User login
SPRINT: Pill burden affects ability to reach systolic BP control
CHICAGO –

“Five or more medications” meant total drug burden, both for hypertension and comorbidities. “When you are treating a patient for hypertension, you care about blood pressure, but you also need to care about” what else they are on, and their total drug burden, “because it affects their ability to get to their blood pressure goal, especially if you’re targeting intensive control,” said lead investigator Catherine Derington, PharmD, a postdoctoral fellow at the University of Colorado, Aurora.
Good old-fashioned exercise and weight loss remain potent non–pill options, she noted at the joint scientific sessions of AHA Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, and American Society of Hypertension.

The take-home message is to use more combination drugs for hypertension “to get patients on fewer pills.” Also, “eliminate drugs [patients] don’t need,” said SPRINT investigator William C. Cushman, MD, professor of medicine and physiology at the University of Tennessee, Memphis, when asked what he thought of the new findings.
SPRINT [Systolic Blood Pressure Intervention Trial] found that targeting systolic blood pressures below 120 mm Hg, as compared with less than 140 mm Hg, led to lower rates of MI, stroke, heart failure, and death from any cause.
Medication burden had no effect on patients in the 140 mm Hg group; they achieved a mean systolic blood pressure (SBP) of 136 mm Hg at 1 year, whether they were on fewer than five drugs a day or more. Hitting that target took an average of 1.8 hypertension medications.
Reaching 120 mm Hg generally took one extra drug (an average of 2.8), and the overall pill burden did matter; 2,463 patients in the intensive arm on fewer than five medications dropped their mean SBP 20 mm Hg at one year, while 1,698 taking more than five had a 15–mm Hg reduction. The group with the lower pill burden had a mean SBP of 120.6 mm Hg and those taking five or more had a mean SBP of 122.5 mm Hg. It’s a small difference but likely important.
Comorbidities in SPRINT included kidney and cardiovascular disease, among others, but the trial excluded patients with diabetes. Many patients were on statins and aspirin.
Pharmacy review is especially a good idea in the elderly.
Patients on five or more medications had higher rates of significant adverse events in the post hoc analysis (about 50% versus about 30%), including hypotension, syncope, electrolyte abnormalities, and acute kidney injury, regardless if they were in the 120–mm Hg or 140–mm Hg group.
About 45% in the intensive arm reported high adherence (Morisky Medication Adherence Scale-8 score greater than 8 at 12 months); medication burden made no difference. Oddly, in the standard-treatment arm, more patients on five or more drugs reported high adherence, 44.5% versus 38.1% among patients taking fewer,
The post hoc analysis was based on pill bottle review at baseline. Medication count did not affect hypertension treatment satisfaction, which was about 84% in the intensive and 77% in the standard arm.
There was no industry funding for the work. Dr. Derington didn’t have any disclosures.
CHICAGO –
“Five or more medications” meant total drug burden, both for hypertension and comorbidities. “When you are treating a patient for hypertension, you care about blood pressure, but you also need to care about” what else they are on, and their total drug burden, “because it affects their ability to get to their blood pressure goal, especially if you’re targeting intensive control,” said lead investigator Catherine Derington, PharmD, a postdoctoral fellow at the University of Colorado, Aurora.
Good old-fashioned exercise and weight loss remain potent non–pill options, she noted at the joint scientific sessions of AHA Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, and American Society of Hypertension.
The take-home message is to use more combination drugs for hypertension “to get patients on fewer pills.” Also, “eliminate drugs [patients] don’t need,” said SPRINT investigator William C. Cushman, MD, professor of medicine and physiology at the University of Tennessee, Memphis, when asked what he thought of the new findings.
SPRINT [Systolic Blood Pressure Intervention Trial] found that targeting systolic blood pressures below 120 mm Hg, as compared with less than 140 mm Hg, led to lower rates of MI, stroke, heart failure, and death from any cause.
Medication burden had no effect on patients in the 140 mm Hg group; they achieved a mean systolic blood pressure (SBP) of 136 mm Hg at 1 year, whether they were on fewer than five drugs a day or more. Hitting that target took an average of 1.8 hypertension medications.
Reaching 120 mm Hg generally took one extra drug (an average of 2.8), and the overall pill burden did matter; 2,463 patients in the intensive arm on fewer than five medications dropped their mean SBP 20 mm Hg at one year, while 1,698 taking more than five had a 15–mm Hg reduction. The group with the lower pill burden had a mean SBP of 120.6 mm Hg and those taking five or more had a mean SBP of 122.5 mm Hg. It’s a small difference but likely important.
Comorbidities in SPRINT included kidney and cardiovascular disease, among others, but the trial excluded patients with diabetes. Many patients were on statins and aspirin.
Pharmacy review is especially a good idea in the elderly.
Patients on five or more medications had higher rates of significant adverse events in the post hoc analysis (about 50% versus about 30%), including hypotension, syncope, electrolyte abnormalities, and acute kidney injury, regardless if they were in the 120–mm Hg or 140–mm Hg group.
About 45% in the intensive arm reported high adherence (Morisky Medication Adherence Scale-8 score greater than 8 at 12 months); medication burden made no difference. Oddly, in the standard-treatment arm, more patients on five or more drugs reported high adherence, 44.5% versus 38.1% among patients taking fewer,
The post hoc analysis was based on pill bottle review at baseline. Medication count did not affect hypertension treatment satisfaction, which was about 84% in the intensive and 77% in the standard arm.
There was no industry funding for the work. Dr. Derington didn’t have any disclosures.
CHICAGO –
“Five or more medications” meant total drug burden, both for hypertension and comorbidities. “When you are treating a patient for hypertension, you care about blood pressure, but you also need to care about” what else they are on, and their total drug burden, “because it affects their ability to get to their blood pressure goal, especially if you’re targeting intensive control,” said lead investigator Catherine Derington, PharmD, a postdoctoral fellow at the University of Colorado, Aurora.
Good old-fashioned exercise and weight loss remain potent non–pill options, she noted at the joint scientific sessions of AHA Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, and American Society of Hypertension.
The take-home message is to use more combination drugs for hypertension “to get patients on fewer pills.” Also, “eliminate drugs [patients] don’t need,” said SPRINT investigator William C. Cushman, MD, professor of medicine and physiology at the University of Tennessee, Memphis, when asked what he thought of the new findings.
SPRINT [Systolic Blood Pressure Intervention Trial] found that targeting systolic blood pressures below 120 mm Hg, as compared with less than 140 mm Hg, led to lower rates of MI, stroke, heart failure, and death from any cause.
Medication burden had no effect on patients in the 140 mm Hg group; they achieved a mean systolic blood pressure (SBP) of 136 mm Hg at 1 year, whether they were on fewer than five drugs a day or more. Hitting that target took an average of 1.8 hypertension medications.
Reaching 120 mm Hg generally took one extra drug (an average of 2.8), and the overall pill burden did matter; 2,463 patients in the intensive arm on fewer than five medications dropped their mean SBP 20 mm Hg at one year, while 1,698 taking more than five had a 15–mm Hg reduction. The group with the lower pill burden had a mean SBP of 120.6 mm Hg and those taking five or more had a mean SBP of 122.5 mm Hg. It’s a small difference but likely important.
Comorbidities in SPRINT included kidney and cardiovascular disease, among others, but the trial excluded patients with diabetes. Many patients were on statins and aspirin.
Pharmacy review is especially a good idea in the elderly.
Patients on five or more medications had higher rates of significant adverse events in the post hoc analysis (about 50% versus about 30%), including hypotension, syncope, electrolyte abnormalities, and acute kidney injury, regardless if they were in the 120–mm Hg or 140–mm Hg group.
About 45% in the intensive arm reported high adherence (Morisky Medication Adherence Scale-8 score greater than 8 at 12 months); medication burden made no difference. Oddly, in the standard-treatment arm, more patients on five or more drugs reported high adherence, 44.5% versus 38.1% among patients taking fewer,
The post hoc analysis was based on pill bottle review at baseline. Medication count did not affect hypertension treatment satisfaction, which was about 84% in the intensive and 77% in the standard arm.
There was no industry funding for the work. Dr. Derington didn’t have any disclosures.
REPORTING FROM JOINT HYPERTENSION 2018
Key clinical point: Consider overall pill burden when managing hypertension.
Major finding: Patients in the SPRINT trial were less likely to meet the intensive treatment goal – systolic blood pressure below 120 mm Hg – if they were taking five or more medications a day.
Study details: Post hoc analysis of the SPRINT trial
Disclosures: There was no industry funding, and the lead investigator didn’t have any disclosures.
Source: Derington C et al. Joint Hypertension 2018, Abstract P208.
Breast cancer risk in type 2 diabetes related to adiposity
ORLANDO – findings from meta-analyses suggest.

In one meta-analysis of data from 21 prospective studies with a total of nearly 15.2 million women, 325,117 breast cancer cases, and a mean follow-up time of 8 years (nearly 33 million person-years), the risk of breast cancer was significantly greater among patients with diabetes than it was among patients without diabetes (summary relative risk, 1.11), Maria Bota reported at the annual scientific sessions of the American Diabetes Association.
However, there was substantial unexplained heterogeneity of results across the individual studies (I2 = 82%), said Ms. Bota, a faculty member at the International Prevention Research Institute, Lyon, France.
“When the analysis was restricted to the 12 studies that adjusted for [body mass index], the summary relative risk decreased to 1.09 and the heterogeneity also decreased to a moderate value of 32%; when the analysis was restricted to the 9 studies that did not adjust for BMI, the summary relative risk increased to 1.14 again, and the heterogeneity increased even more to 91%,” she said.
In an analysis that combined the results of the nine studies that did not adjust for BMI along with crude relative risks from studies that reported both crude and BMI-adjusted relative risks (17 studies in all), the SRR was 1.12, and heterogeneity among the studies was high at 84%.
Additionally, an analysis by menopausal status based on four studies that reported breast cancer in both pre- and postmenopausal women showed SRRs for breast cancer of 0.97 (a 3% decrease in risk) and 1.14 among diabetic vs. nondiabetic premenopausal women and postmenopausal women, respectively, she said, noting that heterogeneity was low (I2 = 0%) among the premenopausal breast cancer study groups and high (I2 = 70%) among the postmenopausal study groups.
The findings provide evidence for a moderately increased risk of breast cancer in women with T2DM, Ms. Bota said.
“However, the effect of the adjustment or lack of adjustment on the heterogeneity suggests that the higher risk of breast cancer in women with diabetes may not be due to diabetes itself, but to adiposity,” she said, adding that “this hypothesis is equally supported by our subgroup analysis according to menopausal status because we saw that the risk of breast cancer was only associated with diabetes in postmenopausal women and this pattern resembles the pattern of the risk of breast cancer associated with adiposity, which is also only increased in postmenopausal women.”
This study was limited by insufficient data for investigating the sources of heterogeneity, she said.
“Therefore we propose ... future pooled analyses based on individual data from good quality prospective studies in order to increase the study power and to do some detailed analysis of the links between adiposity, diabetes, and breast cancer,” she concluded, adding that new studies to examine those relationships only in premenopausal women are also needed
In a separate meta-analysis, she and her colleagues, including Peter Boyle, PhD, president of the International Prevention Research Institute, assessed the association between insulin treatment and breast cancer risk in patients with diabetes.
“The long-acting insulin analogues glargine and detemir have been shown in some studies to be associated with increased risk of breast cancer, and other studies have shown no association between the use of these two compounds and the risk of breast cancer,” Dr. Boyle said in a separate presentation at the ADA meeting. “It was important to sort out a little bit what was going on in the literature.”
Overall, the meta-analysis of data from 12 longitudinal cohort studies – including more than 6,000 cases of breast cancer – showed a slight increase in breast cancer risk in patients taking long-acting insulin (SRR, 1.13) with “a relatively reasonable” level of heterogeneity (I2 = 23%).
“But we see that the story is not that simple,” he said.
For example, some studies included only patients who were prescribed insulin for the first time after the study began (new users), some included only patients who were prescribed insulin before the study began (prevalent users), and some included both (ever users), which may have introduced bias in the results, he explained.
Studies of glargine included 4,168 breast cancer cases over a total of 1,418,743 person-years of observation, and studies of detemir included fewer than 2,047 breast cancer cases (not all studies reported case numbers). Among both new users of glargine and detemir, the SRR was 1.12, suggesting no real association between either glargine or detemir use and breast cancer, he said.
“One important take-home message is that you have to be careful that these pharmaco-epidemiological studies, even when working with the same database, may have conflicting results ... so we still need more robust standards in methods for [such] studies,” he concluded.
Ms. Bota reported having no disclosures. The study presented by Dr. Boyle was funded by Sanofi. Dr. Boyle is president of a charity that has received donations from Pfizer, Roche, Novartis, and Lilly.
SOURCE: Bota M. ADA 2018, Abstract 180-OR; Boyle P. ADA 2018, Abstract 133-OR.
ORLANDO – findings from meta-analyses suggest.
In one meta-analysis of data from 21 prospective studies with a total of nearly 15.2 million women, 325,117 breast cancer cases, and a mean follow-up time of 8 years (nearly 33 million person-years), the risk of breast cancer was significantly greater among patients with diabetes than it was among patients without diabetes (summary relative risk, 1.11), Maria Bota reported at the annual scientific sessions of the American Diabetes Association.
However, there was substantial unexplained heterogeneity of results across the individual studies (I2 = 82%), said Ms. Bota, a faculty member at the International Prevention Research Institute, Lyon, France.
“When the analysis was restricted to the 12 studies that adjusted for [body mass index], the summary relative risk decreased to 1.09 and the heterogeneity also decreased to a moderate value of 32%; when the analysis was restricted to the 9 studies that did not adjust for BMI, the summary relative risk increased to 1.14 again, and the heterogeneity increased even more to 91%,” she said.
In an analysis that combined the results of the nine studies that did not adjust for BMI along with crude relative risks from studies that reported both crude and BMI-adjusted relative risks (17 studies in all), the SRR was 1.12, and heterogeneity among the studies was high at 84%.
Additionally, an analysis by menopausal status based on four studies that reported breast cancer in both pre- and postmenopausal women showed SRRs for breast cancer of 0.97 (a 3% decrease in risk) and 1.14 among diabetic vs. nondiabetic premenopausal women and postmenopausal women, respectively, she said, noting that heterogeneity was low (I2 = 0%) among the premenopausal breast cancer study groups and high (I2 = 70%) among the postmenopausal study groups.
The findings provide evidence for a moderately increased risk of breast cancer in women with T2DM, Ms. Bota said.
“However, the effect of the adjustment or lack of adjustment on the heterogeneity suggests that the higher risk of breast cancer in women with diabetes may not be due to diabetes itself, but to adiposity,” she said, adding that “this hypothesis is equally supported by our subgroup analysis according to menopausal status because we saw that the risk of breast cancer was only associated with diabetes in postmenopausal women and this pattern resembles the pattern of the risk of breast cancer associated with adiposity, which is also only increased in postmenopausal women.”
This study was limited by insufficient data for investigating the sources of heterogeneity, she said.
“Therefore we propose ... future pooled analyses based on individual data from good quality prospective studies in order to increase the study power and to do some detailed analysis of the links between adiposity, diabetes, and breast cancer,” she concluded, adding that new studies to examine those relationships only in premenopausal women are also needed
In a separate meta-analysis, she and her colleagues, including Peter Boyle, PhD, president of the International Prevention Research Institute, assessed the association between insulin treatment and breast cancer risk in patients with diabetes.
“The long-acting insulin analogues glargine and detemir have been shown in some studies to be associated with increased risk of breast cancer, and other studies have shown no association between the use of these two compounds and the risk of breast cancer,” Dr. Boyle said in a separate presentation at the ADA meeting. “It was important to sort out a little bit what was going on in the literature.”
Overall, the meta-analysis of data from 12 longitudinal cohort studies – including more than 6,000 cases of breast cancer – showed a slight increase in breast cancer risk in patients taking long-acting insulin (SRR, 1.13) with “a relatively reasonable” level of heterogeneity (I2 = 23%).
“But we see that the story is not that simple,” he said.
For example, some studies included only patients who were prescribed insulin for the first time after the study began (new users), some included only patients who were prescribed insulin before the study began (prevalent users), and some included both (ever users), which may have introduced bias in the results, he explained.
Studies of glargine included 4,168 breast cancer cases over a total of 1,418,743 person-years of observation, and studies of detemir included fewer than 2,047 breast cancer cases (not all studies reported case numbers). Among both new users of glargine and detemir, the SRR was 1.12, suggesting no real association between either glargine or detemir use and breast cancer, he said.
“One important take-home message is that you have to be careful that these pharmaco-epidemiological studies, even when working with the same database, may have conflicting results ... so we still need more robust standards in methods for [such] studies,” he concluded.
Ms. Bota reported having no disclosures. The study presented by Dr. Boyle was funded by Sanofi. Dr. Boyle is president of a charity that has received donations from Pfizer, Roche, Novartis, and Lilly.
SOURCE: Bota M. ADA 2018, Abstract 180-OR; Boyle P. ADA 2018, Abstract 133-OR.
ORLANDO – findings from meta-analyses suggest.
In one meta-analysis of data from 21 prospective studies with a total of nearly 15.2 million women, 325,117 breast cancer cases, and a mean follow-up time of 8 years (nearly 33 million person-years), the risk of breast cancer was significantly greater among patients with diabetes than it was among patients without diabetes (summary relative risk, 1.11), Maria Bota reported at the annual scientific sessions of the American Diabetes Association.
However, there was substantial unexplained heterogeneity of results across the individual studies (I2 = 82%), said Ms. Bota, a faculty member at the International Prevention Research Institute, Lyon, France.
“When the analysis was restricted to the 12 studies that adjusted for [body mass index], the summary relative risk decreased to 1.09 and the heterogeneity also decreased to a moderate value of 32%; when the analysis was restricted to the 9 studies that did not adjust for BMI, the summary relative risk increased to 1.14 again, and the heterogeneity increased even more to 91%,” she said.
In an analysis that combined the results of the nine studies that did not adjust for BMI along with crude relative risks from studies that reported both crude and BMI-adjusted relative risks (17 studies in all), the SRR was 1.12, and heterogeneity among the studies was high at 84%.
Additionally, an analysis by menopausal status based on four studies that reported breast cancer in both pre- and postmenopausal women showed SRRs for breast cancer of 0.97 (a 3% decrease in risk) and 1.14 among diabetic vs. nondiabetic premenopausal women and postmenopausal women, respectively, she said, noting that heterogeneity was low (I2 = 0%) among the premenopausal breast cancer study groups and high (I2 = 70%) among the postmenopausal study groups.
The findings provide evidence for a moderately increased risk of breast cancer in women with T2DM, Ms. Bota said.
“However, the effect of the adjustment or lack of adjustment on the heterogeneity suggests that the higher risk of breast cancer in women with diabetes may not be due to diabetes itself, but to adiposity,” she said, adding that “this hypothesis is equally supported by our subgroup analysis according to menopausal status because we saw that the risk of breast cancer was only associated with diabetes in postmenopausal women and this pattern resembles the pattern of the risk of breast cancer associated with adiposity, which is also only increased in postmenopausal women.”
This study was limited by insufficient data for investigating the sources of heterogeneity, she said.
“Therefore we propose ... future pooled analyses based on individual data from good quality prospective studies in order to increase the study power and to do some detailed analysis of the links between adiposity, diabetes, and breast cancer,” she concluded, adding that new studies to examine those relationships only in premenopausal women are also needed
In a separate meta-analysis, she and her colleagues, including Peter Boyle, PhD, president of the International Prevention Research Institute, assessed the association between insulin treatment and breast cancer risk in patients with diabetes.
“The long-acting insulin analogues glargine and detemir have been shown in some studies to be associated with increased risk of breast cancer, and other studies have shown no association between the use of these two compounds and the risk of breast cancer,” Dr. Boyle said in a separate presentation at the ADA meeting. “It was important to sort out a little bit what was going on in the literature.”
Overall, the meta-analysis of data from 12 longitudinal cohort studies – including more than 6,000 cases of breast cancer – showed a slight increase in breast cancer risk in patients taking long-acting insulin (SRR, 1.13) with “a relatively reasonable” level of heterogeneity (I2 = 23%).
“But we see that the story is not that simple,” he said.
For example, some studies included only patients who were prescribed insulin for the first time after the study began (new users), some included only patients who were prescribed insulin before the study began (prevalent users), and some included both (ever users), which may have introduced bias in the results, he explained.
Studies of glargine included 4,168 breast cancer cases over a total of 1,418,743 person-years of observation, and studies of detemir included fewer than 2,047 breast cancer cases (not all studies reported case numbers). Among both new users of glargine and detemir, the SRR was 1.12, suggesting no real association between either glargine or detemir use and breast cancer, he said.
“One important take-home message is that you have to be careful that these pharmaco-epidemiological studies, even when working with the same database, may have conflicting results ... so we still need more robust standards in methods for [such] studies,” he concluded.
Ms. Bota reported having no disclosures. The study presented by Dr. Boyle was funded by Sanofi. Dr. Boyle is president of a charity that has received donations from Pfizer, Roche, Novartis, and Lilly.
SOURCE: Bota M. ADA 2018, Abstract 180-OR; Boyle P. ADA 2018, Abstract 133-OR.
REPORTING FROM ADA 2018
Key clinical point: Adiposity accounts for the increased risk of breast cancer among women with diabetes.
Major finding: An analysis of 12 studies that adjusted for BMI showed a summary relative risk for breast cancer of 1.09 in diabetic versus nondiabetic women, with moderate study heterogeneity.
Study details: Meta-analyses including 21 and 12 studies, respectively.
Disclosures: Ms. Bota reported having no disclosures. The study presented by Dr. Boyle was funded by Sanofi. Dr. Boyle is president of a charity that has received donations from Pfizer, Roche, Novartis, and Lilly.
Source: Bota M. ADA 2018, Abstract 180-OR; Boyle P. ADA 2018, Abstract 133-OR.
CKD children need office blood pressures below the 75th percentile
CHICAGO – according to a review of 690 pediatric patients in the Chronic Kidney Disease in Children Cohort Study.

Hypertensive children with CKD were less likely to progress to dialysis or kidney transplant or have a 30% decline in estimated glomerular filtration rate (eGFR) when kept in that range, compared with children above or below it. “Achieved office [blood pressure] between the 50th and 75th percentiles appeared to offer the greatest protection against CKD progression in this cohort,” said investigators led by Joseph Flynn, MD, chief of the nephrology division at Seattle Children’s Hospital.
“We needed to have some [evidence] on what to do based on office blood pressure,” something that had been missing in the literature until now. “I think this is going to be very impactful on the care of children with CKD. Right now, the guidelines say to keep” pressures below the 90th percentile for age and height. “The guidelines [might] need to be changed,” said Dr. Flynn, also the lead author of the American Academy of Pediatrics 2017 blood pressure guidelines for children and adolescents (Pediatrics. 2017 Aug 21. doi: 10.1542/peds.2017-1904).
There was “no evidence to go below the 50th percentile; the 50th-75th seems to be the sweet spot for office blood pressure,” he said at the joint scientific sessions of the AHA Council on Hypertension, the AHA Council on Kidney in Cardiovascular Disease, and the American Society of Hypertension.
The 476 children with nonglomerular CKD actually did worse when their blood pressures were pushed below the 50th percentile, perhaps because of renal hypoperfusion. The 476 children with glomerular CKD did no better or worse below the 50th percentile than they did in the 50th-75th.
Children at or above the 90th percentile had the highest risk of progression, with about 80% needing renal replacement therapy or having a 30% drop in eGFR at 5-8 years of follow-up. Compared with children with glomerular CKD who were in the 90th percentile, the risk hazard (RH) for progression over 3 years was 0.10-0.30 (P less than 0.001) among those children with glomerular CKD who were kept in the 50th-75th percentile. Compared with children with nonglomerular CKD who were in the 90th percentile, the RH over 8 years among those in the 50th-75th percentile was 0.48 (P less than 0.001). Risk of progression in both glomerular and nonglomerular patients in the sweet spot was less than 50% at 5-8 years of follow-up.
When glomerular and nonglomerular patients were considered together, those with pressures below the 50th percentile were less likely to progress than children with pressures between the 75th and 90th, but they were more likely to progress than the 50th-75th percentile group.
Research nurses took three blood pressures by auscultation a minute apart after 5 minutes of rest. Most of the children were treated at first with angiotensin-converting enzyme (ACE) inhibitors/angiotensin receptor blockers, per recommendations. Dihydropyridine calcium channel blockers (for example, amlodipine and nifedipine) were most likely to be used next.
It was a curious finding because dihydropyridines have been show to worsen proteinuria in adults with CKD. The team is investigating to see whether they have the same effect in children. If so, “we’ll be able to tell people to stop using them,” Dr. Flynn said.
The median age in the study was 11.3 years, and almost two-thirds of the subjects were boys. The median duration of disease was 8 years. Some children in the ongoing cohort have been followed for almost 10 years. The analysis is based on children who entered the cohort with hypertension or developed it after enrollment.
The nationwide Chronic Kidney Disease in Children Cohort Study is funded by the National Institutes of Health. Dr. Flynn is an advisor for Ultragenyx and Silvergate Pharmaceuticals.
CHICAGO – according to a review of 690 pediatric patients in the Chronic Kidney Disease in Children Cohort Study.
Hypertensive children with CKD were less likely to progress to dialysis or kidney transplant or have a 30% decline in estimated glomerular filtration rate (eGFR) when kept in that range, compared with children above or below it. “Achieved office [blood pressure] between the 50th and 75th percentiles appeared to offer the greatest protection against CKD progression in this cohort,” said investigators led by Joseph Flynn, MD, chief of the nephrology division at Seattle Children’s Hospital.
“We needed to have some [evidence] on what to do based on office blood pressure,” something that had been missing in the literature until now. “I think this is going to be very impactful on the care of children with CKD. Right now, the guidelines say to keep” pressures below the 90th percentile for age and height. “The guidelines [might] need to be changed,” said Dr. Flynn, also the lead author of the American Academy of Pediatrics 2017 blood pressure guidelines for children and adolescents (Pediatrics. 2017 Aug 21. doi: 10.1542/peds.2017-1904).
There was “no evidence to go below the 50th percentile; the 50th-75th seems to be the sweet spot for office blood pressure,” he said at the joint scientific sessions of the AHA Council on Hypertension, the AHA Council on Kidney in Cardiovascular Disease, and the American Society of Hypertension.
The 476 children with nonglomerular CKD actually did worse when their blood pressures were pushed below the 50th percentile, perhaps because of renal hypoperfusion. The 476 children with glomerular CKD did no better or worse below the 50th percentile than they did in the 50th-75th.
Children at or above the 90th percentile had the highest risk of progression, with about 80% needing renal replacement therapy or having a 30% drop in eGFR at 5-8 years of follow-up. Compared with children with glomerular CKD who were in the 90th percentile, the risk hazard (RH) for progression over 3 years was 0.10-0.30 (P less than 0.001) among those children with glomerular CKD who were kept in the 50th-75th percentile. Compared with children with nonglomerular CKD who were in the 90th percentile, the RH over 8 years among those in the 50th-75th percentile was 0.48 (P less than 0.001). Risk of progression in both glomerular and nonglomerular patients in the sweet spot was less than 50% at 5-8 years of follow-up.
When glomerular and nonglomerular patients were considered together, those with pressures below the 50th percentile were less likely to progress than children with pressures between the 75th and 90th, but they were more likely to progress than the 50th-75th percentile group.
Research nurses took three blood pressures by auscultation a minute apart after 5 minutes of rest. Most of the children were treated at first with angiotensin-converting enzyme (ACE) inhibitors/angiotensin receptor blockers, per recommendations. Dihydropyridine calcium channel blockers (for example, amlodipine and nifedipine) were most likely to be used next.
It was a curious finding because dihydropyridines have been show to worsen proteinuria in adults with CKD. The team is investigating to see whether they have the same effect in children. If so, “we’ll be able to tell people to stop using them,” Dr. Flynn said.
The median age in the study was 11.3 years, and almost two-thirds of the subjects were boys. The median duration of disease was 8 years. Some children in the ongoing cohort have been followed for almost 10 years. The analysis is based on children who entered the cohort with hypertension or developed it after enrollment.
The nationwide Chronic Kidney Disease in Children Cohort Study is funded by the National Institutes of Health. Dr. Flynn is an advisor for Ultragenyx and Silvergate Pharmaceuticals.
CHICAGO – according to a review of 690 pediatric patients in the Chronic Kidney Disease in Children Cohort Study.
Hypertensive children with CKD were less likely to progress to dialysis or kidney transplant or have a 30% decline in estimated glomerular filtration rate (eGFR) when kept in that range, compared with children above or below it. “Achieved office [blood pressure] between the 50th and 75th percentiles appeared to offer the greatest protection against CKD progression in this cohort,” said investigators led by Joseph Flynn, MD, chief of the nephrology division at Seattle Children’s Hospital.
“We needed to have some [evidence] on what to do based on office blood pressure,” something that had been missing in the literature until now. “I think this is going to be very impactful on the care of children with CKD. Right now, the guidelines say to keep” pressures below the 90th percentile for age and height. “The guidelines [might] need to be changed,” said Dr. Flynn, also the lead author of the American Academy of Pediatrics 2017 blood pressure guidelines for children and adolescents (Pediatrics. 2017 Aug 21. doi: 10.1542/peds.2017-1904).
There was “no evidence to go below the 50th percentile; the 50th-75th seems to be the sweet spot for office blood pressure,” he said at the joint scientific sessions of the AHA Council on Hypertension, the AHA Council on Kidney in Cardiovascular Disease, and the American Society of Hypertension.
The 476 children with nonglomerular CKD actually did worse when their blood pressures were pushed below the 50th percentile, perhaps because of renal hypoperfusion. The 476 children with glomerular CKD did no better or worse below the 50th percentile than they did in the 50th-75th.
Children at or above the 90th percentile had the highest risk of progression, with about 80% needing renal replacement therapy or having a 30% drop in eGFR at 5-8 years of follow-up. Compared with children with glomerular CKD who were in the 90th percentile, the risk hazard (RH) for progression over 3 years was 0.10-0.30 (P less than 0.001) among those children with glomerular CKD who were kept in the 50th-75th percentile. Compared with children with nonglomerular CKD who were in the 90th percentile, the RH over 8 years among those in the 50th-75th percentile was 0.48 (P less than 0.001). Risk of progression in both glomerular and nonglomerular patients in the sweet spot was less than 50% at 5-8 years of follow-up.
When glomerular and nonglomerular patients were considered together, those with pressures below the 50th percentile were less likely to progress than children with pressures between the 75th and 90th, but they were more likely to progress than the 50th-75th percentile group.
Research nurses took three blood pressures by auscultation a minute apart after 5 minutes of rest. Most of the children were treated at first with angiotensin-converting enzyme (ACE) inhibitors/angiotensin receptor blockers, per recommendations. Dihydropyridine calcium channel blockers (for example, amlodipine and nifedipine) were most likely to be used next.
It was a curious finding because dihydropyridines have been show to worsen proteinuria in adults with CKD. The team is investigating to see whether they have the same effect in children. If so, “we’ll be able to tell people to stop using them,” Dr. Flynn said.
The median age in the study was 11.3 years, and almost two-thirds of the subjects were boys. The median duration of disease was 8 years. Some children in the ongoing cohort have been followed for almost 10 years. The analysis is based on children who entered the cohort with hypertension or developed it after enrollment.
The nationwide Chronic Kidney Disease in Children Cohort Study is funded by the National Institutes of Health. Dr. Flynn is an advisor for Ultragenyx and Silvergate Pharmaceuticals.
REPORTING FROM JOINT HYPERTENSION 2018
Key clinical point: It’s best to aim for an office blood pressure between the 50th and 75th percentiles in children with CKD.
Major finding: The risk of CKD progression was less than 50% in children kept in that range, which was better than children both above or below it.
Study details: Review of 690 pediatric patients in the Chronic Kidney Disease in Children Cohort Study.
Disclosures: The nationwide Chronic Kidney Disease in Children Cohort Study is funded by the National Institutes of Health. Dr. Joseph Flynn is an advisor for Ultragenyx and Silvergate Pharmaceuticals.
Abortion, the travel ban, and other top Supreme Court rulings affecting your practice
The 2017−2018 term of the Supreme Court of the United States (SCOTUS) was momentous. Justice Anthony Kennedy, who had been the deciding vote in most of the 5 to 4 cases for a generation, announced his retirement as of July 31, 2018. In addition, the Court decided a number of cases of interest to ObGyns. In this article we review some of those cases, as well as consider the future of the Court without Justice Kennedy. In selecting cases, we have given special attention to those in which national medical organizations filed amicus briefs. These “amicus curiae” or “friend of the court” briefs are filed by an entity who is not party to a case but wants to provide information or views to the court.

1. Abortion rulings
The Court decided 2 abortion cases and rejected a request to hear a third.
National Institute of Family and Life Advocates v Becerra
In this case,1 the Court struck down a California law that required pregnancy crisis centers not offering abortions (generally operated by pro-life groups) to provide special notices to clients.2
At stake. These notices would inform clients that California provides free or low-cost services, including abortions, and provide a phone number to call for those services.
There were many amicus briefs filed in this case, including those by the American College of Obstetricians and Gynecologists (ACOG) and other specialty boards,3 as well as the American Association of Pro-Life Obstetricians and Gynecologists and other pro-life organizations.4 ACOG’s brief argued that the California-required notice facilitates the goal of allowing women to receive medical services without harmful delay.
Final ruling. The Court held that the law required clinics to engage in speech with which the clinics disagreed (known as “compelled speech”). It also noted that California disclosure requirements were “wildly underinclusive” because they apply only to some clinics. The majority felt that there was no strong state interest in compelling this speech because there were other alternatives for the state to provide information about the availability of abortion and other services. The Court found that the clinics were likely to succeed on the merits of their claims of a First Amendment (free speech) violation.
Right to abortion for illegal immigrants in custody
A very unusual abortion case involved “Jane Doe,” a minor who was at 8 weeks’ gestation when she illegally crossed the border into the United States.5 She was placed in a federally-funded shelter where she requested an abortion. The facility denied that request.
At stake. Legal argument ensued about releasing her to another facility for an abortion, as the argument was made that pregnant minors who are apprehended crossing into the United States illegally and placed into the custody of federal officials should have abortion access. A lower Court of Appeals ruled against the Trump Administration’s policy of denying abortions to undocumented minors in federal custody. During the process of the federal government taking the case to the Supreme Court, the attorneys for Doe moved appointments around and, without notice, the abortion was performed. Government attorneys said that Doe’s attorneys made “what appear to be material misrepresentations and omissions” designed to “thwart [the Supreme Court’s] review” of the case.5 The government requested that the Court vacate the order of the Court of Appeals so that it could not be used as precedent.
Final ruling. The Court granted the governments request to vacate the lower court’s order because the minor was no longer pregnant and the order was therefore moot. The basic issue in this case (the right of in-custody minors to access abortions) remains unresolved. It is likely to appear before the Court in the future.
Continue to: Access to medical abortions
Access to medical abortions
An Arkansas law requires that a physician administering medical abortions contract with a physician who has admitting privileges at a hospital (a “contracted physician”).
At stake. Planned Parenthood filed suit challenging the requirement as unnecessary and harmful because it would result in the closure of 2 of the 3 abortion providers in Arkansas. ACOG filed an amicus brief urging the Supreme Court to consider the case.6 (Technically this was a petition for a Writ of Certiorari, the procedure by which the Court accepts cases. It accepts only about 1% of applications.) ACOG argued that there was no medical reason for the contracted physician requirement, and noted the harm it would do to women who would not have access to abortions.
Final ruling. On May 29, 2018, the Court declined to hear the case. This case is still active in the lower courts and may eventually return to the Supreme Court.

2. The patent system
The medical profession depends on the patent system to encourage the discovery of new patents efficiently and effectively. In 2012, Congress passed the America Invents Act7 that authorizes a petition by anyone other than the patent holder to the Patent and Trademark Office (PTO) for an “inter partes review” to assess a challenge to the patent’s legitimacy. If the PTO determines that there may be merit to the claim, the Patent Trial and Appeal Board undertakes a trial-like review process that may validate, invalidate, or amend the patent. The Board’s decision is subject to appellate court review.
At stake. This term, the inter partes review was challenged as unconstitutional on technical bases.8
Final ruling. The Court rejected this claim and approved the current administrative inter partes review process. The Court determined that once the Patent Office takes a petition challenging a patent, it must decide all of the claims against the patent, not pick and choose which elements of the challenge to evaluate.9 The Court’s decision upheld patent-review reform, but will require the Patent Office to tweak its procedures.

3. The travel ban
ACOG, the American Medical Association (AMA), the Association of American Medical Colleges, and more than 30 other health care and specialty associations filed an amicus brief regarding one of the most anticipated cases of the term—the “travel ban.”10
At stake. The essential argument of these organizations was that the US health care system depends on professionals from other countries. An efficient and fair immigration program is, therefore, important to advance the nation’s “health security.” During the 2016−2017 term, the Court considered but then removed the issue from its calendar when the Trump Administration issued a revised travel ban.11
In September 2017, President Trump’s proclamation imposed a range of entry restrictions on the citizens of 8 countries, most (but not all) of which are predominantly Muslim. The government indicated that, in a study by Homeland Security and the State Department, these countries were identified as having especially deficient information-sharing practices and presented national security concerns. Trump v Hawaii12 challenged this proclamation.
Final ruling. The majority of the Court upheld the travel ban. For the 5-Justice majority led by Chief Justice Roberts, the case came down to 3 things:
- The Constitution and the laws passed by Congress of necessity give the President great authority to engage in foreign policy, including policies regarding entry into the country.
- The courts are very reluctant to get into the substance of foreign affairs—they are not equipped to know in detail what the facts are, and things change very fast.
- If courts start tinkering with foreign policy and things turn bad, it will appear that the courts are to blame and were interfering in an area about which they are not competent.
Continue to: 4. Did a credit card case add risk to health insurance markets?

It was just a credit card case, but one in which the AMA saw a real risk to regulation of the health insurance markets.
At stake. Technically, Ohio v American Express concerned a claim that American Express (AmEx) violated antitrust laws when it prohibited merchants taking its credit card from “steering” customers to cards with lower fees.13 AmEx maintained that, because credit cards were a special kind of “2-sided” market (connecting merchants on one side and customers on the other), antitrust laws should not be strictly enforced.
The AMA noticed that special rules regarding 2-sided markets might apply to health insurance, and it submitted an amicus brief14 that noted: “dominant health insurance networks … have imposed and could further impose rules or effectively erect barriers that prohibit physicians from referring patients to certain specialists, particularly out-of-network specialists, for innovative and even necessary medical tests.”14 It concluded that the antitrust rule AmEx was suggesting would make it nearly impossible to challenge these unfair provisions in health insurance arrangements.
Final ruling. The Court, however, accepted the AmEx position, making it very difficult to develop an antitrust case against 2-sided markets. It remains to be seen the degree to which the AMA concern about health insurance markets will be realized.

5. Gay wedding and a bakeshop
At stake. In Masterpiece Cakeshop v Colorado, a cakemaker declined to design a cake for a gay wedding and had been disciplined under Colorado law for discriminating against the couple based on sexual orientation.15
Final ruling. The Court, however, found that the Colorado regulators had, ironically, shown such religious animus in the way they treated the baker that the regulators themselves had discriminated on the basis of religion. As a result, the Court reversed the sanctions against the baker.
This decision was fairly narrow. It does not, for example, stand for the proposition that there may be a general religious exception to antidiscrimination laws. The question of broader religious or free-speech objections to antidiscrimination laws remains for another time.
Amicus brief. It was interesting that the American College of Pediatricians, American Association of Pro-Life Obstetricians and Gynecologists, and others, filed an amicus brief to report with concern the “demands that individual medical professionals must perform, assist with, or facilitate abortions, without regard to the teachings of their own faiths, consciences, and convictions.”16 The brief also noted that “issues in the present case implicate the fundamental rights of health care professionals, and to respectfully urge that the Court should by no means permit any weakening or qualification of well-established protections against compelled speech, and of free exercise” of religion.16
Arbitration. The Court upheld, as it has in most recent terms, another arbitration agreement.1 This case concerned an employment agreement in which employees consented to submit to arbitration rather than file lawsuits and not use class action claims.
Search of cell-phone location. Cell phones, whenever turned on, connect with cell towers that record the phone’s location several times a minute. Cell companies store this information, creating a virtual map of where the owner is at all times. The Federal Bureau of Investigation asked a cell company for location information for several people during a 127-day period in which robberies were committed.2 The Court held that the search was illegal in the absence of a warrant.
Public employee unions. The Court held that agency (fair share) fees, in which public employees who are not union members can be required to pay dues for the bargaining and grievance activities (from which they generally benefit), violate the First Amendment. The majority held that forcing public employees to pay fees to unions requires the employees, through those fees, to engage in political activities with which they disagree.3 This is a form of compelled speech, which the Court found violates the First Amendment. Health care professionals who are public employees in positions that have union representation will probably have the opportunity to opt out of agency agreements.
Internet sales tax. The Court permitted states to charge sales tax on out-of-state Internet purchases.4 In doing so, a state may require out-of-state companies to collect taxes on sales to its residents.
References
- Epic Systems Corp. v Lewis, 584 US 16 285 (2018).
- Carpenter v United States, 585 US 16 402 (2018).
- Janus v State, County, and Municipal Employees, 585 US 16 1466 (2018).
- South Dakota v Wayfair, Inc, 585 US 17 494 (2018).
Clues to the future
During the term that ran from October 2, 2017, through June 27, 2018, the Court issued 72 decisions. An unusually high proportion of cases (26%; 19 cases) were decided on a 5 to 4 vote. Last term, the rate of 5 to 4 decisions was 10%; the 6-year average was 18%. The unanimous decision rate was 39% this term, compared with 59% last term, and 50% on average.
The rate of 5 to 4 cases provides a clue about the Court’s general direction. The number of times each Justice was in the majority in those nineteen 5 to 4 decisions included: Chief Justice Roberts, 17; and Justices Kennedy, 16; Gorsuch, 16; Thomas, 15; and Alito, 15; compared with Justices Ginsburg, 5; Breyer, 4; Sotomayor, 4; and Kagan, 3.
The Court convened on October 1, 2018. At this writing, whether the new term starts with 8 or 9 justices remains a question. President Trump nominated Brett Kavanaugh, JD, to take Justice Kennedy’s place on the Court. His professional qualifications and experience appear to make him qualified for a position on the Court, but as we have seen, there are many other elements that go into confirming a Justice’s nomination.
Justice Anthony Kennedy was the deciding vote in the overwhelming majority of the 5 to 4 decisions in 20 of his 30 years on the Court. The areas in which he had an especially important impact include1:
- Gay rights. Justice Kennedy wrote the opinions (usually 5 to 4 decisions) in a number of groundbreaking gay-rights cases, including decriminalizing homosexual conduct, striking down the Defense of Marriage Act, and finding that the Constitution requires states to recognize gay marriage.
- The death penalty. Justice Kennedy wrote decisions that prohibited states from imposing the death penalty for any crime other than murder, for defendants who were under 18 when they committed the crime, and for defendants with serious developmental disabilities. He expressed reservations about long-term solitary confinement, but did not have a case that allowed him to decide its constitutionality.
- The First Amendment. Early in his service on the Court, he held that the First Amendment protected flag burning as a form of speech. He decided many important freespeech and freedom-of-religion cases that have set a standard for protecting those fundamental freedoms.
- Use of health and social science data. Justice Kennedy was more open to mental health information and cited it more often than most other Justices.
- Abortion rights? Many commentators would add protecting the right to choose to have an abortion to the above list. Justice Kennedy was a central figure in one case that declined to back away from Roe v Wade, and joined a more recent decision that struck down a Texas law that created an undue burden on women seeking abortion. Plus, he also voted to uphold abortion restrictions, such as “partial-birth-abortion laws.” So there is a good argument for including abortion rights on the list, although he did not break new ground.
Justice Kennedy as a person
Outside the courtroom, Justice Kennedy is a person of great warmth and compassion. He is a natural teacher and spends a great deal of time with students. When asked how he would like to be remembered, Justice Kennedy once replied, “Somebody who’s decent, and honest, and fair, and who’s absolutely committed to the proposition that freedom is America’s gift to the rest of the world.” I agree with that assessment.
STEVEN R. SMITH, MS, JD
Reference
- South Dakota v Wayfair, Inc, 585 US (2018)
Next term, the Court is scheduled to hear cases regarding pharmaceutical liability, double jeopardy, sex-offender registration, expert witnesses, Social Security disability benefits, and the Age Discrimination in Employment Act. There will be at least 3 arbitration cases. Health care and reproductive rights will continue to be an important part of the Court’s docket.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- National Institute of Family and Life Advocates v Becerra, 585 US 16 1140 (2018).
- California Reproductive Freedom, Accountability, Comprehensive Care, and Transparency Act (FACT Act), Cal. Health & Safety Code Ann. §123470 et seq. (West 2018).
- Brief amici curiae of American Academy of Pediatrics, et al. in National Institute of Family and Life Advocates v Becerra, February 27, 2018.
- Brief amici curiae of American Association of Pro-Life Obstetricians & Gynecologists, et al. in National Institute of Family and Life Advocates v Becerra, January 16, 2018.
- Azar v Garza, 584 US 17 654 (2018).
- Brief amici curiae of American College of Obstetricians and Gynecologists and American Public Health Association in Planned Parenthood of Arkansas and Eastern Oklahoma v Jegley, February 1, 2018.
- Chapter 31, Inter Partes Review. United States Code. Title 35: Patents. Part III, Patents and protection of patents. 2012 Ed. 35 USC 311–319.
- Oil States Energy Services, LLC v Greene’s Energy Group, LLC, 584 US 16 712 (2018).
- SAS Institute Inc. v Iancu, 584 US 16 969 (2018).
- Brief for Association of American Medical Colleges and Others as Amici Curiae Supporting Respondents, Trump v Hawaii. https://www.supremecourt.gov/Docket PDF/17/ 17-965/40128/20180327105855912_17-965%20Amicus%20Br.%20Proclamation.pdf. Accessed September 21, 2018.
- Smith SR, Sanfilippo JS. Supreme Court decisions in 2017 that affected your practice. OBG Manag. 2017;29(12)44–47.
- Trump v Hawaii, 585 US 17 965 (2018).
- Ohio v American Express Co, 585 US 16 1454 (2018).
- Brief amici curiae of American Medical Association and Ohio State Medical Association in Ohio v American Express, December 24, 2017.
- Masterpiece Cakeshop, Ltd. v Colorado Civil Rights Commission, 584 US 16 111 (2018).
- Brief amici curiae of American College of Pediatricians, et al. in Masterpiece Cakeshop v Colorado Civil Rights Commission, September 7, 2017.

Mr. Smith is Professor Emeritus and Dean Emeritus at California Western School of Law, San Diego, California.

Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG MANAGEMENT Board of Editors.
Mr. Smith is Professor Emeritus and Dean Emeritus at California Western School of Law, San Diego, California.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG MANAGEMENT Board of Editors.
Mr. Smith is Professor Emeritus and Dean Emeritus at California Western School of Law, San Diego, California.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG MANAGEMENT Board of Editors.
The 2017−2018 term of the Supreme Court of the United States (SCOTUS) was momentous. Justice Anthony Kennedy, who had been the deciding vote in most of the 5 to 4 cases for a generation, announced his retirement as of July 31, 2018. In addition, the Court decided a number of cases of interest to ObGyns. In this article we review some of those cases, as well as consider the future of the Court without Justice Kennedy. In selecting cases, we have given special attention to those in which national medical organizations filed amicus briefs. These “amicus curiae” or “friend of the court” briefs are filed by an entity who is not party to a case but wants to provide information or views to the court.
1. Abortion rulings
The Court decided 2 abortion cases and rejected a request to hear a third.
National Institute of Family and Life Advocates v Becerra
In this case,1 the Court struck down a California law that required pregnancy crisis centers not offering abortions (generally operated by pro-life groups) to provide special notices to clients.2
At stake. These notices would inform clients that California provides free or low-cost services, including abortions, and provide a phone number to call for those services.
There were many amicus briefs filed in this case, including those by the American College of Obstetricians and Gynecologists (ACOG) and other specialty boards,3 as well as the American Association of Pro-Life Obstetricians and Gynecologists and other pro-life organizations.4 ACOG’s brief argued that the California-required notice facilitates the goal of allowing women to receive medical services without harmful delay.
Final ruling. The Court held that the law required clinics to engage in speech with which the clinics disagreed (known as “compelled speech”). It also noted that California disclosure requirements were “wildly underinclusive” because they apply only to some clinics. The majority felt that there was no strong state interest in compelling this speech because there were other alternatives for the state to provide information about the availability of abortion and other services. The Court found that the clinics were likely to succeed on the merits of their claims of a First Amendment (free speech) violation.
Right to abortion for illegal immigrants in custody
A very unusual abortion case involved “Jane Doe,” a minor who was at 8 weeks’ gestation when she illegally crossed the border into the United States.5 She was placed in a federally-funded shelter where she requested an abortion. The facility denied that request.
At stake. Legal argument ensued about releasing her to another facility for an abortion, as the argument was made that pregnant minors who are apprehended crossing into the United States illegally and placed into the custody of federal officials should have abortion access. A lower Court of Appeals ruled against the Trump Administration’s policy of denying abortions to undocumented minors in federal custody. During the process of the federal government taking the case to the Supreme Court, the attorneys for Doe moved appointments around and, without notice, the abortion was performed. Government attorneys said that Doe’s attorneys made “what appear to be material misrepresentations and omissions” designed to “thwart [the Supreme Court’s] review” of the case.5 The government requested that the Court vacate the order of the Court of Appeals so that it could not be used as precedent.
Final ruling. The Court granted the governments request to vacate the lower court’s order because the minor was no longer pregnant and the order was therefore moot. The basic issue in this case (the right of in-custody minors to access abortions) remains unresolved. It is likely to appear before the Court in the future.
Continue to: Access to medical abortions
Access to medical abortions
An Arkansas law requires that a physician administering medical abortions contract with a physician who has admitting privileges at a hospital (a “contracted physician”).
At stake. Planned Parenthood filed suit challenging the requirement as unnecessary and harmful because it would result in the closure of 2 of the 3 abortion providers in Arkansas. ACOG filed an amicus brief urging the Supreme Court to consider the case.6 (Technically this was a petition for a Writ of Certiorari, the procedure by which the Court accepts cases. It accepts only about 1% of applications.) ACOG argued that there was no medical reason for the contracted physician requirement, and noted the harm it would do to women who would not have access to abortions.
Final ruling. On May 29, 2018, the Court declined to hear the case. This case is still active in the lower courts and may eventually return to the Supreme Court.
2. The patent system
The medical profession depends on the patent system to encourage the discovery of new patents efficiently and effectively. In 2012, Congress passed the America Invents Act7 that authorizes a petition by anyone other than the patent holder to the Patent and Trademark Office (PTO) for an “inter partes review” to assess a challenge to the patent’s legitimacy. If the PTO determines that there may be merit to the claim, the Patent Trial and Appeal Board undertakes a trial-like review process that may validate, invalidate, or amend the patent. The Board’s decision is subject to appellate court review.
At stake. This term, the inter partes review was challenged as unconstitutional on technical bases.8
Final ruling. The Court rejected this claim and approved the current administrative inter partes review process. The Court determined that once the Patent Office takes a petition challenging a patent, it must decide all of the claims against the patent, not pick and choose which elements of the challenge to evaluate.9 The Court’s decision upheld patent-review reform, but will require the Patent Office to tweak its procedures.
3. The travel ban
ACOG, the American Medical Association (AMA), the Association of American Medical Colleges, and more than 30 other health care and specialty associations filed an amicus brief regarding one of the most anticipated cases of the term—the “travel ban.”10
At stake. The essential argument of these organizations was that the US health care system depends on professionals from other countries. An efficient and fair immigration program is, therefore, important to advance the nation’s “health security.” During the 2016−2017 term, the Court considered but then removed the issue from its calendar when the Trump Administration issued a revised travel ban.11
In September 2017, President Trump’s proclamation imposed a range of entry restrictions on the citizens of 8 countries, most (but not all) of which are predominantly Muslim. The government indicated that, in a study by Homeland Security and the State Department, these countries were identified as having especially deficient information-sharing practices and presented national security concerns. Trump v Hawaii12 challenged this proclamation.
Final ruling. The majority of the Court upheld the travel ban. For the 5-Justice majority led by Chief Justice Roberts, the case came down to 3 things:
- The Constitution and the laws passed by Congress of necessity give the President great authority to engage in foreign policy, including policies regarding entry into the country.
- The courts are very reluctant to get into the substance of foreign affairs—they are not equipped to know in detail what the facts are, and things change very fast.
- If courts start tinkering with foreign policy and things turn bad, it will appear that the courts are to blame and were interfering in an area about which they are not competent.
Continue to: 4. Did a credit card case add risk to health insurance markets?
It was just a credit card case, but one in which the AMA saw a real risk to regulation of the health insurance markets.
At stake. Technically, Ohio v American Express concerned a claim that American Express (AmEx) violated antitrust laws when it prohibited merchants taking its credit card from “steering” customers to cards with lower fees.13 AmEx maintained that, because credit cards were a special kind of “2-sided” market (connecting merchants on one side and customers on the other), antitrust laws should not be strictly enforced.
The AMA noticed that special rules regarding 2-sided markets might apply to health insurance, and it submitted an amicus brief14 that noted: “dominant health insurance networks … have imposed and could further impose rules or effectively erect barriers that prohibit physicians from referring patients to certain specialists, particularly out-of-network specialists, for innovative and even necessary medical tests.”14 It concluded that the antitrust rule AmEx was suggesting would make it nearly impossible to challenge these unfair provisions in health insurance arrangements.
Final ruling. The Court, however, accepted the AmEx position, making it very difficult to develop an antitrust case against 2-sided markets. It remains to be seen the degree to which the AMA concern about health insurance markets will be realized.
5. Gay wedding and a bakeshop
At stake. In Masterpiece Cakeshop v Colorado, a cakemaker declined to design a cake for a gay wedding and had been disciplined under Colorado law for discriminating against the couple based on sexual orientation.15
Final ruling. The Court, however, found that the Colorado regulators had, ironically, shown such religious animus in the way they treated the baker that the regulators themselves had discriminated on the basis of religion. As a result, the Court reversed the sanctions against the baker.
This decision was fairly narrow. It does not, for example, stand for the proposition that there may be a general religious exception to antidiscrimination laws. The question of broader religious or free-speech objections to antidiscrimination laws remains for another time.
Amicus brief. It was interesting that the American College of Pediatricians, American Association of Pro-Life Obstetricians and Gynecologists, and others, filed an amicus brief to report with concern the “demands that individual medical professionals must perform, assist with, or facilitate abortions, without regard to the teachings of their own faiths, consciences, and convictions.”16 The brief also noted that “issues in the present case implicate the fundamental rights of health care professionals, and to respectfully urge that the Court should by no means permit any weakening or qualification of well-established protections against compelled speech, and of free exercise” of religion.16
Arbitration. The Court upheld, as it has in most recent terms, another arbitration agreement.1 This case concerned an employment agreement in which employees consented to submit to arbitration rather than file lawsuits and not use class action claims.
Search of cell-phone location. Cell phones, whenever turned on, connect with cell towers that record the phone’s location several times a minute. Cell companies store this information, creating a virtual map of where the owner is at all times. The Federal Bureau of Investigation asked a cell company for location information for several people during a 127-day period in which robberies were committed.2 The Court held that the search was illegal in the absence of a warrant.
Public employee unions. The Court held that agency (fair share) fees, in which public employees who are not union members can be required to pay dues for the bargaining and grievance activities (from which they generally benefit), violate the First Amendment. The majority held that forcing public employees to pay fees to unions requires the employees, through those fees, to engage in political activities with which they disagree.3 This is a form of compelled speech, which the Court found violates the First Amendment. Health care professionals who are public employees in positions that have union representation will probably have the opportunity to opt out of agency agreements.
Internet sales tax. The Court permitted states to charge sales tax on out-of-state Internet purchases.4 In doing so, a state may require out-of-state companies to collect taxes on sales to its residents.
References
- Epic Systems Corp. v Lewis, 584 US 16 285 (2018).
- Carpenter v United States, 585 US 16 402 (2018).
- Janus v State, County, and Municipal Employees, 585 US 16 1466 (2018).
- South Dakota v Wayfair, Inc, 585 US 17 494 (2018).
Clues to the future
During the term that ran from October 2, 2017, through June 27, 2018, the Court issued 72 decisions. An unusually high proportion of cases (26%; 19 cases) were decided on a 5 to 4 vote. Last term, the rate of 5 to 4 decisions was 10%; the 6-year average was 18%. The unanimous decision rate was 39% this term, compared with 59% last term, and 50% on average.
The rate of 5 to 4 cases provides a clue about the Court’s general direction. The number of times each Justice was in the majority in those nineteen 5 to 4 decisions included: Chief Justice Roberts, 17; and Justices Kennedy, 16; Gorsuch, 16; Thomas, 15; and Alito, 15; compared with Justices Ginsburg, 5; Breyer, 4; Sotomayor, 4; and Kagan, 3.
The Court convened on October 1, 2018. At this writing, whether the new term starts with 8 or 9 justices remains a question. President Trump nominated Brett Kavanaugh, JD, to take Justice Kennedy’s place on the Court. His professional qualifications and experience appear to make him qualified for a position on the Court, but as we have seen, there are many other elements that go into confirming a Justice’s nomination.
Justice Anthony Kennedy was the deciding vote in the overwhelming majority of the 5 to 4 decisions in 20 of his 30 years on the Court. The areas in which he had an especially important impact include1:
- Gay rights. Justice Kennedy wrote the opinions (usually 5 to 4 decisions) in a number of groundbreaking gay-rights cases, including decriminalizing homosexual conduct, striking down the Defense of Marriage Act, and finding that the Constitution requires states to recognize gay marriage.
- The death penalty. Justice Kennedy wrote decisions that prohibited states from imposing the death penalty for any crime other than murder, for defendants who were under 18 when they committed the crime, and for defendants with serious developmental disabilities. He expressed reservations about long-term solitary confinement, but did not have a case that allowed him to decide its constitutionality.
- The First Amendment. Early in his service on the Court, he held that the First Amendment protected flag burning as a form of speech. He decided many important freespeech and freedom-of-religion cases that have set a standard for protecting those fundamental freedoms.
- Use of health and social science data. Justice Kennedy was more open to mental health information and cited it more often than most other Justices.
- Abortion rights? Many commentators would add protecting the right to choose to have an abortion to the above list. Justice Kennedy was a central figure in one case that declined to back away from Roe v Wade, and joined a more recent decision that struck down a Texas law that created an undue burden on women seeking abortion. Plus, he also voted to uphold abortion restrictions, such as “partial-birth-abortion laws.” So there is a good argument for including abortion rights on the list, although he did not break new ground.
Justice Kennedy as a person
Outside the courtroom, Justice Kennedy is a person of great warmth and compassion. He is a natural teacher and spends a great deal of time with students. When asked how he would like to be remembered, Justice Kennedy once replied, “Somebody who’s decent, and honest, and fair, and who’s absolutely committed to the proposition that freedom is America’s gift to the rest of the world.” I agree with that assessment.
STEVEN R. SMITH, MS, JD
Reference
- South Dakota v Wayfair, Inc, 585 US (2018)
Next term, the Court is scheduled to hear cases regarding pharmaceutical liability, double jeopardy, sex-offender registration, expert witnesses, Social Security disability benefits, and the Age Discrimination in Employment Act. There will be at least 3 arbitration cases. Health care and reproductive rights will continue to be an important part of the Court’s docket.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The 2017−2018 term of the Supreme Court of the United States (SCOTUS) was momentous. Justice Anthony Kennedy, who had been the deciding vote in most of the 5 to 4 cases for a generation, announced his retirement as of July 31, 2018. In addition, the Court decided a number of cases of interest to ObGyns. In this article we review some of those cases, as well as consider the future of the Court without Justice Kennedy. In selecting cases, we have given special attention to those in which national medical organizations filed amicus briefs. These “amicus curiae” or “friend of the court” briefs are filed by an entity who is not party to a case but wants to provide information or views to the court.
1. Abortion rulings
The Court decided 2 abortion cases and rejected a request to hear a third.
National Institute of Family and Life Advocates v Becerra
In this case,1 the Court struck down a California law that required pregnancy crisis centers not offering abortions (generally operated by pro-life groups) to provide special notices to clients.2
At stake. These notices would inform clients that California provides free or low-cost services, including abortions, and provide a phone number to call for those services.
There were many amicus briefs filed in this case, including those by the American College of Obstetricians and Gynecologists (ACOG) and other specialty boards,3 as well as the American Association of Pro-Life Obstetricians and Gynecologists and other pro-life organizations.4 ACOG’s brief argued that the California-required notice facilitates the goal of allowing women to receive medical services without harmful delay.
Final ruling. The Court held that the law required clinics to engage in speech with which the clinics disagreed (known as “compelled speech”). It also noted that California disclosure requirements were “wildly underinclusive” because they apply only to some clinics. The majority felt that there was no strong state interest in compelling this speech because there were other alternatives for the state to provide information about the availability of abortion and other services. The Court found that the clinics were likely to succeed on the merits of their claims of a First Amendment (free speech) violation.
Right to abortion for illegal immigrants in custody
A very unusual abortion case involved “Jane Doe,” a minor who was at 8 weeks’ gestation when she illegally crossed the border into the United States.5 She was placed in a federally-funded shelter where she requested an abortion. The facility denied that request.
At stake. Legal argument ensued about releasing her to another facility for an abortion, as the argument was made that pregnant minors who are apprehended crossing into the United States illegally and placed into the custody of federal officials should have abortion access. A lower Court of Appeals ruled against the Trump Administration’s policy of denying abortions to undocumented minors in federal custody. During the process of the federal government taking the case to the Supreme Court, the attorneys for Doe moved appointments around and, without notice, the abortion was performed. Government attorneys said that Doe’s attorneys made “what appear to be material misrepresentations and omissions” designed to “thwart [the Supreme Court’s] review” of the case.5 The government requested that the Court vacate the order of the Court of Appeals so that it could not be used as precedent.
Final ruling. The Court granted the governments request to vacate the lower court’s order because the minor was no longer pregnant and the order was therefore moot. The basic issue in this case (the right of in-custody minors to access abortions) remains unresolved. It is likely to appear before the Court in the future.
Continue to: Access to medical abortions
Access to medical abortions
An Arkansas law requires that a physician administering medical abortions contract with a physician who has admitting privileges at a hospital (a “contracted physician”).
At stake. Planned Parenthood filed suit challenging the requirement as unnecessary and harmful because it would result in the closure of 2 of the 3 abortion providers in Arkansas. ACOG filed an amicus brief urging the Supreme Court to consider the case.6 (Technically this was a petition for a Writ of Certiorari, the procedure by which the Court accepts cases. It accepts only about 1% of applications.) ACOG argued that there was no medical reason for the contracted physician requirement, and noted the harm it would do to women who would not have access to abortions.
Final ruling. On May 29, 2018, the Court declined to hear the case. This case is still active in the lower courts and may eventually return to the Supreme Court.
2. The patent system
The medical profession depends on the patent system to encourage the discovery of new patents efficiently and effectively. In 2012, Congress passed the America Invents Act7 that authorizes a petition by anyone other than the patent holder to the Patent and Trademark Office (PTO) for an “inter partes review” to assess a challenge to the patent’s legitimacy. If the PTO determines that there may be merit to the claim, the Patent Trial and Appeal Board undertakes a trial-like review process that may validate, invalidate, or amend the patent. The Board’s decision is subject to appellate court review.
At stake. This term, the inter partes review was challenged as unconstitutional on technical bases.8
Final ruling. The Court rejected this claim and approved the current administrative inter partes review process. The Court determined that once the Patent Office takes a petition challenging a patent, it must decide all of the claims against the patent, not pick and choose which elements of the challenge to evaluate.9 The Court’s decision upheld patent-review reform, but will require the Patent Office to tweak its procedures.
3. The travel ban
ACOG, the American Medical Association (AMA), the Association of American Medical Colleges, and more than 30 other health care and specialty associations filed an amicus brief regarding one of the most anticipated cases of the term—the “travel ban.”10
At stake. The essential argument of these organizations was that the US health care system depends on professionals from other countries. An efficient and fair immigration program is, therefore, important to advance the nation’s “health security.” During the 2016−2017 term, the Court considered but then removed the issue from its calendar when the Trump Administration issued a revised travel ban.11
In September 2017, President Trump’s proclamation imposed a range of entry restrictions on the citizens of 8 countries, most (but not all) of which are predominantly Muslim. The government indicated that, in a study by Homeland Security and the State Department, these countries were identified as having especially deficient information-sharing practices and presented national security concerns. Trump v Hawaii12 challenged this proclamation.
Final ruling. The majority of the Court upheld the travel ban. For the 5-Justice majority led by Chief Justice Roberts, the case came down to 3 things:
- The Constitution and the laws passed by Congress of necessity give the President great authority to engage in foreign policy, including policies regarding entry into the country.
- The courts are very reluctant to get into the substance of foreign affairs—they are not equipped to know in detail what the facts are, and things change very fast.
- If courts start tinkering with foreign policy and things turn bad, it will appear that the courts are to blame and were interfering in an area about which they are not competent.
Continue to: 4. Did a credit card case add risk to health insurance markets?
It was just a credit card case, but one in which the AMA saw a real risk to regulation of the health insurance markets.
At stake. Technically, Ohio v American Express concerned a claim that American Express (AmEx) violated antitrust laws when it prohibited merchants taking its credit card from “steering” customers to cards with lower fees.13 AmEx maintained that, because credit cards were a special kind of “2-sided” market (connecting merchants on one side and customers on the other), antitrust laws should not be strictly enforced.
The AMA noticed that special rules regarding 2-sided markets might apply to health insurance, and it submitted an amicus brief14 that noted: “dominant health insurance networks … have imposed and could further impose rules or effectively erect barriers that prohibit physicians from referring patients to certain specialists, particularly out-of-network specialists, for innovative and even necessary medical tests.”14 It concluded that the antitrust rule AmEx was suggesting would make it nearly impossible to challenge these unfair provisions in health insurance arrangements.
Final ruling. The Court, however, accepted the AmEx position, making it very difficult to develop an antitrust case against 2-sided markets. It remains to be seen the degree to which the AMA concern about health insurance markets will be realized.
5. Gay wedding and a bakeshop
At stake. In Masterpiece Cakeshop v Colorado, a cakemaker declined to design a cake for a gay wedding and had been disciplined under Colorado law for discriminating against the couple based on sexual orientation.15
Final ruling. The Court, however, found that the Colorado regulators had, ironically, shown such religious animus in the way they treated the baker that the regulators themselves had discriminated on the basis of religion. As a result, the Court reversed the sanctions against the baker.
This decision was fairly narrow. It does not, for example, stand for the proposition that there may be a general religious exception to antidiscrimination laws. The question of broader religious or free-speech objections to antidiscrimination laws remains for another time.
Amicus brief. It was interesting that the American College of Pediatricians, American Association of Pro-Life Obstetricians and Gynecologists, and others, filed an amicus brief to report with concern the “demands that individual medical professionals must perform, assist with, or facilitate abortions, without regard to the teachings of their own faiths, consciences, and convictions.”16 The brief also noted that “issues in the present case implicate the fundamental rights of health care professionals, and to respectfully urge that the Court should by no means permit any weakening or qualification of well-established protections against compelled speech, and of free exercise” of religion.16
Arbitration. The Court upheld, as it has in most recent terms, another arbitration agreement.1 This case concerned an employment agreement in which employees consented to submit to arbitration rather than file lawsuits and not use class action claims.
Search of cell-phone location. Cell phones, whenever turned on, connect with cell towers that record the phone’s location several times a minute. Cell companies store this information, creating a virtual map of where the owner is at all times. The Federal Bureau of Investigation asked a cell company for location information for several people during a 127-day period in which robberies were committed.2 The Court held that the search was illegal in the absence of a warrant.
Public employee unions. The Court held that agency (fair share) fees, in which public employees who are not union members can be required to pay dues for the bargaining and grievance activities (from which they generally benefit), violate the First Amendment. The majority held that forcing public employees to pay fees to unions requires the employees, through those fees, to engage in political activities with which they disagree.3 This is a form of compelled speech, which the Court found violates the First Amendment. Health care professionals who are public employees in positions that have union representation will probably have the opportunity to opt out of agency agreements.
Internet sales tax. The Court permitted states to charge sales tax on out-of-state Internet purchases.4 In doing so, a state may require out-of-state companies to collect taxes on sales to its residents.
References
- Epic Systems Corp. v Lewis, 584 US 16 285 (2018).
- Carpenter v United States, 585 US 16 402 (2018).
- Janus v State, County, and Municipal Employees, 585 US 16 1466 (2018).
- South Dakota v Wayfair, Inc, 585 US 17 494 (2018).
Clues to the future
During the term that ran from October 2, 2017, through June 27, 2018, the Court issued 72 decisions. An unusually high proportion of cases (26%; 19 cases) were decided on a 5 to 4 vote. Last term, the rate of 5 to 4 decisions was 10%; the 6-year average was 18%. The unanimous decision rate was 39% this term, compared with 59% last term, and 50% on average.
The rate of 5 to 4 cases provides a clue about the Court’s general direction. The number of times each Justice was in the majority in those nineteen 5 to 4 decisions included: Chief Justice Roberts, 17; and Justices Kennedy, 16; Gorsuch, 16; Thomas, 15; and Alito, 15; compared with Justices Ginsburg, 5; Breyer, 4; Sotomayor, 4; and Kagan, 3.
The Court convened on October 1, 2018. At this writing, whether the new term starts with 8 or 9 justices remains a question. President Trump nominated Brett Kavanaugh, JD, to take Justice Kennedy’s place on the Court. His professional qualifications and experience appear to make him qualified for a position on the Court, but as we have seen, there are many other elements that go into confirming a Justice’s nomination.
Justice Anthony Kennedy was the deciding vote in the overwhelming majority of the 5 to 4 decisions in 20 of his 30 years on the Court. The areas in which he had an especially important impact include1:
- Gay rights. Justice Kennedy wrote the opinions (usually 5 to 4 decisions) in a number of groundbreaking gay-rights cases, including decriminalizing homosexual conduct, striking down the Defense of Marriage Act, and finding that the Constitution requires states to recognize gay marriage.
- The death penalty. Justice Kennedy wrote decisions that prohibited states from imposing the death penalty for any crime other than murder, for defendants who were under 18 when they committed the crime, and for defendants with serious developmental disabilities. He expressed reservations about long-term solitary confinement, but did not have a case that allowed him to decide its constitutionality.
- The First Amendment. Early in his service on the Court, he held that the First Amendment protected flag burning as a form of speech. He decided many important freespeech and freedom-of-religion cases that have set a standard for protecting those fundamental freedoms.
- Use of health and social science data. Justice Kennedy was more open to mental health information and cited it more often than most other Justices.
- Abortion rights? Many commentators would add protecting the right to choose to have an abortion to the above list. Justice Kennedy was a central figure in one case that declined to back away from Roe v Wade, and joined a more recent decision that struck down a Texas law that created an undue burden on women seeking abortion. Plus, he also voted to uphold abortion restrictions, such as “partial-birth-abortion laws.” So there is a good argument for including abortion rights on the list, although he did not break new ground.
Justice Kennedy as a person
Outside the courtroom, Justice Kennedy is a person of great warmth and compassion. He is a natural teacher and spends a great deal of time with students. When asked how he would like to be remembered, Justice Kennedy once replied, “Somebody who’s decent, and honest, and fair, and who’s absolutely committed to the proposition that freedom is America’s gift to the rest of the world.” I agree with that assessment.
STEVEN R. SMITH, MS, JD
Reference
- South Dakota v Wayfair, Inc, 585 US (2018)
Next term, the Court is scheduled to hear cases regarding pharmaceutical liability, double jeopardy, sex-offender registration, expert witnesses, Social Security disability benefits, and the Age Discrimination in Employment Act. There will be at least 3 arbitration cases. Health care and reproductive rights will continue to be an important part of the Court’s docket.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- National Institute of Family and Life Advocates v Becerra, 585 US 16 1140 (2018).
- California Reproductive Freedom, Accountability, Comprehensive Care, and Transparency Act (FACT Act), Cal. Health & Safety Code Ann. §123470 et seq. (West 2018).
- Brief amici curiae of American Academy of Pediatrics, et al. in National Institute of Family and Life Advocates v Becerra, February 27, 2018.
- Brief amici curiae of American Association of Pro-Life Obstetricians & Gynecologists, et al. in National Institute of Family and Life Advocates v Becerra, January 16, 2018.
- Azar v Garza, 584 US 17 654 (2018).
- Brief amici curiae of American College of Obstetricians and Gynecologists and American Public Health Association in Planned Parenthood of Arkansas and Eastern Oklahoma v Jegley, February 1, 2018.
- Chapter 31, Inter Partes Review. United States Code. Title 35: Patents. Part III, Patents and protection of patents. 2012 Ed. 35 USC 311–319.
- Oil States Energy Services, LLC v Greene’s Energy Group, LLC, 584 US 16 712 (2018).
- SAS Institute Inc. v Iancu, 584 US 16 969 (2018).
- Brief for Association of American Medical Colleges and Others as Amici Curiae Supporting Respondents, Trump v Hawaii. https://www.supremecourt.gov/Docket PDF/17/ 17-965/40128/20180327105855912_17-965%20Amicus%20Br.%20Proclamation.pdf. Accessed September 21, 2018.
- Smith SR, Sanfilippo JS. Supreme Court decisions in 2017 that affected your practice. OBG Manag. 2017;29(12)44–47.
- Trump v Hawaii, 585 US 17 965 (2018).
- Ohio v American Express Co, 585 US 16 1454 (2018).
- Brief amici curiae of American Medical Association and Ohio State Medical Association in Ohio v American Express, December 24, 2017.
- Masterpiece Cakeshop, Ltd. v Colorado Civil Rights Commission, 584 US 16 111 (2018).
- Brief amici curiae of American College of Pediatricians, et al. in Masterpiece Cakeshop v Colorado Civil Rights Commission, September 7, 2017.
- National Institute of Family and Life Advocates v Becerra, 585 US 16 1140 (2018).
- California Reproductive Freedom, Accountability, Comprehensive Care, and Transparency Act (FACT Act), Cal. Health & Safety Code Ann. §123470 et seq. (West 2018).
- Brief amici curiae of American Academy of Pediatrics, et al. in National Institute of Family and Life Advocates v Becerra, February 27, 2018.
- Brief amici curiae of American Association of Pro-Life Obstetricians & Gynecologists, et al. in National Institute of Family and Life Advocates v Becerra, January 16, 2018.
- Azar v Garza, 584 US 17 654 (2018).
- Brief amici curiae of American College of Obstetricians and Gynecologists and American Public Health Association in Planned Parenthood of Arkansas and Eastern Oklahoma v Jegley, February 1, 2018.
- Chapter 31, Inter Partes Review. United States Code. Title 35: Patents. Part III, Patents and protection of patents. 2012 Ed. 35 USC 311–319.
- Oil States Energy Services, LLC v Greene’s Energy Group, LLC, 584 US 16 712 (2018).
- SAS Institute Inc. v Iancu, 584 US 16 969 (2018).
- Brief for Association of American Medical Colleges and Others as Amici Curiae Supporting Respondents, Trump v Hawaii. https://www.supremecourt.gov/Docket PDF/17/ 17-965/40128/20180327105855912_17-965%20Amicus%20Br.%20Proclamation.pdf. Accessed September 21, 2018.
- Smith SR, Sanfilippo JS. Supreme Court decisions in 2017 that affected your practice. OBG Manag. 2017;29(12)44–47.
- Trump v Hawaii, 585 US 17 965 (2018).
- Ohio v American Express Co, 585 US 16 1454 (2018).
- Brief amici curiae of American Medical Association and Ohio State Medical Association in Ohio v American Express, December 24, 2017.
- Masterpiece Cakeshop, Ltd. v Colorado Civil Rights Commission, 584 US 16 111 (2018).
- Brief amici curiae of American College of Pediatricians, et al. in Masterpiece Cakeshop v Colorado Civil Rights Commission, September 7, 2017.

ED visits related to psychiatric complaints are up 20% among elderly
SAN DIEGO – Between 2011 and 2015, the proportion of elderly patients presenting to the emergency department with psychiatric complaints increased by 20%, according to a retrospective analysis of national hospital data.
In addition, 10-year increases in age, male sex, nursing home status, and Medicare insurance were associated with an increased likelihood of hospital admission.
“The growing geriatric patient population is a well-known phenomenon across every developed country,” lead researcher Derrick Huang said in an interview in advance of the annual meeting of the American College of Emergency Physicians. “A potent mix of increasing life expectancy, greater disease severity and comorbidities in the elderly, and large-scale demographic shifts has placed a significant strain on both our financial and health care resources. This study corroborates these trends in the emergency department and is a preliminary exploration of potential, newly evolving clinical challenges that the ED team will increasingly face into the future.”
For the study, Mr. Huang, a fourth-year medical student at Oakland University William Beaumont School of Medicine in Rochester, Mich., and his colleagues examined National Ambulatory Medical Care Survey (NAMCS) data between 2011 and 2015. They limited the analysis to emergency department visits with patients in the age groups of 65-74, 75-84, and 85 or older. For the primary outcome of interest, the researchers evaluated demographic variables of age group, sex, residential status, race and ethnicity, and insurance for association with hospital admission. For the secondary outcome of interest, they evaluated presenting ED complaints related to the clinical domains of cardiopulmonary disease, psychiatric disease, and fractures and dislocations for potential trends in the ED geriatric age group between 2011 and 2015.
Mr. Huang and his associates found that, as a percentage of total ED visits, those among patients aged 65 or older rose from 14.9% in 2011 to 15.6% in 2015, an increase of 4.7%. By age group, the proportion of visits during the study period was highest for those aged 65-74 years (43.8%), followed by those aged 75-84 years (34.7%) and those aged 85 and older (21.5%). On multivariate analysis, the 75-84 and age-85-and-older groups were 1.30 and 1.71 times more likely to be admitted to the hospital, compared with the 65-74 group, respectively (P less than .000 for both).
Men were 1.19 times more likely than were women to be admitted (P less than .000). In addition, elderly patients who reside in nursing homes were 1.70 times more likely to be admitted to the hospital, compared with those who lived in private homes (P less than .000), while those with Medicare insurance were 1.57 more likely to be admitted, compared with those who did not have insurance (P = .004).
On trend analysis, ED psychiatric complaints rose incrementally during the study period, from 3.9% in 2011 to 4.7% in 2015, a relative increase of 20.5%. The researchers identified no consistent trend with visit complaints related to cardiopulmonary disease, and fractures and dislocations.
“This was not too surprising, because these difficulties with older patients are not new and many investigators have sought out solutions,” Mr. Huang said. “For example, there have been many interventions both in the ED as well as in the primary care setting designed to identify risk factors and facilitate postdischarge care to prevent falls. These approaches are constantly evolving and will be of increasing importance.”
“For example, we may be seeing a larger of proportion of patients with acute mental health complaints,” Mr. Huang said. “We will need to continue developing our multidisciplinary approach to care by improving coordination with different specialties – and especially outpatient and community health care providers.”
The study’s senior author was Jason Wasserman, PhD of Oakland University William Beaumont School of Medicine. The researchers reported having no financial disclosures.
SOURCE: Huang D et al. Ann Emerg Med. 2018 Oct. doi: 10.1016/j.annemergmed.2018.08.212.
SAN DIEGO – Between 2011 and 2015, the proportion of elderly patients presenting to the emergency department with psychiatric complaints increased by 20%, according to a retrospective analysis of national hospital data.
In addition, 10-year increases in age, male sex, nursing home status, and Medicare insurance were associated with an increased likelihood of hospital admission.
“The growing geriatric patient population is a well-known phenomenon across every developed country,” lead researcher Derrick Huang said in an interview in advance of the annual meeting of the American College of Emergency Physicians. “A potent mix of increasing life expectancy, greater disease severity and comorbidities in the elderly, and large-scale demographic shifts has placed a significant strain on both our financial and health care resources. This study corroborates these trends in the emergency department and is a preliminary exploration of potential, newly evolving clinical challenges that the ED team will increasingly face into the future.”
For the study, Mr. Huang, a fourth-year medical student at Oakland University William Beaumont School of Medicine in Rochester, Mich., and his colleagues examined National Ambulatory Medical Care Survey (NAMCS) data between 2011 and 2015. They limited the analysis to emergency department visits with patients in the age groups of 65-74, 75-84, and 85 or older. For the primary outcome of interest, the researchers evaluated demographic variables of age group, sex, residential status, race and ethnicity, and insurance for association with hospital admission. For the secondary outcome of interest, they evaluated presenting ED complaints related to the clinical domains of cardiopulmonary disease, psychiatric disease, and fractures and dislocations for potential trends in the ED geriatric age group between 2011 and 2015.
Mr. Huang and his associates found that, as a percentage of total ED visits, those among patients aged 65 or older rose from 14.9% in 2011 to 15.6% in 2015, an increase of 4.7%. By age group, the proportion of visits during the study period was highest for those aged 65-74 years (43.8%), followed by those aged 75-84 years (34.7%) and those aged 85 and older (21.5%). On multivariate analysis, the 75-84 and age-85-and-older groups were 1.30 and 1.71 times more likely to be admitted to the hospital, compared with the 65-74 group, respectively (P less than .000 for both).
Men were 1.19 times more likely than were women to be admitted (P less than .000). In addition, elderly patients who reside in nursing homes were 1.70 times more likely to be admitted to the hospital, compared with those who lived in private homes (P less than .000), while those with Medicare insurance were 1.57 more likely to be admitted, compared with those who did not have insurance (P = .004).
On trend analysis, ED psychiatric complaints rose incrementally during the study period, from 3.9% in 2011 to 4.7% in 2015, a relative increase of 20.5%. The researchers identified no consistent trend with visit complaints related to cardiopulmonary disease, and fractures and dislocations.
“This was not too surprising, because these difficulties with older patients are not new and many investigators have sought out solutions,” Mr. Huang said. “For example, there have been many interventions both in the ED as well as in the primary care setting designed to identify risk factors and facilitate postdischarge care to prevent falls. These approaches are constantly evolving and will be of increasing importance.”
“For example, we may be seeing a larger of proportion of patients with acute mental health complaints,” Mr. Huang said. “We will need to continue developing our multidisciplinary approach to care by improving coordination with different specialties – and especially outpatient and community health care providers.”
The study’s senior author was Jason Wasserman, PhD of Oakland University William Beaumont School of Medicine. The researchers reported having no financial disclosures.
SOURCE: Huang D et al. Ann Emerg Med. 2018 Oct. doi: 10.1016/j.annemergmed.2018.08.212.
SAN DIEGO – Between 2011 and 2015, the proportion of elderly patients presenting to the emergency department with psychiatric complaints increased by 20%, according to a retrospective analysis of national hospital data.
In addition, 10-year increases in age, male sex, nursing home status, and Medicare insurance were associated with an increased likelihood of hospital admission.
“The growing geriatric patient population is a well-known phenomenon across every developed country,” lead researcher Derrick Huang said in an interview in advance of the annual meeting of the American College of Emergency Physicians. “A potent mix of increasing life expectancy, greater disease severity and comorbidities in the elderly, and large-scale demographic shifts has placed a significant strain on both our financial and health care resources. This study corroborates these trends in the emergency department and is a preliminary exploration of potential, newly evolving clinical challenges that the ED team will increasingly face into the future.”
For the study, Mr. Huang, a fourth-year medical student at Oakland University William Beaumont School of Medicine in Rochester, Mich., and his colleagues examined National Ambulatory Medical Care Survey (NAMCS) data between 2011 and 2015. They limited the analysis to emergency department visits with patients in the age groups of 65-74, 75-84, and 85 or older. For the primary outcome of interest, the researchers evaluated demographic variables of age group, sex, residential status, race and ethnicity, and insurance for association with hospital admission. For the secondary outcome of interest, they evaluated presenting ED complaints related to the clinical domains of cardiopulmonary disease, psychiatric disease, and fractures and dislocations for potential trends in the ED geriatric age group between 2011 and 2015.
Mr. Huang and his associates found that, as a percentage of total ED visits, those among patients aged 65 or older rose from 14.9% in 2011 to 15.6% in 2015, an increase of 4.7%. By age group, the proportion of visits during the study period was highest for those aged 65-74 years (43.8%), followed by those aged 75-84 years (34.7%) and those aged 85 and older (21.5%). On multivariate analysis, the 75-84 and age-85-and-older groups were 1.30 and 1.71 times more likely to be admitted to the hospital, compared with the 65-74 group, respectively (P less than .000 for both).
Men were 1.19 times more likely than were women to be admitted (P less than .000). In addition, elderly patients who reside in nursing homes were 1.70 times more likely to be admitted to the hospital, compared with those who lived in private homes (P less than .000), while those with Medicare insurance were 1.57 more likely to be admitted, compared with those who did not have insurance (P = .004).
On trend analysis, ED psychiatric complaints rose incrementally during the study period, from 3.9% in 2011 to 4.7% in 2015, a relative increase of 20.5%. The researchers identified no consistent trend with visit complaints related to cardiopulmonary disease, and fractures and dislocations.
“This was not too surprising, because these difficulties with older patients are not new and many investigators have sought out solutions,” Mr. Huang said. “For example, there have been many interventions both in the ED as well as in the primary care setting designed to identify risk factors and facilitate postdischarge care to prevent falls. These approaches are constantly evolving and will be of increasing importance.”
“For example, we may be seeing a larger of proportion of patients with acute mental health complaints,” Mr. Huang said. “We will need to continue developing our multidisciplinary approach to care by improving coordination with different specialties – and especially outpatient and community health care providers.”
The study’s senior author was Jason Wasserman, PhD of Oakland University William Beaumont School of Medicine. The researchers reported having no financial disclosures.
SOURCE: Huang D et al. Ann Emerg Med. 2018 Oct. doi: 10.1016/j.annemergmed.2018.08.212.
REPORTING FROM ACEP18
Key clinical point: An increasing proportion of elderly patients are presenting to the emergency department with mental health complaints.
Major finding: Emergency department psychiatric complaints among elderly patients rose from 3.9% in 2011, to 4.7% in 2015, a relative increase of 20.5%.
Study details: A retrospective analysis of National Ambulatory Medical Care Survey data between 2011 and 2015.
Disclosures: The researchers reported having no financial disclosures.
Source: Huang D et al. Ann Emerg Med. 2018 Oct. doi: 10.1016/j.annemergmed.2018.08.212.
The In Vivo Impact of Leukocyte Injections on Normal Rat Achilles Tendons: Potential Detriment to Tendon Morphology, Cellularity, and Vascularity
ABSTRACT
In this study, we determine the in vivo effects of injecting sub-populations of leukocytes into normal rat Achilles tendons via a controlled laboratory study. Allogenic monocytes, granulocytes, or plasma were injected into 24 healthy rat Achilles tendons. Treated and contralateral un-treated control tendons then assessed for cellularity, histologic morphology, and vascularity after 7 and 14 days. Significant increases of 221% and 249% in cellularity (P = 0.014) were seen on day 14 within Achilles tendons injected with granulocytes as compared to plasma and monocytes, respectively. Also, significant improvement in morphology (P = 0.029) between days 7 and 14 was seen for the granulocyte injected Achilles tendons. Significant increases in cellularity after an injection of granulocytes, compared to monocytes and plasma, corresponds to a significant increase in inflammation within the tissue, suggesting that leukocyte-rich platelet-rich plasma (PRP) preparations are proinflammatory and potentially catabolic when injected into tendon tissue. The concentration and composition of white blood cells within PRP preparations is variable and needs to be better understood in order to optimize clinical utility of PRP injections.
Continue to: Tendinopathies are debilitating conditions...
Tendinopathies are debilitating conditions affecting patients worldwide every day. They arise most frequently from tendon overuse resulting in pathology.1 There are 2 major subtypes of tendinopathy: tendinosis and tendinitis. Tendinosis, the more common condition, is characterized by long-term, chronic degradation of tendon tissue resulting in fibrosis from infiltrating fibroblasts.2 Tendinitis, the less common condition, is characterized by an acute inflammatory response and inflammatory cell infiltrate.2 Both conditions are common, with Achilles tendinopathy affecting 11% of runners and lateral epicondylitis affecting 1% to 3% of the general population.3,4 Many sports-related overuse injuries, such as tendinopathies, go undiagnosed for extended periods of time because medical attention is avoided in order to prevent time loss from training or competing.5 These delays could be eliminated if a non-surgical option for treating tendon pathology was available.
Tendinopathies are believed to result from tendon overuse that causes micro-damage to collagen, as well as from significant changes in protein and enzyme composition within the tendon.6 The damage accumulates over time and eventually leads to chronic inflammation or fibrotic change within tendons, in both cases weakening the tendon and causing pain. Currently, accepted treatments for tendinopathies include: nonsteroidal anti-inflammatory drugs, physical therapy, ultrasound, laser-therapy, corticosteroids, glyceryl trinitrate patches, extracorporeal shock wave therapy, sclerotherapy, and surgery.7 Recently, platelet-rich plasma (PRP) therapy has emerged as a promising treatment for tendinopathies, as well as a variety of other orthopedic indications.
PRP consists of autologous blood from the patient, centrifuged to increase the amount of platelets in the sample above baseline, and subsequently injected around an affected tendon or joint.8 PRP is used to treat tendinopathy because it can supply injured tendons with blood components that aid in healing, which tendons do not receive due to poor vascularity.9 These components include growth factors, such as platelet derived growth factor (PDGF), transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), endothelial growth factor, and leukocytes that can stimulate an inflammatory response within the injured tissue.10 The inflammatory response from the PRP induces a more robust reconstruction and revascularization of the injured tissue, stimulating proliferation, and remodeling.11,12However, significant variability exists within the platelets, leukocytes, and growth factors that comprise PRP. This is attributed to 3 major causes. First, current commercial preparations of PRP result in differing platelet concentrations, as well as leukocyte-rich and leukocyte-poor compositions.13,14 Variability in platelet concentrations results in unreliable amounts of growth factors, including cytokines, TGF-β, PDGF, VEGF and basic fibroblast growth factor in each preparation, while leukocyte levels affect inflammation, all leading to variable effects for each preparation.15,16Second, despite sex and age of the PRP donor not being significant factors influencing variation in growth factor concentrations, the existence of an unexplained variation in concentrations of growth factors between different donors has been observed.17 Third, the selection of activating agents, bovine thrombin or calcium chloride, and their application, whether to the elbow, shoulder, or knee, produces variability.18
While the effects of platelets and growth factors in PRP have been well studied, less is known about the effects of differing cell types. Recently it was reported that the concentrations of leukocytes directly affect the outcomes of PRP injections. McCarrel and colleagues19,20 found that as the number of leukocytes increased, there was a concomitant increase in the expression of inflammatory cytokines and catabolic activity. This effect may result in inferior healing of injured tissues and is attributed to the release of pro-inflammatory cytokines such as interleukin-1β from the leukocytes.21 There is also evidence that minimizing the catabolic effect of leukocytes may be just as important to tissue healing as the maximizing anabolic effect of platelets and growth factors.22
The use of PRP has been highly disputed in recent years due to conflicting reports of its success in treating orthopedic conditions. Numerous favorable studies have shown benefit for treating chronic and acute orthopedic injuries including; rotator cuff tear repair, chronic refractory patellar tendinopathy, and chronic lateral tendinosis/epicondylitis.23-26 Concurrently, articles demonstrating no significant effects from PRP have also been published. One study claiming that PRP injections did not improve outcomes of chronic Achilles tendinopathy did not differentiate whether patients had tendinosis or tendinitis, and did not consider leukocyte concentration in their PRP preparations27 Another study that determined PRP is not beneficial to the healing of ruptured Achilles tendons after surgical repair also failed to consider the concentration of leukocytes in their PRP preparations.28 One of the difficulties in comparing these studies is their heterogeneous nature. This arises from the use of different conditions in each study that makes the studies incomparable. Variations in PRP preparations lead to different concentrations of growth factors, platelets, and leukocyte concentrations. Additionally, tendinopathy models were not specified as tendinosis and tendonitis, and models or patients were not controlled for age, sex, or comorbidities. Given that leukocyte-rich and leukocyte-poor PRP preparations are currently widely used in clinical practice, the discovery of which type of preparation is indicated in which setting is paramount to evidence-based use of this treatment modality. Due to reports suggesting that leukocytes may be detrimental to tendon healing, determining which types of leukocytes are responsible for these effects is vital. As such, the purpose of this study is to determine the in vivo effects of sub-populations of leukocytes on normal rat tendons. This study design allowed us to isolate the effects of the injections to induce a response and remove confounding effects of normal healing response to a damaged tendon and effects from the injection itself. Our hypothesis was that the injection of leukocytes would cause an inflammatory response in rat tendons, leading to catabolic outcomes.
Continue to: METHODS...
METHODS
This was a prospective, in vivo, placebo controlled, randomized animal study. The University’s Institutional Animal Care and Use Committee approved all procedures prior to initiation. Twenty-four male Sprague-Dawley rats were randomized to 3 treatment groups (n = 8): monocytes; granulocytes, and; plasma, as a negative control.
Allogenic blood from 6 additional rats was collected into K2EDTA tubes via cardiac puncture. Allogenic, as opposed to autogenic, blood is commonly used in rat models because of low immunogenic response to blood from rats of the same strain and litter.29,30 The blood was then pooled and the red cells lysed by incubation with Red Blood Cell Lysis Buffer (Roche). The samples were then sorted into fractions containing monocytes and granulocytes using fluorescence activated cell sorting (FACS) using a FACSAria (BD Biosciences). Cells were sorted using Purified PE Mouse Anti-Rat CD11b/c antibodies (BD Pharmingen) specific to monocytes, granulocytes, macrophages, dendritic cells, and microglia, APC-Cy7 Mouse Anti-Rat CD45 antibodies (BD Pharmingen) specific to all hematopoietic cells except erythrocytes, and FITC Mouse Anti-Rat CD42d antibodies (BD Pharmingen) specific to megakaryocytes and platelets. 20 μL of 0.2 mg/mL CD11b/c, 20 μL of 0.2 mg/mL CD 45, and 10 μL of 0.5 mg/mL CD42d antibodies were added to 1 mL of condensed non-red cells collected from the 6 rats and incubated at room temperature in the dark for 15 minutes. A fraction containing only platelet-poor plasma was also collected. For all treatments the injection volume was 75 μL. Rats in the monocyte group were injected with 200,000 cells in platelet-poor plasma, those in the granulocyte group were injected with 900,000 cells in platelet-poor plasma, and rats in the plasma control group received only platelet-poor plasma. The cell concentrations were based on previous studies that documented these concentrations that are found in typical leukocyte-rich PRP preparations.13
The animals were anesthetized with isoflurane gas and then injected aseptically once into their right Achilles tendon. The left Achilles tendon was used as an un-injected control, giving a total of 48 total Achilles tendons studied. At days 7 and 14 post-injection, 4 rats from each group were euthanized and the Achilles tendons were harvested.
The tendons were fixed in neutral buffered formalin for 24 hours and then embedded in paraffin and sectioned sagittally at 12 μm. The tendons were then stained with hematoxylin and eosin (H&E) using standard histological protocols and examined by 3 individuals trained to assess cellularity and morphology. All samples were assigned unrecognizable numbers and randomized prior to examination by individuals. Cell counts were based on the number of nuclei present in 3 mid-tendon high-power fields (400x) per sample. Morphology was graded on a scale of 1 to 3, with 1 being a normal tendon and 3 having severe pathology with total loss of alignment and crimping on 3 low-power fields (100x) per sample (Figures 1A-1G).
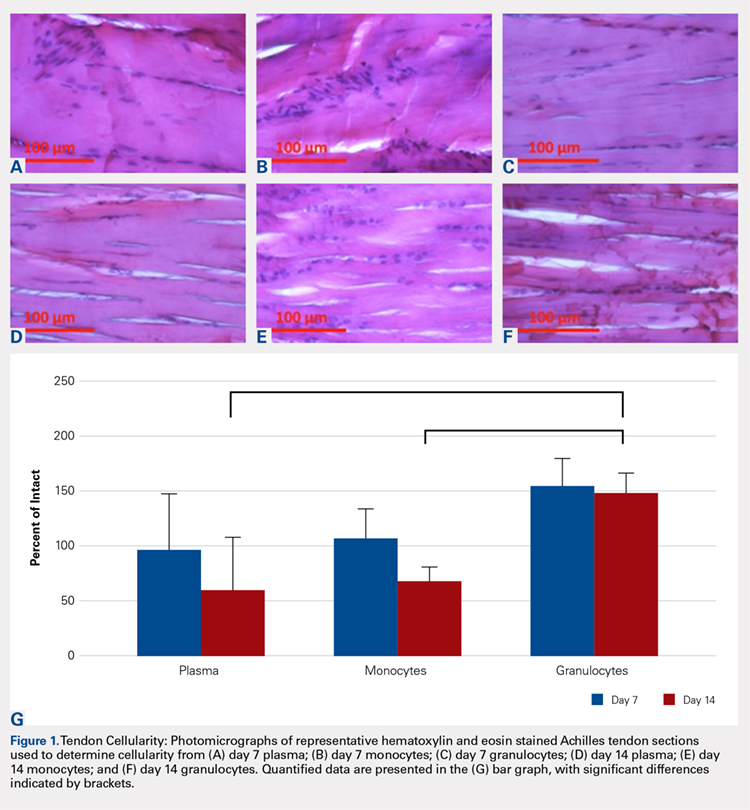
Vascularity was assessed by immunohistochemical staining using Rabbit Polyclonal Anti-CD31 antibodies (Abcam), a marker for vascular endothelial cells, using a Vectastain ABC Kit (Vector Laboratories) system and the ImmPACT AEC Peroxidase (HRP) Substrate (Vector Laboratories). Following staining, automated image analysis was performed (Bioquant). Briefly, all areas that did not contain tendon were masked. CD31 positive areas were then quantified using global thresholding. Vascularity was then calculated as ratio of CD31 positive area to total tendon area. Analyses were performed on 3 mid-tendon medium-power (200x) fields per sample.
For cellularity and morphology, the results for the injected tendons were normalized to those of their contralateral untreated controls and reported as a percentage. Results for vascularity were compared directly between treated tendons. Differences were assessed between groups at each time-point using Independent Samples Median Tests. When significant differences were identified, pairwise comparisons were performed to identify the source of the differences. All analyses were conducted using SPSS (V22, SAS Institute) with significant differences determined for values of P < 0.05.
RESULTS
No significant differences in cellularity between groups were seen at day 7 (P = 0.368) (Figures 1A-1G). However, a significant difference in cellularity between groups was seen at day 14 (P = 0.014). Pairwise tests showed there to be a significant increase in the number of cells in the tendons treated with granulocytes from 221% and 249% in cellularity (P = 0.014) on day 14, as compared to both monocytes and plasma, respectively. Morphologically, no significant differences were seen between groups at either time-point (P = 0.091 for day 7 and P = 1.000 for day 14) (Figures 2A-2G). However, a significant improvement in morphology was observed from day 7 to day 14 in the granulocyte group from 60% to 165% (P = 0.029). Finally, no differences were seen in vascularity between treatment groups at either time-point (P = 0.368 for day 7 and P = 0.535 for day 14) (Figures 3A-3G).
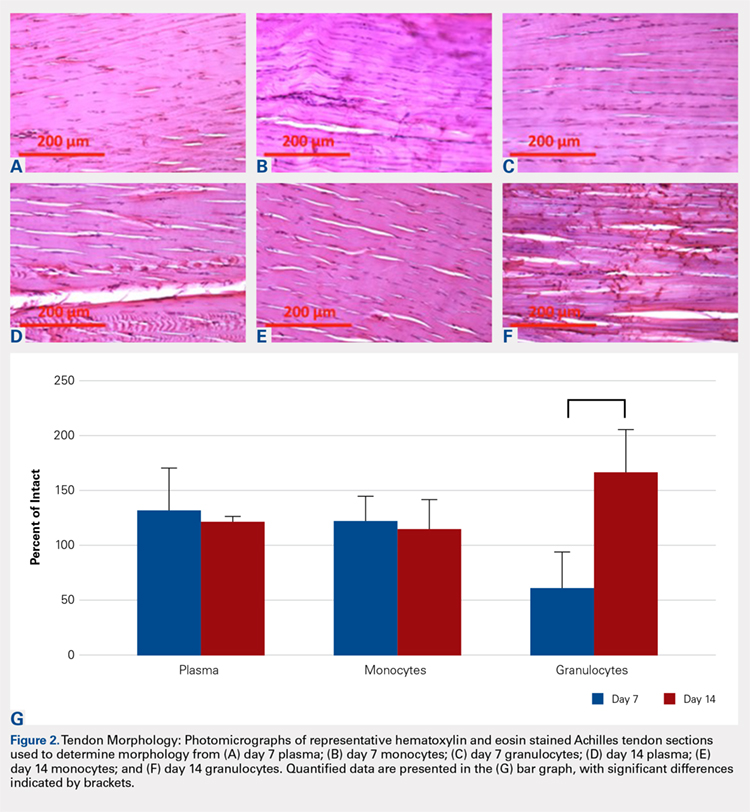
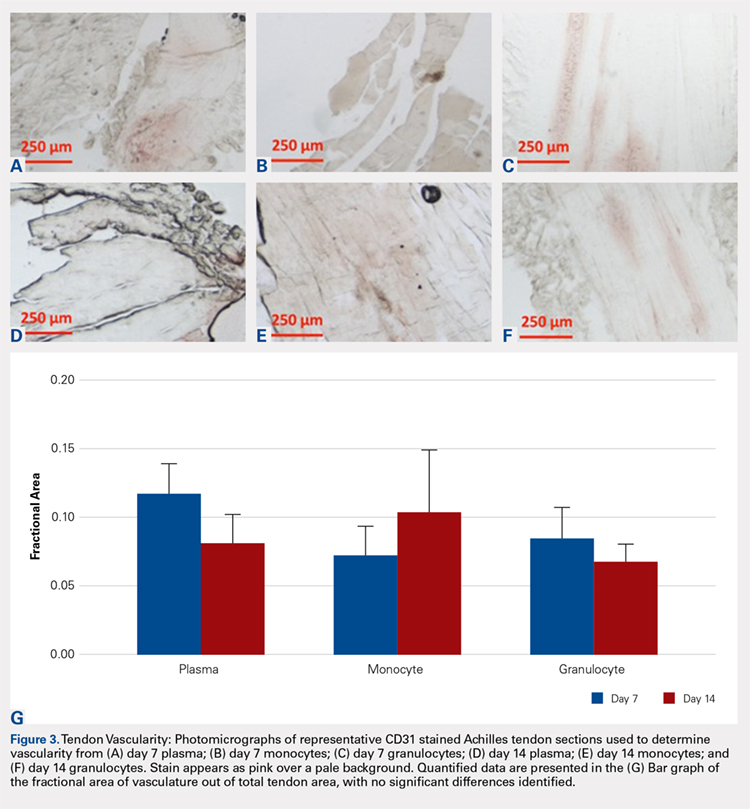
Continue to: DISCUSSION...
DISCUSSION
Our hypothesis that the injection of leukocytes would cause an inflammatory response in rat tendons leading to catabolic outcomes was confirmed in the granulocyte group. It should be noted that prior to the catabolic outcome, there was a transient anabolic effect in the granulocyte group during the second week. Deterioration in morphology was observed in the tendons injected with granulocytes on day 7, which subsequently recovered in the following week. We found that injecting granulocytes into normal tendons resulted in an increase in inflammatory cellularity, when compared to monocytes and plasma injections.
Limitations inherent in this study are those similar to other in vivo studies. To begin with, the results of injections into rat tendons may not be translatable to human tendons. Despite this limitation, the rat is a common model for tendon research.31 Another limitation is that this study injected healthy Achilles tendons, rather than tendons with preexisting tendinopathy. In a naturally occurring tendinopathy, there may be other factors present that interact with PRP, and this model negates the contribution of these factors. Finally, while the immunohistochemistry (IHC) and morphological data are clear, the cellularity data are not clear in identifying the type of cells that were increased by granulocyte injection. However, the cells appeared rounded, resembling inflammatory infiltrate; a common cell type seen in tendons.2 While fibroblasts are also a common infiltrate during chronic tendinopathy, they are generally flat and appear on H&E as long spindle shaped cells. Thus, we believe the increased cellularity of the tendons after granulocyte injections is representative of an increase in inflammation. The increased cellularity could be due to the increased number of cells injected into the tendon; however, our conclusions are consistent with the increased inflammation previously reported linking leukocytes to tendon inflammation.20,22,32
In terms of morphology, we hypothesized that degenerative changes would be seen in the tendons that were injected with granulocytes due to the inflammatory action of these cells. As part of the granulocyte response, neutrophils release proteases and macrophages can stimulate collagen synthesis via fibroblasts, both causing change within the extracellular matrix.33,34 Indeed, we observed a significant change in tissue morphology in the granulocyte group over the course of 14 days. As the degenerative and regenerative effects of granulocytes take time to present, this is likely what we observed to occur between day 7 and 14 after treatment. These observations are also consistent with prior observations that leukocyte-rich PRP injections can be detrimental to tendon healing, but beneficial to tissue degeneration in the setting of chronic tendonitis.20
We hypothesized that the vascularity of the tendons would be similar in all preparations. This was based on previous studies demonstrating that the lack of platelets in the platelet-poor plasma fraction is sufficient to deplete VEGF, the angiogenic agent in PRP.35 In this study, there were no observable differences in vascularity of platelet-poor plasma, monocyte, and granulocyte injections. We attribute this to the lack of VEGF in any of these preparations. The aforementioned study also showed that the lack of platelets in injection was enough to prevent the angiogenic effect of this treatment.35
Continue to: The goal of this study was...
The goal of this study was to assess the morphology, cellularity, and vascularity of normal tendons after injections of different leukocyte populations. This is clinically important because of the potential to tailor future PRP injections on a patient-by-patient basis. In patients requiring an anabolic response, leukocyte-poor PRP may be the best option. In contrast, when patient pathology requires an inflammatory response to improve healing36 or breakdown fibrotic tissue, as seen in tendinosis, leukocyte-rich PRP may be warranted. Further, properly controlled clinical studies are needed to validate these recommendations.
Limitations inherent in this study are those similar to other in vivo studies. First, the results of injections into rat tendons may not be translatable to human tendons. Despite this limitation, the rat is a common model for tendon research.31 A second limitation is that this study injected healthy Achilles tendons, rather than tendons with preexisting tendinopathy. In a naturally occurring tendinopathy, there may be other factors present that interact with PRP, and this model negates the contribution of these factors. Finally, while the IHC and morphological data show clear changes, the cellularity data are not clear in identifying the type of cells that were increased by granulocyte injection. However, the cells appeared rounded, resembling inflammatory infiltrate; a common cell type seen in tendons.2 While fibroblasts are also a common infiltrate during chronic tendinopathy, they are generally flat and appear on H&E as long spindle shaped cells. The last limitation of this study is the lack of functional mechanical testing since, clinically, healing of the tendon is also related to the strength of the tendon. Thus, we believe the increased cellularity of the tendons after granulocyte injections is representative of an increase in inflammation. Moreover, our results are consistent with the increased inflammation previously reported linking leukocytes to tendon inflammation.20,22,32 It is interesting to note that the increase in inflammation does not lead to an increase in vascularity as could be expected.
CONCLUSION
We found that the injection of leukocytes into healthy rat Achilles tendons increases inflammation, as evidenced by increased cellularity and disrupted morphology, which suggests that leukocyte-rich PRP preparations may be contraindicated in settings of acute tendonitis. However, these preparations may be useful for a specific subset of tendinopathies, including chronic tendinosis.
1. Herring SA, Nilson KL. Introduction to overuse injuries. Clin Sports Med. 1987;6(2):225-239.
2. Bass E. Tendinopathy: why the difference between tendinitis and tendinosis matters. Int J Ther Massage Bodywork. 2012;5(1):14-17.
3. James SL, Bates BT, Osternig LR. Injuries to runners. Am J Sports Med. 1978;6(2):40-50.
4. Allander E. Prevalence, incidence, and remission rates of some common rheumatic diseases or syndromes. Scand J Rheumatol. 1974;3(3):145-153.
5. Bahr R. No injuries, but plenty of pain? On the methodology for recording overuse symptoms in sports. Br J Sports Med. 2009;43(13):966-972.
6. Rees JD, Maffulli N, Cook J. Management of tendinopathy. Am J Sports Med. 2009;37(9):1855-1867.
7. Andres BM, Murrell GA. Treatment of tendinopathy: what works, what does not, and what is on the horizon. Clin Orthop Relat Res. 2008;466(7):1539-1554.
8. Hall MP, Band PA, Meislin RJ, Jazrawi LM, Cardone DA. Platelet-rich plasma: current concepts and application in sports medicine. J Am Acad Orthop Surg. 2009;17(10):602-608.
9. Smith JW. Blood Supply of Tendons. Am J Surg. 1965;109:272-276.
10. Wu PI, Diaz R, Borg-Stein J. Platelet-rich plasma. Phys Med Rehabil Clin N Am. 2016;27(4):825-853.
11. Nguyen RT, Borg-Stein J, McInnis K. Applications of platelet-rich plasma in musculoskeletal and sports medicine: an evidence-based approach. PM R. 2011;3(3):226-250.
12. Broughton G 2nd, Janis JE, Attinger CE. Wound healing: an overview. Plast Reconstr Surg. 2006;117(7 Suppl):1e-S-32e-S.
13. Mazzocca AD, McCarthy MB, Chowaniec DM, et al. Platelet-rich plasma differs according to preparation method and human variability. J Bone Joint Surg Am. 2012;94(4):308-316.
14. Mazzocca AD, McCarthy MB, Chowaniec DM, et al. The positive effects of different platelet-rich plasma methods on human muscle, bone, and tendon cells. Am J Sports Med. 2012;40(8):1742-1749.
15. Castillo TN, Pouliot MA, Kim HJ, Dragoo JL. Comparison of growth factor and platelet concentration from commercial platelet-rich plasma separation systems. Am J Sports Med. 2011;39(2):266-271.
16. Cho HS, Song IH, Park SY, Sung MC, Ahn MW, Song KE. Individual variation in growth factor concentrations in platelet-rich plasma and its influence on human mesenchymal stem cells. Korean J Lab Med. 2011;31(3):212-218.
17. Weibrich G, Kleis WK, Hafner G, Hitzler WE. Growth factor levels in platelet-rich plasma and correlations with donor age, sex, and platelet count. J Craniomaxillofac Surg. 2002;30(2):97-102.
18. Taylor DW, Petrera M, Hendry M, Theodoropoulos JS. A systematic review of the use of platelet-rich plasma in sports medicine as a new treatment for tendon and ligament injuries. Clin J Sport Med. 2011;21(4):344-352.
19. McCarrel T, Fortier L. Temporal growth factor release from platelet-rich plasma, trehalose lyophilized platelets, and bone marrow aspirate and their effect on tendon and ligament gene expression. J Orthop Res. 2009;27(8):1033-1042.
20. McCarrel TM, Minas T, Fortier LA. Optimization of leukocyte concentration in platelet-rich plasma for the treatment of tendinopathy. J Bone Joint Surg Am. 2012;94(19):e143(141-148).
21. Pillitteri D, Bassus S, Boller K, et al. Thrombin-induced interleukin 1beta synthesis in platelet suspensions: impact of contaminating leukocytes. Platelets. 2007;18(2):119-127.
22. Boswell SG, Schnabel LV, Mohammed HO, Sundman EA, Minas T, Fortier LA. Increasing platelet concentrations in leukocyte-reduced platelet-rich plasma decrease collagen gene synthesis in tendons. Am J Sports Med. 2014;42(1):42-49.
23. Mishra A, Pavelko T. Treatment of chronic elbow tendinosis with buffered platelet-rich plasma. Am J Sports Med. 2006;34(11):1774-1778.
24. Maniscalco P, Gambera D, Lunati A, et al. The "Cascade" membrane: a new PRP device for tendon ruptures. Description and case report on rotator cuff tendon. Acta Biomed. 2008;79(3):223-226.
25. Filardo G, Kon E, Della Villa S, Vincentelli F, Fornasari PM, Marcacci M. Use of platelet-rich plasma for the treatment of refractory jumper's knee. Int Orthop. 2010;34(6):909-915.
26. Peerbooms JC, Sluimer J, Bruijn DJ, Gosens T. Positive effect of an autologous platelet concentrate in lateral epicondylitis in a double-blind randomized controlled trial: platelet-rich plasma versus corticosteroid injection with a 1-year follow-up. Am J Sports Med. 2010;38(2):255-262.
27. de Vos RJ, Weir A, van Schie HT, et al. Platelet-rich plasma injection for chronic Achilles tendinopathy: a randomized controlled trial. JAMA. 2010;303(2):144-149.
28. Schepull T, Kvist J, Norrman H, Trinks M, Berlin G, Aspenberg P. Autologous platelets have no effect on the healing of human achilles tendon ruptures: a randomized single-blind study. Am J Sports Med. 2011;39(1):38-47.
29. Welsh KI, Burgos H, Batchelor JR. The immune response to allogeneic rat platelets; Ag-B antigens in matrix form lacking Ia. Eur J Immunol. 1977;7(5):267-272.
30. Xue M, Del Bigio MR. Intracortical hemorrhage injury in rats : relationship between blood fractions and brain cell death. Stroke. 2000;31(7):1721-1727.
31. Voleti PB, Buckley MR, Soslowsky LJ. Tendon healing: repair and regeneration. Annu Rev Biomed Eng. 2012;14:47-71.
32. Sundman EA, Cole BJ, Fortier LA. Growth factor and catabolic cytokine concentrations are influenced by the cellular composition of platelet-rich plasma. Am J Sports Med. 2011;39(10):2135-2140.
33. Palmgren MS, deShazo RD, Carter RM, Zimny ML, Shah SV. Mechanisms of neutrophil damage to human alveolar extracellular matrix: the role of serine and metalloproteases. J Allergy Clin Immunol. 1992;89(4):905-915.
34. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. 1989;170(3):727-737.
35. Zhou Y, Zhang J, Wu H, Hogan MV, Wang JH. The differential effects of leukocyte-containing and pure platelet-rich plasma (PRP) on tendon stem/progenitor cells - implications of PRP application for the clinical treatment of tendon injuries. Stem Cell Res Ther. 2015;6:173.
36. Su B, O'Connor JP. NSAID therapy effects on healing of bone, tendon, and the enthesis. J Appl Physiol (1985). 2013;115(6):892-899.
ABSTRACT
In this study, we determine the in vivo effects of injecting sub-populations of leukocytes into normal rat Achilles tendons via a controlled laboratory study. Allogenic monocytes, granulocytes, or plasma were injected into 24 healthy rat Achilles tendons. Treated and contralateral un-treated control tendons then assessed for cellularity, histologic morphology, and vascularity after 7 and 14 days. Significant increases of 221% and 249% in cellularity (P = 0.014) were seen on day 14 within Achilles tendons injected with granulocytes as compared to plasma and monocytes, respectively. Also, significant improvement in morphology (P = 0.029) between days 7 and 14 was seen for the granulocyte injected Achilles tendons. Significant increases in cellularity after an injection of granulocytes, compared to monocytes and plasma, corresponds to a significant increase in inflammation within the tissue, suggesting that leukocyte-rich platelet-rich plasma (PRP) preparations are proinflammatory and potentially catabolic when injected into tendon tissue. The concentration and composition of white blood cells within PRP preparations is variable and needs to be better understood in order to optimize clinical utility of PRP injections.
Continue to: Tendinopathies are debilitating conditions...
Tendinopathies are debilitating conditions affecting patients worldwide every day. They arise most frequently from tendon overuse resulting in pathology.1 There are 2 major subtypes of tendinopathy: tendinosis and tendinitis. Tendinosis, the more common condition, is characterized by long-term, chronic degradation of tendon tissue resulting in fibrosis from infiltrating fibroblasts.2 Tendinitis, the less common condition, is characterized by an acute inflammatory response and inflammatory cell infiltrate.2 Both conditions are common, with Achilles tendinopathy affecting 11% of runners and lateral epicondylitis affecting 1% to 3% of the general population.3,4 Many sports-related overuse injuries, such as tendinopathies, go undiagnosed for extended periods of time because medical attention is avoided in order to prevent time loss from training or competing.5 These delays could be eliminated if a non-surgical option for treating tendon pathology was available.
Tendinopathies are believed to result from tendon overuse that causes micro-damage to collagen, as well as from significant changes in protein and enzyme composition within the tendon.6 The damage accumulates over time and eventually leads to chronic inflammation or fibrotic change within tendons, in both cases weakening the tendon and causing pain. Currently, accepted treatments for tendinopathies include: nonsteroidal anti-inflammatory drugs, physical therapy, ultrasound, laser-therapy, corticosteroids, glyceryl trinitrate patches, extracorporeal shock wave therapy, sclerotherapy, and surgery.7 Recently, platelet-rich plasma (PRP) therapy has emerged as a promising treatment for tendinopathies, as well as a variety of other orthopedic indications.
PRP consists of autologous blood from the patient, centrifuged to increase the amount of platelets in the sample above baseline, and subsequently injected around an affected tendon or joint.8 PRP is used to treat tendinopathy because it can supply injured tendons with blood components that aid in healing, which tendons do not receive due to poor vascularity.9 These components include growth factors, such as platelet derived growth factor (PDGF), transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), endothelial growth factor, and leukocytes that can stimulate an inflammatory response within the injured tissue.10 The inflammatory response from the PRP induces a more robust reconstruction and revascularization of the injured tissue, stimulating proliferation, and remodeling.11,12However, significant variability exists within the platelets, leukocytes, and growth factors that comprise PRP. This is attributed to 3 major causes. First, current commercial preparations of PRP result in differing platelet concentrations, as well as leukocyte-rich and leukocyte-poor compositions.13,14 Variability in platelet concentrations results in unreliable amounts of growth factors, including cytokines, TGF-β, PDGF, VEGF and basic fibroblast growth factor in each preparation, while leukocyte levels affect inflammation, all leading to variable effects for each preparation.15,16Second, despite sex and age of the PRP donor not being significant factors influencing variation in growth factor concentrations, the existence of an unexplained variation in concentrations of growth factors between different donors has been observed.17 Third, the selection of activating agents, bovine thrombin or calcium chloride, and their application, whether to the elbow, shoulder, or knee, produces variability.18
While the effects of platelets and growth factors in PRP have been well studied, less is known about the effects of differing cell types. Recently it was reported that the concentrations of leukocytes directly affect the outcomes of PRP injections. McCarrel and colleagues19,20 found that as the number of leukocytes increased, there was a concomitant increase in the expression of inflammatory cytokines and catabolic activity. This effect may result in inferior healing of injured tissues and is attributed to the release of pro-inflammatory cytokines such as interleukin-1β from the leukocytes.21 There is also evidence that minimizing the catabolic effect of leukocytes may be just as important to tissue healing as the maximizing anabolic effect of platelets and growth factors.22
The use of PRP has been highly disputed in recent years due to conflicting reports of its success in treating orthopedic conditions. Numerous favorable studies have shown benefit for treating chronic and acute orthopedic injuries including; rotator cuff tear repair, chronic refractory patellar tendinopathy, and chronic lateral tendinosis/epicondylitis.23-26 Concurrently, articles demonstrating no significant effects from PRP have also been published. One study claiming that PRP injections did not improve outcomes of chronic Achilles tendinopathy did not differentiate whether patients had tendinosis or tendinitis, and did not consider leukocyte concentration in their PRP preparations27 Another study that determined PRP is not beneficial to the healing of ruptured Achilles tendons after surgical repair also failed to consider the concentration of leukocytes in their PRP preparations.28 One of the difficulties in comparing these studies is their heterogeneous nature. This arises from the use of different conditions in each study that makes the studies incomparable. Variations in PRP preparations lead to different concentrations of growth factors, platelets, and leukocyte concentrations. Additionally, tendinopathy models were not specified as tendinosis and tendonitis, and models or patients were not controlled for age, sex, or comorbidities. Given that leukocyte-rich and leukocyte-poor PRP preparations are currently widely used in clinical practice, the discovery of which type of preparation is indicated in which setting is paramount to evidence-based use of this treatment modality. Due to reports suggesting that leukocytes may be detrimental to tendon healing, determining which types of leukocytes are responsible for these effects is vital. As such, the purpose of this study is to determine the in vivo effects of sub-populations of leukocytes on normal rat tendons. This study design allowed us to isolate the effects of the injections to induce a response and remove confounding effects of normal healing response to a damaged tendon and effects from the injection itself. Our hypothesis was that the injection of leukocytes would cause an inflammatory response in rat tendons, leading to catabolic outcomes.
Continue to: METHODS...
METHODS
This was a prospective, in vivo, placebo controlled, randomized animal study. The University’s Institutional Animal Care and Use Committee approved all procedures prior to initiation. Twenty-four male Sprague-Dawley rats were randomized to 3 treatment groups (n = 8): monocytes; granulocytes, and; plasma, as a negative control.
Allogenic blood from 6 additional rats was collected into K2EDTA tubes via cardiac puncture. Allogenic, as opposed to autogenic, blood is commonly used in rat models because of low immunogenic response to blood from rats of the same strain and litter.29,30 The blood was then pooled and the red cells lysed by incubation with Red Blood Cell Lysis Buffer (Roche). The samples were then sorted into fractions containing monocytes and granulocytes using fluorescence activated cell sorting (FACS) using a FACSAria (BD Biosciences). Cells were sorted using Purified PE Mouse Anti-Rat CD11b/c antibodies (BD Pharmingen) specific to monocytes, granulocytes, macrophages, dendritic cells, and microglia, APC-Cy7 Mouse Anti-Rat CD45 antibodies (BD Pharmingen) specific to all hematopoietic cells except erythrocytes, and FITC Mouse Anti-Rat CD42d antibodies (BD Pharmingen) specific to megakaryocytes and platelets. 20 μL of 0.2 mg/mL CD11b/c, 20 μL of 0.2 mg/mL CD 45, and 10 μL of 0.5 mg/mL CD42d antibodies were added to 1 mL of condensed non-red cells collected from the 6 rats and incubated at room temperature in the dark for 15 minutes. A fraction containing only platelet-poor plasma was also collected. For all treatments the injection volume was 75 μL. Rats in the monocyte group were injected with 200,000 cells in platelet-poor plasma, those in the granulocyte group were injected with 900,000 cells in platelet-poor plasma, and rats in the plasma control group received only platelet-poor plasma. The cell concentrations were based on previous studies that documented these concentrations that are found in typical leukocyte-rich PRP preparations.13
The animals were anesthetized with isoflurane gas and then injected aseptically once into their right Achilles tendon. The left Achilles tendon was used as an un-injected control, giving a total of 48 total Achilles tendons studied. At days 7 and 14 post-injection, 4 rats from each group were euthanized and the Achilles tendons were harvested.
The tendons were fixed in neutral buffered formalin for 24 hours and then embedded in paraffin and sectioned sagittally at 12 μm. The tendons were then stained with hematoxylin and eosin (H&E) using standard histological protocols and examined by 3 individuals trained to assess cellularity and morphology. All samples were assigned unrecognizable numbers and randomized prior to examination by individuals. Cell counts were based on the number of nuclei present in 3 mid-tendon high-power fields (400x) per sample. Morphology was graded on a scale of 1 to 3, with 1 being a normal tendon and 3 having severe pathology with total loss of alignment and crimping on 3 low-power fields (100x) per sample (Figures 1A-1G).
Vascularity was assessed by immunohistochemical staining using Rabbit Polyclonal Anti-CD31 antibodies (Abcam), a marker for vascular endothelial cells, using a Vectastain ABC Kit (Vector Laboratories) system and the ImmPACT AEC Peroxidase (HRP) Substrate (Vector Laboratories). Following staining, automated image analysis was performed (Bioquant). Briefly, all areas that did not contain tendon were masked. CD31 positive areas were then quantified using global thresholding. Vascularity was then calculated as ratio of CD31 positive area to total tendon area. Analyses were performed on 3 mid-tendon medium-power (200x) fields per sample.
For cellularity and morphology, the results for the injected tendons were normalized to those of their contralateral untreated controls and reported as a percentage. Results for vascularity were compared directly between treated tendons. Differences were assessed between groups at each time-point using Independent Samples Median Tests. When significant differences were identified, pairwise comparisons were performed to identify the source of the differences. All analyses were conducted using SPSS (V22, SAS Institute) with significant differences determined for values of P < 0.05.
RESULTS
No significant differences in cellularity between groups were seen at day 7 (P = 0.368) (Figures 1A-1G). However, a significant difference in cellularity between groups was seen at day 14 (P = 0.014). Pairwise tests showed there to be a significant increase in the number of cells in the tendons treated with granulocytes from 221% and 249% in cellularity (P = 0.014) on day 14, as compared to both monocytes and plasma, respectively. Morphologically, no significant differences were seen between groups at either time-point (P = 0.091 for day 7 and P = 1.000 for day 14) (Figures 2A-2G). However, a significant improvement in morphology was observed from day 7 to day 14 in the granulocyte group from 60% to 165% (P = 0.029). Finally, no differences were seen in vascularity between treatment groups at either time-point (P = 0.368 for day 7 and P = 0.535 for day 14) (Figures 3A-3G).
Continue to: DISCUSSION...
DISCUSSION
Our hypothesis that the injection of leukocytes would cause an inflammatory response in rat tendons leading to catabolic outcomes was confirmed in the granulocyte group. It should be noted that prior to the catabolic outcome, there was a transient anabolic effect in the granulocyte group during the second week. Deterioration in morphology was observed in the tendons injected with granulocytes on day 7, which subsequently recovered in the following week. We found that injecting granulocytes into normal tendons resulted in an increase in inflammatory cellularity, when compared to monocytes and plasma injections.
Limitations inherent in this study are those similar to other in vivo studies. To begin with, the results of injections into rat tendons may not be translatable to human tendons. Despite this limitation, the rat is a common model for tendon research.31 Another limitation is that this study injected healthy Achilles tendons, rather than tendons with preexisting tendinopathy. In a naturally occurring tendinopathy, there may be other factors present that interact with PRP, and this model negates the contribution of these factors. Finally, while the immunohistochemistry (IHC) and morphological data are clear, the cellularity data are not clear in identifying the type of cells that were increased by granulocyte injection. However, the cells appeared rounded, resembling inflammatory infiltrate; a common cell type seen in tendons.2 While fibroblasts are also a common infiltrate during chronic tendinopathy, they are generally flat and appear on H&E as long spindle shaped cells. Thus, we believe the increased cellularity of the tendons after granulocyte injections is representative of an increase in inflammation. The increased cellularity could be due to the increased number of cells injected into the tendon; however, our conclusions are consistent with the increased inflammation previously reported linking leukocytes to tendon inflammation.20,22,32
In terms of morphology, we hypothesized that degenerative changes would be seen in the tendons that were injected with granulocytes due to the inflammatory action of these cells. As part of the granulocyte response, neutrophils release proteases and macrophages can stimulate collagen synthesis via fibroblasts, both causing change within the extracellular matrix.33,34 Indeed, we observed a significant change in tissue morphology in the granulocyte group over the course of 14 days. As the degenerative and regenerative effects of granulocytes take time to present, this is likely what we observed to occur between day 7 and 14 after treatment. These observations are also consistent with prior observations that leukocyte-rich PRP injections can be detrimental to tendon healing, but beneficial to tissue degeneration in the setting of chronic tendonitis.20
We hypothesized that the vascularity of the tendons would be similar in all preparations. This was based on previous studies demonstrating that the lack of platelets in the platelet-poor plasma fraction is sufficient to deplete VEGF, the angiogenic agent in PRP.35 In this study, there were no observable differences in vascularity of platelet-poor plasma, monocyte, and granulocyte injections. We attribute this to the lack of VEGF in any of these preparations. The aforementioned study also showed that the lack of platelets in injection was enough to prevent the angiogenic effect of this treatment.35
Continue to: The goal of this study was...
The goal of this study was to assess the morphology, cellularity, and vascularity of normal tendons after injections of different leukocyte populations. This is clinically important because of the potential to tailor future PRP injections on a patient-by-patient basis. In patients requiring an anabolic response, leukocyte-poor PRP may be the best option. In contrast, when patient pathology requires an inflammatory response to improve healing36 or breakdown fibrotic tissue, as seen in tendinosis, leukocyte-rich PRP may be warranted. Further, properly controlled clinical studies are needed to validate these recommendations.
Limitations inherent in this study are those similar to other in vivo studies. First, the results of injections into rat tendons may not be translatable to human tendons. Despite this limitation, the rat is a common model for tendon research.31 A second limitation is that this study injected healthy Achilles tendons, rather than tendons with preexisting tendinopathy. In a naturally occurring tendinopathy, there may be other factors present that interact with PRP, and this model negates the contribution of these factors. Finally, while the IHC and morphological data show clear changes, the cellularity data are not clear in identifying the type of cells that were increased by granulocyte injection. However, the cells appeared rounded, resembling inflammatory infiltrate; a common cell type seen in tendons.2 While fibroblasts are also a common infiltrate during chronic tendinopathy, they are generally flat and appear on H&E as long spindle shaped cells. The last limitation of this study is the lack of functional mechanical testing since, clinically, healing of the tendon is also related to the strength of the tendon. Thus, we believe the increased cellularity of the tendons after granulocyte injections is representative of an increase in inflammation. Moreover, our results are consistent with the increased inflammation previously reported linking leukocytes to tendon inflammation.20,22,32 It is interesting to note that the increase in inflammation does not lead to an increase in vascularity as could be expected.
CONCLUSION
We found that the injection of leukocytes into healthy rat Achilles tendons increases inflammation, as evidenced by increased cellularity and disrupted morphology, which suggests that leukocyte-rich PRP preparations may be contraindicated in settings of acute tendonitis. However, these preparations may be useful for a specific subset of tendinopathies, including chronic tendinosis.
ABSTRACT
In this study, we determine the in vivo effects of injecting sub-populations of leukocytes into normal rat Achilles tendons via a controlled laboratory study. Allogenic monocytes, granulocytes, or plasma were injected into 24 healthy rat Achilles tendons. Treated and contralateral un-treated control tendons then assessed for cellularity, histologic morphology, and vascularity after 7 and 14 days. Significant increases of 221% and 249% in cellularity (P = 0.014) were seen on day 14 within Achilles tendons injected with granulocytes as compared to plasma and monocytes, respectively. Also, significant improvement in morphology (P = 0.029) between days 7 and 14 was seen for the granulocyte injected Achilles tendons. Significant increases in cellularity after an injection of granulocytes, compared to monocytes and plasma, corresponds to a significant increase in inflammation within the tissue, suggesting that leukocyte-rich platelet-rich plasma (PRP) preparations are proinflammatory and potentially catabolic when injected into tendon tissue. The concentration and composition of white blood cells within PRP preparations is variable and needs to be better understood in order to optimize clinical utility of PRP injections.
Continue to: Tendinopathies are debilitating conditions...
Tendinopathies are debilitating conditions affecting patients worldwide every day. They arise most frequently from tendon overuse resulting in pathology.1 There are 2 major subtypes of tendinopathy: tendinosis and tendinitis. Tendinosis, the more common condition, is characterized by long-term, chronic degradation of tendon tissue resulting in fibrosis from infiltrating fibroblasts.2 Tendinitis, the less common condition, is characterized by an acute inflammatory response and inflammatory cell infiltrate.2 Both conditions are common, with Achilles tendinopathy affecting 11% of runners and lateral epicondylitis affecting 1% to 3% of the general population.3,4 Many sports-related overuse injuries, such as tendinopathies, go undiagnosed for extended periods of time because medical attention is avoided in order to prevent time loss from training or competing.5 These delays could be eliminated if a non-surgical option for treating tendon pathology was available.
Tendinopathies are believed to result from tendon overuse that causes micro-damage to collagen, as well as from significant changes in protein and enzyme composition within the tendon.6 The damage accumulates over time and eventually leads to chronic inflammation or fibrotic change within tendons, in both cases weakening the tendon and causing pain. Currently, accepted treatments for tendinopathies include: nonsteroidal anti-inflammatory drugs, physical therapy, ultrasound, laser-therapy, corticosteroids, glyceryl trinitrate patches, extracorporeal shock wave therapy, sclerotherapy, and surgery.7 Recently, platelet-rich plasma (PRP) therapy has emerged as a promising treatment for tendinopathies, as well as a variety of other orthopedic indications.
PRP consists of autologous blood from the patient, centrifuged to increase the amount of platelets in the sample above baseline, and subsequently injected around an affected tendon or joint.8 PRP is used to treat tendinopathy because it can supply injured tendons with blood components that aid in healing, which tendons do not receive due to poor vascularity.9 These components include growth factors, such as platelet derived growth factor (PDGF), transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), endothelial growth factor, and leukocytes that can stimulate an inflammatory response within the injured tissue.10 The inflammatory response from the PRP induces a more robust reconstruction and revascularization of the injured tissue, stimulating proliferation, and remodeling.11,12However, significant variability exists within the platelets, leukocytes, and growth factors that comprise PRP. This is attributed to 3 major causes. First, current commercial preparations of PRP result in differing platelet concentrations, as well as leukocyte-rich and leukocyte-poor compositions.13,14 Variability in platelet concentrations results in unreliable amounts of growth factors, including cytokines, TGF-β, PDGF, VEGF and basic fibroblast growth factor in each preparation, while leukocyte levels affect inflammation, all leading to variable effects for each preparation.15,16Second, despite sex and age of the PRP donor not being significant factors influencing variation in growth factor concentrations, the existence of an unexplained variation in concentrations of growth factors between different donors has been observed.17 Third, the selection of activating agents, bovine thrombin or calcium chloride, and their application, whether to the elbow, shoulder, or knee, produces variability.18
While the effects of platelets and growth factors in PRP have been well studied, less is known about the effects of differing cell types. Recently it was reported that the concentrations of leukocytes directly affect the outcomes of PRP injections. McCarrel and colleagues19,20 found that as the number of leukocytes increased, there was a concomitant increase in the expression of inflammatory cytokines and catabolic activity. This effect may result in inferior healing of injured tissues and is attributed to the release of pro-inflammatory cytokines such as interleukin-1β from the leukocytes.21 There is also evidence that minimizing the catabolic effect of leukocytes may be just as important to tissue healing as the maximizing anabolic effect of platelets and growth factors.22
The use of PRP has been highly disputed in recent years due to conflicting reports of its success in treating orthopedic conditions. Numerous favorable studies have shown benefit for treating chronic and acute orthopedic injuries including; rotator cuff tear repair, chronic refractory patellar tendinopathy, and chronic lateral tendinosis/epicondylitis.23-26 Concurrently, articles demonstrating no significant effects from PRP have also been published. One study claiming that PRP injections did not improve outcomes of chronic Achilles tendinopathy did not differentiate whether patients had tendinosis or tendinitis, and did not consider leukocyte concentration in their PRP preparations27 Another study that determined PRP is not beneficial to the healing of ruptured Achilles tendons after surgical repair also failed to consider the concentration of leukocytes in their PRP preparations.28 One of the difficulties in comparing these studies is their heterogeneous nature. This arises from the use of different conditions in each study that makes the studies incomparable. Variations in PRP preparations lead to different concentrations of growth factors, platelets, and leukocyte concentrations. Additionally, tendinopathy models were not specified as tendinosis and tendonitis, and models or patients were not controlled for age, sex, or comorbidities. Given that leukocyte-rich and leukocyte-poor PRP preparations are currently widely used in clinical practice, the discovery of which type of preparation is indicated in which setting is paramount to evidence-based use of this treatment modality. Due to reports suggesting that leukocytes may be detrimental to tendon healing, determining which types of leukocytes are responsible for these effects is vital. As such, the purpose of this study is to determine the in vivo effects of sub-populations of leukocytes on normal rat tendons. This study design allowed us to isolate the effects of the injections to induce a response and remove confounding effects of normal healing response to a damaged tendon and effects from the injection itself. Our hypothesis was that the injection of leukocytes would cause an inflammatory response in rat tendons, leading to catabolic outcomes.
Continue to: METHODS...
METHODS
This was a prospective, in vivo, placebo controlled, randomized animal study. The University’s Institutional Animal Care and Use Committee approved all procedures prior to initiation. Twenty-four male Sprague-Dawley rats were randomized to 3 treatment groups (n = 8): monocytes; granulocytes, and; plasma, as a negative control.
Allogenic blood from 6 additional rats was collected into K2EDTA tubes via cardiac puncture. Allogenic, as opposed to autogenic, blood is commonly used in rat models because of low immunogenic response to blood from rats of the same strain and litter.29,30 The blood was then pooled and the red cells lysed by incubation with Red Blood Cell Lysis Buffer (Roche). The samples were then sorted into fractions containing monocytes and granulocytes using fluorescence activated cell sorting (FACS) using a FACSAria (BD Biosciences). Cells were sorted using Purified PE Mouse Anti-Rat CD11b/c antibodies (BD Pharmingen) specific to monocytes, granulocytes, macrophages, dendritic cells, and microglia, APC-Cy7 Mouse Anti-Rat CD45 antibodies (BD Pharmingen) specific to all hematopoietic cells except erythrocytes, and FITC Mouse Anti-Rat CD42d antibodies (BD Pharmingen) specific to megakaryocytes and platelets. 20 μL of 0.2 mg/mL CD11b/c, 20 μL of 0.2 mg/mL CD 45, and 10 μL of 0.5 mg/mL CD42d antibodies were added to 1 mL of condensed non-red cells collected from the 6 rats and incubated at room temperature in the dark for 15 minutes. A fraction containing only platelet-poor plasma was also collected. For all treatments the injection volume was 75 μL. Rats in the monocyte group were injected with 200,000 cells in platelet-poor plasma, those in the granulocyte group were injected with 900,000 cells in platelet-poor plasma, and rats in the plasma control group received only platelet-poor plasma. The cell concentrations were based on previous studies that documented these concentrations that are found in typical leukocyte-rich PRP preparations.13
The animals were anesthetized with isoflurane gas and then injected aseptically once into their right Achilles tendon. The left Achilles tendon was used as an un-injected control, giving a total of 48 total Achilles tendons studied. At days 7 and 14 post-injection, 4 rats from each group were euthanized and the Achilles tendons were harvested.
The tendons were fixed in neutral buffered formalin for 24 hours and then embedded in paraffin and sectioned sagittally at 12 μm. The tendons were then stained with hematoxylin and eosin (H&E) using standard histological protocols and examined by 3 individuals trained to assess cellularity and morphology. All samples were assigned unrecognizable numbers and randomized prior to examination by individuals. Cell counts were based on the number of nuclei present in 3 mid-tendon high-power fields (400x) per sample. Morphology was graded on a scale of 1 to 3, with 1 being a normal tendon and 3 having severe pathology with total loss of alignment and crimping on 3 low-power fields (100x) per sample (Figures 1A-1G).
Vascularity was assessed by immunohistochemical staining using Rabbit Polyclonal Anti-CD31 antibodies (Abcam), a marker for vascular endothelial cells, using a Vectastain ABC Kit (Vector Laboratories) system and the ImmPACT AEC Peroxidase (HRP) Substrate (Vector Laboratories). Following staining, automated image analysis was performed (Bioquant). Briefly, all areas that did not contain tendon were masked. CD31 positive areas were then quantified using global thresholding. Vascularity was then calculated as ratio of CD31 positive area to total tendon area. Analyses were performed on 3 mid-tendon medium-power (200x) fields per sample.
For cellularity and morphology, the results for the injected tendons were normalized to those of their contralateral untreated controls and reported as a percentage. Results for vascularity were compared directly between treated tendons. Differences were assessed between groups at each time-point using Independent Samples Median Tests. When significant differences were identified, pairwise comparisons were performed to identify the source of the differences. All analyses were conducted using SPSS (V22, SAS Institute) with significant differences determined for values of P < 0.05.
RESULTS
No significant differences in cellularity between groups were seen at day 7 (P = 0.368) (Figures 1A-1G). However, a significant difference in cellularity between groups was seen at day 14 (P = 0.014). Pairwise tests showed there to be a significant increase in the number of cells in the tendons treated with granulocytes from 221% and 249% in cellularity (P = 0.014) on day 14, as compared to both monocytes and plasma, respectively. Morphologically, no significant differences were seen between groups at either time-point (P = 0.091 for day 7 and P = 1.000 for day 14) (Figures 2A-2G). However, a significant improvement in morphology was observed from day 7 to day 14 in the granulocyte group from 60% to 165% (P = 0.029). Finally, no differences were seen in vascularity between treatment groups at either time-point (P = 0.368 for day 7 and P = 0.535 for day 14) (Figures 3A-3G).
Continue to: DISCUSSION...
DISCUSSION
Our hypothesis that the injection of leukocytes would cause an inflammatory response in rat tendons leading to catabolic outcomes was confirmed in the granulocyte group. It should be noted that prior to the catabolic outcome, there was a transient anabolic effect in the granulocyte group during the second week. Deterioration in morphology was observed in the tendons injected with granulocytes on day 7, which subsequently recovered in the following week. We found that injecting granulocytes into normal tendons resulted in an increase in inflammatory cellularity, when compared to monocytes and plasma injections.
Limitations inherent in this study are those similar to other in vivo studies. To begin with, the results of injections into rat tendons may not be translatable to human tendons. Despite this limitation, the rat is a common model for tendon research.31 Another limitation is that this study injected healthy Achilles tendons, rather than tendons with preexisting tendinopathy. In a naturally occurring tendinopathy, there may be other factors present that interact with PRP, and this model negates the contribution of these factors. Finally, while the immunohistochemistry (IHC) and morphological data are clear, the cellularity data are not clear in identifying the type of cells that were increased by granulocyte injection. However, the cells appeared rounded, resembling inflammatory infiltrate; a common cell type seen in tendons.2 While fibroblasts are also a common infiltrate during chronic tendinopathy, they are generally flat and appear on H&E as long spindle shaped cells. Thus, we believe the increased cellularity of the tendons after granulocyte injections is representative of an increase in inflammation. The increased cellularity could be due to the increased number of cells injected into the tendon; however, our conclusions are consistent with the increased inflammation previously reported linking leukocytes to tendon inflammation.20,22,32
In terms of morphology, we hypothesized that degenerative changes would be seen in the tendons that were injected with granulocytes due to the inflammatory action of these cells. As part of the granulocyte response, neutrophils release proteases and macrophages can stimulate collagen synthesis via fibroblasts, both causing change within the extracellular matrix.33,34 Indeed, we observed a significant change in tissue morphology in the granulocyte group over the course of 14 days. As the degenerative and regenerative effects of granulocytes take time to present, this is likely what we observed to occur between day 7 and 14 after treatment. These observations are also consistent with prior observations that leukocyte-rich PRP injections can be detrimental to tendon healing, but beneficial to tissue degeneration in the setting of chronic tendonitis.20
We hypothesized that the vascularity of the tendons would be similar in all preparations. This was based on previous studies demonstrating that the lack of platelets in the platelet-poor plasma fraction is sufficient to deplete VEGF, the angiogenic agent in PRP.35 In this study, there were no observable differences in vascularity of platelet-poor plasma, monocyte, and granulocyte injections. We attribute this to the lack of VEGF in any of these preparations. The aforementioned study also showed that the lack of platelets in injection was enough to prevent the angiogenic effect of this treatment.35
Continue to: The goal of this study was...
The goal of this study was to assess the morphology, cellularity, and vascularity of normal tendons after injections of different leukocyte populations. This is clinically important because of the potential to tailor future PRP injections on a patient-by-patient basis. In patients requiring an anabolic response, leukocyte-poor PRP may be the best option. In contrast, when patient pathology requires an inflammatory response to improve healing36 or breakdown fibrotic tissue, as seen in tendinosis, leukocyte-rich PRP may be warranted. Further, properly controlled clinical studies are needed to validate these recommendations.
Limitations inherent in this study are those similar to other in vivo studies. First, the results of injections into rat tendons may not be translatable to human tendons. Despite this limitation, the rat is a common model for tendon research.31 A second limitation is that this study injected healthy Achilles tendons, rather than tendons with preexisting tendinopathy. In a naturally occurring tendinopathy, there may be other factors present that interact with PRP, and this model negates the contribution of these factors. Finally, while the IHC and morphological data show clear changes, the cellularity data are not clear in identifying the type of cells that were increased by granulocyte injection. However, the cells appeared rounded, resembling inflammatory infiltrate; a common cell type seen in tendons.2 While fibroblasts are also a common infiltrate during chronic tendinopathy, they are generally flat and appear on H&E as long spindle shaped cells. The last limitation of this study is the lack of functional mechanical testing since, clinically, healing of the tendon is also related to the strength of the tendon. Thus, we believe the increased cellularity of the tendons after granulocyte injections is representative of an increase in inflammation. Moreover, our results are consistent with the increased inflammation previously reported linking leukocytes to tendon inflammation.20,22,32 It is interesting to note that the increase in inflammation does not lead to an increase in vascularity as could be expected.
CONCLUSION
We found that the injection of leukocytes into healthy rat Achilles tendons increases inflammation, as evidenced by increased cellularity and disrupted morphology, which suggests that leukocyte-rich PRP preparations may be contraindicated in settings of acute tendonitis. However, these preparations may be useful for a specific subset of tendinopathies, including chronic tendinosis.
1. Herring SA, Nilson KL. Introduction to overuse injuries. Clin Sports Med. 1987;6(2):225-239.
2. Bass E. Tendinopathy: why the difference between tendinitis and tendinosis matters. Int J Ther Massage Bodywork. 2012;5(1):14-17.
3. James SL, Bates BT, Osternig LR. Injuries to runners. Am J Sports Med. 1978;6(2):40-50.
4. Allander E. Prevalence, incidence, and remission rates of some common rheumatic diseases or syndromes. Scand J Rheumatol. 1974;3(3):145-153.
5. Bahr R. No injuries, but plenty of pain? On the methodology for recording overuse symptoms in sports. Br J Sports Med. 2009;43(13):966-972.
6. Rees JD, Maffulli N, Cook J. Management of tendinopathy. Am J Sports Med. 2009;37(9):1855-1867.
7. Andres BM, Murrell GA. Treatment of tendinopathy: what works, what does not, and what is on the horizon. Clin Orthop Relat Res. 2008;466(7):1539-1554.
8. Hall MP, Band PA, Meislin RJ, Jazrawi LM, Cardone DA. Platelet-rich plasma: current concepts and application in sports medicine. J Am Acad Orthop Surg. 2009;17(10):602-608.
9. Smith JW. Blood Supply of Tendons. Am J Surg. 1965;109:272-276.
10. Wu PI, Diaz R, Borg-Stein J. Platelet-rich plasma. Phys Med Rehabil Clin N Am. 2016;27(4):825-853.
11. Nguyen RT, Borg-Stein J, McInnis K. Applications of platelet-rich plasma in musculoskeletal and sports medicine: an evidence-based approach. PM R. 2011;3(3):226-250.
12. Broughton G 2nd, Janis JE, Attinger CE. Wound healing: an overview. Plast Reconstr Surg. 2006;117(7 Suppl):1e-S-32e-S.
13. Mazzocca AD, McCarthy MB, Chowaniec DM, et al. Platelet-rich plasma differs according to preparation method and human variability. J Bone Joint Surg Am. 2012;94(4):308-316.
14. Mazzocca AD, McCarthy MB, Chowaniec DM, et al. The positive effects of different platelet-rich plasma methods on human muscle, bone, and tendon cells. Am J Sports Med. 2012;40(8):1742-1749.
15. Castillo TN, Pouliot MA, Kim HJ, Dragoo JL. Comparison of growth factor and platelet concentration from commercial platelet-rich plasma separation systems. Am J Sports Med. 2011;39(2):266-271.
16. Cho HS, Song IH, Park SY, Sung MC, Ahn MW, Song KE. Individual variation in growth factor concentrations in platelet-rich plasma and its influence on human mesenchymal stem cells. Korean J Lab Med. 2011;31(3):212-218.
17. Weibrich G, Kleis WK, Hafner G, Hitzler WE. Growth factor levels in platelet-rich plasma and correlations with donor age, sex, and platelet count. J Craniomaxillofac Surg. 2002;30(2):97-102.
18. Taylor DW, Petrera M, Hendry M, Theodoropoulos JS. A systematic review of the use of platelet-rich plasma in sports medicine as a new treatment for tendon and ligament injuries. Clin J Sport Med. 2011;21(4):344-352.
19. McCarrel T, Fortier L. Temporal growth factor release from platelet-rich plasma, trehalose lyophilized platelets, and bone marrow aspirate and their effect on tendon and ligament gene expression. J Orthop Res. 2009;27(8):1033-1042.
20. McCarrel TM, Minas T, Fortier LA. Optimization of leukocyte concentration in platelet-rich plasma for the treatment of tendinopathy. J Bone Joint Surg Am. 2012;94(19):e143(141-148).
21. Pillitteri D, Bassus S, Boller K, et al. Thrombin-induced interleukin 1beta synthesis in platelet suspensions: impact of contaminating leukocytes. Platelets. 2007;18(2):119-127.
22. Boswell SG, Schnabel LV, Mohammed HO, Sundman EA, Minas T, Fortier LA. Increasing platelet concentrations in leukocyte-reduced platelet-rich plasma decrease collagen gene synthesis in tendons. Am J Sports Med. 2014;42(1):42-49.
23. Mishra A, Pavelko T. Treatment of chronic elbow tendinosis with buffered platelet-rich plasma. Am J Sports Med. 2006;34(11):1774-1778.
24. Maniscalco P, Gambera D, Lunati A, et al. The "Cascade" membrane: a new PRP device for tendon ruptures. Description and case report on rotator cuff tendon. Acta Biomed. 2008;79(3):223-226.
25. Filardo G, Kon E, Della Villa S, Vincentelli F, Fornasari PM, Marcacci M. Use of platelet-rich plasma for the treatment of refractory jumper's knee. Int Orthop. 2010;34(6):909-915.
26. Peerbooms JC, Sluimer J, Bruijn DJ, Gosens T. Positive effect of an autologous platelet concentrate in lateral epicondylitis in a double-blind randomized controlled trial: platelet-rich plasma versus corticosteroid injection with a 1-year follow-up. Am J Sports Med. 2010;38(2):255-262.
27. de Vos RJ, Weir A, van Schie HT, et al. Platelet-rich plasma injection for chronic Achilles tendinopathy: a randomized controlled trial. JAMA. 2010;303(2):144-149.
28. Schepull T, Kvist J, Norrman H, Trinks M, Berlin G, Aspenberg P. Autologous platelets have no effect on the healing of human achilles tendon ruptures: a randomized single-blind study. Am J Sports Med. 2011;39(1):38-47.
29. Welsh KI, Burgos H, Batchelor JR. The immune response to allogeneic rat platelets; Ag-B antigens in matrix form lacking Ia. Eur J Immunol. 1977;7(5):267-272.
30. Xue M, Del Bigio MR. Intracortical hemorrhage injury in rats : relationship between blood fractions and brain cell death. Stroke. 2000;31(7):1721-1727.
31. Voleti PB, Buckley MR, Soslowsky LJ. Tendon healing: repair and regeneration. Annu Rev Biomed Eng. 2012;14:47-71.
32. Sundman EA, Cole BJ, Fortier LA. Growth factor and catabolic cytokine concentrations are influenced by the cellular composition of platelet-rich plasma. Am J Sports Med. 2011;39(10):2135-2140.
33. Palmgren MS, deShazo RD, Carter RM, Zimny ML, Shah SV. Mechanisms of neutrophil damage to human alveolar extracellular matrix: the role of serine and metalloproteases. J Allergy Clin Immunol. 1992;89(4):905-915.
34. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. 1989;170(3):727-737.
35. Zhou Y, Zhang J, Wu H, Hogan MV, Wang JH. The differential effects of leukocyte-containing and pure platelet-rich plasma (PRP) on tendon stem/progenitor cells - implications of PRP application for the clinical treatment of tendon injuries. Stem Cell Res Ther. 2015;6:173.
36. Su B, O'Connor JP. NSAID therapy effects on healing of bone, tendon, and the enthesis. J Appl Physiol (1985). 2013;115(6):892-899.
1. Herring SA, Nilson KL. Introduction to overuse injuries. Clin Sports Med. 1987;6(2):225-239.
2. Bass E. Tendinopathy: why the difference between tendinitis and tendinosis matters. Int J Ther Massage Bodywork. 2012;5(1):14-17.
3. James SL, Bates BT, Osternig LR. Injuries to runners. Am J Sports Med. 1978;6(2):40-50.
4. Allander E. Prevalence, incidence, and remission rates of some common rheumatic diseases or syndromes. Scand J Rheumatol. 1974;3(3):145-153.
5. Bahr R. No injuries, but plenty of pain? On the methodology for recording overuse symptoms in sports. Br J Sports Med. 2009;43(13):966-972.
6. Rees JD, Maffulli N, Cook J. Management of tendinopathy. Am J Sports Med. 2009;37(9):1855-1867.
7. Andres BM, Murrell GA. Treatment of tendinopathy: what works, what does not, and what is on the horizon. Clin Orthop Relat Res. 2008;466(7):1539-1554.
8. Hall MP, Band PA, Meislin RJ, Jazrawi LM, Cardone DA. Platelet-rich plasma: current concepts and application in sports medicine. J Am Acad Orthop Surg. 2009;17(10):602-608.
9. Smith JW. Blood Supply of Tendons. Am J Surg. 1965;109:272-276.
10. Wu PI, Diaz R, Borg-Stein J. Platelet-rich plasma. Phys Med Rehabil Clin N Am. 2016;27(4):825-853.
11. Nguyen RT, Borg-Stein J, McInnis K. Applications of platelet-rich plasma in musculoskeletal and sports medicine: an evidence-based approach. PM R. 2011;3(3):226-250.
12. Broughton G 2nd, Janis JE, Attinger CE. Wound healing: an overview. Plast Reconstr Surg. 2006;117(7 Suppl):1e-S-32e-S.
13. Mazzocca AD, McCarthy MB, Chowaniec DM, et al. Platelet-rich plasma differs according to preparation method and human variability. J Bone Joint Surg Am. 2012;94(4):308-316.
14. Mazzocca AD, McCarthy MB, Chowaniec DM, et al. The positive effects of different platelet-rich plasma methods on human muscle, bone, and tendon cells. Am J Sports Med. 2012;40(8):1742-1749.
15. Castillo TN, Pouliot MA, Kim HJ, Dragoo JL. Comparison of growth factor and platelet concentration from commercial platelet-rich plasma separation systems. Am J Sports Med. 2011;39(2):266-271.
16. Cho HS, Song IH, Park SY, Sung MC, Ahn MW, Song KE. Individual variation in growth factor concentrations in platelet-rich plasma and its influence on human mesenchymal stem cells. Korean J Lab Med. 2011;31(3):212-218.
17. Weibrich G, Kleis WK, Hafner G, Hitzler WE. Growth factor levels in platelet-rich plasma and correlations with donor age, sex, and platelet count. J Craniomaxillofac Surg. 2002;30(2):97-102.
18. Taylor DW, Petrera M, Hendry M, Theodoropoulos JS. A systematic review of the use of platelet-rich plasma in sports medicine as a new treatment for tendon and ligament injuries. Clin J Sport Med. 2011;21(4):344-352.
19. McCarrel T, Fortier L. Temporal growth factor release from platelet-rich plasma, trehalose lyophilized platelets, and bone marrow aspirate and their effect on tendon and ligament gene expression. J Orthop Res. 2009;27(8):1033-1042.
20. McCarrel TM, Minas T, Fortier LA. Optimization of leukocyte concentration in platelet-rich plasma for the treatment of tendinopathy. J Bone Joint Surg Am. 2012;94(19):e143(141-148).
21. Pillitteri D, Bassus S, Boller K, et al. Thrombin-induced interleukin 1beta synthesis in platelet suspensions: impact of contaminating leukocytes. Platelets. 2007;18(2):119-127.
22. Boswell SG, Schnabel LV, Mohammed HO, Sundman EA, Minas T, Fortier LA. Increasing platelet concentrations in leukocyte-reduced platelet-rich plasma decrease collagen gene synthesis in tendons. Am J Sports Med. 2014;42(1):42-49.
23. Mishra A, Pavelko T. Treatment of chronic elbow tendinosis with buffered platelet-rich plasma. Am J Sports Med. 2006;34(11):1774-1778.
24. Maniscalco P, Gambera D, Lunati A, et al. The "Cascade" membrane: a new PRP device for tendon ruptures. Description and case report on rotator cuff tendon. Acta Biomed. 2008;79(3):223-226.
25. Filardo G, Kon E, Della Villa S, Vincentelli F, Fornasari PM, Marcacci M. Use of platelet-rich plasma for the treatment of refractory jumper's knee. Int Orthop. 2010;34(6):909-915.
26. Peerbooms JC, Sluimer J, Bruijn DJ, Gosens T. Positive effect of an autologous platelet concentrate in lateral epicondylitis in a double-blind randomized controlled trial: platelet-rich plasma versus corticosteroid injection with a 1-year follow-up. Am J Sports Med. 2010;38(2):255-262.
27. de Vos RJ, Weir A, van Schie HT, et al. Platelet-rich plasma injection for chronic Achilles tendinopathy: a randomized controlled trial. JAMA. 2010;303(2):144-149.
28. Schepull T, Kvist J, Norrman H, Trinks M, Berlin G, Aspenberg P. Autologous platelets have no effect on the healing of human achilles tendon ruptures: a randomized single-blind study. Am J Sports Med. 2011;39(1):38-47.
29. Welsh KI, Burgos H, Batchelor JR. The immune response to allogeneic rat platelets; Ag-B antigens in matrix form lacking Ia. Eur J Immunol. 1977;7(5):267-272.
30. Xue M, Del Bigio MR. Intracortical hemorrhage injury in rats : relationship between blood fractions and brain cell death. Stroke. 2000;31(7):1721-1727.
31. Voleti PB, Buckley MR, Soslowsky LJ. Tendon healing: repair and regeneration. Annu Rev Biomed Eng. 2012;14:47-71.
32. Sundman EA, Cole BJ, Fortier LA. Growth factor and catabolic cytokine concentrations are influenced by the cellular composition of platelet-rich plasma. Am J Sports Med. 2011;39(10):2135-2140.
33. Palmgren MS, deShazo RD, Carter RM, Zimny ML, Shah SV. Mechanisms of neutrophil damage to human alveolar extracellular matrix: the role of serine and metalloproteases. J Allergy Clin Immunol. 1992;89(4):905-915.
34. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. 1989;170(3):727-737.
35. Zhou Y, Zhang J, Wu H, Hogan MV, Wang JH. The differential effects of leukocyte-containing and pure platelet-rich plasma (PRP) on tendon stem/progenitor cells - implications of PRP application for the clinical treatment of tendon injuries. Stem Cell Res Ther. 2015;6:173.
36. Su B, O'Connor JP. NSAID therapy effects on healing of bone, tendon, and the enthesis. J Appl Physiol (1985). 2013;115(6):892-899.
TAKE-HOME POINTS
- Injection of leukocytes into healthy rat Achilles tendons increases inflammation.
- Injection of leukocytes into healthy rat Achilles tendons does not affect vascularity.
- Leukocyte-rich PRP preparations may be contraindicated in settings of acute tendonitis.
- Leukocyte-rich PRP preparations may be useful for chronic tendinosis.
- The concentration and composition of white blood cells within PRP preparations is variable and needs to be better understood in order to optimize clinical utility of PRP injections.
Drug-coated balloons shown noninferior to DES in thin coronaries
MUNICH – for preventing the clinical consequences of restenosis during 12 months following coronary intervention, according to results from a prospective, randomized, multicenter trial.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Drug-coated balloons are already used to treat in-stent coronary restenosis. The findings of the current study establish the tested DCB as noninferior to a DES for treating coronary stenoses in narrow arteries less than 3 mm in diameter, Raban V. Jeger, MD, said at the annual congress of the European Society of Cardiology. The DCB approach avoids placing a metal stent in a narrow coronary and thus has no long-term risk for in-stent thrombosis, said Dr. Jeger, a professor of cardiology at Basel (Switzerland) University Hospital. Dr. Jeger acknowledged that the tested DCB is more expensive than the second-generation DES used as the comparator in most of the control patients, “but I think the benefit to patients is worth” the added cost, he said when discussing his report.
The BASKET-SMALL 2 (NCT01574534) study enrolled 758 patients at 14 centers in Switzerland, Germany, and Austria. The trial limited enrollment to patients who were scheduled to undergo percutaneous coronary intervention for stenosis in a coronary artery that was at least 2.0 mm and less than 3.0 mm in diameter and had first undergone successful predilatation without any flow-limiting dissections or residual stenosis, a step in the DCB procedure that adds to the procedure’s cost.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The study randomized patients to treatment with either a balloon coated with paclitaxel/iopromide (SeQuent Please) or a DES. The first quarter of patients randomized into the DES arm received a first-generation, paclitaxel-eluting DES (Taxus Element); the remaining patients in the comparator arm received a second-generation everolimus-eluting DES (Xience). The DCB tested is not approved for U.S. marketing.
The primary endpoint was the combined rate of cardiac death, nonfatal MI, or target vessel revascularization during 12 months of follow-up. In the intention-to-treat analysis, this occurred in 7.33% of the DCB patients and in 7.45% of the DES patients, a difference that was not statistically significant and that met the prespecified criterion for noninferiority of the DCB. Concurrently with Dr. Jeger’s report at the congress, the results also appeared in an article published in The Lancet (Lancet. 2018 Sep 8;392[10190]:849-56).
One limitation of the study was that the first 25% of patients enrolled into the DES arm received a first-generation DES, while the remaining 75% received a second-generation device. Analysis of the primary endpoint by DES type showed that events occurred more than twice as often in the patients who received a first-generation DES, and their inclusion may have affected the comparator group’s results.
Coronary arteries that need percutaneous intervention and are less than 3 mm in diameter constitute about a third of all target vessels, and they are especially common among women and in patients with diabetes, Dr. Jeger said. Despite this, women made up about a quarter of the study enrollment, and about a third had diabetes. He also noted that a key aspect of adopting the DCB approach into routine practice is that operators would need to have the “courage” to accept some amount of recoil and “minor” dissections after DCB treatment and not feel compelled to correct these with a stent.

Other features of the BASKET-SMALL 2 trial also have raised concerns about the immediate clinical implications of the results, said Roxana Mehran, MD, a professor of medicine at Icahn School of Medicine at Mount Sinai, New York, and the congress’s designated discussant for the report.
The study began in 2012, which means it took more than 5 years to enroll and suggests that the study may have a selection bias. Dr. Mehran also questioned whether it was really a small vessel study, with an enrollment criterion of less than 3 mm in diameter. A future study should be done in “truly” small vessels, those thinner than 2.5 mm, she said.
Dr. Mehran agreed it’s attractive to speculate that, by using a DCB and avoiding stent placement, fewer patients will eventually have very-late adverse events, but this must be proven with longer follow-up and in larger numbers of patients, she said.
Treating thin coronary arteries is a problem because they have a higher risk for in-stent restenosis, although usually we will put a stent in arteries that are at least 2.5 mm wide and sometimes in coronaries as narrow as 2.25 mm. That’s using the narrowest stent we have available. Sometimes in vessels this size, if the result from initial balloon angioplasty looks good on angiography, we accept that outcome and do not place a stent.

Steen Dalby Kristensen, MD , is a professor of cardiology at Aarhus University in Skejby, Denmark. He had no relevant disclosures. He made these comments in a video interview.
Treating thin coronary arteries is a problem because they have a higher risk for in-stent restenosis, although usually we will put a stent in arteries that are at least 2.5 mm wide and sometimes in coronaries as narrow as 2.25 mm. That’s using the narrowest stent we have available. Sometimes in vessels this size, if the result from initial balloon angioplasty looks good on angiography, we accept that outcome and do not place a stent.
Steen Dalby Kristensen, MD , is a professor of cardiology at Aarhus University in Skejby, Denmark. He had no relevant disclosures. He made these comments in a video interview.
Treating thin coronary arteries is a problem because they have a higher risk for in-stent restenosis, although usually we will put a stent in arteries that are at least 2.5 mm wide and sometimes in coronaries as narrow as 2.25 mm. That’s using the narrowest stent we have available. Sometimes in vessels this size, if the result from initial balloon angioplasty looks good on angiography, we accept that outcome and do not place a stent.
Steen Dalby Kristensen, MD , is a professor of cardiology at Aarhus University in Skejby, Denmark. He had no relevant disclosures. He made these comments in a video interview.
MUNICH – for preventing the clinical consequences of restenosis during 12 months following coronary intervention, according to results from a prospective, randomized, multicenter trial.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Drug-coated balloons are already used to treat in-stent coronary restenosis. The findings of the current study establish the tested DCB as noninferior to a DES for treating coronary stenoses in narrow arteries less than 3 mm in diameter, Raban V. Jeger, MD, said at the annual congress of the European Society of Cardiology. The DCB approach avoids placing a metal stent in a narrow coronary and thus has no long-term risk for in-stent thrombosis, said Dr. Jeger, a professor of cardiology at Basel (Switzerland) University Hospital. Dr. Jeger acknowledged that the tested DCB is more expensive than the second-generation DES used as the comparator in most of the control patients, “but I think the benefit to patients is worth” the added cost, he said when discussing his report.
The BASKET-SMALL 2 (NCT01574534) study enrolled 758 patients at 14 centers in Switzerland, Germany, and Austria. The trial limited enrollment to patients who were scheduled to undergo percutaneous coronary intervention for stenosis in a coronary artery that was at least 2.0 mm and less than 3.0 mm in diameter and had first undergone successful predilatation without any flow-limiting dissections or residual stenosis, a step in the DCB procedure that adds to the procedure’s cost.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The study randomized patients to treatment with either a balloon coated with paclitaxel/iopromide (SeQuent Please) or a DES. The first quarter of patients randomized into the DES arm received a first-generation, paclitaxel-eluting DES (Taxus Element); the remaining patients in the comparator arm received a second-generation everolimus-eluting DES (Xience). The DCB tested is not approved for U.S. marketing.
The primary endpoint was the combined rate of cardiac death, nonfatal MI, or target vessel revascularization during 12 months of follow-up. In the intention-to-treat analysis, this occurred in 7.33% of the DCB patients and in 7.45% of the DES patients, a difference that was not statistically significant and that met the prespecified criterion for noninferiority of the DCB. Concurrently with Dr. Jeger’s report at the congress, the results also appeared in an article published in The Lancet (Lancet. 2018 Sep 8;392[10190]:849-56).
One limitation of the study was that the first 25% of patients enrolled into the DES arm received a first-generation DES, while the remaining 75% received a second-generation device. Analysis of the primary endpoint by DES type showed that events occurred more than twice as often in the patients who received a first-generation DES, and their inclusion may have affected the comparator group’s results.
Coronary arteries that need percutaneous intervention and are less than 3 mm in diameter constitute about a third of all target vessels, and they are especially common among women and in patients with diabetes, Dr. Jeger said. Despite this, women made up about a quarter of the study enrollment, and about a third had diabetes. He also noted that a key aspect of adopting the DCB approach into routine practice is that operators would need to have the “courage” to accept some amount of recoil and “minor” dissections after DCB treatment and not feel compelled to correct these with a stent.
Other features of the BASKET-SMALL 2 trial also have raised concerns about the immediate clinical implications of the results, said Roxana Mehran, MD, a professor of medicine at Icahn School of Medicine at Mount Sinai, New York, and the congress’s designated discussant for the report.
The study began in 2012, which means it took more than 5 years to enroll and suggests that the study may have a selection bias. Dr. Mehran also questioned whether it was really a small vessel study, with an enrollment criterion of less than 3 mm in diameter. A future study should be done in “truly” small vessels, those thinner than 2.5 mm, she said.
Dr. Mehran agreed it’s attractive to speculate that, by using a DCB and avoiding stent placement, fewer patients will eventually have very-late adverse events, but this must be proven with longer follow-up and in larger numbers of patients, she said.
MUNICH – for preventing the clinical consequences of restenosis during 12 months following coronary intervention, according to results from a prospective, randomized, multicenter trial.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Drug-coated balloons are already used to treat in-stent coronary restenosis. The findings of the current study establish the tested DCB as noninferior to a DES for treating coronary stenoses in narrow arteries less than 3 mm in diameter, Raban V. Jeger, MD, said at the annual congress of the European Society of Cardiology. The DCB approach avoids placing a metal stent in a narrow coronary and thus has no long-term risk for in-stent thrombosis, said Dr. Jeger, a professor of cardiology at Basel (Switzerland) University Hospital. Dr. Jeger acknowledged that the tested DCB is more expensive than the second-generation DES used as the comparator in most of the control patients, “but I think the benefit to patients is worth” the added cost, he said when discussing his report.
The BASKET-SMALL 2 (NCT01574534) study enrolled 758 patients at 14 centers in Switzerland, Germany, and Austria. The trial limited enrollment to patients who were scheduled to undergo percutaneous coronary intervention for stenosis in a coronary artery that was at least 2.0 mm and less than 3.0 mm in diameter and had first undergone successful predilatation without any flow-limiting dissections or residual stenosis, a step in the DCB procedure that adds to the procedure’s cost.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The study randomized patients to treatment with either a balloon coated with paclitaxel/iopromide (SeQuent Please) or a DES. The first quarter of patients randomized into the DES arm received a first-generation, paclitaxel-eluting DES (Taxus Element); the remaining patients in the comparator arm received a second-generation everolimus-eluting DES (Xience). The DCB tested is not approved for U.S. marketing.
The primary endpoint was the combined rate of cardiac death, nonfatal MI, or target vessel revascularization during 12 months of follow-up. In the intention-to-treat analysis, this occurred in 7.33% of the DCB patients and in 7.45% of the DES patients, a difference that was not statistically significant and that met the prespecified criterion for noninferiority of the DCB. Concurrently with Dr. Jeger’s report at the congress, the results also appeared in an article published in The Lancet (Lancet. 2018 Sep 8;392[10190]:849-56).
One limitation of the study was that the first 25% of patients enrolled into the DES arm received a first-generation DES, while the remaining 75% received a second-generation device. Analysis of the primary endpoint by DES type showed that events occurred more than twice as often in the patients who received a first-generation DES, and their inclusion may have affected the comparator group’s results.
Coronary arteries that need percutaneous intervention and are less than 3 mm in diameter constitute about a third of all target vessels, and they are especially common among women and in patients with diabetes, Dr. Jeger said. Despite this, women made up about a quarter of the study enrollment, and about a third had diabetes. He also noted that a key aspect of adopting the DCB approach into routine practice is that operators would need to have the “courage” to accept some amount of recoil and “minor” dissections after DCB treatment and not feel compelled to correct these with a stent.
Other features of the BASKET-SMALL 2 trial also have raised concerns about the immediate clinical implications of the results, said Roxana Mehran, MD, a professor of medicine at Icahn School of Medicine at Mount Sinai, New York, and the congress’s designated discussant for the report.
The study began in 2012, which means it took more than 5 years to enroll and suggests that the study may have a selection bias. Dr. Mehran also questioned whether it was really a small vessel study, with an enrollment criterion of less than 3 mm in diameter. A future study should be done in “truly” small vessels, those thinner than 2.5 mm, she said.
Dr. Mehran agreed it’s attractive to speculate that, by using a DCB and avoiding stent placement, fewer patients will eventually have very-late adverse events, but this must be proven with longer follow-up and in larger numbers of patients, she said.
REPORTING FROM THE ESC CONGRESS 2018
Key clinical point: Drug-coated balloon treatment worked as well as drug-eluting stents in thin coronaries.
Major finding: Twelve-month MACE occurred in 7.33% of balloon-treated patients and in 7.45% of stent-treated patients.
Study details: BASKET-SMALL 2, an international, multicenter randomized trial with 758 patients.
Disclosures: The investigator-initiated study received partial funding from B. Braun, the company that markets the drug-coated balloon (SeQuent Please) tested in the study. Dr. Jeger has received research funding from B. Braun. Dr. Mehran has been a consultant to Abbott, Bayer, BSC, and CSL Behring and has received research funding from Abbott, Astra Zeneca, Bayer, BCC, DSI, and Janssen.

The hidden cost of excellence
SAN DIEGO – Readiness and excellence in trauma services come at a high cost.

Level I trauma centers in Georgia lay out an average of $10 million each year to maintain the essential services necessary to operate at full throttle 24 hours a day, 7 days a week. At the Medical Center of Central Georgia, Macon, that amounts to about $4,480 per patient, Dennis W. Ashley, MD, said at the annual meeting of American Association for the Surgery of Trauma. The yearly tab comes to about $5 million for level II centers.
Medical staff pay was the biggest single driver of that cost, accounting for an average $5.5 million annually for level I centers and $3 million for level II centers.
“These readiness costs are incurred well before the first patient is even treated,” said Dr. Ashley, director of trauma and adult critical care at the Medical Center of Central Georgia. “These are costs required by trauma center regulations to maintain essential infrastructure – nonpatient care costs that the hospital would not have to pay if it were not a trauma center.”
Hospitals incur these costs to comply with standards outlined in “Resources for Optimal Care of the Injured Patient,” otherwise known as the “Orange Book.”
To assess these by center, the Georgia Trauma Commission created a cost-reporting survey that was distributed to the state’s 6 level I centers and 10 level II centers in 2017. The surveys examined 2016 costs, and were carried out in conjunction with independent financial reviews to guarantee accurate reporting. Data were gathered in four general areas: administrative/program support staff, clinical medical staff, in-house operating room, and outreach and education.
During 2016, the 16 centers treated a total of 24,488 trauma patients: 15,660 in level I centers and 8,828 in level II centers.
Overall, the six level I centers spent a total of about $60.5 million in 2017 on resource readiness expense. The 10 level II centers spent a total of about $51 million. In all, trauma center status costs these institutions more than $112 million that year.
Salary and benefits for clinical and medical staff were the biggest cost driver for both types of trauma centers. Level I centers laid out a total of $33.2 million – about $5.5 million for each center. Level II centers paid a total of $31.7 million – about $3.1 million each.
Administrative and program support staff comprised the next largest expense. Level I centers paid abut $21.6 million altogether – $3.6 million each. Level II centers paid $13.9 million – about $1.4 million each.
The cost of maintaining constant operating room coverage accounted for $4.9 million in the level I centers ($830,000 each), and $2.5 million in level II centers (about $252,000 each).
Outreach and education comprised the smallest portion of spending. This accounted for a total of about $700,000 for level I centers and $1 million for level II centers ($115,000 and $109,000, respectively).
Dr. Ashley further broke down each general category. In the administrative and program support bucket, trauma program managers and trauma coordinators were the main cost drivers for both types of center. Trauma managers cost a grand total of $1.56 million: $113,000 for each level I center and $88,169 for level II centers. Trauma coordinators cost a grand total of $921,800 and senior administrative support accounted for a total of about $590,000.
Program support staff consisted of physical, occupational, and speech therapists, as well as research coordinators and others. The big-ticket items here were case managers, and staff providing physical, occupational, and speech therapy. Each of those services cost a total of about $6 million for all centers (around $600,000 for each level I center and $240,000 for each level II center).
Staffing trauma surgery made up the bulk of costs for clinical medical staff (a total of almost $19 million), followed by orthopedics ($13.8 million) and neurosurgery ($6.5 million).
Education and outreach were comparatively poorly funded, Dr. Ashley said. “We should do a better job on this,” he noted. All the centers combined spent about $340,000 on injury prevention, for example. Educational programs for emergency department staff made up about $590,000, and for intensive care unit staff, the total tab was about $174,000.
The survey plainly shows the financial commitment necessary to maintain high-quality trauma care, he said. “The significant cost of trauma center readiness highlights the need for additional trauma funding.”
He had no financial disclosures.
[email protected]
SAN DIEGO – Readiness and excellence in trauma services come at a high cost.
Level I trauma centers in Georgia lay out an average of $10 million each year to maintain the essential services necessary to operate at full throttle 24 hours a day, 7 days a week. At the Medical Center of Central Georgia, Macon, that amounts to about $4,480 per patient, Dennis W. Ashley, MD, said at the annual meeting of American Association for the Surgery of Trauma. The yearly tab comes to about $5 million for level II centers.
Medical staff pay was the biggest single driver of that cost, accounting for an average $5.5 million annually for level I centers and $3 million for level II centers.
“These readiness costs are incurred well before the first patient is even treated,” said Dr. Ashley, director of trauma and adult critical care at the Medical Center of Central Georgia. “These are costs required by trauma center regulations to maintain essential infrastructure – nonpatient care costs that the hospital would not have to pay if it were not a trauma center.”
Hospitals incur these costs to comply with standards outlined in “Resources for Optimal Care of the Injured Patient,” otherwise known as the “Orange Book.”
To assess these by center, the Georgia Trauma Commission created a cost-reporting survey that was distributed to the state’s 6 level I centers and 10 level II centers in 2017. The surveys examined 2016 costs, and were carried out in conjunction with independent financial reviews to guarantee accurate reporting. Data were gathered in four general areas: administrative/program support staff, clinical medical staff, in-house operating room, and outreach and education.
During 2016, the 16 centers treated a total of 24,488 trauma patients: 15,660 in level I centers and 8,828 in level II centers.
Overall, the six level I centers spent a total of about $60.5 million in 2017 on resource readiness expense. The 10 level II centers spent a total of about $51 million. In all, trauma center status costs these institutions more than $112 million that year.
Salary and benefits for clinical and medical staff were the biggest cost driver for both types of trauma centers. Level I centers laid out a total of $33.2 million – about $5.5 million for each center. Level II centers paid a total of $31.7 million – about $3.1 million each.
Administrative and program support staff comprised the next largest expense. Level I centers paid abut $21.6 million altogether – $3.6 million each. Level II centers paid $13.9 million – about $1.4 million each.
The cost of maintaining constant operating room coverage accounted for $4.9 million in the level I centers ($830,000 each), and $2.5 million in level II centers (about $252,000 each).
Outreach and education comprised the smallest portion of spending. This accounted for a total of about $700,000 for level I centers and $1 million for level II centers ($115,000 and $109,000, respectively).
Dr. Ashley further broke down each general category. In the administrative and program support bucket, trauma program managers and trauma coordinators were the main cost drivers for both types of center. Trauma managers cost a grand total of $1.56 million: $113,000 for each level I center and $88,169 for level II centers. Trauma coordinators cost a grand total of $921,800 and senior administrative support accounted for a total of about $590,000.
Program support staff consisted of physical, occupational, and speech therapists, as well as research coordinators and others. The big-ticket items here were case managers, and staff providing physical, occupational, and speech therapy. Each of those services cost a total of about $6 million for all centers (around $600,000 for each level I center and $240,000 for each level II center).
Staffing trauma surgery made up the bulk of costs for clinical medical staff (a total of almost $19 million), followed by orthopedics ($13.8 million) and neurosurgery ($6.5 million).
Education and outreach were comparatively poorly funded, Dr. Ashley said. “We should do a better job on this,” he noted. All the centers combined spent about $340,000 on injury prevention, for example. Educational programs for emergency department staff made up about $590,000, and for intensive care unit staff, the total tab was about $174,000.
The survey plainly shows the financial commitment necessary to maintain high-quality trauma care, he said. “The significant cost of trauma center readiness highlights the need for additional trauma funding.”
He had no financial disclosures.
[email protected]
SAN DIEGO – Readiness and excellence in trauma services come at a high cost.
Level I trauma centers in Georgia lay out an average of $10 million each year to maintain the essential services necessary to operate at full throttle 24 hours a day, 7 days a week. At the Medical Center of Central Georgia, Macon, that amounts to about $4,480 per patient, Dennis W. Ashley, MD, said at the annual meeting of American Association for the Surgery of Trauma. The yearly tab comes to about $5 million for level II centers.
Medical staff pay was the biggest single driver of that cost, accounting for an average $5.5 million annually for level I centers and $3 million for level II centers.
“These readiness costs are incurred well before the first patient is even treated,” said Dr. Ashley, director of trauma and adult critical care at the Medical Center of Central Georgia. “These are costs required by trauma center regulations to maintain essential infrastructure – nonpatient care costs that the hospital would not have to pay if it were not a trauma center.”
Hospitals incur these costs to comply with standards outlined in “Resources for Optimal Care of the Injured Patient,” otherwise known as the “Orange Book.”
To assess these by center, the Georgia Trauma Commission created a cost-reporting survey that was distributed to the state’s 6 level I centers and 10 level II centers in 2017. The surveys examined 2016 costs, and were carried out in conjunction with independent financial reviews to guarantee accurate reporting. Data were gathered in four general areas: administrative/program support staff, clinical medical staff, in-house operating room, and outreach and education.
During 2016, the 16 centers treated a total of 24,488 trauma patients: 15,660 in level I centers and 8,828 in level II centers.
Overall, the six level I centers spent a total of about $60.5 million in 2017 on resource readiness expense. The 10 level II centers spent a total of about $51 million. In all, trauma center status costs these institutions more than $112 million that year.
Salary and benefits for clinical and medical staff were the biggest cost driver for both types of trauma centers. Level I centers laid out a total of $33.2 million – about $5.5 million for each center. Level II centers paid a total of $31.7 million – about $3.1 million each.
Administrative and program support staff comprised the next largest expense. Level I centers paid abut $21.6 million altogether – $3.6 million each. Level II centers paid $13.9 million – about $1.4 million each.
The cost of maintaining constant operating room coverage accounted for $4.9 million in the level I centers ($830,000 each), and $2.5 million in level II centers (about $252,000 each).
Outreach and education comprised the smallest portion of spending. This accounted for a total of about $700,000 for level I centers and $1 million for level II centers ($115,000 and $109,000, respectively).
Dr. Ashley further broke down each general category. In the administrative and program support bucket, trauma program managers and trauma coordinators were the main cost drivers for both types of center. Trauma managers cost a grand total of $1.56 million: $113,000 for each level I center and $88,169 for level II centers. Trauma coordinators cost a grand total of $921,800 and senior administrative support accounted for a total of about $590,000.
Program support staff consisted of physical, occupational, and speech therapists, as well as research coordinators and others. The big-ticket items here were case managers, and staff providing physical, occupational, and speech therapy. Each of those services cost a total of about $6 million for all centers (around $600,000 for each level I center and $240,000 for each level II center).
Staffing trauma surgery made up the bulk of costs for clinical medical staff (a total of almost $19 million), followed by orthopedics ($13.8 million) and neurosurgery ($6.5 million).
Education and outreach were comparatively poorly funded, Dr. Ashley said. “We should do a better job on this,” he noted. All the centers combined spent about $340,000 on injury prevention, for example. Educational programs for emergency department staff made up about $590,000, and for intensive care unit staff, the total tab was about $174,000.
The survey plainly shows the financial commitment necessary to maintain high-quality trauma care, he said. “The significant cost of trauma center readiness highlights the need for additional trauma funding.”
He had no financial disclosures.
[email protected]
REPORTING FROM THE AAST ANNUAL MEETING
Key clinical point: Meeting excellence requirements costs trauma centers millions of dollars.
Major finding: A level I center invests about $10 million annually to meet readiness requirements.
Study details: The Georgia survey queried six level I centers and 10 level II centers.
Disclosures: Dr. Ashley had no financial disclosures.
Source: Ashely DW et al. AAST 2018. Abstract 18.
Eat, toke or vape: Teens not too picky when it comes to pot’s potpourri
There is no doubt that some high school students will try to get high. However, the ways they’re doing it might be changing.

A survey of more than 3,000 10th-graders from 10 high schools in Los Angeles showed that, while traditional combustible marijuana is still the most popular method, kids are turning to edible and vaporized weed, according to a study published in JAMA Network Open Sept. 28.
Because of L.A.’s size and diversity, the researchers said, the patterns of cannabis use they tracked provide insights about “a wide cross-section” of American teens. They found that 62% of students who had ever tried marijuana had used multiple kinds and around 8% tried all three forms.
Those findings were consistent with the impressions of Andrew G., a 15-year-old from the Washington metropolitan area. (Kaiser Health News is not fully identifying him because he is a minor.) While he doesn’t use marijuana, he guessed that more than half of his peers have tried it. He also knows people who have used it in all three forms.
“I feel like, because it’s being incorporated in new ways – edibles, vaping – the stigma has been broken,” he said.
This attitude of wider acceptance and greater access worries researchers. “We are concerned about the developing teen brain, the potential effects on cognitive development, mood and exposure to cannabinoids and chemicals in these various products,” said Adam Leventhal, PhD, an author on the study and a professor at the University of Southern California, Los Angeles.
Dr. Leventhal called this “polyproduct use” alarming for a number of reasons.
A major one is that the body processes these forms of cannabis differently. Smoking or vaping likely has a more immediate effect, while eating it takes a longer time for the body to process. Teens might not realize how much they’re ingesting, Dr. Leventhal said.
Also, “these novel products could be drawing in youth who could be otherwise be deterred,” he added, referring to a range of commercially available cannabinoid-infused products such as gummy bears and energy drinks.
If kids are consuming many different products, he said, it increases the potential risk for addiction.
“It’s a parallel issue. We definitely do see teens who use more different forms of nicotine and tobacco products are at more risk of addiction,” Dr. Leventhal said.
The study was a cross-sectional survey of 10th-graders in the Los Angeles area. It was conducted during Jan. 2, 2015–Oct. 6, 2015. The location and timing of the survey was important, noted the researchers, because California legalized medical cannabis in 1996. In 2018, it became one of nine states to allow the sale for recreational use.
The findings, Dr. Leventhal said, could be an early warning for a trend that will only increase as more states legalize marijuana. Specifically, cannabis products appear to be gaining ground among teens, compared with other substances.
Teen alcohol and nicotine use, for example, has been on the decline for years, according to Monitoring the Future, a multidecade survey conducted by the University of Michigan, Ann Arbor.
“It’s really a public health and policy success story,” Richard Miech, PhD, the survey’s principal investigator, wrote in an email. “In contrast, marijuana use hasn’t declined much at all in the past two decades.” He was not involved in the JAMA study.
Dr. Leventhal also highlighted a socioeconomic element in the findings.
Traditionally, kids who were in a higher socioeconomic bracket were at a lower risk of using marijuana, he said. That’s still true for traditional and edible cannabis, but wealthier kids were more likely to use vaporized weed, according to the study.
Leventhal thinks the popularity of vaping is pulling more kids and teenagers into marijuana use. It removes some of the common barriers that keeps kids away from drugs: There’s no telltale smell that would alert parents, no harshness in their lungs, and a perception that it’s safer than traditional smoke.
“The same vaporizer could be used, teens can load in a liquid on one day with nicotine and the next day a liquid that has THC or another cannabinoid,” Dr. Leventhal said.
And then it might be harder for parents or teachers to detect kids’ drug use.
Getting caught with a bag of marijuana plants could immediately get a kid in trouble. But if a parent finds a vape that looks like a normal e-cigarette or a package of gummy bears laced with THC, they might not realize what they’re seeing.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
There is no doubt that some high school students will try to get high. However, the ways they’re doing it might be changing.
A survey of more than 3,000 10th-graders from 10 high schools in Los Angeles showed that, while traditional combustible marijuana is still the most popular method, kids are turning to edible and vaporized weed, according to a study published in JAMA Network Open Sept. 28.
Because of L.A.’s size and diversity, the researchers said, the patterns of cannabis use they tracked provide insights about “a wide cross-section” of American teens. They found that 62% of students who had ever tried marijuana had used multiple kinds and around 8% tried all three forms.
Those findings were consistent with the impressions of Andrew G., a 15-year-old from the Washington metropolitan area. (Kaiser Health News is not fully identifying him because he is a minor.) While he doesn’t use marijuana, he guessed that more than half of his peers have tried it. He also knows people who have used it in all three forms.
“I feel like, because it’s being incorporated in new ways – edibles, vaping – the stigma has been broken,” he said.
This attitude of wider acceptance and greater access worries researchers. “We are concerned about the developing teen brain, the potential effects on cognitive development, mood and exposure to cannabinoids and chemicals in these various products,” said Adam Leventhal, PhD, an author on the study and a professor at the University of Southern California, Los Angeles.
Dr. Leventhal called this “polyproduct use” alarming for a number of reasons.
A major one is that the body processes these forms of cannabis differently. Smoking or vaping likely has a more immediate effect, while eating it takes a longer time for the body to process. Teens might not realize how much they’re ingesting, Dr. Leventhal said.
Also, “these novel products could be drawing in youth who could be otherwise be deterred,” he added, referring to a range of commercially available cannabinoid-infused products such as gummy bears and energy drinks.
If kids are consuming many different products, he said, it increases the potential risk for addiction.
“It’s a parallel issue. We definitely do see teens who use more different forms of nicotine and tobacco products are at more risk of addiction,” Dr. Leventhal said.
The study was a cross-sectional survey of 10th-graders in the Los Angeles area. It was conducted during Jan. 2, 2015–Oct. 6, 2015. The location and timing of the survey was important, noted the researchers, because California legalized medical cannabis in 1996. In 2018, it became one of nine states to allow the sale for recreational use.
The findings, Dr. Leventhal said, could be an early warning for a trend that will only increase as more states legalize marijuana. Specifically, cannabis products appear to be gaining ground among teens, compared with other substances.
Teen alcohol and nicotine use, for example, has been on the decline for years, according to Monitoring the Future, a multidecade survey conducted by the University of Michigan, Ann Arbor.
“It’s really a public health and policy success story,” Richard Miech, PhD, the survey’s principal investigator, wrote in an email. “In contrast, marijuana use hasn’t declined much at all in the past two decades.” He was not involved in the JAMA study.
Dr. Leventhal also highlighted a socioeconomic element in the findings.
Traditionally, kids who were in a higher socioeconomic bracket were at a lower risk of using marijuana, he said. That’s still true for traditional and edible cannabis, but wealthier kids were more likely to use vaporized weed, according to the study.
Leventhal thinks the popularity of vaping is pulling more kids and teenagers into marijuana use. It removes some of the common barriers that keeps kids away from drugs: There’s no telltale smell that would alert parents, no harshness in their lungs, and a perception that it’s safer than traditional smoke.
“The same vaporizer could be used, teens can load in a liquid on one day with nicotine and the next day a liquid that has THC or another cannabinoid,” Dr. Leventhal said.
And then it might be harder for parents or teachers to detect kids’ drug use.
Getting caught with a bag of marijuana plants could immediately get a kid in trouble. But if a parent finds a vape that looks like a normal e-cigarette or a package of gummy bears laced with THC, they might not realize what they’re seeing.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
There is no doubt that some high school students will try to get high. However, the ways they’re doing it might be changing.
A survey of more than 3,000 10th-graders from 10 high schools in Los Angeles showed that, while traditional combustible marijuana is still the most popular method, kids are turning to edible and vaporized weed, according to a study published in JAMA Network Open Sept. 28.
Because of L.A.’s size and diversity, the researchers said, the patterns of cannabis use they tracked provide insights about “a wide cross-section” of American teens. They found that 62% of students who had ever tried marijuana had used multiple kinds and around 8% tried all three forms.
Those findings were consistent with the impressions of Andrew G., a 15-year-old from the Washington metropolitan area. (Kaiser Health News is not fully identifying him because he is a minor.) While he doesn’t use marijuana, he guessed that more than half of his peers have tried it. He also knows people who have used it in all three forms.
“I feel like, because it’s being incorporated in new ways – edibles, vaping – the stigma has been broken,” he said.
This attitude of wider acceptance and greater access worries researchers. “We are concerned about the developing teen brain, the potential effects on cognitive development, mood and exposure to cannabinoids and chemicals in these various products,” said Adam Leventhal, PhD, an author on the study and a professor at the University of Southern California, Los Angeles.
Dr. Leventhal called this “polyproduct use” alarming for a number of reasons.
A major one is that the body processes these forms of cannabis differently. Smoking or vaping likely has a more immediate effect, while eating it takes a longer time for the body to process. Teens might not realize how much they’re ingesting, Dr. Leventhal said.
Also, “these novel products could be drawing in youth who could be otherwise be deterred,” he added, referring to a range of commercially available cannabinoid-infused products such as gummy bears and energy drinks.
If kids are consuming many different products, he said, it increases the potential risk for addiction.
“It’s a parallel issue. We definitely do see teens who use more different forms of nicotine and tobacco products are at more risk of addiction,” Dr. Leventhal said.
The study was a cross-sectional survey of 10th-graders in the Los Angeles area. It was conducted during Jan. 2, 2015–Oct. 6, 2015. The location and timing of the survey was important, noted the researchers, because California legalized medical cannabis in 1996. In 2018, it became one of nine states to allow the sale for recreational use.
The findings, Dr. Leventhal said, could be an early warning for a trend that will only increase as more states legalize marijuana. Specifically, cannabis products appear to be gaining ground among teens, compared with other substances.
Teen alcohol and nicotine use, for example, has been on the decline for years, according to Monitoring the Future, a multidecade survey conducted by the University of Michigan, Ann Arbor.
“It’s really a public health and policy success story,” Richard Miech, PhD, the survey’s principal investigator, wrote in an email. “In contrast, marijuana use hasn’t declined much at all in the past two decades.” He was not involved in the JAMA study.
Dr. Leventhal also highlighted a socioeconomic element in the findings.
Traditionally, kids who were in a higher socioeconomic bracket were at a lower risk of using marijuana, he said. That’s still true for traditional and edible cannabis, but wealthier kids were more likely to use vaporized weed, according to the study.
Leventhal thinks the popularity of vaping is pulling more kids and teenagers into marijuana use. It removes some of the common barriers that keeps kids away from drugs: There’s no telltale smell that would alert parents, no harshness in their lungs, and a perception that it’s safer than traditional smoke.
“The same vaporizer could be used, teens can load in a liquid on one day with nicotine and the next day a liquid that has THC or another cannabinoid,” Dr. Leventhal said.
And then it might be harder for parents or teachers to detect kids’ drug use.
Getting caught with a bag of marijuana plants could immediately get a kid in trouble. But if a parent finds a vape that looks like a normal e-cigarette or a package of gummy bears laced with THC, they might not realize what they’re seeing.
KHN’s coverage of children’s health care issues is supported in part by the Heising-Simons Foundation. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Postpartum hemorrhage: Aortic compression to reduce pelvic bleeding
You are performing a repeat cesarean delivery on a 37-year-old G3P2 woman with placenta previa. Immediately after delivery, a postpartum hemorrhage occurs. You order additional uterotonic medications and blood products and prepare for standard surgical interventions including uterine devascularization, uterine compression sutures, and intrauterine balloon tamponade. As the hemorrhage continues, you begin to consider the need to perform a hysterectomy.
Suddenly the anesthesiologist reports that the patient’s blood pressure and heart rate have decreased. She asks you to initiate aortic compression to slow the pelvic bleeding and permit initiation of interventions to restore intravascular volume and optimize cardiovascular status. You have not previously performed this maneuver, and you wonder how to respond to her request.
Preoperative preparation
Anticipating possible adverse outcomes is a key task for every clinician. In the above case, in the setting of a repeat cesarean delivery in a woman with placenta previa, there is an increased risk of postpartum hemorrhage. Therefore, appropriate blood products and equipment should be made available before the operation is initiated. It also may be helpful to review the sequential steps you have found most useful in managing a postpartum hemorrhage prior to starting the procedure.
Rapid response to obstetric hemorrhage
When postpartum hemorrhage occurs during a cesarean delivery, there are many interventions that may successfully control the excessive blood loss, including uterotonics, massive transfusion of blood products, uterine massage, tranexamic acid, uterine devascularization, uterine compression sutures, intrauterine balloon tamponade, uterine artery embolization, uterine tourniquet, internal iliac artery ligation, hysterectomy, and pelvic packing.1 Rapid response to obstetric hemorrhage is important to avoid depletion of coagulation factors and subsequent development of a coagulation disorder. Once a coagulation disorder occurs, it can be very difficult to resolve the problem and complete the surgery.
Abdominal compression
The potentially benefial role of abdominal compression to help reduce blood loss caused by trauma or obstetric hemorrhage has been studied extensively in healthy volunteers. The theory is that abdominal compression will decrease blood flow in the distal aorta, helping to control bleeding in the pelvis and extremities. In one report, 80 to 140 lb of pressure applied to the epigastrium in 9 healthy male participants in a supine position on a rigid surface resulted in decreased blood flow in the common femoral artery as determined by pulsed-wave Doppler ultrasound.2 Abdominal pressure applied above the umbilicus also has been reported to reduce blood pressure in the legs.3 Abdominal compression and tourniquets used on the extremities are not meant to be definitive treatments for traumatic hemorrhages but rather are used to stabilize severely injured patients during transport to emergency surgical care facilities.4
One approach to performing manual abdominal aortic compression involves first gaining a mechanical advantage by positioning yourself above the epigastric area with arms extended. Using one closed fist with the opposite hand providing additional pressure, the equivalent of 80 to 140 lb can be applied to the patient’s upper abdomen.4 To estimate the pressure you can achieve using this method, cover a scale with a towel and use your arms to exert maximum pressure on the scale. What equivalent weight can you reach when applying maximum pressure? What weight can you sustain for a few minutes? Using manual compression, it is difficult for a clinician to exert the equivalent of 140 lb on the epigastrium for the extended period of time needed to transport an injured person to an emergency facility.5 Therefore, mechanical devices such as the abdominal aortic tourniquet (AAT) and the nonpneumatic antishock garment (NASG) have been developed to aid in providing continuous abdominal compression.
Continue to: Abdominal aortic tourniquet
Abdominal aortic tourniquet. The AAT is a corset-like device with an interior pneumatic bladder that is designed to provide sustained compression over the abdomen, therefore compressing the abdominal aorta and reducing blood flow to the pelvis and extremities. In one study with human volunteers, a median pressure of 180 mm Hg (range, 150–230 mm Hg) was associated with cessation of blood flow in the common femoral artery in 7 of 9 volunteers and a decrease in blood flow in all participants as determined by pulsed-wave Doppler ultrasound.6 Participants reported moderate to severe discomfort when the AAT was inflated to a pressure sufficient to stop blood flow in the femoral artery. The AAT device may not be as effective in individuals with an elevated body mass index and excessive abdominal girth.7 In obstetric postpartum hemorrhage, abdominal pressure also has been reported to reduce hemorrhage and femoral artery blood flow. Using a corset-like abdominal binder with an internal spring to provide continuous pressure over the anterior abdomen, Soltan and Sadekreported a beneficial effect of abdominal pressure in the management of severe postpartum hemorrhage in a large observational study in Egypt.8,9
Nonpneumatic antishock garment. The NASG has been studied extensively as a method to help safely transport a woman with severe postpartum hemorrhage to an emergency facility. The NASG is a neoprene and Velcro device with panels for the lower extremities, pelvis, and abdomen (FIGURE 1). The device also has an abdominal segment that includes a compression ball to provide continuous abdominal pressure. When the panels are closed, blood flow to the extremities and pelvis is reduced. In a study of 10 postpartum volunteers, application of the NASG caused decreased blood flow in the internal iliac artery as measured by Doppler ultrasound, but blood flow did not stop completely.10 In an observational study of women with postpartum hemorrhage, use of the NASG device in combination with usual interventions resulted in a decrease in blood loss.11
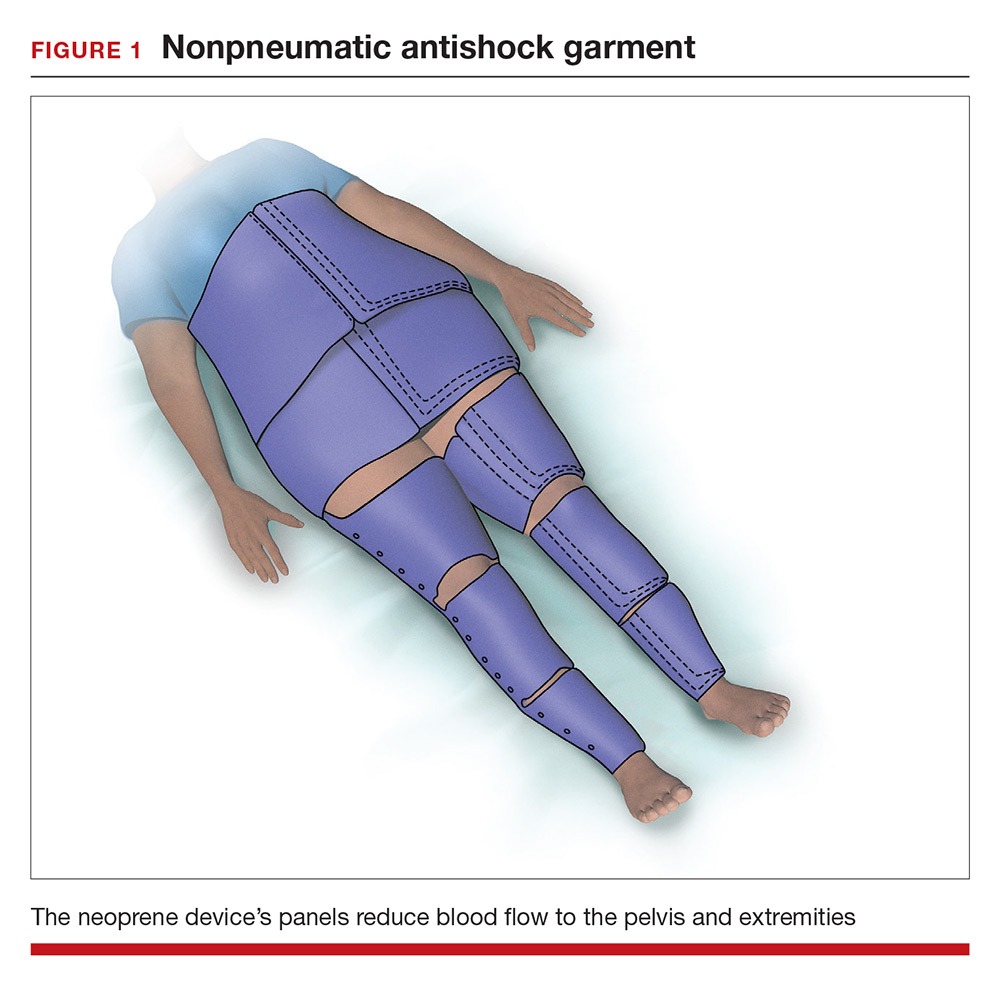
In a cluster randomized trial, 38 birth centers in Africa were randomly assigned to standard management of obstetric hemorrhage or the same protocol plus use of the NASG prior to transport to a regional emergency surgical center. Compared with the group receiving standard management alone, the women who received standard management plus the NASG device had a nonsignificant reduction in maternal mortality (odds ratio, 0.54; 95% confidence interval [CI], 0.14–2.05; P = .37) and a significantly more rapid recovery from hypovolemic shock (hazard ratio, 1.25; 95% CI, 1.02–1.52; P = .03).12 The International Federation of Gynecology and Obstetrics has issued a guideline supporting the use of the device in the management of obstetric hemorrhage in appropriate settings.13
Aortic compression in the setting of an open abdominal incision
During cesarean delivery, the surgeon has access to the abdominal aorta via the open abdominal incision and can directly apply pressure to the aorta at sites ranging from above the sacral promontory to the subdiaphragmatic aorta. Although aortic compression is occasionally noted as a potential intervention to help with the management of postpartum hemorrhage, there is very little literature on this intervention.1 In one case report of an emergency laparotomy in a Jehovah’s Witness patient with a placenta previa, uterine rupture, massive hemorrhage (hematocrit nadir of 6%), and hypovolemic shock, direct pressure applied to the infradiaphragmatic aorta and pelvic organs permitted the anesthesiologist to stabilize the patient’s cardiovascular status, facilitating the patient’s recovery from shock.14 The authors of the case concluded that compression of the aorta and pelvic organs can be lifesaving and is underutilized in the management of uncontrolled obstetric hemorrhage. Other case reports also recommend considering the use of aortic compression to permit the anesthesia team to resuscitate a bleeding patient.15
There is very little published guidance on how to perform aortic compression at cesarean delivery. Techniques for aortic compression include using a closed fist or the heel of the hand to compress the aorta against the lumbosacral spine. Alternatively, use a moist rolled-up surgical towel or laparotomy sponge to compress the aorta against the lumbosacral spine. With a low transverse abdominal incision, the aorta just above the lumbosacral promontory is closest to the surgeon (aorta zone III) (FIGURE 2). If a vertical abdominal incision has been made, the subdiaphragmatic aorta may be within reach of the surgeon (aorta zone II). If an anesthesiologist asks you to apply aortic compression, it is likely that the patient is hypotensive. In this setting, reducing blood flow through the aorta can be achieved with less pressure than required for successful aortic compression in a healthy volunteer.
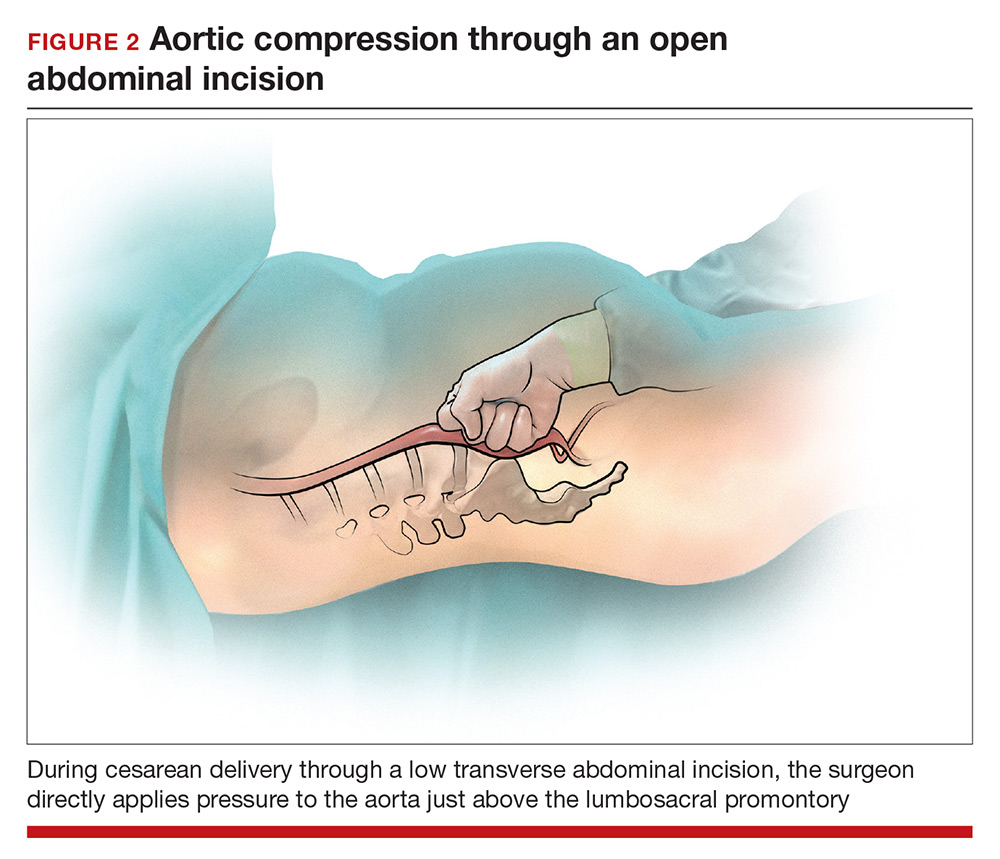
Prolonged aortic compression that completely obstructs blood flow may result in downstream ischemia. This is illustrated by leg ischemia and amputation that have occurred following the use of the resuscitative endovascular balloon occlusion of the aorta (REBOA) occlusion device.16 Another strategy that has been used in the management of massive hemorrhage, when immediate replacement of clotting factors is not possible, is damage control surgery, a technique in which capillary and venous bleeding is controlled by placing pelvic packs or a pelvic umbrella pressure pack and sending the patient to the intensive care unit for resuscitation.17 With damage control surgery, a second procedure is planned to remove the packs after the patient has been stabilized.
With knowledge and practice comes preparedness
Hopefully you will never be asked by an anesthesiologist to stop operating and initiate aortic compression. With effective preprocedure preparation and rapid institution of standard postpartum hemorrhage techniques, it is unlikely aortic compression ever will be needed. If an unusually difficult case triggers a request for aortic compression, you have the knowledge and skills to provide that service.
- Hofmeyr GJ, Qureshi Z. Preventing deaths due to haemorrhage. Best Pract Res Clin Obstet Gynaecol. 2016;36:68–82.
- Blaivas M, Shiver S, Lyon M, et al. Control of hemorrhage in critical femoral or inguinal penetrating wounds—an ultrasound evaluation. Prehosp Disast Med. 2006;21(6):379–382.
- Riley DP, Burgess RW. External abdominal aortic compression: a study of a resuscitation manoeuvre for postpartum hemorrhage. Anaesth Intensive Care. 1994;22(5):571–575.
- Douma M, Smith KE, Brindley PG. Temporization of penetrating abdominal-pelvic trauma with manual external aortic compression: a novel case report. Ann Emerg Med. 2014;64(1):79–81.
- Douma M, Brindley PG. Abdominal aortic and iliac artery compression following penetrating trauma: a study of feasibility. Prehosp Disaster Med. 2014;29:299–302.
- Lyon M, Shiver SA, Greenfield EM, et al. Use of a novel abdominal aortic tourniquet to reduce or eliminate flow in the common femoral artery in human subjects. J Trauma Acute Care Surg. 2012;73(2 suppl 1):S103–S105.
- Taylor DM, Coleman M, Parker PJ. The evaluation of an abdominal aortic tourniquet for the control of pelvic and lower limb hemorrhage. Mil Med. 2013;178(11):1196–1201.
- Soltan MH, Sadek RR. Experience managing postpartum hemorrhage at Minia University Maternity Hospital, Egypt: no mortality using aortic compression. J Obstet Gynaecol Res. 2011;37(11):1557–1563.
- Soltan MH, Faragallah MF, Mosabah MH, et al. External aortic compression device: the first aid for postpartum hemorrhage control. J Obstet Gynaecol Res. 2009;35(3):453–458.
- Lester F, Stenson A, Meyer C, et al. Impact of the non-pneumatic antishock garment on pelvic blood flow in healthy postpartum women. Am J Obstet Gynecol. 2011; 204(5): 409.e1–5.
- Miller S, Hamza S, Bray EH, et al. First aid for obstetric haemorrhage: the pilot study of the non-pneumatic anti-shock garment in Egypt. BJOG. 2006;113(4):424–429.
- Miller S, Bergel EF, El Ayadi AM, et al. Non-pneumatic anti-shock garment (NASG), a first-aid device to decrease maternal mortality from obstetric hemorrhage: a cluster randomized trial. PLoS One. 2013;8(10):e76477.
- FIGO Safe Motherhoood and Newborn Health Committee; International Federation of Gynecology and Obstetrics. Non-pneumatic anti-shock garment to stabilize women with hypovolemic shock secondary to obstetric hemorrhage. Int J Gynaecol Obstet. 2015;128(3):194–195.
- Belfort M, Kofford S, Varner M. Massive obstetric hemorrhage in a Jehovah’s Witness: intraoperative strategies and high-dose erythropoietin use. Am J Perinatol. 2011;28(3):207–210.
- Keogh J, Tsokos N. Aortic compression in massive postpartum hemorrhage—an old but lifesaving technique. Aust N Z J Obstet Gyencol. 1997;37(2):237–238.
- Ribeiro MAF, Feng CYD, Nguyen ATM, et al. The complications associated with resuscitative endovascular balloon occlusion of the aorta (REBOA). World J Emerg Surg. 2018;13:20.
- Pacheco LD, Lozada MJ, Saade GR, et al. Damage-control surgery for obstetric hemorrhage. Obstet Gynecol 2018;132(2):423–427.

Dr. Barbieri is Editor in Chief, OBG Management, and Chair, Obstetrics and Gynecology, Brigham and Women's Hospital, Boston, Massachusetts, and Kate Macy Ladd Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston.
Dr. Barbieri reports no financial relationships relevant to this article.
Dr. Barbieri is Editor in Chief, OBG Management, and Chair, Obstetrics and Gynecology, Brigham and Women's Hospital, Boston, Massachusetts, and Kate Macy Ladd Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston.
Dr. Barbieri reports no financial relationships relevant to this article.
Dr. Barbieri is Editor in Chief, OBG Management, and Chair, Obstetrics and Gynecology, Brigham and Women's Hospital, Boston, Massachusetts, and Kate Macy Ladd Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston.
Dr. Barbieri reports no financial relationships relevant to this article.
You are performing a repeat cesarean delivery on a 37-year-old G3P2 woman with placenta previa. Immediately after delivery, a postpartum hemorrhage occurs. You order additional uterotonic medications and blood products and prepare for standard surgical interventions including uterine devascularization, uterine compression sutures, and intrauterine balloon tamponade. As the hemorrhage continues, you begin to consider the need to perform a hysterectomy.
Suddenly the anesthesiologist reports that the patient’s blood pressure and heart rate have decreased. She asks you to initiate aortic compression to slow the pelvic bleeding and permit initiation of interventions to restore intravascular volume and optimize cardiovascular status. You have not previously performed this maneuver, and you wonder how to respond to her request.
Preoperative preparation
Anticipating possible adverse outcomes is a key task for every clinician. In the above case, in the setting of a repeat cesarean delivery in a woman with placenta previa, there is an increased risk of postpartum hemorrhage. Therefore, appropriate blood products and equipment should be made available before the operation is initiated. It also may be helpful to review the sequential steps you have found most useful in managing a postpartum hemorrhage prior to starting the procedure.
Rapid response to obstetric hemorrhage
When postpartum hemorrhage occurs during a cesarean delivery, there are many interventions that may successfully control the excessive blood loss, including uterotonics, massive transfusion of blood products, uterine massage, tranexamic acid, uterine devascularization, uterine compression sutures, intrauterine balloon tamponade, uterine artery embolization, uterine tourniquet, internal iliac artery ligation, hysterectomy, and pelvic packing.1 Rapid response to obstetric hemorrhage is important to avoid depletion of coagulation factors and subsequent development of a coagulation disorder. Once a coagulation disorder occurs, it can be very difficult to resolve the problem and complete the surgery.
Abdominal compression
The potentially benefial role of abdominal compression to help reduce blood loss caused by trauma or obstetric hemorrhage has been studied extensively in healthy volunteers. The theory is that abdominal compression will decrease blood flow in the distal aorta, helping to control bleeding in the pelvis and extremities. In one report, 80 to 140 lb of pressure applied to the epigastrium in 9 healthy male participants in a supine position on a rigid surface resulted in decreased blood flow in the common femoral artery as determined by pulsed-wave Doppler ultrasound.2 Abdominal pressure applied above the umbilicus also has been reported to reduce blood pressure in the legs.3 Abdominal compression and tourniquets used on the extremities are not meant to be definitive treatments for traumatic hemorrhages but rather are used to stabilize severely injured patients during transport to emergency surgical care facilities.4
One approach to performing manual abdominal aortic compression involves first gaining a mechanical advantage by positioning yourself above the epigastric area with arms extended. Using one closed fist with the opposite hand providing additional pressure, the equivalent of 80 to 140 lb can be applied to the patient’s upper abdomen.4 To estimate the pressure you can achieve using this method, cover a scale with a towel and use your arms to exert maximum pressure on the scale. What equivalent weight can you reach when applying maximum pressure? What weight can you sustain for a few minutes? Using manual compression, it is difficult for a clinician to exert the equivalent of 140 lb on the epigastrium for the extended period of time needed to transport an injured person to an emergency facility.5 Therefore, mechanical devices such as the abdominal aortic tourniquet (AAT) and the nonpneumatic antishock garment (NASG) have been developed to aid in providing continuous abdominal compression.
Continue to: Abdominal aortic tourniquet
Abdominal aortic tourniquet. The AAT is a corset-like device with an interior pneumatic bladder that is designed to provide sustained compression over the abdomen, therefore compressing the abdominal aorta and reducing blood flow to the pelvis and extremities. In one study with human volunteers, a median pressure of 180 mm Hg (range, 150–230 mm Hg) was associated with cessation of blood flow in the common femoral artery in 7 of 9 volunteers and a decrease in blood flow in all participants as determined by pulsed-wave Doppler ultrasound.6 Participants reported moderate to severe discomfort when the AAT was inflated to a pressure sufficient to stop blood flow in the femoral artery. The AAT device may not be as effective in individuals with an elevated body mass index and excessive abdominal girth.7 In obstetric postpartum hemorrhage, abdominal pressure also has been reported to reduce hemorrhage and femoral artery blood flow. Using a corset-like abdominal binder with an internal spring to provide continuous pressure over the anterior abdomen, Soltan and Sadekreported a beneficial effect of abdominal pressure in the management of severe postpartum hemorrhage in a large observational study in Egypt.8,9
Nonpneumatic antishock garment. The NASG has been studied extensively as a method to help safely transport a woman with severe postpartum hemorrhage to an emergency facility. The NASG is a neoprene and Velcro device with panels for the lower extremities, pelvis, and abdomen (FIGURE 1). The device also has an abdominal segment that includes a compression ball to provide continuous abdominal pressure. When the panels are closed, blood flow to the extremities and pelvis is reduced. In a study of 10 postpartum volunteers, application of the NASG caused decreased blood flow in the internal iliac artery as measured by Doppler ultrasound, but blood flow did not stop completely.10 In an observational study of women with postpartum hemorrhage, use of the NASG device in combination with usual interventions resulted in a decrease in blood loss.11
In a cluster randomized trial, 38 birth centers in Africa were randomly assigned to standard management of obstetric hemorrhage or the same protocol plus use of the NASG prior to transport to a regional emergency surgical center. Compared with the group receiving standard management alone, the women who received standard management plus the NASG device had a nonsignificant reduction in maternal mortality (odds ratio, 0.54; 95% confidence interval [CI], 0.14–2.05; P = .37) and a significantly more rapid recovery from hypovolemic shock (hazard ratio, 1.25; 95% CI, 1.02–1.52; P = .03).12 The International Federation of Gynecology and Obstetrics has issued a guideline supporting the use of the device in the management of obstetric hemorrhage in appropriate settings.13
Aortic compression in the setting of an open abdominal incision
During cesarean delivery, the surgeon has access to the abdominal aorta via the open abdominal incision and can directly apply pressure to the aorta at sites ranging from above the sacral promontory to the subdiaphragmatic aorta. Although aortic compression is occasionally noted as a potential intervention to help with the management of postpartum hemorrhage, there is very little literature on this intervention.1 In one case report of an emergency laparotomy in a Jehovah’s Witness patient with a placenta previa, uterine rupture, massive hemorrhage (hematocrit nadir of 6%), and hypovolemic shock, direct pressure applied to the infradiaphragmatic aorta and pelvic organs permitted the anesthesiologist to stabilize the patient’s cardiovascular status, facilitating the patient’s recovery from shock.14 The authors of the case concluded that compression of the aorta and pelvic organs can be lifesaving and is underutilized in the management of uncontrolled obstetric hemorrhage. Other case reports also recommend considering the use of aortic compression to permit the anesthesia team to resuscitate a bleeding patient.15
There is very little published guidance on how to perform aortic compression at cesarean delivery. Techniques for aortic compression include using a closed fist or the heel of the hand to compress the aorta against the lumbosacral spine. Alternatively, use a moist rolled-up surgical towel or laparotomy sponge to compress the aorta against the lumbosacral spine. With a low transverse abdominal incision, the aorta just above the lumbosacral promontory is closest to the surgeon (aorta zone III) (FIGURE 2). If a vertical abdominal incision has been made, the subdiaphragmatic aorta may be within reach of the surgeon (aorta zone II). If an anesthesiologist asks you to apply aortic compression, it is likely that the patient is hypotensive. In this setting, reducing blood flow through the aorta can be achieved with less pressure than required for successful aortic compression in a healthy volunteer.
Prolonged aortic compression that completely obstructs blood flow may result in downstream ischemia. This is illustrated by leg ischemia and amputation that have occurred following the use of the resuscitative endovascular balloon occlusion of the aorta (REBOA) occlusion device.16 Another strategy that has been used in the management of massive hemorrhage, when immediate replacement of clotting factors is not possible, is damage control surgery, a technique in which capillary and venous bleeding is controlled by placing pelvic packs or a pelvic umbrella pressure pack and sending the patient to the intensive care unit for resuscitation.17 With damage control surgery, a second procedure is planned to remove the packs after the patient has been stabilized.
With knowledge and practice comes preparedness
Hopefully you will never be asked by an anesthesiologist to stop operating and initiate aortic compression. With effective preprocedure preparation and rapid institution of standard postpartum hemorrhage techniques, it is unlikely aortic compression ever will be needed. If an unusually difficult case triggers a request for aortic compression, you have the knowledge and skills to provide that service.
You are performing a repeat cesarean delivery on a 37-year-old G3P2 woman with placenta previa. Immediately after delivery, a postpartum hemorrhage occurs. You order additional uterotonic medications and blood products and prepare for standard surgical interventions including uterine devascularization, uterine compression sutures, and intrauterine balloon tamponade. As the hemorrhage continues, you begin to consider the need to perform a hysterectomy.
Suddenly the anesthesiologist reports that the patient’s blood pressure and heart rate have decreased. She asks you to initiate aortic compression to slow the pelvic bleeding and permit initiation of interventions to restore intravascular volume and optimize cardiovascular status. You have not previously performed this maneuver, and you wonder how to respond to her request.
Preoperative preparation
Anticipating possible adverse outcomes is a key task for every clinician. In the above case, in the setting of a repeat cesarean delivery in a woman with placenta previa, there is an increased risk of postpartum hemorrhage. Therefore, appropriate blood products and equipment should be made available before the operation is initiated. It also may be helpful to review the sequential steps you have found most useful in managing a postpartum hemorrhage prior to starting the procedure.
Rapid response to obstetric hemorrhage
When postpartum hemorrhage occurs during a cesarean delivery, there are many interventions that may successfully control the excessive blood loss, including uterotonics, massive transfusion of blood products, uterine massage, tranexamic acid, uterine devascularization, uterine compression sutures, intrauterine balloon tamponade, uterine artery embolization, uterine tourniquet, internal iliac artery ligation, hysterectomy, and pelvic packing.1 Rapid response to obstetric hemorrhage is important to avoid depletion of coagulation factors and subsequent development of a coagulation disorder. Once a coagulation disorder occurs, it can be very difficult to resolve the problem and complete the surgery.
Abdominal compression
The potentially benefial role of abdominal compression to help reduce blood loss caused by trauma or obstetric hemorrhage has been studied extensively in healthy volunteers. The theory is that abdominal compression will decrease blood flow in the distal aorta, helping to control bleeding in the pelvis and extremities. In one report, 80 to 140 lb of pressure applied to the epigastrium in 9 healthy male participants in a supine position on a rigid surface resulted in decreased blood flow in the common femoral artery as determined by pulsed-wave Doppler ultrasound.2 Abdominal pressure applied above the umbilicus also has been reported to reduce blood pressure in the legs.3 Abdominal compression and tourniquets used on the extremities are not meant to be definitive treatments for traumatic hemorrhages but rather are used to stabilize severely injured patients during transport to emergency surgical care facilities.4
One approach to performing manual abdominal aortic compression involves first gaining a mechanical advantage by positioning yourself above the epigastric area with arms extended. Using one closed fist with the opposite hand providing additional pressure, the equivalent of 80 to 140 lb can be applied to the patient’s upper abdomen.4 To estimate the pressure you can achieve using this method, cover a scale with a towel and use your arms to exert maximum pressure on the scale. What equivalent weight can you reach when applying maximum pressure? What weight can you sustain for a few minutes? Using manual compression, it is difficult for a clinician to exert the equivalent of 140 lb on the epigastrium for the extended period of time needed to transport an injured person to an emergency facility.5 Therefore, mechanical devices such as the abdominal aortic tourniquet (AAT) and the nonpneumatic antishock garment (NASG) have been developed to aid in providing continuous abdominal compression.
Continue to: Abdominal aortic tourniquet
Abdominal aortic tourniquet. The AAT is a corset-like device with an interior pneumatic bladder that is designed to provide sustained compression over the abdomen, therefore compressing the abdominal aorta and reducing blood flow to the pelvis and extremities. In one study with human volunteers, a median pressure of 180 mm Hg (range, 150–230 mm Hg) was associated with cessation of blood flow in the common femoral artery in 7 of 9 volunteers and a decrease in blood flow in all participants as determined by pulsed-wave Doppler ultrasound.6 Participants reported moderate to severe discomfort when the AAT was inflated to a pressure sufficient to stop blood flow in the femoral artery. The AAT device may not be as effective in individuals with an elevated body mass index and excessive abdominal girth.7 In obstetric postpartum hemorrhage, abdominal pressure also has been reported to reduce hemorrhage and femoral artery blood flow. Using a corset-like abdominal binder with an internal spring to provide continuous pressure over the anterior abdomen, Soltan and Sadekreported a beneficial effect of abdominal pressure in the management of severe postpartum hemorrhage in a large observational study in Egypt.8,9
Nonpneumatic antishock garment. The NASG has been studied extensively as a method to help safely transport a woman with severe postpartum hemorrhage to an emergency facility. The NASG is a neoprene and Velcro device with panels for the lower extremities, pelvis, and abdomen (FIGURE 1). The device also has an abdominal segment that includes a compression ball to provide continuous abdominal pressure. When the panels are closed, blood flow to the extremities and pelvis is reduced. In a study of 10 postpartum volunteers, application of the NASG caused decreased blood flow in the internal iliac artery as measured by Doppler ultrasound, but blood flow did not stop completely.10 In an observational study of women with postpartum hemorrhage, use of the NASG device in combination with usual interventions resulted in a decrease in blood loss.11
In a cluster randomized trial, 38 birth centers in Africa were randomly assigned to standard management of obstetric hemorrhage or the same protocol plus use of the NASG prior to transport to a regional emergency surgical center. Compared with the group receiving standard management alone, the women who received standard management plus the NASG device had a nonsignificant reduction in maternal mortality (odds ratio, 0.54; 95% confidence interval [CI], 0.14–2.05; P = .37) and a significantly more rapid recovery from hypovolemic shock (hazard ratio, 1.25; 95% CI, 1.02–1.52; P = .03).12 The International Federation of Gynecology and Obstetrics has issued a guideline supporting the use of the device in the management of obstetric hemorrhage in appropriate settings.13
Aortic compression in the setting of an open abdominal incision
During cesarean delivery, the surgeon has access to the abdominal aorta via the open abdominal incision and can directly apply pressure to the aorta at sites ranging from above the sacral promontory to the subdiaphragmatic aorta. Although aortic compression is occasionally noted as a potential intervention to help with the management of postpartum hemorrhage, there is very little literature on this intervention.1 In one case report of an emergency laparotomy in a Jehovah’s Witness patient with a placenta previa, uterine rupture, massive hemorrhage (hematocrit nadir of 6%), and hypovolemic shock, direct pressure applied to the infradiaphragmatic aorta and pelvic organs permitted the anesthesiologist to stabilize the patient’s cardiovascular status, facilitating the patient’s recovery from shock.14 The authors of the case concluded that compression of the aorta and pelvic organs can be lifesaving and is underutilized in the management of uncontrolled obstetric hemorrhage. Other case reports also recommend considering the use of aortic compression to permit the anesthesia team to resuscitate a bleeding patient.15
There is very little published guidance on how to perform aortic compression at cesarean delivery. Techniques for aortic compression include using a closed fist or the heel of the hand to compress the aorta against the lumbosacral spine. Alternatively, use a moist rolled-up surgical towel or laparotomy sponge to compress the aorta against the lumbosacral spine. With a low transverse abdominal incision, the aorta just above the lumbosacral promontory is closest to the surgeon (aorta zone III) (FIGURE 2). If a vertical abdominal incision has been made, the subdiaphragmatic aorta may be within reach of the surgeon (aorta zone II). If an anesthesiologist asks you to apply aortic compression, it is likely that the patient is hypotensive. In this setting, reducing blood flow through the aorta can be achieved with less pressure than required for successful aortic compression in a healthy volunteer.
Prolonged aortic compression that completely obstructs blood flow may result in downstream ischemia. This is illustrated by leg ischemia and amputation that have occurred following the use of the resuscitative endovascular balloon occlusion of the aorta (REBOA) occlusion device.16 Another strategy that has been used in the management of massive hemorrhage, when immediate replacement of clotting factors is not possible, is damage control surgery, a technique in which capillary and venous bleeding is controlled by placing pelvic packs or a pelvic umbrella pressure pack and sending the patient to the intensive care unit for resuscitation.17 With damage control surgery, a second procedure is planned to remove the packs after the patient has been stabilized.
With knowledge and practice comes preparedness
Hopefully you will never be asked by an anesthesiologist to stop operating and initiate aortic compression. With effective preprocedure preparation and rapid institution of standard postpartum hemorrhage techniques, it is unlikely aortic compression ever will be needed. If an unusually difficult case triggers a request for aortic compression, you have the knowledge and skills to provide that service.
- Hofmeyr GJ, Qureshi Z. Preventing deaths due to haemorrhage. Best Pract Res Clin Obstet Gynaecol. 2016;36:68–82.
- Blaivas M, Shiver S, Lyon M, et al. Control of hemorrhage in critical femoral or inguinal penetrating wounds—an ultrasound evaluation. Prehosp Disast Med. 2006;21(6):379–382.
- Riley DP, Burgess RW. External abdominal aortic compression: a study of a resuscitation manoeuvre for postpartum hemorrhage. Anaesth Intensive Care. 1994;22(5):571–575.
- Douma M, Smith KE, Brindley PG. Temporization of penetrating abdominal-pelvic trauma with manual external aortic compression: a novel case report. Ann Emerg Med. 2014;64(1):79–81.
- Douma M, Brindley PG. Abdominal aortic and iliac artery compression following penetrating trauma: a study of feasibility. Prehosp Disaster Med. 2014;29:299–302.
- Lyon M, Shiver SA, Greenfield EM, et al. Use of a novel abdominal aortic tourniquet to reduce or eliminate flow in the common femoral artery in human subjects. J Trauma Acute Care Surg. 2012;73(2 suppl 1):S103–S105.
- Taylor DM, Coleman M, Parker PJ. The evaluation of an abdominal aortic tourniquet for the control of pelvic and lower limb hemorrhage. Mil Med. 2013;178(11):1196–1201.
- Soltan MH, Sadek RR. Experience managing postpartum hemorrhage at Minia University Maternity Hospital, Egypt: no mortality using aortic compression. J Obstet Gynaecol Res. 2011;37(11):1557–1563.
- Soltan MH, Faragallah MF, Mosabah MH, et al. External aortic compression device: the first aid for postpartum hemorrhage control. J Obstet Gynaecol Res. 2009;35(3):453–458.
- Lester F, Stenson A, Meyer C, et al. Impact of the non-pneumatic antishock garment on pelvic blood flow in healthy postpartum women. Am J Obstet Gynecol. 2011; 204(5): 409.e1–5.
- Miller S, Hamza S, Bray EH, et al. First aid for obstetric haemorrhage: the pilot study of the non-pneumatic anti-shock garment in Egypt. BJOG. 2006;113(4):424–429.
- Miller S, Bergel EF, El Ayadi AM, et al. Non-pneumatic anti-shock garment (NASG), a first-aid device to decrease maternal mortality from obstetric hemorrhage: a cluster randomized trial. PLoS One. 2013;8(10):e76477.
- FIGO Safe Motherhoood and Newborn Health Committee; International Federation of Gynecology and Obstetrics. Non-pneumatic anti-shock garment to stabilize women with hypovolemic shock secondary to obstetric hemorrhage. Int J Gynaecol Obstet. 2015;128(3):194–195.
- Belfort M, Kofford S, Varner M. Massive obstetric hemorrhage in a Jehovah’s Witness: intraoperative strategies and high-dose erythropoietin use. Am J Perinatol. 2011;28(3):207–210.
- Keogh J, Tsokos N. Aortic compression in massive postpartum hemorrhage—an old but lifesaving technique. Aust N Z J Obstet Gyencol. 1997;37(2):237–238.
- Ribeiro MAF, Feng CYD, Nguyen ATM, et al. The complications associated with resuscitative endovascular balloon occlusion of the aorta (REBOA). World J Emerg Surg. 2018;13:20.
- Pacheco LD, Lozada MJ, Saade GR, et al. Damage-control surgery for obstetric hemorrhage. Obstet Gynecol 2018;132(2):423–427.
- Hofmeyr GJ, Qureshi Z. Preventing deaths due to haemorrhage. Best Pract Res Clin Obstet Gynaecol. 2016;36:68–82.
- Blaivas M, Shiver S, Lyon M, et al. Control of hemorrhage in critical femoral or inguinal penetrating wounds—an ultrasound evaluation. Prehosp Disast Med. 2006;21(6):379–382.
- Riley DP, Burgess RW. External abdominal aortic compression: a study of a resuscitation manoeuvre for postpartum hemorrhage. Anaesth Intensive Care. 1994;22(5):571–575.
- Douma M, Smith KE, Brindley PG. Temporization of penetrating abdominal-pelvic trauma with manual external aortic compression: a novel case report. Ann Emerg Med. 2014;64(1):79–81.
- Douma M, Brindley PG. Abdominal aortic and iliac artery compression following penetrating trauma: a study of feasibility. Prehosp Disaster Med. 2014;29:299–302.
- Lyon M, Shiver SA, Greenfield EM, et al. Use of a novel abdominal aortic tourniquet to reduce or eliminate flow in the common femoral artery in human subjects. J Trauma Acute Care Surg. 2012;73(2 suppl 1):S103–S105.
- Taylor DM, Coleman M, Parker PJ. The evaluation of an abdominal aortic tourniquet for the control of pelvic and lower limb hemorrhage. Mil Med. 2013;178(11):1196–1201.
- Soltan MH, Sadek RR. Experience managing postpartum hemorrhage at Minia University Maternity Hospital, Egypt: no mortality using aortic compression. J Obstet Gynaecol Res. 2011;37(11):1557–1563.
- Soltan MH, Faragallah MF, Mosabah MH, et al. External aortic compression device: the first aid for postpartum hemorrhage control. J Obstet Gynaecol Res. 2009;35(3):453–458.
- Lester F, Stenson A, Meyer C, et al. Impact of the non-pneumatic antishock garment on pelvic blood flow in healthy postpartum women. Am J Obstet Gynecol. 2011; 204(5): 409.e1–5.
- Miller S, Hamza S, Bray EH, et al. First aid for obstetric haemorrhage: the pilot study of the non-pneumatic anti-shock garment in Egypt. BJOG. 2006;113(4):424–429.
- Miller S, Bergel EF, El Ayadi AM, et al. Non-pneumatic anti-shock garment (NASG), a first-aid device to decrease maternal mortality from obstetric hemorrhage: a cluster randomized trial. PLoS One. 2013;8(10):e76477.
- FIGO Safe Motherhoood and Newborn Health Committee; International Federation of Gynecology and Obstetrics. Non-pneumatic anti-shock garment to stabilize women with hypovolemic shock secondary to obstetric hemorrhage. Int J Gynaecol Obstet. 2015;128(3):194–195.
- Belfort M, Kofford S, Varner M. Massive obstetric hemorrhage in a Jehovah’s Witness: intraoperative strategies and high-dose erythropoietin use. Am J Perinatol. 2011;28(3):207–210.
- Keogh J, Tsokos N. Aortic compression in massive postpartum hemorrhage—an old but lifesaving technique. Aust N Z J Obstet Gyencol. 1997;37(2):237–238.
- Ribeiro MAF, Feng CYD, Nguyen ATM, et al. The complications associated with resuscitative endovascular balloon occlusion of the aorta (REBOA). World J Emerg Surg. 2018;13:20.
- Pacheco LD, Lozada MJ, Saade GR, et al. Damage-control surgery for obstetric hemorrhage. Obstet Gynecol 2018;132(2):423–427.