finding of the filler material in the subcutaneous tissue on histopathology, probably due to embolization. Moreover, trichoscopic findings were all similar to those described after HA embolization.11 The features found in our patient due to the PLLA local reaction were similar to those seen in other conditions such as alopecia areata, pressure alopecia, and chemotherapy-induced alopecia; therefore, histopathology confirmation is mandatory in cases of hair loss associated with PLLA.
The emergence of a secondary patch of alopecia prompts consideration of an intrinsic late inflammatory propensity of PLLA. Immune cells recognize PLLA as a foreign body, and subclinical inflammatory foreign body reactions can cause PLLA-induced collagen synthesis.12 This phenomenon underscores the need for further investigation into the immunologic implications of PLLA in alopecia pathogenesis.
The angiogenic properties of the anagen phase require an adequate blood supply for effective hair growth; therefore, the lack of blood and nutrient supply to the hair bulb triggers miniaturization, a possible explanation for the hair thinning found in the alopecic patch.13
Conclusion
Alopecia as an adverse effect of cosmetic procedures can be distressing for patients, even when reversible. A detailed understanding of scalp anatomy is critical for satisfactory outcomes with aesthetic procedures. Physicians must pay attention to the amount and area of material injected in order to avoid possible mechanisms of ischemia—embolization and/or extravascular compression—especially in highly vascularized areas.
We present a rare report of alopecia as an adverse event of PLLA injection. Dermatologists must be aware of this rare condition, and trichoscopy combined with histopathologic analysis are encouraged for early recognition and proper management.
- Issa NT, Kaiser M, Martinez-Velasco A, et al. Alopecia after cosmetic injection procedures: a review. Dermatol Surg. 2022;48:855-861.
- Alopecia with foreign body granulomas induced by Radiesse injection: a case report. J Cosmet Laser Ther. 2018;20:462-464.
- Munia C, Parada M, de Alvarenga Morais MH. Changes in facial morphology using poly-L-lactic acid application according to vector technique: a case series. J Clin Aesthet Dermatol. 2022;15:38-42.
- Attenello NH, Maas CS. Injectable fillers: review of material and properties. Facial Plast Surg. 2015;31:29-34.
- Mastrokalos DS, Paessler HH. Allergic reaction to biodegradable interference poly-L-lactic acid screws after anterior cruciate ligament reconstruction with bone-patellar tendon-bone graft. Arthroscopy. 2008;24:732-733.
- Wu CW, Wu HJ. Retinal artery occlusion following cosmetic injection of poly-L-lactic acid. Taiwan J Ophthalmol. 2021;11:317-320.
- Yuan JT, Chang TW, Yu SS, et al. Mental artery occlusion from poly-L-lactic acid injection at the lateral chin. Dermatol Surg. 2017;43:1402-1405.
- Ragam A, Agemy SA, Dave SB, et al. Ipsilateral ophthalmic and cerebral infarctions after cosmetic polylactic acid injection into the forehead. J Neuroophthalmol. 2017;37:77-80.
- Witmanowski H, Błochowiak K. Another face of dermal fillers. Postepy Dermatol Alergol. 2020;37:651-659.
- Yang Q, Qiu L, Yi C, et al. Reversible alopecia with localized scalp necrosis after accidental embolization of the parietal artery with hyaluronic acid. Aesthetic Plast Surg. 2017;41:695-699.
- Asz-Sigall D, Iñigo-Gomez K, Ortega-Springall MF, et al. Alopecia secondary to hyaluronic acid embolization: trichoscopic findings. Skin Appendage Disord. 2019;5:396-400.
- Oh S, Lee JH, Kim HM, et al. Poly-L-lactic acid fillers improved dermal collagen synthesis by modulating M2 macrophage polarization in aged animal skin. Cells. 2023;12:1320. doi:10.3390/cells12091320
- Natarelli N, Gahoonia N, Sivamani RK. Integrative and mechanistic approach to the hair growth cycle and hair loss. J Clin Med. 2023;12:893.2. Liu RF, Kuo TT, Chao YY, et al.
Cosmetic procedures carry inherent risks of adverse events. Transient and permanent alopecia are rare complications of these procedures. Although they have not been fully elucidated, several pathologic mechanisms for hair loss following cosmetic procedures have been proposed, including extravascular compression (a phenomenon that has been well documented in bedridden patients) as well as intravascular occlusion leading to inflammation and necrosis, which has been associated with hyaluronic acid (HA) fillers.¹ Cases of alopecia also have been reported following mesotherapy and calcium hydroxyapatite, deoxycholic acid, and botulinum toxin injections.² We report a case of alopecia resulting from poly-L-lactic acid (PLLA) injection in a 35-year-old woman with the intent to raise awareness of this rare adverse event.
Case Report
A healthy 35-year-old woman received aesthetic PLLA injections on the face and frontal hairline performed by an outside dermatologist using the vector technique. During the procedure, the patient experienced intense itchiness at the right temporal artery vascular territory and reported a substantial headache the next day. She also presented with erythema and edema of the frontal and right parietal scalp with a well-delimited livedoid vascular area along the temporal artery territory on the right side of the head 1 day after the procedure (Figure 1). These signs were reported to the outside dermatologist who performed the procedure, but they were not assumed to be adverse events at that time.
The condition persisted for 4 days followed by the development of an irregular 3×2-cm patch of alopecia on the right parietal scalp. A 3-day course of self-administered oral prednisolone 0.2 mg/kg/d was prescribed.
Twenty-seven days after the procedure, the patient presented to our trichology clinic for evaluation of a single patch of nonscarring alopecia on the right parietal scalp. Trichoscopy showed multiple yellow and black dots, broken hairs, pigment deposits, and an erythematous background mainly composed of linear telangiectatic vessels (Figure 2). Histopathologic analysis revealed a lymphocytic inflammatory infiltrate surrounding the follicular units that was compatible with an alopecia areata–like pattern as well as PLLA deposits in the subcutaneous tissue forming foreign body granulomas (Figure 3). The diagnosis of PLLA-induced alopecia was made based on the detection of PLLA at the biopsy site within the patchy alopecia.
Intralesional triamcinolone acetonide 5 mg/mL was administered at 1-cm intervals in the subdermal space (0.1 mL/puncture site). After 14 days, the patient developed an additional patch of alopecia in the same vascular territory as the right temporal artery, positioned just beneath the initial patch, with similar trichoscopy findings. The patches were treated with intralesional triamcinolone acetonide for 3 additional sessions, administered every 4 weeks. Long-term monitoring of the patient revealed regrowth with comparable hair count to the unaffected contralateral scalp, indicative of a nonscarring alopecia.
Comment
Poly-L-lactic acid is a biostimulator synthesized from the α-hydroxy acid family in 1954 that has been safely used in suture materials, resorbable plates, and orthopedic screws.4 Alopecia has been reported as a systemic allergic reaction to biodegradable screws following an orthopedic procedure.5 Prior reports of embolization and retinal ischemia with PLLA have raised concerns regarding its occlusive potential.6-9
Approved by the US Food and Drug Administration in 2004 for soft tissue restoration in HIV-related lipoatrophy, PLLA was expanded to cosmetic applications in 2009. As previously reported with HA fillers, we hypothesize that extravascular compression resulting from the placement of the filler material (due to the volume injected in the scalp area) contributes to the development of alopecia plus PLLA embolism–induced ischemic alopecia in the affected areas.10 In our case, the diagnosis of PLLA-induced alopecia was confirmed based on the finding of the filler material in the subcutaneous tissue on histopathology, probably due to embolization. Moreover, trichoscopic findings were all similar to those described after HA embolization.11 The features found in our patient due to the PLLA local reaction were similar to those seen in other conditions such as alopecia areata, pressure alopecia, and chemotherapy-induced alopecia; therefore, histopathology confirmation is mandatory in cases of hair loss associated with PLLA.
The emergence of a secondary patch of alopecia prompts consideration of an intrinsic late inflammatory propensity of PLLA. Immune cells recognize PLLA as a foreign body, and subclinical inflammatory foreign body reactions can cause PLLA-induced collagen synthesis.12 This phenomenon underscores the need for further investigation into the immunologic implications of PLLA in alopecia pathogenesis.
The angiogenic properties of the anagen phase require an adequate blood supply for effective hair growth; therefore, the lack of blood and nutrient supply to the hair bulb triggers miniaturization, a possible explanation for the hair thinning found in the alopecic patch.13
Conclusion
Alopecia as an adverse effect of cosmetic procedures can be distressing for patients, even when reversible. A detailed understanding of scalp anatomy is critical for satisfactory outcomes with aesthetic procedures. Physicians must pay attention to the amount and area of material injected in order to avoid possible mechanisms of ischemia—embolization and/or extravascular compression—especially in highly vascularized areas.
We present a rare report of alopecia as an adverse event of PLLA injection. Dermatologists must be aware of this rare condition, and trichoscopy combined with histopathologic analysis are encouraged for early recognition and proper management.
Cosmetic procedures carry inherent risks of adverse events. Transient and permanent alopecia are rare complications of these procedures. Although they have not been fully elucidated, several pathologic mechanisms for hair loss following cosmetic procedures have been proposed, including extravascular compression (a phenomenon that has been well documented in bedridden patients) as well as intravascular occlusion leading to inflammation and necrosis, which has been associated with hyaluronic acid (HA) fillers.¹ Cases of alopecia also have been reported following mesotherapy and calcium hydroxyapatite, deoxycholic acid, and botulinum toxin injections.² We report a case of alopecia resulting from poly-L-lactic acid (PLLA) injection in a 35-year-old woman with the intent to raise awareness of this rare adverse event.
Case Report
A healthy 35-year-old woman received aesthetic PLLA injections on the face and frontal hairline performed by an outside dermatologist using the vector technique. During the procedure, the patient experienced intense itchiness at the right temporal artery vascular territory and reported a substantial headache the next day. She also presented with erythema and edema of the frontal and right parietal scalp with a well-delimited livedoid vascular area along the temporal artery territory on the right side of the head 1 day after the procedure (Figure 1). These signs were reported to the outside dermatologist who performed the procedure, but they were not assumed to be adverse events at that time.
The condition persisted for 4 days followed by the development of an irregular 3×2-cm patch of alopecia on the right parietal scalp. A 3-day course of self-administered oral prednisolone 0.2 mg/kg/d was prescribed.
Twenty-seven days after the procedure, the patient presented to our trichology clinic for evaluation of a single patch of nonscarring alopecia on the right parietal scalp. Trichoscopy showed multiple yellow and black dots, broken hairs, pigment deposits, and an erythematous background mainly composed of linear telangiectatic vessels (Figure 2). Histopathologic analysis revealed a lymphocytic inflammatory infiltrate surrounding the follicular units that was compatible with an alopecia areata–like pattern as well as PLLA deposits in the subcutaneous tissue forming foreign body granulomas (Figure 3). The diagnosis of PLLA-induced alopecia was made based on the detection of PLLA at the biopsy site within the patchy alopecia.
Intralesional triamcinolone acetonide 5 mg/mL was administered at 1-cm intervals in the subdermal space (0.1 mL/puncture site). After 14 days, the patient developed an additional patch of alopecia in the same vascular territory as the right temporal artery, positioned just beneath the initial patch, with similar trichoscopy findings. The patches were treated with intralesional triamcinolone acetonide for 3 additional sessions, administered every 4 weeks. Long-term monitoring of the patient revealed regrowth with comparable hair count to the unaffected contralateral scalp, indicative of a nonscarring alopecia.
Comment
Poly-L-lactic acid is a biostimulator synthesized from the α-hydroxy acid family in 1954 that has been safely used in suture materials, resorbable plates, and orthopedic screws.4 Alopecia has been reported as a systemic allergic reaction to biodegradable screws following an orthopedic procedure.5 Prior reports of embolization and retinal ischemia with PLLA have raised concerns regarding its occlusive potential.6-9
Approved by the US Food and Drug Administration in 2004 for soft tissue restoration in HIV-related lipoatrophy, PLLA was expanded to cosmetic applications in 2009. As previously reported with HA fillers, we hypothesize that extravascular compression resulting from the placement of the filler material (due to the volume injected in the scalp area) contributes to the development of alopecia plus PLLA embolism–induced ischemic alopecia in the affected areas.10 In our case, the diagnosis of PLLA-induced alopecia was confirmed based on the finding of the filler material in the subcutaneous tissue on histopathology, probably due to embolization. Moreover, trichoscopic findings were all similar to those described after HA embolization.11 The features found in our patient due to the PLLA local reaction were similar to those seen in other conditions such as alopecia areata, pressure alopecia, and chemotherapy-induced alopecia; therefore, histopathology confirmation is mandatory in cases of hair loss associated with PLLA.
The emergence of a secondary patch of alopecia prompts consideration of an intrinsic late inflammatory propensity of PLLA. Immune cells recognize PLLA as a foreign body, and subclinical inflammatory foreign body reactions can cause PLLA-induced collagen synthesis.12 This phenomenon underscores the need for further investigation into the immunologic implications of PLLA in alopecia pathogenesis.
The angiogenic properties of the anagen phase require an adequate blood supply for effective hair growth; therefore, the lack of blood and nutrient supply to the hair bulb triggers miniaturization, a possible explanation for the hair thinning found in the alopecic patch.13
Conclusion
Alopecia as an adverse effect of cosmetic procedures can be distressing for patients, even when reversible. A detailed understanding of scalp anatomy is critical for satisfactory outcomes with aesthetic procedures. Physicians must pay attention to the amount and area of material injected in order to avoid possible mechanisms of ischemia—embolization and/or extravascular compression—especially in highly vascularized areas.
We present a rare report of alopecia as an adverse event of PLLA injection. Dermatologists must be aware of this rare condition, and trichoscopy combined with histopathologic analysis are encouraged for early recognition and proper management.
- Issa NT, Kaiser M, Martinez-Velasco A, et al. Alopecia after cosmetic injection procedures: a review. Dermatol Surg. 2022;48:855-861.
- Alopecia with foreign body granulomas induced by Radiesse injection: a case report. J Cosmet Laser Ther. 2018;20:462-464.
- Munia C, Parada M, de Alvarenga Morais MH. Changes in facial morphology using poly-L-lactic acid application according to vector technique: a case series. J Clin Aesthet Dermatol. 2022;15:38-42.
- Attenello NH, Maas CS. Injectable fillers: review of material and properties. Facial Plast Surg. 2015;31:29-34.
- Mastrokalos DS, Paessler HH. Allergic reaction to biodegradable interference poly-L-lactic acid screws after anterior cruciate ligament reconstruction with bone-patellar tendon-bone graft. Arthroscopy. 2008;24:732-733.
- Wu CW, Wu HJ. Retinal artery occlusion following cosmetic injection of poly-L-lactic acid. Taiwan J Ophthalmol. 2021;11:317-320.
- Yuan JT, Chang TW, Yu SS, et al. Mental artery occlusion from poly-L-lactic acid injection at the lateral chin. Dermatol Surg. 2017;43:1402-1405.
- Ragam A, Agemy SA, Dave SB, et al. Ipsilateral ophthalmic and cerebral infarctions after cosmetic polylactic acid injection into the forehead. J Neuroophthalmol. 2017;37:77-80.
- Witmanowski H, Błochowiak K. Another face of dermal fillers. Postepy Dermatol Alergol. 2020;37:651-659.
- Yang Q, Qiu L, Yi C, et al. Reversible alopecia with localized scalp necrosis after accidental embolization of the parietal artery with hyaluronic acid. Aesthetic Plast Surg. 2017;41:695-699.
- Asz-Sigall D, Iñigo-Gomez K, Ortega-Springall MF, et al. Alopecia secondary to hyaluronic acid embolization: trichoscopic findings. Skin Appendage Disord. 2019;5:396-400.
- Oh S, Lee JH, Kim HM, et al. Poly-L-lactic acid fillers improved dermal collagen synthesis by modulating M2 macrophage polarization in aged animal skin. Cells. 2023;12:1320. doi:10.3390/cells12091320
- Natarelli N, Gahoonia N, Sivamani RK. Integrative and mechanistic approach to the hair growth cycle and hair loss. J Clin Med. 2023;12:893.2. Liu RF, Kuo TT, Chao YY, et al.
- Issa NT, Kaiser M, Martinez-Velasco A, et al. Alopecia after cosmetic injection procedures: a review. Dermatol Surg. 2022;48:855-861.
- Alopecia with foreign body granulomas induced by Radiesse injection: a case report. J Cosmet Laser Ther. 2018;20:462-464.
- Munia C, Parada M, de Alvarenga Morais MH. Changes in facial morphology using poly-L-lactic acid application according to vector technique: a case series. J Clin Aesthet Dermatol. 2022;15:38-42.
- Attenello NH, Maas CS. Injectable fillers: review of material and properties. Facial Plast Surg. 2015;31:29-34.
- Mastrokalos DS, Paessler HH. Allergic reaction to biodegradable interference poly-L-lactic acid screws after anterior cruciate ligament reconstruction with bone-patellar tendon-bone graft. Arthroscopy. 2008;24:732-733.
- Wu CW, Wu HJ. Retinal artery occlusion following cosmetic injection of poly-L-lactic acid. Taiwan J Ophthalmol. 2021;11:317-320.
- Yuan JT, Chang TW, Yu SS, et al. Mental artery occlusion from poly-L-lactic acid injection at the lateral chin. Dermatol Surg. 2017;43:1402-1405.
- Ragam A, Agemy SA, Dave SB, et al. Ipsilateral ophthalmic and cerebral infarctions after cosmetic polylactic acid injection into the forehead. J Neuroophthalmol. 2017;37:77-80.
- Witmanowski H, Błochowiak K. Another face of dermal fillers. Postepy Dermatol Alergol. 2020;37:651-659.
- Yang Q, Qiu L, Yi C, et al. Reversible alopecia with localized scalp necrosis after accidental embolization of the parietal artery with hyaluronic acid. Aesthetic Plast Surg. 2017;41:695-699.
- Asz-Sigall D, Iñigo-Gomez K, Ortega-Springall MF, et al. Alopecia secondary to hyaluronic acid embolization: trichoscopic findings. Skin Appendage Disord. 2019;5:396-400.
- Oh S, Lee JH, Kim HM, et al. Poly-L-lactic acid fillers improved dermal collagen synthesis by modulating M2 macrophage polarization in aged animal skin. Cells. 2023;12:1320. doi:10.3390/cells12091320
- Natarelli N, Gahoonia N, Sivamani RK. Integrative and mechanistic approach to the hair growth cycle and hair loss. J Clin Med. 2023;12:893.2. Liu RF, Kuo TT, Chao YY, et al.
Practice Points
- Alopecia is a potential adverse event of poly-L-lactic acid (PLLA) injection, and prior reports of embolization and retinal ischemia with PLLA use raise the concern of its occlusive potential.
- The combination of extravascular compression due to the presence of the filler material in the subcutaneous tissue as well as intravascular PLLA embolism may contribute to tissue ischemia–induced alopecia in the affected areas.
- Poly-L-lactic acid also may cause a local inflammatory reaction that is alopecia areata–like, which would explain its similar trichoscopy findings.
Beware the Manchineel: A Case of Irritant Contact Dermatitis
What is the world’s most dangerous tree? According to Guinness World Records1 (and one unlucky contestant on the wilderness survival reality show Naked and Afraid,2 who got its sap in his eyes and needed to be evacuated for treatment), the manchineel tree (Hippomane mancinella) has earned this designation.1-3 Manchineel trees are part of the strand vegetation of islands in the West Indies and along the Caribbean coasts of South and Central America, where their copious root systems help reduce coastal erosion. In the United States, this poisonous tree grows along the southern edge of Florida’s Everglades National Park; the Florida Keys; and the US Virgin Islands, especially Virgin Islands National Park. Although the manchineel tree appears on several endangered species lists,4-6 there are places within its distribution where it is locally abundant and thus poses a risk to residents and visitors.
The first European description of manchineel toxicity was by Peter Martyr d’Anghiera, a court historian and geographer of Christopher Columbus’s patroness, Isabella I, Queen of Castile and Léon. In the early 1500s, Peter Martyr wrote that on Columbus’s second New World voyage in 1493, the crew encountered a mysterious tree that burned the skin and eyes of anyone who had contact with it.7 Columbus called the tree’s fruit manzanilla de la muerte (“little apple of death”) after several sailors became severely ill from eating the fruit.8,9 Manchineel lore is rife with tales of agonizing death after eating the applelike fruit, and several contemporaneous accounts describe indigenous Caribbean islanders using manchineel’s toxic sap as an arrow poison.10
Eating manchineel fruit is known to cause abdominal pain, burning sensations in the oropharynx, and esophageal spasms.11 Several case reports mention that consuming the fruit can create an exaggerated
Case Report
A 64-year-old physician (S.A.N.) came across a stand of manchineel trees while camping in the Virgin Islands National Park on St. John in the US Virgin Islands (Figure 1). The patient—who was knowledgeable about tropical ecology and was familiar with the tree—was curious about its purported cutaneous toxicity and applied the viscous white sap of a broken branchlet (Figure 2) to a patch of skin measuring 4 cm in diameter on the medial left calf. He took serial photographs of the site on days 2, 4 (Figure 3), 6, and 10 (Figure 4), showing the onset of erythema and the subsequent development of follicular pustules. On day 6, a 4-mm punch biopsy specimen was taken of the most prominent pustule. Histopathology showed a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which was consistent with irritant contact dermatitis (Figure 5). On day 8, the region became indurated and tender to pressure; however, there was no warmth, edema, purulent drainage, lymphangitic streaks, or other signs of infection. The region was never itchy; it was uncomfortable only with firm direct pressure. The patient applied hot compresses to the site for 10 minutes 1 to 2 times daily for roughly 2 weeks, and the affected area healed fully (without any additional intervention) in approximately 6 weeks.


Comment
Manchineel is a member of the Euphorbiaceae (also known as the euphorb or spurge) family, a mainly tropical or subtropical plant family that includes many useful as well as many toxic species. Examples of useful plants include cassava (Manihot esculenta) and the rubber tree (Hevea brasiliensis). Many euphorbs have well-described toxicities, and many (eg, castor bean, Ricinus communis) are useful in some circumstances and toxic in others.6,12-14 Many euphorbs are known to cause skin reactions, usually due to toxins in the milky sap that directly irritate the skin or to latex compounds that can induce IgE-mediated contact dermatitis.9,14
Manchineel contains a complex mix of toxins, though no specific one has been identified as the main cause of the associated irritant contact dermatitis. Manchineel sap (and sap of many other euphorbs) contains phorbol esters that may cause direct pH-induced cytotoxicity leading to keratinocyte necrosis. Diterpenes may augment this cytotoxic effect via induction of proinflammatory cytokines.12 Pitts et al5 pointed to a mixture of oxygenated diterpene esters as the primary cause of toxicity and suggested that their water solubility explained occurrences of keratoconjunctivitis after contact with rainwater or dew from the manchineel tree.
All parts of the manchineel tree—fruit, leaves, wood, and sap—are poisonous. In a retrospective series of 97 cases of manchineel fruit ingestion, the most common symptoms were oropharyngeal pain (68% [66/97]), abdominal pain (42% [41/97]), and diarrhea (37% [36/97]). The same series identified 1 (1%) case of bradycardia and hypotension.3 Contact with the wood, exposure to sawdust, and inhalation of smoke from burning the wood can irritate the skin, conjunctivae, or nasopharynx. Rainwater or dew dripping from the leaves onto the skin can cause dermatitis and ophthalmitis, even without direct contact with the tree.4,5
Management—There is no specific treatment for manchineel dermatitis. Because it is an irritant reaction and not a type IV hypersensitivity reaction, topical corticosteroids have minimal benefit. A regimen consisting of a thorough cleansing, wet compresses, and observation, as most symptoms resolve spontaneously within a few days, has been recommended.4 Our patient used hot compresses, which he believes helped heal the site, although his symptoms lasted for several weeks.
Given that there is no specific treatment for manchineel dermatitis, the wisest approach is strict avoidance. On many Caribbean islands, visitors are warned about the manchineel tree, advised to avoid direct contact, and reminded to avoid standing beneath it during a rainstorm (Figure 6).

Conclusion
This article begins with a question: “What is the world’s most dangerous tree?” Many sources from the indexed medical literature as well as the popular press and social media state that it is the manchineel. Although all parts of the manchineel tree are highly toxic, human exposures are uncommon, and deaths are more apocryphal than actual.
- Most dangerous tree. Guinness World Records. Accessed October 14, 2024. https://www.guinnessworldrecords.com/world-records/most-dangerous-tree
- Naked and Afraid: Garden of Evil (S4E9). Discovery Channel. June 21, 2015. Accessed October 14, 2024. https://go.discovery.com/video/naked-and-afraid-discovery/garden-of-evil
- Boucaud-Maitre D, Cachet X, Bouzidi C, et al. Severity of manchineel fruit (Hippomane mancinella) poisoning: a retrospective case series of 97 patients from French Poison Control Centers. Toxicon. 2019;161:28-32. doi:10.1016/j.toxicon.2019.02.014
- Blue LM, Sailing C, Denapoles C, et al. Manchineel dermatitis in North American students in the Caribbean. J Travel Medicine. 2011;18:422-424. doi:10.1111/j.1708-8305.2011.00568.x
- Pitts JF, Barker NH, Gibbons DC, et al. Manchineel keratoconjunctivitis. Br J Ophthalmol. 1993;77:284-288. doi:10.1136/bjo.77.5.284
- Lauter WM, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella, L. I. historical review. J Pharm Sci. 1952;41:199-201. https://doi.org/10.1002/jps.3030410412
- Martyr P. De Orbe Novo: the Eight Decades of Peter Martyr d’Anghera. Vol 1. FA MacNutt (translator). GP Putnam’s Sons; 1912. Accessed October 14, 2024. https://gutenberg.org/cache/epub/12425/pg12425.txt
- Fernandez de Ybarra AM. A forgotten medical worthy, Dr. Diego Alvarex Chanca, of Seville, Spain, and his letter describing the second voyage of Christopher Columbus to America. Med Library Hist J. 1906;4:246-263.
- Muscat MK. Manchineel apple of death. EJIFCC. 2019;30:346-348.
- Handler JS. Aspects of Amerindian ethnography in 17th century Barbados. Caribbean Studies. 1970;9:50-72.
- Howard RA. Three experiences with the manchineel (Hippomane spp., Euphorbiaceae). Biotropica. 1981;13:224-227. https://doi.org/10.2307/2388129
- Rao KV. Toxic principles of Hippomane mancinella. Planta Med. 1974;25:166-171. doi:10.1055/s-0028-1097927
- Lauter WM, Foote PA. Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc. 1955;44:361-363. doi:10.1002/jps.3030440616
- Carroll MN Jr, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella L. III. Toxic actions of extracts of Hippomane mancinella L. J Am Pharm Assoc. 1957;46:93-97. doi:10.1002/jps.3030460206
What is the world’s most dangerous tree? According to Guinness World Records1 (and one unlucky contestant on the wilderness survival reality show Naked and Afraid,2 who got its sap in his eyes and needed to be evacuated for treatment), the manchineel tree (Hippomane mancinella) has earned this designation.1-3 Manchineel trees are part of the strand vegetation of islands in the West Indies and along the Caribbean coasts of South and Central America, where their copious root systems help reduce coastal erosion. In the United States, this poisonous tree grows along the southern edge of Florida’s Everglades National Park; the Florida Keys; and the US Virgin Islands, especially Virgin Islands National Park. Although the manchineel tree appears on several endangered species lists,4-6 there are places within its distribution where it is locally abundant and thus poses a risk to residents and visitors.
The first European description of manchineel toxicity was by Peter Martyr d’Anghiera, a court historian and geographer of Christopher Columbus’s patroness, Isabella I, Queen of Castile and Léon. In the early 1500s, Peter Martyr wrote that on Columbus’s second New World voyage in 1493, the crew encountered a mysterious tree that burned the skin and eyes of anyone who had contact with it.7 Columbus called the tree’s fruit manzanilla de la muerte (“little apple of death”) after several sailors became severely ill from eating the fruit.8,9 Manchineel lore is rife with tales of agonizing death after eating the applelike fruit, and several contemporaneous accounts describe indigenous Caribbean islanders using manchineel’s toxic sap as an arrow poison.10
Eating manchineel fruit is known to cause abdominal pain, burning sensations in the oropharynx, and esophageal spasms.11 Several case reports mention that consuming the fruit can create an exaggerated
Case Report
A 64-year-old physician (S.A.N.) came across a stand of manchineel trees while camping in the Virgin Islands National Park on St. John in the US Virgin Islands (Figure 1). The patient—who was knowledgeable about tropical ecology and was familiar with the tree—was curious about its purported cutaneous toxicity and applied the viscous white sap of a broken branchlet (Figure 2) to a patch of skin measuring 4 cm in diameter on the medial left calf. He took serial photographs of the site on days 2, 4 (Figure 3), 6, and 10 (Figure 4), showing the onset of erythema and the subsequent development of follicular pustules. On day 6, a 4-mm punch biopsy specimen was taken of the most prominent pustule. Histopathology showed a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which was consistent with irritant contact dermatitis (Figure 5). On day 8, the region became indurated and tender to pressure; however, there was no warmth, edema, purulent drainage, lymphangitic streaks, or other signs of infection. The region was never itchy; it was uncomfortable only with firm direct pressure. The patient applied hot compresses to the site for 10 minutes 1 to 2 times daily for roughly 2 weeks, and the affected area healed fully (without any additional intervention) in approximately 6 weeks.
Comment
Manchineel is a member of the Euphorbiaceae (also known as the euphorb or spurge) family, a mainly tropical or subtropical plant family that includes many useful as well as many toxic species. Examples of useful plants include cassava (Manihot esculenta) and the rubber tree (Hevea brasiliensis). Many euphorbs have well-described toxicities, and many (eg, castor bean, Ricinus communis) are useful in some circumstances and toxic in others.6,12-14 Many euphorbs are known to cause skin reactions, usually due to toxins in the milky sap that directly irritate the skin or to latex compounds that can induce IgE-mediated contact dermatitis.9,14
Manchineel contains a complex mix of toxins, though no specific one has been identified as the main cause of the associated irritant contact dermatitis. Manchineel sap (and sap of many other euphorbs) contains phorbol esters that may cause direct pH-induced cytotoxicity leading to keratinocyte necrosis. Diterpenes may augment this cytotoxic effect via induction of proinflammatory cytokines.12 Pitts et al5 pointed to a mixture of oxygenated diterpene esters as the primary cause of toxicity and suggested that their water solubility explained occurrences of keratoconjunctivitis after contact with rainwater or dew from the manchineel tree.
All parts of the manchineel tree—fruit, leaves, wood, and sap—are poisonous. In a retrospective series of 97 cases of manchineel fruit ingestion, the most common symptoms were oropharyngeal pain (68% [66/97]), abdominal pain (42% [41/97]), and diarrhea (37% [36/97]). The same series identified 1 (1%) case of bradycardia and hypotension.3 Contact with the wood, exposure to sawdust, and inhalation of smoke from burning the wood can irritate the skin, conjunctivae, or nasopharynx. Rainwater or dew dripping from the leaves onto the skin can cause dermatitis and ophthalmitis, even without direct contact with the tree.4,5
Management—There is no specific treatment for manchineel dermatitis. Because it is an irritant reaction and not a type IV hypersensitivity reaction, topical corticosteroids have minimal benefit. A regimen consisting of a thorough cleansing, wet compresses, and observation, as most symptoms resolve spontaneously within a few days, has been recommended.4 Our patient used hot compresses, which he believes helped heal the site, although his symptoms lasted for several weeks.
Given that there is no specific treatment for manchineel dermatitis, the wisest approach is strict avoidance. On many Caribbean islands, visitors are warned about the manchineel tree, advised to avoid direct contact, and reminded to avoid standing beneath it during a rainstorm (Figure 6).
Conclusion
This article begins with a question: “What is the world’s most dangerous tree?” Many sources from the indexed medical literature as well as the popular press and social media state that it is the manchineel. Although all parts of the manchineel tree are highly toxic, human exposures are uncommon, and deaths are more apocryphal than actual.
What is the world’s most dangerous tree? According to Guinness World Records1 (and one unlucky contestant on the wilderness survival reality show Naked and Afraid,2 who got its sap in his eyes and needed to be evacuated for treatment), the manchineel tree (Hippomane mancinella) has earned this designation.1-3 Manchineel trees are part of the strand vegetation of islands in the West Indies and along the Caribbean coasts of South and Central America, where their copious root systems help reduce coastal erosion. In the United States, this poisonous tree grows along the southern edge of Florida’s Everglades National Park; the Florida Keys; and the US Virgin Islands, especially Virgin Islands National Park. Although the manchineel tree appears on several endangered species lists,4-6 there are places within its distribution where it is locally abundant and thus poses a risk to residents and visitors.
The first European description of manchineel toxicity was by Peter Martyr d’Anghiera, a court historian and geographer of Christopher Columbus’s patroness, Isabella I, Queen of Castile and Léon. In the early 1500s, Peter Martyr wrote that on Columbus’s second New World voyage in 1493, the crew encountered a mysterious tree that burned the skin and eyes of anyone who had contact with it.7 Columbus called the tree’s fruit manzanilla de la muerte (“little apple of death”) after several sailors became severely ill from eating the fruit.8,9 Manchineel lore is rife with tales of agonizing death after eating the applelike fruit, and several contemporaneous accounts describe indigenous Caribbean islanders using manchineel’s toxic sap as an arrow poison.10
Eating manchineel fruit is known to cause abdominal pain, burning sensations in the oropharynx, and esophageal spasms.11 Several case reports mention that consuming the fruit can create an exaggerated
Case Report
A 64-year-old physician (S.A.N.) came across a stand of manchineel trees while camping in the Virgin Islands National Park on St. John in the US Virgin Islands (Figure 1). The patient—who was knowledgeable about tropical ecology and was familiar with the tree—was curious about its purported cutaneous toxicity and applied the viscous white sap of a broken branchlet (Figure 2) to a patch of skin measuring 4 cm in diameter on the medial left calf. He took serial photographs of the site on days 2, 4 (Figure 3), 6, and 10 (Figure 4), showing the onset of erythema and the subsequent development of follicular pustules. On day 6, a 4-mm punch biopsy specimen was taken of the most prominent pustule. Histopathology showed a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which was consistent with irritant contact dermatitis (Figure 5). On day 8, the region became indurated and tender to pressure; however, there was no warmth, edema, purulent drainage, lymphangitic streaks, or other signs of infection. The region was never itchy; it was uncomfortable only with firm direct pressure. The patient applied hot compresses to the site for 10 minutes 1 to 2 times daily for roughly 2 weeks, and the affected area healed fully (without any additional intervention) in approximately 6 weeks.
Comment
Manchineel is a member of the Euphorbiaceae (also known as the euphorb or spurge) family, a mainly tropical or subtropical plant family that includes many useful as well as many toxic species. Examples of useful plants include cassava (Manihot esculenta) and the rubber tree (Hevea brasiliensis). Many euphorbs have well-described toxicities, and many (eg, castor bean, Ricinus communis) are useful in some circumstances and toxic in others.6,12-14 Many euphorbs are known to cause skin reactions, usually due to toxins in the milky sap that directly irritate the skin or to latex compounds that can induce IgE-mediated contact dermatitis.9,14
Manchineel contains a complex mix of toxins, though no specific one has been identified as the main cause of the associated irritant contact dermatitis. Manchineel sap (and sap of many other euphorbs) contains phorbol esters that may cause direct pH-induced cytotoxicity leading to keratinocyte necrosis. Diterpenes may augment this cytotoxic effect via induction of proinflammatory cytokines.12 Pitts et al5 pointed to a mixture of oxygenated diterpene esters as the primary cause of toxicity and suggested that their water solubility explained occurrences of keratoconjunctivitis after contact with rainwater or dew from the manchineel tree.
All parts of the manchineel tree—fruit, leaves, wood, and sap—are poisonous. In a retrospective series of 97 cases of manchineel fruit ingestion, the most common symptoms were oropharyngeal pain (68% [66/97]), abdominal pain (42% [41/97]), and diarrhea (37% [36/97]). The same series identified 1 (1%) case of bradycardia and hypotension.3 Contact with the wood, exposure to sawdust, and inhalation of smoke from burning the wood can irritate the skin, conjunctivae, or nasopharynx. Rainwater or dew dripping from the leaves onto the skin can cause dermatitis and ophthalmitis, even without direct contact with the tree.4,5
Management—There is no specific treatment for manchineel dermatitis. Because it is an irritant reaction and not a type IV hypersensitivity reaction, topical corticosteroids have minimal benefit. A regimen consisting of a thorough cleansing, wet compresses, and observation, as most symptoms resolve spontaneously within a few days, has been recommended.4 Our patient used hot compresses, which he believes helped heal the site, although his symptoms lasted for several weeks.
Given that there is no specific treatment for manchineel dermatitis, the wisest approach is strict avoidance. On many Caribbean islands, visitors are warned about the manchineel tree, advised to avoid direct contact, and reminded to avoid standing beneath it during a rainstorm (Figure 6).
Conclusion
This article begins with a question: “What is the world’s most dangerous tree?” Many sources from the indexed medical literature as well as the popular press and social media state that it is the manchineel. Although all parts of the manchineel tree are highly toxic, human exposures are uncommon, and deaths are more apocryphal than actual.
- Most dangerous tree. Guinness World Records. Accessed October 14, 2024. https://www.guinnessworldrecords.com/world-records/most-dangerous-tree
- Naked and Afraid: Garden of Evil (S4E9). Discovery Channel. June 21, 2015. Accessed October 14, 2024. https://go.discovery.com/video/naked-and-afraid-discovery/garden-of-evil
- Boucaud-Maitre D, Cachet X, Bouzidi C, et al. Severity of manchineel fruit (Hippomane mancinella) poisoning: a retrospective case series of 97 patients from French Poison Control Centers. Toxicon. 2019;161:28-32. doi:10.1016/j.toxicon.2019.02.014
- Blue LM, Sailing C, Denapoles C, et al. Manchineel dermatitis in North American students in the Caribbean. J Travel Medicine. 2011;18:422-424. doi:10.1111/j.1708-8305.2011.00568.x
- Pitts JF, Barker NH, Gibbons DC, et al. Manchineel keratoconjunctivitis. Br J Ophthalmol. 1993;77:284-288. doi:10.1136/bjo.77.5.284
- Lauter WM, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella, L. I. historical review. J Pharm Sci. 1952;41:199-201. https://doi.org/10.1002/jps.3030410412
- Martyr P. De Orbe Novo: the Eight Decades of Peter Martyr d’Anghera. Vol 1. FA MacNutt (translator). GP Putnam’s Sons; 1912. Accessed October 14, 2024. https://gutenberg.org/cache/epub/12425/pg12425.txt
- Fernandez de Ybarra AM. A forgotten medical worthy, Dr. Diego Alvarex Chanca, of Seville, Spain, and his letter describing the second voyage of Christopher Columbus to America. Med Library Hist J. 1906;4:246-263.
- Muscat MK. Manchineel apple of death. EJIFCC. 2019;30:346-348.
- Handler JS. Aspects of Amerindian ethnography in 17th century Barbados. Caribbean Studies. 1970;9:50-72.
- Howard RA. Three experiences with the manchineel (Hippomane spp., Euphorbiaceae). Biotropica. 1981;13:224-227. https://doi.org/10.2307/2388129
- Rao KV. Toxic principles of Hippomane mancinella. Planta Med. 1974;25:166-171. doi:10.1055/s-0028-1097927
- Lauter WM, Foote PA. Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc. 1955;44:361-363. doi:10.1002/jps.3030440616
- Carroll MN Jr, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella L. III. Toxic actions of extracts of Hippomane mancinella L. J Am Pharm Assoc. 1957;46:93-97. doi:10.1002/jps.3030460206
- Most dangerous tree. Guinness World Records. Accessed October 14, 2024. https://www.guinnessworldrecords.com/world-records/most-dangerous-tree
- Naked and Afraid: Garden of Evil (S4E9). Discovery Channel. June 21, 2015. Accessed October 14, 2024. https://go.discovery.com/video/naked-and-afraid-discovery/garden-of-evil
- Boucaud-Maitre D, Cachet X, Bouzidi C, et al. Severity of manchineel fruit (Hippomane mancinella) poisoning: a retrospective case series of 97 patients from French Poison Control Centers. Toxicon. 2019;161:28-32. doi:10.1016/j.toxicon.2019.02.014
- Blue LM, Sailing C, Denapoles C, et al. Manchineel dermatitis in North American students in the Caribbean. J Travel Medicine. 2011;18:422-424. doi:10.1111/j.1708-8305.2011.00568.x
- Pitts JF, Barker NH, Gibbons DC, et al. Manchineel keratoconjunctivitis. Br J Ophthalmol. 1993;77:284-288. doi:10.1136/bjo.77.5.284
- Lauter WM, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella, L. I. historical review. J Pharm Sci. 1952;41:199-201. https://doi.org/10.1002/jps.3030410412
- Martyr P. De Orbe Novo: the Eight Decades of Peter Martyr d’Anghera. Vol 1. FA MacNutt (translator). GP Putnam’s Sons; 1912. Accessed October 14, 2024. https://gutenberg.org/cache/epub/12425/pg12425.txt
- Fernandez de Ybarra AM. A forgotten medical worthy, Dr. Diego Alvarex Chanca, of Seville, Spain, and his letter describing the second voyage of Christopher Columbus to America. Med Library Hist J. 1906;4:246-263.
- Muscat MK. Manchineel apple of death. EJIFCC. 2019;30:346-348.
- Handler JS. Aspects of Amerindian ethnography in 17th century Barbados. Caribbean Studies. 1970;9:50-72.
- Howard RA. Three experiences with the manchineel (Hippomane spp., Euphorbiaceae). Biotropica. 1981;13:224-227. https://doi.org/10.2307/2388129
- Rao KV. Toxic principles of Hippomane mancinella. Planta Med. 1974;25:166-171. doi:10.1055/s-0028-1097927
- Lauter WM, Foote PA. Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc. 1955;44:361-363. doi:10.1002/jps.3030440616
- Carroll MN Jr, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella L. III. Toxic actions of extracts of Hippomane mancinella L. J Am Pharm Assoc. 1957;46:93-97. doi:10.1002/jps.3030460206
PRACTICE POINTS
- Sap from the manchineel tree—found on the coasts of Caribbean islands, the Atlantic coastline of Central and northern South America, and parts of southernmost Florida—can cause severe dermatologic and ophthalmologic injuries. Eating its fruit can lead to oropharyngeal pain and diarrhea.
- Histopathology of manchineel dermatitis reveals a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which is consistent with irritant contact dermatitis.
- There is no specific treatment for manchineel dermatitis. Case reports advocate a thorough cleansing, application of wet compresses, and observation.