User login
2023 Update on genetics in fetal growth
Whole exome sequencing’s role in diagnosing genetic causes of FGR with and without associated anomalies
Mone F, Mellis R, Gabriel H, et al. Should we offer prenatal exome sequencing for intrauterine growth restriction or short long bones? A systematic review and meta-analysis. Am J Obstet Gynecol. Published online October 7, 2022. doi:10.1016/j.ajog.2022.09.045
Multiple factors can play a role in FGR, including inherent maternal, placental, or fetal factors; the environment; and/or nutrition. However, prenatal diagnosis is an important consideration when exploring the underlying etiology for a growth-restricted fetus, especially in severe or early-onset cases. Many genetic conditions do not result in structural anomalies but can disrupt overall growth. Additionally, phenotyping in the prenatal period is limited and can miss more subtle physical differences that could point to a genetic cause.
When compared with karyotype, chromosomal microarray (CMA) has been shown to increase the diagnostic yield in cases of isolated early FGR by 5%,1,2 and the incidence of chromosomal abnormalities has been reported to be as high as 19% in this population. Let’s explore the data on exome sequencing for prenatal diagnosis in cases of isolated FGR.
Meta-analysis details
In this meta-analysis, the authors reviewed 19 cohort studies or case series that investigated the yield of prenatal sequencing in fetuses with intrauterine growth restriction (IUGR) or short long bones, both in association with and without additional anomalies. All cases had nondiagnostic cytogenetic results. Fetal DNA in most cases was obtained through amniocentesis. Variants classified as likely pathogenic and pathogenic were considered diagnostic. The authors then calculated the incremental yield of prenatal sequencing over cytogenetic studies as a pooled value, comparing the following groups:
- isolated FGR
- growth restriction with associated anomalies
- isolated short long bones
- short long bones with additional skeletal features.
Study outcomes
The total number of cases were as follows: isolated IUGR (n = 71), IUGR associated with additional anomalies (n = 45), isolated short long bones (n = 84), and short long bones associated with additional skeletal findings (n = 252). Causative pathogenic or likely pathogenic variants were identified in 224 (50%) cases. Apparent incremental yields with prenatal sequencing were as follows for the 4 groups (as illustrated in the FIGURE):

- 4% in isolated IUGR (95% confidence interval [CI], -5%–12%)
- 30% in IUGR with additional anomalies (95% CI, 13%–47%)
- 48% in isolated short long bones (95% CI, 26%–70%)
- 68% in short long bones with additional skeletal changes (95% CI, 58%–77%).
Overall, the authors concluded that prenatal sequencing does not improve prenatal diagnosis in cases of isolated IUGR. The majority of these cases were thought to be related to placental insufficiency.
Strengths and limitations
The main limitation of this study with regard to our discussion is the small study populationof isolated growth restriction. The authors indicate that the number of cases of isolated IUGR were too small to draw firm conclusions. Another limitation was the heterogeneity of the isolated FGR population, which was not limited to severe or early-onset cases. However, the authors did demonstrate that growth restriction in association with fetal anomalies has very high genetic yield rates with prenatal sequencing.
Not surprisingly, there is a high yield of diagnosing genetic conditions in pregnancies complicated by isolated or nonisolated short long bones or in cases of growth restriction with multisystem abnormalities. Based on the results of this study, the authors advise against sending for exome sequencing in cases of isolated growth restriction with coexisting evidence of placental insufficiency.
Continue to: Can whole exome sequencing diagnose genetic causes in cases of severe isolated FGR?...
Can whole exome sequencing diagnose genetic causes in cases of severe isolated FGR?
Zhou H, Fu F, Wang Y, et al. Genetic causes of isolated and severe fetal growth restriction in normal chromosomal microarray analysis. Int J Gynaecol Obstet. Published online December 10, 2022. doi:10.1002/ijgo.14620
Severe FGR is diagnosed based on an estimated fetal weight (EFW) or abdominal circumference (AC) below the third percentile. As we discussed in the above study by Mone and colleagues, it does not appear that prenatal sequencing significantly improves the diagnostic yield in all isolated FGR cases. However, this has not been previously explored in isolated severe FGR or cases of early-onset FGR (<32 weeks’ gestation). We know that several monogenic conditions are associated with severe and early-onset isolated fetal growth impairment, including but not limited to Cornelia de Lange syndrome, Smith-Lemli-Opitz syndrome, and Meier-Gorlin syndrome. Often, these syndromes can present in the prenatal period without other phenotypic findings. Therefore, this study explored the possibility that prenatal sequencing plays an important role for severe cases of FGR with nondiagnostic CMA and/or karyotype.
Retrospective study details
Zhou and colleagues retrospectively analyzed 51 cases of severe (EFW or AC below the third percentile) isolated FGR with negative CMA who underwent trio whole exome sequencing, which includes submitting fetal cells as well as both parental samples for testing. Patients with abnormal toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus (TORCH) tests; structural anomalies; and multiple gestation were excluded from the analysis. As in the study by Mone et al, variants classified as likely pathogenic and pathogenic were categorized as diagnostic.
Results
Eight of 51 cases (15.7%) with severe isolated FGR had diagnostic findings on trio whole exome sequencing as shown in the TABLE. Another 8 cases (15.7%) were found to have variants of unknown significance, of which 2 were later determined to be novel pathogenic variants. Genetic conditions uncovered in this cohort include Cornelia de Lange syndrome, pyruvate dehydrogenase deficiency, Dent disease, trichohepaticenteric syndrome, achondroplasia, osteogenesis imperfecta, Pendred syndrome, and both autosomal dominant type 3A and autosomal recessive type 1A deafness. All 10 cases with diagnostic whole exome sequencing or identified novel pathogenic variants were affected by early-onset FGR (<32 weeks’ gestation). Of these 10 cases, 7 patients underwent pregnancy termination.

To summarize, a total of 10 cases (19.6%) of severe isolated early-onset FGR with negative cytogenetic studies were subsequently diagnosed with an underlying genetic condition using prenatal trio whole exome sequencing.
Strengths and limitations
This study is retrospective and has a small sample size (n = 51) that was mostly limited to early-onset isolated severe FGR. However, the diagnostic yield (19.6%) of whole exome sequencing after negative CMA testing was noteworthy and shows that monogenic conditions are an important consideration in the evaluation of severe early-onset FGR, even in the absence of structural abnormalities.
As indications for exome sequencing during pregnancy continue to evolve, severe isolated FGR is emerging as a high-yield condition in which a subset of patients may benefit from the described testing strategy. We learned from our look at the prior study (Mone et al) that unselected isolated growth restriction with evident placental insufficiency may not benefit from exome sequencing, but this study differs in its selection of early-onset, severe cases—defined by diagnosis before 32 weeks’ gestation and an EFW or AC below the third percentile. Almost 20% of cases who met the aforementioned criteria received a genetic diagnosis from exome sequencing. We should remember to offer genetic counseling and diagnostic testing to our patients with severe growth restriction, even in the absence of additional structural anomalies.
Could epigenetic mechanisms of placental dysregulation explain low birthweight and future cardiometabolic disease?
Tekola-Ayele F, Zeng X, Chatterjee S, et al. Placental multi-omics integration identifies candidate functional genes for birthweight. Nat Commun. 2022;13:2384.
FGR has been linked to greater mortality in childhood and increased risk for cardiometabolic disease in adulthood. While genomewide associations studies (GWAS) have defined areas of interest linking genetic variants to low birthweight, their relationship to epigenetic changes in the placenta as well as biologic and functional mechanisms are not yet well understood.
Multiomics used to identify candidate functional genes for birthweight
This study analyzed the methylation and gene expression patterns of 291 placental samples, integrating findings into pathways of previously defined GWAS variants. Patient samples were obtained from participants in the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Fetal Growth Studies–Singleton cohort. The cohort is ethnically diverse, with 97 Hispanic, 74 White, 71 Black, and 49 Asian participants. Of 286 single nucleotide polymorphisms (SNPs) found to be associated with birthweight, 273 were analyzed as part of the authors’ data set. These were found to have 7,901 unique protein-coding mRNAs (expression quantitative trait loci [eQTL]) and more than 100,000 nearby (within 1 Mb) CpG islands thought to be involved in changes in DNA methylation (methylation quantitative trait loci [mQTL]). Each functionally connected GWAS-eQTL-mQTL association is referred to as a triplet.
The next arm of the study investigatedthe connections and pathways within each triplet. Three possible scenarios were explored for birthweight GWAS SNPs using a causal interference test (CIT):
- the SNP alters placental DNA methylation, which then influences gene expression
- the SNP first alters placental DNA expression, which then influences methylation
- the SNP influences placental DNA expression and methylation independently, with no notable crossover between their pathways.
Triplets were investigated using the Mendelian randomization (MR) Steiger directionality test to validate the directionality of the pathways found by CIT. Lastly, the possibility of linkage disequilibrium was also studied using the moloc test.
Results
Using CIT, a causal relationship was predicted in 88 of 197 triplets, in which 84 (95.5%) indicated DNA methylation influences gene expression, and 4 (4.5%) indicated gene expression influences DNA methylation. The authors also used the MR Steiger test to investigate triplets to identify possible causal pathways. Using the MR Steiger test, only 3 of 45 (7%) triplets were found to have independent gene expression and methylation pathways. Thirty-eight of 45 (84%) triplets indicated that gene expression influences DNA methylation, and 7 (15%)triplets demonstrated that DNA methylation influences gene expression. Consistent predictions between CIT and the MR Steiger test revealed 3 triplets in which DNA methylation influences gene expression for the following genes: WNT3A, CTDNEP1, and RANBP2. Additionally, a strong colocalization signal was found among birthweight, DNA methylation, and gene expression for the following genes: PLEKHA1, FES, PRMT7, and CTDNEP1. Gene set enrichment analysis was performed as well and found that low birthweight is associated in substantial upregulation of genes associated with oxidative stress, immune response, adipogenesis, myogenesis, and the production of pancreatic ß cells.
Study strengths and limitations
The study is one of the first to identify regulatory targets for placental DNA methylation and gene expression in previously identified GWAS loci associated with low birthweight. For example, DNA methylation was found to influence gene expression of WNT3A, CTDNEP1, and RANBP2, which have previously been shown in animal studies to impact the vascularization and development of the placenta, embryogenesis, and fetal growth. The study also identified 4 genes (PLEKHA1, FES, PRMT7, and CTDNEP1) thought to have direct regulatory influence on placental DNA methylation and gene expression.
A limitation of the study is that it could not distinguish between whether the epigenetic changes we outlined have a maternal or fetal origin. Another limitation is that tissue used by the authors for analysis was a small placental biopsy, which does not accurately reflect the genetic heterogeneity of the placenta. Finally, this study does not establish causality between the identified epigenetic pathways and low birthweight. ●
We know that the placenta is critical to in utero development. This study begins to explore the genetic changes and programming in the placenta that may have profound effects on health and well-being both early and later in life.
- Li LS, Li DZ. A genetic approach to the etiologic investigation of isolated intrauterine growth restriction. Am J Obstet Gynecol. 2021;225:695-696. doi: 10.1016/j.ajog.2021 .07.021.
- Borrell A, Grande M, Pauta M, et al. Chromosomal microarray analysis in fetuses with growth restriction and normal karyotype: a systematic review and meta-analysis. Fetal Diagn Ther. 2018;44:1-9. doi: 10.1159/000479506.
Whole exome sequencing’s role in diagnosing genetic causes of FGR with and without associated anomalies
Mone F, Mellis R, Gabriel H, et al. Should we offer prenatal exome sequencing for intrauterine growth restriction or short long bones? A systematic review and meta-analysis. Am J Obstet Gynecol. Published online October 7, 2022. doi:10.1016/j.ajog.2022.09.045
Multiple factors can play a role in FGR, including inherent maternal, placental, or fetal factors; the environment; and/or nutrition. However, prenatal diagnosis is an important consideration when exploring the underlying etiology for a growth-restricted fetus, especially in severe or early-onset cases. Many genetic conditions do not result in structural anomalies but can disrupt overall growth. Additionally, phenotyping in the prenatal period is limited and can miss more subtle physical differences that could point to a genetic cause.
When compared with karyotype, chromosomal microarray (CMA) has been shown to increase the diagnostic yield in cases of isolated early FGR by 5%,1,2 and the incidence of chromosomal abnormalities has been reported to be as high as 19% in this population. Let’s explore the data on exome sequencing for prenatal diagnosis in cases of isolated FGR.
Meta-analysis details
In this meta-analysis, the authors reviewed 19 cohort studies or case series that investigated the yield of prenatal sequencing in fetuses with intrauterine growth restriction (IUGR) or short long bones, both in association with and without additional anomalies. All cases had nondiagnostic cytogenetic results. Fetal DNA in most cases was obtained through amniocentesis. Variants classified as likely pathogenic and pathogenic were considered diagnostic. The authors then calculated the incremental yield of prenatal sequencing over cytogenetic studies as a pooled value, comparing the following groups:
- isolated FGR
- growth restriction with associated anomalies
- isolated short long bones
- short long bones with additional skeletal features.
Study outcomes
The total number of cases were as follows: isolated IUGR (n = 71), IUGR associated with additional anomalies (n = 45), isolated short long bones (n = 84), and short long bones associated with additional skeletal findings (n = 252). Causative pathogenic or likely pathogenic variants were identified in 224 (50%) cases. Apparent incremental yields with prenatal sequencing were as follows for the 4 groups (as illustrated in the FIGURE):
- 4% in isolated IUGR (95% confidence interval [CI], -5%–12%)
- 30% in IUGR with additional anomalies (95% CI, 13%–47%)
- 48% in isolated short long bones (95% CI, 26%–70%)
- 68% in short long bones with additional skeletal changes (95% CI, 58%–77%).
Overall, the authors concluded that prenatal sequencing does not improve prenatal diagnosis in cases of isolated IUGR. The majority of these cases were thought to be related to placental insufficiency.
Strengths and limitations
The main limitation of this study with regard to our discussion is the small study populationof isolated growth restriction. The authors indicate that the number of cases of isolated IUGR were too small to draw firm conclusions. Another limitation was the heterogeneity of the isolated FGR population, which was not limited to severe or early-onset cases. However, the authors did demonstrate that growth restriction in association with fetal anomalies has very high genetic yield rates with prenatal sequencing.
Not surprisingly, there is a high yield of diagnosing genetic conditions in pregnancies complicated by isolated or nonisolated short long bones or in cases of growth restriction with multisystem abnormalities. Based on the results of this study, the authors advise against sending for exome sequencing in cases of isolated growth restriction with coexisting evidence of placental insufficiency.
Continue to: Can whole exome sequencing diagnose genetic causes in cases of severe isolated FGR?...
Can whole exome sequencing diagnose genetic causes in cases of severe isolated FGR?
Zhou H, Fu F, Wang Y, et al. Genetic causes of isolated and severe fetal growth restriction in normal chromosomal microarray analysis. Int J Gynaecol Obstet. Published online December 10, 2022. doi:10.1002/ijgo.14620
Severe FGR is diagnosed based on an estimated fetal weight (EFW) or abdominal circumference (AC) below the third percentile. As we discussed in the above study by Mone and colleagues, it does not appear that prenatal sequencing significantly improves the diagnostic yield in all isolated FGR cases. However, this has not been previously explored in isolated severe FGR or cases of early-onset FGR (<32 weeks’ gestation). We know that several monogenic conditions are associated with severe and early-onset isolated fetal growth impairment, including but not limited to Cornelia de Lange syndrome, Smith-Lemli-Opitz syndrome, and Meier-Gorlin syndrome. Often, these syndromes can present in the prenatal period without other phenotypic findings. Therefore, this study explored the possibility that prenatal sequencing plays an important role for severe cases of FGR with nondiagnostic CMA and/or karyotype.
Retrospective study details
Zhou and colleagues retrospectively analyzed 51 cases of severe (EFW or AC below the third percentile) isolated FGR with negative CMA who underwent trio whole exome sequencing, which includes submitting fetal cells as well as both parental samples for testing. Patients with abnormal toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus (TORCH) tests; structural anomalies; and multiple gestation were excluded from the analysis. As in the study by Mone et al, variants classified as likely pathogenic and pathogenic were categorized as diagnostic.
Results
Eight of 51 cases (15.7%) with severe isolated FGR had diagnostic findings on trio whole exome sequencing as shown in the TABLE. Another 8 cases (15.7%) were found to have variants of unknown significance, of which 2 were later determined to be novel pathogenic variants. Genetic conditions uncovered in this cohort include Cornelia de Lange syndrome, pyruvate dehydrogenase deficiency, Dent disease, trichohepaticenteric syndrome, achondroplasia, osteogenesis imperfecta, Pendred syndrome, and both autosomal dominant type 3A and autosomal recessive type 1A deafness. All 10 cases with diagnostic whole exome sequencing or identified novel pathogenic variants were affected by early-onset FGR (<32 weeks’ gestation). Of these 10 cases, 7 patients underwent pregnancy termination.
To summarize, a total of 10 cases (19.6%) of severe isolated early-onset FGR with negative cytogenetic studies were subsequently diagnosed with an underlying genetic condition using prenatal trio whole exome sequencing.
Strengths and limitations
This study is retrospective and has a small sample size (n = 51) that was mostly limited to early-onset isolated severe FGR. However, the diagnostic yield (19.6%) of whole exome sequencing after negative CMA testing was noteworthy and shows that monogenic conditions are an important consideration in the evaluation of severe early-onset FGR, even in the absence of structural abnormalities.
As indications for exome sequencing during pregnancy continue to evolve, severe isolated FGR is emerging as a high-yield condition in which a subset of patients may benefit from the described testing strategy. We learned from our look at the prior study (Mone et al) that unselected isolated growth restriction with evident placental insufficiency may not benefit from exome sequencing, but this study differs in its selection of early-onset, severe cases—defined by diagnosis before 32 weeks’ gestation and an EFW or AC below the third percentile. Almost 20% of cases who met the aforementioned criteria received a genetic diagnosis from exome sequencing. We should remember to offer genetic counseling and diagnostic testing to our patients with severe growth restriction, even in the absence of additional structural anomalies.
Could epigenetic mechanisms of placental dysregulation explain low birthweight and future cardiometabolic disease?
Tekola-Ayele F, Zeng X, Chatterjee S, et al. Placental multi-omics integration identifies candidate functional genes for birthweight. Nat Commun. 2022;13:2384.
FGR has been linked to greater mortality in childhood and increased risk for cardiometabolic disease in adulthood. While genomewide associations studies (GWAS) have defined areas of interest linking genetic variants to low birthweight, their relationship to epigenetic changes in the placenta as well as biologic and functional mechanisms are not yet well understood.
Multiomics used to identify candidate functional genes for birthweight
This study analyzed the methylation and gene expression patterns of 291 placental samples, integrating findings into pathways of previously defined GWAS variants. Patient samples were obtained from participants in the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Fetal Growth Studies–Singleton cohort. The cohort is ethnically diverse, with 97 Hispanic, 74 White, 71 Black, and 49 Asian participants. Of 286 single nucleotide polymorphisms (SNPs) found to be associated with birthweight, 273 were analyzed as part of the authors’ data set. These were found to have 7,901 unique protein-coding mRNAs (expression quantitative trait loci [eQTL]) and more than 100,000 nearby (within 1 Mb) CpG islands thought to be involved in changes in DNA methylation (methylation quantitative trait loci [mQTL]). Each functionally connected GWAS-eQTL-mQTL association is referred to as a triplet.
The next arm of the study investigatedthe connections and pathways within each triplet. Three possible scenarios were explored for birthweight GWAS SNPs using a causal interference test (CIT):
- the SNP alters placental DNA methylation, which then influences gene expression
- the SNP first alters placental DNA expression, which then influences methylation
- the SNP influences placental DNA expression and methylation independently, with no notable crossover between their pathways.
Triplets were investigated using the Mendelian randomization (MR) Steiger directionality test to validate the directionality of the pathways found by CIT. Lastly, the possibility of linkage disequilibrium was also studied using the moloc test.
Results
Using CIT, a causal relationship was predicted in 88 of 197 triplets, in which 84 (95.5%) indicated DNA methylation influences gene expression, and 4 (4.5%) indicated gene expression influences DNA methylation. The authors also used the MR Steiger test to investigate triplets to identify possible causal pathways. Using the MR Steiger test, only 3 of 45 (7%) triplets were found to have independent gene expression and methylation pathways. Thirty-eight of 45 (84%) triplets indicated that gene expression influences DNA methylation, and 7 (15%)triplets demonstrated that DNA methylation influences gene expression. Consistent predictions between CIT and the MR Steiger test revealed 3 triplets in which DNA methylation influences gene expression for the following genes: WNT3A, CTDNEP1, and RANBP2. Additionally, a strong colocalization signal was found among birthweight, DNA methylation, and gene expression for the following genes: PLEKHA1, FES, PRMT7, and CTDNEP1. Gene set enrichment analysis was performed as well and found that low birthweight is associated in substantial upregulation of genes associated with oxidative stress, immune response, adipogenesis, myogenesis, and the production of pancreatic ß cells.
Study strengths and limitations
The study is one of the first to identify regulatory targets for placental DNA methylation and gene expression in previously identified GWAS loci associated with low birthweight. For example, DNA methylation was found to influence gene expression of WNT3A, CTDNEP1, and RANBP2, which have previously been shown in animal studies to impact the vascularization and development of the placenta, embryogenesis, and fetal growth. The study also identified 4 genes (PLEKHA1, FES, PRMT7, and CTDNEP1) thought to have direct regulatory influence on placental DNA methylation and gene expression.
A limitation of the study is that it could not distinguish between whether the epigenetic changes we outlined have a maternal or fetal origin. Another limitation is that tissue used by the authors for analysis was a small placental biopsy, which does not accurately reflect the genetic heterogeneity of the placenta. Finally, this study does not establish causality between the identified epigenetic pathways and low birthweight. ●
We know that the placenta is critical to in utero development. This study begins to explore the genetic changes and programming in the placenta that may have profound effects on health and well-being both early and later in life.
Whole exome sequencing’s role in diagnosing genetic causes of FGR with and without associated anomalies
Mone F, Mellis R, Gabriel H, et al. Should we offer prenatal exome sequencing for intrauterine growth restriction or short long bones? A systematic review and meta-analysis. Am J Obstet Gynecol. Published online October 7, 2022. doi:10.1016/j.ajog.2022.09.045
Multiple factors can play a role in FGR, including inherent maternal, placental, or fetal factors; the environment; and/or nutrition. However, prenatal diagnosis is an important consideration when exploring the underlying etiology for a growth-restricted fetus, especially in severe or early-onset cases. Many genetic conditions do not result in structural anomalies but can disrupt overall growth. Additionally, phenotyping in the prenatal period is limited and can miss more subtle physical differences that could point to a genetic cause.
When compared with karyotype, chromosomal microarray (CMA) has been shown to increase the diagnostic yield in cases of isolated early FGR by 5%,1,2 and the incidence of chromosomal abnormalities has been reported to be as high as 19% in this population. Let’s explore the data on exome sequencing for prenatal diagnosis in cases of isolated FGR.
Meta-analysis details
In this meta-analysis, the authors reviewed 19 cohort studies or case series that investigated the yield of prenatal sequencing in fetuses with intrauterine growth restriction (IUGR) or short long bones, both in association with and without additional anomalies. All cases had nondiagnostic cytogenetic results. Fetal DNA in most cases was obtained through amniocentesis. Variants classified as likely pathogenic and pathogenic were considered diagnostic. The authors then calculated the incremental yield of prenatal sequencing over cytogenetic studies as a pooled value, comparing the following groups:
- isolated FGR
- growth restriction with associated anomalies
- isolated short long bones
- short long bones with additional skeletal features.
Study outcomes
The total number of cases were as follows: isolated IUGR (n = 71), IUGR associated with additional anomalies (n = 45), isolated short long bones (n = 84), and short long bones associated with additional skeletal findings (n = 252). Causative pathogenic or likely pathogenic variants were identified in 224 (50%) cases. Apparent incremental yields with prenatal sequencing were as follows for the 4 groups (as illustrated in the FIGURE):
- 4% in isolated IUGR (95% confidence interval [CI], -5%–12%)
- 30% in IUGR with additional anomalies (95% CI, 13%–47%)
- 48% in isolated short long bones (95% CI, 26%–70%)
- 68% in short long bones with additional skeletal changes (95% CI, 58%–77%).
Overall, the authors concluded that prenatal sequencing does not improve prenatal diagnosis in cases of isolated IUGR. The majority of these cases were thought to be related to placental insufficiency.
Strengths and limitations
The main limitation of this study with regard to our discussion is the small study populationof isolated growth restriction. The authors indicate that the number of cases of isolated IUGR were too small to draw firm conclusions. Another limitation was the heterogeneity of the isolated FGR population, which was not limited to severe or early-onset cases. However, the authors did demonstrate that growth restriction in association with fetal anomalies has very high genetic yield rates with prenatal sequencing.
Not surprisingly, there is a high yield of diagnosing genetic conditions in pregnancies complicated by isolated or nonisolated short long bones or in cases of growth restriction with multisystem abnormalities. Based on the results of this study, the authors advise against sending for exome sequencing in cases of isolated growth restriction with coexisting evidence of placental insufficiency.
Continue to: Can whole exome sequencing diagnose genetic causes in cases of severe isolated FGR?...
Can whole exome sequencing diagnose genetic causes in cases of severe isolated FGR?
Zhou H, Fu F, Wang Y, et al. Genetic causes of isolated and severe fetal growth restriction in normal chromosomal microarray analysis. Int J Gynaecol Obstet. Published online December 10, 2022. doi:10.1002/ijgo.14620
Severe FGR is diagnosed based on an estimated fetal weight (EFW) or abdominal circumference (AC) below the third percentile. As we discussed in the above study by Mone and colleagues, it does not appear that prenatal sequencing significantly improves the diagnostic yield in all isolated FGR cases. However, this has not been previously explored in isolated severe FGR or cases of early-onset FGR (<32 weeks’ gestation). We know that several monogenic conditions are associated with severe and early-onset isolated fetal growth impairment, including but not limited to Cornelia de Lange syndrome, Smith-Lemli-Opitz syndrome, and Meier-Gorlin syndrome. Often, these syndromes can present in the prenatal period without other phenotypic findings. Therefore, this study explored the possibility that prenatal sequencing plays an important role for severe cases of FGR with nondiagnostic CMA and/or karyotype.
Retrospective study details
Zhou and colleagues retrospectively analyzed 51 cases of severe (EFW or AC below the third percentile) isolated FGR with negative CMA who underwent trio whole exome sequencing, which includes submitting fetal cells as well as both parental samples for testing. Patients with abnormal toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus (TORCH) tests; structural anomalies; and multiple gestation were excluded from the analysis. As in the study by Mone et al, variants classified as likely pathogenic and pathogenic were categorized as diagnostic.
Results
Eight of 51 cases (15.7%) with severe isolated FGR had diagnostic findings on trio whole exome sequencing as shown in the TABLE. Another 8 cases (15.7%) were found to have variants of unknown significance, of which 2 were later determined to be novel pathogenic variants. Genetic conditions uncovered in this cohort include Cornelia de Lange syndrome, pyruvate dehydrogenase deficiency, Dent disease, trichohepaticenteric syndrome, achondroplasia, osteogenesis imperfecta, Pendred syndrome, and both autosomal dominant type 3A and autosomal recessive type 1A deafness. All 10 cases with diagnostic whole exome sequencing or identified novel pathogenic variants were affected by early-onset FGR (<32 weeks’ gestation). Of these 10 cases, 7 patients underwent pregnancy termination.
To summarize, a total of 10 cases (19.6%) of severe isolated early-onset FGR with negative cytogenetic studies were subsequently diagnosed with an underlying genetic condition using prenatal trio whole exome sequencing.
Strengths and limitations
This study is retrospective and has a small sample size (n = 51) that was mostly limited to early-onset isolated severe FGR. However, the diagnostic yield (19.6%) of whole exome sequencing after negative CMA testing was noteworthy and shows that monogenic conditions are an important consideration in the evaluation of severe early-onset FGR, even in the absence of structural abnormalities.
As indications for exome sequencing during pregnancy continue to evolve, severe isolated FGR is emerging as a high-yield condition in which a subset of patients may benefit from the described testing strategy. We learned from our look at the prior study (Mone et al) that unselected isolated growth restriction with evident placental insufficiency may not benefit from exome sequencing, but this study differs in its selection of early-onset, severe cases—defined by diagnosis before 32 weeks’ gestation and an EFW or AC below the third percentile. Almost 20% of cases who met the aforementioned criteria received a genetic diagnosis from exome sequencing. We should remember to offer genetic counseling and diagnostic testing to our patients with severe growth restriction, even in the absence of additional structural anomalies.
Could epigenetic mechanisms of placental dysregulation explain low birthweight and future cardiometabolic disease?
Tekola-Ayele F, Zeng X, Chatterjee S, et al. Placental multi-omics integration identifies candidate functional genes for birthweight. Nat Commun. 2022;13:2384.
FGR has been linked to greater mortality in childhood and increased risk for cardiometabolic disease in adulthood. While genomewide associations studies (GWAS) have defined areas of interest linking genetic variants to low birthweight, their relationship to epigenetic changes in the placenta as well as biologic and functional mechanisms are not yet well understood.
Multiomics used to identify candidate functional genes for birthweight
This study analyzed the methylation and gene expression patterns of 291 placental samples, integrating findings into pathways of previously defined GWAS variants. Patient samples were obtained from participants in the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Fetal Growth Studies–Singleton cohort. The cohort is ethnically diverse, with 97 Hispanic, 74 White, 71 Black, and 49 Asian participants. Of 286 single nucleotide polymorphisms (SNPs) found to be associated with birthweight, 273 were analyzed as part of the authors’ data set. These were found to have 7,901 unique protein-coding mRNAs (expression quantitative trait loci [eQTL]) and more than 100,000 nearby (within 1 Mb) CpG islands thought to be involved in changes in DNA methylation (methylation quantitative trait loci [mQTL]). Each functionally connected GWAS-eQTL-mQTL association is referred to as a triplet.
The next arm of the study investigatedthe connections and pathways within each triplet. Three possible scenarios were explored for birthweight GWAS SNPs using a causal interference test (CIT):
- the SNP alters placental DNA methylation, which then influences gene expression
- the SNP first alters placental DNA expression, which then influences methylation
- the SNP influences placental DNA expression and methylation independently, with no notable crossover between their pathways.
Triplets were investigated using the Mendelian randomization (MR) Steiger directionality test to validate the directionality of the pathways found by CIT. Lastly, the possibility of linkage disequilibrium was also studied using the moloc test.
Results
Using CIT, a causal relationship was predicted in 88 of 197 triplets, in which 84 (95.5%) indicated DNA methylation influences gene expression, and 4 (4.5%) indicated gene expression influences DNA methylation. The authors also used the MR Steiger test to investigate triplets to identify possible causal pathways. Using the MR Steiger test, only 3 of 45 (7%) triplets were found to have independent gene expression and methylation pathways. Thirty-eight of 45 (84%) triplets indicated that gene expression influences DNA methylation, and 7 (15%)triplets demonstrated that DNA methylation influences gene expression. Consistent predictions between CIT and the MR Steiger test revealed 3 triplets in which DNA methylation influences gene expression for the following genes: WNT3A, CTDNEP1, and RANBP2. Additionally, a strong colocalization signal was found among birthweight, DNA methylation, and gene expression for the following genes: PLEKHA1, FES, PRMT7, and CTDNEP1. Gene set enrichment analysis was performed as well and found that low birthweight is associated in substantial upregulation of genes associated with oxidative stress, immune response, adipogenesis, myogenesis, and the production of pancreatic ß cells.
Study strengths and limitations
The study is one of the first to identify regulatory targets for placental DNA methylation and gene expression in previously identified GWAS loci associated with low birthweight. For example, DNA methylation was found to influence gene expression of WNT3A, CTDNEP1, and RANBP2, which have previously been shown in animal studies to impact the vascularization and development of the placenta, embryogenesis, and fetal growth. The study also identified 4 genes (PLEKHA1, FES, PRMT7, and CTDNEP1) thought to have direct regulatory influence on placental DNA methylation and gene expression.
A limitation of the study is that it could not distinguish between whether the epigenetic changes we outlined have a maternal or fetal origin. Another limitation is that tissue used by the authors for analysis was a small placental biopsy, which does not accurately reflect the genetic heterogeneity of the placenta. Finally, this study does not establish causality between the identified epigenetic pathways and low birthweight. ●
We know that the placenta is critical to in utero development. This study begins to explore the genetic changes and programming in the placenta that may have profound effects on health and well-being both early and later in life.
- Li LS, Li DZ. A genetic approach to the etiologic investigation of isolated intrauterine growth restriction. Am J Obstet Gynecol. 2021;225:695-696. doi: 10.1016/j.ajog.2021 .07.021.
- Borrell A, Grande M, Pauta M, et al. Chromosomal microarray analysis in fetuses with growth restriction and normal karyotype: a systematic review and meta-analysis. Fetal Diagn Ther. 2018;44:1-9. doi: 10.1159/000479506.
- Li LS, Li DZ. A genetic approach to the etiologic investigation of isolated intrauterine growth restriction. Am J Obstet Gynecol. 2021;225:695-696. doi: 10.1016/j.ajog.2021 .07.021.
- Borrell A, Grande M, Pauta M, et al. Chromosomal microarray analysis in fetuses with growth restriction and normal karyotype: a systematic review and meta-analysis. Fetal Diagn Ther. 2018;44:1-9. doi: 10.1159/000479506.

The perimenopausal period and the benefits of progestin IUDs
Intrauterine devices (IUDs) are now used by more than 15% of US contraceptors. The majority of these IUDs release the progestin levonorgestrel, and with now longer extended use of the IUDs approved by the US Food and Drug Administration (FDA),1-3 they become even more attractive for use for contraception,control of menorrhagia or heavy menstrual bleeding (HMB) during reproductive years and perimenopause, and potentially, although not FDA approved for this purpose, postmenopause for endometrial protection in estrogen users. In this roundtable discussion, we will look at some of the benefits of the IUD for contraception effectiveness and control of bleeding, as well as the potential risks if used for postmenopausal women.
Progestin IUDs and contraception
JoAnn V. Pinkerton, MD, NCMP: Dr. Kaunitz, what are the contraceptive benefits of progestin IUDs during perimenopause?
Andrew M. Kaunitz, MD, NCMP: We know fertility declines as women approach menopause. However, when pregnancy occurs in older reproductive-age women, the pregnancies are often unintended, as reflected by high rates of induced abortion in this population. In addition, the prevalence of maternal comorbidities (during pregnancy and delivery) is higher in older reproductive-age women, with the maternal mortality rate more than 5 times higher compared with that of younger women.4 Two recently published clinical trials assessed the extended use of full-size IUDs containing 52 mg of levonor-gestrel (LNG), with the brand names Mirena and Liletta.1,2 The data from these trials confirmed that both IUDs remain highly effective for up to 8 years of use, and currently, both devices are approved for up to 8 years of use. One caveat is that, in the unusual occurrence of a pregnancy being diagnosed in a woman using an IUD, we as clinicians, must be alert to the high prevalence of ectopic pregnancies in this setting.
Progestin IUDs and HMB
Dr. Pinkerton: Dr. Goldstein, can you comment on how well progestin IUDs work for HMB?
Steven R. Goldstein, MD, NCMP, CCD: Many women who need contraception will use these devices for suppressing HMB, and they can be quite effective, if the diagnosis truly is HMB, at reducing bleeding.5 But that efficacy in bleeding reduction may not be quite as long as the efficacy in pregnancy prevention.6 In my experience, among women using IUDs specifically for their HMB, good bleeding control may require changing the IUD at 3 to 5 years.
Barbara S. Levy, MD: When inserting a LNG-IUD for menorrhagia in the perimenopausal time frame, sometimes I will do a progestin withdrawal first, which will thin the endometrium and induce withdrawal bleeding because, in my experience, if you place an IUD in someone with perimenopausal bleeding, you may end up with a lot of breakthrough bleeding.
Perimenopause and hot flashes
Dr. Pinkerton: Dr. Kaunitz, we have learned that hot flashes often occur and become bothersome to women during perimenopause. Many women have IUDs placed during perimenopause for bleeding. Can you comment about IUD use during perimenopause and postmenopause?
Dr. Kaunitz: In older reproductive-age women who already have a progestin-releasing IUD placed, as they get closer to menopause when vasomotor symptoms (VMS) might occur, if these symptoms are bothersome, the presence or placement of a progestin-releasing IUD can facilitate treatment of perimenopausal VMS with estrogen therapy.
Progestin IUDs cause profound endometrial suppression, reduce bleeding and often, over time, cause users to become amenorrheic.7
The Mirena package insert states, “Amenorrhea develops in about 20% of users by one year.”2 By year 3 and continuing through year 8, the prevalence of amenorrhea with the 52-mg LNG-IUD is 35% to 40%.8 From a study by Nanette Santoro, MD, and colleagues, we know that, in perimenopausal women with a progestin-releasing IUD in place, who are experiencing bothersome VMS, adding transdermal estrogen is very effective in treating and suppressing those hot flashes. In her small clinical trial, among participants with perimenopausal bothersome VMS with an IUD in place, half were randomized to use of transdermal estradiol and then compared with women who did not get the estradiol patch. There was excellent relief of perimenopausal hot flashes with the combination of the progestin IUD for endometrial suppression and transdermal estrogen to relieve hot flashes.9
Dr. Pinkerton: Which women would not be good candidates for the use of this combination?
Dr. Kaunitz: We know that, as women age, the prevalence of conditions that are contraindications to combination contraceptives (estrogen-progestin pills, patches, or rings) starts to increase. Specifically, we see more: hypertension, diabetes, and high body mass index (BMI), or obesity. We also know that migraine headaches in women older than age 35 years is another condition in which ACOG and the Centers for Disease Control and Prevention (CDC) would not recommend use of combination contraceptives.10,11 These older perimenopausal women may be excellent candidates for a progestin-only releasing IUD combined with use of transdermal menopausal doses of estradiol if needed for VMS.
Dr. Goldstein: I do want to add that, in those patients who don’t have these comorbidities, combination estrogen-progestin contraceptives do a very nice job of ovarian suppression and will prevent the erratic production of estradiol, which, in my experience, often results in not only irregular bleeding but also possible exacerbation of perimenopausal mood symptoms.
Dr. Kaunitz: I agree, Steve. The ideal older reproductive-age candidate for combination pills, patch, or ring would be a slender, healthy, nonsmoking woman with normal blood pressure. Such women would be a fairly small subgroup of my practice, but they can safely continue combination contraceptives right through menopause. Consistent with CDC and ACOG guidance, rather than checking gonadotropins to “determine when menopause has occurred,” (which is, in fact, not an evidence-based approach to diagnosing menopause in this setting), such women can continue the combination contraceptive right up until age 55—the likelihood that women are still going to be ovulating or at risk for pregnancy becomes vanishingly small at that age.11,12 Women in their mid-50s can either seamlessly transition to use of systemic estrogen-progestin menopausal therapy or go off hormones completely.
Continue to: The IUD and HMB...
The IUD and HMB
Dr. Pinkerton: Dr. Goldstein, there’s been some good literature on the best management options for women with HMB. What is the most current evidence?
Dr. Goldstein: I think that the retiring of the terms menorrhagia and metrorrhagia may have been premature because HMB implies cyclical bleeding, and this population of women with HMB will typically do quite well. Women who have what we used to call metrorrhagia or irregular bleeding, by definition, need endometrial evaluation to be sure they don’t have some sort of organic pathology. It would be a mistake for clinicians to use an LNG-IUD in patients with abnormal uterine bleeding (AUB) that has not been appropriately evaluated.
If we understand that we are discussing HMB, a Cochrane Review from 202213 suggests that an LNG intrauterine system is the best first-line treatment for reducing menstrual blood loss in perimenopausal women with HMB. Antifibrinolytics appeared second best, while long-cycle progestogens came in third place. Evidence on perception of improvement in satisfaction was ranked as low certainty. That same review found that hysterectomy was the best treatment for reducing bleeding, obviously, followed by resectoscopic endometrial ablation or a nonresectoscopic global endometrial ablation.
The evidence rating was low certainty regarding the likelihood that placing an LNG-IUD in women with HMB will result in amenorrhea, and I think that’s a very important point. The expectation of patients should be reduced or a significantly reduced amount of their HMB, not necessarily amenorrhea. Certainly, minimally invasive hysterectomy will result in total amenorrhea and may have a larger increase in satisfaction, but it has its own set of other kinds of possible complications.
Dr. Kaunitz: In an industry-funded, international multicenter trial,14 women with documented HMB (hemoglobin was eluted from soiled sanitary products), with menstrual blood loss of 80 mL or more per cycle, were randomized to placement of an LNG 52-mg IUD (Mirena) or cyclical medroxyprogesterone acetate (MPA)—oral progestin use.
Although menstrual blood loss declined in both groups, it declined dramatically more in women with an IUD placed, and specifically with the IUD, menstrual blood loss declined by 129 mL on average, whereas the decline in menstrual blood loss with cyclical MPA was 18 mL. This data, along with earlier European data,15 which showed similar findings in women with HMB led to the approval of the Mirena progestin IUD for a second indication to treat HMB in 2009.
I also want to point out that, in the May 2023 issue of Obstetrics & Gynecology, Creinin and colleagues published a similar trial in women with HMB showing, once again, that progestin IUDs (52-mg LNG-IUD, Liletta) are extremely effective in reducing HMB.16 There is crystal clear evidence from randomized trials that both 52-mg LNG-IUDs, Mirena and Liletta, are very effective in reducing HMB and, in fact, are contributing to many women who in the past would have proceeded with surgery, such as ablation or hysterectomy, to control their HMB.
Oral contraception
Dr. Pinkerton: What about using low-dose continuous oral contraceptives noncyclically for women with HMB?
Dr. Goldstein: I do that all the time. It is interesting that Dr. Kaunitz mentions his patient population. It’s why we understand that one size does not fit all. You need to see patients one at a time, and if they are good candidates for a combined estrogen-progestin contraception, whether it’s pills, patches, or rings, giving that continuously does a very nice job in reducing HMB and straightening out some of the other symptoms that these perimenopausal women will have.
IUD risks
Dr. Pinkerton: We all know about use of low-dose oral contraceptives for management of AUB, and we use them, although we worry a little bit about breast cancer risk. Dr. Levy, please comment on the risks with IUDs of expulsions and perforations. What are the downsides of IUDs?
Dr. Levy: Beyond the cost, although it is a minimally invasive procedure, IUD insertion can be an invasive procedure for a patient to undergo; expulsions can occur.17 We know that a substantial percentage of perimenopausal women will have fibroids. Although many fibroids are not located in the uterine cavity, the expulsion rate with HMB for an LNG-IUD can be higher,13,16,18,19 perhaps because of local prostaglandin release with an increase in uterine contractility. There is a low incidence of perforations, but they do happen, particularly among women with scars in the uterus or who have a severely anteflexed or retroflexed uterus, and women with cervical stenosis, for example, if they have had a LEEP procedure, etc. Even though progestin IUDs are outstanding tools in our toolbox, they are invasive to some extent, and they do have the possibility of complications.
Dr. Kaunitz: As Dr. Levy points out, although placement of an IUD may be considered an invasive procedure, it is also an office-based procedure, so women can drive home or drive back to work afterwards without the disruption in their life and the potential complications associated with surgery and anesthesia.
Continue to: Concerns with malpositioning...
Concerns with malpositioning
Dr. Pinkerton: After placement of an IUD, during a follow-up visit, sometimes you can’t visualize the string. The ultrasonography report may reveal, “IUD appears to be in the right place within the endometrium.” Dr. Goldstein, can you comment on how we should use ultrasound when we can’t visualize or find the IUD string, or if the patient complains of abdominal pain, lower abdominal discomfort, or irregular bleeding or spotting and we become concerned about IUD malposition?
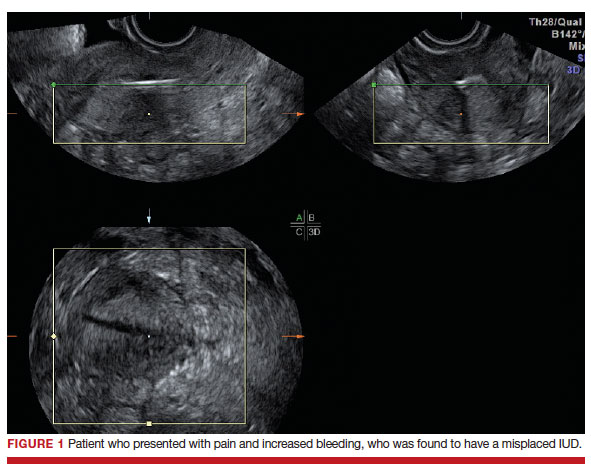
Dr. Goldstein: Ultrasound is not really required after an uncomplicated placement of an IUD or during routine management of women who have no problems who are using an IUD. In patients who present with pain or some abnormal bleeding, however, sometimes it is the IUD being malpositioned. A very interesting study by the late great Beryl Benacerraf20 showed that there was a statistically significant higher incidence of the IUD being poorly positioned when patients have pain or bleeding (FIGURE 1). It was not always apparent on 2D ultrasonography. Using a standard transvaginal ultrasound of the long access plane, the IUD may appear to be very centrally located. However, if you do a 3D coronal section, not infrequently in these patients with any pain or bleeding, one of the arms has pierced the myometrium (FIGURE 2). This can actually be a source of pain and bleeding.
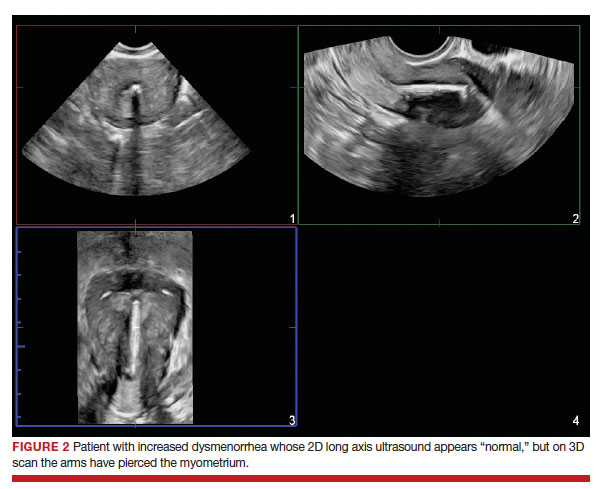
It’s also very interesting when you talk about perforation. I became aware of a big to-do in the medical/legal world about the possibility of the IUD migrating through the uterine cavity.21 This just does not exist, as was already pointed out. If the IUD is really going to go anywhere, if it’s properly placed, it’s going to be expelled through an open cervix. I do believe that, if you have pierced the myometrium through uterine contractility over time, some of these IUDs could work their way through the myometrium and somehow come out of the uterus either totally or partially. I think ultrasound is invaluable in patients with pain and bleeding, but I think you need to have an ultrasound lab capable of doing a 3D coronal section.
Progestin IUDs for HT replacement: Benefits/risks
Dr. Pinkerton: Many clinicians are excited that they can use essentially estrogen alone for women who have a progestin IUD in place. What about the possible off-label use of the progestin IUD to replace oral progestogen for hormone therapy (HT)? Dr. Kaunitz, are there any studies using this for postmenopausal HT (with a reminder that the IUD is not FDA approved for this purpose)?
Dr. Kaunitz: We have data from Europe indicating that, in menopausal women using systemic estrogen, the full-size LNG 52 IUD—Mirena or Liletta—provides excellent endometrial suppression.22 Where we don’t have data is with the smaller IUDs, which would be Kyleena and Skyla, which release smaller amounts of progestin each day into the endometrial cavity.
I have a number of patients, most of them women who started use of a progestin IUD as older reproductive-age women and then started systemic estrogen for treatment of perimenopausal hot flashes and then continued the use of their IUD plus systemic estrogen in treating postmenopausal hot flashes. The IUD is very useful in this setting, but as you pointed out, Dr. Pinkerton, this does represent off-label use.
Dr. Pinkerton: I know this use does not affect plasma lipids or cardiovascular risk markers, although users seem to report that the IUD has improved their quality of life. The question comes up, what are the benefits on cancer risk for using an IUD?
Dr. Levy: It’s such a great question because, as we talk about the balance of risks and benefits for anything that we are offering to our patients, it is really important to focus on some of the benefits. For both the copper and the LNG-IUD, there is a reduction in endometrial cancer,22 as well as pretty good data with the copper IUD about a reduction in cervical cancer.23 Those data are a little bit less clear for the LNG-IUD.
Interestingly, at least one meta-analysis published in 2020 shows about a 30% reduction in ovarian cancer risk with the LNG-IUD.24 We need to focus our patients on these other benefits. They tend to focus on the risks, and, of course, the media blows up the risks, but the benefits are quite substantial beyond just reducing HMB and providing contraception.
Dr. Pinkerton: As Dr. Kaunitz said, when you use this IUD, with its primarily local uterine progestin effects, it’s more like using estrogen alone without as much systemic progestin. Recently I wrote an editorial on the benefits of estrogen alone on the risk of breast cancer, primarily based on the Women’s Health Initiative (WHI) observational long-term 18-year cumulative follow-up. When estrogen alone was prescribed to women after a hysterectomy, estrogen therapy used at menopause did not increase the risk of invasive breast cancer, and was associated with decreased mortality.25 However, the nurse’s health study has suggested that longer-term use may be increased with estrogen alone.26 For women in the WHI with an intact uterus who used estrogen, oral MPA slightly increased the risk for breast cancer, and this elevated risk persisted even after discontinuation. This leads us to the question, what are the risks of breast cancer with progestin IUD use?
I recently reviewed the literature, and the answer is, it’s mixed. The FDA has put language into the package label that acknowledges a potential breast cancer risk for women who use a progestin IUD,27 and that warning states, “Women who currently have or have had breast cancer or suspect breast cancer should not use hormonal contraception because some breast cancers are hormone sensitive.” The label goes on to say, “Observational studies of the risk of breast cancer with the use of a levonorgestrel-releasing IUS don’t provide conclusive evidence of increased risk.” Thus, there is no conclusive answer as to whether there is a possible link of progestin IUDs to breast cancer.
What I tell my patients is that research is inconclusive. However, it’s unlikely for a 52-mg LNG-IUD to significantly increase a woman’s breast cancer risk, except possibly in those already at high risk from other risk factors. I tell them that breast cancer is listed in the package insert as a potential risk. I could not find any data on whether adding a low-dose estradiol patch would further increase that risk. So I counsel women about potential risk, but tell them that I don’t have any strong evidence of risk.
Continue to: Dr. Goldstein...
Dr. Goldstein: If you look in the package insert for Mirena,2 similar to Liletta, certainly the serum levels of LNG are lower than that for combination oral contraceptives. For the IUD progestins, they are not localized only to the uterus, and LNG levels range from about 150 to 200 µg/mL up to 60 months. It’s greater at 12 months, at about 180 µg/mL,at 24 months it was 192 µg/mL, and by 60 months it was 159 µg/mL. It’s important to realize that there is some systemic absorption of progestin with progestin IUDs, and it is not completely a local effect.
JoAnn, you mentioned the WHI data,25 and just to specify, it was not the estrogen-only arm, it was the conjugated equine estrogen-only arm of the WHI. I don’t think that estradiol alone increases breast cancer risk (although there are no good prospective, follow-through, 18-year study data, like the WHI), but I think readers need to understand the difference in the estrogen type.
Endometrial evaluation. My question for the panel is as follows. I agree that the use of the progestin-releasing IUD is very nice for that transition to menopause. I do believe it provides endometrial protection, but we know from other studies that, when we give continuous combined HT, about 21% to 26% of patients will experience some bleeding/staining, responding in the first 4-week cycles, and it can be as high as 9% at 1 year. If I have a patient who bleeds on continuous combined HT, I will evaluate her endometrium, usually just with a simple transvaginal ultrasound. If an IUD is in place, and the patient now begins to have some irregular bleeding, how do you evaluate her with the IUD in place?
Dr. Levy: That is a huge challenge. We know from a recent paper,28 that the endometrial thickness, while an excellent measure for Caucasian and European women, may be a poor marker for endometrial pathology in African-American women. What we thought we knew, which was, if the stripe is 4 mL or less, we can forget about it, I think in our more recent research that is not so true. So you bring up a great point, what do you do? The most reliable evaluation will be with an office hysteroscopy, where you can really look at the entire cavity and for tiny, little polyps and other things. But then we are off label because the use of hysteroscopy with an IUD in place is off label. So we are really in a conundrum.
Dr. Pinkerton: Also, if you do an endometrial biopsy, you might dislodge the IUD. If you think that you are going to take the IUD out, it may not matter if you dislodge it. I will often obtain a transvaginal ultrasound to help me figure out the next step, and maybe look at the dosing of the estrogen and progestin—but you can’t monitor an IUD with blood levels. You are in a vacuum of trying to figure out the best thing to do.
Dr. Kaunitz: One of the hats I wear here in Jacksonville is Director of GYN Ultrasound. I have a fair amount of experience doing endometrial biopsies in women with progestin IUDs in place under abdominal ultrasound guidance and keeping a close eye on the position of the IUD. In the first dozen or so such procedures I did, I was quite concerned about dislodging the IUD. It hasn’t happened yet, and it gives me some reassurance to be able to image the IUD and your endometrial suction curette inside the cavity as you are obtaining endometrial sampling. I have substantial experience now doing that, and so far, no problems. I do counsel all such women in advance that there is some chance I could dislodge their IUD.
Dr. Goldstein: In addition to dislodging the IUD, are you not concerned that, if the pathology is not global, that a blind endometrial sampling may be fraught with some error?
Dr. Kaunitz: The endometrium in women with a progestin-releasing IUD in place tends to be very well suppressed. Although one might occasionally find, for instance, a polyp in that setting, I have not run into, and I don’t expect to encounter going forward, endometrial hyperplasia or cancer in women with current use of a progestin IUD. It’s possible but unlikely.
Dr. Levy: The progestin IUD will counterbalance a type-1 endometrial cancer—an endometrial cancer related to hyperstimulation by estrogen. It will not do anything, to my knowledge, to counterbalance a type 2. I think the art of medicine is, you do the best you can with the first episode of bleeding, and if she persists in her bleeding, we have to persevere and continue to evaluate her.
Dr. Goldstein: I agree 100%.
Dr. Pinkerton: We all agree with you. That’s a really good point.
Continue to: Case examinations...
Case examinations
CASE 1 Woman with intramural fibroids
Dr. Pinkerton: Dr. Goldstein, you have a 48-year-old Black woman who has heavy but regular menstrual bleeding with multiple fibroids (the largest is about 4 to 5 cm, they look intramural, with some distortion of the cavity but not a submucous myoma, and the endometrial depth is 9 cm). Would you insert an IUD, and would you recommend an endometrial biopsy first?
Dr. Goldstein: I am not a huge fan of blind endometrial sampling, and I do think that we use the “biopsy” somewhat inappropriately since sampling is not a directed biopsy. This became obvious in the landmark paper by Guido et al in 1995 and was adopted by ACOG only in 2012.29 Cancers that occupy less than 50% of the endometrial surface area are often missed with such blind sampling. Thus I would not perform an endometrial biopsy first, but would rather rely on properly timed and performed transvaginal ultrasound to rule out any concurrent endometrial disease. I think a lot of patients who have HMB, not only because of their fibroids but also often just due to the surface area of their uterine cavity being increased—so essentially there is more blood volume when they bleed. However, you said that in this case the patient has regular menstrual bleeding, so I am assuming that she is still ovulatory. She may have some adenomyosis. She may have a large uterine cavity. I think she is an excellent candidate for an LNG-releasing IUD to reduce menstrual blood flow significantly. It will not necessarily give her amenorrhea, and it may give her some irregular bleeding. Then at some distant point, say in 5 or 6 months if she does have some irregular staining or bleeding, I would feel much better about the fact that nothing has developed as long as I knew that the endometrium was devoid of pathology when I started.
CASE 2 Woman with family history of breast cancer
Dr. Pinkerton: Dr. Levy, a 44-year-old woman has a family history of breast cancer in her mother at age 72, but she still needs contraceptionbecause of that unintended pregnancy risk in the 40s, and she wants something that is not going to increase her risk of breast cancer. What would you use, and how would you counsel her if you decided to use a progestin IUD?
Dr. Levy: The data are mixed,30-33 but whatever the risk, it is miniscule, and I would bring up the CDC Medical Eligibility Criteria.11 For a patient with a family history of breast cancer, for use of the progestin IUD, it is a 1—no contraindications. What I tend to tell my patients is, if you are worried about breast cancer, watch how much alcohol you are drinking and maintain regular exercise. There are so many preventive things that we can do to reduce risk of breast cancer when she needs contraception. If there is any increase in risk, it is so miniscule that I would very strongly recommend a progestin IUD for her.
Dr. Pinkerton: In addition, in recognizing the different densities of breast, dense breast density could lead to supplemental screening, which also could give her some reassurance that we are adequately screening for breast cancer.
CASE 3 Woman with IUD and VMS
Dr. Pinkerton: Dr. Kaunitz, you have a 52-year-old overweight female. She has been using a progestin IUD for 4 years, is amenorrheic, but now she is having moderate to severe vasomotor symptoms despite the IUD in place. You have talked to her about risks and benefits of HT, and she is interested in starting it. I know we talked about the studies, but I want to know what you are going to tell her. How do you counsel her about off-label use?
Dr. Kaunitz: The most important issue related to treating vasomotor symptoms in this patient is the route of systemic estrogen. Understandably, women’s biggest concern regarding the risks of systemic estrogen-progestin therapy is breast cancer. However, statistically, by far the biggest risk associated with oral estrogen-progestogen therapy, is elevated risk of venous thrombosis and pulmonary embolism. We have seen this, with a number of studies, and the WHI made it crystal clear with risks of oral conjugated equine estrogen at the dose of 0.625 mg daily. Oral estradiol 1 mg daily is also associated with a similar elevated risk of venous thrombosis. We also know that age and BMI are both independent risk factors for thrombosis. So, for a woman in her 50s who has a BMI > 30 mg/kg2, I don’t want to further elevate her risk of thrombosis by giving her oral estrogen, whether it is estradiol or conjugated equine estrogen. This is a patient in whom I would be comfortable using transdermal (patch) estradiol, perhaps starting with a standard dose of 0.05 mg weekly or twice weekly patch, keeping in mind that 0.05 mg in the setting of transdermal estrogen refer to the daily or to the 24-hour release rate. The 1.0 mg of oral estradiol and 0.625 mg of conjugated equine estrogen refers to the mg quantity of estrogen in each tablet. This is a source of great confusion for clinicians.
If, during follow-up, the 0.05 mg estradiol patch is not sufficient to substantially reduce symptoms, we could go up, for instance, to a 0.075 mg estradiol patch. We know very clearly from a variety of observational studies, including a very large UK study,34 that in contrast with oral estrogen, transdermal estradiol is safer from the perspective of thrombosis.
Insurance coverage for IUDs
Dr. Pinkerton: Dr. Levy: Can you discuss IUDs and the Affordable Care Act’s requirement to cover contraceptive services?
Dr. Levy: Unfortunately, we do not know whether this benefit will continue based on a very recent finding from a judge in Texas that ruled the preventive benefits of the ACA were illegal.35 We don’t know what will happen going forward. What I will say is that, unfortunately, many insurance companies have not preserved the meaning of “cover all things,” so what we are finding is that, for example, they only have to cover one type in a class. The FDA defined 18 classes of contraceptives, and a hormonal IUD is one class, so they can decide that they are only going to cover one of the four IUDS. And then women don’t have access to the other three, some of which might be more appropriate for them than another.
The other thing very relevant to this conversation is that, if you use an ICD-10 code for menorrhagia, for HMB, it no longer lives within that ACA preventive care requirement of coverage for contraceptives, and now she is going to owe a big deductible or a copay. If you are practicing in an institution that does not allow the use of IUDs for contraception, like a Catholic institution where I used to practice, you will want to use that ICD-10 code for HMB. But if you want it offered with no out-of-pocket cost for the patient, you need to use the preventive medicine codes and the contraception code. These little nuances for us can make a huge difference for our patients.
Dr. Pinkerton: Thank you for that reminder. I want to thank our panelists, Dr. Levy, Dr. Goldstein, and Dr. Kaunitz, for providing us with such a great mix of evidence and expert opinion and also giving a benefit of their vast experience as award-winning gynecologists. Hopefully, today you have learned the benefits of the progestin IUD not only for contraception in reproductive years and perimenopause but also for treatment of HMB, and the potential benefit due to the more prolonged effectiveness of the IUDs for endometrial protection in postmenopause. This allows less progestin risk, essentially estrogen alone for postmenopausal HT. Unsolved questions remain about whether there is a risk of breast cancer with their use, but there is a clear benefit of protecting against pregnancy and endometrial cancer. ●
- Liletta [package insert]. Allergan; Irvine, California. November 2022.
- Mirena [package insert]. Bayer; Whippany, New Jersey. 2000.
- Kaunitz AM. Safe extended use of levonorgestrel 52-mg IUDs. November 11, 2022. https://www.medscape.com/ viewarticle/983680. Accessed May 8, 2023.
- Kaunitz AM. Clinical practice. Hormonal contraception in women of older reproductive age. N Engl J Med. 2008;358:1262-1270. doi: 10.1056/NEJMcp0708481.
- Tucker ME. IUD-released levonorgestrel eases heavy menstrual periods. Medscape. April 10, 2023. https://www .medscape.com/viewarticle/777406. Accessed May 2, 2023.
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice; Long-Acting Reversible Contraception Working Group. ACOG Committee Opinion No. 450: Increasing use of contraceptive implants and intrauterine devices to reduce unintended pregnancy. Obstet Gynecol. 2009;114:1434-1438.
- Critchley HO, Wang H, Jones RL, et al. Morphological and functional features of endometrial decidualization following long-term intrauterine levonorgestrel delivery. Hum Reprod. 1998;13:1218-1224. doi:10.1093/humrep/13.5.1218.
- Creinin MD, Schreiber CA, Turok DK, et al. Levonorgestrel 52 mg intrauterine system efficacy and safety through 8 years of use. Am J Obstet Gynecol. 2022;227:871.e1-871.e7. doi: 10.1016/j.ajog.2022.05.022.
- Santoro N, Teal S, Gavito C, et al. Use of a levonorgestrelcontaining intrauterine system with supplemental estrogen improves symptoms in perimenopausal women: a pilot study. Menopause. 2015;22:1301-1307. doi: 10.1097 /GME.0000000000000557.
- ACOG Committee on Practice Bulletins-Gynecology ACOG Practice Bulletin. The use of hormonal contraception in women with coexisting medical conditions. Number 18, July 2000. Int J Gynaecol Obstet. 2001;75:93-106. doi: 10.1016 /s0020-7292(01)00520-3.
- Curtis KM, Tepper NK, Jatlaoui TC, Berry-Bibee E, Horton LG, Zapata LB, Simmons KB, Pagano HP, Jamieson DJ, Whiteman MK. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. 2016;65:1-103. doi: 10.15585 /mmwr.rr6503a1.
- ACOG Practice Bulletin No. 206: use of hormonal contraception in women with coexisting medical conditions [published correction appears in: Obstet Gynecol. 2019;133:1288.] Obstet Gynecol. 2019;133:e128-e150. doi:10.1097/AOG.0000000000003072.
- Bofill Rodriguez M, Dias S, Jordan V, et al. Interventions for heavy menstrual bleeding; overview of Cochrane reviews and network meta-analysis. Cochrane Database Syst Rev. 2022;5:CD013180. doi: 10.1002/14651858.CD013180.pub2.
- Kaunitz AM, Bissonnette F, Monteiro I, et al. Levonorgestrelreleasing intrauterine system or medroxyprogesterone for heavy menstrual bleeding: a randomized controlled trial [published correction appears in: Obstet Gynecol. 2010;116:999]. Obstet Gynecol. 2010;116:625-632. doi: 10.1097 /AOG.0b013e3181ec622b.
- Milsom I, Andersson K, Andersch B, et al. A comparison of flurbiprofen, tranexamic acid, and a levonorgestrel-releasing intrauterine contraceptive device in the treatment of idiopathic menorrhagia. Am J Obstet Gynecol. 1991;164:879883. doi: 10.1016/s0002-9378(11)90533-x.
- Creinin MD, Barnhart KT, Gawron LM, et al. Heavy menstrual bleeding treatment with a levonorgestrel 52-mg intrauterine device. Obstet Gynecol. 2023;141:971-978. doi: 10.1097 /AOG.0000000000005137.
- 1Madden T. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014:718-726. doi:10.1097 /aog.0000000000000475.
- Kaunitz AM, Stern L, Doyle J, et al. Use of the levonorgestrelIUD in the treatment of menorrhagia: improving patient outcomes while reducing the need for surgical management. Manag Care Interface. 2007;20:47-50.
- Getahun D, Fassett MJ, Gatz J, et al. Association between menorrhagia and risk of intrauterine device-related uterine perforation and device expulsion: results from the Association of Uterine Perforation and Expulsion of Intrauterine Device study. Am J Obstet Gynecol. 2022;227:59.e1-59.e9.
- Benacerraf BR, Shipp TD, Bromley B. Three-dimensional ultrasound detection of abnormally located intrauterine contraceptive devices that are a source of pelvic pain and abnormal bleeding. Ultrasound Obstet Gynecol. 2009;34:110115.
- Shipp TD, Bromley B, Benacerraf BR. The width of the uterine cavity is narrower in patients with an embedded intrauterine device (IUD) compared to a normally positioned IUD. J Ultrasound Med. 2010;29:1453-1456.
- Depypere H, Inki P. The levonorgestrel-releasing intrauterine system for endometrial protection during estrogen replacement therapy: a clinical review. Climacteric. 2015;18:470-482.
- Minalt N, Caldwell A, Yedlicka GM, et al. Association of intrauterine device use and endometrial, cervical, and ovarian cancer: an expert review. Am J Obstet Gynecol. 2023:S0002-9378(23)00224-7.
- Balayla J, Gil Y, Lasry A, et al. Ever-use of the intra-uterine device and the risk of ovarian cancer. J Obstet Gynaecol. 2021;41:848-853. doi: 10.1080/01443615.2020.1789960.
- Manson JE, Aragaki AK, Rossouw JE, et al. Menopausal hormone therapy and long-term all-cause and cause-specific mortality: the Women’s Health Initiative randomized trials. JAMA. 2017;318:927-938. doi:10.1001/jama.2017.11217.
- Chen WY, Manson JE, Hankinson SE, et al. Unopposed estrogen therapy and the risk of invasive breast cancer. Arch Intern Med. 2006;166:1027-1032. doi: 10.1001 /archinte.166.9.1027.
- Pinkerton JV, Wilson CS, Kaunitz AM. Reassuring data regarding the use of hormone therapy at menopause and risk of breast cancer. Menopause. 2022;29:1001-1004.doi:10.1097 /GME.0000000000002057.
- Romano SS, Doll KM. The impact of fibroids and histologic subtype on the performance of US clinical guidelines for the diagnosis of endometrial cancer among Black women. Ethn Dis. 2020;30:543-552. doi: 10.18865/ed.30.4.543.
- ACOG Committee on Practice Bulletins—Gynecology. Practice bulletin no. 128: diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206. doi: 10.1097/AOG.0b013e318262e320.
- Backman T, Rauramo I, Jaakkola Kimmo, et al. Use of the levonorgestrel-releasing intrauterine system and breast cancer. Obstet Gynecol. 2005;106:813-817.
- Conz L, Mota BS, Bahamondes L, et al. Levonorgestrelreleasing intrauterine system and breast cancer risk: A systematic review and meta-analysis. Acta Obstet Gynecol Scand. 2020;99:970-982.
- Al Kiyumi MH, Al Battashi K, Al-Riyami HA. Levonorgestrelreleasing intrauterine system and breast cancer. Is there an association? Acta Obstet Gynecol Scand. 2021;100:1749.
- Marsden J. Hormonal contraception and breast cancer, what more do we need to know? Post Reprod Health. 2017;23:116127. doi: 10.1177/2053369117715370.
- Vinogradova Y, Coupland C, Hippisley-Cox J. Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ. 2019;364:k4810 doi:10.1136/bmj.k4810.
- Levitt L, Cox C, Dawson L. Q&A: implications of the ruling on the ACA’s preventive services requirement. KFF.org. https://www .kff.org/policy-watch/qa-implications-of-the-ruling-on -the-acas-preventive-services-requirement/#:~:text=On%20 March%2030%2C%202023%2C%20a,cost%2Dsharing%20 for%20their%20enrollees. Accessed May 2, 2023.
MODERATOR

JoAnn V. Pinkerton, MD, NCMP, is Professor, Obstetrics and Gynecology, Division Director of Midlife Health, University of Virginia; Emeritus Executive Director and Past President, North American Menopause Society and recipient of SAAOG 2022 Lifetime Achievement Award.
PARTICIPANTS

Barbara S. Levy, MD, Clinical Professor, Obstetrics and Gynecology, George Washington University School of Medicine and Health Sciences; prior Vice President, Health Policy for the American College of Obstetrics and Gynecology (ACOG); Modern Healthcare Magazine’s 1 of the 50 most influential physician executives and leaders, 2015; 2013 recipient, Lifetime Achievement Award, OBG Management.

Andrew M. Kaunitz, MD, NCMP, Professor and Associate Chair, Obstetrics and Gynecology, University of Florida College of Medicine, Jacksonville. Associate Chair, and recipient, American College of Obstetrics and Gynecology Distinguished Service Award.

Steven R. Goldstein, MD, NCMP, CCD Immediate Past President, International Menopause Society; Past President, NAMS; recipient, NAMS Thomas Clarkson Outstanding Clinical and Basic Science Award; Past President, American Institute of Ultrasound in Medicine (AIUM); recipient, Joseph Holmes Pioneer Award.
The authors report no financial relationships relevant to this article.
MODERATOR
JoAnn V. Pinkerton, MD, NCMP, is Professor, Obstetrics and Gynecology, Division Director of Midlife Health, University of Virginia; Emeritus Executive Director and Past President, North American Menopause Society and recipient of SAAOG 2022 Lifetime Achievement Award.
PARTICIPANTS
Barbara S. Levy, MD, Clinical Professor, Obstetrics and Gynecology, George Washington University School of Medicine and Health Sciences; prior Vice President, Health Policy for the American College of Obstetrics and Gynecology (ACOG); Modern Healthcare Magazine’s 1 of the 50 most influential physician executives and leaders, 2015; 2013 recipient, Lifetime Achievement Award, OBG Management.
Andrew M. Kaunitz, MD, NCMP, Professor and Associate Chair, Obstetrics and Gynecology, University of Florida College of Medicine, Jacksonville. Associate Chair, and recipient, American College of Obstetrics and Gynecology Distinguished Service Award.
Steven R. Goldstein, MD, NCMP, CCD Immediate Past President, International Menopause Society; Past President, NAMS; recipient, NAMS Thomas Clarkson Outstanding Clinical and Basic Science Award; Past President, American Institute of Ultrasound in Medicine (AIUM); recipient, Joseph Holmes Pioneer Award.
The authors report no financial relationships relevant to this article.
MODERATOR
JoAnn V. Pinkerton, MD, NCMP, is Professor, Obstetrics and Gynecology, Division Director of Midlife Health, University of Virginia; Emeritus Executive Director and Past President, North American Menopause Society and recipient of SAAOG 2022 Lifetime Achievement Award.
PARTICIPANTS
Barbara S. Levy, MD, Clinical Professor, Obstetrics and Gynecology, George Washington University School of Medicine and Health Sciences; prior Vice President, Health Policy for the American College of Obstetrics and Gynecology (ACOG); Modern Healthcare Magazine’s 1 of the 50 most influential physician executives and leaders, 2015; 2013 recipient, Lifetime Achievement Award, OBG Management.
Andrew M. Kaunitz, MD, NCMP, Professor and Associate Chair, Obstetrics and Gynecology, University of Florida College of Medicine, Jacksonville. Associate Chair, and recipient, American College of Obstetrics and Gynecology Distinguished Service Award.
Steven R. Goldstein, MD, NCMP, CCD Immediate Past President, International Menopause Society; Past President, NAMS; recipient, NAMS Thomas Clarkson Outstanding Clinical and Basic Science Award; Past President, American Institute of Ultrasound in Medicine (AIUM); recipient, Joseph Holmes Pioneer Award.
The authors report no financial relationships relevant to this article.
Intrauterine devices (IUDs) are now used by more than 15% of US contraceptors. The majority of these IUDs release the progestin levonorgestrel, and with now longer extended use of the IUDs approved by the US Food and Drug Administration (FDA),1-3 they become even more attractive for use for contraception,control of menorrhagia or heavy menstrual bleeding (HMB) during reproductive years and perimenopause, and potentially, although not FDA approved for this purpose, postmenopause for endometrial protection in estrogen users. In this roundtable discussion, we will look at some of the benefits of the IUD for contraception effectiveness and control of bleeding, as well as the potential risks if used for postmenopausal women.
Progestin IUDs and contraception
JoAnn V. Pinkerton, MD, NCMP: Dr. Kaunitz, what are the contraceptive benefits of progestin IUDs during perimenopause?
Andrew M. Kaunitz, MD, NCMP: We know fertility declines as women approach menopause. However, when pregnancy occurs in older reproductive-age women, the pregnancies are often unintended, as reflected by high rates of induced abortion in this population. In addition, the prevalence of maternal comorbidities (during pregnancy and delivery) is higher in older reproductive-age women, with the maternal mortality rate more than 5 times higher compared with that of younger women.4 Two recently published clinical trials assessed the extended use of full-size IUDs containing 52 mg of levonor-gestrel (LNG), with the brand names Mirena and Liletta.1,2 The data from these trials confirmed that both IUDs remain highly effective for up to 8 years of use, and currently, both devices are approved for up to 8 years of use. One caveat is that, in the unusual occurrence of a pregnancy being diagnosed in a woman using an IUD, we as clinicians, must be alert to the high prevalence of ectopic pregnancies in this setting.
Progestin IUDs and HMB
Dr. Pinkerton: Dr. Goldstein, can you comment on how well progestin IUDs work for HMB?
Steven R. Goldstein, MD, NCMP, CCD: Many women who need contraception will use these devices for suppressing HMB, and they can be quite effective, if the diagnosis truly is HMB, at reducing bleeding.5 But that efficacy in bleeding reduction may not be quite as long as the efficacy in pregnancy prevention.6 In my experience, among women using IUDs specifically for their HMB, good bleeding control may require changing the IUD at 3 to 5 years.
Barbara S. Levy, MD: When inserting a LNG-IUD for menorrhagia in the perimenopausal time frame, sometimes I will do a progestin withdrawal first, which will thin the endometrium and induce withdrawal bleeding because, in my experience, if you place an IUD in someone with perimenopausal bleeding, you may end up with a lot of breakthrough bleeding.
Perimenopause and hot flashes
Dr. Pinkerton: Dr. Kaunitz, we have learned that hot flashes often occur and become bothersome to women during perimenopause. Many women have IUDs placed during perimenopause for bleeding. Can you comment about IUD use during perimenopause and postmenopause?
Dr. Kaunitz: In older reproductive-age women who already have a progestin-releasing IUD placed, as they get closer to menopause when vasomotor symptoms (VMS) might occur, if these symptoms are bothersome, the presence or placement of a progestin-releasing IUD can facilitate treatment of perimenopausal VMS with estrogen therapy.
Progestin IUDs cause profound endometrial suppression, reduce bleeding and often, over time, cause users to become amenorrheic.7
The Mirena package insert states, “Amenorrhea develops in about 20% of users by one year.”2 By year 3 and continuing through year 8, the prevalence of amenorrhea with the 52-mg LNG-IUD is 35% to 40%.8 From a study by Nanette Santoro, MD, and colleagues, we know that, in perimenopausal women with a progestin-releasing IUD in place, who are experiencing bothersome VMS, adding transdermal estrogen is very effective in treating and suppressing those hot flashes. In her small clinical trial, among participants with perimenopausal bothersome VMS with an IUD in place, half were randomized to use of transdermal estradiol and then compared with women who did not get the estradiol patch. There was excellent relief of perimenopausal hot flashes with the combination of the progestin IUD for endometrial suppression and transdermal estrogen to relieve hot flashes.9
Dr. Pinkerton: Which women would not be good candidates for the use of this combination?
Dr. Kaunitz: We know that, as women age, the prevalence of conditions that are contraindications to combination contraceptives (estrogen-progestin pills, patches, or rings) starts to increase. Specifically, we see more: hypertension, diabetes, and high body mass index (BMI), or obesity. We also know that migraine headaches in women older than age 35 years is another condition in which ACOG and the Centers for Disease Control and Prevention (CDC) would not recommend use of combination contraceptives.10,11 These older perimenopausal women may be excellent candidates for a progestin-only releasing IUD combined with use of transdermal menopausal doses of estradiol if needed for VMS.
Dr. Goldstein: I do want to add that, in those patients who don’t have these comorbidities, combination estrogen-progestin contraceptives do a very nice job of ovarian suppression and will prevent the erratic production of estradiol, which, in my experience, often results in not only irregular bleeding but also possible exacerbation of perimenopausal mood symptoms.
Dr. Kaunitz: I agree, Steve. The ideal older reproductive-age candidate for combination pills, patch, or ring would be a slender, healthy, nonsmoking woman with normal blood pressure. Such women would be a fairly small subgroup of my practice, but they can safely continue combination contraceptives right through menopause. Consistent with CDC and ACOG guidance, rather than checking gonadotropins to “determine when menopause has occurred,” (which is, in fact, not an evidence-based approach to diagnosing menopause in this setting), such women can continue the combination contraceptive right up until age 55—the likelihood that women are still going to be ovulating or at risk for pregnancy becomes vanishingly small at that age.11,12 Women in their mid-50s can either seamlessly transition to use of systemic estrogen-progestin menopausal therapy or go off hormones completely.
Continue to: The IUD and HMB...
The IUD and HMB
Dr. Pinkerton: Dr. Goldstein, there’s been some good literature on the best management options for women with HMB. What is the most current evidence?
Dr. Goldstein: I think that the retiring of the terms menorrhagia and metrorrhagia may have been premature because HMB implies cyclical bleeding, and this population of women with HMB will typically do quite well. Women who have what we used to call metrorrhagia or irregular bleeding, by definition, need endometrial evaluation to be sure they don’t have some sort of organic pathology. It would be a mistake for clinicians to use an LNG-IUD in patients with abnormal uterine bleeding (AUB) that has not been appropriately evaluated.
If we understand that we are discussing HMB, a Cochrane Review from 202213 suggests that an LNG intrauterine system is the best first-line treatment for reducing menstrual blood loss in perimenopausal women with HMB. Antifibrinolytics appeared second best, while long-cycle progestogens came in third place. Evidence on perception of improvement in satisfaction was ranked as low certainty. That same review found that hysterectomy was the best treatment for reducing bleeding, obviously, followed by resectoscopic endometrial ablation or a nonresectoscopic global endometrial ablation.
The evidence rating was low certainty regarding the likelihood that placing an LNG-IUD in women with HMB will result in amenorrhea, and I think that’s a very important point. The expectation of patients should be reduced or a significantly reduced amount of their HMB, not necessarily amenorrhea. Certainly, minimally invasive hysterectomy will result in total amenorrhea and may have a larger increase in satisfaction, but it has its own set of other kinds of possible complications.
Dr. Kaunitz: In an industry-funded, international multicenter trial,14 women with documented HMB (hemoglobin was eluted from soiled sanitary products), with menstrual blood loss of 80 mL or more per cycle, were randomized to placement of an LNG 52-mg IUD (Mirena) or cyclical medroxyprogesterone acetate (MPA)—oral progestin use.
Although menstrual blood loss declined in both groups, it declined dramatically more in women with an IUD placed, and specifically with the IUD, menstrual blood loss declined by 129 mL on average, whereas the decline in menstrual blood loss with cyclical MPA was 18 mL. This data, along with earlier European data,15 which showed similar findings in women with HMB led to the approval of the Mirena progestin IUD for a second indication to treat HMB in 2009.
I also want to point out that, in the May 2023 issue of Obstetrics & Gynecology, Creinin and colleagues published a similar trial in women with HMB showing, once again, that progestin IUDs (52-mg LNG-IUD, Liletta) are extremely effective in reducing HMB.16 There is crystal clear evidence from randomized trials that both 52-mg LNG-IUDs, Mirena and Liletta, are very effective in reducing HMB and, in fact, are contributing to many women who in the past would have proceeded with surgery, such as ablation or hysterectomy, to control their HMB.
Oral contraception
Dr. Pinkerton: What about using low-dose continuous oral contraceptives noncyclically for women with HMB?
Dr. Goldstein: I do that all the time. It is interesting that Dr. Kaunitz mentions his patient population. It’s why we understand that one size does not fit all. You need to see patients one at a time, and if they are good candidates for a combined estrogen-progestin contraception, whether it’s pills, patches, or rings, giving that continuously does a very nice job in reducing HMB and straightening out some of the other symptoms that these perimenopausal women will have.
IUD risks
Dr. Pinkerton: We all know about use of low-dose oral contraceptives for management of AUB, and we use them, although we worry a little bit about breast cancer risk. Dr. Levy, please comment on the risks with IUDs of expulsions and perforations. What are the downsides of IUDs?
Dr. Levy: Beyond the cost, although it is a minimally invasive procedure, IUD insertion can be an invasive procedure for a patient to undergo; expulsions can occur.17 We know that a substantial percentage of perimenopausal women will have fibroids. Although many fibroids are not located in the uterine cavity, the expulsion rate with HMB for an LNG-IUD can be higher,13,16,18,19 perhaps because of local prostaglandin release with an increase in uterine contractility. There is a low incidence of perforations, but they do happen, particularly among women with scars in the uterus or who have a severely anteflexed or retroflexed uterus, and women with cervical stenosis, for example, if they have had a LEEP procedure, etc. Even though progestin IUDs are outstanding tools in our toolbox, they are invasive to some extent, and they do have the possibility of complications.
Dr. Kaunitz: As Dr. Levy points out, although placement of an IUD may be considered an invasive procedure, it is also an office-based procedure, so women can drive home or drive back to work afterwards without the disruption in their life and the potential complications associated with surgery and anesthesia.
Continue to: Concerns with malpositioning...
Concerns with malpositioning
Dr. Pinkerton: After placement of an IUD, during a follow-up visit, sometimes you can’t visualize the string. The ultrasonography report may reveal, “IUD appears to be in the right place within the endometrium.” Dr. Goldstein, can you comment on how we should use ultrasound when we can’t visualize or find the IUD string, or if the patient complains of abdominal pain, lower abdominal discomfort, or irregular bleeding or spotting and we become concerned about IUD malposition?
Dr. Goldstein: Ultrasound is not really required after an uncomplicated placement of an IUD or during routine management of women who have no problems who are using an IUD. In patients who present with pain or some abnormal bleeding, however, sometimes it is the IUD being malpositioned. A very interesting study by the late great Beryl Benacerraf20 showed that there was a statistically significant higher incidence of the IUD being poorly positioned when patients have pain or bleeding (FIGURE 1). It was not always apparent on 2D ultrasonography. Using a standard transvaginal ultrasound of the long access plane, the IUD may appear to be very centrally located. However, if you do a 3D coronal section, not infrequently in these patients with any pain or bleeding, one of the arms has pierced the myometrium (FIGURE 2). This can actually be a source of pain and bleeding.
It’s also very interesting when you talk about perforation. I became aware of a big to-do in the medical/legal world about the possibility of the IUD migrating through the uterine cavity.21 This just does not exist, as was already pointed out. If the IUD is really going to go anywhere, if it’s properly placed, it’s going to be expelled through an open cervix. I do believe that, if you have pierced the myometrium through uterine contractility over time, some of these IUDs could work their way through the myometrium and somehow come out of the uterus either totally or partially. I think ultrasound is invaluable in patients with pain and bleeding, but I think you need to have an ultrasound lab capable of doing a 3D coronal section.
Progestin IUDs for HT replacement: Benefits/risks
Dr. Pinkerton: Many clinicians are excited that they can use essentially estrogen alone for women who have a progestin IUD in place. What about the possible off-label use of the progestin IUD to replace oral progestogen for hormone therapy (HT)? Dr. Kaunitz, are there any studies using this for postmenopausal HT (with a reminder that the IUD is not FDA approved for this purpose)?
Dr. Kaunitz: We have data from Europe indicating that, in menopausal women using systemic estrogen, the full-size LNG 52 IUD—Mirena or Liletta—provides excellent endometrial suppression.22 Where we don’t have data is with the smaller IUDs, which would be Kyleena and Skyla, which release smaller amounts of progestin each day into the endometrial cavity.
I have a number of patients, most of them women who started use of a progestin IUD as older reproductive-age women and then started systemic estrogen for treatment of perimenopausal hot flashes and then continued the use of their IUD plus systemic estrogen in treating postmenopausal hot flashes. The IUD is very useful in this setting, but as you pointed out, Dr. Pinkerton, this does represent off-label use.
Dr. Pinkerton: I know this use does not affect plasma lipids or cardiovascular risk markers, although users seem to report that the IUD has improved their quality of life. The question comes up, what are the benefits on cancer risk for using an IUD?
Dr. Levy: It’s such a great question because, as we talk about the balance of risks and benefits for anything that we are offering to our patients, it is really important to focus on some of the benefits. For both the copper and the LNG-IUD, there is a reduction in endometrial cancer,22 as well as pretty good data with the copper IUD about a reduction in cervical cancer.23 Those data are a little bit less clear for the LNG-IUD.
Interestingly, at least one meta-analysis published in 2020 shows about a 30% reduction in ovarian cancer risk with the LNG-IUD.24 We need to focus our patients on these other benefits. They tend to focus on the risks, and, of course, the media blows up the risks, but the benefits are quite substantial beyond just reducing HMB and providing contraception.
Dr. Pinkerton: As Dr. Kaunitz said, when you use this IUD, with its primarily local uterine progestin effects, it’s more like using estrogen alone without as much systemic progestin. Recently I wrote an editorial on the benefits of estrogen alone on the risk of breast cancer, primarily based on the Women’s Health Initiative (WHI) observational long-term 18-year cumulative follow-up. When estrogen alone was prescribed to women after a hysterectomy, estrogen therapy used at menopause did not increase the risk of invasive breast cancer, and was associated with decreased mortality.25 However, the nurse’s health study has suggested that longer-term use may be increased with estrogen alone.26 For women in the WHI with an intact uterus who used estrogen, oral MPA slightly increased the risk for breast cancer, and this elevated risk persisted even after discontinuation. This leads us to the question, what are the risks of breast cancer with progestin IUD use?
I recently reviewed the literature, and the answer is, it’s mixed. The FDA has put language into the package label that acknowledges a potential breast cancer risk for women who use a progestin IUD,27 and that warning states, “Women who currently have or have had breast cancer or suspect breast cancer should not use hormonal contraception because some breast cancers are hormone sensitive.” The label goes on to say, “Observational studies of the risk of breast cancer with the use of a levonorgestrel-releasing IUS don’t provide conclusive evidence of increased risk.” Thus, there is no conclusive answer as to whether there is a possible link of progestin IUDs to breast cancer.
What I tell my patients is that research is inconclusive. However, it’s unlikely for a 52-mg LNG-IUD to significantly increase a woman’s breast cancer risk, except possibly in those already at high risk from other risk factors. I tell them that breast cancer is listed in the package insert as a potential risk. I could not find any data on whether adding a low-dose estradiol patch would further increase that risk. So I counsel women about potential risk, but tell them that I don’t have any strong evidence of risk.
Continue to: Dr. Goldstein...
Dr. Goldstein: If you look in the package insert for Mirena,2 similar to Liletta, certainly the serum levels of LNG are lower than that for combination oral contraceptives. For the IUD progestins, they are not localized only to the uterus, and LNG levels range from about 150 to 200 µg/mL up to 60 months. It’s greater at 12 months, at about 180 µg/mL,at 24 months it was 192 µg/mL, and by 60 months it was 159 µg/mL. It’s important to realize that there is some systemic absorption of progestin with progestin IUDs, and it is not completely a local effect.
JoAnn, you mentioned the WHI data,25 and just to specify, it was not the estrogen-only arm, it was the conjugated equine estrogen-only arm of the WHI. I don’t think that estradiol alone increases breast cancer risk (although there are no good prospective, follow-through, 18-year study data, like the WHI), but I think readers need to understand the difference in the estrogen type.
Endometrial evaluation. My question for the panel is as follows. I agree that the use of the progestin-releasing IUD is very nice for that transition to menopause. I do believe it provides endometrial protection, but we know from other studies that, when we give continuous combined HT, about 21% to 26% of patients will experience some bleeding/staining, responding in the first 4-week cycles, and it can be as high as 9% at 1 year. If I have a patient who bleeds on continuous combined HT, I will evaluate her endometrium, usually just with a simple transvaginal ultrasound. If an IUD is in place, and the patient now begins to have some irregular bleeding, how do you evaluate her with the IUD in place?
Dr. Levy: That is a huge challenge. We know from a recent paper,28 that the endometrial thickness, while an excellent measure for Caucasian and European women, may be a poor marker for endometrial pathology in African-American women. What we thought we knew, which was, if the stripe is 4 mL or less, we can forget about it, I think in our more recent research that is not so true. So you bring up a great point, what do you do? The most reliable evaluation will be with an office hysteroscopy, where you can really look at the entire cavity and for tiny, little polyps and other things. But then we are off label because the use of hysteroscopy with an IUD in place is off label. So we are really in a conundrum.
Dr. Pinkerton: Also, if you do an endometrial biopsy, you might dislodge the IUD. If you think that you are going to take the IUD out, it may not matter if you dislodge it. I will often obtain a transvaginal ultrasound to help me figure out the next step, and maybe look at the dosing of the estrogen and progestin—but you can’t monitor an IUD with blood levels. You are in a vacuum of trying to figure out the best thing to do.
Dr. Kaunitz: One of the hats I wear here in Jacksonville is Director of GYN Ultrasound. I have a fair amount of experience doing endometrial biopsies in women with progestin IUDs in place under abdominal ultrasound guidance and keeping a close eye on the position of the IUD. In the first dozen or so such procedures I did, I was quite concerned about dislodging the IUD. It hasn’t happened yet, and it gives me some reassurance to be able to image the IUD and your endometrial suction curette inside the cavity as you are obtaining endometrial sampling. I have substantial experience now doing that, and so far, no problems. I do counsel all such women in advance that there is some chance I could dislodge their IUD.
Dr. Goldstein: In addition to dislodging the IUD, are you not concerned that, if the pathology is not global, that a blind endometrial sampling may be fraught with some error?
Dr. Kaunitz: The endometrium in women with a progestin-releasing IUD in place tends to be very well suppressed. Although one might occasionally find, for instance, a polyp in that setting, I have not run into, and I don’t expect to encounter going forward, endometrial hyperplasia or cancer in women with current use of a progestin IUD. It’s possible but unlikely.
Dr. Levy: The progestin IUD will counterbalance a type-1 endometrial cancer—an endometrial cancer related to hyperstimulation by estrogen. It will not do anything, to my knowledge, to counterbalance a type 2. I think the art of medicine is, you do the best you can with the first episode of bleeding, and if she persists in her bleeding, we have to persevere and continue to evaluate her.
Dr. Goldstein: I agree 100%.
Dr. Pinkerton: We all agree with you. That’s a really good point.
Continue to: Case examinations...
Case examinations
CASE 1 Woman with intramural fibroids
Dr. Pinkerton: Dr. Goldstein, you have a 48-year-old Black woman who has heavy but regular menstrual bleeding with multiple fibroids (the largest is about 4 to 5 cm, they look intramural, with some distortion of the cavity but not a submucous myoma, and the endometrial depth is 9 cm). Would you insert an IUD, and would you recommend an endometrial biopsy first?
Dr. Goldstein: I am not a huge fan of blind endometrial sampling, and I do think that we use the “biopsy” somewhat inappropriately since sampling is not a directed biopsy. This became obvious in the landmark paper by Guido et al in 1995 and was adopted by ACOG only in 2012.29 Cancers that occupy less than 50% of the endometrial surface area are often missed with such blind sampling. Thus I would not perform an endometrial biopsy first, but would rather rely on properly timed and performed transvaginal ultrasound to rule out any concurrent endometrial disease. I think a lot of patients who have HMB, not only because of their fibroids but also often just due to the surface area of their uterine cavity being increased—so essentially there is more blood volume when they bleed. However, you said that in this case the patient has regular menstrual bleeding, so I am assuming that she is still ovulatory. She may have some adenomyosis. She may have a large uterine cavity. I think she is an excellent candidate for an LNG-releasing IUD to reduce menstrual blood flow significantly. It will not necessarily give her amenorrhea, and it may give her some irregular bleeding. Then at some distant point, say in 5 or 6 months if she does have some irregular staining or bleeding, I would feel much better about the fact that nothing has developed as long as I knew that the endometrium was devoid of pathology when I started.
CASE 2 Woman with family history of breast cancer
Dr. Pinkerton: Dr. Levy, a 44-year-old woman has a family history of breast cancer in her mother at age 72, but she still needs contraceptionbecause of that unintended pregnancy risk in the 40s, and she wants something that is not going to increase her risk of breast cancer. What would you use, and how would you counsel her if you decided to use a progestin IUD?
Dr. Levy: The data are mixed,30-33 but whatever the risk, it is miniscule, and I would bring up the CDC Medical Eligibility Criteria.11 For a patient with a family history of breast cancer, for use of the progestin IUD, it is a 1—no contraindications. What I tend to tell my patients is, if you are worried about breast cancer, watch how much alcohol you are drinking and maintain regular exercise. There are so many preventive things that we can do to reduce risk of breast cancer when she needs contraception. If there is any increase in risk, it is so miniscule that I would very strongly recommend a progestin IUD for her.
Dr. Pinkerton: In addition, in recognizing the different densities of breast, dense breast density could lead to supplemental screening, which also could give her some reassurance that we are adequately screening for breast cancer.
CASE 3 Woman with IUD and VMS
Dr. Pinkerton: Dr. Kaunitz, you have a 52-year-old overweight female. She has been using a progestin IUD for 4 years, is amenorrheic, but now she is having moderate to severe vasomotor symptoms despite the IUD in place. You have talked to her about risks and benefits of HT, and she is interested in starting it. I know we talked about the studies, but I want to know what you are going to tell her. How do you counsel her about off-label use?
Dr. Kaunitz: The most important issue related to treating vasomotor symptoms in this patient is the route of systemic estrogen. Understandably, women’s biggest concern regarding the risks of systemic estrogen-progestin therapy is breast cancer. However, statistically, by far the biggest risk associated with oral estrogen-progestogen therapy, is elevated risk of venous thrombosis and pulmonary embolism. We have seen this, with a number of studies, and the WHI made it crystal clear with risks of oral conjugated equine estrogen at the dose of 0.625 mg daily. Oral estradiol 1 mg daily is also associated with a similar elevated risk of venous thrombosis. We also know that age and BMI are both independent risk factors for thrombosis. So, for a woman in her 50s who has a BMI > 30 mg/kg2, I don’t want to further elevate her risk of thrombosis by giving her oral estrogen, whether it is estradiol or conjugated equine estrogen. This is a patient in whom I would be comfortable using transdermal (patch) estradiol, perhaps starting with a standard dose of 0.05 mg weekly or twice weekly patch, keeping in mind that 0.05 mg in the setting of transdermal estrogen refer to the daily or to the 24-hour release rate. The 1.0 mg of oral estradiol and 0.625 mg of conjugated equine estrogen refers to the mg quantity of estrogen in each tablet. This is a source of great confusion for clinicians.
If, during follow-up, the 0.05 mg estradiol patch is not sufficient to substantially reduce symptoms, we could go up, for instance, to a 0.075 mg estradiol patch. We know very clearly from a variety of observational studies, including a very large UK study,34 that in contrast with oral estrogen, transdermal estradiol is safer from the perspective of thrombosis.
Insurance coverage for IUDs
Dr. Pinkerton: Dr. Levy: Can you discuss IUDs and the Affordable Care Act’s requirement to cover contraceptive services?
Dr. Levy: Unfortunately, we do not know whether this benefit will continue based on a very recent finding from a judge in Texas that ruled the preventive benefits of the ACA were illegal.35 We don’t know what will happen going forward. What I will say is that, unfortunately, many insurance companies have not preserved the meaning of “cover all things,” so what we are finding is that, for example, they only have to cover one type in a class. The FDA defined 18 classes of contraceptives, and a hormonal IUD is one class, so they can decide that they are only going to cover one of the four IUDS. And then women don’t have access to the other three, some of which might be more appropriate for them than another.
The other thing very relevant to this conversation is that, if you use an ICD-10 code for menorrhagia, for HMB, it no longer lives within that ACA preventive care requirement of coverage for contraceptives, and now she is going to owe a big deductible or a copay. If you are practicing in an institution that does not allow the use of IUDs for contraception, like a Catholic institution where I used to practice, you will want to use that ICD-10 code for HMB. But if you want it offered with no out-of-pocket cost for the patient, you need to use the preventive medicine codes and the contraception code. These little nuances for us can make a huge difference for our patients.
Dr. Pinkerton: Thank you for that reminder. I want to thank our panelists, Dr. Levy, Dr. Goldstein, and Dr. Kaunitz, for providing us with such a great mix of evidence and expert opinion and also giving a benefit of their vast experience as award-winning gynecologists. Hopefully, today you have learned the benefits of the progestin IUD not only for contraception in reproductive years and perimenopause but also for treatment of HMB, and the potential benefit due to the more prolonged effectiveness of the IUDs for endometrial protection in postmenopause. This allows less progestin risk, essentially estrogen alone for postmenopausal HT. Unsolved questions remain about whether there is a risk of breast cancer with their use, but there is a clear benefit of protecting against pregnancy and endometrial cancer. ●
Intrauterine devices (IUDs) are now used by more than 15% of US contraceptors. The majority of these IUDs release the progestin levonorgestrel, and with now longer extended use of the IUDs approved by the US Food and Drug Administration (FDA),1-3 they become even more attractive for use for contraception,control of menorrhagia or heavy menstrual bleeding (HMB) during reproductive years and perimenopause, and potentially, although not FDA approved for this purpose, postmenopause for endometrial protection in estrogen users. In this roundtable discussion, we will look at some of the benefits of the IUD for contraception effectiveness and control of bleeding, as well as the potential risks if used for postmenopausal women.
Progestin IUDs and contraception
JoAnn V. Pinkerton, MD, NCMP: Dr. Kaunitz, what are the contraceptive benefits of progestin IUDs during perimenopause?
Andrew M. Kaunitz, MD, NCMP: We know fertility declines as women approach menopause. However, when pregnancy occurs in older reproductive-age women, the pregnancies are often unintended, as reflected by high rates of induced abortion in this population. In addition, the prevalence of maternal comorbidities (during pregnancy and delivery) is higher in older reproductive-age women, with the maternal mortality rate more than 5 times higher compared with that of younger women.4 Two recently published clinical trials assessed the extended use of full-size IUDs containing 52 mg of levonor-gestrel (LNG), with the brand names Mirena and Liletta.1,2 The data from these trials confirmed that both IUDs remain highly effective for up to 8 years of use, and currently, both devices are approved for up to 8 years of use. One caveat is that, in the unusual occurrence of a pregnancy being diagnosed in a woman using an IUD, we as clinicians, must be alert to the high prevalence of ectopic pregnancies in this setting.
Progestin IUDs and HMB
Dr. Pinkerton: Dr. Goldstein, can you comment on how well progestin IUDs work for HMB?
Steven R. Goldstein, MD, NCMP, CCD: Many women who need contraception will use these devices for suppressing HMB, and they can be quite effective, if the diagnosis truly is HMB, at reducing bleeding.5 But that efficacy in bleeding reduction may not be quite as long as the efficacy in pregnancy prevention.6 In my experience, among women using IUDs specifically for their HMB, good bleeding control may require changing the IUD at 3 to 5 years.
Barbara S. Levy, MD: When inserting a LNG-IUD for menorrhagia in the perimenopausal time frame, sometimes I will do a progestin withdrawal first, which will thin the endometrium and induce withdrawal bleeding because, in my experience, if you place an IUD in someone with perimenopausal bleeding, you may end up with a lot of breakthrough bleeding.
Perimenopause and hot flashes
Dr. Pinkerton: Dr. Kaunitz, we have learned that hot flashes often occur and become bothersome to women during perimenopause. Many women have IUDs placed during perimenopause for bleeding. Can you comment about IUD use during perimenopause and postmenopause?
Dr. Kaunitz: In older reproductive-age women who already have a progestin-releasing IUD placed, as they get closer to menopause when vasomotor symptoms (VMS) might occur, if these symptoms are bothersome, the presence or placement of a progestin-releasing IUD can facilitate treatment of perimenopausal VMS with estrogen therapy.
Progestin IUDs cause profound endometrial suppression, reduce bleeding and often, over time, cause users to become amenorrheic.7
The Mirena package insert states, “Amenorrhea develops in about 20% of users by one year.”2 By year 3 and continuing through year 8, the prevalence of amenorrhea with the 52-mg LNG-IUD is 35% to 40%.8 From a study by Nanette Santoro, MD, and colleagues, we know that, in perimenopausal women with a progestin-releasing IUD in place, who are experiencing bothersome VMS, adding transdermal estrogen is very effective in treating and suppressing those hot flashes. In her small clinical trial, among participants with perimenopausal bothersome VMS with an IUD in place, half were randomized to use of transdermal estradiol and then compared with women who did not get the estradiol patch. There was excellent relief of perimenopausal hot flashes with the combination of the progestin IUD for endometrial suppression and transdermal estrogen to relieve hot flashes.9
Dr. Pinkerton: Which women would not be good candidates for the use of this combination?
Dr. Kaunitz: We know that, as women age, the prevalence of conditions that are contraindications to combination contraceptives (estrogen-progestin pills, patches, or rings) starts to increase. Specifically, we see more: hypertension, diabetes, and high body mass index (BMI), or obesity. We also know that migraine headaches in women older than age 35 years is another condition in which ACOG and the Centers for Disease Control and Prevention (CDC) would not recommend use of combination contraceptives.10,11 These older perimenopausal women may be excellent candidates for a progestin-only releasing IUD combined with use of transdermal menopausal doses of estradiol if needed for VMS.
Dr. Goldstein: I do want to add that, in those patients who don’t have these comorbidities, combination estrogen-progestin contraceptives do a very nice job of ovarian suppression and will prevent the erratic production of estradiol, which, in my experience, often results in not only irregular bleeding but also possible exacerbation of perimenopausal mood symptoms.
Dr. Kaunitz: I agree, Steve. The ideal older reproductive-age candidate for combination pills, patch, or ring would be a slender, healthy, nonsmoking woman with normal blood pressure. Such women would be a fairly small subgroup of my practice, but they can safely continue combination contraceptives right through menopause. Consistent with CDC and ACOG guidance, rather than checking gonadotropins to “determine when menopause has occurred,” (which is, in fact, not an evidence-based approach to diagnosing menopause in this setting), such women can continue the combination contraceptive right up until age 55—the likelihood that women are still going to be ovulating or at risk for pregnancy becomes vanishingly small at that age.11,12 Women in their mid-50s can either seamlessly transition to use of systemic estrogen-progestin menopausal therapy or go off hormones completely.
Continue to: The IUD and HMB...
The IUD and HMB
Dr. Pinkerton: Dr. Goldstein, there’s been some good literature on the best management options for women with HMB. What is the most current evidence?
Dr. Goldstein: I think that the retiring of the terms menorrhagia and metrorrhagia may have been premature because HMB implies cyclical bleeding, and this population of women with HMB will typically do quite well. Women who have what we used to call metrorrhagia or irregular bleeding, by definition, need endometrial evaluation to be sure they don’t have some sort of organic pathology. It would be a mistake for clinicians to use an LNG-IUD in patients with abnormal uterine bleeding (AUB) that has not been appropriately evaluated.
If we understand that we are discussing HMB, a Cochrane Review from 202213 suggests that an LNG intrauterine system is the best first-line treatment for reducing menstrual blood loss in perimenopausal women with HMB. Antifibrinolytics appeared second best, while long-cycle progestogens came in third place. Evidence on perception of improvement in satisfaction was ranked as low certainty. That same review found that hysterectomy was the best treatment for reducing bleeding, obviously, followed by resectoscopic endometrial ablation or a nonresectoscopic global endometrial ablation.
The evidence rating was low certainty regarding the likelihood that placing an LNG-IUD in women with HMB will result in amenorrhea, and I think that’s a very important point. The expectation of patients should be reduced or a significantly reduced amount of their HMB, not necessarily amenorrhea. Certainly, minimally invasive hysterectomy will result in total amenorrhea and may have a larger increase in satisfaction, but it has its own set of other kinds of possible complications.
Dr. Kaunitz: In an industry-funded, international multicenter trial,14 women with documented HMB (hemoglobin was eluted from soiled sanitary products), with menstrual blood loss of 80 mL or more per cycle, were randomized to placement of an LNG 52-mg IUD (Mirena) or cyclical medroxyprogesterone acetate (MPA)—oral progestin use.
Although menstrual blood loss declined in both groups, it declined dramatically more in women with an IUD placed, and specifically with the IUD, menstrual blood loss declined by 129 mL on average, whereas the decline in menstrual blood loss with cyclical MPA was 18 mL. This data, along with earlier European data,15 which showed similar findings in women with HMB led to the approval of the Mirena progestin IUD for a second indication to treat HMB in 2009.
I also want to point out that, in the May 2023 issue of Obstetrics & Gynecology, Creinin and colleagues published a similar trial in women with HMB showing, once again, that progestin IUDs (52-mg LNG-IUD, Liletta) are extremely effective in reducing HMB.16 There is crystal clear evidence from randomized trials that both 52-mg LNG-IUDs, Mirena and Liletta, are very effective in reducing HMB and, in fact, are contributing to many women who in the past would have proceeded with surgery, such as ablation or hysterectomy, to control their HMB.
Oral contraception
Dr. Pinkerton: What about using low-dose continuous oral contraceptives noncyclically for women with HMB?
Dr. Goldstein: I do that all the time. It is interesting that Dr. Kaunitz mentions his patient population. It’s why we understand that one size does not fit all. You need to see patients one at a time, and if they are good candidates for a combined estrogen-progestin contraception, whether it’s pills, patches, or rings, giving that continuously does a very nice job in reducing HMB and straightening out some of the other symptoms that these perimenopausal women will have.
IUD risks
Dr. Pinkerton: We all know about use of low-dose oral contraceptives for management of AUB, and we use them, although we worry a little bit about breast cancer risk. Dr. Levy, please comment on the risks with IUDs of expulsions and perforations. What are the downsides of IUDs?
Dr. Levy: Beyond the cost, although it is a minimally invasive procedure, IUD insertion can be an invasive procedure for a patient to undergo; expulsions can occur.17 We know that a substantial percentage of perimenopausal women will have fibroids. Although many fibroids are not located in the uterine cavity, the expulsion rate with HMB for an LNG-IUD can be higher,13,16,18,19 perhaps because of local prostaglandin release with an increase in uterine contractility. There is a low incidence of perforations, but they do happen, particularly among women with scars in the uterus or who have a severely anteflexed or retroflexed uterus, and women with cervical stenosis, for example, if they have had a LEEP procedure, etc. Even though progestin IUDs are outstanding tools in our toolbox, they are invasive to some extent, and they do have the possibility of complications.
Dr. Kaunitz: As Dr. Levy points out, although placement of an IUD may be considered an invasive procedure, it is also an office-based procedure, so women can drive home or drive back to work afterwards without the disruption in their life and the potential complications associated with surgery and anesthesia.
Continue to: Concerns with malpositioning...
Concerns with malpositioning
Dr. Pinkerton: After placement of an IUD, during a follow-up visit, sometimes you can’t visualize the string. The ultrasonography report may reveal, “IUD appears to be in the right place within the endometrium.” Dr. Goldstein, can you comment on how we should use ultrasound when we can’t visualize or find the IUD string, or if the patient complains of abdominal pain, lower abdominal discomfort, or irregular bleeding or spotting and we become concerned about IUD malposition?
Dr. Goldstein: Ultrasound is not really required after an uncomplicated placement of an IUD or during routine management of women who have no problems who are using an IUD. In patients who present with pain or some abnormal bleeding, however, sometimes it is the IUD being malpositioned. A very interesting study by the late great Beryl Benacerraf20 showed that there was a statistically significant higher incidence of the IUD being poorly positioned when patients have pain or bleeding (FIGURE 1). It was not always apparent on 2D ultrasonography. Using a standard transvaginal ultrasound of the long access plane, the IUD may appear to be very centrally located. However, if you do a 3D coronal section, not infrequently in these patients with any pain or bleeding, one of the arms has pierced the myometrium (FIGURE 2). This can actually be a source of pain and bleeding.
It’s also very interesting when you talk about perforation. I became aware of a big to-do in the medical/legal world about the possibility of the IUD migrating through the uterine cavity.21 This just does not exist, as was already pointed out. If the IUD is really going to go anywhere, if it’s properly placed, it’s going to be expelled through an open cervix. I do believe that, if you have pierced the myometrium through uterine contractility over time, some of these IUDs could work their way through the myometrium and somehow come out of the uterus either totally or partially. I think ultrasound is invaluable in patients with pain and bleeding, but I think you need to have an ultrasound lab capable of doing a 3D coronal section.
Progestin IUDs for HT replacement: Benefits/risks
Dr. Pinkerton: Many clinicians are excited that they can use essentially estrogen alone for women who have a progestin IUD in place. What about the possible off-label use of the progestin IUD to replace oral progestogen for hormone therapy (HT)? Dr. Kaunitz, are there any studies using this for postmenopausal HT (with a reminder that the IUD is not FDA approved for this purpose)?
Dr. Kaunitz: We have data from Europe indicating that, in menopausal women using systemic estrogen, the full-size LNG 52 IUD—Mirena or Liletta—provides excellent endometrial suppression.22 Where we don’t have data is with the smaller IUDs, which would be Kyleena and Skyla, which release smaller amounts of progestin each day into the endometrial cavity.
I have a number of patients, most of them women who started use of a progestin IUD as older reproductive-age women and then started systemic estrogen for treatment of perimenopausal hot flashes and then continued the use of their IUD plus systemic estrogen in treating postmenopausal hot flashes. The IUD is very useful in this setting, but as you pointed out, Dr. Pinkerton, this does represent off-label use.
Dr. Pinkerton: I know this use does not affect plasma lipids or cardiovascular risk markers, although users seem to report that the IUD has improved their quality of life. The question comes up, what are the benefits on cancer risk for using an IUD?
Dr. Levy: It’s such a great question because, as we talk about the balance of risks and benefits for anything that we are offering to our patients, it is really important to focus on some of the benefits. For both the copper and the LNG-IUD, there is a reduction in endometrial cancer,22 as well as pretty good data with the copper IUD about a reduction in cervical cancer.23 Those data are a little bit less clear for the LNG-IUD.
Interestingly, at least one meta-analysis published in 2020 shows about a 30% reduction in ovarian cancer risk with the LNG-IUD.24 We need to focus our patients on these other benefits. They tend to focus on the risks, and, of course, the media blows up the risks, but the benefits are quite substantial beyond just reducing HMB and providing contraception.
Dr. Pinkerton: As Dr. Kaunitz said, when you use this IUD, with its primarily local uterine progestin effects, it’s more like using estrogen alone without as much systemic progestin. Recently I wrote an editorial on the benefits of estrogen alone on the risk of breast cancer, primarily based on the Women’s Health Initiative (WHI) observational long-term 18-year cumulative follow-up. When estrogen alone was prescribed to women after a hysterectomy, estrogen therapy used at menopause did not increase the risk of invasive breast cancer, and was associated with decreased mortality.25 However, the nurse’s health study has suggested that longer-term use may be increased with estrogen alone.26 For women in the WHI with an intact uterus who used estrogen, oral MPA slightly increased the risk for breast cancer, and this elevated risk persisted even after discontinuation. This leads us to the question, what are the risks of breast cancer with progestin IUD use?
I recently reviewed the literature, and the answer is, it’s mixed. The FDA has put language into the package label that acknowledges a potential breast cancer risk for women who use a progestin IUD,27 and that warning states, “Women who currently have or have had breast cancer or suspect breast cancer should not use hormonal contraception because some breast cancers are hormone sensitive.” The label goes on to say, “Observational studies of the risk of breast cancer with the use of a levonorgestrel-releasing IUS don’t provide conclusive evidence of increased risk.” Thus, there is no conclusive answer as to whether there is a possible link of progestin IUDs to breast cancer.
What I tell my patients is that research is inconclusive. However, it’s unlikely for a 52-mg LNG-IUD to significantly increase a woman’s breast cancer risk, except possibly in those already at high risk from other risk factors. I tell them that breast cancer is listed in the package insert as a potential risk. I could not find any data on whether adding a low-dose estradiol patch would further increase that risk. So I counsel women about potential risk, but tell them that I don’t have any strong evidence of risk.
Continue to: Dr. Goldstein...
Dr. Goldstein: If you look in the package insert for Mirena,2 similar to Liletta, certainly the serum levels of LNG are lower than that for combination oral contraceptives. For the IUD progestins, they are not localized only to the uterus, and LNG levels range from about 150 to 200 µg/mL up to 60 months. It’s greater at 12 months, at about 180 µg/mL,at 24 months it was 192 µg/mL, and by 60 months it was 159 µg/mL. It’s important to realize that there is some systemic absorption of progestin with progestin IUDs, and it is not completely a local effect.
JoAnn, you mentioned the WHI data,25 and just to specify, it was not the estrogen-only arm, it was the conjugated equine estrogen-only arm of the WHI. I don’t think that estradiol alone increases breast cancer risk (although there are no good prospective, follow-through, 18-year study data, like the WHI), but I think readers need to understand the difference in the estrogen type.
Endometrial evaluation. My question for the panel is as follows. I agree that the use of the progestin-releasing IUD is very nice for that transition to menopause. I do believe it provides endometrial protection, but we know from other studies that, when we give continuous combined HT, about 21% to 26% of patients will experience some bleeding/staining, responding in the first 4-week cycles, and it can be as high as 9% at 1 year. If I have a patient who bleeds on continuous combined HT, I will evaluate her endometrium, usually just with a simple transvaginal ultrasound. If an IUD is in place, and the patient now begins to have some irregular bleeding, how do you evaluate her with the IUD in place?
Dr. Levy: That is a huge challenge. We know from a recent paper,28 that the endometrial thickness, while an excellent measure for Caucasian and European women, may be a poor marker for endometrial pathology in African-American women. What we thought we knew, which was, if the stripe is 4 mL or less, we can forget about it, I think in our more recent research that is not so true. So you bring up a great point, what do you do? The most reliable evaluation will be with an office hysteroscopy, where you can really look at the entire cavity and for tiny, little polyps and other things. But then we are off label because the use of hysteroscopy with an IUD in place is off label. So we are really in a conundrum.
Dr. Pinkerton: Also, if you do an endometrial biopsy, you might dislodge the IUD. If you think that you are going to take the IUD out, it may not matter if you dislodge it. I will often obtain a transvaginal ultrasound to help me figure out the next step, and maybe look at the dosing of the estrogen and progestin—but you can’t monitor an IUD with blood levels. You are in a vacuum of trying to figure out the best thing to do.
Dr. Kaunitz: One of the hats I wear here in Jacksonville is Director of GYN Ultrasound. I have a fair amount of experience doing endometrial biopsies in women with progestin IUDs in place under abdominal ultrasound guidance and keeping a close eye on the position of the IUD. In the first dozen or so such procedures I did, I was quite concerned about dislodging the IUD. It hasn’t happened yet, and it gives me some reassurance to be able to image the IUD and your endometrial suction curette inside the cavity as you are obtaining endometrial sampling. I have substantial experience now doing that, and so far, no problems. I do counsel all such women in advance that there is some chance I could dislodge their IUD.
Dr. Goldstein: In addition to dislodging the IUD, are you not concerned that, if the pathology is not global, that a blind endometrial sampling may be fraught with some error?
Dr. Kaunitz: The endometrium in women with a progestin-releasing IUD in place tends to be very well suppressed. Although one might occasionally find, for instance, a polyp in that setting, I have not run into, and I don’t expect to encounter going forward, endometrial hyperplasia or cancer in women with current use of a progestin IUD. It’s possible but unlikely.
Dr. Levy: The progestin IUD will counterbalance a type-1 endometrial cancer—an endometrial cancer related to hyperstimulation by estrogen. It will not do anything, to my knowledge, to counterbalance a type 2. I think the art of medicine is, you do the best you can with the first episode of bleeding, and if she persists in her bleeding, we have to persevere and continue to evaluate her.
Dr. Goldstein: I agree 100%.
Dr. Pinkerton: We all agree with you. That’s a really good point.
Continue to: Case examinations...
Case examinations
CASE 1 Woman with intramural fibroids
Dr. Pinkerton: Dr. Goldstein, you have a 48-year-old Black woman who has heavy but regular menstrual bleeding with multiple fibroids (the largest is about 4 to 5 cm, they look intramural, with some distortion of the cavity but not a submucous myoma, and the endometrial depth is 9 cm). Would you insert an IUD, and would you recommend an endometrial biopsy first?
Dr. Goldstein: I am not a huge fan of blind endometrial sampling, and I do think that we use the “biopsy” somewhat inappropriately since sampling is not a directed biopsy. This became obvious in the landmark paper by Guido et al in 1995 and was adopted by ACOG only in 2012.29 Cancers that occupy less than 50% of the endometrial surface area are often missed with such blind sampling. Thus I would not perform an endometrial biopsy first, but would rather rely on properly timed and performed transvaginal ultrasound to rule out any concurrent endometrial disease. I think a lot of patients who have HMB, not only because of their fibroids but also often just due to the surface area of their uterine cavity being increased—so essentially there is more blood volume when they bleed. However, you said that in this case the patient has regular menstrual bleeding, so I am assuming that she is still ovulatory. She may have some adenomyosis. She may have a large uterine cavity. I think she is an excellent candidate for an LNG-releasing IUD to reduce menstrual blood flow significantly. It will not necessarily give her amenorrhea, and it may give her some irregular bleeding. Then at some distant point, say in 5 or 6 months if she does have some irregular staining or bleeding, I would feel much better about the fact that nothing has developed as long as I knew that the endometrium was devoid of pathology when I started.
CASE 2 Woman with family history of breast cancer
Dr. Pinkerton: Dr. Levy, a 44-year-old woman has a family history of breast cancer in her mother at age 72, but she still needs contraceptionbecause of that unintended pregnancy risk in the 40s, and she wants something that is not going to increase her risk of breast cancer. What would you use, and how would you counsel her if you decided to use a progestin IUD?
Dr. Levy: The data are mixed,30-33 but whatever the risk, it is miniscule, and I would bring up the CDC Medical Eligibility Criteria.11 For a patient with a family history of breast cancer, for use of the progestin IUD, it is a 1—no contraindications. What I tend to tell my patients is, if you are worried about breast cancer, watch how much alcohol you are drinking and maintain regular exercise. There are so many preventive things that we can do to reduce risk of breast cancer when she needs contraception. If there is any increase in risk, it is so miniscule that I would very strongly recommend a progestin IUD for her.
Dr. Pinkerton: In addition, in recognizing the different densities of breast, dense breast density could lead to supplemental screening, which also could give her some reassurance that we are adequately screening for breast cancer.
CASE 3 Woman with IUD and VMS
Dr. Pinkerton: Dr. Kaunitz, you have a 52-year-old overweight female. She has been using a progestin IUD for 4 years, is amenorrheic, but now she is having moderate to severe vasomotor symptoms despite the IUD in place. You have talked to her about risks and benefits of HT, and she is interested in starting it. I know we talked about the studies, but I want to know what you are going to tell her. How do you counsel her about off-label use?
Dr. Kaunitz: The most important issue related to treating vasomotor symptoms in this patient is the route of systemic estrogen. Understandably, women’s biggest concern regarding the risks of systemic estrogen-progestin therapy is breast cancer. However, statistically, by far the biggest risk associated with oral estrogen-progestogen therapy, is elevated risk of venous thrombosis and pulmonary embolism. We have seen this, with a number of studies, and the WHI made it crystal clear with risks of oral conjugated equine estrogen at the dose of 0.625 mg daily. Oral estradiol 1 mg daily is also associated with a similar elevated risk of venous thrombosis. We also know that age and BMI are both independent risk factors for thrombosis. So, for a woman in her 50s who has a BMI > 30 mg/kg2, I don’t want to further elevate her risk of thrombosis by giving her oral estrogen, whether it is estradiol or conjugated equine estrogen. This is a patient in whom I would be comfortable using transdermal (patch) estradiol, perhaps starting with a standard dose of 0.05 mg weekly or twice weekly patch, keeping in mind that 0.05 mg in the setting of transdermal estrogen refer to the daily or to the 24-hour release rate. The 1.0 mg of oral estradiol and 0.625 mg of conjugated equine estrogen refers to the mg quantity of estrogen in each tablet. This is a source of great confusion for clinicians.
If, during follow-up, the 0.05 mg estradiol patch is not sufficient to substantially reduce symptoms, we could go up, for instance, to a 0.075 mg estradiol patch. We know very clearly from a variety of observational studies, including a very large UK study,34 that in contrast with oral estrogen, transdermal estradiol is safer from the perspective of thrombosis.
Insurance coverage for IUDs
Dr. Pinkerton: Dr. Levy: Can you discuss IUDs and the Affordable Care Act’s requirement to cover contraceptive services?
Dr. Levy: Unfortunately, we do not know whether this benefit will continue based on a very recent finding from a judge in Texas that ruled the preventive benefits of the ACA were illegal.35 We don’t know what will happen going forward. What I will say is that, unfortunately, many insurance companies have not preserved the meaning of “cover all things,” so what we are finding is that, for example, they only have to cover one type in a class. The FDA defined 18 classes of contraceptives, and a hormonal IUD is one class, so they can decide that they are only going to cover one of the four IUDS. And then women don’t have access to the other three, some of which might be more appropriate for them than another.
The other thing very relevant to this conversation is that, if you use an ICD-10 code for menorrhagia, for HMB, it no longer lives within that ACA preventive care requirement of coverage for contraceptives, and now she is going to owe a big deductible or a copay. If you are practicing in an institution that does not allow the use of IUDs for contraception, like a Catholic institution where I used to practice, you will want to use that ICD-10 code for HMB. But if you want it offered with no out-of-pocket cost for the patient, you need to use the preventive medicine codes and the contraception code. These little nuances for us can make a huge difference for our patients.
Dr. Pinkerton: Thank you for that reminder. I want to thank our panelists, Dr. Levy, Dr. Goldstein, and Dr. Kaunitz, for providing us with such a great mix of evidence and expert opinion and also giving a benefit of their vast experience as award-winning gynecologists. Hopefully, today you have learned the benefits of the progestin IUD not only for contraception in reproductive years and perimenopause but also for treatment of HMB, and the potential benefit due to the more prolonged effectiveness of the IUDs for endometrial protection in postmenopause. This allows less progestin risk, essentially estrogen alone for postmenopausal HT. Unsolved questions remain about whether there is a risk of breast cancer with their use, but there is a clear benefit of protecting against pregnancy and endometrial cancer. ●
- Liletta [package insert]. Allergan; Irvine, California. November 2022.
- Mirena [package insert]. Bayer; Whippany, New Jersey. 2000.
- Kaunitz AM. Safe extended use of levonorgestrel 52-mg IUDs. November 11, 2022. https://www.medscape.com/ viewarticle/983680. Accessed May 8, 2023.
- Kaunitz AM. Clinical practice. Hormonal contraception in women of older reproductive age. N Engl J Med. 2008;358:1262-1270. doi: 10.1056/NEJMcp0708481.
- Tucker ME. IUD-released levonorgestrel eases heavy menstrual periods. Medscape. April 10, 2023. https://www .medscape.com/viewarticle/777406. Accessed May 2, 2023.
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice; Long-Acting Reversible Contraception Working Group. ACOG Committee Opinion No. 450: Increasing use of contraceptive implants and intrauterine devices to reduce unintended pregnancy. Obstet Gynecol. 2009;114:1434-1438.
- Critchley HO, Wang H, Jones RL, et al. Morphological and functional features of endometrial decidualization following long-term intrauterine levonorgestrel delivery. Hum Reprod. 1998;13:1218-1224. doi:10.1093/humrep/13.5.1218.
- Creinin MD, Schreiber CA, Turok DK, et al. Levonorgestrel 52 mg intrauterine system efficacy and safety through 8 years of use. Am J Obstet Gynecol. 2022;227:871.e1-871.e7. doi: 10.1016/j.ajog.2022.05.022.
- Santoro N, Teal S, Gavito C, et al. Use of a levonorgestrelcontaining intrauterine system with supplemental estrogen improves symptoms in perimenopausal women: a pilot study. Menopause. 2015;22:1301-1307. doi: 10.1097 /GME.0000000000000557.
- ACOG Committee on Practice Bulletins-Gynecology ACOG Practice Bulletin. The use of hormonal contraception in women with coexisting medical conditions. Number 18, July 2000. Int J Gynaecol Obstet. 2001;75:93-106. doi: 10.1016 /s0020-7292(01)00520-3.
- Curtis KM, Tepper NK, Jatlaoui TC, Berry-Bibee E, Horton LG, Zapata LB, Simmons KB, Pagano HP, Jamieson DJ, Whiteman MK. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. 2016;65:1-103. doi: 10.15585 /mmwr.rr6503a1.
- ACOG Practice Bulletin No. 206: use of hormonal contraception in women with coexisting medical conditions [published correction appears in: Obstet Gynecol. 2019;133:1288.] Obstet Gynecol. 2019;133:e128-e150. doi:10.1097/AOG.0000000000003072.
- Bofill Rodriguez M, Dias S, Jordan V, et al. Interventions for heavy menstrual bleeding; overview of Cochrane reviews and network meta-analysis. Cochrane Database Syst Rev. 2022;5:CD013180. doi: 10.1002/14651858.CD013180.pub2.
- Kaunitz AM, Bissonnette F, Monteiro I, et al. Levonorgestrelreleasing intrauterine system or medroxyprogesterone for heavy menstrual bleeding: a randomized controlled trial [published correction appears in: Obstet Gynecol. 2010;116:999]. Obstet Gynecol. 2010;116:625-632. doi: 10.1097 /AOG.0b013e3181ec622b.
- Milsom I, Andersson K, Andersch B, et al. A comparison of flurbiprofen, tranexamic acid, and a levonorgestrel-releasing intrauterine contraceptive device in the treatment of idiopathic menorrhagia. Am J Obstet Gynecol. 1991;164:879883. doi: 10.1016/s0002-9378(11)90533-x.
- Creinin MD, Barnhart KT, Gawron LM, et al. Heavy menstrual bleeding treatment with a levonorgestrel 52-mg intrauterine device. Obstet Gynecol. 2023;141:971-978. doi: 10.1097 /AOG.0000000000005137.
- 1Madden T. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014:718-726. doi:10.1097 /aog.0000000000000475.
- Kaunitz AM, Stern L, Doyle J, et al. Use of the levonorgestrelIUD in the treatment of menorrhagia: improving patient outcomes while reducing the need for surgical management. Manag Care Interface. 2007;20:47-50.
- Getahun D, Fassett MJ, Gatz J, et al. Association between menorrhagia and risk of intrauterine device-related uterine perforation and device expulsion: results from the Association of Uterine Perforation and Expulsion of Intrauterine Device study. Am J Obstet Gynecol. 2022;227:59.e1-59.e9.
- Benacerraf BR, Shipp TD, Bromley B. Three-dimensional ultrasound detection of abnormally located intrauterine contraceptive devices that are a source of pelvic pain and abnormal bleeding. Ultrasound Obstet Gynecol. 2009;34:110115.
- Shipp TD, Bromley B, Benacerraf BR. The width of the uterine cavity is narrower in patients with an embedded intrauterine device (IUD) compared to a normally positioned IUD. J Ultrasound Med. 2010;29:1453-1456.
- Depypere H, Inki P. The levonorgestrel-releasing intrauterine system for endometrial protection during estrogen replacement therapy: a clinical review. Climacteric. 2015;18:470-482.
- Minalt N, Caldwell A, Yedlicka GM, et al. Association of intrauterine device use and endometrial, cervical, and ovarian cancer: an expert review. Am J Obstet Gynecol. 2023:S0002-9378(23)00224-7.
- Balayla J, Gil Y, Lasry A, et al. Ever-use of the intra-uterine device and the risk of ovarian cancer. J Obstet Gynaecol. 2021;41:848-853. doi: 10.1080/01443615.2020.1789960.
- Manson JE, Aragaki AK, Rossouw JE, et al. Menopausal hormone therapy and long-term all-cause and cause-specific mortality: the Women’s Health Initiative randomized trials. JAMA. 2017;318:927-938. doi:10.1001/jama.2017.11217.
- Chen WY, Manson JE, Hankinson SE, et al. Unopposed estrogen therapy and the risk of invasive breast cancer. Arch Intern Med. 2006;166:1027-1032. doi: 10.1001 /archinte.166.9.1027.
- Pinkerton JV, Wilson CS, Kaunitz AM. Reassuring data regarding the use of hormone therapy at menopause and risk of breast cancer. Menopause. 2022;29:1001-1004.doi:10.1097 /GME.0000000000002057.
- Romano SS, Doll KM. The impact of fibroids and histologic subtype on the performance of US clinical guidelines for the diagnosis of endometrial cancer among Black women. Ethn Dis. 2020;30:543-552. doi: 10.18865/ed.30.4.543.
- ACOG Committee on Practice Bulletins—Gynecology. Practice bulletin no. 128: diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206. doi: 10.1097/AOG.0b013e318262e320.
- Backman T, Rauramo I, Jaakkola Kimmo, et al. Use of the levonorgestrel-releasing intrauterine system and breast cancer. Obstet Gynecol. 2005;106:813-817.
- Conz L, Mota BS, Bahamondes L, et al. Levonorgestrelreleasing intrauterine system and breast cancer risk: A systematic review and meta-analysis. Acta Obstet Gynecol Scand. 2020;99:970-982.
- Al Kiyumi MH, Al Battashi K, Al-Riyami HA. Levonorgestrelreleasing intrauterine system and breast cancer. Is there an association? Acta Obstet Gynecol Scand. 2021;100:1749.
- Marsden J. Hormonal contraception and breast cancer, what more do we need to know? Post Reprod Health. 2017;23:116127. doi: 10.1177/2053369117715370.
- Vinogradova Y, Coupland C, Hippisley-Cox J. Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ. 2019;364:k4810 doi:10.1136/bmj.k4810.
- Levitt L, Cox C, Dawson L. Q&A: implications of the ruling on the ACA’s preventive services requirement. KFF.org. https://www .kff.org/policy-watch/qa-implications-of-the-ruling-on -the-acas-preventive-services-requirement/#:~:text=On%20 March%2030%2C%202023%2C%20a,cost%2Dsharing%20 for%20their%20enrollees. Accessed May 2, 2023.
- Liletta [package insert]. Allergan; Irvine, California. November 2022.
- Mirena [package insert]. Bayer; Whippany, New Jersey. 2000.
- Kaunitz AM. Safe extended use of levonorgestrel 52-mg IUDs. November 11, 2022. https://www.medscape.com/ viewarticle/983680. Accessed May 8, 2023.
- Kaunitz AM. Clinical practice. Hormonal contraception in women of older reproductive age. N Engl J Med. 2008;358:1262-1270. doi: 10.1056/NEJMcp0708481.
- Tucker ME. IUD-released levonorgestrel eases heavy menstrual periods. Medscape. April 10, 2023. https://www .medscape.com/viewarticle/777406. Accessed May 2, 2023.
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice; Long-Acting Reversible Contraception Working Group. ACOG Committee Opinion No. 450: Increasing use of contraceptive implants and intrauterine devices to reduce unintended pregnancy. Obstet Gynecol. 2009;114:1434-1438.
- Critchley HO, Wang H, Jones RL, et al. Morphological and functional features of endometrial decidualization following long-term intrauterine levonorgestrel delivery. Hum Reprod. 1998;13:1218-1224. doi:10.1093/humrep/13.5.1218.
- Creinin MD, Schreiber CA, Turok DK, et al. Levonorgestrel 52 mg intrauterine system efficacy and safety through 8 years of use. Am J Obstet Gynecol. 2022;227:871.e1-871.e7. doi: 10.1016/j.ajog.2022.05.022.
- Santoro N, Teal S, Gavito C, et al. Use of a levonorgestrelcontaining intrauterine system with supplemental estrogen improves symptoms in perimenopausal women: a pilot study. Menopause. 2015;22:1301-1307. doi: 10.1097 /GME.0000000000000557.
- ACOG Committee on Practice Bulletins-Gynecology ACOG Practice Bulletin. The use of hormonal contraception in women with coexisting medical conditions. Number 18, July 2000. Int J Gynaecol Obstet. 2001;75:93-106. doi: 10.1016 /s0020-7292(01)00520-3.
- Curtis KM, Tepper NK, Jatlaoui TC, Berry-Bibee E, Horton LG, Zapata LB, Simmons KB, Pagano HP, Jamieson DJ, Whiteman MK. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. 2016;65:1-103. doi: 10.15585 /mmwr.rr6503a1.
- ACOG Practice Bulletin No. 206: use of hormonal contraception in women with coexisting medical conditions [published correction appears in: Obstet Gynecol. 2019;133:1288.] Obstet Gynecol. 2019;133:e128-e150. doi:10.1097/AOG.0000000000003072.
- Bofill Rodriguez M, Dias S, Jordan V, et al. Interventions for heavy menstrual bleeding; overview of Cochrane reviews and network meta-analysis. Cochrane Database Syst Rev. 2022;5:CD013180. doi: 10.1002/14651858.CD013180.pub2.
- Kaunitz AM, Bissonnette F, Monteiro I, et al. Levonorgestrelreleasing intrauterine system or medroxyprogesterone for heavy menstrual bleeding: a randomized controlled trial [published correction appears in: Obstet Gynecol. 2010;116:999]. Obstet Gynecol. 2010;116:625-632. doi: 10.1097 /AOG.0b013e3181ec622b.
- Milsom I, Andersson K, Andersch B, et al. A comparison of flurbiprofen, tranexamic acid, and a levonorgestrel-releasing intrauterine contraceptive device in the treatment of idiopathic menorrhagia. Am J Obstet Gynecol. 1991;164:879883. doi: 10.1016/s0002-9378(11)90533-x.
- Creinin MD, Barnhart KT, Gawron LM, et al. Heavy menstrual bleeding treatment with a levonorgestrel 52-mg intrauterine device. Obstet Gynecol. 2023;141:971-978. doi: 10.1097 /AOG.0000000000005137.
- 1Madden T. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014:718-726. doi:10.1097 /aog.0000000000000475.
- Kaunitz AM, Stern L, Doyle J, et al. Use of the levonorgestrelIUD in the treatment of menorrhagia: improving patient outcomes while reducing the need for surgical management. Manag Care Interface. 2007;20:47-50.
- Getahun D, Fassett MJ, Gatz J, et al. Association between menorrhagia and risk of intrauterine device-related uterine perforation and device expulsion: results from the Association of Uterine Perforation and Expulsion of Intrauterine Device study. Am J Obstet Gynecol. 2022;227:59.e1-59.e9.
- Benacerraf BR, Shipp TD, Bromley B. Three-dimensional ultrasound detection of abnormally located intrauterine contraceptive devices that are a source of pelvic pain and abnormal bleeding. Ultrasound Obstet Gynecol. 2009;34:110115.
- Shipp TD, Bromley B, Benacerraf BR. The width of the uterine cavity is narrower in patients with an embedded intrauterine device (IUD) compared to a normally positioned IUD. J Ultrasound Med. 2010;29:1453-1456.
- Depypere H, Inki P. The levonorgestrel-releasing intrauterine system for endometrial protection during estrogen replacement therapy: a clinical review. Climacteric. 2015;18:470-482.
- Minalt N, Caldwell A, Yedlicka GM, et al. Association of intrauterine device use and endometrial, cervical, and ovarian cancer: an expert review. Am J Obstet Gynecol. 2023:S0002-9378(23)00224-7.
- Balayla J, Gil Y, Lasry A, et al. Ever-use of the intra-uterine device and the risk of ovarian cancer. J Obstet Gynaecol. 2021;41:848-853. doi: 10.1080/01443615.2020.1789960.
- Manson JE, Aragaki AK, Rossouw JE, et al. Menopausal hormone therapy and long-term all-cause and cause-specific mortality: the Women’s Health Initiative randomized trials. JAMA. 2017;318:927-938. doi:10.1001/jama.2017.11217.
- Chen WY, Manson JE, Hankinson SE, et al. Unopposed estrogen therapy and the risk of invasive breast cancer. Arch Intern Med. 2006;166:1027-1032. doi: 10.1001 /archinte.166.9.1027.
- Pinkerton JV, Wilson CS, Kaunitz AM. Reassuring data regarding the use of hormone therapy at menopause and risk of breast cancer. Menopause. 2022;29:1001-1004.doi:10.1097 /GME.0000000000002057.
- Romano SS, Doll KM. The impact of fibroids and histologic subtype on the performance of US clinical guidelines for the diagnosis of endometrial cancer among Black women. Ethn Dis. 2020;30:543-552. doi: 10.18865/ed.30.4.543.
- ACOG Committee on Practice Bulletins—Gynecology. Practice bulletin no. 128: diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206. doi: 10.1097/AOG.0b013e318262e320.
- Backman T, Rauramo I, Jaakkola Kimmo, et al. Use of the levonorgestrel-releasing intrauterine system and breast cancer. Obstet Gynecol. 2005;106:813-817.
- Conz L, Mota BS, Bahamondes L, et al. Levonorgestrelreleasing intrauterine system and breast cancer risk: A systematic review and meta-analysis. Acta Obstet Gynecol Scand. 2020;99:970-982.
- Al Kiyumi MH, Al Battashi K, Al-Riyami HA. Levonorgestrelreleasing intrauterine system and breast cancer. Is there an association? Acta Obstet Gynecol Scand. 2021;100:1749.
- Marsden J. Hormonal contraception and breast cancer, what more do we need to know? Post Reprod Health. 2017;23:116127. doi: 10.1177/2053369117715370.
- Vinogradova Y, Coupland C, Hippisley-Cox J. Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ. 2019;364:k4810 doi:10.1136/bmj.k4810.
- Levitt L, Cox C, Dawson L. Q&A: implications of the ruling on the ACA’s preventive services requirement. KFF.org. https://www .kff.org/policy-watch/qa-implications-of-the-ruling-on -the-acas-preventive-services-requirement/#:~:text=On%20 March%2030%2C%202023%2C%20a,cost%2Dsharing%20 for%20their%20enrollees. Accessed May 2, 2023.
Youth-led sexual health program improves teen knowledge, autonomy
BALTIMORE – , according to research presented at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists.
While the small pilot study focused primarily on assessing feasibility and effectiveness, the results suggest potential for scaling the program up to reach a larger audience and assessing the knowledge disseminated from direct youth participants.

“The good thing about this subject is that not a lot of it has to be context-specific,” Saumya Sao, a clinical researcher in gynecology and obstetrics at the Johns Hopkins University, Baltimore, and the study’s lead author, said in an interview. “A lot of it is just baseline information that everybody needs and doesn’t get.”
Jaime Friedman, MD, a pediatrician and director of marketing at Children’s Primary Care Medical Group in San Diego, was not involved in the study but was impressed with the program’s objectives and results so far.

“While education is massively important, teens don’t always want to hear it from their parents or other adults,” Dr. Friedman said in an interview. “Learning from their peers is one way to overcome this hurdle.”
Given the high rate of sexually transmitted infections and unintended pregnancies in youth, paired with low sexual and reproductive health literacy in this population, the researchers sought to learn whether a program focused on peer-to-peer health education on these topics was feasible. The goal was to increase youth sexual and reproductive health knowledge, self-efficacy, and autonomy using a youth-led intervention.
The researchers hosted nine monthly, interactive, youth-led sessions that lasted 2 hours over Zoom or in person. Incorporated into the meetings were principles from Youth Participatory Action Research (YPAR) and Positive Youth Development (PYD).
The major topics included the following: Use of social media, values and goal-setting, anatomy and menstrual health, risk factors of sexual activities , STI and HIV prevention, contraceptive methods, healthy relationships and consent, practice responding to unhealthy behavior, gender and sexuality, and social media and body image.
The 24 participants were provided with transportation to the study site at the researchers’ institution and received financial compensation for their participation. They were an average 15.8 years old, lived in the greater Baltimore area, and mostly self-identified as female. Eight percent identified as non-binary and half (50%) identified as LGBTQIA+. Just over half the participants (52%) were Black/African American, 28% were Asian/Asian American, 12% were White, and 8% were Hispanic. The participants attended an average 88% of the sessions throughout the full intervention.
For each of the nine sessions, more than 50% of participants reported that they “learned a lot,” and only one participant reported for one session (session 5) that they “didn’t learn” anything. The researchers assessed participants’ knowledge, self-efficacy, and sense of autonomy at baseline and after completion of the intervention. Significant improvements occurred across all areas.
The average score improved by 31% in sexual and reproductive health knowledge (P < .001), 33% in sexual and reproductive health services awareness (P = .002), 46% in advocacy and empowerment (P < .001), 16% in general perceived efficacy (P = .002), and 22% personal sexuality empowerment (P = .006).
Ms. Sao said she was very pleased to see that the improvements were significant in every domain they measured, which she attributed largely to the incorporation of YPAR and PYD into the program.
“We approached it using these two frameworks that really do focus on involving youth in the teaching themselves, so I think that’s what increased their general perceived efficacy and advocacy empowerment without us necessarily having to emphasize, ‘You are advocates,’” Ms. Sao said. “Those frameworks ask the youth for their opinions and then give the youth an opportunity in every single session to be teachers themselves, and I think that lends itself well to all of the domains.”
Ms. Sao was also pleasantly surprised at the high level of retention across the 9 months.
“Every single session was slotted for 2 hours, but they would want to stay for 3 hours. Eventually, we actually started meeting with them twice a month, just adding an extra session,” she said. “As they gained confidence, they were so excited to be peer educators and realized, ‘I can really do this. I can teach my peers. We’re not getting this from anywhere else.’ ”
Ms. Sao and another study author, Maclaine Barré-Quick, an undergraduate research assistant at Johns Hopkins University, said the participants quickly discovered how easy it was to have a non-stigmatizing conversation about many of the topics once a subject was brought up.
“They’re actively looking for that opportunity,” Ms. Barré-Quick said in an interview.
Dr. Friedman agreed that this type of program provides what many adolescents need in a way that they may welcome more than through other methods.
“Adolescents’ bodies are approaching adulthood and function like adults, but their brains are still developing. They don’t have the worldly experience and education of adults, but they think they know everything,” Dr. Friedman said. “They are a population known for their high risk behavior due to their natural impulsivity. This can be a scary combination, especially when it comes to sexual health.”
But if teens don’t want to hear some of the information they need from adults, they may be more open to hearing it from other teens, Dr. Friedman said.
“Using an evidence-based approach ensures the desired outcome of healthier habits, decreased STIs and decreased teen pregnancy,” Dr. Friedman said. “It also adds weight to the argument against abstinence-only education. Teens deserve accurate and evidence-based education about their own bodies.”
Ms. Sao said the next steps will be exploring ways to scale the program up, such as putting the curriculum resources into a bundle available to other educators. They’re also looking at ways to put it into an online platform that’s self-paced, though that requires solving the challenge of having synchronous meetings for youth-led discussion.
“There are certain kinks that we have to work out because there were some activities where I think the students really benefited from having those open discussions with each other, so [we need to determine] how to replicate that in an online format,” Ms. Sao said.
Dr. Friedman agreed that scalability appears to be the biggest challenge, along with funding programs. But if those obstacles can be overcome, such programs would complement and expand on the education she does currently with families.
“I don’t have time for a full sex ed course at each visit,” Dr. Friedman said. “I would like to be able to direct them to a program that I know works and would be easy for them to complete. Even better, this would be an amazing program to ‘sell’ to practices interested in hosting these sessions themselves.”
Ms. Sao said they also hope to assess the impact of the intervention on the participants’ peers to see how well the knowledge and self-efficacy spread through the youths’ teaching.
No external funding was noted. One author reported research support from Hologic and Merck. Dr. Friedman had no disclosures.
BALTIMORE – , according to research presented at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists.
While the small pilot study focused primarily on assessing feasibility and effectiveness, the results suggest potential for scaling the program up to reach a larger audience and assessing the knowledge disseminated from direct youth participants.
“The good thing about this subject is that not a lot of it has to be context-specific,” Saumya Sao, a clinical researcher in gynecology and obstetrics at the Johns Hopkins University, Baltimore, and the study’s lead author, said in an interview. “A lot of it is just baseline information that everybody needs and doesn’t get.”
Jaime Friedman, MD, a pediatrician and director of marketing at Children’s Primary Care Medical Group in San Diego, was not involved in the study but was impressed with the program’s objectives and results so far.
“While education is massively important, teens don’t always want to hear it from their parents or other adults,” Dr. Friedman said in an interview. “Learning from their peers is one way to overcome this hurdle.”
Given the high rate of sexually transmitted infections and unintended pregnancies in youth, paired with low sexual and reproductive health literacy in this population, the researchers sought to learn whether a program focused on peer-to-peer health education on these topics was feasible. The goal was to increase youth sexual and reproductive health knowledge, self-efficacy, and autonomy using a youth-led intervention.
The researchers hosted nine monthly, interactive, youth-led sessions that lasted 2 hours over Zoom or in person. Incorporated into the meetings were principles from Youth Participatory Action Research (YPAR) and Positive Youth Development (PYD).
The major topics included the following: Use of social media, values and goal-setting, anatomy and menstrual health, risk factors of sexual activities , STI and HIV prevention, contraceptive methods, healthy relationships and consent, practice responding to unhealthy behavior, gender and sexuality, and social media and body image.
The 24 participants were provided with transportation to the study site at the researchers’ institution and received financial compensation for their participation. They were an average 15.8 years old, lived in the greater Baltimore area, and mostly self-identified as female. Eight percent identified as non-binary and half (50%) identified as LGBTQIA+. Just over half the participants (52%) were Black/African American, 28% were Asian/Asian American, 12% were White, and 8% were Hispanic. The participants attended an average 88% of the sessions throughout the full intervention.
For each of the nine sessions, more than 50% of participants reported that they “learned a lot,” and only one participant reported for one session (session 5) that they “didn’t learn” anything. The researchers assessed participants’ knowledge, self-efficacy, and sense of autonomy at baseline and after completion of the intervention. Significant improvements occurred across all areas.
The average score improved by 31% in sexual and reproductive health knowledge (P < .001), 33% in sexual and reproductive health services awareness (P = .002), 46% in advocacy and empowerment (P < .001), 16% in general perceived efficacy (P = .002), and 22% personal sexuality empowerment (P = .006).
Ms. Sao said she was very pleased to see that the improvements were significant in every domain they measured, which she attributed largely to the incorporation of YPAR and PYD into the program.
“We approached it using these two frameworks that really do focus on involving youth in the teaching themselves, so I think that’s what increased their general perceived efficacy and advocacy empowerment without us necessarily having to emphasize, ‘You are advocates,’” Ms. Sao said. “Those frameworks ask the youth for their opinions and then give the youth an opportunity in every single session to be teachers themselves, and I think that lends itself well to all of the domains.”
Ms. Sao was also pleasantly surprised at the high level of retention across the 9 months.
“Every single session was slotted for 2 hours, but they would want to stay for 3 hours. Eventually, we actually started meeting with them twice a month, just adding an extra session,” she said. “As they gained confidence, they were so excited to be peer educators and realized, ‘I can really do this. I can teach my peers. We’re not getting this from anywhere else.’ ”
Ms. Sao and another study author, Maclaine Barré-Quick, an undergraduate research assistant at Johns Hopkins University, said the participants quickly discovered how easy it was to have a non-stigmatizing conversation about many of the topics once a subject was brought up.
“They’re actively looking for that opportunity,” Ms. Barré-Quick said in an interview.
Dr. Friedman agreed that this type of program provides what many adolescents need in a way that they may welcome more than through other methods.
“Adolescents’ bodies are approaching adulthood and function like adults, but their brains are still developing. They don’t have the worldly experience and education of adults, but they think they know everything,” Dr. Friedman said. “They are a population known for their high risk behavior due to their natural impulsivity. This can be a scary combination, especially when it comes to sexual health.”
But if teens don’t want to hear some of the information they need from adults, they may be more open to hearing it from other teens, Dr. Friedman said.
“Using an evidence-based approach ensures the desired outcome of healthier habits, decreased STIs and decreased teen pregnancy,” Dr. Friedman said. “It also adds weight to the argument against abstinence-only education. Teens deserve accurate and evidence-based education about their own bodies.”
Ms. Sao said the next steps will be exploring ways to scale the program up, such as putting the curriculum resources into a bundle available to other educators. They’re also looking at ways to put it into an online platform that’s self-paced, though that requires solving the challenge of having synchronous meetings for youth-led discussion.
“There are certain kinks that we have to work out because there were some activities where I think the students really benefited from having those open discussions with each other, so [we need to determine] how to replicate that in an online format,” Ms. Sao said.
Dr. Friedman agreed that scalability appears to be the biggest challenge, along with funding programs. But if those obstacles can be overcome, such programs would complement and expand on the education she does currently with families.
“I don’t have time for a full sex ed course at each visit,” Dr. Friedman said. “I would like to be able to direct them to a program that I know works and would be easy for them to complete. Even better, this would be an amazing program to ‘sell’ to practices interested in hosting these sessions themselves.”
Ms. Sao said they also hope to assess the impact of the intervention on the participants’ peers to see how well the knowledge and self-efficacy spread through the youths’ teaching.
No external funding was noted. One author reported research support from Hologic and Merck. Dr. Friedman had no disclosures.
BALTIMORE – , according to research presented at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists.
While the small pilot study focused primarily on assessing feasibility and effectiveness, the results suggest potential for scaling the program up to reach a larger audience and assessing the knowledge disseminated from direct youth participants.
“The good thing about this subject is that not a lot of it has to be context-specific,” Saumya Sao, a clinical researcher in gynecology and obstetrics at the Johns Hopkins University, Baltimore, and the study’s lead author, said in an interview. “A lot of it is just baseline information that everybody needs and doesn’t get.”
Jaime Friedman, MD, a pediatrician and director of marketing at Children’s Primary Care Medical Group in San Diego, was not involved in the study but was impressed with the program’s objectives and results so far.
“While education is massively important, teens don’t always want to hear it from their parents or other adults,” Dr. Friedman said in an interview. “Learning from their peers is one way to overcome this hurdle.”
Given the high rate of sexually transmitted infections and unintended pregnancies in youth, paired with low sexual and reproductive health literacy in this population, the researchers sought to learn whether a program focused on peer-to-peer health education on these topics was feasible. The goal was to increase youth sexual and reproductive health knowledge, self-efficacy, and autonomy using a youth-led intervention.
The researchers hosted nine monthly, interactive, youth-led sessions that lasted 2 hours over Zoom or in person. Incorporated into the meetings were principles from Youth Participatory Action Research (YPAR) and Positive Youth Development (PYD).
The major topics included the following: Use of social media, values and goal-setting, anatomy and menstrual health, risk factors of sexual activities , STI and HIV prevention, contraceptive methods, healthy relationships and consent, practice responding to unhealthy behavior, gender and sexuality, and social media and body image.
The 24 participants were provided with transportation to the study site at the researchers’ institution and received financial compensation for their participation. They were an average 15.8 years old, lived in the greater Baltimore area, and mostly self-identified as female. Eight percent identified as non-binary and half (50%) identified as LGBTQIA+. Just over half the participants (52%) were Black/African American, 28% were Asian/Asian American, 12% were White, and 8% were Hispanic. The participants attended an average 88% of the sessions throughout the full intervention.
For each of the nine sessions, more than 50% of participants reported that they “learned a lot,” and only one participant reported for one session (session 5) that they “didn’t learn” anything. The researchers assessed participants’ knowledge, self-efficacy, and sense of autonomy at baseline and after completion of the intervention. Significant improvements occurred across all areas.
The average score improved by 31% in sexual and reproductive health knowledge (P < .001), 33% in sexual and reproductive health services awareness (P = .002), 46% in advocacy and empowerment (P < .001), 16% in general perceived efficacy (P = .002), and 22% personal sexuality empowerment (P = .006).
Ms. Sao said she was very pleased to see that the improvements were significant in every domain they measured, which she attributed largely to the incorporation of YPAR and PYD into the program.
“We approached it using these two frameworks that really do focus on involving youth in the teaching themselves, so I think that’s what increased their general perceived efficacy and advocacy empowerment without us necessarily having to emphasize, ‘You are advocates,’” Ms. Sao said. “Those frameworks ask the youth for their opinions and then give the youth an opportunity in every single session to be teachers themselves, and I think that lends itself well to all of the domains.”
Ms. Sao was also pleasantly surprised at the high level of retention across the 9 months.
“Every single session was slotted for 2 hours, but they would want to stay for 3 hours. Eventually, we actually started meeting with them twice a month, just adding an extra session,” she said. “As they gained confidence, they were so excited to be peer educators and realized, ‘I can really do this. I can teach my peers. We’re not getting this from anywhere else.’ ”
Ms. Sao and another study author, Maclaine Barré-Quick, an undergraduate research assistant at Johns Hopkins University, said the participants quickly discovered how easy it was to have a non-stigmatizing conversation about many of the topics once a subject was brought up.
“They’re actively looking for that opportunity,” Ms. Barré-Quick said in an interview.
Dr. Friedman agreed that this type of program provides what many adolescents need in a way that they may welcome more than through other methods.
“Adolescents’ bodies are approaching adulthood and function like adults, but their brains are still developing. They don’t have the worldly experience and education of adults, but they think they know everything,” Dr. Friedman said. “They are a population known for their high risk behavior due to their natural impulsivity. This can be a scary combination, especially when it comes to sexual health.”
But if teens don’t want to hear some of the information they need from adults, they may be more open to hearing it from other teens, Dr. Friedman said.
“Using an evidence-based approach ensures the desired outcome of healthier habits, decreased STIs and decreased teen pregnancy,” Dr. Friedman said. “It also adds weight to the argument against abstinence-only education. Teens deserve accurate and evidence-based education about their own bodies.”
Ms. Sao said the next steps will be exploring ways to scale the program up, such as putting the curriculum resources into a bundle available to other educators. They’re also looking at ways to put it into an online platform that’s self-paced, though that requires solving the challenge of having synchronous meetings for youth-led discussion.
“There are certain kinks that we have to work out because there were some activities where I think the students really benefited from having those open discussions with each other, so [we need to determine] how to replicate that in an online format,” Ms. Sao said.
Dr. Friedman agreed that scalability appears to be the biggest challenge, along with funding programs. But if those obstacles can be overcome, such programs would complement and expand on the education she does currently with families.
“I don’t have time for a full sex ed course at each visit,” Dr. Friedman said. “I would like to be able to direct them to a program that I know works and would be easy for them to complete. Even better, this would be an amazing program to ‘sell’ to practices interested in hosting these sessions themselves.”
Ms. Sao said they also hope to assess the impact of the intervention on the participants’ peers to see how well the knowledge and self-efficacy spread through the youths’ teaching.
No external funding was noted. One author reported research support from Hologic and Merck. Dr. Friedman had no disclosures.
AT ACOG 2023
Circulatory support for RV failure caused by pulmonary embolism
A new review article highlights approaches for mechanical circulatory support in patients with high-risk acute pulmonary embolism (PE).
Pulmonary embolism with hemodynamic significance is widely underdiagnosed, and the mortality rate can be as high as 30%, but new therapeutic developments offer promise. “Over the past few years, a renewed interest in mechanical circulatory support (MCS; both percutaneous and surgical) for acute RVF has emerged, increasing viable treatment options for high-risk acute PE,” wrote the authors of the review, which was published online in Interventional Cardiology Clinics.
Poor outcomes are often driven by RVF, which is tricky to diagnose and manage, and it stems from a sudden increase in pulmonary vascular resistance (PVR) following PE. “The mechanism for increased PVR in acute PE is multifactorial, including direct blood flow impedance, local hypoxia-induced vasoconstriction, and platelet/thrombin-induced release of vasoactive peptides. The cascade of events that then leads to RVF includes decreased RV stoke volume, increased RV wall tension, and RV dilation,” the authors wrote.
The authors noted that diuretics help to correct changes to RV geometry and can improve left ventricle filling, which improves hemodynamics. Diuretics can be used in patients who are hypotensive and volume overloaded, but vasopressors should be employed to support blood pressure.
When using mechanical ventilation, strategies such as low tidal volumes, minimization of positive end expiratory pressure, and prevention of hypoxemia and acidemia should be employed to prevent an increase of pulmonary vascular resistance, which can worsen RV failure.
Pulmonary vasodilators aren’t recommended for acute PE, but inhaled pulmonary vasodilators may be considered in hemodynamically unstable patients.
Surgically implanted right ventricle assistance device are generally not used for acute RV failure in high-risk PE, unless the patient has not improved after medical management.
Percutaneous devices
Percutaneous mechanical circulatory support devices can be used for patients experiencing refractory shock. The review highlighted three such devices, including the Impella RP, tandem-heart right ventricular assist devices (TH-RVAD) or Protek Duo, and venoarterial extracorporeal membrane oxygenation (VA-ECMO), but they are not without limitations. “Challenges to using these devices in patients with acute PE include clot dislodgement, vascular complications, infections, device migration, and fracture of individual elements,” the authors wrote.
The Impella RP is easy to deploy and bypasses the RV, but it can’t provide blood oxygenation and may cause bleeding or hemolysis. TH-RVAD oxygenates the blood and bypasses the RV, but suffers from a large sheath size. VA-ECMO oxygenates the blood but may cause bleeding.
There are important differences among the mechanical support devices, according to Jonathan Ludmir, MD, who was asked to comment. “In reality, if someone has a large pulmonary embolism burden, to put in the Impella RP or the Protek Duo would be a little bit risky, because you’d be sometimes putting the device right where the clot is. At least what we do in our institution, when someone is in extremis despite using [intravenous] medications like vasopressors or inotropes, VA-ECMO is kind of the go to. This is both the quickest and probably most effective way to support the patient. I say the quickest because this is a procedure you can do at the bedside.”
Benefits of PERT
One message that the review only briefly mentions, but Dr. Ludmir believes is key, is employing a pulmonary embolism response team. “That’s been looked at extensively, and it’s a really key part of any decision-making. If someone presents to the emergency room or someone inside the hospital has an acute pulmonary embolism, you have a team of people that can respond and help assess the next step. Typically, that involves a cardiologist or an interventional cardiologist, a hematologist, vascular surgeon, often a cardiac surgeon, so it’s a whole slew of people. Based on the patient assessment they can quickly decide, can this patient just be okay with a blood thinner like heparin? Does this patient need something more aggressive, like a thrombectomy? Or is this a serious case where you involve the shock team or the ECMO team, and you have to stabilize the patient on mechanical circulatory support, so you can accomplish what you need to do to get rid of the pulmonary embolism,” said Dr. Ludmir, who is an assistant professor of medicine at Corrigan Minehan Heart Center at Massachusetts General Hospital and Harvard Medical School, both in Boston.
“Every case is individualized, hence the importance of having a team of a variety of different backgrounds and thoughts to approach it. And I think that’s kind of like the key takeaway. Yes, you have to be familiar with all the therapies, but at the end of the day, not every patient is going to fit into the algorithm for how you approach pulmonary embolism,” said Dr. Ludmir.
Dr. Ludmir has no relevant conflicts of interest.
A new review article highlights approaches for mechanical circulatory support in patients with high-risk acute pulmonary embolism (PE).
Pulmonary embolism with hemodynamic significance is widely underdiagnosed, and the mortality rate can be as high as 30%, but new therapeutic developments offer promise. “Over the past few years, a renewed interest in mechanical circulatory support (MCS; both percutaneous and surgical) for acute RVF has emerged, increasing viable treatment options for high-risk acute PE,” wrote the authors of the review, which was published online in Interventional Cardiology Clinics.
Poor outcomes are often driven by RVF, which is tricky to diagnose and manage, and it stems from a sudden increase in pulmonary vascular resistance (PVR) following PE. “The mechanism for increased PVR in acute PE is multifactorial, including direct blood flow impedance, local hypoxia-induced vasoconstriction, and platelet/thrombin-induced release of vasoactive peptides. The cascade of events that then leads to RVF includes decreased RV stoke volume, increased RV wall tension, and RV dilation,” the authors wrote.
The authors noted that diuretics help to correct changes to RV geometry and can improve left ventricle filling, which improves hemodynamics. Diuretics can be used in patients who are hypotensive and volume overloaded, but vasopressors should be employed to support blood pressure.
When using mechanical ventilation, strategies such as low tidal volumes, minimization of positive end expiratory pressure, and prevention of hypoxemia and acidemia should be employed to prevent an increase of pulmonary vascular resistance, which can worsen RV failure.
Pulmonary vasodilators aren’t recommended for acute PE, but inhaled pulmonary vasodilators may be considered in hemodynamically unstable patients.
Surgically implanted right ventricle assistance device are generally not used for acute RV failure in high-risk PE, unless the patient has not improved after medical management.
Percutaneous devices
Percutaneous mechanical circulatory support devices can be used for patients experiencing refractory shock. The review highlighted three such devices, including the Impella RP, tandem-heart right ventricular assist devices (TH-RVAD) or Protek Duo, and venoarterial extracorporeal membrane oxygenation (VA-ECMO), but they are not without limitations. “Challenges to using these devices in patients with acute PE include clot dislodgement, vascular complications, infections, device migration, and fracture of individual elements,” the authors wrote.
The Impella RP is easy to deploy and bypasses the RV, but it can’t provide blood oxygenation and may cause bleeding or hemolysis. TH-RVAD oxygenates the blood and bypasses the RV, but suffers from a large sheath size. VA-ECMO oxygenates the blood but may cause bleeding.
There are important differences among the mechanical support devices, according to Jonathan Ludmir, MD, who was asked to comment. “In reality, if someone has a large pulmonary embolism burden, to put in the Impella RP or the Protek Duo would be a little bit risky, because you’d be sometimes putting the device right where the clot is. At least what we do in our institution, when someone is in extremis despite using [intravenous] medications like vasopressors or inotropes, VA-ECMO is kind of the go to. This is both the quickest and probably most effective way to support the patient. I say the quickest because this is a procedure you can do at the bedside.”
Benefits of PERT
One message that the review only briefly mentions, but Dr. Ludmir believes is key, is employing a pulmonary embolism response team. “That’s been looked at extensively, and it’s a really key part of any decision-making. If someone presents to the emergency room or someone inside the hospital has an acute pulmonary embolism, you have a team of people that can respond and help assess the next step. Typically, that involves a cardiologist or an interventional cardiologist, a hematologist, vascular surgeon, often a cardiac surgeon, so it’s a whole slew of people. Based on the patient assessment they can quickly decide, can this patient just be okay with a blood thinner like heparin? Does this patient need something more aggressive, like a thrombectomy? Or is this a serious case where you involve the shock team or the ECMO team, and you have to stabilize the patient on mechanical circulatory support, so you can accomplish what you need to do to get rid of the pulmonary embolism,” said Dr. Ludmir, who is an assistant professor of medicine at Corrigan Minehan Heart Center at Massachusetts General Hospital and Harvard Medical School, both in Boston.
“Every case is individualized, hence the importance of having a team of a variety of different backgrounds and thoughts to approach it. And I think that’s kind of like the key takeaway. Yes, you have to be familiar with all the therapies, but at the end of the day, not every patient is going to fit into the algorithm for how you approach pulmonary embolism,” said Dr. Ludmir.
Dr. Ludmir has no relevant conflicts of interest.
A new review article highlights approaches for mechanical circulatory support in patients with high-risk acute pulmonary embolism (PE).
Pulmonary embolism with hemodynamic significance is widely underdiagnosed, and the mortality rate can be as high as 30%, but new therapeutic developments offer promise. “Over the past few years, a renewed interest in mechanical circulatory support (MCS; both percutaneous and surgical) for acute RVF has emerged, increasing viable treatment options for high-risk acute PE,” wrote the authors of the review, which was published online in Interventional Cardiology Clinics.
Poor outcomes are often driven by RVF, which is tricky to diagnose and manage, and it stems from a sudden increase in pulmonary vascular resistance (PVR) following PE. “The mechanism for increased PVR in acute PE is multifactorial, including direct blood flow impedance, local hypoxia-induced vasoconstriction, and platelet/thrombin-induced release of vasoactive peptides. The cascade of events that then leads to RVF includes decreased RV stoke volume, increased RV wall tension, and RV dilation,” the authors wrote.
The authors noted that diuretics help to correct changes to RV geometry and can improve left ventricle filling, which improves hemodynamics. Diuretics can be used in patients who are hypotensive and volume overloaded, but vasopressors should be employed to support blood pressure.
When using mechanical ventilation, strategies such as low tidal volumes, minimization of positive end expiratory pressure, and prevention of hypoxemia and acidemia should be employed to prevent an increase of pulmonary vascular resistance, which can worsen RV failure.
Pulmonary vasodilators aren’t recommended for acute PE, but inhaled pulmonary vasodilators may be considered in hemodynamically unstable patients.
Surgically implanted right ventricle assistance device are generally not used for acute RV failure in high-risk PE, unless the patient has not improved after medical management.
Percutaneous devices
Percutaneous mechanical circulatory support devices can be used for patients experiencing refractory shock. The review highlighted three such devices, including the Impella RP, tandem-heart right ventricular assist devices (TH-RVAD) or Protek Duo, and venoarterial extracorporeal membrane oxygenation (VA-ECMO), but they are not without limitations. “Challenges to using these devices in patients with acute PE include clot dislodgement, vascular complications, infections, device migration, and fracture of individual elements,” the authors wrote.
The Impella RP is easy to deploy and bypasses the RV, but it can’t provide blood oxygenation and may cause bleeding or hemolysis. TH-RVAD oxygenates the blood and bypasses the RV, but suffers from a large sheath size. VA-ECMO oxygenates the blood but may cause bleeding.
There are important differences among the mechanical support devices, according to Jonathan Ludmir, MD, who was asked to comment. “In reality, if someone has a large pulmonary embolism burden, to put in the Impella RP or the Protek Duo would be a little bit risky, because you’d be sometimes putting the device right where the clot is. At least what we do in our institution, when someone is in extremis despite using [intravenous] medications like vasopressors or inotropes, VA-ECMO is kind of the go to. This is both the quickest and probably most effective way to support the patient. I say the quickest because this is a procedure you can do at the bedside.”
Benefits of PERT
One message that the review only briefly mentions, but Dr. Ludmir believes is key, is employing a pulmonary embolism response team. “That’s been looked at extensively, and it’s a really key part of any decision-making. If someone presents to the emergency room or someone inside the hospital has an acute pulmonary embolism, you have a team of people that can respond and help assess the next step. Typically, that involves a cardiologist or an interventional cardiologist, a hematologist, vascular surgeon, often a cardiac surgeon, so it’s a whole slew of people. Based on the patient assessment they can quickly decide, can this patient just be okay with a blood thinner like heparin? Does this patient need something more aggressive, like a thrombectomy? Or is this a serious case where you involve the shock team or the ECMO team, and you have to stabilize the patient on mechanical circulatory support, so you can accomplish what you need to do to get rid of the pulmonary embolism,” said Dr. Ludmir, who is an assistant professor of medicine at Corrigan Minehan Heart Center at Massachusetts General Hospital and Harvard Medical School, both in Boston.
“Every case is individualized, hence the importance of having a team of a variety of different backgrounds and thoughts to approach it. And I think that’s kind of like the key takeaway. Yes, you have to be familiar with all the therapies, but at the end of the day, not every patient is going to fit into the algorithm for how you approach pulmonary embolism,” said Dr. Ludmir.
Dr. Ludmir has no relevant conflicts of interest.
FROM INTERVENTIONAL CARDIOLOGY CLINICS
Leadless dual-chamber pacemaker clears early safety, performance hurdles
Cardiology, well into the age of leadless pacemakers, could be headed for an age of leadless pacemaker systems in which various pacing functions are achieved by multiple implants that “talk” to each other.
Even now, a leadless two-part pacemaker system has shown it can safely achieve atrioventricular (AV) synchrony in patients with standard indications for a dual-chamber device, at least over the short term, suggests a prospective observational study. Currently available leadless pacemakers can stimulate only the right ventricle.
Experienced operators achieved a 98% implantation success rate in 300 patients who received an investigational dual-chamber leadless system, the AVEIR DR i2i (Abbott).
Its two separately implanted miniature pulse generators achieve AV synchrony via “beat-to-beat wireless bidirectional communication,” Daniel J. Cantillon, MD, said when presenting the study at the annual scientific sessions of the Heart Rhythm Societyin New Orleans.
The system seemed to work well regardless of the patient’s body orientation. “Sitting, supine, left lateral, right lateral, standing, normal walk, fast walk – we demonstrated robust AV synchrony in all of those positions and with movement,” said Dr. Cantillon, of the Cleveland Clinic.
Should the device be approved, it could “expand the use case for leadless cardiac pacing” to include atrial-only, ventricular-only, fully functional dual-chamber pacing scenarios.”
Dr. Cantillon is senior author on the study’s online publication in the New England Journal of Medicine, timed to coincide with his HRS presentation, with first author Reinoud E. Knops, MD, PhD, Amsterdam University Medical Center.
“The electrical performance of both the atrial and ventricular leadless pacemakers appears to be similar to that of transvenous dual-chamber pacemakers,” the published report states.
More data needed
The study is important and has “significant implications for our pacing field,” Jonathan P. Piccini, MD, MHS, said in an interview. It suggests that “dual-chamber pacing can be achieved with leadless technology” and “with a very high degree” of AV synchrony.
“Obviously, more data as the technology moves into clinical practice will be critical,” said Dr. Piccini, who directs cardiac electrophysiology at Duke University Medical Center, Durham, N.C. “We will also need to understand which patients are best served by leadless technology and which will be better served with traditional transvenous devices.”
The AVEIR DR i2i system consists of two leadless pulse generators for percutaneous implantation in the right atrium and right ventricle, respectively. They link like components of a wireless network to coordinate their separate sensing and rate-adaptive, AV-synchronous pacing functions.
The right ventricular implant “is physically identical to a commercially available single-chamber leadless pacemaker” from Abbott, the published report states.
Leadless pacemaker systems inherently avoid the two main sources of transvenous devices’ major complication – infection – by not requiring such leads or surgery for creating a pulse-generator subcutaneous pocket.
The first such systems consisted of one implant that could provide single-chamber ventricular pacing but not atrial pacing or AV synchronous pacing. The transcatheter single-chamber leadless Micra (Medtronic) for example, was approved in the United States in April 2016 for ventricular-only pacing.
A successor, the Micra AV, approved in 2020, was designed to simulate AV-synchronous pacing by stimulating the ventricle in sequence with mechanically sensed atrial contractions, as described by Dr. Cantillon and associates. But it could not directly pace the atrium, “rendering it inappropriate for patients with sinus-node dysfunction.”
The AVEIR DR i2i system doesn’t have those limitations. It was, however, associated with 35 device- or procedure-related complications in the study, of which the most common was procedural arrhythmia, “namely atrial fibrillation,” Dr. Cantillon said.
Atrial fibrillation can develop during implantation of pacemakers with transvenous leads but is generally terminated without being considered an important event. Yet the study classified it as a serious complication, inflating the complication rate, because “the patients had to be restored to sinus rhythm so we could assess the AV synchrony and also the atrial electrical performance,” he said.
Some of the devices dislodged from their implantation site within a month of the procedure, but “all of those patients were successfully managed percutaneously,” said Dr. Cantillon.
“The 1.7% dislodgement rate is something that we will need to keep an eye on, as embolization of devices is always a significant concern,” Dr. Piccini said. Still, the observed total complication rate “was certainly in line” with rates associated with conventional pacemaker implantation.
Reliable AV synchrony
Fred M. Kusumoto, MD, Mayo Clinic, Jacksonville, Fla., lauded what seems to be the system’s “incredibly reliable AV synchrony in different conditions, albeit in a very controlled environment.”
Of interest will be whether its performance, including maintenance of AV synchrony, holds up in “a more long-term evaluation in the outpatient setting,” said Dr. Kusumoto, speaking as the invited discussant for Dr. Cantillon’s presentation.
Also missing or in short supply from the study, he observed, are insights about long-term efficacy and complications, battery longevity, effectiveness of its rate-responsive capability, and any effect on clinical outcomes.
Local body network
Of the study’s 300 patients (mean age 69 years; 38% female) at 55 sites in Canada, Europe, and the United States, 63.3% had sinus-node dysfunction and 33.3% had AV block as their primary dual-chamber pacing indication; 298 were successfully implanted with both devices.
About 45% had a history of supraventricular arrhythmia, 4.3% had prior ventricular arrhythmia, and 20% had a history of arrhythmia ablation.
By 3 months, the group reported, the primary safety endpoint (freedom from device- or procedure-related serious adverse events) occurred in 90.3%, compared with the performance goal of 78% (P < .001).
The first of two primary performance endpoints (adequate atrial capture threshold and sensing amplitude by predefined criteria) was met in 90.2%, surpassing the 82.5% performance goal (P < .001).
The second primary performance goal (at least 70% AV synchrony with the patient sitting) was seen in 97.3% against the performance goal of 83% (P < .001).
What shouldn’t be “glossed over” from the study, Dr. Kusumoto offered, is that it’s possible to achieve a wireless connection “between two devices that are actually intracardiac.” That raises the prospect of a “local body network” that could be “expanded even more dramatically with other types of devices. I mean, think of the paradigm shift.”
The AVEIR DR i2i trial was funded by Abbott. Dr. Cantillon discloses receiving honoraria or fees for speaking or consulting from Abbott Laboratories, Boston Scientific, Biosense Webster, and Shockwave Medical, as well as holding royalty rights with AirStrip. Dr. Piccini has disclosed relationships with Abbott, Medtronic, Biotronik, Boston Scientific, and other drug and medical device companies. Dr. Kusumoto reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Cardiology, well into the age of leadless pacemakers, could be headed for an age of leadless pacemaker systems in which various pacing functions are achieved by multiple implants that “talk” to each other.
Even now, a leadless two-part pacemaker system has shown it can safely achieve atrioventricular (AV) synchrony in patients with standard indications for a dual-chamber device, at least over the short term, suggests a prospective observational study. Currently available leadless pacemakers can stimulate only the right ventricle.
Experienced operators achieved a 98% implantation success rate in 300 patients who received an investigational dual-chamber leadless system, the AVEIR DR i2i (Abbott).
Its two separately implanted miniature pulse generators achieve AV synchrony via “beat-to-beat wireless bidirectional communication,” Daniel J. Cantillon, MD, said when presenting the study at the annual scientific sessions of the Heart Rhythm Societyin New Orleans.
The system seemed to work well regardless of the patient’s body orientation. “Sitting, supine, left lateral, right lateral, standing, normal walk, fast walk – we demonstrated robust AV synchrony in all of those positions and with movement,” said Dr. Cantillon, of the Cleveland Clinic.
Should the device be approved, it could “expand the use case for leadless cardiac pacing” to include atrial-only, ventricular-only, fully functional dual-chamber pacing scenarios.”
Dr. Cantillon is senior author on the study’s online publication in the New England Journal of Medicine, timed to coincide with his HRS presentation, with first author Reinoud E. Knops, MD, PhD, Amsterdam University Medical Center.
“The electrical performance of both the atrial and ventricular leadless pacemakers appears to be similar to that of transvenous dual-chamber pacemakers,” the published report states.
More data needed
The study is important and has “significant implications for our pacing field,” Jonathan P. Piccini, MD, MHS, said in an interview. It suggests that “dual-chamber pacing can be achieved with leadless technology” and “with a very high degree” of AV synchrony.
“Obviously, more data as the technology moves into clinical practice will be critical,” said Dr. Piccini, who directs cardiac electrophysiology at Duke University Medical Center, Durham, N.C. “We will also need to understand which patients are best served by leadless technology and which will be better served with traditional transvenous devices.”
The AVEIR DR i2i system consists of two leadless pulse generators for percutaneous implantation in the right atrium and right ventricle, respectively. They link like components of a wireless network to coordinate their separate sensing and rate-adaptive, AV-synchronous pacing functions.
The right ventricular implant “is physically identical to a commercially available single-chamber leadless pacemaker” from Abbott, the published report states.
Leadless pacemaker systems inherently avoid the two main sources of transvenous devices’ major complication – infection – by not requiring such leads or surgery for creating a pulse-generator subcutaneous pocket.
The first such systems consisted of one implant that could provide single-chamber ventricular pacing but not atrial pacing or AV synchronous pacing. The transcatheter single-chamber leadless Micra (Medtronic) for example, was approved in the United States in April 2016 for ventricular-only pacing.
A successor, the Micra AV, approved in 2020, was designed to simulate AV-synchronous pacing by stimulating the ventricle in sequence with mechanically sensed atrial contractions, as described by Dr. Cantillon and associates. But it could not directly pace the atrium, “rendering it inappropriate for patients with sinus-node dysfunction.”
The AVEIR DR i2i system doesn’t have those limitations. It was, however, associated with 35 device- or procedure-related complications in the study, of which the most common was procedural arrhythmia, “namely atrial fibrillation,” Dr. Cantillon said.
Atrial fibrillation can develop during implantation of pacemakers with transvenous leads but is generally terminated without being considered an important event. Yet the study classified it as a serious complication, inflating the complication rate, because “the patients had to be restored to sinus rhythm so we could assess the AV synchrony and also the atrial electrical performance,” he said.
Some of the devices dislodged from their implantation site within a month of the procedure, but “all of those patients were successfully managed percutaneously,” said Dr. Cantillon.
“The 1.7% dislodgement rate is something that we will need to keep an eye on, as embolization of devices is always a significant concern,” Dr. Piccini said. Still, the observed total complication rate “was certainly in line” with rates associated with conventional pacemaker implantation.
Reliable AV synchrony
Fred M. Kusumoto, MD, Mayo Clinic, Jacksonville, Fla., lauded what seems to be the system’s “incredibly reliable AV synchrony in different conditions, albeit in a very controlled environment.”
Of interest will be whether its performance, including maintenance of AV synchrony, holds up in “a more long-term evaluation in the outpatient setting,” said Dr. Kusumoto, speaking as the invited discussant for Dr. Cantillon’s presentation.
Also missing or in short supply from the study, he observed, are insights about long-term efficacy and complications, battery longevity, effectiveness of its rate-responsive capability, and any effect on clinical outcomes.
Local body network
Of the study’s 300 patients (mean age 69 years; 38% female) at 55 sites in Canada, Europe, and the United States, 63.3% had sinus-node dysfunction and 33.3% had AV block as their primary dual-chamber pacing indication; 298 were successfully implanted with both devices.
About 45% had a history of supraventricular arrhythmia, 4.3% had prior ventricular arrhythmia, and 20% had a history of arrhythmia ablation.
By 3 months, the group reported, the primary safety endpoint (freedom from device- or procedure-related serious adverse events) occurred in 90.3%, compared with the performance goal of 78% (P < .001).
The first of two primary performance endpoints (adequate atrial capture threshold and sensing amplitude by predefined criteria) was met in 90.2%, surpassing the 82.5% performance goal (P < .001).
The second primary performance goal (at least 70% AV synchrony with the patient sitting) was seen in 97.3% against the performance goal of 83% (P < .001).
What shouldn’t be “glossed over” from the study, Dr. Kusumoto offered, is that it’s possible to achieve a wireless connection “between two devices that are actually intracardiac.” That raises the prospect of a “local body network” that could be “expanded even more dramatically with other types of devices. I mean, think of the paradigm shift.”
The AVEIR DR i2i trial was funded by Abbott. Dr. Cantillon discloses receiving honoraria or fees for speaking or consulting from Abbott Laboratories, Boston Scientific, Biosense Webster, and Shockwave Medical, as well as holding royalty rights with AirStrip. Dr. Piccini has disclosed relationships with Abbott, Medtronic, Biotronik, Boston Scientific, and other drug and medical device companies. Dr. Kusumoto reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Cardiology, well into the age of leadless pacemakers, could be headed for an age of leadless pacemaker systems in which various pacing functions are achieved by multiple implants that “talk” to each other.
Even now, a leadless two-part pacemaker system has shown it can safely achieve atrioventricular (AV) synchrony in patients with standard indications for a dual-chamber device, at least over the short term, suggests a prospective observational study. Currently available leadless pacemakers can stimulate only the right ventricle.
Experienced operators achieved a 98% implantation success rate in 300 patients who received an investigational dual-chamber leadless system, the AVEIR DR i2i (Abbott).
Its two separately implanted miniature pulse generators achieve AV synchrony via “beat-to-beat wireless bidirectional communication,” Daniel J. Cantillon, MD, said when presenting the study at the annual scientific sessions of the Heart Rhythm Societyin New Orleans.
The system seemed to work well regardless of the patient’s body orientation. “Sitting, supine, left lateral, right lateral, standing, normal walk, fast walk – we demonstrated robust AV synchrony in all of those positions and with movement,” said Dr. Cantillon, of the Cleveland Clinic.
Should the device be approved, it could “expand the use case for leadless cardiac pacing” to include atrial-only, ventricular-only, fully functional dual-chamber pacing scenarios.”
Dr. Cantillon is senior author on the study’s online publication in the New England Journal of Medicine, timed to coincide with his HRS presentation, with first author Reinoud E. Knops, MD, PhD, Amsterdam University Medical Center.
“The electrical performance of both the atrial and ventricular leadless pacemakers appears to be similar to that of transvenous dual-chamber pacemakers,” the published report states.
More data needed
The study is important and has “significant implications for our pacing field,” Jonathan P. Piccini, MD, MHS, said in an interview. It suggests that “dual-chamber pacing can be achieved with leadless technology” and “with a very high degree” of AV synchrony.
“Obviously, more data as the technology moves into clinical practice will be critical,” said Dr. Piccini, who directs cardiac electrophysiology at Duke University Medical Center, Durham, N.C. “We will also need to understand which patients are best served by leadless technology and which will be better served with traditional transvenous devices.”
The AVEIR DR i2i system consists of two leadless pulse generators for percutaneous implantation in the right atrium and right ventricle, respectively. They link like components of a wireless network to coordinate their separate sensing and rate-adaptive, AV-synchronous pacing functions.
The right ventricular implant “is physically identical to a commercially available single-chamber leadless pacemaker” from Abbott, the published report states.
Leadless pacemaker systems inherently avoid the two main sources of transvenous devices’ major complication – infection – by not requiring such leads or surgery for creating a pulse-generator subcutaneous pocket.
The first such systems consisted of one implant that could provide single-chamber ventricular pacing but not atrial pacing or AV synchronous pacing. The transcatheter single-chamber leadless Micra (Medtronic) for example, was approved in the United States in April 2016 for ventricular-only pacing.
A successor, the Micra AV, approved in 2020, was designed to simulate AV-synchronous pacing by stimulating the ventricle in sequence with mechanically sensed atrial contractions, as described by Dr. Cantillon and associates. But it could not directly pace the atrium, “rendering it inappropriate for patients with sinus-node dysfunction.”
The AVEIR DR i2i system doesn’t have those limitations. It was, however, associated with 35 device- or procedure-related complications in the study, of which the most common was procedural arrhythmia, “namely atrial fibrillation,” Dr. Cantillon said.
Atrial fibrillation can develop during implantation of pacemakers with transvenous leads but is generally terminated without being considered an important event. Yet the study classified it as a serious complication, inflating the complication rate, because “the patients had to be restored to sinus rhythm so we could assess the AV synchrony and also the atrial electrical performance,” he said.
Some of the devices dislodged from their implantation site within a month of the procedure, but “all of those patients were successfully managed percutaneously,” said Dr. Cantillon.
“The 1.7% dislodgement rate is something that we will need to keep an eye on, as embolization of devices is always a significant concern,” Dr. Piccini said. Still, the observed total complication rate “was certainly in line” with rates associated with conventional pacemaker implantation.
Reliable AV synchrony
Fred M. Kusumoto, MD, Mayo Clinic, Jacksonville, Fla., lauded what seems to be the system’s “incredibly reliable AV synchrony in different conditions, albeit in a very controlled environment.”
Of interest will be whether its performance, including maintenance of AV synchrony, holds up in “a more long-term evaluation in the outpatient setting,” said Dr. Kusumoto, speaking as the invited discussant for Dr. Cantillon’s presentation.
Also missing or in short supply from the study, he observed, are insights about long-term efficacy and complications, battery longevity, effectiveness of its rate-responsive capability, and any effect on clinical outcomes.
Local body network
Of the study’s 300 patients (mean age 69 years; 38% female) at 55 sites in Canada, Europe, and the United States, 63.3% had sinus-node dysfunction and 33.3% had AV block as their primary dual-chamber pacing indication; 298 were successfully implanted with both devices.
About 45% had a history of supraventricular arrhythmia, 4.3% had prior ventricular arrhythmia, and 20% had a history of arrhythmia ablation.
By 3 months, the group reported, the primary safety endpoint (freedom from device- or procedure-related serious adverse events) occurred in 90.3%, compared with the performance goal of 78% (P < .001).
The first of two primary performance endpoints (adequate atrial capture threshold and sensing amplitude by predefined criteria) was met in 90.2%, surpassing the 82.5% performance goal (P < .001).
The second primary performance goal (at least 70% AV synchrony with the patient sitting) was seen in 97.3% against the performance goal of 83% (P < .001).
What shouldn’t be “glossed over” from the study, Dr. Kusumoto offered, is that it’s possible to achieve a wireless connection “between two devices that are actually intracardiac.” That raises the prospect of a “local body network” that could be “expanded even more dramatically with other types of devices. I mean, think of the paradigm shift.”
The AVEIR DR i2i trial was funded by Abbott. Dr. Cantillon discloses receiving honoraria or fees for speaking or consulting from Abbott Laboratories, Boston Scientific, Biosense Webster, and Shockwave Medical, as well as holding royalty rights with AirStrip. Dr. Piccini has disclosed relationships with Abbott, Medtronic, Biotronik, Boston Scientific, and other drug and medical device companies. Dr. Kusumoto reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM HRS 2023
Researchers locate signals in brain related to chronic pain
a new study in Nature Neuroscience concluded.
The researchers used the devices on four patients who had felt endless nerve pain for more than a year. The devices recorded several times a day, which could pave “the way for implanted devices to one day predict pain signals or even short-circuit them,” The New York Times reported.
The study says the pain “was associated with electrical fluctuations in the orbitofrontal cortex, an area involved in emotion regulation, self-evaluation, and decision-making,” The Times reported. “The research suggests that such patterns of brain activity could serve as biomarkers to guide diagnosis and treatment for millions of people with shooting or burning chronic pain linked to a damaged nervous system.”
Ajay Wasan, MD, and a pain specialist at the University of Pittsburgh who was not involved in the study praised it to the Times.
“The study really advances a whole generation of research that has shown that the functioning of the brain is really important to processing and perceiving pain,” he said.
Chronic pain is defined as persistent or recurring and lasting more than three months. The Centers for Disease Control and Prevention says about 20% of Americans experience it. It has been linked with depression, Alzheimer’s disease and other dementias, suicide, and substance use.
Yet, the study’s authors noted, “pain severity is often measured through subjective report, while objective biomarkers that may guide diagnosis and treatment are lacking.”
Medtronic provided devices for the study. The study authors reported no conflicts of interest.
A version of this article first appeared on WebMD.com.
a new study in Nature Neuroscience concluded.
The researchers used the devices on four patients who had felt endless nerve pain for more than a year. The devices recorded several times a day, which could pave “the way for implanted devices to one day predict pain signals or even short-circuit them,” The New York Times reported.
The study says the pain “was associated with electrical fluctuations in the orbitofrontal cortex, an area involved in emotion regulation, self-evaluation, and decision-making,” The Times reported. “The research suggests that such patterns of brain activity could serve as biomarkers to guide diagnosis and treatment for millions of people with shooting or burning chronic pain linked to a damaged nervous system.”
Ajay Wasan, MD, and a pain specialist at the University of Pittsburgh who was not involved in the study praised it to the Times.
“The study really advances a whole generation of research that has shown that the functioning of the brain is really important to processing and perceiving pain,” he said.
Chronic pain is defined as persistent or recurring and lasting more than three months. The Centers for Disease Control and Prevention says about 20% of Americans experience it. It has been linked with depression, Alzheimer’s disease and other dementias, suicide, and substance use.
Yet, the study’s authors noted, “pain severity is often measured through subjective report, while objective biomarkers that may guide diagnosis and treatment are lacking.”
Medtronic provided devices for the study. The study authors reported no conflicts of interest.
A version of this article first appeared on WebMD.com.
a new study in Nature Neuroscience concluded.
The researchers used the devices on four patients who had felt endless nerve pain for more than a year. The devices recorded several times a day, which could pave “the way for implanted devices to one day predict pain signals or even short-circuit them,” The New York Times reported.
The study says the pain “was associated with electrical fluctuations in the orbitofrontal cortex, an area involved in emotion regulation, self-evaluation, and decision-making,” The Times reported. “The research suggests that such patterns of brain activity could serve as biomarkers to guide diagnosis and treatment for millions of people with shooting or burning chronic pain linked to a damaged nervous system.”
Ajay Wasan, MD, and a pain specialist at the University of Pittsburgh who was not involved in the study praised it to the Times.
“The study really advances a whole generation of research that has shown that the functioning of the brain is really important to processing and perceiving pain,” he said.
Chronic pain is defined as persistent or recurring and lasting more than three months. The Centers for Disease Control and Prevention says about 20% of Americans experience it. It has been linked with depression, Alzheimer’s disease and other dementias, suicide, and substance use.
Yet, the study’s authors noted, “pain severity is often measured through subjective report, while objective biomarkers that may guide diagnosis and treatment are lacking.”
Medtronic provided devices for the study. The study authors reported no conflicts of interest.
A version of this article first appeared on WebMD.com.
FROM NATURE NEUROSCIENCE
ARNI bests ARB to reduce NT-proBNP in stabilized preserved-EF HF
Patients with an ejection fraction (EF) greater than 40% who were stabilized after recent worsening or de novo heart failure (HF) had a greater reduction in natriuretic peptides and less worsening renal function, but a higher rate of hypotension over 8 weeks with sacubitril-valsartan (Entresto) versus valsartan (Diovan) in the PARAGLIDE-HF trial.
A subgroup analysis showed evidence of a larger treatment effect among those with an EF of 60% or less, said Robert Mentz, MD, of the Duke Clinical Research Institute, Durham, N.C.

Dr. Mentz presented the findings at the Heart Failure Association of the European Society of Cardiology (HFA-ESC) scientific sessions. The study was also published online simultaneously in the Journal of the American College of Cardiology.
“Next steps will involve further assessment of the cardiovascular and renal benefits, as well as further exploration of the symptomatic hypotension that we observed,” Dr. Mentz said in an interview.
Meanwhile, he said, “clinicians should be aware of these new data – specifically, the incremental reduction in natriuretic peptide level, compared with valsartan, and potential benefits on cardiovascular and renal events,” particularly in those with an EF greater than 40% to 60% or less.
Larger benefit for EF > 40% to < 60%
PARAGLIDE-HF was a double-blind, randomized controlled trial with 466 patients with EF greater than 40% enrolled within 30 days of a worsening HF event. The median age was 71 years, 52% were women, and 22% were Black.
The trial was a follow-up to PARAGON-HF, which had shown that, in patients with an EF of at least 45%, sacubitril-valsartan did not result in a significantly lower rate of total hospitalizations for HF or death from cardiovascular causes, compared with valsartan.
The primary endpoint for PARAGLIDE was the time-averaged proportional change in N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) from baseline through weeks 4 and 8, as in the PIONEER-HF trial. That trial showed that among patients hospitalized for acute decompensated HF with reduced EF (< 40%), the angiotensin receptor/neprilysin inhibitor (ARNI) led to a greater reduction in NT-proBNP concentration than the angiotensin receptor blocker (ARB).
Similarly, for PARAGLIDE, the time-averaged reduction in NT-proBNP was greater with sacubitril-valsartan, with a change ratio of 0.85 (15% greater reduction).
A secondary hierarchical outcome for PARAGLIDE, using the win ratio, consisted of time to cardiovascular death, number and timing of HF hospitalizations, number and timing of urgent HF visits, and time-averaged proportional change in NT-proBNP from baseline to weeks 4 and 8.
The hierarchical outcome favored sacubitril-valsartan, but was not significant (unmatched win ratio, 1.19).
As noted, sacubitril-valsartan reduced worsening renal function, compared with valsartan (odds ratio, 0.61), but increased symptomatic hypotension (OR, 1.73).
“We will work to better characterize the hypotension events that were observed to help identify those patients at greater risk and to provide further clarity around the timing and implications of these events,” Dr. Mentz said in an interview.
The team hypothesized that such events may be prevented by optimizing volume status and background therapies commonly used to treat hypertension in these patients.
“For instance,” Dr. Mentz suggested, “calcium channel blockers like amlodipine could be dose reduced or discontinued in patients with lower baseline blood pressures to better support sacubitril/valsartan initiation and titration.”
He highlighted the subgroup analysis showing evidence of a larger treatment effect in study patients with an EF of 60% or less for the NT-proBNP change (0.78) and the hierarchical outcome (win ratio, 1.46).
“These data may influence future guidance for sacubitril-valsartan in HF with EF greater than 40%, regardless of HF chronicity [acute or chronic vs. de novo] and treatment setting [hospital vs. clinic],” Dr. Mentz concluded.
Data ‘far from conclusive’
In a comment, Sean Pinney, MD, chief of cardiology at Mount Sinai Morningside, New York, said that the study results “help expand the current evidence base supporting the use of an ARNI in patients” with an EF greater than 40% up to 60%, and “provide confidence that ARNIs help to lower natriuretic peptides.
“It comes as little surprise that not everyone was able to tolerate these medications due to intolerable side effects like dizziness or hypotension,” he said.
Nevertheless, he added, “hopefully, these trial data help strengthen clinicians’ resolve to prescribe sacubitril/valsartan to a growing population of vulnerable patients.”

In a related editorial, Hector O. Ventura, MD, of the Ochsner Clinical School–University of Queensland, New Orleans, and colleagues express several concerns about the study.
Although the trial achieved significance for the primary endpoint, the margin of benefit was less than expected and the magnitude of the NT-proBNP reduction may not have been enough to reach the threshold for clinical benefit, they wrote.
Diuretic dosing in the two groups was not reported, and between-group differences may have contributed to both the differential NT-proBNP reduction and the rates of hypotension.
Furthermore, the sacubitril-valsartan group had a higher proportion of missing NT-proBNP data, which may have biased the results.
“Clinicians who elect to use sacubitril-valsartan in this population should be mindful of the risk for hypotension and select patients carefully, while providing close ambulatory follow up to ensure stability and adherence,” they noted.
“This important trial provides some wins that support selective use of sacubitril-valsartan in HFpEF [as well as] observed losses, which too may help to define better implementation strategies in appropriately selected patients,” the editorialists concluded.
The study was funded by Novartis. Dr. Mentz and other coauthors have received fees from Novartis. Dr. Pinney, Dr. Ventura, and the other editorialists disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Patients with an ejection fraction (EF) greater than 40% who were stabilized after recent worsening or de novo heart failure (HF) had a greater reduction in natriuretic peptides and less worsening renal function, but a higher rate of hypotension over 8 weeks with sacubitril-valsartan (Entresto) versus valsartan (Diovan) in the PARAGLIDE-HF trial.
A subgroup analysis showed evidence of a larger treatment effect among those with an EF of 60% or less, said Robert Mentz, MD, of the Duke Clinical Research Institute, Durham, N.C.
Dr. Mentz presented the findings at the Heart Failure Association of the European Society of Cardiology (HFA-ESC) scientific sessions. The study was also published online simultaneously in the Journal of the American College of Cardiology.
“Next steps will involve further assessment of the cardiovascular and renal benefits, as well as further exploration of the symptomatic hypotension that we observed,” Dr. Mentz said in an interview.
Meanwhile, he said, “clinicians should be aware of these new data – specifically, the incremental reduction in natriuretic peptide level, compared with valsartan, and potential benefits on cardiovascular and renal events,” particularly in those with an EF greater than 40% to 60% or less.
Larger benefit for EF > 40% to < 60%
PARAGLIDE-HF was a double-blind, randomized controlled trial with 466 patients with EF greater than 40% enrolled within 30 days of a worsening HF event. The median age was 71 years, 52% were women, and 22% were Black.
The trial was a follow-up to PARAGON-HF, which had shown that, in patients with an EF of at least 45%, sacubitril-valsartan did not result in a significantly lower rate of total hospitalizations for HF or death from cardiovascular causes, compared with valsartan.
The primary endpoint for PARAGLIDE was the time-averaged proportional change in N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) from baseline through weeks 4 and 8, as in the PIONEER-HF trial. That trial showed that among patients hospitalized for acute decompensated HF with reduced EF (< 40%), the angiotensin receptor/neprilysin inhibitor (ARNI) led to a greater reduction in NT-proBNP concentration than the angiotensin receptor blocker (ARB).
Similarly, for PARAGLIDE, the time-averaged reduction in NT-proBNP was greater with sacubitril-valsartan, with a change ratio of 0.85 (15% greater reduction).
A secondary hierarchical outcome for PARAGLIDE, using the win ratio, consisted of time to cardiovascular death, number and timing of HF hospitalizations, number and timing of urgent HF visits, and time-averaged proportional change in NT-proBNP from baseline to weeks 4 and 8.
The hierarchical outcome favored sacubitril-valsartan, but was not significant (unmatched win ratio, 1.19).
As noted, sacubitril-valsartan reduced worsening renal function, compared with valsartan (odds ratio, 0.61), but increased symptomatic hypotension (OR, 1.73).
“We will work to better characterize the hypotension events that were observed to help identify those patients at greater risk and to provide further clarity around the timing and implications of these events,” Dr. Mentz said in an interview.
The team hypothesized that such events may be prevented by optimizing volume status and background therapies commonly used to treat hypertension in these patients.
“For instance,” Dr. Mentz suggested, “calcium channel blockers like amlodipine could be dose reduced or discontinued in patients with lower baseline blood pressures to better support sacubitril/valsartan initiation and titration.”
He highlighted the subgroup analysis showing evidence of a larger treatment effect in study patients with an EF of 60% or less for the NT-proBNP change (0.78) and the hierarchical outcome (win ratio, 1.46).
“These data may influence future guidance for sacubitril-valsartan in HF with EF greater than 40%, regardless of HF chronicity [acute or chronic vs. de novo] and treatment setting [hospital vs. clinic],” Dr. Mentz concluded.
Data ‘far from conclusive’
In a comment, Sean Pinney, MD, chief of cardiology at Mount Sinai Morningside, New York, said that the study results “help expand the current evidence base supporting the use of an ARNI in patients” with an EF greater than 40% up to 60%, and “provide confidence that ARNIs help to lower natriuretic peptides.
“It comes as little surprise that not everyone was able to tolerate these medications due to intolerable side effects like dizziness or hypotension,” he said.
Nevertheless, he added, “hopefully, these trial data help strengthen clinicians’ resolve to prescribe sacubitril/valsartan to a growing population of vulnerable patients.”
In a related editorial, Hector O. Ventura, MD, of the Ochsner Clinical School–University of Queensland, New Orleans, and colleagues express several concerns about the study.
Although the trial achieved significance for the primary endpoint, the margin of benefit was less than expected and the magnitude of the NT-proBNP reduction may not have been enough to reach the threshold for clinical benefit, they wrote.
Diuretic dosing in the two groups was not reported, and between-group differences may have contributed to both the differential NT-proBNP reduction and the rates of hypotension.
Furthermore, the sacubitril-valsartan group had a higher proportion of missing NT-proBNP data, which may have biased the results.
“Clinicians who elect to use sacubitril-valsartan in this population should be mindful of the risk for hypotension and select patients carefully, while providing close ambulatory follow up to ensure stability and adherence,” they noted.
“This important trial provides some wins that support selective use of sacubitril-valsartan in HFpEF [as well as] observed losses, which too may help to define better implementation strategies in appropriately selected patients,” the editorialists concluded.
The study was funded by Novartis. Dr. Mentz and other coauthors have received fees from Novartis. Dr. Pinney, Dr. Ventura, and the other editorialists disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Patients with an ejection fraction (EF) greater than 40% who were stabilized after recent worsening or de novo heart failure (HF) had a greater reduction in natriuretic peptides and less worsening renal function, but a higher rate of hypotension over 8 weeks with sacubitril-valsartan (Entresto) versus valsartan (Diovan) in the PARAGLIDE-HF trial.
A subgroup analysis showed evidence of a larger treatment effect among those with an EF of 60% or less, said Robert Mentz, MD, of the Duke Clinical Research Institute, Durham, N.C.
Dr. Mentz presented the findings at the Heart Failure Association of the European Society of Cardiology (HFA-ESC) scientific sessions. The study was also published online simultaneously in the Journal of the American College of Cardiology.
“Next steps will involve further assessment of the cardiovascular and renal benefits, as well as further exploration of the symptomatic hypotension that we observed,” Dr. Mentz said in an interview.
Meanwhile, he said, “clinicians should be aware of these new data – specifically, the incremental reduction in natriuretic peptide level, compared with valsartan, and potential benefits on cardiovascular and renal events,” particularly in those with an EF greater than 40% to 60% or less.
Larger benefit for EF > 40% to < 60%
PARAGLIDE-HF was a double-blind, randomized controlled trial with 466 patients with EF greater than 40% enrolled within 30 days of a worsening HF event. The median age was 71 years, 52% were women, and 22% were Black.
The trial was a follow-up to PARAGON-HF, which had shown that, in patients with an EF of at least 45%, sacubitril-valsartan did not result in a significantly lower rate of total hospitalizations for HF or death from cardiovascular causes, compared with valsartan.
The primary endpoint for PARAGLIDE was the time-averaged proportional change in N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) from baseline through weeks 4 and 8, as in the PIONEER-HF trial. That trial showed that among patients hospitalized for acute decompensated HF with reduced EF (< 40%), the angiotensin receptor/neprilysin inhibitor (ARNI) led to a greater reduction in NT-proBNP concentration than the angiotensin receptor blocker (ARB).
Similarly, for PARAGLIDE, the time-averaged reduction in NT-proBNP was greater with sacubitril-valsartan, with a change ratio of 0.85 (15% greater reduction).
A secondary hierarchical outcome for PARAGLIDE, using the win ratio, consisted of time to cardiovascular death, number and timing of HF hospitalizations, number and timing of urgent HF visits, and time-averaged proportional change in NT-proBNP from baseline to weeks 4 and 8.
The hierarchical outcome favored sacubitril-valsartan, but was not significant (unmatched win ratio, 1.19).
As noted, sacubitril-valsartan reduced worsening renal function, compared with valsartan (odds ratio, 0.61), but increased symptomatic hypotension (OR, 1.73).
“We will work to better characterize the hypotension events that were observed to help identify those patients at greater risk and to provide further clarity around the timing and implications of these events,” Dr. Mentz said in an interview.
The team hypothesized that such events may be prevented by optimizing volume status and background therapies commonly used to treat hypertension in these patients.
“For instance,” Dr. Mentz suggested, “calcium channel blockers like amlodipine could be dose reduced or discontinued in patients with lower baseline blood pressures to better support sacubitril/valsartan initiation and titration.”
He highlighted the subgroup analysis showing evidence of a larger treatment effect in study patients with an EF of 60% or less for the NT-proBNP change (0.78) and the hierarchical outcome (win ratio, 1.46).
“These data may influence future guidance for sacubitril-valsartan in HF with EF greater than 40%, regardless of HF chronicity [acute or chronic vs. de novo] and treatment setting [hospital vs. clinic],” Dr. Mentz concluded.
Data ‘far from conclusive’
In a comment, Sean Pinney, MD, chief of cardiology at Mount Sinai Morningside, New York, said that the study results “help expand the current evidence base supporting the use of an ARNI in patients” with an EF greater than 40% up to 60%, and “provide confidence that ARNIs help to lower natriuretic peptides.
“It comes as little surprise that not everyone was able to tolerate these medications due to intolerable side effects like dizziness or hypotension,” he said.
Nevertheless, he added, “hopefully, these trial data help strengthen clinicians’ resolve to prescribe sacubitril/valsartan to a growing population of vulnerable patients.”
In a related editorial, Hector O. Ventura, MD, of the Ochsner Clinical School–University of Queensland, New Orleans, and colleagues express several concerns about the study.
Although the trial achieved significance for the primary endpoint, the margin of benefit was less than expected and the magnitude of the NT-proBNP reduction may not have been enough to reach the threshold for clinical benefit, they wrote.
Diuretic dosing in the two groups was not reported, and between-group differences may have contributed to both the differential NT-proBNP reduction and the rates of hypotension.
Furthermore, the sacubitril-valsartan group had a higher proportion of missing NT-proBNP data, which may have biased the results.
“Clinicians who elect to use sacubitril-valsartan in this population should be mindful of the risk for hypotension and select patients carefully, while providing close ambulatory follow up to ensure stability and adherence,” they noted.
“This important trial provides some wins that support selective use of sacubitril-valsartan in HFpEF [as well as] observed losses, which too may help to define better implementation strategies in appropriately selected patients,” the editorialists concluded.
The study was funded by Novartis. Dr. Mentz and other coauthors have received fees from Novartis. Dr. Pinney, Dr. Ventura, and the other editorialists disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM ESC HEART FAILURE 2023

Adult tonsillectomies work and they’re cost effective
A new randomized trial offers rare insight into outcomes in adult tonsillectomy, a surgical procedure that’s commonly performed in the United States yet falling out of favor. Tonsillectomies are both clinically effective and cost-effective in adult patients with recurrent acute tonsillitis, a British team reports.
The researchers declined to weigh in on whether the procedure is actually better than nonsurgical management. Still, “here at last, we have a substantial piece of scientific evidence which shows that, compared with nonsurgical management, removal of tonsils has a significant impact on the number of sore throat days and on the cost of managing sore throat disease in adults,” said study lead author Janet A. Wilson, MBChB, MD, an emerita professor of otolaryngology at Newcastle University (England), in an interview.
The study was published in The Lancet.
Tonsillectomies have become much less common over the past several decades as questions have arisen about their value. In the United States, the number of procedures performed each year plunged from a high of 1.4 million in 1959 to an estimated 286,000 tonsillectomies performed in children under 15 and 120,000 in people aged 15 in 2010.
It’s harder for adults to tolerate tonsillectomies than children, Dr. Wilson said. In children, surgeons can easily remove tonsils by scraping them off the throat’s side walls. But, she said, “an adult tonsillectomy is more akin to taking off the skin of an unripe orange, so it’s harder work for the surgeon and more traumatizing for the wall of the adult patient’s pharynx. We can only assume that this greater amount of fibrous tissue reflects the cumulative effect of infections over a period of years.”
While tonsillectomies are still performed hundreds of times a day in adults in the United States, a 2014 Cochrane Library review found there’s “insufficient information “to support them versus nonsurgical care as treatments to reduce sore throats.”
For the new multicenter, open-label, randomized study, researchers randomly assigned patients aged 16 and older with recurrent acute tonsillitis to immediate tonsillectomy or nonsurgical management, which Dr. Wilson said can include cold fluids, honey, analgesics/anti-inflammatories. and anesthetic throat lozenges. The study was conducted between 2015 and 2018.
Ultimately, there were 224 and 204 patients, respectively, in the two groups (average age = 23, [19-30], 78% female, 90% White).
Patients who underwent tonsillectomies versus nonsurgical treatment had fewer sore throats over 2 years (median 23 days [IQR 11-46 days] vs. 30 days [14-65 days]) with an incident rate ratio of 0.53 (95% confidence interval, 0.43-0.65, P < 0.0001) after adjustment for clinic site and baseline severity.
The study also shows that “adults who have severe recurrent throat infections with a frequency of seven episodes within 1 year, five or more for 2 consecutive years, or three or more in 3 consecutive years will suffer fewer days of sore throat in the 2 years following tonsillectomy than if they had kept their tonsils,” Dr. Wilson said.
The study doesn’t examine longer-term consequences. A 2018 study of children linked tonsillectomies to “significantly increased relative risk of later respiratory, allergic, and infectious diseases.”
In the new study, nearly 4 in 10 (39%) of the tonsillectomy patients had adverse events linked to the surgeries, and bleeding (19%) was the most common adverse effect. The researchers also estimated that “tonsillectomy has a high probability of being considered cost-effective.”
“Whichever way the results were analyzed and confounding variables allowed for, the result always seems to be the same: Tonsillectomy applied using current qualifying criteria was a worthwhile procedure,” Dr. Wilson said.
Dr. Wilson noted that tonsillectomy patients will suffer a persistent sore throat after surgery, “about the same as a bad episode of tonsillitis.” And she said patients will need to adjust their diet for a few days and take 1-2 weeks off work.
In an interview, internal medicine physician Noel Deep, MD, of Antigo, Wisc., said antibiotics are a common treatment for tonsillitis in primary care clinics. According to him, the United States doesn’t have guidelines for tonsillectomies in adults. He believes they can be considered if tonsillitis keeps recurring three to five times a year and disrupts quality of life.
Dr. Deep said the new study “reinforces the benefit of tonsillectomies. Several studies from Germany, Sweden, Finland, and the United Kingdom have demonstrated benefits of tonsillectomies, but they were only for short periods of less than a year and lacked long-term data.”
He noted that “there is no clear evidence as to when to recommend tonsillectomies.” Clinicians should talk to patients about the potential that tonsillectomies will reduce sore throat episodes and cost the patient less in the long run, he said. It’s also important, he said, to make sure tonsillitis is bacterial before prescribing antibiotics.
The United Kingdom’s National Institute for Health Research funded the study. Dr. Wilson disclosed support for meetings/travel from ENT Scotland, and the other authors report various disclosures, including grants and contracts. Dr. Deep serves on the editorial advisory board of Internal Medicine News and is chair of the American Medical Association Council on Science and Public Health.
A new randomized trial offers rare insight into outcomes in adult tonsillectomy, a surgical procedure that’s commonly performed in the United States yet falling out of favor. Tonsillectomies are both clinically effective and cost-effective in adult patients with recurrent acute tonsillitis, a British team reports.
The researchers declined to weigh in on whether the procedure is actually better than nonsurgical management. Still, “here at last, we have a substantial piece of scientific evidence which shows that, compared with nonsurgical management, removal of tonsils has a significant impact on the number of sore throat days and on the cost of managing sore throat disease in adults,” said study lead author Janet A. Wilson, MBChB, MD, an emerita professor of otolaryngology at Newcastle University (England), in an interview.
The study was published in The Lancet.
Tonsillectomies have become much less common over the past several decades as questions have arisen about their value. In the United States, the number of procedures performed each year plunged from a high of 1.4 million in 1959 to an estimated 286,000 tonsillectomies performed in children under 15 and 120,000 in people aged 15 in 2010.
It’s harder for adults to tolerate tonsillectomies than children, Dr. Wilson said. In children, surgeons can easily remove tonsils by scraping them off the throat’s side walls. But, she said, “an adult tonsillectomy is more akin to taking off the skin of an unripe orange, so it’s harder work for the surgeon and more traumatizing for the wall of the adult patient’s pharynx. We can only assume that this greater amount of fibrous tissue reflects the cumulative effect of infections over a period of years.”
While tonsillectomies are still performed hundreds of times a day in adults in the United States, a 2014 Cochrane Library review found there’s “insufficient information “to support them versus nonsurgical care as treatments to reduce sore throats.”
For the new multicenter, open-label, randomized study, researchers randomly assigned patients aged 16 and older with recurrent acute tonsillitis to immediate tonsillectomy or nonsurgical management, which Dr. Wilson said can include cold fluids, honey, analgesics/anti-inflammatories. and anesthetic throat lozenges. The study was conducted between 2015 and 2018.
Ultimately, there were 224 and 204 patients, respectively, in the two groups (average age = 23, [19-30], 78% female, 90% White).
Patients who underwent tonsillectomies versus nonsurgical treatment had fewer sore throats over 2 years (median 23 days [IQR 11-46 days] vs. 30 days [14-65 days]) with an incident rate ratio of 0.53 (95% confidence interval, 0.43-0.65, P < 0.0001) after adjustment for clinic site and baseline severity.
The study also shows that “adults who have severe recurrent throat infections with a frequency of seven episodes within 1 year, five or more for 2 consecutive years, or three or more in 3 consecutive years will suffer fewer days of sore throat in the 2 years following tonsillectomy than if they had kept their tonsils,” Dr. Wilson said.
The study doesn’t examine longer-term consequences. A 2018 study of children linked tonsillectomies to “significantly increased relative risk of later respiratory, allergic, and infectious diseases.”
In the new study, nearly 4 in 10 (39%) of the tonsillectomy patients had adverse events linked to the surgeries, and bleeding (19%) was the most common adverse effect. The researchers also estimated that “tonsillectomy has a high probability of being considered cost-effective.”
“Whichever way the results were analyzed and confounding variables allowed for, the result always seems to be the same: Tonsillectomy applied using current qualifying criteria was a worthwhile procedure,” Dr. Wilson said.
Dr. Wilson noted that tonsillectomy patients will suffer a persistent sore throat after surgery, “about the same as a bad episode of tonsillitis.” And she said patients will need to adjust their diet for a few days and take 1-2 weeks off work.
In an interview, internal medicine physician Noel Deep, MD, of Antigo, Wisc., said antibiotics are a common treatment for tonsillitis in primary care clinics. According to him, the United States doesn’t have guidelines for tonsillectomies in adults. He believes they can be considered if tonsillitis keeps recurring three to five times a year and disrupts quality of life.
Dr. Deep said the new study “reinforces the benefit of tonsillectomies. Several studies from Germany, Sweden, Finland, and the United Kingdom have demonstrated benefits of tonsillectomies, but they were only for short periods of less than a year and lacked long-term data.”
He noted that “there is no clear evidence as to when to recommend tonsillectomies.” Clinicians should talk to patients about the potential that tonsillectomies will reduce sore throat episodes and cost the patient less in the long run, he said. It’s also important, he said, to make sure tonsillitis is bacterial before prescribing antibiotics.
The United Kingdom’s National Institute for Health Research funded the study. Dr. Wilson disclosed support for meetings/travel from ENT Scotland, and the other authors report various disclosures, including grants and contracts. Dr. Deep serves on the editorial advisory board of Internal Medicine News and is chair of the American Medical Association Council on Science and Public Health.
A new randomized trial offers rare insight into outcomes in adult tonsillectomy, a surgical procedure that’s commonly performed in the United States yet falling out of favor. Tonsillectomies are both clinically effective and cost-effective in adult patients with recurrent acute tonsillitis, a British team reports.
The researchers declined to weigh in on whether the procedure is actually better than nonsurgical management. Still, “here at last, we have a substantial piece of scientific evidence which shows that, compared with nonsurgical management, removal of tonsils has a significant impact on the number of sore throat days and on the cost of managing sore throat disease in adults,” said study lead author Janet A. Wilson, MBChB, MD, an emerita professor of otolaryngology at Newcastle University (England), in an interview.
The study was published in The Lancet.
Tonsillectomies have become much less common over the past several decades as questions have arisen about their value. In the United States, the number of procedures performed each year plunged from a high of 1.4 million in 1959 to an estimated 286,000 tonsillectomies performed in children under 15 and 120,000 in people aged 15 in 2010.
It’s harder for adults to tolerate tonsillectomies than children, Dr. Wilson said. In children, surgeons can easily remove tonsils by scraping them off the throat’s side walls. But, she said, “an adult tonsillectomy is more akin to taking off the skin of an unripe orange, so it’s harder work for the surgeon and more traumatizing for the wall of the adult patient’s pharynx. We can only assume that this greater amount of fibrous tissue reflects the cumulative effect of infections over a period of years.”
While tonsillectomies are still performed hundreds of times a day in adults in the United States, a 2014 Cochrane Library review found there’s “insufficient information “to support them versus nonsurgical care as treatments to reduce sore throats.”
For the new multicenter, open-label, randomized study, researchers randomly assigned patients aged 16 and older with recurrent acute tonsillitis to immediate tonsillectomy or nonsurgical management, which Dr. Wilson said can include cold fluids, honey, analgesics/anti-inflammatories. and anesthetic throat lozenges. The study was conducted between 2015 and 2018.
Ultimately, there were 224 and 204 patients, respectively, in the two groups (average age = 23, [19-30], 78% female, 90% White).
Patients who underwent tonsillectomies versus nonsurgical treatment had fewer sore throats over 2 years (median 23 days [IQR 11-46 days] vs. 30 days [14-65 days]) with an incident rate ratio of 0.53 (95% confidence interval, 0.43-0.65, P < 0.0001) after adjustment for clinic site and baseline severity.
The study also shows that “adults who have severe recurrent throat infections with a frequency of seven episodes within 1 year, five or more for 2 consecutive years, or three or more in 3 consecutive years will suffer fewer days of sore throat in the 2 years following tonsillectomy than if they had kept their tonsils,” Dr. Wilson said.
The study doesn’t examine longer-term consequences. A 2018 study of children linked tonsillectomies to “significantly increased relative risk of later respiratory, allergic, and infectious diseases.”
In the new study, nearly 4 in 10 (39%) of the tonsillectomy patients had adverse events linked to the surgeries, and bleeding (19%) was the most common adverse effect. The researchers also estimated that “tonsillectomy has a high probability of being considered cost-effective.”
“Whichever way the results were analyzed and confounding variables allowed for, the result always seems to be the same: Tonsillectomy applied using current qualifying criteria was a worthwhile procedure,” Dr. Wilson said.
Dr. Wilson noted that tonsillectomy patients will suffer a persistent sore throat after surgery, “about the same as a bad episode of tonsillitis.” And she said patients will need to adjust their diet for a few days and take 1-2 weeks off work.
In an interview, internal medicine physician Noel Deep, MD, of Antigo, Wisc., said antibiotics are a common treatment for tonsillitis in primary care clinics. According to him, the United States doesn’t have guidelines for tonsillectomies in adults. He believes they can be considered if tonsillitis keeps recurring three to five times a year and disrupts quality of life.
Dr. Deep said the new study “reinforces the benefit of tonsillectomies. Several studies from Germany, Sweden, Finland, and the United Kingdom have demonstrated benefits of tonsillectomies, but they were only for short periods of less than a year and lacked long-term data.”
He noted that “there is no clear evidence as to when to recommend tonsillectomies.” Clinicians should talk to patients about the potential that tonsillectomies will reduce sore throat episodes and cost the patient less in the long run, he said. It’s also important, he said, to make sure tonsillitis is bacterial before prescribing antibiotics.
The United Kingdom’s National Institute for Health Research funded the study. Dr. Wilson disclosed support for meetings/travel from ENT Scotland, and the other authors report various disclosures, including grants and contracts. Dr. Deep serves on the editorial advisory board of Internal Medicine News and is chair of the American Medical Association Council on Science and Public Health.
FROM THE LANCET
The weird world of hydrogels: How they’ll change health care
Imagine a day when a simple injection prompts a broken bone to heal. When tiny, ingestible devices linger in the body, unnoticed, tracking our health or delivering life-saving medications. When brain and heart implants mesh with flesh so seamlessly that the body thinks they’ve been there all along.
These are the dreams of materials scientists who have toiled for decades to mimic the complex architecture of the human body in hopes of replacing broken parts or treating disease.
The problem, say bioengineers, is that most replacement and corrective parts – from prosthetics to pacemakers – are made of hard, dry, lifeless materials, like metal or plastic, while biological tissue is soft, wet, and living.
The body knows the difference and tends to reject imitations.
Enter hydrogels, three-dimensional networks of molecules swollen with – by definition – water.
First described in 1960 by creators of soft contact lenses, these weird, shape-shifting substances are able to morph from liquid to solid to a squishy in-between. (Early, simple uses include hair gel or Jell-O.). Slow to gain attention, growing to just 1,000 studies published by 1982, they’ve become the subject of intense study recently, with 100,000 papers published by 2020, and 3,800 already this year alone.
As chemists, biologists, and engineers begin to work more with one another and with medical doctors,
“We are, essentially, hydrogels,” said Benjamin Wiley, PhD, a chemistry professor at Duke University in Durham, N.C. “As people develop new hydrogels that more closely match the tissues in our body, we’ll be able to treat a whole host of ailments we couldn’t treat before.”
From contact lenses to brain implants
Put simply, a hydrogel is like a mesh bag of water.
The mesh is made of polymers, or spaghetti-like strands of molecules, stitched together in a repeating pattern and swollen with H2O, much like the way 3D matrixes in our body surround, support, and give structure to our cells and tissues.
“Imagine a soccer net, with all of these long fibers woven together to create the net,” said Eric Appel, PhD, associate professor of materials science and engineering at Stanford (Calif.) University.
While the broader category of “gels” could be filled with anything, including chemical solvents, water is the key ingredient that sets hydrogels apart, making them ideal for, as some scientists put it, “merging humans and machines.”
Human bones are about 25% water, while muscles hover around 70% and the brain is 85%. The precious liquid plays a host of critical roles, from shuttling nutrients in and waste out to helping cells talk to each other.
Lab-made hydrogels can be loaded with cargo (like a ball in the net), including cells or drugs that help mimic some of those functions.
Hydrogels are soft and pliable like flesh. So, if used in implants, they may be less likely to damage surrounding tissue.
“Think about a metal spoon in your bowl of pudding. As you’re shaking the bowl, the spoon doesn’t stay in place, and you get scarring around the spoon,” said Christina Tringides, PhD, a materials scientist who studies neural engineering. That, she says, is exactly what happens to brain implants when patients breathe or move. “It’s a mechanical mismatch. But with hydrogels, you could get perfect mechanical matching.”
Hydrogels also tend to be nontoxic, so the immune system may be less likely to attack them as foreign bodies.
All this has made hydrogels the new darling of the bioengineering world.
“There has been an absolute explosion of interest in these materials,” Dr. Appel said.
Smarter drug delivery and ingestible electronics
Early versions of hydrogels were thick and gooey, making it hard to get them inside the body.
“Think of a block of Jell-O. You couldn’t inject something like that,” Dr. Appel said.
But Dr. Appel, whose lab develops new drug delivery systems, has been tinkering with gel formulas for years in hopes that these high-tech globs could someday ferry timed-release drugs to just the right spot in the body.
His new hydrogels start as fully formed gels (which help preserve the drug contents) inside a syringe. But once the plunger is pushed, they magically shape-shift to a liquid thin enough to flow easily through a standard needle. Upon exit, they immediately reform into gels, protecting the inherent cargo from degrading.
This could be a game changer at a time when many cutting-edge drugs – think Humira for arthritis or Ozempic for type 2 diabetes – are made of quickly degrading proteins too large and complex to simply jam into a pill. Instead, they must be injected, often frequently.
“Because the gel takes months to dissolve, it slowly delivers the drug over time,” Dr. Appel said. “You could conceivably go from a shot once a week to once every 4 months.”
Such slow-release hydrogels could make vaccines last longer, in turn teaching the body to better resist emerging virus variants, and deliver tumor-busting therapies more precisely, said Dr. Appel, who has formed a startup and hopes to fast-track the first hydrogel drug delivery system to clinical trials within a few years.
Meanwhile, another team at the Massachusetts Institute of Technology has taken a different approach, developing a standard-sized ingestible hydrogel pill that swells up like a puffer fish in the stomach, lasting a month and slowly releasing drugs all the while. To remove the pill, a patient simply drinks a salt-based solution that shrivels the ping-pong ball–sized device so it can be passed out of the body.
In a paper in Nature Communications, the scientists showed the puffer fish pill could also be loaded with tiny cameras or monitors to track conditions like ulcers or cancer.
“The dream is to have a Jell-O-like smart pill that, once swallowed, stays in the stomach and monitors the patient’s health,” said Xuanhe Zhao, PhD, a researcher on the project and associate professor of mechanical engineering at MIT.
Building joints and regrowing bones
Since the 1970s, researchers have mulled using hydrogels to replace human cartilage, a remarkably strong and flexible tissue made of about 90% water but able to withstand the weight of a car on an area about the size of a coin.
Until recently, those efforts have largely failed. Meaning when knee cartilage wears down, things like cartilage transplants, drilling holes to stimulate new growth, or total joint replacements – all of which require lengthy rehab – are the only options.
But that may be about to change.
Dr. Wiley and his colleagues at Duke recently reported that they’d developed the first gel-based cartilage substitute even stronger and more durable than the real thing.
By attaching their hydrogel to a titanium backing to help stick it in place, they hope to repair damaged cartilage “much like a dentist fills a cavity” long before surgery is necessary.
They too have partnered with industry to bring their hydrogel to market – starting with knees.
“Ultimately, the goal is to do any joint – hips, ankles, fingers, and toes,” Dr. Wiley said.
At the University of Toronto, chemist Karina Carneiro, PhD, and dentist Christopher McCulloch, DDS, are also thinking big.
In a recent paper in Proceedings of the National Academy of Sciences, they describe a hydrogel, designed by Dr. Carneiro and made of DNA, that can be injected, migrate to a defect in bone – an irreparable break, hole from surgery, or jawbone withered by age – and fill in the gap like putty. But not only does it patch the hole, it prompts the bone to regenerate.
In rats with holes in their skulls due to surgery, they found that the treatment did not work as well as the existing gold standard for repairing holes in bone – grafting bone from elsewhere in the body. But it did work.
“These are very early days for DNA hydrogels,” cautioned Dr. McCulloch, a study coauthor and professor in the Faculty of Dentistry, noting that it will likely be a decade or more before such technology could be available to patients. “But there is the potential that DNA hydrogel could someday grow bone without having to have highly invasive surgical procedures. That’s a significant advancement.”
A sci-fi future
Perhaps the wildest, and weirdest, potential applications of hydrogels come in the realm of human-machine interaction.
Numerous companies are already dabbling in neural prosthetic or brain computer interfaces that might someday, for instance, let someone who is paralyzed and can’t speak write on a laptop using their thoughts.
The spoon-in-the-Jell-O problem has been a major stumbling block.
But Dr. Tringides, who recently earned her PhD in biophysics from Harvard, is working on it.
She and her team have developed a seaweed-based hydrogel loaded with tiny flecks of nanomaterials that can not only meld nicely into squishy brain tissue but also conduct electricity.
Within a decade, she says, this could replace the clunky platinum metal discs used for electrocorticography – recording electrical activity in the brain to identify where seizures start or doing precise brain surgery.
In 30 to 50 years? Let your imagination run wild.
“I’m a skeptic. I like to take research step by step,” she said. “But things are definitely progressing in an interesting direction.”
A version of this article first appeared on WebMD.com.
Imagine a day when a simple injection prompts a broken bone to heal. When tiny, ingestible devices linger in the body, unnoticed, tracking our health or delivering life-saving medications. When brain and heart implants mesh with flesh so seamlessly that the body thinks they’ve been there all along.
These are the dreams of materials scientists who have toiled for decades to mimic the complex architecture of the human body in hopes of replacing broken parts or treating disease.
The problem, say bioengineers, is that most replacement and corrective parts – from prosthetics to pacemakers – are made of hard, dry, lifeless materials, like metal or plastic, while biological tissue is soft, wet, and living.
The body knows the difference and tends to reject imitations.
Enter hydrogels, three-dimensional networks of molecules swollen with – by definition – water.
First described in 1960 by creators of soft contact lenses, these weird, shape-shifting substances are able to morph from liquid to solid to a squishy in-between. (Early, simple uses include hair gel or Jell-O.). Slow to gain attention, growing to just 1,000 studies published by 1982, they’ve become the subject of intense study recently, with 100,000 papers published by 2020, and 3,800 already this year alone.
As chemists, biologists, and engineers begin to work more with one another and with medical doctors,
“We are, essentially, hydrogels,” said Benjamin Wiley, PhD, a chemistry professor at Duke University in Durham, N.C. “As people develop new hydrogels that more closely match the tissues in our body, we’ll be able to treat a whole host of ailments we couldn’t treat before.”
From contact lenses to brain implants
Put simply, a hydrogel is like a mesh bag of water.
The mesh is made of polymers, or spaghetti-like strands of molecules, stitched together in a repeating pattern and swollen with H2O, much like the way 3D matrixes in our body surround, support, and give structure to our cells and tissues.
“Imagine a soccer net, with all of these long fibers woven together to create the net,” said Eric Appel, PhD, associate professor of materials science and engineering at Stanford (Calif.) University.
While the broader category of “gels” could be filled with anything, including chemical solvents, water is the key ingredient that sets hydrogels apart, making them ideal for, as some scientists put it, “merging humans and machines.”
Human bones are about 25% water, while muscles hover around 70% and the brain is 85%. The precious liquid plays a host of critical roles, from shuttling nutrients in and waste out to helping cells talk to each other.
Lab-made hydrogels can be loaded with cargo (like a ball in the net), including cells or drugs that help mimic some of those functions.
Hydrogels are soft and pliable like flesh. So, if used in implants, they may be less likely to damage surrounding tissue.
“Think about a metal spoon in your bowl of pudding. As you’re shaking the bowl, the spoon doesn’t stay in place, and you get scarring around the spoon,” said Christina Tringides, PhD, a materials scientist who studies neural engineering. That, she says, is exactly what happens to brain implants when patients breathe or move. “It’s a mechanical mismatch. But with hydrogels, you could get perfect mechanical matching.”
Hydrogels also tend to be nontoxic, so the immune system may be less likely to attack them as foreign bodies.
All this has made hydrogels the new darling of the bioengineering world.
“There has been an absolute explosion of interest in these materials,” Dr. Appel said.
Smarter drug delivery and ingestible electronics
Early versions of hydrogels were thick and gooey, making it hard to get them inside the body.
“Think of a block of Jell-O. You couldn’t inject something like that,” Dr. Appel said.
But Dr. Appel, whose lab develops new drug delivery systems, has been tinkering with gel formulas for years in hopes that these high-tech globs could someday ferry timed-release drugs to just the right spot in the body.
His new hydrogels start as fully formed gels (which help preserve the drug contents) inside a syringe. But once the plunger is pushed, they magically shape-shift to a liquid thin enough to flow easily through a standard needle. Upon exit, they immediately reform into gels, protecting the inherent cargo from degrading.
This could be a game changer at a time when many cutting-edge drugs – think Humira for arthritis or Ozempic for type 2 diabetes – are made of quickly degrading proteins too large and complex to simply jam into a pill. Instead, they must be injected, often frequently.
“Because the gel takes months to dissolve, it slowly delivers the drug over time,” Dr. Appel said. “You could conceivably go from a shot once a week to once every 4 months.”
Such slow-release hydrogels could make vaccines last longer, in turn teaching the body to better resist emerging virus variants, and deliver tumor-busting therapies more precisely, said Dr. Appel, who has formed a startup and hopes to fast-track the first hydrogel drug delivery system to clinical trials within a few years.
Meanwhile, another team at the Massachusetts Institute of Technology has taken a different approach, developing a standard-sized ingestible hydrogel pill that swells up like a puffer fish in the stomach, lasting a month and slowly releasing drugs all the while. To remove the pill, a patient simply drinks a salt-based solution that shrivels the ping-pong ball–sized device so it can be passed out of the body.
In a paper in Nature Communications, the scientists showed the puffer fish pill could also be loaded with tiny cameras or monitors to track conditions like ulcers or cancer.
“The dream is to have a Jell-O-like smart pill that, once swallowed, stays in the stomach and monitors the patient’s health,” said Xuanhe Zhao, PhD, a researcher on the project and associate professor of mechanical engineering at MIT.
Building joints and regrowing bones
Since the 1970s, researchers have mulled using hydrogels to replace human cartilage, a remarkably strong and flexible tissue made of about 90% water but able to withstand the weight of a car on an area about the size of a coin.
Until recently, those efforts have largely failed. Meaning when knee cartilage wears down, things like cartilage transplants, drilling holes to stimulate new growth, or total joint replacements – all of which require lengthy rehab – are the only options.
But that may be about to change.
Dr. Wiley and his colleagues at Duke recently reported that they’d developed the first gel-based cartilage substitute even stronger and more durable than the real thing.
By attaching their hydrogel to a titanium backing to help stick it in place, they hope to repair damaged cartilage “much like a dentist fills a cavity” long before surgery is necessary.
They too have partnered with industry to bring their hydrogel to market – starting with knees.
“Ultimately, the goal is to do any joint – hips, ankles, fingers, and toes,” Dr. Wiley said.
At the University of Toronto, chemist Karina Carneiro, PhD, and dentist Christopher McCulloch, DDS, are also thinking big.
In a recent paper in Proceedings of the National Academy of Sciences, they describe a hydrogel, designed by Dr. Carneiro and made of DNA, that can be injected, migrate to a defect in bone – an irreparable break, hole from surgery, or jawbone withered by age – and fill in the gap like putty. But not only does it patch the hole, it prompts the bone to regenerate.
In rats with holes in their skulls due to surgery, they found that the treatment did not work as well as the existing gold standard for repairing holes in bone – grafting bone from elsewhere in the body. But it did work.
“These are very early days for DNA hydrogels,” cautioned Dr. McCulloch, a study coauthor and professor in the Faculty of Dentistry, noting that it will likely be a decade or more before such technology could be available to patients. “But there is the potential that DNA hydrogel could someday grow bone without having to have highly invasive surgical procedures. That’s a significant advancement.”
A sci-fi future
Perhaps the wildest, and weirdest, potential applications of hydrogels come in the realm of human-machine interaction.
Numerous companies are already dabbling in neural prosthetic or brain computer interfaces that might someday, for instance, let someone who is paralyzed and can’t speak write on a laptop using their thoughts.
The spoon-in-the-Jell-O problem has been a major stumbling block.
But Dr. Tringides, who recently earned her PhD in biophysics from Harvard, is working on it.
She and her team have developed a seaweed-based hydrogel loaded with tiny flecks of nanomaterials that can not only meld nicely into squishy brain tissue but also conduct electricity.
Within a decade, she says, this could replace the clunky platinum metal discs used for electrocorticography – recording electrical activity in the brain to identify where seizures start or doing precise brain surgery.
In 30 to 50 years? Let your imagination run wild.
“I’m a skeptic. I like to take research step by step,” she said. “But things are definitely progressing in an interesting direction.”
A version of this article first appeared on WebMD.com.
Imagine a day when a simple injection prompts a broken bone to heal. When tiny, ingestible devices linger in the body, unnoticed, tracking our health or delivering life-saving medications. When brain and heart implants mesh with flesh so seamlessly that the body thinks they’ve been there all along.
These are the dreams of materials scientists who have toiled for decades to mimic the complex architecture of the human body in hopes of replacing broken parts or treating disease.
The problem, say bioengineers, is that most replacement and corrective parts – from prosthetics to pacemakers – are made of hard, dry, lifeless materials, like metal or plastic, while biological tissue is soft, wet, and living.
The body knows the difference and tends to reject imitations.
Enter hydrogels, three-dimensional networks of molecules swollen with – by definition – water.
First described in 1960 by creators of soft contact lenses, these weird, shape-shifting substances are able to morph from liquid to solid to a squishy in-between. (Early, simple uses include hair gel or Jell-O.). Slow to gain attention, growing to just 1,000 studies published by 1982, they’ve become the subject of intense study recently, with 100,000 papers published by 2020, and 3,800 already this year alone.
As chemists, biologists, and engineers begin to work more with one another and with medical doctors,
“We are, essentially, hydrogels,” said Benjamin Wiley, PhD, a chemistry professor at Duke University in Durham, N.C. “As people develop new hydrogels that more closely match the tissues in our body, we’ll be able to treat a whole host of ailments we couldn’t treat before.”
From contact lenses to brain implants
Put simply, a hydrogel is like a mesh bag of water.
The mesh is made of polymers, or spaghetti-like strands of molecules, stitched together in a repeating pattern and swollen with H2O, much like the way 3D matrixes in our body surround, support, and give structure to our cells and tissues.
“Imagine a soccer net, with all of these long fibers woven together to create the net,” said Eric Appel, PhD, associate professor of materials science and engineering at Stanford (Calif.) University.
While the broader category of “gels” could be filled with anything, including chemical solvents, water is the key ingredient that sets hydrogels apart, making them ideal for, as some scientists put it, “merging humans and machines.”
Human bones are about 25% water, while muscles hover around 70% and the brain is 85%. The precious liquid plays a host of critical roles, from shuttling nutrients in and waste out to helping cells talk to each other.
Lab-made hydrogels can be loaded with cargo (like a ball in the net), including cells or drugs that help mimic some of those functions.
Hydrogels are soft and pliable like flesh. So, if used in implants, they may be less likely to damage surrounding tissue.
“Think about a metal spoon in your bowl of pudding. As you’re shaking the bowl, the spoon doesn’t stay in place, and you get scarring around the spoon,” said Christina Tringides, PhD, a materials scientist who studies neural engineering. That, she says, is exactly what happens to brain implants when patients breathe or move. “It’s a mechanical mismatch. But with hydrogels, you could get perfect mechanical matching.”
Hydrogels also tend to be nontoxic, so the immune system may be less likely to attack them as foreign bodies.
All this has made hydrogels the new darling of the bioengineering world.
“There has been an absolute explosion of interest in these materials,” Dr. Appel said.
Smarter drug delivery and ingestible electronics
Early versions of hydrogels were thick and gooey, making it hard to get them inside the body.
“Think of a block of Jell-O. You couldn’t inject something like that,” Dr. Appel said.
But Dr. Appel, whose lab develops new drug delivery systems, has been tinkering with gel formulas for years in hopes that these high-tech globs could someday ferry timed-release drugs to just the right spot in the body.
His new hydrogels start as fully formed gels (which help preserve the drug contents) inside a syringe. But once the plunger is pushed, they magically shape-shift to a liquid thin enough to flow easily through a standard needle. Upon exit, they immediately reform into gels, protecting the inherent cargo from degrading.
This could be a game changer at a time when many cutting-edge drugs – think Humira for arthritis or Ozempic for type 2 diabetes – are made of quickly degrading proteins too large and complex to simply jam into a pill. Instead, they must be injected, often frequently.
“Because the gel takes months to dissolve, it slowly delivers the drug over time,” Dr. Appel said. “You could conceivably go from a shot once a week to once every 4 months.”
Such slow-release hydrogels could make vaccines last longer, in turn teaching the body to better resist emerging virus variants, and deliver tumor-busting therapies more precisely, said Dr. Appel, who has formed a startup and hopes to fast-track the first hydrogel drug delivery system to clinical trials within a few years.
Meanwhile, another team at the Massachusetts Institute of Technology has taken a different approach, developing a standard-sized ingestible hydrogel pill that swells up like a puffer fish in the stomach, lasting a month and slowly releasing drugs all the while. To remove the pill, a patient simply drinks a salt-based solution that shrivels the ping-pong ball–sized device so it can be passed out of the body.
In a paper in Nature Communications, the scientists showed the puffer fish pill could also be loaded with tiny cameras or monitors to track conditions like ulcers or cancer.
“The dream is to have a Jell-O-like smart pill that, once swallowed, stays in the stomach and monitors the patient’s health,” said Xuanhe Zhao, PhD, a researcher on the project and associate professor of mechanical engineering at MIT.
Building joints and regrowing bones
Since the 1970s, researchers have mulled using hydrogels to replace human cartilage, a remarkably strong and flexible tissue made of about 90% water but able to withstand the weight of a car on an area about the size of a coin.
Until recently, those efforts have largely failed. Meaning when knee cartilage wears down, things like cartilage transplants, drilling holes to stimulate new growth, or total joint replacements – all of which require lengthy rehab – are the only options.
But that may be about to change.
Dr. Wiley and his colleagues at Duke recently reported that they’d developed the first gel-based cartilage substitute even stronger and more durable than the real thing.
By attaching their hydrogel to a titanium backing to help stick it in place, they hope to repair damaged cartilage “much like a dentist fills a cavity” long before surgery is necessary.
They too have partnered with industry to bring their hydrogel to market – starting with knees.
“Ultimately, the goal is to do any joint – hips, ankles, fingers, and toes,” Dr. Wiley said.
At the University of Toronto, chemist Karina Carneiro, PhD, and dentist Christopher McCulloch, DDS, are also thinking big.
In a recent paper in Proceedings of the National Academy of Sciences, they describe a hydrogel, designed by Dr. Carneiro and made of DNA, that can be injected, migrate to a defect in bone – an irreparable break, hole from surgery, or jawbone withered by age – and fill in the gap like putty. But not only does it patch the hole, it prompts the bone to regenerate.
In rats with holes in their skulls due to surgery, they found that the treatment did not work as well as the existing gold standard for repairing holes in bone – grafting bone from elsewhere in the body. But it did work.
“These are very early days for DNA hydrogels,” cautioned Dr. McCulloch, a study coauthor and professor in the Faculty of Dentistry, noting that it will likely be a decade or more before such technology could be available to patients. “But there is the potential that DNA hydrogel could someday grow bone without having to have highly invasive surgical procedures. That’s a significant advancement.”
A sci-fi future
Perhaps the wildest, and weirdest, potential applications of hydrogels come in the realm of human-machine interaction.
Numerous companies are already dabbling in neural prosthetic or brain computer interfaces that might someday, for instance, let someone who is paralyzed and can’t speak write on a laptop using their thoughts.
The spoon-in-the-Jell-O problem has been a major stumbling block.
But Dr. Tringides, who recently earned her PhD in biophysics from Harvard, is working on it.
She and her team have developed a seaweed-based hydrogel loaded with tiny flecks of nanomaterials that can not only meld nicely into squishy brain tissue but also conduct electricity.
Within a decade, she says, this could replace the clunky platinum metal discs used for electrocorticography – recording electrical activity in the brain to identify where seizures start or doing precise brain surgery.
In 30 to 50 years? Let your imagination run wild.
“I’m a skeptic. I like to take research step by step,” she said. “But things are definitely progressing in an interesting direction.”
A version of this article first appeared on WebMD.com.
Should you prescribe bioidentical hormones for menopause?
BALTIMORE – according to an expert at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists (ACOG).
Clinicians write an estimated 26 to 33 million prescriptions for compounded bioidentical hormone therapy (cBHT) every year, and almost 41% of menopausal women who need treatment try cBHT during their lives. But these drugs lack the approval for this indication from the Food and Drug Administration.
“There is a public perception that this is natural, safer, and anti-aging,” said Robert Kauffman, MD, a professor of obstetrics and gynecology and assistant dean for research at Texas Tech University Health Sciences Center in Amarillo.
Following the 2002 Women’s Health Initiative report showing a link between hormone therapy (HT) and an increase in the incidence of breast cancer, medical schools have slowed or paused instructing trainees on the traditional treatment, Dr. Kauffman said. The association was later determined to be spurious: HT is not associated with a risk for all-cause mortality or deaths from cardiovascular disease or cancer. However, HT still is largely ignored by younger physicians, Dr. Kauffman said, because of unsubstantiated “dangers” such as heart attack, stroke, and deep vein thrombosis.
The lack of education on HT for medical school students and residents has “opened the door to unsubstantiated marketing claims and practices” for cBHT, Dr. Kauffman said. “Hence, the use of compounded bioidentical hormone therapy has increased” as clinicians look for alternatives.
Groups including ACOG, the North American Menopause Society (NAMS), and the U.S. Preventive Services Task Force recommend against the use of Non–FDA-approved therapies such as cBHT, except for narrow indications. Dr. Kauffman said that drug manufacturers have not conducted randomized controlled trials or observational studies on cBHT in treating menopause.
He cited studies showing quality problems with the compounding process of these drugs, and wide variations in the amount of actual ingredients from product labels. One 2021 study published in Menopause comparing patients taking cBHT or FDA-approved HT found that side effects were significantly higher in the cBHT group (57.6% vs. 14.8%; P < .0001).
But manufacturers of cBHT claim that their products prevent cardiovascular disease and Alzheimer’s disease and decrease the risk for breast cancer and stroke – assertions that are at best unproven, according to Dr. Kauffman.
The National Academies of Sciences, Engineering, and Medicine in 2020 said that clinicians have a duty to inform patients of the insufficient evidence to support clinical use of cBHT and should prescribe the products only to patients with documented allergies to an active ingredient in an FDA-approved agent or who require an alternative dosage.
Patients may also have to pay much more out of pocket for cBHT products because they often are not covered by insurance. Generic HT products, meanwhile, are relatively inexpensive and typically are covered, he noted.
“We have to be careful to avoid financial harm to patients by prescribing things, which are much more expensive than those which are usually available,” Dr. Kauffman said.
Prescribing any non–FDA-approved product, especially when biosimilars are available, places physicians at legal risk, Dr. Kauffman said. Physicians who recommend cBHT should inform patients that the products are not FDA approved and carefully document this discussion in the patient’s electronic health record. State boards of medicine can sanction physicians for “coercion” for prescribing cBHT products without mentioning alternatives, he added.
JoAnn Pinkerton, MD, professor of obstetrics and gynecology at the University of Virginia, Charlottesville, and executive director emeritus of NAMS, who attended the session, praised Dr. Kauffman for providing a balanced and evidence-based overview of the subject.

“There are issues concerning safety, contaminants, and not knowing exactly what dose you’re getting,” with compounded hormones, Dr. Pinkerton said. “They’re being hyped as safer and more effective when in reality, we don’t have any studies that show that information.”
Dr. Pinkerton noted that while a compounded form of physiological testosterone might be relatively reliable, “if you’re using something like a pellet that is super physiologic with incredibly high doses, that you really don’t have any information to stand on that it’s safe or effective ... it might be putting your license at risk.”
A version of this article first appeared on Medscape.com.
BALTIMORE – according to an expert at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists (ACOG).
Clinicians write an estimated 26 to 33 million prescriptions for compounded bioidentical hormone therapy (cBHT) every year, and almost 41% of menopausal women who need treatment try cBHT during their lives. But these drugs lack the approval for this indication from the Food and Drug Administration.
“There is a public perception that this is natural, safer, and anti-aging,” said Robert Kauffman, MD, a professor of obstetrics and gynecology and assistant dean for research at Texas Tech University Health Sciences Center in Amarillo.
Following the 2002 Women’s Health Initiative report showing a link between hormone therapy (HT) and an increase in the incidence of breast cancer, medical schools have slowed or paused instructing trainees on the traditional treatment, Dr. Kauffman said. The association was later determined to be spurious: HT is not associated with a risk for all-cause mortality or deaths from cardiovascular disease or cancer. However, HT still is largely ignored by younger physicians, Dr. Kauffman said, because of unsubstantiated “dangers” such as heart attack, stroke, and deep vein thrombosis.
The lack of education on HT for medical school students and residents has “opened the door to unsubstantiated marketing claims and practices” for cBHT, Dr. Kauffman said. “Hence, the use of compounded bioidentical hormone therapy has increased” as clinicians look for alternatives.
Groups including ACOG, the North American Menopause Society (NAMS), and the U.S. Preventive Services Task Force recommend against the use of Non–FDA-approved therapies such as cBHT, except for narrow indications. Dr. Kauffman said that drug manufacturers have not conducted randomized controlled trials or observational studies on cBHT in treating menopause.
He cited studies showing quality problems with the compounding process of these drugs, and wide variations in the amount of actual ingredients from product labels. One 2021 study published in Menopause comparing patients taking cBHT or FDA-approved HT found that side effects were significantly higher in the cBHT group (57.6% vs. 14.8%; P < .0001).
But manufacturers of cBHT claim that their products prevent cardiovascular disease and Alzheimer’s disease and decrease the risk for breast cancer and stroke – assertions that are at best unproven, according to Dr. Kauffman.
The National Academies of Sciences, Engineering, and Medicine in 2020 said that clinicians have a duty to inform patients of the insufficient evidence to support clinical use of cBHT and should prescribe the products only to patients with documented allergies to an active ingredient in an FDA-approved agent or who require an alternative dosage.
Patients may also have to pay much more out of pocket for cBHT products because they often are not covered by insurance. Generic HT products, meanwhile, are relatively inexpensive and typically are covered, he noted.
“We have to be careful to avoid financial harm to patients by prescribing things, which are much more expensive than those which are usually available,” Dr. Kauffman said.
Prescribing any non–FDA-approved product, especially when biosimilars are available, places physicians at legal risk, Dr. Kauffman said. Physicians who recommend cBHT should inform patients that the products are not FDA approved and carefully document this discussion in the patient’s electronic health record. State boards of medicine can sanction physicians for “coercion” for prescribing cBHT products without mentioning alternatives, he added.
JoAnn Pinkerton, MD, professor of obstetrics and gynecology at the University of Virginia, Charlottesville, and executive director emeritus of NAMS, who attended the session, praised Dr. Kauffman for providing a balanced and evidence-based overview of the subject.
“There are issues concerning safety, contaminants, and not knowing exactly what dose you’re getting,” with compounded hormones, Dr. Pinkerton said. “They’re being hyped as safer and more effective when in reality, we don’t have any studies that show that information.”
Dr. Pinkerton noted that while a compounded form of physiological testosterone might be relatively reliable, “if you’re using something like a pellet that is super physiologic with incredibly high doses, that you really don’t have any information to stand on that it’s safe or effective ... it might be putting your license at risk.”
A version of this article first appeared on Medscape.com.
BALTIMORE – according to an expert at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists (ACOG).
Clinicians write an estimated 26 to 33 million prescriptions for compounded bioidentical hormone therapy (cBHT) every year, and almost 41% of menopausal women who need treatment try cBHT during their lives. But these drugs lack the approval for this indication from the Food and Drug Administration.
“There is a public perception that this is natural, safer, and anti-aging,” said Robert Kauffman, MD, a professor of obstetrics and gynecology and assistant dean for research at Texas Tech University Health Sciences Center in Amarillo.
Following the 2002 Women’s Health Initiative report showing a link between hormone therapy (HT) and an increase in the incidence of breast cancer, medical schools have slowed or paused instructing trainees on the traditional treatment, Dr. Kauffman said. The association was later determined to be spurious: HT is not associated with a risk for all-cause mortality or deaths from cardiovascular disease or cancer. However, HT still is largely ignored by younger physicians, Dr. Kauffman said, because of unsubstantiated “dangers” such as heart attack, stroke, and deep vein thrombosis.
The lack of education on HT for medical school students and residents has “opened the door to unsubstantiated marketing claims and practices” for cBHT, Dr. Kauffman said. “Hence, the use of compounded bioidentical hormone therapy has increased” as clinicians look for alternatives.
Groups including ACOG, the North American Menopause Society (NAMS), and the U.S. Preventive Services Task Force recommend against the use of Non–FDA-approved therapies such as cBHT, except for narrow indications. Dr. Kauffman said that drug manufacturers have not conducted randomized controlled trials or observational studies on cBHT in treating menopause.
He cited studies showing quality problems with the compounding process of these drugs, and wide variations in the amount of actual ingredients from product labels. One 2021 study published in Menopause comparing patients taking cBHT or FDA-approved HT found that side effects were significantly higher in the cBHT group (57.6% vs. 14.8%; P < .0001).
But manufacturers of cBHT claim that their products prevent cardiovascular disease and Alzheimer’s disease and decrease the risk for breast cancer and stroke – assertions that are at best unproven, according to Dr. Kauffman.
The National Academies of Sciences, Engineering, and Medicine in 2020 said that clinicians have a duty to inform patients of the insufficient evidence to support clinical use of cBHT and should prescribe the products only to patients with documented allergies to an active ingredient in an FDA-approved agent or who require an alternative dosage.
Patients may also have to pay much more out of pocket for cBHT products because they often are not covered by insurance. Generic HT products, meanwhile, are relatively inexpensive and typically are covered, he noted.
“We have to be careful to avoid financial harm to patients by prescribing things, which are much more expensive than those which are usually available,” Dr. Kauffman said.
Prescribing any non–FDA-approved product, especially when biosimilars are available, places physicians at legal risk, Dr. Kauffman said. Physicians who recommend cBHT should inform patients that the products are not FDA approved and carefully document this discussion in the patient’s electronic health record. State boards of medicine can sanction physicians for “coercion” for prescribing cBHT products without mentioning alternatives, he added.
JoAnn Pinkerton, MD, professor of obstetrics and gynecology at the University of Virginia, Charlottesville, and executive director emeritus of NAMS, who attended the session, praised Dr. Kauffman for providing a balanced and evidence-based overview of the subject.
“There are issues concerning safety, contaminants, and not knowing exactly what dose you’re getting,” with compounded hormones, Dr. Pinkerton said. “They’re being hyped as safer and more effective when in reality, we don’t have any studies that show that information.”
Dr. Pinkerton noted that while a compounded form of physiological testosterone might be relatively reliable, “if you’re using something like a pellet that is super physiologic with incredibly high doses, that you really don’t have any information to stand on that it’s safe or effective ... it might be putting your license at risk.”
A version of this article first appeared on Medscape.com.
AT ACOG 2023