User login
Multiple Eruptive Dermatofibromas Associated With Down Syndrome
To the Editor:
Dermatofibromas (also known as fibrous histiocytomas) are benign fibrous nodules that most often arise as solitary lesions on the lower extremities. Multiple eruptive dermatofibromas (MEDFs) are uncommon and have been defined as more than 15 in number1 or 5 to 8 dermatofibromas appearing within 4 months.2 They have been reported in association with a number of conditions of immune dysregulation such as systemic lupus erythematosus, Sjögren syndrome, HIV infection, and leukemia.3 Multiple eruptive dermatofibromas also have been described in patients with Down syndrome (DS).4-7 We report a case of MEDFs in a patient with DS and review the literature on the association between MEDFs and DS.
A 38-year-old woman with DS, hidradenitis suppurativa, and hypothyroidism presented with multiple cutaneous lesions developing over the last year. The lesions continued to increase in number but were otherwise asymptomatic. Physical examination revealed approximately 20 rubbery, pink-tan papules measuring less than 1 cm in diameter that were scattered along the trunk (Figure, A), arms, and legs (Figure, B).
 consistent with dermatofibromas on the upper back and left leg")
The patient had no known history of immunosuppression or rheumatologic disease and was otherwise healthy. Basic laboratory tests including a complete blood cell count and antinuclear antibody titer were within reference range. The lesions were clinically consistent with dermatofibromas, but due to their increasing number within a short period of time, a biopsy of a representative lesion was performed to confirm the diagnosis.
The exact incidence of MEDFs is unknown, but they are rare, with one review finding only 50 cases reported from 1960 to 2002.8 They are increasingly recognized as a sign of potential immune dysregulation. Approximately 56% to 70% of cases are seen in patients with an underlying disease state; 80% are immune mediated.8,9 Interestingly, DS has long been associated with notable immune dysfunction,10,11 with evidence suggesting that trisomy 21 may result in widespread changes in gene expression that can lead to interferon activation.12
A PubMed search of articles indexed for MEDLINE using the terms dermatofibroma and Down, dermatofibroma and Down syndrome, eruptive dermatofibroma and Down syndrome, and multiple dermatofibroma and Down syndrome revealed 6 cases of MEDFs in patients with DS that have been reported since 2005.4-7 An additional report by Honda et al13 described a patient with DS who developed 7 dermatofibromas, but no time frame of development was specified. We reviewed the characteristics of 8 patients with DS with MEDFs, which included our patient (Table). The average age at time of presentation was 39 years (median age, 40 years). Six patients (75%) were female and 2 (25%) were male. Dermatofibromas were reported to appear over the course of months to years. Comorbidities included psoriatic arthritis (treated with methotrexate),6 thyroid disorders (ie, Graves disease),6 hypercholesterolemia,6 hidradenitis suppurativa, long-standing mild lymphopenia (1.4×109/L [reference range, 1.5−4.0×109/L]),4 and acute megakaryoblastic leukemia13 treated 15 years before the appearance of dermatofibromas.

Many dermatologic conditions have been reported at increased rates in individuals with DS, including seborrheic dermatitis, alopecia areata, syringomas, elastosis perforans serpiginosa, cutis marmorata, xerosis, and palmoplantar hyperkeratosis.14,15 Although drawing conclusions about associations between MEDFs and DS is limited by our small sample size, we have reported this case and reviewed existing cases of MEDFs in DS to highlight a potential association that may be underrecognized or underreported. More evidence is needed to determine the strength of the association between MEDFs and DS, but dermatologists should be aware that MEDFs may be an additional skin finding associated with DS that is related to the syndrome’s immune dysregulation.
- Baraf CS, Shapiro L. Multiple histiocytomas: report of a case. Arch Dermatol. 1970;101:588-590.
- Ammirati CT, Mann C, Hornstra IK. Multiple eruptive dermatofibromas in three men with HIV infection. Dermatology. 1997;4:344-348.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Lamb RC, Gangopadhyay M, MacDonald A. Multiple dermatofibromas in Down syndrome. Int J Dermatol. 2014;53:E274-E275.
- Monteagudo B, Álvarez-Fernández JC, Iglesias B, et al. Multiple eruptive dermatofibromas in a patient with Down’s syndrome [article in Spanish]. Actas Dermosifiliogr. 2005;96:199.
- Monteagudo B, Suárez-Amor O, Cabanillas M, et al. Down syndrome: another cause of immunosuppression associated with multiple eruptive dermatofibroma? [article in Spanish]. Dermatol Online J. 2009;15:15.
- Tanaka M, Hoashi T, Serizawa N, et al. Multiple unilaterally localized dermatofibromas in a patient with Down syndrome. J Dermatol. 2017;44:1074-1076.
- Niiyama S, Katsuoka K, Happle R, et al. Multiple eruptive dermatofibromas: a review of the literature. Acta Derm Venereol. 2002;82:241-244.
- Her Y, Ku SH, Kim KH. A case of multiple eruptive dermatofibromas in a healthy adult. Ann Dermatol. 2014;26:539-540.
- Bertotto A, Arcangeli C, Crupi S, et al. T cell response to anti-CD3 antibody in Down’s syndrome. Arch Dis Child. 1987;62:1148-1151.
- Kusters MA, Verstegen RH, Gemen EF, et al. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol. 2009;156:189-193.
- Sullivan KD, Evans D, Pandey A, et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic inflammation. Sci Rep. 2017;7:14818.
- Honda M, Tomimura S, de Vega S, et al. Multiple dermatofibromas in a patient with Down syndrome. J Dermatol. 2016;43:346-348.
- Daneshpazhooh M, Nazemi TM, Bigdeloo L, et al. Mucocutaneous findings in 100 children with Down syndrome. Pediatr Dermatol. 2007;24:317-320.
- Madan V, Williams J, Lear JT. Dermatological manifestations of Down’s syndrome. Clin Exp Dermatol. 2006;31:623-629.
To the Editor:
Dermatofibromas (also known as fibrous histiocytomas) are benign fibrous nodules that most often arise as solitary lesions on the lower extremities. Multiple eruptive dermatofibromas (MEDFs) are uncommon and have been defined as more than 15 in number1 or 5 to 8 dermatofibromas appearing within 4 months.2 They have been reported in association with a number of conditions of immune dysregulation such as systemic lupus erythematosus, Sjögren syndrome, HIV infection, and leukemia.3 Multiple eruptive dermatofibromas also have been described in patients with Down syndrome (DS).4-7 We report a case of MEDFs in a patient with DS and review the literature on the association between MEDFs and DS.
A 38-year-old woman with DS, hidradenitis suppurativa, and hypothyroidism presented with multiple cutaneous lesions developing over the last year. The lesions continued to increase in number but were otherwise asymptomatic. Physical examination revealed approximately 20 rubbery, pink-tan papules measuring less than 1 cm in diameter that were scattered along the trunk (Figure, A), arms, and legs (Figure, B).
The patient had no known history of immunosuppression or rheumatologic disease and was otherwise healthy. Basic laboratory tests including a complete blood cell count and antinuclear antibody titer were within reference range. The lesions were clinically consistent with dermatofibromas, but due to their increasing number within a short period of time, a biopsy of a representative lesion was performed to confirm the diagnosis.
The exact incidence of MEDFs is unknown, but they are rare, with one review finding only 50 cases reported from 1960 to 2002.8 They are increasingly recognized as a sign of potential immune dysregulation. Approximately 56% to 70% of cases are seen in patients with an underlying disease state; 80% are immune mediated.8,9 Interestingly, DS has long been associated with notable immune dysfunction,10,11 with evidence suggesting that trisomy 21 may result in widespread changes in gene expression that can lead to interferon activation.12
A PubMed search of articles indexed for MEDLINE using the terms dermatofibroma and Down, dermatofibroma and Down syndrome, eruptive dermatofibroma and Down syndrome, and multiple dermatofibroma and Down syndrome revealed 6 cases of MEDFs in patients with DS that have been reported since 2005.4-7 An additional report by Honda et al13 described a patient with DS who developed 7 dermatofibromas, but no time frame of development was specified. We reviewed the characteristics of 8 patients with DS with MEDFs, which included our patient (Table). The average age at time of presentation was 39 years (median age, 40 years). Six patients (75%) were female and 2 (25%) were male. Dermatofibromas were reported to appear over the course of months to years. Comorbidities included psoriatic arthritis (treated with methotrexate),6 thyroid disorders (ie, Graves disease),6 hypercholesterolemia,6 hidradenitis suppurativa, long-standing mild lymphopenia (1.4×109/L [reference range, 1.5−4.0×109/L]),4 and acute megakaryoblastic leukemia13 treated 15 years before the appearance of dermatofibromas.
Many dermatologic conditions have been reported at increased rates in individuals with DS, including seborrheic dermatitis, alopecia areata, syringomas, elastosis perforans serpiginosa, cutis marmorata, xerosis, and palmoplantar hyperkeratosis.14,15 Although drawing conclusions about associations between MEDFs and DS is limited by our small sample size, we have reported this case and reviewed existing cases of MEDFs in DS to highlight a potential association that may be underrecognized or underreported. More evidence is needed to determine the strength of the association between MEDFs and DS, but dermatologists should be aware that MEDFs may be an additional skin finding associated with DS that is related to the syndrome’s immune dysregulation.
To the Editor:
Dermatofibromas (also known as fibrous histiocytomas) are benign fibrous nodules that most often arise as solitary lesions on the lower extremities. Multiple eruptive dermatofibromas (MEDFs) are uncommon and have been defined as more than 15 in number1 or 5 to 8 dermatofibromas appearing within 4 months.2 They have been reported in association with a number of conditions of immune dysregulation such as systemic lupus erythematosus, Sjögren syndrome, HIV infection, and leukemia.3 Multiple eruptive dermatofibromas also have been described in patients with Down syndrome (DS).4-7 We report a case of MEDFs in a patient with DS and review the literature on the association between MEDFs and DS.
A 38-year-old woman with DS, hidradenitis suppurativa, and hypothyroidism presented with multiple cutaneous lesions developing over the last year. The lesions continued to increase in number but were otherwise asymptomatic. Physical examination revealed approximately 20 rubbery, pink-tan papules measuring less than 1 cm in diameter that were scattered along the trunk (Figure, A), arms, and legs (Figure, B).
The patient had no known history of immunosuppression or rheumatologic disease and was otherwise healthy. Basic laboratory tests including a complete blood cell count and antinuclear antibody titer were within reference range. The lesions were clinically consistent with dermatofibromas, but due to their increasing number within a short period of time, a biopsy of a representative lesion was performed to confirm the diagnosis.
The exact incidence of MEDFs is unknown, but they are rare, with one review finding only 50 cases reported from 1960 to 2002.8 They are increasingly recognized as a sign of potential immune dysregulation. Approximately 56% to 70% of cases are seen in patients with an underlying disease state; 80% are immune mediated.8,9 Interestingly, DS has long been associated with notable immune dysfunction,10,11 with evidence suggesting that trisomy 21 may result in widespread changes in gene expression that can lead to interferon activation.12
A PubMed search of articles indexed for MEDLINE using the terms dermatofibroma and Down, dermatofibroma and Down syndrome, eruptive dermatofibroma and Down syndrome, and multiple dermatofibroma and Down syndrome revealed 6 cases of MEDFs in patients with DS that have been reported since 2005.4-7 An additional report by Honda et al13 described a patient with DS who developed 7 dermatofibromas, but no time frame of development was specified. We reviewed the characteristics of 8 patients with DS with MEDFs, which included our patient (Table). The average age at time of presentation was 39 years (median age, 40 years). Six patients (75%) were female and 2 (25%) were male. Dermatofibromas were reported to appear over the course of months to years. Comorbidities included psoriatic arthritis (treated with methotrexate),6 thyroid disorders (ie, Graves disease),6 hypercholesterolemia,6 hidradenitis suppurativa, long-standing mild lymphopenia (1.4×109/L [reference range, 1.5−4.0×109/L]),4 and acute megakaryoblastic leukemia13 treated 15 years before the appearance of dermatofibromas.
Many dermatologic conditions have been reported at increased rates in individuals with DS, including seborrheic dermatitis, alopecia areata, syringomas, elastosis perforans serpiginosa, cutis marmorata, xerosis, and palmoplantar hyperkeratosis.14,15 Although drawing conclusions about associations between MEDFs and DS is limited by our small sample size, we have reported this case and reviewed existing cases of MEDFs in DS to highlight a potential association that may be underrecognized or underreported. More evidence is needed to determine the strength of the association between MEDFs and DS, but dermatologists should be aware that MEDFs may be an additional skin finding associated with DS that is related to the syndrome’s immune dysregulation.
- Baraf CS, Shapiro L. Multiple histiocytomas: report of a case. Arch Dermatol. 1970;101:588-590.
- Ammirati CT, Mann C, Hornstra IK. Multiple eruptive dermatofibromas in three men with HIV infection. Dermatology. 1997;4:344-348.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Lamb RC, Gangopadhyay M, MacDonald A. Multiple dermatofibromas in Down syndrome. Int J Dermatol. 2014;53:E274-E275.
- Monteagudo B, Álvarez-Fernández JC, Iglesias B, et al. Multiple eruptive dermatofibromas in a patient with Down’s syndrome [article in Spanish]. Actas Dermosifiliogr. 2005;96:199.
- Monteagudo B, Suárez-Amor O, Cabanillas M, et al. Down syndrome: another cause of immunosuppression associated with multiple eruptive dermatofibroma? [article in Spanish]. Dermatol Online J. 2009;15:15.
- Tanaka M, Hoashi T, Serizawa N, et al. Multiple unilaterally localized dermatofibromas in a patient with Down syndrome. J Dermatol. 2017;44:1074-1076.
- Niiyama S, Katsuoka K, Happle R, et al. Multiple eruptive dermatofibromas: a review of the literature. Acta Derm Venereol. 2002;82:241-244.
- Her Y, Ku SH, Kim KH. A case of multiple eruptive dermatofibromas in a healthy adult. Ann Dermatol. 2014;26:539-540.
- Bertotto A, Arcangeli C, Crupi S, et al. T cell response to anti-CD3 antibody in Down’s syndrome. Arch Dis Child. 1987;62:1148-1151.
- Kusters MA, Verstegen RH, Gemen EF, et al. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol. 2009;156:189-193.
- Sullivan KD, Evans D, Pandey A, et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic inflammation. Sci Rep. 2017;7:14818.
- Honda M, Tomimura S, de Vega S, et al. Multiple dermatofibromas in a patient with Down syndrome. J Dermatol. 2016;43:346-348.
- Daneshpazhooh M, Nazemi TM, Bigdeloo L, et al. Mucocutaneous findings in 100 children with Down syndrome. Pediatr Dermatol. 2007;24:317-320.
- Madan V, Williams J, Lear JT. Dermatological manifestations of Down’s syndrome. Clin Exp Dermatol. 2006;31:623-629.
- Baraf CS, Shapiro L. Multiple histiocytomas: report of a case. Arch Dermatol. 1970;101:588-590.
- Ammirati CT, Mann C, Hornstra IK. Multiple eruptive dermatofibromas in three men with HIV infection. Dermatology. 1997;4:344-348.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Lamb RC, Gangopadhyay M, MacDonald A. Multiple dermatofibromas in Down syndrome. Int J Dermatol. 2014;53:E274-E275.
- Monteagudo B, Álvarez-Fernández JC, Iglesias B, et al. Multiple eruptive dermatofibromas in a patient with Down’s syndrome [article in Spanish]. Actas Dermosifiliogr. 2005;96:199.
- Monteagudo B, Suárez-Amor O, Cabanillas M, et al. Down syndrome: another cause of immunosuppression associated with multiple eruptive dermatofibroma? [article in Spanish]. Dermatol Online J. 2009;15:15.
- Tanaka M, Hoashi T, Serizawa N, et al. Multiple unilaterally localized dermatofibromas in a patient with Down syndrome. J Dermatol. 2017;44:1074-1076.
- Niiyama S, Katsuoka K, Happle R, et al. Multiple eruptive dermatofibromas: a review of the literature. Acta Derm Venereol. 2002;82:241-244.
- Her Y, Ku SH, Kim KH. A case of multiple eruptive dermatofibromas in a healthy adult. Ann Dermatol. 2014;26:539-540.
- Bertotto A, Arcangeli C, Crupi S, et al. T cell response to anti-CD3 antibody in Down’s syndrome. Arch Dis Child. 1987;62:1148-1151.
- Kusters MA, Verstegen RH, Gemen EF, et al. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol. 2009;156:189-193.
- Sullivan KD, Evans D, Pandey A, et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic inflammation. Sci Rep. 2017;7:14818.
- Honda M, Tomimura S, de Vega S, et al. Multiple dermatofibromas in a patient with Down syndrome. J Dermatol. 2016;43:346-348.
- Daneshpazhooh M, Nazemi TM, Bigdeloo L, et al. Mucocutaneous findings in 100 children with Down syndrome. Pediatr Dermatol. 2007;24:317-320.
- Madan V, Williams J, Lear JT. Dermatological manifestations of Down’s syndrome. Clin Exp Dermatol. 2006;31:623-629.
Practice Points
- Although dermatofibromas are common and benign skin lesions, multiple eruptive dermatofibromas have been associated with a number of underlying conditions, particularly those associated with immune dysregulation.
- The immune dysregulation reported in Down syndrome may explain the appearance of multiple dermatofibromas.
 consistent with dermatofibromas")
Sun Protection Factor Testing: A Call for an In Vitro Method
The sun protection factor (SPF) value indicates to consumers the level of protection that a given sunscreen formulation provides against erythemally effective UV radiation (UVR). 1 In vivo SPF testing, the gold standard for determining SPF, yields highly variable results and can harm human test participants. 2 In vitro SPF testing methodologies have been under development for years but none have (yet) replaced the in vivo test required by national and international regulatory agencies.
Recent European studies have shown strong data to support a highly standardized in vitro method,1 now under development by the International Organization for Standardization (ISO)—potentially to serve as a new SPF determination standard.1,3 Academia and industry should follow this example and actively take steps to develop and validate a suitable replacement for in vivo SPF testing.
In Vivo SPF Testing
The in vivo SPF test involves comparing doses of UVR necessary to induce erythema in human participants with and without sunscreen applied.2 Although this method has long been the standard for SPF determination, it is associated with the following major disadvantages:
- Cost: The in vivo test is expensive.
- Variability: Results of the test are subject to high interlaboratory variability due to the inherent subjectivity of identifying erythema, the variable skin types of human participants, and other laboratory-dependent factors.2 A study found that the average coefficient of variation for SPF values obtained from 3 or 4 laboratories to be 20%—with values exceeding 50% in some cases. With that level of variability, the same sunscreen may be labeled SPF 30, SPF 50, or SPF 50+, thereby posing a health risk to consumers who rely on the accuracy of such claims. In fact, Miksa et al2 concluded that “the largest obstacle to a reliable SPF assessment for consumer health is the in vivo SPF test itself.”
- Ethical concerns: Human participants are intentionally exposed to harmful UVR until sunburn is achieved. For that reason, there have been calls to abandon the practice of in vivo testing.1
Alternatives to In Vivo SPF Testing
There has been international interest in developing in silico and in vitro alternatives to the in vivo SPF test. These options are attractive because they are relatively inexpensive; avoid exposing human participants to harmful UVR; and have the potential to be more accurate and more reproducible than in vivo tests.
In Vitro Protocols—Many such in vitro tests exist; all generally involve applying a layer of sunscreen to an artificial substrate, exposing it to UVR from a solar simulator, and measuring the UVR transmittance through the product and film by spectrophotometry.1 Prior shortcomings of this method have included suboptimal reproducibility, lack of data on substrate and product properties, and lack of demonstrated equivalency to in vivo SPF testing.4
In Silico Protocols—These tests use data on the UV spectra of sunscreen filters, physical characteristics of sunscreen films on skin, and the unique photoinstability of filters to calculate expected UVR transmittance and SPF of sunscreens based on their ingredients.5 Reports have shown high correlation with in vivo values. Results are not subject to random error; reproducibility is theoretically perfect.5
Regulatory Agencies and In Vitro Testing
In the United States, sunscreens are regulated as over-the-counter drugs. In vivo testing is the only US Food and Drug Administration (FDA)–approved method for determining SPF for labeling purposes.1 In a 2007 Proposed Rule and a 2011 Final Rule, the FDA stated that in vitro SPF tests were an inadequate alternative to in vivo tests because of their shortcomings.4,6
Acknowledging the potential benefits of in vitro testing, the FDA wrote that it would consider in vitro alternatives if equivalency to the in vivo test could be proved.6 The agency has not published an official stance on in vitro SPF testing since those statements in 2007 and 2011. Of note, the FDA deems in vitro testing sufficient for making claims of broad-spectrum coverage.4
In contrast to the regulatory scenario in the United States, Europe regulates sunscreens as cosmetics, and the European Union (EU) has banned animal testing of cosmetics,7 which poses a problem for the development of new sunscreens. It is not surprising, therefore, that in 2006 the European Commission (the executive arm of the EU) published a mandate that in vitro SPF testing methods be actively developed due to ethical concerns associated with in vivo methods.8 In 2017, the International Organization for Standardization released specific validation criteria for proposed in vitro tests to facilitate the eventual approval of such methods.1
Progress of In Vitro Methods
In recent years, advances in in vitro SPF testing methods have addressed shortcomings noted previously by the FDA, which has led to notably improved reproducibility of results and correlation with in vivo values, in large part due to strict standardization of protocols,1 such as tight temperature control of samples, a multisubstrate approach, robotic product application to ensure even distribution, and pre-irradiation of sunscreen samples.
With these improvements, a 2018 study demonstrated an in vitro SPF testing methodology that exceeded published ISO validation criteria for emulsion-type products.1 This method was found to have low interlaboratory variability and high correlation with in vivo SPF values (Pearson r=0.88). Importantly, the authors noted that the consistency and reliability of in vitro SPF testing requires broad institution of a single unified method.1
The method described in the 2018 study1 has been accepted by the ISO Technical Committee and is undergoing further development3
Final Thoughts and Future Steps
Recent data confirm the potential viability of in vitro testing as a primary method of determining SPF values.1 Although ISO has moved forward with development of this method, the FDA has been quiet on in vitro SPF testing since 2011.4 The agency has, however, acknowledged the disadvantages of in vivo broad-spectrum testing, including exposure of human participants to harmful UVR and poor interlaboratory reproducibility.6
Given the technical developments and substantial potential benefits of in vitro testing, we believe that it is time for the FDA to revisit this matter. We propose that the FDA take 2 steps toward in vitro testing. First, publish specific validation criteria that would be deemed necessary for approval of such a test, similar to what ISO published in 2017. Second, thoroughly assess new data supporting the viability of available in vitro testing to determine if the FDA’s stated position that in vitro testing is inadequate remains true.
Although these 2 steps will be important to the process, adoption of an in vitro standard will require more than statements from the FDA. Additional funding should be allocated to researchers who are studying in vitro methodologies, and companies that profit from the multibillion-dollar sunscreen industry should be encouraged to invest in the development of more accurate and more ethical alternatives to in vivo SPF testing.
In vitro SPF testing is inexpensive, avoids the moral quandary of intentionally sunburning human participants, and is more reliable than in vivo testing. It is time for the FDA to facilitate the efforts of academia and industry in taking concrete steps toward approval of an in vitro alternative to in vivo SPF testing.
- Pissavini M, Tricaud C, Wiener G, et al. Validation of an in vitro sun protection factor (SPF) method in blinded ring-testing. Int J Cosmet Sci. 2018;40:263-268. doi:10.1111/ics.12459
- Miksa S, Lutz D, Guy C, et al. Sunscreen sun protection factor claim based on in vivo interlaboratory variability. Int J Cosmet Sci. 2016;38:541-549. doi:10.1111/ics.12333
- ISO/CD 23675: Cosmetics—sun protection test methods—in vitro determination of sun protection factor. International Organization for Standardization (ISO). July 25, 2020. Accessed May 17, 2022. https://www.iso.org/standard/76616.html
- US Food and Drug Administration. Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use. Fed Regist. 2011;76(117):35620-35665. Accessed August 9, 2022. https://www.govinfo.gov/content/pkg/FR-2011-06-17/pdf/2011-14766.pdf
- Herzog B, Osterwalder U. Simulation of sunscreen performance. Pure Appl Chem. 2015;87:937-951. doi:10.1515/pac-2015-0401
- US Food and Drug Administration. Sunscreen drug products for over-the-counter human use; proposed amendment of final monograph. Fed Regist. 2007;72(165):49070-49122. Published August 27, 2007. Accessed August 9, 2022. https://www.govinfo.gov/content/pkg/FR-2007-08-27/pdf/07-4131.pdf
- Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products. November 30, 2009. Accessed August 10, 2022. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02009R1223-20190813
- European Commission Recommendation 2006/647/EC. Published September 22, 2006. Accessed August 10, 2022. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32006H0647
The sun protection factor (SPF) value indicates to consumers the level of protection that a given sunscreen formulation provides against erythemally effective UV radiation (UVR). 1 In vivo SPF testing, the gold standard for determining SPF, yields highly variable results and can harm human test participants. 2 In vitro SPF testing methodologies have been under development for years but none have (yet) replaced the in vivo test required by national and international regulatory agencies.
Recent European studies have shown strong data to support a highly standardized in vitro method,1 now under development by the International Organization for Standardization (ISO)—potentially to serve as a new SPF determination standard.1,3 Academia and industry should follow this example and actively take steps to develop and validate a suitable replacement for in vivo SPF testing.
In Vivo SPF Testing
The in vivo SPF test involves comparing doses of UVR necessary to induce erythema in human participants with and without sunscreen applied.2 Although this method has long been the standard for SPF determination, it is associated with the following major disadvantages:
- Cost: The in vivo test is expensive.
- Variability: Results of the test are subject to high interlaboratory variability due to the inherent subjectivity of identifying erythema, the variable skin types of human participants, and other laboratory-dependent factors.2 A study found that the average coefficient of variation for SPF values obtained from 3 or 4 laboratories to be 20%—with values exceeding 50% in some cases. With that level of variability, the same sunscreen may be labeled SPF 30, SPF 50, or SPF 50+, thereby posing a health risk to consumers who rely on the accuracy of such claims. In fact, Miksa et al2 concluded that “the largest obstacle to a reliable SPF assessment for consumer health is the in vivo SPF test itself.”
- Ethical concerns: Human participants are intentionally exposed to harmful UVR until sunburn is achieved. For that reason, there have been calls to abandon the practice of in vivo testing.1
Alternatives to In Vivo SPF Testing
There has been international interest in developing in silico and in vitro alternatives to the in vivo SPF test. These options are attractive because they are relatively inexpensive; avoid exposing human participants to harmful UVR; and have the potential to be more accurate and more reproducible than in vivo tests.
In Vitro Protocols—Many such in vitro tests exist; all generally involve applying a layer of sunscreen to an artificial substrate, exposing it to UVR from a solar simulator, and measuring the UVR transmittance through the product and film by spectrophotometry.1 Prior shortcomings of this method have included suboptimal reproducibility, lack of data on substrate and product properties, and lack of demonstrated equivalency to in vivo SPF testing.4
In Silico Protocols—These tests use data on the UV spectra of sunscreen filters, physical characteristics of sunscreen films on skin, and the unique photoinstability of filters to calculate expected UVR transmittance and SPF of sunscreens based on their ingredients.5 Reports have shown high correlation with in vivo values. Results are not subject to random error; reproducibility is theoretically perfect.5
Regulatory Agencies and In Vitro Testing
In the United States, sunscreens are regulated as over-the-counter drugs. In vivo testing is the only US Food and Drug Administration (FDA)–approved method for determining SPF for labeling purposes.1 In a 2007 Proposed Rule and a 2011 Final Rule, the FDA stated that in vitro SPF tests were an inadequate alternative to in vivo tests because of their shortcomings.4,6
Acknowledging the potential benefits of in vitro testing, the FDA wrote that it would consider in vitro alternatives if equivalency to the in vivo test could be proved.6 The agency has not published an official stance on in vitro SPF testing since those statements in 2007 and 2011. Of note, the FDA deems in vitro testing sufficient for making claims of broad-spectrum coverage.4
In contrast to the regulatory scenario in the United States, Europe regulates sunscreens as cosmetics, and the European Union (EU) has banned animal testing of cosmetics,7 which poses a problem for the development of new sunscreens. It is not surprising, therefore, that in 2006 the European Commission (the executive arm of the EU) published a mandate that in vitro SPF testing methods be actively developed due to ethical concerns associated with in vivo methods.8 In 2017, the International Organization for Standardization released specific validation criteria for proposed in vitro tests to facilitate the eventual approval of such methods.1
Progress of In Vitro Methods
In recent years, advances in in vitro SPF testing methods have addressed shortcomings noted previously by the FDA, which has led to notably improved reproducibility of results and correlation with in vivo values, in large part due to strict standardization of protocols,1 such as tight temperature control of samples, a multisubstrate approach, robotic product application to ensure even distribution, and pre-irradiation of sunscreen samples.
With these improvements, a 2018 study demonstrated an in vitro SPF testing methodology that exceeded published ISO validation criteria for emulsion-type products.1 This method was found to have low interlaboratory variability and high correlation with in vivo SPF values (Pearson r=0.88). Importantly, the authors noted that the consistency and reliability of in vitro SPF testing requires broad institution of a single unified method.1
The method described in the 2018 study1 has been accepted by the ISO Technical Committee and is undergoing further development3
Final Thoughts and Future Steps
Recent data confirm the potential viability of in vitro testing as a primary method of determining SPF values.1 Although ISO has moved forward with development of this method, the FDA has been quiet on in vitro SPF testing since 2011.4 The agency has, however, acknowledged the disadvantages of in vivo broad-spectrum testing, including exposure of human participants to harmful UVR and poor interlaboratory reproducibility.6
Given the technical developments and substantial potential benefits of in vitro testing, we believe that it is time for the FDA to revisit this matter. We propose that the FDA take 2 steps toward in vitro testing. First, publish specific validation criteria that would be deemed necessary for approval of such a test, similar to what ISO published in 2017. Second, thoroughly assess new data supporting the viability of available in vitro testing to determine if the FDA’s stated position that in vitro testing is inadequate remains true.
Although these 2 steps will be important to the process, adoption of an in vitro standard will require more than statements from the FDA. Additional funding should be allocated to researchers who are studying in vitro methodologies, and companies that profit from the multibillion-dollar sunscreen industry should be encouraged to invest in the development of more accurate and more ethical alternatives to in vivo SPF testing.
In vitro SPF testing is inexpensive, avoids the moral quandary of intentionally sunburning human participants, and is more reliable than in vivo testing. It is time for the FDA to facilitate the efforts of academia and industry in taking concrete steps toward approval of an in vitro alternative to in vivo SPF testing.
The sun protection factor (SPF) value indicates to consumers the level of protection that a given sunscreen formulation provides against erythemally effective UV radiation (UVR). 1 In vivo SPF testing, the gold standard for determining SPF, yields highly variable results and can harm human test participants. 2 In vitro SPF testing methodologies have been under development for years but none have (yet) replaced the in vivo test required by national and international regulatory agencies.
Recent European studies have shown strong data to support a highly standardized in vitro method,1 now under development by the International Organization for Standardization (ISO)—potentially to serve as a new SPF determination standard.1,3 Academia and industry should follow this example and actively take steps to develop and validate a suitable replacement for in vivo SPF testing.
In Vivo SPF Testing
The in vivo SPF test involves comparing doses of UVR necessary to induce erythema in human participants with and without sunscreen applied.2 Although this method has long been the standard for SPF determination, it is associated with the following major disadvantages:
- Cost: The in vivo test is expensive.
- Variability: Results of the test are subject to high interlaboratory variability due to the inherent subjectivity of identifying erythema, the variable skin types of human participants, and other laboratory-dependent factors.2 A study found that the average coefficient of variation for SPF values obtained from 3 or 4 laboratories to be 20%—with values exceeding 50% in some cases. With that level of variability, the same sunscreen may be labeled SPF 30, SPF 50, or SPF 50+, thereby posing a health risk to consumers who rely on the accuracy of such claims. In fact, Miksa et al2 concluded that “the largest obstacle to a reliable SPF assessment for consumer health is the in vivo SPF test itself.”
- Ethical concerns: Human participants are intentionally exposed to harmful UVR until sunburn is achieved. For that reason, there have been calls to abandon the practice of in vivo testing.1
Alternatives to In Vivo SPF Testing
There has been international interest in developing in silico and in vitro alternatives to the in vivo SPF test. These options are attractive because they are relatively inexpensive; avoid exposing human participants to harmful UVR; and have the potential to be more accurate and more reproducible than in vivo tests.
In Vitro Protocols—Many such in vitro tests exist; all generally involve applying a layer of sunscreen to an artificial substrate, exposing it to UVR from a solar simulator, and measuring the UVR transmittance through the product and film by spectrophotometry.1 Prior shortcomings of this method have included suboptimal reproducibility, lack of data on substrate and product properties, and lack of demonstrated equivalency to in vivo SPF testing.4
In Silico Protocols—These tests use data on the UV spectra of sunscreen filters, physical characteristics of sunscreen films on skin, and the unique photoinstability of filters to calculate expected UVR transmittance and SPF of sunscreens based on their ingredients.5 Reports have shown high correlation with in vivo values. Results are not subject to random error; reproducibility is theoretically perfect.5
Regulatory Agencies and In Vitro Testing
In the United States, sunscreens are regulated as over-the-counter drugs. In vivo testing is the only US Food and Drug Administration (FDA)–approved method for determining SPF for labeling purposes.1 In a 2007 Proposed Rule and a 2011 Final Rule, the FDA stated that in vitro SPF tests were an inadequate alternative to in vivo tests because of their shortcomings.4,6
Acknowledging the potential benefits of in vitro testing, the FDA wrote that it would consider in vitro alternatives if equivalency to the in vivo test could be proved.6 The agency has not published an official stance on in vitro SPF testing since those statements in 2007 and 2011. Of note, the FDA deems in vitro testing sufficient for making claims of broad-spectrum coverage.4
In contrast to the regulatory scenario in the United States, Europe regulates sunscreens as cosmetics, and the European Union (EU) has banned animal testing of cosmetics,7 which poses a problem for the development of new sunscreens. It is not surprising, therefore, that in 2006 the European Commission (the executive arm of the EU) published a mandate that in vitro SPF testing methods be actively developed due to ethical concerns associated with in vivo methods.8 In 2017, the International Organization for Standardization released specific validation criteria for proposed in vitro tests to facilitate the eventual approval of such methods.1
Progress of In Vitro Methods
In recent years, advances in in vitro SPF testing methods have addressed shortcomings noted previously by the FDA, which has led to notably improved reproducibility of results and correlation with in vivo values, in large part due to strict standardization of protocols,1 such as tight temperature control of samples, a multisubstrate approach, robotic product application to ensure even distribution, and pre-irradiation of sunscreen samples.
With these improvements, a 2018 study demonstrated an in vitro SPF testing methodology that exceeded published ISO validation criteria for emulsion-type products.1 This method was found to have low interlaboratory variability and high correlation with in vivo SPF values (Pearson r=0.88). Importantly, the authors noted that the consistency and reliability of in vitro SPF testing requires broad institution of a single unified method.1
The method described in the 2018 study1 has been accepted by the ISO Technical Committee and is undergoing further development3
Final Thoughts and Future Steps
Recent data confirm the potential viability of in vitro testing as a primary method of determining SPF values.1 Although ISO has moved forward with development of this method, the FDA has been quiet on in vitro SPF testing since 2011.4 The agency has, however, acknowledged the disadvantages of in vivo broad-spectrum testing, including exposure of human participants to harmful UVR and poor interlaboratory reproducibility.6
Given the technical developments and substantial potential benefits of in vitro testing, we believe that it is time for the FDA to revisit this matter. We propose that the FDA take 2 steps toward in vitro testing. First, publish specific validation criteria that would be deemed necessary for approval of such a test, similar to what ISO published in 2017. Second, thoroughly assess new data supporting the viability of available in vitro testing to determine if the FDA’s stated position that in vitro testing is inadequate remains true.
Although these 2 steps will be important to the process, adoption of an in vitro standard will require more than statements from the FDA. Additional funding should be allocated to researchers who are studying in vitro methodologies, and companies that profit from the multibillion-dollar sunscreen industry should be encouraged to invest in the development of more accurate and more ethical alternatives to in vivo SPF testing.
In vitro SPF testing is inexpensive, avoids the moral quandary of intentionally sunburning human participants, and is more reliable than in vivo testing. It is time for the FDA to facilitate the efforts of academia and industry in taking concrete steps toward approval of an in vitro alternative to in vivo SPF testing.
- Pissavini M, Tricaud C, Wiener G, et al. Validation of an in vitro sun protection factor (SPF) method in blinded ring-testing. Int J Cosmet Sci. 2018;40:263-268. doi:10.1111/ics.12459
- Miksa S, Lutz D, Guy C, et al. Sunscreen sun protection factor claim based on in vivo interlaboratory variability. Int J Cosmet Sci. 2016;38:541-549. doi:10.1111/ics.12333
- ISO/CD 23675: Cosmetics—sun protection test methods—in vitro determination of sun protection factor. International Organization for Standardization (ISO). July 25, 2020. Accessed May 17, 2022. https://www.iso.org/standard/76616.html
- US Food and Drug Administration. Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use. Fed Regist. 2011;76(117):35620-35665. Accessed August 9, 2022. https://www.govinfo.gov/content/pkg/FR-2011-06-17/pdf/2011-14766.pdf
- Herzog B, Osterwalder U. Simulation of sunscreen performance. Pure Appl Chem. 2015;87:937-951. doi:10.1515/pac-2015-0401
- US Food and Drug Administration. Sunscreen drug products for over-the-counter human use; proposed amendment of final monograph. Fed Regist. 2007;72(165):49070-49122. Published August 27, 2007. Accessed August 9, 2022. https://www.govinfo.gov/content/pkg/FR-2007-08-27/pdf/07-4131.pdf
- Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products. November 30, 2009. Accessed August 10, 2022. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02009R1223-20190813
- European Commission Recommendation 2006/647/EC. Published September 22, 2006. Accessed August 10, 2022. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32006H0647
- Pissavini M, Tricaud C, Wiener G, et al. Validation of an in vitro sun protection factor (SPF) method in blinded ring-testing. Int J Cosmet Sci. 2018;40:263-268. doi:10.1111/ics.12459
- Miksa S, Lutz D, Guy C, et al. Sunscreen sun protection factor claim based on in vivo interlaboratory variability. Int J Cosmet Sci. 2016;38:541-549. doi:10.1111/ics.12333
- ISO/CD 23675: Cosmetics—sun protection test methods—in vitro determination of sun protection factor. International Organization for Standardization (ISO). July 25, 2020. Accessed May 17, 2022. https://www.iso.org/standard/76616.html
- US Food and Drug Administration. Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use. Fed Regist. 2011;76(117):35620-35665. Accessed August 9, 2022. https://www.govinfo.gov/content/pkg/FR-2011-06-17/pdf/2011-14766.pdf
- Herzog B, Osterwalder U. Simulation of sunscreen performance. Pure Appl Chem. 2015;87:937-951. doi:10.1515/pac-2015-0401
- US Food and Drug Administration. Sunscreen drug products for over-the-counter human use; proposed amendment of final monograph. Fed Regist. 2007;72(165):49070-49122. Published August 27, 2007. Accessed August 9, 2022. https://www.govinfo.gov/content/pkg/FR-2007-08-27/pdf/07-4131.pdf
- Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products. November 30, 2009. Accessed August 10, 2022. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02009R1223-20190813
- European Commission Recommendation 2006/647/EC. Published September 22, 2006. Accessed August 10, 2022. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32006H0647
Practice Points
- The methodology for determining sun protection factor (SPF) that currently is accepted by the US Food and Drug Administration is an expensive and imprecise in vivo test that exposes human participants to harmful UV radiation.
- In vitro tests for determining SPF may be viable alternatives to the current in vivo gold standard.
- Researchers and the sunscreen industry should actively develop these in vitro methodologies to adopt a more accurate and less harmful test for SPF.
Intralesional Human Papillomavirus Vaccine Therapy for Recalcitrant Plantar Wart Triggers Gout Flare
To the Editor:
There is increasing evidence supporting the use of the human papillomavirus (HPV) vaccine in the treatment of recalcitrant common warts.1 We describe a potential complication associated with HPV vaccine treatment of warts that would be of interest to dermatologists.
A 70-year-old woman presented with a plantar wart measuring 6 mm in diameter at the base of the right hallux of 5 years’ duration. Prior failed therapies for wart removal included multiple paring treatments, cryotherapy, and topical salicylic acid 40% to 60%. The patient had no notable comorbidities; no history of gout; and no known risk factors for gout, such as hypertension, renal insufficiency, diuretic use, obesity, family history, or trauma.
Prior reports cited effective treatment of recalcitrant warts with recombinant HPV vaccines, both intralesionally1 and intramuscularly.2,3 With this knowledge in mind, we administered an intralesional injection with 0.1-mL recombinant HPV 9-valent vaccine to the patient’s plantar wart. Gradual erythema and swelling of the right first metatarsophalangeal joint developed over the next 7 days. Synovial fluid analysis demonstrated negatively birefringent crystals. The patient commenced treatment with colchicine and indomethacin and improved over the next 5 days. The wart resolved 3 months later and required no further treatment.
Prophylactic quadrivalent HPV vaccines have shown efficacy in treating HPV-associated precancerous and cancerous lesions.4 Case reports have suggested that HPV vaccines may be an effective treatment option for recalcitrant warts,1-3,5 especially in cases that do not respond to traditional treatment. It is possible that the mechanism of wart treatment involves overlap in the antigenic epitopes of the HPV types targeted by the vaccine vs the HPV types responsible for causing warts.2 Papillomaviruslike particles, based on the L1 capsid protein, can induce a specific CD8+ activation signal, leading to a vaccine-induced cytotoxic T-cell response that targets the wart cells with HPV-like antigens.6 The HPV vaccine contains aluminium, which has been shown to activate NLRP3 inflammasome,5 which may trigger gout by increasing monosodium urate crystal deposition via IL-1β production.7 This may lead to an increased risk for gout flares, an adverse effect of the HPV vaccine. This finding is supported by other studies of aluminium-containing vaccines that show an association with gout.6 It is noted that these vaccines are mostly delivered intramuscularly or subcutaneously in some cases.
We reported a case of gout triggered by intralesional HPV vaccine treatment of warts. It is unclear whether the gout was induced by the vaccine itself or whether it was due to trauma caused by the intralesional injection near the joint space. Based on our findings, we recommend that patients receiving intralesional injections for wart treatment be advised of this potential adverse effect, especially if they have risk factors for gout or have a history of gout.
- Nofal A, Marei A, Ibrahim AM et al. Intralesional versus intramuscular bivalent human papillomavirus vaccine in the treatment of recalcitrant common warts. J Am Acad Dermatol. 2020;82:94-100.
- Venugopal SS, Murrell DF. Recalcitrant cutaneous warts treated with recombinant quadrivalent human papillomavirus vaccine (types 6, 11, 16, and 18) in a developmentally delayed, 31-year-old white man. Arch Dermatol. 2010;146:475-477.
- Daniel BS, Murrell DF. Complete resolution of chronic multiple verruca vulgaris treated with quadrivalent human papillomavirus vaccine. JAMA Dermatol. 2013;149:370-372.
- Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838-1847.
- Eisenbarth SC, Colegio OR, O’Connor W, et al. Crucial role for the NALP3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122-1166.
- Bellone S, El-Sahwi K, Cocco E, et al. Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8+ cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8+ CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: implications for L1 dendritic cell-based therapeutic vaccines. J Virol. 2009;83:6779-6789.
- Yokose C, McCormick N, Chen C, et al. Risk of gout flares after vaccination: a prospective case cross-over study. Ann Rheum Dis. 2019;78:1601-1604.
To the Editor:
There is increasing evidence supporting the use of the human papillomavirus (HPV) vaccine in the treatment of recalcitrant common warts.1 We describe a potential complication associated with HPV vaccine treatment of warts that would be of interest to dermatologists.
A 70-year-old woman presented with a plantar wart measuring 6 mm in diameter at the base of the right hallux of 5 years’ duration. Prior failed therapies for wart removal included multiple paring treatments, cryotherapy, and topical salicylic acid 40% to 60%. The patient had no notable comorbidities; no history of gout; and no known risk factors for gout, such as hypertension, renal insufficiency, diuretic use, obesity, family history, or trauma.
Prior reports cited effective treatment of recalcitrant warts with recombinant HPV vaccines, both intralesionally1 and intramuscularly.2,3 With this knowledge in mind, we administered an intralesional injection with 0.1-mL recombinant HPV 9-valent vaccine to the patient’s plantar wart. Gradual erythema and swelling of the right first metatarsophalangeal joint developed over the next 7 days. Synovial fluid analysis demonstrated negatively birefringent crystals. The patient commenced treatment with colchicine and indomethacin and improved over the next 5 days. The wart resolved 3 months later and required no further treatment.
Prophylactic quadrivalent HPV vaccines have shown efficacy in treating HPV-associated precancerous and cancerous lesions.4 Case reports have suggested that HPV vaccines may be an effective treatment option for recalcitrant warts,1-3,5 especially in cases that do not respond to traditional treatment. It is possible that the mechanism of wart treatment involves overlap in the antigenic epitopes of the HPV types targeted by the vaccine vs the HPV types responsible for causing warts.2 Papillomaviruslike particles, based on the L1 capsid protein, can induce a specific CD8+ activation signal, leading to a vaccine-induced cytotoxic T-cell response that targets the wart cells with HPV-like antigens.6 The HPV vaccine contains aluminium, which has been shown to activate NLRP3 inflammasome,5 which may trigger gout by increasing monosodium urate crystal deposition via IL-1β production.7 This may lead to an increased risk for gout flares, an adverse effect of the HPV vaccine. This finding is supported by other studies of aluminium-containing vaccines that show an association with gout.6 It is noted that these vaccines are mostly delivered intramuscularly or subcutaneously in some cases.
We reported a case of gout triggered by intralesional HPV vaccine treatment of warts. It is unclear whether the gout was induced by the vaccine itself or whether it was due to trauma caused by the intralesional injection near the joint space. Based on our findings, we recommend that patients receiving intralesional injections for wart treatment be advised of this potential adverse effect, especially if they have risk factors for gout or have a history of gout.
To the Editor:
There is increasing evidence supporting the use of the human papillomavirus (HPV) vaccine in the treatment of recalcitrant common warts.1 We describe a potential complication associated with HPV vaccine treatment of warts that would be of interest to dermatologists.
A 70-year-old woman presented with a plantar wart measuring 6 mm in diameter at the base of the right hallux of 5 years’ duration. Prior failed therapies for wart removal included multiple paring treatments, cryotherapy, and topical salicylic acid 40% to 60%. The patient had no notable comorbidities; no history of gout; and no known risk factors for gout, such as hypertension, renal insufficiency, diuretic use, obesity, family history, or trauma.
Prior reports cited effective treatment of recalcitrant warts with recombinant HPV vaccines, both intralesionally1 and intramuscularly.2,3 With this knowledge in mind, we administered an intralesional injection with 0.1-mL recombinant HPV 9-valent vaccine to the patient’s plantar wart. Gradual erythema and swelling of the right first metatarsophalangeal joint developed over the next 7 days. Synovial fluid analysis demonstrated negatively birefringent crystals. The patient commenced treatment with colchicine and indomethacin and improved over the next 5 days. The wart resolved 3 months later and required no further treatment.
Prophylactic quadrivalent HPV vaccines have shown efficacy in treating HPV-associated precancerous and cancerous lesions.4 Case reports have suggested that HPV vaccines may be an effective treatment option for recalcitrant warts,1-3,5 especially in cases that do not respond to traditional treatment. It is possible that the mechanism of wart treatment involves overlap in the antigenic epitopes of the HPV types targeted by the vaccine vs the HPV types responsible for causing warts.2 Papillomaviruslike particles, based on the L1 capsid protein, can induce a specific CD8+ activation signal, leading to a vaccine-induced cytotoxic T-cell response that targets the wart cells with HPV-like antigens.6 The HPV vaccine contains aluminium, which has been shown to activate NLRP3 inflammasome,5 which may trigger gout by increasing monosodium urate crystal deposition via IL-1β production.7 This may lead to an increased risk for gout flares, an adverse effect of the HPV vaccine. This finding is supported by other studies of aluminium-containing vaccines that show an association with gout.6 It is noted that these vaccines are mostly delivered intramuscularly or subcutaneously in some cases.
We reported a case of gout triggered by intralesional HPV vaccine treatment of warts. It is unclear whether the gout was induced by the vaccine itself or whether it was due to trauma caused by the intralesional injection near the joint space. Based on our findings, we recommend that patients receiving intralesional injections for wart treatment be advised of this potential adverse effect, especially if they have risk factors for gout or have a history of gout.
- Nofal A, Marei A, Ibrahim AM et al. Intralesional versus intramuscular bivalent human papillomavirus vaccine in the treatment of recalcitrant common warts. J Am Acad Dermatol. 2020;82:94-100.
- Venugopal SS, Murrell DF. Recalcitrant cutaneous warts treated with recombinant quadrivalent human papillomavirus vaccine (types 6, 11, 16, and 18) in a developmentally delayed, 31-year-old white man. Arch Dermatol. 2010;146:475-477.
- Daniel BS, Murrell DF. Complete resolution of chronic multiple verruca vulgaris treated with quadrivalent human papillomavirus vaccine. JAMA Dermatol. 2013;149:370-372.
- Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838-1847.
- Eisenbarth SC, Colegio OR, O’Connor W, et al. Crucial role for the NALP3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122-1166.
- Bellone S, El-Sahwi K, Cocco E, et al. Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8+ cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8+ CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: implications for L1 dendritic cell-based therapeutic vaccines. J Virol. 2009;83:6779-6789.
- Yokose C, McCormick N, Chen C, et al. Risk of gout flares after vaccination: a prospective case cross-over study. Ann Rheum Dis. 2019;78:1601-1604.
- Nofal A, Marei A, Ibrahim AM et al. Intralesional versus intramuscular bivalent human papillomavirus vaccine in the treatment of recalcitrant common warts. J Am Acad Dermatol. 2020;82:94-100.
- Venugopal SS, Murrell DF. Recalcitrant cutaneous warts treated with recombinant quadrivalent human papillomavirus vaccine (types 6, 11, 16, and 18) in a developmentally delayed, 31-year-old white man. Arch Dermatol. 2010;146:475-477.
- Daniel BS, Murrell DF. Complete resolution of chronic multiple verruca vulgaris treated with quadrivalent human papillomavirus vaccine. JAMA Dermatol. 2013;149:370-372.
- Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838-1847.
- Eisenbarth SC, Colegio OR, O’Connor W, et al. Crucial role for the NALP3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122-1166.
- Bellone S, El-Sahwi K, Cocco E, et al. Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8+ cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8+ CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: implications for L1 dendritic cell-based therapeutic vaccines. J Virol. 2009;83:6779-6789.
- Yokose C, McCormick N, Chen C, et al. Risk of gout flares after vaccination: a prospective case cross-over study. Ann Rheum Dis. 2019;78:1601-1604.
Practice Points
- Human papillomavirus (HPV) vaccines are increasingly used for recalcitrant warts.
- We describe an unreported adverse effect of gout flare following HPV vaccine treatment of plantar wart.
Calcinosis Cutis Associated With Subcutaneous Glatiramer Acetate
To the Editor:
Calcinosis cutis is a condition characterized by the deposition of insoluble calcium salts in the skin. Dystrophic calcinosis cutis is the most common type, occurring in previously traumatized skin in the absence of abnormal blood calcium levels. It commonly is seen in patients with connective tissue diseases and is thought to be precipitated by chronic inflammation and vascular hypoxia.1 Herein, we describe a case of calcinosis cutis arising after treatment with subcutaneous glatiramer acetate, an agent that is effective for the treatment of relapsing-remitting multiple sclerosis (MS). Diagnostic workup and treatment modalities for calcinosis cutis in this patient population should be considered in the context of minimizing interruption or discontinuation of this disease-modifying agent.
A 53-year-old woman with a history of relapsing-remitting MS and systemic lupus erythematosus (SLE) presented with multiple firm asymptomatic subcutaneous nodules on the thighs of 1 year’s duration that were increasing in number. The involved areas were the injection sites of subcutaneous glatiramer acetate, an immunomodulator for the treatment of MS, which our patient self-administered 3 times weekly. Physical examination revealed multiple flesh-colored to white, firm, and nontender nodules on the thighs (Figure). There was no epidermal change, and she had no other skin involvement. A punch biopsy of one of the nodules revealed calcium deposits in collagen bundles of the deep dermis. Calcium, phosphorus, parathyroid hormone, and vitamin D levels were within reference range. She declined further treatment for the calcinosis cutis and opted to continue treatment with glatiramer acetate, as her MS was well controlled on this medication.

Glatiramer acetate is an immunogenic polypeptide injectable that is approved by the US Food and Drug Administration for the treatment of relapsing-remitting MS.2 It is composed of synthetic polypeptides and contains 4 naturally occurring amino acids. Glatiramer acetate is administered subcutaneously as 20 mg/mL/d or 40 mg/mL 3 times weekly. Transient injection-site reactions are the most common cutaneous adverse events and include localized edema, induration, erythema, pain, and pruritus.3 There have been multiple reports of lobular panniculitis and skin necrosis as well as embolia cutis medicamentosa (Nicolau syndrome).4,5 Our case of calcinosis cutis related to glatiramer acetate is unique. The mechanism of calcinosis cutis in our patient likely was dystrophic due to tissue damage, rather than due to the injection of a calcium-containing substance. Our patient’s history of SLE is a notable risk factor for the development of calcinosis cutis, likely incited by the trauma occurring with subcutaneous injections.6
The mainstay of treatment for localized calcinosis cutis in the setting of connective tissue disease is surgical excision as well as treatment of the underlying disorder. Potential therapies include calcium channel blockers, warfarin, bisphosphonates, intravenous immunoglobulin, minocycline, colchicine, anti–tumor necrosis factor agents, intralesional corticosteroids, intravenous sodium thiosulfate, and CO2 laser.1,6 Our patient was already on intravenous immunoglobulin for MS and hydroxychloroquine for SLE. In select cases where the patient is asymptomatic and prefers not to pursue treatment, no treatment is necessary.
Although calcinosis cutis may occur in SLE alone, it is uncommon and usually is seen in chronic severe SLE, where calcification usually occurs in the setting of pre-existing cutaneous lupus.4 This case report of calcinosis cutis following treatment with glatiramer acetate highlights some of the cutaneous side effects associated with glatiramer acetate injections and should prompt practitioners to consider dystrophic calcinosis cutis in patients requiring subcutaneous medications, particularly in those with pre-existing connective tissue disease.
- Valenzuela A, Chung L. Calcinosis: pathophysiology and management. Curr Opin Rheumatol. 2015;27:542-548.
- Copaxone. Prescribing information. Teva Neuroscience, Inc; 2022. Accessed July 15, 2022. https://www.copaxone.com/globalassets/copaxone/prescribing-information.pdf
- McKeage K. Glatiramer acetate 40 mg/mL in relapsing-remitting multiple sclerosis: a review. CNS Drugs. 2015;29:425-432.
- Balak DMW, Hengstman GJD, Çakmak A, et al. Cutaneous adverse events associated with disease-modifying treatment in multiple sclerosis: a systematic review. Mult Scler. 2012;18:1705-1717.
- Watkins CE, Litchfield J, Youngberg G, et al. Glatiramer acetate-induced lobular panniculitis and skin necrosis. Cutis. 2015;95:E26-E30.
- Reiter N, El-Shabrawi L, Leinweber B, et al. Calcinosis cutis. J Am Acad Dermatol. 2011;65:1-12.
To the Editor:
Calcinosis cutis is a condition characterized by the deposition of insoluble calcium salts in the skin. Dystrophic calcinosis cutis is the most common type, occurring in previously traumatized skin in the absence of abnormal blood calcium levels. It commonly is seen in patients with connective tissue diseases and is thought to be precipitated by chronic inflammation and vascular hypoxia.1 Herein, we describe a case of calcinosis cutis arising after treatment with subcutaneous glatiramer acetate, an agent that is effective for the treatment of relapsing-remitting multiple sclerosis (MS). Diagnostic workup and treatment modalities for calcinosis cutis in this patient population should be considered in the context of minimizing interruption or discontinuation of this disease-modifying agent.
A 53-year-old woman with a history of relapsing-remitting MS and systemic lupus erythematosus (SLE) presented with multiple firm asymptomatic subcutaneous nodules on the thighs of 1 year’s duration that were increasing in number. The involved areas were the injection sites of subcutaneous glatiramer acetate, an immunomodulator for the treatment of MS, which our patient self-administered 3 times weekly. Physical examination revealed multiple flesh-colored to white, firm, and nontender nodules on the thighs (Figure). There was no epidermal change, and she had no other skin involvement. A punch biopsy of one of the nodules revealed calcium deposits in collagen bundles of the deep dermis. Calcium, phosphorus, parathyroid hormone, and vitamin D levels were within reference range. She declined further treatment for the calcinosis cutis and opted to continue treatment with glatiramer acetate, as her MS was well controlled on this medication.
Glatiramer acetate is an immunogenic polypeptide injectable that is approved by the US Food and Drug Administration for the treatment of relapsing-remitting MS.2 It is composed of synthetic polypeptides and contains 4 naturally occurring amino acids. Glatiramer acetate is administered subcutaneously as 20 mg/mL/d or 40 mg/mL 3 times weekly. Transient injection-site reactions are the most common cutaneous adverse events and include localized edema, induration, erythema, pain, and pruritus.3 There have been multiple reports of lobular panniculitis and skin necrosis as well as embolia cutis medicamentosa (Nicolau syndrome).4,5 Our case of calcinosis cutis related to glatiramer acetate is unique. The mechanism of calcinosis cutis in our patient likely was dystrophic due to tissue damage, rather than due to the injection of a calcium-containing substance. Our patient’s history of SLE is a notable risk factor for the development of calcinosis cutis, likely incited by the trauma occurring with subcutaneous injections.6
The mainstay of treatment for localized calcinosis cutis in the setting of connective tissue disease is surgical excision as well as treatment of the underlying disorder. Potential therapies include calcium channel blockers, warfarin, bisphosphonates, intravenous immunoglobulin, minocycline, colchicine, anti–tumor necrosis factor agents, intralesional corticosteroids, intravenous sodium thiosulfate, and CO2 laser.1,6 Our patient was already on intravenous immunoglobulin for MS and hydroxychloroquine for SLE. In select cases where the patient is asymptomatic and prefers not to pursue treatment, no treatment is necessary.
Although calcinosis cutis may occur in SLE alone, it is uncommon and usually is seen in chronic severe SLE, where calcification usually occurs in the setting of pre-existing cutaneous lupus.4 This case report of calcinosis cutis following treatment with glatiramer acetate highlights some of the cutaneous side effects associated with glatiramer acetate injections and should prompt practitioners to consider dystrophic calcinosis cutis in patients requiring subcutaneous medications, particularly in those with pre-existing connective tissue disease.
To the Editor:
Calcinosis cutis is a condition characterized by the deposition of insoluble calcium salts in the skin. Dystrophic calcinosis cutis is the most common type, occurring in previously traumatized skin in the absence of abnormal blood calcium levels. It commonly is seen in patients with connective tissue diseases and is thought to be precipitated by chronic inflammation and vascular hypoxia.1 Herein, we describe a case of calcinosis cutis arising after treatment with subcutaneous glatiramer acetate, an agent that is effective for the treatment of relapsing-remitting multiple sclerosis (MS). Diagnostic workup and treatment modalities for calcinosis cutis in this patient population should be considered in the context of minimizing interruption or discontinuation of this disease-modifying agent.
A 53-year-old woman with a history of relapsing-remitting MS and systemic lupus erythematosus (SLE) presented with multiple firm asymptomatic subcutaneous nodules on the thighs of 1 year’s duration that were increasing in number. The involved areas were the injection sites of subcutaneous glatiramer acetate, an immunomodulator for the treatment of MS, which our patient self-administered 3 times weekly. Physical examination revealed multiple flesh-colored to white, firm, and nontender nodules on the thighs (Figure). There was no epidermal change, and she had no other skin involvement. A punch biopsy of one of the nodules revealed calcium deposits in collagen bundles of the deep dermis. Calcium, phosphorus, parathyroid hormone, and vitamin D levels were within reference range. She declined further treatment for the calcinosis cutis and opted to continue treatment with glatiramer acetate, as her MS was well controlled on this medication.
Glatiramer acetate is an immunogenic polypeptide injectable that is approved by the US Food and Drug Administration for the treatment of relapsing-remitting MS.2 It is composed of synthetic polypeptides and contains 4 naturally occurring amino acids. Glatiramer acetate is administered subcutaneously as 20 mg/mL/d or 40 mg/mL 3 times weekly. Transient injection-site reactions are the most common cutaneous adverse events and include localized edema, induration, erythema, pain, and pruritus.3 There have been multiple reports of lobular panniculitis and skin necrosis as well as embolia cutis medicamentosa (Nicolau syndrome).4,5 Our case of calcinosis cutis related to glatiramer acetate is unique. The mechanism of calcinosis cutis in our patient likely was dystrophic due to tissue damage, rather than due to the injection of a calcium-containing substance. Our patient’s history of SLE is a notable risk factor for the development of calcinosis cutis, likely incited by the trauma occurring with subcutaneous injections.6
The mainstay of treatment for localized calcinosis cutis in the setting of connective tissue disease is surgical excision as well as treatment of the underlying disorder. Potential therapies include calcium channel blockers, warfarin, bisphosphonates, intravenous immunoglobulin, minocycline, colchicine, anti–tumor necrosis factor agents, intralesional corticosteroids, intravenous sodium thiosulfate, and CO2 laser.1,6 Our patient was already on intravenous immunoglobulin for MS and hydroxychloroquine for SLE. In select cases where the patient is asymptomatic and prefers not to pursue treatment, no treatment is necessary.
Although calcinosis cutis may occur in SLE alone, it is uncommon and usually is seen in chronic severe SLE, where calcification usually occurs in the setting of pre-existing cutaneous lupus.4 This case report of calcinosis cutis following treatment with glatiramer acetate highlights some of the cutaneous side effects associated with glatiramer acetate injections and should prompt practitioners to consider dystrophic calcinosis cutis in patients requiring subcutaneous medications, particularly in those with pre-existing connective tissue disease.
- Valenzuela A, Chung L. Calcinosis: pathophysiology and management. Curr Opin Rheumatol. 2015;27:542-548.
- Copaxone. Prescribing information. Teva Neuroscience, Inc; 2022. Accessed July 15, 2022. https://www.copaxone.com/globalassets/copaxone/prescribing-information.pdf
- McKeage K. Glatiramer acetate 40 mg/mL in relapsing-remitting multiple sclerosis: a review. CNS Drugs. 2015;29:425-432.
- Balak DMW, Hengstman GJD, Çakmak A, et al. Cutaneous adverse events associated with disease-modifying treatment in multiple sclerosis: a systematic review. Mult Scler. 2012;18:1705-1717.
- Watkins CE, Litchfield J, Youngberg G, et al. Glatiramer acetate-induced lobular panniculitis and skin necrosis. Cutis. 2015;95:E26-E30.
- Reiter N, El-Shabrawi L, Leinweber B, et al. Calcinosis cutis. J Am Acad Dermatol. 2011;65:1-12.
- Valenzuela A, Chung L. Calcinosis: pathophysiology and management. Curr Opin Rheumatol. 2015;27:542-548.
- Copaxone. Prescribing information. Teva Neuroscience, Inc; 2022. Accessed July 15, 2022. https://www.copaxone.com/globalassets/copaxone/prescribing-information.pdf
- McKeage K. Glatiramer acetate 40 mg/mL in relapsing-remitting multiple sclerosis: a review. CNS Drugs. 2015;29:425-432.
- Balak DMW, Hengstman GJD, Çakmak A, et al. Cutaneous adverse events associated with disease-modifying treatment in multiple sclerosis: a systematic review. Mult Scler. 2012;18:1705-1717.
- Watkins CE, Litchfield J, Youngberg G, et al. Glatiramer acetate-induced lobular panniculitis and skin necrosis. Cutis. 2015;95:E26-E30.
- Reiter N, El-Shabrawi L, Leinweber B, et al. Calcinosis cutis. J Am Acad Dermatol. 2011;65:1-12.
Practice Points
- Glatiramer acetate is a subcutaneous injection utilized for relapsing-remitting multiple sclerosis, and common adverse effects include injection-site reactions such as calcinosis cutis.
- Development of calcinosis cutis in association with glatiramer acetate is not an indication for medication discontinuation.
- Dermatologists should be aware of this potential association, and treatment should be considered in cases of symptomatic calcinosis cutis.

Rituximab for Acquired Hemophilia A in the Setting of Bullous Pemphigoid
To the Editor:
Bullous pemphigoid (BP) is an autoimmune blistering disease characterized by the formation of antihemidesmosomal antibodies, resulting in tense bullae concentrated on the extremities and trunk that often are preceded by a pruritic urticarial phase.1 A rare complication of BP is the subsequent development of acquired hemophilia A. We report a case of BP with associated factor VIII–neutralizing antibodies in a patient who improved with prednisone and rituximab therapy.
A 78-year-old woman presented with red-orange pruritic plaques on the right heel that spread to involve the arms and legs, abdomen, and trunk with new-onset bullae over the course of 2 weeks (Figure 1). Dermatology was consulted, and a diagnosis of BP was confirmed via biopsy and direct immunofluorescence.

Despite treatment with prednisone 40 mg/d and clobetasol ointment 0.05%, she continued to develop extensive cutaneous bullae and new hemorrhagic bullae on the buccal mucosae (Figure 2), necessitating hospital admission. She clinically improved after prednisone was increased to 60 mg/d and mycophenolate mofetil 500 mg twice daily was added; however, she returned 8 days after discharge from the hospital with altered mental status, new-onset hematomas of the abdomen and right leg, and a hemoglobin level of 5.8 g/dL (reference range, 14.0–17.5 g/dL). Activated prothrombin time was prolonged without correction on mixing studies, raising concern for coagulation factor inhibition. Factor VIII activity was diminished to 9% and then 1% three days later. Mycophenolate mofetil was discontinued, and the patient was acutely stabilized with blood transfusions, intravenous immunoglobulin, tranexamic acid, and aminocaproic acid. Rituximab was initiated at 1000 mg and then administered again 2 weeks later. At 7-week follow-up, coagulation studies normalized, and there was no evidence of blistering dermatosis on examination.

Bullous pemphigoid generally is seen in patients older than 60 years, and the incidence increases with age. The disease course follows formation of IgG antibodies against BP180 or BP230, leading to localized activation of the complement cascade at the basement membrane zone.1 Medications, vaccinations, UV radiation, and burns have been implicated in disease induction.2
Identification of antihemidesmosomal antibodies on lesional biopsy via direct immunofluorescence is the gold standard for diagnosis, though indirect antibodies measured via enzyme-linked immunosorbent assay may provide information regarding disease severity.1 Patients with milder disease may be treated with topical corticosteroids, doxycycline, and nicotinamide; however, severe disease requires treatment with systemic glucocorticoids and steroid-sparing agents.3 Rituximab initially was approved by the US Food and Drug Administration for the treatment of pemphigus vulgaris, and mounting evidence for the use of rituximab in BP is promising. Although data are limited to retrospective studies, rituximab has shown notable remission rates and steroid-sparing effects in those with moderate to severe BP.4
Acquired hemophilia A (AHA) is caused by the production of IgG autoantibodies, which block physiologic interactions between factor VIII and factor IX, phospholipids, and von Willebrand factor.5 Acquired hemophilia A often is diagnosed by prolonged activated prothrombin time and decreased factor VIII activity after a previously unaffected patient develops severe bleeding. Treatment involves re-establishing hemostasis and the use of corticosteroids and immunosuppressive agents to diminish autoantibody production.4
Bullous pemphigoid–associated AHA likely is due to antigenic similarity between BP180 and factor VIII, leading to concomitant neutralization of factor VIII with the production of BP-associated autoantibodies.5 Bullous pemphigoid–associated AHA has been reported with manifestations of bleeding concurrent with or after the development of dermatologic disease. Rituximab use has been reported with clinical efficacy in several cases, including our patient.6 Continued hematologic monitoring is recommended, as recurrences are common within the first 2 years.5
- Bağcı IS, Horváth ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Schiavo AL, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol 2013;31:391-399.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-332.
- Cho Y, Chu C, Wang L. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. 2015;173:302-304.
- Zdziarska J, Musial J. Acquired hemophilia A: an underdiagnosed severe bleeding disorder. Pol Arch Med Wewn. 2014;124:200-206.
- Binet Q, Lambert C, Sacré L, et al. Successful management of acquired hemophilia associated with bullous pemphigoid: a case report and review of the literature [published online March 28, 2017]. Case Rep Hematol. 2017;2017:2057019.
To the Editor:
Bullous pemphigoid (BP) is an autoimmune blistering disease characterized by the formation of antihemidesmosomal antibodies, resulting in tense bullae concentrated on the extremities and trunk that often are preceded by a pruritic urticarial phase.1 A rare complication of BP is the subsequent development of acquired hemophilia A. We report a case of BP with associated factor VIII–neutralizing antibodies in a patient who improved with prednisone and rituximab therapy.
A 78-year-old woman presented with red-orange pruritic plaques on the right heel that spread to involve the arms and legs, abdomen, and trunk with new-onset bullae over the course of 2 weeks (Figure 1). Dermatology was consulted, and a diagnosis of BP was confirmed via biopsy and direct immunofluorescence.
Despite treatment with prednisone 40 mg/d and clobetasol ointment 0.05%, she continued to develop extensive cutaneous bullae and new hemorrhagic bullae on the buccal mucosae (Figure 2), necessitating hospital admission. She clinically improved after prednisone was increased to 60 mg/d and mycophenolate mofetil 500 mg twice daily was added; however, she returned 8 days after discharge from the hospital with altered mental status, new-onset hematomas of the abdomen and right leg, and a hemoglobin level of 5.8 g/dL (reference range, 14.0–17.5 g/dL). Activated prothrombin time was prolonged without correction on mixing studies, raising concern for coagulation factor inhibition. Factor VIII activity was diminished to 9% and then 1% three days later. Mycophenolate mofetil was discontinued, and the patient was acutely stabilized with blood transfusions, intravenous immunoglobulin, tranexamic acid, and aminocaproic acid. Rituximab was initiated at 1000 mg and then administered again 2 weeks later. At 7-week follow-up, coagulation studies normalized, and there was no evidence of blistering dermatosis on examination.
Bullous pemphigoid generally is seen in patients older than 60 years, and the incidence increases with age. The disease course follows formation of IgG antibodies against BP180 or BP230, leading to localized activation of the complement cascade at the basement membrane zone.1 Medications, vaccinations, UV radiation, and burns have been implicated in disease induction.2
Identification of antihemidesmosomal antibodies on lesional biopsy via direct immunofluorescence is the gold standard for diagnosis, though indirect antibodies measured via enzyme-linked immunosorbent assay may provide information regarding disease severity.1 Patients with milder disease may be treated with topical corticosteroids, doxycycline, and nicotinamide; however, severe disease requires treatment with systemic glucocorticoids and steroid-sparing agents.3 Rituximab initially was approved by the US Food and Drug Administration for the treatment of pemphigus vulgaris, and mounting evidence for the use of rituximab in BP is promising. Although data are limited to retrospective studies, rituximab has shown notable remission rates and steroid-sparing effects in those with moderate to severe BP.4
Acquired hemophilia A (AHA) is caused by the production of IgG autoantibodies, which block physiologic interactions between factor VIII and factor IX, phospholipids, and von Willebrand factor.5 Acquired hemophilia A often is diagnosed by prolonged activated prothrombin time and decreased factor VIII activity after a previously unaffected patient develops severe bleeding. Treatment involves re-establishing hemostasis and the use of corticosteroids and immunosuppressive agents to diminish autoantibody production.4
Bullous pemphigoid–associated AHA likely is due to antigenic similarity between BP180 and factor VIII, leading to concomitant neutralization of factor VIII with the production of BP-associated autoantibodies.5 Bullous pemphigoid–associated AHA has been reported with manifestations of bleeding concurrent with or after the development of dermatologic disease. Rituximab use has been reported with clinical efficacy in several cases, including our patient.6 Continued hematologic monitoring is recommended, as recurrences are common within the first 2 years.5
To the Editor:
Bullous pemphigoid (BP) is an autoimmune blistering disease characterized by the formation of antihemidesmosomal antibodies, resulting in tense bullae concentrated on the extremities and trunk that often are preceded by a pruritic urticarial phase.1 A rare complication of BP is the subsequent development of acquired hemophilia A. We report a case of BP with associated factor VIII–neutralizing antibodies in a patient who improved with prednisone and rituximab therapy.
A 78-year-old woman presented with red-orange pruritic plaques on the right heel that spread to involve the arms and legs, abdomen, and trunk with new-onset bullae over the course of 2 weeks (Figure 1). Dermatology was consulted, and a diagnosis of BP was confirmed via biopsy and direct immunofluorescence.
Despite treatment with prednisone 40 mg/d and clobetasol ointment 0.05%, she continued to develop extensive cutaneous bullae and new hemorrhagic bullae on the buccal mucosae (Figure 2), necessitating hospital admission. She clinically improved after prednisone was increased to 60 mg/d and mycophenolate mofetil 500 mg twice daily was added; however, she returned 8 days after discharge from the hospital with altered mental status, new-onset hematomas of the abdomen and right leg, and a hemoglobin level of 5.8 g/dL (reference range, 14.0–17.5 g/dL). Activated prothrombin time was prolonged without correction on mixing studies, raising concern for coagulation factor inhibition. Factor VIII activity was diminished to 9% and then 1% three days later. Mycophenolate mofetil was discontinued, and the patient was acutely stabilized with blood transfusions, intravenous immunoglobulin, tranexamic acid, and aminocaproic acid. Rituximab was initiated at 1000 mg and then administered again 2 weeks later. At 7-week follow-up, coagulation studies normalized, and there was no evidence of blistering dermatosis on examination.
Bullous pemphigoid generally is seen in patients older than 60 years, and the incidence increases with age. The disease course follows formation of IgG antibodies against BP180 or BP230, leading to localized activation of the complement cascade at the basement membrane zone.1 Medications, vaccinations, UV radiation, and burns have been implicated in disease induction.2
Identification of antihemidesmosomal antibodies on lesional biopsy via direct immunofluorescence is the gold standard for diagnosis, though indirect antibodies measured via enzyme-linked immunosorbent assay may provide information regarding disease severity.1 Patients with milder disease may be treated with topical corticosteroids, doxycycline, and nicotinamide; however, severe disease requires treatment with systemic glucocorticoids and steroid-sparing agents.3 Rituximab initially was approved by the US Food and Drug Administration for the treatment of pemphigus vulgaris, and mounting evidence for the use of rituximab in BP is promising. Although data are limited to retrospective studies, rituximab has shown notable remission rates and steroid-sparing effects in those with moderate to severe BP.4
Acquired hemophilia A (AHA) is caused by the production of IgG autoantibodies, which block physiologic interactions between factor VIII and factor IX, phospholipids, and von Willebrand factor.5 Acquired hemophilia A often is diagnosed by prolonged activated prothrombin time and decreased factor VIII activity after a previously unaffected patient develops severe bleeding. Treatment involves re-establishing hemostasis and the use of corticosteroids and immunosuppressive agents to diminish autoantibody production.4
Bullous pemphigoid–associated AHA likely is due to antigenic similarity between BP180 and factor VIII, leading to concomitant neutralization of factor VIII with the production of BP-associated autoantibodies.5 Bullous pemphigoid–associated AHA has been reported with manifestations of bleeding concurrent with or after the development of dermatologic disease. Rituximab use has been reported with clinical efficacy in several cases, including our patient.6 Continued hematologic monitoring is recommended, as recurrences are common within the first 2 years.5
- Bağcı IS, Horváth ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Schiavo AL, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol 2013;31:391-399.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-332.
- Cho Y, Chu C, Wang L. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. 2015;173:302-304.
- Zdziarska J, Musial J. Acquired hemophilia A: an underdiagnosed severe bleeding disorder. Pol Arch Med Wewn. 2014;124:200-206.
- Binet Q, Lambert C, Sacré L, et al. Successful management of acquired hemophilia associated with bullous pemphigoid: a case report and review of the literature [published online March 28, 2017]. Case Rep Hematol. 2017;2017:2057019.
- Bağcı IS, Horváth ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Schiavo AL, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol 2013;31:391-399.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-332.
- Cho Y, Chu C, Wang L. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. 2015;173:302-304.
- Zdziarska J, Musial J. Acquired hemophilia A: an underdiagnosed severe bleeding disorder. Pol Arch Med Wewn. 2014;124:200-206.
- Binet Q, Lambert C, Sacré L, et al. Successful management of acquired hemophilia associated with bullous pemphigoid: a case report and review of the literature [published online March 28, 2017]. Case Rep Hematol. 2017;2017:2057019.
Practice Points
- Physicians must be aware of the potential for acquired hemophilia A in patients with bullous pemphigoid (BP).
- Rituximab is an effective therapy for BP and should be considered for patients in this cohort.

Peristomal Pyoderma Gangrenosum at an Ileostomy Site
To the Editor:
Peristomal pyoderma gangrenosum (PPG) is a rare entity first described in 1984.1 Lesions usually begin as pustules that coalesce into an erythematous skin ulceration that contains purulent material. The lesion appears on the skin that surrounds an abdominal stoma. Peristomal pyoderma gangrenosum typically is associated with Crohn disease and ulcerative colitis, cancer, blood dyscrasia, diabetes mellitus, and hepatitis.2 We describe a case of PPG following an ileostomy in a patient with colon cancer and a related history of Crohn disease.
A 32-year-old woman presented to a dermatology office with a spontaneously painful, 3.2-cm ulceration that was extremely tender to palpation, located immediately adjacent to the site of an ileostomy (Figure). The patient had a history of refractory constipation that failed to respond to standard conservative measures 4 years prior. She underwent a colonoscopy, which revealed a 6.5-cm, irregularly shaped, exophytic mass in the rectosigmoid portion of the colon. Histopathologic examination of several biopsies confirmed the diagnosis of moderately well-differentiated adenocarcinoma, and additional evaluation determined the cancer to be stage IIB. She had a medical history of pancolonic Crohn disease since high school that was treated with periodic infusions of infliximab at the standard dose of 5 mg/kg. Colon cancer treatment consisted of preoperative radiotherapy, complete colectomy with ileoanal anastomosis, and creation of a J-pouch and formation of a temporary ileostomy, along with postoperative capecitabine chemotherapy.

The ileostomy eventually was reversed, and the patient did well for 3 years. When the patient developed severe abdominal pain, the J-pouch was examined and found to be remarkably involved with Crohn disease. However, during the colonoscopy, the J-pouch was inadvertently punctured, leading to the formation of a large pelvic abscess. The latter necessitated diversion of stool, and the patient had the original ileostomy recreated.
Prior to presentation to dermatology, various consultants suspected the ulceration was possibly a deep fungal infection, cutaneous Crohn disease, a factitious ulceration, or acute allergic contact dermatitis related to some element of ostomy care. However, dermatologic consultation suggested that the troublesome lesion was classic PPG and recommended administration of a tumor necrosis factor (TNF) α–blocking agent and concomitant intralesional injections of dilute triamcinolone acetonide.
The patient was treated with subcutaneous adalimumab 40 mg once weekly, and received near weekly subcutaneous injections of triamcinolone acetonide 10 mg/mL. After 2 months, the discomfort subsided, and the ulceration gradually resolved into a depressed scar. Eighteen months later, the scar was barely perceptible as a minimally erythematous depression. Adalimumab ultimately was discontinued, as the residual J-pouch was removed, and the biologic drug was associated with extensive alopecia areata–like hair loss. There has been no recurrence of PPG in the 40 months since clinical resolution.
Peristomal pyoderma gangrenosum is an uncommon subtype of pyoderma gangrenosum, which is characterized by chronic, persistent, or recurrent painful ulceration(s) close to an abdominal stoma. In total, fewer than 100 cases of PPG have been reported thus far in the readily available medical literature.3 Inflammatory bowel disease (IBD) is the most frequently diagnosed systemic condition associated with PPG, though other associated conditions include diverticular disease, abdominal malignancy, and neurologic dysfunction. Approximately 2% to 4.3% of all patients who have stoma creation surgery related to underlying IBD develop PPG. It is estimated that the yearly incidence rate of PPG in all abdominal stomas is quite low (approximately 0.6%).4
Peristomal pyoderma gangrenosum can occur at any age, but it tends to predominate in young to middle-aged adults, with a slight female predilection. The etiology and pathogenesis of PPG are largely unknown, though studies have shown that an abnormal immune response may be critical to its development. Risk factors for PPG are not well defined but potentially include autoimmune disorders, a high body mass index, and females or African Americans with IBD.4 Because PPG does not have characteristic histopathologic features, it is a diagnosis of exclusion that is based on the clinical examination and histologic findings that rule out other potential disorders.
There are 4 types of PPG based on the clinical and histopathologic characteristics: ulcerative, pustular, bullous, and vegetative. Peristomal pyoderma gangrenosum tends to be either ulcerative or vegetative, with ulcerative being by far the predominant type. The onset of PPG is quite variable, occurring a few weeks to several years after stoma formation.5 Ulcer size can range from less than 3 cm to 30 cm.4 Lesions begin as deep painful nodules or as superficial hemorrhagic pustules, either idiopathic or following ostensibly minimal trauma. Subsequently, they become necrotic and form an ulceration. The ulcers can be single or multiple lesions, typically with erythematous raised borders and purulent discharge. The ulcers are extremely painful and rapidly progressive. After the ulcers heal, they often leave a characteristic weblike atrophic scar that can break down further following any form of irritation or trauma.5
A prompt diagnosis of PPG is important. A diagnosis of PPG should be considered when dealing with a noninfectious ulcer surrounding a stoma in patients with IBD or other autoimmune conditions.6 Because PPG is a rare skin disorder, it is likely to be missed and lead to unnecessary diagnostic workup and a delay in proper therapy. In our patient, a diagnosis of PPG was overlooked for other infectious and autoimmune causes. The diagnostic evaluation of a patient with PPG is based on 3 principles: (1) ruling out other causes of a peristomal ulcer, such as an abscess, contact dermatitis, or wound infection; (2) determining whether there is an underlying intestinal bowel disease in the stoma; and (3) identifying associated systemic disorders such as vasculitis, erythema nodosum, or similar processes.4 The differential diagnosis depends on the type and stage of PPG and can include malignancy, vasculitis, extraintestinal IBD, infectious disease, and insect bites. A review of the history of the ulcer is helpful in ruling out other diseases, and a colonoscopy or ileoscopy can identify if patients have an underlying active IBD. Swabs for smear and both bacterial and fungal cultures should be taken from the exudate and directly from the ulcer base. Biopsy of the ulcer also helps to exclude alternative diagnoses.6
The primary goals of treating PPG include to reduce pain and the risk for secondary infection, increase pouch adherence, and decrease purulent exudate.7 Although there is not one well-defined optimal therapeutic intervention, there are a variety of effective approaches that may be considered and used. In mild cases, management methods such as dressings, topical agents, or intralesional steroids may be capable of controlling the disease. Daily wound care is important. Moisture-retentive dressings can control pain, induce collagen formation, promote angiogenesis, and prevent contamination. Cleaning the wound with sterile saline and applying an anti-infective agent also may be effective. Application of ultrapotent topical steroids and tacrolimus ointment 0.3% can be used in patients without concomitant secondary infection. In patients who are in remission, human platelet-derived growth factor may be used. Intralesional injections of dilute triamcinolone acetonide or cyclosporine solution also can be helpful. Cyclosporin A was used as a systemic monotherapy to treat a 48-year-old man and 50-year-old woman with the idiopathic form of PPG. After 3 months of treatment, PPG had completely resolved and there were no major side effects.8 Other potential topical therapies that control inflammation and promote wound healing include benzoyl peroxide, chlormethine (topical alkylating agent and nitrogen mustard that has anti-inflammatory properties), nicotine, and 5-aminosalicylic acid. If an ulcer becomes infected, empiric antibiotic therapy should be given immediately and adjusted based on culture and sensitivity results.4
Systemic therapy should be considered in patients who do not respond to topical or local interventions, have a rapid and severe course, or have an active underlying bowel disease. Oral prednisone (1 mg/kg/d) has proved to be one of the most successful drugs used to treat PPG. Treatment should be continued until complete lesion healing, and low-dose maintenance therapy should be administered in recurrent cases. Intravenous corticosteroid therapy—hydrocortisone 100 mg 4 times daily or pulse therapy with intravenous methylprednisolone 1 g/d)—can be used for up to 5 days and may be effective. Oral minocycline 100 mg twice daily may be helpful as an adjunctive therapy to corticosteroids. When corticosteroids fail, oral cyclosporine 3 to 5 mg/kg/d often is prescribed. Studies have shown that patients demonstrate clinical improvement within 3 weeks of cyclosporine initiation, and it has been shown further to be more effective than either azathioprine or methotrexate.4,8
Infliximab, a chimeric antibody that binds both circulating and tissue-bound TNF-α, has been shown to effectively treat PPG. A clinical trial conducted by Brooklyn et al9 found that 46% of patients (6/13) treated with infliximab responded compared with only 6% in a placebo control group (1/17). Although infliximab may result in sepsis, the benefits far outweigh the risks, especially for patients with steroid-refractory PPG.4 Adalimumab is a human monoclonal IgG1 antibody to TNF-α that neutralizes its function by blocking the interaction between the molecule and its receptor. Many clinical studies have shown that adalimumab induces and maintains a clinical response in patients with active Crohn disease. The biologic proved to be effective in our patient, but it is associated with potential side effects that should be monitored including injection-site reactions, pruritus, leukopenia, urticaria, and rare instances of alopecia.10 Etanercept is another potentially effective biologic agent.7 Plasma exchange, immunoglobulin infusion, and interferon-alfa therapy also can be used in refractory PPG cases, though data on these treatments are very limited.4
Unlike routine pyoderma gangrenosum—for which surgical intervention is contraindicated—surgical intervention may be appropriate for the peristomal variant. Surgical treatment options include stoma revision and/or relocation; however, both of these procedures are accompanied by failure rates ranging from 40% to 100%.5 Removal of a diseased intestinal segment, especially one with active IBD, may result in healing of the skin lesion. In our patient, removal of the residual and diseased J-pouch was part of the management plan. However,it generally is recommended that any surgical intervention be accompanied by medical therapy including oral metronidazole 500 mg/d and concomitant administration of an immunosuppressant.1,3
Because PPG tends to recur, long-term maintenance therapy should always be considered. Pain reduction, anemia correction, proper nutrition, and management of associated and underlying diseases should be performed. Meticulous care of the stoma and prevention of leaks also should be emphasized. Overall, if PPG is detected and diagnosed early as well as treated appropriately and aggressively, the patient likely will have a good prognosis.4
- Sheldon DG, Sawchuk LL, Kozarek RA, et al. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000;135:564-569.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000;284:1546-1548.
- Afifi L, Sanchez IM, Wallace MM, et al. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78:1195-1204.
- Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1:1-8.
- Javed A, Pal S, Ahuja V, et al. Management of peristomal pyoderma gangrenosum: two different approaches for the same clinical problem. Trop Gastroenterol. 2011;32:153-156.
- Toh JW, Whiteley I. Devastating peristomal pyoderma gangrenosum: challenges in diagnosis and management. Clin Gastroenterol Hepatol. 2017;15:A19-A20.
- DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60:50-54.
- V’lckova-Laskoska MT, Laskoski DS, Caca-Biljanovska NG, et al. Pyoderma gangrenosum successfully treated with cyclosporin A.Adv Exp Med Biol. 1999;455:541-555.
- Brooklyn TN, Dunnill MGS, Shetty A, at al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn’s disease. Inflamm Bowel Dis. 2009;15:803-806.
To the Editor:
Peristomal pyoderma gangrenosum (PPG) is a rare entity first described in 1984.1 Lesions usually begin as pustules that coalesce into an erythematous skin ulceration that contains purulent material. The lesion appears on the skin that surrounds an abdominal stoma. Peristomal pyoderma gangrenosum typically is associated with Crohn disease and ulcerative colitis, cancer, blood dyscrasia, diabetes mellitus, and hepatitis.2 We describe a case of PPG following an ileostomy in a patient with colon cancer and a related history of Crohn disease.
A 32-year-old woman presented to a dermatology office with a spontaneously painful, 3.2-cm ulceration that was extremely tender to palpation, located immediately adjacent to the site of an ileostomy (Figure). The patient had a history of refractory constipation that failed to respond to standard conservative measures 4 years prior. She underwent a colonoscopy, which revealed a 6.5-cm, irregularly shaped, exophytic mass in the rectosigmoid portion of the colon. Histopathologic examination of several biopsies confirmed the diagnosis of moderately well-differentiated adenocarcinoma, and additional evaluation determined the cancer to be stage IIB. She had a medical history of pancolonic Crohn disease since high school that was treated with periodic infusions of infliximab at the standard dose of 5 mg/kg. Colon cancer treatment consisted of preoperative radiotherapy, complete colectomy with ileoanal anastomosis, and creation of a J-pouch and formation of a temporary ileostomy, along with postoperative capecitabine chemotherapy.
The ileostomy eventually was reversed, and the patient did well for 3 years. When the patient developed severe abdominal pain, the J-pouch was examined and found to be remarkably involved with Crohn disease. However, during the colonoscopy, the J-pouch was inadvertently punctured, leading to the formation of a large pelvic abscess. The latter necessitated diversion of stool, and the patient had the original ileostomy recreated.
Prior to presentation to dermatology, various consultants suspected the ulceration was possibly a deep fungal infection, cutaneous Crohn disease, a factitious ulceration, or acute allergic contact dermatitis related to some element of ostomy care. However, dermatologic consultation suggested that the troublesome lesion was classic PPG and recommended administration of a tumor necrosis factor (TNF) α–blocking agent and concomitant intralesional injections of dilute triamcinolone acetonide.
The patient was treated with subcutaneous adalimumab 40 mg once weekly, and received near weekly subcutaneous injections of triamcinolone acetonide 10 mg/mL. After 2 months, the discomfort subsided, and the ulceration gradually resolved into a depressed scar. Eighteen months later, the scar was barely perceptible as a minimally erythematous depression. Adalimumab ultimately was discontinued, as the residual J-pouch was removed, and the biologic drug was associated with extensive alopecia areata–like hair loss. There has been no recurrence of PPG in the 40 months since clinical resolution.
Peristomal pyoderma gangrenosum is an uncommon subtype of pyoderma gangrenosum, which is characterized by chronic, persistent, or recurrent painful ulceration(s) close to an abdominal stoma. In total, fewer than 100 cases of PPG have been reported thus far in the readily available medical literature.3 Inflammatory bowel disease (IBD) is the most frequently diagnosed systemic condition associated with PPG, though other associated conditions include diverticular disease, abdominal malignancy, and neurologic dysfunction. Approximately 2% to 4.3% of all patients who have stoma creation surgery related to underlying IBD develop PPG. It is estimated that the yearly incidence rate of PPG in all abdominal stomas is quite low (approximately 0.6%).4
Peristomal pyoderma gangrenosum can occur at any age, but it tends to predominate in young to middle-aged adults, with a slight female predilection. The etiology and pathogenesis of PPG are largely unknown, though studies have shown that an abnormal immune response may be critical to its development. Risk factors for PPG are not well defined but potentially include autoimmune disorders, a high body mass index, and females or African Americans with IBD.4 Because PPG does not have characteristic histopathologic features, it is a diagnosis of exclusion that is based on the clinical examination and histologic findings that rule out other potential disorders.
There are 4 types of PPG based on the clinical and histopathologic characteristics: ulcerative, pustular, bullous, and vegetative. Peristomal pyoderma gangrenosum tends to be either ulcerative or vegetative, with ulcerative being by far the predominant type. The onset of PPG is quite variable, occurring a few weeks to several years after stoma formation.5 Ulcer size can range from less than 3 cm to 30 cm.4 Lesions begin as deep painful nodules or as superficial hemorrhagic pustules, either idiopathic or following ostensibly minimal trauma. Subsequently, they become necrotic and form an ulceration. The ulcers can be single or multiple lesions, typically with erythematous raised borders and purulent discharge. The ulcers are extremely painful and rapidly progressive. After the ulcers heal, they often leave a characteristic weblike atrophic scar that can break down further following any form of irritation or trauma.5
A prompt diagnosis of PPG is important. A diagnosis of PPG should be considered when dealing with a noninfectious ulcer surrounding a stoma in patients with IBD or other autoimmune conditions.6 Because PPG is a rare skin disorder, it is likely to be missed and lead to unnecessary diagnostic workup and a delay in proper therapy. In our patient, a diagnosis of PPG was overlooked for other infectious and autoimmune causes. The diagnostic evaluation of a patient with PPG is based on 3 principles: (1) ruling out other causes of a peristomal ulcer, such as an abscess, contact dermatitis, or wound infection; (2) determining whether there is an underlying intestinal bowel disease in the stoma; and (3) identifying associated systemic disorders such as vasculitis, erythema nodosum, or similar processes.4 The differential diagnosis depends on the type and stage of PPG and can include malignancy, vasculitis, extraintestinal IBD, infectious disease, and insect bites. A review of the history of the ulcer is helpful in ruling out other diseases, and a colonoscopy or ileoscopy can identify if patients have an underlying active IBD. Swabs for smear and both bacterial and fungal cultures should be taken from the exudate and directly from the ulcer base. Biopsy of the ulcer also helps to exclude alternative diagnoses.6
The primary goals of treating PPG include to reduce pain and the risk for secondary infection, increase pouch adherence, and decrease purulent exudate.7 Although there is not one well-defined optimal therapeutic intervention, there are a variety of effective approaches that may be considered and used. In mild cases, management methods such as dressings, topical agents, or intralesional steroids may be capable of controlling the disease. Daily wound care is important. Moisture-retentive dressings can control pain, induce collagen formation, promote angiogenesis, and prevent contamination. Cleaning the wound with sterile saline and applying an anti-infective agent also may be effective. Application of ultrapotent topical steroids and tacrolimus ointment 0.3% can be used in patients without concomitant secondary infection. In patients who are in remission, human platelet-derived growth factor may be used. Intralesional injections of dilute triamcinolone acetonide or cyclosporine solution also can be helpful. Cyclosporin A was used as a systemic monotherapy to treat a 48-year-old man and 50-year-old woman with the idiopathic form of PPG. After 3 months of treatment, PPG had completely resolved and there were no major side effects.8 Other potential topical therapies that control inflammation and promote wound healing include benzoyl peroxide, chlormethine (topical alkylating agent and nitrogen mustard that has anti-inflammatory properties), nicotine, and 5-aminosalicylic acid. If an ulcer becomes infected, empiric antibiotic therapy should be given immediately and adjusted based on culture and sensitivity results.4
Systemic therapy should be considered in patients who do not respond to topical or local interventions, have a rapid and severe course, or have an active underlying bowel disease. Oral prednisone (1 mg/kg/d) has proved to be one of the most successful drugs used to treat PPG. Treatment should be continued until complete lesion healing, and low-dose maintenance therapy should be administered in recurrent cases. Intravenous corticosteroid therapy—hydrocortisone 100 mg 4 times daily or pulse therapy with intravenous methylprednisolone 1 g/d)—can be used for up to 5 days and may be effective. Oral minocycline 100 mg twice daily may be helpful as an adjunctive therapy to corticosteroids. When corticosteroids fail, oral cyclosporine 3 to 5 mg/kg/d often is prescribed. Studies have shown that patients demonstrate clinical improvement within 3 weeks of cyclosporine initiation, and it has been shown further to be more effective than either azathioprine or methotrexate.4,8
Infliximab, a chimeric antibody that binds both circulating and tissue-bound TNF-α, has been shown to effectively treat PPG. A clinical trial conducted by Brooklyn et al9 found that 46% of patients (6/13) treated with infliximab responded compared with only 6% in a placebo control group (1/17). Although infliximab may result in sepsis, the benefits far outweigh the risks, especially for patients with steroid-refractory PPG.4 Adalimumab is a human monoclonal IgG1 antibody to TNF-α that neutralizes its function by blocking the interaction between the molecule and its receptor. Many clinical studies have shown that adalimumab induces and maintains a clinical response in patients with active Crohn disease. The biologic proved to be effective in our patient, but it is associated with potential side effects that should be monitored including injection-site reactions, pruritus, leukopenia, urticaria, and rare instances of alopecia.10 Etanercept is another potentially effective biologic agent.7 Plasma exchange, immunoglobulin infusion, and interferon-alfa therapy also can be used in refractory PPG cases, though data on these treatments are very limited.4
Unlike routine pyoderma gangrenosum—for which surgical intervention is contraindicated—surgical intervention may be appropriate for the peristomal variant. Surgical treatment options include stoma revision and/or relocation; however, both of these procedures are accompanied by failure rates ranging from 40% to 100%.5 Removal of a diseased intestinal segment, especially one with active IBD, may result in healing of the skin lesion. In our patient, removal of the residual and diseased J-pouch was part of the management plan. However,it generally is recommended that any surgical intervention be accompanied by medical therapy including oral metronidazole 500 mg/d and concomitant administration of an immunosuppressant.1,3
Because PPG tends to recur, long-term maintenance therapy should always be considered. Pain reduction, anemia correction, proper nutrition, and management of associated and underlying diseases should be performed. Meticulous care of the stoma and prevention of leaks also should be emphasized. Overall, if PPG is detected and diagnosed early as well as treated appropriately and aggressively, the patient likely will have a good prognosis.4
To the Editor:
Peristomal pyoderma gangrenosum (PPG) is a rare entity first described in 1984.1 Lesions usually begin as pustules that coalesce into an erythematous skin ulceration that contains purulent material. The lesion appears on the skin that surrounds an abdominal stoma. Peristomal pyoderma gangrenosum typically is associated with Crohn disease and ulcerative colitis, cancer, blood dyscrasia, diabetes mellitus, and hepatitis.2 We describe a case of PPG following an ileostomy in a patient with colon cancer and a related history of Crohn disease.
A 32-year-old woman presented to a dermatology office with a spontaneously painful, 3.2-cm ulceration that was extremely tender to palpation, located immediately adjacent to the site of an ileostomy (Figure). The patient had a history of refractory constipation that failed to respond to standard conservative measures 4 years prior. She underwent a colonoscopy, which revealed a 6.5-cm, irregularly shaped, exophytic mass in the rectosigmoid portion of the colon. Histopathologic examination of several biopsies confirmed the diagnosis of moderately well-differentiated adenocarcinoma, and additional evaluation determined the cancer to be stage IIB. She had a medical history of pancolonic Crohn disease since high school that was treated with periodic infusions of infliximab at the standard dose of 5 mg/kg. Colon cancer treatment consisted of preoperative radiotherapy, complete colectomy with ileoanal anastomosis, and creation of a J-pouch and formation of a temporary ileostomy, along with postoperative capecitabine chemotherapy.
The ileostomy eventually was reversed, and the patient did well for 3 years. When the patient developed severe abdominal pain, the J-pouch was examined and found to be remarkably involved with Crohn disease. However, during the colonoscopy, the J-pouch was inadvertently punctured, leading to the formation of a large pelvic abscess. The latter necessitated diversion of stool, and the patient had the original ileostomy recreated.
Prior to presentation to dermatology, various consultants suspected the ulceration was possibly a deep fungal infection, cutaneous Crohn disease, a factitious ulceration, or acute allergic contact dermatitis related to some element of ostomy care. However, dermatologic consultation suggested that the troublesome lesion was classic PPG and recommended administration of a tumor necrosis factor (TNF) α–blocking agent and concomitant intralesional injections of dilute triamcinolone acetonide.
The patient was treated with subcutaneous adalimumab 40 mg once weekly, and received near weekly subcutaneous injections of triamcinolone acetonide 10 mg/mL. After 2 months, the discomfort subsided, and the ulceration gradually resolved into a depressed scar. Eighteen months later, the scar was barely perceptible as a minimally erythematous depression. Adalimumab ultimately was discontinued, as the residual J-pouch was removed, and the biologic drug was associated with extensive alopecia areata–like hair loss. There has been no recurrence of PPG in the 40 months since clinical resolution.
Peristomal pyoderma gangrenosum is an uncommon subtype of pyoderma gangrenosum, which is characterized by chronic, persistent, or recurrent painful ulceration(s) close to an abdominal stoma. In total, fewer than 100 cases of PPG have been reported thus far in the readily available medical literature.3 Inflammatory bowel disease (IBD) is the most frequently diagnosed systemic condition associated with PPG, though other associated conditions include diverticular disease, abdominal malignancy, and neurologic dysfunction. Approximately 2% to 4.3% of all patients who have stoma creation surgery related to underlying IBD develop PPG. It is estimated that the yearly incidence rate of PPG in all abdominal stomas is quite low (approximately 0.6%).4
Peristomal pyoderma gangrenosum can occur at any age, but it tends to predominate in young to middle-aged adults, with a slight female predilection. The etiology and pathogenesis of PPG are largely unknown, though studies have shown that an abnormal immune response may be critical to its development. Risk factors for PPG are not well defined but potentially include autoimmune disorders, a high body mass index, and females or African Americans with IBD.4 Because PPG does not have characteristic histopathologic features, it is a diagnosis of exclusion that is based on the clinical examination and histologic findings that rule out other potential disorders.
There are 4 types of PPG based on the clinical and histopathologic characteristics: ulcerative, pustular, bullous, and vegetative. Peristomal pyoderma gangrenosum tends to be either ulcerative or vegetative, with ulcerative being by far the predominant type. The onset of PPG is quite variable, occurring a few weeks to several years after stoma formation.5 Ulcer size can range from less than 3 cm to 30 cm.4 Lesions begin as deep painful nodules or as superficial hemorrhagic pustules, either idiopathic or following ostensibly minimal trauma. Subsequently, they become necrotic and form an ulceration. The ulcers can be single or multiple lesions, typically with erythematous raised borders and purulent discharge. The ulcers are extremely painful and rapidly progressive. After the ulcers heal, they often leave a characteristic weblike atrophic scar that can break down further following any form of irritation or trauma.5
A prompt diagnosis of PPG is important. A diagnosis of PPG should be considered when dealing with a noninfectious ulcer surrounding a stoma in patients with IBD or other autoimmune conditions.6 Because PPG is a rare skin disorder, it is likely to be missed and lead to unnecessary diagnostic workup and a delay in proper therapy. In our patient, a diagnosis of PPG was overlooked for other infectious and autoimmune causes. The diagnostic evaluation of a patient with PPG is based on 3 principles: (1) ruling out other causes of a peristomal ulcer, such as an abscess, contact dermatitis, or wound infection; (2) determining whether there is an underlying intestinal bowel disease in the stoma; and (3) identifying associated systemic disorders such as vasculitis, erythema nodosum, or similar processes.4 The differential diagnosis depends on the type and stage of PPG and can include malignancy, vasculitis, extraintestinal IBD, infectious disease, and insect bites. A review of the history of the ulcer is helpful in ruling out other diseases, and a colonoscopy or ileoscopy can identify if patients have an underlying active IBD. Swabs for smear and both bacterial and fungal cultures should be taken from the exudate and directly from the ulcer base. Biopsy of the ulcer also helps to exclude alternative diagnoses.6
The primary goals of treating PPG include to reduce pain and the risk for secondary infection, increase pouch adherence, and decrease purulent exudate.7 Although there is not one well-defined optimal therapeutic intervention, there are a variety of effective approaches that may be considered and used. In mild cases, management methods such as dressings, topical agents, or intralesional steroids may be capable of controlling the disease. Daily wound care is important. Moisture-retentive dressings can control pain, induce collagen formation, promote angiogenesis, and prevent contamination. Cleaning the wound with sterile saline and applying an anti-infective agent also may be effective. Application of ultrapotent topical steroids and tacrolimus ointment 0.3% can be used in patients without concomitant secondary infection. In patients who are in remission, human platelet-derived growth factor may be used. Intralesional injections of dilute triamcinolone acetonide or cyclosporine solution also can be helpful. Cyclosporin A was used as a systemic monotherapy to treat a 48-year-old man and 50-year-old woman with the idiopathic form of PPG. After 3 months of treatment, PPG had completely resolved and there were no major side effects.8 Other potential topical therapies that control inflammation and promote wound healing include benzoyl peroxide, chlormethine (topical alkylating agent and nitrogen mustard that has anti-inflammatory properties), nicotine, and 5-aminosalicylic acid. If an ulcer becomes infected, empiric antibiotic therapy should be given immediately and adjusted based on culture and sensitivity results.4
Systemic therapy should be considered in patients who do not respond to topical or local interventions, have a rapid and severe course, or have an active underlying bowel disease. Oral prednisone (1 mg/kg/d) has proved to be one of the most successful drugs used to treat PPG. Treatment should be continued until complete lesion healing, and low-dose maintenance therapy should be administered in recurrent cases. Intravenous corticosteroid therapy—hydrocortisone 100 mg 4 times daily or pulse therapy with intravenous methylprednisolone 1 g/d)—can be used for up to 5 days and may be effective. Oral minocycline 100 mg twice daily may be helpful as an adjunctive therapy to corticosteroids. When corticosteroids fail, oral cyclosporine 3 to 5 mg/kg/d often is prescribed. Studies have shown that patients demonstrate clinical improvement within 3 weeks of cyclosporine initiation, and it has been shown further to be more effective than either azathioprine or methotrexate.4,8
Infliximab, a chimeric antibody that binds both circulating and tissue-bound TNF-α, has been shown to effectively treat PPG. A clinical trial conducted by Brooklyn et al9 found that 46% of patients (6/13) treated with infliximab responded compared with only 6% in a placebo control group (1/17). Although infliximab may result in sepsis, the benefits far outweigh the risks, especially for patients with steroid-refractory PPG.4 Adalimumab is a human monoclonal IgG1 antibody to TNF-α that neutralizes its function by blocking the interaction between the molecule and its receptor. Many clinical studies have shown that adalimumab induces and maintains a clinical response in patients with active Crohn disease. The biologic proved to be effective in our patient, but it is associated with potential side effects that should be monitored including injection-site reactions, pruritus, leukopenia, urticaria, and rare instances of alopecia.10 Etanercept is another potentially effective biologic agent.7 Plasma exchange, immunoglobulin infusion, and interferon-alfa therapy also can be used in refractory PPG cases, though data on these treatments are very limited.4
Unlike routine pyoderma gangrenosum—for which surgical intervention is contraindicated—surgical intervention may be appropriate for the peristomal variant. Surgical treatment options include stoma revision and/or relocation; however, both of these procedures are accompanied by failure rates ranging from 40% to 100%.5 Removal of a diseased intestinal segment, especially one with active IBD, may result in healing of the skin lesion. In our patient, removal of the residual and diseased J-pouch was part of the management plan. However,it generally is recommended that any surgical intervention be accompanied by medical therapy including oral metronidazole 500 mg/d and concomitant administration of an immunosuppressant.1,3
Because PPG tends to recur, long-term maintenance therapy should always be considered. Pain reduction, anemia correction, proper nutrition, and management of associated and underlying diseases should be performed. Meticulous care of the stoma and prevention of leaks also should be emphasized. Overall, if PPG is detected and diagnosed early as well as treated appropriately and aggressively, the patient likely will have a good prognosis.4
- Sheldon DG, Sawchuk LL, Kozarek RA, et al. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000;135:564-569.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000;284:1546-1548.
- Afifi L, Sanchez IM, Wallace MM, et al. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78:1195-1204.
- Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1:1-8.
- Javed A, Pal S, Ahuja V, et al. Management of peristomal pyoderma gangrenosum: two different approaches for the same clinical problem. Trop Gastroenterol. 2011;32:153-156.
- Toh JW, Whiteley I. Devastating peristomal pyoderma gangrenosum: challenges in diagnosis and management. Clin Gastroenterol Hepatol. 2017;15:A19-A20.
- DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60:50-54.
- V’lckova-Laskoska MT, Laskoski DS, Caca-Biljanovska NG, et al. Pyoderma gangrenosum successfully treated with cyclosporin A.Adv Exp Med Biol. 1999;455:541-555.
- Brooklyn TN, Dunnill MGS, Shetty A, at al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn’s disease. Inflamm Bowel Dis. 2009;15:803-806.
- Sheldon DG, Sawchuk LL, Kozarek RA, et al. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000;135:564-569.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000;284:1546-1548.
- Afifi L, Sanchez IM, Wallace MM, et al. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78:1195-1204.
- Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1:1-8.
- Javed A, Pal S, Ahuja V, et al. Management of peristomal pyoderma gangrenosum: two different approaches for the same clinical problem. Trop Gastroenterol. 2011;32:153-156.
- Toh JW, Whiteley I. Devastating peristomal pyoderma gangrenosum: challenges in diagnosis and management. Clin Gastroenterol Hepatol. 2017;15:A19-A20.
- DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60:50-54.
- V’lckova-Laskoska MT, Laskoski DS, Caca-Biljanovska NG, et al. Pyoderma gangrenosum successfully treated with cyclosporin A.Adv Exp Med Biol. 1999;455:541-555.
- Brooklyn TN, Dunnill MGS, Shetty A, at al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn’s disease. Inflamm Bowel Dis. 2009;15:803-806.
Practice Points
- A pyoderma gangrenosum subtype occurs in close proximity to an abdominal stoma.
- Peristomal pyoderma gangrenosum is a diagnosis of exclusion.
- Peristomal pyoderma gangrenosum typically responds best to tumor necrosis factor α blockers and corticosteroid therapy (intralesional and systemic).

Focal Palmoplantar Keratoderma and Gingival Keratosis Caused by a KRT16 Mutation
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
 showing focal palmoplantar keratoderma and gingival keratosis in those heterozygous for KRT16 mutation p.R127H")
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.

Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.

Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
Practice Points
- Focal palmoplantar keratoderma and gingival keratosis (FPGK) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma (PPK) and gingival hyperkeratosis presenting as leukokeratosis.
- Focal pressure-related PPK and oral hyperkeratosis also are seen in pachyonychia congenita (PC), which is caused by mutations in keratin genes and is distinguished from FPGK by characteristic nail changes.
- A shared causative gene suggests that FPGK should be considered part of the PC spectrum.

Acute Generalized Exanthematous Pustulosis Induced by the Second-Generation Antipsychotic Cariprazine
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.

A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.

Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.
A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.
Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.
A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.
Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
Practice Points
- The second-generation antipsychotic cariprazine has been shown to be a potential causative agent in acute generalized exanthematous pustulosis (AGEP).
- Treatment of AGEP is mainly supportive and consists of discontinuation of the causative agent as well as symptom control using cold compresses and topical corticosteroids.
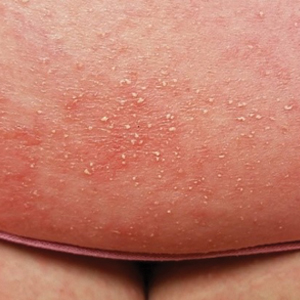
Nevus Lipomatosis Deemed Suspicious by Airport Security
To the Editor:
A 47-year-old man presented at the dermatology clinic with a growing lesion on the left medial thigh.
Physical examination revealed a 5-cm, pedunculated, fatty nodule on the left medial thigh that was clinically consistent with nevus lipomatosis (NL)(Figure). Although benign, trouble traveling through airport security prompted the patient to request shave removal, which subsequently was performed. Histology showed a large pedunculated nodule with prominent adipose tissue, consistent with NL. At 3-month follow-up, the patient reported getting through airport security multiple times without incident.

Nevus lipomatosis is a benign fatty lesion most commonly found on the medial thighs or trunk of adults. The lesion usually is asymptomatic but can become irritated by rubbing or catching on clothing. Our patient had symptomatic NL that caused delays getting through airport security; he experienced full resolution after simple shave removal. In rare instances, both benign and malignant skin conditions have been seen on airport scanning devices since the introduction of increased security measures following September 11, 2001. In 2016, Heymann1 reported a man with a 1.5-cm epidermal inclusion cyst detected by airport security scanners, prompting the traveler to request and carry a medically explanatory letter used to get through security. In 2015 Mayer and Adams2 described a case of nodular melanoma that was detected 20 times over a period of 2 months by airport scanners, and in 2016, Caine et al3 reported a case of desmoplastic melanoma that was detected by airport security, but after its removal was not identified by security for the next 40 flights. Noncutaneous pathology also can be detected by airport scanners. In 2013, Naraynsingh et al4 reported a man with a large left reducible inguinal hernia who was stopped by airport security and subjected to an invasive physical examination of the area. These instances demonstrate the breadth of conditions that can be cumbersome when individuals are traveling by airplane in our current security climate.
Our patient had to go through the trouble of having the benign NL lesion removed to avoid the hassle of repeatedly being stopped by airport security. The patient had the lesion removed and is doing well, but the procedure could have been avoided if systems existed to help patients with dermatologic and medical conditions at airport security. Our patient likely will never be stopped again for the suspicious lump on the left inner thigh, but many others will be stopped for similar reasons.
- Heymann WR. A cyst misinterpreted on airport scan as security threat. JAMA Dermatol. 2016;152:1388. doi:10.1001/jamadermatol.2016.3329
- Mayer JE, Adams BB. Nodular melanoma serendipitously detected by airport full body scanners. Dermatology. 2015;230:16-17. doi:10.1159/000368045
- Caine P, Javed MU, Karoo ROS. A desmoplastic melanoma detected by an airport security scanner. J Plast Reconstr Aesthet Surg. 2016;69:874-876. doi:10.1016/j.bjps.2016.02.022
- Naraynsingh V, Cawich SO, Maharaj R, et al. Inguinal hernia and airport scanners: an emerging indication for repair? 2013;2013:952835. Case Rep Med. doi:10.1155/2013/952835
To the Editor:
A 47-year-old man presented at the dermatology clinic with a growing lesion on the left medial thigh.
Physical examination revealed a 5-cm, pedunculated, fatty nodule on the left medial thigh that was clinically consistent with nevus lipomatosis (NL)(Figure). Although benign, trouble traveling through airport security prompted the patient to request shave removal, which subsequently was performed. Histology showed a large pedunculated nodule with prominent adipose tissue, consistent with NL. At 3-month follow-up, the patient reported getting through airport security multiple times without incident.
Nevus lipomatosis is a benign fatty lesion most commonly found on the medial thighs or trunk of adults. The lesion usually is asymptomatic but can become irritated by rubbing or catching on clothing. Our patient had symptomatic NL that caused delays getting through airport security; he experienced full resolution after simple shave removal. In rare instances, both benign and malignant skin conditions have been seen on airport scanning devices since the introduction of increased security measures following September 11, 2001. In 2016, Heymann1 reported a man with a 1.5-cm epidermal inclusion cyst detected by airport security scanners, prompting the traveler to request and carry a medically explanatory letter used to get through security. In 2015 Mayer and Adams2 described a case of nodular melanoma that was detected 20 times over a period of 2 months by airport scanners, and in 2016, Caine et al3 reported a case of desmoplastic melanoma that was detected by airport security, but after its removal was not identified by security for the next 40 flights. Noncutaneous pathology also can be detected by airport scanners. In 2013, Naraynsingh et al4 reported a man with a large left reducible inguinal hernia who was stopped by airport security and subjected to an invasive physical examination of the area. These instances demonstrate the breadth of conditions that can be cumbersome when individuals are traveling by airplane in our current security climate.
Our patient had to go through the trouble of having the benign NL lesion removed to avoid the hassle of repeatedly being stopped by airport security. The patient had the lesion removed and is doing well, but the procedure could have been avoided if systems existed to help patients with dermatologic and medical conditions at airport security. Our patient likely will never be stopped again for the suspicious lump on the left inner thigh, but many others will be stopped for similar reasons.
To the Editor:
A 47-year-old man presented at the dermatology clinic with a growing lesion on the left medial thigh.
Physical examination revealed a 5-cm, pedunculated, fatty nodule on the left medial thigh that was clinically consistent with nevus lipomatosis (NL)(Figure). Although benign, trouble traveling through airport security prompted the patient to request shave removal, which subsequently was performed. Histology showed a large pedunculated nodule with prominent adipose tissue, consistent with NL. At 3-month follow-up, the patient reported getting through airport security multiple times without incident.
Nevus lipomatosis is a benign fatty lesion most commonly found on the medial thighs or trunk of adults. The lesion usually is asymptomatic but can become irritated by rubbing or catching on clothing. Our patient had symptomatic NL that caused delays getting through airport security; he experienced full resolution after simple shave removal. In rare instances, both benign and malignant skin conditions have been seen on airport scanning devices since the introduction of increased security measures following September 11, 2001. In 2016, Heymann1 reported a man with a 1.5-cm epidermal inclusion cyst detected by airport security scanners, prompting the traveler to request and carry a medically explanatory letter used to get through security. In 2015 Mayer and Adams2 described a case of nodular melanoma that was detected 20 times over a period of 2 months by airport scanners, and in 2016, Caine et al3 reported a case of desmoplastic melanoma that was detected by airport security, but after its removal was not identified by security for the next 40 flights. Noncutaneous pathology also can be detected by airport scanners. In 2013, Naraynsingh et al4 reported a man with a large left reducible inguinal hernia who was stopped by airport security and subjected to an invasive physical examination of the area. These instances demonstrate the breadth of conditions that can be cumbersome when individuals are traveling by airplane in our current security climate.
Our patient had to go through the trouble of having the benign NL lesion removed to avoid the hassle of repeatedly being stopped by airport security. The patient had the lesion removed and is doing well, but the procedure could have been avoided if systems existed to help patients with dermatologic and medical conditions at airport security. Our patient likely will never be stopped again for the suspicious lump on the left inner thigh, but many others will be stopped for similar reasons.
- Heymann WR. A cyst misinterpreted on airport scan as security threat. JAMA Dermatol. 2016;152:1388. doi:10.1001/jamadermatol.2016.3329
- Mayer JE, Adams BB. Nodular melanoma serendipitously detected by airport full body scanners. Dermatology. 2015;230:16-17. doi:10.1159/000368045
- Caine P, Javed MU, Karoo ROS. A desmoplastic melanoma detected by an airport security scanner. J Plast Reconstr Aesthet Surg. 2016;69:874-876. doi:10.1016/j.bjps.2016.02.022
- Naraynsingh V, Cawich SO, Maharaj R, et al. Inguinal hernia and airport scanners: an emerging indication for repair? 2013;2013:952835. Case Rep Med. doi:10.1155/2013/952835
- Heymann WR. A cyst misinterpreted on airport scan as security threat. JAMA Dermatol. 2016;152:1388. doi:10.1001/jamadermatol.2016.3329
- Mayer JE, Adams BB. Nodular melanoma serendipitously detected by airport full body scanners. Dermatology. 2015;230:16-17. doi:10.1159/000368045
- Caine P, Javed MU, Karoo ROS. A desmoplastic melanoma detected by an airport security scanner. J Plast Reconstr Aesthet Surg. 2016;69:874-876. doi:10.1016/j.bjps.2016.02.022
- Naraynsingh V, Cawich SO, Maharaj R, et al. Inguinal hernia and airport scanners: an emerging indication for repair? 2013;2013:952835. Case Rep Med. doi:10.1155/2013/952835
Practice Points
- Nevus lipomatosis is a benign fatty lesion that most commonly is found on the medial thighs or trunk of adults.
- Both benign and malignant skin conditions have been detected on airport scanning devices.
- At times, patients must go through the hassle of having the benign lesions removed to avoid repeated problems at airport security.

Chronic Retiform Purpura of the Abdomen and Thighs: A Fatal Case of Intravascular Large Cell Lymphoma
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).

A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
. B, CD20 immunohistochemical staining highlighted atypical B cells")
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).
A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).
A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
Practice Points
- Intravascular large cell lymphoma (ILCL) is a life-threatening malignancy that can present with retiform purpura and other symptoms of vascular occlusion.
- The diagnosis of ILCL can be challenging because of the presence of distractors, and multiple biopsies may be required to establish pathology.
