User login
Ibrutinib ‘treatment of choice’ in rel/ref MCL

Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—The BTK inhibitor ibrutinib should be considered the treatment of choice for patients with relapsed or refractory mantle cell lymphoma (MCL), according to a speaker at the 2015 ASH Annual Meeting.
Results of the phase 3 RAY trial showed that ibrutinib can prolong progression-free survival (PFS) when compared to the mTOR inhibitor temsirolimus.
There was no significant difference in overall survival (OS) between the treatment arms, but this outcome was influenced by the fact that patients were allowed to cross over from the temsirolimus arm to the ibrutinib arm after they progressed.
A majority of patients in both arms experienced adverse events (AEs), and the incidence of grade 3 or higher AEs was high—about 70% with ibrutinib and 90% with temsirolimus.
Simon Rule, MD, of Derriford Hospital in Plymouth, UK, presented these results at the meeting as abstract 469. The study has been published in The Lancet as well.
The research was sponsored by Janssen Biotech, Inc., which is jointly developing and commercializing ibrutinib with Pharmacyclics LLC, an AbbVie company.
Study design
The trial included 280 patients with relapsed or refractory MCL. They were enrolled from December 2012 to November 2013.
The patients were randomized to receive oral ibrutinib (n=139) at 560 mg or intravenous temsirolimus (n=141) at 175 mg on days 1, 8, and 15 of cycle 1 and 75 mg on days 1, 8, and 15 of all subsequent 21-day cycles until disease progression or unacceptable toxicity.
Starting July 2014, patients were allowed to cross over from the ibrutinib arm to the temsirolimus arm if they had progressive disease, as confirmed by an independent review committee. Thirty-two patients ultimately crossed over.
Patient characteristics
Baseline characteristics were similar between the treatment arms. The median age was 67 (range, 39-84) in the ibrutinib arm and 68 (range, 34-88) in the temsirolimus arm. Most patients had an ECOG performance status of 0 (48.2% and 47.5%, respectively) or 1 (51.1% in both arms).
The median number of prior therapies was 2 in both arms (range, 1-9). A majority of patients had 1 to 2 prior lines of therapy—68.3% in the ibrutinib arm and 66% in the temsirolimus arm.
The median time from the end of last therapy was 8.25 months for the ibrutinib arm and 7.03 months for the temsirolimus arm. And about 30% of patients in each arm were refractory to their last therapy—25.9% and 33.3%, respectively.
About half of patients in each arm had intermediate-risk disease (46.8% in the ibrutinib arm and 48.9% in the temsirolimus arm), followed by low-risk (31.7% and 29.8%, respectively) and high-risk disease (21.6% and 21.3%, respectively).
Most patients had stage IV disease—80.6% in the ibrutinib arm and 85.1% in the temsirolimus arm.
PFS
The study’s primary endpoint was PFS, as assessed by an independent review committee.
At a median follow-up of 20 months, the median PFS was 14.6 months for patients in the ibrutinib arm and 6.2 months for patients in the temsirolimus arm (hazard ratio=0.43, P<0.0001). At 2 years, the PFS was 41% in the ibrutinib arm and 7% in the temsirolimus arm.
Dr Rule noted that, looking at these data, people might question the validity of temsirolimus as a comparator to ibrutinib for this patient population.
“If you look at the median PFS for temsirolimus here, it’s 6.2 months,” he said. “In the registration study for Velcade—bortezomib—in the US, PFS was 6.5 months. If you look at the median PFS in the lenalidomide study that got registration, it was 4 months. So [the PFS for temsirolimus] is very representative of an oral novel agent in the context of mantle cell lymphoma.”
Dr Rule also pointed out that the improvement in PFS with ibrutinib was consistent across subgroups (ie, older age, risk score, tumor bulk, refractory disease). The only exception was patients with blastoid histology, but this was a very small group.
Secondary endpoints
The median OS was not reached in the ibrutinib arm but was 21.3 months in the temsirolimus arm.
This difference was not statistically significant, but Dr Rule noted that the trial was not powered for OS, and the analysis is confounded by the crossover. Twenty-three percent of patients in the temsirolimus arm ultimately received ibrutinib.
The overall response rate (ORR) was 71.9% in the ibrutinib arm and 40.4% in the temsirolimus arm (P<0.0001), according to the independent review committee. The complete response rates were 18.7% (n=26) and 1.4% (n=2), respectively.
The median duration of response was not reached with ibrutinib but was 7 months for temsirolimus. The median time to next treatment was not reached with ibrutinib, but it was 11.6 months in the temsirolimus arm (P<0.0001).
And the median duration of study treatment was 14.4 months in the ibrutinib arm and 3 months in the temsirolimus arm.
Timing counts
Dr Rule also presented response and PFS data according to the number of prior therapies patients received.
He noted that patients were more likely to respond to temsirolimus if they had received fewer prior therapies, but this was not the case with ibrutinib. Ibrutinib produced consistent ORRs regardless of when it was given.
In the ibrutinib arm, the ORR was 71.9% for patients who had received 1 prior line of therapy, 68.4% for those who received 2 prior therapies, and 75% for those who received 3 prior therapies. In the temsirolimus arm, the ORRs were 48%, 39.5%, and 33.3%, respectively.
Conversely, patients had a greater PFS benefit if they received ibrutinib earlier in their treatment course, but this was not true for temsirolimus.
At the median follow-up of 20 months, PFS was more than 60% for ibrutinib-treated patients who had received 1 prior line of therapy and less than 30% for ibrutinib-treated patients who received 2 or more prior lines of therapy. PFS was less than 15% for patients in the temsirolimus arm, regardless of their number of prior therapies.
“So that’s perhaps the first hint that, if we’re going to be using [ibrutinib], we should be using it earlier on,” Dr Rule said. “And I also suspect that, with further follow-up with this study, if this holds up, there will be, indeed, a survival benefit observed.”
Safety
“Despite patients on the ibrutinib arm being exposed to drug more than 4 times longer than those with temsirolimus, the frequency of most cumulative adverse events was lower in the ibrutinib arm,” Dr Rule said.
Still, he noted that most patients had some adverse events. And grade 3 or higher adverse events were reported in 67.6% of patients on ibrutinib and 87.1% of patients on temsirolimus.
Grade 3 or higher AEs included atrial fibrillation (AFib) and major bleeding. AFib occurred in 4.3% of patients in the ibrutinib arm and 1.4% in the temsirolimus arm. Major bleeding occurred in 10.1% and 6.5%, respectively.
Five of the 6 patients with AFib in the ibrutinib arm and all 3 patients who developed AFib in the temsirolimus arm had risk factors for AFib prior to treatment. None of these patients discontinued treatment due to AFib.
Dr Rule said there was no evidence to suggest that either drug increases the risk of second primary malignancies, although 3.6% of patients in the ibrutinib arm and 2.9% in the temsirolimus arm were diagnosed with second primary malignancies (mostly non-melanoma skin cancers).
The most common treatment-emergent AEs (≥20%) of any grade for the ibrutinib arm were diarrhea (28.8%), cough (22.3%), and fatigue (22.3%).
The most common treatment-emergent AEs (>20%) of any grade for the temsirolimus arm were thrombocytopenia (56.1%), anemia (43.2%), diarrhea (30.9%), fatigue (28.8%), neutropenia (25.9%), epistaxis (23.7%), cough (22.3%), peripheral edema (22.3%), nausea (21.6%), pyrexia (20.9%), and stomatitis (20.9%).
The most common hematologic AEs (≥10%) in the ibrutinib and temsirolimus arms, respectively, were thrombocytopenia (18% vs 56.1%), anemia (18% vs 43.2%), and neutropenia (15.8% vs 25.9%).
Six percent of patients in the ibrutinib arm and 26% in the temsirolimus arm discontinued treatment due to AEs.
At a median follow-up of 20 months, 42% of patients in the ibrutinib arm and 45% in the temsirolimus arm had died. The most common cause of death associated with ibrutinib was disease progression, and deaths in the temsirolimus arm were primarily attributed to AEs. ![]()
Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—The BTK inhibitor ibrutinib should be considered the treatment of choice for patients with relapsed or refractory mantle cell lymphoma (MCL), according to a speaker at the 2015 ASH Annual Meeting.
Results of the phase 3 RAY trial showed that ibrutinib can prolong progression-free survival (PFS) when compared to the mTOR inhibitor temsirolimus.
There was no significant difference in overall survival (OS) between the treatment arms, but this outcome was influenced by the fact that patients were allowed to cross over from the temsirolimus arm to the ibrutinib arm after they progressed.
A majority of patients in both arms experienced adverse events (AEs), and the incidence of grade 3 or higher AEs was high—about 70% with ibrutinib and 90% with temsirolimus.
Simon Rule, MD, of Derriford Hospital in Plymouth, UK, presented these results at the meeting as abstract 469. The study has been published in The Lancet as well.
The research was sponsored by Janssen Biotech, Inc., which is jointly developing and commercializing ibrutinib with Pharmacyclics LLC, an AbbVie company.
Study design
The trial included 280 patients with relapsed or refractory MCL. They were enrolled from December 2012 to November 2013.
The patients were randomized to receive oral ibrutinib (n=139) at 560 mg or intravenous temsirolimus (n=141) at 175 mg on days 1, 8, and 15 of cycle 1 and 75 mg on days 1, 8, and 15 of all subsequent 21-day cycles until disease progression or unacceptable toxicity.
Starting July 2014, patients were allowed to cross over from the ibrutinib arm to the temsirolimus arm if they had progressive disease, as confirmed by an independent review committee. Thirty-two patients ultimately crossed over.
Patient characteristics
Baseline characteristics were similar between the treatment arms. The median age was 67 (range, 39-84) in the ibrutinib arm and 68 (range, 34-88) in the temsirolimus arm. Most patients had an ECOG performance status of 0 (48.2% and 47.5%, respectively) or 1 (51.1% in both arms).
The median number of prior therapies was 2 in both arms (range, 1-9). A majority of patients had 1 to 2 prior lines of therapy—68.3% in the ibrutinib arm and 66% in the temsirolimus arm.
The median time from the end of last therapy was 8.25 months for the ibrutinib arm and 7.03 months for the temsirolimus arm. And about 30% of patients in each arm were refractory to their last therapy—25.9% and 33.3%, respectively.
About half of patients in each arm had intermediate-risk disease (46.8% in the ibrutinib arm and 48.9% in the temsirolimus arm), followed by low-risk (31.7% and 29.8%, respectively) and high-risk disease (21.6% and 21.3%, respectively).
Most patients had stage IV disease—80.6% in the ibrutinib arm and 85.1% in the temsirolimus arm.
PFS
The study’s primary endpoint was PFS, as assessed by an independent review committee.
At a median follow-up of 20 months, the median PFS was 14.6 months for patients in the ibrutinib arm and 6.2 months for patients in the temsirolimus arm (hazard ratio=0.43, P<0.0001). At 2 years, the PFS was 41% in the ibrutinib arm and 7% in the temsirolimus arm.
Dr Rule noted that, looking at these data, people might question the validity of temsirolimus as a comparator to ibrutinib for this patient population.
“If you look at the median PFS for temsirolimus here, it’s 6.2 months,” he said. “In the registration study for Velcade—bortezomib—in the US, PFS was 6.5 months. If you look at the median PFS in the lenalidomide study that got registration, it was 4 months. So [the PFS for temsirolimus] is very representative of an oral novel agent in the context of mantle cell lymphoma.”
Dr Rule also pointed out that the improvement in PFS with ibrutinib was consistent across subgroups (ie, older age, risk score, tumor bulk, refractory disease). The only exception was patients with blastoid histology, but this was a very small group.
Secondary endpoints
The median OS was not reached in the ibrutinib arm but was 21.3 months in the temsirolimus arm.
This difference was not statistically significant, but Dr Rule noted that the trial was not powered for OS, and the analysis is confounded by the crossover. Twenty-three percent of patients in the temsirolimus arm ultimately received ibrutinib.
The overall response rate (ORR) was 71.9% in the ibrutinib arm and 40.4% in the temsirolimus arm (P<0.0001), according to the independent review committee. The complete response rates were 18.7% (n=26) and 1.4% (n=2), respectively.
The median duration of response was not reached with ibrutinib but was 7 months for temsirolimus. The median time to next treatment was not reached with ibrutinib, but it was 11.6 months in the temsirolimus arm (P<0.0001).
And the median duration of study treatment was 14.4 months in the ibrutinib arm and 3 months in the temsirolimus arm.
Timing counts
Dr Rule also presented response and PFS data according to the number of prior therapies patients received.
He noted that patients were more likely to respond to temsirolimus if they had received fewer prior therapies, but this was not the case with ibrutinib. Ibrutinib produced consistent ORRs regardless of when it was given.
In the ibrutinib arm, the ORR was 71.9% for patients who had received 1 prior line of therapy, 68.4% for those who received 2 prior therapies, and 75% for those who received 3 prior therapies. In the temsirolimus arm, the ORRs were 48%, 39.5%, and 33.3%, respectively.
Conversely, patients had a greater PFS benefit if they received ibrutinib earlier in their treatment course, but this was not true for temsirolimus.
At the median follow-up of 20 months, PFS was more than 60% for ibrutinib-treated patients who had received 1 prior line of therapy and less than 30% for ibrutinib-treated patients who received 2 or more prior lines of therapy. PFS was less than 15% for patients in the temsirolimus arm, regardless of their number of prior therapies.
“So that’s perhaps the first hint that, if we’re going to be using [ibrutinib], we should be using it earlier on,” Dr Rule said. “And I also suspect that, with further follow-up with this study, if this holds up, there will be, indeed, a survival benefit observed.”
Safety
“Despite patients on the ibrutinib arm being exposed to drug more than 4 times longer than those with temsirolimus, the frequency of most cumulative adverse events was lower in the ibrutinib arm,” Dr Rule said.
Still, he noted that most patients had some adverse events. And grade 3 or higher adverse events were reported in 67.6% of patients on ibrutinib and 87.1% of patients on temsirolimus.
Grade 3 or higher AEs included atrial fibrillation (AFib) and major bleeding. AFib occurred in 4.3% of patients in the ibrutinib arm and 1.4% in the temsirolimus arm. Major bleeding occurred in 10.1% and 6.5%, respectively.
Five of the 6 patients with AFib in the ibrutinib arm and all 3 patients who developed AFib in the temsirolimus arm had risk factors for AFib prior to treatment. None of these patients discontinued treatment due to AFib.
Dr Rule said there was no evidence to suggest that either drug increases the risk of second primary malignancies, although 3.6% of patients in the ibrutinib arm and 2.9% in the temsirolimus arm were diagnosed with second primary malignancies (mostly non-melanoma skin cancers).
The most common treatment-emergent AEs (≥20%) of any grade for the ibrutinib arm were diarrhea (28.8%), cough (22.3%), and fatigue (22.3%).
The most common treatment-emergent AEs (>20%) of any grade for the temsirolimus arm were thrombocytopenia (56.1%), anemia (43.2%), diarrhea (30.9%), fatigue (28.8%), neutropenia (25.9%), epistaxis (23.7%), cough (22.3%), peripheral edema (22.3%), nausea (21.6%), pyrexia (20.9%), and stomatitis (20.9%).
The most common hematologic AEs (≥10%) in the ibrutinib and temsirolimus arms, respectively, were thrombocytopenia (18% vs 56.1%), anemia (18% vs 43.2%), and neutropenia (15.8% vs 25.9%).
Six percent of patients in the ibrutinib arm and 26% in the temsirolimus arm discontinued treatment due to AEs.
At a median follow-up of 20 months, 42% of patients in the ibrutinib arm and 45% in the temsirolimus arm had died. The most common cause of death associated with ibrutinib was disease progression, and deaths in the temsirolimus arm were primarily attributed to AEs. ![]()
Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—The BTK inhibitor ibrutinib should be considered the treatment of choice for patients with relapsed or refractory mantle cell lymphoma (MCL), according to a speaker at the 2015 ASH Annual Meeting.
Results of the phase 3 RAY trial showed that ibrutinib can prolong progression-free survival (PFS) when compared to the mTOR inhibitor temsirolimus.
There was no significant difference in overall survival (OS) between the treatment arms, but this outcome was influenced by the fact that patients were allowed to cross over from the temsirolimus arm to the ibrutinib arm after they progressed.
A majority of patients in both arms experienced adverse events (AEs), and the incidence of grade 3 or higher AEs was high—about 70% with ibrutinib and 90% with temsirolimus.
Simon Rule, MD, of Derriford Hospital in Plymouth, UK, presented these results at the meeting as abstract 469. The study has been published in The Lancet as well.
The research was sponsored by Janssen Biotech, Inc., which is jointly developing and commercializing ibrutinib with Pharmacyclics LLC, an AbbVie company.
Study design
The trial included 280 patients with relapsed or refractory MCL. They were enrolled from December 2012 to November 2013.
The patients were randomized to receive oral ibrutinib (n=139) at 560 mg or intravenous temsirolimus (n=141) at 175 mg on days 1, 8, and 15 of cycle 1 and 75 mg on days 1, 8, and 15 of all subsequent 21-day cycles until disease progression or unacceptable toxicity.
Starting July 2014, patients were allowed to cross over from the ibrutinib arm to the temsirolimus arm if they had progressive disease, as confirmed by an independent review committee. Thirty-two patients ultimately crossed over.
Patient characteristics
Baseline characteristics were similar between the treatment arms. The median age was 67 (range, 39-84) in the ibrutinib arm and 68 (range, 34-88) in the temsirolimus arm. Most patients had an ECOG performance status of 0 (48.2% and 47.5%, respectively) or 1 (51.1% in both arms).
The median number of prior therapies was 2 in both arms (range, 1-9). A majority of patients had 1 to 2 prior lines of therapy—68.3% in the ibrutinib arm and 66% in the temsirolimus arm.
The median time from the end of last therapy was 8.25 months for the ibrutinib arm and 7.03 months for the temsirolimus arm. And about 30% of patients in each arm were refractory to their last therapy—25.9% and 33.3%, respectively.
About half of patients in each arm had intermediate-risk disease (46.8% in the ibrutinib arm and 48.9% in the temsirolimus arm), followed by low-risk (31.7% and 29.8%, respectively) and high-risk disease (21.6% and 21.3%, respectively).
Most patients had stage IV disease—80.6% in the ibrutinib arm and 85.1% in the temsirolimus arm.
PFS
The study’s primary endpoint was PFS, as assessed by an independent review committee.
At a median follow-up of 20 months, the median PFS was 14.6 months for patients in the ibrutinib arm and 6.2 months for patients in the temsirolimus arm (hazard ratio=0.43, P<0.0001). At 2 years, the PFS was 41% in the ibrutinib arm and 7% in the temsirolimus arm.
Dr Rule noted that, looking at these data, people might question the validity of temsirolimus as a comparator to ibrutinib for this patient population.
“If you look at the median PFS for temsirolimus here, it’s 6.2 months,” he said. “In the registration study for Velcade—bortezomib—in the US, PFS was 6.5 months. If you look at the median PFS in the lenalidomide study that got registration, it was 4 months. So [the PFS for temsirolimus] is very representative of an oral novel agent in the context of mantle cell lymphoma.”
Dr Rule also pointed out that the improvement in PFS with ibrutinib was consistent across subgroups (ie, older age, risk score, tumor bulk, refractory disease). The only exception was patients with blastoid histology, but this was a very small group.
Secondary endpoints
The median OS was not reached in the ibrutinib arm but was 21.3 months in the temsirolimus arm.
This difference was not statistically significant, but Dr Rule noted that the trial was not powered for OS, and the analysis is confounded by the crossover. Twenty-three percent of patients in the temsirolimus arm ultimately received ibrutinib.
The overall response rate (ORR) was 71.9% in the ibrutinib arm and 40.4% in the temsirolimus arm (P<0.0001), according to the independent review committee. The complete response rates were 18.7% (n=26) and 1.4% (n=2), respectively.
The median duration of response was not reached with ibrutinib but was 7 months for temsirolimus. The median time to next treatment was not reached with ibrutinib, but it was 11.6 months in the temsirolimus arm (P<0.0001).
And the median duration of study treatment was 14.4 months in the ibrutinib arm and 3 months in the temsirolimus arm.
Timing counts
Dr Rule also presented response and PFS data according to the number of prior therapies patients received.
He noted that patients were more likely to respond to temsirolimus if they had received fewer prior therapies, but this was not the case with ibrutinib. Ibrutinib produced consistent ORRs regardless of when it was given.
In the ibrutinib arm, the ORR was 71.9% for patients who had received 1 prior line of therapy, 68.4% for those who received 2 prior therapies, and 75% for those who received 3 prior therapies. In the temsirolimus arm, the ORRs were 48%, 39.5%, and 33.3%, respectively.
Conversely, patients had a greater PFS benefit if they received ibrutinib earlier in their treatment course, but this was not true for temsirolimus.
At the median follow-up of 20 months, PFS was more than 60% for ibrutinib-treated patients who had received 1 prior line of therapy and less than 30% for ibrutinib-treated patients who received 2 or more prior lines of therapy. PFS was less than 15% for patients in the temsirolimus arm, regardless of their number of prior therapies.
“So that’s perhaps the first hint that, if we’re going to be using [ibrutinib], we should be using it earlier on,” Dr Rule said. “And I also suspect that, with further follow-up with this study, if this holds up, there will be, indeed, a survival benefit observed.”
Safety
“Despite patients on the ibrutinib arm being exposed to drug more than 4 times longer than those with temsirolimus, the frequency of most cumulative adverse events was lower in the ibrutinib arm,” Dr Rule said.
Still, he noted that most patients had some adverse events. And grade 3 or higher adverse events were reported in 67.6% of patients on ibrutinib and 87.1% of patients on temsirolimus.
Grade 3 or higher AEs included atrial fibrillation (AFib) and major bleeding. AFib occurred in 4.3% of patients in the ibrutinib arm and 1.4% in the temsirolimus arm. Major bleeding occurred in 10.1% and 6.5%, respectively.
Five of the 6 patients with AFib in the ibrutinib arm and all 3 patients who developed AFib in the temsirolimus arm had risk factors for AFib prior to treatment. None of these patients discontinued treatment due to AFib.
Dr Rule said there was no evidence to suggest that either drug increases the risk of second primary malignancies, although 3.6% of patients in the ibrutinib arm and 2.9% in the temsirolimus arm were diagnosed with second primary malignancies (mostly non-melanoma skin cancers).
The most common treatment-emergent AEs (≥20%) of any grade for the ibrutinib arm were diarrhea (28.8%), cough (22.3%), and fatigue (22.3%).
The most common treatment-emergent AEs (>20%) of any grade for the temsirolimus arm were thrombocytopenia (56.1%), anemia (43.2%), diarrhea (30.9%), fatigue (28.8%), neutropenia (25.9%), epistaxis (23.7%), cough (22.3%), peripheral edema (22.3%), nausea (21.6%), pyrexia (20.9%), and stomatitis (20.9%).
The most common hematologic AEs (≥10%) in the ibrutinib and temsirolimus arms, respectively, were thrombocytopenia (18% vs 56.1%), anemia (18% vs 43.2%), and neutropenia (15.8% vs 25.9%).
Six percent of patients in the ibrutinib arm and 26% in the temsirolimus arm discontinued treatment due to AEs.
At a median follow-up of 20 months, 42% of patients in the ibrutinib arm and 45% in the temsirolimus arm had died. The most common cause of death associated with ibrutinib was disease progression, and deaths in the temsirolimus arm were primarily attributed to AEs. ![]()
ALL patients over-report their 6MP compliance, researchers say

Photo courtesy of ASH
ORLANDO, FL—A study comparing subjective versus objective reporting of treatment compliance in patients with acute lymphoblastic leukemia (ALL) has shown that about a fourth of patients over-report how compliant they are with taking 6-mercaptopurine (6MP) as part of their maintenance therapy.
An earlier analysis of the Children’s Oncology Group (COG) AALL03N1 compliance study showed that adherence rates of less than 95% were associated with a 3.7-fold increased risk of relapse.
And about 40% of patients were non-adherent. Yet patients indicate they are taking their medication when questioned.
“We ask our patients if they are taking their meds,” said Wendy Landier, PhD, “and they tell us they are.”
“Even in this cohort who were being closely monitored and knew that they were being closely monitored electronically and were asked to self-report, we found over-reporting.”
Dr Landier, of the University of Alabama at Birmingham, reported these findings comparing self-reported adherence with electronic monitoring of 6MP intake at the 2015 ASH Annual Meeting (abstract 82).
The investigators collected data over 6 months from 416 ALL patients who were 21 years at diagnosis or younger and were receiving 6MP as part of their maintenance therapy.
Investigators measured subjective self-reporting by a patient questionnaire, which included patient demographic information in addition to the number of days the patient took 6MP over the past month.
For the objective medication event-monitoring system (MEMS), patients received a 6MP bottle that was fitted with a TrackCapTM. The cap had a microprocessor chip that recorded the date and time of each bottle opening.
Investigators downloaded the data at the end of the study. They then compared the MEMS with the self-reported data.
The investigators classified perfect reporters as those whose self-report corresponded to their MEMS.
They classified over-reporters as those whose self-report was greater than their MEMS data for 5 days or more and 50% of the months.
The rest they classified as others.
Patients were a median age of 6.0 years, and 277 (66.6%) were male. Parents completed the survey for patients younger than 12.
Two hundred forty-two patients (60.9%) had fathers whose education was less than some college, 159 (38.4%) had NCI high-risk disease, and 168 (40.4%) were non-adherent to 6MP as determined by earlier analysis of the COG AALL03N1 study.
Thirty-six percent were non-Hispanic white, 37% were Hispanic, 14% Asian, and 13% African American.
The investigators monitored the patients’ 6MP intake for a total of 1344 patient-months at 87 COG sites.
And the correlation between subjective and objective reporting was moderate, Dr Landier said, with the correlation ranging from 0.36 to 0.58.
Twelve percent of the patients were perfect reporters, with no difference between the reporting methods.
Twenty-four percent over-reported their intake, 1% under-reported their intake, and 64% were other.
The investigators analyzed variables associated with over-reporting and found that age 12 years or older (P=0.02), being Hispanic (P=0.02), Asian (P=0.02), or African American (P<0.001), paternal education less than college (P=0.02), and being classified as 6MP non-adherent (P<0.001) were all significant.
“Over-reporting of 6MP ingestion is common,” Dr Landier said, with 88% of patients or parents over-reporting the number of days 6MP was taken.
“What we’ve learned from this study is that we cannot rely on patients’ self-report in the clinic,” she said. “What we found is that only 12% of our patients are perfect, so to speak, and that the others mainly over-estimate, and I don’t believe intentionally.”
“[W]e need to have a better way of identifying which patients are at risk for over-reporting their intake, whether they’re aware of it or not,” she added. ![]()
Photo courtesy of ASH
ORLANDO, FL—A study comparing subjective versus objective reporting of treatment compliance in patients with acute lymphoblastic leukemia (ALL) has shown that about a fourth of patients over-report how compliant they are with taking 6-mercaptopurine (6MP) as part of their maintenance therapy.
An earlier analysis of the Children’s Oncology Group (COG) AALL03N1 compliance study showed that adherence rates of less than 95% were associated with a 3.7-fold increased risk of relapse.
And about 40% of patients were non-adherent. Yet patients indicate they are taking their medication when questioned.
“We ask our patients if they are taking their meds,” said Wendy Landier, PhD, “and they tell us they are.”
“Even in this cohort who were being closely monitored and knew that they were being closely monitored electronically and were asked to self-report, we found over-reporting.”
Dr Landier, of the University of Alabama at Birmingham, reported these findings comparing self-reported adherence with electronic monitoring of 6MP intake at the 2015 ASH Annual Meeting (abstract 82).
The investigators collected data over 6 months from 416 ALL patients who were 21 years at diagnosis or younger and were receiving 6MP as part of their maintenance therapy.
Investigators measured subjective self-reporting by a patient questionnaire, which included patient demographic information in addition to the number of days the patient took 6MP over the past month.
For the objective medication event-monitoring system (MEMS), patients received a 6MP bottle that was fitted with a TrackCapTM. The cap had a microprocessor chip that recorded the date and time of each bottle opening.
Investigators downloaded the data at the end of the study. They then compared the MEMS with the self-reported data.
The investigators classified perfect reporters as those whose self-report corresponded to their MEMS.
They classified over-reporters as those whose self-report was greater than their MEMS data for 5 days or more and 50% of the months.
The rest they classified as others.
Patients were a median age of 6.0 years, and 277 (66.6%) were male. Parents completed the survey for patients younger than 12.
Two hundred forty-two patients (60.9%) had fathers whose education was less than some college, 159 (38.4%) had NCI high-risk disease, and 168 (40.4%) were non-adherent to 6MP as determined by earlier analysis of the COG AALL03N1 study.
Thirty-six percent were non-Hispanic white, 37% were Hispanic, 14% Asian, and 13% African American.
The investigators monitored the patients’ 6MP intake for a total of 1344 patient-months at 87 COG sites.
And the correlation between subjective and objective reporting was moderate, Dr Landier said, with the correlation ranging from 0.36 to 0.58.
Twelve percent of the patients were perfect reporters, with no difference between the reporting methods.
Twenty-four percent over-reported their intake, 1% under-reported their intake, and 64% were other.
The investigators analyzed variables associated with over-reporting and found that age 12 years or older (P=0.02), being Hispanic (P=0.02), Asian (P=0.02), or African American (P<0.001), paternal education less than college (P=0.02), and being classified as 6MP non-adherent (P<0.001) were all significant.
“Over-reporting of 6MP ingestion is common,” Dr Landier said, with 88% of patients or parents over-reporting the number of days 6MP was taken.
“What we’ve learned from this study is that we cannot rely on patients’ self-report in the clinic,” she said. “What we found is that only 12% of our patients are perfect, so to speak, and that the others mainly over-estimate, and I don’t believe intentionally.”
“[W]e need to have a better way of identifying which patients are at risk for over-reporting their intake, whether they’re aware of it or not,” she added. ![]()
Photo courtesy of ASH
ORLANDO, FL—A study comparing subjective versus objective reporting of treatment compliance in patients with acute lymphoblastic leukemia (ALL) has shown that about a fourth of patients over-report how compliant they are with taking 6-mercaptopurine (6MP) as part of their maintenance therapy.
An earlier analysis of the Children’s Oncology Group (COG) AALL03N1 compliance study showed that adherence rates of less than 95% were associated with a 3.7-fold increased risk of relapse.
And about 40% of patients were non-adherent. Yet patients indicate they are taking their medication when questioned.
“We ask our patients if they are taking their meds,” said Wendy Landier, PhD, “and they tell us they are.”
“Even in this cohort who were being closely monitored and knew that they were being closely monitored electronically and were asked to self-report, we found over-reporting.”
Dr Landier, of the University of Alabama at Birmingham, reported these findings comparing self-reported adherence with electronic monitoring of 6MP intake at the 2015 ASH Annual Meeting (abstract 82).
The investigators collected data over 6 months from 416 ALL patients who were 21 years at diagnosis or younger and were receiving 6MP as part of their maintenance therapy.
Investigators measured subjective self-reporting by a patient questionnaire, which included patient demographic information in addition to the number of days the patient took 6MP over the past month.
For the objective medication event-monitoring system (MEMS), patients received a 6MP bottle that was fitted with a TrackCapTM. The cap had a microprocessor chip that recorded the date and time of each bottle opening.
Investigators downloaded the data at the end of the study. They then compared the MEMS with the self-reported data.
The investigators classified perfect reporters as those whose self-report corresponded to their MEMS.
They classified over-reporters as those whose self-report was greater than their MEMS data for 5 days or more and 50% of the months.
The rest they classified as others.
Patients were a median age of 6.0 years, and 277 (66.6%) were male. Parents completed the survey for patients younger than 12.
Two hundred forty-two patients (60.9%) had fathers whose education was less than some college, 159 (38.4%) had NCI high-risk disease, and 168 (40.4%) were non-adherent to 6MP as determined by earlier analysis of the COG AALL03N1 study.
Thirty-six percent were non-Hispanic white, 37% were Hispanic, 14% Asian, and 13% African American.
The investigators monitored the patients’ 6MP intake for a total of 1344 patient-months at 87 COG sites.
And the correlation between subjective and objective reporting was moderate, Dr Landier said, with the correlation ranging from 0.36 to 0.58.
Twelve percent of the patients were perfect reporters, with no difference between the reporting methods.
Twenty-four percent over-reported their intake, 1% under-reported their intake, and 64% were other.
The investigators analyzed variables associated with over-reporting and found that age 12 years or older (P=0.02), being Hispanic (P=0.02), Asian (P=0.02), or African American (P<0.001), paternal education less than college (P=0.02), and being classified as 6MP non-adherent (P<0.001) were all significant.
“Over-reporting of 6MP ingestion is common,” Dr Landier said, with 88% of patients or parents over-reporting the number of days 6MP was taken.
“What we’ve learned from this study is that we cannot rely on patients’ self-report in the clinic,” she said. “What we found is that only 12% of our patients are perfect, so to speak, and that the others mainly over-estimate, and I don’t believe intentionally.”
“[W]e need to have a better way of identifying which patients are at risk for over-reporting their intake, whether they’re aware of it or not,” she added. ![]()
Engineers create ‘smart wound dressing’

a matrix of polymer islands
(red) that can encapsulate
electronic components
Photo by Melanie Gonick/MIT
Engineers say they have designed “smart wound dressing,” a sticky, stretchy, gel-like material that can incorporate temperature sensors, LED lights, and other electronics, as well as tiny, drug-delivering reservoirs and channels.
The dressing releases medicine in response to changes in skin temperature and can be designed to light up if, say, medicine is running low.
When the dressing is applied to a highly flexible area, such as the elbow or knee, it stretches with the body, keeping the embedded electronics functional and intact.
The key to the design is a hydrogel matrix designed by Xuanhe Zhao, PhD, of the Massachusetts Institute of Technology in Cambridge.
The hydrogel, which was describe in Nature Materials last month, is a rubbery material, mostly composed of water, designed to bond strongly to surfaces such as gold, titanium, aluminum, silicon, glass, and ceramic.
In a paper published in Advanced Materials, Dr Zhao and his colleagues described embedding various electronics within the hydrogel, such as conductive wires, semiconductor chips, LED lights, and temperature sensors.
Dr Zhao said electronics coated in hydrogel may be used not just on the surface of the skin but also inside the body; for example, as implanted, biocompatible glucose sensors, or even soft, compliant neural probes.
“Electronics are usually hard and dry, but the human body is soft and wet,” Dr Zhao said. “These two systems have drastically different properties. If you want to put electronics in close contact with the human body for applications such as healthcare monitoring and drug delivery, it is highly desirable to make the electronic devices soft and stretchable to fit the environment of the human body. That’s the motivation for stretchable hydrogel electronics.”
A strong and stretchy bond
Typical synthetic hydrogels are brittle, barely stretchable, and adhere weakly to other surfaces.
“They’re often used as degradable biomaterials at the current stage,” Dr Zhao said. “If you want to make an electronic device out of hydrogels, you need to think of long-term stability of the hydrogels and interfaces.”
To get around these challenges, his team came up with a design strategy for robust hydrogels, mixing water with a small amount of selected biopolymers to create soft, stretchy materials with a stiffness of 10 to 100 kilopascals—about the range of human soft tissues. The researchers also devised a method to strongly bond the hydrogel to various nonporous surfaces.
In the new study, the researchers applied their techniques to demonstrate several uses for the hydrogel, including encapsulating a titanium wire to form a transparent, stretchable conductor. In experiments, they stretched the encapsulated wire multiple times and found it maintained constant electrical conductivity.
Dr Zhao also created an array of LED lights embedded in a sheet of hydrogel. When attached to different regions of the body, the array continued working, even when stretched across highly deformable areas such as the knee and elbow.
A versatile matrix
Finally, the group embedded various electronic components within a sheet of hydrogel to create a “smart wound dressing,” comprising regularly spaced temperature sensors and tiny drug reservoirs.
The researchers also created pathways for drugs to flow through the hydrogel, by either inserting patterned tubes or drilling tiny holes through the matrix. They placed the dressing over various regions of the body and found that, even when highly stretched, the dressing continued to monitor skin temperature and release drugs according to the sensor readings.
An immediate application of the technology may be as a stretchable, on-demand treatment for burns or other skin conditions, said Hyunwoo Yuk, a graduate student at MIT.
“It’s a very versatile matrix,” Yuk said. “The unique capability here is, when a sensor senses something different, like an abnormal increase in temperature, the device can, on demand, release drugs to that specific location and select a specific drug from one of the reservoirs, which can diffuse in the hydrogel matrix for sustained release over time.”
Delving deeper, Dr Zhao envisions hydrogel to be an ideal, biocompatible vehicle for delivering electronics inside the body. He is currently exploring hydrogel’s potential as a carrier for glucose sensors as well as neural probes. ![]()
a matrix of polymer islands
(red) that can encapsulate
electronic components
Photo by Melanie Gonick/MIT
Engineers say they have designed “smart wound dressing,” a sticky, stretchy, gel-like material that can incorporate temperature sensors, LED lights, and other electronics, as well as tiny, drug-delivering reservoirs and channels.
The dressing releases medicine in response to changes in skin temperature and can be designed to light up if, say, medicine is running low.
When the dressing is applied to a highly flexible area, such as the elbow or knee, it stretches with the body, keeping the embedded electronics functional and intact.
The key to the design is a hydrogel matrix designed by Xuanhe Zhao, PhD, of the Massachusetts Institute of Technology in Cambridge.
The hydrogel, which was describe in Nature Materials last month, is a rubbery material, mostly composed of water, designed to bond strongly to surfaces such as gold, titanium, aluminum, silicon, glass, and ceramic.
In a paper published in Advanced Materials, Dr Zhao and his colleagues described embedding various electronics within the hydrogel, such as conductive wires, semiconductor chips, LED lights, and temperature sensors.
Dr Zhao said electronics coated in hydrogel may be used not just on the surface of the skin but also inside the body; for example, as implanted, biocompatible glucose sensors, or even soft, compliant neural probes.
“Electronics are usually hard and dry, but the human body is soft and wet,” Dr Zhao said. “These two systems have drastically different properties. If you want to put electronics in close contact with the human body for applications such as healthcare monitoring and drug delivery, it is highly desirable to make the electronic devices soft and stretchable to fit the environment of the human body. That’s the motivation for stretchable hydrogel electronics.”
A strong and stretchy bond
Typical synthetic hydrogels are brittle, barely stretchable, and adhere weakly to other surfaces.
“They’re often used as degradable biomaterials at the current stage,” Dr Zhao said. “If you want to make an electronic device out of hydrogels, you need to think of long-term stability of the hydrogels and interfaces.”
To get around these challenges, his team came up with a design strategy for robust hydrogels, mixing water with a small amount of selected biopolymers to create soft, stretchy materials with a stiffness of 10 to 100 kilopascals—about the range of human soft tissues. The researchers also devised a method to strongly bond the hydrogel to various nonporous surfaces.
In the new study, the researchers applied their techniques to demonstrate several uses for the hydrogel, including encapsulating a titanium wire to form a transparent, stretchable conductor. In experiments, they stretched the encapsulated wire multiple times and found it maintained constant electrical conductivity.
Dr Zhao also created an array of LED lights embedded in a sheet of hydrogel. When attached to different regions of the body, the array continued working, even when stretched across highly deformable areas such as the knee and elbow.
A versatile matrix
Finally, the group embedded various electronic components within a sheet of hydrogel to create a “smart wound dressing,” comprising regularly spaced temperature sensors and tiny drug reservoirs.
The researchers also created pathways for drugs to flow through the hydrogel, by either inserting patterned tubes or drilling tiny holes through the matrix. They placed the dressing over various regions of the body and found that, even when highly stretched, the dressing continued to monitor skin temperature and release drugs according to the sensor readings.
An immediate application of the technology may be as a stretchable, on-demand treatment for burns or other skin conditions, said Hyunwoo Yuk, a graduate student at MIT.
“It’s a very versatile matrix,” Yuk said. “The unique capability here is, when a sensor senses something different, like an abnormal increase in temperature, the device can, on demand, release drugs to that specific location and select a specific drug from one of the reservoirs, which can diffuse in the hydrogel matrix for sustained release over time.”
Delving deeper, Dr Zhao envisions hydrogel to be an ideal, biocompatible vehicle for delivering electronics inside the body. He is currently exploring hydrogel’s potential as a carrier for glucose sensors as well as neural probes. ![]()
a matrix of polymer islands
(red) that can encapsulate
electronic components
Photo by Melanie Gonick/MIT
Engineers say they have designed “smart wound dressing,” a sticky, stretchy, gel-like material that can incorporate temperature sensors, LED lights, and other electronics, as well as tiny, drug-delivering reservoirs and channels.
The dressing releases medicine in response to changes in skin temperature and can be designed to light up if, say, medicine is running low.
When the dressing is applied to a highly flexible area, such as the elbow or knee, it stretches with the body, keeping the embedded electronics functional and intact.
The key to the design is a hydrogel matrix designed by Xuanhe Zhao, PhD, of the Massachusetts Institute of Technology in Cambridge.
The hydrogel, which was describe in Nature Materials last month, is a rubbery material, mostly composed of water, designed to bond strongly to surfaces such as gold, titanium, aluminum, silicon, glass, and ceramic.
In a paper published in Advanced Materials, Dr Zhao and his colleagues described embedding various electronics within the hydrogel, such as conductive wires, semiconductor chips, LED lights, and temperature sensors.
Dr Zhao said electronics coated in hydrogel may be used not just on the surface of the skin but also inside the body; for example, as implanted, biocompatible glucose sensors, or even soft, compliant neural probes.
“Electronics are usually hard and dry, but the human body is soft and wet,” Dr Zhao said. “These two systems have drastically different properties. If you want to put electronics in close contact with the human body for applications such as healthcare monitoring and drug delivery, it is highly desirable to make the electronic devices soft and stretchable to fit the environment of the human body. That’s the motivation for stretchable hydrogel electronics.”
A strong and stretchy bond
Typical synthetic hydrogels are brittle, barely stretchable, and adhere weakly to other surfaces.
“They’re often used as degradable biomaterials at the current stage,” Dr Zhao said. “If you want to make an electronic device out of hydrogels, you need to think of long-term stability of the hydrogels and interfaces.”
To get around these challenges, his team came up with a design strategy for robust hydrogels, mixing water with a small amount of selected biopolymers to create soft, stretchy materials with a stiffness of 10 to 100 kilopascals—about the range of human soft tissues. The researchers also devised a method to strongly bond the hydrogel to various nonporous surfaces.
In the new study, the researchers applied their techniques to demonstrate several uses for the hydrogel, including encapsulating a titanium wire to form a transparent, stretchable conductor. In experiments, they stretched the encapsulated wire multiple times and found it maintained constant electrical conductivity.
Dr Zhao also created an array of LED lights embedded in a sheet of hydrogel. When attached to different regions of the body, the array continued working, even when stretched across highly deformable areas such as the knee and elbow.
A versatile matrix
Finally, the group embedded various electronic components within a sheet of hydrogel to create a “smart wound dressing,” comprising regularly spaced temperature sensors and tiny drug reservoirs.
The researchers also created pathways for drugs to flow through the hydrogel, by either inserting patterned tubes or drilling tiny holes through the matrix. They placed the dressing over various regions of the body and found that, even when highly stretched, the dressing continued to monitor skin temperature and release drugs according to the sensor readings.
An immediate application of the technology may be as a stretchable, on-demand treatment for burns or other skin conditions, said Hyunwoo Yuk, a graduate student at MIT.
“It’s a very versatile matrix,” Yuk said. “The unique capability here is, when a sensor senses something different, like an abnormal increase in temperature, the device can, on demand, release drugs to that specific location and select a specific drug from one of the reservoirs, which can diffuse in the hydrogel matrix for sustained release over time.”
Delving deeper, Dr Zhao envisions hydrogel to be an ideal, biocompatible vehicle for delivering electronics inside the body. He is currently exploring hydrogel’s potential as a carrier for glucose sensors as well as neural probes. ![]()
VIDEO: Top-line results from Tourmaline in multiple myeloma, plus ongoing trials and treatment selection
ORLANDO – The combination of the oral proteasome inhibitor ixazomib (Ninlaro, recently approved by the Food and Drug Administration) with lenalidomide and dexamethasone was associated with a 35% improvement in progression free survival in the Tourmaline trial.
In a video interview, Tourmaline investigator Dr. Shaji Kumar, professor of medicine at the Mayo Clinic, Rochester, Minn., discussed the top-line study results, the status of ongoing trials with ixazomib in other combination regimens, and the decision rationales that will need to be considered in selecting one of the newly approved multiple myeloma therapies.
Dr. Kumar has received funding from Takeda, the makers of ixazomib; he has also received funding from Celgene, Onyx, Janssen, and Sanofi.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – The combination of the oral proteasome inhibitor ixazomib (Ninlaro, recently approved by the Food and Drug Administration) with lenalidomide and dexamethasone was associated with a 35% improvement in progression free survival in the Tourmaline trial.
In a video interview, Tourmaline investigator Dr. Shaji Kumar, professor of medicine at the Mayo Clinic, Rochester, Minn., discussed the top-line study results, the status of ongoing trials with ixazomib in other combination regimens, and the decision rationales that will need to be considered in selecting one of the newly approved multiple myeloma therapies.
Dr. Kumar has received funding from Takeda, the makers of ixazomib; he has also received funding from Celgene, Onyx, Janssen, and Sanofi.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – The combination of the oral proteasome inhibitor ixazomib (Ninlaro, recently approved by the Food and Drug Administration) with lenalidomide and dexamethasone was associated with a 35% improvement in progression free survival in the Tourmaline trial.
In a video interview, Tourmaline investigator Dr. Shaji Kumar, professor of medicine at the Mayo Clinic, Rochester, Minn., discussed the top-line study results, the status of ongoing trials with ixazomib in other combination regimens, and the decision rationales that will need to be considered in selecting one of the newly approved multiple myeloma therapies.
Dr. Kumar has received funding from Takeda, the makers of ixazomib; he has also received funding from Celgene, Onyx, Janssen, and Sanofi.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2015

ASH: Rituximab add-on therapy ‘new standard’ in BCP-ALL
ORLANDO – Adding rituximab to standard intensive chemotherapy significantly improved event-free survival in adults with Philadelphia-negative, CD20-positive B-cell precursor acute lymphoblastic leukemia in the phase III GRAALL-R 2005 study.
Rituximab (Rituxan) is already being used to improve outcomes in patients with lymphoma, and non-randomized data support addition of the anti-CD20 monoclonal antibody to chemotherapy in B-cell precursor (BCP) ALL, where the CD20 antigen is expressed in 30% to 40% of patients at diagnosis.
In the randomized GRAALL-R 2005, 2-year event-free survival (EFS) was 65% in the rituximab arm vs. 52% in the control arm (hazard ratio, 0.66; P = .038).

This difference is not explained by the early response rates, which were very close in both arms after one or two induction courses (92% vs. 90%; P = .63), Dr. Sébastien Maury, Hôpital Henri Mondor in Créteil, France, said during the plenary session at the annual meeting of the American Society of Hematology (Ab. 1).
The beneficial effect of rituximab, however, was clearly related to the cumulative incidence of relapse at 18% with vs. 32% without rituximab (HR, 0.52; P = .017).
Despite this advantage, overall survival was similar between patients given chemotherapy with and without rituximab (71% vs. 64%; HR, 0.70; P = .095), he said.
After censoring for patients not receiving allogeneic stem cell transplant in first complete remission, however, rituximab significantly prolonged 2-year EFS (HR, 0.59; P = .021) as well as overall survival (HR, 0.55; P = .018), Dr. Maury said.
“We thus recommend that the addition of rituximab become a new standard of care for these patients, although some aspects including the definition of the optimal dose needs to be determined in further studies,” he concluded.
Dr. Adele Fielding of University College London, who introduced the study at the meeting, said, “For me, the prior knowledge that the drug can be safely already added to chemotherapy in other settings provides profound comfort in a disease in which so many people are already damaged by the current therapies we offer.”
Also, of importance is the potential relevance of rituximab in patients in whom CD20 is present on fewer than 20% of blasts at diagnosis.
Key questions that remain in rituximab therapy of ALL beyond overall benefit include early and late toxicities and how best to judge response and when, she said. Synergies with other agents and the mechanism of action will also require clarification, especially which effector cells are relevant to ensure that agents are not used with rituximab that destroy the optimal chance for response.
“Finally, the cost of introducing novel agents cannot be ignored, even in well-developed economies, and I am hopeful that the cost of this drug will be variable for many countries and many patients.” Dr. Fielding said.
Study details
A total of 220 patients, aged 18-59 years, with newly diagnosed CD20-positive Ph-negative BCP-ALL were randomized to the pediatric-inspired Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) chemotherapy protocol with or without rituximab 375 mg/m2 given during induction (day 1 and 7), salvage reinduction when needed (day 1 and 7), consolidation blocks (6 infusions), late intensification (day 1 and 7, and the first year of maintenance [6 infusions], for a total of 16-18 infusions. Allogeneic stem cell transplant (SCT) was offered after consolidation blocks 1 or 2 to patients with one or more high-risk criteria and an available donor. CD20-positivity was defined as expression of CD20 in more than 20% of leukemia blasts.
Eleven patients were excluded from the analysis because of non–ineligibility criteria, leaving 209 patients in the modified intent-to-treat analysis. Their median age was 40.2 years and 67% had high-risk ALL.
Rates of postinduction minimal residual disease less than 10–4 were 65% and 61% in the rituximab and control arms among 85 evaluable patients(P = .82), and rates of postconsolidation minimal residual disease less than 10–4 were 91% and 82% among 80 evaluable patients (P = .31), Dr. Maury reported.
Notably, more patients in the rituximab arm received allogeneic SCT in their first complete remission (34% vs. 20%; P = .029).
The cumulative incidence of death in first complete remission was 12% in both arms.
In multivariate analysis, rituximab impacted EFS, together with age, central nervous system involvement, white blood cells, or CD20 expression at diagnosis. A preferential effect with rituximab was seen in patients with high CD20 levels that deserves further evaluation, he observed.
There was no difference in the incidence of adverse events between the rituximab and control arms, although there was a trend for more infectious events with rituximab (71 events vs. 55 events), Dr. Maury said.
Allergic events – all but one from aspergillosis – were significantly more common in the control arm (2 events vs. 14 events; P = .002).
The Group for Research in Adult Acute Lymphoblastic Leukemia sponsored the study. Dr. Maury reported having no disclosures.
ORLANDO – Adding rituximab to standard intensive chemotherapy significantly improved event-free survival in adults with Philadelphia-negative, CD20-positive B-cell precursor acute lymphoblastic leukemia in the phase III GRAALL-R 2005 study.
Rituximab (Rituxan) is already being used to improve outcomes in patients with lymphoma, and non-randomized data support addition of the anti-CD20 monoclonal antibody to chemotherapy in B-cell precursor (BCP) ALL, where the CD20 antigen is expressed in 30% to 40% of patients at diagnosis.
In the randomized GRAALL-R 2005, 2-year event-free survival (EFS) was 65% in the rituximab arm vs. 52% in the control arm (hazard ratio, 0.66; P = .038).
This difference is not explained by the early response rates, which were very close in both arms after one or two induction courses (92% vs. 90%; P = .63), Dr. Sébastien Maury, Hôpital Henri Mondor in Créteil, France, said during the plenary session at the annual meeting of the American Society of Hematology (Ab. 1).
The beneficial effect of rituximab, however, was clearly related to the cumulative incidence of relapse at 18% with vs. 32% without rituximab (HR, 0.52; P = .017).
Despite this advantage, overall survival was similar between patients given chemotherapy with and without rituximab (71% vs. 64%; HR, 0.70; P = .095), he said.
After censoring for patients not receiving allogeneic stem cell transplant in first complete remission, however, rituximab significantly prolonged 2-year EFS (HR, 0.59; P = .021) as well as overall survival (HR, 0.55; P = .018), Dr. Maury said.
“We thus recommend that the addition of rituximab become a new standard of care for these patients, although some aspects including the definition of the optimal dose needs to be determined in further studies,” he concluded.
Dr. Adele Fielding of University College London, who introduced the study at the meeting, said, “For me, the prior knowledge that the drug can be safely already added to chemotherapy in other settings provides profound comfort in a disease in which so many people are already damaged by the current therapies we offer.”
Also, of importance is the potential relevance of rituximab in patients in whom CD20 is present on fewer than 20% of blasts at diagnosis.
Key questions that remain in rituximab therapy of ALL beyond overall benefit include early and late toxicities and how best to judge response and when, she said. Synergies with other agents and the mechanism of action will also require clarification, especially which effector cells are relevant to ensure that agents are not used with rituximab that destroy the optimal chance for response.
“Finally, the cost of introducing novel agents cannot be ignored, even in well-developed economies, and I am hopeful that the cost of this drug will be variable for many countries and many patients.” Dr. Fielding said.
Study details
A total of 220 patients, aged 18-59 years, with newly diagnosed CD20-positive Ph-negative BCP-ALL were randomized to the pediatric-inspired Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) chemotherapy protocol with or without rituximab 375 mg/m2 given during induction (day 1 and 7), salvage reinduction when needed (day 1 and 7), consolidation blocks (6 infusions), late intensification (day 1 and 7, and the first year of maintenance [6 infusions], for a total of 16-18 infusions. Allogeneic stem cell transplant (SCT) was offered after consolidation blocks 1 or 2 to patients with one or more high-risk criteria and an available donor. CD20-positivity was defined as expression of CD20 in more than 20% of leukemia blasts.
Eleven patients were excluded from the analysis because of non–ineligibility criteria, leaving 209 patients in the modified intent-to-treat analysis. Their median age was 40.2 years and 67% had high-risk ALL.
Rates of postinduction minimal residual disease less than 10–4 were 65% and 61% in the rituximab and control arms among 85 evaluable patients(P = .82), and rates of postconsolidation minimal residual disease less than 10–4 were 91% and 82% among 80 evaluable patients (P = .31), Dr. Maury reported.
Notably, more patients in the rituximab arm received allogeneic SCT in their first complete remission (34% vs. 20%; P = .029).
The cumulative incidence of death in first complete remission was 12% in both arms.
In multivariate analysis, rituximab impacted EFS, together with age, central nervous system involvement, white blood cells, or CD20 expression at diagnosis. A preferential effect with rituximab was seen in patients with high CD20 levels that deserves further evaluation, he observed.
There was no difference in the incidence of adverse events between the rituximab and control arms, although there was a trend for more infectious events with rituximab (71 events vs. 55 events), Dr. Maury said.
Allergic events – all but one from aspergillosis – were significantly more common in the control arm (2 events vs. 14 events; P = .002).
The Group for Research in Adult Acute Lymphoblastic Leukemia sponsored the study. Dr. Maury reported having no disclosures.
ORLANDO – Adding rituximab to standard intensive chemotherapy significantly improved event-free survival in adults with Philadelphia-negative, CD20-positive B-cell precursor acute lymphoblastic leukemia in the phase III GRAALL-R 2005 study.
Rituximab (Rituxan) is already being used to improve outcomes in patients with lymphoma, and non-randomized data support addition of the anti-CD20 monoclonal antibody to chemotherapy in B-cell precursor (BCP) ALL, where the CD20 antigen is expressed in 30% to 40% of patients at diagnosis.
In the randomized GRAALL-R 2005, 2-year event-free survival (EFS) was 65% in the rituximab arm vs. 52% in the control arm (hazard ratio, 0.66; P = .038).
This difference is not explained by the early response rates, which were very close in both arms after one or two induction courses (92% vs. 90%; P = .63), Dr. Sébastien Maury, Hôpital Henri Mondor in Créteil, France, said during the plenary session at the annual meeting of the American Society of Hematology (Ab. 1).
The beneficial effect of rituximab, however, was clearly related to the cumulative incidence of relapse at 18% with vs. 32% without rituximab (HR, 0.52; P = .017).
Despite this advantage, overall survival was similar between patients given chemotherapy with and without rituximab (71% vs. 64%; HR, 0.70; P = .095), he said.
After censoring for patients not receiving allogeneic stem cell transplant in first complete remission, however, rituximab significantly prolonged 2-year EFS (HR, 0.59; P = .021) as well as overall survival (HR, 0.55; P = .018), Dr. Maury said.
“We thus recommend that the addition of rituximab become a new standard of care for these patients, although some aspects including the definition of the optimal dose needs to be determined in further studies,” he concluded.
Dr. Adele Fielding of University College London, who introduced the study at the meeting, said, “For me, the prior knowledge that the drug can be safely already added to chemotherapy in other settings provides profound comfort in a disease in which so many people are already damaged by the current therapies we offer.”
Also, of importance is the potential relevance of rituximab in patients in whom CD20 is present on fewer than 20% of blasts at diagnosis.
Key questions that remain in rituximab therapy of ALL beyond overall benefit include early and late toxicities and how best to judge response and when, she said. Synergies with other agents and the mechanism of action will also require clarification, especially which effector cells are relevant to ensure that agents are not used with rituximab that destroy the optimal chance for response.
“Finally, the cost of introducing novel agents cannot be ignored, even in well-developed economies, and I am hopeful that the cost of this drug will be variable for many countries and many patients.” Dr. Fielding said.
Study details
A total of 220 patients, aged 18-59 years, with newly diagnosed CD20-positive Ph-negative BCP-ALL were randomized to the pediatric-inspired Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) chemotherapy protocol with or without rituximab 375 mg/m2 given during induction (day 1 and 7), salvage reinduction when needed (day 1 and 7), consolidation blocks (6 infusions), late intensification (day 1 and 7, and the first year of maintenance [6 infusions], for a total of 16-18 infusions. Allogeneic stem cell transplant (SCT) was offered after consolidation blocks 1 or 2 to patients with one or more high-risk criteria and an available donor. CD20-positivity was defined as expression of CD20 in more than 20% of leukemia blasts.
Eleven patients were excluded from the analysis because of non–ineligibility criteria, leaving 209 patients in the modified intent-to-treat analysis. Their median age was 40.2 years and 67% had high-risk ALL.
Rates of postinduction minimal residual disease less than 10–4 were 65% and 61% in the rituximab and control arms among 85 evaluable patients(P = .82), and rates of postconsolidation minimal residual disease less than 10–4 were 91% and 82% among 80 evaluable patients (P = .31), Dr. Maury reported.
Notably, more patients in the rituximab arm received allogeneic SCT in their first complete remission (34% vs. 20%; P = .029).
The cumulative incidence of death in first complete remission was 12% in both arms.
In multivariate analysis, rituximab impacted EFS, together with age, central nervous system involvement, white blood cells, or CD20 expression at diagnosis. A preferential effect with rituximab was seen in patients with high CD20 levels that deserves further evaluation, he observed.
There was no difference in the incidence of adverse events between the rituximab and control arms, although there was a trend for more infectious events with rituximab (71 events vs. 55 events), Dr. Maury said.
Allergic events – all but one from aspergillosis – were significantly more common in the control arm (2 events vs. 14 events; P = .002).
The Group for Research in Adult Acute Lymphoblastic Leukemia sponsored the study. Dr. Maury reported having no disclosures.
AT ASH 2015
Key clinical point: Adding rituximab to standard intensive chemotherapy is a new standard for CD20-positive, Philadelphia-negative, B-cell precursor acute lymphoblastic leukemia.
Major finding: Event-free survival was 65% in the rituximab arm vs. 52% in the control arm (HR, 0.66; P = .038).
Data source: Phase III study in 220 adults with CD20-positive, Ph-negative, BCP-ALL.
Disclosures: Group for Research in Adult Acute Lymphoblastic Leukemia sponsored the study. Dr. Maury reported having no disclosures.

The Changing Landscape of Orthopedic Practice: Challenges and Opportunities
Orthopedic surgery is going through a time of remarkable change. Health care reform, heightened public scrutiny, shifting population demographics, increased reliance on the Internet for information, ongoing metamorphosis of our profession into a business, and lack of consistent high-quality clinical evidence have created a new frontier of challenges and opportunities. At heart are the needs to deliver high-quality education that is in line with new technological media, to reclaim our ability to guide musculoskeletal care at the policymaking level, to fortify our long-held tradition of ethical responsibility, to invest in research and the training of physician-scientists, to maintain unity among the different subspecialties, and to increase female and minority representation. Never before has understanding and applying the key tenets of our philosophy as orthopedic surgeons been more crucial.
The changing landscape of orthopedic practice has been an unsettling topic in many of the American Academy of Orthopaedic Surgeons (AAOS) presidential addresses in recent years.1-11 What are the challenges and what can we learn moving forward? In this article, we seek to answer these questions by drawing insights from the combined experience and wisdom of past AAOS presidents since the turn of the 21st century.
Education
Education is the cornerstone of providing quality musculoskeletal care12 and staying up to date with technological advances.13 The modes of education delivery, however, have changed. No longer is orthopedic education confined to tangible textbooks and journal articles, nor is it limited to those of us in the profession. Instead, orthopedic education has shifted toward online learning14 and is available to patients and nonorthopedic providers.12 With more patients gaining access to rapidly growing online resources, a unique challenge has arisen: an abundance of data with variable quality of evidence influencing the decision-making process. This has created what Richard Kyle15 described as the “trap of the new technology war,” where patient misinformation and direct-to-consumer marketing can lead to dangerous musculoskeletal care delivery, including unrealistic patient expectations.3 To compound the problem, our ability to provide orthopedic education in formats compatible with the new learning mediums has not been up to the demand, with issues of cost, accessibility, and efficacy plaguing the current process.3,5 Also, we have yet to unlock the benefits of surgical simulation, which has the potential to provide more effective training at no risk to the patient.4,13 By adapting to the new learning formats, we can provide numerous new opportunities for keeping up to date on evolving practice management principles, which, with added accessibility, will be used more often by orthopedic surgeons and the public.13
Research
Research is vital for quality improvement and the continuation of excellence.5 It is only with research that we can provide groundbreaking advances and measure the outcomes of our interventions.2 Unfortunately, orthopedic research funding continues to be disproportionately low, especially given that musculoskeletal ailments are the leading cause of both physician visits and chronic impairment in the United States.2 For example, the National Institute of Arthritis and Musculoskeletal and Skin Diseases receives only 10% of what our country spends on cancer research and 15% of what is spent on heart- and lung-disease research.2 To compound the problem of limited funding, the number of physician-scientists has been dropping at an alarming rate.2 As a result, we must not only refocus our research efforts so that they are efficient and effective, but we must also invest in the training of orthopedic physician-scientists to ensure a continuous stream of groundbreaking discoveries. We owe it to our patients to provide them with proven, effective, and high-quality care.
Industry Relationships
Local and national attention will continue to focus on our relationships with industry. The challenge is twofold: mitigating the negative portrayal of industry relationships and navigating the changes applied to industry funding for research and education.9 Our collaboration with industry is important for the development and advancement of orthopedics,15 but it must be guided by the professional and ethical guidelines established by the AAOS, ensuring that the best interest of patients remains a top priority.8,15 We must maintain the public’s trust by using every opportunity to convey our lone goal in collaborating with industry, ie, improving patient care.9 According to James Beaty,7 any relationship with industry should be “so ethical that it could be printed on the front page of the newspaper and we could face our neighbors with our heads held high.”
Gender and Minority Representation
The racial and ethnic makeup of the United States is undergoing a rapid change. Over the next 4 decades, the white population is projected to become the minority, while women will continue to outnumber men.16 Despite the rapidly changing demographics of the United States, health care disparities persist. As of 2011, minorities and women made up only 22.55% and 14.52%, respectively, of all orthopedic surgery residents.17 This limited diversity in orthopedic training programs is alarming and may lead to suboptimal physician–patient relationships, because patients tend to be more comfortable with and respond better to the care provided by physicians of similar background.3 In addition, if we do not integrate women into orthopedics, the number of female medical students applying to orthopedic residency programs might decline.3
Equating excellent medical care with diversity and cultural competence requires that we bridge the gap that has prevented patients from obtaining high-quality care.8 To achieve this goal, we need to continue recruiting orthopedic surgeons from all segments of our population. Ultimately, health care disparities can be effectively reduced through the delivery of culturally competent care.8
Physician–Patient Relationship
Medical liability has resulted in the development of damaging attitudes among physicians, with many viewing patients as potential adversaries and even avoiding high-risk procedures altogether.6 This deterioration of the physician–patient relationship has been another troubling consequence of managed care that emphasizes quantity and speed.1 As a result, we are perceived by the public as impersonal, poor listeners, and difficult to see on short notice.1
The poor perception of orthopedic surgeons by the general public is not acceptable for a field that places such a high value on excellence. Patient-centered care is at the core of quality improvement, and improving patient relationships starts and ends with us and with each patient we treat.6 In a health care environment in which the average orthopedic surgeon cares for thousands of patients each year, we must make certain to use each opportunity to engage our patients and enhance our relationships with them.6 The basic necessities of patient-centered care include empowerment of the patient through education, better communication, and transparency; providing accurate and evidence-based information; and cooperation among physicians.3,6 The benefits of improving personal relationships with patients are multifold and could have lasting positive effects: increased physician and patient satisfaction, better patient compliance, greater practice efficiency, and fewer malpractice lawsuits.1 We can also benefit from mobilizing a greater constituency to advocate alongside us.6
Unity
Despite accounting for less than 3% of all physicians, orthopedic surgeons have assumed an influential voice in the field of medicine.13 This is attributed not only to the success of our interventions but, more importantly, to the fact that we have “stuck together.”13 The concept of “sticking together” may seem a cliché and facile but will certainly be a pressing need as we move ahead. We draw strength from the breadth and diversity of our subspecialties, but this strength may become a weakness if we do not join in promoting the betterment of our profession as a whole.14 To avoid duplications and bring synergy to all our efforts, we should continue to develop new partnerships in our specialty societies6 and speak with one voice to our patients and to the public.15 Joshua Jacobs11 reminds us of the warning Benjamin Franklin imparted to the signers of the Declaration of Independence, “We must hang together, or most assuredly, we will all hang separately.” To ensure the continued strength of the house of orthopedics, we must live by this tenet.
Advocacy
The federal government has become increasingly involved in regulating the practice of medicine.9 Orthopedic surgery has been hit especially hard, because the cost of implants and continued innovation has fueled the belief that our profession is a major contributor to unsustainable health care costs.11 We now face multiple legislative regulations related to physician reimbursement, ownership, self-referral, medical liability, and mandates of the Affordable Care Act.9 As a result, there has been a decreasing role for orthopedic surgeons as independent practitioners, with more orthopedists forgoing physician-owned practices for large hospital corporations. We are also in increasing competition for limited resources.10 This is compounded by the fact that those regulating health care, paying for health care, and allocating research funding fail to comprehend the high societal needs for treatment of musculoskeletal diseases and injuries,6 which will only increase in the coming decades.14
The aforementioned challenges make our involvement at all levels of the political process more necessary than ever before.5,9 E. Anthony Rankin8 reminds us, “As physicians, we cannot in good conscience allow our patients’ access to quality orthopedic care to be determined solely by the government, the insurance companies, the trial lawyers, or others…. Either we will have a voice in defining the future of health care, or it will be defined by others for us.” Our advocacy approach, however, should be very careful. Joshua Jacobs11 cautions that “we will be most effective if our advocacy message is presented as a potential solution to the current health care crisis, not just as a demand for fair reimbursement.” Instead, we can achieve this goal with what Richard Gelberman2 summarized as “doing what we do best: accumulating knowledge, positioning ourselves as the authorities that we are, and using what we learn to advocate for better patient care and research.”
Value Medicine
Orthopedic surgeons are now expected to provide not just high-quality care but low-cost care. In line with the emerging emphasis on value, sharp focus has been placed on the assessment of physician performance and treatment outcomes as quality-of-care measures.6 But how have we measured the quality of the care we provide? We have not, or, at least, we have not had valid or reliable means of doing so.6 Gone are the days of telling the world of the excellence of our profession in the treatment of musculoskeletal disease. We now must prove to our patients, the government, and payers that what we do works.12,13 If we fail to communicate the cost effectiveness of our interventions, our new knowledge and technologies will not be accepted or funded.10 We should, however, not be discouraged by the new “value equation,” but use it as an incentive to provide evidence-based care and to improve the efficiency of resource utilization.14 Today, we are urged to be leaders in quality improvement, both in our individual orthopedic practices and as a profession.10,12,13
Conclusion
Meeting increasingly higher demands for musculoskeletal care in an evolving medical landscape will bring a new set of challenges that will be more frequent and more intense than those in the past.14 Today, we are tasked with providing fiscally efficient, culturally competent, high-quality, evidence-based, and compassionate care. We are also tasked with reclaiming our ability to shape the future of our profession at the policymaking level. In doing so, the need for unity, advocacy, commitment to education and research, women and minority representation, and open communication with the public has never been more relevant. As we continue to advance as a profession, we must resist the temptation to look back in defiance of change but move forward, confident in our ability to evolve. ◾
1. Canale ST. The orthopaedic forum. Falling in love again. J Bone Joint Surg Am. 2000;82(5):739-742.
2. Gelberman RH. The Academy on the edge: taking charge of our future. J Bone Joint Surg Am. 2001;83(6):946-950.
3. Tolo VT. The challenges of change: is orthopaedics ready? J Bone Joint Surg Am. 2002;84(9):1707-1713.
4. Herndon JH. One more turn of the wrench. J Bone Joint Surg Am. 2003;85(10):2036-2048.
5. Bucholz RW. Knowledge is our business. J Bone Joint Surg Am. 2004;86(7):1575-1578.
6. Weinstein SL. Nothing about you...without you. J Bone Joint Surg Am. 2005;87(7):1648-1652.
7. Beaty JH. Presidential address: “Building the best . . . Lifelong learning”. J Am Acad Orthop Surg. 2007;15(9):515-518.
8. Rankin EA. Presidential Address: advocacy now... for our patients and our profession. J Am Acad Orthop Surg. 2008;16(6):303-305.
9. Zuckerman JD. Silk purses, sows’ ears, and heap ash—turning challenges into opportunities. J Am Acad Orthop Surg. 2009;17(5):271-275.
10. Tongue JR. Strong on vision, flexible on details. J Am Acad Orthop Surg. 2012;20(4):187-189.
11. Jacobs JJ. Moving forward: from curses to blessings. J Am Acad Orthop Surg. 2013;21(5):261-265.
12. Callaghan JJ. Quality of care: getting from good to great. J Am Acad Orthop Surg. 2010;8(9):516-519.
13. Berry DJ. Informed by our past, building our future. J Am Acad Orthop Surg. 2011;19(4):187-190.
14. Azar FM. Building a bigger box. J Am Acad Orthop Surg. 2014;22(6):341-345.
15. Kyle RF. Presidential Address: Together we are one. J Am Acad Orthop Surg. 2006;14(5):261-264.
16. Vincent GK, Velkoff VA. The Next Four Decades: The Older Population in the United States: 2010 to 2050. Washington, DC: Economics and Statistics Administration, US Census Bureau, US Dept of Commerce; 2010.
17. American Academy of Orthopaedic Surgeons Department of Research and Scientific Affairs. 1998-2011 Resident Diversity Survey Report. American Academy of Orthopaedic Surgeons website. http://www3.aaos.org/about/diversity/pdfs/resident_trend.pdf. Published March 9, 2012. Accessed October 26, 2015.
Orthopedic surgery is going through a time of remarkable change. Health care reform, heightened public scrutiny, shifting population demographics, increased reliance on the Internet for information, ongoing metamorphosis of our profession into a business, and lack of consistent high-quality clinical evidence have created a new frontier of challenges and opportunities. At heart are the needs to deliver high-quality education that is in line with new technological media, to reclaim our ability to guide musculoskeletal care at the policymaking level, to fortify our long-held tradition of ethical responsibility, to invest in research and the training of physician-scientists, to maintain unity among the different subspecialties, and to increase female and minority representation. Never before has understanding and applying the key tenets of our philosophy as orthopedic surgeons been more crucial.
The changing landscape of orthopedic practice has been an unsettling topic in many of the American Academy of Orthopaedic Surgeons (AAOS) presidential addresses in recent years.1-11 What are the challenges and what can we learn moving forward? In this article, we seek to answer these questions by drawing insights from the combined experience and wisdom of past AAOS presidents since the turn of the 21st century.
Education
Education is the cornerstone of providing quality musculoskeletal care12 and staying up to date with technological advances.13 The modes of education delivery, however, have changed. No longer is orthopedic education confined to tangible textbooks and journal articles, nor is it limited to those of us in the profession. Instead, orthopedic education has shifted toward online learning14 and is available to patients and nonorthopedic providers.12 With more patients gaining access to rapidly growing online resources, a unique challenge has arisen: an abundance of data with variable quality of evidence influencing the decision-making process. This has created what Richard Kyle15 described as the “trap of the new technology war,” where patient misinformation and direct-to-consumer marketing can lead to dangerous musculoskeletal care delivery, including unrealistic patient expectations.3 To compound the problem, our ability to provide orthopedic education in formats compatible with the new learning mediums has not been up to the demand, with issues of cost, accessibility, and efficacy plaguing the current process.3,5 Also, we have yet to unlock the benefits of surgical simulation, which has the potential to provide more effective training at no risk to the patient.4,13 By adapting to the new learning formats, we can provide numerous new opportunities for keeping up to date on evolving practice management principles, which, with added accessibility, will be used more often by orthopedic surgeons and the public.13
Research
Research is vital for quality improvement and the continuation of excellence.5 It is only with research that we can provide groundbreaking advances and measure the outcomes of our interventions.2 Unfortunately, orthopedic research funding continues to be disproportionately low, especially given that musculoskeletal ailments are the leading cause of both physician visits and chronic impairment in the United States.2 For example, the National Institute of Arthritis and Musculoskeletal and Skin Diseases receives only 10% of what our country spends on cancer research and 15% of what is spent on heart- and lung-disease research.2 To compound the problem of limited funding, the number of physician-scientists has been dropping at an alarming rate.2 As a result, we must not only refocus our research efforts so that they are efficient and effective, but we must also invest in the training of orthopedic physician-scientists to ensure a continuous stream of groundbreaking discoveries. We owe it to our patients to provide them with proven, effective, and high-quality care.
Industry Relationships
Local and national attention will continue to focus on our relationships with industry. The challenge is twofold: mitigating the negative portrayal of industry relationships and navigating the changes applied to industry funding for research and education.9 Our collaboration with industry is important for the development and advancement of orthopedics,15 but it must be guided by the professional and ethical guidelines established by the AAOS, ensuring that the best interest of patients remains a top priority.8,15 We must maintain the public’s trust by using every opportunity to convey our lone goal in collaborating with industry, ie, improving patient care.9 According to James Beaty,7 any relationship with industry should be “so ethical that it could be printed on the front page of the newspaper and we could face our neighbors with our heads held high.”
Gender and Minority Representation
The racial and ethnic makeup of the United States is undergoing a rapid change. Over the next 4 decades, the white population is projected to become the minority, while women will continue to outnumber men.16 Despite the rapidly changing demographics of the United States, health care disparities persist. As of 2011, minorities and women made up only 22.55% and 14.52%, respectively, of all orthopedic surgery residents.17 This limited diversity in orthopedic training programs is alarming and may lead to suboptimal physician–patient relationships, because patients tend to be more comfortable with and respond better to the care provided by physicians of similar background.3 In addition, if we do not integrate women into orthopedics, the number of female medical students applying to orthopedic residency programs might decline.3
Equating excellent medical care with diversity and cultural competence requires that we bridge the gap that has prevented patients from obtaining high-quality care.8 To achieve this goal, we need to continue recruiting orthopedic surgeons from all segments of our population. Ultimately, health care disparities can be effectively reduced through the delivery of culturally competent care.8
Physician–Patient Relationship
Medical liability has resulted in the development of damaging attitudes among physicians, with many viewing patients as potential adversaries and even avoiding high-risk procedures altogether.6 This deterioration of the physician–patient relationship has been another troubling consequence of managed care that emphasizes quantity and speed.1 As a result, we are perceived by the public as impersonal, poor listeners, and difficult to see on short notice.1
The poor perception of orthopedic surgeons by the general public is not acceptable for a field that places such a high value on excellence. Patient-centered care is at the core of quality improvement, and improving patient relationships starts and ends with us and with each patient we treat.6 In a health care environment in which the average orthopedic surgeon cares for thousands of patients each year, we must make certain to use each opportunity to engage our patients and enhance our relationships with them.6 The basic necessities of patient-centered care include empowerment of the patient through education, better communication, and transparency; providing accurate and evidence-based information; and cooperation among physicians.3,6 The benefits of improving personal relationships with patients are multifold and could have lasting positive effects: increased physician and patient satisfaction, better patient compliance, greater practice efficiency, and fewer malpractice lawsuits.1 We can also benefit from mobilizing a greater constituency to advocate alongside us.6
Unity
Despite accounting for less than 3% of all physicians, orthopedic surgeons have assumed an influential voice in the field of medicine.13 This is attributed not only to the success of our interventions but, more importantly, to the fact that we have “stuck together.”13 The concept of “sticking together” may seem a cliché and facile but will certainly be a pressing need as we move ahead. We draw strength from the breadth and diversity of our subspecialties, but this strength may become a weakness if we do not join in promoting the betterment of our profession as a whole.14 To avoid duplications and bring synergy to all our efforts, we should continue to develop new partnerships in our specialty societies6 and speak with one voice to our patients and to the public.15 Joshua Jacobs11 reminds us of the warning Benjamin Franklin imparted to the signers of the Declaration of Independence, “We must hang together, or most assuredly, we will all hang separately.” To ensure the continued strength of the house of orthopedics, we must live by this tenet.
Advocacy
The federal government has become increasingly involved in regulating the practice of medicine.9 Orthopedic surgery has been hit especially hard, because the cost of implants and continued innovation has fueled the belief that our profession is a major contributor to unsustainable health care costs.11 We now face multiple legislative regulations related to physician reimbursement, ownership, self-referral, medical liability, and mandates of the Affordable Care Act.9 As a result, there has been a decreasing role for orthopedic surgeons as independent practitioners, with more orthopedists forgoing physician-owned practices for large hospital corporations. We are also in increasing competition for limited resources.10 This is compounded by the fact that those regulating health care, paying for health care, and allocating research funding fail to comprehend the high societal needs for treatment of musculoskeletal diseases and injuries,6 which will only increase in the coming decades.14
The aforementioned challenges make our involvement at all levels of the political process more necessary than ever before.5,9 E. Anthony Rankin8 reminds us, “As physicians, we cannot in good conscience allow our patients’ access to quality orthopedic care to be determined solely by the government, the insurance companies, the trial lawyers, or others…. Either we will have a voice in defining the future of health care, or it will be defined by others for us.” Our advocacy approach, however, should be very careful. Joshua Jacobs11 cautions that “we will be most effective if our advocacy message is presented as a potential solution to the current health care crisis, not just as a demand for fair reimbursement.” Instead, we can achieve this goal with what Richard Gelberman2 summarized as “doing what we do best: accumulating knowledge, positioning ourselves as the authorities that we are, and using what we learn to advocate for better patient care and research.”
Value Medicine
Orthopedic surgeons are now expected to provide not just high-quality care but low-cost care. In line with the emerging emphasis on value, sharp focus has been placed on the assessment of physician performance and treatment outcomes as quality-of-care measures.6 But how have we measured the quality of the care we provide? We have not, or, at least, we have not had valid or reliable means of doing so.6 Gone are the days of telling the world of the excellence of our profession in the treatment of musculoskeletal disease. We now must prove to our patients, the government, and payers that what we do works.12,13 If we fail to communicate the cost effectiveness of our interventions, our new knowledge and technologies will not be accepted or funded.10 We should, however, not be discouraged by the new “value equation,” but use it as an incentive to provide evidence-based care and to improve the efficiency of resource utilization.14 Today, we are urged to be leaders in quality improvement, both in our individual orthopedic practices and as a profession.10,12,13
Conclusion
Meeting increasingly higher demands for musculoskeletal care in an evolving medical landscape will bring a new set of challenges that will be more frequent and more intense than those in the past.14 Today, we are tasked with providing fiscally efficient, culturally competent, high-quality, evidence-based, and compassionate care. We are also tasked with reclaiming our ability to shape the future of our profession at the policymaking level. In doing so, the need for unity, advocacy, commitment to education and research, women and minority representation, and open communication with the public has never been more relevant. As we continue to advance as a profession, we must resist the temptation to look back in defiance of change but move forward, confident in our ability to evolve. ◾
Orthopedic surgery is going through a time of remarkable change. Health care reform, heightened public scrutiny, shifting population demographics, increased reliance on the Internet for information, ongoing metamorphosis of our profession into a business, and lack of consistent high-quality clinical evidence have created a new frontier of challenges and opportunities. At heart are the needs to deliver high-quality education that is in line with new technological media, to reclaim our ability to guide musculoskeletal care at the policymaking level, to fortify our long-held tradition of ethical responsibility, to invest in research and the training of physician-scientists, to maintain unity among the different subspecialties, and to increase female and minority representation. Never before has understanding and applying the key tenets of our philosophy as orthopedic surgeons been more crucial.
The changing landscape of orthopedic practice has been an unsettling topic in many of the American Academy of Orthopaedic Surgeons (AAOS) presidential addresses in recent years.1-11 What are the challenges and what can we learn moving forward? In this article, we seek to answer these questions by drawing insights from the combined experience and wisdom of past AAOS presidents since the turn of the 21st century.
Education
Education is the cornerstone of providing quality musculoskeletal care12 and staying up to date with technological advances.13 The modes of education delivery, however, have changed. No longer is orthopedic education confined to tangible textbooks and journal articles, nor is it limited to those of us in the profession. Instead, orthopedic education has shifted toward online learning14 and is available to patients and nonorthopedic providers.12 With more patients gaining access to rapidly growing online resources, a unique challenge has arisen: an abundance of data with variable quality of evidence influencing the decision-making process. This has created what Richard Kyle15 described as the “trap of the new technology war,” where patient misinformation and direct-to-consumer marketing can lead to dangerous musculoskeletal care delivery, including unrealistic patient expectations.3 To compound the problem, our ability to provide orthopedic education in formats compatible with the new learning mediums has not been up to the demand, with issues of cost, accessibility, and efficacy plaguing the current process.3,5 Also, we have yet to unlock the benefits of surgical simulation, which has the potential to provide more effective training at no risk to the patient.4,13 By adapting to the new learning formats, we can provide numerous new opportunities for keeping up to date on evolving practice management principles, which, with added accessibility, will be used more often by orthopedic surgeons and the public.13
Research
Research is vital for quality improvement and the continuation of excellence.5 It is only with research that we can provide groundbreaking advances and measure the outcomes of our interventions.2 Unfortunately, orthopedic research funding continues to be disproportionately low, especially given that musculoskeletal ailments are the leading cause of both physician visits and chronic impairment in the United States.2 For example, the National Institute of Arthritis and Musculoskeletal and Skin Diseases receives only 10% of what our country spends on cancer research and 15% of what is spent on heart- and lung-disease research.2 To compound the problem of limited funding, the number of physician-scientists has been dropping at an alarming rate.2 As a result, we must not only refocus our research efforts so that they are efficient and effective, but we must also invest in the training of orthopedic physician-scientists to ensure a continuous stream of groundbreaking discoveries. We owe it to our patients to provide them with proven, effective, and high-quality care.
Industry Relationships
Local and national attention will continue to focus on our relationships with industry. The challenge is twofold: mitigating the negative portrayal of industry relationships and navigating the changes applied to industry funding for research and education.9 Our collaboration with industry is important for the development and advancement of orthopedics,15 but it must be guided by the professional and ethical guidelines established by the AAOS, ensuring that the best interest of patients remains a top priority.8,15 We must maintain the public’s trust by using every opportunity to convey our lone goal in collaborating with industry, ie, improving patient care.9 According to James Beaty,7 any relationship with industry should be “so ethical that it could be printed on the front page of the newspaper and we could face our neighbors with our heads held high.”
Gender and Minority Representation
The racial and ethnic makeup of the United States is undergoing a rapid change. Over the next 4 decades, the white population is projected to become the minority, while women will continue to outnumber men.16 Despite the rapidly changing demographics of the United States, health care disparities persist. As of 2011, minorities and women made up only 22.55% and 14.52%, respectively, of all orthopedic surgery residents.17 This limited diversity in orthopedic training programs is alarming and may lead to suboptimal physician–patient relationships, because patients tend to be more comfortable with and respond better to the care provided by physicians of similar background.3 In addition, if we do not integrate women into orthopedics, the number of female medical students applying to orthopedic residency programs might decline.3
Equating excellent medical care with diversity and cultural competence requires that we bridge the gap that has prevented patients from obtaining high-quality care.8 To achieve this goal, we need to continue recruiting orthopedic surgeons from all segments of our population. Ultimately, health care disparities can be effectively reduced through the delivery of culturally competent care.8
Physician–Patient Relationship
Medical liability has resulted in the development of damaging attitudes among physicians, with many viewing patients as potential adversaries and even avoiding high-risk procedures altogether.6 This deterioration of the physician–patient relationship has been another troubling consequence of managed care that emphasizes quantity and speed.1 As a result, we are perceived by the public as impersonal, poor listeners, and difficult to see on short notice.1
The poor perception of orthopedic surgeons by the general public is not acceptable for a field that places such a high value on excellence. Patient-centered care is at the core of quality improvement, and improving patient relationships starts and ends with us and with each patient we treat.6 In a health care environment in which the average orthopedic surgeon cares for thousands of patients each year, we must make certain to use each opportunity to engage our patients and enhance our relationships with them.6 The basic necessities of patient-centered care include empowerment of the patient through education, better communication, and transparency; providing accurate and evidence-based information; and cooperation among physicians.3,6 The benefits of improving personal relationships with patients are multifold and could have lasting positive effects: increased physician and patient satisfaction, better patient compliance, greater practice efficiency, and fewer malpractice lawsuits.1 We can also benefit from mobilizing a greater constituency to advocate alongside us.6
Unity
Despite accounting for less than 3% of all physicians, orthopedic surgeons have assumed an influential voice in the field of medicine.13 This is attributed not only to the success of our interventions but, more importantly, to the fact that we have “stuck together.”13 The concept of “sticking together” may seem a cliché and facile but will certainly be a pressing need as we move ahead. We draw strength from the breadth and diversity of our subspecialties, but this strength may become a weakness if we do not join in promoting the betterment of our profession as a whole.14 To avoid duplications and bring synergy to all our efforts, we should continue to develop new partnerships in our specialty societies6 and speak with one voice to our patients and to the public.15 Joshua Jacobs11 reminds us of the warning Benjamin Franklin imparted to the signers of the Declaration of Independence, “We must hang together, or most assuredly, we will all hang separately.” To ensure the continued strength of the house of orthopedics, we must live by this tenet.
Advocacy
The federal government has become increasingly involved in regulating the practice of medicine.9 Orthopedic surgery has been hit especially hard, because the cost of implants and continued innovation has fueled the belief that our profession is a major contributor to unsustainable health care costs.11 We now face multiple legislative regulations related to physician reimbursement, ownership, self-referral, medical liability, and mandates of the Affordable Care Act.9 As a result, there has been a decreasing role for orthopedic surgeons as independent practitioners, with more orthopedists forgoing physician-owned practices for large hospital corporations. We are also in increasing competition for limited resources.10 This is compounded by the fact that those regulating health care, paying for health care, and allocating research funding fail to comprehend the high societal needs for treatment of musculoskeletal diseases and injuries,6 which will only increase in the coming decades.14
The aforementioned challenges make our involvement at all levels of the political process more necessary than ever before.5,9 E. Anthony Rankin8 reminds us, “As physicians, we cannot in good conscience allow our patients’ access to quality orthopedic care to be determined solely by the government, the insurance companies, the trial lawyers, or others…. Either we will have a voice in defining the future of health care, or it will be defined by others for us.” Our advocacy approach, however, should be very careful. Joshua Jacobs11 cautions that “we will be most effective if our advocacy message is presented as a potential solution to the current health care crisis, not just as a demand for fair reimbursement.” Instead, we can achieve this goal with what Richard Gelberman2 summarized as “doing what we do best: accumulating knowledge, positioning ourselves as the authorities that we are, and using what we learn to advocate for better patient care and research.”
Value Medicine
Orthopedic surgeons are now expected to provide not just high-quality care but low-cost care. In line with the emerging emphasis on value, sharp focus has been placed on the assessment of physician performance and treatment outcomes as quality-of-care measures.6 But how have we measured the quality of the care we provide? We have not, or, at least, we have not had valid or reliable means of doing so.6 Gone are the days of telling the world of the excellence of our profession in the treatment of musculoskeletal disease. We now must prove to our patients, the government, and payers that what we do works.12,13 If we fail to communicate the cost effectiveness of our interventions, our new knowledge and technologies will not be accepted or funded.10 We should, however, not be discouraged by the new “value equation,” but use it as an incentive to provide evidence-based care and to improve the efficiency of resource utilization.14 Today, we are urged to be leaders in quality improvement, both in our individual orthopedic practices and as a profession.10,12,13
Conclusion
Meeting increasingly higher demands for musculoskeletal care in an evolving medical landscape will bring a new set of challenges that will be more frequent and more intense than those in the past.14 Today, we are tasked with providing fiscally efficient, culturally competent, high-quality, evidence-based, and compassionate care. We are also tasked with reclaiming our ability to shape the future of our profession at the policymaking level. In doing so, the need for unity, advocacy, commitment to education and research, women and minority representation, and open communication with the public has never been more relevant. As we continue to advance as a profession, we must resist the temptation to look back in defiance of change but move forward, confident in our ability to evolve. ◾
1. Canale ST. The orthopaedic forum. Falling in love again. J Bone Joint Surg Am. 2000;82(5):739-742.
2. Gelberman RH. The Academy on the edge: taking charge of our future. J Bone Joint Surg Am. 2001;83(6):946-950.
3. Tolo VT. The challenges of change: is orthopaedics ready? J Bone Joint Surg Am. 2002;84(9):1707-1713.
4. Herndon JH. One more turn of the wrench. J Bone Joint Surg Am. 2003;85(10):2036-2048.
5. Bucholz RW. Knowledge is our business. J Bone Joint Surg Am. 2004;86(7):1575-1578.
6. Weinstein SL. Nothing about you...without you. J Bone Joint Surg Am. 2005;87(7):1648-1652.
7. Beaty JH. Presidential address: “Building the best . . . Lifelong learning”. J Am Acad Orthop Surg. 2007;15(9):515-518.
8. Rankin EA. Presidential Address: advocacy now... for our patients and our profession. J Am Acad Orthop Surg. 2008;16(6):303-305.
9. Zuckerman JD. Silk purses, sows’ ears, and heap ash—turning challenges into opportunities. J Am Acad Orthop Surg. 2009;17(5):271-275.
10. Tongue JR. Strong on vision, flexible on details. J Am Acad Orthop Surg. 2012;20(4):187-189.
11. Jacobs JJ. Moving forward: from curses to blessings. J Am Acad Orthop Surg. 2013;21(5):261-265.
12. Callaghan JJ. Quality of care: getting from good to great. J Am Acad Orthop Surg. 2010;8(9):516-519.
13. Berry DJ. Informed by our past, building our future. J Am Acad Orthop Surg. 2011;19(4):187-190.
14. Azar FM. Building a bigger box. J Am Acad Orthop Surg. 2014;22(6):341-345.
15. Kyle RF. Presidential Address: Together we are one. J Am Acad Orthop Surg. 2006;14(5):261-264.
16. Vincent GK, Velkoff VA. The Next Four Decades: The Older Population in the United States: 2010 to 2050. Washington, DC: Economics and Statistics Administration, US Census Bureau, US Dept of Commerce; 2010.
17. American Academy of Orthopaedic Surgeons Department of Research and Scientific Affairs. 1998-2011 Resident Diversity Survey Report. American Academy of Orthopaedic Surgeons website. http://www3.aaos.org/about/diversity/pdfs/resident_trend.pdf. Published March 9, 2012. Accessed October 26, 2015.
1. Canale ST. The orthopaedic forum. Falling in love again. J Bone Joint Surg Am. 2000;82(5):739-742.
2. Gelberman RH. The Academy on the edge: taking charge of our future. J Bone Joint Surg Am. 2001;83(6):946-950.
3. Tolo VT. The challenges of change: is orthopaedics ready? J Bone Joint Surg Am. 2002;84(9):1707-1713.
4. Herndon JH. One more turn of the wrench. J Bone Joint Surg Am. 2003;85(10):2036-2048.
5. Bucholz RW. Knowledge is our business. J Bone Joint Surg Am. 2004;86(7):1575-1578.
6. Weinstein SL. Nothing about you...without you. J Bone Joint Surg Am. 2005;87(7):1648-1652.
7. Beaty JH. Presidential address: “Building the best . . . Lifelong learning”. J Am Acad Orthop Surg. 2007;15(9):515-518.
8. Rankin EA. Presidential Address: advocacy now... for our patients and our profession. J Am Acad Orthop Surg. 2008;16(6):303-305.
9. Zuckerman JD. Silk purses, sows’ ears, and heap ash—turning challenges into opportunities. J Am Acad Orthop Surg. 2009;17(5):271-275.
10. Tongue JR. Strong on vision, flexible on details. J Am Acad Orthop Surg. 2012;20(4):187-189.
11. Jacobs JJ. Moving forward: from curses to blessings. J Am Acad Orthop Surg. 2013;21(5):261-265.
12. Callaghan JJ. Quality of care: getting from good to great. J Am Acad Orthop Surg. 2010;8(9):516-519.
13. Berry DJ. Informed by our past, building our future. J Am Acad Orthop Surg. 2011;19(4):187-190.
14. Azar FM. Building a bigger box. J Am Acad Orthop Surg. 2014;22(6):341-345.
15. Kyle RF. Presidential Address: Together we are one. J Am Acad Orthop Surg. 2006;14(5):261-264.
16. Vincent GK, Velkoff VA. The Next Four Decades: The Older Population in the United States: 2010 to 2050. Washington, DC: Economics and Statistics Administration, US Census Bureau, US Dept of Commerce; 2010.
17. American Academy of Orthopaedic Surgeons Department of Research and Scientific Affairs. 1998-2011 Resident Diversity Survey Report. American Academy of Orthopaedic Surgeons website. http://www3.aaos.org/about/diversity/pdfs/resident_trend.pdf. Published March 9, 2012. Accessed October 26, 2015.

Nonconsecutive Pars Interarticularis Defects
Spondylolysis is a bone defect of the pars interarticularis. It is usually seen in adolescents who participate in sporting activities that involve repetitive axial loads to a hyperextended lower back, such as football offensive lineman, throwing athletes, and gymnasts. It occurs frequently in the L5 pars and can be unilateral or bilateral. The majority of reported multiple-level spondylolysis is at consecutive lumbar segments.1-6 Rarely, it affects noncontiguous levels. Most patients respond well to conservative treatment in the form of activity modification and orthosis.7 Surgical intervention is considered if 6 months of conservative management fails, spondylolisthesis develops, or intractable neurologic symptoms arise.
This case report presents an 18-year-old man with noncontiguous spondylolysis at L2 and L5 who was successfully treated with a 1-level pars repair at L2 after failed conservative management. This unique case highlights the importance of using single-photon emission computed tomography (SPECT) scan and diagnostic pars block when planning for surgical treatment in the rare cases of noncontiguous spondylolysis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
An 18-year-old man presented to the clinic with worsening lower back pain for the previous 4 weeks. He was playing high school baseball and stated the pain was worse when he swung his bat. He had no history of trauma or back pain. Rest was the only alleviating factor, and he was beginning to experience pain when he stood after sitting. He denied any radicular pain. On examination, he had midline tenderness along the upper lumbar spine and pain with lumbar spine extension. His neurologic examination showed normal sensation with 5/5 strength in all key muscle groups. Plain radiograph of the lumbar spine showed an L5 pars defect (Figures 1A, 1B). A SPECT scan showed increased uptake at L2 pars bilaterally; the L5 pars did not show increased uptake (Figures 2A, 2B). A computed tomography (CT) scan confirmed bilateral L2 pars fractures and a left L5 pars fracture (Figures 3A, 3B). Bony changes in the form of marginal sclerosis at the L5, but not the L2, pars suggested that the L2 fracture was acute while the L5 fracture was chronic (Figures 4A, 4B).
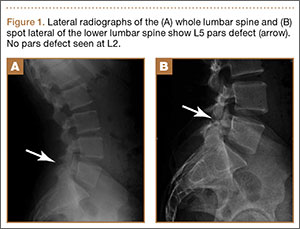

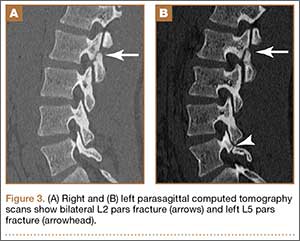

The patient had conservative management for 6 months in the form of lumbosacral orthosis (LSO), cessation of sports activities, and physical therapy. The patient was initially treated with an LSO brace for 3 months, after which he had physical therapy. At 6 month follow-up, he reported continuing, significant back pain. A repeat CT scan of the lumbar spine showed no interval healing of the bilateral L2 or the unilateral L5 pars fractures. As a result of the patient’s noncontiguous pars fractures, a diagnostic CT-guided block of L2 pars was performed to identify which level was his main pain generator (Figure 5). He reported a brief period of complete pain relief after the L2 pars block. With failure of 6 months’ conservative management and positive SPECT scan and diagnostic block, surgical treatment was recommended. Prior to surgical intervention, magnetic resonance imaging was obtained to rule out pathology (eg, disc degeneration, infection, or tumor) other than the pars defect that could require fusion instead of pars repair (Figures 6A, 6B). Because of the patient’s young age, bilateral L2 pars repair rather than fusion was indicated. After 8 months of persistent back pain, he underwent bilateral L2 pars repair with iliac crest autograft, pedicle screws, and sublaminar hook fixation (Figures 7A, 7B). The patient had an uneventful immediate postoperative course. A 6-month postoperative CT scan showed bridging callus at the L2 pars; however, the left L5 pars fracture was still visible (Figures 8A-8C). Over a 6-month postoperative period, the patient had continued improvement in his back pain, advanced his activity as tolerated without problem, and was allowed to resume his sports activities. At 2-year follow-up, he was playing baseball and basketball, and denied any back pain.




Discussion
Lumbar spondylolysis is commonly seen at the fourth and fifth lumbar vertebrae, and accounts for more than 95% of spondylolysis cases.8 Multiple-level spondylolysis is a relatively rare finding with an incidence varying between 1.2% and 5.6%. The majority of the reported multiple-level cases are adjacent.1-3,6 Adolescents often present with a history of insidious-onset low back pain without radicular symptoms that is exacerbated by activity. Occasionally, an acute injury may elicit the onset of pain. A thorough history with emphasis on pain in relation to activity and sports involvement is beneficial. The patient in the current study was a throwing athlete and presented with 4 weeks of back pain that worsened with activity; he had no history of trauma.
Radiographic assessment using standing anteroposterior, lateral, and oblique radiographs of the thoracolumbar spine is useful in the initial assessment. A SPECT scan of the lumbosacral spine is highly sensitive for identifying spondylolytic defects when plain radiographs are within normal limits, yet a high index of suspicion remains given the patient’s history and physical examination findings.9,10 Increased radionuclide uptake within the pars indicates a stress reaction and, possibly, a more acute pathology. The plain radiographs of the patient showed only L5 spondylolysis. However, a SPECT scan showed only increased uptake in L2 pars on both sides. These findings suggested chronic L5 and acute L2 pars defects. A thin-cut CT scan gives the best visualization of pars defect and can help in differentiating chronic defect with sclerotic margins versus acute defect with hazy irregular margins. In the current case, the CT scan showed changes consistent with unilateral chronic L5 and bilateral acute L2 pars defects.
The origin of the pain in spondylolysis is from the tissues rich in nociceptive nerve endings in the loose posterior arch. A CT-guided pars block is a very useful diagnostic preoperative tool that confirms the symptomatic level in cases of multilevel pars defect, especially if they are noncontiguous. In this case, the diagnostic preoperative bilateral L2 pars block confirmed that the pain generator was the acute L2 rather than the chronic L5 pars defect. This step assured that surgical treatment involving only the L2 level would be beneficial in alleviating the patient’s back pain after the failure of 6 months of conservative treatment.
Most patients with single-level spondylolysis respond to conservative treatment, especially after early diagnosis and treatment. The traditional nonoperative treatment of children and adolescents with a symptomatic spondylolytic lesion was a period of rest and progressive increased activity with physical therapy. Immobilization with an LSO was reserved for individuals who did not respond to rest and physical therapy.11 However, multiple studies revealed early immobilization achieved results superior to activity restriction alone, and individuals who underwent a period of activity restriction prior to bracing were more likely to experience persistent symptoms.12-14 Our patient underwent conservative treatment for 6 months, in the form of LSO, cessation of sport activities, and physical therapy, which failed to give him relief of his back pain.
Surgical intervention is warranted for adolescents with persistent, debilitating pain intractable to at least a 6-month period of nonoperative management. Additional indications for surgical management are those individuals who present with neurologic deficits and isthmic spondylolisthesis. Surgical treatment involves direct pars repair with iliac crest bone graft and use of a sublaminar hook/pedicle screw construct, cerclage wire, or pars screw.15-18
In contrast to single-level pars defects that respond well to conservative treatment, there are conflicting reports regarding the management of multiple-level pars fractures; a few reports suggest good outcome with conservative management, but the majority state that surgery is often required and conservative measures are rarely useful.1-4,6 Nayeemuddin and colleagues19 reported a case of a 16-year-old football player who presented with a 4-month history of constant low back pain related to bilateral L3 and L5 pars defects that responded to 1 year of conservative management, when the more acute fractures at L3 showed complete bony union and the patient had symptomatic pain relief and was able to return to full sporting activity.
Chang and colleagues2 reported 10 patients with adjacent 2-level bilateral spondylolysis treated successfully using a pedicle screw–hook construct with autogenous bone grafting. Ogawa and colleagues5 reported adjacent 2-level spondylolysis in 5 patients and 3-level spondylolysis in 2 patients, who were treated successfully by segmental wire fixation and bone grafting. Ivanic and colleagues15 retrospectively reviewed 113 patients with spondylolysis who were treated with direct repair using a hook-screw construct and showed a pseudoarthrosis rate of 13.3%. Superior fusion rates were observed in patients 14 years and younger compared with older patients, particularly those 20 years and older.15 Roca and colleagues16 prospectively analyzed 19 consecutive cases of spondylolysis that were repaired using a hook-screw construct. Twelve of 13 patients (92%) who were 20 years or younger at the time of the study (average age, 17.2 years) had fusion, whereas, in 6 patients 21 years and older (average age, 27.5 years), no cases of fusion were observed. The patients 20 years or younger had significantly better clinical results than those obtained in the patients 21 years and older. The authors concluded that pedicle screw–hook fixation is a useful alternative in the treatment of spondylolysis in adolescents, but did not recommend this procedure in patients older than 20 years.16
Conclusion
The current case demonstrates a unique example of rare noncontiguous pars defects successfully treated with primary repair of 1 level when conservative management failed and the symptomatic defect was isolated. It also highlights the importance of investigating the entirety of the lumbar spine when diagnosis of L5 spondylolysis rules out noncontiguous pars defects. The treatment of noncontiguous pars defects is not well defined; this case showed the importance of using a SPECT scan and a diagnostic pars block to help isolate the symptomatic level when surgical management is considered after a failure of conservative treatment. This case shows 2 possible results: the chronic unilateral L5 defect responded to nonsurgical treatment with asymptomatic fibrous nonunion, while the more acute bilateral L2 defect responded to pars repair with pedicle screw–hook fixation and iliac crest bone graft.
1. Al-Sebai MW, Al-Khawashki H. Spondyloptosis and multiple-level spondylolysis. Eur Spine J. 1999;8(1):75-77.
2. Chang JH, Lee CH, Wu SS, Lin LC, et al. Management of multiple level spondylolysis of the lumbar spine in young males: a report of six cases. J Formos Med Assoc. 2001;100(7)2:497-502.
3. Eingorn D, Pizzutillo PD. Pars interarticularis fusion of multiple levels of lumbar spondylolysis. A case report. Spine. 1985;10(3):250-252.
4. Nozawa S, Shimizu K, Miyamoto K, Tanaka M. Repair of pars interarticularis defect by segmental wire fixation in young athletes with spondylolysis. Am J Sports Med. 2003;31(3):359-364.
5. Ogawa H, Nishimoto H, Hosoe H, Suzuki N, Kanamori Y, Shimizu K. Clinical outcome after segmental wire fixation and bone grafting for repair of the defects in multiple level lumbar spondylolysis. J Spinal Disord Tech. 2007;20(7):521-525.
6. Ravichandran G. Multiple lumbar spondylolyses. Spine. 1980;5(6):552-557.
7. Sys J, Michielsen J, Bracke P, Martens M, Verstreken J. Nonoperative treatment of active spondylolysis in elite athletes with normal X-ray findings: literature review and results of conservative treatment. Eur Spine J. 2001;10(6):498-504.
8. Saraste H. Spondylolysis and spondylolisthesis. Acta Orthop Scand Suppl. 1993;251:84-86.
9. Anderson K, Sarwark JF, Conway JJ, Logue ES, Schafer MS. Quantitative assessment with SPECT imaging of stress injuries of the pars interarticularis and response to bracing. J Pediatr Orthop. 2000;20(1):28-33.
10. Bodner RJ, Heyman S, Drummond DS, Gregg JR. The use of single photon emission computed tomography (SPECT) in the diagnosis of low-back pain in young patients. Spine. 1988;13(10):1155-1160.
11. Steiner ME, Micheli LJ. Treatment of symptomatic spondylolysis and spondylolisthesis with the modified Boston brace. Spine. 1985;10(10):937-943.
12. Blanda J, Bethem D, Moats W, Lew M. Defects of pars interarticularis in athletes: a protocol for nonoperative treatment. J Spinal Disord. 1993;6(5):406-411.
13. Kurd MF, Patel D, Norton R, Picetti G, Friel B, Vaccaro AR. Nonoperative treatment of symptomatic spondylolysis. J Spinal Disord Tech. 2007;20(8):560-564.
14. Pizzutillo PD, Hummer CD 3rd. Nonoperative treatment for painful adolescent spondylolysis or spondylolisthesis. J Pediatr Orthop. 1989;9(5):538-540.
15. Ivanic GM, Pink TP, Achatz W, Ward JC, Homann NC, May M. Direct stabilization of lumbar spondylolysis with a hook screw: mean 11-year follow-up period for 113 patients. Spine. 2003;28(3):255-259.
16. Roca J, Iborra M, Cavanilles-Walker JM, Alberti G. Direct repair of spondylolysis using a new pedicle screw hook fixation: clinical and CT-assessed study: an analysis of 19 patients. J Spinal Disord Tech. 2005;18(suppl):S82-S89.
17. Schlenzka D, Remes V, Helenius I, et al. Direct repair for treatment of symptomatic spondylolysis and low-grade isthmic spondylolisthesis in young patients: no benefit in comparison to segmental fusion after a mean follow-up of 14.8 years. Eur Spine J. 2006;15(10):1437-1447.
18. Buck JE. Direct repair of the defect in spondylolisthesis. Preliminary report. J Bone Joint Surg Br. 1970;52(3):432-437.
19. Nayeemuddin M, Richards PJ, Ahmed EB. The imaging and management of nonconsecutive pars interarticularis defects: a case report and review of literature. Spine J. 2011;11(12):1157-1163.
Spondylolysis is a bone defect of the pars interarticularis. It is usually seen in adolescents who participate in sporting activities that involve repetitive axial loads to a hyperextended lower back, such as football offensive lineman, throwing athletes, and gymnasts. It occurs frequently in the L5 pars and can be unilateral or bilateral. The majority of reported multiple-level spondylolysis is at consecutive lumbar segments.1-6 Rarely, it affects noncontiguous levels. Most patients respond well to conservative treatment in the form of activity modification and orthosis.7 Surgical intervention is considered if 6 months of conservative management fails, spondylolisthesis develops, or intractable neurologic symptoms arise.
This case report presents an 18-year-old man with noncontiguous spondylolysis at L2 and L5 who was successfully treated with a 1-level pars repair at L2 after failed conservative management. This unique case highlights the importance of using single-photon emission computed tomography (SPECT) scan and diagnostic pars block when planning for surgical treatment in the rare cases of noncontiguous spondylolysis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
An 18-year-old man presented to the clinic with worsening lower back pain for the previous 4 weeks. He was playing high school baseball and stated the pain was worse when he swung his bat. He had no history of trauma or back pain. Rest was the only alleviating factor, and he was beginning to experience pain when he stood after sitting. He denied any radicular pain. On examination, he had midline tenderness along the upper lumbar spine and pain with lumbar spine extension. His neurologic examination showed normal sensation with 5/5 strength in all key muscle groups. Plain radiograph of the lumbar spine showed an L5 pars defect (Figures 1A, 1B). A SPECT scan showed increased uptake at L2 pars bilaterally; the L5 pars did not show increased uptake (Figures 2A, 2B). A computed tomography (CT) scan confirmed bilateral L2 pars fractures and a left L5 pars fracture (Figures 3A, 3B). Bony changes in the form of marginal sclerosis at the L5, but not the L2, pars suggested that the L2 fracture was acute while the L5 fracture was chronic (Figures 4A, 4B).
The patient had conservative management for 6 months in the form of lumbosacral orthosis (LSO), cessation of sports activities, and physical therapy. The patient was initially treated with an LSO brace for 3 months, after which he had physical therapy. At 6 month follow-up, he reported continuing, significant back pain. A repeat CT scan of the lumbar spine showed no interval healing of the bilateral L2 or the unilateral L5 pars fractures. As a result of the patient’s noncontiguous pars fractures, a diagnostic CT-guided block of L2 pars was performed to identify which level was his main pain generator (Figure 5). He reported a brief period of complete pain relief after the L2 pars block. With failure of 6 months’ conservative management and positive SPECT scan and diagnostic block, surgical treatment was recommended. Prior to surgical intervention, magnetic resonance imaging was obtained to rule out pathology (eg, disc degeneration, infection, or tumor) other than the pars defect that could require fusion instead of pars repair (Figures 6A, 6B). Because of the patient’s young age, bilateral L2 pars repair rather than fusion was indicated. After 8 months of persistent back pain, he underwent bilateral L2 pars repair with iliac crest autograft, pedicle screws, and sublaminar hook fixation (Figures 7A, 7B). The patient had an uneventful immediate postoperative course. A 6-month postoperative CT scan showed bridging callus at the L2 pars; however, the left L5 pars fracture was still visible (Figures 8A-8C). Over a 6-month postoperative period, the patient had continued improvement in his back pain, advanced his activity as tolerated without problem, and was allowed to resume his sports activities. At 2-year follow-up, he was playing baseball and basketball, and denied any back pain.
Discussion
Lumbar spondylolysis is commonly seen at the fourth and fifth lumbar vertebrae, and accounts for more than 95% of spondylolysis cases.8 Multiple-level spondylolysis is a relatively rare finding with an incidence varying between 1.2% and 5.6%. The majority of the reported multiple-level cases are adjacent.1-3,6 Adolescents often present with a history of insidious-onset low back pain without radicular symptoms that is exacerbated by activity. Occasionally, an acute injury may elicit the onset of pain. A thorough history with emphasis on pain in relation to activity and sports involvement is beneficial. The patient in the current study was a throwing athlete and presented with 4 weeks of back pain that worsened with activity; he had no history of trauma.
Radiographic assessment using standing anteroposterior, lateral, and oblique radiographs of the thoracolumbar spine is useful in the initial assessment. A SPECT scan of the lumbosacral spine is highly sensitive for identifying spondylolytic defects when plain radiographs are within normal limits, yet a high index of suspicion remains given the patient’s history and physical examination findings.9,10 Increased radionuclide uptake within the pars indicates a stress reaction and, possibly, a more acute pathology. The plain radiographs of the patient showed only L5 spondylolysis. However, a SPECT scan showed only increased uptake in L2 pars on both sides. These findings suggested chronic L5 and acute L2 pars defects. A thin-cut CT scan gives the best visualization of pars defect and can help in differentiating chronic defect with sclerotic margins versus acute defect with hazy irregular margins. In the current case, the CT scan showed changes consistent with unilateral chronic L5 and bilateral acute L2 pars defects.
The origin of the pain in spondylolysis is from the tissues rich in nociceptive nerve endings in the loose posterior arch. A CT-guided pars block is a very useful diagnostic preoperative tool that confirms the symptomatic level in cases of multilevel pars defect, especially if they are noncontiguous. In this case, the diagnostic preoperative bilateral L2 pars block confirmed that the pain generator was the acute L2 rather than the chronic L5 pars defect. This step assured that surgical treatment involving only the L2 level would be beneficial in alleviating the patient’s back pain after the failure of 6 months of conservative treatment.
Most patients with single-level spondylolysis respond to conservative treatment, especially after early diagnosis and treatment. The traditional nonoperative treatment of children and adolescents with a symptomatic spondylolytic lesion was a period of rest and progressive increased activity with physical therapy. Immobilization with an LSO was reserved for individuals who did not respond to rest and physical therapy.11 However, multiple studies revealed early immobilization achieved results superior to activity restriction alone, and individuals who underwent a period of activity restriction prior to bracing were more likely to experience persistent symptoms.12-14 Our patient underwent conservative treatment for 6 months, in the form of LSO, cessation of sport activities, and physical therapy, which failed to give him relief of his back pain.
Surgical intervention is warranted for adolescents with persistent, debilitating pain intractable to at least a 6-month period of nonoperative management. Additional indications for surgical management are those individuals who present with neurologic deficits and isthmic spondylolisthesis. Surgical treatment involves direct pars repair with iliac crest bone graft and use of a sublaminar hook/pedicle screw construct, cerclage wire, or pars screw.15-18
In contrast to single-level pars defects that respond well to conservative treatment, there are conflicting reports regarding the management of multiple-level pars fractures; a few reports suggest good outcome with conservative management, but the majority state that surgery is often required and conservative measures are rarely useful.1-4,6 Nayeemuddin and colleagues19 reported a case of a 16-year-old football player who presented with a 4-month history of constant low back pain related to bilateral L3 and L5 pars defects that responded to 1 year of conservative management, when the more acute fractures at L3 showed complete bony union and the patient had symptomatic pain relief and was able to return to full sporting activity.
Chang and colleagues2 reported 10 patients with adjacent 2-level bilateral spondylolysis treated successfully using a pedicle screw–hook construct with autogenous bone grafting. Ogawa and colleagues5 reported adjacent 2-level spondylolysis in 5 patients and 3-level spondylolysis in 2 patients, who were treated successfully by segmental wire fixation and bone grafting. Ivanic and colleagues15 retrospectively reviewed 113 patients with spondylolysis who were treated with direct repair using a hook-screw construct and showed a pseudoarthrosis rate of 13.3%. Superior fusion rates were observed in patients 14 years and younger compared with older patients, particularly those 20 years and older.15 Roca and colleagues16 prospectively analyzed 19 consecutive cases of spondylolysis that were repaired using a hook-screw construct. Twelve of 13 patients (92%) who were 20 years or younger at the time of the study (average age, 17.2 years) had fusion, whereas, in 6 patients 21 years and older (average age, 27.5 years), no cases of fusion were observed. The patients 20 years or younger had significantly better clinical results than those obtained in the patients 21 years and older. The authors concluded that pedicle screw–hook fixation is a useful alternative in the treatment of spondylolysis in adolescents, but did not recommend this procedure in patients older than 20 years.16
Conclusion
The current case demonstrates a unique example of rare noncontiguous pars defects successfully treated with primary repair of 1 level when conservative management failed and the symptomatic defect was isolated. It also highlights the importance of investigating the entirety of the lumbar spine when diagnosis of L5 spondylolysis rules out noncontiguous pars defects. The treatment of noncontiguous pars defects is not well defined; this case showed the importance of using a SPECT scan and a diagnostic pars block to help isolate the symptomatic level when surgical management is considered after a failure of conservative treatment. This case shows 2 possible results: the chronic unilateral L5 defect responded to nonsurgical treatment with asymptomatic fibrous nonunion, while the more acute bilateral L2 defect responded to pars repair with pedicle screw–hook fixation and iliac crest bone graft.
Spondylolysis is a bone defect of the pars interarticularis. It is usually seen in adolescents who participate in sporting activities that involve repetitive axial loads to a hyperextended lower back, such as football offensive lineman, throwing athletes, and gymnasts. It occurs frequently in the L5 pars and can be unilateral or bilateral. The majority of reported multiple-level spondylolysis is at consecutive lumbar segments.1-6 Rarely, it affects noncontiguous levels. Most patients respond well to conservative treatment in the form of activity modification and orthosis.7 Surgical intervention is considered if 6 months of conservative management fails, spondylolisthesis develops, or intractable neurologic symptoms arise.
This case report presents an 18-year-old man with noncontiguous spondylolysis at L2 and L5 who was successfully treated with a 1-level pars repair at L2 after failed conservative management. This unique case highlights the importance of using single-photon emission computed tomography (SPECT) scan and diagnostic pars block when planning for surgical treatment in the rare cases of noncontiguous spondylolysis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
An 18-year-old man presented to the clinic with worsening lower back pain for the previous 4 weeks. He was playing high school baseball and stated the pain was worse when he swung his bat. He had no history of trauma or back pain. Rest was the only alleviating factor, and he was beginning to experience pain when he stood after sitting. He denied any radicular pain. On examination, he had midline tenderness along the upper lumbar spine and pain with lumbar spine extension. His neurologic examination showed normal sensation with 5/5 strength in all key muscle groups. Plain radiograph of the lumbar spine showed an L5 pars defect (Figures 1A, 1B). A SPECT scan showed increased uptake at L2 pars bilaterally; the L5 pars did not show increased uptake (Figures 2A, 2B). A computed tomography (CT) scan confirmed bilateral L2 pars fractures and a left L5 pars fracture (Figures 3A, 3B). Bony changes in the form of marginal sclerosis at the L5, but not the L2, pars suggested that the L2 fracture was acute while the L5 fracture was chronic (Figures 4A, 4B).
The patient had conservative management for 6 months in the form of lumbosacral orthosis (LSO), cessation of sports activities, and physical therapy. The patient was initially treated with an LSO brace for 3 months, after which he had physical therapy. At 6 month follow-up, he reported continuing, significant back pain. A repeat CT scan of the lumbar spine showed no interval healing of the bilateral L2 or the unilateral L5 pars fractures. As a result of the patient’s noncontiguous pars fractures, a diagnostic CT-guided block of L2 pars was performed to identify which level was his main pain generator (Figure 5). He reported a brief period of complete pain relief after the L2 pars block. With failure of 6 months’ conservative management and positive SPECT scan and diagnostic block, surgical treatment was recommended. Prior to surgical intervention, magnetic resonance imaging was obtained to rule out pathology (eg, disc degeneration, infection, or tumor) other than the pars defect that could require fusion instead of pars repair (Figures 6A, 6B). Because of the patient’s young age, bilateral L2 pars repair rather than fusion was indicated. After 8 months of persistent back pain, he underwent bilateral L2 pars repair with iliac crest autograft, pedicle screws, and sublaminar hook fixation (Figures 7A, 7B). The patient had an uneventful immediate postoperative course. A 6-month postoperative CT scan showed bridging callus at the L2 pars; however, the left L5 pars fracture was still visible (Figures 8A-8C). Over a 6-month postoperative period, the patient had continued improvement in his back pain, advanced his activity as tolerated without problem, and was allowed to resume his sports activities. At 2-year follow-up, he was playing baseball and basketball, and denied any back pain.
Discussion
Lumbar spondylolysis is commonly seen at the fourth and fifth lumbar vertebrae, and accounts for more than 95% of spondylolysis cases.8 Multiple-level spondylolysis is a relatively rare finding with an incidence varying between 1.2% and 5.6%. The majority of the reported multiple-level cases are adjacent.1-3,6 Adolescents often present with a history of insidious-onset low back pain without radicular symptoms that is exacerbated by activity. Occasionally, an acute injury may elicit the onset of pain. A thorough history with emphasis on pain in relation to activity and sports involvement is beneficial. The patient in the current study was a throwing athlete and presented with 4 weeks of back pain that worsened with activity; he had no history of trauma.
Radiographic assessment using standing anteroposterior, lateral, and oblique radiographs of the thoracolumbar spine is useful in the initial assessment. A SPECT scan of the lumbosacral spine is highly sensitive for identifying spondylolytic defects when plain radiographs are within normal limits, yet a high index of suspicion remains given the patient’s history and physical examination findings.9,10 Increased radionuclide uptake within the pars indicates a stress reaction and, possibly, a more acute pathology. The plain radiographs of the patient showed only L5 spondylolysis. However, a SPECT scan showed only increased uptake in L2 pars on both sides. These findings suggested chronic L5 and acute L2 pars defects. A thin-cut CT scan gives the best visualization of pars defect and can help in differentiating chronic defect with sclerotic margins versus acute defect with hazy irregular margins. In the current case, the CT scan showed changes consistent with unilateral chronic L5 and bilateral acute L2 pars defects.
The origin of the pain in spondylolysis is from the tissues rich in nociceptive nerve endings in the loose posterior arch. A CT-guided pars block is a very useful diagnostic preoperative tool that confirms the symptomatic level in cases of multilevel pars defect, especially if they are noncontiguous. In this case, the diagnostic preoperative bilateral L2 pars block confirmed that the pain generator was the acute L2 rather than the chronic L5 pars defect. This step assured that surgical treatment involving only the L2 level would be beneficial in alleviating the patient’s back pain after the failure of 6 months of conservative treatment.
Most patients with single-level spondylolysis respond to conservative treatment, especially after early diagnosis and treatment. The traditional nonoperative treatment of children and adolescents with a symptomatic spondylolytic lesion was a period of rest and progressive increased activity with physical therapy. Immobilization with an LSO was reserved for individuals who did not respond to rest and physical therapy.11 However, multiple studies revealed early immobilization achieved results superior to activity restriction alone, and individuals who underwent a period of activity restriction prior to bracing were more likely to experience persistent symptoms.12-14 Our patient underwent conservative treatment for 6 months, in the form of LSO, cessation of sport activities, and physical therapy, which failed to give him relief of his back pain.
Surgical intervention is warranted for adolescents with persistent, debilitating pain intractable to at least a 6-month period of nonoperative management. Additional indications for surgical management are those individuals who present with neurologic deficits and isthmic spondylolisthesis. Surgical treatment involves direct pars repair with iliac crest bone graft and use of a sublaminar hook/pedicle screw construct, cerclage wire, or pars screw.15-18
In contrast to single-level pars defects that respond well to conservative treatment, there are conflicting reports regarding the management of multiple-level pars fractures; a few reports suggest good outcome with conservative management, but the majority state that surgery is often required and conservative measures are rarely useful.1-4,6 Nayeemuddin and colleagues19 reported a case of a 16-year-old football player who presented with a 4-month history of constant low back pain related to bilateral L3 and L5 pars defects that responded to 1 year of conservative management, when the more acute fractures at L3 showed complete bony union and the patient had symptomatic pain relief and was able to return to full sporting activity.
Chang and colleagues2 reported 10 patients with adjacent 2-level bilateral spondylolysis treated successfully using a pedicle screw–hook construct with autogenous bone grafting. Ogawa and colleagues5 reported adjacent 2-level spondylolysis in 5 patients and 3-level spondylolysis in 2 patients, who were treated successfully by segmental wire fixation and bone grafting. Ivanic and colleagues15 retrospectively reviewed 113 patients with spondylolysis who were treated with direct repair using a hook-screw construct and showed a pseudoarthrosis rate of 13.3%. Superior fusion rates were observed in patients 14 years and younger compared with older patients, particularly those 20 years and older.15 Roca and colleagues16 prospectively analyzed 19 consecutive cases of spondylolysis that were repaired using a hook-screw construct. Twelve of 13 patients (92%) who were 20 years or younger at the time of the study (average age, 17.2 years) had fusion, whereas, in 6 patients 21 years and older (average age, 27.5 years), no cases of fusion were observed. The patients 20 years or younger had significantly better clinical results than those obtained in the patients 21 years and older. The authors concluded that pedicle screw–hook fixation is a useful alternative in the treatment of spondylolysis in adolescents, but did not recommend this procedure in patients older than 20 years.16
Conclusion
The current case demonstrates a unique example of rare noncontiguous pars defects successfully treated with primary repair of 1 level when conservative management failed and the symptomatic defect was isolated. It also highlights the importance of investigating the entirety of the lumbar spine when diagnosis of L5 spondylolysis rules out noncontiguous pars defects. The treatment of noncontiguous pars defects is not well defined; this case showed the importance of using a SPECT scan and a diagnostic pars block to help isolate the symptomatic level when surgical management is considered after a failure of conservative treatment. This case shows 2 possible results: the chronic unilateral L5 defect responded to nonsurgical treatment with asymptomatic fibrous nonunion, while the more acute bilateral L2 defect responded to pars repair with pedicle screw–hook fixation and iliac crest bone graft.
1. Al-Sebai MW, Al-Khawashki H. Spondyloptosis and multiple-level spondylolysis. Eur Spine J. 1999;8(1):75-77.
2. Chang JH, Lee CH, Wu SS, Lin LC, et al. Management of multiple level spondylolysis of the lumbar spine in young males: a report of six cases. J Formos Med Assoc. 2001;100(7)2:497-502.
3. Eingorn D, Pizzutillo PD. Pars interarticularis fusion of multiple levels of lumbar spondylolysis. A case report. Spine. 1985;10(3):250-252.
4. Nozawa S, Shimizu K, Miyamoto K, Tanaka M. Repair of pars interarticularis defect by segmental wire fixation in young athletes with spondylolysis. Am J Sports Med. 2003;31(3):359-364.
5. Ogawa H, Nishimoto H, Hosoe H, Suzuki N, Kanamori Y, Shimizu K. Clinical outcome after segmental wire fixation and bone grafting for repair of the defects in multiple level lumbar spondylolysis. J Spinal Disord Tech. 2007;20(7):521-525.
6. Ravichandran G. Multiple lumbar spondylolyses. Spine. 1980;5(6):552-557.
7. Sys J, Michielsen J, Bracke P, Martens M, Verstreken J. Nonoperative treatment of active spondylolysis in elite athletes with normal X-ray findings: literature review and results of conservative treatment. Eur Spine J. 2001;10(6):498-504.
8. Saraste H. Spondylolysis and spondylolisthesis. Acta Orthop Scand Suppl. 1993;251:84-86.
9. Anderson K, Sarwark JF, Conway JJ, Logue ES, Schafer MS. Quantitative assessment with SPECT imaging of stress injuries of the pars interarticularis and response to bracing. J Pediatr Orthop. 2000;20(1):28-33.
10. Bodner RJ, Heyman S, Drummond DS, Gregg JR. The use of single photon emission computed tomography (SPECT) in the diagnosis of low-back pain in young patients. Spine. 1988;13(10):1155-1160.
11. Steiner ME, Micheli LJ. Treatment of symptomatic spondylolysis and spondylolisthesis with the modified Boston brace. Spine. 1985;10(10):937-943.
12. Blanda J, Bethem D, Moats W, Lew M. Defects of pars interarticularis in athletes: a protocol for nonoperative treatment. J Spinal Disord. 1993;6(5):406-411.
13. Kurd MF, Patel D, Norton R, Picetti G, Friel B, Vaccaro AR. Nonoperative treatment of symptomatic spondylolysis. J Spinal Disord Tech. 2007;20(8):560-564.
14. Pizzutillo PD, Hummer CD 3rd. Nonoperative treatment for painful adolescent spondylolysis or spondylolisthesis. J Pediatr Orthop. 1989;9(5):538-540.
15. Ivanic GM, Pink TP, Achatz W, Ward JC, Homann NC, May M. Direct stabilization of lumbar spondylolysis with a hook screw: mean 11-year follow-up period for 113 patients. Spine. 2003;28(3):255-259.
16. Roca J, Iborra M, Cavanilles-Walker JM, Alberti G. Direct repair of spondylolysis using a new pedicle screw hook fixation: clinical and CT-assessed study: an analysis of 19 patients. J Spinal Disord Tech. 2005;18(suppl):S82-S89.
17. Schlenzka D, Remes V, Helenius I, et al. Direct repair for treatment of symptomatic spondylolysis and low-grade isthmic spondylolisthesis in young patients: no benefit in comparison to segmental fusion after a mean follow-up of 14.8 years. Eur Spine J. 2006;15(10):1437-1447.
18. Buck JE. Direct repair of the defect in spondylolisthesis. Preliminary report. J Bone Joint Surg Br. 1970;52(3):432-437.
19. Nayeemuddin M, Richards PJ, Ahmed EB. The imaging and management of nonconsecutive pars interarticularis defects: a case report and review of literature. Spine J. 2011;11(12):1157-1163.
1. Al-Sebai MW, Al-Khawashki H. Spondyloptosis and multiple-level spondylolysis. Eur Spine J. 1999;8(1):75-77.
2. Chang JH, Lee CH, Wu SS, Lin LC, et al. Management of multiple level spondylolysis of the lumbar spine in young males: a report of six cases. J Formos Med Assoc. 2001;100(7)2:497-502.
3. Eingorn D, Pizzutillo PD. Pars interarticularis fusion of multiple levels of lumbar spondylolysis. A case report. Spine. 1985;10(3):250-252.
4. Nozawa S, Shimizu K, Miyamoto K, Tanaka M. Repair of pars interarticularis defect by segmental wire fixation in young athletes with spondylolysis. Am J Sports Med. 2003;31(3):359-364.
5. Ogawa H, Nishimoto H, Hosoe H, Suzuki N, Kanamori Y, Shimizu K. Clinical outcome after segmental wire fixation and bone grafting for repair of the defects in multiple level lumbar spondylolysis. J Spinal Disord Tech. 2007;20(7):521-525.
6. Ravichandran G. Multiple lumbar spondylolyses. Spine. 1980;5(6):552-557.
7. Sys J, Michielsen J, Bracke P, Martens M, Verstreken J. Nonoperative treatment of active spondylolysis in elite athletes with normal X-ray findings: literature review and results of conservative treatment. Eur Spine J. 2001;10(6):498-504.
8. Saraste H. Spondylolysis and spondylolisthesis. Acta Orthop Scand Suppl. 1993;251:84-86.
9. Anderson K, Sarwark JF, Conway JJ, Logue ES, Schafer MS. Quantitative assessment with SPECT imaging of stress injuries of the pars interarticularis and response to bracing. J Pediatr Orthop. 2000;20(1):28-33.
10. Bodner RJ, Heyman S, Drummond DS, Gregg JR. The use of single photon emission computed tomography (SPECT) in the diagnosis of low-back pain in young patients. Spine. 1988;13(10):1155-1160.
11. Steiner ME, Micheli LJ. Treatment of symptomatic spondylolysis and spondylolisthesis with the modified Boston brace. Spine. 1985;10(10):937-943.
12. Blanda J, Bethem D, Moats W, Lew M. Defects of pars interarticularis in athletes: a protocol for nonoperative treatment. J Spinal Disord. 1993;6(5):406-411.
13. Kurd MF, Patel D, Norton R, Picetti G, Friel B, Vaccaro AR. Nonoperative treatment of symptomatic spondylolysis. J Spinal Disord Tech. 2007;20(8):560-564.
14. Pizzutillo PD, Hummer CD 3rd. Nonoperative treatment for painful adolescent spondylolysis or spondylolisthesis. J Pediatr Orthop. 1989;9(5):538-540.
15. Ivanic GM, Pink TP, Achatz W, Ward JC, Homann NC, May M. Direct stabilization of lumbar spondylolysis with a hook screw: mean 11-year follow-up period for 113 patients. Spine. 2003;28(3):255-259.
16. Roca J, Iborra M, Cavanilles-Walker JM, Alberti G. Direct repair of spondylolysis using a new pedicle screw hook fixation: clinical and CT-assessed study: an analysis of 19 patients. J Spinal Disord Tech. 2005;18(suppl):S82-S89.
17. Schlenzka D, Remes V, Helenius I, et al. Direct repair for treatment of symptomatic spondylolysis and low-grade isthmic spondylolisthesis in young patients: no benefit in comparison to segmental fusion after a mean follow-up of 14.8 years. Eur Spine J. 2006;15(10):1437-1447.
18. Buck JE. Direct repair of the defect in spondylolisthesis. Preliminary report. J Bone Joint Surg Br. 1970;52(3):432-437.
19. Nayeemuddin M, Richards PJ, Ahmed EB. The imaging and management of nonconsecutive pars interarticularis defects: a case report and review of literature. Spine J. 2011;11(12):1157-1163.

AES: Hormonal contraceptives can boost seizures in epileptics
PHILADELPHIA – Women with epilepsy often reported having an increased number of seizures when taking a hormonal contraceptive, according to data collected from 1,144 women with epilepsy who completed an online survey.
The data showed that women who used hormonal contraception reported having an increased number of seizures while on the contraceptive about 4.5-fold more often than did women who used nonhormonal contraception. The risk for an increased number of seizures with hormonal contraception seemed greatest for women treated with valproate.
Until now, “valproate was generally accepted as okay to use” by women also taking a hormonal contraceptive, but the new findings suggest that if a woman of childbearing age with epilepsy needs valproate for seizure control she would be better off using a nonhormonal form of contraception such as an intrauterine device, Dr. Andrew G. Herzog said while presenting a poster at the annual meeting of the American Epilepsy Society.

Dr. Herzog highlighted the need for some form of contraception for most younger women on valproate because of the drug’s potential teratogenic effects, but he also stressed that the risk for increased seizures does not appear to affect a majority of women. The survey results showed that overall only 28% of women with epilepsy reported an increased seizure frequency when using a hormonal contraceptive.
“The first goal of a neurologist is to get seizures under control, and you go with the [antiepileptic drugs] that work,” Dr. Herzog said in an interview. Once an effective regimen is found, the physician can then deal with other issues, such as adverse effects as well as the potential for an adverse interaction with a hormonal contraceptive. Valproate can be the antiepileptic drug of choice as it is one of the most effective agents for controlling seizures in patients with primary generalized epilepsy, said Dr. Herzog, professor of neurology at Harvard Medical School, Boston, and director of the neuroendocrine unit of Beth Israel Deaconess Medical Center in Wellesley, Mass.
The new data come from an Internet-based survey, which is subject to biases and appeared to attract a preponderance of responses from women who were better educated and had higher incomes than did the general population. In addition, the researchers collected the data retrospectively. Despite these limitations, the results are notable because they represent the only data set yet reported from a community-based source large enough to allow analysis of the many clinical variables that play into the potential interactions between various contraceptive types, various antiepileptic drug classes, and the diverse number of epilepsy subtypes, he said. Dr. Herzog and his associates are planning a study to collect similar data prospectively, but the results would likely not be available for at least about 5 years, he noted.
The Epilepsy Birth Control Registry enrolled women with epilepsy aged 18-47 years who had a history of using at least one form of contraception while on antiepileptic treatment, and the 1,144 women who completed the survey reported a total of 2,712 contraceptive experiences. The survey asked women, “Do you think this method of birth control changed how often you had seizures?” with the option to reply that their contraceptive method seemed to increase, decrease, or not change their seizure number.
One of the analyses done by Dr. Herzog and his associates compared the responses by women on any form of hormonal contraceptive (combined or progestin pill, hormonal patch, vaginal ring, depot medroxyprogesterone acetate, or implanted hormone) with women on any form of nonhormonal contraception (withdrawal, male or female condom, copper or progestin intrauterine device, or tubal ligation).
The results showed that 72% of women on any hormonal contraceptive and 91% of women on any form of nonhormonal contraceptive reported no change in their seizure frequency. The rates of reporting an increased number of seizures were 19% with hormonal contraceptives and 4% with nonhormonal contraceptives, which computed to a relative risk of about 4.5-fold for an increased number of seizures while on hormonal contraception, compared with nonhormonal contraception, the researchers reported.
Barrier contraception (male or female condoms) had the lowest rate of seizure increase among any of the nonhormonal methods. The risk for greater seizure frequency on hormonal contraceptives of all types was 6.75-fold higher when compared specifically with barrier contraception.
In analyses of specific types of hormonal contraceptives, women using a hormonal patch reported a 68% greater incidence of seizure increases, compared with women using combined oral contraceptive pills (the hormonal method that produced the fewest episodes of seizure increases). Those using a progestin-only pill had a 62% higher rate of seizure increases.
More women on hormonal contraceptives also reported having a decrease in seizures after starting contraception, compared with those starting on a nonhormonal method (9.5% vs. 5.2%, respectively), which calculated to a 85% relative rate increase for decreased seizures. Depot medroxyprogesterone acetate was the only specific hormonal contraceptive that linked with a higher rate of seizure decreases, compared with combined oral pills, a 95% higher rate.
A second analysis of the results by Dr. Herzog and his associates examined the frequencies of seizure outcomes on hormonal and nonhormonal contraceptives stratifying by type of antiepileptic drug women used when starting a particular contraceptive method. This analysis broke down antiepileptic drugs into four types: enzyme inducing (29%), glucuronidated (such as lamotrigine; 27%), nonenzyme inducing (such as levetiracetam; 22%), enzyme inhibiting (valproate; 8%), and a fifth category that included women who were not on any antiepileptic drug (14%).
This analysis showed that the frequency of seizure increases was significantly greater with hormonal contraceptive use, compared with nonhormonal methods, across all five subgroups of antiepileptic drug type. In addition, the frequency of seizure increases with hormonal contraceptives differed significantly, depending on which antiepileptic drug type women used, but these significant differences among the antiepileptic drug types also occurred among women using nonhormonal contraception.
Women receiving a nonenzyme-inducing drug when starting a hormonal contraceptive reported the lowest frequency of seizure increases, a 12% rate. In contrast, women on an enzyme-inhibiting drug, valproate, had the highest rate of increased seizures when starting a hormonal contraceptive, 29%. This calculated out to about a 2.5-fold relative risk increase for having more seizures when starting hormonal contraception while on valproate, compared with women on a nonenzyme-inducing drug, Dr. Herzog reported.
Physicians “need to be on the lookout for the possibility that seizures could increase when women start a hormonal contraceptive,” he concluded.
On Twitter @mitchelzoler
PHILADELPHIA – Women with epilepsy often reported having an increased number of seizures when taking a hormonal contraceptive, according to data collected from 1,144 women with epilepsy who completed an online survey.
The data showed that women who used hormonal contraception reported having an increased number of seizures while on the contraceptive about 4.5-fold more often than did women who used nonhormonal contraception. The risk for an increased number of seizures with hormonal contraception seemed greatest for women treated with valproate.
Until now, “valproate was generally accepted as okay to use” by women also taking a hormonal contraceptive, but the new findings suggest that if a woman of childbearing age with epilepsy needs valproate for seizure control she would be better off using a nonhormonal form of contraception such as an intrauterine device, Dr. Andrew G. Herzog said while presenting a poster at the annual meeting of the American Epilepsy Society.
Dr. Herzog highlighted the need for some form of contraception for most younger women on valproate because of the drug’s potential teratogenic effects, but he also stressed that the risk for increased seizures does not appear to affect a majority of women. The survey results showed that overall only 28% of women with epilepsy reported an increased seizure frequency when using a hormonal contraceptive.
“The first goal of a neurologist is to get seizures under control, and you go with the [antiepileptic drugs] that work,” Dr. Herzog said in an interview. Once an effective regimen is found, the physician can then deal with other issues, such as adverse effects as well as the potential for an adverse interaction with a hormonal contraceptive. Valproate can be the antiepileptic drug of choice as it is one of the most effective agents for controlling seizures in patients with primary generalized epilepsy, said Dr. Herzog, professor of neurology at Harvard Medical School, Boston, and director of the neuroendocrine unit of Beth Israel Deaconess Medical Center in Wellesley, Mass.
The new data come from an Internet-based survey, which is subject to biases and appeared to attract a preponderance of responses from women who were better educated and had higher incomes than did the general population. In addition, the researchers collected the data retrospectively. Despite these limitations, the results are notable because they represent the only data set yet reported from a community-based source large enough to allow analysis of the many clinical variables that play into the potential interactions between various contraceptive types, various antiepileptic drug classes, and the diverse number of epilepsy subtypes, he said. Dr. Herzog and his associates are planning a study to collect similar data prospectively, but the results would likely not be available for at least about 5 years, he noted.
The Epilepsy Birth Control Registry enrolled women with epilepsy aged 18-47 years who had a history of using at least one form of contraception while on antiepileptic treatment, and the 1,144 women who completed the survey reported a total of 2,712 contraceptive experiences. The survey asked women, “Do you think this method of birth control changed how often you had seizures?” with the option to reply that their contraceptive method seemed to increase, decrease, or not change their seizure number.
One of the analyses done by Dr. Herzog and his associates compared the responses by women on any form of hormonal contraceptive (combined or progestin pill, hormonal patch, vaginal ring, depot medroxyprogesterone acetate, or implanted hormone) with women on any form of nonhormonal contraception (withdrawal, male or female condom, copper or progestin intrauterine device, or tubal ligation).
The results showed that 72% of women on any hormonal contraceptive and 91% of women on any form of nonhormonal contraceptive reported no change in their seizure frequency. The rates of reporting an increased number of seizures were 19% with hormonal contraceptives and 4% with nonhormonal contraceptives, which computed to a relative risk of about 4.5-fold for an increased number of seizures while on hormonal contraception, compared with nonhormonal contraception, the researchers reported.
Barrier contraception (male or female condoms) had the lowest rate of seizure increase among any of the nonhormonal methods. The risk for greater seizure frequency on hormonal contraceptives of all types was 6.75-fold higher when compared specifically with barrier contraception.
In analyses of specific types of hormonal contraceptives, women using a hormonal patch reported a 68% greater incidence of seizure increases, compared with women using combined oral contraceptive pills (the hormonal method that produced the fewest episodes of seizure increases). Those using a progestin-only pill had a 62% higher rate of seizure increases.
More women on hormonal contraceptives also reported having a decrease in seizures after starting contraception, compared with those starting on a nonhormonal method (9.5% vs. 5.2%, respectively), which calculated to a 85% relative rate increase for decreased seizures. Depot medroxyprogesterone acetate was the only specific hormonal contraceptive that linked with a higher rate of seizure decreases, compared with combined oral pills, a 95% higher rate.
A second analysis of the results by Dr. Herzog and his associates examined the frequencies of seizure outcomes on hormonal and nonhormonal contraceptives stratifying by type of antiepileptic drug women used when starting a particular contraceptive method. This analysis broke down antiepileptic drugs into four types: enzyme inducing (29%), glucuronidated (such as lamotrigine; 27%), nonenzyme inducing (such as levetiracetam; 22%), enzyme inhibiting (valproate; 8%), and a fifth category that included women who were not on any antiepileptic drug (14%).
This analysis showed that the frequency of seizure increases was significantly greater with hormonal contraceptive use, compared with nonhormonal methods, across all five subgroups of antiepileptic drug type. In addition, the frequency of seizure increases with hormonal contraceptives differed significantly, depending on which antiepileptic drug type women used, but these significant differences among the antiepileptic drug types also occurred among women using nonhormonal contraception.
Women receiving a nonenzyme-inducing drug when starting a hormonal contraceptive reported the lowest frequency of seizure increases, a 12% rate. In contrast, women on an enzyme-inhibiting drug, valproate, had the highest rate of increased seizures when starting a hormonal contraceptive, 29%. This calculated out to about a 2.5-fold relative risk increase for having more seizures when starting hormonal contraception while on valproate, compared with women on a nonenzyme-inducing drug, Dr. Herzog reported.
Physicians “need to be on the lookout for the possibility that seizures could increase when women start a hormonal contraceptive,” he concluded.
On Twitter @mitchelzoler
PHILADELPHIA – Women with epilepsy often reported having an increased number of seizures when taking a hormonal contraceptive, according to data collected from 1,144 women with epilepsy who completed an online survey.
The data showed that women who used hormonal contraception reported having an increased number of seizures while on the contraceptive about 4.5-fold more often than did women who used nonhormonal contraception. The risk for an increased number of seizures with hormonal contraception seemed greatest for women treated with valproate.
Until now, “valproate was generally accepted as okay to use” by women also taking a hormonal contraceptive, but the new findings suggest that if a woman of childbearing age with epilepsy needs valproate for seizure control she would be better off using a nonhormonal form of contraception such as an intrauterine device, Dr. Andrew G. Herzog said while presenting a poster at the annual meeting of the American Epilepsy Society.
Dr. Herzog highlighted the need for some form of contraception for most younger women on valproate because of the drug’s potential teratogenic effects, but he also stressed that the risk for increased seizures does not appear to affect a majority of women. The survey results showed that overall only 28% of women with epilepsy reported an increased seizure frequency when using a hormonal contraceptive.
“The first goal of a neurologist is to get seizures under control, and you go with the [antiepileptic drugs] that work,” Dr. Herzog said in an interview. Once an effective regimen is found, the physician can then deal with other issues, such as adverse effects as well as the potential for an adverse interaction with a hormonal contraceptive. Valproate can be the antiepileptic drug of choice as it is one of the most effective agents for controlling seizures in patients with primary generalized epilepsy, said Dr. Herzog, professor of neurology at Harvard Medical School, Boston, and director of the neuroendocrine unit of Beth Israel Deaconess Medical Center in Wellesley, Mass.
The new data come from an Internet-based survey, which is subject to biases and appeared to attract a preponderance of responses from women who were better educated and had higher incomes than did the general population. In addition, the researchers collected the data retrospectively. Despite these limitations, the results are notable because they represent the only data set yet reported from a community-based source large enough to allow analysis of the many clinical variables that play into the potential interactions between various contraceptive types, various antiepileptic drug classes, and the diverse number of epilepsy subtypes, he said. Dr. Herzog and his associates are planning a study to collect similar data prospectively, but the results would likely not be available for at least about 5 years, he noted.
The Epilepsy Birth Control Registry enrolled women with epilepsy aged 18-47 years who had a history of using at least one form of contraception while on antiepileptic treatment, and the 1,144 women who completed the survey reported a total of 2,712 contraceptive experiences. The survey asked women, “Do you think this method of birth control changed how often you had seizures?” with the option to reply that their contraceptive method seemed to increase, decrease, or not change their seizure number.
One of the analyses done by Dr. Herzog and his associates compared the responses by women on any form of hormonal contraceptive (combined or progestin pill, hormonal patch, vaginal ring, depot medroxyprogesterone acetate, or implanted hormone) with women on any form of nonhormonal contraception (withdrawal, male or female condom, copper or progestin intrauterine device, or tubal ligation).
The results showed that 72% of women on any hormonal contraceptive and 91% of women on any form of nonhormonal contraceptive reported no change in their seizure frequency. The rates of reporting an increased number of seizures were 19% with hormonal contraceptives and 4% with nonhormonal contraceptives, which computed to a relative risk of about 4.5-fold for an increased number of seizures while on hormonal contraception, compared with nonhormonal contraception, the researchers reported.
Barrier contraception (male or female condoms) had the lowest rate of seizure increase among any of the nonhormonal methods. The risk for greater seizure frequency on hormonal contraceptives of all types was 6.75-fold higher when compared specifically with barrier contraception.
In analyses of specific types of hormonal contraceptives, women using a hormonal patch reported a 68% greater incidence of seizure increases, compared with women using combined oral contraceptive pills (the hormonal method that produced the fewest episodes of seizure increases). Those using a progestin-only pill had a 62% higher rate of seizure increases.
More women on hormonal contraceptives also reported having a decrease in seizures after starting contraception, compared with those starting on a nonhormonal method (9.5% vs. 5.2%, respectively), which calculated to a 85% relative rate increase for decreased seizures. Depot medroxyprogesterone acetate was the only specific hormonal contraceptive that linked with a higher rate of seizure decreases, compared with combined oral pills, a 95% higher rate.
A second analysis of the results by Dr. Herzog and his associates examined the frequencies of seizure outcomes on hormonal and nonhormonal contraceptives stratifying by type of antiepileptic drug women used when starting a particular contraceptive method. This analysis broke down antiepileptic drugs into four types: enzyme inducing (29%), glucuronidated (such as lamotrigine; 27%), nonenzyme inducing (such as levetiracetam; 22%), enzyme inhibiting (valproate; 8%), and a fifth category that included women who were not on any antiepileptic drug (14%).
This analysis showed that the frequency of seizure increases was significantly greater with hormonal contraceptive use, compared with nonhormonal methods, across all five subgroups of antiepileptic drug type. In addition, the frequency of seizure increases with hormonal contraceptives differed significantly, depending on which antiepileptic drug type women used, but these significant differences among the antiepileptic drug types also occurred among women using nonhormonal contraception.
Women receiving a nonenzyme-inducing drug when starting a hormonal contraceptive reported the lowest frequency of seizure increases, a 12% rate. In contrast, women on an enzyme-inhibiting drug, valproate, had the highest rate of increased seizures when starting a hormonal contraceptive, 29%. This calculated out to about a 2.5-fold relative risk increase for having more seizures when starting hormonal contraception while on valproate, compared with women on a nonenzyme-inducing drug, Dr. Herzog reported.
Physicians “need to be on the lookout for the possibility that seizures could increase when women start a hormonal contraceptive,” he concluded.
On Twitter @mitchelzoler
AT AES 2015
Key clinical point: Women with epilepsy often reported having more seizures while taking a hormonal contraceptive, compared with women using nonhormonal contraception.
Major finding: Epileptic women reported a 4.5-fold higher rate of increased seizures when using hormonal contraception, compared with nonhormonal contraception.
Data source: Internet-based survey completed by 1,144 women with epilepsy.
Disclosures: The study received partial support from Lundbeck. Dr. Herzog had no personal disclosures.

Acute Onset of Vancomycin Anaphylaxis With Disseminated Intravascular Coagulation in an Orthopedic Patient Despite Prior Repeated Exposure
Vancomycin is a glycopeptide antibiotic that exhibits bactericidal activity against gram-positive cocci. It is commonly recommended for surgical prophylaxis in cases of suspected bacterial resistance or penicillin allergy.1 Two main types of hypersensitivity reactions associated with vancomycin can have similar presentations. Red man syndrome is an anaphylactoid reaction caused by direct release of histamine from mast cells via a nonimmunologic mechanism, and is the more common of the 2 reactions. The second type is an anaphylactic reaction, which is an immunoglobulin E (IgE)–mediated systemic event and requires exposure to become sensitized.2,3
We present a patient who had received vancomycin on at least 12 occasions without incident. On this occasion, however, she developed a true anaphylactic reaction causing acute hemodynamic collapse that she survived after extensive resuscitation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 55-year-old woman had a history of metastatic giant cell tumor of the right proximal tibia. She was originally treated 27 years ago for proximal tibial resection and reconstruction with a custom proximal tibial prosthesis. Four months later, she underwent resection of multiple pulmonary metastases via bilateral thoracotomies in a single surgical setting. After this, the patient had no evidence of recurrent metastatic disease. In subsequent years, the patient underwent multiple revision surgeries for problems such as hardware failure, patellar maltracking, and infection. The patient underwent 19 operations, including several nonorthopedic procedures. Because the patient had a rash after receiving penicillin as a child, she was thought to be allergic to penicillin. Consequently, she received vancomycin as antibiotic prophylaxis for the majority of these procedures. She also received extended courses of vancomycin of at least 6 weeks on 2 separate occasions. During her most recent revision procedure, 6 weeks prior to the procedure under discussion, the patient took vancomycin without incident. She was then found to have a prosthetic infection with Staphylococcus epidermidis, the same organism isolated in her previous infections, and she was advised to undergo a staged revision.
After a preoperative medical evaluation by her primary care physician, the patient was taken to the operating room for prosthesis removal and antibiotic spacer placement. She was anemic with a hemoglobin level of 8.8 g/dL; her erythrocyte sedimentation rate (ESR) was 102 mm/h (normal, <22 mm/h) and her C-reactive protein (CRP) was 38 mg/L (normal, <3 mg/L), but, otherwise, her laboratory values were normal, including a white blood cell count (WBC) of 8100/µL. Her electrocardiogram showed a normal sinus rhythm with nonspecific ST- and T-wave changes. Antibiotics were held until after cultures were taken. General endotracheal tube anesthesia was induced with 2 mg midazolam, 100 µg fentanyl, 180 mg propofol, and 140 mg succinylcholine, followed by 10 mg vecuronium, and maintained with desflurane. A tourniquet was not used per the surgeon’s routine. Dissection was carried down to the prosthesis and showed a small amount of purulent fluid. Transfusion of 1 unit of packed red blood cells (pRBC) was started during the approach owing to relatively low preoperative hemoglobin and significant blood loss. Approximately 500 mL of blood was lost during the approach secondary to the extensive dissection and the local inflammatory response from infection and recent surgery. After cultures were taken, and approximately 10 minutes after blood transfusion began, infusion of 1 g vancomycin in 250 mL normal saline was started via an infusion pump to run over 1 hour.
After infusion of 5 mL vancomycin, the patient’s blood pressure dropped from 117/63 mm Hg to 63/30 mm Hg; her pulse concurrently dropped from 90 to 50 beats/min. Vancomycin infusion was immediately stopped, anesthesia gasses were turned off, and patient received a bolus of normal saline with a second unit of pRBC. Patient received boluses of 0.5 mg to 1.0 mg epinephrine and 100 µg phenylephrine without sustained increase in blood pressure, which had dropped to 54/24 mm Hg, although the patient became tachycardic to ~120 beats/min after epinephrine. A sudden drop in end-tidal CO2 from 40s mm Hg to 20s mm Hg was also noted, indicating continuous but significantly decreased perfusion of the lungs.
We elected to abort the procedure, and a vacuum-assisted closure (VAC) dressing was applied to the open wound. After 15 minutes, the patient’s pulses, which had been faint, became impalpable, and cardiopulmonary resuscitation was initiated for about 7 minutes. The patient received 40 units vasopressin with repeated boluses of 0.5 mg epinephrine; a norepinephrine continuous infusion was started with the return of pulses. The patient also received 50 mg diphenhydramine, 125 mg methylprednisolone, and 20 mg famotidine for suspected anaphylaxis. A central venous line and arterial line were placed, and blood was drawn for laboratory analysis. The patient was noted to have clear breath sounds with no obvious rash, and her urine remained clear. Blood gas showed a profound metabolic acidosis, with pH of 7.09, base deficit of 5.9, and lactate of 8.9. The patient was treated with bicarbonate infusion. The patient was noted to ooze significantly during central venous line and arterial line placement, despite apparently normal coagulation during the surgical approach. Coagulation values were consistent with disseminated intravascular coagulation (DIC): prothrombin time, 57 s (international normalized ratio, 6.7); partial thromboplastin time, >200 s; thrombin time, 110 s; D-dimer, >10,000 ng/mL (normal, 0-200 ng/mL); and fibrinogen, <60 mg/dL (normal, 222-475 mg/dL). The patient’s thromboelastogram showed a flat line indicating an absence of clotting. Interestingly, the platelet count remained near the preoperative level at 338×103/µL. The patient’s blood pressure remained labile and was responsive primarily to epinephrine boluses, of which she received a total of 5 mg. After 1 hour of resuscitation, during which time the patient received a total of 5 L crystalloid and 3 units pRBC, the patient was transferred to the intensive care unit (ICU), intubated, and started on a titrated epinephrine infusion.
Upon arrival in the ICU, the patient quickly stabilized hemodynamically. She was weaned from all inotropic support within 2 hours of arrival. The patient lost 800 mL of blood through wound VAC over the first 12 hours postoperatively and required a total of 11 units of pRBC, 6 units fresh frozen plasma, and 3 units of pooled cryoprecipitate, all of which were compatible. Laboratory values, including arterial pH, lactic acid, and coagulation studies, normalized on the evening of surgery, and, by the next morning, the patient was alert and was extubated without difficulty. Steroids were tapered without hemodynamic compromise while the patient was in the ICU. Cardiology examination revealed no abnormalities. Because of the temporal association of blood transfusion with cardiovascular collapse, pRBC units were retested for antibodies and cultured. Both of these investigations were negative. Wound cultures again were positive for Staphylococcus epidermidis, and blood cultures were negative. The patient was started on daptomycin based on susceptibility profiles. Serum histamine levels taken during initial resuscitation in the operating room were normal. The serum tryptase level obtained at the same time was markedly elevated at >700 ng/mL (normal, <11.5 ng/mL), although this information was not available until several days later.
The patient underwent 2 additional surgeries during the same admission, including the prosthesis removal and tobramycin cement spacer placement, without incident. She was discharged home, again without incident. The patient was later evaluated by an outside allergist and underwent skin puncture and intradermal allergy testing. The results were consistent with a strong IgE-mediated hypersensitivity. Interestingly, she was found not to have a penicillin allergy.
Discussion
Vancomycin hypersensitivity reactions include the anaphylactoid reaction red man syndrome and a true IgE-mediated anaphylactic reaction. Red man syndrome is much more common, with reported rates in infected patients from 3.7% to 47%,4,5 when vancomycin is given at the suggested rate of 1 g over 1 hour. The reaction occurs because of histamine release from mast cells and basophils, and does not require previous sensitization.3 The rate of infusion is directly related to the development of symptoms, with 100% of patients developing symptoms in 1 study with rapid infusion (1 g over 10 min).6 Red man syndrome can typically be prevented by slowing the rate of infusion or by giving an H1 blocker.3 Anaphylaxis is more rare but can occur.7 Anaphylaxis is mediated by vancomycin-specific IgE, which requires previous exposure, as was the case with our patient. Interestingly, the patient had received vancomycin many times without any signs of a hypersensitivity reaction. Antihistamines are not effective in treating anaphylaxis, and epinephrine is the first-line agent.3 This was clearly demonstrated in this case, as there was a significant hemodynamic response to epinephrine and a negligible response to other vasopressors, specifically norepinephrine and vasopressin.
Most hypersensitivity reactions during the course of a surgical procedure occur with induction of anesthesia, with neuromuscular blocking agents and antibiotics being the most common causes.8 In our case, antibiotics were held until after deep cultures were taken. Given the time from induction to the anaphylactic reaction, it is unlikely the reaction resulted from the induction agents or the neuromuscular blocking agent. The possibility of a transfusion reaction was also investigated, since a unit of pRBC was still being transfused when symptoms began. An acute hemolytic transfusion reaction has the classic triad of fever, flank pain, and hemoglobinuria, and can also present as DIC.9 Under anesthesia, DIC can often be the presenting sign. In this case, a hemolytic transfusion reaction appeared very unlikely. All of the blood components the patient received were rechecked and found to be compatible, posttransfusion analysis showed no evidence of hemolysis in any sample, and the direct antiglobulin test was negative in all components.
To our knowledge, there are no reported cases of vancomycin-induced anaphylaxis with concomitant DIC. Symptoms of anaphylaxis after exposure to a possible antigen include rapid onset of hypotension or rapid onset of signs in at least 2 organ systems, including cutaneous, gastrointestinal, respiratory, and cardiovascular.10 Anaphylaxis with DIC is rare after exposure to any substance but has been reported.11 In fact, induction of systemic anaphylaxis in mice is known to cause DIC, with platelet-activating factor suggested as an important common mediator. A similar mechanism is suspected in humans.12
Confirmation of, and, certainly, prediction of, a vancomycin hypersensitivity reaction is difficult. Histamine levels can be used as a measure of mast-cell degranulation, but serum levels peak within 5 minutes and quickly return to baseline, limiting its diagnostic usefulness.3 Tryptase is an enzyme found in the secretory granules of mast cells. It has become an accepted marker of acute anaphylaxis, and, in vancomycin hypersensitivity reactions, can also distinguish between anaphylactic and anaphylactoid reactions.13 Tryptase levels peak 1 to 2 hours after the reaction, making this easier to measure than histamine, but results may not be available for several days, making it useful only in retrospect, as in our case. Skin testing is probably the best way to confirm a hypersensitivity reaction, although even this has been questioned with vancomycin because some find a high false-positive rate3, while others think the false-negative rate is likely too high.7 In this case, we were able to confirm our initial clinical suspicion with both an elevated tryptase level and a positive skin test.
Conclusion
We present a rare case of vancomycin anaphylaxis with DIC after repeated and prolonged previous exposure, which was treated acutely with hemodynamic resuscitation, replacement of blood components, steroids, and, most importantly, repeated boluses of epinephrine. Although several papers have described successful vancomycin desensitization7, this was fortunately not necessary in this case because the causative organism was sensitive to other acceptable antibiotics. The patient has been treated with systemic daptomycin and a tobramycin cement spacer without further incident.
1. Recommendation for the use of intravenous antibiotic prophylaxis in primary total joint arthroplasty. AAOS Information Statement 1027. American Academy of Orthopaedic Surgeons website. http://www.aaos.org/about/papers/advistmt/1027.asp. Published June 2004. Accessed October 28, 2015.
2. Duffy BL. Vancomycin reaction during spinal anesthesia. Anaesth Intensive Case. 2002;30(3):364-366.
3. Wazny LD, Daghigh B. Desensitization protocols for vancomycin hypersensitivity. Ann Pharmacother. 2001;35(11):1458-1464.
4. O’Sullivan TL, Ruffing MJ, Lamp KC, Warbasse LH, Rybak MJ. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773-776.
5. Wallace MR, Mascola JR, Oldfield EC 3rd. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180-1185.
6. Renz CL, Thurn JD, Finn HA, Lynch JP, Moss J. Antihistamine prophylaxis permits rapid vancomycin infusion. Crit Care Med. 1999;27(9):1732-1737.
7. Kupstaite R, Baranauskaite A, Pileckyte M, Sveikata A, Kadusevicius E, Muckiene G. Severe vancomycin-induced anaphylactic reaction. Medicina (Kaunas). 2010;46(1):30-33.
8. Lobera T, Audicana MT, Pozo MD, et al. Study of hypersensitivity reactions and anaphylaxis during anesthesia in Spain. J Investig Allergol Clin Immunol. 2008;18(5):350-356.
9. Berséus O, Boman K, Nessen SC, Westerberg LA. Risks of hemolysis due to anti-A and anti-B caused by the transfusion of blood or blood components containing ABO-incompatible plasma. Transfusion. 2013;53(suppl 1):114S-123S.
10. Schwartz LB. Systemic anaphylaxis, food allergy, and insect sting allergy. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier; 2011:1633-1638.
11. Jung JW, Jeon EJ, Kim JW, et al. A fatal case of intravascular coagulation after bee sting acupuncture. Allergy Asthma Immunol Res. 2012;4(2):107-109.
12. Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100(3):390-394.
13. Renz CL, Laroche D, Thurn JD, et al. Tryptase levels are not increased during vancomycin-induced anaphylactoid reactions. Anesthesiology. 1998;89(3):620-625.
Vancomycin is a glycopeptide antibiotic that exhibits bactericidal activity against gram-positive cocci. It is commonly recommended for surgical prophylaxis in cases of suspected bacterial resistance or penicillin allergy.1 Two main types of hypersensitivity reactions associated with vancomycin can have similar presentations. Red man syndrome is an anaphylactoid reaction caused by direct release of histamine from mast cells via a nonimmunologic mechanism, and is the more common of the 2 reactions. The second type is an anaphylactic reaction, which is an immunoglobulin E (IgE)–mediated systemic event and requires exposure to become sensitized.2,3
We present a patient who had received vancomycin on at least 12 occasions without incident. On this occasion, however, she developed a true anaphylactic reaction causing acute hemodynamic collapse that she survived after extensive resuscitation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 55-year-old woman had a history of metastatic giant cell tumor of the right proximal tibia. She was originally treated 27 years ago for proximal tibial resection and reconstruction with a custom proximal tibial prosthesis. Four months later, she underwent resection of multiple pulmonary metastases via bilateral thoracotomies in a single surgical setting. After this, the patient had no evidence of recurrent metastatic disease. In subsequent years, the patient underwent multiple revision surgeries for problems such as hardware failure, patellar maltracking, and infection. The patient underwent 19 operations, including several nonorthopedic procedures. Because the patient had a rash after receiving penicillin as a child, she was thought to be allergic to penicillin. Consequently, she received vancomycin as antibiotic prophylaxis for the majority of these procedures. She also received extended courses of vancomycin of at least 6 weeks on 2 separate occasions. During her most recent revision procedure, 6 weeks prior to the procedure under discussion, the patient took vancomycin without incident. She was then found to have a prosthetic infection with Staphylococcus epidermidis, the same organism isolated in her previous infections, and she was advised to undergo a staged revision.
After a preoperative medical evaluation by her primary care physician, the patient was taken to the operating room for prosthesis removal and antibiotic spacer placement. She was anemic with a hemoglobin level of 8.8 g/dL; her erythrocyte sedimentation rate (ESR) was 102 mm/h (normal, <22 mm/h) and her C-reactive protein (CRP) was 38 mg/L (normal, <3 mg/L), but, otherwise, her laboratory values were normal, including a white blood cell count (WBC) of 8100/µL. Her electrocardiogram showed a normal sinus rhythm with nonspecific ST- and T-wave changes. Antibiotics were held until after cultures were taken. General endotracheal tube anesthesia was induced with 2 mg midazolam, 100 µg fentanyl, 180 mg propofol, and 140 mg succinylcholine, followed by 10 mg vecuronium, and maintained with desflurane. A tourniquet was not used per the surgeon’s routine. Dissection was carried down to the prosthesis and showed a small amount of purulent fluid. Transfusion of 1 unit of packed red blood cells (pRBC) was started during the approach owing to relatively low preoperative hemoglobin and significant blood loss. Approximately 500 mL of blood was lost during the approach secondary to the extensive dissection and the local inflammatory response from infection and recent surgery. After cultures were taken, and approximately 10 minutes after blood transfusion began, infusion of 1 g vancomycin in 250 mL normal saline was started via an infusion pump to run over 1 hour.
After infusion of 5 mL vancomycin, the patient’s blood pressure dropped from 117/63 mm Hg to 63/30 mm Hg; her pulse concurrently dropped from 90 to 50 beats/min. Vancomycin infusion was immediately stopped, anesthesia gasses were turned off, and patient received a bolus of normal saline with a second unit of pRBC. Patient received boluses of 0.5 mg to 1.0 mg epinephrine and 100 µg phenylephrine without sustained increase in blood pressure, which had dropped to 54/24 mm Hg, although the patient became tachycardic to ~120 beats/min after epinephrine. A sudden drop in end-tidal CO2 from 40s mm Hg to 20s mm Hg was also noted, indicating continuous but significantly decreased perfusion of the lungs.
We elected to abort the procedure, and a vacuum-assisted closure (VAC) dressing was applied to the open wound. After 15 minutes, the patient’s pulses, which had been faint, became impalpable, and cardiopulmonary resuscitation was initiated for about 7 minutes. The patient received 40 units vasopressin with repeated boluses of 0.5 mg epinephrine; a norepinephrine continuous infusion was started with the return of pulses. The patient also received 50 mg diphenhydramine, 125 mg methylprednisolone, and 20 mg famotidine for suspected anaphylaxis. A central venous line and arterial line were placed, and blood was drawn for laboratory analysis. The patient was noted to have clear breath sounds with no obvious rash, and her urine remained clear. Blood gas showed a profound metabolic acidosis, with pH of 7.09, base deficit of 5.9, and lactate of 8.9. The patient was treated with bicarbonate infusion. The patient was noted to ooze significantly during central venous line and arterial line placement, despite apparently normal coagulation during the surgical approach. Coagulation values were consistent with disseminated intravascular coagulation (DIC): prothrombin time, 57 s (international normalized ratio, 6.7); partial thromboplastin time, >200 s; thrombin time, 110 s; D-dimer, >10,000 ng/mL (normal, 0-200 ng/mL); and fibrinogen, <60 mg/dL (normal, 222-475 mg/dL). The patient’s thromboelastogram showed a flat line indicating an absence of clotting. Interestingly, the platelet count remained near the preoperative level at 338×103/µL. The patient’s blood pressure remained labile and was responsive primarily to epinephrine boluses, of which she received a total of 5 mg. After 1 hour of resuscitation, during which time the patient received a total of 5 L crystalloid and 3 units pRBC, the patient was transferred to the intensive care unit (ICU), intubated, and started on a titrated epinephrine infusion.
Upon arrival in the ICU, the patient quickly stabilized hemodynamically. She was weaned from all inotropic support within 2 hours of arrival. The patient lost 800 mL of blood through wound VAC over the first 12 hours postoperatively and required a total of 11 units of pRBC, 6 units fresh frozen plasma, and 3 units of pooled cryoprecipitate, all of which were compatible. Laboratory values, including arterial pH, lactic acid, and coagulation studies, normalized on the evening of surgery, and, by the next morning, the patient was alert and was extubated without difficulty. Steroids were tapered without hemodynamic compromise while the patient was in the ICU. Cardiology examination revealed no abnormalities. Because of the temporal association of blood transfusion with cardiovascular collapse, pRBC units were retested for antibodies and cultured. Both of these investigations were negative. Wound cultures again were positive for Staphylococcus epidermidis, and blood cultures were negative. The patient was started on daptomycin based on susceptibility profiles. Serum histamine levels taken during initial resuscitation in the operating room were normal. The serum tryptase level obtained at the same time was markedly elevated at >700 ng/mL (normal, <11.5 ng/mL), although this information was not available until several days later.
The patient underwent 2 additional surgeries during the same admission, including the prosthesis removal and tobramycin cement spacer placement, without incident. She was discharged home, again without incident. The patient was later evaluated by an outside allergist and underwent skin puncture and intradermal allergy testing. The results were consistent with a strong IgE-mediated hypersensitivity. Interestingly, she was found not to have a penicillin allergy.
Discussion
Vancomycin hypersensitivity reactions include the anaphylactoid reaction red man syndrome and a true IgE-mediated anaphylactic reaction. Red man syndrome is much more common, with reported rates in infected patients from 3.7% to 47%,4,5 when vancomycin is given at the suggested rate of 1 g over 1 hour. The reaction occurs because of histamine release from mast cells and basophils, and does not require previous sensitization.3 The rate of infusion is directly related to the development of symptoms, with 100% of patients developing symptoms in 1 study with rapid infusion (1 g over 10 min).6 Red man syndrome can typically be prevented by slowing the rate of infusion or by giving an H1 blocker.3 Anaphylaxis is more rare but can occur.7 Anaphylaxis is mediated by vancomycin-specific IgE, which requires previous exposure, as was the case with our patient. Interestingly, the patient had received vancomycin many times without any signs of a hypersensitivity reaction. Antihistamines are not effective in treating anaphylaxis, and epinephrine is the first-line agent.3 This was clearly demonstrated in this case, as there was a significant hemodynamic response to epinephrine and a negligible response to other vasopressors, specifically norepinephrine and vasopressin.
Most hypersensitivity reactions during the course of a surgical procedure occur with induction of anesthesia, with neuromuscular blocking agents and antibiotics being the most common causes.8 In our case, antibiotics were held until after deep cultures were taken. Given the time from induction to the anaphylactic reaction, it is unlikely the reaction resulted from the induction agents or the neuromuscular blocking agent. The possibility of a transfusion reaction was also investigated, since a unit of pRBC was still being transfused when symptoms began. An acute hemolytic transfusion reaction has the classic triad of fever, flank pain, and hemoglobinuria, and can also present as DIC.9 Under anesthesia, DIC can often be the presenting sign. In this case, a hemolytic transfusion reaction appeared very unlikely. All of the blood components the patient received were rechecked and found to be compatible, posttransfusion analysis showed no evidence of hemolysis in any sample, and the direct antiglobulin test was negative in all components.
To our knowledge, there are no reported cases of vancomycin-induced anaphylaxis with concomitant DIC. Symptoms of anaphylaxis after exposure to a possible antigen include rapid onset of hypotension or rapid onset of signs in at least 2 organ systems, including cutaneous, gastrointestinal, respiratory, and cardiovascular.10 Anaphylaxis with DIC is rare after exposure to any substance but has been reported.11 In fact, induction of systemic anaphylaxis in mice is known to cause DIC, with platelet-activating factor suggested as an important common mediator. A similar mechanism is suspected in humans.12
Confirmation of, and, certainly, prediction of, a vancomycin hypersensitivity reaction is difficult. Histamine levels can be used as a measure of mast-cell degranulation, but serum levels peak within 5 minutes and quickly return to baseline, limiting its diagnostic usefulness.3 Tryptase is an enzyme found in the secretory granules of mast cells. It has become an accepted marker of acute anaphylaxis, and, in vancomycin hypersensitivity reactions, can also distinguish between anaphylactic and anaphylactoid reactions.13 Tryptase levels peak 1 to 2 hours after the reaction, making this easier to measure than histamine, but results may not be available for several days, making it useful only in retrospect, as in our case. Skin testing is probably the best way to confirm a hypersensitivity reaction, although even this has been questioned with vancomycin because some find a high false-positive rate3, while others think the false-negative rate is likely too high.7 In this case, we were able to confirm our initial clinical suspicion with both an elevated tryptase level and a positive skin test.
Conclusion
We present a rare case of vancomycin anaphylaxis with DIC after repeated and prolonged previous exposure, which was treated acutely with hemodynamic resuscitation, replacement of blood components, steroids, and, most importantly, repeated boluses of epinephrine. Although several papers have described successful vancomycin desensitization7, this was fortunately not necessary in this case because the causative organism was sensitive to other acceptable antibiotics. The patient has been treated with systemic daptomycin and a tobramycin cement spacer without further incident.
Vancomycin is a glycopeptide antibiotic that exhibits bactericidal activity against gram-positive cocci. It is commonly recommended for surgical prophylaxis in cases of suspected bacterial resistance or penicillin allergy.1 Two main types of hypersensitivity reactions associated with vancomycin can have similar presentations. Red man syndrome is an anaphylactoid reaction caused by direct release of histamine from mast cells via a nonimmunologic mechanism, and is the more common of the 2 reactions. The second type is an anaphylactic reaction, which is an immunoglobulin E (IgE)–mediated systemic event and requires exposure to become sensitized.2,3
We present a patient who had received vancomycin on at least 12 occasions without incident. On this occasion, however, she developed a true anaphylactic reaction causing acute hemodynamic collapse that she survived after extensive resuscitation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 55-year-old woman had a history of metastatic giant cell tumor of the right proximal tibia. She was originally treated 27 years ago for proximal tibial resection and reconstruction with a custom proximal tibial prosthesis. Four months later, she underwent resection of multiple pulmonary metastases via bilateral thoracotomies in a single surgical setting. After this, the patient had no evidence of recurrent metastatic disease. In subsequent years, the patient underwent multiple revision surgeries for problems such as hardware failure, patellar maltracking, and infection. The patient underwent 19 operations, including several nonorthopedic procedures. Because the patient had a rash after receiving penicillin as a child, she was thought to be allergic to penicillin. Consequently, she received vancomycin as antibiotic prophylaxis for the majority of these procedures. She also received extended courses of vancomycin of at least 6 weeks on 2 separate occasions. During her most recent revision procedure, 6 weeks prior to the procedure under discussion, the patient took vancomycin without incident. She was then found to have a prosthetic infection with Staphylococcus epidermidis, the same organism isolated in her previous infections, and she was advised to undergo a staged revision.
After a preoperative medical evaluation by her primary care physician, the patient was taken to the operating room for prosthesis removal and antibiotic spacer placement. She was anemic with a hemoglobin level of 8.8 g/dL; her erythrocyte sedimentation rate (ESR) was 102 mm/h (normal, <22 mm/h) and her C-reactive protein (CRP) was 38 mg/L (normal, <3 mg/L), but, otherwise, her laboratory values were normal, including a white blood cell count (WBC) of 8100/µL. Her electrocardiogram showed a normal sinus rhythm with nonspecific ST- and T-wave changes. Antibiotics were held until after cultures were taken. General endotracheal tube anesthesia was induced with 2 mg midazolam, 100 µg fentanyl, 180 mg propofol, and 140 mg succinylcholine, followed by 10 mg vecuronium, and maintained with desflurane. A tourniquet was not used per the surgeon’s routine. Dissection was carried down to the prosthesis and showed a small amount of purulent fluid. Transfusion of 1 unit of packed red blood cells (pRBC) was started during the approach owing to relatively low preoperative hemoglobin and significant blood loss. Approximately 500 mL of blood was lost during the approach secondary to the extensive dissection and the local inflammatory response from infection and recent surgery. After cultures were taken, and approximately 10 minutes after blood transfusion began, infusion of 1 g vancomycin in 250 mL normal saline was started via an infusion pump to run over 1 hour.
After infusion of 5 mL vancomycin, the patient’s blood pressure dropped from 117/63 mm Hg to 63/30 mm Hg; her pulse concurrently dropped from 90 to 50 beats/min. Vancomycin infusion was immediately stopped, anesthesia gasses were turned off, and patient received a bolus of normal saline with a second unit of pRBC. Patient received boluses of 0.5 mg to 1.0 mg epinephrine and 100 µg phenylephrine without sustained increase in blood pressure, which had dropped to 54/24 mm Hg, although the patient became tachycardic to ~120 beats/min after epinephrine. A sudden drop in end-tidal CO2 from 40s mm Hg to 20s mm Hg was also noted, indicating continuous but significantly decreased perfusion of the lungs.
We elected to abort the procedure, and a vacuum-assisted closure (VAC) dressing was applied to the open wound. After 15 minutes, the patient’s pulses, which had been faint, became impalpable, and cardiopulmonary resuscitation was initiated for about 7 minutes. The patient received 40 units vasopressin with repeated boluses of 0.5 mg epinephrine; a norepinephrine continuous infusion was started with the return of pulses. The patient also received 50 mg diphenhydramine, 125 mg methylprednisolone, and 20 mg famotidine for suspected anaphylaxis. A central venous line and arterial line were placed, and blood was drawn for laboratory analysis. The patient was noted to have clear breath sounds with no obvious rash, and her urine remained clear. Blood gas showed a profound metabolic acidosis, with pH of 7.09, base deficit of 5.9, and lactate of 8.9. The patient was treated with bicarbonate infusion. The patient was noted to ooze significantly during central venous line and arterial line placement, despite apparently normal coagulation during the surgical approach. Coagulation values were consistent with disseminated intravascular coagulation (DIC): prothrombin time, 57 s (international normalized ratio, 6.7); partial thromboplastin time, >200 s; thrombin time, 110 s; D-dimer, >10,000 ng/mL (normal, 0-200 ng/mL); and fibrinogen, <60 mg/dL (normal, 222-475 mg/dL). The patient’s thromboelastogram showed a flat line indicating an absence of clotting. Interestingly, the platelet count remained near the preoperative level at 338×103/µL. The patient’s blood pressure remained labile and was responsive primarily to epinephrine boluses, of which she received a total of 5 mg. After 1 hour of resuscitation, during which time the patient received a total of 5 L crystalloid and 3 units pRBC, the patient was transferred to the intensive care unit (ICU), intubated, and started on a titrated epinephrine infusion.
Upon arrival in the ICU, the patient quickly stabilized hemodynamically. She was weaned from all inotropic support within 2 hours of arrival. The patient lost 800 mL of blood through wound VAC over the first 12 hours postoperatively and required a total of 11 units of pRBC, 6 units fresh frozen plasma, and 3 units of pooled cryoprecipitate, all of which were compatible. Laboratory values, including arterial pH, lactic acid, and coagulation studies, normalized on the evening of surgery, and, by the next morning, the patient was alert and was extubated without difficulty. Steroids were tapered without hemodynamic compromise while the patient was in the ICU. Cardiology examination revealed no abnormalities. Because of the temporal association of blood transfusion with cardiovascular collapse, pRBC units were retested for antibodies and cultured. Both of these investigations were negative. Wound cultures again were positive for Staphylococcus epidermidis, and blood cultures were negative. The patient was started on daptomycin based on susceptibility profiles. Serum histamine levels taken during initial resuscitation in the operating room were normal. The serum tryptase level obtained at the same time was markedly elevated at >700 ng/mL (normal, <11.5 ng/mL), although this information was not available until several days later.
The patient underwent 2 additional surgeries during the same admission, including the prosthesis removal and tobramycin cement spacer placement, without incident. She was discharged home, again without incident. The patient was later evaluated by an outside allergist and underwent skin puncture and intradermal allergy testing. The results were consistent with a strong IgE-mediated hypersensitivity. Interestingly, she was found not to have a penicillin allergy.
Discussion
Vancomycin hypersensitivity reactions include the anaphylactoid reaction red man syndrome and a true IgE-mediated anaphylactic reaction. Red man syndrome is much more common, with reported rates in infected patients from 3.7% to 47%,4,5 when vancomycin is given at the suggested rate of 1 g over 1 hour. The reaction occurs because of histamine release from mast cells and basophils, and does not require previous sensitization.3 The rate of infusion is directly related to the development of symptoms, with 100% of patients developing symptoms in 1 study with rapid infusion (1 g over 10 min).6 Red man syndrome can typically be prevented by slowing the rate of infusion or by giving an H1 blocker.3 Anaphylaxis is more rare but can occur.7 Anaphylaxis is mediated by vancomycin-specific IgE, which requires previous exposure, as was the case with our patient. Interestingly, the patient had received vancomycin many times without any signs of a hypersensitivity reaction. Antihistamines are not effective in treating anaphylaxis, and epinephrine is the first-line agent.3 This was clearly demonstrated in this case, as there was a significant hemodynamic response to epinephrine and a negligible response to other vasopressors, specifically norepinephrine and vasopressin.
Most hypersensitivity reactions during the course of a surgical procedure occur with induction of anesthesia, with neuromuscular blocking agents and antibiotics being the most common causes.8 In our case, antibiotics were held until after deep cultures were taken. Given the time from induction to the anaphylactic reaction, it is unlikely the reaction resulted from the induction agents or the neuromuscular blocking agent. The possibility of a transfusion reaction was also investigated, since a unit of pRBC was still being transfused when symptoms began. An acute hemolytic transfusion reaction has the classic triad of fever, flank pain, and hemoglobinuria, and can also present as DIC.9 Under anesthesia, DIC can often be the presenting sign. In this case, a hemolytic transfusion reaction appeared very unlikely. All of the blood components the patient received were rechecked and found to be compatible, posttransfusion analysis showed no evidence of hemolysis in any sample, and the direct antiglobulin test was negative in all components.
To our knowledge, there are no reported cases of vancomycin-induced anaphylaxis with concomitant DIC. Symptoms of anaphylaxis after exposure to a possible antigen include rapid onset of hypotension or rapid onset of signs in at least 2 organ systems, including cutaneous, gastrointestinal, respiratory, and cardiovascular.10 Anaphylaxis with DIC is rare after exposure to any substance but has been reported.11 In fact, induction of systemic anaphylaxis in mice is known to cause DIC, with platelet-activating factor suggested as an important common mediator. A similar mechanism is suspected in humans.12
Confirmation of, and, certainly, prediction of, a vancomycin hypersensitivity reaction is difficult. Histamine levels can be used as a measure of mast-cell degranulation, but serum levels peak within 5 minutes and quickly return to baseline, limiting its diagnostic usefulness.3 Tryptase is an enzyme found in the secretory granules of mast cells. It has become an accepted marker of acute anaphylaxis, and, in vancomycin hypersensitivity reactions, can also distinguish between anaphylactic and anaphylactoid reactions.13 Tryptase levels peak 1 to 2 hours after the reaction, making this easier to measure than histamine, but results may not be available for several days, making it useful only in retrospect, as in our case. Skin testing is probably the best way to confirm a hypersensitivity reaction, although even this has been questioned with vancomycin because some find a high false-positive rate3, while others think the false-negative rate is likely too high.7 In this case, we were able to confirm our initial clinical suspicion with both an elevated tryptase level and a positive skin test.
Conclusion
We present a rare case of vancomycin anaphylaxis with DIC after repeated and prolonged previous exposure, which was treated acutely with hemodynamic resuscitation, replacement of blood components, steroids, and, most importantly, repeated boluses of epinephrine. Although several papers have described successful vancomycin desensitization7, this was fortunately not necessary in this case because the causative organism was sensitive to other acceptable antibiotics. The patient has been treated with systemic daptomycin and a tobramycin cement spacer without further incident.
1. Recommendation for the use of intravenous antibiotic prophylaxis in primary total joint arthroplasty. AAOS Information Statement 1027. American Academy of Orthopaedic Surgeons website. http://www.aaos.org/about/papers/advistmt/1027.asp. Published June 2004. Accessed October 28, 2015.
2. Duffy BL. Vancomycin reaction during spinal anesthesia. Anaesth Intensive Case. 2002;30(3):364-366.
3. Wazny LD, Daghigh B. Desensitization protocols for vancomycin hypersensitivity. Ann Pharmacother. 2001;35(11):1458-1464.
4. O’Sullivan TL, Ruffing MJ, Lamp KC, Warbasse LH, Rybak MJ. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773-776.
5. Wallace MR, Mascola JR, Oldfield EC 3rd. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180-1185.
6. Renz CL, Thurn JD, Finn HA, Lynch JP, Moss J. Antihistamine prophylaxis permits rapid vancomycin infusion. Crit Care Med. 1999;27(9):1732-1737.
7. Kupstaite R, Baranauskaite A, Pileckyte M, Sveikata A, Kadusevicius E, Muckiene G. Severe vancomycin-induced anaphylactic reaction. Medicina (Kaunas). 2010;46(1):30-33.
8. Lobera T, Audicana MT, Pozo MD, et al. Study of hypersensitivity reactions and anaphylaxis during anesthesia in Spain. J Investig Allergol Clin Immunol. 2008;18(5):350-356.
9. Berséus O, Boman K, Nessen SC, Westerberg LA. Risks of hemolysis due to anti-A and anti-B caused by the transfusion of blood or blood components containing ABO-incompatible plasma. Transfusion. 2013;53(suppl 1):114S-123S.
10. Schwartz LB. Systemic anaphylaxis, food allergy, and insect sting allergy. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier; 2011:1633-1638.
11. Jung JW, Jeon EJ, Kim JW, et al. A fatal case of intravascular coagulation after bee sting acupuncture. Allergy Asthma Immunol Res. 2012;4(2):107-109.
12. Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100(3):390-394.
13. Renz CL, Laroche D, Thurn JD, et al. Tryptase levels are not increased during vancomycin-induced anaphylactoid reactions. Anesthesiology. 1998;89(3):620-625.
1. Recommendation for the use of intravenous antibiotic prophylaxis in primary total joint arthroplasty. AAOS Information Statement 1027. American Academy of Orthopaedic Surgeons website. http://www.aaos.org/about/papers/advistmt/1027.asp. Published June 2004. Accessed October 28, 2015.
2. Duffy BL. Vancomycin reaction during spinal anesthesia. Anaesth Intensive Case. 2002;30(3):364-366.
3. Wazny LD, Daghigh B. Desensitization protocols for vancomycin hypersensitivity. Ann Pharmacother. 2001;35(11):1458-1464.
4. O’Sullivan TL, Ruffing MJ, Lamp KC, Warbasse LH, Rybak MJ. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773-776.
5. Wallace MR, Mascola JR, Oldfield EC 3rd. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180-1185.
6. Renz CL, Thurn JD, Finn HA, Lynch JP, Moss J. Antihistamine prophylaxis permits rapid vancomycin infusion. Crit Care Med. 1999;27(9):1732-1737.
7. Kupstaite R, Baranauskaite A, Pileckyte M, Sveikata A, Kadusevicius E, Muckiene G. Severe vancomycin-induced anaphylactic reaction. Medicina (Kaunas). 2010;46(1):30-33.
8. Lobera T, Audicana MT, Pozo MD, et al. Study of hypersensitivity reactions and anaphylaxis during anesthesia in Spain. J Investig Allergol Clin Immunol. 2008;18(5):350-356.
9. Berséus O, Boman K, Nessen SC, Westerberg LA. Risks of hemolysis due to anti-A and anti-B caused by the transfusion of blood or blood components containing ABO-incompatible plasma. Transfusion. 2013;53(suppl 1):114S-123S.
10. Schwartz LB. Systemic anaphylaxis, food allergy, and insect sting allergy. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier; 2011:1633-1638.
11. Jung JW, Jeon EJ, Kim JW, et al. A fatal case of intravascular coagulation after bee sting acupuncture. Allergy Asthma Immunol Res. 2012;4(2):107-109.
12. Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100(3):390-394.
13. Renz CL, Laroche D, Thurn JD, et al. Tryptase levels are not increased during vancomycin-induced anaphylactoid reactions. Anesthesiology. 1998;89(3):620-625.
Treatment delay linked with worse outcome for head and neck cancer
An analysis of more than 50,000 patients with head and neck squamous cell carcinoma (HNSCC) found that prolonged time to treatment initiation (TTI) was an independent predictor of worse mortality.
Median overall survival for TTI of 67 days or fewer was 71 months compared with 49 months for TTI less than 67 days (P less than .001). For TTI of 46 to 52 days, median overall survival (OS) was 72 months; for TTI of 53 to 67 days, 61 months, and for TTI of greater than 67 days, 47 months (P less than .001).
The results provide strong evidence that “TTI greater than 67 days is too long and should be considered unacceptable,” wrote Dr. Colin Murphy, a radiation oncologist at Fox Chase Cancer Center, Philadelphia, and his colleagues.
“The current analysis suggests that increasing TTI beyond the threshold established in this monograph alters HNSCC survival and represents a public health issue,” the researchers stated (J Clin Onc. 2015 Dec 2. doi: 10.1200/JCO.2015.5906).
Data from the National Cancer Data Base pertained to 51,655 patients with head and neck squamous cell carcinoma, including oral tongue, oropharynx, larynx, and hypopharynx, during 1998-2011; median follow-up time was 84 months.
Academic institutions had significantly higher median TTI (28 days), compared with community programs (22-23 days), probably because of patients transitioning care, which was an independently associated factor in higher TTI. Despite higher median TTI, academic institutions were associated with improved overall survival, compared with community hospitals, as were care transitions.
Despite rapid tumor proliferation in HNSCC that can result in stage progression, 9.6% of all patients in 2011 had TTI of greater than 67 days, and 25% (29% at academic institutions) had TTI of greater than 46 days, another benchmark level identified in the study.
Mortality risk, according to TTI, was greater for patients with stage I or II disease, compared with stage III or IV disease, a finding that may be because of lymph node involvement. Development of nodal disease at stage III is a significant risk factor for mortality.
The investigators note that health systems elsewhere, in Denmark for example, have addressed the problem of prolonged TTI. Such efforts require coordination among providers and mandate expedited appointments for a patients with a new cancer diagnosis.
“Recently piloted programs offering next-day appointments with cancer specialists address this reversible predictor of mortality and may partially alleviate increasing TTI. Without such reforms, it is conceivable that outcomes will continue to worsen because of prolonged TTI,” they wrote.
An analysis of more than 50,000 patients with head and neck squamous cell carcinoma (HNSCC) found that prolonged time to treatment initiation (TTI) was an independent predictor of worse mortality.
Median overall survival for TTI of 67 days or fewer was 71 months compared with 49 months for TTI less than 67 days (P less than .001). For TTI of 46 to 52 days, median overall survival (OS) was 72 months; for TTI of 53 to 67 days, 61 months, and for TTI of greater than 67 days, 47 months (P less than .001).
The results provide strong evidence that “TTI greater than 67 days is too long and should be considered unacceptable,” wrote Dr. Colin Murphy, a radiation oncologist at Fox Chase Cancer Center, Philadelphia, and his colleagues.
“The current analysis suggests that increasing TTI beyond the threshold established in this monograph alters HNSCC survival and represents a public health issue,” the researchers stated (J Clin Onc. 2015 Dec 2. doi: 10.1200/JCO.2015.5906).
Data from the National Cancer Data Base pertained to 51,655 patients with head and neck squamous cell carcinoma, including oral tongue, oropharynx, larynx, and hypopharynx, during 1998-2011; median follow-up time was 84 months.
Academic institutions had significantly higher median TTI (28 days), compared with community programs (22-23 days), probably because of patients transitioning care, which was an independently associated factor in higher TTI. Despite higher median TTI, academic institutions were associated with improved overall survival, compared with community hospitals, as were care transitions.
Despite rapid tumor proliferation in HNSCC that can result in stage progression, 9.6% of all patients in 2011 had TTI of greater than 67 days, and 25% (29% at academic institutions) had TTI of greater than 46 days, another benchmark level identified in the study.
Mortality risk, according to TTI, was greater for patients with stage I or II disease, compared with stage III or IV disease, a finding that may be because of lymph node involvement. Development of nodal disease at stage III is a significant risk factor for mortality.
The investigators note that health systems elsewhere, in Denmark for example, have addressed the problem of prolonged TTI. Such efforts require coordination among providers and mandate expedited appointments for a patients with a new cancer diagnosis.
“Recently piloted programs offering next-day appointments with cancer specialists address this reversible predictor of mortality and may partially alleviate increasing TTI. Without such reforms, it is conceivable that outcomes will continue to worsen because of prolonged TTI,” they wrote.
An analysis of more than 50,000 patients with head and neck squamous cell carcinoma (HNSCC) found that prolonged time to treatment initiation (TTI) was an independent predictor of worse mortality.
Median overall survival for TTI of 67 days or fewer was 71 months compared with 49 months for TTI less than 67 days (P less than .001). For TTI of 46 to 52 days, median overall survival (OS) was 72 months; for TTI of 53 to 67 days, 61 months, and for TTI of greater than 67 days, 47 months (P less than .001).
The results provide strong evidence that “TTI greater than 67 days is too long and should be considered unacceptable,” wrote Dr. Colin Murphy, a radiation oncologist at Fox Chase Cancer Center, Philadelphia, and his colleagues.
“The current analysis suggests that increasing TTI beyond the threshold established in this monograph alters HNSCC survival and represents a public health issue,” the researchers stated (J Clin Onc. 2015 Dec 2. doi: 10.1200/JCO.2015.5906).
Data from the National Cancer Data Base pertained to 51,655 patients with head and neck squamous cell carcinoma, including oral tongue, oropharynx, larynx, and hypopharynx, during 1998-2011; median follow-up time was 84 months.
Academic institutions had significantly higher median TTI (28 days), compared with community programs (22-23 days), probably because of patients transitioning care, which was an independently associated factor in higher TTI. Despite higher median TTI, academic institutions were associated with improved overall survival, compared with community hospitals, as were care transitions.
Despite rapid tumor proliferation in HNSCC that can result in stage progression, 9.6% of all patients in 2011 had TTI of greater than 67 days, and 25% (29% at academic institutions) had TTI of greater than 46 days, another benchmark level identified in the study.
Mortality risk, according to TTI, was greater for patients with stage I or II disease, compared with stage III or IV disease, a finding that may be because of lymph node involvement. Development of nodal disease at stage III is a significant risk factor for mortality.
The investigators note that health systems elsewhere, in Denmark for example, have addressed the problem of prolonged TTI. Such efforts require coordination among providers and mandate expedited appointments for a patients with a new cancer diagnosis.
“Recently piloted programs offering next-day appointments with cancer specialists address this reversible predictor of mortality and may partially alleviate increasing TTI. Without such reforms, it is conceivable that outcomes will continue to worsen because of prolonged TTI,” they wrote.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Patients with time to treatment initiation (TTI) greater than 46-52 days had increased risk of mortality, with greatest risk increases for early-stage disease.
Major finding: Median overall survival was 72 months for TTI of 46-52 days, 61 months for 53-67 days, and 47 months for greater than 67 days (P less than .001).
Data source: Data from the National Cancer Data Base pertained to 51,655 patients with had and neck squamous cell carcinoma, including oral tongue, oropharynx, larynx, and hypopharynx, from 1998 to 2011; median follow-up time was 84 months.
Disclosures: Dr. Murphy reported having no disclosures. Several of his coauthors reported ties to industry.