User login
Hospitalists Can Be Good Stewards of Healthcare Dollars

Are hospitals going to be allowed to start “patient profiling” in order to reduce costs? Hospitals already frequently operate their own state departments and perform “extraordinary medical renditions” on uninsured, critically ill noncitizens. Do you really expect us to believe they are identifying these patients in order to provide them more appropriate services? My impression is that since this group has been identified as a cost driver, the aim of any intervention is saving money for the hospital rather than rendering appropriate care.
—Tom Horiagon
Dr. Hospitalist responds:
As hospitalists, we are best positioned to manage the balance among medical, social, and fiduciary responsibilities. The article addresses the data that shows what most of us already know: Most patients who have multiple readmissions have many co-morbid conditions and/or psychiatric and social issues. Hospitalists have the opportunity to use everything in the patient’s history and profile to prescribe the appropriate treatment plan. When we find the right solutions, it would be helpful to us all if they were not only cost effective but also right for the patient.
Although I prefer to not use the term “patient profiling” because of the associated negative connotations, I do believe there are occasions we face with our patients when the most “appropriate service” may not be clinically relevant at all.
For example, we recently began a quality initiative project in our hospital to identify those with acute or chronic pain and the most frequent admissions (greater than 10) in a calendar year. We identified a patient who had a total of 43 admissions across four different hospitals in one calendar year. Clearly, the best care for this individual would be to get him an apartment.
We know that many of these “frequent flyers” tend to absorb vast amounts of our healthcare dollars with multiple imaging studies, lab work, and time taken away from other patients, not to mention the emotional toll some of these patients take on the clinical staff.
Discussions on such matters as tort reform, futile care, and patient nonadherence (many factors and much more complex) have been going on for some time. I don’t see our politicians developing the intestinal fortitude to address these problems any time soon. With our national healthcare expenditures reaching $2.9 trillion (or $9,255 per person in 2013, per cms.gov), who is best situated to make ground level changes than hospitalists? It really doesn’t matter whether these patients are insured or uninsured, whether they are citizens or noncitizens, or whether “an intervention is saving money for the hospital.” In the end, many are utilizing more than their share of medical allocations, and we as taxpayers get to cover that cost.
I believe we can be good doctors and, at the same time, good stewards of our nation’s healthcare dollars.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? Email your questions.
Are hospitals going to be allowed to start “patient profiling” in order to reduce costs? Hospitals already frequently operate their own state departments and perform “extraordinary medical renditions” on uninsured, critically ill noncitizens. Do you really expect us to believe they are identifying these patients in order to provide them more appropriate services? My impression is that since this group has been identified as a cost driver, the aim of any intervention is saving money for the hospital rather than rendering appropriate care.
—Tom Horiagon
Dr. Hospitalist responds:
As hospitalists, we are best positioned to manage the balance among medical, social, and fiduciary responsibilities. The article addresses the data that shows what most of us already know: Most patients who have multiple readmissions have many co-morbid conditions and/or psychiatric and social issues. Hospitalists have the opportunity to use everything in the patient’s history and profile to prescribe the appropriate treatment plan. When we find the right solutions, it would be helpful to us all if they were not only cost effective but also right for the patient.
Although I prefer to not use the term “patient profiling” because of the associated negative connotations, I do believe there are occasions we face with our patients when the most “appropriate service” may not be clinically relevant at all.
For example, we recently began a quality initiative project in our hospital to identify those with acute or chronic pain and the most frequent admissions (greater than 10) in a calendar year. We identified a patient who had a total of 43 admissions across four different hospitals in one calendar year. Clearly, the best care for this individual would be to get him an apartment.
We know that many of these “frequent flyers” tend to absorb vast amounts of our healthcare dollars with multiple imaging studies, lab work, and time taken away from other patients, not to mention the emotional toll some of these patients take on the clinical staff.
Discussions on such matters as tort reform, futile care, and patient nonadherence (many factors and much more complex) have been going on for some time. I don’t see our politicians developing the intestinal fortitude to address these problems any time soon. With our national healthcare expenditures reaching $2.9 trillion (or $9,255 per person in 2013, per cms.gov), who is best situated to make ground level changes than hospitalists? It really doesn’t matter whether these patients are insured or uninsured, whether they are citizens or noncitizens, or whether “an intervention is saving money for the hospital.” In the end, many are utilizing more than their share of medical allocations, and we as taxpayers get to cover that cost.
I believe we can be good doctors and, at the same time, good stewards of our nation’s healthcare dollars.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? Email your questions.
Are hospitals going to be allowed to start “patient profiling” in order to reduce costs? Hospitals already frequently operate their own state departments and perform “extraordinary medical renditions” on uninsured, critically ill noncitizens. Do you really expect us to believe they are identifying these patients in order to provide them more appropriate services? My impression is that since this group has been identified as a cost driver, the aim of any intervention is saving money for the hospital rather than rendering appropriate care.
—Tom Horiagon
Dr. Hospitalist responds:
As hospitalists, we are best positioned to manage the balance among medical, social, and fiduciary responsibilities. The article addresses the data that shows what most of us already know: Most patients who have multiple readmissions have many co-morbid conditions and/or psychiatric and social issues. Hospitalists have the opportunity to use everything in the patient’s history and profile to prescribe the appropriate treatment plan. When we find the right solutions, it would be helpful to us all if they were not only cost effective but also right for the patient.
Although I prefer to not use the term “patient profiling” because of the associated negative connotations, I do believe there are occasions we face with our patients when the most “appropriate service” may not be clinically relevant at all.
For example, we recently began a quality initiative project in our hospital to identify those with acute or chronic pain and the most frequent admissions (greater than 10) in a calendar year. We identified a patient who had a total of 43 admissions across four different hospitals in one calendar year. Clearly, the best care for this individual would be to get him an apartment.
We know that many of these “frequent flyers” tend to absorb vast amounts of our healthcare dollars with multiple imaging studies, lab work, and time taken away from other patients, not to mention the emotional toll some of these patients take on the clinical staff.
Discussions on such matters as tort reform, futile care, and patient nonadherence (many factors and much more complex) have been going on for some time. I don’t see our politicians developing the intestinal fortitude to address these problems any time soon. With our national healthcare expenditures reaching $2.9 trillion (or $9,255 per person in 2013, per cms.gov), who is best situated to make ground level changes than hospitalists? It really doesn’t matter whether these patients are insured or uninsured, whether they are citizens or noncitizens, or whether “an intervention is saving money for the hospital.” In the end, many are utilizing more than their share of medical allocations, and we as taxpayers get to cover that cost.
I believe we can be good doctors and, at the same time, good stewards of our nation’s healthcare dollars.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? Email your questions.
Self-propelled particles stop bleeding

carbonate microparticle
in acidic solution
Image by James Baylis
Researchers say they’ve created self-propelled particles that can travel against the flow of blood to treat severe bleeding.
These calcium carbonate microparticles, which are applied as a powder, release carbon dioxide gas to propel them toward the source of bleeding.
They can be loaded with thrombin and transport the clotting protein through wounds and into damaged tissue in animals.
The researchers described the particles in Science Advances.
“People have developed hundreds of agents that can clot blood, but the issue is that it’s hard to push these therapies against severe blood flow, especially far enough upstream to reach the leaking vessels,” said study author Christian Kastrup, PhD, of the University of British Columbia in Vancouver, Canada.
“Here, for the first time, we’ve come up with an agent that can do that.”
After studying and modeling the movement of their microparticles in vitro, the researchers loaded the particles with thrombin and tested them in mouse and pig models of hemorrhage.
The particles helped clot blood and stopped hemorrhaging in both models. In fact, the gas-generating, thrombin-loaded particles stopped bleeding better than topical thrombin or thrombin-loaded particles that did not produce gas.
The researchers believe that, after more testing and development, their microparticles could have a wide range of uses. And they would be particularly useful for treating bleeding that originates internally, such as in the uterus, sinus, gastrointestinal tract, or abdomen, where traditional topical drugs are less effective.
“The area we’re really focusing on is postpartum hemorrhage: in the uterus, after childbirth, where you can’t see the damaged vessels but you can put the powder into that area and the particles can propel and find those damaged vessels,” Dr Kastrup said.
The researchers also believe the microparticles could be used to deliver a range of therapeutics to wound and hemorrhage sites. ![]()
carbonate microparticle
in acidic solution
Image by James Baylis
Researchers say they’ve created self-propelled particles that can travel against the flow of blood to treat severe bleeding.
These calcium carbonate microparticles, which are applied as a powder, release carbon dioxide gas to propel them toward the source of bleeding.
They can be loaded with thrombin and transport the clotting protein through wounds and into damaged tissue in animals.
The researchers described the particles in Science Advances.
“People have developed hundreds of agents that can clot blood, but the issue is that it’s hard to push these therapies against severe blood flow, especially far enough upstream to reach the leaking vessels,” said study author Christian Kastrup, PhD, of the University of British Columbia in Vancouver, Canada.
“Here, for the first time, we’ve come up with an agent that can do that.”
After studying and modeling the movement of their microparticles in vitro, the researchers loaded the particles with thrombin and tested them in mouse and pig models of hemorrhage.
The particles helped clot blood and stopped hemorrhaging in both models. In fact, the gas-generating, thrombin-loaded particles stopped bleeding better than topical thrombin or thrombin-loaded particles that did not produce gas.
The researchers believe that, after more testing and development, their microparticles could have a wide range of uses. And they would be particularly useful for treating bleeding that originates internally, such as in the uterus, sinus, gastrointestinal tract, or abdomen, where traditional topical drugs are less effective.
“The area we’re really focusing on is postpartum hemorrhage: in the uterus, after childbirth, where you can’t see the damaged vessels but you can put the powder into that area and the particles can propel and find those damaged vessels,” Dr Kastrup said.
The researchers also believe the microparticles could be used to deliver a range of therapeutics to wound and hemorrhage sites. ![]()
carbonate microparticle
in acidic solution
Image by James Baylis
Researchers say they’ve created self-propelled particles that can travel against the flow of blood to treat severe bleeding.
These calcium carbonate microparticles, which are applied as a powder, release carbon dioxide gas to propel them toward the source of bleeding.
They can be loaded with thrombin and transport the clotting protein through wounds and into damaged tissue in animals.
The researchers described the particles in Science Advances.
“People have developed hundreds of agents that can clot blood, but the issue is that it’s hard to push these therapies against severe blood flow, especially far enough upstream to reach the leaking vessels,” said study author Christian Kastrup, PhD, of the University of British Columbia in Vancouver, Canada.
“Here, for the first time, we’ve come up with an agent that can do that.”
After studying and modeling the movement of their microparticles in vitro, the researchers loaded the particles with thrombin and tested them in mouse and pig models of hemorrhage.
The particles helped clot blood and stopped hemorrhaging in both models. In fact, the gas-generating, thrombin-loaded particles stopped bleeding better than topical thrombin or thrombin-loaded particles that did not produce gas.
The researchers believe that, after more testing and development, their microparticles could have a wide range of uses. And they would be particularly useful for treating bleeding that originates internally, such as in the uterus, sinus, gastrointestinal tract, or abdomen, where traditional topical drugs are less effective.
“The area we’re really focusing on is postpartum hemorrhage: in the uterus, after childbirth, where you can’t see the damaged vessels but you can put the powder into that area and the particles can propel and find those damaged vessels,” Dr Kastrup said.
The researchers also believe the microparticles could be used to deliver a range of therapeutics to wound and hemorrhage sites. ![]()
Physician Predictions of Discharge
Hospital discharge planning is a complex process requiring efficient coordination of many different medical and social support services. For this reason, multidisciplinary teams work together to develop individualized discharge plans in an attempt to reduce preventable adverse events related to hospital discharge.[1, 2, 3, 4, 5] Despite these ongoing efforts, optimal discharge strategies have yet to be realized.[1, 4, 5, 6, 7, 8, 9]
One factor that may improve the discharge process is the early identification of patients who are approaching discharge.[10] Multidisciplinary teams cannot fully deploy comprehensive discharge plans until a physician deems a patient to be approaching discharge readiness.[8]
To our knowledge, no studies have examined the performance of physician predictions of upcoming discharge. Instead, prior studies have found that physicians have difficulty predicting the length of stay for patients seen in the emergency room and for elderly patients newly admitted to general medicine floor.[11, 12] The purpose of this study was to evaluate the ability of inpatient general medicine physicians to predict next or same‐day hospital discharges to help inform the timing of discharge planning.
METHODS
We collected daily in‐person predictions from all senior residents and attendings separately on the inpatient general medicine teams (5 resident/attending services and 4 attending‐only services) at a single 950‐bed academic medical center. We asked these physicians to predict whether each patient under their care had a greater than or equal to 80% chance of being discharged the next day, the same day, or neither (ie, no discharge on the next or same day).
Physician predictions of discharge occurred Monday through Friday at 1 of 3 time points: morning (79 am), midday (122 pm), or afternoon (57 pm). Data collection focused on 1 time point per week during 2 different weeks in November 2013 and 1 week in February 2014. Predictions of same‐day discharge could only be made at the morning and midday time points. Each patient could have multiple predictions if they remained hospitalized during subsequent assessments. For each physician making a prediction, we recorded the physician training level (resident or attending).
This protocol was deemed exempt by our university's institutional review board.
Outcomes
We measured the sensitivity (SN), specificity (SP), positive predictive value (PPV), and negative predictive value (NPV) for each type of physician prediction (next day, same day, or no discharge by the end of the next day). We also calculated these measurements for each time point in the time of day subgroup: morning, midday, and afternoon.
Statistical Analyses
Using a normal approximation to the binomial distribution, point estimates and 95% confidence intervals for SN, SP, PPV, and NPV for the group of all patients and for the time of day subgroup are reported. The Cochran‐Armitage trend test was used to examine trends in SN, SP, PPV, and NPV as time to discharge decreased. No adjustments were made for multiple comparisons. A 2‐sided significance level was prespecified at 0.05 for all tests.
For the subset of patients who had discharge predictions made by both a resident and an attending, agreement was examined using the kappa statistic. All analyses were conducted using SAS version 9.3 (SAS Institute, Cary, NC).
RESULTS
A total of 2660 predictions were made by 24 attendings and 15 residents. Nineteen predictions were excluded because of missing prediction type or date of discharge, leaving 2641 predictions for analysis. Table 1 summarizes the total number of predictions within subgroups.
| No. of Predictions | |
|---|---|
| All predictions | 2,641 |
| Day of the week | |
| Monday | 596 |
| Tuesday | 503 |
| Wednesday | 525 |
| Thursday | 551 |
| Friday | 466 |
| Physician training level | |
| Resident | 871 |
| Attending | 1,770 |
| Time of day | |
| Morning (7 am9 am) | 906 |
| Midday (12 pm2 pm) | 832 |
| Afternoon (5 pm7 pm) | 903 |
The overall daily discharge rate in our population was 22.3% (see Supporting Table 1 in the online version of this article for the raw values). The SN and PPV of physician predictions of next‐day discharge were 48% (95% confidence interval [CI]: 43%‐52%) and 51% (95% CI: 46%‐56%), respectively. The SN and PPV for same‐day discharge predictions were 73% (95% CI: 68%‐78%) and 69% (95% CI: 64%‐73%), respectively. The SP for next and same‐day discharge predictions was 90% (95% CI: 89%‐91%) and 95% (95% CI: 94%‐96%), whereas the NPV was 89% (95% CI: 88%‐90%) and 96% (95% CI: 95%‐97%), respectively.
Outcome measures for each prediction type are stratified by time of day and summarized in Table 2. For next‐day discharge predictions, the SN and PPV were lowest in the morning (SN 27%, PPV 33%) and peaked by the afternoon (SN 67%, PPV 69%). Similarly, for same‐day discharges, SN and PPV were highest later in the day (midday SN 88%, PPV 79%). This trend is also demonstrated in the SP and NPV, which increased as time to actual discharge approached, although the trends are not as pronounced as for SN and PPV.
| Validity Measure | Next‐Day Discharge Predictions | Trend P Value | Same‐Day Discharge Predictions | Trend P Value | ||||
|---|---|---|---|---|---|---|---|---|
| Morning | Midday | Afternoon | Morning | Midday | Afternoon | |||
| ||||||||
| Sensitivity | 0.27 (0.210.35) | 0.50 (0.410.59) | 0.67 (0.590.74) | 0.001 | 0.66 (0.590.73) | 0.88 (0.810.93) | 0.001 | |
| Specificity | 0.87 (0.850.90) | 0.90 (0.880.92) | 0.93 (0.910.95) | 0.001 | 0.88 (0.850.90) | 0.95 (0.930.97) | 0.001 | |
| PPV | 0.33 (0.250.41) | 0.48 (0.400.57) | 0.69 (0.610.76) | 0.001 | 0.62 (0.550.68) | 0.79 (0.710.85) | 0.001 | |
| NPV | 0.84 (0.810.87) | 0.91 (0.880.93) | 0.93 (0.910.94) | 0.001 | 0.90 (0.880.92) | 0.98 (0.960.99) | 0.001 | |
The overall agreement between resident and attending predictions was measured and found to have kappa values of 0.51 (P 0.001) for next‐day predictions and 0.73 (P 0.001) for same‐day predictions, indicating moderate and substantial agreement, respectively (see Supporting Table 2 in the online version of this article).[13]
DISCUSSION
This is the first study, to our knowledge, to examine the ability of physicians to predict upcoming discharge during the course of routine general medicine inpatient care. We found that although physicians are poor predictors of discharge in the morning prior to the day of expected discharge, their ability to correctly predict inpatient discharges showed continual improvement as the difference between the prediction time and time of actual discharge shortened.
For next‐day predictions, the most accurate time point was the afternoon, when physicians correctly predicted more than two‐thirds of actual next‐day discharges. This finding suggests that physicians can provide meaningful discharge estimates as early as the afternoon prior to expected discharge. This may be an optimal time for physicians to meet with the multidisciplinary discharge teams, as many preparations hinge on timely and accurate predictions of discharge (eg, arranging patient transportation, postdischarge visits by a home health company). Multidisciplinary teams will also be reassured that an afternoon prediction of next‐day discharge would only prematurely activate discharge resources in roughly 3 out of every 10 occurrences. Even in these instances, patients may benefit from the extra time for disease counselling, medication teaching, and arrangement of home services.[4, 5, 6, 7, 8, 9]
This investigation has several limitations. Our study was conducted at a large tertiary care center over brief time periods, with an overall discharge rate of about 1 in 5 patients per day. Thus, the results may not be generalizable to hospitals with different patient populations, volume, or turnover, or when predictions are made at different times throughout the year. Furthermore, we were unable to determine if the outcome measures were affected by prolonged lengths of stay or excessive predictions on relatively few patients. However, we sought to mitigate these constraints by surveying many different respondents with varying experience levels, caring for a heterogeneous patient population at nonconsecutive time points during the year. A review of our hospital's administrative data suggests that the bed occupancy and average length of stay during our surveys were similar with most other time points during the year, and therefore representative of a typical inpatient general medicine service.
Our investigation was a novel investigation into the performance of physician discharge predictions, which are daily predictions made either explicitly or implicitly by physicians caring for patients on a general medicine ward. By utilizing a simple, subjective survey without bulky calculations, this approach closely mirrors real‐world practice patterns, and if further validated, could be easily assimilated into the normal workflow of a wide range of busy clinicians to more effectively activate individualized discharge plans.[1, 2, 3, 4, 5]
Future work could capture additional patient information, such as functional status, diagnosis, and current length of stay, which would allow identification of certain subsets of patients in which physicians are more or less accurate in predicting hospital discharge. Additionally, the outcomes of incorrect predictions, particularly the surprise discharges that left even though they were predicted to stay, could be assessed. If patients were discharged prematurely, this may be reflected by a higher 30‐day readmission rate, lower clinic follow‐up rate, and/or lower patient satisfaction scores.
CONCLUSION
Although physicians are poor predictors of discharge in the morning prior to the day of expected discharge, their ability to correctly predict inpatient discharges steadily improves as the difference between the prediction time and time of actual discharge shortened. It remains to be determined if systematic incorporation of physician discharge predictions into standard workflows will improve the effectiveness of transition of care interventions.
Disclosure: Nothing to report.
- , , , et al. Care transitions project team: Association between quality improvement for care transition in communities and rehospitalizations among Medicare beneficiaries. JAMA. 2013;309(4):381–391.
- , , , , The care transitions intervention: results of a randomized controlled trial. Arch Intern Med. 2006;166:1822–1828.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281:613–620.
- , , , et al. Hospital‐initiated transitional care interventions as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158:433–440.
- , , , et al. Discharge planning from hospital to home. Cochrane Database Syst Rev. 2013;1:CD000313.
- , , . A prospective study of reasons for prolonged hospitalizations on a general medicine teaching service. J General Intern Med. 2005;20:108–115.
- , , , . The epidemiology of delays in a teaching hospital. Med Care. 1989;27:112–129.
- , , , et al. Development of a checklist of safe discharge practices for hospital patients. J Hosp Med. 2013;8:444–449.
- , , , , Medication reconciliation during transition of care as a patient safety strategy. Ann Intern Med. 2013;158:397–403.
- , , Making effective use of predicted discharge dates to reduce the length of stay in hospital. Nurs Times. 2009;105(15):12–13.
- . Physicians' outcome predictions for elderly patients: Survival, hospital discharge, and length of stay in a department of internal medicine. Scand J Soc Med. 1986;14(3):127–132.
- , , , . Physicians' ability to predict hospital length of stay for patients admitted to the hospital from the emergency department. Emerg Med Int. 2012;2012:824674.
- , . Understanding interobserver agreement: the kappa statistic. Fam Med. 2005;37(5):360–363.
Hospital discharge planning is a complex process requiring efficient coordination of many different medical and social support services. For this reason, multidisciplinary teams work together to develop individualized discharge plans in an attempt to reduce preventable adverse events related to hospital discharge.[1, 2, 3, 4, 5] Despite these ongoing efforts, optimal discharge strategies have yet to be realized.[1, 4, 5, 6, 7, 8, 9]
One factor that may improve the discharge process is the early identification of patients who are approaching discharge.[10] Multidisciplinary teams cannot fully deploy comprehensive discharge plans until a physician deems a patient to be approaching discharge readiness.[8]
To our knowledge, no studies have examined the performance of physician predictions of upcoming discharge. Instead, prior studies have found that physicians have difficulty predicting the length of stay for patients seen in the emergency room and for elderly patients newly admitted to general medicine floor.[11, 12] The purpose of this study was to evaluate the ability of inpatient general medicine physicians to predict next or same‐day hospital discharges to help inform the timing of discharge planning.
METHODS
We collected daily in‐person predictions from all senior residents and attendings separately on the inpatient general medicine teams (5 resident/attending services and 4 attending‐only services) at a single 950‐bed academic medical center. We asked these physicians to predict whether each patient under their care had a greater than or equal to 80% chance of being discharged the next day, the same day, or neither (ie, no discharge on the next or same day).
Physician predictions of discharge occurred Monday through Friday at 1 of 3 time points: morning (79 am), midday (122 pm), or afternoon (57 pm). Data collection focused on 1 time point per week during 2 different weeks in November 2013 and 1 week in February 2014. Predictions of same‐day discharge could only be made at the morning and midday time points. Each patient could have multiple predictions if they remained hospitalized during subsequent assessments. For each physician making a prediction, we recorded the physician training level (resident or attending).
This protocol was deemed exempt by our university's institutional review board.
Outcomes
We measured the sensitivity (SN), specificity (SP), positive predictive value (PPV), and negative predictive value (NPV) for each type of physician prediction (next day, same day, or no discharge by the end of the next day). We also calculated these measurements for each time point in the time of day subgroup: morning, midday, and afternoon.
Statistical Analyses
Using a normal approximation to the binomial distribution, point estimates and 95% confidence intervals for SN, SP, PPV, and NPV for the group of all patients and for the time of day subgroup are reported. The Cochran‐Armitage trend test was used to examine trends in SN, SP, PPV, and NPV as time to discharge decreased. No adjustments were made for multiple comparisons. A 2‐sided significance level was prespecified at 0.05 for all tests.
For the subset of patients who had discharge predictions made by both a resident and an attending, agreement was examined using the kappa statistic. All analyses were conducted using SAS version 9.3 (SAS Institute, Cary, NC).
RESULTS
A total of 2660 predictions were made by 24 attendings and 15 residents. Nineteen predictions were excluded because of missing prediction type or date of discharge, leaving 2641 predictions for analysis. Table 1 summarizes the total number of predictions within subgroups.
| No. of Predictions | |
|---|---|
| All predictions | 2,641 |
| Day of the week | |
| Monday | 596 |
| Tuesday | 503 |
| Wednesday | 525 |
| Thursday | 551 |
| Friday | 466 |
| Physician training level | |
| Resident | 871 |
| Attending | 1,770 |
| Time of day | |
| Morning (7 am9 am) | 906 |
| Midday (12 pm2 pm) | 832 |
| Afternoon (5 pm7 pm) | 903 |
The overall daily discharge rate in our population was 22.3% (see Supporting Table 1 in the online version of this article for the raw values). The SN and PPV of physician predictions of next‐day discharge were 48% (95% confidence interval [CI]: 43%‐52%) and 51% (95% CI: 46%‐56%), respectively. The SN and PPV for same‐day discharge predictions were 73% (95% CI: 68%‐78%) and 69% (95% CI: 64%‐73%), respectively. The SP for next and same‐day discharge predictions was 90% (95% CI: 89%‐91%) and 95% (95% CI: 94%‐96%), whereas the NPV was 89% (95% CI: 88%‐90%) and 96% (95% CI: 95%‐97%), respectively.
Outcome measures for each prediction type are stratified by time of day and summarized in Table 2. For next‐day discharge predictions, the SN and PPV were lowest in the morning (SN 27%, PPV 33%) and peaked by the afternoon (SN 67%, PPV 69%). Similarly, for same‐day discharges, SN and PPV were highest later in the day (midday SN 88%, PPV 79%). This trend is also demonstrated in the SP and NPV, which increased as time to actual discharge approached, although the trends are not as pronounced as for SN and PPV.
| Validity Measure | Next‐Day Discharge Predictions | Trend P Value | Same‐Day Discharge Predictions | Trend P Value | ||||
|---|---|---|---|---|---|---|---|---|
| Morning | Midday | Afternoon | Morning | Midday | Afternoon | |||
| ||||||||
| Sensitivity | 0.27 (0.210.35) | 0.50 (0.410.59) | 0.67 (0.590.74) | 0.001 | 0.66 (0.590.73) | 0.88 (0.810.93) | 0.001 | |
| Specificity | 0.87 (0.850.90) | 0.90 (0.880.92) | 0.93 (0.910.95) | 0.001 | 0.88 (0.850.90) | 0.95 (0.930.97) | 0.001 | |
| PPV | 0.33 (0.250.41) | 0.48 (0.400.57) | 0.69 (0.610.76) | 0.001 | 0.62 (0.550.68) | 0.79 (0.710.85) | 0.001 | |
| NPV | 0.84 (0.810.87) | 0.91 (0.880.93) | 0.93 (0.910.94) | 0.001 | 0.90 (0.880.92) | 0.98 (0.960.99) | 0.001 | |
The overall agreement between resident and attending predictions was measured and found to have kappa values of 0.51 (P 0.001) for next‐day predictions and 0.73 (P 0.001) for same‐day predictions, indicating moderate and substantial agreement, respectively (see Supporting Table 2 in the online version of this article).[13]
DISCUSSION
This is the first study, to our knowledge, to examine the ability of physicians to predict upcoming discharge during the course of routine general medicine inpatient care. We found that although physicians are poor predictors of discharge in the morning prior to the day of expected discharge, their ability to correctly predict inpatient discharges showed continual improvement as the difference between the prediction time and time of actual discharge shortened.
For next‐day predictions, the most accurate time point was the afternoon, when physicians correctly predicted more than two‐thirds of actual next‐day discharges. This finding suggests that physicians can provide meaningful discharge estimates as early as the afternoon prior to expected discharge. This may be an optimal time for physicians to meet with the multidisciplinary discharge teams, as many preparations hinge on timely and accurate predictions of discharge (eg, arranging patient transportation, postdischarge visits by a home health company). Multidisciplinary teams will also be reassured that an afternoon prediction of next‐day discharge would only prematurely activate discharge resources in roughly 3 out of every 10 occurrences. Even in these instances, patients may benefit from the extra time for disease counselling, medication teaching, and arrangement of home services.[4, 5, 6, 7, 8, 9]
This investigation has several limitations. Our study was conducted at a large tertiary care center over brief time periods, with an overall discharge rate of about 1 in 5 patients per day. Thus, the results may not be generalizable to hospitals with different patient populations, volume, or turnover, or when predictions are made at different times throughout the year. Furthermore, we were unable to determine if the outcome measures were affected by prolonged lengths of stay or excessive predictions on relatively few patients. However, we sought to mitigate these constraints by surveying many different respondents with varying experience levels, caring for a heterogeneous patient population at nonconsecutive time points during the year. A review of our hospital's administrative data suggests that the bed occupancy and average length of stay during our surveys were similar with most other time points during the year, and therefore representative of a typical inpatient general medicine service.
Our investigation was a novel investigation into the performance of physician discharge predictions, which are daily predictions made either explicitly or implicitly by physicians caring for patients on a general medicine ward. By utilizing a simple, subjective survey without bulky calculations, this approach closely mirrors real‐world practice patterns, and if further validated, could be easily assimilated into the normal workflow of a wide range of busy clinicians to more effectively activate individualized discharge plans.[1, 2, 3, 4, 5]
Future work could capture additional patient information, such as functional status, diagnosis, and current length of stay, which would allow identification of certain subsets of patients in which physicians are more or less accurate in predicting hospital discharge. Additionally, the outcomes of incorrect predictions, particularly the surprise discharges that left even though they were predicted to stay, could be assessed. If patients were discharged prematurely, this may be reflected by a higher 30‐day readmission rate, lower clinic follow‐up rate, and/or lower patient satisfaction scores.
CONCLUSION
Although physicians are poor predictors of discharge in the morning prior to the day of expected discharge, their ability to correctly predict inpatient discharges steadily improves as the difference between the prediction time and time of actual discharge shortened. It remains to be determined if systematic incorporation of physician discharge predictions into standard workflows will improve the effectiveness of transition of care interventions.
Disclosure: Nothing to report.
Hospital discharge planning is a complex process requiring efficient coordination of many different medical and social support services. For this reason, multidisciplinary teams work together to develop individualized discharge plans in an attempt to reduce preventable adverse events related to hospital discharge.[1, 2, 3, 4, 5] Despite these ongoing efforts, optimal discharge strategies have yet to be realized.[1, 4, 5, 6, 7, 8, 9]
One factor that may improve the discharge process is the early identification of patients who are approaching discharge.[10] Multidisciplinary teams cannot fully deploy comprehensive discharge plans until a physician deems a patient to be approaching discharge readiness.[8]
To our knowledge, no studies have examined the performance of physician predictions of upcoming discharge. Instead, prior studies have found that physicians have difficulty predicting the length of stay for patients seen in the emergency room and for elderly patients newly admitted to general medicine floor.[11, 12] The purpose of this study was to evaluate the ability of inpatient general medicine physicians to predict next or same‐day hospital discharges to help inform the timing of discharge planning.
METHODS
We collected daily in‐person predictions from all senior residents and attendings separately on the inpatient general medicine teams (5 resident/attending services and 4 attending‐only services) at a single 950‐bed academic medical center. We asked these physicians to predict whether each patient under their care had a greater than or equal to 80% chance of being discharged the next day, the same day, or neither (ie, no discharge on the next or same day).
Physician predictions of discharge occurred Monday through Friday at 1 of 3 time points: morning (79 am), midday (122 pm), or afternoon (57 pm). Data collection focused on 1 time point per week during 2 different weeks in November 2013 and 1 week in February 2014. Predictions of same‐day discharge could only be made at the morning and midday time points. Each patient could have multiple predictions if they remained hospitalized during subsequent assessments. For each physician making a prediction, we recorded the physician training level (resident or attending).
This protocol was deemed exempt by our university's institutional review board.
Outcomes
We measured the sensitivity (SN), specificity (SP), positive predictive value (PPV), and negative predictive value (NPV) for each type of physician prediction (next day, same day, or no discharge by the end of the next day). We also calculated these measurements for each time point in the time of day subgroup: morning, midday, and afternoon.
Statistical Analyses
Using a normal approximation to the binomial distribution, point estimates and 95% confidence intervals for SN, SP, PPV, and NPV for the group of all patients and for the time of day subgroup are reported. The Cochran‐Armitage trend test was used to examine trends in SN, SP, PPV, and NPV as time to discharge decreased. No adjustments were made for multiple comparisons. A 2‐sided significance level was prespecified at 0.05 for all tests.
For the subset of patients who had discharge predictions made by both a resident and an attending, agreement was examined using the kappa statistic. All analyses were conducted using SAS version 9.3 (SAS Institute, Cary, NC).
RESULTS
A total of 2660 predictions were made by 24 attendings and 15 residents. Nineteen predictions were excluded because of missing prediction type or date of discharge, leaving 2641 predictions for analysis. Table 1 summarizes the total number of predictions within subgroups.
| No. of Predictions | |
|---|---|
| All predictions | 2,641 |
| Day of the week | |
| Monday | 596 |
| Tuesday | 503 |
| Wednesday | 525 |
| Thursday | 551 |
| Friday | 466 |
| Physician training level | |
| Resident | 871 |
| Attending | 1,770 |
| Time of day | |
| Morning (7 am9 am) | 906 |
| Midday (12 pm2 pm) | 832 |
| Afternoon (5 pm7 pm) | 903 |
The overall daily discharge rate in our population was 22.3% (see Supporting Table 1 in the online version of this article for the raw values). The SN and PPV of physician predictions of next‐day discharge were 48% (95% confidence interval [CI]: 43%‐52%) and 51% (95% CI: 46%‐56%), respectively. The SN and PPV for same‐day discharge predictions were 73% (95% CI: 68%‐78%) and 69% (95% CI: 64%‐73%), respectively. The SP for next and same‐day discharge predictions was 90% (95% CI: 89%‐91%) and 95% (95% CI: 94%‐96%), whereas the NPV was 89% (95% CI: 88%‐90%) and 96% (95% CI: 95%‐97%), respectively.
Outcome measures for each prediction type are stratified by time of day and summarized in Table 2. For next‐day discharge predictions, the SN and PPV were lowest in the morning (SN 27%, PPV 33%) and peaked by the afternoon (SN 67%, PPV 69%). Similarly, for same‐day discharges, SN and PPV were highest later in the day (midday SN 88%, PPV 79%). This trend is also demonstrated in the SP and NPV, which increased as time to actual discharge approached, although the trends are not as pronounced as for SN and PPV.
| Validity Measure | Next‐Day Discharge Predictions | Trend P Value | Same‐Day Discharge Predictions | Trend P Value | ||||
|---|---|---|---|---|---|---|---|---|
| Morning | Midday | Afternoon | Morning | Midday | Afternoon | |||
| ||||||||
| Sensitivity | 0.27 (0.210.35) | 0.50 (0.410.59) | 0.67 (0.590.74) | 0.001 | 0.66 (0.590.73) | 0.88 (0.810.93) | 0.001 | |
| Specificity | 0.87 (0.850.90) | 0.90 (0.880.92) | 0.93 (0.910.95) | 0.001 | 0.88 (0.850.90) | 0.95 (0.930.97) | 0.001 | |
| PPV | 0.33 (0.250.41) | 0.48 (0.400.57) | 0.69 (0.610.76) | 0.001 | 0.62 (0.550.68) | 0.79 (0.710.85) | 0.001 | |
| NPV | 0.84 (0.810.87) | 0.91 (0.880.93) | 0.93 (0.910.94) | 0.001 | 0.90 (0.880.92) | 0.98 (0.960.99) | 0.001 | |
The overall agreement between resident and attending predictions was measured and found to have kappa values of 0.51 (P 0.001) for next‐day predictions and 0.73 (P 0.001) for same‐day predictions, indicating moderate and substantial agreement, respectively (see Supporting Table 2 in the online version of this article).[13]
DISCUSSION
This is the first study, to our knowledge, to examine the ability of physicians to predict upcoming discharge during the course of routine general medicine inpatient care. We found that although physicians are poor predictors of discharge in the morning prior to the day of expected discharge, their ability to correctly predict inpatient discharges showed continual improvement as the difference between the prediction time and time of actual discharge shortened.
For next‐day predictions, the most accurate time point was the afternoon, when physicians correctly predicted more than two‐thirds of actual next‐day discharges. This finding suggests that physicians can provide meaningful discharge estimates as early as the afternoon prior to expected discharge. This may be an optimal time for physicians to meet with the multidisciplinary discharge teams, as many preparations hinge on timely and accurate predictions of discharge (eg, arranging patient transportation, postdischarge visits by a home health company). Multidisciplinary teams will also be reassured that an afternoon prediction of next‐day discharge would only prematurely activate discharge resources in roughly 3 out of every 10 occurrences. Even in these instances, patients may benefit from the extra time for disease counselling, medication teaching, and arrangement of home services.[4, 5, 6, 7, 8, 9]
This investigation has several limitations. Our study was conducted at a large tertiary care center over brief time periods, with an overall discharge rate of about 1 in 5 patients per day. Thus, the results may not be generalizable to hospitals with different patient populations, volume, or turnover, or when predictions are made at different times throughout the year. Furthermore, we were unable to determine if the outcome measures were affected by prolonged lengths of stay or excessive predictions on relatively few patients. However, we sought to mitigate these constraints by surveying many different respondents with varying experience levels, caring for a heterogeneous patient population at nonconsecutive time points during the year. A review of our hospital's administrative data suggests that the bed occupancy and average length of stay during our surveys were similar with most other time points during the year, and therefore representative of a typical inpatient general medicine service.
Our investigation was a novel investigation into the performance of physician discharge predictions, which are daily predictions made either explicitly or implicitly by physicians caring for patients on a general medicine ward. By utilizing a simple, subjective survey without bulky calculations, this approach closely mirrors real‐world practice patterns, and if further validated, could be easily assimilated into the normal workflow of a wide range of busy clinicians to more effectively activate individualized discharge plans.[1, 2, 3, 4, 5]
Future work could capture additional patient information, such as functional status, diagnosis, and current length of stay, which would allow identification of certain subsets of patients in which physicians are more or less accurate in predicting hospital discharge. Additionally, the outcomes of incorrect predictions, particularly the surprise discharges that left even though they were predicted to stay, could be assessed. If patients were discharged prematurely, this may be reflected by a higher 30‐day readmission rate, lower clinic follow‐up rate, and/or lower patient satisfaction scores.
CONCLUSION
Although physicians are poor predictors of discharge in the morning prior to the day of expected discharge, their ability to correctly predict inpatient discharges steadily improves as the difference between the prediction time and time of actual discharge shortened. It remains to be determined if systematic incorporation of physician discharge predictions into standard workflows will improve the effectiveness of transition of care interventions.
Disclosure: Nothing to report.
- , , , et al. Care transitions project team: Association between quality improvement for care transition in communities and rehospitalizations among Medicare beneficiaries. JAMA. 2013;309(4):381–391.
- , , , , The care transitions intervention: results of a randomized controlled trial. Arch Intern Med. 2006;166:1822–1828.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281:613–620.
- , , , et al. Hospital‐initiated transitional care interventions as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158:433–440.
- , , , et al. Discharge planning from hospital to home. Cochrane Database Syst Rev. 2013;1:CD000313.
- , , . A prospective study of reasons for prolonged hospitalizations on a general medicine teaching service. J General Intern Med. 2005;20:108–115.
- , , , . The epidemiology of delays in a teaching hospital. Med Care. 1989;27:112–129.
- , , , et al. Development of a checklist of safe discharge practices for hospital patients. J Hosp Med. 2013;8:444–449.
- , , , , Medication reconciliation during transition of care as a patient safety strategy. Ann Intern Med. 2013;158:397–403.
- , , Making effective use of predicted discharge dates to reduce the length of stay in hospital. Nurs Times. 2009;105(15):12–13.
- . Physicians' outcome predictions for elderly patients: Survival, hospital discharge, and length of stay in a department of internal medicine. Scand J Soc Med. 1986;14(3):127–132.
- , , , . Physicians' ability to predict hospital length of stay for patients admitted to the hospital from the emergency department. Emerg Med Int. 2012;2012:824674.
- , . Understanding interobserver agreement: the kappa statistic. Fam Med. 2005;37(5):360–363.
- , , , et al. Care transitions project team: Association between quality improvement for care transition in communities and rehospitalizations among Medicare beneficiaries. JAMA. 2013;309(4):381–391.
- , , , , The care transitions intervention: results of a randomized controlled trial. Arch Intern Med. 2006;166:1822–1828.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281:613–620.
- , , , et al. Hospital‐initiated transitional care interventions as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158:433–440.
- , , , et al. Discharge planning from hospital to home. Cochrane Database Syst Rev. 2013;1:CD000313.
- , , . A prospective study of reasons for prolonged hospitalizations on a general medicine teaching service. J General Intern Med. 2005;20:108–115.
- , , , . The epidemiology of delays in a teaching hospital. Med Care. 1989;27:112–129.
- , , , et al. Development of a checklist of safe discharge practices for hospital patients. J Hosp Med. 2013;8:444–449.
- , , , , Medication reconciliation during transition of care as a patient safety strategy. Ann Intern Med. 2013;158:397–403.
- , , Making effective use of predicted discharge dates to reduce the length of stay in hospital. Nurs Times. 2009;105(15):12–13.
- . Physicians' outcome predictions for elderly patients: Survival, hospital discharge, and length of stay in a department of internal medicine. Scand J Soc Med. 1986;14(3):127–132.
- , , , . Physicians' ability to predict hospital length of stay for patients admitted to the hospital from the emergency department. Emerg Med Int. 2012;2012:824674.
- , . Understanding interobserver agreement: the kappa statistic. Fam Med. 2005;37(5):360–363.
Assessing Discharge Readiness
Widespread evidence suggests that the period around hospitalization remains a vulnerable time for patients. Nearly 20% of patients experience adverse events, including medication errors and hospital readmissions, within 3 weeks of discharge.[1] Multiple factors contribute to adverse events, including the overwhelming volume of information patients receive on their last day in the hospital and fragmented interdisciplinary communication, both among hospital‐based providers and with community providers.[2, 3, 4] A growing body of literature suggests that to ensure patient understanding and a safe transition, discharge planning should start at time of admission. Yet, in the context of high patient volumes and competing priorities, clinicians often postpone discharge planning until they perceive a patient's discharge is imminent. Discharge bundles, designed to improve the safety of hospital discharge, such as those developed by Project BOOST (Better Outcomes by Optimizing Safe Transitions) or Project RED (Re‐Engineered Discharge), are not designed to help providers determine when a patient might be approaching discharge.[5, 6] Early identification of a patient's probable discharge date can provide vital information to inpatient and outpatient teams as they establish comprehensive discharge plans. Accurate discharge‐date predictions allow for effective discharge planning, serving to reduce length of stay (LOS) and consequently improving patient satisfaction and patient safety.[7] However, in the complex world of internal medicine, can clinicians accurately predict the timing of discharge?
A study by Sullivan and colleagues[8] in this issue of the Journal of Hospital Medicine explores a physician's ability to predict hospital discharge. Trainees and attending physicians on general internal medicine wards were asked to predict whether each patient under their care would be discharged on the next day, on the same day, or neither. Discharge predictions were recorded at 3 time points: mornings (79 am), midday (122 pm), or afternoons (57 pm). For predictions of next‐day discharges, the sensitivity (SN) and positive predictive value (PPV) were highest in the afternoon (SN 67%, PPV 69%), whereas for same‐day discharges, accuracy was highest midday (SN 88%, PPV 79%). The authors note that physicians' ability to correctly predict discharges continually improved as time to actual discharge fell.
This study is novel; to our knowledge, no other studies have evaluated the accuracy with which physicians can predict the actual day of discharge. Although this study is particular to a trainee setting and more specific to a single academic medical center, the results are thought provoking. Why are attendings and trainees unable to predict next‐day discharges more accurately? Can we do better? The majority of medical patients are not electively admitted and therefore may have complex and unpredictable courses compared to elective or surgical admissions. Subspecialty consultants may be guiding clinical care and potentially even determining readiness for discharge. Furthermore, the additional responsibilities of teaching and supervising trainees in academic medical centers may further delay discussions and decisions about patient discharges. Another plausible hypothesis, however, is that determination of barriers to discharge and discharge readiness is a clinical skill that is underappreciated and not taught or modeled sufficiently.
If we are to do better at predicting and planning for discharge, we need to build prompts for discharge readiness assessment into our daily work and education of trainees. Although interdisciplinary rounds are typically held in the morning, Wertheimer and colleagues show that additional afternoon interdisciplinary rounds can help identify patients who might be discharged before noon the next day.[9] In their study, identifying such patients in advance improved the overall early discharge rate, moved the average discharge time to earlier in the day, and decreased the observed‐to‐expected LOS, all without any adverse effects on readmissions. We also need more communication between members of the physician care team, especially with subspecialists helping manage care. The authors describe moderate agreement with next‐day and substantial agreement with same‐day discharges between trainees and attendings. Although the authors do not reveal whether trainees or attendings were more accurate, the discrepancy with next‐day discharges is notable. The disagreement suggests a lack of communication between team members about discharge barriers that can hinder planning efforts. Assessing a patient's readiness for and needs upon discharge, and anticipating a patient's disease trajectory, are important clinical skills. Trainees may lack clinical judgment and experience to accurately predict a patient's clinical evolution. As hospitalists, we can role model how to continuously assess patients' discharge needs throughout hospitalization by discussing discharge barriers during daily rounds. As part of transitions of care curricula, in addition to learning about best practices in discharge planning (eg, medication reconciliation, teach back, follow‐up appointments, effective discharge summaries), trainees should be encouraged to conduct structured, daily assessment of discharge readiness and anticipated day of discharge.
Starting the discharge planning process earlier in an admission has the potential to create more thoughtful, efficient, and ultimately safer discharges for our patients. By building discharge readiness assessments into the daily workflow and education curricula, we can prompt trainees and attendings to communicate with interdisciplinary team members and address potential challenges that patients may face in managing their health after discharge. Adequately preparing patients for safe discharges has readmission implications. With Centers for Medicare and Medicaid Services reducing payments to facilities with high rates of readmissions, reducing avoidable readmissions is a priority for all institutions.[10]
We can accomplish safe and early discharges. However, we must get better at accurately assessing our patients' readiness for discharge if we are to take the first step.
Disclosure
Nothing to report.
Widespread evidence suggests that the period around hospitalization remains a vulnerable time for patients. Nearly 20% of patients experience adverse events, including medication errors and hospital readmissions, within 3 weeks of discharge.[1] Multiple factors contribute to adverse events, including the overwhelming volume of information patients receive on their last day in the hospital and fragmented interdisciplinary communication, both among hospital‐based providers and with community providers.[2, 3, 4] A growing body of literature suggests that to ensure patient understanding and a safe transition, discharge planning should start at time of admission. Yet, in the context of high patient volumes and competing priorities, clinicians often postpone discharge planning until they perceive a patient's discharge is imminent. Discharge bundles, designed to improve the safety of hospital discharge, such as those developed by Project BOOST (Better Outcomes by Optimizing Safe Transitions) or Project RED (Re‐Engineered Discharge), are not designed to help providers determine when a patient might be approaching discharge.[5, 6] Early identification of a patient's probable discharge date can provide vital information to inpatient and outpatient teams as they establish comprehensive discharge plans. Accurate discharge‐date predictions allow for effective discharge planning, serving to reduce length of stay (LOS) and consequently improving patient satisfaction and patient safety.[7] However, in the complex world of internal medicine, can clinicians accurately predict the timing of discharge?
A study by Sullivan and colleagues[8] in this issue of the Journal of Hospital Medicine explores a physician's ability to predict hospital discharge. Trainees and attending physicians on general internal medicine wards were asked to predict whether each patient under their care would be discharged on the next day, on the same day, or neither. Discharge predictions were recorded at 3 time points: mornings (79 am), midday (122 pm), or afternoons (57 pm). For predictions of next‐day discharges, the sensitivity (SN) and positive predictive value (PPV) were highest in the afternoon (SN 67%, PPV 69%), whereas for same‐day discharges, accuracy was highest midday (SN 88%, PPV 79%). The authors note that physicians' ability to correctly predict discharges continually improved as time to actual discharge fell.
This study is novel; to our knowledge, no other studies have evaluated the accuracy with which physicians can predict the actual day of discharge. Although this study is particular to a trainee setting and more specific to a single academic medical center, the results are thought provoking. Why are attendings and trainees unable to predict next‐day discharges more accurately? Can we do better? The majority of medical patients are not electively admitted and therefore may have complex and unpredictable courses compared to elective or surgical admissions. Subspecialty consultants may be guiding clinical care and potentially even determining readiness for discharge. Furthermore, the additional responsibilities of teaching and supervising trainees in academic medical centers may further delay discussions and decisions about patient discharges. Another plausible hypothesis, however, is that determination of barriers to discharge and discharge readiness is a clinical skill that is underappreciated and not taught or modeled sufficiently.
If we are to do better at predicting and planning for discharge, we need to build prompts for discharge readiness assessment into our daily work and education of trainees. Although interdisciplinary rounds are typically held in the morning, Wertheimer and colleagues show that additional afternoon interdisciplinary rounds can help identify patients who might be discharged before noon the next day.[9] In their study, identifying such patients in advance improved the overall early discharge rate, moved the average discharge time to earlier in the day, and decreased the observed‐to‐expected LOS, all without any adverse effects on readmissions. We also need more communication between members of the physician care team, especially with subspecialists helping manage care. The authors describe moderate agreement with next‐day and substantial agreement with same‐day discharges between trainees and attendings. Although the authors do not reveal whether trainees or attendings were more accurate, the discrepancy with next‐day discharges is notable. The disagreement suggests a lack of communication between team members about discharge barriers that can hinder planning efforts. Assessing a patient's readiness for and needs upon discharge, and anticipating a patient's disease trajectory, are important clinical skills. Trainees may lack clinical judgment and experience to accurately predict a patient's clinical evolution. As hospitalists, we can role model how to continuously assess patients' discharge needs throughout hospitalization by discussing discharge barriers during daily rounds. As part of transitions of care curricula, in addition to learning about best practices in discharge planning (eg, medication reconciliation, teach back, follow‐up appointments, effective discharge summaries), trainees should be encouraged to conduct structured, daily assessment of discharge readiness and anticipated day of discharge.
Starting the discharge planning process earlier in an admission has the potential to create more thoughtful, efficient, and ultimately safer discharges for our patients. By building discharge readiness assessments into the daily workflow and education curricula, we can prompt trainees and attendings to communicate with interdisciplinary team members and address potential challenges that patients may face in managing their health after discharge. Adequately preparing patients for safe discharges has readmission implications. With Centers for Medicare and Medicaid Services reducing payments to facilities with high rates of readmissions, reducing avoidable readmissions is a priority for all institutions.[10]
We can accomplish safe and early discharges. However, we must get better at accurately assessing our patients' readiness for discharge if we are to take the first step.
Disclosure
Nothing to report.
Widespread evidence suggests that the period around hospitalization remains a vulnerable time for patients. Nearly 20% of patients experience adverse events, including medication errors and hospital readmissions, within 3 weeks of discharge.[1] Multiple factors contribute to adverse events, including the overwhelming volume of information patients receive on their last day in the hospital and fragmented interdisciplinary communication, both among hospital‐based providers and with community providers.[2, 3, 4] A growing body of literature suggests that to ensure patient understanding and a safe transition, discharge planning should start at time of admission. Yet, in the context of high patient volumes and competing priorities, clinicians often postpone discharge planning until they perceive a patient's discharge is imminent. Discharge bundles, designed to improve the safety of hospital discharge, such as those developed by Project BOOST (Better Outcomes by Optimizing Safe Transitions) or Project RED (Re‐Engineered Discharge), are not designed to help providers determine when a patient might be approaching discharge.[5, 6] Early identification of a patient's probable discharge date can provide vital information to inpatient and outpatient teams as they establish comprehensive discharge plans. Accurate discharge‐date predictions allow for effective discharge planning, serving to reduce length of stay (LOS) and consequently improving patient satisfaction and patient safety.[7] However, in the complex world of internal medicine, can clinicians accurately predict the timing of discharge?
A study by Sullivan and colleagues[8] in this issue of the Journal of Hospital Medicine explores a physician's ability to predict hospital discharge. Trainees and attending physicians on general internal medicine wards were asked to predict whether each patient under their care would be discharged on the next day, on the same day, or neither. Discharge predictions were recorded at 3 time points: mornings (79 am), midday (122 pm), or afternoons (57 pm). For predictions of next‐day discharges, the sensitivity (SN) and positive predictive value (PPV) were highest in the afternoon (SN 67%, PPV 69%), whereas for same‐day discharges, accuracy was highest midday (SN 88%, PPV 79%). The authors note that physicians' ability to correctly predict discharges continually improved as time to actual discharge fell.
This study is novel; to our knowledge, no other studies have evaluated the accuracy with which physicians can predict the actual day of discharge. Although this study is particular to a trainee setting and more specific to a single academic medical center, the results are thought provoking. Why are attendings and trainees unable to predict next‐day discharges more accurately? Can we do better? The majority of medical patients are not electively admitted and therefore may have complex and unpredictable courses compared to elective or surgical admissions. Subspecialty consultants may be guiding clinical care and potentially even determining readiness for discharge. Furthermore, the additional responsibilities of teaching and supervising trainees in academic medical centers may further delay discussions and decisions about patient discharges. Another plausible hypothesis, however, is that determination of barriers to discharge and discharge readiness is a clinical skill that is underappreciated and not taught or modeled sufficiently.
If we are to do better at predicting and planning for discharge, we need to build prompts for discharge readiness assessment into our daily work and education of trainees. Although interdisciplinary rounds are typically held in the morning, Wertheimer and colleagues show that additional afternoon interdisciplinary rounds can help identify patients who might be discharged before noon the next day.[9] In their study, identifying such patients in advance improved the overall early discharge rate, moved the average discharge time to earlier in the day, and decreased the observed‐to‐expected LOS, all without any adverse effects on readmissions. We also need more communication between members of the physician care team, especially with subspecialists helping manage care. The authors describe moderate agreement with next‐day and substantial agreement with same‐day discharges between trainees and attendings. Although the authors do not reveal whether trainees or attendings were more accurate, the discrepancy with next‐day discharges is notable. The disagreement suggests a lack of communication between team members about discharge barriers that can hinder planning efforts. Assessing a patient's readiness for and needs upon discharge, and anticipating a patient's disease trajectory, are important clinical skills. Trainees may lack clinical judgment and experience to accurately predict a patient's clinical evolution. As hospitalists, we can role model how to continuously assess patients' discharge needs throughout hospitalization by discussing discharge barriers during daily rounds. As part of transitions of care curricula, in addition to learning about best practices in discharge planning (eg, medication reconciliation, teach back, follow‐up appointments, effective discharge summaries), trainees should be encouraged to conduct structured, daily assessment of discharge readiness and anticipated day of discharge.
Starting the discharge planning process earlier in an admission has the potential to create more thoughtful, efficient, and ultimately safer discharges for our patients. By building discharge readiness assessments into the daily workflow and education curricula, we can prompt trainees and attendings to communicate with interdisciplinary team members and address potential challenges that patients may face in managing their health after discharge. Adequately preparing patients for safe discharges has readmission implications. With Centers for Medicare and Medicaid Services reducing payments to facilities with high rates of readmissions, reducing avoidable readmissions is a priority for all institutions.[10]
We can accomplish safe and early discharges. However, we must get better at accurately assessing our patients' readiness for discharge if we are to take the first step.
Disclosure
Nothing to report.
Pharmacist Impact on Transitional Care
Hospital readmissions have a significant impact on the healthcare system. Medicare data suggest a 19% all‐cause 30‐day readmission rate, of which 47% may be preventable.[1, 2] The Centers for Medicare & Medicaid Services continue to expand their criteria of disease states that will be penalized for readmissions, now reducing hospital reimbursement rates up to 3%. Pharmacists, by optimizing patient utilization of medications, can play a valuable role in contributing to preventing readmissions.[3]
Lack of acceptable transitional care is a serious problem that is consistently identified in the literature.[4] Transitional care involves 3 domains of transfer: information, education, and destination. A breakdown in any of these components can negatively impact patients and their caregivers.
Prior studies consistently demonstrated a high likelihood of adverse drug events (ADEs) and patients' lack of knowledge regarding medications postdischarge, both of which can lead to readmission. Forster and colleagues found that 19% to 23% of patients experienced an ADE within 5 weeks of discharge from an inpatient visit, 66% to 72% of which were drug related, and approximately one‐third were deemed preventable.[5, 6] One survey found that less than 60% of patients knew the indication for a new medication prescribed at discharge, whereas only 12% reported knowledge of an anticipated ADE.[7]
Pharmacists can play a large role in the information and education aspect of transitional care. Previous studies demonstrate that pharmacist involvement in the discharge process can reduce the incidence of ADEs and have a positive impact on patient satisfaction. There are conflicting data regarding the effect of comprehensive medication education and follow‐up calls by pharmacy team members on ADEs and medication errors (MEs).[3, 8, 9] Although overall pharmacist participation has shown positive patient‐related outcomes, the impact of pharmacists' involvement on readmissions has not been consistently demonstrated.[10, 11, 12, 13, 14]
Our study evaluated the impact of the pharmacy team in the transitions‐of‐care settings in a unique combination utilizing the pharmacist during medication reconciliation, discharge, and with 3 follow‐up phone call interactions postdischarge. Our study was designed to evaluate the impact of intensive pharmacist involvement during the acute care admission as well as for a 30‐day time period postdischarge on both ADEs and readmissions.
METHODS
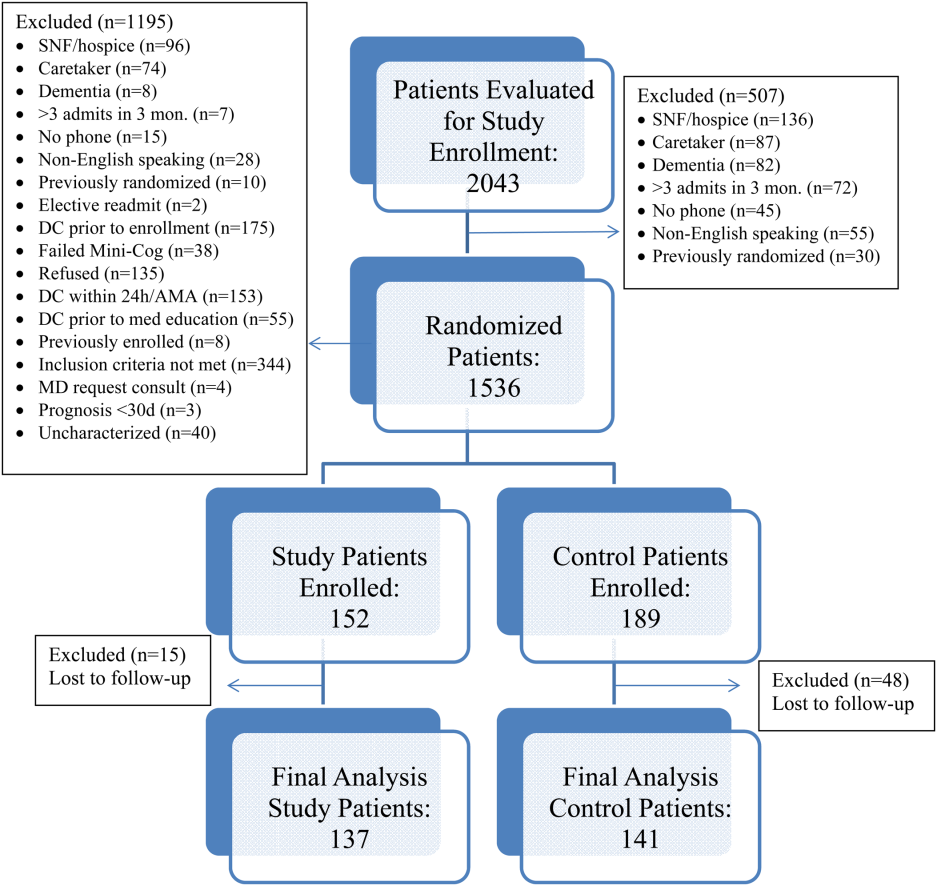
All patients were admitted to hospitalist‐based internal medicine units at Northwestern Memorial Hospital, an 894‐bed academic medical center located in Chicago, Illinois. Patients were randomized by study investigators using a random number generator to either the usual care or intervention arms and then evaluated each day for eligibility to participate in the study. Patients remained blinded throughout the study. Patients met inclusion criteria if they were discharged to home and either discharged on greater than 3 scheduled prescription medications or discharged with at least 1 high‐risk medication. High‐risk medications were classified as anticoagulants, antiplatelets (eg, aspirin and clopidogrel), hypoglycemic agents (eg, insulin), immunosuppressants, or anti‐infectives. Patients also needed to participate in a minimum of 1 postdischarge phone call or experience an emergency department (ED) visit or readmission within 30 days of discharge to meet inclusion criteria. Exclusion criteria included: impaired cognition based on Mini‐Cog screening assessment scale, unable or unwilling to provide informed consent, lack of a personal phone number, nonEnglish speaking, subsequent elective readmission within 30 days of initial visit, more than 3 previous hospital admissions in the past 2 months, palliative care or home/skilled nursing hospice, anticipated length of survival less than 3 months, discharged within 24 hours of admission, discharged against medical advice, or discharged before medication education was conducted (Figure 1). Patients who met inclusion criteria provided informed consent, received a Mini‐Cog screening assessment, and were given the Rapid Estimate of Adult Literacy in Medicine revised (REALM‐R) assessment to evaluate health literacy. The REALM‐R is a word recognition test designed to identify patients at risk for poor health literacy skills. Patients with REALM‐R scores of 6 or less are considered to have low health literacy.[15] Patients were randomized to receive either the usual care or pharmacist‐directed medication evaluation and management as described in Table 1. Patients included in the study were contacted by phone postdischarge, with 3 attempts on consecutive days. Patients who were readmitted as an inpatient or had an ED visit were not contacted for the study after that point.
| Admission Medication Reconciliation | Hospitalist (Confirmation by Pharmacist Reviewing the History and Physical Note in Electronic Medical Record) | Performed by Pharmacy Team Member Face to Face |
|---|---|---|
| ||
| Discharge medication reconciliation | Hospitalist | Pharmacy team member |
| Discharge medication education | Hospitalist and/or nurse | Pharmacy team member |
| Individualized medication plan | No | Yes |
| Postdischarge callback day 3 | No | Yes |
| Postdischarge callback day 14 | No | Yes |
| Postdischarge callback day 30 | Yes | Yes |
| Postdischarge call assessment topic(s) | ADEs/MEs, ED visits, inpatient readmissions | ADEs/MEs, ED visits, inpatient readmissions clarify pharmacy/discharge plan, resolve medication‐related issues, identify/overcome adherence barriers |
Patients enrolled in the control group received the usual standard of care by a clinical pharmacist. This included a medication reconciliation completed from the admitting physician's patient history and physical and medication counseling provided by the physician or nursing staff at discharge. Patients were not interviewed face‐to‐face on admission and did not receive discharge counseling by a pharmacy team member. Patients were assessed daily by the pharmacist for evaluation of the pharmacotherapy plans and presence of MEs or safety‐related concerns. The control group received 1 postdischarge phone call from a pharmacist at day 30 to assess for study endpoints of ADEs, MEs, ED visit, and readmission only. The endpoints of ADEs and MEs were determined by professional judgment by the clinical pharmacist based on an algorithm similar to National Coordinating Council for Medication Error Reporting and Prevention, although a specific tool was not utilized.
The study group received face‐to‐face medication reconciliation on admission by a pharmacist or a pharmacy student. Prior to discharge, a personalized medication plan was created by the pharmacist and discussed with the physician. Medication discrepancies were addressed prior to the discharge instructions being given and discussed with the patient. Medication counseling was performed at discharge by the pharmacist or pharmacy student. Patients received 3 phone calls at 3, 14, and 30 days postdischarge. The presence of ADEs and MEs were evaluated during each phone call. The patients were asked to confirm their medication regimens including drug, indication, dose, route, and frequency. They were also asked questions regarding possible side effects, new symptoms, and any changes to their current therapy. The calls focused on clarifying the pharmacy discharge plan, resolving any unanswered questions or medication‐related issues, identifying and overcoming any barriers to adherence, and assistance with providing patients access to medications by contacting pharmacies and physicians to resolve and troubleshoot further prescription claims and clarifications. Pharmacists performed all postdischarge phone calls. Pharmacy students were able to provide face‐to‐face medication reconciliation upon admission and discharge counseling under the supervision of the pharmacist for the intervention arm.
The patient Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) responses to the medication domain question, Did you clearly understand the purpose for taking each of your medications at the time of discharge? were collected for the 2 designated hospitalist units for both the control and study groups. HCAHPS scores were collected at the 6 months point prior to the study initiation and throughout the 6‐month study period for the control and intervention groups. A physician and 2 pharmacists, who were blinded to the study randomization and results, assessed all Northwestern Memorial Hospital readmissions to determine if the readmissions were medication‐related or not.
This study obtained institutional review board approval from Northwestern University.
Data Collected
Data collected from all patients included demographics (age, sex), payer, reason for admission, number of medications at time of discharge, Charlson Comorbidity Index score, number of high‐risk medications prescribed at time of discharge, length of stay, REALM‐R score, ADEs, inpatient readmission or ED visit, and the reason for readmission or ED visit. Only the first occurrence was counted for patients with both an ED visit and an inpatient readmission. It was estimated that a sample size of 150 patients in each group would provide 80% power to demonstrate a 20% improvement in ADE rates in the study group. Data were analyzed utilizing Fischer exact, 2, and Student t tests, and multivariate logistic regression as appropriate. Analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC).
RESULTS
Over the course of 7 months, 341 patients were enrolled in the study, 189 in the control arm and 152 in the study arm. Forty‐eight patients in the control group and 15 patients in the study group were lost to follow‐up. The final analysis included 278 patients, 141 in the control group and 137 in the study group. Patients were eligible for study inclusion if they received at least 1 phone call, which resulted in more patients being lost to follow‐up in the control arm due to fewer total phone call attempts. Demographic and disposition data for the control and study groups are shown in Table 2. Baseline characteristics between the 2 groups were similar with the exception of total medications at time of discharge. The control group had more total medications at discharge compared to the study group (7.2 vs 6.4, P=0.04). The number of high‐risk medications and the number of scheduled medications were similar between both groups. During medication reconciliation, 380 discrepancies (46.2%) were found in the study group compared to 205 (19.9%) in the control group (P<0.0001). The higher number of identified discrepancies in the study group was expected due to the fact that the pharmacist did not complete a face‐to‐face medication history in the control patients. The average length of stay, REALM‐R scores, and reason for admissions were similar between the 2 groups (Table 2).
| Study, N=137 | Control, N=141 | P Value | |
|---|---|---|---|
| |||
| Sex, male | 52 (37.95%) | 59 (41.8%) | 0.54 |
| Average age, y | 55.4 | 55.8 | 0.87 |
| Average length of stay, d | 5.4 (range, 1104) | 4.6 (range, 028) | 0.67 |
| Average REALM‐R score (range, 08) | 6.8 | 6.7 | 0.67 |
| Average total no. of medications | 6.4 | 7.2 | 0.04 |
| Average no. of scheduled medications | 5.7 | 6.2 | 0.15 |
| Average no. of high‐risk category medications | 2.2 | 2.3 | 0.64 |
| Reason for admission | |||
| Cardiovascular disease | 5 (3.4%) | 15 (8.3%) | 0.035 |
| Pneumonia | 11 (7.5%) | 8 (4.4%) | 0.48 |
| Respiratory | 11 (7.5%) | 9 (5%) | 0.65 |
| Infectious disease | 39 (26.5%) | 53 (29.3%) | 0.13 |
| Gastrointestinal | 25 (17%) | 28 (15.5%) | 0.13 |
| Endocrine | 20 (13.6%) | 34 (18.8%) | 0.76 |
| Genitourinary | 0 (0%) | 0 (0%) | 0.05 |
| Hematological | 19 (12.9%) | 20 (11%) | 1 |
| Injury | 10 (6.8%) | 14 (7.7%) | 1 |
| Neurological | 2 (1.4%) | 0 (0%) | 0.52 |
| Heart failure | 4 (2.7%) | 0 (0%) | 0.24 |
| Myocardial infarction | 0 (0%) | 0 (0%) | 0.58 |
| Mental/substance abuse | 1 (0.7%) | 0 (0%) | 1 |
A total of 55 patients (39%) in the control arm were readmitted to an inpatient hospital or had an ED visit within 30‐days postdischarge compared to 34 patients (24.8%) in the study group (P=0.001) (Table 3). Of the patients readmitted to the ED, 21 were enrolled in the control arm (14.8%) compared to only 6 patients in the study arm (4.4%) (P=0.005). Reviewers concluded that 24% of the control group readmissions were medication‐related versus 23% of the study group (P=1.0). In total, 78 out of 89 readmissions were to Northwestern Memorial Hospital. Medication‐related causes to outside institutions were not evaluated. The causes for all readmissions were not evaluated.
| Study Group, n=137 | Control Group, n=141 | P Value | |
|---|---|---|---|
| |||
| Composite inpatient readmission and ED visit | 34 (24.8%) | 55 (38.7%) | 0.001 |
| ED visits | 6 (4.4%) | 21 (14.8%) | 0.005 |
| Inpatient readmissions | 28 (20.4%) | 34 (23.9%) | 0.43 |
| Medication‐related readmissions | 8 (23.5%) | 13 (23.6%) | 1.0 |
| ADEs/MEs reported at 30‐day phone call | 11/84 patients | 18/86 patients | 0.22 |
| Days to readmission/ED visit | 7.9 (SD 12.5) | 13.2 (SD 9.61) | 0.03 |
| Preintervention: HCAHPS scores pertaining to knowledge of indication of medication question preintervention | 47% | ||
| Postintervention: HCAHPS scores pertaining to knowledge of indication of medication question postintervention | 56% | ||
A sensitivity analysis was undertaken to understand the impact of the lost to follow‐up rate in both the control and study groups. Undertaking an assumption that all 15 patients lost to follow‐up in the study group were readmitted and that 15 of 48 patients lost to follow‐up in the control group were readmitted, the intervention continued to show a significant benefit in reduction of composite ED and inpatient readmissions (35.7% study group vs 49.6% control group, P=0.022)
Multivariate logistic regression analysis that controlled for Charlson Comorbidity Index score, length of stay, total number of medications on discharge, and payer type showed an adjusted odds ratio of 0.55 (95% confidence interval [CI]: 0.32‐0.94) in the intervention cohort compared to controls for the combined endpoint of readmission and ED visit within 30‐days postdischarge. The adjusted odds ratio for 30‐day readmission alone was 0.88 (95% CI: 0.49‐1.61).
Eighteen of the 86 control patients who received a 30‐day postdischarge phone call experienced an ADE or ME compared to 11 of the 83 study patients (P=0.22). Patient satisfaction scores of both designated units as represented by the HCAHPS score in the medication knowledge domain increased from the prestudy period. Patients selected agree or strongly agree only 47% of the time at the 6‐month prestudy point compared to 56% of the time during the 6‐month study period.
DISCUSSION
Although previous studies show conflicting results regarding the impact of pharmacist interventions on readmissions, our study demonstrated a decrease in the composite measure of inpatient readmissions and ED visits. Its success stresses the need for a comprehensive approach that contains continuity of care by healthcare providers to reconcile and manage medications throughout the hospital stay, extending up to a full month postdischarge with multiple phone calls. This included (1) face‐to‐face medication reconciliation on admission, (2) development of a personalized medication plan discussed with the patient's physician, (3) addressing any medication discrepancies to the discharge instructions being given to the patient, (4) medication counseling performed at discharge, and (5) 3 postdischarge phone calls at 3, 14, and 30 days.
A study conducted in 2001 analyzed the Medicare Current Beneficiary Survey (MCBS) and found that living alone, having limited education, and lack of self‐management skills have significant associations with early readmission.[16] Approximately 80 million Americans have limited health literacy and are associated with poor health outcomes and healthcare utilization as seen in a review completed by Berkman and colleagues.[17] Because no difference was found between both groups, it would suggest health literacy did not influence or bias the study group. Additionally, no statistically different medication issues, such as total number of medications or rates of ADEs and MEs, were identified in the patients of this study. This may be explained by the small, final population size at the 30‐day period or that the impact of the pharmacist intervention did not reach the threshold that this study was powered to detect. Also, a lack of statistical significance may be due to the subjective nature of ADEs/MEs and the prevention of ADEs/MEs throughout all patients' hospitalizations from the clinical pharmacist's involvement in care, which was not collected. Although a combined endpoint collecting readmission to either the ED or rehospitalization was lower in the intervention cohort, the isolated rehospitalization endpoint was not significantly different between the 2 groups. ED utilization was markedly decreased, but we may have lacked the power to show a statistically significant decrease in rehospitalization. These results mirror those of the Project RED (Re‐Engineered Discharge) intervention.[17]
HCAHPS surveys are sent to only a small percent of randomly selected patients who are discharged from the hospital. Thus, respondents may or may not have been included in the study, indicating a possible greater impact of the intervention on individual patients than collected. Importantly, the described interventions appeared to improve patients' perception of understanding the purpose of their medications. We found that HCAHPS scores across the 2 units improved, though the intervention only impacted 16.8% of all patients discharged from these units due to the nature of the survey distribution.
The pharmacists' abilities to educate all eligible patients prior to discharge from 7:30 am to 4:00 pm each day of the week was a limitation of this study, as some patients were discharged outside of the duty hours. This may have allowed for a differential exclusion and could have led to selection bias. Another limitation is that a large number of patients were lost to follow‐up in the control group, likely because the first postdischarge contact with patients was not until the day 30 phone calls. The extensive exclusion criteria caused many patients not to be enrolled. Though the intervention arm received postdischarge phone calls at days 3 and 14, only postdischarge call‐backs at day 30 of the intervention arm were compared to the control arm, which could have led to bias in the 30‐day analysis of the intervention arm, as patients may have not reported previous issues that were resolved in earlier phone calls. Medication‐related readmissions were not statistically different between the groups, which could suggest that the difference in readmissions were not solely due to the intervention, and a decrease in healthcare utilization may be due to chance. The subjective nature of how ADEs and MEs were collected also serves as a limitation, as they were only screened for presence or absence and not classified by severity or category. This study was at a single‐center academic institution, which may limit the ability to apply the results to other institutions. Last, outcome assessments relied on participant report, including ADE and ME occurrence and presentation at outside hospitals. Future study evaluation conducted as a multicenter design while continuing to strengthen the continuity of the healthcare provider and patient relationship at each intervention would be ideal. Also, having an objective measure of ADEs and MEs with severity categorization would be beneficial.
Compared to previous literature, our study design was unique in the number of phone calls made to patients postdischarge and its prospective, randomized design. In the previously mentioned study by Walker et al., phone calls were made only at days 3 and 30.[13] Although the majority of readmissions occurred within the first 14 days of discharge, additional visits to the ED and readmissions may have been avoided by contacting patients twice within the critical 14‐day period. Another distinction of this study design was the expansion of a rather limited and peripheral pharmacist role in transitions of care to a much more integrated participation. We believe the relationship developed between patients and their pharmacy care team provided coordination and the continuity of communication regarding their care. Additionally, our study was unique through the use of pharmacy extenders via fourth‐year pharmacy students who were completing their advanced pharmacy practice rotations. Pharmacy extenders can also be certified and trained pharmacy technicians, which many hospitals utilize to perform medication reconciliations at a lower cost than pharmacists. As hospitals face increased demands to shrink budgets due to decreasing reimbursements, healthcare systems will be forced to find creative new ways to use existing resources.
In conclusion, transition of care is a high‐risk situation for many patients. A comprehensive approach by healthcare providers, including pharmacists and pharmacy extenders, may have a positive impact in reducing or preventing ADEs/MEs, inpatient admissions, and ED visits. Although our study focused directly on the impact of a pharmacy care team on transitions‐of‐care, we cannot conclude this applies strictly to pharmacists. Across the nation, the role of various disciplines of healthcare providers in admission, hospitalization, discharge, and postdischarge is not standardized and varies significantly by institution. Importantly, no mechanism currently exists to directly reimburse for such efforts, but demonstration of cost effectiveness through reduced posthospital utilization may justify this investment for accountable care organizations.[18]
- , , , , , . Medicare readmission rates show meaningful decline in 2012. Medicare Medicaid Res Rev. 2013;3(2):E1‐E11.
- , , , et al. Factors contributing to all‐cause 30‐day readmissions: a structured case series across 18 hospitals. Med Care. 2012:50(7):599–605.
- , , , et al. Role of pharmacist counseling in preventing adverse events after hospitalization. Arch Intern Med. 2006;66:565–571.
- , , . Optimizing transitions of care to reduce rehospitalizations. Cleve Clin J Med. 2014;81(5):1–9.
- , , , , . The incidence and severity of adverse events affecting patients following discharge from the hospital. Ann Intern Med. 2003;138:161–167.
- , . Adverse drug events occurring following hospital discharge. J Gen Intern Med. 2005;20:317–323.
- . What do discharged patients know about their medications? Patient Educ Couns. 2005;56:276–282.
- , , , . The impact of telephone calls to patients after hospitalization. Dis Mon. 2002;48:239–248.
- , , , et al. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med. 2012;157:1–10.
- , , , et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150(3):178–187.
- , , , , . The value of inpatient pharmaceutical counselling to elderly patients prior to discharge. Br J Clin Pharmacol. 2002;54:657–664.
- , , , . Postdischarge pharmacist medication reconciliation: impact on readmission rates and financial savings. J Am Pharm Assoc (2003). 2013;53(1):78–84.
- , , , et al. Impact of pharmacist‐facilitated hospital discharge program. Arch Intern Med. 2009;169:2003–2010.
- , , , et al. Does pharmacist‐led medication review help to reduce hospital admissions and deaths in older people? A systematic review and meta‐analysis. Br J Clin Pharmacol. 2008;65(3):303–316.
- . The meaning and the measure of health literacy. J Gen Intern Med. 2006;21(8):878–883.
- , , , , , . Postdischarge environmental and socioeconomic factors and the likelihood of early hospital readmission among community‐dwelling Medicare beneficiaries. Gerontologist. 2008;48(4):495–504.
- , , , , . Low health literacy and health outcomes: an updated systematic review. Ann Intern Med. 2011;155(2):97–107.
- , , , et al. Fostering accountable health care: moving forward in Medicare. Health Affairs. 2009;28(2):219–231.
Hospital readmissions have a significant impact on the healthcare system. Medicare data suggest a 19% all‐cause 30‐day readmission rate, of which 47% may be preventable.[1, 2] The Centers for Medicare & Medicaid Services continue to expand their criteria of disease states that will be penalized for readmissions, now reducing hospital reimbursement rates up to 3%. Pharmacists, by optimizing patient utilization of medications, can play a valuable role in contributing to preventing readmissions.[3]
Lack of acceptable transitional care is a serious problem that is consistently identified in the literature.[4] Transitional care involves 3 domains of transfer: information, education, and destination. A breakdown in any of these components can negatively impact patients and their caregivers.
Prior studies consistently demonstrated a high likelihood of adverse drug events (ADEs) and patients' lack of knowledge regarding medications postdischarge, both of which can lead to readmission. Forster and colleagues found that 19% to 23% of patients experienced an ADE within 5 weeks of discharge from an inpatient visit, 66% to 72% of which were drug related, and approximately one‐third were deemed preventable.[5, 6] One survey found that less than 60% of patients knew the indication for a new medication prescribed at discharge, whereas only 12% reported knowledge of an anticipated ADE.[7]
Pharmacists can play a large role in the information and education aspect of transitional care. Previous studies demonstrate that pharmacist involvement in the discharge process can reduce the incidence of ADEs and have a positive impact on patient satisfaction. There are conflicting data regarding the effect of comprehensive medication education and follow‐up calls by pharmacy team members on ADEs and medication errors (MEs).[3, 8, 9] Although overall pharmacist participation has shown positive patient‐related outcomes, the impact of pharmacists' involvement on readmissions has not been consistently demonstrated.[10, 11, 12, 13, 14]
Our study evaluated the impact of the pharmacy team in the transitions‐of‐care settings in a unique combination utilizing the pharmacist during medication reconciliation, discharge, and with 3 follow‐up phone call interactions postdischarge. Our study was designed to evaluate the impact of intensive pharmacist involvement during the acute care admission as well as for a 30‐day time period postdischarge on both ADEs and readmissions.
METHODS
All patients were admitted to hospitalist‐based internal medicine units at Northwestern Memorial Hospital, an 894‐bed academic medical center located in Chicago, Illinois. Patients were randomized by study investigators using a random number generator to either the usual care or intervention arms and then evaluated each day for eligibility to participate in the study. Patients remained blinded throughout the study. Patients met inclusion criteria if they were discharged to home and either discharged on greater than 3 scheduled prescription medications or discharged with at least 1 high‐risk medication. High‐risk medications were classified as anticoagulants, antiplatelets (eg, aspirin and clopidogrel), hypoglycemic agents (eg, insulin), immunosuppressants, or anti‐infectives. Patients also needed to participate in a minimum of 1 postdischarge phone call or experience an emergency department (ED) visit or readmission within 30 days of discharge to meet inclusion criteria. Exclusion criteria included: impaired cognition based on Mini‐Cog screening assessment scale, unable or unwilling to provide informed consent, lack of a personal phone number, nonEnglish speaking, subsequent elective readmission within 30 days of initial visit, more than 3 previous hospital admissions in the past 2 months, palliative care or home/skilled nursing hospice, anticipated length of survival less than 3 months, discharged within 24 hours of admission, discharged against medical advice, or discharged before medication education was conducted (Figure 1). Patients who met inclusion criteria provided informed consent, received a Mini‐Cog screening assessment, and were given the Rapid Estimate of Adult Literacy in Medicine revised (REALM‐R) assessment to evaluate health literacy. The REALM‐R is a word recognition test designed to identify patients at risk for poor health literacy skills. Patients with REALM‐R scores of 6 or less are considered to have low health literacy.[15] Patients were randomized to receive either the usual care or pharmacist‐directed medication evaluation and management as described in Table 1. Patients included in the study were contacted by phone postdischarge, with 3 attempts on consecutive days. Patients who were readmitted as an inpatient or had an ED visit were not contacted for the study after that point.
| Admission Medication Reconciliation | Hospitalist (Confirmation by Pharmacist Reviewing the History and Physical Note in Electronic Medical Record) | Performed by Pharmacy Team Member Face to Face |
|---|---|---|
| ||
| Discharge medication reconciliation | Hospitalist | Pharmacy team member |
| Discharge medication education | Hospitalist and/or nurse | Pharmacy team member |
| Individualized medication plan | No | Yes |
| Postdischarge callback day 3 | No | Yes |
| Postdischarge callback day 14 | No | Yes |
| Postdischarge callback day 30 | Yes | Yes |
| Postdischarge call assessment topic(s) | ADEs/MEs, ED visits, inpatient readmissions | ADEs/MEs, ED visits, inpatient readmissions clarify pharmacy/discharge plan, resolve medication‐related issues, identify/overcome adherence barriers |
Patients enrolled in the control group received the usual standard of care by a clinical pharmacist. This included a medication reconciliation completed from the admitting physician's patient history and physical and medication counseling provided by the physician or nursing staff at discharge. Patients were not interviewed face‐to‐face on admission and did not receive discharge counseling by a pharmacy team member. Patients were assessed daily by the pharmacist for evaluation of the pharmacotherapy plans and presence of MEs or safety‐related concerns. The control group received 1 postdischarge phone call from a pharmacist at day 30 to assess for study endpoints of ADEs, MEs, ED visit, and readmission only. The endpoints of ADEs and MEs were determined by professional judgment by the clinical pharmacist based on an algorithm similar to National Coordinating Council for Medication Error Reporting and Prevention, although a specific tool was not utilized.
The study group received face‐to‐face medication reconciliation on admission by a pharmacist or a pharmacy student. Prior to discharge, a personalized medication plan was created by the pharmacist and discussed with the physician. Medication discrepancies were addressed prior to the discharge instructions being given and discussed with the patient. Medication counseling was performed at discharge by the pharmacist or pharmacy student. Patients received 3 phone calls at 3, 14, and 30 days postdischarge. The presence of ADEs and MEs were evaluated during each phone call. The patients were asked to confirm their medication regimens including drug, indication, dose, route, and frequency. They were also asked questions regarding possible side effects, new symptoms, and any changes to their current therapy. The calls focused on clarifying the pharmacy discharge plan, resolving any unanswered questions or medication‐related issues, identifying and overcoming any barriers to adherence, and assistance with providing patients access to medications by contacting pharmacies and physicians to resolve and troubleshoot further prescription claims and clarifications. Pharmacists performed all postdischarge phone calls. Pharmacy students were able to provide face‐to‐face medication reconciliation upon admission and discharge counseling under the supervision of the pharmacist for the intervention arm.
The patient Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) responses to the medication domain question, Did you clearly understand the purpose for taking each of your medications at the time of discharge? were collected for the 2 designated hospitalist units for both the control and study groups. HCAHPS scores were collected at the 6 months point prior to the study initiation and throughout the 6‐month study period for the control and intervention groups. A physician and 2 pharmacists, who were blinded to the study randomization and results, assessed all Northwestern Memorial Hospital readmissions to determine if the readmissions were medication‐related or not.
This study obtained institutional review board approval from Northwestern University.
Data Collected
Data collected from all patients included demographics (age, sex), payer, reason for admission, number of medications at time of discharge, Charlson Comorbidity Index score, number of high‐risk medications prescribed at time of discharge, length of stay, REALM‐R score, ADEs, inpatient readmission or ED visit, and the reason for readmission or ED visit. Only the first occurrence was counted for patients with both an ED visit and an inpatient readmission. It was estimated that a sample size of 150 patients in each group would provide 80% power to demonstrate a 20% improvement in ADE rates in the study group. Data were analyzed utilizing Fischer exact, 2, and Student t tests, and multivariate logistic regression as appropriate. Analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC).
RESULTS
Over the course of 7 months, 341 patients were enrolled in the study, 189 in the control arm and 152 in the study arm. Forty‐eight patients in the control group and 15 patients in the study group were lost to follow‐up. The final analysis included 278 patients, 141 in the control group and 137 in the study group. Patients were eligible for study inclusion if they received at least 1 phone call, which resulted in more patients being lost to follow‐up in the control arm due to fewer total phone call attempts. Demographic and disposition data for the control and study groups are shown in Table 2. Baseline characteristics between the 2 groups were similar with the exception of total medications at time of discharge. The control group had more total medications at discharge compared to the study group (7.2 vs 6.4, P=0.04). The number of high‐risk medications and the number of scheduled medications were similar between both groups. During medication reconciliation, 380 discrepancies (46.2%) were found in the study group compared to 205 (19.9%) in the control group (P<0.0001). The higher number of identified discrepancies in the study group was expected due to the fact that the pharmacist did not complete a face‐to‐face medication history in the control patients. The average length of stay, REALM‐R scores, and reason for admissions were similar between the 2 groups (Table 2).
| Study, N=137 | Control, N=141 | P Value | |
|---|---|---|---|
| |||
| Sex, male | 52 (37.95%) | 59 (41.8%) | 0.54 |
| Average age, y | 55.4 | 55.8 | 0.87 |
| Average length of stay, d | 5.4 (range, 1104) | 4.6 (range, 028) | 0.67 |
| Average REALM‐R score (range, 08) | 6.8 | 6.7 | 0.67 |
| Average total no. of medications | 6.4 | 7.2 | 0.04 |
| Average no. of scheduled medications | 5.7 | 6.2 | 0.15 |
| Average no. of high‐risk category medications | 2.2 | 2.3 | 0.64 |
| Reason for admission | |||
| Cardiovascular disease | 5 (3.4%) | 15 (8.3%) | 0.035 |
| Pneumonia | 11 (7.5%) | 8 (4.4%) | 0.48 |
| Respiratory | 11 (7.5%) | 9 (5%) | 0.65 |
| Infectious disease | 39 (26.5%) | 53 (29.3%) | 0.13 |
| Gastrointestinal | 25 (17%) | 28 (15.5%) | 0.13 |
| Endocrine | 20 (13.6%) | 34 (18.8%) | 0.76 |
| Genitourinary | 0 (0%) | 0 (0%) | 0.05 |
| Hematological | 19 (12.9%) | 20 (11%) | 1 |
| Injury | 10 (6.8%) | 14 (7.7%) | 1 |
| Neurological | 2 (1.4%) | 0 (0%) | 0.52 |
| Heart failure | 4 (2.7%) | 0 (0%) | 0.24 |
| Myocardial infarction | 0 (0%) | 0 (0%) | 0.58 |
| Mental/substance abuse | 1 (0.7%) | 0 (0%) | 1 |
A total of 55 patients (39%) in the control arm were readmitted to an inpatient hospital or had an ED visit within 30‐days postdischarge compared to 34 patients (24.8%) in the study group (P=0.001) (Table 3). Of the patients readmitted to the ED, 21 were enrolled in the control arm (14.8%) compared to only 6 patients in the study arm (4.4%) (P=0.005). Reviewers concluded that 24% of the control group readmissions were medication‐related versus 23% of the study group (P=1.0). In total, 78 out of 89 readmissions were to Northwestern Memorial Hospital. Medication‐related causes to outside institutions were not evaluated. The causes for all readmissions were not evaluated.
| Study Group, n=137 | Control Group, n=141 | P Value | |
|---|---|---|---|
| |||
| Composite inpatient readmission and ED visit | 34 (24.8%) | 55 (38.7%) | 0.001 |
| ED visits | 6 (4.4%) | 21 (14.8%) | 0.005 |
| Inpatient readmissions | 28 (20.4%) | 34 (23.9%) | 0.43 |
| Medication‐related readmissions | 8 (23.5%) | 13 (23.6%) | 1.0 |
| ADEs/MEs reported at 30‐day phone call | 11/84 patients | 18/86 patients | 0.22 |
| Days to readmission/ED visit | 7.9 (SD 12.5) | 13.2 (SD 9.61) | 0.03 |
| Preintervention: HCAHPS scores pertaining to knowledge of indication of medication question preintervention | 47% | ||
| Postintervention: HCAHPS scores pertaining to knowledge of indication of medication question postintervention | 56% | ||
A sensitivity analysis was undertaken to understand the impact of the lost to follow‐up rate in both the control and study groups. Undertaking an assumption that all 15 patients lost to follow‐up in the study group were readmitted and that 15 of 48 patients lost to follow‐up in the control group were readmitted, the intervention continued to show a significant benefit in reduction of composite ED and inpatient readmissions (35.7% study group vs 49.6% control group, P=0.022)
Multivariate logistic regression analysis that controlled for Charlson Comorbidity Index score, length of stay, total number of medications on discharge, and payer type showed an adjusted odds ratio of 0.55 (95% confidence interval [CI]: 0.32‐0.94) in the intervention cohort compared to controls for the combined endpoint of readmission and ED visit within 30‐days postdischarge. The adjusted odds ratio for 30‐day readmission alone was 0.88 (95% CI: 0.49‐1.61).
Eighteen of the 86 control patients who received a 30‐day postdischarge phone call experienced an ADE or ME compared to 11 of the 83 study patients (P=0.22). Patient satisfaction scores of both designated units as represented by the HCAHPS score in the medication knowledge domain increased from the prestudy period. Patients selected agree or strongly agree only 47% of the time at the 6‐month prestudy point compared to 56% of the time during the 6‐month study period.
DISCUSSION
Although previous studies show conflicting results regarding the impact of pharmacist interventions on readmissions, our study demonstrated a decrease in the composite measure of inpatient readmissions and ED visits. Its success stresses the need for a comprehensive approach that contains continuity of care by healthcare providers to reconcile and manage medications throughout the hospital stay, extending up to a full month postdischarge with multiple phone calls. This included (1) face‐to‐face medication reconciliation on admission, (2) development of a personalized medication plan discussed with the patient's physician, (3) addressing any medication discrepancies to the discharge instructions being given to the patient, (4) medication counseling performed at discharge, and (5) 3 postdischarge phone calls at 3, 14, and 30 days.
A study conducted in 2001 analyzed the Medicare Current Beneficiary Survey (MCBS) and found that living alone, having limited education, and lack of self‐management skills have significant associations with early readmission.[16] Approximately 80 million Americans have limited health literacy and are associated with poor health outcomes and healthcare utilization as seen in a review completed by Berkman and colleagues.[17] Because no difference was found between both groups, it would suggest health literacy did not influence or bias the study group. Additionally, no statistically different medication issues, such as total number of medications or rates of ADEs and MEs, were identified in the patients of this study. This may be explained by the small, final population size at the 30‐day period or that the impact of the pharmacist intervention did not reach the threshold that this study was powered to detect. Also, a lack of statistical significance may be due to the subjective nature of ADEs/MEs and the prevention of ADEs/MEs throughout all patients' hospitalizations from the clinical pharmacist's involvement in care, which was not collected. Although a combined endpoint collecting readmission to either the ED or rehospitalization was lower in the intervention cohort, the isolated rehospitalization endpoint was not significantly different between the 2 groups. ED utilization was markedly decreased, but we may have lacked the power to show a statistically significant decrease in rehospitalization. These results mirror those of the Project RED (Re‐Engineered Discharge) intervention.[17]
HCAHPS surveys are sent to only a small percent of randomly selected patients who are discharged from the hospital. Thus, respondents may or may not have been included in the study, indicating a possible greater impact of the intervention on individual patients than collected. Importantly, the described interventions appeared to improve patients' perception of understanding the purpose of their medications. We found that HCAHPS scores across the 2 units improved, though the intervention only impacted 16.8% of all patients discharged from these units due to the nature of the survey distribution.
The pharmacists' abilities to educate all eligible patients prior to discharge from 7:30 am to 4:00 pm each day of the week was a limitation of this study, as some patients were discharged outside of the duty hours. This may have allowed for a differential exclusion and could have led to selection bias. Another limitation is that a large number of patients were lost to follow‐up in the control group, likely because the first postdischarge contact with patients was not until the day 30 phone calls. The extensive exclusion criteria caused many patients not to be enrolled. Though the intervention arm received postdischarge phone calls at days 3 and 14, only postdischarge call‐backs at day 30 of the intervention arm were compared to the control arm, which could have led to bias in the 30‐day analysis of the intervention arm, as patients may have not reported previous issues that were resolved in earlier phone calls. Medication‐related readmissions were not statistically different between the groups, which could suggest that the difference in readmissions were not solely due to the intervention, and a decrease in healthcare utilization may be due to chance. The subjective nature of how ADEs and MEs were collected also serves as a limitation, as they were only screened for presence or absence and not classified by severity or category. This study was at a single‐center academic institution, which may limit the ability to apply the results to other institutions. Last, outcome assessments relied on participant report, including ADE and ME occurrence and presentation at outside hospitals. Future study evaluation conducted as a multicenter design while continuing to strengthen the continuity of the healthcare provider and patient relationship at each intervention would be ideal. Also, having an objective measure of ADEs and MEs with severity categorization would be beneficial.
Compared to previous literature, our study design was unique in the number of phone calls made to patients postdischarge and its prospective, randomized design. In the previously mentioned study by Walker et al., phone calls were made only at days 3 and 30.[13] Although the majority of readmissions occurred within the first 14 days of discharge, additional visits to the ED and readmissions may have been avoided by contacting patients twice within the critical 14‐day period. Another distinction of this study design was the expansion of a rather limited and peripheral pharmacist role in transitions of care to a much more integrated participation. We believe the relationship developed between patients and their pharmacy care team provided coordination and the continuity of communication regarding their care. Additionally, our study was unique through the use of pharmacy extenders via fourth‐year pharmacy students who were completing their advanced pharmacy practice rotations. Pharmacy extenders can also be certified and trained pharmacy technicians, which many hospitals utilize to perform medication reconciliations at a lower cost than pharmacists. As hospitals face increased demands to shrink budgets due to decreasing reimbursements, healthcare systems will be forced to find creative new ways to use existing resources.
In conclusion, transition of care is a high‐risk situation for many patients. A comprehensive approach by healthcare providers, including pharmacists and pharmacy extenders, may have a positive impact in reducing or preventing ADEs/MEs, inpatient admissions, and ED visits. Although our study focused directly on the impact of a pharmacy care team on transitions‐of‐care, we cannot conclude this applies strictly to pharmacists. Across the nation, the role of various disciplines of healthcare providers in admission, hospitalization, discharge, and postdischarge is not standardized and varies significantly by institution. Importantly, no mechanism currently exists to directly reimburse for such efforts, but demonstration of cost effectiveness through reduced posthospital utilization may justify this investment for accountable care organizations.[18]
Hospital readmissions have a significant impact on the healthcare system. Medicare data suggest a 19% all‐cause 30‐day readmission rate, of which 47% may be preventable.[1, 2] The Centers for Medicare & Medicaid Services continue to expand their criteria of disease states that will be penalized for readmissions, now reducing hospital reimbursement rates up to 3%. Pharmacists, by optimizing patient utilization of medications, can play a valuable role in contributing to preventing readmissions.[3]
Lack of acceptable transitional care is a serious problem that is consistently identified in the literature.[4] Transitional care involves 3 domains of transfer: information, education, and destination. A breakdown in any of these components can negatively impact patients and their caregivers.
Prior studies consistently demonstrated a high likelihood of adverse drug events (ADEs) and patients' lack of knowledge regarding medications postdischarge, both of which can lead to readmission. Forster and colleagues found that 19% to 23% of patients experienced an ADE within 5 weeks of discharge from an inpatient visit, 66% to 72% of which were drug related, and approximately one‐third were deemed preventable.[5, 6] One survey found that less than 60% of patients knew the indication for a new medication prescribed at discharge, whereas only 12% reported knowledge of an anticipated ADE.[7]
Pharmacists can play a large role in the information and education aspect of transitional care. Previous studies demonstrate that pharmacist involvement in the discharge process can reduce the incidence of ADEs and have a positive impact on patient satisfaction. There are conflicting data regarding the effect of comprehensive medication education and follow‐up calls by pharmacy team members on ADEs and medication errors (MEs).[3, 8, 9] Although overall pharmacist participation has shown positive patient‐related outcomes, the impact of pharmacists' involvement on readmissions has not been consistently demonstrated.[10, 11, 12, 13, 14]
Our study evaluated the impact of the pharmacy team in the transitions‐of‐care settings in a unique combination utilizing the pharmacist during medication reconciliation, discharge, and with 3 follow‐up phone call interactions postdischarge. Our study was designed to evaluate the impact of intensive pharmacist involvement during the acute care admission as well as for a 30‐day time period postdischarge on both ADEs and readmissions.
METHODS
All patients were admitted to hospitalist‐based internal medicine units at Northwestern Memorial Hospital, an 894‐bed academic medical center located in Chicago, Illinois. Patients were randomized by study investigators using a random number generator to either the usual care or intervention arms and then evaluated each day for eligibility to participate in the study. Patients remained blinded throughout the study. Patients met inclusion criteria if they were discharged to home and either discharged on greater than 3 scheduled prescription medications or discharged with at least 1 high‐risk medication. High‐risk medications were classified as anticoagulants, antiplatelets (eg, aspirin and clopidogrel), hypoglycemic agents (eg, insulin), immunosuppressants, or anti‐infectives. Patients also needed to participate in a minimum of 1 postdischarge phone call or experience an emergency department (ED) visit or readmission within 30 days of discharge to meet inclusion criteria. Exclusion criteria included: impaired cognition based on Mini‐Cog screening assessment scale, unable or unwilling to provide informed consent, lack of a personal phone number, nonEnglish speaking, subsequent elective readmission within 30 days of initial visit, more than 3 previous hospital admissions in the past 2 months, palliative care or home/skilled nursing hospice, anticipated length of survival less than 3 months, discharged within 24 hours of admission, discharged against medical advice, or discharged before medication education was conducted (Figure 1). Patients who met inclusion criteria provided informed consent, received a Mini‐Cog screening assessment, and were given the Rapid Estimate of Adult Literacy in Medicine revised (REALM‐R) assessment to evaluate health literacy. The REALM‐R is a word recognition test designed to identify patients at risk for poor health literacy skills. Patients with REALM‐R scores of 6 or less are considered to have low health literacy.[15] Patients were randomized to receive either the usual care or pharmacist‐directed medication evaluation and management as described in Table 1. Patients included in the study were contacted by phone postdischarge, with 3 attempts on consecutive days. Patients who were readmitted as an inpatient or had an ED visit were not contacted for the study after that point.
| Admission Medication Reconciliation | Hospitalist (Confirmation by Pharmacist Reviewing the History and Physical Note in Electronic Medical Record) | Performed by Pharmacy Team Member Face to Face |
|---|---|---|
| ||
| Discharge medication reconciliation | Hospitalist | Pharmacy team member |
| Discharge medication education | Hospitalist and/or nurse | Pharmacy team member |
| Individualized medication plan | No | Yes |
| Postdischarge callback day 3 | No | Yes |
| Postdischarge callback day 14 | No | Yes |
| Postdischarge callback day 30 | Yes | Yes |
| Postdischarge call assessment topic(s) | ADEs/MEs, ED visits, inpatient readmissions | ADEs/MEs, ED visits, inpatient readmissions clarify pharmacy/discharge plan, resolve medication‐related issues, identify/overcome adherence barriers |
Patients enrolled in the control group received the usual standard of care by a clinical pharmacist. This included a medication reconciliation completed from the admitting physician's patient history and physical and medication counseling provided by the physician or nursing staff at discharge. Patients were not interviewed face‐to‐face on admission and did not receive discharge counseling by a pharmacy team member. Patients were assessed daily by the pharmacist for evaluation of the pharmacotherapy plans and presence of MEs or safety‐related concerns. The control group received 1 postdischarge phone call from a pharmacist at day 30 to assess for study endpoints of ADEs, MEs, ED visit, and readmission only. The endpoints of ADEs and MEs were determined by professional judgment by the clinical pharmacist based on an algorithm similar to National Coordinating Council for Medication Error Reporting and Prevention, although a specific tool was not utilized.
The study group received face‐to‐face medication reconciliation on admission by a pharmacist or a pharmacy student. Prior to discharge, a personalized medication plan was created by the pharmacist and discussed with the physician. Medication discrepancies were addressed prior to the discharge instructions being given and discussed with the patient. Medication counseling was performed at discharge by the pharmacist or pharmacy student. Patients received 3 phone calls at 3, 14, and 30 days postdischarge. The presence of ADEs and MEs were evaluated during each phone call. The patients were asked to confirm their medication regimens including drug, indication, dose, route, and frequency. They were also asked questions regarding possible side effects, new symptoms, and any changes to their current therapy. The calls focused on clarifying the pharmacy discharge plan, resolving any unanswered questions or medication‐related issues, identifying and overcoming any barriers to adherence, and assistance with providing patients access to medications by contacting pharmacies and physicians to resolve and troubleshoot further prescription claims and clarifications. Pharmacists performed all postdischarge phone calls. Pharmacy students were able to provide face‐to‐face medication reconciliation upon admission and discharge counseling under the supervision of the pharmacist for the intervention arm.
The patient Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) responses to the medication domain question, Did you clearly understand the purpose for taking each of your medications at the time of discharge? were collected for the 2 designated hospitalist units for both the control and study groups. HCAHPS scores were collected at the 6 months point prior to the study initiation and throughout the 6‐month study period for the control and intervention groups. A physician and 2 pharmacists, who were blinded to the study randomization and results, assessed all Northwestern Memorial Hospital readmissions to determine if the readmissions were medication‐related or not.
This study obtained institutional review board approval from Northwestern University.
Data Collected
Data collected from all patients included demographics (age, sex), payer, reason for admission, number of medications at time of discharge, Charlson Comorbidity Index score, number of high‐risk medications prescribed at time of discharge, length of stay, REALM‐R score, ADEs, inpatient readmission or ED visit, and the reason for readmission or ED visit. Only the first occurrence was counted for patients with both an ED visit and an inpatient readmission. It was estimated that a sample size of 150 patients in each group would provide 80% power to demonstrate a 20% improvement in ADE rates in the study group. Data were analyzed utilizing Fischer exact, 2, and Student t tests, and multivariate logistic regression as appropriate. Analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC).
RESULTS
Over the course of 7 months, 341 patients were enrolled in the study, 189 in the control arm and 152 in the study arm. Forty‐eight patients in the control group and 15 patients in the study group were lost to follow‐up. The final analysis included 278 patients, 141 in the control group and 137 in the study group. Patients were eligible for study inclusion if they received at least 1 phone call, which resulted in more patients being lost to follow‐up in the control arm due to fewer total phone call attempts. Demographic and disposition data for the control and study groups are shown in Table 2. Baseline characteristics between the 2 groups were similar with the exception of total medications at time of discharge. The control group had more total medications at discharge compared to the study group (7.2 vs 6.4, P=0.04). The number of high‐risk medications and the number of scheduled medications were similar between both groups. During medication reconciliation, 380 discrepancies (46.2%) were found in the study group compared to 205 (19.9%) in the control group (P<0.0001). The higher number of identified discrepancies in the study group was expected due to the fact that the pharmacist did not complete a face‐to‐face medication history in the control patients. The average length of stay, REALM‐R scores, and reason for admissions were similar between the 2 groups (Table 2).
| Study, N=137 | Control, N=141 | P Value | |
|---|---|---|---|
| |||
| Sex, male | 52 (37.95%) | 59 (41.8%) | 0.54 |
| Average age, y | 55.4 | 55.8 | 0.87 |
| Average length of stay, d | 5.4 (range, 1104) | 4.6 (range, 028) | 0.67 |
| Average REALM‐R score (range, 08) | 6.8 | 6.7 | 0.67 |
| Average total no. of medications | 6.4 | 7.2 | 0.04 |
| Average no. of scheduled medications | 5.7 | 6.2 | 0.15 |
| Average no. of high‐risk category medications | 2.2 | 2.3 | 0.64 |
| Reason for admission | |||
| Cardiovascular disease | 5 (3.4%) | 15 (8.3%) | 0.035 |
| Pneumonia | 11 (7.5%) | 8 (4.4%) | 0.48 |
| Respiratory | 11 (7.5%) | 9 (5%) | 0.65 |
| Infectious disease | 39 (26.5%) | 53 (29.3%) | 0.13 |
| Gastrointestinal | 25 (17%) | 28 (15.5%) | 0.13 |
| Endocrine | 20 (13.6%) | 34 (18.8%) | 0.76 |
| Genitourinary | 0 (0%) | 0 (0%) | 0.05 |
| Hematological | 19 (12.9%) | 20 (11%) | 1 |
| Injury | 10 (6.8%) | 14 (7.7%) | 1 |
| Neurological | 2 (1.4%) | 0 (0%) | 0.52 |
| Heart failure | 4 (2.7%) | 0 (0%) | 0.24 |
| Myocardial infarction | 0 (0%) | 0 (0%) | 0.58 |
| Mental/substance abuse | 1 (0.7%) | 0 (0%) | 1 |
A total of 55 patients (39%) in the control arm were readmitted to an inpatient hospital or had an ED visit within 30‐days postdischarge compared to 34 patients (24.8%) in the study group (P=0.001) (Table 3). Of the patients readmitted to the ED, 21 were enrolled in the control arm (14.8%) compared to only 6 patients in the study arm (4.4%) (P=0.005). Reviewers concluded that 24% of the control group readmissions were medication‐related versus 23% of the study group (P=1.0). In total, 78 out of 89 readmissions were to Northwestern Memorial Hospital. Medication‐related causes to outside institutions were not evaluated. The causes for all readmissions were not evaluated.
| Study Group, n=137 | Control Group, n=141 | P Value | |
|---|---|---|---|
| |||
| Composite inpatient readmission and ED visit | 34 (24.8%) | 55 (38.7%) | 0.001 |
| ED visits | 6 (4.4%) | 21 (14.8%) | 0.005 |
| Inpatient readmissions | 28 (20.4%) | 34 (23.9%) | 0.43 |
| Medication‐related readmissions | 8 (23.5%) | 13 (23.6%) | 1.0 |
| ADEs/MEs reported at 30‐day phone call | 11/84 patients | 18/86 patients | 0.22 |
| Days to readmission/ED visit | 7.9 (SD 12.5) | 13.2 (SD 9.61) | 0.03 |
| Preintervention: HCAHPS scores pertaining to knowledge of indication of medication question preintervention | 47% | ||
| Postintervention: HCAHPS scores pertaining to knowledge of indication of medication question postintervention | 56% | ||
A sensitivity analysis was undertaken to understand the impact of the lost to follow‐up rate in both the control and study groups. Undertaking an assumption that all 15 patients lost to follow‐up in the study group were readmitted and that 15 of 48 patients lost to follow‐up in the control group were readmitted, the intervention continued to show a significant benefit in reduction of composite ED and inpatient readmissions (35.7% study group vs 49.6% control group, P=0.022)
Multivariate logistic regression analysis that controlled for Charlson Comorbidity Index score, length of stay, total number of medications on discharge, and payer type showed an adjusted odds ratio of 0.55 (95% confidence interval [CI]: 0.32‐0.94) in the intervention cohort compared to controls for the combined endpoint of readmission and ED visit within 30‐days postdischarge. The adjusted odds ratio for 30‐day readmission alone was 0.88 (95% CI: 0.49‐1.61).
Eighteen of the 86 control patients who received a 30‐day postdischarge phone call experienced an ADE or ME compared to 11 of the 83 study patients (P=0.22). Patient satisfaction scores of both designated units as represented by the HCAHPS score in the medication knowledge domain increased from the prestudy period. Patients selected agree or strongly agree only 47% of the time at the 6‐month prestudy point compared to 56% of the time during the 6‐month study period.
DISCUSSION
Although previous studies show conflicting results regarding the impact of pharmacist interventions on readmissions, our study demonstrated a decrease in the composite measure of inpatient readmissions and ED visits. Its success stresses the need for a comprehensive approach that contains continuity of care by healthcare providers to reconcile and manage medications throughout the hospital stay, extending up to a full month postdischarge with multiple phone calls. This included (1) face‐to‐face medication reconciliation on admission, (2) development of a personalized medication plan discussed with the patient's physician, (3) addressing any medication discrepancies to the discharge instructions being given to the patient, (4) medication counseling performed at discharge, and (5) 3 postdischarge phone calls at 3, 14, and 30 days.
A study conducted in 2001 analyzed the Medicare Current Beneficiary Survey (MCBS) and found that living alone, having limited education, and lack of self‐management skills have significant associations with early readmission.[16] Approximately 80 million Americans have limited health literacy and are associated with poor health outcomes and healthcare utilization as seen in a review completed by Berkman and colleagues.[17] Because no difference was found between both groups, it would suggest health literacy did not influence or bias the study group. Additionally, no statistically different medication issues, such as total number of medications or rates of ADEs and MEs, were identified in the patients of this study. This may be explained by the small, final population size at the 30‐day period or that the impact of the pharmacist intervention did not reach the threshold that this study was powered to detect. Also, a lack of statistical significance may be due to the subjective nature of ADEs/MEs and the prevention of ADEs/MEs throughout all patients' hospitalizations from the clinical pharmacist's involvement in care, which was not collected. Although a combined endpoint collecting readmission to either the ED or rehospitalization was lower in the intervention cohort, the isolated rehospitalization endpoint was not significantly different between the 2 groups. ED utilization was markedly decreased, but we may have lacked the power to show a statistically significant decrease in rehospitalization. These results mirror those of the Project RED (Re‐Engineered Discharge) intervention.[17]
HCAHPS surveys are sent to only a small percent of randomly selected patients who are discharged from the hospital. Thus, respondents may or may not have been included in the study, indicating a possible greater impact of the intervention on individual patients than collected. Importantly, the described interventions appeared to improve patients' perception of understanding the purpose of their medications. We found that HCAHPS scores across the 2 units improved, though the intervention only impacted 16.8% of all patients discharged from these units due to the nature of the survey distribution.
The pharmacists' abilities to educate all eligible patients prior to discharge from 7:30 am to 4:00 pm each day of the week was a limitation of this study, as some patients were discharged outside of the duty hours. This may have allowed for a differential exclusion and could have led to selection bias. Another limitation is that a large number of patients were lost to follow‐up in the control group, likely because the first postdischarge contact with patients was not until the day 30 phone calls. The extensive exclusion criteria caused many patients not to be enrolled. Though the intervention arm received postdischarge phone calls at days 3 and 14, only postdischarge call‐backs at day 30 of the intervention arm were compared to the control arm, which could have led to bias in the 30‐day analysis of the intervention arm, as patients may have not reported previous issues that were resolved in earlier phone calls. Medication‐related readmissions were not statistically different between the groups, which could suggest that the difference in readmissions were not solely due to the intervention, and a decrease in healthcare utilization may be due to chance. The subjective nature of how ADEs and MEs were collected also serves as a limitation, as they were only screened for presence or absence and not classified by severity or category. This study was at a single‐center academic institution, which may limit the ability to apply the results to other institutions. Last, outcome assessments relied on participant report, including ADE and ME occurrence and presentation at outside hospitals. Future study evaluation conducted as a multicenter design while continuing to strengthen the continuity of the healthcare provider and patient relationship at each intervention would be ideal. Also, having an objective measure of ADEs and MEs with severity categorization would be beneficial.
Compared to previous literature, our study design was unique in the number of phone calls made to patients postdischarge and its prospective, randomized design. In the previously mentioned study by Walker et al., phone calls were made only at days 3 and 30.[13] Although the majority of readmissions occurred within the first 14 days of discharge, additional visits to the ED and readmissions may have been avoided by contacting patients twice within the critical 14‐day period. Another distinction of this study design was the expansion of a rather limited and peripheral pharmacist role in transitions of care to a much more integrated participation. We believe the relationship developed between patients and their pharmacy care team provided coordination and the continuity of communication regarding their care. Additionally, our study was unique through the use of pharmacy extenders via fourth‐year pharmacy students who were completing their advanced pharmacy practice rotations. Pharmacy extenders can also be certified and trained pharmacy technicians, which many hospitals utilize to perform medication reconciliations at a lower cost than pharmacists. As hospitals face increased demands to shrink budgets due to decreasing reimbursements, healthcare systems will be forced to find creative new ways to use existing resources.
In conclusion, transition of care is a high‐risk situation for many patients. A comprehensive approach by healthcare providers, including pharmacists and pharmacy extenders, may have a positive impact in reducing or preventing ADEs/MEs, inpatient admissions, and ED visits. Although our study focused directly on the impact of a pharmacy care team on transitions‐of‐care, we cannot conclude this applies strictly to pharmacists. Across the nation, the role of various disciplines of healthcare providers in admission, hospitalization, discharge, and postdischarge is not standardized and varies significantly by institution. Importantly, no mechanism currently exists to directly reimburse for such efforts, but demonstration of cost effectiveness through reduced posthospital utilization may justify this investment for accountable care organizations.[18]
- , , , , , . Medicare readmission rates show meaningful decline in 2012. Medicare Medicaid Res Rev. 2013;3(2):E1‐E11.
- , , , et al. Factors contributing to all‐cause 30‐day readmissions: a structured case series across 18 hospitals. Med Care. 2012:50(7):599–605.
- , , , et al. Role of pharmacist counseling in preventing adverse events after hospitalization. Arch Intern Med. 2006;66:565–571.
- , , . Optimizing transitions of care to reduce rehospitalizations. Cleve Clin J Med. 2014;81(5):1–9.
- , , , , . The incidence and severity of adverse events affecting patients following discharge from the hospital. Ann Intern Med. 2003;138:161–167.
- , . Adverse drug events occurring following hospital discharge. J Gen Intern Med. 2005;20:317–323.
- . What do discharged patients know about their medications? Patient Educ Couns. 2005;56:276–282.
- , , , . The impact of telephone calls to patients after hospitalization. Dis Mon. 2002;48:239–248.
- , , , et al. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med. 2012;157:1–10.
- , , , et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150(3):178–187.
- , , , , . The value of inpatient pharmaceutical counselling to elderly patients prior to discharge. Br J Clin Pharmacol. 2002;54:657–664.
- , , , . Postdischarge pharmacist medication reconciliation: impact on readmission rates and financial savings. J Am Pharm Assoc (2003). 2013;53(1):78–84.
- , , , et al. Impact of pharmacist‐facilitated hospital discharge program. Arch Intern Med. 2009;169:2003–2010.
- , , , et al. Does pharmacist‐led medication review help to reduce hospital admissions and deaths in older people? A systematic review and meta‐analysis. Br J Clin Pharmacol. 2008;65(3):303–316.
- . The meaning and the measure of health literacy. J Gen Intern Med. 2006;21(8):878–883.
- , , , , , . Postdischarge environmental and socioeconomic factors and the likelihood of early hospital readmission among community‐dwelling Medicare beneficiaries. Gerontologist. 2008;48(4):495–504.
- , , , , . Low health literacy and health outcomes: an updated systematic review. Ann Intern Med. 2011;155(2):97–107.
- , , , et al. Fostering accountable health care: moving forward in Medicare. Health Affairs. 2009;28(2):219–231.
- , , , , , . Medicare readmission rates show meaningful decline in 2012. Medicare Medicaid Res Rev. 2013;3(2):E1‐E11.
- , , , et al. Factors contributing to all‐cause 30‐day readmissions: a structured case series across 18 hospitals. Med Care. 2012:50(7):599–605.
- , , , et al. Role of pharmacist counseling in preventing adverse events after hospitalization. Arch Intern Med. 2006;66:565–571.
- , , . Optimizing transitions of care to reduce rehospitalizations. Cleve Clin J Med. 2014;81(5):1–9.
- , , , , . The incidence and severity of adverse events affecting patients following discharge from the hospital. Ann Intern Med. 2003;138:161–167.
- , . Adverse drug events occurring following hospital discharge. J Gen Intern Med. 2005;20:317–323.
- . What do discharged patients know about their medications? Patient Educ Couns. 2005;56:276–282.
- , , , . The impact of telephone calls to patients after hospitalization. Dis Mon. 2002;48:239–248.
- , , , et al. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med. 2012;157:1–10.
- , , , et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150(3):178–187.
- , , , , . The value of inpatient pharmaceutical counselling to elderly patients prior to discharge. Br J Clin Pharmacol. 2002;54:657–664.
- , , , . Postdischarge pharmacist medication reconciliation: impact on readmission rates and financial savings. J Am Pharm Assoc (2003). 2013;53(1):78–84.
- , , , et al. Impact of pharmacist‐facilitated hospital discharge program. Arch Intern Med. 2009;169:2003–2010.
- , , , et al. Does pharmacist‐led medication review help to reduce hospital admissions and deaths in older people? A systematic review and meta‐analysis. Br J Clin Pharmacol. 2008;65(3):303–316.
- . The meaning and the measure of health literacy. J Gen Intern Med. 2006;21(8):878–883.
- , , , , , . Postdischarge environmental and socioeconomic factors and the likelihood of early hospital readmission among community‐dwelling Medicare beneficiaries. Gerontologist. 2008;48(4):495–504.
- , , , , . Low health literacy and health outcomes: an updated systematic review. Ann Intern Med. 2011;155(2):97–107.
- , , , et al. Fostering accountable health care: moving forward in Medicare. Health Affairs. 2009;28(2):219–231.
© 2015 Society of Hospital Medicine
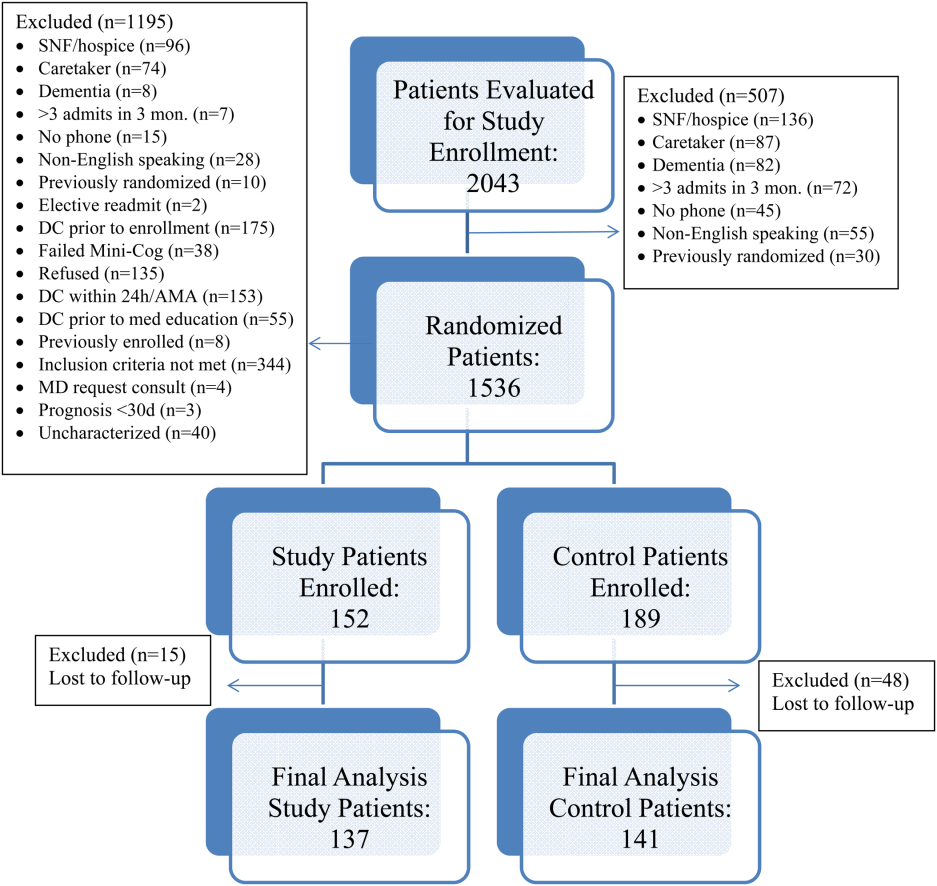
Group issues guideline for iliofemoral DVT

Image by Andre E.X. Brown
A new guideline aims to help physicians identify and manage blood clots, specifically iliofemoral deep vein thrombosis (DVT), in the groin and thigh.
The guideline states that anticoagulant therapy remains the cornerstone of management, but certain patients with iliofemoral DVT may benefit from alternative strategies, such as inferior vena cava filters, compression therapy, and clot removal or reduction strategies.
The guideline, which is based on the latest evidence, was published in CMAJ.
It was developed by a team of hematologists, interventional radiologists, vascular surgeons, emergency department physicians, and primary care physicians.
“We think this clinical practice guideline fills an important gap in knowledge for care providers by providing a practical approach to a common problem that can have serious implications for patients,” said author David Liu, MD, of Vancouver General Hospital in British Columbia, Canada.
“Complications associated with DVT can occur years after the presentation of DVT if it is not managed at onset. DVT is a life-threatening condition in the short term, with long-term implications to the patient and society if not managed properly.”
The guideline team has created a summary of recommendations and a decision tool to help physicians. Highlights include:
- All hospital staff must have the tools to diagnose and determine the severity of iliofemoral DVT.
- Anticoagulants are recommended for all patients with iliofemoral DVT, but the type and length of treatment will vary according to presentation.
- For patients not able to take anticoagulants, use of inferior vena cava filters is recommended with regular follow-up. The filters should be removed as soon as possible.
- Immediate intervention with clot removal is recommended in patients with phlegmasia cerulea dolens to reduce the associated risks of amputation and death.
- Clot removal intervention can also be considered for patients who are at low risk of bleeding to minimize possible long-term complications from iliofemoral DVT that may decrease quality of life (such as post-thrombotic syndrome).
- To manage post-thrombotic syndrome, the use of compression stockings is recommended, although the evidence that this intervention is effective is weak.
- Patient follow-up by the primary care physician is important.

Image by Andre E.X. Brown
A new guideline aims to help physicians identify and manage blood clots, specifically iliofemoral deep vein thrombosis (DVT), in the groin and thigh.
The guideline states that anticoagulant therapy remains the cornerstone of management, but certain patients with iliofemoral DVT may benefit from alternative strategies, such as inferior vena cava filters, compression therapy, and clot removal or reduction strategies.
The guideline, which is based on the latest evidence, was published in CMAJ.
It was developed by a team of hematologists, interventional radiologists, vascular surgeons, emergency department physicians, and primary care physicians.
“We think this clinical practice guideline fills an important gap in knowledge for care providers by providing a practical approach to a common problem that can have serious implications for patients,” said author David Liu, MD, of Vancouver General Hospital in British Columbia, Canada.
“Complications associated with DVT can occur years after the presentation of DVT if it is not managed at onset. DVT is a life-threatening condition in the short term, with long-term implications to the patient and society if not managed properly.”
The guideline team has created a summary of recommendations and a decision tool to help physicians. Highlights include:
- All hospital staff must have the tools to diagnose and determine the severity of iliofemoral DVT.
- Anticoagulants are recommended for all patients with iliofemoral DVT, but the type and length of treatment will vary according to presentation.
- For patients not able to take anticoagulants, use of inferior vena cava filters is recommended with regular follow-up. The filters should be removed as soon as possible.
- Immediate intervention with clot removal is recommended in patients with phlegmasia cerulea dolens to reduce the associated risks of amputation and death.
- Clot removal intervention can also be considered for patients who are at low risk of bleeding to minimize possible long-term complications from iliofemoral DVT that may decrease quality of life (such as post-thrombotic syndrome).
- To manage post-thrombotic syndrome, the use of compression stockings is recommended, although the evidence that this intervention is effective is weak.
- Patient follow-up by the primary care physician is important.
Image by Andre E.X. Brown
A new guideline aims to help physicians identify and manage blood clots, specifically iliofemoral deep vein thrombosis (DVT), in the groin and thigh.
The guideline states that anticoagulant therapy remains the cornerstone of management, but certain patients with iliofemoral DVT may benefit from alternative strategies, such as inferior vena cava filters, compression therapy, and clot removal or reduction strategies.
The guideline, which is based on the latest evidence, was published in CMAJ.
It was developed by a team of hematologists, interventional radiologists, vascular surgeons, emergency department physicians, and primary care physicians.
“We think this clinical practice guideline fills an important gap in knowledge for care providers by providing a practical approach to a common problem that can have serious implications for patients,” said author David Liu, MD, of Vancouver General Hospital in British Columbia, Canada.
“Complications associated with DVT can occur years after the presentation of DVT if it is not managed at onset. DVT is a life-threatening condition in the short term, with long-term implications to the patient and society if not managed properly.”
The guideline team has created a summary of recommendations and a decision tool to help physicians. Highlights include:
- All hospital staff must have the tools to diagnose and determine the severity of iliofemoral DVT.
- Anticoagulants are recommended for all patients with iliofemoral DVT, but the type and length of treatment will vary according to presentation.
- For patients not able to take anticoagulants, use of inferior vena cava filters is recommended with regular follow-up. The filters should be removed as soon as possible.
- Immediate intervention with clot removal is recommended in patients with phlegmasia cerulea dolens to reduce the associated risks of amputation and death.
- Clot removal intervention can also be considered for patients who are at low risk of bleeding to minimize possible long-term complications from iliofemoral DVT that may decrease quality of life (such as post-thrombotic syndrome).
- To manage post-thrombotic syndrome, the use of compression stockings is recommended, although the evidence that this intervention is effective is weak.
- Patient follow-up by the primary care physician is important.
Gout treatment lore doesn’t hold up to evidence
LAS VEGAS – It’s okay to start urate lowering therapy during a gout attack, according to Dr. Brian Mandell, a rheumatology professor at the Cleveland Clinic.
“I was taught that you don’t treat a gout attack with urate lowering therapy” because it could trigger a subsequent mobilization attack. “This has been the paradigm for years,” he said at the annual Perspectives in Rheumatic Diseases held by the Global Academy for Medical Education.
But a 2012 study casts doubt on that. In the study, 26 men hospitalized for gout attack were randomized to 300 mg allopurinol daily, while 25 were randomized to placebo. All of the patients were on indomethacin and colchicine. Urate levels dropped rapidly to below 6 mg/dL in the allopurinol group, but there were only two flares. Placebo subjects, meanwhile, had three flares (P= 0.60). There was no significant difference in daily pain scores (Am J Med. 2012 Nov;125(11):1126-1134.e7.).
“The bottom line is the groups did essentially the same. If you are using prophylaxis to treat an acute attack, you are probably perfectly fine to use allopurinol; you don’t need to wait,” Dr. Mandell said. “Does this mean you should start everybody in the hospital on full-dose allopurinol? No, but I think this really broadens our thought processes in terms of handling urate lowering therapy.”
It’s also time to rethink allopurinol dose adjustments in patients with chronic kidney disease (CKD). The thought has been that these patients should be on lower doses to prevent metabolite buildup in the kidneys and hypersensitivity reactions, but “if you follow those guidelines, less than 20% of patients will lower their serum urate, so you will be giving the drug for nothing,” Dr. Mandell said.
“It is true that allopurinol hypersensitivity is more common in patients with CKD,” but it happens in a few per thousand people, and hypersensitivity might have nothing to do with allopurinol dose. “There’s no repeated documentation between the levels and reactions, and in a number of small studies there is no evidence that dose adjustment decreases the frequency of hypersensitivity reactions. We have no data to say this is the right route to take,” he said.
In fact, a study that included 45 CKD patients found that increasing allopurinol beyond creatinine clearance-based dosing doesn’t cause problems. “Toxicity was not increased in patients receiving higher doses of allopurinol, including those with renal impairment,” the authors concluded (Arthritis Rheum. 2011 Feb;63(2):412-21.).
Given the rarity of hypersensitivity reactions, it’s not surprising they didn’t occur in the study’s small group of CKD patients, Dr. Mandell said. “If you are really ultra conservative, you are [still] not going to use allopurinol in CKD,” he said.
But Dr. Mandell said he starts patients on 50 mg of allopurinol. “I educate them about allergic reactions and increase the dose every 1-2 weeks until I hit” a serum uric acid below 6 mg/dL, which often takes more than 300 mg per day. “Alternatively, you can start febuxostat, and if I were to do that I would start at 20 mg” – breaking the lowest dose pill of 40 mg in half – “and then titrate up,” he said.
“It just doesn’t make sense,” Dr. Mandell said, that allopurinol hypersensitivity is related to dose. Instead, there seems to be a genetic predisposition for toxicity in, for instance, Asian patients with the HLA-B*5801 genotype, he noted.
Pegloticase is a bit more problematic than allopurinol. It’s highly effective – patients can drop their serum uric acid from 14 to 0.1 mg/dL within hours – but that comes at the cost of a high mobilization flair rate and infusion reactions in about a quarter of patients, Dr. Mandell said.
There’s a partial workaround. Dr. Mandell said he checks the uric acid level before the second infusion. If it’s not below 6 mg/dL after the first infusion, patients have antibodies against pegloticase. “The drug isn’t going to work, and patients are more likely to get an allergic reaction, so stop the drug,” he said.
Dr. Mandell is a consultant for AstraZeneca and Crealta Pharmaceuticals. The Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – It’s okay to start urate lowering therapy during a gout attack, according to Dr. Brian Mandell, a rheumatology professor at the Cleveland Clinic.
“I was taught that you don’t treat a gout attack with urate lowering therapy” because it could trigger a subsequent mobilization attack. “This has been the paradigm for years,” he said at the annual Perspectives in Rheumatic Diseases held by the Global Academy for Medical Education.
But a 2012 study casts doubt on that. In the study, 26 men hospitalized for gout attack were randomized to 300 mg allopurinol daily, while 25 were randomized to placebo. All of the patients were on indomethacin and colchicine. Urate levels dropped rapidly to below 6 mg/dL in the allopurinol group, but there were only two flares. Placebo subjects, meanwhile, had three flares (P= 0.60). There was no significant difference in daily pain scores (Am J Med. 2012 Nov;125(11):1126-1134.e7.).
“The bottom line is the groups did essentially the same. If you are using prophylaxis to treat an acute attack, you are probably perfectly fine to use allopurinol; you don’t need to wait,” Dr. Mandell said. “Does this mean you should start everybody in the hospital on full-dose allopurinol? No, but I think this really broadens our thought processes in terms of handling urate lowering therapy.”
It’s also time to rethink allopurinol dose adjustments in patients with chronic kidney disease (CKD). The thought has been that these patients should be on lower doses to prevent metabolite buildup in the kidneys and hypersensitivity reactions, but “if you follow those guidelines, less than 20% of patients will lower their serum urate, so you will be giving the drug for nothing,” Dr. Mandell said.
“It is true that allopurinol hypersensitivity is more common in patients with CKD,” but it happens in a few per thousand people, and hypersensitivity might have nothing to do with allopurinol dose. “There’s no repeated documentation between the levels and reactions, and in a number of small studies there is no evidence that dose adjustment decreases the frequency of hypersensitivity reactions. We have no data to say this is the right route to take,” he said.
In fact, a study that included 45 CKD patients found that increasing allopurinol beyond creatinine clearance-based dosing doesn’t cause problems. “Toxicity was not increased in patients receiving higher doses of allopurinol, including those with renal impairment,” the authors concluded (Arthritis Rheum. 2011 Feb;63(2):412-21.).
Given the rarity of hypersensitivity reactions, it’s not surprising they didn’t occur in the study’s small group of CKD patients, Dr. Mandell said. “If you are really ultra conservative, you are [still] not going to use allopurinol in CKD,” he said.
But Dr. Mandell said he starts patients on 50 mg of allopurinol. “I educate them about allergic reactions and increase the dose every 1-2 weeks until I hit” a serum uric acid below 6 mg/dL, which often takes more than 300 mg per day. “Alternatively, you can start febuxostat, and if I were to do that I would start at 20 mg” – breaking the lowest dose pill of 40 mg in half – “and then titrate up,” he said.
“It just doesn’t make sense,” Dr. Mandell said, that allopurinol hypersensitivity is related to dose. Instead, there seems to be a genetic predisposition for toxicity in, for instance, Asian patients with the HLA-B*5801 genotype, he noted.
Pegloticase is a bit more problematic than allopurinol. It’s highly effective – patients can drop their serum uric acid from 14 to 0.1 mg/dL within hours – but that comes at the cost of a high mobilization flair rate and infusion reactions in about a quarter of patients, Dr. Mandell said.
There’s a partial workaround. Dr. Mandell said he checks the uric acid level before the second infusion. If it’s not below 6 mg/dL after the first infusion, patients have antibodies against pegloticase. “The drug isn’t going to work, and patients are more likely to get an allergic reaction, so stop the drug,” he said.
Dr. Mandell is a consultant for AstraZeneca and Crealta Pharmaceuticals. The Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – It’s okay to start urate lowering therapy during a gout attack, according to Dr. Brian Mandell, a rheumatology professor at the Cleveland Clinic.
“I was taught that you don’t treat a gout attack with urate lowering therapy” because it could trigger a subsequent mobilization attack. “This has been the paradigm for years,” he said at the annual Perspectives in Rheumatic Diseases held by the Global Academy for Medical Education.
But a 2012 study casts doubt on that. In the study, 26 men hospitalized for gout attack were randomized to 300 mg allopurinol daily, while 25 were randomized to placebo. All of the patients were on indomethacin and colchicine. Urate levels dropped rapidly to below 6 mg/dL in the allopurinol group, but there were only two flares. Placebo subjects, meanwhile, had three flares (P= 0.60). There was no significant difference in daily pain scores (Am J Med. 2012 Nov;125(11):1126-1134.e7.).
“The bottom line is the groups did essentially the same. If you are using prophylaxis to treat an acute attack, you are probably perfectly fine to use allopurinol; you don’t need to wait,” Dr. Mandell said. “Does this mean you should start everybody in the hospital on full-dose allopurinol? No, but I think this really broadens our thought processes in terms of handling urate lowering therapy.”
It’s also time to rethink allopurinol dose adjustments in patients with chronic kidney disease (CKD). The thought has been that these patients should be on lower doses to prevent metabolite buildup in the kidneys and hypersensitivity reactions, but “if you follow those guidelines, less than 20% of patients will lower their serum urate, so you will be giving the drug for nothing,” Dr. Mandell said.
“It is true that allopurinol hypersensitivity is more common in patients with CKD,” but it happens in a few per thousand people, and hypersensitivity might have nothing to do with allopurinol dose. “There’s no repeated documentation between the levels and reactions, and in a number of small studies there is no evidence that dose adjustment decreases the frequency of hypersensitivity reactions. We have no data to say this is the right route to take,” he said.
In fact, a study that included 45 CKD patients found that increasing allopurinol beyond creatinine clearance-based dosing doesn’t cause problems. “Toxicity was not increased in patients receiving higher doses of allopurinol, including those with renal impairment,” the authors concluded (Arthritis Rheum. 2011 Feb;63(2):412-21.).
Given the rarity of hypersensitivity reactions, it’s not surprising they didn’t occur in the study’s small group of CKD patients, Dr. Mandell said. “If you are really ultra conservative, you are [still] not going to use allopurinol in CKD,” he said.
But Dr. Mandell said he starts patients on 50 mg of allopurinol. “I educate them about allergic reactions and increase the dose every 1-2 weeks until I hit” a serum uric acid below 6 mg/dL, which often takes more than 300 mg per day. “Alternatively, you can start febuxostat, and if I were to do that I would start at 20 mg” – breaking the lowest dose pill of 40 mg in half – “and then titrate up,” he said.
“It just doesn’t make sense,” Dr. Mandell said, that allopurinol hypersensitivity is related to dose. Instead, there seems to be a genetic predisposition for toxicity in, for instance, Asian patients with the HLA-B*5801 genotype, he noted.
Pegloticase is a bit more problematic than allopurinol. It’s highly effective – patients can drop their serum uric acid from 14 to 0.1 mg/dL within hours – but that comes at the cost of a high mobilization flair rate and infusion reactions in about a quarter of patients, Dr. Mandell said.
There’s a partial workaround. Dr. Mandell said he checks the uric acid level before the second infusion. If it’s not below 6 mg/dL after the first infusion, patients have antibodies against pegloticase. “The drug isn’t going to work, and patients are more likely to get an allergic reaction, so stop the drug,” he said.
Dr. Mandell is a consultant for AstraZeneca and Crealta Pharmaceuticals. The Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM PERSPECTIVES IN RHEUMATIC DISEASES
Chronic insomnia afflicts one in three perimenopausal women
LAS VEGAS – Sleeplessness is both common and distressing for women in midlife, according to a new analysis of data from a large observational study.
One-third of women at all stages of the menopausal transition reported sleep disturbance that met the clinical criteria for insomnia, regardless of many markers of health and socioeconomic status, Colleen L. Ciano, Ph.D., reported at the NAMS 2015 annual meeting.

Acute insomnia is prevalent in adults, and women are known to suffer more than men from chronic insomnia. Perimenopausal women, in particular, may have increased sleep disturbances.
Dr. Ciano, a postdoctoral fellow at the University of Pennsylvania, Philadelphia, and her colleagues used data from the large, longitudinal Study of Women’s Health Across the Nation (SWAN) to ascertain insomnia’s prevalence in perimenopausal and surgically menopausal women. Using SWAN study data, she also attempted to tease out factors that might increase a woman’s risk for chronic insomnia.
SWAN, said Dr. Ciano, was designed to examine the health of midlife women, gathering data from 3,302 women with a mean age of 45.9 years. Ten years of study data are now available from an ethnically diverse sample of women participating at seven sites across the United States.
Four questions from the SWAN questionnaire were identified for analysis as dependent variables. The questions related to trouble falling asleep, nighttime wakings, early-morning wakings, and a subjective rating of sleep quality over 2 weeks. Women were stratified into early or late menopause, or surgical menopause.
More than one-third of women at all stages of the midlife transition, or who were in surgical menopause, reported diagnosable insomnia. On the whole, insomnia increased for women over the study period. Waking after sleep onset also increased for perimenopausal women as they moved through menopause, from 26% to 36%. Sleep quality, as a bifurcated measure of sleep quality derived from a four-point scale, also dropped over the study period.
During the SWAN data collection period, the number of perimenopausal women who met at least one of the American Academy of Sleep Medicine criteria for insomnia rose from just over 30% to just over 40%, while the number of women who reported no insomnia symptoms fell steeply, dropping from just under 70% to approximately 50%.

When Dr. Ciano analyzed the prevalence of insomnia by perimenopausal stage, she found more insomnia in late-perimenopausal women (prevalence 31%-48%), compared with those in early menopause (31%-59%).
Women in late perimenopause had an odds ratio of 1.3 of reporting any symptom of insomnia, compared with those in early menopause, according to Dr. Ciano.
Women who were heavier, older, or depressed, as well as those having night sweats, were all more likely to suffer from insomnia on multivariable analysis (all P=0.002 or less). Factors that did not influence insomnia included marital status, tobacco or alcohol use, race/ethnicity, socioeconomic status, and many comorbidities including diabetes, hypertension, and cardiovascular disease.
Dr. Ciano advised clinicians to pay attention to these findings and initiate insomnia screenings. Routine evaluation of perimenopausal women should include careful questioning and an assessment of sleep patterns and problems. This is especially important since obesity, diabetes, and cardiovascular disease are all prevalent chronic illnesses that can be impacted by insomnia, she said.
Future studies should include physiologic measures and objective sleep data, which could be correlated with subjective reports, said Dr. Ciano. Sleep diaries can be an adjunct to observational and objective data, and can be correlated with such objective measures as endogenous hormone levels and objective vasomotor symptom monitoring.
Dr. Ciano’s research was supported by the National Institute for Nursing Research, Pennsylvana State University, and the Beta Sigma chapter of Sigma Theta Tau. She reported having no relevant financial disclosures.
On Twitter @karioakes
LAS VEGAS – Sleeplessness is both common and distressing for women in midlife, according to a new analysis of data from a large observational study.
One-third of women at all stages of the menopausal transition reported sleep disturbance that met the clinical criteria for insomnia, regardless of many markers of health and socioeconomic status, Colleen L. Ciano, Ph.D., reported at the NAMS 2015 annual meeting.
Acute insomnia is prevalent in adults, and women are known to suffer more than men from chronic insomnia. Perimenopausal women, in particular, may have increased sleep disturbances.
Dr. Ciano, a postdoctoral fellow at the University of Pennsylvania, Philadelphia, and her colleagues used data from the large, longitudinal Study of Women’s Health Across the Nation (SWAN) to ascertain insomnia’s prevalence in perimenopausal and surgically menopausal women. Using SWAN study data, she also attempted to tease out factors that might increase a woman’s risk for chronic insomnia.
SWAN, said Dr. Ciano, was designed to examine the health of midlife women, gathering data from 3,302 women with a mean age of 45.9 years. Ten years of study data are now available from an ethnically diverse sample of women participating at seven sites across the United States.
Four questions from the SWAN questionnaire were identified for analysis as dependent variables. The questions related to trouble falling asleep, nighttime wakings, early-morning wakings, and a subjective rating of sleep quality over 2 weeks. Women were stratified into early or late menopause, or surgical menopause.
More than one-third of women at all stages of the midlife transition, or who were in surgical menopause, reported diagnosable insomnia. On the whole, insomnia increased for women over the study period. Waking after sleep onset also increased for perimenopausal women as they moved through menopause, from 26% to 36%. Sleep quality, as a bifurcated measure of sleep quality derived from a four-point scale, also dropped over the study period.
During the SWAN data collection period, the number of perimenopausal women who met at least one of the American Academy of Sleep Medicine criteria for insomnia rose from just over 30% to just over 40%, while the number of women who reported no insomnia symptoms fell steeply, dropping from just under 70% to approximately 50%.
When Dr. Ciano analyzed the prevalence of insomnia by perimenopausal stage, she found more insomnia in late-perimenopausal women (prevalence 31%-48%), compared with those in early menopause (31%-59%).
Women in late perimenopause had an odds ratio of 1.3 of reporting any symptom of insomnia, compared with those in early menopause, according to Dr. Ciano.
Women who were heavier, older, or depressed, as well as those having night sweats, were all more likely to suffer from insomnia on multivariable analysis (all P=0.002 or less). Factors that did not influence insomnia included marital status, tobacco or alcohol use, race/ethnicity, socioeconomic status, and many comorbidities including diabetes, hypertension, and cardiovascular disease.
Dr. Ciano advised clinicians to pay attention to these findings and initiate insomnia screenings. Routine evaluation of perimenopausal women should include careful questioning and an assessment of sleep patterns and problems. This is especially important since obesity, diabetes, and cardiovascular disease are all prevalent chronic illnesses that can be impacted by insomnia, she said.
Future studies should include physiologic measures and objective sleep data, which could be correlated with subjective reports, said Dr. Ciano. Sleep diaries can be an adjunct to observational and objective data, and can be correlated with such objective measures as endogenous hormone levels and objective vasomotor symptom monitoring.
Dr. Ciano’s research was supported by the National Institute for Nursing Research, Pennsylvana State University, and the Beta Sigma chapter of Sigma Theta Tau. She reported having no relevant financial disclosures.
On Twitter @karioakes
LAS VEGAS – Sleeplessness is both common and distressing for women in midlife, according to a new analysis of data from a large observational study.
One-third of women at all stages of the menopausal transition reported sleep disturbance that met the clinical criteria for insomnia, regardless of many markers of health and socioeconomic status, Colleen L. Ciano, Ph.D., reported at the NAMS 2015 annual meeting.
Acute insomnia is prevalent in adults, and women are known to suffer more than men from chronic insomnia. Perimenopausal women, in particular, may have increased sleep disturbances.
Dr. Ciano, a postdoctoral fellow at the University of Pennsylvania, Philadelphia, and her colleagues used data from the large, longitudinal Study of Women’s Health Across the Nation (SWAN) to ascertain insomnia’s prevalence in perimenopausal and surgically menopausal women. Using SWAN study data, she also attempted to tease out factors that might increase a woman’s risk for chronic insomnia.
SWAN, said Dr. Ciano, was designed to examine the health of midlife women, gathering data from 3,302 women with a mean age of 45.9 years. Ten years of study data are now available from an ethnically diverse sample of women participating at seven sites across the United States.
Four questions from the SWAN questionnaire were identified for analysis as dependent variables. The questions related to trouble falling asleep, nighttime wakings, early-morning wakings, and a subjective rating of sleep quality over 2 weeks. Women were stratified into early or late menopause, or surgical menopause.
More than one-third of women at all stages of the midlife transition, or who were in surgical menopause, reported diagnosable insomnia. On the whole, insomnia increased for women over the study period. Waking after sleep onset also increased for perimenopausal women as they moved through menopause, from 26% to 36%. Sleep quality, as a bifurcated measure of sleep quality derived from a four-point scale, also dropped over the study period.
During the SWAN data collection period, the number of perimenopausal women who met at least one of the American Academy of Sleep Medicine criteria for insomnia rose from just over 30% to just over 40%, while the number of women who reported no insomnia symptoms fell steeply, dropping from just under 70% to approximately 50%.
When Dr. Ciano analyzed the prevalence of insomnia by perimenopausal stage, she found more insomnia in late-perimenopausal women (prevalence 31%-48%), compared with those in early menopause (31%-59%).
Women in late perimenopause had an odds ratio of 1.3 of reporting any symptom of insomnia, compared with those in early menopause, according to Dr. Ciano.
Women who were heavier, older, or depressed, as well as those having night sweats, were all more likely to suffer from insomnia on multivariable analysis (all P=0.002 or less). Factors that did not influence insomnia included marital status, tobacco or alcohol use, race/ethnicity, socioeconomic status, and many comorbidities including diabetes, hypertension, and cardiovascular disease.
Dr. Ciano advised clinicians to pay attention to these findings and initiate insomnia screenings. Routine evaluation of perimenopausal women should include careful questioning and an assessment of sleep patterns and problems. This is especially important since obesity, diabetes, and cardiovascular disease are all prevalent chronic illnesses that can be impacted by insomnia, she said.
Future studies should include physiologic measures and objective sleep data, which could be correlated with subjective reports, said Dr. Ciano. Sleep diaries can be an adjunct to observational and objective data, and can be correlated with such objective measures as endogenous hormone levels and objective vasomotor symptom monitoring.
Dr. Ciano’s research was supported by the National Institute for Nursing Research, Pennsylvana State University, and the Beta Sigma chapter of Sigma Theta Tau. She reported having no relevant financial disclosures.
On Twitter @karioakes
AT THE NAMS 2015 ANNUAL MEETING
Key clinical point: One in three perimenopausal women has persistent insomnia.
Major finding: An average of one-third of perimenopausal women have significant symptoms of insomnia, higher than age-matched prevalence in the general population.
Data source: Analysis of the Study of Women’s Health Across the Nation (SWAN), a multi-site longitudinal study of 3,302 ethnically diverse midlife-aged women.
Disclosures: The research was supported by the National Institute for Nursing Research, Pennsylvana State University, and the Beta Sigma chapter of Sigma Theta Tau. Dr. Ciano reported having no relevant financial disclosures.

NICE can’t recommend new sepsis tests
Photo by William Weinert
The UK’s National Institute for Health and Care Excellence (NICE) has said there is not enough evidence to recommend 3 new blood tests for routine use in the National Health Service.
The tests are designed to identify bacteria and fungi in the bloodstream more rapidly than current tests.
According to NICE, there is too much uncertainty regarding the accuracy of the new tests and the size of benefit they might confer for patients with suspected sepsis.
The tests in question are LightCycler SeptiFast Test MGRADE (Roche Diagnostics), SepsiTest (Molzym Molecular Diagnostics), and IRIDICA BAC BSI assay (Abbott Diagnostics).
They are used to analyze whole blood samples for bacterial and fungal DNA, which may identify pathogens earlier than microbiology techniques. Microbiology techniques require blood samples to be incubated and cultured before pathogens can be identified.
The new tests are designed to enable earlier targeted treatment for patients with sepsis and reduce the use of broad-spectrum antimicrobials, which could help reduce future antimicrobial resistance.
“Rapid molecular tests that can identify which pathogens are the cause of an infection in hours rather than the days typically needed for traditional microbiology tests could ensure the right antibiotics are used much earlier in treatment,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“This, in turn, could improve outcomes for patients with suspected sepsis as well as help to reduce the spread of resistant microbes. However, the committee [advising NICE] concluded that the tests may offer clinical benefit, but there is too much uncertainty in the size of the benefit to determine the effect of introducing the tests into clinical practice.”
“The committee also concluded that, although the rapid molecular tests might provide results more quickly, there was too much uncertainty in the accuracy of the tests for clinicians to be able to base a decision on whether to withdraw or continue antibiotics. The committee therefore decided that further research should be encouraged to determine the clinical scenarios in which the tests may offer most benefit.”
NICE’s draft diagnostics guidance on the LightCycler SeptiFast Test MGRADE, SepsiTest, and IRIDICA BAC BSI assay is available on the NICE website. The closing date for comments on this draft guidance is October 21, 2015. ![]()
Photo by William Weinert
The UK’s National Institute for Health and Care Excellence (NICE) has said there is not enough evidence to recommend 3 new blood tests for routine use in the National Health Service.
The tests are designed to identify bacteria and fungi in the bloodstream more rapidly than current tests.
According to NICE, there is too much uncertainty regarding the accuracy of the new tests and the size of benefit they might confer for patients with suspected sepsis.
The tests in question are LightCycler SeptiFast Test MGRADE (Roche Diagnostics), SepsiTest (Molzym Molecular Diagnostics), and IRIDICA BAC BSI assay (Abbott Diagnostics).
They are used to analyze whole blood samples for bacterial and fungal DNA, which may identify pathogens earlier than microbiology techniques. Microbiology techniques require blood samples to be incubated and cultured before pathogens can be identified.
The new tests are designed to enable earlier targeted treatment for patients with sepsis and reduce the use of broad-spectrum antimicrobials, which could help reduce future antimicrobial resistance.
“Rapid molecular tests that can identify which pathogens are the cause of an infection in hours rather than the days typically needed for traditional microbiology tests could ensure the right antibiotics are used much earlier in treatment,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“This, in turn, could improve outcomes for patients with suspected sepsis as well as help to reduce the spread of resistant microbes. However, the committee [advising NICE] concluded that the tests may offer clinical benefit, but there is too much uncertainty in the size of the benefit to determine the effect of introducing the tests into clinical practice.”
“The committee also concluded that, although the rapid molecular tests might provide results more quickly, there was too much uncertainty in the accuracy of the tests for clinicians to be able to base a decision on whether to withdraw or continue antibiotics. The committee therefore decided that further research should be encouraged to determine the clinical scenarios in which the tests may offer most benefit.”
NICE’s draft diagnostics guidance on the LightCycler SeptiFast Test MGRADE, SepsiTest, and IRIDICA BAC BSI assay is available on the NICE website. The closing date for comments on this draft guidance is October 21, 2015. ![]()
Photo by William Weinert
The UK’s National Institute for Health and Care Excellence (NICE) has said there is not enough evidence to recommend 3 new blood tests for routine use in the National Health Service.
The tests are designed to identify bacteria and fungi in the bloodstream more rapidly than current tests.
According to NICE, there is too much uncertainty regarding the accuracy of the new tests and the size of benefit they might confer for patients with suspected sepsis.
The tests in question are LightCycler SeptiFast Test MGRADE (Roche Diagnostics), SepsiTest (Molzym Molecular Diagnostics), and IRIDICA BAC BSI assay (Abbott Diagnostics).
They are used to analyze whole blood samples for bacterial and fungal DNA, which may identify pathogens earlier than microbiology techniques. Microbiology techniques require blood samples to be incubated and cultured before pathogens can be identified.
The new tests are designed to enable earlier targeted treatment for patients with sepsis and reduce the use of broad-spectrum antimicrobials, which could help reduce future antimicrobial resistance.
“Rapid molecular tests that can identify which pathogens are the cause of an infection in hours rather than the days typically needed for traditional microbiology tests could ensure the right antibiotics are used much earlier in treatment,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“This, in turn, could improve outcomes for patients with suspected sepsis as well as help to reduce the spread of resistant microbes. However, the committee [advising NICE] concluded that the tests may offer clinical benefit, but there is too much uncertainty in the size of the benefit to determine the effect of introducing the tests into clinical practice.”
“The committee also concluded that, although the rapid molecular tests might provide results more quickly, there was too much uncertainty in the accuracy of the tests for clinicians to be able to base a decision on whether to withdraw or continue antibiotics. The committee therefore decided that further research should be encouraged to determine the clinical scenarios in which the tests may offer most benefit.”
NICE’s draft diagnostics guidance on the LightCycler SeptiFast Test MGRADE, SepsiTest, and IRIDICA BAC BSI assay is available on the NICE website. The closing date for comments on this draft guidance is October 21, 2015. ![]()
Progress in treating diabetic foot osteomyelitis
SAN DIEGO – The evidence-based treatment of diabetic foot osteomyelitis has jumped up to the next level as a result of two recent randomized clinical trials, the first-ever to address a couple of key contentious issues, experts agreed recently at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
Dr. Eric Senneville presented highlights of the two randomized trials, one of which examined the optimal duration of antibiotic therapy in patients with nonsurgically treated diabetic foot osteomyelitis. The other study was the first-ever head-to-head randomized comparison of antibiotics versus conservative surgery.

He also touched on another new development in the treatment of diabetic foot osteomyelitis: surgically implanted topical antibiotics, which show promise in specific situations.
Dr. Senneville of Gustave Dron Hospital in Tourcoing, France, was senior investigator in the prospective, randomized, multicenter comparison of outcomes with 6 versus 12 weeks of open-label antibiotic therapy in 40 patients. All participants had bone biopsy–confirmed osteomyelitis with no ischemia, and none underwent any bone resection during the treatment period.
The 6-week regimen proved to be the winning strategy. It resulted in remission in 12 of 20 patients, a result not significantly different from the 14 of 20 remission rate with 12 weeks of treatment. Moreover, significant drug-related gastrointestinal side effects occurred in only three patients in the 6-week-treatment arm, compared with nine patients treated for 12 weeks. There was no difference between the two groups in rates of relapse, need for later bone resection, or spread of osteomyelitis (Diabetes Care. 2015 Feb;38[2]:302-7).
In the surgery-versus-antibiotics trial, investigators at the University of Madrid prospectively randomized 52 patients with diabetic foot osteomyelitis to 90 days of antibiotics with no surgery or to conservative surgery with 10 days of postoperative antibiotics. Dr. Senneville emphasized that this was a select patient population and at this point the results apply only to similar groups; that is, all participants had forefoot osteomyelitis without ischemia or necrosis. There were six dropouts: one in the medically treated arm and five in the surgical group.
The key finding: At 12 weeks of follow-up, main outcomes were similar in the two groups. Eighteen of 24 patients in the medically managed group achieved primary healing, for a 75% cure rate, not significantly different from the 86% rate – 19 of 22 patients cured – in the surgical group. Median time to healing was 7 weeks with antibiotics only and similar at 6 weeks with surgery. Four patients in the antibiotic group worsened and required surgery, while three in the surgery group required reoperation (Diabetes Care. 2014 Mar;37[3]:789-95).
Dr. Senneville noted that the reulceration rate was 10% in the medically treated group and twice that in the surgical group. A higher reulceration rate has also been seen in retrospective studies. It’s thought to result from what has been termed pressure transfer syndrome, which is particularly common among patients who undergo surgery on the first metatarsal head.
Dr. Edgar J. G. Peters of VU Academic Medical Center, Amsterdam, commented that the Spanish randomized trial of antibiotics versus surgery in diabetic osteomyelitis was sorely needed. All too often, he observed, earlier retrospective studies conducted by surgeons concluded that surgery was best, while those carried out by infectious disease specialists found the antiobiotics-only strategy to be superior. Skeptical unbiased physicians were left in the dark.
He noted that this important clinical trial as well as the major randomized trial of 6 versus 12 weeks of antibiotic therapy were published too late for inclusion in the recently released systematic review of treatments for diabetic foot infections conducted by the International Working Group on the Diabetic Foot (Diabetes Metab Res Rev. 2015 Sep 7. doi: 10.1002/dmrr.2706), for which both Dr. Peters and Dr. Senneville were coauthors.
Turning to the novel use of topical antibiotics in patients with diabetic foot osteomyelitis, Dr. Senneville described several potential advantages, including the attainment of optimal drug levels in the presence of peripheral vascular disease and in avascular spaces.
The idea is to place antibiotic-impregnated beads or bone cement in the space created by debridement and removal of infected bone. By filling the dead space, the antibiotic-impregnated cement may control the infection simmering in any areas of infected bone unintentionally left behind, while also reducing the risk of pressure transfer syndrome. The use of antibiotic-eluting bone cement has recently been shown to reduce the need for reoperation (J Foot Ankle Surg. 2015 Jul-Aug;54[4]:536-40).
Dr. Senneville reported serving on speakers’ bureaus for Novartis and Merck and as an advisor to Pfizer.
SAN DIEGO – The evidence-based treatment of diabetic foot osteomyelitis has jumped up to the next level as a result of two recent randomized clinical trials, the first-ever to address a couple of key contentious issues, experts agreed recently at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
Dr. Eric Senneville presented highlights of the two randomized trials, one of which examined the optimal duration of antibiotic therapy in patients with nonsurgically treated diabetic foot osteomyelitis. The other study was the first-ever head-to-head randomized comparison of antibiotics versus conservative surgery.
He also touched on another new development in the treatment of diabetic foot osteomyelitis: surgically implanted topical antibiotics, which show promise in specific situations.
Dr. Senneville of Gustave Dron Hospital in Tourcoing, France, was senior investigator in the prospective, randomized, multicenter comparison of outcomes with 6 versus 12 weeks of open-label antibiotic therapy in 40 patients. All participants had bone biopsy–confirmed osteomyelitis with no ischemia, and none underwent any bone resection during the treatment period.
The 6-week regimen proved to be the winning strategy. It resulted in remission in 12 of 20 patients, a result not significantly different from the 14 of 20 remission rate with 12 weeks of treatment. Moreover, significant drug-related gastrointestinal side effects occurred in only three patients in the 6-week-treatment arm, compared with nine patients treated for 12 weeks. There was no difference between the two groups in rates of relapse, need for later bone resection, or spread of osteomyelitis (Diabetes Care. 2015 Feb;38[2]:302-7).
In the surgery-versus-antibiotics trial, investigators at the University of Madrid prospectively randomized 52 patients with diabetic foot osteomyelitis to 90 days of antibiotics with no surgery or to conservative surgery with 10 days of postoperative antibiotics. Dr. Senneville emphasized that this was a select patient population and at this point the results apply only to similar groups; that is, all participants had forefoot osteomyelitis without ischemia or necrosis. There were six dropouts: one in the medically treated arm and five in the surgical group.
The key finding: At 12 weeks of follow-up, main outcomes were similar in the two groups. Eighteen of 24 patients in the medically managed group achieved primary healing, for a 75% cure rate, not significantly different from the 86% rate – 19 of 22 patients cured – in the surgical group. Median time to healing was 7 weeks with antibiotics only and similar at 6 weeks with surgery. Four patients in the antibiotic group worsened and required surgery, while three in the surgery group required reoperation (Diabetes Care. 2014 Mar;37[3]:789-95).
Dr. Senneville noted that the reulceration rate was 10% in the medically treated group and twice that in the surgical group. A higher reulceration rate has also been seen in retrospective studies. It’s thought to result from what has been termed pressure transfer syndrome, which is particularly common among patients who undergo surgery on the first metatarsal head.
Dr. Edgar J. G. Peters of VU Academic Medical Center, Amsterdam, commented that the Spanish randomized trial of antibiotics versus surgery in diabetic osteomyelitis was sorely needed. All too often, he observed, earlier retrospective studies conducted by surgeons concluded that surgery was best, while those carried out by infectious disease specialists found the antiobiotics-only strategy to be superior. Skeptical unbiased physicians were left in the dark.
He noted that this important clinical trial as well as the major randomized trial of 6 versus 12 weeks of antibiotic therapy were published too late for inclusion in the recently released systematic review of treatments for diabetic foot infections conducted by the International Working Group on the Diabetic Foot (Diabetes Metab Res Rev. 2015 Sep 7. doi: 10.1002/dmrr.2706), for which both Dr. Peters and Dr. Senneville were coauthors.
Turning to the novel use of topical antibiotics in patients with diabetic foot osteomyelitis, Dr. Senneville described several potential advantages, including the attainment of optimal drug levels in the presence of peripheral vascular disease and in avascular spaces.
The idea is to place antibiotic-impregnated beads or bone cement in the space created by debridement and removal of infected bone. By filling the dead space, the antibiotic-impregnated cement may control the infection simmering in any areas of infected bone unintentionally left behind, while also reducing the risk of pressure transfer syndrome. The use of antibiotic-eluting bone cement has recently been shown to reduce the need for reoperation (J Foot Ankle Surg. 2015 Jul-Aug;54[4]:536-40).
Dr. Senneville reported serving on speakers’ bureaus for Novartis and Merck and as an advisor to Pfizer.
SAN DIEGO – The evidence-based treatment of diabetic foot osteomyelitis has jumped up to the next level as a result of two recent randomized clinical trials, the first-ever to address a couple of key contentious issues, experts agreed recently at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
Dr. Eric Senneville presented highlights of the two randomized trials, one of which examined the optimal duration of antibiotic therapy in patients with nonsurgically treated diabetic foot osteomyelitis. The other study was the first-ever head-to-head randomized comparison of antibiotics versus conservative surgery.
He also touched on another new development in the treatment of diabetic foot osteomyelitis: surgically implanted topical antibiotics, which show promise in specific situations.
Dr. Senneville of Gustave Dron Hospital in Tourcoing, France, was senior investigator in the prospective, randomized, multicenter comparison of outcomes with 6 versus 12 weeks of open-label antibiotic therapy in 40 patients. All participants had bone biopsy–confirmed osteomyelitis with no ischemia, and none underwent any bone resection during the treatment period.
The 6-week regimen proved to be the winning strategy. It resulted in remission in 12 of 20 patients, a result not significantly different from the 14 of 20 remission rate with 12 weeks of treatment. Moreover, significant drug-related gastrointestinal side effects occurred in only three patients in the 6-week-treatment arm, compared with nine patients treated for 12 weeks. There was no difference between the two groups in rates of relapse, need for later bone resection, or spread of osteomyelitis (Diabetes Care. 2015 Feb;38[2]:302-7).
In the surgery-versus-antibiotics trial, investigators at the University of Madrid prospectively randomized 52 patients with diabetic foot osteomyelitis to 90 days of antibiotics with no surgery or to conservative surgery with 10 days of postoperative antibiotics. Dr. Senneville emphasized that this was a select patient population and at this point the results apply only to similar groups; that is, all participants had forefoot osteomyelitis without ischemia or necrosis. There were six dropouts: one in the medically treated arm and five in the surgical group.
The key finding: At 12 weeks of follow-up, main outcomes were similar in the two groups. Eighteen of 24 patients in the medically managed group achieved primary healing, for a 75% cure rate, not significantly different from the 86% rate – 19 of 22 patients cured – in the surgical group. Median time to healing was 7 weeks with antibiotics only and similar at 6 weeks with surgery. Four patients in the antibiotic group worsened and required surgery, while three in the surgery group required reoperation (Diabetes Care. 2014 Mar;37[3]:789-95).
Dr. Senneville noted that the reulceration rate was 10% in the medically treated group and twice that in the surgical group. A higher reulceration rate has also been seen in retrospective studies. It’s thought to result from what has been termed pressure transfer syndrome, which is particularly common among patients who undergo surgery on the first metatarsal head.
Dr. Edgar J. G. Peters of VU Academic Medical Center, Amsterdam, commented that the Spanish randomized trial of antibiotics versus surgery in diabetic osteomyelitis was sorely needed. All too often, he observed, earlier retrospective studies conducted by surgeons concluded that surgery was best, while those carried out by infectious disease specialists found the antiobiotics-only strategy to be superior. Skeptical unbiased physicians were left in the dark.
He noted that this important clinical trial as well as the major randomized trial of 6 versus 12 weeks of antibiotic therapy were published too late for inclusion in the recently released systematic review of treatments for diabetic foot infections conducted by the International Working Group on the Diabetic Foot (Diabetes Metab Res Rev. 2015 Sep 7. doi: 10.1002/dmrr.2706), for which both Dr. Peters and Dr. Senneville were coauthors.
Turning to the novel use of topical antibiotics in patients with diabetic foot osteomyelitis, Dr. Senneville described several potential advantages, including the attainment of optimal drug levels in the presence of peripheral vascular disease and in avascular spaces.
The idea is to place antibiotic-impregnated beads or bone cement in the space created by debridement and removal of infected bone. By filling the dead space, the antibiotic-impregnated cement may control the infection simmering in any areas of infected bone unintentionally left behind, while also reducing the risk of pressure transfer syndrome. The use of antibiotic-eluting bone cement has recently been shown to reduce the need for reoperation (J Foot Ankle Surg. 2015 Jul-Aug;54[4]:536-40).
Dr. Senneville reported serving on speakers’ bureaus for Novartis and Merck and as an advisor to Pfizer.
EXPERT ANALYSIS FROM ICAAC 2015
