User login
The rising cost of the pneumococcal vaccine: What gives?
Every November, like clockwork, she gets the same letter, said Lindsay Irvin, MD, a pediatrician in San Antonio.
It’s from the drug company Pfizer, and it informs her that the price tag for the pneumococcal vaccine Prevnar 13 is going up. Again.
And it makes her angry.
“They’re the only ones who make it,” she said. “It’s like buying gas in a hurricane – or Coke in an airport. They charge what they want to.”
The Advisory Committee on Immunization Practices (ACIP) recommends Prevnar 13 for all children younger than 2 years – given at 2, 4, 6, and 15 months – as well as for adults aged 65 years and older.
The vaccine’s formulation has remained mostly unchanged since its 2010 federal approval, but its price continues creeping up, increasing by about 5%-6% most years. In just 8 years, its cost has climbed by more than 50%.
It is among the most expensive vaccines Dr. Irvin provides her young patients.
Doctors and clinics purchase the vaccine and then, once they inject patients, they typically recoup the cost through patients’ insurance coverage. In most cases there are no out-of-pocket costs.
But the steady rise in prices for branded drugs contributes indirectly to rises in premiums, deductibles, and government health spending, analysts say.
A full pediatric course of the vaccine typically involves four shots. In 2010, a single shot cost about $109, according to pricing archives kept by the CDC. It currently costs about $170, according to those archives. Next year, Pfizer says, a shot will cost almost $180.
“Pfizer and other drug companies are raising their prices because they can,” said Gerard Anderson, PhD, a health policy professor at Johns Hopkins University who studies drug pricing. “They have a patent, and they have a CDC recommendation, which is a double whammy – and a strong incentive for price increases.”
The company disagrees – arguing vaccine pricing supports research for new immunizations, along with ongoing efforts to keep products safe and to improve effectiveness. For instance, Prevnar 13’s shelf life was extended from 2 years to 3 years this year. Pricing also doesn’t affect access.
“Thanks to comprehensive health authority guidelines, Prevnar 13 is one of the most widely available public health interventions, supported by broad insurance coverage and innovative federal programs that guarantee access to vulnerable populations,” Pfizer spokeswoman Sally Beatty said in an interview.
But such arguments don’t justify the pattern of “consistent price increases,” suggested Ameet Sarpatwari, PhD, an epidemiologist and lawyer at Harvard Medical School, Boston, who studies drug policy.
“Does that explain what’s going on? Probably not,” he said. “The onus should be on them to show us why this is needed.”
Consumers are not likely to feel a pinch from these increases directly. The Affordable Care Act requires that ACIP-recommended vaccines are covered by insurance, with no cost sharing.
There are other implications, though.
Higher vaccine prices make it harder for physicians to stock up, noted Michael Munger, MD, a family physician in Overland Park, Kan., and president of the American Academy of Family Physicians.
They have to buy immunizations in advance to provide them for patients. Insurance will eventually reimburse them – typically at cost – but it can take months for that to come through, which is an especially tough proposition for small practices on tight budgets.
“You’ve got to keep track of your inventory, and make sure you don’t have any waste, and are going to get adequate reimbursement,” he said. “The cost of vaccines is definitely something in primary care we worry about, because we’re on thin margins. ... You don’t want to provide a service you lose money on, even if it’s as important as immunization.”
Gardasil, a human papillomavirus vaccine, has also seen its price climbing. And, in a similar response, ob.gyns. are providing it in smaller numbers.
A vaccine like Prevnar 13 is harder to make than older vaccines that are much cheaper, said William Moss, MD, a professor at Johns Hopkins Bloomberg School of Public Health, Baltimore, who specializes in vaccines and global children’s health. It provides immunization for 13 different variations of pneumococcal infection. That makes it a more effective vaccine, but also one that requires greater investment.
Critics, however, note that those investments were made by another company, Wyeth Pharmaceuticals. Pfizer bought Wyeth in 2009, along with the rights to the vaccine.
KHN’s coverage of prescription drug development, costs, and pricing is supported by the Laura and John Arnold Foundation. Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Every November, like clockwork, she gets the same letter, said Lindsay Irvin, MD, a pediatrician in San Antonio.
It’s from the drug company Pfizer, and it informs her that the price tag for the pneumococcal vaccine Prevnar 13 is going up. Again.
And it makes her angry.
“They’re the only ones who make it,” she said. “It’s like buying gas in a hurricane – or Coke in an airport. They charge what they want to.”
The Advisory Committee on Immunization Practices (ACIP) recommends Prevnar 13 for all children younger than 2 years – given at 2, 4, 6, and 15 months – as well as for adults aged 65 years and older.
The vaccine’s formulation has remained mostly unchanged since its 2010 federal approval, but its price continues creeping up, increasing by about 5%-6% most years. In just 8 years, its cost has climbed by more than 50%.
It is among the most expensive vaccines Dr. Irvin provides her young patients.
Doctors and clinics purchase the vaccine and then, once they inject patients, they typically recoup the cost through patients’ insurance coverage. In most cases there are no out-of-pocket costs.
But the steady rise in prices for branded drugs contributes indirectly to rises in premiums, deductibles, and government health spending, analysts say.
A full pediatric course of the vaccine typically involves four shots. In 2010, a single shot cost about $109, according to pricing archives kept by the CDC. It currently costs about $170, according to those archives. Next year, Pfizer says, a shot will cost almost $180.
“Pfizer and other drug companies are raising their prices because they can,” said Gerard Anderson, PhD, a health policy professor at Johns Hopkins University who studies drug pricing. “They have a patent, and they have a CDC recommendation, which is a double whammy – and a strong incentive for price increases.”
The company disagrees – arguing vaccine pricing supports research for new immunizations, along with ongoing efforts to keep products safe and to improve effectiveness. For instance, Prevnar 13’s shelf life was extended from 2 years to 3 years this year. Pricing also doesn’t affect access.
“Thanks to comprehensive health authority guidelines, Prevnar 13 is one of the most widely available public health interventions, supported by broad insurance coverage and innovative federal programs that guarantee access to vulnerable populations,” Pfizer spokeswoman Sally Beatty said in an interview.
But such arguments don’t justify the pattern of “consistent price increases,” suggested Ameet Sarpatwari, PhD, an epidemiologist and lawyer at Harvard Medical School, Boston, who studies drug policy.
“Does that explain what’s going on? Probably not,” he said. “The onus should be on them to show us why this is needed.”
Consumers are not likely to feel a pinch from these increases directly. The Affordable Care Act requires that ACIP-recommended vaccines are covered by insurance, with no cost sharing.
There are other implications, though.
Higher vaccine prices make it harder for physicians to stock up, noted Michael Munger, MD, a family physician in Overland Park, Kan., and president of the American Academy of Family Physicians.
They have to buy immunizations in advance to provide them for patients. Insurance will eventually reimburse them – typically at cost – but it can take months for that to come through, which is an especially tough proposition for small practices on tight budgets.
“You’ve got to keep track of your inventory, and make sure you don’t have any waste, and are going to get adequate reimbursement,” he said. “The cost of vaccines is definitely something in primary care we worry about, because we’re on thin margins. ... You don’t want to provide a service you lose money on, even if it’s as important as immunization.”
Gardasil, a human papillomavirus vaccine, has also seen its price climbing. And, in a similar response, ob.gyns. are providing it in smaller numbers.
A vaccine like Prevnar 13 is harder to make than older vaccines that are much cheaper, said William Moss, MD, a professor at Johns Hopkins Bloomberg School of Public Health, Baltimore, who specializes in vaccines and global children’s health. It provides immunization for 13 different variations of pneumococcal infection. That makes it a more effective vaccine, but also one that requires greater investment.
Critics, however, note that those investments were made by another company, Wyeth Pharmaceuticals. Pfizer bought Wyeth in 2009, along with the rights to the vaccine.
KHN’s coverage of prescription drug development, costs, and pricing is supported by the Laura and John Arnold Foundation. Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Every November, like clockwork, she gets the same letter, said Lindsay Irvin, MD, a pediatrician in San Antonio.
It’s from the drug company Pfizer, and it informs her that the price tag for the pneumococcal vaccine Prevnar 13 is going up. Again.
And it makes her angry.
“They’re the only ones who make it,” she said. “It’s like buying gas in a hurricane – or Coke in an airport. They charge what they want to.”
The Advisory Committee on Immunization Practices (ACIP) recommends Prevnar 13 for all children younger than 2 years – given at 2, 4, 6, and 15 months – as well as for adults aged 65 years and older.
The vaccine’s formulation has remained mostly unchanged since its 2010 federal approval, but its price continues creeping up, increasing by about 5%-6% most years. In just 8 years, its cost has climbed by more than 50%.
It is among the most expensive vaccines Dr. Irvin provides her young patients.
Doctors and clinics purchase the vaccine and then, once they inject patients, they typically recoup the cost through patients’ insurance coverage. In most cases there are no out-of-pocket costs.
But the steady rise in prices for branded drugs contributes indirectly to rises in premiums, deductibles, and government health spending, analysts say.
A full pediatric course of the vaccine typically involves four shots. In 2010, a single shot cost about $109, according to pricing archives kept by the CDC. It currently costs about $170, according to those archives. Next year, Pfizer says, a shot will cost almost $180.
“Pfizer and other drug companies are raising their prices because they can,” said Gerard Anderson, PhD, a health policy professor at Johns Hopkins University who studies drug pricing. “They have a patent, and they have a CDC recommendation, which is a double whammy – and a strong incentive for price increases.”
The company disagrees – arguing vaccine pricing supports research for new immunizations, along with ongoing efforts to keep products safe and to improve effectiveness. For instance, Prevnar 13’s shelf life was extended from 2 years to 3 years this year. Pricing also doesn’t affect access.
“Thanks to comprehensive health authority guidelines, Prevnar 13 is one of the most widely available public health interventions, supported by broad insurance coverage and innovative federal programs that guarantee access to vulnerable populations,” Pfizer spokeswoman Sally Beatty said in an interview.
But such arguments don’t justify the pattern of “consistent price increases,” suggested Ameet Sarpatwari, PhD, an epidemiologist and lawyer at Harvard Medical School, Boston, who studies drug policy.
“Does that explain what’s going on? Probably not,” he said. “The onus should be on them to show us why this is needed.”
Consumers are not likely to feel a pinch from these increases directly. The Affordable Care Act requires that ACIP-recommended vaccines are covered by insurance, with no cost sharing.
There are other implications, though.
Higher vaccine prices make it harder for physicians to stock up, noted Michael Munger, MD, a family physician in Overland Park, Kan., and president of the American Academy of Family Physicians.
They have to buy immunizations in advance to provide them for patients. Insurance will eventually reimburse them – typically at cost – but it can take months for that to come through, which is an especially tough proposition for small practices on tight budgets.
“You’ve got to keep track of your inventory, and make sure you don’t have any waste, and are going to get adequate reimbursement,” he said. “The cost of vaccines is definitely something in primary care we worry about, because we’re on thin margins. ... You don’t want to provide a service you lose money on, even if it’s as important as immunization.”
Gardasil, a human papillomavirus vaccine, has also seen its price climbing. And, in a similar response, ob.gyns. are providing it in smaller numbers.
A vaccine like Prevnar 13 is harder to make than older vaccines that are much cheaper, said William Moss, MD, a professor at Johns Hopkins Bloomberg School of Public Health, Baltimore, who specializes in vaccines and global children’s health. It provides immunization for 13 different variations of pneumococcal infection. That makes it a more effective vaccine, but also one that requires greater investment.
Critics, however, note that those investments were made by another company, Wyeth Pharmaceuticals. Pfizer bought Wyeth in 2009, along with the rights to the vaccine.
KHN’s coverage of prescription drug development, costs, and pricing is supported by the Laura and John Arnold Foundation. Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Can Antihypertensive Medication Reduce Dementia Risk in Older Adults?
LONDON—Calcium channel blockers and angiotensin receptor blockers are independently associated with a decreased risk of dementia in older patients, according to a study presented at the 2017 Alzheimer’s Association International Conference and published in the October issue of Journal of Hypertension.
“Calcium channel blockers regulate calcium influx, which may prevent neuronal cell death, inhibit the production of amyloid beta and neurofibrillary tangles, and improve cerebrovascular perfusion. Angiotensin receptor blockers may improve the cerebral blood flow, decrease levels of amyloid beta, and have an anti-inflammatory effect,” said Tessa van Middelaar, MD, a PhD student in the Department of Neurology at the Academic Medical Center in Amsterdam and the Radboud University Medical Center in Nijmegen.

Epidemiologic evidence has suggested that hypertension is an important risk factor for dementia. Trials studying the effect of antihypertensive medication on the incidence of dementia have had inconclusive results, however.
Post Hoc Analysis of Data
Dr. van Middelaar and colleagues conducted a post hoc analysis of data from the Prevention of Dementia by Intensive Vascular Care (preDIVA) trial. In this randomized controlled trial, 3,526 community-dwelling adults ranging in age from 70 to 78 received either intensive vascular care or standard care.
At baseline and during follow-up, data on medication use and medical history were collected every two years. Researchers identified five classes of antihypertensive medication used by participants (ie, beta-blockers, diuretics, ACE inhibitors, calcium channel blockers, and angiotensin receptor blockers).
The investigators used the Mini-Mental State Examination to measure cognition and defined dementia using the Diagnostic and Statistical Manual of Mental Disorders IV criteria. Analyses were restricted to participants who were using antihypertensive medications at baseline. Dr. van Middelaar and colleagues compared incident dementia rates associated with the use of different antihypertensive medication classes, including monotherapy and combination therapy, with those associ
Two Classes Reduced Dementia Risk
At baseline, 1,951 patients used antihypertensive medication. The study population’s mean age was 74.4, and 46.2% of participants were men. In all, 986 patients used beta-blockers, 798 used diuretics, 623 used ACE inhibitors, 522 used calcium channel blockers, and 402 used angiotensin receptor blockers. After a median of 6.7 years of follow-up, 136 participants developed dementia.
The use of calcium channel blockers and the use of angiotensin receptor blockers were associated with a lower incidence of dementia, compared with the use of other antihypertensive medications (hazard ratios, 0.56 and 0.60, respectively). The reduced risk of dementia associated with calcium channel blockers was the most evident in participants without a history of cardiovascular disease and those with uncontrolled hypertension. “A possible explanation for this increased benefit in older people without a history of cardiovascular disease could be related to less pronounced vascular lesions and, therefore, more brain reserve and capacity for functional resilience to cognitive decline,” said Dr. van Middelaar and colleagues. Researchers also found that systolic blood pressure was not significantly lower in participants using calcium channel blockers or angiotensin receptor blockers.
—Erica Tricarico
Suggested Reading
van Middelaar T, van Vught LA, Moll van Charante EP, et al. Lower dementia risk with different classes of antihypertensive medication in older patients. J Hypertens. 2017;35(10):2095-2101.
LONDON—Calcium channel blockers and angiotensin receptor blockers are independently associated with a decreased risk of dementia in older patients, according to a study presented at the 2017 Alzheimer’s Association International Conference and published in the October issue of Journal of Hypertension.
“Calcium channel blockers regulate calcium influx, which may prevent neuronal cell death, inhibit the production of amyloid beta and neurofibrillary tangles, and improve cerebrovascular perfusion. Angiotensin receptor blockers may improve the cerebral blood flow, decrease levels of amyloid beta, and have an anti-inflammatory effect,” said Tessa van Middelaar, MD, a PhD student in the Department of Neurology at the Academic Medical Center in Amsterdam and the Radboud University Medical Center in Nijmegen.
Epidemiologic evidence has suggested that hypertension is an important risk factor for dementia. Trials studying the effect of antihypertensive medication on the incidence of dementia have had inconclusive results, however.
Post Hoc Analysis of Data
Dr. van Middelaar and colleagues conducted a post hoc analysis of data from the Prevention of Dementia by Intensive Vascular Care (preDIVA) trial. In this randomized controlled trial, 3,526 community-dwelling adults ranging in age from 70 to 78 received either intensive vascular care or standard care.
At baseline and during follow-up, data on medication use and medical history were collected every two years. Researchers identified five classes of antihypertensive medication used by participants (ie, beta-blockers, diuretics, ACE inhibitors, calcium channel blockers, and angiotensin receptor blockers).
The investigators used the Mini-Mental State Examination to measure cognition and defined dementia using the Diagnostic and Statistical Manual of Mental Disorders IV criteria. Analyses were restricted to participants who were using antihypertensive medications at baseline. Dr. van Middelaar and colleagues compared incident dementia rates associated with the use of different antihypertensive medication classes, including monotherapy and combination therapy, with those associ
Two Classes Reduced Dementia Risk
At baseline, 1,951 patients used antihypertensive medication. The study population’s mean age was 74.4, and 46.2% of participants were men. In all, 986 patients used beta-blockers, 798 used diuretics, 623 used ACE inhibitors, 522 used calcium channel blockers, and 402 used angiotensin receptor blockers. After a median of 6.7 years of follow-up, 136 participants developed dementia.
The use of calcium channel blockers and the use of angiotensin receptor blockers were associated with a lower incidence of dementia, compared with the use of other antihypertensive medications (hazard ratios, 0.56 and 0.60, respectively). The reduced risk of dementia associated with calcium channel blockers was the most evident in participants without a history of cardiovascular disease and those with uncontrolled hypertension. “A possible explanation for this increased benefit in older people without a history of cardiovascular disease could be related to less pronounced vascular lesions and, therefore, more brain reserve and capacity for functional resilience to cognitive decline,” said Dr. van Middelaar and colleagues. Researchers also found that systolic blood pressure was not significantly lower in participants using calcium channel blockers or angiotensin receptor blockers.
—Erica Tricarico
Suggested Reading
van Middelaar T, van Vught LA, Moll van Charante EP, et al. Lower dementia risk with different classes of antihypertensive medication in older patients. J Hypertens. 2017;35(10):2095-2101.
LONDON—Calcium channel blockers and angiotensin receptor blockers are independently associated with a decreased risk of dementia in older patients, according to a study presented at the 2017 Alzheimer’s Association International Conference and published in the October issue of Journal of Hypertension.
“Calcium channel blockers regulate calcium influx, which may prevent neuronal cell death, inhibit the production of amyloid beta and neurofibrillary tangles, and improve cerebrovascular perfusion. Angiotensin receptor blockers may improve the cerebral blood flow, decrease levels of amyloid beta, and have an anti-inflammatory effect,” said Tessa van Middelaar, MD, a PhD student in the Department of Neurology at the Academic Medical Center in Amsterdam and the Radboud University Medical Center in Nijmegen.
Epidemiologic evidence has suggested that hypertension is an important risk factor for dementia. Trials studying the effect of antihypertensive medication on the incidence of dementia have had inconclusive results, however.
Post Hoc Analysis of Data
Dr. van Middelaar and colleagues conducted a post hoc analysis of data from the Prevention of Dementia by Intensive Vascular Care (preDIVA) trial. In this randomized controlled trial, 3,526 community-dwelling adults ranging in age from 70 to 78 received either intensive vascular care or standard care.
At baseline and during follow-up, data on medication use and medical history were collected every two years. Researchers identified five classes of antihypertensive medication used by participants (ie, beta-blockers, diuretics, ACE inhibitors, calcium channel blockers, and angiotensin receptor blockers).
The investigators used the Mini-Mental State Examination to measure cognition and defined dementia using the Diagnostic and Statistical Manual of Mental Disorders IV criteria. Analyses were restricted to participants who were using antihypertensive medications at baseline. Dr. van Middelaar and colleagues compared incident dementia rates associated with the use of different antihypertensive medication classes, including monotherapy and combination therapy, with those associ
Two Classes Reduced Dementia Risk
At baseline, 1,951 patients used antihypertensive medication. The study population’s mean age was 74.4, and 46.2% of participants were men. In all, 986 patients used beta-blockers, 798 used diuretics, 623 used ACE inhibitors, 522 used calcium channel blockers, and 402 used angiotensin receptor blockers. After a median of 6.7 years of follow-up, 136 participants developed dementia.
The use of calcium channel blockers and the use of angiotensin receptor blockers were associated with a lower incidence of dementia, compared with the use of other antihypertensive medications (hazard ratios, 0.56 and 0.60, respectively). The reduced risk of dementia associated with calcium channel blockers was the most evident in participants without a history of cardiovascular disease and those with uncontrolled hypertension. “A possible explanation for this increased benefit in older people without a history of cardiovascular disease could be related to less pronounced vascular lesions and, therefore, more brain reserve and capacity for functional resilience to cognitive decline,” said Dr. van Middelaar and colleagues. Researchers also found that systolic blood pressure was not significantly lower in participants using calcium channel blockers or angiotensin receptor blockers.
—Erica Tricarico
Suggested Reading
van Middelaar T, van Vught LA, Moll van Charante EP, et al. Lower dementia risk with different classes of antihypertensive medication in older patients. J Hypertens. 2017;35(10):2095-2101.
Real-world data support extended cervical cancer screening interval
A cervical cancer screening interval of 5 years or longer may be safe for women with one or more negative cotests using the high-risk human papillomavirus (HPV) test and cervical cytology, according to the results of a large observational study.
The findings also suggest that one or two negative HPV tests, regardless of cytology, may be enough to extend the screening interval, the researchers noted.
The 5-year risk decreased with each successive HPV-negative and cytology-negative result, falling from 0.098% in the first cotesting round, to 0.052% in the second round, and 0.035% in the third round. The 5-year risks were similar with an HPV-negative cotest result, regardless of the outcome of cytology: 0.114%, 0.061%, and 0.041%.
The results are based on data from more than 990,000 women in the Kaiser Permanente Northern California health care system who had one or more cotests from 2003 to 2014.
These findings come as the U.S. Preventive Services Task Force (USPSTF) has issued draft recommendations that would call for either HPV testing alone every 5 years starting at age 30 years or cytology every 3 years, but no cotesting. The American College of Obstetricians and Gynecologists currently recommends cotesting with cytology and HPV testing every 5 years or cytology alone every 3 years in women aged 30-65 years (Obstet Gynecol. 2016;128[4]:e111-30).
The study was partially funded by the National Cancer Institute. Some of the researchers are employees of the National Cancer Institute. The researchers reported receiving cervical screening tests and diagnostics at reduced or no cost from Roche, BD Biosciences, Cepheid, Arbor Vita, and Hologic.
The study by Castle et al provides important insight into the effects of repeated testing of HPV, cervical cytology, and testing both together. The authors are to be commended for performing their analysis, which involved almost 1 million women.
There are several important observations that can be made. One is that the risk of detecting cancer is decreased after cotesting HPV and cervical cytology both show a negative result, and that this risk is further reduced after the second and third negative HPV/cytology test. This could mean less frequent screening could be safely adopted in women who are HPV and cytology negative, which would lead to fewer women referred for unnecessary colposcopy and treatment, reducing their stress and discomfort.
These data could alter how women are triaged on the basis of the HPV test. In HPV-positive women, for example, current practices are appropriate for the first and perhaps second round of HPV testing, but perhaps subsequent rounds could become less stringent. Conversely, when there is an HPV-negative result, even in the presence of a positive or equivocal cytology, screening intervals may still be safe to lengthen as there is only a small increase in risk versus being both HPV and cytology negative.
Guglielmo Ronco, MD, PhD, is formerly of the City of Health and Science University Hospital of Turin (Italy). Silvia Franceschi, MD, PhD, is with the International Agency for Research on Cancer in Lyon, France. They reported having no relevant financial disclosures. Their comments were adapted from an accompanying editorial (Ann Intern Med. 2017. doi: 10.7326/M17-2872).
The study by Castle et al provides important insight into the effects of repeated testing of HPV, cervical cytology, and testing both together. The authors are to be commended for performing their analysis, which involved almost 1 million women.
There are several important observations that can be made. One is that the risk of detecting cancer is decreased after cotesting HPV and cervical cytology both show a negative result, and that this risk is further reduced after the second and third negative HPV/cytology test. This could mean less frequent screening could be safely adopted in women who are HPV and cytology negative, which would lead to fewer women referred for unnecessary colposcopy and treatment, reducing their stress and discomfort.
These data could alter how women are triaged on the basis of the HPV test. In HPV-positive women, for example, current practices are appropriate for the first and perhaps second round of HPV testing, but perhaps subsequent rounds could become less stringent. Conversely, when there is an HPV-negative result, even in the presence of a positive or equivocal cytology, screening intervals may still be safe to lengthen as there is only a small increase in risk versus being both HPV and cytology negative.
Guglielmo Ronco, MD, PhD, is formerly of the City of Health and Science University Hospital of Turin (Italy). Silvia Franceschi, MD, PhD, is with the International Agency for Research on Cancer in Lyon, France. They reported having no relevant financial disclosures. Their comments were adapted from an accompanying editorial (Ann Intern Med. 2017. doi: 10.7326/M17-2872).
The study by Castle et al provides important insight into the effects of repeated testing of HPV, cervical cytology, and testing both together. The authors are to be commended for performing their analysis, which involved almost 1 million women.
There are several important observations that can be made. One is that the risk of detecting cancer is decreased after cotesting HPV and cervical cytology both show a negative result, and that this risk is further reduced after the second and third negative HPV/cytology test. This could mean less frequent screening could be safely adopted in women who are HPV and cytology negative, which would lead to fewer women referred for unnecessary colposcopy and treatment, reducing their stress and discomfort.
These data could alter how women are triaged on the basis of the HPV test. In HPV-positive women, for example, current practices are appropriate for the first and perhaps second round of HPV testing, but perhaps subsequent rounds could become less stringent. Conversely, when there is an HPV-negative result, even in the presence of a positive or equivocal cytology, screening intervals may still be safe to lengthen as there is only a small increase in risk versus being both HPV and cytology negative.
Guglielmo Ronco, MD, PhD, is formerly of the City of Health and Science University Hospital of Turin (Italy). Silvia Franceschi, MD, PhD, is with the International Agency for Research on Cancer in Lyon, France. They reported having no relevant financial disclosures. Their comments were adapted from an accompanying editorial (Ann Intern Med. 2017. doi: 10.7326/M17-2872).
A cervical cancer screening interval of 5 years or longer may be safe for women with one or more negative cotests using the high-risk human papillomavirus (HPV) test and cervical cytology, according to the results of a large observational study.
The findings also suggest that one or two negative HPV tests, regardless of cytology, may be enough to extend the screening interval, the researchers noted.
The 5-year risk decreased with each successive HPV-negative and cytology-negative result, falling from 0.098% in the first cotesting round, to 0.052% in the second round, and 0.035% in the third round. The 5-year risks were similar with an HPV-negative cotest result, regardless of the outcome of cytology: 0.114%, 0.061%, and 0.041%.
The results are based on data from more than 990,000 women in the Kaiser Permanente Northern California health care system who had one or more cotests from 2003 to 2014.
These findings come as the U.S. Preventive Services Task Force (USPSTF) has issued draft recommendations that would call for either HPV testing alone every 5 years starting at age 30 years or cytology every 3 years, but no cotesting. The American College of Obstetricians and Gynecologists currently recommends cotesting with cytology and HPV testing every 5 years or cytology alone every 3 years in women aged 30-65 years (Obstet Gynecol. 2016;128[4]:e111-30).
The study was partially funded by the National Cancer Institute. Some of the researchers are employees of the National Cancer Institute. The researchers reported receiving cervical screening tests and diagnostics at reduced or no cost from Roche, BD Biosciences, Cepheid, Arbor Vita, and Hologic.
A cervical cancer screening interval of 5 years or longer may be safe for women with one or more negative cotests using the high-risk human papillomavirus (HPV) test and cervical cytology, according to the results of a large observational study.
The findings also suggest that one or two negative HPV tests, regardless of cytology, may be enough to extend the screening interval, the researchers noted.
The 5-year risk decreased with each successive HPV-negative and cytology-negative result, falling from 0.098% in the first cotesting round, to 0.052% in the second round, and 0.035% in the third round. The 5-year risks were similar with an HPV-negative cotest result, regardless of the outcome of cytology: 0.114%, 0.061%, and 0.041%.
The results are based on data from more than 990,000 women in the Kaiser Permanente Northern California health care system who had one or more cotests from 2003 to 2014.
These findings come as the U.S. Preventive Services Task Force (USPSTF) has issued draft recommendations that would call for either HPV testing alone every 5 years starting at age 30 years or cytology every 3 years, but no cotesting. The American College of Obstetricians and Gynecologists currently recommends cotesting with cytology and HPV testing every 5 years or cytology alone every 3 years in women aged 30-65 years (Obstet Gynecol. 2016;128[4]:e111-30).
The study was partially funded by the National Cancer Institute. Some of the researchers are employees of the National Cancer Institute. The researchers reported receiving cervical screening tests and diagnostics at reduced or no cost from Roche, BD Biosciences, Cepheid, Arbor Vita, and Hologic.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point:
Major finding: The 5-year cumulative detection of invasive cervical cancer, CIN3, and adenocarcinoma in situ decreased with each successive HPV-negative and cytology-negative result (0.098%, 0.052%, and 0.035%).
Data source: Data on more than 990,000 women who received cotesting between 2003 and 2014.
Disclosures: The research was partially supported by the National Cancer Institute. Some of the researchers are employees of the National Cancer Institute. The researchers reported receiving cervical screening tests and diagnostics at reduced or no cost from Roche, BD Biosciences, Cepheid, Arbor Vita, and Hologic.
From the Washington Office: Year Two of MIPS …The song remains the same (largely)
The interim final rule for the second year of Centers for Medicare and Medicaid Services (CMS) Quality Payment Program (QPP) was released on November 2, 2017. This rule will apply to performance and reporting for calendar year 2018 and impact payment in 2020. Below, I have highlighted a few of the key components of the 1,653-page rule with special attention to the Merit-based Incentive Payment System (MIPS).
To briefly review, there are two pathways for participation in the QPP, namely MIPS and the Advanced Alternative Payment Models (A-APMs). For 2018, we still expect that the majority of surgeons eligible to participate in the QPP will do so via the MIPS pathway. That said, and for reasons discussed below, CMS estimates that approximately half of the 1.2 million MIPS-eligible clinicians will be required to submit MIPS data in 2018. In addition, CMS estimates that approximately 200,000 eligible clinicians will participate in the QPP in 2018 via the A-APMs.

1) Quality – For 2018, CMS continues to require reporting on six measures, one of which must be an outcome measure or other high-priority measure. Should surgeons choose to report on more than six measures, CMS will use the six with the highest score for purposes of calculating their score for the Quality component. However, CMS did increase the percentage of patients on which reporting is required, aka the completeness threshold, in 2018 to 60%. Measures submitted that fall below the completeness threshold will receive one point. Small practices will receive three points for measures that fail to meet the completeness threshold. Multiple options remain available for submission of data, i.e., electronic health record (EHR), Medicare claims, a qualified registry or a qualified clinical data registry (QCDR). For 2018, the Quality component will make up 50% of the MIPS final score.
Cost
Those familiar with the 2017 version of MIPS will remember that the Cost component was weighted at zero for the first year of the program. CMS discussed, and indeed, initially proposed, to continue weighing cost at zero for 2018. However, because current law requires CMS to weigh cost at 30% beginning with the 2019 performance period, CMS finalized a 10% weight for cost in 2018 with the goal of making the impact of the transition in 2019 less dramatic. CMS will base its calculation of the cost component on the total per capita costs for all beneficiaries attributed to a provider and the Medicare Spending per Beneficiary measure for the entirety of the 2018 performance period. CMS intends to provide performance feedback on both measures by July 1, 2018. Surgeons are not required to submit data for purposes of cost component.
Advancing Care Information (ACI)
There are no major changes to the scoring policy for 2018 and all the applicable Base Score measures must still be reported in order to receive a score for the ACI component. The performance period requirement remains a minimum of 90 continuous days. For 2018, both 2014 Edition and 2015 Edition certified electronic health record technology (CEHRT) remain acceptable. However, those using only a 2015 Edition will be eligible for a 10% bonus. Regardless of edition used , bonus points are also available for reporting to a public health agency or clinical data registry and for the completion of an Improvement Activity (IA) using CEHRT. A significant hardship exemption remains available for those in small practices. As was the case in 2017, the ACI component represents 25% of the final score. However, as was also the case in 2017, one is not required to have an electronic health record to avoid a penalty in 2018.
Improvement Activities
The weight assigned to the IA component remains at 15%. CMS added 21 new IAs in the final rule, bringing the number of IA available from which to choose up to well over 110. CMS also made changes to 27 activities previously adopted. Reporting remains a simple attestation of participation in the activity for 90 continuous days. To receive full credit for the IA component, most surgeons will be required to attest to having participated in two, three, or four activities depending on whether the activities chosen are of medium value or high value. This is not a change. However, those in small or rural practices must only participate in one or two activities to receive full credit. It should be noted that for 2018, one will be able to avoid a penalty in 2020 solely by fulfillment of the requirements imposed by the Improvement Activities component.
As mentioned above, CMS estimates that only approximately 622,000 providers out of the 1.2 million eligible will be required to submit data under MIPS. Many providers are excluded from MIPS based on the low-volume threshold. For 2018, CMS set this threshold at less than or equal to $90,000 in Medicare Part B charges OR less than or equal to 200 Medicare Part B beneficiaries. The effect of this change, compared to the values set for 2017 low-volume threshold, is to exclude more providers from MIPS reporting.
Lastly, many will remember that for 2017, the performance threshold was set at three points, and thus, required only minimal reporting in either quality, ACI, or IA to avoid a penalty. It was expected that the threshold necessary to avoid a penalty for 2018 performance would be increased and indeed, CMS has set that value at 15. Those scoring above 15 will be eligible for a positive update in their Medicare payments in 2020, while those scoring below 15 will receive a penalty. Those who choose to not participate in 2018 will receive a 5% penalty in 2020. However, two points made above warrant reiteration:
a) By fully participating in the IA component, one can accrue the 15 points necessary to avoid a penalty.
b) An EHR is not required to avoid a penalty.
In the coming weeks, we will be updating the QPP website (www.facs.org/qpp) to reflect the changes in the program for 2018. New videos will be available as will be new electronic and print materials to assist Fellows to participate in the program.
Dr. Bailey is a pediatric surgeon and Medical Director, Advocacy, for the Division of Advocacy and Health Policy in the ACS offices in Washington, DC.
The interim final rule for the second year of Centers for Medicare and Medicaid Services (CMS) Quality Payment Program (QPP) was released on November 2, 2017. This rule will apply to performance and reporting for calendar year 2018 and impact payment in 2020. Below, I have highlighted a few of the key components of the 1,653-page rule with special attention to the Merit-based Incentive Payment System (MIPS).
To briefly review, there are two pathways for participation in the QPP, namely MIPS and the Advanced Alternative Payment Models (A-APMs). For 2018, we still expect that the majority of surgeons eligible to participate in the QPP will do so via the MIPS pathway. That said, and for reasons discussed below, CMS estimates that approximately half of the 1.2 million MIPS-eligible clinicians will be required to submit MIPS data in 2018. In addition, CMS estimates that approximately 200,000 eligible clinicians will participate in the QPP in 2018 via the A-APMs.
1) Quality – For 2018, CMS continues to require reporting on six measures, one of which must be an outcome measure or other high-priority measure. Should surgeons choose to report on more than six measures, CMS will use the six with the highest score for purposes of calculating their score for the Quality component. However, CMS did increase the percentage of patients on which reporting is required, aka the completeness threshold, in 2018 to 60%. Measures submitted that fall below the completeness threshold will receive one point. Small practices will receive three points for measures that fail to meet the completeness threshold. Multiple options remain available for submission of data, i.e., electronic health record (EHR), Medicare claims, a qualified registry or a qualified clinical data registry (QCDR). For 2018, the Quality component will make up 50% of the MIPS final score.
Cost
Those familiar with the 2017 version of MIPS will remember that the Cost component was weighted at zero for the first year of the program. CMS discussed, and indeed, initially proposed, to continue weighing cost at zero for 2018. However, because current law requires CMS to weigh cost at 30% beginning with the 2019 performance period, CMS finalized a 10% weight for cost in 2018 with the goal of making the impact of the transition in 2019 less dramatic. CMS will base its calculation of the cost component on the total per capita costs for all beneficiaries attributed to a provider and the Medicare Spending per Beneficiary measure for the entirety of the 2018 performance period. CMS intends to provide performance feedback on both measures by July 1, 2018. Surgeons are not required to submit data for purposes of cost component.
Advancing Care Information (ACI)
There are no major changes to the scoring policy for 2018 and all the applicable Base Score measures must still be reported in order to receive a score for the ACI component. The performance period requirement remains a minimum of 90 continuous days. For 2018, both 2014 Edition and 2015 Edition certified electronic health record technology (CEHRT) remain acceptable. However, those using only a 2015 Edition will be eligible for a 10% bonus. Regardless of edition used , bonus points are also available for reporting to a public health agency or clinical data registry and for the completion of an Improvement Activity (IA) using CEHRT. A significant hardship exemption remains available for those in small practices. As was the case in 2017, the ACI component represents 25% of the final score. However, as was also the case in 2017, one is not required to have an electronic health record to avoid a penalty in 2018.
Improvement Activities
The weight assigned to the IA component remains at 15%. CMS added 21 new IAs in the final rule, bringing the number of IA available from which to choose up to well over 110. CMS also made changes to 27 activities previously adopted. Reporting remains a simple attestation of participation in the activity for 90 continuous days. To receive full credit for the IA component, most surgeons will be required to attest to having participated in two, three, or four activities depending on whether the activities chosen are of medium value or high value. This is not a change. However, those in small or rural practices must only participate in one or two activities to receive full credit. It should be noted that for 2018, one will be able to avoid a penalty in 2020 solely by fulfillment of the requirements imposed by the Improvement Activities component.
As mentioned above, CMS estimates that only approximately 622,000 providers out of the 1.2 million eligible will be required to submit data under MIPS. Many providers are excluded from MIPS based on the low-volume threshold. For 2018, CMS set this threshold at less than or equal to $90,000 in Medicare Part B charges OR less than or equal to 200 Medicare Part B beneficiaries. The effect of this change, compared to the values set for 2017 low-volume threshold, is to exclude more providers from MIPS reporting.
Lastly, many will remember that for 2017, the performance threshold was set at three points, and thus, required only minimal reporting in either quality, ACI, or IA to avoid a penalty. It was expected that the threshold necessary to avoid a penalty for 2018 performance would be increased and indeed, CMS has set that value at 15. Those scoring above 15 will be eligible for a positive update in their Medicare payments in 2020, while those scoring below 15 will receive a penalty. Those who choose to not participate in 2018 will receive a 5% penalty in 2020. However, two points made above warrant reiteration:
a) By fully participating in the IA component, one can accrue the 15 points necessary to avoid a penalty.
b) An EHR is not required to avoid a penalty.
In the coming weeks, we will be updating the QPP website (www.facs.org/qpp) to reflect the changes in the program for 2018. New videos will be available as will be new electronic and print materials to assist Fellows to participate in the program.
Dr. Bailey is a pediatric surgeon and Medical Director, Advocacy, for the Division of Advocacy and Health Policy in the ACS offices in Washington, DC.
The interim final rule for the second year of Centers for Medicare and Medicaid Services (CMS) Quality Payment Program (QPP) was released on November 2, 2017. This rule will apply to performance and reporting for calendar year 2018 and impact payment in 2020. Below, I have highlighted a few of the key components of the 1,653-page rule with special attention to the Merit-based Incentive Payment System (MIPS).
To briefly review, there are two pathways for participation in the QPP, namely MIPS and the Advanced Alternative Payment Models (A-APMs). For 2018, we still expect that the majority of surgeons eligible to participate in the QPP will do so via the MIPS pathway. That said, and for reasons discussed below, CMS estimates that approximately half of the 1.2 million MIPS-eligible clinicians will be required to submit MIPS data in 2018. In addition, CMS estimates that approximately 200,000 eligible clinicians will participate in the QPP in 2018 via the A-APMs.
1) Quality – For 2018, CMS continues to require reporting on six measures, one of which must be an outcome measure or other high-priority measure. Should surgeons choose to report on more than six measures, CMS will use the six with the highest score for purposes of calculating their score for the Quality component. However, CMS did increase the percentage of patients on which reporting is required, aka the completeness threshold, in 2018 to 60%. Measures submitted that fall below the completeness threshold will receive one point. Small practices will receive three points for measures that fail to meet the completeness threshold. Multiple options remain available for submission of data, i.e., electronic health record (EHR), Medicare claims, a qualified registry or a qualified clinical data registry (QCDR). For 2018, the Quality component will make up 50% of the MIPS final score.
Cost
Those familiar with the 2017 version of MIPS will remember that the Cost component was weighted at zero for the first year of the program. CMS discussed, and indeed, initially proposed, to continue weighing cost at zero for 2018. However, because current law requires CMS to weigh cost at 30% beginning with the 2019 performance period, CMS finalized a 10% weight for cost in 2018 with the goal of making the impact of the transition in 2019 less dramatic. CMS will base its calculation of the cost component on the total per capita costs for all beneficiaries attributed to a provider and the Medicare Spending per Beneficiary measure for the entirety of the 2018 performance period. CMS intends to provide performance feedback on both measures by July 1, 2018. Surgeons are not required to submit data for purposes of cost component.
Advancing Care Information (ACI)
There are no major changes to the scoring policy for 2018 and all the applicable Base Score measures must still be reported in order to receive a score for the ACI component. The performance period requirement remains a minimum of 90 continuous days. For 2018, both 2014 Edition and 2015 Edition certified electronic health record technology (CEHRT) remain acceptable. However, those using only a 2015 Edition will be eligible for a 10% bonus. Regardless of edition used , bonus points are also available for reporting to a public health agency or clinical data registry and for the completion of an Improvement Activity (IA) using CEHRT. A significant hardship exemption remains available for those in small practices. As was the case in 2017, the ACI component represents 25% of the final score. However, as was also the case in 2017, one is not required to have an electronic health record to avoid a penalty in 2018.
Improvement Activities
The weight assigned to the IA component remains at 15%. CMS added 21 new IAs in the final rule, bringing the number of IA available from which to choose up to well over 110. CMS also made changes to 27 activities previously adopted. Reporting remains a simple attestation of participation in the activity for 90 continuous days. To receive full credit for the IA component, most surgeons will be required to attest to having participated in two, three, or four activities depending on whether the activities chosen are of medium value or high value. This is not a change. However, those in small or rural practices must only participate in one or two activities to receive full credit. It should be noted that for 2018, one will be able to avoid a penalty in 2020 solely by fulfillment of the requirements imposed by the Improvement Activities component.
As mentioned above, CMS estimates that only approximately 622,000 providers out of the 1.2 million eligible will be required to submit data under MIPS. Many providers are excluded from MIPS based on the low-volume threshold. For 2018, CMS set this threshold at less than or equal to $90,000 in Medicare Part B charges OR less than or equal to 200 Medicare Part B beneficiaries. The effect of this change, compared to the values set for 2017 low-volume threshold, is to exclude more providers from MIPS reporting.
Lastly, many will remember that for 2017, the performance threshold was set at three points, and thus, required only minimal reporting in either quality, ACI, or IA to avoid a penalty. It was expected that the threshold necessary to avoid a penalty for 2018 performance would be increased and indeed, CMS has set that value at 15. Those scoring above 15 will be eligible for a positive update in their Medicare payments in 2020, while those scoring below 15 will receive a penalty. Those who choose to not participate in 2018 will receive a 5% penalty in 2020. However, two points made above warrant reiteration:
a) By fully participating in the IA component, one can accrue the 15 points necessary to avoid a penalty.
b) An EHR is not required to avoid a penalty.
In the coming weeks, we will be updating the QPP website (www.facs.org/qpp) to reflect the changes in the program for 2018. New videos will be available as will be new electronic and print materials to assist Fellows to participate in the program.
Dr. Bailey is a pediatric surgeon and Medical Director, Advocacy, for the Division of Advocacy and Health Policy in the ACS offices in Washington, DC.
Two Antiamyloid Antibodies Will Undergo Further Study
BOSTON—Solanezumab and gantenerumab, both of which failed in phase III studies, will be examined in further trials at much higher doses, according to lectures presented at the 10th Edition of Clinical Trials on Alzheimer’s Disease.

Gantenerumab will be tested in two new phase III studies at a much higher dose than was used in the Scarlet Road and Marguerite Road studies, according to Gregory Klein, PhD, Principal Scientist and Biomarker Experimental Medicine Leader at Roche in Basel, Switzerland.
The Challenges of Antiamyloid Therapies
Solanezumab and gantenerumab are antiamyloid antibodies. In their prior trials, both effectively cleared amyloid plaques, but neither significantly improved cognition in patients with mild to moderate disease. Other studies of antiamyloid therapies have had similar results. Although these drugs stimulate several mechanisms of plaque removal, none has significantly improved thinking or memory.
Some investigators have questioned whether the drugs’ doses were high enough. Doses had been chosen with caution, partly because antiamyloid antibodies can cause amyloid-related imaging abnormalities (ARIA), an inflammatory response of brain edema or microhemorrhages. Concern over this side effect has decreased with the accumulation of more adverse-event data. Most cases of ARIA have been asymptomatic and resolved spontaneously. New open-label extension data from the Scarlet Road and Marguerite Road trials of gantenerumab, plus a new titration model, have increased confidence that patients will tolerate the antibody at subcutaneous doses as high as 1,200 mg.
Researchers also have examined the question of therapeutic timing. It is increasingly apparent that plaque eradication does not rescue cognition. It is possible that Alzheimer’s disease must be treated before amyloid and tau damage the hippocampus and neocortex.
After evaluating the failures of solanezumab and gantenerumab, researchers hypothesized that higher doses delivered earlier in the disease process might be effective, not at restoring lost cognition, but at preventing cognitive decline.
“One of the greatest advances in this field over the past 10 years is the recognition that Alzheimer’s disease is a continuum that likely begins well before the stage we recognize as dementia, and even before the stages of mild cognitive impairment [MCI] and prodromal Alzheimer’s [disease],” said Dr. Sperling. “Treating in the presymptomatic phase may be the best opportunity to bend this curve back toward the trajectory of normal aging.”
Other investigators hold that tau is the prime concern and the main cause of declines in memory and cognition. Tau is present in the brains of most people experiencing cognitively normal aging, but amyloid deposition may spur tau to spread into the neocortex. Preventing amyloid accumulation may prevent dementia not only by controlling amyloid levels, but also by removing a stimulus for the spread of tau.
Dr. Sperling cited unpublished data showing subtle cognitive decline in cognitively normal patients who have amyloid and tau in the brain. Although the cognitive scores were within the normal range, subjects with both proteins declined over two years on specific measures of memory and were more likely to progress to MCI.
“This [result] is striking to me and made me a little worried about the critical window of intervention,” she said. “What is also striking is that even though we restricted the eligibility criteria of A4 to those with normal memory and normal cognition, we do see that tau positivity at baseline is associated with lower baseline performance. Again, we have this suggestion that amyloid is associated with tau, and tau is associated with poor memory, even in normal people.”
A New Study of Solanezumab
Solanezumab’s failure in the series of EXPEDITION studies has prompted the modification of the A4 protocol. “We think solanezumab has an increased chance of success here [compared with the EXPEDITION trial] because we are employing it 10 to 15 years earlier in the disease. But we also want to maximize its chances.”
Thus, she said, investigators have decided to quadruple the dose administered in A4. Subjects will undergo titration from 600 mg to 800 mg for two months and then to 1,600 mg every four weeks. A safety cohort of 200 patients will be monitored for adverse events, especially hemorrhagic or edematous ARIA. “We are also extending the double-blind phase to 240 weeks, which allows everyone to dose-escalate and increases our power to detect small effect sizes,” said Dr. Sperling.
So far, 1,151 patients have been recruited into the study. Dr. Sperling expects the full 1,200-subject cohort to be randomized by the end of 2017.
Gantenerumab May Reduce Amyloid Burden
Gantenerumab will be examined in further trials, following investigators’ analyses of open-label extension data from the Scarlet Road and Marguerite Road studies, said Dr. Klein. Patients in these studies were randomized to either 105 mg or 225 mg of the antibody. Researchers observed no significant cognitive benefits of therapy, but noted trends toward improvement with the higher dose, as well as dose-dependent plaque clearance. These results encouraged researchers to examine higher doses in 52-week open-label extensions of each study.
Dr. Klein presented new imaging data for these studies. In both studies combined, 40 patients were maintained for six to nine months on the highest doses (ie, from 900 mg to 1,200 mg). Of these participants, 17 had almost total clearance of their amyloid burden. Their scans, Dr. Klein said, appear to show traces of amyloid or to be amyloid-negative. The effect was consistent, regardless of the amount of amyloid at baseline. “These are encouraging biomarker data,” he said. “We are going into our new phase III studies, Graduate I and II, optimistic.”
According to a Roche press release, these studies will target patients with prodromal to mild disease at the higher doses. Emails to Roche and its German partner, MorphoSys, were not returned by press time. Dr. Klein’s comments suggest that studies of gantenerumab will continue.
—Michele G. Sullivan
BOSTON—Solanezumab and gantenerumab, both of which failed in phase III studies, will be examined in further trials at much higher doses, according to lectures presented at the 10th Edition of Clinical Trials on Alzheimer’s Disease.
Gantenerumab will be tested in two new phase III studies at a much higher dose than was used in the Scarlet Road and Marguerite Road studies, according to Gregory Klein, PhD, Principal Scientist and Biomarker Experimental Medicine Leader at Roche in Basel, Switzerland.
The Challenges of Antiamyloid Therapies
Solanezumab and gantenerumab are antiamyloid antibodies. In their prior trials, both effectively cleared amyloid plaques, but neither significantly improved cognition in patients with mild to moderate disease. Other studies of antiamyloid therapies have had similar results. Although these drugs stimulate several mechanisms of plaque removal, none has significantly improved thinking or memory.
Some investigators have questioned whether the drugs’ doses were high enough. Doses had been chosen with caution, partly because antiamyloid antibodies can cause amyloid-related imaging abnormalities (ARIA), an inflammatory response of brain edema or microhemorrhages. Concern over this side effect has decreased with the accumulation of more adverse-event data. Most cases of ARIA have been asymptomatic and resolved spontaneously. New open-label extension data from the Scarlet Road and Marguerite Road trials of gantenerumab, plus a new titration model, have increased confidence that patients will tolerate the antibody at subcutaneous doses as high as 1,200 mg.
Researchers also have examined the question of therapeutic timing. It is increasingly apparent that plaque eradication does not rescue cognition. It is possible that Alzheimer’s disease must be treated before amyloid and tau damage the hippocampus and neocortex.
After evaluating the failures of solanezumab and gantenerumab, researchers hypothesized that higher doses delivered earlier in the disease process might be effective, not at restoring lost cognition, but at preventing cognitive decline.
“One of the greatest advances in this field over the past 10 years is the recognition that Alzheimer’s disease is a continuum that likely begins well before the stage we recognize as dementia, and even before the stages of mild cognitive impairment [MCI] and prodromal Alzheimer’s [disease],” said Dr. Sperling. “Treating in the presymptomatic phase may be the best opportunity to bend this curve back toward the trajectory of normal aging.”
Other investigators hold that tau is the prime concern and the main cause of declines in memory and cognition. Tau is present in the brains of most people experiencing cognitively normal aging, but amyloid deposition may spur tau to spread into the neocortex. Preventing amyloid accumulation may prevent dementia not only by controlling amyloid levels, but also by removing a stimulus for the spread of tau.
Dr. Sperling cited unpublished data showing subtle cognitive decline in cognitively normal patients who have amyloid and tau in the brain. Although the cognitive scores were within the normal range, subjects with both proteins declined over two years on specific measures of memory and were more likely to progress to MCI.
“This [result] is striking to me and made me a little worried about the critical window of intervention,” she said. “What is also striking is that even though we restricted the eligibility criteria of A4 to those with normal memory and normal cognition, we do see that tau positivity at baseline is associated with lower baseline performance. Again, we have this suggestion that amyloid is associated with tau, and tau is associated with poor memory, even in normal people.”
A New Study of Solanezumab
Solanezumab’s failure in the series of EXPEDITION studies has prompted the modification of the A4 protocol. “We think solanezumab has an increased chance of success here [compared with the EXPEDITION trial] because we are employing it 10 to 15 years earlier in the disease. But we also want to maximize its chances.”
Thus, she said, investigators have decided to quadruple the dose administered in A4. Subjects will undergo titration from 600 mg to 800 mg for two months and then to 1,600 mg every four weeks. A safety cohort of 200 patients will be monitored for adverse events, especially hemorrhagic or edematous ARIA. “We are also extending the double-blind phase to 240 weeks, which allows everyone to dose-escalate and increases our power to detect small effect sizes,” said Dr. Sperling.
So far, 1,151 patients have been recruited into the study. Dr. Sperling expects the full 1,200-subject cohort to be randomized by the end of 2017.
Gantenerumab May Reduce Amyloid Burden
Gantenerumab will be examined in further trials, following investigators’ analyses of open-label extension data from the Scarlet Road and Marguerite Road studies, said Dr. Klein. Patients in these studies were randomized to either 105 mg or 225 mg of the antibody. Researchers observed no significant cognitive benefits of therapy, but noted trends toward improvement with the higher dose, as well as dose-dependent plaque clearance. These results encouraged researchers to examine higher doses in 52-week open-label extensions of each study.
Dr. Klein presented new imaging data for these studies. In both studies combined, 40 patients were maintained for six to nine months on the highest doses (ie, from 900 mg to 1,200 mg). Of these participants, 17 had almost total clearance of their amyloid burden. Their scans, Dr. Klein said, appear to show traces of amyloid or to be amyloid-negative. The effect was consistent, regardless of the amount of amyloid at baseline. “These are encouraging biomarker data,” he said. “We are going into our new phase III studies, Graduate I and II, optimistic.”
According to a Roche press release, these studies will target patients with prodromal to mild disease at the higher doses. Emails to Roche and its German partner, MorphoSys, were not returned by press time. Dr. Klein’s comments suggest that studies of gantenerumab will continue.
—Michele G. Sullivan
BOSTON—Solanezumab and gantenerumab, both of which failed in phase III studies, will be examined in further trials at much higher doses, according to lectures presented at the 10th Edition of Clinical Trials on Alzheimer’s Disease.
Gantenerumab will be tested in two new phase III studies at a much higher dose than was used in the Scarlet Road and Marguerite Road studies, according to Gregory Klein, PhD, Principal Scientist and Biomarker Experimental Medicine Leader at Roche in Basel, Switzerland.
The Challenges of Antiamyloid Therapies
Solanezumab and gantenerumab are antiamyloid antibodies. In their prior trials, both effectively cleared amyloid plaques, but neither significantly improved cognition in patients with mild to moderate disease. Other studies of antiamyloid therapies have had similar results. Although these drugs stimulate several mechanisms of plaque removal, none has significantly improved thinking or memory.
Some investigators have questioned whether the drugs’ doses were high enough. Doses had been chosen with caution, partly because antiamyloid antibodies can cause amyloid-related imaging abnormalities (ARIA), an inflammatory response of brain edema or microhemorrhages. Concern over this side effect has decreased with the accumulation of more adverse-event data. Most cases of ARIA have been asymptomatic and resolved spontaneously. New open-label extension data from the Scarlet Road and Marguerite Road trials of gantenerumab, plus a new titration model, have increased confidence that patients will tolerate the antibody at subcutaneous doses as high as 1,200 mg.
Researchers also have examined the question of therapeutic timing. It is increasingly apparent that plaque eradication does not rescue cognition. It is possible that Alzheimer’s disease must be treated before amyloid and tau damage the hippocampus and neocortex.
After evaluating the failures of solanezumab and gantenerumab, researchers hypothesized that higher doses delivered earlier in the disease process might be effective, not at restoring lost cognition, but at preventing cognitive decline.
“One of the greatest advances in this field over the past 10 years is the recognition that Alzheimer’s disease is a continuum that likely begins well before the stage we recognize as dementia, and even before the stages of mild cognitive impairment [MCI] and prodromal Alzheimer’s [disease],” said Dr. Sperling. “Treating in the presymptomatic phase may be the best opportunity to bend this curve back toward the trajectory of normal aging.”
Other investigators hold that tau is the prime concern and the main cause of declines in memory and cognition. Tau is present in the brains of most people experiencing cognitively normal aging, but amyloid deposition may spur tau to spread into the neocortex. Preventing amyloid accumulation may prevent dementia not only by controlling amyloid levels, but also by removing a stimulus for the spread of tau.
Dr. Sperling cited unpublished data showing subtle cognitive decline in cognitively normal patients who have amyloid and tau in the brain. Although the cognitive scores were within the normal range, subjects with both proteins declined over two years on specific measures of memory and were more likely to progress to MCI.
“This [result] is striking to me and made me a little worried about the critical window of intervention,” she said. “What is also striking is that even though we restricted the eligibility criteria of A4 to those with normal memory and normal cognition, we do see that tau positivity at baseline is associated with lower baseline performance. Again, we have this suggestion that amyloid is associated with tau, and tau is associated with poor memory, even in normal people.”
A New Study of Solanezumab
Solanezumab’s failure in the series of EXPEDITION studies has prompted the modification of the A4 protocol. “We think solanezumab has an increased chance of success here [compared with the EXPEDITION trial] because we are employing it 10 to 15 years earlier in the disease. But we also want to maximize its chances.”
Thus, she said, investigators have decided to quadruple the dose administered in A4. Subjects will undergo titration from 600 mg to 800 mg for two months and then to 1,600 mg every four weeks. A safety cohort of 200 patients will be monitored for adverse events, especially hemorrhagic or edematous ARIA. “We are also extending the double-blind phase to 240 weeks, which allows everyone to dose-escalate and increases our power to detect small effect sizes,” said Dr. Sperling.
So far, 1,151 patients have been recruited into the study. Dr. Sperling expects the full 1,200-subject cohort to be randomized by the end of 2017.
Gantenerumab May Reduce Amyloid Burden
Gantenerumab will be examined in further trials, following investigators’ analyses of open-label extension data from the Scarlet Road and Marguerite Road studies, said Dr. Klein. Patients in these studies were randomized to either 105 mg or 225 mg of the antibody. Researchers observed no significant cognitive benefits of therapy, but noted trends toward improvement with the higher dose, as well as dose-dependent plaque clearance. These results encouraged researchers to examine higher doses in 52-week open-label extensions of each study.
Dr. Klein presented new imaging data for these studies. In both studies combined, 40 patients were maintained for six to nine months on the highest doses (ie, from 900 mg to 1,200 mg). Of these participants, 17 had almost total clearance of their amyloid burden. Their scans, Dr. Klein said, appear to show traces of amyloid or to be amyloid-negative. The effect was consistent, regardless of the amount of amyloid at baseline. “These are encouraging biomarker data,” he said. “We are going into our new phase III studies, Graduate I and II, optimistic.”
According to a Roche press release, these studies will target patients with prodromal to mild disease at the higher doses. Emails to Roche and its German partner, MorphoSys, were not returned by press time. Dr. Klein’s comments suggest that studies of gantenerumab will continue.
—Michele G. Sullivan
Bilateral ACP shown similar to unilateral in arch replacement study
What may be the largest study comparing unilateral and bilateral antegrade cerebral perfusion during total arch replacement for type A aortic dissection has reported that outcomes between the two approaches are comparable, although the bilateral approach showed some advantages during the operation itself, investigators from China reported in the Journal of Thoracic and Cardiovascular Surgery (2017;154:767-75).
The effectiveness of bilateral antegrade cerebral perfusion (b-ACP) vs. unilateral antegrade cerebral perfusion (u-ACP) has been the focus of extensive debate, lead study author Guang Tong, MD, of the Guangzhou (China) General Hospital, and coauthors said. They compared outcomes in six different metrics, ranging from cardiopulmonary bypass time to length of stay (LOS) in the ICU and hospital, in 203 patients with type A aortic dissection who had total aortic arch replacement with hypothermic circulatory arrest over an 8-year period ending in August 2014; 121 had b-ACP and 82 had u-ACP. “The issue of u-ACP vs. b-ACP has been examined in aortic arch surgery, but few reports have focused on type A aortic dissection,” Dr. Tong and coauthors wrote.
They acknowledged that some surgeons are reluctant to use b-ACP because of its complexity, but their study found no increase in cross-clamp time, cardiopulmonary bypass time, or surgery time in the b-ACP group. They cited another reason surgeons give for avoiding b-ACP: the risk of embolic injury caused by canulating the left common carotid artery in an atheromatous aorta. “In the present study, this risk was avoided by attaching the left common carotid artery to the four-branched prosthetic graft for left hemisphere perfusion,” Dr. Tong and coauthors wrote.
Key outcomes that the researchers found not statistically significant were:
- Overall 30-day mortality (11.6% for b-ACP vs. 20.7% for u-ACP; P = .075).
- Prevalence of postoperative permanent neurologic dysfunction (8.4% vs. 16.9%; P = .091).
- Average ICU LOS (16 ± 17.75 days vs. 17 ± 11.5 days, P =.454).
- Average hospital LOS (26.5 ± 20.6 days vs. 24.8 ± 10.3 days, P = .434).
However, average ventilation time was lower in the b-ACP group (95.5 hours vs. 147 hours; P less than or equal to.001).
Dr. Tong and coauthors used an aggressive approach, as advocated by Dhaval Trivedi, MD, and colleagues (Ann Thorac Surg. 2016;101:896-903), and had a total arch replacement rate of 57.8%. This rate is higher than most published series in the west but comparable to other studies from China, perhaps because of the relatively young age of this study cohort – an average age of 51 years – compared to data sets other studies have used. Dr. Tong and coauthors used a b-ACP strategy that established both cerebral perfusion routes before circulatory arrest.
Rates of the following complications were also not significantly different across the study population: paraplegia (2.8% for b-ACP vs. 3.1% for u-ACP), temporary neurologic dysfunction (4.7% vs. 9.2%), permanent neurologic dysfunction (8.4% vs. 16.9%), renal failure (18% vs. 23.1%), reoperation for bleeding (2.8% vs. 4.6%), and mediastinal infection (3.7% vs. 6.2%).
While b-ACP patients did not have a statistically significant lower incidence of TND, Dr. Tong and coauthors noted the shorter time on ventilation and significantly lower tracheostomy rates for the b-ACP patients, 3.7% vs. 16.9% for the u-ACP group (P = .003). “In our institute, protocols to wean patients from ventilation were normally initiated as soon as consciousness was regained,” Dr. Tong and coauthors wrote.
Among the study limits Dr. Tong and coauthors acknowledged were its retrospective, nonrandomized, single-center nature, and the fact that the surgeries were performed over an 8-year period representing different eras.
The investigators reported having no relevant financial disclosures.
The study by Dr. Tong and coauthors adds to the discussion between the “bilateralists” and “unilateralists,” as Jean Bachet, MD, called the two prevailing camps on cerebral perfusion strategies in his invited commentary (J Thorac Cardiovasc Surg. 2017;154:765-6). And while most clinical reports find outcomes similar between the two approaches, the evidence favors the bilateral approach for total arch replacement.
Citing how the study implied mortality and neurologic morbidity rates almost half those for unilateral perfusion, but not reaching statistical significance, Dr. Bachet said, “The statisticians would say that this is only a trend and no proof, but some trends might be indicative, and significance might only be a matter of number in each arm of the comparison.”
Dr. Bachet raised a question about the unilateral approach – that once the arch is opened it takes a minute or so to insert the small balloon canula into the origin of the left carotid artery or divided vessel and start bilateral perfusion. “A major question arises,” said Dr. Bachet: “Why should we expose our patients to any undue risk just to avoid a simple maneuver, to spare a little time, or for any other fancy and questionable reason?”
Cardiologists have raised that question for more than 20 years. Said Dr. Bachet, “We still wait for the answer.”
Dr. Bachet is a cardiac surgeon in Surgenes, France. He reported having no financial relationships to disclose.
The study by Dr. Tong and coauthors adds to the discussion between the “bilateralists” and “unilateralists,” as Jean Bachet, MD, called the two prevailing camps on cerebral perfusion strategies in his invited commentary (J Thorac Cardiovasc Surg. 2017;154:765-6). And while most clinical reports find outcomes similar between the two approaches, the evidence favors the bilateral approach for total arch replacement.
Citing how the study implied mortality and neurologic morbidity rates almost half those for unilateral perfusion, but not reaching statistical significance, Dr. Bachet said, “The statisticians would say that this is only a trend and no proof, but some trends might be indicative, and significance might only be a matter of number in each arm of the comparison.”
Dr. Bachet raised a question about the unilateral approach – that once the arch is opened it takes a minute or so to insert the small balloon canula into the origin of the left carotid artery or divided vessel and start bilateral perfusion. “A major question arises,” said Dr. Bachet: “Why should we expose our patients to any undue risk just to avoid a simple maneuver, to spare a little time, or for any other fancy and questionable reason?”
Cardiologists have raised that question for more than 20 years. Said Dr. Bachet, “We still wait for the answer.”
Dr. Bachet is a cardiac surgeon in Surgenes, France. He reported having no financial relationships to disclose.
The study by Dr. Tong and coauthors adds to the discussion between the “bilateralists” and “unilateralists,” as Jean Bachet, MD, called the two prevailing camps on cerebral perfusion strategies in his invited commentary (J Thorac Cardiovasc Surg. 2017;154:765-6). And while most clinical reports find outcomes similar between the two approaches, the evidence favors the bilateral approach for total arch replacement.
Citing how the study implied mortality and neurologic morbidity rates almost half those for unilateral perfusion, but not reaching statistical significance, Dr. Bachet said, “The statisticians would say that this is only a trend and no proof, but some trends might be indicative, and significance might only be a matter of number in each arm of the comparison.”
Dr. Bachet raised a question about the unilateral approach – that once the arch is opened it takes a minute or so to insert the small balloon canula into the origin of the left carotid artery or divided vessel and start bilateral perfusion. “A major question arises,” said Dr. Bachet: “Why should we expose our patients to any undue risk just to avoid a simple maneuver, to spare a little time, or for any other fancy and questionable reason?”
Cardiologists have raised that question for more than 20 years. Said Dr. Bachet, “We still wait for the answer.”
Dr. Bachet is a cardiac surgeon in Surgenes, France. He reported having no financial relationships to disclose.
What may be the largest study comparing unilateral and bilateral antegrade cerebral perfusion during total arch replacement for type A aortic dissection has reported that outcomes between the two approaches are comparable, although the bilateral approach showed some advantages during the operation itself, investigators from China reported in the Journal of Thoracic and Cardiovascular Surgery (2017;154:767-75).
The effectiveness of bilateral antegrade cerebral perfusion (b-ACP) vs. unilateral antegrade cerebral perfusion (u-ACP) has been the focus of extensive debate, lead study author Guang Tong, MD, of the Guangzhou (China) General Hospital, and coauthors said. They compared outcomes in six different metrics, ranging from cardiopulmonary bypass time to length of stay (LOS) in the ICU and hospital, in 203 patients with type A aortic dissection who had total aortic arch replacement with hypothermic circulatory arrest over an 8-year period ending in August 2014; 121 had b-ACP and 82 had u-ACP. “The issue of u-ACP vs. b-ACP has been examined in aortic arch surgery, but few reports have focused on type A aortic dissection,” Dr. Tong and coauthors wrote.
They acknowledged that some surgeons are reluctant to use b-ACP because of its complexity, but their study found no increase in cross-clamp time, cardiopulmonary bypass time, or surgery time in the b-ACP group. They cited another reason surgeons give for avoiding b-ACP: the risk of embolic injury caused by canulating the left common carotid artery in an atheromatous aorta. “In the present study, this risk was avoided by attaching the left common carotid artery to the four-branched prosthetic graft for left hemisphere perfusion,” Dr. Tong and coauthors wrote.
Key outcomes that the researchers found not statistically significant were:
- Overall 30-day mortality (11.6% for b-ACP vs. 20.7% for u-ACP; P = .075).
- Prevalence of postoperative permanent neurologic dysfunction (8.4% vs. 16.9%; P = .091).
- Average ICU LOS (16 ± 17.75 days vs. 17 ± 11.5 days, P =.454).
- Average hospital LOS (26.5 ± 20.6 days vs. 24.8 ± 10.3 days, P = .434).
However, average ventilation time was lower in the b-ACP group (95.5 hours vs. 147 hours; P less than or equal to.001).
Dr. Tong and coauthors used an aggressive approach, as advocated by Dhaval Trivedi, MD, and colleagues (Ann Thorac Surg. 2016;101:896-903), and had a total arch replacement rate of 57.8%. This rate is higher than most published series in the west but comparable to other studies from China, perhaps because of the relatively young age of this study cohort – an average age of 51 years – compared to data sets other studies have used. Dr. Tong and coauthors used a b-ACP strategy that established both cerebral perfusion routes before circulatory arrest.
Rates of the following complications were also not significantly different across the study population: paraplegia (2.8% for b-ACP vs. 3.1% for u-ACP), temporary neurologic dysfunction (4.7% vs. 9.2%), permanent neurologic dysfunction (8.4% vs. 16.9%), renal failure (18% vs. 23.1%), reoperation for bleeding (2.8% vs. 4.6%), and mediastinal infection (3.7% vs. 6.2%).
While b-ACP patients did not have a statistically significant lower incidence of TND, Dr. Tong and coauthors noted the shorter time on ventilation and significantly lower tracheostomy rates for the b-ACP patients, 3.7% vs. 16.9% for the u-ACP group (P = .003). “In our institute, protocols to wean patients from ventilation were normally initiated as soon as consciousness was regained,” Dr. Tong and coauthors wrote.
Among the study limits Dr. Tong and coauthors acknowledged were its retrospective, nonrandomized, single-center nature, and the fact that the surgeries were performed over an 8-year period representing different eras.
The investigators reported having no relevant financial disclosures.
What may be the largest study comparing unilateral and bilateral antegrade cerebral perfusion during total arch replacement for type A aortic dissection has reported that outcomes between the two approaches are comparable, although the bilateral approach showed some advantages during the operation itself, investigators from China reported in the Journal of Thoracic and Cardiovascular Surgery (2017;154:767-75).
The effectiveness of bilateral antegrade cerebral perfusion (b-ACP) vs. unilateral antegrade cerebral perfusion (u-ACP) has been the focus of extensive debate, lead study author Guang Tong, MD, of the Guangzhou (China) General Hospital, and coauthors said. They compared outcomes in six different metrics, ranging from cardiopulmonary bypass time to length of stay (LOS) in the ICU and hospital, in 203 patients with type A aortic dissection who had total aortic arch replacement with hypothermic circulatory arrest over an 8-year period ending in August 2014; 121 had b-ACP and 82 had u-ACP. “The issue of u-ACP vs. b-ACP has been examined in aortic arch surgery, but few reports have focused on type A aortic dissection,” Dr. Tong and coauthors wrote.
They acknowledged that some surgeons are reluctant to use b-ACP because of its complexity, but their study found no increase in cross-clamp time, cardiopulmonary bypass time, or surgery time in the b-ACP group. They cited another reason surgeons give for avoiding b-ACP: the risk of embolic injury caused by canulating the left common carotid artery in an atheromatous aorta. “In the present study, this risk was avoided by attaching the left common carotid artery to the four-branched prosthetic graft for left hemisphere perfusion,” Dr. Tong and coauthors wrote.
Key outcomes that the researchers found not statistically significant were:
- Overall 30-day mortality (11.6% for b-ACP vs. 20.7% for u-ACP; P = .075).
- Prevalence of postoperative permanent neurologic dysfunction (8.4% vs. 16.9%; P = .091).
- Average ICU LOS (16 ± 17.75 days vs. 17 ± 11.5 days, P =.454).
- Average hospital LOS (26.5 ± 20.6 days vs. 24.8 ± 10.3 days, P = .434).
However, average ventilation time was lower in the b-ACP group (95.5 hours vs. 147 hours; P less than or equal to.001).
Dr. Tong and coauthors used an aggressive approach, as advocated by Dhaval Trivedi, MD, and colleagues (Ann Thorac Surg. 2016;101:896-903), and had a total arch replacement rate of 57.8%. This rate is higher than most published series in the west but comparable to other studies from China, perhaps because of the relatively young age of this study cohort – an average age of 51 years – compared to data sets other studies have used. Dr. Tong and coauthors used a b-ACP strategy that established both cerebral perfusion routes before circulatory arrest.
Rates of the following complications were also not significantly different across the study population: paraplegia (2.8% for b-ACP vs. 3.1% for u-ACP), temporary neurologic dysfunction (4.7% vs. 9.2%), permanent neurologic dysfunction (8.4% vs. 16.9%), renal failure (18% vs. 23.1%), reoperation for bleeding (2.8% vs. 4.6%), and mediastinal infection (3.7% vs. 6.2%).
While b-ACP patients did not have a statistically significant lower incidence of TND, Dr. Tong and coauthors noted the shorter time on ventilation and significantly lower tracheostomy rates for the b-ACP patients, 3.7% vs. 16.9% for the u-ACP group (P = .003). “In our institute, protocols to wean patients from ventilation were normally initiated as soon as consciousness was regained,” Dr. Tong and coauthors wrote.
Among the study limits Dr. Tong and coauthors acknowledged were its retrospective, nonrandomized, single-center nature, and the fact that the surgeries were performed over an 8-year period representing different eras.
The investigators reported having no relevant financial disclosures.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Clinical outcomes were comparable between groups who underwent unilateral or bilateral antegrade cerebral perfusion in total arch replacement for type A aortic dissection.
Major finding: Overall 30-day mortality was 11.6% in the bilateral ACP group vs. 20.7% for unilateral ACP (P =.075).
Data source: Population of 203 patients who had aortic arch replacement surgery for type A aortic dissection between September 2006 and August 2014.
Disclosures: Dr. Tong and coauthors reported having no relevant financial disclosures.
Kidney transplant for GPA boosts survival
SAN DIEGO – Receiving a kidney transplant increased the likelihood of survival in patients with end-stage renal disease (ESRD) due to granulomatosis with polyangiitis, a study showed.
“We found that there was a huge survival advantage of having a renal transplant,” lead author Zachary S. Wallace, MD, MS, of Harvard Medical School, Boston, said in an interview. “In addition, this improvement in survival seems to be due to a dramatic reduction in death due to cardiovascular disease.”

An estimated 25% of patients with GPA develop ESRD, according to Dr. Wallace, who also works at the vasculitis and glomerulonephritis center at Massachusetts General Hospital, Boston. “GPA and ANCA [antineutrophil cytoplasmic autoantibody]–associated vasculitis in general have a propensity to affect the kidneys, and the reason for that is not entirely known,” he said during the interview. “In the kidney, it most commonly causes a rapidly progressive glomerulonephritis which can cause irreversible renal failure if not aggressively treated.”
Dr. Wallace and his colleagues launched their study to better understand the impact of kidney transplants. “We know that patients with ESRD from more common causes – such as diabetes and hypertension – benefit in terms of survival and quality of life from transplantation,” he said in the interview. “It was unknown if GPA patients similarly benefit. Often, GPA patients have fewer comorbidities than patients with ESRD due to diabetes or hypertension. Since they may be relatively healthier, one might wonder if the survival benefit would be as great in ESRD patients with GPA.”
Dr. Wallace and his colleagues tracked 2,471 cases of ESRD due to GPA from the U.S. Renal Data System. All were wait-listed for a kidney transplant from 1995 to 2014, and the researchers tracked them as late as Jan. 1, 2016. Of the patients studied, 946 received a transplant. The study’s participants tended to be male (59%) and white (86-87%), and they rarely had comorbidities outside of diabetes (64-67%).
There were 438 deaths in the entire group. Those who received transplants were much less likely to die than those who didn’t (adjusted hazard ratio, 0.30; 95% confidence interval, 0.25-0.37; P less than .001), he reported at the annual meeting of the American College of Rheumatology.
Also, those who received transplants were much less likely than those who didn’t to die of cardiovascular disease (adjusted HR, 0.13; 95% CI, 0.08-0.22; P less than .001) and infection (adjusted HR, 0.61; 95% CI, 0.34-1.08; P = .09). There was no statistically significant difference between the groups in terms of deaths from cancer.
“The improvement in survival seems to be due to a dramatic reduction in death due to cardiovascular disease,” Dr. Wallace said in the interview. “While cardiovascular disease is a common cause of death in GPA and ESRD due to other causes, this was not known specifically in patients with ESRD due to GPA.”
The findings provide the following messages to rheumatologists: Renal transplantation in patients with ESRD due to GPA offers a significant survival benefit, and it is important to refer patients early to a renal transplant center, he noted.
“[Rheumatologists] should work closely with primary care physicians and nephrologists to make sure that the patient’s cardiovascular disease risk is being assessed – checking lipids, A1c, etc. – and addressed as necessary,” Dr. Wallace added.
The study authors reported no relevant financial disclosures. Funding included support from the Rheumatology Research Foundation, the Executive Committee on Research at Massachusetts General, and the National Institutes of Health Loan Repayment Program.
SAN DIEGO – Receiving a kidney transplant increased the likelihood of survival in patients with end-stage renal disease (ESRD) due to granulomatosis with polyangiitis, a study showed.
“We found that there was a huge survival advantage of having a renal transplant,” lead author Zachary S. Wallace, MD, MS, of Harvard Medical School, Boston, said in an interview. “In addition, this improvement in survival seems to be due to a dramatic reduction in death due to cardiovascular disease.”
An estimated 25% of patients with GPA develop ESRD, according to Dr. Wallace, who also works at the vasculitis and glomerulonephritis center at Massachusetts General Hospital, Boston. “GPA and ANCA [antineutrophil cytoplasmic autoantibody]–associated vasculitis in general have a propensity to affect the kidneys, and the reason for that is not entirely known,” he said during the interview. “In the kidney, it most commonly causes a rapidly progressive glomerulonephritis which can cause irreversible renal failure if not aggressively treated.”
Dr. Wallace and his colleagues launched their study to better understand the impact of kidney transplants. “We know that patients with ESRD from more common causes – such as diabetes and hypertension – benefit in terms of survival and quality of life from transplantation,” he said in the interview. “It was unknown if GPA patients similarly benefit. Often, GPA patients have fewer comorbidities than patients with ESRD due to diabetes or hypertension. Since they may be relatively healthier, one might wonder if the survival benefit would be as great in ESRD patients with GPA.”
Dr. Wallace and his colleagues tracked 2,471 cases of ESRD due to GPA from the U.S. Renal Data System. All were wait-listed for a kidney transplant from 1995 to 2014, and the researchers tracked them as late as Jan. 1, 2016. Of the patients studied, 946 received a transplant. The study’s participants tended to be male (59%) and white (86-87%), and they rarely had comorbidities outside of diabetes (64-67%).
There were 438 deaths in the entire group. Those who received transplants were much less likely to die than those who didn’t (adjusted hazard ratio, 0.30; 95% confidence interval, 0.25-0.37; P less than .001), he reported at the annual meeting of the American College of Rheumatology.
Also, those who received transplants were much less likely than those who didn’t to die of cardiovascular disease (adjusted HR, 0.13; 95% CI, 0.08-0.22; P less than .001) and infection (adjusted HR, 0.61; 95% CI, 0.34-1.08; P = .09). There was no statistically significant difference between the groups in terms of deaths from cancer.
“The improvement in survival seems to be due to a dramatic reduction in death due to cardiovascular disease,” Dr. Wallace said in the interview. “While cardiovascular disease is a common cause of death in GPA and ESRD due to other causes, this was not known specifically in patients with ESRD due to GPA.”
The findings provide the following messages to rheumatologists: Renal transplantation in patients with ESRD due to GPA offers a significant survival benefit, and it is important to refer patients early to a renal transplant center, he noted.
“[Rheumatologists] should work closely with primary care physicians and nephrologists to make sure that the patient’s cardiovascular disease risk is being assessed – checking lipids, A1c, etc. – and addressed as necessary,” Dr. Wallace added.
The study authors reported no relevant financial disclosures. Funding included support from the Rheumatology Research Foundation, the Executive Committee on Research at Massachusetts General, and the National Institutes of Health Loan Repayment Program.
SAN DIEGO – Receiving a kidney transplant increased the likelihood of survival in patients with end-stage renal disease (ESRD) due to granulomatosis with polyangiitis, a study showed.
“We found that there was a huge survival advantage of having a renal transplant,” lead author Zachary S. Wallace, MD, MS, of Harvard Medical School, Boston, said in an interview. “In addition, this improvement in survival seems to be due to a dramatic reduction in death due to cardiovascular disease.”
An estimated 25% of patients with GPA develop ESRD, according to Dr. Wallace, who also works at the vasculitis and glomerulonephritis center at Massachusetts General Hospital, Boston. “GPA and ANCA [antineutrophil cytoplasmic autoantibody]–associated vasculitis in general have a propensity to affect the kidneys, and the reason for that is not entirely known,” he said during the interview. “In the kidney, it most commonly causes a rapidly progressive glomerulonephritis which can cause irreversible renal failure if not aggressively treated.”
Dr. Wallace and his colleagues launched their study to better understand the impact of kidney transplants. “We know that patients with ESRD from more common causes – such as diabetes and hypertension – benefit in terms of survival and quality of life from transplantation,” he said in the interview. “It was unknown if GPA patients similarly benefit. Often, GPA patients have fewer comorbidities than patients with ESRD due to diabetes or hypertension. Since they may be relatively healthier, one might wonder if the survival benefit would be as great in ESRD patients with GPA.”
Dr. Wallace and his colleagues tracked 2,471 cases of ESRD due to GPA from the U.S. Renal Data System. All were wait-listed for a kidney transplant from 1995 to 2014, and the researchers tracked them as late as Jan. 1, 2016. Of the patients studied, 946 received a transplant. The study’s participants tended to be male (59%) and white (86-87%), and they rarely had comorbidities outside of diabetes (64-67%).
There were 438 deaths in the entire group. Those who received transplants were much less likely to die than those who didn’t (adjusted hazard ratio, 0.30; 95% confidence interval, 0.25-0.37; P less than .001), he reported at the annual meeting of the American College of Rheumatology.
Also, those who received transplants were much less likely than those who didn’t to die of cardiovascular disease (adjusted HR, 0.13; 95% CI, 0.08-0.22; P less than .001) and infection (adjusted HR, 0.61; 95% CI, 0.34-1.08; P = .09). There was no statistically significant difference between the groups in terms of deaths from cancer.
“The improvement in survival seems to be due to a dramatic reduction in death due to cardiovascular disease,” Dr. Wallace said in the interview. “While cardiovascular disease is a common cause of death in GPA and ESRD due to other causes, this was not known specifically in patients with ESRD due to GPA.”
The findings provide the following messages to rheumatologists: Renal transplantation in patients with ESRD due to GPA offers a significant survival benefit, and it is important to refer patients early to a renal transplant center, he noted.
“[Rheumatologists] should work closely with primary care physicians and nephrologists to make sure that the patient’s cardiovascular disease risk is being assessed – checking lipids, A1c, etc. – and addressed as necessary,” Dr. Wallace added.
The study authors reported no relevant financial disclosures. Funding included support from the Rheumatology Research Foundation, the Executive Committee on Research at Massachusetts General, and the National Institutes of Health Loan Repayment Program.
AT ACR 2017
Key clinical point:
Major finding: Compared with patients who were wait-listed for a transplant but didn’t receive one, those who got transplants were much less likely to die during the study period (adjusted HR, 0.30; 95% CI, 0.25-0.37; P less than .001).
Data source: 2,471 patients with ESRD due to GPA who were wait-listed for a kidney transplant from 1995 to 2014 and tracked as late as 2016; 946 received a transplant.
Disclosures: The study authors reported no relevant financial disclosures. Funding included support from the Rheumatology Research Foundation, the Executive Committee on Research at Massachusetts General, and the National Institutes of Health Loan Repayment Program.
CSF May Reveal Dementia Risk in Lewy Body Disease
SAN DIEGO—In patients with Lewy body disease, CSF biomarkers may predict the extent of Alzheimer’s disease pathology, according to a new postmortem analysis presented at the 142nd Annual Meeting of the American Neurological Association.
In the small, prospective study, David Irwin, MD, Assistant Professor of Neurology at the University of Pennsylvania in Philadelphia, and colleagues found that higher antemortem CSF levels of total tau protein and lower amyloid beta1–42 levels were associated with postmortem evidence of cerebral Alzheimer’s disease and synuclein pathology. The connection between CSF amyloid beta1–42 and tau protein had been shown in Alzheimer’s disease, but not in Lewy body disease.

The findings could have prognostic implications, because about 40% of patients with Lewy body disease have enough amyloid beta1–42 and tau tangle pathology to have a secondary diagnosis of Alzheimer’s disease, and greater Alzheimer’s disease pathology has been linked to shorter survival. The results also could help individualize therapy, because patients with Lewy body disease and Alzheimer’s disease characteristics might respond better to antisynuclein and Alzheimer’s disease therapies, according to Dr. Irwin.
The CSF ratio of total tau to amyloid beta1–42 predicted the presence or absence of Alzheimer’s disease pathology with 90% sensitivity and 100% specificity.
If the results are confirmed, “we can identify who has Alzheimer’s copathology and who has a more aggressive form of Lewy body disease where it is reaching the cortex rather than being in the brainstem, and that correlates with memory loss, dementia, and other things that impair the patient’s functioning,” said Dr. Irwin.
The Udall Center for Parkinson’s Research at the University of Pennsylvania has an active autopsy program, and a significant number of patients with Parkinson’s disease donated their brains for research. “It is rare to have autopsy-confirmed samples where we can validate what we are measuring in the spinal fluid to what is actually accumulating in the brain,” Dr. Irwin said.
The study included 24 patients with autopsy-confirmed Lewy body disease who had undergone CSF testing during life. The researchers calculated ordinal pathology scores of tau tangles, amyloid beta1–42 plaques, and alpha-synuclein pathology in seven regions of the brain, producing a global pathology score for each variable. The subjects were then categorized into a group of participants with Lewy body synuclein stages and medium to high Alzheimer’s copathology (SYN+AD, n = 10) and a group with little or no Alzheimer’s copathology (SYN−AD, n = 14).
The researchers analyzed potential associations involving total tau, phosphorylated tau, and amyloid beta1–42. They found that amyloid beta1–42 levels were significantly different between SYN+AD and SYN−AD patients (147.2 pg/mL vs 231.1 pg/mL, respectively), as were CSF total tau levels (63.9 pg/mL vs 36.9 pg/mL). Phosphorylated tau was not significantly different between groups (20.2 pg/mL vs 15.5 pg/mL), which surprised the researchers, since it “is usually the strongest correlate of tangles in the brain,” said Dr. Irwin.
A ratio of CSF total tau to amyloid beta1–42 higher than 0.30 distinguished between SYN+AD and SYN−AD (area under the curve [AUC], 0.92; 90% sensitivity; 100% specificity), while CSF amyloid beta1–42 less than 185 pg/mL identified the neocortical synuclein stage (AUC, 0.92; 77% sensitivity; 82% specificity).
—Jim Kling
SAN DIEGO—In patients with Lewy body disease, CSF biomarkers may predict the extent of Alzheimer’s disease pathology, according to a new postmortem analysis presented at the 142nd Annual Meeting of the American Neurological Association.
In the small, prospective study, David Irwin, MD, Assistant Professor of Neurology at the University of Pennsylvania in Philadelphia, and colleagues found that higher antemortem CSF levels of total tau protein and lower amyloid beta1–42 levels were associated with postmortem evidence of cerebral Alzheimer’s disease and synuclein pathology. The connection between CSF amyloid beta1–42 and tau protein had been shown in Alzheimer’s disease, but not in Lewy body disease.
The findings could have prognostic implications, because about 40% of patients with Lewy body disease have enough amyloid beta1–42 and tau tangle pathology to have a secondary diagnosis of Alzheimer’s disease, and greater Alzheimer’s disease pathology has been linked to shorter survival. The results also could help individualize therapy, because patients with Lewy body disease and Alzheimer’s disease characteristics might respond better to antisynuclein and Alzheimer’s disease therapies, according to Dr. Irwin.
The CSF ratio of total tau to amyloid beta1–42 predicted the presence or absence of Alzheimer’s disease pathology with 90% sensitivity and 100% specificity.
If the results are confirmed, “we can identify who has Alzheimer’s copathology and who has a more aggressive form of Lewy body disease where it is reaching the cortex rather than being in the brainstem, and that correlates with memory loss, dementia, and other things that impair the patient’s functioning,” said Dr. Irwin.
The Udall Center for Parkinson’s Research at the University of Pennsylvania has an active autopsy program, and a significant number of patients with Parkinson’s disease donated their brains for research. “It is rare to have autopsy-confirmed samples where we can validate what we are measuring in the spinal fluid to what is actually accumulating in the brain,” Dr. Irwin said.
The study included 24 patients with autopsy-confirmed Lewy body disease who had undergone CSF testing during life. The researchers calculated ordinal pathology scores of tau tangles, amyloid beta1–42 plaques, and alpha-synuclein pathology in seven regions of the brain, producing a global pathology score for each variable. The subjects were then categorized into a group of participants with Lewy body synuclein stages and medium to high Alzheimer’s copathology (SYN+AD, n = 10) and a group with little or no Alzheimer’s copathology (SYN−AD, n = 14).
The researchers analyzed potential associations involving total tau, phosphorylated tau, and amyloid beta1–42. They found that amyloid beta1–42 levels were significantly different between SYN+AD and SYN−AD patients (147.2 pg/mL vs 231.1 pg/mL, respectively), as were CSF total tau levels (63.9 pg/mL vs 36.9 pg/mL). Phosphorylated tau was not significantly different between groups (20.2 pg/mL vs 15.5 pg/mL), which surprised the researchers, since it “is usually the strongest correlate of tangles in the brain,” said Dr. Irwin.
A ratio of CSF total tau to amyloid beta1–42 higher than 0.30 distinguished between SYN+AD and SYN−AD (area under the curve [AUC], 0.92; 90% sensitivity; 100% specificity), while CSF amyloid beta1–42 less than 185 pg/mL identified the neocortical synuclein stage (AUC, 0.92; 77% sensitivity; 82% specificity).
—Jim Kling
SAN DIEGO—In patients with Lewy body disease, CSF biomarkers may predict the extent of Alzheimer’s disease pathology, according to a new postmortem analysis presented at the 142nd Annual Meeting of the American Neurological Association.
In the small, prospective study, David Irwin, MD, Assistant Professor of Neurology at the University of Pennsylvania in Philadelphia, and colleagues found that higher antemortem CSF levels of total tau protein and lower amyloid beta1–42 levels were associated with postmortem evidence of cerebral Alzheimer’s disease and synuclein pathology. The connection between CSF amyloid beta1–42 and tau protein had been shown in Alzheimer’s disease, but not in Lewy body disease.
The findings could have prognostic implications, because about 40% of patients with Lewy body disease have enough amyloid beta1–42 and tau tangle pathology to have a secondary diagnosis of Alzheimer’s disease, and greater Alzheimer’s disease pathology has been linked to shorter survival. The results also could help individualize therapy, because patients with Lewy body disease and Alzheimer’s disease characteristics might respond better to antisynuclein and Alzheimer’s disease therapies, according to Dr. Irwin.
The CSF ratio of total tau to amyloid beta1–42 predicted the presence or absence of Alzheimer’s disease pathology with 90% sensitivity and 100% specificity.
If the results are confirmed, “we can identify who has Alzheimer’s copathology and who has a more aggressive form of Lewy body disease where it is reaching the cortex rather than being in the brainstem, and that correlates with memory loss, dementia, and other things that impair the patient’s functioning,” said Dr. Irwin.
The Udall Center for Parkinson’s Research at the University of Pennsylvania has an active autopsy program, and a significant number of patients with Parkinson’s disease donated their brains for research. “It is rare to have autopsy-confirmed samples where we can validate what we are measuring in the spinal fluid to what is actually accumulating in the brain,” Dr. Irwin said.
The study included 24 patients with autopsy-confirmed Lewy body disease who had undergone CSF testing during life. The researchers calculated ordinal pathology scores of tau tangles, amyloid beta1–42 plaques, and alpha-synuclein pathology in seven regions of the brain, producing a global pathology score for each variable. The subjects were then categorized into a group of participants with Lewy body synuclein stages and medium to high Alzheimer’s copathology (SYN+AD, n = 10) and a group with little or no Alzheimer’s copathology (SYN−AD, n = 14).
The researchers analyzed potential associations involving total tau, phosphorylated tau, and amyloid beta1–42. They found that amyloid beta1–42 levels were significantly different between SYN+AD and SYN−AD patients (147.2 pg/mL vs 231.1 pg/mL, respectively), as were CSF total tau levels (63.9 pg/mL vs 36.9 pg/mL). Phosphorylated tau was not significantly different between groups (20.2 pg/mL vs 15.5 pg/mL), which surprised the researchers, since it “is usually the strongest correlate of tangles in the brain,” said Dr. Irwin.
A ratio of CSF total tau to amyloid beta1–42 higher than 0.30 distinguished between SYN+AD and SYN−AD (area under the curve [AUC], 0.92; 90% sensitivity; 100% specificity), while CSF amyloid beta1–42 less than 185 pg/mL identified the neocortical synuclein stage (AUC, 0.92; 77% sensitivity; 82% specificity).
—Jim Kling
Does Midlife Physical Activity Affect Cognitive Health in Later Life?
In contrast with previous research, a study published in the Journal of Alzheimer’s Disease indicates that exercise in midlife is not associated with cognitive fitness in late life. The data suggest that physical activity may not help maintain cognitive function or prevent the onset of Alzheimer’s disease. Physical activity in late life, however, is associated with high cognitive function two years later, according to the researchers.

The literature contains few prospective data on the relationship between midlife and late-life physical activity and late-life cognitive performance. Cross-sectional study designs and short durations of follow-up have made several studies of this relationship unsatisfactory, according to Alden L. Gross, PhD, Assistant Professor of Epidemiology at Johns Hopkins Bloomberg School of Public Health in Baltimore, and colleagues.
A Prospective Study of Medical Graduates
To conduct a prospective analysis of the possible association between exercise in midlife and cognitive performance in later life, Dr. Gross and colleagues examined 30 years of longitudinal data from the Johns Hopkins Precursors study. This study enrolled medical graduates from 1948 to 1964 and followed them annually with health questionnaires. Surveys of physical activity were mailed in 1978, 1986, 1989, 1993, 1995, 1997, 2003, and 2006. Participants underwent cognitive testing in 2005 and 2008.
An adjudication committee consisting of physicians assigned diagnoses of dementia and dates of onset based on participants’ medical records. The investigators assigned a metabolic equivalent value (MET) to each category of activity (ie, vigorous, moderate, light, and sleep). They used linear regression for cognitive tests and Cox proportional hazards models for dementia onset to characterize associations with midlife physical activity. Models were adjusted for age, sex, smoking, diabetes, and hypertension.
Exercise Had No Long-Term Cognitive Effects
The study sample included 646 participants (48 women). In 1978, participants’ mean age was 47, mean BMI was 23.7 kg/m2, the prevalence of hypertension was 8.8%, and the prevalence of diabetes was 0.8%. Approximately 74% of participants reported regular exercise. In 2006, 54% of participants had hypertension, and 12% had diabetes. The incidence of dementia was 4.3%, and mean age at onset was 77.
No measure of activity in 1978, including MET h/day, decile of physical activity, and engagement in regular exercise, was associated with cognitive performance in demographic-adjusted or fully adjusted models. Regular exercise in 2006 and MET h/day expended in 2006, however, were associated with better cognitive performance. In addition, no physical activity exposure in 1978 was associated with onset of dementia. Greater physical activity in 2006, however, had a modest inverse association with risk of dementia.
The strength of the association between cognitive outcomes and physical activity in 1978 did not vary by gender. In 2006, however, greater MET h/day in a typical week and regular exercise were significantly associated with better cognitive performance in women, but not in men.
Homogeneous Sample Reduced Confounders
Previous studies included diverse cross sections of people and thus may have been susceptible to residual confounding by factors such as years of education, said the investigators. The Precursors study represents a highly homogeneous sample, however, which reduces the likelihood of such confounding. Nevertheless, the homogenous sample may limit the results’ generalizability, particularly to women.
“The prevailing notion that higher levels of physical activity protect against cognitive impairment in later life may result from the artifact of reverse causation,” said the authors. “While physical activity is strongly recommended for many reasons, our data suggest cognitive health may not be one of those reasons.”
“These findings have implications for intervention work moving forward,” said Dr. Gross. “We still need to focus on causes and mechanisms of Alzheimer’s [disease] and dementia, since we do not yet know which preventive measures may or may not work. For now, when I speak in the community about Alzheimer’s [disease], I find that people take some relief in understanding that there was not anything that anyone might have done to avoid a loved one developing Alzheimer’s [disease]. Of course, the goal for researchers is to identify factors that may help older people maintain their cognitive function into their later years. More long-term studies like the Precursors study are needed.”
—Erik Greb
Suggested Reading
Gross AL, Lu H, Meoni L, et al. Physical activity in midlife is not associated with cognitive health in later life among cognitively normal older adults. J Alzheimers Dis. 2017;59(4):1349-1358.
In contrast with previous research, a study published in the Journal of Alzheimer’s Disease indicates that exercise in midlife is not associated with cognitive fitness in late life. The data suggest that physical activity may not help maintain cognitive function or prevent the onset of Alzheimer’s disease. Physical activity in late life, however, is associated with high cognitive function two years later, according to the researchers.
The literature contains few prospective data on the relationship between midlife and late-life physical activity and late-life cognitive performance. Cross-sectional study designs and short durations of follow-up have made several studies of this relationship unsatisfactory, according to Alden L. Gross, PhD, Assistant Professor of Epidemiology at Johns Hopkins Bloomberg School of Public Health in Baltimore, and colleagues.
A Prospective Study of Medical Graduates
To conduct a prospective analysis of the possible association between exercise in midlife and cognitive performance in later life, Dr. Gross and colleagues examined 30 years of longitudinal data from the Johns Hopkins Precursors study. This study enrolled medical graduates from 1948 to 1964 and followed them annually with health questionnaires. Surveys of physical activity were mailed in 1978, 1986, 1989, 1993, 1995, 1997, 2003, and 2006. Participants underwent cognitive testing in 2005 and 2008.
An adjudication committee consisting of physicians assigned diagnoses of dementia and dates of onset based on participants’ medical records. The investigators assigned a metabolic equivalent value (MET) to each category of activity (ie, vigorous, moderate, light, and sleep). They used linear regression for cognitive tests and Cox proportional hazards models for dementia onset to characterize associations with midlife physical activity. Models were adjusted for age, sex, smoking, diabetes, and hypertension.
Exercise Had No Long-Term Cognitive Effects
The study sample included 646 participants (48 women). In 1978, participants’ mean age was 47, mean BMI was 23.7 kg/m2, the prevalence of hypertension was 8.8%, and the prevalence of diabetes was 0.8%. Approximately 74% of participants reported regular exercise. In 2006, 54% of participants had hypertension, and 12% had diabetes. The incidence of dementia was 4.3%, and mean age at onset was 77.
No measure of activity in 1978, including MET h/day, decile of physical activity, and engagement in regular exercise, was associated with cognitive performance in demographic-adjusted or fully adjusted models. Regular exercise in 2006 and MET h/day expended in 2006, however, were associated with better cognitive performance. In addition, no physical activity exposure in 1978 was associated with onset of dementia. Greater physical activity in 2006, however, had a modest inverse association with risk of dementia.
The strength of the association between cognitive outcomes and physical activity in 1978 did not vary by gender. In 2006, however, greater MET h/day in a typical week and regular exercise were significantly associated with better cognitive performance in women, but not in men.
Homogeneous Sample Reduced Confounders
Previous studies included diverse cross sections of people and thus may have been susceptible to residual confounding by factors such as years of education, said the investigators. The Precursors study represents a highly homogeneous sample, however, which reduces the likelihood of such confounding. Nevertheless, the homogenous sample may limit the results’ generalizability, particularly to women.
“The prevailing notion that higher levels of physical activity protect against cognitive impairment in later life may result from the artifact of reverse causation,” said the authors. “While physical activity is strongly recommended for many reasons, our data suggest cognitive health may not be one of those reasons.”
“These findings have implications for intervention work moving forward,” said Dr. Gross. “We still need to focus on causes and mechanisms of Alzheimer’s [disease] and dementia, since we do not yet know which preventive measures may or may not work. For now, when I speak in the community about Alzheimer’s [disease], I find that people take some relief in understanding that there was not anything that anyone might have done to avoid a loved one developing Alzheimer’s [disease]. Of course, the goal for researchers is to identify factors that may help older people maintain their cognitive function into their later years. More long-term studies like the Precursors study are needed.”
—Erik Greb
Suggested Reading
Gross AL, Lu H, Meoni L, et al. Physical activity in midlife is not associated with cognitive health in later life among cognitively normal older adults. J Alzheimers Dis. 2017;59(4):1349-1358.
In contrast with previous research, a study published in the Journal of Alzheimer’s Disease indicates that exercise in midlife is not associated with cognitive fitness in late life. The data suggest that physical activity may not help maintain cognitive function or prevent the onset of Alzheimer’s disease. Physical activity in late life, however, is associated with high cognitive function two years later, according to the researchers.
The literature contains few prospective data on the relationship between midlife and late-life physical activity and late-life cognitive performance. Cross-sectional study designs and short durations of follow-up have made several studies of this relationship unsatisfactory, according to Alden L. Gross, PhD, Assistant Professor of Epidemiology at Johns Hopkins Bloomberg School of Public Health in Baltimore, and colleagues.
A Prospective Study of Medical Graduates
To conduct a prospective analysis of the possible association between exercise in midlife and cognitive performance in later life, Dr. Gross and colleagues examined 30 years of longitudinal data from the Johns Hopkins Precursors study. This study enrolled medical graduates from 1948 to 1964 and followed them annually with health questionnaires. Surveys of physical activity were mailed in 1978, 1986, 1989, 1993, 1995, 1997, 2003, and 2006. Participants underwent cognitive testing in 2005 and 2008.
An adjudication committee consisting of physicians assigned diagnoses of dementia and dates of onset based on participants’ medical records. The investigators assigned a metabolic equivalent value (MET) to each category of activity (ie, vigorous, moderate, light, and sleep). They used linear regression for cognitive tests and Cox proportional hazards models for dementia onset to characterize associations with midlife physical activity. Models were adjusted for age, sex, smoking, diabetes, and hypertension.
Exercise Had No Long-Term Cognitive Effects
The study sample included 646 participants (48 women). In 1978, participants’ mean age was 47, mean BMI was 23.7 kg/m2, the prevalence of hypertension was 8.8%, and the prevalence of diabetes was 0.8%. Approximately 74% of participants reported regular exercise. In 2006, 54% of participants had hypertension, and 12% had diabetes. The incidence of dementia was 4.3%, and mean age at onset was 77.
No measure of activity in 1978, including MET h/day, decile of physical activity, and engagement in regular exercise, was associated with cognitive performance in demographic-adjusted or fully adjusted models. Regular exercise in 2006 and MET h/day expended in 2006, however, were associated with better cognitive performance. In addition, no physical activity exposure in 1978 was associated with onset of dementia. Greater physical activity in 2006, however, had a modest inverse association with risk of dementia.
The strength of the association between cognitive outcomes and physical activity in 1978 did not vary by gender. In 2006, however, greater MET h/day in a typical week and regular exercise were significantly associated with better cognitive performance in women, but not in men.
Homogeneous Sample Reduced Confounders
Previous studies included diverse cross sections of people and thus may have been susceptible to residual confounding by factors such as years of education, said the investigators. The Precursors study represents a highly homogeneous sample, however, which reduces the likelihood of such confounding. Nevertheless, the homogenous sample may limit the results’ generalizability, particularly to women.
“The prevailing notion that higher levels of physical activity protect against cognitive impairment in later life may result from the artifact of reverse causation,” said the authors. “While physical activity is strongly recommended for many reasons, our data suggest cognitive health may not be one of those reasons.”
“These findings have implications for intervention work moving forward,” said Dr. Gross. “We still need to focus on causes and mechanisms of Alzheimer’s [disease] and dementia, since we do not yet know which preventive measures may or may not work. For now, when I speak in the community about Alzheimer’s [disease], I find that people take some relief in understanding that there was not anything that anyone might have done to avoid a loved one developing Alzheimer’s [disease]. Of course, the goal for researchers is to identify factors that may help older people maintain their cognitive function into their later years. More long-term studies like the Precursors study are needed.”
—Erik Greb
Suggested Reading
Gross AL, Lu H, Meoni L, et al. Physical activity in midlife is not associated with cognitive health in later life among cognitively normal older adults. J Alzheimers Dis. 2017;59(4):1349-1358.
Two-thirds of abortions occur by 8 weeks’ gestation
, although there was variation by maternal age and race/ethnicity, according to the Centers for Disease Control and Prevention.
That year, 65.3% of abortions were performed at a gestational age of 8 weeks or earlier, with 25.6% occurring at 9-13 weeks. Gestational distribution of the remaining abortions was fairly even: 3.4% at 14-15 weeks, 2.2% at 16-17 weeks, 2.0% at 18-20 weeks, and 1.4% at 21 weeks or later, the CDC investigators reported (MMWR Surveill Summ. 2017 Nov 25;66[24]:1-48).
The percentage of abortions occurring at 8 weeks or earlier was lowest for the youngest age group and increased along with maternal age: 43% for those under 15 years of age and progressing up to 72.5% for women over age 40. That scenario was basically reversed for all of the other gestational periods, as the under-15 group had the highest percentage for 9-13 weeks (34.4%), 14-15 (6.7%), 16-17 (3.6%), 18-20 (5.0%), and 21 weeks and later (7.3%). Those over age 40 had the lowest or almost the lowest percentage in each period, reported Tara C. Jatlaoui, MD, and her associates.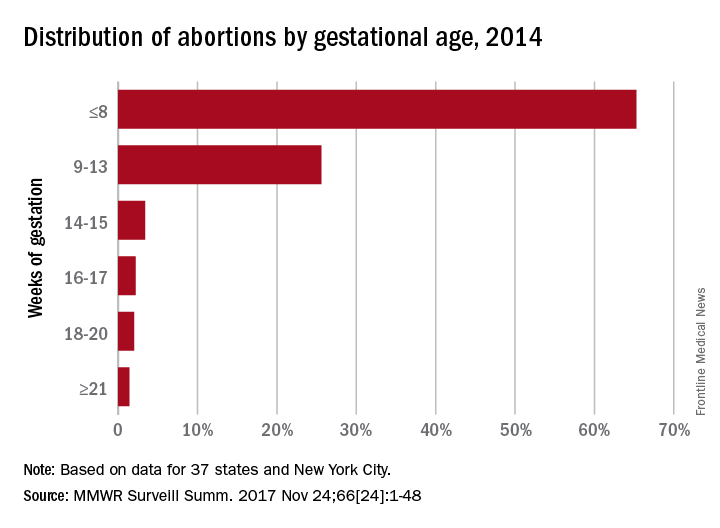
The data for gestational period analysis came from 37 states and New York City. New York State, along with 12 other states, did not report, did not report by gestational age, or did not meet reporting standards.
, although there was variation by maternal age and race/ethnicity, according to the Centers for Disease Control and Prevention.
That year, 65.3% of abortions were performed at a gestational age of 8 weeks or earlier, with 25.6% occurring at 9-13 weeks. Gestational distribution of the remaining abortions was fairly even: 3.4% at 14-15 weeks, 2.2% at 16-17 weeks, 2.0% at 18-20 weeks, and 1.4% at 21 weeks or later, the CDC investigators reported (MMWR Surveill Summ. 2017 Nov 25;66[24]:1-48).
The percentage of abortions occurring at 8 weeks or earlier was lowest for the youngest age group and increased along with maternal age: 43% for those under 15 years of age and progressing up to 72.5% for women over age 40. That scenario was basically reversed for all of the other gestational periods, as the under-15 group had the highest percentage for 9-13 weeks (34.4%), 14-15 (6.7%), 16-17 (3.6%), 18-20 (5.0%), and 21 weeks and later (7.3%). Those over age 40 had the lowest or almost the lowest percentage in each period, reported Tara C. Jatlaoui, MD, and her associates.
The data for gestational period analysis came from 37 states and New York City. New York State, along with 12 other states, did not report, did not report by gestational age, or did not meet reporting standards.
, although there was variation by maternal age and race/ethnicity, according to the Centers for Disease Control and Prevention.
That year, 65.3% of abortions were performed at a gestational age of 8 weeks or earlier, with 25.6% occurring at 9-13 weeks. Gestational distribution of the remaining abortions was fairly even: 3.4% at 14-15 weeks, 2.2% at 16-17 weeks, 2.0% at 18-20 weeks, and 1.4% at 21 weeks or later, the CDC investigators reported (MMWR Surveill Summ. 2017 Nov 25;66[24]:1-48).
The percentage of abortions occurring at 8 weeks or earlier was lowest for the youngest age group and increased along with maternal age: 43% for those under 15 years of age and progressing up to 72.5% for women over age 40. That scenario was basically reversed for all of the other gestational periods, as the under-15 group had the highest percentage for 9-13 weeks (34.4%), 14-15 (6.7%), 16-17 (3.6%), 18-20 (5.0%), and 21 weeks and later (7.3%). Those over age 40 had the lowest or almost the lowest percentage in each period, reported Tara C. Jatlaoui, MD, and her associates.
The data for gestational period analysis came from 37 states and New York City. New York State, along with 12 other states, did not report, did not report by gestational age, or did not meet reporting standards.
FROM MMWR SURVEILLANCE SUMMARIES