User login
Simultaneous equal to sequential treatment for actinic keratoses
Patients with multiple actinic keratoses (AKs) may be treated either sequentially or simultaneously with ingenol mebutate gel, according to the authors of a study that found no difference in outcomes or adverse effects from either treatment approach.
The phase IIIb study conducted in Italy and Spain enrolled 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs. Subjects were randomized to have the two areas (face/scalp and trunk/extremities) treated simultaneously (101 patients) or sequentially (98 patients) with 0.015% and 0.05% ingenol mebutate gel.

There were no significant differences in localized skin responses between the simultaneous and sequential treatment groups, based on the mean composite local skin response scores 3 days after treatment started, which were similar between the groups for the face/scalp and trunk/extremities applications. About 22% of patients in each group experienced adverse events.
At 8 weeks, the complete clearance rates also were not statistically different between the simultaneous and sequential groups (52.7% and 46.9%, respectively; P = .34), and patient satisfaction with treatment was similar for both treatment approaches. At that time, the number of AKs had dropped by a mean of 83.4% among those in the simultaneous group and 79.1% in the sequential group (P = .20).
“The favorable rate of complete clearance in the simultaneous treatment group means that patients can receive their treatment for both areas in one visit, rather than having to return to the clinic for a second cycle of treatment,” wrote Dr. Giovanni Pellacani of the department of dermatology at the University of Modena (Italy) and Reggio Emilia, and his coauthors (J Eur Acad Dermatol Venereol. 2015;29[11]:2192-8).
“Ultimately, the treatment schedule is based on agreement between the physician and the patient; this study helps to support the selection of the most appropriate regimen to treat AK in individual patients,” they commented.
The study was funded by LEO Pharma, the manufacturer of ingenol mebutate gel (Picato). Dr. Pellacani has received consultant fees from the company; three authors are employees of the company; and the other authors declared consultancies, honoraria and, grants from LEO Pharma and other pharmaceutical companies.
Patients with multiple actinic keratoses (AKs) may be treated either sequentially or simultaneously with ingenol mebutate gel, according to the authors of a study that found no difference in outcomes or adverse effects from either treatment approach.
The phase IIIb study conducted in Italy and Spain enrolled 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs. Subjects were randomized to have the two areas (face/scalp and trunk/extremities) treated simultaneously (101 patients) or sequentially (98 patients) with 0.015% and 0.05% ingenol mebutate gel.
There were no significant differences in localized skin responses between the simultaneous and sequential treatment groups, based on the mean composite local skin response scores 3 days after treatment started, which were similar between the groups for the face/scalp and trunk/extremities applications. About 22% of patients in each group experienced adverse events.
At 8 weeks, the complete clearance rates also were not statistically different between the simultaneous and sequential groups (52.7% and 46.9%, respectively; P = .34), and patient satisfaction with treatment was similar for both treatment approaches. At that time, the number of AKs had dropped by a mean of 83.4% among those in the simultaneous group and 79.1% in the sequential group (P = .20).
“The favorable rate of complete clearance in the simultaneous treatment group means that patients can receive their treatment for both areas in one visit, rather than having to return to the clinic for a second cycle of treatment,” wrote Dr. Giovanni Pellacani of the department of dermatology at the University of Modena (Italy) and Reggio Emilia, and his coauthors (J Eur Acad Dermatol Venereol. 2015;29[11]:2192-8).
“Ultimately, the treatment schedule is based on agreement between the physician and the patient; this study helps to support the selection of the most appropriate regimen to treat AK in individual patients,” they commented.
The study was funded by LEO Pharma, the manufacturer of ingenol mebutate gel (Picato). Dr. Pellacani has received consultant fees from the company; three authors are employees of the company; and the other authors declared consultancies, honoraria and, grants from LEO Pharma and other pharmaceutical companies.
Patients with multiple actinic keratoses (AKs) may be treated either sequentially or simultaneously with ingenol mebutate gel, according to the authors of a study that found no difference in outcomes or adverse effects from either treatment approach.
The phase IIIb study conducted in Italy and Spain enrolled 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs. Subjects were randomized to have the two areas (face/scalp and trunk/extremities) treated simultaneously (101 patients) or sequentially (98 patients) with 0.015% and 0.05% ingenol mebutate gel.
There were no significant differences in localized skin responses between the simultaneous and sequential treatment groups, based on the mean composite local skin response scores 3 days after treatment started, which were similar between the groups for the face/scalp and trunk/extremities applications. About 22% of patients in each group experienced adverse events.
At 8 weeks, the complete clearance rates also were not statistically different between the simultaneous and sequential groups (52.7% and 46.9%, respectively; P = .34), and patient satisfaction with treatment was similar for both treatment approaches. At that time, the number of AKs had dropped by a mean of 83.4% among those in the simultaneous group and 79.1% in the sequential group (P = .20).
“The favorable rate of complete clearance in the simultaneous treatment group means that patients can receive their treatment for both areas in one visit, rather than having to return to the clinic for a second cycle of treatment,” wrote Dr. Giovanni Pellacani of the department of dermatology at the University of Modena (Italy) and Reggio Emilia, and his coauthors (J Eur Acad Dermatol Venereol. 2015;29[11]:2192-8).
“Ultimately, the treatment schedule is based on agreement between the physician and the patient; this study helps to support the selection of the most appropriate regimen to treat AK in individual patients,” they commented.
The study was funded by LEO Pharma, the manufacturer of ingenol mebutate gel (Picato). Dr. Pellacani has received consultant fees from the company; three authors are employees of the company; and the other authors declared consultancies, honoraria and, grants from LEO Pharma and other pharmaceutical companies.
FROM THE JOURNAL OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Key clinical point: Patients with multiple AKs may be treated either sequentially or simultaneously with ingenol mebutate gel, with similar efficacy and safety outcomes.
Major finding: The incidence of localized skin responses, complete clearance rates, and patient treatment satisfaction were similar for simultaneous and sequential treatment approaches.
Data source: A phase IIIb randomized, multicenter, open-label, parallel-group study evaluated 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs.
Disclosures: The study was funded by ingenol mebutate gel manufacturer LEO Pharma. Three authors are employees of the company; the other authors declared consultancies, honoraria, and/or grants from LEO Pharma and other pharmaceutical companies.
Posterior Reversible Encephalopathy Syndrome: Temporary Visual Loss After Spinal Deformity Surgery
First described in 1996, posterior reversible encephalopathy syndrome (PRES) exhibits a wide clinical spectrum and is definitively diagnosed through computed tomography (CT) and/or magnetic resonance imaging (MRI) studies of the brain.1 Clinical presentation may include a spectrum of symptoms, including nausea, emesis, seizures, visual loss, paralysis, and headaches.2,3 The most common imaging finding of PRES is bilateral foci of vasogenic edema located in the parieto-occipital white matter.2-6 Other areas of the brain are frequently affected as well, with the frontal and temporal lobes and the basal or cortical ganglia showing signs of distinctly noncytotoxic edema in 12.5% to 54.2% of all cases.3 With the symptom of visual loss being present in 20% to 62.5% of patients with PRES, the syndrome constitutes a rare potential cause for postoperative visual loss (POVL) after spinal surgery, which has a generally good prognosis because most patients will completely regain their eyesight.2,3
We present a unique account of 2 patients who underwent extensive spinal surgery and received a timely diagnosis and treatment of PRES at a single institution. We aim to elucidate the difference in clinical and radiographic presentation of PRES in relation to other known causes of POVL after spinal surgery. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
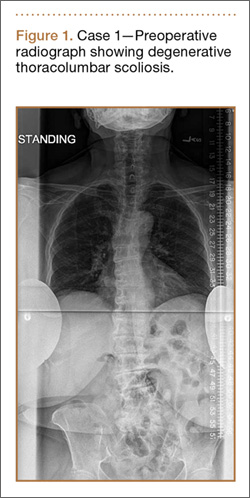
Clinical Presentation. A 78-year-old woman presented to the outpatient clinic with disability due to severe lower back pain. Her surgical history was significant for breast lumpectomy and cataract excision. Her medical history was significant for hypertension, obesity (body mass index, 31.5), hypercholesterolemia, emphysema, and anemia. She had undergone spinal surgery, specifically laminectomies from L2 to S1. The radiographic examination showed degenerative thoracolumbar scoliosis with severe spondylosis, disc space collapse, and ankylosis of L4-L5 (Figure 1).
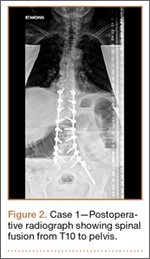
Operative Procedure. The patient underwent transpsoas lumbar interbody fusion (XLIF, NuVasive) from L1 to L4 and posterior spinal fusion from T10 to pelvis (Expedium, Depuy Synthes) (Figure 2). Operative time was 553 minutes; estimated blood loss was 2000 mL due to intraoperative coagulopathy (platelets, 40,000/µL) near the end of the posterior portion of the procedure. Intraoperative hypotension was treated by volume resuscitation and transient use of vasopressor agents. She was transfused with 1700 mL of blood, 150 mL of saline solution, and 420 mL of Lactated Ringer’s solution. No intraoperative complications occurred. The patient was extubated uneventfully on postoperative day 1 and was at baseline neurologically with no visual disturbances.
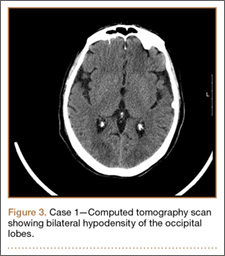
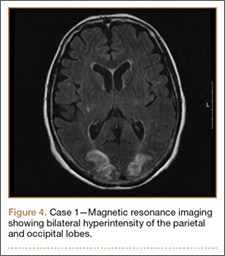
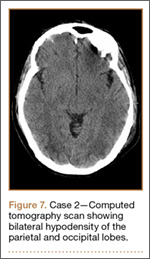
Development and Diagnosis of PRES. The patient made significant progress with physical therapy and developed episodes of hypertension at night on postoperative days 4 to 6. Her mean peak systolic blood pressure was 180 mm Hg. This improved after oral beta-blocker therapy. On postoperative day 6, the patient was ambulating with physical therapy and the aid of a walker. She was found to be neurologically intact, was resting comfortable in a chair reading a book, and was cleared for transfer to a rehabilitation facility the next day. During the morning on postoperative day 7, she developed confusion and visual loss. The patient reported blurry vision followed by complete bilateral painless loss of vision aside from mild light perception. She was unable to identify any objects. She had extinction to double simultaneous stimuli and evidence of agraphesthesia in the left hand. Her neurologic examination was otherwise at baseline. Upon emergent imaging, head CT showed bilateral symmetric areas of hypodensity involving the cortical and subcortical white matter of both occipital lobes (Figure 3). MRI showed extensive bilateral cortical and subcortical signal hyperintensity involving the parietal and occipital lobes (Figure 4). No evidence of petechial or lobar hemorrhage was found.
Treatment and Clinical Course. The patient was transferred to the neurology intensive care unit for neurologic monitoring. She was treated aggressively for recurrent hypertensive episodes. Twenty-four hours after initial blood pressure optimization therapy, she partially recovered her eyesight. She exhibited complete recovery after 48 hours. The patient was discharged to a rehabilitation facility in stable condition on postoperative day 11.
Case 2
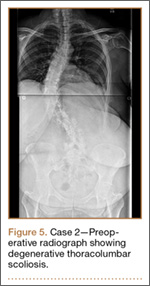
Clinical Presentation. A 51-year-old woman presented to the outpatient clinic with progressive low back pain and decompensation due to degenerative adult scoliosis. Her surgical history was significant for an uneventful Caesarean section. Her medical history was significant for borderline hypertension and obesity (body mass index, 34.4). The radiographic examination showed an S-shaped thoracolumbar curve from T4 to L4 (Figure 5).
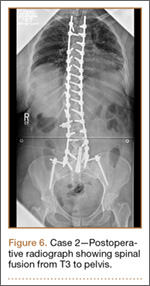
Operative Procedure. After discussions about the risks and benefits of the procedure, the patient underwent posterior spinal fusion from T3 to pelvis (Mesa, K2M) and interbody fusion from L4 to S1 via a presacral approach using the AxiaLIF system (TranS1) (Figure 6). The operation spanned 507 minutes. The patient lost approximately 2200 mL of blood. She was transfused with 1690 mL of blood, 1250 mL of Lactated Ringer’s solution, and 1 unit (50 mL) of albumin. No intraoperative complications occurred.
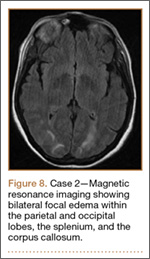
Development and Diagnosis of PRES. The patient was ambulatory with physical therapy and a walker on postoperative day 1. Her albumin levels were noted to be decreased postoperatively (28 mg/mL; normal, >35 mg/mL). She developed intermittent hypertensive episodes and experienced transient peripheral vision loss. After her ophthalmologic symptoms cleared, she was discharged and transferred to a rehabilitation facility on postoperative day 9. Eleven days later, the patient was emergently readmitted for a deep spine wound infection after an onset of wound swelling and fever. She underwent irrigation and débridement of the spine wound with an estimated blood loss of 400 mL. The patient continued to have fevers and was placed on ciprofloxacin and vancomycin, which was changed to levofloxacin on postoperative day 5. Elevated creatinine was noted, and the patient was diagnosed with acute renal failure. On postoperative day 7, oxacillin therapy was commenced. After her cultures grew methicillin-resistant Staphylococcus aureus, a peripherally inserted central catheter line was placed on postoperative day 9. As a result of nausea and constipation, the patient received feeding tubes on postoperative day 11. Additionally, she was diagnosed with a pleural effusion on postoperative day 14. Although her creatinine levels were decreasing, she continued to experience intermittent hypertensive episodes with a mean peak systolic blood pressure of 148 mm Hg. On postoperative day 15, she had a seizure and again developed visual loss. The patient was lethargic and followed only simple commands. She moved all extremities and withdrew symmetrically to noxious stimuli. Upon emergent imaging, head CT showed posterior subcortical white matter hypodensity within the occipital and parietal lobes bilaterally (Figure 7). MRI showed focal regions of symmetric hemispheric edema involving the parietal and occipital lobes in a predominantly subcortical white-matter distribution. Additionally, extensive involvement of the splenium and of the corpus callosum, left greater than right, was observed (Figure 8).
Treatment and Clinical Course. The patient was transferred to the intensive care unit for neuromonitoring. Her hypokalemia and hypertension were treated aggressively to normalize her potassium levels and blood pressure. Her oxacillin therapy was changed to daptomycin. On postoperative day 17, the patient was transferred to another institution for further medical management after achieving full recovery of her eyesight after electrolyte and blood pressure corrections.
Discussion
Posterior reversible encephalopathy syndrome is a rare but frequently devastating complication of spinal surgery, with an estimated incidence of 0.094% to 0.2%.7,8 Pediatric patients, as well as patients undergoing deformity correction surgery and posterior lumbar fusion, which necessitate prone positioning, have a significantly increased risk of POVL after spinal surgery.9 There are several causes of POVL after spinal surgery, each with a unique pathophysiology, clinical presentation, and prognosis.
The most common cause of POVL, accounting for 89% of all cases, is ischemic neuropathy.10 Ischemic neuropathy refers to a hypoperfusion or infarction of the anterior or posterior portion of the optic nerve and presents as painless bilateral vision loss or complete blindness on waking from the surgical procedure.11 Risk factors associated with anterior ischemic neuropathy are primarily diabetes mellitus, prone positioning, nocturnal hypotension, and blood loss.11 Posterior ischemic neuropathy has been most strongly correlated with anemia and hypotension.12 The exact etiology of this complication has not been established, although the prognosis is generally unfavorable, with most vision loss being permanent.10-12
Another potential cause of POVL after spinal surgery is retinal artery occlusion. It is most commonly observed in patients who were improperly positioned, resulting in compression of an orbit on the surface of the headrest or the operating table.13 Retinal artery occlusion characteristically presents as an irreversible unilateral complete loss of vision with a red spot on the macula and an afferent pupillary defect.14
Cortical blindness, another possible common cause of POVL, results from the hypoperfusion of the occipital cortex and has a slightly better prognosis. Cortical blindness generally results from an embolic event that can be visualized through neuroimaging and may be unilateral or bilateral, ranging from mild peripheral vision loss to complete blindness.15
Posterior reversible encephalopathy syndrome, the cause of POVL diagnosed in the 2 patients in this case report, is a neurologic syndrome that differs significantly in its clinical presentation and pathophysiology from the more well-known etiologies. The precise pathophysiologic mechanism of the syndrome is yet to be elucidated. One theory revolves around the failure of cerebral vascular autoregulation. It postulates that intracerebellar hypertension leads to the extravasation of proteins and fluid, resulting in the characteristic vasogenic edema.16,17 The other equally discussed theory postulates that cerebellar vasospasm and subsequent hypoperfusion leading to cellular hypoxemia and ischemia may be responsible.18-20 Posterior reversible encephalopathy syndrome has been reported with increasing frequency, particularly in connection with hypertension, acute renal failure associated with malignancy, cytotoxicity, and corticosteroids, as well as preeclampsia, eclampsia, and autoimmune disorders.1-3,21-23 Traditionally, patients display a combination of different symptoms, including vision changes ranging from slightly decreased perception to complete blindness. Unlike retinal artery occlusion and ischemic optic neuropathy, the onset of vision loss often does not happen immediately after surgery and may occur several hours to days after surgery. Visual disturbance may progressively worsen if the medical cause for the syndrome is not determined and corrected.2,3 In contrast to other known etiologies of POVL, PRES has a relatively favorable prognosis if managed appropriately. Reported case series determined a resolution of the characteristic parieto-occipital vasogenic edema in 83% to 88% of all patients in follow-up neuroimaging after aggressive control of seizures and arterial hypertension.2-3
Both patients undergoing spinal deformity surgery in this report suffered from intermittent hypertensive episodes in the postoperative period. One patient also developed acute renal failure during her hospital stay, and demonstrated low albumin levels postoperatively, which has also been associated with PRES.24 Through the immediate diagnosis and primary control of hypertension, both patients achieved complete neurologic recovery after a mean of 1.5 days (range, 1-2 days); this compares to a recovery period of an average 6.2 days (range, 1-14 days) reported by Ni and colleagues.3 The catastrophic effects of a misdiagnosis and incorrect or untimely treatment were well described in this case report. Several patients who were incorrectly diagnosed with demyelinating disorders or lupus encephalopathy received high doses of immunosuppressants and corticosteroids, known risk factors for the development of PRES.3 The patients subsequently rapidly deteriorated; no patients had a full recovery of their preoperative eyesight, and 1 patient developed complete permanent blindness.3 Optimized multidisciplinary collaboration allowing for a rapid neuro-ophthalmic examination and appropriate neuroimaging will permit an accurate and rapid diagnosis, leading to timely intervention and restoration of vision.
Conclusion
Temporary POVL is a potentially devastating complication of spinal surgery and general anesthesia. The more frequent causes such as ischemic optic neuropathy, retinal artery occlusion, and cortical blindness have very limited effective options for treatment and an overall poor prognosis. The inclusion of PRES in the differential diagnosis of POVL may allow early detection, management, and restoration of vision.
1. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494-500.
2. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427-432.
3. Ni J, Zhou LX, Hao HL, et al. The clinical and radiological spectrum of posterior reversible encephalopathy syndrome: a retrospective series of 24 patients. J Neuroimaging. 2011;21(3):219-224.
4. Stevens CJ, Heran MK. The many faces of posterior reversible encephalopathy syndrome. Br J Radiol. 2012;85(1020):1566-1575.
5. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036-1042.
6. Yoon SD, Cho BM, Oh SM, et al. Clinical and radiological spectrum of posterior reversible encephalopathy syndrome. J Cerebrovasc Endovasc Neurosurg. 2013;15(3):206-213.
7. Patil CG, Lad EM, Lad SP, Ho C, Boakye M. Visual loss after spine surgery: a population-based study. Spine (Phila Pa 1976). 2008;33(13):1491-1496.
8. Stevens WR, Glazer PA, Kelley SD, Lietman TM, Bradford DS. Ophthalmic complications after spinal surgery. Spine (Phila Pa 1976). 1997;22(12):1319-1324.
9. Shen Y, Drum M, Roth S. The prevalence of perioperative visual loss in the United States: a 10-year study from 1996 to 2005 of spinal, orthopedic, cardiac, and general surgery. Anesth Analg. 2009;109(5):1534-1545.
10. Lee LA, Roth S, Posner KL, et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659; quiz 867-868.
11. Hayreh SS. Ischemic optic neuropathies - where are we now? Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1873-1884.
12. Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol. 2005;50(1):15-26.
13. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85.
14. Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87(1):75-78.
15. Berg KT, Harrison AR, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010;4:531-546.
16. Primavera A, Audenino D, Mavilio N, Cocito L. Reversible posterior leucoencephalopathy syndrome in systemic lupus and vasculitis. Ann Rheum Dis. 2001;60(5):534-537.
17. Bartynski WS, Boardman JF. Catheter angiography, MR angiography, and MR perfusion in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008;29(3):447-455.
18. Ito T, Sakai T, Inagawa S, Utsu M, Bun T. MR angiography of cerebral vasospasm in preeclampsia. AJNR Am J Neuroradiol. 1995;16(6):1344-1346.
19. Agarwal R, Davis C, Altinok D, Serajee FJ. Posterior reversible encephalopathy and cerebral vasoconstriction in a patient with hemolytic uremic syndrome. Pediatr Neurol. 2014;50(5):518-521.
20. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29(6):1043-1049.
21. Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205-210.
22. Ekawa Y, Shiota M, Tobiume T, et al. Reversible posterior leukoencephalopathy syndrome accompanying eclampsia: correct diagnosis using preoperative MRI. Tohoku J Exp Med. 2012;226(1):55-58.
23. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome--an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol. 2006;33(11):2178-2183.
24. Pirker A, Kramer L, Voller B, et al. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32(3):527-531.
First described in 1996, posterior reversible encephalopathy syndrome (PRES) exhibits a wide clinical spectrum and is definitively diagnosed through computed tomography (CT) and/or magnetic resonance imaging (MRI) studies of the brain.1 Clinical presentation may include a spectrum of symptoms, including nausea, emesis, seizures, visual loss, paralysis, and headaches.2,3 The most common imaging finding of PRES is bilateral foci of vasogenic edema located in the parieto-occipital white matter.2-6 Other areas of the brain are frequently affected as well, with the frontal and temporal lobes and the basal or cortical ganglia showing signs of distinctly noncytotoxic edema in 12.5% to 54.2% of all cases.3 With the symptom of visual loss being present in 20% to 62.5% of patients with PRES, the syndrome constitutes a rare potential cause for postoperative visual loss (POVL) after spinal surgery, which has a generally good prognosis because most patients will completely regain their eyesight.2,3
We present a unique account of 2 patients who underwent extensive spinal surgery and received a timely diagnosis and treatment of PRES at a single institution. We aim to elucidate the difference in clinical and radiographic presentation of PRES in relation to other known causes of POVL after spinal surgery. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
Clinical Presentation. A 78-year-old woman presented to the outpatient clinic with disability due to severe lower back pain. Her surgical history was significant for breast lumpectomy and cataract excision. Her medical history was significant for hypertension, obesity (body mass index, 31.5), hypercholesterolemia, emphysema, and anemia. She had undergone spinal surgery, specifically laminectomies from L2 to S1. The radiographic examination showed degenerative thoracolumbar scoliosis with severe spondylosis, disc space collapse, and ankylosis of L4-L5 (Figure 1).
Operative Procedure. The patient underwent transpsoas lumbar interbody fusion (XLIF, NuVasive) from L1 to L4 and posterior spinal fusion from T10 to pelvis (Expedium, Depuy Synthes) (Figure 2). Operative time was 553 minutes; estimated blood loss was 2000 mL due to intraoperative coagulopathy (platelets, 40,000/µL) near the end of the posterior portion of the procedure. Intraoperative hypotension was treated by volume resuscitation and transient use of vasopressor agents. She was transfused with 1700 mL of blood, 150 mL of saline solution, and 420 mL of Lactated Ringer’s solution. No intraoperative complications occurred. The patient was extubated uneventfully on postoperative day 1 and was at baseline neurologically with no visual disturbances.
Development and Diagnosis of PRES. The patient made significant progress with physical therapy and developed episodes of hypertension at night on postoperative days 4 to 6. Her mean peak systolic blood pressure was 180 mm Hg. This improved after oral beta-blocker therapy. On postoperative day 6, the patient was ambulating with physical therapy and the aid of a walker. She was found to be neurologically intact, was resting comfortable in a chair reading a book, and was cleared for transfer to a rehabilitation facility the next day. During the morning on postoperative day 7, she developed confusion and visual loss. The patient reported blurry vision followed by complete bilateral painless loss of vision aside from mild light perception. She was unable to identify any objects. She had extinction to double simultaneous stimuli and evidence of agraphesthesia in the left hand. Her neurologic examination was otherwise at baseline. Upon emergent imaging, head CT showed bilateral symmetric areas of hypodensity involving the cortical and subcortical white matter of both occipital lobes (Figure 3). MRI showed extensive bilateral cortical and subcortical signal hyperintensity involving the parietal and occipital lobes (Figure 4). No evidence of petechial or lobar hemorrhage was found.
Treatment and Clinical Course. The patient was transferred to the neurology intensive care unit for neurologic monitoring. She was treated aggressively for recurrent hypertensive episodes. Twenty-four hours after initial blood pressure optimization therapy, she partially recovered her eyesight. She exhibited complete recovery after 48 hours. The patient was discharged to a rehabilitation facility in stable condition on postoperative day 11.
Case 2
Clinical Presentation. A 51-year-old woman presented to the outpatient clinic with progressive low back pain and decompensation due to degenerative adult scoliosis. Her surgical history was significant for an uneventful Caesarean section. Her medical history was significant for borderline hypertension and obesity (body mass index, 34.4). The radiographic examination showed an S-shaped thoracolumbar curve from T4 to L4 (Figure 5).
Operative Procedure. After discussions about the risks and benefits of the procedure, the patient underwent posterior spinal fusion from T3 to pelvis (Mesa, K2M) and interbody fusion from L4 to S1 via a presacral approach using the AxiaLIF system (TranS1) (Figure 6). The operation spanned 507 minutes. The patient lost approximately 2200 mL of blood. She was transfused with 1690 mL of blood, 1250 mL of Lactated Ringer’s solution, and 1 unit (50 mL) of albumin. No intraoperative complications occurred.
Development and Diagnosis of PRES. The patient was ambulatory with physical therapy and a walker on postoperative day 1. Her albumin levels were noted to be decreased postoperatively (28 mg/mL; normal, >35 mg/mL). She developed intermittent hypertensive episodes and experienced transient peripheral vision loss. After her ophthalmologic symptoms cleared, she was discharged and transferred to a rehabilitation facility on postoperative day 9. Eleven days later, the patient was emergently readmitted for a deep spine wound infection after an onset of wound swelling and fever. She underwent irrigation and débridement of the spine wound with an estimated blood loss of 400 mL. The patient continued to have fevers and was placed on ciprofloxacin and vancomycin, which was changed to levofloxacin on postoperative day 5. Elevated creatinine was noted, and the patient was diagnosed with acute renal failure. On postoperative day 7, oxacillin therapy was commenced. After her cultures grew methicillin-resistant Staphylococcus aureus, a peripherally inserted central catheter line was placed on postoperative day 9. As a result of nausea and constipation, the patient received feeding tubes on postoperative day 11. Additionally, she was diagnosed with a pleural effusion on postoperative day 14. Although her creatinine levels were decreasing, she continued to experience intermittent hypertensive episodes with a mean peak systolic blood pressure of 148 mm Hg. On postoperative day 15, she had a seizure and again developed visual loss. The patient was lethargic and followed only simple commands. She moved all extremities and withdrew symmetrically to noxious stimuli. Upon emergent imaging, head CT showed posterior subcortical white matter hypodensity within the occipital and parietal lobes bilaterally (Figure 7). MRI showed focal regions of symmetric hemispheric edema involving the parietal and occipital lobes in a predominantly subcortical white-matter distribution. Additionally, extensive involvement of the splenium and of the corpus callosum, left greater than right, was observed (Figure 8).
Treatment and Clinical Course. The patient was transferred to the intensive care unit for neuromonitoring. Her hypokalemia and hypertension were treated aggressively to normalize her potassium levels and blood pressure. Her oxacillin therapy was changed to daptomycin. On postoperative day 17, the patient was transferred to another institution for further medical management after achieving full recovery of her eyesight after electrolyte and blood pressure corrections.
Discussion
Posterior reversible encephalopathy syndrome is a rare but frequently devastating complication of spinal surgery, with an estimated incidence of 0.094% to 0.2%.7,8 Pediatric patients, as well as patients undergoing deformity correction surgery and posterior lumbar fusion, which necessitate prone positioning, have a significantly increased risk of POVL after spinal surgery.9 There are several causes of POVL after spinal surgery, each with a unique pathophysiology, clinical presentation, and prognosis.
The most common cause of POVL, accounting for 89% of all cases, is ischemic neuropathy.10 Ischemic neuropathy refers to a hypoperfusion or infarction of the anterior or posterior portion of the optic nerve and presents as painless bilateral vision loss or complete blindness on waking from the surgical procedure.11 Risk factors associated with anterior ischemic neuropathy are primarily diabetes mellitus, prone positioning, nocturnal hypotension, and blood loss.11 Posterior ischemic neuropathy has been most strongly correlated with anemia and hypotension.12 The exact etiology of this complication has not been established, although the prognosis is generally unfavorable, with most vision loss being permanent.10-12
Another potential cause of POVL after spinal surgery is retinal artery occlusion. It is most commonly observed in patients who were improperly positioned, resulting in compression of an orbit on the surface of the headrest or the operating table.13 Retinal artery occlusion characteristically presents as an irreversible unilateral complete loss of vision with a red spot on the macula and an afferent pupillary defect.14
Cortical blindness, another possible common cause of POVL, results from the hypoperfusion of the occipital cortex and has a slightly better prognosis. Cortical blindness generally results from an embolic event that can be visualized through neuroimaging and may be unilateral or bilateral, ranging from mild peripheral vision loss to complete blindness.15
Posterior reversible encephalopathy syndrome, the cause of POVL diagnosed in the 2 patients in this case report, is a neurologic syndrome that differs significantly in its clinical presentation and pathophysiology from the more well-known etiologies. The precise pathophysiologic mechanism of the syndrome is yet to be elucidated. One theory revolves around the failure of cerebral vascular autoregulation. It postulates that intracerebellar hypertension leads to the extravasation of proteins and fluid, resulting in the characteristic vasogenic edema.16,17 The other equally discussed theory postulates that cerebellar vasospasm and subsequent hypoperfusion leading to cellular hypoxemia and ischemia may be responsible.18-20 Posterior reversible encephalopathy syndrome has been reported with increasing frequency, particularly in connection with hypertension, acute renal failure associated with malignancy, cytotoxicity, and corticosteroids, as well as preeclampsia, eclampsia, and autoimmune disorders.1-3,21-23 Traditionally, patients display a combination of different symptoms, including vision changes ranging from slightly decreased perception to complete blindness. Unlike retinal artery occlusion and ischemic optic neuropathy, the onset of vision loss often does not happen immediately after surgery and may occur several hours to days after surgery. Visual disturbance may progressively worsen if the medical cause for the syndrome is not determined and corrected.2,3 In contrast to other known etiologies of POVL, PRES has a relatively favorable prognosis if managed appropriately. Reported case series determined a resolution of the characteristic parieto-occipital vasogenic edema in 83% to 88% of all patients in follow-up neuroimaging after aggressive control of seizures and arterial hypertension.2-3
Both patients undergoing spinal deformity surgery in this report suffered from intermittent hypertensive episodes in the postoperative period. One patient also developed acute renal failure during her hospital stay, and demonstrated low albumin levels postoperatively, which has also been associated with PRES.24 Through the immediate diagnosis and primary control of hypertension, both patients achieved complete neurologic recovery after a mean of 1.5 days (range, 1-2 days); this compares to a recovery period of an average 6.2 days (range, 1-14 days) reported by Ni and colleagues.3 The catastrophic effects of a misdiagnosis and incorrect or untimely treatment were well described in this case report. Several patients who were incorrectly diagnosed with demyelinating disorders or lupus encephalopathy received high doses of immunosuppressants and corticosteroids, known risk factors for the development of PRES.3 The patients subsequently rapidly deteriorated; no patients had a full recovery of their preoperative eyesight, and 1 patient developed complete permanent blindness.3 Optimized multidisciplinary collaboration allowing for a rapid neuro-ophthalmic examination and appropriate neuroimaging will permit an accurate and rapid diagnosis, leading to timely intervention and restoration of vision.
Conclusion
Temporary POVL is a potentially devastating complication of spinal surgery and general anesthesia. The more frequent causes such as ischemic optic neuropathy, retinal artery occlusion, and cortical blindness have very limited effective options for treatment and an overall poor prognosis. The inclusion of PRES in the differential diagnosis of POVL may allow early detection, management, and restoration of vision.
First described in 1996, posterior reversible encephalopathy syndrome (PRES) exhibits a wide clinical spectrum and is definitively diagnosed through computed tomography (CT) and/or magnetic resonance imaging (MRI) studies of the brain.1 Clinical presentation may include a spectrum of symptoms, including nausea, emesis, seizures, visual loss, paralysis, and headaches.2,3 The most common imaging finding of PRES is bilateral foci of vasogenic edema located in the parieto-occipital white matter.2-6 Other areas of the brain are frequently affected as well, with the frontal and temporal lobes and the basal or cortical ganglia showing signs of distinctly noncytotoxic edema in 12.5% to 54.2% of all cases.3 With the symptom of visual loss being present in 20% to 62.5% of patients with PRES, the syndrome constitutes a rare potential cause for postoperative visual loss (POVL) after spinal surgery, which has a generally good prognosis because most patients will completely regain their eyesight.2,3
We present a unique account of 2 patients who underwent extensive spinal surgery and received a timely diagnosis and treatment of PRES at a single institution. We aim to elucidate the difference in clinical and radiographic presentation of PRES in relation to other known causes of POVL after spinal surgery. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
Clinical Presentation. A 78-year-old woman presented to the outpatient clinic with disability due to severe lower back pain. Her surgical history was significant for breast lumpectomy and cataract excision. Her medical history was significant for hypertension, obesity (body mass index, 31.5), hypercholesterolemia, emphysema, and anemia. She had undergone spinal surgery, specifically laminectomies from L2 to S1. The radiographic examination showed degenerative thoracolumbar scoliosis with severe spondylosis, disc space collapse, and ankylosis of L4-L5 (Figure 1).
Operative Procedure. The patient underwent transpsoas lumbar interbody fusion (XLIF, NuVasive) from L1 to L4 and posterior spinal fusion from T10 to pelvis (Expedium, Depuy Synthes) (Figure 2). Operative time was 553 minutes; estimated blood loss was 2000 mL due to intraoperative coagulopathy (platelets, 40,000/µL) near the end of the posterior portion of the procedure. Intraoperative hypotension was treated by volume resuscitation and transient use of vasopressor agents. She was transfused with 1700 mL of blood, 150 mL of saline solution, and 420 mL of Lactated Ringer’s solution. No intraoperative complications occurred. The patient was extubated uneventfully on postoperative day 1 and was at baseline neurologically with no visual disturbances.
Development and Diagnosis of PRES. The patient made significant progress with physical therapy and developed episodes of hypertension at night on postoperative days 4 to 6. Her mean peak systolic blood pressure was 180 mm Hg. This improved after oral beta-blocker therapy. On postoperative day 6, the patient was ambulating with physical therapy and the aid of a walker. She was found to be neurologically intact, was resting comfortable in a chair reading a book, and was cleared for transfer to a rehabilitation facility the next day. During the morning on postoperative day 7, she developed confusion and visual loss. The patient reported blurry vision followed by complete bilateral painless loss of vision aside from mild light perception. She was unable to identify any objects. She had extinction to double simultaneous stimuli and evidence of agraphesthesia in the left hand. Her neurologic examination was otherwise at baseline. Upon emergent imaging, head CT showed bilateral symmetric areas of hypodensity involving the cortical and subcortical white matter of both occipital lobes (Figure 3). MRI showed extensive bilateral cortical and subcortical signal hyperintensity involving the parietal and occipital lobes (Figure 4). No evidence of petechial or lobar hemorrhage was found.
Treatment and Clinical Course. The patient was transferred to the neurology intensive care unit for neurologic monitoring. She was treated aggressively for recurrent hypertensive episodes. Twenty-four hours after initial blood pressure optimization therapy, she partially recovered her eyesight. She exhibited complete recovery after 48 hours. The patient was discharged to a rehabilitation facility in stable condition on postoperative day 11.
Case 2
Clinical Presentation. A 51-year-old woman presented to the outpatient clinic with progressive low back pain and decompensation due to degenerative adult scoliosis. Her surgical history was significant for an uneventful Caesarean section. Her medical history was significant for borderline hypertension and obesity (body mass index, 34.4). The radiographic examination showed an S-shaped thoracolumbar curve from T4 to L4 (Figure 5).
Operative Procedure. After discussions about the risks and benefits of the procedure, the patient underwent posterior spinal fusion from T3 to pelvis (Mesa, K2M) and interbody fusion from L4 to S1 via a presacral approach using the AxiaLIF system (TranS1) (Figure 6). The operation spanned 507 minutes. The patient lost approximately 2200 mL of blood. She was transfused with 1690 mL of blood, 1250 mL of Lactated Ringer’s solution, and 1 unit (50 mL) of albumin. No intraoperative complications occurred.
Development and Diagnosis of PRES. The patient was ambulatory with physical therapy and a walker on postoperative day 1. Her albumin levels were noted to be decreased postoperatively (28 mg/mL; normal, >35 mg/mL). She developed intermittent hypertensive episodes and experienced transient peripheral vision loss. After her ophthalmologic symptoms cleared, she was discharged and transferred to a rehabilitation facility on postoperative day 9. Eleven days later, the patient was emergently readmitted for a deep spine wound infection after an onset of wound swelling and fever. She underwent irrigation and débridement of the spine wound with an estimated blood loss of 400 mL. The patient continued to have fevers and was placed on ciprofloxacin and vancomycin, which was changed to levofloxacin on postoperative day 5. Elevated creatinine was noted, and the patient was diagnosed with acute renal failure. On postoperative day 7, oxacillin therapy was commenced. After her cultures grew methicillin-resistant Staphylococcus aureus, a peripherally inserted central catheter line was placed on postoperative day 9. As a result of nausea and constipation, the patient received feeding tubes on postoperative day 11. Additionally, she was diagnosed with a pleural effusion on postoperative day 14. Although her creatinine levels were decreasing, she continued to experience intermittent hypertensive episodes with a mean peak systolic blood pressure of 148 mm Hg. On postoperative day 15, she had a seizure and again developed visual loss. The patient was lethargic and followed only simple commands. She moved all extremities and withdrew symmetrically to noxious stimuli. Upon emergent imaging, head CT showed posterior subcortical white matter hypodensity within the occipital and parietal lobes bilaterally (Figure 7). MRI showed focal regions of symmetric hemispheric edema involving the parietal and occipital lobes in a predominantly subcortical white-matter distribution. Additionally, extensive involvement of the splenium and of the corpus callosum, left greater than right, was observed (Figure 8).
Treatment and Clinical Course. The patient was transferred to the intensive care unit for neuromonitoring. Her hypokalemia and hypertension were treated aggressively to normalize her potassium levels and blood pressure. Her oxacillin therapy was changed to daptomycin. On postoperative day 17, the patient was transferred to another institution for further medical management after achieving full recovery of her eyesight after electrolyte and blood pressure corrections.
Discussion
Posterior reversible encephalopathy syndrome is a rare but frequently devastating complication of spinal surgery, with an estimated incidence of 0.094% to 0.2%.7,8 Pediatric patients, as well as patients undergoing deformity correction surgery and posterior lumbar fusion, which necessitate prone positioning, have a significantly increased risk of POVL after spinal surgery.9 There are several causes of POVL after spinal surgery, each with a unique pathophysiology, clinical presentation, and prognosis.
The most common cause of POVL, accounting for 89% of all cases, is ischemic neuropathy.10 Ischemic neuropathy refers to a hypoperfusion or infarction of the anterior or posterior portion of the optic nerve and presents as painless bilateral vision loss or complete blindness on waking from the surgical procedure.11 Risk factors associated with anterior ischemic neuropathy are primarily diabetes mellitus, prone positioning, nocturnal hypotension, and blood loss.11 Posterior ischemic neuropathy has been most strongly correlated with anemia and hypotension.12 The exact etiology of this complication has not been established, although the prognosis is generally unfavorable, with most vision loss being permanent.10-12
Another potential cause of POVL after spinal surgery is retinal artery occlusion. It is most commonly observed in patients who were improperly positioned, resulting in compression of an orbit on the surface of the headrest or the operating table.13 Retinal artery occlusion characteristically presents as an irreversible unilateral complete loss of vision with a red spot on the macula and an afferent pupillary defect.14
Cortical blindness, another possible common cause of POVL, results from the hypoperfusion of the occipital cortex and has a slightly better prognosis. Cortical blindness generally results from an embolic event that can be visualized through neuroimaging and may be unilateral or bilateral, ranging from mild peripheral vision loss to complete blindness.15
Posterior reversible encephalopathy syndrome, the cause of POVL diagnosed in the 2 patients in this case report, is a neurologic syndrome that differs significantly in its clinical presentation and pathophysiology from the more well-known etiologies. The precise pathophysiologic mechanism of the syndrome is yet to be elucidated. One theory revolves around the failure of cerebral vascular autoregulation. It postulates that intracerebellar hypertension leads to the extravasation of proteins and fluid, resulting in the characteristic vasogenic edema.16,17 The other equally discussed theory postulates that cerebellar vasospasm and subsequent hypoperfusion leading to cellular hypoxemia and ischemia may be responsible.18-20 Posterior reversible encephalopathy syndrome has been reported with increasing frequency, particularly in connection with hypertension, acute renal failure associated with malignancy, cytotoxicity, and corticosteroids, as well as preeclampsia, eclampsia, and autoimmune disorders.1-3,21-23 Traditionally, patients display a combination of different symptoms, including vision changes ranging from slightly decreased perception to complete blindness. Unlike retinal artery occlusion and ischemic optic neuropathy, the onset of vision loss often does not happen immediately after surgery and may occur several hours to days after surgery. Visual disturbance may progressively worsen if the medical cause for the syndrome is not determined and corrected.2,3 In contrast to other known etiologies of POVL, PRES has a relatively favorable prognosis if managed appropriately. Reported case series determined a resolution of the characteristic parieto-occipital vasogenic edema in 83% to 88% of all patients in follow-up neuroimaging after aggressive control of seizures and arterial hypertension.2-3
Both patients undergoing spinal deformity surgery in this report suffered from intermittent hypertensive episodes in the postoperative period. One patient also developed acute renal failure during her hospital stay, and demonstrated low albumin levels postoperatively, which has also been associated with PRES.24 Through the immediate diagnosis and primary control of hypertension, both patients achieved complete neurologic recovery after a mean of 1.5 days (range, 1-2 days); this compares to a recovery period of an average 6.2 days (range, 1-14 days) reported by Ni and colleagues.3 The catastrophic effects of a misdiagnosis and incorrect or untimely treatment were well described in this case report. Several patients who were incorrectly diagnosed with demyelinating disorders or lupus encephalopathy received high doses of immunosuppressants and corticosteroids, known risk factors for the development of PRES.3 The patients subsequently rapidly deteriorated; no patients had a full recovery of their preoperative eyesight, and 1 patient developed complete permanent blindness.3 Optimized multidisciplinary collaboration allowing for a rapid neuro-ophthalmic examination and appropriate neuroimaging will permit an accurate and rapid diagnosis, leading to timely intervention and restoration of vision.
Conclusion
Temporary POVL is a potentially devastating complication of spinal surgery and general anesthesia. The more frequent causes such as ischemic optic neuropathy, retinal artery occlusion, and cortical blindness have very limited effective options for treatment and an overall poor prognosis. The inclusion of PRES in the differential diagnosis of POVL may allow early detection, management, and restoration of vision.
1. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494-500.
2. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427-432.
3. Ni J, Zhou LX, Hao HL, et al. The clinical and radiological spectrum of posterior reversible encephalopathy syndrome: a retrospective series of 24 patients. J Neuroimaging. 2011;21(3):219-224.
4. Stevens CJ, Heran MK. The many faces of posterior reversible encephalopathy syndrome. Br J Radiol. 2012;85(1020):1566-1575.
5. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036-1042.
6. Yoon SD, Cho BM, Oh SM, et al. Clinical and radiological spectrum of posterior reversible encephalopathy syndrome. J Cerebrovasc Endovasc Neurosurg. 2013;15(3):206-213.
7. Patil CG, Lad EM, Lad SP, Ho C, Boakye M. Visual loss after spine surgery: a population-based study. Spine (Phila Pa 1976). 2008;33(13):1491-1496.
8. Stevens WR, Glazer PA, Kelley SD, Lietman TM, Bradford DS. Ophthalmic complications after spinal surgery. Spine (Phila Pa 1976). 1997;22(12):1319-1324.
9. Shen Y, Drum M, Roth S. The prevalence of perioperative visual loss in the United States: a 10-year study from 1996 to 2005 of spinal, orthopedic, cardiac, and general surgery. Anesth Analg. 2009;109(5):1534-1545.
10. Lee LA, Roth S, Posner KL, et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659; quiz 867-868.
11. Hayreh SS. Ischemic optic neuropathies - where are we now? Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1873-1884.
12. Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol. 2005;50(1):15-26.
13. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85.
14. Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87(1):75-78.
15. Berg KT, Harrison AR, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010;4:531-546.
16. Primavera A, Audenino D, Mavilio N, Cocito L. Reversible posterior leucoencephalopathy syndrome in systemic lupus and vasculitis. Ann Rheum Dis. 2001;60(5):534-537.
17. Bartynski WS, Boardman JF. Catheter angiography, MR angiography, and MR perfusion in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008;29(3):447-455.
18. Ito T, Sakai T, Inagawa S, Utsu M, Bun T. MR angiography of cerebral vasospasm in preeclampsia. AJNR Am J Neuroradiol. 1995;16(6):1344-1346.
19. Agarwal R, Davis C, Altinok D, Serajee FJ. Posterior reversible encephalopathy and cerebral vasoconstriction in a patient with hemolytic uremic syndrome. Pediatr Neurol. 2014;50(5):518-521.
20. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29(6):1043-1049.
21. Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205-210.
22. Ekawa Y, Shiota M, Tobiume T, et al. Reversible posterior leukoencephalopathy syndrome accompanying eclampsia: correct diagnosis using preoperative MRI. Tohoku J Exp Med. 2012;226(1):55-58.
23. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome--an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol. 2006;33(11):2178-2183.
24. Pirker A, Kramer L, Voller B, et al. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32(3):527-531.
1. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494-500.
2. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427-432.
3. Ni J, Zhou LX, Hao HL, et al. The clinical and radiological spectrum of posterior reversible encephalopathy syndrome: a retrospective series of 24 patients. J Neuroimaging. 2011;21(3):219-224.
4. Stevens CJ, Heran MK. The many faces of posterior reversible encephalopathy syndrome. Br J Radiol. 2012;85(1020):1566-1575.
5. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036-1042.
6. Yoon SD, Cho BM, Oh SM, et al. Clinical and radiological spectrum of posterior reversible encephalopathy syndrome. J Cerebrovasc Endovasc Neurosurg. 2013;15(3):206-213.
7. Patil CG, Lad EM, Lad SP, Ho C, Boakye M. Visual loss after spine surgery: a population-based study. Spine (Phila Pa 1976). 2008;33(13):1491-1496.
8. Stevens WR, Glazer PA, Kelley SD, Lietman TM, Bradford DS. Ophthalmic complications after spinal surgery. Spine (Phila Pa 1976). 1997;22(12):1319-1324.
9. Shen Y, Drum M, Roth S. The prevalence of perioperative visual loss in the United States: a 10-year study from 1996 to 2005 of spinal, orthopedic, cardiac, and general surgery. Anesth Analg. 2009;109(5):1534-1545.
10. Lee LA, Roth S, Posner KL, et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659; quiz 867-868.
11. Hayreh SS. Ischemic optic neuropathies - where are we now? Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1873-1884.
12. Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol. 2005;50(1):15-26.
13. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85.
14. Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87(1):75-78.
15. Berg KT, Harrison AR, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010;4:531-546.
16. Primavera A, Audenino D, Mavilio N, Cocito L. Reversible posterior leucoencephalopathy syndrome in systemic lupus and vasculitis. Ann Rheum Dis. 2001;60(5):534-537.
17. Bartynski WS, Boardman JF. Catheter angiography, MR angiography, and MR perfusion in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008;29(3):447-455.
18. Ito T, Sakai T, Inagawa S, Utsu M, Bun T. MR angiography of cerebral vasospasm in preeclampsia. AJNR Am J Neuroradiol. 1995;16(6):1344-1346.
19. Agarwal R, Davis C, Altinok D, Serajee FJ. Posterior reversible encephalopathy and cerebral vasoconstriction in a patient with hemolytic uremic syndrome. Pediatr Neurol. 2014;50(5):518-521.
20. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29(6):1043-1049.
21. Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205-210.
22. Ekawa Y, Shiota M, Tobiume T, et al. Reversible posterior leukoencephalopathy syndrome accompanying eclampsia: correct diagnosis using preoperative MRI. Tohoku J Exp Med. 2012;226(1):55-58.
23. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome--an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol. 2006;33(11):2178-2183.
24. Pirker A, Kramer L, Voller B, et al. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32(3):527-531.
Canada may shorten deferral for MSM blood donors

Photo by Charles Haymond
ANAHEIM, CA—Lifting the lifetime ban on blood donations from men who have sex with men (MSM) has not altered the safety of the blood supply in Canada, according to a new study.
The study showed no increase in the rate of HIV-positive blood donations since Canada changed its policy regarding MSM blood donors, allowing MSM to donate if they have not had sexual contact with another man in the last 5 years.
Because of this finding, Canada may shorten the deferral period for MSM blood donors to 1 year, according to Sheila F. O’Brien, PhD, of Canadian Blood Services in Ottawa, Ontario, Canada.
Dr O’Brien mentioned this possibility and presented data from the study at the 2015 AABB Annual Meeting (abstract S35-030E*).
Prior to 2013, MSM in Canada were not allowed to donate blood if they had any sexual contact with another male since 1977. Females were barred from donating if, in the last year, they had sexual contact with a man who had sex with another man after 1977.
On July 22, 2013, Canada changed this policy so that MSM can donate blood if they have abstained from sexual contact with another man for the past 5 years. The deferral period for females is still 12 months if they have had sex with a man who has had sex with another man in the last 5 years, but there is no deferral if the man had sex with another man more than 5 years before.
To evaluate the impact of this policy change, Dr O’Brien and her colleagues assessed compliance with the MSM criteria before and after the change, as well as the number of HIV-positive blood donations before and after the change.
The researchers also assessed the number of donors who would have been deferred according to the old MSM criteria but donated blood under the new criteria.
MSM history
The researchers selected random male donors of whole blood each month from October 2012 to February 2013 (pre-change) and from October 2014 to February 2015 (post-change). These donors were invited to complete an anonymous online survey about their MSM history.
The survey was completed by 9669 donors before the policy change and 6881 donors after the change. There were 77 donors with MSM history before the change (20% first-time donors, 80% repeat) and 75 donors with MSM history after the change (22% first-time, 78% repeat).
Compliance with policy
After the change in policy for MSM blood donors, there was no significant change in the proportion of donors who had recent MSM history but donated anyway (non-compliant). Before the change, 0.37% of blood donors had an MSM partner in the last 5 years, compared to 0.43% after the change (P=0.54).
However, there was a significant change in the proportion of blood donors with MSM history further in the past. Before the MSM policy change, 0.42% of donors had an MSM partner but not in the last 5 years, compared to 0.66% of donors after the change (P=0.04).
“So we have an improvement in compliance, but it’s mainly because the donors are no longer deferrable,” Dr O’Brien explained.
“Donating while ineligible because of MSM history is actually quite rare, and the percentage of donors with MSM history in the last 5 years did not change when we changed the criteria. But we did see a modest increase in newly eligible MSM, so those that had more than 5 years since their last male-to-male sex.”
In all, there were 112 donors who were newly eligible due to the policy change and did, in fact, donate blood between July 22, 2013 and July 21, 2015. Five of these donors were females who had sexual contact with MSMs.
There were 70 “reinstated” donors in the first year after the policy change and 42 in the second year.
HIV-positive donations
The researchers monitored HIV rates in all blood donations from January 2010 to March 2015.
The rates of HIV-positive donations were as follows: 0.20 for 2010 (2/989,916), 0.50 for 2011 (5/995,122), 0.51 for 2012 (5/987,527), 0 (0/525,337) from January 1, 2013 to July 21, 2013 (before the policy change), 0.54 from July 22, 2013 to July 21, 2014 (5/929,656), and 0.22 from July 22, 2014 to July 21, 2015 (2/893,513).
“So absolutely no change in HIV rate following implementation of our 5-year deferral,” Dr O’Brien said.
In all, there were 7 HIV-positive donations after the policy change. Four were from male donors, and 3 were from females.
Three of the male donors (2 first-time donors, 1 repeat) denied having MSM risk factors, and 1 first-time male donor was aware he was HIV-positive at the time of donation. This man said he donated to determine if his HIV medication was working.
Two of the females were repeat donors, and 1 was a first-timer. The first-time donor did not acknowledge any MSM risk factors. One of the repeat donors had a sexual relationship with a bisexual male who was HIV-positive. The other repeat donor had multiple sexual partners, 1 of whom was known to be hepatitis C-positive.
Future policy change
Dr O’Brien noted that the LGBTQ community in Canada has advocated abolishing the deferral period for MSM blood donors or changing to a risk-based policy that would allow more individuals with MSM history to donate blood.
She said the combined blood services in Canada—Canadian Blood Services and Héma-Québec—are now considering a 12-month deferral period for individuals with MSM history.
“We’re pretty sure we’re going to go ahead,” she noted.
However, the groups must submit a policy request to Health Canada, which will ultimately make the decision. ![]()
*Data in the abstract differ from data presented at the meeting.
Photo by Charles Haymond
ANAHEIM, CA—Lifting the lifetime ban on blood donations from men who have sex with men (MSM) has not altered the safety of the blood supply in Canada, according to a new study.
The study showed no increase in the rate of HIV-positive blood donations since Canada changed its policy regarding MSM blood donors, allowing MSM to donate if they have not had sexual contact with another man in the last 5 years.
Because of this finding, Canada may shorten the deferral period for MSM blood donors to 1 year, according to Sheila F. O’Brien, PhD, of Canadian Blood Services in Ottawa, Ontario, Canada.
Dr O’Brien mentioned this possibility and presented data from the study at the 2015 AABB Annual Meeting (abstract S35-030E*).
Prior to 2013, MSM in Canada were not allowed to donate blood if they had any sexual contact with another male since 1977. Females were barred from donating if, in the last year, they had sexual contact with a man who had sex with another man after 1977.
On July 22, 2013, Canada changed this policy so that MSM can donate blood if they have abstained from sexual contact with another man for the past 5 years. The deferral period for females is still 12 months if they have had sex with a man who has had sex with another man in the last 5 years, but there is no deferral if the man had sex with another man more than 5 years before.
To evaluate the impact of this policy change, Dr O’Brien and her colleagues assessed compliance with the MSM criteria before and after the change, as well as the number of HIV-positive blood donations before and after the change.
The researchers also assessed the number of donors who would have been deferred according to the old MSM criteria but donated blood under the new criteria.
MSM history
The researchers selected random male donors of whole blood each month from October 2012 to February 2013 (pre-change) and from October 2014 to February 2015 (post-change). These donors were invited to complete an anonymous online survey about their MSM history.
The survey was completed by 9669 donors before the policy change and 6881 donors after the change. There were 77 donors with MSM history before the change (20% first-time donors, 80% repeat) and 75 donors with MSM history after the change (22% first-time, 78% repeat).
Compliance with policy
After the change in policy for MSM blood donors, there was no significant change in the proportion of donors who had recent MSM history but donated anyway (non-compliant). Before the change, 0.37% of blood donors had an MSM partner in the last 5 years, compared to 0.43% after the change (P=0.54).
However, there was a significant change in the proportion of blood donors with MSM history further in the past. Before the MSM policy change, 0.42% of donors had an MSM partner but not in the last 5 years, compared to 0.66% of donors after the change (P=0.04).
“So we have an improvement in compliance, but it’s mainly because the donors are no longer deferrable,” Dr O’Brien explained.
“Donating while ineligible because of MSM history is actually quite rare, and the percentage of donors with MSM history in the last 5 years did not change when we changed the criteria. But we did see a modest increase in newly eligible MSM, so those that had more than 5 years since their last male-to-male sex.”
In all, there were 112 donors who were newly eligible due to the policy change and did, in fact, donate blood between July 22, 2013 and July 21, 2015. Five of these donors were females who had sexual contact with MSMs.
There were 70 “reinstated” donors in the first year after the policy change and 42 in the second year.
HIV-positive donations
The researchers monitored HIV rates in all blood donations from January 2010 to March 2015.
The rates of HIV-positive donations were as follows: 0.20 for 2010 (2/989,916), 0.50 for 2011 (5/995,122), 0.51 for 2012 (5/987,527), 0 (0/525,337) from January 1, 2013 to July 21, 2013 (before the policy change), 0.54 from July 22, 2013 to July 21, 2014 (5/929,656), and 0.22 from July 22, 2014 to July 21, 2015 (2/893,513).
“So absolutely no change in HIV rate following implementation of our 5-year deferral,” Dr O’Brien said.
In all, there were 7 HIV-positive donations after the policy change. Four were from male donors, and 3 were from females.
Three of the male donors (2 first-time donors, 1 repeat) denied having MSM risk factors, and 1 first-time male donor was aware he was HIV-positive at the time of donation. This man said he donated to determine if his HIV medication was working.
Two of the females were repeat donors, and 1 was a first-timer. The first-time donor did not acknowledge any MSM risk factors. One of the repeat donors had a sexual relationship with a bisexual male who was HIV-positive. The other repeat donor had multiple sexual partners, 1 of whom was known to be hepatitis C-positive.
Future policy change
Dr O’Brien noted that the LGBTQ community in Canada has advocated abolishing the deferral period for MSM blood donors or changing to a risk-based policy that would allow more individuals with MSM history to donate blood.
She said the combined blood services in Canada—Canadian Blood Services and Héma-Québec—are now considering a 12-month deferral period for individuals with MSM history.
“We’re pretty sure we’re going to go ahead,” she noted.
However, the groups must submit a policy request to Health Canada, which will ultimately make the decision. ![]()
*Data in the abstract differ from data presented at the meeting.
Photo by Charles Haymond
ANAHEIM, CA—Lifting the lifetime ban on blood donations from men who have sex with men (MSM) has not altered the safety of the blood supply in Canada, according to a new study.
The study showed no increase in the rate of HIV-positive blood donations since Canada changed its policy regarding MSM blood donors, allowing MSM to donate if they have not had sexual contact with another man in the last 5 years.
Because of this finding, Canada may shorten the deferral period for MSM blood donors to 1 year, according to Sheila F. O’Brien, PhD, of Canadian Blood Services in Ottawa, Ontario, Canada.
Dr O’Brien mentioned this possibility and presented data from the study at the 2015 AABB Annual Meeting (abstract S35-030E*).
Prior to 2013, MSM in Canada were not allowed to donate blood if they had any sexual contact with another male since 1977. Females were barred from donating if, in the last year, they had sexual contact with a man who had sex with another man after 1977.
On July 22, 2013, Canada changed this policy so that MSM can donate blood if they have abstained from sexual contact with another man for the past 5 years. The deferral period for females is still 12 months if they have had sex with a man who has had sex with another man in the last 5 years, but there is no deferral if the man had sex with another man more than 5 years before.
To evaluate the impact of this policy change, Dr O’Brien and her colleagues assessed compliance with the MSM criteria before and after the change, as well as the number of HIV-positive blood donations before and after the change.
The researchers also assessed the number of donors who would have been deferred according to the old MSM criteria but donated blood under the new criteria.
MSM history
The researchers selected random male donors of whole blood each month from October 2012 to February 2013 (pre-change) and from October 2014 to February 2015 (post-change). These donors were invited to complete an anonymous online survey about their MSM history.
The survey was completed by 9669 donors before the policy change and 6881 donors after the change. There were 77 donors with MSM history before the change (20% first-time donors, 80% repeat) and 75 donors with MSM history after the change (22% first-time, 78% repeat).
Compliance with policy
After the change in policy for MSM blood donors, there was no significant change in the proportion of donors who had recent MSM history but donated anyway (non-compliant). Before the change, 0.37% of blood donors had an MSM partner in the last 5 years, compared to 0.43% after the change (P=0.54).
However, there was a significant change in the proportion of blood donors with MSM history further in the past. Before the MSM policy change, 0.42% of donors had an MSM partner but not in the last 5 years, compared to 0.66% of donors after the change (P=0.04).
“So we have an improvement in compliance, but it’s mainly because the donors are no longer deferrable,” Dr O’Brien explained.
“Donating while ineligible because of MSM history is actually quite rare, and the percentage of donors with MSM history in the last 5 years did not change when we changed the criteria. But we did see a modest increase in newly eligible MSM, so those that had more than 5 years since their last male-to-male sex.”
In all, there were 112 donors who were newly eligible due to the policy change and did, in fact, donate blood between July 22, 2013 and July 21, 2015. Five of these donors were females who had sexual contact with MSMs.
There were 70 “reinstated” donors in the first year after the policy change and 42 in the second year.
HIV-positive donations
The researchers monitored HIV rates in all blood donations from January 2010 to March 2015.
The rates of HIV-positive donations were as follows: 0.20 for 2010 (2/989,916), 0.50 for 2011 (5/995,122), 0.51 for 2012 (5/987,527), 0 (0/525,337) from January 1, 2013 to July 21, 2013 (before the policy change), 0.54 from July 22, 2013 to July 21, 2014 (5/929,656), and 0.22 from July 22, 2014 to July 21, 2015 (2/893,513).
“So absolutely no change in HIV rate following implementation of our 5-year deferral,” Dr O’Brien said.
In all, there were 7 HIV-positive donations after the policy change. Four were from male donors, and 3 were from females.
Three of the male donors (2 first-time donors, 1 repeat) denied having MSM risk factors, and 1 first-time male donor was aware he was HIV-positive at the time of donation. This man said he donated to determine if his HIV medication was working.
Two of the females were repeat donors, and 1 was a first-timer. The first-time donor did not acknowledge any MSM risk factors. One of the repeat donors had a sexual relationship with a bisexual male who was HIV-positive. The other repeat donor had multiple sexual partners, 1 of whom was known to be hepatitis C-positive.
Future policy change
Dr O’Brien noted that the LGBTQ community in Canada has advocated abolishing the deferral period for MSM blood donors or changing to a risk-based policy that would allow more individuals with MSM history to donate blood.
She said the combined blood services in Canada—Canadian Blood Services and Héma-Québec—are now considering a 12-month deferral period for individuals with MSM history.
“We’re pretty sure we’re going to go ahead,” she noted.
However, the groups must submit a policy request to Health Canada, which will ultimately make the decision. ![]()
*Data in the abstract differ from data presented at the meeting.
Median DOR, PFS not yet reached for ibrutinib in CLL

Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Alternative splicing enables resistance to CTL019

Photo from Penn Medicine
New research has provided an explanation for resistance to CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy.
Investigators analyzed samples from children with B-cell acute lymphoblastic leukemia (B-ALL) and found evidence to suggest that CTL019 resistance can be caused by CD19 splicing alterations.
These alterations prompt the loss of certain parts of the CD19 protein that are recognized by the CAR T cells.
The team described this work in Cancer Discovery.
They noted that 10% to 20% of B-ALL patients treated with CD19-directed immunotherapy may experience relapse.
“Some of them can be successfully retreated, but, in others, a more pernicious kind of leukemia may emerge, which no longer responds to CTL019,” said study author Andrei Thomas-Tikhonenko, PhD, of the University of Pennsylvania in Philadelphia.
“In some cases, resistance is accompanied by the disappearance of the target CD19 protein from the cell surface . . . . Our goal was to figure out how the CD19 protein manages to vanish and whether it is gone for good or whether it could, under certain circumstances, be coaxed back.”
“Our initial finding from this study was that, in most cases, the CD19 genetic code was not irretrievably lost. We also discovered that the CD19 protein was still being made, but as a shorter version, which escapes detection by the immune system.”
To understand the mechanism of CTL019 resistance, Dr Thomas-Tikhonenko and his colleagues studied multiple tumor samples from 4 children with B-ALL. The samples were collected before the patients were treated with CTL019 and/or after they developed resistance to the therapy.
The investigators found that, in some cases, 1 copy of the gene coding for CD19 (located on chromosome 16) was deleted, and the other copy was damaged as a result of mutations in coding areas of the CD19 gene, most frequently in exon 2.
However, the team also discovered alternatively spliced CD19 messenger RNA species in which exons 2, 5, and 6 were frequently skipped, making mutations in exon 2 largely irrelevant.
Subsequent investigation revealed that deletion of exons 5 and 6 resulted in premature termination of CD19.
Deletion of exon 2 resulted in the production of a modified version of CD19, which was more stable than its standard version. The shortened protein was functional and could perform many of the tasks that CD19 is known to handle, but it cannot be targeted by CTL019.
The importance of exon skipping in CTL019 resistance cannot be overstated, Dr Thomas-Tikhonenko said.
“Without exons 5 and 6, the CD19 protein has no way of being retained on the cell surface,” he explained. “The case of missing exon 2 is more complex. Although the resultant protein can make it to the cell surface, albeit not very efficiently, it can no longer be recognized by CTL019.”
He and his colleagues believe this research can inform future use of CTL019 and immunotherapy in general.
“[A]lternative splicing could be a potent, built-in mechanism of resistance, and it might be better to target proteins that, unlike CD19, are not prone to exon skipping,” Dr Thomas-Tikhonenko said.
“[In addition,] it might be important to preselect patients for CTL019 and similar therapies and make sure that the alternatively spliced CD19 variants are not already present in their leukemias. If they are, resistance could develop very quickly.”
Designing new immunotherapeutics that can recognize the shortened version of CD19 is another approach to overcoming CTL019 resistance, he added.
He and his colleagues noted that this study was limited by the relatively small number of samples analyzed, which might have prevented the investigators from identifying additional mechanisms of resistance. ![]()
Photo from Penn Medicine
New research has provided an explanation for resistance to CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy.
Investigators analyzed samples from children with B-cell acute lymphoblastic leukemia (B-ALL) and found evidence to suggest that CTL019 resistance can be caused by CD19 splicing alterations.
These alterations prompt the loss of certain parts of the CD19 protein that are recognized by the CAR T cells.
The team described this work in Cancer Discovery.
They noted that 10% to 20% of B-ALL patients treated with CD19-directed immunotherapy may experience relapse.
“Some of them can be successfully retreated, but, in others, a more pernicious kind of leukemia may emerge, which no longer responds to CTL019,” said study author Andrei Thomas-Tikhonenko, PhD, of the University of Pennsylvania in Philadelphia.
“In some cases, resistance is accompanied by the disappearance of the target CD19 protein from the cell surface . . . . Our goal was to figure out how the CD19 protein manages to vanish and whether it is gone for good or whether it could, under certain circumstances, be coaxed back.”
“Our initial finding from this study was that, in most cases, the CD19 genetic code was not irretrievably lost. We also discovered that the CD19 protein was still being made, but as a shorter version, which escapes detection by the immune system.”
To understand the mechanism of CTL019 resistance, Dr Thomas-Tikhonenko and his colleagues studied multiple tumor samples from 4 children with B-ALL. The samples were collected before the patients were treated with CTL019 and/or after they developed resistance to the therapy.
The investigators found that, in some cases, 1 copy of the gene coding for CD19 (located on chromosome 16) was deleted, and the other copy was damaged as a result of mutations in coding areas of the CD19 gene, most frequently in exon 2.
However, the team also discovered alternatively spliced CD19 messenger RNA species in which exons 2, 5, and 6 were frequently skipped, making mutations in exon 2 largely irrelevant.
Subsequent investigation revealed that deletion of exons 5 and 6 resulted in premature termination of CD19.
Deletion of exon 2 resulted in the production of a modified version of CD19, which was more stable than its standard version. The shortened protein was functional and could perform many of the tasks that CD19 is known to handle, but it cannot be targeted by CTL019.
The importance of exon skipping in CTL019 resistance cannot be overstated, Dr Thomas-Tikhonenko said.
“Without exons 5 and 6, the CD19 protein has no way of being retained on the cell surface,” he explained. “The case of missing exon 2 is more complex. Although the resultant protein can make it to the cell surface, albeit not very efficiently, it can no longer be recognized by CTL019.”
He and his colleagues believe this research can inform future use of CTL019 and immunotherapy in general.
“[A]lternative splicing could be a potent, built-in mechanism of resistance, and it might be better to target proteins that, unlike CD19, are not prone to exon skipping,” Dr Thomas-Tikhonenko said.
“[In addition,] it might be important to preselect patients for CTL019 and similar therapies and make sure that the alternatively spliced CD19 variants are not already present in their leukemias. If they are, resistance could develop very quickly.”
Designing new immunotherapeutics that can recognize the shortened version of CD19 is another approach to overcoming CTL019 resistance, he added.
He and his colleagues noted that this study was limited by the relatively small number of samples analyzed, which might have prevented the investigators from identifying additional mechanisms of resistance. ![]()
Photo from Penn Medicine
New research has provided an explanation for resistance to CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy.
Investigators analyzed samples from children with B-cell acute lymphoblastic leukemia (B-ALL) and found evidence to suggest that CTL019 resistance can be caused by CD19 splicing alterations.
These alterations prompt the loss of certain parts of the CD19 protein that are recognized by the CAR T cells.
The team described this work in Cancer Discovery.
They noted that 10% to 20% of B-ALL patients treated with CD19-directed immunotherapy may experience relapse.
“Some of them can be successfully retreated, but, in others, a more pernicious kind of leukemia may emerge, which no longer responds to CTL019,” said study author Andrei Thomas-Tikhonenko, PhD, of the University of Pennsylvania in Philadelphia.
“In some cases, resistance is accompanied by the disappearance of the target CD19 protein from the cell surface . . . . Our goal was to figure out how the CD19 protein manages to vanish and whether it is gone for good or whether it could, under certain circumstances, be coaxed back.”
“Our initial finding from this study was that, in most cases, the CD19 genetic code was not irretrievably lost. We also discovered that the CD19 protein was still being made, but as a shorter version, which escapes detection by the immune system.”
To understand the mechanism of CTL019 resistance, Dr Thomas-Tikhonenko and his colleagues studied multiple tumor samples from 4 children with B-ALL. The samples were collected before the patients were treated with CTL019 and/or after they developed resistance to the therapy.
The investigators found that, in some cases, 1 copy of the gene coding for CD19 (located on chromosome 16) was deleted, and the other copy was damaged as a result of mutations in coding areas of the CD19 gene, most frequently in exon 2.
However, the team also discovered alternatively spliced CD19 messenger RNA species in which exons 2, 5, and 6 were frequently skipped, making mutations in exon 2 largely irrelevant.
Subsequent investigation revealed that deletion of exons 5 and 6 resulted in premature termination of CD19.
Deletion of exon 2 resulted in the production of a modified version of CD19, which was more stable than its standard version. The shortened protein was functional and could perform many of the tasks that CD19 is known to handle, but it cannot be targeted by CTL019.
The importance of exon skipping in CTL019 resistance cannot be overstated, Dr Thomas-Tikhonenko said.
“Without exons 5 and 6, the CD19 protein has no way of being retained on the cell surface,” he explained. “The case of missing exon 2 is more complex. Although the resultant protein can make it to the cell surface, albeit not very efficiently, it can no longer be recognized by CTL019.”
He and his colleagues believe this research can inform future use of CTL019 and immunotherapy in general.
“[A]lternative splicing could be a potent, built-in mechanism of resistance, and it might be better to target proteins that, unlike CD19, are not prone to exon skipping,” Dr Thomas-Tikhonenko said.
“[In addition,] it might be important to preselect patients for CTL019 and similar therapies and make sure that the alternatively spliced CD19 variants are not already present in their leukemias. If they are, resistance could develop very quickly.”
Designing new immunotherapeutics that can recognize the shortened version of CD19 is another approach to overcoming CTL019 resistance, he added.
He and his colleagues noted that this study was limited by the relatively small number of samples analyzed, which might have prevented the investigators from identifying additional mechanisms of resistance. ![]()
Drug can block malaria transmission, studies show
Image by Ute Frevert
and Margaret Shear
PHILADELPHIA—A drug used to treat multiple tropical diseases can also inhibit malaria transmission, according to a pair of studies presented at the ASTMH 64th Annual Meeting.
One study suggests the drug, ivermectin, can reduce the transmission of malaria caused by Plasmodium falciparum, the most prevalent malaria parasite in Africa.
The other study indicates that ivermectin can block the transmission of Plasmodium vivax parasites, which are common in Southeast Asia.
Kevin Kobylinski, PhD, of the Armed Forces Research Institute of Medical Sciences in Bangkok, Thailand, presented results of the P vivax study as abstract 1283.
And Brian D. Foy, PhD, of Colorado State University in Fort Collins, presented results of the P falciparum trial as abstract LB-5237.
P falciparum malaria
Dr Foy and his colleagues are assessing the ability of ivermectin to block malaria transmission in 4 villages in Burkina Faso, Africa.
Lab studies have shown that when mosquitoes feed on the blood of people who have taken ivermectin, it interferes with the mosquitoes’ ability to transmit malaria parasites to humans. Sometimes, it kills the mosquitoes outright, but, more often, it weakens them and interferes with their digestive system so they eventually die in the harsh conditions of nature.
“Even if the mosquitoes don’t get enough ivermectin to directly kill them, we think a sublethal dose should be sufficiently toxic to reduce malaria transmission,” Dr Foy said.
But he noted that because the goal is to interrupt malaria transmission, the drug must be taken by a majority of the people in a town or village, who then pass it along to the mosquitoes.
For the last few months, most of the population in the 4 villages in Burkina Faso has been receiving a single dose of ivermectin every 3 weeks. Individuals who are not eligible to take the drug include children less than 90 cm tall, pregnant women, and newly breastfeeding women.
The measure of success is a reduction in malaria incidence among children younger than 5, most of whom will not actually take the drug. But this is the age group most at risk of serious illness and death from the disease.
Thus far, the researchers have observed a roughly 16% reduction in childhood malaria episodes.
“These are preliminary results, but we expect to see further reductions in malaria fevers as we continue with the trial, which is occurring during the rainy season when malaria transmission typically peaks,” Dr Foy said.
“The drop in malaria fevers we’re seeing with the ivermectin treatment is in addition to whatever is being achieved with insecticide-treated bednets, which are in widespread use in all of the villages participating in the study.”
P vivax malaria
Dr Kobylinski and his colleagues found that ivermectin can affect P vivax parasites as well. The researchers tested the drug’s potential to block malaria transmission by feeding blood meals containing ivermectin and P vivax parasites to Anopheles dirus mosquitoes, the predominant malaria vector in Southeast Asia.
Ivermectin effectively killed A dirus mosquitoes, and it inhibited the ability of any surviving mosquitoes to develop P vivax parasites.
Dr Kobylinski noted that malaria experts are considering combining ivermectin with other medications in mass drug administration (MDA) campaigns that would seek to stop the spread of drug-resistant parasites in Southeast Asia by eliminating malaria from the entire region.
He said ivermectin could help increase compliance with an MDA strategy in places like Thailand, where drug-resistant malaria is spreading but overall malaria infection rates are relatively low.
“There is a lot of interest in launching MDA campaigns to fight drug-resistant malaria in Southeast Asia, but it can be hard to convince someone to take malaria medications if they don’t have an active malaria infection,” Dr Kobylinski said.
“But if you put ivermectin into the mix, that could improve participation because many people recognize the benefits of taking ivermectin for more common problems, like scabies.”
About ivermectin
Over the last 3 decades, more than 1 billion doses of ivermectin have been distributed in Africa and Latin America in MDA campaigns that have reduced the burden of lymphatic filariasis, which causes elephantiasis, and onchocerciasis, the disease that causes river blindness.
Ivermectin can also kill several types of debilitating intestinal worms known as soil-transmitted helminths.
Earlier this month, the Nobel Prize in Physiology or Medicine was awarded to a pair of researchers who isolated the precursor of ivermectin, avermectin, from an organism discovered in a single soil sample collected in Japan in the 1970s. ![]()
Image by Ute Frevert
and Margaret Shear
PHILADELPHIA—A drug used to treat multiple tropical diseases can also inhibit malaria transmission, according to a pair of studies presented at the ASTMH 64th Annual Meeting.
One study suggests the drug, ivermectin, can reduce the transmission of malaria caused by Plasmodium falciparum, the most prevalent malaria parasite in Africa.
The other study indicates that ivermectin can block the transmission of Plasmodium vivax parasites, which are common in Southeast Asia.
Kevin Kobylinski, PhD, of the Armed Forces Research Institute of Medical Sciences in Bangkok, Thailand, presented results of the P vivax study as abstract 1283.
And Brian D. Foy, PhD, of Colorado State University in Fort Collins, presented results of the P falciparum trial as abstract LB-5237.
P falciparum malaria
Dr Foy and his colleagues are assessing the ability of ivermectin to block malaria transmission in 4 villages in Burkina Faso, Africa.
Lab studies have shown that when mosquitoes feed on the blood of people who have taken ivermectin, it interferes with the mosquitoes’ ability to transmit malaria parasites to humans. Sometimes, it kills the mosquitoes outright, but, more often, it weakens them and interferes with their digestive system so they eventually die in the harsh conditions of nature.
“Even if the mosquitoes don’t get enough ivermectin to directly kill them, we think a sublethal dose should be sufficiently toxic to reduce malaria transmission,” Dr Foy said.
But he noted that because the goal is to interrupt malaria transmission, the drug must be taken by a majority of the people in a town or village, who then pass it along to the mosquitoes.
For the last few months, most of the population in the 4 villages in Burkina Faso has been receiving a single dose of ivermectin every 3 weeks. Individuals who are not eligible to take the drug include children less than 90 cm tall, pregnant women, and newly breastfeeding women.
The measure of success is a reduction in malaria incidence among children younger than 5, most of whom will not actually take the drug. But this is the age group most at risk of serious illness and death from the disease.
Thus far, the researchers have observed a roughly 16% reduction in childhood malaria episodes.
“These are preliminary results, but we expect to see further reductions in malaria fevers as we continue with the trial, which is occurring during the rainy season when malaria transmission typically peaks,” Dr Foy said.
“The drop in malaria fevers we’re seeing with the ivermectin treatment is in addition to whatever is being achieved with insecticide-treated bednets, which are in widespread use in all of the villages participating in the study.”
P vivax malaria
Dr Kobylinski and his colleagues found that ivermectin can affect P vivax parasites as well. The researchers tested the drug’s potential to block malaria transmission by feeding blood meals containing ivermectin and P vivax parasites to Anopheles dirus mosquitoes, the predominant malaria vector in Southeast Asia.
Ivermectin effectively killed A dirus mosquitoes, and it inhibited the ability of any surviving mosquitoes to develop P vivax parasites.
Dr Kobylinski noted that malaria experts are considering combining ivermectin with other medications in mass drug administration (MDA) campaigns that would seek to stop the spread of drug-resistant parasites in Southeast Asia by eliminating malaria from the entire region.
He said ivermectin could help increase compliance with an MDA strategy in places like Thailand, where drug-resistant malaria is spreading but overall malaria infection rates are relatively low.
“There is a lot of interest in launching MDA campaigns to fight drug-resistant malaria in Southeast Asia, but it can be hard to convince someone to take malaria medications if they don’t have an active malaria infection,” Dr Kobylinski said.
“But if you put ivermectin into the mix, that could improve participation because many people recognize the benefits of taking ivermectin for more common problems, like scabies.”
About ivermectin
Over the last 3 decades, more than 1 billion doses of ivermectin have been distributed in Africa and Latin America in MDA campaigns that have reduced the burden of lymphatic filariasis, which causes elephantiasis, and onchocerciasis, the disease that causes river blindness.
Ivermectin can also kill several types of debilitating intestinal worms known as soil-transmitted helminths.
Earlier this month, the Nobel Prize in Physiology or Medicine was awarded to a pair of researchers who isolated the precursor of ivermectin, avermectin, from an organism discovered in a single soil sample collected in Japan in the 1970s. ![]()
Image by Ute Frevert
and Margaret Shear
PHILADELPHIA—A drug used to treat multiple tropical diseases can also inhibit malaria transmission, according to a pair of studies presented at the ASTMH 64th Annual Meeting.
One study suggests the drug, ivermectin, can reduce the transmission of malaria caused by Plasmodium falciparum, the most prevalent malaria parasite in Africa.
The other study indicates that ivermectin can block the transmission of Plasmodium vivax parasites, which are common in Southeast Asia.
Kevin Kobylinski, PhD, of the Armed Forces Research Institute of Medical Sciences in Bangkok, Thailand, presented results of the P vivax study as abstract 1283.
And Brian D. Foy, PhD, of Colorado State University in Fort Collins, presented results of the P falciparum trial as abstract LB-5237.
P falciparum malaria
Dr Foy and his colleagues are assessing the ability of ivermectin to block malaria transmission in 4 villages in Burkina Faso, Africa.
Lab studies have shown that when mosquitoes feed on the blood of people who have taken ivermectin, it interferes with the mosquitoes’ ability to transmit malaria parasites to humans. Sometimes, it kills the mosquitoes outright, but, more often, it weakens them and interferes with their digestive system so they eventually die in the harsh conditions of nature.
“Even if the mosquitoes don’t get enough ivermectin to directly kill them, we think a sublethal dose should be sufficiently toxic to reduce malaria transmission,” Dr Foy said.
But he noted that because the goal is to interrupt malaria transmission, the drug must be taken by a majority of the people in a town or village, who then pass it along to the mosquitoes.
For the last few months, most of the population in the 4 villages in Burkina Faso has been receiving a single dose of ivermectin every 3 weeks. Individuals who are not eligible to take the drug include children less than 90 cm tall, pregnant women, and newly breastfeeding women.
The measure of success is a reduction in malaria incidence among children younger than 5, most of whom will not actually take the drug. But this is the age group most at risk of serious illness and death from the disease.
Thus far, the researchers have observed a roughly 16% reduction in childhood malaria episodes.
“These are preliminary results, but we expect to see further reductions in malaria fevers as we continue with the trial, which is occurring during the rainy season when malaria transmission typically peaks,” Dr Foy said.
“The drop in malaria fevers we’re seeing with the ivermectin treatment is in addition to whatever is being achieved with insecticide-treated bednets, which are in widespread use in all of the villages participating in the study.”
P vivax malaria
Dr Kobylinski and his colleagues found that ivermectin can affect P vivax parasites as well. The researchers tested the drug’s potential to block malaria transmission by feeding blood meals containing ivermectin and P vivax parasites to Anopheles dirus mosquitoes, the predominant malaria vector in Southeast Asia.
Ivermectin effectively killed A dirus mosquitoes, and it inhibited the ability of any surviving mosquitoes to develop P vivax parasites.
Dr Kobylinski noted that malaria experts are considering combining ivermectin with other medications in mass drug administration (MDA) campaigns that would seek to stop the spread of drug-resistant parasites in Southeast Asia by eliminating malaria from the entire region.
He said ivermectin could help increase compliance with an MDA strategy in places like Thailand, where drug-resistant malaria is spreading but overall malaria infection rates are relatively low.
“There is a lot of interest in launching MDA campaigns to fight drug-resistant malaria in Southeast Asia, but it can be hard to convince someone to take malaria medications if they don’t have an active malaria infection,” Dr Kobylinski said.
“But if you put ivermectin into the mix, that could improve participation because many people recognize the benefits of taking ivermectin for more common problems, like scabies.”
About ivermectin
Over the last 3 decades, more than 1 billion doses of ivermectin have been distributed in Africa and Latin America in MDA campaigns that have reduced the burden of lymphatic filariasis, which causes elephantiasis, and onchocerciasis, the disease that causes river blindness.
Ivermectin can also kill several types of debilitating intestinal worms known as soil-transmitted helminths.
Earlier this month, the Nobel Prize in Physiology or Medicine was awarded to a pair of researchers who isolated the precursor of ivermectin, avermectin, from an organism discovered in a single soil sample collected in Japan in the 1970s. ![]()
Acute Multiple Flexor Tendon Injury and Carpal Tunnel Syndrome After Open Distal Radius Fracture
The literature on extensor tendon rupture and even chronic flexor tendon rupture after volar plating and distal radius fracture malunion is ubiquitous. However, acute and subacute flexor tendon ruptures caused by distal radius fractures have been reported only in limited case reports. These rare injuries may involve multiple tendons and are associated with high-energy mechanisms. This case report details the involvement of multiple flexor tendon injuries associated with a Gustilo-Anderson type II distal radius fracture and the development of acute carpal tunnel syndrome (CTS) after a motor vehicle collision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient is a 46-year-old woman who was involved in a motor vehicle collision. She was triaged as a trauma patient via Advanced Trauma Life Support protocol, and treated with antibiotic and tetanus prophylaxis. Radiographs showed an open, comminuted, displaced intra-articular distal radius fracture on the right side (Figures 1A, 1B). The fracture was closed reduced and splinted in the emergency department (Figures 2A, 2B). On initial examination, the patient had diffuse paresthesias in the digits that were most pronounced in the median nerve distribution. Motor examination was limited secondary to pain; however, she demonstrated gentle flexion and extension of the digits. The hand was well perfused, and a palpable radial pulse was present.
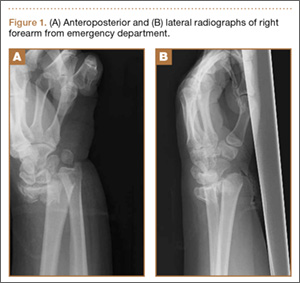
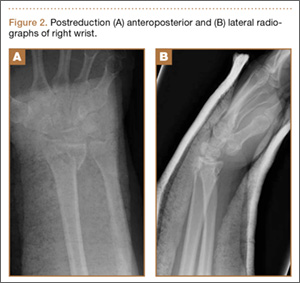
After clearance was obtained, she was taken urgently to the operating room. The wound was volar and transverse, approximately 2 cm in length, and approximately 4 cm proximal to the wrist crease. The wound was extended proximally and distally for a standard volar (Henry) approach. The flexor carpi radialis tendon was found to be partially lacerated, comprising 60% of the tendon. The fracture was readily identified because the deep fascia and the pronator quadratus were disrupted. No deep tendon lacerations were identified. The median nerve was found to be in continuity. After satisfactory débridement of the fracture and the wound, reduction and fixation was achieved with a volar locking plate and a single Kirschner wire. The flexor carpi radialis tendon was repaired with a modified Kessler stitch and epitenon repair. The wound was closed primarily in layers (Figures 3A, 3B).
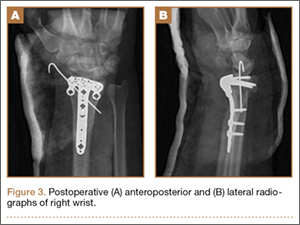
The patient’s immediate postoperative neurologic examination was compromised secondary to the patient having a supraclavicular nerve block for anesthesia. Regional anesthesia was chosen because the patient’s pulmonologist recommended avoiding general anesthesia owing to her history of severe asthma that frequently required corticosteroid treatment. Once the block wore off, she complained of persistent paresthesias in all digits but most pronounced in the median nerve distribution. She was able to flex the interphalangeal joint to the index finger but could not flex the interphalangeal joint to the thumb. Over the course of the night, she was also noted to have worsening pain out of proportion to her injury.
As the paresthesias became denser in the median nerve distribution, she was diagnosed with acute CTS and was taken urgently back to the operating room under general anesthesia. After releasing the carpal tunnel through a separate incision, the original wound was reopened and explored. The median nerve was again visualized and found to be in continuity. All 4 tendons to both the flexor digitorum superficialis and flexor digitorum profundus were identified. The flexor pollicis longus (FPL) was not visualized in the wound. The distal portion of the FPL was retracted in the thumb tendon sheath and retrieved blindly with a tendon passer. The proximal portion was retracted to the mid-forearm. The laceration occurred distal to the musculotendinous junction. The tendon was repaired with a modified Kessler stitch as well as a box suture, resulting in 4 core strands across the tendon. The hand and the wrist were splinted in a thumb spica cast, and the patient was started on a modified Duran protocol 1 week after surgery. Median nerve function improved postoperatively.
Discussion
The rupture of the extensor pollicis longus tendon in nondisplaced distal radius fractures is not uncommon, but occurs in fewer than 5% of nondisplaced distal radius fractures.1 Although less common, chronic complications with flexor tendon rupture after distal radius fracture are well described.1-6 Flexor tendon rupture after distal radius malunion or volar plating is a known complication and is thought to be the result of attritional tendon wear because the flexors rub against protruding bone or plate;3,4,7 however, the initial tendon injury may play a role in those tendons that rupture more quickly.3 When secondary to volar plating, the rupture typically occurs within 1 year of injury,7 but, in both plating and malunion, it has been characterized as a late complication up to 10 years and even 20 years after injury.3,4 Similar to other reports, this rupture was encountered during a volar wrist approach. It has been suggested that, as the incidence of volar plating rises, more acute flexor tendon injuries may be diagnosed because of anatomic exposure,2 but this has not been reported in the literature.
Acute and subacute flexor tendon ruptures are rarely reported in the literature. To our knowledge, there are only 2 other reports of acute flexor tendon rupture2,5 after a distal radius fracture, neither of which involved the FPL. These cases, which involved ruptures of the flexor digitorum superficialis and flexor carpi radialis, were thought to be the result of tendon laceration by a volar bone spike. There is also one report of subacute FPL and flexor digitorum profundus rupture approximately 4 weeks after closed reduction of a distal radius fracture.6 Although sparse, the literature regarding flexor tendon rupture and distal radius fractures suggests that involvement of the flexor digitorum superficialis and the flexor digitorum profundus tendons is most common and that the rupture typically occurs in 1 to 4 months.1
We report a rare case of 2 acute flexor tendon lacerations after a Gustilo-Anderson type II open distal radius fracture, likely caused by the volar spike of bone that created the open injury. This case also was complicated by the development of acute CTS.
To our knowledge, despite a rate of acute CTS reported as high as 5.4% in operatively treated distal radius fractures, there are no established associations between acute CTS and flexor tendon rupture in the setting of distal radius fracture.8,9 In a 2008 retrospective case–control study by Dyer and colleagues,8 fracture translation is the most important risk factor for the development of acute CTS associated with fracture of the distal radius. Although not statistically significant, ipsilateral upper extremity trauma, higher-energy injuries, younger age, and male sex were also associated with the development of acute CTS. Open injuries occurred in only 3 of 50 cases of acute CTS.8
In agreement with published reports, the probability and the timing of tendon rupture are likely related to the severity of the deforming forces applied during the initial insult rather than the resultant stresses.1 Clinicians should have a high suspicion of acute CTS and possible tendon injuries after a high-energy injury with a significantly displaced open distal radius fracture and median nerve paresthesias. A thoughtful and complete preoperative examination of the flexor tendons may prevent the need for reoperation. Concerns for flexor injury and acute CTS should be elevated with the observation of a disrupted pronator. For patients with a volarly displaced fragment after fracture reduction, this concern should be even more elevated.9 Preoperative median nerve symptoms in the setting of the severely displaced fracture should necessitate an acute carpal tunnel release. If 1 flexor tendon is injured, the surgeon should remember that multiple flexor tendons may be involved. We recommend that any injured tendons be repaired primarily, if possible, and the patient started on appropriate rehabilitation.
1. Ashall G. Flexor pollicis longus rupture after fracture of the distal radius. Injury. 1991;22(2):153-155.
2. Dimatteo L, Wolf JM. Flexor carpi radialis tendon rupture as a complication of a closed distal radius fracture: a case report. J Hand Surg Am. 2007;32(6):818-820.
3. Kato N, Nemoto K, Arino H, Ichikawa T, Fujikawa K. Ruptures of flexor tendons at the wrist as a complication of fracture of the distal radius. Scand J Plast Reconstr Surg Hand Surg. 2002;36(4):245-248.
4. Monda MK, Ellis A, Karmani S. Late rupture of flexor pollicis longus tendon 10 years after volar buttress plate fixation of a distal radius fracture: a case report. Acta Orthop Belg. 2010;76(4):549-551.
5. Southmayd WW, Millender LH, Nalebuff EA. Rupture of the flexor tendons of the index finger after Colles’ fracture. Case report. J Bone Joint Surg Am. 1975;57(4):562-563.
6. Wong FY, Pho RW. Median nerve compression, with tendon ruptures, after Colles’ fracture. J Hand Surg Br. 1984;9(2):139-141.
7. Woon CYL, Lee JYL, Ng SW, Teoh LC. Late rupture of flexor pollicis longus tendon after volar distal radius plating: a case report and review of the literature. Inj Extra. 2007;38(7):235-238.
8. Dyer G, Lozano-Calderon S, Gannon C, Baratz M, Ring D. Predictors of acute carpal tunnel syndrome associated with fracture of the distal radius. J Hand Surg Am. 2008;33(8):1309-1313.
9. Paley D, McMurtry RY. Median nerve compression by volarly displaced fragments of the distal radius. Clin Orthop Relat Res. 1987;(215):139-147.
The literature on extensor tendon rupture and even chronic flexor tendon rupture after volar plating and distal radius fracture malunion is ubiquitous. However, acute and subacute flexor tendon ruptures caused by distal radius fractures have been reported only in limited case reports. These rare injuries may involve multiple tendons and are associated with high-energy mechanisms. This case report details the involvement of multiple flexor tendon injuries associated with a Gustilo-Anderson type II distal radius fracture and the development of acute carpal tunnel syndrome (CTS) after a motor vehicle collision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient is a 46-year-old woman who was involved in a motor vehicle collision. She was triaged as a trauma patient via Advanced Trauma Life Support protocol, and treated with antibiotic and tetanus prophylaxis. Radiographs showed an open, comminuted, displaced intra-articular distal radius fracture on the right side (Figures 1A, 1B). The fracture was closed reduced and splinted in the emergency department (Figures 2A, 2B). On initial examination, the patient had diffuse paresthesias in the digits that were most pronounced in the median nerve distribution. Motor examination was limited secondary to pain; however, she demonstrated gentle flexion and extension of the digits. The hand was well perfused, and a palpable radial pulse was present.
After clearance was obtained, she was taken urgently to the operating room. The wound was volar and transverse, approximately 2 cm in length, and approximately 4 cm proximal to the wrist crease. The wound was extended proximally and distally for a standard volar (Henry) approach. The flexor carpi radialis tendon was found to be partially lacerated, comprising 60% of the tendon. The fracture was readily identified because the deep fascia and the pronator quadratus were disrupted. No deep tendon lacerations were identified. The median nerve was found to be in continuity. After satisfactory débridement of the fracture and the wound, reduction and fixation was achieved with a volar locking plate and a single Kirschner wire. The flexor carpi radialis tendon was repaired with a modified Kessler stitch and epitenon repair. The wound was closed primarily in layers (Figures 3A, 3B).
The patient’s immediate postoperative neurologic examination was compromised secondary to the patient having a supraclavicular nerve block for anesthesia. Regional anesthesia was chosen because the patient’s pulmonologist recommended avoiding general anesthesia owing to her history of severe asthma that frequently required corticosteroid treatment. Once the block wore off, she complained of persistent paresthesias in all digits but most pronounced in the median nerve distribution. She was able to flex the interphalangeal joint to the index finger but could not flex the interphalangeal joint to the thumb. Over the course of the night, she was also noted to have worsening pain out of proportion to her injury.
As the paresthesias became denser in the median nerve distribution, she was diagnosed with acute CTS and was taken urgently back to the operating room under general anesthesia. After releasing the carpal tunnel through a separate incision, the original wound was reopened and explored. The median nerve was again visualized and found to be in continuity. All 4 tendons to both the flexor digitorum superficialis and flexor digitorum profundus were identified. The flexor pollicis longus (FPL) was not visualized in the wound. The distal portion of the FPL was retracted in the thumb tendon sheath and retrieved blindly with a tendon passer. The proximal portion was retracted to the mid-forearm. The laceration occurred distal to the musculotendinous junction. The tendon was repaired with a modified Kessler stitch as well as a box suture, resulting in 4 core strands across the tendon. The hand and the wrist were splinted in a thumb spica cast, and the patient was started on a modified Duran protocol 1 week after surgery. Median nerve function improved postoperatively.
Discussion
The rupture of the extensor pollicis longus tendon in nondisplaced distal radius fractures is not uncommon, but occurs in fewer than 5% of nondisplaced distal radius fractures.1 Although less common, chronic complications with flexor tendon rupture after distal radius fracture are well described.1-6 Flexor tendon rupture after distal radius malunion or volar plating is a known complication and is thought to be the result of attritional tendon wear because the flexors rub against protruding bone or plate;3,4,7 however, the initial tendon injury may play a role in those tendons that rupture more quickly.3 When secondary to volar plating, the rupture typically occurs within 1 year of injury,7 but, in both plating and malunion, it has been characterized as a late complication up to 10 years and even 20 years after injury.3,4 Similar to other reports, this rupture was encountered during a volar wrist approach. It has been suggested that, as the incidence of volar plating rises, more acute flexor tendon injuries may be diagnosed because of anatomic exposure,2 but this has not been reported in the literature.
Acute and subacute flexor tendon ruptures are rarely reported in the literature. To our knowledge, there are only 2 other reports of acute flexor tendon rupture2,5 after a distal radius fracture, neither of which involved the FPL. These cases, which involved ruptures of the flexor digitorum superficialis and flexor carpi radialis, were thought to be the result of tendon laceration by a volar bone spike. There is also one report of subacute FPL and flexor digitorum profundus rupture approximately 4 weeks after closed reduction of a distal radius fracture.6 Although sparse, the literature regarding flexor tendon rupture and distal radius fractures suggests that involvement of the flexor digitorum superficialis and the flexor digitorum profundus tendons is most common and that the rupture typically occurs in 1 to 4 months.1
We report a rare case of 2 acute flexor tendon lacerations after a Gustilo-Anderson type II open distal radius fracture, likely caused by the volar spike of bone that created the open injury. This case also was complicated by the development of acute CTS.
To our knowledge, despite a rate of acute CTS reported as high as 5.4% in operatively treated distal radius fractures, there are no established associations between acute CTS and flexor tendon rupture in the setting of distal radius fracture.8,9 In a 2008 retrospective case–control study by Dyer and colleagues,8 fracture translation is the most important risk factor for the development of acute CTS associated with fracture of the distal radius. Although not statistically significant, ipsilateral upper extremity trauma, higher-energy injuries, younger age, and male sex were also associated with the development of acute CTS. Open injuries occurred in only 3 of 50 cases of acute CTS.8
In agreement with published reports, the probability and the timing of tendon rupture are likely related to the severity of the deforming forces applied during the initial insult rather than the resultant stresses.1 Clinicians should have a high suspicion of acute CTS and possible tendon injuries after a high-energy injury with a significantly displaced open distal radius fracture and median nerve paresthesias. A thoughtful and complete preoperative examination of the flexor tendons may prevent the need for reoperation. Concerns for flexor injury and acute CTS should be elevated with the observation of a disrupted pronator. For patients with a volarly displaced fragment after fracture reduction, this concern should be even more elevated.9 Preoperative median nerve symptoms in the setting of the severely displaced fracture should necessitate an acute carpal tunnel release. If 1 flexor tendon is injured, the surgeon should remember that multiple flexor tendons may be involved. We recommend that any injured tendons be repaired primarily, if possible, and the patient started on appropriate rehabilitation.
The literature on extensor tendon rupture and even chronic flexor tendon rupture after volar plating and distal radius fracture malunion is ubiquitous. However, acute and subacute flexor tendon ruptures caused by distal radius fractures have been reported only in limited case reports. These rare injuries may involve multiple tendons and are associated with high-energy mechanisms. This case report details the involvement of multiple flexor tendon injuries associated with a Gustilo-Anderson type II distal radius fracture and the development of acute carpal tunnel syndrome (CTS) after a motor vehicle collision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient is a 46-year-old woman who was involved in a motor vehicle collision. She was triaged as a trauma patient via Advanced Trauma Life Support protocol, and treated with antibiotic and tetanus prophylaxis. Radiographs showed an open, comminuted, displaced intra-articular distal radius fracture on the right side (Figures 1A, 1B). The fracture was closed reduced and splinted in the emergency department (Figures 2A, 2B). On initial examination, the patient had diffuse paresthesias in the digits that were most pronounced in the median nerve distribution. Motor examination was limited secondary to pain; however, she demonstrated gentle flexion and extension of the digits. The hand was well perfused, and a palpable radial pulse was present.
After clearance was obtained, she was taken urgently to the operating room. The wound was volar and transverse, approximately 2 cm in length, and approximately 4 cm proximal to the wrist crease. The wound was extended proximally and distally for a standard volar (Henry) approach. The flexor carpi radialis tendon was found to be partially lacerated, comprising 60% of the tendon. The fracture was readily identified because the deep fascia and the pronator quadratus were disrupted. No deep tendon lacerations were identified. The median nerve was found to be in continuity. After satisfactory débridement of the fracture and the wound, reduction and fixation was achieved with a volar locking plate and a single Kirschner wire. The flexor carpi radialis tendon was repaired with a modified Kessler stitch and epitenon repair. The wound was closed primarily in layers (Figures 3A, 3B).
The patient’s immediate postoperative neurologic examination was compromised secondary to the patient having a supraclavicular nerve block for anesthesia. Regional anesthesia was chosen because the patient’s pulmonologist recommended avoiding general anesthesia owing to her history of severe asthma that frequently required corticosteroid treatment. Once the block wore off, she complained of persistent paresthesias in all digits but most pronounced in the median nerve distribution. She was able to flex the interphalangeal joint to the index finger but could not flex the interphalangeal joint to the thumb. Over the course of the night, she was also noted to have worsening pain out of proportion to her injury.
As the paresthesias became denser in the median nerve distribution, she was diagnosed with acute CTS and was taken urgently back to the operating room under general anesthesia. After releasing the carpal tunnel through a separate incision, the original wound was reopened and explored. The median nerve was again visualized and found to be in continuity. All 4 tendons to both the flexor digitorum superficialis and flexor digitorum profundus were identified. The flexor pollicis longus (FPL) was not visualized in the wound. The distal portion of the FPL was retracted in the thumb tendon sheath and retrieved blindly with a tendon passer. The proximal portion was retracted to the mid-forearm. The laceration occurred distal to the musculotendinous junction. The tendon was repaired with a modified Kessler stitch as well as a box suture, resulting in 4 core strands across the tendon. The hand and the wrist were splinted in a thumb spica cast, and the patient was started on a modified Duran protocol 1 week after surgery. Median nerve function improved postoperatively.
Discussion
The rupture of the extensor pollicis longus tendon in nondisplaced distal radius fractures is not uncommon, but occurs in fewer than 5% of nondisplaced distal radius fractures.1 Although less common, chronic complications with flexor tendon rupture after distal radius fracture are well described.1-6 Flexor tendon rupture after distal radius malunion or volar plating is a known complication and is thought to be the result of attritional tendon wear because the flexors rub against protruding bone or plate;3,4,7 however, the initial tendon injury may play a role in those tendons that rupture more quickly.3 When secondary to volar plating, the rupture typically occurs within 1 year of injury,7 but, in both plating and malunion, it has been characterized as a late complication up to 10 years and even 20 years after injury.3,4 Similar to other reports, this rupture was encountered during a volar wrist approach. It has been suggested that, as the incidence of volar plating rises, more acute flexor tendon injuries may be diagnosed because of anatomic exposure,2 but this has not been reported in the literature.
Acute and subacute flexor tendon ruptures are rarely reported in the literature. To our knowledge, there are only 2 other reports of acute flexor tendon rupture2,5 after a distal radius fracture, neither of which involved the FPL. These cases, which involved ruptures of the flexor digitorum superficialis and flexor carpi radialis, were thought to be the result of tendon laceration by a volar bone spike. There is also one report of subacute FPL and flexor digitorum profundus rupture approximately 4 weeks after closed reduction of a distal radius fracture.6 Although sparse, the literature regarding flexor tendon rupture and distal radius fractures suggests that involvement of the flexor digitorum superficialis and the flexor digitorum profundus tendons is most common and that the rupture typically occurs in 1 to 4 months.1
We report a rare case of 2 acute flexor tendon lacerations after a Gustilo-Anderson type II open distal radius fracture, likely caused by the volar spike of bone that created the open injury. This case also was complicated by the development of acute CTS.
To our knowledge, despite a rate of acute CTS reported as high as 5.4% in operatively treated distal radius fractures, there are no established associations between acute CTS and flexor tendon rupture in the setting of distal radius fracture.8,9 In a 2008 retrospective case–control study by Dyer and colleagues,8 fracture translation is the most important risk factor for the development of acute CTS associated with fracture of the distal radius. Although not statistically significant, ipsilateral upper extremity trauma, higher-energy injuries, younger age, and male sex were also associated with the development of acute CTS. Open injuries occurred in only 3 of 50 cases of acute CTS.8
In agreement with published reports, the probability and the timing of tendon rupture are likely related to the severity of the deforming forces applied during the initial insult rather than the resultant stresses.1 Clinicians should have a high suspicion of acute CTS and possible tendon injuries after a high-energy injury with a significantly displaced open distal radius fracture and median nerve paresthesias. A thoughtful and complete preoperative examination of the flexor tendons may prevent the need for reoperation. Concerns for flexor injury and acute CTS should be elevated with the observation of a disrupted pronator. For patients with a volarly displaced fragment after fracture reduction, this concern should be even more elevated.9 Preoperative median nerve symptoms in the setting of the severely displaced fracture should necessitate an acute carpal tunnel release. If 1 flexor tendon is injured, the surgeon should remember that multiple flexor tendons may be involved. We recommend that any injured tendons be repaired primarily, if possible, and the patient started on appropriate rehabilitation.
1. Ashall G. Flexor pollicis longus rupture after fracture of the distal radius. Injury. 1991;22(2):153-155.
2. Dimatteo L, Wolf JM. Flexor carpi radialis tendon rupture as a complication of a closed distal radius fracture: a case report. J Hand Surg Am. 2007;32(6):818-820.
3. Kato N, Nemoto K, Arino H, Ichikawa T, Fujikawa K. Ruptures of flexor tendons at the wrist as a complication of fracture of the distal radius. Scand J Plast Reconstr Surg Hand Surg. 2002;36(4):245-248.
4. Monda MK, Ellis A, Karmani S. Late rupture of flexor pollicis longus tendon 10 years after volar buttress plate fixation of a distal radius fracture: a case report. Acta Orthop Belg. 2010;76(4):549-551.
5. Southmayd WW, Millender LH, Nalebuff EA. Rupture of the flexor tendons of the index finger after Colles’ fracture. Case report. J Bone Joint Surg Am. 1975;57(4):562-563.
6. Wong FY, Pho RW. Median nerve compression, with tendon ruptures, after Colles’ fracture. J Hand Surg Br. 1984;9(2):139-141.
7. Woon CYL, Lee JYL, Ng SW, Teoh LC. Late rupture of flexor pollicis longus tendon after volar distal radius plating: a case report and review of the literature. Inj Extra. 2007;38(7):235-238.
8. Dyer G, Lozano-Calderon S, Gannon C, Baratz M, Ring D. Predictors of acute carpal tunnel syndrome associated with fracture of the distal radius. J Hand Surg Am. 2008;33(8):1309-1313.
9. Paley D, McMurtry RY. Median nerve compression by volarly displaced fragments of the distal radius. Clin Orthop Relat Res. 1987;(215):139-147.
1. Ashall G. Flexor pollicis longus rupture after fracture of the distal radius. Injury. 1991;22(2):153-155.
2. Dimatteo L, Wolf JM. Flexor carpi radialis tendon rupture as a complication of a closed distal radius fracture: a case report. J Hand Surg Am. 2007;32(6):818-820.
3. Kato N, Nemoto K, Arino H, Ichikawa T, Fujikawa K. Ruptures of flexor tendons at the wrist as a complication of fracture of the distal radius. Scand J Plast Reconstr Surg Hand Surg. 2002;36(4):245-248.
4. Monda MK, Ellis A, Karmani S. Late rupture of flexor pollicis longus tendon 10 years after volar buttress plate fixation of a distal radius fracture: a case report. Acta Orthop Belg. 2010;76(4):549-551.
5. Southmayd WW, Millender LH, Nalebuff EA. Rupture of the flexor tendons of the index finger after Colles’ fracture. Case report. J Bone Joint Surg Am. 1975;57(4):562-563.
6. Wong FY, Pho RW. Median nerve compression, with tendon ruptures, after Colles’ fracture. J Hand Surg Br. 1984;9(2):139-141.
7. Woon CYL, Lee JYL, Ng SW, Teoh LC. Late rupture of flexor pollicis longus tendon after volar distal radius plating: a case report and review of the literature. Inj Extra. 2007;38(7):235-238.
8. Dyer G, Lozano-Calderon S, Gannon C, Baratz M, Ring D. Predictors of acute carpal tunnel syndrome associated with fracture of the distal radius. J Hand Surg Am. 2008;33(8):1309-1313.
9. Paley D, McMurtry RY. Median nerve compression by volarly displaced fragments of the distal radius. Clin Orthop Relat Res. 1987;(215):139-147.
Medicaid Insurance Is Associated With Larger Curves in Patients Who Require Scoliosis Surgery
Rising health care costs have led many health insurers to limit benefits, which may be a problem for children in need of specialty care. Uninsured children have poorer access to specialty care than insured children. Children with public health coverage have better access to specialty care than uninsured children but inferior access compared with privately insured children.1,2 It is well documented that children with government insurance have limited access to orthopedic care for fractures, ligamentous knee injuries, and other injuries.1,3-5 Adolescent idiopathic scoliosis (AIS) differs from many other conditions managed by pediatric orthopedists, as it may be progressive, with management becoming increasingly more complex as the curve magnitude increases.6 The ability to access care earlier in the disease process may allow for earlier nonoperative interventions, such as bracing. For patients who require spinal fusion, earlier diagnosis and referral to a specialist could potentially result in shorter fusions and preserve distal motion segments. The ability to access the health care system in a timely fashion would therefore be of utmost importance for patients with scoliosis.
The literature on AIS is lacking in studies focused on care access based on insurance coverage and the potential impact that this may have on curve progression.7-9 We conducted a study to determine whether there is a difference between patients with and without private insurance who present to a busy urban pediatric orthopedic practice for management of scoliosis that eventually resulted in surgical treatment.
Materials and Methods
After obtaining institutional review board approval for this study, we retrospectively reviewed the medical records of patients (age, 10-18 years) who underwent posterior spinal fusion (PSF) for newly diagnosed AIS between 2008 and 2012. We excluded patients treated with growing spine instrumentation (growing rods), patients younger than 10 years or older than 18 years at presentation, and patients without adequate radiographs or clinical data, including insurance status. To focus on newly diagnosed scoliosis, we also excluded patients who had been seen for second opinions or whose scoliosis had been managed elsewhere in the past. Patients with syndromic, neuromuscular, or congenital scoliosis were also excluded.
Medical records were checked to ascertain time from initial evaluation to decision for surgery, time from recommendation for surgery until actual procedure, and insurance status. Distance traveled was figured from patients’ home addresses. Cobb angles were calculated from initial preoperative and final preoperative posteroanterior (PA) radiographs. Curves as seen on PA, lateral, and maximal effort, supine bending thoracic and lumbar radiographs from the initial preoperative visit were classified using the system of Lenke and colleagues.10 Hospital records were queried to determine number of levels fused at surgery, number of implants placed, and length of stay. Patients were evaluated without prior screening of insurance status and without prior consultation with referring physicians. Surgical procedures were scheduled on a first-come, first-served basis without preference for insurance status.
Results
We identified 135 consecutive patients with newly diagnosed AIS treated with PSF by our group between January 2008 and December 2012 (Table 1). Sixty-one percent had private insurance; 39% had Medicaid. There was no difference in age or ASA (American Society of Anesthesiologists) score between groups. Mean (SD) Cobb angle at initial presentation was 47.5° (14.3°) (range, 18.0°-86.0°) for the private insurance group and 57.2° (15.7°) (range, 23.0°-95.0°) for the Medicaid group (P < .0001). At time of surgery, mean (SD) Cobb angles were 54.6° (11.7°) and 60.6° (13.9°) for the private insurance and Medicaid groups, respectively (P = .008). There was no difference in curve types (Lenke and colleagues10 classification) between groups (Table 2, P = .83). Medicaid patients traveled a shorter mean (SD) distance for care, 56.3 (57.0) miles, versus 73.7 (66.7) miles (P = .05). There was no statistical difference (P = .14) in mean (SD) surgical wait time from surgery recommendation to actual surgery, 103.1 (62.4) days and 128.8 (137.5) days for the private insurance and Medicaid groups, respectively. The difference between patient groups in mean (SD) number of levels fused did not reach statistical significance (P = .16), 10.3 (2.2) levels for the Medicaid group and 9.7 (2.3) levels for the private insurance group. Mean (SD) estimated blood loss was higher for Medicaid patients, 445.7 (415.9) mL versus 335.1 (271.5) mL (P = .06), though there was no difference in use of posterior column osteotomies between groups. There was no difference (P = .11) in mean (SD) length of hospital stay between Medicaid patients, 2.6 (0.8) days, and private insurance patients, 2.4 (0.5) days.
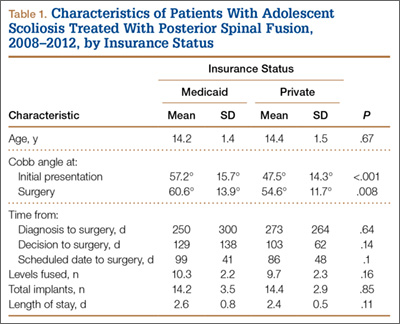
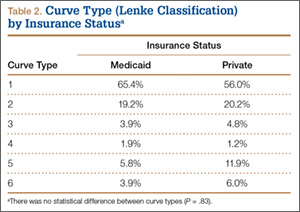
Discussion
According to an extensive body of literature, patients with government insurance have limited access to specialty care.1,11,12 Medicaid-insured children in need of orthopedic care are no exception. Sabharwal and colleagues13 examined a database of pediatric fracture cases and found that 52% of the privately insured patients and 22% of the publicly insured patients received orthopedic care (P = .013).13 When Pierce and colleagues14 called 42 orthopedic practices regarding a fictitious 14-year-old patient with an anterior cruciate ligament tear, 38 offered an appointment within 2 weeks to a privately insured patient, and 6 offered such an appointment to a publicly insured patient. Skaggs and colleagues4 surveyed 230 orthopedic practices nationally and found that Medicaid-insured children had limited access to orthopedic care; 41 practices (18%) would not see a child with Medicaid under any circumstances. Using a fictitious case of a 10-year-old boy with a forearm fracture, Iobst and colleagues3 tried making an appointment at 100 orthopedic offices. Eight gave an appointment within 1 week to a Medicaid-insured patient, and 36 gave an appointment to a privately insured patient.3
There are few data regarding insurance status and scoliosis care in children. Spinal deformity differs from simple fractures and ligamentous injuries, as timely care may result in a less invasive treatment (bracing) if the curvature is caught early. Goldstein and colleagues9 recently evaluated 642 patients who presented for scoliosis evaluation over a 10-year period. There was no difference in curve magnitudes between patients with and without Medicaid insurance. Thirty-two percent of these patients were evaluated for a second opinion, and the authors chose not to subdivide patients on the basis of curve severity and treatment needed, noting only no difference between groups. There was no discussion of the potential difference between patients with and without private insurance with respect to surgically versus nonsurgically treated curves. We wanted to focus specifically on patients who required surgical intervention, as our experience has been that many patients with government insurance present with either very mild scoliosis (10°) or very large curves that were not identified because of lack of primary care access or inadequate school screening. Although summing these 2 groups would result in a similar average, they would represent a different cohort than patients with curves along a bell curve. Furthermore, it is the group of patients who would require surgical intervention that is so critical to identify early in order to intervene.
Our data suggest a difference in presenting curves between patients with and without private insurance. The approximately 10° difference between patient groups in this study could potentially represent the difference between bracing and surgery. Furthermore, Miyanji and colleagues6 evaluated the relationship between Cobb angle and health care consumption and correlated larger curve magnitudes with more levels fused, longer surgeries, and higher rates of transfusion. Specifically, every 10° increase in curve magnitude resulted in 7.8 more minutes of operative time, 0.3 extra levels fused, and 1.5 times increased risk for requiring a blood transfusion.
Cho and Egorova15 recently evaluated insurance status with respect to surgical outcomes using a national inpatient database and found that 42.4% of surgeries for AIS in children with Medicaid had fusions involving 9 or more levels, whereas only 33.6% of privately insured patients had fusions of 9 or more levels. There was no difference in osteotomy or reoperation for pseudarthrosis between groups, but there was a slightly higher rate of infectious (1.1% vs 0.6%) and hemorrhagic (2.5% vs 1.7%) complications in the Medicaid group. Hospital stay was longer in patients with Medicaid, though complications were not different between groups.
The mean difference in the magnitude of the curves treated in our study was not more than 10° between patients with and without Medicaid, perhaps explaining the lack of a statistically significant difference in number of levels fused between groups. Although the groups were similar with respect to the percentage requiring posterior column spinal osteotomies, we noted a difference in estimated blood loss between groups, likely explained by the fact that a junior surgeon was added just before initiation of the study period, potentially skewing the estimated blood loss as this surgeon gained experience. Payer status has been correlated to length of hospital stay in children with scoliosis. Vitale and colleagues8 reviewed the effect of payer status on surgical outcomes in 3606 scoliosis patients from a statewide database in California and concluded that, compared with patients having all other payment sources, Medicaid patients had higher odds for complications and longer hospital stay. Our hospital has adopted a highly coordinated care pathway that allows for discharge on postoperative day 2, likely explaining the lack of any difference in postoperative stay.16
The disparity in curve magnitudes among patients with and without private insurance is striking and probably multifactorial. Very likely, the combination of schools with limited screening programs within urban or rural school systems,17 restricted access to pediatricians,18,19 and longer waits to see orthopedic specialists20 all contribute to this disparity. It should be noted that school screening is mandatory in our state. This discrepancy may be related to a previously established tendency in minority populations toward waiting longer to seek care and refusing surgical recommendations, though we were unable to query socioeconomic factors such as race and household income.21,22 It is clearly important to increase access to care for underinsured patients with scoliosis. A comprehensive approach, including providing better education in the schools, establishing communication with referring primary care providers, and increasing access through more physicians or physician extenders, is likely needed. Orthopedists should perhaps treat scoliosis evaluation with the same sense of urgency given to minor fractures, and primary care providers should try to ensure that appropriate referrals for scoliosis are made. Also curious was the shorter travel distance for Medicaid patients versus private insurance patients in this study. We hypothesize this is related to our urban location and its large Medicaid population.
Our study had several limitations. Our electronic medical records (EMR) system does not store data related to the time a patient calls for an initial appointment, limiting our ability to determine how long patients waited for their initial consultation. Furthermore, the decision to undergo surgery is multifactorial and cannot be simplified into time from initial recommendation to surgery, as some patients delay surgery because of school or other obligations. These data should be reasonably consistent over time, as patients seen in the early spring in both groups may delay surgery until the summer, and those diagnosed in June may prefer earlier surgery.
Summary
Children with AIS are at risk for curve progression. Therefore, delays in providing timely care may result in worsening scoliosis. Compared with private insurance patients, Medicaid patients presented with larger curve magnitudes. Further study is needed to better delineate ways to improve care access for patients with scoliosis in communities with larger Medicaid populations.
1. Skaggs DL. Less access to care for children with Medicaid. Orthopedics. 2003;26(12):1184, 1186.
2. Skinner AC, Mayer ML. Effects of insurance status on children’s access to specialty care: a systematic review of the literature. BMC Health Serv Res. 2007;7:194.
3. Iobst C, King W, Baitner A, Tidwell M, Swirsky S, Skaggs DL. Access to care for children with fractures. J Pediatr Orthop. 2010;30(3):244-247.
4. Skaggs DL, Lehmann CL, Rice C, et al. Access to orthopaedic care for children with Medicaid versus private insurance: results of a national survey. J Pediatr Orthop. 2006;26(3):400-404.
5. Skaggs DL, Oda JE, Lerman L, et al. Insurance status and delay in orthotic treatment in children. J Pediatr Orthop. 2007;27(1):94-97.
6. Miyanji F, Slobogean GP, Samdani AF, et al. Is larger scoliosis curve magnitude associated with increased perioperative health-care resource utilization? A multicenter analysis of 325 adolescent idiopathic scoliosis curves. J Bone Joint Surg Am. 2012;94(9):809-813.
7. Nuno M, Drazin DG, Acosta FL Jr. Differences in treatments and outcomes for idiopathic scoliosis patients treated in the United States from 1998 to 2007: impact of socioeconomic variables and ethnicity. Spine J. 2013;13(2):116-123.
8. Vitale MA, Arons RR, Hyman JE, Skaggs DL, Roye DP, Vitale MG. The contribution of hospital volume, payer status, and other factors on the surgical outcomes of scoliosis patients: a review of 3,606 cases in the state of California. J Pediatr Orthop. 2005;25(3):393-399.
9. Goldstein RY, Joiner ER, Skaggs DL. Insurance status does not predict curve magnitude in adolescent idiopathic scoliosis at first presentation to an orthopaedic surgeon. J Pediatr Orthop. 2015;35(1):39-42.
10. Lenke LG, Betz RR, Harms J, et al. Adolescent idiopathic scoliosis: a new classification to determine extent of spinal arthrodesis. J Bone Joint Surg Am. 2001;83(8):1169-1181.
11. Alosh H, Riley LH 3rd, Skolasky RL. Insurance status, geography, race, and ethnicity as predictors of anterior cervical spine surgery rates and in-hospital mortality: an examination of United States trends from 1992 to 2005. Spine. 2009;34(18):1956-1962.
12. Newacheck PW, Hughes DC, Hung YY, Wong S, Stoddard JJ. The unmet health needs of America’s children. Pediatrics. 2000;105(4 pt 2):989-997.
13. Sabharwal S, Zhao C, McClemens E, Kaufmann A. Pediatric orthopaedic patients presenting to a university emergency department after visiting another emergency department: demographics and health insurance status. J Pediatr Orthop. 2007;27(6):690-694.
14. Pierce TR, Mehlman CT, Tamai J, Skaggs DL. Access to care for the adolescent anterior cruciate ligament patient with Medicaid versus private insurance. J Pediatr Orthop. 2012;32(3):245-248.
15. Cho SK, Egorova NN. The association between insurance status and complications, length of stay, and costs for pediatric idiopathic scoliosis. Spine. 2015;40(4):247-256.
16. Fletcher ND, Shourbaji N, Mitchell PM, Oswald TS, Devito DP, Bruce RW Jr. Clinical and economic implications of early discharge following posterior spinal fusion for adolescent idiopathic scoliosis. J Child Orthop. 2014;8(3):257-263.
17. Kasper MJ, Robbins L, Root L, Peterson MG, Allegrante JP. A musculoskeletal outreach screening, treatment, and education program for urban minority children. Arthritis Care Res. 1993;6(3):126-133.
18. Berman S, Dolins J, Tang SF, Yudkowsky B. Factors that influence the willingness of private primary care pediatricians to accept more Medicaid patients. Pediatrics. 2002;110(2 pt 1):239-248.
19. Sommers BD. Protecting low-income children’s access to care: are physician visits associated with reduced patient dropout from Medicaid and the Children’s Health Insurance Program? Pediatrics. 2006;118(1):e36-e42.
20. Bisgaier J, Polsky D, Rhodes KV. Academic medical centers and equity in specialty care access for children. Arch Pediatr Adolesc Med. 2012;166(4):304-310.
21. Sedlis SP, Fisher VJ, Tice D, Esposito R, Madmon L, Steinberg EH. Racial differences in performance of invasive cardiac procedures in a Department of Veterans Affairs medical center. J Clin Epidemiol. 1997;50(8):899-901.
22. Mitchell JB, McCormack LA. Time trends in late-stage diagnosis of cervical cancer. Differences by race/ethnicity and income. Med Care. 1997;35(12):1220-1224.
Rising health care costs have led many health insurers to limit benefits, which may be a problem for children in need of specialty care. Uninsured children have poorer access to specialty care than insured children. Children with public health coverage have better access to specialty care than uninsured children but inferior access compared with privately insured children.1,2 It is well documented that children with government insurance have limited access to orthopedic care for fractures, ligamentous knee injuries, and other injuries.1,3-5 Adolescent idiopathic scoliosis (AIS) differs from many other conditions managed by pediatric orthopedists, as it may be progressive, with management becoming increasingly more complex as the curve magnitude increases.6 The ability to access care earlier in the disease process may allow for earlier nonoperative interventions, such as bracing. For patients who require spinal fusion, earlier diagnosis and referral to a specialist could potentially result in shorter fusions and preserve distal motion segments. The ability to access the health care system in a timely fashion would therefore be of utmost importance for patients with scoliosis.
The literature on AIS is lacking in studies focused on care access based on insurance coverage and the potential impact that this may have on curve progression.7-9 We conducted a study to determine whether there is a difference between patients with and without private insurance who present to a busy urban pediatric orthopedic practice for management of scoliosis that eventually resulted in surgical treatment.
Materials and Methods
After obtaining institutional review board approval for this study, we retrospectively reviewed the medical records of patients (age, 10-18 years) who underwent posterior spinal fusion (PSF) for newly diagnosed AIS between 2008 and 2012. We excluded patients treated with growing spine instrumentation (growing rods), patients younger than 10 years or older than 18 years at presentation, and patients without adequate radiographs or clinical data, including insurance status. To focus on newly diagnosed scoliosis, we also excluded patients who had been seen for second opinions or whose scoliosis had been managed elsewhere in the past. Patients with syndromic, neuromuscular, or congenital scoliosis were also excluded.
Medical records were checked to ascertain time from initial evaluation to decision for surgery, time from recommendation for surgery until actual procedure, and insurance status. Distance traveled was figured from patients’ home addresses. Cobb angles were calculated from initial preoperative and final preoperative posteroanterior (PA) radiographs. Curves as seen on PA, lateral, and maximal effort, supine bending thoracic and lumbar radiographs from the initial preoperative visit were classified using the system of Lenke and colleagues.10 Hospital records were queried to determine number of levels fused at surgery, number of implants placed, and length of stay. Patients were evaluated without prior screening of insurance status and without prior consultation with referring physicians. Surgical procedures were scheduled on a first-come, first-served basis without preference for insurance status.
Results
We identified 135 consecutive patients with newly diagnosed AIS treated with PSF by our group between January 2008 and December 2012 (Table 1). Sixty-one percent had private insurance; 39% had Medicaid. There was no difference in age or ASA (American Society of Anesthesiologists) score between groups. Mean (SD) Cobb angle at initial presentation was 47.5° (14.3°) (range, 18.0°-86.0°) for the private insurance group and 57.2° (15.7°) (range, 23.0°-95.0°) for the Medicaid group (P < .0001). At time of surgery, mean (SD) Cobb angles were 54.6° (11.7°) and 60.6° (13.9°) for the private insurance and Medicaid groups, respectively (P = .008). There was no difference in curve types (Lenke and colleagues10 classification) between groups (Table 2, P = .83). Medicaid patients traveled a shorter mean (SD) distance for care, 56.3 (57.0) miles, versus 73.7 (66.7) miles (P = .05). There was no statistical difference (P = .14) in mean (SD) surgical wait time from surgery recommendation to actual surgery, 103.1 (62.4) days and 128.8 (137.5) days for the private insurance and Medicaid groups, respectively. The difference between patient groups in mean (SD) number of levels fused did not reach statistical significance (P = .16), 10.3 (2.2) levels for the Medicaid group and 9.7 (2.3) levels for the private insurance group. Mean (SD) estimated blood loss was higher for Medicaid patients, 445.7 (415.9) mL versus 335.1 (271.5) mL (P = .06), though there was no difference in use of posterior column osteotomies between groups. There was no difference (P = .11) in mean (SD) length of hospital stay between Medicaid patients, 2.6 (0.8) days, and private insurance patients, 2.4 (0.5) days.
Discussion
According to an extensive body of literature, patients with government insurance have limited access to specialty care.1,11,12 Medicaid-insured children in need of orthopedic care are no exception. Sabharwal and colleagues13 examined a database of pediatric fracture cases and found that 52% of the privately insured patients and 22% of the publicly insured patients received orthopedic care (P = .013).13 When Pierce and colleagues14 called 42 orthopedic practices regarding a fictitious 14-year-old patient with an anterior cruciate ligament tear, 38 offered an appointment within 2 weeks to a privately insured patient, and 6 offered such an appointment to a publicly insured patient. Skaggs and colleagues4 surveyed 230 orthopedic practices nationally and found that Medicaid-insured children had limited access to orthopedic care; 41 practices (18%) would not see a child with Medicaid under any circumstances. Using a fictitious case of a 10-year-old boy with a forearm fracture, Iobst and colleagues3 tried making an appointment at 100 orthopedic offices. Eight gave an appointment within 1 week to a Medicaid-insured patient, and 36 gave an appointment to a privately insured patient.3
There are few data regarding insurance status and scoliosis care in children. Spinal deformity differs from simple fractures and ligamentous injuries, as timely care may result in a less invasive treatment (bracing) if the curvature is caught early. Goldstein and colleagues9 recently evaluated 642 patients who presented for scoliosis evaluation over a 10-year period. There was no difference in curve magnitudes between patients with and without Medicaid insurance. Thirty-two percent of these patients were evaluated for a second opinion, and the authors chose not to subdivide patients on the basis of curve severity and treatment needed, noting only no difference between groups. There was no discussion of the potential difference between patients with and without private insurance with respect to surgically versus nonsurgically treated curves. We wanted to focus specifically on patients who required surgical intervention, as our experience has been that many patients with government insurance present with either very mild scoliosis (10°) or very large curves that were not identified because of lack of primary care access or inadequate school screening. Although summing these 2 groups would result in a similar average, they would represent a different cohort than patients with curves along a bell curve. Furthermore, it is the group of patients who would require surgical intervention that is so critical to identify early in order to intervene.
Our data suggest a difference in presenting curves between patients with and without private insurance. The approximately 10° difference between patient groups in this study could potentially represent the difference between bracing and surgery. Furthermore, Miyanji and colleagues6 evaluated the relationship between Cobb angle and health care consumption and correlated larger curve magnitudes with more levels fused, longer surgeries, and higher rates of transfusion. Specifically, every 10° increase in curve magnitude resulted in 7.8 more minutes of operative time, 0.3 extra levels fused, and 1.5 times increased risk for requiring a blood transfusion.
Cho and Egorova15 recently evaluated insurance status with respect to surgical outcomes using a national inpatient database and found that 42.4% of surgeries for AIS in children with Medicaid had fusions involving 9 or more levels, whereas only 33.6% of privately insured patients had fusions of 9 or more levels. There was no difference in osteotomy or reoperation for pseudarthrosis between groups, but there was a slightly higher rate of infectious (1.1% vs 0.6%) and hemorrhagic (2.5% vs 1.7%) complications in the Medicaid group. Hospital stay was longer in patients with Medicaid, though complications were not different between groups.
The mean difference in the magnitude of the curves treated in our study was not more than 10° between patients with and without Medicaid, perhaps explaining the lack of a statistically significant difference in number of levels fused between groups. Although the groups were similar with respect to the percentage requiring posterior column spinal osteotomies, we noted a difference in estimated blood loss between groups, likely explained by the fact that a junior surgeon was added just before initiation of the study period, potentially skewing the estimated blood loss as this surgeon gained experience. Payer status has been correlated to length of hospital stay in children with scoliosis. Vitale and colleagues8 reviewed the effect of payer status on surgical outcomes in 3606 scoliosis patients from a statewide database in California and concluded that, compared with patients having all other payment sources, Medicaid patients had higher odds for complications and longer hospital stay. Our hospital has adopted a highly coordinated care pathway that allows for discharge on postoperative day 2, likely explaining the lack of any difference in postoperative stay.16
The disparity in curve magnitudes among patients with and without private insurance is striking and probably multifactorial. Very likely, the combination of schools with limited screening programs within urban or rural school systems,17 restricted access to pediatricians,18,19 and longer waits to see orthopedic specialists20 all contribute to this disparity. It should be noted that school screening is mandatory in our state. This discrepancy may be related to a previously established tendency in minority populations toward waiting longer to seek care and refusing surgical recommendations, though we were unable to query socioeconomic factors such as race and household income.21,22 It is clearly important to increase access to care for underinsured patients with scoliosis. A comprehensive approach, including providing better education in the schools, establishing communication with referring primary care providers, and increasing access through more physicians or physician extenders, is likely needed. Orthopedists should perhaps treat scoliosis evaluation with the same sense of urgency given to minor fractures, and primary care providers should try to ensure that appropriate referrals for scoliosis are made. Also curious was the shorter travel distance for Medicaid patients versus private insurance patients in this study. We hypothesize this is related to our urban location and its large Medicaid population.
Our study had several limitations. Our electronic medical records (EMR) system does not store data related to the time a patient calls for an initial appointment, limiting our ability to determine how long patients waited for their initial consultation. Furthermore, the decision to undergo surgery is multifactorial and cannot be simplified into time from initial recommendation to surgery, as some patients delay surgery because of school or other obligations. These data should be reasonably consistent over time, as patients seen in the early spring in both groups may delay surgery until the summer, and those diagnosed in June may prefer earlier surgery.
Summary
Children with AIS are at risk for curve progression. Therefore, delays in providing timely care may result in worsening scoliosis. Compared with private insurance patients, Medicaid patients presented with larger curve magnitudes. Further study is needed to better delineate ways to improve care access for patients with scoliosis in communities with larger Medicaid populations.
Rising health care costs have led many health insurers to limit benefits, which may be a problem for children in need of specialty care. Uninsured children have poorer access to specialty care than insured children. Children with public health coverage have better access to specialty care than uninsured children but inferior access compared with privately insured children.1,2 It is well documented that children with government insurance have limited access to orthopedic care for fractures, ligamentous knee injuries, and other injuries.1,3-5 Adolescent idiopathic scoliosis (AIS) differs from many other conditions managed by pediatric orthopedists, as it may be progressive, with management becoming increasingly more complex as the curve magnitude increases.6 The ability to access care earlier in the disease process may allow for earlier nonoperative interventions, such as bracing. For patients who require spinal fusion, earlier diagnosis and referral to a specialist could potentially result in shorter fusions and preserve distal motion segments. The ability to access the health care system in a timely fashion would therefore be of utmost importance for patients with scoliosis.
The literature on AIS is lacking in studies focused on care access based on insurance coverage and the potential impact that this may have on curve progression.7-9 We conducted a study to determine whether there is a difference between patients with and without private insurance who present to a busy urban pediatric orthopedic practice for management of scoliosis that eventually resulted in surgical treatment.
Materials and Methods
After obtaining institutional review board approval for this study, we retrospectively reviewed the medical records of patients (age, 10-18 years) who underwent posterior spinal fusion (PSF) for newly diagnosed AIS between 2008 and 2012. We excluded patients treated with growing spine instrumentation (growing rods), patients younger than 10 years or older than 18 years at presentation, and patients without adequate radiographs or clinical data, including insurance status. To focus on newly diagnosed scoliosis, we also excluded patients who had been seen for second opinions or whose scoliosis had been managed elsewhere in the past. Patients with syndromic, neuromuscular, or congenital scoliosis were also excluded.
Medical records were checked to ascertain time from initial evaluation to decision for surgery, time from recommendation for surgery until actual procedure, and insurance status. Distance traveled was figured from patients’ home addresses. Cobb angles were calculated from initial preoperative and final preoperative posteroanterior (PA) radiographs. Curves as seen on PA, lateral, and maximal effort, supine bending thoracic and lumbar radiographs from the initial preoperative visit were classified using the system of Lenke and colleagues.10 Hospital records were queried to determine number of levels fused at surgery, number of implants placed, and length of stay. Patients were evaluated without prior screening of insurance status and without prior consultation with referring physicians. Surgical procedures were scheduled on a first-come, first-served basis without preference for insurance status.
Results
We identified 135 consecutive patients with newly diagnosed AIS treated with PSF by our group between January 2008 and December 2012 (Table 1). Sixty-one percent had private insurance; 39% had Medicaid. There was no difference in age or ASA (American Society of Anesthesiologists) score between groups. Mean (SD) Cobb angle at initial presentation was 47.5° (14.3°) (range, 18.0°-86.0°) for the private insurance group and 57.2° (15.7°) (range, 23.0°-95.0°) for the Medicaid group (P < .0001). At time of surgery, mean (SD) Cobb angles were 54.6° (11.7°) and 60.6° (13.9°) for the private insurance and Medicaid groups, respectively (P = .008). There was no difference in curve types (Lenke and colleagues10 classification) between groups (Table 2, P = .83). Medicaid patients traveled a shorter mean (SD) distance for care, 56.3 (57.0) miles, versus 73.7 (66.7) miles (P = .05). There was no statistical difference (P = .14) in mean (SD) surgical wait time from surgery recommendation to actual surgery, 103.1 (62.4) days and 128.8 (137.5) days for the private insurance and Medicaid groups, respectively. The difference between patient groups in mean (SD) number of levels fused did not reach statistical significance (P = .16), 10.3 (2.2) levels for the Medicaid group and 9.7 (2.3) levels for the private insurance group. Mean (SD) estimated blood loss was higher for Medicaid patients, 445.7 (415.9) mL versus 335.1 (271.5) mL (P = .06), though there was no difference in use of posterior column osteotomies between groups. There was no difference (P = .11) in mean (SD) length of hospital stay between Medicaid patients, 2.6 (0.8) days, and private insurance patients, 2.4 (0.5) days.
Discussion
According to an extensive body of literature, patients with government insurance have limited access to specialty care.1,11,12 Medicaid-insured children in need of orthopedic care are no exception. Sabharwal and colleagues13 examined a database of pediatric fracture cases and found that 52% of the privately insured patients and 22% of the publicly insured patients received orthopedic care (P = .013).13 When Pierce and colleagues14 called 42 orthopedic practices regarding a fictitious 14-year-old patient with an anterior cruciate ligament tear, 38 offered an appointment within 2 weeks to a privately insured patient, and 6 offered such an appointment to a publicly insured patient. Skaggs and colleagues4 surveyed 230 orthopedic practices nationally and found that Medicaid-insured children had limited access to orthopedic care; 41 practices (18%) would not see a child with Medicaid under any circumstances. Using a fictitious case of a 10-year-old boy with a forearm fracture, Iobst and colleagues3 tried making an appointment at 100 orthopedic offices. Eight gave an appointment within 1 week to a Medicaid-insured patient, and 36 gave an appointment to a privately insured patient.3
There are few data regarding insurance status and scoliosis care in children. Spinal deformity differs from simple fractures and ligamentous injuries, as timely care may result in a less invasive treatment (bracing) if the curvature is caught early. Goldstein and colleagues9 recently evaluated 642 patients who presented for scoliosis evaluation over a 10-year period. There was no difference in curve magnitudes between patients with and without Medicaid insurance. Thirty-two percent of these patients were evaluated for a second opinion, and the authors chose not to subdivide patients on the basis of curve severity and treatment needed, noting only no difference between groups. There was no discussion of the potential difference between patients with and without private insurance with respect to surgically versus nonsurgically treated curves. We wanted to focus specifically on patients who required surgical intervention, as our experience has been that many patients with government insurance present with either very mild scoliosis (10°) or very large curves that were not identified because of lack of primary care access or inadequate school screening. Although summing these 2 groups would result in a similar average, they would represent a different cohort than patients with curves along a bell curve. Furthermore, it is the group of patients who would require surgical intervention that is so critical to identify early in order to intervene.
Our data suggest a difference in presenting curves between patients with and without private insurance. The approximately 10° difference between patient groups in this study could potentially represent the difference between bracing and surgery. Furthermore, Miyanji and colleagues6 evaluated the relationship between Cobb angle and health care consumption and correlated larger curve magnitudes with more levels fused, longer surgeries, and higher rates of transfusion. Specifically, every 10° increase in curve magnitude resulted in 7.8 more minutes of operative time, 0.3 extra levels fused, and 1.5 times increased risk for requiring a blood transfusion.
Cho and Egorova15 recently evaluated insurance status with respect to surgical outcomes using a national inpatient database and found that 42.4% of surgeries for AIS in children with Medicaid had fusions involving 9 or more levels, whereas only 33.6% of privately insured patients had fusions of 9 or more levels. There was no difference in osteotomy or reoperation for pseudarthrosis between groups, but there was a slightly higher rate of infectious (1.1% vs 0.6%) and hemorrhagic (2.5% vs 1.7%) complications in the Medicaid group. Hospital stay was longer in patients with Medicaid, though complications were not different between groups.
The mean difference in the magnitude of the curves treated in our study was not more than 10° between patients with and without Medicaid, perhaps explaining the lack of a statistically significant difference in number of levels fused between groups. Although the groups were similar with respect to the percentage requiring posterior column spinal osteotomies, we noted a difference in estimated blood loss between groups, likely explained by the fact that a junior surgeon was added just before initiation of the study period, potentially skewing the estimated blood loss as this surgeon gained experience. Payer status has been correlated to length of hospital stay in children with scoliosis. Vitale and colleagues8 reviewed the effect of payer status on surgical outcomes in 3606 scoliosis patients from a statewide database in California and concluded that, compared with patients having all other payment sources, Medicaid patients had higher odds for complications and longer hospital stay. Our hospital has adopted a highly coordinated care pathway that allows for discharge on postoperative day 2, likely explaining the lack of any difference in postoperative stay.16
The disparity in curve magnitudes among patients with and without private insurance is striking and probably multifactorial. Very likely, the combination of schools with limited screening programs within urban or rural school systems,17 restricted access to pediatricians,18,19 and longer waits to see orthopedic specialists20 all contribute to this disparity. It should be noted that school screening is mandatory in our state. This discrepancy may be related to a previously established tendency in minority populations toward waiting longer to seek care and refusing surgical recommendations, though we were unable to query socioeconomic factors such as race and household income.21,22 It is clearly important to increase access to care for underinsured patients with scoliosis. A comprehensive approach, including providing better education in the schools, establishing communication with referring primary care providers, and increasing access through more physicians or physician extenders, is likely needed. Orthopedists should perhaps treat scoliosis evaluation with the same sense of urgency given to minor fractures, and primary care providers should try to ensure that appropriate referrals for scoliosis are made. Also curious was the shorter travel distance for Medicaid patients versus private insurance patients in this study. We hypothesize this is related to our urban location and its large Medicaid population.
Our study had several limitations. Our electronic medical records (EMR) system does not store data related to the time a patient calls for an initial appointment, limiting our ability to determine how long patients waited for their initial consultation. Furthermore, the decision to undergo surgery is multifactorial and cannot be simplified into time from initial recommendation to surgery, as some patients delay surgery because of school or other obligations. These data should be reasonably consistent over time, as patients seen in the early spring in both groups may delay surgery until the summer, and those diagnosed in June may prefer earlier surgery.
Summary
Children with AIS are at risk for curve progression. Therefore, delays in providing timely care may result in worsening scoliosis. Compared with private insurance patients, Medicaid patients presented with larger curve magnitudes. Further study is needed to better delineate ways to improve care access for patients with scoliosis in communities with larger Medicaid populations.
1. Skaggs DL. Less access to care for children with Medicaid. Orthopedics. 2003;26(12):1184, 1186.
2. Skinner AC, Mayer ML. Effects of insurance status on children’s access to specialty care: a systematic review of the literature. BMC Health Serv Res. 2007;7:194.
3. Iobst C, King W, Baitner A, Tidwell M, Swirsky S, Skaggs DL. Access to care for children with fractures. J Pediatr Orthop. 2010;30(3):244-247.
4. Skaggs DL, Lehmann CL, Rice C, et al. Access to orthopaedic care for children with Medicaid versus private insurance: results of a national survey. J Pediatr Orthop. 2006;26(3):400-404.
5. Skaggs DL, Oda JE, Lerman L, et al. Insurance status and delay in orthotic treatment in children. J Pediatr Orthop. 2007;27(1):94-97.
6. Miyanji F, Slobogean GP, Samdani AF, et al. Is larger scoliosis curve magnitude associated with increased perioperative health-care resource utilization? A multicenter analysis of 325 adolescent idiopathic scoliosis curves. J Bone Joint Surg Am. 2012;94(9):809-813.
7. Nuno M, Drazin DG, Acosta FL Jr. Differences in treatments and outcomes for idiopathic scoliosis patients treated in the United States from 1998 to 2007: impact of socioeconomic variables and ethnicity. Spine J. 2013;13(2):116-123.
8. Vitale MA, Arons RR, Hyman JE, Skaggs DL, Roye DP, Vitale MG. The contribution of hospital volume, payer status, and other factors on the surgical outcomes of scoliosis patients: a review of 3,606 cases in the state of California. J Pediatr Orthop. 2005;25(3):393-399.
9. Goldstein RY, Joiner ER, Skaggs DL. Insurance status does not predict curve magnitude in adolescent idiopathic scoliosis at first presentation to an orthopaedic surgeon. J Pediatr Orthop. 2015;35(1):39-42.
10. Lenke LG, Betz RR, Harms J, et al. Adolescent idiopathic scoliosis: a new classification to determine extent of spinal arthrodesis. J Bone Joint Surg Am. 2001;83(8):1169-1181.
11. Alosh H, Riley LH 3rd, Skolasky RL. Insurance status, geography, race, and ethnicity as predictors of anterior cervical spine surgery rates and in-hospital mortality: an examination of United States trends from 1992 to 2005. Spine. 2009;34(18):1956-1962.
12. Newacheck PW, Hughes DC, Hung YY, Wong S, Stoddard JJ. The unmet health needs of America’s children. Pediatrics. 2000;105(4 pt 2):989-997.
13. Sabharwal S, Zhao C, McClemens E, Kaufmann A. Pediatric orthopaedic patients presenting to a university emergency department after visiting another emergency department: demographics and health insurance status. J Pediatr Orthop. 2007;27(6):690-694.
14. Pierce TR, Mehlman CT, Tamai J, Skaggs DL. Access to care for the adolescent anterior cruciate ligament patient with Medicaid versus private insurance. J Pediatr Orthop. 2012;32(3):245-248.
15. Cho SK, Egorova NN. The association between insurance status and complications, length of stay, and costs for pediatric idiopathic scoliosis. Spine. 2015;40(4):247-256.
16. Fletcher ND, Shourbaji N, Mitchell PM, Oswald TS, Devito DP, Bruce RW Jr. Clinical and economic implications of early discharge following posterior spinal fusion for adolescent idiopathic scoliosis. J Child Orthop. 2014;8(3):257-263.
17. Kasper MJ, Robbins L, Root L, Peterson MG, Allegrante JP. A musculoskeletal outreach screening, treatment, and education program for urban minority children. Arthritis Care Res. 1993;6(3):126-133.
18. Berman S, Dolins J, Tang SF, Yudkowsky B. Factors that influence the willingness of private primary care pediatricians to accept more Medicaid patients. Pediatrics. 2002;110(2 pt 1):239-248.
19. Sommers BD. Protecting low-income children’s access to care: are physician visits associated with reduced patient dropout from Medicaid and the Children’s Health Insurance Program? Pediatrics. 2006;118(1):e36-e42.
20. Bisgaier J, Polsky D, Rhodes KV. Academic medical centers and equity in specialty care access for children. Arch Pediatr Adolesc Med. 2012;166(4):304-310.
21. Sedlis SP, Fisher VJ, Tice D, Esposito R, Madmon L, Steinberg EH. Racial differences in performance of invasive cardiac procedures in a Department of Veterans Affairs medical center. J Clin Epidemiol. 1997;50(8):899-901.
22. Mitchell JB, McCormack LA. Time trends in late-stage diagnosis of cervical cancer. Differences by race/ethnicity and income. Med Care. 1997;35(12):1220-1224.
1. Skaggs DL. Less access to care for children with Medicaid. Orthopedics. 2003;26(12):1184, 1186.
2. Skinner AC, Mayer ML. Effects of insurance status on children’s access to specialty care: a systematic review of the literature. BMC Health Serv Res. 2007;7:194.
3. Iobst C, King W, Baitner A, Tidwell M, Swirsky S, Skaggs DL. Access to care for children with fractures. J Pediatr Orthop. 2010;30(3):244-247.
4. Skaggs DL, Lehmann CL, Rice C, et al. Access to orthopaedic care for children with Medicaid versus private insurance: results of a national survey. J Pediatr Orthop. 2006;26(3):400-404.
5. Skaggs DL, Oda JE, Lerman L, et al. Insurance status and delay in orthotic treatment in children. J Pediatr Orthop. 2007;27(1):94-97.
6. Miyanji F, Slobogean GP, Samdani AF, et al. Is larger scoliosis curve magnitude associated with increased perioperative health-care resource utilization? A multicenter analysis of 325 adolescent idiopathic scoliosis curves. J Bone Joint Surg Am. 2012;94(9):809-813.
7. Nuno M, Drazin DG, Acosta FL Jr. Differences in treatments and outcomes for idiopathic scoliosis patients treated in the United States from 1998 to 2007: impact of socioeconomic variables and ethnicity. Spine J. 2013;13(2):116-123.
8. Vitale MA, Arons RR, Hyman JE, Skaggs DL, Roye DP, Vitale MG. The contribution of hospital volume, payer status, and other factors on the surgical outcomes of scoliosis patients: a review of 3,606 cases in the state of California. J Pediatr Orthop. 2005;25(3):393-399.
9. Goldstein RY, Joiner ER, Skaggs DL. Insurance status does not predict curve magnitude in adolescent idiopathic scoliosis at first presentation to an orthopaedic surgeon. J Pediatr Orthop. 2015;35(1):39-42.
10. Lenke LG, Betz RR, Harms J, et al. Adolescent idiopathic scoliosis: a new classification to determine extent of spinal arthrodesis. J Bone Joint Surg Am. 2001;83(8):1169-1181.
11. Alosh H, Riley LH 3rd, Skolasky RL. Insurance status, geography, race, and ethnicity as predictors of anterior cervical spine surgery rates and in-hospital mortality: an examination of United States trends from 1992 to 2005. Spine. 2009;34(18):1956-1962.
12. Newacheck PW, Hughes DC, Hung YY, Wong S, Stoddard JJ. The unmet health needs of America’s children. Pediatrics. 2000;105(4 pt 2):989-997.
13. Sabharwal S, Zhao C, McClemens E, Kaufmann A. Pediatric orthopaedic patients presenting to a university emergency department after visiting another emergency department: demographics and health insurance status. J Pediatr Orthop. 2007;27(6):690-694.
14. Pierce TR, Mehlman CT, Tamai J, Skaggs DL. Access to care for the adolescent anterior cruciate ligament patient with Medicaid versus private insurance. J Pediatr Orthop. 2012;32(3):245-248.
15. Cho SK, Egorova NN. The association between insurance status and complications, length of stay, and costs for pediatric idiopathic scoliosis. Spine. 2015;40(4):247-256.
16. Fletcher ND, Shourbaji N, Mitchell PM, Oswald TS, Devito DP, Bruce RW Jr. Clinical and economic implications of early discharge following posterior spinal fusion for adolescent idiopathic scoliosis. J Child Orthop. 2014;8(3):257-263.
17. Kasper MJ, Robbins L, Root L, Peterson MG, Allegrante JP. A musculoskeletal outreach screening, treatment, and education program for urban minority children. Arthritis Care Res. 1993;6(3):126-133.
18. Berman S, Dolins J, Tang SF, Yudkowsky B. Factors that influence the willingness of private primary care pediatricians to accept more Medicaid patients. Pediatrics. 2002;110(2 pt 1):239-248.
19. Sommers BD. Protecting low-income children’s access to care: are physician visits associated with reduced patient dropout from Medicaid and the Children’s Health Insurance Program? Pediatrics. 2006;118(1):e36-e42.
20. Bisgaier J, Polsky D, Rhodes KV. Academic medical centers and equity in specialty care access for children. Arch Pediatr Adolesc Med. 2012;166(4):304-310.
21. Sedlis SP, Fisher VJ, Tice D, Esposito R, Madmon L, Steinberg EH. Racial differences in performance of invasive cardiac procedures in a Department of Veterans Affairs medical center. J Clin Epidemiol. 1997;50(8):899-901.
22. Mitchell JB, McCormack LA. Time trends in late-stage diagnosis of cervical cancer. Differences by race/ethnicity and income. Med Care. 1997;35(12):1220-1224.
Risk Factors for Discharge to Rehabilitation Among Hip Fracture Patients
Length of stay (LOS) is a significant driver of costs after hip fracture surgery.1-3 Multiple studies have identified factors associated with increased LOS in hip fracture patients. These factors include admission time, delay to surgery, presence of comorbidities, and older age.4-9
One significant and potentially modifiable factor affecting LOS is delayed transfer to a rehabilitation center after surgery.8-11 Although patients after orthopedic surgeries require additional rehabilitation services or subacute care directly attributable to their injuries, specialized rehabilitation centers may not always have beds readily available.6-11 Studies have shown that delays in transfer to skilled nursing facilities or rehabilitation centers are highly common among orthopedic patients.8 It is therefore imperative that orthopedists have a mechanism for predicting and identifying which patients require rehabilitation services early in the postoperative period. Identifying risk factors and stratifying patients who are most likely to require rehabilitation would facilitate the early transfer of these patients and thereby directly decrease LOS and hospitalization-related costs.
In this article, we report results from prospective, national, multicenter data to identify commonly measured risk factors for discharge to rehabilitation facilities for hip fracture patients. Through multivariate analysis of ACS-NSQIP (American College of Surgeons National Surgical Quality Improvement Program) data, we determined which risk factors significantly predispose patients to discharge to rehabilitation centers versus discharge home. Knowledge of these risk factors allows the practicing orthopedist to be better equipped to identify patients who require additional rehabilitation early in the postoperative course. By mobilizing case managers and social workers to help avoid delays in the transfers of these identified patients, LOS-associated costs may ultimately decrease.
Materials and Methods
After obtaining institutional review board approval for this study from the Office of Research at Vanderbilt University, we prospectively collected 2011 discharge data from the ACS-NSQIP database (these data are unavailable for earlier years). All patients who underwent hip fracture surgery in 2011 were identified by CPT (Current Procedural Terminology) codes. Cases of patients with unknown discharge information and of those who died during their hospitalizations were excluded from analysis. For the remaining patients, discharge information as categorized by ACS-NSQIP included skilled care (eg, subacute hospital, skilled nursing home), unskilled facility (eg, nursing home, assisted facility), separate acute care, and rehabilitation. All other patients were discharged home without additional assistance or to the previous home where they received chronic care, assisted living, or unskilled aid. Patients were dichotomized according to whether they were discharged home or to one of the rehabilitation facilities mentioned.
To determine which risk factors significantly contributed to a patient’s discharge to rehabilitation, we ran univariate analyses using Fisher exact tests for categorical variables and Student t tests for continuous variables on multiple patient factors, including demographics, preoperative comorbidities, and operative factors. Demographics included age and sex. Preoperative comorbidities included 32 conditions: diabetes mellitus, active smoking status, current alcohol use, dyspnea, history of chronic obstructive pulmonary disease, history of congestive heart failure, hypertension requiring medication, history of esophageal varices, history of myocardial infarction, current renal failure, current dialysis dependence, steroid use, recent weight loss, existing bleeding disorder, transfusion before discharge, presence of central nervous system tumor, recent chemotherapy, recent radiation therapy, previous percutaneous coronary intervention, previous percutaneous coronary stenting, history of angina, peripheral vascular disease, cerebrovascular accidents, recent surgery (within 30 days), rest pain, impaired sensorium, history of transient ischemic attacks, current hemiplegia status, current paraplegia status, current quadriplegia status, current ascites, hypertension, and disseminated cancer. Operative factors included wound infection, DNR (do not resuscitate) status, ventilator support, anesthesia type, wound class, ASA (American Society of Anesthesiologists) class, and operative time.
For the univariate analyses, significance was set at P < .05. Demographics, preoperative comorbidities, and operative factors that were significantly associated with discharge to a rehabilitation facility in the univariate analysis were selected as covariates for a multivariate analysis. We incorporated a binary logistic regression to analyze which of these significant risk factors are correlated with a patient’s discharge to a rehabilitation facility after hip fracture surgery.
Results
A total of 4974 patients undergoing surgery for hip fractures in 2011 were identified. Of these patients, 4815 had complete information on discharge location and were included in the analysis.
Table 1 lists the results of the univariate analysis comparing demographics, preoperative comorbidities, and operative factors between the home and rehabilitation groups. Both age (P < .001) and sex (P = .012) were significantly different between groups; the rehabilitation group was older by about 10 years and included significantly more females. In addition to demographic factors, 16 preoperative comorbidities, and 5 surgical factors were significantly associated with discharge to rehabilitation.
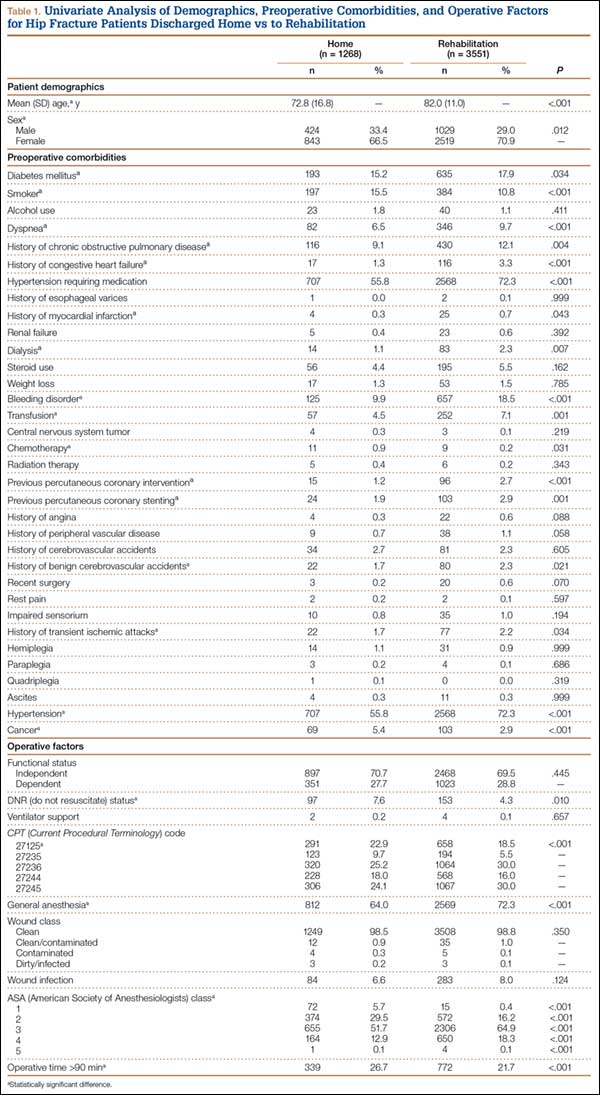
Surgery type significantly affected discharge to rehabilitation (Figure). Patients who were undergoing open plating of a femoral neck fracture or intramedullary nailing of an intertrochanteric, peritrochanteric, or subtrochanteric femoral fracture constituted 30% of all patients discharged to rehabilitation centers. In contrast, patients undergoing percutaneous skeletal fixation of a proximal femoral fracture constituted only 5.5% of all patients discharged to rehabilitation. Based on surgery type, we broke down discharge location further, into categories of skilled nursing facility, unskilled facility (not patient’s previous home), separate acute-care facility, dedicated rehabilitation center, and home. Of all 4815 patients combined, 2102 (43.6%) were discharged to a skilled nursing facility, 31 (0.6%) to an unskilled facility (not home), 106 (2.2%) to separate acute care, 1312 (27.2%) to a dedicated rehabilitation center, and 950 (19.7%) home.
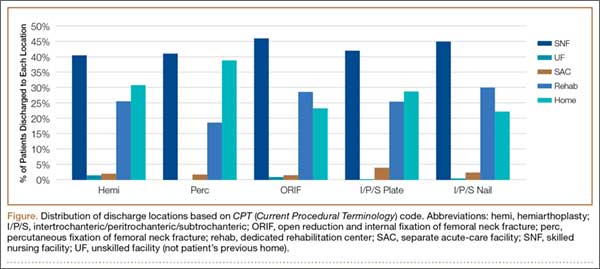
Table 2 lists the significant results from the multivariate logistical analysis comparing discharge to a rehabilitation center and discharge home after controlling for the significant risk factors (Table 1). Current diabetes, history of dyspnea, previous myocardial infarction, history of ischemic attacks, current bleeding disorder, transfusion during hospitalization, previous percutaneous cardiac stenting, chemotherapy, past cerebrovascular accident, presence of cancer, surgery type based on CPT code, history of chronic obstructive pulmonary disease or congestive heart failure, current smoking status, and operative time longer than 90 minutes were not significantly correlated with discharge to rehabilitation in the multivariate analysis. All significant factors were associated with higher odds of discharge to rehabilitation except for DNR status. DNR patients were 2.04 times more likely (95% CI, 1.49-2.78; P < .001) to be discharged home than to rehabilitation centers.
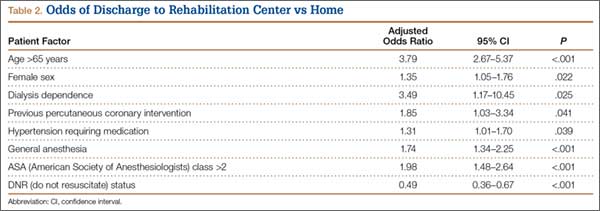
Applying these adjusted odds ratios, we see that an elderly woman (age, >65 years) who underwent general anesthesia with an ASA class higher than 2 was 17.63 times more likely than a patient without these risk factors to be discharged to rehabilitation. If this patient were also dialysis-dependent, she would be 61.52 times more likely than a similar patient without dialysis needs to be discharged to rehabilitation.
Even when controlling for all significant and nonsignificant variables in multivariate logistical analysis, age over 65 years (β = 1.05; P < .001), female sex (β = 1.76; P = .004), dialysis dependence (β = 12.98; P = .036), hypertension requiring medication (β = 1.53; P = .032), and ASA class higher than 2 (β = 1.98; P = .001) were found to be significant risk factors for discharge to rehabilitation.
Discussion
This study was the first to investigate the issue of which patient risk factors allow the practicing orthopedist to identify patients who require rehabilitation after hip fracture surgery. Through our multivariate analysis, which controlled for demographics, comorbidities, and operative factors, we found that older age, female sex, history of percutaneous coronary intervention, dialysis dependence, general anesthesia, and ASA class higher than 2 significantly increased the odds of discharge to a rehabilitation center versus home.
Using our study’s results, we can create a risk stratification model for patients and thereby a means of targeting patients who need rehabilitation and starting the process of finding a rehabilitation bed early in the postoperative course. Our study’s variables are easily measured metrics that may be collected in any hospital setting. Especially for hip fracture patients, early planning and discharge to the appropriate rehabilitation center are important in decreasing LOS and associated hospitalization costs. According to one report,3 about 85% of all hip fracture costs are directly related to LOS, given the unnecessarily long rehabilitation periods in hospitals. Hollingworth and colleagues2 compared costs for patients who remained in the hospital with costs for those discharged with rehabilitation services. Overall costs were significantly lower for patients discharged home with rehabilitation. The authors concluded that 40% of hip fracture patients may be suitable for early discharge.2 In an analysis of Medicare payments for hip fracture treatment, hospital costs including LOS accounted for 60% of all payments.12 The results of these 2 studies suggest that the overall driver of hip fracture costs is prolonged LOS and that, if patients are discharged to rehabilitation, then overall costs may be lowered through a direct reduction in hospital LOS. Given that hip fractures account for almost 350,000 hospital admissions in the United States each year, and using our institution’s average hospital charge per day ($4500), about $1.6 billion may be saved if each patient’s LOS decreased by 1 day.13 Although multiple factors affect LOS, discharge planning is under orthopedists’ direct control. Therefore, early identification of patients who will require rehabilitation may help reduce LOS-associated costs in our health care system.
The patient variables that were significantly associated with discharge to rehabilitation are also associated with increased morbidity and mortality in hip fracture patients, according to the literature,14-20 which provides some external validation of using these risk factors as predictors for rehabilitation. A patient with one of these risk factors may require rehabilitation, given that rehabilitation services are specifically linked to lower morbidity and mortality rates among hip fracture patients. For example, patients with dialysis needs were 3.49 times more likely to be discharged to a rehabilitation center in our study. In a 2000 study by Coco and Rush,16 hip fracture patients on dialysis had a 1-year mortality rate 2.5 times higher than that of patients who were not dialysis-dependent. In 2010, Cameron and colleagues17 found that cardiovascular disease was associated with a 2.68 times higher risk of mortality in hip fracture patients. Similarly in our study, both hypertension and history of percutaneous coronary intervention were associated with discharge to rehabilitation. We found higher odds of discharge to rehabilitation with higher ASA classes, which mirror results from a study by Michel and colleagues,15 who found that higher (vs lower) preoperative ASA classes were associated with higher 1-year mortality in hip fracture patients. Interestingly, DNR status was associated with higher odds of discharge home, which may reflect patients’ desires to forgo noninvasive or lifesaving procedures that may be performed at rehabilitation facilities. Although general anesthesia predisposed patients to discharge to a rehabilitation center, multiple studies have found no association between anesthesia type and postoperative mortality rates for hip fracture patients.18,19 Last, Marcantonio and colleagues20 found delirium specifically had a higher odds ratio for discharge, but our univariate analysis did not find a significant association between impaired sensorium and discharge location. Given the correlation of our risk factors with increased morbidity and mortality in the literature, our study’s results provide the initial groundwork for creating a risk calculator that orthopedists can use to predict discharge to rehabilitation.
Our study had some limitations. Although we analyzed a large number of demographics, preoperative comorbidities, and surgical factors, our univariate analysis was limited to information in the ACS-NSQIP database. We did not incorporate other clinically relevant factors (eg, social factors, including patients’ support networks) that may influence discharge decisions. Furthermore, ACS-NSQIP records patient data only up to 30 days after surgery. Discharge information for the time after that was missing for a subset of hip fracture patients, and these patients had to be excluded, potentially skewing our data. ACS-NSQIP also does not collect cost data for patients based on hospitalization or LOS, so we could not determine whether patients discharged to rehabilitation incurred higher costs because of longer hospitalizations.
Nevertheless, our study identified significant patient and operative variables that are associated with discharge to a rehabilitation center. By identifying hip fracture patients with these risk factors early and mobilizing the appropriate resources, practicing orthopedists should be better equipped to help facilitate the discharge of patients to the appropriate location after surgery. Validation of these risk factors should be prospectively determined with an analysis of LOS and cost implications. Use of a risk calculator may in fact result in decreased LOS and hospital-related costs. Furthermore, using these risk factors in a prospective patient cohort would help validate their use and determine whether there is clinical correlation. The orthopedists in our institution are becoming more aware of these risk factors, but validation is necessary.
1. Garcia AE, Bonnaig JV, Yoneda ZT, et al. Patient variables which may predict length of stay and hospital costs in elderly patients with hip fracture. J Orthop Trauma. 2012;26(11):620-623.
2. Hollingworth W, Todd C, Parker M, Roberts JA, Williams R. Cost analysis of early discharge after hip fracture. BMJ. 1993;307(6909):903-906.
3. Sund R, Riihimäki J, Mäkelä M, et al. Modeling the length of the care episode after hip fracture: does the type of fracture matter? Scand J Surg. 2009;98(3):169-174.
4. Fox KM, Magaziner J, Hebel JR, Kenzora JE, Kashner TM. Intertrochanteric versus femoral neck hip fractures: differential characteristics, treatment, and sequelae. J Gerontol A Biol Sci Med Sci. 1999;54(12):M635-M640.
5. Foss NB, Palm H, Krasheninnikoff M, Kehlet H, Gebuhr P. Impact of surgical complications on length of stay after hip fracture surgery. Injury. 2007;38(7):780-784.
6. Lefaivre KA, Macadam SA, Davidson DJ, Gandhi R, Chan H, Broekhuyse HM. Length of stay, mortality, morbidity and delay to surgery in hip fractures. J Bone Joint Surg Br. 2009;91(7):922-927.
7. Clague JE, Craddock E, Andrew G, Horan MA, Pendleton N. Predictors of outcome following hip fracture. Admission time predicts length of stay and in-hospital mortality. Injury. 2002;33(1):1-6.
8. Parker MJ, Todd CJ, Palmer CR, et al. Inter-hospital variations in length of hospital stay following hip fracture. Age Ageing. 1998;27(31):333-337.
9. Brasel KJ, Rasmussen J, Cauley C, Weigelt JA. Reasons for delayed discharge of trauma patients. J Surg Res. 2002;107(2):223-226.
10. Bonar SK, Tinetti ME, Speechley M, Cooney LM. Factors associated with short- versus long-term skilled nursing facility placement among community-living hip fracture patients. J Am Geriatr Soc. 1990;38(10):1139-1144.
11. Bentler SE, Liu L, Obrizan M, et al. The aftermath of hip fracture: discharge placement, functional status change, and mortality. Am J Epidemiol. 2009;170(10):1290-1299.
12. Birkmeyer JD, Gust C, Baser O, Dimick JB, Sutherland JM, Skinner JS. Medicare payments for common inpatient procedures: implications for episode-based payment bundling. Health Serv Res. 2010;45(6 pt 1):1783-1795.
13. American Academy of Orthopaedic Surgeons. Burden of Musculoskeletal Diseases in the United States: Prevalence, Societal and Economic Cost. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2008.
14. Maciejewski ML, Radcliff A, Henderson WG, et al. Determinants of postsurgical discharge setting for male hip fracture patients. J Rehabil Res Dev. 2013;50(9):1267-1276.
15. Michel JP, Klopfenstein C, Hoffmeyer P, Stern R, Grab B. Hip fracture surgery: is the pre-operative American Society of Anesthesiologists (ASA) score a predictor of functional outcome? Aging Clin Exp Res. 2002;14(5):389-394.
16. Coco M, Rush H. Increased incidence of hip fractures in dialysis patients with low serum parathyroid hormone. Am J Kidney Dis. 2000;36(6):1115-1121.
17. Cameron ID, Chen JS, March LM, et al. Hip fracture causes excess mortality owing to cardiovascular and infectious disease in institutionalized older people: a prospective 5-year study. J Bone Miner Res. 2010;25(4):866-872.
18. White SM, Moppett IK, Griffiths R. Outcome by mode of anaesthesia for hip fracture surgery. An observational audit of 65 535 patients in a national dataset. Anaesthesia. 2014;69(3):224-230.
19. Le-Wendling L, Bihorac A, Baslanti TO, et al. Regional anesthesia as compared with general anesthesia for surgery in geriatric patients with hip fracture: does it decrease morbidity, mortality, and health care costs? Results of a single-centered study. Pain Med. 2012;13(7):948-956.
20. Marcantonio ER, Flacker JM, Michaels M, Resnick NM. Delirium is independently associated with poor functional recovery after hip fracture. J Am Geriatr Soc. 2000;48(6):618-624.
Length of stay (LOS) is a significant driver of costs after hip fracture surgery.1-3 Multiple studies have identified factors associated with increased LOS in hip fracture patients. These factors include admission time, delay to surgery, presence of comorbidities, and older age.4-9
One significant and potentially modifiable factor affecting LOS is delayed transfer to a rehabilitation center after surgery.8-11 Although patients after orthopedic surgeries require additional rehabilitation services or subacute care directly attributable to their injuries, specialized rehabilitation centers may not always have beds readily available.6-11 Studies have shown that delays in transfer to skilled nursing facilities or rehabilitation centers are highly common among orthopedic patients.8 It is therefore imperative that orthopedists have a mechanism for predicting and identifying which patients require rehabilitation services early in the postoperative period. Identifying risk factors and stratifying patients who are most likely to require rehabilitation would facilitate the early transfer of these patients and thereby directly decrease LOS and hospitalization-related costs.
In this article, we report results from prospective, national, multicenter data to identify commonly measured risk factors for discharge to rehabilitation facilities for hip fracture patients. Through multivariate analysis of ACS-NSQIP (American College of Surgeons National Surgical Quality Improvement Program) data, we determined which risk factors significantly predispose patients to discharge to rehabilitation centers versus discharge home. Knowledge of these risk factors allows the practicing orthopedist to be better equipped to identify patients who require additional rehabilitation early in the postoperative course. By mobilizing case managers and social workers to help avoid delays in the transfers of these identified patients, LOS-associated costs may ultimately decrease.
Materials and Methods
After obtaining institutional review board approval for this study from the Office of Research at Vanderbilt University, we prospectively collected 2011 discharge data from the ACS-NSQIP database (these data are unavailable for earlier years). All patients who underwent hip fracture surgery in 2011 were identified by CPT (Current Procedural Terminology) codes. Cases of patients with unknown discharge information and of those who died during their hospitalizations were excluded from analysis. For the remaining patients, discharge information as categorized by ACS-NSQIP included skilled care (eg, subacute hospital, skilled nursing home), unskilled facility (eg, nursing home, assisted facility), separate acute care, and rehabilitation. All other patients were discharged home without additional assistance or to the previous home where they received chronic care, assisted living, or unskilled aid. Patients were dichotomized according to whether they were discharged home or to one of the rehabilitation facilities mentioned.
To determine which risk factors significantly contributed to a patient’s discharge to rehabilitation, we ran univariate analyses using Fisher exact tests for categorical variables and Student t tests for continuous variables on multiple patient factors, including demographics, preoperative comorbidities, and operative factors. Demographics included age and sex. Preoperative comorbidities included 32 conditions: diabetes mellitus, active smoking status, current alcohol use, dyspnea, history of chronic obstructive pulmonary disease, history of congestive heart failure, hypertension requiring medication, history of esophageal varices, history of myocardial infarction, current renal failure, current dialysis dependence, steroid use, recent weight loss, existing bleeding disorder, transfusion before discharge, presence of central nervous system tumor, recent chemotherapy, recent radiation therapy, previous percutaneous coronary intervention, previous percutaneous coronary stenting, history of angina, peripheral vascular disease, cerebrovascular accidents, recent surgery (within 30 days), rest pain, impaired sensorium, history of transient ischemic attacks, current hemiplegia status, current paraplegia status, current quadriplegia status, current ascites, hypertension, and disseminated cancer. Operative factors included wound infection, DNR (do not resuscitate) status, ventilator support, anesthesia type, wound class, ASA (American Society of Anesthesiologists) class, and operative time.
For the univariate analyses, significance was set at P < .05. Demographics, preoperative comorbidities, and operative factors that were significantly associated with discharge to a rehabilitation facility in the univariate analysis were selected as covariates for a multivariate analysis. We incorporated a binary logistic regression to analyze which of these significant risk factors are correlated with a patient’s discharge to a rehabilitation facility after hip fracture surgery.
Results
A total of 4974 patients undergoing surgery for hip fractures in 2011 were identified. Of these patients, 4815 had complete information on discharge location and were included in the analysis.
Table 1 lists the results of the univariate analysis comparing demographics, preoperative comorbidities, and operative factors between the home and rehabilitation groups. Both age (P < .001) and sex (P = .012) were significantly different between groups; the rehabilitation group was older by about 10 years and included significantly more females. In addition to demographic factors, 16 preoperative comorbidities, and 5 surgical factors were significantly associated with discharge to rehabilitation.
Surgery type significantly affected discharge to rehabilitation (Figure). Patients who were undergoing open plating of a femoral neck fracture or intramedullary nailing of an intertrochanteric, peritrochanteric, or subtrochanteric femoral fracture constituted 30% of all patients discharged to rehabilitation centers. In contrast, patients undergoing percutaneous skeletal fixation of a proximal femoral fracture constituted only 5.5% of all patients discharged to rehabilitation. Based on surgery type, we broke down discharge location further, into categories of skilled nursing facility, unskilled facility (not patient’s previous home), separate acute-care facility, dedicated rehabilitation center, and home. Of all 4815 patients combined, 2102 (43.6%) were discharged to a skilled nursing facility, 31 (0.6%) to an unskilled facility (not home), 106 (2.2%) to separate acute care, 1312 (27.2%) to a dedicated rehabilitation center, and 950 (19.7%) home.
Table 2 lists the significant results from the multivariate logistical analysis comparing discharge to a rehabilitation center and discharge home after controlling for the significant risk factors (Table 1). Current diabetes, history of dyspnea, previous myocardial infarction, history of ischemic attacks, current bleeding disorder, transfusion during hospitalization, previous percutaneous cardiac stenting, chemotherapy, past cerebrovascular accident, presence of cancer, surgery type based on CPT code, history of chronic obstructive pulmonary disease or congestive heart failure, current smoking status, and operative time longer than 90 minutes were not significantly correlated with discharge to rehabilitation in the multivariate analysis. All significant factors were associated with higher odds of discharge to rehabilitation except for DNR status. DNR patients were 2.04 times more likely (95% CI, 1.49-2.78; P < .001) to be discharged home than to rehabilitation centers.
Applying these adjusted odds ratios, we see that an elderly woman (age, >65 years) who underwent general anesthesia with an ASA class higher than 2 was 17.63 times more likely than a patient without these risk factors to be discharged to rehabilitation. If this patient were also dialysis-dependent, she would be 61.52 times more likely than a similar patient without dialysis needs to be discharged to rehabilitation.
Even when controlling for all significant and nonsignificant variables in multivariate logistical analysis, age over 65 years (β = 1.05; P < .001), female sex (β = 1.76; P = .004), dialysis dependence (β = 12.98; P = .036), hypertension requiring medication (β = 1.53; P = .032), and ASA class higher than 2 (β = 1.98; P = .001) were found to be significant risk factors for discharge to rehabilitation.
Discussion
This study was the first to investigate the issue of which patient risk factors allow the practicing orthopedist to identify patients who require rehabilitation after hip fracture surgery. Through our multivariate analysis, which controlled for demographics, comorbidities, and operative factors, we found that older age, female sex, history of percutaneous coronary intervention, dialysis dependence, general anesthesia, and ASA class higher than 2 significantly increased the odds of discharge to a rehabilitation center versus home.
Using our study’s results, we can create a risk stratification model for patients and thereby a means of targeting patients who need rehabilitation and starting the process of finding a rehabilitation bed early in the postoperative course. Our study’s variables are easily measured metrics that may be collected in any hospital setting. Especially for hip fracture patients, early planning and discharge to the appropriate rehabilitation center are important in decreasing LOS and associated hospitalization costs. According to one report,3 about 85% of all hip fracture costs are directly related to LOS, given the unnecessarily long rehabilitation periods in hospitals. Hollingworth and colleagues2 compared costs for patients who remained in the hospital with costs for those discharged with rehabilitation services. Overall costs were significantly lower for patients discharged home with rehabilitation. The authors concluded that 40% of hip fracture patients may be suitable for early discharge.2 In an analysis of Medicare payments for hip fracture treatment, hospital costs including LOS accounted for 60% of all payments.12 The results of these 2 studies suggest that the overall driver of hip fracture costs is prolonged LOS and that, if patients are discharged to rehabilitation, then overall costs may be lowered through a direct reduction in hospital LOS. Given that hip fractures account for almost 350,000 hospital admissions in the United States each year, and using our institution’s average hospital charge per day ($4500), about $1.6 billion may be saved if each patient’s LOS decreased by 1 day.13 Although multiple factors affect LOS, discharge planning is under orthopedists’ direct control. Therefore, early identification of patients who will require rehabilitation may help reduce LOS-associated costs in our health care system.
The patient variables that were significantly associated with discharge to rehabilitation are also associated with increased morbidity and mortality in hip fracture patients, according to the literature,14-20 which provides some external validation of using these risk factors as predictors for rehabilitation. A patient with one of these risk factors may require rehabilitation, given that rehabilitation services are specifically linked to lower morbidity and mortality rates among hip fracture patients. For example, patients with dialysis needs were 3.49 times more likely to be discharged to a rehabilitation center in our study. In a 2000 study by Coco and Rush,16 hip fracture patients on dialysis had a 1-year mortality rate 2.5 times higher than that of patients who were not dialysis-dependent. In 2010, Cameron and colleagues17 found that cardiovascular disease was associated with a 2.68 times higher risk of mortality in hip fracture patients. Similarly in our study, both hypertension and history of percutaneous coronary intervention were associated with discharge to rehabilitation. We found higher odds of discharge to rehabilitation with higher ASA classes, which mirror results from a study by Michel and colleagues,15 who found that higher (vs lower) preoperative ASA classes were associated with higher 1-year mortality in hip fracture patients. Interestingly, DNR status was associated with higher odds of discharge home, which may reflect patients’ desires to forgo noninvasive or lifesaving procedures that may be performed at rehabilitation facilities. Although general anesthesia predisposed patients to discharge to a rehabilitation center, multiple studies have found no association between anesthesia type and postoperative mortality rates for hip fracture patients.18,19 Last, Marcantonio and colleagues20 found delirium specifically had a higher odds ratio for discharge, but our univariate analysis did not find a significant association between impaired sensorium and discharge location. Given the correlation of our risk factors with increased morbidity and mortality in the literature, our study’s results provide the initial groundwork for creating a risk calculator that orthopedists can use to predict discharge to rehabilitation.
Our study had some limitations. Although we analyzed a large number of demographics, preoperative comorbidities, and surgical factors, our univariate analysis was limited to information in the ACS-NSQIP database. We did not incorporate other clinically relevant factors (eg, social factors, including patients’ support networks) that may influence discharge decisions. Furthermore, ACS-NSQIP records patient data only up to 30 days after surgery. Discharge information for the time after that was missing for a subset of hip fracture patients, and these patients had to be excluded, potentially skewing our data. ACS-NSQIP also does not collect cost data for patients based on hospitalization or LOS, so we could not determine whether patients discharged to rehabilitation incurred higher costs because of longer hospitalizations.
Nevertheless, our study identified significant patient and operative variables that are associated with discharge to a rehabilitation center. By identifying hip fracture patients with these risk factors early and mobilizing the appropriate resources, practicing orthopedists should be better equipped to help facilitate the discharge of patients to the appropriate location after surgery. Validation of these risk factors should be prospectively determined with an analysis of LOS and cost implications. Use of a risk calculator may in fact result in decreased LOS and hospital-related costs. Furthermore, using these risk factors in a prospective patient cohort would help validate their use and determine whether there is clinical correlation. The orthopedists in our institution are becoming more aware of these risk factors, but validation is necessary.
Length of stay (LOS) is a significant driver of costs after hip fracture surgery.1-3 Multiple studies have identified factors associated with increased LOS in hip fracture patients. These factors include admission time, delay to surgery, presence of comorbidities, and older age.4-9
One significant and potentially modifiable factor affecting LOS is delayed transfer to a rehabilitation center after surgery.8-11 Although patients after orthopedic surgeries require additional rehabilitation services or subacute care directly attributable to their injuries, specialized rehabilitation centers may not always have beds readily available.6-11 Studies have shown that delays in transfer to skilled nursing facilities or rehabilitation centers are highly common among orthopedic patients.8 It is therefore imperative that orthopedists have a mechanism for predicting and identifying which patients require rehabilitation services early in the postoperative period. Identifying risk factors and stratifying patients who are most likely to require rehabilitation would facilitate the early transfer of these patients and thereby directly decrease LOS and hospitalization-related costs.
In this article, we report results from prospective, national, multicenter data to identify commonly measured risk factors for discharge to rehabilitation facilities for hip fracture patients. Through multivariate analysis of ACS-NSQIP (American College of Surgeons National Surgical Quality Improvement Program) data, we determined which risk factors significantly predispose patients to discharge to rehabilitation centers versus discharge home. Knowledge of these risk factors allows the practicing orthopedist to be better equipped to identify patients who require additional rehabilitation early in the postoperative course. By mobilizing case managers and social workers to help avoid delays in the transfers of these identified patients, LOS-associated costs may ultimately decrease.
Materials and Methods
After obtaining institutional review board approval for this study from the Office of Research at Vanderbilt University, we prospectively collected 2011 discharge data from the ACS-NSQIP database (these data are unavailable for earlier years). All patients who underwent hip fracture surgery in 2011 were identified by CPT (Current Procedural Terminology) codes. Cases of patients with unknown discharge information and of those who died during their hospitalizations were excluded from analysis. For the remaining patients, discharge information as categorized by ACS-NSQIP included skilled care (eg, subacute hospital, skilled nursing home), unskilled facility (eg, nursing home, assisted facility), separate acute care, and rehabilitation. All other patients were discharged home without additional assistance or to the previous home where they received chronic care, assisted living, or unskilled aid. Patients were dichotomized according to whether they were discharged home or to one of the rehabilitation facilities mentioned.
To determine which risk factors significantly contributed to a patient’s discharge to rehabilitation, we ran univariate analyses using Fisher exact tests for categorical variables and Student t tests for continuous variables on multiple patient factors, including demographics, preoperative comorbidities, and operative factors. Demographics included age and sex. Preoperative comorbidities included 32 conditions: diabetes mellitus, active smoking status, current alcohol use, dyspnea, history of chronic obstructive pulmonary disease, history of congestive heart failure, hypertension requiring medication, history of esophageal varices, history of myocardial infarction, current renal failure, current dialysis dependence, steroid use, recent weight loss, existing bleeding disorder, transfusion before discharge, presence of central nervous system tumor, recent chemotherapy, recent radiation therapy, previous percutaneous coronary intervention, previous percutaneous coronary stenting, history of angina, peripheral vascular disease, cerebrovascular accidents, recent surgery (within 30 days), rest pain, impaired sensorium, history of transient ischemic attacks, current hemiplegia status, current paraplegia status, current quadriplegia status, current ascites, hypertension, and disseminated cancer. Operative factors included wound infection, DNR (do not resuscitate) status, ventilator support, anesthesia type, wound class, ASA (American Society of Anesthesiologists) class, and operative time.
For the univariate analyses, significance was set at P < .05. Demographics, preoperative comorbidities, and operative factors that were significantly associated with discharge to a rehabilitation facility in the univariate analysis were selected as covariates for a multivariate analysis. We incorporated a binary logistic regression to analyze which of these significant risk factors are correlated with a patient’s discharge to a rehabilitation facility after hip fracture surgery.
Results
A total of 4974 patients undergoing surgery for hip fractures in 2011 were identified. Of these patients, 4815 had complete information on discharge location and were included in the analysis.
Table 1 lists the results of the univariate analysis comparing demographics, preoperative comorbidities, and operative factors between the home and rehabilitation groups. Both age (P < .001) and sex (P = .012) were significantly different between groups; the rehabilitation group was older by about 10 years and included significantly more females. In addition to demographic factors, 16 preoperative comorbidities, and 5 surgical factors were significantly associated with discharge to rehabilitation.
Surgery type significantly affected discharge to rehabilitation (Figure). Patients who were undergoing open plating of a femoral neck fracture or intramedullary nailing of an intertrochanteric, peritrochanteric, or subtrochanteric femoral fracture constituted 30% of all patients discharged to rehabilitation centers. In contrast, patients undergoing percutaneous skeletal fixation of a proximal femoral fracture constituted only 5.5% of all patients discharged to rehabilitation. Based on surgery type, we broke down discharge location further, into categories of skilled nursing facility, unskilled facility (not patient’s previous home), separate acute-care facility, dedicated rehabilitation center, and home. Of all 4815 patients combined, 2102 (43.6%) were discharged to a skilled nursing facility, 31 (0.6%) to an unskilled facility (not home), 106 (2.2%) to separate acute care, 1312 (27.2%) to a dedicated rehabilitation center, and 950 (19.7%) home.
Table 2 lists the significant results from the multivariate logistical analysis comparing discharge to a rehabilitation center and discharge home after controlling for the significant risk factors (Table 1). Current diabetes, history of dyspnea, previous myocardial infarction, history of ischemic attacks, current bleeding disorder, transfusion during hospitalization, previous percutaneous cardiac stenting, chemotherapy, past cerebrovascular accident, presence of cancer, surgery type based on CPT code, history of chronic obstructive pulmonary disease or congestive heart failure, current smoking status, and operative time longer than 90 minutes were not significantly correlated with discharge to rehabilitation in the multivariate analysis. All significant factors were associated with higher odds of discharge to rehabilitation except for DNR status. DNR patients were 2.04 times more likely (95% CI, 1.49-2.78; P < .001) to be discharged home than to rehabilitation centers.
Applying these adjusted odds ratios, we see that an elderly woman (age, >65 years) who underwent general anesthesia with an ASA class higher than 2 was 17.63 times more likely than a patient without these risk factors to be discharged to rehabilitation. If this patient were also dialysis-dependent, she would be 61.52 times more likely than a similar patient without dialysis needs to be discharged to rehabilitation.
Even when controlling for all significant and nonsignificant variables in multivariate logistical analysis, age over 65 years (β = 1.05; P < .001), female sex (β = 1.76; P = .004), dialysis dependence (β = 12.98; P = .036), hypertension requiring medication (β = 1.53; P = .032), and ASA class higher than 2 (β = 1.98; P = .001) were found to be significant risk factors for discharge to rehabilitation.
Discussion
This study was the first to investigate the issue of which patient risk factors allow the practicing orthopedist to identify patients who require rehabilitation after hip fracture surgery. Through our multivariate analysis, which controlled for demographics, comorbidities, and operative factors, we found that older age, female sex, history of percutaneous coronary intervention, dialysis dependence, general anesthesia, and ASA class higher than 2 significantly increased the odds of discharge to a rehabilitation center versus home.
Using our study’s results, we can create a risk stratification model for patients and thereby a means of targeting patients who need rehabilitation and starting the process of finding a rehabilitation bed early in the postoperative course. Our study’s variables are easily measured metrics that may be collected in any hospital setting. Especially for hip fracture patients, early planning and discharge to the appropriate rehabilitation center are important in decreasing LOS and associated hospitalization costs. According to one report,3 about 85% of all hip fracture costs are directly related to LOS, given the unnecessarily long rehabilitation periods in hospitals. Hollingworth and colleagues2 compared costs for patients who remained in the hospital with costs for those discharged with rehabilitation services. Overall costs were significantly lower for patients discharged home with rehabilitation. The authors concluded that 40% of hip fracture patients may be suitable for early discharge.2 In an analysis of Medicare payments for hip fracture treatment, hospital costs including LOS accounted for 60% of all payments.12 The results of these 2 studies suggest that the overall driver of hip fracture costs is prolonged LOS and that, if patients are discharged to rehabilitation, then overall costs may be lowered through a direct reduction in hospital LOS. Given that hip fractures account for almost 350,000 hospital admissions in the United States each year, and using our institution’s average hospital charge per day ($4500), about $1.6 billion may be saved if each patient’s LOS decreased by 1 day.13 Although multiple factors affect LOS, discharge planning is under orthopedists’ direct control. Therefore, early identification of patients who will require rehabilitation may help reduce LOS-associated costs in our health care system.
The patient variables that were significantly associated with discharge to rehabilitation are also associated with increased morbidity and mortality in hip fracture patients, according to the literature,14-20 which provides some external validation of using these risk factors as predictors for rehabilitation. A patient with one of these risk factors may require rehabilitation, given that rehabilitation services are specifically linked to lower morbidity and mortality rates among hip fracture patients. For example, patients with dialysis needs were 3.49 times more likely to be discharged to a rehabilitation center in our study. In a 2000 study by Coco and Rush,16 hip fracture patients on dialysis had a 1-year mortality rate 2.5 times higher than that of patients who were not dialysis-dependent. In 2010, Cameron and colleagues17 found that cardiovascular disease was associated with a 2.68 times higher risk of mortality in hip fracture patients. Similarly in our study, both hypertension and history of percutaneous coronary intervention were associated with discharge to rehabilitation. We found higher odds of discharge to rehabilitation with higher ASA classes, which mirror results from a study by Michel and colleagues,15 who found that higher (vs lower) preoperative ASA classes were associated with higher 1-year mortality in hip fracture patients. Interestingly, DNR status was associated with higher odds of discharge home, which may reflect patients’ desires to forgo noninvasive or lifesaving procedures that may be performed at rehabilitation facilities. Although general anesthesia predisposed patients to discharge to a rehabilitation center, multiple studies have found no association between anesthesia type and postoperative mortality rates for hip fracture patients.18,19 Last, Marcantonio and colleagues20 found delirium specifically had a higher odds ratio for discharge, but our univariate analysis did not find a significant association between impaired sensorium and discharge location. Given the correlation of our risk factors with increased morbidity and mortality in the literature, our study’s results provide the initial groundwork for creating a risk calculator that orthopedists can use to predict discharge to rehabilitation.
Our study had some limitations. Although we analyzed a large number of demographics, preoperative comorbidities, and surgical factors, our univariate analysis was limited to information in the ACS-NSQIP database. We did not incorporate other clinically relevant factors (eg, social factors, including patients’ support networks) that may influence discharge decisions. Furthermore, ACS-NSQIP records patient data only up to 30 days after surgery. Discharge information for the time after that was missing for a subset of hip fracture patients, and these patients had to be excluded, potentially skewing our data. ACS-NSQIP also does not collect cost data for patients based on hospitalization or LOS, so we could not determine whether patients discharged to rehabilitation incurred higher costs because of longer hospitalizations.
Nevertheless, our study identified significant patient and operative variables that are associated with discharge to a rehabilitation center. By identifying hip fracture patients with these risk factors early and mobilizing the appropriate resources, practicing orthopedists should be better equipped to help facilitate the discharge of patients to the appropriate location after surgery. Validation of these risk factors should be prospectively determined with an analysis of LOS and cost implications. Use of a risk calculator may in fact result in decreased LOS and hospital-related costs. Furthermore, using these risk factors in a prospective patient cohort would help validate their use and determine whether there is clinical correlation. The orthopedists in our institution are becoming more aware of these risk factors, but validation is necessary.
1. Garcia AE, Bonnaig JV, Yoneda ZT, et al. Patient variables which may predict length of stay and hospital costs in elderly patients with hip fracture. J Orthop Trauma. 2012;26(11):620-623.
2. Hollingworth W, Todd C, Parker M, Roberts JA, Williams R. Cost analysis of early discharge after hip fracture. BMJ. 1993;307(6909):903-906.
3. Sund R, Riihimäki J, Mäkelä M, et al. Modeling the length of the care episode after hip fracture: does the type of fracture matter? Scand J Surg. 2009;98(3):169-174.
4. Fox KM, Magaziner J, Hebel JR, Kenzora JE, Kashner TM. Intertrochanteric versus femoral neck hip fractures: differential characteristics, treatment, and sequelae. J Gerontol A Biol Sci Med Sci. 1999;54(12):M635-M640.
5. Foss NB, Palm H, Krasheninnikoff M, Kehlet H, Gebuhr P. Impact of surgical complications on length of stay after hip fracture surgery. Injury. 2007;38(7):780-784.
6. Lefaivre KA, Macadam SA, Davidson DJ, Gandhi R, Chan H, Broekhuyse HM. Length of stay, mortality, morbidity and delay to surgery in hip fractures. J Bone Joint Surg Br. 2009;91(7):922-927.
7. Clague JE, Craddock E, Andrew G, Horan MA, Pendleton N. Predictors of outcome following hip fracture. Admission time predicts length of stay and in-hospital mortality. Injury. 2002;33(1):1-6.
8. Parker MJ, Todd CJ, Palmer CR, et al. Inter-hospital variations in length of hospital stay following hip fracture. Age Ageing. 1998;27(31):333-337.
9. Brasel KJ, Rasmussen J, Cauley C, Weigelt JA. Reasons for delayed discharge of trauma patients. J Surg Res. 2002;107(2):223-226.
10. Bonar SK, Tinetti ME, Speechley M, Cooney LM. Factors associated with short- versus long-term skilled nursing facility placement among community-living hip fracture patients. J Am Geriatr Soc. 1990;38(10):1139-1144.
11. Bentler SE, Liu L, Obrizan M, et al. The aftermath of hip fracture: discharge placement, functional status change, and mortality. Am J Epidemiol. 2009;170(10):1290-1299.
12. Birkmeyer JD, Gust C, Baser O, Dimick JB, Sutherland JM, Skinner JS. Medicare payments for common inpatient procedures: implications for episode-based payment bundling. Health Serv Res. 2010;45(6 pt 1):1783-1795.
13. American Academy of Orthopaedic Surgeons. Burden of Musculoskeletal Diseases in the United States: Prevalence, Societal and Economic Cost. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2008.
14. Maciejewski ML, Radcliff A, Henderson WG, et al. Determinants of postsurgical discharge setting for male hip fracture patients. J Rehabil Res Dev. 2013;50(9):1267-1276.
15. Michel JP, Klopfenstein C, Hoffmeyer P, Stern R, Grab B. Hip fracture surgery: is the pre-operative American Society of Anesthesiologists (ASA) score a predictor of functional outcome? Aging Clin Exp Res. 2002;14(5):389-394.
16. Coco M, Rush H. Increased incidence of hip fractures in dialysis patients with low serum parathyroid hormone. Am J Kidney Dis. 2000;36(6):1115-1121.
17. Cameron ID, Chen JS, March LM, et al. Hip fracture causes excess mortality owing to cardiovascular and infectious disease in institutionalized older people: a prospective 5-year study. J Bone Miner Res. 2010;25(4):866-872.
18. White SM, Moppett IK, Griffiths R. Outcome by mode of anaesthesia for hip fracture surgery. An observational audit of 65 535 patients in a national dataset. Anaesthesia. 2014;69(3):224-230.
19. Le-Wendling L, Bihorac A, Baslanti TO, et al. Regional anesthesia as compared with general anesthesia for surgery in geriatric patients with hip fracture: does it decrease morbidity, mortality, and health care costs? Results of a single-centered study. Pain Med. 2012;13(7):948-956.
20. Marcantonio ER, Flacker JM, Michaels M, Resnick NM. Delirium is independently associated with poor functional recovery after hip fracture. J Am Geriatr Soc. 2000;48(6):618-624.
1. Garcia AE, Bonnaig JV, Yoneda ZT, et al. Patient variables which may predict length of stay and hospital costs in elderly patients with hip fracture. J Orthop Trauma. 2012;26(11):620-623.
2. Hollingworth W, Todd C, Parker M, Roberts JA, Williams R. Cost analysis of early discharge after hip fracture. BMJ. 1993;307(6909):903-906.
3. Sund R, Riihimäki J, Mäkelä M, et al. Modeling the length of the care episode after hip fracture: does the type of fracture matter? Scand J Surg. 2009;98(3):169-174.
4. Fox KM, Magaziner J, Hebel JR, Kenzora JE, Kashner TM. Intertrochanteric versus femoral neck hip fractures: differential characteristics, treatment, and sequelae. J Gerontol A Biol Sci Med Sci. 1999;54(12):M635-M640.
5. Foss NB, Palm H, Krasheninnikoff M, Kehlet H, Gebuhr P. Impact of surgical complications on length of stay after hip fracture surgery. Injury. 2007;38(7):780-784.
6. Lefaivre KA, Macadam SA, Davidson DJ, Gandhi R, Chan H, Broekhuyse HM. Length of stay, mortality, morbidity and delay to surgery in hip fractures. J Bone Joint Surg Br. 2009;91(7):922-927.
7. Clague JE, Craddock E, Andrew G, Horan MA, Pendleton N. Predictors of outcome following hip fracture. Admission time predicts length of stay and in-hospital mortality. Injury. 2002;33(1):1-6.
8. Parker MJ, Todd CJ, Palmer CR, et al. Inter-hospital variations in length of hospital stay following hip fracture. Age Ageing. 1998;27(31):333-337.
9. Brasel KJ, Rasmussen J, Cauley C, Weigelt JA. Reasons for delayed discharge of trauma patients. J Surg Res. 2002;107(2):223-226.
10. Bonar SK, Tinetti ME, Speechley M, Cooney LM. Factors associated with short- versus long-term skilled nursing facility placement among community-living hip fracture patients. J Am Geriatr Soc. 1990;38(10):1139-1144.
11. Bentler SE, Liu L, Obrizan M, et al. The aftermath of hip fracture: discharge placement, functional status change, and mortality. Am J Epidemiol. 2009;170(10):1290-1299.
12. Birkmeyer JD, Gust C, Baser O, Dimick JB, Sutherland JM, Skinner JS. Medicare payments for common inpatient procedures: implications for episode-based payment bundling. Health Serv Res. 2010;45(6 pt 1):1783-1795.
13. American Academy of Orthopaedic Surgeons. Burden of Musculoskeletal Diseases in the United States: Prevalence, Societal and Economic Cost. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2008.
14. Maciejewski ML, Radcliff A, Henderson WG, et al. Determinants of postsurgical discharge setting for male hip fracture patients. J Rehabil Res Dev. 2013;50(9):1267-1276.
15. Michel JP, Klopfenstein C, Hoffmeyer P, Stern R, Grab B. Hip fracture surgery: is the pre-operative American Society of Anesthesiologists (ASA) score a predictor of functional outcome? Aging Clin Exp Res. 2002;14(5):389-394.
16. Coco M, Rush H. Increased incidence of hip fractures in dialysis patients with low serum parathyroid hormone. Am J Kidney Dis. 2000;36(6):1115-1121.
17. Cameron ID, Chen JS, March LM, et al. Hip fracture causes excess mortality owing to cardiovascular and infectious disease in institutionalized older people: a prospective 5-year study. J Bone Miner Res. 2010;25(4):866-872.
18. White SM, Moppett IK, Griffiths R. Outcome by mode of anaesthesia for hip fracture surgery. An observational audit of 65 535 patients in a national dataset. Anaesthesia. 2014;69(3):224-230.
19. Le-Wendling L, Bihorac A, Baslanti TO, et al. Regional anesthesia as compared with general anesthesia for surgery in geriatric patients with hip fracture: does it decrease morbidity, mortality, and health care costs? Results of a single-centered study. Pain Med. 2012;13(7):948-956.
20. Marcantonio ER, Flacker JM, Michaels M, Resnick NM. Delirium is independently associated with poor functional recovery after hip fracture. J Am Geriatr Soc. 2000;48(6):618-624.
Reinforcing a Spica Cast With a Fiberglass Bar
Femur fractures (Orthopaedic Trauma Association classes 31, 32, 33)1 are common childhood injuries, occurring at a rate of 19 per 100,000 children in the United States.2 Peak occurrence is bimodal at ages 2 and 17 years. The most common mechanism of injury in children under 6 years is a fall, and hip spica casting is the preferred treatment modality in this group.3-5
A bar connecting the legs of the spica cast has been shown to facilitate patient transport5 and significantly decrease mechanical failure of the spica cast.6 This bar often consists of a broom handle or pipe that must be cut to size during the case and subsequently incorporated into the cast—tasks that are often inconvenient and time-consuming for on-call or emergency department staff unfamiliar with orthopedic tools.
In this article, we review a spica cast application that incorporates a low-cost, lightweight technique for fabricating a connecting bar from existing fiberglass casting material. The Institutional Review Board at Connecticut Children’s Medical Center approved this work.
Technique of Double-Leg Spica Casting With Fiberglass Bar
A spica casting table (Orthopedic Systems) with a well-padded post is placed on the operating room table and adjusted to the length of the patient from perineum to just below the shoulders. With the patient under general anesthesia, folded towels are used to provide 2 to 4 cm of padding on the anterior torso, atop which a waterproof pantaloon is applied. The patient is transferred to the spica table, and the patient’s arms are gently secured to the casting table with cast padding or tape in an abducted position at the shoulders. A surgeon controls the legs by holding the feet with the long fingers just above the heels, the index fingers on the anterior ankle, and the thumbs on the soles of the feet. Cast padding is wrapped from the nipple line to the supramalleolar region on each leg. The bony prominences of the malleoli, patella, fibular head, femoral condyles, iliac crests, and coccyx are well padded.
Fiberglass is then rolled without compression onto the patient, beginning with the torso and perineal areas. The injured leg is wrapped to its final length above the malleoli while the uninjured leg is kept free. Maintaining the position of the injured leg with simultaneous molding at the fracture site, typically to promote valgus, allows fracture reduction. The fracture position is then checked under image intensification. For femur fractures, hip abduction and flexion are set to 45° and 90°, respectively, while knee flexion is between 50° and 90°. The uninjured leg is then wrapped with fiberglass. Additional strips of fiberglass can be used to reinforce weak junctional regions between the torso and the legs, posteriorly over the “intern’s triangle” and anteriorly along the hip crease.
A connecting fiberglass bar is then created using a fiberglass roll once the cast is hardened. A 2-inch fiberglass roll is wrapped around one leg to secure its position (Figure 1A) and then rolled around the second limb (Figure 1B). Fiberglass is then pulled taut and rolled around the bridge that has been created in order to thicken the bar (Figure 2). The roll is again brought around the closest limb, wrapped back across the bridge to the other limb, and rolled out to its full length. Last, the legs are abducted 1 to 2 cm to tension the bar (Figure 3). Although this does not produce enough movement to cause a crease and a resultant ulcer, careful inspection of common pressure points (eg, popliteal fossa) should be performed after the cast is complete.
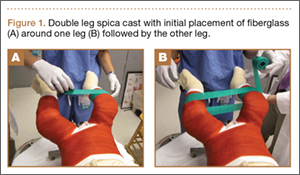
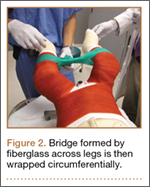

The chest towels are removed, and the final cast is inspected clinically and fluoroscopically at the fracture site before extubation. The cast is trimmed as needed to ensure room for perineal care, as well as full ankle flexion and extension without impingement. Cast edges are further petaled with plastic tape (Hy-Tape International) to provide padding and prevent the waterproof lining from tearing.
Postoperative care involves overnight observation and caregiver practice in perineal care. Frequent rotation from supine to prone is encouraged. Nurses confirm car-seat fit before discharge. If needed, radiographs are obtained 7 to 10 days later to help with wedging adjustment. The cast is removed in the clinic when adequate callus is appreciated on subsequent radiographs.
Case Series
Our experience with this technique in 16 unilateral femur fractures has been favorable (Table). Patient age ranged from 5 months to 3 years. Mean pretreatment angulation was 13° varus and 11° procurvatum. The majority of fractures were femoral shaft fractures; 1 was proximal, 2 distal.
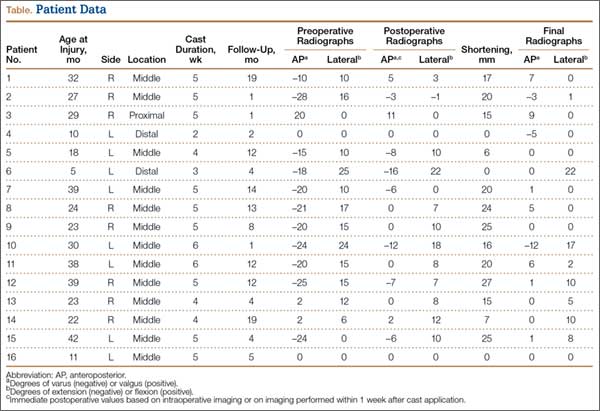
All fractures united without cast revision. Mean cast time was 4.5 weeks (range, 16 days–6 weeks). Immediate postoperative alignment was 2.5° varus (range, 11° valgus to 16° varus) and 7° procurvatum (range, 1° recurvatum to 22° procurvatum). Mean shortening was 1.5 cm (range, 0-2.7 cm). Final alignment was 1° valgus (range, 9° valgus to 12° varus) and 5° procurvatum (range, 0° to 22°). Mean follow-up was 8 months. There were no cases of skin maceration or cast failure. No casts precluded use of a spica car-seat. Figure 4 shows a typical case with a midshaft fracture treated with closed reduction and casting for 4 weeks with good remodeling at final follow-up, 19 months after injury.
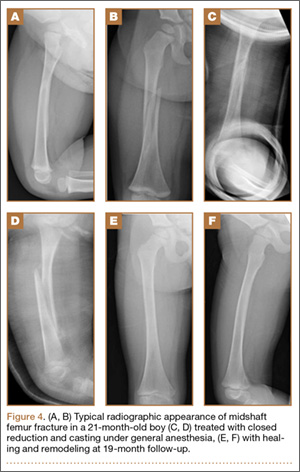
Discussion
Although single-leg walking spica casts have been shown to safely treat low-energy femur fractures in children 1 to 6 years old,7 length-unstable femur fractures, bilateral femur fractures, and patients with hip dysplasia continue to be managed with a double-leg hip spica construct. Cast integrity remains fundamental to the control of most fractures and prevention of cast-related complications, such as skin maceration and ulceration. Surgeons typically use spica cast reinforcement schemes—such as cast augments of the torso–limb junction, with multiple layers of casting material or incorporation of a connecting bar between the legs, typically constructed by overwrapping a wooden dowel in casting material—to improve the mechanical stability of casts.6 The present technique of creating a connecting bar from fiberglass casting material significantly simplifies the standard wooden dowel approach and provided excellent results in our treatment group in terms of cast integrity and fracture alignment. In addition, at our institution, a roll of fiberglass costs $2.10, whereas a wooden dowel costs $3 to $10 and can be difficult to locate if not frequently used. Other tube-shaped materials, such as the disposable material used to package implants and tubes, carry an even lower cost. However, we have found that a single fiberglass roll is most readily available and easiest to apply.
Although proper spica cast application remains important in managing pediatric trauma, it lacks a good technical description in the literature. In this technical report, we have presented our standard spica cast application method, which minimizes the range of cast complications that have been reported, from minor skin irritation to superior mesenteric artery syndrome. Two salient technical highlights are use of waterproof pantaloon liners and cast petaling, which we have found almost eliminate the morbidity of potential skin complications, reported to occur at a rate of 28%.8 In addition, we forgo applying the cast on the injured leg in segments. Application of a short-leg cast on the injured leg to allow traction on the leg during cast application is of dubious utility and may be potentially harmful, with described complications of peroneal nerve palsy and compartment syndrome.9-11 Further, it is important to use an abdominal spacer (eg, a stack of towels) under the cast padding to create room for abdominal expansion and minimize pressure thought to induce superior mesenteric artery syndrome. Plastic or rubber abdominal spacers have also been described.12,13 Last, leg position is important for reduction and maintenance of the fracture, as well as patient care. Literature advocates minimizing hip abduction to just that needed for perineal care and maximizing hip flexion and knee extension to optimize car-seat fit and safety.14
Conclusion
Construction of a spica cast lower limb connecting bar from readily available fiberglass casting material allows a facile and rapid addition to the mechanical stability of a spica cast in the treatment of pediatric femur fractures. The technique is low-cost and obviates the need for additional extraneous materials.
1. Slongo TF, Audigé L; AO Pediatric Classification Group. Fracture and dislocation classification compendium for children: the AO Pediatric Comprehensive Classification of Long Bone Fractures (PCCF). J Orthop Trauma. 2007;21(10):S135-S160.
2. Hinton RY, Lincoln A, Crockett MM, Sponseller P, Smith G. Fractures of the femoral shaft in children. Incidence, mechanisms, and sociodemographic risk factors. J Bone Joint Surg Am. 1999;81(4):500-509.
3. Campbell WC, Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 11th ed. Philadelphia, PA: Mosby Elsevier; 2008.
4. Lovell WW, Winter RB, Morrissy RT, Weinstein SL. Lovell and Winter’s Pediatric Orthopaedics. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.
5. Green NE, Swiontkowski MF, eds. Skeletal Trauma in Children. 4th ed. Philadelphia, PA: Elsevier Health Sciences; 2009.
6. Hosalkar HS, Jones S, Chowdhury M, Chatoo M, Hill RA. Connecting bar for hip spica reinforcement: does it help? J Pediatr Orthop B. 2003;12(2):100-102.
7. Flynn JM, Garner MR, Jones KJ, et al. The treatment of low-energy femoral shaft fractures: a prospective study comparing the “walking spica” with the traditional spica cast. J Bone Joint Surg Am. 2011;93(23):2196-2202.
8. DiFazio R, Vessey J, Zurakowski D, Hresko MT, Matheney T. Incidence of skin complications and associated charges in children treated with hip spica casts for femur fractures. J Pediatr Orthop. 2011;31(1):17-22.
9. Weiss AP, Schenck RC Jr, Sponseller PD, Thompson JD. Peroneal nerve palsy after early cast application for femoral fractures in children. J Pediatr Orthop. 1992;12(1):25-28.
10. Mubarak SJ, Frick S, Sink E, Rathjen K, Noonan KJ. Volkmann contracture and compartment syndromes after femur fractures in children treated with 90/90 spica casts. J Pediatr Orthop. 2006;26(5):567-572.
11. Large TM, Frick SL. Compartment syndrome of the leg after treatment of a femoral fracture with an early sitting spica cast. A report of two cases. J Bone Joint Surg Am. 2003;85(11):2207-2210.
12. Sharma S, Azzopardi T. Reduction of abdominal pressure for prophylaxis of the mesenteric artery syndrome (cast syndrome) in a hip spica—a simple technique. Ann R Coll Surg Engl. 2006;88(3):317.
13. Kiter E, Demirkan F, Kiliç BA, Erkula G. A new technique for creating an abdominal window in a hip spica cast. J Orthop Trauma. 2003;17(6):442-443.
14. Zielinski J, Oliver G, Sybesma J, Walter N, Atkinson P. Casting technique and restraint choice influence child safety during transport of body casted children subjected to a simulated frontal MVA. J Trauma. 2009;66(6):1653-1665.
Femur fractures (Orthopaedic Trauma Association classes 31, 32, 33)1 are common childhood injuries, occurring at a rate of 19 per 100,000 children in the United States.2 Peak occurrence is bimodal at ages 2 and 17 years. The most common mechanism of injury in children under 6 years is a fall, and hip spica casting is the preferred treatment modality in this group.3-5
A bar connecting the legs of the spica cast has been shown to facilitate patient transport5 and significantly decrease mechanical failure of the spica cast.6 This bar often consists of a broom handle or pipe that must be cut to size during the case and subsequently incorporated into the cast—tasks that are often inconvenient and time-consuming for on-call or emergency department staff unfamiliar with orthopedic tools.
In this article, we review a spica cast application that incorporates a low-cost, lightweight technique for fabricating a connecting bar from existing fiberglass casting material. The Institutional Review Board at Connecticut Children’s Medical Center approved this work.
Technique of Double-Leg Spica Casting With Fiberglass Bar
A spica casting table (Orthopedic Systems) with a well-padded post is placed on the operating room table and adjusted to the length of the patient from perineum to just below the shoulders. With the patient under general anesthesia, folded towels are used to provide 2 to 4 cm of padding on the anterior torso, atop which a waterproof pantaloon is applied. The patient is transferred to the spica table, and the patient’s arms are gently secured to the casting table with cast padding or tape in an abducted position at the shoulders. A surgeon controls the legs by holding the feet with the long fingers just above the heels, the index fingers on the anterior ankle, and the thumbs on the soles of the feet. Cast padding is wrapped from the nipple line to the supramalleolar region on each leg. The bony prominences of the malleoli, patella, fibular head, femoral condyles, iliac crests, and coccyx are well padded.
Fiberglass is then rolled without compression onto the patient, beginning with the torso and perineal areas. The injured leg is wrapped to its final length above the malleoli while the uninjured leg is kept free. Maintaining the position of the injured leg with simultaneous molding at the fracture site, typically to promote valgus, allows fracture reduction. The fracture position is then checked under image intensification. For femur fractures, hip abduction and flexion are set to 45° and 90°, respectively, while knee flexion is between 50° and 90°. The uninjured leg is then wrapped with fiberglass. Additional strips of fiberglass can be used to reinforce weak junctional regions between the torso and the legs, posteriorly over the “intern’s triangle” and anteriorly along the hip crease.
A connecting fiberglass bar is then created using a fiberglass roll once the cast is hardened. A 2-inch fiberglass roll is wrapped around one leg to secure its position (Figure 1A) and then rolled around the second limb (Figure 1B). Fiberglass is then pulled taut and rolled around the bridge that has been created in order to thicken the bar (Figure 2). The roll is again brought around the closest limb, wrapped back across the bridge to the other limb, and rolled out to its full length. Last, the legs are abducted 1 to 2 cm to tension the bar (Figure 3). Although this does not produce enough movement to cause a crease and a resultant ulcer, careful inspection of common pressure points (eg, popliteal fossa) should be performed after the cast is complete.
The chest towels are removed, and the final cast is inspected clinically and fluoroscopically at the fracture site before extubation. The cast is trimmed as needed to ensure room for perineal care, as well as full ankle flexion and extension without impingement. Cast edges are further petaled with plastic tape (Hy-Tape International) to provide padding and prevent the waterproof lining from tearing.
Postoperative care involves overnight observation and caregiver practice in perineal care. Frequent rotation from supine to prone is encouraged. Nurses confirm car-seat fit before discharge. If needed, radiographs are obtained 7 to 10 days later to help with wedging adjustment. The cast is removed in the clinic when adequate callus is appreciated on subsequent radiographs.
Case Series
Our experience with this technique in 16 unilateral femur fractures has been favorable (Table). Patient age ranged from 5 months to 3 years. Mean pretreatment angulation was 13° varus and 11° procurvatum. The majority of fractures were femoral shaft fractures; 1 was proximal, 2 distal.
All fractures united without cast revision. Mean cast time was 4.5 weeks (range, 16 days–6 weeks). Immediate postoperative alignment was 2.5° varus (range, 11° valgus to 16° varus) and 7° procurvatum (range, 1° recurvatum to 22° procurvatum). Mean shortening was 1.5 cm (range, 0-2.7 cm). Final alignment was 1° valgus (range, 9° valgus to 12° varus) and 5° procurvatum (range, 0° to 22°). Mean follow-up was 8 months. There were no cases of skin maceration or cast failure. No casts precluded use of a spica car-seat. Figure 4 shows a typical case with a midshaft fracture treated with closed reduction and casting for 4 weeks with good remodeling at final follow-up, 19 months after injury.
Discussion
Although single-leg walking spica casts have been shown to safely treat low-energy femur fractures in children 1 to 6 years old,7 length-unstable femur fractures, bilateral femur fractures, and patients with hip dysplasia continue to be managed with a double-leg hip spica construct. Cast integrity remains fundamental to the control of most fractures and prevention of cast-related complications, such as skin maceration and ulceration. Surgeons typically use spica cast reinforcement schemes—such as cast augments of the torso–limb junction, with multiple layers of casting material or incorporation of a connecting bar between the legs, typically constructed by overwrapping a wooden dowel in casting material—to improve the mechanical stability of casts.6 The present technique of creating a connecting bar from fiberglass casting material significantly simplifies the standard wooden dowel approach and provided excellent results in our treatment group in terms of cast integrity and fracture alignment. In addition, at our institution, a roll of fiberglass costs $2.10, whereas a wooden dowel costs $3 to $10 and can be difficult to locate if not frequently used. Other tube-shaped materials, such as the disposable material used to package implants and tubes, carry an even lower cost. However, we have found that a single fiberglass roll is most readily available and easiest to apply.
Although proper spica cast application remains important in managing pediatric trauma, it lacks a good technical description in the literature. In this technical report, we have presented our standard spica cast application method, which minimizes the range of cast complications that have been reported, from minor skin irritation to superior mesenteric artery syndrome. Two salient technical highlights are use of waterproof pantaloon liners and cast petaling, which we have found almost eliminate the morbidity of potential skin complications, reported to occur at a rate of 28%.8 In addition, we forgo applying the cast on the injured leg in segments. Application of a short-leg cast on the injured leg to allow traction on the leg during cast application is of dubious utility and may be potentially harmful, with described complications of peroneal nerve palsy and compartment syndrome.9-11 Further, it is important to use an abdominal spacer (eg, a stack of towels) under the cast padding to create room for abdominal expansion and minimize pressure thought to induce superior mesenteric artery syndrome. Plastic or rubber abdominal spacers have also been described.12,13 Last, leg position is important for reduction and maintenance of the fracture, as well as patient care. Literature advocates minimizing hip abduction to just that needed for perineal care and maximizing hip flexion and knee extension to optimize car-seat fit and safety.14
Conclusion
Construction of a spica cast lower limb connecting bar from readily available fiberglass casting material allows a facile and rapid addition to the mechanical stability of a spica cast in the treatment of pediatric femur fractures. The technique is low-cost and obviates the need for additional extraneous materials.
Femur fractures (Orthopaedic Trauma Association classes 31, 32, 33)1 are common childhood injuries, occurring at a rate of 19 per 100,000 children in the United States.2 Peak occurrence is bimodal at ages 2 and 17 years. The most common mechanism of injury in children under 6 years is a fall, and hip spica casting is the preferred treatment modality in this group.3-5
A bar connecting the legs of the spica cast has been shown to facilitate patient transport5 and significantly decrease mechanical failure of the spica cast.6 This bar often consists of a broom handle or pipe that must be cut to size during the case and subsequently incorporated into the cast—tasks that are often inconvenient and time-consuming for on-call or emergency department staff unfamiliar with orthopedic tools.
In this article, we review a spica cast application that incorporates a low-cost, lightweight technique for fabricating a connecting bar from existing fiberglass casting material. The Institutional Review Board at Connecticut Children’s Medical Center approved this work.
Technique of Double-Leg Spica Casting With Fiberglass Bar
A spica casting table (Orthopedic Systems) with a well-padded post is placed on the operating room table and adjusted to the length of the patient from perineum to just below the shoulders. With the patient under general anesthesia, folded towels are used to provide 2 to 4 cm of padding on the anterior torso, atop which a waterproof pantaloon is applied. The patient is transferred to the spica table, and the patient’s arms are gently secured to the casting table with cast padding or tape in an abducted position at the shoulders. A surgeon controls the legs by holding the feet with the long fingers just above the heels, the index fingers on the anterior ankle, and the thumbs on the soles of the feet. Cast padding is wrapped from the nipple line to the supramalleolar region on each leg. The bony prominences of the malleoli, patella, fibular head, femoral condyles, iliac crests, and coccyx are well padded.
Fiberglass is then rolled without compression onto the patient, beginning with the torso and perineal areas. The injured leg is wrapped to its final length above the malleoli while the uninjured leg is kept free. Maintaining the position of the injured leg with simultaneous molding at the fracture site, typically to promote valgus, allows fracture reduction. The fracture position is then checked under image intensification. For femur fractures, hip abduction and flexion are set to 45° and 90°, respectively, while knee flexion is between 50° and 90°. The uninjured leg is then wrapped with fiberglass. Additional strips of fiberglass can be used to reinforce weak junctional regions between the torso and the legs, posteriorly over the “intern’s triangle” and anteriorly along the hip crease.
A connecting fiberglass bar is then created using a fiberglass roll once the cast is hardened. A 2-inch fiberglass roll is wrapped around one leg to secure its position (Figure 1A) and then rolled around the second limb (Figure 1B). Fiberglass is then pulled taut and rolled around the bridge that has been created in order to thicken the bar (Figure 2). The roll is again brought around the closest limb, wrapped back across the bridge to the other limb, and rolled out to its full length. Last, the legs are abducted 1 to 2 cm to tension the bar (Figure 3). Although this does not produce enough movement to cause a crease and a resultant ulcer, careful inspection of common pressure points (eg, popliteal fossa) should be performed after the cast is complete.
The chest towels are removed, and the final cast is inspected clinically and fluoroscopically at the fracture site before extubation. The cast is trimmed as needed to ensure room for perineal care, as well as full ankle flexion and extension without impingement. Cast edges are further petaled with plastic tape (Hy-Tape International) to provide padding and prevent the waterproof lining from tearing.
Postoperative care involves overnight observation and caregiver practice in perineal care. Frequent rotation from supine to prone is encouraged. Nurses confirm car-seat fit before discharge. If needed, radiographs are obtained 7 to 10 days later to help with wedging adjustment. The cast is removed in the clinic when adequate callus is appreciated on subsequent radiographs.
Case Series
Our experience with this technique in 16 unilateral femur fractures has been favorable (Table). Patient age ranged from 5 months to 3 years. Mean pretreatment angulation was 13° varus and 11° procurvatum. The majority of fractures were femoral shaft fractures; 1 was proximal, 2 distal.
All fractures united without cast revision. Mean cast time was 4.5 weeks (range, 16 days–6 weeks). Immediate postoperative alignment was 2.5° varus (range, 11° valgus to 16° varus) and 7° procurvatum (range, 1° recurvatum to 22° procurvatum). Mean shortening was 1.5 cm (range, 0-2.7 cm). Final alignment was 1° valgus (range, 9° valgus to 12° varus) and 5° procurvatum (range, 0° to 22°). Mean follow-up was 8 months. There were no cases of skin maceration or cast failure. No casts precluded use of a spica car-seat. Figure 4 shows a typical case with a midshaft fracture treated with closed reduction and casting for 4 weeks with good remodeling at final follow-up, 19 months after injury.
Discussion
Although single-leg walking spica casts have been shown to safely treat low-energy femur fractures in children 1 to 6 years old,7 length-unstable femur fractures, bilateral femur fractures, and patients with hip dysplasia continue to be managed with a double-leg hip spica construct. Cast integrity remains fundamental to the control of most fractures and prevention of cast-related complications, such as skin maceration and ulceration. Surgeons typically use spica cast reinforcement schemes—such as cast augments of the torso–limb junction, with multiple layers of casting material or incorporation of a connecting bar between the legs, typically constructed by overwrapping a wooden dowel in casting material—to improve the mechanical stability of casts.6 The present technique of creating a connecting bar from fiberglass casting material significantly simplifies the standard wooden dowel approach and provided excellent results in our treatment group in terms of cast integrity and fracture alignment. In addition, at our institution, a roll of fiberglass costs $2.10, whereas a wooden dowel costs $3 to $10 and can be difficult to locate if not frequently used. Other tube-shaped materials, such as the disposable material used to package implants and tubes, carry an even lower cost. However, we have found that a single fiberglass roll is most readily available and easiest to apply.
Although proper spica cast application remains important in managing pediatric trauma, it lacks a good technical description in the literature. In this technical report, we have presented our standard spica cast application method, which minimizes the range of cast complications that have been reported, from minor skin irritation to superior mesenteric artery syndrome. Two salient technical highlights are use of waterproof pantaloon liners and cast petaling, which we have found almost eliminate the morbidity of potential skin complications, reported to occur at a rate of 28%.8 In addition, we forgo applying the cast on the injured leg in segments. Application of a short-leg cast on the injured leg to allow traction on the leg during cast application is of dubious utility and may be potentially harmful, with described complications of peroneal nerve palsy and compartment syndrome.9-11 Further, it is important to use an abdominal spacer (eg, a stack of towels) under the cast padding to create room for abdominal expansion and minimize pressure thought to induce superior mesenteric artery syndrome. Plastic or rubber abdominal spacers have also been described.12,13 Last, leg position is important for reduction and maintenance of the fracture, as well as patient care. Literature advocates minimizing hip abduction to just that needed for perineal care and maximizing hip flexion and knee extension to optimize car-seat fit and safety.14
Conclusion
Construction of a spica cast lower limb connecting bar from readily available fiberglass casting material allows a facile and rapid addition to the mechanical stability of a spica cast in the treatment of pediatric femur fractures. The technique is low-cost and obviates the need for additional extraneous materials.
1. Slongo TF, Audigé L; AO Pediatric Classification Group. Fracture and dislocation classification compendium for children: the AO Pediatric Comprehensive Classification of Long Bone Fractures (PCCF). J Orthop Trauma. 2007;21(10):S135-S160.
2. Hinton RY, Lincoln A, Crockett MM, Sponseller P, Smith G. Fractures of the femoral shaft in children. Incidence, mechanisms, and sociodemographic risk factors. J Bone Joint Surg Am. 1999;81(4):500-509.
3. Campbell WC, Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 11th ed. Philadelphia, PA: Mosby Elsevier; 2008.
4. Lovell WW, Winter RB, Morrissy RT, Weinstein SL. Lovell and Winter’s Pediatric Orthopaedics. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.
5. Green NE, Swiontkowski MF, eds. Skeletal Trauma in Children. 4th ed. Philadelphia, PA: Elsevier Health Sciences; 2009.
6. Hosalkar HS, Jones S, Chowdhury M, Chatoo M, Hill RA. Connecting bar for hip spica reinforcement: does it help? J Pediatr Orthop B. 2003;12(2):100-102.
7. Flynn JM, Garner MR, Jones KJ, et al. The treatment of low-energy femoral shaft fractures: a prospective study comparing the “walking spica” with the traditional spica cast. J Bone Joint Surg Am. 2011;93(23):2196-2202.
8. DiFazio R, Vessey J, Zurakowski D, Hresko MT, Matheney T. Incidence of skin complications and associated charges in children treated with hip spica casts for femur fractures. J Pediatr Orthop. 2011;31(1):17-22.
9. Weiss AP, Schenck RC Jr, Sponseller PD, Thompson JD. Peroneal nerve palsy after early cast application for femoral fractures in children. J Pediatr Orthop. 1992;12(1):25-28.
10. Mubarak SJ, Frick S, Sink E, Rathjen K, Noonan KJ. Volkmann contracture and compartment syndromes after femur fractures in children treated with 90/90 spica casts. J Pediatr Orthop. 2006;26(5):567-572.
11. Large TM, Frick SL. Compartment syndrome of the leg after treatment of a femoral fracture with an early sitting spica cast. A report of two cases. J Bone Joint Surg Am. 2003;85(11):2207-2210.
12. Sharma S, Azzopardi T. Reduction of abdominal pressure for prophylaxis of the mesenteric artery syndrome (cast syndrome) in a hip spica—a simple technique. Ann R Coll Surg Engl. 2006;88(3):317.
13. Kiter E, Demirkan F, Kiliç BA, Erkula G. A new technique for creating an abdominal window in a hip spica cast. J Orthop Trauma. 2003;17(6):442-443.
14. Zielinski J, Oliver G, Sybesma J, Walter N, Atkinson P. Casting technique and restraint choice influence child safety during transport of body casted children subjected to a simulated frontal MVA. J Trauma. 2009;66(6):1653-1665.
1. Slongo TF, Audigé L; AO Pediatric Classification Group. Fracture and dislocation classification compendium for children: the AO Pediatric Comprehensive Classification of Long Bone Fractures (PCCF). J Orthop Trauma. 2007;21(10):S135-S160.
2. Hinton RY, Lincoln A, Crockett MM, Sponseller P, Smith G. Fractures of the femoral shaft in children. Incidence, mechanisms, and sociodemographic risk factors. J Bone Joint Surg Am. 1999;81(4):500-509.
3. Campbell WC, Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 11th ed. Philadelphia, PA: Mosby Elsevier; 2008.
4. Lovell WW, Winter RB, Morrissy RT, Weinstein SL. Lovell and Winter’s Pediatric Orthopaedics. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.
5. Green NE, Swiontkowski MF, eds. Skeletal Trauma in Children. 4th ed. Philadelphia, PA: Elsevier Health Sciences; 2009.
6. Hosalkar HS, Jones S, Chowdhury M, Chatoo M, Hill RA. Connecting bar for hip spica reinforcement: does it help? J Pediatr Orthop B. 2003;12(2):100-102.
7. Flynn JM, Garner MR, Jones KJ, et al. The treatment of low-energy femoral shaft fractures: a prospective study comparing the “walking spica” with the traditional spica cast. J Bone Joint Surg Am. 2011;93(23):2196-2202.
8. DiFazio R, Vessey J, Zurakowski D, Hresko MT, Matheney T. Incidence of skin complications and associated charges in children treated with hip spica casts for femur fractures. J Pediatr Orthop. 2011;31(1):17-22.
9. Weiss AP, Schenck RC Jr, Sponseller PD, Thompson JD. Peroneal nerve palsy after early cast application for femoral fractures in children. J Pediatr Orthop. 1992;12(1):25-28.
10. Mubarak SJ, Frick S, Sink E, Rathjen K, Noonan KJ. Volkmann contracture and compartment syndromes after femur fractures in children treated with 90/90 spica casts. J Pediatr Orthop. 2006;26(5):567-572.
11. Large TM, Frick SL. Compartment syndrome of the leg after treatment of a femoral fracture with an early sitting spica cast. A report of two cases. J Bone Joint Surg Am. 2003;85(11):2207-2210.
12. Sharma S, Azzopardi T. Reduction of abdominal pressure for prophylaxis of the mesenteric artery syndrome (cast syndrome) in a hip spica—a simple technique. Ann R Coll Surg Engl. 2006;88(3):317.
13. Kiter E, Demirkan F, Kiliç BA, Erkula G. A new technique for creating an abdominal window in a hip spica cast. J Orthop Trauma. 2003;17(6):442-443.
14. Zielinski J, Oliver G, Sybesma J, Walter N, Atkinson P. Casting technique and restraint choice influence child safety during transport of body casted children subjected to a simulated frontal MVA. J Trauma. 2009;66(6):1653-1665.