User login
New Antiepileptic Medications Expand Patients’ Options
VANCOUVER—None of the seven new antiepileptic drugs (AEDs) approved by the FDA during the past 10 years greatly increase the likelihood of seizure control, relative to previously existing therapies, according to an overview presented at the 68th Annual Meeting of the American Academy of Neurology. The broader array of choices is facilitating the individualization of therapy, however. Several of the newer agents are chemically related to previously existing AEDs, but all of the drugs have distinguishing features.
“We do have more options for the treatment of epilepsy,” confirmed Carl W. Bazil, MD, PhD, Director of the Comprehensive Epilepsy Center at Columbia University College of Physicians and Surgeons in New York City. “Although we do not yet have the perfect therapy, more are coming.”

In his evaluation of how newer agents fit into current clinical practice, Dr. Bazil indicated that relative efficacy among the newer agents has not been well defined in the absence of head-to-head trials. Treatment choice is instead based on their approved indications, side effect profiles, and half-lives.
Yet the emergence of more choices was characterized as a positive development. Since pregabalin received regulatory approval in 2005, the newer therapies have included lacosamide (2008), rufinamide (2009), clobazam (2010), ezogabine (2011), perampanel (2012), eslicarbazepine (2013), and brivaracetam (2016). Of these, Dr. Bazil focused primarily on the latter five and how they fit into routine care. Overall, these agents “have not been all that different” in terms of their associated rates of total seizure control or other meaningful end points.
This similarity does not suggest that these agents are interchangeable. The clinical profiles differ by characteristics such as dosing frequency, relative risk of somnolence and other common side effects, and the potential to ameliorate accompanying symptoms, such as anxiety. Moreover, due to differences in trial design and entry criteria, not all of the newer drugs have been granted the same indication, said Dr. Bazil.
Clobazam for Lennox-Gastaut Syndrome
Clobazam is a benzodiazepine receptor agonist that shares features with other benzodiazepines. One of the key features of this agent is a half-life that approaches 80 hours. This characteristic makes clobazam a “forgiving” drug for those who miss a dose, said Dr. Bazil.
Clobazam’s FDA-approved indication is for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome for individuals age 2 or older. Although this Schedule IV drug is not approved in the United States for partial onset seizures, it is widely used for this indication elsewhere, including in Europe and Canada. Clobazam, like other benzodiazepine-related agents, could have a favorable effect on anxiety or sleep problems, even though it is not indicated for these uses, said Dr. Bazil.
Relative to other commonly used AEDs, the rate of rash associated with clobazam has been relatively low, but dizziness and somnolence are sufficiently common that the once-daily dose should be taken before bedtime, according to Dr. Bazil. Relative to other benzodiazepines, clobazam appears to have a low potential for tolerance and dependence. Initial daily doses of 5 mg to 10 mg are standard, but lower doses should be considered in older patients or those with low body weight, said Dr. Bazil. Titration is performed on a weekly basis to a maximum recommended dose of 60 mg.
Perampanel Has a Long Half-Life
Perampanel, which is a noncompetitive AMPA-receptor inhibitor, has an even longer half-life of 100 hours. The drug’s FDA approval is for the adjunctive treatment of partial epilepsy and primary generalized tonic-clonic seizures in patients age 12 and older. Prudent initial dosing begins at 2 mg daily with a recommended maximum dose of 12 mg, according to Dr. Bazil. Due to its reliance on hepatic metabolism, a relatively slow upward titration of this Schedule III drug is appropriate in patients with impaired liver function, he added.
Dizziness and somnolence are commonly reported side effects for perampanel, as they are for most AEDs, and the drug also has a black box warning about its association with homicidal ideation, irritability and aggression. Yet the drug’s relative efficacy may compensate for these risks in selected patients. Even though researchers have not compared perampanel directly with another AED, the drug had an “impressive” seizure-free rate in a placebo-controlled trial, said Dr. Bazil. In this multicenter study of patients with drug-resistant, primary generalized tonic-clonic seizures, 30.9% of participants on perampanel, versus 12.3% of those on placebo, remained seizure-free during maintenance.
Ezogabine and Risk of Discoloration
Ezogabine, which is FDA-approved as adjunctive therapy for refractory partial epilepsy, is a K-channel opener. Due to a relatively short half-life of seven to 11 hours, thrice-daily dosing is required. In addition, bluish discoloration, primarily of the lips and fingertips, commonly occurs due to an interaction of a drug metabolite with sun exposure, according to Dr. Bazil. Concern that this drug may lead to retinal pigmentation means that eye examinations are recommended at baseline and every six months.
“Clinical use of ezogabine has been limited by the pigmentation and by tid dosing, which patients do not like,” Dr. Bazil said. Although the bluish color resolves over time, patients are likely to discontinue this Schedule V drug, which also is associated with dizziness and somnolence, before this occurs.
Eslicarbazepine, an Unscheduled Agent
Eslicarbazepine, a sodium channel blocker, has been approved as adjunctive and monotherapy for partial epilepsy. Although monotherapy trials with a placebo control are no longer considered ethical in patients with epilepsy, eslicarbazepine and lacosamide have been evaluated and found effective in trials in which other active agents were withdrawn over time to allow response on monotherapy to be monitored, Dr. Bazil explained.
Eslicarbazepine is also distinguished from other newer agents by the fact that it is not a scheduled agent. It is primarily metabolized by the liver and has a half-life of 13 to 20 hours. Due to its structural similarity to carbamazepine and oxcarbazepine, these agents should not be combined, said Dr. Bazil. Eslicarbazepine, like clobazam, also could interfere with the bioavailability of oral contraceptives.
“Eslicarbazepine is essentially a longer-acting carbamazepine, a drug with which we are all familiar,” said Dr. Bazil. Although eslicarbazepine also may cause the dizziness, somnolence, and nausea common to carbamazepine, its longer half-life has the potential to diminish peak effects to improve tolerability.
New and Emerging Therapies
Brivaracetam, which was approved for add-on therapy of refractory partial seizures for patients age 16 or older, “is structurally and functionally related to levetiracetam, but approximately 20 times more potent at the SV2A receptor,” said Dr. Bazil. As this recently approved drug is not yet widely used, its relative clinical utility within the context of other choices is not well established, but the side effects common to AEDs, such as dizziness and somnolence, have been relatively mild on this agent in the clinical trials.
Of treatment strategies on the horizon, Dr. Bazil drew attention to SAGE-547, an experimental agent now in a phase III study that has demonstrated uncommon efficacy for refractory status epilepticus, a condition for which there is a large unmet need for reliably effective treatments. In addition, researchers are conducting ongoing efforts to individualize therapy by identifying targetable underlying genetic mutations responsible for seizure activity.
Overall, the therapies approved during the past 10 years have expanded choice in a way that may increase the likelihood of providing a patient with adequate efficacy and acceptable tolerability. Although newer pharmacologic options have not yet generated a major leap in seizure control, coming therapies or strategies to better match existing therapies to the underlying seizure mechanism may yield greater rates of seizure freedom, said Dr. Bazil.
—Ted Bosworth
Suggested Reading
French JA, Krauss GL, Wechsler RT, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology. 2015;85(11):950-957.
VANCOUVER—None of the seven new antiepileptic drugs (AEDs) approved by the FDA during the past 10 years greatly increase the likelihood of seizure control, relative to previously existing therapies, according to an overview presented at the 68th Annual Meeting of the American Academy of Neurology. The broader array of choices is facilitating the individualization of therapy, however. Several of the newer agents are chemically related to previously existing AEDs, but all of the drugs have distinguishing features.
“We do have more options for the treatment of epilepsy,” confirmed Carl W. Bazil, MD, PhD, Director of the Comprehensive Epilepsy Center at Columbia University College of Physicians and Surgeons in New York City. “Although we do not yet have the perfect therapy, more are coming.”
In his evaluation of how newer agents fit into current clinical practice, Dr. Bazil indicated that relative efficacy among the newer agents has not been well defined in the absence of head-to-head trials. Treatment choice is instead based on their approved indications, side effect profiles, and half-lives.
Yet the emergence of more choices was characterized as a positive development. Since pregabalin received regulatory approval in 2005, the newer therapies have included lacosamide (2008), rufinamide (2009), clobazam (2010), ezogabine (2011), perampanel (2012), eslicarbazepine (2013), and brivaracetam (2016). Of these, Dr. Bazil focused primarily on the latter five and how they fit into routine care. Overall, these agents “have not been all that different” in terms of their associated rates of total seizure control or other meaningful end points.
This similarity does not suggest that these agents are interchangeable. The clinical profiles differ by characteristics such as dosing frequency, relative risk of somnolence and other common side effects, and the potential to ameliorate accompanying symptoms, such as anxiety. Moreover, due to differences in trial design and entry criteria, not all of the newer drugs have been granted the same indication, said Dr. Bazil.
Clobazam for Lennox-Gastaut Syndrome
Clobazam is a benzodiazepine receptor agonist that shares features with other benzodiazepines. One of the key features of this agent is a half-life that approaches 80 hours. This characteristic makes clobazam a “forgiving” drug for those who miss a dose, said Dr. Bazil.
Clobazam’s FDA-approved indication is for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome for individuals age 2 or older. Although this Schedule IV drug is not approved in the United States for partial onset seizures, it is widely used for this indication elsewhere, including in Europe and Canada. Clobazam, like other benzodiazepine-related agents, could have a favorable effect on anxiety or sleep problems, even though it is not indicated for these uses, said Dr. Bazil.
Relative to other commonly used AEDs, the rate of rash associated with clobazam has been relatively low, but dizziness and somnolence are sufficiently common that the once-daily dose should be taken before bedtime, according to Dr. Bazil. Relative to other benzodiazepines, clobazam appears to have a low potential for tolerance and dependence. Initial daily doses of 5 mg to 10 mg are standard, but lower doses should be considered in older patients or those with low body weight, said Dr. Bazil. Titration is performed on a weekly basis to a maximum recommended dose of 60 mg.
Perampanel Has a Long Half-Life
Perampanel, which is a noncompetitive AMPA-receptor inhibitor, has an even longer half-life of 100 hours. The drug’s FDA approval is for the adjunctive treatment of partial epilepsy and primary generalized tonic-clonic seizures in patients age 12 and older. Prudent initial dosing begins at 2 mg daily with a recommended maximum dose of 12 mg, according to Dr. Bazil. Due to its reliance on hepatic metabolism, a relatively slow upward titration of this Schedule III drug is appropriate in patients with impaired liver function, he added.
Dizziness and somnolence are commonly reported side effects for perampanel, as they are for most AEDs, and the drug also has a black box warning about its association with homicidal ideation, irritability and aggression. Yet the drug’s relative efficacy may compensate for these risks in selected patients. Even though researchers have not compared perampanel directly with another AED, the drug had an “impressive” seizure-free rate in a placebo-controlled trial, said Dr. Bazil. In this multicenter study of patients with drug-resistant, primary generalized tonic-clonic seizures, 30.9% of participants on perampanel, versus 12.3% of those on placebo, remained seizure-free during maintenance.
Ezogabine and Risk of Discoloration
Ezogabine, which is FDA-approved as adjunctive therapy for refractory partial epilepsy, is a K-channel opener. Due to a relatively short half-life of seven to 11 hours, thrice-daily dosing is required. In addition, bluish discoloration, primarily of the lips and fingertips, commonly occurs due to an interaction of a drug metabolite with sun exposure, according to Dr. Bazil. Concern that this drug may lead to retinal pigmentation means that eye examinations are recommended at baseline and every six months.
“Clinical use of ezogabine has been limited by the pigmentation and by tid dosing, which patients do not like,” Dr. Bazil said. Although the bluish color resolves over time, patients are likely to discontinue this Schedule V drug, which also is associated with dizziness and somnolence, before this occurs.
Eslicarbazepine, an Unscheduled Agent
Eslicarbazepine, a sodium channel blocker, has been approved as adjunctive and monotherapy for partial epilepsy. Although monotherapy trials with a placebo control are no longer considered ethical in patients with epilepsy, eslicarbazepine and lacosamide have been evaluated and found effective in trials in which other active agents were withdrawn over time to allow response on monotherapy to be monitored, Dr. Bazil explained.
Eslicarbazepine is also distinguished from other newer agents by the fact that it is not a scheduled agent. It is primarily metabolized by the liver and has a half-life of 13 to 20 hours. Due to its structural similarity to carbamazepine and oxcarbazepine, these agents should not be combined, said Dr. Bazil. Eslicarbazepine, like clobazam, also could interfere with the bioavailability of oral contraceptives.
“Eslicarbazepine is essentially a longer-acting carbamazepine, a drug with which we are all familiar,” said Dr. Bazil. Although eslicarbazepine also may cause the dizziness, somnolence, and nausea common to carbamazepine, its longer half-life has the potential to diminish peak effects to improve tolerability.
New and Emerging Therapies
Brivaracetam, which was approved for add-on therapy of refractory partial seizures for patients age 16 or older, “is structurally and functionally related to levetiracetam, but approximately 20 times more potent at the SV2A receptor,” said Dr. Bazil. As this recently approved drug is not yet widely used, its relative clinical utility within the context of other choices is not well established, but the side effects common to AEDs, such as dizziness and somnolence, have been relatively mild on this agent in the clinical trials.
Of treatment strategies on the horizon, Dr. Bazil drew attention to SAGE-547, an experimental agent now in a phase III study that has demonstrated uncommon efficacy for refractory status epilepticus, a condition for which there is a large unmet need for reliably effective treatments. In addition, researchers are conducting ongoing efforts to individualize therapy by identifying targetable underlying genetic mutations responsible for seizure activity.
Overall, the therapies approved during the past 10 years have expanded choice in a way that may increase the likelihood of providing a patient with adequate efficacy and acceptable tolerability. Although newer pharmacologic options have not yet generated a major leap in seizure control, coming therapies or strategies to better match existing therapies to the underlying seizure mechanism may yield greater rates of seizure freedom, said Dr. Bazil.
—Ted Bosworth
VANCOUVER—None of the seven new antiepileptic drugs (AEDs) approved by the FDA during the past 10 years greatly increase the likelihood of seizure control, relative to previously existing therapies, according to an overview presented at the 68th Annual Meeting of the American Academy of Neurology. The broader array of choices is facilitating the individualization of therapy, however. Several of the newer agents are chemically related to previously existing AEDs, but all of the drugs have distinguishing features.
“We do have more options for the treatment of epilepsy,” confirmed Carl W. Bazil, MD, PhD, Director of the Comprehensive Epilepsy Center at Columbia University College of Physicians and Surgeons in New York City. “Although we do not yet have the perfect therapy, more are coming.”
In his evaluation of how newer agents fit into current clinical practice, Dr. Bazil indicated that relative efficacy among the newer agents has not been well defined in the absence of head-to-head trials. Treatment choice is instead based on their approved indications, side effect profiles, and half-lives.
Yet the emergence of more choices was characterized as a positive development. Since pregabalin received regulatory approval in 2005, the newer therapies have included lacosamide (2008), rufinamide (2009), clobazam (2010), ezogabine (2011), perampanel (2012), eslicarbazepine (2013), and brivaracetam (2016). Of these, Dr. Bazil focused primarily on the latter five and how they fit into routine care. Overall, these agents “have not been all that different” in terms of their associated rates of total seizure control or other meaningful end points.
This similarity does not suggest that these agents are interchangeable. The clinical profiles differ by characteristics such as dosing frequency, relative risk of somnolence and other common side effects, and the potential to ameliorate accompanying symptoms, such as anxiety. Moreover, due to differences in trial design and entry criteria, not all of the newer drugs have been granted the same indication, said Dr. Bazil.
Clobazam for Lennox-Gastaut Syndrome
Clobazam is a benzodiazepine receptor agonist that shares features with other benzodiazepines. One of the key features of this agent is a half-life that approaches 80 hours. This characteristic makes clobazam a “forgiving” drug for those who miss a dose, said Dr. Bazil.
Clobazam’s FDA-approved indication is for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome for individuals age 2 or older. Although this Schedule IV drug is not approved in the United States for partial onset seizures, it is widely used for this indication elsewhere, including in Europe and Canada. Clobazam, like other benzodiazepine-related agents, could have a favorable effect on anxiety or sleep problems, even though it is not indicated for these uses, said Dr. Bazil.
Relative to other commonly used AEDs, the rate of rash associated with clobazam has been relatively low, but dizziness and somnolence are sufficiently common that the once-daily dose should be taken before bedtime, according to Dr. Bazil. Relative to other benzodiazepines, clobazam appears to have a low potential for tolerance and dependence. Initial daily doses of 5 mg to 10 mg are standard, but lower doses should be considered in older patients or those with low body weight, said Dr. Bazil. Titration is performed on a weekly basis to a maximum recommended dose of 60 mg.
Perampanel Has a Long Half-Life
Perampanel, which is a noncompetitive AMPA-receptor inhibitor, has an even longer half-life of 100 hours. The drug’s FDA approval is for the adjunctive treatment of partial epilepsy and primary generalized tonic-clonic seizures in patients age 12 and older. Prudent initial dosing begins at 2 mg daily with a recommended maximum dose of 12 mg, according to Dr. Bazil. Due to its reliance on hepatic metabolism, a relatively slow upward titration of this Schedule III drug is appropriate in patients with impaired liver function, he added.
Dizziness and somnolence are commonly reported side effects for perampanel, as they are for most AEDs, and the drug also has a black box warning about its association with homicidal ideation, irritability and aggression. Yet the drug’s relative efficacy may compensate for these risks in selected patients. Even though researchers have not compared perampanel directly with another AED, the drug had an “impressive” seizure-free rate in a placebo-controlled trial, said Dr. Bazil. In this multicenter study of patients with drug-resistant, primary generalized tonic-clonic seizures, 30.9% of participants on perampanel, versus 12.3% of those on placebo, remained seizure-free during maintenance.
Ezogabine and Risk of Discoloration
Ezogabine, which is FDA-approved as adjunctive therapy for refractory partial epilepsy, is a K-channel opener. Due to a relatively short half-life of seven to 11 hours, thrice-daily dosing is required. In addition, bluish discoloration, primarily of the lips and fingertips, commonly occurs due to an interaction of a drug metabolite with sun exposure, according to Dr. Bazil. Concern that this drug may lead to retinal pigmentation means that eye examinations are recommended at baseline and every six months.
“Clinical use of ezogabine has been limited by the pigmentation and by tid dosing, which patients do not like,” Dr. Bazil said. Although the bluish color resolves over time, patients are likely to discontinue this Schedule V drug, which also is associated with dizziness and somnolence, before this occurs.
Eslicarbazepine, an Unscheduled Agent
Eslicarbazepine, a sodium channel blocker, has been approved as adjunctive and monotherapy for partial epilepsy. Although monotherapy trials with a placebo control are no longer considered ethical in patients with epilepsy, eslicarbazepine and lacosamide have been evaluated and found effective in trials in which other active agents were withdrawn over time to allow response on monotherapy to be monitored, Dr. Bazil explained.
Eslicarbazepine is also distinguished from other newer agents by the fact that it is not a scheduled agent. It is primarily metabolized by the liver and has a half-life of 13 to 20 hours. Due to its structural similarity to carbamazepine and oxcarbazepine, these agents should not be combined, said Dr. Bazil. Eslicarbazepine, like clobazam, also could interfere with the bioavailability of oral contraceptives.
“Eslicarbazepine is essentially a longer-acting carbamazepine, a drug with which we are all familiar,” said Dr. Bazil. Although eslicarbazepine also may cause the dizziness, somnolence, and nausea common to carbamazepine, its longer half-life has the potential to diminish peak effects to improve tolerability.
New and Emerging Therapies
Brivaracetam, which was approved for add-on therapy of refractory partial seizures for patients age 16 or older, “is structurally and functionally related to levetiracetam, but approximately 20 times more potent at the SV2A receptor,” said Dr. Bazil. As this recently approved drug is not yet widely used, its relative clinical utility within the context of other choices is not well established, but the side effects common to AEDs, such as dizziness and somnolence, have been relatively mild on this agent in the clinical trials.
Of treatment strategies on the horizon, Dr. Bazil drew attention to SAGE-547, an experimental agent now in a phase III study that has demonstrated uncommon efficacy for refractory status epilepticus, a condition for which there is a large unmet need for reliably effective treatments. In addition, researchers are conducting ongoing efforts to individualize therapy by identifying targetable underlying genetic mutations responsible for seizure activity.
Overall, the therapies approved during the past 10 years have expanded choice in a way that may increase the likelihood of providing a patient with adequate efficacy and acceptable tolerability. Although newer pharmacologic options have not yet generated a major leap in seizure control, coming therapies or strategies to better match existing therapies to the underlying seizure mechanism may yield greater rates of seizure freedom, said Dr. Bazil.
—Ted Bosworth
Suggested Reading
French JA, Krauss GL, Wechsler RT, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology. 2015;85(11):950-957.
Suggested Reading
French JA, Krauss GL, Wechsler RT, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology. 2015;85(11):950-957.

Clinical Challenges - June 2016: Diffuse idiopathic skeletal hyperostosis
What's Your Diagnosis?
The diagnosis
The radiograph (Figure B) and CT scan (Figure C) of the neck revealed severe calcification of soft tissue along the anterior portion of cervical spine starting from C2 to T1. The calcification is pressing on the esophagus and also displaces the trachea forward.
These findings are consistent with diffuse idiopathic skeletal hyperostosis (DISH). Initially, the patient was treated with partial liquid diet and nutritional supplement. Subsequently, the patient underwent orthopedic consultation for surgical consideration.
DISH is a noninflammatory ossification and calcification of ligaments and tendons anterior to the spine. Even though this condition was first described by Forestier et al. in 1950,1 the etiology and pathogenesis of DISH remain unknown. Many risk factors have been postulated to be associated with DISH, such as mechanical factors, diet, and drugs. DISH is more prevalent among the elderly, especially, males; however, it is a rare cause of dysphagia.2 The clinical manifestation varies depending on the location of calcification. There have been case reports of DISH causing dysphagia, difficult airway management, and spinal cord root compression.3
For esophageal involvement, the severity of symptoms is correlated with degree of compression. Initially, DISH may cause only an isolated globus sensation without any significant dysphagia. However, as the compression progresses, it can cause significant progressive dysphagia that mimics the presentation of esophageal cancer. Thus, the effort to rule out esophageal cancer or other structural diseases is needed before the diagnosis of DISH can be established, such as an upper endoscopy or CT of the neck.
Because DISH can resemble benign degenerative osteophytes on a plain neck radiograph, it is sometimes overlooked by physicians. DISH is an uncommon condition; it should be considered in the differential diagnosis of dysphagia, especially in the elderly population.References
1. Forestier, J., Rotes-Querol, J. Senile ankylosing hyperostosis of the spine. Ann Rheum Dis. 1950;9:321-30.
2. Schlapbach, P., Beyeler, C., Gerber, N.J., et al. Diffuse idiopathic skeletal hyperostosis (DISH) of the spine: a cause of back pain? (A controlled study). Br J Rheumatol. 1989;28:299-303.
3. Verlaan, J.J., Boswijk, P.F., de Ru, J.A., et al. Diffuse idiopathic skeletal hyperostosis of the cervical spine: an underestimated cause of dysphagia and airway obstruction. Spine J. 2011;11:1058-67.
The diagnosis
The radiograph (Figure B) and CT scan (Figure C) of the neck revealed severe calcification of soft tissue along the anterior portion of cervical spine starting from C2 to T1. The calcification is pressing on the esophagus and also displaces the trachea forward.
These findings are consistent with diffuse idiopathic skeletal hyperostosis (DISH). Initially, the patient was treated with partial liquid diet and nutritional supplement. Subsequently, the patient underwent orthopedic consultation for surgical consideration.
DISH is a noninflammatory ossification and calcification of ligaments and tendons anterior to the spine. Even though this condition was first described by Forestier et al. in 1950,1 the etiology and pathogenesis of DISH remain unknown. Many risk factors have been postulated to be associated with DISH, such as mechanical factors, diet, and drugs. DISH is more prevalent among the elderly, especially, males; however, it is a rare cause of dysphagia.2 The clinical manifestation varies depending on the location of calcification. There have been case reports of DISH causing dysphagia, difficult airway management, and spinal cord root compression.3
For esophageal involvement, the severity of symptoms is correlated with degree of compression. Initially, DISH may cause only an isolated globus sensation without any significant dysphagia. However, as the compression progresses, it can cause significant progressive dysphagia that mimics the presentation of esophageal cancer. Thus, the effort to rule out esophageal cancer or other structural diseases is needed before the diagnosis of DISH can be established, such as an upper endoscopy or CT of the neck.
Because DISH can resemble benign degenerative osteophytes on a plain neck radiograph, it is sometimes overlooked by physicians. DISH is an uncommon condition; it should be considered in the differential diagnosis of dysphagia, especially in the elderly population.References
1. Forestier, J., Rotes-Querol, J. Senile ankylosing hyperostosis of the spine. Ann Rheum Dis. 1950;9:321-30.
2. Schlapbach, P., Beyeler, C., Gerber, N.J., et al. Diffuse idiopathic skeletal hyperostosis (DISH) of the spine: a cause of back pain? (A controlled study). Br J Rheumatol. 1989;28:299-303.
3. Verlaan, J.J., Boswijk, P.F., de Ru, J.A., et al. Diffuse idiopathic skeletal hyperostosis of the cervical spine: an underestimated cause of dysphagia and airway obstruction. Spine J. 2011;11:1058-67.
The diagnosis
The radiograph (Figure B) and CT scan (Figure C) of the neck revealed severe calcification of soft tissue along the anterior portion of cervical spine starting from C2 to T1. The calcification is pressing on the esophagus and also displaces the trachea forward.
These findings are consistent with diffuse idiopathic skeletal hyperostosis (DISH). Initially, the patient was treated with partial liquid diet and nutritional supplement. Subsequently, the patient underwent orthopedic consultation for surgical consideration.
DISH is a noninflammatory ossification and calcification of ligaments and tendons anterior to the spine. Even though this condition was first described by Forestier et al. in 1950,1 the etiology and pathogenesis of DISH remain unknown. Many risk factors have been postulated to be associated with DISH, such as mechanical factors, diet, and drugs. DISH is more prevalent among the elderly, especially, males; however, it is a rare cause of dysphagia.2 The clinical manifestation varies depending on the location of calcification. There have been case reports of DISH causing dysphagia, difficult airway management, and spinal cord root compression.3
For esophageal involvement, the severity of symptoms is correlated with degree of compression. Initially, DISH may cause only an isolated globus sensation without any significant dysphagia. However, as the compression progresses, it can cause significant progressive dysphagia that mimics the presentation of esophageal cancer. Thus, the effort to rule out esophageal cancer or other structural diseases is needed before the diagnosis of DISH can be established, such as an upper endoscopy or CT of the neck.
Because DISH can resemble benign degenerative osteophytes on a plain neck radiograph, it is sometimes overlooked by physicians. DISH is an uncommon condition; it should be considered in the differential diagnosis of dysphagia, especially in the elderly population.References
1. Forestier, J., Rotes-Querol, J. Senile ankylosing hyperostosis of the spine. Ann Rheum Dis. 1950;9:321-30.
2. Schlapbach, P., Beyeler, C., Gerber, N.J., et al. Diffuse idiopathic skeletal hyperostosis (DISH) of the spine: a cause of back pain? (A controlled study). Br J Rheumatol. 1989;28:299-303.
3. Verlaan, J.J., Boswijk, P.F., de Ru, J.A., et al. Diffuse idiopathic skeletal hyperostosis of the cervical spine: an underestimated cause of dysphagia and airway obstruction. Spine J. 2011;11:1058-67.
What's Your Diagnosis?
What's Your Diagnosis?
What's Your Diagnosis?

Upon arrival, his vital signs were stable. Physical examination was unremarkable. No cervical lymphadenopathy or masses were appreciated in his neck. Hemoglobin, platelets, and white blood cell count were within normal reference ranges. An esophagogram was initially performed, illustrating mildly thickened folds of distal esophagus with no obstruction or ulcer (Figure A).
Upper endoscopy was subsequently performed, revealing normal esophageal mucosa without any strictures. An ENT specialist was consulted because of the concern of oropharyngeal dysphagia. Laryngoscope and video swallowing evaluations were performed; the results were normal. Ultimately, a neck radiograph and computed tomography (CT) of the neck were performed (Figures B and C).
Antibiotic interactions: Answers to 4 common questions
› Avoid preemptive warfarin dose reductions unless you are prescribing trimethoprim/sulfamethoxazole (TMP/SMX) or metronidazole. B
› Recommend a back-up contraceptive method to a woman who is taking a broad-spectrum antibiotic and low-dose OCs—especially if the woman is overweight. C
› Consider using the macrolide, clarithromycin, or the fluoroquinolone, ciprofloxacin, in patients taking medications that prolong QT interval or who are at higher risk for torsades de pointes (TdP). B
› Refrain from cautioning patients taking metronidazole against consuming alcohol. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite encouraging data that antibiotic prescribing is on the decline, patients are still prescribed antibiotics frequently, making these agents the 12th most frequently used drug class.1 At the same time, prescribers are caring for patients with increasingly complex drug regimens that provide fertile ground for drug interactions with these antibiotics. And, of course, lifestyle factors such as alcohol consumption are a consideration when any prescription is written.
As pharmacists, we find that certain questions about antibiotic prescribing and interactions come up with frequency. These questions often pertain to the use of warfarin, oral contraceptives, drugs that prolong the QT interval, and alcohol. But conflicting reports about issues such as monitoring international normalized ratio (INR) in patients taking warfarin and antibiotics, and whether (or which) antibiotics decrease the efficacy of oral contraceptives (OCs) can make decision-making challenging.
This review provides evidence-based answers to questions you may have. It also details some reliable sources of information you can consult (TABLE 12-7) when discussing treatment options with other members of the health care team.
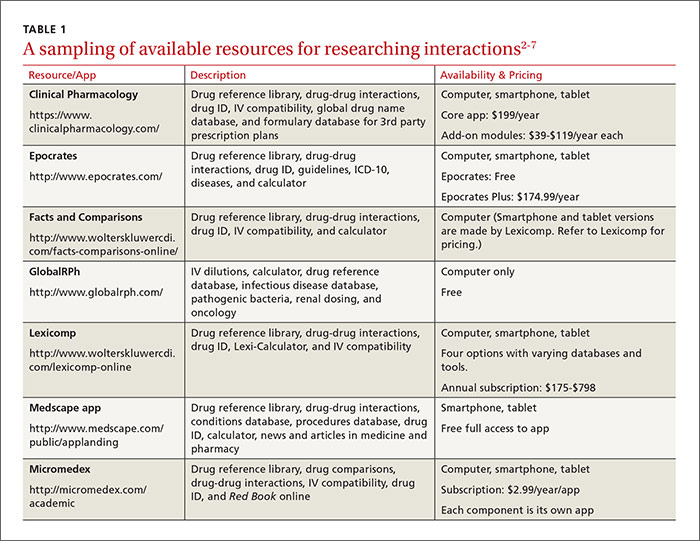
1. Which antibiotics are preferable when a patient is taking warfarin, and are preemptive warfarin dose reductions advisable?
The simple answer is that agents with a lower likelihood of affecting the INR, such as penicillin G, clindamycin, and 1st- and 4th-generation cephalosporins, are a good place to start, and whether to preemptively reduce the warfarin dose hinges on the antibiotic being prescribed.
The more detailed answer. The fundamental mechanisms of interaction between warfarin and antibiotics are two-fold:8
- Antimicrobial agents disrupt gastrointestinal flora that synthesize vitamin K.
- Antimicrobials inhibit cytochrome p450 (CYP450) enzymes (primarily CYP2C9 and 3A4), which are responsible for the metabolism of warfarin.
The antibiotics most likely to interfere with warfarin are TMP/SMX, ciprofloxacin, levofloxacin, metronidazole, fluconazole, azithromycin, and clarithromycin (TABLE 2).9,10 Low-risk agents include clindamycin, cephalexin, and penicillin G. When prescribing an antibiotic for a patient taking warfarin, it is important not only to be aware of the agents that should be avoided, but also the agents that do not require more frequent monitoring of INR.

Preemptive warfarin dose reductions? Some physicians make preemptive warfarin dose reductions in an attempt to avoid supratherapeutic INRs in patients being prescribed antibiotics. But the evidence suggests that this step should be considered only in the presence of the antibiotics TMP/SMX and metronidazole.9,11
A 2008 study investigated the anticoagulation effects of a 10% to 20% preemptive warfarin dose reduction vs no dosing change in patients taking TMP/SMX or levofloxacin. The investigators found that the preemptive warfarin dose reduction (intervention) significantly decreased the number of supratherapeutic INR values above 4 when compared to controls (2 of 8 vs 8 of 9).12
In the dose-reduction group, no patients receiving TMP/SMX developed a subtherapeutic INR, whereas 40% (4 of 10 patients) who received levofloxacin developed a subtherapeutic INR.12 The authors of the study concluded that a prophylactic warfarin dose reduction of 10% to 20% is effective in maintaining therapeutic anticoagulation in patients receiving TMP/SMX. They added that while no change in warfarin dosing is necessary with levofloxacin, short-term INR follow-up is a prudent approach to prevent subtherapeutic INRs. Others recommend INR monitoring when antibiotic therapy is started and stopped and whenever the dose is changed.9
A 2010 retrospective, single-center, cohort study looked at patients who were taking metronidazole and warfarin. Researchers compared those who received a preemptive dose reduction of warfarin (mean reduction was 34.6% ± 13.4%) to those who did not and found a statistically significant mean difference in INR of 1.28 (P=.01).13
Almost half (46%) of the patients who did not receive a warfarin dose reduction had an INR >4, whereas none of the patients in the warfarin dose reduction group did (P=.05). Although this secondary outcome was not statistically significant (most likely due to the small sample population [N=20]), the implication is clinically significant. Two patients who reduced their dose had a subtherapeutic INR compared to none of the patients in the control group, which was also not a statistically significant difference.
The authors concluded that a 30% to 35% reduction in mean daily warfarin dose is effective in maintaining therapeutic anticoagulation in patients started on metronidazole.
Significant bleeding events. A retrospective cohort study of slightly more than 22,000 veterans who were prescribed warfarin for ≥30 uninterrupted days and given antibiotics with either a high or low risk for interaction with warfarin were studied for significant bleeding events for one month.10 Ninety-three significant bleeding events occurred in the high-risk group and 36 occurred in the low-risk group over the course of the study. The agent associated with the greatest increased risk of bleeding was TMP/SMX (hazard ratio [HR]=2.09; 95% CI, 1.45-3.02). Of note, metronidazole was not included in this study endpoint.
The study’s secondary endpoint of INR >4 found that 10% of patients taking metronidazole and 8% of patients taking TMP/SMX in addition to warfarin had INRs >4. Almost 10% (9.7%) of patients prescribed fluconazole had a peak INR value >6. Patients taking low-risk antibiotics (clindamycin or cephalexin) had no increased risk of bleeding. Monitoring INR within 3 to 14 days of starting patients on antibiotics was found to decrease the risk of serious bleeding events (HR=0.61; 95% CI, 0.42-0.88). More frequent INR monitoring by itself (without preemptive warfarin dose reductions) is appropriate for other antibiotics, including macrolides, tetracyclines, and some cephalosporins (2nd and 3rd generation).9
THE BOTTOM LINE When prescribing antibiotics for patients taking warfarin, try to choose agents with a lower likelihood of affecting INR such as penicillin G, clindamycin, and 1st- and 4th-generation cephalosporins. With these agents, there is no need for more frequent INR testing or preemptive reductions in warfarin dose. In patients for whom the use of TMP/SMX or metronidazole can’t be avoided, consider reducing the patient’s warfarin dose by 10% to 35% and rechecking the INR 5 days after starting the antibiotic.9,11,12 When prescribing agents such as fluoroquinolones, macrolides, and tetracyclines, do not reduce the patient’s warfarin dose preemptively and recheck INR 5 days after starting therapy.
2. Do antibiotics decrease the efficacy of oral contraceptives?
It’s unlikely, but antibiotics may reduce the efficacy of OCs.
There have been few, but well documented, reports of women using OCs who became pregnant after taking antimicrobials.14 It is recognized that rifampin, an inducer of enzymes that metabolize estrogens, decreases the efficacy of OCs.15 Ketoconazole’s interaction seems less well documented, but combining the agent with low-estrogen (low-dose) OCs warrants caution.16 What is not well understood is whether more common or broad-spectrum antibiotics also increase the risk of OC failure.
Three mechanisms have been proposed:16
- Antimicrobials affect hepatic enzyme induction, which increases metabolism of hormones.
- Broad-spectrum antibiotics reduce gut bacteria, which alters enterohepatic circulation and reduces plasma hormone concentrations.
- Antibiotics increase gastrointestinal motility, which decreases absorption (and reabsorption) of OCs.
A 2007 study found that when physicians and pharmacists were surveyed and asked if broad-spectrum antibiotics have a clinically significant interaction with OCs, 83% of physicians and 89% of pharmacists answered “Yes;”17 however, a large epidemiologic study performed in the United States showed no association between antibiotic use and OC failure.18
After this report, investigators in the Netherlands completed a similar cross-over analysis and found that there was a relationship between the use of antibiotics and breakthrough pregnancy in a population-based prescription database, but that the results didn’t hold for broad-spectrum antibiotics or in a sensitivity analysis.19 Pharmacokinetic studies are also conflicting, as some have shown an effect on serum hormone levels, while others have not.15,20-22
High- vs low-risk agents. Ciprofloxacin did not affect hormone levels in 2 studies.20,21 Rifampin and voriconazole may enhance systemic exposure to OCs.15,22 And erythromycin and azithromycin may interact with OCs, but the clinical significance of this interaction is still unknown.16

Short-courses of TMP/SMX are generally thought to be safe;16 a small study looked at cotrimoxazole 1 g twice daily in 9 women taking long-term OC steroids and found that short courses of the drug were unlikely to cause any adverse effects on contraceptive control.23 Tetracyclines and penicillins were the antibiotics most frequently involved in case reports of pregnancy from the United Kingdom (TABLE 32).16
It is hypothesized that some women may have a higher risk of OC failure than others due to how they metabolize ethinyl estradiol.24 Another hypothesis is that some women have gut flora that is more susceptible to the antibiotic being used. And still another possibility is that lower doses of hormones are being used in OCs than were studied for this interaction.15 Anything that decreases the concentration of these lower-dose OCs is concerning, especially in patients with a higher body mass index (BMI). The few pharmacokinetic studies that have been conducted show that it takes longer for OCs to reach a steady state in obese women and that they have a lower area under the curve (AUC) and maximum estrogen concentration than women with a normal BMI.25
THE BOTTOM LINE Because the degree of variability between patients is unknown and obesity rates are increasing, concern that low-dose OCs may lose efficacy when combined with antibiotics is warranted. While the absolute risk of breakthrough pregnancy seems small, the most conservative approach is to advise patients to use a back-up method of contraception during times of antibiotic use.
3. Which drugs prolong QT intervals?
Macrolides and fluoroquinolones are 2 classes of antibiotics associated with prolonged QT intervals, but other drugs and risk factors are important to consider, as well.
Physicians often receive phone calls from pharmacists warning about drug-drug interactions when they prescribe macrolides or fluoroquinolones for patients already taking medications known to prolong QT intervals or inhibit cytochrome P450 enzymes. Long QT syndrome increases the risk of TdP, a life-threatening arrhythmia. While TdP is rare, its severity warrants a discussion of risk factors and the likelihood of occurrence.
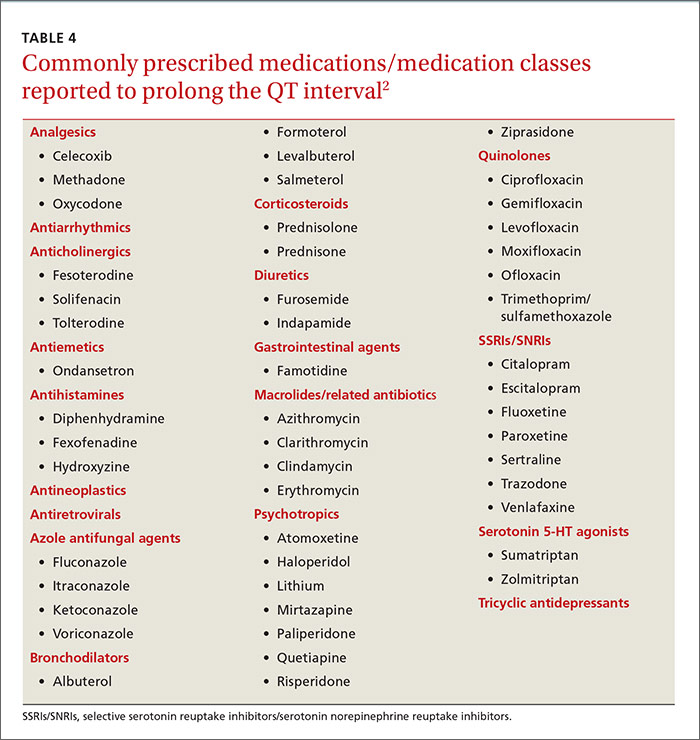
Two QT interval prolonging medications used together in healthy individuals does not warrant a change in therapy. TdP is most likely to occur when 2 or more QT interval prolonging medications are used in a patient who is already at high risk for arrhythmia because of risk factors such as prolonged QT interval at baseline, family history of prolonged QT intervals, female gender, age >60 years, electrolyte abnormalities (hypokalemia, hypomagnesemia, hypocalcemia), underlying comorbid diseases (eg, chronic heart failure, left ventricular hypertrophy, atrial fibrillation), hypertension, bradycardia, and genetic (ion channel) polymorphisms.26,27
Antiarrhythmics and antipsychotics are most commonly associated with drug-induced prolonged QT interval, with most case reports and research being linked to antiarrhythmics (TABLE 42).28 But macrolide and fluoroquinolone antibiotics also have been associated with TdP, although to a lesser extent. In a retrospective analysis of case reports of TdP involving macrolides, erythromycin was present (with or without other medications thought to prolong QT) in 53% of the cases and clarithromycin was involved in 36% of the reports.29
An analysis of 2 studies by the US Food and Drug Administration estimated an occurrence rate of serious cardiac arrhythmias of 46 to 85 per 100,000 users with cardiovascular disease, compared to 5 to 44 per 100,000 users without cardiovascular disease.30 And this may underestimate the actual incidence because spontaneous reporting of adverse effects declines the longer a drug is on the market. Ciprofloxacin is associated with less risk than levofloxacin and gatifloxacin (the latter of which is no longer available in the United States).26
A recent population-based study using data on over 10.6 million people from the Taiwan National Health Insurance Database examined the risk of cardiovascular death among patients using new-generation macrolides, fluoroquinolones, and β-lactam/β-lactamase inhibitors.31 The absolute risk of cardiovascular death per 1000 individuals was 0.06 for clarithromycin, 0.12 for ciprofloxacin, 0.13 for amoxicillin-clavulanate, 0.36 for azithromycin, 0.39 for levofloxacin, and 0.46 for moxifloxacin. The mean interval between first antibiotic use and the adverse cardiac event was <4 days. Not surprisingly, the highest risk was seen in patients with underlying cardiovascular disease.
Another population-based study, this time conducted in Hong Kong, evaluated the cardiovascular safety of clarithromycin compared to that of amoxicillin. Clarithromycin was found to increase the incidence of myocardial infarction, arrhythmia, and cardiac mortality in the short term, with the risk returning to baseline after treatment concluded.32 A binational cohort study of Danish and Swedish adults confirmed that fluoroquinolones (especially ciprofloxacin) do not increase the risk of a serious arrhythmia compared to penicillins.33
THE BOTTOM LINE For patients taking other QT interval prolonging medications or who are at a higher risk for TdP, consider using clarithromycin over erythromycin or azithromycin for a macrolide antibiotic or ciprofloxacin over levofloxacin or moxifloxacin if a fluoroquinolone is warranted. Using 2 drugs that may increase the QT interval is likely safe in the absence of certain risk factors.
4. Should patients avoid alcohol while taking metronidazole?
Probably not.
Warning patients against drinking alcohol while taking metronidazole has been a common practice for years. The mechanism for this theorized interaction was thought to be similar to the interaction between disulfiram and ethanol.34 Disulfiram inhibits hepatic aldehyde dehydrogenase (ALDH) when combined with alcohol, which leads to increased levels of acetaldehyde in the blood and symptoms of flushing, palpitations, nausea, vomiting, headache, and visual disturbances.35 However, multiple studies using rats have found that metronidazole does not inhibit ALDH or increase acetaldehyde concentrations like disulfiram does.34
A 2000 review article discussed 6 cases involving serious metronidazole-ethanol interactions. Ethanol alone was found to explain the reaction in 2 of the cases, and the remaining 4 could be linked to the use of other drugs or disease states.35 A 2002 Finnish study found no statistically significant differences in objective or subjective signs of a disulfiram-like interaction.34 When considering the symptoms associated with the interaction, it is important to remember that many of the symptoms can result from metronidazole therapy alone, regardless of whether other medications or alcohol are used.35
THE BOTTOM LINE Researchers have failed to identify a clinically significant interaction between metronidazole and alcohol. Avoiding alcohol while taking metronidazole does not appear to be necessary.
CORRESPONDENCE
Mary Onysko, PharmD, BCPS, University of Wyoming, School of Pharmacy Health Sciences Center, Room 292, 1000 E. University Avenue, Laramie, WY 82071; [email protected].
1. Kantor ED, Rehm CD, Haas JS, et al. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314:1818-1831.
2. Lexicomp Online. Clinical Drug Information. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed December 8, 2015.
3. GlobalRPh: The Clinician’s Ultimate Reference. Available at: http://www.globalrph.com/. Accessed December 8, 2015.
4. Medscape Apps. Available at: http://www.medscape.com/public/applanding. Accessed December 8, 2015.
5. Micromedex Solutions. Academic Institutions. Available at: http://micromedex.com/academic. Accessed December 8, 2015.
6. Patel A. Clinical Pharmacology Mobile-A mobile web app compatible on all smart phones [review] May 31, 2010. Available at: http://www.imedicalapps.com/2010/05/clinical-pharmocology-app-review/. Accessed December 8, 2015.
7. Epocrates. Available at: http://www.epocrates.com/. Accessed December 8, 2015.
8. Baillargeon J, Holmes HM, Lin Y, et al. Concurrent use of warfarin and antibiotics and the risk of bleeding in older adults. Am J Med. 2012;125:183-189.
9. PL Detail-Document #280806. Antimicrobial drug interactions with warfarin. Pharmacist’s Letter/Prescriber’s Letter. August 2012.
10. Lane M, Zeringue A, McDonald J. Serious bleeding events due to warfarin and antibiotic co-prescription in a cohort of veterans. Am J Med. 2014;127:657-663.e2.
11. Hale SF, Lesar TS. Interaction of vitamin K antagonists and trimethoprim-sulfamethoxazole: ignore at your patient’s risk. Drug Metab Drug Interact. 2014;29:53-60.
12. Ahmed A, Stephens JC, Kaus CA, et al. Impact of preemptive warfarin dose reduction on anticoagulation after initiation of trimethoprim-sulfamethoxazole or levofloxacin. J Thromb Thrombolysis. 2008;26:44-48.
13. Holt RK, Anderson EA, Cantrell MA, et al. Preemptive dose reduction of warfarin in patients initiating metronidazole. Drug Metabol Drug Interact. 2010;25:35-39.
14. Hughes BR, Cunliffe WJ. Interactions between the oral contraceptive pill and antibiotics. Br J Dermatol. 1990;122:717-718.
15. Bolt HM. Interactions between clinically used drugs and oral contraceptives. Environ Health Perspect. 1994;102:35-38.
16. Aronson JK. Meyler’s Side Effects of Drugs. 16th ed. The International Encyclopedia of Adverse Drug Reactions and Interactions. Amsterdam, Netherlands: Elsevier; 2016. Available at: http://ac.els-cdn.com/B978044453717101009X/3-s2.0-B978044453717101009X-main.pdf?_tid=b33f6564-9deb-11e5-a8f0-00000aab0f01&acdnat=1449607315_83f5068fc5105226fcc6d7279c083516. Accessed December 8, 2015.
17. Masters KP, Carr BM. Survey of pharmacists and physicians on drug interactions between combined oral contraceptives and broad-spectrum antibiotics. Pharm Pract (Granada). 2009;7:139-144.
18. Toh S, Mitchell AA, Anderka M, et al; National Birth Defects Prevention Study. Antibiotics and oral contraceptive failure—a case-crossover study. Contraception. 2011;83:418-425.
19. Koopmans PC, Bos JH, de Jong van den Berg LT. Are antibiotics related to oral combination contraceptive failures in the Netherlands? A case-crossover study. Pharmacoepidemiol Drug Saf. 2012;21:865-871.
20. Archer JS, Archer DF. Oral contraceptive efficacy and antibiotic interaction: A myth debunked. J Am Acad Dermatol. 2002;46:917–923.
21. Scholten PC, Droppert RM, Zwinkels MGJ, et al. No interaction between ciprofloxacin and an oral contraceptive. Antimicrob Agents Chemother. 1998;42:3266-3268.
22. Andrews E, Damle BD, Fang A, et al. Pharmacokinetics and tolerability of voriconazole and a combination oral contraceptive co-administered in healthy female subjects. Br J Clin Pharmacol. 2008;65:531-539.
23. Grimmer SF, Allen WL, Back DJ, et al. The effect of cotrimoxazole on oral contraceptive steroids in women. Contraception. 1983;28:53-59.
24. Dickinson BD, Altman RD, Nielsen NH, et al; Council on Scientific Affairs, American Medical Association. Drug interactions between oral contraceptives and antibiotics. Obstet Gynecol. 2001;98:853-860.
25. Edelman AB, Cherala G, Stanczyk FZ. Metabolism and pharmacokinetics of contraceptive steroids in obese women: a review. Contraception. 2010;82:314-323.
26. Owens RC Jr, Ambrose PG. Torsades de pointes associated with fluoroquinolones. Pharmacotherapy. 2002;22:663-668.
27. Letsas KP, Efremidis M, Kounas SP, et al. Clinical characteristics of patients with drug-induced QT interval prolongation and torsade de pointes: identification of risk factors. Clin Res Cardiol. 2009;98:208-212.
28. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363-1372.
29. Shaffer D, Singer S, Korvick J, et al. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration adverse event reporting system. Clin Infect Dis. 2002;35:197-200.
30. FDA Briefing Document. Joint Meeting of the Antimicrobial Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee. November 5, 2015. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM467383.pdf. Accessed June 11, 2016.
31. Chou HW, Wang JL, Chang CH, et al. Risks of cardiac arrhythmia and mortality among patients using new-generation macrolides, fluoroquinolones, and β-lactam/β-lactamase inhibitors: a Taiwanese nationwide study. Clin Infect Dis. 2015;60:566-577.
32. Wong AY, Root A, Douglas IJ, et al. Cardiovascular outcomes associated with use of clarithromycin: population based study. BMJ. 2016;352:h6926.
33. Inghammar M, Svanström H, Melbye M, et al. Oral fluoroquinolone use and serious arrhythmia: bi-national cohort study. BMJ. 2016;352:i843.
34. Visapää JP, Tillonen JS, Kaihovaara PS, et al. Lack of disulfiram-like reaction with metronidazole and ethanol. Ann Pharmacother. 2002;36:971-974. 35. Fjeld H, Raknes G. Is combining metronidazole and alcohol really hazardous? Tidsskr Nor Laegeforen. 2014;134:1661-1663.
› Avoid preemptive warfarin dose reductions unless you are prescribing trimethoprim/sulfamethoxazole (TMP/SMX) or metronidazole. B
› Recommend a back-up contraceptive method to a woman who is taking a broad-spectrum antibiotic and low-dose OCs—especially if the woman is overweight. C
› Consider using the macrolide, clarithromycin, or the fluoroquinolone, ciprofloxacin, in patients taking medications that prolong QT interval or who are at higher risk for torsades de pointes (TdP). B
› Refrain from cautioning patients taking metronidazole against consuming alcohol. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite encouraging data that antibiotic prescribing is on the decline, patients are still prescribed antibiotics frequently, making these agents the 12th most frequently used drug class.1 At the same time, prescribers are caring for patients with increasingly complex drug regimens that provide fertile ground for drug interactions with these antibiotics. And, of course, lifestyle factors such as alcohol consumption are a consideration when any prescription is written.
As pharmacists, we find that certain questions about antibiotic prescribing and interactions come up with frequency. These questions often pertain to the use of warfarin, oral contraceptives, drugs that prolong the QT interval, and alcohol. But conflicting reports about issues such as monitoring international normalized ratio (INR) in patients taking warfarin and antibiotics, and whether (or which) antibiotics decrease the efficacy of oral contraceptives (OCs) can make decision-making challenging.
This review provides evidence-based answers to questions you may have. It also details some reliable sources of information you can consult (TABLE 12-7) when discussing treatment options with other members of the health care team.
1. Which antibiotics are preferable when a patient is taking warfarin, and are preemptive warfarin dose reductions advisable?
The simple answer is that agents with a lower likelihood of affecting the INR, such as penicillin G, clindamycin, and 1st- and 4th-generation cephalosporins, are a good place to start, and whether to preemptively reduce the warfarin dose hinges on the antibiotic being prescribed.
The more detailed answer. The fundamental mechanisms of interaction between warfarin and antibiotics are two-fold:8
- Antimicrobial agents disrupt gastrointestinal flora that synthesize vitamin K.
- Antimicrobials inhibit cytochrome p450 (CYP450) enzymes (primarily CYP2C9 and 3A4), which are responsible for the metabolism of warfarin.
The antibiotics most likely to interfere with warfarin are TMP/SMX, ciprofloxacin, levofloxacin, metronidazole, fluconazole, azithromycin, and clarithromycin (TABLE 2).9,10 Low-risk agents include clindamycin, cephalexin, and penicillin G. When prescribing an antibiotic for a patient taking warfarin, it is important not only to be aware of the agents that should be avoided, but also the agents that do not require more frequent monitoring of INR.
Preemptive warfarin dose reductions? Some physicians make preemptive warfarin dose reductions in an attempt to avoid supratherapeutic INRs in patients being prescribed antibiotics. But the evidence suggests that this step should be considered only in the presence of the antibiotics TMP/SMX and metronidazole.9,11
A 2008 study investigated the anticoagulation effects of a 10% to 20% preemptive warfarin dose reduction vs no dosing change in patients taking TMP/SMX or levofloxacin. The investigators found that the preemptive warfarin dose reduction (intervention) significantly decreased the number of supratherapeutic INR values above 4 when compared to controls (2 of 8 vs 8 of 9).12
In the dose-reduction group, no patients receiving TMP/SMX developed a subtherapeutic INR, whereas 40% (4 of 10 patients) who received levofloxacin developed a subtherapeutic INR.12 The authors of the study concluded that a prophylactic warfarin dose reduction of 10% to 20% is effective in maintaining therapeutic anticoagulation in patients receiving TMP/SMX. They added that while no change in warfarin dosing is necessary with levofloxacin, short-term INR follow-up is a prudent approach to prevent subtherapeutic INRs. Others recommend INR monitoring when antibiotic therapy is started and stopped and whenever the dose is changed.9
A 2010 retrospective, single-center, cohort study looked at patients who were taking metronidazole and warfarin. Researchers compared those who received a preemptive dose reduction of warfarin (mean reduction was 34.6% ± 13.4%) to those who did not and found a statistically significant mean difference in INR of 1.28 (P=.01).13
Almost half (46%) of the patients who did not receive a warfarin dose reduction had an INR >4, whereas none of the patients in the warfarin dose reduction group did (P=.05). Although this secondary outcome was not statistically significant (most likely due to the small sample population [N=20]), the implication is clinically significant. Two patients who reduced their dose had a subtherapeutic INR compared to none of the patients in the control group, which was also not a statistically significant difference.
The authors concluded that a 30% to 35% reduction in mean daily warfarin dose is effective in maintaining therapeutic anticoagulation in patients started on metronidazole.
Significant bleeding events. A retrospective cohort study of slightly more than 22,000 veterans who were prescribed warfarin for ≥30 uninterrupted days and given antibiotics with either a high or low risk for interaction with warfarin were studied for significant bleeding events for one month.10 Ninety-three significant bleeding events occurred in the high-risk group and 36 occurred in the low-risk group over the course of the study. The agent associated with the greatest increased risk of bleeding was TMP/SMX (hazard ratio [HR]=2.09; 95% CI, 1.45-3.02). Of note, metronidazole was not included in this study endpoint.
The study’s secondary endpoint of INR >4 found that 10% of patients taking metronidazole and 8% of patients taking TMP/SMX in addition to warfarin had INRs >4. Almost 10% (9.7%) of patients prescribed fluconazole had a peak INR value >6. Patients taking low-risk antibiotics (clindamycin or cephalexin) had no increased risk of bleeding. Monitoring INR within 3 to 14 days of starting patients on antibiotics was found to decrease the risk of serious bleeding events (HR=0.61; 95% CI, 0.42-0.88). More frequent INR monitoring by itself (without preemptive warfarin dose reductions) is appropriate for other antibiotics, including macrolides, tetracyclines, and some cephalosporins (2nd and 3rd generation).9
THE BOTTOM LINE When prescribing antibiotics for patients taking warfarin, try to choose agents with a lower likelihood of affecting INR such as penicillin G, clindamycin, and 1st- and 4th-generation cephalosporins. With these agents, there is no need for more frequent INR testing or preemptive reductions in warfarin dose. In patients for whom the use of TMP/SMX or metronidazole can’t be avoided, consider reducing the patient’s warfarin dose by 10% to 35% and rechecking the INR 5 days after starting the antibiotic.9,11,12 When prescribing agents such as fluoroquinolones, macrolides, and tetracyclines, do not reduce the patient’s warfarin dose preemptively and recheck INR 5 days after starting therapy.
2. Do antibiotics decrease the efficacy of oral contraceptives?
It’s unlikely, but antibiotics may reduce the efficacy of OCs.
There have been few, but well documented, reports of women using OCs who became pregnant after taking antimicrobials.14 It is recognized that rifampin, an inducer of enzymes that metabolize estrogens, decreases the efficacy of OCs.15 Ketoconazole’s interaction seems less well documented, but combining the agent with low-estrogen (low-dose) OCs warrants caution.16 What is not well understood is whether more common or broad-spectrum antibiotics also increase the risk of OC failure.
Three mechanisms have been proposed:16
- Antimicrobials affect hepatic enzyme induction, which increases metabolism of hormones.
- Broad-spectrum antibiotics reduce gut bacteria, which alters enterohepatic circulation and reduces plasma hormone concentrations.
- Antibiotics increase gastrointestinal motility, which decreases absorption (and reabsorption) of OCs.
A 2007 study found that when physicians and pharmacists were surveyed and asked if broad-spectrum antibiotics have a clinically significant interaction with OCs, 83% of physicians and 89% of pharmacists answered “Yes;”17 however, a large epidemiologic study performed in the United States showed no association between antibiotic use and OC failure.18
After this report, investigators in the Netherlands completed a similar cross-over analysis and found that there was a relationship between the use of antibiotics and breakthrough pregnancy in a population-based prescription database, but that the results didn’t hold for broad-spectrum antibiotics or in a sensitivity analysis.19 Pharmacokinetic studies are also conflicting, as some have shown an effect on serum hormone levels, while others have not.15,20-22
High- vs low-risk agents. Ciprofloxacin did not affect hormone levels in 2 studies.20,21 Rifampin and voriconazole may enhance systemic exposure to OCs.15,22 And erythromycin and azithromycin may interact with OCs, but the clinical significance of this interaction is still unknown.16
Short-courses of TMP/SMX are generally thought to be safe;16 a small study looked at cotrimoxazole 1 g twice daily in 9 women taking long-term OC steroids and found that short courses of the drug were unlikely to cause any adverse effects on contraceptive control.23 Tetracyclines and penicillins were the antibiotics most frequently involved in case reports of pregnancy from the United Kingdom (TABLE 32).16
It is hypothesized that some women may have a higher risk of OC failure than others due to how they metabolize ethinyl estradiol.24 Another hypothesis is that some women have gut flora that is more susceptible to the antibiotic being used. And still another possibility is that lower doses of hormones are being used in OCs than were studied for this interaction.15 Anything that decreases the concentration of these lower-dose OCs is concerning, especially in patients with a higher body mass index (BMI). The few pharmacokinetic studies that have been conducted show that it takes longer for OCs to reach a steady state in obese women and that they have a lower area under the curve (AUC) and maximum estrogen concentration than women with a normal BMI.25
THE BOTTOM LINE Because the degree of variability between patients is unknown and obesity rates are increasing, concern that low-dose OCs may lose efficacy when combined with antibiotics is warranted. While the absolute risk of breakthrough pregnancy seems small, the most conservative approach is to advise patients to use a back-up method of contraception during times of antibiotic use.
3. Which drugs prolong QT intervals?
Macrolides and fluoroquinolones are 2 classes of antibiotics associated with prolonged QT intervals, but other drugs and risk factors are important to consider, as well.
Physicians often receive phone calls from pharmacists warning about drug-drug interactions when they prescribe macrolides or fluoroquinolones for patients already taking medications known to prolong QT intervals or inhibit cytochrome P450 enzymes. Long QT syndrome increases the risk of TdP, a life-threatening arrhythmia. While TdP is rare, its severity warrants a discussion of risk factors and the likelihood of occurrence.
Two QT interval prolonging medications used together in healthy individuals does not warrant a change in therapy. TdP is most likely to occur when 2 or more QT interval prolonging medications are used in a patient who is already at high risk for arrhythmia because of risk factors such as prolonged QT interval at baseline, family history of prolonged QT intervals, female gender, age >60 years, electrolyte abnormalities (hypokalemia, hypomagnesemia, hypocalcemia), underlying comorbid diseases (eg, chronic heart failure, left ventricular hypertrophy, atrial fibrillation), hypertension, bradycardia, and genetic (ion channel) polymorphisms.26,27
Antiarrhythmics and antipsychotics are most commonly associated with drug-induced prolonged QT interval, with most case reports and research being linked to antiarrhythmics (TABLE 42).28 But macrolide and fluoroquinolone antibiotics also have been associated with TdP, although to a lesser extent. In a retrospective analysis of case reports of TdP involving macrolides, erythromycin was present (with or without other medications thought to prolong QT) in 53% of the cases and clarithromycin was involved in 36% of the reports.29
An analysis of 2 studies by the US Food and Drug Administration estimated an occurrence rate of serious cardiac arrhythmias of 46 to 85 per 100,000 users with cardiovascular disease, compared to 5 to 44 per 100,000 users without cardiovascular disease.30 And this may underestimate the actual incidence because spontaneous reporting of adverse effects declines the longer a drug is on the market. Ciprofloxacin is associated with less risk than levofloxacin and gatifloxacin (the latter of which is no longer available in the United States).26
A recent population-based study using data on over 10.6 million people from the Taiwan National Health Insurance Database examined the risk of cardiovascular death among patients using new-generation macrolides, fluoroquinolones, and β-lactam/β-lactamase inhibitors.31 The absolute risk of cardiovascular death per 1000 individuals was 0.06 for clarithromycin, 0.12 for ciprofloxacin, 0.13 for amoxicillin-clavulanate, 0.36 for azithromycin, 0.39 for levofloxacin, and 0.46 for moxifloxacin. The mean interval between first antibiotic use and the adverse cardiac event was <4 days. Not surprisingly, the highest risk was seen in patients with underlying cardiovascular disease.
Another population-based study, this time conducted in Hong Kong, evaluated the cardiovascular safety of clarithromycin compared to that of amoxicillin. Clarithromycin was found to increase the incidence of myocardial infarction, arrhythmia, and cardiac mortality in the short term, with the risk returning to baseline after treatment concluded.32 A binational cohort study of Danish and Swedish adults confirmed that fluoroquinolones (especially ciprofloxacin) do not increase the risk of a serious arrhythmia compared to penicillins.33
THE BOTTOM LINE For patients taking other QT interval prolonging medications or who are at a higher risk for TdP, consider using clarithromycin over erythromycin or azithromycin for a macrolide antibiotic or ciprofloxacin over levofloxacin or moxifloxacin if a fluoroquinolone is warranted. Using 2 drugs that may increase the QT interval is likely safe in the absence of certain risk factors.
4. Should patients avoid alcohol while taking metronidazole?
Probably not.
Warning patients against drinking alcohol while taking metronidazole has been a common practice for years. The mechanism for this theorized interaction was thought to be similar to the interaction between disulfiram and ethanol.34 Disulfiram inhibits hepatic aldehyde dehydrogenase (ALDH) when combined with alcohol, which leads to increased levels of acetaldehyde in the blood and symptoms of flushing, palpitations, nausea, vomiting, headache, and visual disturbances.35 However, multiple studies using rats have found that metronidazole does not inhibit ALDH or increase acetaldehyde concentrations like disulfiram does.34
A 2000 review article discussed 6 cases involving serious metronidazole-ethanol interactions. Ethanol alone was found to explain the reaction in 2 of the cases, and the remaining 4 could be linked to the use of other drugs or disease states.35 A 2002 Finnish study found no statistically significant differences in objective or subjective signs of a disulfiram-like interaction.34 When considering the symptoms associated with the interaction, it is important to remember that many of the symptoms can result from metronidazole therapy alone, regardless of whether other medications or alcohol are used.35
THE BOTTOM LINE Researchers have failed to identify a clinically significant interaction between metronidazole and alcohol. Avoiding alcohol while taking metronidazole does not appear to be necessary.
CORRESPONDENCE
Mary Onysko, PharmD, BCPS, University of Wyoming, School of Pharmacy Health Sciences Center, Room 292, 1000 E. University Avenue, Laramie, WY 82071; [email protected].
› Avoid preemptive warfarin dose reductions unless you are prescribing trimethoprim/sulfamethoxazole (TMP/SMX) or metronidazole. B
› Recommend a back-up contraceptive method to a woman who is taking a broad-spectrum antibiotic and low-dose OCs—especially if the woman is overweight. C
› Consider using the macrolide, clarithromycin, or the fluoroquinolone, ciprofloxacin, in patients taking medications that prolong QT interval or who are at higher risk for torsades de pointes (TdP). B
› Refrain from cautioning patients taking metronidazole against consuming alcohol. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite encouraging data that antibiotic prescribing is on the decline, patients are still prescribed antibiotics frequently, making these agents the 12th most frequently used drug class.1 At the same time, prescribers are caring for patients with increasingly complex drug regimens that provide fertile ground for drug interactions with these antibiotics. And, of course, lifestyle factors such as alcohol consumption are a consideration when any prescription is written.
As pharmacists, we find that certain questions about antibiotic prescribing and interactions come up with frequency. These questions often pertain to the use of warfarin, oral contraceptives, drugs that prolong the QT interval, and alcohol. But conflicting reports about issues such as monitoring international normalized ratio (INR) in patients taking warfarin and antibiotics, and whether (or which) antibiotics decrease the efficacy of oral contraceptives (OCs) can make decision-making challenging.
This review provides evidence-based answers to questions you may have. It also details some reliable sources of information you can consult (TABLE 12-7) when discussing treatment options with other members of the health care team.
1. Which antibiotics are preferable when a patient is taking warfarin, and are preemptive warfarin dose reductions advisable?
The simple answer is that agents with a lower likelihood of affecting the INR, such as penicillin G, clindamycin, and 1st- and 4th-generation cephalosporins, are a good place to start, and whether to preemptively reduce the warfarin dose hinges on the antibiotic being prescribed.
The more detailed answer. The fundamental mechanisms of interaction between warfarin and antibiotics are two-fold:8
- Antimicrobial agents disrupt gastrointestinal flora that synthesize vitamin K.
- Antimicrobials inhibit cytochrome p450 (CYP450) enzymes (primarily CYP2C9 and 3A4), which are responsible for the metabolism of warfarin.
The antibiotics most likely to interfere with warfarin are TMP/SMX, ciprofloxacin, levofloxacin, metronidazole, fluconazole, azithromycin, and clarithromycin (TABLE 2).9,10 Low-risk agents include clindamycin, cephalexin, and penicillin G. When prescribing an antibiotic for a patient taking warfarin, it is important not only to be aware of the agents that should be avoided, but also the agents that do not require more frequent monitoring of INR.
Preemptive warfarin dose reductions? Some physicians make preemptive warfarin dose reductions in an attempt to avoid supratherapeutic INRs in patients being prescribed antibiotics. But the evidence suggests that this step should be considered only in the presence of the antibiotics TMP/SMX and metronidazole.9,11
A 2008 study investigated the anticoagulation effects of a 10% to 20% preemptive warfarin dose reduction vs no dosing change in patients taking TMP/SMX or levofloxacin. The investigators found that the preemptive warfarin dose reduction (intervention) significantly decreased the number of supratherapeutic INR values above 4 when compared to controls (2 of 8 vs 8 of 9).12
In the dose-reduction group, no patients receiving TMP/SMX developed a subtherapeutic INR, whereas 40% (4 of 10 patients) who received levofloxacin developed a subtherapeutic INR.12 The authors of the study concluded that a prophylactic warfarin dose reduction of 10% to 20% is effective in maintaining therapeutic anticoagulation in patients receiving TMP/SMX. They added that while no change in warfarin dosing is necessary with levofloxacin, short-term INR follow-up is a prudent approach to prevent subtherapeutic INRs. Others recommend INR monitoring when antibiotic therapy is started and stopped and whenever the dose is changed.9
A 2010 retrospective, single-center, cohort study looked at patients who were taking metronidazole and warfarin. Researchers compared those who received a preemptive dose reduction of warfarin (mean reduction was 34.6% ± 13.4%) to those who did not and found a statistically significant mean difference in INR of 1.28 (P=.01).13
Almost half (46%) of the patients who did not receive a warfarin dose reduction had an INR >4, whereas none of the patients in the warfarin dose reduction group did (P=.05). Although this secondary outcome was not statistically significant (most likely due to the small sample population [N=20]), the implication is clinically significant. Two patients who reduced their dose had a subtherapeutic INR compared to none of the patients in the control group, which was also not a statistically significant difference.
The authors concluded that a 30% to 35% reduction in mean daily warfarin dose is effective in maintaining therapeutic anticoagulation in patients started on metronidazole.
Significant bleeding events. A retrospective cohort study of slightly more than 22,000 veterans who were prescribed warfarin for ≥30 uninterrupted days and given antibiotics with either a high or low risk for interaction with warfarin were studied for significant bleeding events for one month.10 Ninety-three significant bleeding events occurred in the high-risk group and 36 occurred in the low-risk group over the course of the study. The agent associated with the greatest increased risk of bleeding was TMP/SMX (hazard ratio [HR]=2.09; 95% CI, 1.45-3.02). Of note, metronidazole was not included in this study endpoint.
The study’s secondary endpoint of INR >4 found that 10% of patients taking metronidazole and 8% of patients taking TMP/SMX in addition to warfarin had INRs >4. Almost 10% (9.7%) of patients prescribed fluconazole had a peak INR value >6. Patients taking low-risk antibiotics (clindamycin or cephalexin) had no increased risk of bleeding. Monitoring INR within 3 to 14 days of starting patients on antibiotics was found to decrease the risk of serious bleeding events (HR=0.61; 95% CI, 0.42-0.88). More frequent INR monitoring by itself (without preemptive warfarin dose reductions) is appropriate for other antibiotics, including macrolides, tetracyclines, and some cephalosporins (2nd and 3rd generation).9
THE BOTTOM LINE When prescribing antibiotics for patients taking warfarin, try to choose agents with a lower likelihood of affecting INR such as penicillin G, clindamycin, and 1st- and 4th-generation cephalosporins. With these agents, there is no need for more frequent INR testing or preemptive reductions in warfarin dose. In patients for whom the use of TMP/SMX or metronidazole can’t be avoided, consider reducing the patient’s warfarin dose by 10% to 35% and rechecking the INR 5 days after starting the antibiotic.9,11,12 When prescribing agents such as fluoroquinolones, macrolides, and tetracyclines, do not reduce the patient’s warfarin dose preemptively and recheck INR 5 days after starting therapy.
2. Do antibiotics decrease the efficacy of oral contraceptives?
It’s unlikely, but antibiotics may reduce the efficacy of OCs.
There have been few, but well documented, reports of women using OCs who became pregnant after taking antimicrobials.14 It is recognized that rifampin, an inducer of enzymes that metabolize estrogens, decreases the efficacy of OCs.15 Ketoconazole’s interaction seems less well documented, but combining the agent with low-estrogen (low-dose) OCs warrants caution.16 What is not well understood is whether more common or broad-spectrum antibiotics also increase the risk of OC failure.
Three mechanisms have been proposed:16
- Antimicrobials affect hepatic enzyme induction, which increases metabolism of hormones.
- Broad-spectrum antibiotics reduce gut bacteria, which alters enterohepatic circulation and reduces plasma hormone concentrations.
- Antibiotics increase gastrointestinal motility, which decreases absorption (and reabsorption) of OCs.
A 2007 study found that when physicians and pharmacists were surveyed and asked if broad-spectrum antibiotics have a clinically significant interaction with OCs, 83% of physicians and 89% of pharmacists answered “Yes;”17 however, a large epidemiologic study performed in the United States showed no association between antibiotic use and OC failure.18
After this report, investigators in the Netherlands completed a similar cross-over analysis and found that there was a relationship between the use of antibiotics and breakthrough pregnancy in a population-based prescription database, but that the results didn’t hold for broad-spectrum antibiotics or in a sensitivity analysis.19 Pharmacokinetic studies are also conflicting, as some have shown an effect on serum hormone levels, while others have not.15,20-22
High- vs low-risk agents. Ciprofloxacin did not affect hormone levels in 2 studies.20,21 Rifampin and voriconazole may enhance systemic exposure to OCs.15,22 And erythromycin and azithromycin may interact with OCs, but the clinical significance of this interaction is still unknown.16
Short-courses of TMP/SMX are generally thought to be safe;16 a small study looked at cotrimoxazole 1 g twice daily in 9 women taking long-term OC steroids and found that short courses of the drug were unlikely to cause any adverse effects on contraceptive control.23 Tetracyclines and penicillins were the antibiotics most frequently involved in case reports of pregnancy from the United Kingdom (TABLE 32).16
It is hypothesized that some women may have a higher risk of OC failure than others due to how they metabolize ethinyl estradiol.24 Another hypothesis is that some women have gut flora that is more susceptible to the antibiotic being used. And still another possibility is that lower doses of hormones are being used in OCs than were studied for this interaction.15 Anything that decreases the concentration of these lower-dose OCs is concerning, especially in patients with a higher body mass index (BMI). The few pharmacokinetic studies that have been conducted show that it takes longer for OCs to reach a steady state in obese women and that they have a lower area under the curve (AUC) and maximum estrogen concentration than women with a normal BMI.25
THE BOTTOM LINE Because the degree of variability between patients is unknown and obesity rates are increasing, concern that low-dose OCs may lose efficacy when combined with antibiotics is warranted. While the absolute risk of breakthrough pregnancy seems small, the most conservative approach is to advise patients to use a back-up method of contraception during times of antibiotic use.
3. Which drugs prolong QT intervals?
Macrolides and fluoroquinolones are 2 classes of antibiotics associated with prolonged QT intervals, but other drugs and risk factors are important to consider, as well.
Physicians often receive phone calls from pharmacists warning about drug-drug interactions when they prescribe macrolides or fluoroquinolones for patients already taking medications known to prolong QT intervals or inhibit cytochrome P450 enzymes. Long QT syndrome increases the risk of TdP, a life-threatening arrhythmia. While TdP is rare, its severity warrants a discussion of risk factors and the likelihood of occurrence.
Two QT interval prolonging medications used together in healthy individuals does not warrant a change in therapy. TdP is most likely to occur when 2 or more QT interval prolonging medications are used in a patient who is already at high risk for arrhythmia because of risk factors such as prolonged QT interval at baseline, family history of prolonged QT intervals, female gender, age >60 years, electrolyte abnormalities (hypokalemia, hypomagnesemia, hypocalcemia), underlying comorbid diseases (eg, chronic heart failure, left ventricular hypertrophy, atrial fibrillation), hypertension, bradycardia, and genetic (ion channel) polymorphisms.26,27
Antiarrhythmics and antipsychotics are most commonly associated with drug-induced prolonged QT interval, with most case reports and research being linked to antiarrhythmics (TABLE 42).28 But macrolide and fluoroquinolone antibiotics also have been associated with TdP, although to a lesser extent. In a retrospective analysis of case reports of TdP involving macrolides, erythromycin was present (with or without other medications thought to prolong QT) in 53% of the cases and clarithromycin was involved in 36% of the reports.29
An analysis of 2 studies by the US Food and Drug Administration estimated an occurrence rate of serious cardiac arrhythmias of 46 to 85 per 100,000 users with cardiovascular disease, compared to 5 to 44 per 100,000 users without cardiovascular disease.30 And this may underestimate the actual incidence because spontaneous reporting of adverse effects declines the longer a drug is on the market. Ciprofloxacin is associated with less risk than levofloxacin and gatifloxacin (the latter of which is no longer available in the United States).26
A recent population-based study using data on over 10.6 million people from the Taiwan National Health Insurance Database examined the risk of cardiovascular death among patients using new-generation macrolides, fluoroquinolones, and β-lactam/β-lactamase inhibitors.31 The absolute risk of cardiovascular death per 1000 individuals was 0.06 for clarithromycin, 0.12 for ciprofloxacin, 0.13 for amoxicillin-clavulanate, 0.36 for azithromycin, 0.39 for levofloxacin, and 0.46 for moxifloxacin. The mean interval between first antibiotic use and the adverse cardiac event was <4 days. Not surprisingly, the highest risk was seen in patients with underlying cardiovascular disease.
Another population-based study, this time conducted in Hong Kong, evaluated the cardiovascular safety of clarithromycin compared to that of amoxicillin. Clarithromycin was found to increase the incidence of myocardial infarction, arrhythmia, and cardiac mortality in the short term, with the risk returning to baseline after treatment concluded.32 A binational cohort study of Danish and Swedish adults confirmed that fluoroquinolones (especially ciprofloxacin) do not increase the risk of a serious arrhythmia compared to penicillins.33
THE BOTTOM LINE For patients taking other QT interval prolonging medications or who are at a higher risk for TdP, consider using clarithromycin over erythromycin or azithromycin for a macrolide antibiotic or ciprofloxacin over levofloxacin or moxifloxacin if a fluoroquinolone is warranted. Using 2 drugs that may increase the QT interval is likely safe in the absence of certain risk factors.
4. Should patients avoid alcohol while taking metronidazole?
Probably not.
Warning patients against drinking alcohol while taking metronidazole has been a common practice for years. The mechanism for this theorized interaction was thought to be similar to the interaction between disulfiram and ethanol.34 Disulfiram inhibits hepatic aldehyde dehydrogenase (ALDH) when combined with alcohol, which leads to increased levels of acetaldehyde in the blood and symptoms of flushing, palpitations, nausea, vomiting, headache, and visual disturbances.35 However, multiple studies using rats have found that metronidazole does not inhibit ALDH or increase acetaldehyde concentrations like disulfiram does.34
A 2000 review article discussed 6 cases involving serious metronidazole-ethanol interactions. Ethanol alone was found to explain the reaction in 2 of the cases, and the remaining 4 could be linked to the use of other drugs or disease states.35 A 2002 Finnish study found no statistically significant differences in objective or subjective signs of a disulfiram-like interaction.34 When considering the symptoms associated with the interaction, it is important to remember that many of the symptoms can result from metronidazole therapy alone, regardless of whether other medications or alcohol are used.35
THE BOTTOM LINE Researchers have failed to identify a clinically significant interaction between metronidazole and alcohol. Avoiding alcohol while taking metronidazole does not appear to be necessary.
CORRESPONDENCE
Mary Onysko, PharmD, BCPS, University of Wyoming, School of Pharmacy Health Sciences Center, Room 292, 1000 E. University Avenue, Laramie, WY 82071; [email protected].
1. Kantor ED, Rehm CD, Haas JS, et al. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314:1818-1831.
2. Lexicomp Online. Clinical Drug Information. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed December 8, 2015.
3. GlobalRPh: The Clinician’s Ultimate Reference. Available at: http://www.globalrph.com/. Accessed December 8, 2015.
4. Medscape Apps. Available at: http://www.medscape.com/public/applanding. Accessed December 8, 2015.
5. Micromedex Solutions. Academic Institutions. Available at: http://micromedex.com/academic. Accessed December 8, 2015.
6. Patel A. Clinical Pharmacology Mobile-A mobile web app compatible on all smart phones [review] May 31, 2010. Available at: http://www.imedicalapps.com/2010/05/clinical-pharmocology-app-review/. Accessed December 8, 2015.
7. Epocrates. Available at: http://www.epocrates.com/. Accessed December 8, 2015.
8. Baillargeon J, Holmes HM, Lin Y, et al. Concurrent use of warfarin and antibiotics and the risk of bleeding in older adults. Am J Med. 2012;125:183-189.
9. PL Detail-Document #280806. Antimicrobial drug interactions with warfarin. Pharmacist’s Letter/Prescriber’s Letter. August 2012.
10. Lane M, Zeringue A, McDonald J. Serious bleeding events due to warfarin and antibiotic co-prescription in a cohort of veterans. Am J Med. 2014;127:657-663.e2.
11. Hale SF, Lesar TS. Interaction of vitamin K antagonists and trimethoprim-sulfamethoxazole: ignore at your patient’s risk. Drug Metab Drug Interact. 2014;29:53-60.
12. Ahmed A, Stephens JC, Kaus CA, et al. Impact of preemptive warfarin dose reduction on anticoagulation after initiation of trimethoprim-sulfamethoxazole or levofloxacin. J Thromb Thrombolysis. 2008;26:44-48.
13. Holt RK, Anderson EA, Cantrell MA, et al. Preemptive dose reduction of warfarin in patients initiating metronidazole. Drug Metabol Drug Interact. 2010;25:35-39.
14. Hughes BR, Cunliffe WJ. Interactions between the oral contraceptive pill and antibiotics. Br J Dermatol. 1990;122:717-718.
15. Bolt HM. Interactions between clinically used drugs and oral contraceptives. Environ Health Perspect. 1994;102:35-38.
16. Aronson JK. Meyler’s Side Effects of Drugs. 16th ed. The International Encyclopedia of Adverse Drug Reactions and Interactions. Amsterdam, Netherlands: Elsevier; 2016. Available at: http://ac.els-cdn.com/B978044453717101009X/3-s2.0-B978044453717101009X-main.pdf?_tid=b33f6564-9deb-11e5-a8f0-00000aab0f01&acdnat=1449607315_83f5068fc5105226fcc6d7279c083516. Accessed December 8, 2015.
17. Masters KP, Carr BM. Survey of pharmacists and physicians on drug interactions between combined oral contraceptives and broad-spectrum antibiotics. Pharm Pract (Granada). 2009;7:139-144.
18. Toh S, Mitchell AA, Anderka M, et al; National Birth Defects Prevention Study. Antibiotics and oral contraceptive failure—a case-crossover study. Contraception. 2011;83:418-425.
19. Koopmans PC, Bos JH, de Jong van den Berg LT. Are antibiotics related to oral combination contraceptive failures in the Netherlands? A case-crossover study. Pharmacoepidemiol Drug Saf. 2012;21:865-871.
20. Archer JS, Archer DF. Oral contraceptive efficacy and antibiotic interaction: A myth debunked. J Am Acad Dermatol. 2002;46:917–923.
21. Scholten PC, Droppert RM, Zwinkels MGJ, et al. No interaction between ciprofloxacin and an oral contraceptive. Antimicrob Agents Chemother. 1998;42:3266-3268.
22. Andrews E, Damle BD, Fang A, et al. Pharmacokinetics and tolerability of voriconazole and a combination oral contraceptive co-administered in healthy female subjects. Br J Clin Pharmacol. 2008;65:531-539.
23. Grimmer SF, Allen WL, Back DJ, et al. The effect of cotrimoxazole on oral contraceptive steroids in women. Contraception. 1983;28:53-59.
24. Dickinson BD, Altman RD, Nielsen NH, et al; Council on Scientific Affairs, American Medical Association. Drug interactions between oral contraceptives and antibiotics. Obstet Gynecol. 2001;98:853-860.
25. Edelman AB, Cherala G, Stanczyk FZ. Metabolism and pharmacokinetics of contraceptive steroids in obese women: a review. Contraception. 2010;82:314-323.
26. Owens RC Jr, Ambrose PG. Torsades de pointes associated with fluoroquinolones. Pharmacotherapy. 2002;22:663-668.
27. Letsas KP, Efremidis M, Kounas SP, et al. Clinical characteristics of patients with drug-induced QT interval prolongation and torsade de pointes: identification of risk factors. Clin Res Cardiol. 2009;98:208-212.
28. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363-1372.
29. Shaffer D, Singer S, Korvick J, et al. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration adverse event reporting system. Clin Infect Dis. 2002;35:197-200.
30. FDA Briefing Document. Joint Meeting of the Antimicrobial Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee. November 5, 2015. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM467383.pdf. Accessed June 11, 2016.
31. Chou HW, Wang JL, Chang CH, et al. Risks of cardiac arrhythmia and mortality among patients using new-generation macrolides, fluoroquinolones, and β-lactam/β-lactamase inhibitors: a Taiwanese nationwide study. Clin Infect Dis. 2015;60:566-577.
32. Wong AY, Root A, Douglas IJ, et al. Cardiovascular outcomes associated with use of clarithromycin: population based study. BMJ. 2016;352:h6926.
33. Inghammar M, Svanström H, Melbye M, et al. Oral fluoroquinolone use and serious arrhythmia: bi-national cohort study. BMJ. 2016;352:i843.
34. Visapää JP, Tillonen JS, Kaihovaara PS, et al. Lack of disulfiram-like reaction with metronidazole and ethanol. Ann Pharmacother. 2002;36:971-974. 35. Fjeld H, Raknes G. Is combining metronidazole and alcohol really hazardous? Tidsskr Nor Laegeforen. 2014;134:1661-1663.
1. Kantor ED, Rehm CD, Haas JS, et al. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314:1818-1831.
2. Lexicomp Online. Clinical Drug Information. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed December 8, 2015.
3. GlobalRPh: The Clinician’s Ultimate Reference. Available at: http://www.globalrph.com/. Accessed December 8, 2015.
4. Medscape Apps. Available at: http://www.medscape.com/public/applanding. Accessed December 8, 2015.
5. Micromedex Solutions. Academic Institutions. Available at: http://micromedex.com/academic. Accessed December 8, 2015.
6. Patel A. Clinical Pharmacology Mobile-A mobile web app compatible on all smart phones [review] May 31, 2010. Available at: http://www.imedicalapps.com/2010/05/clinical-pharmocology-app-review/. Accessed December 8, 2015.
7. Epocrates. Available at: http://www.epocrates.com/. Accessed December 8, 2015.
8. Baillargeon J, Holmes HM, Lin Y, et al. Concurrent use of warfarin and antibiotics and the risk of bleeding in older adults. Am J Med. 2012;125:183-189.
9. PL Detail-Document #280806. Antimicrobial drug interactions with warfarin. Pharmacist’s Letter/Prescriber’s Letter. August 2012.
10. Lane M, Zeringue A, McDonald J. Serious bleeding events due to warfarin and antibiotic co-prescription in a cohort of veterans. Am J Med. 2014;127:657-663.e2.
11. Hale SF, Lesar TS. Interaction of vitamin K antagonists and trimethoprim-sulfamethoxazole: ignore at your patient’s risk. Drug Metab Drug Interact. 2014;29:53-60.
12. Ahmed A, Stephens JC, Kaus CA, et al. Impact of preemptive warfarin dose reduction on anticoagulation after initiation of trimethoprim-sulfamethoxazole or levofloxacin. J Thromb Thrombolysis. 2008;26:44-48.
13. Holt RK, Anderson EA, Cantrell MA, et al. Preemptive dose reduction of warfarin in patients initiating metronidazole. Drug Metabol Drug Interact. 2010;25:35-39.
14. Hughes BR, Cunliffe WJ. Interactions between the oral contraceptive pill and antibiotics. Br J Dermatol. 1990;122:717-718.
15. Bolt HM. Interactions between clinically used drugs and oral contraceptives. Environ Health Perspect. 1994;102:35-38.
16. Aronson JK. Meyler’s Side Effects of Drugs. 16th ed. The International Encyclopedia of Adverse Drug Reactions and Interactions. Amsterdam, Netherlands: Elsevier; 2016. Available at: http://ac.els-cdn.com/B978044453717101009X/3-s2.0-B978044453717101009X-main.pdf?_tid=b33f6564-9deb-11e5-a8f0-00000aab0f01&acdnat=1449607315_83f5068fc5105226fcc6d7279c083516. Accessed December 8, 2015.
17. Masters KP, Carr BM. Survey of pharmacists and physicians on drug interactions between combined oral contraceptives and broad-spectrum antibiotics. Pharm Pract (Granada). 2009;7:139-144.
18. Toh S, Mitchell AA, Anderka M, et al; National Birth Defects Prevention Study. Antibiotics and oral contraceptive failure—a case-crossover study. Contraception. 2011;83:418-425.
19. Koopmans PC, Bos JH, de Jong van den Berg LT. Are antibiotics related to oral combination contraceptive failures in the Netherlands? A case-crossover study. Pharmacoepidemiol Drug Saf. 2012;21:865-871.
20. Archer JS, Archer DF. Oral contraceptive efficacy and antibiotic interaction: A myth debunked. J Am Acad Dermatol. 2002;46:917–923.
21. Scholten PC, Droppert RM, Zwinkels MGJ, et al. No interaction between ciprofloxacin and an oral contraceptive. Antimicrob Agents Chemother. 1998;42:3266-3268.
22. Andrews E, Damle BD, Fang A, et al. Pharmacokinetics and tolerability of voriconazole and a combination oral contraceptive co-administered in healthy female subjects. Br J Clin Pharmacol. 2008;65:531-539.
23. Grimmer SF, Allen WL, Back DJ, et al. The effect of cotrimoxazole on oral contraceptive steroids in women. Contraception. 1983;28:53-59.
24. Dickinson BD, Altman RD, Nielsen NH, et al; Council on Scientific Affairs, American Medical Association. Drug interactions between oral contraceptives and antibiotics. Obstet Gynecol. 2001;98:853-860.
25. Edelman AB, Cherala G, Stanczyk FZ. Metabolism and pharmacokinetics of contraceptive steroids in obese women: a review. Contraception. 2010;82:314-323.
26. Owens RC Jr, Ambrose PG. Torsades de pointes associated with fluoroquinolones. Pharmacotherapy. 2002;22:663-668.
27. Letsas KP, Efremidis M, Kounas SP, et al. Clinical characteristics of patients with drug-induced QT interval prolongation and torsade de pointes: identification of risk factors. Clin Res Cardiol. 2009;98:208-212.
28. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363-1372.
29. Shaffer D, Singer S, Korvick J, et al. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration adverse event reporting system. Clin Infect Dis. 2002;35:197-200.
30. FDA Briefing Document. Joint Meeting of the Antimicrobial Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee. November 5, 2015. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM467383.pdf. Accessed June 11, 2016.
31. Chou HW, Wang JL, Chang CH, et al. Risks of cardiac arrhythmia and mortality among patients using new-generation macrolides, fluoroquinolones, and β-lactam/β-lactamase inhibitors: a Taiwanese nationwide study. Clin Infect Dis. 2015;60:566-577.
32. Wong AY, Root A, Douglas IJ, et al. Cardiovascular outcomes associated with use of clarithromycin: population based study. BMJ. 2016;352:h6926.
33. Inghammar M, Svanström H, Melbye M, et al. Oral fluoroquinolone use and serious arrhythmia: bi-national cohort study. BMJ. 2016;352:i843.
34. Visapää JP, Tillonen JS, Kaihovaara PS, et al. Lack of disulfiram-like reaction with metronidazole and ethanol. Ann Pharmacother. 2002;36:971-974. 35. Fjeld H, Raknes G. Is combining metronidazole and alcohol really hazardous? Tidsskr Nor Laegeforen. 2014;134:1661-1663.
Liquid Levodopa Formulation May Simplify Continuous Drug Delivery
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.

Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.

Liquid Levodopa Formulation May Simplify Continuous Drug Delivery
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Sheila Oren, MD, MBA
Until recently, poor levodopa solubility had prevented the development of a liquid formulation of the drug that could be delivered subcutaneously. ND0612L, a novel, proprietary formulation of levodopa and carbidopa, “is the first time that this [drug] is a real liquid,” Dr. Oren said. “Not a suspension or a gel, but a liquid that can be administered parenterally.… In this way, we hope to be able to deliver levodopa continuously in a much simpler way.”
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Sheila Oren, MD, MBA
Until recently, poor levodopa solubility had prevented the development of a liquid formulation of the drug that could be delivered subcutaneously. ND0612L, a novel, proprietary formulation of levodopa and carbidopa, “is the first time that this [drug] is a real liquid,” Dr. Oren said. “Not a suspension or a gel, but a liquid that can be administered parenterally.… In this way, we hope to be able to deliver levodopa continuously in a much simpler way.”
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Sheila Oren, MD, MBA
Until recently, poor levodopa solubility had prevented the development of a liquid formulation of the drug that could be delivered subcutaneously. ND0612L, a novel, proprietary formulation of levodopa and carbidopa, “is the first time that this [drug] is a real liquid,” Dr. Oren said. “Not a suspension or a gel, but a liquid that can be administered parenterally.… In this way, we hope to be able to deliver levodopa continuously in a much simpler way.”
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.

Using a Combination of Therapies to Manage Rosacea

What do your patients need to know at the first visit?
Patients need to understand that rosacea has no gender, age, or race predilection. It is caused by a personal and genetic proinflammatory predisposition. Rosacea patients seem to have a genetic predisposition to overproduce cathelicidins, a small antimicrobial peptide that is produced by the action of stratum corneum tryptic enzyme. They have more production and abnormal forms of cathelicidins produced by high levels of stratum corneum tryptic enzyme. This upregulation of inflammatory cathelicidins on the dermis is associated with vascular instability and exacerbated by triggers such as sunlight, hot drinks, spicy foods, stress, and rapid changing weather.
I divide the condition into proinflammatory predisposition, vascular instability with redness, Demodex infection of the hair follicle, and sebaceous gland overgrowth. More recently, the bacterium Bacillus oleronius was isolated from inside a Demodex mite and was found to produce molecules provoking an immune reaction in rosacea patients (Erbaguci and Ozgöztaşi). Other studies have shown that patients with varying types of rosacea react to the molecules produced by this bacterium, exposing it as a likely trigger for the condition (Li et al). What’s more, this bacterium is sensitive to the antibiotics used to treat rosacea.
What are your go-to treatments? What are the side effects?
For inflammation I prescribe anti-inflammatory (low dose) or antibacterial (high dose) doses of doxycycline and/or anti-inflammatory azelaic acid gel 15% twice daily after application of barrier repair topical hyaluronic acid. For the vascular component I use the temporary relief from the application of brimonidine gel 0.33% in the morning in addition to the topical given for inflammatory rosacea, and the more durable excel V (532 and 1064 nm) laser. Ultimately, topical ivermectin is prescribed for those patients who do not respond to previously mentioned treatments for coverage of Demodex infestation. For rhinophyma I offer a surgical approach and laser treatments; surgical removal of the excess glandular growth is followed by fractional ablative and nonablative treatments for scar reduction after surgery.
All patients should apply an inorganic sun protection factor 50+ sunblock with titanium dioxide and zinc oxide to prevent sunlight from being a trigger. All patients are encouraged to avoid triggers.
I try to prevent the potential side effects associated with rosacea treatments. For example, applying barrier repair hyaluronic acid before azelaic acid to prevent irritation and telling patients they might have vascular rebound phenomena with more redness after brimonidine application wears off. I also explain to patients that laser treatments induce temporary erythema and swelling that may last 3 days.
How do you keep patients compliant with treatment?
In general, my patients are compliant with their treatments, which I ascribe to the simplicity of a twice-daily regimen that is written for them. They understand that I design a treatment regimen for each individual patient based on his/her presentation.
What resources do you recommend to patients for more information?
I recommend web-based resources that can provide further assistance and information, such as the American Academy of Dermatology website (https://www.aad.org/public/diseases/acne-and-rosacea/rosacea), National Rosacea Society (www.rosacea.org), and specific disease foundations (eg, International Rosacea Foundation [www.internationalrosaceafoundation.org]).Suggested Readings
- Erbaguci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425.
- Li J, O’Reilly N, Sheha H, et al. Correlation between ocular Demodex infestation and serum immunoreactivity to Bacillus proteins in patients with facial rosacea. Ophthalmology. 2010;117:870-877.
What do your patients need to know at the first visit?
Patients need to understand that rosacea has no gender, age, or race predilection. It is caused by a personal and genetic proinflammatory predisposition. Rosacea patients seem to have a genetic predisposition to overproduce cathelicidins, a small antimicrobial peptide that is produced by the action of stratum corneum tryptic enzyme. They have more production and abnormal forms of cathelicidins produced by high levels of stratum corneum tryptic enzyme. This upregulation of inflammatory cathelicidins on the dermis is associated with vascular instability and exacerbated by triggers such as sunlight, hot drinks, spicy foods, stress, and rapid changing weather.
I divide the condition into proinflammatory predisposition, vascular instability with redness, Demodex infection of the hair follicle, and sebaceous gland overgrowth. More recently, the bacterium Bacillus oleronius was isolated from inside a Demodex mite and was found to produce molecules provoking an immune reaction in rosacea patients (Erbaguci and Ozgöztaşi). Other studies have shown that patients with varying types of rosacea react to the molecules produced by this bacterium, exposing it as a likely trigger for the condition (Li et al). What’s more, this bacterium is sensitive to the antibiotics used to treat rosacea.
What are your go-to treatments? What are the side effects?
For inflammation I prescribe anti-inflammatory (low dose) or antibacterial (high dose) doses of doxycycline and/or anti-inflammatory azelaic acid gel 15% twice daily after application of barrier repair topical hyaluronic acid. For the vascular component I use the temporary relief from the application of brimonidine gel 0.33% in the morning in addition to the topical given for inflammatory rosacea, and the more durable excel V (532 and 1064 nm) laser. Ultimately, topical ivermectin is prescribed for those patients who do not respond to previously mentioned treatments for coverage of Demodex infestation. For rhinophyma I offer a surgical approach and laser treatments; surgical removal of the excess glandular growth is followed by fractional ablative and nonablative treatments for scar reduction after surgery.
All patients should apply an inorganic sun protection factor 50+ sunblock with titanium dioxide and zinc oxide to prevent sunlight from being a trigger. All patients are encouraged to avoid triggers.
I try to prevent the potential side effects associated with rosacea treatments. For example, applying barrier repair hyaluronic acid before azelaic acid to prevent irritation and telling patients they might have vascular rebound phenomena with more redness after brimonidine application wears off. I also explain to patients that laser treatments induce temporary erythema and swelling that may last 3 days.
How do you keep patients compliant with treatment?
In general, my patients are compliant with their treatments, which I ascribe to the simplicity of a twice-daily regimen that is written for them. They understand that I design a treatment regimen for each individual patient based on his/her presentation.
What resources do you recommend to patients for more information?
I recommend web-based resources that can provide further assistance and information, such as the American Academy of Dermatology website (https://www.aad.org/public/diseases/acne-and-rosacea/rosacea), National Rosacea Society (www.rosacea.org), and specific disease foundations (eg, International Rosacea Foundation [www.internationalrosaceafoundation.org]).Suggested Readings
- Erbaguci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425.
- Li J, O’Reilly N, Sheha H, et al. Correlation between ocular Demodex infestation and serum immunoreactivity to Bacillus proteins in patients with facial rosacea. Ophthalmology. 2010;117:870-877.
What do your patients need to know at the first visit?
Patients need to understand that rosacea has no gender, age, or race predilection. It is caused by a personal and genetic proinflammatory predisposition. Rosacea patients seem to have a genetic predisposition to overproduce cathelicidins, a small antimicrobial peptide that is produced by the action of stratum corneum tryptic enzyme. They have more production and abnormal forms of cathelicidins produced by high levels of stratum corneum tryptic enzyme. This upregulation of inflammatory cathelicidins on the dermis is associated with vascular instability and exacerbated by triggers such as sunlight, hot drinks, spicy foods, stress, and rapid changing weather.
I divide the condition into proinflammatory predisposition, vascular instability with redness, Demodex infection of the hair follicle, and sebaceous gland overgrowth. More recently, the bacterium Bacillus oleronius was isolated from inside a Demodex mite and was found to produce molecules provoking an immune reaction in rosacea patients (Erbaguci and Ozgöztaşi). Other studies have shown that patients with varying types of rosacea react to the molecules produced by this bacterium, exposing it as a likely trigger for the condition (Li et al). What’s more, this bacterium is sensitive to the antibiotics used to treat rosacea.
What are your go-to treatments? What are the side effects?
For inflammation I prescribe anti-inflammatory (low dose) or antibacterial (high dose) doses of doxycycline and/or anti-inflammatory azelaic acid gel 15% twice daily after application of barrier repair topical hyaluronic acid. For the vascular component I use the temporary relief from the application of brimonidine gel 0.33% in the morning in addition to the topical given for inflammatory rosacea, and the more durable excel V (532 and 1064 nm) laser. Ultimately, topical ivermectin is prescribed for those patients who do not respond to previously mentioned treatments for coverage of Demodex infestation. For rhinophyma I offer a surgical approach and laser treatments; surgical removal of the excess glandular growth is followed by fractional ablative and nonablative treatments for scar reduction after surgery.
All patients should apply an inorganic sun protection factor 50+ sunblock with titanium dioxide and zinc oxide to prevent sunlight from being a trigger. All patients are encouraged to avoid triggers.
I try to prevent the potential side effects associated with rosacea treatments. For example, applying barrier repair hyaluronic acid before azelaic acid to prevent irritation and telling patients they might have vascular rebound phenomena with more redness after brimonidine application wears off. I also explain to patients that laser treatments induce temporary erythema and swelling that may last 3 days.
How do you keep patients compliant with treatment?
In general, my patients are compliant with their treatments, which I ascribe to the simplicity of a twice-daily regimen that is written for them. They understand that I design a treatment regimen for each individual patient based on his/her presentation.
What resources do you recommend to patients for more information?
I recommend web-based resources that can provide further assistance and information, such as the American Academy of Dermatology website (https://www.aad.org/public/diseases/acne-and-rosacea/rosacea), National Rosacea Society (www.rosacea.org), and specific disease foundations (eg, International Rosacea Foundation [www.internationalrosaceafoundation.org]).Suggested Readings
- Erbaguci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425.
- Li J, O’Reilly N, Sheha H, et al. Correlation between ocular Demodex infestation and serum immunoreactivity to Bacillus proteins in patients with facial rosacea. Ophthalmology. 2010;117:870-877.

NSAID plus TNFi linked to less ankylosing spondylitis progression
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.

These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
AT THE EULAR 2016 CONGRESS
Key clinical point: Ankylosing spondylitis patients treated with a TNF inhibitor plus an NSAID had the lowest level of new bone formation during treatment for more than 2 years.
Major finding: Patients on a TNF inhibitor and an NSAID had 67% less new bone formation, compared with patients not on an NSAID.
Data source: Cross-sectional cohort study of 527 patients with ankylosing spondylitis enrolled in the Prospective Study of Outcomes in Ankylosing Spondylitis.
Disclosures: Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.

NSAID plus TNFi linked to less ankylosing spondylitis progression
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
AT THE EULAR 2016 CONGRESS
Key clinical point: Ankylosing spondylitis patients treated with a TNF inhibitor plus an NSAID had the lowest level of new bone formation during treatment for more than 2 years.
Major finding: Patients on a TNF inhibitor and an NSAID had 67% less new bone formation, compared with patients not on an NSAID.
Data source: Cross-sectional cohort study of 527 patients with ankylosing spondylitis enrolled in the Prospective Study of Outcomes in Ankylosing Spondylitis.
Disclosures: Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.

Should Medical Marijuana and Cannabinoids Be Used to Treat Epilepsy?
VANCOUVER—“Neurologists are often asked to ‘certify’ patients, but should you recommend or support medical marijuana use for patients with epilepsy?”
Daniel Friedman, MD, MSc, Assistant Professor of Neurology at New York University Langone School of Medicine in New York, addressed this question in his presentation at the 68th Annual Meeting of the American Academy of Neurology.

“There is a disconnect between what the patient, families, and the public understand about cannabinoid therapies for epilepsy and what we as a neurologic community know,” he said. “Much of human evidence for the efficacy of cannabinoids for the treatment of epilepsy is anecdotal” and, of the “few controlled trials, most are small and methodologically flawed.”
A Brief History of Medical Marijuana
Cannabis sativa was first cultivated in approximately 8,000 BCE in China for rope, then medicinally for conditions ranging from menstruation to absentmindedness and, eventually, more than 100 ailments. In the early 19th century, W.B. O’Shaughnessy introduced cannabis to England, where Gowers and other Victorian neurologists used the “Indian hemp” to treat epilepsy.
Medicinal use of cannabis dwindled in the 1930s due to a move to synthetic medicine and Prohibition, which also “coincided with the beginning of the era of rigorous scientific investigation of drug claims,” said Dr. Friedman. Now, “the discovery of the endocannabinoid system in the brain has sparked new research into the therapeutic potential of cannabinoids.”
The Chemicals in Cannabis
The genus Cannabis includes C. sativa, C. indica, and C. ruderalis. A total of 85 phytocannabinoids are found in Cannabis species plants, and cannabidiol (CBD) and delta-9 tetrahydrocannabinol (THC) are the most abundant neuroactive chemicals; others include the terpenes and flavonoids.
CBD-rich oil is a concentrate made from cannabis bred to have a low level of THC and a high level of CBD. GW Pharma manufactures a solution with equal parts purified plant-derived THC and CBD (Sativex). The company also makes an investigational agent, pure CBD (Epidiolex), a 98% purified plant-derived CBD. INSYS Therapeutics is developing a synthetic CBD that is structurally identical to the plant-derived compound.
What Is the Evidence?
Experimental and human studies have provided evidence for the anticonvulsant properties of CBD and THC, and antiseizure effects have also been observed with cannabidivarin (CBDV).
Since 2014, more than 10 US centers have been using Epidiolex under an FDA expanded-access program to treat patients between ages 1 and 30 (median age, 10.5) with severe, treatment-resistant, childhood-onset epilepsy. Patients’ baseline motor seizure rate was 29.5 per month (range, 11.0–96.0 per month).
As of January 2015, 162 patients had completed 12 weeks of observation. Preliminary results showed median motor seizure reduction to be 36.5%. Patients with Dravet syndrome had a 49.8% reduction in seizures, and patients with Lennox-Gastaut syndrome had a 36.8% reduction. Serious adverse events, primarily status epilepticus, were observed in 12% of participants. Only 3% of patients discontinued treatment, however. Commonly reported adverse events included somnolence (25.3%), decreased appetite (19.1%), diarrhea (19.1%), fatigue (13.0%), and convulsion (11.1%).
In addition, children with Dravet syndrome in a phase III trial of Epidiolex had a median reduction in motor seizures of 39% versus 13% for placebo.
“Much of the safety data for chronic cannabinoid use” has come from studies of recreational use, which is “inherently confounded,” Dr. Friedman said. Acute affects include impaired memory, judgment, and motor performance. Chronic use leads to addiction in approximately 9% of users, as well as to cognitive impairment, decreased motivation, and increased risk for psychotic disorders.
Researchers examined pooled data from adults with multiple sclerosis who used cannabinoids for spasticity, pain, and dyskinesias for less than six months. The data encompassed approximately 1,600 exposures to the drugs. About 7% of participants discontinued treatment due to adverse events that included nausea, behavioral and mood changes, suicidality, hallucinations, dizziness, and weakness.
CBD and THC are inhibitors of P450 isozymes, primarily CYP2C and CYP3A, but drug–drug interaction effects are not typically seen in doses used in human studies, said Dr. Friedman. At low micromolar concentrations, CBD inhibits CYP2C19, an enzyme involved in the metabolism of N-desmethylclobazam, phenytoin, diazepam, citalopram, and some tricyclic antidepressants. The CYP2B family may also be induced, thus affecting valproate and clobazam metabolism.
Drugs that affect CBD metabolism include the CYP3A4 inducers, such as carbamazepine and phenytoin, and the CYP3A4 inhibitors such as ketoconazole.
Randomized controlled trials of CBD and CBDV are in progress in patients with Dravet syndrome, Lennox-Gastaut syndrome, and refractory focal epilepsy.
Is Cannabis Right for My Patient?
Since New York State approved the medical use of cannabis, Dr. Friedman has discussed the drug in one of every four of his clinic visits. Deciding whether to recommend medical marijuana for a patient can be difficult, however.
“Would you authorize its use for a 22-year-old patient with treatment-resistant epilepsy who is not a candidate for epilepsy surgery, is having two complex partial seizures a week and one tonic-clonic seizure per month, and is currently on three antiepileptic drugs [AEDs] at high doses,” asked Dr. Friedman. “How about for a 63-year-old woman with focal motor seizures following a meningioma resection who has not been able to tolerate adequate doses of four prior AEDs?”
The answer depends on myriad factors, not the least of which is the legal status of medical marijuana in the neurologist’s state. Although “23 states and the District of Columbia have approved medical marijuana for certain conditions, including epilepsy,” the US Drug Enforcement Administration still considers cannabis and its derivatives schedule I compounds, which means that they have “no accepted medical use in the US,” a high abuse potential, and cannot be prescribed—only “recommended”—by a physician, saidDr. Friedman.
Patients should be evaluated at a comprehensive epilepsy center to determine whether they have been unable to achieve control with conventional therapies due to lack of efficacy or side effects, and whether other proven effective therapies, such as vagus nerve stimulation, a ketogenic diet, or surgery, have been considered, he added.
For patients who do initiate treatment with medical marijuana, the risks and benefits should be carefully weighed, and a treatment plan that includes a timeline for discontinuation should be developed. Laboratory values and clinical status should be monitored regularly.
“Perhaps one day we’ll have [cannabis] in the pharmacy,” Dr. Freidman concluded. Until then, patients should be cautioned against buying cannabinoids on the Internet because, as one FDA study showed, products bought from such sources may have no detectable cannabinoid levels.
—Debra Hughes
VANCOUVER—“Neurologists are often asked to ‘certify’ patients, but should you recommend or support medical marijuana use for patients with epilepsy?”
Daniel Friedman, MD, MSc, Assistant Professor of Neurology at New York University Langone School of Medicine in New York, addressed this question in his presentation at the 68th Annual Meeting of the American Academy of Neurology.
“There is a disconnect between what the patient, families, and the public understand about cannabinoid therapies for epilepsy and what we as a neurologic community know,” he said. “Much of human evidence for the efficacy of cannabinoids for the treatment of epilepsy is anecdotal” and, of the “few controlled trials, most are small and methodologically flawed.”
A Brief History of Medical Marijuana
Cannabis sativa was first cultivated in approximately 8,000 BCE in China for rope, then medicinally for conditions ranging from menstruation to absentmindedness and, eventually, more than 100 ailments. In the early 19th century, W.B. O’Shaughnessy introduced cannabis to England, where Gowers and other Victorian neurologists used the “Indian hemp” to treat epilepsy.
Medicinal use of cannabis dwindled in the 1930s due to a move to synthetic medicine and Prohibition, which also “coincided with the beginning of the era of rigorous scientific investigation of drug claims,” said Dr. Friedman. Now, “the discovery of the endocannabinoid system in the brain has sparked new research into the therapeutic potential of cannabinoids.”
The Chemicals in Cannabis
The genus Cannabis includes C. sativa, C. indica, and C. ruderalis. A total of 85 phytocannabinoids are found in Cannabis species plants, and cannabidiol (CBD) and delta-9 tetrahydrocannabinol (THC) are the most abundant neuroactive chemicals; others include the terpenes and flavonoids.
CBD-rich oil is a concentrate made from cannabis bred to have a low level of THC and a high level of CBD. GW Pharma manufactures a solution with equal parts purified plant-derived THC and CBD (Sativex). The company also makes an investigational agent, pure CBD (Epidiolex), a 98% purified plant-derived CBD. INSYS Therapeutics is developing a synthetic CBD that is structurally identical to the plant-derived compound.
What Is the Evidence?
Experimental and human studies have provided evidence for the anticonvulsant properties of CBD and THC, and antiseizure effects have also been observed with cannabidivarin (CBDV).
Since 2014, more than 10 US centers have been using Epidiolex under an FDA expanded-access program to treat patients between ages 1 and 30 (median age, 10.5) with severe, treatment-resistant, childhood-onset epilepsy. Patients’ baseline motor seizure rate was 29.5 per month (range, 11.0–96.0 per month).
As of January 2015, 162 patients had completed 12 weeks of observation. Preliminary results showed median motor seizure reduction to be 36.5%. Patients with Dravet syndrome had a 49.8% reduction in seizures, and patients with Lennox-Gastaut syndrome had a 36.8% reduction. Serious adverse events, primarily status epilepticus, were observed in 12% of participants. Only 3% of patients discontinued treatment, however. Commonly reported adverse events included somnolence (25.3%), decreased appetite (19.1%), diarrhea (19.1%), fatigue (13.0%), and convulsion (11.1%).
In addition, children with Dravet syndrome in a phase III trial of Epidiolex had a median reduction in motor seizures of 39% versus 13% for placebo.
“Much of the safety data for chronic cannabinoid use” has come from studies of recreational use, which is “inherently confounded,” Dr. Friedman said. Acute affects include impaired memory, judgment, and motor performance. Chronic use leads to addiction in approximately 9% of users, as well as to cognitive impairment, decreased motivation, and increased risk for psychotic disorders.
Researchers examined pooled data from adults with multiple sclerosis who used cannabinoids for spasticity, pain, and dyskinesias for less than six months. The data encompassed approximately 1,600 exposures to the drugs. About 7% of participants discontinued treatment due to adverse events that included nausea, behavioral and mood changes, suicidality, hallucinations, dizziness, and weakness.
CBD and THC are inhibitors of P450 isozymes, primarily CYP2C and CYP3A, but drug–drug interaction effects are not typically seen in doses used in human studies, said Dr. Friedman. At low micromolar concentrations, CBD inhibits CYP2C19, an enzyme involved in the metabolism of N-desmethylclobazam, phenytoin, diazepam, citalopram, and some tricyclic antidepressants. The CYP2B family may also be induced, thus affecting valproate and clobazam metabolism.
Drugs that affect CBD metabolism include the CYP3A4 inducers, such as carbamazepine and phenytoin, and the CYP3A4 inhibitors such as ketoconazole.
Randomized controlled trials of CBD and CBDV are in progress in patients with Dravet syndrome, Lennox-Gastaut syndrome, and refractory focal epilepsy.
Is Cannabis Right for My Patient?
Since New York State approved the medical use of cannabis, Dr. Friedman has discussed the drug in one of every four of his clinic visits. Deciding whether to recommend medical marijuana for a patient can be difficult, however.
“Would you authorize its use for a 22-year-old patient with treatment-resistant epilepsy who is not a candidate for epilepsy surgery, is having two complex partial seizures a week and one tonic-clonic seizure per month, and is currently on three antiepileptic drugs [AEDs] at high doses,” asked Dr. Friedman. “How about for a 63-year-old woman with focal motor seizures following a meningioma resection who has not been able to tolerate adequate doses of four prior AEDs?”
The answer depends on myriad factors, not the least of which is the legal status of medical marijuana in the neurologist’s state. Although “23 states and the District of Columbia have approved medical marijuana for certain conditions, including epilepsy,” the US Drug Enforcement Administration still considers cannabis and its derivatives schedule I compounds, which means that they have “no accepted medical use in the US,” a high abuse potential, and cannot be prescribed—only “recommended”—by a physician, saidDr. Friedman.
Patients should be evaluated at a comprehensive epilepsy center to determine whether they have been unable to achieve control with conventional therapies due to lack of efficacy or side effects, and whether other proven effective therapies, such as vagus nerve stimulation, a ketogenic diet, or surgery, have been considered, he added.
For patients who do initiate treatment with medical marijuana, the risks and benefits should be carefully weighed, and a treatment plan that includes a timeline for discontinuation should be developed. Laboratory values and clinical status should be monitored regularly.
“Perhaps one day we’ll have [cannabis] in the pharmacy,” Dr. Freidman concluded. Until then, patients should be cautioned against buying cannabinoids on the Internet because, as one FDA study showed, products bought from such sources may have no detectable cannabinoid levels.
—Debra Hughes
VANCOUVER—“Neurologists are often asked to ‘certify’ patients, but should you recommend or support medical marijuana use for patients with epilepsy?”
Daniel Friedman, MD, MSc, Assistant Professor of Neurology at New York University Langone School of Medicine in New York, addressed this question in his presentation at the 68th Annual Meeting of the American Academy of Neurology.
“There is a disconnect between what the patient, families, and the public understand about cannabinoid therapies for epilepsy and what we as a neurologic community know,” he said. “Much of human evidence for the efficacy of cannabinoids for the treatment of epilepsy is anecdotal” and, of the “few controlled trials, most are small and methodologically flawed.”
A Brief History of Medical Marijuana
Cannabis sativa was first cultivated in approximately 8,000 BCE in China for rope, then medicinally for conditions ranging from menstruation to absentmindedness and, eventually, more than 100 ailments. In the early 19th century, W.B. O’Shaughnessy introduced cannabis to England, where Gowers and other Victorian neurologists used the “Indian hemp” to treat epilepsy.
Medicinal use of cannabis dwindled in the 1930s due to a move to synthetic medicine and Prohibition, which also “coincided with the beginning of the era of rigorous scientific investigation of drug claims,” said Dr. Friedman. Now, “the discovery of the endocannabinoid system in the brain has sparked new research into the therapeutic potential of cannabinoids.”
The Chemicals in Cannabis
The genus Cannabis includes C. sativa, C. indica, and C. ruderalis. A total of 85 phytocannabinoids are found in Cannabis species plants, and cannabidiol (CBD) and delta-9 tetrahydrocannabinol (THC) are the most abundant neuroactive chemicals; others include the terpenes and flavonoids.
CBD-rich oil is a concentrate made from cannabis bred to have a low level of THC and a high level of CBD. GW Pharma manufactures a solution with equal parts purified plant-derived THC and CBD (Sativex). The company also makes an investigational agent, pure CBD (Epidiolex), a 98% purified plant-derived CBD. INSYS Therapeutics is developing a synthetic CBD that is structurally identical to the plant-derived compound.
What Is the Evidence?
Experimental and human studies have provided evidence for the anticonvulsant properties of CBD and THC, and antiseizure effects have also been observed with cannabidivarin (CBDV).
Since 2014, more than 10 US centers have been using Epidiolex under an FDA expanded-access program to treat patients between ages 1 and 30 (median age, 10.5) with severe, treatment-resistant, childhood-onset epilepsy. Patients’ baseline motor seizure rate was 29.5 per month (range, 11.0–96.0 per month).
As of January 2015, 162 patients had completed 12 weeks of observation. Preliminary results showed median motor seizure reduction to be 36.5%. Patients with Dravet syndrome had a 49.8% reduction in seizures, and patients with Lennox-Gastaut syndrome had a 36.8% reduction. Serious adverse events, primarily status epilepticus, were observed in 12% of participants. Only 3% of patients discontinued treatment, however. Commonly reported adverse events included somnolence (25.3%), decreased appetite (19.1%), diarrhea (19.1%), fatigue (13.0%), and convulsion (11.1%).
In addition, children with Dravet syndrome in a phase III trial of Epidiolex had a median reduction in motor seizures of 39% versus 13% for placebo.
“Much of the safety data for chronic cannabinoid use” has come from studies of recreational use, which is “inherently confounded,” Dr. Friedman said. Acute affects include impaired memory, judgment, and motor performance. Chronic use leads to addiction in approximately 9% of users, as well as to cognitive impairment, decreased motivation, and increased risk for psychotic disorders.
Researchers examined pooled data from adults with multiple sclerosis who used cannabinoids for spasticity, pain, and dyskinesias for less than six months. The data encompassed approximately 1,600 exposures to the drugs. About 7% of participants discontinued treatment due to adverse events that included nausea, behavioral and mood changes, suicidality, hallucinations, dizziness, and weakness.
CBD and THC are inhibitors of P450 isozymes, primarily CYP2C and CYP3A, but drug–drug interaction effects are not typically seen in doses used in human studies, said Dr. Friedman. At low micromolar concentrations, CBD inhibits CYP2C19, an enzyme involved in the metabolism of N-desmethylclobazam, phenytoin, diazepam, citalopram, and some tricyclic antidepressants. The CYP2B family may also be induced, thus affecting valproate and clobazam metabolism.
Drugs that affect CBD metabolism include the CYP3A4 inducers, such as carbamazepine and phenytoin, and the CYP3A4 inhibitors such as ketoconazole.
Randomized controlled trials of CBD and CBDV are in progress in patients with Dravet syndrome, Lennox-Gastaut syndrome, and refractory focal epilepsy.
Is Cannabis Right for My Patient?
Since New York State approved the medical use of cannabis, Dr. Friedman has discussed the drug in one of every four of his clinic visits. Deciding whether to recommend medical marijuana for a patient can be difficult, however.
“Would you authorize its use for a 22-year-old patient with treatment-resistant epilepsy who is not a candidate for epilepsy surgery, is having two complex partial seizures a week and one tonic-clonic seizure per month, and is currently on three antiepileptic drugs [AEDs] at high doses,” asked Dr. Friedman. “How about for a 63-year-old woman with focal motor seizures following a meningioma resection who has not been able to tolerate adequate doses of four prior AEDs?”
The answer depends on myriad factors, not the least of which is the legal status of medical marijuana in the neurologist’s state. Although “23 states and the District of Columbia have approved medical marijuana for certain conditions, including epilepsy,” the US Drug Enforcement Administration still considers cannabis and its derivatives schedule I compounds, which means that they have “no accepted medical use in the US,” a high abuse potential, and cannot be prescribed—only “recommended”—by a physician, saidDr. Friedman.
Patients should be evaluated at a comprehensive epilepsy center to determine whether they have been unable to achieve control with conventional therapies due to lack of efficacy or side effects, and whether other proven effective therapies, such as vagus nerve stimulation, a ketogenic diet, or surgery, have been considered, he added.
For patients who do initiate treatment with medical marijuana, the risks and benefits should be carefully weighed, and a treatment plan that includes a timeline for discontinuation should be developed. Laboratory values and clinical status should be monitored regularly.
“Perhaps one day we’ll have [cannabis] in the pharmacy,” Dr. Freidman concluded. Until then, patients should be cautioned against buying cannabinoids on the Internet because, as one FDA study showed, products bought from such sources may have no detectable cannabinoid levels.
—Debra Hughes

Maternal Exposure to Pregabalin May Cause Birth Defects
First-trimester exposure to pregabalin may increase the risk of major birth defects, according to a study published online ahead of print May 18 in Neurology.
Pregabalin is an FDA-approved treatment for seizures and neuropathic pain. It is also a common off-label treatment for restless legs syndrome, cyclic mood disorders, and generalized anxiety disorder.
Ursula Winterfeld, PhD, of the Swiss Teratogen Information Service and Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland, and colleagues conducted a multicenter, observational cohort study in which they compared pregnancy outcomes of 164 women exposed to pregabalin with 656 controls who were not exposed to any known teratogenic medications or antiepileptic drugs. Data for this study were collected from 2004 to 2013 and included data from France, the United Kingdom, Italy, Finland, Switzerland, the Netherlands, and Turkey.
Of the women on the medication, 77% started taking pregabalin before they became pregnant and stopped taking the drug at a median of six weeks into their pregnancies. Of the women taking pregabalin, 13% were also taking another antiepileptic drug.
Pregnancies of the women who took pregabalin during the first trimester of pregnancy were three times more likely to result in major birth defects than those of women who did not take the drug—6.0% versus 2.1%, respectively. The major birth defects included heart defects and structural problems with the CNS or other organ systems. The study also revealed a lower rate of live births in the pregabalin group due to elective and medically indicated pregnancy terminations.
“We can’t draw any definitive conclusions from this study, since many of the women were taking other drugs that could have played a role in the birth defects and because the study was small and the results need to be confirmed with larger studies, but these results do signal that there may be an increased risk for major birth defects after taking pregabalin during the first trimester of pregnancy,” said Dr. Winterfeld.
She suggested that before a woman is prescribed pregabalin, it is important to make sure the benefits outweigh the risks and that she is carefully informed about the use of effective birth control.
—Adaeze Stephanie Onyechi
Suggested Reading
Winterfeld U, Merlob P, Baud D, et al. Pregnancy outcome following maternal exposure to pregabalin may call for concern. Neurology. 2016 May 18 [Epub ahead of print].
Pennell PB, Meador KJ. A common medication for neuropsychiatric illness may cause common problems in pregnancy. Neurology. 2016 May 18 [Epub ahead of print].
First-trimester exposure to pregabalin may increase the risk of major birth defects, according to a study published online ahead of print May 18 in Neurology.
Pregabalin is an FDA-approved treatment for seizures and neuropathic pain. It is also a common off-label treatment for restless legs syndrome, cyclic mood disorders, and generalized anxiety disorder.
Ursula Winterfeld, PhD, of the Swiss Teratogen Information Service and Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland, and colleagues conducted a multicenter, observational cohort study in which they compared pregnancy outcomes of 164 women exposed to pregabalin with 656 controls who were not exposed to any known teratogenic medications or antiepileptic drugs. Data for this study were collected from 2004 to 2013 and included data from France, the United Kingdom, Italy, Finland, Switzerland, the Netherlands, and Turkey.
Of the women on the medication, 77% started taking pregabalin before they became pregnant and stopped taking the drug at a median of six weeks into their pregnancies. Of the women taking pregabalin, 13% were also taking another antiepileptic drug.
Pregnancies of the women who took pregabalin during the first trimester of pregnancy were three times more likely to result in major birth defects than those of women who did not take the drug—6.0% versus 2.1%, respectively. The major birth defects included heart defects and structural problems with the CNS or other organ systems. The study also revealed a lower rate of live births in the pregabalin group due to elective and medically indicated pregnancy terminations.
“We can’t draw any definitive conclusions from this study, since many of the women were taking other drugs that could have played a role in the birth defects and because the study was small and the results need to be confirmed with larger studies, but these results do signal that there may be an increased risk for major birth defects after taking pregabalin during the first trimester of pregnancy,” said Dr. Winterfeld.
She suggested that before a woman is prescribed pregabalin, it is important to make sure the benefits outweigh the risks and that she is carefully informed about the use of effective birth control.
—Adaeze Stephanie Onyechi
First-trimester exposure to pregabalin may increase the risk of major birth defects, according to a study published online ahead of print May 18 in Neurology.
Pregabalin is an FDA-approved treatment for seizures and neuropathic pain. It is also a common off-label treatment for restless legs syndrome, cyclic mood disorders, and generalized anxiety disorder.
Ursula Winterfeld, PhD, of the Swiss Teratogen Information Service and Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland, and colleagues conducted a multicenter, observational cohort study in which they compared pregnancy outcomes of 164 women exposed to pregabalin with 656 controls who were not exposed to any known teratogenic medications or antiepileptic drugs. Data for this study were collected from 2004 to 2013 and included data from France, the United Kingdom, Italy, Finland, Switzerland, the Netherlands, and Turkey.
Of the women on the medication, 77% started taking pregabalin before they became pregnant and stopped taking the drug at a median of six weeks into their pregnancies. Of the women taking pregabalin, 13% were also taking another antiepileptic drug.
Pregnancies of the women who took pregabalin during the first trimester of pregnancy were three times more likely to result in major birth defects than those of women who did not take the drug—6.0% versus 2.1%, respectively. The major birth defects included heart defects and structural problems with the CNS or other organ systems. The study also revealed a lower rate of live births in the pregabalin group due to elective and medically indicated pregnancy terminations.
“We can’t draw any definitive conclusions from this study, since many of the women were taking other drugs that could have played a role in the birth defects and because the study was small and the results need to be confirmed with larger studies, but these results do signal that there may be an increased risk for major birth defects after taking pregabalin during the first trimester of pregnancy,” said Dr. Winterfeld.
She suggested that before a woman is prescribed pregabalin, it is important to make sure the benefits outweigh the risks and that she is carefully informed about the use of effective birth control.
—Adaeze Stephanie Onyechi
Suggested Reading
Winterfeld U, Merlob P, Baud D, et al. Pregnancy outcome following maternal exposure to pregabalin may call for concern. Neurology. 2016 May 18 [Epub ahead of print].
Pennell PB, Meador KJ. A common medication for neuropsychiatric illness may cause common problems in pregnancy. Neurology. 2016 May 18 [Epub ahead of print].
Suggested Reading
Winterfeld U, Merlob P, Baud D, et al. Pregnancy outcome following maternal exposure to pregabalin may call for concern. Neurology. 2016 May 18 [Epub ahead of print].
Pennell PB, Meador KJ. A common medication for neuropsychiatric illness may cause common problems in pregnancy. Neurology. 2016 May 18 [Epub ahead of print].