User login
Tamibarotene shows strong results in high-risk APL patients
ATLANTA – Maintenance therapy with the synthetic retinoid tamibarotene is more effective than all-trans retinoic acid (ATRA), for decreasing the relapse rate in patients with acute promyelocytic leukemia (APL) – a subtype of acute myeloid leukemia, according to 7-year findings from the JALSG-APL204 randomized controlled trial.
The relapse-free survival findings were particularly pronounced among high-risk patients with leukocyte counts of at least 10,000 per microliter, Akihiro Takeshita, MD, PhD, reported at the annual meeting of the American Society of Hematology.
“These results could lead to a new strategy for the treatment of high-risk patients, which is one of the recent priority issues in the treatment of APL,” said Dr. Takeshita of Hamamatsu (Japan) University.
Of 344 eligible patients aged 15-70 years with newly diagnosed APL and documented cytogenetic and/or molecular evidence of chromosomal translocation t(15;17) or PML/RAR-alpha gene expression, 269 entered the maintenance phase of the study after completing three courses of consolidation therapy and were assigned to receive ATRA or tamibarotene. At a mean follow-up of 7 years, the relapse-free survival rate was 84% in the 135 patients in the ATRA arm, compared with 93% among the 134 patients in the tamibarotene arm.
The difference between the groups was statistically significant, but an even greater difference was seen when the analysis was restricted to 52 high-risk patients with an initial leukocyte count of at least 10,000 per microliter (62% vs. 89%).
Both treatments were generally well tolerated, Dr. Takeshita reported.
Study subjects received ATRA at a daily dose of 45 mg/m2 for remission induction. Once complete remission was achieved, they received chemotherapy based on their initial leukocyte and blast count in the peripheral blood. Those who achieved molecular remission after consolidation chemotherapy were included in the current maintenance phase of the study. During this phase, ATRA was given at a daily dose of 45 mg/m2 divided into 3 doses for 14 days, and tamibarotene was given at a daily dose of 6 mg/m2 divided into 2 doses for 14 days. Each cycle of treatment was repeated every 3 months for 2 years.
Adverse events included secondary hematopoietic disorders in 12 cases, malignancies in 9 cases, and late cardiac complications of grade 3 or higher in 5 cases, but no significant difference in the rates of these events was seen between the two treatment groups, Dr. Takeshita noted.
Tamibarotene was studied in this trial because, compared with ATRA, it has been shown to have about a 10-fold increase in potency for inducing in vitro differentiation of NB-4 cells, enhanced chemical stability, and low affinity for cellular RA-binding protein.
“The clinical efficacy of tamibarotene for the treatment of APL has also been reported,” Dr. Takeshita added.
In the initial phases of the trial, no difference was seen between ATRA and tamibarotene with respect to 4-year relapse-free survival, but there did appear to be improved efficacy with tamibarotene in high-risk patients, which warranted further investigation, he said.
The current findings demonstrate the efficacy of tamibarotene vs. ATRA for decreasing the relapse rate at the 7-year observation point, and confirm the benefit in high-risk patients that was seen in earlier analyses, he concluded.
Dr. Takeshita reported receiving research funding from Chugai Pharmaceutical, Astellas Pharma, Pfizer Japan, and Takeda Pharmaceutical.
SOURCE: Takeshita A et al., ASH 2017, abstract 642.
ATLANTA – Maintenance therapy with the synthetic retinoid tamibarotene is more effective than all-trans retinoic acid (ATRA), for decreasing the relapse rate in patients with acute promyelocytic leukemia (APL) – a subtype of acute myeloid leukemia, according to 7-year findings from the JALSG-APL204 randomized controlled trial.
The relapse-free survival findings were particularly pronounced among high-risk patients with leukocyte counts of at least 10,000 per microliter, Akihiro Takeshita, MD, PhD, reported at the annual meeting of the American Society of Hematology.
“These results could lead to a new strategy for the treatment of high-risk patients, which is one of the recent priority issues in the treatment of APL,” said Dr. Takeshita of Hamamatsu (Japan) University.
Of 344 eligible patients aged 15-70 years with newly diagnosed APL and documented cytogenetic and/or molecular evidence of chromosomal translocation t(15;17) or PML/RAR-alpha gene expression, 269 entered the maintenance phase of the study after completing three courses of consolidation therapy and were assigned to receive ATRA or tamibarotene. At a mean follow-up of 7 years, the relapse-free survival rate was 84% in the 135 patients in the ATRA arm, compared with 93% among the 134 patients in the tamibarotene arm.
The difference between the groups was statistically significant, but an even greater difference was seen when the analysis was restricted to 52 high-risk patients with an initial leukocyte count of at least 10,000 per microliter (62% vs. 89%).
Both treatments were generally well tolerated, Dr. Takeshita reported.
Study subjects received ATRA at a daily dose of 45 mg/m2 for remission induction. Once complete remission was achieved, they received chemotherapy based on their initial leukocyte and blast count in the peripheral blood. Those who achieved molecular remission after consolidation chemotherapy were included in the current maintenance phase of the study. During this phase, ATRA was given at a daily dose of 45 mg/m2 divided into 3 doses for 14 days, and tamibarotene was given at a daily dose of 6 mg/m2 divided into 2 doses for 14 days. Each cycle of treatment was repeated every 3 months for 2 years.
Adverse events included secondary hematopoietic disorders in 12 cases, malignancies in 9 cases, and late cardiac complications of grade 3 or higher in 5 cases, but no significant difference in the rates of these events was seen between the two treatment groups, Dr. Takeshita noted.
Tamibarotene was studied in this trial because, compared with ATRA, it has been shown to have about a 10-fold increase in potency for inducing in vitro differentiation of NB-4 cells, enhanced chemical stability, and low affinity for cellular RA-binding protein.
“The clinical efficacy of tamibarotene for the treatment of APL has also been reported,” Dr. Takeshita added.
In the initial phases of the trial, no difference was seen between ATRA and tamibarotene with respect to 4-year relapse-free survival, but there did appear to be improved efficacy with tamibarotene in high-risk patients, which warranted further investigation, he said.
The current findings demonstrate the efficacy of tamibarotene vs. ATRA for decreasing the relapse rate at the 7-year observation point, and confirm the benefit in high-risk patients that was seen in earlier analyses, he concluded.
Dr. Takeshita reported receiving research funding from Chugai Pharmaceutical, Astellas Pharma, Pfizer Japan, and Takeda Pharmaceutical.
SOURCE: Takeshita A et al., ASH 2017, abstract 642.
ATLANTA – Maintenance therapy with the synthetic retinoid tamibarotene is more effective than all-trans retinoic acid (ATRA), for decreasing the relapse rate in patients with acute promyelocytic leukemia (APL) – a subtype of acute myeloid leukemia, according to 7-year findings from the JALSG-APL204 randomized controlled trial.
The relapse-free survival findings were particularly pronounced among high-risk patients with leukocyte counts of at least 10,000 per microliter, Akihiro Takeshita, MD, PhD, reported at the annual meeting of the American Society of Hematology.
“These results could lead to a new strategy for the treatment of high-risk patients, which is one of the recent priority issues in the treatment of APL,” said Dr. Takeshita of Hamamatsu (Japan) University.
Of 344 eligible patients aged 15-70 years with newly diagnosed APL and documented cytogenetic and/or molecular evidence of chromosomal translocation t(15;17) or PML/RAR-alpha gene expression, 269 entered the maintenance phase of the study after completing three courses of consolidation therapy and were assigned to receive ATRA or tamibarotene. At a mean follow-up of 7 years, the relapse-free survival rate was 84% in the 135 patients in the ATRA arm, compared with 93% among the 134 patients in the tamibarotene arm.
The difference between the groups was statistically significant, but an even greater difference was seen when the analysis was restricted to 52 high-risk patients with an initial leukocyte count of at least 10,000 per microliter (62% vs. 89%).
Both treatments were generally well tolerated, Dr. Takeshita reported.
Study subjects received ATRA at a daily dose of 45 mg/m2 for remission induction. Once complete remission was achieved, they received chemotherapy based on their initial leukocyte and blast count in the peripheral blood. Those who achieved molecular remission after consolidation chemotherapy were included in the current maintenance phase of the study. During this phase, ATRA was given at a daily dose of 45 mg/m2 divided into 3 doses for 14 days, and tamibarotene was given at a daily dose of 6 mg/m2 divided into 2 doses for 14 days. Each cycle of treatment was repeated every 3 months for 2 years.
Adverse events included secondary hematopoietic disorders in 12 cases, malignancies in 9 cases, and late cardiac complications of grade 3 or higher in 5 cases, but no significant difference in the rates of these events was seen between the two treatment groups, Dr. Takeshita noted.
Tamibarotene was studied in this trial because, compared with ATRA, it has been shown to have about a 10-fold increase in potency for inducing in vitro differentiation of NB-4 cells, enhanced chemical stability, and low affinity for cellular RA-binding protein.
“The clinical efficacy of tamibarotene for the treatment of APL has also been reported,” Dr. Takeshita added.
In the initial phases of the trial, no difference was seen between ATRA and tamibarotene with respect to 4-year relapse-free survival, but there did appear to be improved efficacy with tamibarotene in high-risk patients, which warranted further investigation, he said.
The current findings demonstrate the efficacy of tamibarotene vs. ATRA for decreasing the relapse rate at the 7-year observation point, and confirm the benefit in high-risk patients that was seen in earlier analyses, he concluded.
Dr. Takeshita reported receiving research funding from Chugai Pharmaceutical, Astellas Pharma, Pfizer Japan, and Takeda Pharmaceutical.
SOURCE: Takeshita A et al., ASH 2017, abstract 642.
REPORTING FROM ASH 2017
Key clinical point:
Major finding: The 7-year relapse-free survival was 62% vs. 89% with ATRA vs. tamibarotene in high-risk patients.
Study details: Long-term maintenance results in 344 patients from a randomized controlled trial.
Disclosures: Dr. Takeshita reported receiving research funding from Chugai Pharmaceutical, Astellas Pharma, Pfizer Japan, and Takeda Pharmaceutical.
Source: Takeshita A et al. ASH 2017, abstract 642.
RF-positive polyarticular JIA looks like adult RA
New evidence suggests the rheumatoid factor–positive polyarticular subtype of juvenile idiopathic arthritis bears a close genetic resemblance to adult rheumatoid arthritis, lending support to the growing suspicion that in arthritis, biological underpinnings are more important than age of onset when it comes to characterizing and, potentially, choosing treatments.
Previous work had shown that rheumatoid factor (RF)–negative patients have genetic risks similar to those of adults with RF-negative disease. “If the RF-negative patients in adult and childhood are similar, then maybe the RF-positive patients are similar in their genetic background as well. That’s what this study was testing,” study coauthor Anne M. Stevens, MD, PhD, division chief of rheumatology at the University of Washington, Seattle, said in an interview. The study was published online Feb. 9 in Arthritis & Rheumatology.

There are seven recognized categories of juvenile idiopathic arthritis (JIA), and all are believed to have genetic risk factors. Previously, the researchers used the Immunochip custom microarray to map 186 autoimmune disease-associated loci from 11 autoimmune phenotypes, including adult rheumatoid arthritis (RA). In the current work, the researchers analyzed 340 RF-positive polyarticular JIA cases (292 females) and 14,412 controls (8,002 females) from the United States, United Kingdom, Germany, Canada, and Norway. RF-positive polyarticular disease accounts for about 5% of JIA cases, and its symptoms and presentations resemble adult RA.
The researchers found associations in the human leukocyte antigen (HLA) region. The most significant was found at rs3129769, near HLA-DRB1 (P = 5.51 x 10-31). This single nucleotide polymorphism (SNP) was in strong linkage disequilibrium (LD, r2 = 0.88) with the rs660895 HLA-DRB1 SNP that has been reported in adult RA (P = 2.14 x 10-29).
The researchers examined links between RF-positive polyarticular JIA and the 27 SNPs that had been identified in the previous study of oligoarticular/RF-negative polyarticular JIA. Just 6 of those 27 SNPs were significantly associated with RF-positive polyarticular JIA (P less than .05). On the other hand, of 44 SNPs most strongly associated with RA, 19 were associated with RF-positive polyarticular JIA (P less than .05).
That suggests that RF-positive polyarticular JIA cases are different from other JIA cases. “They’re more like adult patients than they’re like child patients,” said Dr. Stevens.
The researchers also compared the weighted genetic risk scores (wGRS) produced from the top RA loci to wGRS produced from the top oligoarticular/RF-negative polyarticular JIA loci. The wGRS from the top RA loci was a better predictor of RF-positive polyarticular JIA cases (area under the curve [AUC] = 0.71 versus AUC = 0.58; P = 8.26 x 10-33).
The wGRS from RA had similar success in predicting RF-positive polyarticular JIA and early-onset RA cases (AUC = 0.75; P = .25), but it fared worse in predicting late-onset RA (at 70 years or older, AUC = 0.62), compared with the wGRS from RF-positive polyarticular JIA (P = 1.65 x 10-5).
Those results suggest that RF-positive polyarticular JIA more closely resembles younger RA cases than older RA cases.
“If you consider early-onset RA patients, less than 29 years old when they develop RA, they look like JIA patients. But older RA patients, who are over 70 when they develop RA, they look like they totally have a different genetic background,” Dr. Stevens said.
The study could have clinical implications. The lead author, Anne Hinks, PhD, is a research fellow at the University of Manchester (England) and has led the charge to characterize JIA. The wGRS score she developed has the potential to help physicians diagnose classify and treat JIA patients. Currently, they must rely on the International League of Associations for Rheumatology criteria, which can take months to work through and may lead to misclassification diagnoses.
And in any case, the emerging genetic research suggests that the underlying genetics of JIA may be a better way to classify patients. “There’s a lot of overlap and risk of misclassifying patients with the current system. This weighted genetic risk score that Dr. Hinks developed could be used to classify patients with one DNA sample. This is the kind of clinical test we need,” Dr. Stevens said.
The study received funding from a range of government and private sources. Dr. Stevens has a patent licensed to Quest Diagnostics, is conducting research collaborations with Seattle Genetics and Kineta, and has received fellowship support from Pfizer.
SOURCE: Hinks A et al. Arthritis Rheumatol. 2018 Feb 9. doi: 10.1002/art.40443
New evidence suggests the rheumatoid factor–positive polyarticular subtype of juvenile idiopathic arthritis bears a close genetic resemblance to adult rheumatoid arthritis, lending support to the growing suspicion that in arthritis, biological underpinnings are more important than age of onset when it comes to characterizing and, potentially, choosing treatments.
Previous work had shown that rheumatoid factor (RF)–negative patients have genetic risks similar to those of adults with RF-negative disease. “If the RF-negative patients in adult and childhood are similar, then maybe the RF-positive patients are similar in their genetic background as well. That’s what this study was testing,” study coauthor Anne M. Stevens, MD, PhD, division chief of rheumatology at the University of Washington, Seattle, said in an interview. The study was published online Feb. 9 in Arthritis & Rheumatology.
There are seven recognized categories of juvenile idiopathic arthritis (JIA), and all are believed to have genetic risk factors. Previously, the researchers used the Immunochip custom microarray to map 186 autoimmune disease-associated loci from 11 autoimmune phenotypes, including adult rheumatoid arthritis (RA). In the current work, the researchers analyzed 340 RF-positive polyarticular JIA cases (292 females) and 14,412 controls (8,002 females) from the United States, United Kingdom, Germany, Canada, and Norway. RF-positive polyarticular disease accounts for about 5% of JIA cases, and its symptoms and presentations resemble adult RA.
The researchers found associations in the human leukocyte antigen (HLA) region. The most significant was found at rs3129769, near HLA-DRB1 (P = 5.51 x 10-31). This single nucleotide polymorphism (SNP) was in strong linkage disequilibrium (LD, r2 = 0.88) with the rs660895 HLA-DRB1 SNP that has been reported in adult RA (P = 2.14 x 10-29).
The researchers examined links between RF-positive polyarticular JIA and the 27 SNPs that had been identified in the previous study of oligoarticular/RF-negative polyarticular JIA. Just 6 of those 27 SNPs were significantly associated with RF-positive polyarticular JIA (P less than .05). On the other hand, of 44 SNPs most strongly associated with RA, 19 were associated with RF-positive polyarticular JIA (P less than .05).
That suggests that RF-positive polyarticular JIA cases are different from other JIA cases. “They’re more like adult patients than they’re like child patients,” said Dr. Stevens.
The researchers also compared the weighted genetic risk scores (wGRS) produced from the top RA loci to wGRS produced from the top oligoarticular/RF-negative polyarticular JIA loci. The wGRS from the top RA loci was a better predictor of RF-positive polyarticular JIA cases (area under the curve [AUC] = 0.71 versus AUC = 0.58; P = 8.26 x 10-33).
The wGRS from RA had similar success in predicting RF-positive polyarticular JIA and early-onset RA cases (AUC = 0.75; P = .25), but it fared worse in predicting late-onset RA (at 70 years or older, AUC = 0.62), compared with the wGRS from RF-positive polyarticular JIA (P = 1.65 x 10-5).
Those results suggest that RF-positive polyarticular JIA more closely resembles younger RA cases than older RA cases.
“If you consider early-onset RA patients, less than 29 years old when they develop RA, they look like JIA patients. But older RA patients, who are over 70 when they develop RA, they look like they totally have a different genetic background,” Dr. Stevens said.
The study could have clinical implications. The lead author, Anne Hinks, PhD, is a research fellow at the University of Manchester (England) and has led the charge to characterize JIA. The wGRS score she developed has the potential to help physicians diagnose classify and treat JIA patients. Currently, they must rely on the International League of Associations for Rheumatology criteria, which can take months to work through and may lead to misclassification diagnoses.
And in any case, the emerging genetic research suggests that the underlying genetics of JIA may be a better way to classify patients. “There’s a lot of overlap and risk of misclassifying patients with the current system. This weighted genetic risk score that Dr. Hinks developed could be used to classify patients with one DNA sample. This is the kind of clinical test we need,” Dr. Stevens said.
The study received funding from a range of government and private sources. Dr. Stevens has a patent licensed to Quest Diagnostics, is conducting research collaborations with Seattle Genetics and Kineta, and has received fellowship support from Pfizer.
SOURCE: Hinks A et al. Arthritis Rheumatol. 2018 Feb 9. doi: 10.1002/art.40443
New evidence suggests the rheumatoid factor–positive polyarticular subtype of juvenile idiopathic arthritis bears a close genetic resemblance to adult rheumatoid arthritis, lending support to the growing suspicion that in arthritis, biological underpinnings are more important than age of onset when it comes to characterizing and, potentially, choosing treatments.
Previous work had shown that rheumatoid factor (RF)–negative patients have genetic risks similar to those of adults with RF-negative disease. “If the RF-negative patients in adult and childhood are similar, then maybe the RF-positive patients are similar in their genetic background as well. That’s what this study was testing,” study coauthor Anne M. Stevens, MD, PhD, division chief of rheumatology at the University of Washington, Seattle, said in an interview. The study was published online Feb. 9 in Arthritis & Rheumatology.
There are seven recognized categories of juvenile idiopathic arthritis (JIA), and all are believed to have genetic risk factors. Previously, the researchers used the Immunochip custom microarray to map 186 autoimmune disease-associated loci from 11 autoimmune phenotypes, including adult rheumatoid arthritis (RA). In the current work, the researchers analyzed 340 RF-positive polyarticular JIA cases (292 females) and 14,412 controls (8,002 females) from the United States, United Kingdom, Germany, Canada, and Norway. RF-positive polyarticular disease accounts for about 5% of JIA cases, and its symptoms and presentations resemble adult RA.
The researchers found associations in the human leukocyte antigen (HLA) region. The most significant was found at rs3129769, near HLA-DRB1 (P = 5.51 x 10-31). This single nucleotide polymorphism (SNP) was in strong linkage disequilibrium (LD, r2 = 0.88) with the rs660895 HLA-DRB1 SNP that has been reported in adult RA (P = 2.14 x 10-29).
The researchers examined links between RF-positive polyarticular JIA and the 27 SNPs that had been identified in the previous study of oligoarticular/RF-negative polyarticular JIA. Just 6 of those 27 SNPs were significantly associated with RF-positive polyarticular JIA (P less than .05). On the other hand, of 44 SNPs most strongly associated with RA, 19 were associated with RF-positive polyarticular JIA (P less than .05).
That suggests that RF-positive polyarticular JIA cases are different from other JIA cases. “They’re more like adult patients than they’re like child patients,” said Dr. Stevens.
The researchers also compared the weighted genetic risk scores (wGRS) produced from the top RA loci to wGRS produced from the top oligoarticular/RF-negative polyarticular JIA loci. The wGRS from the top RA loci was a better predictor of RF-positive polyarticular JIA cases (area under the curve [AUC] = 0.71 versus AUC = 0.58; P = 8.26 x 10-33).
The wGRS from RA had similar success in predicting RF-positive polyarticular JIA and early-onset RA cases (AUC = 0.75; P = .25), but it fared worse in predicting late-onset RA (at 70 years or older, AUC = 0.62), compared with the wGRS from RF-positive polyarticular JIA (P = 1.65 x 10-5).
Those results suggest that RF-positive polyarticular JIA more closely resembles younger RA cases than older RA cases.
“If you consider early-onset RA patients, less than 29 years old when they develop RA, they look like JIA patients. But older RA patients, who are over 70 when they develop RA, they look like they totally have a different genetic background,” Dr. Stevens said.
The study could have clinical implications. The lead author, Anne Hinks, PhD, is a research fellow at the University of Manchester (England) and has led the charge to characterize JIA. The wGRS score she developed has the potential to help physicians diagnose classify and treat JIA patients. Currently, they must rely on the International League of Associations for Rheumatology criteria, which can take months to work through and may lead to misclassification diagnoses.
And in any case, the emerging genetic research suggests that the underlying genetics of JIA may be a better way to classify patients. “There’s a lot of overlap and risk of misclassifying patients with the current system. This weighted genetic risk score that Dr. Hinks developed could be used to classify patients with one DNA sample. This is the kind of clinical test we need,” Dr. Stevens said.
The study received funding from a range of government and private sources. Dr. Stevens has a patent licensed to Quest Diagnostics, is conducting research collaborations with Seattle Genetics and Kineta, and has received fellowship support from Pfizer.
SOURCE: Hinks A et al. Arthritis Rheumatol. 2018 Feb 9. doi: 10.1002/art.40443
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: Genetics underlying arthritis may be more important than age of onset.
Major finding: The weighted genetic risk scores produced from the top RA loci was a better predictor of RF-positive polyarticular JIA than that generated from the top oligoarticular/RF-negative polyarticular JIA loci.
Data source: Case-control analysis of 340 cases and 14,412 controls.
Disclosures: The study received funding from a range of government and private sources. Dr. Stevens has a patent licensed to Quest Diagnostics, is conducting research collaborations with Seattle Genetics and Kineta, and has received fellowship support from Pfizer.
Source: Hinks A et al. Arthritis Rheumatol. 2018 Feb 9. doi: 10.1002/art.40443.
Topical imiquimod helps clear blurred lines in lentigo maligna excision
according to a study from the University of Utah.
Lentigo maligna is a subtype of melanoma in situ, usually occurring in the head and neck regions, the researchers said.
“Neoadjuvant topical imiquimod 5% cream applied 5 times weekly for 8 weeks was associated with decreased MDCs in LM treatment sites compared with the MDCs of negative control sites,” wrote Shadai Flores of the University of Utah, Salt Lake City, and her colleagues.
Previously, the ability to distinguish between the surgical border and surrounding background melanocytic hyperplasia was uncertain. Because of this uncertainty, LM removal required an average margin of 7.2 mm. Another study showed that topical imiquimod 5% cream enabled the removal of most LM tumors with 2-mm margins. This study “sought to evaluate MDCs in imiquimod-treated LM and negative control biopsy specimens to determine if there was a measurable difference in melanocyte density,” the researchers wrote in a research letter published in JAMA Dermatology.
The study prospectively followed 52 cases of LM treated with imiquimod 5% topical cream 5 days per week for 8 weeks followed by conservative staged excisions with 2-mm margins. Treatment with imiquimod 5% of LM was followed by a 2- to 4-month recuperation period before surgery could be performed. All patients in the study were treated by one Mohs surgeon at the Huntsman Cancer Institute at the university.
To establish an MDC baseline, a 10-mm long fusiform biopsy was taken as a negative control. The negative control sample site and the LM site were separated by approximately 6 cm, found on the same side of the body, and showed similar color changes. After a negative control was taken, an LM lesion was resected and subsequently quadrisected. The MDCs then were concurrently counted by the researchers and compared with the negative controls.
Of the 52 LM specimens, 44 (85%) exhibited decreases in MDCs, compared with the negative controls. The median MDC from post–imiquimod-treated sites was 14.4, with a range of 0.5-26.6. This showed marked improvement over the negative controls, which had a median MDC of 20.0 (range of 9.0-36.7). A 2-tailed paired t test revealed that the results displayed statistical significance (P less than .001). Residual LM was seen in the central areas of 9 (17%) specimens, but 43 (83%) had no indication of residual LM.
“The decreased melanocytic hyperplasia in imiquimod-treated sites reduced ambiguity in making a distinction between the border of the excised LM and background melanocytic hyperplasia,” noted Ms. Flores and her colleagues.
The authors had no conflicts of interest.
SOURCE: Flores S et al. JAMA Dermatol. 2018 Feb 16. doi: 10.1001/jamadermatol.2017.5632.
according to a study from the University of Utah.
Lentigo maligna is a subtype of melanoma in situ, usually occurring in the head and neck regions, the researchers said.
“Neoadjuvant topical imiquimod 5% cream applied 5 times weekly for 8 weeks was associated with decreased MDCs in LM treatment sites compared with the MDCs of negative control sites,” wrote Shadai Flores of the University of Utah, Salt Lake City, and her colleagues.
Previously, the ability to distinguish between the surgical border and surrounding background melanocytic hyperplasia was uncertain. Because of this uncertainty, LM removal required an average margin of 7.2 mm. Another study showed that topical imiquimod 5% cream enabled the removal of most LM tumors with 2-mm margins. This study “sought to evaluate MDCs in imiquimod-treated LM and negative control biopsy specimens to determine if there was a measurable difference in melanocyte density,” the researchers wrote in a research letter published in JAMA Dermatology.
The study prospectively followed 52 cases of LM treated with imiquimod 5% topical cream 5 days per week for 8 weeks followed by conservative staged excisions with 2-mm margins. Treatment with imiquimod 5% of LM was followed by a 2- to 4-month recuperation period before surgery could be performed. All patients in the study were treated by one Mohs surgeon at the Huntsman Cancer Institute at the university.
To establish an MDC baseline, a 10-mm long fusiform biopsy was taken as a negative control. The negative control sample site and the LM site were separated by approximately 6 cm, found on the same side of the body, and showed similar color changes. After a negative control was taken, an LM lesion was resected and subsequently quadrisected. The MDCs then were concurrently counted by the researchers and compared with the negative controls.
Of the 52 LM specimens, 44 (85%) exhibited decreases in MDCs, compared with the negative controls. The median MDC from post–imiquimod-treated sites was 14.4, with a range of 0.5-26.6. This showed marked improvement over the negative controls, which had a median MDC of 20.0 (range of 9.0-36.7). A 2-tailed paired t test revealed that the results displayed statistical significance (P less than .001). Residual LM was seen in the central areas of 9 (17%) specimens, but 43 (83%) had no indication of residual LM.
“The decreased melanocytic hyperplasia in imiquimod-treated sites reduced ambiguity in making a distinction between the border of the excised LM and background melanocytic hyperplasia,” noted Ms. Flores and her colleagues.
The authors had no conflicts of interest.
SOURCE: Flores S et al. JAMA Dermatol. 2018 Feb 16. doi: 10.1001/jamadermatol.2017.5632.
according to a study from the University of Utah.
Lentigo maligna is a subtype of melanoma in situ, usually occurring in the head and neck regions, the researchers said.
“Neoadjuvant topical imiquimod 5% cream applied 5 times weekly for 8 weeks was associated with decreased MDCs in LM treatment sites compared with the MDCs of negative control sites,” wrote Shadai Flores of the University of Utah, Salt Lake City, and her colleagues.
Previously, the ability to distinguish between the surgical border and surrounding background melanocytic hyperplasia was uncertain. Because of this uncertainty, LM removal required an average margin of 7.2 mm. Another study showed that topical imiquimod 5% cream enabled the removal of most LM tumors with 2-mm margins. This study “sought to evaluate MDCs in imiquimod-treated LM and negative control biopsy specimens to determine if there was a measurable difference in melanocyte density,” the researchers wrote in a research letter published in JAMA Dermatology.
The study prospectively followed 52 cases of LM treated with imiquimod 5% topical cream 5 days per week for 8 weeks followed by conservative staged excisions with 2-mm margins. Treatment with imiquimod 5% of LM was followed by a 2- to 4-month recuperation period before surgery could be performed. All patients in the study were treated by one Mohs surgeon at the Huntsman Cancer Institute at the university.
To establish an MDC baseline, a 10-mm long fusiform biopsy was taken as a negative control. The negative control sample site and the LM site were separated by approximately 6 cm, found on the same side of the body, and showed similar color changes. After a negative control was taken, an LM lesion was resected and subsequently quadrisected. The MDCs then were concurrently counted by the researchers and compared with the negative controls.
Of the 52 LM specimens, 44 (85%) exhibited decreases in MDCs, compared with the negative controls. The median MDC from post–imiquimod-treated sites was 14.4, with a range of 0.5-26.6. This showed marked improvement over the negative controls, which had a median MDC of 20.0 (range of 9.0-36.7). A 2-tailed paired t test revealed that the results displayed statistical significance (P less than .001). Residual LM was seen in the central areas of 9 (17%) specimens, but 43 (83%) had no indication of residual LM.
“The decreased melanocytic hyperplasia in imiquimod-treated sites reduced ambiguity in making a distinction between the border of the excised LM and background melanocytic hyperplasia,” noted Ms. Flores and her colleagues.
The authors had no conflicts of interest.
SOURCE: Flores S et al. JAMA Dermatol. 2018 Feb 16. doi: 10.1001/jamadermatol.2017.5632.
FROM JAMA DERMATOLOGY
Key clinical point: Neoadjuvant, topical imiquimod 5% cream is associated with a decrease in melanocyte density counts (MDC).
Major finding: Of 52 patient specimens, 44 (85%) exhibited decreases in MDCs, compared with the negative controls.
Study details: A prospective study of 52 cases of lentigo maligna treated with imiquimod 5% topical cream 5 days per week for 8 weeks, followed by conservative staged excisions with 2-mm margins.
Disclosures: The authors had no conflicts of interest.
Source: Flores S et al. JAMA Dermatol. 2018 Feb 16. doi: 10.1001/jamadermatol.2017.5632.
How to advise adolescents ISO drugs on the ‘dark web’
There was a time, not so long ago, when in the popular imagination, a drug deal involved an aging hippie in a tie-dyed shirt and love beads, copping a joint at a Dead concert. Today, however, in the age of the Internet and smartphones, a teenager in his bedroom can select, order, and have delivered to his door illicit drugs via the marketplaces on the “dark web.”
As the name implies, the dark web is a subterranean layer of the Internet that is mysterious, ominous, and sometimes lawless. It lies “beneath” the surface web – the layer where grandmothers post on Facebook and purchase on Amazon.
The dark web largely was the brainchild of three mathematicians at the Naval Research Laboratory as a means of encrypting messages exchanged by the intelligence community. They dubbed their project “Tor,” for “The Onion Router,” as the system consists of layer after layer of random relays, permitting anonymity on the Internet with little risk of tracking or surveillance.
This online underworld first came to my attention several years ago. The father of a teen who was being treated for disruptive mood dysregulation, attention-deficit disorder, and alcohol and cannabis use disorder called to inform me that his son had been arrested at Lollapalooza with hundreds of Adderall tablets and Xanax bars. Several weeks later, in session, the young man disclosed to me that he had found simple instructions online about installing Tor, creating a VPN (a virtual private network), accessing the dark web, transacting with bitcoin, and identifying drug marketplaces. He also demonstrated a detailed knowledge of chemical manufacturing in China, pill pressing in Canada, and money laundering in Switzerland.
With the air of an insider sharing “trade secrets,” this young man described how dealers on the dark web avoid detection and ensure secure delivery of the goods: latex gloves, vacuum sealing, and bleach dipping to obviate fingerprints – human and chemical. He said that dealers will send “dummy” packages to throw off the authorities and that buyers often will use the address of a clueless or absent neighbor. In his case, however, the parcels were delivered to his doorstep.
. For one thing, acquiring drugs in this way can seem less risky, as there is no chance of being robbed at gunpoint in a sketchy neighborhood or being busted for possession during a routine traffic stop.
Crossing over to the world of the dark web also can give an adolescent a sense of being clever and rebellious and of pulling a “fast one” on the parents – and on us, the clinicians who are treating them. And a teen who is interested in using illicit substances and plays “Call of Duty” from the comfort of his family’s basement without actual injury or death might assume that he can attain illicit drugs that are safe and inexpensive.
There are counterarguments to misinformation about the dark web. For example, contrary to the notion that buying drugs on the dark web minimizes interdiction or arrest, clinicians should point out that since international law enforcement shut down Silk Road and incarcerated its founder, Ross Ulbricht (also known as “Dread Pirate Roberts”) in 2013, hundreds of other dark web marketplaces such as AlphaBay and Hansa have been silenced and their operators prosecuted.
Moreover, the U.S. Department of Justice recently launched the Joint Criminal Opioid Darknet Enforcement (J-CODE) group, and the U.S. Postal Service Inspection Service reportedly has been hiring cybercrime and dark web specialists to combat drug trafficking. It might come as a surprise to a teen that, in a state where recreational marijuana is legal, transport and delivery of cannabis by the postal service elevates purchase and possession to the level of a violation of federal law. Informing even the most oblivious or oppositional adolescent that a drug felony can disqualify him for college grants and loans, impede his search for gainful employment, or prohibit him from obtaining a professional license, might give him a moment’s pause.
Adolescents seeking to buy drugs on the dark web should brace themselves for another shock. Whether lulled by custom after years of shopping on Amazon or using PayPal, or simply dulled by addiction, they might have a blind trust that bitcoin tumblers and “dark” escrow accounts will secure their payments. It will be rude awakening when they learn that the transfer and holding of currency on the dark web is vulnerable to hackers and to operators of the marketplaces – who are known to simply abscond with funds.
Currently, the drug marketplaces on the dark web represent a thin slice of the total illicit drug trade. These marketplaces, however, are growing quickly and offer buyers a virtual smorgasbord: the leading prescription drugs bought are Xanax and OxyContin, whereas 3,4-methylenedioxymethamphetamine (MDMA)/ecstasy and cannabis are the most commonly purchased controlled Schedule I substances, according to an article in The Economist. The estimated annual sales for 2016 ranged from $100 million to $200 million, but even more alarming is the percentage of substance abusers who have purchased drugs on the dark web: A recent article in The Independent reported that 13.2% of U.S. respondents self-reported making at least one purchase online, whereas in the United Kingdom, the self-reported percentage was 25.3 %, and in Finland, it was 41.4%.
Evidence abounds that drugs purchased on the dark web often are counterfeit and sometimes “dirty.” There is a report of “Viagra” containing cement dust, of “Ambien” containing haloperidol, and “Xanax” laced with fentanyl, the latter having been linked to several deaths and hospital admissions in the Bay Area in 2016. (Big Pharma, in fact, is engaged in surveillance and investigation of the sale of knockoffs online, The Telegraph reported in article about the proliferation of fake drugs available on the surface web).Unfortunately, attempting to reduce teen substance abuse using law enforcement measures directed at dark web marketplaces might be a game of whack-a-mole: As soon as one supply source is staunched, another surfaces. Indeed, addiction should be viewed not simply in terms of the biopsychosocial model, but also as an economic activity. Thus, it might be more beneficial for clinicians to concentrate our resources and efforts on curtailing the demand through education and treatment.

Dr. Marseille is a psychiatrist who works on the staff at a clinic in Winfield, Ill. His special interests include adolescent and addiction medicine, eating disorders, trauma, bipolar disorder, and the psychiatric manifestations of acute and chronic medical conditions.
There was a time, not so long ago, when in the popular imagination, a drug deal involved an aging hippie in a tie-dyed shirt and love beads, copping a joint at a Dead concert. Today, however, in the age of the Internet and smartphones, a teenager in his bedroom can select, order, and have delivered to his door illicit drugs via the marketplaces on the “dark web.”
As the name implies, the dark web is a subterranean layer of the Internet that is mysterious, ominous, and sometimes lawless. It lies “beneath” the surface web – the layer where grandmothers post on Facebook and purchase on Amazon.
The dark web largely was the brainchild of three mathematicians at the Naval Research Laboratory as a means of encrypting messages exchanged by the intelligence community. They dubbed their project “Tor,” for “The Onion Router,” as the system consists of layer after layer of random relays, permitting anonymity on the Internet with little risk of tracking or surveillance.
This online underworld first came to my attention several years ago. The father of a teen who was being treated for disruptive mood dysregulation, attention-deficit disorder, and alcohol and cannabis use disorder called to inform me that his son had been arrested at Lollapalooza with hundreds of Adderall tablets and Xanax bars. Several weeks later, in session, the young man disclosed to me that he had found simple instructions online about installing Tor, creating a VPN (a virtual private network), accessing the dark web, transacting with bitcoin, and identifying drug marketplaces. He also demonstrated a detailed knowledge of chemical manufacturing in China, pill pressing in Canada, and money laundering in Switzerland.
With the air of an insider sharing “trade secrets,” this young man described how dealers on the dark web avoid detection and ensure secure delivery of the goods: latex gloves, vacuum sealing, and bleach dipping to obviate fingerprints – human and chemical. He said that dealers will send “dummy” packages to throw off the authorities and that buyers often will use the address of a clueless or absent neighbor. In his case, however, the parcels were delivered to his doorstep.
. For one thing, acquiring drugs in this way can seem less risky, as there is no chance of being robbed at gunpoint in a sketchy neighborhood or being busted for possession during a routine traffic stop.
Crossing over to the world of the dark web also can give an adolescent a sense of being clever and rebellious and of pulling a “fast one” on the parents – and on us, the clinicians who are treating them. And a teen who is interested in using illicit substances and plays “Call of Duty” from the comfort of his family’s basement without actual injury or death might assume that he can attain illicit drugs that are safe and inexpensive.
There are counterarguments to misinformation about the dark web. For example, contrary to the notion that buying drugs on the dark web minimizes interdiction or arrest, clinicians should point out that since international law enforcement shut down Silk Road and incarcerated its founder, Ross Ulbricht (also known as “Dread Pirate Roberts”) in 2013, hundreds of other dark web marketplaces such as AlphaBay and Hansa have been silenced and their operators prosecuted.
Moreover, the U.S. Department of Justice recently launched the Joint Criminal Opioid Darknet Enforcement (J-CODE) group, and the U.S. Postal Service Inspection Service reportedly has been hiring cybercrime and dark web specialists to combat drug trafficking. It might come as a surprise to a teen that, in a state where recreational marijuana is legal, transport and delivery of cannabis by the postal service elevates purchase and possession to the level of a violation of federal law. Informing even the most oblivious or oppositional adolescent that a drug felony can disqualify him for college grants and loans, impede his search for gainful employment, or prohibit him from obtaining a professional license, might give him a moment’s pause.
Adolescents seeking to buy drugs on the dark web should brace themselves for another shock. Whether lulled by custom after years of shopping on Amazon or using PayPal, or simply dulled by addiction, they might have a blind trust that bitcoin tumblers and “dark” escrow accounts will secure their payments. It will be rude awakening when they learn that the transfer and holding of currency on the dark web is vulnerable to hackers and to operators of the marketplaces – who are known to simply abscond with funds.
Currently, the drug marketplaces on the dark web represent a thin slice of the total illicit drug trade. These marketplaces, however, are growing quickly and offer buyers a virtual smorgasbord: the leading prescription drugs bought are Xanax and OxyContin, whereas 3,4-methylenedioxymethamphetamine (MDMA)/ecstasy and cannabis are the most commonly purchased controlled Schedule I substances, according to an article in The Economist. The estimated annual sales for 2016 ranged from $100 million to $200 million, but even more alarming is the percentage of substance abusers who have purchased drugs on the dark web: A recent article in The Independent reported that 13.2% of U.S. respondents self-reported making at least one purchase online, whereas in the United Kingdom, the self-reported percentage was 25.3 %, and in Finland, it was 41.4%.
Evidence abounds that drugs purchased on the dark web often are counterfeit and sometimes “dirty.” There is a report of “Viagra” containing cement dust, of “Ambien” containing haloperidol, and “Xanax” laced with fentanyl, the latter having been linked to several deaths and hospital admissions in the Bay Area in 2016. (Big Pharma, in fact, is engaged in surveillance and investigation of the sale of knockoffs online, The Telegraph reported in article about the proliferation of fake drugs available on the surface web).Unfortunately, attempting to reduce teen substance abuse using law enforcement measures directed at dark web marketplaces might be a game of whack-a-mole: As soon as one supply source is staunched, another surfaces. Indeed, addiction should be viewed not simply in terms of the biopsychosocial model, but also as an economic activity. Thus, it might be more beneficial for clinicians to concentrate our resources and efforts on curtailing the demand through education and treatment.
Dr. Marseille is a psychiatrist who works on the staff at a clinic in Winfield, Ill. His special interests include adolescent and addiction medicine, eating disorders, trauma, bipolar disorder, and the psychiatric manifestations of acute and chronic medical conditions.
There was a time, not so long ago, when in the popular imagination, a drug deal involved an aging hippie in a tie-dyed shirt and love beads, copping a joint at a Dead concert. Today, however, in the age of the Internet and smartphones, a teenager in his bedroom can select, order, and have delivered to his door illicit drugs via the marketplaces on the “dark web.”
As the name implies, the dark web is a subterranean layer of the Internet that is mysterious, ominous, and sometimes lawless. It lies “beneath” the surface web – the layer where grandmothers post on Facebook and purchase on Amazon.
The dark web largely was the brainchild of three mathematicians at the Naval Research Laboratory as a means of encrypting messages exchanged by the intelligence community. They dubbed their project “Tor,” for “The Onion Router,” as the system consists of layer after layer of random relays, permitting anonymity on the Internet with little risk of tracking or surveillance.
This online underworld first came to my attention several years ago. The father of a teen who was being treated for disruptive mood dysregulation, attention-deficit disorder, and alcohol and cannabis use disorder called to inform me that his son had been arrested at Lollapalooza with hundreds of Adderall tablets and Xanax bars. Several weeks later, in session, the young man disclosed to me that he had found simple instructions online about installing Tor, creating a VPN (a virtual private network), accessing the dark web, transacting with bitcoin, and identifying drug marketplaces. He also demonstrated a detailed knowledge of chemical manufacturing in China, pill pressing in Canada, and money laundering in Switzerland.
With the air of an insider sharing “trade secrets,” this young man described how dealers on the dark web avoid detection and ensure secure delivery of the goods: latex gloves, vacuum sealing, and bleach dipping to obviate fingerprints – human and chemical. He said that dealers will send “dummy” packages to throw off the authorities and that buyers often will use the address of a clueless or absent neighbor. In his case, however, the parcels were delivered to his doorstep.
. For one thing, acquiring drugs in this way can seem less risky, as there is no chance of being robbed at gunpoint in a sketchy neighborhood or being busted for possession during a routine traffic stop.
Crossing over to the world of the dark web also can give an adolescent a sense of being clever and rebellious and of pulling a “fast one” on the parents – and on us, the clinicians who are treating them. And a teen who is interested in using illicit substances and plays “Call of Duty” from the comfort of his family’s basement without actual injury or death might assume that he can attain illicit drugs that are safe and inexpensive.
There are counterarguments to misinformation about the dark web. For example, contrary to the notion that buying drugs on the dark web minimizes interdiction or arrest, clinicians should point out that since international law enforcement shut down Silk Road and incarcerated its founder, Ross Ulbricht (also known as “Dread Pirate Roberts”) in 2013, hundreds of other dark web marketplaces such as AlphaBay and Hansa have been silenced and their operators prosecuted.
Moreover, the U.S. Department of Justice recently launched the Joint Criminal Opioid Darknet Enforcement (J-CODE) group, and the U.S. Postal Service Inspection Service reportedly has been hiring cybercrime and dark web specialists to combat drug trafficking. It might come as a surprise to a teen that, in a state where recreational marijuana is legal, transport and delivery of cannabis by the postal service elevates purchase and possession to the level of a violation of federal law. Informing even the most oblivious or oppositional adolescent that a drug felony can disqualify him for college grants and loans, impede his search for gainful employment, or prohibit him from obtaining a professional license, might give him a moment’s pause.
Adolescents seeking to buy drugs on the dark web should brace themselves for another shock. Whether lulled by custom after years of shopping on Amazon or using PayPal, or simply dulled by addiction, they might have a blind trust that bitcoin tumblers and “dark” escrow accounts will secure their payments. It will be rude awakening when they learn that the transfer and holding of currency on the dark web is vulnerable to hackers and to operators of the marketplaces – who are known to simply abscond with funds.
Currently, the drug marketplaces on the dark web represent a thin slice of the total illicit drug trade. These marketplaces, however, are growing quickly and offer buyers a virtual smorgasbord: the leading prescription drugs bought are Xanax and OxyContin, whereas 3,4-methylenedioxymethamphetamine (MDMA)/ecstasy and cannabis are the most commonly purchased controlled Schedule I substances, according to an article in The Economist. The estimated annual sales for 2016 ranged from $100 million to $200 million, but even more alarming is the percentage of substance abusers who have purchased drugs on the dark web: A recent article in The Independent reported that 13.2% of U.S. respondents self-reported making at least one purchase online, whereas in the United Kingdom, the self-reported percentage was 25.3 %, and in Finland, it was 41.4%.
Evidence abounds that drugs purchased on the dark web often are counterfeit and sometimes “dirty.” There is a report of “Viagra” containing cement dust, of “Ambien” containing haloperidol, and “Xanax” laced with fentanyl, the latter having been linked to several deaths and hospital admissions in the Bay Area in 2016. (Big Pharma, in fact, is engaged in surveillance and investigation of the sale of knockoffs online, The Telegraph reported in article about the proliferation of fake drugs available on the surface web).Unfortunately, attempting to reduce teen substance abuse using law enforcement measures directed at dark web marketplaces might be a game of whack-a-mole: As soon as one supply source is staunched, another surfaces. Indeed, addiction should be viewed not simply in terms of the biopsychosocial model, but also as an economic activity. Thus, it might be more beneficial for clinicians to concentrate our resources and efforts on curtailing the demand through education and treatment.
Dr. Marseille is a psychiatrist who works on the staff at a clinic in Winfield, Ill. His special interests include adolescent and addiction medicine, eating disorders, trauma, bipolar disorder, and the psychiatric manifestations of acute and chronic medical conditions.
The Clinicians’ Role in Building a System of Care: Army Behavioral Health Since 2001
The Military Health System has changed in countless ways during the era of the Global War on Terror, but no clinical area has been challenged as thoroughly and transformed as extensively as U.S. Army behavioral health care. Through 2011, the wars in Iraq and Afghanistan required soldiers to spend more than 1.5 million years in theater and contributed to significant increases in the incidence of posttraumatic stress disorder (PTSD), depression, and other behavioral health conditions in soldiers and family members.1,2
Unfortunately, the Army’s behavioral health treatment system, which had sufficiently met Army beneficiaries’ needs during peacetime, w
as not adequately resourced or structured to provide care on the scale that was required, and major problems in access, quality, continuity, and safety emerged.3 Army behavioral health and primary care providers experienced these challenges within their own practices, and many developed local solutions. Realizing that a near-complete system transformation was necessary, medical leaders turned to clinicians drawn from the field to develop a strategy to identify the best clinical practices and to build a standardized, cohesive system of care around them.
Although many challenges remain, the Army’s system of behavioral health care has substantially improved and now better supports clinicians in the field as they care for soldiers and their family members. Clinicians at the headquarters and local hospital levels played key roles in this transformation; however, very little literature on the topic exists. Clinicians engaging in current or planning for future health care system changes would benefit from a description of the process.
Unprecedented Challenges
As the wars in Iraq and Afghanistan intensified in the mid-2000s and the demand for behavioral health care grew, the full picture of the shortfalls in the Army’s system of behavioral health care came to the forefront. Provider shortages contributed to long waits for initial appointments, disrupted continuity, and reduced effectiveness of care.4 Even after an infusion of more than $1 billion in congressionally directed funding increased the number of staff and multiplied the clinical and nonclinical programs across the force, significant problems remained.5,6
Army hospitals divided behavioral health care between psychiatry, psychology, and social work fiefdoms, whose stovepipes often fractured communication between providers treating the same patient and prevented effective care coordination. Behavioral health clinics on each post offered different clinical services. Confused soldiers, leaders, and family members were forced to navigate various versions of behavioral health care each time they moved. Inaccurate, locally developed information systems produced little data on the effectiveness of local programs and hindered clinicians and leaders seeking to improve care. Without sufficient clinical capacity on the outpatient side, outpatient providers frequently admitted their patients to inpatient settings because it presented the only option to deliver needed services in a safe setting.
The turning point for improving care occurred in 2012 when senior medical leaders designated a behavioral health leadership team of clinicians, administrators, and analysts at the Army’s Office of the Surgeon General (OTSG) as its first service line. The move consolidated authority over behavioral health-related policy, programs, and funding and clearly communicated that Army Medicine was committed to improving behavioral health care. While behavioral health leaders had operated at OTSG for several years prior, they did not have the authority or resources to make widespread changes. Fortunately, the Army Surgeon General also adopted a command philosophy that emphasized the Operating Company Model, which sought to reduce variance by replicating successful practices across the enterprise. The Operating Company Model also limited each hospital commander’s authority to design their own unique clinical structure and shifted that responsibility to the clinicians in each service line at the headquarters level.
Forming a System of Care
The Behavioral Health Service Line partnered with the U.S. Army Public Health Command and systems engineers at the Massachusetts Institute of Technology to map the existing system of care and identify innovative clinical programs that represented best practices. In addition to 2 programs mandated by the DoD (Behavioral Health in Primary Care Medical Homes7 and Family Advocacy Programs),8 other programs (Embedded Behavioral Health, Multi-Disciplinary Behavioral Health clinics, Intensive Outpatient Program, inpatient care, residential care for substance use disorders, Connect Care, Tele-Behavioral Health, and the Child and Family Behavioral Health System) were selected for replication throughout the Army because they successfully demonstrated promising outcomes and filled a critical need.
To run the emerging standardized clinical programs, the Behavioral Health Service Line streamlined the leadership structure by eliminating all departments organized around provider discipline (ie, psychiatry, psychology, and social work) and created a single department of Behavioral Health at each Army hospital. For the first time, staff were organized into clinical teams based on the needs of the patients, not professional background. This change created new leadership opportunities across disciplines, reduced infighting, and eliminated a major source of confusion for patients and line leaders. The group of standard clinical programs, managed through integrated behavioral health departments in each Army hospital, was dubbed the Behavioral Health System of Care.
Change to department organizations and clinical programs created the need to reconfigure administrative data systems to provide accurate and timely information about system performance. Administrative teams revised Medical Expense Reporting and Performance System codes to provide visibility on important data, such as patient encounters and Revenue Value Units, down to the clinic and provider levels. To improve the reliability of existing data, OTSG staff led a multiyear effort to “clean” several data sources, such as those specifying provider type and work center.
Analysts used the refined workload information to build new tools, such as one to display individual provider productivity and one to predict the number and type of clinical staff required to meet the needs of the population at each Army installation. The administrative reorganization better informed clinical leaders at all levels by supplying meaningful data that enabled comparisons of performance within one or more Army hospitals.8
Translating System Changes Into Better Care
Despite transformative changes to Army behavioral health care, clinicians and leaders still had little insight into what mattered most: measuirng patient response to the treatment. To solve this issue, a team of Army clinical and information technology innovators developed the Behavioral Health Data Portal (BHDP), which used validated scales to collect input from patients at each outpatient visit about their current and recent symptoms. Clinicians use the information to complement their assessment, refine diagnoses, individualize treatment plans, and follow their patients’ progress.9
The BHDP also aggregates information on each patient’s progress into clinical outcome metrics that inform leaders about the effectiveness of the care their clinics provide. For the first time, analysts can establish correlations between specific actions at the clinic level and positive clinical outcomes. For example, the relationship between the use of evidence-based psychotherapies, regular follow-up, a strong therapeutic alliance, and provider use of BHDP have been clearly linked to more rapid resolution of PTSD symptoms. These insights provide opportunities to act on specific processes at the clinic level with high confidence that by doing so, clinicians are improving the effectiveness of the care delivered in their clinics.
Conclusion
The transformation of Army behavioral health care has encompassed all aspects of the treatment system, but it has been led by clinicians working at the local and headquarters levels (Figure). Although many challenges remain, today’s outpatient system is more efficient and effective. For example, Army medical facilities are now able to meet more of the total demand for outpatient behavioral health care of its beneficiaries; 77% in September 2017 compared with a low of 59% in January 2013, based on Army Strategic Management System data as of December 1, 2017. With
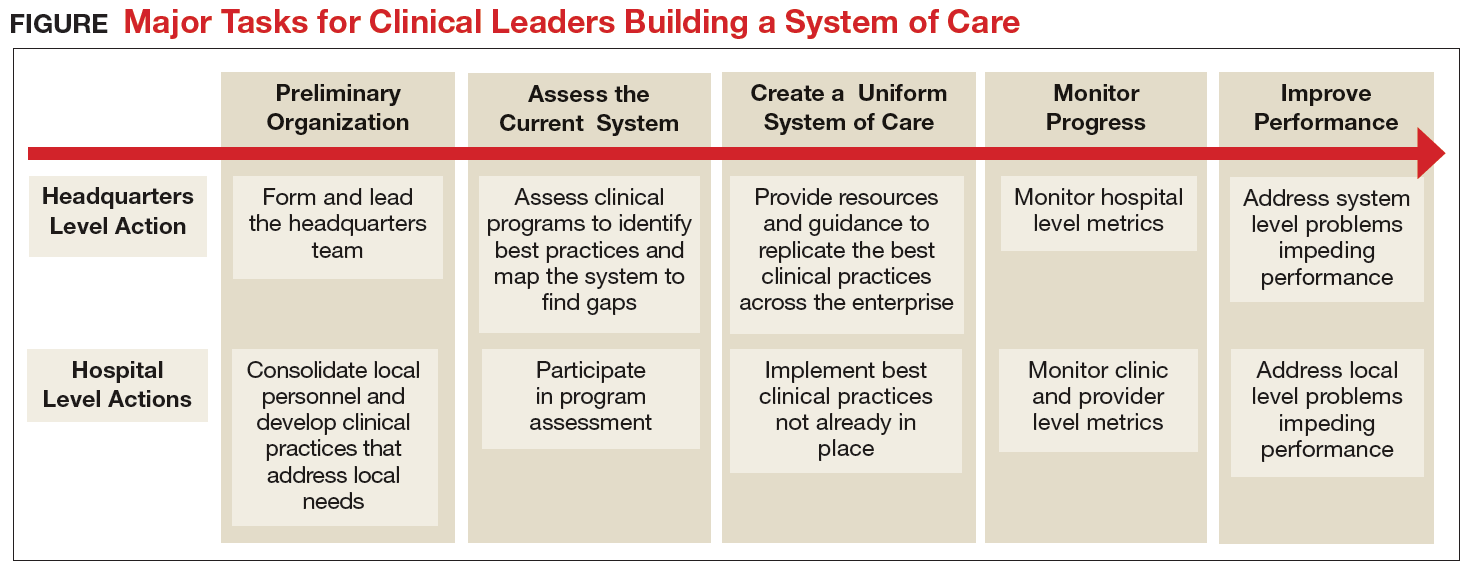
The Army continues to improve its system of care to better inform and enable clinicians to deliver evidence-based care. The clinician-led process that advanced this area of military medicine is applicable to others. A core group of dedicated clinical professionals committed to multiyear processes can implement large-scale changes if they are empowered and resourced by senior medical leaders. A standardized system of care built on clinical best practices and guided by clinicians using accurate data, including clinical outcomes, would benefit any component of the MHS.
1. Baiocchi D. Measuring Army deployments to Iraq and Afghanistan. https://www.rand.org/pubs/research_reports/RR145.html. Published 2013. Accessed December 1, 2017.
2. Hoge C, Castro C, Messer S, McGurk D, Cotting D, Koffman D. Combat duty in Iraq and Afghanistan, mental health problems, and barriers to care. N Engl J Med. 2004;351(1):13-22.
3. Tanielian T, Jaycox LH (eds). Invisible wounds of war. RAND Corporation. https://www.rand.org/pubs/monographs/MG720.html. Published 2008. Accessed December 1, 2017.
4. Prine C. Army’s mental health programs swamped, understaffed. Pittsburgh Tribune-Review. http://triblive .com/x/pittsburghtrib/news/s_721604.html. Published February 7, 2011. Accessed December 1, 2017.
5. Weinick R, Beckjord E, Farmer C, et al. Programs addressing psychological health and traumatic brain injury among U.S. military servicemembers and their families. https://www.rand.org/pubs/technical_reports/TR950 .html. Published 2011. Accessed December 1, 2017.
6. Hoge CW, Ivany CG, Brusher EA, et al. Transformation of mental health care for U.S. soldiers and families during the Iraq and Afghanistan wars: where science and politics intersect. Am J Psychiatry. 2016;173(4):334-343.
7. Hunter CL, Goodie JL. Operational and clinical components for integrated-collaborative behavioral healthcare in the patient-centered medical home. Fam Syst Health. 2010;28(4):308-321.
8. Srinivasan J, Ivany CG, Sarmiento D, Woodson J. How the U.S. Army redesigned its mental health system. Harvard Business Review. https://hbr.org/2017/10/how-the-u-s-army-redesigned-its-mental-health-system. Published October 16, 2017. Accessed December 1, 2017.
9. Srinivasan J, Brown MD, Ivany CG, Woodson J. How the U.S. Army personalized its mental health care. Harvard Business Review. https://hbr.org/2016/12/how-the-u-s-army-personalized-its-mental-health-care. Published December 7, 2016. Accessed December 1, 2017.
The Military Health System has changed in countless ways during the era of the Global War on Terror, but no clinical area has been challenged as thoroughly and transformed as extensively as U.S. Army behavioral health care. Through 2011, the wars in Iraq and Afghanistan required soldiers to spend more than 1.5 million years in theater and contributed to significant increases in the incidence of posttraumatic stress disorder (PTSD), depression, and other behavioral health conditions in soldiers and family members.1,2
Unfortunately, the Army’s behavioral health treatment system, which had sufficiently met Army beneficiaries’ needs during peacetime, w
as not adequately resourced or structured to provide care on the scale that was required, and major problems in access, quality, continuity, and safety emerged.3 Army behavioral health and primary care providers experienced these challenges within their own practices, and many developed local solutions. Realizing that a near-complete system transformation was necessary, medical leaders turned to clinicians drawn from the field to develop a strategy to identify the best clinical practices and to build a standardized, cohesive system of care around them.
Although many challenges remain, the Army’s system of behavioral health care has substantially improved and now better supports clinicians in the field as they care for soldiers and their family members. Clinicians at the headquarters and local hospital levels played key roles in this transformation; however, very little literature on the topic exists. Clinicians engaging in current or planning for future health care system changes would benefit from a description of the process.
Unprecedented Challenges
As the wars in Iraq and Afghanistan intensified in the mid-2000s and the demand for behavioral health care grew, the full picture of the shortfalls in the Army’s system of behavioral health care came to the forefront. Provider shortages contributed to long waits for initial appointments, disrupted continuity, and reduced effectiveness of care.4 Even after an infusion of more than $1 billion in congressionally directed funding increased the number of staff and multiplied the clinical and nonclinical programs across the force, significant problems remained.5,6
Army hospitals divided behavioral health care between psychiatry, psychology, and social work fiefdoms, whose stovepipes often fractured communication between providers treating the same patient and prevented effective care coordination. Behavioral health clinics on each post offered different clinical services. Confused soldiers, leaders, and family members were forced to navigate various versions of behavioral health care each time they moved. Inaccurate, locally developed information systems produced little data on the effectiveness of local programs and hindered clinicians and leaders seeking to improve care. Without sufficient clinical capacity on the outpatient side, outpatient providers frequently admitted their patients to inpatient settings because it presented the only option to deliver needed services in a safe setting.
The turning point for improving care occurred in 2012 when senior medical leaders designated a behavioral health leadership team of clinicians, administrators, and analysts at the Army’s Office of the Surgeon General (OTSG) as its first service line. The move consolidated authority over behavioral health-related policy, programs, and funding and clearly communicated that Army Medicine was committed to improving behavioral health care. While behavioral health leaders had operated at OTSG for several years prior, they did not have the authority or resources to make widespread changes. Fortunately, the Army Surgeon General also adopted a command philosophy that emphasized the Operating Company Model, which sought to reduce variance by replicating successful practices across the enterprise. The Operating Company Model also limited each hospital commander’s authority to design their own unique clinical structure and shifted that responsibility to the clinicians in each service line at the headquarters level.
Forming a System of Care
The Behavioral Health Service Line partnered with the U.S. Army Public Health Command and systems engineers at the Massachusetts Institute of Technology to map the existing system of care and identify innovative clinical programs that represented best practices. In addition to 2 programs mandated by the DoD (Behavioral Health in Primary Care Medical Homes7 and Family Advocacy Programs),8 other programs (Embedded Behavioral Health, Multi-Disciplinary Behavioral Health clinics, Intensive Outpatient Program, inpatient care, residential care for substance use disorders, Connect Care, Tele-Behavioral Health, and the Child and Family Behavioral Health System) were selected for replication throughout the Army because they successfully demonstrated promising outcomes and filled a critical need.
To run the emerging standardized clinical programs, the Behavioral Health Service Line streamlined the leadership structure by eliminating all departments organized around provider discipline (ie, psychiatry, psychology, and social work) and created a single department of Behavioral Health at each Army hospital. For the first time, staff were organized into clinical teams based on the needs of the patients, not professional background. This change created new leadership opportunities across disciplines, reduced infighting, and eliminated a major source of confusion for patients and line leaders. The group of standard clinical programs, managed through integrated behavioral health departments in each Army hospital, was dubbed the Behavioral Health System of Care.
Change to department organizations and clinical programs created the need to reconfigure administrative data systems to provide accurate and timely information about system performance. Administrative teams revised Medical Expense Reporting and Performance System codes to provide visibility on important data, such as patient encounters and Revenue Value Units, down to the clinic and provider levels. To improve the reliability of existing data, OTSG staff led a multiyear effort to “clean” several data sources, such as those specifying provider type and work center.
Analysts used the refined workload information to build new tools, such as one to display individual provider productivity and one to predict the number and type of clinical staff required to meet the needs of the population at each Army installation. The administrative reorganization better informed clinical leaders at all levels by supplying meaningful data that enabled comparisons of performance within one or more Army hospitals.8
Translating System Changes Into Better Care
Despite transformative changes to Army behavioral health care, clinicians and leaders still had little insight into what mattered most: measuirng patient response to the treatment. To solve this issue, a team of Army clinical and information technology innovators developed the Behavioral Health Data Portal (BHDP), which used validated scales to collect input from patients at each outpatient visit about their current and recent symptoms. Clinicians use the information to complement their assessment, refine diagnoses, individualize treatment plans, and follow their patients’ progress.9
The BHDP also aggregates information on each patient’s progress into clinical outcome metrics that inform leaders about the effectiveness of the care their clinics provide. For the first time, analysts can establish correlations between specific actions at the clinic level and positive clinical outcomes. For example, the relationship between the use of evidence-based psychotherapies, regular follow-up, a strong therapeutic alliance, and provider use of BHDP have been clearly linked to more rapid resolution of PTSD symptoms. These insights provide opportunities to act on specific processes at the clinic level with high confidence that by doing so, clinicians are improving the effectiveness of the care delivered in their clinics.
Conclusion
The transformation of Army behavioral health care has encompassed all aspects of the treatment system, but it has been led by clinicians working at the local and headquarters levels (Figure). Although many challenges remain, today’s outpatient system is more efficient and effective. For example, Army medical facilities are now able to meet more of the total demand for outpatient behavioral health care of its beneficiaries; 77% in September 2017 compared with a low of 59% in January 2013, based on Army Strategic Management System data as of December 1, 2017. With
The Army continues to improve its system of care to better inform and enable clinicians to deliver evidence-based care. The clinician-led process that advanced this area of military medicine is applicable to others. A core group of dedicated clinical professionals committed to multiyear processes can implement large-scale changes if they are empowered and resourced by senior medical leaders. A standardized system of care built on clinical best practices and guided by clinicians using accurate data, including clinical outcomes, would benefit any component of the MHS.
The Military Health System has changed in countless ways during the era of the Global War on Terror, but no clinical area has been challenged as thoroughly and transformed as extensively as U.S. Army behavioral health care. Through 2011, the wars in Iraq and Afghanistan required soldiers to spend more than 1.5 million years in theater and contributed to significant increases in the incidence of posttraumatic stress disorder (PTSD), depression, and other behavioral health conditions in soldiers and family members.1,2
Unfortunately, the Army’s behavioral health treatment system, which had sufficiently met Army beneficiaries’ needs during peacetime, w
as not adequately resourced or structured to provide care on the scale that was required, and major problems in access, quality, continuity, and safety emerged.3 Army behavioral health and primary care providers experienced these challenges within their own practices, and many developed local solutions. Realizing that a near-complete system transformation was necessary, medical leaders turned to clinicians drawn from the field to develop a strategy to identify the best clinical practices and to build a standardized, cohesive system of care around them.
Although many challenges remain, the Army’s system of behavioral health care has substantially improved and now better supports clinicians in the field as they care for soldiers and their family members. Clinicians at the headquarters and local hospital levels played key roles in this transformation; however, very little literature on the topic exists. Clinicians engaging in current or planning for future health care system changes would benefit from a description of the process.
Unprecedented Challenges
As the wars in Iraq and Afghanistan intensified in the mid-2000s and the demand for behavioral health care grew, the full picture of the shortfalls in the Army’s system of behavioral health care came to the forefront. Provider shortages contributed to long waits for initial appointments, disrupted continuity, and reduced effectiveness of care.4 Even after an infusion of more than $1 billion in congressionally directed funding increased the number of staff and multiplied the clinical and nonclinical programs across the force, significant problems remained.5,6
Army hospitals divided behavioral health care between psychiatry, psychology, and social work fiefdoms, whose stovepipes often fractured communication between providers treating the same patient and prevented effective care coordination. Behavioral health clinics on each post offered different clinical services. Confused soldiers, leaders, and family members were forced to navigate various versions of behavioral health care each time they moved. Inaccurate, locally developed information systems produced little data on the effectiveness of local programs and hindered clinicians and leaders seeking to improve care. Without sufficient clinical capacity on the outpatient side, outpatient providers frequently admitted their patients to inpatient settings because it presented the only option to deliver needed services in a safe setting.
The turning point for improving care occurred in 2012 when senior medical leaders designated a behavioral health leadership team of clinicians, administrators, and analysts at the Army’s Office of the Surgeon General (OTSG) as its first service line. The move consolidated authority over behavioral health-related policy, programs, and funding and clearly communicated that Army Medicine was committed to improving behavioral health care. While behavioral health leaders had operated at OTSG for several years prior, they did not have the authority or resources to make widespread changes. Fortunately, the Army Surgeon General also adopted a command philosophy that emphasized the Operating Company Model, which sought to reduce variance by replicating successful practices across the enterprise. The Operating Company Model also limited each hospital commander’s authority to design their own unique clinical structure and shifted that responsibility to the clinicians in each service line at the headquarters level.
Forming a System of Care
The Behavioral Health Service Line partnered with the U.S. Army Public Health Command and systems engineers at the Massachusetts Institute of Technology to map the existing system of care and identify innovative clinical programs that represented best practices. In addition to 2 programs mandated by the DoD (Behavioral Health in Primary Care Medical Homes7 and Family Advocacy Programs),8 other programs (Embedded Behavioral Health, Multi-Disciplinary Behavioral Health clinics, Intensive Outpatient Program, inpatient care, residential care for substance use disorders, Connect Care, Tele-Behavioral Health, and the Child and Family Behavioral Health System) were selected for replication throughout the Army because they successfully demonstrated promising outcomes and filled a critical need.
To run the emerging standardized clinical programs, the Behavioral Health Service Line streamlined the leadership structure by eliminating all departments organized around provider discipline (ie, psychiatry, psychology, and social work) and created a single department of Behavioral Health at each Army hospital. For the first time, staff were organized into clinical teams based on the needs of the patients, not professional background. This change created new leadership opportunities across disciplines, reduced infighting, and eliminated a major source of confusion for patients and line leaders. The group of standard clinical programs, managed through integrated behavioral health departments in each Army hospital, was dubbed the Behavioral Health System of Care.
Change to department organizations and clinical programs created the need to reconfigure administrative data systems to provide accurate and timely information about system performance. Administrative teams revised Medical Expense Reporting and Performance System codes to provide visibility on important data, such as patient encounters and Revenue Value Units, down to the clinic and provider levels. To improve the reliability of existing data, OTSG staff led a multiyear effort to “clean” several data sources, such as those specifying provider type and work center.
Analysts used the refined workload information to build new tools, such as one to display individual provider productivity and one to predict the number and type of clinical staff required to meet the needs of the population at each Army installation. The administrative reorganization better informed clinical leaders at all levels by supplying meaningful data that enabled comparisons of performance within one or more Army hospitals.8
Translating System Changes Into Better Care
Despite transformative changes to Army behavioral health care, clinicians and leaders still had little insight into what mattered most: measuirng patient response to the treatment. To solve this issue, a team of Army clinical and information technology innovators developed the Behavioral Health Data Portal (BHDP), which used validated scales to collect input from patients at each outpatient visit about their current and recent symptoms. Clinicians use the information to complement their assessment, refine diagnoses, individualize treatment plans, and follow their patients’ progress.9
The BHDP also aggregates information on each patient’s progress into clinical outcome metrics that inform leaders about the effectiveness of the care their clinics provide. For the first time, analysts can establish correlations between specific actions at the clinic level and positive clinical outcomes. For example, the relationship between the use of evidence-based psychotherapies, regular follow-up, a strong therapeutic alliance, and provider use of BHDP have been clearly linked to more rapid resolution of PTSD symptoms. These insights provide opportunities to act on specific processes at the clinic level with high confidence that by doing so, clinicians are improving the effectiveness of the care delivered in their clinics.
Conclusion
The transformation of Army behavioral health care has encompassed all aspects of the treatment system, but it has been led by clinicians working at the local and headquarters levels (Figure). Although many challenges remain, today’s outpatient system is more efficient and effective. For example, Army medical facilities are now able to meet more of the total demand for outpatient behavioral health care of its beneficiaries; 77% in September 2017 compared with a low of 59% in January 2013, based on Army Strategic Management System data as of December 1, 2017. With
The Army continues to improve its system of care to better inform and enable clinicians to deliver evidence-based care. The clinician-led process that advanced this area of military medicine is applicable to others. A core group of dedicated clinical professionals committed to multiyear processes can implement large-scale changes if they are empowered and resourced by senior medical leaders. A standardized system of care built on clinical best practices and guided by clinicians using accurate data, including clinical outcomes, would benefit any component of the MHS.
1. Baiocchi D. Measuring Army deployments to Iraq and Afghanistan. https://www.rand.org/pubs/research_reports/RR145.html. Published 2013. Accessed December 1, 2017.
2. Hoge C, Castro C, Messer S, McGurk D, Cotting D, Koffman D. Combat duty in Iraq and Afghanistan, mental health problems, and barriers to care. N Engl J Med. 2004;351(1):13-22.
3. Tanielian T, Jaycox LH (eds). Invisible wounds of war. RAND Corporation. https://www.rand.org/pubs/monographs/MG720.html. Published 2008. Accessed December 1, 2017.
4. Prine C. Army’s mental health programs swamped, understaffed. Pittsburgh Tribune-Review. http://triblive .com/x/pittsburghtrib/news/s_721604.html. Published February 7, 2011. Accessed December 1, 2017.
5. Weinick R, Beckjord E, Farmer C, et al. Programs addressing psychological health and traumatic brain injury among U.S. military servicemembers and their families. https://www.rand.org/pubs/technical_reports/TR950 .html. Published 2011. Accessed December 1, 2017.
6. Hoge CW, Ivany CG, Brusher EA, et al. Transformation of mental health care for U.S. soldiers and families during the Iraq and Afghanistan wars: where science and politics intersect. Am J Psychiatry. 2016;173(4):334-343.
7. Hunter CL, Goodie JL. Operational and clinical components for integrated-collaborative behavioral healthcare in the patient-centered medical home. Fam Syst Health. 2010;28(4):308-321.
8. Srinivasan J, Ivany CG, Sarmiento D, Woodson J. How the U.S. Army redesigned its mental health system. Harvard Business Review. https://hbr.org/2017/10/how-the-u-s-army-redesigned-its-mental-health-system. Published October 16, 2017. Accessed December 1, 2017.
9. Srinivasan J, Brown MD, Ivany CG, Woodson J. How the U.S. Army personalized its mental health care. Harvard Business Review. https://hbr.org/2016/12/how-the-u-s-army-personalized-its-mental-health-care. Published December 7, 2016. Accessed December 1, 2017.
1. Baiocchi D. Measuring Army deployments to Iraq and Afghanistan. https://www.rand.org/pubs/research_reports/RR145.html. Published 2013. Accessed December 1, 2017.
2. Hoge C, Castro C, Messer S, McGurk D, Cotting D, Koffman D. Combat duty in Iraq and Afghanistan, mental health problems, and barriers to care. N Engl J Med. 2004;351(1):13-22.
3. Tanielian T, Jaycox LH (eds). Invisible wounds of war. RAND Corporation. https://www.rand.org/pubs/monographs/MG720.html. Published 2008. Accessed December 1, 2017.
4. Prine C. Army’s mental health programs swamped, understaffed. Pittsburgh Tribune-Review. http://triblive .com/x/pittsburghtrib/news/s_721604.html. Published February 7, 2011. Accessed December 1, 2017.
5. Weinick R, Beckjord E, Farmer C, et al. Programs addressing psychological health and traumatic brain injury among U.S. military servicemembers and their families. https://www.rand.org/pubs/technical_reports/TR950 .html. Published 2011. Accessed December 1, 2017.
6. Hoge CW, Ivany CG, Brusher EA, et al. Transformation of mental health care for U.S. soldiers and families during the Iraq and Afghanistan wars: where science and politics intersect. Am J Psychiatry. 2016;173(4):334-343.
7. Hunter CL, Goodie JL. Operational and clinical components for integrated-collaborative behavioral healthcare in the patient-centered medical home. Fam Syst Health. 2010;28(4):308-321.
8. Srinivasan J, Ivany CG, Sarmiento D, Woodson J. How the U.S. Army redesigned its mental health system. Harvard Business Review. https://hbr.org/2017/10/how-the-u-s-army-redesigned-its-mental-health-system. Published October 16, 2017. Accessed December 1, 2017.
9. Srinivasan J, Brown MD, Ivany CG, Woodson J. How the U.S. Army personalized its mental health care. Harvard Business Review. https://hbr.org/2016/12/how-the-u-s-army-personalized-its-mental-health-care. Published December 7, 2016. Accessed December 1, 2017.
Novel Treatment Helps Heal Diabetic Foot Ulcers
About 30 million people in the U.S. have diabetes, and about 25% of those will have a foot ulcer at some point. When circulation is so poor that the ulcer does not heal, or when treatment cannot stop infection, amputation may be necessary. Diabetes is the leading cause of lower limb amputations.
The FDA has approved a new treatment to help heal diabetic foot ulcers. The Dermapace System (Sanuwave, Inc., Suwanee, GA) sends shock waves to mechanically stimulate the wound. The treatment is intended for adult patients with chronic foot ulcers with wound areas no larger than 16 cm2, extending through the epidermis, dermis, tendon, or capsule, but without bone exposure. It is used along with standard diabetic ulcer care.
In 2 multicenter studies of 336 patients, patients were given 1 to 7 treatments over 24 weeks. The shock wave systems increased wound healing by 44%. The patients treated with a sham shock wave therapy had a 30% wound closure rate.
About 30 million people in the U.S. have diabetes, and about 25% of those will have a foot ulcer at some point. When circulation is so poor that the ulcer does not heal, or when treatment cannot stop infection, amputation may be necessary. Diabetes is the leading cause of lower limb amputations.
The FDA has approved a new treatment to help heal diabetic foot ulcers. The Dermapace System (Sanuwave, Inc., Suwanee, GA) sends shock waves to mechanically stimulate the wound. The treatment is intended for adult patients with chronic foot ulcers with wound areas no larger than 16 cm2, extending through the epidermis, dermis, tendon, or capsule, but without bone exposure. It is used along with standard diabetic ulcer care.
In 2 multicenter studies of 336 patients, patients were given 1 to 7 treatments over 24 weeks. The shock wave systems increased wound healing by 44%. The patients treated with a sham shock wave therapy had a 30% wound closure rate.
About 30 million people in the U.S. have diabetes, and about 25% of those will have a foot ulcer at some point. When circulation is so poor that the ulcer does not heal, or when treatment cannot stop infection, amputation may be necessary. Diabetes is the leading cause of lower limb amputations.
The FDA has approved a new treatment to help heal diabetic foot ulcers. The Dermapace System (Sanuwave, Inc., Suwanee, GA) sends shock waves to mechanically stimulate the wound. The treatment is intended for adult patients with chronic foot ulcers with wound areas no larger than 16 cm2, extending through the epidermis, dermis, tendon, or capsule, but without bone exposure. It is used along with standard diabetic ulcer care.
In 2 multicenter studies of 336 patients, patients were given 1 to 7 treatments over 24 weeks. The shock wave systems increased wound healing by 44%. The patients treated with a sham shock wave therapy had a 30% wound closure rate.
AYA cancer patients lost to follow-up

ORLANDO—Many adolescent and young adult (AYA) cancer survivors don’t continue with follow-up care, according to a new study.
Researchers analyzed nearly 2400 AYAs diagnosed with cancer between 2000 and 2015 and found that 37% of these patients had no follow-up visits since 2015.
The proportion of patients with follow-up visits in 2016 did not vary according to cancer type or insurance status, but it did vary according to patients’ time since last cancer treatment.
These findings were presented at the 2018 Cancer Survivorship Symposium (abstract 29).
“Many adolescents and young adults are unaware of what their long-term risks are after they have finished their cancer treatment,” said study investigator Lynda M. Beaupin, MD, of the Roswell Park Cancer Institute in Buffalo, New York.
“Doctors and other healthcare providers need to be more diligent in letting these patients know about future potential side effects and health risks that could occur based on certain aspects of their cancer treatment.”
In a previous study, Dr Beaupin and her colleagues conducted a focus group discussion with 27 AYAs, ages 18 to 39, to query them about the major barriers that prevented them from seeking follow-up care.
Among the factors mentioned were poor communication with their oncologists, ongoing problems in adjusting to life as a cancer survivor, and loss of health insurance.
As a follow-up to that focus group session, the investigators looked at a larger group of AYAs who were treated at Roswell Park Comprehensive Cancer Center.
The team analyzed data from the cancer center’s tumor registry, which included information on a patient’s current age, age at cancer diagnosis, gender, date of diagnosis, date of most recent doctor visit, and type of cancer.
The investigators also looked at patient-provided information on follow-up doctor appointments and insurance status for 2 cohorts of AYA cancer survivors. Patients in cohort A were diagnosed with cancer between 2010 and 2014, and patients in cohort B were diagnosed between 2005 and 2009.
The most common types of cancer were leukemia/lymphoma, melanoma, germ cell tumors, and thyroid and breast cancers.
Findings
There were 2367 patients, ages 18 to 39, who were diagnosed with cancer between 2000 and 2015.
Thirty-seven percent of these patients did not have a follow-up visit since 2015.
There was no difference in follow-up contact according to cancer type or insurance status. However, length of time since patients’ final cancer treatment was a factor in not scheduling a follow-up appointment.
Regardless of insurance status, 33% of cohort A (diagnosed 2010-2014) and 48% of cohort B (diagnosed 2005-2009) did not have a follow-up visit in 2016.
Among insured patients, 33% of those in cohort A and 47% of those in cohort B did not have a visit in 2016. Among uninsured patients, the percentages were 39% and 46%, respectively.
Next steps
The investigators hope to conduct future studies looking at other factors that may affect AYAs’ willingness to seek follow-up care, such as employment status, distance to a cancer center, whether they are getting tested or treated at a facility other than a cancer center, and how AYAs perceive their current quality of life.
“These patients have the potential to live a normal lifespan, and we need to educate them to become their own advocates so they may receive follow-up care on a regular basis,” Dr Beaupin said.
“We hope they continue to receive that follow-up at an established cancer center that has the facilities to assess cardiac health and provide rehabilitation if needed. There are now established survivorship programs nationwide that can provide follow-up care for those who have completed treatment.”
This study was supported by the National Cancer Institute.
ORLANDO—Many adolescent and young adult (AYA) cancer survivors don’t continue with follow-up care, according to a new study.
Researchers analyzed nearly 2400 AYAs diagnosed with cancer between 2000 and 2015 and found that 37% of these patients had no follow-up visits since 2015.
The proportion of patients with follow-up visits in 2016 did not vary according to cancer type or insurance status, but it did vary according to patients’ time since last cancer treatment.
These findings were presented at the 2018 Cancer Survivorship Symposium (abstract 29).
“Many adolescents and young adults are unaware of what their long-term risks are after they have finished their cancer treatment,” said study investigator Lynda M. Beaupin, MD, of the Roswell Park Cancer Institute in Buffalo, New York.
“Doctors and other healthcare providers need to be more diligent in letting these patients know about future potential side effects and health risks that could occur based on certain aspects of their cancer treatment.”
In a previous study, Dr Beaupin and her colleagues conducted a focus group discussion with 27 AYAs, ages 18 to 39, to query them about the major barriers that prevented them from seeking follow-up care.
Among the factors mentioned were poor communication with their oncologists, ongoing problems in adjusting to life as a cancer survivor, and loss of health insurance.
As a follow-up to that focus group session, the investigators looked at a larger group of AYAs who were treated at Roswell Park Comprehensive Cancer Center.
The team analyzed data from the cancer center’s tumor registry, which included information on a patient’s current age, age at cancer diagnosis, gender, date of diagnosis, date of most recent doctor visit, and type of cancer.
The investigators also looked at patient-provided information on follow-up doctor appointments and insurance status for 2 cohorts of AYA cancer survivors. Patients in cohort A were diagnosed with cancer between 2010 and 2014, and patients in cohort B were diagnosed between 2005 and 2009.
The most common types of cancer were leukemia/lymphoma, melanoma, germ cell tumors, and thyroid and breast cancers.
Findings
There were 2367 patients, ages 18 to 39, who were diagnosed with cancer between 2000 and 2015.
Thirty-seven percent of these patients did not have a follow-up visit since 2015.
There was no difference in follow-up contact according to cancer type or insurance status. However, length of time since patients’ final cancer treatment was a factor in not scheduling a follow-up appointment.
Regardless of insurance status, 33% of cohort A (diagnosed 2010-2014) and 48% of cohort B (diagnosed 2005-2009) did not have a follow-up visit in 2016.
Among insured patients, 33% of those in cohort A and 47% of those in cohort B did not have a visit in 2016. Among uninsured patients, the percentages were 39% and 46%, respectively.
Next steps
The investigators hope to conduct future studies looking at other factors that may affect AYAs’ willingness to seek follow-up care, such as employment status, distance to a cancer center, whether they are getting tested or treated at a facility other than a cancer center, and how AYAs perceive their current quality of life.
“These patients have the potential to live a normal lifespan, and we need to educate them to become their own advocates so they may receive follow-up care on a regular basis,” Dr Beaupin said.
“We hope they continue to receive that follow-up at an established cancer center that has the facilities to assess cardiac health and provide rehabilitation if needed. There are now established survivorship programs nationwide that can provide follow-up care for those who have completed treatment.”
This study was supported by the National Cancer Institute.
ORLANDO—Many adolescent and young adult (AYA) cancer survivors don’t continue with follow-up care, according to a new study.
Researchers analyzed nearly 2400 AYAs diagnosed with cancer between 2000 and 2015 and found that 37% of these patients had no follow-up visits since 2015.
The proportion of patients with follow-up visits in 2016 did not vary according to cancer type or insurance status, but it did vary according to patients’ time since last cancer treatment.
These findings were presented at the 2018 Cancer Survivorship Symposium (abstract 29).
“Many adolescents and young adults are unaware of what their long-term risks are after they have finished their cancer treatment,” said study investigator Lynda M. Beaupin, MD, of the Roswell Park Cancer Institute in Buffalo, New York.
“Doctors and other healthcare providers need to be more diligent in letting these patients know about future potential side effects and health risks that could occur based on certain aspects of their cancer treatment.”
In a previous study, Dr Beaupin and her colleagues conducted a focus group discussion with 27 AYAs, ages 18 to 39, to query them about the major barriers that prevented them from seeking follow-up care.
Among the factors mentioned were poor communication with their oncologists, ongoing problems in adjusting to life as a cancer survivor, and loss of health insurance.
As a follow-up to that focus group session, the investigators looked at a larger group of AYAs who were treated at Roswell Park Comprehensive Cancer Center.
The team analyzed data from the cancer center’s tumor registry, which included information on a patient’s current age, age at cancer diagnosis, gender, date of diagnosis, date of most recent doctor visit, and type of cancer.
The investigators also looked at patient-provided information on follow-up doctor appointments and insurance status for 2 cohorts of AYA cancer survivors. Patients in cohort A were diagnosed with cancer between 2010 and 2014, and patients in cohort B were diagnosed between 2005 and 2009.
The most common types of cancer were leukemia/lymphoma, melanoma, germ cell tumors, and thyroid and breast cancers.
Findings
There were 2367 patients, ages 18 to 39, who were diagnosed with cancer between 2000 and 2015.
Thirty-seven percent of these patients did not have a follow-up visit since 2015.
There was no difference in follow-up contact according to cancer type or insurance status. However, length of time since patients’ final cancer treatment was a factor in not scheduling a follow-up appointment.
Regardless of insurance status, 33% of cohort A (diagnosed 2010-2014) and 48% of cohort B (diagnosed 2005-2009) did not have a follow-up visit in 2016.
Among insured patients, 33% of those in cohort A and 47% of those in cohort B did not have a visit in 2016. Among uninsured patients, the percentages were 39% and 46%, respectively.
Next steps
The investigators hope to conduct future studies looking at other factors that may affect AYAs’ willingness to seek follow-up care, such as employment status, distance to a cancer center, whether they are getting tested or treated at a facility other than a cancer center, and how AYAs perceive their current quality of life.
“These patients have the potential to live a normal lifespan, and we need to educate them to become their own advocates so they may receive follow-up care on a regular basis,” Dr Beaupin said.
“We hope they continue to receive that follow-up at an established cancer center that has the facilities to assess cardiac health and provide rehabilitation if needed. There are now established survivorship programs nationwide that can provide follow-up care for those who have completed treatment.”
This study was supported by the National Cancer Institute.
How real is resident burnout?
JACKSONVILLE, FLA. – Burnout is commonly ascribed to surgical residents, but reliable estimates of the numbers involved and a clear, comparable definition of burnout remain elusive.
A large study of general surgery residents has found that almost one in four have at least one symptom of burnout daily and more than two in five report poor psychiatric well-being, according to results of a survey reported at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
“Twenty-two percent of general surgery residents report at least one symptom of burnout daily,” said Daniel “Brock” Hewitt, MD, a research fellow in the Surgical Outcomes and Quality Improvement Center in the department of surgery, Feinberg School of Medicine, Northwestern University, Chicago. “Poor resident wellness is associated with more duty-hour violations, feeling unprepared for residency, and an increase in self-reported medical errors. However, burnout is not associated with worse surgical outcomes.”
The study, undertaken with grant support from The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality, provided three significant insights into resident wellness, Dr. Hewitt said. “When measuring the level of burnout, careful consideration should be given to how burnout is defined,” he said. “Secondly, wellness interventions with attention to preparedness for residency and duty-hour restrictions may help alleviate burnout. And finally, with the significant impact it does have on the individual physician, we believe that burnout needs to be addressed regardless of its impact on patient outcomes.”
The study drew on a questionnaire given to residents immediately following the 2017 American Board of Surgery In-Training Examination (ABSITE), which 3,789 general surgery residents from 115 general surgery programs completed. The survey had a 99.3% response rate. The survey evaluated two factors associated with resident wellness: burnout and poor psychiatric well-being. “Poor wellness is prevalent among physicians and trainees and is associated with depression, suicidal ideation, attrition, and absenteeism,” Dr. Hewitt said.
But to measure burnout, the researchers had to first define it. The instrument Dr. Hewitt and coauthors used is the Maslach Burnout Inventory, named for University of California at Berkley psychology professor Christina Maslach, PhD. It quantifies three different factors for burnout: emotional exhaustion; depersonalization or cynicism; and a low sense of personal accomplishment. This study defined “burnout” as having feelings of both emotional exhaustion and depersonalization, and dismissed the third measure of burnout because physicians rarely posses a low sense of personal accomplishment.
Overall, 22.3% reported one sign or symptom of burnout daily, and 56.5% did so on a weekly basis, Dr. Hewitt said.
However, Dr. Hewitt noted, burnout measurement thresholds can vary “because burnout itself is not an actual diagnosis.” Studies have calculated the burnout rate among surgeons at 28% to 69% (J Am Coll Surg. 2016:222:1230-9). A recent systematic review found up to eight different cutoffs used to define “high burnout” (Cogent Med. 2016 7 Oct; doi.org/10.1080/2331205X.2016.1237605; J Am Coll Surg, 2016:223:440-51)

How studies of physician burnout establish cutoffs and which Maslach Burnout Inventory subscales they measure has an impact on the rates of burnout they report, Dr. Hewitt said. For this study, the researchers used the Maslach scores in the top quartile in both emotional exhaustion and depersonalization to define burnout. “Burnout is probably best defined as a continuum from engagement on the one end and burnout on the other, so there’s some combination of the [Maslach] subscales that lead to burnout,” he said.
Among survey respondents, 19.5% reported high Maslach scores for emotional exhaustion and 9% of depersonalization on a daily basis (6.2% reported both)--22.3% reported at least one daily. On a weekly basis, 54.2% reported high scores for emotional exhaustion and 25.6% high scores for depersonalization, with 56.5% reporting at least one and 22.4% reporting both.
To evaluate residents’ sense of psychiatric well-being, the study used the General Health Questionnaire (Psychol Med. 1998;28:915-21). Among survey respondents, 43% met criteria for poor psychiatric well-being.
Burnout rates among men and women were identical, but a significantly higher percentage of women had poor psychiatric well-being: 48.4% to 39.7% of men.
Burnout and psychiatric well-being scores also varied depending on post-graduate year. Second- and third-year residents were significantly more likely to report burnout, compared to first-year residents, Dr. Hewitt said. Rates of poor psychiatric well-being were lowest for fourth- and fifth-year residents.
Most pronounced was the impact of 80-hour duty week violations had on residents’ sense of burnout and poor psychiatric well-being. Burnout rates were 8.4% for those who reported no monthly duty-hour violations, but doubled and quadrupled with more frequent monthly duty-hour violations: 16.1% for those who reported one to four violations a month; and 35% for those who reported five or more. Dr. Hewitt also noted wellness rates for those in the standard and flexible policy groups in Flexibility In duty hour Requirements for Surgical Trainees Trial (FIRST Trial) programs were similar.
Another factor that contributed to burnout and poor psychiatric well-being was a sense of unpreparedness for residency, Dr. Hewitt said. Burnout rates for those who felt prepared were 8.9% vs. 19.1% for those who didn’t feel prepared. The disparity was less drastic, but nonetheless significant, for poor psychiatric well-being: 37.5% for those who felt prepared and 52.1% for those who didn’t.
With regard to outcomes, Dr. Hewitt said, “Residents in the highest quartile of burnout and the highest quartile of poor psychiatric well-being were significantly more likely to report a near miss or harmful medical error.” Among the burnout group, highest quartile rates were 39.3% for a near miss and 14.4% for a harmful medical error vs. 11.1% and 2.4%, respectively, for the lowest quartile. Among surgical residents who reported poor psychiatric well-being, highest quartile rates were 31.9% for a near miss and 13.2% for a harmful medical error vs. 12.5% and 2.1%, respectively, for those in the lowest quartile.
The study also analyzed outcomes for 134,877 surgical patients and found no association of overall morbidity and death or serious morbidity with resident wellness. “However,” Dr. Hewitt said, “when we look at mortality and failure to rescue, we can see an association with burnout, specifically in programs that have high levels of burnout; these patients have significantly lower odds of mortality and failure to rescue.” Each has an adjusted odds ratios of 0.81.
Among the study limitations Dr. Hewitt noted were its cross-sectional nature that led to inferences of association, not identification of causation; residents completing the survey after the ABSITE may have influenced their answers; and the fact it did not account for certain intermediate factors such as physician or nursing burnout or institutional quality measures.
The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality provided grants to support the research.
SOURCE: Hewitt B et al. abstract 21.01
JACKSONVILLE, FLA. – Burnout is commonly ascribed to surgical residents, but reliable estimates of the numbers involved and a clear, comparable definition of burnout remain elusive.
A large study of general surgery residents has found that almost one in four have at least one symptom of burnout daily and more than two in five report poor psychiatric well-being, according to results of a survey reported at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
“Twenty-two percent of general surgery residents report at least one symptom of burnout daily,” said Daniel “Brock” Hewitt, MD, a research fellow in the Surgical Outcomes and Quality Improvement Center in the department of surgery, Feinberg School of Medicine, Northwestern University, Chicago. “Poor resident wellness is associated with more duty-hour violations, feeling unprepared for residency, and an increase in self-reported medical errors. However, burnout is not associated with worse surgical outcomes.”
The study, undertaken with grant support from The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality, provided three significant insights into resident wellness, Dr. Hewitt said. “When measuring the level of burnout, careful consideration should be given to how burnout is defined,” he said. “Secondly, wellness interventions with attention to preparedness for residency and duty-hour restrictions may help alleviate burnout. And finally, with the significant impact it does have on the individual physician, we believe that burnout needs to be addressed regardless of its impact on patient outcomes.”
The study drew on a questionnaire given to residents immediately following the 2017 American Board of Surgery In-Training Examination (ABSITE), which 3,789 general surgery residents from 115 general surgery programs completed. The survey had a 99.3% response rate. The survey evaluated two factors associated with resident wellness: burnout and poor psychiatric well-being. “Poor wellness is prevalent among physicians and trainees and is associated with depression, suicidal ideation, attrition, and absenteeism,” Dr. Hewitt said.
But to measure burnout, the researchers had to first define it. The instrument Dr. Hewitt and coauthors used is the Maslach Burnout Inventory, named for University of California at Berkley psychology professor Christina Maslach, PhD. It quantifies three different factors for burnout: emotional exhaustion; depersonalization or cynicism; and a low sense of personal accomplishment. This study defined “burnout” as having feelings of both emotional exhaustion and depersonalization, and dismissed the third measure of burnout because physicians rarely posses a low sense of personal accomplishment.
Overall, 22.3% reported one sign or symptom of burnout daily, and 56.5% did so on a weekly basis, Dr. Hewitt said.
However, Dr. Hewitt noted, burnout measurement thresholds can vary “because burnout itself is not an actual diagnosis.” Studies have calculated the burnout rate among surgeons at 28% to 69% (J Am Coll Surg. 2016:222:1230-9). A recent systematic review found up to eight different cutoffs used to define “high burnout” (Cogent Med. 2016 7 Oct; doi.org/10.1080/2331205X.2016.1237605; J Am Coll Surg, 2016:223:440-51)
How studies of physician burnout establish cutoffs and which Maslach Burnout Inventory subscales they measure has an impact on the rates of burnout they report, Dr. Hewitt said. For this study, the researchers used the Maslach scores in the top quartile in both emotional exhaustion and depersonalization to define burnout. “Burnout is probably best defined as a continuum from engagement on the one end and burnout on the other, so there’s some combination of the [Maslach] subscales that lead to burnout,” he said.
Among survey respondents, 19.5% reported high Maslach scores for emotional exhaustion and 9% of depersonalization on a daily basis (6.2% reported both)--22.3% reported at least one daily. On a weekly basis, 54.2% reported high scores for emotional exhaustion and 25.6% high scores for depersonalization, with 56.5% reporting at least one and 22.4% reporting both.
To evaluate residents’ sense of psychiatric well-being, the study used the General Health Questionnaire (Psychol Med. 1998;28:915-21). Among survey respondents, 43% met criteria for poor psychiatric well-being.
Burnout rates among men and women were identical, but a significantly higher percentage of women had poor psychiatric well-being: 48.4% to 39.7% of men.
Burnout and psychiatric well-being scores also varied depending on post-graduate year. Second- and third-year residents were significantly more likely to report burnout, compared to first-year residents, Dr. Hewitt said. Rates of poor psychiatric well-being were lowest for fourth- and fifth-year residents.
Most pronounced was the impact of 80-hour duty week violations had on residents’ sense of burnout and poor psychiatric well-being. Burnout rates were 8.4% for those who reported no monthly duty-hour violations, but doubled and quadrupled with more frequent monthly duty-hour violations: 16.1% for those who reported one to four violations a month; and 35% for those who reported five or more. Dr. Hewitt also noted wellness rates for those in the standard and flexible policy groups in Flexibility In duty hour Requirements for Surgical Trainees Trial (FIRST Trial) programs were similar.
Another factor that contributed to burnout and poor psychiatric well-being was a sense of unpreparedness for residency, Dr. Hewitt said. Burnout rates for those who felt prepared were 8.9% vs. 19.1% for those who didn’t feel prepared. The disparity was less drastic, but nonetheless significant, for poor psychiatric well-being: 37.5% for those who felt prepared and 52.1% for those who didn’t.
With regard to outcomes, Dr. Hewitt said, “Residents in the highest quartile of burnout and the highest quartile of poor psychiatric well-being were significantly more likely to report a near miss or harmful medical error.” Among the burnout group, highest quartile rates were 39.3% for a near miss and 14.4% for a harmful medical error vs. 11.1% and 2.4%, respectively, for the lowest quartile. Among surgical residents who reported poor psychiatric well-being, highest quartile rates were 31.9% for a near miss and 13.2% for a harmful medical error vs. 12.5% and 2.1%, respectively, for those in the lowest quartile.
The study also analyzed outcomes for 134,877 surgical patients and found no association of overall morbidity and death or serious morbidity with resident wellness. “However,” Dr. Hewitt said, “when we look at mortality and failure to rescue, we can see an association with burnout, specifically in programs that have high levels of burnout; these patients have significantly lower odds of mortality and failure to rescue.” Each has an adjusted odds ratios of 0.81.
Among the study limitations Dr. Hewitt noted were its cross-sectional nature that led to inferences of association, not identification of causation; residents completing the survey after the ABSITE may have influenced their answers; and the fact it did not account for certain intermediate factors such as physician or nursing burnout or institutional quality measures.
The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality provided grants to support the research.
SOURCE: Hewitt B et al. abstract 21.01
JACKSONVILLE, FLA. – Burnout is commonly ascribed to surgical residents, but reliable estimates of the numbers involved and a clear, comparable definition of burnout remain elusive.
A large study of general surgery residents has found that almost one in four have at least one symptom of burnout daily and more than two in five report poor psychiatric well-being, according to results of a survey reported at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
“Twenty-two percent of general surgery residents report at least one symptom of burnout daily,” said Daniel “Brock” Hewitt, MD, a research fellow in the Surgical Outcomes and Quality Improvement Center in the department of surgery, Feinberg School of Medicine, Northwestern University, Chicago. “Poor resident wellness is associated with more duty-hour violations, feeling unprepared for residency, and an increase in self-reported medical errors. However, burnout is not associated with worse surgical outcomes.”
The study, undertaken with grant support from The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality, provided three significant insights into resident wellness, Dr. Hewitt said. “When measuring the level of burnout, careful consideration should be given to how burnout is defined,” he said. “Secondly, wellness interventions with attention to preparedness for residency and duty-hour restrictions may help alleviate burnout. And finally, with the significant impact it does have on the individual physician, we believe that burnout needs to be addressed regardless of its impact on patient outcomes.”
The study drew on a questionnaire given to residents immediately following the 2017 American Board of Surgery In-Training Examination (ABSITE), which 3,789 general surgery residents from 115 general surgery programs completed. The survey had a 99.3% response rate. The survey evaluated two factors associated with resident wellness: burnout and poor psychiatric well-being. “Poor wellness is prevalent among physicians and trainees and is associated with depression, suicidal ideation, attrition, and absenteeism,” Dr. Hewitt said.
But to measure burnout, the researchers had to first define it. The instrument Dr. Hewitt and coauthors used is the Maslach Burnout Inventory, named for University of California at Berkley psychology professor Christina Maslach, PhD. It quantifies three different factors for burnout: emotional exhaustion; depersonalization or cynicism; and a low sense of personal accomplishment. This study defined “burnout” as having feelings of both emotional exhaustion and depersonalization, and dismissed the third measure of burnout because physicians rarely posses a low sense of personal accomplishment.
Overall, 22.3% reported one sign or symptom of burnout daily, and 56.5% did so on a weekly basis, Dr. Hewitt said.
However, Dr. Hewitt noted, burnout measurement thresholds can vary “because burnout itself is not an actual diagnosis.” Studies have calculated the burnout rate among surgeons at 28% to 69% (J Am Coll Surg. 2016:222:1230-9). A recent systematic review found up to eight different cutoffs used to define “high burnout” (Cogent Med. 2016 7 Oct; doi.org/10.1080/2331205X.2016.1237605; J Am Coll Surg, 2016:223:440-51)
How studies of physician burnout establish cutoffs and which Maslach Burnout Inventory subscales they measure has an impact on the rates of burnout they report, Dr. Hewitt said. For this study, the researchers used the Maslach scores in the top quartile in both emotional exhaustion and depersonalization to define burnout. “Burnout is probably best defined as a continuum from engagement on the one end and burnout on the other, so there’s some combination of the [Maslach] subscales that lead to burnout,” he said.
Among survey respondents, 19.5% reported high Maslach scores for emotional exhaustion and 9% of depersonalization on a daily basis (6.2% reported both)--22.3% reported at least one daily. On a weekly basis, 54.2% reported high scores for emotional exhaustion and 25.6% high scores for depersonalization, with 56.5% reporting at least one and 22.4% reporting both.
To evaluate residents’ sense of psychiatric well-being, the study used the General Health Questionnaire (Psychol Med. 1998;28:915-21). Among survey respondents, 43% met criteria for poor psychiatric well-being.
Burnout rates among men and women were identical, but a significantly higher percentage of women had poor psychiatric well-being: 48.4% to 39.7% of men.
Burnout and psychiatric well-being scores also varied depending on post-graduate year. Second- and third-year residents were significantly more likely to report burnout, compared to first-year residents, Dr. Hewitt said. Rates of poor psychiatric well-being were lowest for fourth- and fifth-year residents.
Most pronounced was the impact of 80-hour duty week violations had on residents’ sense of burnout and poor psychiatric well-being. Burnout rates were 8.4% for those who reported no monthly duty-hour violations, but doubled and quadrupled with more frequent monthly duty-hour violations: 16.1% for those who reported one to four violations a month; and 35% for those who reported five or more. Dr. Hewitt also noted wellness rates for those in the standard and flexible policy groups in Flexibility In duty hour Requirements for Surgical Trainees Trial (FIRST Trial) programs were similar.
Another factor that contributed to burnout and poor psychiatric well-being was a sense of unpreparedness for residency, Dr. Hewitt said. Burnout rates for those who felt prepared were 8.9% vs. 19.1% for those who didn’t feel prepared. The disparity was less drastic, but nonetheless significant, for poor psychiatric well-being: 37.5% for those who felt prepared and 52.1% for those who didn’t.
With regard to outcomes, Dr. Hewitt said, “Residents in the highest quartile of burnout and the highest quartile of poor psychiatric well-being were significantly more likely to report a near miss or harmful medical error.” Among the burnout group, highest quartile rates were 39.3% for a near miss and 14.4% for a harmful medical error vs. 11.1% and 2.4%, respectively, for the lowest quartile. Among surgical residents who reported poor psychiatric well-being, highest quartile rates were 31.9% for a near miss and 13.2% for a harmful medical error vs. 12.5% and 2.1%, respectively, for those in the lowest quartile.
The study also analyzed outcomes for 134,877 surgical patients and found no association of overall morbidity and death or serious morbidity with resident wellness. “However,” Dr. Hewitt said, “when we look at mortality and failure to rescue, we can see an association with burnout, specifically in programs that have high levels of burnout; these patients have significantly lower odds of mortality and failure to rescue.” Each has an adjusted odds ratios of 0.81.
Among the study limitations Dr. Hewitt noted were its cross-sectional nature that led to inferences of association, not identification of causation; residents completing the survey after the ABSITE may have influenced their answers; and the fact it did not account for certain intermediate factors such as physician or nursing burnout or institutional quality measures.
The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality provided grants to support the research.
SOURCE: Hewitt B et al. abstract 21.01
AT THE ANNUAL ACADEMIC SURGICAL CONGRESS
Key clinical point: Almost one in four surgical residents manifest a daily symptom of burnout.
Major finding: Overall 22.8% of daily and 44.3% had poor psychiatric well-being scores.
Study details: Surveys completed by 3,789 general surgery residents during the American Board of Surgery In-Training Examination in January 2017.
Disclosures: The American Board of Surgery, Accreditation of Graduate Medical Education, American College of Surgeons and Agency for Healthcare Research and Quality provided grants to support the research.
Source: Hewitt B et al. abstract 21.01
Experts review the year in rheumatology ... and what lies ahead
MAUI, HAWAII – Arthur Kavanaugh, MD, program director for the Rheumatology Winter Clinical Symposium, likes to close out the meeting each year in high style by assembling selected conference faculty to offer their personal picks for the top developments in the field during the past year and make predictions about the year to come.
Here’s how they called it:
The top events in rheumatology during the last year
The rise of oral small molecules: The Janus kinase (JAK) inhibitors and other oral small molecules that have begun reaching the marketplace, with many more in development, will bring a paradigm shift in the treatment not only of rheumatic diseases, but in inflammatory bowel disease and skin diseases as well, predicted Alvin F. Wells, MD, PhD, a rheumatologist at Duke University in Durham, N.C., who is also director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
“The challenge is whether Medicare will cover the pills the way they cover the infusions and the other things we do,” according to Dr. Wells.

A bevy of new drugs for psoriatic arthritis and psoriasis: “I think the most important advance in the past year was the approval of a profusion of drugs for psoriatic arthritis and psoriasis. It’s really opened up the landscape for us in terms of treatment options. The downside is it’s going to take us a while to sort through which drugs fit where,” noted Eric J. Ruderman, MD, professor of medicine and associate chief of clinical affairs in the division of rheumatology at Northwestern University in Chicago.
“The drug I was most impressed with was tofacitinib [Xeljanz, an oral JAK inhibitor], not just by its effectiveness but by its potential to change the game, and particularly the data in tumor necrosis factor inhibitor inadequate responders. That was pretty solid data. It really opens the way to oral small molecules for joint diseases,” he added.
Interleukin-18 binding protein for monogenic inflammasome diseases: The biggest recent breakthrough in pediatric rheumatology was the Food and Drug Administration’s April 2017 designation of Breakthrough Therapy status for the recombinant human IL-18 binding protein known as tadekinig alfa for monogenic IL-18-associated autoinflammatory conditions, as well as the biologic’s Orphan Drug Designation for treatment of hemophagocytic lymphohistiocytosis, according to Anne M. Stevens, MD, PhD, professor of pediatrics at the University of Washington, Seattle, and chief of pediatric rheumatology at Seattle Children’s Hospital.

Novel treatment concept emerges in severe SLE: The study that knocked the socks off of Martin J. Bergman, MD, in 2017 was the Dutch SymBiose study, presented at both the European League Against Rheumatism and American College of Rheumatology annual meetings. It included just 14 patients with severe refractory SLE – including 10 with lupus nephritis – and tested a treatment strategy of rituximab (Rituxan) followed a few weeks later by a course of belimumab (Benlysta).
“The results were very dramatic, to say the least,” said Dr. Bergman of Drexel University in Philadelphia. Indeed, this one-two therapeutic punch resulted in sharply reduced levels of pathogenic autoantibodies and immune complex-mediated neutrophil extracellular traps while also knocking down very high baseline Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores to near zero, even while enabling patients to discontinue systemic corticosteroids and mycophenolate mofetil (CellCept). Several much larger clinical trials of this regimen and other similar ones are ongoing in an effort to duplicate the results.
Dr. Kavanaugh said the SymBiose study was one of his own top picks for study of the year as well.

Mainstream use of dupilumab (Dupixent) for moderate to severe atopic dermatitis: “This is a total game changer. It’s really changed a lot of people’s lives,” commented George M. Martin, MD, a dermatologist in private practice on Maui.
“Interestingly, historically drugs that started out in your realm later made their way to dermatology, but now we’re seeing the IL-23 inhibitors starting with us and then making their way into rheumatology and gastroenterology. The IL-23 inhibitors are very powerful drugs; when we’re seeing half of our psoriasis patients achieve PASI 100 responses, it’s very exciting. And these are durable responses,” he noted.
The opioid crisis: What’s the most important recent event in rheumatology?
“That’s easy: The biggest thing in all of medicine is the opioid crisis. Whether you recognize it or not, it’s gigantic. It’s $500 billion of the U.S. economy, every year. Forty percent of rheumatoid arthritis patients and 30% with ankylosing spondylitis are on opioids, and what goes along with that is a lot of ugly stuff,” said John J. Cush, MD, professor of medicine and rheumatology at Baylor University in Dallas.

Moreover, the FDA is now so leery of opioids that the agency has set the bar unrealistically high for approval of newer agents offering reduced abuse potential.
“I’ve been involved with or watched at least six FDA advisory panels looking at new, lower abuse-potential opioids in the last couple years. Only one agent got through,” according to Dr. Cush.
Dr. Troum commented, “I really think this whole opioid epidemic started with the campaign to make pain the fifth vital sign back in the 1990s. Some of the pharmaceutical companies took that concept and really ran with it.”
Rapamycin for inclusion body myositis: Dr. Kavanaugh’s pick for study of the year was what he described as “a brilliant presentation” of a French multicenter, placebo-controlled clinical trial of rapamycin for patients with inclusion body myositis at the 2017 ACR annual meeting.
“The French group considers IBM [inclusion body myositis] to be essentially Alzheimer’s disease of the muscle, marked by amyloid deposition. They chose to study rapamycin, which not only has immunosuppressive properties because it binds to mTOR [the mammalian target of rapamycin], but it also has the ability to inhibit amyloid protein deposition,” he explained.
The investigators reported improved 6-minute-walk distance and pulmonary function in the rapamycin group, whereas placebo-treated controls rapidly deteriorated.
“This is an approved drug for other indications, and we scratch our heads with IBM. It’s super nice to have something like that,” Dr. Kavanaugh observed.
A look at what’s in store
More tele-rheumatology: “I think the biggest thing is going to be more tele-rheumatology, more tele-ultrasound. Kaiser Permanente said 49% of their visits last year were virtual visits; that number is just going to grow,” predicted Dr. Wells.
Especially in medically underserved areas of the country, including large rural expanses, demand for remote tele-rheumatologic consults with high-quality imaging is going to increase, he added.
Here come cannabinoids for pain control: Dr. Troum predicted that in the depths of the national opioid epidemic, in a climate that discourages legitimate prescribing of traditional pain medications, rheumatologists can anticipate growing patient demand for cannabidiol and other cannabinoids for pain relief.
“I have patients coming in their 60s, 70s, and 80s – these are not young people – who are whispering to me, ‘Can I use this for my chronic pain?’ I think there’s going to be a big push for ways other than opioids to treat our patients’ pain,” according to Dr. Troum.
Tipping point nears for JAK inhibitors: In 2018, it will become clear just how seriously the Food and Drug Administration views the signal of possible increased venous thromboembolic risk associated with the oral JAK inhibitors for rheumatoid arthritis. The agency is expected to rule on Eli Lilly and Incyte’s resubmitted application for marketing approval for the JAK inhibitor baricitinib, which was tripped up earlier based in part upon VTE concerns.
“I think the big story in 2018 will be how the JAK story shakes out – whether this VTE thing has legs,” Dr. Ruderman predicted. “A sea change could be coming in our field, and it’s not coming next year or the year after, but 10 years from now: Are we going to move past the era of methotrexate and use generic small molecules instead? We’re going to find out within the next year whether that’s going to happen.”
Phase 3 results coming on tocilizumab for systemic sclerosis: “I think we’re going to see some really exciting systemic sclerosis data coming out this year,” Dr. Stevens said. Based upon the positive phase 2 results presented for tocilizumab (Actemra) last year, she’s optimistic that the ongoing phase 3 randomized trial will demonstrate a significant advantage over placebo in lung function. Also, ongoing separate clinical trials are evaluating an antifibrotic drug and rapamycin for systemic sclerosis.
Dr. Bergman, too, has high hopes for these studies: “I think we may finally be getting to a place where we can see effective drugs in systemic sclerosis.”
Amazon, Berkshire Hathaway, and JPMorgan Chase form a nonprofit to improve employee health care: In a recent press conference, the three CEOs weren’t specific about their plans, but Dr. Martin predicted the companies are likely to self-insure, bypassing Cigna and the other major health insurance companies and then contracting with physicians. He forecast that “probably within the next 5 years, what they do is going to affect everybody in this room.”
Rheumatologists will need to master a new mindset: Many rheumatologists have gotten comfortable with an all-tumor necrosis factor inhibitor treatment menu for their patients with moderate or severe rheumatoid arthritis. That’s got to change, according to Dr. Cush.
“We now have two IL-6 inhibitors, two IL-17 inhibitors, and we’ll soon have two JAK inhibitors. That’s going to be a direct threat to the not right- or left-brain, but the TNF-brain rheumatologist who now writes prescriptions for three TNF inhibitors in a row before questioning the efficacy. The idea is you will now be using drugs with other mechanisms of action first-line, or at the very least, second-line, and that’s going to be a paradigm shift for a lot of people,” he explained.
None of the speakers reported having financial conflicts regarding their comments.
MAUI, HAWAII – Arthur Kavanaugh, MD, program director for the Rheumatology Winter Clinical Symposium, likes to close out the meeting each year in high style by assembling selected conference faculty to offer their personal picks for the top developments in the field during the past year and make predictions about the year to come.
Here’s how they called it:
The top events in rheumatology during the last year
The rise of oral small molecules: The Janus kinase (JAK) inhibitors and other oral small molecules that have begun reaching the marketplace, with many more in development, will bring a paradigm shift in the treatment not only of rheumatic diseases, but in inflammatory bowel disease and skin diseases as well, predicted Alvin F. Wells, MD, PhD, a rheumatologist at Duke University in Durham, N.C., who is also director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
“The challenge is whether Medicare will cover the pills the way they cover the infusions and the other things we do,” according to Dr. Wells.
A bevy of new drugs for psoriatic arthritis and psoriasis: “I think the most important advance in the past year was the approval of a profusion of drugs for psoriatic arthritis and psoriasis. It’s really opened up the landscape for us in terms of treatment options. The downside is it’s going to take us a while to sort through which drugs fit where,” noted Eric J. Ruderman, MD, professor of medicine and associate chief of clinical affairs in the division of rheumatology at Northwestern University in Chicago.
“The drug I was most impressed with was tofacitinib [Xeljanz, an oral JAK inhibitor], not just by its effectiveness but by its potential to change the game, and particularly the data in tumor necrosis factor inhibitor inadequate responders. That was pretty solid data. It really opens the way to oral small molecules for joint diseases,” he added.
Interleukin-18 binding protein for monogenic inflammasome diseases: The biggest recent breakthrough in pediatric rheumatology was the Food and Drug Administration’s April 2017 designation of Breakthrough Therapy status for the recombinant human IL-18 binding protein known as tadekinig alfa for monogenic IL-18-associated autoinflammatory conditions, as well as the biologic’s Orphan Drug Designation for treatment of hemophagocytic lymphohistiocytosis, according to Anne M. Stevens, MD, PhD, professor of pediatrics at the University of Washington, Seattle, and chief of pediatric rheumatology at Seattle Children’s Hospital.
Novel treatment concept emerges in severe SLE: The study that knocked the socks off of Martin J. Bergman, MD, in 2017 was the Dutch SymBiose study, presented at both the European League Against Rheumatism and American College of Rheumatology annual meetings. It included just 14 patients with severe refractory SLE – including 10 with lupus nephritis – and tested a treatment strategy of rituximab (Rituxan) followed a few weeks later by a course of belimumab (Benlysta).
“The results were very dramatic, to say the least,” said Dr. Bergman of Drexel University in Philadelphia. Indeed, this one-two therapeutic punch resulted in sharply reduced levels of pathogenic autoantibodies and immune complex-mediated neutrophil extracellular traps while also knocking down very high baseline Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores to near zero, even while enabling patients to discontinue systemic corticosteroids and mycophenolate mofetil (CellCept). Several much larger clinical trials of this regimen and other similar ones are ongoing in an effort to duplicate the results.
Dr. Kavanaugh said the SymBiose study was one of his own top picks for study of the year as well.
Mainstream use of dupilumab (Dupixent) for moderate to severe atopic dermatitis: “This is a total game changer. It’s really changed a lot of people’s lives,” commented George M. Martin, MD, a dermatologist in private practice on Maui.
“Interestingly, historically drugs that started out in your realm later made their way to dermatology, but now we’re seeing the IL-23 inhibitors starting with us and then making their way into rheumatology and gastroenterology. The IL-23 inhibitors are very powerful drugs; when we’re seeing half of our psoriasis patients achieve PASI 100 responses, it’s very exciting. And these are durable responses,” he noted.
The opioid crisis: What’s the most important recent event in rheumatology?
“That’s easy: The biggest thing in all of medicine is the opioid crisis. Whether you recognize it or not, it’s gigantic. It’s $500 billion of the U.S. economy, every year. Forty percent of rheumatoid arthritis patients and 30% with ankylosing spondylitis are on opioids, and what goes along with that is a lot of ugly stuff,” said John J. Cush, MD, professor of medicine and rheumatology at Baylor University in Dallas.
Moreover, the FDA is now so leery of opioids that the agency has set the bar unrealistically high for approval of newer agents offering reduced abuse potential.
“I’ve been involved with or watched at least six FDA advisory panels looking at new, lower abuse-potential opioids in the last couple years. Only one agent got through,” according to Dr. Cush.
Dr. Troum commented, “I really think this whole opioid epidemic started with the campaign to make pain the fifth vital sign back in the 1990s. Some of the pharmaceutical companies took that concept and really ran with it.”
Rapamycin for inclusion body myositis: Dr. Kavanaugh’s pick for study of the year was what he described as “a brilliant presentation” of a French multicenter, placebo-controlled clinical trial of rapamycin for patients with inclusion body myositis at the 2017 ACR annual meeting.
“The French group considers IBM [inclusion body myositis] to be essentially Alzheimer’s disease of the muscle, marked by amyloid deposition. They chose to study rapamycin, which not only has immunosuppressive properties because it binds to mTOR [the mammalian target of rapamycin], but it also has the ability to inhibit amyloid protein deposition,” he explained.
The investigators reported improved 6-minute-walk distance and pulmonary function in the rapamycin group, whereas placebo-treated controls rapidly deteriorated.
“This is an approved drug for other indications, and we scratch our heads with IBM. It’s super nice to have something like that,” Dr. Kavanaugh observed.
A look at what’s in store
More tele-rheumatology: “I think the biggest thing is going to be more tele-rheumatology, more tele-ultrasound. Kaiser Permanente said 49% of their visits last year were virtual visits; that number is just going to grow,” predicted Dr. Wells.
Especially in medically underserved areas of the country, including large rural expanses, demand for remote tele-rheumatologic consults with high-quality imaging is going to increase, he added.
Here come cannabinoids for pain control: Dr. Troum predicted that in the depths of the national opioid epidemic, in a climate that discourages legitimate prescribing of traditional pain medications, rheumatologists can anticipate growing patient demand for cannabidiol and other cannabinoids for pain relief.
“I have patients coming in their 60s, 70s, and 80s – these are not young people – who are whispering to me, ‘Can I use this for my chronic pain?’ I think there’s going to be a big push for ways other than opioids to treat our patients’ pain,” according to Dr. Troum.
Tipping point nears for JAK inhibitors: In 2018, it will become clear just how seriously the Food and Drug Administration views the signal of possible increased venous thromboembolic risk associated with the oral JAK inhibitors for rheumatoid arthritis. The agency is expected to rule on Eli Lilly and Incyte’s resubmitted application for marketing approval for the JAK inhibitor baricitinib, which was tripped up earlier based in part upon VTE concerns.
“I think the big story in 2018 will be how the JAK story shakes out – whether this VTE thing has legs,” Dr. Ruderman predicted. “A sea change could be coming in our field, and it’s not coming next year or the year after, but 10 years from now: Are we going to move past the era of methotrexate and use generic small molecules instead? We’re going to find out within the next year whether that’s going to happen.”
Phase 3 results coming on tocilizumab for systemic sclerosis: “I think we’re going to see some really exciting systemic sclerosis data coming out this year,” Dr. Stevens said. Based upon the positive phase 2 results presented for tocilizumab (Actemra) last year, she’s optimistic that the ongoing phase 3 randomized trial will demonstrate a significant advantage over placebo in lung function. Also, ongoing separate clinical trials are evaluating an antifibrotic drug and rapamycin for systemic sclerosis.
Dr. Bergman, too, has high hopes for these studies: “I think we may finally be getting to a place where we can see effective drugs in systemic sclerosis.”
Amazon, Berkshire Hathaway, and JPMorgan Chase form a nonprofit to improve employee health care: In a recent press conference, the three CEOs weren’t specific about their plans, but Dr. Martin predicted the companies are likely to self-insure, bypassing Cigna and the other major health insurance companies and then contracting with physicians. He forecast that “probably within the next 5 years, what they do is going to affect everybody in this room.”
Rheumatologists will need to master a new mindset: Many rheumatologists have gotten comfortable with an all-tumor necrosis factor inhibitor treatment menu for their patients with moderate or severe rheumatoid arthritis. That’s got to change, according to Dr. Cush.
“We now have two IL-6 inhibitors, two IL-17 inhibitors, and we’ll soon have two JAK inhibitors. That’s going to be a direct threat to the not right- or left-brain, but the TNF-brain rheumatologist who now writes prescriptions for three TNF inhibitors in a row before questioning the efficacy. The idea is you will now be using drugs with other mechanisms of action first-line, or at the very least, second-line, and that’s going to be a paradigm shift for a lot of people,” he explained.
None of the speakers reported having financial conflicts regarding their comments.
MAUI, HAWAII – Arthur Kavanaugh, MD, program director for the Rheumatology Winter Clinical Symposium, likes to close out the meeting each year in high style by assembling selected conference faculty to offer their personal picks for the top developments in the field during the past year and make predictions about the year to come.
Here’s how they called it:
The top events in rheumatology during the last year
The rise of oral small molecules: The Janus kinase (JAK) inhibitors and other oral small molecules that have begun reaching the marketplace, with many more in development, will bring a paradigm shift in the treatment not only of rheumatic diseases, but in inflammatory bowel disease and skin diseases as well, predicted Alvin F. Wells, MD, PhD, a rheumatologist at Duke University in Durham, N.C., who is also director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
“The challenge is whether Medicare will cover the pills the way they cover the infusions and the other things we do,” according to Dr. Wells.
A bevy of new drugs for psoriatic arthritis and psoriasis: “I think the most important advance in the past year was the approval of a profusion of drugs for psoriatic arthritis and psoriasis. It’s really opened up the landscape for us in terms of treatment options. The downside is it’s going to take us a while to sort through which drugs fit where,” noted Eric J. Ruderman, MD, professor of medicine and associate chief of clinical affairs in the division of rheumatology at Northwestern University in Chicago.
“The drug I was most impressed with was tofacitinib [Xeljanz, an oral JAK inhibitor], not just by its effectiveness but by its potential to change the game, and particularly the data in tumor necrosis factor inhibitor inadequate responders. That was pretty solid data. It really opens the way to oral small molecules for joint diseases,” he added.
Interleukin-18 binding protein for monogenic inflammasome diseases: The biggest recent breakthrough in pediatric rheumatology was the Food and Drug Administration’s April 2017 designation of Breakthrough Therapy status for the recombinant human IL-18 binding protein known as tadekinig alfa for monogenic IL-18-associated autoinflammatory conditions, as well as the biologic’s Orphan Drug Designation for treatment of hemophagocytic lymphohistiocytosis, according to Anne M. Stevens, MD, PhD, professor of pediatrics at the University of Washington, Seattle, and chief of pediatric rheumatology at Seattle Children’s Hospital.
Novel treatment concept emerges in severe SLE: The study that knocked the socks off of Martin J. Bergman, MD, in 2017 was the Dutch SymBiose study, presented at both the European League Against Rheumatism and American College of Rheumatology annual meetings. It included just 14 patients with severe refractory SLE – including 10 with lupus nephritis – and tested a treatment strategy of rituximab (Rituxan) followed a few weeks later by a course of belimumab (Benlysta).
“The results were very dramatic, to say the least,” said Dr. Bergman of Drexel University in Philadelphia. Indeed, this one-two therapeutic punch resulted in sharply reduced levels of pathogenic autoantibodies and immune complex-mediated neutrophil extracellular traps while also knocking down very high baseline Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores to near zero, even while enabling patients to discontinue systemic corticosteroids and mycophenolate mofetil (CellCept). Several much larger clinical trials of this regimen and other similar ones are ongoing in an effort to duplicate the results.
Dr. Kavanaugh said the SymBiose study was one of his own top picks for study of the year as well.
Mainstream use of dupilumab (Dupixent) for moderate to severe atopic dermatitis: “This is a total game changer. It’s really changed a lot of people’s lives,” commented George M. Martin, MD, a dermatologist in private practice on Maui.
“Interestingly, historically drugs that started out in your realm later made their way to dermatology, but now we’re seeing the IL-23 inhibitors starting with us and then making their way into rheumatology and gastroenterology. The IL-23 inhibitors are very powerful drugs; when we’re seeing half of our psoriasis patients achieve PASI 100 responses, it’s very exciting. And these are durable responses,” he noted.
The opioid crisis: What’s the most important recent event in rheumatology?
“That’s easy: The biggest thing in all of medicine is the opioid crisis. Whether you recognize it or not, it’s gigantic. It’s $500 billion of the U.S. economy, every year. Forty percent of rheumatoid arthritis patients and 30% with ankylosing spondylitis are on opioids, and what goes along with that is a lot of ugly stuff,” said John J. Cush, MD, professor of medicine and rheumatology at Baylor University in Dallas.
Moreover, the FDA is now so leery of opioids that the agency has set the bar unrealistically high for approval of newer agents offering reduced abuse potential.
“I’ve been involved with or watched at least six FDA advisory panels looking at new, lower abuse-potential opioids in the last couple years. Only one agent got through,” according to Dr. Cush.
Dr. Troum commented, “I really think this whole opioid epidemic started with the campaign to make pain the fifth vital sign back in the 1990s. Some of the pharmaceutical companies took that concept and really ran with it.”
Rapamycin for inclusion body myositis: Dr. Kavanaugh’s pick for study of the year was what he described as “a brilliant presentation” of a French multicenter, placebo-controlled clinical trial of rapamycin for patients with inclusion body myositis at the 2017 ACR annual meeting.
“The French group considers IBM [inclusion body myositis] to be essentially Alzheimer’s disease of the muscle, marked by amyloid deposition. They chose to study rapamycin, which not only has immunosuppressive properties because it binds to mTOR [the mammalian target of rapamycin], but it also has the ability to inhibit amyloid protein deposition,” he explained.
The investigators reported improved 6-minute-walk distance and pulmonary function in the rapamycin group, whereas placebo-treated controls rapidly deteriorated.
“This is an approved drug for other indications, and we scratch our heads with IBM. It’s super nice to have something like that,” Dr. Kavanaugh observed.
A look at what’s in store
More tele-rheumatology: “I think the biggest thing is going to be more tele-rheumatology, more tele-ultrasound. Kaiser Permanente said 49% of their visits last year were virtual visits; that number is just going to grow,” predicted Dr. Wells.
Especially in medically underserved areas of the country, including large rural expanses, demand for remote tele-rheumatologic consults with high-quality imaging is going to increase, he added.
Here come cannabinoids for pain control: Dr. Troum predicted that in the depths of the national opioid epidemic, in a climate that discourages legitimate prescribing of traditional pain medications, rheumatologists can anticipate growing patient demand for cannabidiol and other cannabinoids for pain relief.
“I have patients coming in their 60s, 70s, and 80s – these are not young people – who are whispering to me, ‘Can I use this for my chronic pain?’ I think there’s going to be a big push for ways other than opioids to treat our patients’ pain,” according to Dr. Troum.
Tipping point nears for JAK inhibitors: In 2018, it will become clear just how seriously the Food and Drug Administration views the signal of possible increased venous thromboembolic risk associated with the oral JAK inhibitors for rheumatoid arthritis. The agency is expected to rule on Eli Lilly and Incyte’s resubmitted application for marketing approval for the JAK inhibitor baricitinib, which was tripped up earlier based in part upon VTE concerns.
“I think the big story in 2018 will be how the JAK story shakes out – whether this VTE thing has legs,” Dr. Ruderman predicted. “A sea change could be coming in our field, and it’s not coming next year or the year after, but 10 years from now: Are we going to move past the era of methotrexate and use generic small molecules instead? We’re going to find out within the next year whether that’s going to happen.”
Phase 3 results coming on tocilizumab for systemic sclerosis: “I think we’re going to see some really exciting systemic sclerosis data coming out this year,” Dr. Stevens said. Based upon the positive phase 2 results presented for tocilizumab (Actemra) last year, she’s optimistic that the ongoing phase 3 randomized trial will demonstrate a significant advantage over placebo in lung function. Also, ongoing separate clinical trials are evaluating an antifibrotic drug and rapamycin for systemic sclerosis.
Dr. Bergman, too, has high hopes for these studies: “I think we may finally be getting to a place where we can see effective drugs in systemic sclerosis.”
Amazon, Berkshire Hathaway, and JPMorgan Chase form a nonprofit to improve employee health care: In a recent press conference, the three CEOs weren’t specific about their plans, but Dr. Martin predicted the companies are likely to self-insure, bypassing Cigna and the other major health insurance companies and then contracting with physicians. He forecast that “probably within the next 5 years, what they do is going to affect everybody in this room.”
Rheumatologists will need to master a new mindset: Many rheumatologists have gotten comfortable with an all-tumor necrosis factor inhibitor treatment menu for their patients with moderate or severe rheumatoid arthritis. That’s got to change, according to Dr. Cush.
“We now have two IL-6 inhibitors, two IL-17 inhibitors, and we’ll soon have two JAK inhibitors. That’s going to be a direct threat to the not right- or left-brain, but the TNF-brain rheumatologist who now writes prescriptions for three TNF inhibitors in a row before questioning the efficacy. The idea is you will now be using drugs with other mechanisms of action first-line, or at the very least, second-line, and that’s going to be a paradigm shift for a lot of people,” he explained.
None of the speakers reported having financial conflicts regarding their comments.
EXPERT ANALYSIS FROM RWCS 2018

Supplemental oxygen use for suspected myocardial infarction without hypoxemia
Background: Clinical guidelines recommend supplemental oxygen in patients with suspected acute myocardial infarction but data to support its use in patients without hypoxemia are limited.
Study design: Open-label, registry based randomized clinical trial.
Setting: Thirty-five hospitals in Sweden with acute cardiac care facilities.
Synopsis: Authors included 6,629 patients aged 30 and older who presented with symptoms suggestive of myocardial infarction. Patients with oxygen saturations 90% or greater were enrolled in the study and randomly assigned to either 6 liters per minute of face mask oxygen for 6-12 hours or ambient air. Median oxygen saturation was 99% in the treatment group and 97% in the ambient air group (P less than .0001). In an intention-to-treat model, 1 year after randomization there was no significant difference in all-cause mortality between the oxygen (5.0%) and ambient air (5.1%) groups (P = .80). There was no difference in the rate of rehospitalization with myocardial infarction or the composite endpoint of all-cause mortality and rehospitalizations for myocardial infarction at 30 days and 1 year. Limitations of this study include lower power than anticipated since calculations were based on a higher mortality rate than observed in this study, and the open-label protocol.
Bottom line: In patients who present with a suspected myocardial infarction without hypoxemia, oxygen therapy is not associated with improved all-cause mortality or decreased rehospitalizations for myocardial infarction.
Citation: Hofmann R, Jernberg T, Erlinge D, et al. Oxygen therapy in suspected acute myocardial infarction. N Engl J Med. 2017;377:1240-9.

Dr. Gala is a hospitalist, Beth Israel Deaconess Medical Center, and instructor in medicine, Harvard Medical School, Boston.
Background: Clinical guidelines recommend supplemental oxygen in patients with suspected acute myocardial infarction but data to support its use in patients without hypoxemia are limited.
Study design: Open-label, registry based randomized clinical trial.
Setting: Thirty-five hospitals in Sweden with acute cardiac care facilities.
Synopsis: Authors included 6,629 patients aged 30 and older who presented with symptoms suggestive of myocardial infarction. Patients with oxygen saturations 90% or greater were enrolled in the study and randomly assigned to either 6 liters per minute of face mask oxygen for 6-12 hours or ambient air. Median oxygen saturation was 99% in the treatment group and 97% in the ambient air group (P less than .0001). In an intention-to-treat model, 1 year after randomization there was no significant difference in all-cause mortality between the oxygen (5.0%) and ambient air (5.1%) groups (P = .80). There was no difference in the rate of rehospitalization with myocardial infarction or the composite endpoint of all-cause mortality and rehospitalizations for myocardial infarction at 30 days and 1 year. Limitations of this study include lower power than anticipated since calculations were based on a higher mortality rate than observed in this study, and the open-label protocol.
Bottom line: In patients who present with a suspected myocardial infarction without hypoxemia, oxygen therapy is not associated with improved all-cause mortality or decreased rehospitalizations for myocardial infarction.
Citation: Hofmann R, Jernberg T, Erlinge D, et al. Oxygen therapy in suspected acute myocardial infarction. N Engl J Med. 2017;377:1240-9.
Dr. Gala is a hospitalist, Beth Israel Deaconess Medical Center, and instructor in medicine, Harvard Medical School, Boston.
Background: Clinical guidelines recommend supplemental oxygen in patients with suspected acute myocardial infarction but data to support its use in patients without hypoxemia are limited.
Study design: Open-label, registry based randomized clinical trial.
Setting: Thirty-five hospitals in Sweden with acute cardiac care facilities.
Synopsis: Authors included 6,629 patients aged 30 and older who presented with symptoms suggestive of myocardial infarction. Patients with oxygen saturations 90% or greater were enrolled in the study and randomly assigned to either 6 liters per minute of face mask oxygen for 6-12 hours or ambient air. Median oxygen saturation was 99% in the treatment group and 97% in the ambient air group (P less than .0001). In an intention-to-treat model, 1 year after randomization there was no significant difference in all-cause mortality between the oxygen (5.0%) and ambient air (5.1%) groups (P = .80). There was no difference in the rate of rehospitalization with myocardial infarction or the composite endpoint of all-cause mortality and rehospitalizations for myocardial infarction at 30 days and 1 year. Limitations of this study include lower power than anticipated since calculations were based on a higher mortality rate than observed in this study, and the open-label protocol.
Bottom line: In patients who present with a suspected myocardial infarction without hypoxemia, oxygen therapy is not associated with improved all-cause mortality or decreased rehospitalizations for myocardial infarction.
Citation: Hofmann R, Jernberg T, Erlinge D, et al. Oxygen therapy in suspected acute myocardial infarction. N Engl J Med. 2017;377:1240-9.
Dr. Gala is a hospitalist, Beth Israel Deaconess Medical Center, and instructor in medicine, Harvard Medical School, Boston.