User login
Intralymphatic Histiocytosis Associated With an Orthopedic Metal Implant
To the Editor:
|
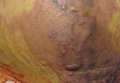
| Figure 1. A 30-cm pink and violaceous, asymmetric, reticulated patch on the lateral aspect of the right thigh. |
|
|
|
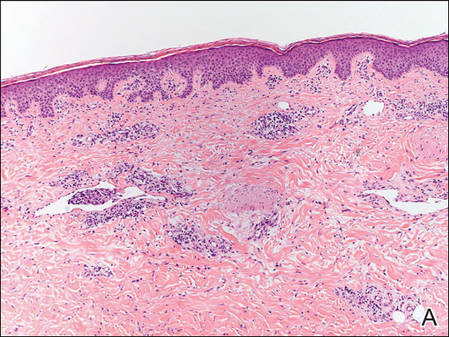
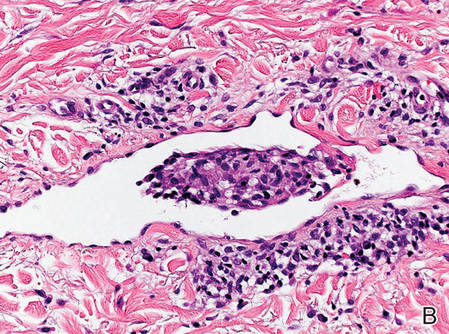
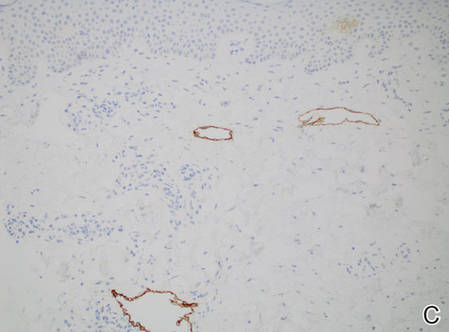
Figure 2. Histopathology revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis with adjacent features of chronic lymphedema (A)(H&E, original magnification ×10) as well as a collection of histiocytes in a dilated lymphatic channel (B)(H&E, original magnification ×40). D2-40 staining demonstrated ectatic lymphatic vessels in the upper dermins (C)(original magnification ×20).
|
A 70-year-old white man presented with an asymptomatic patch on the lateral aspect of the right thigh of 15 months’ duration. The patient believed the patch correlated with a hip replacement 3 years prior; however, it was 6 inches inferior to the incision site. Physical examination revealed a 30-cm pink and violaceous, asymmetric, reticulated patch (Figure 1). The patch was unresponsive to topical corticosteroids as well as a short course of oral prednisone. The patient’s medical history was notable for type 2 diabetes mellitus. Histopathologic examination revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis. In addition, adjacent features of chronic lymphedema were present, namely interstitial fibroplasia with dilated lymphatic vessels and a lymphoplasmacytic infiltrate (Figure 2). These findings were consistent with intralymphatic histiocytosis, a rare disease most commonly associated with rheumatoid arthritis. Our patient did not have a history or clinical symptoms of rheumatoid arthritis.
Intralymphatic histiocytosis is a rare cutaneous condition reported by O’Grady et al1 in 1994. This condition has been most frequently associated with rheumatoid arthritis2; however, there has been an emerging association in patients with orthopedic metal implants, with and without a concomitant diagnosis of rheumatoid arthritis. Cases associated with metal implants are rare.2-7
The condition presents as asymptomatic red, brown, or violaceous patches, plaques, papules, or nodules that are ill defined and tend to demonstrate a livedo reticularis–like pattern. The lesions typically are overlying or in close proximity to a joint. Histopathologic findings include dilated vascular structures in the reticular dermis, some with empty lumina and others containing collections of mononuclear histiocytes. There also may be an inflammatory infiltrate in the adjacent dermis composed of a mix of lymphocytes, plasma cells, and/or histiocytes. Endothelial cells lining the dilated lumina express immunoreactivity for CD31, CD34, D2-40, Lyve-1, and Prox-1. Intravascular histiocytes are positive for CD68 and CD31.6
The pathogenesis of intralymphatic histiocytosis remains undefined. Some hypothesize that intralymphatic histiocytosis could be the early stage of reactive angioendotheliomatosis, as these conditions share clinical and histological features.8 Reactive angioendotheliomatosis also is a rare condition that may present as erythematous to violaceous patches or plaques. The lesions are commonly found on the limbs and may be associated with constitutional symptoms. Histologic findings of reactive angioendotheliomatosis include a proliferation of epithelioid, round, or spindle-shaped cells within the lumina of dermal blood vessels, which show positivity for CD31 and CD34.9 Others suggest the lesions of intralymphatic histiocytosis arise from lymphangiectasia; obstruction of lymphatic drainage due to congenital abnormalities; or acquired damage from infection, trauma, surgery, or radiation.2 Due to the common association with rheumatoid arthritis and orthopedic implants, it is likely that lymphatic stasis secondary to chronic inflammation plays a notable role.
Therapies such as topical and systemic corticosteroids, local radiotherapy, cyclophosphamide, pentoxifylline, and arthrocentesis have been attempted without evidence of efficacy.2 Although intralymphatic histiocytosis is chronic and resistant to therapy, patients can be reassured that the condition runs a benign course.
1. O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
2. Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
3. Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
4. Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
5. Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants. J Cutan Pathol. 2011;38:534-535.
6. Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
7. Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants. Br J Dermatol. 2008;158:402-404.
8. Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
9. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
To the Editor:
|
|
| Figure 1. A 30-cm pink and violaceous, asymmetric, reticulated patch on the lateral aspect of the right thigh. |
|
|
|
|
|
|
Figure 2. Histopathology revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis with adjacent features of chronic lymphedema (A)(H&E, original magnification ×10) as well as a collection of histiocytes in a dilated lymphatic channel (B)(H&E, original magnification ×40). D2-40 staining demonstrated ectatic lymphatic vessels in the upper dermins (C)(original magnification ×20).
|
A 70-year-old white man presented with an asymptomatic patch on the lateral aspect of the right thigh of 15 months’ duration. The patient believed the patch correlated with a hip replacement 3 years prior; however, it was 6 inches inferior to the incision site. Physical examination revealed a 30-cm pink and violaceous, asymmetric, reticulated patch (Figure 1). The patch was unresponsive to topical corticosteroids as well as a short course of oral prednisone. The patient’s medical history was notable for type 2 diabetes mellitus. Histopathologic examination revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis. In addition, adjacent features of chronic lymphedema were present, namely interstitial fibroplasia with dilated lymphatic vessels and a lymphoplasmacytic infiltrate (Figure 2). These findings were consistent with intralymphatic histiocytosis, a rare disease most commonly associated with rheumatoid arthritis. Our patient did not have a history or clinical symptoms of rheumatoid arthritis.
Intralymphatic histiocytosis is a rare cutaneous condition reported by O’Grady et al1 in 1994. This condition has been most frequently associated with rheumatoid arthritis2; however, there has been an emerging association in patients with orthopedic metal implants, with and without a concomitant diagnosis of rheumatoid arthritis. Cases associated with metal implants are rare.2-7
The condition presents as asymptomatic red, brown, or violaceous patches, plaques, papules, or nodules that are ill defined and tend to demonstrate a livedo reticularis–like pattern. The lesions typically are overlying or in close proximity to a joint. Histopathologic findings include dilated vascular structures in the reticular dermis, some with empty lumina and others containing collections of mononuclear histiocytes. There also may be an inflammatory infiltrate in the adjacent dermis composed of a mix of lymphocytes, plasma cells, and/or histiocytes. Endothelial cells lining the dilated lumina express immunoreactivity for CD31, CD34, D2-40, Lyve-1, and Prox-1. Intravascular histiocytes are positive for CD68 and CD31.6
The pathogenesis of intralymphatic histiocytosis remains undefined. Some hypothesize that intralymphatic histiocytosis could be the early stage of reactive angioendotheliomatosis, as these conditions share clinical and histological features.8 Reactive angioendotheliomatosis also is a rare condition that may present as erythematous to violaceous patches or plaques. The lesions are commonly found on the limbs and may be associated with constitutional symptoms. Histologic findings of reactive angioendotheliomatosis include a proliferation of epithelioid, round, or spindle-shaped cells within the lumina of dermal blood vessels, which show positivity for CD31 and CD34.9 Others suggest the lesions of intralymphatic histiocytosis arise from lymphangiectasia; obstruction of lymphatic drainage due to congenital abnormalities; or acquired damage from infection, trauma, surgery, or radiation.2 Due to the common association with rheumatoid arthritis and orthopedic implants, it is likely that lymphatic stasis secondary to chronic inflammation plays a notable role.
Therapies such as topical and systemic corticosteroids, local radiotherapy, cyclophosphamide, pentoxifylline, and arthrocentesis have been attempted without evidence of efficacy.2 Although intralymphatic histiocytosis is chronic and resistant to therapy, patients can be reassured that the condition runs a benign course.
To the Editor:
|
|
| Figure 1. A 30-cm pink and violaceous, asymmetric, reticulated patch on the lateral aspect of the right thigh. |
|
|
|
|
|
|
Figure 2. Histopathology revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis with adjacent features of chronic lymphedema (A)(H&E, original magnification ×10) as well as a collection of histiocytes in a dilated lymphatic channel (B)(H&E, original magnification ×40). D2-40 staining demonstrated ectatic lymphatic vessels in the upper dermins (C)(original magnification ×20).
|
A 70-year-old white man presented with an asymptomatic patch on the lateral aspect of the right thigh of 15 months’ duration. The patient believed the patch correlated with a hip replacement 3 years prior; however, it was 6 inches inferior to the incision site. Physical examination revealed a 30-cm pink and violaceous, asymmetric, reticulated patch (Figure 1). The patch was unresponsive to topical corticosteroids as well as a short course of oral prednisone. The patient’s medical history was notable for type 2 diabetes mellitus. Histopathologic examination revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis. In addition, adjacent features of chronic lymphedema were present, namely interstitial fibroplasia with dilated lymphatic vessels and a lymphoplasmacytic infiltrate (Figure 2). These findings were consistent with intralymphatic histiocytosis, a rare disease most commonly associated with rheumatoid arthritis. Our patient did not have a history or clinical symptoms of rheumatoid arthritis.
Intralymphatic histiocytosis is a rare cutaneous condition reported by O’Grady et al1 in 1994. This condition has been most frequently associated with rheumatoid arthritis2; however, there has been an emerging association in patients with orthopedic metal implants, with and without a concomitant diagnosis of rheumatoid arthritis. Cases associated with metal implants are rare.2-7
The condition presents as asymptomatic red, brown, or violaceous patches, plaques, papules, or nodules that are ill defined and tend to demonstrate a livedo reticularis–like pattern. The lesions typically are overlying or in close proximity to a joint. Histopathologic findings include dilated vascular structures in the reticular dermis, some with empty lumina and others containing collections of mononuclear histiocytes. There also may be an inflammatory infiltrate in the adjacent dermis composed of a mix of lymphocytes, plasma cells, and/or histiocytes. Endothelial cells lining the dilated lumina express immunoreactivity for CD31, CD34, D2-40, Lyve-1, and Prox-1. Intravascular histiocytes are positive for CD68 and CD31.6
The pathogenesis of intralymphatic histiocytosis remains undefined. Some hypothesize that intralymphatic histiocytosis could be the early stage of reactive angioendotheliomatosis, as these conditions share clinical and histological features.8 Reactive angioendotheliomatosis also is a rare condition that may present as erythematous to violaceous patches or plaques. The lesions are commonly found on the limbs and may be associated with constitutional symptoms. Histologic findings of reactive angioendotheliomatosis include a proliferation of epithelioid, round, or spindle-shaped cells within the lumina of dermal blood vessels, which show positivity for CD31 and CD34.9 Others suggest the lesions of intralymphatic histiocytosis arise from lymphangiectasia; obstruction of lymphatic drainage due to congenital abnormalities; or acquired damage from infection, trauma, surgery, or radiation.2 Due to the common association with rheumatoid arthritis and orthopedic implants, it is likely that lymphatic stasis secondary to chronic inflammation plays a notable role.
Therapies such as topical and systemic corticosteroids, local radiotherapy, cyclophosphamide, pentoxifylline, and arthrocentesis have been attempted without evidence of efficacy.2 Although intralymphatic histiocytosis is chronic and resistant to therapy, patients can be reassured that the condition runs a benign course.
1. O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
2. Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
3. Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
4. Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
5. Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants. J Cutan Pathol. 2011;38:534-535.
6. Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
7. Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants. Br J Dermatol. 2008;158:402-404.
8. Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
9. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
1. O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
2. Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
3. Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
4. Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
5. Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants. J Cutan Pathol. 2011;38:534-535.
6. Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
7. Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants. Br J Dermatol. 2008;158:402-404.
8. Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
9. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
Practice Points
- Consider intralymphatic histiocytosis in the differential diagnosis of an asymptomatic skin lesion overlying a joint, particularly in patients with orthopedic metal implants or rheumatoid arthritis.
- Biopsy is essential for the diagnosis of intralymphatic histiocytosis; special stains highlighting dilated lymphatic vessels and intravascular histiocytes may be necessary.
- Intralymphatic histiocytosis is chronic and resistant to therapy; however, patients can be reassured that the condition runs a benign course.
Transition From Lichen Sclerosus to Squamous Cell Carcinoma in a Single Tissue Section
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
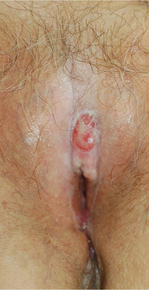
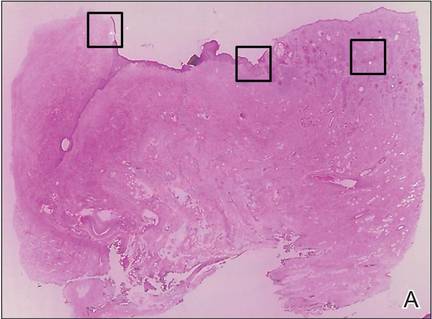
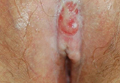
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
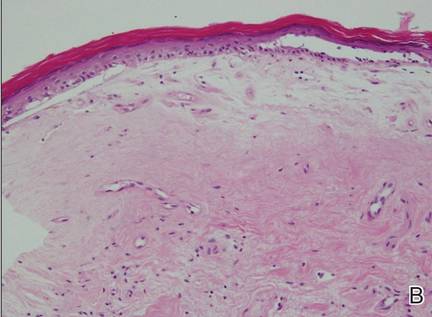
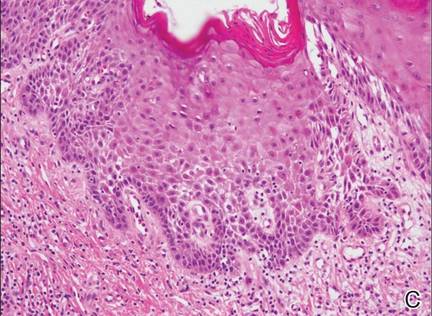

Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
Practice Points
- Lichen sclerosus has a considerable risk for malignant transformation and requires continuous follow-up in all patients.
- Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
Non–Drug-Induced Pemphigus Foliaceus in a Patient With Rheumatoid Arthritis
To the Editor:
The term pemphigus describes a group of autoimmune blistering diseases that are histologically characterized by intraepidermal blisters caused by acantholysis. There are several types of pemphigus foliaceus, such as classic and endemic pemphigus foliaceus, pemphigus erythematosus, pemphigus herpetiformis, and drug-induced pemphigus foliaceus.1

|
| Figure 1. Multiple erosions and crusted lesions were present on the back. |

|
| Figure 2. Subcorneal bulla containing acantholytic keratinocytes and neutrophils (H&E, original magnification ×100). |
Drug-induced pemphigus foliaceus in patients treated with penicillamine for rheumatoid arthritis (RA) is well documented in the literature.2 An association between pemphigus foliaceus and RA without penicillamine therapy is rare. We present a case of a patient with a history of RA who developed this bullous disease.
A 67-year-old woman with a 15-year history of seropositive RA presented with widespread skin lesions of 4 weeks’ duration. Confluent scaly crusted erosions on an erythematous base were present on the back (Figure 1), chest, and abdomen. There was no alteration of the mucous membranes. Medical treatment consisted of methotrexate (10 mg weekly), folic acid (5 mg twice weekly), prednisolone (5 mg daily), and ketoprofen (50 mg daily). Routine blood analysis was unremarkable, except for a positive rheumatoid factor. Histologic examination showed a subcorneal bulla containing acantholytic keratinocytes and neutrophils. There was a mild lymphocytic and eosinophilic infiltrate in the papillary dermis (Figure 2).
Determination of anti-desmoglein 1 and 3 antibodies was performed by a commercial enzyme-linked immunosorbent assay. Desmoglein 1 antibodies were positive with titers of 30 U/mL (positive, ≥20 U/mL), whereas desmoglein 3 antibody was negative. Thus, a diagnosis of pemphigus foliaceus was established. The polymerase chain reaction ligation-based typing method and the nucleotide sequence was used to examine the protein drought-repressed 4 gene complex, DR4, which tested positive.
Based on a diagnosis of pemphigus foliaceus, the patient’s corticosteroid treatment was changed from 5 mg daily of prednisolone to 40 mg daily of methylprednisolone, leading to marked improvement of the cutaneous lesions. After tapering the steroid dosage over a period of 3 months, no relapse occurred.
Pemphigus foliaceus is a rare autoimmune blistering disease. It can be induced by drugs, such as penicillamine and captopril.2,3 Captopril, an angiotensin-converting enzyme inhibitor, is closely related to penicillamine structurally. Both drugs have highly active thiol groups capable of reducing disulfide bonds and inducing acantholysis.4 The drugs taken by our patient typically are not known to induce pemphigus foliaceus.
The association of pemphigus foliaceus with RA in the absence of penicillamine therapy was first described by Wilkinson et al.4 Since then, additional cases have been published.5,6 Pemphigus foliaceus also has been described with other autoimmune conditions such as autoimmune thyroid disease.7
Rheumatoid arthritis has been genetically linked to the HLA-DR4 gene complex, which also was found in our patient. Patients with pemphigus foliaceus and RA have an increased frequency of the class II major histocompatibility complex, serologically defined HLA-DR4, and HLA-DRw6 haplotypes.4 Therefore, we believe that the association of pemphigus foliaceus and RA in our patient might not be fortuitous.
1. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol. 2005;44:470-476.
2. Sugita K, Hirokawa H, Izu K, et al. D-penicillamine-induced pemphigus successfully treated with combination therapy of mizoribine and prednisolone. J Dermatolog Treat. 2004;15:214-217.
3. Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26(2, pt 2):364-366.
4. Wilkinson SM, Smith AG, Davis MJ, et al. Rheumatoid arthritis: an association with pemphigus foliaceus. Acta Derm Venereol. 1992;72:289-291.
5. Sáez-de-Ocariz M, Granados J, Yamamoto-Furusho JK, et al. Rheumatoid arthritis associated with pemphigus foliaceus in a patient not taking penicillamine. Skinmed. 2007;6:252-254.
6. Gürcan HM, Ahmed RA. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-731.
7. Leshem YA, Katzenelson V, Yosipovitch G, et al. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol. 2011;50:827-831.
To the Editor:
The term pemphigus describes a group of autoimmune blistering diseases that are histologically characterized by intraepidermal blisters caused by acantholysis. There are several types of pemphigus foliaceus, such as classic and endemic pemphigus foliaceus, pemphigus erythematosus, pemphigus herpetiformis, and drug-induced pemphigus foliaceus.1
|
|
| Figure 1. Multiple erosions and crusted lesions were present on the back. |
|
|
| Figure 2. Subcorneal bulla containing acantholytic keratinocytes and neutrophils (H&E, original magnification ×100). |
Drug-induced pemphigus foliaceus in patients treated with penicillamine for rheumatoid arthritis (RA) is well documented in the literature.2 An association between pemphigus foliaceus and RA without penicillamine therapy is rare. We present a case of a patient with a history of RA who developed this bullous disease.
A 67-year-old woman with a 15-year history of seropositive RA presented with widespread skin lesions of 4 weeks’ duration. Confluent scaly crusted erosions on an erythematous base were present on the back (Figure 1), chest, and abdomen. There was no alteration of the mucous membranes. Medical treatment consisted of methotrexate (10 mg weekly), folic acid (5 mg twice weekly), prednisolone (5 mg daily), and ketoprofen (50 mg daily). Routine blood analysis was unremarkable, except for a positive rheumatoid factor. Histologic examination showed a subcorneal bulla containing acantholytic keratinocytes and neutrophils. There was a mild lymphocytic and eosinophilic infiltrate in the papillary dermis (Figure 2).
Determination of anti-desmoglein 1 and 3 antibodies was performed by a commercial enzyme-linked immunosorbent assay. Desmoglein 1 antibodies were positive with titers of 30 U/mL (positive, ≥20 U/mL), whereas desmoglein 3 antibody was negative. Thus, a diagnosis of pemphigus foliaceus was established. The polymerase chain reaction ligation-based typing method and the nucleotide sequence was used to examine the protein drought-repressed 4 gene complex, DR4, which tested positive.
Based on a diagnosis of pemphigus foliaceus, the patient’s corticosteroid treatment was changed from 5 mg daily of prednisolone to 40 mg daily of methylprednisolone, leading to marked improvement of the cutaneous lesions. After tapering the steroid dosage over a period of 3 months, no relapse occurred.
Pemphigus foliaceus is a rare autoimmune blistering disease. It can be induced by drugs, such as penicillamine and captopril.2,3 Captopril, an angiotensin-converting enzyme inhibitor, is closely related to penicillamine structurally. Both drugs have highly active thiol groups capable of reducing disulfide bonds and inducing acantholysis.4 The drugs taken by our patient typically are not known to induce pemphigus foliaceus.
The association of pemphigus foliaceus with RA in the absence of penicillamine therapy was first described by Wilkinson et al.4 Since then, additional cases have been published.5,6 Pemphigus foliaceus also has been described with other autoimmune conditions such as autoimmune thyroid disease.7
Rheumatoid arthritis has been genetically linked to the HLA-DR4 gene complex, which also was found in our patient. Patients with pemphigus foliaceus and RA have an increased frequency of the class II major histocompatibility complex, serologically defined HLA-DR4, and HLA-DRw6 haplotypes.4 Therefore, we believe that the association of pemphigus foliaceus and RA in our patient might not be fortuitous.
To the Editor:
The term pemphigus describes a group of autoimmune blistering diseases that are histologically characterized by intraepidermal blisters caused by acantholysis. There are several types of pemphigus foliaceus, such as classic and endemic pemphigus foliaceus, pemphigus erythematosus, pemphigus herpetiformis, and drug-induced pemphigus foliaceus.1
|
|
| Figure 1. Multiple erosions and crusted lesions were present on the back. |
|
|
| Figure 2. Subcorneal bulla containing acantholytic keratinocytes and neutrophils (H&E, original magnification ×100). |
Drug-induced pemphigus foliaceus in patients treated with penicillamine for rheumatoid arthritis (RA) is well documented in the literature.2 An association between pemphigus foliaceus and RA without penicillamine therapy is rare. We present a case of a patient with a history of RA who developed this bullous disease.
A 67-year-old woman with a 15-year history of seropositive RA presented with widespread skin lesions of 4 weeks’ duration. Confluent scaly crusted erosions on an erythematous base were present on the back (Figure 1), chest, and abdomen. There was no alteration of the mucous membranes. Medical treatment consisted of methotrexate (10 mg weekly), folic acid (5 mg twice weekly), prednisolone (5 mg daily), and ketoprofen (50 mg daily). Routine blood analysis was unremarkable, except for a positive rheumatoid factor. Histologic examination showed a subcorneal bulla containing acantholytic keratinocytes and neutrophils. There was a mild lymphocytic and eosinophilic infiltrate in the papillary dermis (Figure 2).
Determination of anti-desmoglein 1 and 3 antibodies was performed by a commercial enzyme-linked immunosorbent assay. Desmoglein 1 antibodies were positive with titers of 30 U/mL (positive, ≥20 U/mL), whereas desmoglein 3 antibody was negative. Thus, a diagnosis of pemphigus foliaceus was established. The polymerase chain reaction ligation-based typing method and the nucleotide sequence was used to examine the protein drought-repressed 4 gene complex, DR4, which tested positive.
Based on a diagnosis of pemphigus foliaceus, the patient’s corticosteroid treatment was changed from 5 mg daily of prednisolone to 40 mg daily of methylprednisolone, leading to marked improvement of the cutaneous lesions. After tapering the steroid dosage over a period of 3 months, no relapse occurred.
Pemphigus foliaceus is a rare autoimmune blistering disease. It can be induced by drugs, such as penicillamine and captopril.2,3 Captopril, an angiotensin-converting enzyme inhibitor, is closely related to penicillamine structurally. Both drugs have highly active thiol groups capable of reducing disulfide bonds and inducing acantholysis.4 The drugs taken by our patient typically are not known to induce pemphigus foliaceus.
The association of pemphigus foliaceus with RA in the absence of penicillamine therapy was first described by Wilkinson et al.4 Since then, additional cases have been published.5,6 Pemphigus foliaceus also has been described with other autoimmune conditions such as autoimmune thyroid disease.7
Rheumatoid arthritis has been genetically linked to the HLA-DR4 gene complex, which also was found in our patient. Patients with pemphigus foliaceus and RA have an increased frequency of the class II major histocompatibility complex, serologically defined HLA-DR4, and HLA-DRw6 haplotypes.4 Therefore, we believe that the association of pemphigus foliaceus and RA in our patient might not be fortuitous.
1. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol. 2005;44:470-476.
2. Sugita K, Hirokawa H, Izu K, et al. D-penicillamine-induced pemphigus successfully treated with combination therapy of mizoribine and prednisolone. J Dermatolog Treat. 2004;15:214-217.
3. Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26(2, pt 2):364-366.
4. Wilkinson SM, Smith AG, Davis MJ, et al. Rheumatoid arthritis: an association with pemphigus foliaceus. Acta Derm Venereol. 1992;72:289-291.
5. Sáez-de-Ocariz M, Granados J, Yamamoto-Furusho JK, et al. Rheumatoid arthritis associated with pemphigus foliaceus in a patient not taking penicillamine. Skinmed. 2007;6:252-254.
6. Gürcan HM, Ahmed RA. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-731.
7. Leshem YA, Katzenelson V, Yosipovitch G, et al. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol. 2011;50:827-831.
1. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol. 2005;44:470-476.
2. Sugita K, Hirokawa H, Izu K, et al. D-penicillamine-induced pemphigus successfully treated with combination therapy of mizoribine and prednisolone. J Dermatolog Treat. 2004;15:214-217.
3. Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26(2, pt 2):364-366.
4. Wilkinson SM, Smith AG, Davis MJ, et al. Rheumatoid arthritis: an association with pemphigus foliaceus. Acta Derm Venereol. 1992;72:289-291.
5. Sáez-de-Ocariz M, Granados J, Yamamoto-Furusho JK, et al. Rheumatoid arthritis associated with pemphigus foliaceus in a patient not taking penicillamine. Skinmed. 2007;6:252-254.
6. Gürcan HM, Ahmed RA. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-731.
7. Leshem YA, Katzenelson V, Yosipovitch G, et al. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol. 2011;50:827-831.
Practice Points
- Physicians should consider pemphigus foliaceus in the differential diagnosis in patients with rheumatoid arthritis and blistering eruptions.
- Appropriate analyses should be performed, including skin biopsy for histologic and immunohistochemical examination as well as search for circulating antibodies.
Concomitant Herpes Zoster and Herpes Simplex Infection
To the Editor:
Infections caused by herpes simplex (HS) and herpes zoster (HZ) usually can be recognized by clinical findings; however, laboratory confirmation sometimes is required. Polymerase chain reaction (PCR) laboratory tests detect HS or HZ in a sensible and specific manner. New PCR systems such as real-time PCR (RT-PCR) give faster and more precise results. We report a case of recurrent concomitant HZ and HS diagnosed by RT-PCR.
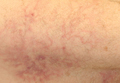
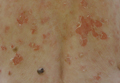
A 62-year-old woman presented with recurrent painful cutaneous lesions on the left buttock and thigh of 9 years’ duration. This eruption was preceded by a burning sensation of 1 week’s duration that extended toward the heel. Cutaneous lesions normally were sparse and persisted for a few days. She also had annular erythematous lesions of 3 years’ duration on the upper trunk and shoulders after sun exposure. On physical examination, an atrophic hypopigmented patch was seen with a few vesicles located on the thigh. Whitish atrophic patches also were found in a linear distribution on the left buttock and thigh (Figure).
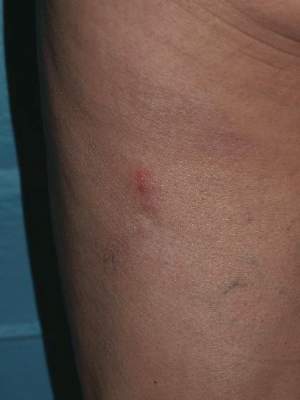
Laboratory results included the following: antinuclear antibody, 1:400 on a nuclear dotted pattern; extractable nuclear antigens (anti-Ro60 and anti-Ro52) were positive (reference range, >15); and rheumatoid factor was 24.1 U/mL (reference range, 0–15 U/mL). The patient did not meet any other American College of Rheumatology criteria1,2 of systemic lupus erythematosus apart from photosensitivity. The rest of the analysis—complete blood cell count, liver enzymes, and biochemistry—was normal or negative. Human immunodeficiency virus, herpes simplex virus types 1 and 2 (HHV-1 and HHV-2), and varicella-zoster virus (VZV) IgM serologies were negative, whereas IgG VZV serology was positive.
The microbiological study via swab obtained from the roof and fluid from the vesicles showed an indeterminate result from the rapid direct antigen detection with immunofluorescent antibodies. Viral cultures were HHV-2 positive and VZV negative. Conventional PCR showed positive results, both for HHV-2 and VZV. A second analysis, performed with RT-PCR from a new sample taken 2 months later, showed the same results, which led to the diagnosis of recurrent concomitant HS and HZ with a recurrent HZ clinical pattern. The patient was started on valacyclovir 1 g daily, and the number and intensity of flares diminished in the months following treatment.
Concomitant HS and HZ on the same dermatome has been described in the literature.3,4 In a retrospective series of 20 immunocompetent patients, HZ was the main presumed diagnosis before laboratory confirmation of diagnosis, and only 1 case corresponded to recurrent HZ.3 Other cases of simultaneous HS and HZ have been described, but they did not occur on the same dermatome. Half of these reported cases were in immunosuppressed patients.5,6
The recurrent nature of HS is well known; however, recurrent cases of HZ are rare. Nevertheless, in a population-based cohort study of patients with a confirmed prior episode of HZ (N=1669), recurrences were found in 6% of patients.7 Recurrence was more common if the patient was immunosuppressed, was female and 50 years or older, and had pain for more than 30 days.7
The recurrence rate was high in our case, but no immunosuppressive factor could be found apart from probable subacute cutaneous lupus erythematosus. Systemic lupus erythematosus has been associated with a high risk for developing HZ secondary to cell-mediated immunosuppression. The annual incidence of HZ can reach 32 of 1000 patients with systemic lupus erythematosus, while in the general population the incidence is only 1.5 to 3 of 1000 patients.8-10
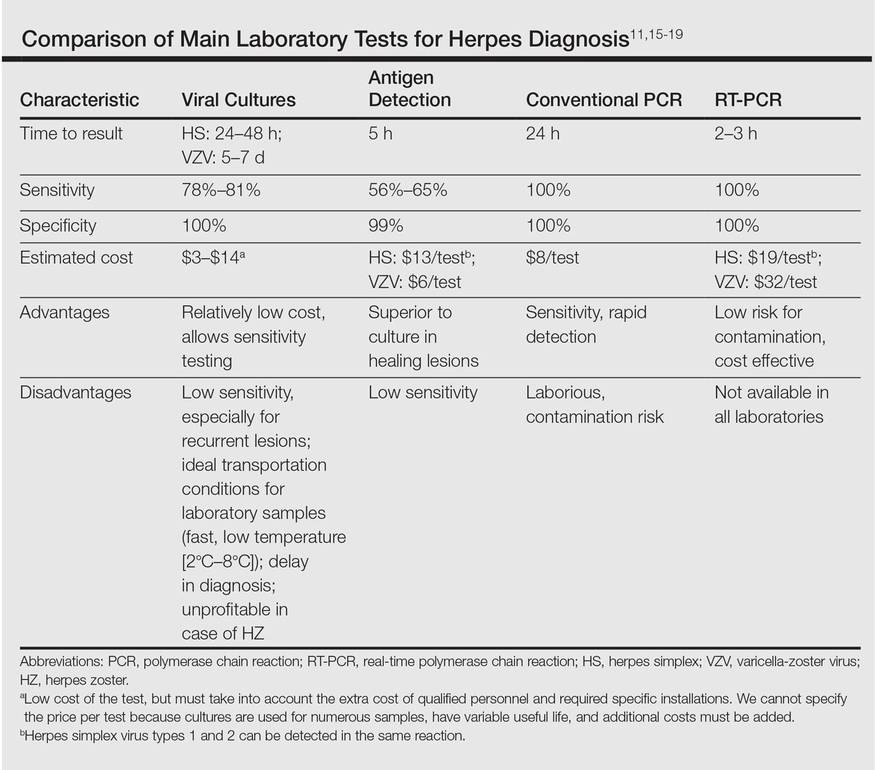
Direct detection of antigens of HS and HZ is a fast and inexpensive technique but lacks the sensitivity of viral cultures. Viral cultures used to be considered the gold standard; however, they are less sensitive than PCR.11 Furthermore, VZV detection is more difficult than HS, leading to a notable percentage of false-negative results.12 Polymerase chain reaction is a fast, reliable, and sensitive laboratory technique. Real-time PCR permits faster results than conventional PCR, specifically for HHV-1, HHV-2, and HZ detection. It also has minimal risk for contamination.13,14 In our opinion, PCR should be the gold standard instead of viral cultures. It has proven its superiority as a rapid method for detection, it is the most sensitive test, it is easier to perform, and it is cost effective (Table).11,15-19 However, viral cultures can allow sensitivity testing and are still an option for determination of susceptibility to antivirals.
In our case, a false-positive was excluded because no sign of possible contamination was found, repeated internal analysis from the same sample confirmed the results, and a new analysis from a new flare showed the same results 2 months later. However, we cannot rule out that the positivity for HZ of the second sample was due to the high sensitivity of the test and a virus latency in nerves.
We propose the use of PCR as a method of choice. Presumably more cases of recurrent HZ and concomitant HS and HZ will be seen with PCR use. In the case of a concomitant infection of HS and HZ, it is reasonable to use an antiviral dosage as in HZ treatment. No literature regarding outcomes from therapy could be found.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271-1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Giehl KA, Müller-Sander E, Rottenkolber M, et al. Identification and characterization of 20 immunocompetent patients with simultaneous varicella zoster and herpes simplex virus infection. JEADV. 2008;22:722-728.
- De Vivo C, Bansal MG, Olarte M, et al. Concurrent herpes simplex type 1 and varicella-zoster in the V2 dermatome in an immunocompetent patient. Cutis. 2001;68:120-122.
- Hyun-Ho P, Mu-Hyoung L. Concurrent reactivation of varicella zoster virus and herpes simplex virus in an immunocompetent child. J Korean Med Sci. 2004;19:598-600.
- Godet C, Beby-Defaux A, Landron C, et al. Concomitant disseminated herpes simplex virus type 2 infection and varicella zoster virus primoinfection in a pregnant woman. Scand J Infect Dis. 2005;37:774-776.
- Yawn BP, Wollan PC, Kurland MJ, et al. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc. 2011;86:88-93.
- Borba EF, Ribeiro AC, Martin P, et al. Incidence, risk factors, and outcome or herpes systemic lupus erythematosus. JCR. 2010;16:119-122.
- Nagasawa K, Yamauchi Y, Tada Y, et al. High incidence of herpes zoster in patients with systemic lupus erythematosus: an immunological analysis. Ann Rheumatic Dis. 1990;49:630-633.
- Kang TY, Lee HS, Kim TH, et al. Clinical and genetic risk factors of herpes zoster in patients with systemic lupus erythematosus. Rheumatol Int. 2005;25:97-102.
- Slomka MJ, Emery L, Munday PE, et al. A comparison of PCR with virus isolation and direct antigen detection for diagnosis and typing of genital herpes. J Med Virol. 1998;55:177-183.
- Nahass GT, Goldstein BA, Zhu WY, et al. Comparison of Tzanck smear, viral culture, and DNA diagnostic methods in detection of herpes simplex and varicella-zoster infection. JAMA. 1992;268:2541-2544.
- Burrows J, Nitsche A, Bayly B, et al. Detection and subtyping of herpes simplex virus in clinical samples by LightCycler PCR, enzyme immunoassay and cell culture. BMC Microbiology. 2002;2:12.
- Bezold GD, Lange ME, Gall H, et al. Detection of cutaneous varicella zoster virus infections by immunofluorescence versus PCR. Eur J Dermatol. 2001;11:108-111.
- Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect. 2004;80:406-410.
- Wald A, Huang ML, Carrell D, et al. Polymerase chain reaction for detection of herpes simplex virus DNA on mucosal surfaces: comparison with HSV isolation in cell culture. J Infect Dis. 2003;188:1345-1351.
- Marshall DS, Linfert DR, Draghi A, et al. Identification of herpes simplex virus genital infection: comparison of a multiplex PCR assay and traditional viral isolation techniques. Mod Pathol. 2001;14:152-156.
- Koening M, Reynolds KS, Aldous W, et al. Comparison of Light-Cycler PCR, enzyme immunoassay, and tissue culture for detection of herpes simplex virus. Diagn Microbiol Infect Dis. 2001;40:107-110.
- Coyle PV, Desai A, Wyatt D, et al. A comparison of virus isolation, indirect immunofluorescence and nested multiplex polymerase chain reaction for the diagnosis of primary and recurrent herpes simplex type 1 and type 2 infections. J Virol Methods. 1999;83:75-82.
To the Editor:
Infections caused by herpes simplex (HS) and herpes zoster (HZ) usually can be recognized by clinical findings; however, laboratory confirmation sometimes is required. Polymerase chain reaction (PCR) laboratory tests detect HS or HZ in a sensible and specific manner. New PCR systems such as real-time PCR (RT-PCR) give faster and more precise results. We report a case of recurrent concomitant HZ and HS diagnosed by RT-PCR.
A 62-year-old woman presented with recurrent painful cutaneous lesions on the left buttock and thigh of 9 years’ duration. This eruption was preceded by a burning sensation of 1 week’s duration that extended toward the heel. Cutaneous lesions normally were sparse and persisted for a few days. She also had annular erythematous lesions of 3 years’ duration on the upper trunk and shoulders after sun exposure. On physical examination, an atrophic hypopigmented patch was seen with a few vesicles located on the thigh. Whitish atrophic patches also were found in a linear distribution on the left buttock and thigh (Figure).
Laboratory results included the following: antinuclear antibody, 1:400 on a nuclear dotted pattern; extractable nuclear antigens (anti-Ro60 and anti-Ro52) were positive (reference range, >15); and rheumatoid factor was 24.1 U/mL (reference range, 0–15 U/mL). The patient did not meet any other American College of Rheumatology criteria1,2 of systemic lupus erythematosus apart from photosensitivity. The rest of the analysis—complete blood cell count, liver enzymes, and biochemistry—was normal or negative. Human immunodeficiency virus, herpes simplex virus types 1 and 2 (HHV-1 and HHV-2), and varicella-zoster virus (VZV) IgM serologies were negative, whereas IgG VZV serology was positive.
The microbiological study via swab obtained from the roof and fluid from the vesicles showed an indeterminate result from the rapid direct antigen detection with immunofluorescent antibodies. Viral cultures were HHV-2 positive and VZV negative. Conventional PCR showed positive results, both for HHV-2 and VZV. A second analysis, performed with RT-PCR from a new sample taken 2 months later, showed the same results, which led to the diagnosis of recurrent concomitant HS and HZ with a recurrent HZ clinical pattern. The patient was started on valacyclovir 1 g daily, and the number and intensity of flares diminished in the months following treatment.
Concomitant HS and HZ on the same dermatome has been described in the literature.3,4 In a retrospective series of 20 immunocompetent patients, HZ was the main presumed diagnosis before laboratory confirmation of diagnosis, and only 1 case corresponded to recurrent HZ.3 Other cases of simultaneous HS and HZ have been described, but they did not occur on the same dermatome. Half of these reported cases were in immunosuppressed patients.5,6
The recurrent nature of HS is well known; however, recurrent cases of HZ are rare. Nevertheless, in a population-based cohort study of patients with a confirmed prior episode of HZ (N=1669), recurrences were found in 6% of patients.7 Recurrence was more common if the patient was immunosuppressed, was female and 50 years or older, and had pain for more than 30 days.7
The recurrence rate was high in our case, but no immunosuppressive factor could be found apart from probable subacute cutaneous lupus erythematosus. Systemic lupus erythematosus has been associated with a high risk for developing HZ secondary to cell-mediated immunosuppression. The annual incidence of HZ can reach 32 of 1000 patients with systemic lupus erythematosus, while in the general population the incidence is only 1.5 to 3 of 1000 patients.8-10
Direct detection of antigens of HS and HZ is a fast and inexpensive technique but lacks the sensitivity of viral cultures. Viral cultures used to be considered the gold standard; however, they are less sensitive than PCR.11 Furthermore, VZV detection is more difficult than HS, leading to a notable percentage of false-negative results.12 Polymerase chain reaction is a fast, reliable, and sensitive laboratory technique. Real-time PCR permits faster results than conventional PCR, specifically for HHV-1, HHV-2, and HZ detection. It also has minimal risk for contamination.13,14 In our opinion, PCR should be the gold standard instead of viral cultures. It has proven its superiority as a rapid method for detection, it is the most sensitive test, it is easier to perform, and it is cost effective (Table).11,15-19 However, viral cultures can allow sensitivity testing and are still an option for determination of susceptibility to antivirals.
In our case, a false-positive was excluded because no sign of possible contamination was found, repeated internal analysis from the same sample confirmed the results, and a new analysis from a new flare showed the same results 2 months later. However, we cannot rule out that the positivity for HZ of the second sample was due to the high sensitivity of the test and a virus latency in nerves.
We propose the use of PCR as a method of choice. Presumably more cases of recurrent HZ and concomitant HS and HZ will be seen with PCR use. In the case of a concomitant infection of HS and HZ, it is reasonable to use an antiviral dosage as in HZ treatment. No literature regarding outcomes from therapy could be found.
To the Editor:
Infections caused by herpes simplex (HS) and herpes zoster (HZ) usually can be recognized by clinical findings; however, laboratory confirmation sometimes is required. Polymerase chain reaction (PCR) laboratory tests detect HS or HZ in a sensible and specific manner. New PCR systems such as real-time PCR (RT-PCR) give faster and more precise results. We report a case of recurrent concomitant HZ and HS diagnosed by RT-PCR.
A 62-year-old woman presented with recurrent painful cutaneous lesions on the left buttock and thigh of 9 years’ duration. This eruption was preceded by a burning sensation of 1 week’s duration that extended toward the heel. Cutaneous lesions normally were sparse and persisted for a few days. She also had annular erythematous lesions of 3 years’ duration on the upper trunk and shoulders after sun exposure. On physical examination, an atrophic hypopigmented patch was seen with a few vesicles located on the thigh. Whitish atrophic patches also were found in a linear distribution on the left buttock and thigh (Figure).
Laboratory results included the following: antinuclear antibody, 1:400 on a nuclear dotted pattern; extractable nuclear antigens (anti-Ro60 and anti-Ro52) were positive (reference range, >15); and rheumatoid factor was 24.1 U/mL (reference range, 0–15 U/mL). The patient did not meet any other American College of Rheumatology criteria1,2 of systemic lupus erythematosus apart from photosensitivity. The rest of the analysis—complete blood cell count, liver enzymes, and biochemistry—was normal or negative. Human immunodeficiency virus, herpes simplex virus types 1 and 2 (HHV-1 and HHV-2), and varicella-zoster virus (VZV) IgM serologies were negative, whereas IgG VZV serology was positive.
The microbiological study via swab obtained from the roof and fluid from the vesicles showed an indeterminate result from the rapid direct antigen detection with immunofluorescent antibodies. Viral cultures were HHV-2 positive and VZV negative. Conventional PCR showed positive results, both for HHV-2 and VZV. A second analysis, performed with RT-PCR from a new sample taken 2 months later, showed the same results, which led to the diagnosis of recurrent concomitant HS and HZ with a recurrent HZ clinical pattern. The patient was started on valacyclovir 1 g daily, and the number and intensity of flares diminished in the months following treatment.
Concomitant HS and HZ on the same dermatome has been described in the literature.3,4 In a retrospective series of 20 immunocompetent patients, HZ was the main presumed diagnosis before laboratory confirmation of diagnosis, and only 1 case corresponded to recurrent HZ.3 Other cases of simultaneous HS and HZ have been described, but they did not occur on the same dermatome. Half of these reported cases were in immunosuppressed patients.5,6
The recurrent nature of HS is well known; however, recurrent cases of HZ are rare. Nevertheless, in a population-based cohort study of patients with a confirmed prior episode of HZ (N=1669), recurrences were found in 6% of patients.7 Recurrence was more common if the patient was immunosuppressed, was female and 50 years or older, and had pain for more than 30 days.7
The recurrence rate was high in our case, but no immunosuppressive factor could be found apart from probable subacute cutaneous lupus erythematosus. Systemic lupus erythematosus has been associated with a high risk for developing HZ secondary to cell-mediated immunosuppression. The annual incidence of HZ can reach 32 of 1000 patients with systemic lupus erythematosus, while in the general population the incidence is only 1.5 to 3 of 1000 patients.8-10
Direct detection of antigens of HS and HZ is a fast and inexpensive technique but lacks the sensitivity of viral cultures. Viral cultures used to be considered the gold standard; however, they are less sensitive than PCR.11 Furthermore, VZV detection is more difficult than HS, leading to a notable percentage of false-negative results.12 Polymerase chain reaction is a fast, reliable, and sensitive laboratory technique. Real-time PCR permits faster results than conventional PCR, specifically for HHV-1, HHV-2, and HZ detection. It also has minimal risk for contamination.13,14 In our opinion, PCR should be the gold standard instead of viral cultures. It has proven its superiority as a rapid method for detection, it is the most sensitive test, it is easier to perform, and it is cost effective (Table).11,15-19 However, viral cultures can allow sensitivity testing and are still an option for determination of susceptibility to antivirals.
In our case, a false-positive was excluded because no sign of possible contamination was found, repeated internal analysis from the same sample confirmed the results, and a new analysis from a new flare showed the same results 2 months later. However, we cannot rule out that the positivity for HZ of the second sample was due to the high sensitivity of the test and a virus latency in nerves.
We propose the use of PCR as a method of choice. Presumably more cases of recurrent HZ and concomitant HS and HZ will be seen with PCR use. In the case of a concomitant infection of HS and HZ, it is reasonable to use an antiviral dosage as in HZ treatment. No literature regarding outcomes from therapy could be found.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271-1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Giehl KA, Müller-Sander E, Rottenkolber M, et al. Identification and characterization of 20 immunocompetent patients with simultaneous varicella zoster and herpes simplex virus infection. JEADV. 2008;22:722-728.
- De Vivo C, Bansal MG, Olarte M, et al. Concurrent herpes simplex type 1 and varicella-zoster in the V2 dermatome in an immunocompetent patient. Cutis. 2001;68:120-122.
- Hyun-Ho P, Mu-Hyoung L. Concurrent reactivation of varicella zoster virus and herpes simplex virus in an immunocompetent child. J Korean Med Sci. 2004;19:598-600.
- Godet C, Beby-Defaux A, Landron C, et al. Concomitant disseminated herpes simplex virus type 2 infection and varicella zoster virus primoinfection in a pregnant woman. Scand J Infect Dis. 2005;37:774-776.
- Yawn BP, Wollan PC, Kurland MJ, et al. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc. 2011;86:88-93.
- Borba EF, Ribeiro AC, Martin P, et al. Incidence, risk factors, and outcome or herpes systemic lupus erythematosus. JCR. 2010;16:119-122.
- Nagasawa K, Yamauchi Y, Tada Y, et al. High incidence of herpes zoster in patients with systemic lupus erythematosus: an immunological analysis. Ann Rheumatic Dis. 1990;49:630-633.
- Kang TY, Lee HS, Kim TH, et al. Clinical and genetic risk factors of herpes zoster in patients with systemic lupus erythematosus. Rheumatol Int. 2005;25:97-102.
- Slomka MJ, Emery L, Munday PE, et al. A comparison of PCR with virus isolation and direct antigen detection for diagnosis and typing of genital herpes. J Med Virol. 1998;55:177-183.
- Nahass GT, Goldstein BA, Zhu WY, et al. Comparison of Tzanck smear, viral culture, and DNA diagnostic methods in detection of herpes simplex and varicella-zoster infection. JAMA. 1992;268:2541-2544.
- Burrows J, Nitsche A, Bayly B, et al. Detection and subtyping of herpes simplex virus in clinical samples by LightCycler PCR, enzyme immunoassay and cell culture. BMC Microbiology. 2002;2:12.
- Bezold GD, Lange ME, Gall H, et al. Detection of cutaneous varicella zoster virus infections by immunofluorescence versus PCR. Eur J Dermatol. 2001;11:108-111.
- Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect. 2004;80:406-410.
- Wald A, Huang ML, Carrell D, et al. Polymerase chain reaction for detection of herpes simplex virus DNA on mucosal surfaces: comparison with HSV isolation in cell culture. J Infect Dis. 2003;188:1345-1351.
- Marshall DS, Linfert DR, Draghi A, et al. Identification of herpes simplex virus genital infection: comparison of a multiplex PCR assay and traditional viral isolation techniques. Mod Pathol. 2001;14:152-156.
- Koening M, Reynolds KS, Aldous W, et al. Comparison of Light-Cycler PCR, enzyme immunoassay, and tissue culture for detection of herpes simplex virus. Diagn Microbiol Infect Dis. 2001;40:107-110.
- Coyle PV, Desai A, Wyatt D, et al. A comparison of virus isolation, indirect immunofluorescence and nested multiplex polymerase chain reaction for the diagnosis of primary and recurrent herpes simplex type 1 and type 2 infections. J Virol Methods. 1999;83:75-82.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271-1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Giehl KA, Müller-Sander E, Rottenkolber M, et al. Identification and characterization of 20 immunocompetent patients with simultaneous varicella zoster and herpes simplex virus infection. JEADV. 2008;22:722-728.
- De Vivo C, Bansal MG, Olarte M, et al. Concurrent herpes simplex type 1 and varicella-zoster in the V2 dermatome in an immunocompetent patient. Cutis. 2001;68:120-122.
- Hyun-Ho P, Mu-Hyoung L. Concurrent reactivation of varicella zoster virus and herpes simplex virus in an immunocompetent child. J Korean Med Sci. 2004;19:598-600.
- Godet C, Beby-Defaux A, Landron C, et al. Concomitant disseminated herpes simplex virus type 2 infection and varicella zoster virus primoinfection in a pregnant woman. Scand J Infect Dis. 2005;37:774-776.
- Yawn BP, Wollan PC, Kurland MJ, et al. Herpes zoster recurrences more frequent than previously reported. Mayo Clin Proc. 2011;86:88-93.
- Borba EF, Ribeiro AC, Martin P, et al. Incidence, risk factors, and outcome or herpes systemic lupus erythematosus. JCR. 2010;16:119-122.
- Nagasawa K, Yamauchi Y, Tada Y, et al. High incidence of herpes zoster in patients with systemic lupus erythematosus: an immunological analysis. Ann Rheumatic Dis. 1990;49:630-633.
- Kang TY, Lee HS, Kim TH, et al. Clinical and genetic risk factors of herpes zoster in patients with systemic lupus erythematosus. Rheumatol Int. 2005;25:97-102.
- Slomka MJ, Emery L, Munday PE, et al. A comparison of PCR with virus isolation and direct antigen detection for diagnosis and typing of genital herpes. J Med Virol. 1998;55:177-183.
- Nahass GT, Goldstein BA, Zhu WY, et al. Comparison of Tzanck smear, viral culture, and DNA diagnostic methods in detection of herpes simplex and varicella-zoster infection. JAMA. 1992;268:2541-2544.
- Burrows J, Nitsche A, Bayly B, et al. Detection and subtyping of herpes simplex virus in clinical samples by LightCycler PCR, enzyme immunoassay and cell culture. BMC Microbiology. 2002;2:12.
- Bezold GD, Lange ME, Gall H, et al. Detection of cutaneous varicella zoster virus infections by immunofluorescence versus PCR. Eur J Dermatol. 2001;11:108-111.
- Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect. 2004;80:406-410.
- Wald A, Huang ML, Carrell D, et al. Polymerase chain reaction for detection of herpes simplex virus DNA on mucosal surfaces: comparison with HSV isolation in cell culture. J Infect Dis. 2003;188:1345-1351.
- Marshall DS, Linfert DR, Draghi A, et al. Identification of herpes simplex virus genital infection: comparison of a multiplex PCR assay and traditional viral isolation techniques. Mod Pathol. 2001;14:152-156.
- Koening M, Reynolds KS, Aldous W, et al. Comparison of Light-Cycler PCR, enzyme immunoassay, and tissue culture for detection of herpes simplex virus. Diagn Microbiol Infect Dis. 2001;40:107-110.
- Coyle PV, Desai A, Wyatt D, et al. A comparison of virus isolation, indirect immunofluorescence and nested multiplex polymerase chain reaction for the diagnosis of primary and recurrent herpes simplex type 1 and type 2 infections. J Virol Methods. 1999;83:75-82.
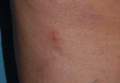
Bowel-Associated Dermatosis-Arthritis Syndrome in a Patient With Crohn Disease
To the Editor:
A 42-year-old woman with Crohn disease of 10 years’ duration presented to the clinic with a chief concern of nonpruritic pustular lesions on the bilateral arms. Physical examination revealed several pustules on the arms with secondary excoriation. She also had a warm tender nodule on the left upper shin and subungual hemorrhages under the fingernails (Figure 1). The patient had previously undergone infliximab therapy, which was discontinued 10 months prior to presentation in anticipation of a partial colectomy and temporary ileostomy that was performed 8 months prior to presentation. She recently had developed bilateral, radiating, sharp lower extremity pain extending from the feet to the hips over the last 2 weeks and swelling of the bilateral legs that impaired her ability to ambulate. Additionally, she had recently traveled to Colorado and a Lyme disease workup was initiated at an outside hospital in Colorado; however, the results were pending. The outside hospital also performed a spinal tap that was negative. At our clinic, biopsies were performed on the shin nodule and a right palmar pustule (Figure 2). There was clinical suspicion of erythema nodosum and subcorneal pustular dermatosis or a vesiculopustular skin manifestation of the patient’s Crohn disease. The patient was switched from generic doxycycline to a brand name variant 150 mg every night at bedtime for 2 weeks. She subsequently was admitted to the inpatient rheumatology service for a complete systemic workup.

The punch biopsy of the left upper shin demonstrated operative hemorrhage and periadnexal lymphocytic inflammation without evidence of fungal or bacterial elements by Gram or Gomori methenamine-silver stain. Clinically, the diagnosis was most likely erythema nodosum, though insufficient hypodermis was present to make the diagnosis with pathology. The shave biopsy of the right medial palm was nondiagnostic but showed a transected pustule with no bacterial or fungal elements by Gram or Gomori methenamine-silver stain (Figure 3). Given the clinical context, the likely pathologic diagnosis was vesiculopustular Crohn disease.
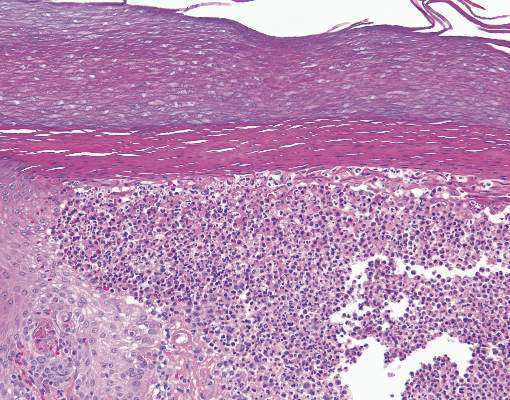
Our patient was started on an empiric steroid trial with rapid improvement of the arthralgia and rash. The presumed diagnosis was a Crohn disease flare and the patient was discharged on an 8-week steroid taper. Three weeks later at a follow-up appointment, the patient’s skin lesions had nearly resolved. The swelling of the legs and feet had substantially decreased, but the joint pain, primarily in the ankles, persisted.
Routine laboratory studies showed a hemoglobin level of 11.6 g/dL (reference range, 12–15 g/dL), white blood cell count of 9.1 K/μL (reference range, 4.5–11.0 K/μL), C-reactive protein level of 20.15 mg/dL (reference range, <1.0 mg/dL), and an antinuclear antibody titer of 160 (<80). Serology for Lyme disease was negative. Serum chemistries were all within reference range and an echocardiogram was normal.
Up to one-third of patients with inflammatory bowel disease (IBD) experience extraintestinal manifestations of their condition. Of these patients, nearly one-third will develop cutaneous manifestations.1 The most common skin diseases associated with IBD are pyoderma gangrenosum and erythema nodosum.2 The differential diagnoses considered in this unique case included early pyoderma gangrenosum, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), and vesiculopustular Crohn disease. Vesiculopustular Crohn disease is a rare component of IBD and also can be present in bowel-associated dermatosis-arthritis syndrome (BADAS). In BADAS, symptoms often include arthritis and systemic symptoms such as fever and malaise. The skin manifestations typically involve the arms and trunk. It often is seen after intestinal bypass surgery but also can be present in patients with gastrointestinal diseases such as IBD.3 Due to its early association with bypass surgery, BADAS previously was referred to as bowel bypass syndrome but has since been seen in relation to other intestinal surgeries and IBD.4 Patients with BADAS often present with episodes of fever, fatigue, and malaise, in addition to arthralgia and cutaneous eruptions. Cases of BADAS related to IBD instead of bypass surgery often can be less severe in nature. Unlike many of these previously reported cases, our patient’s joint pain primarily was in the knees and ankles, whereas typical cases of BADAS cause upper extremity (ie, shoulder, elbow) arthralgia. Our patient occasionally experienced upper extremity pain, but it was less frequent and less severe than the knee and ankle pain. The vesiculopustular lesions in BADAS usually begin as 3- to 10-mm painful macules that then develop into aseptic pustular lesions. These manifestations arise on the upper arms and chest or trunk and can be accompanied by erythema nodosum on the legs.4
It has been hypothesized that BADAS occurs as an immune reaction to bacterial overgrowth in the bowel from IBD, infection, or surgery. The reaction is in response to a bacterial antigen and manifests cutaneously.5 This same pathogenesis is thought to cause various other manifestations of Crohn disease such as erythema nodosum. Bacteria that incite this immune response include Bacteroides fragilis, Escherichia coli, and Streptococcus.
Resolution of both vesiculopustular Crohn disease and of BADAS often occurs with treatment of the underlying IBD but also can be improved with steroids and antibiotics. However, response to antibiotics often is variable.5,6 The mainstay for treatment remains steroids and management of underlying bowel disease.
Bowel-associated dermatosis-arthritis syndrome often is overlooked when compiling differential diagnoses for neutrophilic dermatoses but should be considered in patients with bowel disease or recent surgery. Because the syndrome can be recurrent, early diagnosis can help to prevent and treat relapsing courses of BADAS.
- Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J. 2005;81:580-585.
- Havemann BD. A pustular skin rash in a woman with 2 weeks of diarrhea. MedGenMed. 2005;7:11.
- Bolognia JL, Jorizzo J, Rapini RP. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2008.
- Huang B, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13.
- Truchuelo MT, Alcántara J, Vano-Galván S, et al. Bowel associated dermatosis-arthritis syndrome: another cutaneous manifestation of inflammatory intestinal disease. Int J Dermatol. 2013;52:1596-1598.
- Ashok D, Kiely P. Bowel associated dermatosis-arthritis syndrome: a case report. J Med Case Rep. 2007;1:81.
To the Editor:
A 42-year-old woman with Crohn disease of 10 years’ duration presented to the clinic with a chief concern of nonpruritic pustular lesions on the bilateral arms. Physical examination revealed several pustules on the arms with secondary excoriation. She also had a warm tender nodule on the left upper shin and subungual hemorrhages under the fingernails (Figure 1). The patient had previously undergone infliximab therapy, which was discontinued 10 months prior to presentation in anticipation of a partial colectomy and temporary ileostomy that was performed 8 months prior to presentation. She recently had developed bilateral, radiating, sharp lower extremity pain extending from the feet to the hips over the last 2 weeks and swelling of the bilateral legs that impaired her ability to ambulate. Additionally, she had recently traveled to Colorado and a Lyme disease workup was initiated at an outside hospital in Colorado; however, the results were pending. The outside hospital also performed a spinal tap that was negative. At our clinic, biopsies were performed on the shin nodule and a right palmar pustule (Figure 2). There was clinical suspicion of erythema nodosum and subcorneal pustular dermatosis or a vesiculopustular skin manifestation of the patient’s Crohn disease. The patient was switched from generic doxycycline to a brand name variant 150 mg every night at bedtime for 2 weeks. She subsequently was admitted to the inpatient rheumatology service for a complete systemic workup.
The punch biopsy of the left upper shin demonstrated operative hemorrhage and periadnexal lymphocytic inflammation without evidence of fungal or bacterial elements by Gram or Gomori methenamine-silver stain. Clinically, the diagnosis was most likely erythema nodosum, though insufficient hypodermis was present to make the diagnosis with pathology. The shave biopsy of the right medial palm was nondiagnostic but showed a transected pustule with no bacterial or fungal elements by Gram or Gomori methenamine-silver stain (Figure 3). Given the clinical context, the likely pathologic diagnosis was vesiculopustular Crohn disease.
Our patient was started on an empiric steroid trial with rapid improvement of the arthralgia and rash. The presumed diagnosis was a Crohn disease flare and the patient was discharged on an 8-week steroid taper. Three weeks later at a follow-up appointment, the patient’s skin lesions had nearly resolved. The swelling of the legs and feet had substantially decreased, but the joint pain, primarily in the ankles, persisted.
Routine laboratory studies showed a hemoglobin level of 11.6 g/dL (reference range, 12–15 g/dL), white blood cell count of 9.1 K/μL (reference range, 4.5–11.0 K/μL), C-reactive protein level of 20.15 mg/dL (reference range, <1.0 mg/dL), and an antinuclear antibody titer of 160 (<80). Serology for Lyme disease was negative. Serum chemistries were all within reference range and an echocardiogram was normal.
Up to one-third of patients with inflammatory bowel disease (IBD) experience extraintestinal manifestations of their condition. Of these patients, nearly one-third will develop cutaneous manifestations.1 The most common skin diseases associated with IBD are pyoderma gangrenosum and erythema nodosum.2 The differential diagnoses considered in this unique case included early pyoderma gangrenosum, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), and vesiculopustular Crohn disease. Vesiculopustular Crohn disease is a rare component of IBD and also can be present in bowel-associated dermatosis-arthritis syndrome (BADAS). In BADAS, symptoms often include arthritis and systemic symptoms such as fever and malaise. The skin manifestations typically involve the arms and trunk. It often is seen after intestinal bypass surgery but also can be present in patients with gastrointestinal diseases such as IBD.3 Due to its early association with bypass surgery, BADAS previously was referred to as bowel bypass syndrome but has since been seen in relation to other intestinal surgeries and IBD.4 Patients with BADAS often present with episodes of fever, fatigue, and malaise, in addition to arthralgia and cutaneous eruptions. Cases of BADAS related to IBD instead of bypass surgery often can be less severe in nature. Unlike many of these previously reported cases, our patient’s joint pain primarily was in the knees and ankles, whereas typical cases of BADAS cause upper extremity (ie, shoulder, elbow) arthralgia. Our patient occasionally experienced upper extremity pain, but it was less frequent and less severe than the knee and ankle pain. The vesiculopustular lesions in BADAS usually begin as 3- to 10-mm painful macules that then develop into aseptic pustular lesions. These manifestations arise on the upper arms and chest or trunk and can be accompanied by erythema nodosum on the legs.4
It has been hypothesized that BADAS occurs as an immune reaction to bacterial overgrowth in the bowel from IBD, infection, or surgery. The reaction is in response to a bacterial antigen and manifests cutaneously.5 This same pathogenesis is thought to cause various other manifestations of Crohn disease such as erythema nodosum. Bacteria that incite this immune response include Bacteroides fragilis, Escherichia coli, and Streptococcus.
Resolution of both vesiculopustular Crohn disease and of BADAS often occurs with treatment of the underlying IBD but also can be improved with steroids and antibiotics. However, response to antibiotics often is variable.5,6 The mainstay for treatment remains steroids and management of underlying bowel disease.
Bowel-associated dermatosis-arthritis syndrome often is overlooked when compiling differential diagnoses for neutrophilic dermatoses but should be considered in patients with bowel disease or recent surgery. Because the syndrome can be recurrent, early diagnosis can help to prevent and treat relapsing courses of BADAS.
To the Editor:
A 42-year-old woman with Crohn disease of 10 years’ duration presented to the clinic with a chief concern of nonpruritic pustular lesions on the bilateral arms. Physical examination revealed several pustules on the arms with secondary excoriation. She also had a warm tender nodule on the left upper shin and subungual hemorrhages under the fingernails (Figure 1). The patient had previously undergone infliximab therapy, which was discontinued 10 months prior to presentation in anticipation of a partial colectomy and temporary ileostomy that was performed 8 months prior to presentation. She recently had developed bilateral, radiating, sharp lower extremity pain extending from the feet to the hips over the last 2 weeks and swelling of the bilateral legs that impaired her ability to ambulate. Additionally, she had recently traveled to Colorado and a Lyme disease workup was initiated at an outside hospital in Colorado; however, the results were pending. The outside hospital also performed a spinal tap that was negative. At our clinic, biopsies were performed on the shin nodule and a right palmar pustule (Figure 2). There was clinical suspicion of erythema nodosum and subcorneal pustular dermatosis or a vesiculopustular skin manifestation of the patient’s Crohn disease. The patient was switched from generic doxycycline to a brand name variant 150 mg every night at bedtime for 2 weeks. She subsequently was admitted to the inpatient rheumatology service for a complete systemic workup.
The punch biopsy of the left upper shin demonstrated operative hemorrhage and periadnexal lymphocytic inflammation without evidence of fungal or bacterial elements by Gram or Gomori methenamine-silver stain. Clinically, the diagnosis was most likely erythema nodosum, though insufficient hypodermis was present to make the diagnosis with pathology. The shave biopsy of the right medial palm was nondiagnostic but showed a transected pustule with no bacterial or fungal elements by Gram or Gomori methenamine-silver stain (Figure 3). Given the clinical context, the likely pathologic diagnosis was vesiculopustular Crohn disease.
Our patient was started on an empiric steroid trial with rapid improvement of the arthralgia and rash. The presumed diagnosis was a Crohn disease flare and the patient was discharged on an 8-week steroid taper. Three weeks later at a follow-up appointment, the patient’s skin lesions had nearly resolved. The swelling of the legs and feet had substantially decreased, but the joint pain, primarily in the ankles, persisted.
Routine laboratory studies showed a hemoglobin level of 11.6 g/dL (reference range, 12–15 g/dL), white blood cell count of 9.1 K/μL (reference range, 4.5–11.0 K/μL), C-reactive protein level of 20.15 mg/dL (reference range, <1.0 mg/dL), and an antinuclear antibody titer of 160 (<80). Serology for Lyme disease was negative. Serum chemistries were all within reference range and an echocardiogram was normal.
Up to one-third of patients with inflammatory bowel disease (IBD) experience extraintestinal manifestations of their condition. Of these patients, nearly one-third will develop cutaneous manifestations.1 The most common skin diseases associated with IBD are pyoderma gangrenosum and erythema nodosum.2 The differential diagnoses considered in this unique case included early pyoderma gangrenosum, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), and vesiculopustular Crohn disease. Vesiculopustular Crohn disease is a rare component of IBD and also can be present in bowel-associated dermatosis-arthritis syndrome (BADAS). In BADAS, symptoms often include arthritis and systemic symptoms such as fever and malaise. The skin manifestations typically involve the arms and trunk. It often is seen after intestinal bypass surgery but also can be present in patients with gastrointestinal diseases such as IBD.3 Due to its early association with bypass surgery, BADAS previously was referred to as bowel bypass syndrome but has since been seen in relation to other intestinal surgeries and IBD.4 Patients with BADAS often present with episodes of fever, fatigue, and malaise, in addition to arthralgia and cutaneous eruptions. Cases of BADAS related to IBD instead of bypass surgery often can be less severe in nature. Unlike many of these previously reported cases, our patient’s joint pain primarily was in the knees and ankles, whereas typical cases of BADAS cause upper extremity (ie, shoulder, elbow) arthralgia. Our patient occasionally experienced upper extremity pain, but it was less frequent and less severe than the knee and ankle pain. The vesiculopustular lesions in BADAS usually begin as 3- to 10-mm painful macules that then develop into aseptic pustular lesions. These manifestations arise on the upper arms and chest or trunk and can be accompanied by erythema nodosum on the legs.4
It has been hypothesized that BADAS occurs as an immune reaction to bacterial overgrowth in the bowel from IBD, infection, or surgery. The reaction is in response to a bacterial antigen and manifests cutaneously.5 This same pathogenesis is thought to cause various other manifestations of Crohn disease such as erythema nodosum. Bacteria that incite this immune response include Bacteroides fragilis, Escherichia coli, and Streptococcus.
Resolution of both vesiculopustular Crohn disease and of BADAS often occurs with treatment of the underlying IBD but also can be improved with steroids and antibiotics. However, response to antibiotics often is variable.5,6 The mainstay for treatment remains steroids and management of underlying bowel disease.
Bowel-associated dermatosis-arthritis syndrome often is overlooked when compiling differential diagnoses for neutrophilic dermatoses but should be considered in patients with bowel disease or recent surgery. Because the syndrome can be recurrent, early diagnosis can help to prevent and treat relapsing courses of BADAS.
- Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J. 2005;81:580-585.
- Havemann BD. A pustular skin rash in a woman with 2 weeks of diarrhea. MedGenMed. 2005;7:11.
- Bolognia JL, Jorizzo J, Rapini RP. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2008.
- Huang B, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13.
- Truchuelo MT, Alcántara J, Vano-Galván S, et al. Bowel associated dermatosis-arthritis syndrome: another cutaneous manifestation of inflammatory intestinal disease. Int J Dermatol. 2013;52:1596-1598.
- Ashok D, Kiely P. Bowel associated dermatosis-arthritis syndrome: a case report. J Med Case Rep. 2007;1:81.
- Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J. 2005;81:580-585.
- Havemann BD. A pustular skin rash in a woman with 2 weeks of diarrhea. MedGenMed. 2005;7:11.
- Bolognia JL, Jorizzo J, Rapini RP. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2008.
- Huang B, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13.
- Truchuelo MT, Alcántara J, Vano-Galván S, et al. Bowel associated dermatosis-arthritis syndrome: another cutaneous manifestation of inflammatory intestinal disease. Int J Dermatol. 2013;52:1596-1598.
- Ashok D, Kiely P. Bowel associated dermatosis-arthritis syndrome: a case report. J Med Case Rep. 2007;1:81.
Pruritic Dermatitis Caused by Bird Mite Infestation
To the Editor:
There are a wide variety of zoonotic diseases that can be transmitted from birds to humans. Pigeons, chickens, starlings, canaries, and parakeets are known reservoirs of one particular zoonotic infection caused by the parasitic arthropod Dermanyssus gallinae.1 Dermanyssus gallinae (chicken mite) and Ornithonyssus sylviarum (northern fowl mite) are collectively referred to as bird mites. When these mites are unable to take blood meals from birds, they search out alternative hosts2; in humans, this often leads to the development of pruritic dermatitis.3
A 30-year-old woman presented to our clinic for evaluation of severe generalized pruritus accompanied by a sensation of “bugs on the skin” of 2 weeks’ duration. She noted the pruritus worsened when she was sitting outside on her porch. A few days prior to presentation, she noticed a small, “pinpoint-sized bug” on her arm (<1 mm in size), which she brought in for identification (Figure).

The bug was identified as a bird mite (Dermanyssus gallinae) on light microscopy, which was later confirmed by a medical entomologist. After the diagnosis of bird mite dermatitis was made, the patient noted there was a nest of starlings above the light on her porch. When she later investigated the nest following the current presentation, she noted many small mites crawling around the nest. The nest was removed and her symptoms resolved completely within 2 weeks without treatment.
Bird mites belong to the Arachnida class, under the order Acari. In 1958, Williams4 noted D gallinae’s ability to feed on human blood. Bird mites have 5 stages of development: egg, larva, protonymph, deutonymph, and adult. Protonymphs, deutonymphs, and adults can bite humans for a blood meal.5 Bird mites range from 0.3 to 1 mm in length and have nonsegmented, egg-shaped bodies with 4 pairs of legs. Before taking a blood meal, bird mites generally are a translucent brown color, and appear red when engorged with blood.2 Their small size makes them barely visible to the unaided eye. Of note, D gallinae and O sylviarum can be distinguished from each other based on subtle differences in morphology; for instance, the posterior genitoventral shield of O sylviarum is narrowly rounded, whereas it is broadly rounded in D gallinae. The dorsal shield of O sylviarum abruptly narrows posteriorly but is more smoothly narrowed in D gallinae.6 Additionally, O sylviarum tends to cause more irritating dermatitis in humans than D gallinae.3
Although they can be found worldwide, D gallinae and O sylviarum undergo optimal development at 20°C to 25°C and 70% humidity.3,5,7 Bird mites generally develop over the course of 5 to 12 days; thus, the population of bird mites in a single nest may grow to the tens of thousands before young birds permanently leave. Dermanyssus gallinae can survive for months in abandoned nests without a blood meal, while O sylviarum can survive for several weeks.8 It is important to note that humans are not ideal hosts for bird mites, as they are unable to survive for extended periods of time or reproduce on human hosts.9
When bird mites are no longer able to obtain blood meals from nesting birds, they begin their nocturnal migration to find suitable hosts. Bird nests generally are abandoned in late spring; thus, most patients with bird mite dermatitis present to clinics with bird mite dermatitis in late spring and early summer.10 Mites often travel through cracks in doors, floors, walls, and ceilings but also can gain access to living areas through ventilation ducts and air conditioning units.1 The mite’s bite and crawling on the skin is sometimes noticed by the patient. In general, however, intense itching is not observed until about 1 to 3 days after the mite makes contact with the skin. Patients often report that pruritus is worst at night.9 Papules and vesicles (bite reactions) may accompany the pruritus, and physicians commonly find bloody crust and excoriations in particularly pruritic areas.5 Urticarial plaques and diffuse erythema occasionally also may be present.9 Bird mites sometimes can be scraped from the skin and observed under light microscopy.11 Blood eosinophilia is not found in bird mite dermatitis. On histologic examination, perivascular eosinophilic infiltration can be seen in the upper part of the dermis.12
The differential diagnosis in patients with pruritic dermatitis of unknown origin generally includes scabies, pediculosis, and dermatitis caused by other types of infestation. However, unlike scabies, bird mites do not cause burrows to form on the skin.9 The presence of a bird’s nest near the area where the patient lives places bird mite dermatitis higher in the differential.
Dermanyssus gallinae is a known vector of bacteria (eg, Salmonella, Shigella, Staphylococcus, Spirochaete, Rickettsia, Pasteurella, Chlamydia psittaci, Erysipelothrix rhusiopathiae) as well as the viruses that cause Eastern and Western equine encephalitis and St. Louis encephalitis. Transmission of these bacteria and viruses is known in birds, but transmission to humans has not been reported.2,5,9,13
The management of bird mite dermatitis is straightforward. Usually mites can be successfully removed from the skin simply by bathing. Symptomatic treatment for bites with antihistamines and topical corticosteroids is sometimes but not always necessary.2 Unlike scabies or lice, there is no need for treatment with lindane.1 In terms of the prevention of additional bites, any bird nests located near living areas should be removed. Because bird mites often retreat back to nests between blood meals, insecticide sprays generally are unnecessary in interior spaces. Synthetic pyrethroids (eg, bifenthrin, cyfluthrin, cypermethrin, deltamethrin, cyhalothrin) can be used outside and in attics where nests may be located.2,14,15 However, the ability of bird mites to develop resistance to repeated chemical control could become a future concern.16
Research regarding the true incidence of bird mite dermatitis is lacking. Some researchers believe that the condition is underreported, possibly due to its uncommon environmental origin.3 Reports of bird mite dermatitis in the literature also are scarce. Our case demonstrates the importance of taking a thorough patient history to rule out exposure to bird mites. All patients with pruritic dermatitis of unknown origin should be questioned about possible contact or proximity to bird nests. These simple questions can lead to the correct diagnosis and a treatment plan that will quickly and effectively resolve the pruritic skin eruption.
- Regan AM, Metersky ML, Craven DE. Nosocomial dermatitis and pruritus caused by pigeon mite infestation. Arch Intern Med. 1987;147:2185-2187.
- Collgros H, Iglesias-Sancho M, Aldunce MJ, et al. Dermanyssus gallinae (chicken mite): an underdiagnosed environmental infestation. Clin Exp Dermatol. 2013;38:374-377.
- Bellanger AP, Boris C, Foulet F, et al. Nosocomial dermatitis caused by Dermanyssus gallinae. Infect Cont Hosp Ep. 2008;29:282-283.
- Williams RW. An infestation of a human habitation by Dermanyssus gallinae (de Geer, 1778) (Acarina: Dermanyssidae) in New York resulting in sanguisugent attacks upon the occupants. Am J Trop Med Hyg. 1958;7:627-629.
- Akdemir C, Gülcan E, Tanritanir P. Case report: Dermanyssus gallinae in a patient with pruritus and skin lesions. Turkiye Parazitol Derg. 2009;33:242-244.
- DiPalma A, Giangaspero A, Cafiero MA, et al. A gallery of the key characteristics to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae). Parasites and Vectors. 2012;5:104.
- Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae). Exp Appl Acarol. 1992;15:27-40.
- Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exper Dermatol. 2000;25:129-131.
- Auger P, Nantel J, Meunier N, et al. Skin acariasis caused by Dermanyssus gallinae (de Geer): an in-hospital outbreak. Can Med Assoc J. 1979;120:700-703.
- Kong TK, To WK. Bird mite infestation. N Engl J Med. 2006;354:1728.
- Koh WL, Liu TT, Tay YK. Formication due to true parasitic infection: bird mites. Arch Dermatol. 2011;147:508-509.
- Hidano A, Asanuma K. Letter: Acariasis caused by bird mites. Arch Dermatol. 1976;112:881-882.
- Valiente Moro C, Chauve C, Zenner L. Experimental infection of Salmonella Enteritidis by the poultry red mite, Dermanyssus gallinae. Vet Parasitol. 2007;146:329-336.
- Fletcher MG, Axtell RC. Susceptibilities of northern fowl mite, Ornithonyssus sylviarum (Acarina: Macronyssidae),and chicken mite, Dermanyssus gallinae (Acarina: Dermanyssidae), to selected acaricides. Exp Appl Acarol. 1991;13:137-142.
- Thind BB, Ford HL. Assessment of susceptibility of the poultry red mite Dermanyssus gallinae (Acari: Dermanyssidae) to some acaricides using an adapted filter paper based bioassay. Vet Parasitol. 2007;144:344-348.
- Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239-245.
To the Editor:
There are a wide variety of zoonotic diseases that can be transmitted from birds to humans. Pigeons, chickens, starlings, canaries, and parakeets are known reservoirs of one particular zoonotic infection caused by the parasitic arthropod Dermanyssus gallinae.1 Dermanyssus gallinae (chicken mite) and Ornithonyssus sylviarum (northern fowl mite) are collectively referred to as bird mites. When these mites are unable to take blood meals from birds, they search out alternative hosts2; in humans, this often leads to the development of pruritic dermatitis.3
A 30-year-old woman presented to our clinic for evaluation of severe generalized pruritus accompanied by a sensation of “bugs on the skin” of 2 weeks’ duration. She noted the pruritus worsened when she was sitting outside on her porch. A few days prior to presentation, she noticed a small, “pinpoint-sized bug” on her arm (<1 mm in size), which she brought in for identification (Figure).
The bug was identified as a bird mite (Dermanyssus gallinae) on light microscopy, which was later confirmed by a medical entomologist. After the diagnosis of bird mite dermatitis was made, the patient noted there was a nest of starlings above the light on her porch. When she later investigated the nest following the current presentation, she noted many small mites crawling around the nest. The nest was removed and her symptoms resolved completely within 2 weeks without treatment.
Bird mites belong to the Arachnida class, under the order Acari. In 1958, Williams4 noted D gallinae’s ability to feed on human blood. Bird mites have 5 stages of development: egg, larva, protonymph, deutonymph, and adult. Protonymphs, deutonymphs, and adults can bite humans for a blood meal.5 Bird mites range from 0.3 to 1 mm in length and have nonsegmented, egg-shaped bodies with 4 pairs of legs. Before taking a blood meal, bird mites generally are a translucent brown color, and appear red when engorged with blood.2 Their small size makes them barely visible to the unaided eye. Of note, D gallinae and O sylviarum can be distinguished from each other based on subtle differences in morphology; for instance, the posterior genitoventral shield of O sylviarum is narrowly rounded, whereas it is broadly rounded in D gallinae. The dorsal shield of O sylviarum abruptly narrows posteriorly but is more smoothly narrowed in D gallinae.6 Additionally, O sylviarum tends to cause more irritating dermatitis in humans than D gallinae.3
Although they can be found worldwide, D gallinae and O sylviarum undergo optimal development at 20°C to 25°C and 70% humidity.3,5,7 Bird mites generally develop over the course of 5 to 12 days; thus, the population of bird mites in a single nest may grow to the tens of thousands before young birds permanently leave. Dermanyssus gallinae can survive for months in abandoned nests without a blood meal, while O sylviarum can survive for several weeks.8 It is important to note that humans are not ideal hosts for bird mites, as they are unable to survive for extended periods of time or reproduce on human hosts.9
When bird mites are no longer able to obtain blood meals from nesting birds, they begin their nocturnal migration to find suitable hosts. Bird nests generally are abandoned in late spring; thus, most patients with bird mite dermatitis present to clinics with bird mite dermatitis in late spring and early summer.10 Mites often travel through cracks in doors, floors, walls, and ceilings but also can gain access to living areas through ventilation ducts and air conditioning units.1 The mite’s bite and crawling on the skin is sometimes noticed by the patient. In general, however, intense itching is not observed until about 1 to 3 days after the mite makes contact with the skin. Patients often report that pruritus is worst at night.9 Papules and vesicles (bite reactions) may accompany the pruritus, and physicians commonly find bloody crust and excoriations in particularly pruritic areas.5 Urticarial plaques and diffuse erythema occasionally also may be present.9 Bird mites sometimes can be scraped from the skin and observed under light microscopy.11 Blood eosinophilia is not found in bird mite dermatitis. On histologic examination, perivascular eosinophilic infiltration can be seen in the upper part of the dermis.12
The differential diagnosis in patients with pruritic dermatitis of unknown origin generally includes scabies, pediculosis, and dermatitis caused by other types of infestation. However, unlike scabies, bird mites do not cause burrows to form on the skin.9 The presence of a bird’s nest near the area where the patient lives places bird mite dermatitis higher in the differential.
Dermanyssus gallinae is a known vector of bacteria (eg, Salmonella, Shigella, Staphylococcus, Spirochaete, Rickettsia, Pasteurella, Chlamydia psittaci, Erysipelothrix rhusiopathiae) as well as the viruses that cause Eastern and Western equine encephalitis and St. Louis encephalitis. Transmission of these bacteria and viruses is known in birds, but transmission to humans has not been reported.2,5,9,13
The management of bird mite dermatitis is straightforward. Usually mites can be successfully removed from the skin simply by bathing. Symptomatic treatment for bites with antihistamines and topical corticosteroids is sometimes but not always necessary.2 Unlike scabies or lice, there is no need for treatment with lindane.1 In terms of the prevention of additional bites, any bird nests located near living areas should be removed. Because bird mites often retreat back to nests between blood meals, insecticide sprays generally are unnecessary in interior spaces. Synthetic pyrethroids (eg, bifenthrin, cyfluthrin, cypermethrin, deltamethrin, cyhalothrin) can be used outside and in attics where nests may be located.2,14,15 However, the ability of bird mites to develop resistance to repeated chemical control could become a future concern.16
Research regarding the true incidence of bird mite dermatitis is lacking. Some researchers believe that the condition is underreported, possibly due to its uncommon environmental origin.3 Reports of bird mite dermatitis in the literature also are scarce. Our case demonstrates the importance of taking a thorough patient history to rule out exposure to bird mites. All patients with pruritic dermatitis of unknown origin should be questioned about possible contact or proximity to bird nests. These simple questions can lead to the correct diagnosis and a treatment plan that will quickly and effectively resolve the pruritic skin eruption.
To the Editor:
There are a wide variety of zoonotic diseases that can be transmitted from birds to humans. Pigeons, chickens, starlings, canaries, and parakeets are known reservoirs of one particular zoonotic infection caused by the parasitic arthropod Dermanyssus gallinae.1 Dermanyssus gallinae (chicken mite) and Ornithonyssus sylviarum (northern fowl mite) are collectively referred to as bird mites. When these mites are unable to take blood meals from birds, they search out alternative hosts2; in humans, this often leads to the development of pruritic dermatitis.3
A 30-year-old woman presented to our clinic for evaluation of severe generalized pruritus accompanied by a sensation of “bugs on the skin” of 2 weeks’ duration. She noted the pruritus worsened when she was sitting outside on her porch. A few days prior to presentation, she noticed a small, “pinpoint-sized bug” on her arm (<1 mm in size), which she brought in for identification (Figure).
The bug was identified as a bird mite (Dermanyssus gallinae) on light microscopy, which was later confirmed by a medical entomologist. After the diagnosis of bird mite dermatitis was made, the patient noted there was a nest of starlings above the light on her porch. When she later investigated the nest following the current presentation, she noted many small mites crawling around the nest. The nest was removed and her symptoms resolved completely within 2 weeks without treatment.
Bird mites belong to the Arachnida class, under the order Acari. In 1958, Williams4 noted D gallinae’s ability to feed on human blood. Bird mites have 5 stages of development: egg, larva, protonymph, deutonymph, and adult. Protonymphs, deutonymphs, and adults can bite humans for a blood meal.5 Bird mites range from 0.3 to 1 mm in length and have nonsegmented, egg-shaped bodies with 4 pairs of legs. Before taking a blood meal, bird mites generally are a translucent brown color, and appear red when engorged with blood.2 Their small size makes them barely visible to the unaided eye. Of note, D gallinae and O sylviarum can be distinguished from each other based on subtle differences in morphology; for instance, the posterior genitoventral shield of O sylviarum is narrowly rounded, whereas it is broadly rounded in D gallinae. The dorsal shield of O sylviarum abruptly narrows posteriorly but is more smoothly narrowed in D gallinae.6 Additionally, O sylviarum tends to cause more irritating dermatitis in humans than D gallinae.3
Although they can be found worldwide, D gallinae and O sylviarum undergo optimal development at 20°C to 25°C and 70% humidity.3,5,7 Bird mites generally develop over the course of 5 to 12 days; thus, the population of bird mites in a single nest may grow to the tens of thousands before young birds permanently leave. Dermanyssus gallinae can survive for months in abandoned nests without a blood meal, while O sylviarum can survive for several weeks.8 It is important to note that humans are not ideal hosts for bird mites, as they are unable to survive for extended periods of time or reproduce on human hosts.9
When bird mites are no longer able to obtain blood meals from nesting birds, they begin their nocturnal migration to find suitable hosts. Bird nests generally are abandoned in late spring; thus, most patients with bird mite dermatitis present to clinics with bird mite dermatitis in late spring and early summer.10 Mites often travel through cracks in doors, floors, walls, and ceilings but also can gain access to living areas through ventilation ducts and air conditioning units.1 The mite’s bite and crawling on the skin is sometimes noticed by the patient. In general, however, intense itching is not observed until about 1 to 3 days after the mite makes contact with the skin. Patients often report that pruritus is worst at night.9 Papules and vesicles (bite reactions) may accompany the pruritus, and physicians commonly find bloody crust and excoriations in particularly pruritic areas.5 Urticarial plaques and diffuse erythema occasionally also may be present.9 Bird mites sometimes can be scraped from the skin and observed under light microscopy.11 Blood eosinophilia is not found in bird mite dermatitis. On histologic examination, perivascular eosinophilic infiltration can be seen in the upper part of the dermis.12
The differential diagnosis in patients with pruritic dermatitis of unknown origin generally includes scabies, pediculosis, and dermatitis caused by other types of infestation. However, unlike scabies, bird mites do not cause burrows to form on the skin.9 The presence of a bird’s nest near the area where the patient lives places bird mite dermatitis higher in the differential.
Dermanyssus gallinae is a known vector of bacteria (eg, Salmonella, Shigella, Staphylococcus, Spirochaete, Rickettsia, Pasteurella, Chlamydia psittaci, Erysipelothrix rhusiopathiae) as well as the viruses that cause Eastern and Western equine encephalitis and St. Louis encephalitis. Transmission of these bacteria and viruses is known in birds, but transmission to humans has not been reported.2,5,9,13
The management of bird mite dermatitis is straightforward. Usually mites can be successfully removed from the skin simply by bathing. Symptomatic treatment for bites with antihistamines and topical corticosteroids is sometimes but not always necessary.2 Unlike scabies or lice, there is no need for treatment with lindane.1 In terms of the prevention of additional bites, any bird nests located near living areas should be removed. Because bird mites often retreat back to nests between blood meals, insecticide sprays generally are unnecessary in interior spaces. Synthetic pyrethroids (eg, bifenthrin, cyfluthrin, cypermethrin, deltamethrin, cyhalothrin) can be used outside and in attics where nests may be located.2,14,15 However, the ability of bird mites to develop resistance to repeated chemical control could become a future concern.16
Research regarding the true incidence of bird mite dermatitis is lacking. Some researchers believe that the condition is underreported, possibly due to its uncommon environmental origin.3 Reports of bird mite dermatitis in the literature also are scarce. Our case demonstrates the importance of taking a thorough patient history to rule out exposure to bird mites. All patients with pruritic dermatitis of unknown origin should be questioned about possible contact or proximity to bird nests. These simple questions can lead to the correct diagnosis and a treatment plan that will quickly and effectively resolve the pruritic skin eruption.
- Regan AM, Metersky ML, Craven DE. Nosocomial dermatitis and pruritus caused by pigeon mite infestation. Arch Intern Med. 1987;147:2185-2187.
- Collgros H, Iglesias-Sancho M, Aldunce MJ, et al. Dermanyssus gallinae (chicken mite): an underdiagnosed environmental infestation. Clin Exp Dermatol. 2013;38:374-377.
- Bellanger AP, Boris C, Foulet F, et al. Nosocomial dermatitis caused by Dermanyssus gallinae. Infect Cont Hosp Ep. 2008;29:282-283.
- Williams RW. An infestation of a human habitation by Dermanyssus gallinae (de Geer, 1778) (Acarina: Dermanyssidae) in New York resulting in sanguisugent attacks upon the occupants. Am J Trop Med Hyg. 1958;7:627-629.
- Akdemir C, Gülcan E, Tanritanir P. Case report: Dermanyssus gallinae in a patient with pruritus and skin lesions. Turkiye Parazitol Derg. 2009;33:242-244.
- DiPalma A, Giangaspero A, Cafiero MA, et al. A gallery of the key characteristics to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae). Parasites and Vectors. 2012;5:104.
- Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae). Exp Appl Acarol. 1992;15:27-40.
- Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exper Dermatol. 2000;25:129-131.
- Auger P, Nantel J, Meunier N, et al. Skin acariasis caused by Dermanyssus gallinae (de Geer): an in-hospital outbreak. Can Med Assoc J. 1979;120:700-703.
- Kong TK, To WK. Bird mite infestation. N Engl J Med. 2006;354:1728.
- Koh WL, Liu TT, Tay YK. Formication due to true parasitic infection: bird mites. Arch Dermatol. 2011;147:508-509.
- Hidano A, Asanuma K. Letter: Acariasis caused by bird mites. Arch Dermatol. 1976;112:881-882.
- Valiente Moro C, Chauve C, Zenner L. Experimental infection of Salmonella Enteritidis by the poultry red mite, Dermanyssus gallinae. Vet Parasitol. 2007;146:329-336.
- Fletcher MG, Axtell RC. Susceptibilities of northern fowl mite, Ornithonyssus sylviarum (Acarina: Macronyssidae),and chicken mite, Dermanyssus gallinae (Acarina: Dermanyssidae), to selected acaricides. Exp Appl Acarol. 1991;13:137-142.
- Thind BB, Ford HL. Assessment of susceptibility of the poultry red mite Dermanyssus gallinae (Acari: Dermanyssidae) to some acaricides using an adapted filter paper based bioassay. Vet Parasitol. 2007;144:344-348.
- Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239-245.
- Regan AM, Metersky ML, Craven DE. Nosocomial dermatitis and pruritus caused by pigeon mite infestation. Arch Intern Med. 1987;147:2185-2187.
- Collgros H, Iglesias-Sancho M, Aldunce MJ, et al. Dermanyssus gallinae (chicken mite): an underdiagnosed environmental infestation. Clin Exp Dermatol. 2013;38:374-377.
- Bellanger AP, Boris C, Foulet F, et al. Nosocomial dermatitis caused by Dermanyssus gallinae. Infect Cont Hosp Ep. 2008;29:282-283.
- Williams RW. An infestation of a human habitation by Dermanyssus gallinae (de Geer, 1778) (Acarina: Dermanyssidae) in New York resulting in sanguisugent attacks upon the occupants. Am J Trop Med Hyg. 1958;7:627-629.
- Akdemir C, Gülcan E, Tanritanir P. Case report: Dermanyssus gallinae in a patient with pruritus and skin lesions. Turkiye Parazitol Derg. 2009;33:242-244.
- DiPalma A, Giangaspero A, Cafiero MA, et al. A gallery of the key characteristics to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae). Parasites and Vectors. 2012;5:104.
- Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae). Exp Appl Acarol. 1992;15:27-40.
- Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exper Dermatol. 2000;25:129-131.
- Auger P, Nantel J, Meunier N, et al. Skin acariasis caused by Dermanyssus gallinae (de Geer): an in-hospital outbreak. Can Med Assoc J. 1979;120:700-703.
- Kong TK, To WK. Bird mite infestation. N Engl J Med. 2006;354:1728.
- Koh WL, Liu TT, Tay YK. Formication due to true parasitic infection: bird mites. Arch Dermatol. 2011;147:508-509.
- Hidano A, Asanuma K. Letter: Acariasis caused by bird mites. Arch Dermatol. 1976;112:881-882.
- Valiente Moro C, Chauve C, Zenner L. Experimental infection of Salmonella Enteritidis by the poultry red mite, Dermanyssus gallinae. Vet Parasitol. 2007;146:329-336.
- Fletcher MG, Axtell RC. Susceptibilities of northern fowl mite, Ornithonyssus sylviarum (Acarina: Macronyssidae),and chicken mite, Dermanyssus gallinae (Acarina: Dermanyssidae), to selected acaricides. Exp Appl Acarol. 1991;13:137-142.
- Thind BB, Ford HL. Assessment of susceptibility of the poultry red mite Dermanyssus gallinae (Acari: Dermanyssidae) to some acaricides using an adapted filter paper based bioassay. Vet Parasitol. 2007;144:344-348.
- Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239-245.

Oral Leukoedema with Mucosal Desquamation Caused by Toothpaste Containing Sodium Lauryl Sulfate
To the Editor:
A 34-year-old woman presented for evaluation of dry mouth and painless peeling of the oral mucosa of 2 months’ duration. She denied any other skin eruptions, dry eyes, vulvar or vaginal pain, or recent hair loss. A recent antinuclear antibodies test was negative. The patient’s medical history was otherwise unremarkable and her current medications included multivitamins only.
Oral examination revealed peeling gray-white tissue on the buccal mucosa and mouth floor (Figure 1). After the tissue was manually removed with a tongue blade, the mucosal base was normal in color and texture. The patient denied bruxism, biting of the mucosa or other oral trauma, or use of tobacco or nonsteroidal anti-inflammatory drugs.
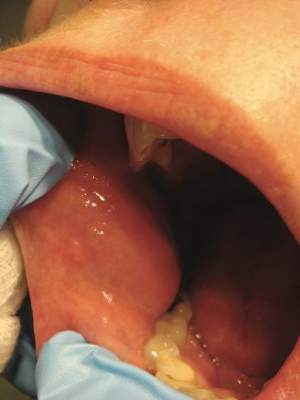
Biopsies from the buccal mucosa were performed to rule out erosive lichen planus and autoimmune blistering disorders. Microscopy revealed parakeratosis and intracellular edema of the mucosa. An intraepithelial cleft at the parakeratotic surface also was present (Figure 2). Minimal inflammation was noted. Fungal staining and direct immunofluorescence were negative.
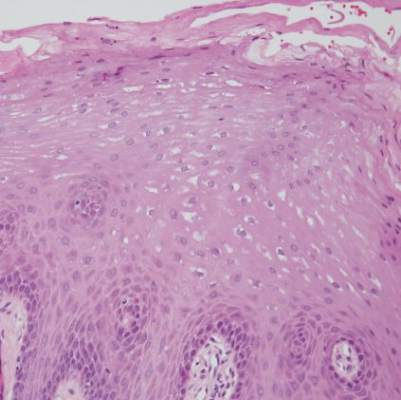
The gray-white clinical appearance of the oral mucosa resembled leukoedema, but the peeling phenomenon was uncharacteristic. Histologically, leukoedema typically has a parakeratotic and acanthotic epithelium with marked intracellular edema of the spinous layer.1,2 Our patient demonstrated intracellular edema with the additional finding of a superficial intraepithelial cleft. These features were consistent with the observed mucosal sloughing and normal tissue base and led to our diagnosis of leukoedema with mucosal desquamation. This clinical and histologic picture was previously described in another report, but a causative agent could not be identified.2
Because leukoedema can be secondary to chemical or mechanical trauma,3 we hypothesized that the patient’s toothpaste may be the causative agent. After discontinuing use of her regular toothpaste and keeping the rest of her oral hygiene routine unchanged, the patient’s condition resolved within 2 days. The patient could not identify how long she had been using the toothpaste before symptoms began.
Our case as well as a report in the literature suggest that leukoedema with mucosal desquamation may be the result of contact mucositis to dental hygiene products.3 Reports in the dental literature suggest that a possible cause for oral mucosal desquamation is sensitivity to sodium lauryl sulfate (SLS),1,4 an ingredient used in some toothpastes, including the one used by our patient. The patient has since switched to a non–SLS-containing toothpaste and has remained asymptomatic. She was unwilling to reintroduce an SLS-containing product for further evaluation.
Sodium lauryl sulfate is a strong anionic detergent that is commonly used as a foaming agent in dentifrices.4 In products with higher concentrations of SLS, the incidence of oral epithelial desquamation increases. Triclosan has been shown to protect against this irritant phenomenon.5 Interestingly, the SLS-containing toothpaste used by our patient did not contain triclosan.
Although leukoedema and mucosal desquamation induced by oral care products are well-described in the dental literature, it is important for dermatologists to be aware of this phenomenon, as the differential diagnosis includes autoimmune blistering disorders and erosive lichen planus, for which dermatology referral may be requested. Further studies of SLS and other toothpaste ingredients are needed to establish if sloughing of the oral mucosa is primarily caused by SLS or another ingredient.
- Shafer WG, Hine MK, Levy BM. A Textbook of Oral Pathology. Philadelphia, PA: WB Saunders; 1983.
- Zegarelli DJ, Silvers DN. Shedding oral mucosa. Cutis. 1994;54:323-326.
- Archard HO, Carlson KP, Stanley HR. Leukoedema of the human oral mucosa. Oral Surg Oral Med Oral Pathol. 1971;25:717-728.
- Herlofson BB, Barkvoll P. Desquamative effect of sodium lauryl sulfate on oral mucosa. a preliminary study. Acta Odontol Scand. 1993;51:39-43.
- Skaare A, Eide G, Herlofson B, et al. The effect of toothpaste containing triclosan on oral mucosal desquamation. a model study. J Clin Periodontology. 1996;23:1100-1103.
To the Editor:
A 34-year-old woman presented for evaluation of dry mouth and painless peeling of the oral mucosa of 2 months’ duration. She denied any other skin eruptions, dry eyes, vulvar or vaginal pain, or recent hair loss. A recent antinuclear antibodies test was negative. The patient’s medical history was otherwise unremarkable and her current medications included multivitamins only.
Oral examination revealed peeling gray-white tissue on the buccal mucosa and mouth floor (Figure 1). After the tissue was manually removed with a tongue blade, the mucosal base was normal in color and texture. The patient denied bruxism, biting of the mucosa or other oral trauma, or use of tobacco or nonsteroidal anti-inflammatory drugs.
Biopsies from the buccal mucosa were performed to rule out erosive lichen planus and autoimmune blistering disorders. Microscopy revealed parakeratosis and intracellular edema of the mucosa. An intraepithelial cleft at the parakeratotic surface also was present (Figure 2). Minimal inflammation was noted. Fungal staining and direct immunofluorescence were negative.
The gray-white clinical appearance of the oral mucosa resembled leukoedema, but the peeling phenomenon was uncharacteristic. Histologically, leukoedema typically has a parakeratotic and acanthotic epithelium with marked intracellular edema of the spinous layer.1,2 Our patient demonstrated intracellular edema with the additional finding of a superficial intraepithelial cleft. These features were consistent with the observed mucosal sloughing and normal tissue base and led to our diagnosis of leukoedema with mucosal desquamation. This clinical and histologic picture was previously described in another report, but a causative agent could not be identified.2
Because leukoedema can be secondary to chemical or mechanical trauma,3 we hypothesized that the patient’s toothpaste may be the causative agent. After discontinuing use of her regular toothpaste and keeping the rest of her oral hygiene routine unchanged, the patient’s condition resolved within 2 days. The patient could not identify how long she had been using the toothpaste before symptoms began.
Our case as well as a report in the literature suggest that leukoedema with mucosal desquamation may be the result of contact mucositis to dental hygiene products.3 Reports in the dental literature suggest that a possible cause for oral mucosal desquamation is sensitivity to sodium lauryl sulfate (SLS),1,4 an ingredient used in some toothpastes, including the one used by our patient. The patient has since switched to a non–SLS-containing toothpaste and has remained asymptomatic. She was unwilling to reintroduce an SLS-containing product for further evaluation.
Sodium lauryl sulfate is a strong anionic detergent that is commonly used as a foaming agent in dentifrices.4 In products with higher concentrations of SLS, the incidence of oral epithelial desquamation increases. Triclosan has been shown to protect against this irritant phenomenon.5 Interestingly, the SLS-containing toothpaste used by our patient did not contain triclosan.
Although leukoedema and mucosal desquamation induced by oral care products are well-described in the dental literature, it is important for dermatologists to be aware of this phenomenon, as the differential diagnosis includes autoimmune blistering disorders and erosive lichen planus, for which dermatology referral may be requested. Further studies of SLS and other toothpaste ingredients are needed to establish if sloughing of the oral mucosa is primarily caused by SLS or another ingredient.
To the Editor:
A 34-year-old woman presented for evaluation of dry mouth and painless peeling of the oral mucosa of 2 months’ duration. She denied any other skin eruptions, dry eyes, vulvar or vaginal pain, or recent hair loss. A recent antinuclear antibodies test was negative. The patient’s medical history was otherwise unremarkable and her current medications included multivitamins only.
Oral examination revealed peeling gray-white tissue on the buccal mucosa and mouth floor (Figure 1). After the tissue was manually removed with a tongue blade, the mucosal base was normal in color and texture. The patient denied bruxism, biting of the mucosa or other oral trauma, or use of tobacco or nonsteroidal anti-inflammatory drugs.
Biopsies from the buccal mucosa were performed to rule out erosive lichen planus and autoimmune blistering disorders. Microscopy revealed parakeratosis and intracellular edema of the mucosa. An intraepithelial cleft at the parakeratotic surface also was present (Figure 2). Minimal inflammation was noted. Fungal staining and direct immunofluorescence were negative.
The gray-white clinical appearance of the oral mucosa resembled leukoedema, but the peeling phenomenon was uncharacteristic. Histologically, leukoedema typically has a parakeratotic and acanthotic epithelium with marked intracellular edema of the spinous layer.1,2 Our patient demonstrated intracellular edema with the additional finding of a superficial intraepithelial cleft. These features were consistent with the observed mucosal sloughing and normal tissue base and led to our diagnosis of leukoedema with mucosal desquamation. This clinical and histologic picture was previously described in another report, but a causative agent could not be identified.2
Because leukoedema can be secondary to chemical or mechanical trauma,3 we hypothesized that the patient’s toothpaste may be the causative agent. After discontinuing use of her regular toothpaste and keeping the rest of her oral hygiene routine unchanged, the patient’s condition resolved within 2 days. The patient could not identify how long she had been using the toothpaste before symptoms began.
Our case as well as a report in the literature suggest that leukoedema with mucosal desquamation may be the result of contact mucositis to dental hygiene products.3 Reports in the dental literature suggest that a possible cause for oral mucosal desquamation is sensitivity to sodium lauryl sulfate (SLS),1,4 an ingredient used in some toothpastes, including the one used by our patient. The patient has since switched to a non–SLS-containing toothpaste and has remained asymptomatic. She was unwilling to reintroduce an SLS-containing product for further evaluation.
Sodium lauryl sulfate is a strong anionic detergent that is commonly used as a foaming agent in dentifrices.4 In products with higher concentrations of SLS, the incidence of oral epithelial desquamation increases. Triclosan has been shown to protect against this irritant phenomenon.5 Interestingly, the SLS-containing toothpaste used by our patient did not contain triclosan.
Although leukoedema and mucosal desquamation induced by oral care products are well-described in the dental literature, it is important for dermatologists to be aware of this phenomenon, as the differential diagnosis includes autoimmune blistering disorders and erosive lichen planus, for which dermatology referral may be requested. Further studies of SLS and other toothpaste ingredients are needed to establish if sloughing of the oral mucosa is primarily caused by SLS or another ingredient.
- Shafer WG, Hine MK, Levy BM. A Textbook of Oral Pathology. Philadelphia, PA: WB Saunders; 1983.
- Zegarelli DJ, Silvers DN. Shedding oral mucosa. Cutis. 1994;54:323-326.
- Archard HO, Carlson KP, Stanley HR. Leukoedema of the human oral mucosa. Oral Surg Oral Med Oral Pathol. 1971;25:717-728.
- Herlofson BB, Barkvoll P. Desquamative effect of sodium lauryl sulfate on oral mucosa. a preliminary study. Acta Odontol Scand. 1993;51:39-43.
- Skaare A, Eide G, Herlofson B, et al. The effect of toothpaste containing triclosan on oral mucosal desquamation. a model study. J Clin Periodontology. 1996;23:1100-1103.
- Shafer WG, Hine MK, Levy BM. A Textbook of Oral Pathology. Philadelphia, PA: WB Saunders; 1983.
- Zegarelli DJ, Silvers DN. Shedding oral mucosa. Cutis. 1994;54:323-326.
- Archard HO, Carlson KP, Stanley HR. Leukoedema of the human oral mucosa. Oral Surg Oral Med Oral Pathol. 1971;25:717-728.
- Herlofson BB, Barkvoll P. Desquamative effect of sodium lauryl sulfate on oral mucosa. a preliminary study. Acta Odontol Scand. 1993;51:39-43.
- Skaare A, Eide G, Herlofson B, et al. The effect of toothpaste containing triclosan on oral mucosal desquamation. a model study. J Clin Periodontology. 1996;23:1100-1103.
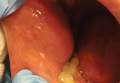
Recalcitrant Hailey-Hailey Disease Responds to Oral Tacrolimus and Botulinum Toxin Type A
To the Editor:
Hailey-Hailey disease, also known as familial benign pemphigus, is a chronic blistering skin disorder that typically presents as a recurrent vesicular or bullous dermatitis found predominantly in the intertriginous regions of the body. Because current treatment regimens for Hailey-Hailey disease are fairly limited, novel treatments may be explored in intractable cases.
A 71-year-old woman presented with well-demarcated erythematous plaques with erosions and white verrucous regions in the perivulvar, vaginal, and perianal areas of 1 month’s duration (Figure 1). The lesions were excruciatingly pruritic and excoriated. The patient reported no personal or family history of similar lesions.

Histopathologic examination of multiple biopsies from the periphery of the plaques showed acantholysis of the epidermis and surface necrosis with negative direct immunofluorescence. A diagnosis of Hailey-Hailey disease was made. Over several months following the initial presentation, the patient was treated with regimens of corticosteroids, antibiotics, antifungals, acitretin, and topical tacrolimus (which showed minimal response), but the condition continued to progress and thus warranted a more aggressive approach. After a 4-week course of oral cyclosporine 1.25 mg/kg twice daily, some healthy granulation tissue had formed, but new erosions continued to develop on the vulva, labia, and intergluteal cleft (Figure 2). Subsequently, a 2-month course of methotrexate yielded similar results with minimal healing and new necrotic areas (Figure 3).
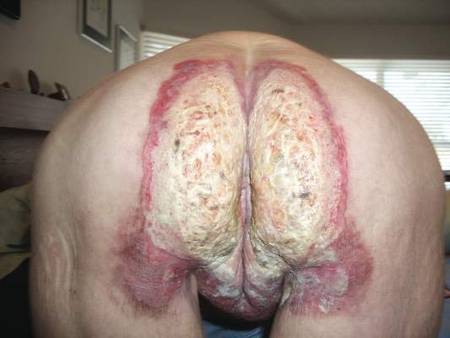
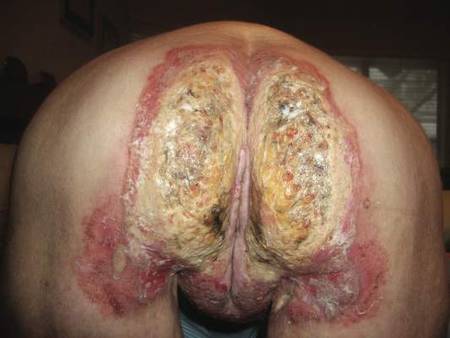
Following methotrexate therapy, treatment with several other oral agents was considered such as azathioprine, mycophenolate mofetil, and oral tacrolimus. Since the patient had previously shown minimal response to topical tacrolimus, a course of oral treatment (0.05 mg/kg twice daily) was initiated. At 4 weeks’ follow-up, extensive healing of the lesions was noted (Figure 4) and the patient reported that the painful pruritus had improved.
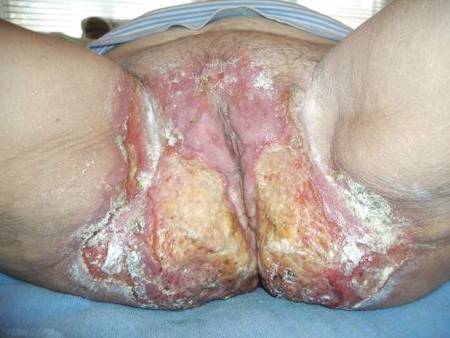
After 6 weeks of treatment, there still were a few small lesions in the intergluteal cleft, which were treated with botulinum toxin type A (100 U diluted with 5 cc of bacteriostatic saline per cm2). After 3 separate injections with botulinum toxin type A and oral tacrolimus for 9 months, complete resolution of the lesions was obtained (Figure 5). After more than 6 months of remission, oral tacrolimus slowly was decreased by 1 mg every 6 weeks until treatment was stopped completely. She has remained in remission without oral treatment. After tapering the tacrolimus, botulinum toxin A injections were continued every 6 months for maintenance with the use of topical tacrolimus 3 to 4 times weekly (Figure 6).
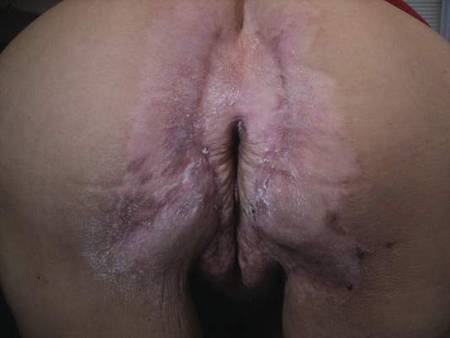
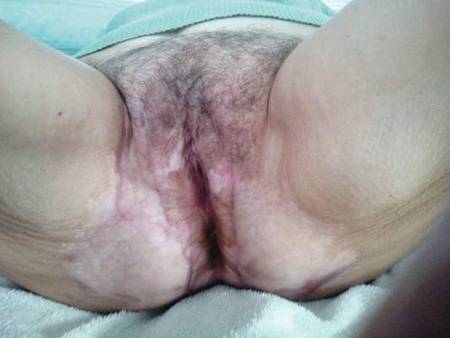
The inheritance of Hailey-Hailey disease is autosomal dominant with incomplete penetrance, although spontaneous mutations are implicated in up to 30% of patients.1,2 The pathogenesis of Hailey-Hailey disease involves a mutation in the ATPase, Ca++ transporting, type 2C, member 1 gene (ATP2C1), which encodes for the hSPCA1 protein.1-2 The malfunction of this ATPase leads to inadequacy of the keratinocyte adhesive barrier resulting in acantholysis and intraepidermal vesicle formation. Macerated plaques also may be found instead of intact vesicles.3
Initial presentation of lesions in Hailey-Hailey disease typically occurs in the third or fourth decades of life but may present at any age.4 Areas exposed to increased amounts of friction (eg, axillae, groin, neck, perineum) commonly are involved.5 Lesions may occur in a relapsing and remitting course and usually are more prominent in summer months because they are exacerbated by sunburn and frictional trauma. Secondary bacterial or fungal infections are common and antibiotics and antifungals often are necessary to prevent progression of the lesions.5,6
Various treatment regimens for Hailey-Hailey disease include topical and oral corticosteroids and antibiotics.5 Topical and oral retinoids, calcitriol, topical tacrolimus, cyclosporin, methotrexate, and even botulinum toxin A have been reported to be effective for refractory cases.7-12 Our case describes a novel regimen of oral tacrolimus in conjunction with botulinum toxin A used in the successful treatment of recalcitrant Hailey-Hailey disease.
Tacrolimus binds to the immunophilin FK506 binding protein, which inhibits calcineurin. Calcineurin, a protein phosphatase, is necessary for T-cell activation through the nuclear factor of activated T cells.This inhibition of calcineurin blocks the expression of several cytokines.13 The efficacy of oral tacrolimus demonstrates that cellular immunity could play a role in the pathogenic mechanism of Hailey-Hailey disease.
Contraindications to oral tacrolimus therapy include renal or hepatic impairment, breast-feeding, pregnancy, and certain neoplastic diseases. There also is an increased risk of patients developing malignancies such as lymphoma or skin cancer due to immunosuppression. Use of oral tacrolimus also requires routine laboratory monitoring of renal and hepatic function, potassium, and blood glucose levels.13
Botulinum toxin A injections augmented the therapeutic approach in our patient possibly by controlling secretions of sweat and mucous, which may cause maceration and lead to exacerbation of Hailey-Hailey disease. Control of secretions may help in creating an environment that is less prone to exacerbation of lesions and secondary infection.14 The combination of oral tacrolimus and botulinum toxin A injections provided a safe therapeutic option for recalcitrant Hailey-Hailey disease in our patient.
- Fairclough RJ, Dode L, Vanoevelen J, et al. Effect of Hailey-Hailey Disease mutations on the function of a new variant of human secretory pathway Ca2+/Mn2+-ATPase (hSPCA1). J Biol Chem. 2003; 278:24721-24730.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Warycha M, Patel R, Meehan S, et al. Familial benign chronic pemphigus (Hailey-Hailey disease). Dermatol Online J. 2009;15:15.
- Tchernev GJ, Cardosa C. Familial benign chronic pemphigus (Hailey-Hailey Disease): use of topical immunomodulators as a modern treatment option. Rev Med Chil. 2011;139:633-637.
- Hunt R, O’Reilly K, Ralston J, et al. Familial benign chronic pemphigus (Hailey-Hailey disease). Dermatol Online J. 2010;16:14.
- Berger EM, Galadari HI, Gottlieb AB. Successful treatment of Hailey-Hailey with acitretin. J Drugs Dermatol. 2007;6:734-736.
- Rabeni EJ, Cunningham NM. Effective treatment of Hailey-Hailey disease with topical tacrolimus. J Am Acad Dermatol. 2002;47:797-798.
- Sand C, Thomsen HK. Topical tacrolimus ointment is an effective therapy for Hailey-Hailey disease. Arch Dermatol. 2003;139:1401-1402.
- Bianchi L, Chimenti MS, Giunta A. Treatment of Hailey-Hailey with topical calcitriol. J Am Acad of Dermatol. 2004;51:475-476.
- Berth-Jones J, Smith SG, Graham-Brown RA. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Vilarinho CF, Ventura F, Brito C. Methotrexate for refractory Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2010;24:106.
- Koeyers WJ, Van Der Geer S, Krekels G. Botulinum toxin type A as an adjuvant treatment modality for extensive Hailey-Hailey disease. J Dermatolog Treatment. 2008;19:251-254.
- Katzung BG, Masters SB, Trevor AJ, eds. Basic and Clinical Pharmacology. 11th ed. New York, NY: McGraw-Hill; 2009.14. Lapiere JC, Hirsh A, Gordon KB, et al. Botulinum toxin type A for the treatment of axillary Hailey-Hailey disease. Dermatol Surg. 2000;26:371-374.
To the Editor:
Hailey-Hailey disease, also known as familial benign pemphigus, is a chronic blistering skin disorder that typically presents as a recurrent vesicular or bullous dermatitis found predominantly in the intertriginous regions of the body. Because current treatment regimens for Hailey-Hailey disease are fairly limited, novel treatments may be explored in intractable cases.
A 71-year-old woman presented with well-demarcated erythematous plaques with erosions and white verrucous regions in the perivulvar, vaginal, and perianal areas of 1 month’s duration (Figure 1). The lesions were excruciatingly pruritic and excoriated. The patient reported no personal or family history of similar lesions.
Histopathologic examination of multiple biopsies from the periphery of the plaques showed acantholysis of the epidermis and surface necrosis with negative direct immunofluorescence. A diagnosis of Hailey-Hailey disease was made. Over several months following the initial presentation, the patient was treated with regimens of corticosteroids, antibiotics, antifungals, acitretin, and topical tacrolimus (which showed minimal response), but the condition continued to progress and thus warranted a more aggressive approach. After a 4-week course of oral cyclosporine 1.25 mg/kg twice daily, some healthy granulation tissue had formed, but new erosions continued to develop on the vulva, labia, and intergluteal cleft (Figure 2). Subsequently, a 2-month course of methotrexate yielded similar results with minimal healing and new necrotic areas (Figure 3).
Following methotrexate therapy, treatment with several other oral agents was considered such as azathioprine, mycophenolate mofetil, and oral tacrolimus. Since the patient had previously shown minimal response to topical tacrolimus, a course of oral treatment (0.05 mg/kg twice daily) was initiated. At 4 weeks’ follow-up, extensive healing of the lesions was noted (Figure 4) and the patient reported that the painful pruritus had improved.
After 6 weeks of treatment, there still were a few small lesions in the intergluteal cleft, which were treated with botulinum toxin type A (100 U diluted with 5 cc of bacteriostatic saline per cm2). After 3 separate injections with botulinum toxin type A and oral tacrolimus for 9 months, complete resolution of the lesions was obtained (Figure 5). After more than 6 months of remission, oral tacrolimus slowly was decreased by 1 mg every 6 weeks until treatment was stopped completely. She has remained in remission without oral treatment. After tapering the tacrolimus, botulinum toxin A injections were continued every 6 months for maintenance with the use of topical tacrolimus 3 to 4 times weekly (Figure 6).
The inheritance of Hailey-Hailey disease is autosomal dominant with incomplete penetrance, although spontaneous mutations are implicated in up to 30% of patients.1,2 The pathogenesis of Hailey-Hailey disease involves a mutation in the ATPase, Ca++ transporting, type 2C, member 1 gene (ATP2C1), which encodes for the hSPCA1 protein.1-2 The malfunction of this ATPase leads to inadequacy of the keratinocyte adhesive barrier resulting in acantholysis and intraepidermal vesicle formation. Macerated plaques also may be found instead of intact vesicles.3
Initial presentation of lesions in Hailey-Hailey disease typically occurs in the third or fourth decades of life but may present at any age.4 Areas exposed to increased amounts of friction (eg, axillae, groin, neck, perineum) commonly are involved.5 Lesions may occur in a relapsing and remitting course and usually are more prominent in summer months because they are exacerbated by sunburn and frictional trauma. Secondary bacterial or fungal infections are common and antibiotics and antifungals often are necessary to prevent progression of the lesions.5,6
Various treatment regimens for Hailey-Hailey disease include topical and oral corticosteroids and antibiotics.5 Topical and oral retinoids, calcitriol, topical tacrolimus, cyclosporin, methotrexate, and even botulinum toxin A have been reported to be effective for refractory cases.7-12 Our case describes a novel regimen of oral tacrolimus in conjunction with botulinum toxin A used in the successful treatment of recalcitrant Hailey-Hailey disease.
Tacrolimus binds to the immunophilin FK506 binding protein, which inhibits calcineurin. Calcineurin, a protein phosphatase, is necessary for T-cell activation through the nuclear factor of activated T cells.This inhibition of calcineurin blocks the expression of several cytokines.13 The efficacy of oral tacrolimus demonstrates that cellular immunity could play a role in the pathogenic mechanism of Hailey-Hailey disease.
Contraindications to oral tacrolimus therapy include renal or hepatic impairment, breast-feeding, pregnancy, and certain neoplastic diseases. There also is an increased risk of patients developing malignancies such as lymphoma or skin cancer due to immunosuppression. Use of oral tacrolimus also requires routine laboratory monitoring of renal and hepatic function, potassium, and blood glucose levels.13
Botulinum toxin A injections augmented the therapeutic approach in our patient possibly by controlling secretions of sweat and mucous, which may cause maceration and lead to exacerbation of Hailey-Hailey disease. Control of secretions may help in creating an environment that is less prone to exacerbation of lesions and secondary infection.14 The combination of oral tacrolimus and botulinum toxin A injections provided a safe therapeutic option for recalcitrant Hailey-Hailey disease in our patient.
To the Editor:
Hailey-Hailey disease, also known as familial benign pemphigus, is a chronic blistering skin disorder that typically presents as a recurrent vesicular or bullous dermatitis found predominantly in the intertriginous regions of the body. Because current treatment regimens for Hailey-Hailey disease are fairly limited, novel treatments may be explored in intractable cases.
A 71-year-old woman presented with well-demarcated erythematous plaques with erosions and white verrucous regions in the perivulvar, vaginal, and perianal areas of 1 month’s duration (Figure 1). The lesions were excruciatingly pruritic and excoriated. The patient reported no personal or family history of similar lesions.
Histopathologic examination of multiple biopsies from the periphery of the plaques showed acantholysis of the epidermis and surface necrosis with negative direct immunofluorescence. A diagnosis of Hailey-Hailey disease was made. Over several months following the initial presentation, the patient was treated with regimens of corticosteroids, antibiotics, antifungals, acitretin, and topical tacrolimus (which showed minimal response), but the condition continued to progress and thus warranted a more aggressive approach. After a 4-week course of oral cyclosporine 1.25 mg/kg twice daily, some healthy granulation tissue had formed, but new erosions continued to develop on the vulva, labia, and intergluteal cleft (Figure 2). Subsequently, a 2-month course of methotrexate yielded similar results with minimal healing and new necrotic areas (Figure 3).
Following methotrexate therapy, treatment with several other oral agents was considered such as azathioprine, mycophenolate mofetil, and oral tacrolimus. Since the patient had previously shown minimal response to topical tacrolimus, a course of oral treatment (0.05 mg/kg twice daily) was initiated. At 4 weeks’ follow-up, extensive healing of the lesions was noted (Figure 4) and the patient reported that the painful pruritus had improved.
After 6 weeks of treatment, there still were a few small lesions in the intergluteal cleft, which were treated with botulinum toxin type A (100 U diluted with 5 cc of bacteriostatic saline per cm2). After 3 separate injections with botulinum toxin type A and oral tacrolimus for 9 months, complete resolution of the lesions was obtained (Figure 5). After more than 6 months of remission, oral tacrolimus slowly was decreased by 1 mg every 6 weeks until treatment was stopped completely. She has remained in remission without oral treatment. After tapering the tacrolimus, botulinum toxin A injections were continued every 6 months for maintenance with the use of topical tacrolimus 3 to 4 times weekly (Figure 6).
The inheritance of Hailey-Hailey disease is autosomal dominant with incomplete penetrance, although spontaneous mutations are implicated in up to 30% of patients.1,2 The pathogenesis of Hailey-Hailey disease involves a mutation in the ATPase, Ca++ transporting, type 2C, member 1 gene (ATP2C1), which encodes for the hSPCA1 protein.1-2 The malfunction of this ATPase leads to inadequacy of the keratinocyte adhesive barrier resulting in acantholysis and intraepidermal vesicle formation. Macerated plaques also may be found instead of intact vesicles.3
Initial presentation of lesions in Hailey-Hailey disease typically occurs in the third or fourth decades of life but may present at any age.4 Areas exposed to increased amounts of friction (eg, axillae, groin, neck, perineum) commonly are involved.5 Lesions may occur in a relapsing and remitting course and usually are more prominent in summer months because they are exacerbated by sunburn and frictional trauma. Secondary bacterial or fungal infections are common and antibiotics and antifungals often are necessary to prevent progression of the lesions.5,6
Various treatment regimens for Hailey-Hailey disease include topical and oral corticosteroids and antibiotics.5 Topical and oral retinoids, calcitriol, topical tacrolimus, cyclosporin, methotrexate, and even botulinum toxin A have been reported to be effective for refractory cases.7-12 Our case describes a novel regimen of oral tacrolimus in conjunction with botulinum toxin A used in the successful treatment of recalcitrant Hailey-Hailey disease.
Tacrolimus binds to the immunophilin FK506 binding protein, which inhibits calcineurin. Calcineurin, a protein phosphatase, is necessary for T-cell activation through the nuclear factor of activated T cells.This inhibition of calcineurin blocks the expression of several cytokines.13 The efficacy of oral tacrolimus demonstrates that cellular immunity could play a role in the pathogenic mechanism of Hailey-Hailey disease.
Contraindications to oral tacrolimus therapy include renal or hepatic impairment, breast-feeding, pregnancy, and certain neoplastic diseases. There also is an increased risk of patients developing malignancies such as lymphoma or skin cancer due to immunosuppression. Use of oral tacrolimus also requires routine laboratory monitoring of renal and hepatic function, potassium, and blood glucose levels.13
Botulinum toxin A injections augmented the therapeutic approach in our patient possibly by controlling secretions of sweat and mucous, which may cause maceration and lead to exacerbation of Hailey-Hailey disease. Control of secretions may help in creating an environment that is less prone to exacerbation of lesions and secondary infection.14 The combination of oral tacrolimus and botulinum toxin A injections provided a safe therapeutic option for recalcitrant Hailey-Hailey disease in our patient.
- Fairclough RJ, Dode L, Vanoevelen J, et al. Effect of Hailey-Hailey Disease mutations on the function of a new variant of human secretory pathway Ca2+/Mn2+-ATPase (hSPCA1). J Biol Chem. 2003; 278:24721-24730.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Warycha M, Patel R, Meehan S, et al. Familial benign chronic pemphigus (Hailey-Hailey disease). Dermatol Online J. 2009;15:15.
- Tchernev GJ, Cardosa C. Familial benign chronic pemphigus (Hailey-Hailey Disease): use of topical immunomodulators as a modern treatment option. Rev Med Chil. 2011;139:633-637.
- Hunt R, O’Reilly K, Ralston J, et al. Familial benign chronic pemphigus (Hailey-Hailey disease). Dermatol Online J. 2010;16:14.
- Berger EM, Galadari HI, Gottlieb AB. Successful treatment of Hailey-Hailey with acitretin. J Drugs Dermatol. 2007;6:734-736.
- Rabeni EJ, Cunningham NM. Effective treatment of Hailey-Hailey disease with topical tacrolimus. J Am Acad Dermatol. 2002;47:797-798.
- Sand C, Thomsen HK. Topical tacrolimus ointment is an effective therapy for Hailey-Hailey disease. Arch Dermatol. 2003;139:1401-1402.
- Bianchi L, Chimenti MS, Giunta A. Treatment of Hailey-Hailey with topical calcitriol. J Am Acad of Dermatol. 2004;51:475-476.
- Berth-Jones J, Smith SG, Graham-Brown RA. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Vilarinho CF, Ventura F, Brito C. Methotrexate for refractory Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2010;24:106.
- Koeyers WJ, Van Der Geer S, Krekels G. Botulinum toxin type A as an adjuvant treatment modality for extensive Hailey-Hailey disease. J Dermatolog Treatment. 2008;19:251-254.
- Katzung BG, Masters SB, Trevor AJ, eds. Basic and Clinical Pharmacology. 11th ed. New York, NY: McGraw-Hill; 2009.14. Lapiere JC, Hirsh A, Gordon KB, et al. Botulinum toxin type A for the treatment of axillary Hailey-Hailey disease. Dermatol Surg. 2000;26:371-374.
- Fairclough RJ, Dode L, Vanoevelen J, et al. Effect of Hailey-Hailey Disease mutations on the function of a new variant of human secretory pathway Ca2+/Mn2+-ATPase (hSPCA1). J Biol Chem. 2003; 278:24721-24730.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Warycha M, Patel R, Meehan S, et al. Familial benign chronic pemphigus (Hailey-Hailey disease). Dermatol Online J. 2009;15:15.
- Tchernev GJ, Cardosa C. Familial benign chronic pemphigus (Hailey-Hailey Disease): use of topical immunomodulators as a modern treatment option. Rev Med Chil. 2011;139:633-637.
- Hunt R, O’Reilly K, Ralston J, et al. Familial benign chronic pemphigus (Hailey-Hailey disease). Dermatol Online J. 2010;16:14.
- Berger EM, Galadari HI, Gottlieb AB. Successful treatment of Hailey-Hailey with acitretin. J Drugs Dermatol. 2007;6:734-736.
- Rabeni EJ, Cunningham NM. Effective treatment of Hailey-Hailey disease with topical tacrolimus. J Am Acad Dermatol. 2002;47:797-798.
- Sand C, Thomsen HK. Topical tacrolimus ointment is an effective therapy for Hailey-Hailey disease. Arch Dermatol. 2003;139:1401-1402.
- Bianchi L, Chimenti MS, Giunta A. Treatment of Hailey-Hailey with topical calcitriol. J Am Acad of Dermatol. 2004;51:475-476.
- Berth-Jones J, Smith SG, Graham-Brown RA. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Vilarinho CF, Ventura F, Brito C. Methotrexate for refractory Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2010;24:106.
- Koeyers WJ, Van Der Geer S, Krekels G. Botulinum toxin type A as an adjuvant treatment modality for extensive Hailey-Hailey disease. J Dermatolog Treatment. 2008;19:251-254.
- Katzung BG, Masters SB, Trevor AJ, eds. Basic and Clinical Pharmacology. 11th ed. New York, NY: McGraw-Hill; 2009.14. Lapiere JC, Hirsh A, Gordon KB, et al. Botulinum toxin type A for the treatment of axillary Hailey-Hailey disease. Dermatol Surg. 2000;26:371-374.
Histologic Correlation of Dermoscopy Findings in a Sebaceous Nevus
To the Editor:
Sebaceous nevus (SN) is a relatively common hamartoma that presents most often as a single congenital hairless plaque on the scalp. After puberty, histologic features characteristically include papillomatous hyperplasia of the epidermis, a large number of mature or nearly mature sebaceous glands, and a lack of terminally differentiated hair follicles; however, histologic findings can be misleading during childhood when sebaceous glands are still underdeveloped. Bright yellow dots, which are thought to indicate the presence of sebaceous glands, may be seen on dermoscopy and can be useful in differentiating SN from aplasia cutis congenita in newborns.
We report a case of an SN in an 18-year-old woman and discuss how the histology findings correlated with features seen on dermoscopy.
An 18-year-old woman presented to our dermatology clinic with an asymptomatic, hairless plaque on the right parietal scalp that had been present since birth. The patient noted that the plaque had recently become larger in size. On physical examination, an 8×3-cm plaque with a smooth, flesh-colored surface was noted with central comedolike structures and an erythematous, verrucous periphery (Figure 1).
Dermoscopy (handheld dermoscope using polarized light) revealed 3 distinct types of round structures within the lesion: (1) comedolike openings (similar to those seen in seborrheic keratosis) that appeared as brownish-yellow, sharply circumscribed structures; (2) milialike cysts (also found in acanthotic seborrheic keratosis), which appeared as bright yellow structures; and (3) multiple whitish structures that were irregular in shape and size and covered the surface of the lesion where there were no other dermoscopic findings (Figure 2). The affected skin was pale to red in color and the verrucous aspect of the surface was better visualized at the edge of the lesion.
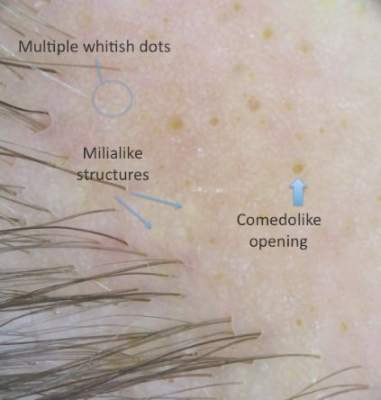
Two 4-mm punch biopsies were performed following dermoscopy: one for horizontal sectioning and one for vertical sectioning. Histologic analysis showed an acanthotic epidermis with an anastomosing network of elongated rete ridges in the superficial dermis. Numerous hyperplasic sebaceous glands were found in the mid dermis, with some also located above this level. Immature hair follicles were present and sebaceous gland ducts communicated directly with the epidermis through dilated hyperkeratinized pathways. Eccrine glands were normal, but no apocrine glands were present. A lymphocytic infiltrate was noted around the sebaceous glands and immature hair follicles and also around dilated capillaries in the superficial dermis. Moderate spongiosis and lymphocytic exocytosis were noted in the glandular epithelium and in the basal layer of the hair follicles and the epidermis. Superficial slides of horizontal sections of the biopsy specimen showed a correlation between the histology findings and dermoscopy images: multiple normal-appearing papilla surrounded by a network of anastomosing rete ridges correlated with multiple whitish structures, keratotic cysts with compact keratin corresponded to bright yellow dots, and larger conglomerates of loose lamelar keratin correlated with comedolike openings. Due to the presence of eczematous changes (eg, epithelial spongiosis, inflammatory cells) observed on histology, a diagnosis of an irritated sebaceous nevus was made, which explained the recent enlargement of the congenital lesion.
Sebaceous nevus is a benign, epidermal appendageal tumor with differentiation towards sebaceous glands that is composed of mature or nearly mature skin structures. Histologically, it is classified as a hamartoma.1 It commonly arises on the scalp as a yellowish or flesh-colored, hairless plaque of variable size. At birth, its surface is smooth and the differential diagnoses include aplasia cutis congenita, congenital triangular alopecia, and alopecia areata.2 As the patient ages, hormones stimulate the proliferation of sebaceous glands and the epidermis, and the lesion gradually acquires a verrucous, waxy surface.3 Benign appendageal tumors often develop inside SN. Basal cell epitheliomas are rarely found.4 Surgical excision is recommended for aesthetic purposes or to prevent the development of tumors.
Histology also varies with the patient’s age and can be misleading in childhood because the sebaceous glands are underdeveloped.5,6 After adrenarche, histology becomes more diagnostic, showing a dermis almost completely filled with sebaceous glands with varying degrees of maturity.2 The presence of incompletely differentiated follicles without hair shafts can be found in newborns and children and may be helpful for the correct histological diagnosis before puberty.1,5 The epidermis presents no abnormalities at birth but develops acanthosis and papillomatosis as the patient ages. Ectopic dilated apocrine glands sometimes can be found deeper in the dermis in the late stage of the lesion.5
In a report by Neri et al,7 multiple bright yellow dots were noted on dermoscopy in 2 children with SN. The investigators concluded that this characteristic feature, which was thought to represent the sebaceous glands, can be useful in differentiating SN from aplasia cutis congenita in early infancy, but no histologic analyses were performed.7 In our patient, we identified 3 different dermoscopic features that correlated with histologic findings. Comedolike openings correlated with the accumulated keratin (ie, keratotic plugs) inside dilated sebaceous gland ducts directly connected to the epidermis. The brownish-yellow color of these openings observed on dermoscopy may be due to the oxidation of kerat-inous material, such as those in seborrheic keratosis lesions (Figure 3). We also noted bright yellow dots similar to those reported by Neri et al7; however, histologic analysis in our patient showed these dots more closely correlated with keratotic cysts similar to milialike structures seen in acanthotic seborrheic keratosis. The material remained lightly colored because no oxidation process had occurred (Figure 4). The third structure found on dermoscopy in our patient was multiple whitish structures that were irregular in shape and size. According to our comparison of superficial horizontal histology slides with dermoscopy images, we hypothesized this finding was the result of epidermal papillomatosis over a dermis filled with enlarged sebaceous glands (Figure 5). This finding was likely absent in the cases previously reported by Neri et al7 because epidermal and glandular changes occur later in the evolution of SN and the patients in these cases were younger than 4 months old.
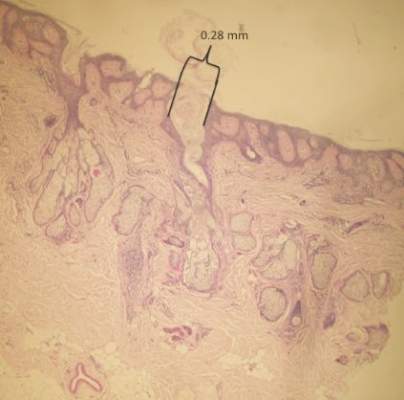
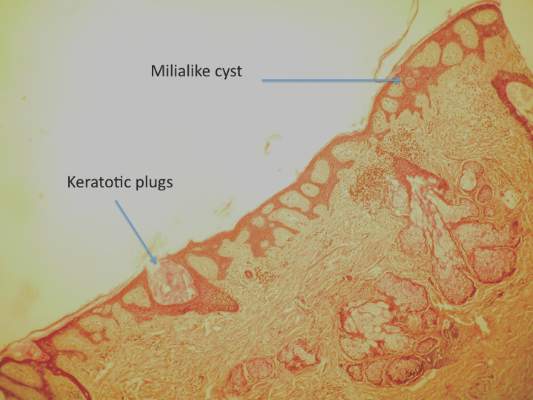
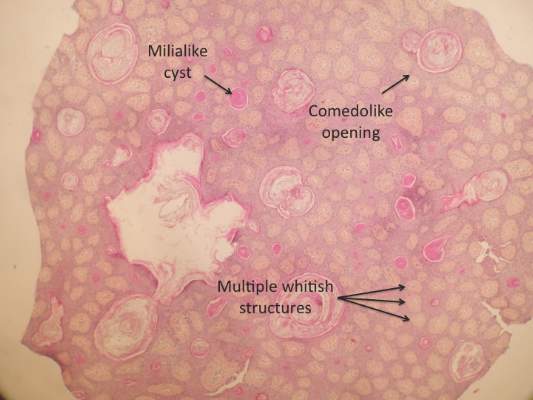
Our correlation of dermoscopic features with histology findings in an 18-year-old woman with an irritated SN highlights the need for more studies needed in order to establish the prevalence of certain dermoscopic findings in this setting, particularly considering the important morphological changes that occur in these lesions as patients age as well as the histological variation among different hamartomas. Over the last decade, dermoscopy has proven to be a useful tool in the diagnosis of various hair and scalp diseases.8 Histologic correlation of dermoscopy findings is essential for more precise understanding of this new imaging technique and should be conducted whenever possible.
- Lever WF, Schaumburg-Lever G. Tumors of the epidermal appendages. In: Lever WF, Schaumburg-Lever G, eds. Histopathology of the Skin. 5th ed. Philadelphia, PA: Lippincott Co; 1975:498-502.
- Civatte J. Tumeurs du cuir chevelu. In: Bouhanna P, Reygagne P, eds. Pathologie du Cheveu et du Cuir Cheveulu. Paris, France: Masson Co; 1999:208-209.
- Gruβendorf-Conen E-I. Adnexal cysts and tumors of the scalp. In: Orfanos CE, Happle R, eds. Hair and Hair Diseases. 1st ed. Berlin Germany: Springer-Verlag Berlin Heidelberg Co; 1990:710-711.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceous: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268.
- Camacho F. Tumeurs du cuir chevelu. In: Camacho F, Montagna W, eds. Trichologie: Maladie du Follicule Pilosébacé. Madrid, Spain: Grupo Aula Medica; 1997:515-516.
- Wechsler J. Hamartome sebace. In: Wechsler J, Fraitag S, Moulonguet I, eds. Pathologie Cutanee Tumorale. Montpelier, France: Sauramps Medical Co; 2009:100-102.
- Neri I, Savoia F, Giacomini F, et al. Usefulness of dermatoscopy for the early diagnosis of sebaceous naevus and differentiation from aplasia cutis congenita [published online ahead of print May 5, 2009]. Clin Exp Dermatol. 2009;34:e50-e52.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
To the Editor:
Sebaceous nevus (SN) is a relatively common hamartoma that presents most often as a single congenital hairless plaque on the scalp. After puberty, histologic features characteristically include papillomatous hyperplasia of the epidermis, a large number of mature or nearly mature sebaceous glands, and a lack of terminally differentiated hair follicles; however, histologic findings can be misleading during childhood when sebaceous glands are still underdeveloped. Bright yellow dots, which are thought to indicate the presence of sebaceous glands, may be seen on dermoscopy and can be useful in differentiating SN from aplasia cutis congenita in newborns.
We report a case of an SN in an 18-year-old woman and discuss how the histology findings correlated with features seen on dermoscopy.
An 18-year-old woman presented to our dermatology clinic with an asymptomatic, hairless plaque on the right parietal scalp that had been present since birth. The patient noted that the plaque had recently become larger in size. On physical examination, an 8×3-cm plaque with a smooth, flesh-colored surface was noted with central comedolike structures and an erythematous, verrucous periphery (Figure 1).
Dermoscopy (handheld dermoscope using polarized light) revealed 3 distinct types of round structures within the lesion: (1) comedolike openings (similar to those seen in seborrheic keratosis) that appeared as brownish-yellow, sharply circumscribed structures; (2) milialike cysts (also found in acanthotic seborrheic keratosis), which appeared as bright yellow structures; and (3) multiple whitish structures that were irregular in shape and size and covered the surface of the lesion where there were no other dermoscopic findings (Figure 2). The affected skin was pale to red in color and the verrucous aspect of the surface was better visualized at the edge of the lesion.
Two 4-mm punch biopsies were performed following dermoscopy: one for horizontal sectioning and one for vertical sectioning. Histologic analysis showed an acanthotic epidermis with an anastomosing network of elongated rete ridges in the superficial dermis. Numerous hyperplasic sebaceous glands were found in the mid dermis, with some also located above this level. Immature hair follicles were present and sebaceous gland ducts communicated directly with the epidermis through dilated hyperkeratinized pathways. Eccrine glands were normal, but no apocrine glands were present. A lymphocytic infiltrate was noted around the sebaceous glands and immature hair follicles and also around dilated capillaries in the superficial dermis. Moderate spongiosis and lymphocytic exocytosis were noted in the glandular epithelium and in the basal layer of the hair follicles and the epidermis. Superficial slides of horizontal sections of the biopsy specimen showed a correlation between the histology findings and dermoscopy images: multiple normal-appearing papilla surrounded by a network of anastomosing rete ridges correlated with multiple whitish structures, keratotic cysts with compact keratin corresponded to bright yellow dots, and larger conglomerates of loose lamelar keratin correlated with comedolike openings. Due to the presence of eczematous changes (eg, epithelial spongiosis, inflammatory cells) observed on histology, a diagnosis of an irritated sebaceous nevus was made, which explained the recent enlargement of the congenital lesion.
Sebaceous nevus is a benign, epidermal appendageal tumor with differentiation towards sebaceous glands that is composed of mature or nearly mature skin structures. Histologically, it is classified as a hamartoma.1 It commonly arises on the scalp as a yellowish or flesh-colored, hairless plaque of variable size. At birth, its surface is smooth and the differential diagnoses include aplasia cutis congenita, congenital triangular alopecia, and alopecia areata.2 As the patient ages, hormones stimulate the proliferation of sebaceous glands and the epidermis, and the lesion gradually acquires a verrucous, waxy surface.3 Benign appendageal tumors often develop inside SN. Basal cell epitheliomas are rarely found.4 Surgical excision is recommended for aesthetic purposes or to prevent the development of tumors.
Histology also varies with the patient’s age and can be misleading in childhood because the sebaceous glands are underdeveloped.5,6 After adrenarche, histology becomes more diagnostic, showing a dermis almost completely filled with sebaceous glands with varying degrees of maturity.2 The presence of incompletely differentiated follicles without hair shafts can be found in newborns and children and may be helpful for the correct histological diagnosis before puberty.1,5 The epidermis presents no abnormalities at birth but develops acanthosis and papillomatosis as the patient ages. Ectopic dilated apocrine glands sometimes can be found deeper in the dermis in the late stage of the lesion.5
In a report by Neri et al,7 multiple bright yellow dots were noted on dermoscopy in 2 children with SN. The investigators concluded that this characteristic feature, which was thought to represent the sebaceous glands, can be useful in differentiating SN from aplasia cutis congenita in early infancy, but no histologic analyses were performed.7 In our patient, we identified 3 different dermoscopic features that correlated with histologic findings. Comedolike openings correlated with the accumulated keratin (ie, keratotic plugs) inside dilated sebaceous gland ducts directly connected to the epidermis. The brownish-yellow color of these openings observed on dermoscopy may be due to the oxidation of kerat-inous material, such as those in seborrheic keratosis lesions (Figure 3). We also noted bright yellow dots similar to those reported by Neri et al7; however, histologic analysis in our patient showed these dots more closely correlated with keratotic cysts similar to milialike structures seen in acanthotic seborrheic keratosis. The material remained lightly colored because no oxidation process had occurred (Figure 4). The third structure found on dermoscopy in our patient was multiple whitish structures that were irregular in shape and size. According to our comparison of superficial horizontal histology slides with dermoscopy images, we hypothesized this finding was the result of epidermal papillomatosis over a dermis filled with enlarged sebaceous glands (Figure 5). This finding was likely absent in the cases previously reported by Neri et al7 because epidermal and glandular changes occur later in the evolution of SN and the patients in these cases were younger than 4 months old.
Our correlation of dermoscopic features with histology findings in an 18-year-old woman with an irritated SN highlights the need for more studies needed in order to establish the prevalence of certain dermoscopic findings in this setting, particularly considering the important morphological changes that occur in these lesions as patients age as well as the histological variation among different hamartomas. Over the last decade, dermoscopy has proven to be a useful tool in the diagnosis of various hair and scalp diseases.8 Histologic correlation of dermoscopy findings is essential for more precise understanding of this new imaging technique and should be conducted whenever possible.
To the Editor:
Sebaceous nevus (SN) is a relatively common hamartoma that presents most often as a single congenital hairless plaque on the scalp. After puberty, histologic features characteristically include papillomatous hyperplasia of the epidermis, a large number of mature or nearly mature sebaceous glands, and a lack of terminally differentiated hair follicles; however, histologic findings can be misleading during childhood when sebaceous glands are still underdeveloped. Bright yellow dots, which are thought to indicate the presence of sebaceous glands, may be seen on dermoscopy and can be useful in differentiating SN from aplasia cutis congenita in newborns.
We report a case of an SN in an 18-year-old woman and discuss how the histology findings correlated with features seen on dermoscopy.
An 18-year-old woman presented to our dermatology clinic with an asymptomatic, hairless plaque on the right parietal scalp that had been present since birth. The patient noted that the plaque had recently become larger in size. On physical examination, an 8×3-cm plaque with a smooth, flesh-colored surface was noted with central comedolike structures and an erythematous, verrucous periphery (Figure 1).
Dermoscopy (handheld dermoscope using polarized light) revealed 3 distinct types of round structures within the lesion: (1) comedolike openings (similar to those seen in seborrheic keratosis) that appeared as brownish-yellow, sharply circumscribed structures; (2) milialike cysts (also found in acanthotic seborrheic keratosis), which appeared as bright yellow structures; and (3) multiple whitish structures that were irregular in shape and size and covered the surface of the lesion where there were no other dermoscopic findings (Figure 2). The affected skin was pale to red in color and the verrucous aspect of the surface was better visualized at the edge of the lesion.
Two 4-mm punch biopsies were performed following dermoscopy: one for horizontal sectioning and one for vertical sectioning. Histologic analysis showed an acanthotic epidermis with an anastomosing network of elongated rete ridges in the superficial dermis. Numerous hyperplasic sebaceous glands were found in the mid dermis, with some also located above this level. Immature hair follicles were present and sebaceous gland ducts communicated directly with the epidermis through dilated hyperkeratinized pathways. Eccrine glands were normal, but no apocrine glands were present. A lymphocytic infiltrate was noted around the sebaceous glands and immature hair follicles and also around dilated capillaries in the superficial dermis. Moderate spongiosis and lymphocytic exocytosis were noted in the glandular epithelium and in the basal layer of the hair follicles and the epidermis. Superficial slides of horizontal sections of the biopsy specimen showed a correlation between the histology findings and dermoscopy images: multiple normal-appearing papilla surrounded by a network of anastomosing rete ridges correlated with multiple whitish structures, keratotic cysts with compact keratin corresponded to bright yellow dots, and larger conglomerates of loose lamelar keratin correlated with comedolike openings. Due to the presence of eczematous changes (eg, epithelial spongiosis, inflammatory cells) observed on histology, a diagnosis of an irritated sebaceous nevus was made, which explained the recent enlargement of the congenital lesion.
Sebaceous nevus is a benign, epidermal appendageal tumor with differentiation towards sebaceous glands that is composed of mature or nearly mature skin structures. Histologically, it is classified as a hamartoma.1 It commonly arises on the scalp as a yellowish or flesh-colored, hairless plaque of variable size. At birth, its surface is smooth and the differential diagnoses include aplasia cutis congenita, congenital triangular alopecia, and alopecia areata.2 As the patient ages, hormones stimulate the proliferation of sebaceous glands and the epidermis, and the lesion gradually acquires a verrucous, waxy surface.3 Benign appendageal tumors often develop inside SN. Basal cell epitheliomas are rarely found.4 Surgical excision is recommended for aesthetic purposes or to prevent the development of tumors.
Histology also varies with the patient’s age and can be misleading in childhood because the sebaceous glands are underdeveloped.5,6 After adrenarche, histology becomes more diagnostic, showing a dermis almost completely filled with sebaceous glands with varying degrees of maturity.2 The presence of incompletely differentiated follicles without hair shafts can be found in newborns and children and may be helpful for the correct histological diagnosis before puberty.1,5 The epidermis presents no abnormalities at birth but develops acanthosis and papillomatosis as the patient ages. Ectopic dilated apocrine glands sometimes can be found deeper in the dermis in the late stage of the lesion.5
In a report by Neri et al,7 multiple bright yellow dots were noted on dermoscopy in 2 children with SN. The investigators concluded that this characteristic feature, which was thought to represent the sebaceous glands, can be useful in differentiating SN from aplasia cutis congenita in early infancy, but no histologic analyses were performed.7 In our patient, we identified 3 different dermoscopic features that correlated with histologic findings. Comedolike openings correlated with the accumulated keratin (ie, keratotic plugs) inside dilated sebaceous gland ducts directly connected to the epidermis. The brownish-yellow color of these openings observed on dermoscopy may be due to the oxidation of kerat-inous material, such as those in seborrheic keratosis lesions (Figure 3). We also noted bright yellow dots similar to those reported by Neri et al7; however, histologic analysis in our patient showed these dots more closely correlated with keratotic cysts similar to milialike structures seen in acanthotic seborrheic keratosis. The material remained lightly colored because no oxidation process had occurred (Figure 4). The third structure found on dermoscopy in our patient was multiple whitish structures that were irregular in shape and size. According to our comparison of superficial horizontal histology slides with dermoscopy images, we hypothesized this finding was the result of epidermal papillomatosis over a dermis filled with enlarged sebaceous glands (Figure 5). This finding was likely absent in the cases previously reported by Neri et al7 because epidermal and glandular changes occur later in the evolution of SN and the patients in these cases were younger than 4 months old.
Our correlation of dermoscopic features with histology findings in an 18-year-old woman with an irritated SN highlights the need for more studies needed in order to establish the prevalence of certain dermoscopic findings in this setting, particularly considering the important morphological changes that occur in these lesions as patients age as well as the histological variation among different hamartomas. Over the last decade, dermoscopy has proven to be a useful tool in the diagnosis of various hair and scalp diseases.8 Histologic correlation of dermoscopy findings is essential for more precise understanding of this new imaging technique and should be conducted whenever possible.
- Lever WF, Schaumburg-Lever G. Tumors of the epidermal appendages. In: Lever WF, Schaumburg-Lever G, eds. Histopathology of the Skin. 5th ed. Philadelphia, PA: Lippincott Co; 1975:498-502.
- Civatte J. Tumeurs du cuir chevelu. In: Bouhanna P, Reygagne P, eds. Pathologie du Cheveu et du Cuir Cheveulu. Paris, France: Masson Co; 1999:208-209.
- Gruβendorf-Conen E-I. Adnexal cysts and tumors of the scalp. In: Orfanos CE, Happle R, eds. Hair and Hair Diseases. 1st ed. Berlin Germany: Springer-Verlag Berlin Heidelberg Co; 1990:710-711.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceous: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268.
- Camacho F. Tumeurs du cuir chevelu. In: Camacho F, Montagna W, eds. Trichologie: Maladie du Follicule Pilosébacé. Madrid, Spain: Grupo Aula Medica; 1997:515-516.
- Wechsler J. Hamartome sebace. In: Wechsler J, Fraitag S, Moulonguet I, eds. Pathologie Cutanee Tumorale. Montpelier, France: Sauramps Medical Co; 2009:100-102.
- Neri I, Savoia F, Giacomini F, et al. Usefulness of dermatoscopy for the early diagnosis of sebaceous naevus and differentiation from aplasia cutis congenita [published online ahead of print May 5, 2009]. Clin Exp Dermatol. 2009;34:e50-e52.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Lever WF, Schaumburg-Lever G. Tumors of the epidermal appendages. In: Lever WF, Schaumburg-Lever G, eds. Histopathology of the Skin. 5th ed. Philadelphia, PA: Lippincott Co; 1975:498-502.
- Civatte J. Tumeurs du cuir chevelu. In: Bouhanna P, Reygagne P, eds. Pathologie du Cheveu et du Cuir Cheveulu. Paris, France: Masson Co; 1999:208-209.
- Gruβendorf-Conen E-I. Adnexal cysts and tumors of the scalp. In: Orfanos CE, Happle R, eds. Hair and Hair Diseases. 1st ed. Berlin Germany: Springer-Verlag Berlin Heidelberg Co; 1990:710-711.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceous: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268.
- Camacho F. Tumeurs du cuir chevelu. In: Camacho F, Montagna W, eds. Trichologie: Maladie du Follicule Pilosébacé. Madrid, Spain: Grupo Aula Medica; 1997:515-516.
- Wechsler J. Hamartome sebace. In: Wechsler J, Fraitag S, Moulonguet I, eds. Pathologie Cutanee Tumorale. Montpelier, France: Sauramps Medical Co; 2009:100-102.
- Neri I, Savoia F, Giacomini F, et al. Usefulness of dermatoscopy for the early diagnosis of sebaceous naevus and differentiation from aplasia cutis congenita [published online ahead of print May 5, 2009]. Clin Exp Dermatol. 2009;34:e50-e52.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
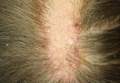
Purpura Fulminans in the Setting of Escherichia coli Septicemia
To the Editor:
Purpura fulminans is a severe and rapidly fatal thrombotic disorder that can occur in association with either hereditary or acquired deficiencies of the natural anticoagulants protein C and protein S.1 It most commonly results from the acute inflammatory response and subsequent disseminated intravascular coagulation (DIC) seen in severe bacterial septicemia. Excessive bleeding, retiform purpura, and skin necrosis may develop as a result of the coagulopathies of typical DIC.1Neisseria meningitidis, Streptococcus, and Staphylococcus frequently are implicated as pathogens, but Escherichia coli–associated purpura fulminans in adults is rare.2,3 We report a case of purpura fulminans in the setting of E coli septicemia.
A 62-year-old woman with a history of end-stage liver disease secondary to alcoholic liver cirrhosis diagnosed 13 years prior complicated by ascites and esophageal varices presented to a primary care clinic for evaluation of a recent-onset nontender lesion on the left buttock. She was hypotensive with a blood pressure of 62/48 mmHg. The patient was prescribed ciprofloxacin 250 mg twice daily and hydrocodone/acetominophen 5 mg/325 mg twice daily as needed for pain management and was discharged. Six hours later, the patient presented to the emergency department with new onset symptoms of confusion and dark-colored spots on the abdomen and lower legs, which her family members noted had developed shortly after the patient took ciprofloxacin. In the emergency department, the patient was noted to be hypotensive and febrile with a severe metabolic acidosis. She was intubated for respiratory failure and received intravenous fluid resuscitation, broad-spectrum antibiotics, and vasopressors. Blood cultures were obtained, and the dermatology department was consulted.
On physical examination, extensive purpuric, reticulated, and stellate plaques with central necrosis and hemorrhagic bullae were noted on the abdomen (Figure, A) and bilateral lower legs (Figure, B) extending onto the thighs. The patient was coagulopathic with persistent sanguineous oozing at intravenous sites and bilateral nares. A small erythematous ulcer with overlying black eschar was noted on the left medial buttock.
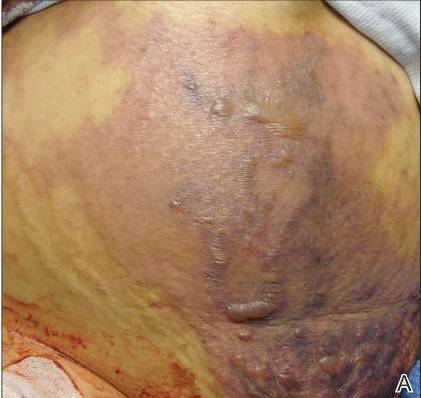
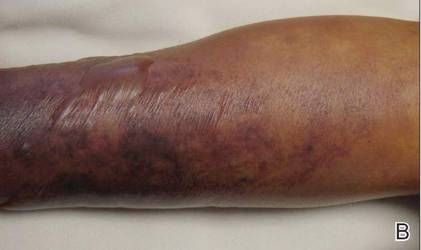
Laboratory test results showed new-onset thrombocytopenia, prolonged prothrombin time/international normalized ratio and partial thromboplastin time, and low fibrinogen levels, which confirmed a diagnosis of acute DIC. Blood cultures were positive for gram-negative rods in 4 out of 4 bottles within 12 hours of being drawn. Further testing identified the microorganism as E coli, and antibiotic susceptibility testing revealed it was sensitive to most antibiotics.
The patient was clinically diagnosed with purpura fulminans secondary to severe E coli septicemia and DIC. This life-threatening disorder is considered a medical emergency with a high mortality rate. Laboratory findings supporting DIC include the presence of schistocytes on a peripheral blood smear, thrombocytopenia, positive plasma protamine paracoagulation test, low fibrinogen levels, and positive fibrin degradation products. Reported cases of purpura fulminans in the setting of E coli septicemia are rare, and meningococcemia is the most common presentation.2,3 Bacterial components (eg, lipopolysaccharides found in the cell walls of gram-negative bacteria) may contribute to the progression of septicemia. Increased levels of endotoxin lipopolysaccharide can lead to septic shock and organ dysfunction.4 However, the release of lipooligosaccharides is associated with the development of meningococcal septicemia, and the lipopolysaccharide levels are directly correlated with prognosis in patients without meningitis.5-7
Human activated protein C concentrate (and its precursor, protein C concentrate) replacement therapy has been shown to improve outcomes in patients with meningococcemia-associated–purpura fulminans and severe sepsis, respectively.8 Heparin may be considered in the treatment of patients with purpura fulminans in addition to the replacement of any missing clotting factors or blood products.9 The international guidelines for the management of severe sepsis and septic shock include early quantitative resuscitation of the patient during the first 6 hours after recognition of sepsis, performing blood cultures before antibiotic therapy, and administering broad-spectrum antimicrobial therapy within 1 hour of recognition of septic shock.10 The elapsed time from triage to the actual administration of appropriate antimicrobials are primary determinants of patient mortality.11 Therefore, physicians must act quickly to stabilize the patient.
Gram-positive bacteria and gram-negative diplococci are common infectious agents implicated in purpura fulminans. Escherichia coli rarely has been identified as the inciting agent for purpura fulminans in adults. The increasing frequency of E coli strains that produce extended-spectrum β-lactamases—enzymes that mediate resistance to extended-spectrum (third generation) cephalosporins (eg, ceftazidime, cefotaxime, ceftriaxone) and monobactams (eg, aztreonam)—complicates matters further when deciding on appropriate antibiotics. Patients who have infections from extended-spectrum β-lactamase strains will require more potent carbapenems (eg, meropenem, imipenem) for treatment of infections. Despite undergoing treatment for septicemia, our patient went into cardiac arrest within 24 hours of presentation to the emergency department and died a few hours later. Physicians should consider E coli as an inciting agent of purpura fulminans and consider appropriate empiric antibiotics with gram-negative coverage to include E coli.
- Madden RM, Gill JC, Marlar RA. Protein C and protein S levels in two patients with acquired purpura fulminans. Br J Haematol. 1990;75:112-117.
- Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001;86:581-586.
- Huemer GM, Bonatti H, Dunst KM. Purpura fulminans due to E. coli septicemia. Wien Klin Wochenschr. 2004;116:82.
- Pugin J. Recognition of bacteria and bacterial products by host immune cells in sepsis. In: Vincent JL, ed. Yearbook of Intensive Care and Emergency Medicine. Berlin, Germany: Springer-Verlag; 1997:11-12.
- Brandtzaeg P, Oktedalen O, Kierulf P, et al. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37-44.
- Brandtzaeg P, Kierulf P, Gaustad P, et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis. 1989;159:195-204.
- Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650-652.
- Hodgson A, Ryan T, Moriarty J, et al. Plasma exchange as a source of protein C for acute onset protein C pathway failure. Br J Haematol. 2002;116:905-908.
- Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60:284-287.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign guidelines committee including the pediatric subgroup. Crit Care Med. 2013;41:580-637.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045-1053.
To the Editor:
Purpura fulminans is a severe and rapidly fatal thrombotic disorder that can occur in association with either hereditary or acquired deficiencies of the natural anticoagulants protein C and protein S.1 It most commonly results from the acute inflammatory response and subsequent disseminated intravascular coagulation (DIC) seen in severe bacterial septicemia. Excessive bleeding, retiform purpura, and skin necrosis may develop as a result of the coagulopathies of typical DIC.1Neisseria meningitidis, Streptococcus, and Staphylococcus frequently are implicated as pathogens, but Escherichia coli–associated purpura fulminans in adults is rare.2,3 We report a case of purpura fulminans in the setting of E coli septicemia.
A 62-year-old woman with a history of end-stage liver disease secondary to alcoholic liver cirrhosis diagnosed 13 years prior complicated by ascites and esophageal varices presented to a primary care clinic for evaluation of a recent-onset nontender lesion on the left buttock. She was hypotensive with a blood pressure of 62/48 mmHg. The patient was prescribed ciprofloxacin 250 mg twice daily and hydrocodone/acetominophen 5 mg/325 mg twice daily as needed for pain management and was discharged. Six hours later, the patient presented to the emergency department with new onset symptoms of confusion and dark-colored spots on the abdomen and lower legs, which her family members noted had developed shortly after the patient took ciprofloxacin. In the emergency department, the patient was noted to be hypotensive and febrile with a severe metabolic acidosis. She was intubated for respiratory failure and received intravenous fluid resuscitation, broad-spectrum antibiotics, and vasopressors. Blood cultures were obtained, and the dermatology department was consulted.
On physical examination, extensive purpuric, reticulated, and stellate plaques with central necrosis and hemorrhagic bullae were noted on the abdomen (Figure, A) and bilateral lower legs (Figure, B) extending onto the thighs. The patient was coagulopathic with persistent sanguineous oozing at intravenous sites and bilateral nares. A small erythematous ulcer with overlying black eschar was noted on the left medial buttock.
Laboratory test results showed new-onset thrombocytopenia, prolonged prothrombin time/international normalized ratio and partial thromboplastin time, and low fibrinogen levels, which confirmed a diagnosis of acute DIC. Blood cultures were positive for gram-negative rods in 4 out of 4 bottles within 12 hours of being drawn. Further testing identified the microorganism as E coli, and antibiotic susceptibility testing revealed it was sensitive to most antibiotics.
The patient was clinically diagnosed with purpura fulminans secondary to severe E coli septicemia and DIC. This life-threatening disorder is considered a medical emergency with a high mortality rate. Laboratory findings supporting DIC include the presence of schistocytes on a peripheral blood smear, thrombocytopenia, positive plasma protamine paracoagulation test, low fibrinogen levels, and positive fibrin degradation products. Reported cases of purpura fulminans in the setting of E coli septicemia are rare, and meningococcemia is the most common presentation.2,3 Bacterial components (eg, lipopolysaccharides found in the cell walls of gram-negative bacteria) may contribute to the progression of septicemia. Increased levels of endotoxin lipopolysaccharide can lead to septic shock and organ dysfunction.4 However, the release of lipooligosaccharides is associated with the development of meningococcal septicemia, and the lipopolysaccharide levels are directly correlated with prognosis in patients without meningitis.5-7
Human activated protein C concentrate (and its precursor, protein C concentrate) replacement therapy has been shown to improve outcomes in patients with meningococcemia-associated–purpura fulminans and severe sepsis, respectively.8 Heparin may be considered in the treatment of patients with purpura fulminans in addition to the replacement of any missing clotting factors or blood products.9 The international guidelines for the management of severe sepsis and septic shock include early quantitative resuscitation of the patient during the first 6 hours after recognition of sepsis, performing blood cultures before antibiotic therapy, and administering broad-spectrum antimicrobial therapy within 1 hour of recognition of septic shock.10 The elapsed time from triage to the actual administration of appropriate antimicrobials are primary determinants of patient mortality.11 Therefore, physicians must act quickly to stabilize the patient.
Gram-positive bacteria and gram-negative diplococci are common infectious agents implicated in purpura fulminans. Escherichia coli rarely has been identified as the inciting agent for purpura fulminans in adults. The increasing frequency of E coli strains that produce extended-spectrum β-lactamases—enzymes that mediate resistance to extended-spectrum (third generation) cephalosporins (eg, ceftazidime, cefotaxime, ceftriaxone) and monobactams (eg, aztreonam)—complicates matters further when deciding on appropriate antibiotics. Patients who have infections from extended-spectrum β-lactamase strains will require more potent carbapenems (eg, meropenem, imipenem) for treatment of infections. Despite undergoing treatment for septicemia, our patient went into cardiac arrest within 24 hours of presentation to the emergency department and died a few hours later. Physicians should consider E coli as an inciting agent of purpura fulminans and consider appropriate empiric antibiotics with gram-negative coverage to include E coli.
To the Editor:
Purpura fulminans is a severe and rapidly fatal thrombotic disorder that can occur in association with either hereditary or acquired deficiencies of the natural anticoagulants protein C and protein S.1 It most commonly results from the acute inflammatory response and subsequent disseminated intravascular coagulation (DIC) seen in severe bacterial septicemia. Excessive bleeding, retiform purpura, and skin necrosis may develop as a result of the coagulopathies of typical DIC.1Neisseria meningitidis, Streptococcus, and Staphylococcus frequently are implicated as pathogens, but Escherichia coli–associated purpura fulminans in adults is rare.2,3 We report a case of purpura fulminans in the setting of E coli septicemia.
A 62-year-old woman with a history of end-stage liver disease secondary to alcoholic liver cirrhosis diagnosed 13 years prior complicated by ascites and esophageal varices presented to a primary care clinic for evaluation of a recent-onset nontender lesion on the left buttock. She was hypotensive with a blood pressure of 62/48 mmHg. The patient was prescribed ciprofloxacin 250 mg twice daily and hydrocodone/acetominophen 5 mg/325 mg twice daily as needed for pain management and was discharged. Six hours later, the patient presented to the emergency department with new onset symptoms of confusion and dark-colored spots on the abdomen and lower legs, which her family members noted had developed shortly after the patient took ciprofloxacin. In the emergency department, the patient was noted to be hypotensive and febrile with a severe metabolic acidosis. She was intubated for respiratory failure and received intravenous fluid resuscitation, broad-spectrum antibiotics, and vasopressors. Blood cultures were obtained, and the dermatology department was consulted.
On physical examination, extensive purpuric, reticulated, and stellate plaques with central necrosis and hemorrhagic bullae were noted on the abdomen (Figure, A) and bilateral lower legs (Figure, B) extending onto the thighs. The patient was coagulopathic with persistent sanguineous oozing at intravenous sites and bilateral nares. A small erythematous ulcer with overlying black eschar was noted on the left medial buttock.
Laboratory test results showed new-onset thrombocytopenia, prolonged prothrombin time/international normalized ratio and partial thromboplastin time, and low fibrinogen levels, which confirmed a diagnosis of acute DIC. Blood cultures were positive for gram-negative rods in 4 out of 4 bottles within 12 hours of being drawn. Further testing identified the microorganism as E coli, and antibiotic susceptibility testing revealed it was sensitive to most antibiotics.
The patient was clinically diagnosed with purpura fulminans secondary to severe E coli septicemia and DIC. This life-threatening disorder is considered a medical emergency with a high mortality rate. Laboratory findings supporting DIC include the presence of schistocytes on a peripheral blood smear, thrombocytopenia, positive plasma protamine paracoagulation test, low fibrinogen levels, and positive fibrin degradation products. Reported cases of purpura fulminans in the setting of E coli septicemia are rare, and meningococcemia is the most common presentation.2,3 Bacterial components (eg, lipopolysaccharides found in the cell walls of gram-negative bacteria) may contribute to the progression of septicemia. Increased levels of endotoxin lipopolysaccharide can lead to septic shock and organ dysfunction.4 However, the release of lipooligosaccharides is associated with the development of meningococcal septicemia, and the lipopolysaccharide levels are directly correlated with prognosis in patients without meningitis.5-7
Human activated protein C concentrate (and its precursor, protein C concentrate) replacement therapy has been shown to improve outcomes in patients with meningococcemia-associated–purpura fulminans and severe sepsis, respectively.8 Heparin may be considered in the treatment of patients with purpura fulminans in addition to the replacement of any missing clotting factors or blood products.9 The international guidelines for the management of severe sepsis and septic shock include early quantitative resuscitation of the patient during the first 6 hours after recognition of sepsis, performing blood cultures before antibiotic therapy, and administering broad-spectrum antimicrobial therapy within 1 hour of recognition of septic shock.10 The elapsed time from triage to the actual administration of appropriate antimicrobials are primary determinants of patient mortality.11 Therefore, physicians must act quickly to stabilize the patient.
Gram-positive bacteria and gram-negative diplococci are common infectious agents implicated in purpura fulminans. Escherichia coli rarely has been identified as the inciting agent for purpura fulminans in adults. The increasing frequency of E coli strains that produce extended-spectrum β-lactamases—enzymes that mediate resistance to extended-spectrum (third generation) cephalosporins (eg, ceftazidime, cefotaxime, ceftriaxone) and monobactams (eg, aztreonam)—complicates matters further when deciding on appropriate antibiotics. Patients who have infections from extended-spectrum β-lactamase strains will require more potent carbapenems (eg, meropenem, imipenem) for treatment of infections. Despite undergoing treatment for septicemia, our patient went into cardiac arrest within 24 hours of presentation to the emergency department and died a few hours later. Physicians should consider E coli as an inciting agent of purpura fulminans and consider appropriate empiric antibiotics with gram-negative coverage to include E coli.
- Madden RM, Gill JC, Marlar RA. Protein C and protein S levels in two patients with acquired purpura fulminans. Br J Haematol. 1990;75:112-117.
- Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001;86:581-586.
- Huemer GM, Bonatti H, Dunst KM. Purpura fulminans due to E. coli septicemia. Wien Klin Wochenschr. 2004;116:82.
- Pugin J. Recognition of bacteria and bacterial products by host immune cells in sepsis. In: Vincent JL, ed. Yearbook of Intensive Care and Emergency Medicine. Berlin, Germany: Springer-Verlag; 1997:11-12.
- Brandtzaeg P, Oktedalen O, Kierulf P, et al. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37-44.
- Brandtzaeg P, Kierulf P, Gaustad P, et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis. 1989;159:195-204.
- Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650-652.
- Hodgson A, Ryan T, Moriarty J, et al. Plasma exchange as a source of protein C for acute onset protein C pathway failure. Br J Haematol. 2002;116:905-908.
- Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60:284-287.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign guidelines committee including the pediatric subgroup. Crit Care Med. 2013;41:580-637.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045-1053.
- Madden RM, Gill JC, Marlar RA. Protein C and protein S levels in two patients with acquired purpura fulminans. Br J Haematol. 1990;75:112-117.
- Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001;86:581-586.
- Huemer GM, Bonatti H, Dunst KM. Purpura fulminans due to E. coli septicemia. Wien Klin Wochenschr. 2004;116:82.
- Pugin J. Recognition of bacteria and bacterial products by host immune cells in sepsis. In: Vincent JL, ed. Yearbook of Intensive Care and Emergency Medicine. Berlin, Germany: Springer-Verlag; 1997:11-12.
- Brandtzaeg P, Oktedalen O, Kierulf P, et al. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37-44.
- Brandtzaeg P, Kierulf P, Gaustad P, et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis. 1989;159:195-204.
- Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650-652.
- Hodgson A, Ryan T, Moriarty J, et al. Plasma exchange as a source of protein C for acute onset protein C pathway failure. Br J Haematol. 2002;116:905-908.
- Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60:284-287.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign guidelines committee including the pediatric subgroup. Crit Care Med. 2013;41:580-637.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045-1053.