User login
Brentuximab Vedotin with Chemotherapy Improves Progression-Free Survival in Advanced-Stage Hodgkin’s Lymphoma
Study Overview
Objective. To compare the efficacy of brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) with that of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) in patients with stage III or IV classic Hodgkin’s lymphoma.
Design. The ECHELON-1 trial, an international, openlabel, randomized phase 3 trial.
Setting and participants. In this multicenter international trial, a total of 1334 patients underwent randomization from November 2012 through January
2016. Eligible patients were 18 years of age older and had newly diagnosed and histologically proven classic Hodgkin’s lymphoma, Ann Arbor stage III or IV. Patients were eligible only if they had not received prior systemic chemotherapy or radiotherapy. All patients were required to have an ECOG performance status of ≤ 2 and adequate hematologic parameters (hemoglobin ≥ 8, ANC ≥ 1500, and platelet count ≥ 75,000). Patients with nodular lymphocyte predominant Hodgkin’s lymphoma, pre-existing peripheral sensory neuropathy, or known cerebral or meningeal disease were excluded.
Intervention. Patients were randomized in a 1:1 fashion to receive A+AVD (brentuximab vedotin 1.2 mg/kg, doxorubicin 25 mg/m2, vinblastine 6 mg/m2 and dacarbazine 375 mg/m2) or ABVD (doxorubicin 25 mg/m2, bleomycin 10 units/m2, vinblastine 6 mg/m2 and dacarbazine 375 mg/m2) IV on days 1 and 15 of each 28-day cycle for up to 6 cycles. A PET scan was done at the end of the second cycle (PET2) and if this showed increased uptake at any site or uptake at a new site of disease (Deauville score 5) patients could be switched to an alternative frontline therapy at the treating physician’s discretion.
Main outcome measures. The primary endpoint of this study was modified progression-free survival (mPFS), defined as time to disease progression, death, or modified progression (noncomplete response after completion of frontline therapy—Deauville score 3, 4, or 5 on PET). Modified progression was incorporated as an endpoint in order to assess the effectiveness of frontline therapy. A secondary endpoint of the study was overall survival (OS).
Results. The baseline characteristics were well balanced between the treatment arms. 58% of the patients were male and 64% had stage IV disease. The median age was 36 years and 9% in each group were over the age of 65. After a median follow-up of 24.9 months, the independently assessed 2-year mPFS was 82.1% and 77.2% in the A+AVD and ABVD groups, respectively (hazard ratio [HR] 0.77; 95% confidence interval [CI] 0.6–0.98). The 2-year mPFS rate according to investigator assessment was 81% and 74.4% in the A+AVD and ABVD groups, respectively. Modified progression (failure to achieve a complete response after completion of frontline therapy resulting in treatment with subsequent therapy) occurred in 9 and 22 patients in the
A+AVD and ABVD groups, respectively. A pre-specified subgroup analysis showed that patients from North America, male patients, patients with involvement of more than 1 extranodal site, patients with a high IPSS score (4–7), patients < 60 years old and those with stage IV disease appeared to benefit more from A+AVD. The rate of PET2 negativity was 89% with A+AVD and 86% with ABVD. The 2-year overall survival was 96.6% in the A+AVD group and 94.9% in the ABVD group (HR 0.72; 95% CI 0.44–1.17). Fewer patients in the A+AVD group received subsequent cancer-directed therapy.
Neutropenia was more commonly reported in the A+AVD group (58% vs. 45%). Moreover, febrile neutropenia was reported in 19% and 8% of patients in the A+AVD and ABVD groups, respectively. Discontinuation rates in either arm for febrile neutropenia was ≤ 1%. The rate of infections was 55% in the A+AVD group and 50% in the ABVD group (grade 3 or higher: 18% and 10%, respectively). After review of the rates of febrile neutropenia, the safety monitoring committee recommended that primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) be used for patients who were yet to be enrolled. The rate of febrile neutropenia in the 83 patients in the A+AVD group who received primary prophylaxis was lower than those who did not (11% vs. 18%). Peripheral neuropathy occurred in 67% of patients in the A+AVD group and 42% in the ABVD group (grade 3 or higher: 11% vs 2%, respectively). Neuropathy lead to discontinuation of a study drug in 10% of patients in the A+AVD group. 67% of patients with peripheral neuropathy in the A+AVD group had resolution or improvement by one grade of their neuropathy at the time of last follow up. Pulmonary toxicity was reported in 2% of patients in the A+AVD group and 7% of the ABVD group (grade 3 or higher: < 1% vs. 3%, respectively). During treatment, 9 deaths were reported in the A+AVD group and 13 deaths in the ABVD group. Of the deaths in the ABVD group, 11 were associated with pulmonary toxicity.
Conclusion. A+AVD had superior efficacy to ABVD in the treatment of patients with advanced-stage Hodgkin’s lymphoma.
Commentary
Hodgkin’s lymphoma (HL) accounts for approximately 10% of all lymphomas in the world annually [1]. While outcomes with frontline therapy for patients with HL have dramatically improved with ABVD, up to 30% of patients have either refractory disease or relapse after initial therapy [2,3]. One particular area of concern in the current treatment of HL with ABVD is the associated pulmonary toxicity of bleomycin. Pulmonary toxicity from bleomycin occurs in approximately 20%–30% of patients and can lead to long-term morbidity [4,5]. In addition, approximately 15% or more of HL patients are elderly and may have co-existing pulmonary disease. In the previously published E2496 trial, the risk of bleomycin lung toxicity in the elderly was 24% [3]. Although the risk of clinically relevant lung toxicity remains low, there is considerable concern about this amongst clinicians. Recent data has challenged the benefit of bleomycin as a component of ABVD. For example, Johnson and colleagues have shown that in patients with a negtive PET scan after 2 cycles of ABVD, the omission of bleomycin (ie, continuation of AVD) resulted in only a 1.6% reduction in 3-year progression-free survival with a decrease in pulmonary toxicity [6].
Recently, there have been notable advances in the treatment of patients with relapsed or refractory HL, including the incorporation of the PD-1 inhibitor
nivolumab as well as the immunotoxin conjugated CD30 monoclonal antibody brentuximab vedotin (BV). Given the activity of such agents in relapsed and refractory patients, there has been much enthusiasm about incorporation of such agents into the frontline setting. In the current ECHELON-1 trial, Connors and colleagues present the results of a randomized phase 3 trial comparing ABVD, the current standard of care, to A+AVD, which replaces bleomycin with BV. The trial used a primary endpoint of modified progression-free survival, where a noncomplete response and after primary therapy and subsequent treatment with anticancer therapy was considered disease progression. Notably, this trial did meet its primary endpoint of improved
modified PFS, with a 4.9% lower risk of progression, death, or noncomplete response and subsequent need for treatment at 2 years. Overall survival was not significantly different at the time of analysis.
There are some noteworthy findings in addition to this. First, A+AVD was associated with a higher risk of febrile neutropenia and infectious complications; however, following the incorporation of G-CSF prophylaxis this risk was lowered. The pulmonary toxicity was lower in the A+AVD group (2% vs. 7%). A+AVD was associated with an increased risk of peripheral neuropathy, which appeared to improve or resolve following discontinuation of therapy. The neuropathy was mainly low grade with only 11% being grade 3 or higher. Although it remains early and follow-up short, A+AVD did appear to have superior efficacy with a decrease in the risk of pulmonary toxicity in this study. It is worth noting that the risk of neurotoxicity was higher, albeit reversible with drug discontinuation. Given these results, A+AVD warrants consideration as frontline therapy in newly diagnosed patients with advanced stage classic Hodgkin’s lymphoma.
Applications for Clinical Practice
The results of this trial suggest that A+AVD with G-CSF support compares favorably to ABVD and may represent an acceptable first-line treatment strategy, particularly for patients at higher risk for pulmonary toxicity, although follow-up remains short at this time.
—Daniel Isaac, DO, MS
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
3. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: An intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
4. Martin WG, Ristow KM, Habermann TM, et al. Bleomycin pulmonary toxicity has a negative impact on the outcome of patients with Hodgkin’s lymphoma. J Clin Oncol 2005;23:7614–20.
5. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the Stanford V regimen and ABVD in the treatment of advanced Hodgkin’s lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
6. Johnson P, Federico M, Kirkwood A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N Engl J Med 2016;374:2419–29.
Study Overview
Objective. To compare the efficacy of brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) with that of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) in patients with stage III or IV classic Hodgkin’s lymphoma.
Design. The ECHELON-1 trial, an international, openlabel, randomized phase 3 trial.
Setting and participants. In this multicenter international trial, a total of 1334 patients underwent randomization from November 2012 through January
2016. Eligible patients were 18 years of age older and had newly diagnosed and histologically proven classic Hodgkin’s lymphoma, Ann Arbor stage III or IV. Patients were eligible only if they had not received prior systemic chemotherapy or radiotherapy. All patients were required to have an ECOG performance status of ≤ 2 and adequate hematologic parameters (hemoglobin ≥ 8, ANC ≥ 1500, and platelet count ≥ 75,000). Patients with nodular lymphocyte predominant Hodgkin’s lymphoma, pre-existing peripheral sensory neuropathy, or known cerebral or meningeal disease were excluded.
Intervention. Patients were randomized in a 1:1 fashion to receive A+AVD (brentuximab vedotin 1.2 mg/kg, doxorubicin 25 mg/m2, vinblastine 6 mg/m2 and dacarbazine 375 mg/m2) or ABVD (doxorubicin 25 mg/m2, bleomycin 10 units/m2, vinblastine 6 mg/m2 and dacarbazine 375 mg/m2) IV on days 1 and 15 of each 28-day cycle for up to 6 cycles. A PET scan was done at the end of the second cycle (PET2) and if this showed increased uptake at any site or uptake at a new site of disease (Deauville score 5) patients could be switched to an alternative frontline therapy at the treating physician’s discretion.
Main outcome measures. The primary endpoint of this study was modified progression-free survival (mPFS), defined as time to disease progression, death, or modified progression (noncomplete response after completion of frontline therapy—Deauville score 3, 4, or 5 on PET). Modified progression was incorporated as an endpoint in order to assess the effectiveness of frontline therapy. A secondary endpoint of the study was overall survival (OS).
Results. The baseline characteristics were well balanced between the treatment arms. 58% of the patients were male and 64% had stage IV disease. The median age was 36 years and 9% in each group were over the age of 65. After a median follow-up of 24.9 months, the independently assessed 2-year mPFS was 82.1% and 77.2% in the A+AVD and ABVD groups, respectively (hazard ratio [HR] 0.77; 95% confidence interval [CI] 0.6–0.98). The 2-year mPFS rate according to investigator assessment was 81% and 74.4% in the A+AVD and ABVD groups, respectively. Modified progression (failure to achieve a complete response after completion of frontline therapy resulting in treatment with subsequent therapy) occurred in 9 and 22 patients in the
A+AVD and ABVD groups, respectively. A pre-specified subgroup analysis showed that patients from North America, male patients, patients with involvement of more than 1 extranodal site, patients with a high IPSS score (4–7), patients < 60 years old and those with stage IV disease appeared to benefit more from A+AVD. The rate of PET2 negativity was 89% with A+AVD and 86% with ABVD. The 2-year overall survival was 96.6% in the A+AVD group and 94.9% in the ABVD group (HR 0.72; 95% CI 0.44–1.17). Fewer patients in the A+AVD group received subsequent cancer-directed therapy.
Neutropenia was more commonly reported in the A+AVD group (58% vs. 45%). Moreover, febrile neutropenia was reported in 19% and 8% of patients in the A+AVD and ABVD groups, respectively. Discontinuation rates in either arm for febrile neutropenia was ≤ 1%. The rate of infections was 55% in the A+AVD group and 50% in the ABVD group (grade 3 or higher: 18% and 10%, respectively). After review of the rates of febrile neutropenia, the safety monitoring committee recommended that primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) be used for patients who were yet to be enrolled. The rate of febrile neutropenia in the 83 patients in the A+AVD group who received primary prophylaxis was lower than those who did not (11% vs. 18%). Peripheral neuropathy occurred in 67% of patients in the A+AVD group and 42% in the ABVD group (grade 3 or higher: 11% vs 2%, respectively). Neuropathy lead to discontinuation of a study drug in 10% of patients in the A+AVD group. 67% of patients with peripheral neuropathy in the A+AVD group had resolution or improvement by one grade of their neuropathy at the time of last follow up. Pulmonary toxicity was reported in 2% of patients in the A+AVD group and 7% of the ABVD group (grade 3 or higher: < 1% vs. 3%, respectively). During treatment, 9 deaths were reported in the A+AVD group and 13 deaths in the ABVD group. Of the deaths in the ABVD group, 11 were associated with pulmonary toxicity.
Conclusion. A+AVD had superior efficacy to ABVD in the treatment of patients with advanced-stage Hodgkin’s lymphoma.
Commentary
Hodgkin’s lymphoma (HL) accounts for approximately 10% of all lymphomas in the world annually [1]. While outcomes with frontline therapy for patients with HL have dramatically improved with ABVD, up to 30% of patients have either refractory disease or relapse after initial therapy [2,3]. One particular area of concern in the current treatment of HL with ABVD is the associated pulmonary toxicity of bleomycin. Pulmonary toxicity from bleomycin occurs in approximately 20%–30% of patients and can lead to long-term morbidity [4,5]. In addition, approximately 15% or more of HL patients are elderly and may have co-existing pulmonary disease. In the previously published E2496 trial, the risk of bleomycin lung toxicity in the elderly was 24% [3]. Although the risk of clinically relevant lung toxicity remains low, there is considerable concern about this amongst clinicians. Recent data has challenged the benefit of bleomycin as a component of ABVD. For example, Johnson and colleagues have shown that in patients with a negtive PET scan after 2 cycles of ABVD, the omission of bleomycin (ie, continuation of AVD) resulted in only a 1.6% reduction in 3-year progression-free survival with a decrease in pulmonary toxicity [6].
Recently, there have been notable advances in the treatment of patients with relapsed or refractory HL, including the incorporation of the PD-1 inhibitor
nivolumab as well as the immunotoxin conjugated CD30 monoclonal antibody brentuximab vedotin (BV). Given the activity of such agents in relapsed and refractory patients, there has been much enthusiasm about incorporation of such agents into the frontline setting. In the current ECHELON-1 trial, Connors and colleagues present the results of a randomized phase 3 trial comparing ABVD, the current standard of care, to A+AVD, which replaces bleomycin with BV. The trial used a primary endpoint of modified progression-free survival, where a noncomplete response and after primary therapy and subsequent treatment with anticancer therapy was considered disease progression. Notably, this trial did meet its primary endpoint of improved
modified PFS, with a 4.9% lower risk of progression, death, or noncomplete response and subsequent need for treatment at 2 years. Overall survival was not significantly different at the time of analysis.
There are some noteworthy findings in addition to this. First, A+AVD was associated with a higher risk of febrile neutropenia and infectious complications; however, following the incorporation of G-CSF prophylaxis this risk was lowered. The pulmonary toxicity was lower in the A+AVD group (2% vs. 7%). A+AVD was associated with an increased risk of peripheral neuropathy, which appeared to improve or resolve following discontinuation of therapy. The neuropathy was mainly low grade with only 11% being grade 3 or higher. Although it remains early and follow-up short, A+AVD did appear to have superior efficacy with a decrease in the risk of pulmonary toxicity in this study. It is worth noting that the risk of neurotoxicity was higher, albeit reversible with drug discontinuation. Given these results, A+AVD warrants consideration as frontline therapy in newly diagnosed patients with advanced stage classic Hodgkin’s lymphoma.
Applications for Clinical Practice
The results of this trial suggest that A+AVD with G-CSF support compares favorably to ABVD and may represent an acceptable first-line treatment strategy, particularly for patients at higher risk for pulmonary toxicity, although follow-up remains short at this time.
—Daniel Isaac, DO, MS
Study Overview
Objective. To compare the efficacy of brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) with that of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) in patients with stage III or IV classic Hodgkin’s lymphoma.
Design. The ECHELON-1 trial, an international, openlabel, randomized phase 3 trial.
Setting and participants. In this multicenter international trial, a total of 1334 patients underwent randomization from November 2012 through January
2016. Eligible patients were 18 years of age older and had newly diagnosed and histologically proven classic Hodgkin’s lymphoma, Ann Arbor stage III or IV. Patients were eligible only if they had not received prior systemic chemotherapy or radiotherapy. All patients were required to have an ECOG performance status of ≤ 2 and adequate hematologic parameters (hemoglobin ≥ 8, ANC ≥ 1500, and platelet count ≥ 75,000). Patients with nodular lymphocyte predominant Hodgkin’s lymphoma, pre-existing peripheral sensory neuropathy, or known cerebral or meningeal disease were excluded.
Intervention. Patients were randomized in a 1:1 fashion to receive A+AVD (brentuximab vedotin 1.2 mg/kg, doxorubicin 25 mg/m2, vinblastine 6 mg/m2 and dacarbazine 375 mg/m2) or ABVD (doxorubicin 25 mg/m2, bleomycin 10 units/m2, vinblastine 6 mg/m2 and dacarbazine 375 mg/m2) IV on days 1 and 15 of each 28-day cycle for up to 6 cycles. A PET scan was done at the end of the second cycle (PET2) and if this showed increased uptake at any site or uptake at a new site of disease (Deauville score 5) patients could be switched to an alternative frontline therapy at the treating physician’s discretion.
Main outcome measures. The primary endpoint of this study was modified progression-free survival (mPFS), defined as time to disease progression, death, or modified progression (noncomplete response after completion of frontline therapy—Deauville score 3, 4, or 5 on PET). Modified progression was incorporated as an endpoint in order to assess the effectiveness of frontline therapy. A secondary endpoint of the study was overall survival (OS).
Results. The baseline characteristics were well balanced between the treatment arms. 58% of the patients were male and 64% had stage IV disease. The median age was 36 years and 9% in each group were over the age of 65. After a median follow-up of 24.9 months, the independently assessed 2-year mPFS was 82.1% and 77.2% in the A+AVD and ABVD groups, respectively (hazard ratio [HR] 0.77; 95% confidence interval [CI] 0.6–0.98). The 2-year mPFS rate according to investigator assessment was 81% and 74.4% in the A+AVD and ABVD groups, respectively. Modified progression (failure to achieve a complete response after completion of frontline therapy resulting in treatment with subsequent therapy) occurred in 9 and 22 patients in the
A+AVD and ABVD groups, respectively. A pre-specified subgroup analysis showed that patients from North America, male patients, patients with involvement of more than 1 extranodal site, patients with a high IPSS score (4–7), patients < 60 years old and those with stage IV disease appeared to benefit more from A+AVD. The rate of PET2 negativity was 89% with A+AVD and 86% with ABVD. The 2-year overall survival was 96.6% in the A+AVD group and 94.9% in the ABVD group (HR 0.72; 95% CI 0.44–1.17). Fewer patients in the A+AVD group received subsequent cancer-directed therapy.
Neutropenia was more commonly reported in the A+AVD group (58% vs. 45%). Moreover, febrile neutropenia was reported in 19% and 8% of patients in the A+AVD and ABVD groups, respectively. Discontinuation rates in either arm for febrile neutropenia was ≤ 1%. The rate of infections was 55% in the A+AVD group and 50% in the ABVD group (grade 3 or higher: 18% and 10%, respectively). After review of the rates of febrile neutropenia, the safety monitoring committee recommended that primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) be used for patients who were yet to be enrolled. The rate of febrile neutropenia in the 83 patients in the A+AVD group who received primary prophylaxis was lower than those who did not (11% vs. 18%). Peripheral neuropathy occurred in 67% of patients in the A+AVD group and 42% in the ABVD group (grade 3 or higher: 11% vs 2%, respectively). Neuropathy lead to discontinuation of a study drug in 10% of patients in the A+AVD group. 67% of patients with peripheral neuropathy in the A+AVD group had resolution or improvement by one grade of their neuropathy at the time of last follow up. Pulmonary toxicity was reported in 2% of patients in the A+AVD group and 7% of the ABVD group (grade 3 or higher: < 1% vs. 3%, respectively). During treatment, 9 deaths were reported in the A+AVD group and 13 deaths in the ABVD group. Of the deaths in the ABVD group, 11 were associated with pulmonary toxicity.
Conclusion. A+AVD had superior efficacy to ABVD in the treatment of patients with advanced-stage Hodgkin’s lymphoma.
Commentary
Hodgkin’s lymphoma (HL) accounts for approximately 10% of all lymphomas in the world annually [1]. While outcomes with frontline therapy for patients with HL have dramatically improved with ABVD, up to 30% of patients have either refractory disease or relapse after initial therapy [2,3]. One particular area of concern in the current treatment of HL with ABVD is the associated pulmonary toxicity of bleomycin. Pulmonary toxicity from bleomycin occurs in approximately 20%–30% of patients and can lead to long-term morbidity [4,5]. In addition, approximately 15% or more of HL patients are elderly and may have co-existing pulmonary disease. In the previously published E2496 trial, the risk of bleomycin lung toxicity in the elderly was 24% [3]. Although the risk of clinically relevant lung toxicity remains low, there is considerable concern about this amongst clinicians. Recent data has challenged the benefit of bleomycin as a component of ABVD. For example, Johnson and colleagues have shown that in patients with a negtive PET scan after 2 cycles of ABVD, the omission of bleomycin (ie, continuation of AVD) resulted in only a 1.6% reduction in 3-year progression-free survival with a decrease in pulmonary toxicity [6].
Recently, there have been notable advances in the treatment of patients with relapsed or refractory HL, including the incorporation of the PD-1 inhibitor
nivolumab as well as the immunotoxin conjugated CD30 monoclonal antibody brentuximab vedotin (BV). Given the activity of such agents in relapsed and refractory patients, there has been much enthusiasm about incorporation of such agents into the frontline setting. In the current ECHELON-1 trial, Connors and colleagues present the results of a randomized phase 3 trial comparing ABVD, the current standard of care, to A+AVD, which replaces bleomycin with BV. The trial used a primary endpoint of modified progression-free survival, where a noncomplete response and after primary therapy and subsequent treatment with anticancer therapy was considered disease progression. Notably, this trial did meet its primary endpoint of improved
modified PFS, with a 4.9% lower risk of progression, death, or noncomplete response and subsequent need for treatment at 2 years. Overall survival was not significantly different at the time of analysis.
There are some noteworthy findings in addition to this. First, A+AVD was associated with a higher risk of febrile neutropenia and infectious complications; however, following the incorporation of G-CSF prophylaxis this risk was lowered. The pulmonary toxicity was lower in the A+AVD group (2% vs. 7%). A+AVD was associated with an increased risk of peripheral neuropathy, which appeared to improve or resolve following discontinuation of therapy. The neuropathy was mainly low grade with only 11% being grade 3 or higher. Although it remains early and follow-up short, A+AVD did appear to have superior efficacy with a decrease in the risk of pulmonary toxicity in this study. It is worth noting that the risk of neurotoxicity was higher, albeit reversible with drug discontinuation. Given these results, A+AVD warrants consideration as frontline therapy in newly diagnosed patients with advanced stage classic Hodgkin’s lymphoma.
Applications for Clinical Practice
The results of this trial suggest that A+AVD with G-CSF support compares favorably to ABVD and may represent an acceptable first-line treatment strategy, particularly for patients at higher risk for pulmonary toxicity, although follow-up remains short at this time.
—Daniel Isaac, DO, MS
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
3. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: An intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
4. Martin WG, Ristow KM, Habermann TM, et al. Bleomycin pulmonary toxicity has a negative impact on the outcome of patients with Hodgkin’s lymphoma. J Clin Oncol 2005;23:7614–20.
5. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the Stanford V regimen and ABVD in the treatment of advanced Hodgkin’s lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
6. Johnson P, Federico M, Kirkwood A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N Engl J Med 2016;374:2419–29.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
3. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: An intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
4. Martin WG, Ristow KM, Habermann TM, et al. Bleomycin pulmonary toxicity has a negative impact on the outcome of patients with Hodgkin’s lymphoma. J Clin Oncol 2005;23:7614–20.
5. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the Stanford V regimen and ABVD in the treatment of advanced Hodgkin’s lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
6. Johnson P, Federico M, Kirkwood A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N Engl J Med 2016;374:2419–29.
Self-Reported Cognitive Impairment Is Rising
More people are reporting cognitive impairment, according to CDC researchers. Overall, the rate of self-reported cognitive impairment rose from 5.7% in 1997 to 6.7% in 2015. Among non-Hispanic white respondents, the rate went from 5.2% to 6.1%. The researchers found no significant trends in cognitive impairment among non-Hispanic black, Native American, Hispanic, or Asian respondents.
Respondents to the National Health Survey were asked whether any family member was “limited in any way because of difficulty remembering or because of experiencing periods of confusion.” The rate of cognitive impairment increased with age in all 5 racial/ethnic groups. The rate was lowest among non-Hispanic white respondents until the 1943-1947 birth cohort. The data are “interesting,” the researchers say, because other recent studies that used data from cognitive tests and clinical assessments found a declining trend in dementia in the U.S. Direct comparisons among studies is inappropriate, however, they note, because of different study designs. Their own findings “might suggest that awareness of cognitive impairment has improved in the United States, especially in recent years,” in part due to heightened public attention to Alzheimer disease.
More public education may be needed to promote awareness, the researchers say, especially among the minority groups. Minorities had lower rates of self-reporting, perhaps because of different cultural beliefs about disease and aging, or because they are less likely to seek treatment for depression, which can contribute to cognitive decline.
More people are reporting cognitive impairment, according to CDC researchers. Overall, the rate of self-reported cognitive impairment rose from 5.7% in 1997 to 6.7% in 2015. Among non-Hispanic white respondents, the rate went from 5.2% to 6.1%. The researchers found no significant trends in cognitive impairment among non-Hispanic black, Native American, Hispanic, or Asian respondents.
Respondents to the National Health Survey were asked whether any family member was “limited in any way because of difficulty remembering or because of experiencing periods of confusion.” The rate of cognitive impairment increased with age in all 5 racial/ethnic groups. The rate was lowest among non-Hispanic white respondents until the 1943-1947 birth cohort. The data are “interesting,” the researchers say, because other recent studies that used data from cognitive tests and clinical assessments found a declining trend in dementia in the U.S. Direct comparisons among studies is inappropriate, however, they note, because of different study designs. Their own findings “might suggest that awareness of cognitive impairment has improved in the United States, especially in recent years,” in part due to heightened public attention to Alzheimer disease.
More public education may be needed to promote awareness, the researchers say, especially among the minority groups. Minorities had lower rates of self-reporting, perhaps because of different cultural beliefs about disease and aging, or because they are less likely to seek treatment for depression, which can contribute to cognitive decline.
More people are reporting cognitive impairment, according to CDC researchers. Overall, the rate of self-reported cognitive impairment rose from 5.7% in 1997 to 6.7% in 2015. Among non-Hispanic white respondents, the rate went from 5.2% to 6.1%. The researchers found no significant trends in cognitive impairment among non-Hispanic black, Native American, Hispanic, or Asian respondents.
Respondents to the National Health Survey were asked whether any family member was “limited in any way because of difficulty remembering or because of experiencing periods of confusion.” The rate of cognitive impairment increased with age in all 5 racial/ethnic groups. The rate was lowest among non-Hispanic white respondents until the 1943-1947 birth cohort. The data are “interesting,” the researchers say, because other recent studies that used data from cognitive tests and clinical assessments found a declining trend in dementia in the U.S. Direct comparisons among studies is inappropriate, however, they note, because of different study designs. Their own findings “might suggest that awareness of cognitive impairment has improved in the United States, especially in recent years,” in part due to heightened public attention to Alzheimer disease.
More public education may be needed to promote awareness, the researchers say, especially among the minority groups. Minorities had lower rates of self-reporting, perhaps because of different cultural beliefs about disease and aging, or because they are less likely to seek treatment for depression, which can contribute to cognitive decline.
Avoiding Inappropriate Medication Prescription in Older Intensive Care Survivors
From the Division of Allergy, Pulmonary, and Critical Care Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN (Dr. Marra), Division of Anesthesiology Critical Care Medicine, Vanderbilt University Medical Center, Nashville, TN (Dr. Hayhurst, Dr. Hughes, Dr. Pandharipande), Department of Clinical and Experimental Science, University of Brescia, Brescia, Italy (Dr. Marengoni), School of Medicine and Surgery,
University of Milano-Bicocca, Milan, Italy (Dr. Bellelli), and Rehabilitation and Aged Care Unit Hospital Ancelle, Cremona, Italy (Dr. Morandi).
Abstract
- Objective: To present an overview of the phenomenon of inappropriate medication prescription in older critically ill patients and examine possible strategies of intervention.
- Methods: Review of the literature.
- Results: Polypharmacy and inappropriate prescribing of medications in older persons may lead to a significant risk of adverse drug-related events and mortality. The intensive care unit (ICU) is often the place where potentially inappropriate medications (PIMs) are first prescribed. Common PIMs at ICU discharge are antipsychotics, benzodiazepines, opioids, anticholinergic medications, antidepressants, and drugs causing orthostatic hypotension. Different classes of medications, typically intended for short-term use, are sometimes inappropriately continued after discharge from the hospital. At admission, potential risk factors for PIM are multiple morbidities, polypharmacy, frailty and cognitive decline; at discharge, a high number of pre-admission PIMs, discharge to a location other than home, discharge from a surgical service, longer length of ICU and hospital stay, and mechanical ventilation. Inappropriate prescribing in older patients can be detected through either the use of explicit criteria, drug utilization reviews, and multidisciplinary teams, including a geriatrician and/or the involvement of a clinical pharmacist.
- Conclusion: Use of PIMs may be common in critical patients, both on admission and at discharge from ICU. Therapeutic reconciliation is recommended at every transition of care (eg, at hospital or ICU admission and discharge) in order to improve appropriateness of prescription.
Key words: elderly; intensive care unit; inappropriate medications; antipsychotics.
Since older persons are often affected by multiple chronic diseases and are prescribed several medications, the quality and safety of prescribing these medications has become a global health care issue [1–4]. Polypharmacy and inappropriate prescribing of medications among the elderly is receiving significant attention in the medical literature [5,6]. Inappropriate medications in the elderly can lead to falls, cognitive impairment and delirium, poorer health status, and higher mortality [7–10]. Medications are considered potentially inappropriate when (a) the risks of treatment outweigh the benefits [11], (b) they are prescribed for periods longer than clinically indicated or without any clear indication, (c) they are not prescribed when indicated [12], and (d) they are likely to interact with other drugs and diseases. Medications included in this category are often referred to as potentially inappropriate medications (PIMs), as in some situations their use is justified; however, if the risk of harm from the drug is judged to outweigh the potential clinical benefit after an individual patient’s clinical circumstances are considered, these drugs are considered “actually inappropriate medications” (AIMs) [6].
Advancing age is associated with substantial pharmacokinetic and pharmacodynamics changes, such as altered distribution volumes and altered permeability of the blood-brain barrier, impaired liver metabolism and renal capacity, up- and down-regulation of target receptors, transmitters, and signaling pathways changes, impaired homeostasis, and increased risk of adverse drug reactions (ADRs) that lead to increased mortality and morbidity and higher health care costs [2,11,13–19]. Studies show that ADRs cause approximately 5% of hospital admissions in the general population, but the percentage rises to 10% in older persons [20].
Avoiding PIMs represents a strategy aimed at reducing drug-related mortality and morbidity. This article provides an overview of the phenomenon of inappropriate medication prescription in older critically ill patients and examines available strategies of intervention.
Inappropriate Medications at ICU Discharge
Though PIMs and AIMs may be identified at the time of hospital discharge, the intensive care unit (ICU) is often the place where these medications are first prescribed [21]. Acute hospitalization may increase PIM prescribing because of newly prescribed medications, the presence of multiple prescribers, inadequate medication reconciliation, and a lack of care coordination among inpatient providers or in the transition back to outpatient care [22)].
A known complication of critical illness and ICU stay is a significant increase in psychological symptoms, sleep cycle alterations, delirium, and cognitive impairment, which may be associated with increased prescription of specific PIMs, such as antipsychotics or benzodiazepines [6,23,24]. Despite the lack of reliable evidence supporting their use in the ICU, antipsychotic agents are used routinely in ICU patients [25] to treat a variety of conditions, such as substance withdrawal, agitation not responding to other therapies, or delirium. Results from a multicenter study of 164,996 hospitalizations across 71 academic medical centers in the US showed that 1 out of 10 ICU patients received an antipsychotic during their hospital stay [25]. Jasiak et al estimated that one-third of patients initiated on an atypical antipsychotic therapy for ICU delirium received a hospital discharge prescription for these medications, with a potential annual outpatient medication cost of approximately $2255 per patient [26].
One potential consequence of antipsychotic use in the ICU is their continuation after the transition to other clinical settings, including discharge from the hospital [27] (Table 1). 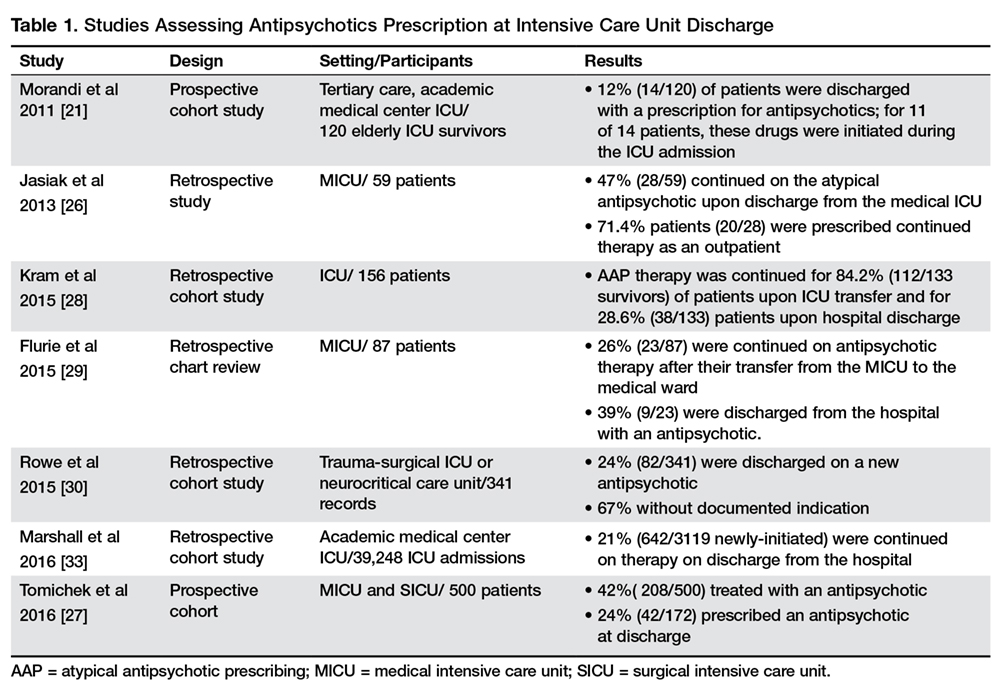
When examining the specific factors that may contribute to a patient being discharged on an antipsychotic, authors found that the specific antipsychotic used correlated with risk of continuation [27,30], with atypical antipsychotics having a greater likelihood of being continued than haloperidol [27,30]. Possible explanation for these results could be that physicians perceive less long-term risk from atypical agents, so may be more likely to continue them on discharge [30]. However, such an approach is not always safe. Indeed, although atypical antipsychotic agents tend to cause less tardive dyskinesia, they are known to be associated with similar rates of other adverse events compared with typical agents and have been linked to an increased risk of sudden cardiac death and pneumonia in the elderly [31,32].
Other factors independently associated with being discharged on a new antipsychotic medication were the severity of the acute illness as measured with the Acute Physiology and Chronic Health Evaluation II score at ICU admission (odds ratio [OR] 1 [95% confidence interval {CI}, 1.0–1.1]) and days treated with benzodiazepines (OR 1.1 [95% CI, 1.0–1.14]) [30]. Conversely, perhaps due to different practice patterns, Tomichek et al did not find an association between benzodiazepines administration and antipsychotic prescription at discharge in post hoc analyses [27].
Another possible reason for antipsychotic continuation may reside in the indication chosen [33]. Antipsychotic agents have sedative properties and they might be used to optimize sleep during hospitalization, despite the lack of evidence to support this indication [34]. Other factors potentially contributing to continuation of antipsychotics may include persistent delirium and agitation, newly diagnosed psychiatric illness, and difficulties experienced by physicians in deprescribing [35] with improper/incomplete medication reconciliation [33].
The continuation of antipsychotic therapy increased 30-day readmission rates in patients compared to those who had therapy stopped before discharge [33]. In addition to the well-described cardiac effects (prolonged QT interval), neuroleptic malignant syndrome and extrapyramidal symptoms may also occur, and longer-term use can predispose patients to metabolic disturbances, falls, and increase the risk of death in elderly patients with dementia [31].
Benzodiazepines and sedative hypnotics are commonly used to treat insomnia and agitation in older adults despite significant risk. Benzodiazepine administration was found to be an independent risk factor for a daily transition to delirium [36,37]. Pandharipande et al reported that every unit dose of lorazepam was associated with a higher risk for daily transition to delirium (OR 1.2, 95% CI 1.1–1.4, P = 0.003) [36] in critically ill patients. A more recent analysis found for every 5 mg of midazolam administered to a patient who is awake and without delirium, there is a 4% chance that this patient will develop delirium the next ICU day [37].
Given that the risk for benzodiazepine-associated delirium is dose-dependent, clinicians should use strategies known to reduce the daily number of benzodiazepines administered that often includes the use of a sedative associated with less delirium occurrence, such as dexmedetomidine or propofol [38]. Evidence has shown that long-term use of benzodiazepines has little benefit with many risks, including an increased susceptibility to spontaneous bacterial infection [39,40] and mortality in the setting of infection [41]. Nakafero et al showed that exposure to benzodiazepines was associated with increased occurrence of both influenza-like-illness–related pneumonia and mortality. Benzodiazepine use was associated also with increased occurrence of asthma exacerbation and with increased all-cause mortality during a median follow-up of 2 years in a cohort of asthmatic patients [42] as well with an increased risk of pneumonia and long-term mortality in patients with a prior diagnosis of community- acquired pneumonia [40]. Long-term use of benzodiazepines is also associated with increased risk of falls [43–45], cognitive impairment [46–48] and disability [49,50].
Other common types of PIMs at ICU discharge were opioids, anticholinergic medications, antidepressants, and drugs causing orthostatic hypotension [6]. Of the anticholinergic AIMs, H2 blockers (61%) and promethazine (15%) were the most common [6]. Only 16% of opioids, 23% of antidepressants, and 10% of drugs causing orthostatic hypotension were found to be actually inappropriate after the patient’s circumstances were considered (eg, postoperative pain control, a new diagnosis of major depressive disorder) [6].
Inappropriate Medications at Hospital Discharge
Medications typically intended for short-term use during acute illness are sometimes continued after discharge without documented indication [51]. Poudel et al found that in 206 patients 70 years of age and older discharged to residential aged care facilities from acute care, at least 1 PIM was identified in 112 (54.4%) patients on admission and 102 (49.5%) patients on discharge [11]. Commonly prescribed PIM categories, at both admission and discharge, were central nervous system, cardiovascular, gastrointestinal, and respiratory drugs and analgesics [6,11,52,53]. Of all medications prescribed at admission (1728), 10.8% were PIMs, and at discharge, of 1759 medications, 9.6% were PIMs. Of the total 187 PIMs on admission, 56 (30%) were stopped, and 131 (70%) were continued; 32 new PIMs were introduced [11].
Morandi et al in 2011 conducted a prospective cohort study including 120 patients age ≥ 60 who were discharged after receiving care in a medical, surgical, or cardiovascular ICU for shock or respiratory failure. The percentage of patients prescribed at least 1 PIM increased from 66% at pre-admission to 85% at discharge. The number of patients with 0 PIMs dropped from 34% at preadmission to 14% at discharge, and the number of patients with 3 or more PIMS increased from 16% at preadmission to 37% at discharge. While it is possible that these drugs may be appropriate when started during an acute illness in the ICU (eg, stress ulcer prophylaxis with H2-antagonists in mechanically ventilated patients), most should have been discontinued at ICU and/or hospital discharge [21].
Inappropriate prescriptions of proton pump inhibitors (PPIs) in hospital and primary care have been widely reported [54,55]. In a study conducted by Ahrens et al in 31 primary care practices, for 58% (263/506) of patients discharged from 35 hospitals with a PPI recommendation in hospital discharge letters, an appropriate indication was missing. In 57% of these cases general practitioners followed this recommendation and continued the prescription for more than 1 month [54]. The strongest factor associated with appropriate and inappropriate continuation of PPI after discharge was PPI prescription prior to hospitalization [54]. Although PPIs are safe, they can cause adverse effects. PPI intake has been found to have a significant association with risk of community-acquired pneumonia [56,57], hip fractures [58], Clostridium difficile-associated diarrhea [55,61,62], and to reduce the therapeutic effects of bisphosphonates [59] and low-dose aspirin [60].
Unintentional medication continuation is not a problem isolated to a single drug class or disease [63]. Scales et al evaluated rates of and risk factors for potentially unintentional medication continuation following hospitalization in a population of elderly patients (≥ 66 years) [51]. They created distinct cohorts by identifying seniors not previously receiving four classes of medications typically used to treat or prevent complications of acute illness: antipsychotic medications; gastric acid suppressants (ie, histamine-2 blockers and proton pump inhibitors); benzodiazepines; and inhaled bronchodilators and steroids [51]. Prescription without documented indication occurred across all medication classes, from 12,209 patients (1.4 %) for antipsychotic medications to 34,140 patients (6.1 %) for gastric acid suppressants [51].
Several potential risk factors were considered. The relationship between multimorbidity and polypharmacy is well described in the literature, and several studies have identified a positive association between the number of drugs and the use of PIMs [64–66]. Conversely, Poudel et al did not find any association between polypharmacy and PIM use [11]. Associations were found between the use of PIMs, frailty status, and cognitive decline of patients at admission and at discharge [11], while no association was observed with age, gender, in-hospital falls, delirium, and functional decline [11,67]. Other potential risk factors of a high number of PIMs at discharge were a high number of pre-admission PIMs, discharge to a location other than home, and discharge from a surgical service [1,6,68,69]. Length of ICU stay and mechanical ventilation had a positive influence on the number of PIMs used by acutely ill older patients [11,63,69]. In the study of Scales et al, the greatest absolute risk factor across all medication groups was longer hospitalization. The increased OR for medication continuation after a hospitalization lasting more than 7 days ranged from 2.03 (95% CI 1.94–2.11) for respiratory inhalers to 6.35 (95% CI 5.91–6.82) for antipsychotic medications [51].
Inappropriate Medications: Where and How to Intervene?
Early detection of PIMs may prevent adverse drug events and improve geriatric care in older adults [13,70]. PIM prevalence can often be a useful indicator of prescribing quality [2]. Appropriate interventions and an improved quality of prescribed medications require appropriate assessment tools to decrease the number of patients discharged on these medications [71,72]. Medication reconciliation is the process of avoiding inadvertent inconsistencies within a patient’s drug regimen, which can occur during transitions in different setting of care [73]. A multidisciplinary team should be involved in the medication reconciliation at each care transition to reevaluate medications use according to the clinical conditions, cognitive/functional status and the coexistence of geriatric syndromes (eg, dementia, malnutrition, delirium, urinary incontinence, frailty) (Figure).
Criteria for the Evaluation of Inappropriate Medications Prescription
Explicit criteria derived from expert reports or published reviews are available (Table 2).

Beers criteria PIMs have been found to be associated with poor health outcomes, including confusion, falls, and mortality [7,75,78]. The STOPP (Screening Tool of Older Person’s potentially inappropriate Prescriptions) and START (Screening Tool to Alert doctors to the Right Treatment) are evidence-based sets of criteria that were developed in Ireland and updated in October 2014, including some of the new criteria for direct oral anticoagulants, drugs affecting or affected by renal system and anti-muscarinic/anticholinergic agents [79]. 
Several other sets of criteria have been published to identify PIMs, such as the FORTA (Fit for the Aged) and the PRISCUS [86] criteria. FORTA allows a disease-related evaluation revealing over-treatment and under-treatment, and medications are graded as follows: A, indispensable drug, clear-cut benefit in terms of efficacy/safety ratio proven in elderly patients for a given indication; B, drugs with proven or obvious efficacy in the elderly, but limited extent of effect or safety concerns; C, drugs with questionable efficacy/safety profiles in the elderly which should be avoided or omitted in the presence of too many drugs or side effects; D, avoid in the elderly, omit first, refer also to negative listings. Negative lists such as PRISCUS, which provide an explicit listing of drugs, independent of the diagnosis, are easy to use. On the other hand, constant updates are needed, and such lists carry the risk of an assumption that drugs not listed would be appropriate in every case [87]. Both sets of criteria have in common that they refer to long-term medication and drugs frequently used during the inpatient stay, such as antibiotics, are hardly taken into account [87].
The Medication Appropriateness Index measures overall prescribing quality through 10 separate but interrelated domains [8]. Three components are used to detect PIMs: indication, effectiveness, and duplication. However, it does not give any precise guidance in relation to specific medicines and therefore has limited application for objectively defining PIMs.
Another prescribing quality assessment tool is the Inappropriate Prescribing in the Elderly Tool (IPET), which consists of a list of the 14 most prevalent prescription errors identified from an extensive list of inappropriate prescription instances drawn up by an expert Canadian Consensus Panel [88,89].
Another approach to assess the appropriateness of drugs prescribed for older people is the use of Drug Utilization Reviews (DURs) [16]. DURs use consensus opinion by drug therapy experts to define standards or explicit criteria for a single drug, class of drugs, or group of drugs [16]. DURs typically use retrospective information from large, nonclinical administrative databases to identify problems such as dosage range, duration, therapeutic duplication, and drug interactions [90, 91]. Monane et al [92] evaluated a program designed to decrease the use of PIMs among the elderly through a computerized online DUR database. Computer alerts triggered telephone calls to physicians by pharmacists to discuss a potential problem and any therapeutic substitution options. From a total of 43,007 telepharmacy calls generated by the alerts, they were able to reach 19,368 physicians regarding 24,266 alerts (56%). The rate of change to a more appropriate therapeutic agent was 24% (5860), but ranged from 40% for long half-life benzodiazepines to 2% to 7% for drugs that theoretically were contraindicated by patients’ self-reported history [92].
Computerized Support Systems to Reduce Inappropriate Prescribing in the Elderly
Other potential solutions for reducing inappropriate medications may include continuing medical education, electronic medical records surveillance, routine clinical evaluation, and/or improved hand-off communication between discharging and accepting providers. Incorporating this assessment of medication appropriateness into the medication reconciliation process when patients are discharged or transferred out of the ICU has the potential to enhance patient safety [21,93]. A randomized controlled trial conducted by Raebel et al [94] reported the effectiveness of a computerized pharmacy alert system plus collaboration between health care professionals for decreasing potentially inappropriate medication dispensing in elderly patients. Another study showed that computer-based access to complete drug profiles and alerts about potential prescribing problems reduced the occurrence of potentially inappropriate prescriptions [95]. A summary of these studies is shown in Table 3. 
Interdisciplinary Teams to Reduce Inappropriate Prescribing in the Elderly
Some studies evaluated the effect of multidisciplinary teamwork in improving inappropriate medication prescribing in the elderly (Table 4). 

Pharmacists in hospitals can play a significant role in the initiation of changes to patient’s therapy and management [11] (Table 5).
Mattison et al recently emphasized that studies of PIMs should determine scenarios in which it is appropriate to prescribe PIMs, moving beyond simply labeling some medications as “potentially inappropriate,” since some PIMs are appropriately prescribed in specific clinical situations [109]. Morandi et al showed that the positive predictive value (PPV) depends on the drug type. Thus, when developing a screening system, one cannot be concerned only with high negative predictive value (NPV), one must consider PPV as well [6]. Screening tools that include medication classes with low PPV will generate false positive “flags” or warnings, which could lead to misguided clinical decisions [6]. The fact that many PIMs are not AIMs also reveals the value of using a multidisciplinary team to identify AIMs from lists of PIMs generated when discharge medication lists are screened [6,110]. Thus, a multidisciplinary team is needed to consider the clinical context to distinguish PIMs from AIMs [6]. Of course, such a team is not available in some settings; when resources are limited, knowledge of which PIMs are most likely AIMs (ie, have high PPVs) could guide the development of computer-based decision support systems or other surveillance approaches that are efficient in that particular setting [6].
Approaches for optimizing prescribing in this population mainly depend on patient needs and comorbidities and most available data are derived from randomized controlled trials involving a single drug. Such trials do not take into account the confounding effects of multiple comorbidities and patient preferences. Therefore, approaches for optimizing prescription management that are available for and validated in younger patients are not applicable to elderly subjects [3,111].
Conclusion
Clinicians should seek to identify and discontinue AIMs at 3 important transitions during a critically ill elderly patient’s hospital course: at the time of hospital or ICU admission; at ICU discharge; and at hospital discharge. The patient’s clinical situation should be reviewed at every transition points, ideally by a multidisciplinary team of clinicians, to judge the appropriateness of each PIM [6]. After the hospital discharge, patient’s medications should be then reviewed by a multidisciplinary team and/or by the primary care physician according to the final discharge destination (ie, home, nursing home, rehabilitation) by using any of the validated tools. Regardless of the approach, it is clear that standardized care processes, including enhanced clinical decision support, are necessary to ensure that physicians do not continue exposing our patients to unnecessary medications and harm after discharge.
Corresponding author: Alessandro Morandi, MD, MPH, [email protected].
Funding/support: Dr. Pandiharipande is supported by National Institutes of Health HL111111 (Bethesda, MD) and by the VA Clinical Science Research and Development Service (Washington, DC) and the National Institutes of Health AG027472 and AG035117 (Bethesda, MD).
Financial disclosures: Dr. Pratik Pandharipande has received a research grant from Hospira Inc in collaboration with the NIH.
1. Lang PO, Hasso Y, Drame M, et al. Potentially inappropriate prescribing including under-use amongst older patients with cognitive or psychiatric co-morbidities. Age Ageing 2010;39:373–81.
2. Spinewine A, Schmader KE, Barber N, et al. Appropriate prescribing in elderly people: how well can it be measured and optimised? Lancet 2007;370:173–84.
3. Lang PO, Vogt-Ferrier N, Hasso Y, et al. Interdisciplinary geriatric and psychiatric care reduces potentially inappropriate prescribing in the hospital: interventional study in 150 acutely ill elderly patients with mental and somatic comorbid conditions. J Am Med Dir Assoc 2012;13:406 e1–7.
4. Cecile M, Seux V, Pauly V, et al. [Adverse drug events in hospitalized elderly patients in a geriatric medicine unit: study of prevalence and risk factors]. Rev Med Interne 2009;30:393–400.
5. Tosato M, Landi F, Martone AM, et al. Potentially inappropriate drug use among hospitalised older adults: results from the CRIME study. Age Ageing 2014;43:767–73.
6. Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013;61:1128–34.
7. Fick DM, Mion LC, Beers MH, Waller JL. Health outcomes associated with potentially inappropriate medication use in older adults. Res Nurs Health 2008;31:42–51.
8. Hanlon JT, Schmader KE, Samsa GP, et al. A method for assessing drug therapy appropriateness. J Clin Epidemiol 1992;45:1045–51.
9. Lau DT, Kasper JD, Potter DEB, et al. Hospitalization and death associated with potentially inappropriate medication prescriptions among elderly nursing home residents. Arch Intern Med 2005;165:68–74.
10. Wright RM, Roumani YF, Boudreau R, et al. Effect of central nervous system medication use on decline in cognition in community-dwelling older adults: findings from the Health, Aging and Body Composition Study. J Am Geriatr Soc 2009;57:243–50.
11. Poudel A, Peel NM, Nissen L, et al. Potentially inappropriate prescribing in older patients discharged from acute care hospitals to residential aged care facilities. Ann Pharmacother 2014;48:1425–33.
12. Wahab MS, Nyfort-Hansen K, Kowalski SR. Inappropriate prescribing in hospitalised Australian elderly as determined by the STOPP criteria. Int J Clin Pharm 2012;34:855–62.
13. O’Mahony D, Gallagher PF. Inappropriate prescribing in the older population: need for new criteria. Age Ageing 2008;37:138–41.
14. Mangoni AA, Jansen PA, Jackson SH. Under-representation of older adults in pharmacokinetic and pharmacodynamic studies: a solvable problem? Exp Rev Clin Pharmacol 2013;6:35–9.
15. Klotz U. The elderly--a challenge for appropriate drug treatment. Eur J Clin Pharmacol 2008;64225–6.
16. Hanlon JT, Schmader KE, Ruby CM, Weinberger M. Suboptimal prescribing in older inpatients and outpatients. J Am Geriatr Soc 2001;49:200–9.
17. Hubbard RE, O’Mahony MS, Woodhouse KW. Medication prescribing in frail older people. Eur J Clin Pharmacol 2013;69:319–26.
18. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012;60:616–31.
19. Page RL, Ruscin JM. The risk of adverse drug events and hospital-related morbidity and mortality among older adults with potentially inappropriate medication use. Am J Geriatr Pharmacother 2006;4:297–305.
20. Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ 2004;329:15–9.
21. Morandi A, Vasilevskis EE, Pandharipande PP, et al. Inappropriate medications in elderly ICU survivors: where to intervene? Arch Intern Med 2011;171:1032–4.
22. Page RL 2nd, Linnebur SA, Bryant LL, Ruscin JM. Inappropriate prescribing in the hospitalized elderly patient: defining the problem, evaluation tools, and possible solutions. Clin Interv Aging 2010;5:75–87.
23. Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med 2013;369:1306–16.
24. Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA 2010;303:763–70.
25. Swan JT, Fitousis K, Hall JB, et al. Antipsychotic use and diagnosis of delirium in the intensive care unit. Crit Care 2012;16:R84.
26. Jasiak KD, Middleton EA, Camamo JM, et al. Evaluation of discontinuation of atypical antipsychotics prescribed for ICU delirium. J Pharm Pract 2013;26:253–6.
27. Tomichek JE, Stollings JL, Pandharipande PP, et al. Antipsychotic prescribing patterns during and after critical illness: a prospective cohort study. Crit Care 2016;20:378.
28. Kram BL, Kram SJ, Brooks KR. Implications of atypical antipsychotic prescribing in the intensive care unit. J Crit Care 2015;30:814–8.
29. Flurie RW, Gonzales JP, Tata AL, et al. Hospital delirium treatment: Continuation of antipsychotic therapy from the intensive care unit to discharge. Am J Health Syst Pharm 2015;72(23 Suppl 3):S133–9.
30. Rowe AS, Hamilton LA, Curtis RA, et al. Risk factors for discharge on a new antipsychotic medication after admission to an intensive care unit. J Crit Care 2015;30:1283–6.
31. Ray WA, Chung CP, Murray KT, et al. Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med 2009;360:225–35.
32. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med 2005;353:2335–41.
33. Marshall J, Herzig SJ, Howell MD, et al. Antipsychotic utilization in the intensive care unit and in transitions of care. J Crit Care 2016;33:119–24.
34. NIH State-of-the-science conference statement on manifestations and management of chronic insomnia in adults. NIH Consens State Sci Statements 2005;22:1–30.
35. Farrell B, Tsang C, Raman-Wilms L, et a;. What are priorities for deprescribing for elderly patients? Capturing the voice of practitioners: a modified delphi process. PLoS One 2015;10:e0122246.
36. Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006;104:21–6.
37. Zaal IJ, Devlin JW, Hazelbag M, et al. Benzodiazepine-associated delirium in critically ill adults. Intensive Care Med 2015;41:2130–7.
38. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306.
39. Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009;301:489–99.
40. Obiora E, Hubbard R, Sanders RD, Myles PR. The impact of benzodiazepines on occurrence of pneumonia and mortality from pneumonia: a nested case-control and survival analysis in a population-based cohort. Thorax 2013;68:163–70.
41. Sanders RD, Godlee A, Fujimori T, et al. Benzodiazepine augmented gamma-amino-butyric acid signaling increases mortality from pneumonia in mice. Crit Care Med 2013;41:1627–36.
42. Nakafero G, Sanders RD, Nguyen-Van-Tam JS, Myles PR. Association between benzodiazepine use and exacerbations and mortality in patients with asthma: a matched case-control and survival analysis using the United Kingdom Clinical Practice Research Datalink. Pharmacoepidemiol Drug Saf 2015;24:793–802.
43. Hartikainen S, Lonnroos E, Louhivuori K. Medication as a risk factor for falls: critical systematic review. J Gerontol A Biol Sci Med Sci 2007;62:1172–81.
44. Pierfitte C, Macouillard G, Thicoipe M, et al. Benzodiazepines and hip fractures in elderly people: case-control study. BMJ 2001;322:704–8.
45. Landi F, Onder G, Cesari M, et al. Psychotropic medications and risk for falls among community-dwelling frail older people: an observational study. J Gerontol A Biol Sci Med Sci 2005;60:622–6.
46. Hanlon JT, Horner RD, Schmader KE, et al. Benzodiazepine use and cognitive function among community-dwelling elderly. Clin Pharmacol Ther 1998;64:684–92.
47. Greenblatt DJ, Harmatz JS, Shapiro L, et al. Sensitivity to triazolam in the elderly. N Engl J Med 1991;324:1691–8.
48. Bertz RJ, Kroboth PD, Kroboth FJ, et al. Alprazolam in young and elderly men: sensitivity and tolerance to psychomotor, sedative and memory effects. J Pharmacol Exp Ther 1997;281:1317–29.
49. Gray SL, LaCroix AZ, Blough D, et al. Is the use of benzodiazepines associated with incident disability? J Am Geriatr Soc 2002;50:1012–8.
50. Gray SL, LaCroix AZ, Hanlon JT, et al. Benzodiazepine use and physical disability in community-dwelling older adults. J Am Geriatr Soc 2006;54:224–30.
51. Scales DC, Fischer HD, Li P, et al. Unintentional continuation of medications intended for acute illness after hospital discharge: a population-based cohort study. J Gen Intern Med 2016;31:196–202.
52. Hamilton H, Gallagher P, Ryan C, et al. Potentially inappropriate medications defined by STOPP criteria and the risk of adverse drug events in older hospitalized patients. Arch Intern Med 2011;171:1013–9.
53. Hanlon JT, Artz MB, Pieper CF, et al. Inappropriate medication use among frail elderly inpatients. Ann Pharmacother 2004;38:9–14.
54. Ahrens D, Behrens G, Himmel W, et al. Appropriateness of proton pump inhibitor recommendations at hospital discharge and continuation in primary care. Int J Clin Pract 2012;66:767–73.
55. McDonald EG, Milligan J, Frenette C, Lee TC. Continuous proton pump inhibitor therapy and the associated risk of recurrent Clostridium difficile infection. JAMA Intern Med 2015;175:784–91.
56. Gulmez SE, Holm A, Frederiksen H, et al. Use of proton pump inhibitors and the risk of community-acquired pneumonia: a population-based case-control study. Arch Intern Med 2007;167:950–5.
57. Laheij RJ, Sturkenboom MC, Hassing RJ, et al. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004;292:1955–60.
58. Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006;296:2947–53.
59. Abrahamsen B, Eiken P, Eastell R. Proton pump inhibitor use and the antifracture efficacy of alendronate. Arch Intern Med 2011;171:998–1004.
60. Charlot M, Grove EL, Hansen PR, et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study. BMJ 2011;342:d2690.
61. Dial S, Delaney JA, Barkun AN, Suissa S. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA 2005;294:2989–95.
62. Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol 2007;102:2047–56; quiz 57.
63. Pavlov A, Muravyev R, Amoateng-Adjepong Y, Manthous CA. Inappropriate discharge on bronchodilators and acid-blocking medications after ICU admission: importance of medication reconciliation. Respir Care 2014;59:1524–9.
64. Baldoni Ade O, Ayres LR, Martinez EZ, et al. Factors associated with potentially inappropriate medications use by the elderly according to Beers criteria 2003 and 2012. Int J Clin Pharm 2014;36:316–24.
65. Gallagher PF, Barry PJ, Ryan C, et al. Inappropriate prescribing in an acutely ill population of elderly patients as determined by Beers’ Criteria. Age Ageing 2008;37:96–101.
66. Montastruc F, Duguet C, Rousseau V, et al. Potentially inappropriate medications and adverse drug reactions in the elderly: a study in a PharmacoVigilance database. Eur J Clin Pharmacol 2014;70:1123–7.
67. Ruggiero C, Dell’Aquila G, Gasperini B, et al. Potentially inappropriate drug prescriptions and risk of hospitalization among older, Italian, nursing home residents: the ULISSE project. Drugs Aging 2010;27:747–58.
68. Onder G, Landi F, Cesari M, et al. Inappropriate medication use among hospitalized older adults in Italy: results from the Italian Group of Pharmacoepidemiology in the Elderly. Eur J Clin Pharmacol 2003;59:157–62.
69. Harugeri A, Joseph J, Parthasarathi G, et al. Potentially inappropriate medication use in elderly patients: A study of prevalence and predictors in two teaching hospitals. J Postgrad Med 2010;56:186–91.
70. Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 2010;170:1648–54.
71. Dimitrow MS, Airaksinen MS, Kivela SL, et al. Comparison of prescribing criteria to evaluate the appropriateness of drug treatment in individuals aged 65 and older: a systematic review. J Am Geriatr Soc 2011;59:1521–30.
72. Levy HB, Marcus EL, Christen C. Beyond the beers criteria: A comparative overview of explicit criteria. Ann Pharmacother 2010;44:1968–75.
73. Marengoni A, Nobili A, Onder G. Best practices for drug prescribing in older adults: a call for action. Drugs Aging 2015;32:887–90.
74. Poudel A, Hubbard RE, Nissen L, Mitchell C. Frailty: a key indicator to minimize inappropriate medication in older people. QJM 2013;106:969–75.
75. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015;63:2227–46.
76. Beers MH, Ouslander JG, Rollingher I, et al. Explicit criteria for determining inappropriate medication use in nursing home residents. UCLA Division of Geriatric Medicine. Arch Intern Med 1991;151:1825–32.
77. Blanco-Reina E, Ariza-Zafra G, Ocana-Riola R, Leon-Ortiz M. 2012 American Geriatrics Society Beers criteria: enhanced applicability for detecting potentially inappropriate medications in European older adults? A comparison with the Screening Tool of Older Person’s Potentially Inappropriate Prescriptions. J Am Geriatr Soc 2014;62:1217–23.
78. Stockl KM, Le L, Zhang S, Harada AS. Clinical and economic outcomes associated with potentially inappropriate prescribing in the elderly. Am J Manag Care 2010;16:e1–10.
79. Hill-Taylor B, Sketris I, Hayden J, et al. Application of the STOPP/START criteria: a systematic review of the prevalence of potentially inappropriate prescribing in older adults, and evidence of clinical, humanistic and economic impact. J Clin Pharm Ther 2013;38:360–72.
80. Barry PJ, Gallagher P, Ryan C, O’Mahony D. START (screening tool to alert doctors to the right treatment)--an evidence-based screening tool to detect prescribing omissions in elderly patients. Age Ageing 2007;36:632–8.
81. Gallagher P, Ryan C, Byrne S, Kennedy J, O’Mahony D. STOPP (Screening Tool of Older Person’s Prescriptions) and START (Screening Tool to Alert doctors to Right Treatment). Consensus validation. Int J Clin Pharmacol Ther 2008;46:72–83.
82. Haag JD, Davis AZ, Hoel RW, et al. Impact of pharmacist-provided medication therapy management on healthcare quality and utilization in recently discharged elderly patients. Am Health Drug Benefits 2016;9:259–68.
83. Gillespie U, Alassaad A, Hammarlund-Udenaes M, et al. Effects of pharmacists’ interventions on appropriateness of prescribing and evaluation of the instruments’ (MAI, STOPP and STARTs’) ability to predict hospitalization--analyses from a randomized controlled trial. PLoS One 2013;8:e62401.
84. Petrarca AM, Lengel AJ, Mangan MN. Inappropriate medication use in the elderly. Consult Pharm 2012;27:583–6.
85. Lavan AH, Gallagher P, Parsons C, O’Mahony D. STOPPFrail (Screening Tool of Older Persons Prescriptions in Frail adults with limited life expectancy): consensus validation. Age Ageing 2017;46:600–7.
86. Holt S, Schmiedl S, Thurmann PA. Potentially inappropriate medications in the elderly: the PRISCUS list. Dtsch Arztebl Int 2010;107:543–51.
87. Wickop B, Harterich S, Sommer C, et al. Potentially inappropriate medication use in multimorbid elderly inpatients: differences between the FORTA, PRISCUS and STOPP ratings. Drugs Real World Outcome 2016;3:317–25.
88. Naugler CT, Brymer C, Stolee P, Arcese ZA. Development and validation of an improving prescribing in the elderly tool. Can J Clin Pharmacol 2000;7:103–7.
89. Barry PJ, O’Keefe N, O’Connor KA, O’Mahony D. Inappropriate prescribing in the elderly: a comparison of the Beers criteria and the improved prescribing in the elderly tool (IPET) in acutely ill elderly hospitalized patients. J Clin Pharm Ther 2006;31:617–26.
90. Knapp DA. Development of criteria for drug utilization review. Clin Pharmacol Ther 1991;50(5 Pt 2):600–2.
91. Lipton HL, Bird JA. Drug utilization review in ambulatory settings: state of the science and directions for outcomes research. Med Care 1993;31:1069–82.
92. Monane M, Matthias DM, Nagle BA, Kelly MA. Improving prescribing patterns for the elderly through an online drug utilization review intervention: a system linking the physician, pharmacist, and computer. JAMA 1998;280:1249–52.
93. Kaur S, Mitchell G, Vitetta L, Roberts MS. Interventions that can reduce inappropriate prescribing in the elderly: a systematic review. Drugs Aging 2009;26:1013–28.
94. Raebel MA, Charles J, Dugan J, et al. Randomized trial to improve prescribing safety in ambulatory elderly patients. J Am Geriatr Soc 2007;55:977–85.
95. Tamblyn R, Huang A, Perreault R, et al. The medical office of the 21st century (MOXXI): effectiveness of computerized decision-making support in reducing inappropriate prescribing in primary care. CMAJ 2003;169:549–56.
96. Dalleur O, Boland B, Losseau C, et al. Reduction of potentially inappropriate medications using the STOPP criteria in frail older inpatients: a randomised controlled study. Drugs Aging 2014;31:291–8.
97. Schmader KE, Hanlon JT, Pieper CF, et al. Effects of geriatric evaluation and management on adverse drug reactions and suboptimal prescribing in the frail elderly. Am J Med 2004;116:394–401.
98. Crotty M, Halbert J, Rowett D, et al. An outreach geriatric medication advisory service in residential aged care: a randomised controlled trial of case conferencing. Age Ageing 2004;33:612–7.
99. Allard J, Hebert R, Rioux M, et al. Efficacy of a clinical medication review on the number of potentially inappropriate prescriptions prescribed for community-dwelling elderly people. CMAJ 2001;164:1291–6.
100. Spinewine A, Swine C, Dhillon S, et al. Effect of a collaborative approach on the quality of prescribing for geriatric inpatients: a randomized, controlled trial. J Am Geriatr Soc 2007;55:658–65.
101. Elliott RA, Woodward MC, Oborne CA. Improving benzodiazepine prescribing for elderly hospital inpatients using audit and multidisciplinary feedback. Intern Med J 2001;31:529–35.
102. Saltvedt I, Spigset O, Ruths S, et al. Patterns of drug prescription in a geriatric evaluation and management unit as compared with the general medical wards: a randomised study. Eur J Clin Pharmacol 2005;61:921–8.
103. Hanlon JT, Weinberger M, Samsa GP, et al. A randomized, controlled trial of a clinical pharmacist intervention to improve inappropriate prescribing in elderly outpatients with polypharmacy. Am J Med 1996;100:428–37.
104. Lipton HL, Bero LA, Bird JA, McPhee SJ. The impact of clinical pharmacists’ consultations on physicians’ geriatric drug prescribing. A randomized controlled trial. Med Care 1992;30:646–58.
105. Krska J, Cromarty JA, Arris F, et al. Pharmacist-led medication review in patients over 65: a randomized, controlled trial in primary care. Age Ageing 2001;30:205–11.
106. Brown BK, Earnhart J. Pharmacists and their effectiveness in ensuring the appropriateness of the chronic medication regimens of geriatric inpatients. Consult Pharm 2004;19:432–6.
107. Belfield KD, Kuyumjian AG, Teran R, et al. Impact of a collaborative strategy to reduce the inappropriate use of acid suppressive therapy in non-intensive care unit patients. Ann Pharmacother 2017;51:577–83.
108. Crotty M, Rowett D, Spurling L, et al. Does the addition of a pharmacist transition coordinator improve evidence-based medication management and health outcomes in older adults moving from the hospital to a long-term care facility? Results of a randomized, controlled trial. Am J Geriatr Pharmacother 2004;2:257–64.
109. Mattison MLP, Afonso KA, Ngo LH, Mukamal KJ. Preventing potentially inappropriate medication use in hospitalized older patients with a computerized provider order entry warning system. Arch Intern Med 2010;170:1331–6.
110. Kaboli PJ, Hoth AB, McClimon BJ, Schnipper JL. Clinical pharmacists and inpatient medical care: a systematic review. Arch Intern Med 2006;166:955–64.
111. Tinetti ME, Bogardus ST Jr, Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004;351:2870–4.
112. Gokula M, Holmes HM. Tools to reduce polypharmacy. Clin Geriatr Med 2012;28:323–41.
From the Division of Allergy, Pulmonary, and Critical Care Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN (Dr. Marra), Division of Anesthesiology Critical Care Medicine, Vanderbilt University Medical Center, Nashville, TN (Dr. Hayhurst, Dr. Hughes, Dr. Pandharipande), Department of Clinical and Experimental Science, University of Brescia, Brescia, Italy (Dr. Marengoni), School of Medicine and Surgery,
University of Milano-Bicocca, Milan, Italy (Dr. Bellelli), and Rehabilitation and Aged Care Unit Hospital Ancelle, Cremona, Italy (Dr. Morandi).
Abstract
- Objective: To present an overview of the phenomenon of inappropriate medication prescription in older critically ill patients and examine possible strategies of intervention.
- Methods: Review of the literature.
- Results: Polypharmacy and inappropriate prescribing of medications in older persons may lead to a significant risk of adverse drug-related events and mortality. The intensive care unit (ICU) is often the place where potentially inappropriate medications (PIMs) are first prescribed. Common PIMs at ICU discharge are antipsychotics, benzodiazepines, opioids, anticholinergic medications, antidepressants, and drugs causing orthostatic hypotension. Different classes of medications, typically intended for short-term use, are sometimes inappropriately continued after discharge from the hospital. At admission, potential risk factors for PIM are multiple morbidities, polypharmacy, frailty and cognitive decline; at discharge, a high number of pre-admission PIMs, discharge to a location other than home, discharge from a surgical service, longer length of ICU and hospital stay, and mechanical ventilation. Inappropriate prescribing in older patients can be detected through either the use of explicit criteria, drug utilization reviews, and multidisciplinary teams, including a geriatrician and/or the involvement of a clinical pharmacist.
- Conclusion: Use of PIMs may be common in critical patients, both on admission and at discharge from ICU. Therapeutic reconciliation is recommended at every transition of care (eg, at hospital or ICU admission and discharge) in order to improve appropriateness of prescription.
Key words: elderly; intensive care unit; inappropriate medications; antipsychotics.
Since older persons are often affected by multiple chronic diseases and are prescribed several medications, the quality and safety of prescribing these medications has become a global health care issue [1–4]. Polypharmacy and inappropriate prescribing of medications among the elderly is receiving significant attention in the medical literature [5,6]. Inappropriate medications in the elderly can lead to falls, cognitive impairment and delirium, poorer health status, and higher mortality [7–10]. Medications are considered potentially inappropriate when (a) the risks of treatment outweigh the benefits [11], (b) they are prescribed for periods longer than clinically indicated or without any clear indication, (c) they are not prescribed when indicated [12], and (d) they are likely to interact with other drugs and diseases. Medications included in this category are often referred to as potentially inappropriate medications (PIMs), as in some situations their use is justified; however, if the risk of harm from the drug is judged to outweigh the potential clinical benefit after an individual patient’s clinical circumstances are considered, these drugs are considered “actually inappropriate medications” (AIMs) [6].
Advancing age is associated with substantial pharmacokinetic and pharmacodynamics changes, such as altered distribution volumes and altered permeability of the blood-brain barrier, impaired liver metabolism and renal capacity, up- and down-regulation of target receptors, transmitters, and signaling pathways changes, impaired homeostasis, and increased risk of adverse drug reactions (ADRs) that lead to increased mortality and morbidity and higher health care costs [2,11,13–19]. Studies show that ADRs cause approximately 5% of hospital admissions in the general population, but the percentage rises to 10% in older persons [20].
Avoiding PIMs represents a strategy aimed at reducing drug-related mortality and morbidity. This article provides an overview of the phenomenon of inappropriate medication prescription in older critically ill patients and examines available strategies of intervention.
Inappropriate Medications at ICU Discharge
Though PIMs and AIMs may be identified at the time of hospital discharge, the intensive care unit (ICU) is often the place where these medications are first prescribed [21]. Acute hospitalization may increase PIM prescribing because of newly prescribed medications, the presence of multiple prescribers, inadequate medication reconciliation, and a lack of care coordination among inpatient providers or in the transition back to outpatient care [22)].
A known complication of critical illness and ICU stay is a significant increase in psychological symptoms, sleep cycle alterations, delirium, and cognitive impairment, which may be associated with increased prescription of specific PIMs, such as antipsychotics or benzodiazepines [6,23,24]. Despite the lack of reliable evidence supporting their use in the ICU, antipsychotic agents are used routinely in ICU patients [25] to treat a variety of conditions, such as substance withdrawal, agitation not responding to other therapies, or delirium. Results from a multicenter study of 164,996 hospitalizations across 71 academic medical centers in the US showed that 1 out of 10 ICU patients received an antipsychotic during their hospital stay [25]. Jasiak et al estimated that one-third of patients initiated on an atypical antipsychotic therapy for ICU delirium received a hospital discharge prescription for these medications, with a potential annual outpatient medication cost of approximately $2255 per patient [26].
One potential consequence of antipsychotic use in the ICU is their continuation after the transition to other clinical settings, including discharge from the hospital [27] (Table 1).
When examining the specific factors that may contribute to a patient being discharged on an antipsychotic, authors found that the specific antipsychotic used correlated with risk of continuation [27,30], with atypical antipsychotics having a greater likelihood of being continued than haloperidol [27,30]. Possible explanation for these results could be that physicians perceive less long-term risk from atypical agents, so may be more likely to continue them on discharge [30]. However, such an approach is not always safe. Indeed, although atypical antipsychotic agents tend to cause less tardive dyskinesia, they are known to be associated with similar rates of other adverse events compared with typical agents and have been linked to an increased risk of sudden cardiac death and pneumonia in the elderly [31,32].
Other factors independently associated with being discharged on a new antipsychotic medication were the severity of the acute illness as measured with the Acute Physiology and Chronic Health Evaluation II score at ICU admission (odds ratio [OR] 1 [95% confidence interval {CI}, 1.0–1.1]) and days treated with benzodiazepines (OR 1.1 [95% CI, 1.0–1.14]) [30]. Conversely, perhaps due to different practice patterns, Tomichek et al did not find an association between benzodiazepines administration and antipsychotic prescription at discharge in post hoc analyses [27].
Another possible reason for antipsychotic continuation may reside in the indication chosen [33]. Antipsychotic agents have sedative properties and they might be used to optimize sleep during hospitalization, despite the lack of evidence to support this indication [34]. Other factors potentially contributing to continuation of antipsychotics may include persistent delirium and agitation, newly diagnosed psychiatric illness, and difficulties experienced by physicians in deprescribing [35] with improper/incomplete medication reconciliation [33].
The continuation of antipsychotic therapy increased 30-day readmission rates in patients compared to those who had therapy stopped before discharge [33]. In addition to the well-described cardiac effects (prolonged QT interval), neuroleptic malignant syndrome and extrapyramidal symptoms may also occur, and longer-term use can predispose patients to metabolic disturbances, falls, and increase the risk of death in elderly patients with dementia [31].
Benzodiazepines and sedative hypnotics are commonly used to treat insomnia and agitation in older adults despite significant risk. Benzodiazepine administration was found to be an independent risk factor for a daily transition to delirium [36,37]. Pandharipande et al reported that every unit dose of lorazepam was associated with a higher risk for daily transition to delirium (OR 1.2, 95% CI 1.1–1.4, P = 0.003) [36] in critically ill patients. A more recent analysis found for every 5 mg of midazolam administered to a patient who is awake and without delirium, there is a 4% chance that this patient will develop delirium the next ICU day [37].
Given that the risk for benzodiazepine-associated delirium is dose-dependent, clinicians should use strategies known to reduce the daily number of benzodiazepines administered that often includes the use of a sedative associated with less delirium occurrence, such as dexmedetomidine or propofol [38]. Evidence has shown that long-term use of benzodiazepines has little benefit with many risks, including an increased susceptibility to spontaneous bacterial infection [39,40] and mortality in the setting of infection [41]. Nakafero et al showed that exposure to benzodiazepines was associated with increased occurrence of both influenza-like-illness–related pneumonia and mortality. Benzodiazepine use was associated also with increased occurrence of asthma exacerbation and with increased all-cause mortality during a median follow-up of 2 years in a cohort of asthmatic patients [42] as well with an increased risk of pneumonia and long-term mortality in patients with a prior diagnosis of community- acquired pneumonia [40]. Long-term use of benzodiazepines is also associated with increased risk of falls [43–45], cognitive impairment [46–48] and disability [49,50].
Other common types of PIMs at ICU discharge were opioids, anticholinergic medications, antidepressants, and drugs causing orthostatic hypotension [6]. Of the anticholinergic AIMs, H2 blockers (61%) and promethazine (15%) were the most common [6]. Only 16% of opioids, 23% of antidepressants, and 10% of drugs causing orthostatic hypotension were found to be actually inappropriate after the patient’s circumstances were considered (eg, postoperative pain control, a new diagnosis of major depressive disorder) [6].
Inappropriate Medications at Hospital Discharge
Medications typically intended for short-term use during acute illness are sometimes continued after discharge without documented indication [51]. Poudel et al found that in 206 patients 70 years of age and older discharged to residential aged care facilities from acute care, at least 1 PIM was identified in 112 (54.4%) patients on admission and 102 (49.5%) patients on discharge [11]. Commonly prescribed PIM categories, at both admission and discharge, were central nervous system, cardiovascular, gastrointestinal, and respiratory drugs and analgesics [6,11,52,53]. Of all medications prescribed at admission (1728), 10.8% were PIMs, and at discharge, of 1759 medications, 9.6% were PIMs. Of the total 187 PIMs on admission, 56 (30%) were stopped, and 131 (70%) were continued; 32 new PIMs were introduced [11].
Morandi et al in 2011 conducted a prospective cohort study including 120 patients age ≥ 60 who were discharged after receiving care in a medical, surgical, or cardiovascular ICU for shock or respiratory failure. The percentage of patients prescribed at least 1 PIM increased from 66% at pre-admission to 85% at discharge. The number of patients with 0 PIMs dropped from 34% at preadmission to 14% at discharge, and the number of patients with 3 or more PIMS increased from 16% at preadmission to 37% at discharge. While it is possible that these drugs may be appropriate when started during an acute illness in the ICU (eg, stress ulcer prophylaxis with H2-antagonists in mechanically ventilated patients), most should have been discontinued at ICU and/or hospital discharge [21].
Inappropriate prescriptions of proton pump inhibitors (PPIs) in hospital and primary care have been widely reported [54,55]. In a study conducted by Ahrens et al in 31 primary care practices, for 58% (263/506) of patients discharged from 35 hospitals with a PPI recommendation in hospital discharge letters, an appropriate indication was missing. In 57% of these cases general practitioners followed this recommendation and continued the prescription for more than 1 month [54]. The strongest factor associated with appropriate and inappropriate continuation of PPI after discharge was PPI prescription prior to hospitalization [54]. Although PPIs are safe, they can cause adverse effects. PPI intake has been found to have a significant association with risk of community-acquired pneumonia [56,57], hip fractures [58], Clostridium difficile-associated diarrhea [55,61,62], and to reduce the therapeutic effects of bisphosphonates [59] and low-dose aspirin [60].
Unintentional medication continuation is not a problem isolated to a single drug class or disease [63]. Scales et al evaluated rates of and risk factors for potentially unintentional medication continuation following hospitalization in a population of elderly patients (≥ 66 years) [51]. They created distinct cohorts by identifying seniors not previously receiving four classes of medications typically used to treat or prevent complications of acute illness: antipsychotic medications; gastric acid suppressants (ie, histamine-2 blockers and proton pump inhibitors); benzodiazepines; and inhaled bronchodilators and steroids [51]. Prescription without documented indication occurred across all medication classes, from 12,209 patients (1.4 %) for antipsychotic medications to 34,140 patients (6.1 %) for gastric acid suppressants [51].
Several potential risk factors were considered. The relationship between multimorbidity and polypharmacy is well described in the literature, and several studies have identified a positive association between the number of drugs and the use of PIMs [64–66]. Conversely, Poudel et al did not find any association between polypharmacy and PIM use [11]. Associations were found between the use of PIMs, frailty status, and cognitive decline of patients at admission and at discharge [11], while no association was observed with age, gender, in-hospital falls, delirium, and functional decline [11,67]. Other potential risk factors of a high number of PIMs at discharge were a high number of pre-admission PIMs, discharge to a location other than home, and discharge from a surgical service [1,6,68,69]. Length of ICU stay and mechanical ventilation had a positive influence on the number of PIMs used by acutely ill older patients [11,63,69]. In the study of Scales et al, the greatest absolute risk factor across all medication groups was longer hospitalization. The increased OR for medication continuation after a hospitalization lasting more than 7 days ranged from 2.03 (95% CI 1.94–2.11) for respiratory inhalers to 6.35 (95% CI 5.91–6.82) for antipsychotic medications [51].
Inappropriate Medications: Where and How to Intervene?
Early detection of PIMs may prevent adverse drug events and improve geriatric care in older adults [13,70]. PIM prevalence can often be a useful indicator of prescribing quality [2]. Appropriate interventions and an improved quality of prescribed medications require appropriate assessment tools to decrease the number of patients discharged on these medications [71,72]. Medication reconciliation is the process of avoiding inadvertent inconsistencies within a patient’s drug regimen, which can occur during transitions in different setting of care [73]. A multidisciplinary team should be involved in the medication reconciliation at each care transition to reevaluate medications use according to the clinical conditions, cognitive/functional status and the coexistence of geriatric syndromes (eg, dementia, malnutrition, delirium, urinary incontinence, frailty) (Figure).
Criteria for the Evaluation of Inappropriate Medications Prescription
Explicit criteria derived from expert reports or published reviews are available (Table 2).
Beers criteria PIMs have been found to be associated with poor health outcomes, including confusion, falls, and mortality [7,75,78]. The STOPP (Screening Tool of Older Person’s potentially inappropriate Prescriptions) and START (Screening Tool to Alert doctors to the Right Treatment) are evidence-based sets of criteria that were developed in Ireland and updated in October 2014, including some of the new criteria for direct oral anticoagulants, drugs affecting or affected by renal system and anti-muscarinic/anticholinergic agents [79].
Several other sets of criteria have been published to identify PIMs, such as the FORTA (Fit for the Aged) and the PRISCUS [86] criteria. FORTA allows a disease-related evaluation revealing over-treatment and under-treatment, and medications are graded as follows: A, indispensable drug, clear-cut benefit in terms of efficacy/safety ratio proven in elderly patients for a given indication; B, drugs with proven or obvious efficacy in the elderly, but limited extent of effect or safety concerns; C, drugs with questionable efficacy/safety profiles in the elderly which should be avoided or omitted in the presence of too many drugs or side effects; D, avoid in the elderly, omit first, refer also to negative listings. Negative lists such as PRISCUS, which provide an explicit listing of drugs, independent of the diagnosis, are easy to use. On the other hand, constant updates are needed, and such lists carry the risk of an assumption that drugs not listed would be appropriate in every case [87]. Both sets of criteria have in common that they refer to long-term medication and drugs frequently used during the inpatient stay, such as antibiotics, are hardly taken into account [87].
The Medication Appropriateness Index measures overall prescribing quality through 10 separate but interrelated domains [8]. Three components are used to detect PIMs: indication, effectiveness, and duplication. However, it does not give any precise guidance in relation to specific medicines and therefore has limited application for objectively defining PIMs.
Another prescribing quality assessment tool is the Inappropriate Prescribing in the Elderly Tool (IPET), which consists of a list of the 14 most prevalent prescription errors identified from an extensive list of inappropriate prescription instances drawn up by an expert Canadian Consensus Panel [88,89].
Another approach to assess the appropriateness of drugs prescribed for older people is the use of Drug Utilization Reviews (DURs) [16]. DURs use consensus opinion by drug therapy experts to define standards or explicit criteria for a single drug, class of drugs, or group of drugs [16]. DURs typically use retrospective information from large, nonclinical administrative databases to identify problems such as dosage range, duration, therapeutic duplication, and drug interactions [90, 91]. Monane et al [92] evaluated a program designed to decrease the use of PIMs among the elderly through a computerized online DUR database. Computer alerts triggered telephone calls to physicians by pharmacists to discuss a potential problem and any therapeutic substitution options. From a total of 43,007 telepharmacy calls generated by the alerts, they were able to reach 19,368 physicians regarding 24,266 alerts (56%). The rate of change to a more appropriate therapeutic agent was 24% (5860), but ranged from 40% for long half-life benzodiazepines to 2% to 7% for drugs that theoretically were contraindicated by patients’ self-reported history [92].
Computerized Support Systems to Reduce Inappropriate Prescribing in the Elderly
Other potential solutions for reducing inappropriate medications may include continuing medical education, electronic medical records surveillance, routine clinical evaluation, and/or improved hand-off communication between discharging and accepting providers. Incorporating this assessment of medication appropriateness into the medication reconciliation process when patients are discharged or transferred out of the ICU has the potential to enhance patient safety [21,93]. A randomized controlled trial conducted by Raebel et al [94] reported the effectiveness of a computerized pharmacy alert system plus collaboration between health care professionals for decreasing potentially inappropriate medication dispensing in elderly patients. Another study showed that computer-based access to complete drug profiles and alerts about potential prescribing problems reduced the occurrence of potentially inappropriate prescriptions [95]. A summary of these studies is shown in Table 3.
Interdisciplinary Teams to Reduce Inappropriate Prescribing in the Elderly
Some studies evaluated the effect of multidisciplinary teamwork in improving inappropriate medication prescribing in the elderly (Table 4).
Pharmacists in hospitals can play a significant role in the initiation of changes to patient’s therapy and management [11] (Table 5).
Mattison et al recently emphasized that studies of PIMs should determine scenarios in which it is appropriate to prescribe PIMs, moving beyond simply labeling some medications as “potentially inappropriate,” since some PIMs are appropriately prescribed in specific clinical situations [109]. Morandi et al showed that the positive predictive value (PPV) depends on the drug type. Thus, when developing a screening system, one cannot be concerned only with high negative predictive value (NPV), one must consider PPV as well [6]. Screening tools that include medication classes with low PPV will generate false positive “flags” or warnings, which could lead to misguided clinical decisions [6]. The fact that many PIMs are not AIMs also reveals the value of using a multidisciplinary team to identify AIMs from lists of PIMs generated when discharge medication lists are screened [6,110]. Thus, a multidisciplinary team is needed to consider the clinical context to distinguish PIMs from AIMs [6]. Of course, such a team is not available in some settings; when resources are limited, knowledge of which PIMs are most likely AIMs (ie, have high PPVs) could guide the development of computer-based decision support systems or other surveillance approaches that are efficient in that particular setting [6].
Approaches for optimizing prescribing in this population mainly depend on patient needs and comorbidities and most available data are derived from randomized controlled trials involving a single drug. Such trials do not take into account the confounding effects of multiple comorbidities and patient preferences. Therefore, approaches for optimizing prescription management that are available for and validated in younger patients are not applicable to elderly subjects [3,111].
Conclusion
Clinicians should seek to identify and discontinue AIMs at 3 important transitions during a critically ill elderly patient’s hospital course: at the time of hospital or ICU admission; at ICU discharge; and at hospital discharge. The patient’s clinical situation should be reviewed at every transition points, ideally by a multidisciplinary team of clinicians, to judge the appropriateness of each PIM [6]. After the hospital discharge, patient’s medications should be then reviewed by a multidisciplinary team and/or by the primary care physician according to the final discharge destination (ie, home, nursing home, rehabilitation) by using any of the validated tools. Regardless of the approach, it is clear that standardized care processes, including enhanced clinical decision support, are necessary to ensure that physicians do not continue exposing our patients to unnecessary medications and harm after discharge.
Corresponding author: Alessandro Morandi, MD, MPH, [email protected].
Funding/support: Dr. Pandiharipande is supported by National Institutes of Health HL111111 (Bethesda, MD) and by the VA Clinical Science Research and Development Service (Washington, DC) and the National Institutes of Health AG027472 and AG035117 (Bethesda, MD).
Financial disclosures: Dr. Pratik Pandharipande has received a research grant from Hospira Inc in collaboration with the NIH.
From the Division of Allergy, Pulmonary, and Critical Care Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN (Dr. Marra), Division of Anesthesiology Critical Care Medicine, Vanderbilt University Medical Center, Nashville, TN (Dr. Hayhurst, Dr. Hughes, Dr. Pandharipande), Department of Clinical and Experimental Science, University of Brescia, Brescia, Italy (Dr. Marengoni), School of Medicine and Surgery,
University of Milano-Bicocca, Milan, Italy (Dr. Bellelli), and Rehabilitation and Aged Care Unit Hospital Ancelle, Cremona, Italy (Dr. Morandi).
Abstract
- Objective: To present an overview of the phenomenon of inappropriate medication prescription in older critically ill patients and examine possible strategies of intervention.
- Methods: Review of the literature.
- Results: Polypharmacy and inappropriate prescribing of medications in older persons may lead to a significant risk of adverse drug-related events and mortality. The intensive care unit (ICU) is often the place where potentially inappropriate medications (PIMs) are first prescribed. Common PIMs at ICU discharge are antipsychotics, benzodiazepines, opioids, anticholinergic medications, antidepressants, and drugs causing orthostatic hypotension. Different classes of medications, typically intended for short-term use, are sometimes inappropriately continued after discharge from the hospital. At admission, potential risk factors for PIM are multiple morbidities, polypharmacy, frailty and cognitive decline; at discharge, a high number of pre-admission PIMs, discharge to a location other than home, discharge from a surgical service, longer length of ICU and hospital stay, and mechanical ventilation. Inappropriate prescribing in older patients can be detected through either the use of explicit criteria, drug utilization reviews, and multidisciplinary teams, including a geriatrician and/or the involvement of a clinical pharmacist.
- Conclusion: Use of PIMs may be common in critical patients, both on admission and at discharge from ICU. Therapeutic reconciliation is recommended at every transition of care (eg, at hospital or ICU admission and discharge) in order to improve appropriateness of prescription.
Key words: elderly; intensive care unit; inappropriate medications; antipsychotics.
Since older persons are often affected by multiple chronic diseases and are prescribed several medications, the quality and safety of prescribing these medications has become a global health care issue [1–4]. Polypharmacy and inappropriate prescribing of medications among the elderly is receiving significant attention in the medical literature [5,6]. Inappropriate medications in the elderly can lead to falls, cognitive impairment and delirium, poorer health status, and higher mortality [7–10]. Medications are considered potentially inappropriate when (a) the risks of treatment outweigh the benefits [11], (b) they are prescribed for periods longer than clinically indicated or without any clear indication, (c) they are not prescribed when indicated [12], and (d) they are likely to interact with other drugs and diseases. Medications included in this category are often referred to as potentially inappropriate medications (PIMs), as in some situations their use is justified; however, if the risk of harm from the drug is judged to outweigh the potential clinical benefit after an individual patient’s clinical circumstances are considered, these drugs are considered “actually inappropriate medications” (AIMs) [6].
Advancing age is associated with substantial pharmacokinetic and pharmacodynamics changes, such as altered distribution volumes and altered permeability of the blood-brain barrier, impaired liver metabolism and renal capacity, up- and down-regulation of target receptors, transmitters, and signaling pathways changes, impaired homeostasis, and increased risk of adverse drug reactions (ADRs) that lead to increased mortality and morbidity and higher health care costs [2,11,13–19]. Studies show that ADRs cause approximately 5% of hospital admissions in the general population, but the percentage rises to 10% in older persons [20].
Avoiding PIMs represents a strategy aimed at reducing drug-related mortality and morbidity. This article provides an overview of the phenomenon of inappropriate medication prescription in older critically ill patients and examines available strategies of intervention.
Inappropriate Medications at ICU Discharge
Though PIMs and AIMs may be identified at the time of hospital discharge, the intensive care unit (ICU) is often the place where these medications are first prescribed [21]. Acute hospitalization may increase PIM prescribing because of newly prescribed medications, the presence of multiple prescribers, inadequate medication reconciliation, and a lack of care coordination among inpatient providers or in the transition back to outpatient care [22)].
A known complication of critical illness and ICU stay is a significant increase in psychological symptoms, sleep cycle alterations, delirium, and cognitive impairment, which may be associated with increased prescription of specific PIMs, such as antipsychotics or benzodiazepines [6,23,24]. Despite the lack of reliable evidence supporting their use in the ICU, antipsychotic agents are used routinely in ICU patients [25] to treat a variety of conditions, such as substance withdrawal, agitation not responding to other therapies, or delirium. Results from a multicenter study of 164,996 hospitalizations across 71 academic medical centers in the US showed that 1 out of 10 ICU patients received an antipsychotic during their hospital stay [25]. Jasiak et al estimated that one-third of patients initiated on an atypical antipsychotic therapy for ICU delirium received a hospital discharge prescription for these medications, with a potential annual outpatient medication cost of approximately $2255 per patient [26].
One potential consequence of antipsychotic use in the ICU is their continuation after the transition to other clinical settings, including discharge from the hospital [27] (Table 1).
When examining the specific factors that may contribute to a patient being discharged on an antipsychotic, authors found that the specific antipsychotic used correlated with risk of continuation [27,30], with atypical antipsychotics having a greater likelihood of being continued than haloperidol [27,30]. Possible explanation for these results could be that physicians perceive less long-term risk from atypical agents, so may be more likely to continue them on discharge [30]. However, such an approach is not always safe. Indeed, although atypical antipsychotic agents tend to cause less tardive dyskinesia, they are known to be associated with similar rates of other adverse events compared with typical agents and have been linked to an increased risk of sudden cardiac death and pneumonia in the elderly [31,32].
Other factors independently associated with being discharged on a new antipsychotic medication were the severity of the acute illness as measured with the Acute Physiology and Chronic Health Evaluation II score at ICU admission (odds ratio [OR] 1 [95% confidence interval {CI}, 1.0–1.1]) and days treated with benzodiazepines (OR 1.1 [95% CI, 1.0–1.14]) [30]. Conversely, perhaps due to different practice patterns, Tomichek et al did not find an association between benzodiazepines administration and antipsychotic prescription at discharge in post hoc analyses [27].
Another possible reason for antipsychotic continuation may reside in the indication chosen [33]. Antipsychotic agents have sedative properties and they might be used to optimize sleep during hospitalization, despite the lack of evidence to support this indication [34]. Other factors potentially contributing to continuation of antipsychotics may include persistent delirium and agitation, newly diagnosed psychiatric illness, and difficulties experienced by physicians in deprescribing [35] with improper/incomplete medication reconciliation [33].
The continuation of antipsychotic therapy increased 30-day readmission rates in patients compared to those who had therapy stopped before discharge [33]. In addition to the well-described cardiac effects (prolonged QT interval), neuroleptic malignant syndrome and extrapyramidal symptoms may also occur, and longer-term use can predispose patients to metabolic disturbances, falls, and increase the risk of death in elderly patients with dementia [31].
Benzodiazepines and sedative hypnotics are commonly used to treat insomnia and agitation in older adults despite significant risk. Benzodiazepine administration was found to be an independent risk factor for a daily transition to delirium [36,37]. Pandharipande et al reported that every unit dose of lorazepam was associated with a higher risk for daily transition to delirium (OR 1.2, 95% CI 1.1–1.4, P = 0.003) [36] in critically ill patients. A more recent analysis found for every 5 mg of midazolam administered to a patient who is awake and without delirium, there is a 4% chance that this patient will develop delirium the next ICU day [37].
Given that the risk for benzodiazepine-associated delirium is dose-dependent, clinicians should use strategies known to reduce the daily number of benzodiazepines administered that often includes the use of a sedative associated with less delirium occurrence, such as dexmedetomidine or propofol [38]. Evidence has shown that long-term use of benzodiazepines has little benefit with many risks, including an increased susceptibility to spontaneous bacterial infection [39,40] and mortality in the setting of infection [41]. Nakafero et al showed that exposure to benzodiazepines was associated with increased occurrence of both influenza-like-illness–related pneumonia and mortality. Benzodiazepine use was associated also with increased occurrence of asthma exacerbation and with increased all-cause mortality during a median follow-up of 2 years in a cohort of asthmatic patients [42] as well with an increased risk of pneumonia and long-term mortality in patients with a prior diagnosis of community- acquired pneumonia [40]. Long-term use of benzodiazepines is also associated with increased risk of falls [43–45], cognitive impairment [46–48] and disability [49,50].
Other common types of PIMs at ICU discharge were opioids, anticholinergic medications, antidepressants, and drugs causing orthostatic hypotension [6]. Of the anticholinergic AIMs, H2 blockers (61%) and promethazine (15%) were the most common [6]. Only 16% of opioids, 23% of antidepressants, and 10% of drugs causing orthostatic hypotension were found to be actually inappropriate after the patient’s circumstances were considered (eg, postoperative pain control, a new diagnosis of major depressive disorder) [6].
Inappropriate Medications at Hospital Discharge
Medications typically intended for short-term use during acute illness are sometimes continued after discharge without documented indication [51]. Poudel et al found that in 206 patients 70 years of age and older discharged to residential aged care facilities from acute care, at least 1 PIM was identified in 112 (54.4%) patients on admission and 102 (49.5%) patients on discharge [11]. Commonly prescribed PIM categories, at both admission and discharge, were central nervous system, cardiovascular, gastrointestinal, and respiratory drugs and analgesics [6,11,52,53]. Of all medications prescribed at admission (1728), 10.8% were PIMs, and at discharge, of 1759 medications, 9.6% were PIMs. Of the total 187 PIMs on admission, 56 (30%) were stopped, and 131 (70%) were continued; 32 new PIMs were introduced [11].
Morandi et al in 2011 conducted a prospective cohort study including 120 patients age ≥ 60 who were discharged after receiving care in a medical, surgical, or cardiovascular ICU for shock or respiratory failure. The percentage of patients prescribed at least 1 PIM increased from 66% at pre-admission to 85% at discharge. The number of patients with 0 PIMs dropped from 34% at preadmission to 14% at discharge, and the number of patients with 3 or more PIMS increased from 16% at preadmission to 37% at discharge. While it is possible that these drugs may be appropriate when started during an acute illness in the ICU (eg, stress ulcer prophylaxis with H2-antagonists in mechanically ventilated patients), most should have been discontinued at ICU and/or hospital discharge [21].
Inappropriate prescriptions of proton pump inhibitors (PPIs) in hospital and primary care have been widely reported [54,55]. In a study conducted by Ahrens et al in 31 primary care practices, for 58% (263/506) of patients discharged from 35 hospitals with a PPI recommendation in hospital discharge letters, an appropriate indication was missing. In 57% of these cases general practitioners followed this recommendation and continued the prescription for more than 1 month [54]. The strongest factor associated with appropriate and inappropriate continuation of PPI after discharge was PPI prescription prior to hospitalization [54]. Although PPIs are safe, they can cause adverse effects. PPI intake has been found to have a significant association with risk of community-acquired pneumonia [56,57], hip fractures [58], Clostridium difficile-associated diarrhea [55,61,62], and to reduce the therapeutic effects of bisphosphonates [59] and low-dose aspirin [60].
Unintentional medication continuation is not a problem isolated to a single drug class or disease [63]. Scales et al evaluated rates of and risk factors for potentially unintentional medication continuation following hospitalization in a population of elderly patients (≥ 66 years) [51]. They created distinct cohorts by identifying seniors not previously receiving four classes of medications typically used to treat or prevent complications of acute illness: antipsychotic medications; gastric acid suppressants (ie, histamine-2 blockers and proton pump inhibitors); benzodiazepines; and inhaled bronchodilators and steroids [51]. Prescription without documented indication occurred across all medication classes, from 12,209 patients (1.4 %) for antipsychotic medications to 34,140 patients (6.1 %) for gastric acid suppressants [51].
Several potential risk factors were considered. The relationship between multimorbidity and polypharmacy is well described in the literature, and several studies have identified a positive association between the number of drugs and the use of PIMs [64–66]. Conversely, Poudel et al did not find any association between polypharmacy and PIM use [11]. Associations were found between the use of PIMs, frailty status, and cognitive decline of patients at admission and at discharge [11], while no association was observed with age, gender, in-hospital falls, delirium, and functional decline [11,67]. Other potential risk factors of a high number of PIMs at discharge were a high number of pre-admission PIMs, discharge to a location other than home, and discharge from a surgical service [1,6,68,69]. Length of ICU stay and mechanical ventilation had a positive influence on the number of PIMs used by acutely ill older patients [11,63,69]. In the study of Scales et al, the greatest absolute risk factor across all medication groups was longer hospitalization. The increased OR for medication continuation after a hospitalization lasting more than 7 days ranged from 2.03 (95% CI 1.94–2.11) for respiratory inhalers to 6.35 (95% CI 5.91–6.82) for antipsychotic medications [51].
Inappropriate Medications: Where and How to Intervene?
Early detection of PIMs may prevent adverse drug events and improve geriatric care in older adults [13,70]. PIM prevalence can often be a useful indicator of prescribing quality [2]. Appropriate interventions and an improved quality of prescribed medications require appropriate assessment tools to decrease the number of patients discharged on these medications [71,72]. Medication reconciliation is the process of avoiding inadvertent inconsistencies within a patient’s drug regimen, which can occur during transitions in different setting of care [73]. A multidisciplinary team should be involved in the medication reconciliation at each care transition to reevaluate medications use according to the clinical conditions, cognitive/functional status and the coexistence of geriatric syndromes (eg, dementia, malnutrition, delirium, urinary incontinence, frailty) (Figure).
Criteria for the Evaluation of Inappropriate Medications Prescription
Explicit criteria derived from expert reports or published reviews are available (Table 2).
Beers criteria PIMs have been found to be associated with poor health outcomes, including confusion, falls, and mortality [7,75,78]. The STOPP (Screening Tool of Older Person’s potentially inappropriate Prescriptions) and START (Screening Tool to Alert doctors to the Right Treatment) are evidence-based sets of criteria that were developed in Ireland and updated in October 2014, including some of the new criteria for direct oral anticoagulants, drugs affecting or affected by renal system and anti-muscarinic/anticholinergic agents [79].
Several other sets of criteria have been published to identify PIMs, such as the FORTA (Fit for the Aged) and the PRISCUS [86] criteria. FORTA allows a disease-related evaluation revealing over-treatment and under-treatment, and medications are graded as follows: A, indispensable drug, clear-cut benefit in terms of efficacy/safety ratio proven in elderly patients for a given indication; B, drugs with proven or obvious efficacy in the elderly, but limited extent of effect or safety concerns; C, drugs with questionable efficacy/safety profiles in the elderly which should be avoided or omitted in the presence of too many drugs or side effects; D, avoid in the elderly, omit first, refer also to negative listings. Negative lists such as PRISCUS, which provide an explicit listing of drugs, independent of the diagnosis, are easy to use. On the other hand, constant updates are needed, and such lists carry the risk of an assumption that drugs not listed would be appropriate in every case [87]. Both sets of criteria have in common that they refer to long-term medication and drugs frequently used during the inpatient stay, such as antibiotics, are hardly taken into account [87].
The Medication Appropriateness Index measures overall prescribing quality through 10 separate but interrelated domains [8]. Three components are used to detect PIMs: indication, effectiveness, and duplication. However, it does not give any precise guidance in relation to specific medicines and therefore has limited application for objectively defining PIMs.
Another prescribing quality assessment tool is the Inappropriate Prescribing in the Elderly Tool (IPET), which consists of a list of the 14 most prevalent prescription errors identified from an extensive list of inappropriate prescription instances drawn up by an expert Canadian Consensus Panel [88,89].
Another approach to assess the appropriateness of drugs prescribed for older people is the use of Drug Utilization Reviews (DURs) [16]. DURs use consensus opinion by drug therapy experts to define standards or explicit criteria for a single drug, class of drugs, or group of drugs [16]. DURs typically use retrospective information from large, nonclinical administrative databases to identify problems such as dosage range, duration, therapeutic duplication, and drug interactions [90, 91]. Monane et al [92] evaluated a program designed to decrease the use of PIMs among the elderly through a computerized online DUR database. Computer alerts triggered telephone calls to physicians by pharmacists to discuss a potential problem and any therapeutic substitution options. From a total of 43,007 telepharmacy calls generated by the alerts, they were able to reach 19,368 physicians regarding 24,266 alerts (56%). The rate of change to a more appropriate therapeutic agent was 24% (5860), but ranged from 40% for long half-life benzodiazepines to 2% to 7% for drugs that theoretically were contraindicated by patients’ self-reported history [92].
Computerized Support Systems to Reduce Inappropriate Prescribing in the Elderly
Other potential solutions for reducing inappropriate medications may include continuing medical education, electronic medical records surveillance, routine clinical evaluation, and/or improved hand-off communication between discharging and accepting providers. Incorporating this assessment of medication appropriateness into the medication reconciliation process when patients are discharged or transferred out of the ICU has the potential to enhance patient safety [21,93]. A randomized controlled trial conducted by Raebel et al [94] reported the effectiveness of a computerized pharmacy alert system plus collaboration between health care professionals for decreasing potentially inappropriate medication dispensing in elderly patients. Another study showed that computer-based access to complete drug profiles and alerts about potential prescribing problems reduced the occurrence of potentially inappropriate prescriptions [95]. A summary of these studies is shown in Table 3.
Interdisciplinary Teams to Reduce Inappropriate Prescribing in the Elderly
Some studies evaluated the effect of multidisciplinary teamwork in improving inappropriate medication prescribing in the elderly (Table 4).
Pharmacists in hospitals can play a significant role in the initiation of changes to patient’s therapy and management [11] (Table 5).
Mattison et al recently emphasized that studies of PIMs should determine scenarios in which it is appropriate to prescribe PIMs, moving beyond simply labeling some medications as “potentially inappropriate,” since some PIMs are appropriately prescribed in specific clinical situations [109]. Morandi et al showed that the positive predictive value (PPV) depends on the drug type. Thus, when developing a screening system, one cannot be concerned only with high negative predictive value (NPV), one must consider PPV as well [6]. Screening tools that include medication classes with low PPV will generate false positive “flags” or warnings, which could lead to misguided clinical decisions [6]. The fact that many PIMs are not AIMs also reveals the value of using a multidisciplinary team to identify AIMs from lists of PIMs generated when discharge medication lists are screened [6,110]. Thus, a multidisciplinary team is needed to consider the clinical context to distinguish PIMs from AIMs [6]. Of course, such a team is not available in some settings; when resources are limited, knowledge of which PIMs are most likely AIMs (ie, have high PPVs) could guide the development of computer-based decision support systems or other surveillance approaches that are efficient in that particular setting [6].
Approaches for optimizing prescribing in this population mainly depend on patient needs and comorbidities and most available data are derived from randomized controlled trials involving a single drug. Such trials do not take into account the confounding effects of multiple comorbidities and patient preferences. Therefore, approaches for optimizing prescription management that are available for and validated in younger patients are not applicable to elderly subjects [3,111].
Conclusion
Clinicians should seek to identify and discontinue AIMs at 3 important transitions during a critically ill elderly patient’s hospital course: at the time of hospital or ICU admission; at ICU discharge; and at hospital discharge. The patient’s clinical situation should be reviewed at every transition points, ideally by a multidisciplinary team of clinicians, to judge the appropriateness of each PIM [6]. After the hospital discharge, patient’s medications should be then reviewed by a multidisciplinary team and/or by the primary care physician according to the final discharge destination (ie, home, nursing home, rehabilitation) by using any of the validated tools. Regardless of the approach, it is clear that standardized care processes, including enhanced clinical decision support, are necessary to ensure that physicians do not continue exposing our patients to unnecessary medications and harm after discharge.
Corresponding author: Alessandro Morandi, MD, MPH, [email protected].
Funding/support: Dr. Pandiharipande is supported by National Institutes of Health HL111111 (Bethesda, MD) and by the VA Clinical Science Research and Development Service (Washington, DC) and the National Institutes of Health AG027472 and AG035117 (Bethesda, MD).
Financial disclosures: Dr. Pratik Pandharipande has received a research grant from Hospira Inc in collaboration with the NIH.
1. Lang PO, Hasso Y, Drame M, et al. Potentially inappropriate prescribing including under-use amongst older patients with cognitive or psychiatric co-morbidities. Age Ageing 2010;39:373–81.
2. Spinewine A, Schmader KE, Barber N, et al. Appropriate prescribing in elderly people: how well can it be measured and optimised? Lancet 2007;370:173–84.
3. Lang PO, Vogt-Ferrier N, Hasso Y, et al. Interdisciplinary geriatric and psychiatric care reduces potentially inappropriate prescribing in the hospital: interventional study in 150 acutely ill elderly patients with mental and somatic comorbid conditions. J Am Med Dir Assoc 2012;13:406 e1–7.
4. Cecile M, Seux V, Pauly V, et al. [Adverse drug events in hospitalized elderly patients in a geriatric medicine unit: study of prevalence and risk factors]. Rev Med Interne 2009;30:393–400.
5. Tosato M, Landi F, Martone AM, et al. Potentially inappropriate drug use among hospitalised older adults: results from the CRIME study. Age Ageing 2014;43:767–73.
6. Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013;61:1128–34.
7. Fick DM, Mion LC, Beers MH, Waller JL. Health outcomes associated with potentially inappropriate medication use in older adults. Res Nurs Health 2008;31:42–51.
8. Hanlon JT, Schmader KE, Samsa GP, et al. A method for assessing drug therapy appropriateness. J Clin Epidemiol 1992;45:1045–51.
9. Lau DT, Kasper JD, Potter DEB, et al. Hospitalization and death associated with potentially inappropriate medication prescriptions among elderly nursing home residents. Arch Intern Med 2005;165:68–74.
10. Wright RM, Roumani YF, Boudreau R, et al. Effect of central nervous system medication use on decline in cognition in community-dwelling older adults: findings from the Health, Aging and Body Composition Study. J Am Geriatr Soc 2009;57:243–50.
11. Poudel A, Peel NM, Nissen L, et al. Potentially inappropriate prescribing in older patients discharged from acute care hospitals to residential aged care facilities. Ann Pharmacother 2014;48:1425–33.
12. Wahab MS, Nyfort-Hansen K, Kowalski SR. Inappropriate prescribing in hospitalised Australian elderly as determined by the STOPP criteria. Int J Clin Pharm 2012;34:855–62.
13. O’Mahony D, Gallagher PF. Inappropriate prescribing in the older population: need for new criteria. Age Ageing 2008;37:138–41.
14. Mangoni AA, Jansen PA, Jackson SH. Under-representation of older adults in pharmacokinetic and pharmacodynamic studies: a solvable problem? Exp Rev Clin Pharmacol 2013;6:35–9.
15. Klotz U. The elderly--a challenge for appropriate drug treatment. Eur J Clin Pharmacol 2008;64225–6.
16. Hanlon JT, Schmader KE, Ruby CM, Weinberger M. Suboptimal prescribing in older inpatients and outpatients. J Am Geriatr Soc 2001;49:200–9.
17. Hubbard RE, O’Mahony MS, Woodhouse KW. Medication prescribing in frail older people. Eur J Clin Pharmacol 2013;69:319–26.
18. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012;60:616–31.
19. Page RL, Ruscin JM. The risk of adverse drug events and hospital-related morbidity and mortality among older adults with potentially inappropriate medication use. Am J Geriatr Pharmacother 2006;4:297–305.
20. Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ 2004;329:15–9.
21. Morandi A, Vasilevskis EE, Pandharipande PP, et al. Inappropriate medications in elderly ICU survivors: where to intervene? Arch Intern Med 2011;171:1032–4.
22. Page RL 2nd, Linnebur SA, Bryant LL, Ruscin JM. Inappropriate prescribing in the hospitalized elderly patient: defining the problem, evaluation tools, and possible solutions. Clin Interv Aging 2010;5:75–87.
23. Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med 2013;369:1306–16.
24. Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA 2010;303:763–70.
25. Swan JT, Fitousis K, Hall JB, et al. Antipsychotic use and diagnosis of delirium in the intensive care unit. Crit Care 2012;16:R84.
26. Jasiak KD, Middleton EA, Camamo JM, et al. Evaluation of discontinuation of atypical antipsychotics prescribed for ICU delirium. J Pharm Pract 2013;26:253–6.
27. Tomichek JE, Stollings JL, Pandharipande PP, et al. Antipsychotic prescribing patterns during and after critical illness: a prospective cohort study. Crit Care 2016;20:378.
28. Kram BL, Kram SJ, Brooks KR. Implications of atypical antipsychotic prescribing in the intensive care unit. J Crit Care 2015;30:814–8.
29. Flurie RW, Gonzales JP, Tata AL, et al. Hospital delirium treatment: Continuation of antipsychotic therapy from the intensive care unit to discharge. Am J Health Syst Pharm 2015;72(23 Suppl 3):S133–9.
30. Rowe AS, Hamilton LA, Curtis RA, et al. Risk factors for discharge on a new antipsychotic medication after admission to an intensive care unit. J Crit Care 2015;30:1283–6.
31. Ray WA, Chung CP, Murray KT, et al. Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med 2009;360:225–35.
32. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med 2005;353:2335–41.
33. Marshall J, Herzig SJ, Howell MD, et al. Antipsychotic utilization in the intensive care unit and in transitions of care. J Crit Care 2016;33:119–24.
34. NIH State-of-the-science conference statement on manifestations and management of chronic insomnia in adults. NIH Consens State Sci Statements 2005;22:1–30.
35. Farrell B, Tsang C, Raman-Wilms L, et a;. What are priorities for deprescribing for elderly patients? Capturing the voice of practitioners: a modified delphi process. PLoS One 2015;10:e0122246.
36. Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006;104:21–6.
37. Zaal IJ, Devlin JW, Hazelbag M, et al. Benzodiazepine-associated delirium in critically ill adults. Intensive Care Med 2015;41:2130–7.
38. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306.
39. Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009;301:489–99.
40. Obiora E, Hubbard R, Sanders RD, Myles PR. The impact of benzodiazepines on occurrence of pneumonia and mortality from pneumonia: a nested case-control and survival analysis in a population-based cohort. Thorax 2013;68:163–70.
41. Sanders RD, Godlee A, Fujimori T, et al. Benzodiazepine augmented gamma-amino-butyric acid signaling increases mortality from pneumonia in mice. Crit Care Med 2013;41:1627–36.
42. Nakafero G, Sanders RD, Nguyen-Van-Tam JS, Myles PR. Association between benzodiazepine use and exacerbations and mortality in patients with asthma: a matched case-control and survival analysis using the United Kingdom Clinical Practice Research Datalink. Pharmacoepidemiol Drug Saf 2015;24:793–802.
43. Hartikainen S, Lonnroos E, Louhivuori K. Medication as a risk factor for falls: critical systematic review. J Gerontol A Biol Sci Med Sci 2007;62:1172–81.
44. Pierfitte C, Macouillard G, Thicoipe M, et al. Benzodiazepines and hip fractures in elderly people: case-control study. BMJ 2001;322:704–8.
45. Landi F, Onder G, Cesari M, et al. Psychotropic medications and risk for falls among community-dwelling frail older people: an observational study. J Gerontol A Biol Sci Med Sci 2005;60:622–6.
46. Hanlon JT, Horner RD, Schmader KE, et al. Benzodiazepine use and cognitive function among community-dwelling elderly. Clin Pharmacol Ther 1998;64:684–92.
47. Greenblatt DJ, Harmatz JS, Shapiro L, et al. Sensitivity to triazolam in the elderly. N Engl J Med 1991;324:1691–8.
48. Bertz RJ, Kroboth PD, Kroboth FJ, et al. Alprazolam in young and elderly men: sensitivity and tolerance to psychomotor, sedative and memory effects. J Pharmacol Exp Ther 1997;281:1317–29.
49. Gray SL, LaCroix AZ, Blough D, et al. Is the use of benzodiazepines associated with incident disability? J Am Geriatr Soc 2002;50:1012–8.
50. Gray SL, LaCroix AZ, Hanlon JT, et al. Benzodiazepine use and physical disability in community-dwelling older adults. J Am Geriatr Soc 2006;54:224–30.
51. Scales DC, Fischer HD, Li P, et al. Unintentional continuation of medications intended for acute illness after hospital discharge: a population-based cohort study. J Gen Intern Med 2016;31:196–202.
52. Hamilton H, Gallagher P, Ryan C, et al. Potentially inappropriate medications defined by STOPP criteria and the risk of adverse drug events in older hospitalized patients. Arch Intern Med 2011;171:1013–9.
53. Hanlon JT, Artz MB, Pieper CF, et al. Inappropriate medication use among frail elderly inpatients. Ann Pharmacother 2004;38:9–14.
54. Ahrens D, Behrens G, Himmel W, et al. Appropriateness of proton pump inhibitor recommendations at hospital discharge and continuation in primary care. Int J Clin Pract 2012;66:767–73.
55. McDonald EG, Milligan J, Frenette C, Lee TC. Continuous proton pump inhibitor therapy and the associated risk of recurrent Clostridium difficile infection. JAMA Intern Med 2015;175:784–91.
56. Gulmez SE, Holm A, Frederiksen H, et al. Use of proton pump inhibitors and the risk of community-acquired pneumonia: a population-based case-control study. Arch Intern Med 2007;167:950–5.
57. Laheij RJ, Sturkenboom MC, Hassing RJ, et al. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004;292:1955–60.
58. Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006;296:2947–53.
59. Abrahamsen B, Eiken P, Eastell R. Proton pump inhibitor use and the antifracture efficacy of alendronate. Arch Intern Med 2011;171:998–1004.
60. Charlot M, Grove EL, Hansen PR, et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study. BMJ 2011;342:d2690.
61. Dial S, Delaney JA, Barkun AN, Suissa S. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA 2005;294:2989–95.
62. Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol 2007;102:2047–56; quiz 57.
63. Pavlov A, Muravyev R, Amoateng-Adjepong Y, Manthous CA. Inappropriate discharge on bronchodilators and acid-blocking medications after ICU admission: importance of medication reconciliation. Respir Care 2014;59:1524–9.
64. Baldoni Ade O, Ayres LR, Martinez EZ, et al. Factors associated with potentially inappropriate medications use by the elderly according to Beers criteria 2003 and 2012. Int J Clin Pharm 2014;36:316–24.
65. Gallagher PF, Barry PJ, Ryan C, et al. Inappropriate prescribing in an acutely ill population of elderly patients as determined by Beers’ Criteria. Age Ageing 2008;37:96–101.
66. Montastruc F, Duguet C, Rousseau V, et al. Potentially inappropriate medications and adverse drug reactions in the elderly: a study in a PharmacoVigilance database. Eur J Clin Pharmacol 2014;70:1123–7.
67. Ruggiero C, Dell’Aquila G, Gasperini B, et al. Potentially inappropriate drug prescriptions and risk of hospitalization among older, Italian, nursing home residents: the ULISSE project. Drugs Aging 2010;27:747–58.
68. Onder G, Landi F, Cesari M, et al. Inappropriate medication use among hospitalized older adults in Italy: results from the Italian Group of Pharmacoepidemiology in the Elderly. Eur J Clin Pharmacol 2003;59:157–62.
69. Harugeri A, Joseph J, Parthasarathi G, et al. Potentially inappropriate medication use in elderly patients: A study of prevalence and predictors in two teaching hospitals. J Postgrad Med 2010;56:186–91.
70. Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 2010;170:1648–54.
71. Dimitrow MS, Airaksinen MS, Kivela SL, et al. Comparison of prescribing criteria to evaluate the appropriateness of drug treatment in individuals aged 65 and older: a systematic review. J Am Geriatr Soc 2011;59:1521–30.
72. Levy HB, Marcus EL, Christen C. Beyond the beers criteria: A comparative overview of explicit criteria. Ann Pharmacother 2010;44:1968–75.
73. Marengoni A, Nobili A, Onder G. Best practices for drug prescribing in older adults: a call for action. Drugs Aging 2015;32:887–90.
74. Poudel A, Hubbard RE, Nissen L, Mitchell C. Frailty: a key indicator to minimize inappropriate medication in older people. QJM 2013;106:969–75.
75. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015;63:2227–46.
76. Beers MH, Ouslander JG, Rollingher I, et al. Explicit criteria for determining inappropriate medication use in nursing home residents. UCLA Division of Geriatric Medicine. Arch Intern Med 1991;151:1825–32.
77. Blanco-Reina E, Ariza-Zafra G, Ocana-Riola R, Leon-Ortiz M. 2012 American Geriatrics Society Beers criteria: enhanced applicability for detecting potentially inappropriate medications in European older adults? A comparison with the Screening Tool of Older Person’s Potentially Inappropriate Prescriptions. J Am Geriatr Soc 2014;62:1217–23.
78. Stockl KM, Le L, Zhang S, Harada AS. Clinical and economic outcomes associated with potentially inappropriate prescribing in the elderly. Am J Manag Care 2010;16:e1–10.
79. Hill-Taylor B, Sketris I, Hayden J, et al. Application of the STOPP/START criteria: a systematic review of the prevalence of potentially inappropriate prescribing in older adults, and evidence of clinical, humanistic and economic impact. J Clin Pharm Ther 2013;38:360–72.
80. Barry PJ, Gallagher P, Ryan C, O’Mahony D. START (screening tool to alert doctors to the right treatment)--an evidence-based screening tool to detect prescribing omissions in elderly patients. Age Ageing 2007;36:632–8.
81. Gallagher P, Ryan C, Byrne S, Kennedy J, O’Mahony D. STOPP (Screening Tool of Older Person’s Prescriptions) and START (Screening Tool to Alert doctors to Right Treatment). Consensus validation. Int J Clin Pharmacol Ther 2008;46:72–83.
82. Haag JD, Davis AZ, Hoel RW, et al. Impact of pharmacist-provided medication therapy management on healthcare quality and utilization in recently discharged elderly patients. Am Health Drug Benefits 2016;9:259–68.
83. Gillespie U, Alassaad A, Hammarlund-Udenaes M, et al. Effects of pharmacists’ interventions on appropriateness of prescribing and evaluation of the instruments’ (MAI, STOPP and STARTs’) ability to predict hospitalization--analyses from a randomized controlled trial. PLoS One 2013;8:e62401.
84. Petrarca AM, Lengel AJ, Mangan MN. Inappropriate medication use in the elderly. Consult Pharm 2012;27:583–6.
85. Lavan AH, Gallagher P, Parsons C, O’Mahony D. STOPPFrail (Screening Tool of Older Persons Prescriptions in Frail adults with limited life expectancy): consensus validation. Age Ageing 2017;46:600–7.
86. Holt S, Schmiedl S, Thurmann PA. Potentially inappropriate medications in the elderly: the PRISCUS list. Dtsch Arztebl Int 2010;107:543–51.
87. Wickop B, Harterich S, Sommer C, et al. Potentially inappropriate medication use in multimorbid elderly inpatients: differences between the FORTA, PRISCUS and STOPP ratings. Drugs Real World Outcome 2016;3:317–25.
88. Naugler CT, Brymer C, Stolee P, Arcese ZA. Development and validation of an improving prescribing in the elderly tool. Can J Clin Pharmacol 2000;7:103–7.
89. Barry PJ, O’Keefe N, O’Connor KA, O’Mahony D. Inappropriate prescribing in the elderly: a comparison of the Beers criteria and the improved prescribing in the elderly tool (IPET) in acutely ill elderly hospitalized patients. J Clin Pharm Ther 2006;31:617–26.
90. Knapp DA. Development of criteria for drug utilization review. Clin Pharmacol Ther 1991;50(5 Pt 2):600–2.
91. Lipton HL, Bird JA. Drug utilization review in ambulatory settings: state of the science and directions for outcomes research. Med Care 1993;31:1069–82.
92. Monane M, Matthias DM, Nagle BA, Kelly MA. Improving prescribing patterns for the elderly through an online drug utilization review intervention: a system linking the physician, pharmacist, and computer. JAMA 1998;280:1249–52.
93. Kaur S, Mitchell G, Vitetta L, Roberts MS. Interventions that can reduce inappropriate prescribing in the elderly: a systematic review. Drugs Aging 2009;26:1013–28.
94. Raebel MA, Charles J, Dugan J, et al. Randomized trial to improve prescribing safety in ambulatory elderly patients. J Am Geriatr Soc 2007;55:977–85.
95. Tamblyn R, Huang A, Perreault R, et al. The medical office of the 21st century (MOXXI): effectiveness of computerized decision-making support in reducing inappropriate prescribing in primary care. CMAJ 2003;169:549–56.
96. Dalleur O, Boland B, Losseau C, et al. Reduction of potentially inappropriate medications using the STOPP criteria in frail older inpatients: a randomised controlled study. Drugs Aging 2014;31:291–8.
97. Schmader KE, Hanlon JT, Pieper CF, et al. Effects of geriatric evaluation and management on adverse drug reactions and suboptimal prescribing in the frail elderly. Am J Med 2004;116:394–401.
98. Crotty M, Halbert J, Rowett D, et al. An outreach geriatric medication advisory service in residential aged care: a randomised controlled trial of case conferencing. Age Ageing 2004;33:612–7.
99. Allard J, Hebert R, Rioux M, et al. Efficacy of a clinical medication review on the number of potentially inappropriate prescriptions prescribed for community-dwelling elderly people. CMAJ 2001;164:1291–6.
100. Spinewine A, Swine C, Dhillon S, et al. Effect of a collaborative approach on the quality of prescribing for geriatric inpatients: a randomized, controlled trial. J Am Geriatr Soc 2007;55:658–65.
101. Elliott RA, Woodward MC, Oborne CA. Improving benzodiazepine prescribing for elderly hospital inpatients using audit and multidisciplinary feedback. Intern Med J 2001;31:529–35.
102. Saltvedt I, Spigset O, Ruths S, et al. Patterns of drug prescription in a geriatric evaluation and management unit as compared with the general medical wards: a randomised study. Eur J Clin Pharmacol 2005;61:921–8.
103. Hanlon JT, Weinberger M, Samsa GP, et al. A randomized, controlled trial of a clinical pharmacist intervention to improve inappropriate prescribing in elderly outpatients with polypharmacy. Am J Med 1996;100:428–37.
104. Lipton HL, Bero LA, Bird JA, McPhee SJ. The impact of clinical pharmacists’ consultations on physicians’ geriatric drug prescribing. A randomized controlled trial. Med Care 1992;30:646–58.
105. Krska J, Cromarty JA, Arris F, et al. Pharmacist-led medication review in patients over 65: a randomized, controlled trial in primary care. Age Ageing 2001;30:205–11.
106. Brown BK, Earnhart J. Pharmacists and their effectiveness in ensuring the appropriateness of the chronic medication regimens of geriatric inpatients. Consult Pharm 2004;19:432–6.
107. Belfield KD, Kuyumjian AG, Teran R, et al. Impact of a collaborative strategy to reduce the inappropriate use of acid suppressive therapy in non-intensive care unit patients. Ann Pharmacother 2017;51:577–83.
108. Crotty M, Rowett D, Spurling L, et al. Does the addition of a pharmacist transition coordinator improve evidence-based medication management and health outcomes in older adults moving from the hospital to a long-term care facility? Results of a randomized, controlled trial. Am J Geriatr Pharmacother 2004;2:257–64.
109. Mattison MLP, Afonso KA, Ngo LH, Mukamal KJ. Preventing potentially inappropriate medication use in hospitalized older patients with a computerized provider order entry warning system. Arch Intern Med 2010;170:1331–6.
110. Kaboli PJ, Hoth AB, McClimon BJ, Schnipper JL. Clinical pharmacists and inpatient medical care: a systematic review. Arch Intern Med 2006;166:955–64.
111. Tinetti ME, Bogardus ST Jr, Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004;351:2870–4.
112. Gokula M, Holmes HM. Tools to reduce polypharmacy. Clin Geriatr Med 2012;28:323–41.
1. Lang PO, Hasso Y, Drame M, et al. Potentially inappropriate prescribing including under-use amongst older patients with cognitive or psychiatric co-morbidities. Age Ageing 2010;39:373–81.
2. Spinewine A, Schmader KE, Barber N, et al. Appropriate prescribing in elderly people: how well can it be measured and optimised? Lancet 2007;370:173–84.
3. Lang PO, Vogt-Ferrier N, Hasso Y, et al. Interdisciplinary geriatric and psychiatric care reduces potentially inappropriate prescribing in the hospital: interventional study in 150 acutely ill elderly patients with mental and somatic comorbid conditions. J Am Med Dir Assoc 2012;13:406 e1–7.
4. Cecile M, Seux V, Pauly V, et al. [Adverse drug events in hospitalized elderly patients in a geriatric medicine unit: study of prevalence and risk factors]. Rev Med Interne 2009;30:393–400.
5. Tosato M, Landi F, Martone AM, et al. Potentially inappropriate drug use among hospitalised older adults: results from the CRIME study. Age Ageing 2014;43:767–73.
6. Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013;61:1128–34.
7. Fick DM, Mion LC, Beers MH, Waller JL. Health outcomes associated with potentially inappropriate medication use in older adults. Res Nurs Health 2008;31:42–51.
8. Hanlon JT, Schmader KE, Samsa GP, et al. A method for assessing drug therapy appropriateness. J Clin Epidemiol 1992;45:1045–51.
9. Lau DT, Kasper JD, Potter DEB, et al. Hospitalization and death associated with potentially inappropriate medication prescriptions among elderly nursing home residents. Arch Intern Med 2005;165:68–74.
10. Wright RM, Roumani YF, Boudreau R, et al. Effect of central nervous system medication use on decline in cognition in community-dwelling older adults: findings from the Health, Aging and Body Composition Study. J Am Geriatr Soc 2009;57:243–50.
11. Poudel A, Peel NM, Nissen L, et al. Potentially inappropriate prescribing in older patients discharged from acute care hospitals to residential aged care facilities. Ann Pharmacother 2014;48:1425–33.
12. Wahab MS, Nyfort-Hansen K, Kowalski SR. Inappropriate prescribing in hospitalised Australian elderly as determined by the STOPP criteria. Int J Clin Pharm 2012;34:855–62.
13. O’Mahony D, Gallagher PF. Inappropriate prescribing in the older population: need for new criteria. Age Ageing 2008;37:138–41.
14. Mangoni AA, Jansen PA, Jackson SH. Under-representation of older adults in pharmacokinetic and pharmacodynamic studies: a solvable problem? Exp Rev Clin Pharmacol 2013;6:35–9.
15. Klotz U. The elderly--a challenge for appropriate drug treatment. Eur J Clin Pharmacol 2008;64225–6.
16. Hanlon JT, Schmader KE, Ruby CM, Weinberger M. Suboptimal prescribing in older inpatients and outpatients. J Am Geriatr Soc 2001;49:200–9.
17. Hubbard RE, O’Mahony MS, Woodhouse KW. Medication prescribing in frail older people. Eur J Clin Pharmacol 2013;69:319–26.
18. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012;60:616–31.
19. Page RL, Ruscin JM. The risk of adverse drug events and hospital-related morbidity and mortality among older adults with potentially inappropriate medication use. Am J Geriatr Pharmacother 2006;4:297–305.
20. Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ 2004;329:15–9.
21. Morandi A, Vasilevskis EE, Pandharipande PP, et al. Inappropriate medications in elderly ICU survivors: where to intervene? Arch Intern Med 2011;171:1032–4.
22. Page RL 2nd, Linnebur SA, Bryant LL, Ruscin JM. Inappropriate prescribing in the hospitalized elderly patient: defining the problem, evaluation tools, and possible solutions. Clin Interv Aging 2010;5:75–87.
23. Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med 2013;369:1306–16.
24. Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA 2010;303:763–70.
25. Swan JT, Fitousis K, Hall JB, et al. Antipsychotic use and diagnosis of delirium in the intensive care unit. Crit Care 2012;16:R84.
26. Jasiak KD, Middleton EA, Camamo JM, et al. Evaluation of discontinuation of atypical antipsychotics prescribed for ICU delirium. J Pharm Pract 2013;26:253–6.
27. Tomichek JE, Stollings JL, Pandharipande PP, et al. Antipsychotic prescribing patterns during and after critical illness: a prospective cohort study. Crit Care 2016;20:378.
28. Kram BL, Kram SJ, Brooks KR. Implications of atypical antipsychotic prescribing in the intensive care unit. J Crit Care 2015;30:814–8.
29. Flurie RW, Gonzales JP, Tata AL, et al. Hospital delirium treatment: Continuation of antipsychotic therapy from the intensive care unit to discharge. Am J Health Syst Pharm 2015;72(23 Suppl 3):S133–9.
30. Rowe AS, Hamilton LA, Curtis RA, et al. Risk factors for discharge on a new antipsychotic medication after admission to an intensive care unit. J Crit Care 2015;30:1283–6.
31. Ray WA, Chung CP, Murray KT, et al. Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med 2009;360:225–35.
32. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med 2005;353:2335–41.
33. Marshall J, Herzig SJ, Howell MD, et al. Antipsychotic utilization in the intensive care unit and in transitions of care. J Crit Care 2016;33:119–24.
34. NIH State-of-the-science conference statement on manifestations and management of chronic insomnia in adults. NIH Consens State Sci Statements 2005;22:1–30.
35. Farrell B, Tsang C, Raman-Wilms L, et a;. What are priorities for deprescribing for elderly patients? Capturing the voice of practitioners: a modified delphi process. PLoS One 2015;10:e0122246.
36. Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006;104:21–6.
37. Zaal IJ, Devlin JW, Hazelbag M, et al. Benzodiazepine-associated delirium in critically ill adults. Intensive Care Med 2015;41:2130–7.
38. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306.
39. Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009;301:489–99.
40. Obiora E, Hubbard R, Sanders RD, Myles PR. The impact of benzodiazepines on occurrence of pneumonia and mortality from pneumonia: a nested case-control and survival analysis in a population-based cohort. Thorax 2013;68:163–70.
41. Sanders RD, Godlee A, Fujimori T, et al. Benzodiazepine augmented gamma-amino-butyric acid signaling increases mortality from pneumonia in mice. Crit Care Med 2013;41:1627–36.
42. Nakafero G, Sanders RD, Nguyen-Van-Tam JS, Myles PR. Association between benzodiazepine use and exacerbations and mortality in patients with asthma: a matched case-control and survival analysis using the United Kingdom Clinical Practice Research Datalink. Pharmacoepidemiol Drug Saf 2015;24:793–802.
43. Hartikainen S, Lonnroos E, Louhivuori K. Medication as a risk factor for falls: critical systematic review. J Gerontol A Biol Sci Med Sci 2007;62:1172–81.
44. Pierfitte C, Macouillard G, Thicoipe M, et al. Benzodiazepines and hip fractures in elderly people: case-control study. BMJ 2001;322:704–8.
45. Landi F, Onder G, Cesari M, et al. Psychotropic medications and risk for falls among community-dwelling frail older people: an observational study. J Gerontol A Biol Sci Med Sci 2005;60:622–6.
46. Hanlon JT, Horner RD, Schmader KE, et al. Benzodiazepine use and cognitive function among community-dwelling elderly. Clin Pharmacol Ther 1998;64:684–92.
47. Greenblatt DJ, Harmatz JS, Shapiro L, et al. Sensitivity to triazolam in the elderly. N Engl J Med 1991;324:1691–8.
48. Bertz RJ, Kroboth PD, Kroboth FJ, et al. Alprazolam in young and elderly men: sensitivity and tolerance to psychomotor, sedative and memory effects. J Pharmacol Exp Ther 1997;281:1317–29.
49. Gray SL, LaCroix AZ, Blough D, et al. Is the use of benzodiazepines associated with incident disability? J Am Geriatr Soc 2002;50:1012–8.
50. Gray SL, LaCroix AZ, Hanlon JT, et al. Benzodiazepine use and physical disability in community-dwelling older adults. J Am Geriatr Soc 2006;54:224–30.
51. Scales DC, Fischer HD, Li P, et al. Unintentional continuation of medications intended for acute illness after hospital discharge: a population-based cohort study. J Gen Intern Med 2016;31:196–202.
52. Hamilton H, Gallagher P, Ryan C, et al. Potentially inappropriate medications defined by STOPP criteria and the risk of adverse drug events in older hospitalized patients. Arch Intern Med 2011;171:1013–9.
53. Hanlon JT, Artz MB, Pieper CF, et al. Inappropriate medication use among frail elderly inpatients. Ann Pharmacother 2004;38:9–14.
54. Ahrens D, Behrens G, Himmel W, et al. Appropriateness of proton pump inhibitor recommendations at hospital discharge and continuation in primary care. Int J Clin Pract 2012;66:767–73.
55. McDonald EG, Milligan J, Frenette C, Lee TC. Continuous proton pump inhibitor therapy and the associated risk of recurrent Clostridium difficile infection. JAMA Intern Med 2015;175:784–91.
56. Gulmez SE, Holm A, Frederiksen H, et al. Use of proton pump inhibitors and the risk of community-acquired pneumonia: a population-based case-control study. Arch Intern Med 2007;167:950–5.
57. Laheij RJ, Sturkenboom MC, Hassing RJ, et al. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004;292:1955–60.
58. Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006;296:2947–53.
59. Abrahamsen B, Eiken P, Eastell R. Proton pump inhibitor use and the antifracture efficacy of alendronate. Arch Intern Med 2011;171:998–1004.
60. Charlot M, Grove EL, Hansen PR, et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study. BMJ 2011;342:d2690.
61. Dial S, Delaney JA, Barkun AN, Suissa S. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA 2005;294:2989–95.
62. Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol 2007;102:2047–56; quiz 57.
63. Pavlov A, Muravyev R, Amoateng-Adjepong Y, Manthous CA. Inappropriate discharge on bronchodilators and acid-blocking medications after ICU admission: importance of medication reconciliation. Respir Care 2014;59:1524–9.
64. Baldoni Ade O, Ayres LR, Martinez EZ, et al. Factors associated with potentially inappropriate medications use by the elderly according to Beers criteria 2003 and 2012. Int J Clin Pharm 2014;36:316–24.
65. Gallagher PF, Barry PJ, Ryan C, et al. Inappropriate prescribing in an acutely ill population of elderly patients as determined by Beers’ Criteria. Age Ageing 2008;37:96–101.
66. Montastruc F, Duguet C, Rousseau V, et al. Potentially inappropriate medications and adverse drug reactions in the elderly: a study in a PharmacoVigilance database. Eur J Clin Pharmacol 2014;70:1123–7.
67. Ruggiero C, Dell’Aquila G, Gasperini B, et al. Potentially inappropriate drug prescriptions and risk of hospitalization among older, Italian, nursing home residents: the ULISSE project. Drugs Aging 2010;27:747–58.
68. Onder G, Landi F, Cesari M, et al. Inappropriate medication use among hospitalized older adults in Italy: results from the Italian Group of Pharmacoepidemiology in the Elderly. Eur J Clin Pharmacol 2003;59:157–62.
69. Harugeri A, Joseph J, Parthasarathi G, et al. Potentially inappropriate medication use in elderly patients: A study of prevalence and predictors in two teaching hospitals. J Postgrad Med 2010;56:186–91.
70. Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 2010;170:1648–54.
71. Dimitrow MS, Airaksinen MS, Kivela SL, et al. Comparison of prescribing criteria to evaluate the appropriateness of drug treatment in individuals aged 65 and older: a systematic review. J Am Geriatr Soc 2011;59:1521–30.
72. Levy HB, Marcus EL, Christen C. Beyond the beers criteria: A comparative overview of explicit criteria. Ann Pharmacother 2010;44:1968–75.
73. Marengoni A, Nobili A, Onder G. Best practices for drug prescribing in older adults: a call for action. Drugs Aging 2015;32:887–90.
74. Poudel A, Hubbard RE, Nissen L, Mitchell C. Frailty: a key indicator to minimize inappropriate medication in older people. QJM 2013;106:969–75.
75. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015;63:2227–46.
76. Beers MH, Ouslander JG, Rollingher I, et al. Explicit criteria for determining inappropriate medication use in nursing home residents. UCLA Division of Geriatric Medicine. Arch Intern Med 1991;151:1825–32.
77. Blanco-Reina E, Ariza-Zafra G, Ocana-Riola R, Leon-Ortiz M. 2012 American Geriatrics Society Beers criteria: enhanced applicability for detecting potentially inappropriate medications in European older adults? A comparison with the Screening Tool of Older Person’s Potentially Inappropriate Prescriptions. J Am Geriatr Soc 2014;62:1217–23.
78. Stockl KM, Le L, Zhang S, Harada AS. Clinical and economic outcomes associated with potentially inappropriate prescribing in the elderly. Am J Manag Care 2010;16:e1–10.
79. Hill-Taylor B, Sketris I, Hayden J, et al. Application of the STOPP/START criteria: a systematic review of the prevalence of potentially inappropriate prescribing in older adults, and evidence of clinical, humanistic and economic impact. J Clin Pharm Ther 2013;38:360–72.
80. Barry PJ, Gallagher P, Ryan C, O’Mahony D. START (screening tool to alert doctors to the right treatment)--an evidence-based screening tool to detect prescribing omissions in elderly patients. Age Ageing 2007;36:632–8.
81. Gallagher P, Ryan C, Byrne S, Kennedy J, O’Mahony D. STOPP (Screening Tool of Older Person’s Prescriptions) and START (Screening Tool to Alert doctors to Right Treatment). Consensus validation. Int J Clin Pharmacol Ther 2008;46:72–83.
82. Haag JD, Davis AZ, Hoel RW, et al. Impact of pharmacist-provided medication therapy management on healthcare quality and utilization in recently discharged elderly patients. Am Health Drug Benefits 2016;9:259–68.
83. Gillespie U, Alassaad A, Hammarlund-Udenaes M, et al. Effects of pharmacists’ interventions on appropriateness of prescribing and evaluation of the instruments’ (MAI, STOPP and STARTs’) ability to predict hospitalization--analyses from a randomized controlled trial. PLoS One 2013;8:e62401.
84. Petrarca AM, Lengel AJ, Mangan MN. Inappropriate medication use in the elderly. Consult Pharm 2012;27:583–6.
85. Lavan AH, Gallagher P, Parsons C, O’Mahony D. STOPPFrail (Screening Tool of Older Persons Prescriptions in Frail adults with limited life expectancy): consensus validation. Age Ageing 2017;46:600–7.
86. Holt S, Schmiedl S, Thurmann PA. Potentially inappropriate medications in the elderly: the PRISCUS list. Dtsch Arztebl Int 2010;107:543–51.
87. Wickop B, Harterich S, Sommer C, et al. Potentially inappropriate medication use in multimorbid elderly inpatients: differences between the FORTA, PRISCUS and STOPP ratings. Drugs Real World Outcome 2016;3:317–25.
88. Naugler CT, Brymer C, Stolee P, Arcese ZA. Development and validation of an improving prescribing in the elderly tool. Can J Clin Pharmacol 2000;7:103–7.
89. Barry PJ, O’Keefe N, O’Connor KA, O’Mahony D. Inappropriate prescribing in the elderly: a comparison of the Beers criteria and the improved prescribing in the elderly tool (IPET) in acutely ill elderly hospitalized patients. J Clin Pharm Ther 2006;31:617–26.
90. Knapp DA. Development of criteria for drug utilization review. Clin Pharmacol Ther 1991;50(5 Pt 2):600–2.
91. Lipton HL, Bird JA. Drug utilization review in ambulatory settings: state of the science and directions for outcomes research. Med Care 1993;31:1069–82.
92. Monane M, Matthias DM, Nagle BA, Kelly MA. Improving prescribing patterns for the elderly through an online drug utilization review intervention: a system linking the physician, pharmacist, and computer. JAMA 1998;280:1249–52.
93. Kaur S, Mitchell G, Vitetta L, Roberts MS. Interventions that can reduce inappropriate prescribing in the elderly: a systematic review. Drugs Aging 2009;26:1013–28.
94. Raebel MA, Charles J, Dugan J, et al. Randomized trial to improve prescribing safety in ambulatory elderly patients. J Am Geriatr Soc 2007;55:977–85.
95. Tamblyn R, Huang A, Perreault R, et al. The medical office of the 21st century (MOXXI): effectiveness of computerized decision-making support in reducing inappropriate prescribing in primary care. CMAJ 2003;169:549–56.
96. Dalleur O, Boland B, Losseau C, et al. Reduction of potentially inappropriate medications using the STOPP criteria in frail older inpatients: a randomised controlled study. Drugs Aging 2014;31:291–8.
97. Schmader KE, Hanlon JT, Pieper CF, et al. Effects of geriatric evaluation and management on adverse drug reactions and suboptimal prescribing in the frail elderly. Am J Med 2004;116:394–401.
98. Crotty M, Halbert J, Rowett D, et al. An outreach geriatric medication advisory service in residential aged care: a randomised controlled trial of case conferencing. Age Ageing 2004;33:612–7.
99. Allard J, Hebert R, Rioux M, et al. Efficacy of a clinical medication review on the number of potentially inappropriate prescriptions prescribed for community-dwelling elderly people. CMAJ 2001;164:1291–6.
100. Spinewine A, Swine C, Dhillon S, et al. Effect of a collaborative approach on the quality of prescribing for geriatric inpatients: a randomized, controlled trial. J Am Geriatr Soc 2007;55:658–65.
101. Elliott RA, Woodward MC, Oborne CA. Improving benzodiazepine prescribing for elderly hospital inpatients using audit and multidisciplinary feedback. Intern Med J 2001;31:529–35.
102. Saltvedt I, Spigset O, Ruths S, et al. Patterns of drug prescription in a geriatric evaluation and management unit as compared with the general medical wards: a randomised study. Eur J Clin Pharmacol 2005;61:921–8.
103. Hanlon JT, Weinberger M, Samsa GP, et al. A randomized, controlled trial of a clinical pharmacist intervention to improve inappropriate prescribing in elderly outpatients with polypharmacy. Am J Med 1996;100:428–37.
104. Lipton HL, Bero LA, Bird JA, McPhee SJ. The impact of clinical pharmacists’ consultations on physicians’ geriatric drug prescribing. A randomized controlled trial. Med Care 1992;30:646–58.
105. Krska J, Cromarty JA, Arris F, et al. Pharmacist-led medication review in patients over 65: a randomized, controlled trial in primary care. Age Ageing 2001;30:205–11.
106. Brown BK, Earnhart J. Pharmacists and their effectiveness in ensuring the appropriateness of the chronic medication regimens of geriatric inpatients. Consult Pharm 2004;19:432–6.
107. Belfield KD, Kuyumjian AG, Teran R, et al. Impact of a collaborative strategy to reduce the inappropriate use of acid suppressive therapy in non-intensive care unit patients. Ann Pharmacother 2017;51:577–83.
108. Crotty M, Rowett D, Spurling L, et al. Does the addition of a pharmacist transition coordinator improve evidence-based medication management and health outcomes in older adults moving from the hospital to a long-term care facility? Results of a randomized, controlled trial. Am J Geriatr Pharmacother 2004;2:257–64.
109. Mattison MLP, Afonso KA, Ngo LH, Mukamal KJ. Preventing potentially inappropriate medication use in hospitalized older patients with a computerized provider order entry warning system. Arch Intern Med 2010;170:1331–6.
110. Kaboli PJ, Hoth AB, McClimon BJ, Schnipper JL. Clinical pharmacists and inpatient medical care: a systematic review. Arch Intern Med 2006;166:955–64.
111. Tinetti ME, Bogardus ST Jr, Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004;351:2870–4.
112. Gokula M, Holmes HM. Tools to reduce polypharmacy. Clin Geriatr Med 2012;28:323–41.
FDG PET can’t replace BM biopsy, study suggests

LA JOLLA, CA—Fluorodeoxyglucose positron emission tomography (FDG PET) cannot replace bone marrow (BM) biopsy in T-cell lymphomas, according to a speaker at the 10th Annual T-cell Lymphoma Forum.
Researchers found that FDG PET results did not exactly correlate with BM biopsy results relating to tumor involvement in patients with T-cell lymphomas.
However, results from FDG PET were found to be an independent prognostic factor for progression-free survival (PFS) and overall survival (OS).
Youngil Koh, MD, of Seoul National University Hospital in Seoul, South Korea, presented this research in a poster and oral presentation at this year’s T-cell Lymphoma Forum.
He and his colleagues set out to investigate the clinical value of FDG PET for evaluating BM tumor involvement and prognosis in T-cell lymphoma patients.
The team analyzed 109 patients who underwent staging with FDG PET and BM biopsy. Most patients had extranodal natural killer/T-cell lymphoma, nasal type (NKTCL, n=46), or angioimmunoblastic T-cell lymphoma (AITL, n=41).
Patients also had peripheral T-cell lymphoma not otherwise specified (n=12), anaplastic large-cell lymphoma (n=4), enteropathy-associated T-cell lymphoma (n=4), and subcutaneous panniculitis-like T-cell lymphoma (n=2).
Most patients (87.2%) received chemotherapy as first-line treatment. Fifty percent were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, 48.1% were IMEP (ifosphamide, methotrexate, etoposide, and prednisolone) or IMEP-like regimens, and 1.9% were “other” regimens.
Other first-line treatments included radiotherapy followed by chemotherapy (10.1%), excision (0.9%), and no treatment (1.8%).
The patients’ median OS was 60.03 months, and the median PFS was 15.7 months.
BM involvement
The researchers analyzed PET BM uptake both visually and quantitatively using the marrow-to-liver ratio (MLR), and they compared these results to BM biopsy results.
According to BM biopsy, 35.8% of patients had tumor involvement.
By visual analysis, the sensitivity of PET for diagnosing positive BM biopsy was 58.5%, and the specificity was 77.9%. By MLR, the sensitivity was 64.1%, and the specificity was 72.9%.
The diagnostic performance of PET for BM involvement was not different across the lymphoma subtypes, Dr Koh said.
Prognosis
“Although FDG PET did not correlate very well with bone marrow biopsy, it had prognostic value, especially MLR,” Dr Koh noted. “And most importantly, in bone marrow biopsy-negative patients, it [MLR] had prognostic value.”
MLR was a significant prognostic factor for PFS (P=0.005) and OS (P<0.001). The same was true for BM biopsy (P=0.009 for PFS and P<0.001 for OS), while visual PET analysis was a significant prognostic factor for OS (P=0.015) but not PFS (P=0.476).
In patients negative by BM biopsy, MLR was a significant prognostic factor for PFS (P=0.001) and OS (P=0.005).
Dr Koh and his colleagues also analyzed the prognostic value of PET and BM biopsy specifically in patients with NKTCL and AITL.
In AITL patients, BM biopsy was a significant prognostic factor for OS (P=0.002) but not PFS (P=0.246). Visual PET analysis was not significant for PFS (P=0.910) or OS (P=0.581), and neither was MLR (P=0.053 for PFS and P=0.156 for OS).
In patients with NKTCL, BM biopsy was a significant prognostic factor for PFS (P=0.008) and OS (P<0.001). Visual PET analysis was not significant for PFS (P=0.469) or OS (P=0.092). And MLR was significant for PFS (P=0.004) and OS (P=0.012).
“Bone marrow findings of FDG PET are an independent prognostic factor in these tumors,” Dr Koh said, “suggesting the biologic relevance of FDG PET findings for aggressiveness or covert bone marrow involvement of tumor cells.” ![]()
LA JOLLA, CA—Fluorodeoxyglucose positron emission tomography (FDG PET) cannot replace bone marrow (BM) biopsy in T-cell lymphomas, according to a speaker at the 10th Annual T-cell Lymphoma Forum.
Researchers found that FDG PET results did not exactly correlate with BM biopsy results relating to tumor involvement in patients with T-cell lymphomas.
However, results from FDG PET were found to be an independent prognostic factor for progression-free survival (PFS) and overall survival (OS).
Youngil Koh, MD, of Seoul National University Hospital in Seoul, South Korea, presented this research in a poster and oral presentation at this year’s T-cell Lymphoma Forum.
He and his colleagues set out to investigate the clinical value of FDG PET for evaluating BM tumor involvement and prognosis in T-cell lymphoma patients.
The team analyzed 109 patients who underwent staging with FDG PET and BM biopsy. Most patients had extranodal natural killer/T-cell lymphoma, nasal type (NKTCL, n=46), or angioimmunoblastic T-cell lymphoma (AITL, n=41).
Patients also had peripheral T-cell lymphoma not otherwise specified (n=12), anaplastic large-cell lymphoma (n=4), enteropathy-associated T-cell lymphoma (n=4), and subcutaneous panniculitis-like T-cell lymphoma (n=2).
Most patients (87.2%) received chemotherapy as first-line treatment. Fifty percent were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, 48.1% were IMEP (ifosphamide, methotrexate, etoposide, and prednisolone) or IMEP-like regimens, and 1.9% were “other” regimens.
Other first-line treatments included radiotherapy followed by chemotherapy (10.1%), excision (0.9%), and no treatment (1.8%).
The patients’ median OS was 60.03 months, and the median PFS was 15.7 months.
BM involvement
The researchers analyzed PET BM uptake both visually and quantitatively using the marrow-to-liver ratio (MLR), and they compared these results to BM biopsy results.
According to BM biopsy, 35.8% of patients had tumor involvement.
By visual analysis, the sensitivity of PET for diagnosing positive BM biopsy was 58.5%, and the specificity was 77.9%. By MLR, the sensitivity was 64.1%, and the specificity was 72.9%.
The diagnostic performance of PET for BM involvement was not different across the lymphoma subtypes, Dr Koh said.
Prognosis
“Although FDG PET did not correlate very well with bone marrow biopsy, it had prognostic value, especially MLR,” Dr Koh noted. “And most importantly, in bone marrow biopsy-negative patients, it [MLR] had prognostic value.”
MLR was a significant prognostic factor for PFS (P=0.005) and OS (P<0.001). The same was true for BM biopsy (P=0.009 for PFS and P<0.001 for OS), while visual PET analysis was a significant prognostic factor for OS (P=0.015) but not PFS (P=0.476).
In patients negative by BM biopsy, MLR was a significant prognostic factor for PFS (P=0.001) and OS (P=0.005).
Dr Koh and his colleagues also analyzed the prognostic value of PET and BM biopsy specifically in patients with NKTCL and AITL.
In AITL patients, BM biopsy was a significant prognostic factor for OS (P=0.002) but not PFS (P=0.246). Visual PET analysis was not significant for PFS (P=0.910) or OS (P=0.581), and neither was MLR (P=0.053 for PFS and P=0.156 for OS).
In patients with NKTCL, BM biopsy was a significant prognostic factor for PFS (P=0.008) and OS (P<0.001). Visual PET analysis was not significant for PFS (P=0.469) or OS (P=0.092). And MLR was significant for PFS (P=0.004) and OS (P=0.012).
“Bone marrow findings of FDG PET are an independent prognostic factor in these tumors,” Dr Koh said, “suggesting the biologic relevance of FDG PET findings for aggressiveness or covert bone marrow involvement of tumor cells.” ![]()
LA JOLLA, CA—Fluorodeoxyglucose positron emission tomography (FDG PET) cannot replace bone marrow (BM) biopsy in T-cell lymphomas, according to a speaker at the 10th Annual T-cell Lymphoma Forum.
Researchers found that FDG PET results did not exactly correlate with BM biopsy results relating to tumor involvement in patients with T-cell lymphomas.
However, results from FDG PET were found to be an independent prognostic factor for progression-free survival (PFS) and overall survival (OS).
Youngil Koh, MD, of Seoul National University Hospital in Seoul, South Korea, presented this research in a poster and oral presentation at this year’s T-cell Lymphoma Forum.
He and his colleagues set out to investigate the clinical value of FDG PET for evaluating BM tumor involvement and prognosis in T-cell lymphoma patients.
The team analyzed 109 patients who underwent staging with FDG PET and BM biopsy. Most patients had extranodal natural killer/T-cell lymphoma, nasal type (NKTCL, n=46), or angioimmunoblastic T-cell lymphoma (AITL, n=41).
Patients also had peripheral T-cell lymphoma not otherwise specified (n=12), anaplastic large-cell lymphoma (n=4), enteropathy-associated T-cell lymphoma (n=4), and subcutaneous panniculitis-like T-cell lymphoma (n=2).
Most patients (87.2%) received chemotherapy as first-line treatment. Fifty percent were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, 48.1% were IMEP (ifosphamide, methotrexate, etoposide, and prednisolone) or IMEP-like regimens, and 1.9% were “other” regimens.
Other first-line treatments included radiotherapy followed by chemotherapy (10.1%), excision (0.9%), and no treatment (1.8%).
The patients’ median OS was 60.03 months, and the median PFS was 15.7 months.
BM involvement
The researchers analyzed PET BM uptake both visually and quantitatively using the marrow-to-liver ratio (MLR), and they compared these results to BM biopsy results.
According to BM biopsy, 35.8% of patients had tumor involvement.
By visual analysis, the sensitivity of PET for diagnosing positive BM biopsy was 58.5%, and the specificity was 77.9%. By MLR, the sensitivity was 64.1%, and the specificity was 72.9%.
The diagnostic performance of PET for BM involvement was not different across the lymphoma subtypes, Dr Koh said.
Prognosis
“Although FDG PET did not correlate very well with bone marrow biopsy, it had prognostic value, especially MLR,” Dr Koh noted. “And most importantly, in bone marrow biopsy-negative patients, it [MLR] had prognostic value.”
MLR was a significant prognostic factor for PFS (P=0.005) and OS (P<0.001). The same was true for BM biopsy (P=0.009 for PFS and P<0.001 for OS), while visual PET analysis was a significant prognostic factor for OS (P=0.015) but not PFS (P=0.476).
In patients negative by BM biopsy, MLR was a significant prognostic factor for PFS (P=0.001) and OS (P=0.005).
Dr Koh and his colleagues also analyzed the prognostic value of PET and BM biopsy specifically in patients with NKTCL and AITL.
In AITL patients, BM biopsy was a significant prognostic factor for OS (P=0.002) but not PFS (P=0.246). Visual PET analysis was not significant for PFS (P=0.910) or OS (P=0.581), and neither was MLR (P=0.053 for PFS and P=0.156 for OS).
In patients with NKTCL, BM biopsy was a significant prognostic factor for PFS (P=0.008) and OS (P<0.001). Visual PET analysis was not significant for PFS (P=0.469) or OS (P=0.092). And MLR was significant for PFS (P=0.004) and OS (P=0.012).
“Bone marrow findings of FDG PET are an independent prognostic factor in these tumors,” Dr Koh said, “suggesting the biologic relevance of FDG PET findings for aggressiveness or covert bone marrow involvement of tumor cells.” ![]()
Inhibitor provides clinical improvement in MF

LA JOLLA, CA—Results of a phase 1 trial suggest MRG-106 can provide clinical improvement in patients with mycosis fungoides (MF), whether the drug is given alone or in conjunction with other therapies.
MRG-106 is an inhibitor of microRNA-155, which is upregulated in MF.
In this ongoing trial, 90% of patients who received MRG-106 have experienced an improvement in mSWAT score, and 59% of patients who received the drug for at least 1 month had a partial response.
The most common adverse events (AEs) attributed to MRG-106 were neutropenia, injection site pain, and fatigue.
Christiane Querfeld, MD, PhD, of the City of Hope in Duarte, California, presented these results at the 10th Annual T-cell Lymphoma Forum. The research is sponsored by miRagen Therapeutics, Inc., the company developing MRG-106.
The trial has enrolled 36 MF patients, 69% of whom are male. Their median age at enrollment was 63 (range, 21-85).
Patients had received a median of 4 prior systemic therapies (range, 1-13) and a median of 3 prior skin-directed therapies (range, 1-8).
At baseline, patients had a median mSWAT score of 43 (range, 2-180). The modified Severity Weighted Assessment Tool (mSWAT) measures the severity of skin disease over a patient’s body.
Part A
In part A of the study, 6 patients received MRG-106 via intralesional injection. A 75 mg dose of the drug was found to be well-tolerated, producing generally minor injection site reactions.
In addition, intralesional injection of MRG-106 produced improvements in CAILS score. The Composite Assessment of Index Lesion Severity (CAILS) score is obtained by adding the severity score of erythema, scaling, plaque elevation, and surface area for up to 5 index lesions.
Part B
In part B, 30 patients received MRG-106 via subcutaneous (SQ) injection, intravenous (IV) infusion, or IV bolus injection.
Patients who received SQ injection or IV infusion received doses of 300 mg, 600 mg, or 900 mg. Those who received an IV bolus only received the 300 mg dose.
Twenty-nine of the 30 patients in part B were evaluable for efficacy. Twenty-six of these patients—90%—had an improvement in mSWAT score from baseline.
“Twenty-six patients had at least stable disease to partial response,” Dr Querfeld noted. “No complete responses yet, but we’re close.”
Twelve patients were still receiving MRG-106 at last follow-up.
Ten of the 17 patients (59%) who had received MRG-106 for more than 1 month had at least a 50% improvement in mSWAT score, or a partial response. Once this was achieved, responses were durable.
One patient was still in response at roughly 470 days of follow-up.
Concomitant therapies
Dr Querfeld and her colleagues looked at patient outcomes in the context of concomitant therapies as well. They analyzed data from 26 patients who had received at least 6 doses of MRG-106.
Half of these patients were receiving MRG-106 alone, and the other half were receiving concomitant therapies, including bexarotene (n=7), interferon-alfa (n=2), methotrexate (n=1), vorinostat (n=1), and “other” treatments (n=2). Patients had been receiving these therapies for anywhere from 4 months to 45 months.
Outcomes were similar in the monotherapy and combination treatment groups. Seven patients in each group had at least a 50% improvement in mSWAT score.
Dosing and administration
“It appears the infusion is superior to the subcutaneous administration,” Dr Querfeld said.
She noted that durable partial responses have been achieved at all dose levels, but the 300 mg and 600 mg IV infusions had the best efficacy and tolerability profiles.
With the 300 mg IV bolus, fewer patients remained on treatment for more than 1 cycle, as compared to the other dosing cohorts. Dr Querfeld said this may be a result of lower total exposure or tolerability due to higher plasma Cmax.
She also noted that patients who received MRG-106 SQ at 600 mg or higher had a higher incidence of injection site reactions.
Safety
AEs of any grade that were attributed to MRG-106 include neutropenia (16%), injection site pain (16%), fatigue (14%), nausea (5%), pruritus (5%), and headache (5%).
Grade 3/4 AEs attributed to MRG-106 were neutropenia (5%) and pruritus (5%).
There were no serious AEs attributed to MRG-106, but there were 2 dose-limiting toxicities. One was a grade 3 tumor flare in a patient receiving the 300 mg IV bolus.
The other dose-limiting toxicity was grade 3 worsening pruritus and possible tumor flare, which occurred twice in 1 patient—with the 900 mg SQ dose and with the 300 mg IV infusion.
The 300 mg IV infusion is the anticipated phase 2 dose. ![]()
LA JOLLA, CA—Results of a phase 1 trial suggest MRG-106 can provide clinical improvement in patients with mycosis fungoides (MF), whether the drug is given alone or in conjunction with other therapies.
MRG-106 is an inhibitor of microRNA-155, which is upregulated in MF.
In this ongoing trial, 90% of patients who received MRG-106 have experienced an improvement in mSWAT score, and 59% of patients who received the drug for at least 1 month had a partial response.
The most common adverse events (AEs) attributed to MRG-106 were neutropenia, injection site pain, and fatigue.
Christiane Querfeld, MD, PhD, of the City of Hope in Duarte, California, presented these results at the 10th Annual T-cell Lymphoma Forum. The research is sponsored by miRagen Therapeutics, Inc., the company developing MRG-106.
The trial has enrolled 36 MF patients, 69% of whom are male. Their median age at enrollment was 63 (range, 21-85).
Patients had received a median of 4 prior systemic therapies (range, 1-13) and a median of 3 prior skin-directed therapies (range, 1-8).
At baseline, patients had a median mSWAT score of 43 (range, 2-180). The modified Severity Weighted Assessment Tool (mSWAT) measures the severity of skin disease over a patient’s body.
Part A
In part A of the study, 6 patients received MRG-106 via intralesional injection. A 75 mg dose of the drug was found to be well-tolerated, producing generally minor injection site reactions.
In addition, intralesional injection of MRG-106 produced improvements in CAILS score. The Composite Assessment of Index Lesion Severity (CAILS) score is obtained by adding the severity score of erythema, scaling, plaque elevation, and surface area for up to 5 index lesions.
Part B
In part B, 30 patients received MRG-106 via subcutaneous (SQ) injection, intravenous (IV) infusion, or IV bolus injection.
Patients who received SQ injection or IV infusion received doses of 300 mg, 600 mg, or 900 mg. Those who received an IV bolus only received the 300 mg dose.
Twenty-nine of the 30 patients in part B were evaluable for efficacy. Twenty-six of these patients—90%—had an improvement in mSWAT score from baseline.
“Twenty-six patients had at least stable disease to partial response,” Dr Querfeld noted. “No complete responses yet, but we’re close.”
Twelve patients were still receiving MRG-106 at last follow-up.
Ten of the 17 patients (59%) who had received MRG-106 for more than 1 month had at least a 50% improvement in mSWAT score, or a partial response. Once this was achieved, responses were durable.
One patient was still in response at roughly 470 days of follow-up.
Concomitant therapies
Dr Querfeld and her colleagues looked at patient outcomes in the context of concomitant therapies as well. They analyzed data from 26 patients who had received at least 6 doses of MRG-106.
Half of these patients were receiving MRG-106 alone, and the other half were receiving concomitant therapies, including bexarotene (n=7), interferon-alfa (n=2), methotrexate (n=1), vorinostat (n=1), and “other” treatments (n=2). Patients had been receiving these therapies for anywhere from 4 months to 45 months.
Outcomes were similar in the monotherapy and combination treatment groups. Seven patients in each group had at least a 50% improvement in mSWAT score.
Dosing and administration
“It appears the infusion is superior to the subcutaneous administration,” Dr Querfeld said.
She noted that durable partial responses have been achieved at all dose levels, but the 300 mg and 600 mg IV infusions had the best efficacy and tolerability profiles.
With the 300 mg IV bolus, fewer patients remained on treatment for more than 1 cycle, as compared to the other dosing cohorts. Dr Querfeld said this may be a result of lower total exposure or tolerability due to higher plasma Cmax.
She also noted that patients who received MRG-106 SQ at 600 mg or higher had a higher incidence of injection site reactions.
Safety
AEs of any grade that were attributed to MRG-106 include neutropenia (16%), injection site pain (16%), fatigue (14%), nausea (5%), pruritus (5%), and headache (5%).
Grade 3/4 AEs attributed to MRG-106 were neutropenia (5%) and pruritus (5%).
There were no serious AEs attributed to MRG-106, but there were 2 dose-limiting toxicities. One was a grade 3 tumor flare in a patient receiving the 300 mg IV bolus.
The other dose-limiting toxicity was grade 3 worsening pruritus and possible tumor flare, which occurred twice in 1 patient—with the 900 mg SQ dose and with the 300 mg IV infusion.
The 300 mg IV infusion is the anticipated phase 2 dose. ![]()
LA JOLLA, CA—Results of a phase 1 trial suggest MRG-106 can provide clinical improvement in patients with mycosis fungoides (MF), whether the drug is given alone or in conjunction with other therapies.
MRG-106 is an inhibitor of microRNA-155, which is upregulated in MF.
In this ongoing trial, 90% of patients who received MRG-106 have experienced an improvement in mSWAT score, and 59% of patients who received the drug for at least 1 month had a partial response.
The most common adverse events (AEs) attributed to MRG-106 were neutropenia, injection site pain, and fatigue.
Christiane Querfeld, MD, PhD, of the City of Hope in Duarte, California, presented these results at the 10th Annual T-cell Lymphoma Forum. The research is sponsored by miRagen Therapeutics, Inc., the company developing MRG-106.
The trial has enrolled 36 MF patients, 69% of whom are male. Their median age at enrollment was 63 (range, 21-85).
Patients had received a median of 4 prior systemic therapies (range, 1-13) and a median of 3 prior skin-directed therapies (range, 1-8).
At baseline, patients had a median mSWAT score of 43 (range, 2-180). The modified Severity Weighted Assessment Tool (mSWAT) measures the severity of skin disease over a patient’s body.
Part A
In part A of the study, 6 patients received MRG-106 via intralesional injection. A 75 mg dose of the drug was found to be well-tolerated, producing generally minor injection site reactions.
In addition, intralesional injection of MRG-106 produced improvements in CAILS score. The Composite Assessment of Index Lesion Severity (CAILS) score is obtained by adding the severity score of erythema, scaling, plaque elevation, and surface area for up to 5 index lesions.
Part B
In part B, 30 patients received MRG-106 via subcutaneous (SQ) injection, intravenous (IV) infusion, or IV bolus injection.
Patients who received SQ injection or IV infusion received doses of 300 mg, 600 mg, or 900 mg. Those who received an IV bolus only received the 300 mg dose.
Twenty-nine of the 30 patients in part B were evaluable for efficacy. Twenty-six of these patients—90%—had an improvement in mSWAT score from baseline.
“Twenty-six patients had at least stable disease to partial response,” Dr Querfeld noted. “No complete responses yet, but we’re close.”
Twelve patients were still receiving MRG-106 at last follow-up.
Ten of the 17 patients (59%) who had received MRG-106 for more than 1 month had at least a 50% improvement in mSWAT score, or a partial response. Once this was achieved, responses were durable.
One patient was still in response at roughly 470 days of follow-up.
Concomitant therapies
Dr Querfeld and her colleagues looked at patient outcomes in the context of concomitant therapies as well. They analyzed data from 26 patients who had received at least 6 doses of MRG-106.
Half of these patients were receiving MRG-106 alone, and the other half were receiving concomitant therapies, including bexarotene (n=7), interferon-alfa (n=2), methotrexate (n=1), vorinostat (n=1), and “other” treatments (n=2). Patients had been receiving these therapies for anywhere from 4 months to 45 months.
Outcomes were similar in the monotherapy and combination treatment groups. Seven patients in each group had at least a 50% improvement in mSWAT score.
Dosing and administration
“It appears the infusion is superior to the subcutaneous administration,” Dr Querfeld said.
She noted that durable partial responses have been achieved at all dose levels, but the 300 mg and 600 mg IV infusions had the best efficacy and tolerability profiles.
With the 300 mg IV bolus, fewer patients remained on treatment for more than 1 cycle, as compared to the other dosing cohorts. Dr Querfeld said this may be a result of lower total exposure or tolerability due to higher plasma Cmax.
She also noted that patients who received MRG-106 SQ at 600 mg or higher had a higher incidence of injection site reactions.
Safety
AEs of any grade that were attributed to MRG-106 include neutropenia (16%), injection site pain (16%), fatigue (14%), nausea (5%), pruritus (5%), and headache (5%).
Grade 3/4 AEs attributed to MRG-106 were neutropenia (5%) and pruritus (5%).
There were no serious AEs attributed to MRG-106, but there were 2 dose-limiting toxicities. One was a grade 3 tumor flare in a patient receiving the 300 mg IV bolus.
The other dose-limiting toxicity was grade 3 worsening pruritus and possible tumor flare, which occurred twice in 1 patient—with the 900 mg SQ dose and with the 300 mg IV infusion.
The 300 mg IV infusion is the anticipated phase 2 dose. ![]()
Drugs appear comparable for delaying SREs in MM
In a phase 3 trial, denosumab proved non-inferior to zoledronic acid for delaying skeletal-related events (SREs) in patients with multiple myeloma (MM).
The median time to first on-study SRE was 23 months in the denosumab arm and 24 months in the zoledronic acid arm.
There were fewer renal adverse events (AEs) but more hypocalcemia AEs in the denosumab arm.
“Until recently, treatment options for the prevention of skeletal-related events in multiple myeloma were limited to bisphosphonates, which are cleared through the kidneys and can be associated with increased renal impairment,” said Noopur Raje, MD, of Massachusetts General Hospital Cancer Center in Boston.
“Denosumab, which is not cleared through the kidneys, provides a new treatment option for the prevention of skeletal-related events in patients with multiple myeloma.”
Dr Raje and her colleagues conducted this phase 3 trial of denosumab and reported the results in The Lancet Oncology. The trial was sponsored by Amgen, the company developing denosumab.
Denosumab is the first fully human monoclonal antibody that binds to and neutralizes RANK ligand—a protein essential for the formation, function, and survival of osteoclasts—thereby inhibiting osteoclast-mediated bone destruction.
In this trial, researchers compared denosumab to zoledronic acid for the prevention of SREs in adults with newly diagnosed MM and bone disease.
The team randomized 1718 patients to receive subcutaneous denosumab at 120 mg and intravenous placebo every 4 weeks (n=859) or intravenous zoledronic acid at 4 mg (adjusted for renal function at baseline) and subcutaneous placebo every 4 weeks (n=859). All patients also received investigators’ choice of first-line MM therapy.
Skeletal surveys using conventional radiography were obtained every 12 to 24 weeks per protocol. The primary endpoint of the study was non-inferiority of denosumab to zoledronic acid for time to first on-study SRE (pathologic fracture, radiation to bone, surgery to bone, or spinal cord compression).
The primary endpoint was met. The median time to first on-study SRE was 22.8 months for patients in the denosumab arm and 24 months for those in the zoledronic acid arm (hazard ratio [HR]=0.98; 95% confidence interval [CI]: 0.85-1.14; P non-inferiority=0.010).
Approximately 60% of all first SREs occurred within the first 3 months, and 81% occurred within the first 6 months.
Overall survival, a secondary endpoint, was similar between the denosumab and zoledronic acid arms (HR=0.90; 95% CI: 0.70-1.16; P=0.41).
There were fewer renal treatment-emergent AEs in the denosumab arm than the zoledronic acid arm—10% and 17%, respectively. There were more hypocalcemia AEs in the denosumab arm than the zoledronic acid arm—17% and 12%, respectively.
The incidence of osteonecrosis of the jaw was 4% in the denosumab arm and 3% in the zoledronic acid arm.
The most common grade 3 or higher treatment-emergent AEs (in the denosumab and zoledronic acid arms, respectively) were neutropenia (15% in both arms), thrombocytopenia (14% and 12%), anemia (12% and 10%), febrile neutropenia (11% and 10%), and pneumonia (8% in both arms).
The most common serious AE was pneumonia (8% in both arms).
Treatment-emergent AEs led to study drug discontinuation in 13% of patients in the denosumab arm and 12% in the zoledronic acid arm.
One patient in the zoledronic acid arm died of cardiac arrest that was deemed treatment-related. No other deaths were considered treatment-related. ![]()
In a phase 3 trial, denosumab proved non-inferior to zoledronic acid for delaying skeletal-related events (SREs) in patients with multiple myeloma (MM).
The median time to first on-study SRE was 23 months in the denosumab arm and 24 months in the zoledronic acid arm.
There were fewer renal adverse events (AEs) but more hypocalcemia AEs in the denosumab arm.
“Until recently, treatment options for the prevention of skeletal-related events in multiple myeloma were limited to bisphosphonates, which are cleared through the kidneys and can be associated with increased renal impairment,” said Noopur Raje, MD, of Massachusetts General Hospital Cancer Center in Boston.
“Denosumab, which is not cleared through the kidneys, provides a new treatment option for the prevention of skeletal-related events in patients with multiple myeloma.”
Dr Raje and her colleagues conducted this phase 3 trial of denosumab and reported the results in The Lancet Oncology. The trial was sponsored by Amgen, the company developing denosumab.
Denosumab is the first fully human monoclonal antibody that binds to and neutralizes RANK ligand—a protein essential for the formation, function, and survival of osteoclasts—thereby inhibiting osteoclast-mediated bone destruction.
In this trial, researchers compared denosumab to zoledronic acid for the prevention of SREs in adults with newly diagnosed MM and bone disease.
The team randomized 1718 patients to receive subcutaneous denosumab at 120 mg and intravenous placebo every 4 weeks (n=859) or intravenous zoledronic acid at 4 mg (adjusted for renal function at baseline) and subcutaneous placebo every 4 weeks (n=859). All patients also received investigators’ choice of first-line MM therapy.
Skeletal surveys using conventional radiography were obtained every 12 to 24 weeks per protocol. The primary endpoint of the study was non-inferiority of denosumab to zoledronic acid for time to first on-study SRE (pathologic fracture, radiation to bone, surgery to bone, or spinal cord compression).
The primary endpoint was met. The median time to first on-study SRE was 22.8 months for patients in the denosumab arm and 24 months for those in the zoledronic acid arm (hazard ratio [HR]=0.98; 95% confidence interval [CI]: 0.85-1.14; P non-inferiority=0.010).
Approximately 60% of all first SREs occurred within the first 3 months, and 81% occurred within the first 6 months.
Overall survival, a secondary endpoint, was similar between the denosumab and zoledronic acid arms (HR=0.90; 95% CI: 0.70-1.16; P=0.41).
There were fewer renal treatment-emergent AEs in the denosumab arm than the zoledronic acid arm—10% and 17%, respectively. There were more hypocalcemia AEs in the denosumab arm than the zoledronic acid arm—17% and 12%, respectively.
The incidence of osteonecrosis of the jaw was 4% in the denosumab arm and 3% in the zoledronic acid arm.
The most common grade 3 or higher treatment-emergent AEs (in the denosumab and zoledronic acid arms, respectively) were neutropenia (15% in both arms), thrombocytopenia (14% and 12%), anemia (12% and 10%), febrile neutropenia (11% and 10%), and pneumonia (8% in both arms).
The most common serious AE was pneumonia (8% in both arms).
Treatment-emergent AEs led to study drug discontinuation in 13% of patients in the denosumab arm and 12% in the zoledronic acid arm.
One patient in the zoledronic acid arm died of cardiac arrest that was deemed treatment-related. No other deaths were considered treatment-related. ![]()
In a phase 3 trial, denosumab proved non-inferior to zoledronic acid for delaying skeletal-related events (SREs) in patients with multiple myeloma (MM).
The median time to first on-study SRE was 23 months in the denosumab arm and 24 months in the zoledronic acid arm.
There were fewer renal adverse events (AEs) but more hypocalcemia AEs in the denosumab arm.
“Until recently, treatment options for the prevention of skeletal-related events in multiple myeloma were limited to bisphosphonates, which are cleared through the kidneys and can be associated with increased renal impairment,” said Noopur Raje, MD, of Massachusetts General Hospital Cancer Center in Boston.
“Denosumab, which is not cleared through the kidneys, provides a new treatment option for the prevention of skeletal-related events in patients with multiple myeloma.”
Dr Raje and her colleagues conducted this phase 3 trial of denosumab and reported the results in The Lancet Oncology. The trial was sponsored by Amgen, the company developing denosumab.
Denosumab is the first fully human monoclonal antibody that binds to and neutralizes RANK ligand—a protein essential for the formation, function, and survival of osteoclasts—thereby inhibiting osteoclast-mediated bone destruction.
In this trial, researchers compared denosumab to zoledronic acid for the prevention of SREs in adults with newly diagnosed MM and bone disease.
The team randomized 1718 patients to receive subcutaneous denosumab at 120 mg and intravenous placebo every 4 weeks (n=859) or intravenous zoledronic acid at 4 mg (adjusted for renal function at baseline) and subcutaneous placebo every 4 weeks (n=859). All patients also received investigators’ choice of first-line MM therapy.
Skeletal surveys using conventional radiography were obtained every 12 to 24 weeks per protocol. The primary endpoint of the study was non-inferiority of denosumab to zoledronic acid for time to first on-study SRE (pathologic fracture, radiation to bone, surgery to bone, or spinal cord compression).
The primary endpoint was met. The median time to first on-study SRE was 22.8 months for patients in the denosumab arm and 24 months for those in the zoledronic acid arm (hazard ratio [HR]=0.98; 95% confidence interval [CI]: 0.85-1.14; P non-inferiority=0.010).
Approximately 60% of all first SREs occurred within the first 3 months, and 81% occurred within the first 6 months.
Overall survival, a secondary endpoint, was similar between the denosumab and zoledronic acid arms (HR=0.90; 95% CI: 0.70-1.16; P=0.41).
There were fewer renal treatment-emergent AEs in the denosumab arm than the zoledronic acid arm—10% and 17%, respectively. There were more hypocalcemia AEs in the denosumab arm than the zoledronic acid arm—17% and 12%, respectively.
The incidence of osteonecrosis of the jaw was 4% in the denosumab arm and 3% in the zoledronic acid arm.
The most common grade 3 or higher treatment-emergent AEs (in the denosumab and zoledronic acid arms, respectively) were neutropenia (15% in both arms), thrombocytopenia (14% and 12%), anemia (12% and 10%), febrile neutropenia (11% and 10%), and pneumonia (8% in both arms).
The most common serious AE was pneumonia (8% in both arms).
Treatment-emergent AEs led to study drug discontinuation in 13% of patients in the denosumab arm and 12% in the zoledronic acid arm.
One patient in the zoledronic acid arm died of cardiac arrest that was deemed treatment-related. No other deaths were considered treatment-related. ![]()
Tamsulosin for Patients With Ureteral Stones?

A 54-year-old man presents to the emergency department (ED) with acute-onset left flank pain that radiates to the groin. CT of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed an appropriate candidate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8%, with a self-reported prevalence of 10.6% in men and 7.1% in women.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with NSAIDs as firstline treatment and opioids as a second-line option.3
In addition, α-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the α-blocker tamsulosin. Two systematic reviews (limited by heterogeneity because some of the studies lacked a placebo control and blinding) concluded that α-blockers increased stone passage within one to six weeks when compared with placebo or no additional therapy.4,5 However, a recent large, multicenter RCT revealed no difference between tamsulosin and nifedipine, or either one compared with placebo, at decreasing the need for further treatment to achieve stone passage within four weeks.6
STUDY SUMMARY
Results broken down by stone size
This meta-analysis, comprising eight double-blind RCTs, examined the effect of oral tamsulosin (0.4 mg/d; average course, 28 d) on distal ureteral stone passage in adult patients (N = 1,384).1 A subgroup analysis comparing stone size (< 5 mm and 5-10 mm) was also conducted to determine whether size modified the effect of tamsulosin.
The eight selected studies were published between 2009 and 2015; the trials were conducted in multiple countries, in ED and outpatient urology settings. The main outcome measure was the risk difference (RD) in stone passage between the tamsulosin group and placebo group after follow-up imaging at three weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk for stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; RD, 17%), but significant heterogeneity existed across the trials (I2, 80.2%). Subgroup analysis by stone size (< 5 mm vs 5-10 mm) revealed that, compared to placebo, tamsulosin was beneficial for larger stones (6 trials, N = 514; RD, 22%; number needed to treat, 5) but not for smaller stones (4 trials, N = 533; RD, –0.3%). The 5-to-10–mm subgroup had a less heterogeneous population of studies than did the < 5-mm subgroup (I2, 33% and 0% respectively).
In terms of adverse events, tamsulosin did not increase the risk for dizziness (RD, 0.2%) or postural hypotension (RD, 0.1%), compared with placebo.
WHAT’S NEW
Increased passage of larger stones
This meta-analysis included only double-blind RCTs; prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Clinician Reviews. 2016;26[4]:20,44), which recommended against the use of α-blockers tamsulosin and nifedipine for ureteral stones measuring < 10 mm.6,7
But the subgroup analysis in this review went one step further by examining passage rates by stone size (< 5 mm vs 5-10 mm) and revealing that passage of larger stones increased with tamsulosin use. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
What about proximal or XL stones?
Only distal stones were included in seven of the eight trials in this analysis. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones > 10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[1]:37-38).
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69(3):353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62(1): 160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015; 386(9991):341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65(2): 118-120.
A 54-year-old man presents to the emergency department (ED) with acute-onset left flank pain that radiates to the groin. CT of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed an appropriate candidate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8%, with a self-reported prevalence of 10.6% in men and 7.1% in women.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with NSAIDs as firstline treatment and opioids as a second-line option.3
In addition, α-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the α-blocker tamsulosin. Two systematic reviews (limited by heterogeneity because some of the studies lacked a placebo control and blinding) concluded that α-blockers increased stone passage within one to six weeks when compared with placebo or no additional therapy.4,5 However, a recent large, multicenter RCT revealed no difference between tamsulosin and nifedipine, or either one compared with placebo, at decreasing the need for further treatment to achieve stone passage within four weeks.6
STUDY SUMMARY
Results broken down by stone size
This meta-analysis, comprising eight double-blind RCTs, examined the effect of oral tamsulosin (0.4 mg/d; average course, 28 d) on distal ureteral stone passage in adult patients (N = 1,384).1 A subgroup analysis comparing stone size (< 5 mm and 5-10 mm) was also conducted to determine whether size modified the effect of tamsulosin.
The eight selected studies were published between 2009 and 2015; the trials were conducted in multiple countries, in ED and outpatient urology settings. The main outcome measure was the risk difference (RD) in stone passage between the tamsulosin group and placebo group after follow-up imaging at three weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk for stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; RD, 17%), but significant heterogeneity existed across the trials (I2, 80.2%). Subgroup analysis by stone size (< 5 mm vs 5-10 mm) revealed that, compared to placebo, tamsulosin was beneficial for larger stones (6 trials, N = 514; RD, 22%; number needed to treat, 5) but not for smaller stones (4 trials, N = 533; RD, –0.3%). The 5-to-10–mm subgroup had a less heterogeneous population of studies than did the < 5-mm subgroup (I2, 33% and 0% respectively).
In terms of adverse events, tamsulosin did not increase the risk for dizziness (RD, 0.2%) or postural hypotension (RD, 0.1%), compared with placebo.
WHAT’S NEW
Increased passage of larger stones
This meta-analysis included only double-blind RCTs; prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Clinician Reviews. 2016;26[4]:20,44), which recommended against the use of α-blockers tamsulosin and nifedipine for ureteral stones measuring < 10 mm.6,7
But the subgroup analysis in this review went one step further by examining passage rates by stone size (< 5 mm vs 5-10 mm) and revealing that passage of larger stones increased with tamsulosin use. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
What about proximal or XL stones?
Only distal stones were included in seven of the eight trials in this analysis. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones > 10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[1]:37-38).
A 54-year-old man presents to the emergency department (ED) with acute-onset left flank pain that radiates to the groin. CT of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed an appropriate candidate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8%, with a self-reported prevalence of 10.6% in men and 7.1% in women.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with NSAIDs as firstline treatment and opioids as a second-line option.3
In addition, α-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the α-blocker tamsulosin. Two systematic reviews (limited by heterogeneity because some of the studies lacked a placebo control and blinding) concluded that α-blockers increased stone passage within one to six weeks when compared with placebo or no additional therapy.4,5 However, a recent large, multicenter RCT revealed no difference between tamsulosin and nifedipine, or either one compared with placebo, at decreasing the need for further treatment to achieve stone passage within four weeks.6
STUDY SUMMARY
Results broken down by stone size
This meta-analysis, comprising eight double-blind RCTs, examined the effect of oral tamsulosin (0.4 mg/d; average course, 28 d) on distal ureteral stone passage in adult patients (N = 1,384).1 A subgroup analysis comparing stone size (< 5 mm and 5-10 mm) was also conducted to determine whether size modified the effect of tamsulosin.
The eight selected studies were published between 2009 and 2015; the trials were conducted in multiple countries, in ED and outpatient urology settings. The main outcome measure was the risk difference (RD) in stone passage between the tamsulosin group and placebo group after follow-up imaging at three weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk for stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; RD, 17%), but significant heterogeneity existed across the trials (I2, 80.2%). Subgroup analysis by stone size (< 5 mm vs 5-10 mm) revealed that, compared to placebo, tamsulosin was beneficial for larger stones (6 trials, N = 514; RD, 22%; number needed to treat, 5) but not for smaller stones (4 trials, N = 533; RD, –0.3%). The 5-to-10–mm subgroup had a less heterogeneous population of studies than did the < 5-mm subgroup (I2, 33% and 0% respectively).
In terms of adverse events, tamsulosin did not increase the risk for dizziness (RD, 0.2%) or postural hypotension (RD, 0.1%), compared with placebo.
WHAT’S NEW
Increased passage of larger stones
This meta-analysis included only double-blind RCTs; prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Clinician Reviews. 2016;26[4]:20,44), which recommended against the use of α-blockers tamsulosin and nifedipine for ureteral stones measuring < 10 mm.6,7
But the subgroup analysis in this review went one step further by examining passage rates by stone size (< 5 mm vs 5-10 mm) and revealing that passage of larger stones increased with tamsulosin use. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
What about proximal or XL stones?
Only distal stones were included in seven of the eight trials in this analysis. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones > 10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[1]:37-38).
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69(3):353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62(1): 160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015; 386(9991):341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65(2): 118-120.
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69(3):353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62(1): 160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015; 386(9991):341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65(2): 118-120.

Numb Toes and Other Woes: Diabetic Peripheral and Autonomic Neuropathies
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.

Study identifies characteristics that may constitute the MS prodrome
SAN DIEGO – Compared with controls, patients who developed MS were more frequently admitted to the hospital or visited a physician for problems related to the nervous system, sensory organs, musculoskeletal system, and genitourinary system in the 5 years prior to MS onset, a multicenter matched cohort study found.
“People are seeking help for different conditions that are most likely related to their MS,” one of the study authors, Elaine Kingwell, PhD, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “It suggests there could be opportunity to recognize and possibly diagnose and treat MS earlier.”

In a previously published study (Lancet Neurol. 2017;16[6]:445-51) led by Helen Tremlett, PhD, and first author Jose Wijnands, PhD, the team found increased health care utilization in people with MS across all health sectors – physician, hospital, and pharmacy (prescriptions filled).
For the current study, the team set out to identify early signs and symptoms that could define the MS prodrome. “We wanted to know why people went to the hospital, why people went to their physician, what kind of drugs they were prescribed, and what kind of specialists they saw,” explained Dr. Kingwell, an epidemiologist in the department of neurology at the University of British Columbia, Vancouver.
The researchers used health administrative and MS-specific data from four Canadian provinces to conduct a multicenter matched cohort study. Individuals were required to be in the province from 5 years pre-index date, measured as either the first demyelinating event in health administrative data or MS onset as determined by the treating neurologist, until the MS cases’ third demyelinating claim or diagnosis date. The potential for a prodromal period was examined in the 5 years pre-index date, and outcomes of interest were number of physician and hospital encounters per ICD-10 chapter, number of physician encounters per physician specialty, and percentage of people with one or more filled prescription per drug class.
The researchers used rate ratios and 95% confidence intervals to compare the rates of physician and hospital encounters between MS cases and controls and the proportion test to compare the percentage of people with one or more filled prescriptions between MS cases and controls. They used random-effects meta-analyses to pool results across Canadian provinces.
The population consisted of 13,951 MS patients and 66,940 controls derived from a health administration cohort and 3,202 MS patients and 16,006 controls derived from MS clinics. Compared with controls, in the 5 years before the first demyelinating claim or symptom onset, cases had more physician and hospital encounters for the nervous system (rate ratio = 1.70-4.75), sensory organs (RR = 1.40-2.28), musculoskeletal system (RR = 1.19-1.70), and genitourinary system (RR = 1.17-1.59), Dr. Kingwell and her associates reported.

Cases also had more encounters with psychiatrists (RR = 1.48-1.66) and urologists (RR = 1.49-1.80), and a higher proportion of filled prescriptions for hormonal preparations and drugs related to the musculoskeletal or genitourinary systems (ranging from 1.1 to 1.5 times higher; P less than .02 for all associations). “While we did not examine each individual drug, we did group drugs by their therapeutic class,” Dr. Wijnands noted in an interview after the meeting. “As for the increased number of visits to psychiatrists, it is intriguing. We don’t necessarily know all the reasons why. It opens up a lot of questions for people to follow up on.”
In contrast, MS cases had fewer pregnancy-related encounters, compared with controls (RR = 0.78-0.88).
Dr. Wijnands acknowledged certain limitations of the study, including its reliance on administrative data to measure the prodromal period. “When we described the prodrome itself, we relied on ICD codes from the physician and hospital data as well as prescriptions filled. Issues or problems for which individuals do not seek medical attention, for instance, would not be captured in our study,” she said.
The study was funded by the National MS Society. Dr. Wijnands receives salary support from the Michael Smith Foundation for Health Research /The Koehle Family Foundation, coauthor Ruth Ann Marrie, MD, PhD, holds the Waugh Family Chair in Multiple Sclerosis, and Dr. Tremlett is funded by the Canada Research Chair Program. Dr. Kingwell reported having no financial disclosures.
[email protected]
SOURCE: Kingwell E et al. ACTRIMS Forum 2018, late-breaking poster 254.
SAN DIEGO – Compared with controls, patients who developed MS were more frequently admitted to the hospital or visited a physician for problems related to the nervous system, sensory organs, musculoskeletal system, and genitourinary system in the 5 years prior to MS onset, a multicenter matched cohort study found.
“People are seeking help for different conditions that are most likely related to their MS,” one of the study authors, Elaine Kingwell, PhD, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “It suggests there could be opportunity to recognize and possibly diagnose and treat MS earlier.”
In a previously published study (Lancet Neurol. 2017;16[6]:445-51) led by Helen Tremlett, PhD, and first author Jose Wijnands, PhD, the team found increased health care utilization in people with MS across all health sectors – physician, hospital, and pharmacy (prescriptions filled).
For the current study, the team set out to identify early signs and symptoms that could define the MS prodrome. “We wanted to know why people went to the hospital, why people went to their physician, what kind of drugs they were prescribed, and what kind of specialists they saw,” explained Dr. Kingwell, an epidemiologist in the department of neurology at the University of British Columbia, Vancouver.
The researchers used health administrative and MS-specific data from four Canadian provinces to conduct a multicenter matched cohort study. Individuals were required to be in the province from 5 years pre-index date, measured as either the first demyelinating event in health administrative data or MS onset as determined by the treating neurologist, until the MS cases’ third demyelinating claim or diagnosis date. The potential for a prodromal period was examined in the 5 years pre-index date, and outcomes of interest were number of physician and hospital encounters per ICD-10 chapter, number of physician encounters per physician specialty, and percentage of people with one or more filled prescription per drug class.
The researchers used rate ratios and 95% confidence intervals to compare the rates of physician and hospital encounters between MS cases and controls and the proportion test to compare the percentage of people with one or more filled prescriptions between MS cases and controls. They used random-effects meta-analyses to pool results across Canadian provinces.
The population consisted of 13,951 MS patients and 66,940 controls derived from a health administration cohort and 3,202 MS patients and 16,006 controls derived from MS clinics. Compared with controls, in the 5 years before the first demyelinating claim or symptom onset, cases had more physician and hospital encounters for the nervous system (rate ratio = 1.70-4.75), sensory organs (RR = 1.40-2.28), musculoskeletal system (RR = 1.19-1.70), and genitourinary system (RR = 1.17-1.59), Dr. Kingwell and her associates reported.
Cases also had more encounters with psychiatrists (RR = 1.48-1.66) and urologists (RR = 1.49-1.80), and a higher proportion of filled prescriptions for hormonal preparations and drugs related to the musculoskeletal or genitourinary systems (ranging from 1.1 to 1.5 times higher; P less than .02 for all associations). “While we did not examine each individual drug, we did group drugs by their therapeutic class,” Dr. Wijnands noted in an interview after the meeting. “As for the increased number of visits to psychiatrists, it is intriguing. We don’t necessarily know all the reasons why. It opens up a lot of questions for people to follow up on.”
In contrast, MS cases had fewer pregnancy-related encounters, compared with controls (RR = 0.78-0.88).
Dr. Wijnands acknowledged certain limitations of the study, including its reliance on administrative data to measure the prodromal period. “When we described the prodrome itself, we relied on ICD codes from the physician and hospital data as well as prescriptions filled. Issues or problems for which individuals do not seek medical attention, for instance, would not be captured in our study,” she said.
The study was funded by the National MS Society. Dr. Wijnands receives salary support from the Michael Smith Foundation for Health Research /The Koehle Family Foundation, coauthor Ruth Ann Marrie, MD, PhD, holds the Waugh Family Chair in Multiple Sclerosis, and Dr. Tremlett is funded by the Canada Research Chair Program. Dr. Kingwell reported having no financial disclosures.
[email protected]
SOURCE: Kingwell E et al. ACTRIMS Forum 2018, late-breaking poster 254.
SAN DIEGO – Compared with controls, patients who developed MS were more frequently admitted to the hospital or visited a physician for problems related to the nervous system, sensory organs, musculoskeletal system, and genitourinary system in the 5 years prior to MS onset, a multicenter matched cohort study found.
“People are seeking help for different conditions that are most likely related to their MS,” one of the study authors, Elaine Kingwell, PhD, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “It suggests there could be opportunity to recognize and possibly diagnose and treat MS earlier.”
In a previously published study (Lancet Neurol. 2017;16[6]:445-51) led by Helen Tremlett, PhD, and first author Jose Wijnands, PhD, the team found increased health care utilization in people with MS across all health sectors – physician, hospital, and pharmacy (prescriptions filled).
For the current study, the team set out to identify early signs and symptoms that could define the MS prodrome. “We wanted to know why people went to the hospital, why people went to their physician, what kind of drugs they were prescribed, and what kind of specialists they saw,” explained Dr. Kingwell, an epidemiologist in the department of neurology at the University of British Columbia, Vancouver.
The researchers used health administrative and MS-specific data from four Canadian provinces to conduct a multicenter matched cohort study. Individuals were required to be in the province from 5 years pre-index date, measured as either the first demyelinating event in health administrative data or MS onset as determined by the treating neurologist, until the MS cases’ third demyelinating claim or diagnosis date. The potential for a prodromal period was examined in the 5 years pre-index date, and outcomes of interest were number of physician and hospital encounters per ICD-10 chapter, number of physician encounters per physician specialty, and percentage of people with one or more filled prescription per drug class.
The researchers used rate ratios and 95% confidence intervals to compare the rates of physician and hospital encounters between MS cases and controls and the proportion test to compare the percentage of people with one or more filled prescriptions between MS cases and controls. They used random-effects meta-analyses to pool results across Canadian provinces.
The population consisted of 13,951 MS patients and 66,940 controls derived from a health administration cohort and 3,202 MS patients and 16,006 controls derived from MS clinics. Compared with controls, in the 5 years before the first demyelinating claim or symptom onset, cases had more physician and hospital encounters for the nervous system (rate ratio = 1.70-4.75), sensory organs (RR = 1.40-2.28), musculoskeletal system (RR = 1.19-1.70), and genitourinary system (RR = 1.17-1.59), Dr. Kingwell and her associates reported.
Cases also had more encounters with psychiatrists (RR = 1.48-1.66) and urologists (RR = 1.49-1.80), and a higher proportion of filled prescriptions for hormonal preparations and drugs related to the musculoskeletal or genitourinary systems (ranging from 1.1 to 1.5 times higher; P less than .02 for all associations). “While we did not examine each individual drug, we did group drugs by their therapeutic class,” Dr. Wijnands noted in an interview after the meeting. “As for the increased number of visits to psychiatrists, it is intriguing. We don’t necessarily know all the reasons why. It opens up a lot of questions for people to follow up on.”
In contrast, MS cases had fewer pregnancy-related encounters, compared with controls (RR = 0.78-0.88).
Dr. Wijnands acknowledged certain limitations of the study, including its reliance on administrative data to measure the prodromal period. “When we described the prodrome itself, we relied on ICD codes from the physician and hospital data as well as prescriptions filled. Issues or problems for which individuals do not seek medical attention, for instance, would not be captured in our study,” she said.
The study was funded by the National MS Society. Dr. Wijnands receives salary support from the Michael Smith Foundation for Health Research /The Koehle Family Foundation, coauthor Ruth Ann Marrie, MD, PhD, holds the Waugh Family Chair in Multiple Sclerosis, and Dr. Tremlett is funded by the Canada Research Chair Program. Dr. Kingwell reported having no financial disclosures.
[email protected]
SOURCE: Kingwell E et al. ACTRIMS Forum 2018, late-breaking poster 254.
REPORTING FROM ACTRIMS FORUM 2018
Key clinical point: Several phenotypic characteristics may constitute the MS prodrome.
Major finding: Compared with controls, in the 5 years before the first demyelinating claim or symptom onset, MS cases had more physician and hospital encounters for the nervous system (rate ratio = 1.70-4.75), sensory organs (RR = 1.40-2.28), and musculoskeletal system (RR = 1.19-1.70).
Study details: A multicenter matched cohort study of 13,951 MS patients and 66,940 controls derived from a health administration cohort and 3,202 MS patients and 16,006 controls derived from MS clinics.
Disclosures: The study was funded by the National MS Society. Dr. Wijnands receives salary support from the Michael Smith Foundation for Health Research /The Koehle Family Foundation, coauthor Ruth Ann Marrie, MD, PhD, holds the Waugh Family Chair in Multiple Sclerosis, and Dr. Tremlett is funded by the Canada Research Chair Program. Dr. Kingwell reported having no financial disclosures.
Source: Kingwell E et al. ACTRIMS Forum 2018, late-breaking poster 254.
Physicians often bypass cognition, depression screening in MS
SAN DIEGO – A new study finds that physicians at two . Physicians who did perform screening hardly ever used validated tools and often didn’t refer appropriate patients for higher-level care.
In addition, researchers interviewed 13 leading MS specialists from coast to coast and “found that about half reported not using formal screening tools to assess cognitive impairment and depression,” said study coauthor Tamar Sapir, PhD, chief scientific officer with Prime Education, a firm based in Fort Lauderdale, Fla., that provides a variety of health-related services such as training and research.

The study findings were presented at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. Three of the study authors spoke in interviews.
The researchers sought to understand how frequently MS patients are screened for cognitive problems and depression.
“Cognitive impairment is experienced by approximately half of patients with multiple sclerosis, yet many are never screened or treated, which can impact their daily activities, their ability to work, and overall quality of life,” Dr. Sapir said.
Depression, meanwhile, is believed to be much more common in patients with MS than in the general population, with one recent meta-analysis of 58 studies finding that the average prevalence was 31%. Other research suggests depression is underdiagnosed and undertreated in this population (J Neurol Sci. 2017 Jan 15;372:331-41; ISRN Neurology. 2012, Article ID 427102. doi: 10.5402/2012/427102).
For the current study, researchers tracked 300 patients at two unidentified MS clinics via their charts over a 2-year period from 2014 to 2016. Their median age was 52 years, 76% were women, and 15 had experienced at least one relapse within the previous 24 months.

“Screening for cognitive impairment and depression was documented for only 52% and 63% of MS patients, respectively, and only about a quarter of patients diagnosed with these conditions were referred to a higher level of care,” said lead author Guy J. Buckle, MD, MPH, of the Andrew C. Carlos MS Institute at Shepherd Center in Atlanta.
Among all 300 patients, just 2% and 4% were screened using a validated tool for cognitive impairment and depression, respectively.
The screening often turned up evidence of the conditions: Physicians saw signs of cognitive impairment in 69% and 78% of those screened aged under 65 years and aged 65 and older, respectively, and they detected depression in 71% and 54% of those screened in those two age groups, respectively.
Researchers also noted several disparities. “Cognitive screening was conducted more frequently in older, employed, or white patients, while the presence of cognitive impairment was documented more often in black, nonworking, and those on Medicare or Medicaid,” Dr. Buckle said. “Depression screening was performed most frequently in older or white patients, yet depression was more common in younger, nonworking patients and those on Medicare/Medicaid.”
In another part of their study, researchers surveyed 13 unidentified “national leaders” in MS research and treatment. Just seven said they used validated tools to screen for cognitive impairment and six said they used them to screen for depression.
“We hear from MS specialists that they want to be measuring for cognition but don’t know how to efficiently work it into their routine, how to approach the patient, and what tools to use,” said study coauthor Derrick S. Robertson, MD, of the University of South Florida, Tampa. “In addition, there is no one tool that is accepted in the MS treatment community.”

MS specialists who didn’t use the screening tests also pointed to factors like lack of reimbursement and lack of integration into electronic medical records. “Doubt very much that neurologists have time to use any of these tests,” one respondent said, referring to cognitive impairment screening.
What’s next? “There are several new exciting developments in clinical trials demonstrating efficacy of disease-modifying therapies in maintaining or improving cognition in patients with relapsing MS,” Dr. Robertson said. “This highlights the urgent need to overcome barriers to use of formal cognitive screening tools in clinical practice to identify patients who need a higher level of care, and perhaps even a change in treatment with the ultimate goal to improve quality of life and overall outcomes.”
Genentech funded the study through an educational grant. Dr. Sapir and three other study authors reported no relevant disclosures. Dr. Buckle and Dr. Robertson reported multiple disclosures, including principle investigator and advisory board/panel member work.
SOURCE: Buckle GJ et al. ACTRIMS Forum 2018, abstract No. P161.
SAN DIEGO – A new study finds that physicians at two . Physicians who did perform screening hardly ever used validated tools and often didn’t refer appropriate patients for higher-level care.
In addition, researchers interviewed 13 leading MS specialists from coast to coast and “found that about half reported not using formal screening tools to assess cognitive impairment and depression,” said study coauthor Tamar Sapir, PhD, chief scientific officer with Prime Education, a firm based in Fort Lauderdale, Fla., that provides a variety of health-related services such as training and research.
The study findings were presented at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. Three of the study authors spoke in interviews.
The researchers sought to understand how frequently MS patients are screened for cognitive problems and depression.
“Cognitive impairment is experienced by approximately half of patients with multiple sclerosis, yet many are never screened or treated, which can impact their daily activities, their ability to work, and overall quality of life,” Dr. Sapir said.
Depression, meanwhile, is believed to be much more common in patients with MS than in the general population, with one recent meta-analysis of 58 studies finding that the average prevalence was 31%. Other research suggests depression is underdiagnosed and undertreated in this population (J Neurol Sci. 2017 Jan 15;372:331-41; ISRN Neurology. 2012, Article ID 427102. doi: 10.5402/2012/427102).
For the current study, researchers tracked 300 patients at two unidentified MS clinics via their charts over a 2-year period from 2014 to 2016. Their median age was 52 years, 76% were women, and 15 had experienced at least one relapse within the previous 24 months.
“Screening for cognitive impairment and depression was documented for only 52% and 63% of MS patients, respectively, and only about a quarter of patients diagnosed with these conditions were referred to a higher level of care,” said lead author Guy J. Buckle, MD, MPH, of the Andrew C. Carlos MS Institute at Shepherd Center in Atlanta.
Among all 300 patients, just 2% and 4% were screened using a validated tool for cognitive impairment and depression, respectively.
The screening often turned up evidence of the conditions: Physicians saw signs of cognitive impairment in 69% and 78% of those screened aged under 65 years and aged 65 and older, respectively, and they detected depression in 71% and 54% of those screened in those two age groups, respectively.
Researchers also noted several disparities. “Cognitive screening was conducted more frequently in older, employed, or white patients, while the presence of cognitive impairment was documented more often in black, nonworking, and those on Medicare or Medicaid,” Dr. Buckle said. “Depression screening was performed most frequently in older or white patients, yet depression was more common in younger, nonworking patients and those on Medicare/Medicaid.”
In another part of their study, researchers surveyed 13 unidentified “national leaders” in MS research and treatment. Just seven said they used validated tools to screen for cognitive impairment and six said they used them to screen for depression.
“We hear from MS specialists that they want to be measuring for cognition but don’t know how to efficiently work it into their routine, how to approach the patient, and what tools to use,” said study coauthor Derrick S. Robertson, MD, of the University of South Florida, Tampa. “In addition, there is no one tool that is accepted in the MS treatment community.”
MS specialists who didn’t use the screening tests also pointed to factors like lack of reimbursement and lack of integration into electronic medical records. “Doubt very much that neurologists have time to use any of these tests,” one respondent said, referring to cognitive impairment screening.
What’s next? “There are several new exciting developments in clinical trials demonstrating efficacy of disease-modifying therapies in maintaining or improving cognition in patients with relapsing MS,” Dr. Robertson said. “This highlights the urgent need to overcome barriers to use of formal cognitive screening tools in clinical practice to identify patients who need a higher level of care, and perhaps even a change in treatment with the ultimate goal to improve quality of life and overall outcomes.”
Genentech funded the study through an educational grant. Dr. Sapir and three other study authors reported no relevant disclosures. Dr. Buckle and Dr. Robertson reported multiple disclosures, including principle investigator and advisory board/panel member work.
SOURCE: Buckle GJ et al. ACTRIMS Forum 2018, abstract No. P161.
SAN DIEGO – A new study finds that physicians at two . Physicians who did perform screening hardly ever used validated tools and often didn’t refer appropriate patients for higher-level care.
In addition, researchers interviewed 13 leading MS specialists from coast to coast and “found that about half reported not using formal screening tools to assess cognitive impairment and depression,” said study coauthor Tamar Sapir, PhD, chief scientific officer with Prime Education, a firm based in Fort Lauderdale, Fla., that provides a variety of health-related services such as training and research.
The study findings were presented at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. Three of the study authors spoke in interviews.
The researchers sought to understand how frequently MS patients are screened for cognitive problems and depression.
“Cognitive impairment is experienced by approximately half of patients with multiple sclerosis, yet many are never screened or treated, which can impact their daily activities, their ability to work, and overall quality of life,” Dr. Sapir said.
Depression, meanwhile, is believed to be much more common in patients with MS than in the general population, with one recent meta-analysis of 58 studies finding that the average prevalence was 31%. Other research suggests depression is underdiagnosed and undertreated in this population (J Neurol Sci. 2017 Jan 15;372:331-41; ISRN Neurology. 2012, Article ID 427102. doi: 10.5402/2012/427102).
For the current study, researchers tracked 300 patients at two unidentified MS clinics via their charts over a 2-year period from 2014 to 2016. Their median age was 52 years, 76% were women, and 15 had experienced at least one relapse within the previous 24 months.
“Screening for cognitive impairment and depression was documented for only 52% and 63% of MS patients, respectively, and only about a quarter of patients diagnosed with these conditions were referred to a higher level of care,” said lead author Guy J. Buckle, MD, MPH, of the Andrew C. Carlos MS Institute at Shepherd Center in Atlanta.
Among all 300 patients, just 2% and 4% were screened using a validated tool for cognitive impairment and depression, respectively.
The screening often turned up evidence of the conditions: Physicians saw signs of cognitive impairment in 69% and 78% of those screened aged under 65 years and aged 65 and older, respectively, and they detected depression in 71% and 54% of those screened in those two age groups, respectively.
Researchers also noted several disparities. “Cognitive screening was conducted more frequently in older, employed, or white patients, while the presence of cognitive impairment was documented more often in black, nonworking, and those on Medicare or Medicaid,” Dr. Buckle said. “Depression screening was performed most frequently in older or white patients, yet depression was more common in younger, nonworking patients and those on Medicare/Medicaid.”
In another part of their study, researchers surveyed 13 unidentified “national leaders” in MS research and treatment. Just seven said they used validated tools to screen for cognitive impairment and six said they used them to screen for depression.
“We hear from MS specialists that they want to be measuring for cognition but don’t know how to efficiently work it into their routine, how to approach the patient, and what tools to use,” said study coauthor Derrick S. Robertson, MD, of the University of South Florida, Tampa. “In addition, there is no one tool that is accepted in the MS treatment community.”
MS specialists who didn’t use the screening tests also pointed to factors like lack of reimbursement and lack of integration into electronic medical records. “Doubt very much that neurologists have time to use any of these tests,” one respondent said, referring to cognitive impairment screening.
What’s next? “There are several new exciting developments in clinical trials demonstrating efficacy of disease-modifying therapies in maintaining or improving cognition in patients with relapsing MS,” Dr. Robertson said. “This highlights the urgent need to overcome barriers to use of formal cognitive screening tools in clinical practice to identify patients who need a higher level of care, and perhaps even a change in treatment with the ultimate goal to improve quality of life and overall outcomes.”
Genentech funded the study through an educational grant. Dr. Sapir and three other study authors reported no relevant disclosures. Dr. Buckle and Dr. Robertson reported multiple disclosures, including principle investigator and advisory board/panel member work.
SOURCE: Buckle GJ et al. ACTRIMS Forum 2018, abstract No. P161.
REPORTING FROM ACTRIMS FORUM 2018
Key clinical point: Screening often turns up signs of trouble, but many MS patients are not screened annually for depression and cognitive impairment.
Major finding: 52% and 63% of patients with MS were screened for cognitive impairment and depression, respectively, over a 1-year period. Study details: 2-year analysis of medical records from two MS clinics in the Southeast.
Disclosures: Genentech funded the study through an educational grant. Some of the study authors reported various disclosures.
Source: Buckle GJ et al. ACTRIMS Forum 2018, abstract No. P161.