User login
Incidence of HPV-associated cancers on the rise
The incidence of human papillomavirus–associated cancers is increasing in the United States, but many of these cases could be prevented by the HPV vaccine, investigators from the Centers for Disease Control and Prevention report.
Between 2008 and 2012 the incidence of HPV-associated cancers was 11.7/100,000 persons, up from 10.8/100,000 persons during the previous 4-year period, according to data from population-based cancer registries. An average of 38,793 HPV-associated cancers were diagnosed in the most recent period, 30,700 (79%) of which can be attributed to HPV. Of those, 28,500 cancers are of HPV types that are preventable with the 9-valent HPV vaccine, reported Laura J. Viens, MD, and her associates.
Among the HPV-associated cancers, an average of 11,7771 cervical cancer cases were diagnosed each year. Rates of cervical cancer per 100,000 persons were higher among blacks (9.2) than among whites (7.1), and among Hispanics (9.7) than non-Hispanics (7.1).
“Increasing [HPV] vaccination coverage could decrease the cancer incidence and disparities in the United States,” the investigators concluded.
Find the full report from Morbidity and Mortality Weekly Report here
The incidence of human papillomavirus–associated cancers is increasing in the United States, but many of these cases could be prevented by the HPV vaccine, investigators from the Centers for Disease Control and Prevention report.
Between 2008 and 2012 the incidence of HPV-associated cancers was 11.7/100,000 persons, up from 10.8/100,000 persons during the previous 4-year period, according to data from population-based cancer registries. An average of 38,793 HPV-associated cancers were diagnosed in the most recent period, 30,700 (79%) of which can be attributed to HPV. Of those, 28,500 cancers are of HPV types that are preventable with the 9-valent HPV vaccine, reported Laura J. Viens, MD, and her associates.
Among the HPV-associated cancers, an average of 11,7771 cervical cancer cases were diagnosed each year. Rates of cervical cancer per 100,000 persons were higher among blacks (9.2) than among whites (7.1), and among Hispanics (9.7) than non-Hispanics (7.1).
“Increasing [HPV] vaccination coverage could decrease the cancer incidence and disparities in the United States,” the investigators concluded.
Find the full report from Morbidity and Mortality Weekly Report here
The incidence of human papillomavirus–associated cancers is increasing in the United States, but many of these cases could be prevented by the HPV vaccine, investigators from the Centers for Disease Control and Prevention report.
Between 2008 and 2012 the incidence of HPV-associated cancers was 11.7/100,000 persons, up from 10.8/100,000 persons during the previous 4-year period, according to data from population-based cancer registries. An average of 38,793 HPV-associated cancers were diagnosed in the most recent period, 30,700 (79%) of which can be attributed to HPV. Of those, 28,500 cancers are of HPV types that are preventable with the 9-valent HPV vaccine, reported Laura J. Viens, MD, and her associates.
Among the HPV-associated cancers, an average of 11,7771 cervical cancer cases were diagnosed each year. Rates of cervical cancer per 100,000 persons were higher among blacks (9.2) than among whites (7.1), and among Hispanics (9.7) than non-Hispanics (7.1).
“Increasing [HPV] vaccination coverage could decrease the cancer incidence and disparities in the United States,” the investigators concluded.
Find the full report from Morbidity and Mortality Weekly Report here
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
No difference between chemo regimens for ovarian clear cell carcinoma
Irinotecan plus cisplatin is no better than paclitaxel plus carboplatin for women with ovarian clear cell carcinoma, according to an international study led by Japanese investigators.
Clear cell carcinoma (CCC) accounts for approximately 10% of epithelial ovarian cancers (EOCs) in Europe and the United States, but is more prevalent in Asia; In Japan CCC accounts for 24% of all EOCs.
In the first phase III, CCC-specific clinical trial, 667 women with stage I to IV CCC were randomized to receive one of two treatment regimens: irinotecan at a dose of 60 mg/m2 on days 1, 8, and 15 plus cisplatin at 60 mg/m2 on day 1 every 4 weeks for six cycles (n = 332) or paclitaxel at a dose of 175 mg/m2 plus carboplatin on day 1 every 3 weeks for six cycles (n = 335).
Japanese women made up 93.5% of the study population, the median age was 53 years, and 6.4% of patients had stage I cancer. Median follow-up time was 44.3 months.
No difference was found between the groups in progression-free or overall survival, Toru Sugiyama, MD, of Iwate Medical University, Morioka, Japan, and associates reported (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.66.9010).
Both regimens were well tolerated, though the toxicity profiles were different, investigators wrote.
Overall, 72% of patients received all six cycles of their respective chemotherapy treatments. Two-year progression-free survival rates were 73.0% (95% confidence interval, 67.7%-77.5%) among patients receiving irinotecan plus cisplatin, 77.6% (95% CI, 72.4%-81.9%) among patients receiving paclitaxel plus carboplatin, and were not significantly different (HR, 1.17; 95% CI, 0.87-1.58; P = .30).
There was also no significant difference in overall survival between the two treatment arms.
In subgroup analyses, there was no significant difference in progression-free or overall survival by disease stage or tumor size.
Grade 3 or 4 anorexia, diarrhea, nausea, vomiting, and febrile neutropenia were the most common adverse events among women receiving irinotecan plus cisplatin. Grade 3 or 4 leukopenia, neutropenia, thrombocytopenia, peripheral sensory neuropathy, and joint pain were the most common adverse events among women receiving paclitaxel plus carboplatin. No treatment-related deaths occurred in either treatment arm.
This study was funded by the Japanese Gynecologic Oncology Group. Dr. Sugiyama and 18 other investigators had no disclosures to report. Six investigators reported serving in advisory roles for or receiving financial compensation or honoraria from multiple companies.
On Twitter @jessnicolecraig
Irinotecan plus cisplatin is no better than paclitaxel plus carboplatin for women with ovarian clear cell carcinoma, according to an international study led by Japanese investigators.
Clear cell carcinoma (CCC) accounts for approximately 10% of epithelial ovarian cancers (EOCs) in Europe and the United States, but is more prevalent in Asia; In Japan CCC accounts for 24% of all EOCs.
In the first phase III, CCC-specific clinical trial, 667 women with stage I to IV CCC were randomized to receive one of two treatment regimens: irinotecan at a dose of 60 mg/m2 on days 1, 8, and 15 plus cisplatin at 60 mg/m2 on day 1 every 4 weeks for six cycles (n = 332) or paclitaxel at a dose of 175 mg/m2 plus carboplatin on day 1 every 3 weeks for six cycles (n = 335).
Japanese women made up 93.5% of the study population, the median age was 53 years, and 6.4% of patients had stage I cancer. Median follow-up time was 44.3 months.
No difference was found between the groups in progression-free or overall survival, Toru Sugiyama, MD, of Iwate Medical University, Morioka, Japan, and associates reported (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.66.9010).
Both regimens were well tolerated, though the toxicity profiles were different, investigators wrote.
Overall, 72% of patients received all six cycles of their respective chemotherapy treatments. Two-year progression-free survival rates were 73.0% (95% confidence interval, 67.7%-77.5%) among patients receiving irinotecan plus cisplatin, 77.6% (95% CI, 72.4%-81.9%) among patients receiving paclitaxel plus carboplatin, and were not significantly different (HR, 1.17; 95% CI, 0.87-1.58; P = .30).
There was also no significant difference in overall survival between the two treatment arms.
In subgroup analyses, there was no significant difference in progression-free or overall survival by disease stage or tumor size.
Grade 3 or 4 anorexia, diarrhea, nausea, vomiting, and febrile neutropenia were the most common adverse events among women receiving irinotecan plus cisplatin. Grade 3 or 4 leukopenia, neutropenia, thrombocytopenia, peripheral sensory neuropathy, and joint pain were the most common adverse events among women receiving paclitaxel plus carboplatin. No treatment-related deaths occurred in either treatment arm.
This study was funded by the Japanese Gynecologic Oncology Group. Dr. Sugiyama and 18 other investigators had no disclosures to report. Six investigators reported serving in advisory roles for or receiving financial compensation or honoraria from multiple companies.
On Twitter @jessnicolecraig
Irinotecan plus cisplatin is no better than paclitaxel plus carboplatin for women with ovarian clear cell carcinoma, according to an international study led by Japanese investigators.
Clear cell carcinoma (CCC) accounts for approximately 10% of epithelial ovarian cancers (EOCs) in Europe and the United States, but is more prevalent in Asia; In Japan CCC accounts for 24% of all EOCs.
In the first phase III, CCC-specific clinical trial, 667 women with stage I to IV CCC were randomized to receive one of two treatment regimens: irinotecan at a dose of 60 mg/m2 on days 1, 8, and 15 plus cisplatin at 60 mg/m2 on day 1 every 4 weeks for six cycles (n = 332) or paclitaxel at a dose of 175 mg/m2 plus carboplatin on day 1 every 3 weeks for six cycles (n = 335).
Japanese women made up 93.5% of the study population, the median age was 53 years, and 6.4% of patients had stage I cancer. Median follow-up time was 44.3 months.
No difference was found between the groups in progression-free or overall survival, Toru Sugiyama, MD, of Iwate Medical University, Morioka, Japan, and associates reported (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.66.9010).
Both regimens were well tolerated, though the toxicity profiles were different, investigators wrote.
Overall, 72% of patients received all six cycles of their respective chemotherapy treatments. Two-year progression-free survival rates were 73.0% (95% confidence interval, 67.7%-77.5%) among patients receiving irinotecan plus cisplatin, 77.6% (95% CI, 72.4%-81.9%) among patients receiving paclitaxel plus carboplatin, and were not significantly different (HR, 1.17; 95% CI, 0.87-1.58; P = .30).
There was also no significant difference in overall survival between the two treatment arms.
In subgroup analyses, there was no significant difference in progression-free or overall survival by disease stage or tumor size.
Grade 3 or 4 anorexia, diarrhea, nausea, vomiting, and febrile neutropenia were the most common adverse events among women receiving irinotecan plus cisplatin. Grade 3 or 4 leukopenia, neutropenia, thrombocytopenia, peripheral sensory neuropathy, and joint pain were the most common adverse events among women receiving paclitaxel plus carboplatin. No treatment-related deaths occurred in either treatment arm.
This study was funded by the Japanese Gynecologic Oncology Group. Dr. Sugiyama and 18 other investigators had no disclosures to report. Six investigators reported serving in advisory roles for or receiving financial compensation or honoraria from multiple companies.
On Twitter @jessnicolecraig
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Irinotecan plus cisplatin is no better than paclitaxel plus carboplatin for women with ovarian clear cell carcinoma
Major finding: Among patients receiving irinotecan plus cisplatin, the 2-year progression-free survival rate was 73.0% (95% confidence interval, 67.7%-77.5%), compared with 77.6% (95% CI, 72.4%-81.9%) for patients receiving paclitaxel plus carboplatin (HR, 1.17; 95% CI, 0.87-1.58; P = .30).
Data source: A randomized, multicenter, phase III trial of 667 patients with ovarian clear cell carcinoma.
Disclosures: This study was funded by the Japanese Gynecologic Oncology Group. Dr. Sugiyama and 18 other investigators had no disclosures to report. Six investigators reported serving in advisory roles for or receiving financial compensation or honoraria from multiple companies.
ATRA-arsenic ‘new standard of care’ for APL
The combination of all-trans-retinoic acid (ATRA) and arsenic trioxide continues to show advantages over ATRA and chemotherapy as first-line therapy for patients with low- or intermediate-risk acute promyelocytic leukemia (APL), investigators report.
The final analysis of theAPL046 study, a noninferiority trial of ATRA plus arsenic trioxide (ATRA-ATO) vs. ATRA and standard chemotherapy, showed that event-free survival (EFS) and overall survival (OS) were significantly better among patients assigned to ATRA-ATO. Patients in this group also had a significantly lower cumulative incidence of relapse, reported Francesco Lo-Coco, MD, of University Tor Vergata in Rome and colleagues.
“Our results support the use of ATRA-ATO in patients with newly diagnosed APL and point to this strategy as the new standard of care for low- or intermediate-risk patients. Studies exploring the role of ATRA-ATO are warranted in other APL subsets including high-risk, pediatric, and elderly patients,” they wrote (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.67.1982).
In the trial, 276 patients with newly diagnosed APL were randomly assigned to receive either ATRA-ATO or ATRA plus chemotherapy with idarubicin, mercaptopurine, and methotrexate.
A total of 263 patients were evaluable for response to induction, including 127 assigned to ATRA-ATO and 136 assigned to ATRA-chemotherapy. Following induction, all patients assigned to ATRA-ATO and 127 assigned to chemotherapy (97%) achieved a complete remission (CR).
After a median follow-up of 40.6 months, the rate of EFS, the primary outcome, was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy (P less than .001). The cumulative incidences of relapse were 1.9% and 13.9%, respectively (P = .0013), and overall survival rates at 50 months were 99.2% vs. 92.6% (P = . 0073).
Hematologic toxicities were more frequent among patients assigned to chemotherapy, while liver function abnormalities occurred more often among those assigned to ATO.
One patient assigned to ATRA-ATO died while in CR, from bronchopneumonia caused by the H1N1 (influenza) virus. Five patients assigned to chemotherapy died in CR, from hemorrhagic shock, pulmonary embolism, bronchopneumonia (two patients) and secondary myelodysplastic syndrome.
Of the 17 patients who experienced relapses during follow-up, 2 assigned to ATRA-ATO had relapses at 22 and 27 months. The remaining 15 relapses were among patients assigned to ATRA-chemotherapy, occurring at a median of 14 months.
The authors noted that the superior EFS and cumulative incidence of relapse with ATRA-ATO emerged only after longer follow-up, indicating that the advantage of ATRA-ATO over ATRA-chemotherapy increases over time and that “the inclusion of ATO in the treatment of low- or intermediate-risk APL not only reduces mortality and hematologic toxicity, but also results in improved and sustained antileukemic activity.”
The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other coauthors disclosed similar relationships.
The combination of all-trans-retinoic acid (ATRA) and arsenic trioxide continues to show advantages over ATRA and chemotherapy as first-line therapy for patients with low- or intermediate-risk acute promyelocytic leukemia (APL), investigators report.
The final analysis of theAPL046 study, a noninferiority trial of ATRA plus arsenic trioxide (ATRA-ATO) vs. ATRA and standard chemotherapy, showed that event-free survival (EFS) and overall survival (OS) were significantly better among patients assigned to ATRA-ATO. Patients in this group also had a significantly lower cumulative incidence of relapse, reported Francesco Lo-Coco, MD, of University Tor Vergata in Rome and colleagues.
“Our results support the use of ATRA-ATO in patients with newly diagnosed APL and point to this strategy as the new standard of care for low- or intermediate-risk patients. Studies exploring the role of ATRA-ATO are warranted in other APL subsets including high-risk, pediatric, and elderly patients,” they wrote (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.67.1982).
In the trial, 276 patients with newly diagnosed APL were randomly assigned to receive either ATRA-ATO or ATRA plus chemotherapy with idarubicin, mercaptopurine, and methotrexate.
A total of 263 patients were evaluable for response to induction, including 127 assigned to ATRA-ATO and 136 assigned to ATRA-chemotherapy. Following induction, all patients assigned to ATRA-ATO and 127 assigned to chemotherapy (97%) achieved a complete remission (CR).
After a median follow-up of 40.6 months, the rate of EFS, the primary outcome, was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy (P less than .001). The cumulative incidences of relapse were 1.9% and 13.9%, respectively (P = .0013), and overall survival rates at 50 months were 99.2% vs. 92.6% (P = . 0073).
Hematologic toxicities were more frequent among patients assigned to chemotherapy, while liver function abnormalities occurred more often among those assigned to ATO.
One patient assigned to ATRA-ATO died while in CR, from bronchopneumonia caused by the H1N1 (influenza) virus. Five patients assigned to chemotherapy died in CR, from hemorrhagic shock, pulmonary embolism, bronchopneumonia (two patients) and secondary myelodysplastic syndrome.
Of the 17 patients who experienced relapses during follow-up, 2 assigned to ATRA-ATO had relapses at 22 and 27 months. The remaining 15 relapses were among patients assigned to ATRA-chemotherapy, occurring at a median of 14 months.
The authors noted that the superior EFS and cumulative incidence of relapse with ATRA-ATO emerged only after longer follow-up, indicating that the advantage of ATRA-ATO over ATRA-chemotherapy increases over time and that “the inclusion of ATO in the treatment of low- or intermediate-risk APL not only reduces mortality and hematologic toxicity, but also results in improved and sustained antileukemic activity.”
The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other coauthors disclosed similar relationships.
The combination of all-trans-retinoic acid (ATRA) and arsenic trioxide continues to show advantages over ATRA and chemotherapy as first-line therapy for patients with low- or intermediate-risk acute promyelocytic leukemia (APL), investigators report.
The final analysis of theAPL046 study, a noninferiority trial of ATRA plus arsenic trioxide (ATRA-ATO) vs. ATRA and standard chemotherapy, showed that event-free survival (EFS) and overall survival (OS) were significantly better among patients assigned to ATRA-ATO. Patients in this group also had a significantly lower cumulative incidence of relapse, reported Francesco Lo-Coco, MD, of University Tor Vergata in Rome and colleagues.
“Our results support the use of ATRA-ATO in patients with newly diagnosed APL and point to this strategy as the new standard of care for low- or intermediate-risk patients. Studies exploring the role of ATRA-ATO are warranted in other APL subsets including high-risk, pediatric, and elderly patients,” they wrote (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.67.1982).
In the trial, 276 patients with newly diagnosed APL were randomly assigned to receive either ATRA-ATO or ATRA plus chemotherapy with idarubicin, mercaptopurine, and methotrexate.
A total of 263 patients were evaluable for response to induction, including 127 assigned to ATRA-ATO and 136 assigned to ATRA-chemotherapy. Following induction, all patients assigned to ATRA-ATO and 127 assigned to chemotherapy (97%) achieved a complete remission (CR).
After a median follow-up of 40.6 months, the rate of EFS, the primary outcome, was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy (P less than .001). The cumulative incidences of relapse were 1.9% and 13.9%, respectively (P = .0013), and overall survival rates at 50 months were 99.2% vs. 92.6% (P = . 0073).
Hematologic toxicities were more frequent among patients assigned to chemotherapy, while liver function abnormalities occurred more often among those assigned to ATO.
One patient assigned to ATRA-ATO died while in CR, from bronchopneumonia caused by the H1N1 (influenza) virus. Five patients assigned to chemotherapy died in CR, from hemorrhagic shock, pulmonary embolism, bronchopneumonia (two patients) and secondary myelodysplastic syndrome.
Of the 17 patients who experienced relapses during follow-up, 2 assigned to ATRA-ATO had relapses at 22 and 27 months. The remaining 15 relapses were among patients assigned to ATRA-chemotherapy, occurring at a median of 14 months.
The authors noted that the superior EFS and cumulative incidence of relapse with ATRA-ATO emerged only after longer follow-up, indicating that the advantage of ATRA-ATO over ATRA-chemotherapy increases over time and that “the inclusion of ATO in the treatment of low- or intermediate-risk APL not only reduces mortality and hematologic toxicity, but also results in improved and sustained antileukemic activity.”
The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other coauthors disclosed similar relationships.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: ATRA-ATO was superior to ATRA-chemotherapy as first-line therapy for acute promyelocytic leukemia.
Major finding: Event-free survival after a median 40.6 months was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy
Data source: Randomized controlled trial comparing ATRA-ATO with ATRA-chemotherapy in 263 patients with newly diagnosed APL.
Disclosures: The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other co-authors disclosed similar relationships.
Flumazenil effective in refractory hypersomnolence
DENVER – Transdermal or sublingual flumazenil is well worth considering for treatment of carefully selected patients with hypersomnolence refractory to conventional wake-promoting medications, Lynn Marie Trotti, MD, said at the annual meeting of the Associated Professional Sleep Societies.
Her retrospective chart review of 153 consecutive patients treated with flumazenil (Romazicon) showed that 63% derived symptomatic benefit, and 39% of patients remained on the drug at the end of the review period, which averaged 6.8 months, reported Dr. Trotti, a neurologist and sleep scientist at Emory University in Atlanta.

Make no mistake: This is off-label therapy. Flumazenil’s approved indication is as intravenous therapy for benzodiazepine sedation. Given orally, the drug is almost entirely eliminated on first-pass metabolism, so she and her coinvestigators had flumazenil compounded at a pharmacy in two formulations: a 6-mg sublingual lozenge and a transcutaneous lotion in a dispenser providing 3 mg of flumazenil per click of the device.
This novel therapy is supported by a plausible mechanistic rationale. Dr. Trotti and colleagues have previously shown that most patients with central hypersomnolence have abnormal potentiation of GABA-A receptors in their cerebrospinal fluid (CSF).
“Functionally speaking, it’s as though they’re producing an endogenous benzodiazepinelike substance,” she explained.
The investigators showed further that the macrolide antibiotic clarithromycin, a negative allosteric modulator of GABA-A receptors, reduced sleepiness in patients with central hypersomnolence syndromes in a randomized, double-blind, crossover trial (Ann Neurol. 2015 Sep;78[3]:454-65).
“It’s not great to use an antibiotic to treat people who don’t have a bacterial infection, so it would be really lovely if we had a GABA antagonist or negative allosteric modulator of GABA-A receptors that was not clarithromycin. And that’s where flumazenil comes into the picture,” according to Dr. Trotti.
As a requirement for receiving flumazenil at the Emory sleep disorders center, patients had to have been refractory to all conventional therapies. Indeed, they had been refractory to an average of 4.6 medications for excessive sleepiness. Also, their hypersomnolence had to have a serious impact on their daily life, for example a job at risk because of inability to drive to and from work safely. Initially, it was further required as a condition for treatment that patients had to show abnormal CSF potentiation of GABA-A receptors; however, the investigators quit requiring a CSF sample after roughly the first 100 candidates proved positive.
Dosing of the sublingual flumazenil lozenges began at one 6-mg lozenge four times daily, titrating up by adding an extra lozenge every 4-5 days until reaching a maximum of 12 per day. The lotion is rubbed on the inside of the forearm at a dose of 3 mg on each arm up to four times per day.
Among the 40 treatment responders who completed the Epworth Sleepiness Scale at baseline and after an average of 6.8 months on flumazenil, the average score improved from 15 to 10.3.
Fifty-seven patients obtained no benefit from flumazenil, 10 stopped the drug because of side effects, 3 quit because they preferred clarithromycin, 8 stopped owing to the cost, and the rest dropped the drug for an assortment of reasons, including planned pregnancy.
The most common treatment-emergent adverse events included dizziness in 13% of subjects, anxiety in 7%, and other mood disturbances in 6%.
Two serious adverse events occurred. One patient with a history of atrial fibrillation experienced a transient ischemic attack. And a patient with systemic lupus erythematous developed asymptomatic CNS lupus vasculopathy. It’s not possible to say whether these events were treatment related because the experience with chronic flumazenil therapy is so limited. After all, the drug is given for its approved indication for no longer than a few days, Dr. Trotti observed.
“This is all clinical data. Obviously I’m not suggesting that everybody should start prescribing flumazenil. That being said, we really need better treatment options,” she said.
The long-term treatment continuation rate with modafinil for hypersomnolence is about 50%, anywhere from 29% to 66% for amphetamines, 38% for clarithromycin, and 37% for pitolisant.
“If you look globally at how likely people with idiopathic hypersomnolence or narcolepsy are to get a satisfactory response to conventional therapies, probably 15%-20% do not get satisfactorily treated with what we currently have available. Prospective controlled studies of flumazenil for treatment of hypersomnolence are certainly needed, but in the absence of those data there’s at least a rationale for people who are severely affected and have nowhere else to turn to consider flumazenil,” Dr. Trotti concluded.
She reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
DENVER – Transdermal or sublingual flumazenil is well worth considering for treatment of carefully selected patients with hypersomnolence refractory to conventional wake-promoting medications, Lynn Marie Trotti, MD, said at the annual meeting of the Associated Professional Sleep Societies.
Her retrospective chart review of 153 consecutive patients treated with flumazenil (Romazicon) showed that 63% derived symptomatic benefit, and 39% of patients remained on the drug at the end of the review period, which averaged 6.8 months, reported Dr. Trotti, a neurologist and sleep scientist at Emory University in Atlanta.
Make no mistake: This is off-label therapy. Flumazenil’s approved indication is as intravenous therapy for benzodiazepine sedation. Given orally, the drug is almost entirely eliminated on first-pass metabolism, so she and her coinvestigators had flumazenil compounded at a pharmacy in two formulations: a 6-mg sublingual lozenge and a transcutaneous lotion in a dispenser providing 3 mg of flumazenil per click of the device.
This novel therapy is supported by a plausible mechanistic rationale. Dr. Trotti and colleagues have previously shown that most patients with central hypersomnolence have abnormal potentiation of GABA-A receptors in their cerebrospinal fluid (CSF).
“Functionally speaking, it’s as though they’re producing an endogenous benzodiazepinelike substance,” she explained.
The investigators showed further that the macrolide antibiotic clarithromycin, a negative allosteric modulator of GABA-A receptors, reduced sleepiness in patients with central hypersomnolence syndromes in a randomized, double-blind, crossover trial (Ann Neurol. 2015 Sep;78[3]:454-65).
“It’s not great to use an antibiotic to treat people who don’t have a bacterial infection, so it would be really lovely if we had a GABA antagonist or negative allosteric modulator of GABA-A receptors that was not clarithromycin. And that’s where flumazenil comes into the picture,” according to Dr. Trotti.
As a requirement for receiving flumazenil at the Emory sleep disorders center, patients had to have been refractory to all conventional therapies. Indeed, they had been refractory to an average of 4.6 medications for excessive sleepiness. Also, their hypersomnolence had to have a serious impact on their daily life, for example a job at risk because of inability to drive to and from work safely. Initially, it was further required as a condition for treatment that patients had to show abnormal CSF potentiation of GABA-A receptors; however, the investigators quit requiring a CSF sample after roughly the first 100 candidates proved positive.
Dosing of the sublingual flumazenil lozenges began at one 6-mg lozenge four times daily, titrating up by adding an extra lozenge every 4-5 days until reaching a maximum of 12 per day. The lotion is rubbed on the inside of the forearm at a dose of 3 mg on each arm up to four times per day.
Among the 40 treatment responders who completed the Epworth Sleepiness Scale at baseline and after an average of 6.8 months on flumazenil, the average score improved from 15 to 10.3.
Fifty-seven patients obtained no benefit from flumazenil, 10 stopped the drug because of side effects, 3 quit because they preferred clarithromycin, 8 stopped owing to the cost, and the rest dropped the drug for an assortment of reasons, including planned pregnancy.
The most common treatment-emergent adverse events included dizziness in 13% of subjects, anxiety in 7%, and other mood disturbances in 6%.
Two serious adverse events occurred. One patient with a history of atrial fibrillation experienced a transient ischemic attack. And a patient with systemic lupus erythematous developed asymptomatic CNS lupus vasculopathy. It’s not possible to say whether these events were treatment related because the experience with chronic flumazenil therapy is so limited. After all, the drug is given for its approved indication for no longer than a few days, Dr. Trotti observed.
“This is all clinical data. Obviously I’m not suggesting that everybody should start prescribing flumazenil. That being said, we really need better treatment options,” she said.
The long-term treatment continuation rate with modafinil for hypersomnolence is about 50%, anywhere from 29% to 66% for amphetamines, 38% for clarithromycin, and 37% for pitolisant.
“If you look globally at how likely people with idiopathic hypersomnolence or narcolepsy are to get a satisfactory response to conventional therapies, probably 15%-20% do not get satisfactorily treated with what we currently have available. Prospective controlled studies of flumazenil for treatment of hypersomnolence are certainly needed, but in the absence of those data there’s at least a rationale for people who are severely affected and have nowhere else to turn to consider flumazenil,” Dr. Trotti concluded.
She reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
DENVER – Transdermal or sublingual flumazenil is well worth considering for treatment of carefully selected patients with hypersomnolence refractory to conventional wake-promoting medications, Lynn Marie Trotti, MD, said at the annual meeting of the Associated Professional Sleep Societies.
Her retrospective chart review of 153 consecutive patients treated with flumazenil (Romazicon) showed that 63% derived symptomatic benefit, and 39% of patients remained on the drug at the end of the review period, which averaged 6.8 months, reported Dr. Trotti, a neurologist and sleep scientist at Emory University in Atlanta.
Make no mistake: This is off-label therapy. Flumazenil’s approved indication is as intravenous therapy for benzodiazepine sedation. Given orally, the drug is almost entirely eliminated on first-pass metabolism, so she and her coinvestigators had flumazenil compounded at a pharmacy in two formulations: a 6-mg sublingual lozenge and a transcutaneous lotion in a dispenser providing 3 mg of flumazenil per click of the device.
This novel therapy is supported by a plausible mechanistic rationale. Dr. Trotti and colleagues have previously shown that most patients with central hypersomnolence have abnormal potentiation of GABA-A receptors in their cerebrospinal fluid (CSF).
“Functionally speaking, it’s as though they’re producing an endogenous benzodiazepinelike substance,” she explained.
The investigators showed further that the macrolide antibiotic clarithromycin, a negative allosteric modulator of GABA-A receptors, reduced sleepiness in patients with central hypersomnolence syndromes in a randomized, double-blind, crossover trial (Ann Neurol. 2015 Sep;78[3]:454-65).
“It’s not great to use an antibiotic to treat people who don’t have a bacterial infection, so it would be really lovely if we had a GABA antagonist or negative allosteric modulator of GABA-A receptors that was not clarithromycin. And that’s where flumazenil comes into the picture,” according to Dr. Trotti.
As a requirement for receiving flumazenil at the Emory sleep disorders center, patients had to have been refractory to all conventional therapies. Indeed, they had been refractory to an average of 4.6 medications for excessive sleepiness. Also, their hypersomnolence had to have a serious impact on their daily life, for example a job at risk because of inability to drive to and from work safely. Initially, it was further required as a condition for treatment that patients had to show abnormal CSF potentiation of GABA-A receptors; however, the investigators quit requiring a CSF sample after roughly the first 100 candidates proved positive.
Dosing of the sublingual flumazenil lozenges began at one 6-mg lozenge four times daily, titrating up by adding an extra lozenge every 4-5 days until reaching a maximum of 12 per day. The lotion is rubbed on the inside of the forearm at a dose of 3 mg on each arm up to four times per day.
Among the 40 treatment responders who completed the Epworth Sleepiness Scale at baseline and after an average of 6.8 months on flumazenil, the average score improved from 15 to 10.3.
Fifty-seven patients obtained no benefit from flumazenil, 10 stopped the drug because of side effects, 3 quit because they preferred clarithromycin, 8 stopped owing to the cost, and the rest dropped the drug for an assortment of reasons, including planned pregnancy.
The most common treatment-emergent adverse events included dizziness in 13% of subjects, anxiety in 7%, and other mood disturbances in 6%.
Two serious adverse events occurred. One patient with a history of atrial fibrillation experienced a transient ischemic attack. And a patient with systemic lupus erythematous developed asymptomatic CNS lupus vasculopathy. It’s not possible to say whether these events were treatment related because the experience with chronic flumazenil therapy is so limited. After all, the drug is given for its approved indication for no longer than a few days, Dr. Trotti observed.
“This is all clinical data. Obviously I’m not suggesting that everybody should start prescribing flumazenil. That being said, we really need better treatment options,” she said.
The long-term treatment continuation rate with modafinil for hypersomnolence is about 50%, anywhere from 29% to 66% for amphetamines, 38% for clarithromycin, and 37% for pitolisant.
“If you look globally at how likely people with idiopathic hypersomnolence or narcolepsy are to get a satisfactory response to conventional therapies, probably 15%-20% do not get satisfactorily treated with what we currently have available. Prospective controlled studies of flumazenil for treatment of hypersomnolence are certainly needed, but in the absence of those data there’s at least a rationale for people who are severely affected and have nowhere else to turn to consider flumazenil,” Dr. Trotti concluded.
She reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
AT SLEEP 2016
Key clinical point: Flumazenil offers a new option for refractory hypersomnolence.
Major finding: Sixty-three percent of patients with highly refractory hypersomnolence derived symptomatic benefit from flumazenil, and 39% of treated patients remained on the drug at a mean 6.8 months of follow-up.
Data source: This was a retrospective chart review of 153 consecutive patients with severe hypersomnolence refractory to all standard therapies who were treated with sublingual or topical flumazenil.
Disclosures: The presenter reported having no financial conflicts regarding her study, supported by the National Institutes of Health.

GERD and Sleep Disorders Often Go Hand in Glove
DENVER – Gastroesophageal reflux disease is fertile soil for medical and psychiatric comorbid conditions, with sleep disorders leading the way, Maurice M. Ohayon, MD, reported at the annual meeting of the Associated Professional Sleep Societies.
He presented a very large, longitudinal, population-based study of gastroesophageal reflux disease (GERD) and its fellow travelers. The study entailed telephone interviews with 12,218 nationally representative subjects in 8 states at time 1, and reinterviews with 10,830 of them 3 years later. The interviews were guided by Sleep-EVAL, a validated computerized diagnostic interview system.

At time 1, 10.6% of subjects reported that they were told by a physician they have GERD and/or they were taking a medication for it. At time 2 (3 years later), the prevalence had increased to 12.4%, according to Dr. Ohayon, professor of psychiatry and behavioral sciences, chief of the division of public mental health and population sciences, and director of the sleep epidemiology research center at Stanford (Calif.) University.
Chronic GERD – that is, GERD at both time points – was present in 3.9% of subjects; 6% of subjects were remitters, meaning that they had GERD at time 1 but not at time 2. Another 8.5% of the total sample had incident GERD, meaning they had GERD at time 2 but not time 1.
The prevalence of GERD rose stepwise with increasing age, from the low single digits in 18- to 34-year-olds to a peak in the 55- to 64-year-old age group, where the prevalence was 13.5% at time 1 and 17% at time 2.

GERD was significantly more common in women. At time 2, the prevalence was 10% in men and 14.4% in women.
Nocturnal awakening was the sleep disorder symptom most commonly associated with GERD. It was a complaint in 28% of subjects who never had GERD, 46% of those with GERD at time 1, and 40% of those with GERD at time 2.
Insomnia, obstructive sleep apnea, and restless legs syndrome were also significantly more common among subjects with GERD. So were musculoskeletal and connective tissue diseases, hypertension, hypercholesterolemia, and major depressive disorder.
The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. Dr. Ohayon reported no financial conflicts of interest.
DENVER – Gastroesophageal reflux disease is fertile soil for medical and psychiatric comorbid conditions, with sleep disorders leading the way, Maurice M. Ohayon, MD, reported at the annual meeting of the Associated Professional Sleep Societies.
He presented a very large, longitudinal, population-based study of gastroesophageal reflux disease (GERD) and its fellow travelers. The study entailed telephone interviews with 12,218 nationally representative subjects in 8 states at time 1, and reinterviews with 10,830 of them 3 years later. The interviews were guided by Sleep-EVAL, a validated computerized diagnostic interview system.
At time 1, 10.6% of subjects reported that they were told by a physician they have GERD and/or they were taking a medication for it. At time 2 (3 years later), the prevalence had increased to 12.4%, according to Dr. Ohayon, professor of psychiatry and behavioral sciences, chief of the division of public mental health and population sciences, and director of the sleep epidemiology research center at Stanford (Calif.) University.
Chronic GERD – that is, GERD at both time points – was present in 3.9% of subjects; 6% of subjects were remitters, meaning that they had GERD at time 1 but not at time 2. Another 8.5% of the total sample had incident GERD, meaning they had GERD at time 2 but not time 1.
The prevalence of GERD rose stepwise with increasing age, from the low single digits in 18- to 34-year-olds to a peak in the 55- to 64-year-old age group, where the prevalence was 13.5% at time 1 and 17% at time 2.
GERD was significantly more common in women. At time 2, the prevalence was 10% in men and 14.4% in women.
Nocturnal awakening was the sleep disorder symptom most commonly associated with GERD. It was a complaint in 28% of subjects who never had GERD, 46% of those with GERD at time 1, and 40% of those with GERD at time 2.
Insomnia, obstructive sleep apnea, and restless legs syndrome were also significantly more common among subjects with GERD. So were musculoskeletal and connective tissue diseases, hypertension, hypercholesterolemia, and major depressive disorder.
The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. Dr. Ohayon reported no financial conflicts of interest.
DENVER – Gastroesophageal reflux disease is fertile soil for medical and psychiatric comorbid conditions, with sleep disorders leading the way, Maurice M. Ohayon, MD, reported at the annual meeting of the Associated Professional Sleep Societies.
He presented a very large, longitudinal, population-based study of gastroesophageal reflux disease (GERD) and its fellow travelers. The study entailed telephone interviews with 12,218 nationally representative subjects in 8 states at time 1, and reinterviews with 10,830 of them 3 years later. The interviews were guided by Sleep-EVAL, a validated computerized diagnostic interview system.
At time 1, 10.6% of subjects reported that they were told by a physician they have GERD and/or they were taking a medication for it. At time 2 (3 years later), the prevalence had increased to 12.4%, according to Dr. Ohayon, professor of psychiatry and behavioral sciences, chief of the division of public mental health and population sciences, and director of the sleep epidemiology research center at Stanford (Calif.) University.
Chronic GERD – that is, GERD at both time points – was present in 3.9% of subjects; 6% of subjects were remitters, meaning that they had GERD at time 1 but not at time 2. Another 8.5% of the total sample had incident GERD, meaning they had GERD at time 2 but not time 1.
The prevalence of GERD rose stepwise with increasing age, from the low single digits in 18- to 34-year-olds to a peak in the 55- to 64-year-old age group, where the prevalence was 13.5% at time 1 and 17% at time 2.
GERD was significantly more common in women. At time 2, the prevalence was 10% in men and 14.4% in women.
Nocturnal awakening was the sleep disorder symptom most commonly associated with GERD. It was a complaint in 28% of subjects who never had GERD, 46% of those with GERD at time 1, and 40% of those with GERD at time 2.
Insomnia, obstructive sleep apnea, and restless legs syndrome were also significantly more common among subjects with GERD. So were musculoskeletal and connective tissue diseases, hypertension, hypercholesterolemia, and major depressive disorder.
The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. Dr. Ohayon reported no financial conflicts of interest.
AT SLEEP 2016

GERD and sleep disorders often go hand in glove
DENVER – Gastroesophageal reflux disease is fertile soil for medical and psychiatric comorbid conditions, with sleep disorders leading the way, Maurice M. Ohayon, MD, reported at the annual meeting of the Associated Professional Sleep Societies.
He presented a very large, longitudinal, population-based study of gastroesophageal reflux disease (GERD) and its fellow travelers. The study entailed telephone interviews with 12,218 nationally representative subjects in 8 states at time 1, and reinterviews with 10,830 of them 3 years later. The interviews were guided by Sleep-EVAL, a validated computerized diagnostic interview system.

At time 1, 10.6% of subjects reported that they were told by a physician they have GERD and/or they were taking a medication for it. At time 2 (3 years later), the prevalence had increased to 12.4%, according to Dr. Ohayon, professor of psychiatry and behavioral sciences, chief of the division of public mental health and population sciences, and director of the sleep epidemiology research center at Stanford (Calif.) University.
Chronic GERD – that is, GERD at both time points – was present in 3.9% of subjects; 6% of subjects were remitters, meaning that they had GERD at time 1 but not at time 2. Another 8.5% of the total sample had incident GERD, meaning they had GERD at time 2 but not time 1.
The prevalence of GERD rose stepwise with increasing age, from the low single digits in 18- to 34-year-olds to a peak in the 55- to 64-year-old age group, where the prevalence was 13.5% at time 1 and 17% at time 2.

GERD was significantly more common in women. At time 2, the prevalence was 10% in men and 14.4% in women.
Nocturnal awakening was the sleep disorder symptom most commonly associated with GERD. It was a complaint in 28% of subjects who never had GERD, 46% of those with GERD at time 1, and 40% of those with GERD at time 2.
Insomnia, obstructive sleep apnea, and restless legs syndrome were also significantly more common among subjects with GERD. So were musculoskeletal and connective tissue diseases, hypertension, hypercholesterolemia, and major depressive disorder.
The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. Dr. Ohayon reported no financial conflicts of interest.
DENVER – Gastroesophageal reflux disease is fertile soil for medical and psychiatric comorbid conditions, with sleep disorders leading the way, Maurice M. Ohayon, MD, reported at the annual meeting of the Associated Professional Sleep Societies.
He presented a very large, longitudinal, population-based study of gastroesophageal reflux disease (GERD) and its fellow travelers. The study entailed telephone interviews with 12,218 nationally representative subjects in 8 states at time 1, and reinterviews with 10,830 of them 3 years later. The interviews were guided by Sleep-EVAL, a validated computerized diagnostic interview system.
At time 1, 10.6% of subjects reported that they were told by a physician they have GERD and/or they were taking a medication for it. At time 2 (3 years later), the prevalence had increased to 12.4%, according to Dr. Ohayon, professor of psychiatry and behavioral sciences, chief of the division of public mental health and population sciences, and director of the sleep epidemiology research center at Stanford (Calif.) University.
Chronic GERD – that is, GERD at both time points – was present in 3.9% of subjects; 6% of subjects were remitters, meaning that they had GERD at time 1 but not at time 2. Another 8.5% of the total sample had incident GERD, meaning they had GERD at time 2 but not time 1.
The prevalence of GERD rose stepwise with increasing age, from the low single digits in 18- to 34-year-olds to a peak in the 55- to 64-year-old age group, where the prevalence was 13.5% at time 1 and 17% at time 2.
GERD was significantly more common in women. At time 2, the prevalence was 10% in men and 14.4% in women.
Nocturnal awakening was the sleep disorder symptom most commonly associated with GERD. It was a complaint in 28% of subjects who never had GERD, 46% of those with GERD at time 1, and 40% of those with GERD at time 2.
Insomnia, obstructive sleep apnea, and restless legs syndrome were also significantly more common among subjects with GERD. So were musculoskeletal and connective tissue diseases, hypertension, hypercholesterolemia, and major depressive disorder.
The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. Dr. Ohayon reported no financial conflicts of interest.
DENVER – Gastroesophageal reflux disease is fertile soil for medical and psychiatric comorbid conditions, with sleep disorders leading the way, Maurice M. Ohayon, MD, reported at the annual meeting of the Associated Professional Sleep Societies.
He presented a very large, longitudinal, population-based study of gastroesophageal reflux disease (GERD) and its fellow travelers. The study entailed telephone interviews with 12,218 nationally representative subjects in 8 states at time 1, and reinterviews with 10,830 of them 3 years later. The interviews were guided by Sleep-EVAL, a validated computerized diagnostic interview system.
At time 1, 10.6% of subjects reported that they were told by a physician they have GERD and/or they were taking a medication for it. At time 2 (3 years later), the prevalence had increased to 12.4%, according to Dr. Ohayon, professor of psychiatry and behavioral sciences, chief of the division of public mental health and population sciences, and director of the sleep epidemiology research center at Stanford (Calif.) University.
Chronic GERD – that is, GERD at both time points – was present in 3.9% of subjects; 6% of subjects were remitters, meaning that they had GERD at time 1 but not at time 2. Another 8.5% of the total sample had incident GERD, meaning they had GERD at time 2 but not time 1.
The prevalence of GERD rose stepwise with increasing age, from the low single digits in 18- to 34-year-olds to a peak in the 55- to 64-year-old age group, where the prevalence was 13.5% at time 1 and 17% at time 2.
GERD was significantly more common in women. At time 2, the prevalence was 10% in men and 14.4% in women.
Nocturnal awakening was the sleep disorder symptom most commonly associated with GERD. It was a complaint in 28% of subjects who never had GERD, 46% of those with GERD at time 1, and 40% of those with GERD at time 2.
Insomnia, obstructive sleep apnea, and restless legs syndrome were also significantly more common among subjects with GERD. So were musculoskeletal and connective tissue diseases, hypertension, hypercholesterolemia, and major depressive disorder.
The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. Dr. Ohayon reported no financial conflicts of interest.
AT SLEEP 2016
Key clinical point: Inquire about potentially treatable sleep disturbances in patients with GERD.
Major finding: Nocturnal awakenings were reported to be a problem in 28% of subjects with no history of gastroesophageal reflux disease, but in up to 46% of those with the digestive disease.
Data source: A longitudinal, population-based study of 12,218 nationally representative adults interviewed regarding the prevalence of GERD as well as comorbid sleep, medical, and psychiatric conditions.
Disclosures: The study was supported by the John Arrillaga Foundation and Takeda Pharmaceuticals. The presenter reported having no financial conflicts of interest.

How practice changes us
I have learned a few things in my almost half-century of hurtling through space with all of you on this fragile blue-green rock. My most recent enlightenment is that the universe has a way of flipping our certitude on its head. Mental acrobatics requires flexibility, requiring us to bend so we don’t break.
On July 1, Minnesota began allowing clinicians to certify patients for “intractable pain” as a qualifying condition for the Minnesota Medical Cannabis Program. Recall that medical marijuana is a Schedule I drug, and we cannot prescribe it. These programs are set up in such a way that the only role a clinician plays is to certify patients with qualifying conditions. This allows a patient to pay a registration fee and visit a cannabis patient center, where a pharmacist will recommend cannabis dose and type.

Months before this, I was waxing professorial about our lack of certainty about dosing and efficacy of medical marijuana. Then I met a 30-year-old with chronic back pain.
She had been evaluated by every subspecialist. This patient was taking and failing supertherapeutic doses of NSAIDs, acetaminophen, and gabapentin. No more surgical options existed. She had been removed from opioid contracts for aberrant behavior. She had had a hysterectomy for severe bleeding. She was in pain and asking for help.
She relates to you that street marijuana has helped with the pain, but she is worried about being arrested and losing her job. Do we put her on another opioid contract? Do we throw up our hands in defeat, apologize, and show her the door?
Serendipitously, I ran across a study evaluating the relationship between cannabis use over a 20-year period and health conditions. The study by Madeline Meier, Ph.D., and her colleagues evaluated 1,037 New Zealanders followed into their late 30s. Laboratory measures were available, and tobacco use was determined (JAMA Psychiatry. 2016 Jul 1;73[7]:731-40).
Cannabis was associated with poorer periodontal health, but with no other health conditions in early midlife. In contrast, tobacco use was associated with significant adverse health consequences in multiple domains.
This study looked at smoked cannabis. In contrast, the cannabis that my patient would take is an oil, negating any potential respiratory health issues from by-products of burning. Furthermore, products with higher concentrations of or consisting exclusively of cannabidiol can be selected. Cannabidiol is proposed to possess health benefits and is not psychoactive.
As the opioid crisis rages, solutions are not readily presenting themselves. Will we be on the wrong side of medical history by providing patients with chronic pain access to medical marijuana? Perhaps we can avoid, at least for a short time, the all-too inevitable outcome of chronic pain patients in their 30s: ever-increasing opioid doses with the same amount of pain, frequent emergency department visits, a fractured patient-physician relationship, and drug overdose.
For our patients’ sake, I hope we can bend before we break.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
I have learned a few things in my almost half-century of hurtling through space with all of you on this fragile blue-green rock. My most recent enlightenment is that the universe has a way of flipping our certitude on its head. Mental acrobatics requires flexibility, requiring us to bend so we don’t break.
On July 1, Minnesota began allowing clinicians to certify patients for “intractable pain” as a qualifying condition for the Minnesota Medical Cannabis Program. Recall that medical marijuana is a Schedule I drug, and we cannot prescribe it. These programs are set up in such a way that the only role a clinician plays is to certify patients with qualifying conditions. This allows a patient to pay a registration fee and visit a cannabis patient center, where a pharmacist will recommend cannabis dose and type.
Months before this, I was waxing professorial about our lack of certainty about dosing and efficacy of medical marijuana. Then I met a 30-year-old with chronic back pain.
She had been evaluated by every subspecialist. This patient was taking and failing supertherapeutic doses of NSAIDs, acetaminophen, and gabapentin. No more surgical options existed. She had been removed from opioid contracts for aberrant behavior. She had had a hysterectomy for severe bleeding. She was in pain and asking for help.
She relates to you that street marijuana has helped with the pain, but she is worried about being arrested and losing her job. Do we put her on another opioid contract? Do we throw up our hands in defeat, apologize, and show her the door?
Serendipitously, I ran across a study evaluating the relationship between cannabis use over a 20-year period and health conditions. The study by Madeline Meier, Ph.D., and her colleagues evaluated 1,037 New Zealanders followed into their late 30s. Laboratory measures were available, and tobacco use was determined (JAMA Psychiatry. 2016 Jul 1;73[7]:731-40).
Cannabis was associated with poorer periodontal health, but with no other health conditions in early midlife. In contrast, tobacco use was associated with significant adverse health consequences in multiple domains.
This study looked at smoked cannabis. In contrast, the cannabis that my patient would take is an oil, negating any potential respiratory health issues from by-products of burning. Furthermore, products with higher concentrations of or consisting exclusively of cannabidiol can be selected. Cannabidiol is proposed to possess health benefits and is not psychoactive.
As the opioid crisis rages, solutions are not readily presenting themselves. Will we be on the wrong side of medical history by providing patients with chronic pain access to medical marijuana? Perhaps we can avoid, at least for a short time, the all-too inevitable outcome of chronic pain patients in their 30s: ever-increasing opioid doses with the same amount of pain, frequent emergency department visits, a fractured patient-physician relationship, and drug overdose.
For our patients’ sake, I hope we can bend before we break.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
I have learned a few things in my almost half-century of hurtling through space with all of you on this fragile blue-green rock. My most recent enlightenment is that the universe has a way of flipping our certitude on its head. Mental acrobatics requires flexibility, requiring us to bend so we don’t break.
On July 1, Minnesota began allowing clinicians to certify patients for “intractable pain” as a qualifying condition for the Minnesota Medical Cannabis Program. Recall that medical marijuana is a Schedule I drug, and we cannot prescribe it. These programs are set up in such a way that the only role a clinician plays is to certify patients with qualifying conditions. This allows a patient to pay a registration fee and visit a cannabis patient center, where a pharmacist will recommend cannabis dose and type.
Months before this, I was waxing professorial about our lack of certainty about dosing and efficacy of medical marijuana. Then I met a 30-year-old with chronic back pain.
She had been evaluated by every subspecialist. This patient was taking and failing supertherapeutic doses of NSAIDs, acetaminophen, and gabapentin. No more surgical options existed. She had been removed from opioid contracts for aberrant behavior. She had had a hysterectomy for severe bleeding. She was in pain and asking for help.
She relates to you that street marijuana has helped with the pain, but she is worried about being arrested and losing her job. Do we put her on another opioid contract? Do we throw up our hands in defeat, apologize, and show her the door?
Serendipitously, I ran across a study evaluating the relationship between cannabis use over a 20-year period and health conditions. The study by Madeline Meier, Ph.D., and her colleagues evaluated 1,037 New Zealanders followed into their late 30s. Laboratory measures were available, and tobacco use was determined (JAMA Psychiatry. 2016 Jul 1;73[7]:731-40).
Cannabis was associated with poorer periodontal health, but with no other health conditions in early midlife. In contrast, tobacco use was associated with significant adverse health consequences in multiple domains.
This study looked at smoked cannabis. In contrast, the cannabis that my patient would take is an oil, negating any potential respiratory health issues from by-products of burning. Furthermore, products with higher concentrations of or consisting exclusively of cannabidiol can be selected. Cannabidiol is proposed to possess health benefits and is not psychoactive.
As the opioid crisis rages, solutions are not readily presenting themselves. Will we be on the wrong side of medical history by providing patients with chronic pain access to medical marijuana? Perhaps we can avoid, at least for a short time, the all-too inevitable outcome of chronic pain patients in their 30s: ever-increasing opioid doses with the same amount of pain, frequent emergency department visits, a fractured patient-physician relationship, and drug overdose.
For our patients’ sake, I hope we can bend before we break.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.

Radiographic vertebral fractures common in ankylosing spondylitis patients
Radiographic vertebral fractures occur frequently in patients with ankylosing spondylitis, according to Dr. Fiona Maas and her associates.
At baseline, 59 (20%) of the 292 patients included in a prospective cohort study had radiographic vertebral fractures. During a 2-year follow-up period, 15 patients developed new fractures and 7 patients experienced an increase in severity of current fractures. A significant majority of fractures were defined as mild, and most were located in the mid-thoracic and thoracolumbar regions of the spine.
Fractures were more likely in patients who were older, had a higher body mass index, had smoked for long durations, had larger occiput-to-wall distances, had more spinal radiographic damage, and had lower hip bone mineral density (BMD). Patients who developed new fractures during the study period were older and had lower hip BMD. Incidence and prevalence of fractures was reduced in patients who were receiving nonsteroidal anti-inflammatory drugs (NSAIDs) at baseline.
“The influence of anti-inflammatory drugs (e.g. NSAIDs, TNF-alpha inhibitors), anti-osteoporotic treatment (e.g. calcium/vitamin D supplements, bisphosphonates), and lifestyle changes (e.g. smoking cessation, body weight control, physical exercise) on the development of vertebral fractures in AS should be further investigated in a large study population with long-term follow-up,” the investigators concluded.
Find the full study in Arthritis Care & Research (doi: 10.1002/acr.22980).
Radiographic vertebral fractures occur frequently in patients with ankylosing spondylitis, according to Dr. Fiona Maas and her associates.
At baseline, 59 (20%) of the 292 patients included in a prospective cohort study had radiographic vertebral fractures. During a 2-year follow-up period, 15 patients developed new fractures and 7 patients experienced an increase in severity of current fractures. A significant majority of fractures were defined as mild, and most were located in the mid-thoracic and thoracolumbar regions of the spine.
Fractures were more likely in patients who were older, had a higher body mass index, had smoked for long durations, had larger occiput-to-wall distances, had more spinal radiographic damage, and had lower hip bone mineral density (BMD). Patients who developed new fractures during the study period were older and had lower hip BMD. Incidence and prevalence of fractures was reduced in patients who were receiving nonsteroidal anti-inflammatory drugs (NSAIDs) at baseline.
“The influence of anti-inflammatory drugs (e.g. NSAIDs, TNF-alpha inhibitors), anti-osteoporotic treatment (e.g. calcium/vitamin D supplements, bisphosphonates), and lifestyle changes (e.g. smoking cessation, body weight control, physical exercise) on the development of vertebral fractures in AS should be further investigated in a large study population with long-term follow-up,” the investigators concluded.
Find the full study in Arthritis Care & Research (doi: 10.1002/acr.22980).
Radiographic vertebral fractures occur frequently in patients with ankylosing spondylitis, according to Dr. Fiona Maas and her associates.
At baseline, 59 (20%) of the 292 patients included in a prospective cohort study had radiographic vertebral fractures. During a 2-year follow-up period, 15 patients developed new fractures and 7 patients experienced an increase in severity of current fractures. A significant majority of fractures were defined as mild, and most were located in the mid-thoracic and thoracolumbar regions of the spine.
Fractures were more likely in patients who were older, had a higher body mass index, had smoked for long durations, had larger occiput-to-wall distances, had more spinal radiographic damage, and had lower hip bone mineral density (BMD). Patients who developed new fractures during the study period were older and had lower hip BMD. Incidence and prevalence of fractures was reduced in patients who were receiving nonsteroidal anti-inflammatory drugs (NSAIDs) at baseline.
“The influence of anti-inflammatory drugs (e.g. NSAIDs, TNF-alpha inhibitors), anti-osteoporotic treatment (e.g. calcium/vitamin D supplements, bisphosphonates), and lifestyle changes (e.g. smoking cessation, body weight control, physical exercise) on the development of vertebral fractures in AS should be further investigated in a large study population with long-term follow-up,” the investigators concluded.
Find the full study in Arthritis Care & Research (doi: 10.1002/acr.22980).
FROM ARTHRITIS CARE & RESEARCH
Retiring Baby Boomers leave fewer workers to pay for Medicare
The influx of aging Baby Boomers into the ranks of the retired will reduce the ratio of workers available to pay for each Medicare part A beneficiary by 40% from 2000 to 2030, according to the 2016 report of the Medicare Trustees.
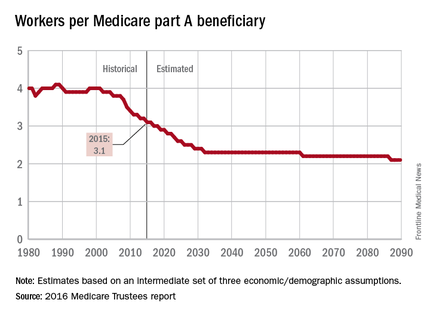
In 2015, there were 3.1 workers for each part A beneficiary, putting the United States in the middle of a projected drop from 4.0 workers per beneficiary in 2000 down to 2.4 in 2030. The Boomer-induced drop will largely be over by then, but the decline will continue until there are about 2.1 workers for each part A beneficiary by 2090, the report said.
“This reduction implies an increase in the [Medicare part A] cost rate of about 50% by 2090, relative to its current level, solely due to this demographic factor,” the trustees noted.
The projections are done using three sets – low-cost, intermediate, and high-cost – of economic and demographic assumptions. The figures presented here are from the intermediate assumption.
The influx of aging Baby Boomers into the ranks of the retired will reduce the ratio of workers available to pay for each Medicare part A beneficiary by 40% from 2000 to 2030, according to the 2016 report of the Medicare Trustees.
In 2015, there were 3.1 workers for each part A beneficiary, putting the United States in the middle of a projected drop from 4.0 workers per beneficiary in 2000 down to 2.4 in 2030. The Boomer-induced drop will largely be over by then, but the decline will continue until there are about 2.1 workers for each part A beneficiary by 2090, the report said.
“This reduction implies an increase in the [Medicare part A] cost rate of about 50% by 2090, relative to its current level, solely due to this demographic factor,” the trustees noted.
The projections are done using three sets – low-cost, intermediate, and high-cost – of economic and demographic assumptions. The figures presented here are from the intermediate assumption.
The influx of aging Baby Boomers into the ranks of the retired will reduce the ratio of workers available to pay for each Medicare part A beneficiary by 40% from 2000 to 2030, according to the 2016 report of the Medicare Trustees.
In 2015, there were 3.1 workers for each part A beneficiary, putting the United States in the middle of a projected drop from 4.0 workers per beneficiary in 2000 down to 2.4 in 2030. The Boomer-induced drop will largely be over by then, but the decline will continue until there are about 2.1 workers for each part A beneficiary by 2090, the report said.
“This reduction implies an increase in the [Medicare part A] cost rate of about 50% by 2090, relative to its current level, solely due to this demographic factor,” the trustees noted.
The projections are done using three sets – low-cost, intermediate, and high-cost – of economic and demographic assumptions. The figures presented here are from the intermediate assumption.
Trump Releases 10-Point Plan for VA Reform
Donald Trump, the presumptive Republican Party nominee focused on the VA and veterans’ health issues in a speech Monday in Virginia Beach, Virginia. In the speech, Mr. Trump called for civil service reform, access to community care for all veterans regardless of Choice program eligibility, and more VA mental health care providers.
“It is the job of the next President to make America safe again, for everyone,” Mr. Trump said in a prepared speech. “That promise of protection must include taking care of every last veteran.”
Echoing the recent recommendations of the Commission on Care, Mr. Trump argued that access to care in the community should be guaranteed for all veterans. “We must extend this right to all veterans, not just those who can’t get an appointment in 30 days, or who live more than 40 miles from a VA hospital, which is the current policy.”
Hillary Clinton, the presumptive Democratic Party nominee, did not directly respond to Mr. Trump’s proposal or those of the Commission on Care. However she has previously stated that she opposes efforts to privatize veteran care. Mrs. Clinton has touted her role in increasing aid to families of veterans and active- duty service members and expanding coverage to include National Guard and reservists as a member of the Armed Services Committee.
Charging that the VA was “corrupt,” Mr. Trump vowed to begin an investigation immediately if he is elected president. “I will instruct my staff that if a valid complaint is not addressed, that the issue be brought directly to me,” he said. “I will pick up the phone and fix it myself if I have to.” The Republican candidate called for civil service reform so that the VA can more easily fire employees.
The Republican candidate’s remarks also drew on the pre-release of information from the VA’s research into suicide rates. Noting that the rate of veteran suicide is lower among patients in the VA system compared with the rate of those who do not use VA services, Mr. Trump called for expansion of mental health services at the VA. “A shocking 20 veterans are committing suicide each day, especially our older veterans,” Mr. Trump said. “This is a national tragedy. The evidence shows that if veterans are in the system, receiving care, they are much less likely to take their own lives than veterans outside the system. That is why we must increase the number of mental health care professionals inside the VA— while ensuring that veterans can access private mental health care as well.”
In his speech, Mr. Trump vowed to enact the following VA reform proposals:
1. I will appoint a Secretary of Veterans Affairs who will make it his or her personal mission to clean up the VA.
2. I am going to use every lawful authority to remove and discipline federal employees or managers who fail our veterans or breach the public trust.
3. I am going to ask Congress to pass legislation that ensures the Secretary of Veterans Affairs has the authority to remove or discipline any employee who risks the health, safety, or well-being of any veteran.
4. I am going to appoint a commission to investigate all the wrongdoing at the VA and then present those findings to Congress as the basis for bold legislative reform.
5. I am going to make sure the honest and dedicated people in the VA have their jobs protected and are put in line for promotions.
6. I will create a private White House Hotline that is answered by a real person 24 hours a day to ensure that no valid complaint about VA wrongdoing falls through the cracks.
7. We are going to stop giving bonuses to people for wasting money and start giving bonuses to people for improving service, saving lives, and cutting waste.
8. We are going to reform our visa programs to ensure American veterans are in the front, not back, of the line.
9 We are going to increase the number of mental health care professionals and increase outreach to veterans outside of the system.
10. We are going to ensure every veteran in America has the choice to seek care at the VA or to seek private medical care.
Donald Trump, the presumptive Republican Party nominee focused on the VA and veterans’ health issues in a speech Monday in Virginia Beach, Virginia. In the speech, Mr. Trump called for civil service reform, access to community care for all veterans regardless of Choice program eligibility, and more VA mental health care providers.
“It is the job of the next President to make America safe again, for everyone,” Mr. Trump said in a prepared speech. “That promise of protection must include taking care of every last veteran.”
Echoing the recent recommendations of the Commission on Care, Mr. Trump argued that access to care in the community should be guaranteed for all veterans. “We must extend this right to all veterans, not just those who can’t get an appointment in 30 days, or who live more than 40 miles from a VA hospital, which is the current policy.”
Hillary Clinton, the presumptive Democratic Party nominee, did not directly respond to Mr. Trump’s proposal or those of the Commission on Care. However she has previously stated that she opposes efforts to privatize veteran care. Mrs. Clinton has touted her role in increasing aid to families of veterans and active- duty service members and expanding coverage to include National Guard and reservists as a member of the Armed Services Committee.
Charging that the VA was “corrupt,” Mr. Trump vowed to begin an investigation immediately if he is elected president. “I will instruct my staff that if a valid complaint is not addressed, that the issue be brought directly to me,” he said. “I will pick up the phone and fix it myself if I have to.” The Republican candidate called for civil service reform so that the VA can more easily fire employees.
The Republican candidate’s remarks also drew on the pre-release of information from the VA’s research into suicide rates. Noting that the rate of veteran suicide is lower among patients in the VA system compared with the rate of those who do not use VA services, Mr. Trump called for expansion of mental health services at the VA. “A shocking 20 veterans are committing suicide each day, especially our older veterans,” Mr. Trump said. “This is a national tragedy. The evidence shows that if veterans are in the system, receiving care, they are much less likely to take their own lives than veterans outside the system. That is why we must increase the number of mental health care professionals inside the VA— while ensuring that veterans can access private mental health care as well.”
In his speech, Mr. Trump vowed to enact the following VA reform proposals:
1. I will appoint a Secretary of Veterans Affairs who will make it his or her personal mission to clean up the VA.
2. I am going to use every lawful authority to remove and discipline federal employees or managers who fail our veterans or breach the public trust.
3. I am going to ask Congress to pass legislation that ensures the Secretary of Veterans Affairs has the authority to remove or discipline any employee who risks the health, safety, or well-being of any veteran.
4. I am going to appoint a commission to investigate all the wrongdoing at the VA and then present those findings to Congress as the basis for bold legislative reform.
5. I am going to make sure the honest and dedicated people in the VA have their jobs protected and are put in line for promotions.
6. I will create a private White House Hotline that is answered by a real person 24 hours a day to ensure that no valid complaint about VA wrongdoing falls through the cracks.
7. We are going to stop giving bonuses to people for wasting money and start giving bonuses to people for improving service, saving lives, and cutting waste.
8. We are going to reform our visa programs to ensure American veterans are in the front, not back, of the line.
9 We are going to increase the number of mental health care professionals and increase outreach to veterans outside of the system.
10. We are going to ensure every veteran in America has the choice to seek care at the VA or to seek private medical care.
Donald Trump, the presumptive Republican Party nominee focused on the VA and veterans’ health issues in a speech Monday in Virginia Beach, Virginia. In the speech, Mr. Trump called for civil service reform, access to community care for all veterans regardless of Choice program eligibility, and more VA mental health care providers.
“It is the job of the next President to make America safe again, for everyone,” Mr. Trump said in a prepared speech. “That promise of protection must include taking care of every last veteran.”
Echoing the recent recommendations of the Commission on Care, Mr. Trump argued that access to care in the community should be guaranteed for all veterans. “We must extend this right to all veterans, not just those who can’t get an appointment in 30 days, or who live more than 40 miles from a VA hospital, which is the current policy.”
Hillary Clinton, the presumptive Democratic Party nominee, did not directly respond to Mr. Trump’s proposal or those of the Commission on Care. However she has previously stated that she opposes efforts to privatize veteran care. Mrs. Clinton has touted her role in increasing aid to families of veterans and active- duty service members and expanding coverage to include National Guard and reservists as a member of the Armed Services Committee.
Charging that the VA was “corrupt,” Mr. Trump vowed to begin an investigation immediately if he is elected president. “I will instruct my staff that if a valid complaint is not addressed, that the issue be brought directly to me,” he said. “I will pick up the phone and fix it myself if I have to.” The Republican candidate called for civil service reform so that the VA can more easily fire employees.
The Republican candidate’s remarks also drew on the pre-release of information from the VA’s research into suicide rates. Noting that the rate of veteran suicide is lower among patients in the VA system compared with the rate of those who do not use VA services, Mr. Trump called for expansion of mental health services at the VA. “A shocking 20 veterans are committing suicide each day, especially our older veterans,” Mr. Trump said. “This is a national tragedy. The evidence shows that if veterans are in the system, receiving care, they are much less likely to take their own lives than veterans outside the system. That is why we must increase the number of mental health care professionals inside the VA— while ensuring that veterans can access private mental health care as well.”
In his speech, Mr. Trump vowed to enact the following VA reform proposals:
1. I will appoint a Secretary of Veterans Affairs who will make it his or her personal mission to clean up the VA.
2. I am going to use every lawful authority to remove and discipline federal employees or managers who fail our veterans or breach the public trust.
3. I am going to ask Congress to pass legislation that ensures the Secretary of Veterans Affairs has the authority to remove or discipline any employee who risks the health, safety, or well-being of any veteran.
4. I am going to appoint a commission to investigate all the wrongdoing at the VA and then present those findings to Congress as the basis for bold legislative reform.
5. I am going to make sure the honest and dedicated people in the VA have their jobs protected and are put in line for promotions.
6. I will create a private White House Hotline that is answered by a real person 24 hours a day to ensure that no valid complaint about VA wrongdoing falls through the cracks.
7. We are going to stop giving bonuses to people for wasting money and start giving bonuses to people for improving service, saving lives, and cutting waste.
8. We are going to reform our visa programs to ensure American veterans are in the front, not back, of the line.
9 We are going to increase the number of mental health care professionals and increase outreach to veterans outside of the system.
10. We are going to ensure every veteran in America has the choice to seek care at the VA or to seek private medical care.