User login
Weakness and pain in arms and legs • dark urine • history of vertebral osteomyelitis • Dx?
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20

Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20
Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20
Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
ACIP vaccine update, 2017
The Advisory Committee on Immunization Practices (ACIP) met 3 times in 2016 and introduced or revised recommendations on influenza, meningococcal, human papillomavirus (HPV), cholera, and hepatitis B vaccines. This Practice Alert highlights the most important new recommendations, except those for influenza vaccines, which were described in a previous Practice Alert.1 (See the summary of how this year’s flu season compares to last year’s.)
SIDEBAR
PRACTICE ALERT UPDATE
How this year's flu season compares to last yearThe 2016-2017 influenza season has been relatively mild, with activity nationwide picking up in late January and continuing to increase in February. As of February 16, 90% of the infections typed were type A, and most of those cases (more than 90%) were H3N1. Not surprisingly, the age group most heavily affected has been the elderly.
The hospitalization rate among those ≥65 years as of early February was 113.5/100,000, which is about half the rate of the same week during the 2014-2015 flu season. The hospitalization rate among those ages 50 to 64 years was 23.5/100,000—about 40% lower than the rate during the same week last flu season. At press time, 20 pediatric deaths had occurred, which is less than one-quarter of the number that occurred during the same time last year, and resistance to oseltamivir had not yet been detected in any isolates.
Source: Centers for Disease Control and Prevention. Situation update: summary of weekly FluView report. Available at: https://www.cdc.gov/flu/weekly/summary.htm. Accessed February 16, 2017.
Meningococcal vaccine: Now recommended for HIV-positive patients
Meningococcal conjugate vaccine (serogroups A, C, W, and Y) is recommended for all adolescents ages 11 to 12 as a single dose with a booster at age 16.2 It is also recommended for adults and for children (starting at age 2 months) who have high-risk conditions such as functional or anatomic asplenia or complement deficiencies. Others at high risk include microbiologists routinely exposed to isolates of Neisseria meningitidis and those traveling to areas of high meningococcal incidence. ACIP recently added human immunodeficiency virus (HIV) infection to the list of high-risk conditions.3
Two meningococcal conjugate vaccines are available in the United States: Menactra, (Sanofi Pasteur), licensed for use in individuals ages 9 months to 55 years; and Menveo (GlaxoSmithKline), licensed for use in individuals ages 2 months to 55 years. Menveo is the preferred vaccine for children younger than 2 years infected with HIV. However, if Menactra is used, give it at least 4 weeks after completing all pneumococcal conjugate vaccine doses and either before or concomitantly with diphtheria and tetanus toxoid and acellular pertussis vaccine (DTaP). All individuals who are HIV positive should receive a multi-dose primary series and booster doses. The number of primary doses and timing of boosters depends on the product used and the ages of those vaccinated (TABLE3).

Although neither meningococcal conjugate vaccine product is licensed for use in individuals 56 years or older, ACIP recommends using one of the products for HIV-infected individuals in this age group because the only meningococcal vaccine licensed for use in adults 56 or older, meningococcal polysaccharide vaccine (MPSV4, Menomune, Sanofi Pasteur), has not been studied in patients with HIV infection.
Serogroup B. Two vaccine products provide short-term protection against meningococcal serogroup B: MenB-FHbp (Trumenba, Wyeth Pharmaceuticals, Inc.) and MenB-4C (Bexsero, GlaxoSmithKline). In 2015, ACIP made a “B” recommendation for the use of these vaccines in individuals 16 to 23 years of age, with the preferred age range being 16 to 18.4 A “B” recommendation means that while ACIP does not advise routine use of the vaccines in this age group, the vaccines can be administered to those who desire them. ACIP has recommended routine use of these products only for individuals 10 years and older who are at high risk for meningococcal disease.5
Trumenba was approved as a 3-dose vaccine, administered at 0, 2, and 6 months. Bexsero requires 2 doses given at least one month apart. At its October 2016 meeting, ACIP approved a 2-dose Trumenba schedule, at 0 and 6 months, when administered to those not at risk for meningococcal disease.6 However, during an outbreak, and for those at high risk for meningococcal disease, adhere to the original 3-dose schedule.
HPV vaccine: Now a 2-dose schedule for younger patients
The only HPV vaccine available in the United States is the 9-valent HPV vaccine (9vHPV), Gardasil 9. It is approved for both males and females ages 9 to 26 years. ACIP recommends it for both sexes at ages 11 or 12, and advises catch-up doses for men through age 21 and women through age 26. It also recommends vaccination through age 26 for men who have sex with men and men
The HPV vaccine is approved for a 3-dose schedule at 0, 1 to 2, and 6 months. At its October 2016 meeting, ACIP approved a 2-dose schedule (0, 6-12 months) for those starting the vaccine before their 15th birthday.7 Those starting the vaccine after their 15th birthday, and individuals at any age with an immune-compromising condition, should receive 3 doses. It is hoped that a 2-dose schedule will help to increase the uptake of this safe, effective, and underused vaccine.
Cholera: A new vaccine is available
In June 2016, the FDA approved a live, attenuated, single-dose, oral vaccine (Vaxchora, PaxVax, Inc.) for the prevention of cholera in adults ages 18 to 64 years. It is the only cholera vaccine approved in the United States.
Cholera occurs at low rates among travelers to areas where the disease is endemic. The key to prevention is food and water precautions, and thus the vaccine is not recommended for most travelers—only for those who are at increased risk of exposure to cholera or who have a medical condition that predisposes them to a poor response to medical care if cholera is contracted.8 Risk increases with long-term or frequent travel to endemic areas where safe food and water is not always available. Examples of compromising medical conditions include a blood type O, low gastric acidity, and heart or kidney disease.
Duration of the vaccine’s effectiveness is unknown, given a lack of data beyond 6 months. No recommendation for revaccination has been made, and this issue will be assessed as more data are collected. Other unknowns about the vaccine include its effectiveness among immune-suppressed individuals and pregnant women, as well as for those who live in cholera endemic areas or were previously vaccinated with another cholera vaccine.
Hepatitis B: Vaccinate newborns sooner
The incidence of hepatitis B virus (HBV) infection has declined by more than 90% since the introduction of a vaccine in 1982.9
Current recommendations for the prevention of HBV include:9
- Screen all pregnant women for hepatitis B surface antigen (HBsAg), and use HBIG and hepatitis B vaccines within 12 hours of birth for all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.
- Administer the 3-dose hepatitis B vaccine to all other infants.
- Routinely vaccinate previously unvaccinated children and adolescents.
- Routinely vaccinate adults who are non-immune and at risk for HBV infection.
At its October 2016 meeting, ACIP adopted a comprehensive update of all HBV prevention recommendations. (This will be the subject of a future Practice Alert.) Included was a revision of a previously permissive recommendation that allowed the first dose of hepatitis B vaccine for newborns to be given within 2 months of hospital discharge. The new recommendation9 states that newborns of mothers known to be HBsAg negative should be vaccinated within 24 hours (if weight is ≥2000 g) or at age one month or at hospital discharge (if weight is <2000 g).
The first dose should be given within 12 hours of birth to all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.9
Immunization schedules
Every year ACIP updates the adult and child immunization schedules to incorporate the changes from the previous year. These can be found on the ACIP Web site at https://www.cdc.gov/vaccines/schedules/hcp/index.html. This Web site remains the most authoritative and accurate source of information on vaccines and immunizations for both professionals and the public.
1. Campos-Outcalt D. Need-to-know information for the 2016-2017 flu season. J Fam Pract. 2016;65:613-617.
2. Cohn AC, MacNeil JR, Clark TA, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1-28.
3. MacNeil JR, Rubin LG, Patton M, et al. Recommendations for use of meningococcal conjugate vaccines in HIV-infected persons— Advisory Committee on Immunization Practices, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1189-1194.
4. MacNeil JR, Rubin LG, Folaranmi T, et al. Use of serogroup B meningococcal vaccines in adolescents and young adults: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1171-1176.
5. Folaranmi T, Rubin L, Martin SW, et al. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:608-612.
6. MacNeil J. Considerations for Use of 2- and 3-Dose Schedules of MenB-FHbp (Trumenba). Presentation at: Advisory Committee on Immunization Practices; October 19, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/meningococcal-05-macneil.pdf. Accessed February 6, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65:1405-1408.
8. Wong KW. Cholera vaccine update and proposed recommendations. Presentation at: Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/cholera-02-wong.pdf. Accessed January 27, 2017.
9. Schillie S. Revised ACIP Hepatitis B (HepB) vaccine recommendations. Presentation at: Advisory Committee on Immunization Practices; October 19, 2016. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/hepatitis-02-schillie-october-2016.pdf. Accessed January 27, 2017.
10.
11. Ko SC, Fan L, Smith EA, et al. Estimated annual perinatal hepatitis B virus infections in the United States, 2000-2009. J Pediatric Infect Dis Soc. 2016;5:114-121.
12. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. MMWR Morb Mortal Wkly Rep. 2006;55:1-25.
13. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099-1102.
The Advisory Committee on Immunization Practices (ACIP) met 3 times in 2016 and introduced or revised recommendations on influenza, meningococcal, human papillomavirus (HPV), cholera, and hepatitis B vaccines. This Practice Alert highlights the most important new recommendations, except those for influenza vaccines, which were described in a previous Practice Alert.1 (See the summary of how this year’s flu season compares to last year’s.)
SIDEBAR
PRACTICE ALERT UPDATE
How this year's flu season compares to last yearThe 2016-2017 influenza season has been relatively mild, with activity nationwide picking up in late January and continuing to increase in February. As of February 16, 90% of the infections typed were type A, and most of those cases (more than 90%) were H3N1. Not surprisingly, the age group most heavily affected has been the elderly.
The hospitalization rate among those ≥65 years as of early February was 113.5/100,000, which is about half the rate of the same week during the 2014-2015 flu season. The hospitalization rate among those ages 50 to 64 years was 23.5/100,000—about 40% lower than the rate during the same week last flu season. At press time, 20 pediatric deaths had occurred, which is less than one-quarter of the number that occurred during the same time last year, and resistance to oseltamivir had not yet been detected in any isolates.
Source: Centers for Disease Control and Prevention. Situation update: summary of weekly FluView report. Available at: https://www.cdc.gov/flu/weekly/summary.htm. Accessed February 16, 2017.
Meningococcal vaccine: Now recommended for HIV-positive patients
Meningococcal conjugate vaccine (serogroups A, C, W, and Y) is recommended for all adolescents ages 11 to 12 as a single dose with a booster at age 16.2 It is also recommended for adults and for children (starting at age 2 months) who have high-risk conditions such as functional or anatomic asplenia or complement deficiencies. Others at high risk include microbiologists routinely exposed to isolates of Neisseria meningitidis and those traveling to areas of high meningococcal incidence. ACIP recently added human immunodeficiency virus (HIV) infection to the list of high-risk conditions.3
Two meningococcal conjugate vaccines are available in the United States: Menactra, (Sanofi Pasteur), licensed for use in individuals ages 9 months to 55 years; and Menveo (GlaxoSmithKline), licensed for use in individuals ages 2 months to 55 years. Menveo is the preferred vaccine for children younger than 2 years infected with HIV. However, if Menactra is used, give it at least 4 weeks after completing all pneumococcal conjugate vaccine doses and either before or concomitantly with diphtheria and tetanus toxoid and acellular pertussis vaccine (DTaP). All individuals who are HIV positive should receive a multi-dose primary series and booster doses. The number of primary doses and timing of boosters depends on the product used and the ages of those vaccinated (TABLE3).
Although neither meningococcal conjugate vaccine product is licensed for use in individuals 56 years or older, ACIP recommends using one of the products for HIV-infected individuals in this age group because the only meningococcal vaccine licensed for use in adults 56 or older, meningococcal polysaccharide vaccine (MPSV4, Menomune, Sanofi Pasteur), has not been studied in patients with HIV infection.
Serogroup B. Two vaccine products provide short-term protection against meningococcal serogroup B: MenB-FHbp (Trumenba, Wyeth Pharmaceuticals, Inc.) and MenB-4C (Bexsero, GlaxoSmithKline). In 2015, ACIP made a “B” recommendation for the use of these vaccines in individuals 16 to 23 years of age, with the preferred age range being 16 to 18.4 A “B” recommendation means that while ACIP does not advise routine use of the vaccines in this age group, the vaccines can be administered to those who desire them. ACIP has recommended routine use of these products only for individuals 10 years and older who are at high risk for meningococcal disease.5
Trumenba was approved as a 3-dose vaccine, administered at 0, 2, and 6 months. Bexsero requires 2 doses given at least one month apart. At its October 2016 meeting, ACIP approved a 2-dose Trumenba schedule, at 0 and 6 months, when administered to those not at risk for meningococcal disease.6 However, during an outbreak, and for those at high risk for meningococcal disease, adhere to the original 3-dose schedule.
HPV vaccine: Now a 2-dose schedule for younger patients
The only HPV vaccine available in the United States is the 9-valent HPV vaccine (9vHPV), Gardasil 9. It is approved for both males and females ages 9 to 26 years. ACIP recommends it for both sexes at ages 11 or 12, and advises catch-up doses for men through age 21 and women through age 26. It also recommends vaccination through age 26 for men who have sex with men and men
The HPV vaccine is approved for a 3-dose schedule at 0, 1 to 2, and 6 months. At its October 2016 meeting, ACIP approved a 2-dose schedule (0, 6-12 months) for those starting the vaccine before their 15th birthday.7 Those starting the vaccine after their 15th birthday, and individuals at any age with an immune-compromising condition, should receive 3 doses. It is hoped that a 2-dose schedule will help to increase the uptake of this safe, effective, and underused vaccine.
Cholera: A new vaccine is available
In June 2016, the FDA approved a live, attenuated, single-dose, oral vaccine (Vaxchora, PaxVax, Inc.) for the prevention of cholera in adults ages 18 to 64 years. It is the only cholera vaccine approved in the United States.
Cholera occurs at low rates among travelers to areas where the disease is endemic. The key to prevention is food and water precautions, and thus the vaccine is not recommended for most travelers—only for those who are at increased risk of exposure to cholera or who have a medical condition that predisposes them to a poor response to medical care if cholera is contracted.8 Risk increases with long-term or frequent travel to endemic areas where safe food and water is not always available. Examples of compromising medical conditions include a blood type O, low gastric acidity, and heart or kidney disease.
Duration of the vaccine’s effectiveness is unknown, given a lack of data beyond 6 months. No recommendation for revaccination has been made, and this issue will be assessed as more data are collected. Other unknowns about the vaccine include its effectiveness among immune-suppressed individuals and pregnant women, as well as for those who live in cholera endemic areas or were previously vaccinated with another cholera vaccine.
Hepatitis B: Vaccinate newborns sooner
The incidence of hepatitis B virus (HBV) infection has declined by more than 90% since the introduction of a vaccine in 1982.9
Current recommendations for the prevention of HBV include:9
- Screen all pregnant women for hepatitis B surface antigen (HBsAg), and use HBIG and hepatitis B vaccines within 12 hours of birth for all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.
- Administer the 3-dose hepatitis B vaccine to all other infants.
- Routinely vaccinate previously unvaccinated children and adolescents.
- Routinely vaccinate adults who are non-immune and at risk for HBV infection.
At its October 2016 meeting, ACIP adopted a comprehensive update of all HBV prevention recommendations. (This will be the subject of a future Practice Alert.) Included was a revision of a previously permissive recommendation that allowed the first dose of hepatitis B vaccine for newborns to be given within 2 months of hospital discharge. The new recommendation9 states that newborns of mothers known to be HBsAg negative should be vaccinated within 24 hours (if weight is ≥2000 g) or at age one month or at hospital discharge (if weight is <2000 g).
The first dose should be given within 12 hours of birth to all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.9
Immunization schedules
Every year ACIP updates the adult and child immunization schedules to incorporate the changes from the previous year. These can be found on the ACIP Web site at https://www.cdc.gov/vaccines/schedules/hcp/index.html. This Web site remains the most authoritative and accurate source of information on vaccines and immunizations for both professionals and the public.
The Advisory Committee on Immunization Practices (ACIP) met 3 times in 2016 and introduced or revised recommendations on influenza, meningococcal, human papillomavirus (HPV), cholera, and hepatitis B vaccines. This Practice Alert highlights the most important new recommendations, except those for influenza vaccines, which were described in a previous Practice Alert.1 (See the summary of how this year’s flu season compares to last year’s.)
SIDEBAR
PRACTICE ALERT UPDATE
How this year's flu season compares to last yearThe 2016-2017 influenza season has been relatively mild, with activity nationwide picking up in late January and continuing to increase in February. As of February 16, 90% of the infections typed were type A, and most of those cases (more than 90%) were H3N1. Not surprisingly, the age group most heavily affected has been the elderly.
The hospitalization rate among those ≥65 years as of early February was 113.5/100,000, which is about half the rate of the same week during the 2014-2015 flu season. The hospitalization rate among those ages 50 to 64 years was 23.5/100,000—about 40% lower than the rate during the same week last flu season. At press time, 20 pediatric deaths had occurred, which is less than one-quarter of the number that occurred during the same time last year, and resistance to oseltamivir had not yet been detected in any isolates.
Source: Centers for Disease Control and Prevention. Situation update: summary of weekly FluView report. Available at: https://www.cdc.gov/flu/weekly/summary.htm. Accessed February 16, 2017.
Meningococcal vaccine: Now recommended for HIV-positive patients
Meningococcal conjugate vaccine (serogroups A, C, W, and Y) is recommended for all adolescents ages 11 to 12 as a single dose with a booster at age 16.2 It is also recommended for adults and for children (starting at age 2 months) who have high-risk conditions such as functional or anatomic asplenia or complement deficiencies. Others at high risk include microbiologists routinely exposed to isolates of Neisseria meningitidis and those traveling to areas of high meningococcal incidence. ACIP recently added human immunodeficiency virus (HIV) infection to the list of high-risk conditions.3
Two meningococcal conjugate vaccines are available in the United States: Menactra, (Sanofi Pasteur), licensed for use in individuals ages 9 months to 55 years; and Menveo (GlaxoSmithKline), licensed for use in individuals ages 2 months to 55 years. Menveo is the preferred vaccine for children younger than 2 years infected with HIV. However, if Menactra is used, give it at least 4 weeks after completing all pneumococcal conjugate vaccine doses and either before or concomitantly with diphtheria and tetanus toxoid and acellular pertussis vaccine (DTaP). All individuals who are HIV positive should receive a multi-dose primary series and booster doses. The number of primary doses and timing of boosters depends on the product used and the ages of those vaccinated (TABLE3).
Although neither meningococcal conjugate vaccine product is licensed for use in individuals 56 years or older, ACIP recommends using one of the products for HIV-infected individuals in this age group because the only meningococcal vaccine licensed for use in adults 56 or older, meningococcal polysaccharide vaccine (MPSV4, Menomune, Sanofi Pasteur), has not been studied in patients with HIV infection.
Serogroup B. Two vaccine products provide short-term protection against meningococcal serogroup B: MenB-FHbp (Trumenba, Wyeth Pharmaceuticals, Inc.) and MenB-4C (Bexsero, GlaxoSmithKline). In 2015, ACIP made a “B” recommendation for the use of these vaccines in individuals 16 to 23 years of age, with the preferred age range being 16 to 18.4 A “B” recommendation means that while ACIP does not advise routine use of the vaccines in this age group, the vaccines can be administered to those who desire them. ACIP has recommended routine use of these products only for individuals 10 years and older who are at high risk for meningococcal disease.5
Trumenba was approved as a 3-dose vaccine, administered at 0, 2, and 6 months. Bexsero requires 2 doses given at least one month apart. At its October 2016 meeting, ACIP approved a 2-dose Trumenba schedule, at 0 and 6 months, when administered to those not at risk for meningococcal disease.6 However, during an outbreak, and for those at high risk for meningococcal disease, adhere to the original 3-dose schedule.
HPV vaccine: Now a 2-dose schedule for younger patients
The only HPV vaccine available in the United States is the 9-valent HPV vaccine (9vHPV), Gardasil 9. It is approved for both males and females ages 9 to 26 years. ACIP recommends it for both sexes at ages 11 or 12, and advises catch-up doses for men through age 21 and women through age 26. It also recommends vaccination through age 26 for men who have sex with men and men
The HPV vaccine is approved for a 3-dose schedule at 0, 1 to 2, and 6 months. At its October 2016 meeting, ACIP approved a 2-dose schedule (0, 6-12 months) for those starting the vaccine before their 15th birthday.7 Those starting the vaccine after their 15th birthday, and individuals at any age with an immune-compromising condition, should receive 3 doses. It is hoped that a 2-dose schedule will help to increase the uptake of this safe, effective, and underused vaccine.
Cholera: A new vaccine is available
In June 2016, the FDA approved a live, attenuated, single-dose, oral vaccine (Vaxchora, PaxVax, Inc.) for the prevention of cholera in adults ages 18 to 64 years. It is the only cholera vaccine approved in the United States.
Cholera occurs at low rates among travelers to areas where the disease is endemic. The key to prevention is food and water precautions, and thus the vaccine is not recommended for most travelers—only for those who are at increased risk of exposure to cholera or who have a medical condition that predisposes them to a poor response to medical care if cholera is contracted.8 Risk increases with long-term or frequent travel to endemic areas where safe food and water is not always available. Examples of compromising medical conditions include a blood type O, low gastric acidity, and heart or kidney disease.
Duration of the vaccine’s effectiveness is unknown, given a lack of data beyond 6 months. No recommendation for revaccination has been made, and this issue will be assessed as more data are collected. Other unknowns about the vaccine include its effectiveness among immune-suppressed individuals and pregnant women, as well as for those who live in cholera endemic areas or were previously vaccinated with another cholera vaccine.
Hepatitis B: Vaccinate newborns sooner
The incidence of hepatitis B virus (HBV) infection has declined by more than 90% since the introduction of a vaccine in 1982.9
Current recommendations for the prevention of HBV include:9
- Screen all pregnant women for hepatitis B surface antigen (HBsAg), and use HBIG and hepatitis B vaccines within 12 hours of birth for all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.
- Administer the 3-dose hepatitis B vaccine to all other infants.
- Routinely vaccinate previously unvaccinated children and adolescents.
- Routinely vaccinate adults who are non-immune and at risk for HBV infection.
At its October 2016 meeting, ACIP adopted a comprehensive update of all HBV prevention recommendations. (This will be the subject of a future Practice Alert.) Included was a revision of a previously permissive recommendation that allowed the first dose of hepatitis B vaccine for newborns to be given within 2 months of hospital discharge. The new recommendation9 states that newborns of mothers known to be HBsAg negative should be vaccinated within 24 hours (if weight is ≥2000 g) or at age one month or at hospital discharge (if weight is <2000 g).
The first dose should be given within 12 hours of birth to all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.9
Immunization schedules
Every year ACIP updates the adult and child immunization schedules to incorporate the changes from the previous year. These can be found on the ACIP Web site at https://www.cdc.gov/vaccines/schedules/hcp/index.html. This Web site remains the most authoritative and accurate source of information on vaccines and immunizations for both professionals and the public.
1. Campos-Outcalt D. Need-to-know information for the 2016-2017 flu season. J Fam Pract. 2016;65:613-617.
2. Cohn AC, MacNeil JR, Clark TA, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1-28.
3. MacNeil JR, Rubin LG, Patton M, et al. Recommendations for use of meningococcal conjugate vaccines in HIV-infected persons— Advisory Committee on Immunization Practices, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1189-1194.
4. MacNeil JR, Rubin LG, Folaranmi T, et al. Use of serogroup B meningococcal vaccines in adolescents and young adults: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1171-1176.
5. Folaranmi T, Rubin L, Martin SW, et al. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:608-612.
6. MacNeil J. Considerations for Use of 2- and 3-Dose Schedules of MenB-FHbp (Trumenba). Presentation at: Advisory Committee on Immunization Practices; October 19, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/meningococcal-05-macneil.pdf. Accessed February 6, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65:1405-1408.
8. Wong KW. Cholera vaccine update and proposed recommendations. Presentation at: Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/cholera-02-wong.pdf. Accessed January 27, 2017.
9. Schillie S. Revised ACIP Hepatitis B (HepB) vaccine recommendations. Presentation at: Advisory Committee on Immunization Practices; October 19, 2016. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/hepatitis-02-schillie-october-2016.pdf. Accessed January 27, 2017.
10.
11. Ko SC, Fan L, Smith EA, et al. Estimated annual perinatal hepatitis B virus infections in the United States, 2000-2009. J Pediatric Infect Dis Soc. 2016;5:114-121.
12. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. MMWR Morb Mortal Wkly Rep. 2006;55:1-25.
13. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099-1102.
1. Campos-Outcalt D. Need-to-know information for the 2016-2017 flu season. J Fam Pract. 2016;65:613-617.
2. Cohn AC, MacNeil JR, Clark TA, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1-28.
3. MacNeil JR, Rubin LG, Patton M, et al. Recommendations for use of meningococcal conjugate vaccines in HIV-infected persons— Advisory Committee on Immunization Practices, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1189-1194.
4. MacNeil JR, Rubin LG, Folaranmi T, et al. Use of serogroup B meningococcal vaccines in adolescents and young adults: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1171-1176.
5. Folaranmi T, Rubin L, Martin SW, et al. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:608-612.
6. MacNeil J. Considerations for Use of 2- and 3-Dose Schedules of MenB-FHbp (Trumenba). Presentation at: Advisory Committee on Immunization Practices; October 19, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/meningococcal-05-macneil.pdf. Accessed February 6, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65:1405-1408.
8. Wong KW. Cholera vaccine update and proposed recommendations. Presentation at: Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/cholera-02-wong.pdf. Accessed January 27, 2017.
9. Schillie S. Revised ACIP Hepatitis B (HepB) vaccine recommendations. Presentation at: Advisory Committee on Immunization Practices; October 19, 2016. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/hepatitis-02-schillie-october-2016.pdf. Accessed January 27, 2017.
10.
11. Ko SC, Fan L, Smith EA, et al. Estimated annual perinatal hepatitis B virus infections in the United States, 2000-2009. J Pediatric Infect Dis Soc. 2016;5:114-121.
12. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. MMWR Morb Mortal Wkly Rep. 2006;55:1-25.
13. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099-1102.
Dupuytren’s disease: How to recognize its early signs
CASE › A 52-year-old right-hand-dominant white man arrived at our clinic complaining that he was unable to straighten his right ring finger. He had no associated pain or numbness, and had not injured his hand. The patient had type 2 diabetes that was controlled with metformin. He had no history of surgery or drug allergies, did not smoke, and said he drank 2 to 3 alcoholic beverages per day. He was a car salesman and was self-conscious when shaking hands with customers. On physical examination, we noted that he held his right ring finger at roughly 45 degrees of flexion at the metacarpophalangeal joint; a painless cord-like structure was palpable on the palmar surface of that joint. His left hand had no abnormalities.
If this were your patient, how would you proceed?
Dupuytren’s disease (DD) is a disabling fibroproliferative disorder of the hand for which there is no cure. While the exact cause of DD is unknown, it has been linked to a number of risk factors, including smoking, alcohol consumption, and diabetes. It affects about 5% of the US population, and up to 70% of affected individuals may initially seek treatment from a primary care physician.1 The disease is also referred to as Dupuytren’s contracture, which describes the flexion contractures of fingers at the end stage of the disease. Palmar fibromatosis is yet another name for the disorder.
DD refers to a spectrum of presentations ranging from nodules to cords to discernible contractures, and it is not known which patients with early Dupuytren changes will progress to severe contracture. With recognition of early changes, nonsurgical intervention is possible, such as collagenase injection or percutaneous fasciotomy, and can slow the progression of DD, restore function, and avoid or delay surgical intervention. DD is a clinically challenging disorder. Treatment for an affected area may resolve symptoms, only to have them recur in that location or another.
How underlying pathology correlates with clinical findings
DD affects the palmar fascia, a thick triangular-shaped sheet of dense fibrous collagenous connective tissue that lies deep to the dermis and superficial to the flexor tendons of the hand with fibers extending both into the skin and into the deep tissue. The palmar fascia secures the skin during gripping and twisting motions, and it bifurcates into distal extensions, called pretendinous bands, that overlay and mimic the flexor tendons.
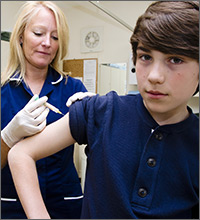
Clinical findings reflect the progression of underlying pathology. The earliest manifestation of DD is painless dimpling of the skin on the palmar surface of the hand.2 Over time, the underlying fibrosis with increased collagen deposition can progress, leading to development of nodules and eventually, cords, which are sometimes mistaken for flexor tendons. Dupuytren-like fibrotic tissue can occur on the sole of the foot (Ledderhose disease) and penis (Peyronie’s disease).2,3 In patients with these coexisting conditions, prognosis is generally worse.4 With the hands, bilateral involvement is common, although it is usually more severe in one hand. The ring finger is the digit most frequently involved, followed by the little finger, middle finger, and index finger. The thumb is rarely affected.3
As the disease progresses and cords contract, the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints develop flexion contractures. The distal interphalangeal (DIP) joint, rarely involved, instead exhibits a hyperextension contracture. Digital flexion contractures are often disabling, interfering with daily activities such as picking up a glass, shaking hands, or putting one's hand in a pants pocket. Many patients seek medical attention only after a palpable nodule, cord, or flexion contracture becomes apparent (FIGURE 1).
In a study of 326 patients who reported Dupuytren’s symptoms, the most common symptoms that led them to seek treatment were, in descending order, a hard bump (48%), a ropelike growth (12%), dimpling (11%), and finger contractures (10%). Only 9% of patients seeking treatment for hand symptoms associated with DD had received a diagnosis of DD from their initial medical encounter, causing an unnecessary delay in treatment.1
Who is at increased risk for DD?
DD is most often seen in elderly white men of European descent.5 In the United States, the prevalence of the disease is roughly 5%,1 compared with 4% to 39% in northwestern Europe (eg, Iceland).6 The male-to-female ratio of DD ranges from 6:1 to 15:1.7 The prevalence of DD appears to increase with age. The prevalence in men and women is similar up to age 45 years, after which the rate is much greater in males.7
DD is associated with many risk factors including smoking, alcohol consumption, vascular insufficiency, epilepsy, hyperlipidemia, manual labor, occupations with exposure to vibration, hand trauma, and even hand surgery such as carpal tunnel release or trigger finger release.8-11 It is also associated with diabetes; particularly type 1 insulin-dependent diabetes.12 There may also be an association with frozen shoulder.12,13
The need for surgical treatment of DD becomes more likely with a history of cigarette smoking and heavy alcohol consumption. There seems to be an association with epilepsy, most likely from anti-epileptic drugs.14 Rheumatoid arthritis is the only condition that has been associated with a lower incidence of DD, possibly because of the use of anti-inflammatory drugs.15 There are genetic differences between patients with and without DD, although a “Dupuytren’s gene” has not been identified.
Rule out possible DD mimics
The differential diagnosis for flexion contracture of the MCP or PIP joints seen in Dupuytren’s disease includes stenosing flexor tenosynovitis, or trigger finger. Other conditions that can mimic DD are ulnar claw hand, trauma scars, intrinsic joint diseases such as degenerative or rheumatoid arthritis (RA), diabetic cheiroarthropathy, camptodactyly, and Volkmann’s contracture. Trauma scars—especially longitudinal scars—have a tendency to contract and develop keloid formation.
Stenosing flexor tenosynovitis and ulnar claw hand are distinguishable from DD by full or nearly full active or passive extension of the affected digit, whereas DD is a true contracture of the joint that does not allow full extension. A careful history can rule out previous injury to the area. Although intrinsic joint disease, such as RA, can cause finger contractures, the joints are usually enlarged, painful, and associated with characteristic radiologic findings.
Unlike DD, diabetic cheiroarthropathy often involves all of the digits except for the thumb, and is often associated with a waxy appearance of the skin. Camptodactyly is an autosomal dominant disorder that more often presents in childhood and can be caused by a number of congenital syndromes. Volkmann’s contracture can manifest as a claw-like deformity of the hand caused by undiagnosed compartment syndrome of the forearm.
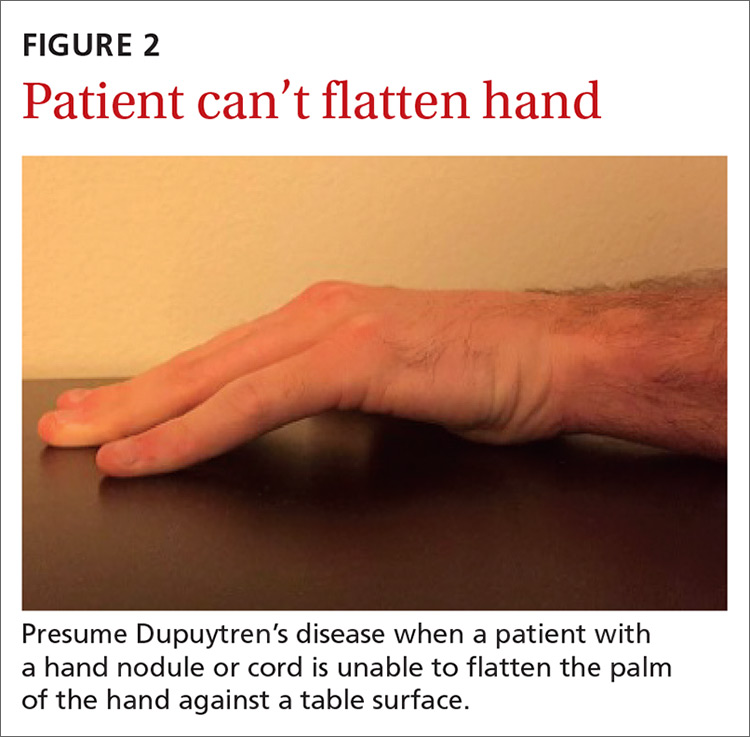
Assess the severity of DD
In your evaluation, first identify palmar or digital fibromatosis presenting as a nodule or a cord. Second, estimate the degree of MCP and PIP joint contractures. A common measure of contracture is the Hueston tabletop test. Ask the patient to place the palm of the hand on a flat surface. If the patient is unable to completely flatten the hand against the surface, presume a positive result (FIGURE 2).16

An accurate measure of the degree of flexion can be accomplished with a goniometer. For a simple assessment of severity, have the patient place each affected finger along the convexity of a spoon. If adjoining surfaces are flush, assume that the contracture is at least 30 degrees (FIGURE 3). The severity of DD can also be graded according to the Tubiana classification system (TABLE17), wherein the total deformity or contracture score is the sum of the angles of all 3 digital joints of the finger.

A look at the treatment options
Once the diagnosis of DD is made, reassure the patient that the disease is not cancerous, although it may be progressive. Advise patients, too, that several treatment options (detailed below) are available, but that surgery is the mainstay of treatment. (Of note: Despite what would seem logical, stretching a cord to straighten the finger is not recommended, as it may actually worsen the condition.18)
Options for early DD. Corticosteroids such as triamcinolone may have a role as an adjunct for early DD by reducing the size and firmness of the Dupuytren’s nodules and possibly slowing the progression of the disease. However, in one study of 63 patients, half of the individuals experienced reactivation of disease between one and 3 years.19
Radiotherapy has been studied as a potential treatment for early DD and for patients unable to undergo surgery. Radiation does have a biologic effect on nodular DD, but has no effect on the cords that cause the joint contracture. In a long-term follow-up study of 135 patients treated with radiotherapy, disease remained stable in 59% of patients, improved in 10%, and progressed in 31%.20 Results were better for early stage disease. This modality is not used for more advanced cases.
Surgery is the primary method by which to restore function and minimize complications. In the past, referral was recommended for MCP joint contractures >30 degrees or for any involvment of the PIP joint, because involvement of the latter carries a worse prognosis and may not be fully correctable, even with surgery.21 However, treatment may also be warranted in cases of functional disability—regardless of the degree of contracture (eg, for pain associated with a prominent nodule or cord when gripping objects).
Depending on disease progression and the surgeon’s preference, the most popular procedures are fasciotomies and fasciectomies. Surgical options for contractures—in increasing order of invasiveness and amounts of fascia removed—are fasciotomy (needle, open), fasciectomy (partial, radical), and dermofasciectomy.
Needle fasciotomy is an outpatient procedure performed under local block. The surgeon uses a needle bevel to transect the diseased cord at multiple puncture sites.
Open fasciotomy is also an outpatient procedure involving an incision that allows direct visualization of the cord and internal structures before dividing the fascia. Percutaneous and open fasciotomies are more successful for MCP joint contractures and less successful for PIP joint contractures.
With partial fasciectomy, only the grossly affected fascia is removed.
With radical fasciectomy, the entire palmar fascia is removed, regardless of which areas are grossly diseased.
Dermofasciectomy removes diseased fascia and the overlying skin, with a skin graft applied to heal the wound.
Another option: Collagenase injection. In February 2010, the US Food and Drug Administration approved collagenase clostridium histolyticum (Xiaflex) as the first drug specifically indicated for the treatment of Dupuytren’s contracture. Collagenase is injected directly into the fascial cord, cleaving its collagen component and progressively softening and breaking down the cord. If not properly injected, collagenase may injure neurovascular structures and may rupture tendons.
So how do the treatment options compare?
We found no randomized prospective trials comparing treatment options for DD, but there are a number of trials that shed light on a variety of the available options.
Treatment success. In general, partial fasciectomies have shown the greatest success in reducing contractures and maintaining the lowest recurrence rates. Correction of contracture to ≤5 degrees was seen in 94% of MCP joint contractures treated with partial fasciectomy, in 77% of MCP joints treated with collagenase, and in 55% of MCP joints treated with percutaneous needle fasciotomies.22,23 PIP joint contracture correction was not as successful: Correction of contracture to ≤5 degrees was seen in 47% of those treated with partial fasciectomy, 40% treated with collagenase, and 26% treated with needle fasciotomy.22,23
Recurrence rates. When recurrence was defined as loss of passive extension >30 degrees, the recurrence rate for MCP joint contractures with partial fasciectomy was 21%, compared with 85% for needle fasciotomy.22 In a similar review, recurrence rates for partial fasciectomy ranged from 12% to 39% compared with 50% to 58% for needle fasciotomy.24 With collagenase injection, 5-year data have shown an overall recurrence rate of 32%, with a recurrence of 26% at the MCP joint and 46% at the PIP joint.25 In this trial, recurrence was
Complications. Although fasciectomies have shown the best efficacy and lowest recurrence rates, complications such as infection, neurapraxia, and digital nerve injury are more likely with these procedures.26,27
Early recognition and referral to a hand specialist for the treatment of DD may allow the use of less invasive techniques and improved functional results.
CASE › Absent a history of trauma and features typical of other hand/digit disorders, we diagnosed Dupuytren’s disease in this patient. Potential treatments included partial fasciectomy and collagenase injection. After discussing the risks and benefits of each of these procedures, the patient elected to undergo a partial fasciectomy. The surgery was uncomplicated and the abnormality was corrected to within 5 degrees of full extension. At the patient’s one-year follow-up visit, there was no evidence of recurrence.
1. DiBenedetti DB, Nguyen D, Zografos L, et al. Prevalence, incidence, and treatments of Dupuytren’s disease in the United States: results from a population-based study. Hand. 2011;6:149-158.
2. Townley WA, Baker R, Sheppard N, et al. Dupuytren’s contracture unfolded. BMJ. 2006;332:397-400.
3. Rayan GM. Clinical presentation and types of Dupuytren’s disease. Hand Clin. 1999;15:87-96.
4. Hindocha S, Stanley JK, Watson S, et al. Dupuytren’s diathesis revisited: Evaluation of prognostic indicators for risk of disease recurrence. J Hand Surg Am. 2006;31:1626-1634.
5. Bayat A, McGrouther DA. Management of Dupuytren’s disease—clear advice for an elusive condition. Ann R Coll Surg Engl. 2006;88:3-8.
6. Gudmundsson KG, Arngrimsson R, Sigfússon M, et al. Epidemiology of Dupuytren’s disease: clinical, serological, and social assessment. The Reykjavik Study. J Clin Epidemiol. 2000;53:291-296.
7. Wilbrand S, Ekbom A, Gerdin B. The sex ratio and rate of reoperation for Dupuytren’s contracture in men and women. J Hand Surg Br. 1999;24:456-459.
8. Noble J, Arafa M, Royle SG, et al. The association between alcohol, hepatic pathology and Dupuytren’s disease. J Hand Surg Br. 1992;17:71–74.
9. Mikkelsen OA. Dupuytren’s disease—initial symptoms, age of onset and spontaneous course. Hand. 1977;9:11-15.
10. Arkkila PE, Kantola IM, Viikari JS. Dupuytren’s disease: association with chronic diabetic complications. J Rheumatol. 1997;24:153-159.
11. Liss GM, Stock SR. Can Dupuytren’s contracture be work related? review of the evidence. Am J Ind Med. 1996;29:521-532.
12. Geoghegan JM, Forbes J, Clark DI, et al. Dupuytren’s disease risk factors. J Hand Surg Br. 2004;29:423-426.
13. Smith SP, Devaraj VS, Bunker TD. The association between frozen shoulder and Dupuytren’s disease. J Shoulder Elbow Surg. 2001;10:149-151.
14. Nunn AC, Schreuder FB. Dupuytren’s contracture: emerging insight into a Viking disease. J Hand Surg. 2014;19:481-490.
15. Arafa M, Steingold R F, Noble J. The incidence of Dupuytren’s disease in patients with rheumatoid arthritis. J Hand Surg Br. 1984;9:165-166.
16. Hueston J. Lessons in Dupuytren’s disease. Ann Chir Main Memb Super. 1992;11:349-354.
17. Tubiana R. Prognosis and treatment of Dupuytren’s contracture. J Bone and Joint Surg AM. 1955;37:1155-1168.
18. Howard JC, Varallo VM, Ross DC, et al. Elevated levels of ß-catenin and fibronectin in three-dimensional collagen cultures of Dupuytren’s disease cells are regulated by tension in vitro. BMC Musculoskelet Disord. 2003;4:16.
19. Ketchum LD, Donahue TK. The injection of nodules of Dupuytren’s disease with triamcinolone acetonide. J Hand Surg Am. 2000;25:1157-1162.
20. Betz N, Ott OJ, Adamietz B, et al. Radiotherapy in early-stage Dupuytren’s contracture. Long-term results after 13 years. Strahlenther Onkol. 2010;186:82-90.
21. Smith AC. Diagnosis and indications for surgical treatment. Hand Clin. 1991;7:635-642.
22. van Rijssen AL, ter Linden H, Werker PM. Five-year results of a randomized clinical trial on treatment in Dupuytren’s disease: percutaneous needle fasciotomy versus limited fasciectomy. Plast Reconstr Surg. 2012;129:469-477.
23. Hurst LC, Badalamente MA, Hentz VR, et al. Injectable collagenase clostridium histolyticum for Dupuytren’s contracture. N Engl J Med. 2009;361:968-979.
24. Chen NC, Srinivasan RC, Shauver MJ, et al. A systematic review of outcomes of fasciotomy, aponeurotomy, and collagenase treatments for Dupuytren’s contracture. Hand. 2011;6:250-255.
25. Peimer CA, Blazar P, Coleman S, et al. Dupuytren contracture recurrence following treatment with collagenase Clostridium histolyticum (CORDLESS [Collagenase Option for Reduction of Dupuytren Long-Term Evaluation of Safety Study]): 5-year data. J Hand Surg Am. 2015;40:1597-1605.
26. Toppi JT, Trompf L, Smoll NR, et al. Dupuytren’s contracture: an analysis of outcomes of percutaneous needle fasciotomy versus open fasciectomy. ANZ J Surg. 2015;85:639-643.
27. Cheung K, Walley KC, Rozental TD. Management of complications of Dupuytren contracture. Hand Clini. 2015;31:345-354.
CASE › A 52-year-old right-hand-dominant white man arrived at our clinic complaining that he was unable to straighten his right ring finger. He had no associated pain or numbness, and had not injured his hand. The patient had type 2 diabetes that was controlled with metformin. He had no history of surgery or drug allergies, did not smoke, and said he drank 2 to 3 alcoholic beverages per day. He was a car salesman and was self-conscious when shaking hands with customers. On physical examination, we noted that he held his right ring finger at roughly 45 degrees of flexion at the metacarpophalangeal joint; a painless cord-like structure was palpable on the palmar surface of that joint. His left hand had no abnormalities.
If this were your patient, how would you proceed?
Dupuytren’s disease (DD) is a disabling fibroproliferative disorder of the hand for which there is no cure. While the exact cause of DD is unknown, it has been linked to a number of risk factors, including smoking, alcohol consumption, and diabetes. It affects about 5% of the US population, and up to 70% of affected individuals may initially seek treatment from a primary care physician.1 The disease is also referred to as Dupuytren’s contracture, which describes the flexion contractures of fingers at the end stage of the disease. Palmar fibromatosis is yet another name for the disorder.
DD refers to a spectrum of presentations ranging from nodules to cords to discernible contractures, and it is not known which patients with early Dupuytren changes will progress to severe contracture. With recognition of early changes, nonsurgical intervention is possible, such as collagenase injection or percutaneous fasciotomy, and can slow the progression of DD, restore function, and avoid or delay surgical intervention. DD is a clinically challenging disorder. Treatment for an affected area may resolve symptoms, only to have them recur in that location or another.
How underlying pathology correlates with clinical findings
DD affects the palmar fascia, a thick triangular-shaped sheet of dense fibrous collagenous connective tissue that lies deep to the dermis and superficial to the flexor tendons of the hand with fibers extending both into the skin and into the deep tissue. The palmar fascia secures the skin during gripping and twisting motions, and it bifurcates into distal extensions, called pretendinous bands, that overlay and mimic the flexor tendons.
Clinical findings reflect the progression of underlying pathology. The earliest manifestation of DD is painless dimpling of the skin on the palmar surface of the hand.2 Over time, the underlying fibrosis with increased collagen deposition can progress, leading to development of nodules and eventually, cords, which are sometimes mistaken for flexor tendons. Dupuytren-like fibrotic tissue can occur on the sole of the foot (Ledderhose disease) and penis (Peyronie’s disease).2,3 In patients with these coexisting conditions, prognosis is generally worse.4 With the hands, bilateral involvement is common, although it is usually more severe in one hand. The ring finger is the digit most frequently involved, followed by the little finger, middle finger, and index finger. The thumb is rarely affected.3
As the disease progresses and cords contract, the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints develop flexion contractures. The distal interphalangeal (DIP) joint, rarely involved, instead exhibits a hyperextension contracture. Digital flexion contractures are often disabling, interfering with daily activities such as picking up a glass, shaking hands, or putting one's hand in a pants pocket. Many patients seek medical attention only after a palpable nodule, cord, or flexion contracture becomes apparent (FIGURE 1).
In a study of 326 patients who reported Dupuytren’s symptoms, the most common symptoms that led them to seek treatment were, in descending order, a hard bump (48%), a ropelike growth (12%), dimpling (11%), and finger contractures (10%). Only 9% of patients seeking treatment for hand symptoms associated with DD had received a diagnosis of DD from their initial medical encounter, causing an unnecessary delay in treatment.1
Who is at increased risk for DD?
DD is most often seen in elderly white men of European descent.5 In the United States, the prevalence of the disease is roughly 5%,1 compared with 4% to 39% in northwestern Europe (eg, Iceland).6 The male-to-female ratio of DD ranges from 6:1 to 15:1.7 The prevalence of DD appears to increase with age. The prevalence in men and women is similar up to age 45 years, after which the rate is much greater in males.7
DD is associated with many risk factors including smoking, alcohol consumption, vascular insufficiency, epilepsy, hyperlipidemia, manual labor, occupations with exposure to vibration, hand trauma, and even hand surgery such as carpal tunnel release or trigger finger release.8-11 It is also associated with diabetes; particularly type 1 insulin-dependent diabetes.12 There may also be an association with frozen shoulder.12,13
The need for surgical treatment of DD becomes more likely with a history of cigarette smoking and heavy alcohol consumption. There seems to be an association with epilepsy, most likely from anti-epileptic drugs.14 Rheumatoid arthritis is the only condition that has been associated with a lower incidence of DD, possibly because of the use of anti-inflammatory drugs.15 There are genetic differences between patients with and without DD, although a “Dupuytren’s gene” has not been identified.
Rule out possible DD mimics
The differential diagnosis for flexion contracture of the MCP or PIP joints seen in Dupuytren’s disease includes stenosing flexor tenosynovitis, or trigger finger. Other conditions that can mimic DD are ulnar claw hand, trauma scars, intrinsic joint diseases such as degenerative or rheumatoid arthritis (RA), diabetic cheiroarthropathy, camptodactyly, and Volkmann’s contracture. Trauma scars—especially longitudinal scars—have a tendency to contract and develop keloid formation.
Stenosing flexor tenosynovitis and ulnar claw hand are distinguishable from DD by full or nearly full active or passive extension of the affected digit, whereas DD is a true contracture of the joint that does not allow full extension. A careful history can rule out previous injury to the area. Although intrinsic joint disease, such as RA, can cause finger contractures, the joints are usually enlarged, painful, and associated with characteristic radiologic findings.
Unlike DD, diabetic cheiroarthropathy often involves all of the digits except for the thumb, and is often associated with a waxy appearance of the skin. Camptodactyly is an autosomal dominant disorder that more often presents in childhood and can be caused by a number of congenital syndromes. Volkmann’s contracture can manifest as a claw-like deformity of the hand caused by undiagnosed compartment syndrome of the forearm.
Assess the severity of DD
In your evaluation, first identify palmar or digital fibromatosis presenting as a nodule or a cord. Second, estimate the degree of MCP and PIP joint contractures. A common measure of contracture is the Hueston tabletop test. Ask the patient to place the palm of the hand on a flat surface. If the patient is unable to completely flatten the hand against the surface, presume a positive result (FIGURE 2).16
An accurate measure of the degree of flexion can be accomplished with a goniometer. For a simple assessment of severity, have the patient place each affected finger along the convexity of a spoon. If adjoining surfaces are flush, assume that the contracture is at least 30 degrees (FIGURE 3). The severity of DD can also be graded according to the Tubiana classification system (TABLE17), wherein the total deformity or contracture score is the sum of the angles of all 3 digital joints of the finger.
A look at the treatment options
Once the diagnosis of DD is made, reassure the patient that the disease is not cancerous, although it may be progressive. Advise patients, too, that several treatment options (detailed below) are available, but that surgery is the mainstay of treatment. (Of note: Despite what would seem logical, stretching a cord to straighten the finger is not recommended, as it may actually worsen the condition.18)
Options for early DD. Corticosteroids such as triamcinolone may have a role as an adjunct for early DD by reducing the size and firmness of the Dupuytren’s nodules and possibly slowing the progression of the disease. However, in one study of 63 patients, half of the individuals experienced reactivation of disease between one and 3 years.19
Radiotherapy has been studied as a potential treatment for early DD and for patients unable to undergo surgery. Radiation does have a biologic effect on nodular DD, but has no effect on the cords that cause the joint contracture. In a long-term follow-up study of 135 patients treated with radiotherapy, disease remained stable in 59% of patients, improved in 10%, and progressed in 31%.20 Results were better for early stage disease. This modality is not used for more advanced cases.
Surgery is the primary method by which to restore function and minimize complications. In the past, referral was recommended for MCP joint contractures >30 degrees or for any involvment of the PIP joint, because involvement of the latter carries a worse prognosis and may not be fully correctable, even with surgery.21 However, treatment may also be warranted in cases of functional disability—regardless of the degree of contracture (eg, for pain associated with a prominent nodule or cord when gripping objects).
Depending on disease progression and the surgeon’s preference, the most popular procedures are fasciotomies and fasciectomies. Surgical options for contractures—in increasing order of invasiveness and amounts of fascia removed—are fasciotomy (needle, open), fasciectomy (partial, radical), and dermofasciectomy.
Needle fasciotomy is an outpatient procedure performed under local block. The surgeon uses a needle bevel to transect the diseased cord at multiple puncture sites.
Open fasciotomy is also an outpatient procedure involving an incision that allows direct visualization of the cord and internal structures before dividing the fascia. Percutaneous and open fasciotomies are more successful for MCP joint contractures and less successful for PIP joint contractures.
With partial fasciectomy, only the grossly affected fascia is removed.
With radical fasciectomy, the entire palmar fascia is removed, regardless of which areas are grossly diseased.
Dermofasciectomy removes diseased fascia and the overlying skin, with a skin graft applied to heal the wound.
Another option: Collagenase injection. In February 2010, the US Food and Drug Administration approved collagenase clostridium histolyticum (Xiaflex) as the first drug specifically indicated for the treatment of Dupuytren’s contracture. Collagenase is injected directly into the fascial cord, cleaving its collagen component and progressively softening and breaking down the cord. If not properly injected, collagenase may injure neurovascular structures and may rupture tendons.
So how do the treatment options compare?
We found no randomized prospective trials comparing treatment options for DD, but there are a number of trials that shed light on a variety of the available options.
Treatment success. In general, partial fasciectomies have shown the greatest success in reducing contractures and maintaining the lowest recurrence rates. Correction of contracture to ≤5 degrees was seen in 94% of MCP joint contractures treated with partial fasciectomy, in 77% of MCP joints treated with collagenase, and in 55% of MCP joints treated with percutaneous needle fasciotomies.22,23 PIP joint contracture correction was not as successful: Correction of contracture to ≤5 degrees was seen in 47% of those treated with partial fasciectomy, 40% treated with collagenase, and 26% treated with needle fasciotomy.22,23
Recurrence rates. When recurrence was defined as loss of passive extension >30 degrees, the recurrence rate for MCP joint contractures with partial fasciectomy was 21%, compared with 85% for needle fasciotomy.22 In a similar review, recurrence rates for partial fasciectomy ranged from 12% to 39% compared with 50% to 58% for needle fasciotomy.24 With collagenase injection, 5-year data have shown an overall recurrence rate of 32%, with a recurrence of 26% at the MCP joint and 46% at the PIP joint.25 In this trial, recurrence was
Complications. Although fasciectomies have shown the best efficacy and lowest recurrence rates, complications such as infection, neurapraxia, and digital nerve injury are more likely with these procedures.26,27
Early recognition and referral to a hand specialist for the treatment of DD may allow the use of less invasive techniques and improved functional results.
CASE › Absent a history of trauma and features typical of other hand/digit disorders, we diagnosed Dupuytren’s disease in this patient. Potential treatments included partial fasciectomy and collagenase injection. After discussing the risks and benefits of each of these procedures, the patient elected to undergo a partial fasciectomy. The surgery was uncomplicated and the abnormality was corrected to within 5 degrees of full extension. At the patient’s one-year follow-up visit, there was no evidence of recurrence.
CASE › A 52-year-old right-hand-dominant white man arrived at our clinic complaining that he was unable to straighten his right ring finger. He had no associated pain or numbness, and had not injured his hand. The patient had type 2 diabetes that was controlled with metformin. He had no history of surgery or drug allergies, did not smoke, and said he drank 2 to 3 alcoholic beverages per day. He was a car salesman and was self-conscious when shaking hands with customers. On physical examination, we noted that he held his right ring finger at roughly 45 degrees of flexion at the metacarpophalangeal joint; a painless cord-like structure was palpable on the palmar surface of that joint. His left hand had no abnormalities.
If this were your patient, how would you proceed?
Dupuytren’s disease (DD) is a disabling fibroproliferative disorder of the hand for which there is no cure. While the exact cause of DD is unknown, it has been linked to a number of risk factors, including smoking, alcohol consumption, and diabetes. It affects about 5% of the US population, and up to 70% of affected individuals may initially seek treatment from a primary care physician.1 The disease is also referred to as Dupuytren’s contracture, which describes the flexion contractures of fingers at the end stage of the disease. Palmar fibromatosis is yet another name for the disorder.
DD refers to a spectrum of presentations ranging from nodules to cords to discernible contractures, and it is not known which patients with early Dupuytren changes will progress to severe contracture. With recognition of early changes, nonsurgical intervention is possible, such as collagenase injection or percutaneous fasciotomy, and can slow the progression of DD, restore function, and avoid or delay surgical intervention. DD is a clinically challenging disorder. Treatment for an affected area may resolve symptoms, only to have them recur in that location or another.
How underlying pathology correlates with clinical findings
DD affects the palmar fascia, a thick triangular-shaped sheet of dense fibrous collagenous connective tissue that lies deep to the dermis and superficial to the flexor tendons of the hand with fibers extending both into the skin and into the deep tissue. The palmar fascia secures the skin during gripping and twisting motions, and it bifurcates into distal extensions, called pretendinous bands, that overlay and mimic the flexor tendons.
Clinical findings reflect the progression of underlying pathology. The earliest manifestation of DD is painless dimpling of the skin on the palmar surface of the hand.2 Over time, the underlying fibrosis with increased collagen deposition can progress, leading to development of nodules and eventually, cords, which are sometimes mistaken for flexor tendons. Dupuytren-like fibrotic tissue can occur on the sole of the foot (Ledderhose disease) and penis (Peyronie’s disease).2,3 In patients with these coexisting conditions, prognosis is generally worse.4 With the hands, bilateral involvement is common, although it is usually more severe in one hand. The ring finger is the digit most frequently involved, followed by the little finger, middle finger, and index finger. The thumb is rarely affected.3
As the disease progresses and cords contract, the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints develop flexion contractures. The distal interphalangeal (DIP) joint, rarely involved, instead exhibits a hyperextension contracture. Digital flexion contractures are often disabling, interfering with daily activities such as picking up a glass, shaking hands, or putting one's hand in a pants pocket. Many patients seek medical attention only after a palpable nodule, cord, or flexion contracture becomes apparent (FIGURE 1).
In a study of 326 patients who reported Dupuytren’s symptoms, the most common symptoms that led them to seek treatment were, in descending order, a hard bump (48%), a ropelike growth (12%), dimpling (11%), and finger contractures (10%). Only 9% of patients seeking treatment for hand symptoms associated with DD had received a diagnosis of DD from their initial medical encounter, causing an unnecessary delay in treatment.1
Who is at increased risk for DD?
DD is most often seen in elderly white men of European descent.5 In the United States, the prevalence of the disease is roughly 5%,1 compared with 4% to 39% in northwestern Europe (eg, Iceland).6 The male-to-female ratio of DD ranges from 6:1 to 15:1.7 The prevalence of DD appears to increase with age. The prevalence in men and women is similar up to age 45 years, after which the rate is much greater in males.7
DD is associated with many risk factors including smoking, alcohol consumption, vascular insufficiency, epilepsy, hyperlipidemia, manual labor, occupations with exposure to vibration, hand trauma, and even hand surgery such as carpal tunnel release or trigger finger release.8-11 It is also associated with diabetes; particularly type 1 insulin-dependent diabetes.12 There may also be an association with frozen shoulder.12,13
The need for surgical treatment of DD becomes more likely with a history of cigarette smoking and heavy alcohol consumption. There seems to be an association with epilepsy, most likely from anti-epileptic drugs.14 Rheumatoid arthritis is the only condition that has been associated with a lower incidence of DD, possibly because of the use of anti-inflammatory drugs.15 There are genetic differences between patients with and without DD, although a “Dupuytren’s gene” has not been identified.
Rule out possible DD mimics
The differential diagnosis for flexion contracture of the MCP or PIP joints seen in Dupuytren’s disease includes stenosing flexor tenosynovitis, or trigger finger. Other conditions that can mimic DD are ulnar claw hand, trauma scars, intrinsic joint diseases such as degenerative or rheumatoid arthritis (RA), diabetic cheiroarthropathy, camptodactyly, and Volkmann’s contracture. Trauma scars—especially longitudinal scars—have a tendency to contract and develop keloid formation.
Stenosing flexor tenosynovitis and ulnar claw hand are distinguishable from DD by full or nearly full active or passive extension of the affected digit, whereas DD is a true contracture of the joint that does not allow full extension. A careful history can rule out previous injury to the area. Although intrinsic joint disease, such as RA, can cause finger contractures, the joints are usually enlarged, painful, and associated with characteristic radiologic findings.
Unlike DD, diabetic cheiroarthropathy often involves all of the digits except for the thumb, and is often associated with a waxy appearance of the skin. Camptodactyly is an autosomal dominant disorder that more often presents in childhood and can be caused by a number of congenital syndromes. Volkmann’s contracture can manifest as a claw-like deformity of the hand caused by undiagnosed compartment syndrome of the forearm.
Assess the severity of DD
In your evaluation, first identify palmar or digital fibromatosis presenting as a nodule or a cord. Second, estimate the degree of MCP and PIP joint contractures. A common measure of contracture is the Hueston tabletop test. Ask the patient to place the palm of the hand on a flat surface. If the patient is unable to completely flatten the hand against the surface, presume a positive result (FIGURE 2).16
An accurate measure of the degree of flexion can be accomplished with a goniometer. For a simple assessment of severity, have the patient place each affected finger along the convexity of a spoon. If adjoining surfaces are flush, assume that the contracture is at least 30 degrees (FIGURE 3). The severity of DD can also be graded according to the Tubiana classification system (TABLE17), wherein the total deformity or contracture score is the sum of the angles of all 3 digital joints of the finger.
A look at the treatment options
Once the diagnosis of DD is made, reassure the patient that the disease is not cancerous, although it may be progressive. Advise patients, too, that several treatment options (detailed below) are available, but that surgery is the mainstay of treatment. (Of note: Despite what would seem logical, stretching a cord to straighten the finger is not recommended, as it may actually worsen the condition.18)
Options for early DD. Corticosteroids such as triamcinolone may have a role as an adjunct for early DD by reducing the size and firmness of the Dupuytren’s nodules and possibly slowing the progression of the disease. However, in one study of 63 patients, half of the individuals experienced reactivation of disease between one and 3 years.19
Radiotherapy has been studied as a potential treatment for early DD and for patients unable to undergo surgery. Radiation does have a biologic effect on nodular DD, but has no effect on the cords that cause the joint contracture. In a long-term follow-up study of 135 patients treated with radiotherapy, disease remained stable in 59% of patients, improved in 10%, and progressed in 31%.20 Results were better for early stage disease. This modality is not used for more advanced cases.
Surgery is the primary method by which to restore function and minimize complications. In the past, referral was recommended for MCP joint contractures >30 degrees or for any involvment of the PIP joint, because involvement of the latter carries a worse prognosis and may not be fully correctable, even with surgery.21 However, treatment may also be warranted in cases of functional disability—regardless of the degree of contracture (eg, for pain associated with a prominent nodule or cord when gripping objects).
Depending on disease progression and the surgeon’s preference, the most popular procedures are fasciotomies and fasciectomies. Surgical options for contractures—in increasing order of invasiveness and amounts of fascia removed—are fasciotomy (needle, open), fasciectomy (partial, radical), and dermofasciectomy.
Needle fasciotomy is an outpatient procedure performed under local block. The surgeon uses a needle bevel to transect the diseased cord at multiple puncture sites.
Open fasciotomy is also an outpatient procedure involving an incision that allows direct visualization of the cord and internal structures before dividing the fascia. Percutaneous and open fasciotomies are more successful for MCP joint contractures and less successful for PIP joint contractures.
With partial fasciectomy, only the grossly affected fascia is removed.
With radical fasciectomy, the entire palmar fascia is removed, regardless of which areas are grossly diseased.
Dermofasciectomy removes diseased fascia and the overlying skin, with a skin graft applied to heal the wound.
Another option: Collagenase injection. In February 2010, the US Food and Drug Administration approved collagenase clostridium histolyticum (Xiaflex) as the first drug specifically indicated for the treatment of Dupuytren’s contracture. Collagenase is injected directly into the fascial cord, cleaving its collagen component and progressively softening and breaking down the cord. If not properly injected, collagenase may injure neurovascular structures and may rupture tendons.
So how do the treatment options compare?
We found no randomized prospective trials comparing treatment options for DD, but there are a number of trials that shed light on a variety of the available options.
Treatment success. In general, partial fasciectomies have shown the greatest success in reducing contractures and maintaining the lowest recurrence rates. Correction of contracture to ≤5 degrees was seen in 94% of MCP joint contractures treated with partial fasciectomy, in 77% of MCP joints treated with collagenase, and in 55% of MCP joints treated with percutaneous needle fasciotomies.22,23 PIP joint contracture correction was not as successful: Correction of contracture to ≤5 degrees was seen in 47% of those treated with partial fasciectomy, 40% treated with collagenase, and 26% treated with needle fasciotomy.22,23
Recurrence rates. When recurrence was defined as loss of passive extension >30 degrees, the recurrence rate for MCP joint contractures with partial fasciectomy was 21%, compared with 85% for needle fasciotomy.22 In a similar review, recurrence rates for partial fasciectomy ranged from 12% to 39% compared with 50% to 58% for needle fasciotomy.24 With collagenase injection, 5-year data have shown an overall recurrence rate of 32%, with a recurrence of 26% at the MCP joint and 46% at the PIP joint.25 In this trial, recurrence was
Complications. Although fasciectomies have shown the best efficacy and lowest recurrence rates, complications such as infection, neurapraxia, and digital nerve injury are more likely with these procedures.26,27
Early recognition and referral to a hand specialist for the treatment of DD may allow the use of less invasive techniques and improved functional results.
CASE › Absent a history of trauma and features typical of other hand/digit disorders, we diagnosed Dupuytren’s disease in this patient. Potential treatments included partial fasciectomy and collagenase injection. After discussing the risks and benefits of each of these procedures, the patient elected to undergo a partial fasciectomy. The surgery was uncomplicated and the abnormality was corrected to within 5 degrees of full extension. At the patient’s one-year follow-up visit, there was no evidence of recurrence.
1. DiBenedetti DB, Nguyen D, Zografos L, et al. Prevalence, incidence, and treatments of Dupuytren’s disease in the United States: results from a population-based study. Hand. 2011;6:149-158.
2. Townley WA, Baker R, Sheppard N, et al. Dupuytren’s contracture unfolded. BMJ. 2006;332:397-400.
3. Rayan GM. Clinical presentation and types of Dupuytren’s disease. Hand Clin. 1999;15:87-96.
4. Hindocha S, Stanley JK, Watson S, et al. Dupuytren’s diathesis revisited: Evaluation of prognostic indicators for risk of disease recurrence. J Hand Surg Am. 2006;31:1626-1634.
5. Bayat A, McGrouther DA. Management of Dupuytren’s disease—clear advice for an elusive condition. Ann R Coll Surg Engl. 2006;88:3-8.
6. Gudmundsson KG, Arngrimsson R, Sigfússon M, et al. Epidemiology of Dupuytren’s disease: clinical, serological, and social assessment. The Reykjavik Study. J Clin Epidemiol. 2000;53:291-296.
7. Wilbrand S, Ekbom A, Gerdin B. The sex ratio and rate of reoperation for Dupuytren’s contracture in men and women. J Hand Surg Br. 1999;24:456-459.
8. Noble J, Arafa M, Royle SG, et al. The association between alcohol, hepatic pathology and Dupuytren’s disease. J Hand Surg Br. 1992;17:71–74.
9. Mikkelsen OA. Dupuytren’s disease—initial symptoms, age of onset and spontaneous course. Hand. 1977;9:11-15.
10. Arkkila PE, Kantola IM, Viikari JS. Dupuytren’s disease: association with chronic diabetic complications. J Rheumatol. 1997;24:153-159.
11. Liss GM, Stock SR. Can Dupuytren’s contracture be work related? review of the evidence. Am J Ind Med. 1996;29:521-532.
12. Geoghegan JM, Forbes J, Clark DI, et al. Dupuytren’s disease risk factors. J Hand Surg Br. 2004;29:423-426.
13. Smith SP, Devaraj VS, Bunker TD. The association between frozen shoulder and Dupuytren’s disease. J Shoulder Elbow Surg. 2001;10:149-151.
14. Nunn AC, Schreuder FB. Dupuytren’s contracture: emerging insight into a Viking disease. J Hand Surg. 2014;19:481-490.
15. Arafa M, Steingold R F, Noble J. The incidence of Dupuytren’s disease in patients with rheumatoid arthritis. J Hand Surg Br. 1984;9:165-166.
16. Hueston J. Lessons in Dupuytren’s disease. Ann Chir Main Memb Super. 1992;11:349-354.
17. Tubiana R. Prognosis and treatment of Dupuytren’s contracture. J Bone and Joint Surg AM. 1955;37:1155-1168.
18. Howard JC, Varallo VM, Ross DC, et al. Elevated levels of ß-catenin and fibronectin in three-dimensional collagen cultures of Dupuytren’s disease cells are regulated by tension in vitro. BMC Musculoskelet Disord. 2003;4:16.
19. Ketchum LD, Donahue TK. The injection of nodules of Dupuytren’s disease with triamcinolone acetonide. J Hand Surg Am. 2000;25:1157-1162.
20. Betz N, Ott OJ, Adamietz B, et al. Radiotherapy in early-stage Dupuytren’s contracture. Long-term results after 13 years. Strahlenther Onkol. 2010;186:82-90.
21. Smith AC. Diagnosis and indications for surgical treatment. Hand Clin. 1991;7:635-642.
22. van Rijssen AL, ter Linden H, Werker PM. Five-year results of a randomized clinical trial on treatment in Dupuytren’s disease: percutaneous needle fasciotomy versus limited fasciectomy. Plast Reconstr Surg. 2012;129:469-477.
23. Hurst LC, Badalamente MA, Hentz VR, et al. Injectable collagenase clostridium histolyticum for Dupuytren’s contracture. N Engl J Med. 2009;361:968-979.
24. Chen NC, Srinivasan RC, Shauver MJ, et al. A systematic review of outcomes of fasciotomy, aponeurotomy, and collagenase treatments for Dupuytren’s contracture. Hand. 2011;6:250-255.
25. Peimer CA, Blazar P, Coleman S, et al. Dupuytren contracture recurrence following treatment with collagenase Clostridium histolyticum (CORDLESS [Collagenase Option for Reduction of Dupuytren Long-Term Evaluation of Safety Study]): 5-year data. J Hand Surg Am. 2015;40:1597-1605.
26. Toppi JT, Trompf L, Smoll NR, et al. Dupuytren’s contracture: an analysis of outcomes of percutaneous needle fasciotomy versus open fasciectomy. ANZ J Surg. 2015;85:639-643.
27. Cheung K, Walley KC, Rozental TD. Management of complications of Dupuytren contracture. Hand Clini. 2015;31:345-354.
1. DiBenedetti DB, Nguyen D, Zografos L, et al. Prevalence, incidence, and treatments of Dupuytren’s disease in the United States: results from a population-based study. Hand. 2011;6:149-158.
2. Townley WA, Baker R, Sheppard N, et al. Dupuytren’s contracture unfolded. BMJ. 2006;332:397-400.
3. Rayan GM. Clinical presentation and types of Dupuytren’s disease. Hand Clin. 1999;15:87-96.
4. Hindocha S, Stanley JK, Watson S, et al. Dupuytren’s diathesis revisited: Evaluation of prognostic indicators for risk of disease recurrence. J Hand Surg Am. 2006;31:1626-1634.
5. Bayat A, McGrouther DA. Management of Dupuytren’s disease—clear advice for an elusive condition. Ann R Coll Surg Engl. 2006;88:3-8.
6. Gudmundsson KG, Arngrimsson R, Sigfússon M, et al. Epidemiology of Dupuytren’s disease: clinical, serological, and social assessment. The Reykjavik Study. J Clin Epidemiol. 2000;53:291-296.
7. Wilbrand S, Ekbom A, Gerdin B. The sex ratio and rate of reoperation for Dupuytren’s contracture in men and women. J Hand Surg Br. 1999;24:456-459.
8. Noble J, Arafa M, Royle SG, et al. The association between alcohol, hepatic pathology and Dupuytren’s disease. J Hand Surg Br. 1992;17:71–74.
9. Mikkelsen OA. Dupuytren’s disease—initial symptoms, age of onset and spontaneous course. Hand. 1977;9:11-15.
10. Arkkila PE, Kantola IM, Viikari JS. Dupuytren’s disease: association with chronic diabetic complications. J Rheumatol. 1997;24:153-159.
11. Liss GM, Stock SR. Can Dupuytren’s contracture be work related? review of the evidence. Am J Ind Med. 1996;29:521-532.
12. Geoghegan JM, Forbes J, Clark DI, et al. Dupuytren’s disease risk factors. J Hand Surg Br. 2004;29:423-426.
13. Smith SP, Devaraj VS, Bunker TD. The association between frozen shoulder and Dupuytren’s disease. J Shoulder Elbow Surg. 2001;10:149-151.
14. Nunn AC, Schreuder FB. Dupuytren’s contracture: emerging insight into a Viking disease. J Hand Surg. 2014;19:481-490.
15. Arafa M, Steingold R F, Noble J. The incidence of Dupuytren’s disease in patients with rheumatoid arthritis. J Hand Surg Br. 1984;9:165-166.
16. Hueston J. Lessons in Dupuytren’s disease. Ann Chir Main Memb Super. 1992;11:349-354.
17. Tubiana R. Prognosis and treatment of Dupuytren’s contracture. J Bone and Joint Surg AM. 1955;37:1155-1168.
18. Howard JC, Varallo VM, Ross DC, et al. Elevated levels of ß-catenin and fibronectin in three-dimensional collagen cultures of Dupuytren’s disease cells are regulated by tension in vitro. BMC Musculoskelet Disord. 2003;4:16.
19. Ketchum LD, Donahue TK. The injection of nodules of Dupuytren’s disease with triamcinolone acetonide. J Hand Surg Am. 2000;25:1157-1162.
20. Betz N, Ott OJ, Adamietz B, et al. Radiotherapy in early-stage Dupuytren’s contracture. Long-term results after 13 years. Strahlenther Onkol. 2010;186:82-90.
21. Smith AC. Diagnosis and indications for surgical treatment. Hand Clin. 1991;7:635-642.
22. van Rijssen AL, ter Linden H, Werker PM. Five-year results of a randomized clinical trial on treatment in Dupuytren’s disease: percutaneous needle fasciotomy versus limited fasciectomy. Plast Reconstr Surg. 2012;129:469-477.
23. Hurst LC, Badalamente MA, Hentz VR, et al. Injectable collagenase clostridium histolyticum for Dupuytren’s contracture. N Engl J Med. 2009;361:968-979.
24. Chen NC, Srinivasan RC, Shauver MJ, et al. A systematic review of outcomes of fasciotomy, aponeurotomy, and collagenase treatments for Dupuytren’s contracture. Hand. 2011;6:250-255.
25. Peimer CA, Blazar P, Coleman S, et al. Dupuytren contracture recurrence following treatment with collagenase Clostridium histolyticum (CORDLESS [Collagenase Option for Reduction of Dupuytren Long-Term Evaluation of Safety Study]): 5-year data. J Hand Surg Am. 2015;40:1597-1605.
26. Toppi JT, Trompf L, Smoll NR, et al. Dupuytren’s contracture: an analysis of outcomes of percutaneous needle fasciotomy versus open fasciectomy. ANZ J Surg. 2015;85:639-643.
27. Cheung K, Walley KC, Rozental TD. Management of complications of Dupuytren contracture. Hand Clini. 2015;31:345-354.
PRACTICE RECOMMENDATIONS
› Evaluate patients with suspected Dupuytren’s disease (DD) for coexisting conditions such as Ledderhose disease and Peyronie’s disease. C
› Use the Hueston tabletop test to assess severity of DD. C
› Do not recommend stretching exercises for DD; they can hasten disease progression. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Chronic pain: How to approach these 3 common conditions
CASE 1 › Lola A is a 28-year-old woman with a history of muscular aches and joint pain throughout her body, fatigue, and mental fogginess. She has been seen by a rheumatologist and has been given a diagnosis of fibromyalgia, but just moved to your town and is establishing care. She is feeling desperate because her pain has worsened and the medication previously prescribed (gabapentin 300 mg tid) is no longer working. She asks to try oxycodone.
CASE 2 › Matt P is a 59-year-old truck driver with severe hip osteoarthritis (OA). His orthopedist recommended against hip replacement at this time because of his young age and a heart condition that makes him high risk. His pain makes sitting for long periods very difficult. He presents to you for help because he is worried he will be unable to continue working.
CASE 3 › Keith B is a 56-year-old construction worker who has been suffering from bouts of back pain for many years. The pain has become more debilitating over time; currently, it is constant, and Mr. B can hardly make it through his work day. He has been getting hydrocodone/acetaminophen from urgent care centers and emergency rooms, but he isn’t sure it is helping and is coming to you to assume his pain management.
Chronic pain (defined as pain >3 months in duration), is a complex, heterogeneous condition affecting an estimated 116 million US adults.1 Much of the management of chronic pain occurs in primary care settings, placing family physicians (FPs) on the front lines of 2 epidemics: that of chronic pain itself and that of the abuse and misuse of opioid pain medications.
In an effort to improve communication about the risks and benefits of opioid therapy and the safety and effectiveness of pain treatments in general, many professional organizations, health care institutions, and recently the Centers for Disease Control and Prevention,2 have published guidelines on the use of opioids for non-malignant chronic pain. With these guidelines in mind—and in light of the latest evidence—we propose the paradigm that follows for the treatment of chronic pain. A critical aspect of this paradigm is determining the pathophysiology underlying a patient’s pain in order to develop a well-rounded, multimodal, evidence-based treatment plan. Detailed here is the application of this approach to the treatment of 3 common chronic pain diagnoses: fibromyalgia, osteoarthritis, and low back pain.
Look to the central and peripheral nervous system
Acute pain begins with activation of peripheral nociceptors at the site of injury. This causes depolarization up the spinal cord and through the brain stem to higher cortical centers where the pain is perceived and localized. Descending neural pathways transport both excitatory and inhibitory information from the brain to the periphery via the spinal cord, which either increases or decreases the perception of pain.3
When damage/injury doesn’t correlate with the perception of pain
Until recently, it was assumed that chronic pain worked much the same way as acute pain and was caused by ongoing nociceptive input in the periphery, but research has shown us that the central nervous system can play a large role in the modulation of nociception. This new understanding comes from the lack of evidence pointing to any pain state in which the degree of nociceptive input correlates with the degree of pain experienced.
For most patients with chronic pain, regardless of their diagnosis, there is some degree of alteration in the processing of nociceptive signals by the central nervous system contributing to the experience of pain.4 This alteration is thought to be the result of peripheral nociceptive signaling persisting past the point of tissue healing, leading to a hypersensitivity of nerve fibers; the fibers then continue to respond to low, or even absent, sensory stimuli.
Central sensitization is the term used when this hypersensitivity develops in the superficial, deep, and ventral cord nerves. When this happens, pain is often accompanied by other systemic symptoms such as fatigue and slowed cognitive processing, often in the setting of little to no actual stimulation of the peripheral nociceptors.3 (For more on this, see “A new paradigm for pain?” J Fam Pract. 2016;65:598-605 or go to http://www.mdedge.com/jfponline/article/111257/pain/new-paradigm-pain.)
TABLE 14 lists the possible mechanisms of pain, which can be broken down into 4 categories: peripheral nociceptive (inflammatory or mechanical), peripheral neuropathic (underlying damage to a peripheral nerve), central (referring to when the central nervous system is the primary entity involved in maintaining the pain), or any combination of the 3.
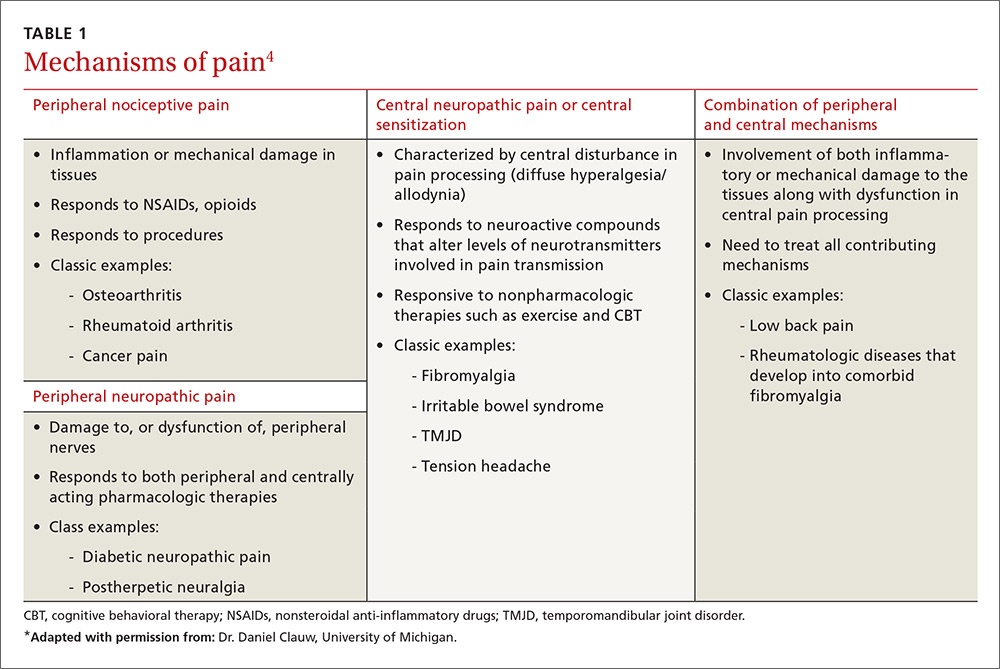
As pain becomes chronic, multiple mechanisms overlap
It is important to remember that for any single pain diagnosis, there is likely to be—at least initially—a principle underlying mechanism generating the pain. But as the pain becomes chronic, an overlap of multiple mechanisms develops, with central sensitization often playing a more dominant role than peripheral stimulation (regardless of the diagnosis).
For example, in a patient with rheumatoid arthritis (RA), peripheral nociceptive input (in the form of inflammation) is likely the initial mechanism at work, but as time goes on, central processing becomes more involved. The patient may then begin to experience pain that is disproportional to what is generally expected with RA and may develop other somatic symptoms. The diagnosis then becomes pain primarily related to RA with central sensitization, and both need to be addressed in a treatment plan. In rheumatic conditions, comorbid fibromyalgia (indicative of central sensitization) is thought to occur in 15% to 30% of patients.5
FPs can utilize the underlying mechanisms to cut across diagnostic labels and tailor treatments to those that are most likely to be effective. For a patient with more prominent peripheral involvement, a procedural intervention such as injections or surgery alone may suffice, whereas a broader approach including psychotherapy, medications, exercise, and other lifestyle interventions may be necessary for a patient with pain caused predominantly by central sensitization.
Addressing both peripheral and central components is essential. One prospective, observational cohort study of more than 600 patients scheduled for unilateral total knee or total hip arthroplasty found that those patients with a higher degree of centralization of pain (as measured by widespread pain index and modified fibromyalgia screening scales6) were less likely to report improvement in the affected body part and in overall body pain following the surgery.7
There is a high degree of overlap among many of the chronic pain syndromes (fibromyalgia, irritable bowel syndrome, interstitial cystitis, chronic headaches) that have been found to have a central sensitization component.8 Providers of primary care are aptly positioned to recognize central sensitization as the underlying pathology and target treatment effectively.
Tailor the treatment plan to the underlying mechanisms of pain
As with any chronic condition, a thorough work-up (complete with history, physical exam, and diagnostic testing, as appropriate) is indicated. In the setting of chronic pain, it’s important to identify both the primary mechanism, as well as secondary factors that may be contributing to the patient’s pain, before developing your treatment plan. These secondary factors may include co-occurring affect disorders,9 a history of trauma,10 poor sleep,11 and tobacco use,12 among others. A history of trauma, for example, co-exists with many pain syndromes. For these patients, central sensitization is responsible for much of their pain. As a result, traditional cognitive behavioral therapy (CBT) may not be the best option because of its focus on accepting pain as a chronic diagnosis; more trauma-focused treatments such as those dealing in emotional awareness and understanding of the central nervous system’s role in chronic pain need to be considered.13
3 common conditions. Below we present evidence-based treatment approaches for 3 conditions that are typically associated with each of the major mechanisms of chronic pain generation: fibromyalgia (a central sensitization cause), OA (a peripheral nociceptive cause), and low back pain (a mixed pain state).
Fibromyalgia: A case of central sensitization
Fibromyalgia is a hallmark diagnosis for those patients in whom central sensitization is the dominant cause of pain. These patients usually present with widespread, diffuse pain, as well as somatic symptoms such as fatigue, memory difficulties, and poor sleep quality.8 When explaining the pain mechanism (ie, central sensitization) to patients, it may be useful to use the analogy of a volume control dial that is stuck in the “high” position and can’t be turned down.
Genes, the environment, and neurotransmitters play a role. The origin of the pain amplification process is believed to be multifactorial.
- Genetic factors are thought to contribute to a predisposition for amplification. To date, 5 sets of genes have been implicated in increased sensitivity to pain leading to increased risk of the development of chronic pain during a patient’s lifetime.14-19
- Environmental factors (eg, early life trauma, physical trauma especially to the trunk, certain infections such as Lyme disease and Epstein-Barr virus, and emotional stress) may trigger or exacerbate symptoms.8 Of note: Only about 5% to 10% of people who experience these triggers actually develop a chronic pain state, while the rest regain their baseline health.4 This raises the question of whether there is a point during an acute pain episode in which one can intervene and prevent the acute pain from becoming chronic in those at higher risk.4
- Imbalances of neurotransmitters (high glutamate;20 low norepinephrine, serotonin,21 and gamma-aminobutyric acid [GABA]22) play a role in central amplification. These substances not only affect sensory transmission, but also control levels of alertness, sleep, mood, and memory.
The diagnostic criteria for fibromyalgia were modified in 2011 to remove the tender point examination and to add in somatic symptoms.6 These criteria can be useful in the clinical setting in identifying not only fibromyalgia itself but also the degree of “fibromyalgianess” a patient has, which is an indicator of how large a role the centralization process plays in the maintenance of chronic pain.23,24
Treatment: Multimodal and patient empowering. Evidence-based treatment options for fibromyalgia, as well as other conditions for which there is a high degree of centralized pain, can be found in TABLE 2.25-36 Multimodal treatment, with an emphasis on patient knowledge and empowerment, is generally thought to be the most beneficial.25,37 Treatment should almost always include CBT and exercise/activity therapies,26,29 which have high degrees of efficacy with few adverse effects.
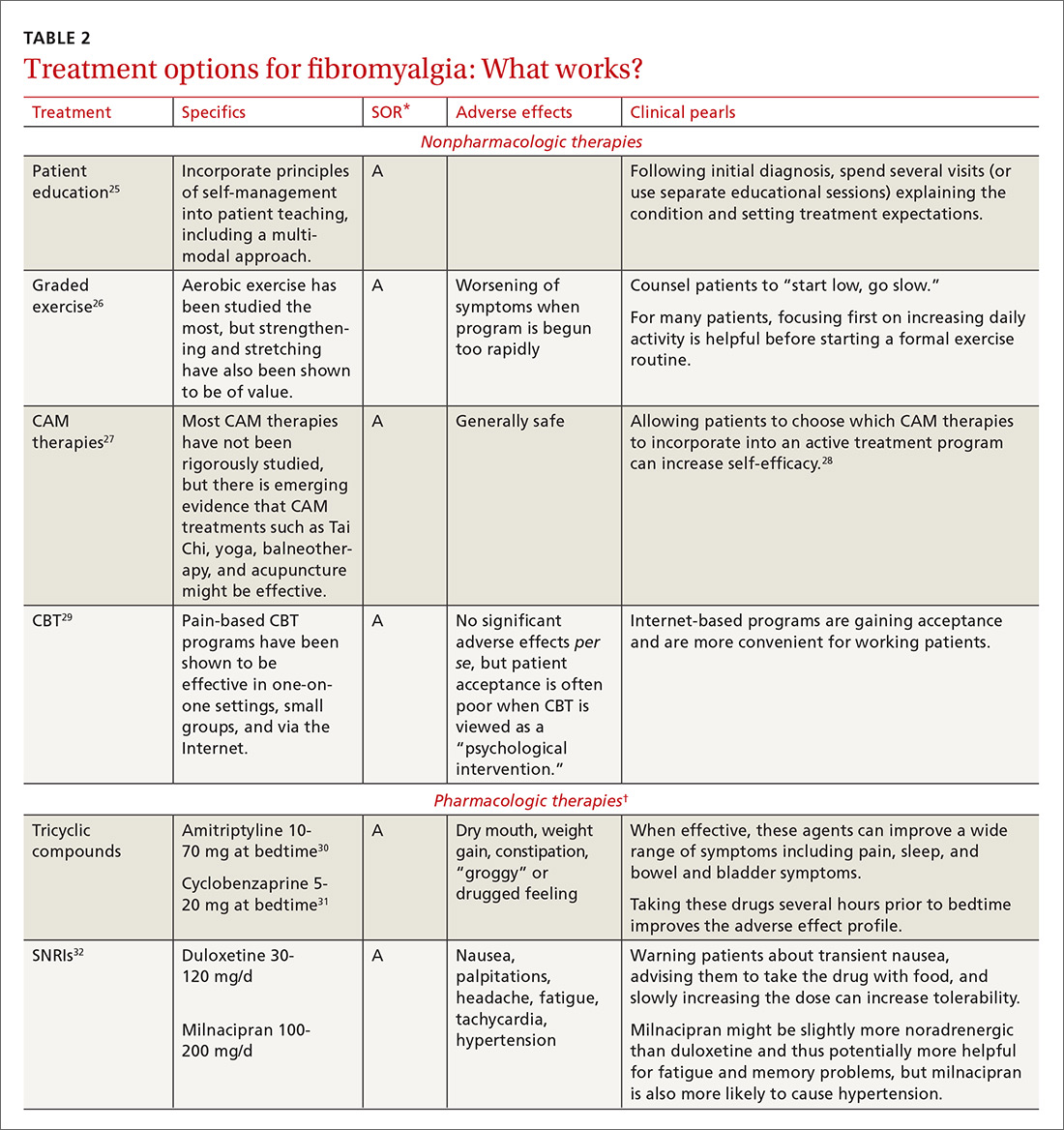

In terms of medication, centrally-acting agents (tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors [SNRIs], and alpha 2 delta ligands) are the most effective. There is little to no data showing benefit from anti-inflammatories or opioids in the setting of fibromyalgia. There is some data to suggest that combination therapy, for example with an SNRI (milnacipran) and an alpha 2 delta ligand (pregabalin), may provide more benefit than treating with pregabalin alone.38
Complementary and alternative therapies (eg, yoga, chiropractic care, acupuncture, massage) are being studied more, and while evidence is only preliminary in terms of efficacy, there is increasing emphasis being placed on the need for patients with chronic pain to shift their treatment expectations to greater acceptance of pain and the need for ongoing self-care.28 (For more advice on managing fibromyalgia, see the related videos at http://bit.ly/2lPEt0f and http://bit.ly/2lmjEcn.)
Osteoarthritis: An example of peripheral nociceptive pain
OA is a condition long thought to be characterized by damage to the cartilage and bone; however, as with many other pain diagnoses, there is frequently little correlation between damage seen on radiographs and the amount of pain that patients experience.
One study analyzed data on almost 7000 patients from the National Health and Nutrition Examination Survey (NHANES I) and found that between 30% and 50% of OA patients with moderate to severe radiographic changes were asymptomatic, and 10% of those with moderate to severe pain had normal radiographs or only mild changes.39 Research is showing that many factors may contribute to this discrepancy, including the typical “wear and tear” of the disease, subacute levels of inflammation that can lead to peripheral sensitization,40 and, in some patients, a centralized pain component. The patients with more centralized pain often have pain that is disproportionate to radiographic evidence, as well as more somatic symptoms such as fatigue, sleep disturbance, and memory issues.41
Treatment should be multimodal and include interventions targeted at halting the progression of damage as well as palliation of pain. All treatment plans for OA should also include exercise, weight reduction, and self-management, in addition to pharmacologic interventions, to reduce both the micro-inflammation and the centralized pain component (when present). Intra-articular injections of various types have been studied with some having more efficacy in pain reduction and functional improvement than others.42-45 See TABLE 342-61 for a summary of evidence-based treatment options.
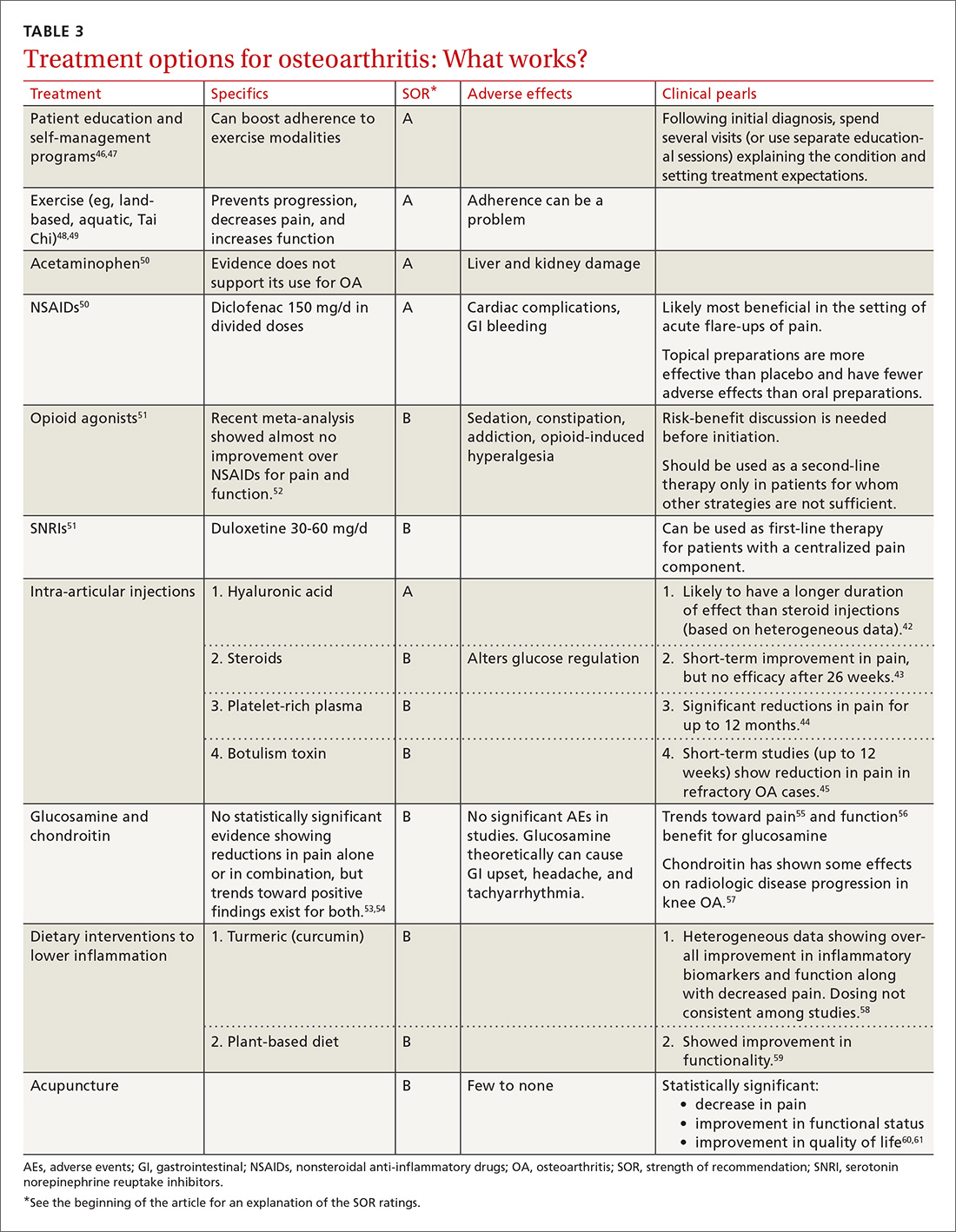
Low back pain—a mixed pain state
Low back pain (LBP) has been recognized as a mixed pain state for quite some time. While some patients may experience purely nociceptive and/or neuropathic pain, most cases are nonspecific with patients experiencing varying degrees of nociceptive (myofascial low back pain), neuropathic (lumbar radiculopathy), and central sensitization pain.62,63 Evidence for centralized pain is demonstrated in studies showing hyperalgesia,64 augmented central pain processing,65 involvement of the emotional brain,66 and delayed recovery influenced by poor coping strategies.67
When developing a treatment plan for a patient with chronic low back pain, remember that the pain derives from a complex combination of pathophysiologic contributors. Identifying where a patient lies on the pain centralization spectrum can help you tailor treatment.
In one study of 548 patients presenting to a tertiary pain clinic with primary spine pain diagnoses, 42% met diagnostic criteria for fibromyalgia.68 Compared to criteria-negative patients, these patients tended to be younger, unemployed, and receiving compensation; they had greater pain intensity, pain interference, and used stronger words to describe their neuropathic pain; they also had higher levels of depression/anxiety and a lower level of physical function.
Because low back pain is a condition with high prevalence and associated disability, many clinical boards have created guidelines for management. These guidelines tend to vary in the strength of evidence used, and the extent to which they are followed in clinical practice remains largely unknown. Recommendations frequently discourage the use of ultrasound/electrotherapy, but many encourage short-term use of medications (see “How effective are opioids for chronic low back pain?” J Fam Pract. 2015;64:584-584), supervised exercise therapy, CBT, and multidisciplinary treatment.
Guidelines tend to differ most widely with regard to recommendations for spinal manipulation and specific drug therapies.69 The classes of drugs that may be most useful when centralized pain is present include the SNRIs and the alpha 2 delta calcium channel ligands.4 See TABLE 470-89 for a summary of evidence-based treatment options.
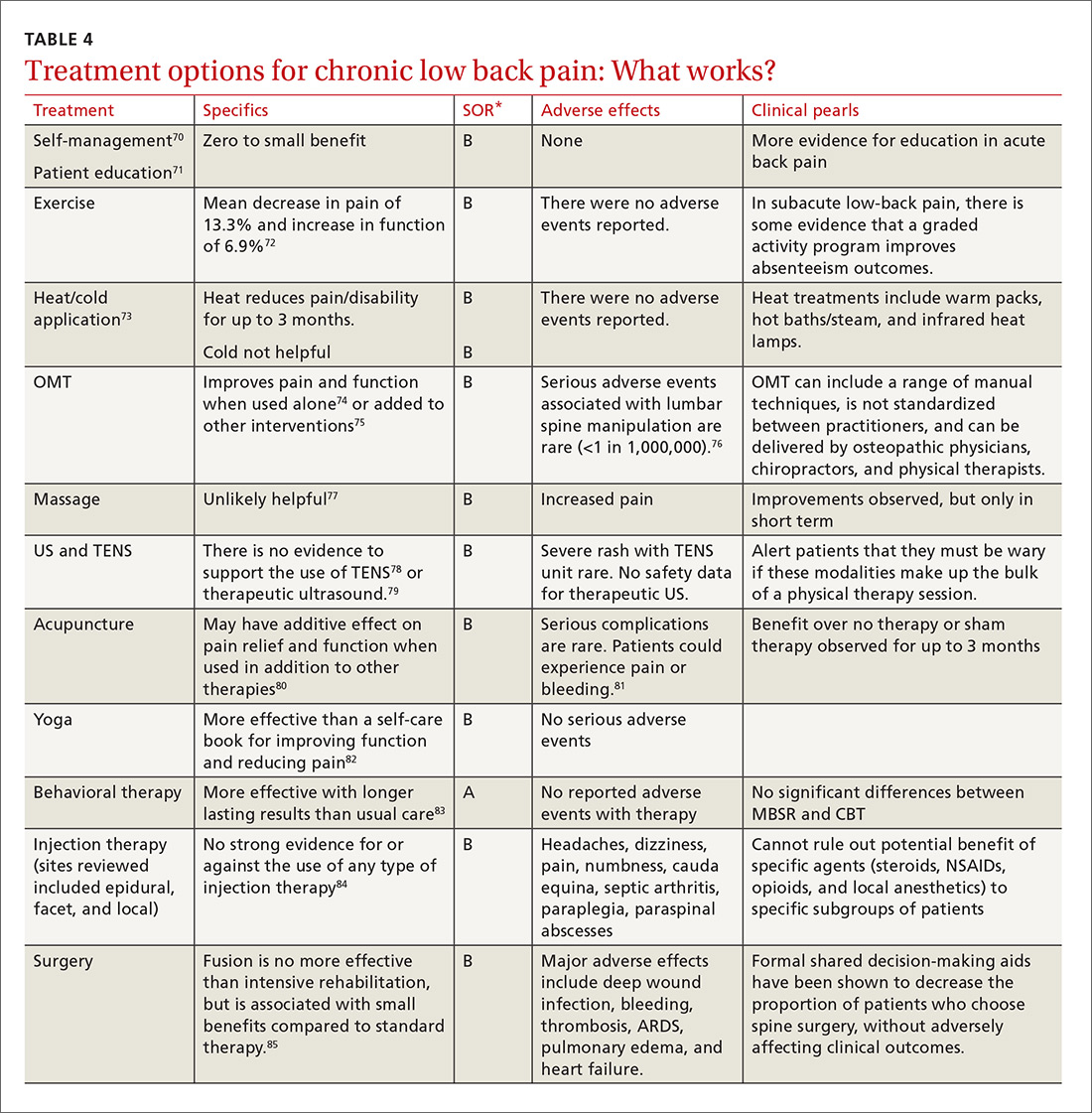
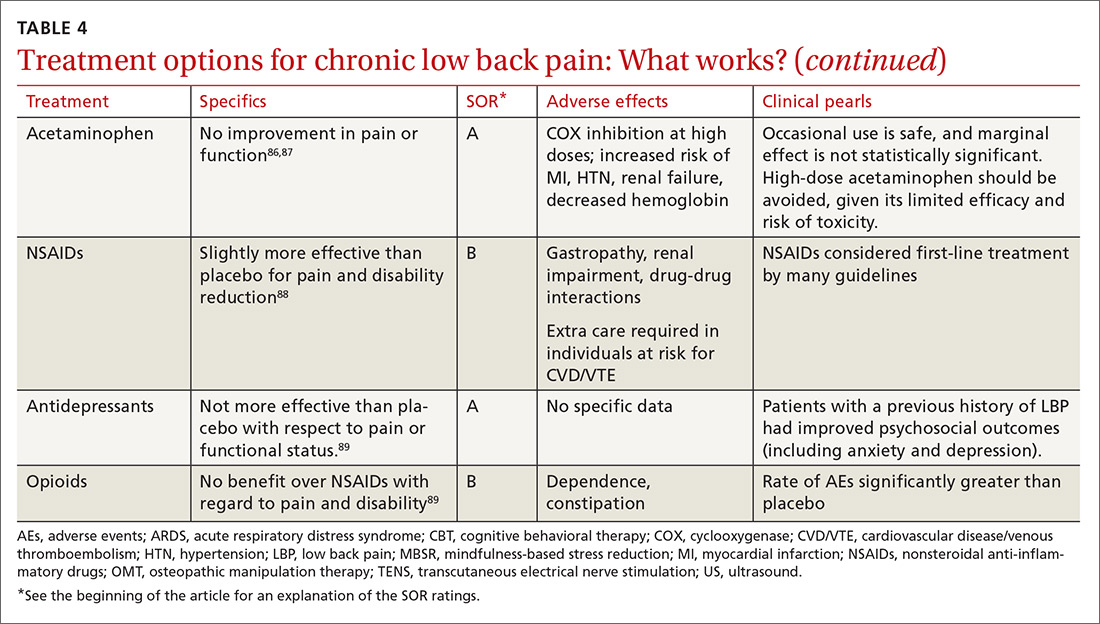
CASE 1 › Ms. A is started on amitriptyline 25 mg at bedtime, which improves her fatigue and cognitive symptoms. During monthly office visits, her FP educates her about the pathophysiology of fibromyalgia and uses motivational interviewing to get her slowly moving and increasing her activity level. She is weaned off the gabapentin previously prescribed, as her symptoms stabilize and improve.
CASE 2 › Mr. P is sent for a steroid injection, which decreases his pain temporarily. During this time, he begins physical therapy; slowly, with increased movement, his function improves. A trial of duloxetine provides pain relief; that combined with intermittent nonsteroidal anti-inflammatory drugs (NSAIDs) has allowed Mr. P to maintain his function and his job.
CASE 3 › Because Mr. B was only taking the narcotics intermittently and wasn’t certain they were helping, CBT was sufficient to wean Mr. B off the medication without any worsening of his pain in the process. By participating in physical therapy, he has learned how to perform certain tasks at his job without pain or injury. He uses NSAIDs as needed for pain.
CORRESPONDENCE
Jill Schneiderhan, MD, 24 Frank Lloyd Wright Dr., Lobby H, Suite 2300, Ann Arbor, MI 48105; [email protected].
ACKNOWLEDGEMENTS
We thank Drs. Daniel Clauw (University of Michigan, Ann Arbor) and Martha Rumschlag (Providence Family Medicine Residency Program, Southfield, Mich), for their valuable contributions to this article.
1. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington (DC): National Academies Press (US); 2011.
2. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recomm Rep. 2016;65:1-49.
3. Aronoff GM. What do we know about the pathophysiology of chronic pain? Implications for treatment considerations. Med Clin North Am. 2016;100:31-42.
4. Clauw DJ. Diagnosing and treating chronic musculoskeletal pain based on the underlying mechanism(s). Best Pract Res Clin Rheumatol. 2015;29:6-19.
5. Clauw DJ, Katz P. The overlap between fibromyalgia and inflammatory rheumatic disease: when and why does it occur? J Clin Rheumatol. 1995;1:335-342.
6. Wolfe F, Clauw DJ, Fitzcharles MA, et al. Fibromyalgia criteria and severity scales for clinical and epidemiological studies: a modification of the ACR Preliminary Diagnostic Criteria for Fibromyalgia. J Rheumatol. 2011;38:1113-1122.
7. Brummett CM, Urquhart AG, Hassett AL, et al. Characteristics of fibromyalgia independently predict poorer long-term analgesic outcomes following total knee and hip arthroplasty. Arthritis Rheumatol. 2015;67:1386-1394.
8. Ablin K, Clauw DJ. From fibrositis to functional somatic syndromes to a bell-shaped curve of pain and sensory sensitivity: evolution of a clinical construct. Rheum Dis Clin North Am. 2009;35:233-251.
9. Giesecke T, Gracely RH, Williams DA, et al. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005;52:1577-1584.
10. Tesarz J, Eich W, Treede RD, et al. Altered pressure pain thresholds and increased wind-up in adult chronic back pain patients with a history of childhood maltreatment: a quantitative sensory testing study. Pain. 2016;157:1799-1809.
11. Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain. 2013;14:1539-1552.
12. Shi Y, Weingarten TN, Mantilla CB, et al. Smoking and pain: pathophysiology and clinical implications. Anesthesiology. 2010;113:977-992.
13. Burger AJ, Lumley MA, Carty JN, et al. The effects of a novel psychological attribution and emotional awareness and expression therapy for chronic musculoskeletal pain: a preliminary, uncontrolled trial. J Psychosom Res. 2016;81:1-8.
14. Zubieta JK, Heitzeg MM, Smith YR, et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240-1243.
15. van Meurs JB, Uitterlinden AG, Stolk L, et al. A functional polymorphism in the catechol-O-methyltransferase gene is associated with osteoarthritis-related pain. Arthritis Rheum. 2009;60:628-629.
16. McLean SA, Diatchenko L, Lee YM, et al. Catechol O-methyltransferase haplotype predicts immediate musculoskeletal neck pain and psychological symptoms after motor vehicle collision. J Pain. 2011;12:101-107.
17. Costigan M, Belfer I, Griffin RS, et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain. 2010;133:2519-2527.
18. Tegeder I, Costigan M, Griffin RS, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269-1277.
19. Amaya F, Wang H, Costigan M, et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852-12860.
20. Harris RE, Napadow V, Huggins JP, et al. Pregabalin rectifies abberrant brain chemistry, connectivity, and functional responses in chronic pain patients. Anesthesiology. 2013;119:1453-1464.
21. Russell IJ, Vaeroy H, Javors M, et al. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35:550-556.
22. Foerster BR, Petrou M, Edden RAE, et al. Reduced insular gamma-aminobutyric acid in fibromyalgia. Arthritis Rheum. 2012;64:579-583.
23. Clauw DJ. Fibromyalgia: a clinical review. JAMA. 2014;311:1547-1555.
26. Hauser W, Klose P, Langhorst J, et al. Efficacy of different types of aerobic exercise in fibromyalgia syndrome: a systematic review and meta-analysis of randomised controlled trials. Arthritis Res Ther. 2010;12:R79.
27. Porter NS, Jason LA, Boulton A, et al. Alternative medical interventions used in the treatment and management of myalgic encephalomyelitis/chronic fatigue syndrome and fibromyalgia. J Altern Complement Med. 2010;16:235-249.
28. Eaves ER, Sherman KJ, Ritenbaugh C, et al. A qualitative study of changes in expectations over time among patients with chronic low back pain seeking four CAM therapies. BMC Complement Altern Med. 2015;15:12.
29. Bernardy K, Fuber N, Kollner V, et al. Efficacy of cognitive-behavioral therapies in fibromyalgia syndrome: a systematic review and metaanalysis of randomized controlled trials. J Rheumatol. 2010;37:1991-2005.
30. Arnold LM, Keck PE Jr, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41:104-113.
31. Moldofsky H, Harris HW, Archambault WT, et al. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: a double-blind randomized placebo-controlled study. J Rheumatol. 2011;38:2653-2663.
32. Arnold LM. Duloxetine and other antidepressants in the treatment of patients with fibromyalgia. Pain Med. 2007;Sep 8 Suppl 2:S63-S74.
33. Häuser W, Bernardy K, Uceyler N, et al. Treatment of fibromyalgia syndrome with gabapentin and pregabalin—a meta-analysis of randomized controlled trials. Pain. 2009;145:69-81.
34. Gaskell H, Moore RA, Derry S, et al. Oxycodone for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2014;Jun 23:CD010692.
35. MacLean AJ, Schwartz TL. Tramadol for the treatment of fibromyalgia. Expert Rev Neurother. 2015;15:469-475.
36. Younger J, Noor N, McCue R, et al. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 2013;65:529-538.
37. Camerini L, Schulz PJ, Nakamoto K. Differential effects of health knowledge and health empowerment over patients’ self-management and health outcomes: a cross-sectional evaluation. Patient Educ Couns. 2012;89:337-344.
38. Mease PJ, Farmer MV, Palmer RH, et al. Milnacipran combined with pregabalin in fibromyalgia: a randomized, open-label study evaluating the safety and efficacy of adding milnacipran in patients with incomplete response to pregabalin. Ther Adv Musculoskeletal Dis. 2013;5:113-126.
39. Hannan MT, Felson DT, Pincus T. Analysis of the discordance between radiographic changes and knee pain in osteoarthritis of the knee. J Rheumatol. 2000;27:1513-1517.
40. Daghestani HN, Kraus VB. Inflammatory biomarkers in osteoarthritis. Osteoarthritis Cartilage. 2015;23:1890-1896.
41. Fingleton C, Smart K, Moloney N, et al. Pain sensitization in people with knee osteoarthritis: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2015;23:1043-1056.
42. Strand V, McIntyre LF, Beach WR, et al. Safety and efficacy of US-approved viscosupplements for knee osteoarthritis: a systematic review and meta-analysis of randomized, saline-controlled trials. J Pain Res. 2015;8:217-228.
43. Jüni P, Hari R, Rutjes AW, et al. Intra-articular corticosteroid for knee osteoarthritis. Cochrane Database Syst Rev. 2015:CD005328.
44. Meheux CJ, McCulloch PC, Lintner DM, et al. Efficacy of intra-articular platelet-rich plasma injections in knee osteoarthritis: a systematic review. Arthroscopy. 2016;32:495-505.
45. Wu T, Song HX, Dong Y, et al. Intra-articular injections of botulinum toxin a for refractory joint pain: a systematic review and meta-analysis. Clin Rehabil. 2016.
46. Jordan JL, Holden MA, Mason EE, et al. Interventions to improve adherence to exercise for chronic musculoskeletal pain in adults. Cochrane Database Syst Rev. 2010:CD005956.
48. Fransen M, McConnell S, Hernandez-Molina G, et al. Exercise for osteoarthritis of the hip. Cochrane Database Syst Rev. 2014:CD007912.
49. Bartels EM, Juhl CB, Christensen R, et al. Aquatic exercise for the treatment of knee and hip osteoarthritis. Cochrane Database Syst Rev. 2016;3:CD005523.
50. da Costa BR, Reichenbach S, Keller N, et al. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet. 2016;387:2093-2105.
51. Myers J, Wielage RC, Han B, et al. The efficacy of duloxetine, non-steroidal anti-inflammatory drugs, and opioids in osteoarthritis: a systematic literature review and meta-analysis. BMC Musculoskelet Disord. 2014;15:76.
52. Berthelot JM, Darrieutort-Lafitte C, Le Goff B, et al. Strong opioids for noncancer pain due to musculoskeletal diseases: not more effective than acetaminophen or NSAIDs. Joint Bone Spine. 2015;82:397-401.
53. Clegg DO, Reda DJ, Harris CL, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med. 2006;354:795-808.
54. Wandel S, Jüni P, Tendal B, et al. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: network meta-analysis. BMJ. 2010;341:c4675.
55. Sawitzke AD, Shi H, Finco MF, et al. Clinical efficacy and safety of glucosamine, chondroitin sulphate, their combination, celecoxib or placebo taken to treat osteoarthritis of the knee: 2-year results from GAIT. Ann Rheum Dis. 2010;69:1459-1464.
56. Wu D, Huang Y, Gu Y, et al. Efficacies of different preparations of glucosamine for the treatment of osteoarthritis: a meta-analysis of randomised, double-blind, placebo-controlled trials. Int J Clin Pract. 2013;67:585-594.
57. Kahan A, Uebelhart D, De Vathaire F, et al. Long-term effects of chondroitins 4 and 6 sulfate on knee osteoarthritis: the study on osteoarthritis progression prevention, a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2009;60:524-533.
58. Perkins K, Sahy W, Beckett RD. Efficacy of curcuma for treatment of osteoarthritis. J Evid Based Complementary Altern Med. 2017;22:156-165.
59. Clinton CM, O’Brien S, Law J, et al. Whole-foods, plant-based diet alleviates the symptoms of osteoarthritis. Arthritis. 2015;2015:708152.
60. Manyanga T, Froese M, Zarychanski R, et al. Pain management with acupuncture in osteoarthritis: a systematic review and meta-analysis. BMC Complement Altern Med. 2014;14:312.
61. Vickers AJ, Cronin AM, Maschino AC, et al. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172:1444-1453.
62. Nijs J, Apeldoorn A, Hallegraeff H, et al. Low back pain: guidelines for the clinical classification of predominant neuropathic, nociceptive, or central sensitization pain. Pain Physician. 2015;18:E333-E346.
63. Fishbain DA, Cole B, Lewis JE, et al. What is the evidence that neuropathic pain is present in chronic low back pain and soft tissue syndromes? An evidence-based structured review. Pain Med. 2014;15:4-15.
64. Hübscher M, Moloney N, Rebbeck T, et al. Contributions of mood, pain catastrophizing, and cold hyperalgesia in acute and chronic low back pain: a comparison with pain-free controls. Clin J Pain. 2014;30:886-893.
65. Giesecke T, Gracely RH, Grant MA, et al. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004;50:613-623.
66. Baliki MN, Chialvo DR, Geha PY, et al. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci. 2006;26:12165-12173.
68. Brummett CM, Goesling J, Tsodikov A, et al. Prevalence of the fibromyalgia phenotype in patients with spine pain presenting to a tertiary care pain clinic and the potential treatment implications. Arthritis Rheum. 2013;65:3285-3292.
69. Koes BW, van Tulder M, Lin CW, et al. An updated overview of clinical guidelines for the management of non-specific low back pain in primary care. Eur Spine J. 2010;19:2075-2094.
70. Oliveira VC, Ferreira PH, Maher CG, et al. Effectiveness of self-management of low back pain: systematic review with meta-analysis. Arthritis Care Res. 2012;64:1739-1748.
71. Engers A, Jellema P, Wensing M, et al. Individual patient education for low back pain. Cochrane Database Syst Rev. 2008:CD004057.
72. Hayden JA, van Tulder MW, Malmivaara A, et al. Exercise therapy for treatment of non-specific low back pain. Cochrane Database Syst Rev. 2005:CD000335.
73. French SD, Cameron M, Walker BF, et al. Superficial heat or cold for low back pain. Cochrane Database Syst Rev. 2006:CD004750.
74. Franke H, Franke JD, Fryer G. Osteopathic manipulative treatment for nonspecific low back pain: a systematic review and meta-analysis. BMC Musculoskeletal Disord. 2014;15:286.
75. Franke H, Fryer G, Ostelo RW, et al. Muscle energy technique for non-specific low back pain. Cochrane Database Syst Rev. 2015:CD009852.
76. Oliphant D. Safety of spinal manipulation in the treatment of lumbar disk herniations: a systematic review and risk assessment. J Manipulative Physiol Ther. 2004:197-210.
77. Furlan AD, Giraldo M, Baskwill A, et al. Massage for low-back pain. Cochrane Database Syst Rev. 2015:CD001929.
78. Khadilkar A, Odebiyi DO, Brosseau L, et al. Transcutaneous electrical nerve stimulation (TENS) versus placebo for chronic low back pain. Cochrane Database Syst Rev. 2008:CD003008.
79. Ebadi S, Henschke N, Nakhostin Ansari N, et al. Therapeutic ultrasound for chronic low back pain. Cochrane Database Syst Rev. 2014:CD009169.
80. Furlan AD, van Tulder MW, Cherkin DC, et al. Acupuncture and dry-needling for low back pain. Cochrane Database Syst Rev. 2005:CD001351.
81. Chou R, Huffman LH. Nonpharmacologic therapies for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med. 2007;147:492-504.
82. Sherman KJ, Cherkin DC, Erro J, et al. Comparing yoga, exercise, and a self-care book for chronic low back pain: a randomized, controlled trial. Ann Intern Med. 2005;143:849-856.
83. Cherkin DC, Sherman KJ, Balderson BH, et al. Effect of mindfulness-based stress reduction vs cognitive behavioral therapy or usual care on back pain and functional limitations in adults with chronic low back pain: a randomized clinical trial. JAMA. 2016;315:1240-1249.
84. Staal JB, de Bie R, de Vet HC, et al. Injection therapy for subacute and chronic low back pain. Cochrane Database Syst Rev. 2008:CD001824.
85. Chou R, Baisden J, Carragee EJ, et al. Surgery for low back pain: a review of the evidence for an American Pain Society Clinical Practice Guideline. Spine. 2009;34:1094-1109.
86. Felson D. Paracetamol is ineffective for spinal pain and knee and hip osteoarthritis. Evid Based Med. 2015;20:205.
87. Machado GC, Maher CG, Ferreira PH, et al. Efficacy and safety of paracetamol for spinal pain and osteoarthritis: systematic review and meta-analysis of randomised placebo controlled trials. BMJ. 2015;350:h1225.
88. Enthoven WT, Roelofs PD, Deyo RA, et al. Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst Rev. 2016;2:CD012087.
89. White AP, Arnold PM, Norvell DC, et al. Pharmacologic management of chronic low back pain: synthesis of the evidence. Spine (Phila Pa 1976). 2011;36:S131-S143.
CASE 1 › Lola A is a 28-year-old woman with a history of muscular aches and joint pain throughout her body, fatigue, and mental fogginess. She has been seen by a rheumatologist and has been given a diagnosis of fibromyalgia, but just moved to your town and is establishing care. She is feeling desperate because her pain has worsened and the medication previously prescribed (gabapentin 300 mg tid) is no longer working. She asks to try oxycodone.
CASE 2 › Matt P is a 59-year-old truck driver with severe hip osteoarthritis (OA). His orthopedist recommended against hip replacement at this time because of his young age and a heart condition that makes him high risk. His pain makes sitting for long periods very difficult. He presents to you for help because he is worried he will be unable to continue working.
CASE 3 › Keith B is a 56-year-old construction worker who has been suffering from bouts of back pain for many years. The pain has become more debilitating over time; currently, it is constant, and Mr. B can hardly make it through his work day. He has been getting hydrocodone/acetaminophen from urgent care centers and emergency rooms, but he isn’t sure it is helping and is coming to you to assume his pain management.
Chronic pain (defined as pain >3 months in duration), is a complex, heterogeneous condition affecting an estimated 116 million US adults.1 Much of the management of chronic pain occurs in primary care settings, placing family physicians (FPs) on the front lines of 2 epidemics: that of chronic pain itself and that of the abuse and misuse of opioid pain medications.
In an effort to improve communication about the risks and benefits of opioid therapy and the safety and effectiveness of pain treatments in general, many professional organizations, health care institutions, and recently the Centers for Disease Control and Prevention,2 have published guidelines on the use of opioids for non-malignant chronic pain. With these guidelines in mind—and in light of the latest evidence—we propose the paradigm that follows for the treatment of chronic pain. A critical aspect of this paradigm is determining the pathophysiology underlying a patient’s pain in order to develop a well-rounded, multimodal, evidence-based treatment plan. Detailed here is the application of this approach to the treatment of 3 common chronic pain diagnoses: fibromyalgia, osteoarthritis, and low back pain.
Look to the central and peripheral nervous system
Acute pain begins with activation of peripheral nociceptors at the site of injury. This causes depolarization up the spinal cord and through the brain stem to higher cortical centers where the pain is perceived and localized. Descending neural pathways transport both excitatory and inhibitory information from the brain to the periphery via the spinal cord, which either increases or decreases the perception of pain.3
When damage/injury doesn’t correlate with the perception of pain
Until recently, it was assumed that chronic pain worked much the same way as acute pain and was caused by ongoing nociceptive input in the periphery, but research has shown us that the central nervous system can play a large role in the modulation of nociception. This new understanding comes from the lack of evidence pointing to any pain state in which the degree of nociceptive input correlates with the degree of pain experienced.
For most patients with chronic pain, regardless of their diagnosis, there is some degree of alteration in the processing of nociceptive signals by the central nervous system contributing to the experience of pain.4 This alteration is thought to be the result of peripheral nociceptive signaling persisting past the point of tissue healing, leading to a hypersensitivity of nerve fibers; the fibers then continue to respond to low, or even absent, sensory stimuli.
Central sensitization is the term used when this hypersensitivity develops in the superficial, deep, and ventral cord nerves. When this happens, pain is often accompanied by other systemic symptoms such as fatigue and slowed cognitive processing, often in the setting of little to no actual stimulation of the peripheral nociceptors.3 (For more on this, see “A new paradigm for pain?” J Fam Pract. 2016;65:598-605 or go to http://www.mdedge.com/jfponline/article/111257/pain/new-paradigm-pain.)
TABLE 14 lists the possible mechanisms of pain, which can be broken down into 4 categories: peripheral nociceptive (inflammatory or mechanical), peripheral neuropathic (underlying damage to a peripheral nerve), central (referring to when the central nervous system is the primary entity involved in maintaining the pain), or any combination of the 3.
As pain becomes chronic, multiple mechanisms overlap
It is important to remember that for any single pain diagnosis, there is likely to be—at least initially—a principle underlying mechanism generating the pain. But as the pain becomes chronic, an overlap of multiple mechanisms develops, with central sensitization often playing a more dominant role than peripheral stimulation (regardless of the diagnosis).
For example, in a patient with rheumatoid arthritis (RA), peripheral nociceptive input (in the form of inflammation) is likely the initial mechanism at work, but as time goes on, central processing becomes more involved. The patient may then begin to experience pain that is disproportional to what is generally expected with RA and may develop other somatic symptoms. The diagnosis then becomes pain primarily related to RA with central sensitization, and both need to be addressed in a treatment plan. In rheumatic conditions, comorbid fibromyalgia (indicative of central sensitization) is thought to occur in 15% to 30% of patients.5
FPs can utilize the underlying mechanisms to cut across diagnostic labels and tailor treatments to those that are most likely to be effective. For a patient with more prominent peripheral involvement, a procedural intervention such as injections or surgery alone may suffice, whereas a broader approach including psychotherapy, medications, exercise, and other lifestyle interventions may be necessary for a patient with pain caused predominantly by central sensitization.
Addressing both peripheral and central components is essential. One prospective, observational cohort study of more than 600 patients scheduled for unilateral total knee or total hip arthroplasty found that those patients with a higher degree of centralization of pain (as measured by widespread pain index and modified fibromyalgia screening scales6) were less likely to report improvement in the affected body part and in overall body pain following the surgery.7
There is a high degree of overlap among many of the chronic pain syndromes (fibromyalgia, irritable bowel syndrome, interstitial cystitis, chronic headaches) that have been found to have a central sensitization component.8 Providers of primary care are aptly positioned to recognize central sensitization as the underlying pathology and target treatment effectively.
Tailor the treatment plan to the underlying mechanisms of pain
As with any chronic condition, a thorough work-up (complete with history, physical exam, and diagnostic testing, as appropriate) is indicated. In the setting of chronic pain, it’s important to identify both the primary mechanism, as well as secondary factors that may be contributing to the patient’s pain, before developing your treatment plan. These secondary factors may include co-occurring affect disorders,9 a history of trauma,10 poor sleep,11 and tobacco use,12 among others. A history of trauma, for example, co-exists with many pain syndromes. For these patients, central sensitization is responsible for much of their pain. As a result, traditional cognitive behavioral therapy (CBT) may not be the best option because of its focus on accepting pain as a chronic diagnosis; more trauma-focused treatments such as those dealing in emotional awareness and understanding of the central nervous system’s role in chronic pain need to be considered.13
3 common conditions. Below we present evidence-based treatment approaches for 3 conditions that are typically associated with each of the major mechanisms of chronic pain generation: fibromyalgia (a central sensitization cause), OA (a peripheral nociceptive cause), and low back pain (a mixed pain state).
Fibromyalgia: A case of central sensitization
Fibromyalgia is a hallmark diagnosis for those patients in whom central sensitization is the dominant cause of pain. These patients usually present with widespread, diffuse pain, as well as somatic symptoms such as fatigue, memory difficulties, and poor sleep quality.8 When explaining the pain mechanism (ie, central sensitization) to patients, it may be useful to use the analogy of a volume control dial that is stuck in the “high” position and can’t be turned down.
Genes, the environment, and neurotransmitters play a role. The origin of the pain amplification process is believed to be multifactorial.
- Genetic factors are thought to contribute to a predisposition for amplification. To date, 5 sets of genes have been implicated in increased sensitivity to pain leading to increased risk of the development of chronic pain during a patient’s lifetime.14-19
- Environmental factors (eg, early life trauma, physical trauma especially to the trunk, certain infections such as Lyme disease and Epstein-Barr virus, and emotional stress) may trigger or exacerbate symptoms.8 Of note: Only about 5% to 10% of people who experience these triggers actually develop a chronic pain state, while the rest regain their baseline health.4 This raises the question of whether there is a point during an acute pain episode in which one can intervene and prevent the acute pain from becoming chronic in those at higher risk.4
- Imbalances of neurotransmitters (high glutamate;20 low norepinephrine, serotonin,21 and gamma-aminobutyric acid [GABA]22) play a role in central amplification. These substances not only affect sensory transmission, but also control levels of alertness, sleep, mood, and memory.
The diagnostic criteria for fibromyalgia were modified in 2011 to remove the tender point examination and to add in somatic symptoms.6 These criteria can be useful in the clinical setting in identifying not only fibromyalgia itself but also the degree of “fibromyalgianess” a patient has, which is an indicator of how large a role the centralization process plays in the maintenance of chronic pain.23,24
Treatment: Multimodal and patient empowering. Evidence-based treatment options for fibromyalgia, as well as other conditions for which there is a high degree of centralized pain, can be found in TABLE 2.25-36 Multimodal treatment, with an emphasis on patient knowledge and empowerment, is generally thought to be the most beneficial.25,37 Treatment should almost always include CBT and exercise/activity therapies,26,29 which have high degrees of efficacy with few adverse effects.
In terms of medication, centrally-acting agents (tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors [SNRIs], and alpha 2 delta ligands) are the most effective. There is little to no data showing benefit from anti-inflammatories or opioids in the setting of fibromyalgia. There is some data to suggest that combination therapy, for example with an SNRI (milnacipran) and an alpha 2 delta ligand (pregabalin), may provide more benefit than treating with pregabalin alone.38
Complementary and alternative therapies (eg, yoga, chiropractic care, acupuncture, massage) are being studied more, and while evidence is only preliminary in terms of efficacy, there is increasing emphasis being placed on the need for patients with chronic pain to shift their treatment expectations to greater acceptance of pain and the need for ongoing self-care.28 (For more advice on managing fibromyalgia, see the related videos at http://bit.ly/2lPEt0f and http://bit.ly/2lmjEcn.)
Osteoarthritis: An example of peripheral nociceptive pain
OA is a condition long thought to be characterized by damage to the cartilage and bone; however, as with many other pain diagnoses, there is frequently little correlation between damage seen on radiographs and the amount of pain that patients experience.
One study analyzed data on almost 7000 patients from the National Health and Nutrition Examination Survey (NHANES I) and found that between 30% and 50% of OA patients with moderate to severe radiographic changes were asymptomatic, and 10% of those with moderate to severe pain had normal radiographs or only mild changes.39 Research is showing that many factors may contribute to this discrepancy, including the typical “wear and tear” of the disease, subacute levels of inflammation that can lead to peripheral sensitization,40 and, in some patients, a centralized pain component. The patients with more centralized pain often have pain that is disproportionate to radiographic evidence, as well as more somatic symptoms such as fatigue, sleep disturbance, and memory issues.41
Treatment should be multimodal and include interventions targeted at halting the progression of damage as well as palliation of pain. All treatment plans for OA should also include exercise, weight reduction, and self-management, in addition to pharmacologic interventions, to reduce both the micro-inflammation and the centralized pain component (when present). Intra-articular injections of various types have been studied with some having more efficacy in pain reduction and functional improvement than others.42-45 See TABLE 342-61 for a summary of evidence-based treatment options.
Low back pain—a mixed pain state
Low back pain (LBP) has been recognized as a mixed pain state for quite some time. While some patients may experience purely nociceptive and/or neuropathic pain, most cases are nonspecific with patients experiencing varying degrees of nociceptive (myofascial low back pain), neuropathic (lumbar radiculopathy), and central sensitization pain.62,63 Evidence for centralized pain is demonstrated in studies showing hyperalgesia,64 augmented central pain processing,65 involvement of the emotional brain,66 and delayed recovery influenced by poor coping strategies.67
When developing a treatment plan for a patient with chronic low back pain, remember that the pain derives from a complex combination of pathophysiologic contributors. Identifying where a patient lies on the pain centralization spectrum can help you tailor treatment.
In one study of 548 patients presenting to a tertiary pain clinic with primary spine pain diagnoses, 42% met diagnostic criteria for fibromyalgia.68 Compared to criteria-negative patients, these patients tended to be younger, unemployed, and receiving compensation; they had greater pain intensity, pain interference, and used stronger words to describe their neuropathic pain; they also had higher levels of depression/anxiety and a lower level of physical function.
Because low back pain is a condition with high prevalence and associated disability, many clinical boards have created guidelines for management. These guidelines tend to vary in the strength of evidence used, and the extent to which they are followed in clinical practice remains largely unknown. Recommendations frequently discourage the use of ultrasound/electrotherapy, but many encourage short-term use of medications (see “How effective are opioids for chronic low back pain?” J Fam Pract. 2015;64:584-584), supervised exercise therapy, CBT, and multidisciplinary treatment.
Guidelines tend to differ most widely with regard to recommendations for spinal manipulation and specific drug therapies.69 The classes of drugs that may be most useful when centralized pain is present include the SNRIs and the alpha 2 delta calcium channel ligands.4 See TABLE 470-89 for a summary of evidence-based treatment options.
CASE 1 › Ms. A is started on amitriptyline 25 mg at bedtime, which improves her fatigue and cognitive symptoms. During monthly office visits, her FP educates her about the pathophysiology of fibromyalgia and uses motivational interviewing to get her slowly moving and increasing her activity level. She is weaned off the gabapentin previously prescribed, as her symptoms stabilize and improve.
CASE 2 › Mr. P is sent for a steroid injection, which decreases his pain temporarily. During this time, he begins physical therapy; slowly, with increased movement, his function improves. A trial of duloxetine provides pain relief; that combined with intermittent nonsteroidal anti-inflammatory drugs (NSAIDs) has allowed Mr. P to maintain his function and his job.
CASE 3 › Because Mr. B was only taking the narcotics intermittently and wasn’t certain they were helping, CBT was sufficient to wean Mr. B off the medication without any worsening of his pain in the process. By participating in physical therapy, he has learned how to perform certain tasks at his job without pain or injury. He uses NSAIDs as needed for pain.
CORRESPONDENCE
Jill Schneiderhan, MD, 24 Frank Lloyd Wright Dr., Lobby H, Suite 2300, Ann Arbor, MI 48105; [email protected].
ACKNOWLEDGEMENTS
We thank Drs. Daniel Clauw (University of Michigan, Ann Arbor) and Martha Rumschlag (Providence Family Medicine Residency Program, Southfield, Mich), for their valuable contributions to this article.
CASE 1 › Lola A is a 28-year-old woman with a history of muscular aches and joint pain throughout her body, fatigue, and mental fogginess. She has been seen by a rheumatologist and has been given a diagnosis of fibromyalgia, but just moved to your town and is establishing care. She is feeling desperate because her pain has worsened and the medication previously prescribed (gabapentin 300 mg tid) is no longer working. She asks to try oxycodone.
CASE 2 › Matt P is a 59-year-old truck driver with severe hip osteoarthritis (OA). His orthopedist recommended against hip replacement at this time because of his young age and a heart condition that makes him high risk. His pain makes sitting for long periods very difficult. He presents to you for help because he is worried he will be unable to continue working.
CASE 3 › Keith B is a 56-year-old construction worker who has been suffering from bouts of back pain for many years. The pain has become more debilitating over time; currently, it is constant, and Mr. B can hardly make it through his work day. He has been getting hydrocodone/acetaminophen from urgent care centers and emergency rooms, but he isn’t sure it is helping and is coming to you to assume his pain management.
Chronic pain (defined as pain >3 months in duration), is a complex, heterogeneous condition affecting an estimated 116 million US adults.1 Much of the management of chronic pain occurs in primary care settings, placing family physicians (FPs) on the front lines of 2 epidemics: that of chronic pain itself and that of the abuse and misuse of opioid pain medications.
In an effort to improve communication about the risks and benefits of opioid therapy and the safety and effectiveness of pain treatments in general, many professional organizations, health care institutions, and recently the Centers for Disease Control and Prevention,2 have published guidelines on the use of opioids for non-malignant chronic pain. With these guidelines in mind—and in light of the latest evidence—we propose the paradigm that follows for the treatment of chronic pain. A critical aspect of this paradigm is determining the pathophysiology underlying a patient’s pain in order to develop a well-rounded, multimodal, evidence-based treatment plan. Detailed here is the application of this approach to the treatment of 3 common chronic pain diagnoses: fibromyalgia, osteoarthritis, and low back pain.
Look to the central and peripheral nervous system
Acute pain begins with activation of peripheral nociceptors at the site of injury. This causes depolarization up the spinal cord and through the brain stem to higher cortical centers where the pain is perceived and localized. Descending neural pathways transport both excitatory and inhibitory information from the brain to the periphery via the spinal cord, which either increases or decreases the perception of pain.3
When damage/injury doesn’t correlate with the perception of pain
Until recently, it was assumed that chronic pain worked much the same way as acute pain and was caused by ongoing nociceptive input in the periphery, but research has shown us that the central nervous system can play a large role in the modulation of nociception. This new understanding comes from the lack of evidence pointing to any pain state in which the degree of nociceptive input correlates with the degree of pain experienced.
For most patients with chronic pain, regardless of their diagnosis, there is some degree of alteration in the processing of nociceptive signals by the central nervous system contributing to the experience of pain.4 This alteration is thought to be the result of peripheral nociceptive signaling persisting past the point of tissue healing, leading to a hypersensitivity of nerve fibers; the fibers then continue to respond to low, or even absent, sensory stimuli.
Central sensitization is the term used when this hypersensitivity develops in the superficial, deep, and ventral cord nerves. When this happens, pain is often accompanied by other systemic symptoms such as fatigue and slowed cognitive processing, often in the setting of little to no actual stimulation of the peripheral nociceptors.3 (For more on this, see “A new paradigm for pain?” J Fam Pract. 2016;65:598-605 or go to http://www.mdedge.com/jfponline/article/111257/pain/new-paradigm-pain.)
TABLE 14 lists the possible mechanisms of pain, which can be broken down into 4 categories: peripheral nociceptive (inflammatory or mechanical), peripheral neuropathic (underlying damage to a peripheral nerve), central (referring to when the central nervous system is the primary entity involved in maintaining the pain), or any combination of the 3.
As pain becomes chronic, multiple mechanisms overlap
It is important to remember that for any single pain diagnosis, there is likely to be—at least initially—a principle underlying mechanism generating the pain. But as the pain becomes chronic, an overlap of multiple mechanisms develops, with central sensitization often playing a more dominant role than peripheral stimulation (regardless of the diagnosis).
For example, in a patient with rheumatoid arthritis (RA), peripheral nociceptive input (in the form of inflammation) is likely the initial mechanism at work, but as time goes on, central processing becomes more involved. The patient may then begin to experience pain that is disproportional to what is generally expected with RA and may develop other somatic symptoms. The diagnosis then becomes pain primarily related to RA with central sensitization, and both need to be addressed in a treatment plan. In rheumatic conditions, comorbid fibromyalgia (indicative of central sensitization) is thought to occur in 15% to 30% of patients.5
FPs can utilize the underlying mechanisms to cut across diagnostic labels and tailor treatments to those that are most likely to be effective. For a patient with more prominent peripheral involvement, a procedural intervention such as injections or surgery alone may suffice, whereas a broader approach including psychotherapy, medications, exercise, and other lifestyle interventions may be necessary for a patient with pain caused predominantly by central sensitization.
Addressing both peripheral and central components is essential. One prospective, observational cohort study of more than 600 patients scheduled for unilateral total knee or total hip arthroplasty found that those patients with a higher degree of centralization of pain (as measured by widespread pain index and modified fibromyalgia screening scales6) were less likely to report improvement in the affected body part and in overall body pain following the surgery.7
There is a high degree of overlap among many of the chronic pain syndromes (fibromyalgia, irritable bowel syndrome, interstitial cystitis, chronic headaches) that have been found to have a central sensitization component.8 Providers of primary care are aptly positioned to recognize central sensitization as the underlying pathology and target treatment effectively.
Tailor the treatment plan to the underlying mechanisms of pain
As with any chronic condition, a thorough work-up (complete with history, physical exam, and diagnostic testing, as appropriate) is indicated. In the setting of chronic pain, it’s important to identify both the primary mechanism, as well as secondary factors that may be contributing to the patient’s pain, before developing your treatment plan. These secondary factors may include co-occurring affect disorders,9 a history of trauma,10 poor sleep,11 and tobacco use,12 among others. A history of trauma, for example, co-exists with many pain syndromes. For these patients, central sensitization is responsible for much of their pain. As a result, traditional cognitive behavioral therapy (CBT) may not be the best option because of its focus on accepting pain as a chronic diagnosis; more trauma-focused treatments such as those dealing in emotional awareness and understanding of the central nervous system’s role in chronic pain need to be considered.13
3 common conditions. Below we present evidence-based treatment approaches for 3 conditions that are typically associated with each of the major mechanisms of chronic pain generation: fibromyalgia (a central sensitization cause), OA (a peripheral nociceptive cause), and low back pain (a mixed pain state).
Fibromyalgia: A case of central sensitization
Fibromyalgia is a hallmark diagnosis for those patients in whom central sensitization is the dominant cause of pain. These patients usually present with widespread, diffuse pain, as well as somatic symptoms such as fatigue, memory difficulties, and poor sleep quality.8 When explaining the pain mechanism (ie, central sensitization) to patients, it may be useful to use the analogy of a volume control dial that is stuck in the “high” position and can’t be turned down.
Genes, the environment, and neurotransmitters play a role. The origin of the pain amplification process is believed to be multifactorial.
- Genetic factors are thought to contribute to a predisposition for amplification. To date, 5 sets of genes have been implicated in increased sensitivity to pain leading to increased risk of the development of chronic pain during a patient’s lifetime.14-19
- Environmental factors (eg, early life trauma, physical trauma especially to the trunk, certain infections such as Lyme disease and Epstein-Barr virus, and emotional stress) may trigger or exacerbate symptoms.8 Of note: Only about 5% to 10% of people who experience these triggers actually develop a chronic pain state, while the rest regain their baseline health.4 This raises the question of whether there is a point during an acute pain episode in which one can intervene and prevent the acute pain from becoming chronic in those at higher risk.4
- Imbalances of neurotransmitters (high glutamate;20 low norepinephrine, serotonin,21 and gamma-aminobutyric acid [GABA]22) play a role in central amplification. These substances not only affect sensory transmission, but also control levels of alertness, sleep, mood, and memory.
The diagnostic criteria for fibromyalgia were modified in 2011 to remove the tender point examination and to add in somatic symptoms.6 These criteria can be useful in the clinical setting in identifying not only fibromyalgia itself but also the degree of “fibromyalgianess” a patient has, which is an indicator of how large a role the centralization process plays in the maintenance of chronic pain.23,24
Treatment: Multimodal and patient empowering. Evidence-based treatment options for fibromyalgia, as well as other conditions for which there is a high degree of centralized pain, can be found in TABLE 2.25-36 Multimodal treatment, with an emphasis on patient knowledge and empowerment, is generally thought to be the most beneficial.25,37 Treatment should almost always include CBT and exercise/activity therapies,26,29 which have high degrees of efficacy with few adverse effects.
In terms of medication, centrally-acting agents (tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors [SNRIs], and alpha 2 delta ligands) are the most effective. There is little to no data showing benefit from anti-inflammatories or opioids in the setting of fibromyalgia. There is some data to suggest that combination therapy, for example with an SNRI (milnacipran) and an alpha 2 delta ligand (pregabalin), may provide more benefit than treating with pregabalin alone.38
Complementary and alternative therapies (eg, yoga, chiropractic care, acupuncture, massage) are being studied more, and while evidence is only preliminary in terms of efficacy, there is increasing emphasis being placed on the need for patients with chronic pain to shift their treatment expectations to greater acceptance of pain and the need for ongoing self-care.28 (For more advice on managing fibromyalgia, see the related videos at http://bit.ly/2lPEt0f and http://bit.ly/2lmjEcn.)
Osteoarthritis: An example of peripheral nociceptive pain
OA is a condition long thought to be characterized by damage to the cartilage and bone; however, as with many other pain diagnoses, there is frequently little correlation between damage seen on radiographs and the amount of pain that patients experience.
One study analyzed data on almost 7000 patients from the National Health and Nutrition Examination Survey (NHANES I) and found that between 30% and 50% of OA patients with moderate to severe radiographic changes were asymptomatic, and 10% of those with moderate to severe pain had normal radiographs or only mild changes.39 Research is showing that many factors may contribute to this discrepancy, including the typical “wear and tear” of the disease, subacute levels of inflammation that can lead to peripheral sensitization,40 and, in some patients, a centralized pain component. The patients with more centralized pain often have pain that is disproportionate to radiographic evidence, as well as more somatic symptoms such as fatigue, sleep disturbance, and memory issues.41
Treatment should be multimodal and include interventions targeted at halting the progression of damage as well as palliation of pain. All treatment plans for OA should also include exercise, weight reduction, and self-management, in addition to pharmacologic interventions, to reduce both the micro-inflammation and the centralized pain component (when present). Intra-articular injections of various types have been studied with some having more efficacy in pain reduction and functional improvement than others.42-45 See TABLE 342-61 for a summary of evidence-based treatment options.
Low back pain—a mixed pain state
Low back pain (LBP) has been recognized as a mixed pain state for quite some time. While some patients may experience purely nociceptive and/or neuropathic pain, most cases are nonspecific with patients experiencing varying degrees of nociceptive (myofascial low back pain), neuropathic (lumbar radiculopathy), and central sensitization pain.62,63 Evidence for centralized pain is demonstrated in studies showing hyperalgesia,64 augmented central pain processing,65 involvement of the emotional brain,66 and delayed recovery influenced by poor coping strategies.67
When developing a treatment plan for a patient with chronic low back pain, remember that the pain derives from a complex combination of pathophysiologic contributors. Identifying where a patient lies on the pain centralization spectrum can help you tailor treatment.
In one study of 548 patients presenting to a tertiary pain clinic with primary spine pain diagnoses, 42% met diagnostic criteria for fibromyalgia.68 Compared to criteria-negative patients, these patients tended to be younger, unemployed, and receiving compensation; they had greater pain intensity, pain interference, and used stronger words to describe their neuropathic pain; they also had higher levels of depression/anxiety and a lower level of physical function.
Because low back pain is a condition with high prevalence and associated disability, many clinical boards have created guidelines for management. These guidelines tend to vary in the strength of evidence used, and the extent to which they are followed in clinical practice remains largely unknown. Recommendations frequently discourage the use of ultrasound/electrotherapy, but many encourage short-term use of medications (see “How effective are opioids for chronic low back pain?” J Fam Pract. 2015;64:584-584), supervised exercise therapy, CBT, and multidisciplinary treatment.
Guidelines tend to differ most widely with regard to recommendations for spinal manipulation and specific drug therapies.69 The classes of drugs that may be most useful when centralized pain is present include the SNRIs and the alpha 2 delta calcium channel ligands.4 See TABLE 470-89 for a summary of evidence-based treatment options.
CASE 1 › Ms. A is started on amitriptyline 25 mg at bedtime, which improves her fatigue and cognitive symptoms. During monthly office visits, her FP educates her about the pathophysiology of fibromyalgia and uses motivational interviewing to get her slowly moving and increasing her activity level. She is weaned off the gabapentin previously prescribed, as her symptoms stabilize and improve.
CASE 2 › Mr. P is sent for a steroid injection, which decreases his pain temporarily. During this time, he begins physical therapy; slowly, with increased movement, his function improves. A trial of duloxetine provides pain relief; that combined with intermittent nonsteroidal anti-inflammatory drugs (NSAIDs) has allowed Mr. P to maintain his function and his job.
CASE 3 › Because Mr. B was only taking the narcotics intermittently and wasn’t certain they were helping, CBT was sufficient to wean Mr. B off the medication without any worsening of his pain in the process. By participating in physical therapy, he has learned how to perform certain tasks at his job without pain or injury. He uses NSAIDs as needed for pain.
CORRESPONDENCE
Jill Schneiderhan, MD, 24 Frank Lloyd Wright Dr., Lobby H, Suite 2300, Ann Arbor, MI 48105; [email protected].
ACKNOWLEDGEMENTS
We thank Drs. Daniel Clauw (University of Michigan, Ann Arbor) and Martha Rumschlag (Providence Family Medicine Residency Program, Southfield, Mich), for their valuable contributions to this article.
1. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington (DC): National Academies Press (US); 2011.
2. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recomm Rep. 2016;65:1-49.
3. Aronoff GM. What do we know about the pathophysiology of chronic pain? Implications for treatment considerations. Med Clin North Am. 2016;100:31-42.
4. Clauw DJ. Diagnosing and treating chronic musculoskeletal pain based on the underlying mechanism(s). Best Pract Res Clin Rheumatol. 2015;29:6-19.
5. Clauw DJ, Katz P. The overlap between fibromyalgia and inflammatory rheumatic disease: when and why does it occur? J Clin Rheumatol. 1995;1:335-342.
6. Wolfe F, Clauw DJ, Fitzcharles MA, et al. Fibromyalgia criteria and severity scales for clinical and epidemiological studies: a modification of the ACR Preliminary Diagnostic Criteria for Fibromyalgia. J Rheumatol. 2011;38:1113-1122.
7. Brummett CM, Urquhart AG, Hassett AL, et al. Characteristics of fibromyalgia independently predict poorer long-term analgesic outcomes following total knee and hip arthroplasty. Arthritis Rheumatol. 2015;67:1386-1394.
8. Ablin K, Clauw DJ. From fibrositis to functional somatic syndromes to a bell-shaped curve of pain and sensory sensitivity: evolution of a clinical construct. Rheum Dis Clin North Am. 2009;35:233-251.
9. Giesecke T, Gracely RH, Williams DA, et al. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005;52:1577-1584.
10. Tesarz J, Eich W, Treede RD, et al. Altered pressure pain thresholds and increased wind-up in adult chronic back pain patients with a history of childhood maltreatment: a quantitative sensory testing study. Pain. 2016;157:1799-1809.
11. Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain. 2013;14:1539-1552.
12. Shi Y, Weingarten TN, Mantilla CB, et al. Smoking and pain: pathophysiology and clinical implications. Anesthesiology. 2010;113:977-992.
13. Burger AJ, Lumley MA, Carty JN, et al. The effects of a novel psychological attribution and emotional awareness and expression therapy for chronic musculoskeletal pain: a preliminary, uncontrolled trial. J Psychosom Res. 2016;81:1-8.
14. Zubieta JK, Heitzeg MM, Smith YR, et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240-1243.
15. van Meurs JB, Uitterlinden AG, Stolk L, et al. A functional polymorphism in the catechol-O-methyltransferase gene is associated with osteoarthritis-related pain. Arthritis Rheum. 2009;60:628-629.
16. McLean SA, Diatchenko L, Lee YM, et al. Catechol O-methyltransferase haplotype predicts immediate musculoskeletal neck pain and psychological symptoms after motor vehicle collision. J Pain. 2011;12:101-107.
17. Costigan M, Belfer I, Griffin RS, et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain. 2010;133:2519-2527.
18. Tegeder I, Costigan M, Griffin RS, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269-1277.
19. Amaya F, Wang H, Costigan M, et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852-12860.
20. Harris RE, Napadow V, Huggins JP, et al. Pregabalin rectifies abberrant brain chemistry, connectivity, and functional responses in chronic pain patients. Anesthesiology. 2013;119:1453-1464.
21. Russell IJ, Vaeroy H, Javors M, et al. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35:550-556.
22. Foerster BR, Petrou M, Edden RAE, et al. Reduced insular gamma-aminobutyric acid in fibromyalgia. Arthritis Rheum. 2012;64:579-583.
23. Clauw DJ. Fibromyalgia: a clinical review. JAMA. 2014;311:1547-1555.
26. Hauser W, Klose P, Langhorst J, et al. Efficacy of different types of aerobic exercise in fibromyalgia syndrome: a systematic review and meta-analysis of randomised controlled trials. Arthritis Res Ther. 2010;12:R79.
27. Porter NS, Jason LA, Boulton A, et al. Alternative medical interventions used in the treatment and management of myalgic encephalomyelitis/chronic fatigue syndrome and fibromyalgia. J Altern Complement Med. 2010;16:235-249.
28. Eaves ER, Sherman KJ, Ritenbaugh C, et al. A qualitative study of changes in expectations over time among patients with chronic low back pain seeking four CAM therapies. BMC Complement Altern Med. 2015;15:12.
29. Bernardy K, Fuber N, Kollner V, et al. Efficacy of cognitive-behavioral therapies in fibromyalgia syndrome: a systematic review and metaanalysis of randomized controlled trials. J Rheumatol. 2010;37:1991-2005.
30. Arnold LM, Keck PE Jr, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41:104-113.
31. Moldofsky H, Harris HW, Archambault WT, et al. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: a double-blind randomized placebo-controlled study. J Rheumatol. 2011;38:2653-2663.
32. Arnold LM. Duloxetine and other antidepressants in the treatment of patients with fibromyalgia. Pain Med. 2007;Sep 8 Suppl 2:S63-S74.
33. Häuser W, Bernardy K, Uceyler N, et al. Treatment of fibromyalgia syndrome with gabapentin and pregabalin—a meta-analysis of randomized controlled trials. Pain. 2009;145:69-81.
34. Gaskell H, Moore RA, Derry S, et al. Oxycodone for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2014;Jun 23:CD010692.
35. MacLean AJ, Schwartz TL. Tramadol for the treatment of fibromyalgia. Expert Rev Neurother. 2015;15:469-475.
36. Younger J, Noor N, McCue R, et al. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 2013;65:529-538.
37. Camerini L, Schulz PJ, Nakamoto K. Differential effects of health knowledge and health empowerment over patients’ self-management and health outcomes: a cross-sectional evaluation. Patient Educ Couns. 2012;89:337-344.
38. Mease PJ, Farmer MV, Palmer RH, et al. Milnacipran combined with pregabalin in fibromyalgia: a randomized, open-label study evaluating the safety and efficacy of adding milnacipran in patients with incomplete response to pregabalin. Ther Adv Musculoskeletal Dis. 2013;5:113-126.
39. Hannan MT, Felson DT, Pincus T. Analysis of the discordance between radiographic changes and knee pain in osteoarthritis of the knee. J Rheumatol. 2000;27:1513-1517.
40. Daghestani HN, Kraus VB. Inflammatory biomarkers in osteoarthritis. Osteoarthritis Cartilage. 2015;23:1890-1896.
41. Fingleton C, Smart K, Moloney N, et al. Pain sensitization in people with knee osteoarthritis: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2015;23:1043-1056.
42. Strand V, McIntyre LF, Beach WR, et al. Safety and efficacy of US-approved viscosupplements for knee osteoarthritis: a systematic review and meta-analysis of randomized, saline-controlled trials. J Pain Res. 2015;8:217-228.
43. Jüni P, Hari R, Rutjes AW, et al. Intra-articular corticosteroid for knee osteoarthritis. Cochrane Database Syst Rev. 2015:CD005328.
44. Meheux CJ, McCulloch PC, Lintner DM, et al. Efficacy of intra-articular platelet-rich plasma injections in knee osteoarthritis: a systematic review. Arthroscopy. 2016;32:495-505.
45. Wu T, Song HX, Dong Y, et al. Intra-articular injections of botulinum toxin a for refractory joint pain: a systematic review and meta-analysis. Clin Rehabil. 2016.
46. Jordan JL, Holden MA, Mason EE, et al. Interventions to improve adherence to exercise for chronic musculoskeletal pain in adults. Cochrane Database Syst Rev. 2010:CD005956.
48. Fransen M, McConnell S, Hernandez-Molina G, et al. Exercise for osteoarthritis of the hip. Cochrane Database Syst Rev. 2014:CD007912.
49. Bartels EM, Juhl CB, Christensen R, et al. Aquatic exercise for the treatment of knee and hip osteoarthritis. Cochrane Database Syst Rev. 2016;3:CD005523.
50. da Costa BR, Reichenbach S, Keller N, et al. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet. 2016;387:2093-2105.
51. Myers J, Wielage RC, Han B, et al. The efficacy of duloxetine, non-steroidal anti-inflammatory drugs, and opioids in osteoarthritis: a systematic literature review and meta-analysis. BMC Musculoskelet Disord. 2014;15:76.
52. Berthelot JM, Darrieutort-Lafitte C, Le Goff B, et al. Strong opioids for noncancer pain due to musculoskeletal diseases: not more effective than acetaminophen or NSAIDs. Joint Bone Spine. 2015;82:397-401.
53. Clegg DO, Reda DJ, Harris CL, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med. 2006;354:795-808.
54. Wandel S, Jüni P, Tendal B, et al. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: network meta-analysis. BMJ. 2010;341:c4675.
55. Sawitzke AD, Shi H, Finco MF, et al. Clinical efficacy and safety of glucosamine, chondroitin sulphate, their combination, celecoxib or placebo taken to treat osteoarthritis of the knee: 2-year results from GAIT. Ann Rheum Dis. 2010;69:1459-1464.
56. Wu D, Huang Y, Gu Y, et al. Efficacies of different preparations of glucosamine for the treatment of osteoarthritis: a meta-analysis of randomised, double-blind, placebo-controlled trials. Int J Clin Pract. 2013;67:585-594.
57. Kahan A, Uebelhart D, De Vathaire F, et al. Long-term effects of chondroitins 4 and 6 sulfate on knee osteoarthritis: the study on osteoarthritis progression prevention, a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2009;60:524-533.
58. Perkins K, Sahy W, Beckett RD. Efficacy of curcuma for treatment of osteoarthritis. J Evid Based Complementary Altern Med. 2017;22:156-165.
59. Clinton CM, O’Brien S, Law J, et al. Whole-foods, plant-based diet alleviates the symptoms of osteoarthritis. Arthritis. 2015;2015:708152.
60. Manyanga T, Froese M, Zarychanski R, et al. Pain management with acupuncture in osteoarthritis: a systematic review and meta-analysis. BMC Complement Altern Med. 2014;14:312.
61. Vickers AJ, Cronin AM, Maschino AC, et al. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172:1444-1453.
62. Nijs J, Apeldoorn A, Hallegraeff H, et al. Low back pain: guidelines for the clinical classification of predominant neuropathic, nociceptive, or central sensitization pain. Pain Physician. 2015;18:E333-E346.
63. Fishbain DA, Cole B, Lewis JE, et al. What is the evidence that neuropathic pain is present in chronic low back pain and soft tissue syndromes? An evidence-based structured review. Pain Med. 2014;15:4-15.
64. Hübscher M, Moloney N, Rebbeck T, et al. Contributions of mood, pain catastrophizing, and cold hyperalgesia in acute and chronic low back pain: a comparison with pain-free controls. Clin J Pain. 2014;30:886-893.
65. Giesecke T, Gracely RH, Grant MA, et al. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004;50:613-623.
66. Baliki MN, Chialvo DR, Geha PY, et al. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci. 2006;26:12165-12173.
68. Brummett CM, Goesling J, Tsodikov A, et al. Prevalence of the fibromyalgia phenotype in patients with spine pain presenting to a tertiary care pain clinic and the potential treatment implications. Arthritis Rheum. 2013;65:3285-3292.
69. Koes BW, van Tulder M, Lin CW, et al. An updated overview of clinical guidelines for the management of non-specific low back pain in primary care. Eur Spine J. 2010;19:2075-2094.
70. Oliveira VC, Ferreira PH, Maher CG, et al. Effectiveness of self-management of low back pain: systematic review with meta-analysis. Arthritis Care Res. 2012;64:1739-1748.
71. Engers A, Jellema P, Wensing M, et al. Individual patient education for low back pain. Cochrane Database Syst Rev. 2008:CD004057.
72. Hayden JA, van Tulder MW, Malmivaara A, et al. Exercise therapy for treatment of non-specific low back pain. Cochrane Database Syst Rev. 2005:CD000335.
73. French SD, Cameron M, Walker BF, et al. Superficial heat or cold for low back pain. Cochrane Database Syst Rev. 2006:CD004750.
74. Franke H, Franke JD, Fryer G. Osteopathic manipulative treatment for nonspecific low back pain: a systematic review and meta-analysis. BMC Musculoskeletal Disord. 2014;15:286.
75. Franke H, Fryer G, Ostelo RW, et al. Muscle energy technique for non-specific low back pain. Cochrane Database Syst Rev. 2015:CD009852.
76. Oliphant D. Safety of spinal manipulation in the treatment of lumbar disk herniations: a systematic review and risk assessment. J Manipulative Physiol Ther. 2004:197-210.
77. Furlan AD, Giraldo M, Baskwill A, et al. Massage for low-back pain. Cochrane Database Syst Rev. 2015:CD001929.
78. Khadilkar A, Odebiyi DO, Brosseau L, et al. Transcutaneous electrical nerve stimulation (TENS) versus placebo for chronic low back pain. Cochrane Database Syst Rev. 2008:CD003008.
79. Ebadi S, Henschke N, Nakhostin Ansari N, et al. Therapeutic ultrasound for chronic low back pain. Cochrane Database Syst Rev. 2014:CD009169.
80. Furlan AD, van Tulder MW, Cherkin DC, et al. Acupuncture and dry-needling for low back pain. Cochrane Database Syst Rev. 2005:CD001351.
81. Chou R, Huffman LH. Nonpharmacologic therapies for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med. 2007;147:492-504.
82. Sherman KJ, Cherkin DC, Erro J, et al. Comparing yoga, exercise, and a self-care book for chronic low back pain: a randomized, controlled trial. Ann Intern Med. 2005;143:849-856.
83. Cherkin DC, Sherman KJ, Balderson BH, et al. Effect of mindfulness-based stress reduction vs cognitive behavioral therapy or usual care on back pain and functional limitations in adults with chronic low back pain: a randomized clinical trial. JAMA. 2016;315:1240-1249.
84. Staal JB, de Bie R, de Vet HC, et al. Injection therapy for subacute and chronic low back pain. Cochrane Database Syst Rev. 2008:CD001824.
85. Chou R, Baisden J, Carragee EJ, et al. Surgery for low back pain: a review of the evidence for an American Pain Society Clinical Practice Guideline. Spine. 2009;34:1094-1109.
86. Felson D. Paracetamol is ineffective for spinal pain and knee and hip osteoarthritis. Evid Based Med. 2015;20:205.
87. Machado GC, Maher CG, Ferreira PH, et al. Efficacy and safety of paracetamol for spinal pain and osteoarthritis: systematic review and meta-analysis of randomised placebo controlled trials. BMJ. 2015;350:h1225.
88. Enthoven WT, Roelofs PD, Deyo RA, et al. Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst Rev. 2016;2:CD012087.
89. White AP, Arnold PM, Norvell DC, et al. Pharmacologic management of chronic low back pain: synthesis of the evidence. Spine (Phila Pa 1976). 2011;36:S131-S143.
1. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington (DC): National Academies Press (US); 2011.
2. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recomm Rep. 2016;65:1-49.
3. Aronoff GM. What do we know about the pathophysiology of chronic pain? Implications for treatment considerations. Med Clin North Am. 2016;100:31-42.
4. Clauw DJ. Diagnosing and treating chronic musculoskeletal pain based on the underlying mechanism(s). Best Pract Res Clin Rheumatol. 2015;29:6-19.
5. Clauw DJ, Katz P. The overlap between fibromyalgia and inflammatory rheumatic disease: when and why does it occur? J Clin Rheumatol. 1995;1:335-342.
6. Wolfe F, Clauw DJ, Fitzcharles MA, et al. Fibromyalgia criteria and severity scales for clinical and epidemiological studies: a modification of the ACR Preliminary Diagnostic Criteria for Fibromyalgia. J Rheumatol. 2011;38:1113-1122.
7. Brummett CM, Urquhart AG, Hassett AL, et al. Characteristics of fibromyalgia independently predict poorer long-term analgesic outcomes following total knee and hip arthroplasty. Arthritis Rheumatol. 2015;67:1386-1394.
8. Ablin K, Clauw DJ. From fibrositis to functional somatic syndromes to a bell-shaped curve of pain and sensory sensitivity: evolution of a clinical construct. Rheum Dis Clin North Am. 2009;35:233-251.
9. Giesecke T, Gracely RH, Williams DA, et al. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005;52:1577-1584.
10. Tesarz J, Eich W, Treede RD, et al. Altered pressure pain thresholds and increased wind-up in adult chronic back pain patients with a history of childhood maltreatment: a quantitative sensory testing study. Pain. 2016;157:1799-1809.
11. Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain. 2013;14:1539-1552.
12. Shi Y, Weingarten TN, Mantilla CB, et al. Smoking and pain: pathophysiology and clinical implications. Anesthesiology. 2010;113:977-992.
13. Burger AJ, Lumley MA, Carty JN, et al. The effects of a novel psychological attribution and emotional awareness and expression therapy for chronic musculoskeletal pain: a preliminary, uncontrolled trial. J Psychosom Res. 2016;81:1-8.
14. Zubieta JK, Heitzeg MM, Smith YR, et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240-1243.
15. van Meurs JB, Uitterlinden AG, Stolk L, et al. A functional polymorphism in the catechol-O-methyltransferase gene is associated with osteoarthritis-related pain. Arthritis Rheum. 2009;60:628-629.
16. McLean SA, Diatchenko L, Lee YM, et al. Catechol O-methyltransferase haplotype predicts immediate musculoskeletal neck pain and psychological symptoms after motor vehicle collision. J Pain. 2011;12:101-107.
17. Costigan M, Belfer I, Griffin RS, et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain. 2010;133:2519-2527.
18. Tegeder I, Costigan M, Griffin RS, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269-1277.
19. Amaya F, Wang H, Costigan M, et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852-12860.
20. Harris RE, Napadow V, Huggins JP, et al. Pregabalin rectifies abberrant brain chemistry, connectivity, and functional responses in chronic pain patients. Anesthesiology. 2013;119:1453-1464.
21. Russell IJ, Vaeroy H, Javors M, et al. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35:550-556.
22. Foerster BR, Petrou M, Edden RAE, et al. Reduced insular gamma-aminobutyric acid in fibromyalgia. Arthritis Rheum. 2012;64:579-583.
23. Clauw DJ. Fibromyalgia: a clinical review. JAMA. 2014;311:1547-1555.
26. Hauser W, Klose P, Langhorst J, et al. Efficacy of different types of aerobic exercise in fibromyalgia syndrome: a systematic review and meta-analysis of randomised controlled trials. Arthritis Res Ther. 2010;12:R79.
27. Porter NS, Jason LA, Boulton A, et al. Alternative medical interventions used in the treatment and management of myalgic encephalomyelitis/chronic fatigue syndrome and fibromyalgia. J Altern Complement Med. 2010;16:235-249.
28. Eaves ER, Sherman KJ, Ritenbaugh C, et al. A qualitative study of changes in expectations over time among patients with chronic low back pain seeking four CAM therapies. BMC Complement Altern Med. 2015;15:12.
29. Bernardy K, Fuber N, Kollner V, et al. Efficacy of cognitive-behavioral therapies in fibromyalgia syndrome: a systematic review and metaanalysis of randomized controlled trials. J Rheumatol. 2010;37:1991-2005.
30. Arnold LM, Keck PE Jr, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41:104-113.
31. Moldofsky H, Harris HW, Archambault WT, et al. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: a double-blind randomized placebo-controlled study. J Rheumatol. 2011;38:2653-2663.
32. Arnold LM. Duloxetine and other antidepressants in the treatment of patients with fibromyalgia. Pain Med. 2007;Sep 8 Suppl 2:S63-S74.
33. Häuser W, Bernardy K, Uceyler N, et al. Treatment of fibromyalgia syndrome with gabapentin and pregabalin—a meta-analysis of randomized controlled trials. Pain. 2009;145:69-81.
34. Gaskell H, Moore RA, Derry S, et al. Oxycodone for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2014;Jun 23:CD010692.
35. MacLean AJ, Schwartz TL. Tramadol for the treatment of fibromyalgia. Expert Rev Neurother. 2015;15:469-475.
36. Younger J, Noor N, McCue R, et al. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 2013;65:529-538.
37. Camerini L, Schulz PJ, Nakamoto K. Differential effects of health knowledge and health empowerment over patients’ self-management and health outcomes: a cross-sectional evaluation. Patient Educ Couns. 2012;89:337-344.
38. Mease PJ, Farmer MV, Palmer RH, et al. Milnacipran combined with pregabalin in fibromyalgia: a randomized, open-label study evaluating the safety and efficacy of adding milnacipran in patients with incomplete response to pregabalin. Ther Adv Musculoskeletal Dis. 2013;5:113-126.
39. Hannan MT, Felson DT, Pincus T. Analysis of the discordance between radiographic changes and knee pain in osteoarthritis of the knee. J Rheumatol. 2000;27:1513-1517.
40. Daghestani HN, Kraus VB. Inflammatory biomarkers in osteoarthritis. Osteoarthritis Cartilage. 2015;23:1890-1896.
41. Fingleton C, Smart K, Moloney N, et al. Pain sensitization in people with knee osteoarthritis: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2015;23:1043-1056.
42. Strand V, McIntyre LF, Beach WR, et al. Safety and efficacy of US-approved viscosupplements for knee osteoarthritis: a systematic review and meta-analysis of randomized, saline-controlled trials. J Pain Res. 2015;8:217-228.
43. Jüni P, Hari R, Rutjes AW, et al. Intra-articular corticosteroid for knee osteoarthritis. Cochrane Database Syst Rev. 2015:CD005328.
44. Meheux CJ, McCulloch PC, Lintner DM, et al. Efficacy of intra-articular platelet-rich plasma injections in knee osteoarthritis: a systematic review. Arthroscopy. 2016;32:495-505.
45. Wu T, Song HX, Dong Y, et al. Intra-articular injections of botulinum toxin a for refractory joint pain: a systematic review and meta-analysis. Clin Rehabil. 2016.
46. Jordan JL, Holden MA, Mason EE, et al. Interventions to improve adherence to exercise for chronic musculoskeletal pain in adults. Cochrane Database Syst Rev. 2010:CD005956.
48. Fransen M, McConnell S, Hernandez-Molina G, et al. Exercise for osteoarthritis of the hip. Cochrane Database Syst Rev. 2014:CD007912.
49. Bartels EM, Juhl CB, Christensen R, et al. Aquatic exercise for the treatment of knee and hip osteoarthritis. Cochrane Database Syst Rev. 2016;3:CD005523.
50. da Costa BR, Reichenbach S, Keller N, et al. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet. 2016;387:2093-2105.
51. Myers J, Wielage RC, Han B, et al. The efficacy of duloxetine, non-steroidal anti-inflammatory drugs, and opioids in osteoarthritis: a systematic literature review and meta-analysis. BMC Musculoskelet Disord. 2014;15:76.
52. Berthelot JM, Darrieutort-Lafitte C, Le Goff B, et al. Strong opioids for noncancer pain due to musculoskeletal diseases: not more effective than acetaminophen or NSAIDs. Joint Bone Spine. 2015;82:397-401.
53. Clegg DO, Reda DJ, Harris CL, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med. 2006;354:795-808.
54. Wandel S, Jüni P, Tendal B, et al. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: network meta-analysis. BMJ. 2010;341:c4675.
55. Sawitzke AD, Shi H, Finco MF, et al. Clinical efficacy and safety of glucosamine, chondroitin sulphate, their combination, celecoxib or placebo taken to treat osteoarthritis of the knee: 2-year results from GAIT. Ann Rheum Dis. 2010;69:1459-1464.
56. Wu D, Huang Y, Gu Y, et al. Efficacies of different preparations of glucosamine for the treatment of osteoarthritis: a meta-analysis of randomised, double-blind, placebo-controlled trials. Int J Clin Pract. 2013;67:585-594.
57. Kahan A, Uebelhart D, De Vathaire F, et al. Long-term effects of chondroitins 4 and 6 sulfate on knee osteoarthritis: the study on osteoarthritis progression prevention, a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2009;60:524-533.
58. Perkins K, Sahy W, Beckett RD. Efficacy of curcuma for treatment of osteoarthritis. J Evid Based Complementary Altern Med. 2017;22:156-165.
59. Clinton CM, O’Brien S, Law J, et al. Whole-foods, plant-based diet alleviates the symptoms of osteoarthritis. Arthritis. 2015;2015:708152.
60. Manyanga T, Froese M, Zarychanski R, et al. Pain management with acupuncture in osteoarthritis: a systematic review and meta-analysis. BMC Complement Altern Med. 2014;14:312.
61. Vickers AJ, Cronin AM, Maschino AC, et al. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172:1444-1453.
62. Nijs J, Apeldoorn A, Hallegraeff H, et al. Low back pain: guidelines for the clinical classification of predominant neuropathic, nociceptive, or central sensitization pain. Pain Physician. 2015;18:E333-E346.
63. Fishbain DA, Cole B, Lewis JE, et al. What is the evidence that neuropathic pain is present in chronic low back pain and soft tissue syndromes? An evidence-based structured review. Pain Med. 2014;15:4-15.
64. Hübscher M, Moloney N, Rebbeck T, et al. Contributions of mood, pain catastrophizing, and cold hyperalgesia in acute and chronic low back pain: a comparison with pain-free controls. Clin J Pain. 2014;30:886-893.
65. Giesecke T, Gracely RH, Grant MA, et al. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004;50:613-623.
66. Baliki MN, Chialvo DR, Geha PY, et al. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci. 2006;26:12165-12173.
68. Brummett CM, Goesling J, Tsodikov A, et al. Prevalence of the fibromyalgia phenotype in patients with spine pain presenting to a tertiary care pain clinic and the potential treatment implications. Arthritis Rheum. 2013;65:3285-3292.
69. Koes BW, van Tulder M, Lin CW, et al. An updated overview of clinical guidelines for the management of non-specific low back pain in primary care. Eur Spine J. 2010;19:2075-2094.
70. Oliveira VC, Ferreira PH, Maher CG, et al. Effectiveness of self-management of low back pain: systematic review with meta-analysis. Arthritis Care Res. 2012;64:1739-1748.
71. Engers A, Jellema P, Wensing M, et al. Individual patient education for low back pain. Cochrane Database Syst Rev. 2008:CD004057.
72. Hayden JA, van Tulder MW, Malmivaara A, et al. Exercise therapy for treatment of non-specific low back pain. Cochrane Database Syst Rev. 2005:CD000335.
73. French SD, Cameron M, Walker BF, et al. Superficial heat or cold for low back pain. Cochrane Database Syst Rev. 2006:CD004750.
74. Franke H, Franke JD, Fryer G. Osteopathic manipulative treatment for nonspecific low back pain: a systematic review and meta-analysis. BMC Musculoskeletal Disord. 2014;15:286.
75. Franke H, Fryer G, Ostelo RW, et al. Muscle energy technique for non-specific low back pain. Cochrane Database Syst Rev. 2015:CD009852.
76. Oliphant D. Safety of spinal manipulation in the treatment of lumbar disk herniations: a systematic review and risk assessment. J Manipulative Physiol Ther. 2004:197-210.
77. Furlan AD, Giraldo M, Baskwill A, et al. Massage for low-back pain. Cochrane Database Syst Rev. 2015:CD001929.
78. Khadilkar A, Odebiyi DO, Brosseau L, et al. Transcutaneous electrical nerve stimulation (TENS) versus placebo for chronic low back pain. Cochrane Database Syst Rev. 2008:CD003008.
79. Ebadi S, Henschke N, Nakhostin Ansari N, et al. Therapeutic ultrasound for chronic low back pain. Cochrane Database Syst Rev. 2014:CD009169.
80. Furlan AD, van Tulder MW, Cherkin DC, et al. Acupuncture and dry-needling for low back pain. Cochrane Database Syst Rev. 2005:CD001351.
81. Chou R, Huffman LH. Nonpharmacologic therapies for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med. 2007;147:492-504.
82. Sherman KJ, Cherkin DC, Erro J, et al. Comparing yoga, exercise, and a self-care book for chronic low back pain: a randomized, controlled trial. Ann Intern Med. 2005;143:849-856.
83. Cherkin DC, Sherman KJ, Balderson BH, et al. Effect of mindfulness-based stress reduction vs cognitive behavioral therapy or usual care on back pain and functional limitations in adults with chronic low back pain: a randomized clinical trial. JAMA. 2016;315:1240-1249.
84. Staal JB, de Bie R, de Vet HC, et al. Injection therapy for subacute and chronic low back pain. Cochrane Database Syst Rev. 2008:CD001824.
85. Chou R, Baisden J, Carragee EJ, et al. Surgery for low back pain: a review of the evidence for an American Pain Society Clinical Practice Guideline. Spine. 2009;34:1094-1109.
86. Felson D. Paracetamol is ineffective for spinal pain and knee and hip osteoarthritis. Evid Based Med. 2015;20:205.
87. Machado GC, Maher CG, Ferreira PH, et al. Efficacy and safety of paracetamol for spinal pain and osteoarthritis: systematic review and meta-analysis of randomised placebo controlled trials. BMJ. 2015;350:h1225.
88. Enthoven WT, Roelofs PD, Deyo RA, et al. Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst Rev. 2016;2:CD012087.
89. White AP, Arnold PM, Norvell DC, et al. Pharmacologic management of chronic low back pain: synthesis of the evidence. Spine (Phila Pa 1976). 2011;36:S131-S143.
PRACTICE RECOMMENDATIONS
› Recommend cognitive behavioral therapy for most patients suffering from chronic pain. A
› Recommend movement and exercise therapies for all patients with chronic pain. A
› Prescribe anti-inflammatory medications for patients with peripheral nociceptive pain and centrally-acting agents, such as tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors, and alpha 2 delta ligands, for patients with centralized pain. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Polio vaccine status unknown? What the CDC recommends

AGA Clinical Practice Update: Using FLIP to assess upper GI tract still murky territory
New clinical practice advice has been issued for use of the functional lumen imaging probe (FLIP) to assess disorders of the upper gastrointestinal tract, with the main takeaway being the device’s potency in diagnosing achalasia.
“Although the strongest data appear to be focused on the management of achalasia, emerging evidence supports the clinical relevance of FLIP in the assessment of disease severity and as an outcome measure in [eosinophilic esophagitis (EoE)] intervention trials,” wrote the authors of the update, led by John E. Pandolfino, MD, of Northwestern University, Chicago. The report is in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.
10.022).

In terms of evaluating the LES, however, FLIP can be used during laparoscopic Heller myotomy or peroral endoscopic myotomy (POEM) as a way of monitoring the LES. Using FLIP this way can help clinicians and surgeons personalize the procedure to each patient, even while it’s ongoing. FLIP also can be used with dilation balloons, with the balloon diameter allowing dilation measurement without the need to also use fluoroscopy.
For treating gastroesophageal reflux disease (GERD), the evidence found in existing literature points with less certainty toward use of FLIP.
“The role of FLIP for physiologic evaluation and management in GERD remains appealing; however, the level of evidence is low and currently FLIP should not be used in routine GERD management,” the authors explained. “Future outcome studies are needed to substantiate the utility of FLIP in GERD and to develop metrics that predict severity and treatment response after antireflux procedures.”
FLIP can be used in managing eosinophilic esophagitis, but is recommended only in certain scenarios. According to the authors, FLIP can be used to measure esophageal narrowing and the overall esophageal body. FLIP also can be used to measure esophageal distensibility, and, in the case of at least one study reviewed by the authors, allows “significantly greater accuracy and precision in estimating the effects of remodeling” in certain patients.
Dr. Pandolfino and his colleagues warned that “current recommendations are limited by the low level of evidence and lack of generalized availability of the analysis paradigms.” They noted the need for “further outcome studies that validate the distensibility plateau threshold and further refinements in software analyses to make this methodology more generalizable.”
Overall, the authors concluded, more study still needs to be done to ascertain exactly what FLIP is capable of and when it can be used to greatest effect. In addition to evaluating its benefit in patients with GERD, research should focus on how to make data obtained via FLIP easier to interpret and put to use.
“More work is needed [that] focuses on optimizing data analysis, standardizing protocols, and defining outcome metrics prior to the widespread adoption [of FLIP] into general clinical practice,” the authors wrote.
Dr. Pandolfino disclosed relationships with Medtronic and Sandhill Scientific. Other coauthors did not report any relevant financial disclosures.
*This story updated on 3/9/2017.
New clinical practice advice has been issued for use of the functional lumen imaging probe (FLIP) to assess disorders of the upper gastrointestinal tract, with the main takeaway being the device’s potency in diagnosing achalasia.
“Although the strongest data appear to be focused on the management of achalasia, emerging evidence supports the clinical relevance of FLIP in the assessment of disease severity and as an outcome measure in [eosinophilic esophagitis (EoE)] intervention trials,” wrote the authors of the update, led by John E. Pandolfino, MD, of Northwestern University, Chicago. The report is in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.
10.022).
In terms of evaluating the LES, however, FLIP can be used during laparoscopic Heller myotomy or peroral endoscopic myotomy (POEM) as a way of monitoring the LES. Using FLIP this way can help clinicians and surgeons personalize the procedure to each patient, even while it’s ongoing. FLIP also can be used with dilation balloons, with the balloon diameter allowing dilation measurement without the need to also use fluoroscopy.
For treating gastroesophageal reflux disease (GERD), the evidence found in existing literature points with less certainty toward use of FLIP.
“The role of FLIP for physiologic evaluation and management in GERD remains appealing; however, the level of evidence is low and currently FLIP should not be used in routine GERD management,” the authors explained. “Future outcome studies are needed to substantiate the utility of FLIP in GERD and to develop metrics that predict severity and treatment response after antireflux procedures.”
FLIP can be used in managing eosinophilic esophagitis, but is recommended only in certain scenarios. According to the authors, FLIP can be used to measure esophageal narrowing and the overall esophageal body. FLIP also can be used to measure esophageal distensibility, and, in the case of at least one study reviewed by the authors, allows “significantly greater accuracy and precision in estimating the effects of remodeling” in certain patients.
Dr. Pandolfino and his colleagues warned that “current recommendations are limited by the low level of evidence and lack of generalized availability of the analysis paradigms.” They noted the need for “further outcome studies that validate the distensibility plateau threshold and further refinements in software analyses to make this methodology more generalizable.”
Overall, the authors concluded, more study still needs to be done to ascertain exactly what FLIP is capable of and when it can be used to greatest effect. In addition to evaluating its benefit in patients with GERD, research should focus on how to make data obtained via FLIP easier to interpret and put to use.
“More work is needed [that] focuses on optimizing data analysis, standardizing protocols, and defining outcome metrics prior to the widespread adoption [of FLIP] into general clinical practice,” the authors wrote.
Dr. Pandolfino disclosed relationships with Medtronic and Sandhill Scientific. Other coauthors did not report any relevant financial disclosures.
*This story updated on 3/9/2017.
New clinical practice advice has been issued for use of the functional lumen imaging probe (FLIP) to assess disorders of the upper gastrointestinal tract, with the main takeaway being the device’s potency in diagnosing achalasia.
“Although the strongest data appear to be focused on the management of achalasia, emerging evidence supports the clinical relevance of FLIP in the assessment of disease severity and as an outcome measure in [eosinophilic esophagitis (EoE)] intervention trials,” wrote the authors of the update, led by John E. Pandolfino, MD, of Northwestern University, Chicago. The report is in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.
10.022).
In terms of evaluating the LES, however, FLIP can be used during laparoscopic Heller myotomy or peroral endoscopic myotomy (POEM) as a way of monitoring the LES. Using FLIP this way can help clinicians and surgeons personalize the procedure to each patient, even while it’s ongoing. FLIP also can be used with dilation balloons, with the balloon diameter allowing dilation measurement without the need to also use fluoroscopy.
For treating gastroesophageal reflux disease (GERD), the evidence found in existing literature points with less certainty toward use of FLIP.
“The role of FLIP for physiologic evaluation and management in GERD remains appealing; however, the level of evidence is low and currently FLIP should not be used in routine GERD management,” the authors explained. “Future outcome studies are needed to substantiate the utility of FLIP in GERD and to develop metrics that predict severity and treatment response after antireflux procedures.”
FLIP can be used in managing eosinophilic esophagitis, but is recommended only in certain scenarios. According to the authors, FLIP can be used to measure esophageal narrowing and the overall esophageal body. FLIP also can be used to measure esophageal distensibility, and, in the case of at least one study reviewed by the authors, allows “significantly greater accuracy and precision in estimating the effects of remodeling” in certain patients.
Dr. Pandolfino and his colleagues warned that “current recommendations are limited by the low level of evidence and lack of generalized availability of the analysis paradigms.” They noted the need for “further outcome studies that validate the distensibility plateau threshold and further refinements in software analyses to make this methodology more generalizable.”
Overall, the authors concluded, more study still needs to be done to ascertain exactly what FLIP is capable of and when it can be used to greatest effect. In addition to evaluating its benefit in patients with GERD, research should focus on how to make data obtained via FLIP easier to interpret and put to use.
“More work is needed [that] focuses on optimizing data analysis, standardizing protocols, and defining outcome metrics prior to the widespread adoption [of FLIP] into general clinical practice,” the authors wrote.
Dr. Pandolfino disclosed relationships with Medtronic and Sandhill Scientific. Other coauthors did not report any relevant financial disclosures.
*This story updated on 3/9/2017.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
AGA Clinical Practice Update: Best practice advice on EBT use released
The AGA Institute has released a series of new best practice statements that gastroenterologists should use when considering a patient for endoscopic bariatric treatments or surgeries (EBTs).
“There is a need for less-invasive weight loss therapies that are more effective and durable than lifestyle interventions alone, less invasive and risky than bariatric surgery, and easily performed at a lower expense than that of surgery, thereby allowing improved access and application to a larger segment of the population with moderate obesity,” wrote the authors of the expert review, led by Barham K. Abu Dayyeh, MD of the Mayo Clinic in Rochester, Minn. The report is in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.035). “[EBTs] potentially meet these criteria and may provide an effective treatment approach to obesity in selected patients.”

The best practice statements come from a review of relevant studies in the Ovid, MEDLINE, EMBASE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus databases, among others, that were published between Jan. 1, 2000, and Sept. 30, 2016.
EBTs should be used on patients who have already been unable to lose weight despite lifestyle interventions and more traditional weight loss methods. However, patients that undergo EBTs should also be placed on a weight loss regimen that includes diet, exercise, and lifestyle changes.

In addition to being used for weight loss, they can also be used to transition a patient to traditional bariatric surgery, or to lower a patient’s weight so that they can undergo a different procedure unrelated to bariatric surgery. Anyone being considered for EBT, or a weight loss regimen involving EBT, should be thoroughly evaluated for comorbidities, behavior, or medical concerns that could lead to adverse effects.
Any patients who are placed on EBT regimens should be followed up regularly by their clinicians, to monitor their progress in terms of weight loss and the development of any adverse effects. Should any adverse outcomes arise, alternative therapies should be implemented as soon as possible. Clinicians are advised to know the ins and outs of risks, contraindications, and potential complications related to EBTs before ever implementing them in their practice, let alone recommending them to a patient.
Finally, it’s imperative that health care institutions with EBT programs make sure there are training protocols clinicians must stringently follow before being allowed to perform EBT procedures.
“Moving ahead, it will be important to better incorporate training in obesity management principles into the GI fellowship curriculum to have a more significant impact,” the authors wrote, adding that it’s important to study the “tandem and sequential use of a combination of EBTs and obesity pharmacotherapies in addition to a comprehensive life-style intervention program.”
Dr. Abu Dayyeh disclosed relationships with Apollo Endosurgery, Metamodix, Aspire Bariatric, and GI Dynamics. Other coauthors also disclosed potential conflicting interests.
The AGA Institute has released a series of new best practice statements that gastroenterologists should use when considering a patient for endoscopic bariatric treatments or surgeries (EBTs).
“There is a need for less-invasive weight loss therapies that are more effective and durable than lifestyle interventions alone, less invasive and risky than bariatric surgery, and easily performed at a lower expense than that of surgery, thereby allowing improved access and application to a larger segment of the population with moderate obesity,” wrote the authors of the expert review, led by Barham K. Abu Dayyeh, MD of the Mayo Clinic in Rochester, Minn. The report is in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.035). “[EBTs] potentially meet these criteria and may provide an effective treatment approach to obesity in selected patients.”
The best practice statements come from a review of relevant studies in the Ovid, MEDLINE, EMBASE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus databases, among others, that were published between Jan. 1, 2000, and Sept. 30, 2016.
EBTs should be used on patients who have already been unable to lose weight despite lifestyle interventions and more traditional weight loss methods. However, patients that undergo EBTs should also be placed on a weight loss regimen that includes diet, exercise, and lifestyle changes.
In addition to being used for weight loss, they can also be used to transition a patient to traditional bariatric surgery, or to lower a patient’s weight so that they can undergo a different procedure unrelated to bariatric surgery. Anyone being considered for EBT, or a weight loss regimen involving EBT, should be thoroughly evaluated for comorbidities, behavior, or medical concerns that could lead to adverse effects.
Any patients who are placed on EBT regimens should be followed up regularly by their clinicians, to monitor their progress in terms of weight loss and the development of any adverse effects. Should any adverse outcomes arise, alternative therapies should be implemented as soon as possible. Clinicians are advised to know the ins and outs of risks, contraindications, and potential complications related to EBTs before ever implementing them in their practice, let alone recommending them to a patient.
Finally, it’s imperative that health care institutions with EBT programs make sure there are training protocols clinicians must stringently follow before being allowed to perform EBT procedures.
“Moving ahead, it will be important to better incorporate training in obesity management principles into the GI fellowship curriculum to have a more significant impact,” the authors wrote, adding that it’s important to study the “tandem and sequential use of a combination of EBTs and obesity pharmacotherapies in addition to a comprehensive life-style intervention program.”
Dr. Abu Dayyeh disclosed relationships with Apollo Endosurgery, Metamodix, Aspire Bariatric, and GI Dynamics. Other coauthors also disclosed potential conflicting interests.
The AGA Institute has released a series of new best practice statements that gastroenterologists should use when considering a patient for endoscopic bariatric treatments or surgeries (EBTs).
“There is a need for less-invasive weight loss therapies that are more effective and durable than lifestyle interventions alone, less invasive and risky than bariatric surgery, and easily performed at a lower expense than that of surgery, thereby allowing improved access and application to a larger segment of the population with moderate obesity,” wrote the authors of the expert review, led by Barham K. Abu Dayyeh, MD of the Mayo Clinic in Rochester, Minn. The report is in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.035). “[EBTs] potentially meet these criteria and may provide an effective treatment approach to obesity in selected patients.”
The best practice statements come from a review of relevant studies in the Ovid, MEDLINE, EMBASE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus databases, among others, that were published between Jan. 1, 2000, and Sept. 30, 2016.
EBTs should be used on patients who have already been unable to lose weight despite lifestyle interventions and more traditional weight loss methods. However, patients that undergo EBTs should also be placed on a weight loss regimen that includes diet, exercise, and lifestyle changes.
In addition to being used for weight loss, they can also be used to transition a patient to traditional bariatric surgery, or to lower a patient’s weight so that they can undergo a different procedure unrelated to bariatric surgery. Anyone being considered for EBT, or a weight loss regimen involving EBT, should be thoroughly evaluated for comorbidities, behavior, or medical concerns that could lead to adverse effects.
Any patients who are placed on EBT regimens should be followed up regularly by their clinicians, to monitor their progress in terms of weight loss and the development of any adverse effects. Should any adverse outcomes arise, alternative therapies should be implemented as soon as possible. Clinicians are advised to know the ins and outs of risks, contraindications, and potential complications related to EBTs before ever implementing them in their practice, let alone recommending them to a patient.
Finally, it’s imperative that health care institutions with EBT programs make sure there are training protocols clinicians must stringently follow before being allowed to perform EBT procedures.
“Moving ahead, it will be important to better incorporate training in obesity management principles into the GI fellowship curriculum to have a more significant impact,” the authors wrote, adding that it’s important to study the “tandem and sequential use of a combination of EBTs and obesity pharmacotherapies in addition to a comprehensive life-style intervention program.”
Dr. Abu Dayyeh disclosed relationships with Apollo Endosurgery, Metamodix, Aspire Bariatric, and GI Dynamics. Other coauthors also disclosed potential conflicting interests.
FROM GASTROENTEROLOGY

AGA Clinical Practice Update: PPIs should be prescribed sparingly, carefully
Updated best practice statements regarding the use of proton pump inhibitors first detail what types of patients should be using short and long-term PPIs.
“When PPIs are appropriately prescribed, their benefits are likely to outweigh their risks [but] when PPIs are inappropriately prescribed, modest risks become important because there is no potential benefit,” wrote the authors of the updated guidance, published in the March issue of Gastroenterology.
“There is currently insufficient evidence to recommend specific strategies for mitigating PPI adverse effects,” noted Daniel E. Freedberg, MD, of Columbia University, New York, and his colleagues.
PPIs should be used on a short-term basis for individuals with gastroesophageal reflux disease (GERD) or conditions such as erosive esophagitis. These patients can also use PPIs for maintenance and occasional symptom management, but those with uncomplicated GERD should be weaned off PPIs if they respond favorably to them.
If a patient is unable to be weaned off PPIs, then ambulatory esophageal pH and impedance monitoring should be done, as this will allow clinicians to determine if the patient has a functional syndrome or GERD. Lifelong PPI treatment should not be considered until this step is taken, according to the new best practice statements.
“Short-term PPIs are highly effective for uncomplicated GERD [but] because patients who cannot reduce PPIs face lifelong therapy, we would consider testing for an acid-related disorder in this situation,” the authors explained. “However, there is no high-quality evidence on which to base this recommendation.”
Patients who have symptomatic GERD or Barrett’s esophagus, either symptomatic or asymptomatic, should be on long-term PPI treatment. Patients who are at a higher risk for NSAID-induced ulcer bleeding should be taking PPIs if they continue to take NSAIDs.
When recommending long-term PPI treatment for a patient, the patient need not use probiotics on a regular basis; there appears to be no need to routinely check the patient’s bone mineral density, serum creatinine, magnesium, or vitamin B12 level on a regular basis. In addition, they need not consume more than the Recommended Dietary Allowance of calcium, magnesium, or vitamin B12.
Finally, the authors state that “specific PPI formulations should not be selected based on potential risks.” This is because no evidence has been found indicating that PPI formulations can be ranked in any way based on risk.
These recommendations come from the AGA’s Clinical Practice Updates Committee, which pored through studies published through July 2016 in the PubMed, EMbase, and Cochrane library databases. Expert opinions and quality assessments on each study contributed to forming these best practice statements.
“In sum, the best current strategies for mitigating the potential risks of long-term PPIs are to avoid prescribing them when they are not indicated and to reduce them to their minimum dose when they are indicated,” Dr. Freedberg and his colleagues concluded.
The researchers did not report any relevant financial disclosures.
Updated best practice statements regarding the use of proton pump inhibitors first detail what types of patients should be using short and long-term PPIs.
“When PPIs are appropriately prescribed, their benefits are likely to outweigh their risks [but] when PPIs are inappropriately prescribed, modest risks become important because there is no potential benefit,” wrote the authors of the updated guidance, published in the March issue of Gastroenterology.
“There is currently insufficient evidence to recommend specific strategies for mitigating PPI adverse effects,” noted Daniel E. Freedberg, MD, of Columbia University, New York, and his colleagues.
PPIs should be used on a short-term basis for individuals with gastroesophageal reflux disease (GERD) or conditions such as erosive esophagitis. These patients can also use PPIs for maintenance and occasional symptom management, but those with uncomplicated GERD should be weaned off PPIs if they respond favorably to them.
If a patient is unable to be weaned off PPIs, then ambulatory esophageal pH and impedance monitoring should be done, as this will allow clinicians to determine if the patient has a functional syndrome or GERD. Lifelong PPI treatment should not be considered until this step is taken, according to the new best practice statements.
“Short-term PPIs are highly effective for uncomplicated GERD [but] because patients who cannot reduce PPIs face lifelong therapy, we would consider testing for an acid-related disorder in this situation,” the authors explained. “However, there is no high-quality evidence on which to base this recommendation.”
Patients who have symptomatic GERD or Barrett’s esophagus, either symptomatic or asymptomatic, should be on long-term PPI treatment. Patients who are at a higher risk for NSAID-induced ulcer bleeding should be taking PPIs if they continue to take NSAIDs.
When recommending long-term PPI treatment for a patient, the patient need not use probiotics on a regular basis; there appears to be no need to routinely check the patient’s bone mineral density, serum creatinine, magnesium, or vitamin B12 level on a regular basis. In addition, they need not consume more than the Recommended Dietary Allowance of calcium, magnesium, or vitamin B12.
Finally, the authors state that “specific PPI formulations should not be selected based on potential risks.” This is because no evidence has been found indicating that PPI formulations can be ranked in any way based on risk.
These recommendations come from the AGA’s Clinical Practice Updates Committee, which pored through studies published through July 2016 in the PubMed, EMbase, and Cochrane library databases. Expert opinions and quality assessments on each study contributed to forming these best practice statements.
“In sum, the best current strategies for mitigating the potential risks of long-term PPIs are to avoid prescribing them when they are not indicated and to reduce them to their minimum dose when they are indicated,” Dr. Freedberg and his colleagues concluded.
The researchers did not report any relevant financial disclosures.
Updated best practice statements regarding the use of proton pump inhibitors first detail what types of patients should be using short and long-term PPIs.
“When PPIs are appropriately prescribed, their benefits are likely to outweigh their risks [but] when PPIs are inappropriately prescribed, modest risks become important because there is no potential benefit,” wrote the authors of the updated guidance, published in the March issue of Gastroenterology.
“There is currently insufficient evidence to recommend specific strategies for mitigating PPI adverse effects,” noted Daniel E. Freedberg, MD, of Columbia University, New York, and his colleagues.
PPIs should be used on a short-term basis for individuals with gastroesophageal reflux disease (GERD) or conditions such as erosive esophagitis. These patients can also use PPIs for maintenance and occasional symptom management, but those with uncomplicated GERD should be weaned off PPIs if they respond favorably to them.
If a patient is unable to be weaned off PPIs, then ambulatory esophageal pH and impedance monitoring should be done, as this will allow clinicians to determine if the patient has a functional syndrome or GERD. Lifelong PPI treatment should not be considered until this step is taken, according to the new best practice statements.
“Short-term PPIs are highly effective for uncomplicated GERD [but] because patients who cannot reduce PPIs face lifelong therapy, we would consider testing for an acid-related disorder in this situation,” the authors explained. “However, there is no high-quality evidence on which to base this recommendation.”
Patients who have symptomatic GERD or Barrett’s esophagus, either symptomatic or asymptomatic, should be on long-term PPI treatment. Patients who are at a higher risk for NSAID-induced ulcer bleeding should be taking PPIs if they continue to take NSAIDs.
When recommending long-term PPI treatment for a patient, the patient need not use probiotics on a regular basis; there appears to be no need to routinely check the patient’s bone mineral density, serum creatinine, magnesium, or vitamin B12 level on a regular basis. In addition, they need not consume more than the Recommended Dietary Allowance of calcium, magnesium, or vitamin B12.
Finally, the authors state that “specific PPI formulations should not be selected based on potential risks.” This is because no evidence has been found indicating that PPI formulations can be ranked in any way based on risk.
These recommendations come from the AGA’s Clinical Practice Updates Committee, which pored through studies published through July 2016 in the PubMed, EMbase, and Cochrane library databases. Expert opinions and quality assessments on each study contributed to forming these best practice statements.
“In sum, the best current strategies for mitigating the potential risks of long-term PPIs are to avoid prescribing them when they are not indicated and to reduce them to their minimum dose when they are indicated,” Dr. Freedberg and his colleagues concluded.
The researchers did not report any relevant financial disclosures.
FROM GASTROENTEROLOGY
March 2017 Quiz 2
Q2: Answer: B
This patient, with no imaging or laboratory findings to suggest cirrhosis, most likely has noncirrhotic portal hypertension (NCPH). There is now a well-described association between HIV and NCPH with the prevalence of NCPH in HIV estimated to be –0.5% to 1%. Patients typically are unaware of any underlying liver disease until presentation with variceal bleeding. Variceal bleeding is a much more common manifestation of NCPH than ascites. Clinical presentation with normal hepatic enzymes and normal hepatic synthetic function is a very typical feature in these patients. Although the exact etiology is not fully understood, NCPH in HIV is likely related to HAART, particularly didanosine use, hypercoagulability, microbial translocation from the gut, and direct effects of HIV. NCPH is a presinusoidal lesion, and liver biopsy may reveal paucity of portal vasculature and focal obliteration of small portal veins. Portal vein thrombosis in patients with HIV and NCPH is common and has been observed in 25%-75% of patients.
Reference
1. Vispo E., Morello J., Rodriguez-Novoa S., Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8.
2. Khanna R., Sarin S.K. Noncirrhotic portal hypertension – Diagnosis and management. J Hepatol. 2014;60:421-41.
Q2: Answer: B
This patient, with no imaging or laboratory findings to suggest cirrhosis, most likely has noncirrhotic portal hypertension (NCPH). There is now a well-described association between HIV and NCPH with the prevalence of NCPH in HIV estimated to be –0.5% to 1%. Patients typically are unaware of any underlying liver disease until presentation with variceal bleeding. Variceal bleeding is a much more common manifestation of NCPH than ascites. Clinical presentation with normal hepatic enzymes and normal hepatic synthetic function is a very typical feature in these patients. Although the exact etiology is not fully understood, NCPH in HIV is likely related to HAART, particularly didanosine use, hypercoagulability, microbial translocation from the gut, and direct effects of HIV. NCPH is a presinusoidal lesion, and liver biopsy may reveal paucity of portal vasculature and focal obliteration of small portal veins. Portal vein thrombosis in patients with HIV and NCPH is common and has been observed in 25%-75% of patients.
Reference
1. Vispo E., Morello J., Rodriguez-Novoa S., Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8.
2. Khanna R., Sarin S.K. Noncirrhotic portal hypertension – Diagnosis and management. J Hepatol. 2014;60:421-41.
Q2: Answer: B
This patient, with no imaging or laboratory findings to suggest cirrhosis, most likely has noncirrhotic portal hypertension (NCPH). There is now a well-described association between HIV and NCPH with the prevalence of NCPH in HIV estimated to be –0.5% to 1%. Patients typically are unaware of any underlying liver disease until presentation with variceal bleeding. Variceal bleeding is a much more common manifestation of NCPH than ascites. Clinical presentation with normal hepatic enzymes and normal hepatic synthetic function is a very typical feature in these patients. Although the exact etiology is not fully understood, NCPH in HIV is likely related to HAART, particularly didanosine use, hypercoagulability, microbial translocation from the gut, and direct effects of HIV. NCPH is a presinusoidal lesion, and liver biopsy may reveal paucity of portal vasculature and focal obliteration of small portal veins. Portal vein thrombosis in patients with HIV and NCPH is common and has been observed in 25%-75% of patients.
Reference
1. Vispo E., Morello J., Rodriguez-Novoa S., Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8.
2. Khanna R., Sarin S.K. Noncirrhotic portal hypertension – Diagnosis and management. J Hepatol. 2014;60:421-41.
Q2: A 52-year-old man with history of recurrent variceal bleeding presents for evaluation. He has an HIV infection that is controlled, with undetectable virus and CD4 count of 423 cells/mcL. He has no known underlying liver disease. He is currently on etravirine, emtricitabine, and tenofovir. He has previously taken didanosine. His physical exam is unremarkable and his laboratory data reveals a normal CBC, normal INR, and normal liver enzymes. Testing for hepatitis B and C and autoimmune liver disease, as well as iron overload and other etiologies of chronic liver disease are all negative. Ultrasound of the abdomen notes a normal-appearing liver and patent portal and hepatic veins. A liver biopsy demonstrates mildly dilated portal veins and mild fibrosis of the portal venous walls. There is no evidence of cirrhosis on the liver biopsy.
March 2017 Quiz 1
Q1: Answer: A
Critique: This is a classic presentation of eosinophilic esophagitis (EoE). As many as half of older children with food impactions suffer from EoE. EoE is characterized by a severe, eosinophilic infiltration of the esophagus that may respond to acid inhibition, systemic or topical steroid therapy, or removal of dietary allergens. Epidemiologic studies suggest a rising incidence in the United States in both children and adults, with at least one case occurring in every 10,000 children each year. Treatment is aimed at alleviating symptoms and healing esophageal inflammation. Allergy testing should be performed at the time of diagnosis; however, radioallergosorbent tests and skin-prick tests are often negative, and only half of affected children have a antecedent history of other allergic symptoms.
A five-food elimination diet can be helpful for many affected children and adults, although adherence to the diet can be difficult. There is a group of affected children who respond to high doses of proton pump inhibitors, and most patients respond to either systemic or topical steroid therapy. Even with therapy, some patients go on to develop esophageal strictures and may need serial or repeated dilatations.
While eosinophilic infiltration and inflammation may be present with gastroesophageal reflux disease and associated esophagitis, the number of eosinophils seen in this boy’s biopsies is much more consistent with EoE. Moreover, stricture formation as a result of peptic esophagitis in a child this age would be extremely rare. While inflammatory bowel disease may be associated with eosinophilic infiltration of the intestinal tract, isolated esophageal Crohn’s disease would be extraordinarily rare.Our patient has no history of any immune deficiency or steroid use that would predispose to fungal esophagitis. Achalasia typically presents with gradually worsening symptoms, and the obstruction would be at the lower esophageal sphincter, not in the mid-esophagus.
Reference
1. Liacouras C., Furuta G., Hirano I., et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3-20.
2. Furuta G., Liacouras C., Collins M., et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
Q1: Answer: A
Critique: This is a classic presentation of eosinophilic esophagitis (EoE). As many as half of older children with food impactions suffer from EoE. EoE is characterized by a severe, eosinophilic infiltration of the esophagus that may respond to acid inhibition, systemic or topical steroid therapy, or removal of dietary allergens. Epidemiologic studies suggest a rising incidence in the United States in both children and adults, with at least one case occurring in every 10,000 children each year. Treatment is aimed at alleviating symptoms and healing esophageal inflammation. Allergy testing should be performed at the time of diagnosis; however, radioallergosorbent tests and skin-prick tests are often negative, and only half of affected children have a antecedent history of other allergic symptoms.
A five-food elimination diet can be helpful for many affected children and adults, although adherence to the diet can be difficult. There is a group of affected children who respond to high doses of proton pump inhibitors, and most patients respond to either systemic or topical steroid therapy. Even with therapy, some patients go on to develop esophageal strictures and may need serial or repeated dilatations.
While eosinophilic infiltration and inflammation may be present with gastroesophageal reflux disease and associated esophagitis, the number of eosinophils seen in this boy’s biopsies is much more consistent with EoE. Moreover, stricture formation as a result of peptic esophagitis in a child this age would be extremely rare. While inflammatory bowel disease may be associated with eosinophilic infiltration of the intestinal tract, isolated esophageal Crohn’s disease would be extraordinarily rare.Our patient has no history of any immune deficiency or steroid use that would predispose to fungal esophagitis. Achalasia typically presents with gradually worsening symptoms, and the obstruction would be at the lower esophageal sphincter, not in the mid-esophagus.
Reference
1. Liacouras C., Furuta G., Hirano I., et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3-20.
2. Furuta G., Liacouras C., Collins M., et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
Q1: Answer: A
Critique: This is a classic presentation of eosinophilic esophagitis (EoE). As many as half of older children with food impactions suffer from EoE. EoE is characterized by a severe, eosinophilic infiltration of the esophagus that may respond to acid inhibition, systemic or topical steroid therapy, or removal of dietary allergens. Epidemiologic studies suggest a rising incidence in the United States in both children and adults, with at least one case occurring in every 10,000 children each year. Treatment is aimed at alleviating symptoms and healing esophageal inflammation. Allergy testing should be performed at the time of diagnosis; however, radioallergosorbent tests and skin-prick tests are often negative, and only half of affected children have a antecedent history of other allergic symptoms.
A five-food elimination diet can be helpful for many affected children and adults, although adherence to the diet can be difficult. There is a group of affected children who respond to high doses of proton pump inhibitors, and most patients respond to either systemic or topical steroid therapy. Even with therapy, some patients go on to develop esophageal strictures and may need serial or repeated dilatations.
While eosinophilic infiltration and inflammation may be present with gastroesophageal reflux disease and associated esophagitis, the number of eosinophils seen in this boy’s biopsies is much more consistent with EoE. Moreover, stricture formation as a result of peptic esophagitis in a child this age would be extremely rare. While inflammatory bowel disease may be associated with eosinophilic infiltration of the intestinal tract, isolated esophageal Crohn’s disease would be extraordinarily rare.Our patient has no history of any immune deficiency or steroid use that would predispose to fungal esophagitis. Achalasia typically presents with gradually worsening symptoms, and the obstruction would be at the lower esophageal sphincter, not in the mid-esophagus.
Reference
1. Liacouras C., Furuta G., Hirano I., et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3-20.
2. Furuta G., Liacouras C., Collins M., et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
Q1: A 14-year-old boy with a history of mild seasonal allergies presents to the emergency room with chest pain and discomfort after eating a steak 2 hours ago. He is having trouble swallowing and feels there is a piece of food stuck in his chest, and he points to his mid-sternum. He tells you this has happened several other times over the past year, and he felt better after he vomited. His physical examination is entirely normal. He is taken to the operating room for emergency endoscopy where a large piece of steak is removed from his mid-esophagus, without complication. Biopsies of the mid-esophagus demonstrate acute and chronic inflammatory changes in the lamina propria with 35 eosinophils per high-powered field.