User login
52-year-old man • erectile dysfunction • insomnia • migraine headaches • disclosure of infidelity
THE CASE
A 52-year-old man requested medicine to help him with erectile dysfunction. After obtaining a medical history and performing a physical exam, the family physician (FP) asked for more details about the patient’s situation. He reported that his wife, who had recently seen the same FP for counseling related to her frustrations with her husband, was uninterested in sex. He then added that he was having an affair with a 32-year-old female co-worker and wanted to improve his sexual function.
He admitted to feeling guilty about this situation and was conflicted about whether to end the affair. He also stated that since the affair, his insomnia had worsened, he was drinking more alcohol, and he was having migraine headaches. As the FP for both patients, and with the knowledge that the wife was worried about possible infidelity, the physician felt some level of conflict about the situation. The following is a discussion of the issues that this patient encounter raised.
DISCUSSION
Issues related to infidelity are common to both men and women. They are also common in same-sex relationships; in general, however, lesbian couples have fewer outside partners, whereas gay men are more likely to seek variety by having multiple partners.1
It is widely understood that successfully committed couples spend quality time together, emphasize each other’s strengths, show respect, accept influence, and nurture their friendship. However, many couples experience infidelity at some time in the course of their marriage. It is difficult to put an exact estimate on rates of infidelity due to problems with research methodology, inaccurate reporting, and a lack of agreement on a definition for infidelity.2 General categories of infidelity include emotional only, sexual only, and combined sexual and emotional infidelity.3,4 In terms of sexual infidelity, one study found that 25% of married men and 15% of married women admitted to having had extramarital sex at least once during their relationship.5 However, other studies suggest that women are closing the “sexual infidelity” gap and engaging in sexual affairs at a similar rate to men.6 There are websites that, in fact, have made it easier for married individuals to engage in affairs.
Reasons for infidelity. Men and women often have different motives in engaging in infidelity. In general, men’s motivations are more often related to sexual dissatisfaction and women’s to emotional dissatisfaction.7,8 However, infidelity may not always be the result of marital unhappiness.
Some studies suggest that the presence of opportunity may override the positive aspects of a relationship.9 Opportunity is heightened in the work environment, as reflected by the finding that 50% of infidelity occurs in the office.10 Research suggests that all relationships may be vulnerable to infidelity if the right opportunities present themselves.11
In general, health care providers are encouraged to use caution in generalizing about infidelity, as the subject is extremely complex, nuanced, and difficult to measure with exactitude.12
Continue to: The impact of infidelity
The impact of infidelity on couples varies due to factors such as the pre-morbid health of the marriage,13 the depth of involvement with the affair partner,14 and pre-existing attitudes about infidelity.13
Infidelity is a common cause of divorce in America. However, in a sample, Schneider et al15 found that despite initial threats to leave the marriage after infidelity, less than one-quarter of partners divorced. Other studies have found that disclosure of the infidelity and a commitment to work on the marriage may be an essential component of healing.16
Emotionally focused couples therapy, with its emphasis on attachment and bonding, may hold promise for helping couples successfully work through the trauma brought on by extramarital relationships.17 Psychologist and infidelity researcher Shirley Glass found that of the two-thirds of couples who chose to stay together after an affair, 80% of them reported a better marriage after treatment.11
Initial steps to take, and questions to ask
Both male and female patients need to feel comfortable surfacing sexual concerns with their clinicians. In this case, the concerns of the husband are interwoven with broader marital issues, which are the source of emotional and psychosomatic distress. His decision regarding his affair carried with it potentially life-altering consequences for his wife, 3 children, and affair partner and her family. It also raised ethical issues for the FP, who was providing care to both the husband and the wife. Appropriate care requires that a physician in this situation
- demonstrate a nonjudgmental approach
- clarify personal ethics in response to patient behaviors
- maintain confidentiality
- apply an ethical framework to resolve value dilemmas
- avoid actions that would be harmful to patients.
Interviewing can help to elicit information that may be clarifying not only to the physician but also to the patient. When interviewing a patient such as the one in this case, it would be wise to ask:
- How long has the affair been going on?
- Why is the patient engaging in the affair?
- Is abuse (emotional or physical) a factor in the marriage?
- Does the patient still have feelings for their spouse? Does the patient want to work on the marriage?
- Has the patient talked to a friend or therapist about the situation?
- Would the patient be willing to talk to a therapist?
Continue to: Ethical and legal considerations
Ethical and legal considerations
Some therapists espouse the view that being “neutral” in the presence of an affair is as much a value judgment as taking one side or the other. In the presence of emotional or physical abuse, it might indeed be best to support a marital separation. However, in other situations when there are young children involved and the patient is undecided about what to do, the FP can discuss the pros and cons of working on a marriage that suffers from more treatable types of disrepair (ie, stress, disconnection, repetitive arguments).
Provision of care. If the patient is unwilling to end the affair, the physician needs to decide whether they feel ethically at ease with prescribing sexually enhancing performance medication, given that the patient’s wife is also a patient. A physician in this situation might feel that they are betraying the wife by providing such medications to the husband. In such cases, it might be appropriate to refer the husband to a colleague.
In all cases of infidelity, however, it is wise to discuss safe-sex practices in order to limit the risk of transmitting a sexually transmitted infection (STI) to the spouse (or affair partner) and offer testing for STIs.
Confidentiality. Despite feelings the physician might have about betraying the wife’s trust by providing the performance-enhancing medicine to the husband, there is very little justification for revealing the affair to the wife. In general, confidentiality can only be broken if there is a high level of imminent danger associated with nondisclosure. The physician needs to realize the serious legal implications of breaking confidentiality in this situation, as such disclosure may prompt the initiation of divorce proceedings.
Real-world recommendations
Check your own biases. Infidelity can trigger a whole host of emotional reactions in physicians based on their own personal and professional history. It is important to be aware of such emotions and if sufficiently triggered, discuss the case with a colleague.
Continue to: Encourage bibliotherapy and marriage therapy
Encourage bibliotherapy and marriage therapy. The conversation might go something like this:
“I would recommend you do some reading about infidelity. If you are interested in working on your marriage, you might want to consider a couples counselor who can help you. Research shows that while such counseling can help couples work through infidelity, disclosure needs to occur as part of that process. Research also indicates that about two-thirds of marriages stay together after the revelation of an affair and that such couples can experience healing if they commit to a therapeutic process. If you are unsure how you want to proceed, it might be helpful for you to explore your situation with an individual therapist. What would you like to do next?”
There are also written resources that the patient might find helpful; see “3 bibliotherapy resources for infidelity” for recommendations.
SIDEBAR
3 bibliotherapy resources for infidelity
Not ‘Just Friends’: Protect Your Relationship from Infidelity and Heal the Trauma of Betrayal (Shirley Glass)
After the Affair: Healing the Pain and Rebuilding Trust When a Partner Has Betrayed You (Janice Abrams-Spring)
How Can I Forgive You: The Courage to Forgive, the Freedom Not To (Janice Abrams-Spring)
Referral to an individual or marriage counselor is warranted if the patient wants to work through the issues alone or with their partner. Disclosure of infidelity may not always be necessary for successful reconciliation if the affair has ended. A marriage therapist to whom you refer needs to be competent in working with infidelity.
Our patient. At the completion of the initial consultation—and after a discussion focused on the issues described, including encouragement to seek counseling—the FP acceded to the patient’s request for sexual performance-enhancing medication.
Continue to: The patient returned a few months...
The patient returned a few months later. His wife had found texts between him and his affair partner and told the patient that they had to enter into couples therapy or she was going to file for divorce. The patient told his physician that he had ended the extramarital relationship and was working on his marriage with a qualified marriage therapist; however, he felt lingering feelings of loss, discomfort in the workplace, and confusion about his choices. The physician was supportive and encouraged him to share these feelings, if possible, with an individual therapist or to find a friend who could listen while being supportive of his marriage. The physician also offered his services as a sounding board.
A year later, the patient had found another job and was still working on his marriage.
THE TAKEAWAY
This case underscores the importance of some basic health care tenets. It reminds us that maintaining patient confidentiality is paramount, and that nonjudgmental interviewing can help us to help our patients navigate challenging situations. The particulars of this case also highlight the importance of referring patients out for individual or marriage counseling and making a referral to a colleague when a situation makes us feel as if we are betraying a patient’s trust.
CORRESPONDENCE
David C. Slawson, MD, 2001 Vail Avenue, Suite 400B, Mercy Medical Plaza, Charlotte, NC 28207; [email protected]
1. Blumstein P, Schwartz P. American Couples: Money, Work, Sex. William Morrow; 1983.
2. Blow A, Hartnett K. Infidelity in committed relationships I: a methodological review. J Marital Fam Ther. 2005;31:183-216. doi: 10.1111/j.1752-0606.2005.tb01555.x
3. Glass S, Wright TL. Sex differences in type of extramarital involvement and marital dissatisfaction. Sex Roles. 1985;12:1101-1120.
4. Thompson AP. Emotional and sexual components of extramarital relations. J Marriage Fam. 1984;46:35-42. doi: 10.2307/351861
5. Laumann EO, Gagnon JH, Michael RT, et al. The Social Organization of Sexuality: Sexual Practices in the United States. University of Chicago Press; 1994.
6. Oliver MB, Hyde JS. Gender differences in sexuality: a meta-analysis. Psychol Bull. 1993;114:29-51. doi: 10.1037/0033-2909.114.1.29
7. Glass SP, Wright TL. Justifications for extramarital relationships: the association between attitudes, behaviors, and gender. J Sex Res. 1992;29:361-387. doi: 10.1080/00224499209551654
8. Spanier GB, Margolis RL. Marital separation and extramarital sexual behavior. J Sex Res. 1983;19:23-48.
9. Atkins DC, Baucom DH, Jacobson NS. Understanding infidelity: correlates in a national random sample. J Fam Psychol. 2001;15:735-749. doi: 10.1037//0893-3200.15.4.735
10. Treas J, Giesen D. Sexual infidelity among married and cohabitating Americans. J Marriage Fam. 2000;62:48-60. doi: 10.1111/j.1741-3737.2000.00048.x
11. Glass SP. Not ‘Just Friends’: Protect Your Relationship From Infidelity and Heal the Trauma of Betrayal. Free Press; 2002.
12. Blow A, Hartnet K. Infidelity in committed relationships II: a substantive review. J Marital Fam Ther. 2005;31:2. doi: 10.1111/j.1752-0606.2005.tb01556.x
13. Buunk B. Conditions that promote breakups as a consequence of extradyadic involvements. J Soc Clin Psychol. 1987;5:271-284. doi: 10.1521/jscp.1987.5.3.271
14. Charn IW, Parnass S. The impact of extramarital relationships on the continuation of marriages. J Sex Marital Therapy. 1995;21:100-115. doi: 10.1080/00926239508404389
15. Schneider JP, Irons RR, Corley MD. Disclosure of extramarital sexual activities by sexually exploitative professionals and other persons with addictive or compulsive sexual disorders. J Sex Edu Therapy. 1999;24:277-287. doi: 10.1080/01614576.1999.11074316
16. Atkins DC, Eldridge KA, Baucom DH, et al. Infidelity and behavioral couple therapy: optimism in the face of betrayal. J Consult Clin Psychol. 2005;73:144-150. doi: 10.1037/0022-006X.73.1.144
17. Johnson SM, Makinen J, Millikin J. Attachment injuries in couple relationships: a new perspective on impasses in emotionally focused marital therapy. J Marital Fam Therapy. 2001;27:145-155. doi: 10.1111/j.1752-0606.2001.tb01152.x
THE CASE
A 52-year-old man requested medicine to help him with erectile dysfunction. After obtaining a medical history and performing a physical exam, the family physician (FP) asked for more details about the patient’s situation. He reported that his wife, who had recently seen the same FP for counseling related to her frustrations with her husband, was uninterested in sex. He then added that he was having an affair with a 32-year-old female co-worker and wanted to improve his sexual function.
He admitted to feeling guilty about this situation and was conflicted about whether to end the affair. He also stated that since the affair, his insomnia had worsened, he was drinking more alcohol, and he was having migraine headaches. As the FP for both patients, and with the knowledge that the wife was worried about possible infidelity, the physician felt some level of conflict about the situation. The following is a discussion of the issues that this patient encounter raised.
DISCUSSION
Issues related to infidelity are common to both men and women. They are also common in same-sex relationships; in general, however, lesbian couples have fewer outside partners, whereas gay men are more likely to seek variety by having multiple partners.1
It is widely understood that successfully committed couples spend quality time together, emphasize each other’s strengths, show respect, accept influence, and nurture their friendship. However, many couples experience infidelity at some time in the course of their marriage. It is difficult to put an exact estimate on rates of infidelity due to problems with research methodology, inaccurate reporting, and a lack of agreement on a definition for infidelity.2 General categories of infidelity include emotional only, sexual only, and combined sexual and emotional infidelity.3,4 In terms of sexual infidelity, one study found that 25% of married men and 15% of married women admitted to having had extramarital sex at least once during their relationship.5 However, other studies suggest that women are closing the “sexual infidelity” gap and engaging in sexual affairs at a similar rate to men.6 There are websites that, in fact, have made it easier for married individuals to engage in affairs.
Reasons for infidelity. Men and women often have different motives in engaging in infidelity. In general, men’s motivations are more often related to sexual dissatisfaction and women’s to emotional dissatisfaction.7,8 However, infidelity may not always be the result of marital unhappiness.
Some studies suggest that the presence of opportunity may override the positive aspects of a relationship.9 Opportunity is heightened in the work environment, as reflected by the finding that 50% of infidelity occurs in the office.10 Research suggests that all relationships may be vulnerable to infidelity if the right opportunities present themselves.11
In general, health care providers are encouraged to use caution in generalizing about infidelity, as the subject is extremely complex, nuanced, and difficult to measure with exactitude.12
Continue to: The impact of infidelity
The impact of infidelity on couples varies due to factors such as the pre-morbid health of the marriage,13 the depth of involvement with the affair partner,14 and pre-existing attitudes about infidelity.13
Infidelity is a common cause of divorce in America. However, in a sample, Schneider et al15 found that despite initial threats to leave the marriage after infidelity, less than one-quarter of partners divorced. Other studies have found that disclosure of the infidelity and a commitment to work on the marriage may be an essential component of healing.16
Emotionally focused couples therapy, with its emphasis on attachment and bonding, may hold promise for helping couples successfully work through the trauma brought on by extramarital relationships.17 Psychologist and infidelity researcher Shirley Glass found that of the two-thirds of couples who chose to stay together after an affair, 80% of them reported a better marriage after treatment.11
Initial steps to take, and questions to ask
Both male and female patients need to feel comfortable surfacing sexual concerns with their clinicians. In this case, the concerns of the husband are interwoven with broader marital issues, which are the source of emotional and psychosomatic distress. His decision regarding his affair carried with it potentially life-altering consequences for his wife, 3 children, and affair partner and her family. It also raised ethical issues for the FP, who was providing care to both the husband and the wife. Appropriate care requires that a physician in this situation
- demonstrate a nonjudgmental approach
- clarify personal ethics in response to patient behaviors
- maintain confidentiality
- apply an ethical framework to resolve value dilemmas
- avoid actions that would be harmful to patients.
Interviewing can help to elicit information that may be clarifying not only to the physician but also to the patient. When interviewing a patient such as the one in this case, it would be wise to ask:
- How long has the affair been going on?
- Why is the patient engaging in the affair?
- Is abuse (emotional or physical) a factor in the marriage?
- Does the patient still have feelings for their spouse? Does the patient want to work on the marriage?
- Has the patient talked to a friend or therapist about the situation?
- Would the patient be willing to talk to a therapist?
Continue to: Ethical and legal considerations
Ethical and legal considerations
Some therapists espouse the view that being “neutral” in the presence of an affair is as much a value judgment as taking one side or the other. In the presence of emotional or physical abuse, it might indeed be best to support a marital separation. However, in other situations when there are young children involved and the patient is undecided about what to do, the FP can discuss the pros and cons of working on a marriage that suffers from more treatable types of disrepair (ie, stress, disconnection, repetitive arguments).
Provision of care. If the patient is unwilling to end the affair, the physician needs to decide whether they feel ethically at ease with prescribing sexually enhancing performance medication, given that the patient’s wife is also a patient. A physician in this situation might feel that they are betraying the wife by providing such medications to the husband. In such cases, it might be appropriate to refer the husband to a colleague.
In all cases of infidelity, however, it is wise to discuss safe-sex practices in order to limit the risk of transmitting a sexually transmitted infection (STI) to the spouse (or affair partner) and offer testing for STIs.
Confidentiality. Despite feelings the physician might have about betraying the wife’s trust by providing the performance-enhancing medicine to the husband, there is very little justification for revealing the affair to the wife. In general, confidentiality can only be broken if there is a high level of imminent danger associated with nondisclosure. The physician needs to realize the serious legal implications of breaking confidentiality in this situation, as such disclosure may prompt the initiation of divorce proceedings.
Real-world recommendations
Check your own biases. Infidelity can trigger a whole host of emotional reactions in physicians based on their own personal and professional history. It is important to be aware of such emotions and if sufficiently triggered, discuss the case with a colleague.
Continue to: Encourage bibliotherapy and marriage therapy
Encourage bibliotherapy and marriage therapy. The conversation might go something like this:
“I would recommend you do some reading about infidelity. If you are interested in working on your marriage, you might want to consider a couples counselor who can help you. Research shows that while such counseling can help couples work through infidelity, disclosure needs to occur as part of that process. Research also indicates that about two-thirds of marriages stay together after the revelation of an affair and that such couples can experience healing if they commit to a therapeutic process. If you are unsure how you want to proceed, it might be helpful for you to explore your situation with an individual therapist. What would you like to do next?”
There are also written resources that the patient might find helpful; see “3 bibliotherapy resources for infidelity” for recommendations.
SIDEBAR
3 bibliotherapy resources for infidelity
Not ‘Just Friends’: Protect Your Relationship from Infidelity and Heal the Trauma of Betrayal (Shirley Glass)
After the Affair: Healing the Pain and Rebuilding Trust When a Partner Has Betrayed You (Janice Abrams-Spring)
How Can I Forgive You: The Courage to Forgive, the Freedom Not To (Janice Abrams-Spring)
Referral to an individual or marriage counselor is warranted if the patient wants to work through the issues alone or with their partner. Disclosure of infidelity may not always be necessary for successful reconciliation if the affair has ended. A marriage therapist to whom you refer needs to be competent in working with infidelity.
Our patient. At the completion of the initial consultation—and after a discussion focused on the issues described, including encouragement to seek counseling—the FP acceded to the patient’s request for sexual performance-enhancing medication.
Continue to: The patient returned a few months...
The patient returned a few months later. His wife had found texts between him and his affair partner and told the patient that they had to enter into couples therapy or she was going to file for divorce. The patient told his physician that he had ended the extramarital relationship and was working on his marriage with a qualified marriage therapist; however, he felt lingering feelings of loss, discomfort in the workplace, and confusion about his choices. The physician was supportive and encouraged him to share these feelings, if possible, with an individual therapist or to find a friend who could listen while being supportive of his marriage. The physician also offered his services as a sounding board.
A year later, the patient had found another job and was still working on his marriage.
THE TAKEAWAY
This case underscores the importance of some basic health care tenets. It reminds us that maintaining patient confidentiality is paramount, and that nonjudgmental interviewing can help us to help our patients navigate challenging situations. The particulars of this case also highlight the importance of referring patients out for individual or marriage counseling and making a referral to a colleague when a situation makes us feel as if we are betraying a patient’s trust.
CORRESPONDENCE
David C. Slawson, MD, 2001 Vail Avenue, Suite 400B, Mercy Medical Plaza, Charlotte, NC 28207; [email protected]
THE CASE
A 52-year-old man requested medicine to help him with erectile dysfunction. After obtaining a medical history and performing a physical exam, the family physician (FP) asked for more details about the patient’s situation. He reported that his wife, who had recently seen the same FP for counseling related to her frustrations with her husband, was uninterested in sex. He then added that he was having an affair with a 32-year-old female co-worker and wanted to improve his sexual function.
He admitted to feeling guilty about this situation and was conflicted about whether to end the affair. He also stated that since the affair, his insomnia had worsened, he was drinking more alcohol, and he was having migraine headaches. As the FP for both patients, and with the knowledge that the wife was worried about possible infidelity, the physician felt some level of conflict about the situation. The following is a discussion of the issues that this patient encounter raised.
DISCUSSION
Issues related to infidelity are common to both men and women. They are also common in same-sex relationships; in general, however, lesbian couples have fewer outside partners, whereas gay men are more likely to seek variety by having multiple partners.1
It is widely understood that successfully committed couples spend quality time together, emphasize each other’s strengths, show respect, accept influence, and nurture their friendship. However, many couples experience infidelity at some time in the course of their marriage. It is difficult to put an exact estimate on rates of infidelity due to problems with research methodology, inaccurate reporting, and a lack of agreement on a definition for infidelity.2 General categories of infidelity include emotional only, sexual only, and combined sexual and emotional infidelity.3,4 In terms of sexual infidelity, one study found that 25% of married men and 15% of married women admitted to having had extramarital sex at least once during their relationship.5 However, other studies suggest that women are closing the “sexual infidelity” gap and engaging in sexual affairs at a similar rate to men.6 There are websites that, in fact, have made it easier for married individuals to engage in affairs.
Reasons for infidelity. Men and women often have different motives in engaging in infidelity. In general, men’s motivations are more often related to sexual dissatisfaction and women’s to emotional dissatisfaction.7,8 However, infidelity may not always be the result of marital unhappiness.
Some studies suggest that the presence of opportunity may override the positive aspects of a relationship.9 Opportunity is heightened in the work environment, as reflected by the finding that 50% of infidelity occurs in the office.10 Research suggests that all relationships may be vulnerable to infidelity if the right opportunities present themselves.11
In general, health care providers are encouraged to use caution in generalizing about infidelity, as the subject is extremely complex, nuanced, and difficult to measure with exactitude.12
Continue to: The impact of infidelity
The impact of infidelity on couples varies due to factors such as the pre-morbid health of the marriage,13 the depth of involvement with the affair partner,14 and pre-existing attitudes about infidelity.13
Infidelity is a common cause of divorce in America. However, in a sample, Schneider et al15 found that despite initial threats to leave the marriage after infidelity, less than one-quarter of partners divorced. Other studies have found that disclosure of the infidelity and a commitment to work on the marriage may be an essential component of healing.16
Emotionally focused couples therapy, with its emphasis on attachment and bonding, may hold promise for helping couples successfully work through the trauma brought on by extramarital relationships.17 Psychologist and infidelity researcher Shirley Glass found that of the two-thirds of couples who chose to stay together after an affair, 80% of them reported a better marriage after treatment.11
Initial steps to take, and questions to ask
Both male and female patients need to feel comfortable surfacing sexual concerns with their clinicians. In this case, the concerns of the husband are interwoven with broader marital issues, which are the source of emotional and psychosomatic distress. His decision regarding his affair carried with it potentially life-altering consequences for his wife, 3 children, and affair partner and her family. It also raised ethical issues for the FP, who was providing care to both the husband and the wife. Appropriate care requires that a physician in this situation
- demonstrate a nonjudgmental approach
- clarify personal ethics in response to patient behaviors
- maintain confidentiality
- apply an ethical framework to resolve value dilemmas
- avoid actions that would be harmful to patients.
Interviewing can help to elicit information that may be clarifying not only to the physician but also to the patient. When interviewing a patient such as the one in this case, it would be wise to ask:
- How long has the affair been going on?
- Why is the patient engaging in the affair?
- Is abuse (emotional or physical) a factor in the marriage?
- Does the patient still have feelings for their spouse? Does the patient want to work on the marriage?
- Has the patient talked to a friend or therapist about the situation?
- Would the patient be willing to talk to a therapist?
Continue to: Ethical and legal considerations
Ethical and legal considerations
Some therapists espouse the view that being “neutral” in the presence of an affair is as much a value judgment as taking one side or the other. In the presence of emotional or physical abuse, it might indeed be best to support a marital separation. However, in other situations when there are young children involved and the patient is undecided about what to do, the FP can discuss the pros and cons of working on a marriage that suffers from more treatable types of disrepair (ie, stress, disconnection, repetitive arguments).
Provision of care. If the patient is unwilling to end the affair, the physician needs to decide whether they feel ethically at ease with prescribing sexually enhancing performance medication, given that the patient’s wife is also a patient. A physician in this situation might feel that they are betraying the wife by providing such medications to the husband. In such cases, it might be appropriate to refer the husband to a colleague.
In all cases of infidelity, however, it is wise to discuss safe-sex practices in order to limit the risk of transmitting a sexually transmitted infection (STI) to the spouse (or affair partner) and offer testing for STIs.
Confidentiality. Despite feelings the physician might have about betraying the wife’s trust by providing the performance-enhancing medicine to the husband, there is very little justification for revealing the affair to the wife. In general, confidentiality can only be broken if there is a high level of imminent danger associated with nondisclosure. The physician needs to realize the serious legal implications of breaking confidentiality in this situation, as such disclosure may prompt the initiation of divorce proceedings.
Real-world recommendations
Check your own biases. Infidelity can trigger a whole host of emotional reactions in physicians based on their own personal and professional history. It is important to be aware of such emotions and if sufficiently triggered, discuss the case with a colleague.
Continue to: Encourage bibliotherapy and marriage therapy
Encourage bibliotherapy and marriage therapy. The conversation might go something like this:
“I would recommend you do some reading about infidelity. If you are interested in working on your marriage, you might want to consider a couples counselor who can help you. Research shows that while such counseling can help couples work through infidelity, disclosure needs to occur as part of that process. Research also indicates that about two-thirds of marriages stay together after the revelation of an affair and that such couples can experience healing if they commit to a therapeutic process. If you are unsure how you want to proceed, it might be helpful for you to explore your situation with an individual therapist. What would you like to do next?”
There are also written resources that the patient might find helpful; see “3 bibliotherapy resources for infidelity” for recommendations.
SIDEBAR
3 bibliotherapy resources for infidelity
Not ‘Just Friends’: Protect Your Relationship from Infidelity and Heal the Trauma of Betrayal (Shirley Glass)
After the Affair: Healing the Pain and Rebuilding Trust When a Partner Has Betrayed You (Janice Abrams-Spring)
How Can I Forgive You: The Courage to Forgive, the Freedom Not To (Janice Abrams-Spring)
Referral to an individual or marriage counselor is warranted if the patient wants to work through the issues alone or with their partner. Disclosure of infidelity may not always be necessary for successful reconciliation if the affair has ended. A marriage therapist to whom you refer needs to be competent in working with infidelity.
Our patient. At the completion of the initial consultation—and after a discussion focused on the issues described, including encouragement to seek counseling—the FP acceded to the patient’s request for sexual performance-enhancing medication.
Continue to: The patient returned a few months...
The patient returned a few months later. His wife had found texts between him and his affair partner and told the patient that they had to enter into couples therapy or she was going to file for divorce. The patient told his physician that he had ended the extramarital relationship and was working on his marriage with a qualified marriage therapist; however, he felt lingering feelings of loss, discomfort in the workplace, and confusion about his choices. The physician was supportive and encouraged him to share these feelings, if possible, with an individual therapist or to find a friend who could listen while being supportive of his marriage. The physician also offered his services as a sounding board.
A year later, the patient had found another job and was still working on his marriage.
THE TAKEAWAY
This case underscores the importance of some basic health care tenets. It reminds us that maintaining patient confidentiality is paramount, and that nonjudgmental interviewing can help us to help our patients navigate challenging situations. The particulars of this case also highlight the importance of referring patients out for individual or marriage counseling and making a referral to a colleague when a situation makes us feel as if we are betraying a patient’s trust.
CORRESPONDENCE
David C. Slawson, MD, 2001 Vail Avenue, Suite 400B, Mercy Medical Plaza, Charlotte, NC 28207; [email protected]
1. Blumstein P, Schwartz P. American Couples: Money, Work, Sex. William Morrow; 1983.
2. Blow A, Hartnett K. Infidelity in committed relationships I: a methodological review. J Marital Fam Ther. 2005;31:183-216. doi: 10.1111/j.1752-0606.2005.tb01555.x
3. Glass S, Wright TL. Sex differences in type of extramarital involvement and marital dissatisfaction. Sex Roles. 1985;12:1101-1120.
4. Thompson AP. Emotional and sexual components of extramarital relations. J Marriage Fam. 1984;46:35-42. doi: 10.2307/351861
5. Laumann EO, Gagnon JH, Michael RT, et al. The Social Organization of Sexuality: Sexual Practices in the United States. University of Chicago Press; 1994.
6. Oliver MB, Hyde JS. Gender differences in sexuality: a meta-analysis. Psychol Bull. 1993;114:29-51. doi: 10.1037/0033-2909.114.1.29
7. Glass SP, Wright TL. Justifications for extramarital relationships: the association between attitudes, behaviors, and gender. J Sex Res. 1992;29:361-387. doi: 10.1080/00224499209551654
8. Spanier GB, Margolis RL. Marital separation and extramarital sexual behavior. J Sex Res. 1983;19:23-48.
9. Atkins DC, Baucom DH, Jacobson NS. Understanding infidelity: correlates in a national random sample. J Fam Psychol. 2001;15:735-749. doi: 10.1037//0893-3200.15.4.735
10. Treas J, Giesen D. Sexual infidelity among married and cohabitating Americans. J Marriage Fam. 2000;62:48-60. doi: 10.1111/j.1741-3737.2000.00048.x
11. Glass SP. Not ‘Just Friends’: Protect Your Relationship From Infidelity and Heal the Trauma of Betrayal. Free Press; 2002.
12. Blow A, Hartnet K. Infidelity in committed relationships II: a substantive review. J Marital Fam Ther. 2005;31:2. doi: 10.1111/j.1752-0606.2005.tb01556.x
13. Buunk B. Conditions that promote breakups as a consequence of extradyadic involvements. J Soc Clin Psychol. 1987;5:271-284. doi: 10.1521/jscp.1987.5.3.271
14. Charn IW, Parnass S. The impact of extramarital relationships on the continuation of marriages. J Sex Marital Therapy. 1995;21:100-115. doi: 10.1080/00926239508404389
15. Schneider JP, Irons RR, Corley MD. Disclosure of extramarital sexual activities by sexually exploitative professionals and other persons with addictive or compulsive sexual disorders. J Sex Edu Therapy. 1999;24:277-287. doi: 10.1080/01614576.1999.11074316
16. Atkins DC, Eldridge KA, Baucom DH, et al. Infidelity and behavioral couple therapy: optimism in the face of betrayal. J Consult Clin Psychol. 2005;73:144-150. doi: 10.1037/0022-006X.73.1.144
17. Johnson SM, Makinen J, Millikin J. Attachment injuries in couple relationships: a new perspective on impasses in emotionally focused marital therapy. J Marital Fam Therapy. 2001;27:145-155. doi: 10.1111/j.1752-0606.2001.tb01152.x
1. Blumstein P, Schwartz P. American Couples: Money, Work, Sex. William Morrow; 1983.
2. Blow A, Hartnett K. Infidelity in committed relationships I: a methodological review. J Marital Fam Ther. 2005;31:183-216. doi: 10.1111/j.1752-0606.2005.tb01555.x
3. Glass S, Wright TL. Sex differences in type of extramarital involvement and marital dissatisfaction. Sex Roles. 1985;12:1101-1120.
4. Thompson AP. Emotional and sexual components of extramarital relations. J Marriage Fam. 1984;46:35-42. doi: 10.2307/351861
5. Laumann EO, Gagnon JH, Michael RT, et al. The Social Organization of Sexuality: Sexual Practices in the United States. University of Chicago Press; 1994.
6. Oliver MB, Hyde JS. Gender differences in sexuality: a meta-analysis. Psychol Bull. 1993;114:29-51. doi: 10.1037/0033-2909.114.1.29
7. Glass SP, Wright TL. Justifications for extramarital relationships: the association between attitudes, behaviors, and gender. J Sex Res. 1992;29:361-387. doi: 10.1080/00224499209551654
8. Spanier GB, Margolis RL. Marital separation and extramarital sexual behavior. J Sex Res. 1983;19:23-48.
9. Atkins DC, Baucom DH, Jacobson NS. Understanding infidelity: correlates in a national random sample. J Fam Psychol. 2001;15:735-749. doi: 10.1037//0893-3200.15.4.735
10. Treas J, Giesen D. Sexual infidelity among married and cohabitating Americans. J Marriage Fam. 2000;62:48-60. doi: 10.1111/j.1741-3737.2000.00048.x
11. Glass SP. Not ‘Just Friends’: Protect Your Relationship From Infidelity and Heal the Trauma of Betrayal. Free Press; 2002.
12. Blow A, Hartnet K. Infidelity in committed relationships II: a substantive review. J Marital Fam Ther. 2005;31:2. doi: 10.1111/j.1752-0606.2005.tb01556.x
13. Buunk B. Conditions that promote breakups as a consequence of extradyadic involvements. J Soc Clin Psychol. 1987;5:271-284. doi: 10.1521/jscp.1987.5.3.271
14. Charn IW, Parnass S. The impact of extramarital relationships on the continuation of marriages. J Sex Marital Therapy. 1995;21:100-115. doi: 10.1080/00926239508404389
15. Schneider JP, Irons RR, Corley MD. Disclosure of extramarital sexual activities by sexually exploitative professionals and other persons with addictive or compulsive sexual disorders. J Sex Edu Therapy. 1999;24:277-287. doi: 10.1080/01614576.1999.11074316
16. Atkins DC, Eldridge KA, Baucom DH, et al. Infidelity and behavioral couple therapy: optimism in the face of betrayal. J Consult Clin Psychol. 2005;73:144-150. doi: 10.1037/0022-006X.73.1.144
17. Johnson SM, Makinen J, Millikin J. Attachment injuries in couple relationships: a new perspective on impasses in emotionally focused marital therapy. J Marital Fam Therapy. 2001;27:145-155. doi: 10.1111/j.1752-0606.2001.tb01152.x

SARS-CoV-2: A Novel Precipitant of Ischemic Priapism
Priapism is a disorder that occurs when the penis maintains a prolonged erection in the absence of appropriate stimulation. The disorder is typically divided into subgroups based on arterial flow: low flow (ischemic) and high flow (nonischemic). Ischemic priapism is the most common form and results from venous congestion due to obstructed outflow and inability of cavernous smooth muscle to contract, resulting in compartment syndrome, tissue hypoxia, hypercapnia, and acidosis.1 Conditions that result in hypercoagulable states and hyperviscosity are associated with ischemic priapism. COVID-19 is well known to cause an acute respiratory illness and systemic inflammatory response and has been increasingly associated with coagulopathy. Studies have shown that 20% to 55% of patients admitted to the hospital for COVID-19 show objective laboratory evidence of a hypercoagulable state.2
To date, there are 6 reported cases of priapism occurring in the setting of COVID-19 with all cases demonstrating the ischemic subtype. The onset of priapism from the beginning of infectious symptoms ranged from 2 days to more than a month. Five of the cases occurred in patients with critical COVID-19 and 1 in the setting of mild disease.3-8 Two critically ill patients did not receive treatment for their ischemic priapism as they were transitioned to expectant management and/or comfort measures.Most were treated with cavernosal blood aspiration and intracavernosal injections of phenylephrine or ethylephrine. Some patients were managed with prophylactic doses of anticoagulation after the identification of priapism; others were transitioned to therapeutic doses. Two patients were followed postdischarge; one patient reported normal nighttime erections with sexual desire 2 weeks postdischarge, and another patient, who underwent a bilateral T-shunt procedure after unsuccessful phenylephrine injections, reported complete erectile dysfunction at 3 months postdischarge.4,7 There was a potentially confounding variable in 2 cases in which propofol infusions were used for sedation management in the setting of mechanical ventilation.6,8 Propofol has been linked to priapism through its blockade of sympathetic activation resulting in persistent relaxation of cavernosal smooth muscle.9 We present a unique case of COVID-19–associated ischemic priapism as our patient had moderate rather than critical COVID-19.
Case Presentation
A 67-year-old male patient presented to the emergency department for a painful erection of 34-hour duration. The patient had been exposed to COVID-19 roughly 2 months prior. Since the exposure, he had experienced headache, nonproductive cough, sore throat, and decreased appetite with weight loss. His medical history included hypertension, thoracic aortic aneurysm, B-cell type chronic lymphocytic leukemia (CLL), and obstructive sleep apnea. Daily outpatient medications included atenolol 100 mg, hydrochlorothiazide 25 mg, and omeprazole 20 mg. The patient stopped tobacco use about 30 years previously. He reported no alcohol consumption or illicit drug use and had no previous episodes of prolonged erection.
The patient was afebrile, hemodynamically stable, and had an oxygen saturation of 92% on room air. Physical examination revealed clear breath sounds and an erect circumcised penis without any lesions, discoloration, or skin necrosis. Laboratory data were remarkable for the following values: 125,660 cells/μL white blood cells (WBCs), 13.82 × 103/ μL neutrophils, 110.58 × 103/μL lymphocytes, 1.26 × 103/μL monocytes, no blasts, 9.4 gm/dL hemoglobin, 100.3 fl mean corpuscular volume, 417,000 cells/μL platelets, 23,671 ng/mL D-dimer, 29.6 seconds activated partial thromboplastin time (aPTT), 16.3 seconds prothrombin time, 743 mg/dL fibrinogen, 474 U/L lactate dehydrogenase, and 202.1 mg/dL haptoglobin. A nasopharyngeal reverse transcription polymerase chain reaction test resulted positive for the SARS-CoV-2 virus, and subsequent chest X-ray revealed bilateral, hazy opacities predominantly in a peripheral distribution. Computed tomography (CT) angiogram of the chest did not reveal pulmonary emboli, pneumothorax, effusions, or lobar consolidation. However, it displayed bilateral ground-glass opacities with interstitial consolidation worst in the upper lobes. Corporal aspiration and blood gas analysis revealed a pH of 7.05, P
Differential Diagnosis
The first consideration in the differential diagnosis of priapism is to differentiate between ischemic and nonischemic. Based on the abnormal blood gas results above, this case clearly falls within the ischemic spectrum. Ischemic priapism secondary to CLL-induced hyperleukocytosis was considered. It has been noted that up to 20% of priapism cases in adults are related to hematologic disorders.10 While it is not uncommon to see hyperleukocytosis (total WBC count > 100 × 109/L) in CLL, leukostasis is rare with most reports demonstrating WBC counts > 1000 × 109/L.11 Hematology, vascular surgery, and urology services were consulted and agreed that ischemic priapism was due to microthrombi or pelvic vein thrombosis secondary to COVID-19–associated coagulopathy (CAC) was the most likely etiology.
Treatment
After corporal aspiration, intracorporal phenylephrine was administered. Diluted phenylephrine (100 ug/mL) was injected every 5 to 10 minutes while intermittently aspirating and irrigating multiple sites along the lateral length of the penile shaft. This initial procedure reduced the erection from 100% to 30% rigidity, with repeat blood gas analysis revealing minimal improvement. CT of the abdomen and pelvis with IV contrast revealed no evidence of pelvic thrombi. A second round of phenylephrine injections were administered, resulting in detumescence. The patient was treated with 2 to 3 L/min of oxygen supplementation via nasal cannula, a 5-day course of remdesivir and low-intensity heparin drip. Following the initial low-intensity heparin drip, the patient transitioned to therapeutic enoxaparin and subsequently was discharged on apixaban for a 3-month course. Since discharge, the patient followed up with hematology. He tolerated and completed the anticoagulation regimen without any recurrences of priapism or residual deficits.
Discussion
Recent studies have overwhelmingly analyzed the incidence and presentation of thrombotic complications in critically ill patients with COVID-19. CAC has been postulated to result from endotheliopathy along with immune cell activation and propagation of coagulation. While COVID-19 has been noted to create lung injury through binding angiotensin-converting enzyme 2 receptors expressed on alveolar pneumocytes, it increasingly has been found to affect endothelial cells throughout the body. Recent postmortem analyses have demonstrated direct viral infection of endothelial cells with consequent diffuse endothelial inflammation, as evidenced by viral inclusions, sequestered immune cells, and endothelial apoptosis.12,13 Manifestations of this endotheliopathy have been delineated through various studies.
An early retrospective study in Wuhan, China, illustrated that 36% of the first 99 patients hospitalized with COVID-19 demonstrated an elevated D-dimer, 6% an elevated aPTT, and 5% an elevated prothrombin time.14 Another retrospective study conducted in Wuhan found a 25% incidence of venous thromboembolic complications in critically ill patients with severe COVID-19.15 In the Netherlands, a study reported the incidence of arterial and venous thrombotic complications to be 31% in 184 critically ill patients with COVID-19, with 81% of these cases involving pulmonary emboli.16
To our knowledge, our patient is the seventh reported case of ischemic priapism occurring in the setting of a COVID-19 infection, and the first to have occurred in its moderate form. Ischemic priapism is often a consequence of penile venous outflow obstruction and resultant stasis of hypoxic blood.7 The prothrombotic state induced by CAC has been proposed to cause the obstruction of small emissary veins in the subtunical space and in turn lead to venous stasis, which propagates the formation of ischemic priapism.8 Furthermore, 4 of the previously reported cases shared laboratory data on their patients, and all demonstrated elevated D-dimer and fibrinogen levels, which strengthens this hypothesis.3,5,7,8 CLL presents a potential confounding variable in this case; however, as we have reviewed earlier, the risk of leukostasis at WBC counts < 1000 × 109/L is very low.11 It is also probable that the patient had some level of immune dysregulation secondary to CLL, leading to his prolonged course and slow clearance of the virus.
Conclusions
Although only a handful of CAC cases leading to ischemic priapism have been reported, the true incidence may be much higher. While our case highlights the importance of considering COVID-19 infection in the differential diagnosis of ischemic priapism, more research is needed to understand incidence and definitively establish a causative relationship.
1. Pryor J, Akkus E, Alter G, et al. Priapism. J Sex Med. 2004;1(1):116-120. doi:10.1111/j.1743-6109.2004.10117.x
2. Lee SG, Fralick M, Sholzberg M. Coagulopathy associated with COVID-19. CMAJ. 2020;192(21):E583. doi:10.1503/cmaj.200685
3. Lam G, McCarthy R, Haider R. A peculiar case of priapism: the hypercoagulable state in patients with severe COVID-19 infection. Eur J Case Rep Intern Med. 2020;7(8):001779. doi:10.12890/2020_001779
4. Addar A, Al Fraidi O, Nazer A, Althonayan N, Ghazwani Y. Priapism for 10 days in a patient with SARS-CoV-2 pneumonia: a case report. J Surg Case Rep. 2021;2021(4):rjab020. doi:10.1093/jscr/rjab020
5. Lamamri M, Chebbi A, Mamane J, et al. Priapism in a patient with coronavirus disease 2019 (COVID-19). Am J Emerg Med. 2021;39:251.e5-251.e7. doi:10.1016/j.ajem.2020.06.027
6. Silverman ML, VanDerVeer SJ, Donnelly TJ. Priapism in COVID-19: a thromboembolic complication. Am J Emerg Med. 2021;45:686.e5-686.e6. doi:10.1016/j.ajem.2020.12.072
7. Giuliano AFM, Vulpi M, Passerini F, et al. SARS-CoV-2 infection as a determining factor to the precipitation of ischemic priapism in a young patient with asymptomatic COVID-19. Case Rep Urol. 2021;2021:9936891. doi:10.1155/2021/9936891
8. Carreno BD, Perez CP, Vasquez D, Oyola JA, Suarez O, Bedoya C. Veno-occlusive priapism in COVID-19 disease. Urol Int. 2021;105(9-10):916-919. doi:10.1159/000514421
9. Senthilkumaran S, Shah S, Ganapathysubramanian, Balamurgan N, Thirumalaikolundusubramanian P. Propofol and priapism. Indian J Pharmacol. 2010;42(4):238-239. doi:10.4103/0253-7613.68430
10. Qu M, Lu X, Wang L, Liu Z, Sun Y, Gao X. Priapism secondary to chronic myeloid leukemia treated by a surgical cavernosa-corpus spongiosum shunt: case report. Asian J Urol. 2019;6(4):373-376. doi:10.1016/j.ajur.2018.12.004
11. Singh N, Singh Lubana S, Dabrowski L, Sidhu G. Leukostasis in chronic lymphocytic leukemia. Am J Case Rep. 2020;21:e924798. doi:10.12659/AJCR.924798
12. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417-1418. doi:10.1016/S0140-6736(20)30937-5
13. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033-2040. doi:10.1182/blood.2020006000
14. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507-513. doi:10.1016/S0140-6736(20)30211-7
15. Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18(6):1421-1424. doi:10.1111/jth.14830
16. Klok FA, Kruip M, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145-147. doi:10.1016/j.thromres.2020.04.013
Priapism is a disorder that occurs when the penis maintains a prolonged erection in the absence of appropriate stimulation. The disorder is typically divided into subgroups based on arterial flow: low flow (ischemic) and high flow (nonischemic). Ischemic priapism is the most common form and results from venous congestion due to obstructed outflow and inability of cavernous smooth muscle to contract, resulting in compartment syndrome, tissue hypoxia, hypercapnia, and acidosis.1 Conditions that result in hypercoagulable states and hyperviscosity are associated with ischemic priapism. COVID-19 is well known to cause an acute respiratory illness and systemic inflammatory response and has been increasingly associated with coagulopathy. Studies have shown that 20% to 55% of patients admitted to the hospital for COVID-19 show objective laboratory evidence of a hypercoagulable state.2
To date, there are 6 reported cases of priapism occurring in the setting of COVID-19 with all cases demonstrating the ischemic subtype. The onset of priapism from the beginning of infectious symptoms ranged from 2 days to more than a month. Five of the cases occurred in patients with critical COVID-19 and 1 in the setting of mild disease.3-8 Two critically ill patients did not receive treatment for their ischemic priapism as they were transitioned to expectant management and/or comfort measures.Most were treated with cavernosal blood aspiration and intracavernosal injections of phenylephrine or ethylephrine. Some patients were managed with prophylactic doses of anticoagulation after the identification of priapism; others were transitioned to therapeutic doses. Two patients were followed postdischarge; one patient reported normal nighttime erections with sexual desire 2 weeks postdischarge, and another patient, who underwent a bilateral T-shunt procedure after unsuccessful phenylephrine injections, reported complete erectile dysfunction at 3 months postdischarge.4,7 There was a potentially confounding variable in 2 cases in which propofol infusions were used for sedation management in the setting of mechanical ventilation.6,8 Propofol has been linked to priapism through its blockade of sympathetic activation resulting in persistent relaxation of cavernosal smooth muscle.9 We present a unique case of COVID-19–associated ischemic priapism as our patient had moderate rather than critical COVID-19.
Case Presentation
A 67-year-old male patient presented to the emergency department for a painful erection of 34-hour duration. The patient had been exposed to COVID-19 roughly 2 months prior. Since the exposure, he had experienced headache, nonproductive cough, sore throat, and decreased appetite with weight loss. His medical history included hypertension, thoracic aortic aneurysm, B-cell type chronic lymphocytic leukemia (CLL), and obstructive sleep apnea. Daily outpatient medications included atenolol 100 mg, hydrochlorothiazide 25 mg, and omeprazole 20 mg. The patient stopped tobacco use about 30 years previously. He reported no alcohol consumption or illicit drug use and had no previous episodes of prolonged erection.
The patient was afebrile, hemodynamically stable, and had an oxygen saturation of 92% on room air. Physical examination revealed clear breath sounds and an erect circumcised penis without any lesions, discoloration, or skin necrosis. Laboratory data were remarkable for the following values: 125,660 cells/μL white blood cells (WBCs), 13.82 × 103/ μL neutrophils, 110.58 × 103/μL lymphocytes, 1.26 × 103/μL monocytes, no blasts, 9.4 gm/dL hemoglobin, 100.3 fl mean corpuscular volume, 417,000 cells/μL platelets, 23,671 ng/mL D-dimer, 29.6 seconds activated partial thromboplastin time (aPTT), 16.3 seconds prothrombin time, 743 mg/dL fibrinogen, 474 U/L lactate dehydrogenase, and 202.1 mg/dL haptoglobin. A nasopharyngeal reverse transcription polymerase chain reaction test resulted positive for the SARS-CoV-2 virus, and subsequent chest X-ray revealed bilateral, hazy opacities predominantly in a peripheral distribution. Computed tomography (CT) angiogram of the chest did not reveal pulmonary emboli, pneumothorax, effusions, or lobar consolidation. However, it displayed bilateral ground-glass opacities with interstitial consolidation worst in the upper lobes. Corporal aspiration and blood gas analysis revealed a pH of 7.05, P
Differential Diagnosis
The first consideration in the differential diagnosis of priapism is to differentiate between ischemic and nonischemic. Based on the abnormal blood gas results above, this case clearly falls within the ischemic spectrum. Ischemic priapism secondary to CLL-induced hyperleukocytosis was considered. It has been noted that up to 20% of priapism cases in adults are related to hematologic disorders.10 While it is not uncommon to see hyperleukocytosis (total WBC count > 100 × 109/L) in CLL, leukostasis is rare with most reports demonstrating WBC counts > 1000 × 109/L.11 Hematology, vascular surgery, and urology services were consulted and agreed that ischemic priapism was due to microthrombi or pelvic vein thrombosis secondary to COVID-19–associated coagulopathy (CAC) was the most likely etiology.
Treatment
After corporal aspiration, intracorporal phenylephrine was administered. Diluted phenylephrine (100 ug/mL) was injected every 5 to 10 minutes while intermittently aspirating and irrigating multiple sites along the lateral length of the penile shaft. This initial procedure reduced the erection from 100% to 30% rigidity, with repeat blood gas analysis revealing minimal improvement. CT of the abdomen and pelvis with IV contrast revealed no evidence of pelvic thrombi. A second round of phenylephrine injections were administered, resulting in detumescence. The patient was treated with 2 to 3 L/min of oxygen supplementation via nasal cannula, a 5-day course of remdesivir and low-intensity heparin drip. Following the initial low-intensity heparin drip, the patient transitioned to therapeutic enoxaparin and subsequently was discharged on apixaban for a 3-month course. Since discharge, the patient followed up with hematology. He tolerated and completed the anticoagulation regimen without any recurrences of priapism or residual deficits.
Discussion
Recent studies have overwhelmingly analyzed the incidence and presentation of thrombotic complications in critically ill patients with COVID-19. CAC has been postulated to result from endotheliopathy along with immune cell activation and propagation of coagulation. While COVID-19 has been noted to create lung injury through binding angiotensin-converting enzyme 2 receptors expressed on alveolar pneumocytes, it increasingly has been found to affect endothelial cells throughout the body. Recent postmortem analyses have demonstrated direct viral infection of endothelial cells with consequent diffuse endothelial inflammation, as evidenced by viral inclusions, sequestered immune cells, and endothelial apoptosis.12,13 Manifestations of this endotheliopathy have been delineated through various studies.
An early retrospective study in Wuhan, China, illustrated that 36% of the first 99 patients hospitalized with COVID-19 demonstrated an elevated D-dimer, 6% an elevated aPTT, and 5% an elevated prothrombin time.14 Another retrospective study conducted in Wuhan found a 25% incidence of venous thromboembolic complications in critically ill patients with severe COVID-19.15 In the Netherlands, a study reported the incidence of arterial and venous thrombotic complications to be 31% in 184 critically ill patients with COVID-19, with 81% of these cases involving pulmonary emboli.16
To our knowledge, our patient is the seventh reported case of ischemic priapism occurring in the setting of a COVID-19 infection, and the first to have occurred in its moderate form. Ischemic priapism is often a consequence of penile venous outflow obstruction and resultant stasis of hypoxic blood.7 The prothrombotic state induced by CAC has been proposed to cause the obstruction of small emissary veins in the subtunical space and in turn lead to venous stasis, which propagates the formation of ischemic priapism.8 Furthermore, 4 of the previously reported cases shared laboratory data on their patients, and all demonstrated elevated D-dimer and fibrinogen levels, which strengthens this hypothesis.3,5,7,8 CLL presents a potential confounding variable in this case; however, as we have reviewed earlier, the risk of leukostasis at WBC counts < 1000 × 109/L is very low.11 It is also probable that the patient had some level of immune dysregulation secondary to CLL, leading to his prolonged course and slow clearance of the virus.
Conclusions
Although only a handful of CAC cases leading to ischemic priapism have been reported, the true incidence may be much higher. While our case highlights the importance of considering COVID-19 infection in the differential diagnosis of ischemic priapism, more research is needed to understand incidence and definitively establish a causative relationship.
Priapism is a disorder that occurs when the penis maintains a prolonged erection in the absence of appropriate stimulation. The disorder is typically divided into subgroups based on arterial flow: low flow (ischemic) and high flow (nonischemic). Ischemic priapism is the most common form and results from venous congestion due to obstructed outflow and inability of cavernous smooth muscle to contract, resulting in compartment syndrome, tissue hypoxia, hypercapnia, and acidosis.1 Conditions that result in hypercoagulable states and hyperviscosity are associated with ischemic priapism. COVID-19 is well known to cause an acute respiratory illness and systemic inflammatory response and has been increasingly associated with coagulopathy. Studies have shown that 20% to 55% of patients admitted to the hospital for COVID-19 show objective laboratory evidence of a hypercoagulable state.2
To date, there are 6 reported cases of priapism occurring in the setting of COVID-19 with all cases demonstrating the ischemic subtype. The onset of priapism from the beginning of infectious symptoms ranged from 2 days to more than a month. Five of the cases occurred in patients with critical COVID-19 and 1 in the setting of mild disease.3-8 Two critically ill patients did not receive treatment for their ischemic priapism as they were transitioned to expectant management and/or comfort measures.Most were treated with cavernosal blood aspiration and intracavernosal injections of phenylephrine or ethylephrine. Some patients were managed with prophylactic doses of anticoagulation after the identification of priapism; others were transitioned to therapeutic doses. Two patients were followed postdischarge; one patient reported normal nighttime erections with sexual desire 2 weeks postdischarge, and another patient, who underwent a bilateral T-shunt procedure after unsuccessful phenylephrine injections, reported complete erectile dysfunction at 3 months postdischarge.4,7 There was a potentially confounding variable in 2 cases in which propofol infusions were used for sedation management in the setting of mechanical ventilation.6,8 Propofol has been linked to priapism through its blockade of sympathetic activation resulting in persistent relaxation of cavernosal smooth muscle.9 We present a unique case of COVID-19–associated ischemic priapism as our patient had moderate rather than critical COVID-19.
Case Presentation
A 67-year-old male patient presented to the emergency department for a painful erection of 34-hour duration. The patient had been exposed to COVID-19 roughly 2 months prior. Since the exposure, he had experienced headache, nonproductive cough, sore throat, and decreased appetite with weight loss. His medical history included hypertension, thoracic aortic aneurysm, B-cell type chronic lymphocytic leukemia (CLL), and obstructive sleep apnea. Daily outpatient medications included atenolol 100 mg, hydrochlorothiazide 25 mg, and omeprazole 20 mg. The patient stopped tobacco use about 30 years previously. He reported no alcohol consumption or illicit drug use and had no previous episodes of prolonged erection.
The patient was afebrile, hemodynamically stable, and had an oxygen saturation of 92% on room air. Physical examination revealed clear breath sounds and an erect circumcised penis without any lesions, discoloration, or skin necrosis. Laboratory data were remarkable for the following values: 125,660 cells/μL white blood cells (WBCs), 13.82 × 103/ μL neutrophils, 110.58 × 103/μL lymphocytes, 1.26 × 103/μL monocytes, no blasts, 9.4 gm/dL hemoglobin, 100.3 fl mean corpuscular volume, 417,000 cells/μL platelets, 23,671 ng/mL D-dimer, 29.6 seconds activated partial thromboplastin time (aPTT), 16.3 seconds prothrombin time, 743 mg/dL fibrinogen, 474 U/L lactate dehydrogenase, and 202.1 mg/dL haptoglobin. A nasopharyngeal reverse transcription polymerase chain reaction test resulted positive for the SARS-CoV-2 virus, and subsequent chest X-ray revealed bilateral, hazy opacities predominantly in a peripheral distribution. Computed tomography (CT) angiogram of the chest did not reveal pulmonary emboli, pneumothorax, effusions, or lobar consolidation. However, it displayed bilateral ground-glass opacities with interstitial consolidation worst in the upper lobes. Corporal aspiration and blood gas analysis revealed a pH of 7.05, P
Differential Diagnosis
The first consideration in the differential diagnosis of priapism is to differentiate between ischemic and nonischemic. Based on the abnormal blood gas results above, this case clearly falls within the ischemic spectrum. Ischemic priapism secondary to CLL-induced hyperleukocytosis was considered. It has been noted that up to 20% of priapism cases in adults are related to hematologic disorders.10 While it is not uncommon to see hyperleukocytosis (total WBC count > 100 × 109/L) in CLL, leukostasis is rare with most reports demonstrating WBC counts > 1000 × 109/L.11 Hematology, vascular surgery, and urology services were consulted and agreed that ischemic priapism was due to microthrombi or pelvic vein thrombosis secondary to COVID-19–associated coagulopathy (CAC) was the most likely etiology.
Treatment
After corporal aspiration, intracorporal phenylephrine was administered. Diluted phenylephrine (100 ug/mL) was injected every 5 to 10 minutes while intermittently aspirating and irrigating multiple sites along the lateral length of the penile shaft. This initial procedure reduced the erection from 100% to 30% rigidity, with repeat blood gas analysis revealing minimal improvement. CT of the abdomen and pelvis with IV contrast revealed no evidence of pelvic thrombi. A second round of phenylephrine injections were administered, resulting in detumescence. The patient was treated with 2 to 3 L/min of oxygen supplementation via nasal cannula, a 5-day course of remdesivir and low-intensity heparin drip. Following the initial low-intensity heparin drip, the patient transitioned to therapeutic enoxaparin and subsequently was discharged on apixaban for a 3-month course. Since discharge, the patient followed up with hematology. He tolerated and completed the anticoagulation regimen without any recurrences of priapism or residual deficits.
Discussion
Recent studies have overwhelmingly analyzed the incidence and presentation of thrombotic complications in critically ill patients with COVID-19. CAC has been postulated to result from endotheliopathy along with immune cell activation and propagation of coagulation. While COVID-19 has been noted to create lung injury through binding angiotensin-converting enzyme 2 receptors expressed on alveolar pneumocytes, it increasingly has been found to affect endothelial cells throughout the body. Recent postmortem analyses have demonstrated direct viral infection of endothelial cells with consequent diffuse endothelial inflammation, as evidenced by viral inclusions, sequestered immune cells, and endothelial apoptosis.12,13 Manifestations of this endotheliopathy have been delineated through various studies.
An early retrospective study in Wuhan, China, illustrated that 36% of the first 99 patients hospitalized with COVID-19 demonstrated an elevated D-dimer, 6% an elevated aPTT, and 5% an elevated prothrombin time.14 Another retrospective study conducted in Wuhan found a 25% incidence of venous thromboembolic complications in critically ill patients with severe COVID-19.15 In the Netherlands, a study reported the incidence of arterial and venous thrombotic complications to be 31% in 184 critically ill patients with COVID-19, with 81% of these cases involving pulmonary emboli.16
To our knowledge, our patient is the seventh reported case of ischemic priapism occurring in the setting of a COVID-19 infection, and the first to have occurred in its moderate form. Ischemic priapism is often a consequence of penile venous outflow obstruction and resultant stasis of hypoxic blood.7 The prothrombotic state induced by CAC has been proposed to cause the obstruction of small emissary veins in the subtunical space and in turn lead to venous stasis, which propagates the formation of ischemic priapism.8 Furthermore, 4 of the previously reported cases shared laboratory data on their patients, and all demonstrated elevated D-dimer and fibrinogen levels, which strengthens this hypothesis.3,5,7,8 CLL presents a potential confounding variable in this case; however, as we have reviewed earlier, the risk of leukostasis at WBC counts < 1000 × 109/L is very low.11 It is also probable that the patient had some level of immune dysregulation secondary to CLL, leading to his prolonged course and slow clearance of the virus.
Conclusions
Although only a handful of CAC cases leading to ischemic priapism have been reported, the true incidence may be much higher. While our case highlights the importance of considering COVID-19 infection in the differential diagnosis of ischemic priapism, more research is needed to understand incidence and definitively establish a causative relationship.
1. Pryor J, Akkus E, Alter G, et al. Priapism. J Sex Med. 2004;1(1):116-120. doi:10.1111/j.1743-6109.2004.10117.x
2. Lee SG, Fralick M, Sholzberg M. Coagulopathy associated with COVID-19. CMAJ. 2020;192(21):E583. doi:10.1503/cmaj.200685
3. Lam G, McCarthy R, Haider R. A peculiar case of priapism: the hypercoagulable state in patients with severe COVID-19 infection. Eur J Case Rep Intern Med. 2020;7(8):001779. doi:10.12890/2020_001779
4. Addar A, Al Fraidi O, Nazer A, Althonayan N, Ghazwani Y. Priapism for 10 days in a patient with SARS-CoV-2 pneumonia: a case report. J Surg Case Rep. 2021;2021(4):rjab020. doi:10.1093/jscr/rjab020
5. Lamamri M, Chebbi A, Mamane J, et al. Priapism in a patient with coronavirus disease 2019 (COVID-19). Am J Emerg Med. 2021;39:251.e5-251.e7. doi:10.1016/j.ajem.2020.06.027
6. Silverman ML, VanDerVeer SJ, Donnelly TJ. Priapism in COVID-19: a thromboembolic complication. Am J Emerg Med. 2021;45:686.e5-686.e6. doi:10.1016/j.ajem.2020.12.072
7. Giuliano AFM, Vulpi M, Passerini F, et al. SARS-CoV-2 infection as a determining factor to the precipitation of ischemic priapism in a young patient with asymptomatic COVID-19. Case Rep Urol. 2021;2021:9936891. doi:10.1155/2021/9936891
8. Carreno BD, Perez CP, Vasquez D, Oyola JA, Suarez O, Bedoya C. Veno-occlusive priapism in COVID-19 disease. Urol Int. 2021;105(9-10):916-919. doi:10.1159/000514421
9. Senthilkumaran S, Shah S, Ganapathysubramanian, Balamurgan N, Thirumalaikolundusubramanian P. Propofol and priapism. Indian J Pharmacol. 2010;42(4):238-239. doi:10.4103/0253-7613.68430
10. Qu M, Lu X, Wang L, Liu Z, Sun Y, Gao X. Priapism secondary to chronic myeloid leukemia treated by a surgical cavernosa-corpus spongiosum shunt: case report. Asian J Urol. 2019;6(4):373-376. doi:10.1016/j.ajur.2018.12.004
11. Singh N, Singh Lubana S, Dabrowski L, Sidhu G. Leukostasis in chronic lymphocytic leukemia. Am J Case Rep. 2020;21:e924798. doi:10.12659/AJCR.924798
12. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417-1418. doi:10.1016/S0140-6736(20)30937-5
13. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033-2040. doi:10.1182/blood.2020006000
14. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507-513. doi:10.1016/S0140-6736(20)30211-7
15. Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18(6):1421-1424. doi:10.1111/jth.14830
16. Klok FA, Kruip M, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145-147. doi:10.1016/j.thromres.2020.04.013
1. Pryor J, Akkus E, Alter G, et al. Priapism. J Sex Med. 2004;1(1):116-120. doi:10.1111/j.1743-6109.2004.10117.x
2. Lee SG, Fralick M, Sholzberg M. Coagulopathy associated with COVID-19. CMAJ. 2020;192(21):E583. doi:10.1503/cmaj.200685
3. Lam G, McCarthy R, Haider R. A peculiar case of priapism: the hypercoagulable state in patients with severe COVID-19 infection. Eur J Case Rep Intern Med. 2020;7(8):001779. doi:10.12890/2020_001779
4. Addar A, Al Fraidi O, Nazer A, Althonayan N, Ghazwani Y. Priapism for 10 days in a patient with SARS-CoV-2 pneumonia: a case report. J Surg Case Rep. 2021;2021(4):rjab020. doi:10.1093/jscr/rjab020
5. Lamamri M, Chebbi A, Mamane J, et al. Priapism in a patient with coronavirus disease 2019 (COVID-19). Am J Emerg Med. 2021;39:251.e5-251.e7. doi:10.1016/j.ajem.2020.06.027
6. Silverman ML, VanDerVeer SJ, Donnelly TJ. Priapism in COVID-19: a thromboembolic complication. Am J Emerg Med. 2021;45:686.e5-686.e6. doi:10.1016/j.ajem.2020.12.072
7. Giuliano AFM, Vulpi M, Passerini F, et al. SARS-CoV-2 infection as a determining factor to the precipitation of ischemic priapism in a young patient with asymptomatic COVID-19. Case Rep Urol. 2021;2021:9936891. doi:10.1155/2021/9936891
8. Carreno BD, Perez CP, Vasquez D, Oyola JA, Suarez O, Bedoya C. Veno-occlusive priapism in COVID-19 disease. Urol Int. 2021;105(9-10):916-919. doi:10.1159/000514421
9. Senthilkumaran S, Shah S, Ganapathysubramanian, Balamurgan N, Thirumalaikolundusubramanian P. Propofol and priapism. Indian J Pharmacol. 2010;42(4):238-239. doi:10.4103/0253-7613.68430
10. Qu M, Lu X, Wang L, Liu Z, Sun Y, Gao X. Priapism secondary to chronic myeloid leukemia treated by a surgical cavernosa-corpus spongiosum shunt: case report. Asian J Urol. 2019;6(4):373-376. doi:10.1016/j.ajur.2018.12.004
11. Singh N, Singh Lubana S, Dabrowski L, Sidhu G. Leukostasis in chronic lymphocytic leukemia. Am J Case Rep. 2020;21:e924798. doi:10.12659/AJCR.924798
12. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417-1418. doi:10.1016/S0140-6736(20)30937-5
13. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033-2040. doi:10.1182/blood.2020006000
14. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507-513. doi:10.1016/S0140-6736(20)30211-7
15. Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18(6):1421-1424. doi:10.1111/jth.14830
16. Klok FA, Kruip M, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145-147. doi:10.1016/j.thromres.2020.04.013
Nodular Sclerosing Hodgkin Lymphoma With Paraneoplastic Cerebellar Degeneration
Paraneoplastic syndrome is a rare disorder involving manifestations of immune dysregulation triggered by malignancy. The immune system develops antibodies to the malignancy, which can cause cross reactivation with various tissues in the body, resulting in an autoimmune response. Paraneoplastic cerebellar degeneration (PCD) is a rare condition caused by immune-mediated damage to the Purkinje cells of the cerebellar tract. Symptoms may include gait instability, double vision, decreased fine motor skills, and ataxia, with progression to brainstem-associated symptoms, such as nystagmus, dysarthria, and dysphagia. Early detection and treatment of the underlying malignancy is critical to halt the progression of autoimmune-mediated destruction. We present a case of a young adult female patient with PCD caused by Purkinje cell cytoplasmic–Tr (PCA-Tr) antibody with Hodgkin lymphoma.
Case Presentation
A 20-year-old previously healthy active-duty female patient presented to the emergency department with acute worsening of chronic intermittent, recurrent episodes of lightheadedness and vertigo. Symptoms persisted for 9 months until acutely worsening over the 2 weeks prior to presentation. She reported left eye double vision but did not report seeing spots, photophobia, tinnitus, or headache. She felt off-balance, leaning on nearby objects to remain standing. Symptoms primarily occurred during ambulation; however, occasionally they happened at rest. Episodes lasted up to several minutes and occurred up to 15 times a day. The patient reported no fever, night sweats, unexplained weight loss, muscle aches, weakness, numbness or tingling, loss of bowel or bladder function, or rash. She had no recent illnesses, changes to medications, or recent travel. Oral intake to include food and water was adequate and unchanged. The patient had a remote history of mild concussions without loss of consciousness while playing sports 4 years previously. She reported no recent trauma. Nine months before, she received treatment for benign paroxysmal positional vertigo (BPPV) with the Epley maneuver with full resolution of symptoms lasting several days. She reported no prescription or over-the-counter medications, herbal remedies, or supplements. She reported no other medical or surgical history and no pertinent social or family history.
Physical examination revealed a nontoxic-appearing female patient with intermittent conversational dysarthria, saccadic pursuits, horizontal nystagmus with lateral gaze, and vertical nystagmus with vertical gaze. The patient exhibited dysdiadochokinesia, or impaired ability to perform rapid alternating hand movements with repetition. Finger-to-nose testing was impaired and heel-to-shin motion remained intact. A Romberg test was positive, and the patient had tandem gait instability. Strength testing, sensation, reflexes, and cranial nerves were otherwise intact. Initial laboratory testing was unremarkable except for mild normocytic anemia. Her infectious workup, including testing for venereal disease, HIV, COVID-19, and Coccidioidies was negative. Heavy metals analysis and urine drug screen were negative. Ophthalmology was consulted and workup revealed small amplitude downbeat nystagmus in primary gaze, sustained gaze evoked lateral beating jerk nystagmus with rebound nystagmus R>L gaze, but there was no evidence of afferent package defect and optic nerve function remained intact. Magnetic resonance imaging of the brain demonstrated cerebellar vermis hypoplasia with prominence of the superior cerebellar folia. Due to concerns for autoimmune encephalitis, a lumbar puncture was performed. Antibody testing revealed PCA-Tr antibodies, which is commonly associated with Hodgkin lymphoma, prompting further evaluation for malignancy.
Computed tomography (CT) of the chest with contrast demonstrated multiple mediastinal masses with a conglomeration of lymph nodes along the right paratracheal region. Further evaluation was performed with a positron emission tomography (PET)–CT, revealing a large conglomeration of hypermetabolic pretracheal, mediastinal, and right supraclavicular lymph that were suggestive of lymphoma. Mediastinoscopy with excisional lymph node biopsy was performed with immunohistochemical staining confirming diagnosis of a nodular sclerosing variant of Hodgkin lymphoma. The patient was treated with IV immunoglobulin at 0.4g/kg daily for 5 days. A central venous catheter was placed into the patient’s right internal jugular vein and a chemotherapy regimen of doxorubicin 46 mg, vinblastine 11 mg, bleomycin 19 units, and dacarbazine 700 mg was initiated. The patient’s symptoms improved with resolution of dysarthria; however, her visual impairment and gait instability persisted. Repeat PET-CT imaging 2 months later revealed interval improvement with decreased intensity and extent of the hypermetabolic lymph nodes and no new hypermetabolic foci.
Discussion
PCA-Tr antibodies affect the delta/notchlike epidermal growth factor–related receptor, expressed on the dendrites of cerebellar Purkinje cells.1 These fibers are the only output neurons of the cerebellar cortex and are critical to the coordination of motor movements, accounting for the ataxia experienced by patients with this subtype of PCD.2 The link between Hodgkin lymphoma and PCA-Tr antibodies has been established; however, most reports involve men with a median age of 61 years with lymphoma-associated symptoms (such as lymphadenopathy) or systemic symptoms (fever, night sweats, or weight loss) preceding neurologic manifestations in 80% of cases.3
Our patient was a young, previously healthy adult female who initially presented with vertigo, a common concern with frequently benign origins. Although there was temporary resolution of symptoms after Epley maneuvers, symptoms recurred and progressed over several months to include brainstem manifestations of nystagmus, diplopia, and dysarthria. Previous reports indicate that after remission of the Hodgkin lymphoma, PCA-Tr antibodies disappear and symptoms can improve or resolve.4,5 Treatment has just begun for our patient and although there has been initial clinical improvement, given the chronicity of symptoms, it is unclear if complete resolution will be achieved.
Conclusions
PCD can result in debilitating neurologic dysfunction and may be associated with malignancy such as Hodgkin lymphoma. This case offers unique insight due to the patient’s demographics and presentation, which involved brainstem pathology typically associated with late-onset disease and preceded by constitutional symptoms. Clinical suspicion of this rare disorder should be considered in all ages, especially if symptoms are progressive or neurologic manifestations arise, as early detection and treatment of the underlying malignancy are paramount to the prevention of significant disability.
1. de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815-824. doi:10.1002/ana.23550
2. MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, et al. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48(4):637-651. doi:10.1016/j.neuroimage.2009.06.073
3. Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234. doi:10.1212/01.wnl.0000041495.87539.98
4. Graus F, Ariño H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin lymphomas. Blood. 2014;123(21):3230-3238. doi:10.1182/blood-2014-03-537506
5. Aly R, Emmady PD. Paraneoplastic cerebellar degeneration. Updated May 8, 2022. Accessed March 30, 2022. https://www.ncbi.nlm.nih.gov/books/NBK560638
Paraneoplastic syndrome is a rare disorder involving manifestations of immune dysregulation triggered by malignancy. The immune system develops antibodies to the malignancy, which can cause cross reactivation with various tissues in the body, resulting in an autoimmune response. Paraneoplastic cerebellar degeneration (PCD) is a rare condition caused by immune-mediated damage to the Purkinje cells of the cerebellar tract. Symptoms may include gait instability, double vision, decreased fine motor skills, and ataxia, with progression to brainstem-associated symptoms, such as nystagmus, dysarthria, and dysphagia. Early detection and treatment of the underlying malignancy is critical to halt the progression of autoimmune-mediated destruction. We present a case of a young adult female patient with PCD caused by Purkinje cell cytoplasmic–Tr (PCA-Tr) antibody with Hodgkin lymphoma.
Case Presentation
A 20-year-old previously healthy active-duty female patient presented to the emergency department with acute worsening of chronic intermittent, recurrent episodes of lightheadedness and vertigo. Symptoms persisted for 9 months until acutely worsening over the 2 weeks prior to presentation. She reported left eye double vision but did not report seeing spots, photophobia, tinnitus, or headache. She felt off-balance, leaning on nearby objects to remain standing. Symptoms primarily occurred during ambulation; however, occasionally they happened at rest. Episodes lasted up to several minutes and occurred up to 15 times a day. The patient reported no fever, night sweats, unexplained weight loss, muscle aches, weakness, numbness or tingling, loss of bowel or bladder function, or rash. She had no recent illnesses, changes to medications, or recent travel. Oral intake to include food and water was adequate and unchanged. The patient had a remote history of mild concussions without loss of consciousness while playing sports 4 years previously. She reported no recent trauma. Nine months before, she received treatment for benign paroxysmal positional vertigo (BPPV) with the Epley maneuver with full resolution of symptoms lasting several days. She reported no prescription or over-the-counter medications, herbal remedies, or supplements. She reported no other medical or surgical history and no pertinent social or family history.
Physical examination revealed a nontoxic-appearing female patient with intermittent conversational dysarthria, saccadic pursuits, horizontal nystagmus with lateral gaze, and vertical nystagmus with vertical gaze. The patient exhibited dysdiadochokinesia, or impaired ability to perform rapid alternating hand movements with repetition. Finger-to-nose testing was impaired and heel-to-shin motion remained intact. A Romberg test was positive, and the patient had tandem gait instability. Strength testing, sensation, reflexes, and cranial nerves were otherwise intact. Initial laboratory testing was unremarkable except for mild normocytic anemia. Her infectious workup, including testing for venereal disease, HIV, COVID-19, and Coccidioidies was negative. Heavy metals analysis and urine drug screen were negative. Ophthalmology was consulted and workup revealed small amplitude downbeat nystagmus in primary gaze, sustained gaze evoked lateral beating jerk nystagmus with rebound nystagmus R>L gaze, but there was no evidence of afferent package defect and optic nerve function remained intact. Magnetic resonance imaging of the brain demonstrated cerebellar vermis hypoplasia with prominence of the superior cerebellar folia. Due to concerns for autoimmune encephalitis, a lumbar puncture was performed. Antibody testing revealed PCA-Tr antibodies, which is commonly associated with Hodgkin lymphoma, prompting further evaluation for malignancy.
Computed tomography (CT) of the chest with contrast demonstrated multiple mediastinal masses with a conglomeration of lymph nodes along the right paratracheal region. Further evaluation was performed with a positron emission tomography (PET)–CT, revealing a large conglomeration of hypermetabolic pretracheal, mediastinal, and right supraclavicular lymph that were suggestive of lymphoma. Mediastinoscopy with excisional lymph node biopsy was performed with immunohistochemical staining confirming diagnosis of a nodular sclerosing variant of Hodgkin lymphoma. The patient was treated with IV immunoglobulin at 0.4g/kg daily for 5 days. A central venous catheter was placed into the patient’s right internal jugular vein and a chemotherapy regimen of doxorubicin 46 mg, vinblastine 11 mg, bleomycin 19 units, and dacarbazine 700 mg was initiated. The patient’s symptoms improved with resolution of dysarthria; however, her visual impairment and gait instability persisted. Repeat PET-CT imaging 2 months later revealed interval improvement with decreased intensity and extent of the hypermetabolic lymph nodes and no new hypermetabolic foci.
Discussion
PCA-Tr antibodies affect the delta/notchlike epidermal growth factor–related receptor, expressed on the dendrites of cerebellar Purkinje cells.1 These fibers are the only output neurons of the cerebellar cortex and are critical to the coordination of motor movements, accounting for the ataxia experienced by patients with this subtype of PCD.2 The link between Hodgkin lymphoma and PCA-Tr antibodies has been established; however, most reports involve men with a median age of 61 years with lymphoma-associated symptoms (such as lymphadenopathy) or systemic symptoms (fever, night sweats, or weight loss) preceding neurologic manifestations in 80% of cases.3
Our patient was a young, previously healthy adult female who initially presented with vertigo, a common concern with frequently benign origins. Although there was temporary resolution of symptoms after Epley maneuvers, symptoms recurred and progressed over several months to include brainstem manifestations of nystagmus, diplopia, and dysarthria. Previous reports indicate that after remission of the Hodgkin lymphoma, PCA-Tr antibodies disappear and symptoms can improve or resolve.4,5 Treatment has just begun for our patient and although there has been initial clinical improvement, given the chronicity of symptoms, it is unclear if complete resolution will be achieved.
Conclusions
PCD can result in debilitating neurologic dysfunction and may be associated with malignancy such as Hodgkin lymphoma. This case offers unique insight due to the patient’s demographics and presentation, which involved brainstem pathology typically associated with late-onset disease and preceded by constitutional symptoms. Clinical suspicion of this rare disorder should be considered in all ages, especially if symptoms are progressive or neurologic manifestations arise, as early detection and treatment of the underlying malignancy are paramount to the prevention of significant disability.
Paraneoplastic syndrome is a rare disorder involving manifestations of immune dysregulation triggered by malignancy. The immune system develops antibodies to the malignancy, which can cause cross reactivation with various tissues in the body, resulting in an autoimmune response. Paraneoplastic cerebellar degeneration (PCD) is a rare condition caused by immune-mediated damage to the Purkinje cells of the cerebellar tract. Symptoms may include gait instability, double vision, decreased fine motor skills, and ataxia, with progression to brainstem-associated symptoms, such as nystagmus, dysarthria, and dysphagia. Early detection and treatment of the underlying malignancy is critical to halt the progression of autoimmune-mediated destruction. We present a case of a young adult female patient with PCD caused by Purkinje cell cytoplasmic–Tr (PCA-Tr) antibody with Hodgkin lymphoma.
Case Presentation
A 20-year-old previously healthy active-duty female patient presented to the emergency department with acute worsening of chronic intermittent, recurrent episodes of lightheadedness and vertigo. Symptoms persisted for 9 months until acutely worsening over the 2 weeks prior to presentation. She reported left eye double vision but did not report seeing spots, photophobia, tinnitus, or headache. She felt off-balance, leaning on nearby objects to remain standing. Symptoms primarily occurred during ambulation; however, occasionally they happened at rest. Episodes lasted up to several minutes and occurred up to 15 times a day. The patient reported no fever, night sweats, unexplained weight loss, muscle aches, weakness, numbness or tingling, loss of bowel or bladder function, or rash. She had no recent illnesses, changes to medications, or recent travel. Oral intake to include food and water was adequate and unchanged. The patient had a remote history of mild concussions without loss of consciousness while playing sports 4 years previously. She reported no recent trauma. Nine months before, she received treatment for benign paroxysmal positional vertigo (BPPV) with the Epley maneuver with full resolution of symptoms lasting several days. She reported no prescription or over-the-counter medications, herbal remedies, or supplements. She reported no other medical or surgical history and no pertinent social or family history.
Physical examination revealed a nontoxic-appearing female patient with intermittent conversational dysarthria, saccadic pursuits, horizontal nystagmus with lateral gaze, and vertical nystagmus with vertical gaze. The patient exhibited dysdiadochokinesia, or impaired ability to perform rapid alternating hand movements with repetition. Finger-to-nose testing was impaired and heel-to-shin motion remained intact. A Romberg test was positive, and the patient had tandem gait instability. Strength testing, sensation, reflexes, and cranial nerves were otherwise intact. Initial laboratory testing was unremarkable except for mild normocytic anemia. Her infectious workup, including testing for venereal disease, HIV, COVID-19, and Coccidioidies was negative. Heavy metals analysis and urine drug screen were negative. Ophthalmology was consulted and workup revealed small amplitude downbeat nystagmus in primary gaze, sustained gaze evoked lateral beating jerk nystagmus with rebound nystagmus R>L gaze, but there was no evidence of afferent package defect and optic nerve function remained intact. Magnetic resonance imaging of the brain demonstrated cerebellar vermis hypoplasia with prominence of the superior cerebellar folia. Due to concerns for autoimmune encephalitis, a lumbar puncture was performed. Antibody testing revealed PCA-Tr antibodies, which is commonly associated with Hodgkin lymphoma, prompting further evaluation for malignancy.
Computed tomography (CT) of the chest with contrast demonstrated multiple mediastinal masses with a conglomeration of lymph nodes along the right paratracheal region. Further evaluation was performed with a positron emission tomography (PET)–CT, revealing a large conglomeration of hypermetabolic pretracheal, mediastinal, and right supraclavicular lymph that were suggestive of lymphoma. Mediastinoscopy with excisional lymph node biopsy was performed with immunohistochemical staining confirming diagnosis of a nodular sclerosing variant of Hodgkin lymphoma. The patient was treated with IV immunoglobulin at 0.4g/kg daily for 5 days. A central venous catheter was placed into the patient’s right internal jugular vein and a chemotherapy regimen of doxorubicin 46 mg, vinblastine 11 mg, bleomycin 19 units, and dacarbazine 700 mg was initiated. The patient’s symptoms improved with resolution of dysarthria; however, her visual impairment and gait instability persisted. Repeat PET-CT imaging 2 months later revealed interval improvement with decreased intensity and extent of the hypermetabolic lymph nodes and no new hypermetabolic foci.
Discussion
PCA-Tr antibodies affect the delta/notchlike epidermal growth factor–related receptor, expressed on the dendrites of cerebellar Purkinje cells.1 These fibers are the only output neurons of the cerebellar cortex and are critical to the coordination of motor movements, accounting for the ataxia experienced by patients with this subtype of PCD.2 The link between Hodgkin lymphoma and PCA-Tr antibodies has been established; however, most reports involve men with a median age of 61 years with lymphoma-associated symptoms (such as lymphadenopathy) or systemic symptoms (fever, night sweats, or weight loss) preceding neurologic manifestations in 80% of cases.3
Our patient was a young, previously healthy adult female who initially presented with vertigo, a common concern with frequently benign origins. Although there was temporary resolution of symptoms after Epley maneuvers, symptoms recurred and progressed over several months to include brainstem manifestations of nystagmus, diplopia, and dysarthria. Previous reports indicate that after remission of the Hodgkin lymphoma, PCA-Tr antibodies disappear and symptoms can improve or resolve.4,5 Treatment has just begun for our patient and although there has been initial clinical improvement, given the chronicity of symptoms, it is unclear if complete resolution will be achieved.
Conclusions
PCD can result in debilitating neurologic dysfunction and may be associated with malignancy such as Hodgkin lymphoma. This case offers unique insight due to the patient’s demographics and presentation, which involved brainstem pathology typically associated with late-onset disease and preceded by constitutional symptoms. Clinical suspicion of this rare disorder should be considered in all ages, especially if symptoms are progressive or neurologic manifestations arise, as early detection and treatment of the underlying malignancy are paramount to the prevention of significant disability.
1. de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815-824. doi:10.1002/ana.23550
2. MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, et al. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48(4):637-651. doi:10.1016/j.neuroimage.2009.06.073
3. Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234. doi:10.1212/01.wnl.0000041495.87539.98
4. Graus F, Ariño H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin lymphomas. Blood. 2014;123(21):3230-3238. doi:10.1182/blood-2014-03-537506
5. Aly R, Emmady PD. Paraneoplastic cerebellar degeneration. Updated May 8, 2022. Accessed March 30, 2022. https://www.ncbi.nlm.nih.gov/books/NBK560638
1. de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815-824. doi:10.1002/ana.23550
2. MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, et al. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48(4):637-651. doi:10.1016/j.neuroimage.2009.06.073
3. Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234. doi:10.1212/01.wnl.0000041495.87539.98
4. Graus F, Ariño H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin lymphomas. Blood. 2014;123(21):3230-3238. doi:10.1182/blood-2014-03-537506
5. Aly R, Emmady PD. Paraneoplastic cerebellar degeneration. Updated May 8, 2022. Accessed March 30, 2022. https://www.ncbi.nlm.nih.gov/books/NBK560638
Insulin Injection-Site Acanthosis Nigricans: Skin Reactions and Clinical Implications
Insulin injection therapy is one of the most widely used health care interventions to manage both type 1 and type 2 diabetes mellitus (T1DM/T2DM). Globally, more than 150 to 200 million people inject insulin into their upper posterior arms, buttocks, anterior and lateral thighs, or abdomen.1,2 In an ideal world, every patient would be using the correct site and rotating their insulin injection sites in accordance with health care professional (HCP) recommendations—systematic injections in one general body location, at least 1 cm away from the previous injection.2 Unfortunately, same-site insulin injection (repeatedly in the same region within 1 cm of previous injections) is a common mistake made by patients with DM—in one study, 63% of participants either did not rotate sites correctly or failed to do so at all.
Insulin-resistant cutaneous complications may occur as a result of same-site insulin injections. The most common is lipohypertrophy, reported in some studies in nearly 50% of patients with DM on insulin therapy.4 Other common cutaneous complications include lipoatrophy and amyloidosis. Injection-site acanthosis nigricans, although uncommon, has been reported in 18 cases in the literature.
Most articles suggest that same-site insulin injections decrease local insulin sensitivity and result in tissue hypertrophy because of the anabolic properties of insulin and increase in insulin binding to insulin-like growth factor-1 (IGF-1) receptor.5-20 The hyperkeratotic growth and varying insulin absorption rates associated with these cutaneous complications increase chances of either hyper- or hypoglycemic episodes in patients.10,11,13 It is the responsibility of the DM care professional to provide proper insulin-injection technique education and perform routine inspection of injection sites to reduce cutaneous complications of insulin therapy. The purpose of this article is to (1) describe a case of acanthosis nigricans resulting from insulin injection at the same site; (2) review case reports
Case Presentation
A 75-year-old patient with an 8-year history of T2DM, as well as stable coronary artery disease, atrial fibrillation, hypertension, hyperlipidemia, chronic obstructive pulmonary disease, and stage 3 chronic kidney disease, presented with 2 discrete abdominal hyperpigmented plaques. At the time of the initial clinic visit, the patient was taking metformin 1000 mg twice daily and insulin glargine 40 units once daily. When insulin was initiated 7 years prior, the patient received
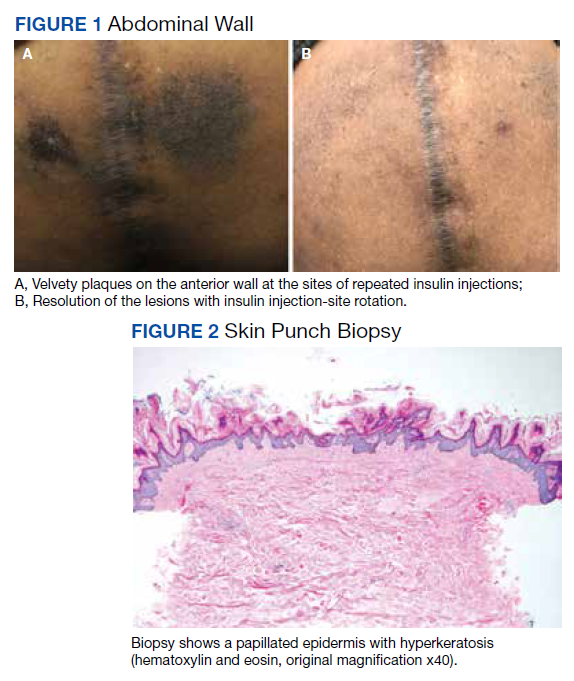
The patient reported 5 years of progressive, asymptomatic hyperpigmentation of the skin surrounding his insulin glargine injection sites and injecting in these same sites daily without rotation. He reported no additional skin changes or symptoms. He had noticed no skin changes while using NPH insulin during his first year of insulin therapy. On examination, the abdominal wall skin demonstrated 2 well-demarcated, nearly black, soft, velvety plaques, measuring 9 × 8 cm on the left side and 4 × 3.5 cm on the right, suggesting acanthosis nigricans (Figure 1A). The remainder of the skin examination, including the flexures, was normal. Of note, the patient received biweekly intramuscular testosterone injections in the gluteal region for secondary hypogonadism with no adverse dermatologic effects. A skin punch biopsy was performed and revealed epidermal papillomatosis and hyperkeratosis, confirming the clinical diagnosis of acanthosis nigricans (Figure 2).
After a review of insulin-injection technique at his clinic visit, the patient started rotating insulin injection sites over his entire abdomen, and the acanthosis nigricans partially improved. A few months later, the patient stopped rotating the insulin injection site, and the acanthosis nigricans worsened again. Because of worsening glycemic control, the patient was then started on insulin aspart. He did not develop any skin changes at the insulin aspart injection site, although he was not rotating its site of injection.
Subsequently, with reeducation and proper injection-site rotation, the patient had resolution of his acanthosis nigricans (Figure 1b).
Discussion
A review of the literature revealed 18 reported cases of acanthosis nigricans at sites of repeated insulin injection (Table).5-20 Acanthosis nigricans at the site of insulin injection afflicts patients of any age, with cases observed in patients aged 14 to 75 years. Sixteen (84%) of 19 cases were male. Fourteen cases (73%) had T2DM; the rest of the patients had T1DM. The duration of insulin injection therapy prior to onset ranged from immediate to 13 years (median 4 years). Fourteen cases (73%) were reported on the abdomen; however, other sites, such as thighs and upper arm, also were reported. Lesions size varied from 12 to 360 cm2. Two cases had associated amyloidosis. The average HbA1c reported at presentation was 10%. Following insulin injection-site rotation, most of the cases reported improvement of both glycemic control and acanthosis nigricans appearance.
In the case described by Kudo and colleagues, a 59-year-old male patient with T2DM had been injecting insulin into the same spot on his abdomen for 10 years. He developed acanthosis nigricans and an amyloidoma so large and firm that it bent the needle when he injected insulin.11
Most of the cases we found in the literature were after 2005 and associated with the use of human or analog insulin. These cases may be related to a bias, as cases may be easier to find in digital archives in the later years, when human or analog insulins have been in common use. Also noteworthy, in cases that reported dosage, most were not very high, and the highest daily dose was 240 IU/d. Ten reports of injection-site acanthosis nigricans were in dermatology journals; only 5 reports were in endocrinology journals and 3 in general medical journals, indicating possible less awareness of this phenomenon in other HCPs who care for patients with DM.
Complications of Same-Site Injections
Acanthosis nigricans. Commonly found in the armpits, neck folds, and groin, acanthosis nigricans is known as one of the calling cards for insulin resistance, obesity, and hyperinsulinemia.21 Acanthosis nigricans can be seen in people with or without DM and is not limited to those on insulin therapy. However, same-site insulin injections for 4 to 6 years also may result in injection-site acanthosis nigricans–like lesions because of factors such as insulin exposure at the local tissue level.16
Acanthosis nigricans development is characterized by hyperpigmented, hyperkeratotic, velvety, and sometimes verrucous plaques.6 Acanthosis nigricans surrounding repeated injection sites is hypothesized to develop as a result of localized hyperinsulinemia secondary to insulin resistance, which increases the stimulation of IGF, thereby causing epidermal hypertrophy.5-20 If insulin injection therapy continues to be administered through the acanthosis nigricans lesion, it results in decreased insulin absorption, leading to poor glycemic control.13
Acanthosis nigricans associated with insulin injection is reversible. After rotation of injection sites, lesions either decrease in size or severity of appearance.5-8,11 Also, by avoiding injection into the hyperkeratotic plaques and using normal subcutaneous tissue for injection, patients’ response to insulin improves, as measured by HbA1c and by decreased daily insulin requirement.6-8,10,12,18-20
Lipohypertrophy. This is characterized by an increase in localized adipose tissue and is the most common cutaneous complication of insulin therapy.2 Lipohypertrophy presents as a firm, rubbery mass in the location of same-site insulin injections.22 Development of lipohypertrophy is suspected to be the result of either (1) anabolic effect of insulin on local adipocytes, promoting fat and protein synthesis; (2) an autoimmune response by immunoglobulin (Ig) G or IgE antibodies to insulin, immune response to insulin of different species, or to insulin injection techniques; or (3) repeated trauma to the injection site from repeated needle usage.4,23
In a study assessing the prevalence of lipohypertrophy and its relation to insulin technique, 49.1% of participants with
Primary prevention measures include injection site inspection and patient education about rotation and abstaining from needle reuse.22 If a patient already has signs of lipohypertrophy, data supports education and insulin injection technique practice as simple and effective means to reduce insulin action variability and increase glycemic control.24
Lipoatrophy. Lipoatrophy is described as a local loss of subcutaneous adipose tissue often in the face, buttocks, legs and arm regions and can be rooted in genetic, immune, or drug-associated etiologies.25 Insulin-induced lipoatrophy is suspected to be the result of tumor necrosis factor-α hyperproduction in reaction to insulin crystal presence at the injection site.26,27 Overall, lipoatrophy development has decreased since the use of recombinant human insulin and analog insulin therapy.28 The decrease is hypothesized to be due to increased subcutaneous tissue absorption rate of human insulin and its analog, decreasing overall adipocyte exposure to localized high insulin concentration.27 Treatments for same-site insulin-derived lipoatrophy include changing injection sites and preparation of insulin.26 When injection into the lipoatrophic site was avoided, glycemic control and lipoatrophy appearance improved.26
Amyloidosis. Amyloidosis indicates the presence of an extracellular bundle of insoluble polymeric protein fibrils in tissues and organs.29 Insulin-induced amyloidosis presents as a hard mass or ball near the injection site.29 Insulin is one of many hormones that can form amyloid fibrils, and there have been several dozen cases reported of amyloid formation at the site of insulin injection.29-31 Although insulin-derived amyloidosis is rare, it may be misdiagnosed as lipohypertrophy due to a lack of histopathologic testing or general awareness of the complication.29
In a case series of 7 patients with amyloidosis, all patients had a mean HbA1c of 9.3% (range, 8.5-10.2%) and averaged 1 IU/kg bodyweight before intervention.30 After the discovery of the mass, participants were instructed to avoid injection into the amyloidoma, and average insulin requirements decreased to 0.48 IU/kg body weight (P = .40).30 Patients with amyloidosis who rotated their injection sites experienced better glycemic control and decreased insulin requirements.30
Pathophysiology of Localized Insulin Resistance
Insulin regulates glucose homeostasis in skeletal muscle and adipose tissue, increases hepatic and adipocyte lipid synthesis, and decreases adipocyte fatty acid release.32 Generalized insulin resistance occurs when target tissues have decreased glucose uptake in response to circulating insulin.32 Insulin resistance increases the amount of free insulin in surrounding tissues. At high concentrations, insulin fosters tissue growth by binding to IGF-1 receptors, stimulating hypertrophy and reproduction of keratinocytes and fibroblasts.33 This pathophysiology helps explain the origin of localized acanthosis nigricans at same-site insulin injections.
Conclusions
Cutaneous complications are a local adverse effect of long-term failure to rotate insulin injection sites. Our case serves as a call to action for HCPs to improve education regarding insulin injection-site rotation, conduct routine injection-site inspection, and actively document cases as they occur to increase public awareness of these important complications.
If a patient with DM presents with unexplained poor glycemic control, consider questioning the patient about injection-site location and how often they are rotating the insulin injection site. Inspect the site for cutaneous complications. Of note, if a patient has a cutaneous complication due to insulin injection, adjust or decrease the insulin dosage when rotating sites to mitigate the risk of hypoglycemic episodes.
Improvement of glycemic control, cosmetic appearance of injection site, and insulin use all begin with skin inspection, injection technique education, and periodic review by a HCP.
1. Foster NC, Beck RW, Miller KM, et al. State of type 1 diabetes management and outcomes from the T1D exchange in 2016-2018. Diabetes Technol Ther. 2019;21(2):66-72. doi:10.1089/dia.2018.0384
2. Frid AH, Kreugel G, Grassi G, et al. New insulin delivery recommendations. Mayo Clin Proc. 2016;91(9):1231-1255. doi:10.1016/j.mayocp.2016.06.010
3. Blanco M, Hernández MT, Strauss KW, Amaya M. Prevalence and risk factors of lipohypertrophy in insulin-injecting patients with diabetes. Diabetes Metab. 2013;39(5):445-453. doi:10.1016/j.diabet.2013.05.006
4. Johansson UB, Amsberg S, Hannerz L, et al. Impaired absorption of insulin aspart from lipohypertrophic injection sites. Diabetes Care. 2005;28(8):2025-2027. doi:10.2337/diacare.28.8.2025
5. Erickson L, Lipschutz DE, Wrigley W, Kearse WO. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209(6):934-935. doi:10.1001/jama.1969.03160190056019
6. Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122(9):1054-1056. doi:10.1001/archderm.1986.01660210104028 7. Gannon D, Ross MW, Mahajan T. Acanthosis nigricans-like plaque and lipohypertrophy in type 1 diabetes. Pract Diabetes International. 2005;22(6).
8. Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144(1):126-127. doi:10.1001/archdermatol.2007.27
9. Pachón Burgos A, Chan Aguilar MP. Visual vignette. Hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14(4):514. doi:10.4158/EP.14.4.514
10. Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94(2):e34-e36. doi:10.1016/j.diabres.2011.07.023
11. Kudo-Watanuki S, Kurihara E, Yamamoto K, Mukai K, Chen KR. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2012;38(1):25-29. doi:10.1111/j.1365-2230.2012.04373.x
12. Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11(12):e85-e87.
13. Kanwar A, Sawatkar G, Dogra S, Bhadada S. Acanthosis nigricans—an uncommon cutaneous adverse effect of a common medication: report of two cases. Indian J Dermatol Venereol Leprol. 2013;79(4):553. doi:10.4103/0378-6323.113112
14. Dhingra M, Garg G, Gupta M, Khurana U, Thami GP. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19(1):9. Published 2013 Jan 15.
15. Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57(1):127-129. doi:10.4103/0377-4929.130920
16. Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39(1):5-9.
17. Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39(12):e163. doi:10.1097/DAD.0000000000000659
18. Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190(47):E1390. doi:10.1503/cmaj.180705
19. Pal R, Bhattacharjee R, Chatterjee D, Bhadada SK, Bhansali A, Dutta P. Exogenous insulin-induced localized acanthosis nigricans: a rare injection site complication. Can J Diabetes. 2020;44(3):219-221. doi:10.1016/j.jcjd.2019.08.010
20. Bomar L, Lewallen R, Jorizzo J. Localized acanthosis nigricans at the site of repetitive insulin injections. Cutis. 2020;105(2);E20-E22.
21. Karadağ AS, You Y, Danarti R, Al-Khuzaei S, Chen W. Acanthosis nigricans and the metabolic syndrome. Clin Dermatol. 2018;36(1):48-53. doi:10.1016/j.clindermatol.2017.09.008
22. Kalra S, Kumar A, Gupta Y. Prevention of lipohypertrophy. J Pak Med Assoc. 2016;66(7):910-911.
23. Singha A, Bhattarcharjee R, Ghosh S, Chakrabarti SK, Baidya A, Chowdhury S. Concurrence of lipoatrophy and lipohypertrophy in children with type 1 diabetes using recombinant human insulin: two case reports. Clin Diabetes. 2016;34(1):51-53. doi:10.2337/diaclin.34.1.51
24. Famulla S, Hövelmann U, Fischer A, et al. Insulin injection into lipohypertrophic tissue: blunted and more variable insulin absorption and action and impaired postprandial glucose control. Diabetes Care. 2016;39(9):1486-1492. doi:10.2337/dc16-0610.
25. Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab. 2000;11(10):410-416. doi:10.1016/s1043-2760(00)00309-x
26. Kondo A, Nakamura A, Takeuchi J, Miyoshi H, Atsumi T. Insulin-Induced Distant Site Lipoatrophy. Diabetes Care. 2017;40(6):e67-e68. doi:10.2337/dc16-2385
27. Jermendy G, Nádas J, Sápi Z. “Lipoblastoma-like” lipoatrophy induced by human insulin: morphological evidence for local dedifferentiation of adipocytes?. Diabetologia. 2000;43(7):955-956. doi:10.1007/s001250051476
28. Mokta JK, Mokta KK, Panda P. Insulin lipodystrophy and lipohypertrophy. Indian J Endocrinol Metab. 2013;17(4):773-774. doi:10.4103/2230-8210.113788
29. Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19(1):174-177. doi:10.4103/2230-8210.146879
30. Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127(5):450-454. doi:10.1016/j.amjmed.2013.10.029
31. Swift B. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabet Med. 2002;19(10):881-882. doi:10.1046/j.1464-5491.2002.07581.x
32. Sesti G. Pathophysiology of insulin resistance. Best Pract Res Clin Endocrinol Metab. 2006;20(4):665-679. doi:10.1016/j.beem.2006.09.007
33. Phiske MM. An approach to acanthosis nigricans. Indian Dermatol Online J. 2014;5(3):239-249. doi:10.4103/2229-5178.137765
Insulin injection therapy is one of the most widely used health care interventions to manage both type 1 and type 2 diabetes mellitus (T1DM/T2DM). Globally, more than 150 to 200 million people inject insulin into their upper posterior arms, buttocks, anterior and lateral thighs, or abdomen.1,2 In an ideal world, every patient would be using the correct site and rotating their insulin injection sites in accordance with health care professional (HCP) recommendations—systematic injections in one general body location, at least 1 cm away from the previous injection.2 Unfortunately, same-site insulin injection (repeatedly in the same region within 1 cm of previous injections) is a common mistake made by patients with DM—in one study, 63% of participants either did not rotate sites correctly or failed to do so at all.
Insulin-resistant cutaneous complications may occur as a result of same-site insulin injections. The most common is lipohypertrophy, reported in some studies in nearly 50% of patients with DM on insulin therapy.4 Other common cutaneous complications include lipoatrophy and amyloidosis. Injection-site acanthosis nigricans, although uncommon, has been reported in 18 cases in the literature.
Most articles suggest that same-site insulin injections decrease local insulin sensitivity and result in tissue hypertrophy because of the anabolic properties of insulin and increase in insulin binding to insulin-like growth factor-1 (IGF-1) receptor.5-20 The hyperkeratotic growth and varying insulin absorption rates associated with these cutaneous complications increase chances of either hyper- or hypoglycemic episodes in patients.10,11,13 It is the responsibility of the DM care professional to provide proper insulin-injection technique education and perform routine inspection of injection sites to reduce cutaneous complications of insulin therapy. The purpose of this article is to (1) describe a case of acanthosis nigricans resulting from insulin injection at the same site; (2) review case reports
Case Presentation
A 75-year-old patient with an 8-year history of T2DM, as well as stable coronary artery disease, atrial fibrillation, hypertension, hyperlipidemia, chronic obstructive pulmonary disease, and stage 3 chronic kidney disease, presented with 2 discrete abdominal hyperpigmented plaques. At the time of the initial clinic visit, the patient was taking metformin 1000 mg twice daily and insulin glargine 40 units once daily. When insulin was initiated 7 years prior, the patient received
The patient reported 5 years of progressive, asymptomatic hyperpigmentation of the skin surrounding his insulin glargine injection sites and injecting in these same sites daily without rotation. He reported no additional skin changes or symptoms. He had noticed no skin changes while using NPH insulin during his first year of insulin therapy. On examination, the abdominal wall skin demonstrated 2 well-demarcated, nearly black, soft, velvety plaques, measuring 9 × 8 cm on the left side and 4 × 3.5 cm on the right, suggesting acanthosis nigricans (Figure 1A). The remainder of the skin examination, including the flexures, was normal. Of note, the patient received biweekly intramuscular testosterone injections in the gluteal region for secondary hypogonadism with no adverse dermatologic effects. A skin punch biopsy was performed and revealed epidermal papillomatosis and hyperkeratosis, confirming the clinical diagnosis of acanthosis nigricans (Figure 2).
After a review of insulin-injection technique at his clinic visit, the patient started rotating insulin injection sites over his entire abdomen, and the acanthosis nigricans partially improved. A few months later, the patient stopped rotating the insulin injection site, and the acanthosis nigricans worsened again. Because of worsening glycemic control, the patient was then started on insulin aspart. He did not develop any skin changes at the insulin aspart injection site, although he was not rotating its site of injection.
Subsequently, with reeducation and proper injection-site rotation, the patient had resolution of his acanthosis nigricans (Figure 1b).
Discussion
A review of the literature revealed 18 reported cases of acanthosis nigricans at sites of repeated insulin injection (Table).5-20 Acanthosis nigricans at the site of insulin injection afflicts patients of any age, with cases observed in patients aged 14 to 75 years. Sixteen (84%) of 19 cases were male. Fourteen cases (73%) had T2DM; the rest of the patients had T1DM. The duration of insulin injection therapy prior to onset ranged from immediate to 13 years (median 4 years). Fourteen cases (73%) were reported on the abdomen; however, other sites, such as thighs and upper arm, also were reported. Lesions size varied from 12 to 360 cm2. Two cases had associated amyloidosis. The average HbA1c reported at presentation was 10%. Following insulin injection-site rotation, most of the cases reported improvement of both glycemic control and acanthosis nigricans appearance.
In the case described by Kudo and colleagues, a 59-year-old male patient with T2DM had been injecting insulin into the same spot on his abdomen for 10 years. He developed acanthosis nigricans and an amyloidoma so large and firm that it bent the needle when he injected insulin.11
Most of the cases we found in the literature were after 2005 and associated with the use of human or analog insulin. These cases may be related to a bias, as cases may be easier to find in digital archives in the later years, when human or analog insulins have been in common use. Also noteworthy, in cases that reported dosage, most were not very high, and the highest daily dose was 240 IU/d. Ten reports of injection-site acanthosis nigricans were in dermatology journals; only 5 reports were in endocrinology journals and 3 in general medical journals, indicating possible less awareness of this phenomenon in other HCPs who care for patients with DM.
Complications of Same-Site Injections
Acanthosis nigricans. Commonly found in the armpits, neck folds, and groin, acanthosis nigricans is known as one of the calling cards for insulin resistance, obesity, and hyperinsulinemia.21 Acanthosis nigricans can be seen in people with or without DM and is not limited to those on insulin therapy. However, same-site insulin injections for 4 to 6 years also may result in injection-site acanthosis nigricans–like lesions because of factors such as insulin exposure at the local tissue level.16
Acanthosis nigricans development is characterized by hyperpigmented, hyperkeratotic, velvety, and sometimes verrucous plaques.6 Acanthosis nigricans surrounding repeated injection sites is hypothesized to develop as a result of localized hyperinsulinemia secondary to insulin resistance, which increases the stimulation of IGF, thereby causing epidermal hypertrophy.5-20 If insulin injection therapy continues to be administered through the acanthosis nigricans lesion, it results in decreased insulin absorption, leading to poor glycemic control.13
Acanthosis nigricans associated with insulin injection is reversible. After rotation of injection sites, lesions either decrease in size or severity of appearance.5-8,11 Also, by avoiding injection into the hyperkeratotic plaques and using normal subcutaneous tissue for injection, patients’ response to insulin improves, as measured by HbA1c and by decreased daily insulin requirement.6-8,10,12,18-20
Lipohypertrophy. This is characterized by an increase in localized adipose tissue and is the most common cutaneous complication of insulin therapy.2 Lipohypertrophy presents as a firm, rubbery mass in the location of same-site insulin injections.22 Development of lipohypertrophy is suspected to be the result of either (1) anabolic effect of insulin on local adipocytes, promoting fat and protein synthesis; (2) an autoimmune response by immunoglobulin (Ig) G or IgE antibodies to insulin, immune response to insulin of different species, or to insulin injection techniques; or (3) repeated trauma to the injection site from repeated needle usage.4,23
In a study assessing the prevalence of lipohypertrophy and its relation to insulin technique, 49.1% of participants with
Primary prevention measures include injection site inspection and patient education about rotation and abstaining from needle reuse.22 If a patient already has signs of lipohypertrophy, data supports education and insulin injection technique practice as simple and effective means to reduce insulin action variability and increase glycemic control.24
Lipoatrophy. Lipoatrophy is described as a local loss of subcutaneous adipose tissue often in the face, buttocks, legs and arm regions and can be rooted in genetic, immune, or drug-associated etiologies.25 Insulin-induced lipoatrophy is suspected to be the result of tumor necrosis factor-α hyperproduction in reaction to insulin crystal presence at the injection site.26,27 Overall, lipoatrophy development has decreased since the use of recombinant human insulin and analog insulin therapy.28 The decrease is hypothesized to be due to increased subcutaneous tissue absorption rate of human insulin and its analog, decreasing overall adipocyte exposure to localized high insulin concentration.27 Treatments for same-site insulin-derived lipoatrophy include changing injection sites and preparation of insulin.26 When injection into the lipoatrophic site was avoided, glycemic control and lipoatrophy appearance improved.26
Amyloidosis. Amyloidosis indicates the presence of an extracellular bundle of insoluble polymeric protein fibrils in tissues and organs.29 Insulin-induced amyloidosis presents as a hard mass or ball near the injection site.29 Insulin is one of many hormones that can form amyloid fibrils, and there have been several dozen cases reported of amyloid formation at the site of insulin injection.29-31 Although insulin-derived amyloidosis is rare, it may be misdiagnosed as lipohypertrophy due to a lack of histopathologic testing or general awareness of the complication.29
In a case series of 7 patients with amyloidosis, all patients had a mean HbA1c of 9.3% (range, 8.5-10.2%) and averaged 1 IU/kg bodyweight before intervention.30 After the discovery of the mass, participants were instructed to avoid injection into the amyloidoma, and average insulin requirements decreased to 0.48 IU/kg body weight (P = .40).30 Patients with amyloidosis who rotated their injection sites experienced better glycemic control and decreased insulin requirements.30
Pathophysiology of Localized Insulin Resistance
Insulin regulates glucose homeostasis in skeletal muscle and adipose tissue, increases hepatic and adipocyte lipid synthesis, and decreases adipocyte fatty acid release.32 Generalized insulin resistance occurs when target tissues have decreased glucose uptake in response to circulating insulin.32 Insulin resistance increases the amount of free insulin in surrounding tissues. At high concentrations, insulin fosters tissue growth by binding to IGF-1 receptors, stimulating hypertrophy and reproduction of keratinocytes and fibroblasts.33 This pathophysiology helps explain the origin of localized acanthosis nigricans at same-site insulin injections.
Conclusions
Cutaneous complications are a local adverse effect of long-term failure to rotate insulin injection sites. Our case serves as a call to action for HCPs to improve education regarding insulin injection-site rotation, conduct routine injection-site inspection, and actively document cases as they occur to increase public awareness of these important complications.
If a patient with DM presents with unexplained poor glycemic control, consider questioning the patient about injection-site location and how often they are rotating the insulin injection site. Inspect the site for cutaneous complications. Of note, if a patient has a cutaneous complication due to insulin injection, adjust or decrease the insulin dosage when rotating sites to mitigate the risk of hypoglycemic episodes.
Improvement of glycemic control, cosmetic appearance of injection site, and insulin use all begin with skin inspection, injection technique education, and periodic review by a HCP.
Insulin injection therapy is one of the most widely used health care interventions to manage both type 1 and type 2 diabetes mellitus (T1DM/T2DM). Globally, more than 150 to 200 million people inject insulin into their upper posterior arms, buttocks, anterior and lateral thighs, or abdomen.1,2 In an ideal world, every patient would be using the correct site and rotating their insulin injection sites in accordance with health care professional (HCP) recommendations—systematic injections in one general body location, at least 1 cm away from the previous injection.2 Unfortunately, same-site insulin injection (repeatedly in the same region within 1 cm of previous injections) is a common mistake made by patients with DM—in one study, 63% of participants either did not rotate sites correctly or failed to do so at all.
Insulin-resistant cutaneous complications may occur as a result of same-site insulin injections. The most common is lipohypertrophy, reported in some studies in nearly 50% of patients with DM on insulin therapy.4 Other common cutaneous complications include lipoatrophy and amyloidosis. Injection-site acanthosis nigricans, although uncommon, has been reported in 18 cases in the literature.
Most articles suggest that same-site insulin injections decrease local insulin sensitivity and result in tissue hypertrophy because of the anabolic properties of insulin and increase in insulin binding to insulin-like growth factor-1 (IGF-1) receptor.5-20 The hyperkeratotic growth and varying insulin absorption rates associated with these cutaneous complications increase chances of either hyper- or hypoglycemic episodes in patients.10,11,13 It is the responsibility of the DM care professional to provide proper insulin-injection technique education and perform routine inspection of injection sites to reduce cutaneous complications of insulin therapy. The purpose of this article is to (1) describe a case of acanthosis nigricans resulting from insulin injection at the same site; (2) review case reports
Case Presentation
A 75-year-old patient with an 8-year history of T2DM, as well as stable coronary artery disease, atrial fibrillation, hypertension, hyperlipidemia, chronic obstructive pulmonary disease, and stage 3 chronic kidney disease, presented with 2 discrete abdominal hyperpigmented plaques. At the time of the initial clinic visit, the patient was taking metformin 1000 mg twice daily and insulin glargine 40 units once daily. When insulin was initiated 7 years prior, the patient received
The patient reported 5 years of progressive, asymptomatic hyperpigmentation of the skin surrounding his insulin glargine injection sites and injecting in these same sites daily without rotation. He reported no additional skin changes or symptoms. He had noticed no skin changes while using NPH insulin during his first year of insulin therapy. On examination, the abdominal wall skin demonstrated 2 well-demarcated, nearly black, soft, velvety plaques, measuring 9 × 8 cm on the left side and 4 × 3.5 cm on the right, suggesting acanthosis nigricans (Figure 1A). The remainder of the skin examination, including the flexures, was normal. Of note, the patient received biweekly intramuscular testosterone injections in the gluteal region for secondary hypogonadism with no adverse dermatologic effects. A skin punch biopsy was performed and revealed epidermal papillomatosis and hyperkeratosis, confirming the clinical diagnosis of acanthosis nigricans (Figure 2).
After a review of insulin-injection technique at his clinic visit, the patient started rotating insulin injection sites over his entire abdomen, and the acanthosis nigricans partially improved. A few months later, the patient stopped rotating the insulin injection site, and the acanthosis nigricans worsened again. Because of worsening glycemic control, the patient was then started on insulin aspart. He did not develop any skin changes at the insulin aspart injection site, although he was not rotating its site of injection.
Subsequently, with reeducation and proper injection-site rotation, the patient had resolution of his acanthosis nigricans (Figure 1b).
Discussion
A review of the literature revealed 18 reported cases of acanthosis nigricans at sites of repeated insulin injection (Table).5-20 Acanthosis nigricans at the site of insulin injection afflicts patients of any age, with cases observed in patients aged 14 to 75 years. Sixteen (84%) of 19 cases were male. Fourteen cases (73%) had T2DM; the rest of the patients had T1DM. The duration of insulin injection therapy prior to onset ranged from immediate to 13 years (median 4 years). Fourteen cases (73%) were reported on the abdomen; however, other sites, such as thighs and upper arm, also were reported. Lesions size varied from 12 to 360 cm2. Two cases had associated amyloidosis. The average HbA1c reported at presentation was 10%. Following insulin injection-site rotation, most of the cases reported improvement of both glycemic control and acanthosis nigricans appearance.
In the case described by Kudo and colleagues, a 59-year-old male patient with T2DM had been injecting insulin into the same spot on his abdomen for 10 years. He developed acanthosis nigricans and an amyloidoma so large and firm that it bent the needle when he injected insulin.11
Most of the cases we found in the literature were after 2005 and associated with the use of human or analog insulin. These cases may be related to a bias, as cases may be easier to find in digital archives in the later years, when human or analog insulins have been in common use. Also noteworthy, in cases that reported dosage, most were not very high, and the highest daily dose was 240 IU/d. Ten reports of injection-site acanthosis nigricans were in dermatology journals; only 5 reports were in endocrinology journals and 3 in general medical journals, indicating possible less awareness of this phenomenon in other HCPs who care for patients with DM.
Complications of Same-Site Injections
Acanthosis nigricans. Commonly found in the armpits, neck folds, and groin, acanthosis nigricans is known as one of the calling cards for insulin resistance, obesity, and hyperinsulinemia.21 Acanthosis nigricans can be seen in people with or without DM and is not limited to those on insulin therapy. However, same-site insulin injections for 4 to 6 years also may result in injection-site acanthosis nigricans–like lesions because of factors such as insulin exposure at the local tissue level.16
Acanthosis nigricans development is characterized by hyperpigmented, hyperkeratotic, velvety, and sometimes verrucous plaques.6 Acanthosis nigricans surrounding repeated injection sites is hypothesized to develop as a result of localized hyperinsulinemia secondary to insulin resistance, which increases the stimulation of IGF, thereby causing epidermal hypertrophy.5-20 If insulin injection therapy continues to be administered through the acanthosis nigricans lesion, it results in decreased insulin absorption, leading to poor glycemic control.13
Acanthosis nigricans associated with insulin injection is reversible. After rotation of injection sites, lesions either decrease in size or severity of appearance.5-8,11 Also, by avoiding injection into the hyperkeratotic plaques and using normal subcutaneous tissue for injection, patients’ response to insulin improves, as measured by HbA1c and by decreased daily insulin requirement.6-8,10,12,18-20
Lipohypertrophy. This is characterized by an increase in localized adipose tissue and is the most common cutaneous complication of insulin therapy.2 Lipohypertrophy presents as a firm, rubbery mass in the location of same-site insulin injections.22 Development of lipohypertrophy is suspected to be the result of either (1) anabolic effect of insulin on local adipocytes, promoting fat and protein synthesis; (2) an autoimmune response by immunoglobulin (Ig) G or IgE antibodies to insulin, immune response to insulin of different species, or to insulin injection techniques; or (3) repeated trauma to the injection site from repeated needle usage.4,23
In a study assessing the prevalence of lipohypertrophy and its relation to insulin technique, 49.1% of participants with
Primary prevention measures include injection site inspection and patient education about rotation and abstaining from needle reuse.22 If a patient already has signs of lipohypertrophy, data supports education and insulin injection technique practice as simple and effective means to reduce insulin action variability and increase glycemic control.24
Lipoatrophy. Lipoatrophy is described as a local loss of subcutaneous adipose tissue often in the face, buttocks, legs and arm regions and can be rooted in genetic, immune, or drug-associated etiologies.25 Insulin-induced lipoatrophy is suspected to be the result of tumor necrosis factor-α hyperproduction in reaction to insulin crystal presence at the injection site.26,27 Overall, lipoatrophy development has decreased since the use of recombinant human insulin and analog insulin therapy.28 The decrease is hypothesized to be due to increased subcutaneous tissue absorption rate of human insulin and its analog, decreasing overall adipocyte exposure to localized high insulin concentration.27 Treatments for same-site insulin-derived lipoatrophy include changing injection sites and preparation of insulin.26 When injection into the lipoatrophic site was avoided, glycemic control and lipoatrophy appearance improved.26
Amyloidosis. Amyloidosis indicates the presence of an extracellular bundle of insoluble polymeric protein fibrils in tissues and organs.29 Insulin-induced amyloidosis presents as a hard mass or ball near the injection site.29 Insulin is one of many hormones that can form amyloid fibrils, and there have been several dozen cases reported of amyloid formation at the site of insulin injection.29-31 Although insulin-derived amyloidosis is rare, it may be misdiagnosed as lipohypertrophy due to a lack of histopathologic testing or general awareness of the complication.29
In a case series of 7 patients with amyloidosis, all patients had a mean HbA1c of 9.3% (range, 8.5-10.2%) and averaged 1 IU/kg bodyweight before intervention.30 After the discovery of the mass, participants were instructed to avoid injection into the amyloidoma, and average insulin requirements decreased to 0.48 IU/kg body weight (P = .40).30 Patients with amyloidosis who rotated their injection sites experienced better glycemic control and decreased insulin requirements.30
Pathophysiology of Localized Insulin Resistance
Insulin regulates glucose homeostasis in skeletal muscle and adipose tissue, increases hepatic and adipocyte lipid synthesis, and decreases adipocyte fatty acid release.32 Generalized insulin resistance occurs when target tissues have decreased glucose uptake in response to circulating insulin.32 Insulin resistance increases the amount of free insulin in surrounding tissues. At high concentrations, insulin fosters tissue growth by binding to IGF-1 receptors, stimulating hypertrophy and reproduction of keratinocytes and fibroblasts.33 This pathophysiology helps explain the origin of localized acanthosis nigricans at same-site insulin injections.
Conclusions
Cutaneous complications are a local adverse effect of long-term failure to rotate insulin injection sites. Our case serves as a call to action for HCPs to improve education regarding insulin injection-site rotation, conduct routine injection-site inspection, and actively document cases as they occur to increase public awareness of these important complications.
If a patient with DM presents with unexplained poor glycemic control, consider questioning the patient about injection-site location and how often they are rotating the insulin injection site. Inspect the site for cutaneous complications. Of note, if a patient has a cutaneous complication due to insulin injection, adjust or decrease the insulin dosage when rotating sites to mitigate the risk of hypoglycemic episodes.
Improvement of glycemic control, cosmetic appearance of injection site, and insulin use all begin with skin inspection, injection technique education, and periodic review by a HCP.
1. Foster NC, Beck RW, Miller KM, et al. State of type 1 diabetes management and outcomes from the T1D exchange in 2016-2018. Diabetes Technol Ther. 2019;21(2):66-72. doi:10.1089/dia.2018.0384
2. Frid AH, Kreugel G, Grassi G, et al. New insulin delivery recommendations. Mayo Clin Proc. 2016;91(9):1231-1255. doi:10.1016/j.mayocp.2016.06.010
3. Blanco M, Hernández MT, Strauss KW, Amaya M. Prevalence and risk factors of lipohypertrophy in insulin-injecting patients with diabetes. Diabetes Metab. 2013;39(5):445-453. doi:10.1016/j.diabet.2013.05.006
4. Johansson UB, Amsberg S, Hannerz L, et al. Impaired absorption of insulin aspart from lipohypertrophic injection sites. Diabetes Care. 2005;28(8):2025-2027. doi:10.2337/diacare.28.8.2025
5. Erickson L, Lipschutz DE, Wrigley W, Kearse WO. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209(6):934-935. doi:10.1001/jama.1969.03160190056019
6. Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122(9):1054-1056. doi:10.1001/archderm.1986.01660210104028 7. Gannon D, Ross MW, Mahajan T. Acanthosis nigricans-like plaque and lipohypertrophy in type 1 diabetes. Pract Diabetes International. 2005;22(6).
8. Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144(1):126-127. doi:10.1001/archdermatol.2007.27
9. Pachón Burgos A, Chan Aguilar MP. Visual vignette. Hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14(4):514. doi:10.4158/EP.14.4.514
10. Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94(2):e34-e36. doi:10.1016/j.diabres.2011.07.023
11. Kudo-Watanuki S, Kurihara E, Yamamoto K, Mukai K, Chen KR. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2012;38(1):25-29. doi:10.1111/j.1365-2230.2012.04373.x
12. Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11(12):e85-e87.
13. Kanwar A, Sawatkar G, Dogra S, Bhadada S. Acanthosis nigricans—an uncommon cutaneous adverse effect of a common medication: report of two cases. Indian J Dermatol Venereol Leprol. 2013;79(4):553. doi:10.4103/0378-6323.113112
14. Dhingra M, Garg G, Gupta M, Khurana U, Thami GP. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19(1):9. Published 2013 Jan 15.
15. Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57(1):127-129. doi:10.4103/0377-4929.130920
16. Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39(1):5-9.
17. Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39(12):e163. doi:10.1097/DAD.0000000000000659
18. Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190(47):E1390. doi:10.1503/cmaj.180705
19. Pal R, Bhattacharjee R, Chatterjee D, Bhadada SK, Bhansali A, Dutta P. Exogenous insulin-induced localized acanthosis nigricans: a rare injection site complication. Can J Diabetes. 2020;44(3):219-221. doi:10.1016/j.jcjd.2019.08.010
20. Bomar L, Lewallen R, Jorizzo J. Localized acanthosis nigricans at the site of repetitive insulin injections. Cutis. 2020;105(2);E20-E22.
21. Karadağ AS, You Y, Danarti R, Al-Khuzaei S, Chen W. Acanthosis nigricans and the metabolic syndrome. Clin Dermatol. 2018;36(1):48-53. doi:10.1016/j.clindermatol.2017.09.008
22. Kalra S, Kumar A, Gupta Y. Prevention of lipohypertrophy. J Pak Med Assoc. 2016;66(7):910-911.
23. Singha A, Bhattarcharjee R, Ghosh S, Chakrabarti SK, Baidya A, Chowdhury S. Concurrence of lipoatrophy and lipohypertrophy in children with type 1 diabetes using recombinant human insulin: two case reports. Clin Diabetes. 2016;34(1):51-53. doi:10.2337/diaclin.34.1.51
24. Famulla S, Hövelmann U, Fischer A, et al. Insulin injection into lipohypertrophic tissue: blunted and more variable insulin absorption and action and impaired postprandial glucose control. Diabetes Care. 2016;39(9):1486-1492. doi:10.2337/dc16-0610.
25. Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab. 2000;11(10):410-416. doi:10.1016/s1043-2760(00)00309-x
26. Kondo A, Nakamura A, Takeuchi J, Miyoshi H, Atsumi T. Insulin-Induced Distant Site Lipoatrophy. Diabetes Care. 2017;40(6):e67-e68. doi:10.2337/dc16-2385
27. Jermendy G, Nádas J, Sápi Z. “Lipoblastoma-like” lipoatrophy induced by human insulin: morphological evidence for local dedifferentiation of adipocytes?. Diabetologia. 2000;43(7):955-956. doi:10.1007/s001250051476
28. Mokta JK, Mokta KK, Panda P. Insulin lipodystrophy and lipohypertrophy. Indian J Endocrinol Metab. 2013;17(4):773-774. doi:10.4103/2230-8210.113788
29. Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19(1):174-177. doi:10.4103/2230-8210.146879
30. Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127(5):450-454. doi:10.1016/j.amjmed.2013.10.029
31. Swift B. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabet Med. 2002;19(10):881-882. doi:10.1046/j.1464-5491.2002.07581.x
32. Sesti G. Pathophysiology of insulin resistance. Best Pract Res Clin Endocrinol Metab. 2006;20(4):665-679. doi:10.1016/j.beem.2006.09.007
33. Phiske MM. An approach to acanthosis nigricans. Indian Dermatol Online J. 2014;5(3):239-249. doi:10.4103/2229-5178.137765
1. Foster NC, Beck RW, Miller KM, et al. State of type 1 diabetes management and outcomes from the T1D exchange in 2016-2018. Diabetes Technol Ther. 2019;21(2):66-72. doi:10.1089/dia.2018.0384
2. Frid AH, Kreugel G, Grassi G, et al. New insulin delivery recommendations. Mayo Clin Proc. 2016;91(9):1231-1255. doi:10.1016/j.mayocp.2016.06.010
3. Blanco M, Hernández MT, Strauss KW, Amaya M. Prevalence and risk factors of lipohypertrophy in insulin-injecting patients with diabetes. Diabetes Metab. 2013;39(5):445-453. doi:10.1016/j.diabet.2013.05.006
4. Johansson UB, Amsberg S, Hannerz L, et al. Impaired absorption of insulin aspart from lipohypertrophic injection sites. Diabetes Care. 2005;28(8):2025-2027. doi:10.2337/diacare.28.8.2025
5. Erickson L, Lipschutz DE, Wrigley W, Kearse WO. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209(6):934-935. doi:10.1001/jama.1969.03160190056019
6. Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122(9):1054-1056. doi:10.1001/archderm.1986.01660210104028 7. Gannon D, Ross MW, Mahajan T. Acanthosis nigricans-like plaque and lipohypertrophy in type 1 diabetes. Pract Diabetes International. 2005;22(6).
8. Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144(1):126-127. doi:10.1001/archdermatol.2007.27
9. Pachón Burgos A, Chan Aguilar MP. Visual vignette. Hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14(4):514. doi:10.4158/EP.14.4.514
10. Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94(2):e34-e36. doi:10.1016/j.diabres.2011.07.023
11. Kudo-Watanuki S, Kurihara E, Yamamoto K, Mukai K, Chen KR. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2012;38(1):25-29. doi:10.1111/j.1365-2230.2012.04373.x
12. Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11(12):e85-e87.
13. Kanwar A, Sawatkar G, Dogra S, Bhadada S. Acanthosis nigricans—an uncommon cutaneous adverse effect of a common medication: report of two cases. Indian J Dermatol Venereol Leprol. 2013;79(4):553. doi:10.4103/0378-6323.113112
14. Dhingra M, Garg G, Gupta M, Khurana U, Thami GP. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19(1):9. Published 2013 Jan 15.
15. Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57(1):127-129. doi:10.4103/0377-4929.130920
16. Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39(1):5-9.
17. Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39(12):e163. doi:10.1097/DAD.0000000000000659
18. Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190(47):E1390. doi:10.1503/cmaj.180705
19. Pal R, Bhattacharjee R, Chatterjee D, Bhadada SK, Bhansali A, Dutta P. Exogenous insulin-induced localized acanthosis nigricans: a rare injection site complication. Can J Diabetes. 2020;44(3):219-221. doi:10.1016/j.jcjd.2019.08.010
20. Bomar L, Lewallen R, Jorizzo J. Localized acanthosis nigricans at the site of repetitive insulin injections. Cutis. 2020;105(2);E20-E22.
21. Karadağ AS, You Y, Danarti R, Al-Khuzaei S, Chen W. Acanthosis nigricans and the metabolic syndrome. Clin Dermatol. 2018;36(1):48-53. doi:10.1016/j.clindermatol.2017.09.008
22. Kalra S, Kumar A, Gupta Y. Prevention of lipohypertrophy. J Pak Med Assoc. 2016;66(7):910-911.
23. Singha A, Bhattarcharjee R, Ghosh S, Chakrabarti SK, Baidya A, Chowdhury S. Concurrence of lipoatrophy and lipohypertrophy in children with type 1 diabetes using recombinant human insulin: two case reports. Clin Diabetes. 2016;34(1):51-53. doi:10.2337/diaclin.34.1.51
24. Famulla S, Hövelmann U, Fischer A, et al. Insulin injection into lipohypertrophic tissue: blunted and more variable insulin absorption and action and impaired postprandial glucose control. Diabetes Care. 2016;39(9):1486-1492. doi:10.2337/dc16-0610.
25. Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab. 2000;11(10):410-416. doi:10.1016/s1043-2760(00)00309-x
26. Kondo A, Nakamura A, Takeuchi J, Miyoshi H, Atsumi T. Insulin-Induced Distant Site Lipoatrophy. Diabetes Care. 2017;40(6):e67-e68. doi:10.2337/dc16-2385
27. Jermendy G, Nádas J, Sápi Z. “Lipoblastoma-like” lipoatrophy induced by human insulin: morphological evidence for local dedifferentiation of adipocytes?. Diabetologia. 2000;43(7):955-956. doi:10.1007/s001250051476
28. Mokta JK, Mokta KK, Panda P. Insulin lipodystrophy and lipohypertrophy. Indian J Endocrinol Metab. 2013;17(4):773-774. doi:10.4103/2230-8210.113788
29. Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19(1):174-177. doi:10.4103/2230-8210.146879
30. Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127(5):450-454. doi:10.1016/j.amjmed.2013.10.029
31. Swift B. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabet Med. 2002;19(10):881-882. doi:10.1046/j.1464-5491.2002.07581.x
32. Sesti G. Pathophysiology of insulin resistance. Best Pract Res Clin Endocrinol Metab. 2006;20(4):665-679. doi:10.1016/j.beem.2006.09.007
33. Phiske MM. An approach to acanthosis nigricans. Indian Dermatol Online J. 2014;5(3):239-249. doi:10.4103/2229-5178.137765
Approach to Pancytopenia in a Deployed Service Member
Pancytopenia is a condition in which all 3 hematologic cell lines are lower than expected in the blood, often representing either an increase in cellular destruction or decrease in bone marrow production. Destruction often occurs in the setting of autoimmune conditions (eg, systemic lupus erythematosus, rheumatoid arthritis) or splenic sequestration, often affecting erythrocytes and platelets more than leukocytes. Decreased production represents central etiologies, which are often due to nutritional deficiencies, infections, drug toxicities, or malabsorption.1 Pancytopenia secondary to vitamin B12 deficiency is rare, accounting for about 5% of the hematologic manifestations of symptomatic vitamin B12 deficient patients.2
Pernicious anemia, named for a once lethal disease, is a form of vitamin B12 (cobalamin) deficiency that results from an autoimmune (type II hypersensitivity) reaction to gastric parietal cells or intrinsic factor. Antibodies bind to gastric parietal cells and reduce gastric acid production, leading to atrophic gastritis, or they bind intrinsic factor and block the binding and absorption of vitamin B12 in the gastrointestinal tract. While first described in the 1820s, it was not until a century later when scientists were studying hematopoiesis in response to the heavy casualty burden from battlefield exsanguination in World War I that dogs fed raw liver were noted to have significantly better blood regeneration response than those fed cooked liver. This discovery led physicians Minot and Murphy to use raw liver to treat pernicious anemia and found that jaundice improved, reticulocyte counts increased, and hemoglobin (Hb) concentration improved, resulting in the duo becoming the first American recipients of the Nobel Prize in physiology or medicine.3 It was ultimately determined in 1948 by chemists Folkers and Todd that the active ingredient in raw liver responsible for this phenomenon was vitamin B12.4

Patients with pernicious anemia typically present with macrocytic anemia, low reticulocyte count, hypersegmented neutrophils, as well as mild leukopenia and/or thrombocytopenia, distinguishable from folate deficiency by an elevated serum methylmalonic acid level. World Health Organization cytopenia thresholds are listed in Table 1.5 Treatment consists of lifelong vitamin B12 supplementation, and endoscopic screening is often recommended after diagnosis due to increased risk of gastrointestinal malignancy.6 Pernicious anemia can be difficult to distinguish from thrombotic thrombocytopenia purpura (TTP), a microangiopathic hemolytic anemia that can cause rapid end-organ failure and death if treatment is delayed.7 While pernicious anemia is not typically hemolytic, case reports of hemolysis in severe deficiency have been reported.7 Adequate bone marrow response to hemolysis in TTP results in an elevated reticulocyte count, which can be useful in differentiating from pernicious anemia where there is typically an inadequate bone marrow response and low reticulocyte count.8,9
The approach to working up pancytopenia begins with a detailed history inquiring about medications, exposures (benzenes, pesticides), alcohol use, and infection history. A thorough physical examination may help point the health care practitioner (HCP) toward a certain etiology, as the differential for pancytopenia is broad. In the deployed soldier downrange, resources are often limited, and the history/physical are crucial in preventing an expensive and unnecessary workup.
Case Presentation
A 24-year-old active-duty female patient presented in late December 2020 to a theater hospital in Djibouti after a witnessed syncopal episode. She had a history of Hashimoto thyroiditis and was taking levothyroxine sodium 75 mcg daily. The patient reported gluten intolerance, which was never formally evaluated. The syncopal episode lasted a few seconds and was not associated with any prodromal or postictal symptoms. No seizure activity was observed, and she had no history of syncopal episodes. She reported that she had been feeling ill 24 to 48 hours prior, with nausea, fatigue, decreased oral intake, decreased urine output, and 2 episodes of nonbilious, nonbloody emesis.
When the patient arrived, she was tachycardic with heart rate in the 130s beats per minute (baseline, 100-110 beats per minute), febrile (103 °F), and had systolic blood pressure (SBP) in the low 100s (baseline, SBP 120s-130s). An electrocardiogram and chest radiographs were unremarkable. Her complete blood count (CBC) could not be processed due to Hb and platelet levels too low to detect on assay (Table 2). Lactate dehydrogenase (LDH) was elevated at > 1000 U/L with mild elevation in liver enzymes (aspartate aminotransferase, 98 U/L; alanine aminotransferase, 51 U/L) and prolonged partial thromboplastin time 70 seconds. She did not report any increased bleeding or bruising. The peripheral blood smear demonstrated pancytopenia, without any schistocytes, and she was started on broad-spectrum antibiotics for presumed sepsis from urinary source and possible TTP.
The patient received 5 units of packed red blood cells, transfusion of platelets, and 2 doses of vitamin B12 in Djibouti with clinical improvement and resolution of orthostasis, hypotension, tachycardia, and fever. Her final posttransfusion CBC showed a Hb level of 11.2 g/dL, white blood cell (WBC) count of 1.7 K/µL, and platelet count of 23 K/µL (Table 3). Two days later her Hb level was 9.0 g/dL, WBC count 1.8 K/µL, and platelet count was 12 K/µL. She was evacuated via air to Landstuhl Regional Medical Center (LRMC) in Germany within 48 hours of presentation, given limited testing capabilities and persistent anemia and thrombocytopenia, refractory to transfusion, concerning for aplastic anemia or acute leukemia.
On arrival at LRMC, she was transfused 1 unit of platelets and given 3 doses of intramuscular vitamin B12 for undetectable levels (< 50 pg/mL) at presentation. An extensive infectious workup was obtained, which did not reveal any viral, bacterial, or parasitic causes. The patient also had a bone marrow biopsy performed at a civilian site, which revealed hypocellular bone marrow. She was transferred to Walter Reed National Military Medical Center (WRNMMC) for further workup and evaluation, given the infectious workup, which was negative. Concern for hematologic malignancy remained. At the time of her arrival, the laboratory values had drastically improved with vitamin supplementation. The patient’s absolute reticulocyte count indicated adequate bone marrow response and because of her improvement, a repeat bone marrow biopsy was not performed.
Intrinsic factor antibodies were elevated (34.5 AU/mL; reference range, 0.0-1.1), which confirmed that this patient’s underlying etiology was secondary to pernicious anemia. The patient continued to improve and repeat vitamin B12 and folate levels revealed that she was responding to therapy. At discharge, intramuscular vitamin B12 injections were planned to continue monthly, indefinitely per guidelines. Oral supplementation is typically avoided due to poor absorption.
Of note, during her inpatient admission at WRNMMC, further evaluation of reported gluten intolerance was performed, which revealed a negative celiac disease panel (IgG/IgA tissue transglutaminase antibodies). On discharge, she was to establish care with gastroenterology for further evaluation, likely including endoscopic evaluation, at her next duty station. She was able to resume full travel and duty functions on discharge from WRNMMC.
Discussion
We highlight a complex case of pancytopenia secondary to pernicious anemia in a deployed service member. With limited resources downrange, the workup of pancytopenia can be resource intensive, expensive, and time sensitive, which can have detrimental impacts on medical readiness. Additionally, undiagnosed coagulopathies can have lethal consequences in a deployed service member where bleeding risk may be elevated depending on the mission. The differential for pancytopenia is vast, and given its relative rarity in pernicious anemia, the HCP must use key components of the history and laboratory results to narrow the differential (eAppendix).10
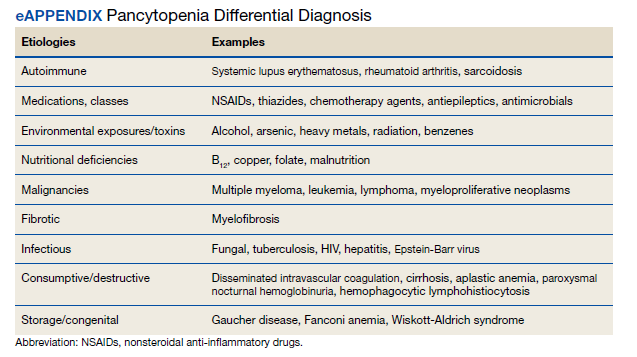
Pernicious anemia commonly presents as an isolated anemia. In a study looking at the hematologic manifestations of 201 cohort patients with well-documented vitamin B12 deficiency, 5% had symptomatic pancytopenia and 1.5% had a hemolytic anemia.2 The majority (> 67%) of hematologic abnormalities were correctable with cobalamin replacement.2 In our case, the solider presented with symptomatic anemia, manifesting as syncope, and was found to have transfusion-resistant pancytopenia.She had a hemolytic anemia with an LDH > 1000 U/L, haptoglobin < 3 mg/dL, and mild transaminitis with hyperbilirubinemia (1.8 mg/dL). No schistocytes were observed on peripheral smear, suggesting intramedullary hemolysis, which is believed to be due to the destruction of megaloblastic cells by macrophages in bone marrow.11 A French study found high LDH levels and low reticulocyte counts to be strongly suggestive of vitamin B12 deficiency and helpful in differentiating pernicious anemia from TTP, given that bone marrow response to anemia in TTP is preserved.8
While vitamin B12 deficiency is not often associated with hemolytic anemia, multiple cases have been reported in the literature.6 Screening for vitamin B12 deficiency may have shortened this patient’s clinical course and limited the need for air evacuation to a stateside quaternary medical center. However, testing for cobalamin levels in overseas deployed environments is difficult, timely, and costly. New technologies, such as optical sensors, can detect vitamin B12 levels in the blood in < 1 minute and offer portable, low-cost options that may be useful in the deployed military setting.12
Diet plays a key role in this case, since the patient had a reported history of gluten intolerance, although it was never documented or evaluated prior to this presentation. Prior to deployment, the patient ate mostly rice, potatoes, and vegetables. While deployed in an austere environment, food options were limited. These conditions forced her to intermittently consume gluten products, which led to gastrointestinal issues, exacerbating her nutritional deficiencies. In the 2 months before her first syncopal episode, she reported worsening fatigue that impacted her ability to exercise. Vitamin B12 stores often take years to deplete, suggesting that she had a chronic nutritional deficiency before deployment. Another possibility was that she developed an autoimmune gastritis that acutely worsened in the setting of poor nutritional intake. Her history of Hashimoto thyroiditis is also important, as up to one-third of patients with autoimmune thyroid disease have been associated with pernicious anemia (range, 3%-32%) with certain shared human leukocyte antigen alleles implicated in autoimmune gastritis.13,14
Conclusions
This rare case of pernicious anemia presenting as pancytopenia illustrates the challenge in working up pancytopenia, especially in austere military environments with limited testing capabilities. Screening for chronic dietary and nutritional deficiency is important in a service member, raising the question of what role predeployment screening may have and what dietary accommodations may be available during overseas deployments, which can potentially dampen inflammation of the gastrointestinal tract, especially for those with preexisting autoimmune gastrointestinal conditions. Also, newer technology allows portable, low-cost testing of cobalamin and may aid in its diagnosis. In patients who are anemic with low vitamin B12, HCPs can begin vitamin B12 supplementation while continuing the workup (eg, antibody testing, endoscopy). If the patient responds appropriately, further workup becomes less urgent, therefore, decreasing resource use and increasing military readiness. When hemolysis is present, a low reticulocyte count can be beneficial to help differentiate this condition from TTP, a life-threatening condition that must also be ruled out or treated. Pernicious anemia should be on the differential in any patients with autoimmune conditions presenting with cytopenias, especially in those with a history of autoimmune thyroid disorders.
1. Takeshima M, Ishikawa H, Kitadate A, et al. Anorexia nervosa-associated pancytopenia mimicking idiopathic aplastic anemia: a case report. BMC Psychiatry. 2018;18(1):150. doi:10.1186/s12888-018-1743-6
2. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56. doi:10.1111/j.1365-2257.2006.00755.x
3. Sinclair L. Recognizing, treating and understanding pernicious anaemia. J R Soc Med. 2008;101(5):262-264. doi:10.1258/jrsm.2008.081006
4. Shampo MA, Kyle RA, Steensma DP. William Murphy—Nobel Prize for the treatment of pernicious anemia. Mayo Clin Proc. 2006;81(6):726. doi:10.4065/81.6.726
5. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139-143. doi:10.1515/jtim-2017-0002
6. Tunio NA, Sheriff MZ, Cooper G. Prevalence of gastric cancer in patients with pernicious anemia: a population-based study. Am J Gastroenterol. 2020;115:S665. doi:10.14309/01.ajg.0000707332.16739.72
7. Bailey M, Maestas T, Betancourt R, Mikhael D, Babiker HM. A rare cause of thrombotic thrombocytopenic purpura- (TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019:1529306. doi:10.1155/2019/1529306
8. Stanley M, Michalski JM. Thrombotic Thrombocytopenic Purpura. StatPearls Publishing LLC; 2021.
9. Noël N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022. doi:10.1093/qjmed/hct142
10. Chiravuri S, De Jesus O. Pancytopenia. StatPearls Publishing LLC; 2021.
11. Gladstone E. Pernicious anemia presenting with pancytopenia and hemolysis: a case report. February 8, 2019. Accessed June 9, 2022. https://www.journalmc.org/index.php/JMC/article/view/3269/2563
12. ScienceDaily. Developing a sensor for vitamin B12 deficiency. October 17, 2016. Accessed June 9, 2022. https://www.sciencedaily.com/releases/2016/10/161017103221.htm
13. Rodriguez NM, Shackelford K. Pernicious Anemia. StatPearls Publishing LLC; 2021.
14. Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024
Pancytopenia is a condition in which all 3 hematologic cell lines are lower than expected in the blood, often representing either an increase in cellular destruction or decrease in bone marrow production. Destruction often occurs in the setting of autoimmune conditions (eg, systemic lupus erythematosus, rheumatoid arthritis) or splenic sequestration, often affecting erythrocytes and platelets more than leukocytes. Decreased production represents central etiologies, which are often due to nutritional deficiencies, infections, drug toxicities, or malabsorption.1 Pancytopenia secondary to vitamin B12 deficiency is rare, accounting for about 5% of the hematologic manifestations of symptomatic vitamin B12 deficient patients.2
Pernicious anemia, named for a once lethal disease, is a form of vitamin B12 (cobalamin) deficiency that results from an autoimmune (type II hypersensitivity) reaction to gastric parietal cells or intrinsic factor. Antibodies bind to gastric parietal cells and reduce gastric acid production, leading to atrophic gastritis, or they bind intrinsic factor and block the binding and absorption of vitamin B12 in the gastrointestinal tract. While first described in the 1820s, it was not until a century later when scientists were studying hematopoiesis in response to the heavy casualty burden from battlefield exsanguination in World War I that dogs fed raw liver were noted to have significantly better blood regeneration response than those fed cooked liver. This discovery led physicians Minot and Murphy to use raw liver to treat pernicious anemia and found that jaundice improved, reticulocyte counts increased, and hemoglobin (Hb) concentration improved, resulting in the duo becoming the first American recipients of the Nobel Prize in physiology or medicine.3 It was ultimately determined in 1948 by chemists Folkers and Todd that the active ingredient in raw liver responsible for this phenomenon was vitamin B12.4
Patients with pernicious anemia typically present with macrocytic anemia, low reticulocyte count, hypersegmented neutrophils, as well as mild leukopenia and/or thrombocytopenia, distinguishable from folate deficiency by an elevated serum methylmalonic acid level. World Health Organization cytopenia thresholds are listed in Table 1.5 Treatment consists of lifelong vitamin B12 supplementation, and endoscopic screening is often recommended after diagnosis due to increased risk of gastrointestinal malignancy.6 Pernicious anemia can be difficult to distinguish from thrombotic thrombocytopenia purpura (TTP), a microangiopathic hemolytic anemia that can cause rapid end-organ failure and death if treatment is delayed.7 While pernicious anemia is not typically hemolytic, case reports of hemolysis in severe deficiency have been reported.7 Adequate bone marrow response to hemolysis in TTP results in an elevated reticulocyte count, which can be useful in differentiating from pernicious anemia where there is typically an inadequate bone marrow response and low reticulocyte count.8,9
The approach to working up pancytopenia begins with a detailed history inquiring about medications, exposures (benzenes, pesticides), alcohol use, and infection history. A thorough physical examination may help point the health care practitioner (HCP) toward a certain etiology, as the differential for pancytopenia is broad. In the deployed soldier downrange, resources are often limited, and the history/physical are crucial in preventing an expensive and unnecessary workup.
Case Presentation
A 24-year-old active-duty female patient presented in late December 2020 to a theater hospital in Djibouti after a witnessed syncopal episode. She had a history of Hashimoto thyroiditis and was taking levothyroxine sodium 75 mcg daily. The patient reported gluten intolerance, which was never formally evaluated. The syncopal episode lasted a few seconds and was not associated with any prodromal or postictal symptoms. No seizure activity was observed, and she had no history of syncopal episodes. She reported that she had been feeling ill 24 to 48 hours prior, with nausea, fatigue, decreased oral intake, decreased urine output, and 2 episodes of nonbilious, nonbloody emesis.
When the patient arrived, she was tachycardic with heart rate in the 130s beats per minute (baseline, 100-110 beats per minute), febrile (103 °F), and had systolic blood pressure (SBP) in the low 100s (baseline, SBP 120s-130s). An electrocardiogram and chest radiographs were unremarkable. Her complete blood count (CBC) could not be processed due to Hb and platelet levels too low to detect on assay (Table 2). Lactate dehydrogenase (LDH) was elevated at > 1000 U/L with mild elevation in liver enzymes (aspartate aminotransferase, 98 U/L; alanine aminotransferase, 51 U/L) and prolonged partial thromboplastin time 70 seconds. She did not report any increased bleeding or bruising. The peripheral blood smear demonstrated pancytopenia, without any schistocytes, and she was started on broad-spectrum antibiotics for presumed sepsis from urinary source and possible TTP.
The patient received 5 units of packed red blood cells, transfusion of platelets, and 2 doses of vitamin B12 in Djibouti with clinical improvement and resolution of orthostasis, hypotension, tachycardia, and fever. Her final posttransfusion CBC showed a Hb level of 11.2 g/dL, white blood cell (WBC) count of 1.7 K/µL, and platelet count of 23 K/µL (Table 3). Two days later her Hb level was 9.0 g/dL, WBC count 1.8 K/µL, and platelet count was 12 K/µL. She was evacuated via air to Landstuhl Regional Medical Center (LRMC) in Germany within 48 hours of presentation, given limited testing capabilities and persistent anemia and thrombocytopenia, refractory to transfusion, concerning for aplastic anemia or acute leukemia.
On arrival at LRMC, she was transfused 1 unit of platelets and given 3 doses of intramuscular vitamin B12 for undetectable levels (< 50 pg/mL) at presentation. An extensive infectious workup was obtained, which did not reveal any viral, bacterial, or parasitic causes. The patient also had a bone marrow biopsy performed at a civilian site, which revealed hypocellular bone marrow. She was transferred to Walter Reed National Military Medical Center (WRNMMC) for further workup and evaluation, given the infectious workup, which was negative. Concern for hematologic malignancy remained. At the time of her arrival, the laboratory values had drastically improved with vitamin supplementation. The patient’s absolute reticulocyte count indicated adequate bone marrow response and because of her improvement, a repeat bone marrow biopsy was not performed.
Intrinsic factor antibodies were elevated (34.5 AU/mL; reference range, 0.0-1.1), which confirmed that this patient’s underlying etiology was secondary to pernicious anemia. The patient continued to improve and repeat vitamin B12 and folate levels revealed that she was responding to therapy. At discharge, intramuscular vitamin B12 injections were planned to continue monthly, indefinitely per guidelines. Oral supplementation is typically avoided due to poor absorption.
Of note, during her inpatient admission at WRNMMC, further evaluation of reported gluten intolerance was performed, which revealed a negative celiac disease panel (IgG/IgA tissue transglutaminase antibodies). On discharge, she was to establish care with gastroenterology for further evaluation, likely including endoscopic evaluation, at her next duty station. She was able to resume full travel and duty functions on discharge from WRNMMC.
Discussion
We highlight a complex case of pancytopenia secondary to pernicious anemia in a deployed service member. With limited resources downrange, the workup of pancytopenia can be resource intensive, expensive, and time sensitive, which can have detrimental impacts on medical readiness. Additionally, undiagnosed coagulopathies can have lethal consequences in a deployed service member where bleeding risk may be elevated depending on the mission. The differential for pancytopenia is vast, and given its relative rarity in pernicious anemia, the HCP must use key components of the history and laboratory results to narrow the differential (eAppendix).10
Pernicious anemia commonly presents as an isolated anemia. In a study looking at the hematologic manifestations of 201 cohort patients with well-documented vitamin B12 deficiency, 5% had symptomatic pancytopenia and 1.5% had a hemolytic anemia.2 The majority (> 67%) of hematologic abnormalities were correctable with cobalamin replacement.2 In our case, the solider presented with symptomatic anemia, manifesting as syncope, and was found to have transfusion-resistant pancytopenia.She had a hemolytic anemia with an LDH > 1000 U/L, haptoglobin < 3 mg/dL, and mild transaminitis with hyperbilirubinemia (1.8 mg/dL). No schistocytes were observed on peripheral smear, suggesting intramedullary hemolysis, which is believed to be due to the destruction of megaloblastic cells by macrophages in bone marrow.11 A French study found high LDH levels and low reticulocyte counts to be strongly suggestive of vitamin B12 deficiency and helpful in differentiating pernicious anemia from TTP, given that bone marrow response to anemia in TTP is preserved.8
While vitamin B12 deficiency is not often associated with hemolytic anemia, multiple cases have been reported in the literature.6 Screening for vitamin B12 deficiency may have shortened this patient’s clinical course and limited the need for air evacuation to a stateside quaternary medical center. However, testing for cobalamin levels in overseas deployed environments is difficult, timely, and costly. New technologies, such as optical sensors, can detect vitamin B12 levels in the blood in < 1 minute and offer portable, low-cost options that may be useful in the deployed military setting.12
Diet plays a key role in this case, since the patient had a reported history of gluten intolerance, although it was never documented or evaluated prior to this presentation. Prior to deployment, the patient ate mostly rice, potatoes, and vegetables. While deployed in an austere environment, food options were limited. These conditions forced her to intermittently consume gluten products, which led to gastrointestinal issues, exacerbating her nutritional deficiencies. In the 2 months before her first syncopal episode, she reported worsening fatigue that impacted her ability to exercise. Vitamin B12 stores often take years to deplete, suggesting that she had a chronic nutritional deficiency before deployment. Another possibility was that she developed an autoimmune gastritis that acutely worsened in the setting of poor nutritional intake. Her history of Hashimoto thyroiditis is also important, as up to one-third of patients with autoimmune thyroid disease have been associated with pernicious anemia (range, 3%-32%) with certain shared human leukocyte antigen alleles implicated in autoimmune gastritis.13,14
Conclusions
This rare case of pernicious anemia presenting as pancytopenia illustrates the challenge in working up pancytopenia, especially in austere military environments with limited testing capabilities. Screening for chronic dietary and nutritional deficiency is important in a service member, raising the question of what role predeployment screening may have and what dietary accommodations may be available during overseas deployments, which can potentially dampen inflammation of the gastrointestinal tract, especially for those with preexisting autoimmune gastrointestinal conditions. Also, newer technology allows portable, low-cost testing of cobalamin and may aid in its diagnosis. In patients who are anemic with low vitamin B12, HCPs can begin vitamin B12 supplementation while continuing the workup (eg, antibody testing, endoscopy). If the patient responds appropriately, further workup becomes less urgent, therefore, decreasing resource use and increasing military readiness. When hemolysis is present, a low reticulocyte count can be beneficial to help differentiate this condition from TTP, a life-threatening condition that must also be ruled out or treated. Pernicious anemia should be on the differential in any patients with autoimmune conditions presenting with cytopenias, especially in those with a history of autoimmune thyroid disorders.
Pancytopenia is a condition in which all 3 hematologic cell lines are lower than expected in the blood, often representing either an increase in cellular destruction or decrease in bone marrow production. Destruction often occurs in the setting of autoimmune conditions (eg, systemic lupus erythematosus, rheumatoid arthritis) or splenic sequestration, often affecting erythrocytes and platelets more than leukocytes. Decreased production represents central etiologies, which are often due to nutritional deficiencies, infections, drug toxicities, or malabsorption.1 Pancytopenia secondary to vitamin B12 deficiency is rare, accounting for about 5% of the hematologic manifestations of symptomatic vitamin B12 deficient patients.2
Pernicious anemia, named for a once lethal disease, is a form of vitamin B12 (cobalamin) deficiency that results from an autoimmune (type II hypersensitivity) reaction to gastric parietal cells or intrinsic factor. Antibodies bind to gastric parietal cells and reduce gastric acid production, leading to atrophic gastritis, or they bind intrinsic factor and block the binding and absorption of vitamin B12 in the gastrointestinal tract. While first described in the 1820s, it was not until a century later when scientists were studying hematopoiesis in response to the heavy casualty burden from battlefield exsanguination in World War I that dogs fed raw liver were noted to have significantly better blood regeneration response than those fed cooked liver. This discovery led physicians Minot and Murphy to use raw liver to treat pernicious anemia and found that jaundice improved, reticulocyte counts increased, and hemoglobin (Hb) concentration improved, resulting in the duo becoming the first American recipients of the Nobel Prize in physiology or medicine.3 It was ultimately determined in 1948 by chemists Folkers and Todd that the active ingredient in raw liver responsible for this phenomenon was vitamin B12.4
Patients with pernicious anemia typically present with macrocytic anemia, low reticulocyte count, hypersegmented neutrophils, as well as mild leukopenia and/or thrombocytopenia, distinguishable from folate deficiency by an elevated serum methylmalonic acid level. World Health Organization cytopenia thresholds are listed in Table 1.5 Treatment consists of lifelong vitamin B12 supplementation, and endoscopic screening is often recommended after diagnosis due to increased risk of gastrointestinal malignancy.6 Pernicious anemia can be difficult to distinguish from thrombotic thrombocytopenia purpura (TTP), a microangiopathic hemolytic anemia that can cause rapid end-organ failure and death if treatment is delayed.7 While pernicious anemia is not typically hemolytic, case reports of hemolysis in severe deficiency have been reported.7 Adequate bone marrow response to hemolysis in TTP results in an elevated reticulocyte count, which can be useful in differentiating from pernicious anemia where there is typically an inadequate bone marrow response and low reticulocyte count.8,9
The approach to working up pancytopenia begins with a detailed history inquiring about medications, exposures (benzenes, pesticides), alcohol use, and infection history. A thorough physical examination may help point the health care practitioner (HCP) toward a certain etiology, as the differential for pancytopenia is broad. In the deployed soldier downrange, resources are often limited, and the history/physical are crucial in preventing an expensive and unnecessary workup.
Case Presentation
A 24-year-old active-duty female patient presented in late December 2020 to a theater hospital in Djibouti after a witnessed syncopal episode. She had a history of Hashimoto thyroiditis and was taking levothyroxine sodium 75 mcg daily. The patient reported gluten intolerance, which was never formally evaluated. The syncopal episode lasted a few seconds and was not associated with any prodromal or postictal symptoms. No seizure activity was observed, and she had no history of syncopal episodes. She reported that she had been feeling ill 24 to 48 hours prior, with nausea, fatigue, decreased oral intake, decreased urine output, and 2 episodes of nonbilious, nonbloody emesis.
When the patient arrived, she was tachycardic with heart rate in the 130s beats per minute (baseline, 100-110 beats per minute), febrile (103 °F), and had systolic blood pressure (SBP) in the low 100s (baseline, SBP 120s-130s). An electrocardiogram and chest radiographs were unremarkable. Her complete blood count (CBC) could not be processed due to Hb and platelet levels too low to detect on assay (Table 2). Lactate dehydrogenase (LDH) was elevated at > 1000 U/L with mild elevation in liver enzymes (aspartate aminotransferase, 98 U/L; alanine aminotransferase, 51 U/L) and prolonged partial thromboplastin time 70 seconds. She did not report any increased bleeding or bruising. The peripheral blood smear demonstrated pancytopenia, without any schistocytes, and she was started on broad-spectrum antibiotics for presumed sepsis from urinary source and possible TTP.
The patient received 5 units of packed red blood cells, transfusion of platelets, and 2 doses of vitamin B12 in Djibouti with clinical improvement and resolution of orthostasis, hypotension, tachycardia, and fever. Her final posttransfusion CBC showed a Hb level of 11.2 g/dL, white blood cell (WBC) count of 1.7 K/µL, and platelet count of 23 K/µL (Table 3). Two days later her Hb level was 9.0 g/dL, WBC count 1.8 K/µL, and platelet count was 12 K/µL. She was evacuated via air to Landstuhl Regional Medical Center (LRMC) in Germany within 48 hours of presentation, given limited testing capabilities and persistent anemia and thrombocytopenia, refractory to transfusion, concerning for aplastic anemia or acute leukemia.
On arrival at LRMC, she was transfused 1 unit of platelets and given 3 doses of intramuscular vitamin B12 for undetectable levels (< 50 pg/mL) at presentation. An extensive infectious workup was obtained, which did not reveal any viral, bacterial, or parasitic causes. The patient also had a bone marrow biopsy performed at a civilian site, which revealed hypocellular bone marrow. She was transferred to Walter Reed National Military Medical Center (WRNMMC) for further workup and evaluation, given the infectious workup, which was negative. Concern for hematologic malignancy remained. At the time of her arrival, the laboratory values had drastically improved with vitamin supplementation. The patient’s absolute reticulocyte count indicated adequate bone marrow response and because of her improvement, a repeat bone marrow biopsy was not performed.
Intrinsic factor antibodies were elevated (34.5 AU/mL; reference range, 0.0-1.1), which confirmed that this patient’s underlying etiology was secondary to pernicious anemia. The patient continued to improve and repeat vitamin B12 and folate levels revealed that she was responding to therapy. At discharge, intramuscular vitamin B12 injections were planned to continue monthly, indefinitely per guidelines. Oral supplementation is typically avoided due to poor absorption.
Of note, during her inpatient admission at WRNMMC, further evaluation of reported gluten intolerance was performed, which revealed a negative celiac disease panel (IgG/IgA tissue transglutaminase antibodies). On discharge, she was to establish care with gastroenterology for further evaluation, likely including endoscopic evaluation, at her next duty station. She was able to resume full travel and duty functions on discharge from WRNMMC.
Discussion
We highlight a complex case of pancytopenia secondary to pernicious anemia in a deployed service member. With limited resources downrange, the workup of pancytopenia can be resource intensive, expensive, and time sensitive, which can have detrimental impacts on medical readiness. Additionally, undiagnosed coagulopathies can have lethal consequences in a deployed service member where bleeding risk may be elevated depending on the mission. The differential for pancytopenia is vast, and given its relative rarity in pernicious anemia, the HCP must use key components of the history and laboratory results to narrow the differential (eAppendix).10
Pernicious anemia commonly presents as an isolated anemia. In a study looking at the hematologic manifestations of 201 cohort patients with well-documented vitamin B12 deficiency, 5% had symptomatic pancytopenia and 1.5% had a hemolytic anemia.2 The majority (> 67%) of hematologic abnormalities were correctable with cobalamin replacement.2 In our case, the solider presented with symptomatic anemia, manifesting as syncope, and was found to have transfusion-resistant pancytopenia.She had a hemolytic anemia with an LDH > 1000 U/L, haptoglobin < 3 mg/dL, and mild transaminitis with hyperbilirubinemia (1.8 mg/dL). No schistocytes were observed on peripheral smear, suggesting intramedullary hemolysis, which is believed to be due to the destruction of megaloblastic cells by macrophages in bone marrow.11 A French study found high LDH levels and low reticulocyte counts to be strongly suggestive of vitamin B12 deficiency and helpful in differentiating pernicious anemia from TTP, given that bone marrow response to anemia in TTP is preserved.8
While vitamin B12 deficiency is not often associated with hemolytic anemia, multiple cases have been reported in the literature.6 Screening for vitamin B12 deficiency may have shortened this patient’s clinical course and limited the need for air evacuation to a stateside quaternary medical center. However, testing for cobalamin levels in overseas deployed environments is difficult, timely, and costly. New technologies, such as optical sensors, can detect vitamin B12 levels in the blood in < 1 minute and offer portable, low-cost options that may be useful in the deployed military setting.12
Diet plays a key role in this case, since the patient had a reported history of gluten intolerance, although it was never documented or evaluated prior to this presentation. Prior to deployment, the patient ate mostly rice, potatoes, and vegetables. While deployed in an austere environment, food options were limited. These conditions forced her to intermittently consume gluten products, which led to gastrointestinal issues, exacerbating her nutritional deficiencies. In the 2 months before her first syncopal episode, she reported worsening fatigue that impacted her ability to exercise. Vitamin B12 stores often take years to deplete, suggesting that she had a chronic nutritional deficiency before deployment. Another possibility was that she developed an autoimmune gastritis that acutely worsened in the setting of poor nutritional intake. Her history of Hashimoto thyroiditis is also important, as up to one-third of patients with autoimmune thyroid disease have been associated with pernicious anemia (range, 3%-32%) with certain shared human leukocyte antigen alleles implicated in autoimmune gastritis.13,14
Conclusions
This rare case of pernicious anemia presenting as pancytopenia illustrates the challenge in working up pancytopenia, especially in austere military environments with limited testing capabilities. Screening for chronic dietary and nutritional deficiency is important in a service member, raising the question of what role predeployment screening may have and what dietary accommodations may be available during overseas deployments, which can potentially dampen inflammation of the gastrointestinal tract, especially for those with preexisting autoimmune gastrointestinal conditions. Also, newer technology allows portable, low-cost testing of cobalamin and may aid in its diagnosis. In patients who are anemic with low vitamin B12, HCPs can begin vitamin B12 supplementation while continuing the workup (eg, antibody testing, endoscopy). If the patient responds appropriately, further workup becomes less urgent, therefore, decreasing resource use and increasing military readiness. When hemolysis is present, a low reticulocyte count can be beneficial to help differentiate this condition from TTP, a life-threatening condition that must also be ruled out or treated. Pernicious anemia should be on the differential in any patients with autoimmune conditions presenting with cytopenias, especially in those with a history of autoimmune thyroid disorders.
1. Takeshima M, Ishikawa H, Kitadate A, et al. Anorexia nervosa-associated pancytopenia mimicking idiopathic aplastic anemia: a case report. BMC Psychiatry. 2018;18(1):150. doi:10.1186/s12888-018-1743-6
2. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56. doi:10.1111/j.1365-2257.2006.00755.x
3. Sinclair L. Recognizing, treating and understanding pernicious anaemia. J R Soc Med. 2008;101(5):262-264. doi:10.1258/jrsm.2008.081006
4. Shampo MA, Kyle RA, Steensma DP. William Murphy—Nobel Prize for the treatment of pernicious anemia. Mayo Clin Proc. 2006;81(6):726. doi:10.4065/81.6.726
5. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139-143. doi:10.1515/jtim-2017-0002
6. Tunio NA, Sheriff MZ, Cooper G. Prevalence of gastric cancer in patients with pernicious anemia: a population-based study. Am J Gastroenterol. 2020;115:S665. doi:10.14309/01.ajg.0000707332.16739.72
7. Bailey M, Maestas T, Betancourt R, Mikhael D, Babiker HM. A rare cause of thrombotic thrombocytopenic purpura- (TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019:1529306. doi:10.1155/2019/1529306
8. Stanley M, Michalski JM. Thrombotic Thrombocytopenic Purpura. StatPearls Publishing LLC; 2021.
9. Noël N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022. doi:10.1093/qjmed/hct142
10. Chiravuri S, De Jesus O. Pancytopenia. StatPearls Publishing LLC; 2021.
11. Gladstone E. Pernicious anemia presenting with pancytopenia and hemolysis: a case report. February 8, 2019. Accessed June 9, 2022. https://www.journalmc.org/index.php/JMC/article/view/3269/2563
12. ScienceDaily. Developing a sensor for vitamin B12 deficiency. October 17, 2016. Accessed June 9, 2022. https://www.sciencedaily.com/releases/2016/10/161017103221.htm
13. Rodriguez NM, Shackelford K. Pernicious Anemia. StatPearls Publishing LLC; 2021.
14. Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024
1. Takeshima M, Ishikawa H, Kitadate A, et al. Anorexia nervosa-associated pancytopenia mimicking idiopathic aplastic anemia: a case report. BMC Psychiatry. 2018;18(1):150. doi:10.1186/s12888-018-1743-6
2. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56. doi:10.1111/j.1365-2257.2006.00755.x
3. Sinclair L. Recognizing, treating and understanding pernicious anaemia. J R Soc Med. 2008;101(5):262-264. doi:10.1258/jrsm.2008.081006
4. Shampo MA, Kyle RA, Steensma DP. William Murphy—Nobel Prize for the treatment of pernicious anemia. Mayo Clin Proc. 2006;81(6):726. doi:10.4065/81.6.726
5. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139-143. doi:10.1515/jtim-2017-0002
6. Tunio NA, Sheriff MZ, Cooper G. Prevalence of gastric cancer in patients with pernicious anemia: a population-based study. Am J Gastroenterol. 2020;115:S665. doi:10.14309/01.ajg.0000707332.16739.72
7. Bailey M, Maestas T, Betancourt R, Mikhael D, Babiker HM. A rare cause of thrombotic thrombocytopenic purpura- (TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019:1529306. doi:10.1155/2019/1529306
8. Stanley M, Michalski JM. Thrombotic Thrombocytopenic Purpura. StatPearls Publishing LLC; 2021.
9. Noël N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022. doi:10.1093/qjmed/hct142
10. Chiravuri S, De Jesus O. Pancytopenia. StatPearls Publishing LLC; 2021.
11. Gladstone E. Pernicious anemia presenting with pancytopenia and hemolysis: a case report. February 8, 2019. Accessed June 9, 2022. https://www.journalmc.org/index.php/JMC/article/view/3269/2563
12. ScienceDaily. Developing a sensor for vitamin B12 deficiency. October 17, 2016. Accessed June 9, 2022. https://www.sciencedaily.com/releases/2016/10/161017103221.htm
13. Rodriguez NM, Shackelford K. Pernicious Anemia. StatPearls Publishing LLC; 2021.
14. Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024
Simultaneous Cases of Carfilzomib-Induced Thrombotic Microangiopathy in 2 Patients With Multiple Myeloma
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
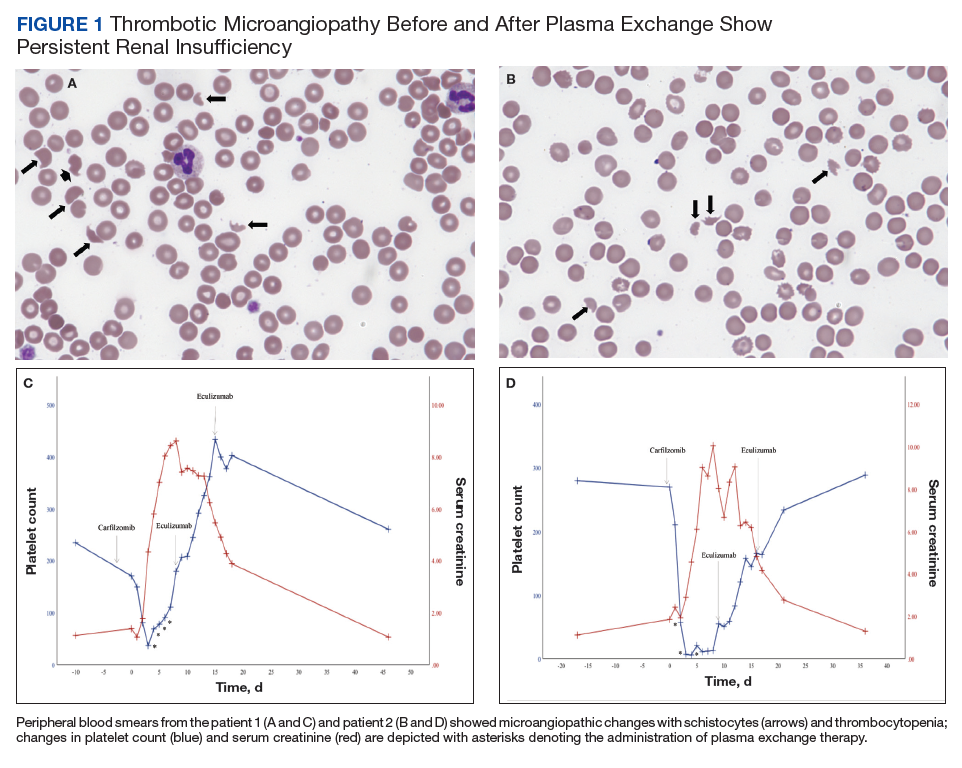
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
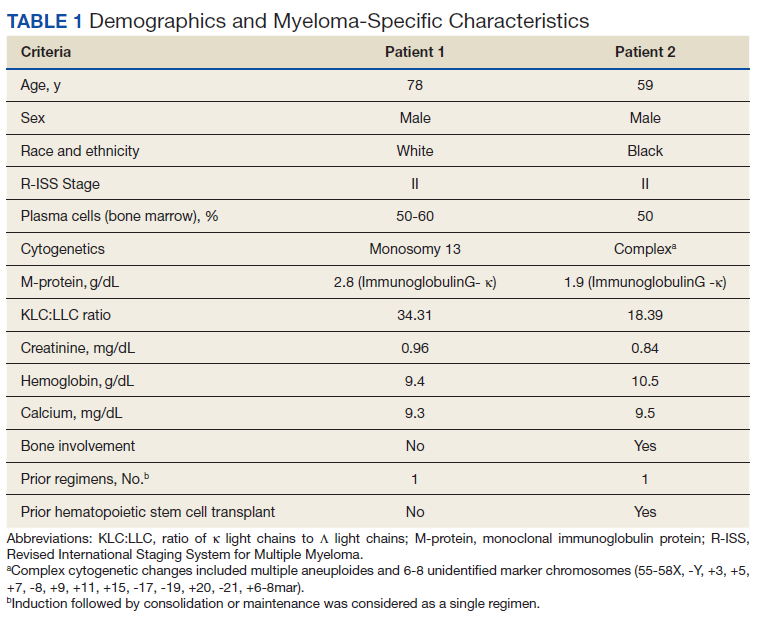
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
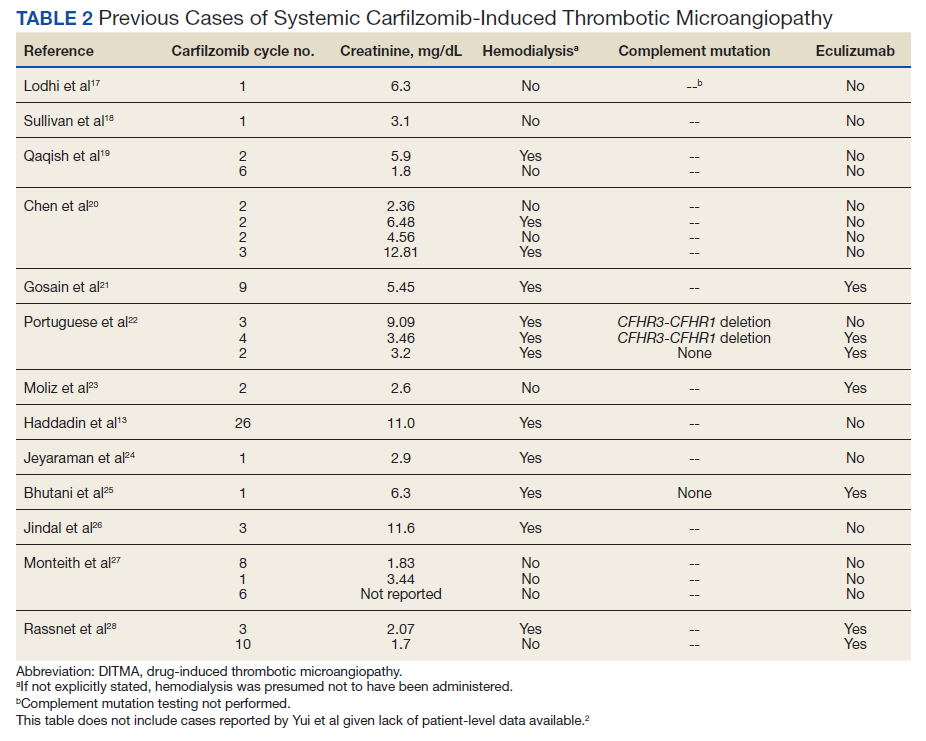
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
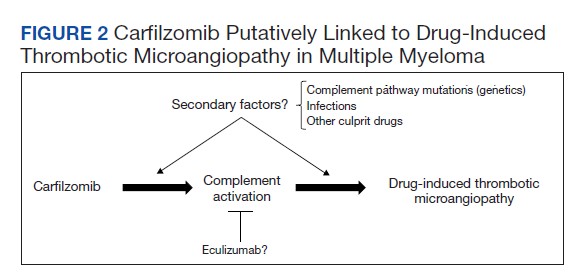
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
Rheumatologic Perspective on Persistent Right-Hand Tenosynovitis Secondary to Mycobacterium marinum Infection
Rheumatologic conditions and infections may imitate each other, often making diagnosis challenging. Therefore, it is imperative to obtain adequate histories and have a keen eye for these potentially confounding differential diagnoses. Immunosuppressants used in managing rheumatologic etiologies have detrimental consequences in undiagnosed underlying infections. Consequently, worsening symptoms with standard therapy should raise awareness to a different diagnosis.
Nontuberculous mycobacteria (NTM) are slow-growing organisms difficult to yield in culture. Initial negative synovial fluid stains and cultures when suspecting NTM infectious arthritis or tenosynovitis should not exclude the diagnosis if there is a strong clinical scenario. The identification of Mycobacterium marinum (M marinum) infection in the hand is of utmost importance given that delayed treatment may cause significant and even permanent disability.
We present the case of a 73-year-old male patient with progressively worsening right-hand tenosynovitis who was evaluated for crystal-induced and sarcoid arthropathies in the setting of negative synovial biopsy cultures but was subsequently diagnosed with M marinum infectious tenosynovitis after a second surgical debridement.
Case Presentation
A 73-year-old male patient with history of type 2 diabetes mellitus, hypertension, hyperlipidemia, hypothyroidism, bilateral knee osteoarthritis, obstructive sleep apnea, and posttraumatic stress disorder presented to the emergency department (ED) with right wrist swelling and pain for 4 days. The patient reported that he was working in his garden when symptoms started. He did not recall any skin abrasions or wounds, insect bites, thorn punctures, trauma, or exposure to swimming pools or fish tanks. Patient was afebrile, and vital signs were within normal range. On physical examination, there was erythema, swelling, and tenderness in the dorsum of the right hand and over the dorsal aspect of the fourth metacarpophalangeal joint (Figure 1). The skin was intact.
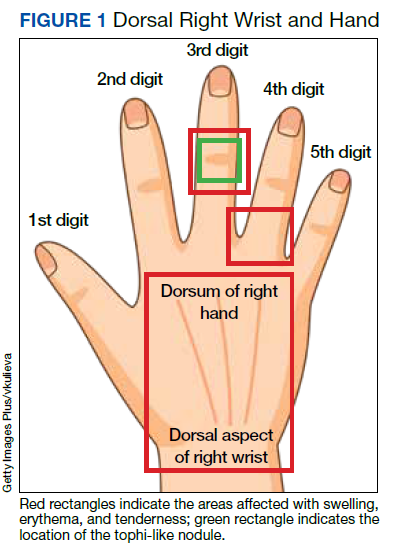
Symptoms had not responded to 7 days of cefalexin nor to a short course of oral steroids. Leukocytosis of 14.35 × 109/L (reference range, 3.90-9.90 × 109/L) with neutrophilia at 11.10 × 109/L (reference range, 1.73-6.37 × 109/L) was noted. Sedimentation rate and C-reactive protein levels were normal. Right-hand X-ray was remarkable for chondrocalcinosis in the triangular fibrocartilage. Right upper extremity magnetic resonance imaging (MRI) revealed diffuse inflammation in the right wrist and hand (Figure 2). There was no evidence of septic arthritis or osteomyelitis. Consequently, orthopedic service recommended no surgical intervention. Additionally, the patient had preserved range of motion that further indicated tenosynovitis, which could be medically managed with antibiotics, rather than a septic joint.
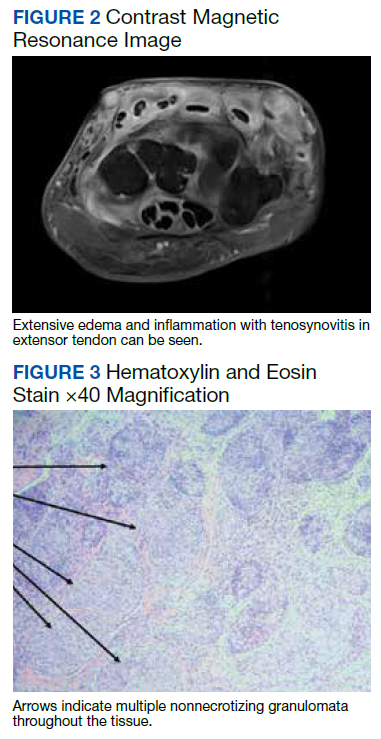
One dose of IV piperacillin/tazobactam was given at the ED, and he was admitted to the internal medicine ward with right hand and wrist cellulitis and indolent suppurative tenosynovitis. Empiric IV ceftriaxone and vancomycin were started as per infectious disease (ID) service with adequate response defined as a reduction of the swelling, erythema, and tenderness of the right hand and wrist. Differential diagnosis included sporotrichosis, nocardia vs NTM infection.
Interventional radiology was consulted for right wrist drainage. However, only 1 mL of fluid was obtained. Synovial fluid was sent for cell count and differential, crystal analysis, bacterial cultures, fungal cultures, and acid-fast bacilli (AFB) stains and culture. Neutrophils were 43% and lymphocytes were 57%. Crystal analysis was negative. Bacterial culture and mycology were negative. AFB stain and culture results were negative after 6 weeks. Based on gardening history and risk of thorn exposure and low suspicion for common bacterial pathogens, ID service switched antibiotics to moxifloxacin, minocycline, and linezolid for broad coverage to complete 3 weeks as outpatient. The patient reported significantly improved pain and handgrip with notable decrease in swelling. Nonetheless, 3 weeks after completing antibiotics, the right-hand pain recurred, raising concern for complex regional syndrome vs crystalline arthropathy.
The patient was referred to rheumatology service for evaluation of crystal-induced arthropathy given chondrocalcinosis. Physical examination revealed right third proximal interphalangeal joint swelling and tenderness with overimposed tophilike nodule. No erythema or palpable effusions were appreciated. Range of motion was preserved. Laboratory workup showed resolved leukocytosis and neutrophilia, and normal sedimentation rate or C-reactive protein levels. Antinuclear antibody panel, rheumatoid factor, and anti–cyclic citrullinated peptide levels were normal. Serum uric acid levels were 5.9 mg/dL. Chlamydia, gonorrhea, and HIV tests were negative. Short course of low-dose oral prednisone starting at 15 mg daily with tapering by 5 mg every 3 days was given for presumptive calcium pyrophosphate deposition vs gout. Nevertheless, right-hand swelling and pain worsened after steroids. Repeat right upper extremity MRI showed persistent soft tissue edema and inflammation along the dorsum of the hand extending to the digits, tenosynovitis, and fluid in the third metacarpophalangeal that could represent a superficial abscess. The patient was hospitalized given concerns of infection.
The relapse of tenosynovitis raised concerns for a persistent infection secondary to a fastidious organism, such as NTM. Thus, inquiries specifically pertaining to any contact with bodies of water were entertained. The patient remembered that he had gone scuba diving in the ocean weeks before symptom onset. This meant scuba diving could then be the inciting event rather than gardening, which placed NTM higher in the differential. ID service did not recommend antibiotics until new cultures were available. Orthopedic service was consulted for surgical debridement. The right dorsal hand, wrist, and distal forearm tendon sheaths were surgically opened to obtain a synovial biopsy.
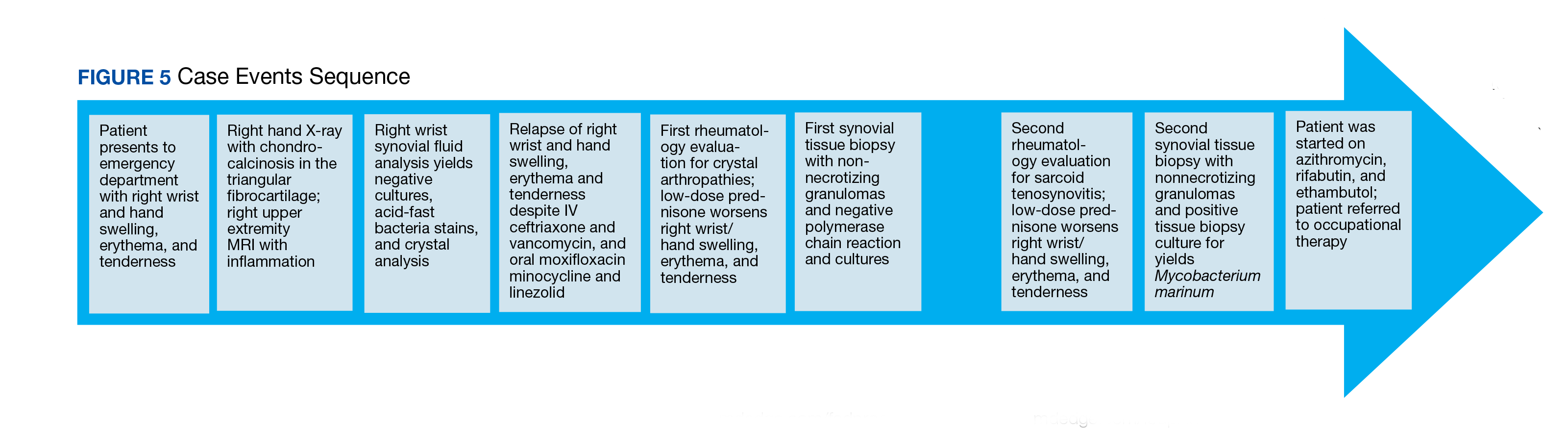
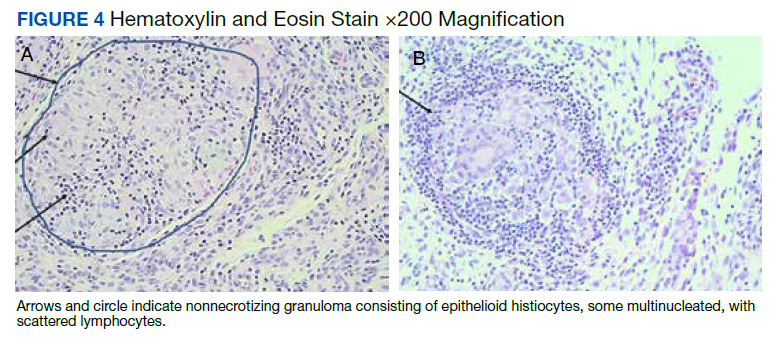
Synovial fluid was sent for fungal, bacterial, and AFB cultures, and synovial biopsy for AFB stains, PCR amplification/sequencing assay, and cultures. Results showed nonnecrotizing granulomas and all cultures were negative (Figures 3, 4). Rheumatology was again consulted for evaluation for sarcoidosis given negative cultures and noncaseating granulomas. Review of systems was completely negative for sarcoidosis. Computed tomography (CT) of the thorax did not show any pulmonary abnormalities, lymphadenopathy, and hilar adenopathy. Serum calcium and angiotensin-converting enzyme levels were normal. ID service recommended against empiric antibiotics given negative culture. Given persistent pain, and reported cases of isolated sarcoid tenosynovitis, low-dose oral prednisone 20 mg daily was given after clearance by ID service. Nonetheless, the right wrist and hand swelling, erythema, and tenderness relapsed with 1 dose of prednisone, leading to a repeat right upper extremity synovial biopsy due to high suspicion for persistent infection with a fastidious organism. New synovial tissue biopsy revealed fibro-adipose tissue with prominent vessels and fibrosis, nonnecrotizing, sarcoidlike granuloma with giant cell granulomatous reaction. The AFB and Grocott methenamine silver stains were negative. PCR was negative for AFB. No crystals were reported. After 5 weeks, the synovial biopsy culture was positive for M marinum. Patient was started on oral azithromycin 500 mg daily, rifabutin 300 mg daily, and ethambutol 15 mg/kg daily. At the time of this report, the patient was still completing antibiotic therapy with adequate response and undergoing occupational therapy rehabilitation (Figure 5).
Discussion
M marinum is an NTM found in bodies of water and marine settings. Infection arises after direct contact of lacerated skin with contaminated water. In a review article of 5 cases of M marinum tenosynovitis, they found that all individuals had wounds with exposure to fish or shrimp while in the water or while handling seafood.1 The incidence of this infection is infrequent, estimated to be 0.04 cases per 100,000, with only about 25% of these cases presenting as tenosynovitis.2 The incubation period ranges from 2 to 4 weeks.3 Late identification of this organism is common because of its slow development. For example, presentation from first exposure to symptom onset may take as long as 32 days.1 In addition, in the same review, surgical intervention occurred in 63 days.1 It has been reported that AFB stains are positive in just 9% of cases, which confounds diagnosis even more.4 After synovial tissue culture is obtained, it takes approximately 6 weeks for the organism to grow. Moreover, diagnosis may take longer if it is not suspected.5
Four types of M marinum infections have been described.5 The status of the immune system plays a role in how the manifestations present. The first type is limited, which is seen in immunocompetent persons, characterized by skin involvement, such as erythematous nodular lesions, that may improve on their own in months or years.4 Conversely, in immunosuppressed patients, the second type of infection may cause sporotrichoid spreading described as following lymphangitic pattern. The third type presents with musculoskeletal findings, such as arthritis, tenosynovitis, bursitis, or osteomyelitis, as seen in our patient. The fourth type consists of systemic manifestations.5 Medications that lower the immune system, such as corticosteroids, chemotherapy, and biologic disease modifying agents, may increase the risk for developing this entity.4 Specifically, antitumor necrosis factor inhibitors have been historically associated with mycobacterium infections.6
Patients are frequently diagnosed with soft tissue infection, such as abscesses or cellulitis, as in our case. They may at times be found to have other musculoskeletal conditions such as trigger finger.1 Other similar presenting entities are psoriatic arthritis, rheumatoid arthritis, and remitting seronegative arthritis.4 These clinical resemblances complicate the scenario, especially when initial cultures are negative, as the treatment for these rheumatic diseases is immunosuppression, which adversely impact the fastidious infection. In our case, the improved swelling and range of motion after the 3-week course of empiric antibiotics for suppurative tenosynovitis was initially reassuring that the previous infection had been successfully treated. Subsequently, the presence of chondrocalcinosis in the triangular fibrocartilage in the right-hand X-rays, persistent pain, and the tophi-like appearance of the right third proximal interphalangeal nodule raised concerns for crystalline arthropathies, such as calcium pyrophosphate deposition vs gout. Nonetheless, given the lack of response to low-dose steroids, an ongoing infectious process was strongly considered.
Sarcoidosis was a concern after the first synovial biopsy revealed noncaseating granulomas and negative stains and cultures. Sarcoid tenosynovitis is rare with only 22 cases described as per a 2015 report.7 Musculoskeletal involvement in sarcoidosis has been reported in 1 to 13% of sarcoid patients.7 Once again, unresponsiveness to steroids led to another synovial biopsy for culture due to potential infection. Akin to other cases, more than one surgical debridement was required to diagnose our patient.
Conclusions
Our case reinforces the vital role of history gathering in establishing diagnoses. It underscores the value of clinical suspicion especially in patients unresponsive to standard treatment for inflammatory arthritis, namely corticosteroids. Tissue biopsy with culture for AFB is crucial for accurate diagnosis in NTM infection, which may imitate rheumatic inflammatory arthritis. Clinicians should be keenly aware of this fastidious, indolent organism in the setting of persistent localized tenosynovitis.
1. Pang HN, Lee JY, Puhaindran ME, Tan SH, Tan AB, Yong FC. Mycobacterium marinum as a cause of chronic granulomatous tenosynovitis in the hand. J Infect. 2007;54(6):584-588. doi:10.1016/j.jinf.2006.11.014
2. Wongworawat MD, Holtom P, Learch TJ, Fedenko A, Stevanovic MV. A prolonged case of Mycobacterium marinum flexor tenosynovitis: radiographic and histological correlation, and review of the literature. Skeletal Radiol. 2003;32(9):542-545. doi:10.1007/s00256-003-0636-y
3. Schubert N, Schill T, Plüß M, Korsten P. Flare or foe? - Mycobacterium marinum infection mimicking rheumatoid arthritis tenosynovitis: case report and literature review. BMC Rheumatol. 2020;4:11. Published 2020 Mar 16. doi:10.1186/s41927-020-0114-3
4. Lam A, Toma W, Schlesinger N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J Rheumatol. 2006;33(4):817-819.
5. Hashish E, Merwad A, Elgaml S, et al. Mycobacterium marinum infection in fish and man: epidemiology, pathophysiology and management; a review. Vet Q. 2018;38(1):35-46. doi:10.1080/01652176.2018.1447171
6. Thanou-Stavraki A, Sawalha AH, Crowson AN, Harley JB. Noodling and Mycobacterium marinum infection mimicking seronegative rheumatoid arthritis complicated by anti-tumor necrosis factor α therapy. Arthritis Care Res (Hoboken). 2011;63(1):160-164. doi:10.1002/acr.20303
7. Al-Ani Z, Oh TC, Macphie E, Woodruff MJ. Sarcoid tenosynovitis, rare presentation of a common disease. Case report and literature review. J Radiol Case Rep. 2015;9(8):16-23. Published 2015 Aug 31. doi:10.3941/jrcr.v9i8.2311
Rheumatologic conditions and infections may imitate each other, often making diagnosis challenging. Therefore, it is imperative to obtain adequate histories and have a keen eye for these potentially confounding differential diagnoses. Immunosuppressants used in managing rheumatologic etiologies have detrimental consequences in undiagnosed underlying infections. Consequently, worsening symptoms with standard therapy should raise awareness to a different diagnosis.
Nontuberculous mycobacteria (NTM) are slow-growing organisms difficult to yield in culture. Initial negative synovial fluid stains and cultures when suspecting NTM infectious arthritis or tenosynovitis should not exclude the diagnosis if there is a strong clinical scenario. The identification of Mycobacterium marinum (M marinum) infection in the hand is of utmost importance given that delayed treatment may cause significant and even permanent disability.
We present the case of a 73-year-old male patient with progressively worsening right-hand tenosynovitis who was evaluated for crystal-induced and sarcoid arthropathies in the setting of negative synovial biopsy cultures but was subsequently diagnosed with M marinum infectious tenosynovitis after a second surgical debridement.
Case Presentation
A 73-year-old male patient with history of type 2 diabetes mellitus, hypertension, hyperlipidemia, hypothyroidism, bilateral knee osteoarthritis, obstructive sleep apnea, and posttraumatic stress disorder presented to the emergency department (ED) with right wrist swelling and pain for 4 days. The patient reported that he was working in his garden when symptoms started. He did not recall any skin abrasions or wounds, insect bites, thorn punctures, trauma, or exposure to swimming pools or fish tanks. Patient was afebrile, and vital signs were within normal range. On physical examination, there was erythema, swelling, and tenderness in the dorsum of the right hand and over the dorsal aspect of the fourth metacarpophalangeal joint (Figure 1). The skin was intact.
Symptoms had not responded to 7 days of cefalexin nor to a short course of oral steroids. Leukocytosis of 14.35 × 109/L (reference range, 3.90-9.90 × 109/L) with neutrophilia at 11.10 × 109/L (reference range, 1.73-6.37 × 109/L) was noted. Sedimentation rate and C-reactive protein levels were normal. Right-hand X-ray was remarkable for chondrocalcinosis in the triangular fibrocartilage. Right upper extremity magnetic resonance imaging (MRI) revealed diffuse inflammation in the right wrist and hand (Figure 2). There was no evidence of septic arthritis or osteomyelitis. Consequently, orthopedic service recommended no surgical intervention. Additionally, the patient had preserved range of motion that further indicated tenosynovitis, which could be medically managed with antibiotics, rather than a septic joint.
One dose of IV piperacillin/tazobactam was given at the ED, and he was admitted to the internal medicine ward with right hand and wrist cellulitis and indolent suppurative tenosynovitis. Empiric IV ceftriaxone and vancomycin were started as per infectious disease (ID) service with adequate response defined as a reduction of the swelling, erythema, and tenderness of the right hand and wrist. Differential diagnosis included sporotrichosis, nocardia vs NTM infection.
Interventional radiology was consulted for right wrist drainage. However, only 1 mL of fluid was obtained. Synovial fluid was sent for cell count and differential, crystal analysis, bacterial cultures, fungal cultures, and acid-fast bacilli (AFB) stains and culture. Neutrophils were 43% and lymphocytes were 57%. Crystal analysis was negative. Bacterial culture and mycology were negative. AFB stain and culture results were negative after 6 weeks. Based on gardening history and risk of thorn exposure and low suspicion for common bacterial pathogens, ID service switched antibiotics to moxifloxacin, minocycline, and linezolid for broad coverage to complete 3 weeks as outpatient. The patient reported significantly improved pain and handgrip with notable decrease in swelling. Nonetheless, 3 weeks after completing antibiotics, the right-hand pain recurred, raising concern for complex regional syndrome vs crystalline arthropathy.
The patient was referred to rheumatology service for evaluation of crystal-induced arthropathy given chondrocalcinosis. Physical examination revealed right third proximal interphalangeal joint swelling and tenderness with overimposed tophilike nodule. No erythema or palpable effusions were appreciated. Range of motion was preserved. Laboratory workup showed resolved leukocytosis and neutrophilia, and normal sedimentation rate or C-reactive protein levels. Antinuclear antibody panel, rheumatoid factor, and anti–cyclic citrullinated peptide levels were normal. Serum uric acid levels were 5.9 mg/dL. Chlamydia, gonorrhea, and HIV tests were negative. Short course of low-dose oral prednisone starting at 15 mg daily with tapering by 5 mg every 3 days was given for presumptive calcium pyrophosphate deposition vs gout. Nevertheless, right-hand swelling and pain worsened after steroids. Repeat right upper extremity MRI showed persistent soft tissue edema and inflammation along the dorsum of the hand extending to the digits, tenosynovitis, and fluid in the third metacarpophalangeal that could represent a superficial abscess. The patient was hospitalized given concerns of infection.
The relapse of tenosynovitis raised concerns for a persistent infection secondary to a fastidious organism, such as NTM. Thus, inquiries specifically pertaining to any contact with bodies of water were entertained. The patient remembered that he had gone scuba diving in the ocean weeks before symptom onset. This meant scuba diving could then be the inciting event rather than gardening, which placed NTM higher in the differential. ID service did not recommend antibiotics until new cultures were available. Orthopedic service was consulted for surgical debridement. The right dorsal hand, wrist, and distal forearm tendon sheaths were surgically opened to obtain a synovial biopsy.
Synovial fluid was sent for fungal, bacterial, and AFB cultures, and synovial biopsy for AFB stains, PCR amplification/sequencing assay, and cultures. Results showed nonnecrotizing granulomas and all cultures were negative (Figures 3, 4). Rheumatology was again consulted for evaluation for sarcoidosis given negative cultures and noncaseating granulomas. Review of systems was completely negative for sarcoidosis. Computed tomography (CT) of the thorax did not show any pulmonary abnormalities, lymphadenopathy, and hilar adenopathy. Serum calcium and angiotensin-converting enzyme levels were normal. ID service recommended against empiric antibiotics given negative culture. Given persistent pain, and reported cases of isolated sarcoid tenosynovitis, low-dose oral prednisone 20 mg daily was given after clearance by ID service. Nonetheless, the right wrist and hand swelling, erythema, and tenderness relapsed with 1 dose of prednisone, leading to a repeat right upper extremity synovial biopsy due to high suspicion for persistent infection with a fastidious organism. New synovial tissue biopsy revealed fibro-adipose tissue with prominent vessels and fibrosis, nonnecrotizing, sarcoidlike granuloma with giant cell granulomatous reaction. The AFB and Grocott methenamine silver stains were negative. PCR was negative for AFB. No crystals were reported. After 5 weeks, the synovial biopsy culture was positive for M marinum. Patient was started on oral azithromycin 500 mg daily, rifabutin 300 mg daily, and ethambutol 15 mg/kg daily. At the time of this report, the patient was still completing antibiotic therapy with adequate response and undergoing occupational therapy rehabilitation (Figure 5).
Discussion
M marinum is an NTM found in bodies of water and marine settings. Infection arises after direct contact of lacerated skin with contaminated water. In a review article of 5 cases of M marinum tenosynovitis, they found that all individuals had wounds with exposure to fish or shrimp while in the water or while handling seafood.1 The incidence of this infection is infrequent, estimated to be 0.04 cases per 100,000, with only about 25% of these cases presenting as tenosynovitis.2 The incubation period ranges from 2 to 4 weeks.3 Late identification of this organism is common because of its slow development. For example, presentation from first exposure to symptom onset may take as long as 32 days.1 In addition, in the same review, surgical intervention occurred in 63 days.1 It has been reported that AFB stains are positive in just 9% of cases, which confounds diagnosis even more.4 After synovial tissue culture is obtained, it takes approximately 6 weeks for the organism to grow. Moreover, diagnosis may take longer if it is not suspected.5
Four types of M marinum infections have been described.5 The status of the immune system plays a role in how the manifestations present. The first type is limited, which is seen in immunocompetent persons, characterized by skin involvement, such as erythematous nodular lesions, that may improve on their own in months or years.4 Conversely, in immunosuppressed patients, the second type of infection may cause sporotrichoid spreading described as following lymphangitic pattern. The third type presents with musculoskeletal findings, such as arthritis, tenosynovitis, bursitis, or osteomyelitis, as seen in our patient. The fourth type consists of systemic manifestations.5 Medications that lower the immune system, such as corticosteroids, chemotherapy, and biologic disease modifying agents, may increase the risk for developing this entity.4 Specifically, antitumor necrosis factor inhibitors have been historically associated with mycobacterium infections.6
Patients are frequently diagnosed with soft tissue infection, such as abscesses or cellulitis, as in our case. They may at times be found to have other musculoskeletal conditions such as trigger finger.1 Other similar presenting entities are psoriatic arthritis, rheumatoid arthritis, and remitting seronegative arthritis.4 These clinical resemblances complicate the scenario, especially when initial cultures are negative, as the treatment for these rheumatic diseases is immunosuppression, which adversely impact the fastidious infection. In our case, the improved swelling and range of motion after the 3-week course of empiric antibiotics for suppurative tenosynovitis was initially reassuring that the previous infection had been successfully treated. Subsequently, the presence of chondrocalcinosis in the triangular fibrocartilage in the right-hand X-rays, persistent pain, and the tophi-like appearance of the right third proximal interphalangeal nodule raised concerns for crystalline arthropathies, such as calcium pyrophosphate deposition vs gout. Nonetheless, given the lack of response to low-dose steroids, an ongoing infectious process was strongly considered.
Sarcoidosis was a concern after the first synovial biopsy revealed noncaseating granulomas and negative stains and cultures. Sarcoid tenosynovitis is rare with only 22 cases described as per a 2015 report.7 Musculoskeletal involvement in sarcoidosis has been reported in 1 to 13% of sarcoid patients.7 Once again, unresponsiveness to steroids led to another synovial biopsy for culture due to potential infection. Akin to other cases, more than one surgical debridement was required to diagnose our patient.
Conclusions
Our case reinforces the vital role of history gathering in establishing diagnoses. It underscores the value of clinical suspicion especially in patients unresponsive to standard treatment for inflammatory arthritis, namely corticosteroids. Tissue biopsy with culture for AFB is crucial for accurate diagnosis in NTM infection, which may imitate rheumatic inflammatory arthritis. Clinicians should be keenly aware of this fastidious, indolent organism in the setting of persistent localized tenosynovitis.
Rheumatologic conditions and infections may imitate each other, often making diagnosis challenging. Therefore, it is imperative to obtain adequate histories and have a keen eye for these potentially confounding differential diagnoses. Immunosuppressants used in managing rheumatologic etiologies have detrimental consequences in undiagnosed underlying infections. Consequently, worsening symptoms with standard therapy should raise awareness to a different diagnosis.
Nontuberculous mycobacteria (NTM) are slow-growing organisms difficult to yield in culture. Initial negative synovial fluid stains and cultures when suspecting NTM infectious arthritis or tenosynovitis should not exclude the diagnosis if there is a strong clinical scenario. The identification of Mycobacterium marinum (M marinum) infection in the hand is of utmost importance given that delayed treatment may cause significant and even permanent disability.
We present the case of a 73-year-old male patient with progressively worsening right-hand tenosynovitis who was evaluated for crystal-induced and sarcoid arthropathies in the setting of negative synovial biopsy cultures but was subsequently diagnosed with M marinum infectious tenosynovitis after a second surgical debridement.
Case Presentation
A 73-year-old male patient with history of type 2 diabetes mellitus, hypertension, hyperlipidemia, hypothyroidism, bilateral knee osteoarthritis, obstructive sleep apnea, and posttraumatic stress disorder presented to the emergency department (ED) with right wrist swelling and pain for 4 days. The patient reported that he was working in his garden when symptoms started. He did not recall any skin abrasions or wounds, insect bites, thorn punctures, trauma, or exposure to swimming pools or fish tanks. Patient was afebrile, and vital signs were within normal range. On physical examination, there was erythema, swelling, and tenderness in the dorsum of the right hand and over the dorsal aspect of the fourth metacarpophalangeal joint (Figure 1). The skin was intact.
Symptoms had not responded to 7 days of cefalexin nor to a short course of oral steroids. Leukocytosis of 14.35 × 109/L (reference range, 3.90-9.90 × 109/L) with neutrophilia at 11.10 × 109/L (reference range, 1.73-6.37 × 109/L) was noted. Sedimentation rate and C-reactive protein levels were normal. Right-hand X-ray was remarkable for chondrocalcinosis in the triangular fibrocartilage. Right upper extremity magnetic resonance imaging (MRI) revealed diffuse inflammation in the right wrist and hand (Figure 2). There was no evidence of septic arthritis or osteomyelitis. Consequently, orthopedic service recommended no surgical intervention. Additionally, the patient had preserved range of motion that further indicated tenosynovitis, which could be medically managed with antibiotics, rather than a septic joint.
One dose of IV piperacillin/tazobactam was given at the ED, and he was admitted to the internal medicine ward with right hand and wrist cellulitis and indolent suppurative tenosynovitis. Empiric IV ceftriaxone and vancomycin were started as per infectious disease (ID) service with adequate response defined as a reduction of the swelling, erythema, and tenderness of the right hand and wrist. Differential diagnosis included sporotrichosis, nocardia vs NTM infection.
Interventional radiology was consulted for right wrist drainage. However, only 1 mL of fluid was obtained. Synovial fluid was sent for cell count and differential, crystal analysis, bacterial cultures, fungal cultures, and acid-fast bacilli (AFB) stains and culture. Neutrophils were 43% and lymphocytes were 57%. Crystal analysis was negative. Bacterial culture and mycology were negative. AFB stain and culture results were negative after 6 weeks. Based on gardening history and risk of thorn exposure and low suspicion for common bacterial pathogens, ID service switched antibiotics to moxifloxacin, minocycline, and linezolid for broad coverage to complete 3 weeks as outpatient. The patient reported significantly improved pain and handgrip with notable decrease in swelling. Nonetheless, 3 weeks after completing antibiotics, the right-hand pain recurred, raising concern for complex regional syndrome vs crystalline arthropathy.
The patient was referred to rheumatology service for evaluation of crystal-induced arthropathy given chondrocalcinosis. Physical examination revealed right third proximal interphalangeal joint swelling and tenderness with overimposed tophilike nodule. No erythema or palpable effusions were appreciated. Range of motion was preserved. Laboratory workup showed resolved leukocytosis and neutrophilia, and normal sedimentation rate or C-reactive protein levels. Antinuclear antibody panel, rheumatoid factor, and anti–cyclic citrullinated peptide levels were normal. Serum uric acid levels were 5.9 mg/dL. Chlamydia, gonorrhea, and HIV tests were negative. Short course of low-dose oral prednisone starting at 15 mg daily with tapering by 5 mg every 3 days was given for presumptive calcium pyrophosphate deposition vs gout. Nevertheless, right-hand swelling and pain worsened after steroids. Repeat right upper extremity MRI showed persistent soft tissue edema and inflammation along the dorsum of the hand extending to the digits, tenosynovitis, and fluid in the third metacarpophalangeal that could represent a superficial abscess. The patient was hospitalized given concerns of infection.
The relapse of tenosynovitis raised concerns for a persistent infection secondary to a fastidious organism, such as NTM. Thus, inquiries specifically pertaining to any contact with bodies of water were entertained. The patient remembered that he had gone scuba diving in the ocean weeks before symptom onset. This meant scuba diving could then be the inciting event rather than gardening, which placed NTM higher in the differential. ID service did not recommend antibiotics until new cultures were available. Orthopedic service was consulted for surgical debridement. The right dorsal hand, wrist, and distal forearm tendon sheaths were surgically opened to obtain a synovial biopsy.
Synovial fluid was sent for fungal, bacterial, and AFB cultures, and synovial biopsy for AFB stains, PCR amplification/sequencing assay, and cultures. Results showed nonnecrotizing granulomas and all cultures were negative (Figures 3, 4). Rheumatology was again consulted for evaluation for sarcoidosis given negative cultures and noncaseating granulomas. Review of systems was completely negative for sarcoidosis. Computed tomography (CT) of the thorax did not show any pulmonary abnormalities, lymphadenopathy, and hilar adenopathy. Serum calcium and angiotensin-converting enzyme levels were normal. ID service recommended against empiric antibiotics given negative culture. Given persistent pain, and reported cases of isolated sarcoid tenosynovitis, low-dose oral prednisone 20 mg daily was given after clearance by ID service. Nonetheless, the right wrist and hand swelling, erythema, and tenderness relapsed with 1 dose of prednisone, leading to a repeat right upper extremity synovial biopsy due to high suspicion for persistent infection with a fastidious organism. New synovial tissue biopsy revealed fibro-adipose tissue with prominent vessels and fibrosis, nonnecrotizing, sarcoidlike granuloma with giant cell granulomatous reaction. The AFB and Grocott methenamine silver stains were negative. PCR was negative for AFB. No crystals were reported. After 5 weeks, the synovial biopsy culture was positive for M marinum. Patient was started on oral azithromycin 500 mg daily, rifabutin 300 mg daily, and ethambutol 15 mg/kg daily. At the time of this report, the patient was still completing antibiotic therapy with adequate response and undergoing occupational therapy rehabilitation (Figure 5).
Discussion
M marinum is an NTM found in bodies of water and marine settings. Infection arises after direct contact of lacerated skin with contaminated water. In a review article of 5 cases of M marinum tenosynovitis, they found that all individuals had wounds with exposure to fish or shrimp while in the water or while handling seafood.1 The incidence of this infection is infrequent, estimated to be 0.04 cases per 100,000, with only about 25% of these cases presenting as tenosynovitis.2 The incubation period ranges from 2 to 4 weeks.3 Late identification of this organism is common because of its slow development. For example, presentation from first exposure to symptom onset may take as long as 32 days.1 In addition, in the same review, surgical intervention occurred in 63 days.1 It has been reported that AFB stains are positive in just 9% of cases, which confounds diagnosis even more.4 After synovial tissue culture is obtained, it takes approximately 6 weeks for the organism to grow. Moreover, diagnosis may take longer if it is not suspected.5
Four types of M marinum infections have been described.5 The status of the immune system plays a role in how the manifestations present. The first type is limited, which is seen in immunocompetent persons, characterized by skin involvement, such as erythematous nodular lesions, that may improve on their own in months or years.4 Conversely, in immunosuppressed patients, the second type of infection may cause sporotrichoid spreading described as following lymphangitic pattern. The third type presents with musculoskeletal findings, such as arthritis, tenosynovitis, bursitis, or osteomyelitis, as seen in our patient. The fourth type consists of systemic manifestations.5 Medications that lower the immune system, such as corticosteroids, chemotherapy, and biologic disease modifying agents, may increase the risk for developing this entity.4 Specifically, antitumor necrosis factor inhibitors have been historically associated with mycobacterium infections.6
Patients are frequently diagnosed with soft tissue infection, such as abscesses or cellulitis, as in our case. They may at times be found to have other musculoskeletal conditions such as trigger finger.1 Other similar presenting entities are psoriatic arthritis, rheumatoid arthritis, and remitting seronegative arthritis.4 These clinical resemblances complicate the scenario, especially when initial cultures are negative, as the treatment for these rheumatic diseases is immunosuppression, which adversely impact the fastidious infection. In our case, the improved swelling and range of motion after the 3-week course of empiric antibiotics for suppurative tenosynovitis was initially reassuring that the previous infection had been successfully treated. Subsequently, the presence of chondrocalcinosis in the triangular fibrocartilage in the right-hand X-rays, persistent pain, and the tophi-like appearance of the right third proximal interphalangeal nodule raised concerns for crystalline arthropathies, such as calcium pyrophosphate deposition vs gout. Nonetheless, given the lack of response to low-dose steroids, an ongoing infectious process was strongly considered.
Sarcoidosis was a concern after the first synovial biopsy revealed noncaseating granulomas and negative stains and cultures. Sarcoid tenosynovitis is rare with only 22 cases described as per a 2015 report.7 Musculoskeletal involvement in sarcoidosis has been reported in 1 to 13% of sarcoid patients.7 Once again, unresponsiveness to steroids led to another synovial biopsy for culture due to potential infection. Akin to other cases, more than one surgical debridement was required to diagnose our patient.
Conclusions
Our case reinforces the vital role of history gathering in establishing diagnoses. It underscores the value of clinical suspicion especially in patients unresponsive to standard treatment for inflammatory arthritis, namely corticosteroids. Tissue biopsy with culture for AFB is crucial for accurate diagnosis in NTM infection, which may imitate rheumatic inflammatory arthritis. Clinicians should be keenly aware of this fastidious, indolent organism in the setting of persistent localized tenosynovitis.
1. Pang HN, Lee JY, Puhaindran ME, Tan SH, Tan AB, Yong FC. Mycobacterium marinum as a cause of chronic granulomatous tenosynovitis in the hand. J Infect. 2007;54(6):584-588. doi:10.1016/j.jinf.2006.11.014
2. Wongworawat MD, Holtom P, Learch TJ, Fedenko A, Stevanovic MV. A prolonged case of Mycobacterium marinum flexor tenosynovitis: radiographic and histological correlation, and review of the literature. Skeletal Radiol. 2003;32(9):542-545. doi:10.1007/s00256-003-0636-y
3. Schubert N, Schill T, Plüß M, Korsten P. Flare or foe? - Mycobacterium marinum infection mimicking rheumatoid arthritis tenosynovitis: case report and literature review. BMC Rheumatol. 2020;4:11. Published 2020 Mar 16. doi:10.1186/s41927-020-0114-3
4. Lam A, Toma W, Schlesinger N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J Rheumatol. 2006;33(4):817-819.
5. Hashish E, Merwad A, Elgaml S, et al. Mycobacterium marinum infection in fish and man: epidemiology, pathophysiology and management; a review. Vet Q. 2018;38(1):35-46. doi:10.1080/01652176.2018.1447171
6. Thanou-Stavraki A, Sawalha AH, Crowson AN, Harley JB. Noodling and Mycobacterium marinum infection mimicking seronegative rheumatoid arthritis complicated by anti-tumor necrosis factor α therapy. Arthritis Care Res (Hoboken). 2011;63(1):160-164. doi:10.1002/acr.20303
7. Al-Ani Z, Oh TC, Macphie E, Woodruff MJ. Sarcoid tenosynovitis, rare presentation of a common disease. Case report and literature review. J Radiol Case Rep. 2015;9(8):16-23. Published 2015 Aug 31. doi:10.3941/jrcr.v9i8.2311
1. Pang HN, Lee JY, Puhaindran ME, Tan SH, Tan AB, Yong FC. Mycobacterium marinum as a cause of chronic granulomatous tenosynovitis in the hand. J Infect. 2007;54(6):584-588. doi:10.1016/j.jinf.2006.11.014
2. Wongworawat MD, Holtom P, Learch TJ, Fedenko A, Stevanovic MV. A prolonged case of Mycobacterium marinum flexor tenosynovitis: radiographic and histological correlation, and review of the literature. Skeletal Radiol. 2003;32(9):542-545. doi:10.1007/s00256-003-0636-y
3. Schubert N, Schill T, Plüß M, Korsten P. Flare or foe? - Mycobacterium marinum infection mimicking rheumatoid arthritis tenosynovitis: case report and literature review. BMC Rheumatol. 2020;4:11. Published 2020 Mar 16. doi:10.1186/s41927-020-0114-3
4. Lam A, Toma W, Schlesinger N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J Rheumatol. 2006;33(4):817-819.
5. Hashish E, Merwad A, Elgaml S, et al. Mycobacterium marinum infection in fish and man: epidemiology, pathophysiology and management; a review. Vet Q. 2018;38(1):35-46. doi:10.1080/01652176.2018.1447171
6. Thanou-Stavraki A, Sawalha AH, Crowson AN, Harley JB. Noodling and Mycobacterium marinum infection mimicking seronegative rheumatoid arthritis complicated by anti-tumor necrosis factor α therapy. Arthritis Care Res (Hoboken). 2011;63(1):160-164. doi:10.1002/acr.20303
7. Al-Ani Z, Oh TC, Macphie E, Woodruff MJ. Sarcoid tenosynovitis, rare presentation of a common disease. Case report and literature review. J Radiol Case Rep. 2015;9(8):16-23. Published 2015 Aug 31. doi:10.3941/jrcr.v9i8.2311
Rhabdomyolysis Occurring After Use of Cocaine Contaminated With Fentanyl Causing Bilateral Brachial Plexopathy
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.

Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
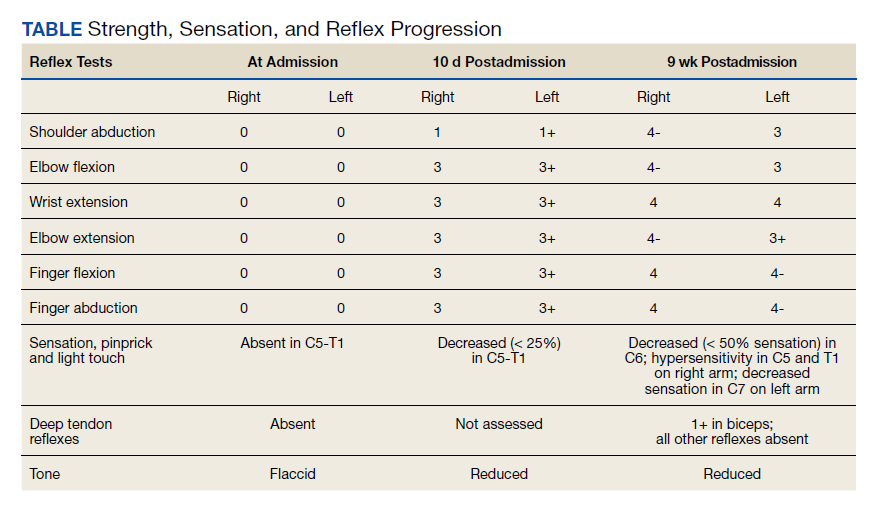
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.
Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.
Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
Benign Pneumatosis Intestinalis: A Case Report and Review of the Literature
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
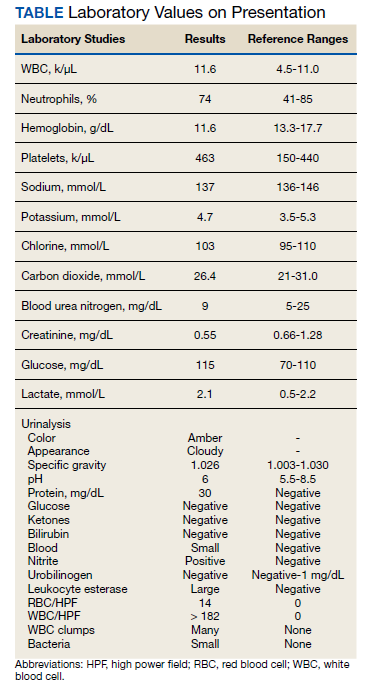
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011