User login
Commentary: Composite risk, not age, is key for timing first colorectal cancer screening
The American Cancer Society’s recent recommendation to lower the age of first screening for colorectal cancer to 45 years does not reflect clear knowledge of risks versus benefits, experts wrote in a recent commentary.

“In the big picture, [the question of whether to start screening at 45 versus 50 years] seems relatively unimportant compared with using individual patient risk for advanced neoplasia in practical, feasible models” that are geared toward adherence, efficiency, and cost-efficacy, wrote Thomas F. Imperiale, MD, of Indiana University, Indianapolis, and his associates. The commentary is in the October issue of Clinical Gastroenterology and Hepatology.
Tailoring age of first screening on an individual level, based on other risk factors and patient preferences, might improve uptake and benefit-risk ratios, balance, they argued.
Rates of colorectal cancer in persons under age 50 years rose by about 22% between 2000 and 2013. However, estimates for the most recent birth cohorts have wide confidence intervals, “indicating imprecision and uncertainty that this trend will continue,” the experts wrote. Furthermore, the absolute risk of colorectal cancer among individuals younger than 50 years has risen only slightly, from 5.9 cases per 100,000 population to 7.2 cases per 100,000 population. “[This] small increase in incidence may represent a true increase or could be due to increased use of colonoscopy in general, and specifically, for diagnosis or high-risk screening of first-degree relatives of persons with colorectal cancer,” the experts wrote.
Implementing the new recommendation could detect earlier-stage (curable) colorectal cancer “in a youthful and productive age group that may be sandwiched between raising children and caring for aging parents,” they continued. Earlier detection could reduce mortality and reduce the costs of treating a disease that often exceeds $100,000 per person annually.
However, the recommendation was based on a modeling study that assumed 100% adherence. In reality, uptake among 45- to 49-year-olds might be 15%-20%, and “who actually shows up for screening could make or break this recommendation,” the experts said. If younger individuals who underwent screening tended to have few risk factors for colorectal cancer, then the new recommendation would lead to many false positives and unnecessary colonoscopies, with the associated fallout of emotional harm and wasted health care resources, they added.
Population-level studies have identified age as the strongest predictor of colorectal cancer, but age “does not perform as well” at patient level, the experts said. They emphasized the role of other risk factors, such as male sex, having a first-degree relative with colorectal cancer, high body mass index, metabolic syndrome, cigarette smoking, diet, adherence to screening, and use of aspirin, nonsteroidal anti-inflammatory drugs, and hormone therapy. “The goal for providers and health systems is to determine whether and how to change screening practice and policy, and how to incorporate this new recommendation into practice, a necessarily complex process that requires knowing patient risk, patient preferences, and the long-term balance of benefits and burdens,” they concluded (Clin Gastroenterol Hepatol. 2018 Aug 13. doi: 10.1016/j.cgh.2018.08.023).
Dr. Imperiale and coauthor Charles J. Kahi, MD, MS, had no disclosures. Coauthor Douglas K. Rex, MD, disclosed ties to Aries Pharmaceutical, Cosmo Pharmaceuticals, Boston Scientific, Sebela, Medtronic, EndoAid Ltd, Olympus, Paion, Braintree, and Medivators. He also chairs the U.S. Multi-Society Task Force on Colorectal Cancer.
* This story was updated on 10/16/2018.
The American Cancer Society’s recent recommendation to lower the age of first screening for colorectal cancer to 45 years does not reflect clear knowledge of risks versus benefits, experts wrote in a recent commentary.
“In the big picture, [the question of whether to start screening at 45 versus 50 years] seems relatively unimportant compared with using individual patient risk for advanced neoplasia in practical, feasible models” that are geared toward adherence, efficiency, and cost-efficacy, wrote Thomas F. Imperiale, MD, of Indiana University, Indianapolis, and his associates. The commentary is in the October issue of Clinical Gastroenterology and Hepatology.
Tailoring age of first screening on an individual level, based on other risk factors and patient preferences, might improve uptake and benefit-risk ratios, balance, they argued.
Rates of colorectal cancer in persons under age 50 years rose by about 22% between 2000 and 2013. However, estimates for the most recent birth cohorts have wide confidence intervals, “indicating imprecision and uncertainty that this trend will continue,” the experts wrote. Furthermore, the absolute risk of colorectal cancer among individuals younger than 50 years has risen only slightly, from 5.9 cases per 100,000 population to 7.2 cases per 100,000 population. “[This] small increase in incidence may represent a true increase or could be due to increased use of colonoscopy in general, and specifically, for diagnosis or high-risk screening of first-degree relatives of persons with colorectal cancer,” the experts wrote.
Implementing the new recommendation could detect earlier-stage (curable) colorectal cancer “in a youthful and productive age group that may be sandwiched between raising children and caring for aging parents,” they continued. Earlier detection could reduce mortality and reduce the costs of treating a disease that often exceeds $100,000 per person annually.
However, the recommendation was based on a modeling study that assumed 100% adherence. In reality, uptake among 45- to 49-year-olds might be 15%-20%, and “who actually shows up for screening could make or break this recommendation,” the experts said. If younger individuals who underwent screening tended to have few risk factors for colorectal cancer, then the new recommendation would lead to many false positives and unnecessary colonoscopies, with the associated fallout of emotional harm and wasted health care resources, they added.
Population-level studies have identified age as the strongest predictor of colorectal cancer, but age “does not perform as well” at patient level, the experts said. They emphasized the role of other risk factors, such as male sex, having a first-degree relative with colorectal cancer, high body mass index, metabolic syndrome, cigarette smoking, diet, adherence to screening, and use of aspirin, nonsteroidal anti-inflammatory drugs, and hormone therapy. “The goal for providers and health systems is to determine whether and how to change screening practice and policy, and how to incorporate this new recommendation into practice, a necessarily complex process that requires knowing patient risk, patient preferences, and the long-term balance of benefits and burdens,” they concluded (Clin Gastroenterol Hepatol. 2018 Aug 13. doi: 10.1016/j.cgh.2018.08.023).
Dr. Imperiale and coauthor Charles J. Kahi, MD, MS, had no disclosures. Coauthor Douglas K. Rex, MD, disclosed ties to Aries Pharmaceutical, Cosmo Pharmaceuticals, Boston Scientific, Sebela, Medtronic, EndoAid Ltd, Olympus, Paion, Braintree, and Medivators. He also chairs the U.S. Multi-Society Task Force on Colorectal Cancer.
* This story was updated on 10/16/2018.
The American Cancer Society’s recent recommendation to lower the age of first screening for colorectal cancer to 45 years does not reflect clear knowledge of risks versus benefits, experts wrote in a recent commentary.
“In the big picture, [the question of whether to start screening at 45 versus 50 years] seems relatively unimportant compared with using individual patient risk for advanced neoplasia in practical, feasible models” that are geared toward adherence, efficiency, and cost-efficacy, wrote Thomas F. Imperiale, MD, of Indiana University, Indianapolis, and his associates. The commentary is in the October issue of Clinical Gastroenterology and Hepatology.
Tailoring age of first screening on an individual level, based on other risk factors and patient preferences, might improve uptake and benefit-risk ratios, balance, they argued.
Rates of colorectal cancer in persons under age 50 years rose by about 22% between 2000 and 2013. However, estimates for the most recent birth cohorts have wide confidence intervals, “indicating imprecision and uncertainty that this trend will continue,” the experts wrote. Furthermore, the absolute risk of colorectal cancer among individuals younger than 50 years has risen only slightly, from 5.9 cases per 100,000 population to 7.2 cases per 100,000 population. “[This] small increase in incidence may represent a true increase or could be due to increased use of colonoscopy in general, and specifically, for diagnosis or high-risk screening of first-degree relatives of persons with colorectal cancer,” the experts wrote.
Implementing the new recommendation could detect earlier-stage (curable) colorectal cancer “in a youthful and productive age group that may be sandwiched between raising children and caring for aging parents,” they continued. Earlier detection could reduce mortality and reduce the costs of treating a disease that often exceeds $100,000 per person annually.
However, the recommendation was based on a modeling study that assumed 100% adherence. In reality, uptake among 45- to 49-year-olds might be 15%-20%, and “who actually shows up for screening could make or break this recommendation,” the experts said. If younger individuals who underwent screening tended to have few risk factors for colorectal cancer, then the new recommendation would lead to many false positives and unnecessary colonoscopies, with the associated fallout of emotional harm and wasted health care resources, they added.
Population-level studies have identified age as the strongest predictor of colorectal cancer, but age “does not perform as well” at patient level, the experts said. They emphasized the role of other risk factors, such as male sex, having a first-degree relative with colorectal cancer, high body mass index, metabolic syndrome, cigarette smoking, diet, adherence to screening, and use of aspirin, nonsteroidal anti-inflammatory drugs, and hormone therapy. “The goal for providers and health systems is to determine whether and how to change screening practice and policy, and how to incorporate this new recommendation into practice, a necessarily complex process that requires knowing patient risk, patient preferences, and the long-term balance of benefits and burdens,” they concluded (Clin Gastroenterol Hepatol. 2018 Aug 13. doi: 10.1016/j.cgh.2018.08.023).
Dr. Imperiale and coauthor Charles J. Kahi, MD, MS, had no disclosures. Coauthor Douglas K. Rex, MD, disclosed ties to Aries Pharmaceutical, Cosmo Pharmaceuticals, Boston Scientific, Sebela, Medtronic, EndoAid Ltd, Olympus, Paion, Braintree, and Medivators. He also chairs the U.S. Multi-Society Task Force on Colorectal Cancer.
* This story was updated on 10/16/2018.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Proinflammatory diet linked to colorectal cancer testing positive for Fusobacterium nucleatum
Diets promoting colonic inflammation were associated with a greater risk of colorectal carcinomas containing Fusobacterium nucleatum bacteria, according to a report in the October issue of Clinical Gastroenterology and Hepatology.
Courtesy American Gastroenterological Association
Proinflammatory diets were not linked to heightened risk for colon cancers without these bacteria, reported Li Liu, MD, PhD, of Dana-Farber Cancer Institute and Harvard Medical School, Boston. “These findings indicate that diet-induced intestinal inflammation alters the gut microbiome to contribute to colorectal carcinogenesis,” they wrote. “Nutritional interventions might be used in precision medicine and cancer prevention.”
Intestinal inflammation, a risk factor for colorectal cancer, is associated with high levels of circulating interleukin 6, C-reactive protein, and tumor necrosis factor–receptor superfamily member 1B. Colonic inflammation impairs the mucosal barrier and alters immune cell responses, which affects the composition of colonic microbiota. Among these, F. nucleatum is known to potentiate colorectal tumors and is associated with proximal tumor location, other tumor features, and cancer progression and chemoresistance.
For the study, the investigators examined self-reported data from more than 124,000 individuals followed for 28 years as part of the Nurses’ Health Study and the Health Professionals Follow-Up Study. They calculated average dietary patterns based on the empiric dietary inflammatory pattern (EDIP) score, which sums weighted intake scores for 18 foods (such as red and processed meat, coffee, tea, and leafy green or dark yellow vegetables) that are known to affect plasma levels of interleukin 6, C-reactive protein, tumor necrosis factor–receptor superfamily member 1B, and tumor necrosis factor alpha–receptor 2. A higher EDIP score denotes a more inflammatory diet.
During the 28-year follow-up period, 951 individuals developed colorectal carcinomas that were tested with a polymerase chain reaction assay for F. nucleatum DNA. A total of 115 tumors tested positive for F. nucleatum. After the researchers controlled for potential confounders, individuals whose EDIP scores were in the highest tertile were significantly more likely to develop F. nucleatum–positive colorectal cancer than were those who scored in the lowest tertile (adjusted hazard ratio, 1.63; 95% confidence interval, 1.03 to 2.58; P = .03). This differential association “appeared to be stronger in proximal colon cancer than in distal colon and rectal cancer,” the researchers said.
More than 90% of individuals in this study were non-Hispanic white, the researchers noted. Tumor tissue was not available from all cases of colorectal cancer and a fairly small number of cases tested positive for tumor F. nucleatum. Nonetheless, the findings suggest that an inflammatory diet could help amplify gut microbiota involved in tumorigenesis, they said. Pending confirmatory studies, they recommended an anti-inflammatory diet with high intake of green leafy vegetables, dark yellow vegetables, coffee, and tea, and with low intake of red meat, processed meat, refined grain, and sugary beverages. They also recommended studying whether F. nucleatum tumor or stool tests could help personalize dietary interventions.
Funders included the National Institutes of Health, Dana Farber Harvard Cancer, Project P. Fund for Colorectal Cancer Research, Friends of the Dana-Farber Cancer Institute, Bennett Family Fund, the Entertainment Industry Foundation, and American Association for Cancer Research, National Natural Science Foundation of China, Chinese Scholarship Council, Huazhong University of Science and Technology, and others. Dr. Liu had no disclosures. One coinvestigator disclosed ties to Genentech/Roche, Lilly, Sanofi, Bayer, and several other biomedical companies.
SOURCE: Liu L et al. Clin Gastroenterol Hepatol. 2018 Apr 24. doi: 10.1016/j.cgh.2018.04.030.
The underlying reasons colorectal cancer (CRC) develops are unknown, but they likely include a complex interaction between genetics and environmental exposures. Recent studies have highlighted important links between diet, the intestinal microbiota, and CRC development and progression.

Liu et al. used the Nurses’ Health Study and Health Professionals Follow-Up Study cohorts to extend our understanding of the relationship between diet, the intestinal microbiota, and CRC. They utilized validated food frequency questionnaires obtained every 4 years and formalin-fixed paraffin embedded CRC tissue samples collected from 951 individuals. They calculated an empiric dietary inflammatory pattern (EDIP) score, which correlates components of the diet with plasma inflammatory markers. After adjusting for confounders, they found high EDIP scores were significantly associated with Fusobacterium nucleatum–positive CRC, but not with F. nucleatum–negative CRC. In addition, they demonstrated this association was stronger for proximal compared with distal CRC. Their findings suggest an inflammatory diet may interact with the intestinal microbiota to promote the development of CRC and they provide a preliminary recommendation to minimize intake of potentially harmful foods (i.e., red meat, processed meat, refined grains, etc.). Despite the intriguing results, the authors do recognize limitations including the small number of cases with F. nucleatum present (n = 115) and the homogeneous cohort (90% non-Hispanic whites), which may limit generalizability.
As clinicians, we should continue strongly advocating for CRC screening and, based on these findings, may consider dietary recommendations to reduce intake of potentially harmful foods. Further research will be needed to confirm these findings in additional cohorts and to clarify the molecular interactions between dietary components, intestinal microbiota, and development of CRC.
Rajesh R. Shah, MD, is assistant professor of gastroenterology, department of internal medicine, Baylor College of Medicine, Houston. He has no conflicts of interest.
The underlying reasons colorectal cancer (CRC) develops are unknown, but they likely include a complex interaction between genetics and environmental exposures. Recent studies have highlighted important links between diet, the intestinal microbiota, and CRC development and progression.
Liu et al. used the Nurses’ Health Study and Health Professionals Follow-Up Study cohorts to extend our understanding of the relationship between diet, the intestinal microbiota, and CRC. They utilized validated food frequency questionnaires obtained every 4 years and formalin-fixed paraffin embedded CRC tissue samples collected from 951 individuals. They calculated an empiric dietary inflammatory pattern (EDIP) score, which correlates components of the diet with plasma inflammatory markers. After adjusting for confounders, they found high EDIP scores were significantly associated with Fusobacterium nucleatum–positive CRC, but not with F. nucleatum–negative CRC. In addition, they demonstrated this association was stronger for proximal compared with distal CRC. Their findings suggest an inflammatory diet may interact with the intestinal microbiota to promote the development of CRC and they provide a preliminary recommendation to minimize intake of potentially harmful foods (i.e., red meat, processed meat, refined grains, etc.). Despite the intriguing results, the authors do recognize limitations including the small number of cases with F. nucleatum present (n = 115) and the homogeneous cohort (90% non-Hispanic whites), which may limit generalizability.
As clinicians, we should continue strongly advocating for CRC screening and, based on these findings, may consider dietary recommendations to reduce intake of potentially harmful foods. Further research will be needed to confirm these findings in additional cohorts and to clarify the molecular interactions between dietary components, intestinal microbiota, and development of CRC.
Rajesh R. Shah, MD, is assistant professor of gastroenterology, department of internal medicine, Baylor College of Medicine, Houston. He has no conflicts of interest.
The underlying reasons colorectal cancer (CRC) develops are unknown, but they likely include a complex interaction between genetics and environmental exposures. Recent studies have highlighted important links between diet, the intestinal microbiota, and CRC development and progression.
Liu et al. used the Nurses’ Health Study and Health Professionals Follow-Up Study cohorts to extend our understanding of the relationship between diet, the intestinal microbiota, and CRC. They utilized validated food frequency questionnaires obtained every 4 years and formalin-fixed paraffin embedded CRC tissue samples collected from 951 individuals. They calculated an empiric dietary inflammatory pattern (EDIP) score, which correlates components of the diet with plasma inflammatory markers. After adjusting for confounders, they found high EDIP scores were significantly associated with Fusobacterium nucleatum–positive CRC, but not with F. nucleatum–negative CRC. In addition, they demonstrated this association was stronger for proximal compared with distal CRC. Their findings suggest an inflammatory diet may interact with the intestinal microbiota to promote the development of CRC and they provide a preliminary recommendation to minimize intake of potentially harmful foods (i.e., red meat, processed meat, refined grains, etc.). Despite the intriguing results, the authors do recognize limitations including the small number of cases with F. nucleatum present (n = 115) and the homogeneous cohort (90% non-Hispanic whites), which may limit generalizability.
As clinicians, we should continue strongly advocating for CRC screening and, based on these findings, may consider dietary recommendations to reduce intake of potentially harmful foods. Further research will be needed to confirm these findings in additional cohorts and to clarify the molecular interactions between dietary components, intestinal microbiota, and development of CRC.
Rajesh R. Shah, MD, is assistant professor of gastroenterology, department of internal medicine, Baylor College of Medicine, Houston. He has no conflicts of interest.
Diets promoting colonic inflammation were associated with a greater risk of colorectal carcinomas containing Fusobacterium nucleatum bacteria, according to a report in the October issue of Clinical Gastroenterology and Hepatology.
Courtesy American Gastroenterological Association
Proinflammatory diets were not linked to heightened risk for colon cancers without these bacteria, reported Li Liu, MD, PhD, of Dana-Farber Cancer Institute and Harvard Medical School, Boston. “These findings indicate that diet-induced intestinal inflammation alters the gut microbiome to contribute to colorectal carcinogenesis,” they wrote. “Nutritional interventions might be used in precision medicine and cancer prevention.”
Intestinal inflammation, a risk factor for colorectal cancer, is associated with high levels of circulating interleukin 6, C-reactive protein, and tumor necrosis factor–receptor superfamily member 1B. Colonic inflammation impairs the mucosal barrier and alters immune cell responses, which affects the composition of colonic microbiota. Among these, F. nucleatum is known to potentiate colorectal tumors and is associated with proximal tumor location, other tumor features, and cancer progression and chemoresistance.
For the study, the investigators examined self-reported data from more than 124,000 individuals followed for 28 years as part of the Nurses’ Health Study and the Health Professionals Follow-Up Study. They calculated average dietary patterns based on the empiric dietary inflammatory pattern (EDIP) score, which sums weighted intake scores for 18 foods (such as red and processed meat, coffee, tea, and leafy green or dark yellow vegetables) that are known to affect plasma levels of interleukin 6, C-reactive protein, tumor necrosis factor–receptor superfamily member 1B, and tumor necrosis factor alpha–receptor 2. A higher EDIP score denotes a more inflammatory diet.
During the 28-year follow-up period, 951 individuals developed colorectal carcinomas that were tested with a polymerase chain reaction assay for F. nucleatum DNA. A total of 115 tumors tested positive for F. nucleatum. After the researchers controlled for potential confounders, individuals whose EDIP scores were in the highest tertile were significantly more likely to develop F. nucleatum–positive colorectal cancer than were those who scored in the lowest tertile (adjusted hazard ratio, 1.63; 95% confidence interval, 1.03 to 2.58; P = .03). This differential association “appeared to be stronger in proximal colon cancer than in distal colon and rectal cancer,” the researchers said.
More than 90% of individuals in this study were non-Hispanic white, the researchers noted. Tumor tissue was not available from all cases of colorectal cancer and a fairly small number of cases tested positive for tumor F. nucleatum. Nonetheless, the findings suggest that an inflammatory diet could help amplify gut microbiota involved in tumorigenesis, they said. Pending confirmatory studies, they recommended an anti-inflammatory diet with high intake of green leafy vegetables, dark yellow vegetables, coffee, and tea, and with low intake of red meat, processed meat, refined grain, and sugary beverages. They also recommended studying whether F. nucleatum tumor or stool tests could help personalize dietary interventions.
Funders included the National Institutes of Health, Dana Farber Harvard Cancer, Project P. Fund for Colorectal Cancer Research, Friends of the Dana-Farber Cancer Institute, Bennett Family Fund, the Entertainment Industry Foundation, and American Association for Cancer Research, National Natural Science Foundation of China, Chinese Scholarship Council, Huazhong University of Science and Technology, and others. Dr. Liu had no disclosures. One coinvestigator disclosed ties to Genentech/Roche, Lilly, Sanofi, Bayer, and several other biomedical companies.
SOURCE: Liu L et al. Clin Gastroenterol Hepatol. 2018 Apr 24. doi: 10.1016/j.cgh.2018.04.030.
Diets promoting colonic inflammation were associated with a greater risk of colorectal carcinomas containing Fusobacterium nucleatum bacteria, according to a report in the October issue of Clinical Gastroenterology and Hepatology.
Courtesy American Gastroenterological Association
Proinflammatory diets were not linked to heightened risk for colon cancers without these bacteria, reported Li Liu, MD, PhD, of Dana-Farber Cancer Institute and Harvard Medical School, Boston. “These findings indicate that diet-induced intestinal inflammation alters the gut microbiome to contribute to colorectal carcinogenesis,” they wrote. “Nutritional interventions might be used in precision medicine and cancer prevention.”
Intestinal inflammation, a risk factor for colorectal cancer, is associated with high levels of circulating interleukin 6, C-reactive protein, and tumor necrosis factor–receptor superfamily member 1B. Colonic inflammation impairs the mucosal barrier and alters immune cell responses, which affects the composition of colonic microbiota. Among these, F. nucleatum is known to potentiate colorectal tumors and is associated with proximal tumor location, other tumor features, and cancer progression and chemoresistance.
For the study, the investigators examined self-reported data from more than 124,000 individuals followed for 28 years as part of the Nurses’ Health Study and the Health Professionals Follow-Up Study. They calculated average dietary patterns based on the empiric dietary inflammatory pattern (EDIP) score, which sums weighted intake scores for 18 foods (such as red and processed meat, coffee, tea, and leafy green or dark yellow vegetables) that are known to affect plasma levels of interleukin 6, C-reactive protein, tumor necrosis factor–receptor superfamily member 1B, and tumor necrosis factor alpha–receptor 2. A higher EDIP score denotes a more inflammatory diet.
During the 28-year follow-up period, 951 individuals developed colorectal carcinomas that were tested with a polymerase chain reaction assay for F. nucleatum DNA. A total of 115 tumors tested positive for F. nucleatum. After the researchers controlled for potential confounders, individuals whose EDIP scores were in the highest tertile were significantly more likely to develop F. nucleatum–positive colorectal cancer than were those who scored in the lowest tertile (adjusted hazard ratio, 1.63; 95% confidence interval, 1.03 to 2.58; P = .03). This differential association “appeared to be stronger in proximal colon cancer than in distal colon and rectal cancer,” the researchers said.
More than 90% of individuals in this study were non-Hispanic white, the researchers noted. Tumor tissue was not available from all cases of colorectal cancer and a fairly small number of cases tested positive for tumor F. nucleatum. Nonetheless, the findings suggest that an inflammatory diet could help amplify gut microbiota involved in tumorigenesis, they said. Pending confirmatory studies, they recommended an anti-inflammatory diet with high intake of green leafy vegetables, dark yellow vegetables, coffee, and tea, and with low intake of red meat, processed meat, refined grain, and sugary beverages. They also recommended studying whether F. nucleatum tumor or stool tests could help personalize dietary interventions.
Funders included the National Institutes of Health, Dana Farber Harvard Cancer, Project P. Fund for Colorectal Cancer Research, Friends of the Dana-Farber Cancer Institute, Bennett Family Fund, the Entertainment Industry Foundation, and American Association for Cancer Research, National Natural Science Foundation of China, Chinese Scholarship Council, Huazhong University of Science and Technology, and others. Dr. Liu had no disclosures. One coinvestigator disclosed ties to Genentech/Roche, Lilly, Sanofi, Bayer, and several other biomedical companies.
SOURCE: Liu L et al. Clin Gastroenterol Hepatol. 2018 Apr 24. doi: 10.1016/j.cgh.2018.04.030.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: A proinflammatory diet was associated with a significantly increased risk for colorectal cancer testing positive for Fusobacterium nucleatum.
Major finding: Dietary scores in the highest inflammatory tertile correlated with significantly increased risk (HR, 1.63; P = .03).
Study details: Longitudinal study of self-reported dietary patterns and cancers among 124,433 individuals with 28 years of follow-up.
Disclosures: Funders included the National Institutes of Health, Dana Farber Harvard Cancer, Project P. Fund for Colorectal Cancer Research, Friends of the Dana-Farber Cancer Institute, Bennett Family Fund, the Entertainment Industry Foundation, and American Association for Cancer Research, National Natural Science Foundation of China, Chinese Scholarship Council, Huazhong University of Science and Technology, and others. Dr. Liu had no disclosures. One coinvestigator disclosed ties to Genentech/Roche, Lilly, Sanofi, Bayer, and several other biomedical companies.
Source: Liu L et al. Clin Gastroenterol Hepatol. 2018 Apr 24. doi: 10.1016/j.cgh.2018.04.030.

Cirrhosis study finds no link between screening, liver cancer mortality
In a case-control study of patients with cirrhosis, screening for hepatocellular carcinoma up to 4 years prior to diagnosis was not associated with lower mortality.

Similar proportions of cases and controls underwent screening with abdominal ultrasonography, serum alpha-fetoprotein (AFP) testing, or both, reported Andrew M. Moon, MD, MPH, of the University of North Carolina at Chapel Hill, and his associates. “There was also no difference in receipt of these screening tests within 1, 2, or 3 years prior to the index date,” they wrote. The report was published in Gastroenterology. The findings “[suggest] that either these screening tests or the currently available treatments [for liver cancer], or both, are suboptimal and need to be improved.”
Because cirrhosis significantly increases the risk of hepatocellular carcinoma, the American Association for the Study of Liver Diseases, the European Association for the Study of the Liver, and the Asian Pacific Association for the Study of the Liver recommend screening cirrhotic patients every 6 months with abdominal ultrasonography with or without concomitant serum AFP. But nonliver societies have not endorsed this approach, citing a lack of high-quality data. One problem is that studies have compared patients whose liver cancer was diagnosed by screening with those diagnosed after they became symptomatic, which creates a lead-time bias that inherently favors screening, Dr. Moon and his associates noted.
To help fill the evidence gap, they identified 238 patients from the Veterans Affairs health care system who had died of hepatocellular carcinoma between 2013 and 2015 and who had been diagnosed with cirrhosis at least 4 years beforehand. They compared these cases with an equal number of patients with cirrhosis who had been in VA care for a similar amount of time and had not died of hepatocellular carcinoma. Cases and controls were matched by etiology of cirrhosis, year that cirrhosis was diagnosed, race, age, sex, Model for End-Stage Liver Disease score, and VA medical center. The researchers identified screening tests by reviewing blinded medical charts.
There were no significant differences in the proportions of cases and controls who underwent screening ultrasonography (52.9% versus 54.2%, respectively), screening serum AFP (74.8% versus 73.5%), either test (81.1% versus 79.4%), or both tests (46.6% versus 48.3%) within 4 years of the index date or the matched control. The result was similar after potential confounders were controlled for and when examining shorter time frames of 1, 2, and 3 years.
It was unlikely that these results reflect delayed diagnosis of liver cancer or a lack of treatment within the VA system, the experts wrote. A total of 51.3% of cases were diagnosed with Milan criteria, which exceeds the proportion in the national Surveillance, Epidemiology, and End Results registry, they noted. None of the fatal cases underwent liver transplantation, but 66.8% received other treatments for liver cancer.
Funders included the National Institutes of Health and the Veterans Affairs Clinical Science Research & Development. The investigators reported having no conflicts of interest.
SOURCE: Moon AM et al. Gastroenterology. 2018 Jul 5. doi: 10.1053/j.gastro.2018.06.079.
In a case-control study of patients with cirrhosis, screening for hepatocellular carcinoma up to 4 years prior to diagnosis was not associated with lower mortality.
Similar proportions of cases and controls underwent screening with abdominal ultrasonography, serum alpha-fetoprotein (AFP) testing, or both, reported Andrew M. Moon, MD, MPH, of the University of North Carolina at Chapel Hill, and his associates. “There was also no difference in receipt of these screening tests within 1, 2, or 3 years prior to the index date,” they wrote. The report was published in Gastroenterology. The findings “[suggest] that either these screening tests or the currently available treatments [for liver cancer], or both, are suboptimal and need to be improved.”
Because cirrhosis significantly increases the risk of hepatocellular carcinoma, the American Association for the Study of Liver Diseases, the European Association for the Study of the Liver, and the Asian Pacific Association for the Study of the Liver recommend screening cirrhotic patients every 6 months with abdominal ultrasonography with or without concomitant serum AFP. But nonliver societies have not endorsed this approach, citing a lack of high-quality data. One problem is that studies have compared patients whose liver cancer was diagnosed by screening with those diagnosed after they became symptomatic, which creates a lead-time bias that inherently favors screening, Dr. Moon and his associates noted.
To help fill the evidence gap, they identified 238 patients from the Veterans Affairs health care system who had died of hepatocellular carcinoma between 2013 and 2015 and who had been diagnosed with cirrhosis at least 4 years beforehand. They compared these cases with an equal number of patients with cirrhosis who had been in VA care for a similar amount of time and had not died of hepatocellular carcinoma. Cases and controls were matched by etiology of cirrhosis, year that cirrhosis was diagnosed, race, age, sex, Model for End-Stage Liver Disease score, and VA medical center. The researchers identified screening tests by reviewing blinded medical charts.
There were no significant differences in the proportions of cases and controls who underwent screening ultrasonography (52.9% versus 54.2%, respectively), screening serum AFP (74.8% versus 73.5%), either test (81.1% versus 79.4%), or both tests (46.6% versus 48.3%) within 4 years of the index date or the matched control. The result was similar after potential confounders were controlled for and when examining shorter time frames of 1, 2, and 3 years.
It was unlikely that these results reflect delayed diagnosis of liver cancer or a lack of treatment within the VA system, the experts wrote. A total of 51.3% of cases were diagnosed with Milan criteria, which exceeds the proportion in the national Surveillance, Epidemiology, and End Results registry, they noted. None of the fatal cases underwent liver transplantation, but 66.8% received other treatments for liver cancer.
Funders included the National Institutes of Health and the Veterans Affairs Clinical Science Research & Development. The investigators reported having no conflicts of interest.
SOURCE: Moon AM et al. Gastroenterology. 2018 Jul 5. doi: 10.1053/j.gastro.2018.06.079.
In a case-control study of patients with cirrhosis, screening for hepatocellular carcinoma up to 4 years prior to diagnosis was not associated with lower mortality.
Similar proportions of cases and controls underwent screening with abdominal ultrasonography, serum alpha-fetoprotein (AFP) testing, or both, reported Andrew M. Moon, MD, MPH, of the University of North Carolina at Chapel Hill, and his associates. “There was also no difference in receipt of these screening tests within 1, 2, or 3 years prior to the index date,” they wrote. The report was published in Gastroenterology. The findings “[suggest] that either these screening tests or the currently available treatments [for liver cancer], or both, are suboptimal and need to be improved.”
Because cirrhosis significantly increases the risk of hepatocellular carcinoma, the American Association for the Study of Liver Diseases, the European Association for the Study of the Liver, and the Asian Pacific Association for the Study of the Liver recommend screening cirrhotic patients every 6 months with abdominal ultrasonography with or without concomitant serum AFP. But nonliver societies have not endorsed this approach, citing a lack of high-quality data. One problem is that studies have compared patients whose liver cancer was diagnosed by screening with those diagnosed after they became symptomatic, which creates a lead-time bias that inherently favors screening, Dr. Moon and his associates noted.
To help fill the evidence gap, they identified 238 patients from the Veterans Affairs health care system who had died of hepatocellular carcinoma between 2013 and 2015 and who had been diagnosed with cirrhosis at least 4 years beforehand. They compared these cases with an equal number of patients with cirrhosis who had been in VA care for a similar amount of time and had not died of hepatocellular carcinoma. Cases and controls were matched by etiology of cirrhosis, year that cirrhosis was diagnosed, race, age, sex, Model for End-Stage Liver Disease score, and VA medical center. The researchers identified screening tests by reviewing blinded medical charts.
There were no significant differences in the proportions of cases and controls who underwent screening ultrasonography (52.9% versus 54.2%, respectively), screening serum AFP (74.8% versus 73.5%), either test (81.1% versus 79.4%), or both tests (46.6% versus 48.3%) within 4 years of the index date or the matched control. The result was similar after potential confounders were controlled for and when examining shorter time frames of 1, 2, and 3 years.
It was unlikely that these results reflect delayed diagnosis of liver cancer or a lack of treatment within the VA system, the experts wrote. A total of 51.3% of cases were diagnosed with Milan criteria, which exceeds the proportion in the national Surveillance, Epidemiology, and End Results registry, they noted. None of the fatal cases underwent liver transplantation, but 66.8% received other treatments for liver cancer.
Funders included the National Institutes of Health and the Veterans Affairs Clinical Science Research & Development. The investigators reported having no conflicts of interest.
SOURCE: Moon AM et al. Gastroenterology. 2018 Jul 5. doi: 10.1053/j.gastro.2018.06.079.
FROM GASTROENTEROLOGY
Key clinical point: Among patients with cirrhosis, screening for hepatocellular carcinoma was not associated with reductions in liver cancer mortality.
Major finding: Similar proportions of cases and controls were screened by abdominal ultrasonography, serum alpha-fetoprotein, or both up to 4 years before the index date and even after researchers controlled for relevant confounders.
Study details: A matched case-control study of 476 patients from the Veterans Affairs health care system.
Disclosures: Funders included the National Institutes of Health and the Veterans Affairs Clinical Science Research & Development. The investigators reported no conflicts of interest.
Source: Moon AM et al. Gastroenterology. 2018 Jul 5. doi: 10.1053/j.gastro.2018.06.079.
Experts update diagnostic guidelines for eosinophilic esophagitis
The diagnosis of eosinophilic esophagitis no longer needs to include a trial of proton pump inhibitor (PPI) therapy, according to an updated international consensus statement published in the October issue of Gastroenterology.
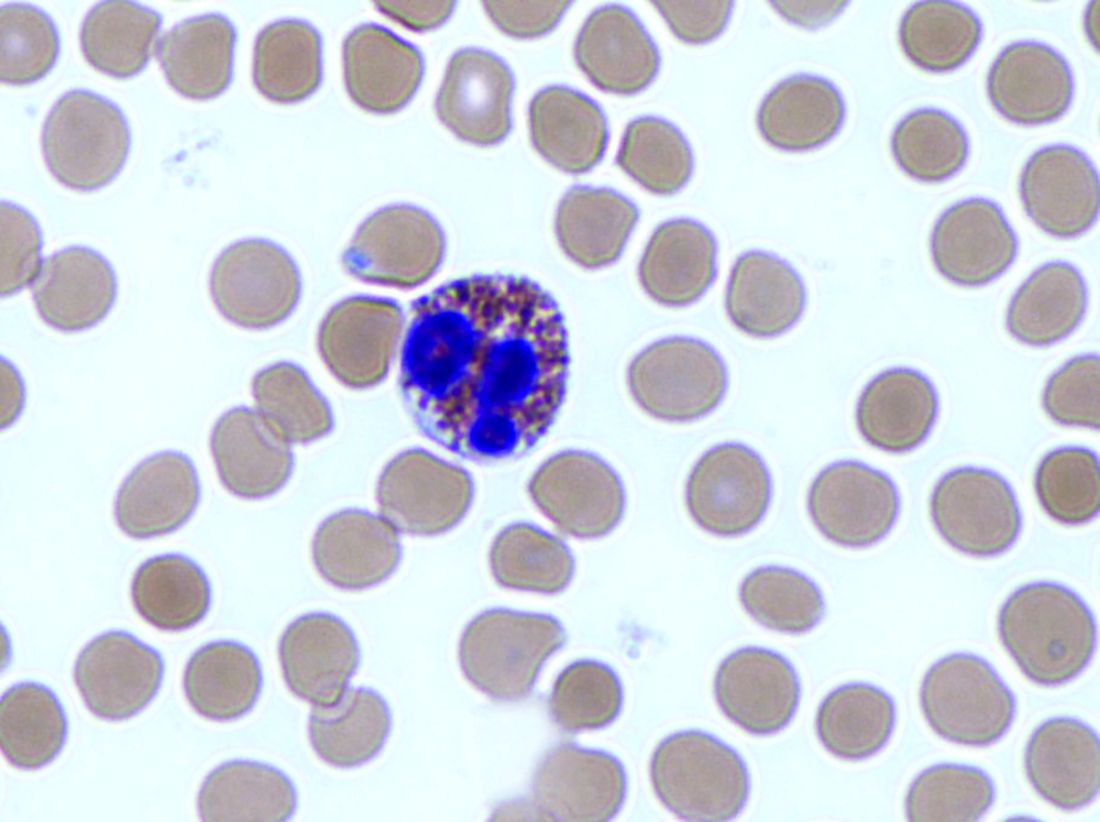
“An initial rationale for the PPI trial was to distinguish eosinophilic esophagitis from gastroesophageal reflux disease, but it is now known that these conditions have a complex relationship and are not necessarily mutually exclusive,” wrote Evan S. Dellon, MD, of the University of North Carolina at Chapel Hill, and his associates. According to current evidence, “PPIs are better classified as a treatment for esophageal eosinophilia that may be due to eosinophilic esophagitis than as a diagnostic criterion,” they said.
Diagnostic guidelines for eosinophilic esophagitis were published first in 2007 and were updated in 2011. The guideline authors recommended either pH monitoring or an 8-week trial of high-dose PPI therapy to rule out inflammation from gastroesophageal reflux disease (GERD).
But subsequent publications described patients with symptomatic esophageal eosinophilia who responded to PPIs and lacked classic GERD symptoms. Guidelines called this condition “PPI-responsive esophageal eosinophilia” and considered it a separate entity from GERD.
However, an “evolving body of research” shows that eosinophilic esophagitis can overlap with GERD, Dr. Dellon and his associates wrote. Furthermore, each of these conditions can trigger the other. Eosinophilic esophagitis can decrease esophageal compliance, leading to secondary reflux, while gastroesophageal reflux can erode the esophageal epithelium, triggering antigen exposure and eosinophilia.
Therefore, Dr. Dellon and his associates recommended defining eosinophilic esophagitis as signs and symptoms of esophageal dysfunction and an esophageal biopsy showing at least 15 eosinophils per high-power field, or approximately 60 eosinophils per millimeter, with infiltration limited to the esophagus. They stressed the importance of esophageal biopsy even if endoscopy shows normal mucosa. “As per prior guidelines, multiple biopsies from two or more esophageal levels, targeting areas of apparent inflammation, are recommended to increase the diagnostic yield,” they added. “Gastric and duodenal biopsies should be obtained as clinically indicated by symptoms, endoscopic findings in the stomach or duodenum, or high index of suspicion for a mucosal process.”
Physicians should increase their suspicion of eosinophilic esophagitis if patients have other types of atopy or endoscopic findings of “rings, furrows, exudates, edema, stricture, narrowing, and crepe-paper mucosa,” they added. In addition to GERD, they recommended looking carefully for other conditions that can trigger esophageal eosinophilia, such as pemphigus, drug hypersensitivity reactions, achalasia, and Crohn’s disease with esophageal involvement.
To create the guideline, Dr. Dellon and his associates searched PubMed for studies of all designs and sizes published from 1966 through December 2016. Teams of experts on specific topics then reviewed and discussed relevant literature. In May 2017, 43 reviewers met for 8 hours to present and discuss conclusions. There was 100% agreement to remove the PPI trial from the diagnostic criteria, the experts noted.
The authors disclosed financial support from the International Gastrointestinal Eosinophilic Diseases Researchers (TIGERS), The David and Denise Bunning Family, and the Rare Disease Clinical Research Network. Dr. Dellon disclosed consulting relationships and receiving research funding from Adare, Celgene/Receptos, Regeneron, and Shire among others. The majority of his coauthors also disclosed relationships with numerous medical companies.
SOURCE: Dellon ES et al. Gastroenterology. 2018 Jul 12. doi: 10.1053/j.gastro.2018.07.009.

Studies in the 1980s linked the presence of esophageal mucosal eosinophils with increased acid exposure on pH monitoring. For the next 2 decades, clinicians viewed eosinophils on esophageal biopsies as diagnostic for GERD such that the initial description of EoE by Attwood in 1993 distinguished EoE from GERD by the presence of esophageal eosinophilia in the absence of either reflux esophagitis or abnormal acid exposure on pH testing. Consequently, the initial diagnostic criteria for EoE in 2007 included a lack of response to PPI and/or normal pH testing to establish the diagnosis of EoE. Reflecting growing uncertainty regarding the ability of PPI therapy to differentiate acid-induced from allergic inflammatory mechanisms, an updated consensus in 2011 introduced the terminology “PPI responsive esophageal eosinophilia (PPIREE)” to describe an increasingly recognized subset of patients with suspected EoE that resolved with PPI. Now, supported by scientific evidence accumulated over the past decade, AGREE has taken a step back by removing the PPI trial from the diagnosis of EoE, thereby abandoning the PPIREE terminology. This step simplifies the diagnosis of EoE and acknowledges that a histologic response to PPI does not “rule in” GERD or “rule out” EoE. It is important to emphasize that the updated criteria still advocate careful consideration of secondary causes of esophageal eosinophilia prior to the diagnosis of EoE.
Ramifications of the updated diagnostic criteria include the opportunities for clinicians to consider use of topical corticosteroids and diet therapies, rather than mandate an up-front PPI trial, in patients with EoE. On a practical level, based on their effectiveness, safety, and ease of administration, PPIs remain positioned as a favorable initial intervention for EoE. Conceptually, however, the paradigm shift highlights the ability of research to improve our understanding of disease pathogenesis and thereby impact clinical management.
Ikuo Hirano, MD, AGAF, is in the division of gastroenterology, Northwestern University, Chicago. He has received grant support from the NIH Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR, U54 AI117804), which is part of the Rare Disease Clinical Research Network. He has received research funding and consulting fees from Celgene, Regeneron, Shire, and others.
Studies in the 1980s linked the presence of esophageal mucosal eosinophils with increased acid exposure on pH monitoring. For the next 2 decades, clinicians viewed eosinophils on esophageal biopsies as diagnostic for GERD such that the initial description of EoE by Attwood in 1993 distinguished EoE from GERD by the presence of esophageal eosinophilia in the absence of either reflux esophagitis or abnormal acid exposure on pH testing. Consequently, the initial diagnostic criteria for EoE in 2007 included a lack of response to PPI and/or normal pH testing to establish the diagnosis of EoE. Reflecting growing uncertainty regarding the ability of PPI therapy to differentiate acid-induced from allergic inflammatory mechanisms, an updated consensus in 2011 introduced the terminology “PPI responsive esophageal eosinophilia (PPIREE)” to describe an increasingly recognized subset of patients with suspected EoE that resolved with PPI. Now, supported by scientific evidence accumulated over the past decade, AGREE has taken a step back by removing the PPI trial from the diagnosis of EoE, thereby abandoning the PPIREE terminology. This step simplifies the diagnosis of EoE and acknowledges that a histologic response to PPI does not “rule in” GERD or “rule out” EoE. It is important to emphasize that the updated criteria still advocate careful consideration of secondary causes of esophageal eosinophilia prior to the diagnosis of EoE.
Ramifications of the updated diagnostic criteria include the opportunities for clinicians to consider use of topical corticosteroids and diet therapies, rather than mandate an up-front PPI trial, in patients with EoE. On a practical level, based on their effectiveness, safety, and ease of administration, PPIs remain positioned as a favorable initial intervention for EoE. Conceptually, however, the paradigm shift highlights the ability of research to improve our understanding of disease pathogenesis and thereby impact clinical management.
Ikuo Hirano, MD, AGAF, is in the division of gastroenterology, Northwestern University, Chicago. He has received grant support from the NIH Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR, U54 AI117804), which is part of the Rare Disease Clinical Research Network. He has received research funding and consulting fees from Celgene, Regeneron, Shire, and others.
Studies in the 1980s linked the presence of esophageal mucosal eosinophils with increased acid exposure on pH monitoring. For the next 2 decades, clinicians viewed eosinophils on esophageal biopsies as diagnostic for GERD such that the initial description of EoE by Attwood in 1993 distinguished EoE from GERD by the presence of esophageal eosinophilia in the absence of either reflux esophagitis or abnormal acid exposure on pH testing. Consequently, the initial diagnostic criteria for EoE in 2007 included a lack of response to PPI and/or normal pH testing to establish the diagnosis of EoE. Reflecting growing uncertainty regarding the ability of PPI therapy to differentiate acid-induced from allergic inflammatory mechanisms, an updated consensus in 2011 introduced the terminology “PPI responsive esophageal eosinophilia (PPIREE)” to describe an increasingly recognized subset of patients with suspected EoE that resolved with PPI. Now, supported by scientific evidence accumulated over the past decade, AGREE has taken a step back by removing the PPI trial from the diagnosis of EoE, thereby abandoning the PPIREE terminology. This step simplifies the diagnosis of EoE and acknowledges that a histologic response to PPI does not “rule in” GERD or “rule out” EoE. It is important to emphasize that the updated criteria still advocate careful consideration of secondary causes of esophageal eosinophilia prior to the diagnosis of EoE.
Ramifications of the updated diagnostic criteria include the opportunities for clinicians to consider use of topical corticosteroids and diet therapies, rather than mandate an up-front PPI trial, in patients with EoE. On a practical level, based on their effectiveness, safety, and ease of administration, PPIs remain positioned as a favorable initial intervention for EoE. Conceptually, however, the paradigm shift highlights the ability of research to improve our understanding of disease pathogenesis and thereby impact clinical management.
Ikuo Hirano, MD, AGAF, is in the division of gastroenterology, Northwestern University, Chicago. He has received grant support from the NIH Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR, U54 AI117804), which is part of the Rare Disease Clinical Research Network. He has received research funding and consulting fees from Celgene, Regeneron, Shire, and others.
The diagnosis of eosinophilic esophagitis no longer needs to include a trial of proton pump inhibitor (PPI) therapy, according to an updated international consensus statement published in the October issue of Gastroenterology.
“An initial rationale for the PPI trial was to distinguish eosinophilic esophagitis from gastroesophageal reflux disease, but it is now known that these conditions have a complex relationship and are not necessarily mutually exclusive,” wrote Evan S. Dellon, MD, of the University of North Carolina at Chapel Hill, and his associates. According to current evidence, “PPIs are better classified as a treatment for esophageal eosinophilia that may be due to eosinophilic esophagitis than as a diagnostic criterion,” they said.
Diagnostic guidelines for eosinophilic esophagitis were published first in 2007 and were updated in 2011. The guideline authors recommended either pH monitoring or an 8-week trial of high-dose PPI therapy to rule out inflammation from gastroesophageal reflux disease (GERD).
But subsequent publications described patients with symptomatic esophageal eosinophilia who responded to PPIs and lacked classic GERD symptoms. Guidelines called this condition “PPI-responsive esophageal eosinophilia” and considered it a separate entity from GERD.
However, an “evolving body of research” shows that eosinophilic esophagitis can overlap with GERD, Dr. Dellon and his associates wrote. Furthermore, each of these conditions can trigger the other. Eosinophilic esophagitis can decrease esophageal compliance, leading to secondary reflux, while gastroesophageal reflux can erode the esophageal epithelium, triggering antigen exposure and eosinophilia.
Therefore, Dr. Dellon and his associates recommended defining eosinophilic esophagitis as signs and symptoms of esophageal dysfunction and an esophageal biopsy showing at least 15 eosinophils per high-power field, or approximately 60 eosinophils per millimeter, with infiltration limited to the esophagus. They stressed the importance of esophageal biopsy even if endoscopy shows normal mucosa. “As per prior guidelines, multiple biopsies from two or more esophageal levels, targeting areas of apparent inflammation, are recommended to increase the diagnostic yield,” they added. “Gastric and duodenal biopsies should be obtained as clinically indicated by symptoms, endoscopic findings in the stomach or duodenum, or high index of suspicion for a mucosal process.”
Physicians should increase their suspicion of eosinophilic esophagitis if patients have other types of atopy or endoscopic findings of “rings, furrows, exudates, edema, stricture, narrowing, and crepe-paper mucosa,” they added. In addition to GERD, they recommended looking carefully for other conditions that can trigger esophageal eosinophilia, such as pemphigus, drug hypersensitivity reactions, achalasia, and Crohn’s disease with esophageal involvement.
To create the guideline, Dr. Dellon and his associates searched PubMed for studies of all designs and sizes published from 1966 through December 2016. Teams of experts on specific topics then reviewed and discussed relevant literature. In May 2017, 43 reviewers met for 8 hours to present and discuss conclusions. There was 100% agreement to remove the PPI trial from the diagnostic criteria, the experts noted.
The authors disclosed financial support from the International Gastrointestinal Eosinophilic Diseases Researchers (TIGERS), The David and Denise Bunning Family, and the Rare Disease Clinical Research Network. Dr. Dellon disclosed consulting relationships and receiving research funding from Adare, Celgene/Receptos, Regeneron, and Shire among others. The majority of his coauthors also disclosed relationships with numerous medical companies.
SOURCE: Dellon ES et al. Gastroenterology. 2018 Jul 12. doi: 10.1053/j.gastro.2018.07.009.
The diagnosis of eosinophilic esophagitis no longer needs to include a trial of proton pump inhibitor (PPI) therapy, according to an updated international consensus statement published in the October issue of Gastroenterology.
“An initial rationale for the PPI trial was to distinguish eosinophilic esophagitis from gastroesophageal reflux disease, but it is now known that these conditions have a complex relationship and are not necessarily mutually exclusive,” wrote Evan S. Dellon, MD, of the University of North Carolina at Chapel Hill, and his associates. According to current evidence, “PPIs are better classified as a treatment for esophageal eosinophilia that may be due to eosinophilic esophagitis than as a diagnostic criterion,” they said.
Diagnostic guidelines for eosinophilic esophagitis were published first in 2007 and were updated in 2011. The guideline authors recommended either pH monitoring or an 8-week trial of high-dose PPI therapy to rule out inflammation from gastroesophageal reflux disease (GERD).
But subsequent publications described patients with symptomatic esophageal eosinophilia who responded to PPIs and lacked classic GERD symptoms. Guidelines called this condition “PPI-responsive esophageal eosinophilia” and considered it a separate entity from GERD.
However, an “evolving body of research” shows that eosinophilic esophagitis can overlap with GERD, Dr. Dellon and his associates wrote. Furthermore, each of these conditions can trigger the other. Eosinophilic esophagitis can decrease esophageal compliance, leading to secondary reflux, while gastroesophageal reflux can erode the esophageal epithelium, triggering antigen exposure and eosinophilia.
Therefore, Dr. Dellon and his associates recommended defining eosinophilic esophagitis as signs and symptoms of esophageal dysfunction and an esophageal biopsy showing at least 15 eosinophils per high-power field, or approximately 60 eosinophils per millimeter, with infiltration limited to the esophagus. They stressed the importance of esophageal biopsy even if endoscopy shows normal mucosa. “As per prior guidelines, multiple biopsies from two or more esophageal levels, targeting areas of apparent inflammation, are recommended to increase the diagnostic yield,” they added. “Gastric and duodenal biopsies should be obtained as clinically indicated by symptoms, endoscopic findings in the stomach or duodenum, or high index of suspicion for a mucosal process.”
Physicians should increase their suspicion of eosinophilic esophagitis if patients have other types of atopy or endoscopic findings of “rings, furrows, exudates, edema, stricture, narrowing, and crepe-paper mucosa,” they added. In addition to GERD, they recommended looking carefully for other conditions that can trigger esophageal eosinophilia, such as pemphigus, drug hypersensitivity reactions, achalasia, and Crohn’s disease with esophageal involvement.
To create the guideline, Dr. Dellon and his associates searched PubMed for studies of all designs and sizes published from 1966 through December 2016. Teams of experts on specific topics then reviewed and discussed relevant literature. In May 2017, 43 reviewers met for 8 hours to present and discuss conclusions. There was 100% agreement to remove the PPI trial from the diagnostic criteria, the experts noted.
The authors disclosed financial support from the International Gastrointestinal Eosinophilic Diseases Researchers (TIGERS), The David and Denise Bunning Family, and the Rare Disease Clinical Research Network. Dr. Dellon disclosed consulting relationships and receiving research funding from Adare, Celgene/Receptos, Regeneron, and Shire among others. The majority of his coauthors also disclosed relationships with numerous medical companies.
SOURCE: Dellon ES et al. Gastroenterology. 2018 Jul 12. doi: 10.1053/j.gastro.2018.07.009.
FROM GASTROENTEROLOGY
Key clinical point: The diagnosis of eosinophilic esophagitis no longer needs to include a trial of proton pump inhibitor therapy.
Major finding: Eosinophilic esophagitis and gastroesophageal reflux disease are not mutually exclusive.
Study details: Review by an international consensus panel of studies published between 1966 and 2016.
Disclosures: The authors disclosed financial support from the International Gastrointestinal Eosinophilic Diseases Researchers (TIGERS), The David and Denise Bunning Family, the Rare Disease Clinical Research Network. Dr. Dellon disclosed consulting relationships with Adare, Allakos, Alivio, Banner, Celgene/Receptos, Enumeral, GSK, Regeneron, and Shire. He also reported receiving research funding from Adare, Celgene/Receptos, Miraca, Meritage, Nutricia, Regeneron, and Shire and educational grants from Banner and Holoclara. The majority of his coauthors disclosed relationships with numerous medical companies.
Source: Dellon ES et al. Gastroenterology. 2018 Jul 12. doi: 10.1053/j.gastro.2018.07.009.
Guidelines offer recommendations for hernia repair in obese patients
and combining repair techniques with bariatric surgery can improve outcomes and lower the rate of complication for select patients, according to recent guidelines released by the American Society for Metabolic and Bariatric Surgery and the American Hernia Society.

Emanuele Lo Menzo, MD, of the Bariatric and Metabolic Institute at Cleveland Clinic Florida in Weston, and his colleagues issued a statement, published in the journal Surgery for Obesity and Related Diseases, based on available evidence from scientific literature on the impact of obesity on hernia surgery and what effect treating obesity has on improving hernia repair outcomes.
The authors noted abdominal wall hernia in obese patients is “a significant and increasingly common challenge for surgeons” and cited recent data from the American College of Surgeons National Surgical Quality Improvement Program that shows 60% of ventral hernia repairs (VHR) are performed on patients with body mass indexes (BMIs) above 30 kg/m2. Overall, they noted that general surgeons perform approximately 350,000 conventional hernia repairs (CHR) and 800,000 incisional hernia (IH) repairs each year.
The literature on the impact of obesity on hernia repair outcomes and the feasibility of a combined operation to address each problem has significant gaps, leaving surgeons to decide on a correct course based on individual patient needs. The guideline offers some recommendations, and notes areas that remain understudied. First, “in patients with severe obesity and [ventral hernia] and both being amenable to laparoscopic repair, combined hernia repair and [metabolic/bariatric surgery] may be safe and associated with good short-term outcomes and low risk of infection.” But the use of synthetic mesh in these patients is not well studied and so the guideline passes on a recommendation of mesh. For those obese patients with symptomatic abdominal wall hernias (AWHs) not amenable to laparoscopy, the guideline notes that metabolic/bariatric surgery first may be the best option.
Risk of hernia in obese patients
Studies suggest there is an increased risk of primary and IH among patients with BMIs greater than 25 kg/m2, with one study finding an 18.2% complication rate after single-incision laparoscopic surgery for patients with BMIs of 40 kg/m2 or higher, compared with a 3.5% complication rate among patients at a normal body weight. Severe BMI also is a risk factor for developing surgical site infection (SSI), which can cause recurrence, the authors said. Evidence from multiple studies further supports BMI as a risk factor for hernia recurrence, and intra-abdominal pressure from obesity increases the risk of developing an AWH.
“While most authors attribute the increased risk for AWH formation in the setting of obesity to BMI alone, others have suggested that abdominal circumference and elevated visceral fat may play a more significant role,” the authors wrote.
However, Dr. Lo Menzo and his colleagues admitted the actual rate of IH is difficult to calculate because some patients may not seek treatment for minimally symptomatic hernias. Patients with higher BMIs may not be aware of or seek treatment for common symptoms of IH such as groin bulge, or when they do seek treatment, it can present with symptoms such as incarceration or strangulation, they said. Patients with higher BMIs also are more likely to be offered “watchful waiting” because of higher complication rates in this patient population, which may contribute to incarceration or strangulation symptoms in these patients, they added.
Complications and recurrence
There is no one recommended repair technique or ideal BMI for hernia repair in obese patients, the authors wrote. One study found laparoscopic VHRs had a complication rate of 1.2% and reoccurrence rate of 5.5% at mean 25-month follow-up in patients with BMIs of 38 kg/m2 or greater, while a different study with a similar design found a 3.8% reoccurrence rate at 18-month follow-up. Degree of obesity can affect complication rate: One study showed that 73% of all complications after laparoscopic VHR occurred in the group of patients with BMIs of 30 kg/m2 or greater; a different study of laparoscopic VHR had an 8.3% hernia reoccurrence rate in patients with BMIs of 40 kg/m2 or greater, compared with patients at a normal weight (2.9%), with time to hernia reoccurrence being shorter in the higher-BMI group. A study of obese patients undergoing retromuscular open repair had a wound complication rate of 16% and a reoccurrence rate of 6%, with another study of patients undergoing umbilical hernia repair showing similar rates of complication and reoccurrence.
Panniculectomy
Among patients who underwent IH repair with panniculectomy, the authors found a 40% complication rate and a 10% reoccurrence rate in patients with BMIs of 40 kg/m2 or greater who received a partial underlay mesh placement hernia repair, while a different study found an increased risk of surgical site occurrences but not SSI in patients with BMIs of 34.3 kg/m2 or greater who underwent open ventral incisional hernia repair with panniculectomy. A third study found BMIs were not linked to a 55% complication rate in patients who underwent open ventral IH repair with and without mesh.
Simultaneous surgery
The authors noted that studies have shown that performing laparoscopic hernia repair and metabolic and bariatric surgery simultaneously is safe and has good short-term results. Specifically, patients who underwent the surgery with synthetic mesh had a low rate of infection or reoccurrence. Patients who underwent simultaneous weight-loss surgery and VHR had an elevated risk of SSI but no increased rate of 30-day mortality or morbidity, according to results from a large-scale registry. However, the authors noted patients with severe obesity may not be good candidates for simultaneous metabolic and bariatric surgery [MBS] and VHR, such as in patients with “large abdominal wall defects, loss of abdominal domain, extensive intestinal adhesive disease, poor quality skin (i.e., attenuated skin, prior skin graft, or ulcerated skin), incarcerated hernias containing bowel, hernias with previous synthetic mesh, hernias with chronic infection, or patients who have already undergone MBS with altered anatomy that is still intact.”
Preop weight loss
There is mixed data on the effect of weight loss prior to VHR through interventions such as very-low-calorie diets, pharmacotherapy, intragastric balloon therapy, and MBS as a first-stage procedure prior to VHR. Many patients treating their obesity with very-low-calorie diets lose approximately 10%-20% of their initial body weight and keep the weight off for at least 18 months, while one study showed patients who underwent intragastric balloon therapy lost approximately 10% of their body weight over 6 months; however, other studies have questioned the efficacy of this therapy, compared with a structured weight loss program or bariatric surgery. The authors also noted the difficulty of coordinating VHR after weight-loss surgery, lack of support from insurers, and they cited reports that cautioned bariatric surgeons to not leave hernias untreated during MBS. There is no current evidence pharmacotherapy through Food and Drug Administration–approved weight-loss drugs prior to hernia repair yield the weight-loss results needed for these patients to improve hernia outcomes, they added.
“Ultimately, there are various appropriate treatment modalities for each patient, and surgeons must use their judgment in selecting from among the different feasible options,” Dr. Lo Menzo and his colleagues wrote in the guidelines.
The authors report no relevant conflicts of interest.
SOURCE: Lo Menzo E et al. Surg Obes Relat Dis. 2018 Jul 19. doi: 10.1016/j.soard.2018.07.005.
and combining repair techniques with bariatric surgery can improve outcomes and lower the rate of complication for select patients, according to recent guidelines released by the American Society for Metabolic and Bariatric Surgery and the American Hernia Society.
Emanuele Lo Menzo, MD, of the Bariatric and Metabolic Institute at Cleveland Clinic Florida in Weston, and his colleagues issued a statement, published in the journal Surgery for Obesity and Related Diseases, based on available evidence from scientific literature on the impact of obesity on hernia surgery and what effect treating obesity has on improving hernia repair outcomes.
The authors noted abdominal wall hernia in obese patients is “a significant and increasingly common challenge for surgeons” and cited recent data from the American College of Surgeons National Surgical Quality Improvement Program that shows 60% of ventral hernia repairs (VHR) are performed on patients with body mass indexes (BMIs) above 30 kg/m2. Overall, they noted that general surgeons perform approximately 350,000 conventional hernia repairs (CHR) and 800,000 incisional hernia (IH) repairs each year.
The literature on the impact of obesity on hernia repair outcomes and the feasibility of a combined operation to address each problem has significant gaps, leaving surgeons to decide on a correct course based on individual patient needs. The guideline offers some recommendations, and notes areas that remain understudied. First, “in patients with severe obesity and [ventral hernia] and both being amenable to laparoscopic repair, combined hernia repair and [metabolic/bariatric surgery] may be safe and associated with good short-term outcomes and low risk of infection.” But the use of synthetic mesh in these patients is not well studied and so the guideline passes on a recommendation of mesh. For those obese patients with symptomatic abdominal wall hernias (AWHs) not amenable to laparoscopy, the guideline notes that metabolic/bariatric surgery first may be the best option.
Risk of hernia in obese patients
Studies suggest there is an increased risk of primary and IH among patients with BMIs greater than 25 kg/m2, with one study finding an 18.2% complication rate after single-incision laparoscopic surgery for patients with BMIs of 40 kg/m2 or higher, compared with a 3.5% complication rate among patients at a normal body weight. Severe BMI also is a risk factor for developing surgical site infection (SSI), which can cause recurrence, the authors said. Evidence from multiple studies further supports BMI as a risk factor for hernia recurrence, and intra-abdominal pressure from obesity increases the risk of developing an AWH.
“While most authors attribute the increased risk for AWH formation in the setting of obesity to BMI alone, others have suggested that abdominal circumference and elevated visceral fat may play a more significant role,” the authors wrote.
However, Dr. Lo Menzo and his colleagues admitted the actual rate of IH is difficult to calculate because some patients may not seek treatment for minimally symptomatic hernias. Patients with higher BMIs may not be aware of or seek treatment for common symptoms of IH such as groin bulge, or when they do seek treatment, it can present with symptoms such as incarceration or strangulation, they said. Patients with higher BMIs also are more likely to be offered “watchful waiting” because of higher complication rates in this patient population, which may contribute to incarceration or strangulation symptoms in these patients, they added.
Complications and recurrence
There is no one recommended repair technique or ideal BMI for hernia repair in obese patients, the authors wrote. One study found laparoscopic VHRs had a complication rate of 1.2% and reoccurrence rate of 5.5% at mean 25-month follow-up in patients with BMIs of 38 kg/m2 or greater, while a different study with a similar design found a 3.8% reoccurrence rate at 18-month follow-up. Degree of obesity can affect complication rate: One study showed that 73% of all complications after laparoscopic VHR occurred in the group of patients with BMIs of 30 kg/m2 or greater; a different study of laparoscopic VHR had an 8.3% hernia reoccurrence rate in patients with BMIs of 40 kg/m2 or greater, compared with patients at a normal weight (2.9%), with time to hernia reoccurrence being shorter in the higher-BMI group. A study of obese patients undergoing retromuscular open repair had a wound complication rate of 16% and a reoccurrence rate of 6%, with another study of patients undergoing umbilical hernia repair showing similar rates of complication and reoccurrence.
Panniculectomy
Among patients who underwent IH repair with panniculectomy, the authors found a 40% complication rate and a 10% reoccurrence rate in patients with BMIs of 40 kg/m2 or greater who received a partial underlay mesh placement hernia repair, while a different study found an increased risk of surgical site occurrences but not SSI in patients with BMIs of 34.3 kg/m2 or greater who underwent open ventral incisional hernia repair with panniculectomy. A third study found BMIs were not linked to a 55% complication rate in patients who underwent open ventral IH repair with and without mesh.
Simultaneous surgery
The authors noted that studies have shown that performing laparoscopic hernia repair and metabolic and bariatric surgery simultaneously is safe and has good short-term results. Specifically, patients who underwent the surgery with synthetic mesh had a low rate of infection or reoccurrence. Patients who underwent simultaneous weight-loss surgery and VHR had an elevated risk of SSI but no increased rate of 30-day mortality or morbidity, according to results from a large-scale registry. However, the authors noted patients with severe obesity may not be good candidates for simultaneous metabolic and bariatric surgery [MBS] and VHR, such as in patients with “large abdominal wall defects, loss of abdominal domain, extensive intestinal adhesive disease, poor quality skin (i.e., attenuated skin, prior skin graft, or ulcerated skin), incarcerated hernias containing bowel, hernias with previous synthetic mesh, hernias with chronic infection, or patients who have already undergone MBS with altered anatomy that is still intact.”
Preop weight loss
There is mixed data on the effect of weight loss prior to VHR through interventions such as very-low-calorie diets, pharmacotherapy, intragastric balloon therapy, and MBS as a first-stage procedure prior to VHR. Many patients treating their obesity with very-low-calorie diets lose approximately 10%-20% of their initial body weight and keep the weight off for at least 18 months, while one study showed patients who underwent intragastric balloon therapy lost approximately 10% of their body weight over 6 months; however, other studies have questioned the efficacy of this therapy, compared with a structured weight loss program or bariatric surgery. The authors also noted the difficulty of coordinating VHR after weight-loss surgery, lack of support from insurers, and they cited reports that cautioned bariatric surgeons to not leave hernias untreated during MBS. There is no current evidence pharmacotherapy through Food and Drug Administration–approved weight-loss drugs prior to hernia repair yield the weight-loss results needed for these patients to improve hernia outcomes, they added.
“Ultimately, there are various appropriate treatment modalities for each patient, and surgeons must use their judgment in selecting from among the different feasible options,” Dr. Lo Menzo and his colleagues wrote in the guidelines.
The authors report no relevant conflicts of interest.
SOURCE: Lo Menzo E et al. Surg Obes Relat Dis. 2018 Jul 19. doi: 10.1016/j.soard.2018.07.005.
and combining repair techniques with bariatric surgery can improve outcomes and lower the rate of complication for select patients, according to recent guidelines released by the American Society for Metabolic and Bariatric Surgery and the American Hernia Society.
Emanuele Lo Menzo, MD, of the Bariatric and Metabolic Institute at Cleveland Clinic Florida in Weston, and his colleagues issued a statement, published in the journal Surgery for Obesity and Related Diseases, based on available evidence from scientific literature on the impact of obesity on hernia surgery and what effect treating obesity has on improving hernia repair outcomes.
The authors noted abdominal wall hernia in obese patients is “a significant and increasingly common challenge for surgeons” and cited recent data from the American College of Surgeons National Surgical Quality Improvement Program that shows 60% of ventral hernia repairs (VHR) are performed on patients with body mass indexes (BMIs) above 30 kg/m2. Overall, they noted that general surgeons perform approximately 350,000 conventional hernia repairs (CHR) and 800,000 incisional hernia (IH) repairs each year.
The literature on the impact of obesity on hernia repair outcomes and the feasibility of a combined operation to address each problem has significant gaps, leaving surgeons to decide on a correct course based on individual patient needs. The guideline offers some recommendations, and notes areas that remain understudied. First, “in patients with severe obesity and [ventral hernia] and both being amenable to laparoscopic repair, combined hernia repair and [metabolic/bariatric surgery] may be safe and associated with good short-term outcomes and low risk of infection.” But the use of synthetic mesh in these patients is not well studied and so the guideline passes on a recommendation of mesh. For those obese patients with symptomatic abdominal wall hernias (AWHs) not amenable to laparoscopy, the guideline notes that metabolic/bariatric surgery first may be the best option.
Risk of hernia in obese patients
Studies suggest there is an increased risk of primary and IH among patients with BMIs greater than 25 kg/m2, with one study finding an 18.2% complication rate after single-incision laparoscopic surgery for patients with BMIs of 40 kg/m2 or higher, compared with a 3.5% complication rate among patients at a normal body weight. Severe BMI also is a risk factor for developing surgical site infection (SSI), which can cause recurrence, the authors said. Evidence from multiple studies further supports BMI as a risk factor for hernia recurrence, and intra-abdominal pressure from obesity increases the risk of developing an AWH.
“While most authors attribute the increased risk for AWH formation in the setting of obesity to BMI alone, others have suggested that abdominal circumference and elevated visceral fat may play a more significant role,” the authors wrote.
However, Dr. Lo Menzo and his colleagues admitted the actual rate of IH is difficult to calculate because some patients may not seek treatment for minimally symptomatic hernias. Patients with higher BMIs may not be aware of or seek treatment for common symptoms of IH such as groin bulge, or when they do seek treatment, it can present with symptoms such as incarceration or strangulation, they said. Patients with higher BMIs also are more likely to be offered “watchful waiting” because of higher complication rates in this patient population, which may contribute to incarceration or strangulation symptoms in these patients, they added.
Complications and recurrence
There is no one recommended repair technique or ideal BMI for hernia repair in obese patients, the authors wrote. One study found laparoscopic VHRs had a complication rate of 1.2% and reoccurrence rate of 5.5% at mean 25-month follow-up in patients with BMIs of 38 kg/m2 or greater, while a different study with a similar design found a 3.8% reoccurrence rate at 18-month follow-up. Degree of obesity can affect complication rate: One study showed that 73% of all complications after laparoscopic VHR occurred in the group of patients with BMIs of 30 kg/m2 or greater; a different study of laparoscopic VHR had an 8.3% hernia reoccurrence rate in patients with BMIs of 40 kg/m2 or greater, compared with patients at a normal weight (2.9%), with time to hernia reoccurrence being shorter in the higher-BMI group. A study of obese patients undergoing retromuscular open repair had a wound complication rate of 16% and a reoccurrence rate of 6%, with another study of patients undergoing umbilical hernia repair showing similar rates of complication and reoccurrence.
Panniculectomy
Among patients who underwent IH repair with panniculectomy, the authors found a 40% complication rate and a 10% reoccurrence rate in patients with BMIs of 40 kg/m2 or greater who received a partial underlay mesh placement hernia repair, while a different study found an increased risk of surgical site occurrences but not SSI in patients with BMIs of 34.3 kg/m2 or greater who underwent open ventral incisional hernia repair with panniculectomy. A third study found BMIs were not linked to a 55% complication rate in patients who underwent open ventral IH repair with and without mesh.
Simultaneous surgery
The authors noted that studies have shown that performing laparoscopic hernia repair and metabolic and bariatric surgery simultaneously is safe and has good short-term results. Specifically, patients who underwent the surgery with synthetic mesh had a low rate of infection or reoccurrence. Patients who underwent simultaneous weight-loss surgery and VHR had an elevated risk of SSI but no increased rate of 30-day mortality or morbidity, according to results from a large-scale registry. However, the authors noted patients with severe obesity may not be good candidates for simultaneous metabolic and bariatric surgery [MBS] and VHR, such as in patients with “large abdominal wall defects, loss of abdominal domain, extensive intestinal adhesive disease, poor quality skin (i.e., attenuated skin, prior skin graft, or ulcerated skin), incarcerated hernias containing bowel, hernias with previous synthetic mesh, hernias with chronic infection, or patients who have already undergone MBS with altered anatomy that is still intact.”
Preop weight loss
There is mixed data on the effect of weight loss prior to VHR through interventions such as very-low-calorie diets, pharmacotherapy, intragastric balloon therapy, and MBS as a first-stage procedure prior to VHR. Many patients treating their obesity with very-low-calorie diets lose approximately 10%-20% of their initial body weight and keep the weight off for at least 18 months, while one study showed patients who underwent intragastric balloon therapy lost approximately 10% of their body weight over 6 months; however, other studies have questioned the efficacy of this therapy, compared with a structured weight loss program or bariatric surgery. The authors also noted the difficulty of coordinating VHR after weight-loss surgery, lack of support from insurers, and they cited reports that cautioned bariatric surgeons to not leave hernias untreated during MBS. There is no current evidence pharmacotherapy through Food and Drug Administration–approved weight-loss drugs prior to hernia repair yield the weight-loss results needed for these patients to improve hernia outcomes, they added.
“Ultimately, there are various appropriate treatment modalities for each patient, and surgeons must use their judgment in selecting from among the different feasible options,” Dr. Lo Menzo and his colleagues wrote in the guidelines.
The authors report no relevant conflicts of interest.
SOURCE: Lo Menzo E et al. Surg Obes Relat Dis. 2018 Jul 19. doi: 10.1016/j.soard.2018.07.005.
FROM SURGERY FOR OBESITY AND RELATED DISEASES

Coagulopathy outbreak underscores danger of synthetic cannabinoids
Synthetic cannabinoids laced with superwarfarin were behind a recent outbreak of severe coagulopathy in Illinois.
In most cases, vitamin K replacement therapy alleviated symptoms, but four patients died after developing intracranial bleeding, said Amar H. Kelkar, MD, of the University of Illinois at Peoria.
Experts continue to look for how and why superwarfarin ended up in synthetic cannabinoids, whose street names include spice and K2, wrote Dr. Kelkar and his associates. Their report is in the New England Journal of Medicine.
Starting in March 2018, more than 150 patients across Illinois presented to hospitals with bleeding diathesis that involved persistent coagulopathy, the investigators explained. Early inquiries revealed that patients had consumed synthetic cannabinoids. Serum tests identified vitamin K antagonists, including brodifacoum, bromadiolone, and difenacoum. During arrests of suspected distributors, police confiscated synthetic cannabinoids that also tested positive for brodifacoum.
To help characterize the outbreak, the investigators reviewed admissions to Saint Francis Medical Center in Peoria, Ill., between March 28 and April 21, 2018. They identified 34 cases in which patients with vitamin K–dependent factor coagulopathy reported recent exposure to synthetic cannabinoids.
Fifteen of these patients underwent confirmatory anticoagulant testing, which universally confirmed superwarfarin poisoning. Brodifacoum was detected in all patients, difenacoum in five patients, bromadiolone in two patients, and warfarin in one patient.
Common presenting symptoms included gross hematuria (56% of patients) and abdominal pain (47%). Computed tomography identified renal abnormalities in 12 patients.
All patients received oral vitamin K1 (phytonadione). Red cell transfusions, fresh-frozen plasma infusions, and 4-factor prothrombin complex concentrate, or a combination of these treatments, were also used in some patients.
Among the four confirmed deaths in this outbreak, one occurred in a patient in this case series. The patient, a 37-year-old woman presenting to the emergency department with markedly reduced consciousness, was reported by her friends to have recently used synthetic cannabinoids and methamphetamine. She had no personal or family history of coagulopathy.
Computed tomography of the head without contrast material revealed severe acute intraparenchymal hemorrhage of the right basal nuclei and insula with intraventricular extension, a 10-mm left-sided midline shift, and herniation.
She met criteria for brain death 15 hours after hospital admission despite treatment with 10 mg of intravenous vitamin K1, four units of fresh frozen plasma, and 2,300 units of Kcentra.
Treating these patients after hospital discharge was difficult because of a lack of consensus guidelines and access to follow-up care, Dr. Kelkar and his associates noted. Some patients were quoted $24,000 to $34,000 per month for oral vitamin K1 therapy, which also made caring for them difficult and highlighted the need for confirmatory laboratory testing of suspected cases of superwarfarin poisoning.
Dr. Kelkar reported having no conflicts of interest. Two coinvestigators reported relationships outside the submitted work with Shire, CSL Behring, HEMA Biologics, and other companies.
SOURCE: Kelkar AH et al. N Engl J Med. 2018;379:1216-23.
Treating patients who are exposed to synthetic cannabinoid and a superwarfarin such as brodifacoum “requires more than the usual ‘treat ’em and street ’em’ approach,” wrote Jean M. Connors, MD.
“Brodifacoum is a successful rodenticide because of its extremely long half-life (approximately 16-36 days in humans),” Dr. Connors noted.
The drug also is lipophilic, causing tissue sequestration. Once exposed, patients often develop coagulopathy lasting 9 months or longer, she said.
Compared with warfarin poisoning, brodifacoum therefore requires substantially higher-dose and longer-term vitamin K1 therapy. Among the patients in this case series, the maximum outpatient dose was 50 mg, three times daily, and one patient was prescribed 25 mg, twice daily for 270 days, Dr. Connors noted.
“[Dr. Kelkar and his associates] highlight the resources and coordination needed for dealing with a public health crisis that has a prolonged duration of effect,” she added. “Because the synthetic cannabinoid market is lucrative, new products with new toxicity profiles are likely to crop up.”
Dr. Connors is with Brigham and Women’s Hospital, Dana-Farber Cancer Institute, and Harvard Medical School, all in Boston. She reported personal fees from Bristol-Myers Squibb, Portola, Dova Pharmaceuticals, and Unum Therapeutics outside the submitted work. These comments are from her accompanying editorial (N Engl J Med. 2018;379:1275-7).
Treating patients who are exposed to synthetic cannabinoid and a superwarfarin such as brodifacoum “requires more than the usual ‘treat ’em and street ’em’ approach,” wrote Jean M. Connors, MD.
“Brodifacoum is a successful rodenticide because of its extremely long half-life (approximately 16-36 days in humans),” Dr. Connors noted.
The drug also is lipophilic, causing tissue sequestration. Once exposed, patients often develop coagulopathy lasting 9 months or longer, she said.
Compared with warfarin poisoning, brodifacoum therefore requires substantially higher-dose and longer-term vitamin K1 therapy. Among the patients in this case series, the maximum outpatient dose was 50 mg, three times daily, and one patient was prescribed 25 mg, twice daily for 270 days, Dr. Connors noted.
“[Dr. Kelkar and his associates] highlight the resources and coordination needed for dealing with a public health crisis that has a prolonged duration of effect,” she added. “Because the synthetic cannabinoid market is lucrative, new products with new toxicity profiles are likely to crop up.”
Dr. Connors is with Brigham and Women’s Hospital, Dana-Farber Cancer Institute, and Harvard Medical School, all in Boston. She reported personal fees from Bristol-Myers Squibb, Portola, Dova Pharmaceuticals, and Unum Therapeutics outside the submitted work. These comments are from her accompanying editorial (N Engl J Med. 2018;379:1275-7).
Treating patients who are exposed to synthetic cannabinoid and a superwarfarin such as brodifacoum “requires more than the usual ‘treat ’em and street ’em’ approach,” wrote Jean M. Connors, MD.
“Brodifacoum is a successful rodenticide because of its extremely long half-life (approximately 16-36 days in humans),” Dr. Connors noted.
The drug also is lipophilic, causing tissue sequestration. Once exposed, patients often develop coagulopathy lasting 9 months or longer, she said.
Compared with warfarin poisoning, brodifacoum therefore requires substantially higher-dose and longer-term vitamin K1 therapy. Among the patients in this case series, the maximum outpatient dose was 50 mg, three times daily, and one patient was prescribed 25 mg, twice daily for 270 days, Dr. Connors noted.
“[Dr. Kelkar and his associates] highlight the resources and coordination needed for dealing with a public health crisis that has a prolonged duration of effect,” she added. “Because the synthetic cannabinoid market is lucrative, new products with new toxicity profiles are likely to crop up.”
Dr. Connors is with Brigham and Women’s Hospital, Dana-Farber Cancer Institute, and Harvard Medical School, all in Boston. She reported personal fees from Bristol-Myers Squibb, Portola, Dova Pharmaceuticals, and Unum Therapeutics outside the submitted work. These comments are from her accompanying editorial (N Engl J Med. 2018;379:1275-7).
Synthetic cannabinoids laced with superwarfarin were behind a recent outbreak of severe coagulopathy in Illinois.
In most cases, vitamin K replacement therapy alleviated symptoms, but four patients died after developing intracranial bleeding, said Amar H. Kelkar, MD, of the University of Illinois at Peoria.
Experts continue to look for how and why superwarfarin ended up in synthetic cannabinoids, whose street names include spice and K2, wrote Dr. Kelkar and his associates. Their report is in the New England Journal of Medicine.
Starting in March 2018, more than 150 patients across Illinois presented to hospitals with bleeding diathesis that involved persistent coagulopathy, the investigators explained. Early inquiries revealed that patients had consumed synthetic cannabinoids. Serum tests identified vitamin K antagonists, including brodifacoum, bromadiolone, and difenacoum. During arrests of suspected distributors, police confiscated synthetic cannabinoids that also tested positive for brodifacoum.
To help characterize the outbreak, the investigators reviewed admissions to Saint Francis Medical Center in Peoria, Ill., between March 28 and April 21, 2018. They identified 34 cases in which patients with vitamin K–dependent factor coagulopathy reported recent exposure to synthetic cannabinoids.
Fifteen of these patients underwent confirmatory anticoagulant testing, which universally confirmed superwarfarin poisoning. Brodifacoum was detected in all patients, difenacoum in five patients, bromadiolone in two patients, and warfarin in one patient.
Common presenting symptoms included gross hematuria (56% of patients) and abdominal pain (47%). Computed tomography identified renal abnormalities in 12 patients.
All patients received oral vitamin K1 (phytonadione). Red cell transfusions, fresh-frozen plasma infusions, and 4-factor prothrombin complex concentrate, or a combination of these treatments, were also used in some patients.
Among the four confirmed deaths in this outbreak, one occurred in a patient in this case series. The patient, a 37-year-old woman presenting to the emergency department with markedly reduced consciousness, was reported by her friends to have recently used synthetic cannabinoids and methamphetamine. She had no personal or family history of coagulopathy.
Computed tomography of the head without contrast material revealed severe acute intraparenchymal hemorrhage of the right basal nuclei and insula with intraventricular extension, a 10-mm left-sided midline shift, and herniation.
She met criteria for brain death 15 hours after hospital admission despite treatment with 10 mg of intravenous vitamin K1, four units of fresh frozen plasma, and 2,300 units of Kcentra.
Treating these patients after hospital discharge was difficult because of a lack of consensus guidelines and access to follow-up care, Dr. Kelkar and his associates noted. Some patients were quoted $24,000 to $34,000 per month for oral vitamin K1 therapy, which also made caring for them difficult and highlighted the need for confirmatory laboratory testing of suspected cases of superwarfarin poisoning.
Dr. Kelkar reported having no conflicts of interest. Two coinvestigators reported relationships outside the submitted work with Shire, CSL Behring, HEMA Biologics, and other companies.
SOURCE: Kelkar AH et al. N Engl J Med. 2018;379:1216-23.
Synthetic cannabinoids laced with superwarfarin were behind a recent outbreak of severe coagulopathy in Illinois.
In most cases, vitamin K replacement therapy alleviated symptoms, but four patients died after developing intracranial bleeding, said Amar H. Kelkar, MD, of the University of Illinois at Peoria.
Experts continue to look for how and why superwarfarin ended up in synthetic cannabinoids, whose street names include spice and K2, wrote Dr. Kelkar and his associates. Their report is in the New England Journal of Medicine.
Starting in March 2018, more than 150 patients across Illinois presented to hospitals with bleeding diathesis that involved persistent coagulopathy, the investigators explained. Early inquiries revealed that patients had consumed synthetic cannabinoids. Serum tests identified vitamin K antagonists, including brodifacoum, bromadiolone, and difenacoum. During arrests of suspected distributors, police confiscated synthetic cannabinoids that also tested positive for brodifacoum.
To help characterize the outbreak, the investigators reviewed admissions to Saint Francis Medical Center in Peoria, Ill., between March 28 and April 21, 2018. They identified 34 cases in which patients with vitamin K–dependent factor coagulopathy reported recent exposure to synthetic cannabinoids.
Fifteen of these patients underwent confirmatory anticoagulant testing, which universally confirmed superwarfarin poisoning. Brodifacoum was detected in all patients, difenacoum in five patients, bromadiolone in two patients, and warfarin in one patient.
Common presenting symptoms included gross hematuria (56% of patients) and abdominal pain (47%). Computed tomography identified renal abnormalities in 12 patients.
All patients received oral vitamin K1 (phytonadione). Red cell transfusions, fresh-frozen plasma infusions, and 4-factor prothrombin complex concentrate, or a combination of these treatments, were also used in some patients.
Among the four confirmed deaths in this outbreak, one occurred in a patient in this case series. The patient, a 37-year-old woman presenting to the emergency department with markedly reduced consciousness, was reported by her friends to have recently used synthetic cannabinoids and methamphetamine. She had no personal or family history of coagulopathy.
Computed tomography of the head without contrast material revealed severe acute intraparenchymal hemorrhage of the right basal nuclei and insula with intraventricular extension, a 10-mm left-sided midline shift, and herniation.
She met criteria for brain death 15 hours after hospital admission despite treatment with 10 mg of intravenous vitamin K1, four units of fresh frozen plasma, and 2,300 units of Kcentra.
Treating these patients after hospital discharge was difficult because of a lack of consensus guidelines and access to follow-up care, Dr. Kelkar and his associates noted. Some patients were quoted $24,000 to $34,000 per month for oral vitamin K1 therapy, which also made caring for them difficult and highlighted the need for confirmatory laboratory testing of suspected cases of superwarfarin poisoning.
Dr. Kelkar reported having no conflicts of interest. Two coinvestigators reported relationships outside the submitted work with Shire, CSL Behring, HEMA Biologics, and other companies.
SOURCE: Kelkar AH et al. N Engl J Med. 2018;379:1216-23.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: There were more than 150 cases in Illinois with four deaths among patients who developed spontaneous intracranial bleeding.
Study details: A single-institution case series of 15 patients.
Disclosures: Dr. Kelkar reported having no conflicts of interest. Two coinvestigators reported relationships outside the submitted work with Shire, CSL Behring, HEMA Biologics, and other companies.
Source: Kelkar AH et al. N Engl J Med. 2018;379:1216-23.
ICYMI: Wearable cardioverter-defibrillator ineffective at reducing arrhythmic death risk
Patients who recently experienced an MI and had an ejection fraction of 35% or less who used a wearable cardioverter-defibrillator had no decreased risk of arrhythmic death, compared with people who did not use a wearable cardioverter-defibrillator, according to an analysis of data from the multicenter, randomized, and controlled VEST trial (NCT01446965). The results were published Sept. 26 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1800781).
We covered this story before it was published in the journal. Find our conference coverage at the links below.
Patients who recently experienced an MI and had an ejection fraction of 35% or less who used a wearable cardioverter-defibrillator had no decreased risk of arrhythmic death, compared with people who did not use a wearable cardioverter-defibrillator, according to an analysis of data from the multicenter, randomized, and controlled VEST trial (NCT01446965). The results were published Sept. 26 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1800781).
We covered this story before it was published in the journal. Find our conference coverage at the links below.
Patients who recently experienced an MI and had an ejection fraction of 35% or less who used a wearable cardioverter-defibrillator had no decreased risk of arrhythmic death, compared with people who did not use a wearable cardioverter-defibrillator, according to an analysis of data from the multicenter, randomized, and controlled VEST trial (NCT01446965). The results were published Sept. 26 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1800781).
We covered this story before it was published in the journal. Find our conference coverage at the links below.
FROM NEW ENGLAND JOURNAL OF MEDICINE
AGA comments on HHS’ drug affordability blueprint
AGA’s new drug affordability principles were put into action in July when AGA Chair Sheila Crowe, MD, AGAF, provided comments on the Department of Health & Human Services (HHS) recent policy statement and Request for Information, “HHS Blueprint to Lower Drug Prices and Reduce Out-of-Pocket Costs” (Blueprint). Comments were limited to four areas of the Blueprint.
Medicare Part B to Part D drug transition
Over the past decade there has been interest in consolidating Part B and Part D drug coverage and payment. AGA urges physician-administered drugs and biologics to remain under Part B due to the complexities surrounding them.
Since Part D does not allow for supplemental coverage and has higher coinsurance, this action would achieve savings by shifting costs to Medicare beneficiaries. Moving them to Part D would also increase the risk of waste leading to unnecessary Medicare spending. AGA urges the administration to avoid policy solutions that achieve Medicare program savings at the expense of Medicare beneficiaries.
Indication-based payments
The Blueprint seems to imply that off-label uses of prescription drugs are inherently less valuable than on-label uses. If the administration moves towards value-based pricing, off-label indications should not automatically be valued less, or priced lower, than on-label indications. Specifically, AGA urges the administration to ensure all medically accepted indications are appropriately valued for a drug or biologic.
Medicare Part B Competitive Acquisition Program (CAP)
AGA does not oppose the idea of a new, voluntary CAP program as it would allow interested physicians and practices to provide Part B drug administration without the burden of high acquisition costs.
AGA strongly opposes a future Part B CAP that includes vendors or Medicare carriers conducting medical reviews or utilization management. Utilization management undermines shared decision-making between physicians and patients, increases physician burden, and often puts patients at risk by delaying access to necessary care.
Reduce patient out-of-pocket spending
As out-of-pocket costs continue to rise, AGA supports the administration’s plans to increase cost transparency in the Medicare program as it increases the efficiency of the shared decision-making process between patient and physician. Drug and biologic manufacturers, health plans, and pharmacy managers should work together to lower out-of-pocket expenses for Medicare beneficiaries and for all people with digestive diseases.
Although AGA shares the Blueprint’s goal of lowering the cost of prescription drugs, lowering out-of-pocket costs for patients, increasing competition and fostering innovation, we are concerned that the recent proposal by the Trump administration to allow Medicare Advantage (MA) plans to utilize step therapy would threaten the aforementioned goals. Step therapy is a utilization management tool used by insurers that requires patients to fail one or more medications before covering the original therapy that is prescribed by the physician. AGA is concerned that the recent announcement by the Trump administration would not provide patients with the necessary protections, would increase the regulatory burden that physicians already face with step therapy and prior authorization, and could hinder innovation by preferring the lowest cost medication which may not necessarily be the most effective. AGA will continue to push for necessary patient protections to ensure that patients have the ability to appeal step therapy protocols when appropriate and are able to receive the medication that their physician thinks is the most effective to manage their condition.
AGA’s new drug affordability principles were put into action in July when AGA Chair Sheila Crowe, MD, AGAF, provided comments on the Department of Health & Human Services (HHS) recent policy statement and Request for Information, “HHS Blueprint to Lower Drug Prices and Reduce Out-of-Pocket Costs” (Blueprint). Comments were limited to four areas of the Blueprint.
Medicare Part B to Part D drug transition
Over the past decade there has been interest in consolidating Part B and Part D drug coverage and payment. AGA urges physician-administered drugs and biologics to remain under Part B due to the complexities surrounding them.
Since Part D does not allow for supplemental coverage and has higher coinsurance, this action would achieve savings by shifting costs to Medicare beneficiaries. Moving them to Part D would also increase the risk of waste leading to unnecessary Medicare spending. AGA urges the administration to avoid policy solutions that achieve Medicare program savings at the expense of Medicare beneficiaries.
Indication-based payments
The Blueprint seems to imply that off-label uses of prescription drugs are inherently less valuable than on-label uses. If the administration moves towards value-based pricing, off-label indications should not automatically be valued less, or priced lower, than on-label indications. Specifically, AGA urges the administration to ensure all medically accepted indications are appropriately valued for a drug or biologic.
Medicare Part B Competitive Acquisition Program (CAP)
AGA does not oppose the idea of a new, voluntary CAP program as it would allow interested physicians and practices to provide Part B drug administration without the burden of high acquisition costs.
AGA strongly opposes a future Part B CAP that includes vendors or Medicare carriers conducting medical reviews or utilization management. Utilization management undermines shared decision-making between physicians and patients, increases physician burden, and often puts patients at risk by delaying access to necessary care.
Reduce patient out-of-pocket spending
As out-of-pocket costs continue to rise, AGA supports the administration’s plans to increase cost transparency in the Medicare program as it increases the efficiency of the shared decision-making process between patient and physician. Drug and biologic manufacturers, health plans, and pharmacy managers should work together to lower out-of-pocket expenses for Medicare beneficiaries and for all people with digestive diseases.
Although AGA shares the Blueprint’s goal of lowering the cost of prescription drugs, lowering out-of-pocket costs for patients, increasing competition and fostering innovation, we are concerned that the recent proposal by the Trump administration to allow Medicare Advantage (MA) plans to utilize step therapy would threaten the aforementioned goals. Step therapy is a utilization management tool used by insurers that requires patients to fail one or more medications before covering the original therapy that is prescribed by the physician. AGA is concerned that the recent announcement by the Trump administration would not provide patients with the necessary protections, would increase the regulatory burden that physicians already face with step therapy and prior authorization, and could hinder innovation by preferring the lowest cost medication which may not necessarily be the most effective. AGA will continue to push for necessary patient protections to ensure that patients have the ability to appeal step therapy protocols when appropriate and are able to receive the medication that their physician thinks is the most effective to manage their condition.
AGA’s new drug affordability principles were put into action in July when AGA Chair Sheila Crowe, MD, AGAF, provided comments on the Department of Health & Human Services (HHS) recent policy statement and Request for Information, “HHS Blueprint to Lower Drug Prices and Reduce Out-of-Pocket Costs” (Blueprint). Comments were limited to four areas of the Blueprint.
Medicare Part B to Part D drug transition
Over the past decade there has been interest in consolidating Part B and Part D drug coverage and payment. AGA urges physician-administered drugs and biologics to remain under Part B due to the complexities surrounding them.
Since Part D does not allow for supplemental coverage and has higher coinsurance, this action would achieve savings by shifting costs to Medicare beneficiaries. Moving them to Part D would also increase the risk of waste leading to unnecessary Medicare spending. AGA urges the administration to avoid policy solutions that achieve Medicare program savings at the expense of Medicare beneficiaries.
Indication-based payments
The Blueprint seems to imply that off-label uses of prescription drugs are inherently less valuable than on-label uses. If the administration moves towards value-based pricing, off-label indications should not automatically be valued less, or priced lower, than on-label indications. Specifically, AGA urges the administration to ensure all medically accepted indications are appropriately valued for a drug or biologic.
Medicare Part B Competitive Acquisition Program (CAP)
AGA does not oppose the idea of a new, voluntary CAP program as it would allow interested physicians and practices to provide Part B drug administration without the burden of high acquisition costs.
AGA strongly opposes a future Part B CAP that includes vendors or Medicare carriers conducting medical reviews or utilization management. Utilization management undermines shared decision-making between physicians and patients, increases physician burden, and often puts patients at risk by delaying access to necessary care.
Reduce patient out-of-pocket spending
As out-of-pocket costs continue to rise, AGA supports the administration’s plans to increase cost transparency in the Medicare program as it increases the efficiency of the shared decision-making process between patient and physician. Drug and biologic manufacturers, health plans, and pharmacy managers should work together to lower out-of-pocket expenses for Medicare beneficiaries and for all people with digestive diseases.
Although AGA shares the Blueprint’s goal of lowering the cost of prescription drugs, lowering out-of-pocket costs for patients, increasing competition and fostering innovation, we are concerned that the recent proposal by the Trump administration to allow Medicare Advantage (MA) plans to utilize step therapy would threaten the aforementioned goals. Step therapy is a utilization management tool used by insurers that requires patients to fail one or more medications before covering the original therapy that is prescribed by the physician. AGA is concerned that the recent announcement by the Trump administration would not provide patients with the necessary protections, would increase the regulatory burden that physicians already face with step therapy and prior authorization, and could hinder innovation by preferring the lowest cost medication which may not necessarily be the most effective. AGA will continue to push for necessary patient protections to ensure that patients have the ability to appeal step therapy protocols when appropriate and are able to receive the medication that their physician thinks is the most effective to manage their condition.
AGA Center for Gut Microbiome Research and Education scientific advisory board welcomes new members
Four leading experts in microbiome research have recently been appointed to the scientific advisory board of the AGA Center for Gut Microbiome Research and Education.
Robert A. Britton, PhD
Baylor College of Medicine, Houston, Texas
Dr. Britton studies the role of microbes in health and diseases, with a focus on identifying microbes with therapeutic properties for a variety of disorders.
Suzanne Devkota, PhD
Cedars-Sinai Medical Center, Los Angeles, California
Dr. Devkota investigates the role of diet in shaping the community of bacteria that live in our intestines (the “gut microbiome”).
Lita M. Proctor, PhD
National Human Genome Research Institute, Rockville, Maryland
Dr. Proctor is responsible for coordination of the Human Microbiome Project (HMP), an eight-year NIH Common Fund initiative to create a toolbox of resources for the emerging field of microbiome research.
Liping Zhao, PhD
Rutgers University, New Brunswick, New Jersey
Dr. Zhao studies the interactions between diet and gut microbiota in the onset and progression of chronic diseases such as obesity and diabetes.
AGA recognizes the outgoing members of the scientific advisory board who have made valuable contributions to the center’s work over their terms: Lee M. Kaplan, MD, PhD, AGAF, Zain Kassam, MD, MPH, and Ece Mutlu, MD, MBA, AGAF.
Four leading experts in microbiome research have recently been appointed to the scientific advisory board of the AGA Center for Gut Microbiome Research and Education.
Robert A. Britton, PhD
Baylor College of Medicine, Houston, Texas
Dr. Britton studies the role of microbes in health and diseases, with a focus on identifying microbes with therapeutic properties for a variety of disorders.
Suzanne Devkota, PhD
Cedars-Sinai Medical Center, Los Angeles, California
Dr. Devkota investigates the role of diet in shaping the community of bacteria that live in our intestines (the “gut microbiome”).
Lita M. Proctor, PhD
National Human Genome Research Institute, Rockville, Maryland
Dr. Proctor is responsible for coordination of the Human Microbiome Project (HMP), an eight-year NIH Common Fund initiative to create a toolbox of resources for the emerging field of microbiome research.
Liping Zhao, PhD
Rutgers University, New Brunswick, New Jersey
Dr. Zhao studies the interactions between diet and gut microbiota in the onset and progression of chronic diseases such as obesity and diabetes.
AGA recognizes the outgoing members of the scientific advisory board who have made valuable contributions to the center’s work over their terms: Lee M. Kaplan, MD, PhD, AGAF, Zain Kassam, MD, MPH, and Ece Mutlu, MD, MBA, AGAF.
Four leading experts in microbiome research have recently been appointed to the scientific advisory board of the AGA Center for Gut Microbiome Research and Education.
Robert A. Britton, PhD
Baylor College of Medicine, Houston, Texas
Dr. Britton studies the role of microbes in health and diseases, with a focus on identifying microbes with therapeutic properties for a variety of disorders.
Suzanne Devkota, PhD
Cedars-Sinai Medical Center, Los Angeles, California
Dr. Devkota investigates the role of diet in shaping the community of bacteria that live in our intestines (the “gut microbiome”).
Lita M. Proctor, PhD
National Human Genome Research Institute, Rockville, Maryland
Dr. Proctor is responsible for coordination of the Human Microbiome Project (HMP), an eight-year NIH Common Fund initiative to create a toolbox of resources for the emerging field of microbiome research.
Liping Zhao, PhD
Rutgers University, New Brunswick, New Jersey
Dr. Zhao studies the interactions between diet and gut microbiota in the onset and progression of chronic diseases such as obesity and diabetes.
AGA recognizes the outgoing members of the scientific advisory board who have made valuable contributions to the center’s work over their terms: Lee M. Kaplan, MD, PhD, AGAF, Zain Kassam, MD, MPH, and Ece Mutlu, MD, MBA, AGAF.
Senate approves $2 billion NIH increase
AGA applauds Congress for recognizing the need to sustain the momentum for NIH funding and to ensure that it has the purchasing power it needs to attract the best and brightest scientists to pursue careers in research.
The Senate approved the fiscal year (FY) 2019 Labor-HHS-Education Appropriations bill that included a $2 billion increase in funding for the National Institutes of Health (NIH). This increase represents a 5.5% increase in NIH funding, on top of the 8.8% increase that NIH received as part of the Omnibus Appropriations bill for FY 2018. The funding also represents the largest increase in funding since the doubling period (FY 1999-FY 2003), and enabled NIH to support 1,149 additional research grants.
The $2 billion increase included in the Senate bill reflects the necessary funding that NIH needs to keep pace with medical research inflation. This increase will enable NIH to continue to fund innovative research, improve the quality of care for millions of Americans, and maintain U.S. global leadership in medical research. The House has recommended a $1.1 billion increase in NIH funding, but AGA will be pushing for the $2 billion increase. House and Senate leaders will be working to negotiate an agreement on funding.
AGA applauds Congress for recognizing the need to sustain the momentum for NIH funding and to ensure that it has the purchasing power it needs to attract the best and brightest scientists to pursue careers in research.
The Senate approved the fiscal year (FY) 2019 Labor-HHS-Education Appropriations bill that included a $2 billion increase in funding for the National Institutes of Health (NIH). This increase represents a 5.5% increase in NIH funding, on top of the 8.8% increase that NIH received as part of the Omnibus Appropriations bill for FY 2018. The funding also represents the largest increase in funding since the doubling period (FY 1999-FY 2003), and enabled NIH to support 1,149 additional research grants.
The $2 billion increase included in the Senate bill reflects the necessary funding that NIH needs to keep pace with medical research inflation. This increase will enable NIH to continue to fund innovative research, improve the quality of care for millions of Americans, and maintain U.S. global leadership in medical research. The House has recommended a $1.1 billion increase in NIH funding, but AGA will be pushing for the $2 billion increase. House and Senate leaders will be working to negotiate an agreement on funding.
AGA applauds Congress for recognizing the need to sustain the momentum for NIH funding and to ensure that it has the purchasing power it needs to attract the best and brightest scientists to pursue careers in research.
The Senate approved the fiscal year (FY) 2019 Labor-HHS-Education Appropriations bill that included a $2 billion increase in funding for the National Institutes of Health (NIH). This increase represents a 5.5% increase in NIH funding, on top of the 8.8% increase that NIH received as part of the Omnibus Appropriations bill for FY 2018. The funding also represents the largest increase in funding since the doubling period (FY 1999-FY 2003), and enabled NIH to support 1,149 additional research grants.
The $2 billion increase included in the Senate bill reflects the necessary funding that NIH needs to keep pace with medical research inflation. This increase will enable NIH to continue to fund innovative research, improve the quality of care for millions of Americans, and maintain U.S. global leadership in medical research. The House has recommended a $1.1 billion increase in NIH funding, but AGA will be pushing for the $2 billion increase. House and Senate leaders will be working to negotiate an agreement on funding.