User login
Binge-Eating Disorder: Prevalence, Predictors, and Management in the Primary Care Setting
From the Department of Psychology, Eastern Michigan University, Ypsilanti, MI.
Abstract
- Objective: To describe the epidemiology, clinical features, clinical course, medical complications, and treatment of binge-eating disorder (BED).
- Methods: Review of the literature.
- Results: BED, the most common eating disorder, is a distinct pattern of binge eating accompanied by a sense of loss of control over eating without inappropriate compensatory behaviors. Because people with BED more commonly seek treatment for the psychological and medical factors that are associated with the disorder, patients’ first point of contact with the medical profession is likely to be the primary care physician (PCP). The PCP’s role includes making efforts to screen for BED symptoms, employing motivational interviewing strategies to enhance likelihood of following through with treatment, providing psychoeducational information about eating and weight control, monitoring eating, weight, and related medical problems at follow-up visits, and making referrals to behavioral health specialists who can deliver empirically supported treatments for BED.
- Conclusion: Proper screening and referral in the primary care setting can optimize the likelihood that patients obtain empirically supported treatment.

BED is the most common eating disorder, but it is one for which many do not seek treatment directly. Rather, those struggling with BED more commonly seek treatment for the psychological and medical factors that are strongly associated with the disorder. As will be reviewed below, these factors include poor social adjustment, functional impairment, psychological distress and psychiatric comorbidity, and myriad medical sequelae due to obesity and weight cycling. As such, the BED patient’s point of first contact with the medical profession is most likely to be with the primary care physician, who has several roles in the treatment of BED. There is a limited evidence base for pharmacological treatment of BED, with some medications yielding short-term reductions in binge eating, but none with strong support for long-term efficacy [4]. However, with the recent FDA approval of lisdexamfetamine dimesylate for the treatment of moderate to severe BED, this picture may change. Nonetheless, pharmacologic interventions for comorbid medical conditions will fall solidly in the bailiwick of the primary care physician. In addition, the primary care physician’s role includes making efforts to screen for BED symptoms; employing motivational interviewing strategies to enhance likelihood of following through with treatment; providing psychoeducational information about eating and weight control; monitoring eating, weight, and related medical problems at follow-up visits; and making referrals to behavioral health specialists who can deliver empirically supported treatments for BED. Finally, because BED is typically associated with weight gain over time [5], the primary care physician is encouraged to reinforce the clinical significance of weight maintenance as opposed to necessarily promoting a goal of weight loss. The rationale for this primary care approach is reviewed below, in consideration of the scientific literature and a case study highlighting common clinical features.
Case Study
Initial Presentation
A 35-year-old Caucasian woman schedules an appointment for her annual physical examination with her primary care physician. She reports generally good health but complains of low mood, joint pain, and difficulties managing her weight. Her blood pressure is managed with 100 mg/day of metoprolol. The only other medication she takes is birth control (ethinyl estradiol 20 mcg).
Physical Examination
During physical examination, it is determined that the patient is 5'6" and weighs 286 lb, with a body mass index (BMI) of 46.2 kg/m2, placing her in WHO obesity class III. The patient’s blood pressure is 130/85 mm Hg (medically managed), and her heart rate is 83 bpm. The patient states that she has been experiencing episodes of low mood off and on most of her life; she recently ended a relationship, which has exacerbated her symptoms. The physician states that the patient has gained a significant amount of weight since her last physical examination. The patient reports that she quit smoking 6 months ago and has since gained approximately 30 lb; she has considered smoking again to manage her weight.
• What are the diagnostic criteria for BED?
BED diagnostic criteria (Table 1) have been closely examined for their validity and clinical utility, and several have been the subject of intense debate in the BED literature. The first BED criterion, recurrent episodes of binge eating, refers to 3 essential components: amount of food, time period, and a subjective experience of loss of control. The majority of debate regarding this criterion revolves around the requirement for consumption of a “large amount of food.” There are 2 primary arguments against this criterion. First, it is inherently subjective and requires the person making the diagnosis to distinguish between normative food intake and excessive food intake [6]. There is also some debate as to whether or not individuals with BED actually consume large amounts of food when they binge. However, research supports that those with BED may consume over 1000 kcal during binge episodes, far more than those without BED who are asked to binge eat in the lab [7,8].
Nonetheless, a distinction has been made between objective binge-eating episodes (OBE) and subjective binge eating episodes (SBE) [9]. OBEs are binge eating episodes that meet the full criteria including a large amount of food and a subjective loss of control. SBEs, in contrast, are binge eating episodes that include a subjective loss of control but not a large quantity of food. If consumption of a large quantity of food is essential to the underlying pathology of BED, one would expect that OBEs and SBEs would be associated with different clinical characteristics. However, several studies have failed to find significant difference between individuals reporting OBEs and SBEs with regard to age, age of BE onset, BE severity, interpersonal problems, depressive symptoms, generalized psychopathology, and ED-related psychopathology [10–13]. Results regarding prognosis are mixed, with some suggesting that SBE more readily responds to placebo, while others suggest that SBEs are slower to remit than OBEs [11,13,14]. With respect to primary care, this literature suggests that it is not necessary for busy primary care physicians to devote time to understanding the amount of food consumed by the patient; if the patient perceives that her eating is out of control and excessive, that can generally be considered valid data in terms of considering a BED diagnosis, particularly when combined with even moderately overweight status.
In contrast to the controversy regarding amount of food, the majority of studies suggest that BED binge eating episodes fall within the 2-hour duration specified by the DSM-5 criteria, although longer durations have been reported [13]. The loss of control (LOC) criterion also appears to be relatively well-supported across studies [13,14]. LOC is a key defining feature of a binge eating episode for individuals with and without BED [15–18].Furthermore, the emotional distress associated with loss of control has been associated with depressive symptoms, appearance dissatisfaction, and poorer mental health-related quality of life [19]. In contrast, one study found that 18.6% of self-reported binges were not associated with loss of control [20]. Of note, there is some concern that the focus on LOC in the diagnostic criteria may lead to under diagnosis of BED among men, as women with BED were more likely than men to identify LOC as a core aspect of a binge eating episode [17].
The second DSM-5 criterion for BED requires that BE episodes be associated with 3 or more of the following: (a) eating more rapidly than normal; (b) eating until uncomfortably full; (c) eating large amounts of food in the absence of hunger; (d) eating alone because of embarrassment about how much one is eating; and (e) feeling disgusted with oneself, depressed, or very guilty after overeating. This criterion is not as controversial as the first, and has correspondingly not received as much attention in the BED literature. However, results from a handful of studies provide some support for their inclusion, particularly in light of the fact that individuals are only required to endorse 3 of the 5 symptoms [13–15,17,21].
The third criteria for BED requires that individuals experience “marked distress” about BE. Only one known study has directly evaluated the distress criterion, and its validity was confirmed by results that suggested individuals with full-threshold BED had significantly greater ED-related psychopathology and depressive symptoms as compared to individuals who met all but the distress criteria for a BED diagnosis [22].
The fourth criteria for BED stipulates that BE occurs an average of once a week for 3 months. Previously, DSM-IV-TR required more frequent episodes, at least 2 days a week for 6 months, but this was criticized as lacking in empirical basis [23]. The current state of the evidence suggests that, with regard to frequency of BE episodes, BED best fits a continuous model rather than a categorical model. That is, symptoms and related impairment exist across a severity spectrum as a function of how often BE episodes occur. For example, in a critical review, Wilson and Sysko noted that individuals with sub-threshold frequency of BE episodes had less severe psychopathology than those meeting criteria for DSM-IV BE frequency (ie, at least 2 days a week for 6 months), but they were still significantly more impaired than those who did not binge eat [24]. The authors asserted that there was no empirical rationale for preserving the criteria of 2 binge days per week for 6 months, and indeed, DSM-5 adopted a more relaxed standard. As is the case with symptoms of many psychological disorders, there does not appear to be a definitive and concrete point at which binge eating becomes pathological [23]. Fortunately, reliability for the new criteria is good and appears superior to the DSM-IV criteria [25].
Finally, the last criteria for BED—which remains unchanged from the provisional criteria in DSM-IV-TR —is essentially a rule-out that states that BE should not be accompanied by the regular use of “inappropriate compensatory behaviors” or exclusively occur during the course of anorexia or bulimia. These criteria have also been criticized as being subjective, particularly in light of the fact that individuals with BED often report a history of infrequent purging behavior and frequently engage in weight-loss attempts [6,13,14]. However, the need for a rule-out is clear given that BE also occurs during the course of bulimia and anorexia, binge-eating/purging type, and it is supported by the low rates of crossover from BED to bulimia and/or anorexia [26].
Remission and severity specifiers are new to DSM-5. With respect to the latter, a recent study observed small but significant elevations in eating pathology among those with moderate severity BED, relative to the eating pathology experienced by those with mild severity, but there were no differences in level of associated depression. Interestingly, a better differentiator of severity of eating pathology and depression among patients with BED was overvaluation of shape/weight [27]. As such, the primary care physician might be better advised to focus on indicators of this important variable by querying the extent to which the patient’s shape and weight have influenced how she feels about (judges/thinks/evaluates) herself as a person, rather than using the number of BED symptoms alone as the indicator of severity.
• What is the epidemiology of BED?
Based on DSM-IV-TR criteria, the overall lifetime prevalence rate for BED has been reported to be 2.8%, and it is more common in women (3.5%) than men (2%) [28]; the overall 12-month prevalence rate is 1.2% (1.6% in women and 0.8% in men) [28]. Using DSM-5 criteria, a recent study observed that lifetime prevalence of BED by age 20 was 3.0% for BED and an additional 3.6% for subthreshold BED, with peak age of onset (for both) between ages 18 to 20 years [29]. Notably, even though prevalence rates are slightly higher using DSM-5 criteria (presumably, due to the relaxed criteria for frequency and duration of binge eating), effect sizes for impairment are also higher, suggesting that the revised criteria are not identifying BED cases marked by less impairment [29]. Although often thought of as a disorder common among young women, BED prevalence among middle-aged women (40–60 years) has a prevalence of at least 1.5%, with additional subthreshold cases being common in this age-range; groups meeting full BED criteria and subthreshold cases are both characterized by high levels of distress and impairment [30].
Gender Differences
Men engage in overeating as much or more than women but are less likely to endorse a loss of control and/or distress associated with BE [28,31], and thus are less likely to meet full BED criteria. However, when men do meet criteria for BED, they experience as much clinical impairment as their female counterparts [32]. Additionally, men’s BE may be more directly affected by body image dissatisfaction than women’s BE, and although it is associated with negative affect, it is less likely to be associated with interactions between negative affect and dietary restraint than seems to be the case for women [33]. In addition, in the primary care setting, men with BED were strikingly similar to their female counterparts on most historical and developmental variables [33]. However, men reported more frequent strenuous exercise, whereas women reported that onset of overweight and dieting occurred earlier in life [34]. That same study observed that men (57%) were more likely than women with BED (31%) to meet criteria for metabolic syndrome, even after controlling for race and BMI. A second study by the same research group again demonstrated that men with BED are more likely to show elevated blood pressure, triglycerides, and meet criteria for metabolic syndrome, whereas women are more likely to have elevated total cholesterol [35].
Race/Ethnicity
The evidence related to rates of BED among ethnic minorities is equivocal, with some studies demonstrating that Caucasian women are more likely to experience clinical levels of BED symptoms [36,37], others finding comparable rates between Caucasian and African-American women [38,39], and still others discussing the possibility of finding the greatest rates of binge eating in ethnic minority samples [40], especially in light of the high rates of obesity observed in some ethnic minority groups [41,42]. Studies that focus on subclinical levels of eating pathology among undergraduate students are most likely to find significant ethnic differences, while studies of nonclinical samples utilizing diagnostic threshold find the fewest differences [43]. There is at least some research demonstrating the highest rates of body image disturbance or eating problems among Asian Americans [44,45]. In addition, Latino individuals with BED may have higher levels of ED-related psychopathology as compared with Caucasian individuals [46]. Finally, Caucasian individuals who experience BED may be more likely to utilize mental health services as compared with other ethnic groups [47].
Age
Lower rates of BED have been documented in elderly individuals relative to their younger counterparts in population-based studies [28]. However, this may be due to recall bias, birth cohort effects, restricted access to studies, and/or increased medical morbidity leading to premature mortality [48]. Guerdjikova et al [48] also noted that many treatment outcomes studies have exclusion criteria related to age. This is unfortunate, as elderly individuals and their younger counterparts appear to exhibit similar levels of BE behavior, distress due to BE, weight and shape concerns, psychiatric comorbidity, and obesity. However, elderly individuals have reported later onset, longer duration of illness, and less medical morbidity [48]. In another study, Mangweth-Matzek et al [30] surveyed women between the ages of 40 and 60; they found that very few respondents met full criteria for an eating disorder. However, when criteria were relaxed (ie, dropping associated symptomology for BED and frequency criteria for bulimia nervosa) an additional 4.8% of the sample met criteria. Notably, women with subthreshold eating disorders reported very similar levels of comorbid psychopathology as women whose symptoms met diagnostic criteria.
• What tools are available for assessment of BED in the primary care setting?
Two of the most commonly used questionnaires in specialty clinics are the Eating Disorders Examination– Questionnaire (EDE-Q [49]), and the Questionnaire on Eating and Weight Patterns – Revised (QEWP-R [50]). In the primary care setting, both appear to be low-cost and time-efficient methods of screening for BED. The EDE-Q, however, may underestimate frequency of binge eating episodes and overestimate the extent of eating-related pathology [51]. Notably, the QEWP has been revised to reflect DSM-5 criteria and is available free of charge (QEWP-5 [52]). The Binge Eating Scale [53] is a 16-item scale often used to assess severity of binge eating; it is free and easily accessible online. Regardless of what measure is used, research indicates that a higher proportion of people agree to having episodes where they ‘‘lose control over eating’’ than when asked about having episodes of ‘‘binge eating’’ [54], so asking about loss of control over eating might be the more advisable way to open the discussion with patients about their eating behavior. In assessing for binge eating, physicians should also be aware of some of the differences in clinical presentation observed for ethnic minorities (eg, lower drive for thinness among African-American women) as well as some research demonstrating that measures such as the Eating Disorder Diagnostic Scale do not assess equivalent constructs in African-American and Caucasian clients [55]. Finally, while self-report measures often serve a practical function of quickly assessing a large group, physicians may want to consider relying on interview-based techniques for clients with lower levels of education attainment and literacy; at least one study has demonstrated problems with readability and comprehensibility with most BED measures [56].

• What are the clinical features of BED?
BED and Obesity
The specific impact of BED on health is difficult to separate from the impact of obesity on health, as the two conditions frequently co-occur and are confounded in many studies. Of relevance to the primary care setting, many BED patients report gaining a substantial amount of weight in the year prior to seeking treatment [57].
Although individuals with BED are often obese, proponents of classifying BED as a separate DSM diagnosis argue that individuals with BED differ from their non-BED obese counterparts in regards to eating patterns, eating disordered psychopathology, and associated features and comorbidities. Individuals with BED consume more calories in laboratory studies than weight-matched controls [6,7,58]. In contrast, studies utilizing ecological momentary assessment (ie, real-time assessments) found no differences between BED obese and non-BED obese participants in the frequency of self-reported binge eating and caloric intake during binge eating episodes [59,60]. BED participants, however, were more likely to report higher stress, desire to binge, negative affect, dietary restraint, and being alone immediately before self-reported binge eating episodes. Furthermore, individuals with BED also demonstrate more ED-related psychopathology than non-BED obese individuals [61–63]. Psychiatric comorbidity is also higher among BED obese individuals as compared their non-BED obese counterparts, and the increased comorbidity is accounted for by the severity of binge eating as opposed to the severity of obesity [6,64–67]. In addition, research demonstrates that obese individuals with BED, as compared with non-obese BED patients, have a poorer quality of life [68].
BED and Bulimia Nervosa
Numerous studies have supported the distinction between bulimia nervosa and BED [69–76]. Diagnostically, bulimia nervosa differs from BED by its requirement of recurrent inappropriate compensatory behaviors in order to prevent weight gain, such as self-induced vomiting; misuse of laxatives, diuretics, or other medications; fasting; or excessive exercise [3]. BED and bulimia nervosa are distinguished by distinct risk factors, prevalence, course, and treatment outcomes [28,67,77]. Individuals with BED are less likely than individuals with bulimia to diet before onset of the disorder, and fewer individuals with BED cross over into other ED diagnostic categories [26,78–81]. Finally, BED and bulimia nervosa are associated with different constellations of ED-related symptoms and associated features [28,63,79]. For example, relative to BE patients, those with bulimia show greater work impairment and psychiatric comorbidity [28], higher dietary restraint and eating concerns [63], and lower rates of obesity [79].
Psychiatric Comorbidity
BED is associated with poor social adjustment, greater functional impairment, and significant psychiatric comorbidity, including overall distress and suicidality [67]. In a study of comorbidity with only selected disorders (mood, anxiety, impulse-control, and substance use disorder), 78.9% of individuals with BED had a lifetime history of at least one comorbidity, 20.2% had one comorbid disorder, 9.8% had two, and 48.9% had three or more [28]. Furthermore, the presence of current psychiatric comorbidity is associated with greater ED-related psychopathology and associated distress [40,41]. The most common comorbidities (lifetime rates) are specific phobia (37.1%), social phobia (31.9%), major depressive disorder (32.3%), post-traumatic stress disorder (PTSD) (26.3%), alcohol abuse/dependence (21.4%), conduct disorder (20%), attention-deficit/ hyperactivity disorder (19.8%), illicit drug use/dependence (19.4%), and oppositional-defiant disorder (18%) [28]. A recent report supports that this level of comorbidity is evident in primary care settings, noting that PTSD in particular is common and associated with a host of other difficulties, including depression, anxiety, drug use disorders, greater eating disorder pathology, and poorer psychological functioning [82]. Personality disorders are also commonly comorbid with BED, with the highest lifetime rates for avoidant (11%), obsessive compulsive (10%), and borderline (9%) personality disorders [83]. Finally, cigarette smoking is also associated with binge eating [83,84], likely evolving out of a weight-control smoking profile [85], and this is of relevance to the primary care setting in that smokers with BED gain more weight upon smoking cessation than do their non-BED counterparts [86].
Further Evaluation
To assess behavioral factors related to obesity and recent weight gain, the physician asks the patient if she ever eats what would be considered an unusually large amount of food for the circumstance. The patient acknowledges that she does so regularly, particularly in response to negative moods. The patient also describes that these episodes contribute to ongoing low mood, such that she feels highly depressed and hopeless following binge episodes. The physician then asks about the patient’s exercise habits and weight management techniques. While the patient denies engaging in compensatory behaviors (eg, vomiting, laxative use) to counteract excessive eating, she does report a history of dieting in which she dramatically restricts her food intake and subsequently loses weight. The patient states that these periods are inevitably followed by a resumption of overeating, and she typically gains back more weight than she originally lost. The patient estimates that she has lost and regained more than 20 lb at least 5 times during her lifetime. In addition, the patient reports difficulty maintaining a regular exercise regimen, especially since the onset of osteoarthritis-related joint pain in the past year. After the evaluation, the physician orders an electrocardiogram (ECG) and blood work. The ECG shows that the P-wave, QRS, and T-wave axes are shifted leftward, but within normal limits. A follow-up appointment is scheduled in 2 weeks.
• What are the medical complications of BED?
BED is associated with numerous negative health sequelae including obesity, sleeping problems, musculoskeletal pain, joint pain, headaches, gastrointestinal problems, menstrual problems, shortness of breath, chest pain, diabetes, low health-related quality of life, and functional health impairments [87–90], with many of these risks persisting even after controlling for BMI [91]. A 5-year follow-up of 134 individuals with BED and 134 individuals with no history of eating disorders, who were frequency-matched for age, sex, and baseline body mass index (BMI), provides further support that BED confers risk of components of metabolic syndrome beyond the risks associated with BMI alone [92]. Specifically, BED cases had higher longitudinal risk of developing dyslipidemia, hypertension, type 2 diabetes, any metabolic syndrome component, and two or more metabolic syndrome components. Alarmingly, these findings even emerge in studies of pediatric samples, wherein BED predicts development of metabolic syndrome, elevated triglycerides, and increases in visceral adiposity [93].
• What are risk factors for BED?
A number of risk factors for BED have been identified, although many are risk factors for a number of psychiatric disorders and not specific to BED. These general risk factors include depression/negative affectivity [94,95], parental mood and substance use disorder, maternal problematic parenting, and separation from parents [95]. A host of risk factors have been identified for disordered eating, in general, including body dissatisfaction [94], early onset of dieting [94], and perfectionism [96]. A number of other variables are risk factors for both BED and bulimia (but not anorexia), including a history of childhood bully and teasing, negative self-evaluation, parental depression, and negative family communication about shape and weight [81,96]. In a study comparing BED cases to psychiatric controls, childhood obesity, familial eating problems, family discord, and high parental demands differentiated the BED cases [95]. In summary, it has been suggested that BED risk is conferred by factors that increase risk of psychiatric disorder in general and those that confer risk for obesity [81]. Of note, the risk factors studied do not appear to differ between black and white women [95].
Genetic risk factors appear to play a strong role in the development of BED. Risk for BED tends to aggregate in families independently of the risk for obesity, although the presence of BED in a first-degree relative does increase risk for obesity [97]. Heritability estimates for BED range from 45% to 57% [98,99], which is greater than the heritability estimate for subthreshold binge eating (ie, overeating with a sense of loss of control, 41%) [100]. In addition, symptom-level analyses support moderate genetic contributions for each BED symptom [98], supporting the integrity of the diagnostic criteria. Finally, shared environment appears to play a very small role in the familial transmission of BED, and the contribution of unique environmental factors in development of BED appears to be substantial [97,101].
With regard to the neurobiological underpinnings of BED, it appears that BED may be associated with hypersensitivity to reward, a phenomenon that is strongly associated with the striatum and dopaminergic mechanisms [102,103]. In support of this hypothesis, Davis et al [102] reported that BED was differentially related to genotypes that reflect a greater density of D2 receptors and higher D2 binding potential as compared to obese controls. Additionally, greater increases in striatal DA and unique activation patterns in the right ventral striatum have been demonstrated in individuals with BED as compared to obese non-BED controls in response to food-related stimuli [103,104]. Other findings have implicated the orbitofrontal cortex (OFC) in BED, which is another brain region responsible for reward processing, particularly as it relates to the hedonic value of food stimuli [103]. Increased volume of grey matter has been documented in individuals with BED and bulimia as compared to normal weight controls, and stronger medial OFC activation while viewing pictures of food was observed in individuals with BED as compared to individuals with bulimia, overweight controls, and normal controls [105].
Difficulties with affect regulation have also been implicated in the development of BED. Two theories that implicate a primary and specific role for affect regulation in BED are cited most frequently in the extant literature: the affect regulation theory and the escape theory. The affect regulation theory [106] posits that BE is a conditioned response to negative affect which is correspondingly negatively reinforced by reductions in negative affect, which could occur during or after BE. Escape theory [107] posits that aversive self-awareness causes negative affect, which in turn triggers BE. BE is then negatively reinforced by reductions in negative affect during a binge via an escape from self-awareness that is accomplished through cognitive narrowing to the immediate stimulus environment. In contrast to the affect regulation theory, escape theory predicts that negative affect will increase after BE when self-awareness is restored. Results regarding changes in affect during BE episodes are conflicting as to whether BE is associated with decreases, no change [108–110], or even increases in negative affect. In particular, a meta-analytic review of 36 studies that examined affect via ecological momentary sampling found moderate increases in negative affect following binge episodes [111]. To some degree, results of this meta-analysis may not generalize to BED, per se, given that it included other binge eating groups, such as those with bulimia nervosa. However, in general, studies suggest that negative affect is an antecedent for BE and increased negative affect may be a consequence of BE, at least among women. More information is needed regarding aversive self-awareness before and after BE, cognitive narrowing, and changes in affect during BE. As such, the current state of the literature provides only partial support for affect regulation models of BED in women. Furthermore, it remains unknown if these results will generalize to men.
Clinical Course
Evidence regarding the course and stability of BED is conflicting and unclear. Several prospective studies have suggested that BED is not a stable disorder, exhibiting high rates of remission over time [26,99,112]. However, the samples have been criticized for being small, completely female, younger than typical individuals with BED, and post-ED treatment. In contrast, a prospective study that included older women and a combination of treated and untreated women suggested remission rates at 1 year that were much lower (7%) [78]. Additionally, a retrospective study [113] reported an average BED duration of 14.4 years. In a review of the studies cited above, Wonderlich et al [6] concluded that “[a]lthough there is variability in the data, it does appear that BED differs from other eating disorders in terms of a greater tendency toward recovery and fluctuation, although this may be embedded in a chronic pattern of remission and relapse.” Viewing BED as a disorder with a chronic pattern of remission and relapse could explain why individuals with BED retrospectively report a longer duration of illness, as they may be more likely to conceptualize their illness as one continuous course punctuated by different periods of severity rather than several distinct bouts of BED. Finally, although diagnostic crossover is a frequent phenomenon among other eating disorders, the crossover rate for BED appears relatively low as compared to anorexia and bulimia [6,26,28,66].
Follow-up
Laboratory examination shows TSH levels within normal limits and cholesterol levels of 48 mg/dL(HDL), 162 mg/dL (LDL), and 270 mg/dL (total). Triglyceride levels are 300 mg/dL and the patient’s fasting glucose level is 115 mg/dL. At the patient’s follow-up appointment, the physician states that a number of laboratory results indicated negative weight-related health consequences, including high cholesterol, high triglycerides, hypertension, and probable pre-diabetes. The patient initially disregards the significance of these results, stating she only gained weight due to her break-up and quitting smoking, and she is motivated to diet to lose weight in the near future. The physician asks for more information about the patient’s eating behavior, in particular asking if she ever feels as if she loses control over her eating. The patient reluctantly admits to this, and the physician provides a referral to a behavioral health specialist. The patient expresses ambivalence and a desire to try to manage her weight on her own. The physician uses motivational interviewing techniques to enhance motivation to follow up on this referral. In addition, the patient is encouraged to make small changes to her diet and slowly increase her exercise by taking walks. Another follow-up appointment is scheduled in 3 months.
• Which treatments are most effective for BED?
Despite the negative sequalae of BED, studies suggest that it often goes untreated [114]. Women with BED, as compared to women with anorexia and bulimia, are less likely to seek treatment for BED and less likely to receive treatment for their eating disorder when they do seek it out [114–116]. Barriers to treatment may include shame and internalized weight stigma, lack of knowledge about where to seek treatment, a belief that willpower should be sufficient to overcome the problem, lack of understanding that BED is a psychiatric disorder, finances/insurance barriers, and lack of BED detection by non-specialist treatment providers [115]. These barriers are particularly concerning, as women with BED report greater health care utilization and comprise a large segment of patients in weight control programs. Therefore, it appears individuals with BED seek help for the negative consequences of the disorder, but they are less likely to seek and receive help for the likely root cause of their concerns. This is a particularly damaging pattern, as the presence of BED may negatively impact the outcome of obesity treatment [117]. There are, however, a number of promising treatments for BED, as described below:
Cognitive Behavioral Therapy
Cognitive behavioral therapy (CBT) is generally considered to be the most well-established and empirically supported treatment for BED [118,119]. The cognitive behavioral conceptualization of BED is based on Fairburn, Cooper, and Shafran’s [120] transdiagnostic model of eating disorders (CBT-E), which is an expanded version of the cognitive behavioral model of bulimia nervosa [121]. CBT-E posits that the core pathology in eating disorders is a dysfunctional system in which self-worth is based on eating habits, shape, or weight, and the individual’s ability to control them. Attempts to maintain self-worth by controlling eating, shape, and weight result in extreme and brittle forms of dietary restraint. Inevitable violations of the individual’s dietary rules are then interpreted as lack of self-control, leading to a temporary abandonment of dietary restraint and consequent BE. These dietary slips and corresponding BE often occur in response to acute changes in mood, and BE is thus negatively reinforced by “neutralizing” negative mood states. Lapses in dietary restraint also result in secondary negative self-evaluation, which serves to further exacerbate a cycle of increased dietary restraint to improve self-worth and then inevitable dietary lapses leading to BE. CBT-E expanded upon CBT-BN by postulating 4 processes that maintain ED: severe perfectionism (clinical perfectionism), unconditional and pervasive low self-esteem (core low self-esteem), difficulties coping with intense mood states (mood intolerance), and developmental interpersonal difficulties (interpersonal difficulties). Of note, the CBT-E model explicitly states that individuals may differ in the extent to which they experience the 4 maintaining processes and not every individual will experience all four.
Overall, treatment is focused on normalizing eating patterns (ie, not weight loss), cognitive restructuring for weight/shape concerns and other triggers for binge eating, and relapse prevention [122]. CBT has produced substantial reductions in binge eating as compared to no treatment [123–125] and supportive therapy [126]. The majority of RCTs have reported remission rates greater than 50% [127]. Unfortunately, CBT has generally not produced meaningful weight loss [118,122,127–129], but this may be a contraindicated goal. CBT has demonstrated improvements in a number of features associated with BED including eating disordered psychopathology [122,124,130,131], depression [122,124,130,132], social adjustment [133], and self-esteem [132]. Treatment gains are generally well-maintained at 1-year to 4-year follow-up [122,123,130,133,134]. Individual and group treatments appear to produce similar results [134], and treatment completion rates have been estimated at approximately 80% across different delivery formats [127]. One strength of the CBT literature is the inclusion of participants with severe psychopathology, which facilitates the generalizability of these findings [127].
A number of factors have been associated with treatment outcome in CBT trials. Poor treatment outcomes have been associated with a history of weight problems during childhood, high levels of emotional eating at baseline, interpersonal dysfunction, and low group cohesion during group CBT [110,124,134]. Overvaluation of weight and shape demonstrated a statistical trend toward negatively impacting outcomes in one study. The presence of a cluster B personality disorder (ie, borderline histrionic, antisocial, and narcissistic personality disorders) predicted higher levels of binge eating at 1-year follow-up in a combined sample of participants treated with group CBT or group interpersonal psychotherapy (IPT) [135].
Alternatively, positive treatment outcomes have been associated with low levels of emotional eating at baseline, older age of onset, weight loss history that is negative for amphetamine use, and decreases in depressive symptoms during treatment [124,134,136,137]. In addition, early response to treatment (defined as a 65%–70% reduction in binge eating within 4 weeks of starting treatment) tends to be associated with greater long-term (ie, 1–2 year) remission from BED and lower eating disorder psychopathology, across a variety of psychological treatment approaches [138–144].
Interpersonal Psychotherapy
IPT for BED was adapted by Wilfley and colleagues [145] from IPT for depression, and the rationale for its use with BED is based on successful outcomes for individuals with bulimia and multiple studies documenting interpersonal deficits in individuals with BED [146]. IPT seeks to address interpersonal problems in 4 areas: interpersonal conflict, grief, role transitions, and interpersonal deficits [135]. While adapting IPT for BED, it was noted that the course of BED tends to be more chronic than the course of depression, thus the focus of IPT for BED was shifted from addressing the interpersonal precipitants of the disorder to the interpersonal factors that maintain the disorder [145]. Fewer studies examining the effectiveness of IPT in treating BED have been published than those examining CBT for BED, but it appears that IPT is as efficacious as CBT immediately post-treatment [130], and at 1- [130] and 4-year follow-up [147]. In addition, at least 2 studies have been published that compare IPT, cognitive behavioral therapy–guided self-help (CBTgsh), and behavioral weight loss [133,141]. Overall, results support the use of both IPT and CBTgsh (discussed in more detail below), with important moderators of treatment effects observed. For example, Wilson et al [133] found that clients with higher levels of psychopathology were better suited for IPT. The authors conclude that these results could inform a model of evidence-based stepped care, where CBTgsh, a low-cost, low-intensity treatment, should be considered as the first line of treatment. Secondarily, IPT, which represents a more specialized and expensive form of treatment, could be considered the next level of care, particularly for clients who are not demonstrating rapid improvement in response to CBTgsh.
Dialectic Behavior Therapy
A small number of studies have investigated the treatment of BED with dialectical behavior therapy (DBT). Originally developed to treat borderline personality disorder [148], DBT is of particular interest given its explicit targeting of emotion regulation. According to the DBT model of BED [149], emotional dysregulation is the core psychopathology in this disorder, and binge eating is viewed as attempts to influence, change, or control painful emotions. Initially, promising results were published showing positive treatment effects in an uncontrolled study [150] as well as wait-list controlled trials [151]. Notably, relative to wait-list controls, participants in a DBT guided self-help program (who received an orientation, DBT manual, and six 20-minute support calls across 13 weeks) reported reduced past-month binge eating, higher binge eating abstinence rates, and over the longer term improved quality of life and reductions in ED psychopathology. However, a comparison of DBT-BED with an active comparison control group (ie, nonspecific supportive therapy) failed to find significant differences between the 2 treatments (defined as effect size greater than 0.5) at 12-month follow-up in binge eating abstinence, binge eating frequency, most ED-related psychopathology, positive affect, depression, and self-esteem [152]. Therefore, DBT may have potential and, at a minimum, is equally efficacious as supportive therapy.
Mindfulness- and Meditation-Based Therapies
Treatment outcome studies utilizing mindfulness-based therapies, including mindfulness-based stress reduction (MBSR) and acceptance and commitment therapy (ACT), make up a small but promising body of literature. Reasoning that negative affect, eating in the absence of hunger, and emotional eating may comprise one pathway to binge eating [153,154], it follows that mindfulness-based therapies may act through their effects on emotion regulation, acceptance strategies for tolerating negative affect, and awareness of bodily cues. A recent review identified 19 studies exploring the effects of mindfulness-based interventions on binge eating severity and frequency as well as a number of related indicators, observing positive effects for this form of treatment [155]. For example, MB-EAT [156] is a group treatment for BED that is primarily based on MBSR. Treatment is targeted at cultivating mindfulness, mindful eating, emotional balance, and self-acceptance[157]. The treatment also places particular emphasis on developing self-awareness of internal hunger and satiety cues. A recent randomized controlled trial of MB-EAT produced significant improvements in binge eating frequency and BE-related psychopathology [158]. Furthermore, process variables including hunger awareness, satiety awareness, and mindfulness were correlated with positive outcomes. In addition, a small study (n = 39) that compared ACT to standard follow-up utilized by a bariatric surgery team demonstrated significantly greater improvements in disordered eating, body satisfaction, and quality of life for clients who participated in ACT [159]. In brief, results suggest that mindfulness-based interventions represent an additional treatment approach with supporting but limited evidence to date.
Self-Help Interventions
Self-help interventions for BED are categorized as pure self-help or guided self-help. In treatment outcome studies, pure self-help is generally conducted with a self-help manual, although several studies have examined more novel formats such as the internet, video, and CD-ROM. GSH also uses a self-help manual (or other format) with the addition of brief sessions with health care providers who have varying degrees of expertise with the type of therapy being utilized. CBT is the most commonly utilized therapeutic modality in treatment outcome studies of self-help interventions, and they most often utilize Fairburn’s Overcoming Binge Eating self-help manual [160].
Two studies have directly compared pure and guided self-help with Fairburn’s manual and produced conflicting results. Carter and Fairburn [161] found that in a sample of primarily white women with BED, pure self-help (CBTsh; n = 24) and guided self-help (CBTgsh; n = 24) were equally effective, and both were superior to wait-list controls at 6-month follow-up in producing BE abstinence (CBTsh = 40%, CBTgsh = 50%), reducing binge eating, ED-related psychopathology, and general psychiatric symptoms. In contrast, a study comparing CBTsh and CBTgsh in 40 primarily white women with recurrent binge eating (82.5% diagnosed with BED), guided self-help was superior to pure self-help at the end of treatment in reducing BE frequency, eating concern, and restraint [162]. CBTgsh and CBTsh were equally effective in producing BE abstinence (50% and 30%, respectively), and reducing shape concern, weight concern, and general psychiatric symptoms [162]. Higher levels of general psychiatric symptoms were predictive of higher BE frequency post-treatment for both treatments. It should be noted that participants in both conditions experienced statistically significant improvements on all variables as compared to baseline.
CBTgsh also performed as well or better than individualized treatments in one study [133]. CBTgsh, IPT, and behavioral weight loss (BWL) were compared in a large study of 205 primarily white, obese or overweight individuals diagnosed with BED. The 3 treatments produced equivalent outcomes for binge eating at post-treatment, but BWL produced significantly greater weight loss. However, at 2-year follow-up, the CBTgsh and IPT groups had maintained treatment gains and were significantly superior to BWL in reductions in binge eating. The 3 groups were equivalent with regard to weight loss at the 2-year follow-up, and none reported clinically significant weight loss. Of note, as compared to the IPT and BWL groups, the CBTgsh group received 10 sessions as opposed to 20, received 25-minute sessions as opposed to 60-minute sessions, and were treated by providers with limited levels of experience as opposed to doctoral-level clinical psychologists.
To summarize, CBT is the most often studied type of self-help treatment. Both CBTsh and CBTgsh produced improvements in binge eating and associated psychopathology as compared to baseline and wait-list controls, and treatment gains were maintained at 6-month follow-up. Conclusions regarding the relative superiority of pure self-help or guided self-help are premature given the small number of studies and conflicting results.
In addition, limited information is available regarding moderators and predictors of guided self-help outcomes. Masheb and Grilo [163] performed a cluster analysis of the sample for the CBTgsh vs. BWLgsh described above [164] and identified 2 clinically significant subtypes: a dietary-negative affect subtype characterized by high restraint, low self-esteem, and depressive symptoms; and an overvaluation of weight and shape subtype. For both the CBTgsh and BWLgsh groups, the dietary-negative affect subtype experienced higher levels of binge eating frequency, and the overvaluation of weight and shape subtype experienced higher levels of ED-related psychopathology. Additionally, individuals receiving BWLgsh who experienced a rapid response to treatment reported lower BE frequency, greater weight loss, and higher restraint than participants without a rapid response [142]. In contrast, rapid response did not appear to affect outcomes for CBTgsh participants. Finally, the combination of low self-esteem and high ED-related psychopathology negatively affected BE remission rates for CBTgsh recipients [133].
Pharmacologic Treatment
Currently only one medication, lisdexamfetamine dimesylate, has been FDA-approved for the treatment of BED. Previously approved for treating both adults and children with attention-deficit hyperactivity disorder, lisdexamfetamine dimesylate is a central nervous system stimulant and has been found to significantly reduce number of binge days, with robust effect sizes [165]. Beyond this medication, the evidence for pharmacologic treatment of BED is limited. A recent review identified only 22 studies exploring the effects of pharmacologic treatment in a methodologically rigorous way (eg, double-blind placebo design) [4]. To date, a number of different medication classes have been evaluated, including antidepressants, anticonvulsants, stimulants, anti-obesity drugs, and others. Overall, there is some evidence that antidepressant and anticonvulsant agents are efficacious at reducing BE frequency [166,167] and sometimes effective regarding statistically significant weight loss [168,169]. However, the majority of results are generally disappointing, both with respect to reductions in binge eating and sustained weight loss [48,170,171]. In addition, there are serious limitations in the literature that must be considered, including the limited number of studies that address the high placebo response observed in clinical samples, limited follow-up windows, and inadequate multiplicitious confirmatory trials. Despite these limitations, the evidence base related to pharmacologic treatment is continuously evolving and represents an important future direction for the treatment of BED.
Treatment
Prior to her next medical follow-up, the patient meets with a psychologist. The patient discloses that she has been binge eating several times per week for over a year; she also discloses a history of prolonged sexual abuse perpetrated by a step-parent during her childhood. When the patient returns to her follow-up medical appointment, she reports that her psychologist has diagnosed her with BED and PTSD. She states that they are using cognitive behavioral techniques to regulate her mood and eating behavior, with a specific aim of avoiding excessive dietary restraint. In addition, they are working together to discuss her unfulfilling romantic history and processing her experiences of trauma. Since her last appointment with the primary care physician, she reports an increased awareness of her eating habits, improvement in mood, and a 10-lb decrease in her weight.
The patient reports that she has continued to meet weekly with her psychologist and has slowly begun reintroducing low-impact exercise to her routine. She continues to lose weight gradually, but with a priority of stabilizing eating behavior and avoiding binge episodes versus aiming for weight loss. She reports that her mood has stabilized. Her cholesterol and triglycerides remain high, but her blood pressure is controlled effectively with medication. Her physician recommends continued psychological treatment, periodic meetings with a nutritionist, and prescribes medication for her cholesterol. A follow-up appointment with her physician is scheduled in 6 months.
Summary

Corresponding author: Karen K. Saules, PhD, Eastern Michigan University, Psychology Clinic, 611 W. Cross St., Ypsilanti, MI 48197, [email protected]
Financial disclosures: None.
1. Stunkard AJ. Eating patterns and obesity. Psychiatr Q 1959;33:284–95.
2. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Text revision. Washington (DC): The Association; 2000.
3. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: The Association; 2013.
4. Reas D, Grilo C. Pharmacological treatment of binge eating disorder: update review and synthesis. Hum Psychopharmacol Clin Exp 2015;23:1463–78.
5. Wilson G. Treatment of binge eating disorder. Psychiatr Clin North Am 2011;34:773–83.
6. Wonderlich SA, Gordon KH, Mitchell JE, et al. The validity and clinical utility of binge eating disorder. Int J Eat Disord 2009;42:687–705.
7. Walsh BT, Boudreau G. Laboratory studies of binge eating disorder. Int J Eat Disord 2003;34 Suppl:S30–8.
8. Guss JL, Kissileff HR, Devlin MJ, et al. Binge size increases with body mass index in women with binge-eating disorder. Obes Res 2002;10:1021–9.
9. Fairburn CG, Cooper Z. The eating disorder examination. In: Fairburn CG, Cooper Z, editors. Binge eating: nature, assessment, and treatment. New York: Guilford Press;1993:317–60.
10. Keel PK, Mayer SA, Harnden-Fischer JH. Importance of size in defining binge eating episodes in bulimia nervosa. Int J Eat Disord 2001;29:294–301.
11. Niego SH, Pratt EM, Agras WS. Subjective or objective binge: is the distinction valid? Int J Eat Disord 1997;22: 291–8.
12. Pratt E, Niego S, Agras W. Does the size of a binge matter? Int J Eat 1998;24:307–12.
13. Wolfe BE, Baker CW, Smith AT, et al. Validity and utility of the current definition of binge eating. Int J Eat Disord 2009;42:674–86.
14. Latner JD, Clyne C. The diagnostic validity of the criteria for binge eating disorder. Int J Eat Disord 2008;41:1–14.
15. Johnson WG, Boutelle KN, Torgrud L. What is a binge? The influence of amount, duration, and loss of control criterial on judgements of binge eating. Int J Eat Disord 2000;27:471–9.
16. Johnson WG, Roberson-NR, Rohan KJ, et al. An experimental investigation of DSM-IV binge-eating criteria. Eat Behav 2003;4:295–303.
17. Reslan S, Saules KK. College students’ definitions of an eating ‘binge’ differ as a function of gender and binge eating disorder status. Eat Behav 2011;12:225–7.
18. Telch CF, Pratt EM, Niego SH. Obese women with binge eating disorder define the term binge. Int J Eat Disord 1998;24:313–7.
19. Colles SL, Dixon JB, O’Brien PE. Loss of control is central to psychological disturbance associated with binge eating disorder. Obesity (Silver Spring) 2008;16:608–14.
20. Stein KF, Kenardy J, Wiseman C V, et al. What’s driving the binge in binge eating disorder? A prospective examination of precursors and consequences. Int J Eat Disord 2007;40:195–203.
21. Rossiter EM, Agras WS, Telch CF, et al. The eating patterns of non-purging bulimic subjects. Int J Eat Disord 1992;11:111–120.
22. Grilo CM, White MA. A controlled evaluation of the distress criterion for binge eating disorder. J Consult Clin Psychol 2011;79:509–14.
23. Trace SE, Thornton LM, Root TL, et al. Effects of reducing the frequency and duration criteria for binge eating on lifetime prevalence of bulimia nervosa and binge eating disorder: implications for DSM-5. Int J Eat Disord 2012;45: 531–6.
24. Wilson GT, Sysko R. Frequency of binge eating episodes in bulimia nervosa and binge eating disorder: diagnostic considerations. Int J Eat Disord 2009;42:603–10.
25. Sysko R, Roberto CA, Barnes RD, et al. Test-retest reliability of the proposed DSM-5 eating disorder diagnostic criteria. Psychiatry Res 2012;196:302–8.
26. Fairburn CG, Cooper Z, Doll HA, et al. The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiatry 2000;57:659–65.
27. Grilo CM, Ivezaj V, White MA. Evaluation of the DSM-5 severity indicator for binge eating disorder in a community sample. Behav Res Ther 2015;66:72–6.
28. Hudson JI, Hiripi E, Pope HG, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry 2007;61:348–58.
29. Stice E, Marti CN, Rohde P. Prevalence, incidence, impairment, and course of the proposed DSM-5 eating disorder diagnoses in an 8-year prospective community study of young women. J Abnorm Psychol 2012;122:445–57.
30. Mangweth-Matzek B, Hoek HW, Rupp CI, et al. Prevalence of eating disorders in middle-aged women. Int J Eat Disord 2014;47:320–4.
31. Ivezaj V, Saules KK, Hoodin F, et al. The relationship between binge eating and weight status on depression, anxiety, and body image among a diverse college sample: a focus on bi/multiracial women. Eat Behav 2010;11:18–24.
32. Striegel-Moore RH, Bedrosian R, Wang C, et al. Why men should be included in research on binge eating: results from a comparison of psychosocial impairment in men and women. Int J Eat Disord 2012;45:233–40.
33. Womble LG, Williamson DA, Martin CK, et al. Psychosocial variables associated with binge eating in obese males and females. Int J Eat Disord 2001;30:217–21.
34. Udo T, McKee SA., White MA., et al. Sex differences in biopsychosocial correlates of binge eating disorder: a study of treatment-seeking obese adults in primary care setting. Gen Hosp Psychiatry 2013;35:587–91.
35. Udo T, McKee SA., White MA, et al. Menopause and metabolic syndrome in obese individuals with binge eating disorder. Eat Behav 2014;15:182–5.
36. Napolitano MA., Himes S. Race, weight, and correlates of binge eating in female college students. Eat Behav 2011;12:29–36.
37. Sorbara M, Geliebter A. Body image disturbance in obese outpatients before and after weight loss in relation to race, gender, binge eating, and age of onset of obesity. Int J Eat Disord 2002;31:416–23.
38. Alegria M, Woo M, Cao Z, et al. Prevalence and correlates of eating disorders in Latinos in the U.S. Int J Eat Disord 2007;40:S15–21.
39. Striegel-Moore RH, Wilfley DE, Pike KM, et al. Recurrent binge eating in black American women. Arch Fam Med 2000;9: 83–7.
40. Reslan S, Saules KK. Assessing the prevalence of and factors associated with overweight, obesity, and binge eating as a function of ethnicity. Eat Weight Disord-St 2013;18:209–19.
41. Grilo CM, White MA, Barnes RD, et al. Psychiatric disorder co-morbidity and correlates in an ethnically diverse sample of obese patients with binge eating disorder in primary care settings. Compr Psychiatry 2012;54:209–16.
42. Smolak L, Striegel Moore RH. Challenging the myth of the golden girl: ethnicity and eating disorders. In: Smolak L, Striegel Moore RH, editors. Eating disorders innovative directions in research and practice. Washington (DC): American Psychological Association;2001:111–32.
43. Wildes JE, Emery RE. The role of ethnicity and culture in the development of eating disturbance and body dissatisfaction: a meta-analutic review. Clin Phychology Rev 2001;21:521–50.
44. George JBE, Franko DL. Cultural issues in eating pathology and body image among children and adolescents. J Pediatr Psychol 2010;35:231–42.
45. Kelly NR, Cotter EW, Tanofsky-Kraff M, et al. Racial variations in binge eating, body image concerns, and compulsive exercise among men. Psychol Men Masc 2014;13:326-36.
46. Franko DL, Thompson-Brenner H, Thompson DR, et al. Racial/ethnic differences in adults in randomized clinical trials of binge eating disorder. J Consult Clin Psychol 2012;80:186–95.
47. Marques L, Alegria M, Becker AE, et al. Comparative prevalence, correlates of impairment, and service utilization for eating disorders across U.S. ethnic groups: Implications for reducing ethnic disparities in health care access for eating disorders. Int J Eat Disord 2012;44:412–20.
48. Guerdjikova AI, O’Melia AM, Mori N, et al. Binge eating disorder in elderly individuals. Int J Eat Disord 2012;45: 905–8.
49. Fairburn CG. Beglin SJ. Assessment of eating disorders: interview or self-report questionnaire? Int J Eat Disord 1994;16:363–70.
50. Spitzer R, Yanovski S, Marcus M. The questionnaire on eating and weight patterns-revised (QEWP-R). New York State Psychiatric Institute, Mew York 1993.
51. Barnes RD, Masheb RM, White MA, et al. Comparison of methods for identifying and assessing obese patients with binge eating disorder in primary care settings. Int J Eat Disord 2011;44:157–63.
52. Yanovski SZ, Marcus MD, Wadden TA, et al. The questionnaire on eating and weight patterns-5: an updated screening instrument for binge eating disorder. Int J Eat Disord 2015;48:259–61.
53. Gormally J, Black S, Daston S, et al. The assessment of binge eating severity among obese persons. Addict Behav 1982;7:47–55.
54. Alfonsson S. Replacing the term ‘binge eating’ with ‘loss of control over eating’ affects eating disorder screening in clinical care. Obes Res Clin Pract 2015;6–7.
55. Kelly NR, Mitchell KS, Gow RW, et al. An evaluation of the reliability and construct validity of eating disorder measures in white and black women. Psychol Assess 2012;24:608–17.
56. Richards LK, McHugh RK, Pratt EM, et al. Readability and comprehension of self-report binge eating measures. Eat Behav 2013;14:167–70.
57. Masheb RM, White MA, Grilo CM. Substantial weight gains are common prior to treatment-seeking in obese patients with binge eating disorder. Compr Psychiatry 2013;54:880–4.
58. Heaner MK, Walsh BT. A history of the identification of the characteristic eating disturbances of Bulimia Nervosa, Binge Eating Disorder and Anorexia Nervosa. Appetite 2013;65:185–8.
59. Greeno CG, Wing RR, Shiffman S. Binge antecedents in obese women with and without binge eating disorder. J Consult Clin Psychol 2000;68:95–102.
60. Le Grange D, Gorin A, Catley D, et al. Does momentary assessment detect binge eating in overweight women that is denied at interview? Eur Eat Disord Rev 2001;9:309–24.
61. Brody ML, Walsh BT, Devlin MJ. Binge eating disorder: reliability and validity of a new diagnostic category. J Consult Clin Psychol 1994;62:381–6.
62. Hsu LKG, Mulliken B, McDonagh B, et al. Binge eating disorder in extreme obesity. Int J Obes Relat Metab Disord 2002;26:1398–403.
63. Wilfley DE, Schwartz MB, Spurrell EB, et al. Using the eating disorder examination to identify the specific psychopathology of binge eating disorder. Int J Eat Disord 2000;27:259–69.
64. Grucza RA, Przybeck TR, Cloninger CR. Prevalence and correlates of binge eating disorder in a community sample. Compr Psychiatry 2007;48:124–31..
65. Telch CF, Stice E. Psychiatric comorbidity in women with binge eating disorder: prevalence rates from a non-treatment-seeking sample. J Consult Clin Psychol 1998;66:768–76.
66. Wilfley DE, Bishop ME, Wilson GT, et al. Classification of eating disorders: toward DSM-V. Int J Eat Disord 2007; 40:s123–9.
67. Wilfley DE, Wilson GT, Agras WS. The clinical significance of binge eating disorder. Int J Eat Disord 2003;34 Suppl:S96–106.
68. Perez M, Warren CS. The relationship between quality of life, binge-eating disorder, and obesity status in an ethnically diverse sample. Obesity (Silver Spring) 2012;20:879–85.
69. Bulik CM, Sullivan PF, Kendler KS. An empirical study of the classification of eating disorders. Am J Psychiatry 2000;157:886–95.
70. Keel PK, Heatherton TF, Dorer DJ, et al. Point prevalence of bulimia nervosa in 1982, 1992, and 2002. Psychol Med 2006;36:119–27.
71. Mitchell JE, Crosby RD, Wonderlich SA, et al. Latent profile analysis of a cohort of patients with eating disorders not otherwise specified. Int J Eat Disord 2007;40:s95–8.
72. Rockert W, Kaplan AS, Olmsted MP. Eating disorder not otherwise specified : the view from a tertiary care treatment center. Int J Eat Disord 2007;40:s99–103.
73. Striegel-Moore RH, Franko DL, Thompson D, et al. An empirical study of the typology of bulimia nervosa and its spectrum variants. Psychol Med 2005;35:1563–72.
74. Sullivan PF, Bulik CM, Kendler KS. The epidemiology and classification of bulimia nervosa. Psychol Med 1998;28:599–610.
75. Wade TD, Crosby RD, Martin NG. Use of latent profile analysis to identify eating disorder phenotypes in an adult Australian twin cohort. Arch Gen Psychiatry 2006;63:1377–84.
76. Striegel-Moore RH, Franko DL. Should binge eating disorder be included in the DSM-V? A critical review and state of the evidence. Annu Rev Clin Psychol 2008;4:305–24.
77. Striegel-Moore RH, Cachelin FM. Etiology of eating disorders in women. Couns Psychol 2001;29:635–61.
78. Crow SJ, Stewart Agras W, Halmi K, et al. Full syndromal versus subthreshold anorexia nervosa, bulimia nervosa, and binge eating disorder: a multicenter study. Int J Eat Disord 2002;32:309–18.
79. Striegel-Moore RH, Cachelin FM, Dohm F a, et al. Comparison of binge eating disorder and bulimia nervosa in a community sample. Int J Eat Disord 2001;29:157–65.
80. Wilfley DE, Schwartz MB, Spurrell EB, et al. Assessing the specific psychopathology of binge eating disorder patients: Interview or self-report? Behav Res Ther 1997;35:1151–9.
81. Fairburn CG, Doll HA, Welch SL, et al. Risk factors for binge eating disorder. Arch Gen Psychiatry 1998;55:425–32.
82. Grilo CM, White M a., Barnes RD, et al. Posttraumatic stress disorder in women with binge eating disorder in primary care. J Psychiatr Pract 2012;18:408–12.
83. Cassin SE, von Ranson KM. Is binge eating experienced as an addiction? Appetite 2007;49:687–90.
84. Kelly-Weeder S, Jennings KM, Wolfe BE. Gender differences in binge eating and behavioral correlates among college students. Eat Weight Disord 2012;17:e200–2.
85. Pomerleau CS, Ehrlich E, Tate JC, et al. The female weight-control smoker: a profile. J Subst Abuse 1993;5:391–400.
86. White MA, Masheb RM, Grilo CM. Function of binge eating behavior. Int J Eat Disord 2010;43:572–75.
87. Bulik CM, Reichborn-Kjennerud T. Medical morbidity in binge eating disorder. Int J Eat Disord 2003;34:S39–46.
88. Johnson JG, Spitzer RL, Williams JBW. Health problems, impairment and illnesses associated with bulimia nervosa and binge eating disorder among primary care and obstetric gynaecology patients. Psychol Med 2001;31.
89. Mond JM, Hay PJ, Rodgers B, et al. Assessing quality of life in eating disorder patients. Qual Life Res 2005;14:171–8.
90. Marchesini G, Natale S, Chierici S, et al. Effects of cognitive-behavioural therapy on health-related quality of life in obese subjects with and without binge eating disorder. Int J Obes Relat Metab Disord 2002;26:1261–7.
91. Reichborn-Kjennerud T, Bulik CM, Sullivan PF, et al. Psychiatric and medical symptoms in binge eating in the absence of compensatory behaviors. Obes Res 2004;12:1445–54.
92. Hudson JI, Lalonde JK, Coit CE, et al. Longitudinal study of the diagnosis of components of the metabolic syndrome in individuals with binge-eating disorder. Am J Clin Nutr 2010;91:1568–73.
93. Tanofsky-Kraff M, Shomaker LB, Stern EA, et al. Children’s binge eating and development of metabolic syndrome. Int J Obes 2012;36:956–62.
94. Stice E, Marti CN, Durant S. Risk factors for onset of eating disorders: evidence of multiple risk pathways from an 8-year prospective study. Behav Res Ther 2011;49:622–7.
95. Striegel-Moore RH, Fairburn CG, Wilfley DE, et al. Toward an understanding of risk factors for binge-eating disorder in black and white women: a community-based case-control study. Psychol Med 2005;35:907–17.
96. Hilbert A, Pike KM, Goldschmidt AB, et al. Risk factors across the eating disorders. Psychiatry Res 2014;220:500–6.
97. Hudson JI, Lalonde JK, Berry JM, et al. Binge-eating disorder as a distinct familial phenotype in obese individuals. Arch Gen Psychiatry 2006;63:313–19.
98. Mitchell KS, Neale MC, Bulik CM, et al. Binge eating disorder: a symptom-level investigation of genetic and environmental influences on liability. Psychol Med 2010;40:1899–906.
99. Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry 2008;69:266–73.
100. Reichborn-Kjennerud T, Bulik CM, Tambs K, et al. Genetic and environmental influences on binge eating in the absence of compensatory behaviors: a population-based twin study. Int J Eat Disord 2004;36:307–14.
101. Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry 2008;69:266–73.
102. Davis C, Levitan RD, Yilmaz Z, et al. Binge eating disorder and the dopamine D2 receptor: genotypes and sub-phenotypes. Prog Neuropsychopharmacol Biol Psychiatry 2012;38:328–35.
103. Weygandt M, Schaefer A, Schienle A, et al. Diagnosing different binge-eating disorders based on reward-related brain activation patterns. Hum Brain Mapp 2012;33:2135–46.
104. Wang G-J, Geliebter A, Volkow ND, et al. Enhanced striatal dopamine release during food stimulation in binge eating disorder. Obesity (Silver Spring) 2011;19:1601–8.
105. Schäfer A, Vaitl D, Schienle A. Regional grey matter volume abnormalities in bulimia nervosa and binge-eating disorder. Neuroimage 2010;50:639–43.
106. Hawkins RC, Clement PF. Binge eating: measurement problems and a conceptual model. In: Fremouw WJ, Clement PF, editors. The binge purge syndrome: diagnosis, treatment, and research. New York: Springer;1984:229–51.
107. Heatherton TF, Baumeister RF. Binge eating as escape from self-awareness. Psychol Bull 1991;110:86–108.
108. Deaver CM, Miltenberger RG, Smyth J, et al. An evaluation of affect and binge eating. Behav Modif 2003;27: 578–99.
109. Stickney MI, Miltenberger RG, Wolff G. A descriptive analysis of factors contributing to binge eating. J Behav Ther Exp Psychiatry 1999;30:177–89.
110. Hilbert A, Saelens BE, Stein RI, et al. Pretreatment and process predictors of outcome in interpersonal and cognitive behavioral psychotherapy for binge eating disorder. J Consult Clin Psychol 2007;75:645–51.
111. Haedt-Matt AA, Keel PK. Revisiting the affect regulation model of binge eating: a meta-analysis of studies using ecological momentary assessment. Psychol Bull 2012;137:660–81.
112. Cachelin FM, Striegel-Moore RH, Elder K a, et al. Natural course of a community sample of women with binge eating disorder. Int J Eat Disord 1999;25:45–54.
113. Pope HG, Lalonde JK, Pindyck LJ, et al. Binge eating disorder: a stable syndrome. Am J Psychiatry 2006;163:2181–3.
114. Pike KM, Dohm F a, Striegel-Moore RH, et al. A comparison of black and white women with binge eating disorder. Am J Psychiatry 2001;158:1455–60.
115. Cachelin FM, Striegel-Moore RH. Help seeking and barriers to treatment in a community sample of Mexican American and European American women with eating disorders. Int J Eat Disord 2006;39:154–61.
116. Cachelin FM, Rebeck R, Veisel C, et al. Barriers to treatment for eating disorders among ethnically diverse women. Int J Eat Disord 2001;30:269–78.
117. Blaine B, Rodman J. Responses to weight loss treatment among obese individuals with and without BED: a matched-study meta-analysis. Eat Weight Disord 2007;12:54–60.
118. Iacovino JM, Gredysa DM, Altman M, et al. Psychological treatments for binge eating disorder. Curr Psychiatry Rep 2012;14:432–46.
119. Wilson GT, Shafran R. Eating disorders guidelines from NICE. Lancet 2005;365:79–81.
120. Fairburn CG, Cooper Z, Shafran R. Cognitive behaviour therapy for eating disorders: a ‘transdiagnostic’ theory and treatment. Behav Res Ther 2003;41:509–28.
121. Fairburn CG, Cooper Z, Cooper PJ. The clinical features and maintenance of bulimia nervosa. In: Brownell KD, Foreyt JP, editors. Handbook of eating disorders: physiology, psychology and treatment of obesity, anorexia and bulimia. New York: Guiliford;1986:389–404.
122. Grilo CM, Masheb RM, Wilson GT, et al. Cognitive-behavioral therapy, behavioral weight loss, and sequential treatment for obese patients with binge-eating disorder: a randomized controlled trial. J Consult Clin Psychol 2011;79:675–85.
123. Agras WS, Telch CF, Arnow B, et al. One-year follow-up of cognitive-behavioral therapy for obese individuals with binge eating disorder. J Consult Clin Psychol 1997;65:343–7.
124. Dingemans AE, Spinhoven P, van Furth EF. Predictors and mediators of treatment outcome in patients with binge eating disorder. Behav Res Ther 2007;45:2551–62.
125. Eldredge KL, Stewart Agras W, Arnow B, et al. The effects of extending cognitive-behavioral therapy for binge eating disorder among initial treatment nonresponders. Int J Eat Disord 1997;21:347–52.
126. Kenardy J, Mensch M, Bowen K, et al. Group therapy for binge eating in type 2 diabetes: a randomized trial. Diabet Med 2002;19:234–9.
127. Wilson GT, Grilo CM, Vitousek KM. Psychological treatment of eating disorders. Am Psychol 2007;62:199–216.
128. Munsch S, Meyer AH, Biedert E. Efficacy and predictors of long-term treatment success for cognitive-behavioral treatment and behavioral weight-loss-treatment in overweight individuals with binge eating disorder. Behav Res Ther 2012;50:775–8.
129. Brown TA, Keel PK. Current and emerging directions in the treatment of eating disorders. Subst Abuse 2012;6: 33–61.
130. Wilfley DE, Welch RR, Stein RI, et al. A randomized comparison of group cognitive-behavioral therapy and group interpersonal psychotherapy for the treatment of overweight individuals with binge-eating disorder. Arch Gen Psychiatry 2002;59:713–21.
131. Grilo CM, Masheb RM, Wilson GT. Efficacy of cognitive behavioral therapy and fluoxetine for the treatment of binge eating disorder: a randomized double-blind placebo-controlled comparison. Biol Psychiatry 2005;57:301–9.
132. Gorin AA, Le Grange D, Stone AA. Effectiveness of spouse involvement in cognitive behavioral therapy for binge eating disorder. Int J Eat Disord 2003; 33: 421–33.
133. Wilson GT, Wilfley DE, Agras WS, et al. Psychological treatments for binge eating disorder. Arch Gen Psychiatry 2010;67:94–101.
134. Ricca V, Castellini G, Mannucci E, et al. Comparison of individual and group cognitive behavioral therapy for binge eating disorder. a randomized, three-year follow-up study. Appetite 2010;55:656–65.
135. Wilfley DE, Friedman MA, Dounchis JZ, et al. Comorbid psychopathology in binge eating disorder: relation to eating disorder severity at baseline and following treatment. J Consult Clin Psychol 2000;68:641–9.
136. Castellini G, Mannucci E, Lo Sauro C, et al. Different moderators of cognitive-behavioral therapy on subjective and objective binge eating in bulimia nervosa and binge eating disorder: a three-year follow-up study. Psychother Psychosom 2012;81:11–20.
137. Grilo CM, Masheb RM, Crosby RD. Predictors and moderators of response to cognitive behavioral therapy and medication for the treatment of binge eating disorder. J Consult Clin Psychol 2012;80:897–906.
138. Grilo CM, Masheb RM, Wilson GT. Rapid response to treatment for binge eating disorder. J Consult Clin Psychol 2006;74:602–13.
139. Grilo CM, Masheb RM. Rapid response predicts binge eating and weight loss in binge eating disorder: findings from a controlled trial of orlistat with guided self-help cognitive behavioral therapy. Behav Res Ther 2007;45:2537–50.
140. Grilo CM, White MA, Wilson GT, et al. Rapid response predicts 12-month post-treatment outcomes in binge-eating disorder: theoretical and clinical implications. Psychol Med 2012;42:807–17.
141. Hilbert A, Hildebrandt T, Agras WS, et al. Rapid response in psychological treatments for binge eating disorder. 2015;83:649–654.
142. Masheb RM, Grilo CM. Rapid response predicts treatment outcomes in binge eating disorder: implications for stepped care. J Consult Clin Psychol 2007;75:639–44.
143. Safer DL, Joyce EE. Does rapid response to two group psychotherapies for binge eating disorder predict abstinence? Behav Res Ther 2011;49:339–45.
144. Zunker C, Peterson CB, Cao L, et al. A receiver operator characteristics analysis of treatment outcome in binge eating disorder to identify patterns of rapid response. Behav Res Ther 2010;48:1227–31.
145. Wilfley DE, Frank M, Welch R, et al. Adapting interpersonal psychotherapy to a group format (IPT-G) for binge eating disorder: toward a model for adapting empirically supported treatments. Psychother Res 1998;8:379–81.
146. Blomquist KK, Ansell EB, White M a, et al. Interpersonal problems and developmental trajectories of binge eating disorder. Compr Psychiatry 2012;53:1088–95.
147. Hilbert A, Bishop ME, Stein RI, et al. Long-term efficacy of psychological treatments for binge eating disorder. Br J Psychiatry 2012;200:232–7.
148. Linehan MM. Cognitive-behavioral treatment of borderline personality disorders. New York: Guilford Press; 1993.
149. Wiser S, Telch CF. Dialectical behavior therapy for Binge-Eating Disorder. J Clin Psychol 1999;55:755–68.
150. Telch CF, Agras WS, Linehan MM. Group dialectical behavior therapy for binge-eating disorder:a preliminary, uncontrolled trial. Behav Ther 2000;31:569–82.
151. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061–5.
152. Safer DL, Robinson AH, JB. Outcome from a randomized controlled trial of group therapy for binge eating disorder: comparing dialectical behavior therapy adapted for binge eating to an active comparison group therapy. Behav Ther 2010;41:106–20.
153. Baer RA. Mindfulness-based treatment approaches: Clinician’s guide to evidence base and applications. San Diego: Academic Press; 2005.
154. Van Strien T Van, Engels RCME, Leeuwe J Van, et al. The Stice model of overeating: Tests in clinical and non-clinical samples. Appetite 2005;45:205–13.
155. Godfrey KM, Gallo LC, Afari N. Mindfulness-based interventions for binge eating: a systematic review and meta-analysis. J Behav Med 2015;38:348–362.
156. Kristeller JL, Hallett CB. An exploratory study of a meditation-based intervention for binge eating disorder. J Health Psychol 1999;4:357–63.
157. Kristeller JL, Wolever RQ. Mindfulness-based eating awareness training for treating binge eating disorder: the conceptual foundation. Eat Disord 2011;19:49–61.
158. Kristeller J, Wolever RQ, Sheets V. Mindfulness-based eating awareness training (MB-EAT) for binge eating: a randomized clinical trial. Mindfulness (N Y) 2014;5:282–97.
159. Weineland S, Arvidsson D, Kakoulidis TP, et al. Acceptance and commitment therapy for bariatric surgery patients, a pilot RCT. Obes Res Clin Pract 2012;6:e21–30.
160. Fairburn CG. Overcoming binge eating. 2nd ed. New York: Guilford Press; 2013.
161. Carter JC, Fairburn CG. Cognitive-behavioral self-help for binge eating disorder: a controlled effectiveness study. J Consult Clin Psychol 1998;66:616–23.
162. Loeb KL, Wilson GT, Gilbert JS, et al. Guided and unguided self-help for binge eating. Behav Res Ther 2000;38:259–72.
163. Masheb RM, Grilo CM. Prognostic significance of two sub-categorization methods for the treatment of binge eating disorder: Negative affect and overvaluation predict, but do not moderate, specific outcomes. Behav Res Ther 2008;46:428–37.
164. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge eating disorder. Behav Res Ther 2005;43:1509–25.
165. Citrome L. Vortioxetine for major depressive disorder: A systematic review of the efficacy and safety profile for this newly approved antidepressant - What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract 2015; 68:60–82.
166. Guerdjikova A, McElroy S, Kotwal R, et al. High-dose escitalopram in the treatment of binge-eating disorder with obesity: a placebo-controlled monotherapy trial. Hum Psychopharmacol 2008;23:1–11.
167. McElroy S, Hudson J, Malhotra JL, et al. Citalopram in the treatment of binge-eating disorder: a placebo-controlled trial. J Clin Psychiatry 2003;64:807–13.
168. McElroy S, Casuto L, Nelson EB, et al. Placebo-controlled trial of sertraline in the treatment of binge eating disorder. Am J Psychiatry 2000;157:1004–6.
169. McElroy S, Hudson J, Mitchell JE, et al. Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry 2015;72:235–46.
170. Pearlstein T, Spurell E, Hohlstein LA, et al. A double-blind, placebo-controlled trial of fluvoxamine in binge eating disorder: a high placebo response. Arch Women’s Ment Health 2003;6:147–51.
171. Hudson J, McElroy S. Raymond NC, et al. Fluvoxamine in the treatment of binge-eating disorder: a multicenter placebo-controlled, double-blind trial. Am J Psychiatry 1998;155:1756–62.
172. Miller W, Rollnick S. Motivational Interviewing: helping people change. 3rd ed. New York: Guilford Press; 2013.
173. Rollnick S, Miller W, Butler C. Motivational interviewing in health care: helping patients change behavior. New York: Guilford Press; 2008.
From the Department of Psychology, Eastern Michigan University, Ypsilanti, MI.
Abstract
- Objective: To describe the epidemiology, clinical features, clinical course, medical complications, and treatment of binge-eating disorder (BED).
- Methods: Review of the literature.
- Results: BED, the most common eating disorder, is a distinct pattern of binge eating accompanied by a sense of loss of control over eating without inappropriate compensatory behaviors. Because people with BED more commonly seek treatment for the psychological and medical factors that are associated with the disorder, patients’ first point of contact with the medical profession is likely to be the primary care physician (PCP). The PCP’s role includes making efforts to screen for BED symptoms, employing motivational interviewing strategies to enhance likelihood of following through with treatment, providing psychoeducational information about eating and weight control, monitoring eating, weight, and related medical problems at follow-up visits, and making referrals to behavioral health specialists who can deliver empirically supported treatments for BED.
- Conclusion: Proper screening and referral in the primary care setting can optimize the likelihood that patients obtain empirically supported treatment.
BED is the most common eating disorder, but it is one for which many do not seek treatment directly. Rather, those struggling with BED more commonly seek treatment for the psychological and medical factors that are strongly associated with the disorder. As will be reviewed below, these factors include poor social adjustment, functional impairment, psychological distress and psychiatric comorbidity, and myriad medical sequelae due to obesity and weight cycling. As such, the BED patient’s point of first contact with the medical profession is most likely to be with the primary care physician, who has several roles in the treatment of BED. There is a limited evidence base for pharmacological treatment of BED, with some medications yielding short-term reductions in binge eating, but none with strong support for long-term efficacy [4]. However, with the recent FDA approval of lisdexamfetamine dimesylate for the treatment of moderate to severe BED, this picture may change. Nonetheless, pharmacologic interventions for comorbid medical conditions will fall solidly in the bailiwick of the primary care physician. In addition, the primary care physician’s role includes making efforts to screen for BED symptoms; employing motivational interviewing strategies to enhance likelihood of following through with treatment; providing psychoeducational information about eating and weight control; monitoring eating, weight, and related medical problems at follow-up visits; and making referrals to behavioral health specialists who can deliver empirically supported treatments for BED. Finally, because BED is typically associated with weight gain over time [5], the primary care physician is encouraged to reinforce the clinical significance of weight maintenance as opposed to necessarily promoting a goal of weight loss. The rationale for this primary care approach is reviewed below, in consideration of the scientific literature and a case study highlighting common clinical features.
Case Study
Initial Presentation
A 35-year-old Caucasian woman schedules an appointment for her annual physical examination with her primary care physician. She reports generally good health but complains of low mood, joint pain, and difficulties managing her weight. Her blood pressure is managed with 100 mg/day of metoprolol. The only other medication she takes is birth control (ethinyl estradiol 20 mcg).
Physical Examination
During physical examination, it is determined that the patient is 5'6" and weighs 286 lb, with a body mass index (BMI) of 46.2 kg/m2, placing her in WHO obesity class III. The patient’s blood pressure is 130/85 mm Hg (medically managed), and her heart rate is 83 bpm. The patient states that she has been experiencing episodes of low mood off and on most of her life; she recently ended a relationship, which has exacerbated her symptoms. The physician states that the patient has gained a significant amount of weight since her last physical examination. The patient reports that she quit smoking 6 months ago and has since gained approximately 30 lb; she has considered smoking again to manage her weight.
• What are the diagnostic criteria for BED?
BED diagnostic criteria (Table 1) have been closely examined for their validity and clinical utility, and several have been the subject of intense debate in the BED literature. The first BED criterion, recurrent episodes of binge eating, refers to 3 essential components: amount of food, time period, and a subjective experience of loss of control. The majority of debate regarding this criterion revolves around the requirement for consumption of a “large amount of food.” There are 2 primary arguments against this criterion. First, it is inherently subjective and requires the person making the diagnosis to distinguish between normative food intake and excessive food intake [6]. There is also some debate as to whether or not individuals with BED actually consume large amounts of food when they binge. However, research supports that those with BED may consume over 1000 kcal during binge episodes, far more than those without BED who are asked to binge eat in the lab [7,8].
Nonetheless, a distinction has been made between objective binge-eating episodes (OBE) and subjective binge eating episodes (SBE) [9]. OBEs are binge eating episodes that meet the full criteria including a large amount of food and a subjective loss of control. SBEs, in contrast, are binge eating episodes that include a subjective loss of control but not a large quantity of food. If consumption of a large quantity of food is essential to the underlying pathology of BED, one would expect that OBEs and SBEs would be associated with different clinical characteristics. However, several studies have failed to find significant difference between individuals reporting OBEs and SBEs with regard to age, age of BE onset, BE severity, interpersonal problems, depressive symptoms, generalized psychopathology, and ED-related psychopathology [10–13]. Results regarding prognosis are mixed, with some suggesting that SBE more readily responds to placebo, while others suggest that SBEs are slower to remit than OBEs [11,13,14]. With respect to primary care, this literature suggests that it is not necessary for busy primary care physicians to devote time to understanding the amount of food consumed by the patient; if the patient perceives that her eating is out of control and excessive, that can generally be considered valid data in terms of considering a BED diagnosis, particularly when combined with even moderately overweight status.
In contrast to the controversy regarding amount of food, the majority of studies suggest that BED binge eating episodes fall within the 2-hour duration specified by the DSM-5 criteria, although longer durations have been reported [13]. The loss of control (LOC) criterion also appears to be relatively well-supported across studies [13,14]. LOC is a key defining feature of a binge eating episode for individuals with and without BED [15–18].Furthermore, the emotional distress associated with loss of control has been associated with depressive symptoms, appearance dissatisfaction, and poorer mental health-related quality of life [19]. In contrast, one study found that 18.6% of self-reported binges were not associated with loss of control [20]. Of note, there is some concern that the focus on LOC in the diagnostic criteria may lead to under diagnosis of BED among men, as women with BED were more likely than men to identify LOC as a core aspect of a binge eating episode [17].
The second DSM-5 criterion for BED requires that BE episodes be associated with 3 or more of the following: (a) eating more rapidly than normal; (b) eating until uncomfortably full; (c) eating large amounts of food in the absence of hunger; (d) eating alone because of embarrassment about how much one is eating; and (e) feeling disgusted with oneself, depressed, or very guilty after overeating. This criterion is not as controversial as the first, and has correspondingly not received as much attention in the BED literature. However, results from a handful of studies provide some support for their inclusion, particularly in light of the fact that individuals are only required to endorse 3 of the 5 symptoms [13–15,17,21].
The third criteria for BED requires that individuals experience “marked distress” about BE. Only one known study has directly evaluated the distress criterion, and its validity was confirmed by results that suggested individuals with full-threshold BED had significantly greater ED-related psychopathology and depressive symptoms as compared to individuals who met all but the distress criteria for a BED diagnosis [22].
The fourth criteria for BED stipulates that BE occurs an average of once a week for 3 months. Previously, DSM-IV-TR required more frequent episodes, at least 2 days a week for 6 months, but this was criticized as lacking in empirical basis [23]. The current state of the evidence suggests that, with regard to frequency of BE episodes, BED best fits a continuous model rather than a categorical model. That is, symptoms and related impairment exist across a severity spectrum as a function of how often BE episodes occur. For example, in a critical review, Wilson and Sysko noted that individuals with sub-threshold frequency of BE episodes had less severe psychopathology than those meeting criteria for DSM-IV BE frequency (ie, at least 2 days a week for 6 months), but they were still significantly more impaired than those who did not binge eat [24]. The authors asserted that there was no empirical rationale for preserving the criteria of 2 binge days per week for 6 months, and indeed, DSM-5 adopted a more relaxed standard. As is the case with symptoms of many psychological disorders, there does not appear to be a definitive and concrete point at which binge eating becomes pathological [23]. Fortunately, reliability for the new criteria is good and appears superior to the DSM-IV criteria [25].
Finally, the last criteria for BED—which remains unchanged from the provisional criteria in DSM-IV-TR —is essentially a rule-out that states that BE should not be accompanied by the regular use of “inappropriate compensatory behaviors” or exclusively occur during the course of anorexia or bulimia. These criteria have also been criticized as being subjective, particularly in light of the fact that individuals with BED often report a history of infrequent purging behavior and frequently engage in weight-loss attempts [6,13,14]. However, the need for a rule-out is clear given that BE also occurs during the course of bulimia and anorexia, binge-eating/purging type, and it is supported by the low rates of crossover from BED to bulimia and/or anorexia [26].
Remission and severity specifiers are new to DSM-5. With respect to the latter, a recent study observed small but significant elevations in eating pathology among those with moderate severity BED, relative to the eating pathology experienced by those with mild severity, but there were no differences in level of associated depression. Interestingly, a better differentiator of severity of eating pathology and depression among patients with BED was overvaluation of shape/weight [27]. As such, the primary care physician might be better advised to focus on indicators of this important variable by querying the extent to which the patient’s shape and weight have influenced how she feels about (judges/thinks/evaluates) herself as a person, rather than using the number of BED symptoms alone as the indicator of severity.
• What is the epidemiology of BED?
Based on DSM-IV-TR criteria, the overall lifetime prevalence rate for BED has been reported to be 2.8%, and it is more common in women (3.5%) than men (2%) [28]; the overall 12-month prevalence rate is 1.2% (1.6% in women and 0.8% in men) [28]. Using DSM-5 criteria, a recent study observed that lifetime prevalence of BED by age 20 was 3.0% for BED and an additional 3.6% for subthreshold BED, with peak age of onset (for both) between ages 18 to 20 years [29]. Notably, even though prevalence rates are slightly higher using DSM-5 criteria (presumably, due to the relaxed criteria for frequency and duration of binge eating), effect sizes for impairment are also higher, suggesting that the revised criteria are not identifying BED cases marked by less impairment [29]. Although often thought of as a disorder common among young women, BED prevalence among middle-aged women (40–60 years) has a prevalence of at least 1.5%, with additional subthreshold cases being common in this age-range; groups meeting full BED criteria and subthreshold cases are both characterized by high levels of distress and impairment [30].
Gender Differences
Men engage in overeating as much or more than women but are less likely to endorse a loss of control and/or distress associated with BE [28,31], and thus are less likely to meet full BED criteria. However, when men do meet criteria for BED, they experience as much clinical impairment as their female counterparts [32]. Additionally, men’s BE may be more directly affected by body image dissatisfaction than women’s BE, and although it is associated with negative affect, it is less likely to be associated with interactions between negative affect and dietary restraint than seems to be the case for women [33]. In addition, in the primary care setting, men with BED were strikingly similar to their female counterparts on most historical and developmental variables [33]. However, men reported more frequent strenuous exercise, whereas women reported that onset of overweight and dieting occurred earlier in life [34]. That same study observed that men (57%) were more likely than women with BED (31%) to meet criteria for metabolic syndrome, even after controlling for race and BMI. A second study by the same research group again demonstrated that men with BED are more likely to show elevated blood pressure, triglycerides, and meet criteria for metabolic syndrome, whereas women are more likely to have elevated total cholesterol [35].
Race/Ethnicity
The evidence related to rates of BED among ethnic minorities is equivocal, with some studies demonstrating that Caucasian women are more likely to experience clinical levels of BED symptoms [36,37], others finding comparable rates between Caucasian and African-American women [38,39], and still others discussing the possibility of finding the greatest rates of binge eating in ethnic minority samples [40], especially in light of the high rates of obesity observed in some ethnic minority groups [41,42]. Studies that focus on subclinical levels of eating pathology among undergraduate students are most likely to find significant ethnic differences, while studies of nonclinical samples utilizing diagnostic threshold find the fewest differences [43]. There is at least some research demonstrating the highest rates of body image disturbance or eating problems among Asian Americans [44,45]. In addition, Latino individuals with BED may have higher levels of ED-related psychopathology as compared with Caucasian individuals [46]. Finally, Caucasian individuals who experience BED may be more likely to utilize mental health services as compared with other ethnic groups [47].
Age
Lower rates of BED have been documented in elderly individuals relative to their younger counterparts in population-based studies [28]. However, this may be due to recall bias, birth cohort effects, restricted access to studies, and/or increased medical morbidity leading to premature mortality [48]. Guerdjikova et al [48] also noted that many treatment outcomes studies have exclusion criteria related to age. This is unfortunate, as elderly individuals and their younger counterparts appear to exhibit similar levels of BE behavior, distress due to BE, weight and shape concerns, psychiatric comorbidity, and obesity. However, elderly individuals have reported later onset, longer duration of illness, and less medical morbidity [48]. In another study, Mangweth-Matzek et al [30] surveyed women between the ages of 40 and 60; they found that very few respondents met full criteria for an eating disorder. However, when criteria were relaxed (ie, dropping associated symptomology for BED and frequency criteria for bulimia nervosa) an additional 4.8% of the sample met criteria. Notably, women with subthreshold eating disorders reported very similar levels of comorbid psychopathology as women whose symptoms met diagnostic criteria.
• What tools are available for assessment of BED in the primary care setting?
Two of the most commonly used questionnaires in specialty clinics are the Eating Disorders Examination– Questionnaire (EDE-Q [49]), and the Questionnaire on Eating and Weight Patterns – Revised (QEWP-R [50]). In the primary care setting, both appear to be low-cost and time-efficient methods of screening for BED. The EDE-Q, however, may underestimate frequency of binge eating episodes and overestimate the extent of eating-related pathology [51]. Notably, the QEWP has been revised to reflect DSM-5 criteria and is available free of charge (QEWP-5 [52]). The Binge Eating Scale [53] is a 16-item scale often used to assess severity of binge eating; it is free and easily accessible online. Regardless of what measure is used, research indicates that a higher proportion of people agree to having episodes where they ‘‘lose control over eating’’ than when asked about having episodes of ‘‘binge eating’’ [54], so asking about loss of control over eating might be the more advisable way to open the discussion with patients about their eating behavior. In assessing for binge eating, physicians should also be aware of some of the differences in clinical presentation observed for ethnic minorities (eg, lower drive for thinness among African-American women) as well as some research demonstrating that measures such as the Eating Disorder Diagnostic Scale do not assess equivalent constructs in African-American and Caucasian clients [55]. Finally, while self-report measures often serve a practical function of quickly assessing a large group, physicians may want to consider relying on interview-based techniques for clients with lower levels of education attainment and literacy; at least one study has demonstrated problems with readability and comprehensibility with most BED measures [56].
• What are the clinical features of BED?
BED and Obesity
The specific impact of BED on health is difficult to separate from the impact of obesity on health, as the two conditions frequently co-occur and are confounded in many studies. Of relevance to the primary care setting, many BED patients report gaining a substantial amount of weight in the year prior to seeking treatment [57].
Although individuals with BED are often obese, proponents of classifying BED as a separate DSM diagnosis argue that individuals with BED differ from their non-BED obese counterparts in regards to eating patterns, eating disordered psychopathology, and associated features and comorbidities. Individuals with BED consume more calories in laboratory studies than weight-matched controls [6,7,58]. In contrast, studies utilizing ecological momentary assessment (ie, real-time assessments) found no differences between BED obese and non-BED obese participants in the frequency of self-reported binge eating and caloric intake during binge eating episodes [59,60]. BED participants, however, were more likely to report higher stress, desire to binge, negative affect, dietary restraint, and being alone immediately before self-reported binge eating episodes. Furthermore, individuals with BED also demonstrate more ED-related psychopathology than non-BED obese individuals [61–63]. Psychiatric comorbidity is also higher among BED obese individuals as compared their non-BED obese counterparts, and the increased comorbidity is accounted for by the severity of binge eating as opposed to the severity of obesity [6,64–67]. In addition, research demonstrates that obese individuals with BED, as compared with non-obese BED patients, have a poorer quality of life [68].
BED and Bulimia Nervosa
Numerous studies have supported the distinction between bulimia nervosa and BED [69–76]. Diagnostically, bulimia nervosa differs from BED by its requirement of recurrent inappropriate compensatory behaviors in order to prevent weight gain, such as self-induced vomiting; misuse of laxatives, diuretics, or other medications; fasting; or excessive exercise [3]. BED and bulimia nervosa are distinguished by distinct risk factors, prevalence, course, and treatment outcomes [28,67,77]. Individuals with BED are less likely than individuals with bulimia to diet before onset of the disorder, and fewer individuals with BED cross over into other ED diagnostic categories [26,78–81]. Finally, BED and bulimia nervosa are associated with different constellations of ED-related symptoms and associated features [28,63,79]. For example, relative to BE patients, those with bulimia show greater work impairment and psychiatric comorbidity [28], higher dietary restraint and eating concerns [63], and lower rates of obesity [79].
Psychiatric Comorbidity
BED is associated with poor social adjustment, greater functional impairment, and significant psychiatric comorbidity, including overall distress and suicidality [67]. In a study of comorbidity with only selected disorders (mood, anxiety, impulse-control, and substance use disorder), 78.9% of individuals with BED had a lifetime history of at least one comorbidity, 20.2% had one comorbid disorder, 9.8% had two, and 48.9% had three or more [28]. Furthermore, the presence of current psychiatric comorbidity is associated with greater ED-related psychopathology and associated distress [40,41]. The most common comorbidities (lifetime rates) are specific phobia (37.1%), social phobia (31.9%), major depressive disorder (32.3%), post-traumatic stress disorder (PTSD) (26.3%), alcohol abuse/dependence (21.4%), conduct disorder (20%), attention-deficit/ hyperactivity disorder (19.8%), illicit drug use/dependence (19.4%), and oppositional-defiant disorder (18%) [28]. A recent report supports that this level of comorbidity is evident in primary care settings, noting that PTSD in particular is common and associated with a host of other difficulties, including depression, anxiety, drug use disorders, greater eating disorder pathology, and poorer psychological functioning [82]. Personality disorders are also commonly comorbid with BED, with the highest lifetime rates for avoidant (11%), obsessive compulsive (10%), and borderline (9%) personality disorders [83]. Finally, cigarette smoking is also associated with binge eating [83,84], likely evolving out of a weight-control smoking profile [85], and this is of relevance to the primary care setting in that smokers with BED gain more weight upon smoking cessation than do their non-BED counterparts [86].
Further Evaluation
To assess behavioral factors related to obesity and recent weight gain, the physician asks the patient if she ever eats what would be considered an unusually large amount of food for the circumstance. The patient acknowledges that she does so regularly, particularly in response to negative moods. The patient also describes that these episodes contribute to ongoing low mood, such that she feels highly depressed and hopeless following binge episodes. The physician then asks about the patient’s exercise habits and weight management techniques. While the patient denies engaging in compensatory behaviors (eg, vomiting, laxative use) to counteract excessive eating, she does report a history of dieting in which she dramatically restricts her food intake and subsequently loses weight. The patient states that these periods are inevitably followed by a resumption of overeating, and she typically gains back more weight than she originally lost. The patient estimates that she has lost and regained more than 20 lb at least 5 times during her lifetime. In addition, the patient reports difficulty maintaining a regular exercise regimen, especially since the onset of osteoarthritis-related joint pain in the past year. After the evaluation, the physician orders an electrocardiogram (ECG) and blood work. The ECG shows that the P-wave, QRS, and T-wave axes are shifted leftward, but within normal limits. A follow-up appointment is scheduled in 2 weeks.
• What are the medical complications of BED?
BED is associated with numerous negative health sequelae including obesity, sleeping problems, musculoskeletal pain, joint pain, headaches, gastrointestinal problems, menstrual problems, shortness of breath, chest pain, diabetes, low health-related quality of life, and functional health impairments [87–90], with many of these risks persisting even after controlling for BMI [91]. A 5-year follow-up of 134 individuals with BED and 134 individuals with no history of eating disorders, who were frequency-matched for age, sex, and baseline body mass index (BMI), provides further support that BED confers risk of components of metabolic syndrome beyond the risks associated with BMI alone [92]. Specifically, BED cases had higher longitudinal risk of developing dyslipidemia, hypertension, type 2 diabetes, any metabolic syndrome component, and two or more metabolic syndrome components. Alarmingly, these findings even emerge in studies of pediatric samples, wherein BED predicts development of metabolic syndrome, elevated triglycerides, and increases in visceral adiposity [93].
• What are risk factors for BED?
A number of risk factors for BED have been identified, although many are risk factors for a number of psychiatric disorders and not specific to BED. These general risk factors include depression/negative affectivity [94,95], parental mood and substance use disorder, maternal problematic parenting, and separation from parents [95]. A host of risk factors have been identified for disordered eating, in general, including body dissatisfaction [94], early onset of dieting [94], and perfectionism [96]. A number of other variables are risk factors for both BED and bulimia (but not anorexia), including a history of childhood bully and teasing, negative self-evaluation, parental depression, and negative family communication about shape and weight [81,96]. In a study comparing BED cases to psychiatric controls, childhood obesity, familial eating problems, family discord, and high parental demands differentiated the BED cases [95]. In summary, it has been suggested that BED risk is conferred by factors that increase risk of psychiatric disorder in general and those that confer risk for obesity [81]. Of note, the risk factors studied do not appear to differ between black and white women [95].
Genetic risk factors appear to play a strong role in the development of BED. Risk for BED tends to aggregate in families independently of the risk for obesity, although the presence of BED in a first-degree relative does increase risk for obesity [97]. Heritability estimates for BED range from 45% to 57% [98,99], which is greater than the heritability estimate for subthreshold binge eating (ie, overeating with a sense of loss of control, 41%) [100]. In addition, symptom-level analyses support moderate genetic contributions for each BED symptom [98], supporting the integrity of the diagnostic criteria. Finally, shared environment appears to play a very small role in the familial transmission of BED, and the contribution of unique environmental factors in development of BED appears to be substantial [97,101].
With regard to the neurobiological underpinnings of BED, it appears that BED may be associated with hypersensitivity to reward, a phenomenon that is strongly associated with the striatum and dopaminergic mechanisms [102,103]. In support of this hypothesis, Davis et al [102] reported that BED was differentially related to genotypes that reflect a greater density of D2 receptors and higher D2 binding potential as compared to obese controls. Additionally, greater increases in striatal DA and unique activation patterns in the right ventral striatum have been demonstrated in individuals with BED as compared to obese non-BED controls in response to food-related stimuli [103,104]. Other findings have implicated the orbitofrontal cortex (OFC) in BED, which is another brain region responsible for reward processing, particularly as it relates to the hedonic value of food stimuli [103]. Increased volume of grey matter has been documented in individuals with BED and bulimia as compared to normal weight controls, and stronger medial OFC activation while viewing pictures of food was observed in individuals with BED as compared to individuals with bulimia, overweight controls, and normal controls [105].
Difficulties with affect regulation have also been implicated in the development of BED. Two theories that implicate a primary and specific role for affect regulation in BED are cited most frequently in the extant literature: the affect regulation theory and the escape theory. The affect regulation theory [106] posits that BE is a conditioned response to negative affect which is correspondingly negatively reinforced by reductions in negative affect, which could occur during or after BE. Escape theory [107] posits that aversive self-awareness causes negative affect, which in turn triggers BE. BE is then negatively reinforced by reductions in negative affect during a binge via an escape from self-awareness that is accomplished through cognitive narrowing to the immediate stimulus environment. In contrast to the affect regulation theory, escape theory predicts that negative affect will increase after BE when self-awareness is restored. Results regarding changes in affect during BE episodes are conflicting as to whether BE is associated with decreases, no change [108–110], or even increases in negative affect. In particular, a meta-analytic review of 36 studies that examined affect via ecological momentary sampling found moderate increases in negative affect following binge episodes [111]. To some degree, results of this meta-analysis may not generalize to BED, per se, given that it included other binge eating groups, such as those with bulimia nervosa. However, in general, studies suggest that negative affect is an antecedent for BE and increased negative affect may be a consequence of BE, at least among women. More information is needed regarding aversive self-awareness before and after BE, cognitive narrowing, and changes in affect during BE. As such, the current state of the literature provides only partial support for affect regulation models of BED in women. Furthermore, it remains unknown if these results will generalize to men.
Clinical Course
Evidence regarding the course and stability of BED is conflicting and unclear. Several prospective studies have suggested that BED is not a stable disorder, exhibiting high rates of remission over time [26,99,112]. However, the samples have been criticized for being small, completely female, younger than typical individuals with BED, and post-ED treatment. In contrast, a prospective study that included older women and a combination of treated and untreated women suggested remission rates at 1 year that were much lower (7%) [78]. Additionally, a retrospective study [113] reported an average BED duration of 14.4 years. In a review of the studies cited above, Wonderlich et al [6] concluded that “[a]lthough there is variability in the data, it does appear that BED differs from other eating disorders in terms of a greater tendency toward recovery and fluctuation, although this may be embedded in a chronic pattern of remission and relapse.” Viewing BED as a disorder with a chronic pattern of remission and relapse could explain why individuals with BED retrospectively report a longer duration of illness, as they may be more likely to conceptualize their illness as one continuous course punctuated by different periods of severity rather than several distinct bouts of BED. Finally, although diagnostic crossover is a frequent phenomenon among other eating disorders, the crossover rate for BED appears relatively low as compared to anorexia and bulimia [6,26,28,66].
Follow-up
Laboratory examination shows TSH levels within normal limits and cholesterol levels of 48 mg/dL(HDL), 162 mg/dL (LDL), and 270 mg/dL (total). Triglyceride levels are 300 mg/dL and the patient’s fasting glucose level is 115 mg/dL. At the patient’s follow-up appointment, the physician states that a number of laboratory results indicated negative weight-related health consequences, including high cholesterol, high triglycerides, hypertension, and probable pre-diabetes. The patient initially disregards the significance of these results, stating she only gained weight due to her break-up and quitting smoking, and she is motivated to diet to lose weight in the near future. The physician asks for more information about the patient’s eating behavior, in particular asking if she ever feels as if she loses control over her eating. The patient reluctantly admits to this, and the physician provides a referral to a behavioral health specialist. The patient expresses ambivalence and a desire to try to manage her weight on her own. The physician uses motivational interviewing techniques to enhance motivation to follow up on this referral. In addition, the patient is encouraged to make small changes to her diet and slowly increase her exercise by taking walks. Another follow-up appointment is scheduled in 3 months.
• Which treatments are most effective for BED?
Despite the negative sequalae of BED, studies suggest that it often goes untreated [114]. Women with BED, as compared to women with anorexia and bulimia, are less likely to seek treatment for BED and less likely to receive treatment for their eating disorder when they do seek it out [114–116]. Barriers to treatment may include shame and internalized weight stigma, lack of knowledge about where to seek treatment, a belief that willpower should be sufficient to overcome the problem, lack of understanding that BED is a psychiatric disorder, finances/insurance barriers, and lack of BED detection by non-specialist treatment providers [115]. These barriers are particularly concerning, as women with BED report greater health care utilization and comprise a large segment of patients in weight control programs. Therefore, it appears individuals with BED seek help for the negative consequences of the disorder, but they are less likely to seek and receive help for the likely root cause of their concerns. This is a particularly damaging pattern, as the presence of BED may negatively impact the outcome of obesity treatment [117]. There are, however, a number of promising treatments for BED, as described below:
Cognitive Behavioral Therapy
Cognitive behavioral therapy (CBT) is generally considered to be the most well-established and empirically supported treatment for BED [118,119]. The cognitive behavioral conceptualization of BED is based on Fairburn, Cooper, and Shafran’s [120] transdiagnostic model of eating disorders (CBT-E), which is an expanded version of the cognitive behavioral model of bulimia nervosa [121]. CBT-E posits that the core pathology in eating disorders is a dysfunctional system in which self-worth is based on eating habits, shape, or weight, and the individual’s ability to control them. Attempts to maintain self-worth by controlling eating, shape, and weight result in extreme and brittle forms of dietary restraint. Inevitable violations of the individual’s dietary rules are then interpreted as lack of self-control, leading to a temporary abandonment of dietary restraint and consequent BE. These dietary slips and corresponding BE often occur in response to acute changes in mood, and BE is thus negatively reinforced by “neutralizing” negative mood states. Lapses in dietary restraint also result in secondary negative self-evaluation, which serves to further exacerbate a cycle of increased dietary restraint to improve self-worth and then inevitable dietary lapses leading to BE. CBT-E expanded upon CBT-BN by postulating 4 processes that maintain ED: severe perfectionism (clinical perfectionism), unconditional and pervasive low self-esteem (core low self-esteem), difficulties coping with intense mood states (mood intolerance), and developmental interpersonal difficulties (interpersonal difficulties). Of note, the CBT-E model explicitly states that individuals may differ in the extent to which they experience the 4 maintaining processes and not every individual will experience all four.
Overall, treatment is focused on normalizing eating patterns (ie, not weight loss), cognitive restructuring for weight/shape concerns and other triggers for binge eating, and relapse prevention [122]. CBT has produced substantial reductions in binge eating as compared to no treatment [123–125] and supportive therapy [126]. The majority of RCTs have reported remission rates greater than 50% [127]. Unfortunately, CBT has generally not produced meaningful weight loss [118,122,127–129], but this may be a contraindicated goal. CBT has demonstrated improvements in a number of features associated with BED including eating disordered psychopathology [122,124,130,131], depression [122,124,130,132], social adjustment [133], and self-esteem [132]. Treatment gains are generally well-maintained at 1-year to 4-year follow-up [122,123,130,133,134]. Individual and group treatments appear to produce similar results [134], and treatment completion rates have been estimated at approximately 80% across different delivery formats [127]. One strength of the CBT literature is the inclusion of participants with severe psychopathology, which facilitates the generalizability of these findings [127].
A number of factors have been associated with treatment outcome in CBT trials. Poor treatment outcomes have been associated with a history of weight problems during childhood, high levels of emotional eating at baseline, interpersonal dysfunction, and low group cohesion during group CBT [110,124,134]. Overvaluation of weight and shape demonstrated a statistical trend toward negatively impacting outcomes in one study. The presence of a cluster B personality disorder (ie, borderline histrionic, antisocial, and narcissistic personality disorders) predicted higher levels of binge eating at 1-year follow-up in a combined sample of participants treated with group CBT or group interpersonal psychotherapy (IPT) [135].
Alternatively, positive treatment outcomes have been associated with low levels of emotional eating at baseline, older age of onset, weight loss history that is negative for amphetamine use, and decreases in depressive symptoms during treatment [124,134,136,137]. In addition, early response to treatment (defined as a 65%–70% reduction in binge eating within 4 weeks of starting treatment) tends to be associated with greater long-term (ie, 1–2 year) remission from BED and lower eating disorder psychopathology, across a variety of psychological treatment approaches [138–144].
Interpersonal Psychotherapy
IPT for BED was adapted by Wilfley and colleagues [145] from IPT for depression, and the rationale for its use with BED is based on successful outcomes for individuals with bulimia and multiple studies documenting interpersonal deficits in individuals with BED [146]. IPT seeks to address interpersonal problems in 4 areas: interpersonal conflict, grief, role transitions, and interpersonal deficits [135]. While adapting IPT for BED, it was noted that the course of BED tends to be more chronic than the course of depression, thus the focus of IPT for BED was shifted from addressing the interpersonal precipitants of the disorder to the interpersonal factors that maintain the disorder [145]. Fewer studies examining the effectiveness of IPT in treating BED have been published than those examining CBT for BED, but it appears that IPT is as efficacious as CBT immediately post-treatment [130], and at 1- [130] and 4-year follow-up [147]. In addition, at least 2 studies have been published that compare IPT, cognitive behavioral therapy–guided self-help (CBTgsh), and behavioral weight loss [133,141]. Overall, results support the use of both IPT and CBTgsh (discussed in more detail below), with important moderators of treatment effects observed. For example, Wilson et al [133] found that clients with higher levels of psychopathology were better suited for IPT. The authors conclude that these results could inform a model of evidence-based stepped care, where CBTgsh, a low-cost, low-intensity treatment, should be considered as the first line of treatment. Secondarily, IPT, which represents a more specialized and expensive form of treatment, could be considered the next level of care, particularly for clients who are not demonstrating rapid improvement in response to CBTgsh.
Dialectic Behavior Therapy
A small number of studies have investigated the treatment of BED with dialectical behavior therapy (DBT). Originally developed to treat borderline personality disorder [148], DBT is of particular interest given its explicit targeting of emotion regulation. According to the DBT model of BED [149], emotional dysregulation is the core psychopathology in this disorder, and binge eating is viewed as attempts to influence, change, or control painful emotions. Initially, promising results were published showing positive treatment effects in an uncontrolled study [150] as well as wait-list controlled trials [151]. Notably, relative to wait-list controls, participants in a DBT guided self-help program (who received an orientation, DBT manual, and six 20-minute support calls across 13 weeks) reported reduced past-month binge eating, higher binge eating abstinence rates, and over the longer term improved quality of life and reductions in ED psychopathology. However, a comparison of DBT-BED with an active comparison control group (ie, nonspecific supportive therapy) failed to find significant differences between the 2 treatments (defined as effect size greater than 0.5) at 12-month follow-up in binge eating abstinence, binge eating frequency, most ED-related psychopathology, positive affect, depression, and self-esteem [152]. Therefore, DBT may have potential and, at a minimum, is equally efficacious as supportive therapy.
Mindfulness- and Meditation-Based Therapies
Treatment outcome studies utilizing mindfulness-based therapies, including mindfulness-based stress reduction (MBSR) and acceptance and commitment therapy (ACT), make up a small but promising body of literature. Reasoning that negative affect, eating in the absence of hunger, and emotional eating may comprise one pathway to binge eating [153,154], it follows that mindfulness-based therapies may act through their effects on emotion regulation, acceptance strategies for tolerating negative affect, and awareness of bodily cues. A recent review identified 19 studies exploring the effects of mindfulness-based interventions on binge eating severity and frequency as well as a number of related indicators, observing positive effects for this form of treatment [155]. For example, MB-EAT [156] is a group treatment for BED that is primarily based on MBSR. Treatment is targeted at cultivating mindfulness, mindful eating, emotional balance, and self-acceptance[157]. The treatment also places particular emphasis on developing self-awareness of internal hunger and satiety cues. A recent randomized controlled trial of MB-EAT produced significant improvements in binge eating frequency and BE-related psychopathology [158]. Furthermore, process variables including hunger awareness, satiety awareness, and mindfulness were correlated with positive outcomes. In addition, a small study (n = 39) that compared ACT to standard follow-up utilized by a bariatric surgery team demonstrated significantly greater improvements in disordered eating, body satisfaction, and quality of life for clients who participated in ACT [159]. In brief, results suggest that mindfulness-based interventions represent an additional treatment approach with supporting but limited evidence to date.
Self-Help Interventions
Self-help interventions for BED are categorized as pure self-help or guided self-help. In treatment outcome studies, pure self-help is generally conducted with a self-help manual, although several studies have examined more novel formats such as the internet, video, and CD-ROM. GSH also uses a self-help manual (or other format) with the addition of brief sessions with health care providers who have varying degrees of expertise with the type of therapy being utilized. CBT is the most commonly utilized therapeutic modality in treatment outcome studies of self-help interventions, and they most often utilize Fairburn’s Overcoming Binge Eating self-help manual [160].
Two studies have directly compared pure and guided self-help with Fairburn’s manual and produced conflicting results. Carter and Fairburn [161] found that in a sample of primarily white women with BED, pure self-help (CBTsh; n = 24) and guided self-help (CBTgsh; n = 24) were equally effective, and both were superior to wait-list controls at 6-month follow-up in producing BE abstinence (CBTsh = 40%, CBTgsh = 50%), reducing binge eating, ED-related psychopathology, and general psychiatric symptoms. In contrast, a study comparing CBTsh and CBTgsh in 40 primarily white women with recurrent binge eating (82.5% diagnosed with BED), guided self-help was superior to pure self-help at the end of treatment in reducing BE frequency, eating concern, and restraint [162]. CBTgsh and CBTsh were equally effective in producing BE abstinence (50% and 30%, respectively), and reducing shape concern, weight concern, and general psychiatric symptoms [162]. Higher levels of general psychiatric symptoms were predictive of higher BE frequency post-treatment for both treatments. It should be noted that participants in both conditions experienced statistically significant improvements on all variables as compared to baseline.
CBTgsh also performed as well or better than individualized treatments in one study [133]. CBTgsh, IPT, and behavioral weight loss (BWL) were compared in a large study of 205 primarily white, obese or overweight individuals diagnosed with BED. The 3 treatments produced equivalent outcomes for binge eating at post-treatment, but BWL produced significantly greater weight loss. However, at 2-year follow-up, the CBTgsh and IPT groups had maintained treatment gains and were significantly superior to BWL in reductions in binge eating. The 3 groups were equivalent with regard to weight loss at the 2-year follow-up, and none reported clinically significant weight loss. Of note, as compared to the IPT and BWL groups, the CBTgsh group received 10 sessions as opposed to 20, received 25-minute sessions as opposed to 60-minute sessions, and were treated by providers with limited levels of experience as opposed to doctoral-level clinical psychologists.
To summarize, CBT is the most often studied type of self-help treatment. Both CBTsh and CBTgsh produced improvements in binge eating and associated psychopathology as compared to baseline and wait-list controls, and treatment gains were maintained at 6-month follow-up. Conclusions regarding the relative superiority of pure self-help or guided self-help are premature given the small number of studies and conflicting results.
In addition, limited information is available regarding moderators and predictors of guided self-help outcomes. Masheb and Grilo [163] performed a cluster analysis of the sample for the CBTgsh vs. BWLgsh described above [164] and identified 2 clinically significant subtypes: a dietary-negative affect subtype characterized by high restraint, low self-esteem, and depressive symptoms; and an overvaluation of weight and shape subtype. For both the CBTgsh and BWLgsh groups, the dietary-negative affect subtype experienced higher levels of binge eating frequency, and the overvaluation of weight and shape subtype experienced higher levels of ED-related psychopathology. Additionally, individuals receiving BWLgsh who experienced a rapid response to treatment reported lower BE frequency, greater weight loss, and higher restraint than participants without a rapid response [142]. In contrast, rapid response did not appear to affect outcomes for CBTgsh participants. Finally, the combination of low self-esteem and high ED-related psychopathology negatively affected BE remission rates for CBTgsh recipients [133].
Pharmacologic Treatment
Currently only one medication, lisdexamfetamine dimesylate, has been FDA-approved for the treatment of BED. Previously approved for treating both adults and children with attention-deficit hyperactivity disorder, lisdexamfetamine dimesylate is a central nervous system stimulant and has been found to significantly reduce number of binge days, with robust effect sizes [165]. Beyond this medication, the evidence for pharmacologic treatment of BED is limited. A recent review identified only 22 studies exploring the effects of pharmacologic treatment in a methodologically rigorous way (eg, double-blind placebo design) [4]. To date, a number of different medication classes have been evaluated, including antidepressants, anticonvulsants, stimulants, anti-obesity drugs, and others. Overall, there is some evidence that antidepressant and anticonvulsant agents are efficacious at reducing BE frequency [166,167] and sometimes effective regarding statistically significant weight loss [168,169]. However, the majority of results are generally disappointing, both with respect to reductions in binge eating and sustained weight loss [48,170,171]. In addition, there are serious limitations in the literature that must be considered, including the limited number of studies that address the high placebo response observed in clinical samples, limited follow-up windows, and inadequate multiplicitious confirmatory trials. Despite these limitations, the evidence base related to pharmacologic treatment is continuously evolving and represents an important future direction for the treatment of BED.
Treatment
Prior to her next medical follow-up, the patient meets with a psychologist. The patient discloses that she has been binge eating several times per week for over a year; she also discloses a history of prolonged sexual abuse perpetrated by a step-parent during her childhood. When the patient returns to her follow-up medical appointment, she reports that her psychologist has diagnosed her with BED and PTSD. She states that they are using cognitive behavioral techniques to regulate her mood and eating behavior, with a specific aim of avoiding excessive dietary restraint. In addition, they are working together to discuss her unfulfilling romantic history and processing her experiences of trauma. Since her last appointment with the primary care physician, she reports an increased awareness of her eating habits, improvement in mood, and a 10-lb decrease in her weight.
The patient reports that she has continued to meet weekly with her psychologist and has slowly begun reintroducing low-impact exercise to her routine. She continues to lose weight gradually, but with a priority of stabilizing eating behavior and avoiding binge episodes versus aiming for weight loss. She reports that her mood has stabilized. Her cholesterol and triglycerides remain high, but her blood pressure is controlled effectively with medication. Her physician recommends continued psychological treatment, periodic meetings with a nutritionist, and prescribes medication for her cholesterol. A follow-up appointment with her physician is scheduled in 6 months.
Summary
Corresponding author: Karen K. Saules, PhD, Eastern Michigan University, Psychology Clinic, 611 W. Cross St., Ypsilanti, MI 48197, [email protected]
Financial disclosures: None.
From the Department of Psychology, Eastern Michigan University, Ypsilanti, MI.
Abstract
- Objective: To describe the epidemiology, clinical features, clinical course, medical complications, and treatment of binge-eating disorder (BED).
- Methods: Review of the literature.
- Results: BED, the most common eating disorder, is a distinct pattern of binge eating accompanied by a sense of loss of control over eating without inappropriate compensatory behaviors. Because people with BED more commonly seek treatment for the psychological and medical factors that are associated with the disorder, patients’ first point of contact with the medical profession is likely to be the primary care physician (PCP). The PCP’s role includes making efforts to screen for BED symptoms, employing motivational interviewing strategies to enhance likelihood of following through with treatment, providing psychoeducational information about eating and weight control, monitoring eating, weight, and related medical problems at follow-up visits, and making referrals to behavioral health specialists who can deliver empirically supported treatments for BED.
- Conclusion: Proper screening and referral in the primary care setting can optimize the likelihood that patients obtain empirically supported treatment.
BED is the most common eating disorder, but it is one for which many do not seek treatment directly. Rather, those struggling with BED more commonly seek treatment for the psychological and medical factors that are strongly associated with the disorder. As will be reviewed below, these factors include poor social adjustment, functional impairment, psychological distress and psychiatric comorbidity, and myriad medical sequelae due to obesity and weight cycling. As such, the BED patient’s point of first contact with the medical profession is most likely to be with the primary care physician, who has several roles in the treatment of BED. There is a limited evidence base for pharmacological treatment of BED, with some medications yielding short-term reductions in binge eating, but none with strong support for long-term efficacy [4]. However, with the recent FDA approval of lisdexamfetamine dimesylate for the treatment of moderate to severe BED, this picture may change. Nonetheless, pharmacologic interventions for comorbid medical conditions will fall solidly in the bailiwick of the primary care physician. In addition, the primary care physician’s role includes making efforts to screen for BED symptoms; employing motivational interviewing strategies to enhance likelihood of following through with treatment; providing psychoeducational information about eating and weight control; monitoring eating, weight, and related medical problems at follow-up visits; and making referrals to behavioral health specialists who can deliver empirically supported treatments for BED. Finally, because BED is typically associated with weight gain over time [5], the primary care physician is encouraged to reinforce the clinical significance of weight maintenance as opposed to necessarily promoting a goal of weight loss. The rationale for this primary care approach is reviewed below, in consideration of the scientific literature and a case study highlighting common clinical features.
Case Study
Initial Presentation
A 35-year-old Caucasian woman schedules an appointment for her annual physical examination with her primary care physician. She reports generally good health but complains of low mood, joint pain, and difficulties managing her weight. Her blood pressure is managed with 100 mg/day of metoprolol. The only other medication she takes is birth control (ethinyl estradiol 20 mcg).
Physical Examination
During physical examination, it is determined that the patient is 5'6" and weighs 286 lb, with a body mass index (BMI) of 46.2 kg/m2, placing her in WHO obesity class III. The patient’s blood pressure is 130/85 mm Hg (medically managed), and her heart rate is 83 bpm. The patient states that she has been experiencing episodes of low mood off and on most of her life; she recently ended a relationship, which has exacerbated her symptoms. The physician states that the patient has gained a significant amount of weight since her last physical examination. The patient reports that she quit smoking 6 months ago and has since gained approximately 30 lb; she has considered smoking again to manage her weight.
• What are the diagnostic criteria for BED?
BED diagnostic criteria (Table 1) have been closely examined for their validity and clinical utility, and several have been the subject of intense debate in the BED literature. The first BED criterion, recurrent episodes of binge eating, refers to 3 essential components: amount of food, time period, and a subjective experience of loss of control. The majority of debate regarding this criterion revolves around the requirement for consumption of a “large amount of food.” There are 2 primary arguments against this criterion. First, it is inherently subjective and requires the person making the diagnosis to distinguish between normative food intake and excessive food intake [6]. There is also some debate as to whether or not individuals with BED actually consume large amounts of food when they binge. However, research supports that those with BED may consume over 1000 kcal during binge episodes, far more than those without BED who are asked to binge eat in the lab [7,8].
Nonetheless, a distinction has been made between objective binge-eating episodes (OBE) and subjective binge eating episodes (SBE) [9]. OBEs are binge eating episodes that meet the full criteria including a large amount of food and a subjective loss of control. SBEs, in contrast, are binge eating episodes that include a subjective loss of control but not a large quantity of food. If consumption of a large quantity of food is essential to the underlying pathology of BED, one would expect that OBEs and SBEs would be associated with different clinical characteristics. However, several studies have failed to find significant difference between individuals reporting OBEs and SBEs with regard to age, age of BE onset, BE severity, interpersonal problems, depressive symptoms, generalized psychopathology, and ED-related psychopathology [10–13]. Results regarding prognosis are mixed, with some suggesting that SBE more readily responds to placebo, while others suggest that SBEs are slower to remit than OBEs [11,13,14]. With respect to primary care, this literature suggests that it is not necessary for busy primary care physicians to devote time to understanding the amount of food consumed by the patient; if the patient perceives that her eating is out of control and excessive, that can generally be considered valid data in terms of considering a BED diagnosis, particularly when combined with even moderately overweight status.
In contrast to the controversy regarding amount of food, the majority of studies suggest that BED binge eating episodes fall within the 2-hour duration specified by the DSM-5 criteria, although longer durations have been reported [13]. The loss of control (LOC) criterion also appears to be relatively well-supported across studies [13,14]. LOC is a key defining feature of a binge eating episode for individuals with and without BED [15–18].Furthermore, the emotional distress associated with loss of control has been associated with depressive symptoms, appearance dissatisfaction, and poorer mental health-related quality of life [19]. In contrast, one study found that 18.6% of self-reported binges were not associated with loss of control [20]. Of note, there is some concern that the focus on LOC in the diagnostic criteria may lead to under diagnosis of BED among men, as women with BED were more likely than men to identify LOC as a core aspect of a binge eating episode [17].
The second DSM-5 criterion for BED requires that BE episodes be associated with 3 or more of the following: (a) eating more rapidly than normal; (b) eating until uncomfortably full; (c) eating large amounts of food in the absence of hunger; (d) eating alone because of embarrassment about how much one is eating; and (e) feeling disgusted with oneself, depressed, or very guilty after overeating. This criterion is not as controversial as the first, and has correspondingly not received as much attention in the BED literature. However, results from a handful of studies provide some support for their inclusion, particularly in light of the fact that individuals are only required to endorse 3 of the 5 symptoms [13–15,17,21].
The third criteria for BED requires that individuals experience “marked distress” about BE. Only one known study has directly evaluated the distress criterion, and its validity was confirmed by results that suggested individuals with full-threshold BED had significantly greater ED-related psychopathology and depressive symptoms as compared to individuals who met all but the distress criteria for a BED diagnosis [22].
The fourth criteria for BED stipulates that BE occurs an average of once a week for 3 months. Previously, DSM-IV-TR required more frequent episodes, at least 2 days a week for 6 months, but this was criticized as lacking in empirical basis [23]. The current state of the evidence suggests that, with regard to frequency of BE episodes, BED best fits a continuous model rather than a categorical model. That is, symptoms and related impairment exist across a severity spectrum as a function of how often BE episodes occur. For example, in a critical review, Wilson and Sysko noted that individuals with sub-threshold frequency of BE episodes had less severe psychopathology than those meeting criteria for DSM-IV BE frequency (ie, at least 2 days a week for 6 months), but they were still significantly more impaired than those who did not binge eat [24]. The authors asserted that there was no empirical rationale for preserving the criteria of 2 binge days per week for 6 months, and indeed, DSM-5 adopted a more relaxed standard. As is the case with symptoms of many psychological disorders, there does not appear to be a definitive and concrete point at which binge eating becomes pathological [23]. Fortunately, reliability for the new criteria is good and appears superior to the DSM-IV criteria [25].
Finally, the last criteria for BED—which remains unchanged from the provisional criteria in DSM-IV-TR —is essentially a rule-out that states that BE should not be accompanied by the regular use of “inappropriate compensatory behaviors” or exclusively occur during the course of anorexia or bulimia. These criteria have also been criticized as being subjective, particularly in light of the fact that individuals with BED often report a history of infrequent purging behavior and frequently engage in weight-loss attempts [6,13,14]. However, the need for a rule-out is clear given that BE also occurs during the course of bulimia and anorexia, binge-eating/purging type, and it is supported by the low rates of crossover from BED to bulimia and/or anorexia [26].
Remission and severity specifiers are new to DSM-5. With respect to the latter, a recent study observed small but significant elevations in eating pathology among those with moderate severity BED, relative to the eating pathology experienced by those with mild severity, but there were no differences in level of associated depression. Interestingly, a better differentiator of severity of eating pathology and depression among patients with BED was overvaluation of shape/weight [27]. As such, the primary care physician might be better advised to focus on indicators of this important variable by querying the extent to which the patient’s shape and weight have influenced how she feels about (judges/thinks/evaluates) herself as a person, rather than using the number of BED symptoms alone as the indicator of severity.
• What is the epidemiology of BED?
Based on DSM-IV-TR criteria, the overall lifetime prevalence rate for BED has been reported to be 2.8%, and it is more common in women (3.5%) than men (2%) [28]; the overall 12-month prevalence rate is 1.2% (1.6% in women and 0.8% in men) [28]. Using DSM-5 criteria, a recent study observed that lifetime prevalence of BED by age 20 was 3.0% for BED and an additional 3.6% for subthreshold BED, with peak age of onset (for both) between ages 18 to 20 years [29]. Notably, even though prevalence rates are slightly higher using DSM-5 criteria (presumably, due to the relaxed criteria for frequency and duration of binge eating), effect sizes for impairment are also higher, suggesting that the revised criteria are not identifying BED cases marked by less impairment [29]. Although often thought of as a disorder common among young women, BED prevalence among middle-aged women (40–60 years) has a prevalence of at least 1.5%, with additional subthreshold cases being common in this age-range; groups meeting full BED criteria and subthreshold cases are both characterized by high levels of distress and impairment [30].
Gender Differences
Men engage in overeating as much or more than women but are less likely to endorse a loss of control and/or distress associated with BE [28,31], and thus are less likely to meet full BED criteria. However, when men do meet criteria for BED, they experience as much clinical impairment as their female counterparts [32]. Additionally, men’s BE may be more directly affected by body image dissatisfaction than women’s BE, and although it is associated with negative affect, it is less likely to be associated with interactions between negative affect and dietary restraint than seems to be the case for women [33]. In addition, in the primary care setting, men with BED were strikingly similar to their female counterparts on most historical and developmental variables [33]. However, men reported more frequent strenuous exercise, whereas women reported that onset of overweight and dieting occurred earlier in life [34]. That same study observed that men (57%) were more likely than women with BED (31%) to meet criteria for metabolic syndrome, even after controlling for race and BMI. A second study by the same research group again demonstrated that men with BED are more likely to show elevated blood pressure, triglycerides, and meet criteria for metabolic syndrome, whereas women are more likely to have elevated total cholesterol [35].
Race/Ethnicity
The evidence related to rates of BED among ethnic minorities is equivocal, with some studies demonstrating that Caucasian women are more likely to experience clinical levels of BED symptoms [36,37], others finding comparable rates between Caucasian and African-American women [38,39], and still others discussing the possibility of finding the greatest rates of binge eating in ethnic minority samples [40], especially in light of the high rates of obesity observed in some ethnic minority groups [41,42]. Studies that focus on subclinical levels of eating pathology among undergraduate students are most likely to find significant ethnic differences, while studies of nonclinical samples utilizing diagnostic threshold find the fewest differences [43]. There is at least some research demonstrating the highest rates of body image disturbance or eating problems among Asian Americans [44,45]. In addition, Latino individuals with BED may have higher levels of ED-related psychopathology as compared with Caucasian individuals [46]. Finally, Caucasian individuals who experience BED may be more likely to utilize mental health services as compared with other ethnic groups [47].
Age
Lower rates of BED have been documented in elderly individuals relative to their younger counterparts in population-based studies [28]. However, this may be due to recall bias, birth cohort effects, restricted access to studies, and/or increased medical morbidity leading to premature mortality [48]. Guerdjikova et al [48] also noted that many treatment outcomes studies have exclusion criteria related to age. This is unfortunate, as elderly individuals and their younger counterparts appear to exhibit similar levels of BE behavior, distress due to BE, weight and shape concerns, psychiatric comorbidity, and obesity. However, elderly individuals have reported later onset, longer duration of illness, and less medical morbidity [48]. In another study, Mangweth-Matzek et al [30] surveyed women between the ages of 40 and 60; they found that very few respondents met full criteria for an eating disorder. However, when criteria were relaxed (ie, dropping associated symptomology for BED and frequency criteria for bulimia nervosa) an additional 4.8% of the sample met criteria. Notably, women with subthreshold eating disorders reported very similar levels of comorbid psychopathology as women whose symptoms met diagnostic criteria.
• What tools are available for assessment of BED in the primary care setting?
Two of the most commonly used questionnaires in specialty clinics are the Eating Disorders Examination– Questionnaire (EDE-Q [49]), and the Questionnaire on Eating and Weight Patterns – Revised (QEWP-R [50]). In the primary care setting, both appear to be low-cost and time-efficient methods of screening for BED. The EDE-Q, however, may underestimate frequency of binge eating episodes and overestimate the extent of eating-related pathology [51]. Notably, the QEWP has been revised to reflect DSM-5 criteria and is available free of charge (QEWP-5 [52]). The Binge Eating Scale [53] is a 16-item scale often used to assess severity of binge eating; it is free and easily accessible online. Regardless of what measure is used, research indicates that a higher proportion of people agree to having episodes where they ‘‘lose control over eating’’ than when asked about having episodes of ‘‘binge eating’’ [54], so asking about loss of control over eating might be the more advisable way to open the discussion with patients about their eating behavior. In assessing for binge eating, physicians should also be aware of some of the differences in clinical presentation observed for ethnic minorities (eg, lower drive for thinness among African-American women) as well as some research demonstrating that measures such as the Eating Disorder Diagnostic Scale do not assess equivalent constructs in African-American and Caucasian clients [55]. Finally, while self-report measures often serve a practical function of quickly assessing a large group, physicians may want to consider relying on interview-based techniques for clients with lower levels of education attainment and literacy; at least one study has demonstrated problems with readability and comprehensibility with most BED measures [56].
• What are the clinical features of BED?
BED and Obesity
The specific impact of BED on health is difficult to separate from the impact of obesity on health, as the two conditions frequently co-occur and are confounded in many studies. Of relevance to the primary care setting, many BED patients report gaining a substantial amount of weight in the year prior to seeking treatment [57].
Although individuals with BED are often obese, proponents of classifying BED as a separate DSM diagnosis argue that individuals with BED differ from their non-BED obese counterparts in regards to eating patterns, eating disordered psychopathology, and associated features and comorbidities. Individuals with BED consume more calories in laboratory studies than weight-matched controls [6,7,58]. In contrast, studies utilizing ecological momentary assessment (ie, real-time assessments) found no differences between BED obese and non-BED obese participants in the frequency of self-reported binge eating and caloric intake during binge eating episodes [59,60]. BED participants, however, were more likely to report higher stress, desire to binge, negative affect, dietary restraint, and being alone immediately before self-reported binge eating episodes. Furthermore, individuals with BED also demonstrate more ED-related psychopathology than non-BED obese individuals [61–63]. Psychiatric comorbidity is also higher among BED obese individuals as compared their non-BED obese counterparts, and the increased comorbidity is accounted for by the severity of binge eating as opposed to the severity of obesity [6,64–67]. In addition, research demonstrates that obese individuals with BED, as compared with non-obese BED patients, have a poorer quality of life [68].
BED and Bulimia Nervosa
Numerous studies have supported the distinction between bulimia nervosa and BED [69–76]. Diagnostically, bulimia nervosa differs from BED by its requirement of recurrent inappropriate compensatory behaviors in order to prevent weight gain, such as self-induced vomiting; misuse of laxatives, diuretics, or other medications; fasting; or excessive exercise [3]. BED and bulimia nervosa are distinguished by distinct risk factors, prevalence, course, and treatment outcomes [28,67,77]. Individuals with BED are less likely than individuals with bulimia to diet before onset of the disorder, and fewer individuals with BED cross over into other ED diagnostic categories [26,78–81]. Finally, BED and bulimia nervosa are associated with different constellations of ED-related symptoms and associated features [28,63,79]. For example, relative to BE patients, those with bulimia show greater work impairment and psychiatric comorbidity [28], higher dietary restraint and eating concerns [63], and lower rates of obesity [79].
Psychiatric Comorbidity
BED is associated with poor social adjustment, greater functional impairment, and significant psychiatric comorbidity, including overall distress and suicidality [67]. In a study of comorbidity with only selected disorders (mood, anxiety, impulse-control, and substance use disorder), 78.9% of individuals with BED had a lifetime history of at least one comorbidity, 20.2% had one comorbid disorder, 9.8% had two, and 48.9% had three or more [28]. Furthermore, the presence of current psychiatric comorbidity is associated with greater ED-related psychopathology and associated distress [40,41]. The most common comorbidities (lifetime rates) are specific phobia (37.1%), social phobia (31.9%), major depressive disorder (32.3%), post-traumatic stress disorder (PTSD) (26.3%), alcohol abuse/dependence (21.4%), conduct disorder (20%), attention-deficit/ hyperactivity disorder (19.8%), illicit drug use/dependence (19.4%), and oppositional-defiant disorder (18%) [28]. A recent report supports that this level of comorbidity is evident in primary care settings, noting that PTSD in particular is common and associated with a host of other difficulties, including depression, anxiety, drug use disorders, greater eating disorder pathology, and poorer psychological functioning [82]. Personality disorders are also commonly comorbid with BED, with the highest lifetime rates for avoidant (11%), obsessive compulsive (10%), and borderline (9%) personality disorders [83]. Finally, cigarette smoking is also associated with binge eating [83,84], likely evolving out of a weight-control smoking profile [85], and this is of relevance to the primary care setting in that smokers with BED gain more weight upon smoking cessation than do their non-BED counterparts [86].
Further Evaluation
To assess behavioral factors related to obesity and recent weight gain, the physician asks the patient if she ever eats what would be considered an unusually large amount of food for the circumstance. The patient acknowledges that she does so regularly, particularly in response to negative moods. The patient also describes that these episodes contribute to ongoing low mood, such that she feels highly depressed and hopeless following binge episodes. The physician then asks about the patient’s exercise habits and weight management techniques. While the patient denies engaging in compensatory behaviors (eg, vomiting, laxative use) to counteract excessive eating, she does report a history of dieting in which she dramatically restricts her food intake and subsequently loses weight. The patient states that these periods are inevitably followed by a resumption of overeating, and she typically gains back more weight than she originally lost. The patient estimates that she has lost and regained more than 20 lb at least 5 times during her lifetime. In addition, the patient reports difficulty maintaining a regular exercise regimen, especially since the onset of osteoarthritis-related joint pain in the past year. After the evaluation, the physician orders an electrocardiogram (ECG) and blood work. The ECG shows that the P-wave, QRS, and T-wave axes are shifted leftward, but within normal limits. A follow-up appointment is scheduled in 2 weeks.
• What are the medical complications of BED?
BED is associated with numerous negative health sequelae including obesity, sleeping problems, musculoskeletal pain, joint pain, headaches, gastrointestinal problems, menstrual problems, shortness of breath, chest pain, diabetes, low health-related quality of life, and functional health impairments [87–90], with many of these risks persisting even after controlling for BMI [91]. A 5-year follow-up of 134 individuals with BED and 134 individuals with no history of eating disorders, who were frequency-matched for age, sex, and baseline body mass index (BMI), provides further support that BED confers risk of components of metabolic syndrome beyond the risks associated with BMI alone [92]. Specifically, BED cases had higher longitudinal risk of developing dyslipidemia, hypertension, type 2 diabetes, any metabolic syndrome component, and two or more metabolic syndrome components. Alarmingly, these findings even emerge in studies of pediatric samples, wherein BED predicts development of metabolic syndrome, elevated triglycerides, and increases in visceral adiposity [93].
• What are risk factors for BED?
A number of risk factors for BED have been identified, although many are risk factors for a number of psychiatric disorders and not specific to BED. These general risk factors include depression/negative affectivity [94,95], parental mood and substance use disorder, maternal problematic parenting, and separation from parents [95]. A host of risk factors have been identified for disordered eating, in general, including body dissatisfaction [94], early onset of dieting [94], and perfectionism [96]. A number of other variables are risk factors for both BED and bulimia (but not anorexia), including a history of childhood bully and teasing, negative self-evaluation, parental depression, and negative family communication about shape and weight [81,96]. In a study comparing BED cases to psychiatric controls, childhood obesity, familial eating problems, family discord, and high parental demands differentiated the BED cases [95]. In summary, it has been suggested that BED risk is conferred by factors that increase risk of psychiatric disorder in general and those that confer risk for obesity [81]. Of note, the risk factors studied do not appear to differ between black and white women [95].
Genetic risk factors appear to play a strong role in the development of BED. Risk for BED tends to aggregate in families independently of the risk for obesity, although the presence of BED in a first-degree relative does increase risk for obesity [97]. Heritability estimates for BED range from 45% to 57% [98,99], which is greater than the heritability estimate for subthreshold binge eating (ie, overeating with a sense of loss of control, 41%) [100]. In addition, symptom-level analyses support moderate genetic contributions for each BED symptom [98], supporting the integrity of the diagnostic criteria. Finally, shared environment appears to play a very small role in the familial transmission of BED, and the contribution of unique environmental factors in development of BED appears to be substantial [97,101].
With regard to the neurobiological underpinnings of BED, it appears that BED may be associated with hypersensitivity to reward, a phenomenon that is strongly associated with the striatum and dopaminergic mechanisms [102,103]. In support of this hypothesis, Davis et al [102] reported that BED was differentially related to genotypes that reflect a greater density of D2 receptors and higher D2 binding potential as compared to obese controls. Additionally, greater increases in striatal DA and unique activation patterns in the right ventral striatum have been demonstrated in individuals with BED as compared to obese non-BED controls in response to food-related stimuli [103,104]. Other findings have implicated the orbitofrontal cortex (OFC) in BED, which is another brain region responsible for reward processing, particularly as it relates to the hedonic value of food stimuli [103]. Increased volume of grey matter has been documented in individuals with BED and bulimia as compared to normal weight controls, and stronger medial OFC activation while viewing pictures of food was observed in individuals with BED as compared to individuals with bulimia, overweight controls, and normal controls [105].
Difficulties with affect regulation have also been implicated in the development of BED. Two theories that implicate a primary and specific role for affect regulation in BED are cited most frequently in the extant literature: the affect regulation theory and the escape theory. The affect regulation theory [106] posits that BE is a conditioned response to negative affect which is correspondingly negatively reinforced by reductions in negative affect, which could occur during or after BE. Escape theory [107] posits that aversive self-awareness causes negative affect, which in turn triggers BE. BE is then negatively reinforced by reductions in negative affect during a binge via an escape from self-awareness that is accomplished through cognitive narrowing to the immediate stimulus environment. In contrast to the affect regulation theory, escape theory predicts that negative affect will increase after BE when self-awareness is restored. Results regarding changes in affect during BE episodes are conflicting as to whether BE is associated with decreases, no change [108–110], or even increases in negative affect. In particular, a meta-analytic review of 36 studies that examined affect via ecological momentary sampling found moderate increases in negative affect following binge episodes [111]. To some degree, results of this meta-analysis may not generalize to BED, per se, given that it included other binge eating groups, such as those with bulimia nervosa. However, in general, studies suggest that negative affect is an antecedent for BE and increased negative affect may be a consequence of BE, at least among women. More information is needed regarding aversive self-awareness before and after BE, cognitive narrowing, and changes in affect during BE. As such, the current state of the literature provides only partial support for affect regulation models of BED in women. Furthermore, it remains unknown if these results will generalize to men.
Clinical Course
Evidence regarding the course and stability of BED is conflicting and unclear. Several prospective studies have suggested that BED is not a stable disorder, exhibiting high rates of remission over time [26,99,112]. However, the samples have been criticized for being small, completely female, younger than typical individuals with BED, and post-ED treatment. In contrast, a prospective study that included older women and a combination of treated and untreated women suggested remission rates at 1 year that were much lower (7%) [78]. Additionally, a retrospective study [113] reported an average BED duration of 14.4 years. In a review of the studies cited above, Wonderlich et al [6] concluded that “[a]lthough there is variability in the data, it does appear that BED differs from other eating disorders in terms of a greater tendency toward recovery and fluctuation, although this may be embedded in a chronic pattern of remission and relapse.” Viewing BED as a disorder with a chronic pattern of remission and relapse could explain why individuals with BED retrospectively report a longer duration of illness, as they may be more likely to conceptualize their illness as one continuous course punctuated by different periods of severity rather than several distinct bouts of BED. Finally, although diagnostic crossover is a frequent phenomenon among other eating disorders, the crossover rate for BED appears relatively low as compared to anorexia and bulimia [6,26,28,66].
Follow-up
Laboratory examination shows TSH levels within normal limits and cholesterol levels of 48 mg/dL(HDL), 162 mg/dL (LDL), and 270 mg/dL (total). Triglyceride levels are 300 mg/dL and the patient’s fasting glucose level is 115 mg/dL. At the patient’s follow-up appointment, the physician states that a number of laboratory results indicated negative weight-related health consequences, including high cholesterol, high triglycerides, hypertension, and probable pre-diabetes. The patient initially disregards the significance of these results, stating she only gained weight due to her break-up and quitting smoking, and she is motivated to diet to lose weight in the near future. The physician asks for more information about the patient’s eating behavior, in particular asking if she ever feels as if she loses control over her eating. The patient reluctantly admits to this, and the physician provides a referral to a behavioral health specialist. The patient expresses ambivalence and a desire to try to manage her weight on her own. The physician uses motivational interviewing techniques to enhance motivation to follow up on this referral. In addition, the patient is encouraged to make small changes to her diet and slowly increase her exercise by taking walks. Another follow-up appointment is scheduled in 3 months.
• Which treatments are most effective for BED?
Despite the negative sequalae of BED, studies suggest that it often goes untreated [114]. Women with BED, as compared to women with anorexia and bulimia, are less likely to seek treatment for BED and less likely to receive treatment for their eating disorder when they do seek it out [114–116]. Barriers to treatment may include shame and internalized weight stigma, lack of knowledge about where to seek treatment, a belief that willpower should be sufficient to overcome the problem, lack of understanding that BED is a psychiatric disorder, finances/insurance barriers, and lack of BED detection by non-specialist treatment providers [115]. These barriers are particularly concerning, as women with BED report greater health care utilization and comprise a large segment of patients in weight control programs. Therefore, it appears individuals with BED seek help for the negative consequences of the disorder, but they are less likely to seek and receive help for the likely root cause of their concerns. This is a particularly damaging pattern, as the presence of BED may negatively impact the outcome of obesity treatment [117]. There are, however, a number of promising treatments for BED, as described below:
Cognitive Behavioral Therapy
Cognitive behavioral therapy (CBT) is generally considered to be the most well-established and empirically supported treatment for BED [118,119]. The cognitive behavioral conceptualization of BED is based on Fairburn, Cooper, and Shafran’s [120] transdiagnostic model of eating disorders (CBT-E), which is an expanded version of the cognitive behavioral model of bulimia nervosa [121]. CBT-E posits that the core pathology in eating disorders is a dysfunctional system in which self-worth is based on eating habits, shape, or weight, and the individual’s ability to control them. Attempts to maintain self-worth by controlling eating, shape, and weight result in extreme and brittle forms of dietary restraint. Inevitable violations of the individual’s dietary rules are then interpreted as lack of self-control, leading to a temporary abandonment of dietary restraint and consequent BE. These dietary slips and corresponding BE often occur in response to acute changes in mood, and BE is thus negatively reinforced by “neutralizing” negative mood states. Lapses in dietary restraint also result in secondary negative self-evaluation, which serves to further exacerbate a cycle of increased dietary restraint to improve self-worth and then inevitable dietary lapses leading to BE. CBT-E expanded upon CBT-BN by postulating 4 processes that maintain ED: severe perfectionism (clinical perfectionism), unconditional and pervasive low self-esteem (core low self-esteem), difficulties coping with intense mood states (mood intolerance), and developmental interpersonal difficulties (interpersonal difficulties). Of note, the CBT-E model explicitly states that individuals may differ in the extent to which they experience the 4 maintaining processes and not every individual will experience all four.
Overall, treatment is focused on normalizing eating patterns (ie, not weight loss), cognitive restructuring for weight/shape concerns and other triggers for binge eating, and relapse prevention [122]. CBT has produced substantial reductions in binge eating as compared to no treatment [123–125] and supportive therapy [126]. The majority of RCTs have reported remission rates greater than 50% [127]. Unfortunately, CBT has generally not produced meaningful weight loss [118,122,127–129], but this may be a contraindicated goal. CBT has demonstrated improvements in a number of features associated with BED including eating disordered psychopathology [122,124,130,131], depression [122,124,130,132], social adjustment [133], and self-esteem [132]. Treatment gains are generally well-maintained at 1-year to 4-year follow-up [122,123,130,133,134]. Individual and group treatments appear to produce similar results [134], and treatment completion rates have been estimated at approximately 80% across different delivery formats [127]. One strength of the CBT literature is the inclusion of participants with severe psychopathology, which facilitates the generalizability of these findings [127].
A number of factors have been associated with treatment outcome in CBT trials. Poor treatment outcomes have been associated with a history of weight problems during childhood, high levels of emotional eating at baseline, interpersonal dysfunction, and low group cohesion during group CBT [110,124,134]. Overvaluation of weight and shape demonstrated a statistical trend toward negatively impacting outcomes in one study. The presence of a cluster B personality disorder (ie, borderline histrionic, antisocial, and narcissistic personality disorders) predicted higher levels of binge eating at 1-year follow-up in a combined sample of participants treated with group CBT or group interpersonal psychotherapy (IPT) [135].
Alternatively, positive treatment outcomes have been associated with low levels of emotional eating at baseline, older age of onset, weight loss history that is negative for amphetamine use, and decreases in depressive symptoms during treatment [124,134,136,137]. In addition, early response to treatment (defined as a 65%–70% reduction in binge eating within 4 weeks of starting treatment) tends to be associated with greater long-term (ie, 1–2 year) remission from BED and lower eating disorder psychopathology, across a variety of psychological treatment approaches [138–144].
Interpersonal Psychotherapy
IPT for BED was adapted by Wilfley and colleagues [145] from IPT for depression, and the rationale for its use with BED is based on successful outcomes for individuals with bulimia and multiple studies documenting interpersonal deficits in individuals with BED [146]. IPT seeks to address interpersonal problems in 4 areas: interpersonal conflict, grief, role transitions, and interpersonal deficits [135]. While adapting IPT for BED, it was noted that the course of BED tends to be more chronic than the course of depression, thus the focus of IPT for BED was shifted from addressing the interpersonal precipitants of the disorder to the interpersonal factors that maintain the disorder [145]. Fewer studies examining the effectiveness of IPT in treating BED have been published than those examining CBT for BED, but it appears that IPT is as efficacious as CBT immediately post-treatment [130], and at 1- [130] and 4-year follow-up [147]. In addition, at least 2 studies have been published that compare IPT, cognitive behavioral therapy–guided self-help (CBTgsh), and behavioral weight loss [133,141]. Overall, results support the use of both IPT and CBTgsh (discussed in more detail below), with important moderators of treatment effects observed. For example, Wilson et al [133] found that clients with higher levels of psychopathology were better suited for IPT. The authors conclude that these results could inform a model of evidence-based stepped care, where CBTgsh, a low-cost, low-intensity treatment, should be considered as the first line of treatment. Secondarily, IPT, which represents a more specialized and expensive form of treatment, could be considered the next level of care, particularly for clients who are not demonstrating rapid improvement in response to CBTgsh.
Dialectic Behavior Therapy
A small number of studies have investigated the treatment of BED with dialectical behavior therapy (DBT). Originally developed to treat borderline personality disorder [148], DBT is of particular interest given its explicit targeting of emotion regulation. According to the DBT model of BED [149], emotional dysregulation is the core psychopathology in this disorder, and binge eating is viewed as attempts to influence, change, or control painful emotions. Initially, promising results were published showing positive treatment effects in an uncontrolled study [150] as well as wait-list controlled trials [151]. Notably, relative to wait-list controls, participants in a DBT guided self-help program (who received an orientation, DBT manual, and six 20-minute support calls across 13 weeks) reported reduced past-month binge eating, higher binge eating abstinence rates, and over the longer term improved quality of life and reductions in ED psychopathology. However, a comparison of DBT-BED with an active comparison control group (ie, nonspecific supportive therapy) failed to find significant differences between the 2 treatments (defined as effect size greater than 0.5) at 12-month follow-up in binge eating abstinence, binge eating frequency, most ED-related psychopathology, positive affect, depression, and self-esteem [152]. Therefore, DBT may have potential and, at a minimum, is equally efficacious as supportive therapy.
Mindfulness- and Meditation-Based Therapies
Treatment outcome studies utilizing mindfulness-based therapies, including mindfulness-based stress reduction (MBSR) and acceptance and commitment therapy (ACT), make up a small but promising body of literature. Reasoning that negative affect, eating in the absence of hunger, and emotional eating may comprise one pathway to binge eating [153,154], it follows that mindfulness-based therapies may act through their effects on emotion regulation, acceptance strategies for tolerating negative affect, and awareness of bodily cues. A recent review identified 19 studies exploring the effects of mindfulness-based interventions on binge eating severity and frequency as well as a number of related indicators, observing positive effects for this form of treatment [155]. For example, MB-EAT [156] is a group treatment for BED that is primarily based on MBSR. Treatment is targeted at cultivating mindfulness, mindful eating, emotional balance, and self-acceptance[157]. The treatment also places particular emphasis on developing self-awareness of internal hunger and satiety cues. A recent randomized controlled trial of MB-EAT produced significant improvements in binge eating frequency and BE-related psychopathology [158]. Furthermore, process variables including hunger awareness, satiety awareness, and mindfulness were correlated with positive outcomes. In addition, a small study (n = 39) that compared ACT to standard follow-up utilized by a bariatric surgery team demonstrated significantly greater improvements in disordered eating, body satisfaction, and quality of life for clients who participated in ACT [159]. In brief, results suggest that mindfulness-based interventions represent an additional treatment approach with supporting but limited evidence to date.
Self-Help Interventions
Self-help interventions for BED are categorized as pure self-help or guided self-help. In treatment outcome studies, pure self-help is generally conducted with a self-help manual, although several studies have examined more novel formats such as the internet, video, and CD-ROM. GSH also uses a self-help manual (or other format) with the addition of brief sessions with health care providers who have varying degrees of expertise with the type of therapy being utilized. CBT is the most commonly utilized therapeutic modality in treatment outcome studies of self-help interventions, and they most often utilize Fairburn’s Overcoming Binge Eating self-help manual [160].
Two studies have directly compared pure and guided self-help with Fairburn’s manual and produced conflicting results. Carter and Fairburn [161] found that in a sample of primarily white women with BED, pure self-help (CBTsh; n = 24) and guided self-help (CBTgsh; n = 24) were equally effective, and both were superior to wait-list controls at 6-month follow-up in producing BE abstinence (CBTsh = 40%, CBTgsh = 50%), reducing binge eating, ED-related psychopathology, and general psychiatric symptoms. In contrast, a study comparing CBTsh and CBTgsh in 40 primarily white women with recurrent binge eating (82.5% diagnosed with BED), guided self-help was superior to pure self-help at the end of treatment in reducing BE frequency, eating concern, and restraint [162]. CBTgsh and CBTsh were equally effective in producing BE abstinence (50% and 30%, respectively), and reducing shape concern, weight concern, and general psychiatric symptoms [162]. Higher levels of general psychiatric symptoms were predictive of higher BE frequency post-treatment for both treatments. It should be noted that participants in both conditions experienced statistically significant improvements on all variables as compared to baseline.
CBTgsh also performed as well or better than individualized treatments in one study [133]. CBTgsh, IPT, and behavioral weight loss (BWL) were compared in a large study of 205 primarily white, obese or overweight individuals diagnosed with BED. The 3 treatments produced equivalent outcomes for binge eating at post-treatment, but BWL produced significantly greater weight loss. However, at 2-year follow-up, the CBTgsh and IPT groups had maintained treatment gains and were significantly superior to BWL in reductions in binge eating. The 3 groups were equivalent with regard to weight loss at the 2-year follow-up, and none reported clinically significant weight loss. Of note, as compared to the IPT and BWL groups, the CBTgsh group received 10 sessions as opposed to 20, received 25-minute sessions as opposed to 60-minute sessions, and were treated by providers with limited levels of experience as opposed to doctoral-level clinical psychologists.
To summarize, CBT is the most often studied type of self-help treatment. Both CBTsh and CBTgsh produced improvements in binge eating and associated psychopathology as compared to baseline and wait-list controls, and treatment gains were maintained at 6-month follow-up. Conclusions regarding the relative superiority of pure self-help or guided self-help are premature given the small number of studies and conflicting results.
In addition, limited information is available regarding moderators and predictors of guided self-help outcomes. Masheb and Grilo [163] performed a cluster analysis of the sample for the CBTgsh vs. BWLgsh described above [164] and identified 2 clinically significant subtypes: a dietary-negative affect subtype characterized by high restraint, low self-esteem, and depressive symptoms; and an overvaluation of weight and shape subtype. For both the CBTgsh and BWLgsh groups, the dietary-negative affect subtype experienced higher levels of binge eating frequency, and the overvaluation of weight and shape subtype experienced higher levels of ED-related psychopathology. Additionally, individuals receiving BWLgsh who experienced a rapid response to treatment reported lower BE frequency, greater weight loss, and higher restraint than participants without a rapid response [142]. In contrast, rapid response did not appear to affect outcomes for CBTgsh participants. Finally, the combination of low self-esteem and high ED-related psychopathology negatively affected BE remission rates for CBTgsh recipients [133].
Pharmacologic Treatment
Currently only one medication, lisdexamfetamine dimesylate, has been FDA-approved for the treatment of BED. Previously approved for treating both adults and children with attention-deficit hyperactivity disorder, lisdexamfetamine dimesylate is a central nervous system stimulant and has been found to significantly reduce number of binge days, with robust effect sizes [165]. Beyond this medication, the evidence for pharmacologic treatment of BED is limited. A recent review identified only 22 studies exploring the effects of pharmacologic treatment in a methodologically rigorous way (eg, double-blind placebo design) [4]. To date, a number of different medication classes have been evaluated, including antidepressants, anticonvulsants, stimulants, anti-obesity drugs, and others. Overall, there is some evidence that antidepressant and anticonvulsant agents are efficacious at reducing BE frequency [166,167] and sometimes effective regarding statistically significant weight loss [168,169]. However, the majority of results are generally disappointing, both with respect to reductions in binge eating and sustained weight loss [48,170,171]. In addition, there are serious limitations in the literature that must be considered, including the limited number of studies that address the high placebo response observed in clinical samples, limited follow-up windows, and inadequate multiplicitious confirmatory trials. Despite these limitations, the evidence base related to pharmacologic treatment is continuously evolving and represents an important future direction for the treatment of BED.
Treatment
Prior to her next medical follow-up, the patient meets with a psychologist. The patient discloses that she has been binge eating several times per week for over a year; she also discloses a history of prolonged sexual abuse perpetrated by a step-parent during her childhood. When the patient returns to her follow-up medical appointment, she reports that her psychologist has diagnosed her with BED and PTSD. She states that they are using cognitive behavioral techniques to regulate her mood and eating behavior, with a specific aim of avoiding excessive dietary restraint. In addition, they are working together to discuss her unfulfilling romantic history and processing her experiences of trauma. Since her last appointment with the primary care physician, she reports an increased awareness of her eating habits, improvement in mood, and a 10-lb decrease in her weight.
The patient reports that she has continued to meet weekly with her psychologist and has slowly begun reintroducing low-impact exercise to her routine. She continues to lose weight gradually, but with a priority of stabilizing eating behavior and avoiding binge episodes versus aiming for weight loss. She reports that her mood has stabilized. Her cholesterol and triglycerides remain high, but her blood pressure is controlled effectively with medication. Her physician recommends continued psychological treatment, periodic meetings with a nutritionist, and prescribes medication for her cholesterol. A follow-up appointment with her physician is scheduled in 6 months.
Summary
Corresponding author: Karen K. Saules, PhD, Eastern Michigan University, Psychology Clinic, 611 W. Cross St., Ypsilanti, MI 48197, [email protected]
Financial disclosures: None.
1. Stunkard AJ. Eating patterns and obesity. Psychiatr Q 1959;33:284–95.
2. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Text revision. Washington (DC): The Association; 2000.
3. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: The Association; 2013.
4. Reas D, Grilo C. Pharmacological treatment of binge eating disorder: update review and synthesis. Hum Psychopharmacol Clin Exp 2015;23:1463–78.
5. Wilson G. Treatment of binge eating disorder. Psychiatr Clin North Am 2011;34:773–83.
6. Wonderlich SA, Gordon KH, Mitchell JE, et al. The validity and clinical utility of binge eating disorder. Int J Eat Disord 2009;42:687–705.
7. Walsh BT, Boudreau G. Laboratory studies of binge eating disorder. Int J Eat Disord 2003;34 Suppl:S30–8.
8. Guss JL, Kissileff HR, Devlin MJ, et al. Binge size increases with body mass index in women with binge-eating disorder. Obes Res 2002;10:1021–9.
9. Fairburn CG, Cooper Z. The eating disorder examination. In: Fairburn CG, Cooper Z, editors. Binge eating: nature, assessment, and treatment. New York: Guilford Press;1993:317–60.
10. Keel PK, Mayer SA, Harnden-Fischer JH. Importance of size in defining binge eating episodes in bulimia nervosa. Int J Eat Disord 2001;29:294–301.
11. Niego SH, Pratt EM, Agras WS. Subjective or objective binge: is the distinction valid? Int J Eat Disord 1997;22: 291–8.
12. Pratt E, Niego S, Agras W. Does the size of a binge matter? Int J Eat 1998;24:307–12.
13. Wolfe BE, Baker CW, Smith AT, et al. Validity and utility of the current definition of binge eating. Int J Eat Disord 2009;42:674–86.
14. Latner JD, Clyne C. The diagnostic validity of the criteria for binge eating disorder. Int J Eat Disord 2008;41:1–14.
15. Johnson WG, Boutelle KN, Torgrud L. What is a binge? The influence of amount, duration, and loss of control criterial on judgements of binge eating. Int J Eat Disord 2000;27:471–9.
16. Johnson WG, Roberson-NR, Rohan KJ, et al. An experimental investigation of DSM-IV binge-eating criteria. Eat Behav 2003;4:295–303.
17. Reslan S, Saules KK. College students’ definitions of an eating ‘binge’ differ as a function of gender and binge eating disorder status. Eat Behav 2011;12:225–7.
18. Telch CF, Pratt EM, Niego SH. Obese women with binge eating disorder define the term binge. Int J Eat Disord 1998;24:313–7.
19. Colles SL, Dixon JB, O’Brien PE. Loss of control is central to psychological disturbance associated with binge eating disorder. Obesity (Silver Spring) 2008;16:608–14.
20. Stein KF, Kenardy J, Wiseman C V, et al. What’s driving the binge in binge eating disorder? A prospective examination of precursors and consequences. Int J Eat Disord 2007;40:195–203.
21. Rossiter EM, Agras WS, Telch CF, et al. The eating patterns of non-purging bulimic subjects. Int J Eat Disord 1992;11:111–120.
22. Grilo CM, White MA. A controlled evaluation of the distress criterion for binge eating disorder. J Consult Clin Psychol 2011;79:509–14.
23. Trace SE, Thornton LM, Root TL, et al. Effects of reducing the frequency and duration criteria for binge eating on lifetime prevalence of bulimia nervosa and binge eating disorder: implications for DSM-5. Int J Eat Disord 2012;45: 531–6.
24. Wilson GT, Sysko R. Frequency of binge eating episodes in bulimia nervosa and binge eating disorder: diagnostic considerations. Int J Eat Disord 2009;42:603–10.
25. Sysko R, Roberto CA, Barnes RD, et al. Test-retest reliability of the proposed DSM-5 eating disorder diagnostic criteria. Psychiatry Res 2012;196:302–8.
26. Fairburn CG, Cooper Z, Doll HA, et al. The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiatry 2000;57:659–65.
27. Grilo CM, Ivezaj V, White MA. Evaluation of the DSM-5 severity indicator for binge eating disorder in a community sample. Behav Res Ther 2015;66:72–6.
28. Hudson JI, Hiripi E, Pope HG, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry 2007;61:348–58.
29. Stice E, Marti CN, Rohde P. Prevalence, incidence, impairment, and course of the proposed DSM-5 eating disorder diagnoses in an 8-year prospective community study of young women. J Abnorm Psychol 2012;122:445–57.
30. Mangweth-Matzek B, Hoek HW, Rupp CI, et al. Prevalence of eating disorders in middle-aged women. Int J Eat Disord 2014;47:320–4.
31. Ivezaj V, Saules KK, Hoodin F, et al. The relationship between binge eating and weight status on depression, anxiety, and body image among a diverse college sample: a focus on bi/multiracial women. Eat Behav 2010;11:18–24.
32. Striegel-Moore RH, Bedrosian R, Wang C, et al. Why men should be included in research on binge eating: results from a comparison of psychosocial impairment in men and women. Int J Eat Disord 2012;45:233–40.
33. Womble LG, Williamson DA, Martin CK, et al. Psychosocial variables associated with binge eating in obese males and females. Int J Eat Disord 2001;30:217–21.
34. Udo T, McKee SA., White MA., et al. Sex differences in biopsychosocial correlates of binge eating disorder: a study of treatment-seeking obese adults in primary care setting. Gen Hosp Psychiatry 2013;35:587–91.
35. Udo T, McKee SA., White MA, et al. Menopause and metabolic syndrome in obese individuals with binge eating disorder. Eat Behav 2014;15:182–5.
36. Napolitano MA., Himes S. Race, weight, and correlates of binge eating in female college students. Eat Behav 2011;12:29–36.
37. Sorbara M, Geliebter A. Body image disturbance in obese outpatients before and after weight loss in relation to race, gender, binge eating, and age of onset of obesity. Int J Eat Disord 2002;31:416–23.
38. Alegria M, Woo M, Cao Z, et al. Prevalence and correlates of eating disorders in Latinos in the U.S. Int J Eat Disord 2007;40:S15–21.
39. Striegel-Moore RH, Wilfley DE, Pike KM, et al. Recurrent binge eating in black American women. Arch Fam Med 2000;9: 83–7.
40. Reslan S, Saules KK. Assessing the prevalence of and factors associated with overweight, obesity, and binge eating as a function of ethnicity. Eat Weight Disord-St 2013;18:209–19.
41. Grilo CM, White MA, Barnes RD, et al. Psychiatric disorder co-morbidity and correlates in an ethnically diverse sample of obese patients with binge eating disorder in primary care settings. Compr Psychiatry 2012;54:209–16.
42. Smolak L, Striegel Moore RH. Challenging the myth of the golden girl: ethnicity and eating disorders. In: Smolak L, Striegel Moore RH, editors. Eating disorders innovative directions in research and practice. Washington (DC): American Psychological Association;2001:111–32.
43. Wildes JE, Emery RE. The role of ethnicity and culture in the development of eating disturbance and body dissatisfaction: a meta-analutic review. Clin Phychology Rev 2001;21:521–50.
44. George JBE, Franko DL. Cultural issues in eating pathology and body image among children and adolescents. J Pediatr Psychol 2010;35:231–42.
45. Kelly NR, Cotter EW, Tanofsky-Kraff M, et al. Racial variations in binge eating, body image concerns, and compulsive exercise among men. Psychol Men Masc 2014;13:326-36.
46. Franko DL, Thompson-Brenner H, Thompson DR, et al. Racial/ethnic differences in adults in randomized clinical trials of binge eating disorder. J Consult Clin Psychol 2012;80:186–95.
47. Marques L, Alegria M, Becker AE, et al. Comparative prevalence, correlates of impairment, and service utilization for eating disorders across U.S. ethnic groups: Implications for reducing ethnic disparities in health care access for eating disorders. Int J Eat Disord 2012;44:412–20.
48. Guerdjikova AI, O’Melia AM, Mori N, et al. Binge eating disorder in elderly individuals. Int J Eat Disord 2012;45: 905–8.
49. Fairburn CG. Beglin SJ. Assessment of eating disorders: interview or self-report questionnaire? Int J Eat Disord 1994;16:363–70.
50. Spitzer R, Yanovski S, Marcus M. The questionnaire on eating and weight patterns-revised (QEWP-R). New York State Psychiatric Institute, Mew York 1993.
51. Barnes RD, Masheb RM, White MA, et al. Comparison of methods for identifying and assessing obese patients with binge eating disorder in primary care settings. Int J Eat Disord 2011;44:157–63.
52. Yanovski SZ, Marcus MD, Wadden TA, et al. The questionnaire on eating and weight patterns-5: an updated screening instrument for binge eating disorder. Int J Eat Disord 2015;48:259–61.
53. Gormally J, Black S, Daston S, et al. The assessment of binge eating severity among obese persons. Addict Behav 1982;7:47–55.
54. Alfonsson S. Replacing the term ‘binge eating’ with ‘loss of control over eating’ affects eating disorder screening in clinical care. Obes Res Clin Pract 2015;6–7.
55. Kelly NR, Mitchell KS, Gow RW, et al. An evaluation of the reliability and construct validity of eating disorder measures in white and black women. Psychol Assess 2012;24:608–17.
56. Richards LK, McHugh RK, Pratt EM, et al. Readability and comprehension of self-report binge eating measures. Eat Behav 2013;14:167–70.
57. Masheb RM, White MA, Grilo CM. Substantial weight gains are common prior to treatment-seeking in obese patients with binge eating disorder. Compr Psychiatry 2013;54:880–4.
58. Heaner MK, Walsh BT. A history of the identification of the characteristic eating disturbances of Bulimia Nervosa, Binge Eating Disorder and Anorexia Nervosa. Appetite 2013;65:185–8.
59. Greeno CG, Wing RR, Shiffman S. Binge antecedents in obese women with and without binge eating disorder. J Consult Clin Psychol 2000;68:95–102.
60. Le Grange D, Gorin A, Catley D, et al. Does momentary assessment detect binge eating in overweight women that is denied at interview? Eur Eat Disord Rev 2001;9:309–24.
61. Brody ML, Walsh BT, Devlin MJ. Binge eating disorder: reliability and validity of a new diagnostic category. J Consult Clin Psychol 1994;62:381–6.
62. Hsu LKG, Mulliken B, McDonagh B, et al. Binge eating disorder in extreme obesity. Int J Obes Relat Metab Disord 2002;26:1398–403.
63. Wilfley DE, Schwartz MB, Spurrell EB, et al. Using the eating disorder examination to identify the specific psychopathology of binge eating disorder. Int J Eat Disord 2000;27:259–69.
64. Grucza RA, Przybeck TR, Cloninger CR. Prevalence and correlates of binge eating disorder in a community sample. Compr Psychiatry 2007;48:124–31..
65. Telch CF, Stice E. Psychiatric comorbidity in women with binge eating disorder: prevalence rates from a non-treatment-seeking sample. J Consult Clin Psychol 1998;66:768–76.
66. Wilfley DE, Bishop ME, Wilson GT, et al. Classification of eating disorders: toward DSM-V. Int J Eat Disord 2007; 40:s123–9.
67. Wilfley DE, Wilson GT, Agras WS. The clinical significance of binge eating disorder. Int J Eat Disord 2003;34 Suppl:S96–106.
68. Perez M, Warren CS. The relationship between quality of life, binge-eating disorder, and obesity status in an ethnically diverse sample. Obesity (Silver Spring) 2012;20:879–85.
69. Bulik CM, Sullivan PF, Kendler KS. An empirical study of the classification of eating disorders. Am J Psychiatry 2000;157:886–95.
70. Keel PK, Heatherton TF, Dorer DJ, et al. Point prevalence of bulimia nervosa in 1982, 1992, and 2002. Psychol Med 2006;36:119–27.
71. Mitchell JE, Crosby RD, Wonderlich SA, et al. Latent profile analysis of a cohort of patients with eating disorders not otherwise specified. Int J Eat Disord 2007;40:s95–8.
72. Rockert W, Kaplan AS, Olmsted MP. Eating disorder not otherwise specified : the view from a tertiary care treatment center. Int J Eat Disord 2007;40:s99–103.
73. Striegel-Moore RH, Franko DL, Thompson D, et al. An empirical study of the typology of bulimia nervosa and its spectrum variants. Psychol Med 2005;35:1563–72.
74. Sullivan PF, Bulik CM, Kendler KS. The epidemiology and classification of bulimia nervosa. Psychol Med 1998;28:599–610.
75. Wade TD, Crosby RD, Martin NG. Use of latent profile analysis to identify eating disorder phenotypes in an adult Australian twin cohort. Arch Gen Psychiatry 2006;63:1377–84.
76. Striegel-Moore RH, Franko DL. Should binge eating disorder be included in the DSM-V? A critical review and state of the evidence. Annu Rev Clin Psychol 2008;4:305–24.
77. Striegel-Moore RH, Cachelin FM. Etiology of eating disorders in women. Couns Psychol 2001;29:635–61.
78. Crow SJ, Stewart Agras W, Halmi K, et al. Full syndromal versus subthreshold anorexia nervosa, bulimia nervosa, and binge eating disorder: a multicenter study. Int J Eat Disord 2002;32:309–18.
79. Striegel-Moore RH, Cachelin FM, Dohm F a, et al. Comparison of binge eating disorder and bulimia nervosa in a community sample. Int J Eat Disord 2001;29:157–65.
80. Wilfley DE, Schwartz MB, Spurrell EB, et al. Assessing the specific psychopathology of binge eating disorder patients: Interview or self-report? Behav Res Ther 1997;35:1151–9.
81. Fairburn CG, Doll HA, Welch SL, et al. Risk factors for binge eating disorder. Arch Gen Psychiatry 1998;55:425–32.
82. Grilo CM, White M a., Barnes RD, et al. Posttraumatic stress disorder in women with binge eating disorder in primary care. J Psychiatr Pract 2012;18:408–12.
83. Cassin SE, von Ranson KM. Is binge eating experienced as an addiction? Appetite 2007;49:687–90.
84. Kelly-Weeder S, Jennings KM, Wolfe BE. Gender differences in binge eating and behavioral correlates among college students. Eat Weight Disord 2012;17:e200–2.
85. Pomerleau CS, Ehrlich E, Tate JC, et al. The female weight-control smoker: a profile. J Subst Abuse 1993;5:391–400.
86. White MA, Masheb RM, Grilo CM. Function of binge eating behavior. Int J Eat Disord 2010;43:572–75.
87. Bulik CM, Reichborn-Kjennerud T. Medical morbidity in binge eating disorder. Int J Eat Disord 2003;34:S39–46.
88. Johnson JG, Spitzer RL, Williams JBW. Health problems, impairment and illnesses associated with bulimia nervosa and binge eating disorder among primary care and obstetric gynaecology patients. Psychol Med 2001;31.
89. Mond JM, Hay PJ, Rodgers B, et al. Assessing quality of life in eating disorder patients. Qual Life Res 2005;14:171–8.
90. Marchesini G, Natale S, Chierici S, et al. Effects of cognitive-behavioural therapy on health-related quality of life in obese subjects with and without binge eating disorder. Int J Obes Relat Metab Disord 2002;26:1261–7.
91. Reichborn-Kjennerud T, Bulik CM, Sullivan PF, et al. Psychiatric and medical symptoms in binge eating in the absence of compensatory behaviors. Obes Res 2004;12:1445–54.
92. Hudson JI, Lalonde JK, Coit CE, et al. Longitudinal study of the diagnosis of components of the metabolic syndrome in individuals with binge-eating disorder. Am J Clin Nutr 2010;91:1568–73.
93. Tanofsky-Kraff M, Shomaker LB, Stern EA, et al. Children’s binge eating and development of metabolic syndrome. Int J Obes 2012;36:956–62.
94. Stice E, Marti CN, Durant S. Risk factors for onset of eating disorders: evidence of multiple risk pathways from an 8-year prospective study. Behav Res Ther 2011;49:622–7.
95. Striegel-Moore RH, Fairburn CG, Wilfley DE, et al. Toward an understanding of risk factors for binge-eating disorder in black and white women: a community-based case-control study. Psychol Med 2005;35:907–17.
96. Hilbert A, Pike KM, Goldschmidt AB, et al. Risk factors across the eating disorders. Psychiatry Res 2014;220:500–6.
97. Hudson JI, Lalonde JK, Berry JM, et al. Binge-eating disorder as a distinct familial phenotype in obese individuals. Arch Gen Psychiatry 2006;63:313–19.
98. Mitchell KS, Neale MC, Bulik CM, et al. Binge eating disorder: a symptom-level investigation of genetic and environmental influences on liability. Psychol Med 2010;40:1899–906.
99. Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry 2008;69:266–73.
100. Reichborn-Kjennerud T, Bulik CM, Tambs K, et al. Genetic and environmental influences on binge eating in the absence of compensatory behaviors: a population-based twin study. Int J Eat Disord 2004;36:307–14.
101. Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry 2008;69:266–73.
102. Davis C, Levitan RD, Yilmaz Z, et al. Binge eating disorder and the dopamine D2 receptor: genotypes and sub-phenotypes. Prog Neuropsychopharmacol Biol Psychiatry 2012;38:328–35.
103. Weygandt M, Schaefer A, Schienle A, et al. Diagnosing different binge-eating disorders based on reward-related brain activation patterns. Hum Brain Mapp 2012;33:2135–46.
104. Wang G-J, Geliebter A, Volkow ND, et al. Enhanced striatal dopamine release during food stimulation in binge eating disorder. Obesity (Silver Spring) 2011;19:1601–8.
105. Schäfer A, Vaitl D, Schienle A. Regional grey matter volume abnormalities in bulimia nervosa and binge-eating disorder. Neuroimage 2010;50:639–43.
106. Hawkins RC, Clement PF. Binge eating: measurement problems and a conceptual model. In: Fremouw WJ, Clement PF, editors. The binge purge syndrome: diagnosis, treatment, and research. New York: Springer;1984:229–51.
107. Heatherton TF, Baumeister RF. Binge eating as escape from self-awareness. Psychol Bull 1991;110:86–108.
108. Deaver CM, Miltenberger RG, Smyth J, et al. An evaluation of affect and binge eating. Behav Modif 2003;27: 578–99.
109. Stickney MI, Miltenberger RG, Wolff G. A descriptive analysis of factors contributing to binge eating. J Behav Ther Exp Psychiatry 1999;30:177–89.
110. Hilbert A, Saelens BE, Stein RI, et al. Pretreatment and process predictors of outcome in interpersonal and cognitive behavioral psychotherapy for binge eating disorder. J Consult Clin Psychol 2007;75:645–51.
111. Haedt-Matt AA, Keel PK. Revisiting the affect regulation model of binge eating: a meta-analysis of studies using ecological momentary assessment. Psychol Bull 2012;137:660–81.
112. Cachelin FM, Striegel-Moore RH, Elder K a, et al. Natural course of a community sample of women with binge eating disorder. Int J Eat Disord 1999;25:45–54.
113. Pope HG, Lalonde JK, Pindyck LJ, et al. Binge eating disorder: a stable syndrome. Am J Psychiatry 2006;163:2181–3.
114. Pike KM, Dohm F a, Striegel-Moore RH, et al. A comparison of black and white women with binge eating disorder. Am J Psychiatry 2001;158:1455–60.
115. Cachelin FM, Striegel-Moore RH. Help seeking and barriers to treatment in a community sample of Mexican American and European American women with eating disorders. Int J Eat Disord 2006;39:154–61.
116. Cachelin FM, Rebeck R, Veisel C, et al. Barriers to treatment for eating disorders among ethnically diverse women. Int J Eat Disord 2001;30:269–78.
117. Blaine B, Rodman J. Responses to weight loss treatment among obese individuals with and without BED: a matched-study meta-analysis. Eat Weight Disord 2007;12:54–60.
118. Iacovino JM, Gredysa DM, Altman M, et al. Psychological treatments for binge eating disorder. Curr Psychiatry Rep 2012;14:432–46.
119. Wilson GT, Shafran R. Eating disorders guidelines from NICE. Lancet 2005;365:79–81.
120. Fairburn CG, Cooper Z, Shafran R. Cognitive behaviour therapy for eating disorders: a ‘transdiagnostic’ theory and treatment. Behav Res Ther 2003;41:509–28.
121. Fairburn CG, Cooper Z, Cooper PJ. The clinical features and maintenance of bulimia nervosa. In: Brownell KD, Foreyt JP, editors. Handbook of eating disorders: physiology, psychology and treatment of obesity, anorexia and bulimia. New York: Guiliford;1986:389–404.
122. Grilo CM, Masheb RM, Wilson GT, et al. Cognitive-behavioral therapy, behavioral weight loss, and sequential treatment for obese patients with binge-eating disorder: a randomized controlled trial. J Consult Clin Psychol 2011;79:675–85.
123. Agras WS, Telch CF, Arnow B, et al. One-year follow-up of cognitive-behavioral therapy for obese individuals with binge eating disorder. J Consult Clin Psychol 1997;65:343–7.
124. Dingemans AE, Spinhoven P, van Furth EF. Predictors and mediators of treatment outcome in patients with binge eating disorder. Behav Res Ther 2007;45:2551–62.
125. Eldredge KL, Stewart Agras W, Arnow B, et al. The effects of extending cognitive-behavioral therapy for binge eating disorder among initial treatment nonresponders. Int J Eat Disord 1997;21:347–52.
126. Kenardy J, Mensch M, Bowen K, et al. Group therapy for binge eating in type 2 diabetes: a randomized trial. Diabet Med 2002;19:234–9.
127. Wilson GT, Grilo CM, Vitousek KM. Psychological treatment of eating disorders. Am Psychol 2007;62:199–216.
128. Munsch S, Meyer AH, Biedert E. Efficacy and predictors of long-term treatment success for cognitive-behavioral treatment and behavioral weight-loss-treatment in overweight individuals with binge eating disorder. Behav Res Ther 2012;50:775–8.
129. Brown TA, Keel PK. Current and emerging directions in the treatment of eating disorders. Subst Abuse 2012;6: 33–61.
130. Wilfley DE, Welch RR, Stein RI, et al. A randomized comparison of group cognitive-behavioral therapy and group interpersonal psychotherapy for the treatment of overweight individuals with binge-eating disorder. Arch Gen Psychiatry 2002;59:713–21.
131. Grilo CM, Masheb RM, Wilson GT. Efficacy of cognitive behavioral therapy and fluoxetine for the treatment of binge eating disorder: a randomized double-blind placebo-controlled comparison. Biol Psychiatry 2005;57:301–9.
132. Gorin AA, Le Grange D, Stone AA. Effectiveness of spouse involvement in cognitive behavioral therapy for binge eating disorder. Int J Eat Disord 2003; 33: 421–33.
133. Wilson GT, Wilfley DE, Agras WS, et al. Psychological treatments for binge eating disorder. Arch Gen Psychiatry 2010;67:94–101.
134. Ricca V, Castellini G, Mannucci E, et al. Comparison of individual and group cognitive behavioral therapy for binge eating disorder. a randomized, three-year follow-up study. Appetite 2010;55:656–65.
135. Wilfley DE, Friedman MA, Dounchis JZ, et al. Comorbid psychopathology in binge eating disorder: relation to eating disorder severity at baseline and following treatment. J Consult Clin Psychol 2000;68:641–9.
136. Castellini G, Mannucci E, Lo Sauro C, et al. Different moderators of cognitive-behavioral therapy on subjective and objective binge eating in bulimia nervosa and binge eating disorder: a three-year follow-up study. Psychother Psychosom 2012;81:11–20.
137. Grilo CM, Masheb RM, Crosby RD. Predictors and moderators of response to cognitive behavioral therapy and medication for the treatment of binge eating disorder. J Consult Clin Psychol 2012;80:897–906.
138. Grilo CM, Masheb RM, Wilson GT. Rapid response to treatment for binge eating disorder. J Consult Clin Psychol 2006;74:602–13.
139. Grilo CM, Masheb RM. Rapid response predicts binge eating and weight loss in binge eating disorder: findings from a controlled trial of orlistat with guided self-help cognitive behavioral therapy. Behav Res Ther 2007;45:2537–50.
140. Grilo CM, White MA, Wilson GT, et al. Rapid response predicts 12-month post-treatment outcomes in binge-eating disorder: theoretical and clinical implications. Psychol Med 2012;42:807–17.
141. Hilbert A, Hildebrandt T, Agras WS, et al. Rapid response in psychological treatments for binge eating disorder. 2015;83:649–654.
142. Masheb RM, Grilo CM. Rapid response predicts treatment outcomes in binge eating disorder: implications for stepped care. J Consult Clin Psychol 2007;75:639–44.
143. Safer DL, Joyce EE. Does rapid response to two group psychotherapies for binge eating disorder predict abstinence? Behav Res Ther 2011;49:339–45.
144. Zunker C, Peterson CB, Cao L, et al. A receiver operator characteristics analysis of treatment outcome in binge eating disorder to identify patterns of rapid response. Behav Res Ther 2010;48:1227–31.
145. Wilfley DE, Frank M, Welch R, et al. Adapting interpersonal psychotherapy to a group format (IPT-G) for binge eating disorder: toward a model for adapting empirically supported treatments. Psychother Res 1998;8:379–81.
146. Blomquist KK, Ansell EB, White M a, et al. Interpersonal problems and developmental trajectories of binge eating disorder. Compr Psychiatry 2012;53:1088–95.
147. Hilbert A, Bishop ME, Stein RI, et al. Long-term efficacy of psychological treatments for binge eating disorder. Br J Psychiatry 2012;200:232–7.
148. Linehan MM. Cognitive-behavioral treatment of borderline personality disorders. New York: Guilford Press; 1993.
149. Wiser S, Telch CF. Dialectical behavior therapy for Binge-Eating Disorder. J Clin Psychol 1999;55:755–68.
150. Telch CF, Agras WS, Linehan MM. Group dialectical behavior therapy for binge-eating disorder:a preliminary, uncontrolled trial. Behav Ther 2000;31:569–82.
151. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061–5.
152. Safer DL, Robinson AH, JB. Outcome from a randomized controlled trial of group therapy for binge eating disorder: comparing dialectical behavior therapy adapted for binge eating to an active comparison group therapy. Behav Ther 2010;41:106–20.
153. Baer RA. Mindfulness-based treatment approaches: Clinician’s guide to evidence base and applications. San Diego: Academic Press; 2005.
154. Van Strien T Van, Engels RCME, Leeuwe J Van, et al. The Stice model of overeating: Tests in clinical and non-clinical samples. Appetite 2005;45:205–13.
155. Godfrey KM, Gallo LC, Afari N. Mindfulness-based interventions for binge eating: a systematic review and meta-analysis. J Behav Med 2015;38:348–362.
156. Kristeller JL, Hallett CB. An exploratory study of a meditation-based intervention for binge eating disorder. J Health Psychol 1999;4:357–63.
157. Kristeller JL, Wolever RQ. Mindfulness-based eating awareness training for treating binge eating disorder: the conceptual foundation. Eat Disord 2011;19:49–61.
158. Kristeller J, Wolever RQ, Sheets V. Mindfulness-based eating awareness training (MB-EAT) for binge eating: a randomized clinical trial. Mindfulness (N Y) 2014;5:282–97.
159. Weineland S, Arvidsson D, Kakoulidis TP, et al. Acceptance and commitment therapy for bariatric surgery patients, a pilot RCT. Obes Res Clin Pract 2012;6:e21–30.
160. Fairburn CG. Overcoming binge eating. 2nd ed. New York: Guilford Press; 2013.
161. Carter JC, Fairburn CG. Cognitive-behavioral self-help for binge eating disorder: a controlled effectiveness study. J Consult Clin Psychol 1998;66:616–23.
162. Loeb KL, Wilson GT, Gilbert JS, et al. Guided and unguided self-help for binge eating. Behav Res Ther 2000;38:259–72.
163. Masheb RM, Grilo CM. Prognostic significance of two sub-categorization methods for the treatment of binge eating disorder: Negative affect and overvaluation predict, but do not moderate, specific outcomes. Behav Res Ther 2008;46:428–37.
164. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge eating disorder. Behav Res Ther 2005;43:1509–25.
165. Citrome L. Vortioxetine for major depressive disorder: A systematic review of the efficacy and safety profile for this newly approved antidepressant - What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract 2015; 68:60–82.
166. Guerdjikova A, McElroy S, Kotwal R, et al. High-dose escitalopram in the treatment of binge-eating disorder with obesity: a placebo-controlled monotherapy trial. Hum Psychopharmacol 2008;23:1–11.
167. McElroy S, Hudson J, Malhotra JL, et al. Citalopram in the treatment of binge-eating disorder: a placebo-controlled trial. J Clin Psychiatry 2003;64:807–13.
168. McElroy S, Casuto L, Nelson EB, et al. Placebo-controlled trial of sertraline in the treatment of binge eating disorder. Am J Psychiatry 2000;157:1004–6.
169. McElroy S, Hudson J, Mitchell JE, et al. Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry 2015;72:235–46.
170. Pearlstein T, Spurell E, Hohlstein LA, et al. A double-blind, placebo-controlled trial of fluvoxamine in binge eating disorder: a high placebo response. Arch Women’s Ment Health 2003;6:147–51.
171. Hudson J, McElroy S. Raymond NC, et al. Fluvoxamine in the treatment of binge-eating disorder: a multicenter placebo-controlled, double-blind trial. Am J Psychiatry 1998;155:1756–62.
172. Miller W, Rollnick S. Motivational Interviewing: helping people change. 3rd ed. New York: Guilford Press; 2013.
173. Rollnick S, Miller W, Butler C. Motivational interviewing in health care: helping patients change behavior. New York: Guilford Press; 2008.
1. Stunkard AJ. Eating patterns and obesity. Psychiatr Q 1959;33:284–95.
2. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Text revision. Washington (DC): The Association; 2000.
3. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: The Association; 2013.
4. Reas D, Grilo C. Pharmacological treatment of binge eating disorder: update review and synthesis. Hum Psychopharmacol Clin Exp 2015;23:1463–78.
5. Wilson G. Treatment of binge eating disorder. Psychiatr Clin North Am 2011;34:773–83.
6. Wonderlich SA, Gordon KH, Mitchell JE, et al. The validity and clinical utility of binge eating disorder. Int J Eat Disord 2009;42:687–705.
7. Walsh BT, Boudreau G. Laboratory studies of binge eating disorder. Int J Eat Disord 2003;34 Suppl:S30–8.
8. Guss JL, Kissileff HR, Devlin MJ, et al. Binge size increases with body mass index in women with binge-eating disorder. Obes Res 2002;10:1021–9.
9. Fairburn CG, Cooper Z. The eating disorder examination. In: Fairburn CG, Cooper Z, editors. Binge eating: nature, assessment, and treatment. New York: Guilford Press;1993:317–60.
10. Keel PK, Mayer SA, Harnden-Fischer JH. Importance of size in defining binge eating episodes in bulimia nervosa. Int J Eat Disord 2001;29:294–301.
11. Niego SH, Pratt EM, Agras WS. Subjective or objective binge: is the distinction valid? Int J Eat Disord 1997;22: 291–8.
12. Pratt E, Niego S, Agras W. Does the size of a binge matter? Int J Eat 1998;24:307–12.
13. Wolfe BE, Baker CW, Smith AT, et al. Validity and utility of the current definition of binge eating. Int J Eat Disord 2009;42:674–86.
14. Latner JD, Clyne C. The diagnostic validity of the criteria for binge eating disorder. Int J Eat Disord 2008;41:1–14.
15. Johnson WG, Boutelle KN, Torgrud L. What is a binge? The influence of amount, duration, and loss of control criterial on judgements of binge eating. Int J Eat Disord 2000;27:471–9.
16. Johnson WG, Roberson-NR, Rohan KJ, et al. An experimental investigation of DSM-IV binge-eating criteria. Eat Behav 2003;4:295–303.
17. Reslan S, Saules KK. College students’ definitions of an eating ‘binge’ differ as a function of gender and binge eating disorder status. Eat Behav 2011;12:225–7.
18. Telch CF, Pratt EM, Niego SH. Obese women with binge eating disorder define the term binge. Int J Eat Disord 1998;24:313–7.
19. Colles SL, Dixon JB, O’Brien PE. Loss of control is central to psychological disturbance associated with binge eating disorder. Obesity (Silver Spring) 2008;16:608–14.
20. Stein KF, Kenardy J, Wiseman C V, et al. What’s driving the binge in binge eating disorder? A prospective examination of precursors and consequences. Int J Eat Disord 2007;40:195–203.
21. Rossiter EM, Agras WS, Telch CF, et al. The eating patterns of non-purging bulimic subjects. Int J Eat Disord 1992;11:111–120.
22. Grilo CM, White MA. A controlled evaluation of the distress criterion for binge eating disorder. J Consult Clin Psychol 2011;79:509–14.
23. Trace SE, Thornton LM, Root TL, et al. Effects of reducing the frequency and duration criteria for binge eating on lifetime prevalence of bulimia nervosa and binge eating disorder: implications for DSM-5. Int J Eat Disord 2012;45: 531–6.
24. Wilson GT, Sysko R. Frequency of binge eating episodes in bulimia nervosa and binge eating disorder: diagnostic considerations. Int J Eat Disord 2009;42:603–10.
25. Sysko R, Roberto CA, Barnes RD, et al. Test-retest reliability of the proposed DSM-5 eating disorder diagnostic criteria. Psychiatry Res 2012;196:302–8.
26. Fairburn CG, Cooper Z, Doll HA, et al. The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiatry 2000;57:659–65.
27. Grilo CM, Ivezaj V, White MA. Evaluation of the DSM-5 severity indicator for binge eating disorder in a community sample. Behav Res Ther 2015;66:72–6.
28. Hudson JI, Hiripi E, Pope HG, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry 2007;61:348–58.
29. Stice E, Marti CN, Rohde P. Prevalence, incidence, impairment, and course of the proposed DSM-5 eating disorder diagnoses in an 8-year prospective community study of young women. J Abnorm Psychol 2012;122:445–57.
30. Mangweth-Matzek B, Hoek HW, Rupp CI, et al. Prevalence of eating disorders in middle-aged women. Int J Eat Disord 2014;47:320–4.
31. Ivezaj V, Saules KK, Hoodin F, et al. The relationship between binge eating and weight status on depression, anxiety, and body image among a diverse college sample: a focus on bi/multiracial women. Eat Behav 2010;11:18–24.
32. Striegel-Moore RH, Bedrosian R, Wang C, et al. Why men should be included in research on binge eating: results from a comparison of psychosocial impairment in men and women. Int J Eat Disord 2012;45:233–40.
33. Womble LG, Williamson DA, Martin CK, et al. Psychosocial variables associated with binge eating in obese males and females. Int J Eat Disord 2001;30:217–21.
34. Udo T, McKee SA., White MA., et al. Sex differences in biopsychosocial correlates of binge eating disorder: a study of treatment-seeking obese adults in primary care setting. Gen Hosp Psychiatry 2013;35:587–91.
35. Udo T, McKee SA., White MA, et al. Menopause and metabolic syndrome in obese individuals with binge eating disorder. Eat Behav 2014;15:182–5.
36. Napolitano MA., Himes S. Race, weight, and correlates of binge eating in female college students. Eat Behav 2011;12:29–36.
37. Sorbara M, Geliebter A. Body image disturbance in obese outpatients before and after weight loss in relation to race, gender, binge eating, and age of onset of obesity. Int J Eat Disord 2002;31:416–23.
38. Alegria M, Woo M, Cao Z, et al. Prevalence and correlates of eating disorders in Latinos in the U.S. Int J Eat Disord 2007;40:S15–21.
39. Striegel-Moore RH, Wilfley DE, Pike KM, et al. Recurrent binge eating in black American women. Arch Fam Med 2000;9: 83–7.
40. Reslan S, Saules KK. Assessing the prevalence of and factors associated with overweight, obesity, and binge eating as a function of ethnicity. Eat Weight Disord-St 2013;18:209–19.
41. Grilo CM, White MA, Barnes RD, et al. Psychiatric disorder co-morbidity and correlates in an ethnically diverse sample of obese patients with binge eating disorder in primary care settings. Compr Psychiatry 2012;54:209–16.
42. Smolak L, Striegel Moore RH. Challenging the myth of the golden girl: ethnicity and eating disorders. In: Smolak L, Striegel Moore RH, editors. Eating disorders innovative directions in research and practice. Washington (DC): American Psychological Association;2001:111–32.
43. Wildes JE, Emery RE. The role of ethnicity and culture in the development of eating disturbance and body dissatisfaction: a meta-analutic review. Clin Phychology Rev 2001;21:521–50.
44. George JBE, Franko DL. Cultural issues in eating pathology and body image among children and adolescents. J Pediatr Psychol 2010;35:231–42.
45. Kelly NR, Cotter EW, Tanofsky-Kraff M, et al. Racial variations in binge eating, body image concerns, and compulsive exercise among men. Psychol Men Masc 2014;13:326-36.
46. Franko DL, Thompson-Brenner H, Thompson DR, et al. Racial/ethnic differences in adults in randomized clinical trials of binge eating disorder. J Consult Clin Psychol 2012;80:186–95.
47. Marques L, Alegria M, Becker AE, et al. Comparative prevalence, correlates of impairment, and service utilization for eating disorders across U.S. ethnic groups: Implications for reducing ethnic disparities in health care access for eating disorders. Int J Eat Disord 2012;44:412–20.
48. Guerdjikova AI, O’Melia AM, Mori N, et al. Binge eating disorder in elderly individuals. Int J Eat Disord 2012;45: 905–8.
49. Fairburn CG. Beglin SJ. Assessment of eating disorders: interview or self-report questionnaire? Int J Eat Disord 1994;16:363–70.
50. Spitzer R, Yanovski S, Marcus M. The questionnaire on eating and weight patterns-revised (QEWP-R). New York State Psychiatric Institute, Mew York 1993.
51. Barnes RD, Masheb RM, White MA, et al. Comparison of methods for identifying and assessing obese patients with binge eating disorder in primary care settings. Int J Eat Disord 2011;44:157–63.
52. Yanovski SZ, Marcus MD, Wadden TA, et al. The questionnaire on eating and weight patterns-5: an updated screening instrument for binge eating disorder. Int J Eat Disord 2015;48:259–61.
53. Gormally J, Black S, Daston S, et al. The assessment of binge eating severity among obese persons. Addict Behav 1982;7:47–55.
54. Alfonsson S. Replacing the term ‘binge eating’ with ‘loss of control over eating’ affects eating disorder screening in clinical care. Obes Res Clin Pract 2015;6–7.
55. Kelly NR, Mitchell KS, Gow RW, et al. An evaluation of the reliability and construct validity of eating disorder measures in white and black women. Psychol Assess 2012;24:608–17.
56. Richards LK, McHugh RK, Pratt EM, et al. Readability and comprehension of self-report binge eating measures. Eat Behav 2013;14:167–70.
57. Masheb RM, White MA, Grilo CM. Substantial weight gains are common prior to treatment-seeking in obese patients with binge eating disorder. Compr Psychiatry 2013;54:880–4.
58. Heaner MK, Walsh BT. A history of the identification of the characteristic eating disturbances of Bulimia Nervosa, Binge Eating Disorder and Anorexia Nervosa. Appetite 2013;65:185–8.
59. Greeno CG, Wing RR, Shiffman S. Binge antecedents in obese women with and without binge eating disorder. J Consult Clin Psychol 2000;68:95–102.
60. Le Grange D, Gorin A, Catley D, et al. Does momentary assessment detect binge eating in overweight women that is denied at interview? Eur Eat Disord Rev 2001;9:309–24.
61. Brody ML, Walsh BT, Devlin MJ. Binge eating disorder: reliability and validity of a new diagnostic category. J Consult Clin Psychol 1994;62:381–6.
62. Hsu LKG, Mulliken B, McDonagh B, et al. Binge eating disorder in extreme obesity. Int J Obes Relat Metab Disord 2002;26:1398–403.
63. Wilfley DE, Schwartz MB, Spurrell EB, et al. Using the eating disorder examination to identify the specific psychopathology of binge eating disorder. Int J Eat Disord 2000;27:259–69.
64. Grucza RA, Przybeck TR, Cloninger CR. Prevalence and correlates of binge eating disorder in a community sample. Compr Psychiatry 2007;48:124–31..
65. Telch CF, Stice E. Psychiatric comorbidity in women with binge eating disorder: prevalence rates from a non-treatment-seeking sample. J Consult Clin Psychol 1998;66:768–76.
66. Wilfley DE, Bishop ME, Wilson GT, et al. Classification of eating disorders: toward DSM-V. Int J Eat Disord 2007; 40:s123–9.
67. Wilfley DE, Wilson GT, Agras WS. The clinical significance of binge eating disorder. Int J Eat Disord 2003;34 Suppl:S96–106.
68. Perez M, Warren CS. The relationship between quality of life, binge-eating disorder, and obesity status in an ethnically diverse sample. Obesity (Silver Spring) 2012;20:879–85.
69. Bulik CM, Sullivan PF, Kendler KS. An empirical study of the classification of eating disorders. Am J Psychiatry 2000;157:886–95.
70. Keel PK, Heatherton TF, Dorer DJ, et al. Point prevalence of bulimia nervosa in 1982, 1992, and 2002. Psychol Med 2006;36:119–27.
71. Mitchell JE, Crosby RD, Wonderlich SA, et al. Latent profile analysis of a cohort of patients with eating disorders not otherwise specified. Int J Eat Disord 2007;40:s95–8.
72. Rockert W, Kaplan AS, Olmsted MP. Eating disorder not otherwise specified : the view from a tertiary care treatment center. Int J Eat Disord 2007;40:s99–103.
73. Striegel-Moore RH, Franko DL, Thompson D, et al. An empirical study of the typology of bulimia nervosa and its spectrum variants. Psychol Med 2005;35:1563–72.
74. Sullivan PF, Bulik CM, Kendler KS. The epidemiology and classification of bulimia nervosa. Psychol Med 1998;28:599–610.
75. Wade TD, Crosby RD, Martin NG. Use of latent profile analysis to identify eating disorder phenotypes in an adult Australian twin cohort. Arch Gen Psychiatry 2006;63:1377–84.
76. Striegel-Moore RH, Franko DL. Should binge eating disorder be included in the DSM-V? A critical review and state of the evidence. Annu Rev Clin Psychol 2008;4:305–24.
77. Striegel-Moore RH, Cachelin FM. Etiology of eating disorders in women. Couns Psychol 2001;29:635–61.
78. Crow SJ, Stewart Agras W, Halmi K, et al. Full syndromal versus subthreshold anorexia nervosa, bulimia nervosa, and binge eating disorder: a multicenter study. Int J Eat Disord 2002;32:309–18.
79. Striegel-Moore RH, Cachelin FM, Dohm F a, et al. Comparison of binge eating disorder and bulimia nervosa in a community sample. Int J Eat Disord 2001;29:157–65.
80. Wilfley DE, Schwartz MB, Spurrell EB, et al. Assessing the specific psychopathology of binge eating disorder patients: Interview or self-report? Behav Res Ther 1997;35:1151–9.
81. Fairburn CG, Doll HA, Welch SL, et al. Risk factors for binge eating disorder. Arch Gen Psychiatry 1998;55:425–32.
82. Grilo CM, White M a., Barnes RD, et al. Posttraumatic stress disorder in women with binge eating disorder in primary care. J Psychiatr Pract 2012;18:408–12.
83. Cassin SE, von Ranson KM. Is binge eating experienced as an addiction? Appetite 2007;49:687–90.
84. Kelly-Weeder S, Jennings KM, Wolfe BE. Gender differences in binge eating and behavioral correlates among college students. Eat Weight Disord 2012;17:e200–2.
85. Pomerleau CS, Ehrlich E, Tate JC, et al. The female weight-control smoker: a profile. J Subst Abuse 1993;5:391–400.
86. White MA, Masheb RM, Grilo CM. Function of binge eating behavior. Int J Eat Disord 2010;43:572–75.
87. Bulik CM, Reichborn-Kjennerud T. Medical morbidity in binge eating disorder. Int J Eat Disord 2003;34:S39–46.
88. Johnson JG, Spitzer RL, Williams JBW. Health problems, impairment and illnesses associated with bulimia nervosa and binge eating disorder among primary care and obstetric gynaecology patients. Psychol Med 2001;31.
89. Mond JM, Hay PJ, Rodgers B, et al. Assessing quality of life in eating disorder patients. Qual Life Res 2005;14:171–8.
90. Marchesini G, Natale S, Chierici S, et al. Effects of cognitive-behavioural therapy on health-related quality of life in obese subjects with and without binge eating disorder. Int J Obes Relat Metab Disord 2002;26:1261–7.
91. Reichborn-Kjennerud T, Bulik CM, Sullivan PF, et al. Psychiatric and medical symptoms in binge eating in the absence of compensatory behaviors. Obes Res 2004;12:1445–54.
92. Hudson JI, Lalonde JK, Coit CE, et al. Longitudinal study of the diagnosis of components of the metabolic syndrome in individuals with binge-eating disorder. Am J Clin Nutr 2010;91:1568–73.
93. Tanofsky-Kraff M, Shomaker LB, Stern EA, et al. Children’s binge eating and development of metabolic syndrome. Int J Obes 2012;36:956–62.
94. Stice E, Marti CN, Durant S. Risk factors for onset of eating disorders: evidence of multiple risk pathways from an 8-year prospective study. Behav Res Ther 2011;49:622–7.
95. Striegel-Moore RH, Fairburn CG, Wilfley DE, et al. Toward an understanding of risk factors for binge-eating disorder in black and white women: a community-based case-control study. Psychol Med 2005;35:907–17.
96. Hilbert A, Pike KM, Goldschmidt AB, et al. Risk factors across the eating disorders. Psychiatry Res 2014;220:500–6.
97. Hudson JI, Lalonde JK, Berry JM, et al. Binge-eating disorder as a distinct familial phenotype in obese individuals. Arch Gen Psychiatry 2006;63:313–19.
98. Mitchell KS, Neale MC, Bulik CM, et al. Binge eating disorder: a symptom-level investigation of genetic and environmental influences on liability. Psychol Med 2010;40:1899–906.
99. Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry 2008;69:266–73.
100. Reichborn-Kjennerud T, Bulik CM, Tambs K, et al. Genetic and environmental influences on binge eating in the absence of compensatory behaviors: a population-based twin study. Int J Eat Disord 2004;36:307–14.
101. Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry 2008;69:266–73.
102. Davis C, Levitan RD, Yilmaz Z, et al. Binge eating disorder and the dopamine D2 receptor: genotypes and sub-phenotypes. Prog Neuropsychopharmacol Biol Psychiatry 2012;38:328–35.
103. Weygandt M, Schaefer A, Schienle A, et al. Diagnosing different binge-eating disorders based on reward-related brain activation patterns. Hum Brain Mapp 2012;33:2135–46.
104. Wang G-J, Geliebter A, Volkow ND, et al. Enhanced striatal dopamine release during food stimulation in binge eating disorder. Obesity (Silver Spring) 2011;19:1601–8.
105. Schäfer A, Vaitl D, Schienle A. Regional grey matter volume abnormalities in bulimia nervosa and binge-eating disorder. Neuroimage 2010;50:639–43.
106. Hawkins RC, Clement PF. Binge eating: measurement problems and a conceptual model. In: Fremouw WJ, Clement PF, editors. The binge purge syndrome: diagnosis, treatment, and research. New York: Springer;1984:229–51.
107. Heatherton TF, Baumeister RF. Binge eating as escape from self-awareness. Psychol Bull 1991;110:86–108.
108. Deaver CM, Miltenberger RG, Smyth J, et al. An evaluation of affect and binge eating. Behav Modif 2003;27: 578–99.
109. Stickney MI, Miltenberger RG, Wolff G. A descriptive analysis of factors contributing to binge eating. J Behav Ther Exp Psychiatry 1999;30:177–89.
110. Hilbert A, Saelens BE, Stein RI, et al. Pretreatment and process predictors of outcome in interpersonal and cognitive behavioral psychotherapy for binge eating disorder. J Consult Clin Psychol 2007;75:645–51.
111. Haedt-Matt AA, Keel PK. Revisiting the affect regulation model of binge eating: a meta-analysis of studies using ecological momentary assessment. Psychol Bull 2012;137:660–81.
112. Cachelin FM, Striegel-Moore RH, Elder K a, et al. Natural course of a community sample of women with binge eating disorder. Int J Eat Disord 1999;25:45–54.
113. Pope HG, Lalonde JK, Pindyck LJ, et al. Binge eating disorder: a stable syndrome. Am J Psychiatry 2006;163:2181–3.
114. Pike KM, Dohm F a, Striegel-Moore RH, et al. A comparison of black and white women with binge eating disorder. Am J Psychiatry 2001;158:1455–60.
115. Cachelin FM, Striegel-Moore RH. Help seeking and barriers to treatment in a community sample of Mexican American and European American women with eating disorders. Int J Eat Disord 2006;39:154–61.
116. Cachelin FM, Rebeck R, Veisel C, et al. Barriers to treatment for eating disorders among ethnically diverse women. Int J Eat Disord 2001;30:269–78.
117. Blaine B, Rodman J. Responses to weight loss treatment among obese individuals with and without BED: a matched-study meta-analysis. Eat Weight Disord 2007;12:54–60.
118. Iacovino JM, Gredysa DM, Altman M, et al. Psychological treatments for binge eating disorder. Curr Psychiatry Rep 2012;14:432–46.
119. Wilson GT, Shafran R. Eating disorders guidelines from NICE. Lancet 2005;365:79–81.
120. Fairburn CG, Cooper Z, Shafran R. Cognitive behaviour therapy for eating disorders: a ‘transdiagnostic’ theory and treatment. Behav Res Ther 2003;41:509–28.
121. Fairburn CG, Cooper Z, Cooper PJ. The clinical features and maintenance of bulimia nervosa. In: Brownell KD, Foreyt JP, editors. Handbook of eating disorders: physiology, psychology and treatment of obesity, anorexia and bulimia. New York: Guiliford;1986:389–404.
122. Grilo CM, Masheb RM, Wilson GT, et al. Cognitive-behavioral therapy, behavioral weight loss, and sequential treatment for obese patients with binge-eating disorder: a randomized controlled trial. J Consult Clin Psychol 2011;79:675–85.
123. Agras WS, Telch CF, Arnow B, et al. One-year follow-up of cognitive-behavioral therapy for obese individuals with binge eating disorder. J Consult Clin Psychol 1997;65:343–7.
124. Dingemans AE, Spinhoven P, van Furth EF. Predictors and mediators of treatment outcome in patients with binge eating disorder. Behav Res Ther 2007;45:2551–62.
125. Eldredge KL, Stewart Agras W, Arnow B, et al. The effects of extending cognitive-behavioral therapy for binge eating disorder among initial treatment nonresponders. Int J Eat Disord 1997;21:347–52.
126. Kenardy J, Mensch M, Bowen K, et al. Group therapy for binge eating in type 2 diabetes: a randomized trial. Diabet Med 2002;19:234–9.
127. Wilson GT, Grilo CM, Vitousek KM. Psychological treatment of eating disorders. Am Psychol 2007;62:199–216.
128. Munsch S, Meyer AH, Biedert E. Efficacy and predictors of long-term treatment success for cognitive-behavioral treatment and behavioral weight-loss-treatment in overweight individuals with binge eating disorder. Behav Res Ther 2012;50:775–8.
129. Brown TA, Keel PK. Current and emerging directions in the treatment of eating disorders. Subst Abuse 2012;6: 33–61.
130. Wilfley DE, Welch RR, Stein RI, et al. A randomized comparison of group cognitive-behavioral therapy and group interpersonal psychotherapy for the treatment of overweight individuals with binge-eating disorder. Arch Gen Psychiatry 2002;59:713–21.
131. Grilo CM, Masheb RM, Wilson GT. Efficacy of cognitive behavioral therapy and fluoxetine for the treatment of binge eating disorder: a randomized double-blind placebo-controlled comparison. Biol Psychiatry 2005;57:301–9.
132. Gorin AA, Le Grange D, Stone AA. Effectiveness of spouse involvement in cognitive behavioral therapy for binge eating disorder. Int J Eat Disord 2003; 33: 421–33.
133. Wilson GT, Wilfley DE, Agras WS, et al. Psychological treatments for binge eating disorder. Arch Gen Psychiatry 2010;67:94–101.
134. Ricca V, Castellini G, Mannucci E, et al. Comparison of individual and group cognitive behavioral therapy for binge eating disorder. a randomized, three-year follow-up study. Appetite 2010;55:656–65.
135. Wilfley DE, Friedman MA, Dounchis JZ, et al. Comorbid psychopathology in binge eating disorder: relation to eating disorder severity at baseline and following treatment. J Consult Clin Psychol 2000;68:641–9.
136. Castellini G, Mannucci E, Lo Sauro C, et al. Different moderators of cognitive-behavioral therapy on subjective and objective binge eating in bulimia nervosa and binge eating disorder: a three-year follow-up study. Psychother Psychosom 2012;81:11–20.
137. Grilo CM, Masheb RM, Crosby RD. Predictors and moderators of response to cognitive behavioral therapy and medication for the treatment of binge eating disorder. J Consult Clin Psychol 2012;80:897–906.
138. Grilo CM, Masheb RM, Wilson GT. Rapid response to treatment for binge eating disorder. J Consult Clin Psychol 2006;74:602–13.
139. Grilo CM, Masheb RM. Rapid response predicts binge eating and weight loss in binge eating disorder: findings from a controlled trial of orlistat with guided self-help cognitive behavioral therapy. Behav Res Ther 2007;45:2537–50.
140. Grilo CM, White MA, Wilson GT, et al. Rapid response predicts 12-month post-treatment outcomes in binge-eating disorder: theoretical and clinical implications. Psychol Med 2012;42:807–17.
141. Hilbert A, Hildebrandt T, Agras WS, et al. Rapid response in psychological treatments for binge eating disorder. 2015;83:649–654.
142. Masheb RM, Grilo CM. Rapid response predicts treatment outcomes in binge eating disorder: implications for stepped care. J Consult Clin Psychol 2007;75:639–44.
143. Safer DL, Joyce EE. Does rapid response to two group psychotherapies for binge eating disorder predict abstinence? Behav Res Ther 2011;49:339–45.
144. Zunker C, Peterson CB, Cao L, et al. A receiver operator characteristics analysis of treatment outcome in binge eating disorder to identify patterns of rapid response. Behav Res Ther 2010;48:1227–31.
145. Wilfley DE, Frank M, Welch R, et al. Adapting interpersonal psychotherapy to a group format (IPT-G) for binge eating disorder: toward a model for adapting empirically supported treatments. Psychother Res 1998;8:379–81.
146. Blomquist KK, Ansell EB, White M a, et al. Interpersonal problems and developmental trajectories of binge eating disorder. Compr Psychiatry 2012;53:1088–95.
147. Hilbert A, Bishop ME, Stein RI, et al. Long-term efficacy of psychological treatments for binge eating disorder. Br J Psychiatry 2012;200:232–7.
148. Linehan MM. Cognitive-behavioral treatment of borderline personality disorders. New York: Guilford Press; 1993.
149. Wiser S, Telch CF. Dialectical behavior therapy for Binge-Eating Disorder. J Clin Psychol 1999;55:755–68.
150. Telch CF, Agras WS, Linehan MM. Group dialectical behavior therapy for binge-eating disorder:a preliminary, uncontrolled trial. Behav Ther 2000;31:569–82.
151. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061–5.
152. Safer DL, Robinson AH, JB. Outcome from a randomized controlled trial of group therapy for binge eating disorder: comparing dialectical behavior therapy adapted for binge eating to an active comparison group therapy. Behav Ther 2010;41:106–20.
153. Baer RA. Mindfulness-based treatment approaches: Clinician’s guide to evidence base and applications. San Diego: Academic Press; 2005.
154. Van Strien T Van, Engels RCME, Leeuwe J Van, et al. The Stice model of overeating: Tests in clinical and non-clinical samples. Appetite 2005;45:205–13.
155. Godfrey KM, Gallo LC, Afari N. Mindfulness-based interventions for binge eating: a systematic review and meta-analysis. J Behav Med 2015;38:348–362.
156. Kristeller JL, Hallett CB. An exploratory study of a meditation-based intervention for binge eating disorder. J Health Psychol 1999;4:357–63.
157. Kristeller JL, Wolever RQ. Mindfulness-based eating awareness training for treating binge eating disorder: the conceptual foundation. Eat Disord 2011;19:49–61.
158. Kristeller J, Wolever RQ, Sheets V. Mindfulness-based eating awareness training (MB-EAT) for binge eating: a randomized clinical trial. Mindfulness (N Y) 2014;5:282–97.
159. Weineland S, Arvidsson D, Kakoulidis TP, et al. Acceptance and commitment therapy for bariatric surgery patients, a pilot RCT. Obes Res Clin Pract 2012;6:e21–30.
160. Fairburn CG. Overcoming binge eating. 2nd ed. New York: Guilford Press; 2013.
161. Carter JC, Fairburn CG. Cognitive-behavioral self-help for binge eating disorder: a controlled effectiveness study. J Consult Clin Psychol 1998;66:616–23.
162. Loeb KL, Wilson GT, Gilbert JS, et al. Guided and unguided self-help for binge eating. Behav Res Ther 2000;38:259–72.
163. Masheb RM, Grilo CM. Prognostic significance of two sub-categorization methods for the treatment of binge eating disorder: Negative affect and overvaluation predict, but do not moderate, specific outcomes. Behav Res Ther 2008;46:428–37.
164. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge eating disorder. Behav Res Ther 2005;43:1509–25.
165. Citrome L. Vortioxetine for major depressive disorder: A systematic review of the efficacy and safety profile for this newly approved antidepressant - What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract 2015; 68:60–82.
166. Guerdjikova A, McElroy S, Kotwal R, et al. High-dose escitalopram in the treatment of binge-eating disorder with obesity: a placebo-controlled monotherapy trial. Hum Psychopharmacol 2008;23:1–11.
167. McElroy S, Hudson J, Malhotra JL, et al. Citalopram in the treatment of binge-eating disorder: a placebo-controlled trial. J Clin Psychiatry 2003;64:807–13.
168. McElroy S, Casuto L, Nelson EB, et al. Placebo-controlled trial of sertraline in the treatment of binge eating disorder. Am J Psychiatry 2000;157:1004–6.
169. McElroy S, Hudson J, Mitchell JE, et al. Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry 2015;72:235–46.
170. Pearlstein T, Spurell E, Hohlstein LA, et al. A double-blind, placebo-controlled trial of fluvoxamine in binge eating disorder: a high placebo response. Arch Women’s Ment Health 2003;6:147–51.
171. Hudson J, McElroy S. Raymond NC, et al. Fluvoxamine in the treatment of binge-eating disorder: a multicenter placebo-controlled, double-blind trial. Am J Psychiatry 1998;155:1756–62.
172. Miller W, Rollnick S. Motivational Interviewing: helping people change. 3rd ed. New York: Guilford Press; 2013.
173. Rollnick S, Miller W, Butler C. Motivational interviewing in health care: helping patients change behavior. New York: Guilford Press; 2008.
Psychosis and catatonia after dancing with a dangerous partner
CASE Rigid, frightened, and mute
Mr. D, age 23, presents for evaluation immediately after discharge from another hospital, where he had been treated for altered mental status.
Ten days earlier, Mr. D’s friends obtained 2C-B (2,5-dimethoxy-4-bromophenethylamine), from the “Darknet,” an underground niche of the Internet. He ingested 20 mg of 2C-B in powder form. Although his friends recovered from a “safe trip,” Mr. D decompensated rapidly over the next few days with persistent psychosis, experiencing both auditory and visual hallucinations. He is “acting strange“ at work, and trying to find “hidden codes” in data. Mr. D also has persistent thought disorganization. He speaks of “connections” between people and things, and says that he is an alien in a spaceship. His friends and family report that he is talking rapidly and is sleeping only 2 or 3 hours each night. Mr. D abruptly quit his job as an analyst a few days after taking the drug.
Mr. D is a single, Ivy League-educated man and is described as hardworking and analytical. His family denies any recent mood changes or life stressors. They report that 1 month ago, Mr. D began smoking marijuana daily. He has no significant medical or psychiatric history, and no family history of psychiatric disorders.
What is your most likely diagnosis for Mr. D?
a) delirium due to a general medical condition
b) substance-induced psychotic disorder
c) catatonia due to a general medical condition
d) schizophrenia
e) bipolar I disorder, currently manic, with psychosis
The authors’ observations
Ring-substituted phenethylamines, commonly known as 2Cs, are designer drugs that are emerging as new substances of abuse.1 2C-B belongs to the phenethylamine subclass of monoamine alkaloids that includes more familiar drugs such as amphetamines, methamphetamines, and 3,4-methylenedioxy-methamphetamine (MDMA).2 It was first synthesized in 1974 by Alexander Shulgin, later described in his book Phenethylamines I Have Known and Loved: A Chemical Love Story, and its hallucinogenic activity is reported to be similar to LSD, mescaline, and psilocybin.3 The literature is scant on the acute effects of 2C intoxication or long-term sequelae of 2C ingestion.1 Most available information regarding the pharmacology of 2C-B comes from users who have reported their drug experiences on blogs, Web sites and forums, and in the media.4
2C-B usually is taken orally in powder or tablet form, in a dose of 10 to 50 mg.4 After an onset period of 20 to 90 minutes, the drug’s effect reaches maximum effect in 15 to 30 minutes, then plateaus for 2 to 7 hours, and comes down within 1 to 2 hours.4 2C-B is known to be orally active, and its hallucinogenic effects are mediated by its actions as a partial serotonin 5HT-2A and 5HT-2C receptor agonist.5 Entactogenic-stimulating effects have been reported at low doses (4 to 10 mg), whereas visual hallucinations with intense colors and object distortion have been reported at moderate doses (10 to 20 mg).4
2C-B, which users often take at parties or raves, appeared on the drug market in the mid 1980s and early 1990s under the names Nexus, Erox, Performax, Toonies, Bromo, Spectrum, and Venus and marketed as a replacement for MDMA after it became a Schedule I drug in the United States.4,6 Some users consume 2C-B in combination with other illicit drugs, including MDMA (called a “party pack”) or LSD (referred to as a “banana split”).6
According to the U.S. Drug Enforcement Agency, law enforcement authorities first seized 2C-B laboratories in California in 1986 and Arizona in 1992.6 Distribution of the drug has been sporadic since it became Schedule I in 1995, and it has been seized from several states, including Virginia, Nevada, Maine, Illinois, Missouri, South Dakota, and Kansas.6
EXAMINATION Passive and mute
On examination, Mr. D is lying in bed with eyes closed and extremities extended in an odd, rigid posture. He is resistant to attempts at passive movement, is nonresponsive to verbal commands, and is mute. A review of vital signs shows tachycardia, 110 beats per minute, but the physical exam is otherwise unremarkable. His Bush-Francis Catatonia Rating Scale (BFCRS) score is 17, indicating a diagnosis of catatonia. Mini-Mental Status Examination cannot be completed because Mr. D is unable to participate.
Laboratory studies reveal an elevated creatinine kinase (CK) level of 356 U/L. Results of a complete blood count, comprehensive metabolic panel, urinalysis, and thyroid-stimulating hormone are normal. Blood alcohol level is <10 mg/dL. Acetaminophen and salicylate levels are normal (<5 mg/dL). Records from his recent hospitalization reveal normal head CT, chest radiography, EEG, and urinalysis, and a negative urine drug screen.
What is the next step in managing Mr. D’s catatonic symptoms?
a) IV normal saline
b) IV lorazepam
c) emergent electroconvulsive therapy (ECT)
d) IM haloperidol
e) IM olanzapine
TREATMENT Saline and psychotropics
While in the emergency room, Mr. D receives 2 L of IV saline. His CK level falls to 137 U/L. A challenge with IV lorazepam, 2 mg, also is performed. Mr. D becomes talkative and follows commands with fluid movements, but his disorganized, delusional thoughts persist. BFCRS score has improved to 9 (Table 1). He is admitted to the psychiatric unit and started on oral lorazepam, 2 mg, 3 times daily, for catatonia, and olanzapine, 10 mg/d, for psychosis.

The differential diagnosis for Mr. D’s psychosis includes substance-induced psychotic disorder, schizophrenia, bipolar disorder, and psychosis with another organic cause (Table 2).7 Further medical workup is completed, including a urine drug screen, testing for HIV, hepatitis B, syphilis, lead and heavy metals, ceruloplasmin, vitamin B12, folate, antinuclear antibody, sedimentation rate, and brain MRI. Cannabinoids are detected in his urine drug screen. Another urine sample is sent to an outside lab to test for several synthetic drugs, including MDMA, 3,4-methylenedioxy- N-ethyl-amphetamine, 2C-B, 2C-C, 2C-I, and 2C-P, results of which also are negative.

By the second day of hospitalization, Mr. D appears less disorganized but continues to complain of “scrambled thoughts” and appears guarded. Despite initial response to IV lorazepam and its continuation in oral form, over the next day Mr. D appears more psychomotor-slowed, with motor stiffness. His score on the BFCRS increases, with significant posturing; vital signs remain stable, however.
What is your next step in managing his catatonic symptoms?
a) increase olanzapine
b) decrease olanzapine
c) decrease lorazepam
d) emergent ECT
e) switch to haloperidol
The authors’ observations
Although catatonia can be associated with a mood or psychotic disorder, it also can be induced by a medication or general medical condition (Table 3).8 It is thought that catatonia is associated with decreased γ-aminobutyric acid (GABA) and dopamine D2 receptor activity, and increased N-methyl-d-aspartate (NMDA) receptor activity.9 Antipsychotics could worsen catatonia through D2 blockade. Benzodiazepines, however, improve catatonia by increasing GABA and decreasing NMDA receptor activity. In this case, Mr. D was naïve to antipsychotics and seemed to be sensitive to them, as evidenced by his worsening symptoms.
Which condition should be considered in the differential diagnosis?
a) parkinsonian-hyperpyrexia syndrome
b) neuroleptic malignant syndrome (NMS)
c) stiff person syndrome
d) serotonin syndrome
e) CNS infection
The authors’ observations
NMS, catatonia, and parkinsonian-hyperpyrexia syndrome are all related to diminished action of dopamine at the D2 receptor. Although the mechanism of catatonia is not completely understood, NMS is thought to be caused by blockade at the D2 receptors by antipsychotics, whereas parkinsonian-hyperpyrexia syndrome is related to withdrawal of dopamine agonists. Because of the similarity in symptoms and proposed mechanisms, some experts hypothesize that NMS is a drug-induced malignant catatonia.10,11 Interestingly, NMS and catatonia respond to withdrawal of antipsychotics, and addition of benzodiazepines and ECT.
Mr. D showed posturing and other behavioral abnormalities, which are less common in NMS. Furthermore, although he had episodes of mild tachycardia, autonomic dysregulation—a hallmark of NMS—was not found. Given the common shared deficiency of activity at the D2 receptor in both NMS and catatonia, antipsychotics could cause or worsen either condition.
TREATMENT ECT
Mr. D’s olanzapine dosage is decreased to 2.5 mg/d. His catatonic symptoms improve with each dosage of oral lorazepam; however, effects seem to lessen and last for shorter periods over the following day. Additionally, Mr. D again becomes more disorganized, stiff, and unable to feed or bathe himself, and develops episodes of mild tachycardia.
Given Mr. D’s partial and poorly sustained response to lorazepam, a trial of ECT is pursued. On the third day of hospitalization, he receives ECT with bi-frontal lead placement at 25% energy. Concurrently, olanzapine is discontinued because of worsening muscle stiffness and concern about neuroleptic sensitivity. His BFCRS score after ECT is 2, and he is noted to be more interactive on the inpatient unit. He continues to receive ECT 3 times a week, with notable improvement, but ongoing psychotic symptoms and catatonic symptoms partially reemerge between ECT treatments. Lead placement is changed to bi-temporal by the third treatment, and the energy setting is increased from 25% to 50%, and to 75% by the sixth treatment. An additional nighttime dose of oral lorazepam, 2 mg, is added after the sixth treatment, in an attempt to reduce “wearing off” by morning.
After the seventh treatment, Mr. D is able to maintain logical conversation without re-emergence of catatonic symptoms over 2 days, signifying a turning point in the treatment course. The ECT energy setting is decreased to 50% to minimize potential memory deficits. His insight into his illness and treatment dramatically improve over the next few days. ECT is discontinued after the tenth treatment and Mr. D is discharged home to the care of his family.
The authors’ observations
Randomized clinical trials studying the effectiveness of ECT for catatonia are limited. Much of what we know about ECT comes from case reports that describe excellent outcomes for a variety of treatment-resistant illnesses, including catatonia in mood disorders, schizophrenia, autism, and other organic brain disease.12
Although benzodiazepines often are the first-line treatment for catatonia caused by any underlying illness, one study showed only 1 of 41 patients achieved remission with benzodiazepines, compared with 100% of those treated with ECT13; another study supported these results with 8 of 9 lorazepam non-responders responding to ECT.14 There are few case reports of substance-induced catatonia in the absence of other chronic mental illness, although none report use of ECT. However, a study showed no significant difference in the effectiveness of ECT for catatonia caused by an affective disorder or schizophrenia.15
Mr. D’s case exemplifies complete remission of catatonia induced by a psychoactive substance.
OUTCOME Steady improvement
Mr. D is followed in the outpatient clinic for 1 month after discharge; lorazepam is tapered successfully. During this time frame, psychotic and catatonic symptoms do not re-emerge. He reports some initial working memory deficits that improve steadily. There is no evidence of any significant psychiatric signs or symptoms, including neurovegetative symptoms of depression, mania or hypomania, perceptual disturbances, or disorganized thoughts or behaviors. He remains abstinent from alcohol, tobacco, and all psychoactive substances.
Bottom Line
Persistent psychosis and catatonia after the use of newer designer drugs such as 2C-B are rare, but these drugs carry serious potential complications that clinicians should be aware of. Benzodiazepines and electroconvulsive therapy have been proved effective for catatonia that is related to a number of psychiatric illnesses, often resulting in good outcomes. However, current evidence on their use is limited, particularly regarding treatment of substance-induced psychosis and catatonia.
Related Resources
• Meyer MR, Maurer HH. Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab. 2010;11(5):468-482.
• Rickli A, Luethi D, Reinisch J, et al. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology. 2015;99:546-553.
Drug Brand Names
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dean BV, Stellpflug SJ, Burnett AM, et al. 2C or not 2C: phenethylamine designer drug review. J Med Toxicol. 2013;9(2):172-178.
2. Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
3. Shulgin A, Shulgin A. PiHKAL: a chemical love story. Berkley, CA: Transform Press; 1991.
4. Papoutsis I, Nikolaou P, Stefanidou M, et al. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicology. 2015;3(1):1-11.
5. Caudevilla-Gálligo F, Riba J, Ventura M, et al. 4-Bromo-2, 5-dimethoxyphenethylamine (2C-B): presence in the recreational drug market in Spain, pattern of use and subjective effects. J Psychopharmacol. 2012;26(7):1026-1035.
6. National Drug Intelligence Center. Information bulletin: 2C-B (Nexus) reappears on the club drug scene. http:// www.Justice.gov/archive/ndic/pubs0/665. Published May 2001. Accessed June 12, 2015.
7. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
8. Masand PS, Levenson JL, et al. Mania, catatonia, and psychosis. In: Levenson JL, ed. The American Psychiatric Publishing textbook of psychosomatic medicine. Washington, DC: American Psychiatric Publishing; 2005: 239-241.
9. Carroll BT. The universal field of hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Lee JW. Neuroleptic-induced catatonia: clinical presentation, response to benzodiazepines, and relationship to neuroleptic malignant syndrome. J Clin Psychopharmacol. 2010;30(1):3-10.
11. Vancaester E, Santens P. Catatonia and neuroleptic malignant syndrome: two sides of a coin? Acta Neurol Belg. 2007;107(2):47-50.
12. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
13. Hatta K, Miyakawa K, Ota T, et al. Maximal response to electroconvulsive therapy for the treatment of catatonic symptoms. J ECT. 2007;23(4):233-235.
14. Payee H, Chandrasekaran R, Raju GV. Catatonic syndrome: treatment response to Lorazepam. Indian J Psychiatry. 1999;41(1):49-53.
15. Rohland BM, Carroll BT, Jacoby RG. ECT in the treatment of the catatonic syndrome. J Affect Disord. 1993;29(4):255-261.
3,4-methylenedioxy-methamphetamine, MDMA, Phenethylamines I Have Known and Loved: A Chemical Love Story, LSD, substance abuse, substance use
CASE Rigid, frightened, and mute
Mr. D, age 23, presents for evaluation immediately after discharge from another hospital, where he had been treated for altered mental status.
Ten days earlier, Mr. D’s friends obtained 2C-B (2,5-dimethoxy-4-bromophenethylamine), from the “Darknet,” an underground niche of the Internet. He ingested 20 mg of 2C-B in powder form. Although his friends recovered from a “safe trip,” Mr. D decompensated rapidly over the next few days with persistent psychosis, experiencing both auditory and visual hallucinations. He is “acting strange“ at work, and trying to find “hidden codes” in data. Mr. D also has persistent thought disorganization. He speaks of “connections” between people and things, and says that he is an alien in a spaceship. His friends and family report that he is talking rapidly and is sleeping only 2 or 3 hours each night. Mr. D abruptly quit his job as an analyst a few days after taking the drug.
Mr. D is a single, Ivy League-educated man and is described as hardworking and analytical. His family denies any recent mood changes or life stressors. They report that 1 month ago, Mr. D began smoking marijuana daily. He has no significant medical or psychiatric history, and no family history of psychiatric disorders.
What is your most likely diagnosis for Mr. D?
a) delirium due to a general medical condition
b) substance-induced psychotic disorder
c) catatonia due to a general medical condition
d) schizophrenia
e) bipolar I disorder, currently manic, with psychosis
The authors’ observations
Ring-substituted phenethylamines, commonly known as 2Cs, are designer drugs that are emerging as new substances of abuse.1 2C-B belongs to the phenethylamine subclass of monoamine alkaloids that includes more familiar drugs such as amphetamines, methamphetamines, and 3,4-methylenedioxy-methamphetamine (MDMA).2 It was first synthesized in 1974 by Alexander Shulgin, later described in his book Phenethylamines I Have Known and Loved: A Chemical Love Story, and its hallucinogenic activity is reported to be similar to LSD, mescaline, and psilocybin.3 The literature is scant on the acute effects of 2C intoxication or long-term sequelae of 2C ingestion.1 Most available information regarding the pharmacology of 2C-B comes from users who have reported their drug experiences on blogs, Web sites and forums, and in the media.4
2C-B usually is taken orally in powder or tablet form, in a dose of 10 to 50 mg.4 After an onset period of 20 to 90 minutes, the drug’s effect reaches maximum effect in 15 to 30 minutes, then plateaus for 2 to 7 hours, and comes down within 1 to 2 hours.4 2C-B is known to be orally active, and its hallucinogenic effects are mediated by its actions as a partial serotonin 5HT-2A and 5HT-2C receptor agonist.5 Entactogenic-stimulating effects have been reported at low doses (4 to 10 mg), whereas visual hallucinations with intense colors and object distortion have been reported at moderate doses (10 to 20 mg).4
2C-B, which users often take at parties or raves, appeared on the drug market in the mid 1980s and early 1990s under the names Nexus, Erox, Performax, Toonies, Bromo, Spectrum, and Venus and marketed as a replacement for MDMA after it became a Schedule I drug in the United States.4,6 Some users consume 2C-B in combination with other illicit drugs, including MDMA (called a “party pack”) or LSD (referred to as a “banana split”).6
According to the U.S. Drug Enforcement Agency, law enforcement authorities first seized 2C-B laboratories in California in 1986 and Arizona in 1992.6 Distribution of the drug has been sporadic since it became Schedule I in 1995, and it has been seized from several states, including Virginia, Nevada, Maine, Illinois, Missouri, South Dakota, and Kansas.6
EXAMINATION Passive and mute
On examination, Mr. D is lying in bed with eyes closed and extremities extended in an odd, rigid posture. He is resistant to attempts at passive movement, is nonresponsive to verbal commands, and is mute. A review of vital signs shows tachycardia, 110 beats per minute, but the physical exam is otherwise unremarkable. His Bush-Francis Catatonia Rating Scale (BFCRS) score is 17, indicating a diagnosis of catatonia. Mini-Mental Status Examination cannot be completed because Mr. D is unable to participate.
Laboratory studies reveal an elevated creatinine kinase (CK) level of 356 U/L. Results of a complete blood count, comprehensive metabolic panel, urinalysis, and thyroid-stimulating hormone are normal. Blood alcohol level is <10 mg/dL. Acetaminophen and salicylate levels are normal (<5 mg/dL). Records from his recent hospitalization reveal normal head CT, chest radiography, EEG, and urinalysis, and a negative urine drug screen.
What is the next step in managing Mr. D’s catatonic symptoms?
a) IV normal saline
b) IV lorazepam
c) emergent electroconvulsive therapy (ECT)
d) IM haloperidol
e) IM olanzapine
TREATMENT Saline and psychotropics
While in the emergency room, Mr. D receives 2 L of IV saline. His CK level falls to 137 U/L. A challenge with IV lorazepam, 2 mg, also is performed. Mr. D becomes talkative and follows commands with fluid movements, but his disorganized, delusional thoughts persist. BFCRS score has improved to 9 (Table 1). He is admitted to the psychiatric unit and started on oral lorazepam, 2 mg, 3 times daily, for catatonia, and olanzapine, 10 mg/d, for psychosis.
The differential diagnosis for Mr. D’s psychosis includes substance-induced psychotic disorder, schizophrenia, bipolar disorder, and psychosis with another organic cause (Table 2).7 Further medical workup is completed, including a urine drug screen, testing for HIV, hepatitis B, syphilis, lead and heavy metals, ceruloplasmin, vitamin B12, folate, antinuclear antibody, sedimentation rate, and brain MRI. Cannabinoids are detected in his urine drug screen. Another urine sample is sent to an outside lab to test for several synthetic drugs, including MDMA, 3,4-methylenedioxy- N-ethyl-amphetamine, 2C-B, 2C-C, 2C-I, and 2C-P, results of which also are negative.
By the second day of hospitalization, Mr. D appears less disorganized but continues to complain of “scrambled thoughts” and appears guarded. Despite initial response to IV lorazepam and its continuation in oral form, over the next day Mr. D appears more psychomotor-slowed, with motor stiffness. His score on the BFCRS increases, with significant posturing; vital signs remain stable, however.
What is your next step in managing his catatonic symptoms?
a) increase olanzapine
b) decrease olanzapine
c) decrease lorazepam
d) emergent ECT
e) switch to haloperidol
The authors’ observations
Although catatonia can be associated with a mood or psychotic disorder, it also can be induced by a medication or general medical condition (Table 3).8 It is thought that catatonia is associated with decreased γ-aminobutyric acid (GABA) and dopamine D2 receptor activity, and increased N-methyl-d-aspartate (NMDA) receptor activity.9 Antipsychotics could worsen catatonia through D2 blockade. Benzodiazepines, however, improve catatonia by increasing GABA and decreasing NMDA receptor activity. In this case, Mr. D was naïve to antipsychotics and seemed to be sensitive to them, as evidenced by his worsening symptoms.
Which condition should be considered in the differential diagnosis?
a) parkinsonian-hyperpyrexia syndrome
b) neuroleptic malignant syndrome (NMS)
c) stiff person syndrome
d) serotonin syndrome
e) CNS infection
The authors’ observations
NMS, catatonia, and parkinsonian-hyperpyrexia syndrome are all related to diminished action of dopamine at the D2 receptor. Although the mechanism of catatonia is not completely understood, NMS is thought to be caused by blockade at the D2 receptors by antipsychotics, whereas parkinsonian-hyperpyrexia syndrome is related to withdrawal of dopamine agonists. Because of the similarity in symptoms and proposed mechanisms, some experts hypothesize that NMS is a drug-induced malignant catatonia.10,11 Interestingly, NMS and catatonia respond to withdrawal of antipsychotics, and addition of benzodiazepines and ECT.
Mr. D showed posturing and other behavioral abnormalities, which are less common in NMS. Furthermore, although he had episodes of mild tachycardia, autonomic dysregulation—a hallmark of NMS—was not found. Given the common shared deficiency of activity at the D2 receptor in both NMS and catatonia, antipsychotics could cause or worsen either condition.
TREATMENT ECT
Mr. D’s olanzapine dosage is decreased to 2.5 mg/d. His catatonic symptoms improve with each dosage of oral lorazepam; however, effects seem to lessen and last for shorter periods over the following day. Additionally, Mr. D again becomes more disorganized, stiff, and unable to feed or bathe himself, and develops episodes of mild tachycardia.
Given Mr. D’s partial and poorly sustained response to lorazepam, a trial of ECT is pursued. On the third day of hospitalization, he receives ECT with bi-frontal lead placement at 25% energy. Concurrently, olanzapine is discontinued because of worsening muscle stiffness and concern about neuroleptic sensitivity. His BFCRS score after ECT is 2, and he is noted to be more interactive on the inpatient unit. He continues to receive ECT 3 times a week, with notable improvement, but ongoing psychotic symptoms and catatonic symptoms partially reemerge between ECT treatments. Lead placement is changed to bi-temporal by the third treatment, and the energy setting is increased from 25% to 50%, and to 75% by the sixth treatment. An additional nighttime dose of oral lorazepam, 2 mg, is added after the sixth treatment, in an attempt to reduce “wearing off” by morning.
After the seventh treatment, Mr. D is able to maintain logical conversation without re-emergence of catatonic symptoms over 2 days, signifying a turning point in the treatment course. The ECT energy setting is decreased to 50% to minimize potential memory deficits. His insight into his illness and treatment dramatically improve over the next few days. ECT is discontinued after the tenth treatment and Mr. D is discharged home to the care of his family.
The authors’ observations
Randomized clinical trials studying the effectiveness of ECT for catatonia are limited. Much of what we know about ECT comes from case reports that describe excellent outcomes for a variety of treatment-resistant illnesses, including catatonia in mood disorders, schizophrenia, autism, and other organic brain disease.12
Although benzodiazepines often are the first-line treatment for catatonia caused by any underlying illness, one study showed only 1 of 41 patients achieved remission with benzodiazepines, compared with 100% of those treated with ECT13; another study supported these results with 8 of 9 lorazepam non-responders responding to ECT.14 There are few case reports of substance-induced catatonia in the absence of other chronic mental illness, although none report use of ECT. However, a study showed no significant difference in the effectiveness of ECT for catatonia caused by an affective disorder or schizophrenia.15
Mr. D’s case exemplifies complete remission of catatonia induced by a psychoactive substance.
OUTCOME Steady improvement
Mr. D is followed in the outpatient clinic for 1 month after discharge; lorazepam is tapered successfully. During this time frame, psychotic and catatonic symptoms do not re-emerge. He reports some initial working memory deficits that improve steadily. There is no evidence of any significant psychiatric signs or symptoms, including neurovegetative symptoms of depression, mania or hypomania, perceptual disturbances, or disorganized thoughts or behaviors. He remains abstinent from alcohol, tobacco, and all psychoactive substances.
Bottom Line
Persistent psychosis and catatonia after the use of newer designer drugs such as 2C-B are rare, but these drugs carry serious potential complications that clinicians should be aware of. Benzodiazepines and electroconvulsive therapy have been proved effective for catatonia that is related to a number of psychiatric illnesses, often resulting in good outcomes. However, current evidence on their use is limited, particularly regarding treatment of substance-induced psychosis and catatonia.
Related Resources
• Meyer MR, Maurer HH. Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab. 2010;11(5):468-482.
• Rickli A, Luethi D, Reinisch J, et al. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology. 2015;99:546-553.
Drug Brand Names
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Rigid, frightened, and mute
Mr. D, age 23, presents for evaluation immediately after discharge from another hospital, where he had been treated for altered mental status.
Ten days earlier, Mr. D’s friends obtained 2C-B (2,5-dimethoxy-4-bromophenethylamine), from the “Darknet,” an underground niche of the Internet. He ingested 20 mg of 2C-B in powder form. Although his friends recovered from a “safe trip,” Mr. D decompensated rapidly over the next few days with persistent psychosis, experiencing both auditory and visual hallucinations. He is “acting strange“ at work, and trying to find “hidden codes” in data. Mr. D also has persistent thought disorganization. He speaks of “connections” between people and things, and says that he is an alien in a spaceship. His friends and family report that he is talking rapidly and is sleeping only 2 or 3 hours each night. Mr. D abruptly quit his job as an analyst a few days after taking the drug.
Mr. D is a single, Ivy League-educated man and is described as hardworking and analytical. His family denies any recent mood changes or life stressors. They report that 1 month ago, Mr. D began smoking marijuana daily. He has no significant medical or psychiatric history, and no family history of psychiatric disorders.
What is your most likely diagnosis for Mr. D?
a) delirium due to a general medical condition
b) substance-induced psychotic disorder
c) catatonia due to a general medical condition
d) schizophrenia
e) bipolar I disorder, currently manic, with psychosis
The authors’ observations
Ring-substituted phenethylamines, commonly known as 2Cs, are designer drugs that are emerging as new substances of abuse.1 2C-B belongs to the phenethylamine subclass of monoamine alkaloids that includes more familiar drugs such as amphetamines, methamphetamines, and 3,4-methylenedioxy-methamphetamine (MDMA).2 It was first synthesized in 1974 by Alexander Shulgin, later described in his book Phenethylamines I Have Known and Loved: A Chemical Love Story, and its hallucinogenic activity is reported to be similar to LSD, mescaline, and psilocybin.3 The literature is scant on the acute effects of 2C intoxication or long-term sequelae of 2C ingestion.1 Most available information regarding the pharmacology of 2C-B comes from users who have reported their drug experiences on blogs, Web sites and forums, and in the media.4
2C-B usually is taken orally in powder or tablet form, in a dose of 10 to 50 mg.4 After an onset period of 20 to 90 minutes, the drug’s effect reaches maximum effect in 15 to 30 minutes, then plateaus for 2 to 7 hours, and comes down within 1 to 2 hours.4 2C-B is known to be orally active, and its hallucinogenic effects are mediated by its actions as a partial serotonin 5HT-2A and 5HT-2C receptor agonist.5 Entactogenic-stimulating effects have been reported at low doses (4 to 10 mg), whereas visual hallucinations with intense colors and object distortion have been reported at moderate doses (10 to 20 mg).4
2C-B, which users often take at parties or raves, appeared on the drug market in the mid 1980s and early 1990s under the names Nexus, Erox, Performax, Toonies, Bromo, Spectrum, and Venus and marketed as a replacement for MDMA after it became a Schedule I drug in the United States.4,6 Some users consume 2C-B in combination with other illicit drugs, including MDMA (called a “party pack”) or LSD (referred to as a “banana split”).6
According to the U.S. Drug Enforcement Agency, law enforcement authorities first seized 2C-B laboratories in California in 1986 and Arizona in 1992.6 Distribution of the drug has been sporadic since it became Schedule I in 1995, and it has been seized from several states, including Virginia, Nevada, Maine, Illinois, Missouri, South Dakota, and Kansas.6
EXAMINATION Passive and mute
On examination, Mr. D is lying in bed with eyes closed and extremities extended in an odd, rigid posture. He is resistant to attempts at passive movement, is nonresponsive to verbal commands, and is mute. A review of vital signs shows tachycardia, 110 beats per minute, but the physical exam is otherwise unremarkable. His Bush-Francis Catatonia Rating Scale (BFCRS) score is 17, indicating a diagnosis of catatonia. Mini-Mental Status Examination cannot be completed because Mr. D is unable to participate.
Laboratory studies reveal an elevated creatinine kinase (CK) level of 356 U/L. Results of a complete blood count, comprehensive metabolic panel, urinalysis, and thyroid-stimulating hormone are normal. Blood alcohol level is <10 mg/dL. Acetaminophen and salicylate levels are normal (<5 mg/dL). Records from his recent hospitalization reveal normal head CT, chest radiography, EEG, and urinalysis, and a negative urine drug screen.
What is the next step in managing Mr. D’s catatonic symptoms?
a) IV normal saline
b) IV lorazepam
c) emergent electroconvulsive therapy (ECT)
d) IM haloperidol
e) IM olanzapine
TREATMENT Saline and psychotropics
While in the emergency room, Mr. D receives 2 L of IV saline. His CK level falls to 137 U/L. A challenge with IV lorazepam, 2 mg, also is performed. Mr. D becomes talkative and follows commands with fluid movements, but his disorganized, delusional thoughts persist. BFCRS score has improved to 9 (Table 1). He is admitted to the psychiatric unit and started on oral lorazepam, 2 mg, 3 times daily, for catatonia, and olanzapine, 10 mg/d, for psychosis.
The differential diagnosis for Mr. D’s psychosis includes substance-induced psychotic disorder, schizophrenia, bipolar disorder, and psychosis with another organic cause (Table 2).7 Further medical workup is completed, including a urine drug screen, testing for HIV, hepatitis B, syphilis, lead and heavy metals, ceruloplasmin, vitamin B12, folate, antinuclear antibody, sedimentation rate, and brain MRI. Cannabinoids are detected in his urine drug screen. Another urine sample is sent to an outside lab to test for several synthetic drugs, including MDMA, 3,4-methylenedioxy- N-ethyl-amphetamine, 2C-B, 2C-C, 2C-I, and 2C-P, results of which also are negative.
By the second day of hospitalization, Mr. D appears less disorganized but continues to complain of “scrambled thoughts” and appears guarded. Despite initial response to IV lorazepam and its continuation in oral form, over the next day Mr. D appears more psychomotor-slowed, with motor stiffness. His score on the BFCRS increases, with significant posturing; vital signs remain stable, however.
What is your next step in managing his catatonic symptoms?
a) increase olanzapine
b) decrease olanzapine
c) decrease lorazepam
d) emergent ECT
e) switch to haloperidol
The authors’ observations
Although catatonia can be associated with a mood or psychotic disorder, it also can be induced by a medication or general medical condition (Table 3).8 It is thought that catatonia is associated with decreased γ-aminobutyric acid (GABA) and dopamine D2 receptor activity, and increased N-methyl-d-aspartate (NMDA) receptor activity.9 Antipsychotics could worsen catatonia through D2 blockade. Benzodiazepines, however, improve catatonia by increasing GABA and decreasing NMDA receptor activity. In this case, Mr. D was naïve to antipsychotics and seemed to be sensitive to them, as evidenced by his worsening symptoms.
Which condition should be considered in the differential diagnosis?
a) parkinsonian-hyperpyrexia syndrome
b) neuroleptic malignant syndrome (NMS)
c) stiff person syndrome
d) serotonin syndrome
e) CNS infection
The authors’ observations
NMS, catatonia, and parkinsonian-hyperpyrexia syndrome are all related to diminished action of dopamine at the D2 receptor. Although the mechanism of catatonia is not completely understood, NMS is thought to be caused by blockade at the D2 receptors by antipsychotics, whereas parkinsonian-hyperpyrexia syndrome is related to withdrawal of dopamine agonists. Because of the similarity in symptoms and proposed mechanisms, some experts hypothesize that NMS is a drug-induced malignant catatonia.10,11 Interestingly, NMS and catatonia respond to withdrawal of antipsychotics, and addition of benzodiazepines and ECT.
Mr. D showed posturing and other behavioral abnormalities, which are less common in NMS. Furthermore, although he had episodes of mild tachycardia, autonomic dysregulation—a hallmark of NMS—was not found. Given the common shared deficiency of activity at the D2 receptor in both NMS and catatonia, antipsychotics could cause or worsen either condition.
TREATMENT ECT
Mr. D’s olanzapine dosage is decreased to 2.5 mg/d. His catatonic symptoms improve with each dosage of oral lorazepam; however, effects seem to lessen and last for shorter periods over the following day. Additionally, Mr. D again becomes more disorganized, stiff, and unable to feed or bathe himself, and develops episodes of mild tachycardia.
Given Mr. D’s partial and poorly sustained response to lorazepam, a trial of ECT is pursued. On the third day of hospitalization, he receives ECT with bi-frontal lead placement at 25% energy. Concurrently, olanzapine is discontinued because of worsening muscle stiffness and concern about neuroleptic sensitivity. His BFCRS score after ECT is 2, and he is noted to be more interactive on the inpatient unit. He continues to receive ECT 3 times a week, with notable improvement, but ongoing psychotic symptoms and catatonic symptoms partially reemerge between ECT treatments. Lead placement is changed to bi-temporal by the third treatment, and the energy setting is increased from 25% to 50%, and to 75% by the sixth treatment. An additional nighttime dose of oral lorazepam, 2 mg, is added after the sixth treatment, in an attempt to reduce “wearing off” by morning.
After the seventh treatment, Mr. D is able to maintain logical conversation without re-emergence of catatonic symptoms over 2 days, signifying a turning point in the treatment course. The ECT energy setting is decreased to 50% to minimize potential memory deficits. His insight into his illness and treatment dramatically improve over the next few days. ECT is discontinued after the tenth treatment and Mr. D is discharged home to the care of his family.
The authors’ observations
Randomized clinical trials studying the effectiveness of ECT for catatonia are limited. Much of what we know about ECT comes from case reports that describe excellent outcomes for a variety of treatment-resistant illnesses, including catatonia in mood disorders, schizophrenia, autism, and other organic brain disease.12
Although benzodiazepines often are the first-line treatment for catatonia caused by any underlying illness, one study showed only 1 of 41 patients achieved remission with benzodiazepines, compared with 100% of those treated with ECT13; another study supported these results with 8 of 9 lorazepam non-responders responding to ECT.14 There are few case reports of substance-induced catatonia in the absence of other chronic mental illness, although none report use of ECT. However, a study showed no significant difference in the effectiveness of ECT for catatonia caused by an affective disorder or schizophrenia.15
Mr. D’s case exemplifies complete remission of catatonia induced by a psychoactive substance.
OUTCOME Steady improvement
Mr. D is followed in the outpatient clinic for 1 month after discharge; lorazepam is tapered successfully. During this time frame, psychotic and catatonic symptoms do not re-emerge. He reports some initial working memory deficits that improve steadily. There is no evidence of any significant psychiatric signs or symptoms, including neurovegetative symptoms of depression, mania or hypomania, perceptual disturbances, or disorganized thoughts or behaviors. He remains abstinent from alcohol, tobacco, and all psychoactive substances.
Bottom Line
Persistent psychosis and catatonia after the use of newer designer drugs such as 2C-B are rare, but these drugs carry serious potential complications that clinicians should be aware of. Benzodiazepines and electroconvulsive therapy have been proved effective for catatonia that is related to a number of psychiatric illnesses, often resulting in good outcomes. However, current evidence on their use is limited, particularly regarding treatment of substance-induced psychosis and catatonia.
Related Resources
• Meyer MR, Maurer HH. Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab. 2010;11(5):468-482.
• Rickli A, Luethi D, Reinisch J, et al. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology. 2015;99:546-553.
Drug Brand Names
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dean BV, Stellpflug SJ, Burnett AM, et al. 2C or not 2C: phenethylamine designer drug review. J Med Toxicol. 2013;9(2):172-178.
2. Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
3. Shulgin A, Shulgin A. PiHKAL: a chemical love story. Berkley, CA: Transform Press; 1991.
4. Papoutsis I, Nikolaou P, Stefanidou M, et al. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicology. 2015;3(1):1-11.
5. Caudevilla-Gálligo F, Riba J, Ventura M, et al. 4-Bromo-2, 5-dimethoxyphenethylamine (2C-B): presence in the recreational drug market in Spain, pattern of use and subjective effects. J Psychopharmacol. 2012;26(7):1026-1035.
6. National Drug Intelligence Center. Information bulletin: 2C-B (Nexus) reappears on the club drug scene. http:// www.Justice.gov/archive/ndic/pubs0/665. Published May 2001. Accessed June 12, 2015.
7. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
8. Masand PS, Levenson JL, et al. Mania, catatonia, and psychosis. In: Levenson JL, ed. The American Psychiatric Publishing textbook of psychosomatic medicine. Washington, DC: American Psychiatric Publishing; 2005: 239-241.
9. Carroll BT. The universal field of hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Lee JW. Neuroleptic-induced catatonia: clinical presentation, response to benzodiazepines, and relationship to neuroleptic malignant syndrome. J Clin Psychopharmacol. 2010;30(1):3-10.
11. Vancaester E, Santens P. Catatonia and neuroleptic malignant syndrome: two sides of a coin? Acta Neurol Belg. 2007;107(2):47-50.
12. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
13. Hatta K, Miyakawa K, Ota T, et al. Maximal response to electroconvulsive therapy for the treatment of catatonic symptoms. J ECT. 2007;23(4):233-235.
14. Payee H, Chandrasekaran R, Raju GV. Catatonic syndrome: treatment response to Lorazepam. Indian J Psychiatry. 1999;41(1):49-53.
15. Rohland BM, Carroll BT, Jacoby RG. ECT in the treatment of the catatonic syndrome. J Affect Disord. 1993;29(4):255-261.
1. Dean BV, Stellpflug SJ, Burnett AM, et al. 2C or not 2C: phenethylamine designer drug review. J Med Toxicol. 2013;9(2):172-178.
2. Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
3. Shulgin A, Shulgin A. PiHKAL: a chemical love story. Berkley, CA: Transform Press; 1991.
4. Papoutsis I, Nikolaou P, Stefanidou M, et al. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicology. 2015;3(1):1-11.
5. Caudevilla-Gálligo F, Riba J, Ventura M, et al. 4-Bromo-2, 5-dimethoxyphenethylamine (2C-B): presence in the recreational drug market in Spain, pattern of use and subjective effects. J Psychopharmacol. 2012;26(7):1026-1035.
6. National Drug Intelligence Center. Information bulletin: 2C-B (Nexus) reappears on the club drug scene. http:// www.Justice.gov/archive/ndic/pubs0/665. Published May 2001. Accessed June 12, 2015.
7. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
8. Masand PS, Levenson JL, et al. Mania, catatonia, and psychosis. In: Levenson JL, ed. The American Psychiatric Publishing textbook of psychosomatic medicine. Washington, DC: American Psychiatric Publishing; 2005: 239-241.
9. Carroll BT. The universal field of hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Lee JW. Neuroleptic-induced catatonia: clinical presentation, response to benzodiazepines, and relationship to neuroleptic malignant syndrome. J Clin Psychopharmacol. 2010;30(1):3-10.
11. Vancaester E, Santens P. Catatonia and neuroleptic malignant syndrome: two sides of a coin? Acta Neurol Belg. 2007;107(2):47-50.
12. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
13. Hatta K, Miyakawa K, Ota T, et al. Maximal response to electroconvulsive therapy for the treatment of catatonic symptoms. J ECT. 2007;23(4):233-235.
14. Payee H, Chandrasekaran R, Raju GV. Catatonic syndrome: treatment response to Lorazepam. Indian J Psychiatry. 1999;41(1):49-53.
15. Rohland BM, Carroll BT, Jacoby RG. ECT in the treatment of the catatonic syndrome. J Affect Disord. 1993;29(4):255-261.
3,4-methylenedioxy-methamphetamine, MDMA, Phenethylamines I Have Known and Loved: A Chemical Love Story, LSD, substance abuse, substance use
3,4-methylenedioxy-methamphetamine, MDMA, Phenethylamines I Have Known and Loved: A Chemical Love Story, LSD, substance abuse, substance use
Malnourished and psychotic, and found incompetent to stand trial
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
Urine drug screens: When might a test result be false-positive?
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.

However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.

There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.
However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.
There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.
However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.
There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.

Evidence-based Management of Newly Diagnosed Chronic Lymphocytic Leukemia
From the Division of Hematology, Ohio State University, Columbus, OH.
Abstract
- Objective: To describe the diagnosis and initial management of chronic lymphocytic leukemia (CLL), including first-line treatment options.
- Methods: Case presentation and review of the literature.
- Results: Most CLL patients demonstrate a chronic, relapsing and remitting course with intervals of months to years between treatments. Recent advances in genetic and molecular markers for risk stratification of CLL significantly impact how clinicians determine prognosis and predict response to treatment for patients with newly diagnosed disease. This information, along with patient factors such as age and health status, should be considered when formulating an initial treatment strategy. Combinations of chemotherapy and immunotherapy offer the longest progression-free survival and overall survival benefit yet reported. For elderly patients or those with significant comorbidities who may not tolerate standard chemoimmunotherapy, less intensive but still effective therapies now exist. Patients with the highest risk disease, such as those with deletions of chromosome 17p, respond poorly to conventional treatment and should be referred to experienced centers where investigational therapies and allogeneic stem cell transplantation are available.
- Conclusion: Both disease characteristics and patient factors should guide the selection among the various effective therapies for CLL. While chemoimmunotherapy is the most effective treatment developed to date, its use may become less prevalent as newer agents are incorporated into initial and relapse treatment algorithms.
Chronic lymphocytic leukemia (CLL) is a chronic malignancy of B-lymphocytes demonstrating a heterogeneous clinical course ranging from indolent to more rapidly progressive. The chief clinical feature is an elevated peripheral blood lymphocyte count, and patients can demonstrate lymphadenopathy, splenomegaly, hepatomegaly, constitutional symptoms, and in late stages bone marrow failure. It is the most common leukemia among adults in the Western world, accounting for between 22% to 30% of new leukemia diagnoses worldwide [1]. Recent incidence rates in the United States are 3.83 cases per 100,000 person-years [2]. The incidence of CLL increases with age, and most new cases are diagnosed in persons 65 years of age or older [1,2]. As reported 5-year survival rates are between 68% and 81% with a median survival of 10 years in some series, the prevalence is significantly higher than the incidence [3]. However, this may even be an underestimate of the population burden of disease, as many cases are not reported to tumor registries [4].
Many patients with CLL are asymptomatic and do not require treatment until years after diagnosis. In these cases a watch and wait approach is taken. The typical natural history of CLL is characterized by periods of effective treatment when required, followed by treatment-free intervals of several years in many cases. However, this can be misleading, as the clinical course for any individual patient is highly variable. Development of cytogenetic and molecular testing has allowed for identification of patients with a higher risk of progression and lower response rates to traditional cytotoxic treatments [5]. For example, depending on chromosomal abnormalities present, median survival can vary from 32 to 133 months [3].
The assessment of underlying disease risk thus provides important information when considering a treatment approach and should be routinely performed for newly diagnosed patients. While the development of highly effective chemoimmunotherapy has allowed most groups of CLL patients to live for many years, some groups do not enjoy the same survival. Recent advances in CLL treatment seek to abrogate such adverse risk factors, thereby improving the survival for all patients with CLL. Given the expected survival of years for most CLL patients, frontline treatment planning must be done in the context of a long-term treatment strategy keeping the risk for late toxicities, such as secondary malignancies, in mind.
Case Study
Initial Presentation
A 50-year-old man is referred for evaluation of cervical lymphadenopathy that had progressed over the prior 6 months. He denies associated symptoms of fatigue, fevers, night sweats, or unintentional weight loss but does report early satiety. On examination there are multiple mobile, enlarged cervical lymph nodes bilaterally. Axillary lymph nodes are likewise enlarged. The liver edge is not palpable, but the spleen is palpable below the belt line. Complete blood count reveals a white blood cell count of 196,000 with 97% lymphocytes. Hemoglobin is 11.0 g/dL and platelet count is 122,000/dL. He recalls being told 3 years previously that his white blood cell count was 48,000 during an emergency department visit for cellulitis.
• How is CLL diagnosed and staged?
CLL is often suspected when patients present with an elevated lymphocyte count. Presenting symptoms of CLL commonly include lymphadenopathy, an enlarged spleen, and constitutional or “B” symptoms such as fatigue, unintentional weight loss, or drenching night sweats. However, only 25% of patients are symptomatic at diagnosis [1]. Many patients with CLL are now diagnosed after a routine blood test, long before the disease is clinically apparent.
The diagnosis of CLL can be made from the peripheral blood and does not require a bone marrow biopsy. According to 2008 guidelines from the International Workshop on Chronic Lymphocytic Leukemia (IWCLL), diagnosis requires at least 5000/uL clonal B-lymphocytes in the peripheral blood. The clonality must be confirmed by immunophenotyping. At time of diagnosis the peripheral blood smear should be examined for the characteristic cells: small mature lymphocytes with a narrow rim of cytoplasm and dense nuclei consisting of clumped chromatin. Larger, atypical cells can be present as long as they do not exceed 55% of the total number of lymphocytes [6].
The immunophenotype of CLL includes aberrant expression of CD5 and a T-cell antigen, along with the characteristic B-cell antigens CD19, CD20, and CD23. The leukemic clone may be either kappa or lambda light chain restricted. Expression of surface immunoglobulin, CD20, and CD79a is typically low compared to that of normal B cells, although there can be some variability in the immunophenotype [6].
![]()
Care should be taken to exclude other malignancies with a similar morphology. Leukemic phase mantle cell lymphoma, other low grade lymphomas, and hairy cell leukemia are commonly mistaken for CLL. Immunophenotyping and cytogenetics are usually sufficient to differentiate these. Testing for a balanced translocation involving chromosomes 11 and 14 to exclude mantle cell lymphoma can be helpful, as both CLL and mantle cell lymphoma can appear morphologically similar and share immunophenotypic features (CD5+/CD19+).

Case Continued
The patient’s peripheral blood is drawn for routine immunophenotyping as well as cytogenetic and molecular testing. When he returns to discuss the results 10 days later, he learns that peripheral blood immunophenotyping demonstrates a dim kappa restricted monoclonal population of B-cells that expressed CD19, CD20(dim), CD23, CD38, CD5, and CD43. The lymphocytes are negative for CD10, FMC7, and CD79b, consistent with a CLL immunophenotype. This patient fulfills diagnostic criteria for CLL and has Rai stage II or intermediate-risk disease. Interphase cytogenetic studies of the peripheral blood demonstrate deletions of chromosomes 11q22.3 and 13q14.3. The immunoglobulin heavy chain gene (IGHV) is unmutated.
• How can a CLL patient’s disease risk be characterized?
Historically, staging at diagnosis, pattern of bone marrow infiltration, and response to therapy were used to gauge prognosis. In more recent years, cytogenetic and molecular testing methods have been developed to augment risk stratification. Testing of prognostic significance that influences clinical management includes IGHV mutational status and interphase cytogenetics using FISH [3,12–14]. Expression of ZAP-70 and CD38 are both independent predictors of poorer prognosis in CLL but are not recommended for routine clinical use. Standardized methodology for the measurement of Zap-70 in particular limits the utility of that test in routine clinical practice [15]. Performed at diagnosis, a time when many patients are asymptomatic, cytogenetic testing with FISH and IGHV mutational analysis can predict time to first treatment and increasingly identify high-risk patients for whom investigational early intervention approaches may be considered [16]. While cytogenetic testing has utility at time of diagnosis, it should be considered necessary prior to deciding on the first-line treatment.

Cytogenetics are also important in predicting response to therapy. For instance, patients with deletion(11q) disease have improved survival when treated with regimens containing an alkylating agent [18]. Deletion(17p) patients respond poorly to traditional cytotoxic agents, and treatments with alternate mechanisms of action should be used [5,19]. The gene for tumor suppressor protein TP53 is encoded in this region of chromosome 17, thus treatment with agents that act independent of pathways involving TP53 are preferred [20].
In addition to cytogenetic testing, quantization of somatic mutations in the gene encoding the variable region of the immune globulin heavy chain gene (IGHV) can help define disease-specific risk. When greater than 98% sequence homology is seen, the gene is considered IGHV unmutated. Patients with an unmutated IGHV have worse overall survival. In one study of Rai stage 0 CLL patients, those with an unmutated IGHV had a survival of only 95 months, compared with 293 months in the mutated group [12].
• When should CLL be treated?
CLL is not curable with current standard therapies, and starting treatment at time of diagnosis for early stage, asymptomatic, CLL patients does not improve overall survival and adds treatment-related toxicities [21,22]. Consequently, the decision to treat is based on treating or preventing complications from the disease, and observation is recommended for most asymptomatic, early-stage patients [6]. Because median survival in CLL is often measured in years, deferring treatment can limit both the short- and long-term complications of therapy, especially the significant risk of secondary malignancies associated with some therapies [23]. However, deferring treatment can significantly impact both a patient’s emotional well-being and quality of life, which should be kept in mind when first discussing the rationale for observation with asymptomatic patients [24].

causes.
For patients with anemia, neutropenia, or thrombocytopenia that is autoimmune in nature, treatment should typically begin with corticosteroids, as it would for non-CLL associated cases of autoimmune cytopenias. If steroids are not effective, second-line treatments appropriate for the situation are generally employed, including intravenous immunoglobulin, cyclosporine, azathioprine, and splenectomy. Rituximab has also been shown to be effective in steroid-refractory cases of autoimmune hemolytic anemia associated with CLL [26]. Only if cytopenias are refractory to appropriate second-line therapy should CLL-directed treatments be considered, assuming there are no other indications to treat the underlying CLL [6]. Bone marrow biopsy can be helpful in differentiating autoimmune cytopenias from marrow failure due to CLL infiltration.
• What treatments are most appropriate for young, fit patients?
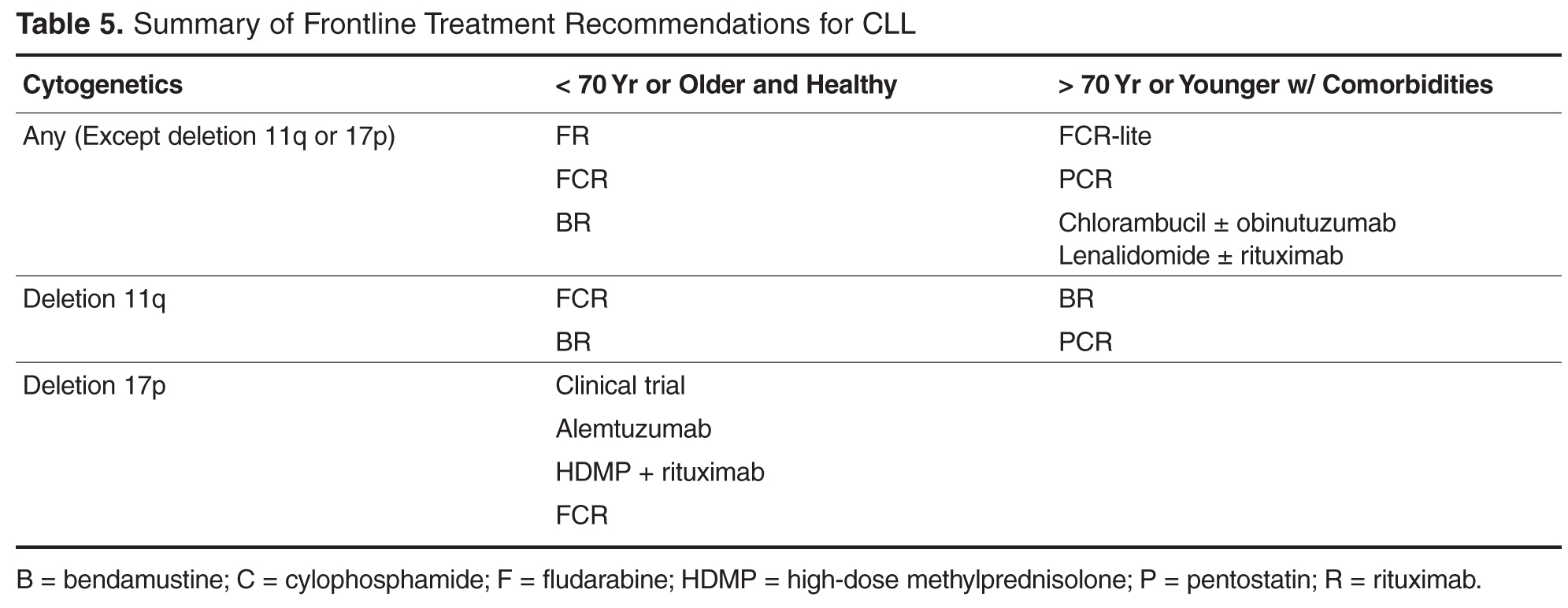
For younger patients who are in good general health, the standard treatment choice is combination chemoimmunotherapy. While single agent therapies can effectively palliate symptoms in most cases, they do not offer a survival benefit. Treatment with chemoimmunotherapy, consisting of cytotoxic chemotherapy given in combination with an anti-CD20 monoclonal antibody (generally rituximab), results in high response rates and conveys an advantage with respect to both progression-free survival (PFS) and overall survival (OS). Several chemoimmunotherapy regimens are commonly used.
As compared to fludarabine alone, frontline therapy with the combination of rituximab and fludarabine (FR) results in both a higher overall response rate (84% compared with 63% with fludarabine alone) and more complete responses (38% compared with 20% with fludarabine alone). The probability of PFS at 2 years is also better with FR: 67% compared to 45% with single agent fludarabine [28,29]. Neutropenia is more common with the combination regimen but does not appear to increase the rate of infection. Rituximab infusion reactions are commonly observed, so a stepped-up dosing schedule was developed to decrease their incidence and severity.
Fludarabine, cyclophosphamide, and rituximab (FCR) is another highly effective regimen. This combination has similar efficacy to FR with a 90% to 95% overall response rate (ORR) and 44% to 70% complete response (CR) rate [19,30]. Long-term results with this regimen are favorable; 6-year OS of 77% and median time to progression of 80 months have been reported in a follow-up study [31]. However, hematologic toxicity, including severe neutropenia, is common, and many patients are unable to complete all planned therapy [19]. The addition of cyclophosphamide does appear to be especially important for patients with a deletion(11q). Several clinical trials have consistently found that measures of response and survival are improved for deletion(11q) patients receiving an alkylating agent in addition to a nucleoside analogue [18,32,33]. Outcomes in patients with deletion(17p) disease remain poor after FCR; this subset demonstrates the shortest PFS at only 11.5 months [19].
A more recently developed chemoimmunotherapy option for younger, fit patients is bendamustine and rituximab (BR). Bendamustine has structural similarities to both alkylating agents and purine analogues, and is significantly more efficacious than chlorambucil as a single agent [34]. The combination is generally well tolerated, and a phase 2 trial of the combination reported an overall response rate (ORR) of 88.0% [32]. Notably, when the results were examined by genetic risk group, the regimen remained effective for deletion(11q) patients, who achieved overall and CR rates of 90% and 40%, respectively. Unfortunately, only 37.5% of deletion(17p) patients responded, and no patients achieved a CR [32].
The risk for therapy-related neoplasms should be taken into account when selecting initial therapy given the expected long-term survival of most CLL patients. About 8 out of 300 FCR-treated patients developed a therapy-related neoplasm in one study [31]. Treatment with FR, which does not include an alkylating agent, does not appear to have the same risk. In a study reporting long-term follow-up on 104 patients treated with FR, none developed a therapy related neoplasm [35]. Risks associated with bendamustine have not been well characterized but appear to be lower than FC. While inclusion of an alkylating agent is important for deletion(11q) patients, it is not clear if other patients similarly benefit, thus meriting the potentially increased risk for second cancers.
Fortunately, the choice among these similarly effective regimens will soon be based on high-quality, comparative data. FCR and BR have now been directly compared as a first-line treatment in the German CLL Study Group CLL10 trial. At interim analysis, both regimens had the same ORR and 2-year OS. However, CRs were less common in the BR group (38.1% versus 47.4% with FCR) and PFS was likewise inferior. Expectedly, the FCR group experienced more myelotoxicity and infections. The rate of severe neutropenia with FCR was higher at 81.7% compared to only 56.8% with BR [36]. This may be an important consideration when selecting a regimen for individual patients. Baseline renal function may influence choice as well. The active metabolite of fludarabine is eliminated through the kidneys and patients with decreased renal function have been excluded from clinical trials of FCR [19,37]. The phase 2 study of BR included patients with impaired renal function and 35% of participants had a creatinine clearance of less than 70 mL/min. It is notable that increased toxicity was seen in this subset, including higher rates of myelosuppression and infection [32]. As few direct comparisons have been done, the choice between effective first-line chemoimmunotherapy regimens can be difficult. The final results of the CLL 10 trial, as well as the now completed CALGB 10404 trial comparing FCR to FR, will provide new evidence regarding the relative risks and benefits of these regimens, particularly for patients without high-risk chromosomal abnormalities.
• What treatments are most effective for patients with deletion(17p) CLL?
As noted above, deletion(17p) CLL responds poorly to standard treatments. This relative lack of durable response to chemoimmunotherapy appears attributable to loss of function of the tumor suppressor protein TP53 which is encoded in the affected area [20,32,38]. In vivo evidence suggests that fludarabine works through a TP53-dependent mechanism, which likely explains the poor results obtained when deletion(17p) patients are treated with fludarabine-based combinations [38]. Patients harboring deletion(17p) or TP53 mutations should thus be referred for participation in clinical trials or allogeneic stem cell transplantation [17,27].
If initial treatment of a patient with deletion(17p) begins outside of a clinical trial, it should ideally be comprised of agents that have a TP53-independent mechanism of action [20]. Alemtuzumab, a humanized monoclonal antibody against the CD52 antigen expressed on the surface of normal and malignant B- and T-lymphocytes, demonstrated ORR of 33% to 50% in studies of patients with relapsed and refractory CLL [39–42]. A retrospective analysis found that similar outcomes were seen in those who had a TP53 mutation or deletion(17p). A subsequent study of previously untreated CLL patients randomized to treatment with 12 weeks of alemtuzumab or chlorambucil found that alemtuzumab-treated deletion(17p) patients had an ORR of 64% and median PFS of 10.7 months [43]. Alemtuzumab is therefore a rational choice for first-line therapy in this population. Hematologic toxicity is frequent, however, and all patients must receive prophylaxis against and monitoring for reactivation of CMV infection [43]. Infusion reactions are common but may be reduced by subcutaneous administration without apparent loss of efficacy [42,44]. While alemtuzumab is no longer marketed in the United States for the indication of CLL, it is available free of charge from the manufacturer [45].
High-dose methylprednisolone with rituximab (HDMP-R) has also been successfully used as both salvage and first-line therapy in this group. As salvage therapy, responses were seen in greater than 90% of patients, including over 50% of deletion(17p) patients [46-48]. In treatment-naïve CLL, the ORR was 96% [49], although data for patients with deletion 17p is limited in the frontline setting. Myelotoxicity attributable to the regimen is modest, but good antimicrobial prophylaxis is warranted, as well as close monitoring for hyperglycemia in at-risk patients.
• How is treatment modified for older or less fit patients?
For patients older than 70, or those who have significant comorbidities, effective therapies are still available. As most new diagnoses of CLL are made in patients older than 65, age is but one important factor determining an individual patient’s ability to tolerate treatment. The German CLL Study Group has usefully classified elderly patients into 3 treatment groups based on fitness and goals of care. The first group of medically fit patients with a normal life expectancy, sometimes referred to as the “go go” group, generally tolerate standard chemoimmunotherapy. A second group of older patients with significant life-limiting comorbid conditions—the so-called “no go” patients —should be offered best supportive care rather than CLL-directed treatment. A third group of “slow go” patients falls in between these two; these patients have comorbidities with variable life expectancy and will likely tolerate and benefit from CLL-directed therapy [50].
While some older patients can safely receive chemoimmunotherapy at standard doses and schedules, FCR can prove intolerable for even the medically fit elderly. Because inferior outcomes have been reported among patients older than 70 [30,31], a reduced-dose FCR regimen (FCR-lite) has been studied. Doses of fludarabine and cyclophosphamide were reduced by 20% and 40% respectively and dosing frequency of rituximab was increased. The CR rate was favorable at 77%, the rate of severe neutropenia was reduced to only 13%, and most patients completed all planned therapy [51]. Alternatively, the combination of pentostatin, cyclophosphamide, and rituximab (PCR) has also been successfully used in older patients. The overall and CR rates, 91% and 63% respectively, were durable at 26 months of follow-up. Importantly, there was no statistically significant difference in response or toxicity among the 28% of patients older than 70 [52,53].
For less fit patients, chlorambucil remains a reasonable option. Chlorambucil, a well-tolerated oral alkylating agent, has been used as a frontline therapy in CLL for decades. Chlorambucil has demonstrated consistent response rates in at least 4 clinical trials and is an appropriate option for patients who cannot tolerate more intensive therapy [54]. When a multicenter phase III trial compared it directly to fludarabine in patients over 65, the PFS and OS were no different despite favorable response rates in fludarabine-treated patients [55]. The effectiveness of single-agent chlorambucil can be improved, and the tolerability maintained, with the addition of a CD20-directed monoclonal antibody [56]. Obinutuzumab, a glycolengineered type II antibody against CD20, has recently been shown to improve treatment efficacy when used in combination with chlorambucil [57]. The CLL11 trial randomized patients with comorbid conditions to 1 of 3 treatments: single-agent chlorambucil, chlorambucil with rituximab (R-Clb), or chlorambucil with obinutuzumab (G-Clb). Both chemoimmunotherapy combinations outperformed chlorambucil alone, but the inclusion of obinutuzumab was associated with higher CR rates and longer PFS than rituximab, although infusion reactions and neutropenia were more common in the obinutuzumab arm [57]. Based on this result, the US Food and Drug Administration has now approved obinutuzumab for use in combination with chlorambucil as frontline therapy. While regulatory approval is without restriction with respect to patient age or fitness, a chlorambucil backbone remains most appropriate for older patients and/or those with significant comorbidities.
• What therapies are currently under development?
Numerous targeted treatments and novel immunotherapies are under active investigation in CLL. With greater specificity for CLL, these emerging agents offer the possibility of more effective yet less toxic treatments that will undoubtedly change the landscape for future CLL therapy. These agents are currently most studied as salvage therapies, and given their targeted mechanism of action can be highly effective in relapsed and refractory patients who frequently harbor poor risk cytogenetic abnormalities such as deletion(17p). Data for these agents as initial treatment is limited. Ongoing clinical trials employing these newer agents will need to be reported before these drugs can be recommended as frontline therapies.
Frontline experience with the oral immunomodulatory agent lenalidomide is more extensive. Lenalidomide offers convenient daily dosing and a favorable toxicity profile. When given on a continuous dosing schedule to patients who were 65 years old or older, the ORR was 65%, and 88% of patients were still alive at 2 years’ follow-up. The quality of response continued to improve beyond 18 months of treatment. Neutropenia, the most common severe toxicity, complicated about a third of cycles. Tumor flare attributable to immune activation was also seen, but in most cases was low-grade and did not require intervention [58,59]. While life-threatening tumor lysis syndrome and tumor flare have been seen with lenalidomide in CLL, such concerns are largely abrogated by a lower starting dose and careful intrapatient dose titration [60]. Lenalidomide has also been combined with rituximab and yielded promising results. Sixty-nine treatment-naïve patients were treated with escalating doses of lenalidomide along with rituximab infusions starting at the end of cycle 1 in a phase 2 study. They achieved an 88% ORR with 16% CRs. Toxicities were generally manageable, but patients over 65 were less likely to reach higher doses of lenalidomide or complete all planned treatment cycles [61]. Unfortunately, the FDA recently halted accrual to a phase 3 frontline clinical trial comparing lenalidomide to chlorambucil due to excess mortality in the lenalidomide arm among patients over the age of 80 [62]. More detailed outcomes from that study should be forthcoming.
Perhaps the most remarkable recent advance in CLL medicine, however, is the advent of orally bioavailable small molecule inhibitors of the B-cell receptor (BCR) signaling pathway. BCR signaling plays a vitally important role in supporting the growth and survival of malignant B-cells, activating a number of downstream kinases (Syk, Btk, PI3K, among others) which are potential therapeutic targets. Proof of principle for this approach was demonstrated with the Syk inhibitor fostamatinib in a phase 1/2 trial enrolling patients with B-cell non-Hodgkin lymphoma and CLL. CLL/SLL patients had the highest response rates of any subgroup in that study, with 6 out of 11 patients responding [63]. In a subsequent phase 1b study of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib, durable partial remissions were reported in more than 70% of multiply relapsed and refractory patients, including genetically high-risk patients [64–66]. Ibrutinib appears safer and better tolerated than traditional chemoimmunotherapy in the relapsed setting; consequently, it is now being studied as a first-line therapy both alone and in combination with other agents [67]. Other BCR signaling agents under study, such as the phosphatidylinositol 3-kinase inhibitor idelalisib, demonstrate similar safety and high response rates across both genetic risk and patient age groups [68].
New targeted drugs are not limited to the BCR signaling pathway. ABT-199 inhibits B-cell leukemia/lymphoma 2 (BCL-2), which is an anti-apoptotic protein in the cell death pathway, and has demonstrated remarkable clinical efficacy in relapsed and refractory CLL patients [69]. As more experience is gained with these targeted agents, it is expected that they will be rapidly incorporated into frontline therapies. However, these agents are just now being studied in comparison to standard initial treatments, such as FCR, and it is not yet clear they will offer an advantage over current chemoimmunotherapy in this setting [70–72]. Since these single agents typically do not induce complete remissions, and require indefinite therapy to maintain response, optimal combination therapies are under intensive investigation.
Case Conclusion
The patient and his physician elect to begin treatment owing to symptomatic cervical lymphadenopathy and massive splenomegaly. Given the presence of a deletion(11q) abnormality, but hoping to limit the risk for both short- and long-term toxicities, this younger, fit patient is treated with 6 cycles of bendamustine and rituximab. At the conclusion of treatment, neither the cervical lymph nodes nor spleen remain palpable. His blood counts have also normalized, with a white blood cell count of 4700 with 8.1% lymphocyotes, hemoglobin of 14.3 gm/dL, and platelets of 151,000/dL.
Summary
CLL follows a chronic course requiring treatment at variable intervals. Both genetic risk features and patient factors should be considered when determining initial therapy. Cytogenetic and molecular testing can characterize the likelihood of treatment success, information useful for treatment planning. Chemoimmunotherapy is highly effective for most patients, including patients with deletion(11q) CLL, where the inclusion of an alkylating agent in frontline therapy alters the natural history of disease. However, patients with deletion(17p) and or TP53-mutated disease respond poorly to standard treatment and should be considered for investigational therapies [73]. Novel approaches to CLL therapy, most notably immunotherapies and BCR-targeted agents, hold the promise to further improve outcomes, particularly for the highest risk patients and those elderly and/or infirm patients who tolerate chemotherapy poorly. Frontline therapy should rapidly evolve as emerging agents enter advanced phase investigation.
Corresponding author: Jeffrey Jones, MD, MPH, Div. of Hematology, Ohio State University, A350B Starling Loving Hall, 320 West 10th Ave., Columbus, OH 43210, [email protected].
Financial disclosures: Dr. Jones disclosed that he is on the advisory boards and has received research support from Genentech, Pharmacyclics, and Gilead.
Author contributions: conception and design, KAR, JAJ; analysis and interpretation of data, KAR, JAJ; drafting of article, KAR, JAJ; critical revision of the article, KAR, JAJ.
1. Redaelli A, Laskin BL, Stephens JM, et al. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur J Cancer Care (Engl) 2004;13:279–87.
2. Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007;139:809–19.
3. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6.
4. Zent CS, Kyasa MJ, Evans R, Schichman SA. Chronic lymphocytic leukemia incidence is substantially higher than estimated from tumor registry data. Cancer 2001;92:1325–30.
5. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006;24:437–43.
6. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56.
7. Rawstron AC, Bennett FL, O'Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83.
8. Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–34.
9. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
10. Binet JL, Lepoprier M, Dighiero G, et al. A clinical staging system for chronic lymphocytic leukemia: prognostic significance. Cancer 1977:40:855–64.
11. Eichhorst BF, Fischer K, Fink AM, et al. Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 2011;117:1817–21.
12. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54.
13. Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:
1764–75.
14. Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–9.
15. Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006;70:209–13.
16. Wierda WG, O'Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–95.
17. Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 2008;26:5094–100.
18. Ding W, Ferrajoli A. Evidence-based mini-review: the role of alkylating agents in the initial treatment of chronic lymphocytic leukemia patients with the 11q deletion. Hematology Am Soc Hematol Educ Program 2010;2010:90–2.
19. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
20. Badoux XC, Keating MJ, Wierda WG.What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011;6:36–46.
21. Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–14.
22. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 1999;91:861–8.
23. Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 2010;28:4935–44.
24. Shanafelt TD, Bowen D, Venkat C, et al. Quality of life in chronic lymphocytic leukemia: an international survey of 1482 patients. Br J Haematol 2007;139:255–64.
25. Baer MR, Stein RS, Dessypris EN. Chronic lymphocytic leukemia with hyperleukocytosis. The hyperviscosity syndrome. Cancer 1985;56:2865–9.
26. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 2002;16:2092–5.
27. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: non-hodgkin's lymphomas. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
28. Byrd JC, Rai K, Peterson BL, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49–53.
29. Byrd JC, Peterson BL, Morrison VA, et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003;101:6–14.
30. Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079–88.
31. Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008;112:975–80.
32. Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16.
33. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9.
34. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–84.
35. Woyach JA, Ruppert AS, Heerema NA, et al. Chemoimmunotherapy with fludarabine and rituximab produces extended overall survival and progression-free survival in chronic lymphocytic leukemia: long-term follow-up of CALGB study 9712. J Clin Oncol 2011;29:1349–55.
36. Fink AM, et al., Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximabversus bendamustine and rituximabin previously untreated and physically fit patientswith advanced chronic lymphocytic leukemia: results of a planned interim analysis of the CLL10 Trial, an international, randomized study of the German CLL Study Group (GCLLSG). Blood 2013;122:526.
37. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet 2002;41:93–103.
38. Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood 2004;104:1428–34.
39. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61.
40. Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81.
41. Osuji NC, Del Giudice I, Matutes E, et al, The efficacy of alemtuzumab for refractory chronic lymphocytic leukemia in relation to cytogenetic abnormalities of p53. Haematologica 2005;90:1435–6.
42. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
43. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–23.
44. Lundin J, Kimby E, Björkholm M, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002;100:768–73.
45. Genzyme. US Campath Distribution Program. Cambridge, MA: Genzyme. Available at http://www.campath.com/.
46. Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65.
47. Bowen DA, Call TG, Jenkins GD, et al. Methylprednisolone-rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007;48:2412–7.
48. Castro JE, Sandoval-Sus JD, Bole J, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of fludarabine refractory high-risk chronic lymphocytic leukemia. Leukemia 2008;22:2048–53.
49. Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89.
50. Eichhorst B, Goede V, Hallek M. Treatment of elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009;50:171–8.
51. Foon KA, Boyiadzis M, Land SR, et al. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498–503.
52. Kay NE, Geyer SM, Call TG, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 2007;109:405–11.
53. Shanafelt TD, Lin T, Geyer SM, et al. Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer 2007;109:2291–8.
54. Catovsky D, Else M, Richards S. Chlorambucil--still not bad: a reappraisal. Clin Lymphoma Myeloma Leuk 2011;11 Suppl 1:S2–6.
55. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009;114:3382–91.
56. Laurenti L, Vannata B, Innocenti I, et al. Chlorambucil plus rituximab as front-line therapy in elderly/unfit patients affected by b-cell chronic lymphocytic leukemia: results of a single-centre experience. Mediterr J Hematol Infect Dis 2013;5:e2013031.
57. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014 Jan 8. [Epub ahead of print].
58. Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood 2011;118:3489–98.
59. Strati P, Keating MJ, Wierda WG, et al. Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 2013;122:734–7.
60. Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol 2007;25:5047.
61. James DF, Brown JR, Werner L, et al. Lenalidomide and rituximab for the initial treatment of patients with chronic lymphocytic leukemia (CLL): a multicenter study of the CLL Research Consortium. ASH Annual Meeting Abstracts 2011;118:291.
62. US Food and Drug Administration. FDA halts clinical trial of drug Revlimid (lenalidomide) for chronic lymphocytic leukemia due to safety concerns. Available at http://www.fda.gov/Drugs/DrugSafety/ucm361444.htm.
63. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010;115:2578–85.
64. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
65. Farooqui M, Aue G, Valdez J, et al. Single agent ibrutinib (PCI-32765) achieves equally good and durable responses in chronic lymphocytic leukemia (CLL) patients with and without deletion 17p. Blood 2013;122:673.
66. Byrd JC, Furman RR, Coutre S, et al. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) monotherapy demonstrates long-term safety and durability of response in chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) patients in an open-label extension study. Blood 2013;122:4163.
67. Brown JR, Barrientos JC, Barr PM, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study. ASH Annual Meeting Abstracts 2013.
68. O'Brien SM, Lamanna N, Kipps TJ, et al. A phase II study of the selective phosphatidylinositol 3-kinase delta (PI3K{delta}) inhibitor idelalisib (GS-1101) in combination with rituximab (R) in treatment-naive patients (pts) ≥ 65 years with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). J Clin Oncol 2013;31(15 Suppl); Abstract 7005.
69. Seymour JF, Davids MS, Pagel JM, et al. Bcl-2 Inhibitor ABT-199 (GDC-0199) monotherapy shows anti-tumor activity including complete remissions in high-risk relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Blood 2013;122:872.
70. Rituxumab and bendamustine hydrochloride, rituxumab and ibrutunib, or ibrutinib alone in treating older patients with previously untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT01886872?term=ibrutinib+cll&rank=8.
71. A multicenter, open-label, phase 3 study of the bruton's tyrosine kinase inhibitor pci-32765 versus chlorambucil in patients 65 years of older with treatment-naive chronic lymphocytic leukemia or small lymphocytic lymphoma (RESONATE-2) Available at http://clinicaltrials.gov/ct2/show/NCT01722487?term=ibrutinib+cll&rank=12.
72. Ibrutinib and rituximab compared with fludarabine phosphate, cyclophosphamide, and rituxumab in treating patients with untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT02048813?term=ibrutinib+cll&rank=2.
73. Strati P, Keating MJ, O'Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia (CLL) with 17p deletion. J Clin Oncol 2013;31(15 suppl): Abstract 7102.
From the Division of Hematology, Ohio State University, Columbus, OH.
Abstract
- Objective: To describe the diagnosis and initial management of chronic lymphocytic leukemia (CLL), including first-line treatment options.
- Methods: Case presentation and review of the literature.
- Results: Most CLL patients demonstrate a chronic, relapsing and remitting course with intervals of months to years between treatments. Recent advances in genetic and molecular markers for risk stratification of CLL significantly impact how clinicians determine prognosis and predict response to treatment for patients with newly diagnosed disease. This information, along with patient factors such as age and health status, should be considered when formulating an initial treatment strategy. Combinations of chemotherapy and immunotherapy offer the longest progression-free survival and overall survival benefit yet reported. For elderly patients or those with significant comorbidities who may not tolerate standard chemoimmunotherapy, less intensive but still effective therapies now exist. Patients with the highest risk disease, such as those with deletions of chromosome 17p, respond poorly to conventional treatment and should be referred to experienced centers where investigational therapies and allogeneic stem cell transplantation are available.
- Conclusion: Both disease characteristics and patient factors should guide the selection among the various effective therapies for CLL. While chemoimmunotherapy is the most effective treatment developed to date, its use may become less prevalent as newer agents are incorporated into initial and relapse treatment algorithms.
Chronic lymphocytic leukemia (CLL) is a chronic malignancy of B-lymphocytes demonstrating a heterogeneous clinical course ranging from indolent to more rapidly progressive. The chief clinical feature is an elevated peripheral blood lymphocyte count, and patients can demonstrate lymphadenopathy, splenomegaly, hepatomegaly, constitutional symptoms, and in late stages bone marrow failure. It is the most common leukemia among adults in the Western world, accounting for between 22% to 30% of new leukemia diagnoses worldwide [1]. Recent incidence rates in the United States are 3.83 cases per 100,000 person-years [2]. The incidence of CLL increases with age, and most new cases are diagnosed in persons 65 years of age or older [1,2]. As reported 5-year survival rates are between 68% and 81% with a median survival of 10 years in some series, the prevalence is significantly higher than the incidence [3]. However, this may even be an underestimate of the population burden of disease, as many cases are not reported to tumor registries [4].
Many patients with CLL are asymptomatic and do not require treatment until years after diagnosis. In these cases a watch and wait approach is taken. The typical natural history of CLL is characterized by periods of effective treatment when required, followed by treatment-free intervals of several years in many cases. However, this can be misleading, as the clinical course for any individual patient is highly variable. Development of cytogenetic and molecular testing has allowed for identification of patients with a higher risk of progression and lower response rates to traditional cytotoxic treatments [5]. For example, depending on chromosomal abnormalities present, median survival can vary from 32 to 133 months [3].
The assessment of underlying disease risk thus provides important information when considering a treatment approach and should be routinely performed for newly diagnosed patients. While the development of highly effective chemoimmunotherapy has allowed most groups of CLL patients to live for many years, some groups do not enjoy the same survival. Recent advances in CLL treatment seek to abrogate such adverse risk factors, thereby improving the survival for all patients with CLL. Given the expected survival of years for most CLL patients, frontline treatment planning must be done in the context of a long-term treatment strategy keeping the risk for late toxicities, such as secondary malignancies, in mind.
Case Study
Initial Presentation
A 50-year-old man is referred for evaluation of cervical lymphadenopathy that had progressed over the prior 6 months. He denies associated symptoms of fatigue, fevers, night sweats, or unintentional weight loss but does report early satiety. On examination there are multiple mobile, enlarged cervical lymph nodes bilaterally. Axillary lymph nodes are likewise enlarged. The liver edge is not palpable, but the spleen is palpable below the belt line. Complete blood count reveals a white blood cell count of 196,000 with 97% lymphocytes. Hemoglobin is 11.0 g/dL and platelet count is 122,000/dL. He recalls being told 3 years previously that his white blood cell count was 48,000 during an emergency department visit for cellulitis.
• How is CLL diagnosed and staged?
CLL is often suspected when patients present with an elevated lymphocyte count. Presenting symptoms of CLL commonly include lymphadenopathy, an enlarged spleen, and constitutional or “B” symptoms such as fatigue, unintentional weight loss, or drenching night sweats. However, only 25% of patients are symptomatic at diagnosis [1]. Many patients with CLL are now diagnosed after a routine blood test, long before the disease is clinically apparent.
The diagnosis of CLL can be made from the peripheral blood and does not require a bone marrow biopsy. According to 2008 guidelines from the International Workshop on Chronic Lymphocytic Leukemia (IWCLL), diagnosis requires at least 5000/uL clonal B-lymphocytes in the peripheral blood. The clonality must be confirmed by immunophenotyping. At time of diagnosis the peripheral blood smear should be examined for the characteristic cells: small mature lymphocytes with a narrow rim of cytoplasm and dense nuclei consisting of clumped chromatin. Larger, atypical cells can be present as long as they do not exceed 55% of the total number of lymphocytes [6].
The immunophenotype of CLL includes aberrant expression of CD5 and a T-cell antigen, along with the characteristic B-cell antigens CD19, CD20, and CD23. The leukemic clone may be either kappa or lambda light chain restricted. Expression of surface immunoglobulin, CD20, and CD79a is typically low compared to that of normal B cells, although there can be some variability in the immunophenotype [6].
![]()
Care should be taken to exclude other malignancies with a similar morphology. Leukemic phase mantle cell lymphoma, other low grade lymphomas, and hairy cell leukemia are commonly mistaken for CLL. Immunophenotyping and cytogenetics are usually sufficient to differentiate these. Testing for a balanced translocation involving chromosomes 11 and 14 to exclude mantle cell lymphoma can be helpful, as both CLL and mantle cell lymphoma can appear morphologically similar and share immunophenotypic features (CD5+/CD19+).
Case Continued
The patient’s peripheral blood is drawn for routine immunophenotyping as well as cytogenetic and molecular testing. When he returns to discuss the results 10 days later, he learns that peripheral blood immunophenotyping demonstrates a dim kappa restricted monoclonal population of B-cells that expressed CD19, CD20(dim), CD23, CD38, CD5, and CD43. The lymphocytes are negative for CD10, FMC7, and CD79b, consistent with a CLL immunophenotype. This patient fulfills diagnostic criteria for CLL and has Rai stage II or intermediate-risk disease. Interphase cytogenetic studies of the peripheral blood demonstrate deletions of chromosomes 11q22.3 and 13q14.3. The immunoglobulin heavy chain gene (IGHV) is unmutated.
• How can a CLL patient’s disease risk be characterized?
Historically, staging at diagnosis, pattern of bone marrow infiltration, and response to therapy were used to gauge prognosis. In more recent years, cytogenetic and molecular testing methods have been developed to augment risk stratification. Testing of prognostic significance that influences clinical management includes IGHV mutational status and interphase cytogenetics using FISH [3,12–14]. Expression of ZAP-70 and CD38 are both independent predictors of poorer prognosis in CLL but are not recommended for routine clinical use. Standardized methodology for the measurement of Zap-70 in particular limits the utility of that test in routine clinical practice [15]. Performed at diagnosis, a time when many patients are asymptomatic, cytogenetic testing with FISH and IGHV mutational analysis can predict time to first treatment and increasingly identify high-risk patients for whom investigational early intervention approaches may be considered [16]. While cytogenetic testing has utility at time of diagnosis, it should be considered necessary prior to deciding on the first-line treatment.
Cytogenetics are also important in predicting response to therapy. For instance, patients with deletion(11q) disease have improved survival when treated with regimens containing an alkylating agent [18]. Deletion(17p) patients respond poorly to traditional cytotoxic agents, and treatments with alternate mechanisms of action should be used [5,19]. The gene for tumor suppressor protein TP53 is encoded in this region of chromosome 17, thus treatment with agents that act independent of pathways involving TP53 are preferred [20].
In addition to cytogenetic testing, quantization of somatic mutations in the gene encoding the variable region of the immune globulin heavy chain gene (IGHV) can help define disease-specific risk. When greater than 98% sequence homology is seen, the gene is considered IGHV unmutated. Patients with an unmutated IGHV have worse overall survival. In one study of Rai stage 0 CLL patients, those with an unmutated IGHV had a survival of only 95 months, compared with 293 months in the mutated group [12].
• When should CLL be treated?
CLL is not curable with current standard therapies, and starting treatment at time of diagnosis for early stage, asymptomatic, CLL patients does not improve overall survival and adds treatment-related toxicities [21,22]. Consequently, the decision to treat is based on treating or preventing complications from the disease, and observation is recommended for most asymptomatic, early-stage patients [6]. Because median survival in CLL is often measured in years, deferring treatment can limit both the short- and long-term complications of therapy, especially the significant risk of secondary malignancies associated with some therapies [23]. However, deferring treatment can significantly impact both a patient’s emotional well-being and quality of life, which should be kept in mind when first discussing the rationale for observation with asymptomatic patients [24].
causes.
For patients with anemia, neutropenia, or thrombocytopenia that is autoimmune in nature, treatment should typically begin with corticosteroids, as it would for non-CLL associated cases of autoimmune cytopenias. If steroids are not effective, second-line treatments appropriate for the situation are generally employed, including intravenous immunoglobulin, cyclosporine, azathioprine, and splenectomy. Rituximab has also been shown to be effective in steroid-refractory cases of autoimmune hemolytic anemia associated with CLL [26]. Only if cytopenias are refractory to appropriate second-line therapy should CLL-directed treatments be considered, assuming there are no other indications to treat the underlying CLL [6]. Bone marrow biopsy can be helpful in differentiating autoimmune cytopenias from marrow failure due to CLL infiltration.
• What treatments are most appropriate for young, fit patients?
For younger patients who are in good general health, the standard treatment choice is combination chemoimmunotherapy. While single agent therapies can effectively palliate symptoms in most cases, they do not offer a survival benefit. Treatment with chemoimmunotherapy, consisting of cytotoxic chemotherapy given in combination with an anti-CD20 monoclonal antibody (generally rituximab), results in high response rates and conveys an advantage with respect to both progression-free survival (PFS) and overall survival (OS). Several chemoimmunotherapy regimens are commonly used.
As compared to fludarabine alone, frontline therapy with the combination of rituximab and fludarabine (FR) results in both a higher overall response rate (84% compared with 63% with fludarabine alone) and more complete responses (38% compared with 20% with fludarabine alone). The probability of PFS at 2 years is also better with FR: 67% compared to 45% with single agent fludarabine [28,29]. Neutropenia is more common with the combination regimen but does not appear to increase the rate of infection. Rituximab infusion reactions are commonly observed, so a stepped-up dosing schedule was developed to decrease their incidence and severity.
Fludarabine, cyclophosphamide, and rituximab (FCR) is another highly effective regimen. This combination has similar efficacy to FR with a 90% to 95% overall response rate (ORR) and 44% to 70% complete response (CR) rate [19,30]. Long-term results with this regimen are favorable; 6-year OS of 77% and median time to progression of 80 months have been reported in a follow-up study [31]. However, hematologic toxicity, including severe neutropenia, is common, and many patients are unable to complete all planned therapy [19]. The addition of cyclophosphamide does appear to be especially important for patients with a deletion(11q). Several clinical trials have consistently found that measures of response and survival are improved for deletion(11q) patients receiving an alkylating agent in addition to a nucleoside analogue [18,32,33]. Outcomes in patients with deletion(17p) disease remain poor after FCR; this subset demonstrates the shortest PFS at only 11.5 months [19].
A more recently developed chemoimmunotherapy option for younger, fit patients is bendamustine and rituximab (BR). Bendamustine has structural similarities to both alkylating agents and purine analogues, and is significantly more efficacious than chlorambucil as a single agent [34]. The combination is generally well tolerated, and a phase 2 trial of the combination reported an overall response rate (ORR) of 88.0% [32]. Notably, when the results were examined by genetic risk group, the regimen remained effective for deletion(11q) patients, who achieved overall and CR rates of 90% and 40%, respectively. Unfortunately, only 37.5% of deletion(17p) patients responded, and no patients achieved a CR [32].
The risk for therapy-related neoplasms should be taken into account when selecting initial therapy given the expected long-term survival of most CLL patients. About 8 out of 300 FCR-treated patients developed a therapy-related neoplasm in one study [31]. Treatment with FR, which does not include an alkylating agent, does not appear to have the same risk. In a study reporting long-term follow-up on 104 patients treated with FR, none developed a therapy related neoplasm [35]. Risks associated with bendamustine have not been well characterized but appear to be lower than FC. While inclusion of an alkylating agent is important for deletion(11q) patients, it is not clear if other patients similarly benefit, thus meriting the potentially increased risk for second cancers.
Fortunately, the choice among these similarly effective regimens will soon be based on high-quality, comparative data. FCR and BR have now been directly compared as a first-line treatment in the German CLL Study Group CLL10 trial. At interim analysis, both regimens had the same ORR and 2-year OS. However, CRs were less common in the BR group (38.1% versus 47.4% with FCR) and PFS was likewise inferior. Expectedly, the FCR group experienced more myelotoxicity and infections. The rate of severe neutropenia with FCR was higher at 81.7% compared to only 56.8% with BR [36]. This may be an important consideration when selecting a regimen for individual patients. Baseline renal function may influence choice as well. The active metabolite of fludarabine is eliminated through the kidneys and patients with decreased renal function have been excluded from clinical trials of FCR [19,37]. The phase 2 study of BR included patients with impaired renal function and 35% of participants had a creatinine clearance of less than 70 mL/min. It is notable that increased toxicity was seen in this subset, including higher rates of myelosuppression and infection [32]. As few direct comparisons have been done, the choice between effective first-line chemoimmunotherapy regimens can be difficult. The final results of the CLL 10 trial, as well as the now completed CALGB 10404 trial comparing FCR to FR, will provide new evidence regarding the relative risks and benefits of these regimens, particularly for patients without high-risk chromosomal abnormalities.
• What treatments are most effective for patients with deletion(17p) CLL?
As noted above, deletion(17p) CLL responds poorly to standard treatments. This relative lack of durable response to chemoimmunotherapy appears attributable to loss of function of the tumor suppressor protein TP53 which is encoded in the affected area [20,32,38]. In vivo evidence suggests that fludarabine works through a TP53-dependent mechanism, which likely explains the poor results obtained when deletion(17p) patients are treated with fludarabine-based combinations [38]. Patients harboring deletion(17p) or TP53 mutations should thus be referred for participation in clinical trials or allogeneic stem cell transplantation [17,27].
If initial treatment of a patient with deletion(17p) begins outside of a clinical trial, it should ideally be comprised of agents that have a TP53-independent mechanism of action [20]. Alemtuzumab, a humanized monoclonal antibody against the CD52 antigen expressed on the surface of normal and malignant B- and T-lymphocytes, demonstrated ORR of 33% to 50% in studies of patients with relapsed and refractory CLL [39–42]. A retrospective analysis found that similar outcomes were seen in those who had a TP53 mutation or deletion(17p). A subsequent study of previously untreated CLL patients randomized to treatment with 12 weeks of alemtuzumab or chlorambucil found that alemtuzumab-treated deletion(17p) patients had an ORR of 64% and median PFS of 10.7 months [43]. Alemtuzumab is therefore a rational choice for first-line therapy in this population. Hematologic toxicity is frequent, however, and all patients must receive prophylaxis against and monitoring for reactivation of CMV infection [43]. Infusion reactions are common but may be reduced by subcutaneous administration without apparent loss of efficacy [42,44]. While alemtuzumab is no longer marketed in the United States for the indication of CLL, it is available free of charge from the manufacturer [45].
High-dose methylprednisolone with rituximab (HDMP-R) has also been successfully used as both salvage and first-line therapy in this group. As salvage therapy, responses were seen in greater than 90% of patients, including over 50% of deletion(17p) patients [46-48]. In treatment-naïve CLL, the ORR was 96% [49], although data for patients with deletion 17p is limited in the frontline setting. Myelotoxicity attributable to the regimen is modest, but good antimicrobial prophylaxis is warranted, as well as close monitoring for hyperglycemia in at-risk patients.
• How is treatment modified for older or less fit patients?
For patients older than 70, or those who have significant comorbidities, effective therapies are still available. As most new diagnoses of CLL are made in patients older than 65, age is but one important factor determining an individual patient’s ability to tolerate treatment. The German CLL Study Group has usefully classified elderly patients into 3 treatment groups based on fitness and goals of care. The first group of medically fit patients with a normal life expectancy, sometimes referred to as the “go go” group, generally tolerate standard chemoimmunotherapy. A second group of older patients with significant life-limiting comorbid conditions—the so-called “no go” patients —should be offered best supportive care rather than CLL-directed treatment. A third group of “slow go” patients falls in between these two; these patients have comorbidities with variable life expectancy and will likely tolerate and benefit from CLL-directed therapy [50].
While some older patients can safely receive chemoimmunotherapy at standard doses and schedules, FCR can prove intolerable for even the medically fit elderly. Because inferior outcomes have been reported among patients older than 70 [30,31], a reduced-dose FCR regimen (FCR-lite) has been studied. Doses of fludarabine and cyclophosphamide were reduced by 20% and 40% respectively and dosing frequency of rituximab was increased. The CR rate was favorable at 77%, the rate of severe neutropenia was reduced to only 13%, and most patients completed all planned therapy [51]. Alternatively, the combination of pentostatin, cyclophosphamide, and rituximab (PCR) has also been successfully used in older patients. The overall and CR rates, 91% and 63% respectively, were durable at 26 months of follow-up. Importantly, there was no statistically significant difference in response or toxicity among the 28% of patients older than 70 [52,53].
For less fit patients, chlorambucil remains a reasonable option. Chlorambucil, a well-tolerated oral alkylating agent, has been used as a frontline therapy in CLL for decades. Chlorambucil has demonstrated consistent response rates in at least 4 clinical trials and is an appropriate option for patients who cannot tolerate more intensive therapy [54]. When a multicenter phase III trial compared it directly to fludarabine in patients over 65, the PFS and OS were no different despite favorable response rates in fludarabine-treated patients [55]. The effectiveness of single-agent chlorambucil can be improved, and the tolerability maintained, with the addition of a CD20-directed monoclonal antibody [56]. Obinutuzumab, a glycolengineered type II antibody against CD20, has recently been shown to improve treatment efficacy when used in combination with chlorambucil [57]. The CLL11 trial randomized patients with comorbid conditions to 1 of 3 treatments: single-agent chlorambucil, chlorambucil with rituximab (R-Clb), or chlorambucil with obinutuzumab (G-Clb). Both chemoimmunotherapy combinations outperformed chlorambucil alone, but the inclusion of obinutuzumab was associated with higher CR rates and longer PFS than rituximab, although infusion reactions and neutropenia were more common in the obinutuzumab arm [57]. Based on this result, the US Food and Drug Administration has now approved obinutuzumab for use in combination with chlorambucil as frontline therapy. While regulatory approval is without restriction with respect to patient age or fitness, a chlorambucil backbone remains most appropriate for older patients and/or those with significant comorbidities.
• What therapies are currently under development?
Numerous targeted treatments and novel immunotherapies are under active investigation in CLL. With greater specificity for CLL, these emerging agents offer the possibility of more effective yet less toxic treatments that will undoubtedly change the landscape for future CLL therapy. These agents are currently most studied as salvage therapies, and given their targeted mechanism of action can be highly effective in relapsed and refractory patients who frequently harbor poor risk cytogenetic abnormalities such as deletion(17p). Data for these agents as initial treatment is limited. Ongoing clinical trials employing these newer agents will need to be reported before these drugs can be recommended as frontline therapies.
Frontline experience with the oral immunomodulatory agent lenalidomide is more extensive. Lenalidomide offers convenient daily dosing and a favorable toxicity profile. When given on a continuous dosing schedule to patients who were 65 years old or older, the ORR was 65%, and 88% of patients were still alive at 2 years’ follow-up. The quality of response continued to improve beyond 18 months of treatment. Neutropenia, the most common severe toxicity, complicated about a third of cycles. Tumor flare attributable to immune activation was also seen, but in most cases was low-grade and did not require intervention [58,59]. While life-threatening tumor lysis syndrome and tumor flare have been seen with lenalidomide in CLL, such concerns are largely abrogated by a lower starting dose and careful intrapatient dose titration [60]. Lenalidomide has also been combined with rituximab and yielded promising results. Sixty-nine treatment-naïve patients were treated with escalating doses of lenalidomide along with rituximab infusions starting at the end of cycle 1 in a phase 2 study. They achieved an 88% ORR with 16% CRs. Toxicities were generally manageable, but patients over 65 were less likely to reach higher doses of lenalidomide or complete all planned treatment cycles [61]. Unfortunately, the FDA recently halted accrual to a phase 3 frontline clinical trial comparing lenalidomide to chlorambucil due to excess mortality in the lenalidomide arm among patients over the age of 80 [62]. More detailed outcomes from that study should be forthcoming.
Perhaps the most remarkable recent advance in CLL medicine, however, is the advent of orally bioavailable small molecule inhibitors of the B-cell receptor (BCR) signaling pathway. BCR signaling plays a vitally important role in supporting the growth and survival of malignant B-cells, activating a number of downstream kinases (Syk, Btk, PI3K, among others) which are potential therapeutic targets. Proof of principle for this approach was demonstrated with the Syk inhibitor fostamatinib in a phase 1/2 trial enrolling patients with B-cell non-Hodgkin lymphoma and CLL. CLL/SLL patients had the highest response rates of any subgroup in that study, with 6 out of 11 patients responding [63]. In a subsequent phase 1b study of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib, durable partial remissions were reported in more than 70% of multiply relapsed and refractory patients, including genetically high-risk patients [64–66]. Ibrutinib appears safer and better tolerated than traditional chemoimmunotherapy in the relapsed setting; consequently, it is now being studied as a first-line therapy both alone and in combination with other agents [67]. Other BCR signaling agents under study, such as the phosphatidylinositol 3-kinase inhibitor idelalisib, demonstrate similar safety and high response rates across both genetic risk and patient age groups [68].
New targeted drugs are not limited to the BCR signaling pathway. ABT-199 inhibits B-cell leukemia/lymphoma 2 (BCL-2), which is an anti-apoptotic protein in the cell death pathway, and has demonstrated remarkable clinical efficacy in relapsed and refractory CLL patients [69]. As more experience is gained with these targeted agents, it is expected that they will be rapidly incorporated into frontline therapies. However, these agents are just now being studied in comparison to standard initial treatments, such as FCR, and it is not yet clear they will offer an advantage over current chemoimmunotherapy in this setting [70–72]. Since these single agents typically do not induce complete remissions, and require indefinite therapy to maintain response, optimal combination therapies are under intensive investigation.
Case Conclusion
The patient and his physician elect to begin treatment owing to symptomatic cervical lymphadenopathy and massive splenomegaly. Given the presence of a deletion(11q) abnormality, but hoping to limit the risk for both short- and long-term toxicities, this younger, fit patient is treated with 6 cycles of bendamustine and rituximab. At the conclusion of treatment, neither the cervical lymph nodes nor spleen remain palpable. His blood counts have also normalized, with a white blood cell count of 4700 with 8.1% lymphocyotes, hemoglobin of 14.3 gm/dL, and platelets of 151,000/dL.
Summary
CLL follows a chronic course requiring treatment at variable intervals. Both genetic risk features and patient factors should be considered when determining initial therapy. Cytogenetic and molecular testing can characterize the likelihood of treatment success, information useful for treatment planning. Chemoimmunotherapy is highly effective for most patients, including patients with deletion(11q) CLL, where the inclusion of an alkylating agent in frontline therapy alters the natural history of disease. However, patients with deletion(17p) and or TP53-mutated disease respond poorly to standard treatment and should be considered for investigational therapies [73]. Novel approaches to CLL therapy, most notably immunotherapies and BCR-targeted agents, hold the promise to further improve outcomes, particularly for the highest risk patients and those elderly and/or infirm patients who tolerate chemotherapy poorly. Frontline therapy should rapidly evolve as emerging agents enter advanced phase investigation.
Corresponding author: Jeffrey Jones, MD, MPH, Div. of Hematology, Ohio State University, A350B Starling Loving Hall, 320 West 10th Ave., Columbus, OH 43210, [email protected].
Financial disclosures: Dr. Jones disclosed that he is on the advisory boards and has received research support from Genentech, Pharmacyclics, and Gilead.
Author contributions: conception and design, KAR, JAJ; analysis and interpretation of data, KAR, JAJ; drafting of article, KAR, JAJ; critical revision of the article, KAR, JAJ.
From the Division of Hematology, Ohio State University, Columbus, OH.
Abstract
- Objective: To describe the diagnosis and initial management of chronic lymphocytic leukemia (CLL), including first-line treatment options.
- Methods: Case presentation and review of the literature.
- Results: Most CLL patients demonstrate a chronic, relapsing and remitting course with intervals of months to years between treatments. Recent advances in genetic and molecular markers for risk stratification of CLL significantly impact how clinicians determine prognosis and predict response to treatment for patients with newly diagnosed disease. This information, along with patient factors such as age and health status, should be considered when formulating an initial treatment strategy. Combinations of chemotherapy and immunotherapy offer the longest progression-free survival and overall survival benefit yet reported. For elderly patients or those with significant comorbidities who may not tolerate standard chemoimmunotherapy, less intensive but still effective therapies now exist. Patients with the highest risk disease, such as those with deletions of chromosome 17p, respond poorly to conventional treatment and should be referred to experienced centers where investigational therapies and allogeneic stem cell transplantation are available.
- Conclusion: Both disease characteristics and patient factors should guide the selection among the various effective therapies for CLL. While chemoimmunotherapy is the most effective treatment developed to date, its use may become less prevalent as newer agents are incorporated into initial and relapse treatment algorithms.
Chronic lymphocytic leukemia (CLL) is a chronic malignancy of B-lymphocytes demonstrating a heterogeneous clinical course ranging from indolent to more rapidly progressive. The chief clinical feature is an elevated peripheral blood lymphocyte count, and patients can demonstrate lymphadenopathy, splenomegaly, hepatomegaly, constitutional symptoms, and in late stages bone marrow failure. It is the most common leukemia among adults in the Western world, accounting for between 22% to 30% of new leukemia diagnoses worldwide [1]. Recent incidence rates in the United States are 3.83 cases per 100,000 person-years [2]. The incidence of CLL increases with age, and most new cases are diagnosed in persons 65 years of age or older [1,2]. As reported 5-year survival rates are between 68% and 81% with a median survival of 10 years in some series, the prevalence is significantly higher than the incidence [3]. However, this may even be an underestimate of the population burden of disease, as many cases are not reported to tumor registries [4].
Many patients with CLL are asymptomatic and do not require treatment until years after diagnosis. In these cases a watch and wait approach is taken. The typical natural history of CLL is characterized by periods of effective treatment when required, followed by treatment-free intervals of several years in many cases. However, this can be misleading, as the clinical course for any individual patient is highly variable. Development of cytogenetic and molecular testing has allowed for identification of patients with a higher risk of progression and lower response rates to traditional cytotoxic treatments [5]. For example, depending on chromosomal abnormalities present, median survival can vary from 32 to 133 months [3].
The assessment of underlying disease risk thus provides important information when considering a treatment approach and should be routinely performed for newly diagnosed patients. While the development of highly effective chemoimmunotherapy has allowed most groups of CLL patients to live for many years, some groups do not enjoy the same survival. Recent advances in CLL treatment seek to abrogate such adverse risk factors, thereby improving the survival for all patients with CLL. Given the expected survival of years for most CLL patients, frontline treatment planning must be done in the context of a long-term treatment strategy keeping the risk for late toxicities, such as secondary malignancies, in mind.
Case Study
Initial Presentation
A 50-year-old man is referred for evaluation of cervical lymphadenopathy that had progressed over the prior 6 months. He denies associated symptoms of fatigue, fevers, night sweats, or unintentional weight loss but does report early satiety. On examination there are multiple mobile, enlarged cervical lymph nodes bilaterally. Axillary lymph nodes are likewise enlarged. The liver edge is not palpable, but the spleen is palpable below the belt line. Complete blood count reveals a white blood cell count of 196,000 with 97% lymphocytes. Hemoglobin is 11.0 g/dL and platelet count is 122,000/dL. He recalls being told 3 years previously that his white blood cell count was 48,000 during an emergency department visit for cellulitis.
• How is CLL diagnosed and staged?
CLL is often suspected when patients present with an elevated lymphocyte count. Presenting symptoms of CLL commonly include lymphadenopathy, an enlarged spleen, and constitutional or “B” symptoms such as fatigue, unintentional weight loss, or drenching night sweats. However, only 25% of patients are symptomatic at diagnosis [1]. Many patients with CLL are now diagnosed after a routine blood test, long before the disease is clinically apparent.
The diagnosis of CLL can be made from the peripheral blood and does not require a bone marrow biopsy. According to 2008 guidelines from the International Workshop on Chronic Lymphocytic Leukemia (IWCLL), diagnosis requires at least 5000/uL clonal B-lymphocytes in the peripheral blood. The clonality must be confirmed by immunophenotyping. At time of diagnosis the peripheral blood smear should be examined for the characteristic cells: small mature lymphocytes with a narrow rim of cytoplasm and dense nuclei consisting of clumped chromatin. Larger, atypical cells can be present as long as they do not exceed 55% of the total number of lymphocytes [6].
The immunophenotype of CLL includes aberrant expression of CD5 and a T-cell antigen, along with the characteristic B-cell antigens CD19, CD20, and CD23. The leukemic clone may be either kappa or lambda light chain restricted. Expression of surface immunoglobulin, CD20, and CD79a is typically low compared to that of normal B cells, although there can be some variability in the immunophenotype [6].
![]()
Care should be taken to exclude other malignancies with a similar morphology. Leukemic phase mantle cell lymphoma, other low grade lymphomas, and hairy cell leukemia are commonly mistaken for CLL. Immunophenotyping and cytogenetics are usually sufficient to differentiate these. Testing for a balanced translocation involving chromosomes 11 and 14 to exclude mantle cell lymphoma can be helpful, as both CLL and mantle cell lymphoma can appear morphologically similar and share immunophenotypic features (CD5+/CD19+).
Case Continued
The patient’s peripheral blood is drawn for routine immunophenotyping as well as cytogenetic and molecular testing. When he returns to discuss the results 10 days later, he learns that peripheral blood immunophenotyping demonstrates a dim kappa restricted monoclonal population of B-cells that expressed CD19, CD20(dim), CD23, CD38, CD5, and CD43. The lymphocytes are negative for CD10, FMC7, and CD79b, consistent with a CLL immunophenotype. This patient fulfills diagnostic criteria for CLL and has Rai stage II or intermediate-risk disease. Interphase cytogenetic studies of the peripheral blood demonstrate deletions of chromosomes 11q22.3 and 13q14.3. The immunoglobulin heavy chain gene (IGHV) is unmutated.
• How can a CLL patient’s disease risk be characterized?
Historically, staging at diagnosis, pattern of bone marrow infiltration, and response to therapy were used to gauge prognosis. In more recent years, cytogenetic and molecular testing methods have been developed to augment risk stratification. Testing of prognostic significance that influences clinical management includes IGHV mutational status and interphase cytogenetics using FISH [3,12–14]. Expression of ZAP-70 and CD38 are both independent predictors of poorer prognosis in CLL but are not recommended for routine clinical use. Standardized methodology for the measurement of Zap-70 in particular limits the utility of that test in routine clinical practice [15]. Performed at diagnosis, a time when many patients are asymptomatic, cytogenetic testing with FISH and IGHV mutational analysis can predict time to first treatment and increasingly identify high-risk patients for whom investigational early intervention approaches may be considered [16]. While cytogenetic testing has utility at time of diagnosis, it should be considered necessary prior to deciding on the first-line treatment.
Cytogenetics are also important in predicting response to therapy. For instance, patients with deletion(11q) disease have improved survival when treated with regimens containing an alkylating agent [18]. Deletion(17p) patients respond poorly to traditional cytotoxic agents, and treatments with alternate mechanisms of action should be used [5,19]. The gene for tumor suppressor protein TP53 is encoded in this region of chromosome 17, thus treatment with agents that act independent of pathways involving TP53 are preferred [20].
In addition to cytogenetic testing, quantization of somatic mutations in the gene encoding the variable region of the immune globulin heavy chain gene (IGHV) can help define disease-specific risk. When greater than 98% sequence homology is seen, the gene is considered IGHV unmutated. Patients with an unmutated IGHV have worse overall survival. In one study of Rai stage 0 CLL patients, those with an unmutated IGHV had a survival of only 95 months, compared with 293 months in the mutated group [12].
• When should CLL be treated?
CLL is not curable with current standard therapies, and starting treatment at time of diagnosis for early stage, asymptomatic, CLL patients does not improve overall survival and adds treatment-related toxicities [21,22]. Consequently, the decision to treat is based on treating or preventing complications from the disease, and observation is recommended for most asymptomatic, early-stage patients [6]. Because median survival in CLL is often measured in years, deferring treatment can limit both the short- and long-term complications of therapy, especially the significant risk of secondary malignancies associated with some therapies [23]. However, deferring treatment can significantly impact both a patient’s emotional well-being and quality of life, which should be kept in mind when first discussing the rationale for observation with asymptomatic patients [24].
causes.
For patients with anemia, neutropenia, or thrombocytopenia that is autoimmune in nature, treatment should typically begin with corticosteroids, as it would for non-CLL associated cases of autoimmune cytopenias. If steroids are not effective, second-line treatments appropriate for the situation are generally employed, including intravenous immunoglobulin, cyclosporine, azathioprine, and splenectomy. Rituximab has also been shown to be effective in steroid-refractory cases of autoimmune hemolytic anemia associated with CLL [26]. Only if cytopenias are refractory to appropriate second-line therapy should CLL-directed treatments be considered, assuming there are no other indications to treat the underlying CLL [6]. Bone marrow biopsy can be helpful in differentiating autoimmune cytopenias from marrow failure due to CLL infiltration.
• What treatments are most appropriate for young, fit patients?
For younger patients who are in good general health, the standard treatment choice is combination chemoimmunotherapy. While single agent therapies can effectively palliate symptoms in most cases, they do not offer a survival benefit. Treatment with chemoimmunotherapy, consisting of cytotoxic chemotherapy given in combination with an anti-CD20 monoclonal antibody (generally rituximab), results in high response rates and conveys an advantage with respect to both progression-free survival (PFS) and overall survival (OS). Several chemoimmunotherapy regimens are commonly used.
As compared to fludarabine alone, frontline therapy with the combination of rituximab and fludarabine (FR) results in both a higher overall response rate (84% compared with 63% with fludarabine alone) and more complete responses (38% compared with 20% with fludarabine alone). The probability of PFS at 2 years is also better with FR: 67% compared to 45% with single agent fludarabine [28,29]. Neutropenia is more common with the combination regimen but does not appear to increase the rate of infection. Rituximab infusion reactions are commonly observed, so a stepped-up dosing schedule was developed to decrease their incidence and severity.
Fludarabine, cyclophosphamide, and rituximab (FCR) is another highly effective regimen. This combination has similar efficacy to FR with a 90% to 95% overall response rate (ORR) and 44% to 70% complete response (CR) rate [19,30]. Long-term results with this regimen are favorable; 6-year OS of 77% and median time to progression of 80 months have been reported in a follow-up study [31]. However, hematologic toxicity, including severe neutropenia, is common, and many patients are unable to complete all planned therapy [19]. The addition of cyclophosphamide does appear to be especially important for patients with a deletion(11q). Several clinical trials have consistently found that measures of response and survival are improved for deletion(11q) patients receiving an alkylating agent in addition to a nucleoside analogue [18,32,33]. Outcomes in patients with deletion(17p) disease remain poor after FCR; this subset demonstrates the shortest PFS at only 11.5 months [19].
A more recently developed chemoimmunotherapy option for younger, fit patients is bendamustine and rituximab (BR). Bendamustine has structural similarities to both alkylating agents and purine analogues, and is significantly more efficacious than chlorambucil as a single agent [34]. The combination is generally well tolerated, and a phase 2 trial of the combination reported an overall response rate (ORR) of 88.0% [32]. Notably, when the results were examined by genetic risk group, the regimen remained effective for deletion(11q) patients, who achieved overall and CR rates of 90% and 40%, respectively. Unfortunately, only 37.5% of deletion(17p) patients responded, and no patients achieved a CR [32].
The risk for therapy-related neoplasms should be taken into account when selecting initial therapy given the expected long-term survival of most CLL patients. About 8 out of 300 FCR-treated patients developed a therapy-related neoplasm in one study [31]. Treatment with FR, which does not include an alkylating agent, does not appear to have the same risk. In a study reporting long-term follow-up on 104 patients treated with FR, none developed a therapy related neoplasm [35]. Risks associated with bendamustine have not been well characterized but appear to be lower than FC. While inclusion of an alkylating agent is important for deletion(11q) patients, it is not clear if other patients similarly benefit, thus meriting the potentially increased risk for second cancers.
Fortunately, the choice among these similarly effective regimens will soon be based on high-quality, comparative data. FCR and BR have now been directly compared as a first-line treatment in the German CLL Study Group CLL10 trial. At interim analysis, both regimens had the same ORR and 2-year OS. However, CRs were less common in the BR group (38.1% versus 47.4% with FCR) and PFS was likewise inferior. Expectedly, the FCR group experienced more myelotoxicity and infections. The rate of severe neutropenia with FCR was higher at 81.7% compared to only 56.8% with BR [36]. This may be an important consideration when selecting a regimen for individual patients. Baseline renal function may influence choice as well. The active metabolite of fludarabine is eliminated through the kidneys and patients with decreased renal function have been excluded from clinical trials of FCR [19,37]. The phase 2 study of BR included patients with impaired renal function and 35% of participants had a creatinine clearance of less than 70 mL/min. It is notable that increased toxicity was seen in this subset, including higher rates of myelosuppression and infection [32]. As few direct comparisons have been done, the choice between effective first-line chemoimmunotherapy regimens can be difficult. The final results of the CLL 10 trial, as well as the now completed CALGB 10404 trial comparing FCR to FR, will provide new evidence regarding the relative risks and benefits of these regimens, particularly for patients without high-risk chromosomal abnormalities.
• What treatments are most effective for patients with deletion(17p) CLL?
As noted above, deletion(17p) CLL responds poorly to standard treatments. This relative lack of durable response to chemoimmunotherapy appears attributable to loss of function of the tumor suppressor protein TP53 which is encoded in the affected area [20,32,38]. In vivo evidence suggests that fludarabine works through a TP53-dependent mechanism, which likely explains the poor results obtained when deletion(17p) patients are treated with fludarabine-based combinations [38]. Patients harboring deletion(17p) or TP53 mutations should thus be referred for participation in clinical trials or allogeneic stem cell transplantation [17,27].
If initial treatment of a patient with deletion(17p) begins outside of a clinical trial, it should ideally be comprised of agents that have a TP53-independent mechanism of action [20]. Alemtuzumab, a humanized monoclonal antibody against the CD52 antigen expressed on the surface of normal and malignant B- and T-lymphocytes, demonstrated ORR of 33% to 50% in studies of patients with relapsed and refractory CLL [39–42]. A retrospective analysis found that similar outcomes were seen in those who had a TP53 mutation or deletion(17p). A subsequent study of previously untreated CLL patients randomized to treatment with 12 weeks of alemtuzumab or chlorambucil found that alemtuzumab-treated deletion(17p) patients had an ORR of 64% and median PFS of 10.7 months [43]. Alemtuzumab is therefore a rational choice for first-line therapy in this population. Hematologic toxicity is frequent, however, and all patients must receive prophylaxis against and monitoring for reactivation of CMV infection [43]. Infusion reactions are common but may be reduced by subcutaneous administration without apparent loss of efficacy [42,44]. While alemtuzumab is no longer marketed in the United States for the indication of CLL, it is available free of charge from the manufacturer [45].
High-dose methylprednisolone with rituximab (HDMP-R) has also been successfully used as both salvage and first-line therapy in this group. As salvage therapy, responses were seen in greater than 90% of patients, including over 50% of deletion(17p) patients [46-48]. In treatment-naïve CLL, the ORR was 96% [49], although data for patients with deletion 17p is limited in the frontline setting. Myelotoxicity attributable to the regimen is modest, but good antimicrobial prophylaxis is warranted, as well as close monitoring for hyperglycemia in at-risk patients.
• How is treatment modified for older or less fit patients?
For patients older than 70, or those who have significant comorbidities, effective therapies are still available. As most new diagnoses of CLL are made in patients older than 65, age is but one important factor determining an individual patient’s ability to tolerate treatment. The German CLL Study Group has usefully classified elderly patients into 3 treatment groups based on fitness and goals of care. The first group of medically fit patients with a normal life expectancy, sometimes referred to as the “go go” group, generally tolerate standard chemoimmunotherapy. A second group of older patients with significant life-limiting comorbid conditions—the so-called “no go” patients —should be offered best supportive care rather than CLL-directed treatment. A third group of “slow go” patients falls in between these two; these patients have comorbidities with variable life expectancy and will likely tolerate and benefit from CLL-directed therapy [50].
While some older patients can safely receive chemoimmunotherapy at standard doses and schedules, FCR can prove intolerable for even the medically fit elderly. Because inferior outcomes have been reported among patients older than 70 [30,31], a reduced-dose FCR regimen (FCR-lite) has been studied. Doses of fludarabine and cyclophosphamide were reduced by 20% and 40% respectively and dosing frequency of rituximab was increased. The CR rate was favorable at 77%, the rate of severe neutropenia was reduced to only 13%, and most patients completed all planned therapy [51]. Alternatively, the combination of pentostatin, cyclophosphamide, and rituximab (PCR) has also been successfully used in older patients. The overall and CR rates, 91% and 63% respectively, were durable at 26 months of follow-up. Importantly, there was no statistically significant difference in response or toxicity among the 28% of patients older than 70 [52,53].
For less fit patients, chlorambucil remains a reasonable option. Chlorambucil, a well-tolerated oral alkylating agent, has been used as a frontline therapy in CLL for decades. Chlorambucil has demonstrated consistent response rates in at least 4 clinical trials and is an appropriate option for patients who cannot tolerate more intensive therapy [54]. When a multicenter phase III trial compared it directly to fludarabine in patients over 65, the PFS and OS were no different despite favorable response rates in fludarabine-treated patients [55]. The effectiveness of single-agent chlorambucil can be improved, and the tolerability maintained, with the addition of a CD20-directed monoclonal antibody [56]. Obinutuzumab, a glycolengineered type II antibody against CD20, has recently been shown to improve treatment efficacy when used in combination with chlorambucil [57]. The CLL11 trial randomized patients with comorbid conditions to 1 of 3 treatments: single-agent chlorambucil, chlorambucil with rituximab (R-Clb), or chlorambucil with obinutuzumab (G-Clb). Both chemoimmunotherapy combinations outperformed chlorambucil alone, but the inclusion of obinutuzumab was associated with higher CR rates and longer PFS than rituximab, although infusion reactions and neutropenia were more common in the obinutuzumab arm [57]. Based on this result, the US Food and Drug Administration has now approved obinutuzumab for use in combination with chlorambucil as frontline therapy. While regulatory approval is without restriction with respect to patient age or fitness, a chlorambucil backbone remains most appropriate for older patients and/or those with significant comorbidities.
• What therapies are currently under development?
Numerous targeted treatments and novel immunotherapies are under active investigation in CLL. With greater specificity for CLL, these emerging agents offer the possibility of more effective yet less toxic treatments that will undoubtedly change the landscape for future CLL therapy. These agents are currently most studied as salvage therapies, and given their targeted mechanism of action can be highly effective in relapsed and refractory patients who frequently harbor poor risk cytogenetic abnormalities such as deletion(17p). Data for these agents as initial treatment is limited. Ongoing clinical trials employing these newer agents will need to be reported before these drugs can be recommended as frontline therapies.
Frontline experience with the oral immunomodulatory agent lenalidomide is more extensive. Lenalidomide offers convenient daily dosing and a favorable toxicity profile. When given on a continuous dosing schedule to patients who were 65 years old or older, the ORR was 65%, and 88% of patients were still alive at 2 years’ follow-up. The quality of response continued to improve beyond 18 months of treatment. Neutropenia, the most common severe toxicity, complicated about a third of cycles. Tumor flare attributable to immune activation was also seen, but in most cases was low-grade and did not require intervention [58,59]. While life-threatening tumor lysis syndrome and tumor flare have been seen with lenalidomide in CLL, such concerns are largely abrogated by a lower starting dose and careful intrapatient dose titration [60]. Lenalidomide has also been combined with rituximab and yielded promising results. Sixty-nine treatment-naïve patients were treated with escalating doses of lenalidomide along with rituximab infusions starting at the end of cycle 1 in a phase 2 study. They achieved an 88% ORR with 16% CRs. Toxicities were generally manageable, but patients over 65 were less likely to reach higher doses of lenalidomide or complete all planned treatment cycles [61]. Unfortunately, the FDA recently halted accrual to a phase 3 frontline clinical trial comparing lenalidomide to chlorambucil due to excess mortality in the lenalidomide arm among patients over the age of 80 [62]. More detailed outcomes from that study should be forthcoming.
Perhaps the most remarkable recent advance in CLL medicine, however, is the advent of orally bioavailable small molecule inhibitors of the B-cell receptor (BCR) signaling pathway. BCR signaling plays a vitally important role in supporting the growth and survival of malignant B-cells, activating a number of downstream kinases (Syk, Btk, PI3K, among others) which are potential therapeutic targets. Proof of principle for this approach was demonstrated with the Syk inhibitor fostamatinib in a phase 1/2 trial enrolling patients with B-cell non-Hodgkin lymphoma and CLL. CLL/SLL patients had the highest response rates of any subgroup in that study, with 6 out of 11 patients responding [63]. In a subsequent phase 1b study of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib, durable partial remissions were reported in more than 70% of multiply relapsed and refractory patients, including genetically high-risk patients [64–66]. Ibrutinib appears safer and better tolerated than traditional chemoimmunotherapy in the relapsed setting; consequently, it is now being studied as a first-line therapy both alone and in combination with other agents [67]. Other BCR signaling agents under study, such as the phosphatidylinositol 3-kinase inhibitor idelalisib, demonstrate similar safety and high response rates across both genetic risk and patient age groups [68].
New targeted drugs are not limited to the BCR signaling pathway. ABT-199 inhibits B-cell leukemia/lymphoma 2 (BCL-2), which is an anti-apoptotic protein in the cell death pathway, and has demonstrated remarkable clinical efficacy in relapsed and refractory CLL patients [69]. As more experience is gained with these targeted agents, it is expected that they will be rapidly incorporated into frontline therapies. However, these agents are just now being studied in comparison to standard initial treatments, such as FCR, and it is not yet clear they will offer an advantage over current chemoimmunotherapy in this setting [70–72]. Since these single agents typically do not induce complete remissions, and require indefinite therapy to maintain response, optimal combination therapies are under intensive investigation.
Case Conclusion
The patient and his physician elect to begin treatment owing to symptomatic cervical lymphadenopathy and massive splenomegaly. Given the presence of a deletion(11q) abnormality, but hoping to limit the risk for both short- and long-term toxicities, this younger, fit patient is treated with 6 cycles of bendamustine and rituximab. At the conclusion of treatment, neither the cervical lymph nodes nor spleen remain palpable. His blood counts have also normalized, with a white blood cell count of 4700 with 8.1% lymphocyotes, hemoglobin of 14.3 gm/dL, and platelets of 151,000/dL.
Summary
CLL follows a chronic course requiring treatment at variable intervals. Both genetic risk features and patient factors should be considered when determining initial therapy. Cytogenetic and molecular testing can characterize the likelihood of treatment success, information useful for treatment planning. Chemoimmunotherapy is highly effective for most patients, including patients with deletion(11q) CLL, where the inclusion of an alkylating agent in frontline therapy alters the natural history of disease. However, patients with deletion(17p) and or TP53-mutated disease respond poorly to standard treatment and should be considered for investigational therapies [73]. Novel approaches to CLL therapy, most notably immunotherapies and BCR-targeted agents, hold the promise to further improve outcomes, particularly for the highest risk patients and those elderly and/or infirm patients who tolerate chemotherapy poorly. Frontline therapy should rapidly evolve as emerging agents enter advanced phase investigation.
Corresponding author: Jeffrey Jones, MD, MPH, Div. of Hematology, Ohio State University, A350B Starling Loving Hall, 320 West 10th Ave., Columbus, OH 43210, [email protected].
Financial disclosures: Dr. Jones disclosed that he is on the advisory boards and has received research support from Genentech, Pharmacyclics, and Gilead.
Author contributions: conception and design, KAR, JAJ; analysis and interpretation of data, KAR, JAJ; drafting of article, KAR, JAJ; critical revision of the article, KAR, JAJ.
1. Redaelli A, Laskin BL, Stephens JM, et al. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur J Cancer Care (Engl) 2004;13:279–87.
2. Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007;139:809–19.
3. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6.
4. Zent CS, Kyasa MJ, Evans R, Schichman SA. Chronic lymphocytic leukemia incidence is substantially higher than estimated from tumor registry data. Cancer 2001;92:1325–30.
5. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006;24:437–43.
6. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56.
7. Rawstron AC, Bennett FL, O'Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83.
8. Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–34.
9. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
10. Binet JL, Lepoprier M, Dighiero G, et al. A clinical staging system for chronic lymphocytic leukemia: prognostic significance. Cancer 1977:40:855–64.
11. Eichhorst BF, Fischer K, Fink AM, et al. Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 2011;117:1817–21.
12. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54.
13. Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:
1764–75.
14. Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–9.
15. Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006;70:209–13.
16. Wierda WG, O'Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–95.
17. Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 2008;26:5094–100.
18. Ding W, Ferrajoli A. Evidence-based mini-review: the role of alkylating agents in the initial treatment of chronic lymphocytic leukemia patients with the 11q deletion. Hematology Am Soc Hematol Educ Program 2010;2010:90–2.
19. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
20. Badoux XC, Keating MJ, Wierda WG.What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011;6:36–46.
21. Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–14.
22. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 1999;91:861–8.
23. Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 2010;28:4935–44.
24. Shanafelt TD, Bowen D, Venkat C, et al. Quality of life in chronic lymphocytic leukemia: an international survey of 1482 patients. Br J Haematol 2007;139:255–64.
25. Baer MR, Stein RS, Dessypris EN. Chronic lymphocytic leukemia with hyperleukocytosis. The hyperviscosity syndrome. Cancer 1985;56:2865–9.
26. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 2002;16:2092–5.
27. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: non-hodgkin's lymphomas. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
28. Byrd JC, Rai K, Peterson BL, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49–53.
29. Byrd JC, Peterson BL, Morrison VA, et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003;101:6–14.
30. Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079–88.
31. Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008;112:975–80.
32. Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16.
33. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9.
34. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–84.
35. Woyach JA, Ruppert AS, Heerema NA, et al. Chemoimmunotherapy with fludarabine and rituximab produces extended overall survival and progression-free survival in chronic lymphocytic leukemia: long-term follow-up of CALGB study 9712. J Clin Oncol 2011;29:1349–55.
36. Fink AM, et al., Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximabversus bendamustine and rituximabin previously untreated and physically fit patientswith advanced chronic lymphocytic leukemia: results of a planned interim analysis of the CLL10 Trial, an international, randomized study of the German CLL Study Group (GCLLSG). Blood 2013;122:526.
37. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet 2002;41:93–103.
38. Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood 2004;104:1428–34.
39. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61.
40. Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81.
41. Osuji NC, Del Giudice I, Matutes E, et al, The efficacy of alemtuzumab for refractory chronic lymphocytic leukemia in relation to cytogenetic abnormalities of p53. Haematologica 2005;90:1435–6.
42. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
43. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–23.
44. Lundin J, Kimby E, Björkholm M, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002;100:768–73.
45. Genzyme. US Campath Distribution Program. Cambridge, MA: Genzyme. Available at http://www.campath.com/.
46. Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65.
47. Bowen DA, Call TG, Jenkins GD, et al. Methylprednisolone-rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007;48:2412–7.
48. Castro JE, Sandoval-Sus JD, Bole J, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of fludarabine refractory high-risk chronic lymphocytic leukemia. Leukemia 2008;22:2048–53.
49. Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89.
50. Eichhorst B, Goede V, Hallek M. Treatment of elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009;50:171–8.
51. Foon KA, Boyiadzis M, Land SR, et al. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498–503.
52. Kay NE, Geyer SM, Call TG, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 2007;109:405–11.
53. Shanafelt TD, Lin T, Geyer SM, et al. Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer 2007;109:2291–8.
54. Catovsky D, Else M, Richards S. Chlorambucil--still not bad: a reappraisal. Clin Lymphoma Myeloma Leuk 2011;11 Suppl 1:S2–6.
55. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009;114:3382–91.
56. Laurenti L, Vannata B, Innocenti I, et al. Chlorambucil plus rituximab as front-line therapy in elderly/unfit patients affected by b-cell chronic lymphocytic leukemia: results of a single-centre experience. Mediterr J Hematol Infect Dis 2013;5:e2013031.
57. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014 Jan 8. [Epub ahead of print].
58. Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood 2011;118:3489–98.
59. Strati P, Keating MJ, Wierda WG, et al. Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 2013;122:734–7.
60. Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol 2007;25:5047.
61. James DF, Brown JR, Werner L, et al. Lenalidomide and rituximab for the initial treatment of patients with chronic lymphocytic leukemia (CLL): a multicenter study of the CLL Research Consortium. ASH Annual Meeting Abstracts 2011;118:291.
62. US Food and Drug Administration. FDA halts clinical trial of drug Revlimid (lenalidomide) for chronic lymphocytic leukemia due to safety concerns. Available at http://www.fda.gov/Drugs/DrugSafety/ucm361444.htm.
63. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010;115:2578–85.
64. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
65. Farooqui M, Aue G, Valdez J, et al. Single agent ibrutinib (PCI-32765) achieves equally good and durable responses in chronic lymphocytic leukemia (CLL) patients with and without deletion 17p. Blood 2013;122:673.
66. Byrd JC, Furman RR, Coutre S, et al. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) monotherapy demonstrates long-term safety and durability of response in chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) patients in an open-label extension study. Blood 2013;122:4163.
67. Brown JR, Barrientos JC, Barr PM, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study. ASH Annual Meeting Abstracts 2013.
68. O'Brien SM, Lamanna N, Kipps TJ, et al. A phase II study of the selective phosphatidylinositol 3-kinase delta (PI3K{delta}) inhibitor idelalisib (GS-1101) in combination with rituximab (R) in treatment-naive patients (pts) ≥ 65 years with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). J Clin Oncol 2013;31(15 Suppl); Abstract 7005.
69. Seymour JF, Davids MS, Pagel JM, et al. Bcl-2 Inhibitor ABT-199 (GDC-0199) monotherapy shows anti-tumor activity including complete remissions in high-risk relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Blood 2013;122:872.
70. Rituxumab and bendamustine hydrochloride, rituxumab and ibrutunib, or ibrutinib alone in treating older patients with previously untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT01886872?term=ibrutinib+cll&rank=8.
71. A multicenter, open-label, phase 3 study of the bruton's tyrosine kinase inhibitor pci-32765 versus chlorambucil in patients 65 years of older with treatment-naive chronic lymphocytic leukemia or small lymphocytic lymphoma (RESONATE-2) Available at http://clinicaltrials.gov/ct2/show/NCT01722487?term=ibrutinib+cll&rank=12.
72. Ibrutinib and rituximab compared with fludarabine phosphate, cyclophosphamide, and rituxumab in treating patients with untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT02048813?term=ibrutinib+cll&rank=2.
73. Strati P, Keating MJ, O'Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia (CLL) with 17p deletion. J Clin Oncol 2013;31(15 suppl): Abstract 7102.
1. Redaelli A, Laskin BL, Stephens JM, et al. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur J Cancer Care (Engl) 2004;13:279–87.
2. Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007;139:809–19.
3. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6.
4. Zent CS, Kyasa MJ, Evans R, Schichman SA. Chronic lymphocytic leukemia incidence is substantially higher than estimated from tumor registry data. Cancer 2001;92:1325–30.
5. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006;24:437–43.
6. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56.
7. Rawstron AC, Bennett FL, O'Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83.
8. Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–34.
9. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
10. Binet JL, Lepoprier M, Dighiero G, et al. A clinical staging system for chronic lymphocytic leukemia: prognostic significance. Cancer 1977:40:855–64.
11. Eichhorst BF, Fischer K, Fink AM, et al. Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 2011;117:1817–21.
12. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54.
13. Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:
1764–75.
14. Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–9.
15. Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006;70:209–13.
16. Wierda WG, O'Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–95.
17. Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 2008;26:5094–100.
18. Ding W, Ferrajoli A. Evidence-based mini-review: the role of alkylating agents in the initial treatment of chronic lymphocytic leukemia patients with the 11q deletion. Hematology Am Soc Hematol Educ Program 2010;2010:90–2.
19. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
20. Badoux XC, Keating MJ, Wierda WG.What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011;6:36–46.
21. Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–14.
22. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 1999;91:861–8.
23. Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 2010;28:4935–44.
24. Shanafelt TD, Bowen D, Venkat C, et al. Quality of life in chronic lymphocytic leukemia: an international survey of 1482 patients. Br J Haematol 2007;139:255–64.
25. Baer MR, Stein RS, Dessypris EN. Chronic lymphocytic leukemia with hyperleukocytosis. The hyperviscosity syndrome. Cancer 1985;56:2865–9.
26. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 2002;16:2092–5.
27. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: non-hodgkin's lymphomas. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
28. Byrd JC, Rai K, Peterson BL, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49–53.
29. Byrd JC, Peterson BL, Morrison VA, et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003;101:6–14.
30. Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079–88.
31. Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008;112:975–80.
32. Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16.
33. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9.
34. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–84.
35. Woyach JA, Ruppert AS, Heerema NA, et al. Chemoimmunotherapy with fludarabine and rituximab produces extended overall survival and progression-free survival in chronic lymphocytic leukemia: long-term follow-up of CALGB study 9712. J Clin Oncol 2011;29:1349–55.
36. Fink AM, et al., Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximabversus bendamustine and rituximabin previously untreated and physically fit patientswith advanced chronic lymphocytic leukemia: results of a planned interim analysis of the CLL10 Trial, an international, randomized study of the German CLL Study Group (GCLLSG). Blood 2013;122:526.
37. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet 2002;41:93–103.
38. Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood 2004;104:1428–34.
39. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61.
40. Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81.
41. Osuji NC, Del Giudice I, Matutes E, et al, The efficacy of alemtuzumab for refractory chronic lymphocytic leukemia in relation to cytogenetic abnormalities of p53. Haematologica 2005;90:1435–6.
42. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
43. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–23.
44. Lundin J, Kimby E, Björkholm M, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002;100:768–73.
45. Genzyme. US Campath Distribution Program. Cambridge, MA: Genzyme. Available at http://www.campath.com/.
46. Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65.
47. Bowen DA, Call TG, Jenkins GD, et al. Methylprednisolone-rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007;48:2412–7.
48. Castro JE, Sandoval-Sus JD, Bole J, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of fludarabine refractory high-risk chronic lymphocytic leukemia. Leukemia 2008;22:2048–53.
49. Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89.
50. Eichhorst B, Goede V, Hallek M. Treatment of elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009;50:171–8.
51. Foon KA, Boyiadzis M, Land SR, et al. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498–503.
52. Kay NE, Geyer SM, Call TG, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 2007;109:405–11.
53. Shanafelt TD, Lin T, Geyer SM, et al. Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer 2007;109:2291–8.
54. Catovsky D, Else M, Richards S. Chlorambucil--still not bad: a reappraisal. Clin Lymphoma Myeloma Leuk 2011;11 Suppl 1:S2–6.
55. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009;114:3382–91.
56. Laurenti L, Vannata B, Innocenti I, et al. Chlorambucil plus rituximab as front-line therapy in elderly/unfit patients affected by b-cell chronic lymphocytic leukemia: results of a single-centre experience. Mediterr J Hematol Infect Dis 2013;5:e2013031.
57. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014 Jan 8. [Epub ahead of print].
58. Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood 2011;118:3489–98.
59. Strati P, Keating MJ, Wierda WG, et al. Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 2013;122:734–7.
60. Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol 2007;25:5047.
61. James DF, Brown JR, Werner L, et al. Lenalidomide and rituximab for the initial treatment of patients with chronic lymphocytic leukemia (CLL): a multicenter study of the CLL Research Consortium. ASH Annual Meeting Abstracts 2011;118:291.
62. US Food and Drug Administration. FDA halts clinical trial of drug Revlimid (lenalidomide) for chronic lymphocytic leukemia due to safety concerns. Available at http://www.fda.gov/Drugs/DrugSafety/ucm361444.htm.
63. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010;115:2578–85.
64. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
65. Farooqui M, Aue G, Valdez J, et al. Single agent ibrutinib (PCI-32765) achieves equally good and durable responses in chronic lymphocytic leukemia (CLL) patients with and without deletion 17p. Blood 2013;122:673.
66. Byrd JC, Furman RR, Coutre S, et al. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) monotherapy demonstrates long-term safety and durability of response in chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) patients in an open-label extension study. Blood 2013;122:4163.
67. Brown JR, Barrientos JC, Barr PM, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study. ASH Annual Meeting Abstracts 2013.
68. O'Brien SM, Lamanna N, Kipps TJ, et al. A phase II study of the selective phosphatidylinositol 3-kinase delta (PI3K{delta}) inhibitor idelalisib (GS-1101) in combination with rituximab (R) in treatment-naive patients (pts) ≥ 65 years with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). J Clin Oncol 2013;31(15 Suppl); Abstract 7005.
69. Seymour JF, Davids MS, Pagel JM, et al. Bcl-2 Inhibitor ABT-199 (GDC-0199) monotherapy shows anti-tumor activity including complete remissions in high-risk relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Blood 2013;122:872.
70. Rituxumab and bendamustine hydrochloride, rituxumab and ibrutunib, or ibrutinib alone in treating older patients with previously untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT01886872?term=ibrutinib+cll&rank=8.
71. A multicenter, open-label, phase 3 study of the bruton's tyrosine kinase inhibitor pci-32765 versus chlorambucil in patients 65 years of older with treatment-naive chronic lymphocytic leukemia or small lymphocytic lymphoma (RESONATE-2) Available at http://clinicaltrials.gov/ct2/show/NCT01722487?term=ibrutinib+cll&rank=12.
72. Ibrutinib and rituximab compared with fludarabine phosphate, cyclophosphamide, and rituxumab in treating patients with untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT02048813?term=ibrutinib+cll&rank=2.
73. Strati P, Keating MJ, O'Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia (CLL) with 17p deletion. J Clin Oncol 2013;31(15 suppl): Abstract 7102.
Management of Locally Advanced Rectal Adenocarcinoma
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
A medication change, then involuntary lip smacking and tongue rolling
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
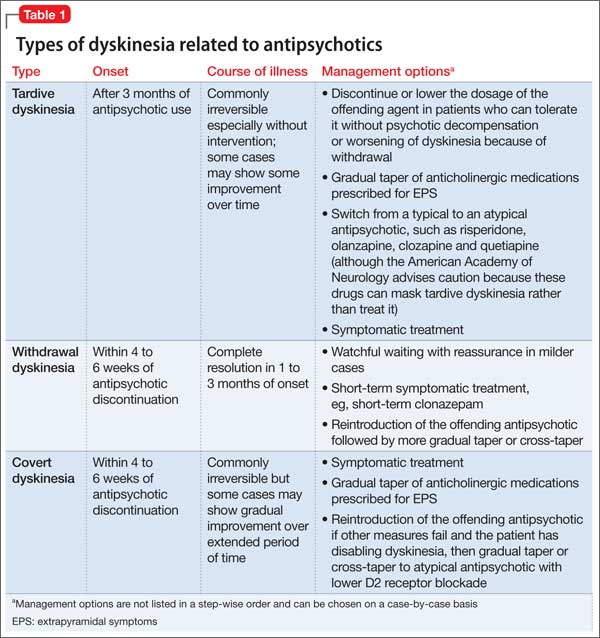
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
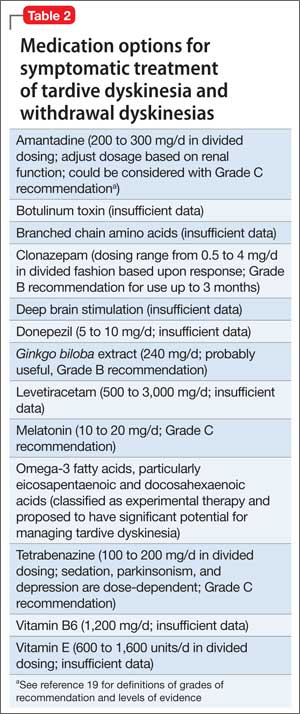
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
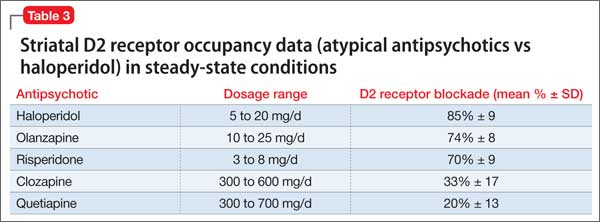
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
A depressed adolescent who won’t eat and reacts slowly
CASE A fainting spell
Ms. A, age 13, is admitted to a pediatric unit after fainting and losing consciousness for 5 minutes in the shower, during which time she was non-responsive. She reports feeling nauseated and having blurry vision before dropping to the floor.
Ms. A reports intentional self-restriction of calories, self-induced vomiting, and other purging behaviors, such as laxative abuse and excessive exercising.
During the mental status examination, Ms. A is lying in bed wearing hospital clothes, legs flexed at the knee, hands on her side, and a fixed gaze at the ceiling with poor eye contact. She is of slender stature and tall, seems slightly older than her stated age, and is poorly groomed.
Throughout the interview, Ms. A has significant psychomotor retardation, reports her mood as tired, and has a blunted affect. She speaks at a low volume and has poverty of speech; she takes deep sighs before answering questions. Her thought process is linear and she cooperates with the interview. She has poor recall, including delayed 3-minute recall and poor sustained attention. Her abstraction capacity is fair and her intellect is average and comparable with her age group. Ms. A is preoccupied that eating will cause weight gain. She denies hallucinations but reports passive death wishes with self-harm by scratching.
What is the differential diagnosis to explain Ms. A’s presentation?
a) syncope
b) seizures
c) dehydration
d) hypotension
HISTORY Preoccupied with weight
Ms. A reports vomiting twice a day, while showering and at night when no one is around, every day for 2 months. She stopped eating and taking in fluids 3 days before admission to the medical unit. Also, she reports restricting her diet to 700 to 1,000 calories a day, skipping lunch at school, and eating minimally at night. Ms. A uses raspberry ketones and green coffee beans, which are advertised to aid weight loss, and laxative pills from her mother’s medicine cabinet once or twice a week when her throat is sore from vomiting. She reports exercising excessively, which includes running, crunches, and lifting weights. She has lost approximately 30 lb in the last 2 months.
Ms. A says she fears gaining weight and feels increased guilt after eating a meal. She said that looking at food induced “anxiety attack” symptoms of increased heart rate, sweaty palms, feeling of choking, nervousness, and shakiness. She adds that she does not want to be “bigger” than her classmates. Her understanding of the consequences of not eating is, “It will get worse, I will shut down and die. I do not fear death, I only fear getting bigger than others.”
She reports that her fixation on avoiding food started when she realized that she was the tallest girl in her class and the only girl in her class running on the track team, after which she quit athletics. She reports that depression symptoms pre-dated her eating disorder symptoms; onset of significant depression likely was precipitated by her grandfather’s death a year earlier, and then exacerbated by the recent death of a family pet.
Ms. A’s depressive symptoms are described as anhedonia (avoiding being outside and not enjoying drawing anymore), decreased energy, tearfulness, sadness, decreased concentration, and passive suicidal thoughts. Her mother is supportive and motivates her daughter to “get better.” Ms. A denies any symptoms of psychosis, other anxiety symptoms, other mood disorder symptoms, substance abuse, or homicidality.
Ms. A’s mother says she felt that, recently, her daughter has been having some difficulty with confused thoughts and significantly delayed responses. However, the mother reports that her daughter always had somewhat delayed responses from what she felt is typical. Her mother adds that Ms. A’s suicidal thoughts have worsened since her daughter started restricting her diet.
Which diagnosis likely accounts for Ms. A’s presentation?
a) major depressive disorder (MDD)
b) eating disorder, not otherwise specified (NOS)
c) anorexia nervosa, purging type
d) catatonia, unspecified
e) anxiety disorder NOS
f) cognitive disorder
g) psychosis NOS
The authors’ observations
There are many reported causes of catatonia in children and adolescents, including those that are psychiatric, medical, or neurological, as well as drugs (Table 1).1,2 Affective disorders have been associated with catatonia in adults, but has not been widely reported in children and adolescents.1,3 Organic and neurologic causes, such as neurological tumors and cerebral hemorrhage, should be ruled out first because, although rare, they can be fatal (Table 2).2 If the cause of catatonia is not recognized quickly (Figure,1,2) effective treatment could be delayed.4



Catatonia involves psychomotor abnormalities, which are listed in Table 3.1,4

Presentation in adults and adolescents is similar.
An eating disorder could be comorbid with another psychiatric disorder, such as MDD, dysthymia, or panic disorder.5 Ms. A’s report of depression before she began restricting food favored a primary diagnosis of MDD. Her depressive symptoms of low appetite or low self-worth could have led to her preoccupation with body image.
There has been evidence that negative self-image and eating disorders are associated, but data are limited and the connection remains unclear.6 Ms. A’s self-esteem was very low. Her fixation on restricting food could have been perpetuated by her self-criticism and by being excluded from her peer group in school. Her weight loss could have brought anxiety symptoms to the forefront because of physiologic changes that accompany extreme weight loss.
The treatment team was concerned about her delayed responses, which could be explained by the catatonic features that reflected the severity of her depression. She had no obvious symptoms of psychosis, but her intrusive thoughts and obsessions with avoiding food did not completely rule out psychosis.
Childhood-onset schizophrenia, although rare, has been associated with catatonia; following up with a catatonia rating scale, such as the Catatonia Rating Scale or the Bush- Francis Catatonia Rating Scale (BFCRS), would be useful for tracking symptom progress. In Ms. A’s case, her mood disorder was primary, but did not rule out psychosis-like prodromal symptoms.7
Ms. A is diagnosed with MDD, single episode, severe, with catatonic features, and without psychosis, and eating disorder, NOS.
EVALUATION Mostly normal
Ms. A does not have a history of mental illness and was not seeing a psychiatrist or therapist, nor did she have any prior psychiatric admissions. She denies suicide attempts, but reports self-injurious behavior involving scratching her skin, which started during the current mood episode. She has never taken any psychotropic medications. Ms. A lives at home with her biological mother and father and 17-year-old brother. She attends middle school with average grades and has no history of disciplinary actions. She has no history of bullying or teasing, although she did report some previous difficulty with relational aggression toward her peers in the 5th grade. Her mother has a history of anorexia nervosa that began when she was a teenager, but these symptoms are stable and under control. There is additionally a family history of bipolar disorder.
Ms. A has a family history of coronary artery disease and diabetes in the mother and maternal relatives. Her grandfather died from liver cancer. She was allergic to sulfa drugs and was taking a multivitamin and minocycline for acne.
Physical examination reveals some superficial scratches but otherwise was within normal limits. Initial lab results reveal a normal complete blood count and differential. Thyroid-stimulating hormone is 1.29 mIU/L and free T4 is 0.96 mg/dL, both within normal limits. Urinalysis is within normal limits and urine pregnancy test is negative. A comprehensive metabolic panel shows mild elevation in aspartate aminotransferase (AST) at 60 U/L and alanine aminotransferase (ALT) at 92 U/L, respectively. Phosphorus level is within normal limits. Prealbumin level is slightly low at 15.1 mg/dL.
Which treatment plan would you recommend for Ms. A?
a) discharge with outpatient psychiatric treatment
b) recommend medical stabilization with follow-up from the psychosomatic team and then outpatient psychiatric follow-up
c) admit her to the psychiatric acute inpatient hospital with psychiatric outpatient discharge follow-up plan
d) discharge her home with follow-up with her primary care physician
e) recommend follow-up from the psychosomatic team while on medical floor with acute inpatient admission and psychiatric outpatient follow-up at discharge
The authors’ observations
Scarcity of data and reporting of cases of adolescent catatonia limits guidance for diagnosis and treatment.8 There are several rating scales with variability in definition, but that overall provide a guiding tool for detecting catatonia. The Brief Cognitive Rating Scale is considered the most versatile because it is more valid, reliable, and requires less time to complete than other rating scales.9
Ms. A’s symptoms were a combination of depressive symptoms with severity defined by catatonic features, eating disorder with worsening course, anxiety symptoms, and genetic loading of eating disorder in her mother. The challenge of this case was making an accurate diagnosis and treating Ms. A, which required continuous observation following an eating disorder protocol, resolution of her catatonia, resuming a normal diet, and decreasing her suicidality. Retrospectively, her scores on the BFCRS were high on screening items 1 to 14, which measure presence or absence and severity of symptoms.
The best option was to admit Ms. A to an inpatient psychiatric facility after she is cleared medically with outpatient services to follow up.
How would you treat Ms. A’s symptoms?
a) aggressively treat catatonia
b) address her eating disorder
c) work to resolve her depression
The authors’ observations
The challenge was to choose the psychotropic medication that would target her depression, obsessive, rigid thoughts, and catatonia. Administering an antidepressant with an antipsychotic would have relieved her depressive and obsessive symptoms but would not have improved her catatonia. The psychosomatic medicine team recommended starting a selective serotonin reuptake inhibitor and a benzodiazepine to target both the depression and the catatonic symptoms. Ms. A received sertraline, 12.5 mg/d, which was increased to 25 mg/d on the third day. IV lorazepam, 1 mg, 3 times a day, was recommended but the pediatric team prescribed an oral formulation. The hospital’s eating disorder protocol was instituted on the day of admission.
Treatment options for catatonia
Benzodiazepines are the first line of treatment for catatonia and other neuroleptics, specifically antipsychotics, have been considered dangerous.10 Benzodiazepine-resistant catatonia, which is sometimes seen in patients with autism, might respond to electroconvulsive therapy (ECT),11 although in some states it cannot be administered to children age <18.12 Benzodiazepines have shown dramatic improvement within hours, as has ECT.8,13 Additionally, if patients do not respond to a benzodiazepine or ECT, consider other options such as zolpidem, olanzapine,14 or sensory integration system (in adolescents with autism).15
Ms. A did not need ECT or an alternative treatment because she responded well to 3 doses of oral lorazepam. Her amotivation, negativism, and rigidity with prolonged posturing improved. Her psychomotor retardation improved overall, although she reported some dizziness and had some postural hypotension, which was attributed to her eating issues and dehydration.
OUTCOME Feeling motivated
Ms. A is transferred to psychiatric inpatient unit. She tolerates sertraline, which is titrated to 50 mg/d. She is placed on the hospital’s standard eating disorder protocol. She continues to eat well with adequate intake of solids and liquid and exhibits only some anxiety associated with meals. During the course of hospitalization, she attends group therapy and her catatonic symptoms completely resolve. She says she thinks that her thoughts are improving and that she is not longer feeling confused. She reports being motivated to continue to improve her eating disorder symptoms.
The treatment team holds a family session during which family dynamic issues that are stressful to Ms. A are discussed, such as some conflict with her parents as well as some negative interactions between Ms. A and her father. Repeat comprehensive metabolic panel on admission to the inpatient psychiatric hospital reveals persistent elevation of AST at 92 U/L and ALT at 143 U/L. Ms. A is discharged home with follow-up with a psychiatrist and a therapist. The treatment team also recommends that she follow up in a program that specializes in eating disorders.
4-month follow-up. Ms. A returns to inpatient psychiatric hospital after overdose of sertraline and aripiprazole, which were started by an outpatient psychiatrist. She reports severe depressive symptoms because of school stressors. She denies any problems eating and did not show any symptoms of catatonia. In her chart, there is a mention of “cloudy thoughts” and quietness. At this admission, her ALT is 17 U/L and AST is 19 U/L. Sertraline is increased to 150 mg/d and aripiprazole is reduced to 2 mg/d and then later increased to 5 mg/d, after which she is discharged home with an outpatient psychiatric follow-up.
1-year follow-up. Ms. A has been following up with an outpatient psychiatrist and is receiving sertraline, 150 mg/d, aripiprazole, 2.5 mg/d, and extended-release methylphenidate, 36 mg/d, along with L-methylfolate, multivitamins, and omega-3 fish oil as adjuvants for her depressive symptoms. Ms. A does not show symptoms of an eating disorder or catatonia, and her depression and psychomotor activity have improved, with better overall functionality, after adding the stimulant and adjunctives to the antidepressant.
The authors’ observations
The importance of including catatonia NOS with its various specifiers, such as medical, metabolic, toxic, affective, etc., has been discussed.16,17 In Ms. A’s case, instead of treating the specific symptoms—affective or eating disorder or obsessive quality of thought content, mimicking psychotic-like symptoms—addressing the catatonia initially had a better outcome. More studies related to chronic and acute catatonia in adolescents are needed because of the risk of increased morbidity and premature death.18 Early recognition of catatonia is needed19 because it often is underdiagnosed.20
Eating disorders often become worse over the first 5 years, and close monitoring and assessment is needed for adolescents.21 Also, prodromal psychotic symptoms require follow-up because techniques for early detection and intervention for children and adolescents are still in their infancy.22
Bottom Line
Catatonia in adolescents should be addressed early, when it is treatable and the outcome is favorable. It is important to recognize catatonia in an emergency department or inpatient medical unit setting in a hospital because it is often underdiagnosed or misdiagnosed. The presentation of catatonia is similar in adolescents and adults. Benzodiazepines are first-line treatment for catatonia; consider electroconvulsive therapy if patients do not respond to drug therapy.
Related Resources
• Roberto AJ, Pinnaka S, Mohan A, et al. Adolescent catatonia successfully treated with lorazepam and aripiprazole. Case Rep Psychiatry. 2014;2014:309517. doi: 10.1155/2014/309517.
• Raffin M, Zugaj-Bensaou L, Bodeau N, et al. Treatment use in a prospective naturalistic cohort of children and adolescents with catatonia. Eur Child Adolesc Psychiatry. 2015;24(4):441-449.
Drug Brand Names
Aripiprazole • Abilify Minocycline • Minocin
L-methylfolate • Deplin Olanzapine • Zyprexa
Lorazepam • Ativan Sertraline • Zoloft
Methylphenidate • Ritalin, Concerta Zolpidem • Ambien, Intermezzo
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dhossche D, Wilson C, Wachtel LE. Catatonia in childhood and adolescence: implications for the DSM-5. Primary Psychiatry. http://primarypsychiatry.com/catatonia-in-childhood-and-adolescence-implications-for-the-dsm-5. Published May 21, 2013. Accessed July 2, 2015.
2. Lahutte B, Cornic F, Bonnot O, et al. Multidisciplinary approach of organic catatonia in children and adolescents may improve treatment decision making. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(6):1393-1398.
3. Brake JA, Abidi S. A case of adolescent catatonia. J Can Acad Child Adolesc Psychiatry. 2010;19(2):138-140.
4. Consoli A, Raffin M, Laurent C, et al. Medical and developmental risk factors of catatonia in children and adolescents: a prospective case-control study. Schizophr Res. 2012;137(1-3):151-158.
5. Zaider TI, Johnson JG, Cockell SJ. Psychiatric comorbidity associated with eating disorder symptomatology among adolescents in the community. Int J Eat Disord. 2000;28(1):58-67.
6. Forsén Mantilla E, Bergsten K, Birgegård A. Self-image and eating disorder symptoms in normal and clinical adolescents. Eat Behav. 2014;15(1):125-131.
7. Bonnot O, Tanguy ML, Consoli A, et al. Does catatonia influence the phenomenology of childhood onset schizophrenia beyond motor symptoms? Psychiatry Res. 2008;158(3):356-362.
8. Singh LK, Praharaj SK. Immediate response to lorazepam in a patient with 17 years of chronic catatonia. J Neuropsychiatry Clin Neurosci. 2013;25(3):E47-E48.
9. Sienaert P, Rooseleer J, De Fruyt J. Measuring catatonia: a systematic review of rating scales. J Affect Disord. 2011;135(1-3):1-9.
10. Cottencin O, Warembourg F, de Chouly de Lenclave MB, et al. Catatonia and consultation-liaison psychiatry study of 12 cases. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1170-1176.
11. Wachtel LE, Hermida A, Dhossche DM. Maintenance electroconvulsive therapy in autistic catatonia: a case series review. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(4):581-587.
12. Wachtel LE, Dhossche DM, Kellner CH. When is electroconvulsive therapy appropriate for children and adolescents? Med Hypotheses. 2011;76(3):395-399.
13. Takaoka K, Takata T. Catatonia in childhood and adolescence. Psychiatry Clin Neurosci. 2003;57(2):129-137.
14. Ceylan MF, Kul M, Kultur SE, et al. Major depression with catatonic features in a child remitted with olanzapine. J Child Adolesc Psychopharmacol. 2010;20(3):225-227.
15. Consoli A, Gheorghiev C, Jutard C, et al. Lorazepam, fluoxetine and packing therapy in an adolescent with pervasive developmental disorder and catatonia. J Physiol Paris. 2010;104(6):309-314.
16. Dhossche D, Cohen D, Ghaziuddin N, et al. The study of pediatric catatonia supports a home of its own for catatonia in DSM-5. Med Hypotheses. 2010;75(6):558-560.
17. Taylor MA, Fink M. Catatonia in psychiatric classification: a home of its own. Am J Psychiatry. 2003;160(7):1233-1241.
18. Cornic F, Consoli A, Tanguy ML, et al. Association of adolescent catatonia with increased mortality and morbidity: evidence from a prospective follow-up study. Schizophr Res. 2009;113(2-3):233-240.
19. Quigley J, Lommel KM, Coffey B. Catatonia in an adolescent with Asperger’s disorder. J Child Adolesc Psychopharmacol. 2009;19(1):93-96.
20. Ghaziuddin N, Dhossche D, Marcotte K. Retrospective chart review of catatonia in child and adolescent psychiatric patients. Acta Psychiatr Scand. 2012;125(1):33-38.
21. Ackard DM, Fulkerson JA, Neumark-Sztainer D. Stability of eating disorder diagnostic classifications in adolescents: five-year longitudinal findings from a population-based study. Eat Disord. 2011;19(4):308-322.
22. Schimmelmann BG, Schultze-Lutter F. Early detection and intervention of psychosis in children and adolescents: urgent need for studies. Eur Child Adolesc Psychiatry. 2012;21(5):239-241.
CASE A fainting spell
Ms. A, age 13, is admitted to a pediatric unit after fainting and losing consciousness for 5 minutes in the shower, during which time she was non-responsive. She reports feeling nauseated and having blurry vision before dropping to the floor.
Ms. A reports intentional self-restriction of calories, self-induced vomiting, and other purging behaviors, such as laxative abuse and excessive exercising.
During the mental status examination, Ms. A is lying in bed wearing hospital clothes, legs flexed at the knee, hands on her side, and a fixed gaze at the ceiling with poor eye contact. She is of slender stature and tall, seems slightly older than her stated age, and is poorly groomed.
Throughout the interview, Ms. A has significant psychomotor retardation, reports her mood as tired, and has a blunted affect. She speaks at a low volume and has poverty of speech; she takes deep sighs before answering questions. Her thought process is linear and she cooperates with the interview. She has poor recall, including delayed 3-minute recall and poor sustained attention. Her abstraction capacity is fair and her intellect is average and comparable with her age group. Ms. A is preoccupied that eating will cause weight gain. She denies hallucinations but reports passive death wishes with self-harm by scratching.
What is the differential diagnosis to explain Ms. A’s presentation?
a) syncope
b) seizures
c) dehydration
d) hypotension
HISTORY Preoccupied with weight
Ms. A reports vomiting twice a day, while showering and at night when no one is around, every day for 2 months. She stopped eating and taking in fluids 3 days before admission to the medical unit. Also, she reports restricting her diet to 700 to 1,000 calories a day, skipping lunch at school, and eating minimally at night. Ms. A uses raspberry ketones and green coffee beans, which are advertised to aid weight loss, and laxative pills from her mother’s medicine cabinet once or twice a week when her throat is sore from vomiting. She reports exercising excessively, which includes running, crunches, and lifting weights. She has lost approximately 30 lb in the last 2 months.
Ms. A says she fears gaining weight and feels increased guilt after eating a meal. She said that looking at food induced “anxiety attack” symptoms of increased heart rate, sweaty palms, feeling of choking, nervousness, and shakiness. She adds that she does not want to be “bigger” than her classmates. Her understanding of the consequences of not eating is, “It will get worse, I will shut down and die. I do not fear death, I only fear getting bigger than others.”
She reports that her fixation on avoiding food started when she realized that she was the tallest girl in her class and the only girl in her class running on the track team, after which she quit athletics. She reports that depression symptoms pre-dated her eating disorder symptoms; onset of significant depression likely was precipitated by her grandfather’s death a year earlier, and then exacerbated by the recent death of a family pet.
Ms. A’s depressive symptoms are described as anhedonia (avoiding being outside and not enjoying drawing anymore), decreased energy, tearfulness, sadness, decreased concentration, and passive suicidal thoughts. Her mother is supportive and motivates her daughter to “get better.” Ms. A denies any symptoms of psychosis, other anxiety symptoms, other mood disorder symptoms, substance abuse, or homicidality.
Ms. A’s mother says she felt that, recently, her daughter has been having some difficulty with confused thoughts and significantly delayed responses. However, the mother reports that her daughter always had somewhat delayed responses from what she felt is typical. Her mother adds that Ms. A’s suicidal thoughts have worsened since her daughter started restricting her diet.
Which diagnosis likely accounts for Ms. A’s presentation?
a) major depressive disorder (MDD)
b) eating disorder, not otherwise specified (NOS)
c) anorexia nervosa, purging type
d) catatonia, unspecified
e) anxiety disorder NOS
f) cognitive disorder
g) psychosis NOS
The authors’ observations
There are many reported causes of catatonia in children and adolescents, including those that are psychiatric, medical, or neurological, as well as drugs (Table 1).1,2 Affective disorders have been associated with catatonia in adults, but has not been widely reported in children and adolescents.1,3 Organic and neurologic causes, such as neurological tumors and cerebral hemorrhage, should be ruled out first because, although rare, they can be fatal (Table 2).2 If the cause of catatonia is not recognized quickly (Figure,1,2) effective treatment could be delayed.4
Catatonia involves psychomotor abnormalities, which are listed in Table 3.1,4
Presentation in adults and adolescents is similar.
An eating disorder could be comorbid with another psychiatric disorder, such as MDD, dysthymia, or panic disorder.5 Ms. A’s report of depression before she began restricting food favored a primary diagnosis of MDD. Her depressive symptoms of low appetite or low self-worth could have led to her preoccupation with body image.
There has been evidence that negative self-image and eating disorders are associated, but data are limited and the connection remains unclear.6 Ms. A’s self-esteem was very low. Her fixation on restricting food could have been perpetuated by her self-criticism and by being excluded from her peer group in school. Her weight loss could have brought anxiety symptoms to the forefront because of physiologic changes that accompany extreme weight loss.
The treatment team was concerned about her delayed responses, which could be explained by the catatonic features that reflected the severity of her depression. She had no obvious symptoms of psychosis, but her intrusive thoughts and obsessions with avoiding food did not completely rule out psychosis.
Childhood-onset schizophrenia, although rare, has been associated with catatonia; following up with a catatonia rating scale, such as the Catatonia Rating Scale or the Bush- Francis Catatonia Rating Scale (BFCRS), would be useful for tracking symptom progress. In Ms. A’s case, her mood disorder was primary, but did not rule out psychosis-like prodromal symptoms.7
Ms. A is diagnosed with MDD, single episode, severe, with catatonic features, and without psychosis, and eating disorder, NOS.
EVALUATION Mostly normal
Ms. A does not have a history of mental illness and was not seeing a psychiatrist or therapist, nor did she have any prior psychiatric admissions. She denies suicide attempts, but reports self-injurious behavior involving scratching her skin, which started during the current mood episode. She has never taken any psychotropic medications. Ms. A lives at home with her biological mother and father and 17-year-old brother. She attends middle school with average grades and has no history of disciplinary actions. She has no history of bullying or teasing, although she did report some previous difficulty with relational aggression toward her peers in the 5th grade. Her mother has a history of anorexia nervosa that began when she was a teenager, but these symptoms are stable and under control. There is additionally a family history of bipolar disorder.
Ms. A has a family history of coronary artery disease and diabetes in the mother and maternal relatives. Her grandfather died from liver cancer. She was allergic to sulfa drugs and was taking a multivitamin and minocycline for acne.
Physical examination reveals some superficial scratches but otherwise was within normal limits. Initial lab results reveal a normal complete blood count and differential. Thyroid-stimulating hormone is 1.29 mIU/L and free T4 is 0.96 mg/dL, both within normal limits. Urinalysis is within normal limits and urine pregnancy test is negative. A comprehensive metabolic panel shows mild elevation in aspartate aminotransferase (AST) at 60 U/L and alanine aminotransferase (ALT) at 92 U/L, respectively. Phosphorus level is within normal limits. Prealbumin level is slightly low at 15.1 mg/dL.
Which treatment plan would you recommend for Ms. A?
a) discharge with outpatient psychiatric treatment
b) recommend medical stabilization with follow-up from the psychosomatic team and then outpatient psychiatric follow-up
c) admit her to the psychiatric acute inpatient hospital with psychiatric outpatient discharge follow-up plan
d) discharge her home with follow-up with her primary care physician
e) recommend follow-up from the psychosomatic team while on medical floor with acute inpatient admission and psychiatric outpatient follow-up at discharge
The authors’ observations
Scarcity of data and reporting of cases of adolescent catatonia limits guidance for diagnosis and treatment.8 There are several rating scales with variability in definition, but that overall provide a guiding tool for detecting catatonia. The Brief Cognitive Rating Scale is considered the most versatile because it is more valid, reliable, and requires less time to complete than other rating scales.9
Ms. A’s symptoms were a combination of depressive symptoms with severity defined by catatonic features, eating disorder with worsening course, anxiety symptoms, and genetic loading of eating disorder in her mother. The challenge of this case was making an accurate diagnosis and treating Ms. A, which required continuous observation following an eating disorder protocol, resolution of her catatonia, resuming a normal diet, and decreasing her suicidality. Retrospectively, her scores on the BFCRS were high on screening items 1 to 14, which measure presence or absence and severity of symptoms.
The best option was to admit Ms. A to an inpatient psychiatric facility after she is cleared medically with outpatient services to follow up.
How would you treat Ms. A’s symptoms?
a) aggressively treat catatonia
b) address her eating disorder
c) work to resolve her depression
The authors’ observations
The challenge was to choose the psychotropic medication that would target her depression, obsessive, rigid thoughts, and catatonia. Administering an antidepressant with an antipsychotic would have relieved her depressive and obsessive symptoms but would not have improved her catatonia. The psychosomatic medicine team recommended starting a selective serotonin reuptake inhibitor and a benzodiazepine to target both the depression and the catatonic symptoms. Ms. A received sertraline, 12.5 mg/d, which was increased to 25 mg/d on the third day. IV lorazepam, 1 mg, 3 times a day, was recommended but the pediatric team prescribed an oral formulation. The hospital’s eating disorder protocol was instituted on the day of admission.
Treatment options for catatonia
Benzodiazepines are the first line of treatment for catatonia and other neuroleptics, specifically antipsychotics, have been considered dangerous.10 Benzodiazepine-resistant catatonia, which is sometimes seen in patients with autism, might respond to electroconvulsive therapy (ECT),11 although in some states it cannot be administered to children age <18.12 Benzodiazepines have shown dramatic improvement within hours, as has ECT.8,13 Additionally, if patients do not respond to a benzodiazepine or ECT, consider other options such as zolpidem, olanzapine,14 or sensory integration system (in adolescents with autism).15
Ms. A did not need ECT or an alternative treatment because she responded well to 3 doses of oral lorazepam. Her amotivation, negativism, and rigidity with prolonged posturing improved. Her psychomotor retardation improved overall, although she reported some dizziness and had some postural hypotension, which was attributed to her eating issues and dehydration.
OUTCOME Feeling motivated
Ms. A is transferred to psychiatric inpatient unit. She tolerates sertraline, which is titrated to 50 mg/d. She is placed on the hospital’s standard eating disorder protocol. She continues to eat well with adequate intake of solids and liquid and exhibits only some anxiety associated with meals. During the course of hospitalization, she attends group therapy and her catatonic symptoms completely resolve. She says she thinks that her thoughts are improving and that she is not longer feeling confused. She reports being motivated to continue to improve her eating disorder symptoms.
The treatment team holds a family session during which family dynamic issues that are stressful to Ms. A are discussed, such as some conflict with her parents as well as some negative interactions between Ms. A and her father. Repeat comprehensive metabolic panel on admission to the inpatient psychiatric hospital reveals persistent elevation of AST at 92 U/L and ALT at 143 U/L. Ms. A is discharged home with follow-up with a psychiatrist and a therapist. The treatment team also recommends that she follow up in a program that specializes in eating disorders.
4-month follow-up. Ms. A returns to inpatient psychiatric hospital after overdose of sertraline and aripiprazole, which were started by an outpatient psychiatrist. She reports severe depressive symptoms because of school stressors. She denies any problems eating and did not show any symptoms of catatonia. In her chart, there is a mention of “cloudy thoughts” and quietness. At this admission, her ALT is 17 U/L and AST is 19 U/L. Sertraline is increased to 150 mg/d and aripiprazole is reduced to 2 mg/d and then later increased to 5 mg/d, after which she is discharged home with an outpatient psychiatric follow-up.
1-year follow-up. Ms. A has been following up with an outpatient psychiatrist and is receiving sertraline, 150 mg/d, aripiprazole, 2.5 mg/d, and extended-release methylphenidate, 36 mg/d, along with L-methylfolate, multivitamins, and omega-3 fish oil as adjuvants for her depressive symptoms. Ms. A does not show symptoms of an eating disorder or catatonia, and her depression and psychomotor activity have improved, with better overall functionality, after adding the stimulant and adjunctives to the antidepressant.
The authors’ observations
The importance of including catatonia NOS with its various specifiers, such as medical, metabolic, toxic, affective, etc., has been discussed.16,17 In Ms. A’s case, instead of treating the specific symptoms—affective or eating disorder or obsessive quality of thought content, mimicking psychotic-like symptoms—addressing the catatonia initially had a better outcome. More studies related to chronic and acute catatonia in adolescents are needed because of the risk of increased morbidity and premature death.18 Early recognition of catatonia is needed19 because it often is underdiagnosed.20
Eating disorders often become worse over the first 5 years, and close monitoring and assessment is needed for adolescents.21 Also, prodromal psychotic symptoms require follow-up because techniques for early detection and intervention for children and adolescents are still in their infancy.22
Bottom Line
Catatonia in adolescents should be addressed early, when it is treatable and the outcome is favorable. It is important to recognize catatonia in an emergency department or inpatient medical unit setting in a hospital because it is often underdiagnosed or misdiagnosed. The presentation of catatonia is similar in adolescents and adults. Benzodiazepines are first-line treatment for catatonia; consider electroconvulsive therapy if patients do not respond to drug therapy.
Related Resources
• Roberto AJ, Pinnaka S, Mohan A, et al. Adolescent catatonia successfully treated with lorazepam and aripiprazole. Case Rep Psychiatry. 2014;2014:309517. doi: 10.1155/2014/309517.
• Raffin M, Zugaj-Bensaou L, Bodeau N, et al. Treatment use in a prospective naturalistic cohort of children and adolescents with catatonia. Eur Child Adolesc Psychiatry. 2015;24(4):441-449.
Drug Brand Names
Aripiprazole • Abilify Minocycline • Minocin
L-methylfolate • Deplin Olanzapine • Zyprexa
Lorazepam • Ativan Sertraline • Zoloft
Methylphenidate • Ritalin, Concerta Zolpidem • Ambien, Intermezzo
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE A fainting spell
Ms. A, age 13, is admitted to a pediatric unit after fainting and losing consciousness for 5 minutes in the shower, during which time she was non-responsive. She reports feeling nauseated and having blurry vision before dropping to the floor.
Ms. A reports intentional self-restriction of calories, self-induced vomiting, and other purging behaviors, such as laxative abuse and excessive exercising.
During the mental status examination, Ms. A is lying in bed wearing hospital clothes, legs flexed at the knee, hands on her side, and a fixed gaze at the ceiling with poor eye contact. She is of slender stature and tall, seems slightly older than her stated age, and is poorly groomed.
Throughout the interview, Ms. A has significant psychomotor retardation, reports her mood as tired, and has a blunted affect. She speaks at a low volume and has poverty of speech; she takes deep sighs before answering questions. Her thought process is linear and she cooperates with the interview. She has poor recall, including delayed 3-minute recall and poor sustained attention. Her abstraction capacity is fair and her intellect is average and comparable with her age group. Ms. A is preoccupied that eating will cause weight gain. She denies hallucinations but reports passive death wishes with self-harm by scratching.
What is the differential diagnosis to explain Ms. A’s presentation?
a) syncope
b) seizures
c) dehydration
d) hypotension
HISTORY Preoccupied with weight
Ms. A reports vomiting twice a day, while showering and at night when no one is around, every day for 2 months. She stopped eating and taking in fluids 3 days before admission to the medical unit. Also, she reports restricting her diet to 700 to 1,000 calories a day, skipping lunch at school, and eating minimally at night. Ms. A uses raspberry ketones and green coffee beans, which are advertised to aid weight loss, and laxative pills from her mother’s medicine cabinet once or twice a week when her throat is sore from vomiting. She reports exercising excessively, which includes running, crunches, and lifting weights. She has lost approximately 30 lb in the last 2 months.
Ms. A says she fears gaining weight and feels increased guilt after eating a meal. She said that looking at food induced “anxiety attack” symptoms of increased heart rate, sweaty palms, feeling of choking, nervousness, and shakiness. She adds that she does not want to be “bigger” than her classmates. Her understanding of the consequences of not eating is, “It will get worse, I will shut down and die. I do not fear death, I only fear getting bigger than others.”
She reports that her fixation on avoiding food started when she realized that she was the tallest girl in her class and the only girl in her class running on the track team, after which she quit athletics. She reports that depression symptoms pre-dated her eating disorder symptoms; onset of significant depression likely was precipitated by her grandfather’s death a year earlier, and then exacerbated by the recent death of a family pet.
Ms. A’s depressive symptoms are described as anhedonia (avoiding being outside and not enjoying drawing anymore), decreased energy, tearfulness, sadness, decreased concentration, and passive suicidal thoughts. Her mother is supportive and motivates her daughter to “get better.” Ms. A denies any symptoms of psychosis, other anxiety symptoms, other mood disorder symptoms, substance abuse, or homicidality.
Ms. A’s mother says she felt that, recently, her daughter has been having some difficulty with confused thoughts and significantly delayed responses. However, the mother reports that her daughter always had somewhat delayed responses from what she felt is typical. Her mother adds that Ms. A’s suicidal thoughts have worsened since her daughter started restricting her diet.
Which diagnosis likely accounts for Ms. A’s presentation?
a) major depressive disorder (MDD)
b) eating disorder, not otherwise specified (NOS)
c) anorexia nervosa, purging type
d) catatonia, unspecified
e) anxiety disorder NOS
f) cognitive disorder
g) psychosis NOS
The authors’ observations
There are many reported causes of catatonia in children and adolescents, including those that are psychiatric, medical, or neurological, as well as drugs (Table 1).1,2 Affective disorders have been associated with catatonia in adults, but has not been widely reported in children and adolescents.1,3 Organic and neurologic causes, such as neurological tumors and cerebral hemorrhage, should be ruled out first because, although rare, they can be fatal (Table 2).2 If the cause of catatonia is not recognized quickly (Figure,1,2) effective treatment could be delayed.4
Catatonia involves psychomotor abnormalities, which are listed in Table 3.1,4
Presentation in adults and adolescents is similar.
An eating disorder could be comorbid with another psychiatric disorder, such as MDD, dysthymia, or panic disorder.5 Ms. A’s report of depression before she began restricting food favored a primary diagnosis of MDD. Her depressive symptoms of low appetite or low self-worth could have led to her preoccupation with body image.
There has been evidence that negative self-image and eating disorders are associated, but data are limited and the connection remains unclear.6 Ms. A’s self-esteem was very low. Her fixation on restricting food could have been perpetuated by her self-criticism and by being excluded from her peer group in school. Her weight loss could have brought anxiety symptoms to the forefront because of physiologic changes that accompany extreme weight loss.
The treatment team was concerned about her delayed responses, which could be explained by the catatonic features that reflected the severity of her depression. She had no obvious symptoms of psychosis, but her intrusive thoughts and obsessions with avoiding food did not completely rule out psychosis.
Childhood-onset schizophrenia, although rare, has been associated with catatonia; following up with a catatonia rating scale, such as the Catatonia Rating Scale or the Bush- Francis Catatonia Rating Scale (BFCRS), would be useful for tracking symptom progress. In Ms. A’s case, her mood disorder was primary, but did not rule out psychosis-like prodromal symptoms.7
Ms. A is diagnosed with MDD, single episode, severe, with catatonic features, and without psychosis, and eating disorder, NOS.
EVALUATION Mostly normal
Ms. A does not have a history of mental illness and was not seeing a psychiatrist or therapist, nor did she have any prior psychiatric admissions. She denies suicide attempts, but reports self-injurious behavior involving scratching her skin, which started during the current mood episode. She has never taken any psychotropic medications. Ms. A lives at home with her biological mother and father and 17-year-old brother. She attends middle school with average grades and has no history of disciplinary actions. She has no history of bullying or teasing, although she did report some previous difficulty with relational aggression toward her peers in the 5th grade. Her mother has a history of anorexia nervosa that began when she was a teenager, but these symptoms are stable and under control. There is additionally a family history of bipolar disorder.
Ms. A has a family history of coronary artery disease and diabetes in the mother and maternal relatives. Her grandfather died from liver cancer. She was allergic to sulfa drugs and was taking a multivitamin and minocycline for acne.
Physical examination reveals some superficial scratches but otherwise was within normal limits. Initial lab results reveal a normal complete blood count and differential. Thyroid-stimulating hormone is 1.29 mIU/L and free T4 is 0.96 mg/dL, both within normal limits. Urinalysis is within normal limits and urine pregnancy test is negative. A comprehensive metabolic panel shows mild elevation in aspartate aminotransferase (AST) at 60 U/L and alanine aminotransferase (ALT) at 92 U/L, respectively. Phosphorus level is within normal limits. Prealbumin level is slightly low at 15.1 mg/dL.
Which treatment plan would you recommend for Ms. A?
a) discharge with outpatient psychiatric treatment
b) recommend medical stabilization with follow-up from the psychosomatic team and then outpatient psychiatric follow-up
c) admit her to the psychiatric acute inpatient hospital with psychiatric outpatient discharge follow-up plan
d) discharge her home with follow-up with her primary care physician
e) recommend follow-up from the psychosomatic team while on medical floor with acute inpatient admission and psychiatric outpatient follow-up at discharge
The authors’ observations
Scarcity of data and reporting of cases of adolescent catatonia limits guidance for diagnosis and treatment.8 There are several rating scales with variability in definition, but that overall provide a guiding tool for detecting catatonia. The Brief Cognitive Rating Scale is considered the most versatile because it is more valid, reliable, and requires less time to complete than other rating scales.9
Ms. A’s symptoms were a combination of depressive symptoms with severity defined by catatonic features, eating disorder with worsening course, anxiety symptoms, and genetic loading of eating disorder in her mother. The challenge of this case was making an accurate diagnosis and treating Ms. A, which required continuous observation following an eating disorder protocol, resolution of her catatonia, resuming a normal diet, and decreasing her suicidality. Retrospectively, her scores on the BFCRS were high on screening items 1 to 14, which measure presence or absence and severity of symptoms.
The best option was to admit Ms. A to an inpatient psychiatric facility after she is cleared medically with outpatient services to follow up.
How would you treat Ms. A’s symptoms?
a) aggressively treat catatonia
b) address her eating disorder
c) work to resolve her depression
The authors’ observations
The challenge was to choose the psychotropic medication that would target her depression, obsessive, rigid thoughts, and catatonia. Administering an antidepressant with an antipsychotic would have relieved her depressive and obsessive symptoms but would not have improved her catatonia. The psychosomatic medicine team recommended starting a selective serotonin reuptake inhibitor and a benzodiazepine to target both the depression and the catatonic symptoms. Ms. A received sertraline, 12.5 mg/d, which was increased to 25 mg/d on the third day. IV lorazepam, 1 mg, 3 times a day, was recommended but the pediatric team prescribed an oral formulation. The hospital’s eating disorder protocol was instituted on the day of admission.
Treatment options for catatonia
Benzodiazepines are the first line of treatment for catatonia and other neuroleptics, specifically antipsychotics, have been considered dangerous.10 Benzodiazepine-resistant catatonia, which is sometimes seen in patients with autism, might respond to electroconvulsive therapy (ECT),11 although in some states it cannot be administered to children age <18.12 Benzodiazepines have shown dramatic improvement within hours, as has ECT.8,13 Additionally, if patients do not respond to a benzodiazepine or ECT, consider other options such as zolpidem, olanzapine,14 or sensory integration system (in adolescents with autism).15
Ms. A did not need ECT or an alternative treatment because she responded well to 3 doses of oral lorazepam. Her amotivation, negativism, and rigidity with prolonged posturing improved. Her psychomotor retardation improved overall, although she reported some dizziness and had some postural hypotension, which was attributed to her eating issues and dehydration.
OUTCOME Feeling motivated
Ms. A is transferred to psychiatric inpatient unit. She tolerates sertraline, which is titrated to 50 mg/d. She is placed on the hospital’s standard eating disorder protocol. She continues to eat well with adequate intake of solids and liquid and exhibits only some anxiety associated with meals. During the course of hospitalization, she attends group therapy and her catatonic symptoms completely resolve. She says she thinks that her thoughts are improving and that she is not longer feeling confused. She reports being motivated to continue to improve her eating disorder symptoms.
The treatment team holds a family session during which family dynamic issues that are stressful to Ms. A are discussed, such as some conflict with her parents as well as some negative interactions between Ms. A and her father. Repeat comprehensive metabolic panel on admission to the inpatient psychiatric hospital reveals persistent elevation of AST at 92 U/L and ALT at 143 U/L. Ms. A is discharged home with follow-up with a psychiatrist and a therapist. The treatment team also recommends that she follow up in a program that specializes in eating disorders.
4-month follow-up. Ms. A returns to inpatient psychiatric hospital after overdose of sertraline and aripiprazole, which were started by an outpatient psychiatrist. She reports severe depressive symptoms because of school stressors. She denies any problems eating and did not show any symptoms of catatonia. In her chart, there is a mention of “cloudy thoughts” and quietness. At this admission, her ALT is 17 U/L and AST is 19 U/L. Sertraline is increased to 150 mg/d and aripiprazole is reduced to 2 mg/d and then later increased to 5 mg/d, after which she is discharged home with an outpatient psychiatric follow-up.
1-year follow-up. Ms. A has been following up with an outpatient psychiatrist and is receiving sertraline, 150 mg/d, aripiprazole, 2.5 mg/d, and extended-release methylphenidate, 36 mg/d, along with L-methylfolate, multivitamins, and omega-3 fish oil as adjuvants for her depressive symptoms. Ms. A does not show symptoms of an eating disorder or catatonia, and her depression and psychomotor activity have improved, with better overall functionality, after adding the stimulant and adjunctives to the antidepressant.
The authors’ observations
The importance of including catatonia NOS with its various specifiers, such as medical, metabolic, toxic, affective, etc., has been discussed.16,17 In Ms. A’s case, instead of treating the specific symptoms—affective or eating disorder or obsessive quality of thought content, mimicking psychotic-like symptoms—addressing the catatonia initially had a better outcome. More studies related to chronic and acute catatonia in adolescents are needed because of the risk of increased morbidity and premature death.18 Early recognition of catatonia is needed19 because it often is underdiagnosed.20
Eating disorders often become worse over the first 5 years, and close monitoring and assessment is needed for adolescents.21 Also, prodromal psychotic symptoms require follow-up because techniques for early detection and intervention for children and adolescents are still in their infancy.22
Bottom Line
Catatonia in adolescents should be addressed early, when it is treatable and the outcome is favorable. It is important to recognize catatonia in an emergency department or inpatient medical unit setting in a hospital because it is often underdiagnosed or misdiagnosed. The presentation of catatonia is similar in adolescents and adults. Benzodiazepines are first-line treatment for catatonia; consider electroconvulsive therapy if patients do not respond to drug therapy.
Related Resources
• Roberto AJ, Pinnaka S, Mohan A, et al. Adolescent catatonia successfully treated with lorazepam and aripiprazole. Case Rep Psychiatry. 2014;2014:309517. doi: 10.1155/2014/309517.
• Raffin M, Zugaj-Bensaou L, Bodeau N, et al. Treatment use in a prospective naturalistic cohort of children and adolescents with catatonia. Eur Child Adolesc Psychiatry. 2015;24(4):441-449.
Drug Brand Names
Aripiprazole • Abilify Minocycline • Minocin
L-methylfolate • Deplin Olanzapine • Zyprexa
Lorazepam • Ativan Sertraline • Zoloft
Methylphenidate • Ritalin, Concerta Zolpidem • Ambien, Intermezzo
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dhossche D, Wilson C, Wachtel LE. Catatonia in childhood and adolescence: implications for the DSM-5. Primary Psychiatry. http://primarypsychiatry.com/catatonia-in-childhood-and-adolescence-implications-for-the-dsm-5. Published May 21, 2013. Accessed July 2, 2015.
2. Lahutte B, Cornic F, Bonnot O, et al. Multidisciplinary approach of organic catatonia in children and adolescents may improve treatment decision making. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(6):1393-1398.
3. Brake JA, Abidi S. A case of adolescent catatonia. J Can Acad Child Adolesc Psychiatry. 2010;19(2):138-140.
4. Consoli A, Raffin M, Laurent C, et al. Medical and developmental risk factors of catatonia in children and adolescents: a prospective case-control study. Schizophr Res. 2012;137(1-3):151-158.
5. Zaider TI, Johnson JG, Cockell SJ. Psychiatric comorbidity associated with eating disorder symptomatology among adolescents in the community. Int J Eat Disord. 2000;28(1):58-67.
6. Forsén Mantilla E, Bergsten K, Birgegård A. Self-image and eating disorder symptoms in normal and clinical adolescents. Eat Behav. 2014;15(1):125-131.
7. Bonnot O, Tanguy ML, Consoli A, et al. Does catatonia influence the phenomenology of childhood onset schizophrenia beyond motor symptoms? Psychiatry Res. 2008;158(3):356-362.
8. Singh LK, Praharaj SK. Immediate response to lorazepam in a patient with 17 years of chronic catatonia. J Neuropsychiatry Clin Neurosci. 2013;25(3):E47-E48.
9. Sienaert P, Rooseleer J, De Fruyt J. Measuring catatonia: a systematic review of rating scales. J Affect Disord. 2011;135(1-3):1-9.
10. Cottencin O, Warembourg F, de Chouly de Lenclave MB, et al. Catatonia and consultation-liaison psychiatry study of 12 cases. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1170-1176.
11. Wachtel LE, Hermida A, Dhossche DM. Maintenance electroconvulsive therapy in autistic catatonia: a case series review. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(4):581-587.
12. Wachtel LE, Dhossche DM, Kellner CH. When is electroconvulsive therapy appropriate for children and adolescents? Med Hypotheses. 2011;76(3):395-399.
13. Takaoka K, Takata T. Catatonia in childhood and adolescence. Psychiatry Clin Neurosci. 2003;57(2):129-137.
14. Ceylan MF, Kul M, Kultur SE, et al. Major depression with catatonic features in a child remitted with olanzapine. J Child Adolesc Psychopharmacol. 2010;20(3):225-227.
15. Consoli A, Gheorghiev C, Jutard C, et al. Lorazepam, fluoxetine and packing therapy in an adolescent with pervasive developmental disorder and catatonia. J Physiol Paris. 2010;104(6):309-314.
16. Dhossche D, Cohen D, Ghaziuddin N, et al. The study of pediatric catatonia supports a home of its own for catatonia in DSM-5. Med Hypotheses. 2010;75(6):558-560.
17. Taylor MA, Fink M. Catatonia in psychiatric classification: a home of its own. Am J Psychiatry. 2003;160(7):1233-1241.
18. Cornic F, Consoli A, Tanguy ML, et al. Association of adolescent catatonia with increased mortality and morbidity: evidence from a prospective follow-up study. Schizophr Res. 2009;113(2-3):233-240.
19. Quigley J, Lommel KM, Coffey B. Catatonia in an adolescent with Asperger’s disorder. J Child Adolesc Psychopharmacol. 2009;19(1):93-96.
20. Ghaziuddin N, Dhossche D, Marcotte K. Retrospective chart review of catatonia in child and adolescent psychiatric patients. Acta Psychiatr Scand. 2012;125(1):33-38.
21. Ackard DM, Fulkerson JA, Neumark-Sztainer D. Stability of eating disorder diagnostic classifications in adolescents: five-year longitudinal findings from a population-based study. Eat Disord. 2011;19(4):308-322.
22. Schimmelmann BG, Schultze-Lutter F. Early detection and intervention of psychosis in children and adolescents: urgent need for studies. Eur Child Adolesc Psychiatry. 2012;21(5):239-241.
1. Dhossche D, Wilson C, Wachtel LE. Catatonia in childhood and adolescence: implications for the DSM-5. Primary Psychiatry. http://primarypsychiatry.com/catatonia-in-childhood-and-adolescence-implications-for-the-dsm-5. Published May 21, 2013. Accessed July 2, 2015.
2. Lahutte B, Cornic F, Bonnot O, et al. Multidisciplinary approach of organic catatonia in children and adolescents may improve treatment decision making. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(6):1393-1398.
3. Brake JA, Abidi S. A case of adolescent catatonia. J Can Acad Child Adolesc Psychiatry. 2010;19(2):138-140.
4. Consoli A, Raffin M, Laurent C, et al. Medical and developmental risk factors of catatonia in children and adolescents: a prospective case-control study. Schizophr Res. 2012;137(1-3):151-158.
5. Zaider TI, Johnson JG, Cockell SJ. Psychiatric comorbidity associated with eating disorder symptomatology among adolescents in the community. Int J Eat Disord. 2000;28(1):58-67.
6. Forsén Mantilla E, Bergsten K, Birgegård A. Self-image and eating disorder symptoms in normal and clinical adolescents. Eat Behav. 2014;15(1):125-131.
7. Bonnot O, Tanguy ML, Consoli A, et al. Does catatonia influence the phenomenology of childhood onset schizophrenia beyond motor symptoms? Psychiatry Res. 2008;158(3):356-362.
8. Singh LK, Praharaj SK. Immediate response to lorazepam in a patient with 17 years of chronic catatonia. J Neuropsychiatry Clin Neurosci. 2013;25(3):E47-E48.
9. Sienaert P, Rooseleer J, De Fruyt J. Measuring catatonia: a systematic review of rating scales. J Affect Disord. 2011;135(1-3):1-9.
10. Cottencin O, Warembourg F, de Chouly de Lenclave MB, et al. Catatonia and consultation-liaison psychiatry study of 12 cases. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1170-1176.
11. Wachtel LE, Hermida A, Dhossche DM. Maintenance electroconvulsive therapy in autistic catatonia: a case series review. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(4):581-587.
12. Wachtel LE, Dhossche DM, Kellner CH. When is electroconvulsive therapy appropriate for children and adolescents? Med Hypotheses. 2011;76(3):395-399.
13. Takaoka K, Takata T. Catatonia in childhood and adolescence. Psychiatry Clin Neurosci. 2003;57(2):129-137.
14. Ceylan MF, Kul M, Kultur SE, et al. Major depression with catatonic features in a child remitted with olanzapine. J Child Adolesc Psychopharmacol. 2010;20(3):225-227.
15. Consoli A, Gheorghiev C, Jutard C, et al. Lorazepam, fluoxetine and packing therapy in an adolescent with pervasive developmental disorder and catatonia. J Physiol Paris. 2010;104(6):309-314.
16. Dhossche D, Cohen D, Ghaziuddin N, et al. The study of pediatric catatonia supports a home of its own for catatonia in DSM-5. Med Hypotheses. 2010;75(6):558-560.
17. Taylor MA, Fink M. Catatonia in psychiatric classification: a home of its own. Am J Psychiatry. 2003;160(7):1233-1241.
18. Cornic F, Consoli A, Tanguy ML, et al. Association of adolescent catatonia with increased mortality and morbidity: evidence from a prospective follow-up study. Schizophr Res. 2009;113(2-3):233-240.
19. Quigley J, Lommel KM, Coffey B. Catatonia in an adolescent with Asperger’s disorder. J Child Adolesc Psychopharmacol. 2009;19(1):93-96.
20. Ghaziuddin N, Dhossche D, Marcotte K. Retrospective chart review of catatonia in child and adolescent psychiatric patients. Acta Psychiatr Scand. 2012;125(1):33-38.
21. Ackard DM, Fulkerson JA, Neumark-Sztainer D. Stability of eating disorder diagnostic classifications in adolescents: five-year longitudinal findings from a population-based study. Eat Disord. 2011;19(4):308-322.
22. Schimmelmann BG, Schultze-Lutter F. Early detection and intervention of psychosis in children and adolescents: urgent need for studies. Eur Child Adolesc Psychiatry. 2012;21(5):239-241.
Depressed and confused, and dizzy while walking the dog
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
Avoiding common drug−drug interactions
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight

hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.