User login
Study eyes mortality among octogenarians after emergency Hartmann’s procedure
LOS ANGELES – Patients over the age of 80 who present with diverticulitis requiring an emergent Hartmann’s procedure have a 30-day mortality rate of 20%, results from a study of national data demonstrated.
“Given the high morbidity and mortality described in this study, further work to elucidate whether an elective surgical therapy should be pursued in the octogenarian population is warranted,” lead study author Dr. Ian C. Bostock said in an interview in advance of the annual meeting of the American Society of Colon and Rectal Surgeons.

In an effort to investigate the 30-day outcomes for patients undergoing emergent Hartmann’s procedures for diverticular disease, Dr. Bostock of the department of general surgery at Dartmouth Hitchcock Medical Center, Lebanon, N.H., and his associates queried the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) database from 2005 to 2013 to identify all patients aged 80 years or older who underwent an open and laparoscopic Hartmann’s procedure in an emergency setting for diverticular disease. They divided patients into two groups: those with 30-day postoperative mortality (expired) and those alive after 30 days (alive), and used univariate analysis to assess the risk of mortality and to identify associated risk factors.
Of the 464 patients who met inclusion criteria, 91 expired within 30 days postoperatively, for a mortality rate of 20%. No statistically significant differences were observed between the expired and alive groups in terms of age, gender distribution, body mass index, smoking status, alcohol use, prior chemotherapy/radiotherapy, comorbid conditions such as diabetes, hypertension, current hemodialysis use, and operative time. Factors identified to be associated with a higher risk for death were congestive heart failure (odds ratio, 3.0), steroid use (OR, 3.0), chronic obstructive pulmonary disease (OR, 2.1), and ASA classification of greater than 3 (OR, 2.9). Additionally, the development of postoperative cardiac arrest (OR, 22.9), MI (OR, 8.7), renal failure (OR, 6.3), respiratory failure (OR, 4.7), and septic shock (OR, 5.6) were associated with death. A laparoscopic procedure was shown to have a protective effect (0.169).
“Interestingly, the most common complication in both groups was respiratory failure,” Dr. Bostock said. “These results suggest that the elderly are more prone to respiratory complications as a whole. These results have been corroborated in prior studies in patients exposed to major abdominal operations.”
Dr. Bostock acknowledged certain limitations of the study, including the fact that ACS-NSQIP is unable to track procedure-specific complications that might occur after surgery. “It mainly helps us to determine the morbidity rate after specific types of procedures,” he said. “Additionally, the exact indication for emergent operation in the patients included in our analysis is unknown since we don’t have any access to specific patient data and/or chart review.”
The researchers reported having no financial disclosures.
LOS ANGELES – Patients over the age of 80 who present with diverticulitis requiring an emergent Hartmann’s procedure have a 30-day mortality rate of 20%, results from a study of national data demonstrated.
“Given the high morbidity and mortality described in this study, further work to elucidate whether an elective surgical therapy should be pursued in the octogenarian population is warranted,” lead study author Dr. Ian C. Bostock said in an interview in advance of the annual meeting of the American Society of Colon and Rectal Surgeons.
In an effort to investigate the 30-day outcomes for patients undergoing emergent Hartmann’s procedures for diverticular disease, Dr. Bostock of the department of general surgery at Dartmouth Hitchcock Medical Center, Lebanon, N.H., and his associates queried the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) database from 2005 to 2013 to identify all patients aged 80 years or older who underwent an open and laparoscopic Hartmann’s procedure in an emergency setting for diverticular disease. They divided patients into two groups: those with 30-day postoperative mortality (expired) and those alive after 30 days (alive), and used univariate analysis to assess the risk of mortality and to identify associated risk factors.
Of the 464 patients who met inclusion criteria, 91 expired within 30 days postoperatively, for a mortality rate of 20%. No statistically significant differences were observed between the expired and alive groups in terms of age, gender distribution, body mass index, smoking status, alcohol use, prior chemotherapy/radiotherapy, comorbid conditions such as diabetes, hypertension, current hemodialysis use, and operative time. Factors identified to be associated with a higher risk for death were congestive heart failure (odds ratio, 3.0), steroid use (OR, 3.0), chronic obstructive pulmonary disease (OR, 2.1), and ASA classification of greater than 3 (OR, 2.9). Additionally, the development of postoperative cardiac arrest (OR, 22.9), MI (OR, 8.7), renal failure (OR, 6.3), respiratory failure (OR, 4.7), and septic shock (OR, 5.6) were associated with death. A laparoscopic procedure was shown to have a protective effect (0.169).
“Interestingly, the most common complication in both groups was respiratory failure,” Dr. Bostock said. “These results suggest that the elderly are more prone to respiratory complications as a whole. These results have been corroborated in prior studies in patients exposed to major abdominal operations.”
Dr. Bostock acknowledged certain limitations of the study, including the fact that ACS-NSQIP is unable to track procedure-specific complications that might occur after surgery. “It mainly helps us to determine the morbidity rate after specific types of procedures,” he said. “Additionally, the exact indication for emergent operation in the patients included in our analysis is unknown since we don’t have any access to specific patient data and/or chart review.”
The researchers reported having no financial disclosures.
LOS ANGELES – Patients over the age of 80 who present with diverticulitis requiring an emergent Hartmann’s procedure have a 30-day mortality rate of 20%, results from a study of national data demonstrated.
“Given the high morbidity and mortality described in this study, further work to elucidate whether an elective surgical therapy should be pursued in the octogenarian population is warranted,” lead study author Dr. Ian C. Bostock said in an interview in advance of the annual meeting of the American Society of Colon and Rectal Surgeons.
In an effort to investigate the 30-day outcomes for patients undergoing emergent Hartmann’s procedures for diverticular disease, Dr. Bostock of the department of general surgery at Dartmouth Hitchcock Medical Center, Lebanon, N.H., and his associates queried the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) database from 2005 to 2013 to identify all patients aged 80 years or older who underwent an open and laparoscopic Hartmann’s procedure in an emergency setting for diverticular disease. They divided patients into two groups: those with 30-day postoperative mortality (expired) and those alive after 30 days (alive), and used univariate analysis to assess the risk of mortality and to identify associated risk factors.
Of the 464 patients who met inclusion criteria, 91 expired within 30 days postoperatively, for a mortality rate of 20%. No statistically significant differences were observed between the expired and alive groups in terms of age, gender distribution, body mass index, smoking status, alcohol use, prior chemotherapy/radiotherapy, comorbid conditions such as diabetes, hypertension, current hemodialysis use, and operative time. Factors identified to be associated with a higher risk for death were congestive heart failure (odds ratio, 3.0), steroid use (OR, 3.0), chronic obstructive pulmonary disease (OR, 2.1), and ASA classification of greater than 3 (OR, 2.9). Additionally, the development of postoperative cardiac arrest (OR, 22.9), MI (OR, 8.7), renal failure (OR, 6.3), respiratory failure (OR, 4.7), and septic shock (OR, 5.6) were associated with death. A laparoscopic procedure was shown to have a protective effect (0.169).
“Interestingly, the most common complication in both groups was respiratory failure,” Dr. Bostock said. “These results suggest that the elderly are more prone to respiratory complications as a whole. These results have been corroborated in prior studies in patients exposed to major abdominal operations.”
Dr. Bostock acknowledged certain limitations of the study, including the fact that ACS-NSQIP is unable to track procedure-specific complications that might occur after surgery. “It mainly helps us to determine the morbidity rate after specific types of procedures,” he said. “Additionally, the exact indication for emergent operation in the patients included in our analysis is unknown since we don’t have any access to specific patient data and/or chart review.”
The researchers reported having no financial disclosures.
AT THE ASCRS ANNUAL MEETING
Key clinical point: One in five octogenarians with diverticulitis who undergo an emergency Hartmann’s procedure die within 30 days postoperatively.
Major finding: The 30-day postoperative mortality rate for octogenarians who underwent an emergency Hartmann’s procedure for diverticular disease was 20%.
Data source: An analysis of American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) data from 464 patients aged 80 and older who underwent an open and laparoscopic Hartmann’s procedure in an emergency setting for diverticular disease.
Disclosures: Dr. Bostock reported having no financial disclosures.

Aspirin lowers bile duct cancer risk
Regular aspirin use is associated with an approximately threefold reduction in risk for the three major subtypes of cholangiocarcinoma, results of a case-control study indicate.
In a study comparing patients with bile duct cancers with matched controls, aspirin use was associated with a 65% reduction in risk for intrahepatic cholangiocarcinoma (CCA), 66% reduction in risk for perihilar CCA, and 71% reduction in risk for distal CCA, reported Dr. Jonggi Choi and colleagues from the Mayo Medical School in Rochester, Minn.
“This is one of the largest hospital-based case-control studies evaluating risk factors for CCA in Western populations. We found that aspirin use had a significant inverse association with CCA development,” they wrote in Hepatology (2016. doi: 10.1002/hep.28529).
They also found that other disorders, including primary sclerosing cholangitis (PSC), non–PSC related cirrhosis, biliary tract diseases, hepatitis B infections, and diabetes, as well as smoking, were associated with varying magnitudes of risk for different CCA subtypes.
“This supports the hypothesis that the three CCA subtypes are distinct diseases and that each subtype thus has its own susceptibility to risk factors,” they wrote.
The investigators conducted a case-control study to look at various risk factors for CCA using data on all patients seen for CCA at the Mayo Clinic in Rochester from 2000 through 2014. Each case was matched by age, race, sex, and residence to two controls, chosen from among patients enrolled in the Mayo Clinic Biobank.
There were a total of 2,395 cases (1,169 with intrahepatic CCA, 995 with perihilar CCA, and 231 with distal CCA) and 4,769 controls. In all, 24.7% of cases and 44.6% of controls had used aspirin.
In multivariate logistic regression analysis controlling for demographic factors, obesity, hypertension, diabetes, stroke, coronary artery disease, peripheral vascular disease, atrial fibrillation, nonalcoholic fatty liver disease/nonalcoholic steatohepatitis, PSC, cirrhosis, irritable bowel disease, and smoking status, aspirin was significantly associated with a reduction in risk for all CCA subtypes with adjusted odds ratios (AOR) of 0.35 for intrahepatic CCA, 0.34 for perihilar cancer, and 0.29 for distal CCA (P for all less than .001).
In addition, they found that PSC was strongly associated with risk for perihilar CCA (AOR 453; P less than .001), intrahepatic CCA (AOR 93.4, P less than .001), and distal CCA (AOR 34.0, P = .002).
Cirrhosis not related to PSC was also associated with intrahepatic and perihilar CCA (AOR 13.8 and 14.1, respectively, P less than .001 for each), but not with distal CCA. Isolated inflammatory bowel disease without PSC was not associated with elevated risk of any CCA subtype.
Regular aspirin use is associated with an approximately threefold reduction in risk for the three major subtypes of cholangiocarcinoma, results of a case-control study indicate.
In a study comparing patients with bile duct cancers with matched controls, aspirin use was associated with a 65% reduction in risk for intrahepatic cholangiocarcinoma (CCA), 66% reduction in risk for perihilar CCA, and 71% reduction in risk for distal CCA, reported Dr. Jonggi Choi and colleagues from the Mayo Medical School in Rochester, Minn.
“This is one of the largest hospital-based case-control studies evaluating risk factors for CCA in Western populations. We found that aspirin use had a significant inverse association with CCA development,” they wrote in Hepatology (2016. doi: 10.1002/hep.28529).
They also found that other disorders, including primary sclerosing cholangitis (PSC), non–PSC related cirrhosis, biliary tract diseases, hepatitis B infections, and diabetes, as well as smoking, were associated with varying magnitudes of risk for different CCA subtypes.
“This supports the hypothesis that the three CCA subtypes are distinct diseases and that each subtype thus has its own susceptibility to risk factors,” they wrote.
The investigators conducted a case-control study to look at various risk factors for CCA using data on all patients seen for CCA at the Mayo Clinic in Rochester from 2000 through 2014. Each case was matched by age, race, sex, and residence to two controls, chosen from among patients enrolled in the Mayo Clinic Biobank.
There were a total of 2,395 cases (1,169 with intrahepatic CCA, 995 with perihilar CCA, and 231 with distal CCA) and 4,769 controls. In all, 24.7% of cases and 44.6% of controls had used aspirin.
In multivariate logistic regression analysis controlling for demographic factors, obesity, hypertension, diabetes, stroke, coronary artery disease, peripheral vascular disease, atrial fibrillation, nonalcoholic fatty liver disease/nonalcoholic steatohepatitis, PSC, cirrhosis, irritable bowel disease, and smoking status, aspirin was significantly associated with a reduction in risk for all CCA subtypes with adjusted odds ratios (AOR) of 0.35 for intrahepatic CCA, 0.34 for perihilar cancer, and 0.29 for distal CCA (P for all less than .001).
In addition, they found that PSC was strongly associated with risk for perihilar CCA (AOR 453; P less than .001), intrahepatic CCA (AOR 93.4, P less than .001), and distal CCA (AOR 34.0, P = .002).
Cirrhosis not related to PSC was also associated with intrahepatic and perihilar CCA (AOR 13.8 and 14.1, respectively, P less than .001 for each), but not with distal CCA. Isolated inflammatory bowel disease without PSC was not associated with elevated risk of any CCA subtype.
Regular aspirin use is associated with an approximately threefold reduction in risk for the three major subtypes of cholangiocarcinoma, results of a case-control study indicate.
In a study comparing patients with bile duct cancers with matched controls, aspirin use was associated with a 65% reduction in risk for intrahepatic cholangiocarcinoma (CCA), 66% reduction in risk for perihilar CCA, and 71% reduction in risk for distal CCA, reported Dr. Jonggi Choi and colleagues from the Mayo Medical School in Rochester, Minn.
“This is one of the largest hospital-based case-control studies evaluating risk factors for CCA in Western populations. We found that aspirin use had a significant inverse association with CCA development,” they wrote in Hepatology (2016. doi: 10.1002/hep.28529).
They also found that other disorders, including primary sclerosing cholangitis (PSC), non–PSC related cirrhosis, biliary tract diseases, hepatitis B infections, and diabetes, as well as smoking, were associated with varying magnitudes of risk for different CCA subtypes.
“This supports the hypothesis that the three CCA subtypes are distinct diseases and that each subtype thus has its own susceptibility to risk factors,” they wrote.
The investigators conducted a case-control study to look at various risk factors for CCA using data on all patients seen for CCA at the Mayo Clinic in Rochester from 2000 through 2014. Each case was matched by age, race, sex, and residence to two controls, chosen from among patients enrolled in the Mayo Clinic Biobank.
There were a total of 2,395 cases (1,169 with intrahepatic CCA, 995 with perihilar CCA, and 231 with distal CCA) and 4,769 controls. In all, 24.7% of cases and 44.6% of controls had used aspirin.
In multivariate logistic regression analysis controlling for demographic factors, obesity, hypertension, diabetes, stroke, coronary artery disease, peripheral vascular disease, atrial fibrillation, nonalcoholic fatty liver disease/nonalcoholic steatohepatitis, PSC, cirrhosis, irritable bowel disease, and smoking status, aspirin was significantly associated with a reduction in risk for all CCA subtypes with adjusted odds ratios (AOR) of 0.35 for intrahepatic CCA, 0.34 for perihilar cancer, and 0.29 for distal CCA (P for all less than .001).
In addition, they found that PSC was strongly associated with risk for perihilar CCA (AOR 453; P less than .001), intrahepatic CCA (AOR 93.4, P less than .001), and distal CCA (AOR 34.0, P = .002).
Cirrhosis not related to PSC was also associated with intrahepatic and perihilar CCA (AOR 13.8 and 14.1, respectively, P less than .001 for each), but not with distal CCA. Isolated inflammatory bowel disease without PSC was not associated with elevated risk of any CCA subtype.
FROM HEPATOLOGY
Key clinical point: Aspirin use is associated with an approximately threefold reduction in risk for the three cholangiocarcinoma (CCA) subtypes.
Major finding: Respective adjusted odds ratios for aspirin and intrahepatic, perihilar, and distal CCA were 0.35, 0.34, and 0.29.
Data source: Case-control study including 2,395 patients with CCA and 4,769 controls.
Disclosures: The study was supported by the National Institutes of Health, Mayo Clinic, Mayo Foundation, and the Cholangiocarcinoma Foundation. The authors reported no conflicts of interest.

How Genetic Epilepsy Testing Can Impact Clinical Care and Practice
PHILADELPHIA—Genetic epilepsy testing can impact clinical care and practice by providing diagnostic certainty and suggesting an approach to medical management, said Annapurna Poduri, MD, MPH, in a lecture at the 69th Annual Meeting of the American Epilepsy Society.

Annapurna Poduri, MD, MPH
With a precise genetic diagnosis, neurologists might be able to give more information about a patient’s prognosis. In addition, a genetic diagnosis can end a patient’s “diagnostic odyssey,” which can entail the burden of blood tests, lumbar punctures, and repeated imaging, said Dr. Poduri, Associate Professor of Neurology at Harvard Medical School in Boston.
“There are a small but growing number of genes associated with specific treatment recommendations,” she said. “We might be able to move toward precision medicine in epilepsy.”
New Evidence
Epilepsy genetics is “not a new topic, but it’s one where we have a lot of new evidence.” Twin studies and family studies helped establish the role of genetics in epilepsy, and in the 1990s, researchers identified several genes associated with epilepsy, including SCN1A, SCN1B, CHRNA4, and GABRB2.
When the human genome was sequenced, large association studies were not initially revealing about new causes of epilepsy. New sequencing technologies and the ability to assess copy number variation, however, have led to further advances. “In 2010, there were three papers showing us the role of copy number variation in epilepsy that had previously not had an identified etiology,” Dr. Poduri said.
Researchers found that as many as 3% of cases of genetic generalized epilepsy, formerly called idiopathic generalized epilepsy, might have deletions in the regions 15q11.2, 15q13.3, or 16p13.11. “You might say … that’s not a lot of our cases in epilepsy. But in fact, there hadn’t been this sort of robust evidence for the role of copy number variation in genetic generalized epilepsy and some of the focal epilepsies until this point,” said Dr. Poduri. “These were seminal discoveries that paved the way for further testing.”
The Epi4K Consortium and Epilepsy Phenome/Genome Project, which involved 26 institutions, undertook one effort that identified additional epilepsy-related mutations and genes. The project initially looked at individuals with infantile spasms and Lennox-Gastaut syndrome. Sequencing the exomes of 264 patients and their parents “allowed us to enlist the power of having a relatively large group of these otherwise rare disorders,” said Dr. Poduri. Among the known epilepsy genes identified by the study was SCN1A. “If we hadn’t already known … that SCN1A is an important gene for epilepsy, this is the sort of study that could tell us.” The study broadened the phenotype associated with mutations in STXBP1, which previously had been associated with Ohtahara syndrome. The investigators also identified a new epilepsy gene: DNM1. This gene did not achieve genome-wide significance in the initial study, but when researchers added about a hundred trios from a European cohort, EuroEPINOMICS, DNM1 rose to significance, said Dr. Poduri. “There were a total of five cases across these 356 trios, and a robust analysis of all of these data together through the Epi4K and EuroEPINOMICS Consortia was able to put this gene robustly on the map.”
The Effect on Practice
Such discoveries can affect clinical practice and patient management. “When you have enough data pointing to a new gene like [DNM1]—this is a synaptic gene, it makes sense, but previously was not on our radar—it means that gene testing panels are going to incorporate genes like this and be able to find more mutations like this,” said Dr. Poduri.
Certain genes are associated with specific phenotypes. The patients with de novo mutations in DNM1 had infantile spasms, and four of the five patients developed Lennox-Gastaut syndrome. “All of them had severe to profound intellectual disability and hypotonia,” said Dr. Poduri. “It gives you some sense of [the] prognosis.” Likewise, if a patient had a mutation that “uniformly was associated with a benign course, that might give us some reassurance that it’s not a progressive disorder,” she said. Findings like this might help to give physicians and patients’ families some diagnostic certainty.
Some genes suggest specific treatments to pursue or avoid. For example, in treating patients with Dravet syndrome and mutations in SCN1A, “we tend to avoid lamotrigine and phenytoin,” Dr. Poduri said. In general, neurologists avoid these sodium channel agents because they have seen patient worsening, although these recommendations are not universal, she said.
According to recent case reports, patients with SCN2A- and SCN8A- associated encephalopathies have responded to high-dose phenytoin. Mutations in SLC2A1 are associated with glucose transporter type 1 deficiency, which can be treated with the ketogenic diet. Mutations in ALDH7A1 are associated with pyridoxine deficiency, and mutations in PNPO are associated with a pyridoxal-5-phosphate deficiency. Rectifying the deficiencies may affect seizures in some cases, Dr. Poduri said.
Pierson et al reported in 2014 the case of a child with a GRIN2A mutation that changed leucine to methionine at amino acid position 812. This change increased the potency of the NMDA agonists glutamate and glycine. Investigators found that, with this mutation, memantine can block the excessive gain of function response. With adjunctive memantine therapy, the child had a dramatic reduction in seizures, although not a dramatic improvement in development, Dr. Poduri said. The child had been receiving treatment with lacosamide, rufinamide, and valproic acid. After starting memantine, lacosamide and rufinamide were tapered off.
A precise genetic diagnosis also might influence the decision between epilepsy surgery and medical treatment. For example, if a patient with infantile spasms and a mutation in SCN1A has a focal lesion, a neurologist may consider surgery, but some studies suggest that outcomes are not always favorable for these patients, Dr. Poduri said.
Need for Awareness
Neurologists should pursue updated genetic testing for their patients to take advantage of the latest genetic discoveries, Dr. Poduri said. They should learn which genes are associated with epilepsy syndromes, and bear in mind that the list of identified genes is growing.
Dr. Poduri hopes clinical practices will engage with clinical researchers and gene discovery efforts, which can lead to clinically relevant models, preclinical trials, and eventually precision medicine. “This may seem like a bit of a dream, but I think it’s really where we’re moving, and we actually have traction now in all of these areas,” she said.
—Jake Remaly
PHILADELPHIA—Genetic epilepsy testing can impact clinical care and practice by providing diagnostic certainty and suggesting an approach to medical management, said Annapurna Poduri, MD, MPH, in a lecture at the 69th Annual Meeting of the American Epilepsy Society.
Annapurna Poduri, MD, MPH
With a precise genetic diagnosis, neurologists might be able to give more information about a patient’s prognosis. In addition, a genetic diagnosis can end a patient’s “diagnostic odyssey,” which can entail the burden of blood tests, lumbar punctures, and repeated imaging, said Dr. Poduri, Associate Professor of Neurology at Harvard Medical School in Boston.
“There are a small but growing number of genes associated with specific treatment recommendations,” she said. “We might be able to move toward precision medicine in epilepsy.”
New Evidence
Epilepsy genetics is “not a new topic, but it’s one where we have a lot of new evidence.” Twin studies and family studies helped establish the role of genetics in epilepsy, and in the 1990s, researchers identified several genes associated with epilepsy, including SCN1A, SCN1B, CHRNA4, and GABRB2.
When the human genome was sequenced, large association studies were not initially revealing about new causes of epilepsy. New sequencing technologies and the ability to assess copy number variation, however, have led to further advances. “In 2010, there were three papers showing us the role of copy number variation in epilepsy that had previously not had an identified etiology,” Dr. Poduri said.
Researchers found that as many as 3% of cases of genetic generalized epilepsy, formerly called idiopathic generalized epilepsy, might have deletions in the regions 15q11.2, 15q13.3, or 16p13.11. “You might say … that’s not a lot of our cases in epilepsy. But in fact, there hadn’t been this sort of robust evidence for the role of copy number variation in genetic generalized epilepsy and some of the focal epilepsies until this point,” said Dr. Poduri. “These were seminal discoveries that paved the way for further testing.”
The Epi4K Consortium and Epilepsy Phenome/Genome Project, which involved 26 institutions, undertook one effort that identified additional epilepsy-related mutations and genes. The project initially looked at individuals with infantile spasms and Lennox-Gastaut syndrome. Sequencing the exomes of 264 patients and their parents “allowed us to enlist the power of having a relatively large group of these otherwise rare disorders,” said Dr. Poduri. Among the known epilepsy genes identified by the study was SCN1A. “If we hadn’t already known … that SCN1A is an important gene for epilepsy, this is the sort of study that could tell us.” The study broadened the phenotype associated with mutations in STXBP1, which previously had been associated with Ohtahara syndrome. The investigators also identified a new epilepsy gene: DNM1. This gene did not achieve genome-wide significance in the initial study, but when researchers added about a hundred trios from a European cohort, EuroEPINOMICS, DNM1 rose to significance, said Dr. Poduri. “There were a total of five cases across these 356 trios, and a robust analysis of all of these data together through the Epi4K and EuroEPINOMICS Consortia was able to put this gene robustly on the map.”
The Effect on Practice
Such discoveries can affect clinical practice and patient management. “When you have enough data pointing to a new gene like [DNM1]—this is a synaptic gene, it makes sense, but previously was not on our radar—it means that gene testing panels are going to incorporate genes like this and be able to find more mutations like this,” said Dr. Poduri.
Certain genes are associated with specific phenotypes. The patients with de novo mutations in DNM1 had infantile spasms, and four of the five patients developed Lennox-Gastaut syndrome. “All of them had severe to profound intellectual disability and hypotonia,” said Dr. Poduri. “It gives you some sense of [the] prognosis.” Likewise, if a patient had a mutation that “uniformly was associated with a benign course, that might give us some reassurance that it’s not a progressive disorder,” she said. Findings like this might help to give physicians and patients’ families some diagnostic certainty.
Some genes suggest specific treatments to pursue or avoid. For example, in treating patients with Dravet syndrome and mutations in SCN1A, “we tend to avoid lamotrigine and phenytoin,” Dr. Poduri said. In general, neurologists avoid these sodium channel agents because they have seen patient worsening, although these recommendations are not universal, she said.
According to recent case reports, patients with SCN2A- and SCN8A- associated encephalopathies have responded to high-dose phenytoin. Mutations in SLC2A1 are associated with glucose transporter type 1 deficiency, which can be treated with the ketogenic diet. Mutations in ALDH7A1 are associated with pyridoxine deficiency, and mutations in PNPO are associated with a pyridoxal-5-phosphate deficiency. Rectifying the deficiencies may affect seizures in some cases, Dr. Poduri said.
Pierson et al reported in 2014 the case of a child with a GRIN2A mutation that changed leucine to methionine at amino acid position 812. This change increased the potency of the NMDA agonists glutamate and glycine. Investigators found that, with this mutation, memantine can block the excessive gain of function response. With adjunctive memantine therapy, the child had a dramatic reduction in seizures, although not a dramatic improvement in development, Dr. Poduri said. The child had been receiving treatment with lacosamide, rufinamide, and valproic acid. After starting memantine, lacosamide and rufinamide were tapered off.
A precise genetic diagnosis also might influence the decision between epilepsy surgery and medical treatment. For example, if a patient with infantile spasms and a mutation in SCN1A has a focal lesion, a neurologist may consider surgery, but some studies suggest that outcomes are not always favorable for these patients, Dr. Poduri said.
Need for Awareness
Neurologists should pursue updated genetic testing for their patients to take advantage of the latest genetic discoveries, Dr. Poduri said. They should learn which genes are associated with epilepsy syndromes, and bear in mind that the list of identified genes is growing.
Dr. Poduri hopes clinical practices will engage with clinical researchers and gene discovery efforts, which can lead to clinically relevant models, preclinical trials, and eventually precision medicine. “This may seem like a bit of a dream, but I think it’s really where we’re moving, and we actually have traction now in all of these areas,” she said.
—Jake Remaly
PHILADELPHIA—Genetic epilepsy testing can impact clinical care and practice by providing diagnostic certainty and suggesting an approach to medical management, said Annapurna Poduri, MD, MPH, in a lecture at the 69th Annual Meeting of the American Epilepsy Society.
Annapurna Poduri, MD, MPH
With a precise genetic diagnosis, neurologists might be able to give more information about a patient’s prognosis. In addition, a genetic diagnosis can end a patient’s “diagnostic odyssey,” which can entail the burden of blood tests, lumbar punctures, and repeated imaging, said Dr. Poduri, Associate Professor of Neurology at Harvard Medical School in Boston.
“There are a small but growing number of genes associated with specific treatment recommendations,” she said. “We might be able to move toward precision medicine in epilepsy.”
New Evidence
Epilepsy genetics is “not a new topic, but it’s one where we have a lot of new evidence.” Twin studies and family studies helped establish the role of genetics in epilepsy, and in the 1990s, researchers identified several genes associated with epilepsy, including SCN1A, SCN1B, CHRNA4, and GABRB2.
When the human genome was sequenced, large association studies were not initially revealing about new causes of epilepsy. New sequencing technologies and the ability to assess copy number variation, however, have led to further advances. “In 2010, there were three papers showing us the role of copy number variation in epilepsy that had previously not had an identified etiology,” Dr. Poduri said.
Researchers found that as many as 3% of cases of genetic generalized epilepsy, formerly called idiopathic generalized epilepsy, might have deletions in the regions 15q11.2, 15q13.3, or 16p13.11. “You might say … that’s not a lot of our cases in epilepsy. But in fact, there hadn’t been this sort of robust evidence for the role of copy number variation in genetic generalized epilepsy and some of the focal epilepsies until this point,” said Dr. Poduri. “These were seminal discoveries that paved the way for further testing.”
The Epi4K Consortium and Epilepsy Phenome/Genome Project, which involved 26 institutions, undertook one effort that identified additional epilepsy-related mutations and genes. The project initially looked at individuals with infantile spasms and Lennox-Gastaut syndrome. Sequencing the exomes of 264 patients and their parents “allowed us to enlist the power of having a relatively large group of these otherwise rare disorders,” said Dr. Poduri. Among the known epilepsy genes identified by the study was SCN1A. “If we hadn’t already known … that SCN1A is an important gene for epilepsy, this is the sort of study that could tell us.” The study broadened the phenotype associated with mutations in STXBP1, which previously had been associated with Ohtahara syndrome. The investigators also identified a new epilepsy gene: DNM1. This gene did not achieve genome-wide significance in the initial study, but when researchers added about a hundred trios from a European cohort, EuroEPINOMICS, DNM1 rose to significance, said Dr. Poduri. “There were a total of five cases across these 356 trios, and a robust analysis of all of these data together through the Epi4K and EuroEPINOMICS Consortia was able to put this gene robustly on the map.”
The Effect on Practice
Such discoveries can affect clinical practice and patient management. “When you have enough data pointing to a new gene like [DNM1]—this is a synaptic gene, it makes sense, but previously was not on our radar—it means that gene testing panels are going to incorporate genes like this and be able to find more mutations like this,” said Dr. Poduri.
Certain genes are associated with specific phenotypes. The patients with de novo mutations in DNM1 had infantile spasms, and four of the five patients developed Lennox-Gastaut syndrome. “All of them had severe to profound intellectual disability and hypotonia,” said Dr. Poduri. “It gives you some sense of [the] prognosis.” Likewise, if a patient had a mutation that “uniformly was associated with a benign course, that might give us some reassurance that it’s not a progressive disorder,” she said. Findings like this might help to give physicians and patients’ families some diagnostic certainty.
Some genes suggest specific treatments to pursue or avoid. For example, in treating patients with Dravet syndrome and mutations in SCN1A, “we tend to avoid lamotrigine and phenytoin,” Dr. Poduri said. In general, neurologists avoid these sodium channel agents because they have seen patient worsening, although these recommendations are not universal, she said.
According to recent case reports, patients with SCN2A- and SCN8A- associated encephalopathies have responded to high-dose phenytoin. Mutations in SLC2A1 are associated with glucose transporter type 1 deficiency, which can be treated with the ketogenic diet. Mutations in ALDH7A1 are associated with pyridoxine deficiency, and mutations in PNPO are associated with a pyridoxal-5-phosphate deficiency. Rectifying the deficiencies may affect seizures in some cases, Dr. Poduri said.
Pierson et al reported in 2014 the case of a child with a GRIN2A mutation that changed leucine to methionine at amino acid position 812. This change increased the potency of the NMDA agonists glutamate and glycine. Investigators found that, with this mutation, memantine can block the excessive gain of function response. With adjunctive memantine therapy, the child had a dramatic reduction in seizures, although not a dramatic improvement in development, Dr. Poduri said. The child had been receiving treatment with lacosamide, rufinamide, and valproic acid. After starting memantine, lacosamide and rufinamide were tapered off.
A precise genetic diagnosis also might influence the decision between epilepsy surgery and medical treatment. For example, if a patient with infantile spasms and a mutation in SCN1A has a focal lesion, a neurologist may consider surgery, but some studies suggest that outcomes are not always favorable for these patients, Dr. Poduri said.
Need for Awareness
Neurologists should pursue updated genetic testing for their patients to take advantage of the latest genetic discoveries, Dr. Poduri said. They should learn which genes are associated with epilepsy syndromes, and bear in mind that the list of identified genes is growing.
Dr. Poduri hopes clinical practices will engage with clinical researchers and gene discovery efforts, which can lead to clinically relevant models, preclinical trials, and eventually precision medicine. “This may seem like a bit of a dream, but I think it’s really where we’re moving, and we actually have traction now in all of these areas,” she said.
—Jake Remaly

Acute heart failure mortality climbs with severity of peripheral edema
CHICAGO – Breathlessness typically results in hospital admission for patients with heart failure, but peripheral edema is what prolongs their stay, according to Dr. John G.F. Cleland.
Moreover, it’s not only hospital length of stay that climbs with increasing severity of peripheral edema on admission. So does mortality, both during the index admission and long term, Dr. Cleland reported at the annual meeting of the American College of Cardiology.

He presented a retrospective analysis of 121,229 patients admitted with a primary diagnosis of heart failure to more than 90% of the hospitals in England and Wales during 2007-2013.
“It turns out that the majority of patients we’re seeing admitted with heart failure at U.K. hospitals are not admitted because of severe breathlessness at rest; they’re admitted for increasing fluid retention. They have lots of peripheral edema, which is actually associated with bad outcome. The patients who are very breathless tend to have the better outcome because they have left heart failure. The ones who are full of peripheral edema have more renal dysfunction and anemia, and they’re more likely to have right heart failure,” said Dr. Cleland, professor of cardiology at Imperial College London.
“I’m not saying the breathless group isn’t a target, but this group with peripheral edema is a bigger target – and we’re not designing trials to address their problems,” he added.
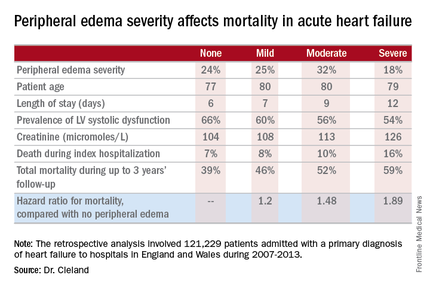
Compared with patients with no peripheral edema on hospital admission, the risk of mortality during that hospitalization and up to 3 years of subsequent follow-up was increased 1.2-fold in those with mild peripheral edema, 1.48-fold with moderate peripheral edema, and 1.89-fold in those with severe peripheral edema.
“We’re designing all the big clinical trials in acute heart failure to capture what I regard as neither fish nor fowl. They’re recruiting patients 6-12 hours after admission for acute heart failure. But by that point they’ve pretty well responded to their intravenous diuretics, and we’re just catching the tail end of their breathlessness. They’re not an emergency. The problem is really their peripheral edema, and that’s a day 2/day 3 problem. The first 6 hours of care really isn’t relevant to this group,” according to the internationally renowned heart failure researcher.
Indeed, he said he’d like to see the term “acute heart failure” laid to rest.
“If you think of acute decompensated heart failure, you think of patients wearing an oxygen mask coming in by an ambulance with blue lights flashing, and it’s an emergency. We need to move the mind-set. We shouldn’t call it acute heart failure at all, we should start talking about hospitalized heart failure, of which some is acute but much of it is subacute in people who have been deteriorating over several weeks and have just gotten to the point where they’re not coping at home anymore. They call a taxi or a friend who takes them to the hospital. Then they take a wheelchair from the taxi to the ER, but they don’t really need an ER at all,” Dr. Cleland said.
Many of these patients could be redirected to a different sort of facility at great cost savings, he added.
“In the United Kingdom and I think in the States, we’re now talking about furosemide lounges where, rather than admit the patient, you can bring them up as a day case, give them intravenous therapy, then [have them] go home at night, perhaps coming back for 3 or 4 days if needed. People are also now looking at home infusion services, and there’s a nice device for giving subcutaneous doses of furosemide as well,” the cardiologist said.
Apart from diuretics, there really aren’t any effective medications at present for peripheral edema in heart failure. But there are novel investigational agents worthy of evaluation, according to Dr. Cleland, including drugs aimed at improving mitochondrial function, agents that inhibit channels that allow edema to gather, and iron therapy.
The study was supported by the British Society for Heart Failure and the National Institute for Cardiovascular Outcomes Research. Dr. Cleland reported having no relevant financial conflicts.
CHICAGO – Breathlessness typically results in hospital admission for patients with heart failure, but peripheral edema is what prolongs their stay, according to Dr. John G.F. Cleland.
Moreover, it’s not only hospital length of stay that climbs with increasing severity of peripheral edema on admission. So does mortality, both during the index admission and long term, Dr. Cleland reported at the annual meeting of the American College of Cardiology.
He presented a retrospective analysis of 121,229 patients admitted with a primary diagnosis of heart failure to more than 90% of the hospitals in England and Wales during 2007-2013.
“It turns out that the majority of patients we’re seeing admitted with heart failure at U.K. hospitals are not admitted because of severe breathlessness at rest; they’re admitted for increasing fluid retention. They have lots of peripheral edema, which is actually associated with bad outcome. The patients who are very breathless tend to have the better outcome because they have left heart failure. The ones who are full of peripheral edema have more renal dysfunction and anemia, and they’re more likely to have right heart failure,” said Dr. Cleland, professor of cardiology at Imperial College London.
“I’m not saying the breathless group isn’t a target, but this group with peripheral edema is a bigger target – and we’re not designing trials to address their problems,” he added.
Compared with patients with no peripheral edema on hospital admission, the risk of mortality during that hospitalization and up to 3 years of subsequent follow-up was increased 1.2-fold in those with mild peripheral edema, 1.48-fold with moderate peripheral edema, and 1.89-fold in those with severe peripheral edema.
“We’re designing all the big clinical trials in acute heart failure to capture what I regard as neither fish nor fowl. They’re recruiting patients 6-12 hours after admission for acute heart failure. But by that point they’ve pretty well responded to their intravenous diuretics, and we’re just catching the tail end of their breathlessness. They’re not an emergency. The problem is really their peripheral edema, and that’s a day 2/day 3 problem. The first 6 hours of care really isn’t relevant to this group,” according to the internationally renowned heart failure researcher.
Indeed, he said he’d like to see the term “acute heart failure” laid to rest.
“If you think of acute decompensated heart failure, you think of patients wearing an oxygen mask coming in by an ambulance with blue lights flashing, and it’s an emergency. We need to move the mind-set. We shouldn’t call it acute heart failure at all, we should start talking about hospitalized heart failure, of which some is acute but much of it is subacute in people who have been deteriorating over several weeks and have just gotten to the point where they’re not coping at home anymore. They call a taxi or a friend who takes them to the hospital. Then they take a wheelchair from the taxi to the ER, but they don’t really need an ER at all,” Dr. Cleland said.
Many of these patients could be redirected to a different sort of facility at great cost savings, he added.
“In the United Kingdom and I think in the States, we’re now talking about furosemide lounges where, rather than admit the patient, you can bring them up as a day case, give them intravenous therapy, then [have them] go home at night, perhaps coming back for 3 or 4 days if needed. People are also now looking at home infusion services, and there’s a nice device for giving subcutaneous doses of furosemide as well,” the cardiologist said.
Apart from diuretics, there really aren’t any effective medications at present for peripheral edema in heart failure. But there are novel investigational agents worthy of evaluation, according to Dr. Cleland, including drugs aimed at improving mitochondrial function, agents that inhibit channels that allow edema to gather, and iron therapy.
The study was supported by the British Society for Heart Failure and the National Institute for Cardiovascular Outcomes Research. Dr. Cleland reported having no relevant financial conflicts.
CHICAGO – Breathlessness typically results in hospital admission for patients with heart failure, but peripheral edema is what prolongs their stay, according to Dr. John G.F. Cleland.
Moreover, it’s not only hospital length of stay that climbs with increasing severity of peripheral edema on admission. So does mortality, both during the index admission and long term, Dr. Cleland reported at the annual meeting of the American College of Cardiology.
He presented a retrospective analysis of 121,229 patients admitted with a primary diagnosis of heart failure to more than 90% of the hospitals in England and Wales during 2007-2013.
“It turns out that the majority of patients we’re seeing admitted with heart failure at U.K. hospitals are not admitted because of severe breathlessness at rest; they’re admitted for increasing fluid retention. They have lots of peripheral edema, which is actually associated with bad outcome. The patients who are very breathless tend to have the better outcome because they have left heart failure. The ones who are full of peripheral edema have more renal dysfunction and anemia, and they’re more likely to have right heart failure,” said Dr. Cleland, professor of cardiology at Imperial College London.
“I’m not saying the breathless group isn’t a target, but this group with peripheral edema is a bigger target – and we’re not designing trials to address their problems,” he added.
Compared with patients with no peripheral edema on hospital admission, the risk of mortality during that hospitalization and up to 3 years of subsequent follow-up was increased 1.2-fold in those with mild peripheral edema, 1.48-fold with moderate peripheral edema, and 1.89-fold in those with severe peripheral edema.
“We’re designing all the big clinical trials in acute heart failure to capture what I regard as neither fish nor fowl. They’re recruiting patients 6-12 hours after admission for acute heart failure. But by that point they’ve pretty well responded to their intravenous diuretics, and we’re just catching the tail end of their breathlessness. They’re not an emergency. The problem is really their peripheral edema, and that’s a day 2/day 3 problem. The first 6 hours of care really isn’t relevant to this group,” according to the internationally renowned heart failure researcher.
Indeed, he said he’d like to see the term “acute heart failure” laid to rest.
“If you think of acute decompensated heart failure, you think of patients wearing an oxygen mask coming in by an ambulance with blue lights flashing, and it’s an emergency. We need to move the mind-set. We shouldn’t call it acute heart failure at all, we should start talking about hospitalized heart failure, of which some is acute but much of it is subacute in people who have been deteriorating over several weeks and have just gotten to the point where they’re not coping at home anymore. They call a taxi or a friend who takes them to the hospital. Then they take a wheelchair from the taxi to the ER, but they don’t really need an ER at all,” Dr. Cleland said.
Many of these patients could be redirected to a different sort of facility at great cost savings, he added.
“In the United Kingdom and I think in the States, we’re now talking about furosemide lounges where, rather than admit the patient, you can bring them up as a day case, give them intravenous therapy, then [have them] go home at night, perhaps coming back for 3 or 4 days if needed. People are also now looking at home infusion services, and there’s a nice device for giving subcutaneous doses of furosemide as well,” the cardiologist said.
Apart from diuretics, there really aren’t any effective medications at present for peripheral edema in heart failure. But there are novel investigational agents worthy of evaluation, according to Dr. Cleland, including drugs aimed at improving mitochondrial function, agents that inhibit channels that allow edema to gather, and iron therapy.
The study was supported by the British Society for Heart Failure and the National Institute for Cardiovascular Outcomes Research. Dr. Cleland reported having no relevant financial conflicts.
AT ACC 16
Key clinical point: Leg swelling warrants greater attention in patients hospitalized for acute heart failure.
Major finding: In-hospital mortality was more than twice as great in patients admitted for acute heart failure with severe peripheral edema, compared with no leg swelling.
Data source: A retrospective study of more than 121,000 patients hospitalized for acute heart failure in England and Wales.
Disclosures: The study was supported by the British Society for Heart Failure and the National Institute for Cardiovascular Outcomes Research. The presenter reported having no relevant financial conflicts.

Cosmetic Corner: Dermatologists Weigh in on Tinted Moisturizers
To improve patient care and outcomes, leading dermatologists offered their recommendations on tinted moisturizers. Consideration must be given to:
- Anthelios 50 Mineral Tinted Ultra Light Sunscreen Fluid
La Roche-Posay Laboratoire Dermatologique
“I love this product because it provides sun protection and gives your skin a bit of color.”—Gary Goldenberg, MD, New York, New York
“I recommend this moisturizer to my patients because it has broad-spectrum UV protection, antioxidants, and a nongreasy texture.”—Shari Lipner, MD, PhD, New York, New York
- Hydra Life BB Creme
Christian Dior Perfumes LLC
“This is a lightweight, tinted cream, which comes in 3 shades for different Fitzpatrick skin types. It also provides broad-spectrum UVA/UVB protection with SPF 30.”—Cherise M. Levi, DO, New York, New York
- Neutrogena Oil-Free Acne Correct & Cover Pink Grapefruit Moisturizer
Johnson & Johnson Consumer Inc.
“It is a tinted moisturizer that also helps to fight acne with salicylic acid.”—Anthony M. Rossi, MD, New York, New York
Cutis invites readers to send us their recommendations. Body scrubs, OTC acne treatments, and cleansing devices will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on tinted moisturizers. Consideration must be given to:
- Anthelios 50 Mineral Tinted Ultra Light Sunscreen Fluid
La Roche-Posay Laboratoire Dermatologique
“I love this product because it provides sun protection and gives your skin a bit of color.”—Gary Goldenberg, MD, New York, New York
“I recommend this moisturizer to my patients because it has broad-spectrum UV protection, antioxidants, and a nongreasy texture.”—Shari Lipner, MD, PhD, New York, New York
- Hydra Life BB Creme
Christian Dior Perfumes LLC
“This is a lightweight, tinted cream, which comes in 3 shades for different Fitzpatrick skin types. It also provides broad-spectrum UVA/UVB protection with SPF 30.”—Cherise M. Levi, DO, New York, New York
- Neutrogena Oil-Free Acne Correct & Cover Pink Grapefruit Moisturizer
Johnson & Johnson Consumer Inc.
“It is a tinted moisturizer that also helps to fight acne with salicylic acid.”—Anthony M. Rossi, MD, New York, New York
Cutis invites readers to send us their recommendations. Body scrubs, OTC acne treatments, and cleansing devices will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on tinted moisturizers. Consideration must be given to:
- Anthelios 50 Mineral Tinted Ultra Light Sunscreen Fluid
La Roche-Posay Laboratoire Dermatologique
“I love this product because it provides sun protection and gives your skin a bit of color.”—Gary Goldenberg, MD, New York, New York
“I recommend this moisturizer to my patients because it has broad-spectrum UV protection, antioxidants, and a nongreasy texture.”—Shari Lipner, MD, PhD, New York, New York
- Hydra Life BB Creme
Christian Dior Perfumes LLC
“This is a lightweight, tinted cream, which comes in 3 shades for different Fitzpatrick skin types. It also provides broad-spectrum UVA/UVB protection with SPF 30.”—Cherise M. Levi, DO, New York, New York
- Neutrogena Oil-Free Acne Correct & Cover Pink Grapefruit Moisturizer
Johnson & Johnson Consumer Inc.
“It is a tinted moisturizer that also helps to fight acne with salicylic acid.”—Anthony M. Rossi, MD, New York, New York
Cutis invites readers to send us their recommendations. Body scrubs, OTC acne treatments, and cleansing devices will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Evidence Builds for Mesenchymal Stem Cell Therapy in Multiple Sclerosis
NEW ORLEANS—In an uncontrolled, prospective study, repeated intrathecal administration of autologous mesenchymal bone marrow–derived stromal stem cells to treat multiple sclerosis was safe and induced accelerated beneficial effects in some patients.
Of 28 patients with either secondary progressive or relapsing-progressive multiple sclerosis who were experiencing severe clinical deterioration and failure to respond to first- and second-line immunomodulatory treatments, 25 experienced either stable or improved Expanded Disability Status Scale (EDSS) scores following autologous mesenchymal stem cell (MSC) injections. The mean score decreased from 6.76 at study entry to 6.57 at a mean follow-up of 3.6 years, Panayiota Petrou, MD, of Hadassah University Hospital in Jerusalem, and her colleagues reported at the ACTRIMS 2016 Forum.
In addition, 17 patients experienced improvements in at least one functional system of the EDSS, including 14 who experienced improved motor function, five who experienced improved speech or bulbar functions, four who experienced improved urinary function, and six who experienced improved cerebellar function. Eight patients remained stable during the entire follow-up period.
In a prior pilot trial, intrathecal administration of MSCs was shown to be safe and provided “some indications of potentially clinically meaningful beneficial effects on the progression of the disease,” said the investigators.
The current study provides further support for those findings. It included patients who experienced severe clinical deterioration of at least 0.5 points on the EDSS during the year prior to study enrollment, or who had at least one major relapse without sufficient recovery following steroid treatment. Study subjects had a mean age of 56 and mean disease duration of 15.4 years. They received at least two courses and up to 10 injections with 1 million cells/kg; most patients received two injections (eight patients) or three injections (nine patients), and the participants were followed for up to six years. No serious side effects were observed during long-term follow-up. Eight patients experienced headaches or fever in the hours and days after injection; two patients experienced symptoms of encephalopathy that resolved within a few hours. One patient experienced back pain, and one patient had neck rigidity. No long-term side effects were reported.
Immunologic follow-up showed a transient up-regulation of regulatory T cells and down-regulation of the proliferative ability of lymphocytes and of several immune activation surface markers for up to three months.
—Sharon Worcester
NEW ORLEANS—In an uncontrolled, prospective study, repeated intrathecal administration of autologous mesenchymal bone marrow–derived stromal stem cells to treat multiple sclerosis was safe and induced accelerated beneficial effects in some patients.
Of 28 patients with either secondary progressive or relapsing-progressive multiple sclerosis who were experiencing severe clinical deterioration and failure to respond to first- and second-line immunomodulatory treatments, 25 experienced either stable or improved Expanded Disability Status Scale (EDSS) scores following autologous mesenchymal stem cell (MSC) injections. The mean score decreased from 6.76 at study entry to 6.57 at a mean follow-up of 3.6 years, Panayiota Petrou, MD, of Hadassah University Hospital in Jerusalem, and her colleagues reported at the ACTRIMS 2016 Forum.
In addition, 17 patients experienced improvements in at least one functional system of the EDSS, including 14 who experienced improved motor function, five who experienced improved speech or bulbar functions, four who experienced improved urinary function, and six who experienced improved cerebellar function. Eight patients remained stable during the entire follow-up period.
In a prior pilot trial, intrathecal administration of MSCs was shown to be safe and provided “some indications of potentially clinically meaningful beneficial effects on the progression of the disease,” said the investigators.
The current study provides further support for those findings. It included patients who experienced severe clinical deterioration of at least 0.5 points on the EDSS during the year prior to study enrollment, or who had at least one major relapse without sufficient recovery following steroid treatment. Study subjects had a mean age of 56 and mean disease duration of 15.4 years. They received at least two courses and up to 10 injections with 1 million cells/kg; most patients received two injections (eight patients) or three injections (nine patients), and the participants were followed for up to six years. No serious side effects were observed during long-term follow-up. Eight patients experienced headaches or fever in the hours and days after injection; two patients experienced symptoms of encephalopathy that resolved within a few hours. One patient experienced back pain, and one patient had neck rigidity. No long-term side effects were reported.
Immunologic follow-up showed a transient up-regulation of regulatory T cells and down-regulation of the proliferative ability of lymphocytes and of several immune activation surface markers for up to three months.
—Sharon Worcester
NEW ORLEANS—In an uncontrolled, prospective study, repeated intrathecal administration of autologous mesenchymal bone marrow–derived stromal stem cells to treat multiple sclerosis was safe and induced accelerated beneficial effects in some patients.
Of 28 patients with either secondary progressive or relapsing-progressive multiple sclerosis who were experiencing severe clinical deterioration and failure to respond to first- and second-line immunomodulatory treatments, 25 experienced either stable or improved Expanded Disability Status Scale (EDSS) scores following autologous mesenchymal stem cell (MSC) injections. The mean score decreased from 6.76 at study entry to 6.57 at a mean follow-up of 3.6 years, Panayiota Petrou, MD, of Hadassah University Hospital in Jerusalem, and her colleagues reported at the ACTRIMS 2016 Forum.
In addition, 17 patients experienced improvements in at least one functional system of the EDSS, including 14 who experienced improved motor function, five who experienced improved speech or bulbar functions, four who experienced improved urinary function, and six who experienced improved cerebellar function. Eight patients remained stable during the entire follow-up period.
In a prior pilot trial, intrathecal administration of MSCs was shown to be safe and provided “some indications of potentially clinically meaningful beneficial effects on the progression of the disease,” said the investigators.
The current study provides further support for those findings. It included patients who experienced severe clinical deterioration of at least 0.5 points on the EDSS during the year prior to study enrollment, or who had at least one major relapse without sufficient recovery following steroid treatment. Study subjects had a mean age of 56 and mean disease duration of 15.4 years. They received at least two courses and up to 10 injections with 1 million cells/kg; most patients received two injections (eight patients) or three injections (nine patients), and the participants were followed for up to six years. No serious side effects were observed during long-term follow-up. Eight patients experienced headaches or fever in the hours and days after injection; two patients experienced symptoms of encephalopathy that resolved within a few hours. One patient experienced back pain, and one patient had neck rigidity. No long-term side effects were reported.
Immunologic follow-up showed a transient up-regulation of regulatory T cells and down-regulation of the proliferative ability of lymphocytes and of several immune activation surface markers for up to three months.
—Sharon Worcester
Remicade to infliximab biosimilar switches fare well in real-life practice
GLASGOW – Switching patients on the anti–tumor necrosis factor drug Remicade to a biosimilar infliximab product resulted in good efficacy and tolerability with substantial cost savings in two real-world studies from the United Kingdom.
A total of 52 (88%) of 59 patients who had switched to the biosimilar infliximab CT-P13 (Inflectra) for the treatment of various indications for which Remicade is approved remained on the biosimilar after 10 months of follow-up and experienced comparable adverse events and similar levels of efficacy before and after switching, Dr. Lucy Parker of University Hospital Southampton NHS Foundation Trust reported at the British Society for Rheumatology annual conference. A second smaller study reported at the conference also showed similar results with the same infliximab biosimilar, which is also marketed as Remsima in Europe.
CT-P13 had efficacy, immunogenicity, and pharmacokinetic and pharmacodynamic parameters comparable to Remicade in 1-year follow-up data from the phase III randomized PLANETRAstudy (Arthritis Res Ther. 2016;18:82. doi: 10.1186/s13075-016-0981-6), but whether these study findings hold in a routine practice setting remains to be determined, Dr. Parker noted.
In Dr. Parker and colleagues’ study of data from the Southampton Biological Therapies Review Service, every patient was initially “seen in clinic and had the opportunity to speak to their consultant rheumatologist about the potential switchover from the originator drug to the new version,” she explained. Patients were then contacted directly by letter to explain the potential switch and given an information leaflet on Inflectra. They were also given access to a dedicated helpline number that could be used to re-explain the switch and discuss any concerns after switching had occurred.
In May 2015, all 59 patients being treated with Remicade were gradually switched over to the biosimilar version. Patients were reviewed after 3 months and then at 6-month intervals.
“Every single patient agreed to switch,” Dr. Parker observed. No patient used the helpline service or asked to talk with the consultant rheumatologist. One delegate noted during discussion that it was impressive that all patients agreed to switch, but Dr. Parker suggested that it was probably important that people were invited to switch rather being told that they would be switched. “If anyone had refused, then they would have been kept on Remicade,” she observed.
Dr. Parker noted that the patients were taking Remicade for various rheumatologic conditions, including 29 with rheumatoid arthritis (RA), 14 with ankylosing spondylitis (AS), 14 with psoriatic arthritis (PsA), and 2 with enteropathic arthritis. The mean age was 58.9 years, 51% of patients were female, mean disease duration was 18 years, and there was a mean of 5.7 years on Remicade before the switch. Patients had been diagnosed an average of 10 years before the first use of a biologic agent, 56% were also taking methotrexate, and 17% were on another disease-modifying antirheumatic drug (DMARD).
There was no significant difference in disease activity before or after switching, with respective mean 28-joint Disease Activity Scores of 3.4 and 3.3 and Bath Ankylosing Spondylitis Disease Activity Index scores of 3.7 and 3.6.
Dr. Parker noted that two 6-month periods before switching were compared to a 6-month period after switching and there were a similarly low number of cases reporting inefficacy, which was defined as any increase in symptoms or disease activity measure. Inefficacy was seen in one patient 12 months before the switch, two patients 6 months before the switch, and three patients after the switch. All three of the latter patients were switched back to Remicade, with one being then further switched to rituximab (Rituxan).
There were four adverse events in patients switched to the biosimilar versus three and four cases in the two 6-month periods before the switch. Adverse events after switching were widespread pain or myalgia and arthralgia after two infusions in two patients with PsA, multiple subjective symptoms such as dizziness and labile blood pressure and forgetfulness in another patient with PsA who also had these symptoms before the switch, and a case of chronic osteomyelitic foot infection in a patient with RA that also predated the switch. Biologic therapy was stopped in the RA patient, one of the PsA patients switched back to Remicade, and the other two were switched to ustekinumab (Stelara).
Dr. Parker reported that switching to the biosimilar has significantly cut the cost of treatment by £197,974 (about $291,000 USD) or 41.5% in their practice.
A team from St. George’s University Hospitals NHS Foundation Trust in London reported in a poster session similar findings after switching 31 patients to Remsima (Rheumatology [Oxford]. 2016;55[suppl 1]:i125-i126). Most switches occurred in RA patients (n = 18), followed by seven with AS and five with PsA. One patient did not switch following a consultant decision after the patient in question developed septic arthritis.
Dr. Ritu Malaiya and associates found equivalent efficacy responses in the vast majority of cases. Only one patient switched back to Remicade. Dr. Malaiya said that their experience of switching was, “on the whole, positive.” Effective planning and education of patients and staff around the switch was again considered vital and “instrumental to our early success,” the team reported. “Overall, patients were keen for others to benefit from more cost-effective drugs.”
Dr. Parker and Dr. Malaiya reported having no financial disclosures. Coauthors of the studies disclosed acting as consultants or receiving research grants from manufacturers of anti-TNF therapies.
GLASGOW – Switching patients on the anti–tumor necrosis factor drug Remicade to a biosimilar infliximab product resulted in good efficacy and tolerability with substantial cost savings in two real-world studies from the United Kingdom.
A total of 52 (88%) of 59 patients who had switched to the biosimilar infliximab CT-P13 (Inflectra) for the treatment of various indications for which Remicade is approved remained on the biosimilar after 10 months of follow-up and experienced comparable adverse events and similar levels of efficacy before and after switching, Dr. Lucy Parker of University Hospital Southampton NHS Foundation Trust reported at the British Society for Rheumatology annual conference. A second smaller study reported at the conference also showed similar results with the same infliximab biosimilar, which is also marketed as Remsima in Europe.
CT-P13 had efficacy, immunogenicity, and pharmacokinetic and pharmacodynamic parameters comparable to Remicade in 1-year follow-up data from the phase III randomized PLANETRAstudy (Arthritis Res Ther. 2016;18:82. doi: 10.1186/s13075-016-0981-6), but whether these study findings hold in a routine practice setting remains to be determined, Dr. Parker noted.
In Dr. Parker and colleagues’ study of data from the Southampton Biological Therapies Review Service, every patient was initially “seen in clinic and had the opportunity to speak to their consultant rheumatologist about the potential switchover from the originator drug to the new version,” she explained. Patients were then contacted directly by letter to explain the potential switch and given an information leaflet on Inflectra. They were also given access to a dedicated helpline number that could be used to re-explain the switch and discuss any concerns after switching had occurred.
In May 2015, all 59 patients being treated with Remicade were gradually switched over to the biosimilar version. Patients were reviewed after 3 months and then at 6-month intervals.
“Every single patient agreed to switch,” Dr. Parker observed. No patient used the helpline service or asked to talk with the consultant rheumatologist. One delegate noted during discussion that it was impressive that all patients agreed to switch, but Dr. Parker suggested that it was probably important that people were invited to switch rather being told that they would be switched. “If anyone had refused, then they would have been kept on Remicade,” she observed.
Dr. Parker noted that the patients were taking Remicade for various rheumatologic conditions, including 29 with rheumatoid arthritis (RA), 14 with ankylosing spondylitis (AS), 14 with psoriatic arthritis (PsA), and 2 with enteropathic arthritis. The mean age was 58.9 years, 51% of patients were female, mean disease duration was 18 years, and there was a mean of 5.7 years on Remicade before the switch. Patients had been diagnosed an average of 10 years before the first use of a biologic agent, 56% were also taking methotrexate, and 17% were on another disease-modifying antirheumatic drug (DMARD).
There was no significant difference in disease activity before or after switching, with respective mean 28-joint Disease Activity Scores of 3.4 and 3.3 and Bath Ankylosing Spondylitis Disease Activity Index scores of 3.7 and 3.6.
Dr. Parker noted that two 6-month periods before switching were compared to a 6-month period after switching and there were a similarly low number of cases reporting inefficacy, which was defined as any increase in symptoms or disease activity measure. Inefficacy was seen in one patient 12 months before the switch, two patients 6 months before the switch, and three patients after the switch. All three of the latter patients were switched back to Remicade, with one being then further switched to rituximab (Rituxan).
There were four adverse events in patients switched to the biosimilar versus three and four cases in the two 6-month periods before the switch. Adverse events after switching were widespread pain or myalgia and arthralgia after two infusions in two patients with PsA, multiple subjective symptoms such as dizziness and labile blood pressure and forgetfulness in another patient with PsA who also had these symptoms before the switch, and a case of chronic osteomyelitic foot infection in a patient with RA that also predated the switch. Biologic therapy was stopped in the RA patient, one of the PsA patients switched back to Remicade, and the other two were switched to ustekinumab (Stelara).
Dr. Parker reported that switching to the biosimilar has significantly cut the cost of treatment by £197,974 (about $291,000 USD) or 41.5% in their practice.
A team from St. George’s University Hospitals NHS Foundation Trust in London reported in a poster session similar findings after switching 31 patients to Remsima (Rheumatology [Oxford]. 2016;55[suppl 1]:i125-i126). Most switches occurred in RA patients (n = 18), followed by seven with AS and five with PsA. One patient did not switch following a consultant decision after the patient in question developed septic arthritis.
Dr. Ritu Malaiya and associates found equivalent efficacy responses in the vast majority of cases. Only one patient switched back to Remicade. Dr. Malaiya said that their experience of switching was, “on the whole, positive.” Effective planning and education of patients and staff around the switch was again considered vital and “instrumental to our early success,” the team reported. “Overall, patients were keen for others to benefit from more cost-effective drugs.”
Dr. Parker and Dr. Malaiya reported having no financial disclosures. Coauthors of the studies disclosed acting as consultants or receiving research grants from manufacturers of anti-TNF therapies.
GLASGOW – Switching patients on the anti–tumor necrosis factor drug Remicade to a biosimilar infliximab product resulted in good efficacy and tolerability with substantial cost savings in two real-world studies from the United Kingdom.
A total of 52 (88%) of 59 patients who had switched to the biosimilar infliximab CT-P13 (Inflectra) for the treatment of various indications for which Remicade is approved remained on the biosimilar after 10 months of follow-up and experienced comparable adverse events and similar levels of efficacy before and after switching, Dr. Lucy Parker of University Hospital Southampton NHS Foundation Trust reported at the British Society for Rheumatology annual conference. A second smaller study reported at the conference also showed similar results with the same infliximab biosimilar, which is also marketed as Remsima in Europe.
CT-P13 had efficacy, immunogenicity, and pharmacokinetic and pharmacodynamic parameters comparable to Remicade in 1-year follow-up data from the phase III randomized PLANETRAstudy (Arthritis Res Ther. 2016;18:82. doi: 10.1186/s13075-016-0981-6), but whether these study findings hold in a routine practice setting remains to be determined, Dr. Parker noted.
In Dr. Parker and colleagues’ study of data from the Southampton Biological Therapies Review Service, every patient was initially “seen in clinic and had the opportunity to speak to their consultant rheumatologist about the potential switchover from the originator drug to the new version,” she explained. Patients were then contacted directly by letter to explain the potential switch and given an information leaflet on Inflectra. They were also given access to a dedicated helpline number that could be used to re-explain the switch and discuss any concerns after switching had occurred.
In May 2015, all 59 patients being treated with Remicade were gradually switched over to the biosimilar version. Patients were reviewed after 3 months and then at 6-month intervals.
“Every single patient agreed to switch,” Dr. Parker observed. No patient used the helpline service or asked to talk with the consultant rheumatologist. One delegate noted during discussion that it was impressive that all patients agreed to switch, but Dr. Parker suggested that it was probably important that people were invited to switch rather being told that they would be switched. “If anyone had refused, then they would have been kept on Remicade,” she observed.
Dr. Parker noted that the patients were taking Remicade for various rheumatologic conditions, including 29 with rheumatoid arthritis (RA), 14 with ankylosing spondylitis (AS), 14 with psoriatic arthritis (PsA), and 2 with enteropathic arthritis. The mean age was 58.9 years, 51% of patients were female, mean disease duration was 18 years, and there was a mean of 5.7 years on Remicade before the switch. Patients had been diagnosed an average of 10 years before the first use of a biologic agent, 56% were also taking methotrexate, and 17% were on another disease-modifying antirheumatic drug (DMARD).
There was no significant difference in disease activity before or after switching, with respective mean 28-joint Disease Activity Scores of 3.4 and 3.3 and Bath Ankylosing Spondylitis Disease Activity Index scores of 3.7 and 3.6.
Dr. Parker noted that two 6-month periods before switching were compared to a 6-month period after switching and there were a similarly low number of cases reporting inefficacy, which was defined as any increase in symptoms or disease activity measure. Inefficacy was seen in one patient 12 months before the switch, two patients 6 months before the switch, and three patients after the switch. All three of the latter patients were switched back to Remicade, with one being then further switched to rituximab (Rituxan).
There were four adverse events in patients switched to the biosimilar versus three and four cases in the two 6-month periods before the switch. Adverse events after switching were widespread pain or myalgia and arthralgia after two infusions in two patients with PsA, multiple subjective symptoms such as dizziness and labile blood pressure and forgetfulness in another patient with PsA who also had these symptoms before the switch, and a case of chronic osteomyelitic foot infection in a patient with RA that also predated the switch. Biologic therapy was stopped in the RA patient, one of the PsA patients switched back to Remicade, and the other two were switched to ustekinumab (Stelara).
Dr. Parker reported that switching to the biosimilar has significantly cut the cost of treatment by £197,974 (about $291,000 USD) or 41.5% in their practice.
A team from St. George’s University Hospitals NHS Foundation Trust in London reported in a poster session similar findings after switching 31 patients to Remsima (Rheumatology [Oxford]. 2016;55[suppl 1]:i125-i126). Most switches occurred in RA patients (n = 18), followed by seven with AS and five with PsA. One patient did not switch following a consultant decision after the patient in question developed septic arthritis.
Dr. Ritu Malaiya and associates found equivalent efficacy responses in the vast majority of cases. Only one patient switched back to Remicade. Dr. Malaiya said that their experience of switching was, “on the whole, positive.” Effective planning and education of patients and staff around the switch was again considered vital and “instrumental to our early success,” the team reported. “Overall, patients were keen for others to benefit from more cost-effective drugs.”
Dr. Parker and Dr. Malaiya reported having no financial disclosures. Coauthors of the studies disclosed acting as consultants or receiving research grants from manufacturers of anti-TNF therapies.
Key clinical point: Both biosimilar versions of infliximab available in Europe appear to be as effective and tolerated as Remicade after switching from it.
Major finding: Of 59 patients who had switched to Inflectra, 88% remained on the biosimilar after 10 months of follow-up.
Data source: Two real-world studies of switching to biosimilar infliximab in the United Kingdom.
Disclosures: Dr. Parker and Dr. Malaiya reported having no financial disclosures. Coauthors of the studies disclosed acting as consultants or receiving research grants from manufacturers of anti-TNF therapies.
QUIZ: Which of the Following Statements Is True Regarding Hospitalists’ Assessments of Patients’ Decision-Making Capacity?
[WpProQuiz 7]
[WpProQuiz_toplist 7]
[WpProQuiz 7]
[WpProQuiz_toplist 7]
[WpProQuiz 7]
[WpProQuiz_toplist 7]
May 2016 Quiz 2
Q2: Answer: A
Campylobacter species are a major cause of diarrheal illness in the world. The organism inhabits the intestinal tracts of a wide range of animal hosts, notably poultry; contamination from these sources can lead to food-borne disease. Given the self-limited nature of most Campylobacter infections and the limited efficacy of routine antimicrobial therapy, treatment is warranted only for patients with features of severe disease or risk for severe disease.
Patients with severe disease include individuals with bloody stools, high fever, extraintestinal infection, worsening or relapsing symptoms, or symptoms lasting longer than 1 week. Those at risk for severe disease include patients who are elderly, pregnant, or immunocompromised. First-line agents for treatment of Campylobacter infection include fluoroquinolones (if sensitive) or azithromycin. Campylobacter is inherently resistant to trimethoprim and beta-lactam antibiotics, including penicillin and most cephalosporins.
In the United States, the rate of resistance to fluoroquinolones also is increasing. The rate of ciprofloxacin resistance among Campylobacter isolated in the United States increased from 0% to 19% between 1989 and 2001. Inappropriate and overprescription of fluoroquinolones in humans, combined with increased fluoroquinolone use in the poultry industry in particular, have contributed to the increased prevalence of fluoroquinolone resistance.
The rate of macrolide resistance among Campylobacter has remained stable at less than 5% in most parts of the world.
References
- Dasti J.I., Tareen A.M., Lugert R., et al. Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int J Med Microbiol. 2010;300:205-11.
- Gupta A., Nelson J.M., Barrett T.J., et al. Antimicrobial resistance among Campylobacter strains, United States, 1997-2001. Emerging infectious diseases. Jun 2004;10:1102-9.
Q2: Answer: A
Campylobacter species are a major cause of diarrheal illness in the world. The organism inhabits the intestinal tracts of a wide range of animal hosts, notably poultry; contamination from these sources can lead to food-borne disease. Given the self-limited nature of most Campylobacter infections and the limited efficacy of routine antimicrobial therapy, treatment is warranted only for patients with features of severe disease or risk for severe disease.
Patients with severe disease include individuals with bloody stools, high fever, extraintestinal infection, worsening or relapsing symptoms, or symptoms lasting longer than 1 week. Those at risk for severe disease include patients who are elderly, pregnant, or immunocompromised. First-line agents for treatment of Campylobacter infection include fluoroquinolones (if sensitive) or azithromycin. Campylobacter is inherently resistant to trimethoprim and beta-lactam antibiotics, including penicillin and most cephalosporins.
In the United States, the rate of resistance to fluoroquinolones also is increasing. The rate of ciprofloxacin resistance among Campylobacter isolated in the United States increased from 0% to 19% between 1989 and 2001. Inappropriate and overprescription of fluoroquinolones in humans, combined with increased fluoroquinolone use in the poultry industry in particular, have contributed to the increased prevalence of fluoroquinolone resistance.
The rate of macrolide resistance among Campylobacter has remained stable at less than 5% in most parts of the world.
References
- Dasti J.I., Tareen A.M., Lugert R., et al. Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int J Med Microbiol. 2010;300:205-11.
- Gupta A., Nelson J.M., Barrett T.J., et al. Antimicrobial resistance among Campylobacter strains, United States, 1997-2001. Emerging infectious diseases. Jun 2004;10:1102-9.
Q2: Answer: A
Campylobacter species are a major cause of diarrheal illness in the world. The organism inhabits the intestinal tracts of a wide range of animal hosts, notably poultry; contamination from these sources can lead to food-borne disease. Given the self-limited nature of most Campylobacter infections and the limited efficacy of routine antimicrobial therapy, treatment is warranted only for patients with features of severe disease or risk for severe disease.
Patients with severe disease include individuals with bloody stools, high fever, extraintestinal infection, worsening or relapsing symptoms, or symptoms lasting longer than 1 week. Those at risk for severe disease include patients who are elderly, pregnant, or immunocompromised. First-line agents for treatment of Campylobacter infection include fluoroquinolones (if sensitive) or azithromycin. Campylobacter is inherently resistant to trimethoprim and beta-lactam antibiotics, including penicillin and most cephalosporins.
In the United States, the rate of resistance to fluoroquinolones also is increasing. The rate of ciprofloxacin resistance among Campylobacter isolated in the United States increased from 0% to 19% between 1989 and 2001. Inappropriate and overprescription of fluoroquinolones in humans, combined with increased fluoroquinolone use in the poultry industry in particular, have contributed to the increased prevalence of fluoroquinolone resistance.
The rate of macrolide resistance among Campylobacter has remained stable at less than 5% in most parts of the world.
References
- Dasti J.I., Tareen A.M., Lugert R., et al. Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int J Med Microbiol. 2010;300:205-11.
- Gupta A., Nelson J.M., Barrett T.J., et al. Antimicrobial resistance among Campylobacter strains, United States, 1997-2001. Emerging infectious diseases. Jun 2004;10:1102-9.
A 37-year-old man presents to the clinic with a 1-week history of diarrhea. He is a poultry farmer. His symptoms started with nausea and abdominal cramps. Subsequently, he developed diarrhea, reported as 10-12 loose stools with passage of blood. He also reported high fever. Abdominal examination revealed right lower quadrant abdominal tenderness. Stool cultures were ordered and came back positive for Campylobacter infection.
May 2016 Quiz 1
Q1: Answer: D
Rationale: The most frequent mechanism for gastroesophageal reflux is transient lower esophageal sphincter relaxation (TLESR). In the setting of a compliant esophagogastric junction, TLESRs allow movement of content from stomach to esophagus. Gastric distension following meals is a prime trigger for TLESRs, and this is why reflux is most frequent after a meal.
Further, there can be an unbuffered layer of acid floating above the ingested meal (acid pocket) in close proximity to the esophagogastric junction, which is immediately available for reflux during TLESRs. All the other mechanisms listed contribute to the pathophysiology of reflux disease. Increased intra-abdominal pressure contributes to the pressure gradient promoting reflux.
Weak lower esophageal sphincter tone and hiatus hernia are structural deficiencies at the esophagogastric junction that add to the compliance of the esophagogastric junction, making it easier for reflux to occur during a TLESR. Ineffective esophageal body motility contributes to prolonged exposure of the esophageal mucosa to refluxed content, as this can impact clearance of the refluxate, particularly in the recumbent position.
References
- Wu J.C., Mui L.M., Cheung C.M., et al. Obesity is associated with increased transient lower esophageal sphincter relaxation. Gastroenterology 2007;132:883-9.
- Mittal R.K., Lange R.C., McCallum R.W. Identification and mechanism of delayed esophageal acid clearance in subjects with hiatus hernia. Gastroenterology 1987;92:130-5.
Q1: Answer: D
Rationale: The most frequent mechanism for gastroesophageal reflux is transient lower esophageal sphincter relaxation (TLESR). In the setting of a compliant esophagogastric junction, TLESRs allow movement of content from stomach to esophagus. Gastric distension following meals is a prime trigger for TLESRs, and this is why reflux is most frequent after a meal.
Further, there can be an unbuffered layer of acid floating above the ingested meal (acid pocket) in close proximity to the esophagogastric junction, which is immediately available for reflux during TLESRs. All the other mechanisms listed contribute to the pathophysiology of reflux disease. Increased intra-abdominal pressure contributes to the pressure gradient promoting reflux.
Weak lower esophageal sphincter tone and hiatus hernia are structural deficiencies at the esophagogastric junction that add to the compliance of the esophagogastric junction, making it easier for reflux to occur during a TLESR. Ineffective esophageal body motility contributes to prolonged exposure of the esophageal mucosa to refluxed content, as this can impact clearance of the refluxate, particularly in the recumbent position.
References
- Wu J.C., Mui L.M., Cheung C.M., et al. Obesity is associated with increased transient lower esophageal sphincter relaxation. Gastroenterology 2007;132:883-9.
- Mittal R.K., Lange R.C., McCallum R.W. Identification and mechanism of delayed esophageal acid clearance in subjects with hiatus hernia. Gastroenterology 1987;92:130-5.
Q1: Answer: D
Rationale: The most frequent mechanism for gastroesophageal reflux is transient lower esophageal sphincter relaxation (TLESR). In the setting of a compliant esophagogastric junction, TLESRs allow movement of content from stomach to esophagus. Gastric distension following meals is a prime trigger for TLESRs, and this is why reflux is most frequent after a meal.
Further, there can be an unbuffered layer of acid floating above the ingested meal (acid pocket) in close proximity to the esophagogastric junction, which is immediately available for reflux during TLESRs. All the other mechanisms listed contribute to the pathophysiology of reflux disease. Increased intra-abdominal pressure contributes to the pressure gradient promoting reflux.
Weak lower esophageal sphincter tone and hiatus hernia are structural deficiencies at the esophagogastric junction that add to the compliance of the esophagogastric junction, making it easier for reflux to occur during a TLESR. Ineffective esophageal body motility contributes to prolonged exposure of the esophageal mucosa to refluxed content, as this can impact clearance of the refluxate, particularly in the recumbent position.
References
- Wu J.C., Mui L.M., Cheung C.M., et al. Obesity is associated with increased transient lower esophageal sphincter relaxation. Gastroenterology 2007;132:883-9.
- Mittal R.K., Lange R.C., McCallum R.W. Identification and mechanism of delayed esophageal acid clearance in subjects with hiatus hernia. Gastroenterology 1987;92:130-5.
A 24-year-old woman presents for continuing management of gastroesophageal reflux disease. Although her heartburn has resolved on twice-a-day omeprazole therapy, she continues to have postprandial regurgitation. She has no nocturnal symptoms. Prior evaluation has demonstrated a 3-cm sliding hiatus hernia, but no erosive esophagitis. She is overweight with a body mass index of 32 kg/m2. Physical examination is normal.