User login
Azathioprine Hypersensitivity Presenting as Neutrophilic Dermatosis and Erythema Nodosum
To the Editor:
Azathioprine (AZA) hypersensitivity is an immunologically mediated reaction that presents within 1 to 4 weeks of drug initiation.1 Its cutaneous manifestations include Sweet syndrome, erythema nodosum (EN), and acute generalized exanthematous pustulosis, with 88% of cases presenting as neutrophilic dermatoses.2 Confirmation with cutaneous biopsy and cessation of medication is essential to prevent life-threatening anaphylactoid reactions.
A 58-year-old man with a history of Crohn disease was admitted with high fevers (>38.9°C); abdominal pain; diarrhea; and a nonpruritic “pimplelike” rash on the face, chest, and back with a tender nodule on the right leg of 5 days’ duration. Eight days prior to admission, he had started AZA for treatment of Crohn disease. In the hospital he received intravenous metronidazole for a presumed bowel infection; however, the lesions and symptoms did not resolve. Other medical history included psoriatic arthritis for which he was taking oral prednisone 50 mg daily; prednisone was continued during hospitalization.
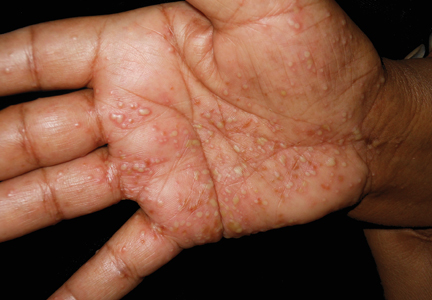
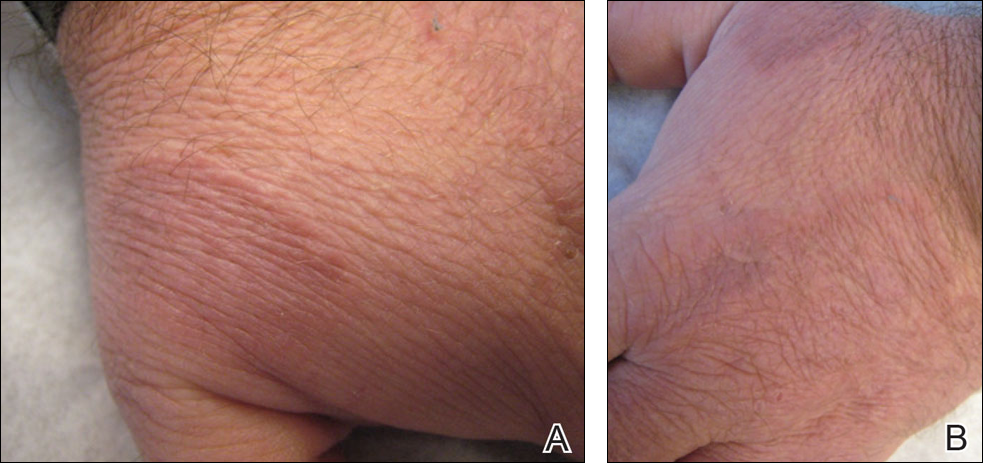
Physical examination showed that the patient was alert and well appearing. On the face, upper chest and back (Figure 1), shoulders, and knees were fewer than 20 sparsely distributed, nontender, 3- to 4-mm pustules. The patient’s scalp, lower back, abdomen, arms, and feet were spared. There also was a solitary 3.5-cm, tender, erythematous nodule on the right lower leg (Figure 2). Blood tests revealed leukocytosis (15,000/mm3 [reference range, 4300–10,300/mm3]) with neutrophilia (90%) and an elevated C-reactive protein level of 173 mg/L (reference range, <10 mg/L). Liver function tests were normal. Thiopurine methyltransferase (TPMT) was on the low end of the reference range. Tissue culture of a shoulder pustule grew only Staphylococcus non-aureus. Blood cultures were negative. A 4-mm punch biopsy specimen from the right leg nodule revealed septal panniculitis with neutrophilic and granulomatous infiltrate consistent with EN.
A clinical diagnosis of AZA hypersensitivity was made. Antibiotics and AZA were discontinued and the patient’s lesions resolved within 6 days. Medication rechallenge was not attempted and the patient is now managed with infliximab.
Azathioprine is a well-known and commonly used drug for inflammatory bowel diseases, rheumatoid arthritis, and prevention of transplant rejection. Hypersensitivity is a lesser-known complication of AZA therapy, with most reactions occurring within 4 weeks of treatment initiation. A PubMed search of articles indexed for MEDLINE using the search terms azathioprine and hypersensitivity found only 67 documented cases of AZA hypersensitivity between 1986 and 2009.2 Common findings include fever, malaise, arthralgia, nausea, vomiting, diarrhea, headache, and neutrophilic dermatoses.
Previously reported cases of AZA hypersensitivity with cutaneous manifestations include Sweet syndrome (17.9%), small vessel vasculitis (10.4%), EN (4.4%), acute generalized exanthematous pustulosis (4.4%), and nonspecific cutaneous findings (11.9%).2 One other case reported AZA hypersensitivity presenting as EN with a neutrophilic pustular dermatosis.3 Although Sweet syndrome–like lesions, EN, and acute generalized exanthematous pustulosis have been reported in the context of inflammatory bowel disease, in this case the appearance of these symptoms within 1 week of AZA initiation and resolution after AZA discontinuation is highly suggestive of AZA hypersensitivity. Also, several reports have documented rapid (within a few hours) recurrence of symptoms on rechallenge with AZA.4-6 Moreover, cases of cutaneous AZA hypersensitivity reactions in patients with no history of inflammatory bowel diseases have been reported.6-8
As in this case, cutaneous AZA hypersensitivity can occur even in the setting of normal TPMT levels, suggesting that this phenomenon is a dose-independent reaction.2 Abnormal metabolism of AZA does not appear to be related to previously reported neutrophilic pustular dermatosis3,4 or EN.4 Although the mechanism of hypersensitivity is unclear, there is a report of a patient who developed AZA hypersensitivity but was able to tolerate 6-mercaptopurine, a metabolite of AZA. The authors suggested that the imidazole component of AZA might be responsible for hypersensitivity reactions.9
The differential diagnosis of a patient with these findings includes infectious, rheumatologic, neurologic, or autoimmune diseases, as well as septic shock. Hence, negative cultures and a failure to respond to antibiotics make infection less likely. An appropriate time course of AZA initiation, the development of rash, and a cutaneous biopsy can lead to prompt diagnosis and cessation of AZA.
Once AZA hypersensitivity is suspected, the drug should be discontinued and the reaction should resolve within 2 to 3 days2 and the skin lesions within 5 to 6 days.2,10 Medication rechallenge is contraindicated because AZA rarely has been associated with shock syndrome and hypotension.11-19
Azathioprine hypersensitivity is a serious yet still underrecognized condition in the dermatologic community. In our case, symptoms appeared rapidly and resolved quickly after AZA was discontinued. Azathioprine-induced neutrophilic dermatosis presenting with EN should be recognized as a potential dermatologic manifestation of AZA hypersensitivity, which is a dose-dependent reaction even with normal TPMT levels. Rechallenge with AZA is not recommended due to the risk of a life-threatening anaphylactoid reaction.
- Meggitt SJ, Anstey AV, Mohd Mustapa MF, et al. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165:711-734.
- Bidinger JJ, Sky K, Battafarano DF, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. J Am Acad Dermatol. 2011;65:184-191.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- De Fonclare AL, Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol. 2007;143:744-748.
- Jeurissen ME, Boerbooms AM, van de Putte LB, et al. Azathioprine induced fever, chills, rash, and hepatotoxicity in rheumatoid arthritis. Ann Rheum Dis. 1990;49:25-27.
- Goldenberg DL, Stor RA. Azathioprine hypersensitivity mimicking an acute exacerbation of dermatomyositis. J Rheumatol. 1975;2:346-349.
- Watts GF, Corston R. Hypersensitivity to azathioprine in myasthenia gravis. Postgrad Med J. 1984;60:362-363.
- El-Azhary RA, Brunner KL, Gibson LE. Sweet syndrome as a manifestation of azathioprine hypersensitivity. Mayo Clin Proc. 2008;83:1026-1030.
- Stetter M, Schmidl M, Krapf R. Azathioprine hypersensitivity mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1994;23:874-877.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Cunningham T, Barraclough D, Muirdin K. Azathioprine induced shock. Br Med J. 1981;283:823-824.
- Elston GE, Johnston GA, Mortimer NJ, et al. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol. 2007;32:52-53.
- Fields CL, Robinson JW, Roy TM, et al. Hypersensitivity reaction to azathioprine. South Med J. 1998;91:471-474.
- Keystone E, Schabas R. Hypotension with oliguria: a side effect of azathioprine. Arthritis Rheum. 1981;24:1453-1454.
- Rosenthal E. Azathioprine shock. Postgrad Med J. 1986;62:677-678.
- Sofat N, Houghton J, McHale J, et al. Azathioprine hypersensitivity. Ann Rheum Dis. 2001;60:719-720.
- Knowles SR, Gupta AK, Shear NH, et al. Azathioprine hypersensitivity-like reactions—a case report and a review of the literature. Clin Exp Dermatol. 1995;20:353-356.
- Demirtaş-Ertan G, Rowshani AT, ten Berge IJ. Azathioprine-induced shock in a patient suffering from undifferentiated erosive oligoarthritis. Neth J Med. 2006;64:124-126.
- Zaltzman M, Kallenbach J, Shapiro T, et al. Life-threatening hypotension associated with azathioprine therapy. a case report. S Afr Med J. 1984;65:306.
To the Editor:
Azathioprine (AZA) hypersensitivity is an immunologically mediated reaction that presents within 1 to 4 weeks of drug initiation.1 Its cutaneous manifestations include Sweet syndrome, erythema nodosum (EN), and acute generalized exanthematous pustulosis, with 88% of cases presenting as neutrophilic dermatoses.2 Confirmation with cutaneous biopsy and cessation of medication is essential to prevent life-threatening anaphylactoid reactions.
A 58-year-old man with a history of Crohn disease was admitted with high fevers (>38.9°C); abdominal pain; diarrhea; and a nonpruritic “pimplelike” rash on the face, chest, and back with a tender nodule on the right leg of 5 days’ duration. Eight days prior to admission, he had started AZA for treatment of Crohn disease. In the hospital he received intravenous metronidazole for a presumed bowel infection; however, the lesions and symptoms did not resolve. Other medical history included psoriatic arthritis for which he was taking oral prednisone 50 mg daily; prednisone was continued during hospitalization.
Physical examination showed that the patient was alert and well appearing. On the face, upper chest and back (Figure 1), shoulders, and knees were fewer than 20 sparsely distributed, nontender, 3- to 4-mm pustules. The patient’s scalp, lower back, abdomen, arms, and feet were spared. There also was a solitary 3.5-cm, tender, erythematous nodule on the right lower leg (Figure 2). Blood tests revealed leukocytosis (15,000/mm3 [reference range, 4300–10,300/mm3]) with neutrophilia (90%) and an elevated C-reactive protein level of 173 mg/L (reference range, <10 mg/L). Liver function tests were normal. Thiopurine methyltransferase (TPMT) was on the low end of the reference range. Tissue culture of a shoulder pustule grew only Staphylococcus non-aureus. Blood cultures were negative. A 4-mm punch biopsy specimen from the right leg nodule revealed septal panniculitis with neutrophilic and granulomatous infiltrate consistent with EN.
A clinical diagnosis of AZA hypersensitivity was made. Antibiotics and AZA were discontinued and the patient’s lesions resolved within 6 days. Medication rechallenge was not attempted and the patient is now managed with infliximab.
Azathioprine is a well-known and commonly used drug for inflammatory bowel diseases, rheumatoid arthritis, and prevention of transplant rejection. Hypersensitivity is a lesser-known complication of AZA therapy, with most reactions occurring within 4 weeks of treatment initiation. A PubMed search of articles indexed for MEDLINE using the search terms azathioprine and hypersensitivity found only 67 documented cases of AZA hypersensitivity between 1986 and 2009.2 Common findings include fever, malaise, arthralgia, nausea, vomiting, diarrhea, headache, and neutrophilic dermatoses.
Previously reported cases of AZA hypersensitivity with cutaneous manifestations include Sweet syndrome (17.9%), small vessel vasculitis (10.4%), EN (4.4%), acute generalized exanthematous pustulosis (4.4%), and nonspecific cutaneous findings (11.9%).2 One other case reported AZA hypersensitivity presenting as EN with a neutrophilic pustular dermatosis.3 Although Sweet syndrome–like lesions, EN, and acute generalized exanthematous pustulosis have been reported in the context of inflammatory bowel disease, in this case the appearance of these symptoms within 1 week of AZA initiation and resolution after AZA discontinuation is highly suggestive of AZA hypersensitivity. Also, several reports have documented rapid (within a few hours) recurrence of symptoms on rechallenge with AZA.4-6 Moreover, cases of cutaneous AZA hypersensitivity reactions in patients with no history of inflammatory bowel diseases have been reported.6-8
As in this case, cutaneous AZA hypersensitivity can occur even in the setting of normal TPMT levels, suggesting that this phenomenon is a dose-independent reaction.2 Abnormal metabolism of AZA does not appear to be related to previously reported neutrophilic pustular dermatosis3,4 or EN.4 Although the mechanism of hypersensitivity is unclear, there is a report of a patient who developed AZA hypersensitivity but was able to tolerate 6-mercaptopurine, a metabolite of AZA. The authors suggested that the imidazole component of AZA might be responsible for hypersensitivity reactions.9
The differential diagnosis of a patient with these findings includes infectious, rheumatologic, neurologic, or autoimmune diseases, as well as septic shock. Hence, negative cultures and a failure to respond to antibiotics make infection less likely. An appropriate time course of AZA initiation, the development of rash, and a cutaneous biopsy can lead to prompt diagnosis and cessation of AZA.
Once AZA hypersensitivity is suspected, the drug should be discontinued and the reaction should resolve within 2 to 3 days2 and the skin lesions within 5 to 6 days.2,10 Medication rechallenge is contraindicated because AZA rarely has been associated with shock syndrome and hypotension.11-19
Azathioprine hypersensitivity is a serious yet still underrecognized condition in the dermatologic community. In our case, symptoms appeared rapidly and resolved quickly after AZA was discontinued. Azathioprine-induced neutrophilic dermatosis presenting with EN should be recognized as a potential dermatologic manifestation of AZA hypersensitivity, which is a dose-dependent reaction even with normal TPMT levels. Rechallenge with AZA is not recommended due to the risk of a life-threatening anaphylactoid reaction.
To the Editor:
Azathioprine (AZA) hypersensitivity is an immunologically mediated reaction that presents within 1 to 4 weeks of drug initiation.1 Its cutaneous manifestations include Sweet syndrome, erythema nodosum (EN), and acute generalized exanthematous pustulosis, with 88% of cases presenting as neutrophilic dermatoses.2 Confirmation with cutaneous biopsy and cessation of medication is essential to prevent life-threatening anaphylactoid reactions.
A 58-year-old man with a history of Crohn disease was admitted with high fevers (>38.9°C); abdominal pain; diarrhea; and a nonpruritic “pimplelike” rash on the face, chest, and back with a tender nodule on the right leg of 5 days’ duration. Eight days prior to admission, he had started AZA for treatment of Crohn disease. In the hospital he received intravenous metronidazole for a presumed bowel infection; however, the lesions and symptoms did not resolve. Other medical history included psoriatic arthritis for which he was taking oral prednisone 50 mg daily; prednisone was continued during hospitalization.
Physical examination showed that the patient was alert and well appearing. On the face, upper chest and back (Figure 1), shoulders, and knees were fewer than 20 sparsely distributed, nontender, 3- to 4-mm pustules. The patient’s scalp, lower back, abdomen, arms, and feet were spared. There also was a solitary 3.5-cm, tender, erythematous nodule on the right lower leg (Figure 2). Blood tests revealed leukocytosis (15,000/mm3 [reference range, 4300–10,300/mm3]) with neutrophilia (90%) and an elevated C-reactive protein level of 173 mg/L (reference range, <10 mg/L). Liver function tests were normal. Thiopurine methyltransferase (TPMT) was on the low end of the reference range. Tissue culture of a shoulder pustule grew only Staphylococcus non-aureus. Blood cultures were negative. A 4-mm punch biopsy specimen from the right leg nodule revealed septal panniculitis with neutrophilic and granulomatous infiltrate consistent with EN.
A clinical diagnosis of AZA hypersensitivity was made. Antibiotics and AZA were discontinued and the patient’s lesions resolved within 6 days. Medication rechallenge was not attempted and the patient is now managed with infliximab.
Azathioprine is a well-known and commonly used drug for inflammatory bowel diseases, rheumatoid arthritis, and prevention of transplant rejection. Hypersensitivity is a lesser-known complication of AZA therapy, with most reactions occurring within 4 weeks of treatment initiation. A PubMed search of articles indexed for MEDLINE using the search terms azathioprine and hypersensitivity found only 67 documented cases of AZA hypersensitivity between 1986 and 2009.2 Common findings include fever, malaise, arthralgia, nausea, vomiting, diarrhea, headache, and neutrophilic dermatoses.
Previously reported cases of AZA hypersensitivity with cutaneous manifestations include Sweet syndrome (17.9%), small vessel vasculitis (10.4%), EN (4.4%), acute generalized exanthematous pustulosis (4.4%), and nonspecific cutaneous findings (11.9%).2 One other case reported AZA hypersensitivity presenting as EN with a neutrophilic pustular dermatosis.3 Although Sweet syndrome–like lesions, EN, and acute generalized exanthematous pustulosis have been reported in the context of inflammatory bowel disease, in this case the appearance of these symptoms within 1 week of AZA initiation and resolution after AZA discontinuation is highly suggestive of AZA hypersensitivity. Also, several reports have documented rapid (within a few hours) recurrence of symptoms on rechallenge with AZA.4-6 Moreover, cases of cutaneous AZA hypersensitivity reactions in patients with no history of inflammatory bowel diseases have been reported.6-8
As in this case, cutaneous AZA hypersensitivity can occur even in the setting of normal TPMT levels, suggesting that this phenomenon is a dose-independent reaction.2 Abnormal metabolism of AZA does not appear to be related to previously reported neutrophilic pustular dermatosis3,4 or EN.4 Although the mechanism of hypersensitivity is unclear, there is a report of a patient who developed AZA hypersensitivity but was able to tolerate 6-mercaptopurine, a metabolite of AZA. The authors suggested that the imidazole component of AZA might be responsible for hypersensitivity reactions.9
The differential diagnosis of a patient with these findings includes infectious, rheumatologic, neurologic, or autoimmune diseases, as well as septic shock. Hence, negative cultures and a failure to respond to antibiotics make infection less likely. An appropriate time course of AZA initiation, the development of rash, and a cutaneous biopsy can lead to prompt diagnosis and cessation of AZA.
Once AZA hypersensitivity is suspected, the drug should be discontinued and the reaction should resolve within 2 to 3 days2 and the skin lesions within 5 to 6 days.2,10 Medication rechallenge is contraindicated because AZA rarely has been associated with shock syndrome and hypotension.11-19
Azathioprine hypersensitivity is a serious yet still underrecognized condition in the dermatologic community. In our case, symptoms appeared rapidly and resolved quickly after AZA was discontinued. Azathioprine-induced neutrophilic dermatosis presenting with EN should be recognized as a potential dermatologic manifestation of AZA hypersensitivity, which is a dose-dependent reaction even with normal TPMT levels. Rechallenge with AZA is not recommended due to the risk of a life-threatening anaphylactoid reaction.
- Meggitt SJ, Anstey AV, Mohd Mustapa MF, et al. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165:711-734.
- Bidinger JJ, Sky K, Battafarano DF, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. J Am Acad Dermatol. 2011;65:184-191.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- De Fonclare AL, Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol. 2007;143:744-748.
- Jeurissen ME, Boerbooms AM, van de Putte LB, et al. Azathioprine induced fever, chills, rash, and hepatotoxicity in rheumatoid arthritis. Ann Rheum Dis. 1990;49:25-27.
- Goldenberg DL, Stor RA. Azathioprine hypersensitivity mimicking an acute exacerbation of dermatomyositis. J Rheumatol. 1975;2:346-349.
- Watts GF, Corston R. Hypersensitivity to azathioprine in myasthenia gravis. Postgrad Med J. 1984;60:362-363.
- El-Azhary RA, Brunner KL, Gibson LE. Sweet syndrome as a manifestation of azathioprine hypersensitivity. Mayo Clin Proc. 2008;83:1026-1030.
- Stetter M, Schmidl M, Krapf R. Azathioprine hypersensitivity mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1994;23:874-877.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Cunningham T, Barraclough D, Muirdin K. Azathioprine induced shock. Br Med J. 1981;283:823-824.
- Elston GE, Johnston GA, Mortimer NJ, et al. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol. 2007;32:52-53.
- Fields CL, Robinson JW, Roy TM, et al. Hypersensitivity reaction to azathioprine. South Med J. 1998;91:471-474.
- Keystone E, Schabas R. Hypotension with oliguria: a side effect of azathioprine. Arthritis Rheum. 1981;24:1453-1454.
- Rosenthal E. Azathioprine shock. Postgrad Med J. 1986;62:677-678.
- Sofat N, Houghton J, McHale J, et al. Azathioprine hypersensitivity. Ann Rheum Dis. 2001;60:719-720.
- Knowles SR, Gupta AK, Shear NH, et al. Azathioprine hypersensitivity-like reactions—a case report and a review of the literature. Clin Exp Dermatol. 1995;20:353-356.
- Demirtaş-Ertan G, Rowshani AT, ten Berge IJ. Azathioprine-induced shock in a patient suffering from undifferentiated erosive oligoarthritis. Neth J Med. 2006;64:124-126.
- Zaltzman M, Kallenbach J, Shapiro T, et al. Life-threatening hypotension associated with azathioprine therapy. a case report. S Afr Med J. 1984;65:306.
- Meggitt SJ, Anstey AV, Mohd Mustapa MF, et al. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165:711-734.
- Bidinger JJ, Sky K, Battafarano DF, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. J Am Acad Dermatol. 2011;65:184-191.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- De Fonclare AL, Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol. 2007;143:744-748.
- Jeurissen ME, Boerbooms AM, van de Putte LB, et al. Azathioprine induced fever, chills, rash, and hepatotoxicity in rheumatoid arthritis. Ann Rheum Dis. 1990;49:25-27.
- Goldenberg DL, Stor RA. Azathioprine hypersensitivity mimicking an acute exacerbation of dermatomyositis. J Rheumatol. 1975;2:346-349.
- Watts GF, Corston R. Hypersensitivity to azathioprine in myasthenia gravis. Postgrad Med J. 1984;60:362-363.
- El-Azhary RA, Brunner KL, Gibson LE. Sweet syndrome as a manifestation of azathioprine hypersensitivity. Mayo Clin Proc. 2008;83:1026-1030.
- Stetter M, Schmidl M, Krapf R. Azathioprine hypersensitivity mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1994;23:874-877.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Cunningham T, Barraclough D, Muirdin K. Azathioprine induced shock. Br Med J. 1981;283:823-824.
- Elston GE, Johnston GA, Mortimer NJ, et al. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol. 2007;32:52-53.
- Fields CL, Robinson JW, Roy TM, et al. Hypersensitivity reaction to azathioprine. South Med J. 1998;91:471-474.
- Keystone E, Schabas R. Hypotension with oliguria: a side effect of azathioprine. Arthritis Rheum. 1981;24:1453-1454.
- Rosenthal E. Azathioprine shock. Postgrad Med J. 1986;62:677-678.
- Sofat N, Houghton J, McHale J, et al. Azathioprine hypersensitivity. Ann Rheum Dis. 2001;60:719-720.
- Knowles SR, Gupta AK, Shear NH, et al. Azathioprine hypersensitivity-like reactions—a case report and a review of the literature. Clin Exp Dermatol. 1995;20:353-356.
- Demirtaş-Ertan G, Rowshani AT, ten Berge IJ. Azathioprine-induced shock in a patient suffering from undifferentiated erosive oligoarthritis. Neth J Med. 2006;64:124-126.
- Zaltzman M, Kallenbach J, Shapiro T, et al. Life-threatening hypotension associated with azathioprine therapy. a case report. S Afr Med J. 1984;65:306.
Practice Points
- Azathioprine is a well-known immunosuppressant for renal transplant recipients and inflammatory bowel disease with several off-label uses in dermatology including immunobullous dermatoses, neutrophilic dermatoses, and autoimmune connective tissue diseases.
- Azathioprine hypersensitivity is rare and can present with systemic symptoms of fever and a neutrophilic dermatosis, which is usually self-limited but can progress to an anaphylactoid reaction with multiorgan failure.
- If a more mild hypersensitivity reaction is appreciated, then a rechallenge is not recommended and should be avoided.
Resolution of Disseminated Granuloma Annulare With Removal of Surgical Hardware
To the Editor:
Disseminated granuloma annulare is a noninfectious granulomatous disease of unknown etiology. Reported precipitating factors include trauma, sun exposure, viral infection, vaccination, and malignancy.1 In contrast to a localized variant, disseminated granuloma annulare is associated with a later age of onset, longer duration, and recalcitrance to therapy.2 Although a variety of therapeutic approaches exist, there are limited efficacy data, which is complicated by the spontaneous, self-limited nature of the disease.3,4
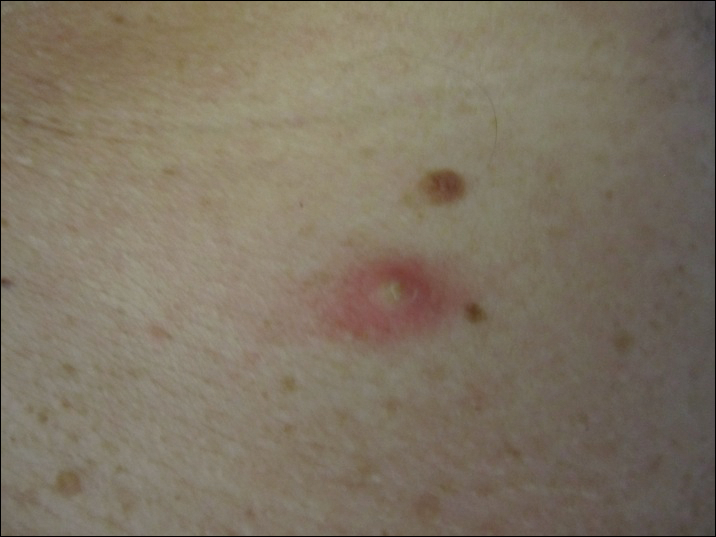
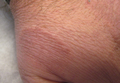
A 47-year-old man presented with an eruption of a thick red plaque on the dorsal aspect of the left hand (Figure). The eruption began 6 weeks following fixation of a Galeazzi fracture of the right radius with a stainless steel volar plate. Subsequent to the initial eruption, similar indurated plaques developed on the left thenar area, bilateral axillae, and bilateral legs. A punch biopsy was conducted to rule out necrobiosis lipoidica diabeticorum and sarcoidosis as well as to histopathologically confirm the clinical diagnosis of disseminated granuloma annulare. Following diagnosis, the patient received topical clobetasol for application to the advancing borders of the plaques. At 4-month follow-up, additional plaques continued to develop. The patient was not interested in pursuing alternative courses of therapy and felt that the implantation of surgical hardware was the cause. To the best of our knowledge, there have been no reports of precipitation of disseminated granuloma annulare in response to surgical hardware. Given the time course of onset of the eruption it was plausible that the hardware was the inciting event. The orthopedist thought that the fracture had healed sufficiently to remove the volar plate. The patient elected to have the hardware removed to potentially resolve or arrest the progression of the plaques. Resolution of the plaques was observed by the patient 2 weeks following surgical removal of the volar plate. At 4 months following hardware removal, the patient only had 2 slightly pink, hyperpigmented lesions on the left hand in the areas most severely affected, with complete resolution of all other plaques. The patient was given topical clobetasol for the residual lesions.
Precipitation and spontaneous resolution of disseminated granuloma annulare following the implantation and removal of surgical hardware is rare. Resolution following hardware removal is consistent with the theory that pathogenesis is due to a delayed-type hypersensitivity reaction to an inciting factor.5 Our case suggests that disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware, which should be considered in the etiology and potential therapeutic options for this disorder.
- Mills A, Chetty R. Auricular granuloma annulare. a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Dicken CH, Carrington SG, Winkelmann RK. Generalized granuloma annulare. Arch Dermatol. 1969;99:556-563.
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea [published online May 31, 2009]. Ann Dermatol. 2009:21:113-119
- Cyr PR. Diagnosis and management of granuloma annulare. Am Fam Physician. 2006;74:1729-1734.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
To the Editor:
Disseminated granuloma annulare is a noninfectious granulomatous disease of unknown etiology. Reported precipitating factors include trauma, sun exposure, viral infection, vaccination, and malignancy.1 In contrast to a localized variant, disseminated granuloma annulare is associated with a later age of onset, longer duration, and recalcitrance to therapy.2 Although a variety of therapeutic approaches exist, there are limited efficacy data, which is complicated by the spontaneous, self-limited nature of the disease.3,4
A 47-year-old man presented with an eruption of a thick red plaque on the dorsal aspect of the left hand (Figure). The eruption began 6 weeks following fixation of a Galeazzi fracture of the right radius with a stainless steel volar plate. Subsequent to the initial eruption, similar indurated plaques developed on the left thenar area, bilateral axillae, and bilateral legs. A punch biopsy was conducted to rule out necrobiosis lipoidica diabeticorum and sarcoidosis as well as to histopathologically confirm the clinical diagnosis of disseminated granuloma annulare. Following diagnosis, the patient received topical clobetasol for application to the advancing borders of the plaques. At 4-month follow-up, additional plaques continued to develop. The patient was not interested in pursuing alternative courses of therapy and felt that the implantation of surgical hardware was the cause. To the best of our knowledge, there have been no reports of precipitation of disseminated granuloma annulare in response to surgical hardware. Given the time course of onset of the eruption it was plausible that the hardware was the inciting event. The orthopedist thought that the fracture had healed sufficiently to remove the volar plate. The patient elected to have the hardware removed to potentially resolve or arrest the progression of the plaques. Resolution of the plaques was observed by the patient 2 weeks following surgical removal of the volar plate. At 4 months following hardware removal, the patient only had 2 slightly pink, hyperpigmented lesions on the left hand in the areas most severely affected, with complete resolution of all other plaques. The patient was given topical clobetasol for the residual lesions.
Precipitation and spontaneous resolution of disseminated granuloma annulare following the implantation and removal of surgical hardware is rare. Resolution following hardware removal is consistent with the theory that pathogenesis is due to a delayed-type hypersensitivity reaction to an inciting factor.5 Our case suggests that disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware, which should be considered in the etiology and potential therapeutic options for this disorder.
To the Editor:
Disseminated granuloma annulare is a noninfectious granulomatous disease of unknown etiology. Reported precipitating factors include trauma, sun exposure, viral infection, vaccination, and malignancy.1 In contrast to a localized variant, disseminated granuloma annulare is associated with a later age of onset, longer duration, and recalcitrance to therapy.2 Although a variety of therapeutic approaches exist, there are limited efficacy data, which is complicated by the spontaneous, self-limited nature of the disease.3,4
A 47-year-old man presented with an eruption of a thick red plaque on the dorsal aspect of the left hand (Figure). The eruption began 6 weeks following fixation of a Galeazzi fracture of the right radius with a stainless steel volar plate. Subsequent to the initial eruption, similar indurated plaques developed on the left thenar area, bilateral axillae, and bilateral legs. A punch biopsy was conducted to rule out necrobiosis lipoidica diabeticorum and sarcoidosis as well as to histopathologically confirm the clinical diagnosis of disseminated granuloma annulare. Following diagnosis, the patient received topical clobetasol for application to the advancing borders of the plaques. At 4-month follow-up, additional plaques continued to develop. The patient was not interested in pursuing alternative courses of therapy and felt that the implantation of surgical hardware was the cause. To the best of our knowledge, there have been no reports of precipitation of disseminated granuloma annulare in response to surgical hardware. Given the time course of onset of the eruption it was plausible that the hardware was the inciting event. The orthopedist thought that the fracture had healed sufficiently to remove the volar plate. The patient elected to have the hardware removed to potentially resolve or arrest the progression of the plaques. Resolution of the plaques was observed by the patient 2 weeks following surgical removal of the volar plate. At 4 months following hardware removal, the patient only had 2 slightly pink, hyperpigmented lesions on the left hand in the areas most severely affected, with complete resolution of all other plaques. The patient was given topical clobetasol for the residual lesions.
Precipitation and spontaneous resolution of disseminated granuloma annulare following the implantation and removal of surgical hardware is rare. Resolution following hardware removal is consistent with the theory that pathogenesis is due to a delayed-type hypersensitivity reaction to an inciting factor.5 Our case suggests that disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware, which should be considered in the etiology and potential therapeutic options for this disorder.
- Mills A, Chetty R. Auricular granuloma annulare. a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Dicken CH, Carrington SG, Winkelmann RK. Generalized granuloma annulare. Arch Dermatol. 1969;99:556-563.
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea [published online May 31, 2009]. Ann Dermatol. 2009:21:113-119
- Cyr PR. Diagnosis and management of granuloma annulare. Am Fam Physician. 2006;74:1729-1734.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
- Mills A, Chetty R. Auricular granuloma annulare. a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Dicken CH, Carrington SG, Winkelmann RK. Generalized granuloma annulare. Arch Dermatol. 1969;99:556-563.
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea [published online May 31, 2009]. Ann Dermatol. 2009:21:113-119
- Cyr PR. Diagnosis and management of granuloma annulare. Am Fam Physician. 2006;74:1729-1734.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
Practice Points
- Disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware.
- Resolution may occur following removal of surgical hardware.
Cutaneous T-Cell Lymphoma in a Patient With Celiac Disease
To the Editor:
Mycosis fungoides (MF) is the most common form of a heterogeneous group of non-Hodgkin lymphomas known as cutaneous T-cell lymphomas. Celiac disease (CD) is associated with increased risk for development of enteropathy-associated T-cell lymphoma and other intraintestinal and extraintestinal non-Hodgkin lymphomas, but a firm association between CD and MF has not been established.1 The first and second cases of concomitant MF and CD were reported in 1985 and 2009 by Coulson and Sanderson2 and Moreira et al,3 respectively. Two other reports of celiac-associated dermatitis herpetiformis and MF exist.4,5 We report a patient with a unique constellation of MF, CD, and Sjögren syndrome (SS).
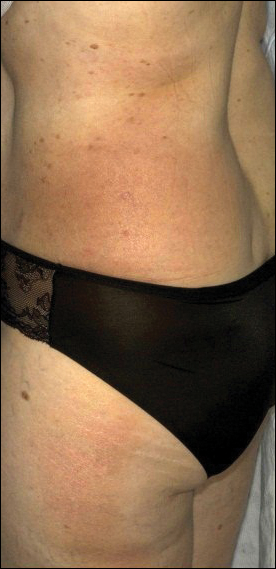
A 54-year-old woman presented with a worsening nonpruritic, slightly tender, eczematous patch on the back of 19 years’ duration. She had a history of SS diagnosed by salivary gland biopsy. She also had a diagnosis of CD confirmed with positive antigliadin IgA antibodies, with a dramatic improvement in symptoms on a gluten-free diet (GFD) after having abdominal pain and diarrhea for many years. She had no evidence of dermatitis herpetiformis. Recently, more red-brown areas of confluent light pink erythema without clear-cut borders had appeared on the axillae, trunk, and thigh (Figure). The patient also noted new lesions and more erythema of the patches when not adhering to a GFD. A biopsy specimen from the left side of the lateral trunk revealed a bandlike lymphocytic infiltrate with irregular nuclear contours displaying epidermotropism with a few Pautrier microabscesses. Immunohistochemistry showed strong CD3 and CD4 positivity with loss of CD7 and scattered CD8 staining. Peripheral blood flow cytometry showed no aberrant cell populations. The patient was diagnosed with MF stage IB and treated with topical corticosteroids and natural light with improvement.
It has been hypothesized that early MF is an autoimmune process caused by dysregulation of a lymphocytic reaction against chronic exogenous or endogenous antigens.4,5 The association of MF with CD supports the possibility of lymphocytic stimulation by a persistent antigen (ie, gluten) in the gastrointestinal tract. Porter et al4 suggested that in susceptible individuals, the resulting clonal T cells may migrate into the epidermis, causing MF. This theory also is supported by the finding that adherence to a GFD leads to decreased risk for malignancy and morbidity.6 In our patient, the chronic autoimmune stimulation in SS could be a factor in the pathogenesis of MF. Additionally, SS, CD, and MF are all strongly associated with increased incidence of specific but different HLA class II antigens. Mycosis fungoides is associated with HLA-DR5 and DQB1*03 alleles, CD with HLA-DQ2 and DQ8, and SS with HLA-DR15 and DR3. We do not know the HLA type of our patient, but she likely possessed multiple alleles, leading to the unique aggregation of diseases.
Furthermore, studies have shown that lymphocytes in CD patients display impaired regulatory T-cell function, causing increased incidence of autoimmune diseases and malignancy.7,8 By this theory, the occurrence of MF in patients is facilitated by the inability of CD lymphocytes to control the abnormal T-cell proliferation in the skin. Interestingly, the finding of SS in our patient supports the possibility of impaired regulatory T-cell function.
Although the occurrence of both MF and CD in our patient could be coincidental, the possibility of correlation must be considered as more cases are documented.
- Catassi C, Fabiani E, Corrao G, et al; Italian Working Group on Coeliac Disease and Non-Hodgkin’s-Lymphoma. Risk of non-Hodgkin Lymphoma in celiac disease. JAMA. 2002;287:1413-1419.
- Coulson IH, Sanderson KV. T-cell lymphoma presenting as tumour d’emblée mycosis fungoides associated with coeliac disease. J R Soc Med. 1985;78(suppl 11):23-24.
- Moreira AI, Menezes N, Varela P, et al. Primary cutaneous peripheral T cell lymphoma and celiac disease [in Portuguese]. Rev Assoc Med Bras. 2009;55:253-256.
- Porter WM, Dawe SA, Bunker CB. Dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2001;26:304-305.
- Sun G, Berthelot C, Duvic M. A second case of dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2008;33:506-507.
- Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten free diet. Gut. 1989;30:333-338.
- Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513-1519.
- Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis [published online June 2, 2013]. Nature. 2013;498:506-510.
To the Editor:
Mycosis fungoides (MF) is the most common form of a heterogeneous group of non-Hodgkin lymphomas known as cutaneous T-cell lymphomas. Celiac disease (CD) is associated with increased risk for development of enteropathy-associated T-cell lymphoma and other intraintestinal and extraintestinal non-Hodgkin lymphomas, but a firm association between CD and MF has not been established.1 The first and second cases of concomitant MF and CD were reported in 1985 and 2009 by Coulson and Sanderson2 and Moreira et al,3 respectively. Two other reports of celiac-associated dermatitis herpetiformis and MF exist.4,5 We report a patient with a unique constellation of MF, CD, and Sjögren syndrome (SS).
A 54-year-old woman presented with a worsening nonpruritic, slightly tender, eczematous patch on the back of 19 years’ duration. She had a history of SS diagnosed by salivary gland biopsy. She also had a diagnosis of CD confirmed with positive antigliadin IgA antibodies, with a dramatic improvement in symptoms on a gluten-free diet (GFD) after having abdominal pain and diarrhea for many years. She had no evidence of dermatitis herpetiformis. Recently, more red-brown areas of confluent light pink erythema without clear-cut borders had appeared on the axillae, trunk, and thigh (Figure). The patient also noted new lesions and more erythema of the patches when not adhering to a GFD. A biopsy specimen from the left side of the lateral trunk revealed a bandlike lymphocytic infiltrate with irregular nuclear contours displaying epidermotropism with a few Pautrier microabscesses. Immunohistochemistry showed strong CD3 and CD4 positivity with loss of CD7 and scattered CD8 staining. Peripheral blood flow cytometry showed no aberrant cell populations. The patient was diagnosed with MF stage IB and treated with topical corticosteroids and natural light with improvement.
It has been hypothesized that early MF is an autoimmune process caused by dysregulation of a lymphocytic reaction against chronic exogenous or endogenous antigens.4,5 The association of MF with CD supports the possibility of lymphocytic stimulation by a persistent antigen (ie, gluten) in the gastrointestinal tract. Porter et al4 suggested that in susceptible individuals, the resulting clonal T cells may migrate into the epidermis, causing MF. This theory also is supported by the finding that adherence to a GFD leads to decreased risk for malignancy and morbidity.6 In our patient, the chronic autoimmune stimulation in SS could be a factor in the pathogenesis of MF. Additionally, SS, CD, and MF are all strongly associated with increased incidence of specific but different HLA class II antigens. Mycosis fungoides is associated with HLA-DR5 and DQB1*03 alleles, CD with HLA-DQ2 and DQ8, and SS with HLA-DR15 and DR3. We do not know the HLA type of our patient, but she likely possessed multiple alleles, leading to the unique aggregation of diseases.
Furthermore, studies have shown that lymphocytes in CD patients display impaired regulatory T-cell function, causing increased incidence of autoimmune diseases and malignancy.7,8 By this theory, the occurrence of MF in patients is facilitated by the inability of CD lymphocytes to control the abnormal T-cell proliferation in the skin. Interestingly, the finding of SS in our patient supports the possibility of impaired regulatory T-cell function.
Although the occurrence of both MF and CD in our patient could be coincidental, the possibility of correlation must be considered as more cases are documented.
To the Editor:
Mycosis fungoides (MF) is the most common form of a heterogeneous group of non-Hodgkin lymphomas known as cutaneous T-cell lymphomas. Celiac disease (CD) is associated with increased risk for development of enteropathy-associated T-cell lymphoma and other intraintestinal and extraintestinal non-Hodgkin lymphomas, but a firm association between CD and MF has not been established.1 The first and second cases of concomitant MF and CD were reported in 1985 and 2009 by Coulson and Sanderson2 and Moreira et al,3 respectively. Two other reports of celiac-associated dermatitis herpetiformis and MF exist.4,5 We report a patient with a unique constellation of MF, CD, and Sjögren syndrome (SS).
A 54-year-old woman presented with a worsening nonpruritic, slightly tender, eczematous patch on the back of 19 years’ duration. She had a history of SS diagnosed by salivary gland biopsy. She also had a diagnosis of CD confirmed with positive antigliadin IgA antibodies, with a dramatic improvement in symptoms on a gluten-free diet (GFD) after having abdominal pain and diarrhea for many years. She had no evidence of dermatitis herpetiformis. Recently, more red-brown areas of confluent light pink erythema without clear-cut borders had appeared on the axillae, trunk, and thigh (Figure). The patient also noted new lesions and more erythema of the patches when not adhering to a GFD. A biopsy specimen from the left side of the lateral trunk revealed a bandlike lymphocytic infiltrate with irregular nuclear contours displaying epidermotropism with a few Pautrier microabscesses. Immunohistochemistry showed strong CD3 and CD4 positivity with loss of CD7 and scattered CD8 staining. Peripheral blood flow cytometry showed no aberrant cell populations. The patient was diagnosed with MF stage IB and treated with topical corticosteroids and natural light with improvement.
It has been hypothesized that early MF is an autoimmune process caused by dysregulation of a lymphocytic reaction against chronic exogenous or endogenous antigens.4,5 The association of MF with CD supports the possibility of lymphocytic stimulation by a persistent antigen (ie, gluten) in the gastrointestinal tract. Porter et al4 suggested that in susceptible individuals, the resulting clonal T cells may migrate into the epidermis, causing MF. This theory also is supported by the finding that adherence to a GFD leads to decreased risk for malignancy and morbidity.6 In our patient, the chronic autoimmune stimulation in SS could be a factor in the pathogenesis of MF. Additionally, SS, CD, and MF are all strongly associated with increased incidence of specific but different HLA class II antigens. Mycosis fungoides is associated with HLA-DR5 and DQB1*03 alleles, CD with HLA-DQ2 and DQ8, and SS with HLA-DR15 and DR3. We do not know the HLA type of our patient, but she likely possessed multiple alleles, leading to the unique aggregation of diseases.
Furthermore, studies have shown that lymphocytes in CD patients display impaired regulatory T-cell function, causing increased incidence of autoimmune diseases and malignancy.7,8 By this theory, the occurrence of MF in patients is facilitated by the inability of CD lymphocytes to control the abnormal T-cell proliferation in the skin. Interestingly, the finding of SS in our patient supports the possibility of impaired regulatory T-cell function.
Although the occurrence of both MF and CD in our patient could be coincidental, the possibility of correlation must be considered as more cases are documented.
- Catassi C, Fabiani E, Corrao G, et al; Italian Working Group on Coeliac Disease and Non-Hodgkin’s-Lymphoma. Risk of non-Hodgkin Lymphoma in celiac disease. JAMA. 2002;287:1413-1419.
- Coulson IH, Sanderson KV. T-cell lymphoma presenting as tumour d’emblée mycosis fungoides associated with coeliac disease. J R Soc Med. 1985;78(suppl 11):23-24.
- Moreira AI, Menezes N, Varela P, et al. Primary cutaneous peripheral T cell lymphoma and celiac disease [in Portuguese]. Rev Assoc Med Bras. 2009;55:253-256.
- Porter WM, Dawe SA, Bunker CB. Dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2001;26:304-305.
- Sun G, Berthelot C, Duvic M. A second case of dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2008;33:506-507.
- Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten free diet. Gut. 1989;30:333-338.
- Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513-1519.
- Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis [published online June 2, 2013]. Nature. 2013;498:506-510.
- Catassi C, Fabiani E, Corrao G, et al; Italian Working Group on Coeliac Disease and Non-Hodgkin’s-Lymphoma. Risk of non-Hodgkin Lymphoma in celiac disease. JAMA. 2002;287:1413-1419.
- Coulson IH, Sanderson KV. T-cell lymphoma presenting as tumour d’emblée mycosis fungoides associated with coeliac disease. J R Soc Med. 1985;78(suppl 11):23-24.
- Moreira AI, Menezes N, Varela P, et al. Primary cutaneous peripheral T cell lymphoma and celiac disease [in Portuguese]. Rev Assoc Med Bras. 2009;55:253-256.
- Porter WM, Dawe SA, Bunker CB. Dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2001;26:304-305.
- Sun G, Berthelot C, Duvic M. A second case of dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2008;33:506-507.
- Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten free diet. Gut. 1989;30:333-338.
- Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513-1519.
- Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis [published online June 2, 2013]. Nature. 2013;498:506-510.
Practice Points
- Mycosis fungoides, the most common type of cutaneous T-cell lymphoma, is an entity for which the pathogenesis is largely unknown.
- Our case and other cases of celiac disease and mycosis fungoides seem to support the immunologic hypothesis of lymphocytic stimulation by a persistent antigen.
Pemphigus Vulgaris Successfully Treated With Doxycycline Monotherapy
To the Editor:
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease with notable morbidity and mortality if not treated appropriately due to loss of epidermal barrier function and subsequent infection and loss of body fluids. Although the use of systemic corticosteroids and immunosuppressive agents has improved the prognosis, these drugs also may have severe adverse effects, especially in elderly patients. Hence, alternative and safer therapies with anti-inflammatory and immunomodulatory agents such as tetracyclines and nicotinamide have been used with variable results. We report a case of PV that was successfully treated with doxycycline.
An 81-year-old man presented with well-demarcated erosions with overlying yellow crust as well as vesicles and pustules on the scalp (Figure 1A), forehead, bilateral cheeks, and upper back (Figure 1B) of 6 months’ duration. He used topical fluorouracil in the month prior to presentation for suspected actinic keratosis but had stopped its use after 2 weeks. At the first visit, a diagnosis of a reaction to topical fluorouracil with secondary bacterial infection was made and he was prescribed doxycycline hyclate 100 mg twice daily. The patient returned 4 weeks later for follow-up and reported initial notable improvement with subsequent worsening of lesions after he ran out of doxycycline. On physical examination the lesions had considerably improved from the last visit, but he still had a few erosions on the scalp and a few in the oral mucosa. A 1-cm shallow erosion with minimal surrounding erythema on the forehead was present, along with fewer scattered, edematous, erythematous plaques on the back and chest. Pemphigus vulgaris was suspected and 2 shave biopsies from the lesions on the back and cheek were obtained for confirmation. Histopathologic examination revealed epidermal hyperplasia and suprabasal acantholysis as well as moderate perivascular and perifollicular lymphocytic infiltrate with several eosinophils and plasma cells, characteristic of PV (Figure 2). Direct immunofluorescence showed moderate intercellular deposition of IgG within the basal layer and to a lesser extent within suprabasal layers as well as moderate intercellular deposition of C3 within the basal layer, characteristic of PV. IgA and IgM were not present. Indirect immunofluorescence using monkey esophagus revealed no antibodies against the intercellular space of the basement membrane zone. Due to the dramatic response, he continued on doxycycline 100 mg twice daily and remained in complete remission. Ten months after initiating treatment he discontinued doxycycline for 2 days and developed a 1-cm lesion on the left cheek. He resumed treatment with clearing of lesions and was slowly tapered to 50 mg of doxycycline once daily, remaining in complete remission (Figure 3). Doxycycline was discontinued 16 months after initiation; he has remained clear at 13 weeks.
The treatment of PV is challenging given the multiple side effects of steroids, especially in elderly patients. Tetracyclines have an advantageous side-effect profile and they have been shown to be efficacious in treating PV when combined with nicotinamide or when used as adjuvant therapy to steroids.1-3 Our case shows a patient who was treated exclusively with doxycycline and achieved complete remission.
Tetracyclines have multiple biological activities in addition to their antimicrobial function that may provide a therapeutic benefit in PV. They possess immunomodulatory and anti-inflammatory effects by inhibiting leukocyte chemotaxis and activation4-8 and inhibiting cytokine release. They inhibit matrix metalloproteinases, which are the major enzymes responsible for breakdown of the extracellular matrix,9 and they indirectly inhibit neutrophil elastase by protecting α1-protease inhibitor from matrix metalloproteinase degradation.10 Additionally, tetracyclines increase the cohesion of the dermoepidermal junction11; whether they increase the adhesion between epidermal cells is unknown. It has been determined that CD4+ T cells play an essential role in the pathogenesis of PV by promoting anti-desmoglein 3 antibody production.12 Szeto et al13 reported that minocycline, a member of the tetracycline family, has suppressive effects on CD4+ T-cell activation by hindering the activation of nuclear factor of activated T cells (NFAT), a key regulatory factor in T-cell activation. We hypothesize that doxycycline exerted what appears to be immunomodulatory properties in our patient by suppressing CD4+ T-cell activity.
In conclusion, tetracyclines can be an effective and promising therapy for PV given their relatively few side effects and immunomodulating properties. However, further randomized controlled trials will be important to support our conclusion.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993;28:998-1000.
- Caelbotta A, Saenz AM, Gonzalez F, et al. Pemphigus vulgaris: benefits of tetracycline as adjuvant therapy in series of thirteen patients. Int J Dermatol. 1999;38:217-221.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus defining a niche for these low toxicity agents. J Am Acad Dermatol. 2014;71:475-479.
- Majeski JA, McClellan MA, Alexander JW. Effect of antibiotics on the in vitro neutrophil chemotactic response. Am Surg. 1976;42:785-788.
- Esterly NB, Furley NL, Flanagan LE. The effect of antimicrobial agents on leukocyte chemotaxis. J Invest Dermatol. 1978;70:51-55.
- Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52-58.
- Esterly NB, Koransky JS, Furey NL, et al. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch Dermatol. 1984;120:1308-1313.
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258-265.
- Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. 2011;63:130-145.
- Golub LM, Evans RT, McNamara TF, et al. A nonantimicrobial tetracycline inhibits gingival matrix metalloproteinases and bone loss in Porphyromonas gingivalis–induced periodontitis in rats. Ann N Y Acad Sci. 1994;732:96-111.
- Humbert P, Treffel P, Chapius JF, et al. The tetracyclines in dermatology. J Am Acad Dermatol. 1991;25:691-697.
- Nishifuji K, Amagai M, Kuwana M, et al. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88-94.
- Szeto GL, Pomerantz JL, Graham DRM, et al. Minocycline suppresses activation of nuclear factor of activated T cells 1 (NFAT1) in human CD4 T Cells. J Biol Chem. 2011;286:11275-11282.
To the Editor:
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease with notable morbidity and mortality if not treated appropriately due to loss of epidermal barrier function and subsequent infection and loss of body fluids. Although the use of systemic corticosteroids and immunosuppressive agents has improved the prognosis, these drugs also may have severe adverse effects, especially in elderly patients. Hence, alternative and safer therapies with anti-inflammatory and immunomodulatory agents such as tetracyclines and nicotinamide have been used with variable results. We report a case of PV that was successfully treated with doxycycline.
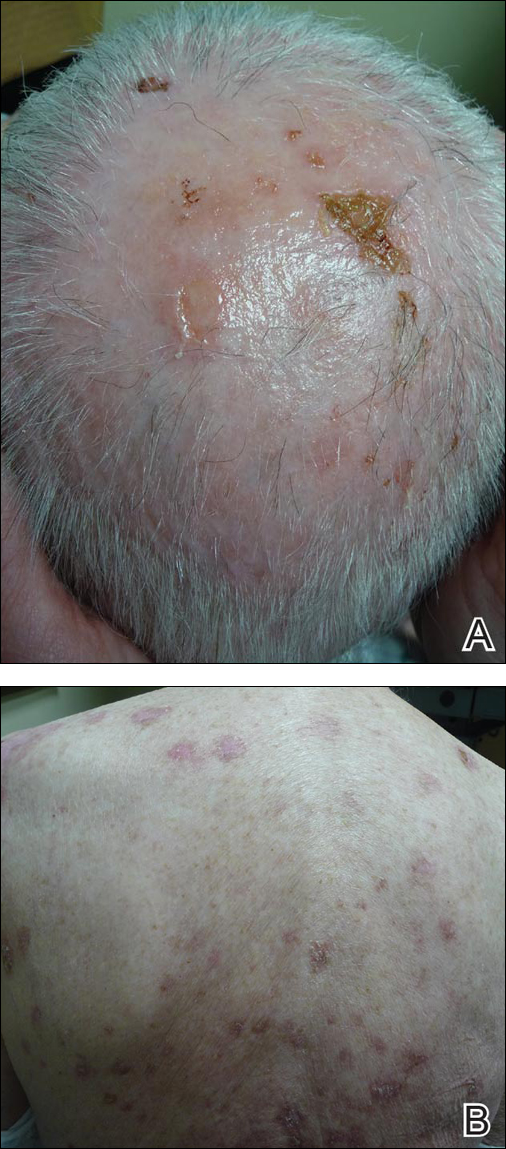
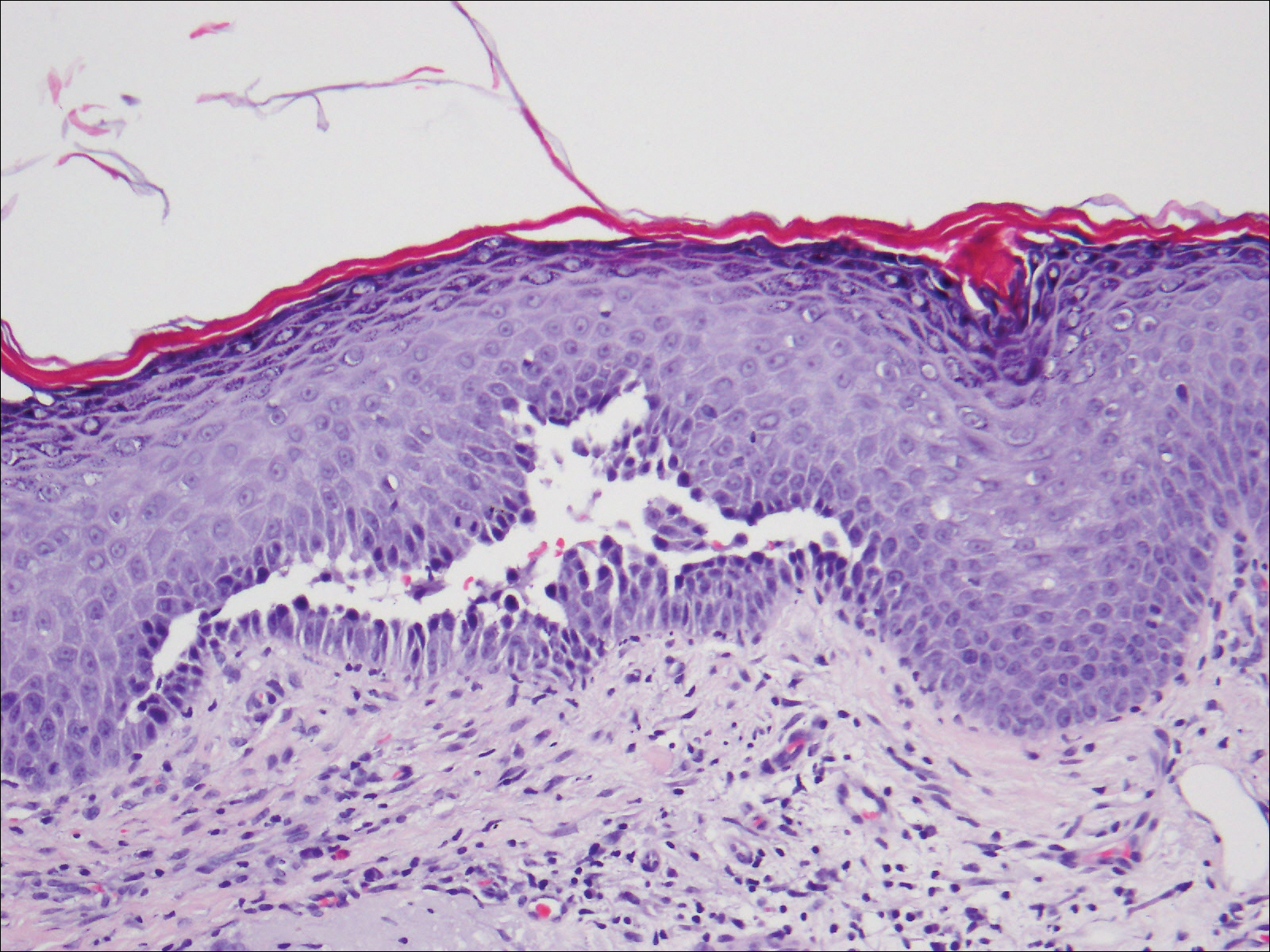
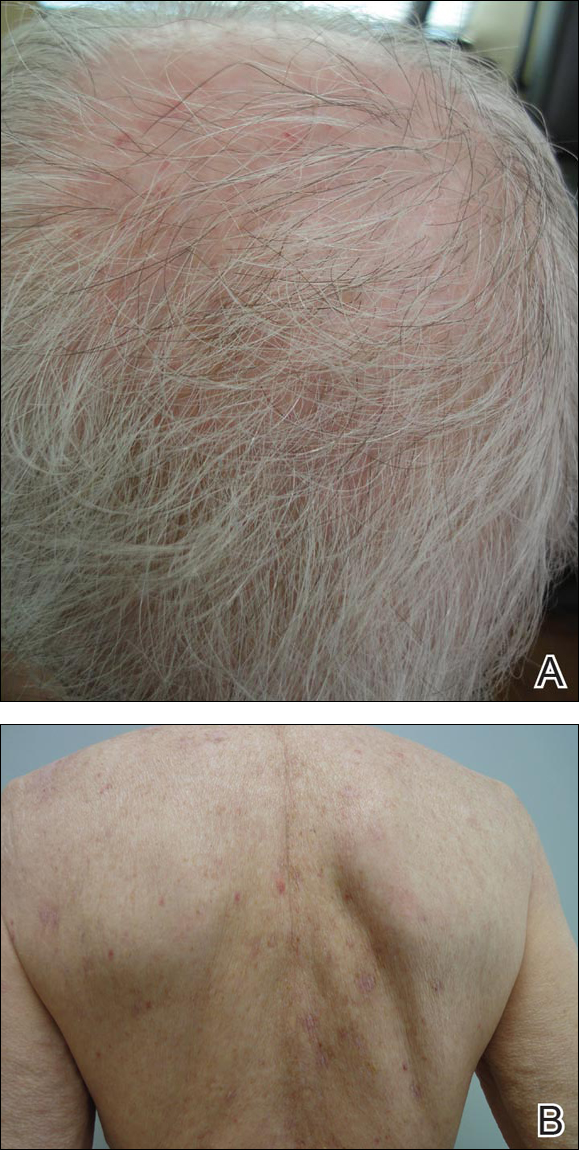
An 81-year-old man presented with well-demarcated erosions with overlying yellow crust as well as vesicles and pustules on the scalp (Figure 1A), forehead, bilateral cheeks, and upper back (Figure 1B) of 6 months’ duration. He used topical fluorouracil in the month prior to presentation for suspected actinic keratosis but had stopped its use after 2 weeks. At the first visit, a diagnosis of a reaction to topical fluorouracil with secondary bacterial infection was made and he was prescribed doxycycline hyclate 100 mg twice daily. The patient returned 4 weeks later for follow-up and reported initial notable improvement with subsequent worsening of lesions after he ran out of doxycycline. On physical examination the lesions had considerably improved from the last visit, but he still had a few erosions on the scalp and a few in the oral mucosa. A 1-cm shallow erosion with minimal surrounding erythema on the forehead was present, along with fewer scattered, edematous, erythematous plaques on the back and chest. Pemphigus vulgaris was suspected and 2 shave biopsies from the lesions on the back and cheek were obtained for confirmation. Histopathologic examination revealed epidermal hyperplasia and suprabasal acantholysis as well as moderate perivascular and perifollicular lymphocytic infiltrate with several eosinophils and plasma cells, characteristic of PV (Figure 2). Direct immunofluorescence showed moderate intercellular deposition of IgG within the basal layer and to a lesser extent within suprabasal layers as well as moderate intercellular deposition of C3 within the basal layer, characteristic of PV. IgA and IgM were not present. Indirect immunofluorescence using monkey esophagus revealed no antibodies against the intercellular space of the basement membrane zone. Due to the dramatic response, he continued on doxycycline 100 mg twice daily and remained in complete remission. Ten months after initiating treatment he discontinued doxycycline for 2 days and developed a 1-cm lesion on the left cheek. He resumed treatment with clearing of lesions and was slowly tapered to 50 mg of doxycycline once daily, remaining in complete remission (Figure 3). Doxycycline was discontinued 16 months after initiation; he has remained clear at 13 weeks.
The treatment of PV is challenging given the multiple side effects of steroids, especially in elderly patients. Tetracyclines have an advantageous side-effect profile and they have been shown to be efficacious in treating PV when combined with nicotinamide or when used as adjuvant therapy to steroids.1-3 Our case shows a patient who was treated exclusively with doxycycline and achieved complete remission.
Tetracyclines have multiple biological activities in addition to their antimicrobial function that may provide a therapeutic benefit in PV. They possess immunomodulatory and anti-inflammatory effects by inhibiting leukocyte chemotaxis and activation4-8 and inhibiting cytokine release. They inhibit matrix metalloproteinases, which are the major enzymes responsible for breakdown of the extracellular matrix,9 and they indirectly inhibit neutrophil elastase by protecting α1-protease inhibitor from matrix metalloproteinase degradation.10 Additionally, tetracyclines increase the cohesion of the dermoepidermal junction11; whether they increase the adhesion between epidermal cells is unknown. It has been determined that CD4+ T cells play an essential role in the pathogenesis of PV by promoting anti-desmoglein 3 antibody production.12 Szeto et al13 reported that minocycline, a member of the tetracycline family, has suppressive effects on CD4+ T-cell activation by hindering the activation of nuclear factor of activated T cells (NFAT), a key regulatory factor in T-cell activation. We hypothesize that doxycycline exerted what appears to be immunomodulatory properties in our patient by suppressing CD4+ T-cell activity.
In conclusion, tetracyclines can be an effective and promising therapy for PV given their relatively few side effects and immunomodulating properties. However, further randomized controlled trials will be important to support our conclusion.
To the Editor:
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease with notable morbidity and mortality if not treated appropriately due to loss of epidermal barrier function and subsequent infection and loss of body fluids. Although the use of systemic corticosteroids and immunosuppressive agents has improved the prognosis, these drugs also may have severe adverse effects, especially in elderly patients. Hence, alternative and safer therapies with anti-inflammatory and immunomodulatory agents such as tetracyclines and nicotinamide have been used with variable results. We report a case of PV that was successfully treated with doxycycline.
An 81-year-old man presented with well-demarcated erosions with overlying yellow crust as well as vesicles and pustules on the scalp (Figure 1A), forehead, bilateral cheeks, and upper back (Figure 1B) of 6 months’ duration. He used topical fluorouracil in the month prior to presentation for suspected actinic keratosis but had stopped its use after 2 weeks. At the first visit, a diagnosis of a reaction to topical fluorouracil with secondary bacterial infection was made and he was prescribed doxycycline hyclate 100 mg twice daily. The patient returned 4 weeks later for follow-up and reported initial notable improvement with subsequent worsening of lesions after he ran out of doxycycline. On physical examination the lesions had considerably improved from the last visit, but he still had a few erosions on the scalp and a few in the oral mucosa. A 1-cm shallow erosion with minimal surrounding erythema on the forehead was present, along with fewer scattered, edematous, erythematous plaques on the back and chest. Pemphigus vulgaris was suspected and 2 shave biopsies from the lesions on the back and cheek were obtained for confirmation. Histopathologic examination revealed epidermal hyperplasia and suprabasal acantholysis as well as moderate perivascular and perifollicular lymphocytic infiltrate with several eosinophils and plasma cells, characteristic of PV (Figure 2). Direct immunofluorescence showed moderate intercellular deposition of IgG within the basal layer and to a lesser extent within suprabasal layers as well as moderate intercellular deposition of C3 within the basal layer, characteristic of PV. IgA and IgM were not present. Indirect immunofluorescence using monkey esophagus revealed no antibodies against the intercellular space of the basement membrane zone. Due to the dramatic response, he continued on doxycycline 100 mg twice daily and remained in complete remission. Ten months after initiating treatment he discontinued doxycycline for 2 days and developed a 1-cm lesion on the left cheek. He resumed treatment with clearing of lesions and was slowly tapered to 50 mg of doxycycline once daily, remaining in complete remission (Figure 3). Doxycycline was discontinued 16 months after initiation; he has remained clear at 13 weeks.
The treatment of PV is challenging given the multiple side effects of steroids, especially in elderly patients. Tetracyclines have an advantageous side-effect profile and they have been shown to be efficacious in treating PV when combined with nicotinamide or when used as adjuvant therapy to steroids.1-3 Our case shows a patient who was treated exclusively with doxycycline and achieved complete remission.
Tetracyclines have multiple biological activities in addition to their antimicrobial function that may provide a therapeutic benefit in PV. They possess immunomodulatory and anti-inflammatory effects by inhibiting leukocyte chemotaxis and activation4-8 and inhibiting cytokine release. They inhibit matrix metalloproteinases, which are the major enzymes responsible for breakdown of the extracellular matrix,9 and they indirectly inhibit neutrophil elastase by protecting α1-protease inhibitor from matrix metalloproteinase degradation.10 Additionally, tetracyclines increase the cohesion of the dermoepidermal junction11; whether they increase the adhesion between epidermal cells is unknown. It has been determined that CD4+ T cells play an essential role in the pathogenesis of PV by promoting anti-desmoglein 3 antibody production.12 Szeto et al13 reported that minocycline, a member of the tetracycline family, has suppressive effects on CD4+ T-cell activation by hindering the activation of nuclear factor of activated T cells (NFAT), a key regulatory factor in T-cell activation. We hypothesize that doxycycline exerted what appears to be immunomodulatory properties in our patient by suppressing CD4+ T-cell activity.
In conclusion, tetracyclines can be an effective and promising therapy for PV given their relatively few side effects and immunomodulating properties. However, further randomized controlled trials will be important to support our conclusion.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993;28:998-1000.
- Caelbotta A, Saenz AM, Gonzalez F, et al. Pemphigus vulgaris: benefits of tetracycline as adjuvant therapy in series of thirteen patients. Int J Dermatol. 1999;38:217-221.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus defining a niche for these low toxicity agents. J Am Acad Dermatol. 2014;71:475-479.
- Majeski JA, McClellan MA, Alexander JW. Effect of antibiotics on the in vitro neutrophil chemotactic response. Am Surg. 1976;42:785-788.
- Esterly NB, Furley NL, Flanagan LE. The effect of antimicrobial agents on leukocyte chemotaxis. J Invest Dermatol. 1978;70:51-55.
- Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52-58.
- Esterly NB, Koransky JS, Furey NL, et al. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch Dermatol. 1984;120:1308-1313.
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258-265.
- Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. 2011;63:130-145.
- Golub LM, Evans RT, McNamara TF, et al. A nonantimicrobial tetracycline inhibits gingival matrix metalloproteinases and bone loss in Porphyromonas gingivalis–induced periodontitis in rats. Ann N Y Acad Sci. 1994;732:96-111.
- Humbert P, Treffel P, Chapius JF, et al. The tetracyclines in dermatology. J Am Acad Dermatol. 1991;25:691-697.
- Nishifuji K, Amagai M, Kuwana M, et al. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88-94.
- Szeto GL, Pomerantz JL, Graham DRM, et al. Minocycline suppresses activation of nuclear factor of activated T cells 1 (NFAT1) in human CD4 T Cells. J Biol Chem. 2011;286:11275-11282.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993;28:998-1000.
- Caelbotta A, Saenz AM, Gonzalez F, et al. Pemphigus vulgaris: benefits of tetracycline as adjuvant therapy in series of thirteen patients. Int J Dermatol. 1999;38:217-221.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus defining a niche for these low toxicity agents. J Am Acad Dermatol. 2014;71:475-479.
- Majeski JA, McClellan MA, Alexander JW. Effect of antibiotics on the in vitro neutrophil chemotactic response. Am Surg. 1976;42:785-788.
- Esterly NB, Furley NL, Flanagan LE. The effect of antimicrobial agents on leukocyte chemotaxis. J Invest Dermatol. 1978;70:51-55.
- Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52-58.
- Esterly NB, Koransky JS, Furey NL, et al. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch Dermatol. 1984;120:1308-1313.
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258-265.
- Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. 2011;63:130-145.
- Golub LM, Evans RT, McNamara TF, et al. A nonantimicrobial tetracycline inhibits gingival matrix metalloproteinases and bone loss in Porphyromonas gingivalis–induced periodontitis in rats. Ann N Y Acad Sci. 1994;732:96-111.
- Humbert P, Treffel P, Chapius JF, et al. The tetracyclines in dermatology. J Am Acad Dermatol. 1991;25:691-697.
- Nishifuji K, Amagai M, Kuwana M, et al. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88-94.
- Szeto GL, Pomerantz JL, Graham DRM, et al. Minocycline suppresses activation of nuclear factor of activated T cells 1 (NFAT1) in human CD4 T Cells. J Biol Chem. 2011;286:11275-11282.
Practice Points
- The treatment of pemphigus vulgaris (PV) is challenging given the side-effect profile of commonly used systemic medications, including steroids, especially in elderly patients.
- Tetracyclines have an advantageous side-effect profile and may be efficacious in treating PV.
Primary Herpes Simplex Virus Infection of the Nipple in a Breastfeeding Woman
To the Editor:
A 33-year-old woman presented with tenderness of the left breast and nipple of 2 weeks’ duration and fever of 2 days’ duration. The pain was so severe it precluded nursing. She rented a hospital-grade electric breast pump to continue lactation but only could produce 1 ounce of milk daily. The mother had been breastfeeding her 13-month-old twins since birth and did not report any prior difficulties with breastfeeding. Both twins had a history of mucosal sores 2 months prior and a recent outbreak of perioral vesicles following an upper respiratory tract illness that was consistent with gingivostomatitis, followed by a cutaneous outbreak secondary to herpes simplex virus (HSV) type 1 infection. The patient had no known history of HSV infection. Prior to presentation the patient was treated with oral dicloxacillin and then cephalexin for suspected bacterial mastitis. She also had used combination clotrimazole-betamethasone cream for possible superficial candidiasis. The patient had no relief with these treatments.
Physical examination revealed approximately 20 microvesicles (<1 mm) on an erythematous base clustered around the left areola (Figure). Erythematous streaks were noted from the medial aspect of the areolar margin extending to the central sternum. The left breast was firm and engorged but without apparent plugged lactiferous ducts. There was no lymphadenopathy. No lesions were present on the palms, soles, and oral mucosa.
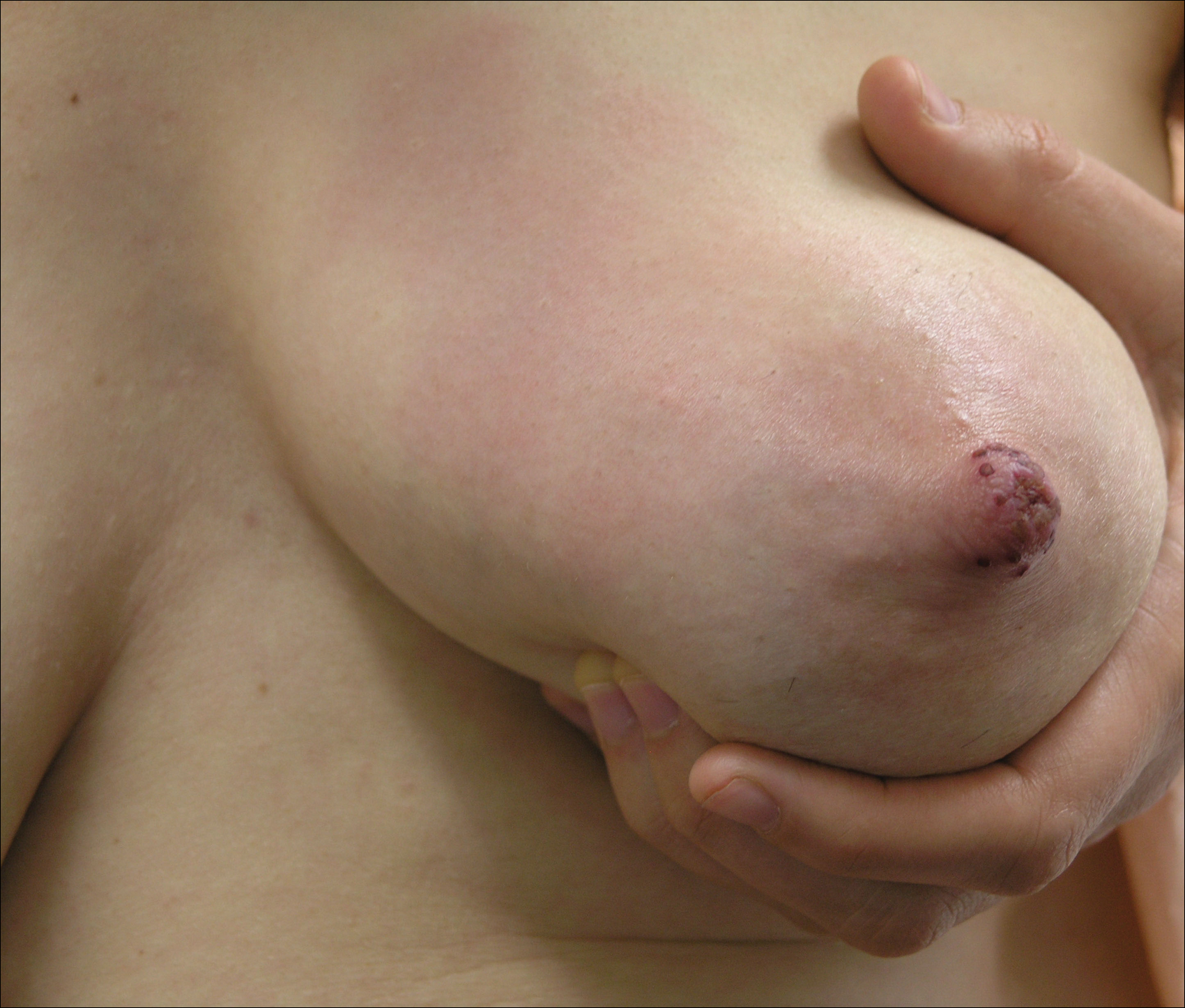
The patient was empirically treated with valacyclovir, trimethoprim-sulfamethoxazole, and nonsteroidal anti-inflammatory drugs while awaiting laboratory results. Bacterial cultures were negative. Viral titers revealed positive combination HSV-1 and HSV-2 IgM (4.64 [<0.91=negative, 0.91–1.09=equivocal, >1.09=positive]) and negative HSV-1 and HSV-2 IgG (<0.91[<0.91=negative, 0.91–1.09=equivocal, >1.09=positive]), which confirmed the diagnosis of primary HSV infection. Two months later viral titers were positive for HSV-1 IgG (1.3) and negative for HSV-2 IgG (<0.91).
At 1-week follow-up the patient reported that the fever had subsided 1 day after initial presentation. After commencement of antiviral therapy, she continued to have some mild residual tenderness, but the vesicles had crusted over and markedly improved. Upon further questioning, the patient’s husband had a history of oral HSV-1 and was likely the primary source for the infection in the infants.
Herpes simplex virus infection primarily is transmitted through direct mucocutaneous contact with either oral or genital lesions of an infected individual. Transmission of HSV from infant to mother rarely is described. A PubMed search of articles indexed for MEDLINE using the terms herpes mastitis, herpes of the breast, infant to maternal transmission, gingivostomatitis, primary herpes, and breastfeeding yielded 4 reported cases of HSV of the nipple in breastfeeding women from children with herpetic gingivostomatitis.1-4
Herpes simplex virus infection is common in neonatal and pediatric populations. In the United States, more than 30% of children (aged <14 years) have evidence of HSV-1 infection on serology. Herpes simplex virus infections in children can range from uncomplicated mucocutaneous diseases to severe life-threatening infections involving the central nervous system. In children, antivirals should be initiated within 72 hours of symptom onset to prevent more serious complications. Diagnostic testing was not performed on the infants in this case because the 72-hour treatment window had passed. In particular, neonates (aged <3 months) will require intravenous antivirals to prevent the development of central nervous system disease, which occurs in 33% of neonatal HSV infections.5 It is critically important to confirm the diagnosis of HSV in a breastfeeding woman, when clinically indicated, with a viral culture, serology, direct immunofluorescence assay, polymerase chain reaction, or Tzanck smear because other conditions such as plugged lactiferous ducts, candidal mastitis, or bacterial mastitis may mimic HSV. Rapid and accurate diagnosis of the breastfeeding woman with HSV of the nipple can help identify children with herpetic gingivostomatitis that is not readily apparent.
- Quinn PT, Lofberg JV. Maternal herpetic breast infection: another hazard of neonatal herpes simplex. Med J Aust. 1978;2:411-412.
- Dekio S, Kawasaki Y, Jidoi J. Herpes simplex on nipples inoculated from herpetic gingivostomatitis of a baby. Clin Exp Dermatol. 1986;11:664-666.
- Sealander JY, Kerr CP. Herpes simplex of the nipple: infant-to-mother transmission. Am Fam Physician. 1989;39:111-113.
- Gupta S, Malhotra AK, Dash SS. Child to mother transmission of herpes simplex virus-1 infection at an unusual site. J Eur Acad Dermatol Venereol. 2008;22:878-879.
- James SH, Whitley RJ. Treatment of herpes simplex virus infections in pediatric patients: current status and future needs. Clin Pharmacol Ther. 2010;88:720-724.
To the Editor:
A 33-year-old woman presented with tenderness of the left breast and nipple of 2 weeks’ duration and fever of 2 days’ duration. The pain was so severe it precluded nursing. She rented a hospital-grade electric breast pump to continue lactation but only could produce 1 ounce of milk daily. The mother had been breastfeeding her 13-month-old twins since birth and did not report any prior difficulties with breastfeeding. Both twins had a history of mucosal sores 2 months prior and a recent outbreak of perioral vesicles following an upper respiratory tract illness that was consistent with gingivostomatitis, followed by a cutaneous outbreak secondary to herpes simplex virus (HSV) type 1 infection. The patient had no known history of HSV infection. Prior to presentation the patient was treated with oral dicloxacillin and then cephalexin for suspected bacterial mastitis. She also had used combination clotrimazole-betamethasone cream for possible superficial candidiasis. The patient had no relief with these treatments.
Physical examination revealed approximately 20 microvesicles (<1 mm) on an erythematous base clustered around the left areola (Figure). Erythematous streaks were noted from the medial aspect of the areolar margin extending to the central sternum. The left breast was firm and engorged but without apparent plugged lactiferous ducts. There was no lymphadenopathy. No lesions were present on the palms, soles, and oral mucosa.
The patient was empirically treated with valacyclovir, trimethoprim-sulfamethoxazole, and nonsteroidal anti-inflammatory drugs while awaiting laboratory results. Bacterial cultures were negative. Viral titers revealed positive combination HSV-1 and HSV-2 IgM (4.64 [<0.91=negative, 0.91–1.09=equivocal, >1.09=positive]) and negative HSV-1 and HSV-2 IgG (<0.91[<0.91=negative, 0.91–1.09=equivocal, >1.09=positive]), which confirmed the diagnosis of primary HSV infection. Two months later viral titers were positive for HSV-1 IgG (1.3) and negative for HSV-2 IgG (<0.91).
At 1-week follow-up the patient reported that the fever had subsided 1 day after initial presentation. After commencement of antiviral therapy, she continued to have some mild residual tenderness, but the vesicles had crusted over and markedly improved. Upon further questioning, the patient’s husband had a history of oral HSV-1 and was likely the primary source for the infection in the infants.
Herpes simplex virus infection primarily is transmitted through direct mucocutaneous contact with either oral or genital lesions of an infected individual. Transmission of HSV from infant to mother rarely is described. A PubMed search of articles indexed for MEDLINE using the terms herpes mastitis, herpes of the breast, infant to maternal transmission, gingivostomatitis, primary herpes, and breastfeeding yielded 4 reported cases of HSV of the nipple in breastfeeding women from children with herpetic gingivostomatitis.1-4
Herpes simplex virus infection is common in neonatal and pediatric populations. In the United States, more than 30% of children (aged <14 years) have evidence of HSV-1 infection on serology. Herpes simplex virus infections in children can range from uncomplicated mucocutaneous diseases to severe life-threatening infections involving the central nervous system. In children, antivirals should be initiated within 72 hours of symptom onset to prevent more serious complications. Diagnostic testing was not performed on the infants in this case because the 72-hour treatment window had passed. In particular, neonates (aged <3 months) will require intravenous antivirals to prevent the development of central nervous system disease, which occurs in 33% of neonatal HSV infections.5 It is critically important to confirm the diagnosis of HSV in a breastfeeding woman, when clinically indicated, with a viral culture, serology, direct immunofluorescence assay, polymerase chain reaction, or Tzanck smear because other conditions such as plugged lactiferous ducts, candidal mastitis, or bacterial mastitis may mimic HSV. Rapid and accurate diagnosis of the breastfeeding woman with HSV of the nipple can help identify children with herpetic gingivostomatitis that is not readily apparent.
To the Editor:
A 33-year-old woman presented with tenderness of the left breast and nipple of 2 weeks’ duration and fever of 2 days’ duration. The pain was so severe it precluded nursing. She rented a hospital-grade electric breast pump to continue lactation but only could produce 1 ounce of milk daily. The mother had been breastfeeding her 13-month-old twins since birth and did not report any prior difficulties with breastfeeding. Both twins had a history of mucosal sores 2 months prior and a recent outbreak of perioral vesicles following an upper respiratory tract illness that was consistent with gingivostomatitis, followed by a cutaneous outbreak secondary to herpes simplex virus (HSV) type 1 infection. The patient had no known history of HSV infection. Prior to presentation the patient was treated with oral dicloxacillin and then cephalexin for suspected bacterial mastitis. She also had used combination clotrimazole-betamethasone cream for possible superficial candidiasis. The patient had no relief with these treatments.
Physical examination revealed approximately 20 microvesicles (<1 mm) on an erythematous base clustered around the left areola (Figure). Erythematous streaks were noted from the medial aspect of the areolar margin extending to the central sternum. The left breast was firm and engorged but without apparent plugged lactiferous ducts. There was no lymphadenopathy. No lesions were present on the palms, soles, and oral mucosa.
The patient was empirically treated with valacyclovir, trimethoprim-sulfamethoxazole, and nonsteroidal anti-inflammatory drugs while awaiting laboratory results. Bacterial cultures were negative. Viral titers revealed positive combination HSV-1 and HSV-2 IgM (4.64 [<0.91=negative, 0.91–1.09=equivocal, >1.09=positive]) and negative HSV-1 and HSV-2 IgG (<0.91[<0.91=negative, 0.91–1.09=equivocal, >1.09=positive]), which confirmed the diagnosis of primary HSV infection. Two months later viral titers were positive for HSV-1 IgG (1.3) and negative for HSV-2 IgG (<0.91).
At 1-week follow-up the patient reported that the fever had subsided 1 day after initial presentation. After commencement of antiviral therapy, she continued to have some mild residual tenderness, but the vesicles had crusted over and markedly improved. Upon further questioning, the patient’s husband had a history of oral HSV-1 and was likely the primary source for the infection in the infants.
Herpes simplex virus infection primarily is transmitted through direct mucocutaneous contact with either oral or genital lesions of an infected individual. Transmission of HSV from infant to mother rarely is described. A PubMed search of articles indexed for MEDLINE using the terms herpes mastitis, herpes of the breast, infant to maternal transmission, gingivostomatitis, primary herpes, and breastfeeding yielded 4 reported cases of HSV of the nipple in breastfeeding women from children with herpetic gingivostomatitis.1-4
Herpes simplex virus infection is common in neonatal and pediatric populations. In the United States, more than 30% of children (aged <14 years) have evidence of HSV-1 infection on serology. Herpes simplex virus infections in children can range from uncomplicated mucocutaneous diseases to severe life-threatening infections involving the central nervous system. In children, antivirals should be initiated within 72 hours of symptom onset to prevent more serious complications. Diagnostic testing was not performed on the infants in this case because the 72-hour treatment window had passed. In particular, neonates (aged <3 months) will require intravenous antivirals to prevent the development of central nervous system disease, which occurs in 33% of neonatal HSV infections.5 It is critically important to confirm the diagnosis of HSV in a breastfeeding woman, when clinically indicated, with a viral culture, serology, direct immunofluorescence assay, polymerase chain reaction, or Tzanck smear because other conditions such as plugged lactiferous ducts, candidal mastitis, or bacterial mastitis may mimic HSV. Rapid and accurate diagnosis of the breastfeeding woman with HSV of the nipple can help identify children with herpetic gingivostomatitis that is not readily apparent.
- Quinn PT, Lofberg JV. Maternal herpetic breast infection: another hazard of neonatal herpes simplex. Med J Aust. 1978;2:411-412.
- Dekio S, Kawasaki Y, Jidoi J. Herpes simplex on nipples inoculated from herpetic gingivostomatitis of a baby. Clin Exp Dermatol. 1986;11:664-666.
- Sealander JY, Kerr CP. Herpes simplex of the nipple: infant-to-mother transmission. Am Fam Physician. 1989;39:111-113.
- Gupta S, Malhotra AK, Dash SS. Child to mother transmission of herpes simplex virus-1 infection at an unusual site. J Eur Acad Dermatol Venereol. 2008;22:878-879.
- James SH, Whitley RJ. Treatment of herpes simplex virus infections in pediatric patients: current status and future needs. Clin Pharmacol Ther. 2010;88:720-724.
- Quinn PT, Lofberg JV. Maternal herpetic breast infection: another hazard of neonatal herpes simplex. Med J Aust. 1978;2:411-412.
- Dekio S, Kawasaki Y, Jidoi J. Herpes simplex on nipples inoculated from herpetic gingivostomatitis of a baby. Clin Exp Dermatol. 1986;11:664-666.
- Sealander JY, Kerr CP. Herpes simplex of the nipple: infant-to-mother transmission. Am Fam Physician. 1989;39:111-113.
- Gupta S, Malhotra AK, Dash SS. Child to mother transmission of herpes simplex virus-1 infection at an unusual site. J Eur Acad Dermatol Venereol. 2008;22:878-879.
- James SH, Whitley RJ. Treatment of herpes simplex virus infections in pediatric patients: current status and future needs. Clin Pharmacol Ther. 2010;88:720-724.
Practice Points
- Herpes mastitis should be included in the differential diagnosis for breast pain during lactation.
- Children of breastfeeding women diagnosed with herpes mastitis require immediate evaluation for a possible source of the infection, as complications of herpes viral infection in infants can be severe and life threatening.
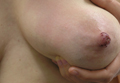
Necrolytic Migratory Erythema With Recalcitrant Dermatitis as the Only Presenting Symptom
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
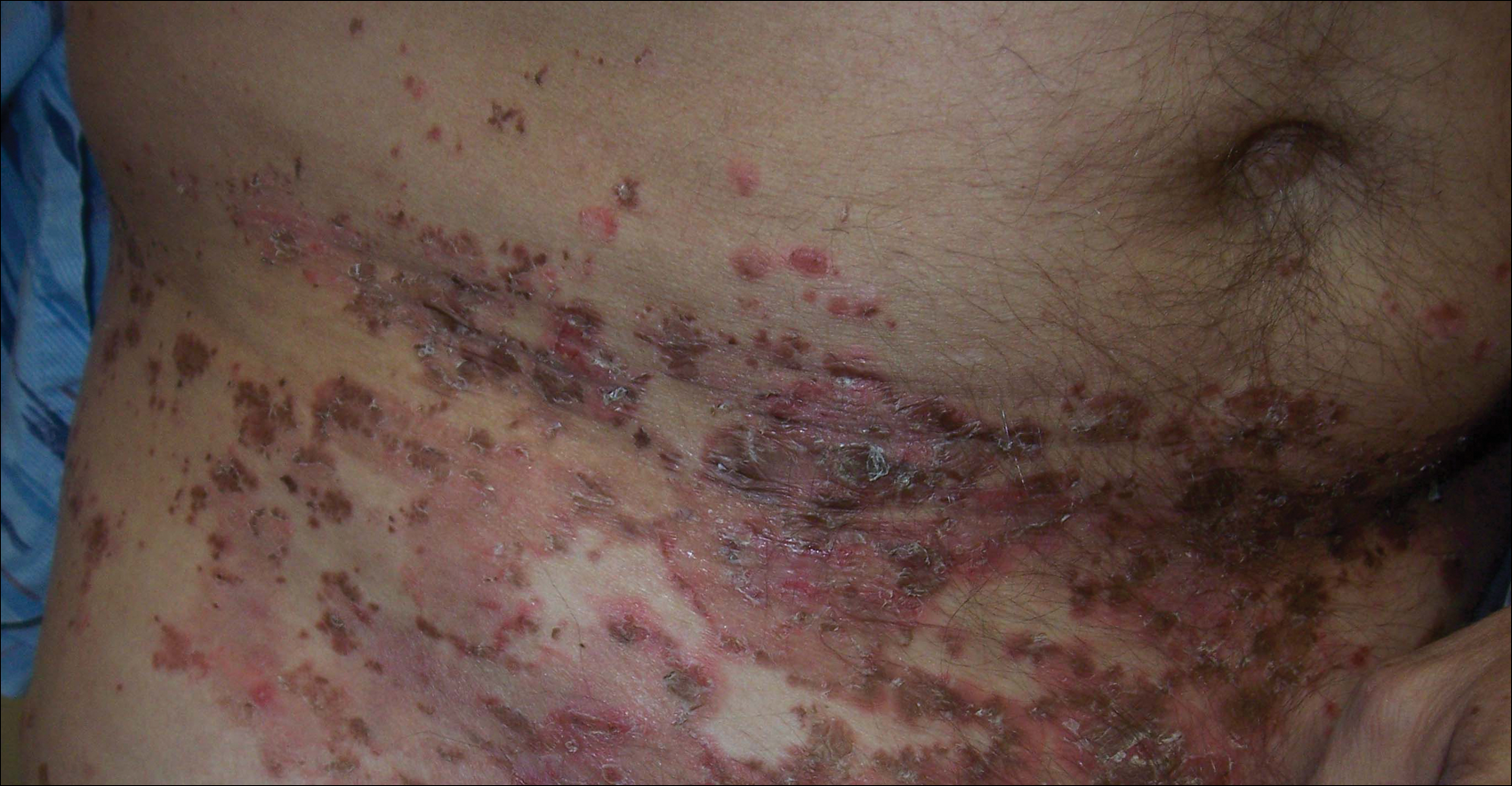
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
Practice Points
- Recalcitrant dermatitis may be a symptom of internal malignancy.
- Glucagon levels are helpful in identifying glucagonomas of the pancreas.
- Although surgical excision is the preferred treatment of glucagonomas, azathioprine also can control dermatitis associated with necrolytic migratory erythema.
If a Chronic Wound Does Not Heal, Biopsy It: A Clinical Lesson on Underlying Malignancies
To the Editor:
Experience, subjective opinion, and relationships with patients are cornerstones of general practice but also can be pitfalls. It is common for a late-presenting patient to offer a seemingly rational explanation for a long-standing lesion. Unless an objective analysis of the clinical problem is undertaken, it can be easy to embark on an incorrect treatment pathway for the patient’s condition.
One of the luxuries of specialist hospital medicine or surgery is the ability to focus on a narrow range of clinical problems, which makes it easier to spot the anomaly, as long as it is within the purview of the practitioner. We report 2 cases of skin malignancies that were assumed to be chronic wounds of benign etiology.
A 63-year-old builder was referred by his general practitioner with a chronic wound on the right forearm of 4 years’ duration. His medical history included psoriasis, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested possible incidental origin following a prior trauma or a psoriatic-related lesion. The patient reported that the lesion did not resemble prior psoriatic lesions and it had deteriorated substantially over the last 2 years. Furthermore, a small ulcer was starting to develop on the left forearm. Further advice was requested by the general practitioner regarding wound dressings. On examination a sloughy ulcer measuring 8.5×7.5 cm had eroded to expose necrotic tendons with surrounding induration and cellulitis (Figure 1A). In addition, a psoriatic lesion was found on the left forearm (Figure 1B). There were no palpable axillary lymph nodes. Clinical suspicion, incision biopsies, and subsequent histology confirmed cutaneous CD4+ T-cell lymphoma. This case was reviewed at a multidisciplinary team meeting and referred to the hematology-oncology department. The patient subsequently underwent chemotherapy with liposomal doxorubicin and radiotherapy over a period of 5 months. An elective right forearm amputation was planned due to erosion of the ulcer through tendons down to bone (Figure 2).
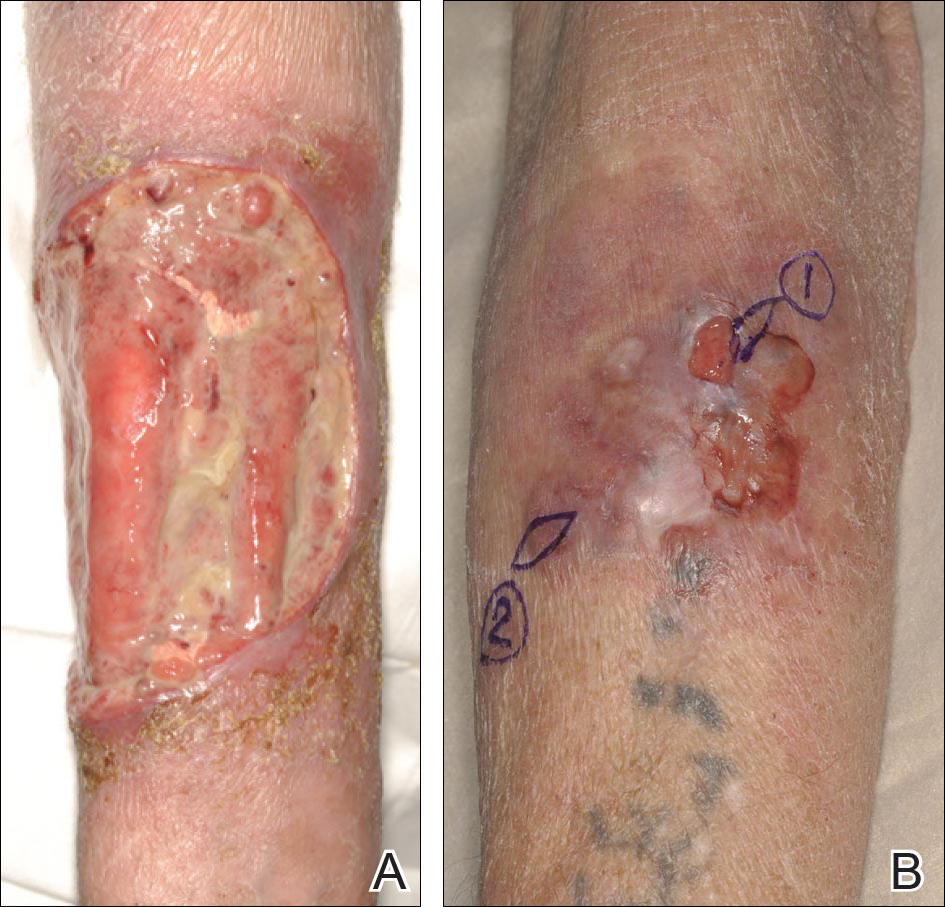
A 48-year-old Latvian lorry driver was referred by his general practitioner with a chronic wound on the left shoulder of 6 years’ duration. His medical history included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested the etiology was a burn from a hot metal rod 6 years prior. Advice was sought regarding dressings and suitability for a possible skin graft. Physical examination showed a 4.5×10-cm ulcer fixed to the underlying tissue on the anterior aspect of the left shoulder with no evidence of infection or presence of a foreign body (Figure 3A). Clinical suspicion, incision biopsies, and subsequent histology confirmed a highly infiltrative/morphoeic, partly nodular, and partly diffuse basal cell carcinoma (BCC) that measured 92 mm in diameter extending to the subcutis with no involvement of muscle or perineural or vascular invasion. The patient underwent wide local excision of the BCC with frozen section control. The BCC had eroded into the deltoid muscle and to the periosteum of the clavicle (Figure 3B). The defect was reconstructed with a pedicled muscle-sparing latissimus dorsi musculocutaneous flap. The patient presented for follow-up months following reconstruction with an uneventful recovery (Figure 3C).
These 2 cases highlight easy pitfalls for an unsuspecting clinician. Although both cases had alternative plausible explanations, they proved to be cutaneous malignancies. The powerful message these cases send is that long-standing chronic wounds should be biopsied to exclude malignancy. Some of the other common underlying causes of wounds that may prevent healing are highlighted in the Table. Vascular insufficiency usually presents in characteristic patterns with a good clinical history and associated signs and findings on investigation. A foreign body, which can be anything from an orthopedic metal implant to a retained stitch from surgery or nonmedical material, may be the culprit and may be identified from a thorough medical history or appropriate imaging.
Infection is another possible explanation of a nonhealing wound. On the face, an underlying dental abscess with a sinus tracking from the root of the tooth to the skin of the cheek or jaw may be the source. Elsewhere on the body, chronic osteomyelitis may be the cause, which may be from any infective origin from Staphylococcus aureus to tuberculosis, and will most commonly present with a discharging sinus but also may present with a nonspecific ulcer.
Chronic wounds also may not heal because of a multitude of patient factors such as poor nutrition, diabetes mellitus, medication (eg, steroids, nonsteroidal anti-inflammatory drugs), other inflammatory causes, and poor mobility. Chronic wounds represent a substantial burden to patients, health care professionals, and the health care system. In the United States alone, they affect 5.7 million patients and cost an estimated $20 billion.1 Approximately 1% of the Western population will present with leg ulceration at some point in their lives.2
Physical examination of ulcers in any clinical setting can be difficult. We postulate that it can be made more difficult at times in primary care because the patient may add confounding elements for consideration or seemingly plausible explanations. However, whenever possible, a physician should ask, “Could there possibly be an underlying malignancy here?” If there is any chance of malignancy despite plausible explanations being offered, the lesion should be biopsied.
- Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing [published online July 7, 2008]. Burns. 2009;35:171-180.
- Callam MJ. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology. 1992;7(suppl 1):6-12.
To the Editor:
Experience, subjective opinion, and relationships with patients are cornerstones of general practice but also can be pitfalls. It is common for a late-presenting patient to offer a seemingly rational explanation for a long-standing lesion. Unless an objective analysis of the clinical problem is undertaken, it can be easy to embark on an incorrect treatment pathway for the patient’s condition.
One of the luxuries of specialist hospital medicine or surgery is the ability to focus on a narrow range of clinical problems, which makes it easier to spot the anomaly, as long as it is within the purview of the practitioner. We report 2 cases of skin malignancies that were assumed to be chronic wounds of benign etiology.
A 63-year-old builder was referred by his general practitioner with a chronic wound on the right forearm of 4 years’ duration. His medical history included psoriasis, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested possible incidental origin following a prior trauma or a psoriatic-related lesion. The patient reported that the lesion did not resemble prior psoriatic lesions and it had deteriorated substantially over the last 2 years. Furthermore, a small ulcer was starting to develop on the left forearm. Further advice was requested by the general practitioner regarding wound dressings. On examination a sloughy ulcer measuring 8.5×7.5 cm had eroded to expose necrotic tendons with surrounding induration and cellulitis (Figure 1A). In addition, a psoriatic lesion was found on the left forearm (Figure 1B). There were no palpable axillary lymph nodes. Clinical suspicion, incision biopsies, and subsequent histology confirmed cutaneous CD4+ T-cell lymphoma. This case was reviewed at a multidisciplinary team meeting and referred to the hematology-oncology department. The patient subsequently underwent chemotherapy with liposomal doxorubicin and radiotherapy over a period of 5 months. An elective right forearm amputation was planned due to erosion of the ulcer through tendons down to bone (Figure 2).
A 48-year-old Latvian lorry driver was referred by his general practitioner with a chronic wound on the left shoulder of 6 years’ duration. His medical history included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested the etiology was a burn from a hot metal rod 6 years prior. Advice was sought regarding dressings and suitability for a possible skin graft. Physical examination showed a 4.5×10-cm ulcer fixed to the underlying tissue on the anterior aspect of the left shoulder with no evidence of infection or presence of a foreign body (Figure 3A). Clinical suspicion, incision biopsies, and subsequent histology confirmed a highly infiltrative/morphoeic, partly nodular, and partly diffuse basal cell carcinoma (BCC) that measured 92 mm in diameter extending to the subcutis with no involvement of muscle or perineural or vascular invasion. The patient underwent wide local excision of the BCC with frozen section control. The BCC had eroded into the deltoid muscle and to the periosteum of the clavicle (Figure 3B). The defect was reconstructed with a pedicled muscle-sparing latissimus dorsi musculocutaneous flap. The patient presented for follow-up months following reconstruction with an uneventful recovery (Figure 3C).
These 2 cases highlight easy pitfalls for an unsuspecting clinician. Although both cases had alternative plausible explanations, they proved to be cutaneous malignancies. The powerful message these cases send is that long-standing chronic wounds should be biopsied to exclude malignancy. Some of the other common underlying causes of wounds that may prevent healing are highlighted in the Table. Vascular insufficiency usually presents in characteristic patterns with a good clinical history and associated signs and findings on investigation. A foreign body, which can be anything from an orthopedic metal implant to a retained stitch from surgery or nonmedical material, may be the culprit and may be identified from a thorough medical history or appropriate imaging.
Infection is another possible explanation of a nonhealing wound. On the face, an underlying dental abscess with a sinus tracking from the root of the tooth to the skin of the cheek or jaw may be the source. Elsewhere on the body, chronic osteomyelitis may be the cause, which may be from any infective origin from Staphylococcus aureus to tuberculosis, and will most commonly present with a discharging sinus but also may present with a nonspecific ulcer.
Chronic wounds also may not heal because of a multitude of patient factors such as poor nutrition, diabetes mellitus, medication (eg, steroids, nonsteroidal anti-inflammatory drugs), other inflammatory causes, and poor mobility. Chronic wounds represent a substantial burden to patients, health care professionals, and the health care system. In the United States alone, they affect 5.7 million patients and cost an estimated $20 billion.1 Approximately 1% of the Western population will present with leg ulceration at some point in their lives.2
Physical examination of ulcers in any clinical setting can be difficult. We postulate that it can be made more difficult at times in primary care because the patient may add confounding elements for consideration or seemingly plausible explanations. However, whenever possible, a physician should ask, “Could there possibly be an underlying malignancy here?” If there is any chance of malignancy despite plausible explanations being offered, the lesion should be biopsied.
To the Editor:
Experience, subjective opinion, and relationships with patients are cornerstones of general practice but also can be pitfalls. It is common for a late-presenting patient to offer a seemingly rational explanation for a long-standing lesion. Unless an objective analysis of the clinical problem is undertaken, it can be easy to embark on an incorrect treatment pathway for the patient’s condition.
One of the luxuries of specialist hospital medicine or surgery is the ability to focus on a narrow range of clinical problems, which makes it easier to spot the anomaly, as long as it is within the purview of the practitioner. We report 2 cases of skin malignancies that were assumed to be chronic wounds of benign etiology.
A 63-year-old builder was referred by his general practitioner with a chronic wound on the right forearm of 4 years’ duration. His medical history included psoriasis, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested possible incidental origin following a prior trauma or a psoriatic-related lesion. The patient reported that the lesion did not resemble prior psoriatic lesions and it had deteriorated substantially over the last 2 years. Furthermore, a small ulcer was starting to develop on the left forearm. Further advice was requested by the general practitioner regarding wound dressings. On examination a sloughy ulcer measuring 8.5×7.5 cm had eroded to expose necrotic tendons with surrounding induration and cellulitis (Figure 1A). In addition, a psoriatic lesion was found on the left forearm (Figure 1B). There were no palpable axillary lymph nodes. Clinical suspicion, incision biopsies, and subsequent histology confirmed cutaneous CD4+ T-cell lymphoma. This case was reviewed at a multidisciplinary team meeting and referred to the hematology-oncology department. The patient subsequently underwent chemotherapy with liposomal doxorubicin and radiotherapy over a period of 5 months. An elective right forearm amputation was planned due to erosion of the ulcer through tendons down to bone (Figure 2).
A 48-year-old Latvian lorry driver was referred by his general practitioner with a chronic wound on the left shoulder of 6 years’ duration. His medical history included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested the etiology was a burn from a hot metal rod 6 years prior. Advice was sought regarding dressings and suitability for a possible skin graft. Physical examination showed a 4.5×10-cm ulcer fixed to the underlying tissue on the anterior aspect of the left shoulder with no evidence of infection or presence of a foreign body (Figure 3A). Clinical suspicion, incision biopsies, and subsequent histology confirmed a highly infiltrative/morphoeic, partly nodular, and partly diffuse basal cell carcinoma (BCC) that measured 92 mm in diameter extending to the subcutis with no involvement of muscle or perineural or vascular invasion. The patient underwent wide local excision of the BCC with frozen section control. The BCC had eroded into the deltoid muscle and to the periosteum of the clavicle (Figure 3B). The defect was reconstructed with a pedicled muscle-sparing latissimus dorsi musculocutaneous flap. The patient presented for follow-up months following reconstruction with an uneventful recovery (Figure 3C).
These 2 cases highlight easy pitfalls for an unsuspecting clinician. Although both cases had alternative plausible explanations, they proved to be cutaneous malignancies. The powerful message these cases send is that long-standing chronic wounds should be biopsied to exclude malignancy. Some of the other common underlying causes of wounds that may prevent healing are highlighted in the Table. Vascular insufficiency usually presents in characteristic patterns with a good clinical history and associated signs and findings on investigation. A foreign body, which can be anything from an orthopedic metal implant to a retained stitch from surgery or nonmedical material, may be the culprit and may be identified from a thorough medical history or appropriate imaging.
Infection is another possible explanation of a nonhealing wound. On the face, an underlying dental abscess with a sinus tracking from the root of the tooth to the skin of the cheek or jaw may be the source. Elsewhere on the body, chronic osteomyelitis may be the cause, which may be from any infective origin from Staphylococcus aureus to tuberculosis, and will most commonly present with a discharging sinus but also may present with a nonspecific ulcer.
Chronic wounds also may not heal because of a multitude of patient factors such as poor nutrition, diabetes mellitus, medication (eg, steroids, nonsteroidal anti-inflammatory drugs), other inflammatory causes, and poor mobility. Chronic wounds represent a substantial burden to patients, health care professionals, and the health care system. In the United States alone, they affect 5.7 million patients and cost an estimated $20 billion.1 Approximately 1% of the Western population will present with leg ulceration at some point in their lives.2
Physical examination of ulcers in any clinical setting can be difficult. We postulate that it can be made more difficult at times in primary care because the patient may add confounding elements for consideration or seemingly plausible explanations. However, whenever possible, a physician should ask, “Could there possibly be an underlying malignancy here?” If there is any chance of malignancy despite plausible explanations being offered, the lesion should be biopsied.
- Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing [published online July 7, 2008]. Burns. 2009;35:171-180.
- Callam MJ. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology. 1992;7(suppl 1):6-12.
- Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing [published online July 7, 2008]. Burns. 2009;35:171-180.
- Callam MJ. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology. 1992;7(suppl 1):6-12.
Practice Points
- Patients with chronic wounds should have a thorough history and examination, appropriate laboratory tests, and purposeful search to determine etiology.
- Long-standing chronic wounds should be biopsied to exclude malignancy.
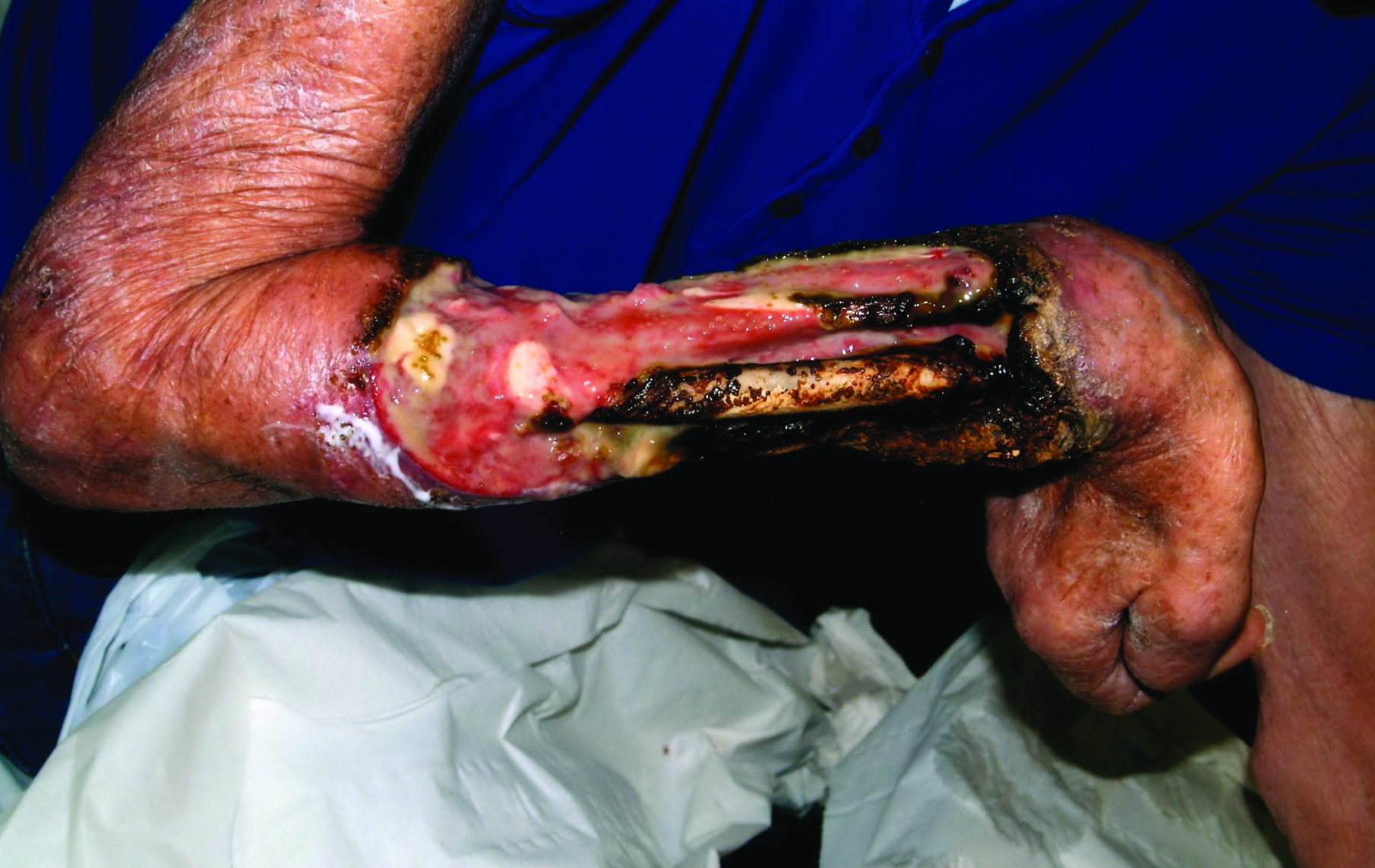
Onychomadesis Following Hand-foot-and-mouth Disease
To the Editor:
Onychomadesis is characterized by separation of the nail plate from the matrix due to a temporary arrest in nail matrix activity. Hand-foot-and-mouth disease (HFMD) is a relatively common viral infection, especially in children. Although the relationship between onychomadesis and HFMD has been noted, there are few reports in the literature.1-9 We present 2 cases of onychomadesis following HFMD in Taiwanese siblings.
A 3-year-old girl presented with proximal nail plate detachment from the proximal nail fold on the bilateral great toenails (Figure 1) and a transverse whole-thickness sulcus on the bilateral thumbnails (Figure 2) of several weeks’ duration. Her 6-year-old sister had similar nail changes. Hand-foot-and-mouth disease was diagnosed about 4 weeks prior to nail changes. The mother reported that only the younger sister experienced fever. There was no history of notable medication intake, nail trauma, periungual erythema, vesicular lesion, or dermatitis. In both patients, the nail changes were temporary with spontaneous normal nail plate regrowth several months later. A diagnosis of onychomadesis was made.
The etiology of onychomadesis includes drug ingestion, especially chemotherapy; severe systemic diseases; high fever; infection, including viral illnesses such as influenza, measles, and HFMD; and idiopathic onychomadesis.1,2,5,10 In 2000, Clementz and Mancini1 reported 5 children with nail matrix arrest following HFMD and suggested an epidemic caused by the same virus strain. Bernier et al2 reported another 4 cases and suggested more than one viral strain may have been implicated in the nail matrix arrest. Although these authors list HFMD as one of the causes of onychomadesis,1,2 the number of cases reported was small; however, studies with a larger number of cases and even outbreak have been reported more recently.3-8 Salazar et al3 reported an onychomadesis outbreak associated with HFMD in Valencia, Spain, in 2008 (N=298). This outbreak primarily was caused by coxsackievirus (CV) A10 (49% of cases).5 Another onychomadesis outbreak occurred in Saragossa, Spain, in 2008, and CV B1, B2, and unidentified nonpoliovirus enterovirus were isolated.6 Outbreaks also occurred in Finland in 2008, and the causative agents were identified as CV A6 and A10.7,8 The latency period for onychomadesis following HFMD was 1 to 2 months (mean, 40 days), and the majority of cases occurred in patients younger than 6 years.1-5 Not all of the nails were involved; in one report, each patient shed only 4 nails on average.6
Although there is a definite relationship between HFMD and onychomadesis, the mechanism is still unclear. Some authors claim that nail matrix arrest is caused by high fever10; however, we found that 40% (2/5)1 to 63% (10/16)4 of reported cases did not have a fever. Additionally, only 1 of our patients had fever. Therefore high fever–induced nail matrix arrest is not a reasonable explanation. Davia et al5 observed no relationship between onychomadesis and the severity of HFMD. In addition, no serious complications of HFMD were mentioned in prior reports.
We propose that HFMD-related onychomadesis is caused by the viral infection itself, rather than by severe systemic disease.1-5,7 Certain viral strains associated with HFMD can induce arrest of nail matrix activity. Osterback et al7 detected CV A6 in shed nail fragments and suggested that virus replication damaged the nail matrix and resulted in temporary nail dystrophy. This hypothesis can explain that only some nails, not all, were involved. In our cases, we noted an incomplete and slanted cleft on the thumbnail (Figure 2). We also found that incomplete onychomadesis appeared in the clinical photograph from a prior report.5 The slanted cleft in our case may be caused by secondary external force after original incomplete onychomadesis or a different rate of nail regrowth because of different intensity of nail matrix damage. The phenomenon of incomplete onychomadesis in the same nail further suggests the mechanism of onychomadesis following HFMD is localized nail matrix damage.
In conclusion, we report 2 cases of onychomadesis associated with HFMD. Our report highlights that there is no racial difference in post-HFMD onychomadesis. These cases highlight that HFMD is an important cause of onychomadesis, especially in children. We suggest that certain viral strains associated with HFMD may specifically arrest nail matrix growth activity, regardless of fever or disease severity.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Bernier V, Labreze C, Bury F, et al. Nail matrix arrest in the course of hand, foot and mouth disease. Eur J Pediatr. 2001;160:649-651.
- Salazar A, Febrer I, Guiral S, et al. Onychomadesis outbreak in Valencia, Spain, June 2008. Euro Surveill. 2008;13:18917.
- Redondo Granado MJ, Torres Hinojal MC, Izquierdo López B. Post viral onychomadesis outbreak in Valladolid [in Spanish]. An Pediatr (Barc). 2009;71:436-439.
- Davia JL, Bel PH, Ninet VZ, et al. Onychomadesis outbreak in Valencia, Spain associated with hand, foot, and mouth disease caused by enteroviruses. Pediatr Dermatol. 2011;28:1-5.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Habif TP. Nail diseases. In: Habif TP, ed. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:947-973.
To the Editor:
Onychomadesis is characterized by separation of the nail plate from the matrix due to a temporary arrest in nail matrix activity. Hand-foot-and-mouth disease (HFMD) is a relatively common viral infection, especially in children. Although the relationship between onychomadesis and HFMD has been noted, there are few reports in the literature.1-9 We present 2 cases of onychomadesis following HFMD in Taiwanese siblings.
A 3-year-old girl presented with proximal nail plate detachment from the proximal nail fold on the bilateral great toenails (Figure 1) and a transverse whole-thickness sulcus on the bilateral thumbnails (Figure 2) of several weeks’ duration. Her 6-year-old sister had similar nail changes. Hand-foot-and-mouth disease was diagnosed about 4 weeks prior to nail changes. The mother reported that only the younger sister experienced fever. There was no history of notable medication intake, nail trauma, periungual erythema, vesicular lesion, or dermatitis. In both patients, the nail changes were temporary with spontaneous normal nail plate regrowth several months later. A diagnosis of onychomadesis was made.
The etiology of onychomadesis includes drug ingestion, especially chemotherapy; severe systemic diseases; high fever; infection, including viral illnesses such as influenza, measles, and HFMD; and idiopathic onychomadesis.1,2,5,10 In 2000, Clementz and Mancini1 reported 5 children with nail matrix arrest following HFMD and suggested an epidemic caused by the same virus strain. Bernier et al2 reported another 4 cases and suggested more than one viral strain may have been implicated in the nail matrix arrest. Although these authors list HFMD as one of the causes of onychomadesis,1,2 the number of cases reported was small; however, studies with a larger number of cases and even outbreak have been reported more recently.3-8 Salazar et al3 reported an onychomadesis outbreak associated with HFMD in Valencia, Spain, in 2008 (N=298). This outbreak primarily was caused by coxsackievirus (CV) A10 (49% of cases).5 Another onychomadesis outbreak occurred in Saragossa, Spain, in 2008, and CV B1, B2, and unidentified nonpoliovirus enterovirus were isolated.6 Outbreaks also occurred in Finland in 2008, and the causative agents were identified as CV A6 and A10.7,8 The latency period for onychomadesis following HFMD was 1 to 2 months (mean, 40 days), and the majority of cases occurred in patients younger than 6 years.1-5 Not all of the nails were involved; in one report, each patient shed only 4 nails on average.6
Although there is a definite relationship between HFMD and onychomadesis, the mechanism is still unclear. Some authors claim that nail matrix arrest is caused by high fever10; however, we found that 40% (2/5)1 to 63% (10/16)4 of reported cases did not have a fever. Additionally, only 1 of our patients had fever. Therefore high fever–induced nail matrix arrest is not a reasonable explanation. Davia et al5 observed no relationship between onychomadesis and the severity of HFMD. In addition, no serious complications of HFMD were mentioned in prior reports.
We propose that HFMD-related onychomadesis is caused by the viral infection itself, rather than by severe systemic disease.1-5,7 Certain viral strains associated with HFMD can induce arrest of nail matrix activity. Osterback et al7 detected CV A6 in shed nail fragments and suggested that virus replication damaged the nail matrix and resulted in temporary nail dystrophy. This hypothesis can explain that only some nails, not all, were involved. In our cases, we noted an incomplete and slanted cleft on the thumbnail (Figure 2). We also found that incomplete onychomadesis appeared in the clinical photograph from a prior report.5 The slanted cleft in our case may be caused by secondary external force after original incomplete onychomadesis or a different rate of nail regrowth because of different intensity of nail matrix damage. The phenomenon of incomplete onychomadesis in the same nail further suggests the mechanism of onychomadesis following HFMD is localized nail matrix damage.
In conclusion, we report 2 cases of onychomadesis associated with HFMD. Our report highlights that there is no racial difference in post-HFMD onychomadesis. These cases highlight that HFMD is an important cause of onychomadesis, especially in children. We suggest that certain viral strains associated with HFMD may specifically arrest nail matrix growth activity, regardless of fever or disease severity.
To the Editor:
Onychomadesis is characterized by separation of the nail plate from the matrix due to a temporary arrest in nail matrix activity. Hand-foot-and-mouth disease (HFMD) is a relatively common viral infection, especially in children. Although the relationship between onychomadesis and HFMD has been noted, there are few reports in the literature.1-9 We present 2 cases of onychomadesis following HFMD in Taiwanese siblings.
A 3-year-old girl presented with proximal nail plate detachment from the proximal nail fold on the bilateral great toenails (Figure 1) and a transverse whole-thickness sulcus on the bilateral thumbnails (Figure 2) of several weeks’ duration. Her 6-year-old sister had similar nail changes. Hand-foot-and-mouth disease was diagnosed about 4 weeks prior to nail changes. The mother reported that only the younger sister experienced fever. There was no history of notable medication intake, nail trauma, periungual erythema, vesicular lesion, or dermatitis. In both patients, the nail changes were temporary with spontaneous normal nail plate regrowth several months later. A diagnosis of onychomadesis was made.
The etiology of onychomadesis includes drug ingestion, especially chemotherapy; severe systemic diseases; high fever; infection, including viral illnesses such as influenza, measles, and HFMD; and idiopathic onychomadesis.1,2,5,10 In 2000, Clementz and Mancini1 reported 5 children with nail matrix arrest following HFMD and suggested an epidemic caused by the same virus strain. Bernier et al2 reported another 4 cases and suggested more than one viral strain may have been implicated in the nail matrix arrest. Although these authors list HFMD as one of the causes of onychomadesis,1,2 the number of cases reported was small; however, studies with a larger number of cases and even outbreak have been reported more recently.3-8 Salazar et al3 reported an onychomadesis outbreak associated with HFMD in Valencia, Spain, in 2008 (N=298). This outbreak primarily was caused by coxsackievirus (CV) A10 (49% of cases).5 Another onychomadesis outbreak occurred in Saragossa, Spain, in 2008, and CV B1, B2, and unidentified nonpoliovirus enterovirus were isolated.6 Outbreaks also occurred in Finland in 2008, and the causative agents were identified as CV A6 and A10.7,8 The latency period for onychomadesis following HFMD was 1 to 2 months (mean, 40 days), and the majority of cases occurred in patients younger than 6 years.1-5 Not all of the nails were involved; in one report, each patient shed only 4 nails on average.6
Although there is a definite relationship between HFMD and onychomadesis, the mechanism is still unclear. Some authors claim that nail matrix arrest is caused by high fever10; however, we found that 40% (2/5)1 to 63% (10/16)4 of reported cases did not have a fever. Additionally, only 1 of our patients had fever. Therefore high fever–induced nail matrix arrest is not a reasonable explanation. Davia et al5 observed no relationship between onychomadesis and the severity of HFMD. In addition, no serious complications of HFMD were mentioned in prior reports.
We propose that HFMD-related onychomadesis is caused by the viral infection itself, rather than by severe systemic disease.1-5,7 Certain viral strains associated with HFMD can induce arrest of nail matrix activity. Osterback et al7 detected CV A6 in shed nail fragments and suggested that virus replication damaged the nail matrix and resulted in temporary nail dystrophy. This hypothesis can explain that only some nails, not all, were involved. In our cases, we noted an incomplete and slanted cleft on the thumbnail (Figure 2). We also found that incomplete onychomadesis appeared in the clinical photograph from a prior report.5 The slanted cleft in our case may be caused by secondary external force after original incomplete onychomadesis or a different rate of nail regrowth because of different intensity of nail matrix damage. The phenomenon of incomplete onychomadesis in the same nail further suggests the mechanism of onychomadesis following HFMD is localized nail matrix damage.
In conclusion, we report 2 cases of onychomadesis associated with HFMD. Our report highlights that there is no racial difference in post-HFMD onychomadesis. These cases highlight that HFMD is an important cause of onychomadesis, especially in children. We suggest that certain viral strains associated with HFMD may specifically arrest nail matrix growth activity, regardless of fever or disease severity.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Bernier V, Labreze C, Bury F, et al. Nail matrix arrest in the course of hand, foot and mouth disease. Eur J Pediatr. 2001;160:649-651.
- Salazar A, Febrer I, Guiral S, et al. Onychomadesis outbreak in Valencia, Spain, June 2008. Euro Surveill. 2008;13:18917.
- Redondo Granado MJ, Torres Hinojal MC, Izquierdo López B. Post viral onychomadesis outbreak in Valladolid [in Spanish]. An Pediatr (Barc). 2009;71:436-439.
- Davia JL, Bel PH, Ninet VZ, et al. Onychomadesis outbreak in Valencia, Spain associated with hand, foot, and mouth disease caused by enteroviruses. Pediatr Dermatol. 2011;28:1-5.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Habif TP. Nail diseases. In: Habif TP, ed. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:947-973.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Bernier V, Labreze C, Bury F, et al. Nail matrix arrest in the course of hand, foot and mouth disease. Eur J Pediatr. 2001;160:649-651.
- Salazar A, Febrer I, Guiral S, et al. Onychomadesis outbreak in Valencia, Spain, June 2008. Euro Surveill. 2008;13:18917.
- Redondo Granado MJ, Torres Hinojal MC, Izquierdo López B. Post viral onychomadesis outbreak in Valladolid [in Spanish]. An Pediatr (Barc). 2009;71:436-439.
- Davia JL, Bel PH, Ninet VZ, et al. Onychomadesis outbreak in Valencia, Spain associated with hand, foot, and mouth disease caused by enteroviruses. Pediatr Dermatol. 2011;28:1-5.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Habif TP. Nail diseases. In: Habif TP, ed. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:947-973.
Practice Points
- Onychomadesis is a late complication of hand-foot-and-mouth disease (HFMD) with a latency period of 1 to 2 months.
- Although the mechanism between onychomadesis and HFMD is still unclear, we propose that it is caused by the viral infection itself rather than severe systemic disease.
The Elongated Dermatofibroma: A New Dermoscopic Variant?
To the Editor:
Dermatofibroma is a common cutaneous lesion that most frequently affects young or middle-aged adults, especially women.1 Clinically, it appears as a firm, pink or brown nodule. It may be painful or show a tendency for scarring. The pathognomonic feature of dermatofibroma, regarded as a fibrohistiocytic tumor, is the so-called button sign caused by skin depression following pressure. We present a unique case of elongated dermatofibroma with a linear, white, scarlike patch with a brownish pigmented network and globules.
A 40-year-old woman presented with a linear elongated lesion localized to the right side of the infrascapular region of 10 years’ duration. The lesion initially was a small brownish plaque. There was no history of trauma or scratching. Over the next 10 years, the lesion slowly progressed, finally becoming a linear, atrophic, brownish plaque that was 2.5-cm long (Figure 1). The button sign was positive. On dermoscopy the central, elongated, white patch was visualized not as a typical round patch but as a scarlike white line (Figure 2A) surrounded by a brownish network that was especially pronounced in the distal parts of the lesion. In the upper part of the lesion, multiple marginally disseminated, dark brown dots were present. Brownish globules within the linear white patch also were observed in the lower central part. Figure 2B presents a dermoscopic picture of the linear variant of dermatofibroma. For cosmetic reasons, the patient underwent total surgical excision of the lesion. Histopathology revealed distinct characteristics of dermatofibroma (Figures 3A and 3B).
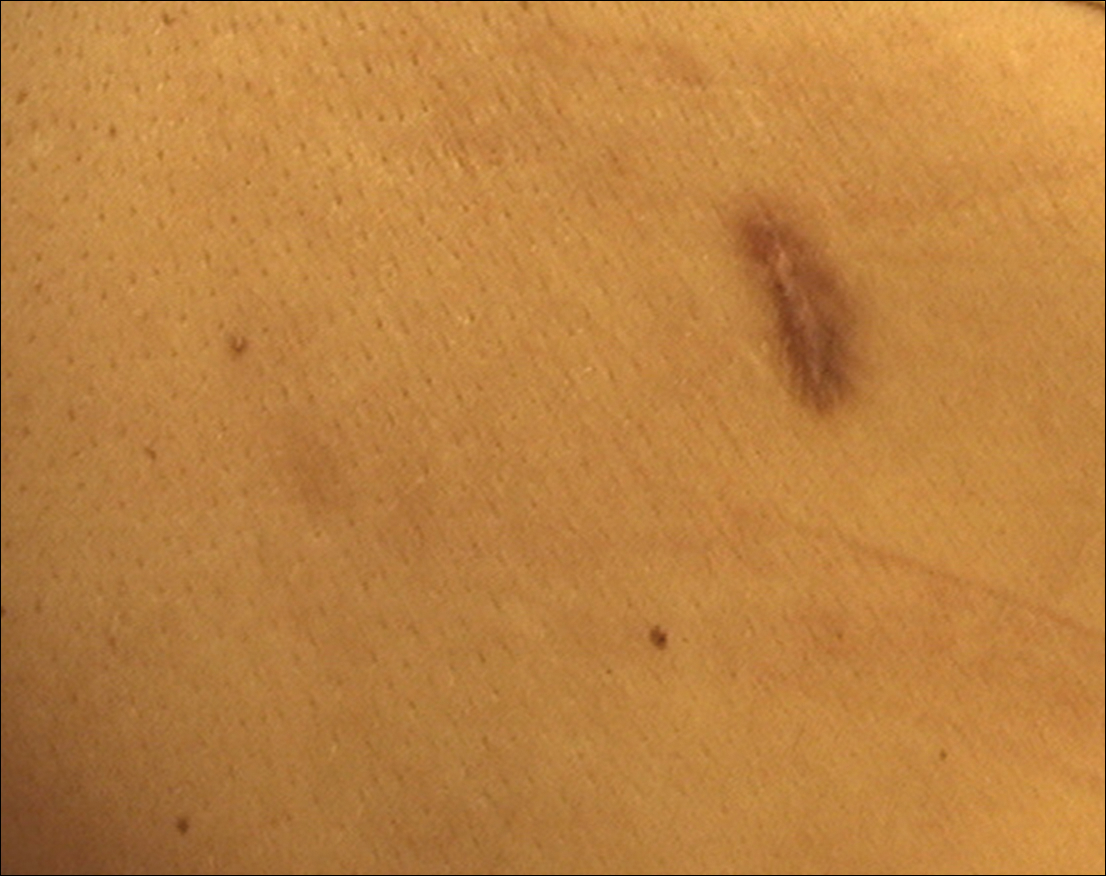
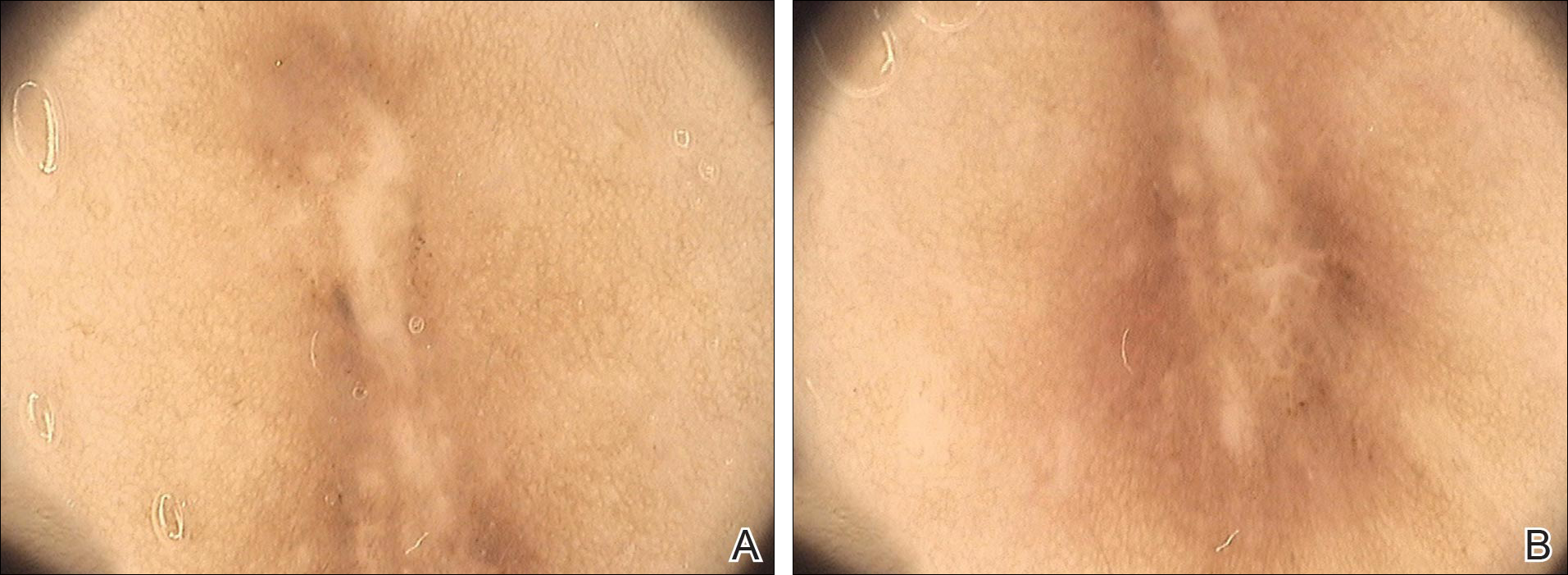
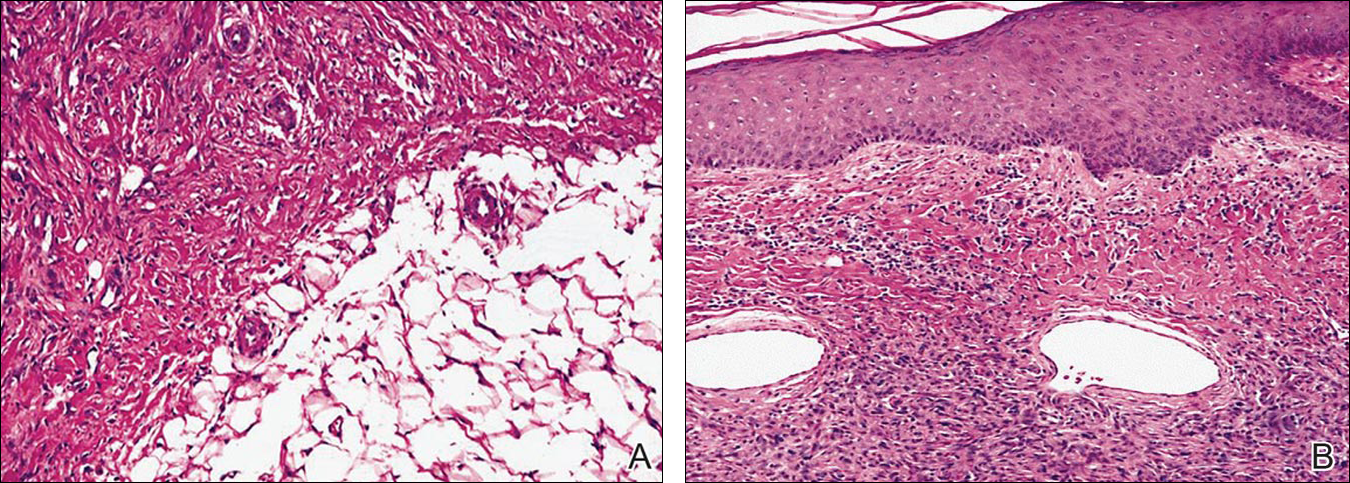
The most common features of dermatofibromas seen in polarized and nonpolarized dermoscopy are central white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.2 Kilinc Karaarslan et al3 described atypical dermatofibromas with linear irregular crypts, which were seen in 26.9% of all studied cases. These irregular crypts were mainly medium in size (10 lesions), with only 2 lesions being tiny and regularly distributed. Only one lesion had atypical clinical and dermoscopic features occurring as an atrophic plaque with multiple small scarlike areas and peripherally distributed pigment network.3 Based on this typology, we believe our patient represents a case of elongated dermatofibroma that could be an atrophic variant of dermatofibroma. This form would not appear as a small scarlike area with pigment network in a somewhat patchy distribution3 but as a scarlike linear chord with a bipolar pigment network. Zaballos et al1 described 10 dermoscopic patterns of dermatofibroma (N=412); the most common was a central white patch and peripheral pigment network in approximately 35% of cases. A white scarlike patch was observed in 57.0% of dermat-ofibromas in 4 variants: (1) a solitary structure located in the center; (2) multiple white scarlike patches; (3) white scarlike patch extending throughout the lesion or irregularly distributed; and (4) white network (central, total, or irregular).1 Agero et al2 first described the new feature as a central white patch characterized by shiny white streaks. The most frequent dermoscopic pattern associated with dermatofibromas is the central white scarlike patch and peripheral delicate pigment network.1,4 Arpaia et al4 observed that dermoscopic patterns may correspond to distinct sequential stages of the formation of dermatofibroma. The linear character we described may be related to a variant of scarring keloid dermatofibroma.5
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
- Kilinc Karaarslan I, Gencoglan G, Akalin T, et al. Different dermoscopic faces of dermatofibromas. J Am Acad Dermatol. 2007;57:401-406.
- Arpaia N, Cassano N, Vena GA. Dermoscopic patterns of dermatofibroma. Dermatol Surg. 2005;31:1336-1339.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564-568.
To the Editor:
Dermatofibroma is a common cutaneous lesion that most frequently affects young or middle-aged adults, especially women.1 Clinically, it appears as a firm, pink or brown nodule. It may be painful or show a tendency for scarring. The pathognomonic feature of dermatofibroma, regarded as a fibrohistiocytic tumor, is the so-called button sign caused by skin depression following pressure. We present a unique case of elongated dermatofibroma with a linear, white, scarlike patch with a brownish pigmented network and globules.
A 40-year-old woman presented with a linear elongated lesion localized to the right side of the infrascapular region of 10 years’ duration. The lesion initially was a small brownish plaque. There was no history of trauma or scratching. Over the next 10 years, the lesion slowly progressed, finally becoming a linear, atrophic, brownish plaque that was 2.5-cm long (Figure 1). The button sign was positive. On dermoscopy the central, elongated, white patch was visualized not as a typical round patch but as a scarlike white line (Figure 2A) surrounded by a brownish network that was especially pronounced in the distal parts of the lesion. In the upper part of the lesion, multiple marginally disseminated, dark brown dots were present. Brownish globules within the linear white patch also were observed in the lower central part. Figure 2B presents a dermoscopic picture of the linear variant of dermatofibroma. For cosmetic reasons, the patient underwent total surgical excision of the lesion. Histopathology revealed distinct characteristics of dermatofibroma (Figures 3A and 3B).
The most common features of dermatofibromas seen in polarized and nonpolarized dermoscopy are central white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.2 Kilinc Karaarslan et al3 described atypical dermatofibromas with linear irregular crypts, which were seen in 26.9% of all studied cases. These irregular crypts were mainly medium in size (10 lesions), with only 2 lesions being tiny and regularly distributed. Only one lesion had atypical clinical and dermoscopic features occurring as an atrophic plaque with multiple small scarlike areas and peripherally distributed pigment network.3 Based on this typology, we believe our patient represents a case of elongated dermatofibroma that could be an atrophic variant of dermatofibroma. This form would not appear as a small scarlike area with pigment network in a somewhat patchy distribution3 but as a scarlike linear chord with a bipolar pigment network. Zaballos et al1 described 10 dermoscopic patterns of dermatofibroma (N=412); the most common was a central white patch and peripheral pigment network in approximately 35% of cases. A white scarlike patch was observed in 57.0% of dermat-ofibromas in 4 variants: (1) a solitary structure located in the center; (2) multiple white scarlike patches; (3) white scarlike patch extending throughout the lesion or irregularly distributed; and (4) white network (central, total, or irregular).1 Agero et al2 first described the new feature as a central white patch characterized by shiny white streaks. The most frequent dermoscopic pattern associated with dermatofibromas is the central white scarlike patch and peripheral delicate pigment network.1,4 Arpaia et al4 observed that dermoscopic patterns may correspond to distinct sequential stages of the formation of dermatofibroma. The linear character we described may be related to a variant of scarring keloid dermatofibroma.5
To the Editor:
Dermatofibroma is a common cutaneous lesion that most frequently affects young or middle-aged adults, especially women.1 Clinically, it appears as a firm, pink or brown nodule. It may be painful or show a tendency for scarring. The pathognomonic feature of dermatofibroma, regarded as a fibrohistiocytic tumor, is the so-called button sign caused by skin depression following pressure. We present a unique case of elongated dermatofibroma with a linear, white, scarlike patch with a brownish pigmented network and globules.
A 40-year-old woman presented with a linear elongated lesion localized to the right side of the infrascapular region of 10 years’ duration. The lesion initially was a small brownish plaque. There was no history of trauma or scratching. Over the next 10 years, the lesion slowly progressed, finally becoming a linear, atrophic, brownish plaque that was 2.5-cm long (Figure 1). The button sign was positive. On dermoscopy the central, elongated, white patch was visualized not as a typical round patch but as a scarlike white line (Figure 2A) surrounded by a brownish network that was especially pronounced in the distal parts of the lesion. In the upper part of the lesion, multiple marginally disseminated, dark brown dots were present. Brownish globules within the linear white patch also were observed in the lower central part. Figure 2B presents a dermoscopic picture of the linear variant of dermatofibroma. For cosmetic reasons, the patient underwent total surgical excision of the lesion. Histopathology revealed distinct characteristics of dermatofibroma (Figures 3A and 3B).
The most common features of dermatofibromas seen in polarized and nonpolarized dermoscopy are central white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.2 Kilinc Karaarslan et al3 described atypical dermatofibromas with linear irregular crypts, which were seen in 26.9% of all studied cases. These irregular crypts were mainly medium in size (10 lesions), with only 2 lesions being tiny and regularly distributed. Only one lesion had atypical clinical and dermoscopic features occurring as an atrophic plaque with multiple small scarlike areas and peripherally distributed pigment network.3 Based on this typology, we believe our patient represents a case of elongated dermatofibroma that could be an atrophic variant of dermatofibroma. This form would not appear as a small scarlike area with pigment network in a somewhat patchy distribution3 but as a scarlike linear chord with a bipolar pigment network. Zaballos et al1 described 10 dermoscopic patterns of dermatofibroma (N=412); the most common was a central white patch and peripheral pigment network in approximately 35% of cases. A white scarlike patch was observed in 57.0% of dermat-ofibromas in 4 variants: (1) a solitary structure located in the center; (2) multiple white scarlike patches; (3) white scarlike patch extending throughout the lesion or irregularly distributed; and (4) white network (central, total, or irregular).1 Agero et al2 first described the new feature as a central white patch characterized by shiny white streaks. The most frequent dermoscopic pattern associated with dermatofibromas is the central white scarlike patch and peripheral delicate pigment network.1,4 Arpaia et al4 observed that dermoscopic patterns may correspond to distinct sequential stages of the formation of dermatofibroma. The linear character we described may be related to a variant of scarring keloid dermatofibroma.5
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
- Kilinc Karaarslan I, Gencoglan G, Akalin T, et al. Different dermoscopic faces of dermatofibromas. J Am Acad Dermatol. 2007;57:401-406.
- Arpaia N, Cassano N, Vena GA. Dermoscopic patterns of dermatofibroma. Dermatol Surg. 2005;31:1336-1339.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564-568.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
- Kilinc Karaarslan I, Gencoglan G, Akalin T, et al. Different dermoscopic faces of dermatofibromas. J Am Acad Dermatol. 2007;57:401-406.
- Arpaia N, Cassano N, Vena GA. Dermoscopic patterns of dermatofibroma. Dermatol Surg. 2005;31:1336-1339.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564-568.
Practice Points
- The most common features of dermatofibromas are white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.
- Dermoscopy may be used in the diagnostic workup of pigmented nonmelanocytic lesions.

Palmoplantar Pustular Eruption Due to Dabigatran
To the Editor:
A 71-year-old woman with hypertension and atrial fibrillation due to thyrotoxicosis was prescribed dabigatran for stroke prevention by her cardiologist. She also was taking pantoprazole, methimazole, and amiodarone at the time of presentation, all managed by her endocrinologist. She had no known drug allergies but reported a remote history of a palmar rash after eating shellfish. She otherwise had never had any problems with her skin and had no family history of psoriasis. She had a history of smoking 50 packs per year but had quit 6 months prior to presentation. After two 150-mg doses of dabigatran, she noticed numerous mildly tender and itchy eruptions on the palmar and plantar surfaces with no associated respiratory, oropharyngeal, or constitutional symptoms. She denied any recent shellfish ingestion. On dermatologic examination, numerous discreet pustules were present on the bilateral palmar and plantar surfaces with minimal erythema of the underlying skin (Figure).
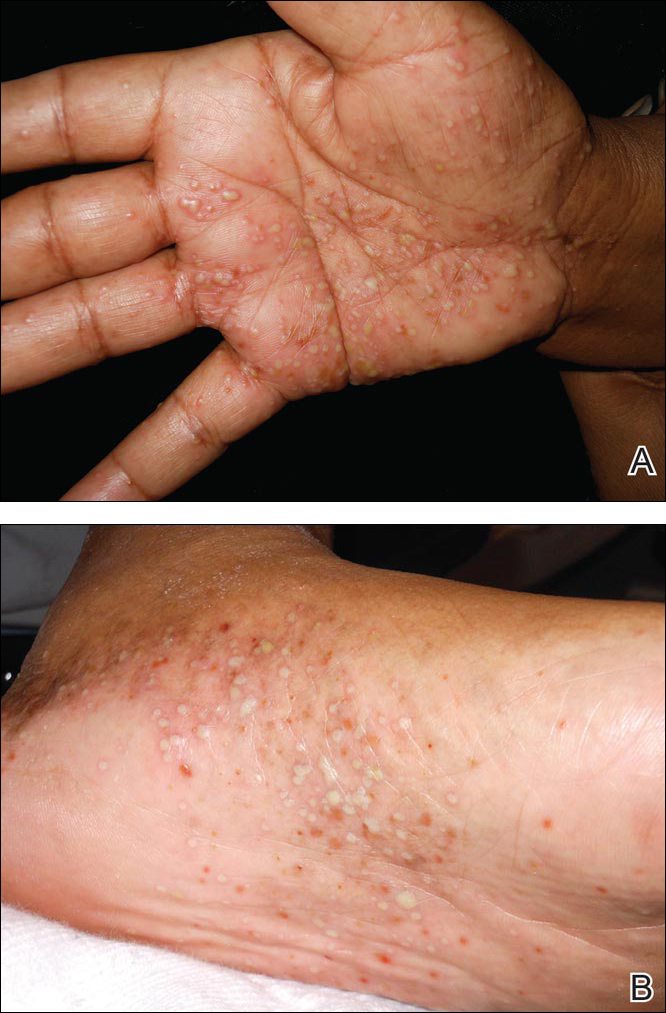
A punch biopsy was taken from a newly forming lesion on the right palm. Histopathology revealed mild hyperkeratosis, spongiosis with lymphocyte exocytosis, intraepidermal vesiculation, and a sparse upper dermal and perivascular lymphohistiocytic infiltration. No neutrophils or microabscesses were seen. Staining with periodic acid–Schiff revealed no fungi, and S-100 staining revealed numerous Langerhans cells in the epidermis. Although the skin lesions clinically appeared pustular, the results were consistent with an eczematous drug reaction. Laboratory values, including a complete blood cell count, iron studies, chemistry panels, liver function, thyroid function, and coagulation studies, were remarkable only for mild anemia. The patient declined any topical or systemic skin treatment. Dabigatran was discontinued, and the lesions began to clear immediately thereafter. Dabigatran was not reintroduced. Enoxaparin subsequently was prescribed for anticoagulation. The diagnosis of a drug reaction due to dabigatran was made, which was supported with a score of 7 on the Naranjo scale (0=doubtful; 1–4=possible; 5–8=probable; ≥9=definite) for determining probability of drug-induced adverse reactions.1 The differential diagnosis for the skin eruption included palmoplantar pustular psoriasis, dyshidrotic eczema, and allergic contact dermatitis, but the clinical history did not support these diagnoses.
Dabigatran is a direct thrombin inhibitor used to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Based on results of the RE-LY (Randomization Evaluation of Long-term Anticoagulation Therapy) trial published in 2009, dabigatran 150 mg twice daily significantly reduced the risk for stroke and systemic emboli in patients with atrial fibrillation compared to warfarin (annual risk, 1.11% vs 1.69%; relative risk, 0.66; 95% CI, 0.53-0.82; P<.001) with the advantage of not requiring frequent monitoring of the international normalized ratio.2 The most common adverse effect of dabigatran in this trial was dyspepsia (11.3% vs 5.8%). Drug hypersensitivity, allergic edema, and anaphylaxis were reported in less than 0.1% of patients taking dabigatran.2
According to a PubMed search of articles indexed for MEDLINE using the search terms dabigatran cutaneous reaction and dabigatran rash, 4 case reports of cutaneous eruption due to dabigatran were identified. In one report, a 20-year-old man with atrial fibrillation developed an eruption similar to our patient on the thigh and forearm after 2 weeks of taking oral dabigatran 150 mg twice daily. It resolved without complication after topical corticosteroid use and discontinuation of dabigatran.3 In another report, a 78-year-old man presented to the emergency department after taking two 150-mg doses of dabigatran with a diffuse, full-body, pruritic rash that resolved with oral diphenhydramine and discontinuation of dabigatran.4 A third case described a 59-year-old man who was taking 150 mg dabigatran twice daily for 5 days before developing a rash.5 The fourth case involved a 74-year-old woman who developed leukocytoclastic vasculitis 1 week after taking dabigatran 150 mg twice daily.6
It is important to monitor for and report hypersensitivity reactions in patients taking dabigatran. Drug exanthems may cause discomfort or even herald more serious hypersensitivity reactions. Patients experiencing these reactions may discontinue therapy without notifying a physician and consequently place themselves at risk for embolism or stroke.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation [published online August 30, 2009]. N Engl J Med. 2009;361:1139-1151.
- Whitehead H, Boyd J, Blais D, et al. Drug induced exanthem following dabigatran. Ann Pharmacother. 2011;45:e53.
- Eid TJ, Shah SA. Dabigatran-induced rash. Am J Health Syst Pharm. 2011;68:1489-1490.
- To K, Reynolds C, Spinler SA. Rash associated with dabigatran etrexilate. Pharmacotherapy. 2013;33:e23-e27.
- Cakmak MA, Sahin S, Cinar N, et al. Adverse skin reaction caused by dabigatran. Eur Rev Med Pharmacol Sci. 2014;18:2595.
To the Editor:
A 71-year-old woman with hypertension and atrial fibrillation due to thyrotoxicosis was prescribed dabigatran for stroke prevention by her cardiologist. She also was taking pantoprazole, methimazole, and amiodarone at the time of presentation, all managed by her endocrinologist. She had no known drug allergies but reported a remote history of a palmar rash after eating shellfish. She otherwise had never had any problems with her skin and had no family history of psoriasis. She had a history of smoking 50 packs per year but had quit 6 months prior to presentation. After two 150-mg doses of dabigatran, she noticed numerous mildly tender and itchy eruptions on the palmar and plantar surfaces with no associated respiratory, oropharyngeal, or constitutional symptoms. She denied any recent shellfish ingestion. On dermatologic examination, numerous discreet pustules were present on the bilateral palmar and plantar surfaces with minimal erythema of the underlying skin (Figure).
A punch biopsy was taken from a newly forming lesion on the right palm. Histopathology revealed mild hyperkeratosis, spongiosis with lymphocyte exocytosis, intraepidermal vesiculation, and a sparse upper dermal and perivascular lymphohistiocytic infiltration. No neutrophils or microabscesses were seen. Staining with periodic acid–Schiff revealed no fungi, and S-100 staining revealed numerous Langerhans cells in the epidermis. Although the skin lesions clinically appeared pustular, the results were consistent with an eczematous drug reaction. Laboratory values, including a complete blood cell count, iron studies, chemistry panels, liver function, thyroid function, and coagulation studies, were remarkable only for mild anemia. The patient declined any topical or systemic skin treatment. Dabigatran was discontinued, and the lesions began to clear immediately thereafter. Dabigatran was not reintroduced. Enoxaparin subsequently was prescribed for anticoagulation. The diagnosis of a drug reaction due to dabigatran was made, which was supported with a score of 7 on the Naranjo scale (0=doubtful; 1–4=possible; 5–8=probable; ≥9=definite) for determining probability of drug-induced adverse reactions.1 The differential diagnosis for the skin eruption included palmoplantar pustular psoriasis, dyshidrotic eczema, and allergic contact dermatitis, but the clinical history did not support these diagnoses.
Dabigatran is a direct thrombin inhibitor used to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Based on results of the RE-LY (Randomization Evaluation of Long-term Anticoagulation Therapy) trial published in 2009, dabigatran 150 mg twice daily significantly reduced the risk for stroke and systemic emboli in patients with atrial fibrillation compared to warfarin (annual risk, 1.11% vs 1.69%; relative risk, 0.66; 95% CI, 0.53-0.82; P<.001) with the advantage of not requiring frequent monitoring of the international normalized ratio.2 The most common adverse effect of dabigatran in this trial was dyspepsia (11.3% vs 5.8%). Drug hypersensitivity, allergic edema, and anaphylaxis were reported in less than 0.1% of patients taking dabigatran.2
According to a PubMed search of articles indexed for MEDLINE using the search terms dabigatran cutaneous reaction and dabigatran rash, 4 case reports of cutaneous eruption due to dabigatran were identified. In one report, a 20-year-old man with atrial fibrillation developed an eruption similar to our patient on the thigh and forearm after 2 weeks of taking oral dabigatran 150 mg twice daily. It resolved without complication after topical corticosteroid use and discontinuation of dabigatran.3 In another report, a 78-year-old man presented to the emergency department after taking two 150-mg doses of dabigatran with a diffuse, full-body, pruritic rash that resolved with oral diphenhydramine and discontinuation of dabigatran.4 A third case described a 59-year-old man who was taking 150 mg dabigatran twice daily for 5 days before developing a rash.5 The fourth case involved a 74-year-old woman who developed leukocytoclastic vasculitis 1 week after taking dabigatran 150 mg twice daily.6
It is important to monitor for and report hypersensitivity reactions in patients taking dabigatran. Drug exanthems may cause discomfort or even herald more serious hypersensitivity reactions. Patients experiencing these reactions may discontinue therapy without notifying a physician and consequently place themselves at risk for embolism or stroke.
To the Editor:
A 71-year-old woman with hypertension and atrial fibrillation due to thyrotoxicosis was prescribed dabigatran for stroke prevention by her cardiologist. She also was taking pantoprazole, methimazole, and amiodarone at the time of presentation, all managed by her endocrinologist. She had no known drug allergies but reported a remote history of a palmar rash after eating shellfish. She otherwise had never had any problems with her skin and had no family history of psoriasis. She had a history of smoking 50 packs per year but had quit 6 months prior to presentation. After two 150-mg doses of dabigatran, she noticed numerous mildly tender and itchy eruptions on the palmar and plantar surfaces with no associated respiratory, oropharyngeal, or constitutional symptoms. She denied any recent shellfish ingestion. On dermatologic examination, numerous discreet pustules were present on the bilateral palmar and plantar surfaces with minimal erythema of the underlying skin (Figure).
A punch biopsy was taken from a newly forming lesion on the right palm. Histopathology revealed mild hyperkeratosis, spongiosis with lymphocyte exocytosis, intraepidermal vesiculation, and a sparse upper dermal and perivascular lymphohistiocytic infiltration. No neutrophils or microabscesses were seen. Staining with periodic acid–Schiff revealed no fungi, and S-100 staining revealed numerous Langerhans cells in the epidermis. Although the skin lesions clinically appeared pustular, the results were consistent with an eczematous drug reaction. Laboratory values, including a complete blood cell count, iron studies, chemistry panels, liver function, thyroid function, and coagulation studies, were remarkable only for mild anemia. The patient declined any topical or systemic skin treatment. Dabigatran was discontinued, and the lesions began to clear immediately thereafter. Dabigatran was not reintroduced. Enoxaparin subsequently was prescribed for anticoagulation. The diagnosis of a drug reaction due to dabigatran was made, which was supported with a score of 7 on the Naranjo scale (0=doubtful; 1–4=possible; 5–8=probable; ≥9=definite) for determining probability of drug-induced adverse reactions.1 The differential diagnosis for the skin eruption included palmoplantar pustular psoriasis, dyshidrotic eczema, and allergic contact dermatitis, but the clinical history did not support these diagnoses.
Dabigatran is a direct thrombin inhibitor used to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Based on results of the RE-LY (Randomization Evaluation of Long-term Anticoagulation Therapy) trial published in 2009, dabigatran 150 mg twice daily significantly reduced the risk for stroke and systemic emboli in patients with atrial fibrillation compared to warfarin (annual risk, 1.11% vs 1.69%; relative risk, 0.66; 95% CI, 0.53-0.82; P<.001) with the advantage of not requiring frequent monitoring of the international normalized ratio.2 The most common adverse effect of dabigatran in this trial was dyspepsia (11.3% vs 5.8%). Drug hypersensitivity, allergic edema, and anaphylaxis were reported in less than 0.1% of patients taking dabigatran.2
According to a PubMed search of articles indexed for MEDLINE using the search terms dabigatran cutaneous reaction and dabigatran rash, 4 case reports of cutaneous eruption due to dabigatran were identified. In one report, a 20-year-old man with atrial fibrillation developed an eruption similar to our patient on the thigh and forearm after 2 weeks of taking oral dabigatran 150 mg twice daily. It resolved without complication after topical corticosteroid use and discontinuation of dabigatran.3 In another report, a 78-year-old man presented to the emergency department after taking two 150-mg doses of dabigatran with a diffuse, full-body, pruritic rash that resolved with oral diphenhydramine and discontinuation of dabigatran.4 A third case described a 59-year-old man who was taking 150 mg dabigatran twice daily for 5 days before developing a rash.5 The fourth case involved a 74-year-old woman who developed leukocytoclastic vasculitis 1 week after taking dabigatran 150 mg twice daily.6
It is important to monitor for and report hypersensitivity reactions in patients taking dabigatran. Drug exanthems may cause discomfort or even herald more serious hypersensitivity reactions. Patients experiencing these reactions may discontinue therapy without notifying a physician and consequently place themselves at risk for embolism or stroke.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation [published online August 30, 2009]. N Engl J Med. 2009;361:1139-1151.
- Whitehead H, Boyd J, Blais D, et al. Drug induced exanthem following dabigatran. Ann Pharmacother. 2011;45:e53.
- Eid TJ, Shah SA. Dabigatran-induced rash. Am J Health Syst Pharm. 2011;68:1489-1490.
- To K, Reynolds C, Spinler SA. Rash associated with dabigatran etrexilate. Pharmacotherapy. 2013;33:e23-e27.
- Cakmak MA, Sahin S, Cinar N, et al. Adverse skin reaction caused by dabigatran. Eur Rev Med Pharmacol Sci. 2014;18:2595.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation [published online August 30, 2009]. N Engl J Med. 2009;361:1139-1151.
- Whitehead H, Boyd J, Blais D, et al. Drug induced exanthem following dabigatran. Ann Pharmacother. 2011;45:e53.
- Eid TJ, Shah SA. Dabigatran-induced rash. Am J Health Syst Pharm. 2011;68:1489-1490.
- To K, Reynolds C, Spinler SA. Rash associated with dabigatran etrexilate. Pharmacotherapy. 2013;33:e23-e27.
- Cakmak MA, Sahin S, Cinar N, et al. Adverse skin reaction caused by dabigatran. Eur Rev Med Pharmacol Sci. 2014;18:2595.
Practice Points
- Dabigatran is a direct thrombin inhibitor used in patients with atrial fibrillation to prevent thromboembolic events.
- Although the most common adverse effects of dabigatran are bleeding and dyspepsia, clinicians also should be aware of the potential for cutaneous hypersensitivity reactions to this drug.