User login
From the Division of Hematology, Ohio State University, Columbus, OH.
Abstract
- Objective: To describe the diagnosis and initial management of chronic lymphocytic leukemia (CLL), including first-line treatment options.
- Methods: Case presentation and review of the literature.
- Results: Most CLL patients demonstrate a chronic, relapsing and remitting course with intervals of months to years between treatments. Recent advances in genetic and molecular markers for risk stratification of CLL significantly impact how clinicians determine prognosis and predict response to treatment for patients with newly diagnosed disease. This information, along with patient factors such as age and health status, should be considered when formulating an initial treatment strategy. Combinations of chemotherapy and immunotherapy offer the longest progression-free survival and overall survival benefit yet reported. For elderly patients or those with significant comorbidities who may not tolerate standard chemoimmunotherapy, less intensive but still effective therapies now exist. Patients with the highest risk disease, such as those with deletions of chromosome 17p, respond poorly to conventional treatment and should be referred to experienced centers where investigational therapies and allogeneic stem cell transplantation are available.
- Conclusion: Both disease characteristics and patient factors should guide the selection among the various effective therapies for CLL. While chemoimmunotherapy is the most effective treatment developed to date, its use may become less prevalent as newer agents are incorporated into initial and relapse treatment algorithms.
Chronic lymphocytic leukemia (CLL) is a chronic malignancy of B-lymphocytes demonstrating a heterogeneous clinical course ranging from indolent to more rapidly progressive. The chief clinical feature is an elevated peripheral blood lymphocyte count, and patients can demonstrate lymphadenopathy, splenomegaly, hepatomegaly, constitutional symptoms, and in late stages bone marrow failure. It is the most common leukemia among adults in the Western world, accounting for between 22% to 30% of new leukemia diagnoses worldwide [1]. Recent incidence rates in the United States are 3.83 cases per 100,000 person-years [2]. The incidence of CLL increases with age, and most new cases are diagnosed in persons 65 years of age or older [1,2]. As reported 5-year survival rates are between 68% and 81% with a median survival of 10 years in some series, the prevalence is significantly higher than the incidence [3]. However, this may even be an underestimate of the population burden of disease, as many cases are not reported to tumor registries [4].
Many patients with CLL are asymptomatic and do not require treatment until years after diagnosis. In these cases a watch and wait approach is taken. The typical natural history of CLL is characterized by periods of effective treatment when required, followed by treatment-free intervals of several years in many cases. However, this can be misleading, as the clinical course for any individual patient is highly variable. Development of cytogenetic and molecular testing has allowed for identification of patients with a higher risk of progression and lower response rates to traditional cytotoxic treatments [5]. For example, depending on chromosomal abnormalities present, median survival can vary from 32 to 133 months [3].
The assessment of underlying disease risk thus provides important information when considering a treatment approach and should be routinely performed for newly diagnosed patients. While the development of highly effective chemoimmunotherapy has allowed most groups of CLL patients to live for many years, some groups do not enjoy the same survival. Recent advances in CLL treatment seek to abrogate such adverse risk factors, thereby improving the survival for all patients with CLL. Given the expected survival of years for most CLL patients, frontline treatment planning must be done in the context of a long-term treatment strategy keeping the risk for late toxicities, such as secondary malignancies, in mind.
Case Study
Initial Presentation
A 50-year-old man is referred for evaluation of cervical lymphadenopathy that had progressed over the prior 6 months. He denies associated symptoms of fatigue, fevers, night sweats, or unintentional weight loss but does report early satiety. On examination there are multiple mobile, enlarged cervical lymph nodes bilaterally. Axillary lymph nodes are likewise enlarged. The liver edge is not palpable, but the spleen is palpable below the belt line. Complete blood count reveals a white blood cell count of 196,000 with 97% lymphocytes. Hemoglobin is 11.0 g/dL and platelet count is 122,000/dL. He recalls being told 3 years previously that his white blood cell count was 48,000 during an emergency department visit for cellulitis.
• How is CLL diagnosed and staged?
CLL is often suspected when patients present with an elevated lymphocyte count. Presenting symptoms of CLL commonly include lymphadenopathy, an enlarged spleen, and constitutional or “B” symptoms such as fatigue, unintentional weight loss, or drenching night sweats. However, only 25% of patients are symptomatic at diagnosis [1]. Many patients with CLL are now diagnosed after a routine blood test, long before the disease is clinically apparent.
The diagnosis of CLL can be made from the peripheral blood and does not require a bone marrow biopsy. According to 2008 guidelines from the International Workshop on Chronic Lymphocytic Leukemia (IWCLL), diagnosis requires at least 5000/uL clonal B-lymphocytes in the peripheral blood. The clonality must be confirmed by immunophenotyping. At time of diagnosis the peripheral blood smear should be examined for the characteristic cells: small mature lymphocytes with a narrow rim of cytoplasm and dense nuclei consisting of clumped chromatin. Larger, atypical cells can be present as long as they do not exceed 55% of the total number of lymphocytes [6].
The immunophenotype of CLL includes aberrant expression of CD5 and a T-cell antigen, along with the characteristic B-cell antigens CD19, CD20, and CD23. The leukemic clone may be either kappa or lambda light chain restricted. Expression of surface immunoglobulin, CD20, and CD79a is typically low compared to that of normal B cells, although there can be some variability in the immunophenotype [6].
![]()
Care should be taken to exclude other malignancies with a similar morphology. Leukemic phase mantle cell lymphoma, other low grade lymphomas, and hairy cell leukemia are commonly mistaken for CLL. Immunophenotyping and cytogenetics are usually sufficient to differentiate these. Testing for a balanced translocation involving chromosomes 11 and 14 to exclude mantle cell lymphoma can be helpful, as both CLL and mantle cell lymphoma can appear morphologically similar and share immunophenotypic features (CD5+/CD19+).

Case Continued
The patient’s peripheral blood is drawn for routine immunophenotyping as well as cytogenetic and molecular testing. When he returns to discuss the results 10 days later, he learns that peripheral blood immunophenotyping demonstrates a dim kappa restricted monoclonal population of B-cells that expressed CD19, CD20(dim), CD23, CD38, CD5, and CD43. The lymphocytes are negative for CD10, FMC7, and CD79b, consistent with a CLL immunophenotype. This patient fulfills diagnostic criteria for CLL and has Rai stage II or intermediate-risk disease. Interphase cytogenetic studies of the peripheral blood demonstrate deletions of chromosomes 11q22.3 and 13q14.3. The immunoglobulin heavy chain gene (IGHV) is unmutated.
• How can a CLL patient’s disease risk be characterized?
Historically, staging at diagnosis, pattern of bone marrow infiltration, and response to therapy were used to gauge prognosis. In more recent years, cytogenetic and molecular testing methods have been developed to augment risk stratification. Testing of prognostic significance that influences clinical management includes IGHV mutational status and interphase cytogenetics using FISH [3,12–14]. Expression of ZAP-70 and CD38 are both independent predictors of poorer prognosis in CLL but are not recommended for routine clinical use. Standardized methodology for the measurement of Zap-70 in particular limits the utility of that test in routine clinical practice [15]. Performed at diagnosis, a time when many patients are asymptomatic, cytogenetic testing with FISH and IGHV mutational analysis can predict time to first treatment and increasingly identify high-risk patients for whom investigational early intervention approaches may be considered [16]. While cytogenetic testing has utility at time of diagnosis, it should be considered necessary prior to deciding on the first-line treatment.

Cytogenetics are also important in predicting response to therapy. For instance, patients with deletion(11q) disease have improved survival when treated with regimens containing an alkylating agent [18]. Deletion(17p) patients respond poorly to traditional cytotoxic agents, and treatments with alternate mechanisms of action should be used [5,19]. The gene for tumor suppressor protein TP53 is encoded in this region of chromosome 17, thus treatment with agents that act independent of pathways involving TP53 are preferred [20].
In addition to cytogenetic testing, quantization of somatic mutations in the gene encoding the variable region of the immune globulin heavy chain gene (IGHV) can help define disease-specific risk. When greater than 98% sequence homology is seen, the gene is considered IGHV unmutated. Patients with an unmutated IGHV have worse overall survival. In one study of Rai stage 0 CLL patients, those with an unmutated IGHV had a survival of only 95 months, compared with 293 months in the mutated group [12].
• When should CLL be treated?
CLL is not curable with current standard therapies, and starting treatment at time of diagnosis for early stage, asymptomatic, CLL patients does not improve overall survival and adds treatment-related toxicities [21,22]. Consequently, the decision to treat is based on treating or preventing complications from the disease, and observation is recommended for most asymptomatic, early-stage patients [6]. Because median survival in CLL is often measured in years, deferring treatment can limit both the short- and long-term complications of therapy, especially the significant risk of secondary malignancies associated with some therapies [23]. However, deferring treatment can significantly impact both a patient’s emotional well-being and quality of life, which should be kept in mind when first discussing the rationale for observation with asymptomatic patients [24].

causes.
For patients with anemia, neutropenia, or thrombocytopenia that is autoimmune in nature, treatment should typically begin with corticosteroids, as it would for non-CLL associated cases of autoimmune cytopenias. If steroids are not effective, second-line treatments appropriate for the situation are generally employed, including intravenous immunoglobulin, cyclosporine, azathioprine, and splenectomy. Rituximab has also been shown to be effective in steroid-refractory cases of autoimmune hemolytic anemia associated with CLL [26]. Only if cytopenias are refractory to appropriate second-line therapy should CLL-directed treatments be considered, assuming there are no other indications to treat the underlying CLL [6]. Bone marrow biopsy can be helpful in differentiating autoimmune cytopenias from marrow failure due to CLL infiltration.
• What treatments are most appropriate for young, fit patients?
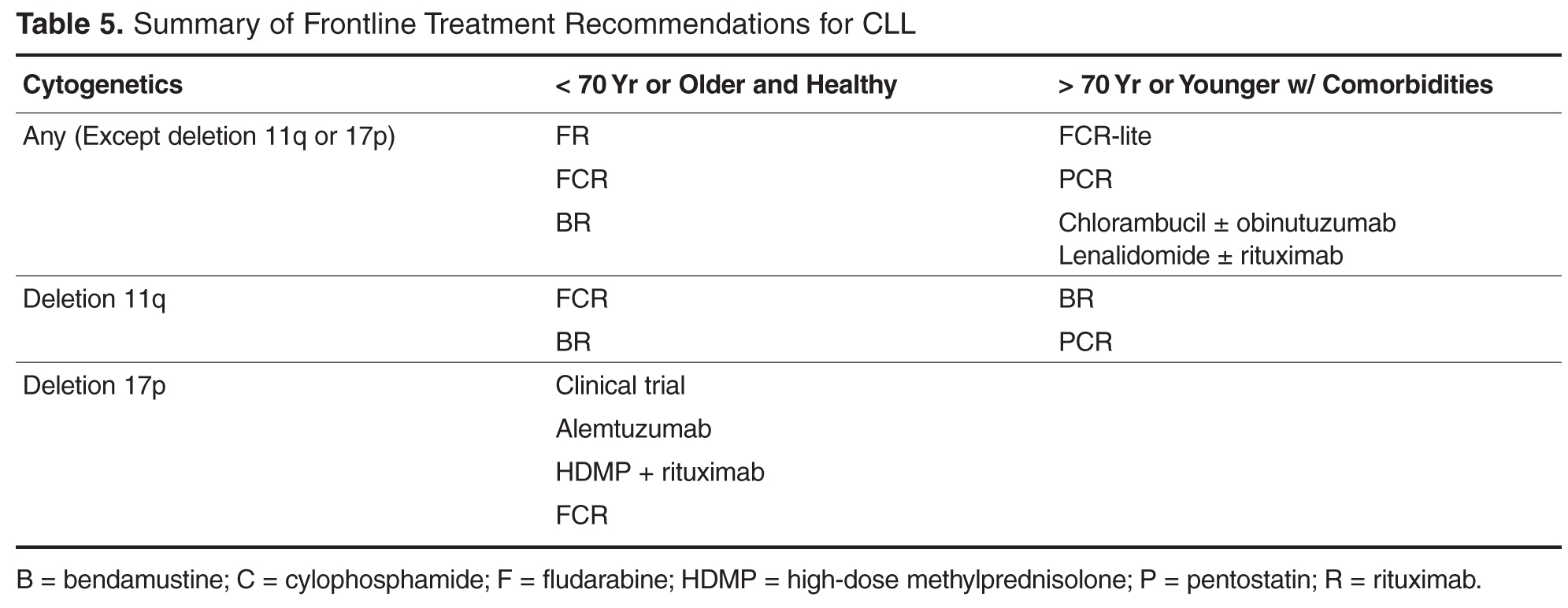
For younger patients who are in good general health, the standard treatment choice is combination chemoimmunotherapy. While single agent therapies can effectively palliate symptoms in most cases, they do not offer a survival benefit. Treatment with chemoimmunotherapy, consisting of cytotoxic chemotherapy given in combination with an anti-CD20 monoclonal antibody (generally rituximab), results in high response rates and conveys an advantage with respect to both progression-free survival (PFS) and overall survival (OS). Several chemoimmunotherapy regimens are commonly used.
As compared to fludarabine alone, frontline therapy with the combination of rituximab and fludarabine (FR) results in both a higher overall response rate (84% compared with 63% with fludarabine alone) and more complete responses (38% compared with 20% with fludarabine alone). The probability of PFS at 2 years is also better with FR: 67% compared to 45% with single agent fludarabine [28,29]. Neutropenia is more common with the combination regimen but does not appear to increase the rate of infection. Rituximab infusion reactions are commonly observed, so a stepped-up dosing schedule was developed to decrease their incidence and severity.
Fludarabine, cyclophosphamide, and rituximab (FCR) is another highly effective regimen. This combination has similar efficacy to FR with a 90% to 95% overall response rate (ORR) and 44% to 70% complete response (CR) rate [19,30]. Long-term results with this regimen are favorable; 6-year OS of 77% and median time to progression of 80 months have been reported in a follow-up study [31]. However, hematologic toxicity, including severe neutropenia, is common, and many patients are unable to complete all planned therapy [19]. The addition of cyclophosphamide does appear to be especially important for patients with a deletion(11q). Several clinical trials have consistently found that measures of response and survival are improved for deletion(11q) patients receiving an alkylating agent in addition to a nucleoside analogue [18,32,33]. Outcomes in patients with deletion(17p) disease remain poor after FCR; this subset demonstrates the shortest PFS at only 11.5 months [19].
A more recently developed chemoimmunotherapy option for younger, fit patients is bendamustine and rituximab (BR). Bendamustine has structural similarities to both alkylating agents and purine analogues, and is significantly more efficacious than chlorambucil as a single agent [34]. The combination is generally well tolerated, and a phase 2 trial of the combination reported an overall response rate (ORR) of 88.0% [32]. Notably, when the results were examined by genetic risk group, the regimen remained effective for deletion(11q) patients, who achieved overall and CR rates of 90% and 40%, respectively. Unfortunately, only 37.5% of deletion(17p) patients responded, and no patients achieved a CR [32].
The risk for therapy-related neoplasms should be taken into account when selecting initial therapy given the expected long-term survival of most CLL patients. About 8 out of 300 FCR-treated patients developed a therapy-related neoplasm in one study [31]. Treatment with FR, which does not include an alkylating agent, does not appear to have the same risk. In a study reporting long-term follow-up on 104 patients treated with FR, none developed a therapy related neoplasm [35]. Risks associated with bendamustine have not been well characterized but appear to be lower than FC. While inclusion of an alkylating agent is important for deletion(11q) patients, it is not clear if other patients similarly benefit, thus meriting the potentially increased risk for second cancers.
Fortunately, the choice among these similarly effective regimens will soon be based on high-quality, comparative data. FCR and BR have now been directly compared as a first-line treatment in the German CLL Study Group CLL10 trial. At interim analysis, both regimens had the same ORR and 2-year OS. However, CRs were less common in the BR group (38.1% versus 47.4% with FCR) and PFS was likewise inferior. Expectedly, the FCR group experienced more myelotoxicity and infections. The rate of severe neutropenia with FCR was higher at 81.7% compared to only 56.8% with BR [36]. This may be an important consideration when selecting a regimen for individual patients. Baseline renal function may influence choice as well. The active metabolite of fludarabine is eliminated through the kidneys and patients with decreased renal function have been excluded from clinical trials of FCR [19,37]. The phase 2 study of BR included patients with impaired renal function and 35% of participants had a creatinine clearance of less than 70 mL/min. It is notable that increased toxicity was seen in this subset, including higher rates of myelosuppression and infection [32]. As few direct comparisons have been done, the choice between effective first-line chemoimmunotherapy regimens can be difficult. The final results of the CLL 10 trial, as well as the now completed CALGB 10404 trial comparing FCR to FR, will provide new evidence regarding the relative risks and benefits of these regimens, particularly for patients without high-risk chromosomal abnormalities.
• What treatments are most effective for patients with deletion(17p) CLL?
As noted above, deletion(17p) CLL responds poorly to standard treatments. This relative lack of durable response to chemoimmunotherapy appears attributable to loss of function of the tumor suppressor protein TP53 which is encoded in the affected area [20,32,38]. In vivo evidence suggests that fludarabine works through a TP53-dependent mechanism, which likely explains the poor results obtained when deletion(17p) patients are treated with fludarabine-based combinations [38]. Patients harboring deletion(17p) or TP53 mutations should thus be referred for participation in clinical trials or allogeneic stem cell transplantation [17,27].
If initial treatment of a patient with deletion(17p) begins outside of a clinical trial, it should ideally be comprised of agents that have a TP53-independent mechanism of action [20]. Alemtuzumab, a humanized monoclonal antibody against the CD52 antigen expressed on the surface of normal and malignant B- and T-lymphocytes, demonstrated ORR of 33% to 50% in studies of patients with relapsed and refractory CLL [39–42]. A retrospective analysis found that similar outcomes were seen in those who had a TP53 mutation or deletion(17p). A subsequent study of previously untreated CLL patients randomized to treatment with 12 weeks of alemtuzumab or chlorambucil found that alemtuzumab-treated deletion(17p) patients had an ORR of 64% and median PFS of 10.7 months [43]. Alemtuzumab is therefore a rational choice for first-line therapy in this population. Hematologic toxicity is frequent, however, and all patients must receive prophylaxis against and monitoring for reactivation of CMV infection [43]. Infusion reactions are common but may be reduced by subcutaneous administration without apparent loss of efficacy [42,44]. While alemtuzumab is no longer marketed in the United States for the indication of CLL, it is available free of charge from the manufacturer [45].
High-dose methylprednisolone with rituximab (HDMP-R) has also been successfully used as both salvage and first-line therapy in this group. As salvage therapy, responses were seen in greater than 90% of patients, including over 50% of deletion(17p) patients [46-48]. In treatment-naïve CLL, the ORR was 96% [49], although data for patients with deletion 17p is limited in the frontline setting. Myelotoxicity attributable to the regimen is modest, but good antimicrobial prophylaxis is warranted, as well as close monitoring for hyperglycemia in at-risk patients.
• How is treatment modified for older or less fit patients?
For patients older than 70, or those who have significant comorbidities, effective therapies are still available. As most new diagnoses of CLL are made in patients older than 65, age is but one important factor determining an individual patient’s ability to tolerate treatment. The German CLL Study Group has usefully classified elderly patients into 3 treatment groups based on fitness and goals of care. The first group of medically fit patients with a normal life expectancy, sometimes referred to as the “go go” group, generally tolerate standard chemoimmunotherapy. A second group of older patients with significant life-limiting comorbid conditions—the so-called “no go” patients —should be offered best supportive care rather than CLL-directed treatment. A third group of “slow go” patients falls in between these two; these patients have comorbidities with variable life expectancy and will likely tolerate and benefit from CLL-directed therapy [50].
While some older patients can safely receive chemoimmunotherapy at standard doses and schedules, FCR can prove intolerable for even the medically fit elderly. Because inferior outcomes have been reported among patients older than 70 [30,31], a reduced-dose FCR regimen (FCR-lite) has been studied. Doses of fludarabine and cyclophosphamide were reduced by 20% and 40% respectively and dosing frequency of rituximab was increased. The CR rate was favorable at 77%, the rate of severe neutropenia was reduced to only 13%, and most patients completed all planned therapy [51]. Alternatively, the combination of pentostatin, cyclophosphamide, and rituximab (PCR) has also been successfully used in older patients. The overall and CR rates, 91% and 63% respectively, were durable at 26 months of follow-up. Importantly, there was no statistically significant difference in response or toxicity among the 28% of patients older than 70 [52,53].
For less fit patients, chlorambucil remains a reasonable option. Chlorambucil, a well-tolerated oral alkylating agent, has been used as a frontline therapy in CLL for decades. Chlorambucil has demonstrated consistent response rates in at least 4 clinical trials and is an appropriate option for patients who cannot tolerate more intensive therapy [54]. When a multicenter phase III trial compared it directly to fludarabine in patients over 65, the PFS and OS were no different despite favorable response rates in fludarabine-treated patients [55]. The effectiveness of single-agent chlorambucil can be improved, and the tolerability maintained, with the addition of a CD20-directed monoclonal antibody [56]. Obinutuzumab, a glycolengineered type II antibody against CD20, has recently been shown to improve treatment efficacy when used in combination with chlorambucil [57]. The CLL11 trial randomized patients with comorbid conditions to 1 of 3 treatments: single-agent chlorambucil, chlorambucil with rituximab (R-Clb), or chlorambucil with obinutuzumab (G-Clb). Both chemoimmunotherapy combinations outperformed chlorambucil alone, but the inclusion of obinutuzumab was associated with higher CR rates and longer PFS than rituximab, although infusion reactions and neutropenia were more common in the obinutuzumab arm [57]. Based on this result, the US Food and Drug Administration has now approved obinutuzumab for use in combination with chlorambucil as frontline therapy. While regulatory approval is without restriction with respect to patient age or fitness, a chlorambucil backbone remains most appropriate for older patients and/or those with significant comorbidities.
• What therapies are currently under development?
Numerous targeted treatments and novel immunotherapies are under active investigation in CLL. With greater specificity for CLL, these emerging agents offer the possibility of more effective yet less toxic treatments that will undoubtedly change the landscape for future CLL therapy. These agents are currently most studied as salvage therapies, and given their targeted mechanism of action can be highly effective in relapsed and refractory patients who frequently harbor poor risk cytogenetic abnormalities such as deletion(17p). Data for these agents as initial treatment is limited. Ongoing clinical trials employing these newer agents will need to be reported before these drugs can be recommended as frontline therapies.
Frontline experience with the oral immunomodulatory agent lenalidomide is more extensive. Lenalidomide offers convenient daily dosing and a favorable toxicity profile. When given on a continuous dosing schedule to patients who were 65 years old or older, the ORR was 65%, and 88% of patients were still alive at 2 years’ follow-up. The quality of response continued to improve beyond 18 months of treatment. Neutropenia, the most common severe toxicity, complicated about a third of cycles. Tumor flare attributable to immune activation was also seen, but in most cases was low-grade and did not require intervention [58,59]. While life-threatening tumor lysis syndrome and tumor flare have been seen with lenalidomide in CLL, such concerns are largely abrogated by a lower starting dose and careful intrapatient dose titration [60]. Lenalidomide has also been combined with rituximab and yielded promising results. Sixty-nine treatment-naïve patients were treated with escalating doses of lenalidomide along with rituximab infusions starting at the end of cycle 1 in a phase 2 study. They achieved an 88% ORR with 16% CRs. Toxicities were generally manageable, but patients over 65 were less likely to reach higher doses of lenalidomide or complete all planned treatment cycles [61]. Unfortunately, the FDA recently halted accrual to a phase 3 frontline clinical trial comparing lenalidomide to chlorambucil due to excess mortality in the lenalidomide arm among patients over the age of 80 [62]. More detailed outcomes from that study should be forthcoming.
Perhaps the most remarkable recent advance in CLL medicine, however, is the advent of orally bioavailable small molecule inhibitors of the B-cell receptor (BCR) signaling pathway. BCR signaling plays a vitally important role in supporting the growth and survival of malignant B-cells, activating a number of downstream kinases (Syk, Btk, PI3K, among others) which are potential therapeutic targets. Proof of principle for this approach was demonstrated with the Syk inhibitor fostamatinib in a phase 1/2 trial enrolling patients with B-cell non-Hodgkin lymphoma and CLL. CLL/SLL patients had the highest response rates of any subgroup in that study, with 6 out of 11 patients responding [63]. In a subsequent phase 1b study of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib, durable partial remissions were reported in more than 70% of multiply relapsed and refractory patients, including genetically high-risk patients [64–66]. Ibrutinib appears safer and better tolerated than traditional chemoimmunotherapy in the relapsed setting; consequently, it is now being studied as a first-line therapy both alone and in combination with other agents [67]. Other BCR signaling agents under study, such as the phosphatidylinositol 3-kinase inhibitor idelalisib, demonstrate similar safety and high response rates across both genetic risk and patient age groups [68].
New targeted drugs are not limited to the BCR signaling pathway. ABT-199 inhibits B-cell leukemia/lymphoma 2 (BCL-2), which is an anti-apoptotic protein in the cell death pathway, and has demonstrated remarkable clinical efficacy in relapsed and refractory CLL patients [69]. As more experience is gained with these targeted agents, it is expected that they will be rapidly incorporated into frontline therapies. However, these agents are just now being studied in comparison to standard initial treatments, such as FCR, and it is not yet clear they will offer an advantage over current chemoimmunotherapy in this setting [70–72]. Since these single agents typically do not induce complete remissions, and require indefinite therapy to maintain response, optimal combination therapies are under intensive investigation.
Case Conclusion
The patient and his physician elect to begin treatment owing to symptomatic cervical lymphadenopathy and massive splenomegaly. Given the presence of a deletion(11q) abnormality, but hoping to limit the risk for both short- and long-term toxicities, this younger, fit patient is treated with 6 cycles of bendamustine and rituximab. At the conclusion of treatment, neither the cervical lymph nodes nor spleen remain palpable. His blood counts have also normalized, with a white blood cell count of 4700 with 8.1% lymphocyotes, hemoglobin of 14.3 gm/dL, and platelets of 151,000/dL.
Summary
CLL follows a chronic course requiring treatment at variable intervals. Both genetic risk features and patient factors should be considered when determining initial therapy. Cytogenetic and molecular testing can characterize the likelihood of treatment success, information useful for treatment planning. Chemoimmunotherapy is highly effective for most patients, including patients with deletion(11q) CLL, where the inclusion of an alkylating agent in frontline therapy alters the natural history of disease. However, patients with deletion(17p) and or TP53-mutated disease respond poorly to standard treatment and should be considered for investigational therapies [73]. Novel approaches to CLL therapy, most notably immunotherapies and BCR-targeted agents, hold the promise to further improve outcomes, particularly for the highest risk patients and those elderly and/or infirm patients who tolerate chemotherapy poorly. Frontline therapy should rapidly evolve as emerging agents enter advanced phase investigation.
Corresponding author: Jeffrey Jones, MD, MPH, Div. of Hematology, Ohio State University, A350B Starling Loving Hall, 320 West 10th Ave., Columbus, OH 43210, [email protected].
Financial disclosures: Dr. Jones disclosed that he is on the advisory boards and has received research support from Genentech, Pharmacyclics, and Gilead.
Author contributions: conception and design, KAR, JAJ; analysis and interpretation of data, KAR, JAJ; drafting of article, KAR, JAJ; critical revision of the article, KAR, JAJ.
1. Redaelli A, Laskin BL, Stephens JM, et al. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur J Cancer Care (Engl) 2004;13:279–87.
2. Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007;139:809–19.
3. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6.
4. Zent CS, Kyasa MJ, Evans R, Schichman SA. Chronic lymphocytic leukemia incidence is substantially higher than estimated from tumor registry data. Cancer 2001;92:1325–30.
5. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006;24:437–43.
6. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56.
7. Rawstron AC, Bennett FL, O'Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83.
8. Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–34.
9. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
10. Binet JL, Lepoprier M, Dighiero G, et al. A clinical staging system for chronic lymphocytic leukemia: prognostic significance. Cancer 1977:40:855–64.
11. Eichhorst BF, Fischer K, Fink AM, et al. Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 2011;117:1817–21.
12. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54.
13. Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:
1764–75.
14. Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–9.
15. Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006;70:209–13.
16. Wierda WG, O'Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–95.
17. Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 2008;26:5094–100.
18. Ding W, Ferrajoli A. Evidence-based mini-review: the role of alkylating agents in the initial treatment of chronic lymphocytic leukemia patients with the 11q deletion. Hematology Am Soc Hematol Educ Program 2010;2010:90–2.
19. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
20. Badoux XC, Keating MJ, Wierda WG.What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011;6:36–46.
21. Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–14.
22. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 1999;91:861–8.
23. Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 2010;28:4935–44.
24. Shanafelt TD, Bowen D, Venkat C, et al. Quality of life in chronic lymphocytic leukemia: an international survey of 1482 patients. Br J Haematol 2007;139:255–64.
25. Baer MR, Stein RS, Dessypris EN. Chronic lymphocytic leukemia with hyperleukocytosis. The hyperviscosity syndrome. Cancer 1985;56:2865–9.
26. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 2002;16:2092–5.
27. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: non-hodgkin's lymphomas. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
28. Byrd JC, Rai K, Peterson BL, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49–53.
29. Byrd JC, Peterson BL, Morrison VA, et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003;101:6–14.
30. Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079–88.
31. Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008;112:975–80.
32. Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16.
33. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9.
34. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–84.
35. Woyach JA, Ruppert AS, Heerema NA, et al. Chemoimmunotherapy with fludarabine and rituximab produces extended overall survival and progression-free survival in chronic lymphocytic leukemia: long-term follow-up of CALGB study 9712. J Clin Oncol 2011;29:1349–55.
36. Fink AM, et al., Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximabversus bendamustine and rituximabin previously untreated and physically fit patientswith advanced chronic lymphocytic leukemia: results of a planned interim analysis of the CLL10 Trial, an international, randomized study of the German CLL Study Group (GCLLSG). Blood 2013;122:526.
37. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet 2002;41:93–103.
38. Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood 2004;104:1428–34.
39. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61.
40. Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81.
41. Osuji NC, Del Giudice I, Matutes E, et al, The efficacy of alemtuzumab for refractory chronic lymphocytic leukemia in relation to cytogenetic abnormalities of p53. Haematologica 2005;90:1435–6.
42. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
43. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–23.
44. Lundin J, Kimby E, Björkholm M, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002;100:768–73.
45. Genzyme. US Campath Distribution Program. Cambridge, MA: Genzyme. Available at http://www.campath.com/.
46. Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65.
47. Bowen DA, Call TG, Jenkins GD, et al. Methylprednisolone-rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007;48:2412–7.
48. Castro JE, Sandoval-Sus JD, Bole J, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of fludarabine refractory high-risk chronic lymphocytic leukemia. Leukemia 2008;22:2048–53.
49. Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89.
50. Eichhorst B, Goede V, Hallek M. Treatment of elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009;50:171–8.
51. Foon KA, Boyiadzis M, Land SR, et al. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498–503.
52. Kay NE, Geyer SM, Call TG, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 2007;109:405–11.
53. Shanafelt TD, Lin T, Geyer SM, et al. Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer 2007;109:2291–8.
54. Catovsky D, Else M, Richards S. Chlorambucil--still not bad: a reappraisal. Clin Lymphoma Myeloma Leuk 2011;11 Suppl 1:S2–6.
55. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009;114:3382–91.
56. Laurenti L, Vannata B, Innocenti I, et al. Chlorambucil plus rituximab as front-line therapy in elderly/unfit patients affected by b-cell chronic lymphocytic leukemia: results of a single-centre experience. Mediterr J Hematol Infect Dis 2013;5:e2013031.
57. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014 Jan 8. [Epub ahead of print].
58. Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood 2011;118:3489–98.
59. Strati P, Keating MJ, Wierda WG, et al. Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 2013;122:734–7.
60. Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol 2007;25:5047.
61. James DF, Brown JR, Werner L, et al. Lenalidomide and rituximab for the initial treatment of patients with chronic lymphocytic leukemia (CLL): a multicenter study of the CLL Research Consortium. ASH Annual Meeting Abstracts 2011;118:291.
62. US Food and Drug Administration. FDA halts clinical trial of drug Revlimid (lenalidomide) for chronic lymphocytic leukemia due to safety concerns. Available at http://www.fda.gov/Drugs/DrugSafety/ucm361444.htm.
63. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010;115:2578–85.
64. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
65. Farooqui M, Aue G, Valdez J, et al. Single agent ibrutinib (PCI-32765) achieves equally good and durable responses in chronic lymphocytic leukemia (CLL) patients with and without deletion 17p. Blood 2013;122:673.
66. Byrd JC, Furman RR, Coutre S, et al. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) monotherapy demonstrates long-term safety and durability of response in chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) patients in an open-label extension study. Blood 2013;122:4163.
67. Brown JR, Barrientos JC, Barr PM, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study. ASH Annual Meeting Abstracts 2013.
68. O'Brien SM, Lamanna N, Kipps TJ, et al. A phase II study of the selective phosphatidylinositol 3-kinase delta (PI3K{delta}) inhibitor idelalisib (GS-1101) in combination with rituximab (R) in treatment-naive patients (pts) ≥ 65 years with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). J Clin Oncol 2013;31(15 Suppl); Abstract 7005.
69. Seymour JF, Davids MS, Pagel JM, et al. Bcl-2 Inhibitor ABT-199 (GDC-0199) monotherapy shows anti-tumor activity including complete remissions in high-risk relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Blood 2013;122:872.
70. Rituxumab and bendamustine hydrochloride, rituxumab and ibrutunib, or ibrutinib alone in treating older patients with previously untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT01886872?term=ibrutinib+cll&rank=8.
71. A multicenter, open-label, phase 3 study of the bruton's tyrosine kinase inhibitor pci-32765 versus chlorambucil in patients 65 years of older with treatment-naive chronic lymphocytic leukemia or small lymphocytic lymphoma (RESONATE-2) Available at http://clinicaltrials.gov/ct2/show/NCT01722487?term=ibrutinib+cll&rank=12.
72. Ibrutinib and rituximab compared with fludarabine phosphate, cyclophosphamide, and rituxumab in treating patients with untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT02048813?term=ibrutinib+cll&rank=2.
73. Strati P, Keating MJ, O'Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia (CLL) with 17p deletion. J Clin Oncol 2013;31(15 suppl): Abstract 7102.
From the Division of Hematology, Ohio State University, Columbus, OH.
Abstract
- Objective: To describe the diagnosis and initial management of chronic lymphocytic leukemia (CLL), including first-line treatment options.
- Methods: Case presentation and review of the literature.
- Results: Most CLL patients demonstrate a chronic, relapsing and remitting course with intervals of months to years between treatments. Recent advances in genetic and molecular markers for risk stratification of CLL significantly impact how clinicians determine prognosis and predict response to treatment for patients with newly diagnosed disease. This information, along with patient factors such as age and health status, should be considered when formulating an initial treatment strategy. Combinations of chemotherapy and immunotherapy offer the longest progression-free survival and overall survival benefit yet reported. For elderly patients or those with significant comorbidities who may not tolerate standard chemoimmunotherapy, less intensive but still effective therapies now exist. Patients with the highest risk disease, such as those with deletions of chromosome 17p, respond poorly to conventional treatment and should be referred to experienced centers where investigational therapies and allogeneic stem cell transplantation are available.
- Conclusion: Both disease characteristics and patient factors should guide the selection among the various effective therapies for CLL. While chemoimmunotherapy is the most effective treatment developed to date, its use may become less prevalent as newer agents are incorporated into initial and relapse treatment algorithms.
Chronic lymphocytic leukemia (CLL) is a chronic malignancy of B-lymphocytes demonstrating a heterogeneous clinical course ranging from indolent to more rapidly progressive. The chief clinical feature is an elevated peripheral blood lymphocyte count, and patients can demonstrate lymphadenopathy, splenomegaly, hepatomegaly, constitutional symptoms, and in late stages bone marrow failure. It is the most common leukemia among adults in the Western world, accounting for between 22% to 30% of new leukemia diagnoses worldwide [1]. Recent incidence rates in the United States are 3.83 cases per 100,000 person-years [2]. The incidence of CLL increases with age, and most new cases are diagnosed in persons 65 years of age or older [1,2]. As reported 5-year survival rates are between 68% and 81% with a median survival of 10 years in some series, the prevalence is significantly higher than the incidence [3]. However, this may even be an underestimate of the population burden of disease, as many cases are not reported to tumor registries [4].
Many patients with CLL are asymptomatic and do not require treatment until years after diagnosis. In these cases a watch and wait approach is taken. The typical natural history of CLL is characterized by periods of effective treatment when required, followed by treatment-free intervals of several years in many cases. However, this can be misleading, as the clinical course for any individual patient is highly variable. Development of cytogenetic and molecular testing has allowed for identification of patients with a higher risk of progression and lower response rates to traditional cytotoxic treatments [5]. For example, depending on chromosomal abnormalities present, median survival can vary from 32 to 133 months [3].
The assessment of underlying disease risk thus provides important information when considering a treatment approach and should be routinely performed for newly diagnosed patients. While the development of highly effective chemoimmunotherapy has allowed most groups of CLL patients to live for many years, some groups do not enjoy the same survival. Recent advances in CLL treatment seek to abrogate such adverse risk factors, thereby improving the survival for all patients with CLL. Given the expected survival of years for most CLL patients, frontline treatment planning must be done in the context of a long-term treatment strategy keeping the risk for late toxicities, such as secondary malignancies, in mind.
Case Study
Initial Presentation
A 50-year-old man is referred for evaluation of cervical lymphadenopathy that had progressed over the prior 6 months. He denies associated symptoms of fatigue, fevers, night sweats, or unintentional weight loss but does report early satiety. On examination there are multiple mobile, enlarged cervical lymph nodes bilaterally. Axillary lymph nodes are likewise enlarged. The liver edge is not palpable, but the spleen is palpable below the belt line. Complete blood count reveals a white blood cell count of 196,000 with 97% lymphocytes. Hemoglobin is 11.0 g/dL and platelet count is 122,000/dL. He recalls being told 3 years previously that his white blood cell count was 48,000 during an emergency department visit for cellulitis.
• How is CLL diagnosed and staged?
CLL is often suspected when patients present with an elevated lymphocyte count. Presenting symptoms of CLL commonly include lymphadenopathy, an enlarged spleen, and constitutional or “B” symptoms such as fatigue, unintentional weight loss, or drenching night sweats. However, only 25% of patients are symptomatic at diagnosis [1]. Many patients with CLL are now diagnosed after a routine blood test, long before the disease is clinically apparent.
The diagnosis of CLL can be made from the peripheral blood and does not require a bone marrow biopsy. According to 2008 guidelines from the International Workshop on Chronic Lymphocytic Leukemia (IWCLL), diagnosis requires at least 5000/uL clonal B-lymphocytes in the peripheral blood. The clonality must be confirmed by immunophenotyping. At time of diagnosis the peripheral blood smear should be examined for the characteristic cells: small mature lymphocytes with a narrow rim of cytoplasm and dense nuclei consisting of clumped chromatin. Larger, atypical cells can be present as long as they do not exceed 55% of the total number of lymphocytes [6].
The immunophenotype of CLL includes aberrant expression of CD5 and a T-cell antigen, along with the characteristic B-cell antigens CD19, CD20, and CD23. The leukemic clone may be either kappa or lambda light chain restricted. Expression of surface immunoglobulin, CD20, and CD79a is typically low compared to that of normal B cells, although there can be some variability in the immunophenotype [6].
![]()
Care should be taken to exclude other malignancies with a similar morphology. Leukemic phase mantle cell lymphoma, other low grade lymphomas, and hairy cell leukemia are commonly mistaken for CLL. Immunophenotyping and cytogenetics are usually sufficient to differentiate these. Testing for a balanced translocation involving chromosomes 11 and 14 to exclude mantle cell lymphoma can be helpful, as both CLL and mantle cell lymphoma can appear morphologically similar and share immunophenotypic features (CD5+/CD19+).
Case Continued
The patient’s peripheral blood is drawn for routine immunophenotyping as well as cytogenetic and molecular testing. When he returns to discuss the results 10 days later, he learns that peripheral blood immunophenotyping demonstrates a dim kappa restricted monoclonal population of B-cells that expressed CD19, CD20(dim), CD23, CD38, CD5, and CD43. The lymphocytes are negative for CD10, FMC7, and CD79b, consistent with a CLL immunophenotype. This patient fulfills diagnostic criteria for CLL and has Rai stage II or intermediate-risk disease. Interphase cytogenetic studies of the peripheral blood demonstrate deletions of chromosomes 11q22.3 and 13q14.3. The immunoglobulin heavy chain gene (IGHV) is unmutated.
• How can a CLL patient’s disease risk be characterized?
Historically, staging at diagnosis, pattern of bone marrow infiltration, and response to therapy were used to gauge prognosis. In more recent years, cytogenetic and molecular testing methods have been developed to augment risk stratification. Testing of prognostic significance that influences clinical management includes IGHV mutational status and interphase cytogenetics using FISH [3,12–14]. Expression of ZAP-70 and CD38 are both independent predictors of poorer prognosis in CLL but are not recommended for routine clinical use. Standardized methodology for the measurement of Zap-70 in particular limits the utility of that test in routine clinical practice [15]. Performed at diagnosis, a time when many patients are asymptomatic, cytogenetic testing with FISH and IGHV mutational analysis can predict time to first treatment and increasingly identify high-risk patients for whom investigational early intervention approaches may be considered [16]. While cytogenetic testing has utility at time of diagnosis, it should be considered necessary prior to deciding on the first-line treatment.
Cytogenetics are also important in predicting response to therapy. For instance, patients with deletion(11q) disease have improved survival when treated with regimens containing an alkylating agent [18]. Deletion(17p) patients respond poorly to traditional cytotoxic agents, and treatments with alternate mechanisms of action should be used [5,19]. The gene for tumor suppressor protein TP53 is encoded in this region of chromosome 17, thus treatment with agents that act independent of pathways involving TP53 are preferred [20].
In addition to cytogenetic testing, quantization of somatic mutations in the gene encoding the variable region of the immune globulin heavy chain gene (IGHV) can help define disease-specific risk. When greater than 98% sequence homology is seen, the gene is considered IGHV unmutated. Patients with an unmutated IGHV have worse overall survival. In one study of Rai stage 0 CLL patients, those with an unmutated IGHV had a survival of only 95 months, compared with 293 months in the mutated group [12].
• When should CLL be treated?
CLL is not curable with current standard therapies, and starting treatment at time of diagnosis for early stage, asymptomatic, CLL patients does not improve overall survival and adds treatment-related toxicities [21,22]. Consequently, the decision to treat is based on treating or preventing complications from the disease, and observation is recommended for most asymptomatic, early-stage patients [6]. Because median survival in CLL is often measured in years, deferring treatment can limit both the short- and long-term complications of therapy, especially the significant risk of secondary malignancies associated with some therapies [23]. However, deferring treatment can significantly impact both a patient’s emotional well-being and quality of life, which should be kept in mind when first discussing the rationale for observation with asymptomatic patients [24].
causes.
For patients with anemia, neutropenia, or thrombocytopenia that is autoimmune in nature, treatment should typically begin with corticosteroids, as it would for non-CLL associated cases of autoimmune cytopenias. If steroids are not effective, second-line treatments appropriate for the situation are generally employed, including intravenous immunoglobulin, cyclosporine, azathioprine, and splenectomy. Rituximab has also been shown to be effective in steroid-refractory cases of autoimmune hemolytic anemia associated with CLL [26]. Only if cytopenias are refractory to appropriate second-line therapy should CLL-directed treatments be considered, assuming there are no other indications to treat the underlying CLL [6]. Bone marrow biopsy can be helpful in differentiating autoimmune cytopenias from marrow failure due to CLL infiltration.
• What treatments are most appropriate for young, fit patients?
For younger patients who are in good general health, the standard treatment choice is combination chemoimmunotherapy. While single agent therapies can effectively palliate symptoms in most cases, they do not offer a survival benefit. Treatment with chemoimmunotherapy, consisting of cytotoxic chemotherapy given in combination with an anti-CD20 monoclonal antibody (generally rituximab), results in high response rates and conveys an advantage with respect to both progression-free survival (PFS) and overall survival (OS). Several chemoimmunotherapy regimens are commonly used.
As compared to fludarabine alone, frontline therapy with the combination of rituximab and fludarabine (FR) results in both a higher overall response rate (84% compared with 63% with fludarabine alone) and more complete responses (38% compared with 20% with fludarabine alone). The probability of PFS at 2 years is also better with FR: 67% compared to 45% with single agent fludarabine [28,29]. Neutropenia is more common with the combination regimen but does not appear to increase the rate of infection. Rituximab infusion reactions are commonly observed, so a stepped-up dosing schedule was developed to decrease their incidence and severity.
Fludarabine, cyclophosphamide, and rituximab (FCR) is another highly effective regimen. This combination has similar efficacy to FR with a 90% to 95% overall response rate (ORR) and 44% to 70% complete response (CR) rate [19,30]. Long-term results with this regimen are favorable; 6-year OS of 77% and median time to progression of 80 months have been reported in a follow-up study [31]. However, hematologic toxicity, including severe neutropenia, is common, and many patients are unable to complete all planned therapy [19]. The addition of cyclophosphamide does appear to be especially important for patients with a deletion(11q). Several clinical trials have consistently found that measures of response and survival are improved for deletion(11q) patients receiving an alkylating agent in addition to a nucleoside analogue [18,32,33]. Outcomes in patients with deletion(17p) disease remain poor after FCR; this subset demonstrates the shortest PFS at only 11.5 months [19].
A more recently developed chemoimmunotherapy option for younger, fit patients is bendamustine and rituximab (BR). Bendamustine has structural similarities to both alkylating agents and purine analogues, and is significantly more efficacious than chlorambucil as a single agent [34]. The combination is generally well tolerated, and a phase 2 trial of the combination reported an overall response rate (ORR) of 88.0% [32]. Notably, when the results were examined by genetic risk group, the regimen remained effective for deletion(11q) patients, who achieved overall and CR rates of 90% and 40%, respectively. Unfortunately, only 37.5% of deletion(17p) patients responded, and no patients achieved a CR [32].
The risk for therapy-related neoplasms should be taken into account when selecting initial therapy given the expected long-term survival of most CLL patients. About 8 out of 300 FCR-treated patients developed a therapy-related neoplasm in one study [31]. Treatment with FR, which does not include an alkylating agent, does not appear to have the same risk. In a study reporting long-term follow-up on 104 patients treated with FR, none developed a therapy related neoplasm [35]. Risks associated with bendamustine have not been well characterized but appear to be lower than FC. While inclusion of an alkylating agent is important for deletion(11q) patients, it is not clear if other patients similarly benefit, thus meriting the potentially increased risk for second cancers.
Fortunately, the choice among these similarly effective regimens will soon be based on high-quality, comparative data. FCR and BR have now been directly compared as a first-line treatment in the German CLL Study Group CLL10 trial. At interim analysis, both regimens had the same ORR and 2-year OS. However, CRs were less common in the BR group (38.1% versus 47.4% with FCR) and PFS was likewise inferior. Expectedly, the FCR group experienced more myelotoxicity and infections. The rate of severe neutropenia with FCR was higher at 81.7% compared to only 56.8% with BR [36]. This may be an important consideration when selecting a regimen for individual patients. Baseline renal function may influence choice as well. The active metabolite of fludarabine is eliminated through the kidneys and patients with decreased renal function have been excluded from clinical trials of FCR [19,37]. The phase 2 study of BR included patients with impaired renal function and 35% of participants had a creatinine clearance of less than 70 mL/min. It is notable that increased toxicity was seen in this subset, including higher rates of myelosuppression and infection [32]. As few direct comparisons have been done, the choice between effective first-line chemoimmunotherapy regimens can be difficult. The final results of the CLL 10 trial, as well as the now completed CALGB 10404 trial comparing FCR to FR, will provide new evidence regarding the relative risks and benefits of these regimens, particularly for patients without high-risk chromosomal abnormalities.
• What treatments are most effective for patients with deletion(17p) CLL?
As noted above, deletion(17p) CLL responds poorly to standard treatments. This relative lack of durable response to chemoimmunotherapy appears attributable to loss of function of the tumor suppressor protein TP53 which is encoded in the affected area [20,32,38]. In vivo evidence suggests that fludarabine works through a TP53-dependent mechanism, which likely explains the poor results obtained when deletion(17p) patients are treated with fludarabine-based combinations [38]. Patients harboring deletion(17p) or TP53 mutations should thus be referred for participation in clinical trials or allogeneic stem cell transplantation [17,27].
If initial treatment of a patient with deletion(17p) begins outside of a clinical trial, it should ideally be comprised of agents that have a TP53-independent mechanism of action [20]. Alemtuzumab, a humanized monoclonal antibody against the CD52 antigen expressed on the surface of normal and malignant B- and T-lymphocytes, demonstrated ORR of 33% to 50% in studies of patients with relapsed and refractory CLL [39–42]. A retrospective analysis found that similar outcomes were seen in those who had a TP53 mutation or deletion(17p). A subsequent study of previously untreated CLL patients randomized to treatment with 12 weeks of alemtuzumab or chlorambucil found that alemtuzumab-treated deletion(17p) patients had an ORR of 64% and median PFS of 10.7 months [43]. Alemtuzumab is therefore a rational choice for first-line therapy in this population. Hematologic toxicity is frequent, however, and all patients must receive prophylaxis against and monitoring for reactivation of CMV infection [43]. Infusion reactions are common but may be reduced by subcutaneous administration without apparent loss of efficacy [42,44]. While alemtuzumab is no longer marketed in the United States for the indication of CLL, it is available free of charge from the manufacturer [45].
High-dose methylprednisolone with rituximab (HDMP-R) has also been successfully used as both salvage and first-line therapy in this group. As salvage therapy, responses were seen in greater than 90% of patients, including over 50% of deletion(17p) patients [46-48]. In treatment-naïve CLL, the ORR was 96% [49], although data for patients with deletion 17p is limited in the frontline setting. Myelotoxicity attributable to the regimen is modest, but good antimicrobial prophylaxis is warranted, as well as close monitoring for hyperglycemia in at-risk patients.
• How is treatment modified for older or less fit patients?
For patients older than 70, or those who have significant comorbidities, effective therapies are still available. As most new diagnoses of CLL are made in patients older than 65, age is but one important factor determining an individual patient’s ability to tolerate treatment. The German CLL Study Group has usefully classified elderly patients into 3 treatment groups based on fitness and goals of care. The first group of medically fit patients with a normal life expectancy, sometimes referred to as the “go go” group, generally tolerate standard chemoimmunotherapy. A second group of older patients with significant life-limiting comorbid conditions—the so-called “no go” patients —should be offered best supportive care rather than CLL-directed treatment. A third group of “slow go” patients falls in between these two; these patients have comorbidities with variable life expectancy and will likely tolerate and benefit from CLL-directed therapy [50].
While some older patients can safely receive chemoimmunotherapy at standard doses and schedules, FCR can prove intolerable for even the medically fit elderly. Because inferior outcomes have been reported among patients older than 70 [30,31], a reduced-dose FCR regimen (FCR-lite) has been studied. Doses of fludarabine and cyclophosphamide were reduced by 20% and 40% respectively and dosing frequency of rituximab was increased. The CR rate was favorable at 77%, the rate of severe neutropenia was reduced to only 13%, and most patients completed all planned therapy [51]. Alternatively, the combination of pentostatin, cyclophosphamide, and rituximab (PCR) has also been successfully used in older patients. The overall and CR rates, 91% and 63% respectively, were durable at 26 months of follow-up. Importantly, there was no statistically significant difference in response or toxicity among the 28% of patients older than 70 [52,53].
For less fit patients, chlorambucil remains a reasonable option. Chlorambucil, a well-tolerated oral alkylating agent, has been used as a frontline therapy in CLL for decades. Chlorambucil has demonstrated consistent response rates in at least 4 clinical trials and is an appropriate option for patients who cannot tolerate more intensive therapy [54]. When a multicenter phase III trial compared it directly to fludarabine in patients over 65, the PFS and OS were no different despite favorable response rates in fludarabine-treated patients [55]. The effectiveness of single-agent chlorambucil can be improved, and the tolerability maintained, with the addition of a CD20-directed monoclonal antibody [56]. Obinutuzumab, a glycolengineered type II antibody against CD20, has recently been shown to improve treatment efficacy when used in combination with chlorambucil [57]. The CLL11 trial randomized patients with comorbid conditions to 1 of 3 treatments: single-agent chlorambucil, chlorambucil with rituximab (R-Clb), or chlorambucil with obinutuzumab (G-Clb). Both chemoimmunotherapy combinations outperformed chlorambucil alone, but the inclusion of obinutuzumab was associated with higher CR rates and longer PFS than rituximab, although infusion reactions and neutropenia were more common in the obinutuzumab arm [57]. Based on this result, the US Food and Drug Administration has now approved obinutuzumab for use in combination with chlorambucil as frontline therapy. While regulatory approval is without restriction with respect to patient age or fitness, a chlorambucil backbone remains most appropriate for older patients and/or those with significant comorbidities.
• What therapies are currently under development?
Numerous targeted treatments and novel immunotherapies are under active investigation in CLL. With greater specificity for CLL, these emerging agents offer the possibility of more effective yet less toxic treatments that will undoubtedly change the landscape for future CLL therapy. These agents are currently most studied as salvage therapies, and given their targeted mechanism of action can be highly effective in relapsed and refractory patients who frequently harbor poor risk cytogenetic abnormalities such as deletion(17p). Data for these agents as initial treatment is limited. Ongoing clinical trials employing these newer agents will need to be reported before these drugs can be recommended as frontline therapies.
Frontline experience with the oral immunomodulatory agent lenalidomide is more extensive. Lenalidomide offers convenient daily dosing and a favorable toxicity profile. When given on a continuous dosing schedule to patients who were 65 years old or older, the ORR was 65%, and 88% of patients were still alive at 2 years’ follow-up. The quality of response continued to improve beyond 18 months of treatment. Neutropenia, the most common severe toxicity, complicated about a third of cycles. Tumor flare attributable to immune activation was also seen, but in most cases was low-grade and did not require intervention [58,59]. While life-threatening tumor lysis syndrome and tumor flare have been seen with lenalidomide in CLL, such concerns are largely abrogated by a lower starting dose and careful intrapatient dose titration [60]. Lenalidomide has also been combined with rituximab and yielded promising results. Sixty-nine treatment-naïve patients were treated with escalating doses of lenalidomide along with rituximab infusions starting at the end of cycle 1 in a phase 2 study. They achieved an 88% ORR with 16% CRs. Toxicities were generally manageable, but patients over 65 were less likely to reach higher doses of lenalidomide or complete all planned treatment cycles [61]. Unfortunately, the FDA recently halted accrual to a phase 3 frontline clinical trial comparing lenalidomide to chlorambucil due to excess mortality in the lenalidomide arm among patients over the age of 80 [62]. More detailed outcomes from that study should be forthcoming.
Perhaps the most remarkable recent advance in CLL medicine, however, is the advent of orally bioavailable small molecule inhibitors of the B-cell receptor (BCR) signaling pathway. BCR signaling plays a vitally important role in supporting the growth and survival of malignant B-cells, activating a number of downstream kinases (Syk, Btk, PI3K, among others) which are potential therapeutic targets. Proof of principle for this approach was demonstrated with the Syk inhibitor fostamatinib in a phase 1/2 trial enrolling patients with B-cell non-Hodgkin lymphoma and CLL. CLL/SLL patients had the highest response rates of any subgroup in that study, with 6 out of 11 patients responding [63]. In a subsequent phase 1b study of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib, durable partial remissions were reported in more than 70% of multiply relapsed and refractory patients, including genetically high-risk patients [64–66]. Ibrutinib appears safer and better tolerated than traditional chemoimmunotherapy in the relapsed setting; consequently, it is now being studied as a first-line therapy both alone and in combination with other agents [67]. Other BCR signaling agents under study, such as the phosphatidylinositol 3-kinase inhibitor idelalisib, demonstrate similar safety and high response rates across both genetic risk and patient age groups [68].
New targeted drugs are not limited to the BCR signaling pathway. ABT-199 inhibits B-cell leukemia/lymphoma 2 (BCL-2), which is an anti-apoptotic protein in the cell death pathway, and has demonstrated remarkable clinical efficacy in relapsed and refractory CLL patients [69]. As more experience is gained with these targeted agents, it is expected that they will be rapidly incorporated into frontline therapies. However, these agents are just now being studied in comparison to standard initial treatments, such as FCR, and it is not yet clear they will offer an advantage over current chemoimmunotherapy in this setting [70–72]. Since these single agents typically do not induce complete remissions, and require indefinite therapy to maintain response, optimal combination therapies are under intensive investigation.
Case Conclusion
The patient and his physician elect to begin treatment owing to symptomatic cervical lymphadenopathy and massive splenomegaly. Given the presence of a deletion(11q) abnormality, but hoping to limit the risk for both short- and long-term toxicities, this younger, fit patient is treated with 6 cycles of bendamustine and rituximab. At the conclusion of treatment, neither the cervical lymph nodes nor spleen remain palpable. His blood counts have also normalized, with a white blood cell count of 4700 with 8.1% lymphocyotes, hemoglobin of 14.3 gm/dL, and platelets of 151,000/dL.
Summary
CLL follows a chronic course requiring treatment at variable intervals. Both genetic risk features and patient factors should be considered when determining initial therapy. Cytogenetic and molecular testing can characterize the likelihood of treatment success, information useful for treatment planning. Chemoimmunotherapy is highly effective for most patients, including patients with deletion(11q) CLL, where the inclusion of an alkylating agent in frontline therapy alters the natural history of disease. However, patients with deletion(17p) and or TP53-mutated disease respond poorly to standard treatment and should be considered for investigational therapies [73]. Novel approaches to CLL therapy, most notably immunotherapies and BCR-targeted agents, hold the promise to further improve outcomes, particularly for the highest risk patients and those elderly and/or infirm patients who tolerate chemotherapy poorly. Frontline therapy should rapidly evolve as emerging agents enter advanced phase investigation.
Corresponding author: Jeffrey Jones, MD, MPH, Div. of Hematology, Ohio State University, A350B Starling Loving Hall, 320 West 10th Ave., Columbus, OH 43210, [email protected].
Financial disclosures: Dr. Jones disclosed that he is on the advisory boards and has received research support from Genentech, Pharmacyclics, and Gilead.
Author contributions: conception and design, KAR, JAJ; analysis and interpretation of data, KAR, JAJ; drafting of article, KAR, JAJ; critical revision of the article, KAR, JAJ.
From the Division of Hematology, Ohio State University, Columbus, OH.
Abstract
- Objective: To describe the diagnosis and initial management of chronic lymphocytic leukemia (CLL), including first-line treatment options.
- Methods: Case presentation and review of the literature.
- Results: Most CLL patients demonstrate a chronic, relapsing and remitting course with intervals of months to years between treatments. Recent advances in genetic and molecular markers for risk stratification of CLL significantly impact how clinicians determine prognosis and predict response to treatment for patients with newly diagnosed disease. This information, along with patient factors such as age and health status, should be considered when formulating an initial treatment strategy. Combinations of chemotherapy and immunotherapy offer the longest progression-free survival and overall survival benefit yet reported. For elderly patients or those with significant comorbidities who may not tolerate standard chemoimmunotherapy, less intensive but still effective therapies now exist. Patients with the highest risk disease, such as those with deletions of chromosome 17p, respond poorly to conventional treatment and should be referred to experienced centers where investigational therapies and allogeneic stem cell transplantation are available.
- Conclusion: Both disease characteristics and patient factors should guide the selection among the various effective therapies for CLL. While chemoimmunotherapy is the most effective treatment developed to date, its use may become less prevalent as newer agents are incorporated into initial and relapse treatment algorithms.
Chronic lymphocytic leukemia (CLL) is a chronic malignancy of B-lymphocytes demonstrating a heterogeneous clinical course ranging from indolent to more rapidly progressive. The chief clinical feature is an elevated peripheral blood lymphocyte count, and patients can demonstrate lymphadenopathy, splenomegaly, hepatomegaly, constitutional symptoms, and in late stages bone marrow failure. It is the most common leukemia among adults in the Western world, accounting for between 22% to 30% of new leukemia diagnoses worldwide [1]. Recent incidence rates in the United States are 3.83 cases per 100,000 person-years [2]. The incidence of CLL increases with age, and most new cases are diagnosed in persons 65 years of age or older [1,2]. As reported 5-year survival rates are between 68% and 81% with a median survival of 10 years in some series, the prevalence is significantly higher than the incidence [3]. However, this may even be an underestimate of the population burden of disease, as many cases are not reported to tumor registries [4].
Many patients with CLL are asymptomatic and do not require treatment until years after diagnosis. In these cases a watch and wait approach is taken. The typical natural history of CLL is characterized by periods of effective treatment when required, followed by treatment-free intervals of several years in many cases. However, this can be misleading, as the clinical course for any individual patient is highly variable. Development of cytogenetic and molecular testing has allowed for identification of patients with a higher risk of progression and lower response rates to traditional cytotoxic treatments [5]. For example, depending on chromosomal abnormalities present, median survival can vary from 32 to 133 months [3].
The assessment of underlying disease risk thus provides important information when considering a treatment approach and should be routinely performed for newly diagnosed patients. While the development of highly effective chemoimmunotherapy has allowed most groups of CLL patients to live for many years, some groups do not enjoy the same survival. Recent advances in CLL treatment seek to abrogate such adverse risk factors, thereby improving the survival for all patients with CLL. Given the expected survival of years for most CLL patients, frontline treatment planning must be done in the context of a long-term treatment strategy keeping the risk for late toxicities, such as secondary malignancies, in mind.
Case Study
Initial Presentation
A 50-year-old man is referred for evaluation of cervical lymphadenopathy that had progressed over the prior 6 months. He denies associated symptoms of fatigue, fevers, night sweats, or unintentional weight loss but does report early satiety. On examination there are multiple mobile, enlarged cervical lymph nodes bilaterally. Axillary lymph nodes are likewise enlarged. The liver edge is not palpable, but the spleen is palpable below the belt line. Complete blood count reveals a white blood cell count of 196,000 with 97% lymphocytes. Hemoglobin is 11.0 g/dL and platelet count is 122,000/dL. He recalls being told 3 years previously that his white blood cell count was 48,000 during an emergency department visit for cellulitis.
• How is CLL diagnosed and staged?
CLL is often suspected when patients present with an elevated lymphocyte count. Presenting symptoms of CLL commonly include lymphadenopathy, an enlarged spleen, and constitutional or “B” symptoms such as fatigue, unintentional weight loss, or drenching night sweats. However, only 25% of patients are symptomatic at diagnosis [1]. Many patients with CLL are now diagnosed after a routine blood test, long before the disease is clinically apparent.
The diagnosis of CLL can be made from the peripheral blood and does not require a bone marrow biopsy. According to 2008 guidelines from the International Workshop on Chronic Lymphocytic Leukemia (IWCLL), diagnosis requires at least 5000/uL clonal B-lymphocytes in the peripheral blood. The clonality must be confirmed by immunophenotyping. At time of diagnosis the peripheral blood smear should be examined for the characteristic cells: small mature lymphocytes with a narrow rim of cytoplasm and dense nuclei consisting of clumped chromatin. Larger, atypical cells can be present as long as they do not exceed 55% of the total number of lymphocytes [6].
The immunophenotype of CLL includes aberrant expression of CD5 and a T-cell antigen, along with the characteristic B-cell antigens CD19, CD20, and CD23. The leukemic clone may be either kappa or lambda light chain restricted. Expression of surface immunoglobulin, CD20, and CD79a is typically low compared to that of normal B cells, although there can be some variability in the immunophenotype [6].
![]()
Care should be taken to exclude other malignancies with a similar morphology. Leukemic phase mantle cell lymphoma, other low grade lymphomas, and hairy cell leukemia are commonly mistaken for CLL. Immunophenotyping and cytogenetics are usually sufficient to differentiate these. Testing for a balanced translocation involving chromosomes 11 and 14 to exclude mantle cell lymphoma can be helpful, as both CLL and mantle cell lymphoma can appear morphologically similar and share immunophenotypic features (CD5+/CD19+).
Case Continued
The patient’s peripheral blood is drawn for routine immunophenotyping as well as cytogenetic and molecular testing. When he returns to discuss the results 10 days later, he learns that peripheral blood immunophenotyping demonstrates a dim kappa restricted monoclonal population of B-cells that expressed CD19, CD20(dim), CD23, CD38, CD5, and CD43. The lymphocytes are negative for CD10, FMC7, and CD79b, consistent with a CLL immunophenotype. This patient fulfills diagnostic criteria for CLL and has Rai stage II or intermediate-risk disease. Interphase cytogenetic studies of the peripheral blood demonstrate deletions of chromosomes 11q22.3 and 13q14.3. The immunoglobulin heavy chain gene (IGHV) is unmutated.
• How can a CLL patient’s disease risk be characterized?
Historically, staging at diagnosis, pattern of bone marrow infiltration, and response to therapy were used to gauge prognosis. In more recent years, cytogenetic and molecular testing methods have been developed to augment risk stratification. Testing of prognostic significance that influences clinical management includes IGHV mutational status and interphase cytogenetics using FISH [3,12–14]. Expression of ZAP-70 and CD38 are both independent predictors of poorer prognosis in CLL but are not recommended for routine clinical use. Standardized methodology for the measurement of Zap-70 in particular limits the utility of that test in routine clinical practice [15]. Performed at diagnosis, a time when many patients are asymptomatic, cytogenetic testing with FISH and IGHV mutational analysis can predict time to first treatment and increasingly identify high-risk patients for whom investigational early intervention approaches may be considered [16]. While cytogenetic testing has utility at time of diagnosis, it should be considered necessary prior to deciding on the first-line treatment.
Cytogenetics are also important in predicting response to therapy. For instance, patients with deletion(11q) disease have improved survival when treated with regimens containing an alkylating agent [18]. Deletion(17p) patients respond poorly to traditional cytotoxic agents, and treatments with alternate mechanisms of action should be used [5,19]. The gene for tumor suppressor protein TP53 is encoded in this region of chromosome 17, thus treatment with agents that act independent of pathways involving TP53 are preferred [20].
In addition to cytogenetic testing, quantization of somatic mutations in the gene encoding the variable region of the immune globulin heavy chain gene (IGHV) can help define disease-specific risk. When greater than 98% sequence homology is seen, the gene is considered IGHV unmutated. Patients with an unmutated IGHV have worse overall survival. In one study of Rai stage 0 CLL patients, those with an unmutated IGHV had a survival of only 95 months, compared with 293 months in the mutated group [12].
• When should CLL be treated?
CLL is not curable with current standard therapies, and starting treatment at time of diagnosis for early stage, asymptomatic, CLL patients does not improve overall survival and adds treatment-related toxicities [21,22]. Consequently, the decision to treat is based on treating or preventing complications from the disease, and observation is recommended for most asymptomatic, early-stage patients [6]. Because median survival in CLL is often measured in years, deferring treatment can limit both the short- and long-term complications of therapy, especially the significant risk of secondary malignancies associated with some therapies [23]. However, deferring treatment can significantly impact both a patient’s emotional well-being and quality of life, which should be kept in mind when first discussing the rationale for observation with asymptomatic patients [24].
causes.
For patients with anemia, neutropenia, or thrombocytopenia that is autoimmune in nature, treatment should typically begin with corticosteroids, as it would for non-CLL associated cases of autoimmune cytopenias. If steroids are not effective, second-line treatments appropriate for the situation are generally employed, including intravenous immunoglobulin, cyclosporine, azathioprine, and splenectomy. Rituximab has also been shown to be effective in steroid-refractory cases of autoimmune hemolytic anemia associated with CLL [26]. Only if cytopenias are refractory to appropriate second-line therapy should CLL-directed treatments be considered, assuming there are no other indications to treat the underlying CLL [6]. Bone marrow biopsy can be helpful in differentiating autoimmune cytopenias from marrow failure due to CLL infiltration.
• What treatments are most appropriate for young, fit patients?
For younger patients who are in good general health, the standard treatment choice is combination chemoimmunotherapy. While single agent therapies can effectively palliate symptoms in most cases, they do not offer a survival benefit. Treatment with chemoimmunotherapy, consisting of cytotoxic chemotherapy given in combination with an anti-CD20 monoclonal antibody (generally rituximab), results in high response rates and conveys an advantage with respect to both progression-free survival (PFS) and overall survival (OS). Several chemoimmunotherapy regimens are commonly used.
As compared to fludarabine alone, frontline therapy with the combination of rituximab and fludarabine (FR) results in both a higher overall response rate (84% compared with 63% with fludarabine alone) and more complete responses (38% compared with 20% with fludarabine alone). The probability of PFS at 2 years is also better with FR: 67% compared to 45% with single agent fludarabine [28,29]. Neutropenia is more common with the combination regimen but does not appear to increase the rate of infection. Rituximab infusion reactions are commonly observed, so a stepped-up dosing schedule was developed to decrease their incidence and severity.
Fludarabine, cyclophosphamide, and rituximab (FCR) is another highly effective regimen. This combination has similar efficacy to FR with a 90% to 95% overall response rate (ORR) and 44% to 70% complete response (CR) rate [19,30]. Long-term results with this regimen are favorable; 6-year OS of 77% and median time to progression of 80 months have been reported in a follow-up study [31]. However, hematologic toxicity, including severe neutropenia, is common, and many patients are unable to complete all planned therapy [19]. The addition of cyclophosphamide does appear to be especially important for patients with a deletion(11q). Several clinical trials have consistently found that measures of response and survival are improved for deletion(11q) patients receiving an alkylating agent in addition to a nucleoside analogue [18,32,33]. Outcomes in patients with deletion(17p) disease remain poor after FCR; this subset demonstrates the shortest PFS at only 11.5 months [19].
A more recently developed chemoimmunotherapy option for younger, fit patients is bendamustine and rituximab (BR). Bendamustine has structural similarities to both alkylating agents and purine analogues, and is significantly more efficacious than chlorambucil as a single agent [34]. The combination is generally well tolerated, and a phase 2 trial of the combination reported an overall response rate (ORR) of 88.0% [32]. Notably, when the results were examined by genetic risk group, the regimen remained effective for deletion(11q) patients, who achieved overall and CR rates of 90% and 40%, respectively. Unfortunately, only 37.5% of deletion(17p) patients responded, and no patients achieved a CR [32].
The risk for therapy-related neoplasms should be taken into account when selecting initial therapy given the expected long-term survival of most CLL patients. About 8 out of 300 FCR-treated patients developed a therapy-related neoplasm in one study [31]. Treatment with FR, which does not include an alkylating agent, does not appear to have the same risk. In a study reporting long-term follow-up on 104 patients treated with FR, none developed a therapy related neoplasm [35]. Risks associated with bendamustine have not been well characterized but appear to be lower than FC. While inclusion of an alkylating agent is important for deletion(11q) patients, it is not clear if other patients similarly benefit, thus meriting the potentially increased risk for second cancers.
Fortunately, the choice among these similarly effective regimens will soon be based on high-quality, comparative data. FCR and BR have now been directly compared as a first-line treatment in the German CLL Study Group CLL10 trial. At interim analysis, both regimens had the same ORR and 2-year OS. However, CRs were less common in the BR group (38.1% versus 47.4% with FCR) and PFS was likewise inferior. Expectedly, the FCR group experienced more myelotoxicity and infections. The rate of severe neutropenia with FCR was higher at 81.7% compared to only 56.8% with BR [36]. This may be an important consideration when selecting a regimen for individual patients. Baseline renal function may influence choice as well. The active metabolite of fludarabine is eliminated through the kidneys and patients with decreased renal function have been excluded from clinical trials of FCR [19,37]. The phase 2 study of BR included patients with impaired renal function and 35% of participants had a creatinine clearance of less than 70 mL/min. It is notable that increased toxicity was seen in this subset, including higher rates of myelosuppression and infection [32]. As few direct comparisons have been done, the choice between effective first-line chemoimmunotherapy regimens can be difficult. The final results of the CLL 10 trial, as well as the now completed CALGB 10404 trial comparing FCR to FR, will provide new evidence regarding the relative risks and benefits of these regimens, particularly for patients without high-risk chromosomal abnormalities.
• What treatments are most effective for patients with deletion(17p) CLL?
As noted above, deletion(17p) CLL responds poorly to standard treatments. This relative lack of durable response to chemoimmunotherapy appears attributable to loss of function of the tumor suppressor protein TP53 which is encoded in the affected area [20,32,38]. In vivo evidence suggests that fludarabine works through a TP53-dependent mechanism, which likely explains the poor results obtained when deletion(17p) patients are treated with fludarabine-based combinations [38]. Patients harboring deletion(17p) or TP53 mutations should thus be referred for participation in clinical trials or allogeneic stem cell transplantation [17,27].
If initial treatment of a patient with deletion(17p) begins outside of a clinical trial, it should ideally be comprised of agents that have a TP53-independent mechanism of action [20]. Alemtuzumab, a humanized monoclonal antibody against the CD52 antigen expressed on the surface of normal and malignant B- and T-lymphocytes, demonstrated ORR of 33% to 50% in studies of patients with relapsed and refractory CLL [39–42]. A retrospective analysis found that similar outcomes were seen in those who had a TP53 mutation or deletion(17p). A subsequent study of previously untreated CLL patients randomized to treatment with 12 weeks of alemtuzumab or chlorambucil found that alemtuzumab-treated deletion(17p) patients had an ORR of 64% and median PFS of 10.7 months [43]. Alemtuzumab is therefore a rational choice for first-line therapy in this population. Hematologic toxicity is frequent, however, and all patients must receive prophylaxis against and monitoring for reactivation of CMV infection [43]. Infusion reactions are common but may be reduced by subcutaneous administration without apparent loss of efficacy [42,44]. While alemtuzumab is no longer marketed in the United States for the indication of CLL, it is available free of charge from the manufacturer [45].
High-dose methylprednisolone with rituximab (HDMP-R) has also been successfully used as both salvage and first-line therapy in this group. As salvage therapy, responses were seen in greater than 90% of patients, including over 50% of deletion(17p) patients [46-48]. In treatment-naïve CLL, the ORR was 96% [49], although data for patients with deletion 17p is limited in the frontline setting. Myelotoxicity attributable to the regimen is modest, but good antimicrobial prophylaxis is warranted, as well as close monitoring for hyperglycemia in at-risk patients.
• How is treatment modified for older or less fit patients?
For patients older than 70, or those who have significant comorbidities, effective therapies are still available. As most new diagnoses of CLL are made in patients older than 65, age is but one important factor determining an individual patient’s ability to tolerate treatment. The German CLL Study Group has usefully classified elderly patients into 3 treatment groups based on fitness and goals of care. The first group of medically fit patients with a normal life expectancy, sometimes referred to as the “go go” group, generally tolerate standard chemoimmunotherapy. A second group of older patients with significant life-limiting comorbid conditions—the so-called “no go” patients —should be offered best supportive care rather than CLL-directed treatment. A third group of “slow go” patients falls in between these two; these patients have comorbidities with variable life expectancy and will likely tolerate and benefit from CLL-directed therapy [50].
While some older patients can safely receive chemoimmunotherapy at standard doses and schedules, FCR can prove intolerable for even the medically fit elderly. Because inferior outcomes have been reported among patients older than 70 [30,31], a reduced-dose FCR regimen (FCR-lite) has been studied. Doses of fludarabine and cyclophosphamide were reduced by 20% and 40% respectively and dosing frequency of rituximab was increased. The CR rate was favorable at 77%, the rate of severe neutropenia was reduced to only 13%, and most patients completed all planned therapy [51]. Alternatively, the combination of pentostatin, cyclophosphamide, and rituximab (PCR) has also been successfully used in older patients. The overall and CR rates, 91% and 63% respectively, were durable at 26 months of follow-up. Importantly, there was no statistically significant difference in response or toxicity among the 28% of patients older than 70 [52,53].
For less fit patients, chlorambucil remains a reasonable option. Chlorambucil, a well-tolerated oral alkylating agent, has been used as a frontline therapy in CLL for decades. Chlorambucil has demonstrated consistent response rates in at least 4 clinical trials and is an appropriate option for patients who cannot tolerate more intensive therapy [54]. When a multicenter phase III trial compared it directly to fludarabine in patients over 65, the PFS and OS were no different despite favorable response rates in fludarabine-treated patients [55]. The effectiveness of single-agent chlorambucil can be improved, and the tolerability maintained, with the addition of a CD20-directed monoclonal antibody [56]. Obinutuzumab, a glycolengineered type II antibody against CD20, has recently been shown to improve treatment efficacy when used in combination with chlorambucil [57]. The CLL11 trial randomized patients with comorbid conditions to 1 of 3 treatments: single-agent chlorambucil, chlorambucil with rituximab (R-Clb), or chlorambucil with obinutuzumab (G-Clb). Both chemoimmunotherapy combinations outperformed chlorambucil alone, but the inclusion of obinutuzumab was associated with higher CR rates and longer PFS than rituximab, although infusion reactions and neutropenia were more common in the obinutuzumab arm [57]. Based on this result, the US Food and Drug Administration has now approved obinutuzumab for use in combination with chlorambucil as frontline therapy. While regulatory approval is without restriction with respect to patient age or fitness, a chlorambucil backbone remains most appropriate for older patients and/or those with significant comorbidities.
• What therapies are currently under development?
Numerous targeted treatments and novel immunotherapies are under active investigation in CLL. With greater specificity for CLL, these emerging agents offer the possibility of more effective yet less toxic treatments that will undoubtedly change the landscape for future CLL therapy. These agents are currently most studied as salvage therapies, and given their targeted mechanism of action can be highly effective in relapsed and refractory patients who frequently harbor poor risk cytogenetic abnormalities such as deletion(17p). Data for these agents as initial treatment is limited. Ongoing clinical trials employing these newer agents will need to be reported before these drugs can be recommended as frontline therapies.
Frontline experience with the oral immunomodulatory agent lenalidomide is more extensive. Lenalidomide offers convenient daily dosing and a favorable toxicity profile. When given on a continuous dosing schedule to patients who were 65 years old or older, the ORR was 65%, and 88% of patients were still alive at 2 years’ follow-up. The quality of response continued to improve beyond 18 months of treatment. Neutropenia, the most common severe toxicity, complicated about a third of cycles. Tumor flare attributable to immune activation was also seen, but in most cases was low-grade and did not require intervention [58,59]. While life-threatening tumor lysis syndrome and tumor flare have been seen with lenalidomide in CLL, such concerns are largely abrogated by a lower starting dose and careful intrapatient dose titration [60]. Lenalidomide has also been combined with rituximab and yielded promising results. Sixty-nine treatment-naïve patients were treated with escalating doses of lenalidomide along with rituximab infusions starting at the end of cycle 1 in a phase 2 study. They achieved an 88% ORR with 16% CRs. Toxicities were generally manageable, but patients over 65 were less likely to reach higher doses of lenalidomide or complete all planned treatment cycles [61]. Unfortunately, the FDA recently halted accrual to a phase 3 frontline clinical trial comparing lenalidomide to chlorambucil due to excess mortality in the lenalidomide arm among patients over the age of 80 [62]. More detailed outcomes from that study should be forthcoming.
Perhaps the most remarkable recent advance in CLL medicine, however, is the advent of orally bioavailable small molecule inhibitors of the B-cell receptor (BCR) signaling pathway. BCR signaling plays a vitally important role in supporting the growth and survival of malignant B-cells, activating a number of downstream kinases (Syk, Btk, PI3K, among others) which are potential therapeutic targets. Proof of principle for this approach was demonstrated with the Syk inhibitor fostamatinib in a phase 1/2 trial enrolling patients with B-cell non-Hodgkin lymphoma and CLL. CLL/SLL patients had the highest response rates of any subgroup in that study, with 6 out of 11 patients responding [63]. In a subsequent phase 1b study of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib, durable partial remissions were reported in more than 70% of multiply relapsed and refractory patients, including genetically high-risk patients [64–66]. Ibrutinib appears safer and better tolerated than traditional chemoimmunotherapy in the relapsed setting; consequently, it is now being studied as a first-line therapy both alone and in combination with other agents [67]. Other BCR signaling agents under study, such as the phosphatidylinositol 3-kinase inhibitor idelalisib, demonstrate similar safety and high response rates across both genetic risk and patient age groups [68].
New targeted drugs are not limited to the BCR signaling pathway. ABT-199 inhibits B-cell leukemia/lymphoma 2 (BCL-2), which is an anti-apoptotic protein in the cell death pathway, and has demonstrated remarkable clinical efficacy in relapsed and refractory CLL patients [69]. As more experience is gained with these targeted agents, it is expected that they will be rapidly incorporated into frontline therapies. However, these agents are just now being studied in comparison to standard initial treatments, such as FCR, and it is not yet clear they will offer an advantage over current chemoimmunotherapy in this setting [70–72]. Since these single agents typically do not induce complete remissions, and require indefinite therapy to maintain response, optimal combination therapies are under intensive investigation.
Case Conclusion
The patient and his physician elect to begin treatment owing to symptomatic cervical lymphadenopathy and massive splenomegaly. Given the presence of a deletion(11q) abnormality, but hoping to limit the risk for both short- and long-term toxicities, this younger, fit patient is treated with 6 cycles of bendamustine and rituximab. At the conclusion of treatment, neither the cervical lymph nodes nor spleen remain palpable. His blood counts have also normalized, with a white blood cell count of 4700 with 8.1% lymphocyotes, hemoglobin of 14.3 gm/dL, and platelets of 151,000/dL.
Summary
CLL follows a chronic course requiring treatment at variable intervals. Both genetic risk features and patient factors should be considered when determining initial therapy. Cytogenetic and molecular testing can characterize the likelihood of treatment success, information useful for treatment planning. Chemoimmunotherapy is highly effective for most patients, including patients with deletion(11q) CLL, where the inclusion of an alkylating agent in frontline therapy alters the natural history of disease. However, patients with deletion(17p) and or TP53-mutated disease respond poorly to standard treatment and should be considered for investigational therapies [73]. Novel approaches to CLL therapy, most notably immunotherapies and BCR-targeted agents, hold the promise to further improve outcomes, particularly for the highest risk patients and those elderly and/or infirm patients who tolerate chemotherapy poorly. Frontline therapy should rapidly evolve as emerging agents enter advanced phase investigation.
Corresponding author: Jeffrey Jones, MD, MPH, Div. of Hematology, Ohio State University, A350B Starling Loving Hall, 320 West 10th Ave., Columbus, OH 43210, [email protected].
Financial disclosures: Dr. Jones disclosed that he is on the advisory boards and has received research support from Genentech, Pharmacyclics, and Gilead.
Author contributions: conception and design, KAR, JAJ; analysis and interpretation of data, KAR, JAJ; drafting of article, KAR, JAJ; critical revision of the article, KAR, JAJ.
1. Redaelli A, Laskin BL, Stephens JM, et al. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur J Cancer Care (Engl) 2004;13:279–87.
2. Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007;139:809–19.
3. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6.
4. Zent CS, Kyasa MJ, Evans R, Schichman SA. Chronic lymphocytic leukemia incidence is substantially higher than estimated from tumor registry data. Cancer 2001;92:1325–30.
5. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006;24:437–43.
6. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56.
7. Rawstron AC, Bennett FL, O'Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83.
8. Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–34.
9. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
10. Binet JL, Lepoprier M, Dighiero G, et al. A clinical staging system for chronic lymphocytic leukemia: prognostic significance. Cancer 1977:40:855–64.
11. Eichhorst BF, Fischer K, Fink AM, et al. Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 2011;117:1817–21.
12. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54.
13. Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:
1764–75.
14. Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–9.
15. Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006;70:209–13.
16. Wierda WG, O'Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–95.
17. Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 2008;26:5094–100.
18. Ding W, Ferrajoli A. Evidence-based mini-review: the role of alkylating agents in the initial treatment of chronic lymphocytic leukemia patients with the 11q deletion. Hematology Am Soc Hematol Educ Program 2010;2010:90–2.
19. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
20. Badoux XC, Keating MJ, Wierda WG.What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011;6:36–46.
21. Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–14.
22. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 1999;91:861–8.
23. Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 2010;28:4935–44.
24. Shanafelt TD, Bowen D, Venkat C, et al. Quality of life in chronic lymphocytic leukemia: an international survey of 1482 patients. Br J Haematol 2007;139:255–64.
25. Baer MR, Stein RS, Dessypris EN. Chronic lymphocytic leukemia with hyperleukocytosis. The hyperviscosity syndrome. Cancer 1985;56:2865–9.
26. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 2002;16:2092–5.
27. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: non-hodgkin's lymphomas. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
28. Byrd JC, Rai K, Peterson BL, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49–53.
29. Byrd JC, Peterson BL, Morrison VA, et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003;101:6–14.
30. Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079–88.
31. Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008;112:975–80.
32. Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16.
33. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9.
34. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–84.
35. Woyach JA, Ruppert AS, Heerema NA, et al. Chemoimmunotherapy with fludarabine and rituximab produces extended overall survival and progression-free survival in chronic lymphocytic leukemia: long-term follow-up of CALGB study 9712. J Clin Oncol 2011;29:1349–55.
36. Fink AM, et al., Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximabversus bendamustine and rituximabin previously untreated and physically fit patientswith advanced chronic lymphocytic leukemia: results of a planned interim analysis of the CLL10 Trial, an international, randomized study of the German CLL Study Group (GCLLSG). Blood 2013;122:526.
37. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet 2002;41:93–103.
38. Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood 2004;104:1428–34.
39. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61.
40. Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81.
41. Osuji NC, Del Giudice I, Matutes E, et al, The efficacy of alemtuzumab for refractory chronic lymphocytic leukemia in relation to cytogenetic abnormalities of p53. Haematologica 2005;90:1435–6.
42. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
43. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–23.
44. Lundin J, Kimby E, Björkholm M, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002;100:768–73.
45. Genzyme. US Campath Distribution Program. Cambridge, MA: Genzyme. Available at http://www.campath.com/.
46. Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65.
47. Bowen DA, Call TG, Jenkins GD, et al. Methylprednisolone-rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007;48:2412–7.
48. Castro JE, Sandoval-Sus JD, Bole J, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of fludarabine refractory high-risk chronic lymphocytic leukemia. Leukemia 2008;22:2048–53.
49. Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89.
50. Eichhorst B, Goede V, Hallek M. Treatment of elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009;50:171–8.
51. Foon KA, Boyiadzis M, Land SR, et al. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498–503.
52. Kay NE, Geyer SM, Call TG, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 2007;109:405–11.
53. Shanafelt TD, Lin T, Geyer SM, et al. Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer 2007;109:2291–8.
54. Catovsky D, Else M, Richards S. Chlorambucil--still not bad: a reappraisal. Clin Lymphoma Myeloma Leuk 2011;11 Suppl 1:S2–6.
55. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009;114:3382–91.
56. Laurenti L, Vannata B, Innocenti I, et al. Chlorambucil plus rituximab as front-line therapy in elderly/unfit patients affected by b-cell chronic lymphocytic leukemia: results of a single-centre experience. Mediterr J Hematol Infect Dis 2013;5:e2013031.
57. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014 Jan 8. [Epub ahead of print].
58. Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood 2011;118:3489–98.
59. Strati P, Keating MJ, Wierda WG, et al. Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 2013;122:734–7.
60. Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol 2007;25:5047.
61. James DF, Brown JR, Werner L, et al. Lenalidomide and rituximab for the initial treatment of patients with chronic lymphocytic leukemia (CLL): a multicenter study of the CLL Research Consortium. ASH Annual Meeting Abstracts 2011;118:291.
62. US Food and Drug Administration. FDA halts clinical trial of drug Revlimid (lenalidomide) for chronic lymphocytic leukemia due to safety concerns. Available at http://www.fda.gov/Drugs/DrugSafety/ucm361444.htm.
63. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010;115:2578–85.
64. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
65. Farooqui M, Aue G, Valdez J, et al. Single agent ibrutinib (PCI-32765) achieves equally good and durable responses in chronic lymphocytic leukemia (CLL) patients with and without deletion 17p. Blood 2013;122:673.
66. Byrd JC, Furman RR, Coutre S, et al. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) monotherapy demonstrates long-term safety and durability of response in chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) patients in an open-label extension study. Blood 2013;122:4163.
67. Brown JR, Barrientos JC, Barr PM, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study. ASH Annual Meeting Abstracts 2013.
68. O'Brien SM, Lamanna N, Kipps TJ, et al. A phase II study of the selective phosphatidylinositol 3-kinase delta (PI3K{delta}) inhibitor idelalisib (GS-1101) in combination with rituximab (R) in treatment-naive patients (pts) ≥ 65 years with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). J Clin Oncol 2013;31(15 Suppl); Abstract 7005.
69. Seymour JF, Davids MS, Pagel JM, et al. Bcl-2 Inhibitor ABT-199 (GDC-0199) monotherapy shows anti-tumor activity including complete remissions in high-risk relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Blood 2013;122:872.
70. Rituxumab and bendamustine hydrochloride, rituxumab and ibrutunib, or ibrutinib alone in treating older patients with previously untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT01886872?term=ibrutinib+cll&rank=8.
71. A multicenter, open-label, phase 3 study of the bruton's tyrosine kinase inhibitor pci-32765 versus chlorambucil in patients 65 years of older with treatment-naive chronic lymphocytic leukemia or small lymphocytic lymphoma (RESONATE-2) Available at http://clinicaltrials.gov/ct2/show/NCT01722487?term=ibrutinib+cll&rank=12.
72. Ibrutinib and rituximab compared with fludarabine phosphate, cyclophosphamide, and rituxumab in treating patients with untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT02048813?term=ibrutinib+cll&rank=2.
73. Strati P, Keating MJ, O'Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia (CLL) with 17p deletion. J Clin Oncol 2013;31(15 suppl): Abstract 7102.
1. Redaelli A, Laskin BL, Stephens JM, et al. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur J Cancer Care (Engl) 2004;13:279–87.
2. Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol 2007;139:809–19.
3. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6.
4. Zent CS, Kyasa MJ, Evans R, Schichman SA. Chronic lymphocytic leukemia incidence is substantially higher than estimated from tumor registry data. Cancer 2001;92:1325–30.
5. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006;24:437–43.
6. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56.
7. Rawstron AC, Bennett FL, O'Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83.
8. Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–34.
9. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
10. Binet JL, Lepoprier M, Dighiero G, et al. A clinical staging system for chronic lymphocytic leukemia: prognostic significance. Cancer 1977:40:855–64.
11. Eichhorst BF, Fischer K, Fink AM, et al. Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 2011;117:1817–21.
12. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54.
13. Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:
1764–75.
14. Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–9.
15. Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom 2006;70:209–13.
16. Wierda WG, O'Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–95.
17. Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 2008;26:5094–100.
18. Ding W, Ferrajoli A. Evidence-based mini-review: the role of alkylating agents in the initial treatment of chronic lymphocytic leukemia patients with the 11q deletion. Hematology Am Soc Hematol Educ Program 2010;2010:90–2.
19. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
20. Badoux XC, Keating MJ, Wierda WG.What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep 2011;6:36–46.
21. Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 1998;338:1506–14.
22. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 1999;91:861–8.
23. Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 2010;28:4935–44.
24. Shanafelt TD, Bowen D, Venkat C, et al. Quality of life in chronic lymphocytic leukemia: an international survey of 1482 patients. Br J Haematol 2007;139:255–64.
25. Baer MR, Stein RS, Dessypris EN. Chronic lymphocytic leukemia with hyperleukocytosis. The hyperviscosity syndrome. Cancer 1985;56:2865–9.
26. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia 2002;16:2092–5.
27. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: non-hodgkin's lymphomas. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
28. Byrd JC, Rai K, Peterson BL, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49–53.
29. Byrd JC, Peterson BL, Morrison VA, et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003;101:6–14.
30. Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079–88.
31. Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008;112:975–80.
32. Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16.
33. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9.
34. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–84.
35. Woyach JA, Ruppert AS, Heerema NA, et al. Chemoimmunotherapy with fludarabine and rituximab produces extended overall survival and progression-free survival in chronic lymphocytic leukemia: long-term follow-up of CALGB study 9712. J Clin Oncol 2011;29:1349–55.
36. Fink AM, et al., Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximabversus bendamustine and rituximabin previously untreated and physically fit patientswith advanced chronic lymphocytic leukemia: results of a planned interim analysis of the CLL10 Trial, an international, randomized study of the German CLL Study Group (GCLLSG). Blood 2013;122:526.
37. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet 2002;41:93–103.
38. Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood 2004;104:1428–34.
39. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–61.
40. Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81.
41. Osuji NC, Del Giudice I, Matutes E, et al, The efficacy of alemtuzumab for refractory chronic lymphocytic leukemia in relation to cytogenetic abnormalities of p53. Haematologica 2005;90:1435–6.
42. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
43. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–23.
44. Lundin J, Kimby E, Björkholm M, et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002;100:768–73.
45. Genzyme. US Campath Distribution Program. Cambridge, MA: Genzyme. Available at http://www.campath.com/.
46. Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65.
47. Bowen DA, Call TG, Jenkins GD, et al. Methylprednisolone-rituximab is an effective salvage therapy for patients with relapsed chronic lymphocytic leukemia including those with unfavorable cytogenetic features. Leuk Lymphoma 2007;48:2412–7.
48. Castro JE, Sandoval-Sus JD, Bole J, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of fludarabine refractory high-risk chronic lymphocytic leukemia. Leukemia 2008;22:2048–53.
49. Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89.
50. Eichhorst B, Goede V, Hallek M. Treatment of elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma 2009;50:171–8.
51. Foon KA, Boyiadzis M, Land SR, et al. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498–503.
52. Kay NE, Geyer SM, Call TG, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 2007;109:405–11.
53. Shanafelt TD, Lin T, Geyer SM, et al. Pentostatin, cyclophosphamide, and rituximab regimen in older patients with chronic lymphocytic leukemia. Cancer 2007;109:2291–8.
54. Catovsky D, Else M, Richards S. Chlorambucil--still not bad: a reappraisal. Clin Lymphoma Myeloma Leuk 2011;11 Suppl 1:S2–6.
55. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood 2009;114:3382–91.
56. Laurenti L, Vannata B, Innocenti I, et al. Chlorambucil plus rituximab as front-line therapy in elderly/unfit patients affected by b-cell chronic lymphocytic leukemia: results of a single-centre experience. Mediterr J Hematol Infect Dis 2013;5:e2013031.
57. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014 Jan 8. [Epub ahead of print].
58. Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood 2011;118:3489–98.
59. Strati P, Keating MJ, Wierda WG, et al. Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 2013;122:734–7.
60. Moutouh-de Parseval LA, Weiss L, DeLap RJ, et al. Tumor lysis syndrome/tumor flare reaction in lenalidomide-treated chronic lymphocytic leukemia. J Clin Oncol 2007;25:5047.
61. James DF, Brown JR, Werner L, et al. Lenalidomide and rituximab for the initial treatment of patients with chronic lymphocytic leukemia (CLL): a multicenter study of the CLL Research Consortium. ASH Annual Meeting Abstracts 2011;118:291.
62. US Food and Drug Administration. FDA halts clinical trial of drug Revlimid (lenalidomide) for chronic lymphocytic leukemia due to safety concerns. Available at http://www.fda.gov/Drugs/DrugSafety/ucm361444.htm.
63. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010;115:2578–85.
64. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
65. Farooqui M, Aue G, Valdez J, et al. Single agent ibrutinib (PCI-32765) achieves equally good and durable responses in chronic lymphocytic leukemia (CLL) patients with and without deletion 17p. Blood 2013;122:673.
66. Byrd JC, Furman RR, Coutre S, et al. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) monotherapy demonstrates long-term safety and durability of response in chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) patients in an open-label extension study. Blood 2013;122:4163.
67. Brown JR, Barrientos JC, Barr PM, et al. Ibrutinib in combination with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL/SLL: final results of a phase 1b study. ASH Annual Meeting Abstracts 2013.
68. O'Brien SM, Lamanna N, Kipps TJ, et al. A phase II study of the selective phosphatidylinositol 3-kinase delta (PI3K{delta}) inhibitor idelalisib (GS-1101) in combination with rituximab (R) in treatment-naive patients (pts) ≥ 65 years with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). J Clin Oncol 2013;31(15 Suppl); Abstract 7005.
69. Seymour JF, Davids MS, Pagel JM, et al. Bcl-2 Inhibitor ABT-199 (GDC-0199) monotherapy shows anti-tumor activity including complete remissions in high-risk relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Blood 2013;122:872.
70. Rituxumab and bendamustine hydrochloride, rituxumab and ibrutunib, or ibrutinib alone in treating older patients with previously untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT01886872?term=ibrutinib+cll&rank=8.
71. A multicenter, open-label, phase 3 study of the bruton's tyrosine kinase inhibitor pci-32765 versus chlorambucil in patients 65 years of older with treatment-naive chronic lymphocytic leukemia or small lymphocytic lymphoma (RESONATE-2) Available at http://clinicaltrials.gov/ct2/show/NCT01722487?term=ibrutinib+cll&rank=12.
72. Ibrutinib and rituximab compared with fludarabine phosphate, cyclophosphamide, and rituxumab in treating patients with untreated chronic lymphocytic leukemia. Available at http://clinicaltrials.gov/ct2/show/NCT02048813?term=ibrutinib+cll&rank=2.
73. Strati P, Keating MJ, O'Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia (CLL) with 17p deletion. J Clin Oncol 2013;31(15 suppl): Abstract 7102.