User login
HDAC inhibition may boost immune therapy efficacy in breast cancer
ORLANDO – The novel combination of entinostat and nivolumab with or without ipilimumab showed encouraging safety, tolerability, and antitumor activity in early results from an ongoing phase 1 trial of patients with advanced breast cancer.
Of 30 patients who were enrolled and treated in the dose-escalation phase of the study as of Feb. 24, 2018, 20 had evaluable responses, and of those, 3 had a partial response for an overall response rate of 15%. An additional 12 had stable disease, and 5 had disease progression, Roisin M. Connolly, MD, reported in a poster at the annual conference of the National Comprehensive Cancer Network.

Responses were seen in all 3 DL1 patients, 12 of 14 DL2 patients, 3 of 4 DL3 patients, and 2 of 9 (with 4 pending first restaging) DL4 patients. Dose-limiting toxicities included one case of pneumonitis at DL2 and an allergic reaction in one DL4 patient, said Dr. Connolly of Johns Hopkins University, Baltimore.
The most common treatment-associated adverse events occurring in 6 or more patients included anemia, fatigue, neutropenia, nausea, and rash, with each occurring in 12 to 22 patients, including grade 3 anemia in 7 patients, grade 3 fatigue in 4 patients, and grade 3 neutropenia in 5 patients. Grade 4 adverse events included lymphopenia in one patient and elevated lipase in one patient, she said.
Possible immune-related adverse events included hypothyroidism in 2 DL2 patients and 3 DL3 patients, hyperthyroidism in 1 DL3 patient, colitis in 1 DL2 and 1 DL3 patient, pneumonitis in 4 DL2 patients, rash in 10 DL2-DL4 patients, and meningoencephalitis and myasthenia gravis in 1 DL3 patient.
Study participants were adults with a mean age of 60 years with metastatic or unresectable solid tumors for which standard treatments did not exist or were no longer effective, or for which treatment with anti–programmed cell death ligand1/cytotoxic T-lymphocyte antigen 4 treatment was appropriate. All had good performance status and adequate organ and pulmonary function, less than 30% liver involvement, and any brain metastases were stable. Those with active autoimmune disease or a history of autoimmune disease that might recur were excluded, as were patients treated within 14 days of enrollment.
“The rationale for the study was based on preclinical work suggesting that epigenetic modifiers might be able to enhance the efficacy of immune therapies, and this would be particularly important for ‘colder’ tumor types like breast cancer that might not have the same sort of responses that we see in other tumor types,” Dr. Connolly explained in an interview. “The lab work suggested, for example, that the [histone deacetylase] inhibitor entinostat might affect myeloid-derived suppressor and regulatory T cells that might prevent cytotoxic T cells from fighting the cancer.”
There may be other mechanisms for this activity as well, she noted.
The run-in period with entinostat alone allowed collection of pre- and posttreatment biopsies to examine the effects on the tissues, such as whether treatment affects T cells, myeloid-derived suppressor cells, or their pathways, she said.
“We’re seeing [the] same types of toxicities seen with combination immune-oncology strategies, and we’re seeing some tumor responses that are of interest. Now we will delve into the tissue biopsies and blood samples we’ve collected to explore the mechanisms in more detail. In the near future we will open our breast cancer expansion cohort to look in more detail at what these drugs might be doing in breast cancer,” she added.
Specifically, she and her colleagues are evaluating the effects of treatment on immune-related biomarkers, measuring tumor-specific mutations and mutant neoantigens recognized by patient T cells in tumor biopsies, evaluating changes in the frequency of T cells recognizing tumor-specific mutant neoantigens in peripheral blood lymphocytes pre- and posttherapy, and looking at epigenetic changes pre- and posttherapy.
These preliminary findings suggest that the combination of entinostat and nivolumab with or without ipilimumab is safe and tolerable, with expected rates of immune-related adverse events, Dr. Connolly said, noting that the recommended phase 2 dose to be used in the dose expansion phase of the study has yet to be determined.
The findings, should they be confirmed as the trial progresses, could have important implications because immune checkpoint inhibitors, which work best in patients with immunogenic cancers that naturally attract T-cell infiltration into their tumor microenvironment, have limited single-agent activity in tumors, such as breast cancer, that are not believed to be immunogenic, she reported. Such cancers have thus far had only modest responses to single-agent immune checkpoint inhibition in advanced triple-negative and HER2+ breast cancer, with overall response rates of 5%-20%.
However, women who do respond to immune checkpoint inhibition tend to have durable and sustainable responses, she said, explaining that suboptimal immune responsiveness is likely a result of a lack of tumor antigen expression and/or recognition, as well as multiple suppressive signals in the tumor microenvironment.
Should the novel strategy tested in this study for converting breast cancers into immune responsive tumors facilitate improved response to immune checkpoint agents, it has the potential to significantly extend survival in breast cancer patients, she concluded.
This study was funded by grants from the National Institutes of Health, Bloomberg Kimmel Institute for Immunotherapy, NCCN, and the Mary Kay Foundation, as well as a V Foundation award. Dr. Connolly reported having no disclosures
SOURCE: Connolly RM et al. NCCN, Poster 3.
ORLANDO – The novel combination of entinostat and nivolumab with or without ipilimumab showed encouraging safety, tolerability, and antitumor activity in early results from an ongoing phase 1 trial of patients with advanced breast cancer.
Of 30 patients who were enrolled and treated in the dose-escalation phase of the study as of Feb. 24, 2018, 20 had evaluable responses, and of those, 3 had a partial response for an overall response rate of 15%. An additional 12 had stable disease, and 5 had disease progression, Roisin M. Connolly, MD, reported in a poster at the annual conference of the National Comprehensive Cancer Network.
Responses were seen in all 3 DL1 patients, 12 of 14 DL2 patients, 3 of 4 DL3 patients, and 2 of 9 (with 4 pending first restaging) DL4 patients. Dose-limiting toxicities included one case of pneumonitis at DL2 and an allergic reaction in one DL4 patient, said Dr. Connolly of Johns Hopkins University, Baltimore.
The most common treatment-associated adverse events occurring in 6 or more patients included anemia, fatigue, neutropenia, nausea, and rash, with each occurring in 12 to 22 patients, including grade 3 anemia in 7 patients, grade 3 fatigue in 4 patients, and grade 3 neutropenia in 5 patients. Grade 4 adverse events included lymphopenia in one patient and elevated lipase in one patient, she said.
Possible immune-related adverse events included hypothyroidism in 2 DL2 patients and 3 DL3 patients, hyperthyroidism in 1 DL3 patient, colitis in 1 DL2 and 1 DL3 patient, pneumonitis in 4 DL2 patients, rash in 10 DL2-DL4 patients, and meningoencephalitis and myasthenia gravis in 1 DL3 patient.
Study participants were adults with a mean age of 60 years with metastatic or unresectable solid tumors for which standard treatments did not exist or were no longer effective, or for which treatment with anti–programmed cell death ligand1/cytotoxic T-lymphocyte antigen 4 treatment was appropriate. All had good performance status and adequate organ and pulmonary function, less than 30% liver involvement, and any brain metastases were stable. Those with active autoimmune disease or a history of autoimmune disease that might recur were excluded, as were patients treated within 14 days of enrollment.
“The rationale for the study was based on preclinical work suggesting that epigenetic modifiers might be able to enhance the efficacy of immune therapies, and this would be particularly important for ‘colder’ tumor types like breast cancer that might not have the same sort of responses that we see in other tumor types,” Dr. Connolly explained in an interview. “The lab work suggested, for example, that the [histone deacetylase] inhibitor entinostat might affect myeloid-derived suppressor and regulatory T cells that might prevent cytotoxic T cells from fighting the cancer.”
There may be other mechanisms for this activity as well, she noted.
The run-in period with entinostat alone allowed collection of pre- and posttreatment biopsies to examine the effects on the tissues, such as whether treatment affects T cells, myeloid-derived suppressor cells, or their pathways, she said.
“We’re seeing [the] same types of toxicities seen with combination immune-oncology strategies, and we’re seeing some tumor responses that are of interest. Now we will delve into the tissue biopsies and blood samples we’ve collected to explore the mechanisms in more detail. In the near future we will open our breast cancer expansion cohort to look in more detail at what these drugs might be doing in breast cancer,” she added.
Specifically, she and her colleagues are evaluating the effects of treatment on immune-related biomarkers, measuring tumor-specific mutations and mutant neoantigens recognized by patient T cells in tumor biopsies, evaluating changes in the frequency of T cells recognizing tumor-specific mutant neoantigens in peripheral blood lymphocytes pre- and posttherapy, and looking at epigenetic changes pre- and posttherapy.
These preliminary findings suggest that the combination of entinostat and nivolumab with or without ipilimumab is safe and tolerable, with expected rates of immune-related adverse events, Dr. Connolly said, noting that the recommended phase 2 dose to be used in the dose expansion phase of the study has yet to be determined.
The findings, should they be confirmed as the trial progresses, could have important implications because immune checkpoint inhibitors, which work best in patients with immunogenic cancers that naturally attract T-cell infiltration into their tumor microenvironment, have limited single-agent activity in tumors, such as breast cancer, that are not believed to be immunogenic, she reported. Such cancers have thus far had only modest responses to single-agent immune checkpoint inhibition in advanced triple-negative and HER2+ breast cancer, with overall response rates of 5%-20%.
However, women who do respond to immune checkpoint inhibition tend to have durable and sustainable responses, she said, explaining that suboptimal immune responsiveness is likely a result of a lack of tumor antigen expression and/or recognition, as well as multiple suppressive signals in the tumor microenvironment.
Should the novel strategy tested in this study for converting breast cancers into immune responsive tumors facilitate improved response to immune checkpoint agents, it has the potential to significantly extend survival in breast cancer patients, she concluded.
This study was funded by grants from the National Institutes of Health, Bloomberg Kimmel Institute for Immunotherapy, NCCN, and the Mary Kay Foundation, as well as a V Foundation award. Dr. Connolly reported having no disclosures
SOURCE: Connolly RM et al. NCCN, Poster 3.
ORLANDO – The novel combination of entinostat and nivolumab with or without ipilimumab showed encouraging safety, tolerability, and antitumor activity in early results from an ongoing phase 1 trial of patients with advanced breast cancer.
Of 30 patients who were enrolled and treated in the dose-escalation phase of the study as of Feb. 24, 2018, 20 had evaluable responses, and of those, 3 had a partial response for an overall response rate of 15%. An additional 12 had stable disease, and 5 had disease progression, Roisin M. Connolly, MD, reported in a poster at the annual conference of the National Comprehensive Cancer Network.
Responses were seen in all 3 DL1 patients, 12 of 14 DL2 patients, 3 of 4 DL3 patients, and 2 of 9 (with 4 pending first restaging) DL4 patients. Dose-limiting toxicities included one case of pneumonitis at DL2 and an allergic reaction in one DL4 patient, said Dr. Connolly of Johns Hopkins University, Baltimore.
The most common treatment-associated adverse events occurring in 6 or more patients included anemia, fatigue, neutropenia, nausea, and rash, with each occurring in 12 to 22 patients, including grade 3 anemia in 7 patients, grade 3 fatigue in 4 patients, and grade 3 neutropenia in 5 patients. Grade 4 adverse events included lymphopenia in one patient and elevated lipase in one patient, she said.
Possible immune-related adverse events included hypothyroidism in 2 DL2 patients and 3 DL3 patients, hyperthyroidism in 1 DL3 patient, colitis in 1 DL2 and 1 DL3 patient, pneumonitis in 4 DL2 patients, rash in 10 DL2-DL4 patients, and meningoencephalitis and myasthenia gravis in 1 DL3 patient.
Study participants were adults with a mean age of 60 years with metastatic or unresectable solid tumors for which standard treatments did not exist or were no longer effective, or for which treatment with anti–programmed cell death ligand1/cytotoxic T-lymphocyte antigen 4 treatment was appropriate. All had good performance status and adequate organ and pulmonary function, less than 30% liver involvement, and any brain metastases were stable. Those with active autoimmune disease or a history of autoimmune disease that might recur were excluded, as were patients treated within 14 days of enrollment.
“The rationale for the study was based on preclinical work suggesting that epigenetic modifiers might be able to enhance the efficacy of immune therapies, and this would be particularly important for ‘colder’ tumor types like breast cancer that might not have the same sort of responses that we see in other tumor types,” Dr. Connolly explained in an interview. “The lab work suggested, for example, that the [histone deacetylase] inhibitor entinostat might affect myeloid-derived suppressor and regulatory T cells that might prevent cytotoxic T cells from fighting the cancer.”
There may be other mechanisms for this activity as well, she noted.
The run-in period with entinostat alone allowed collection of pre- and posttreatment biopsies to examine the effects on the tissues, such as whether treatment affects T cells, myeloid-derived suppressor cells, or their pathways, she said.
“We’re seeing [the] same types of toxicities seen with combination immune-oncology strategies, and we’re seeing some tumor responses that are of interest. Now we will delve into the tissue biopsies and blood samples we’ve collected to explore the mechanisms in more detail. In the near future we will open our breast cancer expansion cohort to look in more detail at what these drugs might be doing in breast cancer,” she added.
Specifically, she and her colleagues are evaluating the effects of treatment on immune-related biomarkers, measuring tumor-specific mutations and mutant neoantigens recognized by patient T cells in tumor biopsies, evaluating changes in the frequency of T cells recognizing tumor-specific mutant neoantigens in peripheral blood lymphocytes pre- and posttherapy, and looking at epigenetic changes pre- and posttherapy.
These preliminary findings suggest that the combination of entinostat and nivolumab with or without ipilimumab is safe and tolerable, with expected rates of immune-related adverse events, Dr. Connolly said, noting that the recommended phase 2 dose to be used in the dose expansion phase of the study has yet to be determined.
The findings, should they be confirmed as the trial progresses, could have important implications because immune checkpoint inhibitors, which work best in patients with immunogenic cancers that naturally attract T-cell infiltration into their tumor microenvironment, have limited single-agent activity in tumors, such as breast cancer, that are not believed to be immunogenic, she reported. Such cancers have thus far had only modest responses to single-agent immune checkpoint inhibition in advanced triple-negative and HER2+ breast cancer, with overall response rates of 5%-20%.
However, women who do respond to immune checkpoint inhibition tend to have durable and sustainable responses, she said, explaining that suboptimal immune responsiveness is likely a result of a lack of tumor antigen expression and/or recognition, as well as multiple suppressive signals in the tumor microenvironment.
Should the novel strategy tested in this study for converting breast cancers into immune responsive tumors facilitate improved response to immune checkpoint agents, it has the potential to significantly extend survival in breast cancer patients, she concluded.
This study was funded by grants from the National Institutes of Health, Bloomberg Kimmel Institute for Immunotherapy, NCCN, and the Mary Kay Foundation, as well as a V Foundation award. Dr. Connolly reported having no disclosures
SOURCE: Connolly RM et al. NCCN, Poster 3.
REPORTING FROM THE NCCN ANNUAL CONFERENCE
Key clinical point:
Major finding: Three patients had a partial response, 12 had stable disease, 5 progressed.
Study details: A phase 1 dose-expansion study involving 30 patients.
Disclosures: This study was funded by grants from the National Institutes of Health, Bloomberg Kimmel Institute for Immunotherapy, NCCN, and the Mary Kay Foundation, and by a V Foundation award. Dr. Connolly reported having no disclosures.
Source: Connolly RM et al. NCCN, Poster 3.
Sling revisions: pain as indication linked with SUI recurrence
ORLANDO – Pain as an indication for midurethral sling revision is associated with an elevated risk of postoperative stress urinary incontinence recurrence, according to a review of 129 cases.
At a mean follow-up of 21 months, the overall rate of recurrent stress urinary incontinence (SUI) among study subjects was 39.5%, which is similar to what has been previously reported in the literature. However, women who underwent revision for the indication of pain, including dyspareunia, pelvic pain, or muscle spasms, had an SUI recurrence rate of 70%, Meagan S. Cramer, MD, reported at the annual scientific meeting of the Society of Gynecologic Surgeons.

Women older than 55 years at the time of revision were significantly less likely to recur (adjusted odds ratio, 0.34 vs. younger women), and those whose original slings were placed less than 1 year prior to revision had a significantly lower rate of SUI recurrence, compared with those whose slings were placed more than 1 year prior (23.1% vs. 46.7%), she noted.
“There was no difference in BMI [body mass index], race, or baseline comorbidities between the recurrence vs. no recurrence group. There was also no difference in estrogen use preoperatively, concurrent replacement with another sling, preoperative [pelvic organ prolapse quantification] exam, length of sling excised, or the original type of sling placed between the recurrence versus non recurrence groups,” she said. “On multivariable regression analysis controlling for age, BMI, race, tobacco use, time since original sling, erosion on exam, technique used for revision, and presenting indication, pain as an indication of revision was still associated with an increased risk for recurrent incontinence, with an adjusted odds ratio of 9.08.”
Study subjects were women aged 18 years and older who underwent mesh sling revision from January 2009 to July 2016 at a single center. Women were excluded if they underwent revision or excision of vaginal mesh, or if they underwent sling adjustment for persistent incontinence if the incontinence had never resolved after the original sling placement. Those without postoperative follow-up were also excluded.
“Approximately 2%-4% of women who undergo midurethral sling surgery for stress incontinence will later require revision or excision of the sling due to erosion, pain, and/or voiding dysfunction. Based on current literature, up to 56% of these women can experience postoperative recurrence of stress incontinence following their revision,” Dr. Cramer said, noting that there is no consensus on which women will require future surgeries for stress incontinence after revision.
In the current study, women with pain were significantly more likely to undergo a complete excision of their sling (45.0% vs. 17.9% with other indications).
“We also discovered that of all women who underwent complete excision of a prior sling, those with pain had a higher rate of recurrent SUI, compared with those without pain [66.7% vs. 31.6%], but the difference was not statistically significant,” Dr. Cramer said in an interview.
She noted, however, that partial sling excision has been shown in at least one prior study “to provide just as much benefit as complete sling excision for patients with pain without leading to an increased risk of SUI.”
Though limited by the retrospective study design, the current findings suggest that pain as an indication for sling revision is associated with a higher risk of postoperative recurrent SUI, regardless of the type of revision, she said.
“Future prospective studies investigating the relationship between the indication for revision and postoperative stress incontinence would help to further define the roles of pain in this process,” she said, adding that in the meantime, given the prior findings regarding the benefits of partial sling excision, it may be reasonable to advise patients with pain that undergoing a partial sling excision could reduce the risk of recurrent SUI.
Dr. Cramer reported having no disclosures.
SOURCE: Cramer MS et al. SGS 2018, Oral Presentation 04.
ORLANDO – Pain as an indication for midurethral sling revision is associated with an elevated risk of postoperative stress urinary incontinence recurrence, according to a review of 129 cases.
At a mean follow-up of 21 months, the overall rate of recurrent stress urinary incontinence (SUI) among study subjects was 39.5%, which is similar to what has been previously reported in the literature. However, women who underwent revision for the indication of pain, including dyspareunia, pelvic pain, or muscle spasms, had an SUI recurrence rate of 70%, Meagan S. Cramer, MD, reported at the annual scientific meeting of the Society of Gynecologic Surgeons.
Women older than 55 years at the time of revision were significantly less likely to recur (adjusted odds ratio, 0.34 vs. younger women), and those whose original slings were placed less than 1 year prior to revision had a significantly lower rate of SUI recurrence, compared with those whose slings were placed more than 1 year prior (23.1% vs. 46.7%), she noted.
“There was no difference in BMI [body mass index], race, or baseline comorbidities between the recurrence vs. no recurrence group. There was also no difference in estrogen use preoperatively, concurrent replacement with another sling, preoperative [pelvic organ prolapse quantification] exam, length of sling excised, or the original type of sling placed between the recurrence versus non recurrence groups,” she said. “On multivariable regression analysis controlling for age, BMI, race, tobacco use, time since original sling, erosion on exam, technique used for revision, and presenting indication, pain as an indication of revision was still associated with an increased risk for recurrent incontinence, with an adjusted odds ratio of 9.08.”
Study subjects were women aged 18 years and older who underwent mesh sling revision from January 2009 to July 2016 at a single center. Women were excluded if they underwent revision or excision of vaginal mesh, or if they underwent sling adjustment for persistent incontinence if the incontinence had never resolved after the original sling placement. Those without postoperative follow-up were also excluded.
“Approximately 2%-4% of women who undergo midurethral sling surgery for stress incontinence will later require revision or excision of the sling due to erosion, pain, and/or voiding dysfunction. Based on current literature, up to 56% of these women can experience postoperative recurrence of stress incontinence following their revision,” Dr. Cramer said, noting that there is no consensus on which women will require future surgeries for stress incontinence after revision.
In the current study, women with pain were significantly more likely to undergo a complete excision of their sling (45.0% vs. 17.9% with other indications).
“We also discovered that of all women who underwent complete excision of a prior sling, those with pain had a higher rate of recurrent SUI, compared with those without pain [66.7% vs. 31.6%], but the difference was not statistically significant,” Dr. Cramer said in an interview.
She noted, however, that partial sling excision has been shown in at least one prior study “to provide just as much benefit as complete sling excision for patients with pain without leading to an increased risk of SUI.”
Though limited by the retrospective study design, the current findings suggest that pain as an indication for sling revision is associated with a higher risk of postoperative recurrent SUI, regardless of the type of revision, she said.
“Future prospective studies investigating the relationship between the indication for revision and postoperative stress incontinence would help to further define the roles of pain in this process,” she said, adding that in the meantime, given the prior findings regarding the benefits of partial sling excision, it may be reasonable to advise patients with pain that undergoing a partial sling excision could reduce the risk of recurrent SUI.
Dr. Cramer reported having no disclosures.
SOURCE: Cramer MS et al. SGS 2018, Oral Presentation 04.
ORLANDO – Pain as an indication for midurethral sling revision is associated with an elevated risk of postoperative stress urinary incontinence recurrence, according to a review of 129 cases.
At a mean follow-up of 21 months, the overall rate of recurrent stress urinary incontinence (SUI) among study subjects was 39.5%, which is similar to what has been previously reported in the literature. However, women who underwent revision for the indication of pain, including dyspareunia, pelvic pain, or muscle spasms, had an SUI recurrence rate of 70%, Meagan S. Cramer, MD, reported at the annual scientific meeting of the Society of Gynecologic Surgeons.
Women older than 55 years at the time of revision were significantly less likely to recur (adjusted odds ratio, 0.34 vs. younger women), and those whose original slings were placed less than 1 year prior to revision had a significantly lower rate of SUI recurrence, compared with those whose slings were placed more than 1 year prior (23.1% vs. 46.7%), she noted.
“There was no difference in BMI [body mass index], race, or baseline comorbidities between the recurrence vs. no recurrence group. There was also no difference in estrogen use preoperatively, concurrent replacement with another sling, preoperative [pelvic organ prolapse quantification] exam, length of sling excised, or the original type of sling placed between the recurrence versus non recurrence groups,” she said. “On multivariable regression analysis controlling for age, BMI, race, tobacco use, time since original sling, erosion on exam, technique used for revision, and presenting indication, pain as an indication of revision was still associated with an increased risk for recurrent incontinence, with an adjusted odds ratio of 9.08.”
Study subjects were women aged 18 years and older who underwent mesh sling revision from January 2009 to July 2016 at a single center. Women were excluded if they underwent revision or excision of vaginal mesh, or if they underwent sling adjustment for persistent incontinence if the incontinence had never resolved after the original sling placement. Those without postoperative follow-up were also excluded.
“Approximately 2%-4% of women who undergo midurethral sling surgery for stress incontinence will later require revision or excision of the sling due to erosion, pain, and/or voiding dysfunction. Based on current literature, up to 56% of these women can experience postoperative recurrence of stress incontinence following their revision,” Dr. Cramer said, noting that there is no consensus on which women will require future surgeries for stress incontinence after revision.
In the current study, women with pain were significantly more likely to undergo a complete excision of their sling (45.0% vs. 17.9% with other indications).
“We also discovered that of all women who underwent complete excision of a prior sling, those with pain had a higher rate of recurrent SUI, compared with those without pain [66.7% vs. 31.6%], but the difference was not statistically significant,” Dr. Cramer said in an interview.
She noted, however, that partial sling excision has been shown in at least one prior study “to provide just as much benefit as complete sling excision for patients with pain without leading to an increased risk of SUI.”
Though limited by the retrospective study design, the current findings suggest that pain as an indication for sling revision is associated with a higher risk of postoperative recurrent SUI, regardless of the type of revision, she said.
“Future prospective studies investigating the relationship between the indication for revision and postoperative stress incontinence would help to further define the roles of pain in this process,” she said, adding that in the meantime, given the prior findings regarding the benefits of partial sling excision, it may be reasonable to advise patients with pain that undergoing a partial sling excision could reduce the risk of recurrent SUI.
Dr. Cramer reported having no disclosures.
SOURCE: Cramer MS et al. SGS 2018, Oral Presentation 04.
REPORTING FROM SGS 2018
Key clinical point: Sling revisions for pain may increase the risk of postoperative stress urinary incontinence recurrence.
Major finding: The recurrent SUI rate after revision was 39.5% overall vs. 70% among those with pain as an indication.
Study details: A retrospective cohort study of 129 women.
Disclosures: Dr. Cramer reported having no disclosures.
Source: Cramer MS et al. SGS 2018, Oral Presentation 04.
Dr. T. Berry Brazelton was a pioneer of child-centered parenting
You may not realize it, but as you navigated through this morning’s hospital rounds and your busy office schedule, some of what you did and how you did it was the result of the pioneering work of Boston-based pediatrician T. Berry Brazelton, MD, who died March 13, 2018, at the age of 99.
You probably found the newborn you needed to examine in his mother’s hospital room. The 3-year-old in the croup tent was sharing his room with his father, who was sleeping on a cot at his crib side, and three out of the first four patients you saw in your office had been breastfed. These scenarios would have been unheard of 50 years ago. But Dr. Brazelton’s voice was the most widely heard, yet gentlest and persuasive in support of rooming-in and breastfeeding.

My fellow house officers and I had been accustomed to picking up infants to assess their tone. However, when Dr. Brazelton picked up a newborn, it was more like a conversation, an interview, and in a sense, it was a meeting of the minds.
It wasn’t that we had been rejecting the notion that a newborn could have a personality. It is just that we hadn’t been taught to look for it or to take it seriously. Dr. Brazelton taught us how to examine the person inside that little body and understand the importance of her temperament. By sharing what we learned from doing a Brazelton-style exam, we hoped to encourage the child’s parents to adopt more realistic expectations, and as a consequence, make parenting less mysterious and stressful.
When I first met Dr. Brazelton, he was in his mid-40s and just beginning on his trajectory toward national prominence. When we were assigned to take care of his hospitalized patients, it was obvious that his patient skills with sick children had taken a back seat to his interest in newborn temperament. He was more than willing to let us make the management decisions. In retrospect, that experience was a warning that I, like many other pediatricians, would face the similar challenge of maintaining my clinical skills in the face of a patient mix that was steadily acquiring a more behavioral and developmental flavor.
It is impossible to quantify the degree to which Dr. Brazelton’s ubiquity contributed to the popularity of a more child-centered parenting style. However, I think it would be unfair to blame him for the unfortunate phenomenon known as “helicopter parenting.”

Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
You may not realize it, but as you navigated through this morning’s hospital rounds and your busy office schedule, some of what you did and how you did it was the result of the pioneering work of Boston-based pediatrician T. Berry Brazelton, MD, who died March 13, 2018, at the age of 99.
You probably found the newborn you needed to examine in his mother’s hospital room. The 3-year-old in the croup tent was sharing his room with his father, who was sleeping on a cot at his crib side, and three out of the first four patients you saw in your office had been breastfed. These scenarios would have been unheard of 50 years ago. But Dr. Brazelton’s voice was the most widely heard, yet gentlest and persuasive in support of rooming-in and breastfeeding.
My fellow house officers and I had been accustomed to picking up infants to assess their tone. However, when Dr. Brazelton picked up a newborn, it was more like a conversation, an interview, and in a sense, it was a meeting of the minds.
It wasn’t that we had been rejecting the notion that a newborn could have a personality. It is just that we hadn’t been taught to look for it or to take it seriously. Dr. Brazelton taught us how to examine the person inside that little body and understand the importance of her temperament. By sharing what we learned from doing a Brazelton-style exam, we hoped to encourage the child’s parents to adopt more realistic expectations, and as a consequence, make parenting less mysterious and stressful.
When I first met Dr. Brazelton, he was in his mid-40s and just beginning on his trajectory toward national prominence. When we were assigned to take care of his hospitalized patients, it was obvious that his patient skills with sick children had taken a back seat to his interest in newborn temperament. He was more than willing to let us make the management decisions. In retrospect, that experience was a warning that I, like many other pediatricians, would face the similar challenge of maintaining my clinical skills in the face of a patient mix that was steadily acquiring a more behavioral and developmental flavor.
It is impossible to quantify the degree to which Dr. Brazelton’s ubiquity contributed to the popularity of a more child-centered parenting style. However, I think it would be unfair to blame him for the unfortunate phenomenon known as “helicopter parenting.”
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
You may not realize it, but as you navigated through this morning’s hospital rounds and your busy office schedule, some of what you did and how you did it was the result of the pioneering work of Boston-based pediatrician T. Berry Brazelton, MD, who died March 13, 2018, at the age of 99.
You probably found the newborn you needed to examine in his mother’s hospital room. The 3-year-old in the croup tent was sharing his room with his father, who was sleeping on a cot at his crib side, and three out of the first four patients you saw in your office had been breastfed. These scenarios would have been unheard of 50 years ago. But Dr. Brazelton’s voice was the most widely heard, yet gentlest and persuasive in support of rooming-in and breastfeeding.
My fellow house officers and I had been accustomed to picking up infants to assess their tone. However, when Dr. Brazelton picked up a newborn, it was more like a conversation, an interview, and in a sense, it was a meeting of the minds.
It wasn’t that we had been rejecting the notion that a newborn could have a personality. It is just that we hadn’t been taught to look for it or to take it seriously. Dr. Brazelton taught us how to examine the person inside that little body and understand the importance of her temperament. By sharing what we learned from doing a Brazelton-style exam, we hoped to encourage the child’s parents to adopt more realistic expectations, and as a consequence, make parenting less mysterious and stressful.
When I first met Dr. Brazelton, he was in his mid-40s and just beginning on his trajectory toward national prominence. When we were assigned to take care of his hospitalized patients, it was obvious that his patient skills with sick children had taken a back seat to his interest in newborn temperament. He was more than willing to let us make the management decisions. In retrospect, that experience was a warning that I, like many other pediatricians, would face the similar challenge of maintaining my clinical skills in the face of a patient mix that was steadily acquiring a more behavioral and developmental flavor.
It is impossible to quantify the degree to which Dr. Brazelton’s ubiquity contributed to the popularity of a more child-centered parenting style. However, I think it would be unfair to blame him for the unfortunate phenomenon known as “helicopter parenting.”
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].

Femoral artery endarterectomy still ‘gold standard’
CHICAGO – Additional higher quality supporting evidence is needed before , Jeffrey J. Siracuse, MD, FACS, asserted at a symposium on vascular surgery sponsored by Northwestern University.
“Open surgery in the CFA [common femoral artery] is probably still the gold standard in most cases,” said Dr. Siracuse, a vascular surgeon at Boston University.

He was quick to note that others would disagree. Stenting and other endovascular interventions in the CFA are booming in popularity, particularly among cardiologists, interventional radiologists, and the patients to whom the clinicians present the option in a favorable light. But this enthusiasm is based almost entirely on small, single-center, retrospective studies conducted in patients with heterogeneous profiles. The one prospective randomized multicenter trial of stenting versus surgery for CFA stenosis published to date – the French TECCO study – has a number of key limitations, flaws, and unanswered questions, which endovascular proponents have overlooked in their enthusiasm to promote an “endo-first” approach in the CFA, according to Dr. Siracuse.
“Everyone’s pretty much jumping on the bandwagon now. I think endovascular therapy of the CFA is here to stay. You’re going to see more people doing it, and potentially doing it incorrectly,” he predicted.
“The biggest thing I worry about with stenting is covering or jailing out the deep femoral artery. On multiple occasions – including a case just 2 weeks ago – I’ve taken out stents placed in the CFA by others that developed in-stent hyperplasia to the extent that the entire stent goes down, the DFA is covered, and now all of a sudden you’ve lost all flow to the leg. That’s my biggest concern with stenting,” he said.
Dr. Siracuse has other reservations as well. The CFA has traditionally been considered a “no-stent zone” because of the unique biomechanical stresses the artery is subjected to as a result of torsion, flexion, and extension at the hip joint. These forces render the area particularly vulnerable to neointimal hyperplasia, acute thrombosis, and stent fracture.
In addition, he noted, CFA endarterectomy for atherosclerotic lesions is a mature, well-established operation with an excellent track record for safety and durability. Dr. Siracuse’s review of procedural safety in 1,513 patients in the American College of Surgeons National Surgical Quality Improvement Project database during 2007-2010 showed a 30-day mortality of 1.5% and a 7.9% rate of major or minor complications (Vasc Endovascular Surg. 2014 Jan;48[1]:27-33).
In contrast, his review of 1,014 patients who underwent nonemergent endovascular CFA interventions for CFA stenosis without acute limb ischemia in the Vascular Quality Initiative registry demonstrated a 1-year patency rate of 85.3%, significantly lower than historically observed patency rates for endarterectomy. The 30-day mortality rate of 1.6% associated with endovascular interventions was essentially the same as in his earlier analysis of endarterectomy in the ACS NSQIP database, and the average 1.5-day hospital length of stay was shorter than with open surgery. Of considerable concern, however, stent implantation, which was performed in 35% of the endovascular interventions, was an independent predictor of amputation or death, with an associated 195% increased risk (J Vasc Surg. 2017 Apr;[4]:1039-46).
The travails of TECCO
The 17-center French TECCO study randomized 117 patients with de novo CFA atherosclerotic lesions to treatment via self-expanding stents or open surgery. A total of 98 participants were Rutherford stage 3, making TECCO primarily a study of claudicants. The primary outcome – the 30-day combined rate of morbidity and mortality – occurred in 26% of the surgical patients, a significantly higher rate than the 12.5% in the stent population. After a median follow-up of 24 months, the rates of primary patency, target lesion and extremity revascularization, and sustained clinical improvement were similar in the two groups (JACC Cardiovasc Interv. 2017 Jul 10;10[13]:1344-54).
The TECCO findings were hailed by endovascular therapy partisans as a big win. However, closer examination tells a different story, according to Dr. Siracuse.
There was no 30-day mortality in this rather small study. All 16 morbidity events occurring in the open surgery group within 30 days were relatively minor: 10 cases of delayed wound healing, 4 cases of postoperative paresthesia requiring medication, and 2 cases of lymphorrhea lasting longer than 3 days. In contrast, the seven morbidity events in the stent group included a complication requiring urgent open surgical repair at the time of stenting, one stent fracture, and a major amputation.
“The investigators didn’t elaborate on that major amputation, but I thought it was a little alarming because you should not have a major amputation with CFA interventions for claudicants,” the vascular surgeon commented. “Really, do people care about a lymphatic leak or do they care about amputation? I think more needs to be fleshed out about what really happened in that case.”
He was also puzzled by the hospital lengths of stay: a mean of 3.2 days in the stent group and 6.3 days in the open surgery group. “I think those lengths of stay are astounding. Very high and unusual,” he observed.
Dr. Siracuse predicted that much-needed high-quality data comparing treatments of the CFA will be provided by the BEST-CLI trial (Best Endovascular versus Surgical Treatment for Critical Limb Ischemia), which has been updated to include both open and endovascular interventions.
He reported having no financial conflicts of interest regarding his presentation.
CHICAGO – Additional higher quality supporting evidence is needed before , Jeffrey J. Siracuse, MD, FACS, asserted at a symposium on vascular surgery sponsored by Northwestern University.
“Open surgery in the CFA [common femoral artery] is probably still the gold standard in most cases,” said Dr. Siracuse, a vascular surgeon at Boston University.
He was quick to note that others would disagree. Stenting and other endovascular interventions in the CFA are booming in popularity, particularly among cardiologists, interventional radiologists, and the patients to whom the clinicians present the option in a favorable light. But this enthusiasm is based almost entirely on small, single-center, retrospective studies conducted in patients with heterogeneous profiles. The one prospective randomized multicenter trial of stenting versus surgery for CFA stenosis published to date – the French TECCO study – has a number of key limitations, flaws, and unanswered questions, which endovascular proponents have overlooked in their enthusiasm to promote an “endo-first” approach in the CFA, according to Dr. Siracuse.
“Everyone’s pretty much jumping on the bandwagon now. I think endovascular therapy of the CFA is here to stay. You’re going to see more people doing it, and potentially doing it incorrectly,” he predicted.
“The biggest thing I worry about with stenting is covering or jailing out the deep femoral artery. On multiple occasions – including a case just 2 weeks ago – I’ve taken out stents placed in the CFA by others that developed in-stent hyperplasia to the extent that the entire stent goes down, the DFA is covered, and now all of a sudden you’ve lost all flow to the leg. That’s my biggest concern with stenting,” he said.
Dr. Siracuse has other reservations as well. The CFA has traditionally been considered a “no-stent zone” because of the unique biomechanical stresses the artery is subjected to as a result of torsion, flexion, and extension at the hip joint. These forces render the area particularly vulnerable to neointimal hyperplasia, acute thrombosis, and stent fracture.
In addition, he noted, CFA endarterectomy for atherosclerotic lesions is a mature, well-established operation with an excellent track record for safety and durability. Dr. Siracuse’s review of procedural safety in 1,513 patients in the American College of Surgeons National Surgical Quality Improvement Project database during 2007-2010 showed a 30-day mortality of 1.5% and a 7.9% rate of major or minor complications (Vasc Endovascular Surg. 2014 Jan;48[1]:27-33).
In contrast, his review of 1,014 patients who underwent nonemergent endovascular CFA interventions for CFA stenosis without acute limb ischemia in the Vascular Quality Initiative registry demonstrated a 1-year patency rate of 85.3%, significantly lower than historically observed patency rates for endarterectomy. The 30-day mortality rate of 1.6% associated with endovascular interventions was essentially the same as in his earlier analysis of endarterectomy in the ACS NSQIP database, and the average 1.5-day hospital length of stay was shorter than with open surgery. Of considerable concern, however, stent implantation, which was performed in 35% of the endovascular interventions, was an independent predictor of amputation or death, with an associated 195% increased risk (J Vasc Surg. 2017 Apr;[4]:1039-46).
The travails of TECCO
The 17-center French TECCO study randomized 117 patients with de novo CFA atherosclerotic lesions to treatment via self-expanding stents or open surgery. A total of 98 participants were Rutherford stage 3, making TECCO primarily a study of claudicants. The primary outcome – the 30-day combined rate of morbidity and mortality – occurred in 26% of the surgical patients, a significantly higher rate than the 12.5% in the stent population. After a median follow-up of 24 months, the rates of primary patency, target lesion and extremity revascularization, and sustained clinical improvement were similar in the two groups (JACC Cardiovasc Interv. 2017 Jul 10;10[13]:1344-54).
The TECCO findings were hailed by endovascular therapy partisans as a big win. However, closer examination tells a different story, according to Dr. Siracuse.
There was no 30-day mortality in this rather small study. All 16 morbidity events occurring in the open surgery group within 30 days were relatively minor: 10 cases of delayed wound healing, 4 cases of postoperative paresthesia requiring medication, and 2 cases of lymphorrhea lasting longer than 3 days. In contrast, the seven morbidity events in the stent group included a complication requiring urgent open surgical repair at the time of stenting, one stent fracture, and a major amputation.
“The investigators didn’t elaborate on that major amputation, but I thought it was a little alarming because you should not have a major amputation with CFA interventions for claudicants,” the vascular surgeon commented. “Really, do people care about a lymphatic leak or do they care about amputation? I think more needs to be fleshed out about what really happened in that case.”
He was also puzzled by the hospital lengths of stay: a mean of 3.2 days in the stent group and 6.3 days in the open surgery group. “I think those lengths of stay are astounding. Very high and unusual,” he observed.
Dr. Siracuse predicted that much-needed high-quality data comparing treatments of the CFA will be provided by the BEST-CLI trial (Best Endovascular versus Surgical Treatment for Critical Limb Ischemia), which has been updated to include both open and endovascular interventions.
He reported having no financial conflicts of interest regarding his presentation.
CHICAGO – Additional higher quality supporting evidence is needed before , Jeffrey J. Siracuse, MD, FACS, asserted at a symposium on vascular surgery sponsored by Northwestern University.
“Open surgery in the CFA [common femoral artery] is probably still the gold standard in most cases,” said Dr. Siracuse, a vascular surgeon at Boston University.
He was quick to note that others would disagree. Stenting and other endovascular interventions in the CFA are booming in popularity, particularly among cardiologists, interventional radiologists, and the patients to whom the clinicians present the option in a favorable light. But this enthusiasm is based almost entirely on small, single-center, retrospective studies conducted in patients with heterogeneous profiles. The one prospective randomized multicenter trial of stenting versus surgery for CFA stenosis published to date – the French TECCO study – has a number of key limitations, flaws, and unanswered questions, which endovascular proponents have overlooked in their enthusiasm to promote an “endo-first” approach in the CFA, according to Dr. Siracuse.
“Everyone’s pretty much jumping on the bandwagon now. I think endovascular therapy of the CFA is here to stay. You’re going to see more people doing it, and potentially doing it incorrectly,” he predicted.
“The biggest thing I worry about with stenting is covering or jailing out the deep femoral artery. On multiple occasions – including a case just 2 weeks ago – I’ve taken out stents placed in the CFA by others that developed in-stent hyperplasia to the extent that the entire stent goes down, the DFA is covered, and now all of a sudden you’ve lost all flow to the leg. That’s my biggest concern with stenting,” he said.
Dr. Siracuse has other reservations as well. The CFA has traditionally been considered a “no-stent zone” because of the unique biomechanical stresses the artery is subjected to as a result of torsion, flexion, and extension at the hip joint. These forces render the area particularly vulnerable to neointimal hyperplasia, acute thrombosis, and stent fracture.
In addition, he noted, CFA endarterectomy for atherosclerotic lesions is a mature, well-established operation with an excellent track record for safety and durability. Dr. Siracuse’s review of procedural safety in 1,513 patients in the American College of Surgeons National Surgical Quality Improvement Project database during 2007-2010 showed a 30-day mortality of 1.5% and a 7.9% rate of major or minor complications (Vasc Endovascular Surg. 2014 Jan;48[1]:27-33).
In contrast, his review of 1,014 patients who underwent nonemergent endovascular CFA interventions for CFA stenosis without acute limb ischemia in the Vascular Quality Initiative registry demonstrated a 1-year patency rate of 85.3%, significantly lower than historically observed patency rates for endarterectomy. The 30-day mortality rate of 1.6% associated with endovascular interventions was essentially the same as in his earlier analysis of endarterectomy in the ACS NSQIP database, and the average 1.5-day hospital length of stay was shorter than with open surgery. Of considerable concern, however, stent implantation, which was performed in 35% of the endovascular interventions, was an independent predictor of amputation or death, with an associated 195% increased risk (J Vasc Surg. 2017 Apr;[4]:1039-46).
The travails of TECCO
The 17-center French TECCO study randomized 117 patients with de novo CFA atherosclerotic lesions to treatment via self-expanding stents or open surgery. A total of 98 participants were Rutherford stage 3, making TECCO primarily a study of claudicants. The primary outcome – the 30-day combined rate of morbidity and mortality – occurred in 26% of the surgical patients, a significantly higher rate than the 12.5% in the stent population. After a median follow-up of 24 months, the rates of primary patency, target lesion and extremity revascularization, and sustained clinical improvement were similar in the two groups (JACC Cardiovasc Interv. 2017 Jul 10;10[13]:1344-54).
The TECCO findings were hailed by endovascular therapy partisans as a big win. However, closer examination tells a different story, according to Dr. Siracuse.
There was no 30-day mortality in this rather small study. All 16 morbidity events occurring in the open surgery group within 30 days were relatively minor: 10 cases of delayed wound healing, 4 cases of postoperative paresthesia requiring medication, and 2 cases of lymphorrhea lasting longer than 3 days. In contrast, the seven morbidity events in the stent group included a complication requiring urgent open surgical repair at the time of stenting, one stent fracture, and a major amputation.
“The investigators didn’t elaborate on that major amputation, but I thought it was a little alarming because you should not have a major amputation with CFA interventions for claudicants,” the vascular surgeon commented. “Really, do people care about a lymphatic leak or do they care about amputation? I think more needs to be fleshed out about what really happened in that case.”
He was also puzzled by the hospital lengths of stay: a mean of 3.2 days in the stent group and 6.3 days in the open surgery group. “I think those lengths of stay are astounding. Very high and unusual,” he observed.
Dr. Siracuse predicted that much-needed high-quality data comparing treatments of the CFA will be provided by the BEST-CLI trial (Best Endovascular versus Surgical Treatment for Critical Limb Ischemia), which has been updated to include both open and endovascular interventions.
He reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE NORTHWESTERN VASCULAR SYMPOSIUM
Anterior discoid resection using a ‘squeeze’ technique
Rectosigmoid endometriosis has been estimated to affect between 4% and 37% of patients with endometriosis and is one of the most advanced and complex forms of the disease. Bowel endometriosis can be asymptomatic but often involves severe dysmenorrhea, dyspareunia, and a spectrum of bowel symptoms such as dyschezia, diarrhea, constipation, bloating, and rectal bleeding. Deep infiltrating rectovaginal endometriosis causes persistent or recurrent pain and is best treated by surgical removal of nodular lesions.
I have found that laparoscopic full-thickness disc resection (anterior discoid resection) with primary two-layer closure is often feasible and avoids the need for a complete bowel reanastomosis. It may not be an option in cases of multifocal rectal involvement (which may affect between one-quarter and one-third of patients with bowel endometriosis) or in cases involving large rectal nodules or luminal stenosis secondary to advanced fibrosis. In these cases, segmental bowel resection (low anterior resection) is often necessary. When anterior discoid resection is feasible, however, patients face significantly less morbidity with comparable outcomes.

Less morbidity
Preoperative evaluation is far from straightforward, and practices vary. Transvaginal ultrasonography is used for diagnosing rectal endometriosis in select centers in certain regions of the world, but there are important limitations; not only is it highly operator dependent, but its limited range does not allow for the detection of endometriosis higher in the sigmoid colon. Endorectal ultrasonography can be an excellent tool for more fully evaluating rectal wall involvement, but it does not usually allow for the evaluation of disease elsewhere in the pelvis.
The preoperative tool we utilize most often along with clinical examination is MRI with vaginal and rectal contrast. MRI provides us with a superior anatomic perspective on the disease. Not only can we assess the depth of bowel wall infiltration and the distribution of the affected areas of the bowel, but we can see the bladder, the uterosacral ligaments, and how the uterus is situated relative to areas of disease. However, there are individualized limits to how high the contrast will travel, even with bowel preparation; disease that occurs significantly above the uterus often cannot be visualized as well as disease that occurs lower.
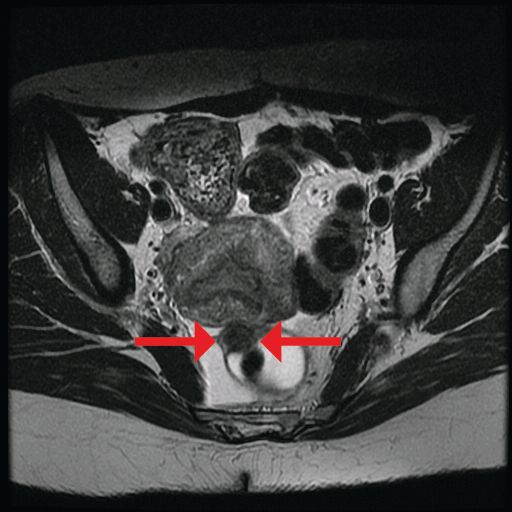
My general surgeon colleague and I have been working together for years, and we both are involved in counseling the patient suspected of having deep infiltrating disease. I typically talk with the patient about the probability of segmental resection based on my exam and preoperative MRI, and my colleague expands on this discussion with further explanation of the risks of bowel surgery.
Segmental resection has been associated with significant postoperative complications. In a single-center series of 436 laparoscopic colorectal resections for deep infiltrating endometriosis, rectovaginal and anastomotic fistula were among the most frequent postoperative complications (3.2% and 1.1%), along with transient urinary retention, which occurred in almost 20% (Surg Endosc. 2010 Jan;24:63-7).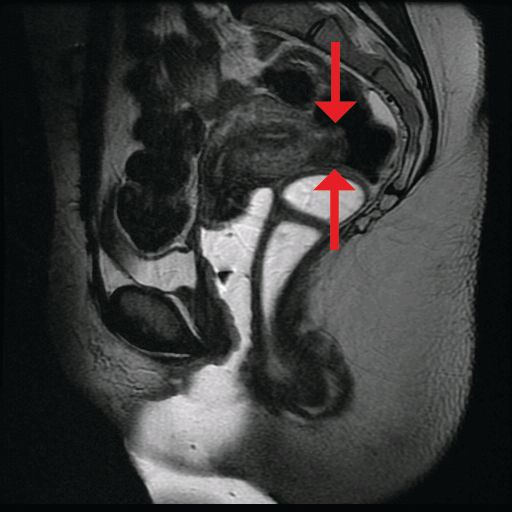
Patients undergoing discoid resection for deep infiltrating endometriosis also had a significantly lower rate of temporary ileostomy (2.1% vs. 9.1%), a reduced rate of postoperative fever, and a reduced rate of gastrointestinal complications, mainly anastomotic leak or rectovaginal fistula (2.1% vs. 5.6%). There were no significant differences in the recurrence rate (13.8% vs. 11.5%).
A retrospective cohort study from our institution similarly showed decreased operative time, blood loss, hospital stay, and a lower rate of anastomotic strictures in patients who underwent laparoscopic anterior discoid resection between 2001 and 2009. The ADR group consistently had higher increments of improvement in bowel symptoms and dyspareunia, compared with patients who were selected to have segmental resection. Patients were followed for a mean of 41 months (JSLS. 2011;15[3]:331-8).
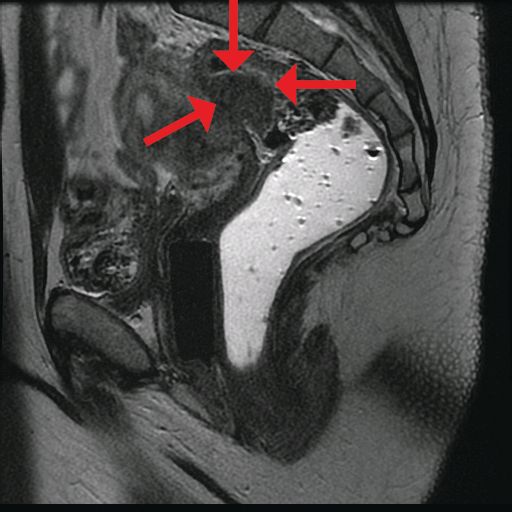
In general, there is agreement among surgeons that for consideration of discoid resection, nodule diameter should not exceed 3 cm, with a maximum of half of the bowel circumference and a maximum of 60% stenosis. I view these numbers as guiding principles, however, and not firm rules. Surgical decisions should be personalized based on the patient, the surgeon’s impression of the extent of the disease, and the ability to perform anterior discoid resection without compromising the rectal lumen with primary closure of the defect.
The technique
Rectosigmoid endometriotic nodules may present within the context of an obliterated posterior cul-de-sac, but the avascular pararectal space can be used to approach the nodules. Detailed knowledge of the avascular planes of this space, as well as the rectovaginal space, is crucial. Development of the rectovaginal space frees the bowel from its attachments to the posterior uterus and vagina. Judicious use of energized instruments in sharp dissection, and frequently sharp cold cutting, should be used near the bowel serosa to prevent thermal injury.
Presurgical imaging usually offers a good assessment of a nodule’s size and location, but intraoperatively, I typically use an atraumatic grasper to further assess size and contour and to determine if the nodule is suitable for discoid resection. If so, a suture is placed through the nodule to improve manipulation, and enucleation of the nodule itself is achieved through a “squeeze” technique in which an advanced bipolar device is used to circumscribe the lesion, dissecting the nodule as the device bounces off the thick endometriotic tissue.
The ENSEAL bipolar device (Ethicon, Somerville, N.J.) was designed as a vessel sealer, but because it will not cut through hard tissue as will other laparoscopic cutting devices, it serves as a useful tool for resecting endometriotic nodules while minimizing the removal of healthy rectal tissue. The device bounces off the nodule because it will avoid cutting through the thick tissue; in the process, it facilitates a fairly complete enucleation of the endometriotic nodule, starting with dissection until an intentional colotomy/enterotomy is made and followed by circumscription of the lesion once the rectum is entered.
Gentle traction and counter-traction increase the efficiency of dissection and minimize the amount of normal rectal tissue removed. Quick cutting with short bursts of energy allows for good hemostasis and minimizes thermal spread, which will maximize tissue healing from subsequent repair.
I then use a rectal probe as a template for repair. The probe is advanced underneath the defect between the distal and proximal portions, and the tissue is moved over the probe to ensure that the repair will be tension free. An ability to reapproximate the defect while keeping the probe in place indicates that the defect can be safely closed. (For a video presentation of the surgery, see www.surgeryu.com/leeobgyn.) If suturing is not feasible, the general surgeon is called to perform segmental resection.
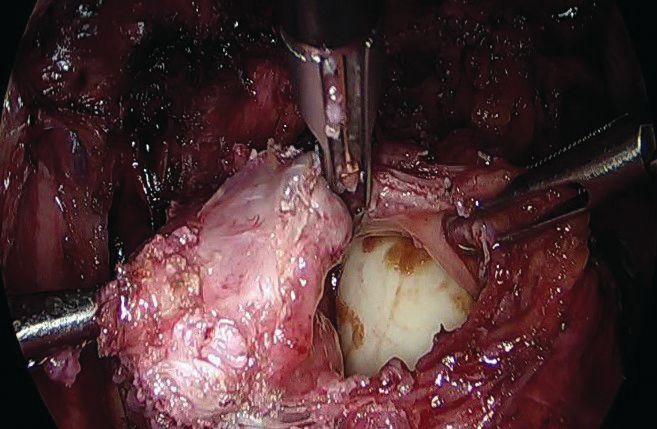
The integrity of the repair is then thoroughly assessed with an air leak test. A bowel clamp is placed across the rectum and the pelvis is filled with sterile saline. Air is placed into the rectum with a rigid proctoscope while the operative field is inspected for evidence of an air leak.
Discoid resection may also be performed with a circular stapler. While this technique is faster than suturing, its use is limited by nodule size and has the potential to compromise complete excision of the nodule.
Dr. Lee is director of minimally invasive gynecologic surgery, Magee-Women’s Hospital of the University of Pittsburgh Medical Center.
Rectosigmoid endometriosis has been estimated to affect between 4% and 37% of patients with endometriosis and is one of the most advanced and complex forms of the disease. Bowel endometriosis can be asymptomatic but often involves severe dysmenorrhea, dyspareunia, and a spectrum of bowel symptoms such as dyschezia, diarrhea, constipation, bloating, and rectal bleeding. Deep infiltrating rectovaginal endometriosis causes persistent or recurrent pain and is best treated by surgical removal of nodular lesions.
I have found that laparoscopic full-thickness disc resection (anterior discoid resection) with primary two-layer closure is often feasible and avoids the need for a complete bowel reanastomosis. It may not be an option in cases of multifocal rectal involvement (which may affect between one-quarter and one-third of patients with bowel endometriosis) or in cases involving large rectal nodules or luminal stenosis secondary to advanced fibrosis. In these cases, segmental bowel resection (low anterior resection) is often necessary. When anterior discoid resection is feasible, however, patients face significantly less morbidity with comparable outcomes.
Less morbidity
Preoperative evaluation is far from straightforward, and practices vary. Transvaginal ultrasonography is used for diagnosing rectal endometriosis in select centers in certain regions of the world, but there are important limitations; not only is it highly operator dependent, but its limited range does not allow for the detection of endometriosis higher in the sigmoid colon. Endorectal ultrasonography can be an excellent tool for more fully evaluating rectal wall involvement, but it does not usually allow for the evaluation of disease elsewhere in the pelvis.
The preoperative tool we utilize most often along with clinical examination is MRI with vaginal and rectal contrast. MRI provides us with a superior anatomic perspective on the disease. Not only can we assess the depth of bowel wall infiltration and the distribution of the affected areas of the bowel, but we can see the bladder, the uterosacral ligaments, and how the uterus is situated relative to areas of disease. However, there are individualized limits to how high the contrast will travel, even with bowel preparation; disease that occurs significantly above the uterus often cannot be visualized as well as disease that occurs lower.
My general surgeon colleague and I have been working together for years, and we both are involved in counseling the patient suspected of having deep infiltrating disease. I typically talk with the patient about the probability of segmental resection based on my exam and preoperative MRI, and my colleague expands on this discussion with further explanation of the risks of bowel surgery.
Segmental resection has been associated with significant postoperative complications. In a single-center series of 436 laparoscopic colorectal resections for deep infiltrating endometriosis, rectovaginal and anastomotic fistula were among the most frequent postoperative complications (3.2% and 1.1%), along with transient urinary retention, which occurred in almost 20% (Surg Endosc. 2010 Jan;24:63-7).
Patients undergoing discoid resection for deep infiltrating endometriosis also had a significantly lower rate of temporary ileostomy (2.1% vs. 9.1%), a reduced rate of postoperative fever, and a reduced rate of gastrointestinal complications, mainly anastomotic leak or rectovaginal fistula (2.1% vs. 5.6%). There were no significant differences in the recurrence rate (13.8% vs. 11.5%).
A retrospective cohort study from our institution similarly showed decreased operative time, blood loss, hospital stay, and a lower rate of anastomotic strictures in patients who underwent laparoscopic anterior discoid resection between 2001 and 2009. The ADR group consistently had higher increments of improvement in bowel symptoms and dyspareunia, compared with patients who were selected to have segmental resection. Patients were followed for a mean of 41 months (JSLS. 2011;15[3]:331-8).
In general, there is agreement among surgeons that for consideration of discoid resection, nodule diameter should not exceed 3 cm, with a maximum of half of the bowel circumference and a maximum of 60% stenosis. I view these numbers as guiding principles, however, and not firm rules. Surgical decisions should be personalized based on the patient, the surgeon’s impression of the extent of the disease, and the ability to perform anterior discoid resection without compromising the rectal lumen with primary closure of the defect.
The technique
Rectosigmoid endometriotic nodules may present within the context of an obliterated posterior cul-de-sac, but the avascular pararectal space can be used to approach the nodules. Detailed knowledge of the avascular planes of this space, as well as the rectovaginal space, is crucial. Development of the rectovaginal space frees the bowel from its attachments to the posterior uterus and vagina. Judicious use of energized instruments in sharp dissection, and frequently sharp cold cutting, should be used near the bowel serosa to prevent thermal injury.
Presurgical imaging usually offers a good assessment of a nodule’s size and location, but intraoperatively, I typically use an atraumatic grasper to further assess size and contour and to determine if the nodule is suitable for discoid resection. If so, a suture is placed through the nodule to improve manipulation, and enucleation of the nodule itself is achieved through a “squeeze” technique in which an advanced bipolar device is used to circumscribe the lesion, dissecting the nodule as the device bounces off the thick endometriotic tissue.
The ENSEAL bipolar device (Ethicon, Somerville, N.J.) was designed as a vessel sealer, but because it will not cut through hard tissue as will other laparoscopic cutting devices, it serves as a useful tool for resecting endometriotic nodules while minimizing the removal of healthy rectal tissue. The device bounces off the nodule because it will avoid cutting through the thick tissue; in the process, it facilitates a fairly complete enucleation of the endometriotic nodule, starting with dissection until an intentional colotomy/enterotomy is made and followed by circumscription of the lesion once the rectum is entered.
Gentle traction and counter-traction increase the efficiency of dissection and minimize the amount of normal rectal tissue removed. Quick cutting with short bursts of energy allows for good hemostasis and minimizes thermal spread, which will maximize tissue healing from subsequent repair.
I then use a rectal probe as a template for repair. The probe is advanced underneath the defect between the distal and proximal portions, and the tissue is moved over the probe to ensure that the repair will be tension free. An ability to reapproximate the defect while keeping the probe in place indicates that the defect can be safely closed. (For a video presentation of the surgery, see www.surgeryu.com/leeobgyn.) If suturing is not feasible, the general surgeon is called to perform segmental resection.
The integrity of the repair is then thoroughly assessed with an air leak test. A bowel clamp is placed across the rectum and the pelvis is filled with sterile saline. Air is placed into the rectum with a rigid proctoscope while the operative field is inspected for evidence of an air leak.
Discoid resection may also be performed with a circular stapler. While this technique is faster than suturing, its use is limited by nodule size and has the potential to compromise complete excision of the nodule.
Dr. Lee is director of minimally invasive gynecologic surgery, Magee-Women’s Hospital of the University of Pittsburgh Medical Center.
Rectosigmoid endometriosis has been estimated to affect between 4% and 37% of patients with endometriosis and is one of the most advanced and complex forms of the disease. Bowel endometriosis can be asymptomatic but often involves severe dysmenorrhea, dyspareunia, and a spectrum of bowel symptoms such as dyschezia, diarrhea, constipation, bloating, and rectal bleeding. Deep infiltrating rectovaginal endometriosis causes persistent or recurrent pain and is best treated by surgical removal of nodular lesions.
I have found that laparoscopic full-thickness disc resection (anterior discoid resection) with primary two-layer closure is often feasible and avoids the need for a complete bowel reanastomosis. It may not be an option in cases of multifocal rectal involvement (which may affect between one-quarter and one-third of patients with bowel endometriosis) or in cases involving large rectal nodules or luminal stenosis secondary to advanced fibrosis. In these cases, segmental bowel resection (low anterior resection) is often necessary. When anterior discoid resection is feasible, however, patients face significantly less morbidity with comparable outcomes.
Less morbidity
Preoperative evaluation is far from straightforward, and practices vary. Transvaginal ultrasonography is used for diagnosing rectal endometriosis in select centers in certain regions of the world, but there are important limitations; not only is it highly operator dependent, but its limited range does not allow for the detection of endometriosis higher in the sigmoid colon. Endorectal ultrasonography can be an excellent tool for more fully evaluating rectal wall involvement, but it does not usually allow for the evaluation of disease elsewhere in the pelvis.
The preoperative tool we utilize most often along with clinical examination is MRI with vaginal and rectal contrast. MRI provides us with a superior anatomic perspective on the disease. Not only can we assess the depth of bowel wall infiltration and the distribution of the affected areas of the bowel, but we can see the bladder, the uterosacral ligaments, and how the uterus is situated relative to areas of disease. However, there are individualized limits to how high the contrast will travel, even with bowel preparation; disease that occurs significantly above the uterus often cannot be visualized as well as disease that occurs lower.
My general surgeon colleague and I have been working together for years, and we both are involved in counseling the patient suspected of having deep infiltrating disease. I typically talk with the patient about the probability of segmental resection based on my exam and preoperative MRI, and my colleague expands on this discussion with further explanation of the risks of bowel surgery.
Segmental resection has been associated with significant postoperative complications. In a single-center series of 436 laparoscopic colorectal resections for deep infiltrating endometriosis, rectovaginal and anastomotic fistula were among the most frequent postoperative complications (3.2% and 1.1%), along with transient urinary retention, which occurred in almost 20% (Surg Endosc. 2010 Jan;24:63-7).
Patients undergoing discoid resection for deep infiltrating endometriosis also had a significantly lower rate of temporary ileostomy (2.1% vs. 9.1%), a reduced rate of postoperative fever, and a reduced rate of gastrointestinal complications, mainly anastomotic leak or rectovaginal fistula (2.1% vs. 5.6%). There were no significant differences in the recurrence rate (13.8% vs. 11.5%).
A retrospective cohort study from our institution similarly showed decreased operative time, blood loss, hospital stay, and a lower rate of anastomotic strictures in patients who underwent laparoscopic anterior discoid resection between 2001 and 2009. The ADR group consistently had higher increments of improvement in bowel symptoms and dyspareunia, compared with patients who were selected to have segmental resection. Patients were followed for a mean of 41 months (JSLS. 2011;15[3]:331-8).
In general, there is agreement among surgeons that for consideration of discoid resection, nodule diameter should not exceed 3 cm, with a maximum of half of the bowel circumference and a maximum of 60% stenosis. I view these numbers as guiding principles, however, and not firm rules. Surgical decisions should be personalized based on the patient, the surgeon’s impression of the extent of the disease, and the ability to perform anterior discoid resection without compromising the rectal lumen with primary closure of the defect.
The technique
Rectosigmoid endometriotic nodules may present within the context of an obliterated posterior cul-de-sac, but the avascular pararectal space can be used to approach the nodules. Detailed knowledge of the avascular planes of this space, as well as the rectovaginal space, is crucial. Development of the rectovaginal space frees the bowel from its attachments to the posterior uterus and vagina. Judicious use of energized instruments in sharp dissection, and frequently sharp cold cutting, should be used near the bowel serosa to prevent thermal injury.
Presurgical imaging usually offers a good assessment of a nodule’s size and location, but intraoperatively, I typically use an atraumatic grasper to further assess size and contour and to determine if the nodule is suitable for discoid resection. If so, a suture is placed through the nodule to improve manipulation, and enucleation of the nodule itself is achieved through a “squeeze” technique in which an advanced bipolar device is used to circumscribe the lesion, dissecting the nodule as the device bounces off the thick endometriotic tissue.
The ENSEAL bipolar device (Ethicon, Somerville, N.J.) was designed as a vessel sealer, but because it will not cut through hard tissue as will other laparoscopic cutting devices, it serves as a useful tool for resecting endometriotic nodules while minimizing the removal of healthy rectal tissue. The device bounces off the nodule because it will avoid cutting through the thick tissue; in the process, it facilitates a fairly complete enucleation of the endometriotic nodule, starting with dissection until an intentional colotomy/enterotomy is made and followed by circumscription of the lesion once the rectum is entered.
Gentle traction and counter-traction increase the efficiency of dissection and minimize the amount of normal rectal tissue removed. Quick cutting with short bursts of energy allows for good hemostasis and minimizes thermal spread, which will maximize tissue healing from subsequent repair.
I then use a rectal probe as a template for repair. The probe is advanced underneath the defect between the distal and proximal portions, and the tissue is moved over the probe to ensure that the repair will be tension free. An ability to reapproximate the defect while keeping the probe in place indicates that the defect can be safely closed. (For a video presentation of the surgery, see www.surgeryu.com/leeobgyn.) If suturing is not feasible, the general surgeon is called to perform segmental resection.
The integrity of the repair is then thoroughly assessed with an air leak test. A bowel clamp is placed across the rectum and the pelvis is filled with sterile saline. Air is placed into the rectum with a rigid proctoscope while the operative field is inspected for evidence of an air leak.
Discoid resection may also be performed with a circular stapler. While this technique is faster than suturing, its use is limited by nodule size and has the potential to compromise complete excision of the nodule.
Dr. Lee is director of minimally invasive gynecologic surgery, Magee-Women’s Hospital of the University of Pittsburgh Medical Center.
Discoid resection of rectal endometriotic nodules
The treatment of the rectovaginal endometriotic nodule continues to be controversial. While proponents of “shaving” the nodule are quick to point out that compared with segmental bowel resection, pelvic pain, dyspareunia, dysmenorrhea, and postoperative pregnancy rates are similarly reduced, most comparative studies are retrospective and are not randomized. That is, patients with larger nodules or multifocal disease with deep infiltration into the muscularis layer of the bowel, or involving more than half of the bowel wall circumference, with surrounding severe fibrosis, invariably are more likely to undergo segmental bowel resection. Even with performance of segmental bowel resection to treat more extensive disease, there is a trend toward greater improvement of pain-related symptoms when compared with the “shaving” technique. Furthermore, the risk of rectal recurrence is acknowledged to be greater in patients undergoing endometriotic rectal nodule shaving.
Concern must be raised with segmental bowel resection. Not only is the risk of temporary ileostomy increased, but subsequent anastomotic leakage and rectovaginal fistula is noted in up to 10% of women. Although reduced with nerve sparing techniques, bladder denervation secondary to damage of the parasympathetic plexus causes urinary retention. In a study of 436 cases of laparoscopic colorectal resection, 9.5% presented after 30 days with persistent urinary retention and 4.2% with constipation (Surg Endosc. 2010 Jan;24:63-7).

For this edition of the Master Class in Gynecologic Surgery, I have invited Ted Lee, MD, director of minimally invasive gynecologic surgery, Magee-Womens Hospital of the University of Pittsburgh Medical Center, to discuss laparoscopic rectosigmoid resection for a deep endometriotic nodule. While many surgeons utilize a single-use curved circular stapler, I appreciate Dr. Lee’s innovative technique, for both its ease of use and its safety.
Dr. Lee has received multiple awards for his efforts, including best surgical video presentation by the AAGL. He is also the only five-time winner of the prestigious Golden Laparoscope Award for best surgical video from the AAGL.
A highly-regarded lecturer and surgeon, Dr. Lee has taught and performed live surgeries around the world.
Dr. Lee’s practice is entirely dedicated to minimally invasive surgical options for women. He is a firm believer that virtually all benign gynecologic surgical conditions should be treated using a minimally invasive approach. Dr. Lee’s clinical expertise includes minimally invasive surgery for treatments of endometriosis (including severe endometriosis involving bowel, bladder, and ureter); fibroids; abnormal uterine bleeding; urinary incontinence; and pelvic organ prolapse.
It is a great honor for the Master Class in Gynecologic Surgery to have Dr. Lee as guest author for this important area of our surgical arena.
Dr. Miller is a minimally invasive gynecologic surgeon in Naperville, Ill., and a past president of the AAGL.
The treatment of the rectovaginal endometriotic nodule continues to be controversial. While proponents of “shaving” the nodule are quick to point out that compared with segmental bowel resection, pelvic pain, dyspareunia, dysmenorrhea, and postoperative pregnancy rates are similarly reduced, most comparative studies are retrospective and are not randomized. That is, patients with larger nodules or multifocal disease with deep infiltration into the muscularis layer of the bowel, or involving more than half of the bowel wall circumference, with surrounding severe fibrosis, invariably are more likely to undergo segmental bowel resection. Even with performance of segmental bowel resection to treat more extensive disease, there is a trend toward greater improvement of pain-related symptoms when compared with the “shaving” technique. Furthermore, the risk of rectal recurrence is acknowledged to be greater in patients undergoing endometriotic rectal nodule shaving.
Concern must be raised with segmental bowel resection. Not only is the risk of temporary ileostomy increased, but subsequent anastomotic leakage and rectovaginal fistula is noted in up to 10% of women. Although reduced with nerve sparing techniques, bladder denervation secondary to damage of the parasympathetic plexus causes urinary retention. In a study of 436 cases of laparoscopic colorectal resection, 9.5% presented after 30 days with persistent urinary retention and 4.2% with constipation (Surg Endosc. 2010 Jan;24:63-7).
For this edition of the Master Class in Gynecologic Surgery, I have invited Ted Lee, MD, director of minimally invasive gynecologic surgery, Magee-Womens Hospital of the University of Pittsburgh Medical Center, to discuss laparoscopic rectosigmoid resection for a deep endometriotic nodule. While many surgeons utilize a single-use curved circular stapler, I appreciate Dr. Lee’s innovative technique, for both its ease of use and its safety.
Dr. Lee has received multiple awards for his efforts, including best surgical video presentation by the AAGL. He is also the only five-time winner of the prestigious Golden Laparoscope Award for best surgical video from the AAGL.
A highly-regarded lecturer and surgeon, Dr. Lee has taught and performed live surgeries around the world.
Dr. Lee’s practice is entirely dedicated to minimally invasive surgical options for women. He is a firm believer that virtually all benign gynecologic surgical conditions should be treated using a minimally invasive approach. Dr. Lee’s clinical expertise includes minimally invasive surgery for treatments of endometriosis (including severe endometriosis involving bowel, bladder, and ureter); fibroids; abnormal uterine bleeding; urinary incontinence; and pelvic organ prolapse.
It is a great honor for the Master Class in Gynecologic Surgery to have Dr. Lee as guest author for this important area of our surgical arena.
Dr. Miller is a minimally invasive gynecologic surgeon in Naperville, Ill., and a past president of the AAGL.
The treatment of the rectovaginal endometriotic nodule continues to be controversial. While proponents of “shaving” the nodule are quick to point out that compared with segmental bowel resection, pelvic pain, dyspareunia, dysmenorrhea, and postoperative pregnancy rates are similarly reduced, most comparative studies are retrospective and are not randomized. That is, patients with larger nodules or multifocal disease with deep infiltration into the muscularis layer of the bowel, or involving more than half of the bowel wall circumference, with surrounding severe fibrosis, invariably are more likely to undergo segmental bowel resection. Even with performance of segmental bowel resection to treat more extensive disease, there is a trend toward greater improvement of pain-related symptoms when compared with the “shaving” technique. Furthermore, the risk of rectal recurrence is acknowledged to be greater in patients undergoing endometriotic rectal nodule shaving.
Concern must be raised with segmental bowel resection. Not only is the risk of temporary ileostomy increased, but subsequent anastomotic leakage and rectovaginal fistula is noted in up to 10% of women. Although reduced with nerve sparing techniques, bladder denervation secondary to damage of the parasympathetic plexus causes urinary retention. In a study of 436 cases of laparoscopic colorectal resection, 9.5% presented after 30 days with persistent urinary retention and 4.2% with constipation (Surg Endosc. 2010 Jan;24:63-7).
For this edition of the Master Class in Gynecologic Surgery, I have invited Ted Lee, MD, director of minimally invasive gynecologic surgery, Magee-Womens Hospital of the University of Pittsburgh Medical Center, to discuss laparoscopic rectosigmoid resection for a deep endometriotic nodule. While many surgeons utilize a single-use curved circular stapler, I appreciate Dr. Lee’s innovative technique, for both its ease of use and its safety.
Dr. Lee has received multiple awards for his efforts, including best surgical video presentation by the AAGL. He is also the only five-time winner of the prestigious Golden Laparoscope Award for best surgical video from the AAGL.
A highly-regarded lecturer and surgeon, Dr. Lee has taught and performed live surgeries around the world.
Dr. Lee’s practice is entirely dedicated to minimally invasive surgical options for women. He is a firm believer that virtually all benign gynecologic surgical conditions should be treated using a minimally invasive approach. Dr. Lee’s clinical expertise includes minimally invasive surgery for treatments of endometriosis (including severe endometriosis involving bowel, bladder, and ureter); fibroids; abnormal uterine bleeding; urinary incontinence; and pelvic organ prolapse.
It is a great honor for the Master Class in Gynecologic Surgery to have Dr. Lee as guest author for this important area of our surgical arena.
Dr. Miller is a minimally invasive gynecologic surgeon in Naperville, Ill., and a past president of the AAGL.

Frankincense extract may reduce disease activity in relapsing-remitting MS
A standardized frankincense extract was safe, well tolerated, and potentially efficacious as an oral treatment in patients with relapsing-remitting multiple sclerosis (RRMS), according to results of a small, nonrandomized study.
Patients receiving the herbal treatment had significantly fewer contrast-enhancing lesions on MRI versus baseline in the study, which was published in the Journal of Neurology, Neurosurgery & Psychiatry.
“Despite our encouraging results, it is difficult to forecast the efficacy of a standardized frankincense extract in RRMS,” Dr. Stürner and her colleagues wrote in their report on the study.
Boswellic acids, believed to be the active compound in frankincense, has been used as an anti-inflammatory substance for thousands of years in Eastern medicine, according to the study authors.
Frankincense extracts were safe and well tolerated in several small, randomized trials including patients with inflammatory or autoimmune diseases, they noted.
Dr. Stürner and her colleagues tested a standardized frankincense extract in the SABA phase 2a trial, an investigator-initiated, open-label, pilot study including 38 patients with RRMS.
Patients underwent observation for 4 months, then were treated with the extract for up to 8 months plus an optional extension phase of up to 36 months. A total of 28 patients completed the initial treatment period, and 18 participated in the extension period, according to the study results.
The median number of monthly contrast-enhancing lesions was significantly reduced from 1.00 at baseline to 0.50 during the initial treatment period (P less than .0001). In addition, significantly less brain atrophy was noted after the treatment phase as compared with the baseline observation phase (P = .0081).
Adverse events, mainly infections or gastrointestinal symptoms, were mild (57.7%) or moderate (38.6%), investigators added.
Treatment significantly increased regulatory CD4-positive T cell markers and decreased interleukin-17A–producing CD8-positive T cells, according to results of mechanistic studies that were also reported.
Other anti-inflammatory drugs that have been licensed act broadly on the immune system, according to investigators, and so those agents require careful monitoring for side effects.
“The right balance between efficacy and safety profile becomes increasingly more important for a young patient population, who require long-term treatment,” Dr. Stürner and her coauthors wrote. “The results of the SABA trial are promising from this perspective.”
The study was funded in part by Alpinia Laudanum Institute, which supplied the standardized frankincense extract at no charge. Individual authors reported competing interests with Alpinia Laudanum, Biogen, Sanofi Genzyme, Novartis, Merck Serono, and others.
SOURCE: Stürner KH et al. J Neurol Neurosurg Psychiatry. 2018 Apr;89(4):330-8.
Results of the SABA phase 2a trial suggest that frankincense could be a new therapeutic agent for mildly disabled young patients with RRMS who require long-term treatment, according to Dr. Francesco Patti.
In the study, administration of standardized oral frankincense extract significantly reduced the median number and volume of contrast-enhancing lesions and the number of new T2 lesions. The treatment also increased brain parenchymal volume and reduced the annualized relapse rate. While disability scores remained unchanged, measures of function and quality of life improved.
The treatment effects also appeared to be durable, based on the extension phase results.
Blood and immunologic findings suggested that the treatment was not toxic and that it exerted immunomodulatory activity by reduction of IL-17–producing CD8-positive T cells, as well as anti-inflammatory properties through inhibitory effects on 5-lipo-oxygenase, microsomial prostaglandin E2 synthase-1, LL-37, and nuclear factor-kB activities.
“This mechanism of action, attributing a role of these enzymes in neuroinflammation, might offer a new therapeutic approach,” he concluded in his editorial.
Francesco Patti, MD , is with the Multiple Sclerosis Hub Center, University of Catania (Italy). These comments are derived from his editorial ( J Neurol Neurosurg Psychiatry. 2018;89:327 ). Dr. Patti declared no competing interests related to the editorial.
Results of the SABA phase 2a trial suggest that frankincense could be a new therapeutic agent for mildly disabled young patients with RRMS who require long-term treatment, according to Dr. Francesco Patti.
In the study, administration of standardized oral frankincense extract significantly reduced the median number and volume of contrast-enhancing lesions and the number of new T2 lesions. The treatment also increased brain parenchymal volume and reduced the annualized relapse rate. While disability scores remained unchanged, measures of function and quality of life improved.
The treatment effects also appeared to be durable, based on the extension phase results.
Blood and immunologic findings suggested that the treatment was not toxic and that it exerted immunomodulatory activity by reduction of IL-17–producing CD8-positive T cells, as well as anti-inflammatory properties through inhibitory effects on 5-lipo-oxygenase, microsomial prostaglandin E2 synthase-1, LL-37, and nuclear factor-kB activities.
“This mechanism of action, attributing a role of these enzymes in neuroinflammation, might offer a new therapeutic approach,” he concluded in his editorial.
Francesco Patti, MD , is with the Multiple Sclerosis Hub Center, University of Catania (Italy). These comments are derived from his editorial ( J Neurol Neurosurg Psychiatry. 2018;89:327 ). Dr. Patti declared no competing interests related to the editorial.
Results of the SABA phase 2a trial suggest that frankincense could be a new therapeutic agent for mildly disabled young patients with RRMS who require long-term treatment, according to Dr. Francesco Patti.
In the study, administration of standardized oral frankincense extract significantly reduced the median number and volume of contrast-enhancing lesions and the number of new T2 lesions. The treatment also increased brain parenchymal volume and reduced the annualized relapse rate. While disability scores remained unchanged, measures of function and quality of life improved.
The treatment effects also appeared to be durable, based on the extension phase results.
Blood and immunologic findings suggested that the treatment was not toxic and that it exerted immunomodulatory activity by reduction of IL-17–producing CD8-positive T cells, as well as anti-inflammatory properties through inhibitory effects on 5-lipo-oxygenase, microsomial prostaglandin E2 synthase-1, LL-37, and nuclear factor-kB activities.
“This mechanism of action, attributing a role of these enzymes in neuroinflammation, might offer a new therapeutic approach,” he concluded in his editorial.
Francesco Patti, MD , is with the Multiple Sclerosis Hub Center, University of Catania (Italy). These comments are derived from his editorial ( J Neurol Neurosurg Psychiatry. 2018;89:327 ). Dr. Patti declared no competing interests related to the editorial.
A standardized frankincense extract was safe, well tolerated, and potentially efficacious as an oral treatment in patients with relapsing-remitting multiple sclerosis (RRMS), according to results of a small, nonrandomized study.
Patients receiving the herbal treatment had significantly fewer contrast-enhancing lesions on MRI versus baseline in the study, which was published in the Journal of Neurology, Neurosurgery & Psychiatry.
“Despite our encouraging results, it is difficult to forecast the efficacy of a standardized frankincense extract in RRMS,” Dr. Stürner and her colleagues wrote in their report on the study.
Boswellic acids, believed to be the active compound in frankincense, has been used as an anti-inflammatory substance for thousands of years in Eastern medicine, according to the study authors.
Frankincense extracts were safe and well tolerated in several small, randomized trials including patients with inflammatory or autoimmune diseases, they noted.
Dr. Stürner and her colleagues tested a standardized frankincense extract in the SABA phase 2a trial, an investigator-initiated, open-label, pilot study including 38 patients with RRMS.
Patients underwent observation for 4 months, then were treated with the extract for up to 8 months plus an optional extension phase of up to 36 months. A total of 28 patients completed the initial treatment period, and 18 participated in the extension period, according to the study results.
The median number of monthly contrast-enhancing lesions was significantly reduced from 1.00 at baseline to 0.50 during the initial treatment period (P less than .0001). In addition, significantly less brain atrophy was noted after the treatment phase as compared with the baseline observation phase (P = .0081).
Adverse events, mainly infections or gastrointestinal symptoms, were mild (57.7%) or moderate (38.6%), investigators added.
Treatment significantly increased regulatory CD4-positive T cell markers and decreased interleukin-17A–producing CD8-positive T cells, according to results of mechanistic studies that were also reported.
Other anti-inflammatory drugs that have been licensed act broadly on the immune system, according to investigators, and so those agents require careful monitoring for side effects.
“The right balance between efficacy and safety profile becomes increasingly more important for a young patient population, who require long-term treatment,” Dr. Stürner and her coauthors wrote. “The results of the SABA trial are promising from this perspective.”
The study was funded in part by Alpinia Laudanum Institute, which supplied the standardized frankincense extract at no charge. Individual authors reported competing interests with Alpinia Laudanum, Biogen, Sanofi Genzyme, Novartis, Merck Serono, and others.
SOURCE: Stürner KH et al. J Neurol Neurosurg Psychiatry. 2018 Apr;89(4):330-8.
A standardized frankincense extract was safe, well tolerated, and potentially efficacious as an oral treatment in patients with relapsing-remitting multiple sclerosis (RRMS), according to results of a small, nonrandomized study.
Patients receiving the herbal treatment had significantly fewer contrast-enhancing lesions on MRI versus baseline in the study, which was published in the Journal of Neurology, Neurosurgery & Psychiatry.
“Despite our encouraging results, it is difficult to forecast the efficacy of a standardized frankincense extract in RRMS,” Dr. Stürner and her colleagues wrote in their report on the study.
Boswellic acids, believed to be the active compound in frankincense, has been used as an anti-inflammatory substance for thousands of years in Eastern medicine, according to the study authors.
Frankincense extracts were safe and well tolerated in several small, randomized trials including patients with inflammatory or autoimmune diseases, they noted.
Dr. Stürner and her colleagues tested a standardized frankincense extract in the SABA phase 2a trial, an investigator-initiated, open-label, pilot study including 38 patients with RRMS.
Patients underwent observation for 4 months, then were treated with the extract for up to 8 months plus an optional extension phase of up to 36 months. A total of 28 patients completed the initial treatment period, and 18 participated in the extension period, according to the study results.
The median number of monthly contrast-enhancing lesions was significantly reduced from 1.00 at baseline to 0.50 during the initial treatment period (P less than .0001). In addition, significantly less brain atrophy was noted after the treatment phase as compared with the baseline observation phase (P = .0081).
Adverse events, mainly infections or gastrointestinal symptoms, were mild (57.7%) or moderate (38.6%), investigators added.
Treatment significantly increased regulatory CD4-positive T cell markers and decreased interleukin-17A–producing CD8-positive T cells, according to results of mechanistic studies that were also reported.
Other anti-inflammatory drugs that have been licensed act broadly on the immune system, according to investigators, and so those agents require careful monitoring for side effects.
“The right balance between efficacy and safety profile becomes increasingly more important for a young patient population, who require long-term treatment,” Dr. Stürner and her coauthors wrote. “The results of the SABA trial are promising from this perspective.”
The study was funded in part by Alpinia Laudanum Institute, which supplied the standardized frankincense extract at no charge. Individual authors reported competing interests with Alpinia Laudanum, Biogen, Sanofi Genzyme, Novartis, Merck Serono, and others.
SOURCE: Stürner KH et al. J Neurol Neurosurg Psychiatry. 2018 Apr;89(4):330-8.
FROM Journal of Neurology, Neurosurgery & Psychiatry
Key clinical point:
Major finding: The median number of monthly contrast-enhancing lesions was significantly reduced from 1.00 at baseline to 0.50 on treatment (P less than .0001).
Study details: The SABA phase 2a trial, an investigator-initiated, open-label, pilot study including 38 patients with RRMS.
Disclosures: The study was funded in part by Alpinia Laudanum Institute, which supplied the standardized frankincense extract at no charge. Individual authors reported competing interests with Alpinia Laudanum, Biogen, Sanofi Genzyme, Novartis, Merck Serono, and others.
Source: Stürner KH et al. J Neurol Neurosurg Psychiatry. 2018;89:330-8.
Sexual harassment, violence is our problem, too
This past year has seen an incredible transformation in our awareness and understanding of the extent of sexual harassment and assault in our society. The #metoo campaign and the brave women throughout the country who have come out and told us their stories have truly made a difference.
For years, I, and many other pediatricians like me, have counseled teenage girls on how to stay safe as they prepare to enter college and universities. So many times, I have mentioned the shocking statistics on rape and sexual assault in college. I have cautioned these young women on how to stay safe, to stay close to their girlfriends, to not accept drinks from other people, and if drinking, not to drink so much that they don’t know what is going on around them … and so on and so forth with many dos and don’ts.
But here is something I only recently noticed: In the last 18 years of practice, my counsel to the boys was limited to how to stay safe, protect themselves against sexually transmitted infections, and how to avoid pregnancy. It did not cross my mind to counsel the teenage boys on their respective dos and don’ts, especially when it comes to their behavior with women. For example, stern advice on how to respect women, that only yes means yes, and frankly how to make sure they don’t become sexual harassers or worse.
I have questioned myself since then, wondering if I was alone with this oversight. I asked several other pediatricians as well to see if they had spoken about sexual harassment and assault in this context with their teenage male patients. The vast majority had the same experience as I did. They had all counseled the girls on how to protect themselves from becoming victims, but somehow not the boys (or girls for that matter) on how to help them not become the aggressors.
It has occurred to me that our focus as physicians has largely been limited to what the victims can do to not become victims.
As pediatricians, we counsel parents on how to keep their children safe, and how to protect them. It is our obligation to help, to guide, and to teach parents. We teach parents about discipline, limit setting, sleeping, feeding, and so many other things. We need to teach them how to talk to their boys and girls about appropriate behavior with whomever they may be interested in, about sexual harassment, and assault, and how not to become part of the problem.

It is our obligation as pediatricians to teach the children we see growing up in front of us how to behave in an adult, sexual world. We can make a bigger difference than we might think. As physicians, we promised to do no harm when we took our oath. It is now our turn also to take proactive steps to stop the harm caused by others.
Dr. Rimawi is a pediatrician in private practice in Atherton, Calif. Email her at [email protected].
This past year has seen an incredible transformation in our awareness and understanding of the extent of sexual harassment and assault in our society. The #metoo campaign and the brave women throughout the country who have come out and told us their stories have truly made a difference.
For years, I, and many other pediatricians like me, have counseled teenage girls on how to stay safe as they prepare to enter college and universities. So many times, I have mentioned the shocking statistics on rape and sexual assault in college. I have cautioned these young women on how to stay safe, to stay close to their girlfriends, to not accept drinks from other people, and if drinking, not to drink so much that they don’t know what is going on around them … and so on and so forth with many dos and don’ts.
But here is something I only recently noticed: In the last 18 years of practice, my counsel to the boys was limited to how to stay safe, protect themselves against sexually transmitted infections, and how to avoid pregnancy. It did not cross my mind to counsel the teenage boys on their respective dos and don’ts, especially when it comes to their behavior with women. For example, stern advice on how to respect women, that only yes means yes, and frankly how to make sure they don’t become sexual harassers or worse.
I have questioned myself since then, wondering if I was alone with this oversight. I asked several other pediatricians as well to see if they had spoken about sexual harassment and assault in this context with their teenage male patients. The vast majority had the same experience as I did. They had all counseled the girls on how to protect themselves from becoming victims, but somehow not the boys (or girls for that matter) on how to help them not become the aggressors.
It has occurred to me that our focus as physicians has largely been limited to what the victims can do to not become victims.
As pediatricians, we counsel parents on how to keep their children safe, and how to protect them. It is our obligation to help, to guide, and to teach parents. We teach parents about discipline, limit setting, sleeping, feeding, and so many other things. We need to teach them how to talk to their boys and girls about appropriate behavior with whomever they may be interested in, about sexual harassment, and assault, and how not to become part of the problem.
It is our obligation as pediatricians to teach the children we see growing up in front of us how to behave in an adult, sexual world. We can make a bigger difference than we might think. As physicians, we promised to do no harm when we took our oath. It is now our turn also to take proactive steps to stop the harm caused by others.
Dr. Rimawi is a pediatrician in private practice in Atherton, Calif. Email her at [email protected].
This past year has seen an incredible transformation in our awareness and understanding of the extent of sexual harassment and assault in our society. The #metoo campaign and the brave women throughout the country who have come out and told us their stories have truly made a difference.
For years, I, and many other pediatricians like me, have counseled teenage girls on how to stay safe as they prepare to enter college and universities. So many times, I have mentioned the shocking statistics on rape and sexual assault in college. I have cautioned these young women on how to stay safe, to stay close to their girlfriends, to not accept drinks from other people, and if drinking, not to drink so much that they don’t know what is going on around them … and so on and so forth with many dos and don’ts.
But here is something I only recently noticed: In the last 18 years of practice, my counsel to the boys was limited to how to stay safe, protect themselves against sexually transmitted infections, and how to avoid pregnancy. It did not cross my mind to counsel the teenage boys on their respective dos and don’ts, especially when it comes to their behavior with women. For example, stern advice on how to respect women, that only yes means yes, and frankly how to make sure they don’t become sexual harassers or worse.
I have questioned myself since then, wondering if I was alone with this oversight. I asked several other pediatricians as well to see if they had spoken about sexual harassment and assault in this context with their teenage male patients. The vast majority had the same experience as I did. They had all counseled the girls on how to protect themselves from becoming victims, but somehow not the boys (or girls for that matter) on how to help them not become the aggressors.
It has occurred to me that our focus as physicians has largely been limited to what the victims can do to not become victims.
As pediatricians, we counsel parents on how to keep their children safe, and how to protect them. It is our obligation to help, to guide, and to teach parents. We teach parents about discipline, limit setting, sleeping, feeding, and so many other things. We need to teach them how to talk to their boys and girls about appropriate behavior with whomever they may be interested in, about sexual harassment, and assault, and how not to become part of the problem.
It is our obligation as pediatricians to teach the children we see growing up in front of us how to behave in an adult, sexual world. We can make a bigger difference than we might think. As physicians, we promised to do no harm when we took our oath. It is now our turn also to take proactive steps to stop the harm caused by others.
Dr. Rimawi is a pediatrician in private practice in Atherton, Calif. Email her at [email protected].
Here’s one issue blue and red states agree on: Preventing deaths of expectant and new mothers
Alarmed that the U.S. is the most dangerous affluent country in which to give birth, state and local lawmakers around the country are adopting a flurry of bipartisan bills aimed at reforming how maternal deaths are identified and investigated.
In Indiana earlier this month, Republican Gov. Eric Holcomb signed a bill creating a maternal mortality review committee to scrutinize deaths and near-deaths among expectant and new mothers and make policy recommendations to improve maternal health.

Oregon’s governor and Washington, D.C.’s mayor, both Democrats, are expected to sign similar legislation in the coming days. Proposals are pending in Pennsylvania, Connecticut, Maryland and New Jersey.
Legislators from several of these states credited the ProPublica/NPR “Lost Mothers” series with raising their awareness and concern about the issue. Maryland Delegate Jheanelle Wilkins, who introduced a bill there, said that the series, especially articles looking at why black mothers are at greatest risk of dying and nearly dying, inspired her and her fellow lawmakers.
“A friend of mine posted one of the stories on Facebook and she challenged her elected officials — Who’s going to do something about it?” Wilkins said.
About 35 states have now established review committees or are in the process of doing so, as well as four cities: New York, Philadelphia, Baltimore and Washington. Two federal bills introduced last year, which would create a grant program to help states introduce or improve review committees, remain stalled in committee.
Between 700 and 900 women die each year in the U.S. from causes related to pregnancy or childbirth, and the rate has risen even as it has declined in other wealthy countries. The rate of life-threatening complications has also soared since the 1990s, endangering more than 50,000 U.S. women a year. A new report by the CDC Foundation — a nonprofit created by Congress to support the Centers for Disease Control and Prevention — suggests that more than 60 percent of pregnancy- and childbirth-related deaths in the U.S. are preventable.
The “Lost Mothers” project highlighted a number of reasons the U.S. has fallen behind other countries, including a greater focus on the health of the baby than of the mother, treatment guidelines that vary from one doctor or hospital to the next, and government failures to collect accurate data and to study maternal deaths and near-deaths to understand how they might be prevented.
Maternal mortality review committees can play a key role in this process, public health experts say, by identifying pregnancy-related deaths that might otherwise be overlooked, analyzing the factors contributing to those deaths, and translating the lessons into policy changes. That’s what happens in Great Britain, where a national committee investigates every maternal death and the findings help set women’s health policy across the country.
As recently as 2016, only about half the states had such panels. The number has been growing quickly, said Andria Cornell, senior program manager for women’s health and maternal health lead at the Association of Maternal & Child Health Programs, a nonprofit advocacy group.
“This is a time of unprecedented political and social will for establishing maternal mortality review committees,” she said. “We’ve definitely come to a tipping point.”
Cornell credited two forces for driving the change: journalism focused on maternal deaths and a national project led by AMCHP, the Centers for Disease Control and the CDC Foundation.
With money from Merck for Mothers, a charitable initiative created by the pharmaceutical giant, the project has funded a web portal that provides information on starting and improving review committees and a tool, the Maternal Mortality Review Information Application, that shows jurisdictions how to standardize data collection from review panels so that it’s comparable from one state to the next.
Review committees do have limitations. Many are understaffed and poorly funded, with limited authority to dig deeply into systemic problems or implement meaningful reforms. It generally takes several years for them to produce their reports, in part because committee members — including doctors, public health experts, medical examiners, and the like — have other demands on their time and aren’t compensated.
Also, committee records and reports are de-identified — stripped of any information that might point to a particular woman, caregiver or hospital. Thus the review is of little use in assigning responsibility for individual deaths, or evaluating whether some hospitals, doctors or nurses are especially prone to error. Still, recommendations and findings from reviews have proven helpful in states such as California in shaping preventive efforts that have reduced maternal mortality rates.
Indiana epitomizes the national movement to use the review committee process to scrutinize maternal deaths. There, the focus had long been on reducing infant mortality: The state has the highest rate of neonatal deaths outside the South. Maternal deaths weren’t on the radar, even though the state’s maternal mortality rate is around 41 women per 100,000 births, according to a new analysis of federal data by United Health Foundation — or double the rate of maternal deaths in neighboring Illinois and Ohio.
“I’ll be honest,” said state Sen. Jean Leising, a Republican from rural southeastern Indiana. “I’m on the Health Committee ... and I had no idea our maternal statistics were so lousy.”
The bill she sponsored — creating a committee for the next five years to study not just maternal mortality but also life-threatening complications, or severe maternal morbidity — sailed through the legislature, in part because of a change in governors. Holcomb, who replaced Mike Pence and is seen as more of a pragmatist, appointed a female obstetrician-gynecologist to be his new health commissioner.
The bill’s supporters drew a connection between maternal and infant mortality, said Dr. Brownsyne Tucker Edmonds, legislative chair for the Indiana chapter of the American Congress of Obstetricians and Gynecologists: “We could bring forth the idea that healthy moms have healthy babies.”
Oregon’s bill, which also passed easily this month, creates a review committee that will start by focusing on maternal deaths; by 2021, it will also begin looking at severe maternal morbidity. Over that period, it will cost the state more than $450,000 — a significant public commitment to a women’s health initiative.
“I did think, wow, that’s more money than I thought it was going to be, but no one blinked an eye,” said Rep. Alissa Keny-Guyer of Portland, the bill’s chief sponsor. “That just shows how much support this idea has.”
In the District of Columbia, concerns about the high maternal mortality rate — in 2014, it stood at about 40.7 deaths per 100,000 births, according to the analysis by United Health Foundation, substantially exceeding the U.S. rate and those of neighboring Virginia and Maryland — have periodically sparked talk of a review committee, but not enough to push a measure through.
Last year, after two hospitals in Northeast and predominantly black Southeast Washington closed maternity units, concerns grew over access to quality care, particularly for low-income and minority women. Nationally, black women have a maternal mortality rate three to four times higher than white women, and the District suspects its gap is even wider.
“Those disparities were the more acute driver of why we felt we needed to take this action,” said Councilmember Charles Allen, who introduced the measure to establish the panel. “You have to know what is driving this wide disparity before you can really have the strategies for how to fix it.”
The D.C. bill still must be signed into law and, like all District legislation, reviewed by Congress before it becomes effective. It calls for one full-time employee to assist the panel’s work, a position that Allen said he expects to be funded in the budget that will be passed later in the year. In addition to health care professionals, a social worker and representatives of community groups that specialize in women’s health, the D.C. committee will also include “one person who has been directly impacted by a maternal mortality or severe maternal morbidity.” Maternal health advocates say listening to such voices is a critical step in addressing how disparities in race, income and education affect outcomes.
That’s what prompted Wilkins, the Maryland delegate, to introduce her bill, which passed the House this month and will be taken up in the Senate in April. Maryland established its review committee in 2000, but in the panel’s most recent report, the participants consisted almost exclusively of medical professionals, mostly doctors and nurse-midwives. Wilkins’s bill would require the committee to meet at least twice a year with a group that includes representatives from the Maryland Office of Minority Health and Health Disparities, the Maryland Patient Safety Center, women’s health advocacy organizations, and a relative of a mother who died, and to incorporate their recommendations into its final report.
“The women who are impacted and the organizations that work with the communities they live in — we need to make sure they are at the table,” Wilkins said.
Other pending proposals would revamp New Jersey’s 80-plus-year-old review process and establish a new review committee in Connecticut.
Pennsylvania, which ranks sixth in the number of births in the U.S., is currently the largest state without a maternal mortality review committee, but lawmakers are advancing a measure to change that. It passed the House in December and recently cleared a Senate committee; it’s now headed for consideration by the full Senate.
State Rep. Ryan Mackenzie, R-Lehigh, introduced the bill last October after doctors from his district showed him grim data on rising maternal mortality rates in the nation and the state. Pennsylvania’s rate has more than doubled since 1994, according to a December 2017 report in the Pittsburgh Post-Gazette. When Mackenzie ran the idea of creating a statewide review process past House colleagues, several responded that media reports about maternal mortality, including our “Lost Mothers” series, had spurred them to consider similar measures.
Mackenzie’s bill calls for a committee of at least 14 members, most of them health care professionals, with a special emphasis on members working in communities most affected by maternal deaths and a lack of access to care. The measure does not include funding, and specifies that committee members would be unpaid, but Mackenzie said the state Department of Health would redirect existing staff to support it.
Another important aim in creating a statewide review process is making sure maternal deaths are being defined and tracked consistently, Mackenzie said. The state health department has tabulated annual totals for years, but counts only deaths that occur up to 42 days after pregnancy and not those that happen within a year, the standard used by the CDC and most review committees. When Philadelphia’s maternal mortality review panel compared the state’s numbers with its own for the city from 2010 to 2012, the state’s count was about 30 percent lower. Mackenzie’s bill would align Pennsylvania’s committee with the one-year standard.
“We’re hoping to save lives,” Mackenzie said in proposing the review committee. “Based on the results in other states, we think this is realistic.”
ProPublica is a Pulitzer Prize-winning investigative newsroom.
Alarmed that the U.S. is the most dangerous affluent country in which to give birth, state and local lawmakers around the country are adopting a flurry of bipartisan bills aimed at reforming how maternal deaths are identified and investigated.
In Indiana earlier this month, Republican Gov. Eric Holcomb signed a bill creating a maternal mortality review committee to scrutinize deaths and near-deaths among expectant and new mothers and make policy recommendations to improve maternal health.
Oregon’s governor and Washington, D.C.’s mayor, both Democrats, are expected to sign similar legislation in the coming days. Proposals are pending in Pennsylvania, Connecticut, Maryland and New Jersey.
Legislators from several of these states credited the ProPublica/NPR “Lost Mothers” series with raising their awareness and concern about the issue. Maryland Delegate Jheanelle Wilkins, who introduced a bill there, said that the series, especially articles looking at why black mothers are at greatest risk of dying and nearly dying, inspired her and her fellow lawmakers.
“A friend of mine posted one of the stories on Facebook and she challenged her elected officials — Who’s going to do something about it?” Wilkins said.
About 35 states have now established review committees or are in the process of doing so, as well as four cities: New York, Philadelphia, Baltimore and Washington. Two federal bills introduced last year, which would create a grant program to help states introduce or improve review committees, remain stalled in committee.
Between 700 and 900 women die each year in the U.S. from causes related to pregnancy or childbirth, and the rate has risen even as it has declined in other wealthy countries. The rate of life-threatening complications has also soared since the 1990s, endangering more than 50,000 U.S. women a year. A new report by the CDC Foundation — a nonprofit created by Congress to support the Centers for Disease Control and Prevention — suggests that more than 60 percent of pregnancy- and childbirth-related deaths in the U.S. are preventable.
The “Lost Mothers” project highlighted a number of reasons the U.S. has fallen behind other countries, including a greater focus on the health of the baby than of the mother, treatment guidelines that vary from one doctor or hospital to the next, and government failures to collect accurate data and to study maternal deaths and near-deaths to understand how they might be prevented.
Maternal mortality review committees can play a key role in this process, public health experts say, by identifying pregnancy-related deaths that might otherwise be overlooked, analyzing the factors contributing to those deaths, and translating the lessons into policy changes. That’s what happens in Great Britain, where a national committee investigates every maternal death and the findings help set women’s health policy across the country.
As recently as 2016, only about half the states had such panels. The number has been growing quickly, said Andria Cornell, senior program manager for women’s health and maternal health lead at the Association of Maternal & Child Health Programs, a nonprofit advocacy group.
“This is a time of unprecedented political and social will for establishing maternal mortality review committees,” she said. “We’ve definitely come to a tipping point.”
Cornell credited two forces for driving the change: journalism focused on maternal deaths and a national project led by AMCHP, the Centers for Disease Control and the CDC Foundation.
With money from Merck for Mothers, a charitable initiative created by the pharmaceutical giant, the project has funded a web portal that provides information on starting and improving review committees and a tool, the Maternal Mortality Review Information Application, that shows jurisdictions how to standardize data collection from review panels so that it’s comparable from one state to the next.
Review committees do have limitations. Many are understaffed and poorly funded, with limited authority to dig deeply into systemic problems or implement meaningful reforms. It generally takes several years for them to produce their reports, in part because committee members — including doctors, public health experts, medical examiners, and the like — have other demands on their time and aren’t compensated.
Also, committee records and reports are de-identified — stripped of any information that might point to a particular woman, caregiver or hospital. Thus the review is of little use in assigning responsibility for individual deaths, or evaluating whether some hospitals, doctors or nurses are especially prone to error. Still, recommendations and findings from reviews have proven helpful in states such as California in shaping preventive efforts that have reduced maternal mortality rates.
Indiana epitomizes the national movement to use the review committee process to scrutinize maternal deaths. There, the focus had long been on reducing infant mortality: The state has the highest rate of neonatal deaths outside the South. Maternal deaths weren’t on the radar, even though the state’s maternal mortality rate is around 41 women per 100,000 births, according to a new analysis of federal data by United Health Foundation — or double the rate of maternal deaths in neighboring Illinois and Ohio.
“I’ll be honest,” said state Sen. Jean Leising, a Republican from rural southeastern Indiana. “I’m on the Health Committee ... and I had no idea our maternal statistics were so lousy.”
The bill she sponsored — creating a committee for the next five years to study not just maternal mortality but also life-threatening complications, or severe maternal morbidity — sailed through the legislature, in part because of a change in governors. Holcomb, who replaced Mike Pence and is seen as more of a pragmatist, appointed a female obstetrician-gynecologist to be his new health commissioner.
The bill’s supporters drew a connection between maternal and infant mortality, said Dr. Brownsyne Tucker Edmonds, legislative chair for the Indiana chapter of the American Congress of Obstetricians and Gynecologists: “We could bring forth the idea that healthy moms have healthy babies.”
Oregon’s bill, which also passed easily this month, creates a review committee that will start by focusing on maternal deaths; by 2021, it will also begin looking at severe maternal morbidity. Over that period, it will cost the state more than $450,000 — a significant public commitment to a women’s health initiative.
“I did think, wow, that’s more money than I thought it was going to be, but no one blinked an eye,” said Rep. Alissa Keny-Guyer of Portland, the bill’s chief sponsor. “That just shows how much support this idea has.”
In the District of Columbia, concerns about the high maternal mortality rate — in 2014, it stood at about 40.7 deaths per 100,000 births, according to the analysis by United Health Foundation, substantially exceeding the U.S. rate and those of neighboring Virginia and Maryland — have periodically sparked talk of a review committee, but not enough to push a measure through.
Last year, after two hospitals in Northeast and predominantly black Southeast Washington closed maternity units, concerns grew over access to quality care, particularly for low-income and minority women. Nationally, black women have a maternal mortality rate three to four times higher than white women, and the District suspects its gap is even wider.
“Those disparities were the more acute driver of why we felt we needed to take this action,” said Councilmember Charles Allen, who introduced the measure to establish the panel. “You have to know what is driving this wide disparity before you can really have the strategies for how to fix it.”
The D.C. bill still must be signed into law and, like all District legislation, reviewed by Congress before it becomes effective. It calls for one full-time employee to assist the panel’s work, a position that Allen said he expects to be funded in the budget that will be passed later in the year. In addition to health care professionals, a social worker and representatives of community groups that specialize in women’s health, the D.C. committee will also include “one person who has been directly impacted by a maternal mortality or severe maternal morbidity.” Maternal health advocates say listening to such voices is a critical step in addressing how disparities in race, income and education affect outcomes.
That’s what prompted Wilkins, the Maryland delegate, to introduce her bill, which passed the House this month and will be taken up in the Senate in April. Maryland established its review committee in 2000, but in the panel’s most recent report, the participants consisted almost exclusively of medical professionals, mostly doctors and nurse-midwives. Wilkins’s bill would require the committee to meet at least twice a year with a group that includes representatives from the Maryland Office of Minority Health and Health Disparities, the Maryland Patient Safety Center, women’s health advocacy organizations, and a relative of a mother who died, and to incorporate their recommendations into its final report.
“The women who are impacted and the organizations that work with the communities they live in — we need to make sure they are at the table,” Wilkins said.
Other pending proposals would revamp New Jersey’s 80-plus-year-old review process and establish a new review committee in Connecticut.
Pennsylvania, which ranks sixth in the number of births in the U.S., is currently the largest state without a maternal mortality review committee, but lawmakers are advancing a measure to change that. It passed the House in December and recently cleared a Senate committee; it’s now headed for consideration by the full Senate.
State Rep. Ryan Mackenzie, R-Lehigh, introduced the bill last October after doctors from his district showed him grim data on rising maternal mortality rates in the nation and the state. Pennsylvania’s rate has more than doubled since 1994, according to a December 2017 report in the Pittsburgh Post-Gazette. When Mackenzie ran the idea of creating a statewide review process past House colleagues, several responded that media reports about maternal mortality, including our “Lost Mothers” series, had spurred them to consider similar measures.
Mackenzie’s bill calls for a committee of at least 14 members, most of them health care professionals, with a special emphasis on members working in communities most affected by maternal deaths and a lack of access to care. The measure does not include funding, and specifies that committee members would be unpaid, but Mackenzie said the state Department of Health would redirect existing staff to support it.
Another important aim in creating a statewide review process is making sure maternal deaths are being defined and tracked consistently, Mackenzie said. The state health department has tabulated annual totals for years, but counts only deaths that occur up to 42 days after pregnancy and not those that happen within a year, the standard used by the CDC and most review committees. When Philadelphia’s maternal mortality review panel compared the state’s numbers with its own for the city from 2010 to 2012, the state’s count was about 30 percent lower. Mackenzie’s bill would align Pennsylvania’s committee with the one-year standard.
“We’re hoping to save lives,” Mackenzie said in proposing the review committee. “Based on the results in other states, we think this is realistic.”
ProPublica is a Pulitzer Prize-winning investigative newsroom.
Alarmed that the U.S. is the most dangerous affluent country in which to give birth, state and local lawmakers around the country are adopting a flurry of bipartisan bills aimed at reforming how maternal deaths are identified and investigated.
In Indiana earlier this month, Republican Gov. Eric Holcomb signed a bill creating a maternal mortality review committee to scrutinize deaths and near-deaths among expectant and new mothers and make policy recommendations to improve maternal health.
Oregon’s governor and Washington, D.C.’s mayor, both Democrats, are expected to sign similar legislation in the coming days. Proposals are pending in Pennsylvania, Connecticut, Maryland and New Jersey.
Legislators from several of these states credited the ProPublica/NPR “Lost Mothers” series with raising their awareness and concern about the issue. Maryland Delegate Jheanelle Wilkins, who introduced a bill there, said that the series, especially articles looking at why black mothers are at greatest risk of dying and nearly dying, inspired her and her fellow lawmakers.
“A friend of mine posted one of the stories on Facebook and she challenged her elected officials — Who’s going to do something about it?” Wilkins said.
About 35 states have now established review committees or are in the process of doing so, as well as four cities: New York, Philadelphia, Baltimore and Washington. Two federal bills introduced last year, which would create a grant program to help states introduce or improve review committees, remain stalled in committee.
Between 700 and 900 women die each year in the U.S. from causes related to pregnancy or childbirth, and the rate has risen even as it has declined in other wealthy countries. The rate of life-threatening complications has also soared since the 1990s, endangering more than 50,000 U.S. women a year. A new report by the CDC Foundation — a nonprofit created by Congress to support the Centers for Disease Control and Prevention — suggests that more than 60 percent of pregnancy- and childbirth-related deaths in the U.S. are preventable.
The “Lost Mothers” project highlighted a number of reasons the U.S. has fallen behind other countries, including a greater focus on the health of the baby than of the mother, treatment guidelines that vary from one doctor or hospital to the next, and government failures to collect accurate data and to study maternal deaths and near-deaths to understand how they might be prevented.
Maternal mortality review committees can play a key role in this process, public health experts say, by identifying pregnancy-related deaths that might otherwise be overlooked, analyzing the factors contributing to those deaths, and translating the lessons into policy changes. That’s what happens in Great Britain, where a national committee investigates every maternal death and the findings help set women’s health policy across the country.
As recently as 2016, only about half the states had such panels. The number has been growing quickly, said Andria Cornell, senior program manager for women’s health and maternal health lead at the Association of Maternal & Child Health Programs, a nonprofit advocacy group.
“This is a time of unprecedented political and social will for establishing maternal mortality review committees,” she said. “We’ve definitely come to a tipping point.”
Cornell credited two forces for driving the change: journalism focused on maternal deaths and a national project led by AMCHP, the Centers for Disease Control and the CDC Foundation.
With money from Merck for Mothers, a charitable initiative created by the pharmaceutical giant, the project has funded a web portal that provides information on starting and improving review committees and a tool, the Maternal Mortality Review Information Application, that shows jurisdictions how to standardize data collection from review panels so that it’s comparable from one state to the next.
Review committees do have limitations. Many are understaffed and poorly funded, with limited authority to dig deeply into systemic problems or implement meaningful reforms. It generally takes several years for them to produce their reports, in part because committee members — including doctors, public health experts, medical examiners, and the like — have other demands on their time and aren’t compensated.
Also, committee records and reports are de-identified — stripped of any information that might point to a particular woman, caregiver or hospital. Thus the review is of little use in assigning responsibility for individual deaths, or evaluating whether some hospitals, doctors or nurses are especially prone to error. Still, recommendations and findings from reviews have proven helpful in states such as California in shaping preventive efforts that have reduced maternal mortality rates.
Indiana epitomizes the national movement to use the review committee process to scrutinize maternal deaths. There, the focus had long been on reducing infant mortality: The state has the highest rate of neonatal deaths outside the South. Maternal deaths weren’t on the radar, even though the state’s maternal mortality rate is around 41 women per 100,000 births, according to a new analysis of federal data by United Health Foundation — or double the rate of maternal deaths in neighboring Illinois and Ohio.
“I’ll be honest,” said state Sen. Jean Leising, a Republican from rural southeastern Indiana. “I’m on the Health Committee ... and I had no idea our maternal statistics were so lousy.”
The bill she sponsored — creating a committee for the next five years to study not just maternal mortality but also life-threatening complications, or severe maternal morbidity — sailed through the legislature, in part because of a change in governors. Holcomb, who replaced Mike Pence and is seen as more of a pragmatist, appointed a female obstetrician-gynecologist to be his new health commissioner.
The bill’s supporters drew a connection between maternal and infant mortality, said Dr. Brownsyne Tucker Edmonds, legislative chair for the Indiana chapter of the American Congress of Obstetricians and Gynecologists: “We could bring forth the idea that healthy moms have healthy babies.”
Oregon’s bill, which also passed easily this month, creates a review committee that will start by focusing on maternal deaths; by 2021, it will also begin looking at severe maternal morbidity. Over that period, it will cost the state more than $450,000 — a significant public commitment to a women’s health initiative.
“I did think, wow, that’s more money than I thought it was going to be, but no one blinked an eye,” said Rep. Alissa Keny-Guyer of Portland, the bill’s chief sponsor. “That just shows how much support this idea has.”
In the District of Columbia, concerns about the high maternal mortality rate — in 2014, it stood at about 40.7 deaths per 100,000 births, according to the analysis by United Health Foundation, substantially exceeding the U.S. rate and those of neighboring Virginia and Maryland — have periodically sparked talk of a review committee, but not enough to push a measure through.
Last year, after two hospitals in Northeast and predominantly black Southeast Washington closed maternity units, concerns grew over access to quality care, particularly for low-income and minority women. Nationally, black women have a maternal mortality rate three to four times higher than white women, and the District suspects its gap is even wider.
“Those disparities were the more acute driver of why we felt we needed to take this action,” said Councilmember Charles Allen, who introduced the measure to establish the panel. “You have to know what is driving this wide disparity before you can really have the strategies for how to fix it.”
The D.C. bill still must be signed into law and, like all District legislation, reviewed by Congress before it becomes effective. It calls for one full-time employee to assist the panel’s work, a position that Allen said he expects to be funded in the budget that will be passed later in the year. In addition to health care professionals, a social worker and representatives of community groups that specialize in women’s health, the D.C. committee will also include “one person who has been directly impacted by a maternal mortality or severe maternal morbidity.” Maternal health advocates say listening to such voices is a critical step in addressing how disparities in race, income and education affect outcomes.
That’s what prompted Wilkins, the Maryland delegate, to introduce her bill, which passed the House this month and will be taken up in the Senate in April. Maryland established its review committee in 2000, but in the panel’s most recent report, the participants consisted almost exclusively of medical professionals, mostly doctors and nurse-midwives. Wilkins’s bill would require the committee to meet at least twice a year with a group that includes representatives from the Maryland Office of Minority Health and Health Disparities, the Maryland Patient Safety Center, women’s health advocacy organizations, and a relative of a mother who died, and to incorporate their recommendations into its final report.
“The women who are impacted and the organizations that work with the communities they live in — we need to make sure they are at the table,” Wilkins said.
Other pending proposals would revamp New Jersey’s 80-plus-year-old review process and establish a new review committee in Connecticut.
Pennsylvania, which ranks sixth in the number of births in the U.S., is currently the largest state without a maternal mortality review committee, but lawmakers are advancing a measure to change that. It passed the House in December and recently cleared a Senate committee; it’s now headed for consideration by the full Senate.
State Rep. Ryan Mackenzie, R-Lehigh, introduced the bill last October after doctors from his district showed him grim data on rising maternal mortality rates in the nation and the state. Pennsylvania’s rate has more than doubled since 1994, according to a December 2017 report in the Pittsburgh Post-Gazette. When Mackenzie ran the idea of creating a statewide review process past House colleagues, several responded that media reports about maternal mortality, including our “Lost Mothers” series, had spurred them to consider similar measures.
Mackenzie’s bill calls for a committee of at least 14 members, most of them health care professionals, with a special emphasis on members working in communities most affected by maternal deaths and a lack of access to care. The measure does not include funding, and specifies that committee members would be unpaid, but Mackenzie said the state Department of Health would redirect existing staff to support it.
Another important aim in creating a statewide review process is making sure maternal deaths are being defined and tracked consistently, Mackenzie said. The state health department has tabulated annual totals for years, but counts only deaths that occur up to 42 days after pregnancy and not those that happen within a year, the standard used by the CDC and most review committees. When Philadelphia’s maternal mortality review panel compared the state’s numbers with its own for the city from 2010 to 2012, the state’s count was about 30 percent lower. Mackenzie’s bill would align Pennsylvania’s committee with the one-year standard.
“We’re hoping to save lives,” Mackenzie said in proposing the review committee. “Based on the results in other states, we think this is realistic.”
ProPublica is a Pulitzer Prize-winning investigative newsroom.
High-dose oral ibuprofen most effective for PDA closure
compared with other pharmacological treatments, according to results of a recent systematic review and meta-analysis.
However, using placebo or no treatment at all did not increase the odds of mortality, necrotizing enterocolitis, or intraventricular hemorrhage in the study, published in JAMA.
PDA is a common cardiovascular issue among prematurely born infants. According to Dr. Mitra and his coauthors, it’s thought that a large proportion of PDAs spontaneously close in a few days and have minimal effect on clinical outcomes.
As a result, treatment is often targeted to PDAs deemed hemodynamically significant based on clinical and echocardiographic parameters, the authors wrote, although there is little guidance on what, if any, treatment to use in this situation.
“The dilemma is whether to use pharmacotherapy at all, and if a decision is made to treat the PDA medically, what should be the ideal choice of pharmacotherapy,” they wrote.
Dr. Mitra and colleagues conducted a systematic review and meta-analysis of 68 randomized clinical trials including 4,802 infants with clinically or echocardiographically diagnosed, hemodynamically significant PDA.
They found that closure of hemodynamically significant PDA was significantly more likely with high-dose oral ibuprofen, compared with the two of the most widely used treatments, namely standard-dose intravenous ibuprofen (odds ratio, 3.59) and standard-dose intravenous indomethacin (odds ratio, 2.35).
Despite that finding, there was no significant difference in the odds of mortality, necrotizing enterocolitis, or intraventricular hemorrhage for use of placebo or no treatment, compared with any of the treatment modalities, the investigators added.
“With increasing emphasis on conservative management of PDA, these results may encourage researchers to revisit placebo-controlled trials against newer pharmacotherapeutic options,” they said.
Study authors reported no relevant potential conflicts of interest.
SOURCE: Mitra S et al. JAMA. 2018;319(12):1221-38.
compared with other pharmacological treatments, according to results of a recent systematic review and meta-analysis.
However, using placebo or no treatment at all did not increase the odds of mortality, necrotizing enterocolitis, or intraventricular hemorrhage in the study, published in JAMA.
PDA is a common cardiovascular issue among prematurely born infants. According to Dr. Mitra and his coauthors, it’s thought that a large proportion of PDAs spontaneously close in a few days and have minimal effect on clinical outcomes.
As a result, treatment is often targeted to PDAs deemed hemodynamically significant based on clinical and echocardiographic parameters, the authors wrote, although there is little guidance on what, if any, treatment to use in this situation.
“The dilemma is whether to use pharmacotherapy at all, and if a decision is made to treat the PDA medically, what should be the ideal choice of pharmacotherapy,” they wrote.
Dr. Mitra and colleagues conducted a systematic review and meta-analysis of 68 randomized clinical trials including 4,802 infants with clinically or echocardiographically diagnosed, hemodynamically significant PDA.
They found that closure of hemodynamically significant PDA was significantly more likely with high-dose oral ibuprofen, compared with the two of the most widely used treatments, namely standard-dose intravenous ibuprofen (odds ratio, 3.59) and standard-dose intravenous indomethacin (odds ratio, 2.35).
Despite that finding, there was no significant difference in the odds of mortality, necrotizing enterocolitis, or intraventricular hemorrhage for use of placebo or no treatment, compared with any of the treatment modalities, the investigators added.
“With increasing emphasis on conservative management of PDA, these results may encourage researchers to revisit placebo-controlled trials against newer pharmacotherapeutic options,” they said.
Study authors reported no relevant potential conflicts of interest.
SOURCE: Mitra S et al. JAMA. 2018;319(12):1221-38.
compared with other pharmacological treatments, according to results of a recent systematic review and meta-analysis.
However, using placebo or no treatment at all did not increase the odds of mortality, necrotizing enterocolitis, or intraventricular hemorrhage in the study, published in JAMA.
PDA is a common cardiovascular issue among prematurely born infants. According to Dr. Mitra and his coauthors, it’s thought that a large proportion of PDAs spontaneously close in a few days and have minimal effect on clinical outcomes.
As a result, treatment is often targeted to PDAs deemed hemodynamically significant based on clinical and echocardiographic parameters, the authors wrote, although there is little guidance on what, if any, treatment to use in this situation.
“The dilemma is whether to use pharmacotherapy at all, and if a decision is made to treat the PDA medically, what should be the ideal choice of pharmacotherapy,” they wrote.
Dr. Mitra and colleagues conducted a systematic review and meta-analysis of 68 randomized clinical trials including 4,802 infants with clinically or echocardiographically diagnosed, hemodynamically significant PDA.
They found that closure of hemodynamically significant PDA was significantly more likely with high-dose oral ibuprofen, compared with the two of the most widely used treatments, namely standard-dose intravenous ibuprofen (odds ratio, 3.59) and standard-dose intravenous indomethacin (odds ratio, 2.35).
Despite that finding, there was no significant difference in the odds of mortality, necrotizing enterocolitis, or intraventricular hemorrhage for use of placebo or no treatment, compared with any of the treatment modalities, the investigators added.
“With increasing emphasis on conservative management of PDA, these results may encourage researchers to revisit placebo-controlled trials against newer pharmacotherapeutic options,” they said.
Study authors reported no relevant potential conflicts of interest.
SOURCE: Mitra S et al. JAMA. 2018;319(12):1221-38.
FROM JAMA
Key clinical point: Compared with other pharmacological treatments, high-dose oral ibuprofen may be most likely to result in closure of hemodynamically significant PDA in preterm infants.
Major finding: Closure of hemodynamically significant PDA was significantly more likely with high-dose oral ibuprofen, compared with standard-dose intravenous ibuprofen (odds ratio, 3.59) and intravenous indomethacin (odds ratio, 2.35).
Study details: A systematic review and meta-analysis of 68 randomized clinical trials including 4,802 infants with clinically or echocardiographically diagnosed, hemodynamically significant PDA.
Disclosures: Study authors reported no relevant potential conflicts of interest.
Source: Mitra S et al. JAMA. 2018;319(12):1221-38.