User login
How Quality Improvement Programs Improve Hospitals, Communities

And it’s now easier than ever to get started, right from your computer.
Online Webinars
Coaching a Quality Improvement Team: Basics for Being Sure Any QI team and Project Are on the Right Track
Presenter: Jordan Messler, MD, SFHM
Date: October 28, 2015
Time: 1:00 p.m. EDT
Elevating Provider Experience to Improve Patient Experience
Presenter: Mark Rudolph, MD
Date: November 11, 2015
Time: 2:00 p.m. EDT
And it’s now easier than ever to get started, right from your computer.
Online Webinars
Coaching a Quality Improvement Team: Basics for Being Sure Any QI team and Project Are on the Right Track
Presenter: Jordan Messler, MD, SFHM
Date: October 28, 2015
Time: 1:00 p.m. EDT
Elevating Provider Experience to Improve Patient Experience
Presenter: Mark Rudolph, MD
Date: November 11, 2015
Time: 2:00 p.m. EDT
And it’s now easier than ever to get started, right from your computer.
Online Webinars
Coaching a Quality Improvement Team: Basics for Being Sure Any QI team and Project Are on the Right Track
Presenter: Jordan Messler, MD, SFHM
Date: October 28, 2015
Time: 1:00 p.m. EDT
Elevating Provider Experience to Improve Patient Experience
Presenter: Mark Rudolph, MD
Date: November 11, 2015
Time: 2:00 p.m. EDT
Start Planning Now for Hospital Medicine 2016

Hospital Medicine 2016 has been updated to meet the educational needs of hospitalists of all stripes, with new sessions and pre-courses, as well as entirely new tracks on the most cutting-edge topics in the movement.
New tracks planned: post-acute care, health IT for hospitalists, co-management/peri-operative medicine.
Recurring tracks: rapid fire, clinical, young hospitalists, practice management, academic/research, quality, pediatric, potpourri, the doctor-patient relationship.
For an updated list of pre-courses and other sessions, visit Hospital Medicine 2016. Register by Jan. 11, 2016, to save $50.
Hospital Medicine 2016 has been updated to meet the educational needs of hospitalists of all stripes, with new sessions and pre-courses, as well as entirely new tracks on the most cutting-edge topics in the movement.
New tracks planned: post-acute care, health IT for hospitalists, co-management/peri-operative medicine.
Recurring tracks: rapid fire, clinical, young hospitalists, practice management, academic/research, quality, pediatric, potpourri, the doctor-patient relationship.
For an updated list of pre-courses and other sessions, visit Hospital Medicine 2016. Register by Jan. 11, 2016, to save $50.
Hospital Medicine 2016 has been updated to meet the educational needs of hospitalists of all stripes, with new sessions and pre-courses, as well as entirely new tracks on the most cutting-edge topics in the movement.
New tracks planned: post-acute care, health IT for hospitalists, co-management/peri-operative medicine.
Recurring tracks: rapid fire, clinical, young hospitalists, practice management, academic/research, quality, pediatric, potpourri, the doctor-patient relationship.
For an updated list of pre-courses and other sessions, visit Hospital Medicine 2016. Register by Jan. 11, 2016, to save $50.
Society of Hospital Medicine Membership Ambassador Program Ends December 2015
You are one of the best representatives of the hospital medicine movement. You can share your enthusiasm for the specialty and for improving the care of hospitalized patients by telling others about SHM.
And, as an added bonus, you can earn credit toward SHM membership dues.
Through the end of the year, all active SHM members can earn 2016-2017 dues credits and special recognition for recruiting new physician, physician assistant, nurse practitioner, pharmacist, or affiliate members.
Active members will be eligible for:
- A $35 credit toward 2016-2017 dues when recruiting one new member;
- A $50 credit toward 2016-2017 dues when recruiting 2-4 new members;
- A $75 credit toward 2016-2017 dues when recruiting 5-9 new members; or
- A $125 credit toward 2016-2017 dues when recruiting 10+ new members.
For every member recruited, individuals will receive one entry into a grand prize drawing to receive complimentary registration to HM16 in San Diego.
Click here for more details.
You are one of the best representatives of the hospital medicine movement. You can share your enthusiasm for the specialty and for improving the care of hospitalized patients by telling others about SHM.
And, as an added bonus, you can earn credit toward SHM membership dues.
Through the end of the year, all active SHM members can earn 2016-2017 dues credits and special recognition for recruiting new physician, physician assistant, nurse practitioner, pharmacist, or affiliate members.
Active members will be eligible for:
- A $35 credit toward 2016-2017 dues when recruiting one new member;
- A $50 credit toward 2016-2017 dues when recruiting 2-4 new members;
- A $75 credit toward 2016-2017 dues when recruiting 5-9 new members; or
- A $125 credit toward 2016-2017 dues when recruiting 10+ new members.
For every member recruited, individuals will receive one entry into a grand prize drawing to receive complimentary registration to HM16 in San Diego.
Click here for more details.
You are one of the best representatives of the hospital medicine movement. You can share your enthusiasm for the specialty and for improving the care of hospitalized patients by telling others about SHM.
And, as an added bonus, you can earn credit toward SHM membership dues.
Through the end of the year, all active SHM members can earn 2016-2017 dues credits and special recognition for recruiting new physician, physician assistant, nurse practitioner, pharmacist, or affiliate members.
Active members will be eligible for:
- A $35 credit toward 2016-2017 dues when recruiting one new member;
- A $50 credit toward 2016-2017 dues when recruiting 2-4 new members;
- A $75 credit toward 2016-2017 dues when recruiting 5-9 new members; or
- A $125 credit toward 2016-2017 dues when recruiting 10+ new members.
For every member recruited, individuals will receive one entry into a grand prize drawing to receive complimentary registration to HM16 in San Diego.
Click here for more details.
Society of Hospital Medicine Awards, Committee, Board Nominations Due October 16

- Nominating yourself or a colleague for one of SHM’s Awards of Excellence, which will be presented at HM16 in San Diego;
- Joining a committee that matches your professional interests or personal passions;
- Applying for SHM’s board of directors; or
- Nominating a colleague for the Master in Hospital Medicine designation, SHM’s most prestigious honor.
For more information, click on the “membership” section of the SHM website.
- Nominating yourself or a colleague for one of SHM’s Awards of Excellence, which will be presented at HM16 in San Diego;
- Joining a committee that matches your professional interests or personal passions;
- Applying for SHM’s board of directors; or
- Nominating a colleague for the Master in Hospital Medicine designation, SHM’s most prestigious honor.
For more information, click on the “membership” section of the SHM website.
- Nominating yourself or a colleague for one of SHM’s Awards of Excellence, which will be presented at HM16 in San Diego;
- Joining a committee that matches your professional interests or personal passions;
- Applying for SHM’s board of directors; or
- Nominating a colleague for the Master in Hospital Medicine designation, SHM’s most prestigious honor.
For more information, click on the “membership” section of the SHM website.
Hospitalist Maintenance of Certification Exam Prep Tool Available Online

SPARK is the only test prep resource designed specifically for hospitalists and the American Board of Internal Medicine Focused Practice in Hospital Medicine MOC exam. Unlike other test prep tools, this focuses on topics unique to the everyday practice of hospital medicine, including:
- Palliative care, medical ethics, and decision-making;
- Peri-operative care and consultative co-management; and
- Quality, safety, and clinical reasoning.
SPARK gives hospitalists the peace of mind that comes with knowing they are ready for the MOC exam; it features 175 vignette-style, single best answer, multiple-choice questions, complete with answers, discussion, reasoning, references, and quizzing capabilities. This new resource provides targeted study areas to supplement other educational material.
SPARK is the only test prep resource designed specifically for hospitalists and the American Board of Internal Medicine Focused Practice in Hospital Medicine MOC exam. Unlike other test prep tools, this focuses on topics unique to the everyday practice of hospital medicine, including:
- Palliative care, medical ethics, and decision-making;
- Peri-operative care and consultative co-management; and
- Quality, safety, and clinical reasoning.
SPARK gives hospitalists the peace of mind that comes with knowing they are ready for the MOC exam; it features 175 vignette-style, single best answer, multiple-choice questions, complete with answers, discussion, reasoning, references, and quizzing capabilities. This new resource provides targeted study areas to supplement other educational material.
SPARK is the only test prep resource designed specifically for hospitalists and the American Board of Internal Medicine Focused Practice in Hospital Medicine MOC exam. Unlike other test prep tools, this focuses on topics unique to the everyday practice of hospital medicine, including:
- Palliative care, medical ethics, and decision-making;
- Peri-operative care and consultative co-management; and
- Quality, safety, and clinical reasoning.
SPARK gives hospitalists the peace of mind that comes with knowing they are ready for the MOC exam; it features 175 vignette-style, single best answer, multiple-choice questions, complete with answers, discussion, reasoning, references, and quizzing capabilities. This new resource provides targeted study areas to supplement other educational material.
Fellow, Senior Fellow in Hospital Medicine Applications Due November 15
Get started today on your application for SHM’s other designations, Fellow in Hospital Medicine (FHM) and Senior Fellow in Hospital Medicine (SFHM). Don’t wait until the last minute; the application can take some time to assemble.
Click here to apply.
Get started today on your application for SHM’s other designations, Fellow in Hospital Medicine (FHM) and Senior Fellow in Hospital Medicine (SFHM). Don’t wait until the last minute; the application can take some time to assemble.
Click here to apply.
Get started today on your application for SHM’s other designations, Fellow in Hospital Medicine (FHM) and Senior Fellow in Hospital Medicine (SFHM). Don’t wait until the last minute; the application can take some time to assemble.
Click here to apply.
Hospitalists Can Earn CME Credits for Acute Coronary Syndrome Performance Improvement
Approximately 1.7 million patients are hospitalized for acute coronary syndrome (ACS), and 600,000 die of an acute myocardial infarction. Although ACS is a major cause of morbidity and mortality, a broad range of clinical strategies can affect outcomes if implemented effectively. In addition, quality improvement (QI) strategies implemented around ACS can improve performance on quality measures.
The ACS PI-CME is a self-directed, web-based activity designed to help you evaluate your practice. Participation is free. Upon completion of the activity, participants will receive 20 CME credits.
The educational interventions will be pragmatic and address the challenges faced by clinicians responsible for managing patient care. They include:
- Etiology and diagnosis of ACS: educating the team on the pathophysiology of atherosclerotic plaque;
- Inpatient treatment of ACS; and
- Transitions of care for ACS patients.
Act today, because spaces are limited for this program. For more information, visit the QI section of SHM’s website.
Brendon Shank is SHM’s associate vice president of communications.ences (CHS) 13-105 10833 Le Conte Ave., Los Angeles, Calif.
Approximately 1.7 million patients are hospitalized for acute coronary syndrome (ACS), and 600,000 die of an acute myocardial infarction. Although ACS is a major cause of morbidity and mortality, a broad range of clinical strategies can affect outcomes if implemented effectively. In addition, quality improvement (QI) strategies implemented around ACS can improve performance on quality measures.
The ACS PI-CME is a self-directed, web-based activity designed to help you evaluate your practice. Participation is free. Upon completion of the activity, participants will receive 20 CME credits.
The educational interventions will be pragmatic and address the challenges faced by clinicians responsible for managing patient care. They include:
- Etiology and diagnosis of ACS: educating the team on the pathophysiology of atherosclerotic plaque;
- Inpatient treatment of ACS; and
- Transitions of care for ACS patients.
Act today, because spaces are limited for this program. For more information, visit the QI section of SHM’s website.
Brendon Shank is SHM’s associate vice president of communications.ences (CHS) 13-105 10833 Le Conte Ave., Los Angeles, Calif.
Approximately 1.7 million patients are hospitalized for acute coronary syndrome (ACS), and 600,000 die of an acute myocardial infarction. Although ACS is a major cause of morbidity and mortality, a broad range of clinical strategies can affect outcomes if implemented effectively. In addition, quality improvement (QI) strategies implemented around ACS can improve performance on quality measures.
The ACS PI-CME is a self-directed, web-based activity designed to help you evaluate your practice. Participation is free. Upon completion of the activity, participants will receive 20 CME credits.
The educational interventions will be pragmatic and address the challenges faced by clinicians responsible for managing patient care. They include:
- Etiology and diagnosis of ACS: educating the team on the pathophysiology of atherosclerotic plaque;
- Inpatient treatment of ACS; and
- Transitions of care for ACS patients.
Act today, because spaces are limited for this program. For more information, visit the QI section of SHM’s website.
Brendon Shank is SHM’s associate vice president of communications.ences (CHS) 13-105 10833 Le Conte Ave., Los Angeles, Calif.
Society of Hospital Medicine Hosts Future of Hospital Medicine Event Series
The Society of Hospital Medicine (SHM) hosts a series of special events for students and residents on campuses throughout the country. Learn more about these networking receptions featuring nationally recognized hospitalists speaking on careers in hospital medicine.
Jefferson University Hospital
Oct. 21, 5-6:30 p.m.
Bluemle Life Sciences Building, Room 101
233 South 10th Street, Philadelphia, Pa.
University of California at Los Angeles
October 22, noon to 1:15 p.m.
David Geffen School of Medicine at UCLA Center for Health Sciences (CHS) 13-105
10833 Le Conte Ave., Los Angeles, Calif.
The Society of Hospital Medicine (SHM) hosts a series of special events for students and residents on campuses throughout the country. Learn more about these networking receptions featuring nationally recognized hospitalists speaking on careers in hospital medicine.
Jefferson University Hospital
Oct. 21, 5-6:30 p.m.
Bluemle Life Sciences Building, Room 101
233 South 10th Street, Philadelphia, Pa.
University of California at Los Angeles
October 22, noon to 1:15 p.m.
David Geffen School of Medicine at UCLA Center for Health Sciences (CHS) 13-105
10833 Le Conte Ave., Los Angeles, Calif.
The Society of Hospital Medicine (SHM) hosts a series of special events for students and residents on campuses throughout the country. Learn more about these networking receptions featuring nationally recognized hospitalists speaking on careers in hospital medicine.
Jefferson University Hospital
Oct. 21, 5-6:30 p.m.
Bluemle Life Sciences Building, Room 101
233 South 10th Street, Philadelphia, Pa.
University of California at Los Angeles
October 22, noon to 1:15 p.m.
David Geffen School of Medicine at UCLA Center for Health Sciences (CHS) 13-105
10833 Le Conte Ave., Los Angeles, Calif.
How Veterans Affairs Healthcare Services Are Like Accountable Care Organizations
According to the Centers for Medicare and Medicaid Services, an accountable care organization (ACO) is defined as a “group of doctors, hospitals, and other healthcare providers, who come together voluntarily to give coordinated high quality care to their Medicare patients.” The goal of an ACO is “to ensure that patients, especially chronically ill, get the right care at the right time, while avoiding unnecessary duplication of services and preventing medical errors.”
In many ways, the Department of Veterans Affairs (VA) is similar to an ACO. While some of the veterans have Medicare, not all of them do. Across the nation, the VA has the infrastructure to deliver high quality care to our patients. Large medical centers that are affiliated with medical schools and academic teaching hospitals teach medical students and resident physicians to provide excellent care to our patients. To meet the needs of our patients in smaller cities or rural areas, community-based outpatient clinics (CBOCs) deliver quality care to patients.
Our electronic medical record, called the Computerized Personal Record System (CPRS), links veterans nationally. A patient can be seen at the CBOC in Mansfield, Ohio, the Palo Alto VA medical center in California, and the Washington, D.C., VA medical center, and not have to worry about the physicians not having access to his medical information. This prevents physicians from ordering unnecessary radiographic studies, and it can decrease the chance of medication errors and polypharmacy.
The use of electronic consults, also known as eConsults, allows for faster access to specialists. After the PCP orders the patient’s chart, the specialist will review the information, provide recommendations to the PCP, and determine how quickly the patient needs to be seen by the specialist. This is important for our rural population, who will then have to make fewer trips to medical centers.
The Specialty Care Access Network-Extension of Community Healthcare Outcomes (SCAN-ECHO) project is another tool designed to assist our rural population. The program targets those who have diabetes, heart failure, and/or chronic pain. Patients travel to their CBOC and interact via the internet with the VA specialist located at a larger medical center, thereby reducing the number of long trips they must make to the medical center and the long waits they would normally have to endure to be seen by specialists.
Telehealth is another way the VA is coordinating high quality care for our veterans. In the comfort of their own homes, veterans upload weight, vitals, and blood glucose levels to assist physicians in monitoring and treating chronic medical conditions.
The VA also delivers highly quality care through its pharmacies. Electronic ordering of outpatient medications for patients is extremely easy; these medications can either be mailed home or made available for same day pick-up. Certain medications are restricted and require approval by specialists; however, when patients fulfill criteria for a nonformulary medication, it is easily accessible. In addition, the approval process is evidence-based, limiting the effect of pharmaceutical companies on patient care.
As a result of using the formulary process for medications, patients share in the savings through lower co-pays. Pharmacists participate in both antibiotic stewardship, as with inpatient vancomycin dosing, and in managing inpatient anticoagulation, which is often more reliable and less expensive than using physicians.
Through these and other programs, the VA ensures that patients receive the services they need in a thoughtful, evidence-based, and timely way.
Dr. Nemeth is a hospitalist at Louis Stokes VA Medical Center in Cleveland, Ohio, and assistant professor of medicine at Case Western Reserve University School of Medicine. He is a member of SHM’s Veterans Affairs Task Force.
According to the Centers for Medicare and Medicaid Services, an accountable care organization (ACO) is defined as a “group of doctors, hospitals, and other healthcare providers, who come together voluntarily to give coordinated high quality care to their Medicare patients.” The goal of an ACO is “to ensure that patients, especially chronically ill, get the right care at the right time, while avoiding unnecessary duplication of services and preventing medical errors.”
In many ways, the Department of Veterans Affairs (VA) is similar to an ACO. While some of the veterans have Medicare, not all of them do. Across the nation, the VA has the infrastructure to deliver high quality care to our patients. Large medical centers that are affiliated with medical schools and academic teaching hospitals teach medical students and resident physicians to provide excellent care to our patients. To meet the needs of our patients in smaller cities or rural areas, community-based outpatient clinics (CBOCs) deliver quality care to patients.
Our electronic medical record, called the Computerized Personal Record System (CPRS), links veterans nationally. A patient can be seen at the CBOC in Mansfield, Ohio, the Palo Alto VA medical center in California, and the Washington, D.C., VA medical center, and not have to worry about the physicians not having access to his medical information. This prevents physicians from ordering unnecessary radiographic studies, and it can decrease the chance of medication errors and polypharmacy.
The use of electronic consults, also known as eConsults, allows for faster access to specialists. After the PCP orders the patient’s chart, the specialist will review the information, provide recommendations to the PCP, and determine how quickly the patient needs to be seen by the specialist. This is important for our rural population, who will then have to make fewer trips to medical centers.
The Specialty Care Access Network-Extension of Community Healthcare Outcomes (SCAN-ECHO) project is another tool designed to assist our rural population. The program targets those who have diabetes, heart failure, and/or chronic pain. Patients travel to their CBOC and interact via the internet with the VA specialist located at a larger medical center, thereby reducing the number of long trips they must make to the medical center and the long waits they would normally have to endure to be seen by specialists.
Telehealth is another way the VA is coordinating high quality care for our veterans. In the comfort of their own homes, veterans upload weight, vitals, and blood glucose levels to assist physicians in monitoring and treating chronic medical conditions.
The VA also delivers highly quality care through its pharmacies. Electronic ordering of outpatient medications for patients is extremely easy; these medications can either be mailed home or made available for same day pick-up. Certain medications are restricted and require approval by specialists; however, when patients fulfill criteria for a nonformulary medication, it is easily accessible. In addition, the approval process is evidence-based, limiting the effect of pharmaceutical companies on patient care.
As a result of using the formulary process for medications, patients share in the savings through lower co-pays. Pharmacists participate in both antibiotic stewardship, as with inpatient vancomycin dosing, and in managing inpatient anticoagulation, which is often more reliable and less expensive than using physicians.
Through these and other programs, the VA ensures that patients receive the services they need in a thoughtful, evidence-based, and timely way.
Dr. Nemeth is a hospitalist at Louis Stokes VA Medical Center in Cleveland, Ohio, and assistant professor of medicine at Case Western Reserve University School of Medicine. He is a member of SHM’s Veterans Affairs Task Force.
According to the Centers for Medicare and Medicaid Services, an accountable care organization (ACO) is defined as a “group of doctors, hospitals, and other healthcare providers, who come together voluntarily to give coordinated high quality care to their Medicare patients.” The goal of an ACO is “to ensure that patients, especially chronically ill, get the right care at the right time, while avoiding unnecessary duplication of services and preventing medical errors.”
In many ways, the Department of Veterans Affairs (VA) is similar to an ACO. While some of the veterans have Medicare, not all of them do. Across the nation, the VA has the infrastructure to deliver high quality care to our patients. Large medical centers that are affiliated with medical schools and academic teaching hospitals teach medical students and resident physicians to provide excellent care to our patients. To meet the needs of our patients in smaller cities or rural areas, community-based outpatient clinics (CBOCs) deliver quality care to patients.
Our electronic medical record, called the Computerized Personal Record System (CPRS), links veterans nationally. A patient can be seen at the CBOC in Mansfield, Ohio, the Palo Alto VA medical center in California, and the Washington, D.C., VA medical center, and not have to worry about the physicians not having access to his medical information. This prevents physicians from ordering unnecessary radiographic studies, and it can decrease the chance of medication errors and polypharmacy.
The use of electronic consults, also known as eConsults, allows for faster access to specialists. After the PCP orders the patient’s chart, the specialist will review the information, provide recommendations to the PCP, and determine how quickly the patient needs to be seen by the specialist. This is important for our rural population, who will then have to make fewer trips to medical centers.
The Specialty Care Access Network-Extension of Community Healthcare Outcomes (SCAN-ECHO) project is another tool designed to assist our rural population. The program targets those who have diabetes, heart failure, and/or chronic pain. Patients travel to their CBOC and interact via the internet with the VA specialist located at a larger medical center, thereby reducing the number of long trips they must make to the medical center and the long waits they would normally have to endure to be seen by specialists.
Telehealth is another way the VA is coordinating high quality care for our veterans. In the comfort of their own homes, veterans upload weight, vitals, and blood glucose levels to assist physicians in monitoring and treating chronic medical conditions.
The VA also delivers highly quality care through its pharmacies. Electronic ordering of outpatient medications for patients is extremely easy; these medications can either be mailed home or made available for same day pick-up. Certain medications are restricted and require approval by specialists; however, when patients fulfill criteria for a nonformulary medication, it is easily accessible. In addition, the approval process is evidence-based, limiting the effect of pharmaceutical companies on patient care.
As a result of using the formulary process for medications, patients share in the savings through lower co-pays. Pharmacists participate in both antibiotic stewardship, as with inpatient vancomycin dosing, and in managing inpatient anticoagulation, which is often more reliable and less expensive than using physicians.
Through these and other programs, the VA ensures that patients receive the services they need in a thoughtful, evidence-based, and timely way.
Dr. Nemeth is a hospitalist at Louis Stokes VA Medical Center in Cleveland, Ohio, and assistant professor of medicine at Case Western Reserve University School of Medicine. He is a member of SHM’s Veterans Affairs Task Force.
Treating EBV-associated lymphomas with VSTs
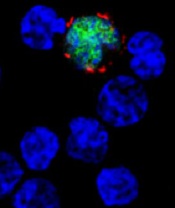
among uninfected cells (blue)
Image by Benjamin
Chaigne-Delalande
NEW YORK—Type 2 Epstein-Barr virus (EBV) tumors, such as Hodgkin and non-Hodgkin lymphomas, are challenging to treat with virus-specific T (VST) cells, according to researchers.
These lymphomas express a more restricted array of EBV antigens that are not particularly immunogenic.
Nevertheless, researchers are devising an approach using peptide mixtures to activate EBV VSTs for use in these patients.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described this work at the inaugural CRI-CIMT-EATI-AACR International Immunotherapy of Cancer Conference. She also described the researchers’ efforts to create off-the-shelf VSTs.
Dr Heslop explained that, in addition to the more restricted array of EBV antigens, EBV-associated tumors often produce inhibitory cytokines that can impede the activity of T cells.
So the researchers devised a strategy to expand these low-frequency clones by stimulating responding T cells with dendritic cells genetically modified with an ADV viral vector to overexpress LMP1 and LMP2. After multiple stimulations, they obtained an autologous product from the patient.
The team then tested the cytotoxic T cells in 21 patients with relapsed disease and in 29 patients as adjuvant therapy after stem cell transplant (n=14) or chemotherapy (n=15).
In the adjuvant arm, all patients but 1 remain in remission up to 5 years later.
In the relapsed arm, 11 had a complete response (CR), 2 had a partial response (PR), and 8 had progressive disease within 2 to 8 weeks.
“Importantly, there was no toxicity,” Dr Heslop said. “[A]ll were heavily pretreated with multiple lines of therapy for lymphoma, so I think the response rate is encouraging.”
An alternative approach: Pepmix-activated EBV VSTs
Although the antitumor effects of the above approach were encouraging, “we had a very complex manufacturing methodology that we didn’t think was sufficiently scaleable and robust for clinical studies,” Dr Heslop said.
“We also thought there would be potential regulatory issues with live EBV virus and the adenoviral vector,” she added.
And the researchers were concerned about the competition from the EBV/Ad-LMP dominant antigens.
So they devised an alternative approach using peptide mixture (pepmix)-activated EBVSTs.
This approach used autologous monocyte-derived dendritic cells as the antigen-presenting cells for the first stimulation.
The researchers pulsed them with overlapping peptides derived from 4 EBV antigens expressed in the tumors (EBV-LMP1, LMP2, EBNA1, and BARF1). They then expanded and opsonized the cells with IL-7 and IL-15.
For the second stimulation, the team used the T cells pulsed with the peptides and a K562 line pulsed with co-stimulatory molecules. And this process took 23 days, as opposed to the 2-3 months with the previous product.
The researchers have treated 9 patients with these EBVSTs as adjuvant therapy after autologous stem cell transplant. All patients remain in remission.
They also treated 6 patients with active disease. Two are in CR, 2 are in PR, and 2 have progressed.
This trial is ongoing, but the researchers believe that targeting the more challenging type 2 latency tumors with autologous cells can overcome T-cell anergy by using IL-7 and IL-15.
“Obviously, we need more numbers to know what the range of response is,” Dr Heslop said, although, at this early stage, it appears pepmix-activated T cells can produce antitumor responses. ![]()
among uninfected cells (blue)
Image by Benjamin
Chaigne-Delalande
NEW YORK—Type 2 Epstein-Barr virus (EBV) tumors, such as Hodgkin and non-Hodgkin lymphomas, are challenging to treat with virus-specific T (VST) cells, according to researchers.
These lymphomas express a more restricted array of EBV antigens that are not particularly immunogenic.
Nevertheless, researchers are devising an approach using peptide mixtures to activate EBV VSTs for use in these patients.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described this work at the inaugural CRI-CIMT-EATI-AACR International Immunotherapy of Cancer Conference. She also described the researchers’ efforts to create off-the-shelf VSTs.
Dr Heslop explained that, in addition to the more restricted array of EBV antigens, EBV-associated tumors often produce inhibitory cytokines that can impede the activity of T cells.
So the researchers devised a strategy to expand these low-frequency clones by stimulating responding T cells with dendritic cells genetically modified with an ADV viral vector to overexpress LMP1 and LMP2. After multiple stimulations, they obtained an autologous product from the patient.
The team then tested the cytotoxic T cells in 21 patients with relapsed disease and in 29 patients as adjuvant therapy after stem cell transplant (n=14) or chemotherapy (n=15).
In the adjuvant arm, all patients but 1 remain in remission up to 5 years later.
In the relapsed arm, 11 had a complete response (CR), 2 had a partial response (PR), and 8 had progressive disease within 2 to 8 weeks.
“Importantly, there was no toxicity,” Dr Heslop said. “[A]ll were heavily pretreated with multiple lines of therapy for lymphoma, so I think the response rate is encouraging.”
An alternative approach: Pepmix-activated EBV VSTs
Although the antitumor effects of the above approach were encouraging, “we had a very complex manufacturing methodology that we didn’t think was sufficiently scaleable and robust for clinical studies,” Dr Heslop said.
“We also thought there would be potential regulatory issues with live EBV virus and the adenoviral vector,” she added.
And the researchers were concerned about the competition from the EBV/Ad-LMP dominant antigens.
So they devised an alternative approach using peptide mixture (pepmix)-activated EBVSTs.
This approach used autologous monocyte-derived dendritic cells as the antigen-presenting cells for the first stimulation.
The researchers pulsed them with overlapping peptides derived from 4 EBV antigens expressed in the tumors (EBV-LMP1, LMP2, EBNA1, and BARF1). They then expanded and opsonized the cells with IL-7 and IL-15.
For the second stimulation, the team used the T cells pulsed with the peptides and a K562 line pulsed with co-stimulatory molecules. And this process took 23 days, as opposed to the 2-3 months with the previous product.
The researchers have treated 9 patients with these EBVSTs as adjuvant therapy after autologous stem cell transplant. All patients remain in remission.
They also treated 6 patients with active disease. Two are in CR, 2 are in PR, and 2 have progressed.
This trial is ongoing, but the researchers believe that targeting the more challenging type 2 latency tumors with autologous cells can overcome T-cell anergy by using IL-7 and IL-15.
“Obviously, we need more numbers to know what the range of response is,” Dr Heslop said, although, at this early stage, it appears pepmix-activated T cells can produce antitumor responses. ![]()
among uninfected cells (blue)
Image by Benjamin
Chaigne-Delalande
NEW YORK—Type 2 Epstein-Barr virus (EBV) tumors, such as Hodgkin and non-Hodgkin lymphomas, are challenging to treat with virus-specific T (VST) cells, according to researchers.
These lymphomas express a more restricted array of EBV antigens that are not particularly immunogenic.
Nevertheless, researchers are devising an approach using peptide mixtures to activate EBV VSTs for use in these patients.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described this work at the inaugural CRI-CIMT-EATI-AACR International Immunotherapy of Cancer Conference. She also described the researchers’ efforts to create off-the-shelf VSTs.
Dr Heslop explained that, in addition to the more restricted array of EBV antigens, EBV-associated tumors often produce inhibitory cytokines that can impede the activity of T cells.
So the researchers devised a strategy to expand these low-frequency clones by stimulating responding T cells with dendritic cells genetically modified with an ADV viral vector to overexpress LMP1 and LMP2. After multiple stimulations, they obtained an autologous product from the patient.
The team then tested the cytotoxic T cells in 21 patients with relapsed disease and in 29 patients as adjuvant therapy after stem cell transplant (n=14) or chemotherapy (n=15).
In the adjuvant arm, all patients but 1 remain in remission up to 5 years later.
In the relapsed arm, 11 had a complete response (CR), 2 had a partial response (PR), and 8 had progressive disease within 2 to 8 weeks.
“Importantly, there was no toxicity,” Dr Heslop said. “[A]ll were heavily pretreated with multiple lines of therapy for lymphoma, so I think the response rate is encouraging.”
An alternative approach: Pepmix-activated EBV VSTs
Although the antitumor effects of the above approach were encouraging, “we had a very complex manufacturing methodology that we didn’t think was sufficiently scaleable and robust for clinical studies,” Dr Heslop said.
“We also thought there would be potential regulatory issues with live EBV virus and the adenoviral vector,” she added.
And the researchers were concerned about the competition from the EBV/Ad-LMP dominant antigens.
So they devised an alternative approach using peptide mixture (pepmix)-activated EBVSTs.
This approach used autologous monocyte-derived dendritic cells as the antigen-presenting cells for the first stimulation.
The researchers pulsed them with overlapping peptides derived from 4 EBV antigens expressed in the tumors (EBV-LMP1, LMP2, EBNA1, and BARF1). They then expanded and opsonized the cells with IL-7 and IL-15.
For the second stimulation, the team used the T cells pulsed with the peptides and a K562 line pulsed with co-stimulatory molecules. And this process took 23 days, as opposed to the 2-3 months with the previous product.
The researchers have treated 9 patients with these EBVSTs as adjuvant therapy after autologous stem cell transplant. All patients remain in remission.
They also treated 6 patients with active disease. Two are in CR, 2 are in PR, and 2 have progressed.
This trial is ongoing, but the researchers believe that targeting the more challenging type 2 latency tumors with autologous cells can overcome T-cell anergy by using IL-7 and IL-15.
“Obviously, we need more numbers to know what the range of response is,” Dr Heslop said, although, at this early stage, it appears pepmix-activated T cells can produce antitumor responses. ![]()