User login
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14 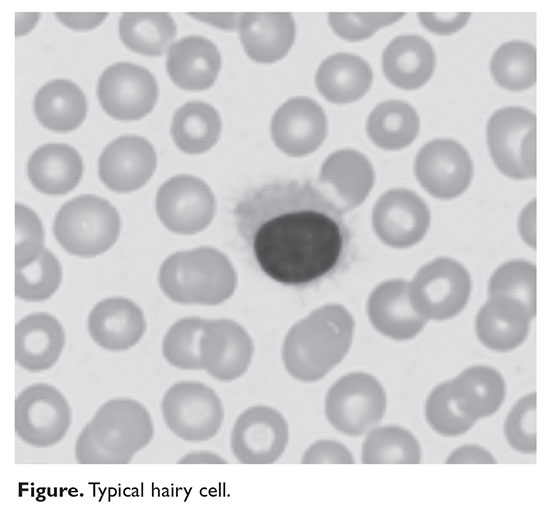
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease. 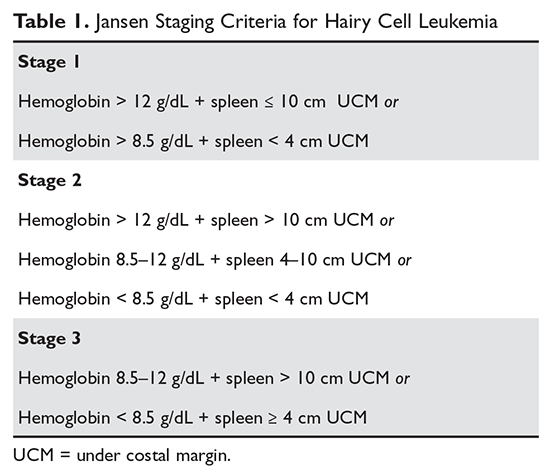
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29 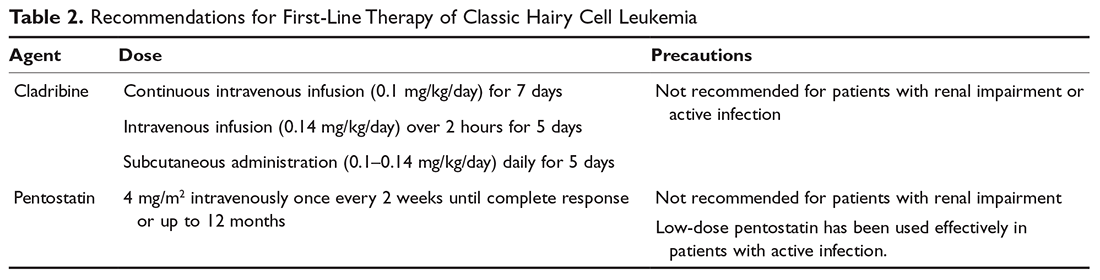
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3. 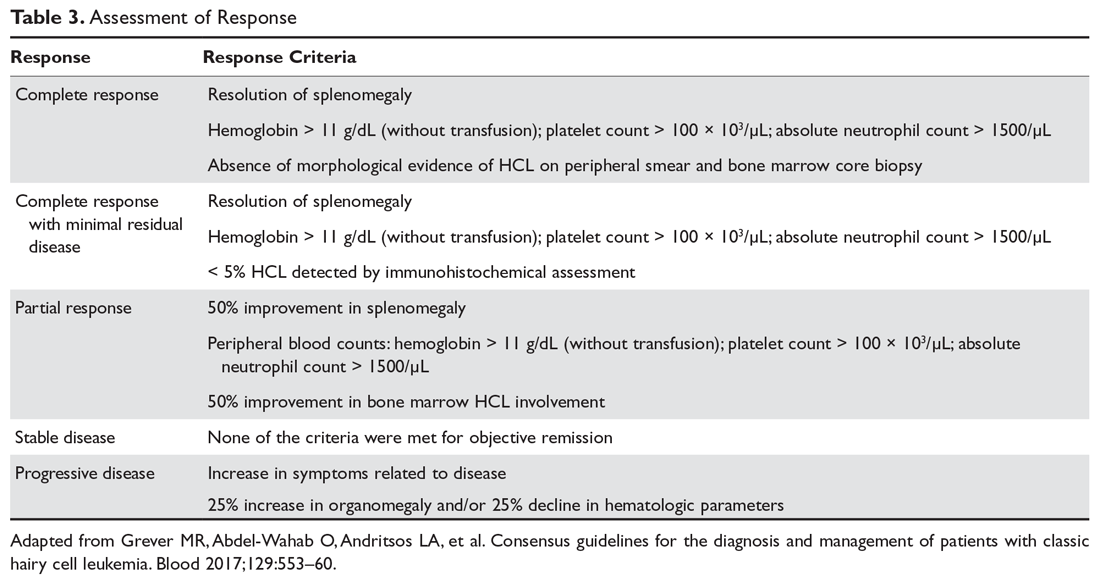
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.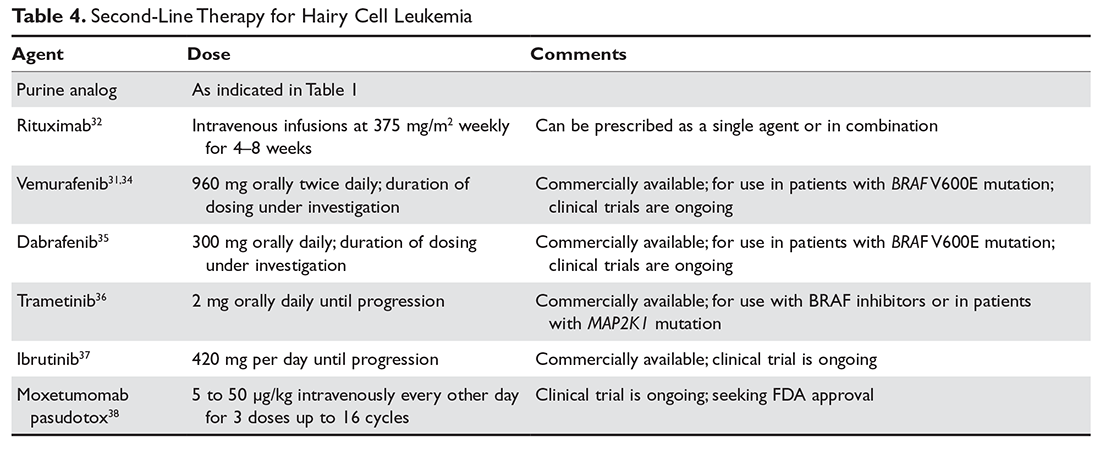
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease.
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3.
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease.
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3.
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.