User login
Advanced heart failure symptoms linked to mortality
ORLANDO – Advanced heart failure patients who are hospitalized for heart failure and have a higher symptom burden at discharge have a significantly increased rate of death or rehospitalization over the next 6 months, based on an analysis of 393 patients enrolled in a heart failure trial.
The strong link between severe symptom burden and poor near-term outcomes persisted despite adjustment for various markers of heart failure severity, suggesting that treatment aimed at reducing symptoms may be able to reduce mortality or heart failure hospitalization in advanced heart failure patients, Ellen K. Hummel, MD, said at the annual scientific meeting of the Heart Failure Society of America.
In her analysis, a severe symptom burden at the time of hospital discharge linked with an adjusted 2.9-fold increased mortality rate and a 2.5-fold increased rate of days dead or hospitalized during the next 6 months, said Dr. Hummel, a geriatric and palliative care specialist at the University of Michigan in Ann Arbor. These elevated rate ratios for patients with severe symptoms at hospital discharge were in comparison to the ratios for advanced heart failure patients in the study with no symptoms at discharge.
Three symptoms contributed to the symptom score she used in her analysis: fatigue, scored on a scale of 0-3; dyspnea, also scored 0-3; and gastrointestinal distress, scored as 0-2, creating a maximum score of 8. Her analysis categorized mild as a total score of 1-4 and severe as 5 or greater. In the study population she used for her analysis, patients enrolled in the multicenter ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) trial, 111 of the 393 evaluable patients (28%) had none of these symptoms, 239 (61%) had mild symptoms, and 43 (11%) had severe symptoms. Scoring was done by patients based on their subjective self-assessment at the time of hospital discharge.
The absolute, observed 6-month mortality rates were roughly 45% among patients with severe symptoms, about 17% in patients with mild symptoms, and about 12% in those with no symptoms.
The primary purpose of ESCAPE was to assess the impact that routine collection of data from a pulmonary artery catheter during hospitalization has on outcomes; the results showed no significant link between improved outcomes and getting these data (JAMA. 2005 Oct 5;294[13]:1625-33). The study ran during 2000-2003 at 26 centers in the United States and Canada. Of the 433 advanced heart failure patients enrolled in ESCAPE, 393 had complete records to allow the current analysis.
The adjustments that Dr. Hummel made in the proportional hazard analysis took into account New York Heart Association class, and severity of disease at the time of hospital discharge measured by the ESCAPE Discharge Risk Score. This score takes into account age, 6-minute walk distance, blood urea nitrogen, brain natriuretic peptide levels, blood pressure, selected drug treatments, sodium level, and history of cardiopulmonary resuscitation or mechanical ventilation.
Dr. Hummel had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
ORLANDO – Advanced heart failure patients who are hospitalized for heart failure and have a higher symptom burden at discharge have a significantly increased rate of death or rehospitalization over the next 6 months, based on an analysis of 393 patients enrolled in a heart failure trial.
The strong link between severe symptom burden and poor near-term outcomes persisted despite adjustment for various markers of heart failure severity, suggesting that treatment aimed at reducing symptoms may be able to reduce mortality or heart failure hospitalization in advanced heart failure patients, Ellen K. Hummel, MD, said at the annual scientific meeting of the Heart Failure Society of America.
In her analysis, a severe symptom burden at the time of hospital discharge linked with an adjusted 2.9-fold increased mortality rate and a 2.5-fold increased rate of days dead or hospitalized during the next 6 months, said Dr. Hummel, a geriatric and palliative care specialist at the University of Michigan in Ann Arbor. These elevated rate ratios for patients with severe symptoms at hospital discharge were in comparison to the ratios for advanced heart failure patients in the study with no symptoms at discharge.
Three symptoms contributed to the symptom score she used in her analysis: fatigue, scored on a scale of 0-3; dyspnea, also scored 0-3; and gastrointestinal distress, scored as 0-2, creating a maximum score of 8. Her analysis categorized mild as a total score of 1-4 and severe as 5 or greater. In the study population she used for her analysis, patients enrolled in the multicenter ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) trial, 111 of the 393 evaluable patients (28%) had none of these symptoms, 239 (61%) had mild symptoms, and 43 (11%) had severe symptoms. Scoring was done by patients based on their subjective self-assessment at the time of hospital discharge.
The absolute, observed 6-month mortality rates were roughly 45% among patients with severe symptoms, about 17% in patients with mild symptoms, and about 12% in those with no symptoms.
The primary purpose of ESCAPE was to assess the impact that routine collection of data from a pulmonary artery catheter during hospitalization has on outcomes; the results showed no significant link between improved outcomes and getting these data (JAMA. 2005 Oct 5;294[13]:1625-33). The study ran during 2000-2003 at 26 centers in the United States and Canada. Of the 433 advanced heart failure patients enrolled in ESCAPE, 393 had complete records to allow the current analysis.
The adjustments that Dr. Hummel made in the proportional hazard analysis took into account New York Heart Association class, and severity of disease at the time of hospital discharge measured by the ESCAPE Discharge Risk Score. This score takes into account age, 6-minute walk distance, blood urea nitrogen, brain natriuretic peptide levels, blood pressure, selected drug treatments, sodium level, and history of cardiopulmonary resuscitation or mechanical ventilation.
Dr. Hummel had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
ORLANDO – Advanced heart failure patients who are hospitalized for heart failure and have a higher symptom burden at discharge have a significantly increased rate of death or rehospitalization over the next 6 months, based on an analysis of 393 patients enrolled in a heart failure trial.
The strong link between severe symptom burden and poor near-term outcomes persisted despite adjustment for various markers of heart failure severity, suggesting that treatment aimed at reducing symptoms may be able to reduce mortality or heart failure hospitalization in advanced heart failure patients, Ellen K. Hummel, MD, said at the annual scientific meeting of the Heart Failure Society of America.
In her analysis, a severe symptom burden at the time of hospital discharge linked with an adjusted 2.9-fold increased mortality rate and a 2.5-fold increased rate of days dead or hospitalized during the next 6 months, said Dr. Hummel, a geriatric and palliative care specialist at the University of Michigan in Ann Arbor. These elevated rate ratios for patients with severe symptoms at hospital discharge were in comparison to the ratios for advanced heart failure patients in the study with no symptoms at discharge.
Three symptoms contributed to the symptom score she used in her analysis: fatigue, scored on a scale of 0-3; dyspnea, also scored 0-3; and gastrointestinal distress, scored as 0-2, creating a maximum score of 8. Her analysis categorized mild as a total score of 1-4 and severe as 5 or greater. In the study population she used for her analysis, patients enrolled in the multicenter ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) trial, 111 of the 393 evaluable patients (28%) had none of these symptoms, 239 (61%) had mild symptoms, and 43 (11%) had severe symptoms. Scoring was done by patients based on their subjective self-assessment at the time of hospital discharge.
The absolute, observed 6-month mortality rates were roughly 45% among patients with severe symptoms, about 17% in patients with mild symptoms, and about 12% in those with no symptoms.
The primary purpose of ESCAPE was to assess the impact that routine collection of data from a pulmonary artery catheter during hospitalization has on outcomes; the results showed no significant link between improved outcomes and getting these data (JAMA. 2005 Oct 5;294[13]:1625-33). The study ran during 2000-2003 at 26 centers in the United States and Canada. Of the 433 advanced heart failure patients enrolled in ESCAPE, 393 had complete records to allow the current analysis.
The adjustments that Dr. Hummel made in the proportional hazard analysis took into account New York Heart Association class, and severity of disease at the time of hospital discharge measured by the ESCAPE Discharge Risk Score. This score takes into account age, 6-minute walk distance, blood urea nitrogen, brain natriuretic peptide levels, blood pressure, selected drug treatments, sodium level, and history of cardiopulmonary resuscitation or mechanical ventilation.
Dr. Hummel had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
AT THE HFSA ANNUAL SCIENTIFIC MEETING
Key clinical point:
Major finding: Patients with severe symptoms at discharge had a 2.9-fold increased rate of death, compared with those with no symptoms.
Data source: A post hoc analysis of data collected from 393 patients enrolled in the ESCAPE trial.
Disclosures: Dr. Hummel had no relevant financial disclosures.
Sensory-related difficulties in children
A Google search of “sensory issues in children” reveals more than 20 million results and a wide range of terminology that can be confusing to parents, providers, and youth themselves. Phenomena such as sensory processing disorder, sensory integration disorder, sensory discrimination disorder, and sensory defensiveness are noted, and autism spectrum disorder (ASD) is a label not uncommonly attached to the former terms.
The fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM) does not include a discrete diagnosis to apply to children who have sensory differences, (meaning that they have difficulties regulating sensory input and such difficulties affect their ability to successfully relate to the world around them), but these differences are now part of the diagnostic criteria for ASD. The literature indicates that a majority of youth with ASD demonstrate features of sensory overresponsiveness (JAMA Psychiatry. 2015 Aug;72[8]:778-86), and providers should rightfully be concerned about the possibility of autism in a youngster who is presenting with severe negative responses to auditory, visual, and/or tactile stimuli.
Notably, however, even though sensory problems are considered a feature of autism, they are not pathognomonic for the disorder, and most children with these problems are, in fact, not autistic. Children with deficits in their ability to modulate sensory stimuli can present with a wide range of emotional-behavioral problems, including externalizing behaviors and internalizing symptoms manifesting with anxiety, attention challenges, mood dysregulation, and overall poor adaptive functioning. The relationship between sensory issues (both underresponsiveness and overresponsiveness) and psychopathology is rather complicated as sensory dysfunction can exist independent of a psychiatric disorder, be a significant risk factor for the development of the disorder (J Abnorm Child Psychol. 2009 Nov;37[8]:1077-87), and have symptom overlap with the disorder.
All in all, in spite of this complexity, since the 1960’s (Am J Occup Ther. 1964 Jan-Feb;18:6-11), it’s been clear that sensory dysfunction in children is associated with impairments in development, learning, and self-regulation. Parents of these children experience elevated levels of stress (J Child Fam Stud. 2013 Oct 1;22[7]:912-21), and early identification of sensory differences, psychoeducation, and referral for treatment are critical to minimize these impacts and foster positive outcomes.
Case Summary

In gathering a history and administering the Autism Diagnostic Observation Schedule (ADOS), it became clear that Sarah did not present with the social-communicative impairments that characterize ASD, but she did demonstrate repetitive hand flapping, troubles tolerating large social get-togethers, hypersensitivities, and a vulnerability to getting stuck when attempting to transition between activities. It is not uncommon for Sarah to use “fight-and-flight” reactions when faced with internal or external discomfort. Child Behavior Checklist data revealed multi-informant endorsement of clinical range symptoms across broad-band and narrow-band domains. Additionally, the Sensory Profile–2 yielded elevated scores in categories measuring sensory seeking and sensory sensitivity. The Sensory Profile is a standardized tool that uses caregiver and teacher-completed questionnaires to examine a child’s sensory processing abilities and provide data regarding the effect of such sensory processing on functional performance. Integrating all the available data, our team certainly appreciated Sarah’s profound sensory overresponsiveness, and a diagnosis of an unspecified anxiety disorder was provided along with consideration for attention-deficit/hyperactivity disorder (ADHD) (with teacher input needed to further investigate this possibility). The family history revealed anxiety disorders occurring both maternally and paternally. Additionally, Sarah’s mother’s acknowledged having her own similar sensory issues as a child.
Discussion
Associations among anxiety, sensory overresponsiveness, and ADHD are recognized in the literature (Am J Occup Ther. 2009 Jul-Aug;63[4]:433-40) and have implications for treatment. Furthermore, there is evidence that there is a heritable aspect to sensory processing abnormalities, and tactile defensiveness is associated with fearful temperament and anxiety (J Abnorm Child Psychol. 2006 Jun;34[3]:393-407). In Sarah’s case, her intense behavioral response to ordinary sensory stimuli was striking, and she had not yet been referred for an occupational therapy evaluation, which was the primary recommendation to further assess and understand her complicated sensory profile. As one component of a comprehensive treatment plan, an occupational therapist (www.aota.org), by using evidence-based practices in a sensory-integration framework, could be helpful in addressing Sarah’s range of challenges and promoting positive outcomes related to socialization, behavioral regulation, and attention. Occupational therapists, with assistance from other team members, also could work with Sarah and her family on developing relaxation skills and use exposure and response prevention–oriented intervention strategies to address anxieties. Families, however, should be counseled about the limited data on the use of sensory-based therapies (Pediatrics. 2012 Jun;129[6]:1186-9); the use of parent-management training/family coaching should also be a treatment consideration to help promote overall regulatory functioning in the household.
Clinical pearl
When encountering youth with sensory-related challenges, a clinician’s diagnostic considerations should be more than just thinking about the possibility of an autism spectrum disorder. Symptoms of sensory overresponsiveness are associated with other emotional-behavioral conditions, but also can be seen without co-occurring psychopathology. With the latter, however, providers should be mindful that family-related impairments still may be quite noteworthy (J Am Acad Child Adolesc Psychiatry. 2011 Dec;50[12]:1210-9) and associated behavior problems could be attributed incorrectly to other diagnoses (which may lead to the recommendation of ineffective and inappropriate treatments). Much more research is needed to help develop a robust framework for diagnosing and labeling sensory issues in children and studying the efficacy of available intervention strategies.
Dr. Dickerson, a child and adolescent psychiatrist, is assistant professor of psychiatry at the University of Vermont, Burlington, where he is director of the autism diagnostic clinic.
A Google search of “sensory issues in children” reveals more than 20 million results and a wide range of terminology that can be confusing to parents, providers, and youth themselves. Phenomena such as sensory processing disorder, sensory integration disorder, sensory discrimination disorder, and sensory defensiveness are noted, and autism spectrum disorder (ASD) is a label not uncommonly attached to the former terms.
The fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM) does not include a discrete diagnosis to apply to children who have sensory differences, (meaning that they have difficulties regulating sensory input and such difficulties affect their ability to successfully relate to the world around them), but these differences are now part of the diagnostic criteria for ASD. The literature indicates that a majority of youth with ASD demonstrate features of sensory overresponsiveness (JAMA Psychiatry. 2015 Aug;72[8]:778-86), and providers should rightfully be concerned about the possibility of autism in a youngster who is presenting with severe negative responses to auditory, visual, and/or tactile stimuli.
Notably, however, even though sensory problems are considered a feature of autism, they are not pathognomonic for the disorder, and most children with these problems are, in fact, not autistic. Children with deficits in their ability to modulate sensory stimuli can present with a wide range of emotional-behavioral problems, including externalizing behaviors and internalizing symptoms manifesting with anxiety, attention challenges, mood dysregulation, and overall poor adaptive functioning. The relationship between sensory issues (both underresponsiveness and overresponsiveness) and psychopathology is rather complicated as sensory dysfunction can exist independent of a psychiatric disorder, be a significant risk factor for the development of the disorder (J Abnorm Child Psychol. 2009 Nov;37[8]:1077-87), and have symptom overlap with the disorder.
All in all, in spite of this complexity, since the 1960’s (Am J Occup Ther. 1964 Jan-Feb;18:6-11), it’s been clear that sensory dysfunction in children is associated with impairments in development, learning, and self-regulation. Parents of these children experience elevated levels of stress (J Child Fam Stud. 2013 Oct 1;22[7]:912-21), and early identification of sensory differences, psychoeducation, and referral for treatment are critical to minimize these impacts and foster positive outcomes.
Case Summary
In gathering a history and administering the Autism Diagnostic Observation Schedule (ADOS), it became clear that Sarah did not present with the social-communicative impairments that characterize ASD, but she did demonstrate repetitive hand flapping, troubles tolerating large social get-togethers, hypersensitivities, and a vulnerability to getting stuck when attempting to transition between activities. It is not uncommon for Sarah to use “fight-and-flight” reactions when faced with internal or external discomfort. Child Behavior Checklist data revealed multi-informant endorsement of clinical range symptoms across broad-band and narrow-band domains. Additionally, the Sensory Profile–2 yielded elevated scores in categories measuring sensory seeking and sensory sensitivity. The Sensory Profile is a standardized tool that uses caregiver and teacher-completed questionnaires to examine a child’s sensory processing abilities and provide data regarding the effect of such sensory processing on functional performance. Integrating all the available data, our team certainly appreciated Sarah’s profound sensory overresponsiveness, and a diagnosis of an unspecified anxiety disorder was provided along with consideration for attention-deficit/hyperactivity disorder (ADHD) (with teacher input needed to further investigate this possibility). The family history revealed anxiety disorders occurring both maternally and paternally. Additionally, Sarah’s mother’s acknowledged having her own similar sensory issues as a child.
Discussion
Associations among anxiety, sensory overresponsiveness, and ADHD are recognized in the literature (Am J Occup Ther. 2009 Jul-Aug;63[4]:433-40) and have implications for treatment. Furthermore, there is evidence that there is a heritable aspect to sensory processing abnormalities, and tactile defensiveness is associated with fearful temperament and anxiety (J Abnorm Child Psychol. 2006 Jun;34[3]:393-407). In Sarah’s case, her intense behavioral response to ordinary sensory stimuli was striking, and she had not yet been referred for an occupational therapy evaluation, which was the primary recommendation to further assess and understand her complicated sensory profile. As one component of a comprehensive treatment plan, an occupational therapist (www.aota.org), by using evidence-based practices in a sensory-integration framework, could be helpful in addressing Sarah’s range of challenges and promoting positive outcomes related to socialization, behavioral regulation, and attention. Occupational therapists, with assistance from other team members, also could work with Sarah and her family on developing relaxation skills and use exposure and response prevention–oriented intervention strategies to address anxieties. Families, however, should be counseled about the limited data on the use of sensory-based therapies (Pediatrics. 2012 Jun;129[6]:1186-9); the use of parent-management training/family coaching should also be a treatment consideration to help promote overall regulatory functioning in the household.
Clinical pearl
When encountering youth with sensory-related challenges, a clinician’s diagnostic considerations should be more than just thinking about the possibility of an autism spectrum disorder. Symptoms of sensory overresponsiveness are associated with other emotional-behavioral conditions, but also can be seen without co-occurring psychopathology. With the latter, however, providers should be mindful that family-related impairments still may be quite noteworthy (J Am Acad Child Adolesc Psychiatry. 2011 Dec;50[12]:1210-9) and associated behavior problems could be attributed incorrectly to other diagnoses (which may lead to the recommendation of ineffective and inappropriate treatments). Much more research is needed to help develop a robust framework for diagnosing and labeling sensory issues in children and studying the efficacy of available intervention strategies.
Dr. Dickerson, a child and adolescent psychiatrist, is assistant professor of psychiatry at the University of Vermont, Burlington, where he is director of the autism diagnostic clinic.
A Google search of “sensory issues in children” reveals more than 20 million results and a wide range of terminology that can be confusing to parents, providers, and youth themselves. Phenomena such as sensory processing disorder, sensory integration disorder, sensory discrimination disorder, and sensory defensiveness are noted, and autism spectrum disorder (ASD) is a label not uncommonly attached to the former terms.
The fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM) does not include a discrete diagnosis to apply to children who have sensory differences, (meaning that they have difficulties regulating sensory input and such difficulties affect their ability to successfully relate to the world around them), but these differences are now part of the diagnostic criteria for ASD. The literature indicates that a majority of youth with ASD demonstrate features of sensory overresponsiveness (JAMA Psychiatry. 2015 Aug;72[8]:778-86), and providers should rightfully be concerned about the possibility of autism in a youngster who is presenting with severe negative responses to auditory, visual, and/or tactile stimuli.
Notably, however, even though sensory problems are considered a feature of autism, they are not pathognomonic for the disorder, and most children with these problems are, in fact, not autistic. Children with deficits in their ability to modulate sensory stimuli can present with a wide range of emotional-behavioral problems, including externalizing behaviors and internalizing symptoms manifesting with anxiety, attention challenges, mood dysregulation, and overall poor adaptive functioning. The relationship between sensory issues (both underresponsiveness and overresponsiveness) and psychopathology is rather complicated as sensory dysfunction can exist independent of a psychiatric disorder, be a significant risk factor for the development of the disorder (J Abnorm Child Psychol. 2009 Nov;37[8]:1077-87), and have symptom overlap with the disorder.
All in all, in spite of this complexity, since the 1960’s (Am J Occup Ther. 1964 Jan-Feb;18:6-11), it’s been clear that sensory dysfunction in children is associated with impairments in development, learning, and self-regulation. Parents of these children experience elevated levels of stress (J Child Fam Stud. 2013 Oct 1;22[7]:912-21), and early identification of sensory differences, psychoeducation, and referral for treatment are critical to minimize these impacts and foster positive outcomes.
Case Summary
In gathering a history and administering the Autism Diagnostic Observation Schedule (ADOS), it became clear that Sarah did not present with the social-communicative impairments that characterize ASD, but she did demonstrate repetitive hand flapping, troubles tolerating large social get-togethers, hypersensitivities, and a vulnerability to getting stuck when attempting to transition between activities. It is not uncommon for Sarah to use “fight-and-flight” reactions when faced with internal or external discomfort. Child Behavior Checklist data revealed multi-informant endorsement of clinical range symptoms across broad-band and narrow-band domains. Additionally, the Sensory Profile–2 yielded elevated scores in categories measuring sensory seeking and sensory sensitivity. The Sensory Profile is a standardized tool that uses caregiver and teacher-completed questionnaires to examine a child’s sensory processing abilities and provide data regarding the effect of such sensory processing on functional performance. Integrating all the available data, our team certainly appreciated Sarah’s profound sensory overresponsiveness, and a diagnosis of an unspecified anxiety disorder was provided along with consideration for attention-deficit/hyperactivity disorder (ADHD) (with teacher input needed to further investigate this possibility). The family history revealed anxiety disorders occurring both maternally and paternally. Additionally, Sarah’s mother’s acknowledged having her own similar sensory issues as a child.
Discussion
Associations among anxiety, sensory overresponsiveness, and ADHD are recognized in the literature (Am J Occup Ther. 2009 Jul-Aug;63[4]:433-40) and have implications for treatment. Furthermore, there is evidence that there is a heritable aspect to sensory processing abnormalities, and tactile defensiveness is associated with fearful temperament and anxiety (J Abnorm Child Psychol. 2006 Jun;34[3]:393-407). In Sarah’s case, her intense behavioral response to ordinary sensory stimuli was striking, and she had not yet been referred for an occupational therapy evaluation, which was the primary recommendation to further assess and understand her complicated sensory profile. As one component of a comprehensive treatment plan, an occupational therapist (www.aota.org), by using evidence-based practices in a sensory-integration framework, could be helpful in addressing Sarah’s range of challenges and promoting positive outcomes related to socialization, behavioral regulation, and attention. Occupational therapists, with assistance from other team members, also could work with Sarah and her family on developing relaxation skills and use exposure and response prevention–oriented intervention strategies to address anxieties. Families, however, should be counseled about the limited data on the use of sensory-based therapies (Pediatrics. 2012 Jun;129[6]:1186-9); the use of parent-management training/family coaching should also be a treatment consideration to help promote overall regulatory functioning in the household.
Clinical pearl
When encountering youth with sensory-related challenges, a clinician’s diagnostic considerations should be more than just thinking about the possibility of an autism spectrum disorder. Symptoms of sensory overresponsiveness are associated with other emotional-behavioral conditions, but also can be seen without co-occurring psychopathology. With the latter, however, providers should be mindful that family-related impairments still may be quite noteworthy (J Am Acad Child Adolesc Psychiatry. 2011 Dec;50[12]:1210-9) and associated behavior problems could be attributed incorrectly to other diagnoses (which may lead to the recommendation of ineffective and inappropriate treatments). Much more research is needed to help develop a robust framework for diagnosing and labeling sensory issues in children and studying the efficacy of available intervention strategies.
Dr. Dickerson, a child and adolescent psychiatrist, is assistant professor of psychiatry at the University of Vermont, Burlington, where he is director of the autism diagnostic clinic.
Obesity risk rises when kids aren’t active 1 hour a day
SAN FRANCISCO – The behavioral characteristic that correlated the most with obesity in school-age children was being active less than 1 hour per day, according to findings from a prospective study.
Meredith Johnston, DO, of Eau Claire Cooperative in Columbia, S.C., performed the study with Nirupma Sharma, MD, at Children’s Hospital of Georgia in Augusta. The prospective, questionnaire-based study was conducted with 103 children, aged 5-18 years, over a 6-month period. Dr. Johnston reported the results in a poster presentation at the annual meeting of the American Academy of Pediatrics.

“Giving a child one modifiable lifestyle factor to incorporate into their lifestyle instead of overwhelming them with multiple changes is more likely to produce significant change to prevent obesity,” Dr. Johnston said in an interview. “And finding an activity that the child enjoys will produce the best adherence and greatest long-term effects.”
Considering the findings of the study, “children should exercise 30 minutes to 1 hour a day to prevent childhood obesity,” she explained.
A recent review in the Annual Review of Public Health has shown a change in teen physical activity patterns due to an increase in screen time and a decrease in opportunities for physical activities at school and in the community. Giving patients tools for exercise such as dancing to YouTube videos or playing active video games might be a good idea, Dr. Johnston said.
Drinking more than five cans of soda in a day also was significantly associated with a higher BMI (P = .001). That lifestyle factor could be addressed at subsequent well-child visits.
Such efforts are critical, she noted, because an estimated 17% of 2- to 19-year-olds are obese, and 15% are overweight.
Dr. Johnston said she had no relevant financial disclosures.
SAN FRANCISCO – The behavioral characteristic that correlated the most with obesity in school-age children was being active less than 1 hour per day, according to findings from a prospective study.
Meredith Johnston, DO, of Eau Claire Cooperative in Columbia, S.C., performed the study with Nirupma Sharma, MD, at Children’s Hospital of Georgia in Augusta. The prospective, questionnaire-based study was conducted with 103 children, aged 5-18 years, over a 6-month period. Dr. Johnston reported the results in a poster presentation at the annual meeting of the American Academy of Pediatrics.
“Giving a child one modifiable lifestyle factor to incorporate into their lifestyle instead of overwhelming them with multiple changes is more likely to produce significant change to prevent obesity,” Dr. Johnston said in an interview. “And finding an activity that the child enjoys will produce the best adherence and greatest long-term effects.”
Considering the findings of the study, “children should exercise 30 minutes to 1 hour a day to prevent childhood obesity,” she explained.
A recent review in the Annual Review of Public Health has shown a change in teen physical activity patterns due to an increase in screen time and a decrease in opportunities for physical activities at school and in the community. Giving patients tools for exercise such as dancing to YouTube videos or playing active video games might be a good idea, Dr. Johnston said.
Drinking more than five cans of soda in a day also was significantly associated with a higher BMI (P = .001). That lifestyle factor could be addressed at subsequent well-child visits.
Such efforts are critical, she noted, because an estimated 17% of 2- to 19-year-olds are obese, and 15% are overweight.
Dr. Johnston said she had no relevant financial disclosures.
SAN FRANCISCO – The behavioral characteristic that correlated the most with obesity in school-age children was being active less than 1 hour per day, according to findings from a prospective study.
Meredith Johnston, DO, of Eau Claire Cooperative in Columbia, S.C., performed the study with Nirupma Sharma, MD, at Children’s Hospital of Georgia in Augusta. The prospective, questionnaire-based study was conducted with 103 children, aged 5-18 years, over a 6-month period. Dr. Johnston reported the results in a poster presentation at the annual meeting of the American Academy of Pediatrics.
“Giving a child one modifiable lifestyle factor to incorporate into their lifestyle instead of overwhelming them with multiple changes is more likely to produce significant change to prevent obesity,” Dr. Johnston said in an interview. “And finding an activity that the child enjoys will produce the best adherence and greatest long-term effects.”
Considering the findings of the study, “children should exercise 30 minutes to 1 hour a day to prevent childhood obesity,” she explained.
A recent review in the Annual Review of Public Health has shown a change in teen physical activity patterns due to an increase in screen time and a decrease in opportunities for physical activities at school and in the community. Giving patients tools for exercise such as dancing to YouTube videos or playing active video games might be a good idea, Dr. Johnston said.
Drinking more than five cans of soda in a day also was significantly associated with a higher BMI (P = .001). That lifestyle factor could be addressed at subsequent well-child visits.
Such efforts are critical, she noted, because an estimated 17% of 2- to 19-year-olds are obese, and 15% are overweight.
Dr. Johnston said she had no relevant financial disclosures.
AT AAP 2016
Key clinical point:
Major finding: Being active less than 1 hour per day was significantly associated with a higher body mass index in children (P = .006 for 30 minutes to 1 hour, and P = .017 for less than 30 minutes).
Data source: A prospective, questionnaire-based study involving 103 children, aged 5-18 years, over a 6-month period.
Disclosures: Dr. Johnston said she had no relevant financial disclosures.
New trials address unresolved issues in endovascular therapy for acute ischemic stroke
BALTIMORE – Endovascular therapy is a bedrock of acute ischemic stroke treatment, but some vexing issues remain unresolved, according to Joseph Broderick, MD, of the University of Cincinnati.
“We have learned a tremendous amount the last few years. We know that thrombectomy can work if initiated in patients within 6 hours of stroke onset with or without TPA [tissue plasminogen activator]. The question is what we don’t know,” Dr. Broderick said.
The good news is that clinical trials are underway or planned to address several of these issues, he said at the annual meeting of the American Neurological Association.
Mechanical thrombectomy beyond 6 hours
Whether the treatment window for thrombectomy can be safely extended in appropriately selected patients beyond 6 hours from onset is “one of the most important issues we face,” Dr. Broderick said.

Along with DEFUSE 3, the DWI or CTP Assessment With Clinical Mismatch in the Triage of Wake-Up and Late Presenting Strokes Undergoing Neurointervention (DAWN) trial will examine whether medical management that includes thrombectomy delivered up to 24 hours after AIS symptoms first appear will yield better clinical outcomes than medical management alone in appropriate patients.
The use of mechanical thrombectomy up to 12 hours after symptom development in AIS patients for whom intravenous TPA can’t be used or doesn’t work will be assessed in the Perfusion Imaging Selection of Ischemic Stroke Patients for Endovascular Therapy (POSITIVE) trial. The primary objective is to show less stroke-related disability and improved good functional outcomes, compared with those who receive best medical therapy.
The first Multicenter Randomized Clinical Trial of Endovascular Treatment in the Netherlands (MR CLEAN) trial, which used stent retrievers in almost all cases, demonstrated the benefit of endovascular therapy up to 6 hours after stroke onset in the proximal anterior circulation when used along with best medical therapy (N Engl J Med. 2015;372:11-20). The MR CLEAN LATE trial involving 500 patients will expand the time window up to 12 hours.
Triage of acute stroke patients
The triage of AIS patients before their arrival at the hospital also is an evolving emergency medical service issue. While the classic triage involving CT takes time, the use of scoring systems like the National Institutes of Health Stroke Scale and the Cincinnati Stroke Triage Assessment Tool, or telemedicine-based consultation, can be done faster and with less training.
Where the patient is taken first could be important. The RACECAT trial being conducted in the Catalan territory will compare transfer of acute stroke patients with suspected large-vessel occlusion to the closest local stroke center versus direct transfer to an endovascular stroke center.
Conscious sedation or general anesthesia
Some patients undergoing thrombectomy may be able to tolerate conscious sedation in place of general anesthesia. For these patients, cost-effectiveness becomes a dominant consideration. Several anesthesia trials are addressing the issue. The Sedation Versus Intubation for Endovascular Stroke Treatment (SIESTA) trial involving 150 German patients has been completed, but the findings have not been reported. The General or Local Anaesthesia in Intra-arterial Therapy (GOLIATH) trial is recruiting patients in the Netherlands, with a target enrollment of 128. Other trials are planned in China and Sweden, but recruitment has not started.
Thrombectomy alone
For patients with AIS caused by basilar artery occlusion, stroke specialists have wondered whether intra-arterial treatment, including thrombolysis and potentially thrombectomy, could be more effective than best medical therapy, including intravenous thrombolysis. The Basilar Artery International Cooperation Study (BASICS) in Europe and the BEST trial in China are two trials in the early stages of assessing this question.
A related issue is whether thrombectomy alone is better than intravenous TPA followed by thrombectomy. This issue is being addressed in another MR CLEAN trial that is designed to compare the two approaches in 500 patients randomized to one or the other treatment.
Other intravenous antithrombotic drugs
Another unresolved issue relates to the use of other antithrombotic medications delivered intravenously in combination with TPA. This could be the sole treatment strategy, or could be used in patients who also undergo endovascular thrombectomy. The Multi-Arm Optimization of Stroke Thrombolysis (MOST) trial will randomize patients to receive argatroban or eptifibatide along with TPA, or TPA alone, within 3 hours of stroke symptom onset. Endovascular therapy will be done as indicated.
Heparin use in endovascular therapy
The proper use of antithrombotics like heparin in patients treated with endovascular therapy is unclear. The MR CLEAN MED trial will assess the benefits of two doses of unfractionated heparin and aspirin alone or in combination in 1,500 patients with AIS who receive intra-arterial treatment for confirmed anterior circulation occlusion.
On the horizon
Whether neuroprotection can improve the outcome in patients who undergo embolectomy is unclear, and will be the subject of a trial in Canada that is not yet recruiting patients. Finally, the best way to manage blood pressure following embolectomy and reperfusion is also unclear.
Dr. Broderick had no relevant financial disclosures.
BALTIMORE – Endovascular therapy is a bedrock of acute ischemic stroke treatment, but some vexing issues remain unresolved, according to Joseph Broderick, MD, of the University of Cincinnati.
“We have learned a tremendous amount the last few years. We know that thrombectomy can work if initiated in patients within 6 hours of stroke onset with or without TPA [tissue plasminogen activator]. The question is what we don’t know,” Dr. Broderick said.
The good news is that clinical trials are underway or planned to address several of these issues, he said at the annual meeting of the American Neurological Association.
Mechanical thrombectomy beyond 6 hours
Whether the treatment window for thrombectomy can be safely extended in appropriately selected patients beyond 6 hours from onset is “one of the most important issues we face,” Dr. Broderick said.
Along with DEFUSE 3, the DWI or CTP Assessment With Clinical Mismatch in the Triage of Wake-Up and Late Presenting Strokes Undergoing Neurointervention (DAWN) trial will examine whether medical management that includes thrombectomy delivered up to 24 hours after AIS symptoms first appear will yield better clinical outcomes than medical management alone in appropriate patients.
The use of mechanical thrombectomy up to 12 hours after symptom development in AIS patients for whom intravenous TPA can’t be used or doesn’t work will be assessed in the Perfusion Imaging Selection of Ischemic Stroke Patients for Endovascular Therapy (POSITIVE) trial. The primary objective is to show less stroke-related disability and improved good functional outcomes, compared with those who receive best medical therapy.
The first Multicenter Randomized Clinical Trial of Endovascular Treatment in the Netherlands (MR CLEAN) trial, which used stent retrievers in almost all cases, demonstrated the benefit of endovascular therapy up to 6 hours after stroke onset in the proximal anterior circulation when used along with best medical therapy (N Engl J Med. 2015;372:11-20). The MR CLEAN LATE trial involving 500 patients will expand the time window up to 12 hours.
Triage of acute stroke patients
The triage of AIS patients before their arrival at the hospital also is an evolving emergency medical service issue. While the classic triage involving CT takes time, the use of scoring systems like the National Institutes of Health Stroke Scale and the Cincinnati Stroke Triage Assessment Tool, or telemedicine-based consultation, can be done faster and with less training.
Where the patient is taken first could be important. The RACECAT trial being conducted in the Catalan territory will compare transfer of acute stroke patients with suspected large-vessel occlusion to the closest local stroke center versus direct transfer to an endovascular stroke center.
Conscious sedation or general anesthesia
Some patients undergoing thrombectomy may be able to tolerate conscious sedation in place of general anesthesia. For these patients, cost-effectiveness becomes a dominant consideration. Several anesthesia trials are addressing the issue. The Sedation Versus Intubation for Endovascular Stroke Treatment (SIESTA) trial involving 150 German patients has been completed, but the findings have not been reported. The General or Local Anaesthesia in Intra-arterial Therapy (GOLIATH) trial is recruiting patients in the Netherlands, with a target enrollment of 128. Other trials are planned in China and Sweden, but recruitment has not started.
Thrombectomy alone
For patients with AIS caused by basilar artery occlusion, stroke specialists have wondered whether intra-arterial treatment, including thrombolysis and potentially thrombectomy, could be more effective than best medical therapy, including intravenous thrombolysis. The Basilar Artery International Cooperation Study (BASICS) in Europe and the BEST trial in China are two trials in the early stages of assessing this question.
A related issue is whether thrombectomy alone is better than intravenous TPA followed by thrombectomy. This issue is being addressed in another MR CLEAN trial that is designed to compare the two approaches in 500 patients randomized to one or the other treatment.
Other intravenous antithrombotic drugs
Another unresolved issue relates to the use of other antithrombotic medications delivered intravenously in combination with TPA. This could be the sole treatment strategy, or could be used in patients who also undergo endovascular thrombectomy. The Multi-Arm Optimization of Stroke Thrombolysis (MOST) trial will randomize patients to receive argatroban or eptifibatide along with TPA, or TPA alone, within 3 hours of stroke symptom onset. Endovascular therapy will be done as indicated.
Heparin use in endovascular therapy
The proper use of antithrombotics like heparin in patients treated with endovascular therapy is unclear. The MR CLEAN MED trial will assess the benefits of two doses of unfractionated heparin and aspirin alone or in combination in 1,500 patients with AIS who receive intra-arterial treatment for confirmed anterior circulation occlusion.
On the horizon
Whether neuroprotection can improve the outcome in patients who undergo embolectomy is unclear, and will be the subject of a trial in Canada that is not yet recruiting patients. Finally, the best way to manage blood pressure following embolectomy and reperfusion is also unclear.
Dr. Broderick had no relevant financial disclosures.
BALTIMORE – Endovascular therapy is a bedrock of acute ischemic stroke treatment, but some vexing issues remain unresolved, according to Joseph Broderick, MD, of the University of Cincinnati.
“We have learned a tremendous amount the last few years. We know that thrombectomy can work if initiated in patients within 6 hours of stroke onset with or without TPA [tissue plasminogen activator]. The question is what we don’t know,” Dr. Broderick said.
The good news is that clinical trials are underway or planned to address several of these issues, he said at the annual meeting of the American Neurological Association.
Mechanical thrombectomy beyond 6 hours
Whether the treatment window for thrombectomy can be safely extended in appropriately selected patients beyond 6 hours from onset is “one of the most important issues we face,” Dr. Broderick said.
Along with DEFUSE 3, the DWI or CTP Assessment With Clinical Mismatch in the Triage of Wake-Up and Late Presenting Strokes Undergoing Neurointervention (DAWN) trial will examine whether medical management that includes thrombectomy delivered up to 24 hours after AIS symptoms first appear will yield better clinical outcomes than medical management alone in appropriate patients.
The use of mechanical thrombectomy up to 12 hours after symptom development in AIS patients for whom intravenous TPA can’t be used or doesn’t work will be assessed in the Perfusion Imaging Selection of Ischemic Stroke Patients for Endovascular Therapy (POSITIVE) trial. The primary objective is to show less stroke-related disability and improved good functional outcomes, compared with those who receive best medical therapy.
The first Multicenter Randomized Clinical Trial of Endovascular Treatment in the Netherlands (MR CLEAN) trial, which used stent retrievers in almost all cases, demonstrated the benefit of endovascular therapy up to 6 hours after stroke onset in the proximal anterior circulation when used along with best medical therapy (N Engl J Med. 2015;372:11-20). The MR CLEAN LATE trial involving 500 patients will expand the time window up to 12 hours.
Triage of acute stroke patients
The triage of AIS patients before their arrival at the hospital also is an evolving emergency medical service issue. While the classic triage involving CT takes time, the use of scoring systems like the National Institutes of Health Stroke Scale and the Cincinnati Stroke Triage Assessment Tool, or telemedicine-based consultation, can be done faster and with less training.
Where the patient is taken first could be important. The RACECAT trial being conducted in the Catalan territory will compare transfer of acute stroke patients with suspected large-vessel occlusion to the closest local stroke center versus direct transfer to an endovascular stroke center.
Conscious sedation or general anesthesia
Some patients undergoing thrombectomy may be able to tolerate conscious sedation in place of general anesthesia. For these patients, cost-effectiveness becomes a dominant consideration. Several anesthesia trials are addressing the issue. The Sedation Versus Intubation for Endovascular Stroke Treatment (SIESTA) trial involving 150 German patients has been completed, but the findings have not been reported. The General or Local Anaesthesia in Intra-arterial Therapy (GOLIATH) trial is recruiting patients in the Netherlands, with a target enrollment of 128. Other trials are planned in China and Sweden, but recruitment has not started.
Thrombectomy alone
For patients with AIS caused by basilar artery occlusion, stroke specialists have wondered whether intra-arterial treatment, including thrombolysis and potentially thrombectomy, could be more effective than best medical therapy, including intravenous thrombolysis. The Basilar Artery International Cooperation Study (BASICS) in Europe and the BEST trial in China are two trials in the early stages of assessing this question.
A related issue is whether thrombectomy alone is better than intravenous TPA followed by thrombectomy. This issue is being addressed in another MR CLEAN trial that is designed to compare the two approaches in 500 patients randomized to one or the other treatment.
Other intravenous antithrombotic drugs
Another unresolved issue relates to the use of other antithrombotic medications delivered intravenously in combination with TPA. This could be the sole treatment strategy, or could be used in patients who also undergo endovascular thrombectomy. The Multi-Arm Optimization of Stroke Thrombolysis (MOST) trial will randomize patients to receive argatroban or eptifibatide along with TPA, or TPA alone, within 3 hours of stroke symptom onset. Endovascular therapy will be done as indicated.
Heparin use in endovascular therapy
The proper use of antithrombotics like heparin in patients treated with endovascular therapy is unclear. The MR CLEAN MED trial will assess the benefits of two doses of unfractionated heparin and aspirin alone or in combination in 1,500 patients with AIS who receive intra-arterial treatment for confirmed anterior circulation occlusion.
On the horizon
Whether neuroprotection can improve the outcome in patients who undergo embolectomy is unclear, and will be the subject of a trial in Canada that is not yet recruiting patients. Finally, the best way to manage blood pressure following embolectomy and reperfusion is also unclear.
Dr. Broderick had no relevant financial disclosures.
United States nears 3,000 Zika-infected pregnancies
There were 54 new cases of pregnant women with laboratory evidence of Zika virus in the 50 states and the District of Columbia reported during the week ending Oct. 20 – the largest weekly increase in a month, according to the Centers for Disease Control and Prevention.
The number of cases reported in the U.S. territories, 100, was lower for the second week in a row, however, so the U.S. total for the week was a fairly average 154. The total number of pregnant women with laboratory evidence of Zika virus infection is now 2,980 for the year: 953 in the states/D.C. and 2,027 in the territories, the CDC reported.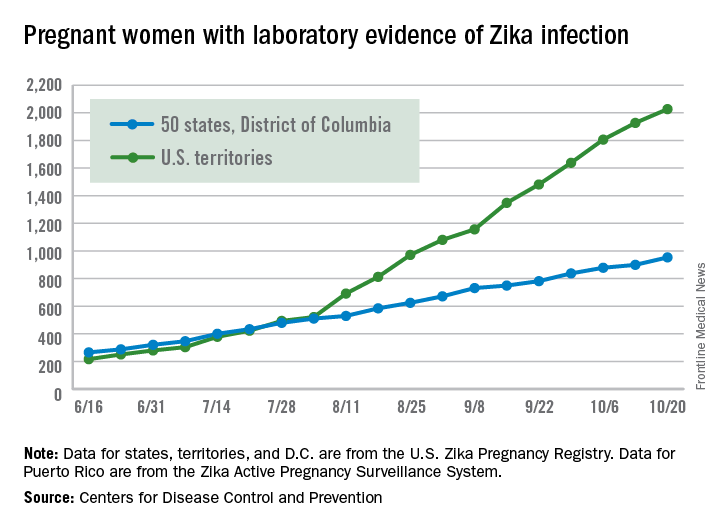
Among all Americans, the number of cases for 2015-2016 is now up to 32,814, with 1,396 new cases reported for the week ending Oct. 26: 75 in the states/D.C. and 1,321 in the territories. Almost all (98%) of the territorial cases have occurred in Puerto Rico, which continues to retroactively report cases, the CDC Arboviral Disease Branch noted.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 54 new cases of pregnant women with laboratory evidence of Zika virus in the 50 states and the District of Columbia reported during the week ending Oct. 20 – the largest weekly increase in a month, according to the Centers for Disease Control and Prevention.
The number of cases reported in the U.S. territories, 100, was lower for the second week in a row, however, so the U.S. total for the week was a fairly average 154. The total number of pregnant women with laboratory evidence of Zika virus infection is now 2,980 for the year: 953 in the states/D.C. and 2,027 in the territories, the CDC reported.
Among all Americans, the number of cases for 2015-2016 is now up to 32,814, with 1,396 new cases reported for the week ending Oct. 26: 75 in the states/D.C. and 1,321 in the territories. Almost all (98%) of the territorial cases have occurred in Puerto Rico, which continues to retroactively report cases, the CDC Arboviral Disease Branch noted.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 54 new cases of pregnant women with laboratory evidence of Zika virus in the 50 states and the District of Columbia reported during the week ending Oct. 20 – the largest weekly increase in a month, according to the Centers for Disease Control and Prevention.
The number of cases reported in the U.S. territories, 100, was lower for the second week in a row, however, so the U.S. total for the week was a fairly average 154. The total number of pregnant women with laboratory evidence of Zika virus infection is now 2,980 for the year: 953 in the states/D.C. and 2,027 in the territories, the CDC reported.
Among all Americans, the number of cases for 2015-2016 is now up to 32,814, with 1,396 new cases reported for the week ending Oct. 26: 75 in the states/D.C. and 1,321 in the territories. Almost all (98%) of the territorial cases have occurred in Puerto Rico, which continues to retroactively report cases, the CDC Arboviral Disease Branch noted.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Updated AAP safe sleep recs for infants reinforce life-saving messages
SAN FRANCISCO – At sleep time, infants should share their parents’ bedroom on a separate sleep surface without bed sharing, should be placed on their backs on a firm surface, and should have a sleep area free of blankets and soft objects, according to updated guidelines from the American Academy of Pediatrics aimed at reducing the risk of sudden infant death syndrome (SIDS) and other sleep-related infant deaths.
Drafted by a multidisciplinary task force, the set of 19 evidence-based recommendations largely reiterate messages that the academy has promoted for years such as “back to sleep for every sleep,” according to task force member Fern R. Hauck, MD, the Spencer P. Bass, MD, Twenty-First Century Professor of Family Medicine at the University of Virginia, Charlottesville. They were unveiled in a press briefing at the academy’s annual meeting and simultaneously published (Pediatrics. 2016;138[5]:e20162938).
Progress, but still a ways to go
Education campaigns that convey these and related messages to new parents and other caregivers have led to a more than halving of the rate of SIDS in recent decades. Yet, 3,500 infants are still lost each year to this syndrome and other sleep-related causes of infant death, such as unintentional suffocation, collectively called sudden unexpected infant death (SUID).

New is a recommendation for skin-to-skin care for at least the first hour of life for healthy newborns, as soon as the mother is alert enough to respond to her infant, according to Dr. Hauck. The aims here are to optimize neurodevelopment and promote temperature regulation.
There is no evidence that swaddling reduces the risk of SIDS, but parents can still use this technique if they wish as long as infants are placed on their back and it is discontinued as soon as they start to show signs of rolling over, she said. Evidence is also lacking for new technologies marketed as protective, for example, crib mattresses designed to reduce re-breathing of carbon dioxide should an infant become prone.
Sleeping in the parents’ room but on a separate surface decreases the risk of SIDS by as much as 50%, according to several studies. Bed sharing is not recommended because of the risk of suffocation, strangulation, and entrapment, the policy states.
The updated recommendations should be followed for every sleep and by every caregiver, until the child reaches 1 year of age, Dr. Hauck stressed. “This includes nap time and bedtime sleep, at home, in day care, or in any other locations where the baby is sleeping.”
“We feel that these messages need to start while the mom’s pregnant because some of the decisions that are made that are not always the best decisions, when the mother is exhausted, can occur spur of the moment,” she added. “As pediatricians, you can set up a prebirth visit to start talking about this, and obstetricians should be doing more as well to bring this up during their prenatal visits.”
Other recommendations include offering a pacifier at nap time and bedtime; avoiding smoke exposure during pregnancy and after birth; and avoiding alcohol and illicit drug use during pregnancy and after birth.
Breastfeeding issues
Although breastfeeding protects against SIDS, it can pose some problems for safe sleep practices, acknowledged Lori B. Feldman-Winter, MD, a liaison from the AAP section on breastfeeding to the task force, as well as head of the division of adolescent medicine and professor of pediatrics at Cooper University Health Care in Camden, N.J.

Bedside sleepers (also called sidecar sleepers) that attach to the parents’ bed may help facilitate the dual aims of breastfeeding and safe sleep, but they have not been formally studied to assess their impact on SIDS risk.
Raising awareness
“As a father and pediatrician, I want parents to know that their baby is safest following the AAP safe sleep recommendations, and spreading this message has become my life’s mission,” said Dr. Samuel P. Hanke, a pediatric cardiologist at the University of Cincinnati, who knows the heartbreak of SIDS firsthand.

“We know practicing safe sleep is hard. We have to be vigilant. We need to start adopting a mentality that safe sleep is not negotiable,” Dr. Hanke asserted. “We cannot emphasize enough that practicing safe sleep for every sleep is as important as buckling your child into a car seat for every drive. And just like car seats, this change won’t occur overnight.”
Federal commitment
Since the 1970s, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) in Bethesda, Md., has been supporting and performing much of the research on which the updated recommendations are based. This research continues to help identify areas where greater efforts are needed, according to acting director Catherine Y. Spong, MD.

NICHD also conducts and collaborates on related education campaigns, such as Safe to Sleep, to disseminate messages such as those in the updated AAP recommendations as widely as possible.
“I encourage all physicians, pediatricians, nurses, and other health care and child care providers to lend their authoritative voices to the Safe to Sleep effort,” Dr. Spong said. “Join us all in sharing safe infant sleep recommendations and in supporting parents and caregivers to make informed decisions that will help keep their baby safe during sleep.”
A closer look at setting
Published in conjunction with the guidelines is a study on risk factors that looked at the role of the setting in which sleep-related infant deaths occur (Pediatrics. 2016 Oct 24:e20161124).
The analysis of nearly 12,000 such deaths found that, relative to counterparts who died in their home, infants who died outside of their home were more likely to be in a stroller or car seat at the time (adjusted odds ratio, 2.6) and in other locations, such as on the floor or a futon (1.9), and to have been placed prone (1.1). They were less likely to have been sharing a bed (0.7).
The groups did not differ in terms of whether the infant was sleeping in an adult bed or on a person, on a couch or chair, or with any objects in their sleep environment.
“Caregivers should be educated on the importance of placing infants to sleep supine in cribs/bassinets to protect against sleep-related deaths, both in and out of the home,” conclude the investigators, one of whom disclosed serving as a paid expert witness in cases of sleep-related infant death.
SAN FRANCISCO – At sleep time, infants should share their parents’ bedroom on a separate sleep surface without bed sharing, should be placed on their backs on a firm surface, and should have a sleep area free of blankets and soft objects, according to updated guidelines from the American Academy of Pediatrics aimed at reducing the risk of sudden infant death syndrome (SIDS) and other sleep-related infant deaths.
Drafted by a multidisciplinary task force, the set of 19 evidence-based recommendations largely reiterate messages that the academy has promoted for years such as “back to sleep for every sleep,” according to task force member Fern R. Hauck, MD, the Spencer P. Bass, MD, Twenty-First Century Professor of Family Medicine at the University of Virginia, Charlottesville. They were unveiled in a press briefing at the academy’s annual meeting and simultaneously published (Pediatrics. 2016;138[5]:e20162938).
Progress, but still a ways to go
Education campaigns that convey these and related messages to new parents and other caregivers have led to a more than halving of the rate of SIDS in recent decades. Yet, 3,500 infants are still lost each year to this syndrome and other sleep-related causes of infant death, such as unintentional suffocation, collectively called sudden unexpected infant death (SUID).
New is a recommendation for skin-to-skin care for at least the first hour of life for healthy newborns, as soon as the mother is alert enough to respond to her infant, according to Dr. Hauck. The aims here are to optimize neurodevelopment and promote temperature regulation.
There is no evidence that swaddling reduces the risk of SIDS, but parents can still use this technique if they wish as long as infants are placed on their back and it is discontinued as soon as they start to show signs of rolling over, she said. Evidence is also lacking for new technologies marketed as protective, for example, crib mattresses designed to reduce re-breathing of carbon dioxide should an infant become prone.
Sleeping in the parents’ room but on a separate surface decreases the risk of SIDS by as much as 50%, according to several studies. Bed sharing is not recommended because of the risk of suffocation, strangulation, and entrapment, the policy states.
The updated recommendations should be followed for every sleep and by every caregiver, until the child reaches 1 year of age, Dr. Hauck stressed. “This includes nap time and bedtime sleep, at home, in day care, or in any other locations where the baby is sleeping.”
“We feel that these messages need to start while the mom’s pregnant because some of the decisions that are made that are not always the best decisions, when the mother is exhausted, can occur spur of the moment,” she added. “As pediatricians, you can set up a prebirth visit to start talking about this, and obstetricians should be doing more as well to bring this up during their prenatal visits.”
Other recommendations include offering a pacifier at nap time and bedtime; avoiding smoke exposure during pregnancy and after birth; and avoiding alcohol and illicit drug use during pregnancy and after birth.
Breastfeeding issues
Although breastfeeding protects against SIDS, it can pose some problems for safe sleep practices, acknowledged Lori B. Feldman-Winter, MD, a liaison from the AAP section on breastfeeding to the task force, as well as head of the division of adolescent medicine and professor of pediatrics at Cooper University Health Care in Camden, N.J.
Bedside sleepers (also called sidecar sleepers) that attach to the parents’ bed may help facilitate the dual aims of breastfeeding and safe sleep, but they have not been formally studied to assess their impact on SIDS risk.
Raising awareness
“As a father and pediatrician, I want parents to know that their baby is safest following the AAP safe sleep recommendations, and spreading this message has become my life’s mission,” said Dr. Samuel P. Hanke, a pediatric cardiologist at the University of Cincinnati, who knows the heartbreak of SIDS firsthand.
“We know practicing safe sleep is hard. We have to be vigilant. We need to start adopting a mentality that safe sleep is not negotiable,” Dr. Hanke asserted. “We cannot emphasize enough that practicing safe sleep for every sleep is as important as buckling your child into a car seat for every drive. And just like car seats, this change won’t occur overnight.”
Federal commitment
Since the 1970s, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) in Bethesda, Md., has been supporting and performing much of the research on which the updated recommendations are based. This research continues to help identify areas where greater efforts are needed, according to acting director Catherine Y. Spong, MD.
NICHD also conducts and collaborates on related education campaigns, such as Safe to Sleep, to disseminate messages such as those in the updated AAP recommendations as widely as possible.
“I encourage all physicians, pediatricians, nurses, and other health care and child care providers to lend their authoritative voices to the Safe to Sleep effort,” Dr. Spong said. “Join us all in sharing safe infant sleep recommendations and in supporting parents and caregivers to make informed decisions that will help keep their baby safe during sleep.”
A closer look at setting
Published in conjunction with the guidelines is a study on risk factors that looked at the role of the setting in which sleep-related infant deaths occur (Pediatrics. 2016 Oct 24:e20161124).
The analysis of nearly 12,000 such deaths found that, relative to counterparts who died in their home, infants who died outside of their home were more likely to be in a stroller or car seat at the time (adjusted odds ratio, 2.6) and in other locations, such as on the floor or a futon (1.9), and to have been placed prone (1.1). They were less likely to have been sharing a bed (0.7).
The groups did not differ in terms of whether the infant was sleeping in an adult bed or on a person, on a couch or chair, or with any objects in their sleep environment.
“Caregivers should be educated on the importance of placing infants to sleep supine in cribs/bassinets to protect against sleep-related deaths, both in and out of the home,” conclude the investigators, one of whom disclosed serving as a paid expert witness in cases of sleep-related infant death.
SAN FRANCISCO – At sleep time, infants should share their parents’ bedroom on a separate sleep surface without bed sharing, should be placed on their backs on a firm surface, and should have a sleep area free of blankets and soft objects, according to updated guidelines from the American Academy of Pediatrics aimed at reducing the risk of sudden infant death syndrome (SIDS) and other sleep-related infant deaths.
Drafted by a multidisciplinary task force, the set of 19 evidence-based recommendations largely reiterate messages that the academy has promoted for years such as “back to sleep for every sleep,” according to task force member Fern R. Hauck, MD, the Spencer P. Bass, MD, Twenty-First Century Professor of Family Medicine at the University of Virginia, Charlottesville. They were unveiled in a press briefing at the academy’s annual meeting and simultaneously published (Pediatrics. 2016;138[5]:e20162938).
Progress, but still a ways to go
Education campaigns that convey these and related messages to new parents and other caregivers have led to a more than halving of the rate of SIDS in recent decades. Yet, 3,500 infants are still lost each year to this syndrome and other sleep-related causes of infant death, such as unintentional suffocation, collectively called sudden unexpected infant death (SUID).
New is a recommendation for skin-to-skin care for at least the first hour of life for healthy newborns, as soon as the mother is alert enough to respond to her infant, according to Dr. Hauck. The aims here are to optimize neurodevelopment and promote temperature regulation.
There is no evidence that swaddling reduces the risk of SIDS, but parents can still use this technique if they wish as long as infants are placed on their back and it is discontinued as soon as they start to show signs of rolling over, she said. Evidence is also lacking for new technologies marketed as protective, for example, crib mattresses designed to reduce re-breathing of carbon dioxide should an infant become prone.
Sleeping in the parents’ room but on a separate surface decreases the risk of SIDS by as much as 50%, according to several studies. Bed sharing is not recommended because of the risk of suffocation, strangulation, and entrapment, the policy states.
The updated recommendations should be followed for every sleep and by every caregiver, until the child reaches 1 year of age, Dr. Hauck stressed. “This includes nap time and bedtime sleep, at home, in day care, or in any other locations where the baby is sleeping.”
“We feel that these messages need to start while the mom’s pregnant because some of the decisions that are made that are not always the best decisions, when the mother is exhausted, can occur spur of the moment,” she added. “As pediatricians, you can set up a prebirth visit to start talking about this, and obstetricians should be doing more as well to bring this up during their prenatal visits.”
Other recommendations include offering a pacifier at nap time and bedtime; avoiding smoke exposure during pregnancy and after birth; and avoiding alcohol and illicit drug use during pregnancy and after birth.
Breastfeeding issues
Although breastfeeding protects against SIDS, it can pose some problems for safe sleep practices, acknowledged Lori B. Feldman-Winter, MD, a liaison from the AAP section on breastfeeding to the task force, as well as head of the division of adolescent medicine and professor of pediatrics at Cooper University Health Care in Camden, N.J.
Bedside sleepers (also called sidecar sleepers) that attach to the parents’ bed may help facilitate the dual aims of breastfeeding and safe sleep, but they have not been formally studied to assess their impact on SIDS risk.
Raising awareness
“As a father and pediatrician, I want parents to know that their baby is safest following the AAP safe sleep recommendations, and spreading this message has become my life’s mission,” said Dr. Samuel P. Hanke, a pediatric cardiologist at the University of Cincinnati, who knows the heartbreak of SIDS firsthand.
“We know practicing safe sleep is hard. We have to be vigilant. We need to start adopting a mentality that safe sleep is not negotiable,” Dr. Hanke asserted. “We cannot emphasize enough that practicing safe sleep for every sleep is as important as buckling your child into a car seat for every drive. And just like car seats, this change won’t occur overnight.”
Federal commitment
Since the 1970s, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) in Bethesda, Md., has been supporting and performing much of the research on which the updated recommendations are based. This research continues to help identify areas where greater efforts are needed, according to acting director Catherine Y. Spong, MD.
NICHD also conducts and collaborates on related education campaigns, such as Safe to Sleep, to disseminate messages such as those in the updated AAP recommendations as widely as possible.
“I encourage all physicians, pediatricians, nurses, and other health care and child care providers to lend their authoritative voices to the Safe to Sleep effort,” Dr. Spong said. “Join us all in sharing safe infant sleep recommendations and in supporting parents and caregivers to make informed decisions that will help keep their baby safe during sleep.”
A closer look at setting
Published in conjunction with the guidelines is a study on risk factors that looked at the role of the setting in which sleep-related infant deaths occur (Pediatrics. 2016 Oct 24:e20161124).
The analysis of nearly 12,000 such deaths found that, relative to counterparts who died in their home, infants who died outside of their home were more likely to be in a stroller or car seat at the time (adjusted odds ratio, 2.6) and in other locations, such as on the floor or a futon (1.9), and to have been placed prone (1.1). They were less likely to have been sharing a bed (0.7).
The groups did not differ in terms of whether the infant was sleeping in an adult bed or on a person, on a couch or chair, or with any objects in their sleep environment.
“Caregivers should be educated on the importance of placing infants to sleep supine in cribs/bassinets to protect against sleep-related deaths, both in and out of the home,” conclude the investigators, one of whom disclosed serving as a paid expert witness in cases of sleep-related infant death.
EXPERT ANALYSIS FROM AAP 16
Medication-assisted treatment in group settings may result in greater job satisfaction, more reimbursements
WASHINGTON – For practices that offer medication-assisted treatment but perhaps are struggling to balance follow-up appointments with new patient inductions, Leah K. Bauer, MD, has a suggestion: group sessions.
“It’s a lot of fun, and makes my practice more dynamic. It gets me out of the grind of ‘see a patient; write a note; repeat,’ ” Dr. Bauer said at the American Psychiatric Association’s Institute on Psychiatric Services.
Compressing 26.5 hours of individual clinical time into 12 hours of monthly group sessions held twice a week for 90 minutes each, Dr. Bauer said, resulted in an additional $41,000 of revenue annually, with inductions doubling from 8 to 16 per month.
One reason is that despite the sessions taking place in a group setting, she and her staff bill for a series of individual appointments using the CPT code 99212. “It is perfectly legal, and not very recognized,” Dr. Bauer said, noting that the sessions are in a group context, but that she does get to have one-on-one interaction with her patients with the added therapeutic value that peer support brings.
Modeling appropriate behavior is easier in the group setting, she said: “Patients don’t all have to test the same limits.” Instead, they can learn from the interaction of another patient with Dr. Bauer as the therapist. The group setting also helps her deliver more consistent care to all her patients, she said. “I am more conscious of what I am saying.”
A hospitalist and psychiatrist, Dr. Bauer leads group MAT with the help of a clinician cofacilitator who she says reinforces what is being said in the group and acts as a scribe, reducing Dr. Bauer’s administrative burden. “This improves my job satisfaction tremendously,” she said.
Patients sign a “check-in” sheet that also serves as their treatment plan that includes their goals and objectives. It includes the patients’ written self-reflections, what their week was like, and other entries about their mood and struggles with their recovery. The information also is recorded in their patient records. “The sheet is problem focused, and has a lot of counseling and coordination of care built in,” Dr. Bauer said.
If a patient comes to the session late, there is no lost time or productivity for the MAT team, because the group meets regardless of who attends. Patients can come as much or as little as they like every 1-4 weeks. “It’s very flexible,” Dr. Bauer said.
She does not have data on her patient outcomes in the group setting vs. the individual one, but Dr. Bauer said in an interview that she believes it is as effective and allows more people who need MAT to receive it, because few clinics in her state offer it.
The group structure does place more demand on the hospital’s pharmacy, she said, in that, after the sessions, patients arrive en masse to fill their buprenorphine prescriptions.
Questions about confidentiality do arise, although each session begins with a reminder to keep private what is shared during the meetings. However, Dr. Bauer said, she thinks some patients remain reluctant to speak their minds for fear of what they say not remaining confidential. “This can limit the depth of what’s discussed,” she said.
Dr. Bauer said she did not have any relevant financial disclosures.
[email protected]
On Twitter @whitneymcknight
WASHINGTON – For practices that offer medication-assisted treatment but perhaps are struggling to balance follow-up appointments with new patient inductions, Leah K. Bauer, MD, has a suggestion: group sessions.
“It’s a lot of fun, and makes my practice more dynamic. It gets me out of the grind of ‘see a patient; write a note; repeat,’ ” Dr. Bauer said at the American Psychiatric Association’s Institute on Psychiatric Services.
Compressing 26.5 hours of individual clinical time into 12 hours of monthly group sessions held twice a week for 90 minutes each, Dr. Bauer said, resulted in an additional $41,000 of revenue annually, with inductions doubling from 8 to 16 per month.
One reason is that despite the sessions taking place in a group setting, she and her staff bill for a series of individual appointments using the CPT code 99212. “It is perfectly legal, and not very recognized,” Dr. Bauer said, noting that the sessions are in a group context, but that she does get to have one-on-one interaction with her patients with the added therapeutic value that peer support brings.
Modeling appropriate behavior is easier in the group setting, she said: “Patients don’t all have to test the same limits.” Instead, they can learn from the interaction of another patient with Dr. Bauer as the therapist. The group setting also helps her deliver more consistent care to all her patients, she said. “I am more conscious of what I am saying.”
A hospitalist and psychiatrist, Dr. Bauer leads group MAT with the help of a clinician cofacilitator who she says reinforces what is being said in the group and acts as a scribe, reducing Dr. Bauer’s administrative burden. “This improves my job satisfaction tremendously,” she said.
Patients sign a “check-in” sheet that also serves as their treatment plan that includes their goals and objectives. It includes the patients’ written self-reflections, what their week was like, and other entries about their mood and struggles with their recovery. The information also is recorded in their patient records. “The sheet is problem focused, and has a lot of counseling and coordination of care built in,” Dr. Bauer said.
If a patient comes to the session late, there is no lost time or productivity for the MAT team, because the group meets regardless of who attends. Patients can come as much or as little as they like every 1-4 weeks. “It’s very flexible,” Dr. Bauer said.
She does not have data on her patient outcomes in the group setting vs. the individual one, but Dr. Bauer said in an interview that she believes it is as effective and allows more people who need MAT to receive it, because few clinics in her state offer it.
The group structure does place more demand on the hospital’s pharmacy, she said, in that, after the sessions, patients arrive en masse to fill their buprenorphine prescriptions.
Questions about confidentiality do arise, although each session begins with a reminder to keep private what is shared during the meetings. However, Dr. Bauer said, she thinks some patients remain reluctant to speak their minds for fear of what they say not remaining confidential. “This can limit the depth of what’s discussed,” she said.
Dr. Bauer said she did not have any relevant financial disclosures.
[email protected]
On Twitter @whitneymcknight
WASHINGTON – For practices that offer medication-assisted treatment but perhaps are struggling to balance follow-up appointments with new patient inductions, Leah K. Bauer, MD, has a suggestion: group sessions.
“It’s a lot of fun, and makes my practice more dynamic. It gets me out of the grind of ‘see a patient; write a note; repeat,’ ” Dr. Bauer said at the American Psychiatric Association’s Institute on Psychiatric Services.
Compressing 26.5 hours of individual clinical time into 12 hours of monthly group sessions held twice a week for 90 minutes each, Dr. Bauer said, resulted in an additional $41,000 of revenue annually, with inductions doubling from 8 to 16 per month.
One reason is that despite the sessions taking place in a group setting, she and her staff bill for a series of individual appointments using the CPT code 99212. “It is perfectly legal, and not very recognized,” Dr. Bauer said, noting that the sessions are in a group context, but that she does get to have one-on-one interaction with her patients with the added therapeutic value that peer support brings.
Modeling appropriate behavior is easier in the group setting, she said: “Patients don’t all have to test the same limits.” Instead, they can learn from the interaction of another patient with Dr. Bauer as the therapist. The group setting also helps her deliver more consistent care to all her patients, she said. “I am more conscious of what I am saying.”
A hospitalist and psychiatrist, Dr. Bauer leads group MAT with the help of a clinician cofacilitator who she says reinforces what is being said in the group and acts as a scribe, reducing Dr. Bauer’s administrative burden. “This improves my job satisfaction tremendously,” she said.
Patients sign a “check-in” sheet that also serves as their treatment plan that includes their goals and objectives. It includes the patients’ written self-reflections, what their week was like, and other entries about their mood and struggles with their recovery. The information also is recorded in their patient records. “The sheet is problem focused, and has a lot of counseling and coordination of care built in,” Dr. Bauer said.
If a patient comes to the session late, there is no lost time or productivity for the MAT team, because the group meets regardless of who attends. Patients can come as much or as little as they like every 1-4 weeks. “It’s very flexible,” Dr. Bauer said.
She does not have data on her patient outcomes in the group setting vs. the individual one, but Dr. Bauer said in an interview that she believes it is as effective and allows more people who need MAT to receive it, because few clinics in her state offer it.
The group structure does place more demand on the hospital’s pharmacy, she said, in that, after the sessions, patients arrive en masse to fill their buprenorphine prescriptions.
Questions about confidentiality do arise, although each session begins with a reminder to keep private what is shared during the meetings. However, Dr. Bauer said, she thinks some patients remain reluctant to speak their minds for fear of what they say not remaining confidential. “This can limit the depth of what’s discussed,” she said.
Dr. Bauer said she did not have any relevant financial disclosures.
[email protected]
On Twitter @whitneymcknight
Verrucous Plaque on the Leg
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
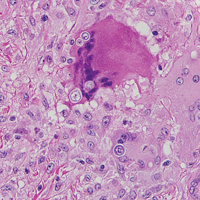
2016 a big year for mental health legislation
• The Comprehensive Addiction and Recovery Act. Signed into law in July, CARA is primarily intended, as its name indicates, to comprehensively address the opioid addiction crisis. The law includes components aimed at prevention, treatment, recovery, law enforcement, criminal justice reform, and overdose treatment. It also has provisions aimed at expanded prescription drug monitoring programs and other drug abuse prevention mechanisms.
Another important part of this law is the expansion of buprenorphine prescribing rights to nurse practitioners and physician assistants who obtain the necessary training. Public comment is being taken through Nov. 1, on what the requirements should be. This provision will be revisited in 2021.
In all, the law allocates $25 million annually from 2017 through 2021 to expand medication-assisted treatment (MAT). An abundance of evidence shows that MAT – the use of opioid agonists and partial opioid agonists such as methadone, naltrexone, and buprenorphine, in combination with psychosocial therapies – is more effective than either modality alone.
• The Helping Families in Mental Health Crisis Act. Originating in 2013 in response to the Newtown, Conn., massacre, this bill (H.R. 2646) is focused on serious mental illness. It passed the House in July with near unanimous support, but awaits the Senate to sort out its own version of the bill. If this bill is passed, HIPAA laws would loosen, allowing practices to share all but psychotherapy notes with caregivers and guardians of people with serious mental illness. Practices also could bill Medicaid and Medicare for medical and mental health services delivered on the same day, and there would be support for the better integration of medical and mental health electronic medical records. The current 30-day limit on inpatient psychiatric facility services paid for by Medicaid would be lifted, and $20 million in grants would be made available for assisted outpatient treatment of this population. Grants for expanded use of telepsychiatry also would be available.
• The Mental Health Reform Act of 2016. Essentially the companion bill to the one passed in the House, this bill (S. 2680) also calls for the expansion of telepsychiatry, especially for pediatric and adolescent mental and behavioral health needs. If it becomes law, grant money would be made available for better integration of primary and behavioral health care services. This bill calls for strengthening current mental health parity laws by requiring additional federal guidance to help insurance plans comply. The bill currently is stalled on the Senate floor and is not expected to be revisited until after the presidential election.
• The Medication Assisted Treatment for Opioid Use Disorders final rule. As of August, the rule allows addiction medicine specialists to treat up to 275 patients using buprenorphine for substance use disorder annually. Previously, they were held to treating no more than 100 such patients per year. For practices not already licensed to provide MAT, this rule doesn’t have much direct impact. However, for practices with patients on their panels who are struggling with substance use disorders, this could expand available referral resources.
• The Quality Payment Program final rule. While this rule – borne of the MACRA (Medicare Access and CHIP Reauthorization Act) law that is now referred to as the Quality Payment Program – does not directly address mental and behavioral health, it directly affects delivery of these services when it goes into effect in 2017. Because mental and behavioral health outcomes of patient panels that include Medicare populations will be assessed as part of overall patient outcomes, providing effective, integrated services will be imperative. How severely a practice will be penalized for poor outcomes will depend upon how that practice chooses to set up its reimbursement structures and quality metrics over the next few years.
[email protected]
On Twitter @whitneymcknight
• The Comprehensive Addiction and Recovery Act. Signed into law in July, CARA is primarily intended, as its name indicates, to comprehensively address the opioid addiction crisis. The law includes components aimed at prevention, treatment, recovery, law enforcement, criminal justice reform, and overdose treatment. It also has provisions aimed at expanded prescription drug monitoring programs and other drug abuse prevention mechanisms.
Another important part of this law is the expansion of buprenorphine prescribing rights to nurse practitioners and physician assistants who obtain the necessary training. Public comment is being taken through Nov. 1, on what the requirements should be. This provision will be revisited in 2021.
In all, the law allocates $25 million annually from 2017 through 2021 to expand medication-assisted treatment (MAT). An abundance of evidence shows that MAT – the use of opioid agonists and partial opioid agonists such as methadone, naltrexone, and buprenorphine, in combination with psychosocial therapies – is more effective than either modality alone.
• The Helping Families in Mental Health Crisis Act. Originating in 2013 in response to the Newtown, Conn., massacre, this bill (H.R. 2646) is focused on serious mental illness. It passed the House in July with near unanimous support, but awaits the Senate to sort out its own version of the bill. If this bill is passed, HIPAA laws would loosen, allowing practices to share all but psychotherapy notes with caregivers and guardians of people with serious mental illness. Practices also could bill Medicaid and Medicare for medical and mental health services delivered on the same day, and there would be support for the better integration of medical and mental health electronic medical records. The current 30-day limit on inpatient psychiatric facility services paid for by Medicaid would be lifted, and $20 million in grants would be made available for assisted outpatient treatment of this population. Grants for expanded use of telepsychiatry also would be available.
• The Mental Health Reform Act of 2016. Essentially the companion bill to the one passed in the House, this bill (S. 2680) also calls for the expansion of telepsychiatry, especially for pediatric and adolescent mental and behavioral health needs. If it becomes law, grant money would be made available for better integration of primary and behavioral health care services. This bill calls for strengthening current mental health parity laws by requiring additional federal guidance to help insurance plans comply. The bill currently is stalled on the Senate floor and is not expected to be revisited until after the presidential election.
• The Medication Assisted Treatment for Opioid Use Disorders final rule. As of August, the rule allows addiction medicine specialists to treat up to 275 patients using buprenorphine for substance use disorder annually. Previously, they were held to treating no more than 100 such patients per year. For practices not already licensed to provide MAT, this rule doesn’t have much direct impact. However, for practices with patients on their panels who are struggling with substance use disorders, this could expand available referral resources.
• The Quality Payment Program final rule. While this rule – borne of the MACRA (Medicare Access and CHIP Reauthorization Act) law that is now referred to as the Quality Payment Program – does not directly address mental and behavioral health, it directly affects delivery of these services when it goes into effect in 2017. Because mental and behavioral health outcomes of patient panels that include Medicare populations will be assessed as part of overall patient outcomes, providing effective, integrated services will be imperative. How severely a practice will be penalized for poor outcomes will depend upon how that practice chooses to set up its reimbursement structures and quality metrics over the next few years.
[email protected]
On Twitter @whitneymcknight
• The Comprehensive Addiction and Recovery Act. Signed into law in July, CARA is primarily intended, as its name indicates, to comprehensively address the opioid addiction crisis. The law includes components aimed at prevention, treatment, recovery, law enforcement, criminal justice reform, and overdose treatment. It also has provisions aimed at expanded prescription drug monitoring programs and other drug abuse prevention mechanisms.
Another important part of this law is the expansion of buprenorphine prescribing rights to nurse practitioners and physician assistants who obtain the necessary training. Public comment is being taken through Nov. 1, on what the requirements should be. This provision will be revisited in 2021.
In all, the law allocates $25 million annually from 2017 through 2021 to expand medication-assisted treatment (MAT). An abundance of evidence shows that MAT – the use of opioid agonists and partial opioid agonists such as methadone, naltrexone, and buprenorphine, in combination with psychosocial therapies – is more effective than either modality alone.
• The Helping Families in Mental Health Crisis Act. Originating in 2013 in response to the Newtown, Conn., massacre, this bill (H.R. 2646) is focused on serious mental illness. It passed the House in July with near unanimous support, but awaits the Senate to sort out its own version of the bill. If this bill is passed, HIPAA laws would loosen, allowing practices to share all but psychotherapy notes with caregivers and guardians of people with serious mental illness. Practices also could bill Medicaid and Medicare for medical and mental health services delivered on the same day, and there would be support for the better integration of medical and mental health electronic medical records. The current 30-day limit on inpatient psychiatric facility services paid for by Medicaid would be lifted, and $20 million in grants would be made available for assisted outpatient treatment of this population. Grants for expanded use of telepsychiatry also would be available.
• The Mental Health Reform Act of 2016. Essentially the companion bill to the one passed in the House, this bill (S. 2680) also calls for the expansion of telepsychiatry, especially for pediatric and adolescent mental and behavioral health needs. If it becomes law, grant money would be made available for better integration of primary and behavioral health care services. This bill calls for strengthening current mental health parity laws by requiring additional federal guidance to help insurance plans comply. The bill currently is stalled on the Senate floor and is not expected to be revisited until after the presidential election.
• The Medication Assisted Treatment for Opioid Use Disorders final rule. As of August, the rule allows addiction medicine specialists to treat up to 275 patients using buprenorphine for substance use disorder annually. Previously, they were held to treating no more than 100 such patients per year. For practices not already licensed to provide MAT, this rule doesn’t have much direct impact. However, for practices with patients on their panels who are struggling with substance use disorders, this could expand available referral resources.
• The Quality Payment Program final rule. While this rule – borne of the MACRA (Medicare Access and CHIP Reauthorization Act) law that is now referred to as the Quality Payment Program – does not directly address mental and behavioral health, it directly affects delivery of these services when it goes into effect in 2017. Because mental and behavioral health outcomes of patient panels that include Medicare populations will be assessed as part of overall patient outcomes, providing effective, integrated services will be imperative. How severely a practice will be penalized for poor outcomes will depend upon how that practice chooses to set up its reimbursement structures and quality metrics over the next few years.
[email protected]
On Twitter @whitneymcknight
Aquatic Antagonists: Cutaneous Sea Urchin Spine Injury
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
Practice Points
- Radiographic imaging may aid in the identification of sea urchin spines, especially if there are no visible or palpable skin nodules.
- Treatment of sea urchin spine injuries typically involves surgical removal of the spines with local anesthesia and cutaneous extraction.
- Prompt extraction of sea urchin spines can improve pain symptoms and decrease the likelihood of granuloma formation, infection, and extracutaneous complications.