User login
Don’t underestimate opioid use in HIV-positive adults
Prescription opioid use in HIV-positive individuals is underestimated, based on data from a 12-week study of medical and nonmedical prescription drug use in HIV patients attending a medical center and community clinic.
Of the 254 participants in the study, 43% reported medical use of prescription opioids, and 11% of the opioid use was reported as nonmedical.
Previous studies of prescription drug use in the HIV-positive population have mainly focused on nonmedical use, so “the existing research likely underestimates overall exposure of HIV-positive individuals to prescription medications,” wrote Abigail Norris Turner, PhD, of Ohio State University in Columbus, and her colleagues. Nonmedical use was defined as “using more medication than prescribed, using medication prescribed to someone else, or using medication for a purpose other than its prescribed use,” the researchers wrote.
The cross-sectional, self-administered survey involved data collection during July and August 2015 at a large, urban medical center (149 patients) and a community-based AIDS service organization (105 patients). Most of the study participants were male (91%), were identified as gay or bisexual (79%), and were at least 40 years old (61%).
Overall, 27% of the participants reported nonmedical use of any prescription drug during the previous year. Of these, 17% reported using one medication, 8% reported using two medications, and 2% reported using three or more medications. During the past month, 14% reported nonmedical use of a prescription drug, with 17% reporting one medication, 8% reporting two medications, and 2% reporting three or more medications. Reports of drug use were similar between the hospital group and clinic group.
The findings were limited by the cross-sectional nature of the study and the reliance on self-reports, the researchers noted; therefore, “we cannot make inferences about the causes and consequences of prescription medication use,” they said.
However, the results suggest that “it would be prudent for HIV providers to regularly review their opioid prescription practices to make sure they are appropriate; this review could also help identify patients at risk for opioid dependence and those who may benefit from referral to a pain medicine specialist or addiction medicine specialist,” they wrote.
The study was supported in part by the National Institutes of Health and the SOLAR Foundation Research Fund at the Ohio State University.
Read the full study here: AIDS Care 2016 Jun 20. doi: 10.1080/09540121.2016.1198746.
Prescription opioid use in HIV-positive individuals is underestimated, based on data from a 12-week study of medical and nonmedical prescription drug use in HIV patients attending a medical center and community clinic.
Of the 254 participants in the study, 43% reported medical use of prescription opioids, and 11% of the opioid use was reported as nonmedical.
Previous studies of prescription drug use in the HIV-positive population have mainly focused on nonmedical use, so “the existing research likely underestimates overall exposure of HIV-positive individuals to prescription medications,” wrote Abigail Norris Turner, PhD, of Ohio State University in Columbus, and her colleagues. Nonmedical use was defined as “using more medication than prescribed, using medication prescribed to someone else, or using medication for a purpose other than its prescribed use,” the researchers wrote.
The cross-sectional, self-administered survey involved data collection during July and August 2015 at a large, urban medical center (149 patients) and a community-based AIDS service organization (105 patients). Most of the study participants were male (91%), were identified as gay or bisexual (79%), and were at least 40 years old (61%).
Overall, 27% of the participants reported nonmedical use of any prescription drug during the previous year. Of these, 17% reported using one medication, 8% reported using two medications, and 2% reported using three or more medications. During the past month, 14% reported nonmedical use of a prescription drug, with 17% reporting one medication, 8% reporting two medications, and 2% reporting three or more medications. Reports of drug use were similar between the hospital group and clinic group.
The findings were limited by the cross-sectional nature of the study and the reliance on self-reports, the researchers noted; therefore, “we cannot make inferences about the causes and consequences of prescription medication use,” they said.
However, the results suggest that “it would be prudent for HIV providers to regularly review their opioid prescription practices to make sure they are appropriate; this review could also help identify patients at risk for opioid dependence and those who may benefit from referral to a pain medicine specialist or addiction medicine specialist,” they wrote.
The study was supported in part by the National Institutes of Health and the SOLAR Foundation Research Fund at the Ohio State University.
Read the full study here: AIDS Care 2016 Jun 20. doi: 10.1080/09540121.2016.1198746.
Prescription opioid use in HIV-positive individuals is underestimated, based on data from a 12-week study of medical and nonmedical prescription drug use in HIV patients attending a medical center and community clinic.
Of the 254 participants in the study, 43% reported medical use of prescription opioids, and 11% of the opioid use was reported as nonmedical.
Previous studies of prescription drug use in the HIV-positive population have mainly focused on nonmedical use, so “the existing research likely underestimates overall exposure of HIV-positive individuals to prescription medications,” wrote Abigail Norris Turner, PhD, of Ohio State University in Columbus, and her colleagues. Nonmedical use was defined as “using more medication than prescribed, using medication prescribed to someone else, or using medication for a purpose other than its prescribed use,” the researchers wrote.
The cross-sectional, self-administered survey involved data collection during July and August 2015 at a large, urban medical center (149 patients) and a community-based AIDS service organization (105 patients). Most of the study participants were male (91%), were identified as gay or bisexual (79%), and were at least 40 years old (61%).
Overall, 27% of the participants reported nonmedical use of any prescription drug during the previous year. Of these, 17% reported using one medication, 8% reported using two medications, and 2% reported using three or more medications. During the past month, 14% reported nonmedical use of a prescription drug, with 17% reporting one medication, 8% reporting two medications, and 2% reporting three or more medications. Reports of drug use were similar between the hospital group and clinic group.
The findings were limited by the cross-sectional nature of the study and the reliance on self-reports, the researchers noted; therefore, “we cannot make inferences about the causes and consequences of prescription medication use,” they said.
However, the results suggest that “it would be prudent for HIV providers to regularly review their opioid prescription practices to make sure they are appropriate; this review could also help identify patients at risk for opioid dependence and those who may benefit from referral to a pain medicine specialist or addiction medicine specialist,” they wrote.
The study was supported in part by the National Institutes of Health and the SOLAR Foundation Research Fund at the Ohio State University.
Read the full study here: AIDS Care 2016 Jun 20. doi: 10.1080/09540121.2016.1198746.
FROM AIDS CARE
Key clinical point: Opioid exposure in HIV-positive adults is more prevalent than previous research suggests.
Major finding: Based on data from 254 individuals, 43% reported medical opioid use, and 11% reported nonmedical opioid use.
Data source: A cross-sectional survey involving 254 HIV-positive adults at two study sites.
Disclosures: The study was supported in part by the National Institutes of Health and the SOLAR Foundation Research Fund at the Ohio State University.
FDA’s arthritis advisory committee unanimously backs biosimilar Humira
In a 26-0 vote, the Food and Drug Administration’s arthritis advisory committee recommended July 12 that the agency license an Amgen product as biosimilar to Humira (adalimumab) for seven distinct indications: rheumatoid arthritis, juvenile idiopathic arthritis (in patients at least 4 years old), psoriatic arthritis, ankylosing spondylitis, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis.
Developing Story
In a 26-0 vote, the Food and Drug Administration’s arthritis advisory committee recommended July 12 that the agency license an Amgen product as biosimilar to Humira (adalimumab) for seven distinct indications: rheumatoid arthritis, juvenile idiopathic arthritis (in patients at least 4 years old), psoriatic arthritis, ankylosing spondylitis, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis.
Developing Story
In a 26-0 vote, the Food and Drug Administration’s arthritis advisory committee recommended July 12 that the agency license an Amgen product as biosimilar to Humira (adalimumab) for seven distinct indications: rheumatoid arthritis, juvenile idiopathic arthritis (in patients at least 4 years old), psoriatic arthritis, ankylosing spondylitis, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis.
Developing Story
Medical Marijuana Cuts Medicare Spending and May Reduce Dependency on Prescription Opioids
Physicians wrote significantly fewer prescriptions for painkillers and other medications for elderly and disabled patients who had legal access to medical marijuana, a new study finds.
In fact, Medicare saved more than $165 million in 2013 on prescription drugs in the District of Columbia and 17 states that allowed cannabis to be used as medicine, researchers calculated. If every state in the nation legalized medical marijuana, the study forecast that the federal program would save more than $468 million a year on pharmaceuticals for disabled Americans and those 65 and older.
No health insurance, including Medicare, will reimburse for the cost of marijuana. Although medical cannabis is legal today in 25 states and the District of Columbia, federal law continues to prohibit its prescription in all circumstances.
The new study, published July 6 in Health Affairs, was the first to ask if there's any evidence that medical marijuana is being used as medicine, said senior author W. David Bradford in a phone interview. The answer is yes, said Bradford, a health economist and a professor at the University of Georgia in Athens.
"When states turned on medical marijuana laws, we did see a rather substantial turn away from FDA-approved medicine," he said.
Researchers analyzed Medicare data from 2010 through 2013 for drugs approved by the U.S. Food and Drug Administration (FDA) to treat nine ailments - from pain to depression and nausea - for which marijuana might be an alternative remedy.
They expected to see fewer prescriptions for FDA-approved drugs that might treat the same conditions as cannabis. Indeed, except for glaucoma, doctors wrote fewer prescriptions for all nine ailments after medical marijuana laws took effect, the study found.
The number of Medicare prescriptions significantly dropped for drugs that treat pain, depression, anxiety, nausea, psychoses, seizures and sleep disorders.
For pain, the annual number of daily doses prescribed per physician fell by more than 11 percent.
"The results show that marijuana might be beneficial with diverting people away from opioids," Bradford said.
A 2014 study found that opioid overdose death rates were on average nearly 25 percent lower in states where medical marijuana was legal compared to states where it remained illegal. Chronic or severe pain is considered a primary indicator for medical marijuana in most states where it is legal.
Nearly two million Americans either abused or were dependent on prescription opioids in 2014, according to the U.S. Centers for Disease Control and Prevention (CDC). Since 1999, more than 165,000 Americans have died from prescription opioid overdoses.
Addiction psychiatrist Dr. Kevin Hill questioned whether medical marijuana patients might in some cases be getting inferior or incorrect treatment, and if so, whether the resulting extra healthcare costs would overshadow the Medicare drug savings. Hill, a professor at Harvard Medical School in Boston, was not involved in the new study.
"Fewer opioid prescriptions in medical marijuana states might be a good thing, but I am concerned about the overall quality of care delivered in medical marijuana specialty clinics," he told Reuters Health in an email.
He criticized the implementation of medical marijuana laws in many states as often leading to "medical care that is of poor quality."
Part of the problem stems from a dearth of research into the efficacy of medical marijuana.
Although California became the first state to legalize medical marijuana in 1996, federal law enacted by Congress in 1970 continues to put cannabis in the same category as heroin, Schedule 1 of the Comprehensive Drug Abuse Prevention and Control Act, and finds it has no medicinal value. Consequently, research has been severely limited.
Sheigla Murphy, a medical sociologist who was not involved in the current study, praised it as a major contribution to the literature on the role of medical marijuana in older adults.
Murphy directs the Center for Substance Abuse Studies in San Francisco and has done prior research on marijuana and baby boomers. She said some older adults prefer marijuana to painkillers and sleeping pills.
"It fits with the problems of older age, problems with sleeping, depression, arthritis, worn-out body parts that begin to hurt. Marijuana can relieve these without the side effects of grogginess and worrying about addiction," she said.
"As we're trying to reduce the number of pain medications, I think marijuana would be a welcome addition to the pharmacopeia," she said. "The one thing we know is no one has ever died of it."
SOURCE: http://bit.ly/1lx2GBv
Health Affairs 2016.
Physicians wrote significantly fewer prescriptions for painkillers and other medications for elderly and disabled patients who had legal access to medical marijuana, a new study finds.
In fact, Medicare saved more than $165 million in 2013 on prescription drugs in the District of Columbia and 17 states that allowed cannabis to be used as medicine, researchers calculated. If every state in the nation legalized medical marijuana, the study forecast that the federal program would save more than $468 million a year on pharmaceuticals for disabled Americans and those 65 and older.
No health insurance, including Medicare, will reimburse for the cost of marijuana. Although medical cannabis is legal today in 25 states and the District of Columbia, federal law continues to prohibit its prescription in all circumstances.
The new study, published July 6 in Health Affairs, was the first to ask if there's any evidence that medical marijuana is being used as medicine, said senior author W. David Bradford in a phone interview. The answer is yes, said Bradford, a health economist and a professor at the University of Georgia in Athens.
"When states turned on medical marijuana laws, we did see a rather substantial turn away from FDA-approved medicine," he said.
Researchers analyzed Medicare data from 2010 through 2013 for drugs approved by the U.S. Food and Drug Administration (FDA) to treat nine ailments - from pain to depression and nausea - for which marijuana might be an alternative remedy.
They expected to see fewer prescriptions for FDA-approved drugs that might treat the same conditions as cannabis. Indeed, except for glaucoma, doctors wrote fewer prescriptions for all nine ailments after medical marijuana laws took effect, the study found.
The number of Medicare prescriptions significantly dropped for drugs that treat pain, depression, anxiety, nausea, psychoses, seizures and sleep disorders.
For pain, the annual number of daily doses prescribed per physician fell by more than 11 percent.
"The results show that marijuana might be beneficial with diverting people away from opioids," Bradford said.
A 2014 study found that opioid overdose death rates were on average nearly 25 percent lower in states where medical marijuana was legal compared to states where it remained illegal. Chronic or severe pain is considered a primary indicator for medical marijuana in most states where it is legal.
Nearly two million Americans either abused or were dependent on prescription opioids in 2014, according to the U.S. Centers for Disease Control and Prevention (CDC). Since 1999, more than 165,000 Americans have died from prescription opioid overdoses.
Addiction psychiatrist Dr. Kevin Hill questioned whether medical marijuana patients might in some cases be getting inferior or incorrect treatment, and if so, whether the resulting extra healthcare costs would overshadow the Medicare drug savings. Hill, a professor at Harvard Medical School in Boston, was not involved in the new study.
"Fewer opioid prescriptions in medical marijuana states might be a good thing, but I am concerned about the overall quality of care delivered in medical marijuana specialty clinics," he told Reuters Health in an email.
He criticized the implementation of medical marijuana laws in many states as often leading to "medical care that is of poor quality."
Part of the problem stems from a dearth of research into the efficacy of medical marijuana.
Although California became the first state to legalize medical marijuana in 1996, federal law enacted by Congress in 1970 continues to put cannabis in the same category as heroin, Schedule 1 of the Comprehensive Drug Abuse Prevention and Control Act, and finds it has no medicinal value. Consequently, research has been severely limited.
Sheigla Murphy, a medical sociologist who was not involved in the current study, praised it as a major contribution to the literature on the role of medical marijuana in older adults.
Murphy directs the Center for Substance Abuse Studies in San Francisco and has done prior research on marijuana and baby boomers. She said some older adults prefer marijuana to painkillers and sleeping pills.
"It fits with the problems of older age, problems with sleeping, depression, arthritis, worn-out body parts that begin to hurt. Marijuana can relieve these without the side effects of grogginess and worrying about addiction," she said.
"As we're trying to reduce the number of pain medications, I think marijuana would be a welcome addition to the pharmacopeia," she said. "The one thing we know is no one has ever died of it."
SOURCE: http://bit.ly/1lx2GBv
Health Affairs 2016.
Physicians wrote significantly fewer prescriptions for painkillers and other medications for elderly and disabled patients who had legal access to medical marijuana, a new study finds.
In fact, Medicare saved more than $165 million in 2013 on prescription drugs in the District of Columbia and 17 states that allowed cannabis to be used as medicine, researchers calculated. If every state in the nation legalized medical marijuana, the study forecast that the federal program would save more than $468 million a year on pharmaceuticals for disabled Americans and those 65 and older.
No health insurance, including Medicare, will reimburse for the cost of marijuana. Although medical cannabis is legal today in 25 states and the District of Columbia, federal law continues to prohibit its prescription in all circumstances.
The new study, published July 6 in Health Affairs, was the first to ask if there's any evidence that medical marijuana is being used as medicine, said senior author W. David Bradford in a phone interview. The answer is yes, said Bradford, a health economist and a professor at the University of Georgia in Athens.
"When states turned on medical marijuana laws, we did see a rather substantial turn away from FDA-approved medicine," he said.
Researchers analyzed Medicare data from 2010 through 2013 for drugs approved by the U.S. Food and Drug Administration (FDA) to treat nine ailments - from pain to depression and nausea - for which marijuana might be an alternative remedy.
They expected to see fewer prescriptions for FDA-approved drugs that might treat the same conditions as cannabis. Indeed, except for glaucoma, doctors wrote fewer prescriptions for all nine ailments after medical marijuana laws took effect, the study found.
The number of Medicare prescriptions significantly dropped for drugs that treat pain, depression, anxiety, nausea, psychoses, seizures and sleep disorders.
For pain, the annual number of daily doses prescribed per physician fell by more than 11 percent.
"The results show that marijuana might be beneficial with diverting people away from opioids," Bradford said.
A 2014 study found that opioid overdose death rates were on average nearly 25 percent lower in states where medical marijuana was legal compared to states where it remained illegal. Chronic or severe pain is considered a primary indicator for medical marijuana in most states where it is legal.
Nearly two million Americans either abused or were dependent on prescription opioids in 2014, according to the U.S. Centers for Disease Control and Prevention (CDC). Since 1999, more than 165,000 Americans have died from prescription opioid overdoses.
Addiction psychiatrist Dr. Kevin Hill questioned whether medical marijuana patients might in some cases be getting inferior or incorrect treatment, and if so, whether the resulting extra healthcare costs would overshadow the Medicare drug savings. Hill, a professor at Harvard Medical School in Boston, was not involved in the new study.
"Fewer opioid prescriptions in medical marijuana states might be a good thing, but I am concerned about the overall quality of care delivered in medical marijuana specialty clinics," he told Reuters Health in an email.
He criticized the implementation of medical marijuana laws in many states as often leading to "medical care that is of poor quality."
Part of the problem stems from a dearth of research into the efficacy of medical marijuana.
Although California became the first state to legalize medical marijuana in 1996, federal law enacted by Congress in 1970 continues to put cannabis in the same category as heroin, Schedule 1 of the Comprehensive Drug Abuse Prevention and Control Act, and finds it has no medicinal value. Consequently, research has been severely limited.
Sheigla Murphy, a medical sociologist who was not involved in the current study, praised it as a major contribution to the literature on the role of medical marijuana in older adults.
Murphy directs the Center for Substance Abuse Studies in San Francisco and has done prior research on marijuana and baby boomers. She said some older adults prefer marijuana to painkillers and sleeping pills.
"It fits with the problems of older age, problems with sleeping, depression, arthritis, worn-out body parts that begin to hurt. Marijuana can relieve these without the side effects of grogginess and worrying about addiction," she said.
"As we're trying to reduce the number of pain medications, I think marijuana would be a welcome addition to the pharmacopeia," she said. "The one thing we know is no one has ever died of it."
SOURCE: http://bit.ly/1lx2GBv
Health Affairs 2016.
Expert: Screen military spouses for sleep problems
DENVER – Sleep problems and their host of deleterious physical and psychosocial consequences are pervasive among the civilian female spouses of U.S. military service members, according to the first large study to examine the issue.
“Our findings suggest that if we’re trying to promote the resilience and adjustment of military spouses and their families – particularly in light of 14 years of protracted overseas combat, where many families are experiencing deployment – sleep might be a really important target,” Wendy M. Troxel, PhD, said while presenting the study results at the annual meeting of the Associated Professional Sleep Societies.

The study population consisted of a nationally representative group of 1,805 female civilian spouses of military service members. Forty-four percent reported short sleep duration, meaning 6 hours or less per night. Another 18% got 5 hours or less. The Centers for Disease Control and Prevention and the American Academy of Sleep Medicine recommend 7 hours or more, noted Dr. Troxel, senior behavioral and social scientist at RAND Corporation in Pittsburgh.
More than half (54%) of military spouses reported that their sleep problems contributed to daytime impairment, such as intolerance of their spouse or children, frequent crying, or suboptimal performance at work or chores. One-third of the spouses reported feeling daytime fatigue three or more times per week, and another 29% experienced daytime fatigue one or two days per week.
As has been observed in other studies, sleep problems in military spouses were associated with psychosocial impairment. In linear regression analyses, the spouses with poor sleep quality, daytime impairment, and/or fatigue had significantly more depressive symptoms on the Patient Health Questionnaire–8, lower marital satisfaction, and poor self-rated health. Women with shorter sleep duration had more depressive symptoms and poorer self-rated health, but not lower marital satisfaction.
The spouses of services members currently deployed in combat zones had significantly worse sleep quality as measured on the Pittsburgh Sleep Quality Index compared with spouses of previously or never-deployed service members. But for the other outcomes of interest – sleep duration, daytime impairment, and fatigue – there were no differences based upon deployment status.
“This shows that it’s not just about the stress of combat deployment, it’s also about the stresses of everyday military life. Military families experience a great deal of stress that could lead to sleep disturbances whether or not a service member has been deployed: frequent residential moves, very long and unpredictable work hours, demanding jobs, threatening training environments,” Dr. Troxel said. “I think we need to be thinking about sleep as an important health indicator of military families in general across the deployment cycle.”
The study results are a call to action, she added.
“These findings highlight the importance of targeted screening, prevention, and intervention methods for military spouses. And primary care is where most people present with sleep problems,” Dr. Troxel said.
There are formal screening instruments for sleep problems, but in her view they really need to be better validated for use in primary care before widespread adoption.
“Simply having providers ask three quick questions about their patients’ sleep and daytime function is a good, practical approach: How’s the quality of your sleep? How much do you sleep on average? Do you have enough energy during the daytime to get through your day-to-day functioning? That’s informative enough to indicate the utility of moving on to longer screening tools or to referral for evidence-based treatments,” she said.
Dr. Troxel noted successful intervention in sleep problems in civilian spouses is a priority for reasons beyond their personal well-being. “Civilian spouses are most often the primary caretakers for service members who return from war with either invisible mental health wounds or physical health wounds,” she said. “And we’re expecting to have need for a lot of caretakers.”
The RAND National Defense Research Institute, the Office of the Surgeon General, the U.S. Army, and the Defense Centers of Excellence for Psychological Health and Traumatic Brain Injury funded the study.
DENVER – Sleep problems and their host of deleterious physical and psychosocial consequences are pervasive among the civilian female spouses of U.S. military service members, according to the first large study to examine the issue.
“Our findings suggest that if we’re trying to promote the resilience and adjustment of military spouses and their families – particularly in light of 14 years of protracted overseas combat, where many families are experiencing deployment – sleep might be a really important target,” Wendy M. Troxel, PhD, said while presenting the study results at the annual meeting of the Associated Professional Sleep Societies.
The study population consisted of a nationally representative group of 1,805 female civilian spouses of military service members. Forty-four percent reported short sleep duration, meaning 6 hours or less per night. Another 18% got 5 hours or less. The Centers for Disease Control and Prevention and the American Academy of Sleep Medicine recommend 7 hours or more, noted Dr. Troxel, senior behavioral and social scientist at RAND Corporation in Pittsburgh.
More than half (54%) of military spouses reported that their sleep problems contributed to daytime impairment, such as intolerance of their spouse or children, frequent crying, or suboptimal performance at work or chores. One-third of the spouses reported feeling daytime fatigue three or more times per week, and another 29% experienced daytime fatigue one or two days per week.
As has been observed in other studies, sleep problems in military spouses were associated with psychosocial impairment. In linear regression analyses, the spouses with poor sleep quality, daytime impairment, and/or fatigue had significantly more depressive symptoms on the Patient Health Questionnaire–8, lower marital satisfaction, and poor self-rated health. Women with shorter sleep duration had more depressive symptoms and poorer self-rated health, but not lower marital satisfaction.
The spouses of services members currently deployed in combat zones had significantly worse sleep quality as measured on the Pittsburgh Sleep Quality Index compared with spouses of previously or never-deployed service members. But for the other outcomes of interest – sleep duration, daytime impairment, and fatigue – there were no differences based upon deployment status.
“This shows that it’s not just about the stress of combat deployment, it’s also about the stresses of everyday military life. Military families experience a great deal of stress that could lead to sleep disturbances whether or not a service member has been deployed: frequent residential moves, very long and unpredictable work hours, demanding jobs, threatening training environments,” Dr. Troxel said. “I think we need to be thinking about sleep as an important health indicator of military families in general across the deployment cycle.”
The study results are a call to action, she added.
“These findings highlight the importance of targeted screening, prevention, and intervention methods for military spouses. And primary care is where most people present with sleep problems,” Dr. Troxel said.
There are formal screening instruments for sleep problems, but in her view they really need to be better validated for use in primary care before widespread adoption.
“Simply having providers ask three quick questions about their patients’ sleep and daytime function is a good, practical approach: How’s the quality of your sleep? How much do you sleep on average? Do you have enough energy during the daytime to get through your day-to-day functioning? That’s informative enough to indicate the utility of moving on to longer screening tools or to referral for evidence-based treatments,” she said.
Dr. Troxel noted successful intervention in sleep problems in civilian spouses is a priority for reasons beyond their personal well-being. “Civilian spouses are most often the primary caretakers for service members who return from war with either invisible mental health wounds or physical health wounds,” she said. “And we’re expecting to have need for a lot of caretakers.”
The RAND National Defense Research Institute, the Office of the Surgeon General, the U.S. Army, and the Defense Centers of Excellence for Psychological Health and Traumatic Brain Injury funded the study.
DENVER – Sleep problems and their host of deleterious physical and psychosocial consequences are pervasive among the civilian female spouses of U.S. military service members, according to the first large study to examine the issue.
“Our findings suggest that if we’re trying to promote the resilience and adjustment of military spouses and their families – particularly in light of 14 years of protracted overseas combat, where many families are experiencing deployment – sleep might be a really important target,” Wendy M. Troxel, PhD, said while presenting the study results at the annual meeting of the Associated Professional Sleep Societies.
The study population consisted of a nationally representative group of 1,805 female civilian spouses of military service members. Forty-four percent reported short sleep duration, meaning 6 hours or less per night. Another 18% got 5 hours or less. The Centers for Disease Control and Prevention and the American Academy of Sleep Medicine recommend 7 hours or more, noted Dr. Troxel, senior behavioral and social scientist at RAND Corporation in Pittsburgh.
More than half (54%) of military spouses reported that their sleep problems contributed to daytime impairment, such as intolerance of their spouse or children, frequent crying, or suboptimal performance at work or chores. One-third of the spouses reported feeling daytime fatigue three or more times per week, and another 29% experienced daytime fatigue one or two days per week.
As has been observed in other studies, sleep problems in military spouses were associated with psychosocial impairment. In linear regression analyses, the spouses with poor sleep quality, daytime impairment, and/or fatigue had significantly more depressive symptoms on the Patient Health Questionnaire–8, lower marital satisfaction, and poor self-rated health. Women with shorter sleep duration had more depressive symptoms and poorer self-rated health, but not lower marital satisfaction.
The spouses of services members currently deployed in combat zones had significantly worse sleep quality as measured on the Pittsburgh Sleep Quality Index compared with spouses of previously or never-deployed service members. But for the other outcomes of interest – sleep duration, daytime impairment, and fatigue – there were no differences based upon deployment status.
“This shows that it’s not just about the stress of combat deployment, it’s also about the stresses of everyday military life. Military families experience a great deal of stress that could lead to sleep disturbances whether or not a service member has been deployed: frequent residential moves, very long and unpredictable work hours, demanding jobs, threatening training environments,” Dr. Troxel said. “I think we need to be thinking about sleep as an important health indicator of military families in general across the deployment cycle.”
The study results are a call to action, she added.
“These findings highlight the importance of targeted screening, prevention, and intervention methods for military spouses. And primary care is where most people present with sleep problems,” Dr. Troxel said.
There are formal screening instruments for sleep problems, but in her view they really need to be better validated for use in primary care before widespread adoption.
“Simply having providers ask three quick questions about their patients’ sleep and daytime function is a good, practical approach: How’s the quality of your sleep? How much do you sleep on average? Do you have enough energy during the daytime to get through your day-to-day functioning? That’s informative enough to indicate the utility of moving on to longer screening tools or to referral for evidence-based treatments,” she said.
Dr. Troxel noted successful intervention in sleep problems in civilian spouses is a priority for reasons beyond their personal well-being. “Civilian spouses are most often the primary caretakers for service members who return from war with either invisible mental health wounds or physical health wounds,” she said. “And we’re expecting to have need for a lot of caretakers.”
The RAND National Defense Research Institute, the Office of the Surgeon General, the U.S. Army, and the Defense Centers of Excellence for Psychological Health and Traumatic Brain Injury funded the study.
AT SLEEP 2016
Key clinical point: A significant portion of civilian female spouses of U.S. military members report short sleep duration and daytime impairment due to sleep problems.
Major finding: Fifty-four percent of a large group of civilian spouses of U.S. military service members reported daytime impairment due to sleep problems.
Data source: A cross-sectional study of the prevalence and consequences of sleep problems in 1,805 civilian female military spouses.
Disclosures: The RAND National Defense Research Institute, the Office of the Surgeon General, the U.S. Army, and the Defense Centers of Excellence for Psychological Health and Traumatic Brain Injury funded the study. Dr. Troxel is employed by RAND Corporation.

Clinical Challenges - July 2016:Angiosarcoma of the colon with multiple liver metastases
What's Your Diagnosis?
The diagnosis
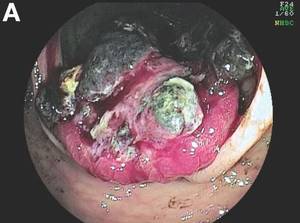
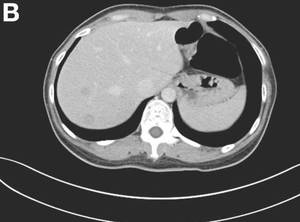
The colonoscopy disclosed an erythematous, ulcerated tumor with spontaneous bleeding in the ascending colon (Figure A). Noncontrast abdominal CT showed multiple hypoattenuating lesions in the liver. These lesions had central enhancement in the early arterial phase and progressive enhancement in the delayed phase (Figure B).
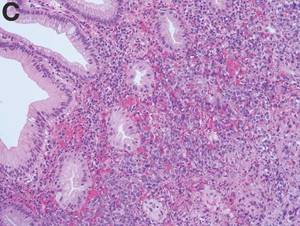
Angiosarcoma is a rare, malignant tumor arising from vascular endothelium. These tumors usually develop in skin and in the soft tissue of the head and neck region.1 Gastrointestinal angiosarcomas are rare with fewer than 20 cases reported in the literature.1,2 Patients with colonic angiosarcoma usually present with abdominal pain, rectal bleeding, or a palpable abdominal mass.1,2 On colonoscopy, the tumor has been described as a submucosal tumor1,2or a hemorrhagic mass with spontaneous bleeding3, as in this case.

1. Lo, T.H., Tsai, M.S., Chen, T.A. Angiosarcoma of sigmoid colon with intraperitoneal bleeding: case report and literature review. Ann R Coll Surg Engl. 2011;93:e91-3.
2. El Chaar, M., McQuay, N. Jr. Sigmoid colon angiosarcoma with intraperitoneal bleeding and early metastasis. J Surg Educ. 2007;64: 54-6.
3. Cammarota, G., Schinzari, G., Larocca, L.M. et al. Duodenal metastasis from a primary angiosarcoma of the colon. Gastrointest Endosc. 2006;63:330.
The diagnosis
The colonoscopy disclosed an erythematous, ulcerated tumor with spontaneous bleeding in the ascending colon (Figure A). Noncontrast abdominal CT showed multiple hypoattenuating lesions in the liver. These lesions had central enhancement in the early arterial phase and progressive enhancement in the delayed phase (Figure B).
Angiosarcoma is a rare, malignant tumor arising from vascular endothelium. These tumors usually develop in skin and in the soft tissue of the head and neck region.1 Gastrointestinal angiosarcomas are rare with fewer than 20 cases reported in the literature.1,2 Patients with colonic angiosarcoma usually present with abdominal pain, rectal bleeding, or a palpable abdominal mass.1,2 On colonoscopy, the tumor has been described as a submucosal tumor1,2or a hemorrhagic mass with spontaneous bleeding3, as in this case.
1. Lo, T.H., Tsai, M.S., Chen, T.A. Angiosarcoma of sigmoid colon with intraperitoneal bleeding: case report and literature review. Ann R Coll Surg Engl. 2011;93:e91-3.
2. El Chaar, M., McQuay, N. Jr. Sigmoid colon angiosarcoma with intraperitoneal bleeding and early metastasis. J Surg Educ. 2007;64: 54-6.
3. Cammarota, G., Schinzari, G., Larocca, L.M. et al. Duodenal metastasis from a primary angiosarcoma of the colon. Gastrointest Endosc. 2006;63:330.
The diagnosis
The colonoscopy disclosed an erythematous, ulcerated tumor with spontaneous bleeding in the ascending colon (Figure A). Noncontrast abdominal CT showed multiple hypoattenuating lesions in the liver. These lesions had central enhancement in the early arterial phase and progressive enhancement in the delayed phase (Figure B).
Angiosarcoma is a rare, malignant tumor arising from vascular endothelium. These tumors usually develop in skin and in the soft tissue of the head and neck region.1 Gastrointestinal angiosarcomas are rare with fewer than 20 cases reported in the literature.1,2 Patients with colonic angiosarcoma usually present with abdominal pain, rectal bleeding, or a palpable abdominal mass.1,2 On colonoscopy, the tumor has been described as a submucosal tumor1,2or a hemorrhagic mass with spontaneous bleeding3, as in this case.
1. Lo, T.H., Tsai, M.S., Chen, T.A. Angiosarcoma of sigmoid colon with intraperitoneal bleeding: case report and literature review. Ann R Coll Surg Engl. 2011;93:e91-3.
2. El Chaar, M., McQuay, N. Jr. Sigmoid colon angiosarcoma with intraperitoneal bleeding and early metastasis. J Surg Educ. 2007;64: 54-6.
3. Cammarota, G., Schinzari, G., Larocca, L.M. et al. Duodenal metastasis from a primary angiosarcoma of the colon. Gastrointest Endosc. 2006;63:330.
What's Your Diagnosis?
What's Your Diagnosis?
What's Your Diagnosis?
By Dr. Maw-Soan Soon, Dr. Chih-Jung Chen, and Dr. Hsu-Heng Yen. Published previously in Gastroenterology (2012:143:1155, 1400).
She denied weight loss or change in bowel habits.
A stool fecal occult test was positive and colonoscopy was performed (Figure A).
Debunking Psoriasis Myths: Is Psoriasis Contagious?
Myth: Psoriasis is contagious
One of the challenges patients with psoriasis face is misinformation among their peers, especially about the cause of psoriasis. Results from a survey conducted in France (N=1005) regarding knowledge of psoriasis and perceptions toward psoriasis patients indicated that approximately 16.5% of respondents believed that psoriasis is contagious; 6.8% believed that the disease is related to personal hygiene and 3.2% believed that it affects more people with low personal hygiene (Halioua et al). Moreover, approximately 62.4% of respondents recognized a lack of information about psoriasis and 19.7% noted that they have misconceptions about the disease.
Another study from Poland on the feelings of stigmatization among psoriasis patients (N=102) found that 72 psoriasis patients reported that they felt other people think their skin disease is contagious; only 30 psoriasis patients indicated that they did not feel this way (Hrehorów et al).
These findings underscore the need for more information on psoriasis in the general public worldwide. It is important to spread the message that psoriasis is a chronic inflammatory disease of the immune system that affects approximately 7.5 million individuals in the United States. One cannot "catch" psoriasis; psoriasis starts or worsens because of a variety of triggers (eg, infections, injury to the skin, stress, cold weather, smoking, heavy alcohol consumption, certain medications). The National Institute of Arthritis and Musculoskeletal and Skin Diseases offers a simple but revealing explanation for the cause of psoriasis: "Normally, T cells help protect the body against infection and disease. In the case of psoriasis, T cells are put into action by mistake and become so active that they trigger other immune responses, which lead to inflammation and to rapid turnover of skin cells." Genetics may play a role in the development of psoriasis, along with environmental factors.
With several new therapies available, psoriasis has gained media attention. Dermatologists can circulate more information about the causes of psoriasis, which may work toward helping patients with psoriasis cope with the social impact of the condition.
Halioua B, Sid-Mohand D, Roussel ME, et al. Extent of misconceptions, negative prejudices and discriminatory behaviour to psoriasis patients in France. J Eur Acad Dermatol Venereol. 2016;30:650-654.
Hrehorów E, Salomon J, Matusiak L, et al. Patients with psoriasis feel stigmatized. Acta Derm Venereol. 2012;92:67-72.
Psoriasis. American Academy of Dermatology website. https://www.aad.org/media/stats/conditions/psoriasis. Accessed July 7, 2016.
Psoriasis causes. Mayo Clinic website. http://www.mayoclinic.org/diseases-conditions/psoriasis/basics/causes/con-20030838. Published June 17, 2015. Accessed July 7, 2016.
Questions and answers about psoriasis. National Institute of Arthritis and Musculoskeletal and Skin Diseases website. http://www.niams.nih.gov/Health_Info/Psoriasis/default.asp. Published October 2013. Accessed July 7, 2016.
Myth: Psoriasis is contagious
One of the challenges patients with psoriasis face is misinformation among their peers, especially about the cause of psoriasis. Results from a survey conducted in France (N=1005) regarding knowledge of psoriasis and perceptions toward psoriasis patients indicated that approximately 16.5% of respondents believed that psoriasis is contagious; 6.8% believed that the disease is related to personal hygiene and 3.2% believed that it affects more people with low personal hygiene (Halioua et al). Moreover, approximately 62.4% of respondents recognized a lack of information about psoriasis and 19.7% noted that they have misconceptions about the disease.
Another study from Poland on the feelings of stigmatization among psoriasis patients (N=102) found that 72 psoriasis patients reported that they felt other people think their skin disease is contagious; only 30 psoriasis patients indicated that they did not feel this way (Hrehorów et al).
These findings underscore the need for more information on psoriasis in the general public worldwide. It is important to spread the message that psoriasis is a chronic inflammatory disease of the immune system that affects approximately 7.5 million individuals in the United States. One cannot "catch" psoriasis; psoriasis starts or worsens because of a variety of triggers (eg, infections, injury to the skin, stress, cold weather, smoking, heavy alcohol consumption, certain medications). The National Institute of Arthritis and Musculoskeletal and Skin Diseases offers a simple but revealing explanation for the cause of psoriasis: "Normally, T cells help protect the body against infection and disease. In the case of psoriasis, T cells are put into action by mistake and become so active that they trigger other immune responses, which lead to inflammation and to rapid turnover of skin cells." Genetics may play a role in the development of psoriasis, along with environmental factors.
With several new therapies available, psoriasis has gained media attention. Dermatologists can circulate more information about the causes of psoriasis, which may work toward helping patients with psoriasis cope with the social impact of the condition.
Myth: Psoriasis is contagious
One of the challenges patients with psoriasis face is misinformation among their peers, especially about the cause of psoriasis. Results from a survey conducted in France (N=1005) regarding knowledge of psoriasis and perceptions toward psoriasis patients indicated that approximately 16.5% of respondents believed that psoriasis is contagious; 6.8% believed that the disease is related to personal hygiene and 3.2% believed that it affects more people with low personal hygiene (Halioua et al). Moreover, approximately 62.4% of respondents recognized a lack of information about psoriasis and 19.7% noted that they have misconceptions about the disease.
Another study from Poland on the feelings of stigmatization among psoriasis patients (N=102) found that 72 psoriasis patients reported that they felt other people think their skin disease is contagious; only 30 psoriasis patients indicated that they did not feel this way (Hrehorów et al).
These findings underscore the need for more information on psoriasis in the general public worldwide. It is important to spread the message that psoriasis is a chronic inflammatory disease of the immune system that affects approximately 7.5 million individuals in the United States. One cannot "catch" psoriasis; psoriasis starts or worsens because of a variety of triggers (eg, infections, injury to the skin, stress, cold weather, smoking, heavy alcohol consumption, certain medications). The National Institute of Arthritis and Musculoskeletal and Skin Diseases offers a simple but revealing explanation for the cause of psoriasis: "Normally, T cells help protect the body against infection and disease. In the case of psoriasis, T cells are put into action by mistake and become so active that they trigger other immune responses, which lead to inflammation and to rapid turnover of skin cells." Genetics may play a role in the development of psoriasis, along with environmental factors.
With several new therapies available, psoriasis has gained media attention. Dermatologists can circulate more information about the causes of psoriasis, which may work toward helping patients with psoriasis cope with the social impact of the condition.
Halioua B, Sid-Mohand D, Roussel ME, et al. Extent of misconceptions, negative prejudices and discriminatory behaviour to psoriasis patients in France. J Eur Acad Dermatol Venereol. 2016;30:650-654.
Hrehorów E, Salomon J, Matusiak L, et al. Patients with psoriasis feel stigmatized. Acta Derm Venereol. 2012;92:67-72.
Psoriasis. American Academy of Dermatology website. https://www.aad.org/media/stats/conditions/psoriasis. Accessed July 7, 2016.
Psoriasis causes. Mayo Clinic website. http://www.mayoclinic.org/diseases-conditions/psoriasis/basics/causes/con-20030838. Published June 17, 2015. Accessed July 7, 2016.
Questions and answers about psoriasis. National Institute of Arthritis and Musculoskeletal and Skin Diseases website. http://www.niams.nih.gov/Health_Info/Psoriasis/default.asp. Published October 2013. Accessed July 7, 2016.
Halioua B, Sid-Mohand D, Roussel ME, et al. Extent of misconceptions, negative prejudices and discriminatory behaviour to psoriasis patients in France. J Eur Acad Dermatol Venereol. 2016;30:650-654.
Hrehorów E, Salomon J, Matusiak L, et al. Patients with psoriasis feel stigmatized. Acta Derm Venereol. 2012;92:67-72.
Psoriasis. American Academy of Dermatology website. https://www.aad.org/media/stats/conditions/psoriasis. Accessed July 7, 2016.
Psoriasis causes. Mayo Clinic website. http://www.mayoclinic.org/diseases-conditions/psoriasis/basics/causes/con-20030838. Published June 17, 2015. Accessed July 7, 2016.
Questions and answers about psoriasis. National Institute of Arthritis and Musculoskeletal and Skin Diseases website. http://www.niams.nih.gov/Health_Info/Psoriasis/default.asp. Published October 2013. Accessed July 7, 2016.

Collaborative depression care model offers promise in rural practice
An ob.gyn. practice in rural Washington state has successfully implemented a collaborative care model for depression management that relies on “depression care managers” and a consulting psychiatrist to aid ob.gyns. in providing effective depression care.
The initiative was a rural test run of the DAWN (Depression Attention for Women Now) intervention that was shown in a randomized controlled trial 2 years ago to improve depression outcomes for women in two urban ob.gyn. clinics affiliated with the University of Washington in Seattle. Both the trial and the pilot project were funded by the National Institute of Mental Health.

Nearly 75% of the 25 women enrolled in the rural pilot project had a significant improvement in their Patient Health Questionnaire-9 (PHQ-9) scores, and more than half improved their depression symptoms by at least 50%, Roger Rowles, MD, of the Generations practice in Yakima, Wash., and Susan D. Reed, MD, a coinvestigator of the original DAWN trial, reported at the annual meeting of the American College of Obstetricians and Gynecologists.
They urged others to consider taking a similar collaborative approach to depression care, especially now that the U.S. Preventive Services Task Force has recommended depression screening for all adults, including pregnant and postpartum women.
Previously, “when we identified someone with perinatal depression, we’d need a 30-minute initial consultation and then 15-20 minutes on a regular basis,” Dr. Rowles said. “We didn’t have the time to do that. ... And I had no training. Most of us felt we weren’t capable [of providing quality depression care].”
In the DAWN model, a social worker, nurse, medical assistant, or other staff member is trained to provide collaborative depression care and takes charge of this care, regularly meeting in-person or by phone with the patient to promote engagement and to closely monitor treatment progress.
Patients choose their initial treatment, including medication and Problem-Solving Treatment in Primary Care (PST-PC) therapy, an evidence-based brief behavioral intervention that helps identify stressors and improve problem solving. The depression care manager delivers the PST-PC therapy, tracks treatment response and compliance, and participates in weekly structured case reviews with the ob.gyn. and a consulting psychiatrist.
Results then and now
The original DAWN trial randomized women at two urban clinics to either 12 months of collaborative depression care or to usual care. Usual care included educational material, access to the clinic social worker, and possible psychiatry referral and prescriptions from the ob.gyn.
At 6 months, the reduction in depression scores as measured by the 20-item Hopkins Symptom Checklist was similar between the two groups, but at 12 months and at 18 months follow-up, the intervention group had significantly lower scores. They were more likely to have at least a 50% decrease in depressive symptoms at 12 months and were significantly more satisfied with their depression care (Obstet Gynecol. 2014 Jun;123[6]:1237-46).
The pilot project in Yakima, a farming community of 80,000 people, was of shorter duration than the randomized trial and focused on women coming for periconception, pregnancy, and postpartum care.
The majority – 74% – had significant improvement in their PHQ scores (a final score of less than 10), and almost one-third had a final score of less than 5. A score of 10 or more indicates the likelihood of having major depression. More than half – 59% – had at least a 50% improvement in depressive symptoms.
Unlike in the original DAWN trial, depression care managers in the Yakima project used text messaging in addition to phone calls to stay engaged with patients and monitor treatment. Almost all of the 25 enrolled women received PST-PC, and approximately 56% received antidepressants, for a mean treatment time of 14 weeks.
“Given that short duration of follow-up, the improvement we saw was very good,” said Amritha Bhat, MD, MBBS, the University of Washington psychiatrist who served as the consulting psychiatrist for the project.
Without a placebo-controlled arm, the researchers don’t know how much of the improvement was due to the collaborative care, Dr. Bhat said in an interview. “But we know now that it’s feasible in a rural setting.”
Depression care needs
Women have two times the rate of major depression as men, with prevalence rates of 13% annually and 21% over a lifetime, and low-income and minority women are at highest risk of depression and are also more likely to seek routine care from ob.gyns., according to Dr. Reed, who is a professor of ob.gyn at the University of Washington, Seattle, and chief of ob.gyn. at Harborview Medical Center.
Dr. Rowles said he was “astounded” that 30% of the screened patients in his practice had positive results. Many “either didn’t want to participate in the project or had exclusions, but even so we enrolled [our cohort] quickly,” he said.
The DAWN model stipulates that depression care managers support women as much as possible with social service interventions, facilitating financial assistance for medications, and serving as a “point person” for assistance with housing, food, domestic abuse, and other issues.
The task of integrating a social service element into depression care is necessary but can be daunting, Dr. Rowles said. In Yakima, he said, the significant need for basic assistance was a “big frustration” to the depression care managers involved in the project. “They wanted to do more, because they’d identified these problems and had a rapport [with the women], but our resources in Yakima are not that great.”
To prepare the Yakima practice for the intervention, a University of Washington team visited the clinic to educate staff about collaborative care, and three depression care managers were trained to deliver PST-PC and manage care. The depression care managers – a nurse employed by the local hospital, a nurse employed by another clinic for farm workers, and a local psychologist – “were the glue for this program,” Dr. Rowles said.
In other settings, depression care managers might more likely be clinic social workers or other members of the practice.
A psychiatrist’s involvement is also crucial, particularly when it comes to prescribing antidepressant medications at their full effective doses, Dr. Reed stressed. “You won’t feel comfortable pushing those doses to the max without [a psychiatrist to consult with],” Dr. Reed said at the ACOG meeting.
In the DAWN trial, similar numbers of patients in the collaborative care group and the usual care group were prescribed antidepressants, but more patients in the intervention group had their doses increased to an effective therapeutic range, and more patients adhered to their medication regimens, she said.
The 12-month DAWN intervention was provided at a cost per patient of up to $1,000.
Dr. Reed’s clinic has sustained funding for the intervention since the National Institute of Mental Health grant expired, and Dr. Rowles said he was seeking funding to continue DAWN at his Yakima clinic. Funding sources for ob.gyn. practices interested in implementing the model may include state funding agencies and organizations such as the March of Dimes, Dr. Reed said, noting that some states offer maternal services support that could be helpful for integrating collaborative depression care.
While the pilot project focused on pregnancy care, Dr. Reed urged ob.gyns. to think broadly. “Do you know when you catch these women? When they come in for their Pap smears and their routine care, before they become pregnant,” she said. “If you help them with their mood disorder early, they’ll do so much better with pregnancy.”
Information about DAWN – including resources on PST-PC and antidepressant medication, and an intervention manual – can be found at www.dawncare.org.
An ob.gyn. practice in rural Washington state has successfully implemented a collaborative care model for depression management that relies on “depression care managers” and a consulting psychiatrist to aid ob.gyns. in providing effective depression care.
The initiative was a rural test run of the DAWN (Depression Attention for Women Now) intervention that was shown in a randomized controlled trial 2 years ago to improve depression outcomes for women in two urban ob.gyn. clinics affiliated with the University of Washington in Seattle. Both the trial and the pilot project were funded by the National Institute of Mental Health.
Nearly 75% of the 25 women enrolled in the rural pilot project had a significant improvement in their Patient Health Questionnaire-9 (PHQ-9) scores, and more than half improved their depression symptoms by at least 50%, Roger Rowles, MD, of the Generations practice in Yakima, Wash., and Susan D. Reed, MD, a coinvestigator of the original DAWN trial, reported at the annual meeting of the American College of Obstetricians and Gynecologists.
They urged others to consider taking a similar collaborative approach to depression care, especially now that the U.S. Preventive Services Task Force has recommended depression screening for all adults, including pregnant and postpartum women.
Previously, “when we identified someone with perinatal depression, we’d need a 30-minute initial consultation and then 15-20 minutes on a regular basis,” Dr. Rowles said. “We didn’t have the time to do that. ... And I had no training. Most of us felt we weren’t capable [of providing quality depression care].”
In the DAWN model, a social worker, nurse, medical assistant, or other staff member is trained to provide collaborative depression care and takes charge of this care, regularly meeting in-person or by phone with the patient to promote engagement and to closely monitor treatment progress.
Patients choose their initial treatment, including medication and Problem-Solving Treatment in Primary Care (PST-PC) therapy, an evidence-based brief behavioral intervention that helps identify stressors and improve problem solving. The depression care manager delivers the PST-PC therapy, tracks treatment response and compliance, and participates in weekly structured case reviews with the ob.gyn. and a consulting psychiatrist.
Results then and now
The original DAWN trial randomized women at two urban clinics to either 12 months of collaborative depression care or to usual care. Usual care included educational material, access to the clinic social worker, and possible psychiatry referral and prescriptions from the ob.gyn.
At 6 months, the reduction in depression scores as measured by the 20-item Hopkins Symptom Checklist was similar between the two groups, but at 12 months and at 18 months follow-up, the intervention group had significantly lower scores. They were more likely to have at least a 50% decrease in depressive symptoms at 12 months and were significantly more satisfied with their depression care (Obstet Gynecol. 2014 Jun;123[6]:1237-46).
The pilot project in Yakima, a farming community of 80,000 people, was of shorter duration than the randomized trial and focused on women coming for periconception, pregnancy, and postpartum care.
The majority – 74% – had significant improvement in their PHQ scores (a final score of less than 10), and almost one-third had a final score of less than 5. A score of 10 or more indicates the likelihood of having major depression. More than half – 59% – had at least a 50% improvement in depressive symptoms.
Unlike in the original DAWN trial, depression care managers in the Yakima project used text messaging in addition to phone calls to stay engaged with patients and monitor treatment. Almost all of the 25 enrolled women received PST-PC, and approximately 56% received antidepressants, for a mean treatment time of 14 weeks.
“Given that short duration of follow-up, the improvement we saw was very good,” said Amritha Bhat, MD, MBBS, the University of Washington psychiatrist who served as the consulting psychiatrist for the project.
Without a placebo-controlled arm, the researchers don’t know how much of the improvement was due to the collaborative care, Dr. Bhat said in an interview. “But we know now that it’s feasible in a rural setting.”
Depression care needs
Women have two times the rate of major depression as men, with prevalence rates of 13% annually and 21% over a lifetime, and low-income and minority women are at highest risk of depression and are also more likely to seek routine care from ob.gyns., according to Dr. Reed, who is a professor of ob.gyn at the University of Washington, Seattle, and chief of ob.gyn. at Harborview Medical Center.
Dr. Rowles said he was “astounded” that 30% of the screened patients in his practice had positive results. Many “either didn’t want to participate in the project or had exclusions, but even so we enrolled [our cohort] quickly,” he said.
The DAWN model stipulates that depression care managers support women as much as possible with social service interventions, facilitating financial assistance for medications, and serving as a “point person” for assistance with housing, food, domestic abuse, and other issues.
The task of integrating a social service element into depression care is necessary but can be daunting, Dr. Rowles said. In Yakima, he said, the significant need for basic assistance was a “big frustration” to the depression care managers involved in the project. “They wanted to do more, because they’d identified these problems and had a rapport [with the women], but our resources in Yakima are not that great.”
To prepare the Yakima practice for the intervention, a University of Washington team visited the clinic to educate staff about collaborative care, and three depression care managers were trained to deliver PST-PC and manage care. The depression care managers – a nurse employed by the local hospital, a nurse employed by another clinic for farm workers, and a local psychologist – “were the glue for this program,” Dr. Rowles said.
In other settings, depression care managers might more likely be clinic social workers or other members of the practice.
A psychiatrist’s involvement is also crucial, particularly when it comes to prescribing antidepressant medications at their full effective doses, Dr. Reed stressed. “You won’t feel comfortable pushing those doses to the max without [a psychiatrist to consult with],” Dr. Reed said at the ACOG meeting.
In the DAWN trial, similar numbers of patients in the collaborative care group and the usual care group were prescribed antidepressants, but more patients in the intervention group had their doses increased to an effective therapeutic range, and more patients adhered to their medication regimens, she said.
The 12-month DAWN intervention was provided at a cost per patient of up to $1,000.
Dr. Reed’s clinic has sustained funding for the intervention since the National Institute of Mental Health grant expired, and Dr. Rowles said he was seeking funding to continue DAWN at his Yakima clinic. Funding sources for ob.gyn. practices interested in implementing the model may include state funding agencies and organizations such as the March of Dimes, Dr. Reed said, noting that some states offer maternal services support that could be helpful for integrating collaborative depression care.
While the pilot project focused on pregnancy care, Dr. Reed urged ob.gyns. to think broadly. “Do you know when you catch these women? When they come in for their Pap smears and their routine care, before they become pregnant,” she said. “If you help them with their mood disorder early, they’ll do so much better with pregnancy.”
Information about DAWN – including resources on PST-PC and antidepressant medication, and an intervention manual – can be found at www.dawncare.org.
An ob.gyn. practice in rural Washington state has successfully implemented a collaborative care model for depression management that relies on “depression care managers” and a consulting psychiatrist to aid ob.gyns. in providing effective depression care.
The initiative was a rural test run of the DAWN (Depression Attention for Women Now) intervention that was shown in a randomized controlled trial 2 years ago to improve depression outcomes for women in two urban ob.gyn. clinics affiliated with the University of Washington in Seattle. Both the trial and the pilot project were funded by the National Institute of Mental Health.
Nearly 75% of the 25 women enrolled in the rural pilot project had a significant improvement in their Patient Health Questionnaire-9 (PHQ-9) scores, and more than half improved their depression symptoms by at least 50%, Roger Rowles, MD, of the Generations practice in Yakima, Wash., and Susan D. Reed, MD, a coinvestigator of the original DAWN trial, reported at the annual meeting of the American College of Obstetricians and Gynecologists.
They urged others to consider taking a similar collaborative approach to depression care, especially now that the U.S. Preventive Services Task Force has recommended depression screening for all adults, including pregnant and postpartum women.
Previously, “when we identified someone with perinatal depression, we’d need a 30-minute initial consultation and then 15-20 minutes on a regular basis,” Dr. Rowles said. “We didn’t have the time to do that. ... And I had no training. Most of us felt we weren’t capable [of providing quality depression care].”
In the DAWN model, a social worker, nurse, medical assistant, or other staff member is trained to provide collaborative depression care and takes charge of this care, regularly meeting in-person or by phone with the patient to promote engagement and to closely monitor treatment progress.
Patients choose their initial treatment, including medication and Problem-Solving Treatment in Primary Care (PST-PC) therapy, an evidence-based brief behavioral intervention that helps identify stressors and improve problem solving. The depression care manager delivers the PST-PC therapy, tracks treatment response and compliance, and participates in weekly structured case reviews with the ob.gyn. and a consulting psychiatrist.
Results then and now
The original DAWN trial randomized women at two urban clinics to either 12 months of collaborative depression care or to usual care. Usual care included educational material, access to the clinic social worker, and possible psychiatry referral and prescriptions from the ob.gyn.
At 6 months, the reduction in depression scores as measured by the 20-item Hopkins Symptom Checklist was similar between the two groups, but at 12 months and at 18 months follow-up, the intervention group had significantly lower scores. They were more likely to have at least a 50% decrease in depressive symptoms at 12 months and were significantly more satisfied with their depression care (Obstet Gynecol. 2014 Jun;123[6]:1237-46).
The pilot project in Yakima, a farming community of 80,000 people, was of shorter duration than the randomized trial and focused on women coming for periconception, pregnancy, and postpartum care.
The majority – 74% – had significant improvement in their PHQ scores (a final score of less than 10), and almost one-third had a final score of less than 5. A score of 10 or more indicates the likelihood of having major depression. More than half – 59% – had at least a 50% improvement in depressive symptoms.
Unlike in the original DAWN trial, depression care managers in the Yakima project used text messaging in addition to phone calls to stay engaged with patients and monitor treatment. Almost all of the 25 enrolled women received PST-PC, and approximately 56% received antidepressants, for a mean treatment time of 14 weeks.
“Given that short duration of follow-up, the improvement we saw was very good,” said Amritha Bhat, MD, MBBS, the University of Washington psychiatrist who served as the consulting psychiatrist for the project.
Without a placebo-controlled arm, the researchers don’t know how much of the improvement was due to the collaborative care, Dr. Bhat said in an interview. “But we know now that it’s feasible in a rural setting.”
Depression care needs
Women have two times the rate of major depression as men, with prevalence rates of 13% annually and 21% over a lifetime, and low-income and minority women are at highest risk of depression and are also more likely to seek routine care from ob.gyns., according to Dr. Reed, who is a professor of ob.gyn at the University of Washington, Seattle, and chief of ob.gyn. at Harborview Medical Center.
Dr. Rowles said he was “astounded” that 30% of the screened patients in his practice had positive results. Many “either didn’t want to participate in the project or had exclusions, but even so we enrolled [our cohort] quickly,” he said.
The DAWN model stipulates that depression care managers support women as much as possible with social service interventions, facilitating financial assistance for medications, and serving as a “point person” for assistance with housing, food, domestic abuse, and other issues.
The task of integrating a social service element into depression care is necessary but can be daunting, Dr. Rowles said. In Yakima, he said, the significant need for basic assistance was a “big frustration” to the depression care managers involved in the project. “They wanted to do more, because they’d identified these problems and had a rapport [with the women], but our resources in Yakima are not that great.”
To prepare the Yakima practice for the intervention, a University of Washington team visited the clinic to educate staff about collaborative care, and three depression care managers were trained to deliver PST-PC and manage care. The depression care managers – a nurse employed by the local hospital, a nurse employed by another clinic for farm workers, and a local psychologist – “were the glue for this program,” Dr. Rowles said.
In other settings, depression care managers might more likely be clinic social workers or other members of the practice.
A psychiatrist’s involvement is also crucial, particularly when it comes to prescribing antidepressant medications at their full effective doses, Dr. Reed stressed. “You won’t feel comfortable pushing those doses to the max without [a psychiatrist to consult with],” Dr. Reed said at the ACOG meeting.
In the DAWN trial, similar numbers of patients in the collaborative care group and the usual care group were prescribed antidepressants, but more patients in the intervention group had their doses increased to an effective therapeutic range, and more patients adhered to their medication regimens, she said.
The 12-month DAWN intervention was provided at a cost per patient of up to $1,000.
Dr. Reed’s clinic has sustained funding for the intervention since the National Institute of Mental Health grant expired, and Dr. Rowles said he was seeking funding to continue DAWN at his Yakima clinic. Funding sources for ob.gyn. practices interested in implementing the model may include state funding agencies and organizations such as the March of Dimes, Dr. Reed said, noting that some states offer maternal services support that could be helpful for integrating collaborative depression care.
While the pilot project focused on pregnancy care, Dr. Reed urged ob.gyns. to think broadly. “Do you know when you catch these women? When they come in for their Pap smears and their routine care, before they become pregnant,” she said. “If you help them with their mood disorder early, they’ll do so much better with pregnancy.”
Information about DAWN – including resources on PST-PC and antidepressant medication, and an intervention manual – can be found at www.dawncare.org.

Method reveals cells of origin in AML

in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
US cancer centers spend millions in advertising

chemotherapy
Photo by Rhoda Baer
US cancer centers have substantially increased spending for consumer-directed advertising in recent years, according to researchers.
The team looked at consumer advertising for 890 cancer centers and found that total advertising spending for these centers increased from $54,229,849 in 2005 to $173,510,900 in 2014.
The researchers reported these findings in a letter to JAMA Internal Medicine.
The team analyzed data from an agency that tracks the content and number of advertisements and calculates expenditures. An advertiser was considered a cancer center if its name contained certain key words.
Advertising expenditure data covered 6 media outlets: television, magazines, radio, newspapers, billboards, and the Internet.
According to these data, from 2005 to 2014, 890 cancer centers advertised to the public. In general, inflation-adjusted spending increased for all of the types of advertising the researchers analyzed.
The team said the greatest relative growth in spending was for Internet display advertisements, which increased from less than 1% of total advertising spending in 2005 ($302,030/$54,229,849) to 5% in 2014 ($8,633,000/$173,510,900).
The researchers also found that, in 2014, 20 cancer centers accounted for 86% of the roughly $174 million total advertising spending.
Cancer Treatment Centers of America spent the most (at $101.7 million), followed by MD Anderson Cancer Center (at $13.9 million) and Memorial Sloan Kettering Cancer Center (at $9.1 million).
Among the 20 centers, 5 were for-profit, 17 were Commission on Cancer-accredited, and 9 were National Cancer Institute-designated.
The researchers said their findings likely underestimate advertising spending because the available data didn’t include all types of advertising, such as ads in cancer-specific magazines.
The team also said the effect of cancer-center advertising on the quality and costs of cancer care should be investigated. ![]()
chemotherapy
Photo by Rhoda Baer
US cancer centers have substantially increased spending for consumer-directed advertising in recent years, according to researchers.
The team looked at consumer advertising for 890 cancer centers and found that total advertising spending for these centers increased from $54,229,849 in 2005 to $173,510,900 in 2014.
The researchers reported these findings in a letter to JAMA Internal Medicine.
The team analyzed data from an agency that tracks the content and number of advertisements and calculates expenditures. An advertiser was considered a cancer center if its name contained certain key words.
Advertising expenditure data covered 6 media outlets: television, magazines, radio, newspapers, billboards, and the Internet.
According to these data, from 2005 to 2014, 890 cancer centers advertised to the public. In general, inflation-adjusted spending increased for all of the types of advertising the researchers analyzed.
The team said the greatest relative growth in spending was for Internet display advertisements, which increased from less than 1% of total advertising spending in 2005 ($302,030/$54,229,849) to 5% in 2014 ($8,633,000/$173,510,900).
The researchers also found that, in 2014, 20 cancer centers accounted for 86% of the roughly $174 million total advertising spending.
Cancer Treatment Centers of America spent the most (at $101.7 million), followed by MD Anderson Cancer Center (at $13.9 million) and Memorial Sloan Kettering Cancer Center (at $9.1 million).
Among the 20 centers, 5 were for-profit, 17 were Commission on Cancer-accredited, and 9 were National Cancer Institute-designated.
The researchers said their findings likely underestimate advertising spending because the available data didn’t include all types of advertising, such as ads in cancer-specific magazines.
The team also said the effect of cancer-center advertising on the quality and costs of cancer care should be investigated. ![]()
chemotherapy
Photo by Rhoda Baer
US cancer centers have substantially increased spending for consumer-directed advertising in recent years, according to researchers.
The team looked at consumer advertising for 890 cancer centers and found that total advertising spending for these centers increased from $54,229,849 in 2005 to $173,510,900 in 2014.
The researchers reported these findings in a letter to JAMA Internal Medicine.
The team analyzed data from an agency that tracks the content and number of advertisements and calculates expenditures. An advertiser was considered a cancer center if its name contained certain key words.
Advertising expenditure data covered 6 media outlets: television, magazines, radio, newspapers, billboards, and the Internet.
According to these data, from 2005 to 2014, 890 cancer centers advertised to the public. In general, inflation-adjusted spending increased for all of the types of advertising the researchers analyzed.
The team said the greatest relative growth in spending was for Internet display advertisements, which increased from less than 1% of total advertising spending in 2005 ($302,030/$54,229,849) to 5% in 2014 ($8,633,000/$173,510,900).
The researchers also found that, in 2014, 20 cancer centers accounted for 86% of the roughly $174 million total advertising spending.
Cancer Treatment Centers of America spent the most (at $101.7 million), followed by MD Anderson Cancer Center (at $13.9 million) and Memorial Sloan Kettering Cancer Center (at $9.1 million).
Among the 20 centers, 5 were for-profit, 17 were Commission on Cancer-accredited, and 9 were National Cancer Institute-designated.
The researchers said their findings likely underestimate advertising spending because the available data didn’t include all types of advertising, such as ads in cancer-specific magazines.
The team also said the effect of cancer-center advertising on the quality and costs of cancer care should be investigated. ![]()
Finding could help treat and prevent MM, team says

Photo by Daniel Sone
Research published in Leukemia suggests bone marrow mesenchymal stem cells (BMMSCs) are altered in monoclonal gammopathy of undetermined significance (MGUS) as well as in multiple myeloma (MM).
This discovery indicates that changes in the bone marrow needed for MM to grow have already taken hold in patients with MGUS, raising the possibility that early medical intervention could prevent the development of MM.
“It is now clear that the bone marrow of patients with MGUS, traditionally thought of as a benign condition, is significantly different to that of healthy individuals,” said study author Daniel Tennant, PhD, of the University of Birmingham in the UK.
“The bone marrow environment in these patients appears capable of supporting cancer growth, even though the majority of patients will not progress to myeloma. While this research is in the early stages, it offers the exciting possibility that early intervention could potentially delay or even prevent cancer development.”
With this study, Dr Tennant and his colleagues found that, early on in MGUS development, BMMSCs change their behavior and become more supportive of cancer growth.
The team found that a key gene, PADI2, becomes particularly overactive in BMMSCs, which leads to overproduction of the signaling molecule interleukin-6 (IL-6).
BMMSCs release IL-6 into the bone marrow, where it binds with receptors on the surface of cancerous plasma cells, instructing them to multiply rapidly and resist cell death signals.
It is already known that high levels of IL-6 in a patient’s bone marrow significantly reduces the effectiveness of the drug bortezomib.
The researchers believe that drugs designed to target the PADI2 gene in MGUS and MM patients could significantly reduce the supportive signaling that MM cells depend on, and such drugs may increase the effectiveness of current treatments.
The team also noted that the PADI2 gene has been linked to other types of cancer, rheumatoid arthritis, Alzheimer’s disease, and autoimmune disease. So any drug developed could have wider applications beyond MM treatment.
“There is an urgent need for new treatments for myeloma, which, as well as being largely incurable, can have a devastating impact on quality of life,” said Alasdair Rankin, director of research at the blood cancer charity Bloodwise, which helped to fund this study.
“With an increasing elderly population, MGUS and myeloma are only going to become more common. Drugs designed to remove the support system myeloma uses to grow could be an effective way of treating the disease, or even preventing it altogether.” ![]()
Photo by Daniel Sone
Research published in Leukemia suggests bone marrow mesenchymal stem cells (BMMSCs) are altered in monoclonal gammopathy of undetermined significance (MGUS) as well as in multiple myeloma (MM).
This discovery indicates that changes in the bone marrow needed for MM to grow have already taken hold in patients with MGUS, raising the possibility that early medical intervention could prevent the development of MM.
“It is now clear that the bone marrow of patients with MGUS, traditionally thought of as a benign condition, is significantly different to that of healthy individuals,” said study author Daniel Tennant, PhD, of the University of Birmingham in the UK.
“The bone marrow environment in these patients appears capable of supporting cancer growth, even though the majority of patients will not progress to myeloma. While this research is in the early stages, it offers the exciting possibility that early intervention could potentially delay or even prevent cancer development.”
With this study, Dr Tennant and his colleagues found that, early on in MGUS development, BMMSCs change their behavior and become more supportive of cancer growth.
The team found that a key gene, PADI2, becomes particularly overactive in BMMSCs, which leads to overproduction of the signaling molecule interleukin-6 (IL-6).
BMMSCs release IL-6 into the bone marrow, where it binds with receptors on the surface of cancerous plasma cells, instructing them to multiply rapidly and resist cell death signals.
It is already known that high levels of IL-6 in a patient’s bone marrow significantly reduces the effectiveness of the drug bortezomib.
The researchers believe that drugs designed to target the PADI2 gene in MGUS and MM patients could significantly reduce the supportive signaling that MM cells depend on, and such drugs may increase the effectiveness of current treatments.
The team also noted that the PADI2 gene has been linked to other types of cancer, rheumatoid arthritis, Alzheimer’s disease, and autoimmune disease. So any drug developed could have wider applications beyond MM treatment.
“There is an urgent need for new treatments for myeloma, which, as well as being largely incurable, can have a devastating impact on quality of life,” said Alasdair Rankin, director of research at the blood cancer charity Bloodwise, which helped to fund this study.
“With an increasing elderly population, MGUS and myeloma are only going to become more common. Drugs designed to remove the support system myeloma uses to grow could be an effective way of treating the disease, or even preventing it altogether.” ![]()
Photo by Daniel Sone
Research published in Leukemia suggests bone marrow mesenchymal stem cells (BMMSCs) are altered in monoclonal gammopathy of undetermined significance (MGUS) as well as in multiple myeloma (MM).
This discovery indicates that changes in the bone marrow needed for MM to grow have already taken hold in patients with MGUS, raising the possibility that early medical intervention could prevent the development of MM.
“It is now clear that the bone marrow of patients with MGUS, traditionally thought of as a benign condition, is significantly different to that of healthy individuals,” said study author Daniel Tennant, PhD, of the University of Birmingham in the UK.
“The bone marrow environment in these patients appears capable of supporting cancer growth, even though the majority of patients will not progress to myeloma. While this research is in the early stages, it offers the exciting possibility that early intervention could potentially delay or even prevent cancer development.”
With this study, Dr Tennant and his colleagues found that, early on in MGUS development, BMMSCs change their behavior and become more supportive of cancer growth.
The team found that a key gene, PADI2, becomes particularly overactive in BMMSCs, which leads to overproduction of the signaling molecule interleukin-6 (IL-6).
BMMSCs release IL-6 into the bone marrow, where it binds with receptors on the surface of cancerous plasma cells, instructing them to multiply rapidly and resist cell death signals.
It is already known that high levels of IL-6 in a patient’s bone marrow significantly reduces the effectiveness of the drug bortezomib.
The researchers believe that drugs designed to target the PADI2 gene in MGUS and MM patients could significantly reduce the supportive signaling that MM cells depend on, and such drugs may increase the effectiveness of current treatments.
The team also noted that the PADI2 gene has been linked to other types of cancer, rheumatoid arthritis, Alzheimer’s disease, and autoimmune disease. So any drug developed could have wider applications beyond MM treatment.
“There is an urgent need for new treatments for myeloma, which, as well as being largely incurable, can have a devastating impact on quality of life,” said Alasdair Rankin, director of research at the blood cancer charity Bloodwise, which helped to fund this study.
“With an increasing elderly population, MGUS and myeloma are only going to become more common. Drugs designed to remove the support system myeloma uses to grow could be an effective way of treating the disease, or even preventing it altogether.” ![]()