User login
Autonomic dysfunction: A guide for FPs
Signs and symptoms of autonomic dysfunction commonly present in the primary care setting. Potential causes of dysfunction include certain medications and age-related changes in physiology, as well as conditions such as diabetes mellitus, multiple sclerosis, and Parkinson’s disease (TABLE1). This evidence-based review details common manifestations of autonomic dysfunction, provides a streamlined approach to patients presenting with symptoms, and reviews appropriate step-wise management.

When a delicate balance is disrupted
The autonomic nervous system provides brisk physiologic adjustments necessary to maintain homeostasis. Physiologic functions impacted by the central nervous system include: heart rate, blood pressure (BP), tone of the bladder sphincter and detrusor muscle, bowel motility, bronchodilation and constriction, pupillary dilation and constriction, sweating, catecholamine release, erection, ejaculation and orgasm, tearing, and salivation.1
Disorders of the autonomic system may result from pathologies of the central or peripheral nervous system or from medications including some antihypertensives, selective serotonin-reuptake inhibitors (SSRIs), and opioids.1 Such disorders tend to be grouped into one of 3 categories: those involving the brain, those involving the spinal cord, and autonomic neuropathies.1
The source of dysautonomia can often be determined by clinical context, coexisting neurologic abnormalities, targeted testing of the autonomic nervous system, and neuroimaging.1
Worrisome symptoms prompt a visit
A thorough history is critical to zeroing in on a patient’s complaints and ultimately providing treatment that will help manage symptoms.
When patient complaints are suggestive of autonomic dysfunction, a review of systems should include inquiry about lightheadedness, abnormal salivation, temperature changes of the extremities, gastrointestinal issues (vomiting, constipation, or diarrhea), and symptoms of presyncope/syncope or urinary or sexual dysfunction.1 The physical exam should include recordings of BP and heart rate in the supine and standing positions and a complete neurologic examination.1 Findings will typically point to one or more common complications.
Common complications of autonomic dysfunction
Complications of autonomic dysfunction include impotence, bladder dysfunction, gastrointestinal (GI) dysfunction, and orthostatic hypotension and vasomotor abnormalities. A less common condition—autonomic dysreflexia, which is a distinct type of autonomic dysfunction, and a true medical emergency—is also important to keep in mind.
Impotence
Autonomic neuropathy is a common cause of impotence and retrograde ejaculation. Loss of early morning erections and complete loss of nocturnal erections often have an etiology related to vascular disease and/or autonomic neuropathy. In addition, poor glycemic control and vascular risk factors appear to be associated with the development of diabetic autonomic neuropathy.2
Development of an erection requires an increase in parasympathetic activity and a decrease in sympathetic output. Nocturnal penile tumescence testing has been used to infer parasympathetic damage to the penis in men with diabetes who do not have vascular disease.3
First- and second-line agents. Phosphodiesterase-5 inhibitors (eg, sildenafil, tadalafil, vardenafil) have demonstrated efficacy in improving the ability to achieve and maintain erections in patients with autonomic dysfunction, including diabetic autonomic neuropathy.4-6 Second-line therapies with proven efficacy include intraurethral application and intracavernosal injections of alprostadil.7,8
Bladder dysfunction
Sympathetic activity increases bladder sphincter tone and inhibits detrusor activity, while the parasympathetic nervous system increases detrusor activity and decreases sphincter tone to aid in voiding.1 Disrupted autonomic activity can lead to urinary frequency, retention, and hesitancy; overactive bladder; and incontinence.1 Brain and spinal cord disease above the level of the lumbar spine results in urinary frequency and small bladder volumes, whereas diseases involving autonomic nerve fibers to and from the bladder result in large bladder volumes and overflow incontinence.9
Patients presenting with lower urinary tract symptoms require a comprehensive evaluation to rule out other pathologies, as the differential for such symptoms is broad and includes infection, malignancies, interstitial cystitis, and bladder stones. The initial evaluation of lower urinary tract symptoms should include a history and physical exam including that of the abdomen, pelvis, and neurologic system. Lab work should assess renal function and blood glucose, and should include urinalysis and culture to rule out infection and/or hematuria. A prostate-specific antigen (PSA) test may be appropriate in men with a life expectancy >10 years, after counseling regarding the risks and benefits of screening.
Anticholinergic drugs with antimuscarinic effects, such as oxybutynin, may be used to treat symptoms of urge incontinence and overactive bladder. They work to suppress involuntary contractions of the bladder’s smooth muscle by blocking the release of acetylcholine. These medications relax the bladder’s outer layer of muscle—the detrusor. Such medications often have a number of anticholinergic adverse effects, such as dry mouth and constipation, sometimes leading to discontinuation. A post-void residual (PVR) test may be helpful in guiding management. For example, caution should be used in patients with elevated PVRs, as anticholinergics can worsen urinary retention.
Beta-3 agonists (eg, mirabegron) are a novel class of medications used to treat overactive bladder. These medications act to increase sympathetic tone in the bladder. Because they have the potential to raise BP, monitor BP in patients taking these agents. In addition, monitor patients taking antimuscarinics or beta-3 agonists for the development of urinary retention.
Other tests, treatments. Urodynamic testing is recommended for patients who fail to respond to treatment. Combining behavioral therapy with medication has been shown to be effective in patients with urge incontinence.10 Botulinum toxin type A, injected directly into the detrusor muscle, can be as effective as medication in patients with urinary urge incontinence.11
Detrusor underactivity is defined as contraction of reduced strength and/or duration, resulting in prolonged bladder emptying and/or a failure to achieve complete bladder emptying within a normal timespan.12 This diagnosis is typically made using urodynamic testing.13 PVRs ≥150 mL are considered evidence of urinary retention. Overflow incontinence can result from detrusor underactivity.
Consider a trial of a cholinergic agonist, such as bethanechol, in patients with urinary retention. Some patients will require intermittent straight catheterization or chronic indwelling foley or suprapubic catheters to void.
Gastrointestinal dysfunction
In patients with diabetes, GI autonomic neuropathy can result in altered esophageal motility leading to gastroesophageal reflux disease (GERD) or dysphagia, gastroparesis, or diabetic enteropathy.14 Gastroparesis often presents as nausea, vomiting, and bloating.1 It may be diagnosed via gastric emptying studies (scintigraphy), and often requires a multidimensional approach to treatment.
Management. Food may be chopped or pureed to aid in digestion. Metoclopramide is the most commonly used prokinetic agent, but avoid its use in patients with parkinsonism. In more severe cases, consider adding domperidone and erythromycin as prokinetic agents. Recommend antiemetics, such as diphenhydramine, ondansetron, and prochlorperazine for management of nausea and vomiting. Severe cases of gastroparesis may merit a venting gastrostomy tube for decompression and/or feeding via a jejunostomy tube.15 Impaired intestinal mobility may lead to stasis syndrome, causing diarrhea.
Hypermobility caused by decreased sympathetic inhibition can also contribute to diarrhea. Altered anal sphincter function tone may contribute to fecal incontinence. Management should focus on balancing electrolytes, maintaining adequate fluid intake, and relieving symptoms. Consider antidiarrheals such as loperamide, but use them with caution to avoid toxic megacolon.16
Constipation. Another common manifestation of autonomic dysfunction in the GI tract is severe constipation.1 This may be managed conservatively with hydration, increased activity, and increased fiber intake. If such measures prove inadequate, consider stool softeners and laxatives.
Patients with constipation due to spinal cord lesions may benefit from a routine bowel regimen. To provide predictable defecation, advise patients to begin by inserting a stimulant rectal suppository. Follow with gentle digital stimulation of the distal rectum for one minute or less. They’ll need to repeat the process every 5 to 10 minutes until stool evacuation is complete. A forward-leaning position may assist with evacuation. It is helpful to perform this routine at the same time each day.17
Orthostatic (postural) hypotension
The autonomic nervous system plays an important role in maintaining BP during positional changes. The sympathetic nervous system adjusts the tone in arteries, veins, and the heart. Baroreceptors located primarily in the carotid arteries and aorta, are highly sensitive to changes in BP. When the baroreceptors sense the slightest drop in pressure, a coordinated increase in sympathetic outflow occurs. Arteries constrict to increase peripheral resistance and BP, and heart rate and contractility increase, all in an attempt to maintain BP and perfusion.18
The most common causes of orthostatic hypotension are not neurologic in origin,9 but rather involve medications, hypovolemia, and impaired autonomic reflexes. The condition is common in the elderly, with one study demonstrating a prevalence of 18.2% in those ≥65 years.19
Orthostatic hypotension may present with dimming or loss of vision, lightheadedness, diaphoresis, diminished hearing, pallor, and weakness. As a result, it is a risk factor for falls. Syncope results when the drop in BP impairs cerebral perfusion. Signs of impaired baroreflexes are supine hypertension, a heart rate that is fixed regardless of posture (the heart rate should increase upon standing), postprandial hypotension, and an excessively high nocturnal BP.1
Orthostatic hypotension is diagnosed when, within 3 minutes of quiet standing after a 5-minute period of supine rest, one or both of the following is present: at least a 20 mm Hg-fall in systolic pressure or at least a 10 mm Hg-fall in diastolic pressure.20 Soysal et al demonstrated that such a drop in BP, measured one minute after standing, is adequate and effective for diagnosing orthostatic hypotension in the elderly.21
Nonpharmacologic management. Recognition and removal of medications that can exacerbate orthostatic hypotension is the first step in managing the condition. Such medications include diuretics, beta-blockers, alpha adrenergic blockers, vasodilators, antipsychotics, antidepressants (SSRIs, trazodone, monoamine oxidase inhibitors, and tricyclic antidepressants), phosphodiesterase inhibitors, narcotics, and antiparkinsonian medications.22
Lifestyle interventions, such as having the patient arise slowly and maintain good hydration, can be helpful. Eating smaller, more frequent meals may also help if the orthostatic hypotension is triggered postprandially. Compressive stockings can help limit venous pooling in the lower extremities and improve venous return. Tensing the legs by crossing them while standing on both feet has been shown to increase cardiac output and BP.23 An aerobic exercise regimen of walking or stair climbing 30 to 45 minutes/day 3 days/week for 6 months was shown to eliminate symptoms of orthostasis on tilt table testing in elderly patients with cardiac deconditioning, as opposed to chronic autonomic failure.24
The reduction in central blood volume associated with autonomic insufficiency (due to increased urinary sodium and water excretion) can be lessened by increasing sodium and water intake.25-27
Pharmacotherapy. Fludrocortisone acetate, a synthetic mineralocorticoid, is the medication of first choice for most patients with orthostatic hypotension whose symptoms are not adequately controlled using nonpharmacologic measures,28 but keep in mind that treating orthostatic hypotension with fludrocortisones is an off-label use of the medication.
Monitor patients taking fludrocortisone for worsened supine hypertension and edema. Also, check their serum potassium levels one to 2 weeks after initiation of therapy and after dose increases. Frequent home monitoring of BP in sitting, standing, and supine positions may be helpful in assessing response to therapy.
If the patient remains symptomatic despite therapy with fludrocortisone, consider adding an alpha-1 adrenergic agonist, such as midodrine. Avoid prescribing midodrine, however, for patients with advanced cardiovascular disease, urinary retention, or uncontrolled hypertension.29
Autonomic dysreflexia: A medical emergency
Autonomic dysreflexia, a medical emergency that must be recognized immediately, is a distinct type of autonomic dysfunction seen in patients with spinal cord injury at or above the T6 level.30 It is a condition of uncontrolled sympathetic response secondary to an underlying condition such as infection, urinary retention, or rectal distention.30
Common symptoms include headache, significant hypertension, flushing of the skin, and diaphoresis above the level of injury.2 In addition, a review of systems should screen for fever, visual changes, abnormalities of the cardiovascular system, syncope, bowel and bladder symptoms, and sexual dysfunction.
Patients demonstrating autonomic dysreflexia should be placed in the upright position to produce an orthostatic decrease in BP.30 Patients should be evaluated to identify any reversible precipitants, such as urinary retention or fecal impaction. Severe attacks involving hypertensive crisis require prompt transfer to the emergency department. Sublingual nifedipine or an intravenous agent, such as hydralazine, may be used to lower BP.31
CORRESPONDENCE
Kristen Thornton, MD, 777 South Clinton Ave., Rochester, NY 14620; [email protected]
1. Low PA, Engstrom JW. Disorders of the autonomic nervous system. In: Kasper D, Fauci A, Hauser S, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015. Available at: http://accessmedicine.mhmedical.com/content.aspx?bookid=1130&Sectionid=79755967. Accessed May 15, 2016.
2. Ko SH, Park SA, Cho JH, et al. Progression of cardiovascular dysfunction in patients with type 2 diabetes: a 7 year follow-up study. Diabetes Care. 2008;31:1832-1836.
3. Brown JS, Wessells H, Chancellor MB, et al. Urologic complications of diabetes. Diabetes Care. 2005;28:177-185.
4. Rendell MS, Rajfer J, Wicker PA, et al. Sildenafil for treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. Sildenafil Diabetes Study Group. JAMA. 1999;281:421-426.
5. Goldstein I, Young JM, Fischer J, et al. Vardenafil, a new phosphodiesterase type 5 inhibitor, in the treatment of erectile dysfunction in men with diabetes: a multicenter double-blind placebo-controlled fixed-dose study. Diabetes Care. 2003;26:777-783.
6. Sáenz de Tejada I, Anglin G, Knight JR, et al. Effects of tadalafil on erectile dysfunction in men with diabetes. Diabetes Care. 2002;25:2159-2164.
7. Padma-Nathan H, Hellstrom WJ, Kaiser FE, et al. Treatment of men with erectile dysfunction with transurethral alprostadil. Medicated Urethral System for Erection (MUSE) Study Group. N Engl J Med. 1997;336:1-7.
8. Linet OI, Ogrinc FG. Efficacy and safety of intracavernosal alprostadil in men with erectile dysfunction. The Alprostadil Study Group. N Engl J Med. 1996;334:873-877.
9. Engstrom JW, Maring JB. Disorders of the autonomic nervous system. In: Braunwald E, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 15th ed. New York, NY: McGraw Hill; 2001.
10. Burgio KL, Locher JL, Goode PS. Combined behavioral and drug therapy for urge incontinence in older women. J Am Geriatr Soc. 2000;48:370-374.
11. Visco AG, Brubaker L, Richter HE, et al. Anticholinergic therapy vs. onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med. 2012;367:1803-1813.
12. Haylen BT, de Ridder D, Freeman RM, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Neurourol Urodyn. 2010;29:4-20.
13. Osman NI, Chapple CR, Abrams P, et al. Detrusor underactivity and the underactive bladder: a new clinical entity? A review of current terminology, definitions, epidemiology, aetiology, and diagnosis. Eur Urol. 2014;65:389-398.
14. Kempler P, Amarenco G, Freeman R, et al. Management strategies for gastrointestinal, erectile, bladder, and sudomotor dysfunction in patients with diabetes. Diabetes Metab Res Rev. 2011;27:665-677.
15. Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med. 2007;356:820-829.
16. Shakil A, Church RJ, Rao SS. Gastrointestinal complications of diabetes. Am Fam Physician. 2008;77:1697-1702.
17. Krassioukov A, Eng JJ, Claxton G, et al. Neurogenic bowel management after spinal cord injury: a systematic review of the evidence. Spinal Cord. 2010;48:718-733.
18. Bradley JG, Davis K. Orthostatic hypotension. Am Fam Physician. 2003;68:2393-2399.
19. Rutan GH, Hermanson B, Bild DE, et al. Orthostatic hypotension in older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Hypertension. 1992;19(6 Pt 1):508-519.
20. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21:69-72.
21. Soysal P, Aydin AE, Koc Okudur S, et al. When should orthostatic BP changes be evaluated in elderly: 1st, 3rd or 5th minute? Arch Gerontol Geriatr. 2016;65:199-203.
22. Perlmuter LC, Sarda G, Casavant V, et al. A review of the etiology, associated comorbidities, and treatment of orthostatic hypotension. Am J Ther. 2013;20:279-291.
23. Ten Harkel ADJ, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci. 1994;87:553-558.
24. Carroll JF, Wood CE, Pollock ML, et al. Hormonal responses in elders experiencing pre-syncopal symptoms during head-up tilt before and after exercise training. J Gerontol A Biol Sci Med Sci. 1995;50:M324-M329.
25. Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med. 2002;112:355-360.
26. Young T, Mathias C. The effects of water ingestion on orthostatic hypotension in two groups of chronic autonomic failure: multiple system atrophy and pure autonomic failure. J Neurol Neurosurg Psychiatry. 2004;75:1737-1741.
27. Humm AM, Mason LM, Mathias CJ. Effects of water drinking on cardiovascular responses to supine exercise and on orthostatic hypotension after exercise in pure autonomic failure. J Neurol Neurosurg Psychiatry. 2008;79:1160-1164.
28. Campbell IW, Ewing DJ, Clarke BF. 9-Alpha-fluorohydrocortisone in the treatment of postural hypotension in diabetic autonomic neuropathy. Diabetes. 1975;24:381-384.
29. Raj SR, Coffin ST. Medical therapy and physical maneuvers in the treatment of the vasovagal syncope and orthostatic hypotension. Prog Cardiovasc Dis. 2013;55:425-433.
30. Karlsson AK. Autonomic dysreflexia. Spinal Cord. 1999;37:383-391.
31. Bycroft J, Shergill IS, Choong EAL, et al. Autonomic dysreflexia: a medical emergency. Postgrad Med J. 2005;81:232-235.
Signs and symptoms of autonomic dysfunction commonly present in the primary care setting. Potential causes of dysfunction include certain medications and age-related changes in physiology, as well as conditions such as diabetes mellitus, multiple sclerosis, and Parkinson’s disease (TABLE1). This evidence-based review details common manifestations of autonomic dysfunction, provides a streamlined approach to patients presenting with symptoms, and reviews appropriate step-wise management.
When a delicate balance is disrupted
The autonomic nervous system provides brisk physiologic adjustments necessary to maintain homeostasis. Physiologic functions impacted by the central nervous system include: heart rate, blood pressure (BP), tone of the bladder sphincter and detrusor muscle, bowel motility, bronchodilation and constriction, pupillary dilation and constriction, sweating, catecholamine release, erection, ejaculation and orgasm, tearing, and salivation.1
Disorders of the autonomic system may result from pathologies of the central or peripheral nervous system or from medications including some antihypertensives, selective serotonin-reuptake inhibitors (SSRIs), and opioids.1 Such disorders tend to be grouped into one of 3 categories: those involving the brain, those involving the spinal cord, and autonomic neuropathies.1
The source of dysautonomia can often be determined by clinical context, coexisting neurologic abnormalities, targeted testing of the autonomic nervous system, and neuroimaging.1
Worrisome symptoms prompt a visit
A thorough history is critical to zeroing in on a patient’s complaints and ultimately providing treatment that will help manage symptoms.
When patient complaints are suggestive of autonomic dysfunction, a review of systems should include inquiry about lightheadedness, abnormal salivation, temperature changes of the extremities, gastrointestinal issues (vomiting, constipation, or diarrhea), and symptoms of presyncope/syncope or urinary or sexual dysfunction.1 The physical exam should include recordings of BP and heart rate in the supine and standing positions and a complete neurologic examination.1 Findings will typically point to one or more common complications.
Common complications of autonomic dysfunction
Complications of autonomic dysfunction include impotence, bladder dysfunction, gastrointestinal (GI) dysfunction, and orthostatic hypotension and vasomotor abnormalities. A less common condition—autonomic dysreflexia, which is a distinct type of autonomic dysfunction, and a true medical emergency—is also important to keep in mind.
Impotence
Autonomic neuropathy is a common cause of impotence and retrograde ejaculation. Loss of early morning erections and complete loss of nocturnal erections often have an etiology related to vascular disease and/or autonomic neuropathy. In addition, poor glycemic control and vascular risk factors appear to be associated with the development of diabetic autonomic neuropathy.2
Development of an erection requires an increase in parasympathetic activity and a decrease in sympathetic output. Nocturnal penile tumescence testing has been used to infer parasympathetic damage to the penis in men with diabetes who do not have vascular disease.3
First- and second-line agents. Phosphodiesterase-5 inhibitors (eg, sildenafil, tadalafil, vardenafil) have demonstrated efficacy in improving the ability to achieve and maintain erections in patients with autonomic dysfunction, including diabetic autonomic neuropathy.4-6 Second-line therapies with proven efficacy include intraurethral application and intracavernosal injections of alprostadil.7,8
Bladder dysfunction
Sympathetic activity increases bladder sphincter tone and inhibits detrusor activity, while the parasympathetic nervous system increases detrusor activity and decreases sphincter tone to aid in voiding.1 Disrupted autonomic activity can lead to urinary frequency, retention, and hesitancy; overactive bladder; and incontinence.1 Brain and spinal cord disease above the level of the lumbar spine results in urinary frequency and small bladder volumes, whereas diseases involving autonomic nerve fibers to and from the bladder result in large bladder volumes and overflow incontinence.9
Patients presenting with lower urinary tract symptoms require a comprehensive evaluation to rule out other pathologies, as the differential for such symptoms is broad and includes infection, malignancies, interstitial cystitis, and bladder stones. The initial evaluation of lower urinary tract symptoms should include a history and physical exam including that of the abdomen, pelvis, and neurologic system. Lab work should assess renal function and blood glucose, and should include urinalysis and culture to rule out infection and/or hematuria. A prostate-specific antigen (PSA) test may be appropriate in men with a life expectancy >10 years, after counseling regarding the risks and benefits of screening.
Anticholinergic drugs with antimuscarinic effects, such as oxybutynin, may be used to treat symptoms of urge incontinence and overactive bladder. They work to suppress involuntary contractions of the bladder’s smooth muscle by blocking the release of acetylcholine. These medications relax the bladder’s outer layer of muscle—the detrusor. Such medications often have a number of anticholinergic adverse effects, such as dry mouth and constipation, sometimes leading to discontinuation. A post-void residual (PVR) test may be helpful in guiding management. For example, caution should be used in patients with elevated PVRs, as anticholinergics can worsen urinary retention.
Beta-3 agonists (eg, mirabegron) are a novel class of medications used to treat overactive bladder. These medications act to increase sympathetic tone in the bladder. Because they have the potential to raise BP, monitor BP in patients taking these agents. In addition, monitor patients taking antimuscarinics or beta-3 agonists for the development of urinary retention.
Other tests, treatments. Urodynamic testing is recommended for patients who fail to respond to treatment. Combining behavioral therapy with medication has been shown to be effective in patients with urge incontinence.10 Botulinum toxin type A, injected directly into the detrusor muscle, can be as effective as medication in patients with urinary urge incontinence.11
Detrusor underactivity is defined as contraction of reduced strength and/or duration, resulting in prolonged bladder emptying and/or a failure to achieve complete bladder emptying within a normal timespan.12 This diagnosis is typically made using urodynamic testing.13 PVRs ≥150 mL are considered evidence of urinary retention. Overflow incontinence can result from detrusor underactivity.
Consider a trial of a cholinergic agonist, such as bethanechol, in patients with urinary retention. Some patients will require intermittent straight catheterization or chronic indwelling foley or suprapubic catheters to void.
Gastrointestinal dysfunction
In patients with diabetes, GI autonomic neuropathy can result in altered esophageal motility leading to gastroesophageal reflux disease (GERD) or dysphagia, gastroparesis, or diabetic enteropathy.14 Gastroparesis often presents as nausea, vomiting, and bloating.1 It may be diagnosed via gastric emptying studies (scintigraphy), and often requires a multidimensional approach to treatment.
Management. Food may be chopped or pureed to aid in digestion. Metoclopramide is the most commonly used prokinetic agent, but avoid its use in patients with parkinsonism. In more severe cases, consider adding domperidone and erythromycin as prokinetic agents. Recommend antiemetics, such as diphenhydramine, ondansetron, and prochlorperazine for management of nausea and vomiting. Severe cases of gastroparesis may merit a venting gastrostomy tube for decompression and/or feeding via a jejunostomy tube.15 Impaired intestinal mobility may lead to stasis syndrome, causing diarrhea.
Hypermobility caused by decreased sympathetic inhibition can also contribute to diarrhea. Altered anal sphincter function tone may contribute to fecal incontinence. Management should focus on balancing electrolytes, maintaining adequate fluid intake, and relieving symptoms. Consider antidiarrheals such as loperamide, but use them with caution to avoid toxic megacolon.16
Constipation. Another common manifestation of autonomic dysfunction in the GI tract is severe constipation.1 This may be managed conservatively with hydration, increased activity, and increased fiber intake. If such measures prove inadequate, consider stool softeners and laxatives.
Patients with constipation due to spinal cord lesions may benefit from a routine bowel regimen. To provide predictable defecation, advise patients to begin by inserting a stimulant rectal suppository. Follow with gentle digital stimulation of the distal rectum for one minute or less. They’ll need to repeat the process every 5 to 10 minutes until stool evacuation is complete. A forward-leaning position may assist with evacuation. It is helpful to perform this routine at the same time each day.17
Orthostatic (postural) hypotension
The autonomic nervous system plays an important role in maintaining BP during positional changes. The sympathetic nervous system adjusts the tone in arteries, veins, and the heart. Baroreceptors located primarily in the carotid arteries and aorta, are highly sensitive to changes in BP. When the baroreceptors sense the slightest drop in pressure, a coordinated increase in sympathetic outflow occurs. Arteries constrict to increase peripheral resistance and BP, and heart rate and contractility increase, all in an attempt to maintain BP and perfusion.18
The most common causes of orthostatic hypotension are not neurologic in origin,9 but rather involve medications, hypovolemia, and impaired autonomic reflexes. The condition is common in the elderly, with one study demonstrating a prevalence of 18.2% in those ≥65 years.19
Orthostatic hypotension may present with dimming or loss of vision, lightheadedness, diaphoresis, diminished hearing, pallor, and weakness. As a result, it is a risk factor for falls. Syncope results when the drop in BP impairs cerebral perfusion. Signs of impaired baroreflexes are supine hypertension, a heart rate that is fixed regardless of posture (the heart rate should increase upon standing), postprandial hypotension, and an excessively high nocturnal BP.1
Orthostatic hypotension is diagnosed when, within 3 minutes of quiet standing after a 5-minute period of supine rest, one or both of the following is present: at least a 20 mm Hg-fall in systolic pressure or at least a 10 mm Hg-fall in diastolic pressure.20 Soysal et al demonstrated that such a drop in BP, measured one minute after standing, is adequate and effective for diagnosing orthostatic hypotension in the elderly.21
Nonpharmacologic management. Recognition and removal of medications that can exacerbate orthostatic hypotension is the first step in managing the condition. Such medications include diuretics, beta-blockers, alpha adrenergic blockers, vasodilators, antipsychotics, antidepressants (SSRIs, trazodone, monoamine oxidase inhibitors, and tricyclic antidepressants), phosphodiesterase inhibitors, narcotics, and antiparkinsonian medications.22
Lifestyle interventions, such as having the patient arise slowly and maintain good hydration, can be helpful. Eating smaller, more frequent meals may also help if the orthostatic hypotension is triggered postprandially. Compressive stockings can help limit venous pooling in the lower extremities and improve venous return. Tensing the legs by crossing them while standing on both feet has been shown to increase cardiac output and BP.23 An aerobic exercise regimen of walking or stair climbing 30 to 45 minutes/day 3 days/week for 6 months was shown to eliminate symptoms of orthostasis on tilt table testing in elderly patients with cardiac deconditioning, as opposed to chronic autonomic failure.24
The reduction in central blood volume associated with autonomic insufficiency (due to increased urinary sodium and water excretion) can be lessened by increasing sodium and water intake.25-27
Pharmacotherapy. Fludrocortisone acetate, a synthetic mineralocorticoid, is the medication of first choice for most patients with orthostatic hypotension whose symptoms are not adequately controlled using nonpharmacologic measures,28 but keep in mind that treating orthostatic hypotension with fludrocortisones is an off-label use of the medication.
Monitor patients taking fludrocortisone for worsened supine hypertension and edema. Also, check their serum potassium levels one to 2 weeks after initiation of therapy and after dose increases. Frequent home monitoring of BP in sitting, standing, and supine positions may be helpful in assessing response to therapy.
If the patient remains symptomatic despite therapy with fludrocortisone, consider adding an alpha-1 adrenergic agonist, such as midodrine. Avoid prescribing midodrine, however, for patients with advanced cardiovascular disease, urinary retention, or uncontrolled hypertension.29
Autonomic dysreflexia: A medical emergency
Autonomic dysreflexia, a medical emergency that must be recognized immediately, is a distinct type of autonomic dysfunction seen in patients with spinal cord injury at or above the T6 level.30 It is a condition of uncontrolled sympathetic response secondary to an underlying condition such as infection, urinary retention, or rectal distention.30
Common symptoms include headache, significant hypertension, flushing of the skin, and diaphoresis above the level of injury.2 In addition, a review of systems should screen for fever, visual changes, abnormalities of the cardiovascular system, syncope, bowel and bladder symptoms, and sexual dysfunction.
Patients demonstrating autonomic dysreflexia should be placed in the upright position to produce an orthostatic decrease in BP.30 Patients should be evaluated to identify any reversible precipitants, such as urinary retention or fecal impaction. Severe attacks involving hypertensive crisis require prompt transfer to the emergency department. Sublingual nifedipine or an intravenous agent, such as hydralazine, may be used to lower BP.31
CORRESPONDENCE
Kristen Thornton, MD, 777 South Clinton Ave., Rochester, NY 14620; [email protected]
Signs and symptoms of autonomic dysfunction commonly present in the primary care setting. Potential causes of dysfunction include certain medications and age-related changes in physiology, as well as conditions such as diabetes mellitus, multiple sclerosis, and Parkinson’s disease (TABLE1). This evidence-based review details common manifestations of autonomic dysfunction, provides a streamlined approach to patients presenting with symptoms, and reviews appropriate step-wise management.
When a delicate balance is disrupted
The autonomic nervous system provides brisk physiologic adjustments necessary to maintain homeostasis. Physiologic functions impacted by the central nervous system include: heart rate, blood pressure (BP), tone of the bladder sphincter and detrusor muscle, bowel motility, bronchodilation and constriction, pupillary dilation and constriction, sweating, catecholamine release, erection, ejaculation and orgasm, tearing, and salivation.1
Disorders of the autonomic system may result from pathologies of the central or peripheral nervous system or from medications including some antihypertensives, selective serotonin-reuptake inhibitors (SSRIs), and opioids.1 Such disorders tend to be grouped into one of 3 categories: those involving the brain, those involving the spinal cord, and autonomic neuropathies.1
The source of dysautonomia can often be determined by clinical context, coexisting neurologic abnormalities, targeted testing of the autonomic nervous system, and neuroimaging.1
Worrisome symptoms prompt a visit
A thorough history is critical to zeroing in on a patient’s complaints and ultimately providing treatment that will help manage symptoms.
When patient complaints are suggestive of autonomic dysfunction, a review of systems should include inquiry about lightheadedness, abnormal salivation, temperature changes of the extremities, gastrointestinal issues (vomiting, constipation, or diarrhea), and symptoms of presyncope/syncope or urinary or sexual dysfunction.1 The physical exam should include recordings of BP and heart rate in the supine and standing positions and a complete neurologic examination.1 Findings will typically point to one or more common complications.
Common complications of autonomic dysfunction
Complications of autonomic dysfunction include impotence, bladder dysfunction, gastrointestinal (GI) dysfunction, and orthostatic hypotension and vasomotor abnormalities. A less common condition—autonomic dysreflexia, which is a distinct type of autonomic dysfunction, and a true medical emergency—is also important to keep in mind.
Impotence
Autonomic neuropathy is a common cause of impotence and retrograde ejaculation. Loss of early morning erections and complete loss of nocturnal erections often have an etiology related to vascular disease and/or autonomic neuropathy. In addition, poor glycemic control and vascular risk factors appear to be associated with the development of diabetic autonomic neuropathy.2
Development of an erection requires an increase in parasympathetic activity and a decrease in sympathetic output. Nocturnal penile tumescence testing has been used to infer parasympathetic damage to the penis in men with diabetes who do not have vascular disease.3
First- and second-line agents. Phosphodiesterase-5 inhibitors (eg, sildenafil, tadalafil, vardenafil) have demonstrated efficacy in improving the ability to achieve and maintain erections in patients with autonomic dysfunction, including diabetic autonomic neuropathy.4-6 Second-line therapies with proven efficacy include intraurethral application and intracavernosal injections of alprostadil.7,8
Bladder dysfunction
Sympathetic activity increases bladder sphincter tone and inhibits detrusor activity, while the parasympathetic nervous system increases detrusor activity and decreases sphincter tone to aid in voiding.1 Disrupted autonomic activity can lead to urinary frequency, retention, and hesitancy; overactive bladder; and incontinence.1 Brain and spinal cord disease above the level of the lumbar spine results in urinary frequency and small bladder volumes, whereas diseases involving autonomic nerve fibers to and from the bladder result in large bladder volumes and overflow incontinence.9
Patients presenting with lower urinary tract symptoms require a comprehensive evaluation to rule out other pathologies, as the differential for such symptoms is broad and includes infection, malignancies, interstitial cystitis, and bladder stones. The initial evaluation of lower urinary tract symptoms should include a history and physical exam including that of the abdomen, pelvis, and neurologic system. Lab work should assess renal function and blood glucose, and should include urinalysis and culture to rule out infection and/or hematuria. A prostate-specific antigen (PSA) test may be appropriate in men with a life expectancy >10 years, after counseling regarding the risks and benefits of screening.
Anticholinergic drugs with antimuscarinic effects, such as oxybutynin, may be used to treat symptoms of urge incontinence and overactive bladder. They work to suppress involuntary contractions of the bladder’s smooth muscle by blocking the release of acetylcholine. These medications relax the bladder’s outer layer of muscle—the detrusor. Such medications often have a number of anticholinergic adverse effects, such as dry mouth and constipation, sometimes leading to discontinuation. A post-void residual (PVR) test may be helpful in guiding management. For example, caution should be used in patients with elevated PVRs, as anticholinergics can worsen urinary retention.
Beta-3 agonists (eg, mirabegron) are a novel class of medications used to treat overactive bladder. These medications act to increase sympathetic tone in the bladder. Because they have the potential to raise BP, monitor BP in patients taking these agents. In addition, monitor patients taking antimuscarinics or beta-3 agonists for the development of urinary retention.
Other tests, treatments. Urodynamic testing is recommended for patients who fail to respond to treatment. Combining behavioral therapy with medication has been shown to be effective in patients with urge incontinence.10 Botulinum toxin type A, injected directly into the detrusor muscle, can be as effective as medication in patients with urinary urge incontinence.11
Detrusor underactivity is defined as contraction of reduced strength and/or duration, resulting in prolonged bladder emptying and/or a failure to achieve complete bladder emptying within a normal timespan.12 This diagnosis is typically made using urodynamic testing.13 PVRs ≥150 mL are considered evidence of urinary retention. Overflow incontinence can result from detrusor underactivity.
Consider a trial of a cholinergic agonist, such as bethanechol, in patients with urinary retention. Some patients will require intermittent straight catheterization or chronic indwelling foley or suprapubic catheters to void.
Gastrointestinal dysfunction
In patients with diabetes, GI autonomic neuropathy can result in altered esophageal motility leading to gastroesophageal reflux disease (GERD) or dysphagia, gastroparesis, or diabetic enteropathy.14 Gastroparesis often presents as nausea, vomiting, and bloating.1 It may be diagnosed via gastric emptying studies (scintigraphy), and often requires a multidimensional approach to treatment.
Management. Food may be chopped or pureed to aid in digestion. Metoclopramide is the most commonly used prokinetic agent, but avoid its use in patients with parkinsonism. In more severe cases, consider adding domperidone and erythromycin as prokinetic agents. Recommend antiemetics, such as diphenhydramine, ondansetron, and prochlorperazine for management of nausea and vomiting. Severe cases of gastroparesis may merit a venting gastrostomy tube for decompression and/or feeding via a jejunostomy tube.15 Impaired intestinal mobility may lead to stasis syndrome, causing diarrhea.
Hypermobility caused by decreased sympathetic inhibition can also contribute to diarrhea. Altered anal sphincter function tone may contribute to fecal incontinence. Management should focus on balancing electrolytes, maintaining adequate fluid intake, and relieving symptoms. Consider antidiarrheals such as loperamide, but use them with caution to avoid toxic megacolon.16
Constipation. Another common manifestation of autonomic dysfunction in the GI tract is severe constipation.1 This may be managed conservatively with hydration, increased activity, and increased fiber intake. If such measures prove inadequate, consider stool softeners and laxatives.
Patients with constipation due to spinal cord lesions may benefit from a routine bowel regimen. To provide predictable defecation, advise patients to begin by inserting a stimulant rectal suppository. Follow with gentle digital stimulation of the distal rectum for one minute or less. They’ll need to repeat the process every 5 to 10 minutes until stool evacuation is complete. A forward-leaning position may assist with evacuation. It is helpful to perform this routine at the same time each day.17
Orthostatic (postural) hypotension
The autonomic nervous system plays an important role in maintaining BP during positional changes. The sympathetic nervous system adjusts the tone in arteries, veins, and the heart. Baroreceptors located primarily in the carotid arteries and aorta, are highly sensitive to changes in BP. When the baroreceptors sense the slightest drop in pressure, a coordinated increase in sympathetic outflow occurs. Arteries constrict to increase peripheral resistance and BP, and heart rate and contractility increase, all in an attempt to maintain BP and perfusion.18
The most common causes of orthostatic hypotension are not neurologic in origin,9 but rather involve medications, hypovolemia, and impaired autonomic reflexes. The condition is common in the elderly, with one study demonstrating a prevalence of 18.2% in those ≥65 years.19
Orthostatic hypotension may present with dimming or loss of vision, lightheadedness, diaphoresis, diminished hearing, pallor, and weakness. As a result, it is a risk factor for falls. Syncope results when the drop in BP impairs cerebral perfusion. Signs of impaired baroreflexes are supine hypertension, a heart rate that is fixed regardless of posture (the heart rate should increase upon standing), postprandial hypotension, and an excessively high nocturnal BP.1
Orthostatic hypotension is diagnosed when, within 3 minutes of quiet standing after a 5-minute period of supine rest, one or both of the following is present: at least a 20 mm Hg-fall in systolic pressure or at least a 10 mm Hg-fall in diastolic pressure.20 Soysal et al demonstrated that such a drop in BP, measured one minute after standing, is adequate and effective for diagnosing orthostatic hypotension in the elderly.21
Nonpharmacologic management. Recognition and removal of medications that can exacerbate orthostatic hypotension is the first step in managing the condition. Such medications include diuretics, beta-blockers, alpha adrenergic blockers, vasodilators, antipsychotics, antidepressants (SSRIs, trazodone, monoamine oxidase inhibitors, and tricyclic antidepressants), phosphodiesterase inhibitors, narcotics, and antiparkinsonian medications.22
Lifestyle interventions, such as having the patient arise slowly and maintain good hydration, can be helpful. Eating smaller, more frequent meals may also help if the orthostatic hypotension is triggered postprandially. Compressive stockings can help limit venous pooling in the lower extremities and improve venous return. Tensing the legs by crossing them while standing on both feet has been shown to increase cardiac output and BP.23 An aerobic exercise regimen of walking or stair climbing 30 to 45 minutes/day 3 days/week for 6 months was shown to eliminate symptoms of orthostasis on tilt table testing in elderly patients with cardiac deconditioning, as opposed to chronic autonomic failure.24
The reduction in central blood volume associated with autonomic insufficiency (due to increased urinary sodium and water excretion) can be lessened by increasing sodium and water intake.25-27
Pharmacotherapy. Fludrocortisone acetate, a synthetic mineralocorticoid, is the medication of first choice for most patients with orthostatic hypotension whose symptoms are not adequately controlled using nonpharmacologic measures,28 but keep in mind that treating orthostatic hypotension with fludrocortisones is an off-label use of the medication.
Monitor patients taking fludrocortisone for worsened supine hypertension and edema. Also, check their serum potassium levels one to 2 weeks after initiation of therapy and after dose increases. Frequent home monitoring of BP in sitting, standing, and supine positions may be helpful in assessing response to therapy.
If the patient remains symptomatic despite therapy with fludrocortisone, consider adding an alpha-1 adrenergic agonist, such as midodrine. Avoid prescribing midodrine, however, for patients with advanced cardiovascular disease, urinary retention, or uncontrolled hypertension.29
Autonomic dysreflexia: A medical emergency
Autonomic dysreflexia, a medical emergency that must be recognized immediately, is a distinct type of autonomic dysfunction seen in patients with spinal cord injury at or above the T6 level.30 It is a condition of uncontrolled sympathetic response secondary to an underlying condition such as infection, urinary retention, or rectal distention.30
Common symptoms include headache, significant hypertension, flushing of the skin, and diaphoresis above the level of injury.2 In addition, a review of systems should screen for fever, visual changes, abnormalities of the cardiovascular system, syncope, bowel and bladder symptoms, and sexual dysfunction.
Patients demonstrating autonomic dysreflexia should be placed in the upright position to produce an orthostatic decrease in BP.30 Patients should be evaluated to identify any reversible precipitants, such as urinary retention or fecal impaction. Severe attacks involving hypertensive crisis require prompt transfer to the emergency department. Sublingual nifedipine or an intravenous agent, such as hydralazine, may be used to lower BP.31
CORRESPONDENCE
Kristen Thornton, MD, 777 South Clinton Ave., Rochester, NY 14620; [email protected]
1. Low PA, Engstrom JW. Disorders of the autonomic nervous system. In: Kasper D, Fauci A, Hauser S, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015. Available at: http://accessmedicine.mhmedical.com/content.aspx?bookid=1130&Sectionid=79755967. Accessed May 15, 2016.
2. Ko SH, Park SA, Cho JH, et al. Progression of cardiovascular dysfunction in patients with type 2 diabetes: a 7 year follow-up study. Diabetes Care. 2008;31:1832-1836.
3. Brown JS, Wessells H, Chancellor MB, et al. Urologic complications of diabetes. Diabetes Care. 2005;28:177-185.
4. Rendell MS, Rajfer J, Wicker PA, et al. Sildenafil for treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. Sildenafil Diabetes Study Group. JAMA. 1999;281:421-426.
5. Goldstein I, Young JM, Fischer J, et al. Vardenafil, a new phosphodiesterase type 5 inhibitor, in the treatment of erectile dysfunction in men with diabetes: a multicenter double-blind placebo-controlled fixed-dose study. Diabetes Care. 2003;26:777-783.
6. Sáenz de Tejada I, Anglin G, Knight JR, et al. Effects of tadalafil on erectile dysfunction in men with diabetes. Diabetes Care. 2002;25:2159-2164.
7. Padma-Nathan H, Hellstrom WJ, Kaiser FE, et al. Treatment of men with erectile dysfunction with transurethral alprostadil. Medicated Urethral System for Erection (MUSE) Study Group. N Engl J Med. 1997;336:1-7.
8. Linet OI, Ogrinc FG. Efficacy and safety of intracavernosal alprostadil in men with erectile dysfunction. The Alprostadil Study Group. N Engl J Med. 1996;334:873-877.
9. Engstrom JW, Maring JB. Disorders of the autonomic nervous system. In: Braunwald E, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 15th ed. New York, NY: McGraw Hill; 2001.
10. Burgio KL, Locher JL, Goode PS. Combined behavioral and drug therapy for urge incontinence in older women. J Am Geriatr Soc. 2000;48:370-374.
11. Visco AG, Brubaker L, Richter HE, et al. Anticholinergic therapy vs. onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med. 2012;367:1803-1813.
12. Haylen BT, de Ridder D, Freeman RM, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Neurourol Urodyn. 2010;29:4-20.
13. Osman NI, Chapple CR, Abrams P, et al. Detrusor underactivity and the underactive bladder: a new clinical entity? A review of current terminology, definitions, epidemiology, aetiology, and diagnosis. Eur Urol. 2014;65:389-398.
14. Kempler P, Amarenco G, Freeman R, et al. Management strategies for gastrointestinal, erectile, bladder, and sudomotor dysfunction in patients with diabetes. Diabetes Metab Res Rev. 2011;27:665-677.
15. Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med. 2007;356:820-829.
16. Shakil A, Church RJ, Rao SS. Gastrointestinal complications of diabetes. Am Fam Physician. 2008;77:1697-1702.
17. Krassioukov A, Eng JJ, Claxton G, et al. Neurogenic bowel management after spinal cord injury: a systematic review of the evidence. Spinal Cord. 2010;48:718-733.
18. Bradley JG, Davis K. Orthostatic hypotension. Am Fam Physician. 2003;68:2393-2399.
19. Rutan GH, Hermanson B, Bild DE, et al. Orthostatic hypotension in older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Hypertension. 1992;19(6 Pt 1):508-519.
20. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21:69-72.
21. Soysal P, Aydin AE, Koc Okudur S, et al. When should orthostatic BP changes be evaluated in elderly: 1st, 3rd or 5th minute? Arch Gerontol Geriatr. 2016;65:199-203.
22. Perlmuter LC, Sarda G, Casavant V, et al. A review of the etiology, associated comorbidities, and treatment of orthostatic hypotension. Am J Ther. 2013;20:279-291.
23. Ten Harkel ADJ, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci. 1994;87:553-558.
24. Carroll JF, Wood CE, Pollock ML, et al. Hormonal responses in elders experiencing pre-syncopal symptoms during head-up tilt before and after exercise training. J Gerontol A Biol Sci Med Sci. 1995;50:M324-M329.
25. Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med. 2002;112:355-360.
26. Young T, Mathias C. The effects of water ingestion on orthostatic hypotension in two groups of chronic autonomic failure: multiple system atrophy and pure autonomic failure. J Neurol Neurosurg Psychiatry. 2004;75:1737-1741.
27. Humm AM, Mason LM, Mathias CJ. Effects of water drinking on cardiovascular responses to supine exercise and on orthostatic hypotension after exercise in pure autonomic failure. J Neurol Neurosurg Psychiatry. 2008;79:1160-1164.
28. Campbell IW, Ewing DJ, Clarke BF. 9-Alpha-fluorohydrocortisone in the treatment of postural hypotension in diabetic autonomic neuropathy. Diabetes. 1975;24:381-384.
29. Raj SR, Coffin ST. Medical therapy and physical maneuvers in the treatment of the vasovagal syncope and orthostatic hypotension. Prog Cardiovasc Dis. 2013;55:425-433.
30. Karlsson AK. Autonomic dysreflexia. Spinal Cord. 1999;37:383-391.
31. Bycroft J, Shergill IS, Choong EAL, et al. Autonomic dysreflexia: a medical emergency. Postgrad Med J. 2005;81:232-235.
1. Low PA, Engstrom JW. Disorders of the autonomic nervous system. In: Kasper D, Fauci A, Hauser S, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015. Available at: http://accessmedicine.mhmedical.com/content.aspx?bookid=1130&Sectionid=79755967. Accessed May 15, 2016.
2. Ko SH, Park SA, Cho JH, et al. Progression of cardiovascular dysfunction in patients with type 2 diabetes: a 7 year follow-up study. Diabetes Care. 2008;31:1832-1836.
3. Brown JS, Wessells H, Chancellor MB, et al. Urologic complications of diabetes. Diabetes Care. 2005;28:177-185.
4. Rendell MS, Rajfer J, Wicker PA, et al. Sildenafil for treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. Sildenafil Diabetes Study Group. JAMA. 1999;281:421-426.
5. Goldstein I, Young JM, Fischer J, et al. Vardenafil, a new phosphodiesterase type 5 inhibitor, in the treatment of erectile dysfunction in men with diabetes: a multicenter double-blind placebo-controlled fixed-dose study. Diabetes Care. 2003;26:777-783.
6. Sáenz de Tejada I, Anglin G, Knight JR, et al. Effects of tadalafil on erectile dysfunction in men with diabetes. Diabetes Care. 2002;25:2159-2164.
7. Padma-Nathan H, Hellstrom WJ, Kaiser FE, et al. Treatment of men with erectile dysfunction with transurethral alprostadil. Medicated Urethral System for Erection (MUSE) Study Group. N Engl J Med. 1997;336:1-7.
8. Linet OI, Ogrinc FG. Efficacy and safety of intracavernosal alprostadil in men with erectile dysfunction. The Alprostadil Study Group. N Engl J Med. 1996;334:873-877.
9. Engstrom JW, Maring JB. Disorders of the autonomic nervous system. In: Braunwald E, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 15th ed. New York, NY: McGraw Hill; 2001.
10. Burgio KL, Locher JL, Goode PS. Combined behavioral and drug therapy for urge incontinence in older women. J Am Geriatr Soc. 2000;48:370-374.
11. Visco AG, Brubaker L, Richter HE, et al. Anticholinergic therapy vs. onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med. 2012;367:1803-1813.
12. Haylen BT, de Ridder D, Freeman RM, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Neurourol Urodyn. 2010;29:4-20.
13. Osman NI, Chapple CR, Abrams P, et al. Detrusor underactivity and the underactive bladder: a new clinical entity? A review of current terminology, definitions, epidemiology, aetiology, and diagnosis. Eur Urol. 2014;65:389-398.
14. Kempler P, Amarenco G, Freeman R, et al. Management strategies for gastrointestinal, erectile, bladder, and sudomotor dysfunction in patients with diabetes. Diabetes Metab Res Rev. 2011;27:665-677.
15. Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med. 2007;356:820-829.
16. Shakil A, Church RJ, Rao SS. Gastrointestinal complications of diabetes. Am Fam Physician. 2008;77:1697-1702.
17. Krassioukov A, Eng JJ, Claxton G, et al. Neurogenic bowel management after spinal cord injury: a systematic review of the evidence. Spinal Cord. 2010;48:718-733.
18. Bradley JG, Davis K. Orthostatic hypotension. Am Fam Physician. 2003;68:2393-2399.
19. Rutan GH, Hermanson B, Bild DE, et al. Orthostatic hypotension in older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Hypertension. 1992;19(6 Pt 1):508-519.
20. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21:69-72.
21. Soysal P, Aydin AE, Koc Okudur S, et al. When should orthostatic BP changes be evaluated in elderly: 1st, 3rd or 5th minute? Arch Gerontol Geriatr. 2016;65:199-203.
22. Perlmuter LC, Sarda G, Casavant V, et al. A review of the etiology, associated comorbidities, and treatment of orthostatic hypotension. Am J Ther. 2013;20:279-291.
23. Ten Harkel ADJ, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci. 1994;87:553-558.
24. Carroll JF, Wood CE, Pollock ML, et al. Hormonal responses in elders experiencing pre-syncopal symptoms during head-up tilt before and after exercise training. J Gerontol A Biol Sci Med Sci. 1995;50:M324-M329.
25. Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med. 2002;112:355-360.
26. Young T, Mathias C. The effects of water ingestion on orthostatic hypotension in two groups of chronic autonomic failure: multiple system atrophy and pure autonomic failure. J Neurol Neurosurg Psychiatry. 2004;75:1737-1741.
27. Humm AM, Mason LM, Mathias CJ. Effects of water drinking on cardiovascular responses to supine exercise and on orthostatic hypotension after exercise in pure autonomic failure. J Neurol Neurosurg Psychiatry. 2008;79:1160-1164.
28. Campbell IW, Ewing DJ, Clarke BF. 9-Alpha-fluorohydrocortisone in the treatment of postural hypotension in diabetic autonomic neuropathy. Diabetes. 1975;24:381-384.
29. Raj SR, Coffin ST. Medical therapy and physical maneuvers in the treatment of the vasovagal syncope and orthostatic hypotension. Prog Cardiovasc Dis. 2013;55:425-433.
30. Karlsson AK. Autonomic dysreflexia. Spinal Cord. 1999;37:383-391.
31. Bycroft J, Shergill IS, Choong EAL, et al. Autonomic dysreflexia: a medical emergency. Postgrad Med J. 2005;81:232-235.
PRACTICE RECOMMENDATIONS
› Begin a trial of an antimuscarinic if initial nonpharmacologic treatment of urge incontinence or overactive bladder is ineffective. B
› Start step-wise treatment beginning with metoclopramide A, followed by domperidone, and, finally, oral erythromycin B in patients with gastroparesis who have failed conservative measures.
› Employ step-wise pharmacologic treatment, starting with fludrocortisone, for patients with disabling symptoms of orthostatic hypotension who fail to respond to nonpharmacologic measures. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
What FPs need to know about naloxone kits

Medical Director,
Office-Based Opioid Therapy
Dept. of Psychiatry and Behavioral Sciences
Mount Sinai Beth Israel, New York, NY
Medical Director,
Office-Based Opioid Therapy
Dept. of Psychiatry and Behavioral Sciences
Mount Sinai Beth Israel, New York, NY
Medical Director,
Office-Based Opioid Therapy
Dept. of Psychiatry and Behavioral Sciences
Mount Sinai Beth Israel, New York, NY
Need an edge with T2DM? The case for team-based care

Preventive upstream therapy prevents progression of atrial fib
BARCELONA – Aggressive treatment of known risk factors for atrial fibrillation resulted in improved 1-year maintenance of sinus rhythm in patients with recent-onset atrial fibrillation and heart failure in the randomized multicenter RACE 3 trial, Isabelle C. van Gelder, MD, reported at the annual congress of the European Society of Cardiology.
“We now screen for AF, making it possible to catch patients early. That’s what we’ve learned from this trial: if we start treating patients after their first episode of AF and aggressively reduce risk factors for AF, it may help the sinus rhythm. I think that’s an important message: do not wait too long, start treatment early,” said Dr. van Gelder, professor of cardiology at the University of Groningen, the Netherlands.
She calls the interventional strategy tested in RACE 3 “risk factor-driven upstream therapy.” The four-pronged strategy consisted of statin therapy, a mineralcorticoid receptor antagonist, an ACE inhibitor and/or an angiotensin receptor blocker, and a 9- to 11-week supervised cardiac rehabilitation program emphasizing lifestyle modification through physical training and dietary changes supported by professional counseling to promote adherence.
“These are interventions designed to improve the atrial substrate,” Dr. van Gelder explained.
RACE 3 (Routine versus Aggressive Upstream Rhythm Control for Prevention of Early Atrial Fibrillation in Heart Failure 3) was a multicenter, randomized, nonblinded clinical trial including 245 patients with, on average, a 3-month history of AF, a 2-month history of persistent AF, and a 2-month history of mild to moderate heart failure, either with preserved or reduced ejection fraction. All participants received guideline-directed rhythm control and heart failure therapies. In addition, half of participants were randomized to the upstream intervention. Three weeks after enrollment, all patients underwent electrical cardioversion.
The primary outcome was maintenance of sinus rhythm at 1 year as determined by 7-day Holter monitoring analyzed in blinded fashion at a central laboratory. The rate was 75% in the upstream intervention group, significantly better than the 63% in controls. This represented a 76% greater likelihood of sinus rhythm at 1 year in the upstream intervention group. They also showed significant reductions in systolic and diastolic blood pressure, N-terminal pro-brain natriuretic peptide, and LDL cholesterol, compared with controls. However, at 1 year, the two groups didn’t differ significantly in body mass index or left atrial volume. The lack of impact on left atrial volume was disappointing, Dr. van Gelder said.
“The remodeling process starts long before the first episode of AF, although we don’t know exactly when. Although we intended to intervene early in the remodeling process, I think we weren’t that early,” according to the cardiologist.
Discussant Josep Brugada, MD, applauded the Dutch investigators for opening the door to evidence-based preventive upstream therapy for AF, which he declared is vital given the worsening AF epidemic.

“In recent years enormous efforts have been put into treating symptoms of AF, but clearly we have failed to control the epidemic of AF in our societies, probably because we’ve been aiming only at treating symptoms, not treating the causes,” observed Dr. Brugada of the University of Barcelona.
He added, however, that the RACE 3 intervention didn’t go far enough.
“It’s a bit of a disappointment that there is no change in BMI seen after 1 year. Zero. That probably means the rehabilitation program wasn’t strong enough. Yet, the study results are positive, so if we used physical training in a stronger way to get a reduction in body weight and BMI, probably the outcome would be even greater,” he said.
To be maximally effective, an upstream intervention for AF should also address two other important risk factors for the arrhythmia: heavy alcohol drinking and obstructive sleep apnea, the electrophysiologist added.
The RACE 3 trial was supported by the Netherlands Heart Foundation and the Netherlands Heart Institute. Dr. van Gelder reported having no relevant financial interests.
Dr. van Gelder discussed the RACE 3 trial and results in a video interview.
BARCELONA – Aggressive treatment of known risk factors for atrial fibrillation resulted in improved 1-year maintenance of sinus rhythm in patients with recent-onset atrial fibrillation and heart failure in the randomized multicenter RACE 3 trial, Isabelle C. van Gelder, MD, reported at the annual congress of the European Society of Cardiology.
“We now screen for AF, making it possible to catch patients early. That’s what we’ve learned from this trial: if we start treating patients after their first episode of AF and aggressively reduce risk factors for AF, it may help the sinus rhythm. I think that’s an important message: do not wait too long, start treatment early,” said Dr. van Gelder, professor of cardiology at the University of Groningen, the Netherlands.
She calls the interventional strategy tested in RACE 3 “risk factor-driven upstream therapy.” The four-pronged strategy consisted of statin therapy, a mineralcorticoid receptor antagonist, an ACE inhibitor and/or an angiotensin receptor blocker, and a 9- to 11-week supervised cardiac rehabilitation program emphasizing lifestyle modification through physical training and dietary changes supported by professional counseling to promote adherence.
“These are interventions designed to improve the atrial substrate,” Dr. van Gelder explained.
RACE 3 (Routine versus Aggressive Upstream Rhythm Control for Prevention of Early Atrial Fibrillation in Heart Failure 3) was a multicenter, randomized, nonblinded clinical trial including 245 patients with, on average, a 3-month history of AF, a 2-month history of persistent AF, and a 2-month history of mild to moderate heart failure, either with preserved or reduced ejection fraction. All participants received guideline-directed rhythm control and heart failure therapies. In addition, half of participants were randomized to the upstream intervention. Three weeks after enrollment, all patients underwent electrical cardioversion.
The primary outcome was maintenance of sinus rhythm at 1 year as determined by 7-day Holter monitoring analyzed in blinded fashion at a central laboratory. The rate was 75% in the upstream intervention group, significantly better than the 63% in controls. This represented a 76% greater likelihood of sinus rhythm at 1 year in the upstream intervention group. They also showed significant reductions in systolic and diastolic blood pressure, N-terminal pro-brain natriuretic peptide, and LDL cholesterol, compared with controls. However, at 1 year, the two groups didn’t differ significantly in body mass index or left atrial volume. The lack of impact on left atrial volume was disappointing, Dr. van Gelder said.
“The remodeling process starts long before the first episode of AF, although we don’t know exactly when. Although we intended to intervene early in the remodeling process, I think we weren’t that early,” according to the cardiologist.
Discussant Josep Brugada, MD, applauded the Dutch investigators for opening the door to evidence-based preventive upstream therapy for AF, which he declared is vital given the worsening AF epidemic.
“In recent years enormous efforts have been put into treating symptoms of AF, but clearly we have failed to control the epidemic of AF in our societies, probably because we’ve been aiming only at treating symptoms, not treating the causes,” observed Dr. Brugada of the University of Barcelona.
He added, however, that the RACE 3 intervention didn’t go far enough.
“It’s a bit of a disappointment that there is no change in BMI seen after 1 year. Zero. That probably means the rehabilitation program wasn’t strong enough. Yet, the study results are positive, so if we used physical training in a stronger way to get a reduction in body weight and BMI, probably the outcome would be even greater,” he said.
To be maximally effective, an upstream intervention for AF should also address two other important risk factors for the arrhythmia: heavy alcohol drinking and obstructive sleep apnea, the electrophysiologist added.
The RACE 3 trial was supported by the Netherlands Heart Foundation and the Netherlands Heart Institute. Dr. van Gelder reported having no relevant financial interests.
Dr. van Gelder discussed the RACE 3 trial and results in a video interview.
BARCELONA – Aggressive treatment of known risk factors for atrial fibrillation resulted in improved 1-year maintenance of sinus rhythm in patients with recent-onset atrial fibrillation and heart failure in the randomized multicenter RACE 3 trial, Isabelle C. van Gelder, MD, reported at the annual congress of the European Society of Cardiology.
“We now screen for AF, making it possible to catch patients early. That’s what we’ve learned from this trial: if we start treating patients after their first episode of AF and aggressively reduce risk factors for AF, it may help the sinus rhythm. I think that’s an important message: do not wait too long, start treatment early,” said Dr. van Gelder, professor of cardiology at the University of Groningen, the Netherlands.
She calls the interventional strategy tested in RACE 3 “risk factor-driven upstream therapy.” The four-pronged strategy consisted of statin therapy, a mineralcorticoid receptor antagonist, an ACE inhibitor and/or an angiotensin receptor blocker, and a 9- to 11-week supervised cardiac rehabilitation program emphasizing lifestyle modification through physical training and dietary changes supported by professional counseling to promote adherence.
“These are interventions designed to improve the atrial substrate,” Dr. van Gelder explained.
RACE 3 (Routine versus Aggressive Upstream Rhythm Control for Prevention of Early Atrial Fibrillation in Heart Failure 3) was a multicenter, randomized, nonblinded clinical trial including 245 patients with, on average, a 3-month history of AF, a 2-month history of persistent AF, and a 2-month history of mild to moderate heart failure, either with preserved or reduced ejection fraction. All participants received guideline-directed rhythm control and heart failure therapies. In addition, half of participants were randomized to the upstream intervention. Three weeks after enrollment, all patients underwent electrical cardioversion.
The primary outcome was maintenance of sinus rhythm at 1 year as determined by 7-day Holter monitoring analyzed in blinded fashion at a central laboratory. The rate was 75% in the upstream intervention group, significantly better than the 63% in controls. This represented a 76% greater likelihood of sinus rhythm at 1 year in the upstream intervention group. They also showed significant reductions in systolic and diastolic blood pressure, N-terminal pro-brain natriuretic peptide, and LDL cholesterol, compared with controls. However, at 1 year, the two groups didn’t differ significantly in body mass index or left atrial volume. The lack of impact on left atrial volume was disappointing, Dr. van Gelder said.
“The remodeling process starts long before the first episode of AF, although we don’t know exactly when. Although we intended to intervene early in the remodeling process, I think we weren’t that early,” according to the cardiologist.
Discussant Josep Brugada, MD, applauded the Dutch investigators for opening the door to evidence-based preventive upstream therapy for AF, which he declared is vital given the worsening AF epidemic.
“In recent years enormous efforts have been put into treating symptoms of AF, but clearly we have failed to control the epidemic of AF in our societies, probably because we’ve been aiming only at treating symptoms, not treating the causes,” observed Dr. Brugada of the University of Barcelona.
He added, however, that the RACE 3 intervention didn’t go far enough.
“It’s a bit of a disappointment that there is no change in BMI seen after 1 year. Zero. That probably means the rehabilitation program wasn’t strong enough. Yet, the study results are positive, so if we used physical training in a stronger way to get a reduction in body weight and BMI, probably the outcome would be even greater,” he said.
To be maximally effective, an upstream intervention for AF should also address two other important risk factors for the arrhythmia: heavy alcohol drinking and obstructive sleep apnea, the electrophysiologist added.
The RACE 3 trial was supported by the Netherlands Heart Foundation and the Netherlands Heart Institute. Dr. van Gelder reported having no relevant financial interests.
Dr. van Gelder discussed the RACE 3 trial and results in a video interview.
AT THE ESC CONGRESS 2017
Key clinical point:
Major finding: At 1 year, 75% of patients with baseline persistent atrial fibrillation who received a four-pronged program of upstream risk factor modification were in sinus rhythm, compared with 63% of controls.
Data source: RACE 3 was a multicenter, randomized, nonblinded clinical trial including 245 patients with a recent history of persistent atrial fibrillation and heart failure.
Disclosures: The RACE 3 trial was supported by the Netherlands Heart Foundation and the Netherlands Heart Institute. The presenter reported having no relevant financial interests.
Individualizing Treatment of Hyperglycemia in Type 2 Diabetes
From the University of Arizona College of Pharmacy and the University of Arizona College of Medicine-Tucson, Tucson, AZ.
Abstract
- Objective: To summarize key issues relevant to managing hyperglycemia in patients with type 2 diabetes mellitus (T2DM) and review a strategy for initiating and intensifying therapy.
- Methods: Review of the literature.
- Results: The 6 most widely used pharmacologic treatment options for hyperglycemia in T2DM are metformin, sulfonylureas, dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter-2 inhibitors, and insulin. Recent guidelines stress the importance of an individualized, patient-centered approach to managing hyperglycemia in T2DM, although sufficient guidance for nonspecialists on how to individualize treatment is often lacking. For patients with no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. Due to the progressive nature of T2DM, glycemic control on metformin monotherapy is likely to deteriorate over time, and there is no consensus as to what the second-line agent should be. A second agent should be selected based on glycemic goal and potential advantages and disadvantages of each agent for any given patient. If the patient progresses to the point where dual therapy does not provide adequate control, either a third non-insulin agent or insulin can be added.
- Conclusion: Although research is increasingly focusing on what the ideal number and sequence of drugs should be when managing T2DM, investigating all possible combinations in diverse patient populations is not feasible. Physicians therefore must continue to rely on clinical judgment to determine how to apply trial data to the treatment of individual patients.
Key words: type 2 diabetes; patient-centered care; antihyper-glycemic drugs; insulin; therapeutic decision-making.
Diabetes mellitus affects approximately 29.1 million people, or 9.3% of the U.S. population [1,2]. The high prevalence of diabetes and its associated multiple complications, including cardiovascular disease (CVD), blindness, renal failure, lower extremity amputations, and premature death, lead to a tremendous overall burden of disease. The financial cost is staggering as well, with more than 1 in 5 health care dollars spent on treating diabetes or its complications [3]. The goal of diabetes treatment is to prevent acute complications and reduce the risk of long-term complications. Interventions that have been shown to improve diabetes outcomes include medications for glycemic control and treatment of cardiovascular risk factors, nutrition and physical activity counseling, smoking cessation, immunizations, psychosocial care, and ongoing surveillance and early treatment for eye, kidney, and foot problems [4].
Glycemic management in type 2 diabetes mellitus (T2DM), the focus of this review, is growing increasingly complex and has been the subject of numerous extensive reviews [5,6] and published guidelines [4,7]. In the context of an increasing array of available pharmacologic options, there are mounting uncertainties regarding the benefits of intensive glycemic control as well as increasing concerns about potential adverse treatment effects, hypoglycemia in particular. While previous guidelines encouraged specific approaches for most patients, more recent guidelines stress the importance of a patient-centered approach with shared decision-making [4]. Less prescriptive guidelines are more appropriate, given the current state of science, but they also may be viewed as providing insufficient guidance to some providers. It can be overwhelming for a non-specialist to try to match the nuances of antihyperglycemic medications to the nuances of each patient’s preferences and medical characteristics.
This article examines key issues faced by primary care providers when managing hyperglycemia in patients with T2DM and outlines a stepwise approach to determining the optimal antihyperglycemic agent(s) (Table 1). 
Confirm Diagnosis of T2DM
It can be difficult to distinguish between type 1 diabetes mellitus and T2DM in some individuals due to overlapping characteristics. However, correctly classifying a patient’s diabetes at the outset is essential, as the classification helps determine the best treatment regimen and is rarely reconsidered [4,8]. Considerable evidence suggests that misclassification of diabetes occurs frequently [9,10], resulting in patients receiving inappropriate treatment. Clinical characteristics suggestive of T2DM include older age and features of insulin resistance such as obesity, hyper-tension, hypertriglyceridemia, and low high-density lipoprotein cholesterol. When these features are not present, an alternate diagnosis should be entertained.

Establish Glycemic Goal
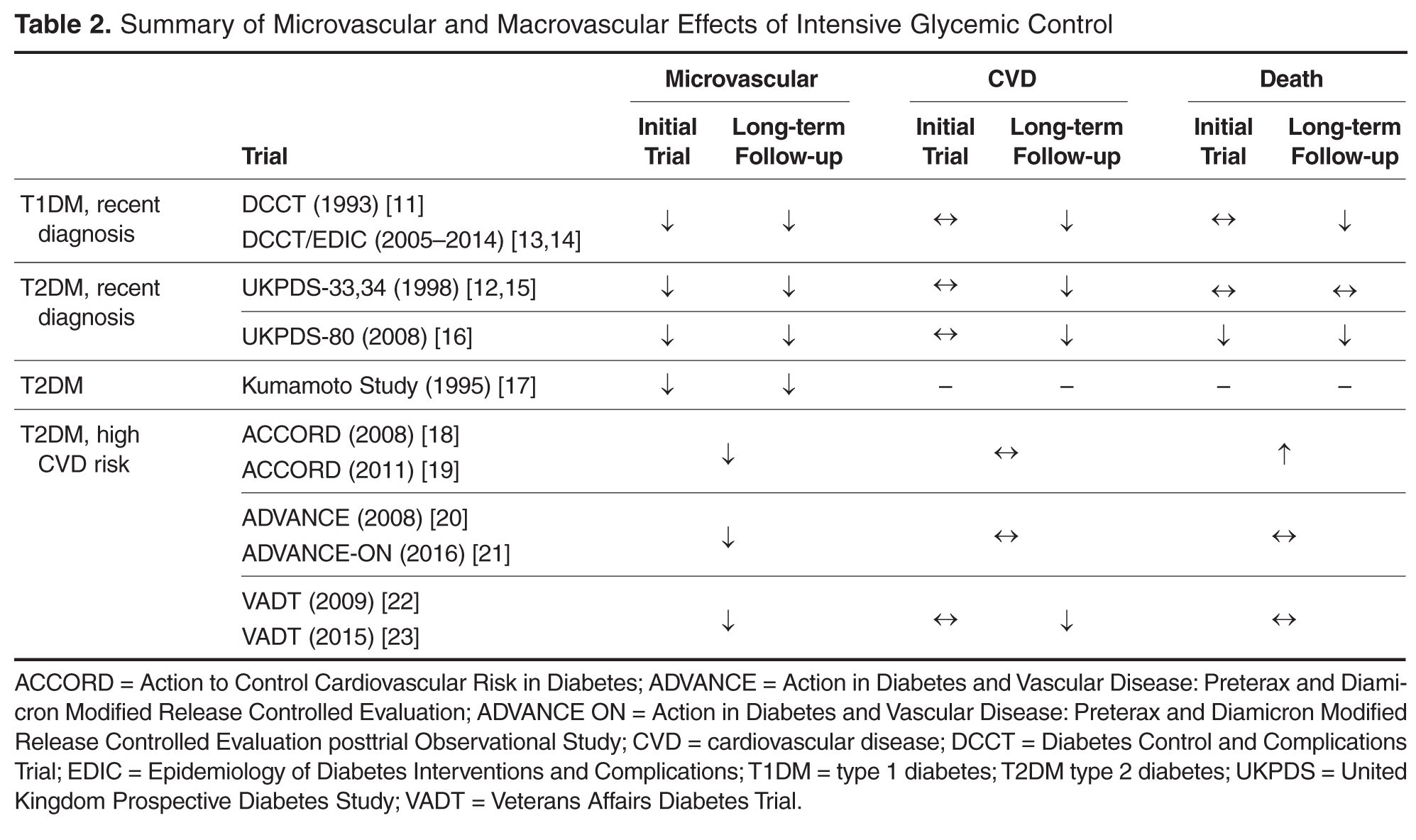
Research over the past decade has led to a growing appreciation of the enormous complexity of hyperglycemia management. During the 1990s, landmark trials such as the Diabetes Control and Complications Trial (DCCT) [11] and UK Prospective Diabetes Study (UKPDS) [12] demonstrated that improving glucose control could reduce the incidence of microvascular complications [11,12], prompting a lower-is-better philosophy regarding glucose targets. Despite limited evidence to support such thinking, this viewpoint was adopted by the developers of many guidelines. During the following decade more research was devoted to determining whether aggressively lowering a patient’s glucose could also improve macrovascular outcomes. Table 2 summarizes microvascular and macrovascular effects of intensive glycemic control seen in major trials [11–23]. After several major trials [20,22] found only mild cardiovascular benefits and even suggested harm [18], experts and policy makers began to reconsider the value of tightly controlling glucose levels [24]. Since then, other studies have demonstrated that the potential benefits and risks of glucose control are strongly related to individual patient factors, such as age and duration of diabetes, and associated comorbidities, such as CVD and impaired renal function [6].
A one-size-fits-all glycemic goal is no longer recommended. Personalization is necessary, balancing the potential benefits and risks of treatments required to achieve that goal. Whereas an A1C of < 7% is an appropriate target for some individuals with diabetes, glycemic targets may be more or less stringent based on patient features including life expectancy, duration of diabetes, comorbidities, and patient attitude and support system (Table 3) [4].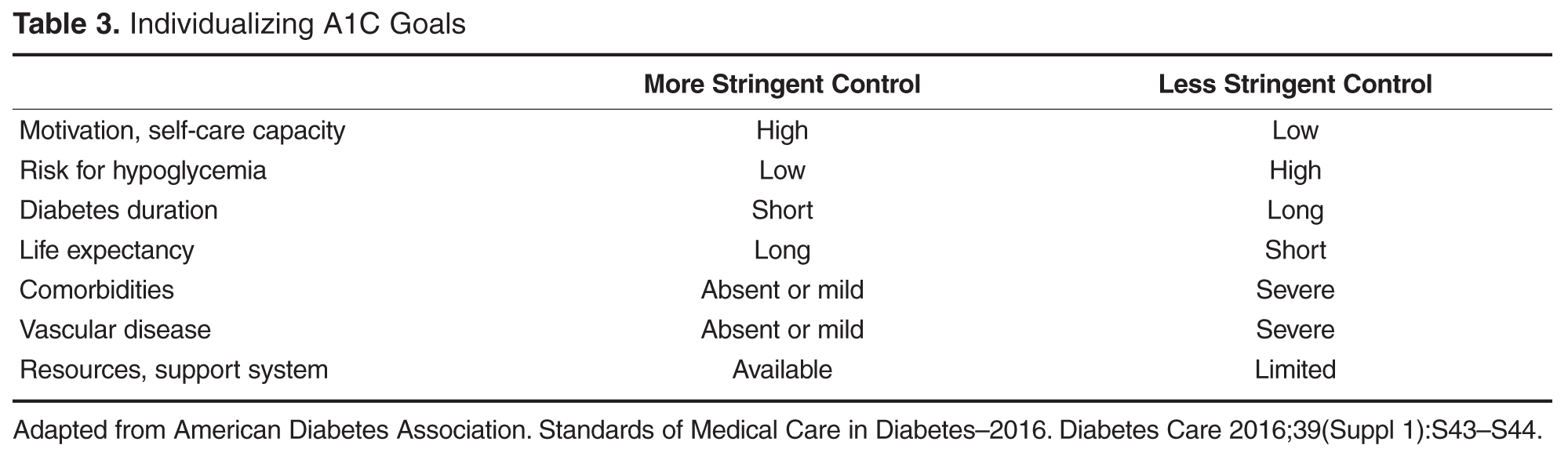
A particular group in which less stringent goals should be considered is older patients, especially those with complex or poor health status [4,25]. The risk of intensive glycemic control may exceed the benefits in these patients, as they are at higher risk of hypoglycemia and polypharmacy [26]. A goal A1C of 7% to 7.5% is now recommended for healthy older adults, and less stringent A1C goals of 7.5% to 8% and 8% to 8.5% should be considered based on the presence and severity of multiple coexisting chronic illnesses, decreased self-care ability, or cognitive impairment [4,25]. Unfortunately, overtreatment is frequently seen in this group. In a recent study of patients over age 65 years, about 40% of those with complex or poor health status had tight glycemic control with A1C below 6.5% [26]. An analysis of U.S. Veterans Affairs administration data showed that only 27% of 12,917 patients older than 65 with very low A1C (< 6%) and about 21% of those with A1C of 6% to 6.5% underwent treatment deintensification [27].
Initiate Treatment with Metformin
There is strong consensus that metformin is the preferred drug for monotherapy due to its long proven safety record, low cost, weight-reduction benefit, and potential cardiovascular advantages [4,16]. As long as there are no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. The recommendation is based on the fact that adherence to diet, weight reduction, and regular exercise is not sustained in most patients, and most patients ultimately will require treatment. Since metformin is usually well-tolerated, does not cause hypoglycemia, has a favorable effect on body weight, and is relatively inexpensive, potential benefits of early initiation of medication appear to outweigh potential risks.
The U.S. Food and Drug Administration (FDA) recently relaxed prescribing polices to extend the use of this important medication to patients who have mild–moderate, but stable, chronic kidney disease (CKD) [28]. Metformin is recommended as first-line therapy and should be used unless it is contraindicated (ie, estimated glomerular filtration rate [eGFR] < 30 mL/min/1.73 m2)[4,7,29].
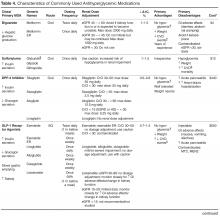
Add Additional Agent(s) as Needed to Achieve Goal
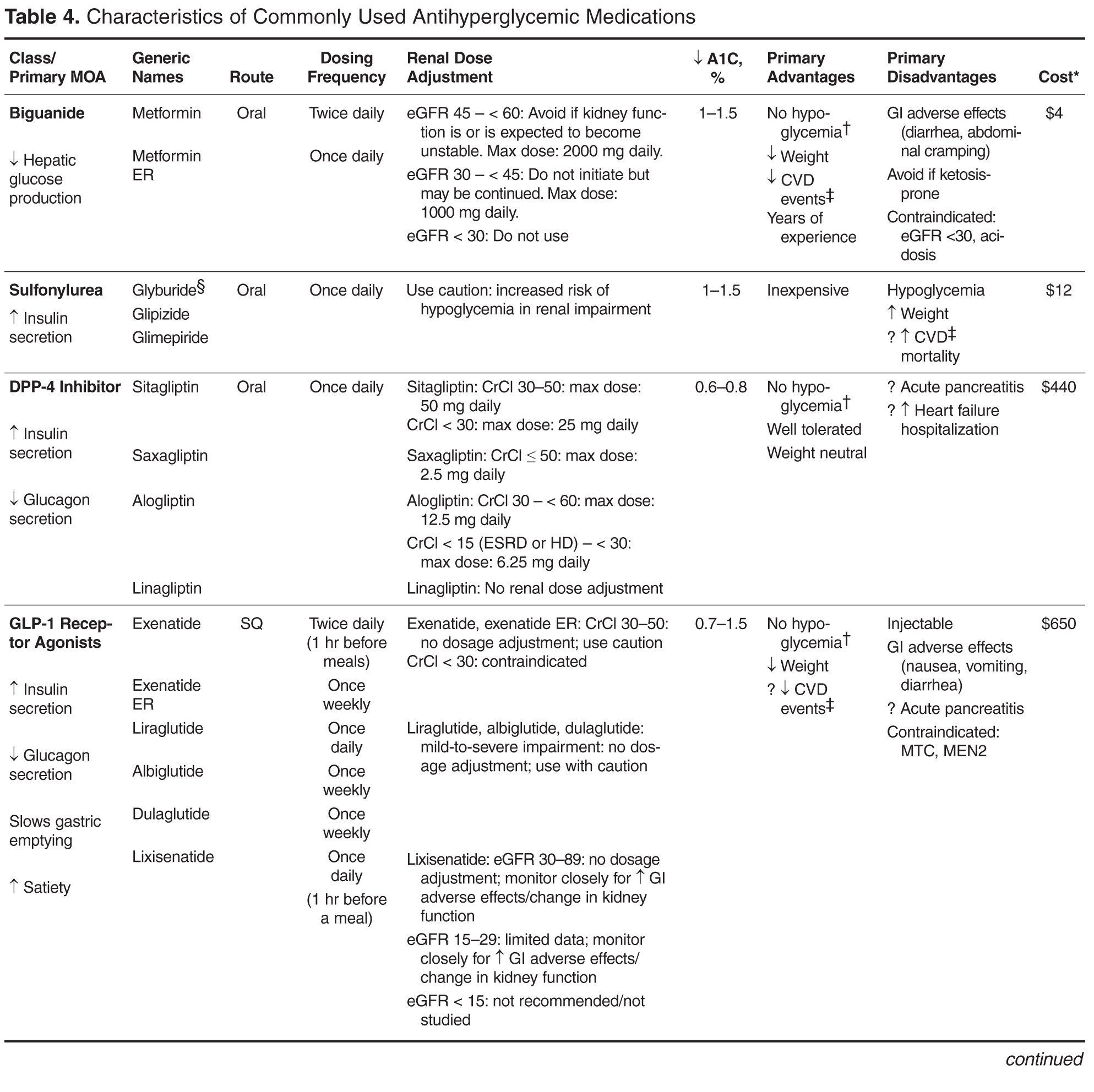
Other than metformin, evidence is limited for the optimal use of the burgeoning array of available agents, especially in dual or triple combinations [6,30]. Research is now starting to focus more on what the ideal number and sequence of drugs should be. The Glycemic Reduction Approach in Diabetes (GRADE) study, which will compare long-term benefits and risks of the 4 most widely used antihyperglycemic medications in combination with metformin, is now underway [31,32]. The 4 classes being studied are sulfonylurea, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, and a basal, 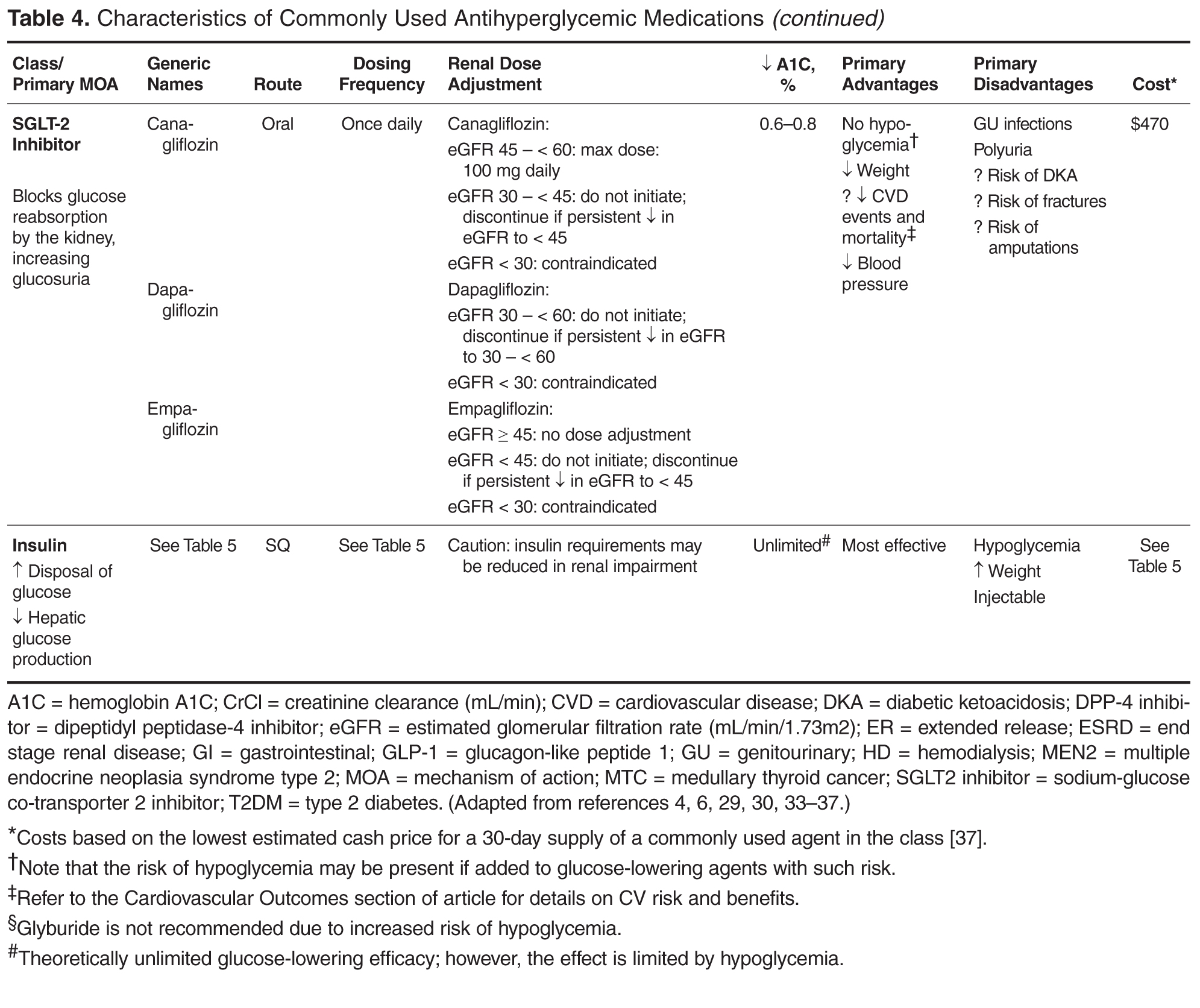
Eleven classes of non-insulin medications are currently approved for treating hyperglycemia in T2DM [4]. Within each class, numerous agents are available. Six of these classes (ie, α-glucosidase inhibitors, colesevelam, bromocriptine, pramlintide, meglitinides, and thiazolidinediones) are not used frequently 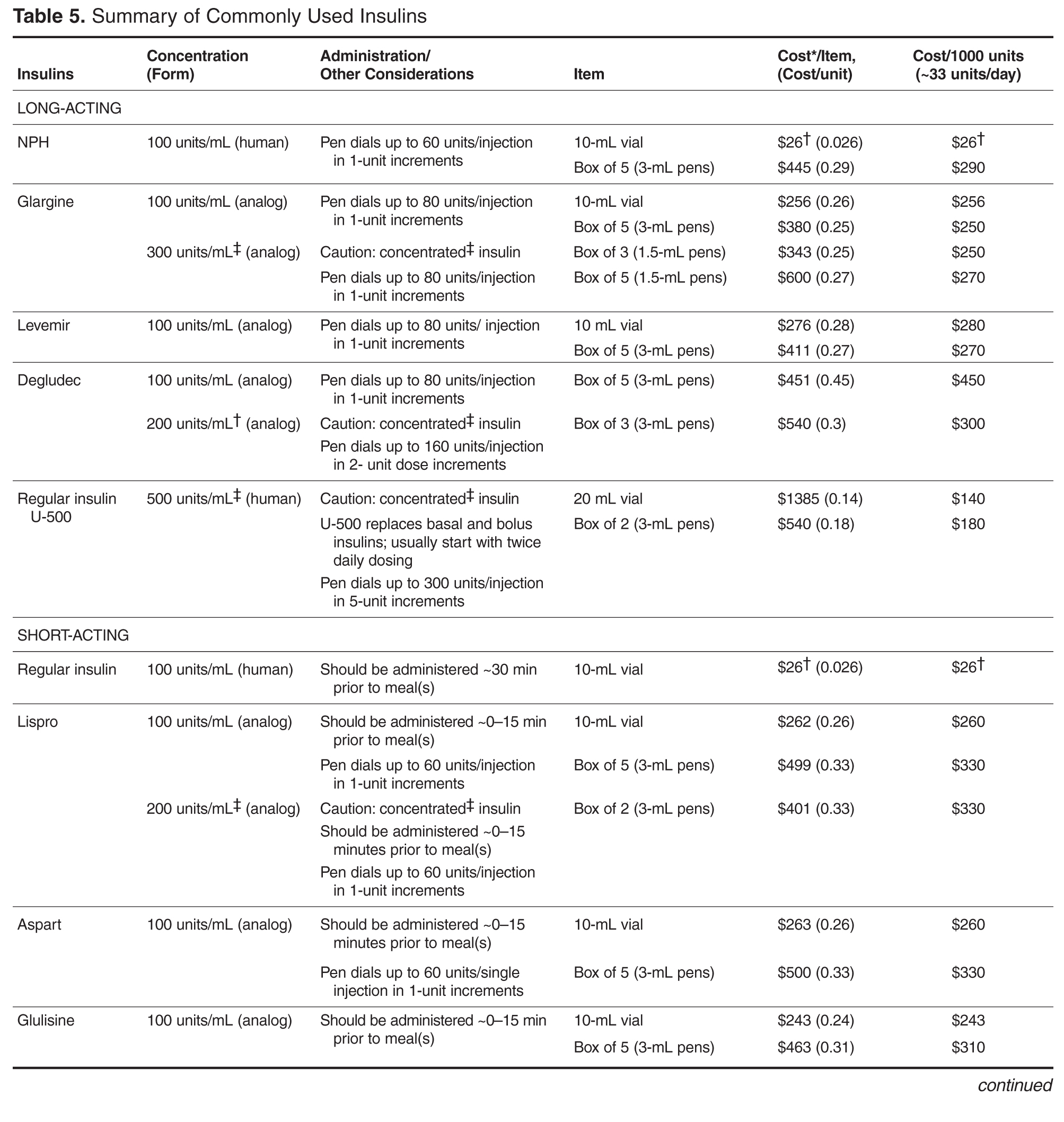
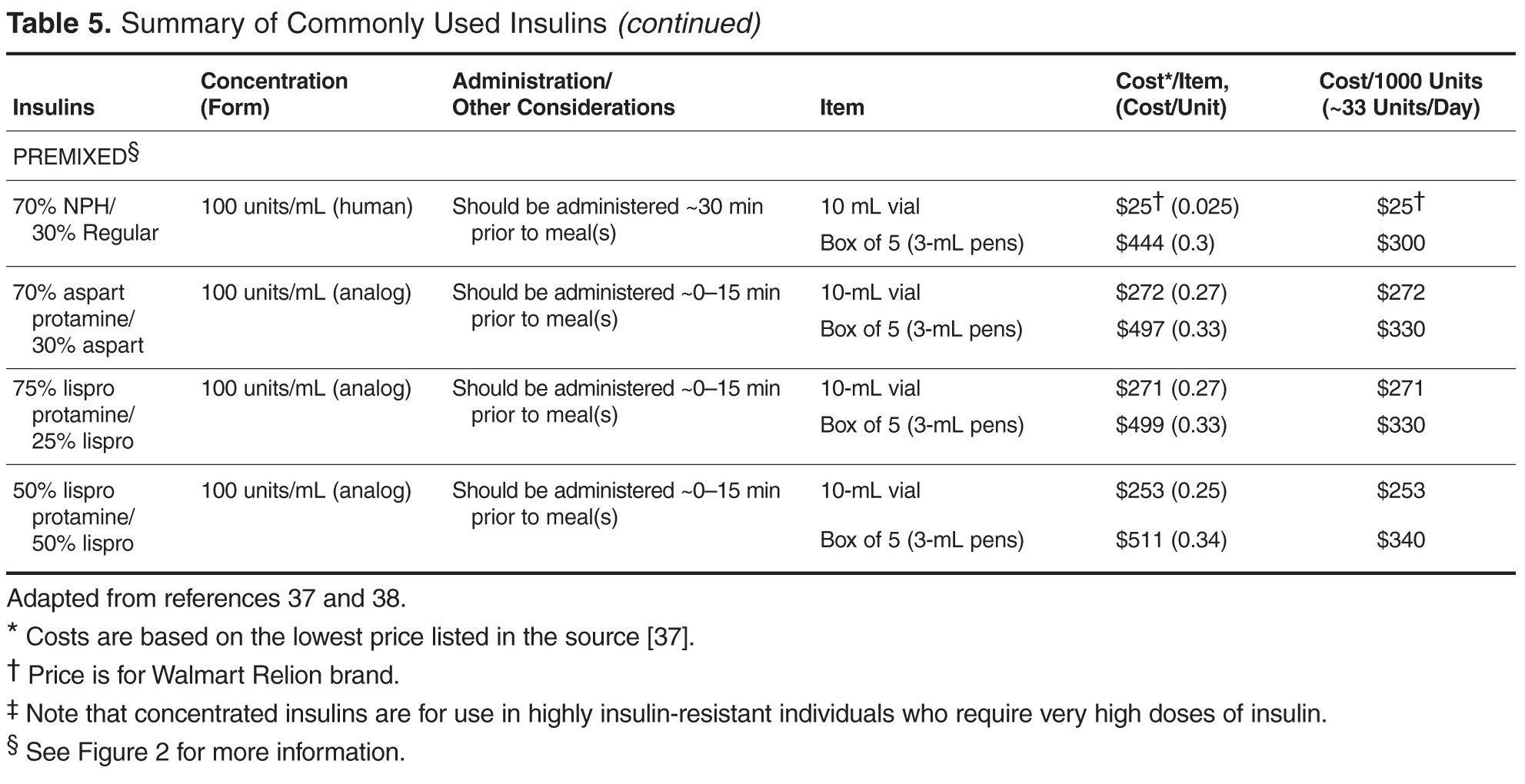
Consider Effects on A1C
There is a paucity of high-quality, head-to-head comparison trials evaluating the ability of available agents to achieve recommended glycemic targets. This is important because the glucose-lowering effectiveness of individual medications is strongly influenced by baseline characteristics such as A1C, duration of diabetes, and previous therapy. With these limitations in mind, the relative glucose-lowering effectiveness of commonly used agents is shown in Table 4. When used as monotherapy, A1C reductions of approximately 1% to 1.5% are achieved with metformin, sulfonylureas, and GLP-1 receptor agonists [6,30,34,35,39]. DPP-4 inhibitors and SGLT-2 inhibitors have more modest glucose-lowering efficacy, with A1C reductions of approximately 0.5% to 1% [6,30,34,35,39]. Larger effects may be seen in individuals with higher baseline A1C and those who are drug naïve. Insulin is the most effective glucose-lowering agent—it can reduce virtually any level of A1C down to the normal range, with hypoglycemia being the only limiting factor. When a patient has uncontrolled hyperglycemia on metformin monotherapy, or if there is a contraindication or intolerance to metformin, clinicians should consider the potential glucose-lowering effects of other available options and should choose an agent that conceivably could bring a patient close to meeting their treatment goal.
Eliminate Options with Unacceptable Adverse Effects
When the pharmacologic options with acceptable A1C-lowering potential have been identified, the ones with contraindications and potential serious adverse effects for the individual patient can immediately be eliminated (Table 4). For example, if a patient has an eGFR < 30 mL/min/1.73 m2, metformin, sulfonylureas, GLP-1 receptor agonists, most DPP-4 inhibitors, and SGLT-2 inhibitors are either contraindicated or should be used with caution. In patients with severe osteoporosis, SGLT-2 inhibitors may not be the best option. In patients with a history of diabetic ketoacidosis (DKA), caution should be used with metformin and SGLT-2 inhibitors. There have been concerns of possible acute pancreatitis and neoplasia with the incretin-based agents, the DPP-4 inhibitors and GLP-1 receptor agonists [40,41], although other clinical trials and observational data have not found increased risk [42–45]. Nevertheless, these agents potentially should be avoided in patients with a history of pancreatitis or neoplasm. SGLT-2 inhibitors may be associated with genitourinary infections and volume depletion [46–48] and probably should be avoided in patients at high risk for these conditions.
If the adverse effects are not serious, changing the way the medication is administered may allow the patient to tolerate agents with high potential benefits. For example, metformin is commonly associated with gastrointestinal (GI) adverse effects, which can be reduced or avoided with slow titration of the dose [6] or by switching to an extended-release formulation [49]. GLP-1 receptor agonists are associated with GI adverse effects [6] and in most cases slow titration is recommended.
Evaluate Potential Risks/Benefits of Remaining Options
Hypoglycemia. The barrier of hypoglycemia generally precludes maintenance of euglycemia and full realization of the long-term benefits of good glucose control over a lifetime. Once considered a trivial issue, concerns about hypoglycemia in T2DM are increasingly being raised [19,50–55]. Clearly, hypoglycemia occurs more often as glycemic targets are lowered to near-normal values, especially in those with advanced age and multiple comorbidities [55]. Various comorbidities frequently encountered particularly as patients age also are associated with increasing propensity for experiencing hypoglycemia and untoward outcomes from it. These include coronary artery disease, heart failure, renal and liver disease, and dementia. Hypoglycemia, when it occurs, may lead to dysrhythmias, dizziness, accidents and falls, work disability, and decreased quality of life. In addition to relaxing blood glucose targets in high-risk patients, drug selection should favor agents that do not precipitate such events (Table 4).
Fortunately, the commonly used non-insulin agents are not associated with hypoglycemia unless they are used in combination with sulfonylureas or insulin. Sulfonylureas should be used with caution and other options considered in patients with high risk for hypoglycemia. When insulin is required, regimens which minimize risk of hypoglycemia should be used. For example, adding a GLP-1 receptor agonist to basal insulin as an alternative to mealtime insulin has been shown to be equally effective with a lower risk of hypoglycemia [4,6]. Also, premixed insulin preparations should be avoided or used cautiously in individuals who miss meals frequently. Additionally, newer basal insulins that exhibit longer duration of action are now available in the United States. Preliminary studies have shown that the newly FDA-approved longer-acting basal insulins, insulin degludec and glargine U-300, may be associated with a reduced risk for hypoglycemia [56,57]. However, it remains unclear how and when these newer agents will best be incorporated into a treatment regimen.
Body weight. Nearly 90% of people living with T2DM are overweight or obese. Given the close tie between obesity and T2DM, treating obesity is an obvious consideration in diabetes treatment. Major trials have shown the effectiveness of lifestyle modifications and weight reduction in delaying, prevention, and management of T2DM [4,58,59].With this in mind, clinicians should consider preferentially using antihyperglycemic agents with weight-lowering or weight-neutral effects. Among commonly used antihyperglycemic agents, metformin, GLP-1 receptor agonists, and SGLT-2 inhibitors have been shown to have weight-reduction benefits, and DPP-4 inhibitors are weight neutral. On the other hand, sulfonylureas and insulin are associated with weight gain. A systematic review and meta-analysis including 204 studies with study durations ranging from 3 months to 8 years showed comparative effects of diabetes medications with a differential effect on weight of up to 5 kg (Table 4) [60].
Metformin is associated with an average weight loss of 1.9 to 3.1 kg that was sustained with long-term use for at least 10 years in the Diabetes Prevention Program Outcomes Study [61].A systematic review of 7 randomized trials showed that in patients with T2DM, the SGLT-2 inhibitors dapagliflozin and canagliflozin were associated with weight loss (mean weighted difference of –1.81 kg and –2.3 kg, respectively) [62]. A systematic review and meta-analysis of 25 randomized controlled trials showed greater weight loss (mean weighted difference of –2.9 kg) in overweight or obese patients with or without T2DM using GLP-1 receptor agonists when compared to placebo, insulin, or oral antihyperglycemic agents [63]. Of note, the GLP-1 receptor agonist liraglutide is now approved for weight loss in patients with or without diabetes [64]. The maximum doses approved for diabetes and obesity treatment are 1.8 and 3.0 mg/day, respectively.
Since weight loss is associated with improved glycemic control, an area of emerging interest is the use of antiobesity medications for managing diabetes. Although most older weight-loss medications were only approved for short-term use, some newer agents are approved for longer-term use. Lorcaserin and the combination drugs topiramate/phentermine and naltrexone/bupropion are approved for chronic therapy, provided certain conditions are met. Patients on weight reduction agents should be monitored regularly. 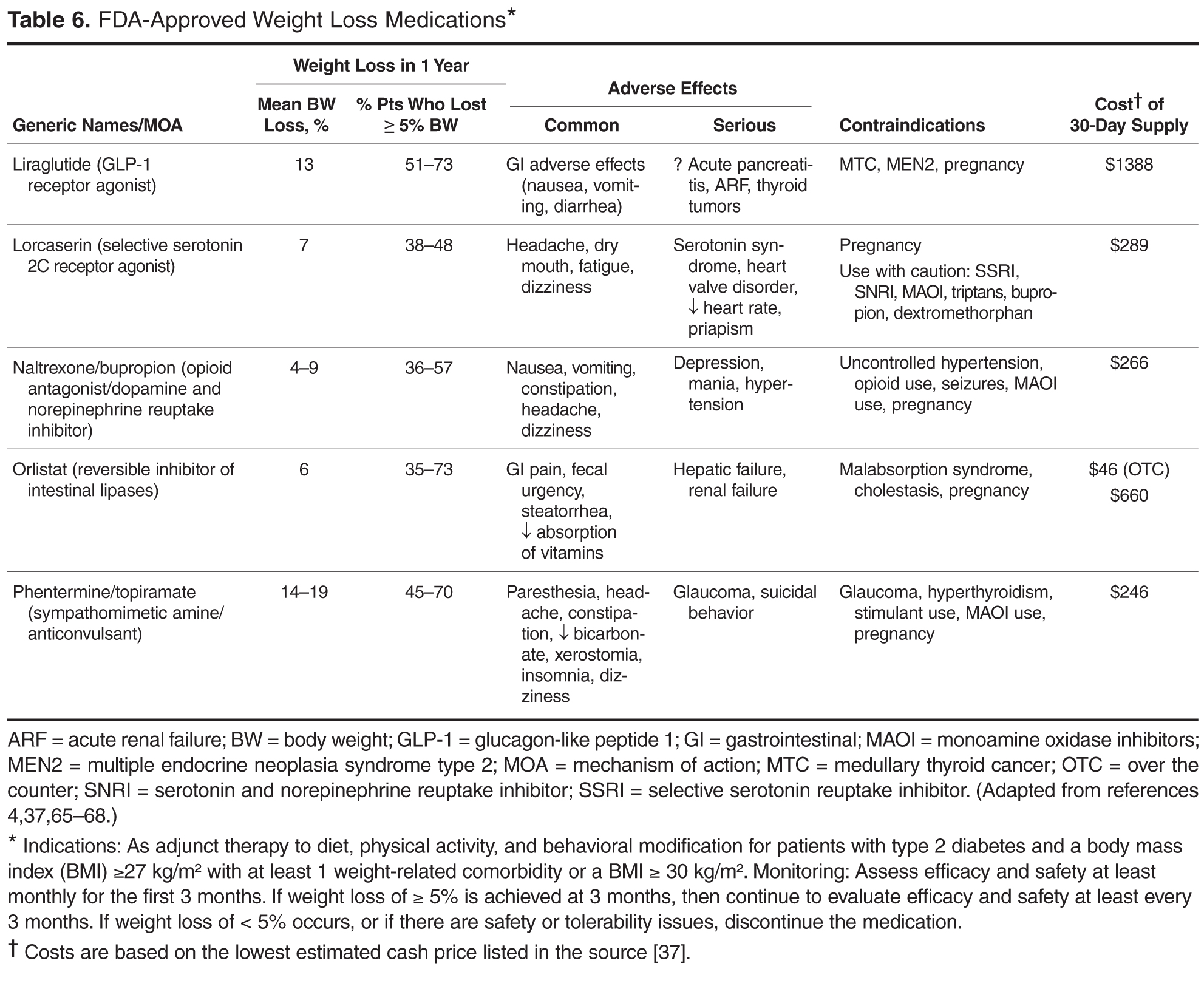
An even more radical departure from conventional therapy for diabetes is the consideration of metabolic, or weight-loss, surgery, which has been found to be associated with rapid and dramatic improvements in blood glucose control. Metabolic surgery has been shown to improve glucose control more effectively than any known pharmaceutical or behavioral approach. For example, in an observational study of obese patients with T2DM, bariatric surgery led to diabetes remission rates of 72.3% 2 years after surgery and 30.4% 15 years after surgery compared to 16.4% and 6.5%, respectively, in control patients [69]. With long-term follow-up, significant decreases in microvascular and macrovascular complications were seen in the surgical group [69]. Compared with medical therapy alone, bariatric surgery plus medical therapy has been associated with more weight loss, better glycemic control, less need for diabetes medications, and improved quality of life [70]. A 2016 joint statement by numerous international diabetes organizations recommends considering metabolic surgery as a treatment for T2DM and obesity [71]. American Diabetes Association guidelines recommend consideration of bariatric surgery in individuals with T2DM who have a body mass index greater than 35 kg/m2,especially if achieving disease control is difficult by means of lifestyle modifications and medications [4].
Cardiovascular outcomes. Cardiovascular risk is about 2 to 4 times higher in patients with diabetes, and about half of patients with this condition develop heart failure [4,72]. CVD is responsible for most of the mortality in T2DM [72]. Therefore, prevention of cardiovascular morbidity and mortality is an important goal for diabetes treatment. Due to concerns about potential cardiovascular risks associated with glucose-lowering medications [73–76], the FDA has issued regulatory requirements for manufacturers to monitor the cardiovascular risk profile for these drugs [77]. Recent trials have led to a better understanding of potential cardiovascular benefits or harms of antihyperglycemic medications.
Metformin, the widely recommended first-line therapy for T2DM, carries a large body of evidence supporting its cardiovascular benefits. For example, the UKPDS found that compared to conventional therapy (mostly diet), metformin reduced cardiovascular events and mortality in obese patients with T2DM [15]. This result was supported in Hyperinsulinemia: the Outcome of its Metabolic Effect (HOME) study where, as an add-on to insulin, metformin decreased macrovascular complications when compared to placebo [78]. Research over the past decade also has assuaged concerns about metformin safety in heart failure [60]. A systematic review of observational studies involving 34,000 patients conducted in 2013 showed that metformin is as safe as other glucose-lowering medications in patients with diabetes and heart failure even in the presence of CKD [4,79]. Furthermore, numerous investigations have found metformin is not associated with increased hospitalizations or risk of lactic acidosis [80]. Metformin can be used safely in patients with diabetes and heart failure [60].
Although sulfonylureas have long been a mainstay of diabetes therapy, concerns about their potential adverse cardiovascular effects have been raised by numerous studies [81]. Tolbutamide, a first-generation sulfonylurea, was removed from the market after the University Group Diabetes Program study found increased CVD deaths with this agent versus placebo. Subsequently, the FDA issued a warning for all sulfonylureas [74]. The increased cardiovascular risk associated with sulfonylureas is thought to be due to their effect on cardiac mitochondrial potassium ATP channels. Sulfonylureas bind to these channels, preventing a protective phenomenon called ischemic preconditioning and resulting in a weakened defense against myocardial injury [76]. A recent study showed an increased risk of coronary heart disease associated with long-term use of sulfonylureas in women with diabetes [81].
GLP-1 receptor agonists have recently received much attention for their potential beneficial effects on cardiovascular outcomes. In a recent trial, lixisenatide was shown to be safe in patients with T2DM and acute coronary syndrome when compared to placebo [82]. More recently, the Liraglutide Effect and Action in Diabetes: Evaluation of cardiovascular outcome Results (LEADER) trial demonstrated significant cardiovascular benefits with liraglutide in patients with T2DM and established or high CVD risk [83]. The composite outcome of the first occurrence of death from cardiovascular causes, nonfatal myocardial infarction (MI), or nonfatal stroke, occurred less frequently in the liraglutide group compared to placebo (13% versus 14.9%, respectively), and there were fewer deaths from cardiovascular causes in the liraglutide group compared to placebo (4.7% and 6.0%, respectively) [83]. Other trials investigating the cardiovascular outcomes of this class [84,85] are in progress.
Another class with potential cardiovascular benefits is the SGLT-2 inhibitors. In a recent cardiovascular outcome study, empagliflozin significantly lowered the composite of cardiovascular death, nonfatal MI, or nonfatal stroke in T2DM patients with high cardiovascular risk compared to placebo (10.5% and 12.1%, respectively) [86]. There are several large ongoing studies evaluating the cardiovascular effects of other SGLT-2 inhibitors [87–89].
DPP-4 inhibitors were examined in recent studies and have shown no cardiovascular benefits [42,44,90].The studies showed mixed results regarding an association between DPP-4 inhibitors and heart failure. In one study, saxagliptin was associated with increased hospitalization for heart failure compared to placebo [44], while 2 noninferiority trials did not show a significant increase in heart failure hospitalizations associated with alogliptin and sitagliptin when compared to placebo [42,90].
Administration Considerations
Many patients with T2DM require multiple agents for glycemic control. Additional medications used for comorbid conditions add to this burden. When choosing antihyperglycemic agents, the route and frequency of administration, as well as the patients’ preferences and ability, should be considered. Either once or twice daily dosing is available for most agents, and once weekly dosing is available for some of the GLP-1 receptor agonists. Once daily or once weekly formulations may improve adherence and be more desirable than preparations that are dosed twice daily. Most of the commonly used medications are dosed orally. Although many patients find this route of administration preferable to insulin or GLP-1 receptor agonists, which require injections, some patients may prefer the risk/benefit of injectable agents. All GLP-1 receptor agonists come in a pen delivery system, which eliminates mixing and provides more convenient administration. Extended-release exenatide also is available as a single-dose tray that requires mixing and may be more cumbersome to inject.
Insulin requires special consideration. There has been an enormous increase in the number of insulin products on the market in the past 2 decades. These products include insulin analogs, concentrated insulins (U-200, U-300, and U-500), premixed insulin preparations, and ultra-long-acting insulin [91]. The availability of insulin options with different concentrations, onsets, and durations of actions has made decision making on which insulin to use difficult. Clinicians need to consider patient preference, dosing frequency, and timing with regard to meals, insulin dose, administration, as well as cost. For example, concentrated insulin is preferred for a patient on high doses of insulin requiring injecting a large volume of insulin. Rapid-acting insulin analogs would be more appropriate for patients who have difficulty administering their regular insulin 20 to 30 minutes before eating. Premixed insulin preparations make it impossible to independently adjust short- and long-acting components. However, these may be good choices in patients who have consistent meal schedules and who want to simplify administration. Despite a prevailing misconception that NPH must be given twice a day, it has long been recognized that in T2DM, a single daily injection of NPH yields improvements in control similar to those achieved with 2 daily injections [92].
Cost Considerations
Treating T2DM imposes a great financial burden on individuals living with diabetes and their families due to the high cost of the medications. Table 4 and Table 5 provide information on the cost of non-insulin and insulin diabetes medications for patients who do not have prescription insurance coverage. From a practical standpoint, choice of diabetes agents is largely influenced by insurance formularies.
The older agents, metformin and the sulfonylureas, are available for a cash (no insurance) price of as little as $4 per month. This is in stark contrast to the SGLT-2 inhibitors, GLP-1 receptor agonists, and DPP-4 inhibitors, which range in cost between $400 and $600 per month. Of recent concern, the cost of insulin has been skyrocketing, with a more than 500% increase in the cost of certain insulins from 2001 to 2015 [93]. According to the Medical Expenditure Panel Survey (MEPS) from 2002 to 2013, the mean price of insulin increased by about 200% (from $4.34/mL to $12.92/mL) during this period, which was significantly higher than increases in the price of non-insulin comparators [94]. The introduction of biosimilar insulins to the market is expected to offer treatment options with lower cost. This will be tested when the biosimilar glargine, the first FDA-approved biosimilar insulin, becomes available in the U.S. market. However, a significant reduction in insulin prices is not expected soon [95].
When insulin is required, most patients with T2DM can be treated with older human insulins, which have similar efficacy and lower costs than the more expensive newer insulin analogs. A Cochrane review comparing basal insulin analogs to NPH showed similar efficacy in glycemic control with minimal clinical benefit in the form of less nocturnal hypoglycemia in the insulin analog arm [96]. Furthermore, similar glycemic control and risk of hypoglycemia was seen when regular insulin was compared with the rapid-acting insulin analogs [97]. The cost of human NPH insulin for a patient on a total daily dose of 60 units is approximately $52 per month. This contrasts with the most widely used insulin, insulin glargine, which has a cash price of about $500 per month for the same amount (Table 5). Insulin pens, which are convenient, are more expensive. Interestingly, human insulins do not require prescriptions, allowing underinsured, underfunded patients ongoing access to them.
Incorporating Patient Preferences
Research evidence is necessary but insufficient for making patient care decisions. Along with the potential benefits, harms, costs, and inconveniences of the management options, patient perspectives, beliefs, expectations, and health-related goals must be considered. Patients will undoubtedly have preferences regarding defining goals and ranking options. Clinicians should discuss therapeutic goals and treatment options and work collaboratively with patients in determining management strategies [98].

Summary
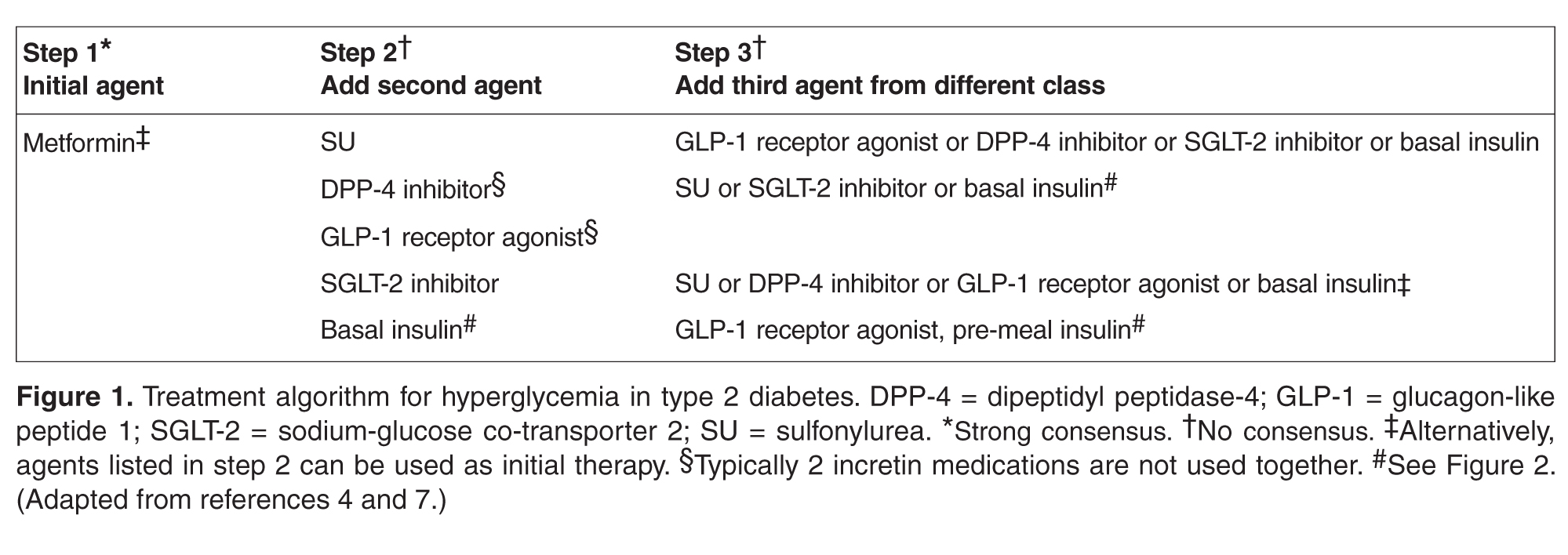
Potential treatment approaches for treating hyperglycemia in T2DM are summarized in Figure 1 and Figure 2 [4,7]. As long as there are no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. Even if metformin monotherapy is initially effective, glycemic control is likely to deteriorate over time due to progressive loss of β-cell function in T2DM.
There is no consensus as to what the second-line agent should be. Selection of a second agent should be made based on potential advantages and disadvantages of each agent for any given patient. A patient-centered approach is preferred over a fixed algorithm. If the patient progresses to the point where dual therapy does not provide adequate control, either a third non-insulin agent or insulin can be added. In patients with modestly elevated A1C (below ~8%), addition of a third non-insulin agent may be equally effective as (but more expensive than) addition of insulin.
Patients with significantly elevated A1C levels on non-insulin agents usually should have insulin added to their regimen. When insulin is added, metformin should be continued. DPP-4 inhibitors and sulfonylureas are typically stopped. If SGLT-2 inhibitors and/or GLP-1 receptor agonists are continued, this may aid with weight maintenance. However, continuing these agents is likely to be expensive and associated with problems associated with polypharmacy.
The most widely recommended strategy for initiating insulin in T2DM is to add a single bedtime injection of basal insulin (ie, NPH, glargine, detemir, or degludec) to the patient’s regimen. This regimen has been found to be effective in numerous studies and controls hyperglycemia in up to 60% of patients [99]. If the patient is treated with a single bedtime injection of insulin and the fasting glucose level is within the target range but the A1C level remains above goal, addition of mealtime insulin injections is likely to be beneficial. Alternatively, addition of a GLP-1 receptor agonist to basal insulin has been shown to be equally beneficial [4,6]. When adding mealtime insulin, a common strategy is to add a single injection of a rapid-acting insulin (eg, lispro, aspart, glulisine) before the patient’s largest meal of the day. Additional premeal injections of rapid-acting insulin may be added as needed, based on self-monitoring blood glucose results. If glycemia remains significantly uncontrolled on more than 200 units of insulin per day, switching to a concentrated form of insulin (eg, U-200, U-300, or U-500) should be considered.
Corresponding author: Maryam Fazel, PharmD, BCPS, BCACP, CDE, 1295 N. Martin Ave. (Room B211B), Tucson, Arizona 85721-0202, [email protected].
Financial disclosures: None.
1. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. Centers for Disease Control and Prevention Web site. www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf. Accessed November 29, 2016.
2. Statistics about diabetes. American Diabetes Association Web site. www.diabetes.org/diabetes-basics/statistics/. Accessed November 29, 2016.
3. American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013;36:1033–46.
4. American Diabetes Association. Standards of medical care in diabetes--2016. Diabetes Care 2016;39(Suppl. 1).
5. Raz I, Riddle MC, Rosenstock J, et al. Personalized management of hyperglycemia in type 2 diabetes: reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 2013;36:1779–88.
6. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015;38:140–9.
7. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm--2016 Executive Summary. Endocr Pract 2016;22:84–113.
8. Steenkamp DW, Alexanian SM, Sternthal E. Approach to the patient with atypical diabetes. CMAJ 2014;186:678–84.
9. de Lusignan S, Sadek N, Mulnier H, et al. Miscoding, misclassification and misdiagnosis of diabetes in primary care. Diabet Med 2012;29:181–9.
10. Tripathi A, Rizvi AA, Knight LM, Jerrell JM. Prevalence and impact of initial misclassification of pediatric type 1 diabetes mellitus. South Med J 2012;105:513–7.
11. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993;329:977–86.
12. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:837–53.
13. Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–53.
14. Nathan DM, DCCT/EDIC Research Group. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 2014;37:9–16.
15. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:854–65.
16. Holman RR, Paul SK, Bethel MA, et al. 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–89.
17. Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995;28:103–17.
18. Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–59.
19. ACCORD Study Group, Gerstein HC, Miller ME, Genuth S, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med 2011;364:818–28.
20. ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008;358:2560–72.
21. Wong MG, Perkovic V, Chalmers J, et al. Long-term Benefits of Intensive Glucose Control for Preventing End-Stage Kidney Disease: ADVANCE-ON. Diabetes Care 2016;39:694–700.
22. Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009;360:129–39.
23. Hayward RA, Reaven PD, Wiitala WL, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;372:2197–206.
24. American Diabetes Association. Standards of medical care in diabetes--2009. Diabetes Care 2009;32 Suppl 1:S13–61.
25. American Geriatrics Society Expert Panel on Care of Older Adults with Diabetes Mellitus, Moreno G, Mangione CM, Kimbro L, Vaisberg E. Guidelines abstracted from the American Geriatrics Society Guidelines for Improving the Care of Older Adults with Diabetes Mellitus: 2013 update. J Am Geriatr Soc 2013;61:2020–6.
26. Lipska KJ, Ross JS, Miao Y, et al. Potential overtreatment of diabetes mellitus in older adults with tight glycemic control. JAMA Intern Med 2015;175:356–62.
27. Sussman JB, Kerr EA, Saini SD, et al. Rates of deintensification of blood pressure and glycemic medication treatment based on levels of control and life expectancy in older patients with diabetes mellitus. JAMA Intern Med 2015;175:1942–9.
28. FDA Drug Safety Communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function. FDA Web site. www.fda.gov/Drugs/DrugSafety/ucm493244.htm. Accessed December 1, 2016.
29. Inzucchi SE, Lipska KJ, Mayo H, et al. Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA 2014;312:2668–75.
30. Nathan DM. Diabetes: advances in diagnosis and treatment. JAMA 2015;314:1052–62.
31. Nathan DM, Buse JB, Kahn SE, et al. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care 2013;36:2254–61.
32. NIH begins recruitment for long-term study of diabetes drug efficacy. NIH Web site. www.nih.gov/news-events/news-releases/nih-begins-recruitment-long-term-study-diabetes-drug-efficacy. Accessed December 1, 2016.
33. Hermayer KL, Dake A. Newer oral and noninsulin therapies to treat type 2 diabetes mellitus. Cleve Clin J Med 2016;83(5 Suppl 1):S18–26.
34. Bolen S, Wilson L, Vassy J, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med 2007;147:386–99.
35. Bolen S, Tseng E, Hutfless S, et al. Oral diabetes medications for adults with type 2 diabetes: an update. Agency for Healthcare Research and Quality (US); 2011 Mar Report No: 11-EHC038-EF. AHRQ Comparative Effectiveness Reviews.
36. Metformin, glyburide, glipizide, glimeperide, sitagliptin, saxagliptin, linagliptin, lixisenatide, alogliptin, exenatide, liraglutide, albiglutide, dulaglutide, canagliflozin, danagliflozin, empagliflozin: drug information. Waltham (MA): UpToDate, Inc.; 2016. Accessed September 23, 2016.
37. GoodRx Web site. http://www.goodrx.com. Accessed August 6, 2016 and December 2016.
38. Insulins available in the United States. Diabetesforcast Web site. Accessed August 6, 2016. www.diabetesforecast.org/2016/mar-apr/images/2016-insulin-chart-new.pdf.
39. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012;55:577–96.
40. Elashoff M, Matveyenko AV, Gier B, et al. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011;141:150–6.
41. Butler AE, Campbell-Thompson M, Gurlo T, et al. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 2013;62:2595–604.
42. Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;373:232–42.
43. White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013;369:1327–35.
44. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013;369:1317–26.
45. Egan AG, Blind E, Dunder K, et al. Pancreatic safety of incretin-based drugs--FDA and EMA assessment. N Engl J Med 2014;370:794–7.
46. Vasilakou D, Karagiannis T, Athanasiadou E, et al. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 2013;159:262–74.
47. Nyirjesy P, Sobel JD, Fung A, et al. Genital mycotic infections with canagliflozin, a sodium glucose co-transporter 2 inhibitor, in patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. Curr Med Res Opin 2014;30:1109–19.
48. Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care 2013;36:2508–15.
49. Blonde L, Dailey G, Jabbour S, et al. Gastrointestinal tolerability of extended-release metformin tablets compared to immediate-release metformin tablets: results of a retrospective cohort study. Curr Med Res Opin 2004;20:562–72.
50. Kalra S, Mukherjee JJ, Venkataraman S, et al. Hypoglycemia: the neglected complication. Indian J Endocrinol Metab 2013;17:819–34.
51. Paty BW. The role of hypoglycemia in cardiovascular outcomes in diabetes. Can J Diabetes 2015;39 Suppl 5:S155–9.
52. Zoungas S, Patel A, Chalmers J, et al. Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010;363:1410–8.
53. Whitmer RA, Karter AJ, Yaffe K, et al. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 2009;301:1565–72.
54. McCoy RG, Van Houten HK, Ziegenfuss JY, et al. Increased mortality of patients with diabetes reporting severe hypoglycemia. Diabetes Care 2012;35:1897–901.
55. McCoy RG, Lipska KJ, Yao X, et al. Intensive treatment and severe hypoglycemia among adults with type 2 diabetes. JAMA Intern Med 2016;176:969–78.
56. Rodbard HW, Gough S, Lane W, et al. Reduced risk of hypoglycemia with insulin degludec versus insulin glargine in patients with type 2 diabetes requiring high doses of basal insulin: a meta-analysis of 5 randomized begin trials. Endocr Pract 2014;20:285–92.
57. Yki-Jarvinen H, Bergenstal R, Ziemen M, et al. New insulin glargine 300 units/mL versus glargine 100 units/mL in people with type 2 diabetes using oral agents and basal insulin: glucose control and hypoglycemia in a 6-month randomized controlled trial (EDITION 2). Diabetes Care 2014;37:3235–43.
58. Tuomilehto J, Lindstrom J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001;344:1343–50.
59. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403.
60. Maruthur NM, Tseng E, Hutfless S, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 2016;164:740–51.
61. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2012;35:731–7.
62. Clar C, Gill JA, Court R, Waugh N. Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2012;2:10.1136/bmjopen,2012-001007.
63. Vilsboll T, Christensen M, Junker AE, et al. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ 2012;344:d7771.
64. FDA approves weight-management drug Saxenda. FDA Web site www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm427913.htm. Accessed September 22, 2016.
65. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015;100:342–62.
66. Liraglutide, lorcaserin, naltrexone/bupropion, orlistat, phentermine/topiramate: drug information. Greenwood Village (CO): Truven Health Analytics; 2016. www.micromedexsolutions.com. Accessed May 13, 2016.
67. Liraglutide, lorcaserin, naltrexone/bupropion, orlistat, phentermine/topiramate: drug information. Waltham (MA): UpToDate, Inc.; 2016. Accessed May 13, 2016.
68. Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014;311:74–86.
69. Sjostrom L, Peltonen M, Jacobson P, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA 2014;311:2297–304.
70. Schauer PR, Bhatt DL, Kirwan JP, et al. Bariatric surgery versus intensive medical therapy for diabetes--3-year outcomes. N Engl J Med 2014;370:2002–13.
71. Rubino F, Nathan DM, Eckel RH, et al. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care 2016;39:861–77.
72. Lathief S, Inzucchi SE. Approach to diabetes management in patients with CVD. Trends Cardiovasc Med 2016;26:165–79.
73. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356:2457–71.
74. Knatterud GL, Klimt CR, Levin ME, et al. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA 1978;240:37–42.
75. Masoudi FA, Inzucchi SE, Wang Y, et al. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation 2005;111:583–90.
76. Klepzig H, Kober G, Matter C, et al. Sulfonylureas and ischaemic preconditioning; a double-blind, placebo-controlled evaluation of glimepiride and glibenclamide. Eur Heart J 1999;20:439–46.
77. FDA announces new recommendations on evaluating cardiovascular risk in drugs intended to treat type 2 diabetes. FDA Web site. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116994.htm. Accessed August 20, 2016.
78. Kooy A, de Jager J, Lehert P, et al. Long-term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch Intern Med 2009;169:616–25.
79. Eurich DT, Weir DL, Majumdar SR, et al. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: systematic review of observational studies involving 34,000 patients. Circ Heart Fail 2013;6:395–402.
80. Tahrani AA, Varughese GI, Scarpello JH, Hanna FW. Metformin, heart failure, and lactic acidosis: is metformin absolutely contraindicated? BMJ 2007;335:508–12.
81. Li Y, Hu Y, Ley SH, et al. Sulfonylurea use and incident cardiovascular disease among patients with type 2 diabetes: prospective cohort study among women. Diabetes Care 2014;37:3106–13.
82. Bentley-Lewis R, Aguilar D, Riddle MC, et al. Rationale, design, and baseline characteristics in Evaluation of LIXisenatide in Acute Coronary Syndrome, a long-term cardiovascular end point trial of lixisenatide versus placebo. Am Heart J 2015;169:631,638.e7.
83. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311–22.
84. Exenatide Study of Cardiovascular Event Lowering Trial (EXSCEL): A Trial To Evaluate Cardiovascular Outcomes After Treatment With Exenatide Once Weekly In Patients With Type 2 Diabetes Mellitus. clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01144338. 2016 Accessed September 23, 2016.
85. Researching Cardiovascular Events With a Weekly Incretin in Diabetes (REWIND). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01394952. Accessed September 23, 2016.
86. Schernthaner G, Schernthaner-Reiter MH, Schernthaner GH. EMPA-REG and other cardiovascular outcome trials of glucose-lowering agents: implications for future treatment strategies in type 2 diabetes mellitus. Clin Ther 2016;38:1288–98.
87. CANVAS--CANagliflozin cardiovascular Assesssment Study (CANVAS). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01032629. Accessed September 23, 2016.
88. Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy (CREDENCE). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT02065791. Accessed September 23, 2016.
89. Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01730534. Accessed September 23, 2016.
90. Zannad F, Cannon CP, Cushman WC, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015;385(9982):2067–76.
91. Van Klompenburg E, Heins JR. New insulin options for diabetic patients. S D Med 2016;69:84–5.
92. Rosenstock J, Schwartz SL, Clark CM Jr, et al. Basal insulin therapy in type 2 diabetes: 28-week comparison of insulin glargine (HOE 901) and NPH insulin. Diabetes Care 2001;24:631–6.
93. Tylee T, Hirsch IB. Costs associated with using different insulin preparations. JAMA 2015;314:665–6.
94. Hua X, Carvalho N, Tew M, et al. Expenditures and prices of antihyperglycemic medications in the United States: 2002-2013. JAMA 2016;315:1400–2.
95. Heinemann L. Biosimilar insulin and costs: what can we expect? J Diabetes Sci Technol 2016;10:457–62.
96. Horvath K, Jeitler K, Berghold A, et al. Long-acting insulin analogues versus NPH insulin (human isophane insulin) for type 2 diabetes mellitus. Cochrane Database Syst Rev 2007;(2)(2):CD005613.
97. Mannucci E, Monami M, Marchionni N. Short-acting insulin analogues vs. regular human insulin in type 2 diabetes: a meta-analysis. Diabetes Obes Metab 2009;11:53–9.
98. Powell PW, Corathers SD, Raymond J, Streisand R. New approaches to providing individualized diabetes care in the 21st century. Curr Diabetes Rev 2015;11:222–30.
99. Riddle MC, Rosenstock J, Gerich J, Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003;26:3080–6.
From the University of Arizona College of Pharmacy and the University of Arizona College of Medicine-Tucson, Tucson, AZ.
Abstract
- Objective: To summarize key issues relevant to managing hyperglycemia in patients with type 2 diabetes mellitus (T2DM) and review a strategy for initiating and intensifying therapy.
- Methods: Review of the literature.
- Results: The 6 most widely used pharmacologic treatment options for hyperglycemia in T2DM are metformin, sulfonylureas, dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter-2 inhibitors, and insulin. Recent guidelines stress the importance of an individualized, patient-centered approach to managing hyperglycemia in T2DM, although sufficient guidance for nonspecialists on how to individualize treatment is often lacking. For patients with no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. Due to the progressive nature of T2DM, glycemic control on metformin monotherapy is likely to deteriorate over time, and there is no consensus as to what the second-line agent should be. A second agent should be selected based on glycemic goal and potential advantages and disadvantages of each agent for any given patient. If the patient progresses to the point where dual therapy does not provide adequate control, either a third non-insulin agent or insulin can be added.
- Conclusion: Although research is increasingly focusing on what the ideal number and sequence of drugs should be when managing T2DM, investigating all possible combinations in diverse patient populations is not feasible. Physicians therefore must continue to rely on clinical judgment to determine how to apply trial data to the treatment of individual patients.
Key words: type 2 diabetes; patient-centered care; antihyper-glycemic drugs; insulin; therapeutic decision-making.
Diabetes mellitus affects approximately 29.1 million people, or 9.3% of the U.S. population [1,2]. The high prevalence of diabetes and its associated multiple complications, including cardiovascular disease (CVD), blindness, renal failure, lower extremity amputations, and premature death, lead to a tremendous overall burden of disease. The financial cost is staggering as well, with more than 1 in 5 health care dollars spent on treating diabetes or its complications [3]. The goal of diabetes treatment is to prevent acute complications and reduce the risk of long-term complications. Interventions that have been shown to improve diabetes outcomes include medications for glycemic control and treatment of cardiovascular risk factors, nutrition and physical activity counseling, smoking cessation, immunizations, psychosocial care, and ongoing surveillance and early treatment for eye, kidney, and foot problems [4].
Glycemic management in type 2 diabetes mellitus (T2DM), the focus of this review, is growing increasingly complex and has been the subject of numerous extensive reviews [5,6] and published guidelines [4,7]. In the context of an increasing array of available pharmacologic options, there are mounting uncertainties regarding the benefits of intensive glycemic control as well as increasing concerns about potential adverse treatment effects, hypoglycemia in particular. While previous guidelines encouraged specific approaches for most patients, more recent guidelines stress the importance of a patient-centered approach with shared decision-making [4]. Less prescriptive guidelines are more appropriate, given the current state of science, but they also may be viewed as providing insufficient guidance to some providers. It can be overwhelming for a non-specialist to try to match the nuances of antihyperglycemic medications to the nuances of each patient’s preferences and medical characteristics.
This article examines key issues faced by primary care providers when managing hyperglycemia in patients with T2DM and outlines a stepwise approach to determining the optimal antihyperglycemic agent(s) (Table 1).
Confirm Diagnosis of T2DM
It can be difficult to distinguish between type 1 diabetes mellitus and T2DM in some individuals due to overlapping characteristics. However, correctly classifying a patient’s diabetes at the outset is essential, as the classification helps determine the best treatment regimen and is rarely reconsidered [4,8]. Considerable evidence suggests that misclassification of diabetes occurs frequently [9,10], resulting in patients receiving inappropriate treatment. Clinical characteristics suggestive of T2DM include older age and features of insulin resistance such as obesity, hyper-tension, hypertriglyceridemia, and low high-density lipoprotein cholesterol. When these features are not present, an alternate diagnosis should be entertained.
Establish Glycemic Goal
Research over the past decade has led to a growing appreciation of the enormous complexity of hyperglycemia management. During the 1990s, landmark trials such as the Diabetes Control and Complications Trial (DCCT) [11] and UK Prospective Diabetes Study (UKPDS) [12] demonstrated that improving glucose control could reduce the incidence of microvascular complications [11,12], prompting a lower-is-better philosophy regarding glucose targets. Despite limited evidence to support such thinking, this viewpoint was adopted by the developers of many guidelines. During the following decade more research was devoted to determining whether aggressively lowering a patient’s glucose could also improve macrovascular outcomes. Table 2 summarizes microvascular and macrovascular effects of intensive glycemic control seen in major trials [11–23]. After several major trials [20,22] found only mild cardiovascular benefits and even suggested harm [18], experts and policy makers began to reconsider the value of tightly controlling glucose levels [24]. Since then, other studies have demonstrated that the potential benefits and risks of glucose control are strongly related to individual patient factors, such as age and duration of diabetes, and associated comorbidities, such as CVD and impaired renal function [6].
A one-size-fits-all glycemic goal is no longer recommended. Personalization is necessary, balancing the potential benefits and risks of treatments required to achieve that goal. Whereas an A1C of < 7% is an appropriate target for some individuals with diabetes, glycemic targets may be more or less stringent based on patient features including life expectancy, duration of diabetes, comorbidities, and patient attitude and support system (Table 3) [4].
A particular group in which less stringent goals should be considered is older patients, especially those with complex or poor health status [4,25]. The risk of intensive glycemic control may exceed the benefits in these patients, as they are at higher risk of hypoglycemia and polypharmacy [26]. A goal A1C of 7% to 7.5% is now recommended for healthy older adults, and less stringent A1C goals of 7.5% to 8% and 8% to 8.5% should be considered based on the presence and severity of multiple coexisting chronic illnesses, decreased self-care ability, or cognitive impairment [4,25]. Unfortunately, overtreatment is frequently seen in this group. In a recent study of patients over age 65 years, about 40% of those with complex or poor health status had tight glycemic control with A1C below 6.5% [26]. An analysis of U.S. Veterans Affairs administration data showed that only 27% of 12,917 patients older than 65 with very low A1C (< 6%) and about 21% of those with A1C of 6% to 6.5% underwent treatment deintensification [27].
Initiate Treatment with Metformin
There is strong consensus that metformin is the preferred drug for monotherapy due to its long proven safety record, low cost, weight-reduction benefit, and potential cardiovascular advantages [4,16]. As long as there are no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. The recommendation is based on the fact that adherence to diet, weight reduction, and regular exercise is not sustained in most patients, and most patients ultimately will require treatment. Since metformin is usually well-tolerated, does not cause hypoglycemia, has a favorable effect on body weight, and is relatively inexpensive, potential benefits of early initiation of medication appear to outweigh potential risks.
The U.S. Food and Drug Administration (FDA) recently relaxed prescribing polices to extend the use of this important medication to patients who have mild–moderate, but stable, chronic kidney disease (CKD) [28]. Metformin is recommended as first-line therapy and should be used unless it is contraindicated (ie, estimated glomerular filtration rate [eGFR] < 30 mL/min/1.73 m2)[4,7,29].
Add Additional Agent(s) as Needed to Achieve Goal
Other than metformin, evidence is limited for the optimal use of the burgeoning array of available agents, especially in dual or triple combinations [6,30]. Research is now starting to focus more on what the ideal number and sequence of drugs should be. The Glycemic Reduction Approach in Diabetes (GRADE) study, which will compare long-term benefits and risks of the 4 most widely used antihyperglycemic medications in combination with metformin, is now underway [31,32]. The 4 classes being studied are sulfonylurea, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, and a basal,
Eleven classes of non-insulin medications are currently approved for treating hyperglycemia in T2DM [4]. Within each class, numerous agents are available. Six of these classes (ie, α-glucosidase inhibitors, colesevelam, bromocriptine, pramlintide, meglitinides, and thiazolidinediones) are not used frequently
Consider Effects on A1C
There is a paucity of high-quality, head-to-head comparison trials evaluating the ability of available agents to achieve recommended glycemic targets. This is important because the glucose-lowering effectiveness of individual medications is strongly influenced by baseline characteristics such as A1C, duration of diabetes, and previous therapy. With these limitations in mind, the relative glucose-lowering effectiveness of commonly used agents is shown in Table 4. When used as monotherapy, A1C reductions of approximately 1% to 1.5% are achieved with metformin, sulfonylureas, and GLP-1 receptor agonists [6,30,34,35,39]. DPP-4 inhibitors and SGLT-2 inhibitors have more modest glucose-lowering efficacy, with A1C reductions of approximately 0.5% to 1% [6,30,34,35,39]. Larger effects may be seen in individuals with higher baseline A1C and those who are drug naïve. Insulin is the most effective glucose-lowering agent—it can reduce virtually any level of A1C down to the normal range, with hypoglycemia being the only limiting factor. When a patient has uncontrolled hyperglycemia on metformin monotherapy, or if there is a contraindication or intolerance to metformin, clinicians should consider the potential glucose-lowering effects of other available options and should choose an agent that conceivably could bring a patient close to meeting their treatment goal.
Eliminate Options with Unacceptable Adverse Effects
When the pharmacologic options with acceptable A1C-lowering potential have been identified, the ones with contraindications and potential serious adverse effects for the individual patient can immediately be eliminated (Table 4). For example, if a patient has an eGFR < 30 mL/min/1.73 m2, metformin, sulfonylureas, GLP-1 receptor agonists, most DPP-4 inhibitors, and SGLT-2 inhibitors are either contraindicated or should be used with caution. In patients with severe osteoporosis, SGLT-2 inhibitors may not be the best option. In patients with a history of diabetic ketoacidosis (DKA), caution should be used with metformin and SGLT-2 inhibitors. There have been concerns of possible acute pancreatitis and neoplasia with the incretin-based agents, the DPP-4 inhibitors and GLP-1 receptor agonists [40,41], although other clinical trials and observational data have not found increased risk [42–45]. Nevertheless, these agents potentially should be avoided in patients with a history of pancreatitis or neoplasm. SGLT-2 inhibitors may be associated with genitourinary infections and volume depletion [46–48] and probably should be avoided in patients at high risk for these conditions.
If the adverse effects are not serious, changing the way the medication is administered may allow the patient to tolerate agents with high potential benefits. For example, metformin is commonly associated with gastrointestinal (GI) adverse effects, which can be reduced or avoided with slow titration of the dose [6] or by switching to an extended-release formulation [49]. GLP-1 receptor agonists are associated with GI adverse effects [6] and in most cases slow titration is recommended.
Evaluate Potential Risks/Benefits of Remaining Options
Hypoglycemia. The barrier of hypoglycemia generally precludes maintenance of euglycemia and full realization of the long-term benefits of good glucose control over a lifetime. Once considered a trivial issue, concerns about hypoglycemia in T2DM are increasingly being raised [19,50–55]. Clearly, hypoglycemia occurs more often as glycemic targets are lowered to near-normal values, especially in those with advanced age and multiple comorbidities [55]. Various comorbidities frequently encountered particularly as patients age also are associated with increasing propensity for experiencing hypoglycemia and untoward outcomes from it. These include coronary artery disease, heart failure, renal and liver disease, and dementia. Hypoglycemia, when it occurs, may lead to dysrhythmias, dizziness, accidents and falls, work disability, and decreased quality of life. In addition to relaxing blood glucose targets in high-risk patients, drug selection should favor agents that do not precipitate such events (Table 4).
Fortunately, the commonly used non-insulin agents are not associated with hypoglycemia unless they are used in combination with sulfonylureas or insulin. Sulfonylureas should be used with caution and other options considered in patients with high risk for hypoglycemia. When insulin is required, regimens which minimize risk of hypoglycemia should be used. For example, adding a GLP-1 receptor agonist to basal insulin as an alternative to mealtime insulin has been shown to be equally effective with a lower risk of hypoglycemia [4,6]. Also, premixed insulin preparations should be avoided or used cautiously in individuals who miss meals frequently. Additionally, newer basal insulins that exhibit longer duration of action are now available in the United States. Preliminary studies have shown that the newly FDA-approved longer-acting basal insulins, insulin degludec and glargine U-300, may be associated with a reduced risk for hypoglycemia [56,57]. However, it remains unclear how and when these newer agents will best be incorporated into a treatment regimen.
Body weight. Nearly 90% of people living with T2DM are overweight or obese. Given the close tie between obesity and T2DM, treating obesity is an obvious consideration in diabetes treatment. Major trials have shown the effectiveness of lifestyle modifications and weight reduction in delaying, prevention, and management of T2DM [4,58,59].With this in mind, clinicians should consider preferentially using antihyperglycemic agents with weight-lowering or weight-neutral effects. Among commonly used antihyperglycemic agents, metformin, GLP-1 receptor agonists, and SGLT-2 inhibitors have been shown to have weight-reduction benefits, and DPP-4 inhibitors are weight neutral. On the other hand, sulfonylureas and insulin are associated with weight gain. A systematic review and meta-analysis including 204 studies with study durations ranging from 3 months to 8 years showed comparative effects of diabetes medications with a differential effect on weight of up to 5 kg (Table 4) [60].
Metformin is associated with an average weight loss of 1.9 to 3.1 kg that was sustained with long-term use for at least 10 years in the Diabetes Prevention Program Outcomes Study [61].A systematic review of 7 randomized trials showed that in patients with T2DM, the SGLT-2 inhibitors dapagliflozin and canagliflozin were associated with weight loss (mean weighted difference of –1.81 kg and –2.3 kg, respectively) [62]. A systematic review and meta-analysis of 25 randomized controlled trials showed greater weight loss (mean weighted difference of –2.9 kg) in overweight or obese patients with or without T2DM using GLP-1 receptor agonists when compared to placebo, insulin, or oral antihyperglycemic agents [63]. Of note, the GLP-1 receptor agonist liraglutide is now approved for weight loss in patients with or without diabetes [64]. The maximum doses approved for diabetes and obesity treatment are 1.8 and 3.0 mg/day, respectively.
Since weight loss is associated with improved glycemic control, an area of emerging interest is the use of antiobesity medications for managing diabetes. Although most older weight-loss medications were only approved for short-term use, some newer agents are approved for longer-term use. Lorcaserin and the combination drugs topiramate/phentermine and naltrexone/bupropion are approved for chronic therapy, provided certain conditions are met. Patients on weight reduction agents should be monitored regularly.
An even more radical departure from conventional therapy for diabetes is the consideration of metabolic, or weight-loss, surgery, which has been found to be associated with rapid and dramatic improvements in blood glucose control. Metabolic surgery has been shown to improve glucose control more effectively than any known pharmaceutical or behavioral approach. For example, in an observational study of obese patients with T2DM, bariatric surgery led to diabetes remission rates of 72.3% 2 years after surgery and 30.4% 15 years after surgery compared to 16.4% and 6.5%, respectively, in control patients [69]. With long-term follow-up, significant decreases in microvascular and macrovascular complications were seen in the surgical group [69]. Compared with medical therapy alone, bariatric surgery plus medical therapy has been associated with more weight loss, better glycemic control, less need for diabetes medications, and improved quality of life [70]. A 2016 joint statement by numerous international diabetes organizations recommends considering metabolic surgery as a treatment for T2DM and obesity [71]. American Diabetes Association guidelines recommend consideration of bariatric surgery in individuals with T2DM who have a body mass index greater than 35 kg/m2,especially if achieving disease control is difficult by means of lifestyle modifications and medications [4].
Cardiovascular outcomes. Cardiovascular risk is about 2 to 4 times higher in patients with diabetes, and about half of patients with this condition develop heart failure [4,72]. CVD is responsible for most of the mortality in T2DM [72]. Therefore, prevention of cardiovascular morbidity and mortality is an important goal for diabetes treatment. Due to concerns about potential cardiovascular risks associated with glucose-lowering medications [73–76], the FDA has issued regulatory requirements for manufacturers to monitor the cardiovascular risk profile for these drugs [77]. Recent trials have led to a better understanding of potential cardiovascular benefits or harms of antihyperglycemic medications.
Metformin, the widely recommended first-line therapy for T2DM, carries a large body of evidence supporting its cardiovascular benefits. For example, the UKPDS found that compared to conventional therapy (mostly diet), metformin reduced cardiovascular events and mortality in obese patients with T2DM [15]. This result was supported in Hyperinsulinemia: the Outcome of its Metabolic Effect (HOME) study where, as an add-on to insulin, metformin decreased macrovascular complications when compared to placebo [78]. Research over the past decade also has assuaged concerns about metformin safety in heart failure [60]. A systematic review of observational studies involving 34,000 patients conducted in 2013 showed that metformin is as safe as other glucose-lowering medications in patients with diabetes and heart failure even in the presence of CKD [4,79]. Furthermore, numerous investigations have found metformin is not associated with increased hospitalizations or risk of lactic acidosis [80]. Metformin can be used safely in patients with diabetes and heart failure [60].
Although sulfonylureas have long been a mainstay of diabetes therapy, concerns about their potential adverse cardiovascular effects have been raised by numerous studies [81]. Tolbutamide, a first-generation sulfonylurea, was removed from the market after the University Group Diabetes Program study found increased CVD deaths with this agent versus placebo. Subsequently, the FDA issued a warning for all sulfonylureas [74]. The increased cardiovascular risk associated with sulfonylureas is thought to be due to their effect on cardiac mitochondrial potassium ATP channels. Sulfonylureas bind to these channels, preventing a protective phenomenon called ischemic preconditioning and resulting in a weakened defense against myocardial injury [76]. A recent study showed an increased risk of coronary heart disease associated with long-term use of sulfonylureas in women with diabetes [81].
GLP-1 receptor agonists have recently received much attention for their potential beneficial effects on cardiovascular outcomes. In a recent trial, lixisenatide was shown to be safe in patients with T2DM and acute coronary syndrome when compared to placebo [82]. More recently, the Liraglutide Effect and Action in Diabetes: Evaluation of cardiovascular outcome Results (LEADER) trial demonstrated significant cardiovascular benefits with liraglutide in patients with T2DM and established or high CVD risk [83]. The composite outcome of the first occurrence of death from cardiovascular causes, nonfatal myocardial infarction (MI), or nonfatal stroke, occurred less frequently in the liraglutide group compared to placebo (13% versus 14.9%, respectively), and there were fewer deaths from cardiovascular causes in the liraglutide group compared to placebo (4.7% and 6.0%, respectively) [83]. Other trials investigating the cardiovascular outcomes of this class [84,85] are in progress.
Another class with potential cardiovascular benefits is the SGLT-2 inhibitors. In a recent cardiovascular outcome study, empagliflozin significantly lowered the composite of cardiovascular death, nonfatal MI, or nonfatal stroke in T2DM patients with high cardiovascular risk compared to placebo (10.5% and 12.1%, respectively) [86]. There are several large ongoing studies evaluating the cardiovascular effects of other SGLT-2 inhibitors [87–89].
DPP-4 inhibitors were examined in recent studies and have shown no cardiovascular benefits [42,44,90].The studies showed mixed results regarding an association between DPP-4 inhibitors and heart failure. In one study, saxagliptin was associated with increased hospitalization for heart failure compared to placebo [44], while 2 noninferiority trials did not show a significant increase in heart failure hospitalizations associated with alogliptin and sitagliptin when compared to placebo [42,90].
Administration Considerations
Many patients with T2DM require multiple agents for glycemic control. Additional medications used for comorbid conditions add to this burden. When choosing antihyperglycemic agents, the route and frequency of administration, as well as the patients’ preferences and ability, should be considered. Either once or twice daily dosing is available for most agents, and once weekly dosing is available for some of the GLP-1 receptor agonists. Once daily or once weekly formulations may improve adherence and be more desirable than preparations that are dosed twice daily. Most of the commonly used medications are dosed orally. Although many patients find this route of administration preferable to insulin or GLP-1 receptor agonists, which require injections, some patients may prefer the risk/benefit of injectable agents. All GLP-1 receptor agonists come in a pen delivery system, which eliminates mixing and provides more convenient administration. Extended-release exenatide also is available as a single-dose tray that requires mixing and may be more cumbersome to inject.
Insulin requires special consideration. There has been an enormous increase in the number of insulin products on the market in the past 2 decades. These products include insulin analogs, concentrated insulins (U-200, U-300, and U-500), premixed insulin preparations, and ultra-long-acting insulin [91]. The availability of insulin options with different concentrations, onsets, and durations of actions has made decision making on which insulin to use difficult. Clinicians need to consider patient preference, dosing frequency, and timing with regard to meals, insulin dose, administration, as well as cost. For example, concentrated insulin is preferred for a patient on high doses of insulin requiring injecting a large volume of insulin. Rapid-acting insulin analogs would be more appropriate for patients who have difficulty administering their regular insulin 20 to 30 minutes before eating. Premixed insulin preparations make it impossible to independently adjust short- and long-acting components. However, these may be good choices in patients who have consistent meal schedules and who want to simplify administration. Despite a prevailing misconception that NPH must be given twice a day, it has long been recognized that in T2DM, a single daily injection of NPH yields improvements in control similar to those achieved with 2 daily injections [92].
Cost Considerations
Treating T2DM imposes a great financial burden on individuals living with diabetes and their families due to the high cost of the medications. Table 4 and Table 5 provide information on the cost of non-insulin and insulin diabetes medications for patients who do not have prescription insurance coverage. From a practical standpoint, choice of diabetes agents is largely influenced by insurance formularies.
The older agents, metformin and the sulfonylureas, are available for a cash (no insurance) price of as little as $4 per month. This is in stark contrast to the SGLT-2 inhibitors, GLP-1 receptor agonists, and DPP-4 inhibitors, which range in cost between $400 and $600 per month. Of recent concern, the cost of insulin has been skyrocketing, with a more than 500% increase in the cost of certain insulins from 2001 to 2015 [93]. According to the Medical Expenditure Panel Survey (MEPS) from 2002 to 2013, the mean price of insulin increased by about 200% (from $4.34/mL to $12.92/mL) during this period, which was significantly higher than increases in the price of non-insulin comparators [94]. The introduction of biosimilar insulins to the market is expected to offer treatment options with lower cost. This will be tested when the biosimilar glargine, the first FDA-approved biosimilar insulin, becomes available in the U.S. market. However, a significant reduction in insulin prices is not expected soon [95].
When insulin is required, most patients with T2DM can be treated with older human insulins, which have similar efficacy and lower costs than the more expensive newer insulin analogs. A Cochrane review comparing basal insulin analogs to NPH showed similar efficacy in glycemic control with minimal clinical benefit in the form of less nocturnal hypoglycemia in the insulin analog arm [96]. Furthermore, similar glycemic control and risk of hypoglycemia was seen when regular insulin was compared with the rapid-acting insulin analogs [97]. The cost of human NPH insulin for a patient on a total daily dose of 60 units is approximately $52 per month. This contrasts with the most widely used insulin, insulin glargine, which has a cash price of about $500 per month for the same amount (Table 5). Insulin pens, which are convenient, are more expensive. Interestingly, human insulins do not require prescriptions, allowing underinsured, underfunded patients ongoing access to them.
Incorporating Patient Preferences
Research evidence is necessary but insufficient for making patient care decisions. Along with the potential benefits, harms, costs, and inconveniences of the management options, patient perspectives, beliefs, expectations, and health-related goals must be considered. Patients will undoubtedly have preferences regarding defining goals and ranking options. Clinicians should discuss therapeutic goals and treatment options and work collaboratively with patients in determining management strategies [98].
Summary
Potential treatment approaches for treating hyperglycemia in T2DM are summarized in Figure 1 and Figure 2 [4,7]. As long as there are no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. Even if metformin monotherapy is initially effective, glycemic control is likely to deteriorate over time due to progressive loss of β-cell function in T2DM.
There is no consensus as to what the second-line agent should be. Selection of a second agent should be made based on potential advantages and disadvantages of each agent for any given patient. A patient-centered approach is preferred over a fixed algorithm. If the patient progresses to the point where dual therapy does not provide adequate control, either a third non-insulin agent or insulin can be added. In patients with modestly elevated A1C (below ~8%), addition of a third non-insulin agent may be equally effective as (but more expensive than) addition of insulin.
Patients with significantly elevated A1C levels on non-insulin agents usually should have insulin added to their regimen. When insulin is added, metformin should be continued. DPP-4 inhibitors and sulfonylureas are typically stopped. If SGLT-2 inhibitors and/or GLP-1 receptor agonists are continued, this may aid with weight maintenance. However, continuing these agents is likely to be expensive and associated with problems associated with polypharmacy.
The most widely recommended strategy for initiating insulin in T2DM is to add a single bedtime injection of basal insulin (ie, NPH, glargine, detemir, or degludec) to the patient’s regimen. This regimen has been found to be effective in numerous studies and controls hyperglycemia in up to 60% of patients [99]. If the patient is treated with a single bedtime injection of insulin and the fasting glucose level is within the target range but the A1C level remains above goal, addition of mealtime insulin injections is likely to be beneficial. Alternatively, addition of a GLP-1 receptor agonist to basal insulin has been shown to be equally beneficial [4,6]. When adding mealtime insulin, a common strategy is to add a single injection of a rapid-acting insulin (eg, lispro, aspart, glulisine) before the patient’s largest meal of the day. Additional premeal injections of rapid-acting insulin may be added as needed, based on self-monitoring blood glucose results. If glycemia remains significantly uncontrolled on more than 200 units of insulin per day, switching to a concentrated form of insulin (eg, U-200, U-300, or U-500) should be considered.
Corresponding author: Maryam Fazel, PharmD, BCPS, BCACP, CDE, 1295 N. Martin Ave. (Room B211B), Tucson, Arizona 85721-0202, [email protected].
Financial disclosures: None.
From the University of Arizona College of Pharmacy and the University of Arizona College of Medicine-Tucson, Tucson, AZ.
Abstract
- Objective: To summarize key issues relevant to managing hyperglycemia in patients with type 2 diabetes mellitus (T2DM) and review a strategy for initiating and intensifying therapy.
- Methods: Review of the literature.
- Results: The 6 most widely used pharmacologic treatment options for hyperglycemia in T2DM are metformin, sulfonylureas, dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter-2 inhibitors, and insulin. Recent guidelines stress the importance of an individualized, patient-centered approach to managing hyperglycemia in T2DM, although sufficient guidance for nonspecialists on how to individualize treatment is often lacking. For patients with no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. Due to the progressive nature of T2DM, glycemic control on metformin monotherapy is likely to deteriorate over time, and there is no consensus as to what the second-line agent should be. A second agent should be selected based on glycemic goal and potential advantages and disadvantages of each agent for any given patient. If the patient progresses to the point where dual therapy does not provide adequate control, either a third non-insulin agent or insulin can be added.
- Conclusion: Although research is increasingly focusing on what the ideal number and sequence of drugs should be when managing T2DM, investigating all possible combinations in diverse patient populations is not feasible. Physicians therefore must continue to rely on clinical judgment to determine how to apply trial data to the treatment of individual patients.
Key words: type 2 diabetes; patient-centered care; antihyper-glycemic drugs; insulin; therapeutic decision-making.
Diabetes mellitus affects approximately 29.1 million people, or 9.3% of the U.S. population [1,2]. The high prevalence of diabetes and its associated multiple complications, including cardiovascular disease (CVD), blindness, renal failure, lower extremity amputations, and premature death, lead to a tremendous overall burden of disease. The financial cost is staggering as well, with more than 1 in 5 health care dollars spent on treating diabetes or its complications [3]. The goal of diabetes treatment is to prevent acute complications and reduce the risk of long-term complications. Interventions that have been shown to improve diabetes outcomes include medications for glycemic control and treatment of cardiovascular risk factors, nutrition and physical activity counseling, smoking cessation, immunizations, psychosocial care, and ongoing surveillance and early treatment for eye, kidney, and foot problems [4].
Glycemic management in type 2 diabetes mellitus (T2DM), the focus of this review, is growing increasingly complex and has been the subject of numerous extensive reviews [5,6] and published guidelines [4,7]. In the context of an increasing array of available pharmacologic options, there are mounting uncertainties regarding the benefits of intensive glycemic control as well as increasing concerns about potential adverse treatment effects, hypoglycemia in particular. While previous guidelines encouraged specific approaches for most patients, more recent guidelines stress the importance of a patient-centered approach with shared decision-making [4]. Less prescriptive guidelines are more appropriate, given the current state of science, but they also may be viewed as providing insufficient guidance to some providers. It can be overwhelming for a non-specialist to try to match the nuances of antihyperglycemic medications to the nuances of each patient’s preferences and medical characteristics.
This article examines key issues faced by primary care providers when managing hyperglycemia in patients with T2DM and outlines a stepwise approach to determining the optimal antihyperglycemic agent(s) (Table 1).
Confirm Diagnosis of T2DM
It can be difficult to distinguish between type 1 diabetes mellitus and T2DM in some individuals due to overlapping characteristics. However, correctly classifying a patient’s diabetes at the outset is essential, as the classification helps determine the best treatment regimen and is rarely reconsidered [4,8]. Considerable evidence suggests that misclassification of diabetes occurs frequently [9,10], resulting in patients receiving inappropriate treatment. Clinical characteristics suggestive of T2DM include older age and features of insulin resistance such as obesity, hyper-tension, hypertriglyceridemia, and low high-density lipoprotein cholesterol. When these features are not present, an alternate diagnosis should be entertained.
Establish Glycemic Goal
Research over the past decade has led to a growing appreciation of the enormous complexity of hyperglycemia management. During the 1990s, landmark trials such as the Diabetes Control and Complications Trial (DCCT) [11] and UK Prospective Diabetes Study (UKPDS) [12] demonstrated that improving glucose control could reduce the incidence of microvascular complications [11,12], prompting a lower-is-better philosophy regarding glucose targets. Despite limited evidence to support such thinking, this viewpoint was adopted by the developers of many guidelines. During the following decade more research was devoted to determining whether aggressively lowering a patient’s glucose could also improve macrovascular outcomes. Table 2 summarizes microvascular and macrovascular effects of intensive glycemic control seen in major trials [11–23]. After several major trials [20,22] found only mild cardiovascular benefits and even suggested harm [18], experts and policy makers began to reconsider the value of tightly controlling glucose levels [24]. Since then, other studies have demonstrated that the potential benefits and risks of glucose control are strongly related to individual patient factors, such as age and duration of diabetes, and associated comorbidities, such as CVD and impaired renal function [6].
A one-size-fits-all glycemic goal is no longer recommended. Personalization is necessary, balancing the potential benefits and risks of treatments required to achieve that goal. Whereas an A1C of < 7% is an appropriate target for some individuals with diabetes, glycemic targets may be more or less stringent based on patient features including life expectancy, duration of diabetes, comorbidities, and patient attitude and support system (Table 3) [4].
A particular group in which less stringent goals should be considered is older patients, especially those with complex or poor health status [4,25]. The risk of intensive glycemic control may exceed the benefits in these patients, as they are at higher risk of hypoglycemia and polypharmacy [26]. A goal A1C of 7% to 7.5% is now recommended for healthy older adults, and less stringent A1C goals of 7.5% to 8% and 8% to 8.5% should be considered based on the presence and severity of multiple coexisting chronic illnesses, decreased self-care ability, or cognitive impairment [4,25]. Unfortunately, overtreatment is frequently seen in this group. In a recent study of patients over age 65 years, about 40% of those with complex or poor health status had tight glycemic control with A1C below 6.5% [26]. An analysis of U.S. Veterans Affairs administration data showed that only 27% of 12,917 patients older than 65 with very low A1C (< 6%) and about 21% of those with A1C of 6% to 6.5% underwent treatment deintensification [27].
Initiate Treatment with Metformin
There is strong consensus that metformin is the preferred drug for monotherapy due to its long proven safety record, low cost, weight-reduction benefit, and potential cardiovascular advantages [4,16]. As long as there are no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. The recommendation is based on the fact that adherence to diet, weight reduction, and regular exercise is not sustained in most patients, and most patients ultimately will require treatment. Since metformin is usually well-tolerated, does not cause hypoglycemia, has a favorable effect on body weight, and is relatively inexpensive, potential benefits of early initiation of medication appear to outweigh potential risks.
The U.S. Food and Drug Administration (FDA) recently relaxed prescribing polices to extend the use of this important medication to patients who have mild–moderate, but stable, chronic kidney disease (CKD) [28]. Metformin is recommended as first-line therapy and should be used unless it is contraindicated (ie, estimated glomerular filtration rate [eGFR] < 30 mL/min/1.73 m2)[4,7,29].
Add Additional Agent(s) as Needed to Achieve Goal
Other than metformin, evidence is limited for the optimal use of the burgeoning array of available agents, especially in dual or triple combinations [6,30]. Research is now starting to focus more on what the ideal number and sequence of drugs should be. The Glycemic Reduction Approach in Diabetes (GRADE) study, which will compare long-term benefits and risks of the 4 most widely used antihyperglycemic medications in combination with metformin, is now underway [31,32]. The 4 classes being studied are sulfonylurea, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, and a basal,
Eleven classes of non-insulin medications are currently approved for treating hyperglycemia in T2DM [4]. Within each class, numerous agents are available. Six of these classes (ie, α-glucosidase inhibitors, colesevelam, bromocriptine, pramlintide, meglitinides, and thiazolidinediones) are not used frequently
Consider Effects on A1C
There is a paucity of high-quality, head-to-head comparison trials evaluating the ability of available agents to achieve recommended glycemic targets. This is important because the glucose-lowering effectiveness of individual medications is strongly influenced by baseline characteristics such as A1C, duration of diabetes, and previous therapy. With these limitations in mind, the relative glucose-lowering effectiveness of commonly used agents is shown in Table 4. When used as monotherapy, A1C reductions of approximately 1% to 1.5% are achieved with metformin, sulfonylureas, and GLP-1 receptor agonists [6,30,34,35,39]. DPP-4 inhibitors and SGLT-2 inhibitors have more modest glucose-lowering efficacy, with A1C reductions of approximately 0.5% to 1% [6,30,34,35,39]. Larger effects may be seen in individuals with higher baseline A1C and those who are drug naïve. Insulin is the most effective glucose-lowering agent—it can reduce virtually any level of A1C down to the normal range, with hypoglycemia being the only limiting factor. When a patient has uncontrolled hyperglycemia on metformin monotherapy, or if there is a contraindication or intolerance to metformin, clinicians should consider the potential glucose-lowering effects of other available options and should choose an agent that conceivably could bring a patient close to meeting their treatment goal.
Eliminate Options with Unacceptable Adverse Effects
When the pharmacologic options with acceptable A1C-lowering potential have been identified, the ones with contraindications and potential serious adverse effects for the individual patient can immediately be eliminated (Table 4). For example, if a patient has an eGFR < 30 mL/min/1.73 m2, metformin, sulfonylureas, GLP-1 receptor agonists, most DPP-4 inhibitors, and SGLT-2 inhibitors are either contraindicated or should be used with caution. In patients with severe osteoporosis, SGLT-2 inhibitors may not be the best option. In patients with a history of diabetic ketoacidosis (DKA), caution should be used with metformin and SGLT-2 inhibitors. There have been concerns of possible acute pancreatitis and neoplasia with the incretin-based agents, the DPP-4 inhibitors and GLP-1 receptor agonists [40,41], although other clinical trials and observational data have not found increased risk [42–45]. Nevertheless, these agents potentially should be avoided in patients with a history of pancreatitis or neoplasm. SGLT-2 inhibitors may be associated with genitourinary infections and volume depletion [46–48] and probably should be avoided in patients at high risk for these conditions.
If the adverse effects are not serious, changing the way the medication is administered may allow the patient to tolerate agents with high potential benefits. For example, metformin is commonly associated with gastrointestinal (GI) adverse effects, which can be reduced or avoided with slow titration of the dose [6] or by switching to an extended-release formulation [49]. GLP-1 receptor agonists are associated with GI adverse effects [6] and in most cases slow titration is recommended.
Evaluate Potential Risks/Benefits of Remaining Options
Hypoglycemia. The barrier of hypoglycemia generally precludes maintenance of euglycemia and full realization of the long-term benefits of good glucose control over a lifetime. Once considered a trivial issue, concerns about hypoglycemia in T2DM are increasingly being raised [19,50–55]. Clearly, hypoglycemia occurs more often as glycemic targets are lowered to near-normal values, especially in those with advanced age and multiple comorbidities [55]. Various comorbidities frequently encountered particularly as patients age also are associated with increasing propensity for experiencing hypoglycemia and untoward outcomes from it. These include coronary artery disease, heart failure, renal and liver disease, and dementia. Hypoglycemia, when it occurs, may lead to dysrhythmias, dizziness, accidents and falls, work disability, and decreased quality of life. In addition to relaxing blood glucose targets in high-risk patients, drug selection should favor agents that do not precipitate such events (Table 4).
Fortunately, the commonly used non-insulin agents are not associated with hypoglycemia unless they are used in combination with sulfonylureas or insulin. Sulfonylureas should be used with caution and other options considered in patients with high risk for hypoglycemia. When insulin is required, regimens which minimize risk of hypoglycemia should be used. For example, adding a GLP-1 receptor agonist to basal insulin as an alternative to mealtime insulin has been shown to be equally effective with a lower risk of hypoglycemia [4,6]. Also, premixed insulin preparations should be avoided or used cautiously in individuals who miss meals frequently. Additionally, newer basal insulins that exhibit longer duration of action are now available in the United States. Preliminary studies have shown that the newly FDA-approved longer-acting basal insulins, insulin degludec and glargine U-300, may be associated with a reduced risk for hypoglycemia [56,57]. However, it remains unclear how and when these newer agents will best be incorporated into a treatment regimen.
Body weight. Nearly 90% of people living with T2DM are overweight or obese. Given the close tie between obesity and T2DM, treating obesity is an obvious consideration in diabetes treatment. Major trials have shown the effectiveness of lifestyle modifications and weight reduction in delaying, prevention, and management of T2DM [4,58,59].With this in mind, clinicians should consider preferentially using antihyperglycemic agents with weight-lowering or weight-neutral effects. Among commonly used antihyperglycemic agents, metformin, GLP-1 receptor agonists, and SGLT-2 inhibitors have been shown to have weight-reduction benefits, and DPP-4 inhibitors are weight neutral. On the other hand, sulfonylureas and insulin are associated with weight gain. A systematic review and meta-analysis including 204 studies with study durations ranging from 3 months to 8 years showed comparative effects of diabetes medications with a differential effect on weight of up to 5 kg (Table 4) [60].
Metformin is associated with an average weight loss of 1.9 to 3.1 kg that was sustained with long-term use for at least 10 years in the Diabetes Prevention Program Outcomes Study [61].A systematic review of 7 randomized trials showed that in patients with T2DM, the SGLT-2 inhibitors dapagliflozin and canagliflozin were associated with weight loss (mean weighted difference of –1.81 kg and –2.3 kg, respectively) [62]. A systematic review and meta-analysis of 25 randomized controlled trials showed greater weight loss (mean weighted difference of –2.9 kg) in overweight or obese patients with or without T2DM using GLP-1 receptor agonists when compared to placebo, insulin, or oral antihyperglycemic agents [63]. Of note, the GLP-1 receptor agonist liraglutide is now approved for weight loss in patients with or without diabetes [64]. The maximum doses approved for diabetes and obesity treatment are 1.8 and 3.0 mg/day, respectively.
Since weight loss is associated with improved glycemic control, an area of emerging interest is the use of antiobesity medications for managing diabetes. Although most older weight-loss medications were only approved for short-term use, some newer agents are approved for longer-term use. Lorcaserin and the combination drugs topiramate/phentermine and naltrexone/bupropion are approved for chronic therapy, provided certain conditions are met. Patients on weight reduction agents should be monitored regularly.
An even more radical departure from conventional therapy for diabetes is the consideration of metabolic, or weight-loss, surgery, which has been found to be associated with rapid and dramatic improvements in blood glucose control. Metabolic surgery has been shown to improve glucose control more effectively than any known pharmaceutical or behavioral approach. For example, in an observational study of obese patients with T2DM, bariatric surgery led to diabetes remission rates of 72.3% 2 years after surgery and 30.4% 15 years after surgery compared to 16.4% and 6.5%, respectively, in control patients [69]. With long-term follow-up, significant decreases in microvascular and macrovascular complications were seen in the surgical group [69]. Compared with medical therapy alone, bariatric surgery plus medical therapy has been associated with more weight loss, better glycemic control, less need for diabetes medications, and improved quality of life [70]. A 2016 joint statement by numerous international diabetes organizations recommends considering metabolic surgery as a treatment for T2DM and obesity [71]. American Diabetes Association guidelines recommend consideration of bariatric surgery in individuals with T2DM who have a body mass index greater than 35 kg/m2,especially if achieving disease control is difficult by means of lifestyle modifications and medications [4].
Cardiovascular outcomes. Cardiovascular risk is about 2 to 4 times higher in patients with diabetes, and about half of patients with this condition develop heart failure [4,72]. CVD is responsible for most of the mortality in T2DM [72]. Therefore, prevention of cardiovascular morbidity and mortality is an important goal for diabetes treatment. Due to concerns about potential cardiovascular risks associated with glucose-lowering medications [73–76], the FDA has issued regulatory requirements for manufacturers to monitor the cardiovascular risk profile for these drugs [77]. Recent trials have led to a better understanding of potential cardiovascular benefits or harms of antihyperglycemic medications.
Metformin, the widely recommended first-line therapy for T2DM, carries a large body of evidence supporting its cardiovascular benefits. For example, the UKPDS found that compared to conventional therapy (mostly diet), metformin reduced cardiovascular events and mortality in obese patients with T2DM [15]. This result was supported in Hyperinsulinemia: the Outcome of its Metabolic Effect (HOME) study where, as an add-on to insulin, metformin decreased macrovascular complications when compared to placebo [78]. Research over the past decade also has assuaged concerns about metformin safety in heart failure [60]. A systematic review of observational studies involving 34,000 patients conducted in 2013 showed that metformin is as safe as other glucose-lowering medications in patients with diabetes and heart failure even in the presence of CKD [4,79]. Furthermore, numerous investigations have found metformin is not associated with increased hospitalizations or risk of lactic acidosis [80]. Metformin can be used safely in patients with diabetes and heart failure [60].
Although sulfonylureas have long been a mainstay of diabetes therapy, concerns about their potential adverse cardiovascular effects have been raised by numerous studies [81]. Tolbutamide, a first-generation sulfonylurea, was removed from the market after the University Group Diabetes Program study found increased CVD deaths with this agent versus placebo. Subsequently, the FDA issued a warning for all sulfonylureas [74]. The increased cardiovascular risk associated with sulfonylureas is thought to be due to their effect on cardiac mitochondrial potassium ATP channels. Sulfonylureas bind to these channels, preventing a protective phenomenon called ischemic preconditioning and resulting in a weakened defense against myocardial injury [76]. A recent study showed an increased risk of coronary heart disease associated with long-term use of sulfonylureas in women with diabetes [81].
GLP-1 receptor agonists have recently received much attention for their potential beneficial effects on cardiovascular outcomes. In a recent trial, lixisenatide was shown to be safe in patients with T2DM and acute coronary syndrome when compared to placebo [82]. More recently, the Liraglutide Effect and Action in Diabetes: Evaluation of cardiovascular outcome Results (LEADER) trial demonstrated significant cardiovascular benefits with liraglutide in patients with T2DM and established or high CVD risk [83]. The composite outcome of the first occurrence of death from cardiovascular causes, nonfatal myocardial infarction (MI), or nonfatal stroke, occurred less frequently in the liraglutide group compared to placebo (13% versus 14.9%, respectively), and there were fewer deaths from cardiovascular causes in the liraglutide group compared to placebo (4.7% and 6.0%, respectively) [83]. Other trials investigating the cardiovascular outcomes of this class [84,85] are in progress.
Another class with potential cardiovascular benefits is the SGLT-2 inhibitors. In a recent cardiovascular outcome study, empagliflozin significantly lowered the composite of cardiovascular death, nonfatal MI, or nonfatal stroke in T2DM patients with high cardiovascular risk compared to placebo (10.5% and 12.1%, respectively) [86]. There are several large ongoing studies evaluating the cardiovascular effects of other SGLT-2 inhibitors [87–89].
DPP-4 inhibitors were examined in recent studies and have shown no cardiovascular benefits [42,44,90].The studies showed mixed results regarding an association between DPP-4 inhibitors and heart failure. In one study, saxagliptin was associated with increased hospitalization for heart failure compared to placebo [44], while 2 noninferiority trials did not show a significant increase in heart failure hospitalizations associated with alogliptin and sitagliptin when compared to placebo [42,90].
Administration Considerations
Many patients with T2DM require multiple agents for glycemic control. Additional medications used for comorbid conditions add to this burden. When choosing antihyperglycemic agents, the route and frequency of administration, as well as the patients’ preferences and ability, should be considered. Either once or twice daily dosing is available for most agents, and once weekly dosing is available for some of the GLP-1 receptor agonists. Once daily or once weekly formulations may improve adherence and be more desirable than preparations that are dosed twice daily. Most of the commonly used medications are dosed orally. Although many patients find this route of administration preferable to insulin or GLP-1 receptor agonists, which require injections, some patients may prefer the risk/benefit of injectable agents. All GLP-1 receptor agonists come in a pen delivery system, which eliminates mixing and provides more convenient administration. Extended-release exenatide also is available as a single-dose tray that requires mixing and may be more cumbersome to inject.
Insulin requires special consideration. There has been an enormous increase in the number of insulin products on the market in the past 2 decades. These products include insulin analogs, concentrated insulins (U-200, U-300, and U-500), premixed insulin preparations, and ultra-long-acting insulin [91]. The availability of insulin options with different concentrations, onsets, and durations of actions has made decision making on which insulin to use difficult. Clinicians need to consider patient preference, dosing frequency, and timing with regard to meals, insulin dose, administration, as well as cost. For example, concentrated insulin is preferred for a patient on high doses of insulin requiring injecting a large volume of insulin. Rapid-acting insulin analogs would be more appropriate for patients who have difficulty administering their regular insulin 20 to 30 minutes before eating. Premixed insulin preparations make it impossible to independently adjust short- and long-acting components. However, these may be good choices in patients who have consistent meal schedules and who want to simplify administration. Despite a prevailing misconception that NPH must be given twice a day, it has long been recognized that in T2DM, a single daily injection of NPH yields improvements in control similar to those achieved with 2 daily injections [92].
Cost Considerations
Treating T2DM imposes a great financial burden on individuals living with diabetes and their families due to the high cost of the medications. Table 4 and Table 5 provide information on the cost of non-insulin and insulin diabetes medications for patients who do not have prescription insurance coverage. From a practical standpoint, choice of diabetes agents is largely influenced by insurance formularies.
The older agents, metformin and the sulfonylureas, are available for a cash (no insurance) price of as little as $4 per month. This is in stark contrast to the SGLT-2 inhibitors, GLP-1 receptor agonists, and DPP-4 inhibitors, which range in cost between $400 and $600 per month. Of recent concern, the cost of insulin has been skyrocketing, with a more than 500% increase in the cost of certain insulins from 2001 to 2015 [93]. According to the Medical Expenditure Panel Survey (MEPS) from 2002 to 2013, the mean price of insulin increased by about 200% (from $4.34/mL to $12.92/mL) during this period, which was significantly higher than increases in the price of non-insulin comparators [94]. The introduction of biosimilar insulins to the market is expected to offer treatment options with lower cost. This will be tested when the biosimilar glargine, the first FDA-approved biosimilar insulin, becomes available in the U.S. market. However, a significant reduction in insulin prices is not expected soon [95].
When insulin is required, most patients with T2DM can be treated with older human insulins, which have similar efficacy and lower costs than the more expensive newer insulin analogs. A Cochrane review comparing basal insulin analogs to NPH showed similar efficacy in glycemic control with minimal clinical benefit in the form of less nocturnal hypoglycemia in the insulin analog arm [96]. Furthermore, similar glycemic control and risk of hypoglycemia was seen when regular insulin was compared with the rapid-acting insulin analogs [97]. The cost of human NPH insulin for a patient on a total daily dose of 60 units is approximately $52 per month. This contrasts with the most widely used insulin, insulin glargine, which has a cash price of about $500 per month for the same amount (Table 5). Insulin pens, which are convenient, are more expensive. Interestingly, human insulins do not require prescriptions, allowing underinsured, underfunded patients ongoing access to them.
Incorporating Patient Preferences
Research evidence is necessary but insufficient for making patient care decisions. Along with the potential benefits, harms, costs, and inconveniences of the management options, patient perspectives, beliefs, expectations, and health-related goals must be considered. Patients will undoubtedly have preferences regarding defining goals and ranking options. Clinicians should discuss therapeutic goals and treatment options and work collaboratively with patients in determining management strategies [98].
Summary
Potential treatment approaches for treating hyperglycemia in T2DM are summarized in Figure 1 and Figure 2 [4,7]. As long as there are no contraindications, metformin should be recommended concurrent with lifestyle intervention at the time of diabetes diagnosis. Even if metformin monotherapy is initially effective, glycemic control is likely to deteriorate over time due to progressive loss of β-cell function in T2DM.
There is no consensus as to what the second-line agent should be. Selection of a second agent should be made based on potential advantages and disadvantages of each agent for any given patient. A patient-centered approach is preferred over a fixed algorithm. If the patient progresses to the point where dual therapy does not provide adequate control, either a third non-insulin agent or insulin can be added. In patients with modestly elevated A1C (below ~8%), addition of a third non-insulin agent may be equally effective as (but more expensive than) addition of insulin.
Patients with significantly elevated A1C levels on non-insulin agents usually should have insulin added to their regimen. When insulin is added, metformin should be continued. DPP-4 inhibitors and sulfonylureas are typically stopped. If SGLT-2 inhibitors and/or GLP-1 receptor agonists are continued, this may aid with weight maintenance. However, continuing these agents is likely to be expensive and associated with problems associated with polypharmacy.
The most widely recommended strategy for initiating insulin in T2DM is to add a single bedtime injection of basal insulin (ie, NPH, glargine, detemir, or degludec) to the patient’s regimen. This regimen has been found to be effective in numerous studies and controls hyperglycemia in up to 60% of patients [99]. If the patient is treated with a single bedtime injection of insulin and the fasting glucose level is within the target range but the A1C level remains above goal, addition of mealtime insulin injections is likely to be beneficial. Alternatively, addition of a GLP-1 receptor agonist to basal insulin has been shown to be equally beneficial [4,6]. When adding mealtime insulin, a common strategy is to add a single injection of a rapid-acting insulin (eg, lispro, aspart, glulisine) before the patient’s largest meal of the day. Additional premeal injections of rapid-acting insulin may be added as needed, based on self-monitoring blood glucose results. If glycemia remains significantly uncontrolled on more than 200 units of insulin per day, switching to a concentrated form of insulin (eg, U-200, U-300, or U-500) should be considered.
Corresponding author: Maryam Fazel, PharmD, BCPS, BCACP, CDE, 1295 N. Martin Ave. (Room B211B), Tucson, Arizona 85721-0202, [email protected].
Financial disclosures: None.
1. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. Centers for Disease Control and Prevention Web site. www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf. Accessed November 29, 2016.
2. Statistics about diabetes. American Diabetes Association Web site. www.diabetes.org/diabetes-basics/statistics/. Accessed November 29, 2016.
3. American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013;36:1033–46.
4. American Diabetes Association. Standards of medical care in diabetes--2016. Diabetes Care 2016;39(Suppl. 1).
5. Raz I, Riddle MC, Rosenstock J, et al. Personalized management of hyperglycemia in type 2 diabetes: reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 2013;36:1779–88.
6. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015;38:140–9.
7. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm--2016 Executive Summary. Endocr Pract 2016;22:84–113.
8. Steenkamp DW, Alexanian SM, Sternthal E. Approach to the patient with atypical diabetes. CMAJ 2014;186:678–84.
9. de Lusignan S, Sadek N, Mulnier H, et al. Miscoding, misclassification and misdiagnosis of diabetes in primary care. Diabet Med 2012;29:181–9.
10. Tripathi A, Rizvi AA, Knight LM, Jerrell JM. Prevalence and impact of initial misclassification of pediatric type 1 diabetes mellitus. South Med J 2012;105:513–7.
11. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993;329:977–86.
12. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:837–53.
13. Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–53.
14. Nathan DM, DCCT/EDIC Research Group. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 2014;37:9–16.
15. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:854–65.
16. Holman RR, Paul SK, Bethel MA, et al. 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–89.
17. Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995;28:103–17.
18. Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–59.
19. ACCORD Study Group, Gerstein HC, Miller ME, Genuth S, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med 2011;364:818–28.
20. ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008;358:2560–72.
21. Wong MG, Perkovic V, Chalmers J, et al. Long-term Benefits of Intensive Glucose Control for Preventing End-Stage Kidney Disease: ADVANCE-ON. Diabetes Care 2016;39:694–700.
22. Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009;360:129–39.
23. Hayward RA, Reaven PD, Wiitala WL, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;372:2197–206.
24. American Diabetes Association. Standards of medical care in diabetes--2009. Diabetes Care 2009;32 Suppl 1:S13–61.
25. American Geriatrics Society Expert Panel on Care of Older Adults with Diabetes Mellitus, Moreno G, Mangione CM, Kimbro L, Vaisberg E. Guidelines abstracted from the American Geriatrics Society Guidelines for Improving the Care of Older Adults with Diabetes Mellitus: 2013 update. J Am Geriatr Soc 2013;61:2020–6.
26. Lipska KJ, Ross JS, Miao Y, et al. Potential overtreatment of diabetes mellitus in older adults with tight glycemic control. JAMA Intern Med 2015;175:356–62.
27. Sussman JB, Kerr EA, Saini SD, et al. Rates of deintensification of blood pressure and glycemic medication treatment based on levels of control and life expectancy in older patients with diabetes mellitus. JAMA Intern Med 2015;175:1942–9.
28. FDA Drug Safety Communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function. FDA Web site. www.fda.gov/Drugs/DrugSafety/ucm493244.htm. Accessed December 1, 2016.
29. Inzucchi SE, Lipska KJ, Mayo H, et al. Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA 2014;312:2668–75.
30. Nathan DM. Diabetes: advances in diagnosis and treatment. JAMA 2015;314:1052–62.
31. Nathan DM, Buse JB, Kahn SE, et al. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care 2013;36:2254–61.
32. NIH begins recruitment for long-term study of diabetes drug efficacy. NIH Web site. www.nih.gov/news-events/news-releases/nih-begins-recruitment-long-term-study-diabetes-drug-efficacy. Accessed December 1, 2016.
33. Hermayer KL, Dake A. Newer oral and noninsulin therapies to treat type 2 diabetes mellitus. Cleve Clin J Med 2016;83(5 Suppl 1):S18–26.
34. Bolen S, Wilson L, Vassy J, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med 2007;147:386–99.
35. Bolen S, Tseng E, Hutfless S, et al. Oral diabetes medications for adults with type 2 diabetes: an update. Agency for Healthcare Research and Quality (US); 2011 Mar Report No: 11-EHC038-EF. AHRQ Comparative Effectiveness Reviews.
36. Metformin, glyburide, glipizide, glimeperide, sitagliptin, saxagliptin, linagliptin, lixisenatide, alogliptin, exenatide, liraglutide, albiglutide, dulaglutide, canagliflozin, danagliflozin, empagliflozin: drug information. Waltham (MA): UpToDate, Inc.; 2016. Accessed September 23, 2016.
37. GoodRx Web site. http://www.goodrx.com. Accessed August 6, 2016 and December 2016.
38. Insulins available in the United States. Diabetesforcast Web site. Accessed August 6, 2016. www.diabetesforecast.org/2016/mar-apr/images/2016-insulin-chart-new.pdf.
39. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012;55:577–96.
40. Elashoff M, Matveyenko AV, Gier B, et al. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011;141:150–6.
41. Butler AE, Campbell-Thompson M, Gurlo T, et al. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 2013;62:2595–604.
42. Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;373:232–42.
43. White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013;369:1327–35.
44. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013;369:1317–26.
45. Egan AG, Blind E, Dunder K, et al. Pancreatic safety of incretin-based drugs--FDA and EMA assessment. N Engl J Med 2014;370:794–7.
46. Vasilakou D, Karagiannis T, Athanasiadou E, et al. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 2013;159:262–74.
47. Nyirjesy P, Sobel JD, Fung A, et al. Genital mycotic infections with canagliflozin, a sodium glucose co-transporter 2 inhibitor, in patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. Curr Med Res Opin 2014;30:1109–19.
48. Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care 2013;36:2508–15.
49. Blonde L, Dailey G, Jabbour S, et al. Gastrointestinal tolerability of extended-release metformin tablets compared to immediate-release metformin tablets: results of a retrospective cohort study. Curr Med Res Opin 2004;20:562–72.
50. Kalra S, Mukherjee JJ, Venkataraman S, et al. Hypoglycemia: the neglected complication. Indian J Endocrinol Metab 2013;17:819–34.
51. Paty BW. The role of hypoglycemia in cardiovascular outcomes in diabetes. Can J Diabetes 2015;39 Suppl 5:S155–9.
52. Zoungas S, Patel A, Chalmers J, et al. Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010;363:1410–8.
53. Whitmer RA, Karter AJ, Yaffe K, et al. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 2009;301:1565–72.
54. McCoy RG, Van Houten HK, Ziegenfuss JY, et al. Increased mortality of patients with diabetes reporting severe hypoglycemia. Diabetes Care 2012;35:1897–901.
55. McCoy RG, Lipska KJ, Yao X, et al. Intensive treatment and severe hypoglycemia among adults with type 2 diabetes. JAMA Intern Med 2016;176:969–78.
56. Rodbard HW, Gough S, Lane W, et al. Reduced risk of hypoglycemia with insulin degludec versus insulin glargine in patients with type 2 diabetes requiring high doses of basal insulin: a meta-analysis of 5 randomized begin trials. Endocr Pract 2014;20:285–92.
57. Yki-Jarvinen H, Bergenstal R, Ziemen M, et al. New insulin glargine 300 units/mL versus glargine 100 units/mL in people with type 2 diabetes using oral agents and basal insulin: glucose control and hypoglycemia in a 6-month randomized controlled trial (EDITION 2). Diabetes Care 2014;37:3235–43.
58. Tuomilehto J, Lindstrom J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001;344:1343–50.
59. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403.
60. Maruthur NM, Tseng E, Hutfless S, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 2016;164:740–51.
61. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2012;35:731–7.
62. Clar C, Gill JA, Court R, Waugh N. Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2012;2:10.1136/bmjopen,2012-001007.
63. Vilsboll T, Christensen M, Junker AE, et al. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ 2012;344:d7771.
64. FDA approves weight-management drug Saxenda. FDA Web site www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm427913.htm. Accessed September 22, 2016.
65. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015;100:342–62.
66. Liraglutide, lorcaserin, naltrexone/bupropion, orlistat, phentermine/topiramate: drug information. Greenwood Village (CO): Truven Health Analytics; 2016. www.micromedexsolutions.com. Accessed May 13, 2016.
67. Liraglutide, lorcaserin, naltrexone/bupropion, orlistat, phentermine/topiramate: drug information. Waltham (MA): UpToDate, Inc.; 2016. Accessed May 13, 2016.
68. Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014;311:74–86.
69. Sjostrom L, Peltonen M, Jacobson P, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA 2014;311:2297–304.
70. Schauer PR, Bhatt DL, Kirwan JP, et al. Bariatric surgery versus intensive medical therapy for diabetes--3-year outcomes. N Engl J Med 2014;370:2002–13.
71. Rubino F, Nathan DM, Eckel RH, et al. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care 2016;39:861–77.
72. Lathief S, Inzucchi SE. Approach to diabetes management in patients with CVD. Trends Cardiovasc Med 2016;26:165–79.
73. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356:2457–71.
74. Knatterud GL, Klimt CR, Levin ME, et al. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA 1978;240:37–42.
75. Masoudi FA, Inzucchi SE, Wang Y, et al. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation 2005;111:583–90.
76. Klepzig H, Kober G, Matter C, et al. Sulfonylureas and ischaemic preconditioning; a double-blind, placebo-controlled evaluation of glimepiride and glibenclamide. Eur Heart J 1999;20:439–46.
77. FDA announces new recommendations on evaluating cardiovascular risk in drugs intended to treat type 2 diabetes. FDA Web site. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116994.htm. Accessed August 20, 2016.
78. Kooy A, de Jager J, Lehert P, et al. Long-term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch Intern Med 2009;169:616–25.
79. Eurich DT, Weir DL, Majumdar SR, et al. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: systematic review of observational studies involving 34,000 patients. Circ Heart Fail 2013;6:395–402.
80. Tahrani AA, Varughese GI, Scarpello JH, Hanna FW. Metformin, heart failure, and lactic acidosis: is metformin absolutely contraindicated? BMJ 2007;335:508–12.
81. Li Y, Hu Y, Ley SH, et al. Sulfonylurea use and incident cardiovascular disease among patients with type 2 diabetes: prospective cohort study among women. Diabetes Care 2014;37:3106–13.
82. Bentley-Lewis R, Aguilar D, Riddle MC, et al. Rationale, design, and baseline characteristics in Evaluation of LIXisenatide in Acute Coronary Syndrome, a long-term cardiovascular end point trial of lixisenatide versus placebo. Am Heart J 2015;169:631,638.e7.
83. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311–22.
84. Exenatide Study of Cardiovascular Event Lowering Trial (EXSCEL): A Trial To Evaluate Cardiovascular Outcomes After Treatment With Exenatide Once Weekly In Patients With Type 2 Diabetes Mellitus. clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01144338. 2016 Accessed September 23, 2016.
85. Researching Cardiovascular Events With a Weekly Incretin in Diabetes (REWIND). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01394952. Accessed September 23, 2016.
86. Schernthaner G, Schernthaner-Reiter MH, Schernthaner GH. EMPA-REG and other cardiovascular outcome trials of glucose-lowering agents: implications for future treatment strategies in type 2 diabetes mellitus. Clin Ther 2016;38:1288–98.
87. CANVAS--CANagliflozin cardiovascular Assesssment Study (CANVAS). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01032629. Accessed September 23, 2016.
88. Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy (CREDENCE). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT02065791. Accessed September 23, 2016.
89. Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01730534. Accessed September 23, 2016.
90. Zannad F, Cannon CP, Cushman WC, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015;385(9982):2067–76.
91. Van Klompenburg E, Heins JR. New insulin options for diabetic patients. S D Med 2016;69:84–5.
92. Rosenstock J, Schwartz SL, Clark CM Jr, et al. Basal insulin therapy in type 2 diabetes: 28-week comparison of insulin glargine (HOE 901) and NPH insulin. Diabetes Care 2001;24:631–6.
93. Tylee T, Hirsch IB. Costs associated with using different insulin preparations. JAMA 2015;314:665–6.
94. Hua X, Carvalho N, Tew M, et al. Expenditures and prices of antihyperglycemic medications in the United States: 2002-2013. JAMA 2016;315:1400–2.
95. Heinemann L. Biosimilar insulin and costs: what can we expect? J Diabetes Sci Technol 2016;10:457–62.
96. Horvath K, Jeitler K, Berghold A, et al. Long-acting insulin analogues versus NPH insulin (human isophane insulin) for type 2 diabetes mellitus. Cochrane Database Syst Rev 2007;(2)(2):CD005613.
97. Mannucci E, Monami M, Marchionni N. Short-acting insulin analogues vs. regular human insulin in type 2 diabetes: a meta-analysis. Diabetes Obes Metab 2009;11:53–9.
98. Powell PW, Corathers SD, Raymond J, Streisand R. New approaches to providing individualized diabetes care in the 21st century. Curr Diabetes Rev 2015;11:222–30.
99. Riddle MC, Rosenstock J, Gerich J, Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003;26:3080–6.
1. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. Centers for Disease Control and Prevention Web site. www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf. Accessed November 29, 2016.
2. Statistics about diabetes. American Diabetes Association Web site. www.diabetes.org/diabetes-basics/statistics/. Accessed November 29, 2016.
3. American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013;36:1033–46.
4. American Diabetes Association. Standards of medical care in diabetes--2016. Diabetes Care 2016;39(Suppl. 1).
5. Raz I, Riddle MC, Rosenstock J, et al. Personalized management of hyperglycemia in type 2 diabetes: reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 2013;36:1779–88.
6. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015;38:140–9.
7. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm--2016 Executive Summary. Endocr Pract 2016;22:84–113.
8. Steenkamp DW, Alexanian SM, Sternthal E. Approach to the patient with atypical diabetes. CMAJ 2014;186:678–84.
9. de Lusignan S, Sadek N, Mulnier H, et al. Miscoding, misclassification and misdiagnosis of diabetes in primary care. Diabet Med 2012;29:181–9.
10. Tripathi A, Rizvi AA, Knight LM, Jerrell JM. Prevalence and impact of initial misclassification of pediatric type 1 diabetes mellitus. South Med J 2012;105:513–7.
11. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993;329:977–86.
12. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:837–53.
13. Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–53.
14. Nathan DM, DCCT/EDIC Research Group. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 2014;37:9–16.
15. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:854–65.
16. Holman RR, Paul SK, Bethel MA, et al. 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–89.
17. Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995;28:103–17.
18. Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–59.
19. ACCORD Study Group, Gerstein HC, Miller ME, Genuth S, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med 2011;364:818–28.
20. ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008;358:2560–72.
21. Wong MG, Perkovic V, Chalmers J, et al. Long-term Benefits of Intensive Glucose Control for Preventing End-Stage Kidney Disease: ADVANCE-ON. Diabetes Care 2016;39:694–700.
22. Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009;360:129–39.
23. Hayward RA, Reaven PD, Wiitala WL, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;372:2197–206.
24. American Diabetes Association. Standards of medical care in diabetes--2009. Diabetes Care 2009;32 Suppl 1:S13–61.
25. American Geriatrics Society Expert Panel on Care of Older Adults with Diabetes Mellitus, Moreno G, Mangione CM, Kimbro L, Vaisberg E. Guidelines abstracted from the American Geriatrics Society Guidelines for Improving the Care of Older Adults with Diabetes Mellitus: 2013 update. J Am Geriatr Soc 2013;61:2020–6.
26. Lipska KJ, Ross JS, Miao Y, et al. Potential overtreatment of diabetes mellitus in older adults with tight glycemic control. JAMA Intern Med 2015;175:356–62.
27. Sussman JB, Kerr EA, Saini SD, et al. Rates of deintensification of blood pressure and glycemic medication treatment based on levels of control and life expectancy in older patients with diabetes mellitus. JAMA Intern Med 2015;175:1942–9.
28. FDA Drug Safety Communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function. FDA Web site. www.fda.gov/Drugs/DrugSafety/ucm493244.htm. Accessed December 1, 2016.
29. Inzucchi SE, Lipska KJ, Mayo H, et al. Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA 2014;312:2668–75.
30. Nathan DM. Diabetes: advances in diagnosis and treatment. JAMA 2015;314:1052–62.
31. Nathan DM, Buse JB, Kahn SE, et al. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care 2013;36:2254–61.
32. NIH begins recruitment for long-term study of diabetes drug efficacy. NIH Web site. www.nih.gov/news-events/news-releases/nih-begins-recruitment-long-term-study-diabetes-drug-efficacy. Accessed December 1, 2016.
33. Hermayer KL, Dake A. Newer oral and noninsulin therapies to treat type 2 diabetes mellitus. Cleve Clin J Med 2016;83(5 Suppl 1):S18–26.
34. Bolen S, Wilson L, Vassy J, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med 2007;147:386–99.
35. Bolen S, Tseng E, Hutfless S, et al. Oral diabetes medications for adults with type 2 diabetes: an update. Agency for Healthcare Research and Quality (US); 2011 Mar Report No: 11-EHC038-EF. AHRQ Comparative Effectiveness Reviews.
36. Metformin, glyburide, glipizide, glimeperide, sitagliptin, saxagliptin, linagliptin, lixisenatide, alogliptin, exenatide, liraglutide, albiglutide, dulaglutide, canagliflozin, danagliflozin, empagliflozin: drug information. Waltham (MA): UpToDate, Inc.; 2016. Accessed September 23, 2016.
37. GoodRx Web site. http://www.goodrx.com. Accessed August 6, 2016 and December 2016.
38. Insulins available in the United States. Diabetesforcast Web site. Accessed August 6, 2016. www.diabetesforecast.org/2016/mar-apr/images/2016-insulin-chart-new.pdf.
39. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012;55:577–96.
40. Elashoff M, Matveyenko AV, Gier B, et al. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011;141:150–6.
41. Butler AE, Campbell-Thompson M, Gurlo T, et al. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 2013;62:2595–604.
42. Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;373:232–42.
43. White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013;369:1327–35.
44. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013;369:1317–26.
45. Egan AG, Blind E, Dunder K, et al. Pancreatic safety of incretin-based drugs--FDA and EMA assessment. N Engl J Med 2014;370:794–7.
46. Vasilakou D, Karagiannis T, Athanasiadou E, et al. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 2013;159:262–74.
47. Nyirjesy P, Sobel JD, Fung A, et al. Genital mycotic infections with canagliflozin, a sodium glucose co-transporter 2 inhibitor, in patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. Curr Med Res Opin 2014;30:1109–19.
48. Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care 2013;36:2508–15.
49. Blonde L, Dailey G, Jabbour S, et al. Gastrointestinal tolerability of extended-release metformin tablets compared to immediate-release metformin tablets: results of a retrospective cohort study. Curr Med Res Opin 2004;20:562–72.
50. Kalra S, Mukherjee JJ, Venkataraman S, et al. Hypoglycemia: the neglected complication. Indian J Endocrinol Metab 2013;17:819–34.
51. Paty BW. The role of hypoglycemia in cardiovascular outcomes in diabetes. Can J Diabetes 2015;39 Suppl 5:S155–9.
52. Zoungas S, Patel A, Chalmers J, et al. Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010;363:1410–8.
53. Whitmer RA, Karter AJ, Yaffe K, et al. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 2009;301:1565–72.
54. McCoy RG, Van Houten HK, Ziegenfuss JY, et al. Increased mortality of patients with diabetes reporting severe hypoglycemia. Diabetes Care 2012;35:1897–901.
55. McCoy RG, Lipska KJ, Yao X, et al. Intensive treatment and severe hypoglycemia among adults with type 2 diabetes. JAMA Intern Med 2016;176:969–78.
56. Rodbard HW, Gough S, Lane W, et al. Reduced risk of hypoglycemia with insulin degludec versus insulin glargine in patients with type 2 diabetes requiring high doses of basal insulin: a meta-analysis of 5 randomized begin trials. Endocr Pract 2014;20:285–92.
57. Yki-Jarvinen H, Bergenstal R, Ziemen M, et al. New insulin glargine 300 units/mL versus glargine 100 units/mL in people with type 2 diabetes using oral agents and basal insulin: glucose control and hypoglycemia in a 6-month randomized controlled trial (EDITION 2). Diabetes Care 2014;37:3235–43.
58. Tuomilehto J, Lindstrom J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001;344:1343–50.
59. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403.
60. Maruthur NM, Tseng E, Hutfless S, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 2016;164:740–51.
61. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2012;35:731–7.
62. Clar C, Gill JA, Court R, Waugh N. Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2012;2:10.1136/bmjopen,2012-001007.
63. Vilsboll T, Christensen M, Junker AE, et al. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ 2012;344:d7771.
64. FDA approves weight-management drug Saxenda. FDA Web site www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm427913.htm. Accessed September 22, 2016.
65. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015;100:342–62.
66. Liraglutide, lorcaserin, naltrexone/bupropion, orlistat, phentermine/topiramate: drug information. Greenwood Village (CO): Truven Health Analytics; 2016. www.micromedexsolutions.com. Accessed May 13, 2016.
67. Liraglutide, lorcaserin, naltrexone/bupropion, orlistat, phentermine/topiramate: drug information. Waltham (MA): UpToDate, Inc.; 2016. Accessed May 13, 2016.
68. Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014;311:74–86.
69. Sjostrom L, Peltonen M, Jacobson P, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA 2014;311:2297–304.
70. Schauer PR, Bhatt DL, Kirwan JP, et al. Bariatric surgery versus intensive medical therapy for diabetes--3-year outcomes. N Engl J Med 2014;370:2002–13.
71. Rubino F, Nathan DM, Eckel RH, et al. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Diabetes Care 2016;39:861–77.
72. Lathief S, Inzucchi SE. Approach to diabetes management in patients with CVD. Trends Cardiovasc Med 2016;26:165–79.
73. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356:2457–71.
74. Knatterud GL, Klimt CR, Levin ME, et al. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA 1978;240:37–42.
75. Masoudi FA, Inzucchi SE, Wang Y, et al. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation 2005;111:583–90.
76. Klepzig H, Kober G, Matter C, et al. Sulfonylureas and ischaemic preconditioning; a double-blind, placebo-controlled evaluation of glimepiride and glibenclamide. Eur Heart J 1999;20:439–46.
77. FDA announces new recommendations on evaluating cardiovascular risk in drugs intended to treat type 2 diabetes. FDA Web site. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116994.htm. Accessed August 20, 2016.
78. Kooy A, de Jager J, Lehert P, et al. Long-term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch Intern Med 2009;169:616–25.
79. Eurich DT, Weir DL, Majumdar SR, et al. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: systematic review of observational studies involving 34,000 patients. Circ Heart Fail 2013;6:395–402.
80. Tahrani AA, Varughese GI, Scarpello JH, Hanna FW. Metformin, heart failure, and lactic acidosis: is metformin absolutely contraindicated? BMJ 2007;335:508–12.
81. Li Y, Hu Y, Ley SH, et al. Sulfonylurea use and incident cardiovascular disease among patients with type 2 diabetes: prospective cohort study among women. Diabetes Care 2014;37:3106–13.
82. Bentley-Lewis R, Aguilar D, Riddle MC, et al. Rationale, design, and baseline characteristics in Evaluation of LIXisenatide in Acute Coronary Syndrome, a long-term cardiovascular end point trial of lixisenatide versus placebo. Am Heart J 2015;169:631,638.e7.
83. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311–22.
84. Exenatide Study of Cardiovascular Event Lowering Trial (EXSCEL): A Trial To Evaluate Cardiovascular Outcomes After Treatment With Exenatide Once Weekly In Patients With Type 2 Diabetes Mellitus. clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01144338. 2016 Accessed September 23, 2016.
85. Researching Cardiovascular Events With a Weekly Incretin in Diabetes (REWIND). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01394952. Accessed September 23, 2016.
86. Schernthaner G, Schernthaner-Reiter MH, Schernthaner GH. EMPA-REG and other cardiovascular outcome trials of glucose-lowering agents: implications for future treatment strategies in type 2 diabetes mellitus. Clin Ther 2016;38:1288–98.
87. CANVAS--CANagliflozin cardiovascular Assesssment Study (CANVAS). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01032629. Accessed September 23, 2016.
88. Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy (CREDENCE). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT02065791. Accessed September 23, 2016.
89. Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58). clinicaltrials.gov Web site. https://clinicaltrials.gov/ct2/show/NCT01730534. Accessed September 23, 2016.
90. Zannad F, Cannon CP, Cushman WC, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015;385(9982):2067–76.
91. Van Klompenburg E, Heins JR. New insulin options for diabetic patients. S D Med 2016;69:84–5.
92. Rosenstock J, Schwartz SL, Clark CM Jr, et al. Basal insulin therapy in type 2 diabetes: 28-week comparison of insulin glargine (HOE 901) and NPH insulin. Diabetes Care 2001;24:631–6.
93. Tylee T, Hirsch IB. Costs associated with using different insulin preparations. JAMA 2015;314:665–6.
94. Hua X, Carvalho N, Tew M, et al. Expenditures and prices of antihyperglycemic medications in the United States: 2002-2013. JAMA 2016;315:1400–2.
95. Heinemann L. Biosimilar insulin and costs: what can we expect? J Diabetes Sci Technol 2016;10:457–62.
96. Horvath K, Jeitler K, Berghold A, et al. Long-acting insulin analogues versus NPH insulin (human isophane insulin) for type 2 diabetes mellitus. Cochrane Database Syst Rev 2007;(2)(2):CD005613.
97. Mannucci E, Monami M, Marchionni N. Short-acting insulin analogues vs. regular human insulin in type 2 diabetes: a meta-analysis. Diabetes Obes Metab 2009;11:53–9.
98. Powell PW, Corathers SD, Raymond J, Streisand R. New approaches to providing individualized diabetes care in the 21st century. Curr Diabetes Rev 2015;11:222–30.
99. Riddle MC, Rosenstock J, Gerich J, Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003;26:3080–6.
Patient Expectations and Total Knee Arthroplasty
From the Department of Physical Therapy, University of Alberta, Edmonton AB (Dr. Jones) and UT MD Anderson Cancer Center, Houston, TX (Dr. Suarez-Almazor).
Abstract
- Objective: To discuss patient expectations of total knee arthroplasty (TKA), instruments used to measure expectations, and the association between expectations, health outcomes, and satisfaction.
- Methods: Review of the literature.
- Results: TKA is an elective surgery for patients with persistent pain and disability caused by knee arthritis. Expectations regarding the surgical procedure and recovery can vary by diagnosis, personal characteristics, functional status, employment status, and trust in physicians. Patients have high overall expectations for recovery, particularly for pain relief and walking. Surgeons’ expectations tend to be more optimistic than patients’, although a subset of patients may have unrealistically high expectations. Although total joint replacement is an effective treatment for advanced arthritis, approximately 30% of potential candidates are unwilling to proceed with surgery. Potential surgical candidates unwilling to proceed with surgery tend to be older, female, and from ethnic minority groups. Several patient-related factors are associated with satisfaction with TKA, including primary diagnosis, preoperative pain and function, and mental health, including depression, but the relationships of satisfaction with gender, age, and comorbid conditions are less certain.
- Conclusion: A better understanding of patient expectations of TKA and recovery can identify knowledge gaps, misconceptions, and communication barriers, and ultimately improve shared decision making. A core set of reliable and valid instruments to measure expectations may encourage their routine use in both clinical and research settings.
Key words: total knee arthroplasty; osteoarthritis; patient expectations; shared decision making; joint replacement.
Total knee arthroplasty (TKA) is an elective surgery for patients with persistent pain and disability caused by knee arthritis. It is viewed as an effective and cost-effective surgical treatment for end-stage osteoarthritis (OA) [1–4]. As the population ages and obesity rates steadily increase, so will the utilization rates for TKA, with projected demand in the United States expected to grow 673% by 2030 [5–7]. The key indicators for receiving primary TKA are end-stage OA and joint pain [8]. Although TKA is a surgical option when conservative management is exhausted, no consensus exists as to the severity of symptoms required to consider surgery [9]. Variation in the utilization of TKA exists with respect to gender, racial/ethnicity, hospital, and geography [10,11]. These differences cannot be explained by prevalence of arthritis or symptoms or by access to health care alone. Increasingly, studies have shown these variations are largely attributable to patients’ preferences, driven by their beliefs, concerns, familiarity with the procedure, and expectations, along with physician opinion [12]. While physician opinions and recommendations clearly influence patients’ decisions, they do so primarily by modulating patients’ beliefs and expectations.
Patient expectations, not only of the effectiveness of the procedure itself but also of the recovery process, influence the decision to undergo an elective surgery such as joint arthroplasty. Ideally, these expectations should be informed by evidence, but often, lack of knowledge, preconceived beliefs, and misconceptions can taint informed decision making. A better understanding of patient expectations of TKA and recovery can identify knowledge gaps, misconceptions, and communication barriers, and ultimately improve shared decision making. Understanding patient expectations and factors that influence expectations provides a fuller appreciation of the outcomes that are meaningful to patients and can guide preoperative education and open dialogue with patients within a shared decision making model of care. In this paper, we discuss patient expectations of TKA, including expectations regarding outcomes and recovery, fulfillment of expectations, and the association of fulfilled expectations with satisfaction.
Measurement of Expectations
The construct of expectation is complex and situational. The ambiguity within the literature occurs most likely because expectations are multifaceted. Expectation involves the notion of expectancy, with respect to health care, that given events are likely to occur as a result of a medical procedure or treatment. This concept is in contrast to wants, which reflects a patient’s desire or wishes that an event will occur [13]. The term patient expectation, however, is commonly confused with patient preference or value. Preference implies a relative valuation or comparison by the patient and, unlike expectation, may not be explicitly expressed by the patient [13]. Different types of health care expectations exist that broadly relate to what patients expect regarding health care structure, process, and outcome [14].
Studies of patient expectations are diverse within the orthopedic research field and reflect differing theoretical underpinnings and lack of standardization. The lack of standardization makes measuring the complex concept of expectations challenging. While a number of conceptual models exist, Bowling and colleagues aptly recognize the multidimensionality of expectations and that no one conceptual model captures patient expectations [14]. The lack of standardization was noted in a systematic review by Haanstra and colleagues who found great variety in the definitions and measurements of expectations in studies examining their relationship with outcomes of total joint arthroplasty [15].
No gold standard measure exists for measuring patient expectations of orthopedic surgery. Zywiel’s systematic review [16] of 66 studies identified 7 validated instruments for measuring patient expectations for orthopedic surgery: of these, 2 were specific to TKA (Hospital for Special Surgery (HSS) Expectation Survey [17] and Expectation Domain of the New Knee Scoring System [18,19]), and 2 were generic to musculoskeletal conditions (Expectation domain of the Musculoskeletal Outcomes Data Evaluation and Management System (MODEMS) Instruments [20] and the Sunnybrook Surgery Expectation Survey [21]). A number of other measures used within the literature were identified; however, the psychometric properties for many of these measures were not reported and any evidence of testing and validation were lacking [16]. Some studies used a single question to measure expectations. As patient expectation is multi-dimensional, using a single item to evaluate expectations is problematic. Zywiel and others have called for a core set of reliable and valid instruments to measure expectations [14,22], which may encourage their routine use in both clinical and research settings.
Patients Expectations for TKA Recovery
Although patient concerns vary in terms of importance and severity [23], pain and physical limitations are primary concerns for patients seeking TKA. Patients have high overall expectations for recovery, particularly for pain relief and walking [24–32]. TKA is an elective surgical procedure that provides substantial pain relief and improvements in function and quality of life, with the largest gains seen within the first 6 months [33,34]. Both short-term and long-term effect sizes for pain relief and functional recovery are large, in excess of 1.0 [34]. Over 70% of patients undergoing TKA expect to be pain-free, and 35% expect to have no limitations with routine activities [24,28,31].
Expectations regarding the surgical procedure and recovery can vary by diagnosis, personal characteristics, functional status [17], employment status, and trust in physicians [32,35]. There is, however, inconsistent evidence on associative preoperative factors of recovery expectations. While some evidence supports an association between higher expectations and younger age and greater preoperative functional limitation [26–28,32,36–38], others have reported no significant association with several preoperative factors including age, gender, and preoperative functional status [24,26,37]. Lower overall expectations [28] and lower expectations for pain relief [21] were also seen for patients with a greater number of comorbid conditions.
It may be that patients with high preoperative expectations are more optimistic, interpret their health-reported quality of life gains more liberally, and are more likely to adhere to rehabilitation treatment [24,25]. Optimism is a generalized expectancy of a positive outcome that is related to indicators of well-being [39]. Presurgical optimism was shown to be associated with less postsurgical pain and anxiety in patients undergoing total hip and knee arthroplasty [40].
In addition to general future-oriented constructs, such as optimism, treatment-specific psychological constructs, such as treatment credibility and treatment expectancy, are seen in patients with total joint arthroplasty. A strong but not redundant association is seen between treatment expectancy and treatment credibility, that is, expectations of a treatment may be related as to how credible the treatment outcomes appear [41,42]. Haanstra and colleagues advocate further clinical work to explore which factor predicts total joint arthroplasty outcomes so that patients who are at a higher risk of poor outcomes can be identified [42].
Others have recognized that perspectives and expectations of surgical outcomes differ between patient and surgeon [43–45]. Overall, surgeons’ expectations tend to be more optimistic than patients expectations of outcomes, although a subset of patients may have unrealistically high expectations [46]. Patients do not always realize that some of their expectations cannot be met by current orthopedic procedures, and this gap in understanding is an important source of discrepancies in expectations and patient dissatisfaction [46]. Ghomrawi and colleagues reported that approximately one-third of 205 patients undergoing primary TKA had higher expectations than their surgeons did. Being male and having lower preoperative pain was associated with having discordantly higher preoperative expectations [44]. For realistic expectations to be set, patients need accurate and understandable information about expected positive outcomes of surgery such as level of function and symptom relief as well as the risk of joint failure, adverse events, complications, and activity limitations. Although little work has explored the alignment of patient and surgeon’s expectations, setting realistic expectations may be aided by using a shared decision making approach that incorporates patient preferences and values, the best available evidence, and the surgeon’s expertise.
Expectations and Willingness to Undergo Surgery
Although total joint replacement is an effective treatment for advanced arthritis, approximately 30% of potential candidates are “unwilling” to proceed with surgery [47,48]. Willingness is a component of the medical decision making process and is influenced by preferences. Potential surgical candidates unwilling to proceed with surgery tend to be older, female, and from ethnic minor-ity groups [12,47–49]. Preference-sensitive medical decisions, such as whether or not to proceed with TKA, are related to patients’ attitudes and perceptions, which can be affected by sociocultural influences. In a cohort of 627 male patients with moderate to severe OA who were viewed as “good” candidates for total joint arthroplasty, more African Americans (24%) than Caucasian Americans (15%) had lower expectations for outcomes of surgery [35]. In particular, African Americans expressed concerns about postoperative pain and walking. Similar findings were also reported in another study in which minority patients were less likely to consider TKA [12]. Determinants of preferences were patients’ beliefs about the efficacy of the procedure and knowing others who had already undergone TKA [12]. Ibrahim and colleagues postulated that outcome expectations mediated or influenced the willingness to undergo total joint arthroplasty surgery [49]. Interventional work that built upon this premise suggested that willingness to proceed with TKA could be modified by educational interventions. In a randomized controlled trial of 639 African American patients attending Veteran’s Affairs primary clinics who received a decision aid with or without brief counseling, willingness to proceed with TKA increased and patient-provider communication improved among the patients who received any intervention [50]. Yet in another randomized trial involving African American patients who received care from an academic center, a combination decision aid and motivational interviewing strategy was no better than an educational pamphlet in improving patients’ preferences toward joint replacement surgery for knee OA [51]. This led the authors to recommend further exploration of patients’ knowledge, beliefs, and attitudes regarding surgical treatments for OA.
Effect of Expectations on Health Outcomes and Satisfaction
Some evidence suggests that better outcomes are seen in patients with higher expectations of recovery and, in turn, expectations that are met influence patient satisfaction. A systematic review of several chronic conditions showed with consistency across studies that positive recovery expectations were associated with better health outcomes [22]. The effect size varied with the condition and measure; however, none of the 16 studies examined arthritis or joint arthroplasty. Conversely, a systematic review of 18 prospective longitudinal cohort studies examining the association between expectation and outcomes (ie, pain, function, stiffness, satisfaction, overall improvement) reported less than convincing evidence of an association between patient preoperative expectations and treatment outcomes for THA and TKA in terms of short- and long-term postoperative pain and functional outcomes [15]. No consistent associations were seen with adjusted analysis of patient expectations and pain or functional outcomes at greater than 6 weeks [15]. Inconsistencies seen among the reviewed articles may be related to a number of issues centred on terminology, construct, expectation measures, and confounding effects.
Although TKA is an effective surgical procedure with large gains reported, 14% to 25% participants report little or no symptom improvement and/or dissatisfaction up to 1 year after surgery [1,52–59]. In a study with 5 years of follow-up, a decline in the satisfaction rate was seen after 1 year, although this decline was seen more so with physical function than with pain [38]. Although dissatisfaction can be attributed to surgical complications, in many cases, no technical or medical reasons can be identified. In a subset of patients who received TKA, surgical intervention does not adequately address patients’ concerns of pain and activity limitation. To compound matters, fair agreement was reported between patient and surgeon regarding satisfaction at 6 and 12 months postoperative. Disagreement between the patient and surgeon was explained by unmet expectations and postoperative complications [60]. When there was discordance, more often than not patients were less satisfied with TKA outcomes than surgeons [60,61].
While several theories explain patient satisfaction [62–65], evidence from total joint arthroplasty studies support the concept that satisfaction is derived from fulfillment of expectations [17,52]. Preoperative expectations are not to be confused with postoperative fulfilment of expectations, which are reflective of whether expectations of treatment have been met. Satisfaction is a value judgment and can be viewed as an affective domain, whereas expectation is a cognitive domain [66]. Patient satisfaction is regarded as the extent of a person’s experience compared to their expectation. As with expectations, a number of theoretical constructs exist concerning patient satisfaction [14,67]. Many dimensions of satisfaction exist, with patient expectations being central to these constructs. Deviation from expectations, however, does not necessarily correspond to dissatisfaction [67].
Several patient-related factors are associated with satisfaction with TKA, including primary diagnosis, preoperative pain and function, and mental health, including depression, but the relationships of satisfaction with gender, age, and comorbid conditions are less certain [33,38,52,55,56,68]. Greater preoperative pain, postoperative complications, lower 1-year WOMAC scores and functional limitations were associated with dissatisfied patients [38,52,53,59]. While no consistent associations were seen with preoperative expectations, consistent evidence has shown that fulfillment of expectations has an impact on satisfaction [31,36,52,58,69].
It should be acknowledged that the concept of fulfillment of expectations is not the same as satisfaction. A patient can be satisfied with TKA even though their expectations have not been met. The fulfillment of expectations is dependent upon the type of expectation and the postoperative time period. Fulfillment of expectations were seen with pain relief, function, walking and health status [25,31,70] while patients expectations were not always met with leisure activities [38].
Shared Decision Making
The shared decision making process, in which the patient and physician share responsibility and actively participate in the clinical decision making process [71], may help in ensuring that patients’ expectations are met. Shared decision making requires eliciting patients’ preferences and values, providing clear information on the processes that will occur during surgery, recovery, rehabilitation, and in the longer phase of recovery, and what realistic outcomes can be expected. While a more “paternalistic” approach predominated in earlier years, the current trends indicate greater patient involvement in decision making with the surgeon, with open discussion of patient goals and expectations [71]. This approach also aids patients in their preparation for the recovery and rehabilitation stages, which can be challenging, especially if they are unaware as to what to expect. Patient expectations are more likely to be met when there is shared decision making and patients have been given relevant information and understand what is a reasonable outcome. While a shared decision making approach is advocated within orthopedics [72], patient expectations are largely not measured in the clinical setting.
Patient education is an integral component of assisting patients to make informed decisions; however, it is unknown whether education alone can modify expectations. Educational approaches can include group classes, videos, and written materials [73]. Limited evidence from a randomized controlled trial suggests that preoperative expectations can be modified by preoperative education classes by decreasing the number of expectations and having more expectations in agreement with the surgeons’ expectations [29]. Mancuso and colleagues, who looked at whether a preoperative education session could modify expectations found that larger changes in expectations were seen with those patients who had greater baseline expectation scores, worse pain and function, and were older [29]. Others have also reported that preoperative education reduces anxiety by providing patients with an understanding of what to expect [74,75]. An assumption is that expectations can be changed by improving knowledge, which underscores the need for relevant meaningful education to increase knowledge and instill realistic expectations. Others have surmised there is a proportion of patients who will continue to have unexpectedly high unrealistic expectations regardless of educational session [31,37]. This would suggest that education is not the only approach to modify expectations but rather different strategies may need to be implemented for a certain subsets of patients with unrealistic expectations.
Conclusion
Patient expectation is an important element to be considered in shared clinical decision making, as it can influ-ence preferences and subsequent satisfaction. Patients considering TKA have specific needs and expectations that they presume will be addressed with the surgery. If these are realistic, they can be met, and will result in greater patient satisfaction and better ongoing adherence to health care recommendations [76]. While much work has been conducted in identifying which patient characteristics may influence health expectations, additional research is needed to further determine how to shape expectations within a realistic, achievable framework. While traditional patient education is an important element to enhance knowledge, the limited available evidence suggests it is not sufficiently effective on its own. Other strategies such as use of individualized decision aids, provision of peer support, and enhanced provider-patient communication have been effective in many areas of health care and warrant evaluation in this field.
Corresponding author: Allyson Jones, PhD, Rm 2-50, Corbett Hall, University of Alberta, Edmonton, Alberta Canada T6G 2G4, [email protected].
Financial disclosures: None.
Author contributions: conception and design, CAJ, MES; analysis and interpretation of data, MES; drafting of article, CAJ, MES; critical revision of the article, CAJ, MES; collection and assembly of data, CAJ.
1. Jones CA, Voaklander DC, Johnston DW, Suarez-Almazor ME. Health related quality of life outcomes after total hip and knee arthroplasties in a community based population. J Rheumatol 2000;27:1745–52.
2. Waimann CA, Fernandez-Mazarambroz RJ, Cantor SB, et al. Cost-effectiveness of total knee replacement: a prospective cohort study. Arthritis Care Res 2014;66:592–9.
3. Jenkins PJ, Clement ND, Hamilton DF, et al. Predicting the cost-effectiveness of total hip and knee replacement: a health economic analysis. Bone Joint J 2013;95:115–21.
4. Losina E, Walensky RP, Kessler CL, et al. Cost-effectiveness of total knee arthroplasty in the United States: patient risk and hospital volume. Arch Intern Med 2009;169:1113–21.
5. Cram P, Lu X, Kates SL, et al. Total knee arthroplasty volume, utilization, and outcomes among Medicare beneficiaries, 1991-2010. JAMA 2012;308:1227–36.
6. Kurtz S, Ong K, Lau E, et al. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007;89:780–5.
7. Jain NB, Higgins LD, Ozumba D, et al. Trends in epidemiology of knee arthroplasty in the United States, 1990-2000. Arthritis Rheum 2005;52:3928–33.
8. Engel C, Hamilton NA, Potter PT, Zautra AJ. Impact of two types of expectancy on recovery from total knee replacement surgery (TKR) in adults with osteoarthritis. Behav Med 2004;30:113–23.
9. Carr AJ, Robertsson O, Graves S, et al. Knee replacement. Lancet 2012;379:1331–40.
10. Skinner J, Weinstein JN, Sporer SM, Wennberg JE. Racial, ethnic, and geographic disparities in rates of knee arthroplasty among Medicare patients. N Engl J Med 2003;349:1350–9.
11. Cobos R, Latorre A, Aizpuru F, et al. Variability of indication criteria in knee and hip replacement: an observational study. BMC Musculoskelet Disord 2010;11:249.
12. Suarez-Almazor ME, Souchek J, Kelly PA, et al. Ethnic variation in knee replacement: patient preferences or uninformed disparity? Arch Intern Med 2005;165:1117–24.
13. Uhlmann RF, Inui TS, Carter WB. Patient requests and expectations. Definitions and clinical applications. Med Care 1984;22:681–5.
14. Bowling A, Rowe G, Lambert N, et al. The measurement of patients’ expectations for health care: a review and psychometric testing of a measure of patients’ expectations. Health Technology Assessment 2012;16:1–515.
15. Haanstra TM, van den Berg T, Ostelo RW, et al. Systematic review: do patient expectations influence treatment outcomes in total knee and total hip arthroplasty? Health Qual Life Outcomes 2012;10:152.
16. Zywiel MG, Mahomed A, Gandhi R, et al. Measuring expectations in orthopaedic surgery: a systematic review. Clin Orthop Rel Res 2013;471:3446–56.
17. Mancuso CA, Sculco TP, Wickiewicz TL, et al. Patients’ expectations of knee surgery. J Bone Joint Surg Am 2001;83A:1005–12.
18. Noble PC, Scuderi GR, Brekke AC, et al. Development of a new Knee Society scoring system. Clin Orthopaed Rel Res 2012;470:20–32.
19. Scuderi GR, Bourne RB, Noble PC, et al. The new Knee Society Knee Scoring System. Clin Orthop Relat Res 2012;470:3–19.
20. Saleh KJ, Bershadsky B, Cheng E, Kane R. Lessons learned from the hip and knee musculoskeletal outcomes data evaluation and management system. Clin Orthop Relat Res 2004; 272–8.
21. Razmjou H, Finkelstein JA, Yee A, et al. Relationship between preoperative patient characteristics and expectations in candidates for total knee arthroplasty. Physiotherapy Canada 2009;61:38–45.
22. Mondloch MV, Cole DC, Frank JW. Does how you do depend on how you think you’ll do? A systematic review of the evidence for a relation between patients’ recovery expectations and health outcomes. CMAJ 2001;165:174–9.
23. Wright JG, Santaguida PL, Young N, et al. Patient preferences before and after total knee arthroplasty. J Clin Epidemiol 2010;63:774–82.
24. Mahomed NN, Liang MH, Cook EF, et al.: The importance of patient expectations in predicting functional outcomes after total joint arthroplasty. J Rheumatology 2002;29:1273–9.
25. Gonzalez Saenz de Tejada M, Escobar A, Herrera C, et al. Patient expectations and health-related quality of life outcomes following total joint replacement. Value Health 2010;13:447–54.
26. Hepinstall MS, Rutledge JR, Bornstein LJ, et al. Factors that impact expectations before total knee arthroplasty. J Arthroplasty 2011;26:870–6.
27. Muniesa JM, Marco E, Tejero M, et al. Analysis of the expectations of elderly patients before undergoing total knee replacement. Arch Gerontol Geriatr 2010;51:E83-E87.
28. Lingard EA, Sledge CB, Learmonth ID. Patient expectations regarding total knee arthroplasty: Differences among the United States, United Kingdom, and Australia. J Bone Joint Surg Am 2006;88:1201–7.
29. Mancuso CA, Graziano S, Briskie LM, et al. Randomized trials to modify patients’ preoperative expectations of hip and knee arthroplasties. Clin Orthopaed Rel Res 2008;466:424–31.
30. de AS, Kallen MA, Amick B, et al. Patients’ expectations about total knee arthroplasty outcomes. Health Expect 2016;19:299–308.
31. Mannion AF, Kampfen S, Munzinger U, Kramers-de Q. The role of patient expectations in predicting outcome after total knee arthroplasty. Arthritis Res Ther 2009;11:R139.
32. Yoo JH, Chang CB, Kang YG, et al. Patient expectations of total knee replacement and their association with sociodemographic factors and functional status. J Bone Joint Surg Br 2011;93:337–44.
33. Ethgen O, Bruyere O, Richy F, et al. Health-related quality of life in total hip and total knee arthroplasty. A qualitative and systematic review of the literature. J Bone Joint Surg Am 2004;86:963–74.
34. Jones CA, Pohar S. Health-related quality of life after total joint arthroplasty: a scoping review. Clin Geriatr Med 2012;28:395–429.
35. Groeneveld PW, Kwoh CK, Mor MK, et al. Racial differences in expectations of joint replacement surgery outcomes. Arthritis Rheum 2008;59:730–7.
36. Scott CEH, Bugler KE, Clement ND, et al. Patient expectations of arthroplasty of the hip and knee. J Bone Joint Surg Br 2012;94:974–81.
37. Smith J, Soon VL, Boyd A, et al. What do Scottish patients expect of their total knee arthroplasty? J Arthroplasty 2016;31:786–92.
38. Nilsdotter AK, Toksvig-Larsen S, Roos EM. Knee arthroplasty: are patients’ expectations fulfilled? A prospective study of pain and function in 102 patients with 5-year follow-up. Acta Orthopaedica 2009;80:55–61.
39. Alarcon GM, Bowling NA, Khazon S. Great expectations: A meta-analytic examination of optimism and hope. Person Ind Diff 2013;54:821–7.
40. Pinto P, McIntyre T, Ferrero R, et al. Predictors of acute postsurgical pain and anxiety following primary total hip and knee arthroplasty. J Pain 2013;14:502–15.
41. Devilly GJ, Borkovec TD. Psychometric properties of the credibility/expectancy questionnaire. J Behav Ther Exp Psychiatry 2000;31:73–86.
42. Haanstra TM, Tilbury C, Kamper SJ, et al. Can optimism, pessimism, hope, treatment credibility and treatment expectancy be distinguished in patients undergoing total hip and total knee arthroplasty? PLoS One 2015;10.
43. Verbeek J, Sengers MJ, Riemens L, Haafkens J.Patient expectations of treatment for back pain: a systematic review of qualitative and quantitative studies. Spine 2004; 29:2309–18.
44. Ghomrawi HM, Mancuso CA, Westrich GH, et al. Discordance in TKA expectations between patients and surgeons. Clin Orthopaed Rel Res 2013;471:175–80.
45. Cordero-Ampuero J, Darder A, Santillana J, et al. Evaluation of patients’ and physicians’ expectations and attributes of osteoarthritis treatment using Kano methodology. Qual Life Res 2012;21:1391–404.
46. Noble PC, Fuller-Lafreniere S, Meftah M, Dwyer MK. Challenges in outcome measurement: discrepancies between patient and provider definitions of success. Clin Orthopaed Rel Res 2013;471:3437–45.
47. Hawker GA, Wright JG, Coyte PC, et al. Determining the need for hip and knee arthroplasty: the role of clinical severity and patients’ preferences. Med Care 2001;39:206–16.
48. Juni P, Dieppe P, Donovan J, et al. Population requirement for primary knee replacement surgery: a cross-sectional study. Rheumatology 2003;42:516–21.
49. Ibrahim SA, Siminoff LA, Burant CJ, Kwoh CK. Differences in expectations of outcome mediate African American/white patient differences in “willingness” to consider joint replacement. Arthritis Rheum 2002;46:2429–35.
50. Ibrahim SA, Hanusa BH, Hannon MJ, et al. Willingness and access to joint replacement among African American patients with knee osteoarthritis: a randomized, controlled intervention. Arthritis Rheum 2013;65:1253–61.
51. Vina ER, Richardson D, Medvedeva E, et al. Does a patient-centered educational intervention affect African-American access to knee replacement? A randomized trial. Clin Orthop Relat Res 2016;474:1755–64.
52. Noble PC, Conditt MA, Cook KF, Mathis KB. The John Insall Award - Patient expectations affect satisfaction with total knee arthroplasty. Clin Orthop Relat Res 2006; 35–43.
53. Robertsson O, Dunbar M, Pehrsson T, et al. Patient satisfaction after knee arthroplasty: a report on 27,372 knees operated on between 1981 and 1995 in Sweden. Acta Orthop Scand 2000;71:262–7.
54. Lau RL, Gandhi R, Mahomed S, Mahomed N. Patient satisfaction after total knee and hip arthroplasty. Clin Geriatr Med 2012;28:349–65.
55. Scott CEH, Howie CR, Macdonald D, Biant LC. Predicting dissatisfaction following total knee replacement. A prospective study of 1217 patients. J Bone Joint Surg Br 2010; 92B:1253–8.
56. Baker PN, van der Meulen JH, Lewsey J, Gregg PJ. The role of pain and function in determining patient satisfaction after total knee replacement. Data from the National Joint Registry for England and Wales. J Bone Joint Surg Br 2007;89:893–900.
57. Khatib Y, Madan A, Naylor JM, Harris IA: Do psychological factors predict poor outcome in patients undergoing TKA? a systematic review. Clin Orthopaed Rel Res 2015;473:2630–8.
58. Adie S, Dao A, Harris IA, et al. Satisfaction with joint replacement in public versus private hospitals: a cohort study. ANZ J Surg 2012;82:616–24.
59. Bourne RB, Chesworth BM, Davis AM, et al. Patient satisfaction after total knee arthroplasty: who is satisfied and who is not? Clin Orthop Relat Res 2010;468:57–63.
60. Harris IA, Harris AM, Naylor et al. Discordance between patient and surgeon satisfaction after total joint arthroplasty. J Arthroplasty 2013;28:722–7.
61. Choi YJ, Ra H. Patient satisfaction after total knee arthroplasty. Knee Surg Relat Res 2016;28:1–15.
62. Williams B. Patient satisfaction: a valid concept? Soc Sci Med 1994;38:509–16.
63. Ware JE Jr, Snyder MK, Wright WR, Davies AR. Defining and measuring patient satisfaction with medical care. Eval Program Plann 1983;6:247–63.
64. Linder-Pelz SU. Toward a theory of patient satisfaction. Soc Sci Med 1982;16:577–82.
65. Hudak PL, Hogg-Johnson S, Bombardier C, et al. Testing a new theory of patient satisfaction with treatment outcome. Med Care 2004;42:726–39.
66. Thompson AG, Sunol R. Expectations as determinants of patient satisfaction: concepts, theory and evidence. Int J Qual Health Care 1995;l7:127–41.
67. Pascoe GC. Patient satisfaction in primary health care: a literature review and analysis. Eval Program Plann 1983;6:185–210.
68. Gandhi R, Davey JR, Mahomed NN. Predicting patient dissatisfaction following joint replacement surgery. J Rheumatol 2008;35:2415–8.
69. Waljee J, McGlinn EP, Sears ED, Chung KC. Patient expectations and patient-reported outcomes in surgery: A systematic review. Surgery 2014;155:799-808.
70. Suda AJ, Seeger JB, Bitsch RG, et al. Are patients’ expectations of hip and knee arthroplasty fulfilled? A prospective study of 130 patients. Orthopedics 2010;33:76.
71. Slover J, Shue J, Koenig K. Shared decision-making in orthopaedic surgery. Clin Orthopaed Rel Res 2012;470:1046–53.
72. Weinstein JN. The missing piece: embracing shared decision making to reform health care. Spine 2000;25:1–4.
73 Aydin D, Klit J, Jacobsen S, et al. No major effects of preoperative education in patients undergoing hip or knee replacement--a systematic review. Dan Med J 2015;62.
74. Spalding NJ. Reducing anxiety by pre-operative education: make the future familiar. Occup Ther Int 2003;10:278–93.
75. Kearney M, Jennrich MK, Lyons S, et al. Effects of preoperative education on patient outcomes after joint replacement surgery. Orthop Nurs 2011;30:391–6.
76. Sherbourne CD, Hays RD, Ordway L, et al. Antecedents of adherence to medical recommendations: results from the Medical Outcomes Study. J Behav Med 1992;15:447–68.
From the Department of Physical Therapy, University of Alberta, Edmonton AB (Dr. Jones) and UT MD Anderson Cancer Center, Houston, TX (Dr. Suarez-Almazor).
Abstract
- Objective: To discuss patient expectations of total knee arthroplasty (TKA), instruments used to measure expectations, and the association between expectations, health outcomes, and satisfaction.
- Methods: Review of the literature.
- Results: TKA is an elective surgery for patients with persistent pain and disability caused by knee arthritis. Expectations regarding the surgical procedure and recovery can vary by diagnosis, personal characteristics, functional status, employment status, and trust in physicians. Patients have high overall expectations for recovery, particularly for pain relief and walking. Surgeons’ expectations tend to be more optimistic than patients’, although a subset of patients may have unrealistically high expectations. Although total joint replacement is an effective treatment for advanced arthritis, approximately 30% of potential candidates are unwilling to proceed with surgery. Potential surgical candidates unwilling to proceed with surgery tend to be older, female, and from ethnic minority groups. Several patient-related factors are associated with satisfaction with TKA, including primary diagnosis, preoperative pain and function, and mental health, including depression, but the relationships of satisfaction with gender, age, and comorbid conditions are less certain.
- Conclusion: A better understanding of patient expectations of TKA and recovery can identify knowledge gaps, misconceptions, and communication barriers, and ultimately improve shared decision making. A core set of reliable and valid instruments to measure expectations may encourage their routine use in both clinical and research settings.
Key words: total knee arthroplasty; osteoarthritis; patient expectations; shared decision making; joint replacement.
Total knee arthroplasty (TKA) is an elective surgery for patients with persistent pain and disability caused by knee arthritis. It is viewed as an effective and cost-effective surgical treatment for end-stage osteoarthritis (OA) [1–4]. As the population ages and obesity rates steadily increase, so will the utilization rates for TKA, with projected demand in the United States expected to grow 673% by 2030 [5–7]. The key indicators for receiving primary TKA are end-stage OA and joint pain [8]. Although TKA is a surgical option when conservative management is exhausted, no consensus exists as to the severity of symptoms required to consider surgery [9]. Variation in the utilization of TKA exists with respect to gender, racial/ethnicity, hospital, and geography [10,11]. These differences cannot be explained by prevalence of arthritis or symptoms or by access to health care alone. Increasingly, studies have shown these variations are largely attributable to patients’ preferences, driven by their beliefs, concerns, familiarity with the procedure, and expectations, along with physician opinion [12]. While physician opinions and recommendations clearly influence patients’ decisions, they do so primarily by modulating patients’ beliefs and expectations.
Patient expectations, not only of the effectiveness of the procedure itself but also of the recovery process, influence the decision to undergo an elective surgery such as joint arthroplasty. Ideally, these expectations should be informed by evidence, but often, lack of knowledge, preconceived beliefs, and misconceptions can taint informed decision making. A better understanding of patient expectations of TKA and recovery can identify knowledge gaps, misconceptions, and communication barriers, and ultimately improve shared decision making. Understanding patient expectations and factors that influence expectations provides a fuller appreciation of the outcomes that are meaningful to patients and can guide preoperative education and open dialogue with patients within a shared decision making model of care. In this paper, we discuss patient expectations of TKA, including expectations regarding outcomes and recovery, fulfillment of expectations, and the association of fulfilled expectations with satisfaction.
Measurement of Expectations
The construct of expectation is complex and situational. The ambiguity within the literature occurs most likely because expectations are multifaceted. Expectation involves the notion of expectancy, with respect to health care, that given events are likely to occur as a result of a medical procedure or treatment. This concept is in contrast to wants, which reflects a patient’s desire or wishes that an event will occur [13]. The term patient expectation, however, is commonly confused with patient preference or value. Preference implies a relative valuation or comparison by the patient and, unlike expectation, may not be explicitly expressed by the patient [13]. Different types of health care expectations exist that broadly relate to what patients expect regarding health care structure, process, and outcome [14].
Studies of patient expectations are diverse within the orthopedic research field and reflect differing theoretical underpinnings and lack of standardization. The lack of standardization makes measuring the complex concept of expectations challenging. While a number of conceptual models exist, Bowling and colleagues aptly recognize the multidimensionality of expectations and that no one conceptual model captures patient expectations [14]. The lack of standardization was noted in a systematic review by Haanstra and colleagues who found great variety in the definitions and measurements of expectations in studies examining their relationship with outcomes of total joint arthroplasty [15].
No gold standard measure exists for measuring patient expectations of orthopedic surgery. Zywiel’s systematic review [16] of 66 studies identified 7 validated instruments for measuring patient expectations for orthopedic surgery: of these, 2 were specific to TKA (Hospital for Special Surgery (HSS) Expectation Survey [17] and Expectation Domain of the New Knee Scoring System [18,19]), and 2 were generic to musculoskeletal conditions (Expectation domain of the Musculoskeletal Outcomes Data Evaluation and Management System (MODEMS) Instruments [20] and the Sunnybrook Surgery Expectation Survey [21]). A number of other measures used within the literature were identified; however, the psychometric properties for many of these measures were not reported and any evidence of testing and validation were lacking [16]. Some studies used a single question to measure expectations. As patient expectation is multi-dimensional, using a single item to evaluate expectations is problematic. Zywiel and others have called for a core set of reliable and valid instruments to measure expectations [14,22], which may encourage their routine use in both clinical and research settings.
Patients Expectations for TKA Recovery
Although patient concerns vary in terms of importance and severity [23], pain and physical limitations are primary concerns for patients seeking TKA. Patients have high overall expectations for recovery, particularly for pain relief and walking [24–32]. TKA is an elective surgical procedure that provides substantial pain relief and improvements in function and quality of life, with the largest gains seen within the first 6 months [33,34]. Both short-term and long-term effect sizes for pain relief and functional recovery are large, in excess of 1.0 [34]. Over 70% of patients undergoing TKA expect to be pain-free, and 35% expect to have no limitations with routine activities [24,28,31].
Expectations regarding the surgical procedure and recovery can vary by diagnosis, personal characteristics, functional status [17], employment status, and trust in physicians [32,35]. There is, however, inconsistent evidence on associative preoperative factors of recovery expectations. While some evidence supports an association between higher expectations and younger age and greater preoperative functional limitation [26–28,32,36–38], others have reported no significant association with several preoperative factors including age, gender, and preoperative functional status [24,26,37]. Lower overall expectations [28] and lower expectations for pain relief [21] were also seen for patients with a greater number of comorbid conditions.
It may be that patients with high preoperative expectations are more optimistic, interpret their health-reported quality of life gains more liberally, and are more likely to adhere to rehabilitation treatment [24,25]. Optimism is a generalized expectancy of a positive outcome that is related to indicators of well-being [39]. Presurgical optimism was shown to be associated with less postsurgical pain and anxiety in patients undergoing total hip and knee arthroplasty [40].
In addition to general future-oriented constructs, such as optimism, treatment-specific psychological constructs, such as treatment credibility and treatment expectancy, are seen in patients with total joint arthroplasty. A strong but not redundant association is seen between treatment expectancy and treatment credibility, that is, expectations of a treatment may be related as to how credible the treatment outcomes appear [41,42]. Haanstra and colleagues advocate further clinical work to explore which factor predicts total joint arthroplasty outcomes so that patients who are at a higher risk of poor outcomes can be identified [42].
Others have recognized that perspectives and expectations of surgical outcomes differ between patient and surgeon [43–45]. Overall, surgeons’ expectations tend to be more optimistic than patients expectations of outcomes, although a subset of patients may have unrealistically high expectations [46]. Patients do not always realize that some of their expectations cannot be met by current orthopedic procedures, and this gap in understanding is an important source of discrepancies in expectations and patient dissatisfaction [46]. Ghomrawi and colleagues reported that approximately one-third of 205 patients undergoing primary TKA had higher expectations than their surgeons did. Being male and having lower preoperative pain was associated with having discordantly higher preoperative expectations [44]. For realistic expectations to be set, patients need accurate and understandable information about expected positive outcomes of surgery such as level of function and symptom relief as well as the risk of joint failure, adverse events, complications, and activity limitations. Although little work has explored the alignment of patient and surgeon’s expectations, setting realistic expectations may be aided by using a shared decision making approach that incorporates patient preferences and values, the best available evidence, and the surgeon’s expertise.
Expectations and Willingness to Undergo Surgery
Although total joint replacement is an effective treatment for advanced arthritis, approximately 30% of potential candidates are “unwilling” to proceed with surgery [47,48]. Willingness is a component of the medical decision making process and is influenced by preferences. Potential surgical candidates unwilling to proceed with surgery tend to be older, female, and from ethnic minor-ity groups [12,47–49]. Preference-sensitive medical decisions, such as whether or not to proceed with TKA, are related to patients’ attitudes and perceptions, which can be affected by sociocultural influences. In a cohort of 627 male patients with moderate to severe OA who were viewed as “good” candidates for total joint arthroplasty, more African Americans (24%) than Caucasian Americans (15%) had lower expectations for outcomes of surgery [35]. In particular, African Americans expressed concerns about postoperative pain and walking. Similar findings were also reported in another study in which minority patients were less likely to consider TKA [12]. Determinants of preferences were patients’ beliefs about the efficacy of the procedure and knowing others who had already undergone TKA [12]. Ibrahim and colleagues postulated that outcome expectations mediated or influenced the willingness to undergo total joint arthroplasty surgery [49]. Interventional work that built upon this premise suggested that willingness to proceed with TKA could be modified by educational interventions. In a randomized controlled trial of 639 African American patients attending Veteran’s Affairs primary clinics who received a decision aid with or without brief counseling, willingness to proceed with TKA increased and patient-provider communication improved among the patients who received any intervention [50]. Yet in another randomized trial involving African American patients who received care from an academic center, a combination decision aid and motivational interviewing strategy was no better than an educational pamphlet in improving patients’ preferences toward joint replacement surgery for knee OA [51]. This led the authors to recommend further exploration of patients’ knowledge, beliefs, and attitudes regarding surgical treatments for OA.
Effect of Expectations on Health Outcomes and Satisfaction
Some evidence suggests that better outcomes are seen in patients with higher expectations of recovery and, in turn, expectations that are met influence patient satisfaction. A systematic review of several chronic conditions showed with consistency across studies that positive recovery expectations were associated with better health outcomes [22]. The effect size varied with the condition and measure; however, none of the 16 studies examined arthritis or joint arthroplasty. Conversely, a systematic review of 18 prospective longitudinal cohort studies examining the association between expectation and outcomes (ie, pain, function, stiffness, satisfaction, overall improvement) reported less than convincing evidence of an association between patient preoperative expectations and treatment outcomes for THA and TKA in terms of short- and long-term postoperative pain and functional outcomes [15]. No consistent associations were seen with adjusted analysis of patient expectations and pain or functional outcomes at greater than 6 weeks [15]. Inconsistencies seen among the reviewed articles may be related to a number of issues centred on terminology, construct, expectation measures, and confounding effects.
Although TKA is an effective surgical procedure with large gains reported, 14% to 25% participants report little or no symptom improvement and/or dissatisfaction up to 1 year after surgery [1,52–59]. In a study with 5 years of follow-up, a decline in the satisfaction rate was seen after 1 year, although this decline was seen more so with physical function than with pain [38]. Although dissatisfaction can be attributed to surgical complications, in many cases, no technical or medical reasons can be identified. In a subset of patients who received TKA, surgical intervention does not adequately address patients’ concerns of pain and activity limitation. To compound matters, fair agreement was reported between patient and surgeon regarding satisfaction at 6 and 12 months postoperative. Disagreement between the patient and surgeon was explained by unmet expectations and postoperative complications [60]. When there was discordance, more often than not patients were less satisfied with TKA outcomes than surgeons [60,61].
While several theories explain patient satisfaction [62–65], evidence from total joint arthroplasty studies support the concept that satisfaction is derived from fulfillment of expectations [17,52]. Preoperative expectations are not to be confused with postoperative fulfilment of expectations, which are reflective of whether expectations of treatment have been met. Satisfaction is a value judgment and can be viewed as an affective domain, whereas expectation is a cognitive domain [66]. Patient satisfaction is regarded as the extent of a person’s experience compared to their expectation. As with expectations, a number of theoretical constructs exist concerning patient satisfaction [14,67]. Many dimensions of satisfaction exist, with patient expectations being central to these constructs. Deviation from expectations, however, does not necessarily correspond to dissatisfaction [67].
Several patient-related factors are associated with satisfaction with TKA, including primary diagnosis, preoperative pain and function, and mental health, including depression, but the relationships of satisfaction with gender, age, and comorbid conditions are less certain [33,38,52,55,56,68]. Greater preoperative pain, postoperative complications, lower 1-year WOMAC scores and functional limitations were associated with dissatisfied patients [38,52,53,59]. While no consistent associations were seen with preoperative expectations, consistent evidence has shown that fulfillment of expectations has an impact on satisfaction [31,36,52,58,69].
It should be acknowledged that the concept of fulfillment of expectations is not the same as satisfaction. A patient can be satisfied with TKA even though their expectations have not been met. The fulfillment of expectations is dependent upon the type of expectation and the postoperative time period. Fulfillment of expectations were seen with pain relief, function, walking and health status [25,31,70] while patients expectations were not always met with leisure activities [38].
Shared Decision Making
The shared decision making process, in which the patient and physician share responsibility and actively participate in the clinical decision making process [71], may help in ensuring that patients’ expectations are met. Shared decision making requires eliciting patients’ preferences and values, providing clear information on the processes that will occur during surgery, recovery, rehabilitation, and in the longer phase of recovery, and what realistic outcomes can be expected. While a more “paternalistic” approach predominated in earlier years, the current trends indicate greater patient involvement in decision making with the surgeon, with open discussion of patient goals and expectations [71]. This approach also aids patients in their preparation for the recovery and rehabilitation stages, which can be challenging, especially if they are unaware as to what to expect. Patient expectations are more likely to be met when there is shared decision making and patients have been given relevant information and understand what is a reasonable outcome. While a shared decision making approach is advocated within orthopedics [72], patient expectations are largely not measured in the clinical setting.
Patient education is an integral component of assisting patients to make informed decisions; however, it is unknown whether education alone can modify expectations. Educational approaches can include group classes, videos, and written materials [73]. Limited evidence from a randomized controlled trial suggests that preoperative expectations can be modified by preoperative education classes by decreasing the number of expectations and having more expectations in agreement with the surgeons’ expectations [29]. Mancuso and colleagues, who looked at whether a preoperative education session could modify expectations found that larger changes in expectations were seen with those patients who had greater baseline expectation scores, worse pain and function, and were older [29]. Others have also reported that preoperative education reduces anxiety by providing patients with an understanding of what to expect [74,75]. An assumption is that expectations can be changed by improving knowledge, which underscores the need for relevant meaningful education to increase knowledge and instill realistic expectations. Others have surmised there is a proportion of patients who will continue to have unexpectedly high unrealistic expectations regardless of educational session [31,37]. This would suggest that education is not the only approach to modify expectations but rather different strategies may need to be implemented for a certain subsets of patients with unrealistic expectations.
Conclusion
Patient expectation is an important element to be considered in shared clinical decision making, as it can influ-ence preferences and subsequent satisfaction. Patients considering TKA have specific needs and expectations that they presume will be addressed with the surgery. If these are realistic, they can be met, and will result in greater patient satisfaction and better ongoing adherence to health care recommendations [76]. While much work has been conducted in identifying which patient characteristics may influence health expectations, additional research is needed to further determine how to shape expectations within a realistic, achievable framework. While traditional patient education is an important element to enhance knowledge, the limited available evidence suggests it is not sufficiently effective on its own. Other strategies such as use of individualized decision aids, provision of peer support, and enhanced provider-patient communication have been effective in many areas of health care and warrant evaluation in this field.
Corresponding author: Allyson Jones, PhD, Rm 2-50, Corbett Hall, University of Alberta, Edmonton, Alberta Canada T6G 2G4, [email protected].
Financial disclosures: None.
Author contributions: conception and design, CAJ, MES; analysis and interpretation of data, MES; drafting of article, CAJ, MES; critical revision of the article, CAJ, MES; collection and assembly of data, CAJ.
From the Department of Physical Therapy, University of Alberta, Edmonton AB (Dr. Jones) and UT MD Anderson Cancer Center, Houston, TX (Dr. Suarez-Almazor).
Abstract
- Objective: To discuss patient expectations of total knee arthroplasty (TKA), instruments used to measure expectations, and the association between expectations, health outcomes, and satisfaction.
- Methods: Review of the literature.
- Results: TKA is an elective surgery for patients with persistent pain and disability caused by knee arthritis. Expectations regarding the surgical procedure and recovery can vary by diagnosis, personal characteristics, functional status, employment status, and trust in physicians. Patients have high overall expectations for recovery, particularly for pain relief and walking. Surgeons’ expectations tend to be more optimistic than patients’, although a subset of patients may have unrealistically high expectations. Although total joint replacement is an effective treatment for advanced arthritis, approximately 30% of potential candidates are unwilling to proceed with surgery. Potential surgical candidates unwilling to proceed with surgery tend to be older, female, and from ethnic minority groups. Several patient-related factors are associated with satisfaction with TKA, including primary diagnosis, preoperative pain and function, and mental health, including depression, but the relationships of satisfaction with gender, age, and comorbid conditions are less certain.
- Conclusion: A better understanding of patient expectations of TKA and recovery can identify knowledge gaps, misconceptions, and communication barriers, and ultimately improve shared decision making. A core set of reliable and valid instruments to measure expectations may encourage their routine use in both clinical and research settings.
Key words: total knee arthroplasty; osteoarthritis; patient expectations; shared decision making; joint replacement.
Total knee arthroplasty (TKA) is an elective surgery for patients with persistent pain and disability caused by knee arthritis. It is viewed as an effective and cost-effective surgical treatment for end-stage osteoarthritis (OA) [1–4]. As the population ages and obesity rates steadily increase, so will the utilization rates for TKA, with projected demand in the United States expected to grow 673% by 2030 [5–7]. The key indicators for receiving primary TKA are end-stage OA and joint pain [8]. Although TKA is a surgical option when conservative management is exhausted, no consensus exists as to the severity of symptoms required to consider surgery [9]. Variation in the utilization of TKA exists with respect to gender, racial/ethnicity, hospital, and geography [10,11]. These differences cannot be explained by prevalence of arthritis or symptoms or by access to health care alone. Increasingly, studies have shown these variations are largely attributable to patients’ preferences, driven by their beliefs, concerns, familiarity with the procedure, and expectations, along with physician opinion [12]. While physician opinions and recommendations clearly influence patients’ decisions, they do so primarily by modulating patients’ beliefs and expectations.
Patient expectations, not only of the effectiveness of the procedure itself but also of the recovery process, influence the decision to undergo an elective surgery such as joint arthroplasty. Ideally, these expectations should be informed by evidence, but often, lack of knowledge, preconceived beliefs, and misconceptions can taint informed decision making. A better understanding of patient expectations of TKA and recovery can identify knowledge gaps, misconceptions, and communication barriers, and ultimately improve shared decision making. Understanding patient expectations and factors that influence expectations provides a fuller appreciation of the outcomes that are meaningful to patients and can guide preoperative education and open dialogue with patients within a shared decision making model of care. In this paper, we discuss patient expectations of TKA, including expectations regarding outcomes and recovery, fulfillment of expectations, and the association of fulfilled expectations with satisfaction.
Measurement of Expectations
The construct of expectation is complex and situational. The ambiguity within the literature occurs most likely because expectations are multifaceted. Expectation involves the notion of expectancy, with respect to health care, that given events are likely to occur as a result of a medical procedure or treatment. This concept is in contrast to wants, which reflects a patient’s desire or wishes that an event will occur [13]. The term patient expectation, however, is commonly confused with patient preference or value. Preference implies a relative valuation or comparison by the patient and, unlike expectation, may not be explicitly expressed by the patient [13]. Different types of health care expectations exist that broadly relate to what patients expect regarding health care structure, process, and outcome [14].
Studies of patient expectations are diverse within the orthopedic research field and reflect differing theoretical underpinnings and lack of standardization. The lack of standardization makes measuring the complex concept of expectations challenging. While a number of conceptual models exist, Bowling and colleagues aptly recognize the multidimensionality of expectations and that no one conceptual model captures patient expectations [14]. The lack of standardization was noted in a systematic review by Haanstra and colleagues who found great variety in the definitions and measurements of expectations in studies examining their relationship with outcomes of total joint arthroplasty [15].
No gold standard measure exists for measuring patient expectations of orthopedic surgery. Zywiel’s systematic review [16] of 66 studies identified 7 validated instruments for measuring patient expectations for orthopedic surgery: of these, 2 were specific to TKA (Hospital for Special Surgery (HSS) Expectation Survey [17] and Expectation Domain of the New Knee Scoring System [18,19]), and 2 were generic to musculoskeletal conditions (Expectation domain of the Musculoskeletal Outcomes Data Evaluation and Management System (MODEMS) Instruments [20] and the Sunnybrook Surgery Expectation Survey [21]). A number of other measures used within the literature were identified; however, the psychometric properties for many of these measures were not reported and any evidence of testing and validation were lacking [16]. Some studies used a single question to measure expectations. As patient expectation is multi-dimensional, using a single item to evaluate expectations is problematic. Zywiel and others have called for a core set of reliable and valid instruments to measure expectations [14,22], which may encourage their routine use in both clinical and research settings.
Patients Expectations for TKA Recovery
Although patient concerns vary in terms of importance and severity [23], pain and physical limitations are primary concerns for patients seeking TKA. Patients have high overall expectations for recovery, particularly for pain relief and walking [24–32]. TKA is an elective surgical procedure that provides substantial pain relief and improvements in function and quality of life, with the largest gains seen within the first 6 months [33,34]. Both short-term and long-term effect sizes for pain relief and functional recovery are large, in excess of 1.0 [34]. Over 70% of patients undergoing TKA expect to be pain-free, and 35% expect to have no limitations with routine activities [24,28,31].
Expectations regarding the surgical procedure and recovery can vary by diagnosis, personal characteristics, functional status [17], employment status, and trust in physicians [32,35]. There is, however, inconsistent evidence on associative preoperative factors of recovery expectations. While some evidence supports an association between higher expectations and younger age and greater preoperative functional limitation [26–28,32,36–38], others have reported no significant association with several preoperative factors including age, gender, and preoperative functional status [24,26,37]. Lower overall expectations [28] and lower expectations for pain relief [21] were also seen for patients with a greater number of comorbid conditions.
It may be that patients with high preoperative expectations are more optimistic, interpret their health-reported quality of life gains more liberally, and are more likely to adhere to rehabilitation treatment [24,25]. Optimism is a generalized expectancy of a positive outcome that is related to indicators of well-being [39]. Presurgical optimism was shown to be associated with less postsurgical pain and anxiety in patients undergoing total hip and knee arthroplasty [40].
In addition to general future-oriented constructs, such as optimism, treatment-specific psychological constructs, such as treatment credibility and treatment expectancy, are seen in patients with total joint arthroplasty. A strong but not redundant association is seen between treatment expectancy and treatment credibility, that is, expectations of a treatment may be related as to how credible the treatment outcomes appear [41,42]. Haanstra and colleagues advocate further clinical work to explore which factor predicts total joint arthroplasty outcomes so that patients who are at a higher risk of poor outcomes can be identified [42].
Others have recognized that perspectives and expectations of surgical outcomes differ between patient and surgeon [43–45]. Overall, surgeons’ expectations tend to be more optimistic than patients expectations of outcomes, although a subset of patients may have unrealistically high expectations [46]. Patients do not always realize that some of their expectations cannot be met by current orthopedic procedures, and this gap in understanding is an important source of discrepancies in expectations and patient dissatisfaction [46]. Ghomrawi and colleagues reported that approximately one-third of 205 patients undergoing primary TKA had higher expectations than their surgeons did. Being male and having lower preoperative pain was associated with having discordantly higher preoperative expectations [44]. For realistic expectations to be set, patients need accurate and understandable information about expected positive outcomes of surgery such as level of function and symptom relief as well as the risk of joint failure, adverse events, complications, and activity limitations. Although little work has explored the alignment of patient and surgeon’s expectations, setting realistic expectations may be aided by using a shared decision making approach that incorporates patient preferences and values, the best available evidence, and the surgeon’s expertise.
Expectations and Willingness to Undergo Surgery
Although total joint replacement is an effective treatment for advanced arthritis, approximately 30% of potential candidates are “unwilling” to proceed with surgery [47,48]. Willingness is a component of the medical decision making process and is influenced by preferences. Potential surgical candidates unwilling to proceed with surgery tend to be older, female, and from ethnic minor-ity groups [12,47–49]. Preference-sensitive medical decisions, such as whether or not to proceed with TKA, are related to patients’ attitudes and perceptions, which can be affected by sociocultural influences. In a cohort of 627 male patients with moderate to severe OA who were viewed as “good” candidates for total joint arthroplasty, more African Americans (24%) than Caucasian Americans (15%) had lower expectations for outcomes of surgery [35]. In particular, African Americans expressed concerns about postoperative pain and walking. Similar findings were also reported in another study in which minority patients were less likely to consider TKA [12]. Determinants of preferences were patients’ beliefs about the efficacy of the procedure and knowing others who had already undergone TKA [12]. Ibrahim and colleagues postulated that outcome expectations mediated or influenced the willingness to undergo total joint arthroplasty surgery [49]. Interventional work that built upon this premise suggested that willingness to proceed with TKA could be modified by educational interventions. In a randomized controlled trial of 639 African American patients attending Veteran’s Affairs primary clinics who received a decision aid with or without brief counseling, willingness to proceed with TKA increased and patient-provider communication improved among the patients who received any intervention [50]. Yet in another randomized trial involving African American patients who received care from an academic center, a combination decision aid and motivational interviewing strategy was no better than an educational pamphlet in improving patients’ preferences toward joint replacement surgery for knee OA [51]. This led the authors to recommend further exploration of patients’ knowledge, beliefs, and attitudes regarding surgical treatments for OA.
Effect of Expectations on Health Outcomes and Satisfaction
Some evidence suggests that better outcomes are seen in patients with higher expectations of recovery and, in turn, expectations that are met influence patient satisfaction. A systematic review of several chronic conditions showed with consistency across studies that positive recovery expectations were associated with better health outcomes [22]. The effect size varied with the condition and measure; however, none of the 16 studies examined arthritis or joint arthroplasty. Conversely, a systematic review of 18 prospective longitudinal cohort studies examining the association between expectation and outcomes (ie, pain, function, stiffness, satisfaction, overall improvement) reported less than convincing evidence of an association between patient preoperative expectations and treatment outcomes for THA and TKA in terms of short- and long-term postoperative pain and functional outcomes [15]. No consistent associations were seen with adjusted analysis of patient expectations and pain or functional outcomes at greater than 6 weeks [15]. Inconsistencies seen among the reviewed articles may be related to a number of issues centred on terminology, construct, expectation measures, and confounding effects.
Although TKA is an effective surgical procedure with large gains reported, 14% to 25% participants report little or no symptom improvement and/or dissatisfaction up to 1 year after surgery [1,52–59]. In a study with 5 years of follow-up, a decline in the satisfaction rate was seen after 1 year, although this decline was seen more so with physical function than with pain [38]. Although dissatisfaction can be attributed to surgical complications, in many cases, no technical or medical reasons can be identified. In a subset of patients who received TKA, surgical intervention does not adequately address patients’ concerns of pain and activity limitation. To compound matters, fair agreement was reported between patient and surgeon regarding satisfaction at 6 and 12 months postoperative. Disagreement between the patient and surgeon was explained by unmet expectations and postoperative complications [60]. When there was discordance, more often than not patients were less satisfied with TKA outcomes than surgeons [60,61].
While several theories explain patient satisfaction [62–65], evidence from total joint arthroplasty studies support the concept that satisfaction is derived from fulfillment of expectations [17,52]. Preoperative expectations are not to be confused with postoperative fulfilment of expectations, which are reflective of whether expectations of treatment have been met. Satisfaction is a value judgment and can be viewed as an affective domain, whereas expectation is a cognitive domain [66]. Patient satisfaction is regarded as the extent of a person’s experience compared to their expectation. As with expectations, a number of theoretical constructs exist concerning patient satisfaction [14,67]. Many dimensions of satisfaction exist, with patient expectations being central to these constructs. Deviation from expectations, however, does not necessarily correspond to dissatisfaction [67].
Several patient-related factors are associated with satisfaction with TKA, including primary diagnosis, preoperative pain and function, and mental health, including depression, but the relationships of satisfaction with gender, age, and comorbid conditions are less certain [33,38,52,55,56,68]. Greater preoperative pain, postoperative complications, lower 1-year WOMAC scores and functional limitations were associated with dissatisfied patients [38,52,53,59]. While no consistent associations were seen with preoperative expectations, consistent evidence has shown that fulfillment of expectations has an impact on satisfaction [31,36,52,58,69].
It should be acknowledged that the concept of fulfillment of expectations is not the same as satisfaction. A patient can be satisfied with TKA even though their expectations have not been met. The fulfillment of expectations is dependent upon the type of expectation and the postoperative time period. Fulfillment of expectations were seen with pain relief, function, walking and health status [25,31,70] while patients expectations were not always met with leisure activities [38].
Shared Decision Making
The shared decision making process, in which the patient and physician share responsibility and actively participate in the clinical decision making process [71], may help in ensuring that patients’ expectations are met. Shared decision making requires eliciting patients’ preferences and values, providing clear information on the processes that will occur during surgery, recovery, rehabilitation, and in the longer phase of recovery, and what realistic outcomes can be expected. While a more “paternalistic” approach predominated in earlier years, the current trends indicate greater patient involvement in decision making with the surgeon, with open discussion of patient goals and expectations [71]. This approach also aids patients in their preparation for the recovery and rehabilitation stages, which can be challenging, especially if they are unaware as to what to expect. Patient expectations are more likely to be met when there is shared decision making and patients have been given relevant information and understand what is a reasonable outcome. While a shared decision making approach is advocated within orthopedics [72], patient expectations are largely not measured in the clinical setting.
Patient education is an integral component of assisting patients to make informed decisions; however, it is unknown whether education alone can modify expectations. Educational approaches can include group classes, videos, and written materials [73]. Limited evidence from a randomized controlled trial suggests that preoperative expectations can be modified by preoperative education classes by decreasing the number of expectations and having more expectations in agreement with the surgeons’ expectations [29]. Mancuso and colleagues, who looked at whether a preoperative education session could modify expectations found that larger changes in expectations were seen with those patients who had greater baseline expectation scores, worse pain and function, and were older [29]. Others have also reported that preoperative education reduces anxiety by providing patients with an understanding of what to expect [74,75]. An assumption is that expectations can be changed by improving knowledge, which underscores the need for relevant meaningful education to increase knowledge and instill realistic expectations. Others have surmised there is a proportion of patients who will continue to have unexpectedly high unrealistic expectations regardless of educational session [31,37]. This would suggest that education is not the only approach to modify expectations but rather different strategies may need to be implemented for a certain subsets of patients with unrealistic expectations.
Conclusion
Patient expectation is an important element to be considered in shared clinical decision making, as it can influ-ence preferences and subsequent satisfaction. Patients considering TKA have specific needs and expectations that they presume will be addressed with the surgery. If these are realistic, they can be met, and will result in greater patient satisfaction and better ongoing adherence to health care recommendations [76]. While much work has been conducted in identifying which patient characteristics may influence health expectations, additional research is needed to further determine how to shape expectations within a realistic, achievable framework. While traditional patient education is an important element to enhance knowledge, the limited available evidence suggests it is not sufficiently effective on its own. Other strategies such as use of individualized decision aids, provision of peer support, and enhanced provider-patient communication have been effective in many areas of health care and warrant evaluation in this field.
Corresponding author: Allyson Jones, PhD, Rm 2-50, Corbett Hall, University of Alberta, Edmonton, Alberta Canada T6G 2G4, [email protected].
Financial disclosures: None.
Author contributions: conception and design, CAJ, MES; analysis and interpretation of data, MES; drafting of article, CAJ, MES; critical revision of the article, CAJ, MES; collection and assembly of data, CAJ.
1. Jones CA, Voaklander DC, Johnston DW, Suarez-Almazor ME. Health related quality of life outcomes after total hip and knee arthroplasties in a community based population. J Rheumatol 2000;27:1745–52.
2. Waimann CA, Fernandez-Mazarambroz RJ, Cantor SB, et al. Cost-effectiveness of total knee replacement: a prospective cohort study. Arthritis Care Res 2014;66:592–9.
3. Jenkins PJ, Clement ND, Hamilton DF, et al. Predicting the cost-effectiveness of total hip and knee replacement: a health economic analysis. Bone Joint J 2013;95:115–21.
4. Losina E, Walensky RP, Kessler CL, et al. Cost-effectiveness of total knee arthroplasty in the United States: patient risk and hospital volume. Arch Intern Med 2009;169:1113–21.
5. Cram P, Lu X, Kates SL, et al. Total knee arthroplasty volume, utilization, and outcomes among Medicare beneficiaries, 1991-2010. JAMA 2012;308:1227–36.
6. Kurtz S, Ong K, Lau E, et al. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007;89:780–5.
7. Jain NB, Higgins LD, Ozumba D, et al. Trends in epidemiology of knee arthroplasty in the United States, 1990-2000. Arthritis Rheum 2005;52:3928–33.
8. Engel C, Hamilton NA, Potter PT, Zautra AJ. Impact of two types of expectancy on recovery from total knee replacement surgery (TKR) in adults with osteoarthritis. Behav Med 2004;30:113–23.
9. Carr AJ, Robertsson O, Graves S, et al. Knee replacement. Lancet 2012;379:1331–40.
10. Skinner J, Weinstein JN, Sporer SM, Wennberg JE. Racial, ethnic, and geographic disparities in rates of knee arthroplasty among Medicare patients. N Engl J Med 2003;349:1350–9.
11. Cobos R, Latorre A, Aizpuru F, et al. Variability of indication criteria in knee and hip replacement: an observational study. BMC Musculoskelet Disord 2010;11:249.
12. Suarez-Almazor ME, Souchek J, Kelly PA, et al. Ethnic variation in knee replacement: patient preferences or uninformed disparity? Arch Intern Med 2005;165:1117–24.
13. Uhlmann RF, Inui TS, Carter WB. Patient requests and expectations. Definitions and clinical applications. Med Care 1984;22:681–5.
14. Bowling A, Rowe G, Lambert N, et al. The measurement of patients’ expectations for health care: a review and psychometric testing of a measure of patients’ expectations. Health Technology Assessment 2012;16:1–515.
15. Haanstra TM, van den Berg T, Ostelo RW, et al. Systematic review: do patient expectations influence treatment outcomes in total knee and total hip arthroplasty? Health Qual Life Outcomes 2012;10:152.
16. Zywiel MG, Mahomed A, Gandhi R, et al. Measuring expectations in orthopaedic surgery: a systematic review. Clin Orthop Rel Res 2013;471:3446–56.
17. Mancuso CA, Sculco TP, Wickiewicz TL, et al. Patients’ expectations of knee surgery. J Bone Joint Surg Am 2001;83A:1005–12.
18. Noble PC, Scuderi GR, Brekke AC, et al. Development of a new Knee Society scoring system. Clin Orthopaed Rel Res 2012;470:20–32.
19. Scuderi GR, Bourne RB, Noble PC, et al. The new Knee Society Knee Scoring System. Clin Orthop Relat Res 2012;470:3–19.
20. Saleh KJ, Bershadsky B, Cheng E, Kane R. Lessons learned from the hip and knee musculoskeletal outcomes data evaluation and management system. Clin Orthop Relat Res 2004; 272–8.
21. Razmjou H, Finkelstein JA, Yee A, et al. Relationship between preoperative patient characteristics and expectations in candidates for total knee arthroplasty. Physiotherapy Canada 2009;61:38–45.
22. Mondloch MV, Cole DC, Frank JW. Does how you do depend on how you think you’ll do? A systematic review of the evidence for a relation between patients’ recovery expectations and health outcomes. CMAJ 2001;165:174–9.
23. Wright JG, Santaguida PL, Young N, et al. Patient preferences before and after total knee arthroplasty. J Clin Epidemiol 2010;63:774–82.
24. Mahomed NN, Liang MH, Cook EF, et al.: The importance of patient expectations in predicting functional outcomes after total joint arthroplasty. J Rheumatology 2002;29:1273–9.
25. Gonzalez Saenz de Tejada M, Escobar A, Herrera C, et al. Patient expectations and health-related quality of life outcomes following total joint replacement. Value Health 2010;13:447–54.
26. Hepinstall MS, Rutledge JR, Bornstein LJ, et al. Factors that impact expectations before total knee arthroplasty. J Arthroplasty 2011;26:870–6.
27. Muniesa JM, Marco E, Tejero M, et al. Analysis of the expectations of elderly patients before undergoing total knee replacement. Arch Gerontol Geriatr 2010;51:E83-E87.
28. Lingard EA, Sledge CB, Learmonth ID. Patient expectations regarding total knee arthroplasty: Differences among the United States, United Kingdom, and Australia. J Bone Joint Surg Am 2006;88:1201–7.
29. Mancuso CA, Graziano S, Briskie LM, et al. Randomized trials to modify patients’ preoperative expectations of hip and knee arthroplasties. Clin Orthopaed Rel Res 2008;466:424–31.
30. de AS, Kallen MA, Amick B, et al. Patients’ expectations about total knee arthroplasty outcomes. Health Expect 2016;19:299–308.
31. Mannion AF, Kampfen S, Munzinger U, Kramers-de Q. The role of patient expectations in predicting outcome after total knee arthroplasty. Arthritis Res Ther 2009;11:R139.
32. Yoo JH, Chang CB, Kang YG, et al. Patient expectations of total knee replacement and their association with sociodemographic factors and functional status. J Bone Joint Surg Br 2011;93:337–44.
33. Ethgen O, Bruyere O, Richy F, et al. Health-related quality of life in total hip and total knee arthroplasty. A qualitative and systematic review of the literature. J Bone Joint Surg Am 2004;86:963–74.
34. Jones CA, Pohar S. Health-related quality of life after total joint arthroplasty: a scoping review. Clin Geriatr Med 2012;28:395–429.
35. Groeneveld PW, Kwoh CK, Mor MK, et al. Racial differences in expectations of joint replacement surgery outcomes. Arthritis Rheum 2008;59:730–7.
36. Scott CEH, Bugler KE, Clement ND, et al. Patient expectations of arthroplasty of the hip and knee. J Bone Joint Surg Br 2012;94:974–81.
37. Smith J, Soon VL, Boyd A, et al. What do Scottish patients expect of their total knee arthroplasty? J Arthroplasty 2016;31:786–92.
38. Nilsdotter AK, Toksvig-Larsen S, Roos EM. Knee arthroplasty: are patients’ expectations fulfilled? A prospective study of pain and function in 102 patients with 5-year follow-up. Acta Orthopaedica 2009;80:55–61.
39. Alarcon GM, Bowling NA, Khazon S. Great expectations: A meta-analytic examination of optimism and hope. Person Ind Diff 2013;54:821–7.
40. Pinto P, McIntyre T, Ferrero R, et al. Predictors of acute postsurgical pain and anxiety following primary total hip and knee arthroplasty. J Pain 2013;14:502–15.
41. Devilly GJ, Borkovec TD. Psychometric properties of the credibility/expectancy questionnaire. J Behav Ther Exp Psychiatry 2000;31:73–86.
42. Haanstra TM, Tilbury C, Kamper SJ, et al. Can optimism, pessimism, hope, treatment credibility and treatment expectancy be distinguished in patients undergoing total hip and total knee arthroplasty? PLoS One 2015;10.
43. Verbeek J, Sengers MJ, Riemens L, Haafkens J.Patient expectations of treatment for back pain: a systematic review of qualitative and quantitative studies. Spine 2004; 29:2309–18.
44. Ghomrawi HM, Mancuso CA, Westrich GH, et al. Discordance in TKA expectations between patients and surgeons. Clin Orthopaed Rel Res 2013;471:175–80.
45. Cordero-Ampuero J, Darder A, Santillana J, et al. Evaluation of patients’ and physicians’ expectations and attributes of osteoarthritis treatment using Kano methodology. Qual Life Res 2012;21:1391–404.
46. Noble PC, Fuller-Lafreniere S, Meftah M, Dwyer MK. Challenges in outcome measurement: discrepancies between patient and provider definitions of success. Clin Orthopaed Rel Res 2013;471:3437–45.
47. Hawker GA, Wright JG, Coyte PC, et al. Determining the need for hip and knee arthroplasty: the role of clinical severity and patients’ preferences. Med Care 2001;39:206–16.
48. Juni P, Dieppe P, Donovan J, et al. Population requirement for primary knee replacement surgery: a cross-sectional study. Rheumatology 2003;42:516–21.
49. Ibrahim SA, Siminoff LA, Burant CJ, Kwoh CK. Differences in expectations of outcome mediate African American/white patient differences in “willingness” to consider joint replacement. Arthritis Rheum 2002;46:2429–35.
50. Ibrahim SA, Hanusa BH, Hannon MJ, et al. Willingness and access to joint replacement among African American patients with knee osteoarthritis: a randomized, controlled intervention. Arthritis Rheum 2013;65:1253–61.
51. Vina ER, Richardson D, Medvedeva E, et al. Does a patient-centered educational intervention affect African-American access to knee replacement? A randomized trial. Clin Orthop Relat Res 2016;474:1755–64.
52. Noble PC, Conditt MA, Cook KF, Mathis KB. The John Insall Award - Patient expectations affect satisfaction with total knee arthroplasty. Clin Orthop Relat Res 2006; 35–43.
53. Robertsson O, Dunbar M, Pehrsson T, et al. Patient satisfaction after knee arthroplasty: a report on 27,372 knees operated on between 1981 and 1995 in Sweden. Acta Orthop Scand 2000;71:262–7.
54. Lau RL, Gandhi R, Mahomed S, Mahomed N. Patient satisfaction after total knee and hip arthroplasty. Clin Geriatr Med 2012;28:349–65.
55. Scott CEH, Howie CR, Macdonald D, Biant LC. Predicting dissatisfaction following total knee replacement. A prospective study of 1217 patients. J Bone Joint Surg Br 2010; 92B:1253–8.
56. Baker PN, van der Meulen JH, Lewsey J, Gregg PJ. The role of pain and function in determining patient satisfaction after total knee replacement. Data from the National Joint Registry for England and Wales. J Bone Joint Surg Br 2007;89:893–900.
57. Khatib Y, Madan A, Naylor JM, Harris IA: Do psychological factors predict poor outcome in patients undergoing TKA? a systematic review. Clin Orthopaed Rel Res 2015;473:2630–8.
58. Adie S, Dao A, Harris IA, et al. Satisfaction with joint replacement in public versus private hospitals: a cohort study. ANZ J Surg 2012;82:616–24.
59. Bourne RB, Chesworth BM, Davis AM, et al. Patient satisfaction after total knee arthroplasty: who is satisfied and who is not? Clin Orthop Relat Res 2010;468:57–63.
60. Harris IA, Harris AM, Naylor et al. Discordance between patient and surgeon satisfaction after total joint arthroplasty. J Arthroplasty 2013;28:722–7.
61. Choi YJ, Ra H. Patient satisfaction after total knee arthroplasty. Knee Surg Relat Res 2016;28:1–15.
62. Williams B. Patient satisfaction: a valid concept? Soc Sci Med 1994;38:509–16.
63. Ware JE Jr, Snyder MK, Wright WR, Davies AR. Defining and measuring patient satisfaction with medical care. Eval Program Plann 1983;6:247–63.
64. Linder-Pelz SU. Toward a theory of patient satisfaction. Soc Sci Med 1982;16:577–82.
65. Hudak PL, Hogg-Johnson S, Bombardier C, et al. Testing a new theory of patient satisfaction with treatment outcome. Med Care 2004;42:726–39.
66. Thompson AG, Sunol R. Expectations as determinants of patient satisfaction: concepts, theory and evidence. Int J Qual Health Care 1995;l7:127–41.
67. Pascoe GC. Patient satisfaction in primary health care: a literature review and analysis. Eval Program Plann 1983;6:185–210.
68. Gandhi R, Davey JR, Mahomed NN. Predicting patient dissatisfaction following joint replacement surgery. J Rheumatol 2008;35:2415–8.
69. Waljee J, McGlinn EP, Sears ED, Chung KC. Patient expectations and patient-reported outcomes in surgery: A systematic review. Surgery 2014;155:799-808.
70. Suda AJ, Seeger JB, Bitsch RG, et al. Are patients’ expectations of hip and knee arthroplasty fulfilled? A prospective study of 130 patients. Orthopedics 2010;33:76.
71. Slover J, Shue J, Koenig K. Shared decision-making in orthopaedic surgery. Clin Orthopaed Rel Res 2012;470:1046–53.
72. Weinstein JN. The missing piece: embracing shared decision making to reform health care. Spine 2000;25:1–4.
73 Aydin D, Klit J, Jacobsen S, et al. No major effects of preoperative education in patients undergoing hip or knee replacement--a systematic review. Dan Med J 2015;62.
74. Spalding NJ. Reducing anxiety by pre-operative education: make the future familiar. Occup Ther Int 2003;10:278–93.
75. Kearney M, Jennrich MK, Lyons S, et al. Effects of preoperative education on patient outcomes after joint replacement surgery. Orthop Nurs 2011;30:391–6.
76. Sherbourne CD, Hays RD, Ordway L, et al. Antecedents of adherence to medical recommendations: results from the Medical Outcomes Study. J Behav Med 1992;15:447–68.
1. Jones CA, Voaklander DC, Johnston DW, Suarez-Almazor ME. Health related quality of life outcomes after total hip and knee arthroplasties in a community based population. J Rheumatol 2000;27:1745–52.
2. Waimann CA, Fernandez-Mazarambroz RJ, Cantor SB, et al. Cost-effectiveness of total knee replacement: a prospective cohort study. Arthritis Care Res 2014;66:592–9.
3. Jenkins PJ, Clement ND, Hamilton DF, et al. Predicting the cost-effectiveness of total hip and knee replacement: a health economic analysis. Bone Joint J 2013;95:115–21.
4. Losina E, Walensky RP, Kessler CL, et al. Cost-effectiveness of total knee arthroplasty in the United States: patient risk and hospital volume. Arch Intern Med 2009;169:1113–21.
5. Cram P, Lu X, Kates SL, et al. Total knee arthroplasty volume, utilization, and outcomes among Medicare beneficiaries, 1991-2010. JAMA 2012;308:1227–36.
6. Kurtz S, Ong K, Lau E, et al. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007;89:780–5.
7. Jain NB, Higgins LD, Ozumba D, et al. Trends in epidemiology of knee arthroplasty in the United States, 1990-2000. Arthritis Rheum 2005;52:3928–33.
8. Engel C, Hamilton NA, Potter PT, Zautra AJ. Impact of two types of expectancy on recovery from total knee replacement surgery (TKR) in adults with osteoarthritis. Behav Med 2004;30:113–23.
9. Carr AJ, Robertsson O, Graves S, et al. Knee replacement. Lancet 2012;379:1331–40.
10. Skinner J, Weinstein JN, Sporer SM, Wennberg JE. Racial, ethnic, and geographic disparities in rates of knee arthroplasty among Medicare patients. N Engl J Med 2003;349:1350–9.
11. Cobos R, Latorre A, Aizpuru F, et al. Variability of indication criteria in knee and hip replacement: an observational study. BMC Musculoskelet Disord 2010;11:249.
12. Suarez-Almazor ME, Souchek J, Kelly PA, et al. Ethnic variation in knee replacement: patient preferences or uninformed disparity? Arch Intern Med 2005;165:1117–24.
13. Uhlmann RF, Inui TS, Carter WB. Patient requests and expectations. Definitions and clinical applications. Med Care 1984;22:681–5.
14. Bowling A, Rowe G, Lambert N, et al. The measurement of patients’ expectations for health care: a review and psychometric testing of a measure of patients’ expectations. Health Technology Assessment 2012;16:1–515.
15. Haanstra TM, van den Berg T, Ostelo RW, et al. Systematic review: do patient expectations influence treatment outcomes in total knee and total hip arthroplasty? Health Qual Life Outcomes 2012;10:152.
16. Zywiel MG, Mahomed A, Gandhi R, et al. Measuring expectations in orthopaedic surgery: a systematic review. Clin Orthop Rel Res 2013;471:3446–56.
17. Mancuso CA, Sculco TP, Wickiewicz TL, et al. Patients’ expectations of knee surgery. J Bone Joint Surg Am 2001;83A:1005–12.
18. Noble PC, Scuderi GR, Brekke AC, et al. Development of a new Knee Society scoring system. Clin Orthopaed Rel Res 2012;470:20–32.
19. Scuderi GR, Bourne RB, Noble PC, et al. The new Knee Society Knee Scoring System. Clin Orthop Relat Res 2012;470:3–19.
20. Saleh KJ, Bershadsky B, Cheng E, Kane R. Lessons learned from the hip and knee musculoskeletal outcomes data evaluation and management system. Clin Orthop Relat Res 2004; 272–8.
21. Razmjou H, Finkelstein JA, Yee A, et al. Relationship between preoperative patient characteristics and expectations in candidates for total knee arthroplasty. Physiotherapy Canada 2009;61:38–45.
22. Mondloch MV, Cole DC, Frank JW. Does how you do depend on how you think you’ll do? A systematic review of the evidence for a relation between patients’ recovery expectations and health outcomes. CMAJ 2001;165:174–9.
23. Wright JG, Santaguida PL, Young N, et al. Patient preferences before and after total knee arthroplasty. J Clin Epidemiol 2010;63:774–82.
24. Mahomed NN, Liang MH, Cook EF, et al.: The importance of patient expectations in predicting functional outcomes after total joint arthroplasty. J Rheumatology 2002;29:1273–9.
25. Gonzalez Saenz de Tejada M, Escobar A, Herrera C, et al. Patient expectations and health-related quality of life outcomes following total joint replacement. Value Health 2010;13:447–54.
26. Hepinstall MS, Rutledge JR, Bornstein LJ, et al. Factors that impact expectations before total knee arthroplasty. J Arthroplasty 2011;26:870–6.
27. Muniesa JM, Marco E, Tejero M, et al. Analysis of the expectations of elderly patients before undergoing total knee replacement. Arch Gerontol Geriatr 2010;51:E83-E87.
28. Lingard EA, Sledge CB, Learmonth ID. Patient expectations regarding total knee arthroplasty: Differences among the United States, United Kingdom, and Australia. J Bone Joint Surg Am 2006;88:1201–7.
29. Mancuso CA, Graziano S, Briskie LM, et al. Randomized trials to modify patients’ preoperative expectations of hip and knee arthroplasties. Clin Orthopaed Rel Res 2008;466:424–31.
30. de AS, Kallen MA, Amick B, et al. Patients’ expectations about total knee arthroplasty outcomes. Health Expect 2016;19:299–308.
31. Mannion AF, Kampfen S, Munzinger U, Kramers-de Q. The role of patient expectations in predicting outcome after total knee arthroplasty. Arthritis Res Ther 2009;11:R139.
32. Yoo JH, Chang CB, Kang YG, et al. Patient expectations of total knee replacement and their association with sociodemographic factors and functional status. J Bone Joint Surg Br 2011;93:337–44.
33. Ethgen O, Bruyere O, Richy F, et al. Health-related quality of life in total hip and total knee arthroplasty. A qualitative and systematic review of the literature. J Bone Joint Surg Am 2004;86:963–74.
34. Jones CA, Pohar S. Health-related quality of life after total joint arthroplasty: a scoping review. Clin Geriatr Med 2012;28:395–429.
35. Groeneveld PW, Kwoh CK, Mor MK, et al. Racial differences in expectations of joint replacement surgery outcomes. Arthritis Rheum 2008;59:730–7.
36. Scott CEH, Bugler KE, Clement ND, et al. Patient expectations of arthroplasty of the hip and knee. J Bone Joint Surg Br 2012;94:974–81.
37. Smith J, Soon VL, Boyd A, et al. What do Scottish patients expect of their total knee arthroplasty? J Arthroplasty 2016;31:786–92.
38. Nilsdotter AK, Toksvig-Larsen S, Roos EM. Knee arthroplasty: are patients’ expectations fulfilled? A prospective study of pain and function in 102 patients with 5-year follow-up. Acta Orthopaedica 2009;80:55–61.
39. Alarcon GM, Bowling NA, Khazon S. Great expectations: A meta-analytic examination of optimism and hope. Person Ind Diff 2013;54:821–7.
40. Pinto P, McIntyre T, Ferrero R, et al. Predictors of acute postsurgical pain and anxiety following primary total hip and knee arthroplasty. J Pain 2013;14:502–15.
41. Devilly GJ, Borkovec TD. Psychometric properties of the credibility/expectancy questionnaire. J Behav Ther Exp Psychiatry 2000;31:73–86.
42. Haanstra TM, Tilbury C, Kamper SJ, et al. Can optimism, pessimism, hope, treatment credibility and treatment expectancy be distinguished in patients undergoing total hip and total knee arthroplasty? PLoS One 2015;10.
43. Verbeek J, Sengers MJ, Riemens L, Haafkens J.Patient expectations of treatment for back pain: a systematic review of qualitative and quantitative studies. Spine 2004; 29:2309–18.
44. Ghomrawi HM, Mancuso CA, Westrich GH, et al. Discordance in TKA expectations between patients and surgeons. Clin Orthopaed Rel Res 2013;471:175–80.
45. Cordero-Ampuero J, Darder A, Santillana J, et al. Evaluation of patients’ and physicians’ expectations and attributes of osteoarthritis treatment using Kano methodology. Qual Life Res 2012;21:1391–404.
46. Noble PC, Fuller-Lafreniere S, Meftah M, Dwyer MK. Challenges in outcome measurement: discrepancies between patient and provider definitions of success. Clin Orthopaed Rel Res 2013;471:3437–45.
47. Hawker GA, Wright JG, Coyte PC, et al. Determining the need for hip and knee arthroplasty: the role of clinical severity and patients’ preferences. Med Care 2001;39:206–16.
48. Juni P, Dieppe P, Donovan J, et al. Population requirement for primary knee replacement surgery: a cross-sectional study. Rheumatology 2003;42:516–21.
49. Ibrahim SA, Siminoff LA, Burant CJ, Kwoh CK. Differences in expectations of outcome mediate African American/white patient differences in “willingness” to consider joint replacement. Arthritis Rheum 2002;46:2429–35.
50. Ibrahim SA, Hanusa BH, Hannon MJ, et al. Willingness and access to joint replacement among African American patients with knee osteoarthritis: a randomized, controlled intervention. Arthritis Rheum 2013;65:1253–61.
51. Vina ER, Richardson D, Medvedeva E, et al. Does a patient-centered educational intervention affect African-American access to knee replacement? A randomized trial. Clin Orthop Relat Res 2016;474:1755–64.
52. Noble PC, Conditt MA, Cook KF, Mathis KB. The John Insall Award - Patient expectations affect satisfaction with total knee arthroplasty. Clin Orthop Relat Res 2006; 35–43.
53. Robertsson O, Dunbar M, Pehrsson T, et al. Patient satisfaction after knee arthroplasty: a report on 27,372 knees operated on between 1981 and 1995 in Sweden. Acta Orthop Scand 2000;71:262–7.
54. Lau RL, Gandhi R, Mahomed S, Mahomed N. Patient satisfaction after total knee and hip arthroplasty. Clin Geriatr Med 2012;28:349–65.
55. Scott CEH, Howie CR, Macdonald D, Biant LC. Predicting dissatisfaction following total knee replacement. A prospective study of 1217 patients. J Bone Joint Surg Br 2010; 92B:1253–8.
56. Baker PN, van der Meulen JH, Lewsey J, Gregg PJ. The role of pain and function in determining patient satisfaction after total knee replacement. Data from the National Joint Registry for England and Wales. J Bone Joint Surg Br 2007;89:893–900.
57. Khatib Y, Madan A, Naylor JM, Harris IA: Do psychological factors predict poor outcome in patients undergoing TKA? a systematic review. Clin Orthopaed Rel Res 2015;473:2630–8.
58. Adie S, Dao A, Harris IA, et al. Satisfaction with joint replacement in public versus private hospitals: a cohort study. ANZ J Surg 2012;82:616–24.
59. Bourne RB, Chesworth BM, Davis AM, et al. Patient satisfaction after total knee arthroplasty: who is satisfied and who is not? Clin Orthop Relat Res 2010;468:57–63.
60. Harris IA, Harris AM, Naylor et al. Discordance between patient and surgeon satisfaction after total joint arthroplasty. J Arthroplasty 2013;28:722–7.
61. Choi YJ, Ra H. Patient satisfaction after total knee arthroplasty. Knee Surg Relat Res 2016;28:1–15.
62. Williams B. Patient satisfaction: a valid concept? Soc Sci Med 1994;38:509–16.
63. Ware JE Jr, Snyder MK, Wright WR, Davies AR. Defining and measuring patient satisfaction with medical care. Eval Program Plann 1983;6:247–63.
64. Linder-Pelz SU. Toward a theory of patient satisfaction. Soc Sci Med 1982;16:577–82.
65. Hudak PL, Hogg-Johnson S, Bombardier C, et al. Testing a new theory of patient satisfaction with treatment outcome. Med Care 2004;42:726–39.
66. Thompson AG, Sunol R. Expectations as determinants of patient satisfaction: concepts, theory and evidence. Int J Qual Health Care 1995;l7:127–41.
67. Pascoe GC. Patient satisfaction in primary health care: a literature review and analysis. Eval Program Plann 1983;6:185–210.
68. Gandhi R, Davey JR, Mahomed NN. Predicting patient dissatisfaction following joint replacement surgery. J Rheumatol 2008;35:2415–8.
69. Waljee J, McGlinn EP, Sears ED, Chung KC. Patient expectations and patient-reported outcomes in surgery: A systematic review. Surgery 2014;155:799-808.
70. Suda AJ, Seeger JB, Bitsch RG, et al. Are patients’ expectations of hip and knee arthroplasty fulfilled? A prospective study of 130 patients. Orthopedics 2010;33:76.
71. Slover J, Shue J, Koenig K. Shared decision-making in orthopaedic surgery. Clin Orthopaed Rel Res 2012;470:1046–53.
72. Weinstein JN. The missing piece: embracing shared decision making to reform health care. Spine 2000;25:1–4.
73 Aydin D, Klit J, Jacobsen S, et al. No major effects of preoperative education in patients undergoing hip or knee replacement--a systematic review. Dan Med J 2015;62.
74. Spalding NJ. Reducing anxiety by pre-operative education: make the future familiar. Occup Ther Int 2003;10:278–93.
75. Kearney M, Jennrich MK, Lyons S, et al. Effects of preoperative education on patient outcomes after joint replacement surgery. Orthop Nurs 2011;30:391–6.
76. Sherbourne CD, Hays RD, Ordway L, et al. Antecedents of adherence to medical recommendations: results from the Medical Outcomes Study. J Behav Med 1992;15:447–68.
Focus on lifestyle to manage menopause symptoms after breast cancer
Lifestyle modification, rather than hormone therapy, should form the basis for managing estrogen-depletion symptoms and associated clinical problems in breast cancer survivors, according to a review of available evidence.
The review, conducted by the writing group for the Endocrine Society’s guidelines on management of menopausal symptoms, was prompted by the paucity of both randomized controlled trials in breast cancer survivors with estrogen deficiency issues and guidelines that sufficiently focus on treatment of this subgroup of women.

“A large proportion of women experience menopausal symptoms or clinical manifestations of estrogen deficiency during treatment of their breast cancer or after completion of therapy. The specific symptoms and clinical challenges differ based on menopausal status prior to initiation of cancer treatment and therapeutic agents used,” the researchers wrote in a report published in the Journal of Clinical Endocrinology & Metabolism (2017 Aug 2. doi: 10.1210/jc.2017-01138).
For instance, among premenopausal women treated with chemotherapy, ovarian insufficiency, severe menopausal symptoms, and infertility can result. Postmenopasual women treated with aromatase inhibitors may experience arthralgia, accelerated bone loss, and osteoporotic fractures, as well as severe vulvovaginal atrophy, they explained, noting that both premenopausal and postmenopausal survivors can experience moderate-to-severe vasomotor symptoms and sleep disturbance with related fatigue, depressive symptoms, and mood changes.
“Less common problems include weight gain, symptomatic osteoarthritis and intervertebral disk degeneration, degenerative skin changes, radiation and chemotherapy-related cardiovascular disease, and reduced quality of life,” the researchers wrote.
Based on a review of randomized controlled clinical trials, observational studies, evidence-based guidelines, and expert opinion from professional societies, the writing group concluded that individualized lifestyle modifications and nonpharmacologic therapies are recommended for the treatment of these symptoms.
Specifically, the writing group recommended smoking cessation, weight loss when indicated, limited alcohol intake, maintenance of adequate vitamin D and calcium levels, a healthy diet, and regular physical activity for all women with prior breast cancer.
They also recommended nonpharmacologic therapies for vasomotor symptoms, and noted that cognitive behavioral therapy, hypnosis, and acupuncture are among the approaches that may be helpful.
Vaginal lubricants and moisturizers can also be helpful for mild vulvovaginal atrophy, they wrote. For women with more severe symptoms or signs of estrogen deficiency, pharmacologic agents are available to relieve vasomotor symptoms and vulvovaginal atrophy, and to prevent and treat fractures, they wrote, adding that “therapy must be individualized based on each woman’s needs and goals for therapy.”
Among emerging approaches to treatment of symptoms are selective estrogen receptor modulators (SERMs), tissue selective estrogen complex (TSEC) therapy, estetrol, and neurokinin B inhibitors, which show promise for expanding options for symptom relief with less breast cancer risk. However, these have not yet been tested in women with prior breast cancer, the researchers noted.
Dr. Santen reported receiving research funding from Panterhei Bioscience. Other authors received research funding from Therapeutics MD and Lawley Pharmaceuticals, and honoraria from Abbott, Besins Health Care, and Pfizer.
Lifestyle modification, rather than hormone therapy, should form the basis for managing estrogen-depletion symptoms and associated clinical problems in breast cancer survivors, according to a review of available evidence.
The review, conducted by the writing group for the Endocrine Society’s guidelines on management of menopausal symptoms, was prompted by the paucity of both randomized controlled trials in breast cancer survivors with estrogen deficiency issues and guidelines that sufficiently focus on treatment of this subgroup of women.
“A large proportion of women experience menopausal symptoms or clinical manifestations of estrogen deficiency during treatment of their breast cancer or after completion of therapy. The specific symptoms and clinical challenges differ based on menopausal status prior to initiation of cancer treatment and therapeutic agents used,” the researchers wrote in a report published in the Journal of Clinical Endocrinology & Metabolism (2017 Aug 2. doi: 10.1210/jc.2017-01138).
For instance, among premenopausal women treated with chemotherapy, ovarian insufficiency, severe menopausal symptoms, and infertility can result. Postmenopasual women treated with aromatase inhibitors may experience arthralgia, accelerated bone loss, and osteoporotic fractures, as well as severe vulvovaginal atrophy, they explained, noting that both premenopausal and postmenopausal survivors can experience moderate-to-severe vasomotor symptoms and sleep disturbance with related fatigue, depressive symptoms, and mood changes.
“Less common problems include weight gain, symptomatic osteoarthritis and intervertebral disk degeneration, degenerative skin changes, radiation and chemotherapy-related cardiovascular disease, and reduced quality of life,” the researchers wrote.
Based on a review of randomized controlled clinical trials, observational studies, evidence-based guidelines, and expert opinion from professional societies, the writing group concluded that individualized lifestyle modifications and nonpharmacologic therapies are recommended for the treatment of these symptoms.
Specifically, the writing group recommended smoking cessation, weight loss when indicated, limited alcohol intake, maintenance of adequate vitamin D and calcium levels, a healthy diet, and regular physical activity for all women with prior breast cancer.
They also recommended nonpharmacologic therapies for vasomotor symptoms, and noted that cognitive behavioral therapy, hypnosis, and acupuncture are among the approaches that may be helpful.
Vaginal lubricants and moisturizers can also be helpful for mild vulvovaginal atrophy, they wrote. For women with more severe symptoms or signs of estrogen deficiency, pharmacologic agents are available to relieve vasomotor symptoms and vulvovaginal atrophy, and to prevent and treat fractures, they wrote, adding that “therapy must be individualized based on each woman’s needs and goals for therapy.”
Among emerging approaches to treatment of symptoms are selective estrogen receptor modulators (SERMs), tissue selective estrogen complex (TSEC) therapy, estetrol, and neurokinin B inhibitors, which show promise for expanding options for symptom relief with less breast cancer risk. However, these have not yet been tested in women with prior breast cancer, the researchers noted.
Dr. Santen reported receiving research funding from Panterhei Bioscience. Other authors received research funding from Therapeutics MD and Lawley Pharmaceuticals, and honoraria from Abbott, Besins Health Care, and Pfizer.
Lifestyle modification, rather than hormone therapy, should form the basis for managing estrogen-depletion symptoms and associated clinical problems in breast cancer survivors, according to a review of available evidence.
The review, conducted by the writing group for the Endocrine Society’s guidelines on management of menopausal symptoms, was prompted by the paucity of both randomized controlled trials in breast cancer survivors with estrogen deficiency issues and guidelines that sufficiently focus on treatment of this subgroup of women.
“A large proportion of women experience menopausal symptoms or clinical manifestations of estrogen deficiency during treatment of their breast cancer or after completion of therapy. The specific symptoms and clinical challenges differ based on menopausal status prior to initiation of cancer treatment and therapeutic agents used,” the researchers wrote in a report published in the Journal of Clinical Endocrinology & Metabolism (2017 Aug 2. doi: 10.1210/jc.2017-01138).
For instance, among premenopausal women treated with chemotherapy, ovarian insufficiency, severe menopausal symptoms, and infertility can result. Postmenopasual women treated with aromatase inhibitors may experience arthralgia, accelerated bone loss, and osteoporotic fractures, as well as severe vulvovaginal atrophy, they explained, noting that both premenopausal and postmenopausal survivors can experience moderate-to-severe vasomotor symptoms and sleep disturbance with related fatigue, depressive symptoms, and mood changes.
“Less common problems include weight gain, symptomatic osteoarthritis and intervertebral disk degeneration, degenerative skin changes, radiation and chemotherapy-related cardiovascular disease, and reduced quality of life,” the researchers wrote.
Based on a review of randomized controlled clinical trials, observational studies, evidence-based guidelines, and expert opinion from professional societies, the writing group concluded that individualized lifestyle modifications and nonpharmacologic therapies are recommended for the treatment of these symptoms.
Specifically, the writing group recommended smoking cessation, weight loss when indicated, limited alcohol intake, maintenance of adequate vitamin D and calcium levels, a healthy diet, and regular physical activity for all women with prior breast cancer.
They also recommended nonpharmacologic therapies for vasomotor symptoms, and noted that cognitive behavioral therapy, hypnosis, and acupuncture are among the approaches that may be helpful.
Vaginal lubricants and moisturizers can also be helpful for mild vulvovaginal atrophy, they wrote. For women with more severe symptoms or signs of estrogen deficiency, pharmacologic agents are available to relieve vasomotor symptoms and vulvovaginal atrophy, and to prevent and treat fractures, they wrote, adding that “therapy must be individualized based on each woman’s needs and goals for therapy.”
Among emerging approaches to treatment of symptoms are selective estrogen receptor modulators (SERMs), tissue selective estrogen complex (TSEC) therapy, estetrol, and neurokinin B inhibitors, which show promise for expanding options for symptom relief with less breast cancer risk. However, these have not yet been tested in women with prior breast cancer, the researchers noted.
Dr. Santen reported receiving research funding from Panterhei Bioscience. Other authors received research funding from Therapeutics MD and Lawley Pharmaceuticals, and honoraria from Abbott, Besins Health Care, and Pfizer.
FROM THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM
Developing vaccines against enterovirus-A71 called a priority
MADRID – Is there a need for an enterovirus-A71 vaccine?
This is a new question for North American and European physicians, but not so new in Asia.
“China says yes, with more than 15 million cases of hand, foot, and mouth disease resulting in 3,500 deaths since surveillance started in 2009,” Heli Harvala, MD, said at the annual meeting of the European Society for Paediatric Infectious Diseases.
Seroconversion rates 28 days after the second dose of these vaccines, both directed specifically against viral subgenotype C-4, are 92%-100%. Vaccine efficacy is 91%-97%, according to Dr. Harvala, a consultant medical virologist at University College London.
It remains a mystery why major outbreaks of severe EV-A71 disease have mostly occurred in Asia, with the notable exception of a Spanish outbreak of EV-A71 encephalitis in 2016. The possibility of much wider spread is concerning.
The Chinese monovalent EV-A71 vaccines, however, are seen as a stopgap. For one thing, recent evidence suggests that it’s probably not the specific EV-A71 C-4 viral subgenotype that accounts for all severe disease.
“I think we have to aim for a multivalent vaccine,” Dr. Harvala said.
Now in clinical trials, investigational bivalent vaccines are directed against other EV-A71 subgenotypes in addition to C-4, and also against another enterovirus, coxsackievirus serotype A16, the most common cause of classic hand, foot, and mouth disease in the United States. But that’s probably not enough, according to Dr. Harvala. She noted that coxsackievirus A6, which was first identified more than 50 years ago, abruptly became the main cause of mild hand, foot, and mouth disease in China in 2013 and again in 2015. Moreover, its role in severe cases is growing, and there have been important outbreaks in the United States in recent years. These severe cases come in three main presentations, resembling either erythema multiforme, chicken pox, or eczema herpeticum.
Dr. Harvala added that a next-generation vaccine probably also should offer protection against enterovirus-D68. In 2014, there were 1,153 laboratory-confirmed EV-D68 infections and 14 deaths in the United States and Canada. This infection poses a diagnostic challenge: while the virus is readily detectable on throat swabs, it’s only rarely present in stool or cerebrospinal fluid samples.
“It’s important to keep in mind that this infection is still underdiagnosed. We are not really looking for it,” she said.
No specific treatment for enterovirus infections is available. Three capsid-binding antiviral agents now are in clinical trials: pleconaril, vapendavir, and pocapavir. In addition, translational studies have demonstrated that the SSRI fluoxetine inhibits enterovirus replication, but there have been no clinical trials as yet.
Although development of antivirals effective against enterovirus is an active area of research, Dr. Harvala thinks drug resistance will be an issue, underscoring the importance of vaccine development.
She reported having no financial conflicts of interest regarding her presentation.
MADRID – Is there a need for an enterovirus-A71 vaccine?
This is a new question for North American and European physicians, but not so new in Asia.
“China says yes, with more than 15 million cases of hand, foot, and mouth disease resulting in 3,500 deaths since surveillance started in 2009,” Heli Harvala, MD, said at the annual meeting of the European Society for Paediatric Infectious Diseases.
Seroconversion rates 28 days after the second dose of these vaccines, both directed specifically against viral subgenotype C-4, are 92%-100%. Vaccine efficacy is 91%-97%, according to Dr. Harvala, a consultant medical virologist at University College London.
It remains a mystery why major outbreaks of severe EV-A71 disease have mostly occurred in Asia, with the notable exception of a Spanish outbreak of EV-A71 encephalitis in 2016. The possibility of much wider spread is concerning.
The Chinese monovalent EV-A71 vaccines, however, are seen as a stopgap. For one thing, recent evidence suggests that it’s probably not the specific EV-A71 C-4 viral subgenotype that accounts for all severe disease.
“I think we have to aim for a multivalent vaccine,” Dr. Harvala said.
Now in clinical trials, investigational bivalent vaccines are directed against other EV-A71 subgenotypes in addition to C-4, and also against another enterovirus, coxsackievirus serotype A16, the most common cause of classic hand, foot, and mouth disease in the United States. But that’s probably not enough, according to Dr. Harvala. She noted that coxsackievirus A6, which was first identified more than 50 years ago, abruptly became the main cause of mild hand, foot, and mouth disease in China in 2013 and again in 2015. Moreover, its role in severe cases is growing, and there have been important outbreaks in the United States in recent years. These severe cases come in three main presentations, resembling either erythema multiforme, chicken pox, or eczema herpeticum.
Dr. Harvala added that a next-generation vaccine probably also should offer protection against enterovirus-D68. In 2014, there were 1,153 laboratory-confirmed EV-D68 infections and 14 deaths in the United States and Canada. This infection poses a diagnostic challenge: while the virus is readily detectable on throat swabs, it’s only rarely present in stool or cerebrospinal fluid samples.
“It’s important to keep in mind that this infection is still underdiagnosed. We are not really looking for it,” she said.
No specific treatment for enterovirus infections is available. Three capsid-binding antiviral agents now are in clinical trials: pleconaril, vapendavir, and pocapavir. In addition, translational studies have demonstrated that the SSRI fluoxetine inhibits enterovirus replication, but there have been no clinical trials as yet.
Although development of antivirals effective against enterovirus is an active area of research, Dr. Harvala thinks drug resistance will be an issue, underscoring the importance of vaccine development.
She reported having no financial conflicts of interest regarding her presentation.
MADRID – Is there a need for an enterovirus-A71 vaccine?
This is a new question for North American and European physicians, but not so new in Asia.
“China says yes, with more than 15 million cases of hand, foot, and mouth disease resulting in 3,500 deaths since surveillance started in 2009,” Heli Harvala, MD, said at the annual meeting of the European Society for Paediatric Infectious Diseases.
Seroconversion rates 28 days after the second dose of these vaccines, both directed specifically against viral subgenotype C-4, are 92%-100%. Vaccine efficacy is 91%-97%, according to Dr. Harvala, a consultant medical virologist at University College London.
It remains a mystery why major outbreaks of severe EV-A71 disease have mostly occurred in Asia, with the notable exception of a Spanish outbreak of EV-A71 encephalitis in 2016. The possibility of much wider spread is concerning.
The Chinese monovalent EV-A71 vaccines, however, are seen as a stopgap. For one thing, recent evidence suggests that it’s probably not the specific EV-A71 C-4 viral subgenotype that accounts for all severe disease.
“I think we have to aim for a multivalent vaccine,” Dr. Harvala said.
Now in clinical trials, investigational bivalent vaccines are directed against other EV-A71 subgenotypes in addition to C-4, and also against another enterovirus, coxsackievirus serotype A16, the most common cause of classic hand, foot, and mouth disease in the United States. But that’s probably not enough, according to Dr. Harvala. She noted that coxsackievirus A6, which was first identified more than 50 years ago, abruptly became the main cause of mild hand, foot, and mouth disease in China in 2013 and again in 2015. Moreover, its role in severe cases is growing, and there have been important outbreaks in the United States in recent years. These severe cases come in three main presentations, resembling either erythema multiforme, chicken pox, or eczema herpeticum.
Dr. Harvala added that a next-generation vaccine probably also should offer protection against enterovirus-D68. In 2014, there were 1,153 laboratory-confirmed EV-D68 infections and 14 deaths in the United States and Canada. This infection poses a diagnostic challenge: while the virus is readily detectable on throat swabs, it’s only rarely present in stool or cerebrospinal fluid samples.
“It’s important to keep in mind that this infection is still underdiagnosed. We are not really looking for it,” she said.
No specific treatment for enterovirus infections is available. Three capsid-binding antiviral agents now are in clinical trials: pleconaril, vapendavir, and pocapavir. In addition, translational studies have demonstrated that the SSRI fluoxetine inhibits enterovirus replication, but there have been no clinical trials as yet.
Although development of antivirals effective against enterovirus is an active area of research, Dr. Harvala thinks drug resistance will be an issue, underscoring the importance of vaccine development.
She reported having no financial conflicts of interest regarding her presentation.
EXPERT ANALYSIS FROM ESPID 2017
DoD Delays Transgender Ban Pending New Study
Following the directive of the “Military Service by Transgender Individuals” Presidential Memorandum to reinstitute a ban on service for transgender service members, the Secretary of Defense has called for a new study of the impact of transgender service members on “military readiness, lethality, and unit cohesion, with due regard for budgetary constraints and consistent with applicable law.” Secretary of Defense Jim Mattis announced that he will “establish a panel of experts serving within the Departments of Defense and Homeland Security to provide advice and recommendations on the implementation of the president’s direction.”
The study follows on the heels of a July 2016 study issued by the RAND Corporation, which found that there are between 1,300 to 6,600 transgender active duty service members. According to the study, the “costs of gender transition-related health care are relatively low” and they had a “minimal” impact on force readiness.
The new study is due to President Trump by February 21, 2018 and is required to include a plan for implementing the ban. The potential ban on transgender service members is still set to go into effect March 23, 2018. However, there is some disagreement over whether the Presidential Memorandum leave the DoD with latitude to protect currently serving transgender service members or not.
The expected health care costs associated with transgender service members remains a significant factor in the policy decision. The RAND study estimated that health care costs would increase by between $2.4 million and $8.4 million annually with transgender service members out of an estimated $6.2 billion spent on active component health care spending, which would represent a 0.04% to 0.13% of the budget.
Following the directive of the “Military Service by Transgender Individuals” Presidential Memorandum to reinstitute a ban on service for transgender service members, the Secretary of Defense has called for a new study of the impact of transgender service members on “military readiness, lethality, and unit cohesion, with due regard for budgetary constraints and consistent with applicable law.” Secretary of Defense Jim Mattis announced that he will “establish a panel of experts serving within the Departments of Defense and Homeland Security to provide advice and recommendations on the implementation of the president’s direction.”
The study follows on the heels of a July 2016 study issued by the RAND Corporation, which found that there are between 1,300 to 6,600 transgender active duty service members. According to the study, the “costs of gender transition-related health care are relatively low” and they had a “minimal” impact on force readiness.
The new study is due to President Trump by February 21, 2018 and is required to include a plan for implementing the ban. The potential ban on transgender service members is still set to go into effect March 23, 2018. However, there is some disagreement over whether the Presidential Memorandum leave the DoD with latitude to protect currently serving transgender service members or not.
The expected health care costs associated with transgender service members remains a significant factor in the policy decision. The RAND study estimated that health care costs would increase by between $2.4 million and $8.4 million annually with transgender service members out of an estimated $6.2 billion spent on active component health care spending, which would represent a 0.04% to 0.13% of the budget.
Following the directive of the “Military Service by Transgender Individuals” Presidential Memorandum to reinstitute a ban on service for transgender service members, the Secretary of Defense has called for a new study of the impact of transgender service members on “military readiness, lethality, and unit cohesion, with due regard for budgetary constraints and consistent with applicable law.” Secretary of Defense Jim Mattis announced that he will “establish a panel of experts serving within the Departments of Defense and Homeland Security to provide advice and recommendations on the implementation of the president’s direction.”
The study follows on the heels of a July 2016 study issued by the RAND Corporation, which found that there are between 1,300 to 6,600 transgender active duty service members. According to the study, the “costs of gender transition-related health care are relatively low” and they had a “minimal” impact on force readiness.
The new study is due to President Trump by February 21, 2018 and is required to include a plan for implementing the ban. The potential ban on transgender service members is still set to go into effect March 23, 2018. However, there is some disagreement over whether the Presidential Memorandum leave the DoD with latitude to protect currently serving transgender service members or not.
The expected health care costs associated with transgender service members remains a significant factor in the policy decision. The RAND study estimated that health care costs would increase by between $2.4 million and $8.4 million annually with transgender service members out of an estimated $6.2 billion spent on active component health care spending, which would represent a 0.04% to 0.13% of the budget.
Waging war against medical misinformation
When Jennifer Gunter, MD, learned that Marie Claire magazine published an article suggesting that the recent solar eclipse affected menstrual cycles, she turned to social media to dismantle that assertion.
“If the new moon were associated with menstruation, then all women all over the world would be menstruating at the same time,” Dr. Gunter wrote in an Aug. 21, 2017 entry on her personal blog, which includes the slogan “Wielding the Lasso of Truth.” “Have none of these people ever considered that? Maybe that happens in the “Mists of Avalon,” but it doesn’t happen on Earth.”

Disputing such claims “keeps me pretty busy,” said Dr. Gunter, who’s become a hero to many in the ob.gyn. community, with more than 55,000 followers on Twitter and about 5,000 subscribers to her blog, which she launched in 2010.
“When I go to medical conferences, I get lots of physicians high-fiving me and thanking me, and that’s really nice,” she said. “They’re just like me. They’ve sat with patients and tried to explain how bioidentical hormones aren’t ground up yams and why salivary hormone testing is not recommended. They tell me that they’ve taken my blog posts and turned them into handouts. That’s the kind of thing that keeps me going: knowing that I’ve written something that’s going to help somebody else.”
Dr. Gunter, who practices gynecology in San Francisco, said that she established a social media presence because she’s compelled to wage war against medical misinformation, especially content marketed to women. “I can’t stand seeing websites that tell people they can lose weight doing this new fad or by taking certain supplements,” the Winnipeg, Canada, native said. “That’s the same snake oil that used to go town to town in the 1800s. It’s not any different; it’s just on a larger scale. Misinformation makes me sad. If people are making informed choices that’s one thing, but if they are hooked in by fancy websites with attractive celebrities and doctors branding supplements with their names, that bothers me. The Internet is only as good as the information you put into it. When you search for something, it’s really a shame that an accurate website isn’t coming up first, but it’s all driven by page clicks. What’s surprised me the most has been that people were ready for a voice like mine to stand up. I think the same feelings I’ve had have been brewing for a lot of people.”
Dr. Gunter devotes about 45 minutes every weekday to social media. Inspiration for her blog posts and her tweets on @DrJenGunter come from a variety of sources, often from followers who send her links to articles about wild medical claims. “I might post on my lunch hour but never during the work day,” she said. “A lot of people like to unwind at the end of the day by watching TV. I’m divorced and my kids are with their dad half the time so on the evenings when I’m by myself, if I’ve read something interesting, I’ll tweet about it.”
She shared the following tips for ob.gyns. looking to establish a presence on social media:
- Be HIPAA compliant. “I don’t think I’ve ever used my work day to inform what I write about because there’s so much going on in the news,” she said. “You have to be HIPAA compliant because your patients may be following you online. In many ways, I hope that people I’m going to care for do read some of the things that I put out there because I’m trying to give good health information.”
- Be authentic. “When people meet me and have probably read some of my tweets and have heard me talk, they say they can match the voice with the person,” Dr. Gunter said. “People can tell when you’re not being authentic. That doesn’t work for me. I don’t have time to invent another persona!”
- Share articles you think are worthwhile. “If I have a friend who is in the tech industry, and if he posts an article on that topic, I figure he curated that, so maybe I’ll read it,” she said. “Likewise, if you’re a doctor and you have people following you on Facebook who are not physicians, they might say, ‘My friend who I went to high school with who’s a doctor thinks this New York Times article is worth reading. Maybe I should read it.’ People can lurk and share things, and that helps. I follow lots of doctors on Twitter that I don’t regularly engage with, but they post articles, and I’ll read them.”
- Make it clear who you’re speaking for. Are you speaking for your employer or for yourself? Are you giving medical advice, or is the content your opinion only? Include an appropriate disclaimer in your profile. Dr. Gunter’s Twitter profile reads: “Appropriately Confident OB/GYN, Canadian Spice, Sexpert, Lasso of Truth, Pegasister, Not medical advice, I speak for no one but me.”
- If you post regularly, expect criticism at some point. “If social media is not for you, you shouldn’t do it,” Dr. Gunter said. “But if you have a small Facebook account, why not share some good quality articles that you found interesting? You’d be surprised. What you might think is a throwaway message might be of real help to someone who doesn’t practice medicine. There’s a real hunger for good quality curated information.”
When Jennifer Gunter, MD, learned that Marie Claire magazine published an article suggesting that the recent solar eclipse affected menstrual cycles, she turned to social media to dismantle that assertion.
“If the new moon were associated with menstruation, then all women all over the world would be menstruating at the same time,” Dr. Gunter wrote in an Aug. 21, 2017 entry on her personal blog, which includes the slogan “Wielding the Lasso of Truth.” “Have none of these people ever considered that? Maybe that happens in the “Mists of Avalon,” but it doesn’t happen on Earth.”
Disputing such claims “keeps me pretty busy,” said Dr. Gunter, who’s become a hero to many in the ob.gyn. community, with more than 55,000 followers on Twitter and about 5,000 subscribers to her blog, which she launched in 2010.
“When I go to medical conferences, I get lots of physicians high-fiving me and thanking me, and that’s really nice,” she said. “They’re just like me. They’ve sat with patients and tried to explain how bioidentical hormones aren’t ground up yams and why salivary hormone testing is not recommended. They tell me that they’ve taken my blog posts and turned them into handouts. That’s the kind of thing that keeps me going: knowing that I’ve written something that’s going to help somebody else.”
Dr. Gunter, who practices gynecology in San Francisco, said that she established a social media presence because she’s compelled to wage war against medical misinformation, especially content marketed to women. “I can’t stand seeing websites that tell people they can lose weight doing this new fad or by taking certain supplements,” the Winnipeg, Canada, native said. “That’s the same snake oil that used to go town to town in the 1800s. It’s not any different; it’s just on a larger scale. Misinformation makes me sad. If people are making informed choices that’s one thing, but if they are hooked in by fancy websites with attractive celebrities and doctors branding supplements with their names, that bothers me. The Internet is only as good as the information you put into it. When you search for something, it’s really a shame that an accurate website isn’t coming up first, but it’s all driven by page clicks. What’s surprised me the most has been that people were ready for a voice like mine to stand up. I think the same feelings I’ve had have been brewing for a lot of people.”
Dr. Gunter devotes about 45 minutes every weekday to social media. Inspiration for her blog posts and her tweets on @DrJenGunter come from a variety of sources, often from followers who send her links to articles about wild medical claims. “I might post on my lunch hour but never during the work day,” she said. “A lot of people like to unwind at the end of the day by watching TV. I’m divorced and my kids are with their dad half the time so on the evenings when I’m by myself, if I’ve read something interesting, I’ll tweet about it.”
She shared the following tips for ob.gyns. looking to establish a presence on social media:
- Be HIPAA compliant. “I don’t think I’ve ever used my work day to inform what I write about because there’s so much going on in the news,” she said. “You have to be HIPAA compliant because your patients may be following you online. In many ways, I hope that people I’m going to care for do read some of the things that I put out there because I’m trying to give good health information.”
- Be authentic. “When people meet me and have probably read some of my tweets and have heard me talk, they say they can match the voice with the person,” Dr. Gunter said. “People can tell when you’re not being authentic. That doesn’t work for me. I don’t have time to invent another persona!”
- Share articles you think are worthwhile. “If I have a friend who is in the tech industry, and if he posts an article on that topic, I figure he curated that, so maybe I’ll read it,” she said. “Likewise, if you’re a doctor and you have people following you on Facebook who are not physicians, they might say, ‘My friend who I went to high school with who’s a doctor thinks this New York Times article is worth reading. Maybe I should read it.’ People can lurk and share things, and that helps. I follow lots of doctors on Twitter that I don’t regularly engage with, but they post articles, and I’ll read them.”
- Make it clear who you’re speaking for. Are you speaking for your employer or for yourself? Are you giving medical advice, or is the content your opinion only? Include an appropriate disclaimer in your profile. Dr. Gunter’s Twitter profile reads: “Appropriately Confident OB/GYN, Canadian Spice, Sexpert, Lasso of Truth, Pegasister, Not medical advice, I speak for no one but me.”
- If you post regularly, expect criticism at some point. “If social media is not for you, you shouldn’t do it,” Dr. Gunter said. “But if you have a small Facebook account, why not share some good quality articles that you found interesting? You’d be surprised. What you might think is a throwaway message might be of real help to someone who doesn’t practice medicine. There’s a real hunger for good quality curated information.”
When Jennifer Gunter, MD, learned that Marie Claire magazine published an article suggesting that the recent solar eclipse affected menstrual cycles, she turned to social media to dismantle that assertion.
“If the new moon were associated with menstruation, then all women all over the world would be menstruating at the same time,” Dr. Gunter wrote in an Aug. 21, 2017 entry on her personal blog, which includes the slogan “Wielding the Lasso of Truth.” “Have none of these people ever considered that? Maybe that happens in the “Mists of Avalon,” but it doesn’t happen on Earth.”
Disputing such claims “keeps me pretty busy,” said Dr. Gunter, who’s become a hero to many in the ob.gyn. community, with more than 55,000 followers on Twitter and about 5,000 subscribers to her blog, which she launched in 2010.
“When I go to medical conferences, I get lots of physicians high-fiving me and thanking me, and that’s really nice,” she said. “They’re just like me. They’ve sat with patients and tried to explain how bioidentical hormones aren’t ground up yams and why salivary hormone testing is not recommended. They tell me that they’ve taken my blog posts and turned them into handouts. That’s the kind of thing that keeps me going: knowing that I’ve written something that’s going to help somebody else.”
Dr. Gunter, who practices gynecology in San Francisco, said that she established a social media presence because she’s compelled to wage war against medical misinformation, especially content marketed to women. “I can’t stand seeing websites that tell people they can lose weight doing this new fad or by taking certain supplements,” the Winnipeg, Canada, native said. “That’s the same snake oil that used to go town to town in the 1800s. It’s not any different; it’s just on a larger scale. Misinformation makes me sad. If people are making informed choices that’s one thing, but if they are hooked in by fancy websites with attractive celebrities and doctors branding supplements with their names, that bothers me. The Internet is only as good as the information you put into it. When you search for something, it’s really a shame that an accurate website isn’t coming up first, but it’s all driven by page clicks. What’s surprised me the most has been that people were ready for a voice like mine to stand up. I think the same feelings I’ve had have been brewing for a lot of people.”
Dr. Gunter devotes about 45 minutes every weekday to social media. Inspiration for her blog posts and her tweets on @DrJenGunter come from a variety of sources, often from followers who send her links to articles about wild medical claims. “I might post on my lunch hour but never during the work day,” she said. “A lot of people like to unwind at the end of the day by watching TV. I’m divorced and my kids are with their dad half the time so on the evenings when I’m by myself, if I’ve read something interesting, I’ll tweet about it.”
She shared the following tips for ob.gyns. looking to establish a presence on social media:
- Be HIPAA compliant. “I don’t think I’ve ever used my work day to inform what I write about because there’s so much going on in the news,” she said. “You have to be HIPAA compliant because your patients may be following you online. In many ways, I hope that people I’m going to care for do read some of the things that I put out there because I’m trying to give good health information.”
- Be authentic. “When people meet me and have probably read some of my tweets and have heard me talk, they say they can match the voice with the person,” Dr. Gunter said. “People can tell when you’re not being authentic. That doesn’t work for me. I don’t have time to invent another persona!”
- Share articles you think are worthwhile. “If I have a friend who is in the tech industry, and if he posts an article on that topic, I figure he curated that, so maybe I’ll read it,” she said. “Likewise, if you’re a doctor and you have people following you on Facebook who are not physicians, they might say, ‘My friend who I went to high school with who’s a doctor thinks this New York Times article is worth reading. Maybe I should read it.’ People can lurk and share things, and that helps. I follow lots of doctors on Twitter that I don’t regularly engage with, but they post articles, and I’ll read them.”
- Make it clear who you’re speaking for. Are you speaking for your employer or for yourself? Are you giving medical advice, or is the content your opinion only? Include an appropriate disclaimer in your profile. Dr. Gunter’s Twitter profile reads: “Appropriately Confident OB/GYN, Canadian Spice, Sexpert, Lasso of Truth, Pegasister, Not medical advice, I speak for no one but me.”
- If you post regularly, expect criticism at some point. “If social media is not for you, you shouldn’t do it,” Dr. Gunter said. “But if you have a small Facebook account, why not share some good quality articles that you found interesting? You’d be surprised. What you might think is a throwaway message might be of real help to someone who doesn’t practice medicine. There’s a real hunger for good quality curated information.”