User login
Clinical presentation, diagnosis, and management of typical and atypical bronchopulmonary carcinoid
Carcinoid lung tumors represent the most indolent form of a spectrum of bronchopulmonary neuroendocrine tumors (NETs) that includes small-cell carcinoma as its most malignant member, as well as several other forms of intermediately aggressive tumors, such as atypical carcinoid.1 Carcinoids represent 1.2% of all primary lung malignancies. Their incidence in the United States has increased rapidly over the last 30 years and is currently about 6% a year. Lung carcinoids are more prevalent in whites compared with blacks, and in Asians compared with non-Asians. They are less common in Hispanics compared with non-Hispanics.1 Typical carcinoids represent 80%-90% of all lung carcinoids and occur more frequently in the fifth and sixth decades of life. They can, however, occur at any age, and are the most common lung tumor in childhood.1
Etiology and risk factors
Unlike carcinoma of the lung, no external environmental toxin or other stimulus has been identified as a causative agent for the development of pulmonary carcinoid tumors. It is not clear if there is an association between bronchial NETs and smoking.1 Nearly all bronchial NETs are sporadic; however, they can rarely occur in the setting of multiple endocrine neoplasia type 1.1
Presentation
About 60% of the patients with bronchial carcinoids are symptomatic at presentation. The most common clinical findings are those associated with bronchial obstruction, such as persistent cough, hemoptysis, and recurrent or obstructive pneumonitis. Wheezing, chest pain, and dyspnea also may be noted.2 Various endocrine or neuroendocrine syndromes can be initial clinical manifestations of either typical or atypical pulmonary carcinoid tumors, but that is not common Cushing syndrome (ectopic production and secretion of adrenocorticotropic hormone [ACTH]) may occur in about 2% of lung carcinoid.3 In cases of malignancy, the presence of metastatic disease can produce weight loss, weakness, and a general feeling of ill health.
Diagnostic work-up
Biochemical test
There is no biochemical study that can be used as a screening test to determine the presence of a carcinoid tumor or to diagnose a known pulmonary mass as a carcinoid tumor. Neuroendocrine cells produce biologically active amines and peptides that can be detected in serum and urine. Although the syndromes associated with lung carcinoids are seen in about 1%-2% of the patients, assays of specific hormones or other circulating neuroendocrine substances, such as ACTH, melanocyte-stimulating hormone, or growth hormone may establish the existence of a clinically suspected syndrome.
Chest radiography
An abnormal finding on chest radiography is present in about 75% of patients with a pulmonary carcinoid tumor.1 Findings include either the presence of the tumor mass itself or indirect evidence of its presence observed as parenchymal changes associated with bronchial obstruction from the mass.
Computed-tomography imaging
High-resolution computed-tomography (CT) imaging is the one of the best types of CT examination for evaluation of a pulmonary carcinoid tumor.4 A CT scan provides excellent resolution of tumor extent, location, and the presence or absence of mediastinal adenopathy. It also aids in morphologic characterization of peripheral (Figure 1) and especially centrally located carcinoids, which may be purely intraluminal (polypoid configuration), exclusively extra luminal, or more frequently, a mixture of intraluminal and extraluminal components.
CT scans may also be helpful for differentiating tumor from postobstructive atelectasis or bronchial obstruction-related mucoid impaction. Intravenous contrast in CT imaging can be useful in differentiating malignant from benign lesions. Because carcinoid tumors are highly vascular, they show greater enhancement on contrast CT than do benign lesions. The sensitivity of CT for detecting metastatic hilar or mediastinal nodes is high, but specificity is as low as 45%.4
Typical carcinoid is rarely metastatic so most patients do not need CT or MRI imaging to evaluate for liver involvement. Liver imaging is appropriate in patients with evidence of mediastinal involvement, relatively high mitotic rate, or clinical evidence of the carcinoid syndrome.8 To evaluate for metastatic spread to the liver, multiphase contrast-enhanced liver CT scans should be performed with arterial and portal-venous phases because carcinoid liver metastases are often hypervascular and appear isodense relative to the liver parenchyma after contrast administration.4 An MRI is often preferred the modality to evaluate for metastatic spread to the liver because of its higher sensitivity.5
Positron-emission tomography
Although carcinoid tumors of the lung are highly vascular, they do not show increased metabolic activity on positron-emission tomography (PET) and would be incorrectly designated as benign lesions on the basis of findings from a PET scan. Fludeoxyglucose F-18 PET has shown utility as a radiologic marker for atypical carcinoids, particularly for those with a higher proliferation index with Ki-67 index of 10%-20%.6
Radionucleotide studies
Somatostatin receptors (SSRs) are present in many tumors of neuroendocrine origin, including carcinoid tumors. These receptors interact with each other and undergo dimerization and internalization. SSTR subtypes (SSTRs) overexpressed in NETs are related to the type, origin, and grade of differentiation of tumor. The overexpression of an SSTR is a characteristic feature of bronchial NETs, which can be used to localize the primary tumor and its metastases by imaging with the radiolabeled SST analogues. Radionucleotide imaging modalities commonly used include single-photon–emission tomography and positron-emission tomography.
With regard to SSR scintigraphy testing, PET using Ga–DOTATATE/TOC is preferable to Octreoscan if it is available, because offers better spatial resolution and has a shorter scanning time. It has sensitivity of 97% and specificity of 92% and hence is preferable over Octreoscan in highly aggressive, atypical bronchial NETs. It also provides an estimate of receptor density and evidence of the functionality of receptors, which helps with selection of suitable treatments that act on these receptors.7
Tumor markers
Serum levels of chromogranin A in bronchial NETs are expressed at a lower rate than are other sites of carcinoid tumors, so its measurement is of limited utility in following disease activity in bronchial NETs.4,8
Bronchoscopy
About 75% of pulmonary carcinoids are visible on bronchoscopy. The bronchoscopic appearance may be characteristic but it is preferable that brushings or biopsy be performed to confirm the diagnosis. For central tumors endobronchial; and for peripheral tumors, CT-guided percutaneous biopsy is the accepted diagnostic approach. Cytologic study of bronchial brushings is more sensitive than sputum cytology, but the diagnostic yield of brushing is low overall (about 40%) and hence fine-needle biopsy is preferred. 8
A negative finding on biopsy should not produce a false sense of confidence. If a suspicion of malignancy exists despite a negative finding on transthoracic biopsy, surgical excision of the nodule and pathologic analysis should be undertaken.
Histological findings
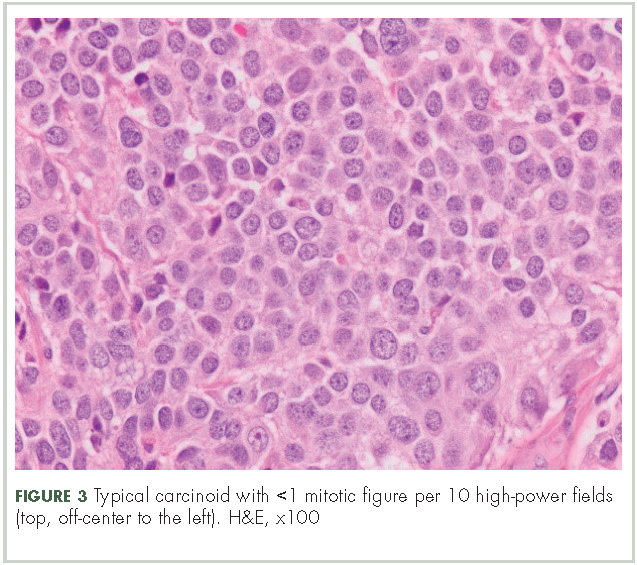
In typical carcinoid tumors, cells tend to group in nests, cords, or broad sheets. Arrangement is orderly, with groups of cells separated by highly vascular septa of connective tissue.9Individual cell features include small and polygonal cells with finely granular eosinophilic cytoplasm (Figure 2). Nuclei are small and round. Mitoses are infrequent (Figure 3).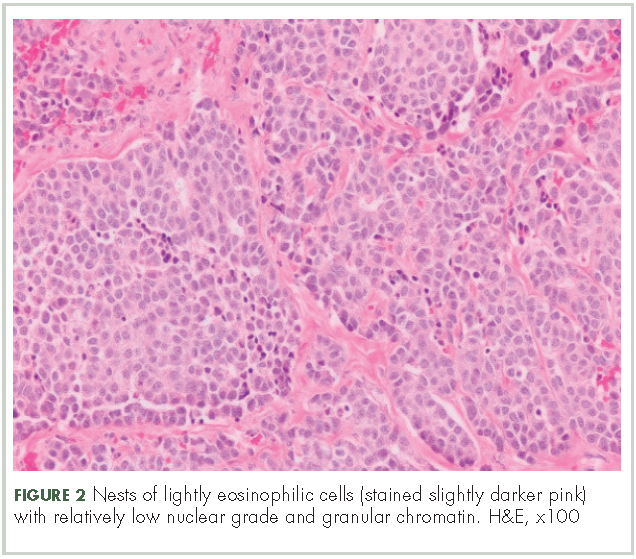
On electron microscopy, well-formed desmosomes and abundant neurosecretory granules are seen. Many pulmonary carcinoid tumors stain positive for a variety of neuroendocrine markers. Electron microscopy is of historical interest but is not used for tissue diagnosis for bronchial carcinoid patients.
Typical vs atypical tumors
In all, 10% of the carcinoid tumors are atypical in nature. They are generally larger than typical carcinoids and are located in the periphery of the lung in about 50% of cases. They have more aggressive behavior and tend to metastasize more commonly.2 Neither location nor size are distinguishing features. The distinction is based on histology and includes one or all of the following features:8,9
n Increased mitotic activity in a tumor with an identifiable carcinoid cellular arrangement with 2-10 mitotic figures per high-power field.9
n Pleomorphism and irregular nuclei with hyperchromatism and prominent nucleoli.
n Areas of increased cellularity with loss of the regular, organized architecture observed in typical carcinoid.
n Areas of necrosis within the tumor.
Ki-67 cell proliferation labeling index can be used to distinguish between high-grade lung NETs (>40%) and carcinoids (<20%), particularly in crushed biopsy specimens in which carcinoids may be mistaken for small-cell lung cancers. However, given overlap in the distribution of Ki-67 labeling index between typical carcinoids (≤5%) and atypical carcinoids (≤20%), Ki-67 expression does not reliably distinguish between well-differentiated lung carcinoids. The utility of Ki-67 to differentiate between typical and atypical carcinoids has yet to be established, and it is not presently recommended.9 Hence, the number of mitotic figures per high-power field of viable tumor area and presence or absence of necrosis continue to be the salient features distinguishing typical and atypical bronchial NETs.
Staging10
Lung NETs are staged using the same tumor, node, metastasis (TNM) classification from the American Joint Committee on Cancer (AJCC) that is used for bronchogenic lung carcinomas (Table).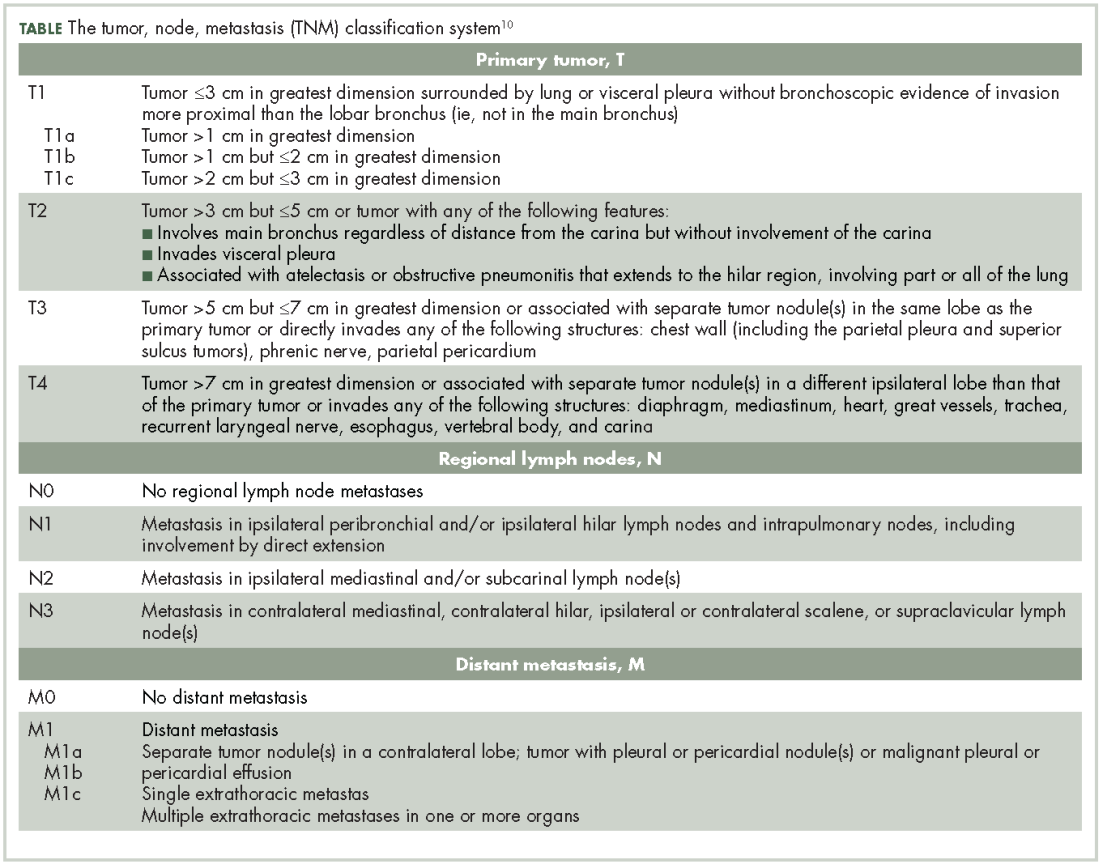
Typical bronchial NETs most commonly present as stage I tumors, whereas more than one-half of atypical tumors are stage II (bronchopulmonary nodal involvement) or III (mediastinal nodal involvement) at presentation.
Treatment
Localized or nonmetastatic and rescetable disease
Surgical treatment. As with other non–small-cell lung cancers (NSCLCs), surgical resection is the treatment of choice for early-stage carcinoid. The long-term prognosis is typically excellent, with a 10-year disease-free survival of 77%-94%.11, 12 The extent of resection is determined by the tumor size, histology, and location. For NSCLC, the standard surgical approach is the minimal anatomic resection (lobectomy, sleeve lobectomy, bilobectomy, or pneumonectomy) needed to get microscopically negative margins, with an associated mediastinal and hilar lymph node dissection for staging.13
Given the indolent nature of typical carcinoids, there has been extensive research to evaluate whether a sublobar resection is oncologically appropriate for these tumors. Although there are no comprehensive randomized studies comparing sublobar resection with lobectomy for typical carcinoids, findings from numerous database reviews and single-center studies suggest that sublobar resections are noninferior.14-17 Due to the higher nodal metastatic rate and the overall poorer prognosis associated with atypical carcinoids, formal anatomic resection is still recommended with atypical histology.18
An adaptive approach must be taken for patients who undergo wedge resection of pulmonary lesions without a known diagnosis. If intraoperative frozen section is consistent with carcinoid and the margins are negative, mediastinal lymph node dissection should be performed. If the patient is node negative, then completion lobectomy is not required. In node-positive patients with adequate pulmonary reserve, lobectomy should be performed regardless of histology. If atypical features are found during pathologic evaluation, then interval completion lobectomy may be patients with adequate pulmonary reserve.19,20
As with other pulmonary malignancies, clinical or radiographic suspicion of mediastinal lymph node involvement requires invasive staging before pulmonary resection is considered. If the patient is proven to have mediastinal metastatic disease, then multimodality treatment should be considered.20
Adjuvant therapy. Postoperative adjuvant therapy for most resected bronchial NETs, even in the setting of positive lymph nodes, is generally not recommended.7 In clinical practice, adjuvant platinum-based chemotherapy with or without radiation therapy (RT) is a reasonable option for patients with histologically aggressive-appearing or poorly differentiated stage III atypical bronchial NETs, although there is only limited evidence to support this. RT is a reasonable option for atypical bronchial NETs if gross residual disease remains after surgery, although it has not been proven that this improves outcomes.7
Nonmetastatic and unresectable disease
For inoperable patients and for those with surgically unresectable but nonmetastatic disease, options for local control of tumor growth include RT with or without concurrent chemotherapy and palliative endobronchial resection of obstructing tumor.21
Metastatic and unresectable disease
Everolimus. In February 2016, everolimus was approved by the US Food and Drug Administration (FDA) as first-line therapy for progressive, well-differentiated, nonfunctional NETs of lung origin that are unresectable, locally advanced, or metastatic. The aApproval was based on the RADIANT-4 trial, in which median progression-free survival was 11 months in the 205 patients allocated to receive everolimus (10 mg/day) and 3.9 months in the 97 patients who received placebo. Everolimus was associated with a 52% reduction in the estimated risk of progression or death.22
Somastatin analogues (SSA). There is lack of comprehensive data on the role of SSA compared with everolimus in lung carcinoid. The National Comprehensive Cancer Network (NCCN) guidelines on NETs and SCLCs recommend the consideration of octreotide or lanreotide as first-line therapies for select patients with symptoms of carcinoid syndrome or octreotide-positive scans.21 Guidelines from the European Neuroendocrine Tumor Society (ENETS)19 also recommend the use of SSAs as a first-line option in patients with: lung carcinoids exhibiting hormone-related symptoms or slowly progressive typical or atypical carcinoid with a low proliferative index (preferably Ki-67 <10%), provided there is a strongly positive SSTR status.
In cases in which metastatic lung NETs are associated with the carcinoid syndrome, initiation of long-acting SSA therapy in combination with everolimus is recommended.
Cytotoxic chemotherapy. According to the NCCN guidelines, cisplatin-etoposide or other cytotoxic regimens (eg, those that are temozolomide based) are recommended for advanced typical and atypical carcinoids, with cisplatin-etoposide being the preferred first-line systemic regimen in stage IV atypical carcinoid.22 ENETS guidelines stipulate that systemic chemotherapy is generally restricted to atypical carcinoid after failure of first-line therapies and only under certain conditions (Ki-67 >15%, rapidly progressive disease, and SSTR-negative disease).19 Based on a summary of NCCN and ENET guidelines:
n For patients with highly aggressive atypical bronchial NETs, a combination of platinum- and etoposide-based regimens such as those used for small-cell lung cancer has shown better response rate and overall survival data.
n For patients with typical or atypical bronchial NETs, temozolomide can be used as monotherapy or combination with capecitabine, although there are no findings from large randomized controlled trials to support this. Capecitabine-temozolomide has recently shown moderate activity in a small, single-institution study of patients with advanced lung carcinoids (N = 19), with 11 of 17 assessable patients (65%) demonstrating stable disease or partial response.23
n The following regimens can also be used for advanced disease after failure of somastatin analogues, although there are limited data for objective responses:24,25fluorouracil plus dacarbazine; epirubicin, capecitabine plus oxaliplatin; and capecitabine plus liposomal doxorubicin.
Participation in a clinical trial should be encouraged for patients with progressive bronchial NETs during any line of therapy. For patients who have a limited, potentially resectable liver-isolated metastatic NET, surgical resection should be pursued. For more extensive unresectable liver-dominant metastatic disease, treatment options include embolization, radiofrequency ablation, and cryoablation.20,22
Posttreatment surveillance
Posttreatment surveillance after resection of node-positive typical bronchial NETs and for all atypical tumors.26 Patients with lymph-node–negative typical bronchial NETs are very unlikely to benefit from postoperative surveillance because of the very low risk of recurrence. CT imaging (including the thorax and abdomen) every 6 months for 2 years, followed by annual scans for a total of 5-10 years are a reasonable surveillance schedule.
Prognosis 18,27
Typical bronchial NETs have an excellent prognosis after surgical resection. Reported 5-year survival rates are 87%-100%; the corresponding rates at 10 years are 82%-87%. Features associated with negative prognostic significance include lymph-node involvement and incomplete resection.
Atypical bronchial NETs have a worse prognosis than do typical tumors. Five-year survival rates range widely, from 30%-95%; the corresponding rates at 10 years are 35%-56%. Atypical tumors have a greater tendency to metastasize (16%-23%) and recur locally (3%-25%). Distant metastases to the liver or bone are more common than local recurrence. Adverse influence of nodal metastases on prognosis is more profound than for typical tumors. Survival rates by stage for patients who underwent surgical resection (including typical and atypical carcinoid27) are: stage I, 93%; stage II, 85%; stage III, 75%; and stage IV, 57%.
1. Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008;113(10):2655-2664.
2. Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119(6):1647-1651.
3. Limper AH, Carpenter PC, Scheithauer B, Staats BA. The Cushing syndrome induced by bronchial carcinoid tumors. Ann Intern Med. 1992;117(3):209-214.
4. Meisinger QC, Klein JS, Butnor KJ, Gentchos G, Leavitt BJ. CT features of peripheral pulmonary carcinoid tumors. AJR Am J Roentgenol. 2011;197(5):1073-1080.
5. Guckel C, Schnabel K, Deimling M, Steinbrich W. Solitary pulmonary nodules: MR evaluation of enhancement patterns with contrast-enhanced dynamic snapshot gradient-echo imaging. Radiology. 1996;200(3):681-686.
6. Jindal T, Kumar A, Venkitaraman B, et al. Evaluation of the role of [18F]FDG-PET/CT and [68Ga]DOTATOC-PET/CT in differentiating typical and atypical pulmonary carcinoids. Cancer Imaging. 2011;11:70-75.
7. Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620.
8. Travis WD. Pathology and diagnosis of neuroendocrine tumors: lung neuroendocrine. Thorac Surg Clin. 2014;24(3):257-266.
9. Warren WH, Memoli VA, Gould VE. Immunohistochemical and ultrastructural analysis of bronchopulmonary neuroendocrine neoplasms. II. Well-differentiated neuroendocrine carcinomas. Ultrastruct Pathol. 1984;7(2-3):185-199.
10. Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: Proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39-51.
11. McCaughan BC, Martini N, Bains MS. Bronchial carcinoids. Review of 124 cases. J Thorac Cardiovasc Surg. 1985;89(1):8-17.
12. Hurt R, Bates M. Carcinoid tumours of the bronchus: a 33-year experience. Thorax. 1984;39(8):617-623.
13. Ettinger DS, Wood DE, Akerley W, et al. Non-small cell lung cancer, version 1.2015. J Natl Compr Cancer Netw. 2014;12(12):1738-1761.
14. Ferguson MK, Landreneau RJ, Hazelrigg SR, et al. Long-term outcome after resection for bronchial carcinoid tumors. Eur J Cardiothorac Surg. 2000;18(2):156-161.
15. Lucchi M, Melfi F, Ribechini A, et al. Sleeve and wedge parenchyma-sparing bronchial resections in low-grade neoplasms of the bronchial airway. J Thorac Cardiovasc Surg. 2007;134(2):373-377.
16. Yendamuri S, Gold D, Jayaprakash V, Dexter E, Nwogu C, Demmy T. Is sublobar resection sufficient for carcinoid tumors? Ann Thorac Surg. 2011;92(5):1774-1778; discussion 8-9.
17. Fox M, Van Berkel V, Bousamra M II, Sloan S, Martin RC II. Surgical management of pulmonary carcinoid tumors: sublobar resection versus lobectomy. Am J Surg. 2013;205(2):200-208.
18. Cardillo G, Sera F, Di Martino M, et al. Bronchial carcinoid tumors: nodal status and long-term survival after resection. Ann Thorac Surg. 2004;77(5):1781-1785.
19. Oberg K, Hellman P, Ferolla P, Papotti M; ESMO Guidelines Working Group. Neuroendocrine bronchial and thymic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7:vii120-3).
20. Filosso PL, Ferolla P, Guerrera F, et al. Multidisciplinary management of advanced lung neuroendocrine tumors. J Thorac Dis. 2015;7(Suppl 2):S163-171.
21. Kulke MH, Shah MH, Benson AB III, et al. Neuroendocrine tumors, version 1.2015. J Natl Compr Cancer Netw. 2015;13(1):78-108.
22. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
23. Ramirez RA, Beyer DT, Chauhan A, Boudreaux JP, Wang YZ, Woltering EA. The role of capecitabine/temozolomide in metastatic neuroendocrine tumors. Oncologist. 2016;21(6):671-675.
24. Bajetta E, Rimassa L, Carnaghi C, et al. 5-Fluorouracil, dacarbazine, and epirubicin in the treatment of patients with neuroendocrine tumors. Cancer. 1998;83(2):372-378.
25. Masi G, Fornaro L, Cupini S, et al. Refractory neuroendocrine tumor-response to liposomal doxorubicin and capecitabine. Nat Rev Clin Oncol. 2009;6(11):670-674.
26. Lou F, Sarkaria I, Pietanza C, et al. Recurrence of pulmonary carcinoid tumors after resection: implications for postoperative surveillance. Ann Thorac Surg. 2013;96(4):1156-1162.
27. Beasley MB, Thunnissen FB, Brambilla E, et al. Pulmonary atypical carcinoid: predictors of survival in 106 cases. Human Pathol. 2000;31(10):1255-1265.
Carcinoid lung tumors represent the most indolent form of a spectrum of bronchopulmonary neuroendocrine tumors (NETs) that includes small-cell carcinoma as its most malignant member, as well as several other forms of intermediately aggressive tumors, such as atypical carcinoid.1 Carcinoids represent 1.2% of all primary lung malignancies. Their incidence in the United States has increased rapidly over the last 30 years and is currently about 6% a year. Lung carcinoids are more prevalent in whites compared with blacks, and in Asians compared with non-Asians. They are less common in Hispanics compared with non-Hispanics.1 Typical carcinoids represent 80%-90% of all lung carcinoids and occur more frequently in the fifth and sixth decades of life. They can, however, occur at any age, and are the most common lung tumor in childhood.1
Etiology and risk factors
Unlike carcinoma of the lung, no external environmental toxin or other stimulus has been identified as a causative agent for the development of pulmonary carcinoid tumors. It is not clear if there is an association between bronchial NETs and smoking.1 Nearly all bronchial NETs are sporadic; however, they can rarely occur in the setting of multiple endocrine neoplasia type 1.1
Presentation
About 60% of the patients with bronchial carcinoids are symptomatic at presentation. The most common clinical findings are those associated with bronchial obstruction, such as persistent cough, hemoptysis, and recurrent or obstructive pneumonitis. Wheezing, chest pain, and dyspnea also may be noted.2 Various endocrine or neuroendocrine syndromes can be initial clinical manifestations of either typical or atypical pulmonary carcinoid tumors, but that is not common Cushing syndrome (ectopic production and secretion of adrenocorticotropic hormone [ACTH]) may occur in about 2% of lung carcinoid.3 In cases of malignancy, the presence of metastatic disease can produce weight loss, weakness, and a general feeling of ill health.
Diagnostic work-up
Biochemical test
There is no biochemical study that can be used as a screening test to determine the presence of a carcinoid tumor or to diagnose a known pulmonary mass as a carcinoid tumor. Neuroendocrine cells produce biologically active amines and peptides that can be detected in serum and urine. Although the syndromes associated with lung carcinoids are seen in about 1%-2% of the patients, assays of specific hormones or other circulating neuroendocrine substances, such as ACTH, melanocyte-stimulating hormone, or growth hormone may establish the existence of a clinically suspected syndrome.
Chest radiography
An abnormal finding on chest radiography is present in about 75% of patients with a pulmonary carcinoid tumor.1 Findings include either the presence of the tumor mass itself or indirect evidence of its presence observed as parenchymal changes associated with bronchial obstruction from the mass.
Computed-tomography imaging
High-resolution computed-tomography (CT) imaging is the one of the best types of CT examination for evaluation of a pulmonary carcinoid tumor.4 A CT scan provides excellent resolution of tumor extent, location, and the presence or absence of mediastinal adenopathy. It also aids in morphologic characterization of peripheral (Figure 1) and especially centrally located carcinoids, which may be purely intraluminal (polypoid configuration), exclusively extra luminal, or more frequently, a mixture of intraluminal and extraluminal components.
CT scans may also be helpful for differentiating tumor from postobstructive atelectasis or bronchial obstruction-related mucoid impaction. Intravenous contrast in CT imaging can be useful in differentiating malignant from benign lesions. Because carcinoid tumors are highly vascular, they show greater enhancement on contrast CT than do benign lesions. The sensitivity of CT for detecting metastatic hilar or mediastinal nodes is high, but specificity is as low as 45%.4
Typical carcinoid is rarely metastatic so most patients do not need CT or MRI imaging to evaluate for liver involvement. Liver imaging is appropriate in patients with evidence of mediastinal involvement, relatively high mitotic rate, or clinical evidence of the carcinoid syndrome.8 To evaluate for metastatic spread to the liver, multiphase contrast-enhanced liver CT scans should be performed with arterial and portal-venous phases because carcinoid liver metastases are often hypervascular and appear isodense relative to the liver parenchyma after contrast administration.4 An MRI is often preferred the modality to evaluate for metastatic spread to the liver because of its higher sensitivity.5
Positron-emission tomography
Although carcinoid tumors of the lung are highly vascular, they do not show increased metabolic activity on positron-emission tomography (PET) and would be incorrectly designated as benign lesions on the basis of findings from a PET scan. Fludeoxyglucose F-18 PET has shown utility as a radiologic marker for atypical carcinoids, particularly for those with a higher proliferation index with Ki-67 index of 10%-20%.6
Radionucleotide studies
Somatostatin receptors (SSRs) are present in many tumors of neuroendocrine origin, including carcinoid tumors. These receptors interact with each other and undergo dimerization and internalization. SSTR subtypes (SSTRs) overexpressed in NETs are related to the type, origin, and grade of differentiation of tumor. The overexpression of an SSTR is a characteristic feature of bronchial NETs, which can be used to localize the primary tumor and its metastases by imaging with the radiolabeled SST analogues. Radionucleotide imaging modalities commonly used include single-photon–emission tomography and positron-emission tomography.
With regard to SSR scintigraphy testing, PET using Ga–DOTATATE/TOC is preferable to Octreoscan if it is available, because offers better spatial resolution and has a shorter scanning time. It has sensitivity of 97% and specificity of 92% and hence is preferable over Octreoscan in highly aggressive, atypical bronchial NETs. It also provides an estimate of receptor density and evidence of the functionality of receptors, which helps with selection of suitable treatments that act on these receptors.7
Tumor markers
Serum levels of chromogranin A in bronchial NETs are expressed at a lower rate than are other sites of carcinoid tumors, so its measurement is of limited utility in following disease activity in bronchial NETs.4,8
Bronchoscopy
About 75% of pulmonary carcinoids are visible on bronchoscopy. The bronchoscopic appearance may be characteristic but it is preferable that brushings or biopsy be performed to confirm the diagnosis. For central tumors endobronchial; and for peripheral tumors, CT-guided percutaneous biopsy is the accepted diagnostic approach. Cytologic study of bronchial brushings is more sensitive than sputum cytology, but the diagnostic yield of brushing is low overall (about 40%) and hence fine-needle biopsy is preferred. 8
A negative finding on biopsy should not produce a false sense of confidence. If a suspicion of malignancy exists despite a negative finding on transthoracic biopsy, surgical excision of the nodule and pathologic analysis should be undertaken.
Histological findings
In typical carcinoid tumors, cells tend to group in nests, cords, or broad sheets. Arrangement is orderly, with groups of cells separated by highly vascular septa of connective tissue.9Individual cell features include small and polygonal cells with finely granular eosinophilic cytoplasm (Figure 2). Nuclei are small and round. Mitoses are infrequent (Figure 3).
On electron microscopy, well-formed desmosomes and abundant neurosecretory granules are seen. Many pulmonary carcinoid tumors stain positive for a variety of neuroendocrine markers. Electron microscopy is of historical interest but is not used for tissue diagnosis for bronchial carcinoid patients.
Typical vs atypical tumors
In all, 10% of the carcinoid tumors are atypical in nature. They are generally larger than typical carcinoids and are located in the periphery of the lung in about 50% of cases. They have more aggressive behavior and tend to metastasize more commonly.2 Neither location nor size are distinguishing features. The distinction is based on histology and includes one or all of the following features:8,9
n Increased mitotic activity in a tumor with an identifiable carcinoid cellular arrangement with 2-10 mitotic figures per high-power field.9
n Pleomorphism and irregular nuclei with hyperchromatism and prominent nucleoli.
n Areas of increased cellularity with loss of the regular, organized architecture observed in typical carcinoid.
n Areas of necrosis within the tumor.
Ki-67 cell proliferation labeling index can be used to distinguish between high-grade lung NETs (>40%) and carcinoids (<20%), particularly in crushed biopsy specimens in which carcinoids may be mistaken for small-cell lung cancers. However, given overlap in the distribution of Ki-67 labeling index between typical carcinoids (≤5%) and atypical carcinoids (≤20%), Ki-67 expression does not reliably distinguish between well-differentiated lung carcinoids. The utility of Ki-67 to differentiate between typical and atypical carcinoids has yet to be established, and it is not presently recommended.9 Hence, the number of mitotic figures per high-power field of viable tumor area and presence or absence of necrosis continue to be the salient features distinguishing typical and atypical bronchial NETs.
Staging10
Lung NETs are staged using the same tumor, node, metastasis (TNM) classification from the American Joint Committee on Cancer (AJCC) that is used for bronchogenic lung carcinomas (Table).
Typical bronchial NETs most commonly present as stage I tumors, whereas more than one-half of atypical tumors are stage II (bronchopulmonary nodal involvement) or III (mediastinal nodal involvement) at presentation.
Treatment
Localized or nonmetastatic and rescetable disease
Surgical treatment. As with other non–small-cell lung cancers (NSCLCs), surgical resection is the treatment of choice for early-stage carcinoid. The long-term prognosis is typically excellent, with a 10-year disease-free survival of 77%-94%.11, 12 The extent of resection is determined by the tumor size, histology, and location. For NSCLC, the standard surgical approach is the minimal anatomic resection (lobectomy, sleeve lobectomy, bilobectomy, or pneumonectomy) needed to get microscopically negative margins, with an associated mediastinal and hilar lymph node dissection for staging.13
Given the indolent nature of typical carcinoids, there has been extensive research to evaluate whether a sublobar resection is oncologically appropriate for these tumors. Although there are no comprehensive randomized studies comparing sublobar resection with lobectomy for typical carcinoids, findings from numerous database reviews and single-center studies suggest that sublobar resections are noninferior.14-17 Due to the higher nodal metastatic rate and the overall poorer prognosis associated with atypical carcinoids, formal anatomic resection is still recommended with atypical histology.18
An adaptive approach must be taken for patients who undergo wedge resection of pulmonary lesions without a known diagnosis. If intraoperative frozen section is consistent with carcinoid and the margins are negative, mediastinal lymph node dissection should be performed. If the patient is node negative, then completion lobectomy is not required. In node-positive patients with adequate pulmonary reserve, lobectomy should be performed regardless of histology. If atypical features are found during pathologic evaluation, then interval completion lobectomy may be patients with adequate pulmonary reserve.19,20
As with other pulmonary malignancies, clinical or radiographic suspicion of mediastinal lymph node involvement requires invasive staging before pulmonary resection is considered. If the patient is proven to have mediastinal metastatic disease, then multimodality treatment should be considered.20
Adjuvant therapy. Postoperative adjuvant therapy for most resected bronchial NETs, even in the setting of positive lymph nodes, is generally not recommended.7 In clinical practice, adjuvant platinum-based chemotherapy with or without radiation therapy (RT) is a reasonable option for patients with histologically aggressive-appearing or poorly differentiated stage III atypical bronchial NETs, although there is only limited evidence to support this. RT is a reasonable option for atypical bronchial NETs if gross residual disease remains after surgery, although it has not been proven that this improves outcomes.7
Nonmetastatic and unresectable disease
For inoperable patients and for those with surgically unresectable but nonmetastatic disease, options for local control of tumor growth include RT with or without concurrent chemotherapy and palliative endobronchial resection of obstructing tumor.21
Metastatic and unresectable disease
Everolimus. In February 2016, everolimus was approved by the US Food and Drug Administration (FDA) as first-line therapy for progressive, well-differentiated, nonfunctional NETs of lung origin that are unresectable, locally advanced, or metastatic. The aApproval was based on the RADIANT-4 trial, in which median progression-free survival was 11 months in the 205 patients allocated to receive everolimus (10 mg/day) and 3.9 months in the 97 patients who received placebo. Everolimus was associated with a 52% reduction in the estimated risk of progression or death.22
Somastatin analogues (SSA). There is lack of comprehensive data on the role of SSA compared with everolimus in lung carcinoid. The National Comprehensive Cancer Network (NCCN) guidelines on NETs and SCLCs recommend the consideration of octreotide or lanreotide as first-line therapies for select patients with symptoms of carcinoid syndrome or octreotide-positive scans.21 Guidelines from the European Neuroendocrine Tumor Society (ENETS)19 also recommend the use of SSAs as a first-line option in patients with: lung carcinoids exhibiting hormone-related symptoms or slowly progressive typical or atypical carcinoid with a low proliferative index (preferably Ki-67 <10%), provided there is a strongly positive SSTR status.
In cases in which metastatic lung NETs are associated with the carcinoid syndrome, initiation of long-acting SSA therapy in combination with everolimus is recommended.
Cytotoxic chemotherapy. According to the NCCN guidelines, cisplatin-etoposide or other cytotoxic regimens (eg, those that are temozolomide based) are recommended for advanced typical and atypical carcinoids, with cisplatin-etoposide being the preferred first-line systemic regimen in stage IV atypical carcinoid.22 ENETS guidelines stipulate that systemic chemotherapy is generally restricted to atypical carcinoid after failure of first-line therapies and only under certain conditions (Ki-67 >15%, rapidly progressive disease, and SSTR-negative disease).19 Based on a summary of NCCN and ENET guidelines:
n For patients with highly aggressive atypical bronchial NETs, a combination of platinum- and etoposide-based regimens such as those used for small-cell lung cancer has shown better response rate and overall survival data.
n For patients with typical or atypical bronchial NETs, temozolomide can be used as monotherapy or combination with capecitabine, although there are no findings from large randomized controlled trials to support this. Capecitabine-temozolomide has recently shown moderate activity in a small, single-institution study of patients with advanced lung carcinoids (N = 19), with 11 of 17 assessable patients (65%) demonstrating stable disease or partial response.23
n The following regimens can also be used for advanced disease after failure of somastatin analogues, although there are limited data for objective responses:24,25fluorouracil plus dacarbazine; epirubicin, capecitabine plus oxaliplatin; and capecitabine plus liposomal doxorubicin.
Participation in a clinical trial should be encouraged for patients with progressive bronchial NETs during any line of therapy. For patients who have a limited, potentially resectable liver-isolated metastatic NET, surgical resection should be pursued. For more extensive unresectable liver-dominant metastatic disease, treatment options include embolization, radiofrequency ablation, and cryoablation.20,22
Posttreatment surveillance
Posttreatment surveillance after resection of node-positive typical bronchial NETs and for all atypical tumors.26 Patients with lymph-node–negative typical bronchial NETs are very unlikely to benefit from postoperative surveillance because of the very low risk of recurrence. CT imaging (including the thorax and abdomen) every 6 months for 2 years, followed by annual scans for a total of 5-10 years are a reasonable surveillance schedule.
Prognosis 18,27
Typical bronchial NETs have an excellent prognosis after surgical resection. Reported 5-year survival rates are 87%-100%; the corresponding rates at 10 years are 82%-87%. Features associated with negative prognostic significance include lymph-node involvement and incomplete resection.
Atypical bronchial NETs have a worse prognosis than do typical tumors. Five-year survival rates range widely, from 30%-95%; the corresponding rates at 10 years are 35%-56%. Atypical tumors have a greater tendency to metastasize (16%-23%) and recur locally (3%-25%). Distant metastases to the liver or bone are more common than local recurrence. Adverse influence of nodal metastases on prognosis is more profound than for typical tumors. Survival rates by stage for patients who underwent surgical resection (including typical and atypical carcinoid27) are: stage I, 93%; stage II, 85%; stage III, 75%; and stage IV, 57%.
Carcinoid lung tumors represent the most indolent form of a spectrum of bronchopulmonary neuroendocrine tumors (NETs) that includes small-cell carcinoma as its most malignant member, as well as several other forms of intermediately aggressive tumors, such as atypical carcinoid.1 Carcinoids represent 1.2% of all primary lung malignancies. Their incidence in the United States has increased rapidly over the last 30 years and is currently about 6% a year. Lung carcinoids are more prevalent in whites compared with blacks, and in Asians compared with non-Asians. They are less common in Hispanics compared with non-Hispanics.1 Typical carcinoids represent 80%-90% of all lung carcinoids and occur more frequently in the fifth and sixth decades of life. They can, however, occur at any age, and are the most common lung tumor in childhood.1
Etiology and risk factors
Unlike carcinoma of the lung, no external environmental toxin or other stimulus has been identified as a causative agent for the development of pulmonary carcinoid tumors. It is not clear if there is an association between bronchial NETs and smoking.1 Nearly all bronchial NETs are sporadic; however, they can rarely occur in the setting of multiple endocrine neoplasia type 1.1
Presentation
About 60% of the patients with bronchial carcinoids are symptomatic at presentation. The most common clinical findings are those associated with bronchial obstruction, such as persistent cough, hemoptysis, and recurrent or obstructive pneumonitis. Wheezing, chest pain, and dyspnea also may be noted.2 Various endocrine or neuroendocrine syndromes can be initial clinical manifestations of either typical or atypical pulmonary carcinoid tumors, but that is not common Cushing syndrome (ectopic production and secretion of adrenocorticotropic hormone [ACTH]) may occur in about 2% of lung carcinoid.3 In cases of malignancy, the presence of metastatic disease can produce weight loss, weakness, and a general feeling of ill health.
Diagnostic work-up
Biochemical test
There is no biochemical study that can be used as a screening test to determine the presence of a carcinoid tumor or to diagnose a known pulmonary mass as a carcinoid tumor. Neuroendocrine cells produce biologically active amines and peptides that can be detected in serum and urine. Although the syndromes associated with lung carcinoids are seen in about 1%-2% of the patients, assays of specific hormones or other circulating neuroendocrine substances, such as ACTH, melanocyte-stimulating hormone, or growth hormone may establish the existence of a clinically suspected syndrome.
Chest radiography
An abnormal finding on chest radiography is present in about 75% of patients with a pulmonary carcinoid tumor.1 Findings include either the presence of the tumor mass itself or indirect evidence of its presence observed as parenchymal changes associated with bronchial obstruction from the mass.
Computed-tomography imaging
High-resolution computed-tomography (CT) imaging is the one of the best types of CT examination for evaluation of a pulmonary carcinoid tumor.4 A CT scan provides excellent resolution of tumor extent, location, and the presence or absence of mediastinal adenopathy. It also aids in morphologic characterization of peripheral (Figure 1) and especially centrally located carcinoids, which may be purely intraluminal (polypoid configuration), exclusively extra luminal, or more frequently, a mixture of intraluminal and extraluminal components.
CT scans may also be helpful for differentiating tumor from postobstructive atelectasis or bronchial obstruction-related mucoid impaction. Intravenous contrast in CT imaging can be useful in differentiating malignant from benign lesions. Because carcinoid tumors are highly vascular, they show greater enhancement on contrast CT than do benign lesions. The sensitivity of CT for detecting metastatic hilar or mediastinal nodes is high, but specificity is as low as 45%.4
Typical carcinoid is rarely metastatic so most patients do not need CT or MRI imaging to evaluate for liver involvement. Liver imaging is appropriate in patients with evidence of mediastinal involvement, relatively high mitotic rate, or clinical evidence of the carcinoid syndrome.8 To evaluate for metastatic spread to the liver, multiphase contrast-enhanced liver CT scans should be performed with arterial and portal-venous phases because carcinoid liver metastases are often hypervascular and appear isodense relative to the liver parenchyma after contrast administration.4 An MRI is often preferred the modality to evaluate for metastatic spread to the liver because of its higher sensitivity.5
Positron-emission tomography
Although carcinoid tumors of the lung are highly vascular, they do not show increased metabolic activity on positron-emission tomography (PET) and would be incorrectly designated as benign lesions on the basis of findings from a PET scan. Fludeoxyglucose F-18 PET has shown utility as a radiologic marker for atypical carcinoids, particularly for those with a higher proliferation index with Ki-67 index of 10%-20%.6
Radionucleotide studies
Somatostatin receptors (SSRs) are present in many tumors of neuroendocrine origin, including carcinoid tumors. These receptors interact with each other and undergo dimerization and internalization. SSTR subtypes (SSTRs) overexpressed in NETs are related to the type, origin, and grade of differentiation of tumor. The overexpression of an SSTR is a characteristic feature of bronchial NETs, which can be used to localize the primary tumor and its metastases by imaging with the radiolabeled SST analogues. Radionucleotide imaging modalities commonly used include single-photon–emission tomography and positron-emission tomography.
With regard to SSR scintigraphy testing, PET using Ga–DOTATATE/TOC is preferable to Octreoscan if it is available, because offers better spatial resolution and has a shorter scanning time. It has sensitivity of 97% and specificity of 92% and hence is preferable over Octreoscan in highly aggressive, atypical bronchial NETs. It also provides an estimate of receptor density and evidence of the functionality of receptors, which helps with selection of suitable treatments that act on these receptors.7
Tumor markers
Serum levels of chromogranin A in bronchial NETs are expressed at a lower rate than are other sites of carcinoid tumors, so its measurement is of limited utility in following disease activity in bronchial NETs.4,8
Bronchoscopy
About 75% of pulmonary carcinoids are visible on bronchoscopy. The bronchoscopic appearance may be characteristic but it is preferable that brushings or biopsy be performed to confirm the diagnosis. For central tumors endobronchial; and for peripheral tumors, CT-guided percutaneous biopsy is the accepted diagnostic approach. Cytologic study of bronchial brushings is more sensitive than sputum cytology, but the diagnostic yield of brushing is low overall (about 40%) and hence fine-needle biopsy is preferred. 8
A negative finding on biopsy should not produce a false sense of confidence. If a suspicion of malignancy exists despite a negative finding on transthoracic biopsy, surgical excision of the nodule and pathologic analysis should be undertaken.
Histological findings
In typical carcinoid tumors, cells tend to group in nests, cords, or broad sheets. Arrangement is orderly, with groups of cells separated by highly vascular septa of connective tissue.9Individual cell features include small and polygonal cells with finely granular eosinophilic cytoplasm (Figure 2). Nuclei are small and round. Mitoses are infrequent (Figure 3).
On electron microscopy, well-formed desmosomes and abundant neurosecretory granules are seen. Many pulmonary carcinoid tumors stain positive for a variety of neuroendocrine markers. Electron microscopy is of historical interest but is not used for tissue diagnosis for bronchial carcinoid patients.
Typical vs atypical tumors
In all, 10% of the carcinoid tumors are atypical in nature. They are generally larger than typical carcinoids and are located in the periphery of the lung in about 50% of cases. They have more aggressive behavior and tend to metastasize more commonly.2 Neither location nor size are distinguishing features. The distinction is based on histology and includes one or all of the following features:8,9
n Increased mitotic activity in a tumor with an identifiable carcinoid cellular arrangement with 2-10 mitotic figures per high-power field.9
n Pleomorphism and irregular nuclei with hyperchromatism and prominent nucleoli.
n Areas of increased cellularity with loss of the regular, organized architecture observed in typical carcinoid.
n Areas of necrosis within the tumor.
Ki-67 cell proliferation labeling index can be used to distinguish between high-grade lung NETs (>40%) and carcinoids (<20%), particularly in crushed biopsy specimens in which carcinoids may be mistaken for small-cell lung cancers. However, given overlap in the distribution of Ki-67 labeling index between typical carcinoids (≤5%) and atypical carcinoids (≤20%), Ki-67 expression does not reliably distinguish between well-differentiated lung carcinoids. The utility of Ki-67 to differentiate between typical and atypical carcinoids has yet to be established, and it is not presently recommended.9 Hence, the number of mitotic figures per high-power field of viable tumor area and presence or absence of necrosis continue to be the salient features distinguishing typical and atypical bronchial NETs.
Staging10
Lung NETs are staged using the same tumor, node, metastasis (TNM) classification from the American Joint Committee on Cancer (AJCC) that is used for bronchogenic lung carcinomas (Table).
Typical bronchial NETs most commonly present as stage I tumors, whereas more than one-half of atypical tumors are stage II (bronchopulmonary nodal involvement) or III (mediastinal nodal involvement) at presentation.
Treatment
Localized or nonmetastatic and rescetable disease
Surgical treatment. As with other non–small-cell lung cancers (NSCLCs), surgical resection is the treatment of choice for early-stage carcinoid. The long-term prognosis is typically excellent, with a 10-year disease-free survival of 77%-94%.11, 12 The extent of resection is determined by the tumor size, histology, and location. For NSCLC, the standard surgical approach is the minimal anatomic resection (lobectomy, sleeve lobectomy, bilobectomy, or pneumonectomy) needed to get microscopically negative margins, with an associated mediastinal and hilar lymph node dissection for staging.13
Given the indolent nature of typical carcinoids, there has been extensive research to evaluate whether a sublobar resection is oncologically appropriate for these tumors. Although there are no comprehensive randomized studies comparing sublobar resection with lobectomy for typical carcinoids, findings from numerous database reviews and single-center studies suggest that sublobar resections are noninferior.14-17 Due to the higher nodal metastatic rate and the overall poorer prognosis associated with atypical carcinoids, formal anatomic resection is still recommended with atypical histology.18
An adaptive approach must be taken for patients who undergo wedge resection of pulmonary lesions without a known diagnosis. If intraoperative frozen section is consistent with carcinoid and the margins are negative, mediastinal lymph node dissection should be performed. If the patient is node negative, then completion lobectomy is not required. In node-positive patients with adequate pulmonary reserve, lobectomy should be performed regardless of histology. If atypical features are found during pathologic evaluation, then interval completion lobectomy may be patients with adequate pulmonary reserve.19,20
As with other pulmonary malignancies, clinical or radiographic suspicion of mediastinal lymph node involvement requires invasive staging before pulmonary resection is considered. If the patient is proven to have mediastinal metastatic disease, then multimodality treatment should be considered.20
Adjuvant therapy. Postoperative adjuvant therapy for most resected bronchial NETs, even in the setting of positive lymph nodes, is generally not recommended.7 In clinical practice, adjuvant platinum-based chemotherapy with or without radiation therapy (RT) is a reasonable option for patients with histologically aggressive-appearing or poorly differentiated stage III atypical bronchial NETs, although there is only limited evidence to support this. RT is a reasonable option for atypical bronchial NETs if gross residual disease remains after surgery, although it has not been proven that this improves outcomes.7
Nonmetastatic and unresectable disease
For inoperable patients and for those with surgically unresectable but nonmetastatic disease, options for local control of tumor growth include RT with or without concurrent chemotherapy and palliative endobronchial resection of obstructing tumor.21
Metastatic and unresectable disease
Everolimus. In February 2016, everolimus was approved by the US Food and Drug Administration (FDA) as first-line therapy for progressive, well-differentiated, nonfunctional NETs of lung origin that are unresectable, locally advanced, or metastatic. The aApproval was based on the RADIANT-4 trial, in which median progression-free survival was 11 months in the 205 patients allocated to receive everolimus (10 mg/day) and 3.9 months in the 97 patients who received placebo. Everolimus was associated with a 52% reduction in the estimated risk of progression or death.22
Somastatin analogues (SSA). There is lack of comprehensive data on the role of SSA compared with everolimus in lung carcinoid. The National Comprehensive Cancer Network (NCCN) guidelines on NETs and SCLCs recommend the consideration of octreotide or lanreotide as first-line therapies for select patients with symptoms of carcinoid syndrome or octreotide-positive scans.21 Guidelines from the European Neuroendocrine Tumor Society (ENETS)19 also recommend the use of SSAs as a first-line option in patients with: lung carcinoids exhibiting hormone-related symptoms or slowly progressive typical or atypical carcinoid with a low proliferative index (preferably Ki-67 <10%), provided there is a strongly positive SSTR status.
In cases in which metastatic lung NETs are associated with the carcinoid syndrome, initiation of long-acting SSA therapy in combination with everolimus is recommended.
Cytotoxic chemotherapy. According to the NCCN guidelines, cisplatin-etoposide or other cytotoxic regimens (eg, those that are temozolomide based) are recommended for advanced typical and atypical carcinoids, with cisplatin-etoposide being the preferred first-line systemic regimen in stage IV atypical carcinoid.22 ENETS guidelines stipulate that systemic chemotherapy is generally restricted to atypical carcinoid after failure of first-line therapies and only under certain conditions (Ki-67 >15%, rapidly progressive disease, and SSTR-negative disease).19 Based on a summary of NCCN and ENET guidelines:
n For patients with highly aggressive atypical bronchial NETs, a combination of platinum- and etoposide-based regimens such as those used for small-cell lung cancer has shown better response rate and overall survival data.
n For patients with typical or atypical bronchial NETs, temozolomide can be used as monotherapy or combination with capecitabine, although there are no findings from large randomized controlled trials to support this. Capecitabine-temozolomide has recently shown moderate activity in a small, single-institution study of patients with advanced lung carcinoids (N = 19), with 11 of 17 assessable patients (65%) demonstrating stable disease or partial response.23
n The following regimens can also be used for advanced disease after failure of somastatin analogues, although there are limited data for objective responses:24,25fluorouracil plus dacarbazine; epirubicin, capecitabine plus oxaliplatin; and capecitabine plus liposomal doxorubicin.
Participation in a clinical trial should be encouraged for patients with progressive bronchial NETs during any line of therapy. For patients who have a limited, potentially resectable liver-isolated metastatic NET, surgical resection should be pursued. For more extensive unresectable liver-dominant metastatic disease, treatment options include embolization, radiofrequency ablation, and cryoablation.20,22
Posttreatment surveillance
Posttreatment surveillance after resection of node-positive typical bronchial NETs and for all atypical tumors.26 Patients with lymph-node–negative typical bronchial NETs are very unlikely to benefit from postoperative surveillance because of the very low risk of recurrence. CT imaging (including the thorax and abdomen) every 6 months for 2 years, followed by annual scans for a total of 5-10 years are a reasonable surveillance schedule.
Prognosis 18,27
Typical bronchial NETs have an excellent prognosis after surgical resection. Reported 5-year survival rates are 87%-100%; the corresponding rates at 10 years are 82%-87%. Features associated with negative prognostic significance include lymph-node involvement and incomplete resection.
Atypical bronchial NETs have a worse prognosis than do typical tumors. Five-year survival rates range widely, from 30%-95%; the corresponding rates at 10 years are 35%-56%. Atypical tumors have a greater tendency to metastasize (16%-23%) and recur locally (3%-25%). Distant metastases to the liver or bone are more common than local recurrence. Adverse influence of nodal metastases on prognosis is more profound than for typical tumors. Survival rates by stage for patients who underwent surgical resection (including typical and atypical carcinoid27) are: stage I, 93%; stage II, 85%; stage III, 75%; and stage IV, 57%.
1. Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008;113(10):2655-2664.
2. Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119(6):1647-1651.
3. Limper AH, Carpenter PC, Scheithauer B, Staats BA. The Cushing syndrome induced by bronchial carcinoid tumors. Ann Intern Med. 1992;117(3):209-214.
4. Meisinger QC, Klein JS, Butnor KJ, Gentchos G, Leavitt BJ. CT features of peripheral pulmonary carcinoid tumors. AJR Am J Roentgenol. 2011;197(5):1073-1080.
5. Guckel C, Schnabel K, Deimling M, Steinbrich W. Solitary pulmonary nodules: MR evaluation of enhancement patterns with contrast-enhanced dynamic snapshot gradient-echo imaging. Radiology. 1996;200(3):681-686.
6. Jindal T, Kumar A, Venkitaraman B, et al. Evaluation of the role of [18F]FDG-PET/CT and [68Ga]DOTATOC-PET/CT in differentiating typical and atypical pulmonary carcinoids. Cancer Imaging. 2011;11:70-75.
7. Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620.
8. Travis WD. Pathology and diagnosis of neuroendocrine tumors: lung neuroendocrine. Thorac Surg Clin. 2014;24(3):257-266.
9. Warren WH, Memoli VA, Gould VE. Immunohistochemical and ultrastructural analysis of bronchopulmonary neuroendocrine neoplasms. II. Well-differentiated neuroendocrine carcinomas. Ultrastruct Pathol. 1984;7(2-3):185-199.
10. Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: Proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39-51.
11. McCaughan BC, Martini N, Bains MS. Bronchial carcinoids. Review of 124 cases. J Thorac Cardiovasc Surg. 1985;89(1):8-17.
12. Hurt R, Bates M. Carcinoid tumours of the bronchus: a 33-year experience. Thorax. 1984;39(8):617-623.
13. Ettinger DS, Wood DE, Akerley W, et al. Non-small cell lung cancer, version 1.2015. J Natl Compr Cancer Netw. 2014;12(12):1738-1761.
14. Ferguson MK, Landreneau RJ, Hazelrigg SR, et al. Long-term outcome after resection for bronchial carcinoid tumors. Eur J Cardiothorac Surg. 2000;18(2):156-161.
15. Lucchi M, Melfi F, Ribechini A, et al. Sleeve and wedge parenchyma-sparing bronchial resections in low-grade neoplasms of the bronchial airway. J Thorac Cardiovasc Surg. 2007;134(2):373-377.
16. Yendamuri S, Gold D, Jayaprakash V, Dexter E, Nwogu C, Demmy T. Is sublobar resection sufficient for carcinoid tumors? Ann Thorac Surg. 2011;92(5):1774-1778; discussion 8-9.
17. Fox M, Van Berkel V, Bousamra M II, Sloan S, Martin RC II. Surgical management of pulmonary carcinoid tumors: sublobar resection versus lobectomy. Am J Surg. 2013;205(2):200-208.
18. Cardillo G, Sera F, Di Martino M, et al. Bronchial carcinoid tumors: nodal status and long-term survival after resection. Ann Thorac Surg. 2004;77(5):1781-1785.
19. Oberg K, Hellman P, Ferolla P, Papotti M; ESMO Guidelines Working Group. Neuroendocrine bronchial and thymic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7:vii120-3).
20. Filosso PL, Ferolla P, Guerrera F, et al. Multidisciplinary management of advanced lung neuroendocrine tumors. J Thorac Dis. 2015;7(Suppl 2):S163-171.
21. Kulke MH, Shah MH, Benson AB III, et al. Neuroendocrine tumors, version 1.2015. J Natl Compr Cancer Netw. 2015;13(1):78-108.
22. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
23. Ramirez RA, Beyer DT, Chauhan A, Boudreaux JP, Wang YZ, Woltering EA. The role of capecitabine/temozolomide in metastatic neuroendocrine tumors. Oncologist. 2016;21(6):671-675.
24. Bajetta E, Rimassa L, Carnaghi C, et al. 5-Fluorouracil, dacarbazine, and epirubicin in the treatment of patients with neuroendocrine tumors. Cancer. 1998;83(2):372-378.
25. Masi G, Fornaro L, Cupini S, et al. Refractory neuroendocrine tumor-response to liposomal doxorubicin and capecitabine. Nat Rev Clin Oncol. 2009;6(11):670-674.
26. Lou F, Sarkaria I, Pietanza C, et al. Recurrence of pulmonary carcinoid tumors after resection: implications for postoperative surveillance. Ann Thorac Surg. 2013;96(4):1156-1162.
27. Beasley MB, Thunnissen FB, Brambilla E, et al. Pulmonary atypical carcinoid: predictors of survival in 106 cases. Human Pathol. 2000;31(10):1255-1265.
1. Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008;113(10):2655-2664.
2. Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119(6):1647-1651.
3. Limper AH, Carpenter PC, Scheithauer B, Staats BA. The Cushing syndrome induced by bronchial carcinoid tumors. Ann Intern Med. 1992;117(3):209-214.
4. Meisinger QC, Klein JS, Butnor KJ, Gentchos G, Leavitt BJ. CT features of peripheral pulmonary carcinoid tumors. AJR Am J Roentgenol. 2011;197(5):1073-1080.
5. Guckel C, Schnabel K, Deimling M, Steinbrich W. Solitary pulmonary nodules: MR evaluation of enhancement patterns with contrast-enhanced dynamic snapshot gradient-echo imaging. Radiology. 1996;200(3):681-686.
6. Jindal T, Kumar A, Venkitaraman B, et al. Evaluation of the role of [18F]FDG-PET/CT and [68Ga]DOTATOC-PET/CT in differentiating typical and atypical pulmonary carcinoids. Cancer Imaging. 2011;11:70-75.
7. Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620.
8. Travis WD. Pathology and diagnosis of neuroendocrine tumors: lung neuroendocrine. Thorac Surg Clin. 2014;24(3):257-266.
9. Warren WH, Memoli VA, Gould VE. Immunohistochemical and ultrastructural analysis of bronchopulmonary neuroendocrine neoplasms. II. Well-differentiated neuroendocrine carcinomas. Ultrastruct Pathol. 1984;7(2-3):185-199.
10. Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: Proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39-51.
11. McCaughan BC, Martini N, Bains MS. Bronchial carcinoids. Review of 124 cases. J Thorac Cardiovasc Surg. 1985;89(1):8-17.
12. Hurt R, Bates M. Carcinoid tumours of the bronchus: a 33-year experience. Thorax. 1984;39(8):617-623.
13. Ettinger DS, Wood DE, Akerley W, et al. Non-small cell lung cancer, version 1.2015. J Natl Compr Cancer Netw. 2014;12(12):1738-1761.
14. Ferguson MK, Landreneau RJ, Hazelrigg SR, et al. Long-term outcome after resection for bronchial carcinoid tumors. Eur J Cardiothorac Surg. 2000;18(2):156-161.
15. Lucchi M, Melfi F, Ribechini A, et al. Sleeve and wedge parenchyma-sparing bronchial resections in low-grade neoplasms of the bronchial airway. J Thorac Cardiovasc Surg. 2007;134(2):373-377.
16. Yendamuri S, Gold D, Jayaprakash V, Dexter E, Nwogu C, Demmy T. Is sublobar resection sufficient for carcinoid tumors? Ann Thorac Surg. 2011;92(5):1774-1778; discussion 8-9.
17. Fox M, Van Berkel V, Bousamra M II, Sloan S, Martin RC II. Surgical management of pulmonary carcinoid tumors: sublobar resection versus lobectomy. Am J Surg. 2013;205(2):200-208.
18. Cardillo G, Sera F, Di Martino M, et al. Bronchial carcinoid tumors: nodal status and long-term survival after resection. Ann Thorac Surg. 2004;77(5):1781-1785.
19. Oberg K, Hellman P, Ferolla P, Papotti M; ESMO Guidelines Working Group. Neuroendocrine bronchial and thymic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7:vii120-3).
20. Filosso PL, Ferolla P, Guerrera F, et al. Multidisciplinary management of advanced lung neuroendocrine tumors. J Thorac Dis. 2015;7(Suppl 2):S163-171.
21. Kulke MH, Shah MH, Benson AB III, et al. Neuroendocrine tumors, version 1.2015. J Natl Compr Cancer Netw. 2015;13(1):78-108.
22. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
23. Ramirez RA, Beyer DT, Chauhan A, Boudreaux JP, Wang YZ, Woltering EA. The role of capecitabine/temozolomide in metastatic neuroendocrine tumors. Oncologist. 2016;21(6):671-675.
24. Bajetta E, Rimassa L, Carnaghi C, et al. 5-Fluorouracil, dacarbazine, and epirubicin in the treatment of patients with neuroendocrine tumors. Cancer. 1998;83(2):372-378.
25. Masi G, Fornaro L, Cupini S, et al. Refractory neuroendocrine tumor-response to liposomal doxorubicin and capecitabine. Nat Rev Clin Oncol. 2009;6(11):670-674.
26. Lou F, Sarkaria I, Pietanza C, et al. Recurrence of pulmonary carcinoid tumors after resection: implications for postoperative surveillance. Ann Thorac Surg. 2013;96(4):1156-1162.
27. Beasley MB, Thunnissen FB, Brambilla E, et al. Pulmonary atypical carcinoid: predictors of survival in 106 cases. Human Pathol. 2000;31(10):1255-1265.
Gene variants associated with high-risk pediatric ALL

New research has revealed germline variations associated with high-risk acute lymphoblastic leukemia (ALL) in children.
Researchers sequenced the TP53 tumor suppressor gene in nearly 4000 children with ALL and identified 22 pathogenic germline variants.
These variants were associated with inferior survival and an increased risk of developing second malignancies.
Jun J. Yang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues reported these findings in the Journal of Clinical Oncology.
The researchers performed targeted sequencing of TP53 coding regions in 3801 children with ALL who were enrolled in 2 Children’s Oncology Group trials (AALL0232 and P9900).
The sequencing revealed 49 unique TP53 coding variants, which were found in 77 children. Twenty-two of the variants were pathogenic, and they were found in 26 children.
The researchers also analyzed data from 60,706 control subjects without ALL and found the 22 pathogenic variants were more likely to be found in the ALL patients than controls. The odds ratio was 5.2 (P<0.001).
Among the ALL patients, those who had the pathogenic variants were significantly older at diagnosis than those without the variants, with median ages of 15.5 years and 7.3 years, respectively (P<0.001).
The pathogenic variants were most common in patients with hypodiploid ALL. About 65% of patients who carried the pathogenic variants had hypodiploid ALL, as did 1.2% of children with wild-type genotype (P<0.001).
The pathogenic variants were associated with inferior event-free survival and overall survival as well. The hazard ratios were 4.2 (P<0.001) and 3.9 (P=0.001), respectively.
And the pathogenic variants were associated with a higher risk of second cancers. The 5-year cumulative incidence of second malignancies was 25.1% among patients with the pathogenic variants and 0.7% among patients without the variants (P<0.001).
“These germline variations are a double whammy for carriers,” Dr Yang said. “Not only is their risk of developing leukemia very high, they are also more likely to relapse or develop a second cancer.”
The association between the pathogenic variants and second cancers has prompted Dr Yang and his colleagues to explore ways to help patients manage their risk.
“Maybe these patients should avoid certain ALL therapies in order to reduce their risk of developing another cancer,” Dr Yang said. “I believe this finding may change treatment and follow-up for these high-risk patients.”
Dr Yang and his colleagues also noted that inherited variations in TP53 are a hallmark of Li-Fraumeni syndrome. And the syndrome might partially explain the high rate of second cancers in ALL patients with the pathogenic TP53 variants.
“The ALL treatment might have added to that risk,” Dr Yang said, “but we do not know for sure.” ![]()
New research has revealed germline variations associated with high-risk acute lymphoblastic leukemia (ALL) in children.
Researchers sequenced the TP53 tumor suppressor gene in nearly 4000 children with ALL and identified 22 pathogenic germline variants.
These variants were associated with inferior survival and an increased risk of developing second malignancies.
Jun J. Yang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues reported these findings in the Journal of Clinical Oncology.
The researchers performed targeted sequencing of TP53 coding regions in 3801 children with ALL who were enrolled in 2 Children’s Oncology Group trials (AALL0232 and P9900).
The sequencing revealed 49 unique TP53 coding variants, which were found in 77 children. Twenty-two of the variants were pathogenic, and they were found in 26 children.
The researchers also analyzed data from 60,706 control subjects without ALL and found the 22 pathogenic variants were more likely to be found in the ALL patients than controls. The odds ratio was 5.2 (P<0.001).
Among the ALL patients, those who had the pathogenic variants were significantly older at diagnosis than those without the variants, with median ages of 15.5 years and 7.3 years, respectively (P<0.001).
The pathogenic variants were most common in patients with hypodiploid ALL. About 65% of patients who carried the pathogenic variants had hypodiploid ALL, as did 1.2% of children with wild-type genotype (P<0.001).
The pathogenic variants were associated with inferior event-free survival and overall survival as well. The hazard ratios were 4.2 (P<0.001) and 3.9 (P=0.001), respectively.
And the pathogenic variants were associated with a higher risk of second cancers. The 5-year cumulative incidence of second malignancies was 25.1% among patients with the pathogenic variants and 0.7% among patients without the variants (P<0.001).
“These germline variations are a double whammy for carriers,” Dr Yang said. “Not only is their risk of developing leukemia very high, they are also more likely to relapse or develop a second cancer.”
The association between the pathogenic variants and second cancers has prompted Dr Yang and his colleagues to explore ways to help patients manage their risk.
“Maybe these patients should avoid certain ALL therapies in order to reduce their risk of developing another cancer,” Dr Yang said. “I believe this finding may change treatment and follow-up for these high-risk patients.”
Dr Yang and his colleagues also noted that inherited variations in TP53 are a hallmark of Li-Fraumeni syndrome. And the syndrome might partially explain the high rate of second cancers in ALL patients with the pathogenic TP53 variants.
“The ALL treatment might have added to that risk,” Dr Yang said, “but we do not know for sure.” ![]()
New research has revealed germline variations associated with high-risk acute lymphoblastic leukemia (ALL) in children.
Researchers sequenced the TP53 tumor suppressor gene in nearly 4000 children with ALL and identified 22 pathogenic germline variants.
These variants were associated with inferior survival and an increased risk of developing second malignancies.
Jun J. Yang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues reported these findings in the Journal of Clinical Oncology.
The researchers performed targeted sequencing of TP53 coding regions in 3801 children with ALL who were enrolled in 2 Children’s Oncology Group trials (AALL0232 and P9900).
The sequencing revealed 49 unique TP53 coding variants, which were found in 77 children. Twenty-two of the variants were pathogenic, and they were found in 26 children.
The researchers also analyzed data from 60,706 control subjects without ALL and found the 22 pathogenic variants were more likely to be found in the ALL patients than controls. The odds ratio was 5.2 (P<0.001).
Among the ALL patients, those who had the pathogenic variants were significantly older at diagnosis than those without the variants, with median ages of 15.5 years and 7.3 years, respectively (P<0.001).
The pathogenic variants were most common in patients with hypodiploid ALL. About 65% of patients who carried the pathogenic variants had hypodiploid ALL, as did 1.2% of children with wild-type genotype (P<0.001).
The pathogenic variants were associated with inferior event-free survival and overall survival as well. The hazard ratios were 4.2 (P<0.001) and 3.9 (P=0.001), respectively.
And the pathogenic variants were associated with a higher risk of second cancers. The 5-year cumulative incidence of second malignancies was 25.1% among patients with the pathogenic variants and 0.7% among patients without the variants (P<0.001).
“These germline variations are a double whammy for carriers,” Dr Yang said. “Not only is their risk of developing leukemia very high, they are also more likely to relapse or develop a second cancer.”
The association between the pathogenic variants and second cancers has prompted Dr Yang and his colleagues to explore ways to help patients manage their risk.
“Maybe these patients should avoid certain ALL therapies in order to reduce their risk of developing another cancer,” Dr Yang said. “I believe this finding may change treatment and follow-up for these high-risk patients.”
Dr Yang and his colleagues also noted that inherited variations in TP53 are a hallmark of Li-Fraumeni syndrome. And the syndrome might partially explain the high rate of second cancers in ALL patients with the pathogenic TP53 variants.
“The ALL treatment might have added to that risk,” Dr Yang said, “but we do not know for sure.” ![]()
Method prolongs lifespan of blood samples

A new blood stabilization method significantly prolongs the lifespan of blood samples for microfluidic sorting and transcriptome profiling of rare circulating tumor cells (CTCs), according to researchers.
The method involves reducing the storage temperature to 4° C to reversibly slow down cellular processes while also counteracting the platelet activation that can occur because of the low temperature.
The researchers said this work overcomes a significant barrier to the translation of liquid biopsy technologies for precision oncology and other applications.
Keith Wong, PhD, of the Massachusetts General Hospital Center for Engineering in Medicine (MGH-CEM), and his colleagues described this work in Nature Communications.
When isolating CTCs from fresh, unprocessed blood, timing is everything. Even minor changes in the quality of a blood sample—such as the breakdown of red cells, leukocyte activation, or clot formation— can greatly affect cell-sorting mechanisms and the quality of the biomolecules isolated for cancer detection.
According to published studies, factors such as the total number of CTCs in a sample and the number with high-quality RNA decrease by around 50% within the first 4 to 5 hours after the sample is collected.
“At Mass. General, we have the luxury of being so integrated with the clinical team that we can process blood specimens in the lab typically within an hour or 2 after they are drawn,” Dr Wong said.
“But to make these liquid biopsy technologies routine lab tests for the rest of the world, we need ways to keep blood alive for much longer than several hours, since these assays are best performed in central laboratories for reasons of cost-effectiveness and reproducibility.”
With this in mind, Dr Wong and his colleagues set out to preserve blood in its native state with minimal alterations.
“We wanted to slow down the biological clock as much as possible by using hypothermia, but that is not as simple as it sounds,” said study author Shannon Tessier, PhD, also of MGH-CEM.
“Low temperature is a powerful means to decrease metabolism, but a host of unwanted side effects occur at the same time. In some ways, these challenges are similar to those we face in organ preservation, where we have to optimize strategies for a very complex mix of cells.”
To achieve these goals, the researchers first analyzed the effects of hypothermic storage conditions.
The team found that hypothermic storage (4° C) of blood anticoagulated with acid citrate dextrose maintained “cellular morphology, integrity, and surface epitope stability of diverse hematologic cell types” over 72 hours.
However, the researchers also observed platelet activation.
“We are preserving the blood very well, including the coagulation function of platelets,” Dr Wong said. “But, unfortunately, cooling causes profound activation of platelets. Now, we need a targeted approach for platelets so they don’t form nasty clots in the microfluidic blood-sorting device.”
Fortunately, the researchers found that glycoprotein IIb/IIIa inhibitors were able to counter cooling-induced platelet aggregation. And ion chelation treatment with ethylenediaminetetraacetic acid removed activated platelets from leukocytes.
The team said these steps—hypothermic storage, treatment with glycoprotein IIb/IIIa inhibitors, and ion chelation—allowed whole blood preserved for 3 days to be processed as if it were freshly drawn, with very high purity and virtually no loss in CTC numbers.
“The critical achievement here is that the isolated tumor cells contain high-quality RNA that is suitable for demanding molecular assays, such as single-cell qPCR, droplet digital PCR, and RNA sequencing,” Dr Tessier said.
To test their blood preservation method, Dr Tessier and her colleagues used blood specimens from a group of 10 patients with metastatic prostate cancer.
The researchers compared CTC analysis in preserved blood samples and paired fresh samples from the same patients.
There was 92% agreement in the detection of 12 cancer-specific gene transcripts between the fresh and preserved blood samples.
In addition, there was 100% agreement in the detection of a transcript called AR-V7. Recently published studies showed that the presence of AR-V7 mRNA in prostate cancer CTCs predicts resistance to androgen receptor inhibitors, indicating that chemotherapy may be a better option for such patients.
“The ability to preserve the blood for several days and still be able to pick up this clinically relevant biomarker is remarkable,” said study author David Miyamoto, MD, PhD, of MGH Cancer Center.
“This is very exciting for clinicians because AR-V7 mRNA can only be detected using CTCs and not with circulating tumor DNA or other cell-free assays.”
The researchers highlighted the universal nature of their blood preservation approach by pointing to its compatibility with the microfluidic CTC-iChip device, which isolates tumor cells by rapid removal of blood cells. The team said this suggests the potential impact of this work extends beyond cancer detection.
“With exciting breakthroughs in immunotherapy, stem cell transplantation, and regenerative medicine—in which peripheral blood is often the source of cells for functional assays or ex vivo expansion—the ability to preserve live cells will greatly ease logistical timelines and reduce the cost of complex cell-based assays,” Dr Wong said. ![]()
A new blood stabilization method significantly prolongs the lifespan of blood samples for microfluidic sorting and transcriptome profiling of rare circulating tumor cells (CTCs), according to researchers.
The method involves reducing the storage temperature to 4° C to reversibly slow down cellular processes while also counteracting the platelet activation that can occur because of the low temperature.
The researchers said this work overcomes a significant barrier to the translation of liquid biopsy technologies for precision oncology and other applications.
Keith Wong, PhD, of the Massachusetts General Hospital Center for Engineering in Medicine (MGH-CEM), and his colleagues described this work in Nature Communications.
When isolating CTCs from fresh, unprocessed blood, timing is everything. Even minor changes in the quality of a blood sample—such as the breakdown of red cells, leukocyte activation, or clot formation— can greatly affect cell-sorting mechanisms and the quality of the biomolecules isolated for cancer detection.
According to published studies, factors such as the total number of CTCs in a sample and the number with high-quality RNA decrease by around 50% within the first 4 to 5 hours after the sample is collected.
“At Mass. General, we have the luxury of being so integrated with the clinical team that we can process blood specimens in the lab typically within an hour or 2 after they are drawn,” Dr Wong said.
“But to make these liquid biopsy technologies routine lab tests for the rest of the world, we need ways to keep blood alive for much longer than several hours, since these assays are best performed in central laboratories for reasons of cost-effectiveness and reproducibility.”
With this in mind, Dr Wong and his colleagues set out to preserve blood in its native state with minimal alterations.
“We wanted to slow down the biological clock as much as possible by using hypothermia, but that is not as simple as it sounds,” said study author Shannon Tessier, PhD, also of MGH-CEM.
“Low temperature is a powerful means to decrease metabolism, but a host of unwanted side effects occur at the same time. In some ways, these challenges are similar to those we face in organ preservation, where we have to optimize strategies for a very complex mix of cells.”
To achieve these goals, the researchers first analyzed the effects of hypothermic storage conditions.
The team found that hypothermic storage (4° C) of blood anticoagulated with acid citrate dextrose maintained “cellular morphology, integrity, and surface epitope stability of diverse hematologic cell types” over 72 hours.
However, the researchers also observed platelet activation.
“We are preserving the blood very well, including the coagulation function of platelets,” Dr Wong said. “But, unfortunately, cooling causes profound activation of platelets. Now, we need a targeted approach for platelets so they don’t form nasty clots in the microfluidic blood-sorting device.”
Fortunately, the researchers found that glycoprotein IIb/IIIa inhibitors were able to counter cooling-induced platelet aggregation. And ion chelation treatment with ethylenediaminetetraacetic acid removed activated platelets from leukocytes.
The team said these steps—hypothermic storage, treatment with glycoprotein IIb/IIIa inhibitors, and ion chelation—allowed whole blood preserved for 3 days to be processed as if it were freshly drawn, with very high purity and virtually no loss in CTC numbers.
“The critical achievement here is that the isolated tumor cells contain high-quality RNA that is suitable for demanding molecular assays, such as single-cell qPCR, droplet digital PCR, and RNA sequencing,” Dr Tessier said.
To test their blood preservation method, Dr Tessier and her colleagues used blood specimens from a group of 10 patients with metastatic prostate cancer.
The researchers compared CTC analysis in preserved blood samples and paired fresh samples from the same patients.
There was 92% agreement in the detection of 12 cancer-specific gene transcripts between the fresh and preserved blood samples.
In addition, there was 100% agreement in the detection of a transcript called AR-V7. Recently published studies showed that the presence of AR-V7 mRNA in prostate cancer CTCs predicts resistance to androgen receptor inhibitors, indicating that chemotherapy may be a better option for such patients.
“The ability to preserve the blood for several days and still be able to pick up this clinically relevant biomarker is remarkable,” said study author David Miyamoto, MD, PhD, of MGH Cancer Center.
“This is very exciting for clinicians because AR-V7 mRNA can only be detected using CTCs and not with circulating tumor DNA or other cell-free assays.”
The researchers highlighted the universal nature of their blood preservation approach by pointing to its compatibility with the microfluidic CTC-iChip device, which isolates tumor cells by rapid removal of blood cells. The team said this suggests the potential impact of this work extends beyond cancer detection.
“With exciting breakthroughs in immunotherapy, stem cell transplantation, and regenerative medicine—in which peripheral blood is often the source of cells for functional assays or ex vivo expansion—the ability to preserve live cells will greatly ease logistical timelines and reduce the cost of complex cell-based assays,” Dr Wong said. ![]()
A new blood stabilization method significantly prolongs the lifespan of blood samples for microfluidic sorting and transcriptome profiling of rare circulating tumor cells (CTCs), according to researchers.
The method involves reducing the storage temperature to 4° C to reversibly slow down cellular processes while also counteracting the platelet activation that can occur because of the low temperature.
The researchers said this work overcomes a significant barrier to the translation of liquid biopsy technologies for precision oncology and other applications.
Keith Wong, PhD, of the Massachusetts General Hospital Center for Engineering in Medicine (MGH-CEM), and his colleagues described this work in Nature Communications.
When isolating CTCs from fresh, unprocessed blood, timing is everything. Even minor changes in the quality of a blood sample—such as the breakdown of red cells, leukocyte activation, or clot formation— can greatly affect cell-sorting mechanisms and the quality of the biomolecules isolated for cancer detection.
According to published studies, factors such as the total number of CTCs in a sample and the number with high-quality RNA decrease by around 50% within the first 4 to 5 hours after the sample is collected.
“At Mass. General, we have the luxury of being so integrated with the clinical team that we can process blood specimens in the lab typically within an hour or 2 after they are drawn,” Dr Wong said.
“But to make these liquid biopsy technologies routine lab tests for the rest of the world, we need ways to keep blood alive for much longer than several hours, since these assays are best performed in central laboratories for reasons of cost-effectiveness and reproducibility.”
With this in mind, Dr Wong and his colleagues set out to preserve blood in its native state with minimal alterations.
“We wanted to slow down the biological clock as much as possible by using hypothermia, but that is not as simple as it sounds,” said study author Shannon Tessier, PhD, also of MGH-CEM.
“Low temperature is a powerful means to decrease metabolism, but a host of unwanted side effects occur at the same time. In some ways, these challenges are similar to those we face in organ preservation, where we have to optimize strategies for a very complex mix of cells.”
To achieve these goals, the researchers first analyzed the effects of hypothermic storage conditions.
The team found that hypothermic storage (4° C) of blood anticoagulated with acid citrate dextrose maintained “cellular morphology, integrity, and surface epitope stability of diverse hematologic cell types” over 72 hours.
However, the researchers also observed platelet activation.
“We are preserving the blood very well, including the coagulation function of platelets,” Dr Wong said. “But, unfortunately, cooling causes profound activation of platelets. Now, we need a targeted approach for platelets so they don’t form nasty clots in the microfluidic blood-sorting device.”
Fortunately, the researchers found that glycoprotein IIb/IIIa inhibitors were able to counter cooling-induced platelet aggregation. And ion chelation treatment with ethylenediaminetetraacetic acid removed activated platelets from leukocytes.
The team said these steps—hypothermic storage, treatment with glycoprotein IIb/IIIa inhibitors, and ion chelation—allowed whole blood preserved for 3 days to be processed as if it were freshly drawn, with very high purity and virtually no loss in CTC numbers.
“The critical achievement here is that the isolated tumor cells contain high-quality RNA that is suitable for demanding molecular assays, such as single-cell qPCR, droplet digital PCR, and RNA sequencing,” Dr Tessier said.
To test their blood preservation method, Dr Tessier and her colleagues used blood specimens from a group of 10 patients with metastatic prostate cancer.
The researchers compared CTC analysis in preserved blood samples and paired fresh samples from the same patients.
There was 92% agreement in the detection of 12 cancer-specific gene transcripts between the fresh and preserved blood samples.
In addition, there was 100% agreement in the detection of a transcript called AR-V7. Recently published studies showed that the presence of AR-V7 mRNA in prostate cancer CTCs predicts resistance to androgen receptor inhibitors, indicating that chemotherapy may be a better option for such patients.
“The ability to preserve the blood for several days and still be able to pick up this clinically relevant biomarker is remarkable,” said study author David Miyamoto, MD, PhD, of MGH Cancer Center.
“This is very exciting for clinicians because AR-V7 mRNA can only be detected using CTCs and not with circulating tumor DNA or other cell-free assays.”
The researchers highlighted the universal nature of their blood preservation approach by pointing to its compatibility with the microfluidic CTC-iChip device, which isolates tumor cells by rapid removal of blood cells. The team said this suggests the potential impact of this work extends beyond cancer detection.
“With exciting breakthroughs in immunotherapy, stem cell transplantation, and regenerative medicine—in which peripheral blood is often the source of cells for functional assays or ex vivo expansion—the ability to preserve live cells will greatly ease logistical timelines and reduce the cost of complex cell-based assays,” Dr Wong said. ![]()
Drug receives breakthrough designation for SCD

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to voxelotor (previously GBT440) for the treatment of sickle cell disease (SCD).
Voxelotor is being developed by Global Blood Therapeutics, Inc., as a potentially disease-modifying therapy for SCD.
The drug works by increasing hemoglobin’s affinity for oxygen. Since oxygenated sickle hemoglobin does not polymerize, it is believed that voxelotor blocks polymerization and the resultant sickling of red blood cells.
If voxelotor can restore normal hemoglobin function and improve oxygen delivery, the therapy may be capable of modifying the progression of SCD.
The FDA previously granted voxelotor fast track designation, orphan drug designation, and rare pediatric disease designation.
The FDA’s decision to grant breakthrough therapy designation to voxelotor was based on clinical data submitted from the following studies:
- Part A of the phase 3 HOPE study (GBT440-031)
- A phase 1/2 study and open-label extension in adults (GBT440-001/GBT440-024), which was presented at the 2016 ASH Annual Meeting
- The ongoing phase 2 HOPE-KIDS 1 study in children age 6 to 17 (GBT440-007), which was presented at the 2017 ASH Annual Meeting (abstract 689)
- The compassionate access experience in adults with severe SCD who were not eligible for the HOPE study, which was presented at the 2017 ASH Annual Meeting (abstract 3545).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to voxelotor (previously GBT440) for the treatment of sickle cell disease (SCD).
Voxelotor is being developed by Global Blood Therapeutics, Inc., as a potentially disease-modifying therapy for SCD.
The drug works by increasing hemoglobin’s affinity for oxygen. Since oxygenated sickle hemoglobin does not polymerize, it is believed that voxelotor blocks polymerization and the resultant sickling of red blood cells.
If voxelotor can restore normal hemoglobin function and improve oxygen delivery, the therapy may be capable of modifying the progression of SCD.
The FDA previously granted voxelotor fast track designation, orphan drug designation, and rare pediatric disease designation.
The FDA’s decision to grant breakthrough therapy designation to voxelotor was based on clinical data submitted from the following studies:
- Part A of the phase 3 HOPE study (GBT440-031)
- A phase 1/2 study and open-label extension in adults (GBT440-001/GBT440-024), which was presented at the 2016 ASH Annual Meeting
- The ongoing phase 2 HOPE-KIDS 1 study in children age 6 to 17 (GBT440-007), which was presented at the 2017 ASH Annual Meeting (abstract 689)
- The compassionate access experience in adults with severe SCD who were not eligible for the HOPE study, which was presented at the 2017 ASH Annual Meeting (abstract 3545).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to voxelotor (previously GBT440) for the treatment of sickle cell disease (SCD).
Voxelotor is being developed by Global Blood Therapeutics, Inc., as a potentially disease-modifying therapy for SCD.
The drug works by increasing hemoglobin’s affinity for oxygen. Since oxygenated sickle hemoglobin does not polymerize, it is believed that voxelotor blocks polymerization and the resultant sickling of red blood cells.
If voxelotor can restore normal hemoglobin function and improve oxygen delivery, the therapy may be capable of modifying the progression of SCD.
The FDA previously granted voxelotor fast track designation, orphan drug designation, and rare pediatric disease designation.
The FDA’s decision to grant breakthrough therapy designation to voxelotor was based on clinical data submitted from the following studies:
- Part A of the phase 3 HOPE study (GBT440-031)
- A phase 1/2 study and open-label extension in adults (GBT440-001/GBT440-024), which was presented at the 2016 ASH Annual Meeting
- The ongoing phase 2 HOPE-KIDS 1 study in children age 6 to 17 (GBT440-007), which was presented at the 2017 ASH Annual Meeting (abstract 689)
- The compassionate access experience in adults with severe SCD who were not eligible for the HOPE study, which was presented at the 2017 ASH Annual Meeting (abstract 3545).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
For Patients With CKD, Don’t Wait—Vaccinate!
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
Huge AWARE study shows chronic spontaneous urticaria is seriously undertreated
GENEVA – Partial results of the first worldwide observational study on the burden of chronic spontaneous urticaria (CSU) and its treatment in real-world clinical practice can be summarized succinctly: “It’s worse than we thought,” Marcus Maurer, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“We knew it was bad, but what comes out of looking at the daily life and treatment patterns in patients with CSU is a nightmare,” said Dr. Maurer, professor of dermatology and allergy and director of research in the department of dermatology at Charite University Hospital in Berlin.

Dr. Maurer presented the year-1 results in 1,550 AWARE participants at 256 sites across Germany. The results, he said, are sobering and reflective of what is being found in the other participating countries. The findings document a low level of adherence to the published treatment guidelines, and a lot of miserable CSU patients.
“My talk here is a clear call to action,” he declared. “The AWARE study shows we have two problems. One, patients don’t get treatment – and we can solve that. And number two, patients don’t get the right treatment. And the right treatment is treatment that controls the disease. In the guidelines we say, ‘Treat the disease until it is gone.’ That’s a great sentence. It means we should give enough treatment to control the disease: as much as needed, as little as possible, and in that order. First we establish control, then we have all the time in the world to find a treatment that gets patients to stay that way in the long run.”
He is a coauthor of the guidelines, a joint initiative of the European Academy of Allergy and Clinical Immunology, the EU-funded Global Allergy and Asthma European Network of centers of excellence, the European Dermatology Forum, and the World Allergy Organization (Allergy. 2014 Jul;69[7]:868-87).
He asked a packed hall of dermatologists how many of them like taking care of patients with CSU. One lonely hand went up. Dr. Maurer was unsurprised.
“, and it’s one where we struggle. No question, we struggle with these patients, maybe more so than with our psoriasis and atopic dermatitis patients. But the good news is there are new concepts of this disease and new treatment approaches,” he said. “We know that in 98% of patients, using the guideline algorithm, we can establish control in CSU.”
He summarized the joint guideline algorithm as follows: Routinely use a simple validated assessment tool such as the patient-reported Urticaria Control Test (UCT) (J Allergy Clin Immunol. 2014 May;133[5]:1365-72) to determine a CSU patient’s burden of disease, prescribe second-generation, nonsedating H1 antihistamines as first-line therapy, updose those agents to up to four times standard dosing as second-line therapy if the follow-up UCT results show the disease is still uncontrolled, and if it remains uncontrolled then move on to third-line treatment with add-on omalizumab (Xolair) or a maximum of 10 days of montelukast (Singulair) or cyclosporine.
When he asked how many audience members use the UCT, which assesses disease control over the previous 4 weeks, a mere handful responded affirmatively. “Please,” he urged his colleagues, “this test is so simple. It’s four questions, it’s free, it’s available online in more than 30 languages. It allows us to identify patients with uncontrolled disease. We have the ability to control the disease now, so let use this test. Patients can fill it out quickly in your waiting room so you can monitor how good your treatment is.”
AWARE study bears bad news
At study enrollment, the 1,550 German participants in the AWARE study had been diagnosed with CSU a mean of 4.9 years previously. Yet fully 78% had uncontrolled CSU as defined by a score below 12 points on the 0-16 UCT. Forty-four percent of patients had experienced angioedema within the previous 6 months, and 56% percent of patients had a baseline Dermatology Life Quality Index (DLQI) score indicating CSU had a moderate, very large, or extremely large effect on their quality of life. Yet 36% of patients weren’t receiving any treatment at baseline, and only 6.5% of patients were receiving guideline-recommended add-on third-line therapy.
In particular, most physicians fail to appreciate the negative impact of angioedema in CSU patients.
“This is not a swollen lip just once a year, this is angioedema weekly or monthly that really impairs quality of life. It’s not just a swollen lip or a swollen eye for the day or for a couple of hours, it’s the fear of going to sleep because in the morning you may wake up looking like that and not be able to go to work,” Dr. Maurer pointed out.
During the first year of the study, the overall situation improved significantly over the course of office visits every 3 months. The prevalence of DLQI scores indicative of a moderate or worse quality of life impact gradually fell from 56% to 28%. The proportion of patients on third-line treatment rose stepwise to 29.5% at 1 year. The rate of uncontrolled CSU as defined by a UCT score below 12 dropped steadily over time to 42%. At 1 year, only 16% of patients reported having an angioedema episode within the prior 12 weeks.
Still, this leaves much room for further improvement, Dr. Maurer commented.
“We’re on the right track, but patients who are in our treatment for a year should have their disease controlled. That’s our job. We have the treatments, and we should provide them. Even after 1 year there are still patients with a DLQI score above 10, and that’s not good,” he said.
Dr. Maurer was first author of the recently published interim results of the German portion of the AWARE study (Clin Exp Allergy. 2017 May;47[5]:684-52). The AWARE study is sponsored by Novartis, manufacturer of omalizumab, which is approved by the Food and Drug Administration for treating CSU. Dr. Maurer reported receiving research grants from and serving as an advisor to and paid speaker for Novartis and numerous other pharmaceutical companies.
[email protected]
GENEVA – Partial results of the first worldwide observational study on the burden of chronic spontaneous urticaria (CSU) and its treatment in real-world clinical practice can be summarized succinctly: “It’s worse than we thought,” Marcus Maurer, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“We knew it was bad, but what comes out of looking at the daily life and treatment patterns in patients with CSU is a nightmare,” said Dr. Maurer, professor of dermatology and allergy and director of research in the department of dermatology at Charite University Hospital in Berlin.
Dr. Maurer presented the year-1 results in 1,550 AWARE participants at 256 sites across Germany. The results, he said, are sobering and reflective of what is being found in the other participating countries. The findings document a low level of adherence to the published treatment guidelines, and a lot of miserable CSU patients.
“My talk here is a clear call to action,” he declared. “The AWARE study shows we have two problems. One, patients don’t get treatment – and we can solve that. And number two, patients don’t get the right treatment. And the right treatment is treatment that controls the disease. In the guidelines we say, ‘Treat the disease until it is gone.’ That’s a great sentence. It means we should give enough treatment to control the disease: as much as needed, as little as possible, and in that order. First we establish control, then we have all the time in the world to find a treatment that gets patients to stay that way in the long run.”
He is a coauthor of the guidelines, a joint initiative of the European Academy of Allergy and Clinical Immunology, the EU-funded Global Allergy and Asthma European Network of centers of excellence, the European Dermatology Forum, and the World Allergy Organization (Allergy. 2014 Jul;69[7]:868-87).
He asked a packed hall of dermatologists how many of them like taking care of patients with CSU. One lonely hand went up. Dr. Maurer was unsurprised.
“, and it’s one where we struggle. No question, we struggle with these patients, maybe more so than with our psoriasis and atopic dermatitis patients. But the good news is there are new concepts of this disease and new treatment approaches,” he said. “We know that in 98% of patients, using the guideline algorithm, we can establish control in CSU.”
He summarized the joint guideline algorithm as follows: Routinely use a simple validated assessment tool such as the patient-reported Urticaria Control Test (UCT) (J Allergy Clin Immunol. 2014 May;133[5]:1365-72) to determine a CSU patient’s burden of disease, prescribe second-generation, nonsedating H1 antihistamines as first-line therapy, updose those agents to up to four times standard dosing as second-line therapy if the follow-up UCT results show the disease is still uncontrolled, and if it remains uncontrolled then move on to third-line treatment with add-on omalizumab (Xolair) or a maximum of 10 days of montelukast (Singulair) or cyclosporine.
When he asked how many audience members use the UCT, which assesses disease control over the previous 4 weeks, a mere handful responded affirmatively. “Please,” he urged his colleagues, “this test is so simple. It’s four questions, it’s free, it’s available online in more than 30 languages. It allows us to identify patients with uncontrolled disease. We have the ability to control the disease now, so let use this test. Patients can fill it out quickly in your waiting room so you can monitor how good your treatment is.”
AWARE study bears bad news
At study enrollment, the 1,550 German participants in the AWARE study had been diagnosed with CSU a mean of 4.9 years previously. Yet fully 78% had uncontrolled CSU as defined by a score below 12 points on the 0-16 UCT. Forty-four percent of patients had experienced angioedema within the previous 6 months, and 56% percent of patients had a baseline Dermatology Life Quality Index (DLQI) score indicating CSU had a moderate, very large, or extremely large effect on their quality of life. Yet 36% of patients weren’t receiving any treatment at baseline, and only 6.5% of patients were receiving guideline-recommended add-on third-line therapy.
In particular, most physicians fail to appreciate the negative impact of angioedema in CSU patients.
“This is not a swollen lip just once a year, this is angioedema weekly or monthly that really impairs quality of life. It’s not just a swollen lip or a swollen eye for the day or for a couple of hours, it’s the fear of going to sleep because in the morning you may wake up looking like that and not be able to go to work,” Dr. Maurer pointed out.
During the first year of the study, the overall situation improved significantly over the course of office visits every 3 months. The prevalence of DLQI scores indicative of a moderate or worse quality of life impact gradually fell from 56% to 28%. The proportion of patients on third-line treatment rose stepwise to 29.5% at 1 year. The rate of uncontrolled CSU as defined by a UCT score below 12 dropped steadily over time to 42%. At 1 year, only 16% of patients reported having an angioedema episode within the prior 12 weeks.
Still, this leaves much room for further improvement, Dr. Maurer commented.
“We’re on the right track, but patients who are in our treatment for a year should have their disease controlled. That’s our job. We have the treatments, and we should provide them. Even after 1 year there are still patients with a DLQI score above 10, and that’s not good,” he said.
Dr. Maurer was first author of the recently published interim results of the German portion of the AWARE study (Clin Exp Allergy. 2017 May;47[5]:684-52). The AWARE study is sponsored by Novartis, manufacturer of omalizumab, which is approved by the Food and Drug Administration for treating CSU. Dr. Maurer reported receiving research grants from and serving as an advisor to and paid speaker for Novartis and numerous other pharmaceutical companies.
[email protected]
GENEVA – Partial results of the first worldwide observational study on the burden of chronic spontaneous urticaria (CSU) and its treatment in real-world clinical practice can be summarized succinctly: “It’s worse than we thought,” Marcus Maurer, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“We knew it was bad, but what comes out of looking at the daily life and treatment patterns in patients with CSU is a nightmare,” said Dr. Maurer, professor of dermatology and allergy and director of research in the department of dermatology at Charite University Hospital in Berlin.
Dr. Maurer presented the year-1 results in 1,550 AWARE participants at 256 sites across Germany. The results, he said, are sobering and reflective of what is being found in the other participating countries. The findings document a low level of adherence to the published treatment guidelines, and a lot of miserable CSU patients.
“My talk here is a clear call to action,” he declared. “The AWARE study shows we have two problems. One, patients don’t get treatment – and we can solve that. And number two, patients don’t get the right treatment. And the right treatment is treatment that controls the disease. In the guidelines we say, ‘Treat the disease until it is gone.’ That’s a great sentence. It means we should give enough treatment to control the disease: as much as needed, as little as possible, and in that order. First we establish control, then we have all the time in the world to find a treatment that gets patients to stay that way in the long run.”
He is a coauthor of the guidelines, a joint initiative of the European Academy of Allergy and Clinical Immunology, the EU-funded Global Allergy and Asthma European Network of centers of excellence, the European Dermatology Forum, and the World Allergy Organization (Allergy. 2014 Jul;69[7]:868-87).
He asked a packed hall of dermatologists how many of them like taking care of patients with CSU. One lonely hand went up. Dr. Maurer was unsurprised.
“, and it’s one where we struggle. No question, we struggle with these patients, maybe more so than with our psoriasis and atopic dermatitis patients. But the good news is there are new concepts of this disease and new treatment approaches,” he said. “We know that in 98% of patients, using the guideline algorithm, we can establish control in CSU.”
He summarized the joint guideline algorithm as follows: Routinely use a simple validated assessment tool such as the patient-reported Urticaria Control Test (UCT) (J Allergy Clin Immunol. 2014 May;133[5]:1365-72) to determine a CSU patient’s burden of disease, prescribe second-generation, nonsedating H1 antihistamines as first-line therapy, updose those agents to up to four times standard dosing as second-line therapy if the follow-up UCT results show the disease is still uncontrolled, and if it remains uncontrolled then move on to third-line treatment with add-on omalizumab (Xolair) or a maximum of 10 days of montelukast (Singulair) or cyclosporine.
When he asked how many audience members use the UCT, which assesses disease control over the previous 4 weeks, a mere handful responded affirmatively. “Please,” he urged his colleagues, “this test is so simple. It’s four questions, it’s free, it’s available online in more than 30 languages. It allows us to identify patients with uncontrolled disease. We have the ability to control the disease now, so let use this test. Patients can fill it out quickly in your waiting room so you can monitor how good your treatment is.”
AWARE study bears bad news
At study enrollment, the 1,550 German participants in the AWARE study had been diagnosed with CSU a mean of 4.9 years previously. Yet fully 78% had uncontrolled CSU as defined by a score below 12 points on the 0-16 UCT. Forty-four percent of patients had experienced angioedema within the previous 6 months, and 56% percent of patients had a baseline Dermatology Life Quality Index (DLQI) score indicating CSU had a moderate, very large, or extremely large effect on their quality of life. Yet 36% of patients weren’t receiving any treatment at baseline, and only 6.5% of patients were receiving guideline-recommended add-on third-line therapy.
In particular, most physicians fail to appreciate the negative impact of angioedema in CSU patients.
“This is not a swollen lip just once a year, this is angioedema weekly or monthly that really impairs quality of life. It’s not just a swollen lip or a swollen eye for the day or for a couple of hours, it’s the fear of going to sleep because in the morning you may wake up looking like that and not be able to go to work,” Dr. Maurer pointed out.
During the first year of the study, the overall situation improved significantly over the course of office visits every 3 months. The prevalence of DLQI scores indicative of a moderate or worse quality of life impact gradually fell from 56% to 28%. The proportion of patients on third-line treatment rose stepwise to 29.5% at 1 year. The rate of uncontrolled CSU as defined by a UCT score below 12 dropped steadily over time to 42%. At 1 year, only 16% of patients reported having an angioedema episode within the prior 12 weeks.
Still, this leaves much room for further improvement, Dr. Maurer commented.
“We’re on the right track, but patients who are in our treatment for a year should have their disease controlled. That’s our job. We have the treatments, and we should provide them. Even after 1 year there are still patients with a DLQI score above 10, and that’s not good,” he said.
Dr. Maurer was first author of the recently published interim results of the German portion of the AWARE study (Clin Exp Allergy. 2017 May;47[5]:684-52). The AWARE study is sponsored by Novartis, manufacturer of omalizumab, which is approved by the Food and Drug Administration for treating CSU. Dr. Maurer reported receiving research grants from and serving as an advisor to and paid speaker for Novartis and numerous other pharmaceutical companies.
[email protected]
EXPERT ANALYSIS FROM THE EADV CONGRESS
HHS nominee Azar open to price negotiations for Part B drugs
Negotiating Medicare Part B drug prices could be on the agenda if Alex Azar, President Trump’s nominee to run the Health and Human Services department, gets confirmed.
before the Senate Finance Committee on Jan. 9. “In Part D, we do significant negotiation through pharmacy benefit managers that get the best rates of any commercial payers. We don’t do that in Part B. ... We basically pay sales price plus 6% or some other number. ... In Part B, we should be looking at those approaches” to price negotiations.
Mr. Azar added that in specific cases, such as when the government buys naloxone for use by first responders, “there is absolutely nothing wrong with the government negotiating that.”
The tone at Mr. Azar’s confirmation hearing generally was more conversational and less adversarial than that of his predecessor, Tom Price, MD, although contentious exchanges did occur, particularly around drug pricing.
“Across the board, drug prices are too high,” said Mr. Azar, who previously was president of Eli Lilly’s U.S. operations. “This is what is so bizarre about the way this system is organized that those price increases happen and ... during that same period, the net realized price by the company stayed flat. So just to cover for increased rebates, the patient who walks into the pharmacy whose insurance may not be paying for that is absorbing that cost and that is what I want to work with you to try and solve.”
Echoing comments made at a courtesy hearing before the Senate Health, Education, Labor and Pensions Committee on Nov. 29, Mr. Azar suggested the need for patent reform to push generic competition as a price-lowering tool.
The cordial tone to the hearing does not guarantee bipartisan support for Mr. Azar’s confirmation.
Sen. Maria Cantwell (D-Wash.) made it explicitly clear that her vote would be based on Mr. Azar’s thoughts about Medicaid expansion and on Mr. Azar’s suggested use of block grants “in the interest of sustainability.”
Regarding Medicaid, Mr. Azar sidestepped questions related to requiring “able-bodied” recipients to work.
“I don’t have a definition [of able-bodied] in hand,” Mr. Azar responded to Sen. Sherrod Brown (D-Ohio). “It would be something we’d work on with Congress.”
“You have no definition of ‘able-bodied adult’ that would be appropriate for differentiating between and among Medicaid recipients that you can share with us?” Sen. Brown replied, to which Mr. Azar had no specific answer.
Mr. Azar testified that said he was “a big supporter of telehealth and alternative means of providing care, especially in rural communities. I think sometimes we can be penny-wise [and] pound-foolish in these areas.”
Mr. Azar also voiced support to wellness incentive programs.
In the area of insurance access and premiums, Mr. Azar said that, as secretary, “I [would] have a very important obligation to make whatever program that I am entrusted with work as well as possible. What we have now is not working for people. It is not working for the 10 million who are in that individual market right now. For many of those people, it can be a false insurance card. It can be insurance, but a very high deductible or not having access to providers, so it’s unaffordable use of care. I want to solve the program for them,” as well as for the 28 million who are not in the individual market, to make it more affordable for all.
Regarding the contentious Center for Medicare and Medicaid Innovation, Mr. Azar voiced support of its use of mandatory participation demonstration projects and said that, depending on the circumstance, sometimes the use of mandatory participation is necessary to test out ideas, and if it is, he said, “so be it.”
Mr. Azar said he was interested in learning more about ensuring access to long-acting reversible contraception. He declined to answer questions on this because of his lack of knowledge but said he wanted more information.
A committee vote on Mr. Azar’s nomination had not been scheduled at press time. Should the committee support him, the full Senate would then consider his nomination.
Negotiating Medicare Part B drug prices could be on the agenda if Alex Azar, President Trump’s nominee to run the Health and Human Services department, gets confirmed.
before the Senate Finance Committee on Jan. 9. “In Part D, we do significant negotiation through pharmacy benefit managers that get the best rates of any commercial payers. We don’t do that in Part B. ... We basically pay sales price plus 6% or some other number. ... In Part B, we should be looking at those approaches” to price negotiations.
Mr. Azar added that in specific cases, such as when the government buys naloxone for use by first responders, “there is absolutely nothing wrong with the government negotiating that.”
The tone at Mr. Azar’s confirmation hearing generally was more conversational and less adversarial than that of his predecessor, Tom Price, MD, although contentious exchanges did occur, particularly around drug pricing.
“Across the board, drug prices are too high,” said Mr. Azar, who previously was president of Eli Lilly’s U.S. operations. “This is what is so bizarre about the way this system is organized that those price increases happen and ... during that same period, the net realized price by the company stayed flat. So just to cover for increased rebates, the patient who walks into the pharmacy whose insurance may not be paying for that is absorbing that cost and that is what I want to work with you to try and solve.”
Echoing comments made at a courtesy hearing before the Senate Health, Education, Labor and Pensions Committee on Nov. 29, Mr. Azar suggested the need for patent reform to push generic competition as a price-lowering tool.
The cordial tone to the hearing does not guarantee bipartisan support for Mr. Azar’s confirmation.
Sen. Maria Cantwell (D-Wash.) made it explicitly clear that her vote would be based on Mr. Azar’s thoughts about Medicaid expansion and on Mr. Azar’s suggested use of block grants “in the interest of sustainability.”
Regarding Medicaid, Mr. Azar sidestepped questions related to requiring “able-bodied” recipients to work.
“I don’t have a definition [of able-bodied] in hand,” Mr. Azar responded to Sen. Sherrod Brown (D-Ohio). “It would be something we’d work on with Congress.”
“You have no definition of ‘able-bodied adult’ that would be appropriate for differentiating between and among Medicaid recipients that you can share with us?” Sen. Brown replied, to which Mr. Azar had no specific answer.
Mr. Azar testified that said he was “a big supporter of telehealth and alternative means of providing care, especially in rural communities. I think sometimes we can be penny-wise [and] pound-foolish in these areas.”
Mr. Azar also voiced support to wellness incentive programs.
In the area of insurance access and premiums, Mr. Azar said that, as secretary, “I [would] have a very important obligation to make whatever program that I am entrusted with work as well as possible. What we have now is not working for people. It is not working for the 10 million who are in that individual market right now. For many of those people, it can be a false insurance card. It can be insurance, but a very high deductible or not having access to providers, so it’s unaffordable use of care. I want to solve the program for them,” as well as for the 28 million who are not in the individual market, to make it more affordable for all.
Regarding the contentious Center for Medicare and Medicaid Innovation, Mr. Azar voiced support of its use of mandatory participation demonstration projects and said that, depending on the circumstance, sometimes the use of mandatory participation is necessary to test out ideas, and if it is, he said, “so be it.”
Mr. Azar said he was interested in learning more about ensuring access to long-acting reversible contraception. He declined to answer questions on this because of his lack of knowledge but said he wanted more information.
A committee vote on Mr. Azar’s nomination had not been scheduled at press time. Should the committee support him, the full Senate would then consider his nomination.
Negotiating Medicare Part B drug prices could be on the agenda if Alex Azar, President Trump’s nominee to run the Health and Human Services department, gets confirmed.
before the Senate Finance Committee on Jan. 9. “In Part D, we do significant negotiation through pharmacy benefit managers that get the best rates of any commercial payers. We don’t do that in Part B. ... We basically pay sales price plus 6% or some other number. ... In Part B, we should be looking at those approaches” to price negotiations.
Mr. Azar added that in specific cases, such as when the government buys naloxone for use by first responders, “there is absolutely nothing wrong with the government negotiating that.”
The tone at Mr. Azar’s confirmation hearing generally was more conversational and less adversarial than that of his predecessor, Tom Price, MD, although contentious exchanges did occur, particularly around drug pricing.
“Across the board, drug prices are too high,” said Mr. Azar, who previously was president of Eli Lilly’s U.S. operations. “This is what is so bizarre about the way this system is organized that those price increases happen and ... during that same period, the net realized price by the company stayed flat. So just to cover for increased rebates, the patient who walks into the pharmacy whose insurance may not be paying for that is absorbing that cost and that is what I want to work with you to try and solve.”
Echoing comments made at a courtesy hearing before the Senate Health, Education, Labor and Pensions Committee on Nov. 29, Mr. Azar suggested the need for patent reform to push generic competition as a price-lowering tool.
The cordial tone to the hearing does not guarantee bipartisan support for Mr. Azar’s confirmation.
Sen. Maria Cantwell (D-Wash.) made it explicitly clear that her vote would be based on Mr. Azar’s thoughts about Medicaid expansion and on Mr. Azar’s suggested use of block grants “in the interest of sustainability.”
Regarding Medicaid, Mr. Azar sidestepped questions related to requiring “able-bodied” recipients to work.
“I don’t have a definition [of able-bodied] in hand,” Mr. Azar responded to Sen. Sherrod Brown (D-Ohio). “It would be something we’d work on with Congress.”
“You have no definition of ‘able-bodied adult’ that would be appropriate for differentiating between and among Medicaid recipients that you can share with us?” Sen. Brown replied, to which Mr. Azar had no specific answer.
Mr. Azar testified that said he was “a big supporter of telehealth and alternative means of providing care, especially in rural communities. I think sometimes we can be penny-wise [and] pound-foolish in these areas.”
Mr. Azar also voiced support to wellness incentive programs.
In the area of insurance access and premiums, Mr. Azar said that, as secretary, “I [would] have a very important obligation to make whatever program that I am entrusted with work as well as possible. What we have now is not working for people. It is not working for the 10 million who are in that individual market right now. For many of those people, it can be a false insurance card. It can be insurance, but a very high deductible or not having access to providers, so it’s unaffordable use of care. I want to solve the program for them,” as well as for the 28 million who are not in the individual market, to make it more affordable for all.
Regarding the contentious Center for Medicare and Medicaid Innovation, Mr. Azar voiced support of its use of mandatory participation demonstration projects and said that, depending on the circumstance, sometimes the use of mandatory participation is necessary to test out ideas, and if it is, he said, “so be it.”
Mr. Azar said he was interested in learning more about ensuring access to long-acting reversible contraception. He declined to answer questions on this because of his lack of knowledge but said he wanted more information.
A committee vote on Mr. Azar’s nomination had not been scheduled at press time. Should the committee support him, the full Senate would then consider his nomination.
REPORTING FROM A SENATE FINANCE COMMITTEE HEARING
Common food additive makes C. difficile more virulent
, a study showed.
“Out of several carbon sources identified that supported CD2015 growth [epidemic RT027 isolate], we found the disaccharide trehalose increased the growth yield of CD2015 by approximately fivefold, compared with a non-RT027 strain,” according to James Collins, PhD, of Baylor University, Houston, and his colleagues. The increased growth of the epidemic strain of C. difficile observed by Dr. Collins and his team demonstrates that trehalose is a robust carbon source for C. difficile bacterium.
In one experiment, mice with humanized microbiota were infected with two strains of RT027, either R20291 (n = 27) or R20291-delta treA (n = 28), a phosphotrehalase enzyme (TreA) deletion mutant that cannot metabolize trehalose. Mice were then given 5 mM of trehalose ad libitum in their drinking water. Researchers observed that the mice infected with R20291-delta treA had much lower mortality rates than the R20291 group (33.3% vs.78.6%). These findings were then reinforced with a second experiment using mice with humanized microbiota, in which trehalose addition increased mortality in RT027 mice, compared with RT027-infected mice that were not given dietary trehalose.
While Dr. Collins and his team demonstrated the effect of trehalose on C. difficile in mice, they also conducted a limited analysis of ileostomy effluent from three human donors. The researchers found that in two of three samples, treA was strongly induced in CD2015, but not in another ribotype, CD2048. This demonstrates that amounts of trehalose found in food are high enough to be metabolized by certain epidemic strains of C. difficile in humans.
Prior to 2000, trehalose use was limited by a relatively high cost of production, approximately $700 per kilogram. A production innovation that utilized a novel enzymatic method that yielded trehalose from starch brought the price of trehalose to approximately $3 per kilogram, making it a commercially viable food supplement. After being considered “generally recognized as safe” by the U.S. Food and Drug Administration in 2000 and approved for use in Europe, the trehalose concentrations in food skyrocketed from around 2% to 11.25%, and trehalose became widely used in several foods, including ice cream, pasta, and ground beef.
Dr. Collins and his associates said that there is considerable evidence that the widespread use of dietary trehalose has contributed to the spread of epidemic C. difficile ribotypes. First, strains RT027 and RT078 have always had the ability to metabolize trehalose, as evidenced by outbreaks of nonepidemic C. difficile in the 1980s. But no epidemic outbreaks were reported until after 2003, several years after trehalose was approved by the FDA. Second, RT027 and RT078 are phylogenetically distant, but independently evolved the ability to metabolize low levels of trehalose. Third, increased severity of the RT027 strain, which metabolizes trehalose in mice, is consistent with increased virulence of RT078 and RT027 in human patients. Fourth, a competitive advantage is conferred to C. difficile being able to metabolize trehalose in low concentrations in a diverse intestinal setting. Finally, the levels of trehalose in ileostomy fluid from patients who eat a normal diet are high enough to be utilized by RT027 strains.
“On the basis of these observations, we propose that the widespread adoption and use of the disaccharide trehalose in the human diet has played a significant role in the emergence of these epidemic and hypervirulent strains,” Dr. Collins and his colleagues wrote in their article in Nature.
The authors of the study had no relevant financial disclosures to report.
SOURCE: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
, a study showed.
“Out of several carbon sources identified that supported CD2015 growth [epidemic RT027 isolate], we found the disaccharide trehalose increased the growth yield of CD2015 by approximately fivefold, compared with a non-RT027 strain,” according to James Collins, PhD, of Baylor University, Houston, and his colleagues. The increased growth of the epidemic strain of C. difficile observed by Dr. Collins and his team demonstrates that trehalose is a robust carbon source for C. difficile bacterium.
In one experiment, mice with humanized microbiota were infected with two strains of RT027, either R20291 (n = 27) or R20291-delta treA (n = 28), a phosphotrehalase enzyme (TreA) deletion mutant that cannot metabolize trehalose. Mice were then given 5 mM of trehalose ad libitum in their drinking water. Researchers observed that the mice infected with R20291-delta treA had much lower mortality rates than the R20291 group (33.3% vs.78.6%). These findings were then reinforced with a second experiment using mice with humanized microbiota, in which trehalose addition increased mortality in RT027 mice, compared with RT027-infected mice that were not given dietary trehalose.
While Dr. Collins and his team demonstrated the effect of trehalose on C. difficile in mice, they also conducted a limited analysis of ileostomy effluent from three human donors. The researchers found that in two of three samples, treA was strongly induced in CD2015, but not in another ribotype, CD2048. This demonstrates that amounts of trehalose found in food are high enough to be metabolized by certain epidemic strains of C. difficile in humans.
Prior to 2000, trehalose use was limited by a relatively high cost of production, approximately $700 per kilogram. A production innovation that utilized a novel enzymatic method that yielded trehalose from starch brought the price of trehalose to approximately $3 per kilogram, making it a commercially viable food supplement. After being considered “generally recognized as safe” by the U.S. Food and Drug Administration in 2000 and approved for use in Europe, the trehalose concentrations in food skyrocketed from around 2% to 11.25%, and trehalose became widely used in several foods, including ice cream, pasta, and ground beef.
Dr. Collins and his associates said that there is considerable evidence that the widespread use of dietary trehalose has contributed to the spread of epidemic C. difficile ribotypes. First, strains RT027 and RT078 have always had the ability to metabolize trehalose, as evidenced by outbreaks of nonepidemic C. difficile in the 1980s. But no epidemic outbreaks were reported until after 2003, several years after trehalose was approved by the FDA. Second, RT027 and RT078 are phylogenetically distant, but independently evolved the ability to metabolize low levels of trehalose. Third, increased severity of the RT027 strain, which metabolizes trehalose in mice, is consistent with increased virulence of RT078 and RT027 in human patients. Fourth, a competitive advantage is conferred to C. difficile being able to metabolize trehalose in low concentrations in a diverse intestinal setting. Finally, the levels of trehalose in ileostomy fluid from patients who eat a normal diet are high enough to be utilized by RT027 strains.
“On the basis of these observations, we propose that the widespread adoption and use of the disaccharide trehalose in the human diet has played a significant role in the emergence of these epidemic and hypervirulent strains,” Dr. Collins and his colleagues wrote in their article in Nature.
The authors of the study had no relevant financial disclosures to report.
SOURCE: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
, a study showed.
“Out of several carbon sources identified that supported CD2015 growth [epidemic RT027 isolate], we found the disaccharide trehalose increased the growth yield of CD2015 by approximately fivefold, compared with a non-RT027 strain,” according to James Collins, PhD, of Baylor University, Houston, and his colleagues. The increased growth of the epidemic strain of C. difficile observed by Dr. Collins and his team demonstrates that trehalose is a robust carbon source for C. difficile bacterium.
In one experiment, mice with humanized microbiota were infected with two strains of RT027, either R20291 (n = 27) or R20291-delta treA (n = 28), a phosphotrehalase enzyme (TreA) deletion mutant that cannot metabolize trehalose. Mice were then given 5 mM of trehalose ad libitum in their drinking water. Researchers observed that the mice infected with R20291-delta treA had much lower mortality rates than the R20291 group (33.3% vs.78.6%). These findings were then reinforced with a second experiment using mice with humanized microbiota, in which trehalose addition increased mortality in RT027 mice, compared with RT027-infected mice that were not given dietary trehalose.
While Dr. Collins and his team demonstrated the effect of trehalose on C. difficile in mice, they also conducted a limited analysis of ileostomy effluent from three human donors. The researchers found that in two of three samples, treA was strongly induced in CD2015, but not in another ribotype, CD2048. This demonstrates that amounts of trehalose found in food are high enough to be metabolized by certain epidemic strains of C. difficile in humans.
Prior to 2000, trehalose use was limited by a relatively high cost of production, approximately $700 per kilogram. A production innovation that utilized a novel enzymatic method that yielded trehalose from starch brought the price of trehalose to approximately $3 per kilogram, making it a commercially viable food supplement. After being considered “generally recognized as safe” by the U.S. Food and Drug Administration in 2000 and approved for use in Europe, the trehalose concentrations in food skyrocketed from around 2% to 11.25%, and trehalose became widely used in several foods, including ice cream, pasta, and ground beef.
Dr. Collins and his associates said that there is considerable evidence that the widespread use of dietary trehalose has contributed to the spread of epidemic C. difficile ribotypes. First, strains RT027 and RT078 have always had the ability to metabolize trehalose, as evidenced by outbreaks of nonepidemic C. difficile in the 1980s. But no epidemic outbreaks were reported until after 2003, several years after trehalose was approved by the FDA. Second, RT027 and RT078 are phylogenetically distant, but independently evolved the ability to metabolize low levels of trehalose. Third, increased severity of the RT027 strain, which metabolizes trehalose in mice, is consistent with increased virulence of RT078 and RT027 in human patients. Fourth, a competitive advantage is conferred to C. difficile being able to metabolize trehalose in low concentrations in a diverse intestinal setting. Finally, the levels of trehalose in ileostomy fluid from patients who eat a normal diet are high enough to be utilized by RT027 strains.
“On the basis of these observations, we propose that the widespread adoption and use of the disaccharide trehalose in the human diet has played a significant role in the emergence of these epidemic and hypervirulent strains,” Dr. Collins and his colleagues wrote in their article in Nature.
The authors of the study had no relevant financial disclosures to report.
SOURCE: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
FROM NATURE
Key clinical point: Metabolizing trehalose increases the virulence and mortality of C. difficile ribotype 027 (RT027).
Major finding: The ability to metabolize trehalose with the phosphotrehalase enzyme (TreA) increases mortality in RT027.
Study details: Experimental mouse models and an analysis of ileostomy effluent from three anonymous donors.
Disclosures: All authors had no financial disclosures to report.
Source: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
The felt pelvic anatomy model: A teaching tool for students and residents

Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
This video is brought to you by![]()
Internet addiction ‘an impairing but treatable problem’
SAN DIEGO – The concept of Internet addiction is imperfect, but it has validity and describes an impairing but treatable problem, according to Diana D. Deister, MD.
“Timely diagnosis can lead to good treatment and symptom improvement,” she said at the annual meeting and scientific symposium of the American Academy of Addiction Psychiatry.
Frameworks for the diagnosis of Internet addiction (IA)/pathological Internet use (PIU) vary but are based mostly on DSM-IV substance abuse and dependency criteria, DSM-IV pathological gambling, or other models. “There are many different checklists and diagnostic instruments you can use, and common synonyms include pathological Internet use, problematic Internet use, and compulsive Internet use,” said Dr. Deister, a child and adolescent psychiatrist at Boston Children’s Hospital. “They may not all refer to the exact same problem you might encounter in clinical practice.”
The Internet Addiction Diagnostic Questionnaire, developed in 1998 by Kimberly S. Young, PsyD, is widely used in research and consists of eight yes or no questions (Cyberpsychol Behav. 1998;1[3]:237-44). Answering “yes” to five out of the eight questions gives you a diagnosis of IA. “There are no preferred symptoms that everybody has to have in order to make the diagnosis, so this is more like the current SUD diagnosis for DSM-5,” Dr. Deister explained. “And there’s no time criteria.”
Clinicians can secure a more detailed assessment of IA symptoms by using the Internet Addiction Test (IAT), which Dr. Young developed in 2013. This tool consists of 20 questions rated on a five-point Likert scale based on frequency, where 0 stands for “not applicable” and 5 stands for “always.” Questions include “How often do you find that you stay online longer than you intended?” “How often do you block out disturbing thoughts about your life with soothing thoughts of the Internet?” And “How often do you feel depressed, moody, or nervous when you are offline, which goes away once you are back online?” A score of 80-100 is considered to meet criteria for IA, a score of 50-79 is considered borderline IA, while a score of below 50 is considered not pathological. Variations of this test can be found in the medical literature, including the Internet Process Addiction Test (Behav Sci. 2015;5:341-52), which contains subscales of Internet use: surfing, online gaming, social networking, and sex.
Dr. Deister noted that imaging studies of brain pathways in IA have demonstrated that mesolimbic dopaminergic projections to the nucleus accumbens from the ventral tegmental area are involved, as well as diminished activity of the ventral medial prefrontal cortex. In a tract-based spatial statistics study, researchers used diffusor tensor imaging (DTI) to study the white matter integrity in 18 adolescents with IA, and 18 age- and gender-matched controls (PLoS ONE. 7[1]:e30253. doi:10.1371/journal.pone.0030253). Compared with controls, the IA group scored higher in the IAT, the Strengths and Difficulties Questionnaire, the Screen for Child Anxiety Related Emotional Disorders scale, and Family Assessment Device measure. DTI scans demonstrated widespread reductions of fractional anisotropy (FA; a measure of white matter health) in major white matter pathways. In addition, significantly negative correlations were found between FA values in the left genu of the corpus callosum and the Screen for Child Anxiety Related Emotional Disorders, and between FA values in the left external capsule and in the IAT.
“The reason these white matter tracts could be important is that there have been previous studies showing that you can improve the health of your white matter tracks through exercise and physical therapy,” Dr. Deister said. “So it may be that this knowledge could lead to new treatments for some of these patients.”
One study of the dose dependence of IA symptoms found that adolescents who met criteria for addictive Internet use, compared with those who met criteria for borderline-addictive Internet use, had higher rates of peer problems (adjusted odds ratio, 7.14 vs. AOR, 5.28); conduct problems (AOR, 22.31 vs. AOR, 4.77); hyperactivity (AOR, 9.49 vs. AOR, 5.58); and emotional symptoms (AOR, 19.06 vs. AOR, 2.85; Int J Adolesc Med. 2014;26[3]:369-75). Multivariate regression analysis revealed that Internet addictive behavior was independently associated with using the Internet for retrieving sexual information (AOR, 1.17) and for participating in games with monetary rewards (AOR, 1.90).
Dr. Deister said IA and aggression go hand in hand. A study of 714 middle school students in Seoul, South Korea, showed a linear association between aggression and IA (Cyberpsychol Behav Soc Netw. 2015;18[5]:260-7). A separate qualitative study of 27 university students in the United States found that the consequences of Internet use led to decreased physical activity, decreased sleep, decreased face-to-face time with other people, and poorer academic performance and concentration (PLoS ONE. 2015;10[2]:e0117372). Another study found that trait anhedonia at baseline predicted greater levels of compulsive Internet use, addiction to online activities, and greater likelihood of addiction to online/offline games (Comput Human Behav. 2016;62:475-9).
Treatment for IA takes all the usual forms, but outcome studies are limited. Cognitive-behavioral therapy for IA was first developed as a 12-week model with a familiar design of behavior modification, cognitive restructuring, harm reduction, and relapse prevention, Dr. Deister said. One study of treatment outcomes that used CBT in 128 IA patients found that 95% of patients were able to manage symptoms at the end of 12 weeks of treatment, while 78% sustained recovery 6 months after treatment (J Behav Addict. 2013;2[4]:209-15). Meanwhile, a meta-analysis of 12 studies related to IA found that medications and psychotherapy were found to be effective for improving IA status, time spent online, and depression and anxiety symptoms (Clin Psychol Rev. 2013;33:317-29). A more recent intention-to-treat analysis of IA disorders in adolescents and adults found that patients referred to treatment showed significant improvements in compulsive Internet use over time (J Behav Addict. 2017;6[4]:579-92). Differential effects were found depending on patients’ compliance, with high compliance generally resulting in significantly higher rates of change.
Dr. Deister reported having no financial disclosures.
SAN DIEGO – The concept of Internet addiction is imperfect, but it has validity and describes an impairing but treatable problem, according to Diana D. Deister, MD.
“Timely diagnosis can lead to good treatment and symptom improvement,” she said at the annual meeting and scientific symposium of the American Academy of Addiction Psychiatry.
Frameworks for the diagnosis of Internet addiction (IA)/pathological Internet use (PIU) vary but are based mostly on DSM-IV substance abuse and dependency criteria, DSM-IV pathological gambling, or other models. “There are many different checklists and diagnostic instruments you can use, and common synonyms include pathological Internet use, problematic Internet use, and compulsive Internet use,” said Dr. Deister, a child and adolescent psychiatrist at Boston Children’s Hospital. “They may not all refer to the exact same problem you might encounter in clinical practice.”
The Internet Addiction Diagnostic Questionnaire, developed in 1998 by Kimberly S. Young, PsyD, is widely used in research and consists of eight yes or no questions (Cyberpsychol Behav. 1998;1[3]:237-44). Answering “yes” to five out of the eight questions gives you a diagnosis of IA. “There are no preferred symptoms that everybody has to have in order to make the diagnosis, so this is more like the current SUD diagnosis for DSM-5,” Dr. Deister explained. “And there’s no time criteria.”
Clinicians can secure a more detailed assessment of IA symptoms by using the Internet Addiction Test (IAT), which Dr. Young developed in 2013. This tool consists of 20 questions rated on a five-point Likert scale based on frequency, where 0 stands for “not applicable” and 5 stands for “always.” Questions include “How often do you find that you stay online longer than you intended?” “How often do you block out disturbing thoughts about your life with soothing thoughts of the Internet?” And “How often do you feel depressed, moody, or nervous when you are offline, which goes away once you are back online?” A score of 80-100 is considered to meet criteria for IA, a score of 50-79 is considered borderline IA, while a score of below 50 is considered not pathological. Variations of this test can be found in the medical literature, including the Internet Process Addiction Test (Behav Sci. 2015;5:341-52), which contains subscales of Internet use: surfing, online gaming, social networking, and sex.
Dr. Deister noted that imaging studies of brain pathways in IA have demonstrated that mesolimbic dopaminergic projections to the nucleus accumbens from the ventral tegmental area are involved, as well as diminished activity of the ventral medial prefrontal cortex. In a tract-based spatial statistics study, researchers used diffusor tensor imaging (DTI) to study the white matter integrity in 18 adolescents with IA, and 18 age- and gender-matched controls (PLoS ONE. 7[1]:e30253. doi:10.1371/journal.pone.0030253). Compared with controls, the IA group scored higher in the IAT, the Strengths and Difficulties Questionnaire, the Screen for Child Anxiety Related Emotional Disorders scale, and Family Assessment Device measure. DTI scans demonstrated widespread reductions of fractional anisotropy (FA; a measure of white matter health) in major white matter pathways. In addition, significantly negative correlations were found between FA values in the left genu of the corpus callosum and the Screen for Child Anxiety Related Emotional Disorders, and between FA values in the left external capsule and in the IAT.
“The reason these white matter tracts could be important is that there have been previous studies showing that you can improve the health of your white matter tracks through exercise and physical therapy,” Dr. Deister said. “So it may be that this knowledge could lead to new treatments for some of these patients.”
One study of the dose dependence of IA symptoms found that adolescents who met criteria for addictive Internet use, compared with those who met criteria for borderline-addictive Internet use, had higher rates of peer problems (adjusted odds ratio, 7.14 vs. AOR, 5.28); conduct problems (AOR, 22.31 vs. AOR, 4.77); hyperactivity (AOR, 9.49 vs. AOR, 5.58); and emotional symptoms (AOR, 19.06 vs. AOR, 2.85; Int J Adolesc Med. 2014;26[3]:369-75). Multivariate regression analysis revealed that Internet addictive behavior was independently associated with using the Internet for retrieving sexual information (AOR, 1.17) and for participating in games with monetary rewards (AOR, 1.90).
Dr. Deister said IA and aggression go hand in hand. A study of 714 middle school students in Seoul, South Korea, showed a linear association between aggression and IA (Cyberpsychol Behav Soc Netw. 2015;18[5]:260-7). A separate qualitative study of 27 university students in the United States found that the consequences of Internet use led to decreased physical activity, decreased sleep, decreased face-to-face time with other people, and poorer academic performance and concentration (PLoS ONE. 2015;10[2]:e0117372). Another study found that trait anhedonia at baseline predicted greater levels of compulsive Internet use, addiction to online activities, and greater likelihood of addiction to online/offline games (Comput Human Behav. 2016;62:475-9).
Treatment for IA takes all the usual forms, but outcome studies are limited. Cognitive-behavioral therapy for IA was first developed as a 12-week model with a familiar design of behavior modification, cognitive restructuring, harm reduction, and relapse prevention, Dr. Deister said. One study of treatment outcomes that used CBT in 128 IA patients found that 95% of patients were able to manage symptoms at the end of 12 weeks of treatment, while 78% sustained recovery 6 months after treatment (J Behav Addict. 2013;2[4]:209-15). Meanwhile, a meta-analysis of 12 studies related to IA found that medications and psychotherapy were found to be effective for improving IA status, time spent online, and depression and anxiety symptoms (Clin Psychol Rev. 2013;33:317-29). A more recent intention-to-treat analysis of IA disorders in adolescents and adults found that patients referred to treatment showed significant improvements in compulsive Internet use over time (J Behav Addict. 2017;6[4]:579-92). Differential effects were found depending on patients’ compliance, with high compliance generally resulting in significantly higher rates of change.
Dr. Deister reported having no financial disclosures.
SAN DIEGO – The concept of Internet addiction is imperfect, but it has validity and describes an impairing but treatable problem, according to Diana D. Deister, MD.
“Timely diagnosis can lead to good treatment and symptom improvement,” she said at the annual meeting and scientific symposium of the American Academy of Addiction Psychiatry.
Frameworks for the diagnosis of Internet addiction (IA)/pathological Internet use (PIU) vary but are based mostly on DSM-IV substance abuse and dependency criteria, DSM-IV pathological gambling, or other models. “There are many different checklists and diagnostic instruments you can use, and common synonyms include pathological Internet use, problematic Internet use, and compulsive Internet use,” said Dr. Deister, a child and adolescent psychiatrist at Boston Children’s Hospital. “They may not all refer to the exact same problem you might encounter in clinical practice.”
The Internet Addiction Diagnostic Questionnaire, developed in 1998 by Kimberly S. Young, PsyD, is widely used in research and consists of eight yes or no questions (Cyberpsychol Behav. 1998;1[3]:237-44). Answering “yes” to five out of the eight questions gives you a diagnosis of IA. “There are no preferred symptoms that everybody has to have in order to make the diagnosis, so this is more like the current SUD diagnosis for DSM-5,” Dr. Deister explained. “And there’s no time criteria.”
Clinicians can secure a more detailed assessment of IA symptoms by using the Internet Addiction Test (IAT), which Dr. Young developed in 2013. This tool consists of 20 questions rated on a five-point Likert scale based on frequency, where 0 stands for “not applicable” and 5 stands for “always.” Questions include “How often do you find that you stay online longer than you intended?” “How often do you block out disturbing thoughts about your life with soothing thoughts of the Internet?” And “How often do you feel depressed, moody, or nervous when you are offline, which goes away once you are back online?” A score of 80-100 is considered to meet criteria for IA, a score of 50-79 is considered borderline IA, while a score of below 50 is considered not pathological. Variations of this test can be found in the medical literature, including the Internet Process Addiction Test (Behav Sci. 2015;5:341-52), which contains subscales of Internet use: surfing, online gaming, social networking, and sex.
Dr. Deister noted that imaging studies of brain pathways in IA have demonstrated that mesolimbic dopaminergic projections to the nucleus accumbens from the ventral tegmental area are involved, as well as diminished activity of the ventral medial prefrontal cortex. In a tract-based spatial statistics study, researchers used diffusor tensor imaging (DTI) to study the white matter integrity in 18 adolescents with IA, and 18 age- and gender-matched controls (PLoS ONE. 7[1]:e30253. doi:10.1371/journal.pone.0030253). Compared with controls, the IA group scored higher in the IAT, the Strengths and Difficulties Questionnaire, the Screen for Child Anxiety Related Emotional Disorders scale, and Family Assessment Device measure. DTI scans demonstrated widespread reductions of fractional anisotropy (FA; a measure of white matter health) in major white matter pathways. In addition, significantly negative correlations were found between FA values in the left genu of the corpus callosum and the Screen for Child Anxiety Related Emotional Disorders, and between FA values in the left external capsule and in the IAT.
“The reason these white matter tracts could be important is that there have been previous studies showing that you can improve the health of your white matter tracks through exercise and physical therapy,” Dr. Deister said. “So it may be that this knowledge could lead to new treatments for some of these patients.”
One study of the dose dependence of IA symptoms found that adolescents who met criteria for addictive Internet use, compared with those who met criteria for borderline-addictive Internet use, had higher rates of peer problems (adjusted odds ratio, 7.14 vs. AOR, 5.28); conduct problems (AOR, 22.31 vs. AOR, 4.77); hyperactivity (AOR, 9.49 vs. AOR, 5.58); and emotional symptoms (AOR, 19.06 vs. AOR, 2.85; Int J Adolesc Med. 2014;26[3]:369-75). Multivariate regression analysis revealed that Internet addictive behavior was independently associated with using the Internet for retrieving sexual information (AOR, 1.17) and for participating in games with monetary rewards (AOR, 1.90).
Dr. Deister said IA and aggression go hand in hand. A study of 714 middle school students in Seoul, South Korea, showed a linear association between aggression and IA (Cyberpsychol Behav Soc Netw. 2015;18[5]:260-7). A separate qualitative study of 27 university students in the United States found that the consequences of Internet use led to decreased physical activity, decreased sleep, decreased face-to-face time with other people, and poorer academic performance and concentration (PLoS ONE. 2015;10[2]:e0117372). Another study found that trait anhedonia at baseline predicted greater levels of compulsive Internet use, addiction to online activities, and greater likelihood of addiction to online/offline games (Comput Human Behav. 2016;62:475-9).
Treatment for IA takes all the usual forms, but outcome studies are limited. Cognitive-behavioral therapy for IA was first developed as a 12-week model with a familiar design of behavior modification, cognitive restructuring, harm reduction, and relapse prevention, Dr. Deister said. One study of treatment outcomes that used CBT in 128 IA patients found that 95% of patients were able to manage symptoms at the end of 12 weeks of treatment, while 78% sustained recovery 6 months after treatment (J Behav Addict. 2013;2[4]:209-15). Meanwhile, a meta-analysis of 12 studies related to IA found that medications and psychotherapy were found to be effective for improving IA status, time spent online, and depression and anxiety symptoms (Clin Psychol Rev. 2013;33:317-29). A more recent intention-to-treat analysis of IA disorders in adolescents and adults found that patients referred to treatment showed significant improvements in compulsive Internet use over time (J Behav Addict. 2017;6[4]:579-92). Differential effects were found depending on patients’ compliance, with high compliance generally resulting in significantly higher rates of change.
Dr. Deister reported having no financial disclosures.
REPORTING FROM AAAP