User login
Obesity is a risk factor for recurrence in aromatase inhibitor-treated HR+ BC
Key clinical point: Obesity increased the risk for recurrence in postmenopausal women with early-stage hormone receptor-positive (HR+) breast cancer (BC) who were treated with aromatase inhibitors (AI).
Major finding: Among patients with AI-treated HR+ BC, the risk for BC recurrence was significantly higher in those with obesity (adjusted hazard ratio [aHR] 1.18; 95% CI 1.01-1.37) and severe obesity (aHR 1.32; 95% CI 1.08-1.62) than in those with a healthy weight.
Study details: Findings are from a nationwide cohort study including 13,230 postmenopausal women with stages I-III HR+ BC who received AI.
Disclosures: This study was supported by the Jeppe Juhl Memorial Foundation, Denmark, and other Danish sources. Jensen MR declared receiving from Novartis meeting expenses and personal fees unrelated to this study.
Source: Harborg S et al. Obesity and risk of recurrence in patients with breast cancer treated with aromatase inhibitors. JAMA Netw Open. 2023;6(10):e2337780 (Oct 13). doi: 10.1001/jamanetworkopen.2023.37780
Key clinical point: Obesity increased the risk for recurrence in postmenopausal women with early-stage hormone receptor-positive (HR+) breast cancer (BC) who were treated with aromatase inhibitors (AI).
Major finding: Among patients with AI-treated HR+ BC, the risk for BC recurrence was significantly higher in those with obesity (adjusted hazard ratio [aHR] 1.18; 95% CI 1.01-1.37) and severe obesity (aHR 1.32; 95% CI 1.08-1.62) than in those with a healthy weight.
Study details: Findings are from a nationwide cohort study including 13,230 postmenopausal women with stages I-III HR+ BC who received AI.
Disclosures: This study was supported by the Jeppe Juhl Memorial Foundation, Denmark, and other Danish sources. Jensen MR declared receiving from Novartis meeting expenses and personal fees unrelated to this study.
Source: Harborg S et al. Obesity and risk of recurrence in patients with breast cancer treated with aromatase inhibitors. JAMA Netw Open. 2023;6(10):e2337780 (Oct 13). doi: 10.1001/jamanetworkopen.2023.37780
Key clinical point: Obesity increased the risk for recurrence in postmenopausal women with early-stage hormone receptor-positive (HR+) breast cancer (BC) who were treated with aromatase inhibitors (AI).
Major finding: Among patients with AI-treated HR+ BC, the risk for BC recurrence was significantly higher in those with obesity (adjusted hazard ratio [aHR] 1.18; 95% CI 1.01-1.37) and severe obesity (aHR 1.32; 95% CI 1.08-1.62) than in those with a healthy weight.
Study details: Findings are from a nationwide cohort study including 13,230 postmenopausal women with stages I-III HR+ BC who received AI.
Disclosures: This study was supported by the Jeppe Juhl Memorial Foundation, Denmark, and other Danish sources. Jensen MR declared receiving from Novartis meeting expenses and personal fees unrelated to this study.
Source: Harborg S et al. Obesity and risk of recurrence in patients with breast cancer treated with aromatase inhibitors. JAMA Netw Open. 2023;6(10):e2337780 (Oct 13). doi: 10.1001/jamanetworkopen.2023.37780
The future of medicine is RNA
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Every once in a while, medicine changes in a fundamental way, and we may not realize it while it’s happening. I wasn’t around in 1928 when Fleming discovered penicillin; or in 1953 when Watson, Crick, and Franklin characterized the double-helical structure of DNA.
But looking at medicine today, there are essentially two places where I think we will see, in retrospect, that we were at a fundamental turning point. One is artificial intelligence, which gets so much attention and hype that I will simply say yes, this will change things, stay tuned.
The other is a bit more obscure, but I suspect it may be just as impactful. That other thing is

I want to start with the idea that many diseases are, fundamentally, a problem of proteins. In some cases, like hypercholesterolemia, the body produces too much protein; in others, like hemophilia, too little.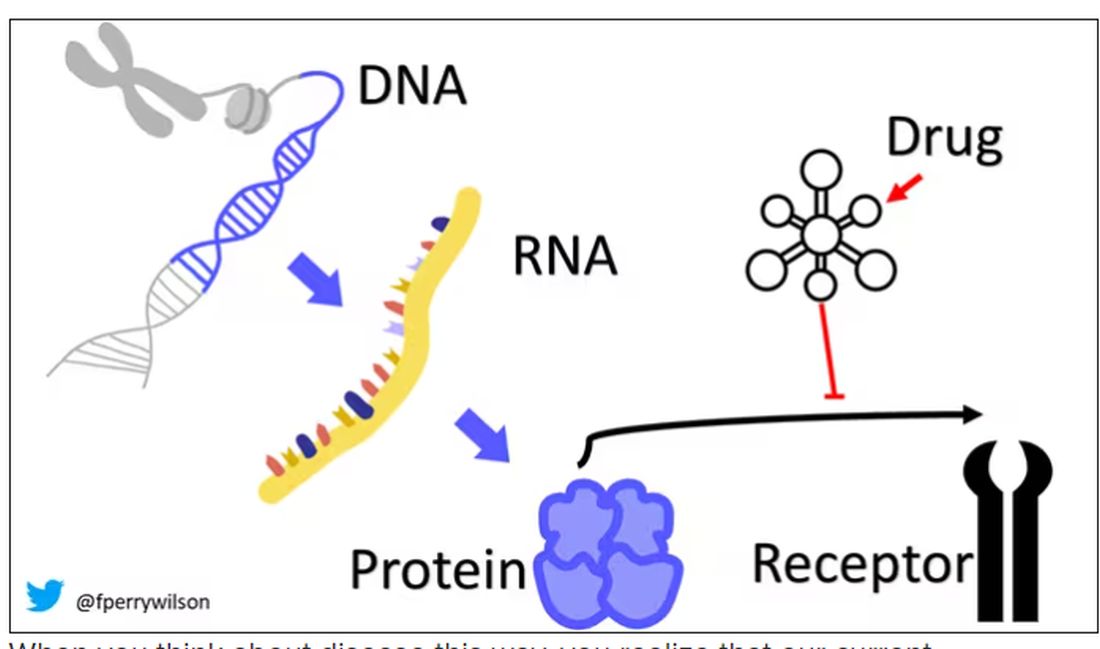
When you think about disease this way, you realize that our current medications take effect late in the disease game. We have these molecules that try to block a protein from its receptor, prevent a protein from cleaving another protein, or increase the rate that a protein is broken down. It’s all distal to the fundamental problem: the production of the bad protein in the first place.
Enter small inhibitory RNAs, or siRNAs for short, discovered in 1998 by Andrew Fire and Craig Mello at UMass Worcester. The two won the Nobel prize in medicine just 8 years later; that’s a really short time, highlighting just how important this discovery was. In contrast, Karikó and Weissman won the Nobel for mRNA vaccines this year, after inventing them 18 years ago.
siRNAs are the body’s way of targeting proteins for destruction before they are ever created. About 20 base pairs long, siRNAs seek out a complementary target mRNA, attach to it, and call in a group of proteins to destroy it. With the target mRNA gone, no protein can be created.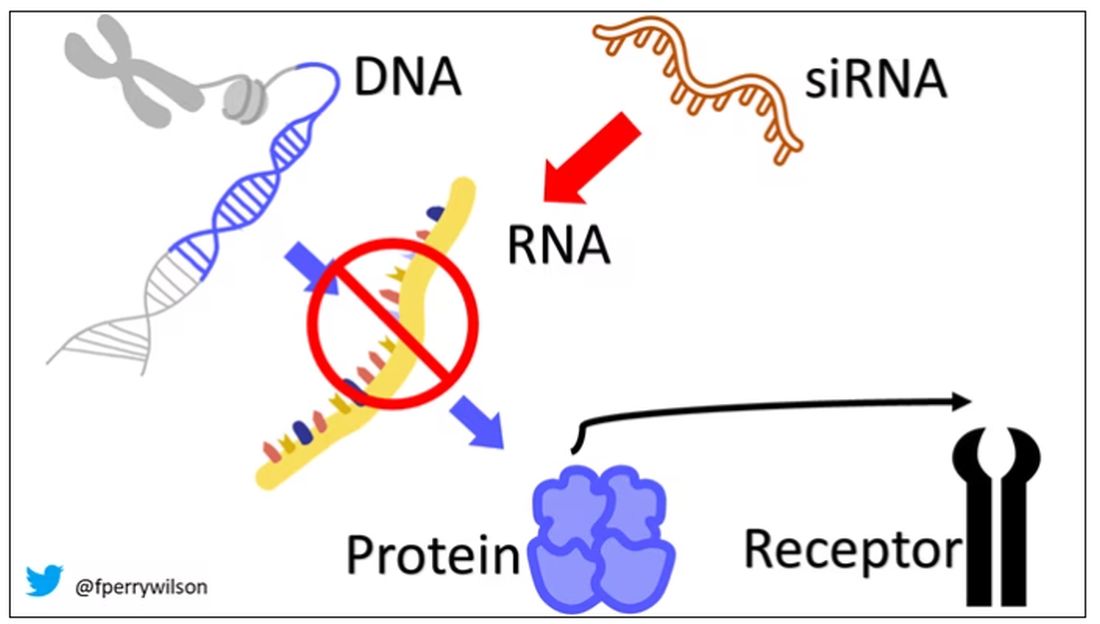
You see where this is going, right? How does high cholesterol kill you? Proteins. How does Staphylococcus aureus kill you? Proteins. Even viruses can’t replicate if their RNA is prevented from being turned into proteins.
So, how do we use siRNAs? A new paper appearing in JAMA describes a fairly impressive use case.
The background here is that higher levels of lipoprotein(a), an LDL-like protein, are associated with cardiovascular disease, heart attack, and stroke. But unfortunately, statins really don’t have any effect on lipoprotein(a) levels. Neither does diet. Your lipoprotein(a) level seems to be more or less hard-coded genetically.
So, what if we stop the genetic machinery from working? Enter lepodisiran, a drug from Eli Lilly. Unlike so many other medications, which are usually found in nature, purified, and synthesized, lepodisiran was created from scratch. It’s not hard. Thanks to the Human Genome Project, we know the genetic code for lipoprotein(a), so inventing an siRNA to target it specifically is trivial. That’s one of the key features of siRNA – you don’t have to find a chemical that binds strongly to some protein receptor, and worry about the off-target effects and all that nonsense. You just pick a protein you want to suppress and you suppress it.
Okay, it’s not that simple. siRNA is broken down very quickly by the body, so it needs to be targeted to the organ of interest – in this case, the liver, since that is where lipoprotein(a) is synthesized. Lepodisiran is targeted to the liver by this special targeting label here.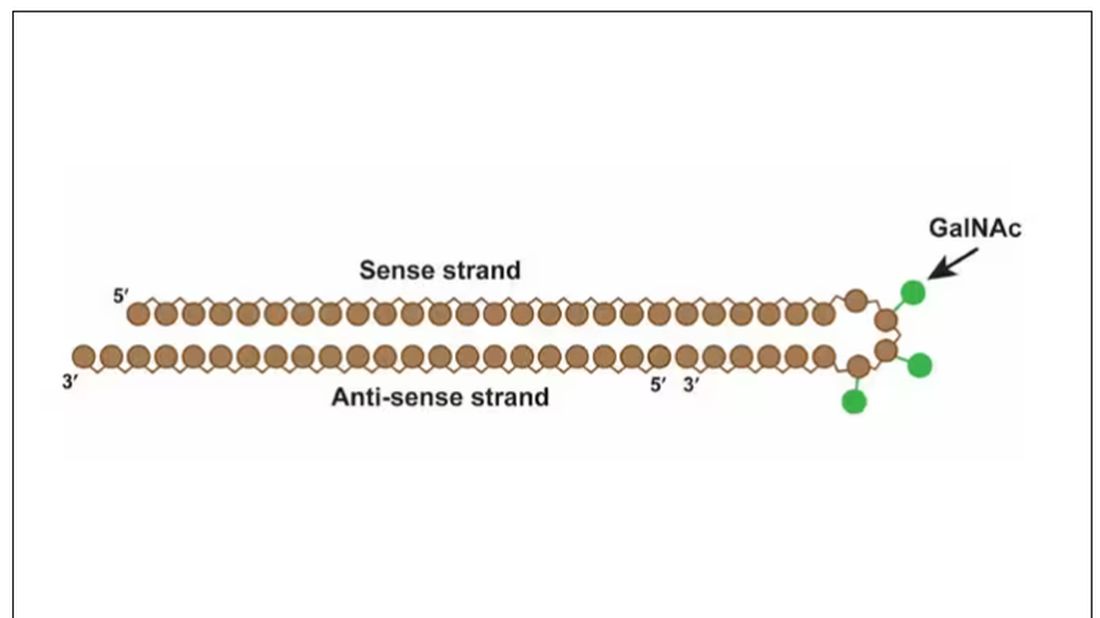
The report is a standard dose-escalation trial. Six patients, all with elevated lipoprotein(a) levels, were started with a 4-mg dose (two additional individuals got placebo). They were intensely monitored, spending 3 days in a research unit for multiple blood draws followed by weekly, and then biweekly outpatient visits. Once they had done well, the next group of six people received a higher dose (two more got placebo), and the process was repeated – six times total – until the highest dose, 608 mg, was reached.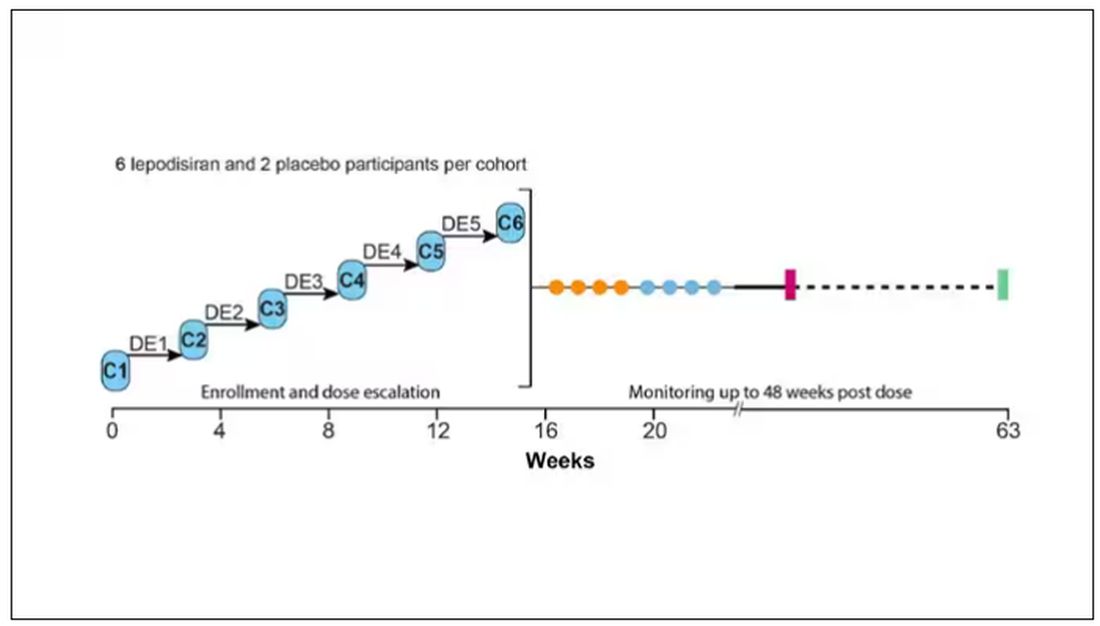
This is an injection, of course; siRNA wouldn’t withstand the harshness of the digestive system. And it’s only one injection. You can see from the blood concentration curves that within about 48 hours, circulating lepodisiran was not detectable.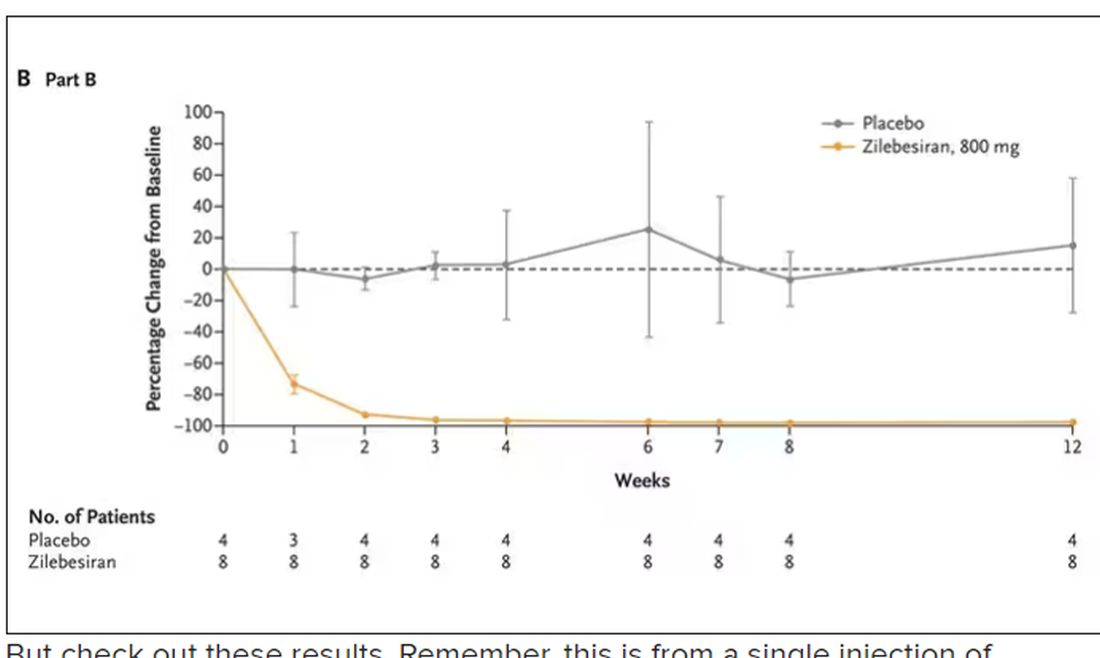
But check out these results. Remember, this is from a single injection of lepodisiran.
Lipoprotein(a) levels start to drop within a week of administration, and they stay down. In the higher-dose groups, levels are nearly undetectable a year after that injection.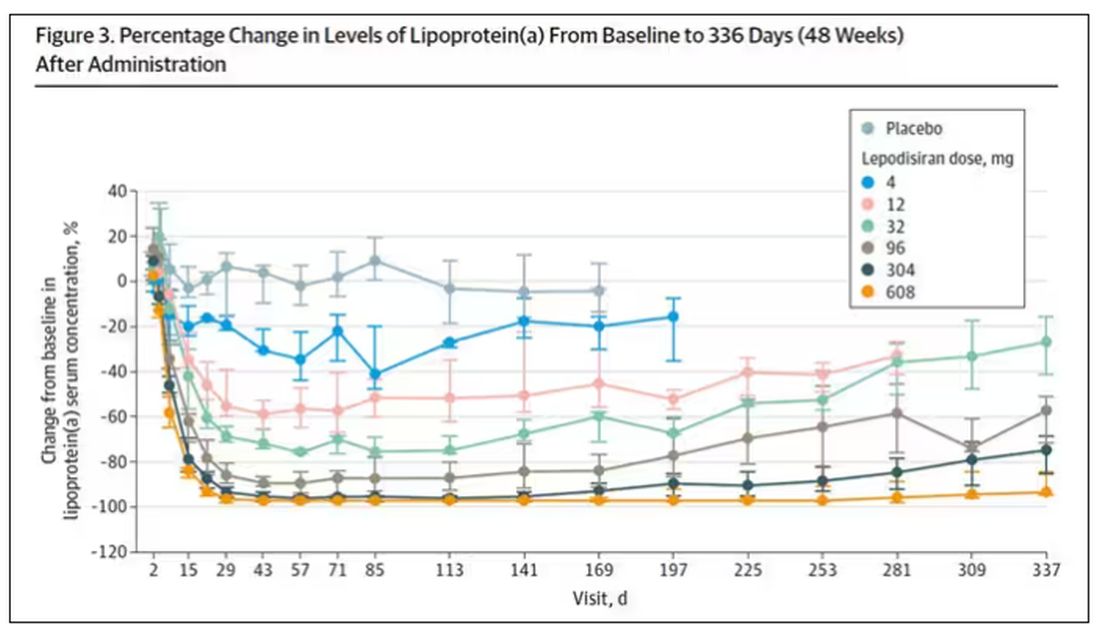
It was this graph that made me sit back and think that there might be something new under the sun. A single injection that can suppress protein synthesis for an entire year? If it really works, it changes the game.
Of course, this study wasn’t powered to look at important outcomes like heart attacks and strokes. It was primarily designed to assess safety, and the drug was pretty well tolerated, with similar rates of adverse events in the drug and placebo groups.
As crazy as it sounds, the real concern here might be that this drug is too good; is it safe to drop your lipoprotein(a) levels to zero for a year? I don’t know. But lower doses don’t have quite as strong an effect.
Trust me, these drugs are going to change things. They already are. In July, The New England Journal of Medicine published a study of zilebesiran, an siRNA that inhibits the production of angiotensinogen, to control blood pressure. Similar story: One injection led to a basically complete suppression of angiotensinogen and a sustained decrease in blood pressure.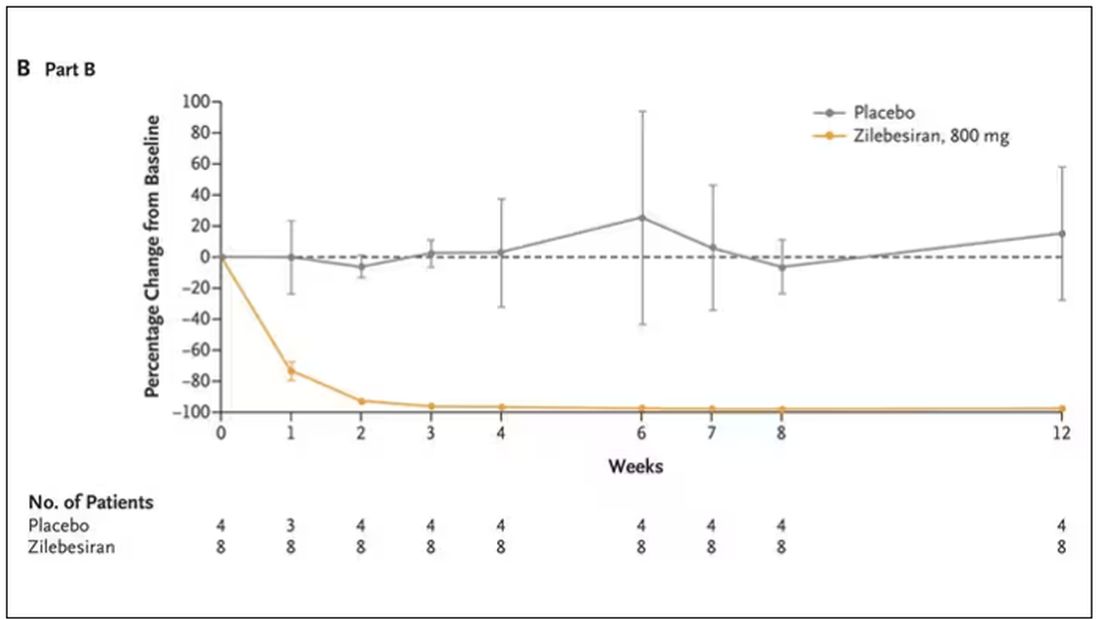
I’m not exaggerating when I say that there may come a time when you go to your doctor once a year, get your RNA shots, and don’t have to take any other medication from that point on. And that time may be, like, 5 years from now. It’s wild.
Seems to me that that rapid Nobel Prize was very well deserved.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships. This transcript has been edited for clarity.
A version of this article appeared on Medscape.com.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Every once in a while, medicine changes in a fundamental way, and we may not realize it while it’s happening. I wasn’t around in 1928 when Fleming discovered penicillin; or in 1953 when Watson, Crick, and Franklin characterized the double-helical structure of DNA.
But looking at medicine today, there are essentially two places where I think we will see, in retrospect, that we were at a fundamental turning point. One is artificial intelligence, which gets so much attention and hype that I will simply say yes, this will change things, stay tuned.
The other is a bit more obscure, but I suspect it may be just as impactful. That other thing is
I want to start with the idea that many diseases are, fundamentally, a problem of proteins. In some cases, like hypercholesterolemia, the body produces too much protein; in others, like hemophilia, too little.
When you think about disease this way, you realize that our current medications take effect late in the disease game. We have these molecules that try to block a protein from its receptor, prevent a protein from cleaving another protein, or increase the rate that a protein is broken down. It’s all distal to the fundamental problem: the production of the bad protein in the first place.
Enter small inhibitory RNAs, or siRNAs for short, discovered in 1998 by Andrew Fire and Craig Mello at UMass Worcester. The two won the Nobel prize in medicine just 8 years later; that’s a really short time, highlighting just how important this discovery was. In contrast, Karikó and Weissman won the Nobel for mRNA vaccines this year, after inventing them 18 years ago.
siRNAs are the body’s way of targeting proteins for destruction before they are ever created. About 20 base pairs long, siRNAs seek out a complementary target mRNA, attach to it, and call in a group of proteins to destroy it. With the target mRNA gone, no protein can be created.
You see where this is going, right? How does high cholesterol kill you? Proteins. How does Staphylococcus aureus kill you? Proteins. Even viruses can’t replicate if their RNA is prevented from being turned into proteins.
So, how do we use siRNAs? A new paper appearing in JAMA describes a fairly impressive use case.
The background here is that higher levels of lipoprotein(a), an LDL-like protein, are associated with cardiovascular disease, heart attack, and stroke. But unfortunately, statins really don’t have any effect on lipoprotein(a) levels. Neither does diet. Your lipoprotein(a) level seems to be more or less hard-coded genetically.
So, what if we stop the genetic machinery from working? Enter lepodisiran, a drug from Eli Lilly. Unlike so many other medications, which are usually found in nature, purified, and synthesized, lepodisiran was created from scratch. It’s not hard. Thanks to the Human Genome Project, we know the genetic code for lipoprotein(a), so inventing an siRNA to target it specifically is trivial. That’s one of the key features of siRNA – you don’t have to find a chemical that binds strongly to some protein receptor, and worry about the off-target effects and all that nonsense. You just pick a protein you want to suppress and you suppress it.
Okay, it’s not that simple. siRNA is broken down very quickly by the body, so it needs to be targeted to the organ of interest – in this case, the liver, since that is where lipoprotein(a) is synthesized. Lepodisiran is targeted to the liver by this special targeting label here.
The report is a standard dose-escalation trial. Six patients, all with elevated lipoprotein(a) levels, were started with a 4-mg dose (two additional individuals got placebo). They were intensely monitored, spending 3 days in a research unit for multiple blood draws followed by weekly, and then biweekly outpatient visits. Once they had done well, the next group of six people received a higher dose (two more got placebo), and the process was repeated – six times total – until the highest dose, 608 mg, was reached.
This is an injection, of course; siRNA wouldn’t withstand the harshness of the digestive system. And it’s only one injection. You can see from the blood concentration curves that within about 48 hours, circulating lepodisiran was not detectable.
But check out these results. Remember, this is from a single injection of lepodisiran.
Lipoprotein(a) levels start to drop within a week of administration, and they stay down. In the higher-dose groups, levels are nearly undetectable a year after that injection.
It was this graph that made me sit back and think that there might be something new under the sun. A single injection that can suppress protein synthesis for an entire year? If it really works, it changes the game.
Of course, this study wasn’t powered to look at important outcomes like heart attacks and strokes. It was primarily designed to assess safety, and the drug was pretty well tolerated, with similar rates of adverse events in the drug and placebo groups.
As crazy as it sounds, the real concern here might be that this drug is too good; is it safe to drop your lipoprotein(a) levels to zero for a year? I don’t know. But lower doses don’t have quite as strong an effect.
Trust me, these drugs are going to change things. They already are. In July, The New England Journal of Medicine published a study of zilebesiran, an siRNA that inhibits the production of angiotensinogen, to control blood pressure. Similar story: One injection led to a basically complete suppression of angiotensinogen and a sustained decrease in blood pressure.
I’m not exaggerating when I say that there may come a time when you go to your doctor once a year, get your RNA shots, and don’t have to take any other medication from that point on. And that time may be, like, 5 years from now. It’s wild.
Seems to me that that rapid Nobel Prize was very well deserved.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships. This transcript has been edited for clarity.
A version of this article appeared on Medscape.com.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Every once in a while, medicine changes in a fundamental way, and we may not realize it while it’s happening. I wasn’t around in 1928 when Fleming discovered penicillin; or in 1953 when Watson, Crick, and Franklin characterized the double-helical structure of DNA.
But looking at medicine today, there are essentially two places where I think we will see, in retrospect, that we were at a fundamental turning point. One is artificial intelligence, which gets so much attention and hype that I will simply say yes, this will change things, stay tuned.
The other is a bit more obscure, but I suspect it may be just as impactful. That other thing is
I want to start with the idea that many diseases are, fundamentally, a problem of proteins. In some cases, like hypercholesterolemia, the body produces too much protein; in others, like hemophilia, too little.
When you think about disease this way, you realize that our current medications take effect late in the disease game. We have these molecules that try to block a protein from its receptor, prevent a protein from cleaving another protein, or increase the rate that a protein is broken down. It’s all distal to the fundamental problem: the production of the bad protein in the first place.
Enter small inhibitory RNAs, or siRNAs for short, discovered in 1998 by Andrew Fire and Craig Mello at UMass Worcester. The two won the Nobel prize in medicine just 8 years later; that’s a really short time, highlighting just how important this discovery was. In contrast, Karikó and Weissman won the Nobel for mRNA vaccines this year, after inventing them 18 years ago.
siRNAs are the body’s way of targeting proteins for destruction before they are ever created. About 20 base pairs long, siRNAs seek out a complementary target mRNA, attach to it, and call in a group of proteins to destroy it. With the target mRNA gone, no protein can be created.
You see where this is going, right? How does high cholesterol kill you? Proteins. How does Staphylococcus aureus kill you? Proteins. Even viruses can’t replicate if their RNA is prevented from being turned into proteins.
So, how do we use siRNAs? A new paper appearing in JAMA describes a fairly impressive use case.
The background here is that higher levels of lipoprotein(a), an LDL-like protein, are associated with cardiovascular disease, heart attack, and stroke. But unfortunately, statins really don’t have any effect on lipoprotein(a) levels. Neither does diet. Your lipoprotein(a) level seems to be more or less hard-coded genetically.
So, what if we stop the genetic machinery from working? Enter lepodisiran, a drug from Eli Lilly. Unlike so many other medications, which are usually found in nature, purified, and synthesized, lepodisiran was created from scratch. It’s not hard. Thanks to the Human Genome Project, we know the genetic code for lipoprotein(a), so inventing an siRNA to target it specifically is trivial. That’s one of the key features of siRNA – you don’t have to find a chemical that binds strongly to some protein receptor, and worry about the off-target effects and all that nonsense. You just pick a protein you want to suppress and you suppress it.
Okay, it’s not that simple. siRNA is broken down very quickly by the body, so it needs to be targeted to the organ of interest – in this case, the liver, since that is where lipoprotein(a) is synthesized. Lepodisiran is targeted to the liver by this special targeting label here.
The report is a standard dose-escalation trial. Six patients, all with elevated lipoprotein(a) levels, were started with a 4-mg dose (two additional individuals got placebo). They were intensely monitored, spending 3 days in a research unit for multiple blood draws followed by weekly, and then biweekly outpatient visits. Once they had done well, the next group of six people received a higher dose (two more got placebo), and the process was repeated – six times total – until the highest dose, 608 mg, was reached.
This is an injection, of course; siRNA wouldn’t withstand the harshness of the digestive system. And it’s only one injection. You can see from the blood concentration curves that within about 48 hours, circulating lepodisiran was not detectable.
But check out these results. Remember, this is from a single injection of lepodisiran.
Lipoprotein(a) levels start to drop within a week of administration, and they stay down. In the higher-dose groups, levels are nearly undetectable a year after that injection.
It was this graph that made me sit back and think that there might be something new under the sun. A single injection that can suppress protein synthesis for an entire year? If it really works, it changes the game.
Of course, this study wasn’t powered to look at important outcomes like heart attacks and strokes. It was primarily designed to assess safety, and the drug was pretty well tolerated, with similar rates of adverse events in the drug and placebo groups.
As crazy as it sounds, the real concern here might be that this drug is too good; is it safe to drop your lipoprotein(a) levels to zero for a year? I don’t know. But lower doses don’t have quite as strong an effect.
Trust me, these drugs are going to change things. They already are. In July, The New England Journal of Medicine published a study of zilebesiran, an siRNA that inhibits the production of angiotensinogen, to control blood pressure. Similar story: One injection led to a basically complete suppression of angiotensinogen and a sustained decrease in blood pressure.
I’m not exaggerating when I say that there may come a time when you go to your doctor once a year, get your RNA shots, and don’t have to take any other medication from that point on. And that time may be, like, 5 years from now. It’s wild.
Seems to me that that rapid Nobel Prize was very well deserved.
Dr. F. Perry Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships. This transcript has been edited for clarity.
A version of this article appeared on Medscape.com.
Meta-analysis shows benefits of capecitabine-based chemo in early TNBC
Key clinical point: Capecitabine-based chemotherapy improved prognostic outcomes in patients with early-stage triple-negative breast cancer (TNBC).
Major finding: Capecitabine-based chemotherapy vs capecitabine-free regimens improved disease-free survival (DFS; hazard ratio [HR] 0.81; P < .001) and overall survival (HR 0.75; P < .001) outcomes. DFS benefits were particularly observed in the adjuvant setting (HR 0.79; P < .001) and in the subgroup of patients with lymph node-negative TNBC (HR 0.68; P = .006) and in those who received capecitabine for ≥ 6 cycles (HR 0.71; P < .001).
Study details: Findings are from a meta-analysis of 12 randomized controlled trials including 5390 patients with TNBC who were treated with capecitabine-based chemotherapy or capecitabine-free regimens.
Disclosures: This study was supported by the National Natural Science Foundation of China and the Natural Science Foundation of Chongqing. The authors declared no conflicts of interest.
Source: Bai J et al. Capecitabine-based chemotherapy in early-stage triple-negative breast cancer: A meta-analysis. Front Oncol. 2023;13:1245650 (Oct 25). doi: 10.3389/fonc.2023.1245650
Key clinical point: Capecitabine-based chemotherapy improved prognostic outcomes in patients with early-stage triple-negative breast cancer (TNBC).
Major finding: Capecitabine-based chemotherapy vs capecitabine-free regimens improved disease-free survival (DFS; hazard ratio [HR] 0.81; P < .001) and overall survival (HR 0.75; P < .001) outcomes. DFS benefits were particularly observed in the adjuvant setting (HR 0.79; P < .001) and in the subgroup of patients with lymph node-negative TNBC (HR 0.68; P = .006) and in those who received capecitabine for ≥ 6 cycles (HR 0.71; P < .001).
Study details: Findings are from a meta-analysis of 12 randomized controlled trials including 5390 patients with TNBC who were treated with capecitabine-based chemotherapy or capecitabine-free regimens.
Disclosures: This study was supported by the National Natural Science Foundation of China and the Natural Science Foundation of Chongqing. The authors declared no conflicts of interest.
Source: Bai J et al. Capecitabine-based chemotherapy in early-stage triple-negative breast cancer: A meta-analysis. Front Oncol. 2023;13:1245650 (Oct 25). doi: 10.3389/fonc.2023.1245650
Key clinical point: Capecitabine-based chemotherapy improved prognostic outcomes in patients with early-stage triple-negative breast cancer (TNBC).
Major finding: Capecitabine-based chemotherapy vs capecitabine-free regimens improved disease-free survival (DFS; hazard ratio [HR] 0.81; P < .001) and overall survival (HR 0.75; P < .001) outcomes. DFS benefits were particularly observed in the adjuvant setting (HR 0.79; P < .001) and in the subgroup of patients with lymph node-negative TNBC (HR 0.68; P = .006) and in those who received capecitabine for ≥ 6 cycles (HR 0.71; P < .001).
Study details: Findings are from a meta-analysis of 12 randomized controlled trials including 5390 patients with TNBC who were treated with capecitabine-based chemotherapy or capecitabine-free regimens.
Disclosures: This study was supported by the National Natural Science Foundation of China and the Natural Science Foundation of Chongqing. The authors declared no conflicts of interest.
Source: Bai J et al. Capecitabine-based chemotherapy in early-stage triple-negative breast cancer: A meta-analysis. Front Oncol. 2023;13:1245650 (Oct 25). doi: 10.3389/fonc.2023.1245650
Regional nodal irradiation may not be needed after preoperative systemic therapy in HER2+ BC
Key clinical point: Patients with human epidermal growth factor receptor 2-positive (HER2+) breast cancer (BC) who received docetaxel/carboplatin/trastuzumab/pertuzumab (TCHP)-based preoperative systemic therapy experienced no extra clinical benefits with postoperative regional nodal irradiation (RNI).
Major finding: Patients who did vs did not receive RNI had comparable locoregional recurrence frequency (2.6% vs 1.0%; P = .651) and disease-free survival outcomes (hazard ratio 0.72; P = .638); however, pathological complete response was achieved by a significantly higher proportion of patients in the no-RNI vs RNI group (72.5% vs 44.4%; P < .001).
Study details: This retrospective study included 255 patients with HER2+ BC who received six cycles of TCHP, of which 60% of patients received RNI.
Disclosures: This study did not declare the source of funding or conflicts of interest.
Source: Kim N, Kim J-Y, et al. Benefit of postoperative regional nodal irradiation in patients receiving preoperative systemic therapy with docetaxel/carboplatin/trastuzumab/pertuzumab for HER2-positive breast cancer. Breast. 2023;72:103594 (Oct 30). doi: 10.1016/j.breast.2023.103594
Key clinical point: Patients with human epidermal growth factor receptor 2-positive (HER2+) breast cancer (BC) who received docetaxel/carboplatin/trastuzumab/pertuzumab (TCHP)-based preoperative systemic therapy experienced no extra clinical benefits with postoperative regional nodal irradiation (RNI).
Major finding: Patients who did vs did not receive RNI had comparable locoregional recurrence frequency (2.6% vs 1.0%; P = .651) and disease-free survival outcomes (hazard ratio 0.72; P = .638); however, pathological complete response was achieved by a significantly higher proportion of patients in the no-RNI vs RNI group (72.5% vs 44.4%; P < .001).
Study details: This retrospective study included 255 patients with HER2+ BC who received six cycles of TCHP, of which 60% of patients received RNI.
Disclosures: This study did not declare the source of funding or conflicts of interest.
Source: Kim N, Kim J-Y, et al. Benefit of postoperative regional nodal irradiation in patients receiving preoperative systemic therapy with docetaxel/carboplatin/trastuzumab/pertuzumab for HER2-positive breast cancer. Breast. 2023;72:103594 (Oct 30). doi: 10.1016/j.breast.2023.103594
Key clinical point: Patients with human epidermal growth factor receptor 2-positive (HER2+) breast cancer (BC) who received docetaxel/carboplatin/trastuzumab/pertuzumab (TCHP)-based preoperative systemic therapy experienced no extra clinical benefits with postoperative regional nodal irradiation (RNI).
Major finding: Patients who did vs did not receive RNI had comparable locoregional recurrence frequency (2.6% vs 1.0%; P = .651) and disease-free survival outcomes (hazard ratio 0.72; P = .638); however, pathological complete response was achieved by a significantly higher proportion of patients in the no-RNI vs RNI group (72.5% vs 44.4%; P < .001).
Study details: This retrospective study included 255 patients with HER2+ BC who received six cycles of TCHP, of which 60% of patients received RNI.
Disclosures: This study did not declare the source of funding or conflicts of interest.
Source: Kim N, Kim J-Y, et al. Benefit of postoperative regional nodal irradiation in patients receiving preoperative systemic therapy with docetaxel/carboplatin/trastuzumab/pertuzumab for HER2-positive breast cancer. Breast. 2023;72:103594 (Oct 30). doi: 10.1016/j.breast.2023.103594
Taxanes followed by PLD show promise in metastatic BC under real-world settings
Key clinical point: First-line treatment with taxanes followed by pegylated liposomal doxorubicin (PLD) was associated with improved prognostic outcomes in patients with human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC) than with PLD followed by taxanes.
Major finding: First-line taxane followed by PLD vs first-line PLD followed by taxane significantly improved time to next chemotherapy (9.9 vs 4.9 months; P = .006) and progression-free survival outcomes (9.0 vs 4.4 months; P = .005).
Study details: Findings are from a retrospective study including 42 patients with HER2− metastatic BC who received first-line PLD and later taxane (n = 23) or first-line taxane and later PLD (n = 19).
Disclosures: This study did not receive any specific grants. The authors declared no conflicts of interest.
Source: Wallrabenstein T et al. Upfront taxane could be superior to pegylated liposomal doxorubicin (PLD): A retrospective real-world analysis of treatment sequence taxane-PLD versus PLD-taxane in patients with metastatic breast cancer. Cancers (Basel). 2023;15(20):4953 (Oct 12). doi: 10.3390/cancers15204953
Key clinical point: First-line treatment with taxanes followed by pegylated liposomal doxorubicin (PLD) was associated with improved prognostic outcomes in patients with human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC) than with PLD followed by taxanes.
Major finding: First-line taxane followed by PLD vs first-line PLD followed by taxane significantly improved time to next chemotherapy (9.9 vs 4.9 months; P = .006) and progression-free survival outcomes (9.0 vs 4.4 months; P = .005).
Study details: Findings are from a retrospective study including 42 patients with HER2− metastatic BC who received first-line PLD and later taxane (n = 23) or first-line taxane and later PLD (n = 19).
Disclosures: This study did not receive any specific grants. The authors declared no conflicts of interest.
Source: Wallrabenstein T et al. Upfront taxane could be superior to pegylated liposomal doxorubicin (PLD): A retrospective real-world analysis of treatment sequence taxane-PLD versus PLD-taxane in patients with metastatic breast cancer. Cancers (Basel). 2023;15(20):4953 (Oct 12). doi: 10.3390/cancers15204953
Key clinical point: First-line treatment with taxanes followed by pegylated liposomal doxorubicin (PLD) was associated with improved prognostic outcomes in patients with human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC) than with PLD followed by taxanes.
Major finding: First-line taxane followed by PLD vs first-line PLD followed by taxane significantly improved time to next chemotherapy (9.9 vs 4.9 months; P = .006) and progression-free survival outcomes (9.0 vs 4.4 months; P = .005).
Study details: Findings are from a retrospective study including 42 patients with HER2− metastatic BC who received first-line PLD and later taxane (n = 23) or first-line taxane and later PLD (n = 19).
Disclosures: This study did not receive any specific grants. The authors declared no conflicts of interest.
Source: Wallrabenstein T et al. Upfront taxane could be superior to pegylated liposomal doxorubicin (PLD): A retrospective real-world analysis of treatment sequence taxane-PLD versus PLD-taxane in patients with metastatic breast cancer. Cancers (Basel). 2023;15(20):4953 (Oct 12). doi: 10.3390/cancers15204953
Better efficacy-safety with 3-week vs 4-week nab-paclitaxel in HER2− metastatic BC
Key clinical point: A 3-week vs 4-week nab-paclitaxel schedule showed more effective anti-tumor activity and a more manageable safety profile in patients with human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC).
Major finding: Compared with a 4-week paclitaxel regimen, a 3-week regimen led to a 56% improvement in progression-free survival outcomes (hazard ratio 0.44; P = .029) and was associated with a lower rate of grade ≥ 3 adverse events (14.9% vs 42.6%).
Study details: Findings are from a phase 2 study including 94 patients with HER2− metastatic BC who were randomly assigned to receive nab-paclitaxel for either a 3-week or 4-week schedule.
Disclosures: This study was sponsored by CSPC Ouyi Pharmaceutical Co., Ltd, China. The authors declared no conflicts of interest.
Source: Liu Y et al. Three-week versus 4-week schedule of nab-paclitaxel in patients with metastatic breast cancer: A randomized phase II study. Oncologist. 2023 (Oct 26). doi: 10.1093/oncolo/oyad288
Key clinical point: A 3-week vs 4-week nab-paclitaxel schedule showed more effective anti-tumor activity and a more manageable safety profile in patients with human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC).
Major finding: Compared with a 4-week paclitaxel regimen, a 3-week regimen led to a 56% improvement in progression-free survival outcomes (hazard ratio 0.44; P = .029) and was associated with a lower rate of grade ≥ 3 adverse events (14.9% vs 42.6%).
Study details: Findings are from a phase 2 study including 94 patients with HER2− metastatic BC who were randomly assigned to receive nab-paclitaxel for either a 3-week or 4-week schedule.
Disclosures: This study was sponsored by CSPC Ouyi Pharmaceutical Co., Ltd, China. The authors declared no conflicts of interest.
Source: Liu Y et al. Three-week versus 4-week schedule of nab-paclitaxel in patients with metastatic breast cancer: A randomized phase II study. Oncologist. 2023 (Oct 26). doi: 10.1093/oncolo/oyad288
Key clinical point: A 3-week vs 4-week nab-paclitaxel schedule showed more effective anti-tumor activity and a more manageable safety profile in patients with human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC).
Major finding: Compared with a 4-week paclitaxel regimen, a 3-week regimen led to a 56% improvement in progression-free survival outcomes (hazard ratio 0.44; P = .029) and was associated with a lower rate of grade ≥ 3 adverse events (14.9% vs 42.6%).
Study details: Findings are from a phase 2 study including 94 patients with HER2− metastatic BC who were randomly assigned to receive nab-paclitaxel for either a 3-week or 4-week schedule.
Disclosures: This study was sponsored by CSPC Ouyi Pharmaceutical Co., Ltd, China. The authors declared no conflicts of interest.
Source: Liu Y et al. Three-week versus 4-week schedule of nab-paclitaxel in patients with metastatic breast cancer: A randomized phase II study. Oncologist. 2023 (Oct 26). doi: 10.1093/oncolo/oyad288
MRI as effective as MRI+mammography for BC screening in women with dense breasts
Key clinical point: In women with dense breasts, who are generally at an intermediate risk for breast cancer (BC), screening with magnetic resonance imaging (MRI) alone or with mammography increased the rates of screen-detected early-stage cancer and false-positive recalls compared with mammography alone.
Major finding: The rate of screen-detected early-stage cancer in women with dense breasts was higher with MRI alone vs MRI + mammography (difference 11.7/1000 examinations; 95% CI 4.6-18.8/1000 examinations) and MR + mammography vs mammography alone (difference 4.0/1000 examinations; 95% CI 1.4-6.7/1000 examinations); however, false-positive recall rates were higher with MRI + mammography vs mammography alone (difference 149.8/1000 examinations; 95% CI 135.7-163.9/1000 examinations) and comparable with both MRI and MRI + mammography.
Study details: This cohort study analyzed the data of women aged 40-79 years who had undergone screening with MRI (2611 screenings), MRI + mammography (6518 screenings), or mammography (65,180 screenings) from the Breast Cancer Surveillance Consortium registry.
Disclosures: This study was funded by the US Patient-Centered Outcomes Research Institute award and other sources. The authors declared no conflicts of interest.
Source: Kerlikowske K et al. Supplemental magnetic resonance imaging plus mammography compared with magnetic resonance imaging or mammography by extent of breast density. J Natl Cancer Inst. 2023 (Oct 27). doi: 10.1093/jnci/djad201
Key clinical point: In women with dense breasts, who are generally at an intermediate risk for breast cancer (BC), screening with magnetic resonance imaging (MRI) alone or with mammography increased the rates of screen-detected early-stage cancer and false-positive recalls compared with mammography alone.
Major finding: The rate of screen-detected early-stage cancer in women with dense breasts was higher with MRI alone vs MRI + mammography (difference 11.7/1000 examinations; 95% CI 4.6-18.8/1000 examinations) and MR + mammography vs mammography alone (difference 4.0/1000 examinations; 95% CI 1.4-6.7/1000 examinations); however, false-positive recall rates were higher with MRI + mammography vs mammography alone (difference 149.8/1000 examinations; 95% CI 135.7-163.9/1000 examinations) and comparable with both MRI and MRI + mammography.
Study details: This cohort study analyzed the data of women aged 40-79 years who had undergone screening with MRI (2611 screenings), MRI + mammography (6518 screenings), or mammography (65,180 screenings) from the Breast Cancer Surveillance Consortium registry.
Disclosures: This study was funded by the US Patient-Centered Outcomes Research Institute award and other sources. The authors declared no conflicts of interest.
Source: Kerlikowske K et al. Supplemental magnetic resonance imaging plus mammography compared with magnetic resonance imaging or mammography by extent of breast density. J Natl Cancer Inst. 2023 (Oct 27). doi: 10.1093/jnci/djad201
Key clinical point: In women with dense breasts, who are generally at an intermediate risk for breast cancer (BC), screening with magnetic resonance imaging (MRI) alone or with mammography increased the rates of screen-detected early-stage cancer and false-positive recalls compared with mammography alone.
Major finding: The rate of screen-detected early-stage cancer in women with dense breasts was higher with MRI alone vs MRI + mammography (difference 11.7/1000 examinations; 95% CI 4.6-18.8/1000 examinations) and MR + mammography vs mammography alone (difference 4.0/1000 examinations; 95% CI 1.4-6.7/1000 examinations); however, false-positive recall rates were higher with MRI + mammography vs mammography alone (difference 149.8/1000 examinations; 95% CI 135.7-163.9/1000 examinations) and comparable with both MRI and MRI + mammography.
Study details: This cohort study analyzed the data of women aged 40-79 years who had undergone screening with MRI (2611 screenings), MRI + mammography (6518 screenings), or mammography (65,180 screenings) from the Breast Cancer Surveillance Consortium registry.
Disclosures: This study was funded by the US Patient-Centered Outcomes Research Institute award and other sources. The authors declared no conflicts of interest.
Source: Kerlikowske K et al. Supplemental magnetic resonance imaging plus mammography compared with magnetic resonance imaging or mammography by extent of breast density. J Natl Cancer Inst. 2023 (Oct 27). doi: 10.1093/jnci/djad201
Trial shows utility of small-volume blood collection tubes
A large Canadian clinical trial has found that using small-volume tubes to collect blood samples for laboratory testing of intensive care unit patients can reduce blood transfusions without affecting lab results.
“We showed in a large pragmatic cluster trial that automatically collect less blood for laboratory testing reduced red blood cell transfusions by about 10 units of red blood cells per 100 patients in the ICU,” lead study author Deborah M. Siegal, MD, associate professor at the University of Ottawa and scientist at the Ottawa Hospital Research Institute, said.
The study was coordinated by the Population Health Research Institute, an affiliate of McMaster University in Hamilton (Ont.) Health Sciences, where Dr. Siegal worked before moving to Ottawa.

The STRATUS randomized clinical trial, published in JAMA, involved 25 adult medical-surgical ICUs across Canada, where 21,201 patients were randomized to either standard-volume or small-volume tubes for collecting blood samples. During the course of the study, each site switched to the small-volume collection tubes.
“We also showed there were no negative effects on lab testing, and by that we measured the sufficiency of the specimens,” Dr. Siegal added. “We were able to show that there wasn’t a problem with the amount of blood that was available for the tests to be done.”
The samples were collected from February 2019 through January 2021, through the period of COVID-19 restrictions. Dr. Siegal explained that 6,210 patients admitted early in the COVID-19 pandemic were excluded from the primary analysis, but were included in secondary analyses.
Study results
While the study found no significant difference in RBC units per patient per ICU stage – a relative risk of .91 (95% confidence interval, 0.79-1.05; P = .19), it did find an absolute reduction of 7.24 RBC units/100 patients per ICU stay.
Findings from the secondary analyses, which included 27,411 patients, were:
- A 12% reduction in RBC units per patient per ICU stay after switching from standard-volume to small-volume tubes (RR, 0.88; 95% CI, 0.77-1; P = .04).
- An absolute reduction of 9.84 RBC units/100 patients per ICU stay (95% CI, 0.24-20.76).
In the primary analysis population, the median transfusion-adjusted hemoglobin was not statistically different between the standard- and small-volume collection tube groups, with an average difference of 0.1 g/dL (95% CI, –0.04 to .23), but it was lower in the secondary population, with a mean difference of .17 g/dL (95% CI, 0.05-0.29).
“Those patients that we analyzed in the secondary analysis population received about 36,000 units of blood, just in 25 ICU units in Canada in less than 2 years,” Dr. Siegal said. “If we saved 10 units per 100 patients, that’s 1,500 units of blood. That really speaks to a small effect at the individual patient level but really potential for widespread effect. We are now in a period of blood product shortage not only in Canada but worldwide.”
First clinical trial for small tubes
Dr. Siegal noted this was the first clinical trial to compare standard- and small-volume blood collection tools, “and also to show there is both a benefit and a lack of harm,” Dr. Siegal said. “We thought that a randomized trial was the best way to move the needle. If we could design a trial of a large population of patients to show benefit and no harm, it would be a win, and that’s in fact what happened.”
She added, “The tubes essentially have the same cost, work the same, and go on the same equipment the same way the standard-volume tubes do, so it wasn’t a practice change for people in the hospital.”
The study also found an identical low rate of unusable specimens did not differ regardless of the type of collection tube: less than .03%.
Dr. Siegal said the study group is collaborating with hematology stakeholders in Canada, including Canadian Blood Services, which provides blood plasma to the country’s provincial and territorial health systems, and is reaching out to the American Society of Hematology.
“We’re going to target both hematologists and critical care providers and, even more broadly than the critical care community, hospitals, because anemia is big problem in hospitals,” Dr. Siegal said. “I think we can think about this more broadly.”
The study received funding from the Hamilton Academic Health Sciences Organization. Dr. Siegal disclosed relationships with Bristol-Myers Squibb-Pfizer, AstraZeneca and Roche.
A large Canadian clinical trial has found that using small-volume tubes to collect blood samples for laboratory testing of intensive care unit patients can reduce blood transfusions without affecting lab results.
“We showed in a large pragmatic cluster trial that automatically collect less blood for laboratory testing reduced red blood cell transfusions by about 10 units of red blood cells per 100 patients in the ICU,” lead study author Deborah M. Siegal, MD, associate professor at the University of Ottawa and scientist at the Ottawa Hospital Research Institute, said.
The study was coordinated by the Population Health Research Institute, an affiliate of McMaster University in Hamilton (Ont.) Health Sciences, where Dr. Siegal worked before moving to Ottawa.
The STRATUS randomized clinical trial, published in JAMA, involved 25 adult medical-surgical ICUs across Canada, where 21,201 patients were randomized to either standard-volume or small-volume tubes for collecting blood samples. During the course of the study, each site switched to the small-volume collection tubes.
“We also showed there were no negative effects on lab testing, and by that we measured the sufficiency of the specimens,” Dr. Siegal added. “We were able to show that there wasn’t a problem with the amount of blood that was available for the tests to be done.”
The samples were collected from February 2019 through January 2021, through the period of COVID-19 restrictions. Dr. Siegal explained that 6,210 patients admitted early in the COVID-19 pandemic were excluded from the primary analysis, but were included in secondary analyses.
Study results
While the study found no significant difference in RBC units per patient per ICU stage – a relative risk of .91 (95% confidence interval, 0.79-1.05; P = .19), it did find an absolute reduction of 7.24 RBC units/100 patients per ICU stay.
Findings from the secondary analyses, which included 27,411 patients, were:
- A 12% reduction in RBC units per patient per ICU stay after switching from standard-volume to small-volume tubes (RR, 0.88; 95% CI, 0.77-1; P = .04).
- An absolute reduction of 9.84 RBC units/100 patients per ICU stay (95% CI, 0.24-20.76).
In the primary analysis population, the median transfusion-adjusted hemoglobin was not statistically different between the standard- and small-volume collection tube groups, with an average difference of 0.1 g/dL (95% CI, –0.04 to .23), but it was lower in the secondary population, with a mean difference of .17 g/dL (95% CI, 0.05-0.29).
“Those patients that we analyzed in the secondary analysis population received about 36,000 units of blood, just in 25 ICU units in Canada in less than 2 years,” Dr. Siegal said. “If we saved 10 units per 100 patients, that’s 1,500 units of blood. That really speaks to a small effect at the individual patient level but really potential for widespread effect. We are now in a period of blood product shortage not only in Canada but worldwide.”
First clinical trial for small tubes
Dr. Siegal noted this was the first clinical trial to compare standard- and small-volume blood collection tools, “and also to show there is both a benefit and a lack of harm,” Dr. Siegal said. “We thought that a randomized trial was the best way to move the needle. If we could design a trial of a large population of patients to show benefit and no harm, it would be a win, and that’s in fact what happened.”
She added, “The tubes essentially have the same cost, work the same, and go on the same equipment the same way the standard-volume tubes do, so it wasn’t a practice change for people in the hospital.”
The study also found an identical low rate of unusable specimens did not differ regardless of the type of collection tube: less than .03%.
Dr. Siegal said the study group is collaborating with hematology stakeholders in Canada, including Canadian Blood Services, which provides blood plasma to the country’s provincial and territorial health systems, and is reaching out to the American Society of Hematology.
“We’re going to target both hematologists and critical care providers and, even more broadly than the critical care community, hospitals, because anemia is big problem in hospitals,” Dr. Siegal said. “I think we can think about this more broadly.”
The study received funding from the Hamilton Academic Health Sciences Organization. Dr. Siegal disclosed relationships with Bristol-Myers Squibb-Pfizer, AstraZeneca and Roche.
A large Canadian clinical trial has found that using small-volume tubes to collect blood samples for laboratory testing of intensive care unit patients can reduce blood transfusions without affecting lab results.
“We showed in a large pragmatic cluster trial that automatically collect less blood for laboratory testing reduced red blood cell transfusions by about 10 units of red blood cells per 100 patients in the ICU,” lead study author Deborah M. Siegal, MD, associate professor at the University of Ottawa and scientist at the Ottawa Hospital Research Institute, said.
The study was coordinated by the Population Health Research Institute, an affiliate of McMaster University in Hamilton (Ont.) Health Sciences, where Dr. Siegal worked before moving to Ottawa.
The STRATUS randomized clinical trial, published in JAMA, involved 25 adult medical-surgical ICUs across Canada, where 21,201 patients were randomized to either standard-volume or small-volume tubes for collecting blood samples. During the course of the study, each site switched to the small-volume collection tubes.
“We also showed there were no negative effects on lab testing, and by that we measured the sufficiency of the specimens,” Dr. Siegal added. “We were able to show that there wasn’t a problem with the amount of blood that was available for the tests to be done.”
The samples were collected from February 2019 through January 2021, through the period of COVID-19 restrictions. Dr. Siegal explained that 6,210 patients admitted early in the COVID-19 pandemic were excluded from the primary analysis, but were included in secondary analyses.
Study results
While the study found no significant difference in RBC units per patient per ICU stage – a relative risk of .91 (95% confidence interval, 0.79-1.05; P = .19), it did find an absolute reduction of 7.24 RBC units/100 patients per ICU stay.
Findings from the secondary analyses, which included 27,411 patients, were:
- A 12% reduction in RBC units per patient per ICU stay after switching from standard-volume to small-volume tubes (RR, 0.88; 95% CI, 0.77-1; P = .04).
- An absolute reduction of 9.84 RBC units/100 patients per ICU stay (95% CI, 0.24-20.76).
In the primary analysis population, the median transfusion-adjusted hemoglobin was not statistically different between the standard- and small-volume collection tube groups, with an average difference of 0.1 g/dL (95% CI, –0.04 to .23), but it was lower in the secondary population, with a mean difference of .17 g/dL (95% CI, 0.05-0.29).
“Those patients that we analyzed in the secondary analysis population received about 36,000 units of blood, just in 25 ICU units in Canada in less than 2 years,” Dr. Siegal said. “If we saved 10 units per 100 patients, that’s 1,500 units of blood. That really speaks to a small effect at the individual patient level but really potential for widespread effect. We are now in a period of blood product shortage not only in Canada but worldwide.”
First clinical trial for small tubes
Dr. Siegal noted this was the first clinical trial to compare standard- and small-volume blood collection tools, “and also to show there is both a benefit and a lack of harm,” Dr. Siegal said. “We thought that a randomized trial was the best way to move the needle. If we could design a trial of a large population of patients to show benefit and no harm, it would be a win, and that’s in fact what happened.”
She added, “The tubes essentially have the same cost, work the same, and go on the same equipment the same way the standard-volume tubes do, so it wasn’t a practice change for people in the hospital.”
The study also found an identical low rate of unusable specimens did not differ regardless of the type of collection tube: less than .03%.
Dr. Siegal said the study group is collaborating with hematology stakeholders in Canada, including Canadian Blood Services, which provides blood plasma to the country’s provincial and territorial health systems, and is reaching out to the American Society of Hematology.
“We’re going to target both hematologists and critical care providers and, even more broadly than the critical care community, hospitals, because anemia is big problem in hospitals,” Dr. Siegal said. “I think we can think about this more broadly.”
The study received funding from the Hamilton Academic Health Sciences Organization. Dr. Siegal disclosed relationships with Bristol-Myers Squibb-Pfizer, AstraZeneca and Roche.
FROM JAMA
Prognosis remains poor in inflammatory BC despite neoadjuvant chemotherapy
Key clinical point: Despite treatment with neoadjuvant chemotherapy (NAC), patients with locally advanced inflammatory breast cancer (BC) showed poorer survival outcomes than those with noninflammatory BC.
Major finding: Patients with inflammatory vs noninflammatory locally advanced BC who received NAC had significantly lower rates of 5-year overall survival (58.9% vs 86.7%; P = .00005), relapse-free survival (53.0% vs 80.3%; P = .0001), and distant relapse-free survival (53.3% vs 80.9%; P = .0001).
Study details: This retrospective analysis included 84 patients with stage III inflammatory BC and 81 matched-control individuals with stage III noninflammatory BC, all of whom received neoadjuvant chemotherapy.
Disclosures: This study did not receive any specific funding. KU Park declared being a consultant with Bayer LLC. The other authors declared no conflicts of interest.
Source: Johnson KCC et al. Survival outcomes seen with neoadjuvant chemotherapy in the management of locally advanced inflammatory breast cancer (IBC) versus matched controls. Breast. 2023;72:103591 (Oct 13). doi: 10.1016/j.breast.2023.103591
Key clinical point: Despite treatment with neoadjuvant chemotherapy (NAC), patients with locally advanced inflammatory breast cancer (BC) showed poorer survival outcomes than those with noninflammatory BC.
Major finding: Patients with inflammatory vs noninflammatory locally advanced BC who received NAC had significantly lower rates of 5-year overall survival (58.9% vs 86.7%; P = .00005), relapse-free survival (53.0% vs 80.3%; P = .0001), and distant relapse-free survival (53.3% vs 80.9%; P = .0001).
Study details: This retrospective analysis included 84 patients with stage III inflammatory BC and 81 matched-control individuals with stage III noninflammatory BC, all of whom received neoadjuvant chemotherapy.
Disclosures: This study did not receive any specific funding. KU Park declared being a consultant with Bayer LLC. The other authors declared no conflicts of interest.
Source: Johnson KCC et al. Survival outcomes seen with neoadjuvant chemotherapy in the management of locally advanced inflammatory breast cancer (IBC) versus matched controls. Breast. 2023;72:103591 (Oct 13). doi: 10.1016/j.breast.2023.103591
Key clinical point: Despite treatment with neoadjuvant chemotherapy (NAC), patients with locally advanced inflammatory breast cancer (BC) showed poorer survival outcomes than those with noninflammatory BC.
Major finding: Patients with inflammatory vs noninflammatory locally advanced BC who received NAC had significantly lower rates of 5-year overall survival (58.9% vs 86.7%; P = .00005), relapse-free survival (53.0% vs 80.3%; P = .0001), and distant relapse-free survival (53.3% vs 80.9%; P = .0001).
Study details: This retrospective analysis included 84 patients with stage III inflammatory BC and 81 matched-control individuals with stage III noninflammatory BC, all of whom received neoadjuvant chemotherapy.
Disclosures: This study did not receive any specific funding. KU Park declared being a consultant with Bayer LLC. The other authors declared no conflicts of interest.
Source: Johnson KCC et al. Survival outcomes seen with neoadjuvant chemotherapy in the management of locally advanced inflammatory breast cancer (IBC) versus matched controls. Breast. 2023;72:103591 (Oct 13). doi: 10.1016/j.breast.2023.103591
Oral SERD improve PFS in ER+/HER2− metastatic BC, shows meta-analysis
Key clinical point: Oral selective estrogen receptor degraders (SERD) improved the progression-free survival (PFS) outcomes in patients with estrogen receptor-positive (ER+) human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC), particularly in those with ESR1 mutations.
Major finding: Compared with endocrine therapies (ET) of the physician’s choice, oral SERD led to a greater improvement in PFS outcomes in the overall population (hazard ratio [HR] 0.783; P < .001) and in the subgroup of patients with ESR1 mutations (HR 0.557; P < .001); however, no PFS benefit was observed in the ESR1 wild-type subgroup (P = .543).
Study details: Findings are from a meta-analysis of individual patient data from four randomized clinical trials including 1290 patients with ER+/HER2− metastatic BC who received oral SERD or ET of physician’s choice.
Disclosures: This study did not receive any specific funding. Some authors declared receiving honoraria, research funding, or travel grants from or serving in advisory or consulting roles for various sources.
Source: Wong NZH et al. Efficacy of oral SERDs in the treatment of ER+, HER2 - metastatic breast cancer, a stratified analysis of the ESR1 wild type and mutant subgroups. Ann Oncol. 2023 (Oct 21). doi: 10.1016/j.annonc.2023.10.122
Key clinical point: Oral selective estrogen receptor degraders (SERD) improved the progression-free survival (PFS) outcomes in patients with estrogen receptor-positive (ER+) human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC), particularly in those with ESR1 mutations.
Major finding: Compared with endocrine therapies (ET) of the physician’s choice, oral SERD led to a greater improvement in PFS outcomes in the overall population (hazard ratio [HR] 0.783; P < .001) and in the subgroup of patients with ESR1 mutations (HR 0.557; P < .001); however, no PFS benefit was observed in the ESR1 wild-type subgroup (P = .543).
Study details: Findings are from a meta-analysis of individual patient data from four randomized clinical trials including 1290 patients with ER+/HER2− metastatic BC who received oral SERD or ET of physician’s choice.
Disclosures: This study did not receive any specific funding. Some authors declared receiving honoraria, research funding, or travel grants from or serving in advisory or consulting roles for various sources.
Source: Wong NZH et al. Efficacy of oral SERDs in the treatment of ER+, HER2 - metastatic breast cancer, a stratified analysis of the ESR1 wild type and mutant subgroups. Ann Oncol. 2023 (Oct 21). doi: 10.1016/j.annonc.2023.10.122
Key clinical point: Oral selective estrogen receptor degraders (SERD) improved the progression-free survival (PFS) outcomes in patients with estrogen receptor-positive (ER+) human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer (BC), particularly in those with ESR1 mutations.
Major finding: Compared with endocrine therapies (ET) of the physician’s choice, oral SERD led to a greater improvement in PFS outcomes in the overall population (hazard ratio [HR] 0.783; P < .001) and in the subgroup of patients with ESR1 mutations (HR 0.557; P < .001); however, no PFS benefit was observed in the ESR1 wild-type subgroup (P = .543).
Study details: Findings are from a meta-analysis of individual patient data from four randomized clinical trials including 1290 patients with ER+/HER2− metastatic BC who received oral SERD or ET of physician’s choice.
Disclosures: This study did not receive any specific funding. Some authors declared receiving honoraria, research funding, or travel grants from or serving in advisory or consulting roles for various sources.
Source: Wong NZH et al. Efficacy of oral SERDs in the treatment of ER+, HER2 - metastatic breast cancer, a stratified analysis of the ESR1 wild type and mutant subgroups. Ann Oncol. 2023 (Oct 21). doi: 10.1016/j.annonc.2023.10.122