User login
How to get surgical-like results using MitraClip
SNOWMASS, COLO. – Achieving optimal surgical-like results via transcatheter repair of primary mitral regurgitation using the MitraClip in prohibitively high-surgical-risk patients becomes much more likely by taking into account key predictive anatomic and procedural features as well as the major comorbidities influencing outcome, Paul Sorajja, MD, said at the Annual Cardiovascular Conference at Snowmass sponsored by the American College of Cardiology.

. Dr. Sorajja was first author of a study of 1-year outcomes in 2,952 patients with MR in the Society of Thoracic Surgery/American College of Cardiology Transcatheter Valve Therapy Registry (STS/ACC TVT) who were commercially treated with the MitraClip, at present the only transcatheter device approved for this indication. Sixty-two percent had a surgical-like result, and their 1-year mortality rate of 21.7% was significantly lower than the 29.2% rate for patients with residual grade 2 MR and the 48.9% mortality in patients left with grade 3 or 4 MR.
It’s noteworthy that even though the acute procedural success rate – defined as residual grade 2 or less MR – was impressively high at 91.8% in this group of nearly 3,000 patients, roughly one in five in the overall series was rehospitalized for heart failure within 1 year, and one in four was dead. So there remains considerable room for improvement in long-term outcomes of transcatheter repair of MR, said Dr. Sorajja, director of the Center of Valve and Structural Heart Disease at the Minneapolis Heart Institute.
Anatomic predictors of good short-term outcome
“There are specific anatomic criteria, but an easier way to think about whether your patient could get an optimal result is to remember the physical limits of this therapy. What I teach is 1-2-5-50: the MitraClip device is about 1 cm tall, about 2 cm wide, and you need about 5 mm of leaflet inside the clip to create coaptation for MR reduction. And if you do that, you will reduce the mitral valve area by about 50%, so you have to be very careful in patients with small valve areas because you can worsen mitral stenosis,” the cardiologist explained.
Only two transechocardiographic views are needed to know if a patient will have a good result with the MitraClip. A bicommissural view traversing the valve medial to lateral shows where the MR jet is; if it’s in the middle of the mitral valve, that’s favorable because it means the interventionalist has a lot of freedom to operate. Then, going orthogonal from the bicommissural view to get an anterior-posterior view allows the operator to get a good look at the valve leaflets and apply the 1-2-5 rule to determine if the leaflet approximation is favorable, he continued.
Some mitral valve anatomy variants make it challenging to get surgical-like results with a transcatheter repair. These include calcified leaflets, large gaps between leaflets, Barlow’s valves, and small leaflets. Be aware of key success-limiting comorbidities.
In the STS/ACC TVT registry study, severe tricuspid regurgitation, present in 10% of patients preprocedurally, virtually doubled the adjusted risk of 1-year mortality in multivariate analyses.
“Tricuspid regurgitation is one of the most common concomitant lesions. And the presence of tricuspid regurgitation is ominous,” Dr. Sorajja said. “Treatment of concomitant lesions such as this is going to be necessary for us to get a truly surgical-like result in outcomes.”
Toward this end, he put in a plug to consider referring patients with severe tricuspid regurgitation for enrollment in the TRILUMINATE II trial, the pivotal U.S. trial for the investigational Tri-Clip device for transcatheter tricuspid valve repair, for which he is coprincipal investigator. The trial, pitting the Tri-Clip against medical therapy, is due to start in the spring of 2019.
In multivariate analyses of the MitraClip registry study, other predictors of the combined endpoint of death and rehospitalization for heart failure at 1 year, in addition to severe tricuspid regurgitation, included dialysis, with an adjusted 2.09-fold increased risk; moderate or severe lung disease, with a 1.28-fold risk; postprocedural residual MR; diminished left ventricular ejection fraction; and advanced age (J Am Coll Cardiol. 2017 Nov 7;70[19]:2315-27).
Case experience counts
In a soon-to-be-published more up-to-date analysis of more than 12,000 MitraClip patients in the STS/ACC TVT registry, which captures all U.S. commercial use of the device for its approved indication, Dr. Sorajja and his coinvestigators documented a procedural truism: The more cases an interventionalist performs, the better the results. However, even inexperienced users of the MitraClip were able to obtain at least moderate results – that is, residual grade 2 or less MR – 90% of the time or more.
“There is some change with greater experience, but it’s actually quite small,” according to Dr. Sorajja.
However, optimal results – grade 0 or 1 MR – are another matter entirely.
“For grade 1 or less, the learning curve is much steeper. It plateaus somewhere between 50 and 75 cases. In other words, in most cases you can get to moderate MR, but getting to grade 1 requires more experience. That relationship between case experience and outcome also applies to complication rates and case time,” he said.
Although at present the MitraClip is the only Food and Drug Administration–approved transcatheter device for MR repair, there are many others in the developmental pipeline, the he noted.
Dr. Sorajja reported receiving research funding from Abbott Structural, Boston Scientific, Edwards Lifesciences, and Medtronic, and serving as a consultant to those companies and several others.
SNOWMASS, COLO. – Achieving optimal surgical-like results via transcatheter repair of primary mitral regurgitation using the MitraClip in prohibitively high-surgical-risk patients becomes much more likely by taking into account key predictive anatomic and procedural features as well as the major comorbidities influencing outcome, Paul Sorajja, MD, said at the Annual Cardiovascular Conference at Snowmass sponsored by the American College of Cardiology.
. Dr. Sorajja was first author of a study of 1-year outcomes in 2,952 patients with MR in the Society of Thoracic Surgery/American College of Cardiology Transcatheter Valve Therapy Registry (STS/ACC TVT) who were commercially treated with the MitraClip, at present the only transcatheter device approved for this indication. Sixty-two percent had a surgical-like result, and their 1-year mortality rate of 21.7% was significantly lower than the 29.2% rate for patients with residual grade 2 MR and the 48.9% mortality in patients left with grade 3 or 4 MR.
It’s noteworthy that even though the acute procedural success rate – defined as residual grade 2 or less MR – was impressively high at 91.8% in this group of nearly 3,000 patients, roughly one in five in the overall series was rehospitalized for heart failure within 1 year, and one in four was dead. So there remains considerable room for improvement in long-term outcomes of transcatheter repair of MR, said Dr. Sorajja, director of the Center of Valve and Structural Heart Disease at the Minneapolis Heart Institute.
Anatomic predictors of good short-term outcome
“There are specific anatomic criteria, but an easier way to think about whether your patient could get an optimal result is to remember the physical limits of this therapy. What I teach is 1-2-5-50: the MitraClip device is about 1 cm tall, about 2 cm wide, and you need about 5 mm of leaflet inside the clip to create coaptation for MR reduction. And if you do that, you will reduce the mitral valve area by about 50%, so you have to be very careful in patients with small valve areas because you can worsen mitral stenosis,” the cardiologist explained.
Only two transechocardiographic views are needed to know if a patient will have a good result with the MitraClip. A bicommissural view traversing the valve medial to lateral shows where the MR jet is; if it’s in the middle of the mitral valve, that’s favorable because it means the interventionalist has a lot of freedom to operate. Then, going orthogonal from the bicommissural view to get an anterior-posterior view allows the operator to get a good look at the valve leaflets and apply the 1-2-5 rule to determine if the leaflet approximation is favorable, he continued.
Some mitral valve anatomy variants make it challenging to get surgical-like results with a transcatheter repair. These include calcified leaflets, large gaps between leaflets, Barlow’s valves, and small leaflets. Be aware of key success-limiting comorbidities.
In the STS/ACC TVT registry study, severe tricuspid regurgitation, present in 10% of patients preprocedurally, virtually doubled the adjusted risk of 1-year mortality in multivariate analyses.
“Tricuspid regurgitation is one of the most common concomitant lesions. And the presence of tricuspid regurgitation is ominous,” Dr. Sorajja said. “Treatment of concomitant lesions such as this is going to be necessary for us to get a truly surgical-like result in outcomes.”
Toward this end, he put in a plug to consider referring patients with severe tricuspid regurgitation for enrollment in the TRILUMINATE II trial, the pivotal U.S. trial for the investigational Tri-Clip device for transcatheter tricuspid valve repair, for which he is coprincipal investigator. The trial, pitting the Tri-Clip against medical therapy, is due to start in the spring of 2019.
In multivariate analyses of the MitraClip registry study, other predictors of the combined endpoint of death and rehospitalization for heart failure at 1 year, in addition to severe tricuspid regurgitation, included dialysis, with an adjusted 2.09-fold increased risk; moderate or severe lung disease, with a 1.28-fold risk; postprocedural residual MR; diminished left ventricular ejection fraction; and advanced age (J Am Coll Cardiol. 2017 Nov 7;70[19]:2315-27).
Case experience counts
In a soon-to-be-published more up-to-date analysis of more than 12,000 MitraClip patients in the STS/ACC TVT registry, which captures all U.S. commercial use of the device for its approved indication, Dr. Sorajja and his coinvestigators documented a procedural truism: The more cases an interventionalist performs, the better the results. However, even inexperienced users of the MitraClip were able to obtain at least moderate results – that is, residual grade 2 or less MR – 90% of the time or more.
“There is some change with greater experience, but it’s actually quite small,” according to Dr. Sorajja.
However, optimal results – grade 0 or 1 MR – are another matter entirely.
“For grade 1 or less, the learning curve is much steeper. It plateaus somewhere between 50 and 75 cases. In other words, in most cases you can get to moderate MR, but getting to grade 1 requires more experience. That relationship between case experience and outcome also applies to complication rates and case time,” he said.
Although at present the MitraClip is the only Food and Drug Administration–approved transcatheter device for MR repair, there are many others in the developmental pipeline, the he noted.
Dr. Sorajja reported receiving research funding from Abbott Structural, Boston Scientific, Edwards Lifesciences, and Medtronic, and serving as a consultant to those companies and several others.
SNOWMASS, COLO. – Achieving optimal surgical-like results via transcatheter repair of primary mitral regurgitation using the MitraClip in prohibitively high-surgical-risk patients becomes much more likely by taking into account key predictive anatomic and procedural features as well as the major comorbidities influencing outcome, Paul Sorajja, MD, said at the Annual Cardiovascular Conference at Snowmass sponsored by the American College of Cardiology.
. Dr. Sorajja was first author of a study of 1-year outcomes in 2,952 patients with MR in the Society of Thoracic Surgery/American College of Cardiology Transcatheter Valve Therapy Registry (STS/ACC TVT) who were commercially treated with the MitraClip, at present the only transcatheter device approved for this indication. Sixty-two percent had a surgical-like result, and their 1-year mortality rate of 21.7% was significantly lower than the 29.2% rate for patients with residual grade 2 MR and the 48.9% mortality in patients left with grade 3 or 4 MR.
It’s noteworthy that even though the acute procedural success rate – defined as residual grade 2 or less MR – was impressively high at 91.8% in this group of nearly 3,000 patients, roughly one in five in the overall series was rehospitalized for heart failure within 1 year, and one in four was dead. So there remains considerable room for improvement in long-term outcomes of transcatheter repair of MR, said Dr. Sorajja, director of the Center of Valve and Structural Heart Disease at the Minneapolis Heart Institute.
Anatomic predictors of good short-term outcome
“There are specific anatomic criteria, but an easier way to think about whether your patient could get an optimal result is to remember the physical limits of this therapy. What I teach is 1-2-5-50: the MitraClip device is about 1 cm tall, about 2 cm wide, and you need about 5 mm of leaflet inside the clip to create coaptation for MR reduction. And if you do that, you will reduce the mitral valve area by about 50%, so you have to be very careful in patients with small valve areas because you can worsen mitral stenosis,” the cardiologist explained.
Only two transechocardiographic views are needed to know if a patient will have a good result with the MitraClip. A bicommissural view traversing the valve medial to lateral shows where the MR jet is; if it’s in the middle of the mitral valve, that’s favorable because it means the interventionalist has a lot of freedom to operate. Then, going orthogonal from the bicommissural view to get an anterior-posterior view allows the operator to get a good look at the valve leaflets and apply the 1-2-5 rule to determine if the leaflet approximation is favorable, he continued.
Some mitral valve anatomy variants make it challenging to get surgical-like results with a transcatheter repair. These include calcified leaflets, large gaps between leaflets, Barlow’s valves, and small leaflets. Be aware of key success-limiting comorbidities.
In the STS/ACC TVT registry study, severe tricuspid regurgitation, present in 10% of patients preprocedurally, virtually doubled the adjusted risk of 1-year mortality in multivariate analyses.
“Tricuspid regurgitation is one of the most common concomitant lesions. And the presence of tricuspid regurgitation is ominous,” Dr. Sorajja said. “Treatment of concomitant lesions such as this is going to be necessary for us to get a truly surgical-like result in outcomes.”
Toward this end, he put in a plug to consider referring patients with severe tricuspid regurgitation for enrollment in the TRILUMINATE II trial, the pivotal U.S. trial for the investigational Tri-Clip device for transcatheter tricuspid valve repair, for which he is coprincipal investigator. The trial, pitting the Tri-Clip against medical therapy, is due to start in the spring of 2019.
In multivariate analyses of the MitraClip registry study, other predictors of the combined endpoint of death and rehospitalization for heart failure at 1 year, in addition to severe tricuspid regurgitation, included dialysis, with an adjusted 2.09-fold increased risk; moderate or severe lung disease, with a 1.28-fold risk; postprocedural residual MR; diminished left ventricular ejection fraction; and advanced age (J Am Coll Cardiol. 2017 Nov 7;70[19]:2315-27).
Case experience counts
In a soon-to-be-published more up-to-date analysis of more than 12,000 MitraClip patients in the STS/ACC TVT registry, which captures all U.S. commercial use of the device for its approved indication, Dr. Sorajja and his coinvestigators documented a procedural truism: The more cases an interventionalist performs, the better the results. However, even inexperienced users of the MitraClip were able to obtain at least moderate results – that is, residual grade 2 or less MR – 90% of the time or more.
“There is some change with greater experience, but it’s actually quite small,” according to Dr. Sorajja.
However, optimal results – grade 0 or 1 MR – are another matter entirely.
“For grade 1 or less, the learning curve is much steeper. It plateaus somewhere between 50 and 75 cases. In other words, in most cases you can get to moderate MR, but getting to grade 1 requires more experience. That relationship between case experience and outcome also applies to complication rates and case time,” he said.
Although at present the MitraClip is the only Food and Drug Administration–approved transcatheter device for MR repair, there are many others in the developmental pipeline, the he noted.
Dr. Sorajja reported receiving research funding from Abbott Structural, Boston Scientific, Edwards Lifesciences, and Medtronic, and serving as a consultant to those companies and several others.
EXPERT ANALYSIS FROM ACC SNOWMASS 2019
Apply for VAM Travel Awards
Applications are due Feb. 27 for travel scholarships to attend the Vascular Annual Meeting. Available is a resident/medical student award and a diversity medical student award. Recipients are eligible to receive complimentary meeting registration plus a travel scholarship. They also get to participate in the Scholarship Program, including the hugely popular simulation training. Students and residents, please apply. And SVS members, urge the students you know to apply, as well.
Applications are due Feb. 27 for travel scholarships to attend the Vascular Annual Meeting. Available is a resident/medical student award and a diversity medical student award. Recipients are eligible to receive complimentary meeting registration plus a travel scholarship. They also get to participate in the Scholarship Program, including the hugely popular simulation training. Students and residents, please apply. And SVS members, urge the students you know to apply, as well.
Applications are due Feb. 27 for travel scholarships to attend the Vascular Annual Meeting. Available is a resident/medical student award and a diversity medical student award. Recipients are eligible to receive complimentary meeting registration plus a travel scholarship. They also get to participate in the Scholarship Program, including the hugely popular simulation training. Students and residents, please apply. And SVS members, urge the students you know to apply, as well.
Flu activity hits seasonal high
Influenza activity increased for the third consecutive week and has now reached its highest point for the 2018-2019 flu season, according to the Centers for Disease Control and Prevention.
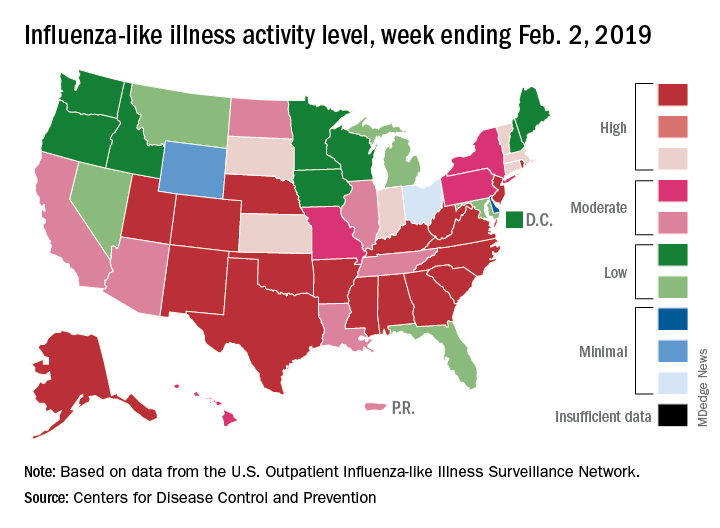
The proportion of outpatient visits for influenza-like illness (ILI) hit 4.3% for the week ending Feb. 2, which topped the previous high of 4.0% that was reached in late December (the national baseline rate is 2.2%). Outpatient ILI visits then dipped down to 3.1% after 2 weeks of decreases before rising again in mid-January, the CDC’s influenza division reported Feb. 8.
Season-high activity also was seen at the state level for the week ending Feb. 2. There were 18 states at level 10 on the CDC’s 1-10 scale of ILI activity, which was up from 16 the week before, and a total of 24 states were in the high range from 8-10, compared with 23 for the previous week. The geographic spread of influenza was reported as widespread in 47 states and Puerto Rico, the CDC said.
Four flu-related pediatric deaths were reported during the week ending Feb. 2, two of which occurred the previous week, which brings the total for the 2018-2019 season to 28, the CDC said.
There were 158 flu-related deaths among all ages during the week ending Jan. 26 – the latest for which such data are available – with reporting almost 75% complete. The previous week saw 177 overall flu deaths, with reporting for that week over 90% complete. During the corresponding weeks of the very severe 2017-2018 flu season, the overall death totals were 1,448 and 1,626, CDC data show.
Influenza activity increased for the third consecutive week and has now reached its highest point for the 2018-2019 flu season, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) hit 4.3% for the week ending Feb. 2, which topped the previous high of 4.0% that was reached in late December (the national baseline rate is 2.2%). Outpatient ILI visits then dipped down to 3.1% after 2 weeks of decreases before rising again in mid-January, the CDC’s influenza division reported Feb. 8.
Season-high activity also was seen at the state level for the week ending Feb. 2. There were 18 states at level 10 on the CDC’s 1-10 scale of ILI activity, which was up from 16 the week before, and a total of 24 states were in the high range from 8-10, compared with 23 for the previous week. The geographic spread of influenza was reported as widespread in 47 states and Puerto Rico, the CDC said.
Four flu-related pediatric deaths were reported during the week ending Feb. 2, two of which occurred the previous week, which brings the total for the 2018-2019 season to 28, the CDC said.
There were 158 flu-related deaths among all ages during the week ending Jan. 26 – the latest for which such data are available – with reporting almost 75% complete. The previous week saw 177 overall flu deaths, with reporting for that week over 90% complete. During the corresponding weeks of the very severe 2017-2018 flu season, the overall death totals were 1,448 and 1,626, CDC data show.
Influenza activity increased for the third consecutive week and has now reached its highest point for the 2018-2019 flu season, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) hit 4.3% for the week ending Feb. 2, which topped the previous high of 4.0% that was reached in late December (the national baseline rate is 2.2%). Outpatient ILI visits then dipped down to 3.1% after 2 weeks of decreases before rising again in mid-January, the CDC’s influenza division reported Feb. 8.
Season-high activity also was seen at the state level for the week ending Feb. 2. There were 18 states at level 10 on the CDC’s 1-10 scale of ILI activity, which was up from 16 the week before, and a total of 24 states were in the high range from 8-10, compared with 23 for the previous week. The geographic spread of influenza was reported as widespread in 47 states and Puerto Rico, the CDC said.
Four flu-related pediatric deaths were reported during the week ending Feb. 2, two of which occurred the previous week, which brings the total for the 2018-2019 season to 28, the CDC said.
There were 158 flu-related deaths among all ages during the week ending Jan. 26 – the latest for which such data are available – with reporting almost 75% complete. The previous week saw 177 overall flu deaths, with reporting for that week over 90% complete. During the corresponding weeks of the very severe 2017-2018 flu season, the overall death totals were 1,448 and 1,626, CDC data show.
In search of an ear
On our way up north to go backcountry skiing with another couple, we stopped at a roadside restaurant/tavern for lunch. We seated ourselves and, after a long 10 minutes, our waitperson arrived like a tornado, looking frazzled. She offered an apology and the first installment of her tale of woe. Before taking our order, she explained it all began when her car wouldn’t start, and then her day care provider called to say that she was sick and our server would have to find some other arrangement for the day. When our meal finally arrived, it looked appetizing but didn’t quite match our order. Again, our waitperson apologized, adding that it has been a particularly hard week because her husband was out of town and not around to help with her three children.

Had we been dining at a high-end restaurant with a white tablecloth and a candle, we would have considered our server’s behavior unprofessional and off-putting. However, we were in no hurry as the light snow had turned to a ski-unfriendly drizzle. While our original intent had been to simply have lunch, we accepted our role as a sympathetic audience for this unfortunate woman. In fact, we asked a few open-ended questions to help the cathartic process along.
The need to share one’s troubles seems to be a universal human trait. Our server had no illusions that we were going to provide any solutions to her problems. Nor was she seeking any expression of sympathy beyond our patience. However, I’m sure that unburdening herself by telling the story made her feel better, at least temporarily. Hopefully, there would be additional understanding diners to help her through the day.
For many people, the workplace serves as a therapeutic outlet where they can share their troubles and concerns. At times, the whining can be annoying to coworkers but in general, woe sharing is a harmless and valuable perk of having a job. Unless, of course, one’s job is primarily serving the public.
As physicians we are accustomed listening to our patients’ troubles. However, our job is not one of those that affords much opportunity to unburden ourselves of our own concerns. The patients assume that we are the problem solvers and don’t have any of our own. Or, if we do have some troubles, their office visit is not the time for us to share them.

The occasional sharing, such as that we are running late because we’ve had a flat on the way to the office, is harmless and can remind patients that we are human. But one must be careful stay off the slippery slope that leads to unprofessional oversharing.
Without that luxury of a workplace that allows for occasional catharsis, physicians have an additional risk for burnout. There are no easy solutions. Sharing with patients is unprofessional. Our peers are as busy as we are and probably don’t have the time to listen. Or at least they don’t seem to have the time. And then there is that ego-vulnerability issue where we are hesitant to reveal to anyone, be they staff or peers, that we have a soft underbelly.
I don’t have any easy answers to the problem beyond the usual suggestion that, Personally, I have to admit that, when my bad day was the result of an accumulation of minor bumps, I would follow our waitperson’s example and share them selectively with patients whom I deluded myself into believing had the time and concern to listen. It probably was unprofessional, but it made me feel better.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
On our way up north to go backcountry skiing with another couple, we stopped at a roadside restaurant/tavern for lunch. We seated ourselves and, after a long 10 minutes, our waitperson arrived like a tornado, looking frazzled. She offered an apology and the first installment of her tale of woe. Before taking our order, she explained it all began when her car wouldn’t start, and then her day care provider called to say that she was sick and our server would have to find some other arrangement for the day. When our meal finally arrived, it looked appetizing but didn’t quite match our order. Again, our waitperson apologized, adding that it has been a particularly hard week because her husband was out of town and not around to help with her three children.
Had we been dining at a high-end restaurant with a white tablecloth and a candle, we would have considered our server’s behavior unprofessional and off-putting. However, we were in no hurry as the light snow had turned to a ski-unfriendly drizzle. While our original intent had been to simply have lunch, we accepted our role as a sympathetic audience for this unfortunate woman. In fact, we asked a few open-ended questions to help the cathartic process along.
The need to share one’s troubles seems to be a universal human trait. Our server had no illusions that we were going to provide any solutions to her problems. Nor was she seeking any expression of sympathy beyond our patience. However, I’m sure that unburdening herself by telling the story made her feel better, at least temporarily. Hopefully, there would be additional understanding diners to help her through the day.
For many people, the workplace serves as a therapeutic outlet where they can share their troubles and concerns. At times, the whining can be annoying to coworkers but in general, woe sharing is a harmless and valuable perk of having a job. Unless, of course, one’s job is primarily serving the public.
As physicians we are accustomed listening to our patients’ troubles. However, our job is not one of those that affords much opportunity to unburden ourselves of our own concerns. The patients assume that we are the problem solvers and don’t have any of our own. Or, if we do have some troubles, their office visit is not the time for us to share them.
The occasional sharing, such as that we are running late because we’ve had a flat on the way to the office, is harmless and can remind patients that we are human. But one must be careful stay off the slippery slope that leads to unprofessional oversharing.
Without that luxury of a workplace that allows for occasional catharsis, physicians have an additional risk for burnout. There are no easy solutions. Sharing with patients is unprofessional. Our peers are as busy as we are and probably don’t have the time to listen. Or at least they don’t seem to have the time. And then there is that ego-vulnerability issue where we are hesitant to reveal to anyone, be they staff or peers, that we have a soft underbelly.
I don’t have any easy answers to the problem beyond the usual suggestion that, Personally, I have to admit that, when my bad day was the result of an accumulation of minor bumps, I would follow our waitperson’s example and share them selectively with patients whom I deluded myself into believing had the time and concern to listen. It probably was unprofessional, but it made me feel better.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
On our way up north to go backcountry skiing with another couple, we stopped at a roadside restaurant/tavern for lunch. We seated ourselves and, after a long 10 minutes, our waitperson arrived like a tornado, looking frazzled. She offered an apology and the first installment of her tale of woe. Before taking our order, she explained it all began when her car wouldn’t start, and then her day care provider called to say that she was sick and our server would have to find some other arrangement for the day. When our meal finally arrived, it looked appetizing but didn’t quite match our order. Again, our waitperson apologized, adding that it has been a particularly hard week because her husband was out of town and not around to help with her three children.
Had we been dining at a high-end restaurant with a white tablecloth and a candle, we would have considered our server’s behavior unprofessional and off-putting. However, we were in no hurry as the light snow had turned to a ski-unfriendly drizzle. While our original intent had been to simply have lunch, we accepted our role as a sympathetic audience for this unfortunate woman. In fact, we asked a few open-ended questions to help the cathartic process along.
The need to share one’s troubles seems to be a universal human trait. Our server had no illusions that we were going to provide any solutions to her problems. Nor was she seeking any expression of sympathy beyond our patience. However, I’m sure that unburdening herself by telling the story made her feel better, at least temporarily. Hopefully, there would be additional understanding diners to help her through the day.
For many people, the workplace serves as a therapeutic outlet where they can share their troubles and concerns. At times, the whining can be annoying to coworkers but in general, woe sharing is a harmless and valuable perk of having a job. Unless, of course, one’s job is primarily serving the public.
As physicians we are accustomed listening to our patients’ troubles. However, our job is not one of those that affords much opportunity to unburden ourselves of our own concerns. The patients assume that we are the problem solvers and don’t have any of our own. Or, if we do have some troubles, their office visit is not the time for us to share them.
The occasional sharing, such as that we are running late because we’ve had a flat on the way to the office, is harmless and can remind patients that we are human. But one must be careful stay off the slippery slope that leads to unprofessional oversharing.
Without that luxury of a workplace that allows for occasional catharsis, physicians have an additional risk for burnout. There are no easy solutions. Sharing with patients is unprofessional. Our peers are as busy as we are and probably don’t have the time to listen. Or at least they don’t seem to have the time. And then there is that ego-vulnerability issue where we are hesitant to reveal to anyone, be they staff or peers, that we have a soft underbelly.
I don’t have any easy answers to the problem beyond the usual suggestion that, Personally, I have to admit that, when my bad day was the result of an accumulation of minor bumps, I would follow our waitperson’s example and share them selectively with patients whom I deluded myself into believing had the time and concern to listen. It probably was unprofessional, but it made me feel better.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
Woman loses hands and feet after cystectomy
Woman loses hands and feet after cystectomy: $109M award
On November 1, a 45-year-old woman underwent laparoscopic excision of a benign ovarian cyst performed by a minimally invasive gynecologic (MIG) surgeon. After surgery, the patient’s blood pressure (BP) declined. She was given fluids, but her BP remained low. The next day, she became incoherent and her BP could not be stabilized. Twenty-seven hours after surgery, the 5-cm umbilical incision opened while the patient was attempting to stand up from the commode. A large amount of bloody discharge drained.
At 11:00
At 4:30
The patient remained unconscious from the time of the exploratory operation until the end of January. She required additional surgeries to control the bacteria as well as amputation of both hands above the wrists and both feet above the ankles due to gangrene. Because she no longer had an abdominal wall, a skin sac was created to hold her intestines outside of her body. When a fistula developed, a colostomy was performed.
She went to a Maryland hospital for rehab, where she learned to walk with prosthetic feet and to use her prosthetic hands. Currently, she has constant abdominal pain, can walk a short distance, and uses a wheelchair. She requires 24/7 assistance for everyday tasks. She can no longer work and is on disability.
PATIENT’S CLAIM: The patient sued the university health system that employed the MIG surgeon. During the cystectomy, he almost completely transected her small intestine, but did not find the injury during surgery. This allowed bacteria to enter the abdominal cavity, causing sepsis and necrotizing fasciitis. The trauma surgeon referred to the injury as an enterotomy, not a tear.
During the procedure, the surgeon used ADEPT, a solution to prevent the formation of adhesions. The patient’s ObGyn expert concluded that ADEPT created an environment that allowed the necrotizing fasciitis to flourish.
The ICU physicians concluded that the patient was stable enough to be transported for a CT scan, but the surgeon repeatedly delayed the procedure and did not call for a surgical consult until 12 hours later. Had the CT scan or exploratory surgery occurred earlier, the diagnosis would have been discovered, and the bacteria would have been prevented from spreading. She would not have required extensive doses of vasopressors, which increase BP by cutting off blood circulation to the 4 extremities. In this case, use of vasopressors led to gangrene and the subsequent amputations.
Continue to: DEFENDANTS’ DEFENSE...
DEFENDANTS’ DEFENSE: The defendants denied all allegations. The expert witness for the defense opined that the surgeon had only nicked the intestine and that the main injury was a tear that had occurred on its own. The defense also claimed that the surgeon did not call for a CT scan because it would not have shown the source of the patient’s condition.
VERDICT: After 2 trials ended with hung juries, a $109 million Florida verdict was returned against the university health system. Under Florida’s sovereign immunity statute, the patient must seek recovery of all but $100,000 of the award through the Florida legislature in a separate claims bill.
Child has hypoxic brain injury: $7.75M settlement
At 41 weeks’ gestation, a mother presented to the emergency department (ED) for delivery after an unremarkable pregnancy. During the last 90 minutes of labor, fetal heart-rate (FHR) monitoring showed nonreassuring findings. After a vaginal delivery, the infant was found to have a hypoxic brain injury.
PARENT’S CLAIM: Even though nonreassuring FHR monitoring findings occurred, the physicians did not offer cesarean delivery (CD). The pediatrician and ED physician were negligent in failing to provide proper neonatal resuscitation and in recognizing a problem with the infants’ intubation. The delay in delivery and poor resuscitation procedure caused the child’s injury.
DEFENDANTS’ DEFENSE: All allegations were denied. There was no deviation from the standard of care.
VERDICT: A $7.75 million Massachusetts settlement was reached.
Kidney failed after hysterectomy
A 46-year-old woman underwent a hysterectomy performed by her ObGyn. Surgery went well but the patient continued to report symptoms. A year later, she underwent an oophorectomy. Two years later, the patient reported blood in her urine and underwent a computed tomography scan, which revealed an obstructed left ureter that had caused injury to the left kidney. Seven months later, the kidney was removed.
PATIENT’S CLAIM: Her kidney loss was a direct result of the ObGyn’s initial surgical procedure. He had placed several clips near the ureter and did not verify their position or protect the ureter. He also failed to address her reported symptoms in a timely manner.
PHYSICIAN’S DEFENSE: The damage to the ureter is a known risk of hysterectomy and oophorectomy. The obstruction developed over time, not as an immediate result of the surgery.
VERDICT: A Kentucky defense verdict was returned.
History of shoulder dystocia, Erb's palsy: $1.2M settlement
An obese mother was admitted to the hospital at 39 weeks’ gestation with signs of labor. She requested a CD and was advised that she had progressed too far for that to be an option, and that vaginal delivery would be safe. During the second stage of labor, shoulder dystocia was encountered. The ObGyn made several attempts to deliver using downward traction, but was unsuccessful. A second ObGyn swept the shoulder with an internal maneuver of his hand and delivered the baby. The child has a severe brachial plexus injury at multiple spinal levels resulting in Erb’s palsy.
PARENT’S CLAIM: A CD should have been performed. The first ObGyn failed to provide a CD and repeatedly applied excessive downward traction, causing the infant’s injury.
Continue to: PHYSICIAN’S DEFENSE...
PHYSICIAN’S DEFENSE: Shoulder dystocia is unpredictable and an unpreventable obstetric emergency. The ObGyn used proper maneuvers to release the shoulder dystocia.
VERDICT: A $1.2 million Virginia settlement was reached.
Ureter injured during hysterectomy
When a patient was found to have multiple, symptomatic fibroids and an enlarged uterus, her gynecologist suggested a total laparoscopic hysterectomy. During the procedure, when he inspected the pelvis and found multiple fibroids in and around the uterus, the gynecologist converted to a supracervical hysterectomy. Surgery was difficult because of a large myoma on the right broad ligament.
The patient tolerated surgery well and was released home the next day. At follow-up one week later, she had no signs or symptoms of ureter injury. Later that same evening, she experienced sharp flank pain and nausea. When she called the gynecologist, he sent her to the emergency department. A computed tomography scan showed extravasation of the right ureter. She underwent months of stent placements and replacements, nephrostomies, and ultimately ureteral reimplantation surgery.
PATIENT’S CLAIM: The gynecologist caused a thermal injury to her right ureter during the hysterectomy by misusing an electrocautery device. There was a delay in timely diagnosis postsurgery.
PHYSICIAN’S DEFENSE: The gynecologist contended that he employed proper surgical technique, and that he reacted properly when the patient reported the pain.
VERDICT: A Virginia defense verdict was returned.
Woman loses hands and feet after cystectomy: $109M award
On November 1, a 45-year-old woman underwent laparoscopic excision of a benign ovarian cyst performed by a minimally invasive gynecologic (MIG) surgeon. After surgery, the patient’s blood pressure (BP) declined. She was given fluids, but her BP remained low. The next day, she became incoherent and her BP could not be stabilized. Twenty-seven hours after surgery, the 5-cm umbilical incision opened while the patient was attempting to stand up from the commode. A large amount of bloody discharge drained.
At 11:00
At 4:30
The patient remained unconscious from the time of the exploratory operation until the end of January. She required additional surgeries to control the bacteria as well as amputation of both hands above the wrists and both feet above the ankles due to gangrene. Because she no longer had an abdominal wall, a skin sac was created to hold her intestines outside of her body. When a fistula developed, a colostomy was performed.
She went to a Maryland hospital for rehab, where she learned to walk with prosthetic feet and to use her prosthetic hands. Currently, she has constant abdominal pain, can walk a short distance, and uses a wheelchair. She requires 24/7 assistance for everyday tasks. She can no longer work and is on disability.
PATIENT’S CLAIM: The patient sued the university health system that employed the MIG surgeon. During the cystectomy, he almost completely transected her small intestine, but did not find the injury during surgery. This allowed bacteria to enter the abdominal cavity, causing sepsis and necrotizing fasciitis. The trauma surgeon referred to the injury as an enterotomy, not a tear.
During the procedure, the surgeon used ADEPT, a solution to prevent the formation of adhesions. The patient’s ObGyn expert concluded that ADEPT created an environment that allowed the necrotizing fasciitis to flourish.
The ICU physicians concluded that the patient was stable enough to be transported for a CT scan, but the surgeon repeatedly delayed the procedure and did not call for a surgical consult until 12 hours later. Had the CT scan or exploratory surgery occurred earlier, the diagnosis would have been discovered, and the bacteria would have been prevented from spreading. She would not have required extensive doses of vasopressors, which increase BP by cutting off blood circulation to the 4 extremities. In this case, use of vasopressors led to gangrene and the subsequent amputations.
Continue to: DEFENDANTS’ DEFENSE...
DEFENDANTS’ DEFENSE: The defendants denied all allegations. The expert witness for the defense opined that the surgeon had only nicked the intestine and that the main injury was a tear that had occurred on its own. The defense also claimed that the surgeon did not call for a CT scan because it would not have shown the source of the patient’s condition.
VERDICT: After 2 trials ended with hung juries, a $109 million Florida verdict was returned against the university health system. Under Florida’s sovereign immunity statute, the patient must seek recovery of all but $100,000 of the award through the Florida legislature in a separate claims bill.
Child has hypoxic brain injury: $7.75M settlement
At 41 weeks’ gestation, a mother presented to the emergency department (ED) for delivery after an unremarkable pregnancy. During the last 90 minutes of labor, fetal heart-rate (FHR) monitoring showed nonreassuring findings. After a vaginal delivery, the infant was found to have a hypoxic brain injury.
PARENT’S CLAIM: Even though nonreassuring FHR monitoring findings occurred, the physicians did not offer cesarean delivery (CD). The pediatrician and ED physician were negligent in failing to provide proper neonatal resuscitation and in recognizing a problem with the infants’ intubation. The delay in delivery and poor resuscitation procedure caused the child’s injury.
DEFENDANTS’ DEFENSE: All allegations were denied. There was no deviation from the standard of care.
VERDICT: A $7.75 million Massachusetts settlement was reached.
Kidney failed after hysterectomy
A 46-year-old woman underwent a hysterectomy performed by her ObGyn. Surgery went well but the patient continued to report symptoms. A year later, she underwent an oophorectomy. Two years later, the patient reported blood in her urine and underwent a computed tomography scan, which revealed an obstructed left ureter that had caused injury to the left kidney. Seven months later, the kidney was removed.
PATIENT’S CLAIM: Her kidney loss was a direct result of the ObGyn’s initial surgical procedure. He had placed several clips near the ureter and did not verify their position or protect the ureter. He also failed to address her reported symptoms in a timely manner.
PHYSICIAN’S DEFENSE: The damage to the ureter is a known risk of hysterectomy and oophorectomy. The obstruction developed over time, not as an immediate result of the surgery.
VERDICT: A Kentucky defense verdict was returned.
History of shoulder dystocia, Erb's palsy: $1.2M settlement
An obese mother was admitted to the hospital at 39 weeks’ gestation with signs of labor. She requested a CD and was advised that she had progressed too far for that to be an option, and that vaginal delivery would be safe. During the second stage of labor, shoulder dystocia was encountered. The ObGyn made several attempts to deliver using downward traction, but was unsuccessful. A second ObGyn swept the shoulder with an internal maneuver of his hand and delivered the baby. The child has a severe brachial plexus injury at multiple spinal levels resulting in Erb’s palsy.
PARENT’S CLAIM: A CD should have been performed. The first ObGyn failed to provide a CD and repeatedly applied excessive downward traction, causing the infant’s injury.
Continue to: PHYSICIAN’S DEFENSE...
PHYSICIAN’S DEFENSE: Shoulder dystocia is unpredictable and an unpreventable obstetric emergency. The ObGyn used proper maneuvers to release the shoulder dystocia.
VERDICT: A $1.2 million Virginia settlement was reached.
Ureter injured during hysterectomy
When a patient was found to have multiple, symptomatic fibroids and an enlarged uterus, her gynecologist suggested a total laparoscopic hysterectomy. During the procedure, when he inspected the pelvis and found multiple fibroids in and around the uterus, the gynecologist converted to a supracervical hysterectomy. Surgery was difficult because of a large myoma on the right broad ligament.
The patient tolerated surgery well and was released home the next day. At follow-up one week later, she had no signs or symptoms of ureter injury. Later that same evening, she experienced sharp flank pain and nausea. When she called the gynecologist, he sent her to the emergency department. A computed tomography scan showed extravasation of the right ureter. She underwent months of stent placements and replacements, nephrostomies, and ultimately ureteral reimplantation surgery.
PATIENT’S CLAIM: The gynecologist caused a thermal injury to her right ureter during the hysterectomy by misusing an electrocautery device. There was a delay in timely diagnosis postsurgery.
PHYSICIAN’S DEFENSE: The gynecologist contended that he employed proper surgical technique, and that he reacted properly when the patient reported the pain.
VERDICT: A Virginia defense verdict was returned.
Woman loses hands and feet after cystectomy: $109M award
On November 1, a 45-year-old woman underwent laparoscopic excision of a benign ovarian cyst performed by a minimally invasive gynecologic (MIG) surgeon. After surgery, the patient’s blood pressure (BP) declined. She was given fluids, but her BP remained low. The next day, she became incoherent and her BP could not be stabilized. Twenty-seven hours after surgery, the 5-cm umbilical incision opened while the patient was attempting to stand up from the commode. A large amount of bloody discharge drained.
At 11:00
At 4:30
The patient remained unconscious from the time of the exploratory operation until the end of January. She required additional surgeries to control the bacteria as well as amputation of both hands above the wrists and both feet above the ankles due to gangrene. Because she no longer had an abdominal wall, a skin sac was created to hold her intestines outside of her body. When a fistula developed, a colostomy was performed.
She went to a Maryland hospital for rehab, where she learned to walk with prosthetic feet and to use her prosthetic hands. Currently, she has constant abdominal pain, can walk a short distance, and uses a wheelchair. She requires 24/7 assistance for everyday tasks. She can no longer work and is on disability.
PATIENT’S CLAIM: The patient sued the university health system that employed the MIG surgeon. During the cystectomy, he almost completely transected her small intestine, but did not find the injury during surgery. This allowed bacteria to enter the abdominal cavity, causing sepsis and necrotizing fasciitis. The trauma surgeon referred to the injury as an enterotomy, not a tear.
During the procedure, the surgeon used ADEPT, a solution to prevent the formation of adhesions. The patient’s ObGyn expert concluded that ADEPT created an environment that allowed the necrotizing fasciitis to flourish.
The ICU physicians concluded that the patient was stable enough to be transported for a CT scan, but the surgeon repeatedly delayed the procedure and did not call for a surgical consult until 12 hours later. Had the CT scan or exploratory surgery occurred earlier, the diagnosis would have been discovered, and the bacteria would have been prevented from spreading. She would not have required extensive doses of vasopressors, which increase BP by cutting off blood circulation to the 4 extremities. In this case, use of vasopressors led to gangrene and the subsequent amputations.
Continue to: DEFENDANTS’ DEFENSE...
DEFENDANTS’ DEFENSE: The defendants denied all allegations. The expert witness for the defense opined that the surgeon had only nicked the intestine and that the main injury was a tear that had occurred on its own. The defense also claimed that the surgeon did not call for a CT scan because it would not have shown the source of the patient’s condition.
VERDICT: After 2 trials ended with hung juries, a $109 million Florida verdict was returned against the university health system. Under Florida’s sovereign immunity statute, the patient must seek recovery of all but $100,000 of the award through the Florida legislature in a separate claims bill.
Child has hypoxic brain injury: $7.75M settlement
At 41 weeks’ gestation, a mother presented to the emergency department (ED) for delivery after an unremarkable pregnancy. During the last 90 minutes of labor, fetal heart-rate (FHR) monitoring showed nonreassuring findings. After a vaginal delivery, the infant was found to have a hypoxic brain injury.
PARENT’S CLAIM: Even though nonreassuring FHR monitoring findings occurred, the physicians did not offer cesarean delivery (CD). The pediatrician and ED physician were negligent in failing to provide proper neonatal resuscitation and in recognizing a problem with the infants’ intubation. The delay in delivery and poor resuscitation procedure caused the child’s injury.
DEFENDANTS’ DEFENSE: All allegations were denied. There was no deviation from the standard of care.
VERDICT: A $7.75 million Massachusetts settlement was reached.
Kidney failed after hysterectomy
A 46-year-old woman underwent a hysterectomy performed by her ObGyn. Surgery went well but the patient continued to report symptoms. A year later, she underwent an oophorectomy. Two years later, the patient reported blood in her urine and underwent a computed tomography scan, which revealed an obstructed left ureter that had caused injury to the left kidney. Seven months later, the kidney was removed.
PATIENT’S CLAIM: Her kidney loss was a direct result of the ObGyn’s initial surgical procedure. He had placed several clips near the ureter and did not verify their position or protect the ureter. He also failed to address her reported symptoms in a timely manner.
PHYSICIAN’S DEFENSE: The damage to the ureter is a known risk of hysterectomy and oophorectomy. The obstruction developed over time, not as an immediate result of the surgery.
VERDICT: A Kentucky defense verdict was returned.
History of shoulder dystocia, Erb's palsy: $1.2M settlement
An obese mother was admitted to the hospital at 39 weeks’ gestation with signs of labor. She requested a CD and was advised that she had progressed too far for that to be an option, and that vaginal delivery would be safe. During the second stage of labor, shoulder dystocia was encountered. The ObGyn made several attempts to deliver using downward traction, but was unsuccessful. A second ObGyn swept the shoulder with an internal maneuver of his hand and delivered the baby. The child has a severe brachial plexus injury at multiple spinal levels resulting in Erb’s palsy.
PARENT’S CLAIM: A CD should have been performed. The first ObGyn failed to provide a CD and repeatedly applied excessive downward traction, causing the infant’s injury.
Continue to: PHYSICIAN’S DEFENSE...
PHYSICIAN’S DEFENSE: Shoulder dystocia is unpredictable and an unpreventable obstetric emergency. The ObGyn used proper maneuvers to release the shoulder dystocia.
VERDICT: A $1.2 million Virginia settlement was reached.
Ureter injured during hysterectomy
When a patient was found to have multiple, symptomatic fibroids and an enlarged uterus, her gynecologist suggested a total laparoscopic hysterectomy. During the procedure, when he inspected the pelvis and found multiple fibroids in and around the uterus, the gynecologist converted to a supracervical hysterectomy. Surgery was difficult because of a large myoma on the right broad ligament.
The patient tolerated surgery well and was released home the next day. At follow-up one week later, she had no signs or symptoms of ureter injury. Later that same evening, she experienced sharp flank pain and nausea. When she called the gynecologist, he sent her to the emergency department. A computed tomography scan showed extravasation of the right ureter. She underwent months of stent placements and replacements, nephrostomies, and ultimately ureteral reimplantation surgery.
PATIENT’S CLAIM: The gynecologist caused a thermal injury to her right ureter during the hysterectomy by misusing an electrocautery device. There was a delay in timely diagnosis postsurgery.
PHYSICIAN’S DEFENSE: The gynecologist contended that he employed proper surgical technique, and that he reacted properly when the patient reported the pain.
VERDICT: A Virginia defense verdict was returned.
Marijuana smoking is an independent risk factor for lung disease in HIV+
Long-term marijuana smoking was associated with lung disease in HIV-infected (HIV+) but not HIV uninfected (HIV–) men who have sex with men (MSM), according to the results of a large, prospective cohort study.

“There were no significant interactions between marijuana and tobacco smoking in any multivariable model tested for HIV+ participants, indicating independent effects of these factors,” wrote David R. Lorenz, PhD, of the Dana-Farber Cancer Institute, Boston, and his colleagues.
These findings are especially important given that the proportion of HIV+ individuals who frequently smoke marijuana is higher than in the general population in the United States, and has increased in recent years, according to the report, published online in EClinicalMedicine.
The study examined 2,704 MSM who met eligibility criteria (1,352 HIV+ and 1,352 HIV− individuals), with a median age of 44 years at baseline and a median follow-up of 10.5 years. A total of 27% of HIV+ participants reported daily or weekly marijuana smoking for 1 year or more during follow-up, compared with 18% of the HIV− participants.
HIV+ participants who smoked marijuana were more likely to report one or more pulmonary diagnoses, versus nonsmoking HIV+ individuals during follow-up (41.0% vs. 30.0% infectious, and 24.8% vs. 19.0% noninfectious), according to the authors. In contrast, there was no association between marijuana smoking and either an infectious or noninfectious pulmonary diagnosis among HIV− participants (24.2% vs. 20.9%, and 14.8% vs. 17.7%, respectively).
For HIV+ individuals, each 10 days/month increase in marijuana smoking in the prior 2-year period was found to be associated with a 6% increased risk of infectious pulmonary diagnosis (hazard risk 1.06 [95% confidence interval 1.00-1.11]; P = .041). Overall, they found that from the 53,000 person-visits in the study, marijuana smoking was associated with increased risk of both infectious and noninfectious pulmonary diagnoses among the 1,352 HIV-infected participants independent of CD4 count, antiretroviral therapy (ART) adherence, and demographic factors as well.
In particular, viral suppression did not seem to interfere with this association between marijuana smoking and infectious pulmonary diagnoses, as it remained significant in models restricted to those person-visits with suppressed HIV viral load (HR 1.41 [1.03-1.91], P = .029).
The authors suggested that HIV-specific factors such as lung immune cell depletion and dysfunction, persistent immune cell activation, systemic inflammation, respiratory microbiome alterations, and oxidative stress, or a combination of these effects, may interact with the alveolar macrophage dysfunction seen in both humans and mouse models exposed to marijuana smoke. Thus, “a potential additive risk of marijuana smoking and HIV disease may explain the increased prevalence of infectious pulmonary diagnoses in our adjusted analyses,” Dr. Lorenz and his colleagues stated.
“These findings suggest that marijuana smoking is a modifiable risk factor that healthcare providers should consider when seeking to prevent or treat lung disease in people infected with HIV, particularly those with other known risk factors including heavy tobacco smoking, and low CD4 T cell count or advanced HIV disease,” they concluded.
The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
SOURCE: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
Long-term marijuana smoking was associated with lung disease in HIV-infected (HIV+) but not HIV uninfected (HIV–) men who have sex with men (MSM), according to the results of a large, prospective cohort study.
“There were no significant interactions between marijuana and tobacco smoking in any multivariable model tested for HIV+ participants, indicating independent effects of these factors,” wrote David R. Lorenz, PhD, of the Dana-Farber Cancer Institute, Boston, and his colleagues.
These findings are especially important given that the proportion of HIV+ individuals who frequently smoke marijuana is higher than in the general population in the United States, and has increased in recent years, according to the report, published online in EClinicalMedicine.
The study examined 2,704 MSM who met eligibility criteria (1,352 HIV+ and 1,352 HIV− individuals), with a median age of 44 years at baseline and a median follow-up of 10.5 years. A total of 27% of HIV+ participants reported daily or weekly marijuana smoking for 1 year or more during follow-up, compared with 18% of the HIV− participants.
HIV+ participants who smoked marijuana were more likely to report one or more pulmonary diagnoses, versus nonsmoking HIV+ individuals during follow-up (41.0% vs. 30.0% infectious, and 24.8% vs. 19.0% noninfectious), according to the authors. In contrast, there was no association between marijuana smoking and either an infectious or noninfectious pulmonary diagnosis among HIV− participants (24.2% vs. 20.9%, and 14.8% vs. 17.7%, respectively).
For HIV+ individuals, each 10 days/month increase in marijuana smoking in the prior 2-year period was found to be associated with a 6% increased risk of infectious pulmonary diagnosis (hazard risk 1.06 [95% confidence interval 1.00-1.11]; P = .041). Overall, they found that from the 53,000 person-visits in the study, marijuana smoking was associated with increased risk of both infectious and noninfectious pulmonary diagnoses among the 1,352 HIV-infected participants independent of CD4 count, antiretroviral therapy (ART) adherence, and demographic factors as well.
In particular, viral suppression did not seem to interfere with this association between marijuana smoking and infectious pulmonary diagnoses, as it remained significant in models restricted to those person-visits with suppressed HIV viral load (HR 1.41 [1.03-1.91], P = .029).
The authors suggested that HIV-specific factors such as lung immune cell depletion and dysfunction, persistent immune cell activation, systemic inflammation, respiratory microbiome alterations, and oxidative stress, or a combination of these effects, may interact with the alveolar macrophage dysfunction seen in both humans and mouse models exposed to marijuana smoke. Thus, “a potential additive risk of marijuana smoking and HIV disease may explain the increased prevalence of infectious pulmonary diagnoses in our adjusted analyses,” Dr. Lorenz and his colleagues stated.
“These findings suggest that marijuana smoking is a modifiable risk factor that healthcare providers should consider when seeking to prevent or treat lung disease in people infected with HIV, particularly those with other known risk factors including heavy tobacco smoking, and low CD4 T cell count or advanced HIV disease,” they concluded.
The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
SOURCE: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
Long-term marijuana smoking was associated with lung disease in HIV-infected (HIV+) but not HIV uninfected (HIV–) men who have sex with men (MSM), according to the results of a large, prospective cohort study.
“There were no significant interactions between marijuana and tobacco smoking in any multivariable model tested for HIV+ participants, indicating independent effects of these factors,” wrote David R. Lorenz, PhD, of the Dana-Farber Cancer Institute, Boston, and his colleagues.
These findings are especially important given that the proportion of HIV+ individuals who frequently smoke marijuana is higher than in the general population in the United States, and has increased in recent years, according to the report, published online in EClinicalMedicine.
The study examined 2,704 MSM who met eligibility criteria (1,352 HIV+ and 1,352 HIV− individuals), with a median age of 44 years at baseline and a median follow-up of 10.5 years. A total of 27% of HIV+ participants reported daily or weekly marijuana smoking for 1 year or more during follow-up, compared with 18% of the HIV− participants.
HIV+ participants who smoked marijuana were more likely to report one or more pulmonary diagnoses, versus nonsmoking HIV+ individuals during follow-up (41.0% vs. 30.0% infectious, and 24.8% vs. 19.0% noninfectious), according to the authors. In contrast, there was no association between marijuana smoking and either an infectious or noninfectious pulmonary diagnosis among HIV− participants (24.2% vs. 20.9%, and 14.8% vs. 17.7%, respectively).
For HIV+ individuals, each 10 days/month increase in marijuana smoking in the prior 2-year period was found to be associated with a 6% increased risk of infectious pulmonary diagnosis (hazard risk 1.06 [95% confidence interval 1.00-1.11]; P = .041). Overall, they found that from the 53,000 person-visits in the study, marijuana smoking was associated with increased risk of both infectious and noninfectious pulmonary diagnoses among the 1,352 HIV-infected participants independent of CD4 count, antiretroviral therapy (ART) adherence, and demographic factors as well.
In particular, viral suppression did not seem to interfere with this association between marijuana smoking and infectious pulmonary diagnoses, as it remained significant in models restricted to those person-visits with suppressed HIV viral load (HR 1.41 [1.03-1.91], P = .029).
The authors suggested that HIV-specific factors such as lung immune cell depletion and dysfunction, persistent immune cell activation, systemic inflammation, respiratory microbiome alterations, and oxidative stress, or a combination of these effects, may interact with the alveolar macrophage dysfunction seen in both humans and mouse models exposed to marijuana smoke. Thus, “a potential additive risk of marijuana smoking and HIV disease may explain the increased prevalence of infectious pulmonary diagnoses in our adjusted analyses,” Dr. Lorenz and his colleagues stated.
“These findings suggest that marijuana smoking is a modifiable risk factor that healthcare providers should consider when seeking to prevent or treat lung disease in people infected with HIV, particularly those with other known risk factors including heavy tobacco smoking, and low CD4 T cell count or advanced HIV disease,” they concluded.
The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
SOURCE: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
FROM ECLINICALMEDICINE
Key clinical point: HIV+ but not HIV– marijuana smokers had an increased rate of pulmonary diagnoses.
Major finding: HIV+ marijuana smokers were more likely to report one or more infectious or noninfectious pulmonary diagnoses, compared with nonsmoking HIV+ individuals (41.0% vs. 30.0%, and 24.8% vs. 19.0%, respectively).
Study details: A prospective cohort study of 1,352 HIV+ vs. 1,352 HIV– men who have sex with men.
Disclosures: The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
Source: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
Supreme Court halts Louisiana abortion law from taking effect
The U.S. Supreme Court has temporarily barred a Louisiana law that would require stricter requirements for physicians who provide abortion care, the first abortion-related decision for the current conservative-leaning high court.

In a Feb. 7, 2019, order, Supreme Court justices stopped the law from moving forward until they can decide whether to accept the case for oral argument. The law in question would require Louisiana physicians who perform abortions to have admitting privileges at a hospital within 30 miles of the clinic where they offer abortion care.
A group of health professionals sued over the Louisiana statute after it was enacted in 2014, arguing that the requirement was unconstitutional because it placed an undue burden on women seeking abortions. A federal court agreed, concluding that the law would leave a significant number of Louisiana women unable to get an abortion. The state appealed to the 5th U.S. Circuit Court of Appeals, which reversed the decision in January 2019. The physician plaintiffs then urged the Supreme Court to stop the law, scheduled to take effect on Feb. 4 while the case continued through the courts. The health professionals argue that no physicians in Louisiana would be available to perform abortions after 17 weeks of pregnancy if the law proceeds and that only one physician in the state would be available to provide an abortion in the earlier stages of pregnancy. Attorneys for the state countered that health providers are overestimating the law’s effect and requested that the measure be allowed to go forward.
In a Feb. 1 order, the Supreme Court provided the plaintiffs a short stay while they reviewed briefs in the case. Then, in a 5-4 decision on Feb. 7, the majority court halted the legal challenge indefinitely until they can decide whether to take up the case.
Four justices – Clarence Thomas, Samuel Alito, Neil Gorsuch, and Brett Kavanaugh – dissented from the majority, writing that they would have allowed Louisiana to enforce the law. Chief Justice John Roberts joined the high court’s four more liberal justices in stopping the law’s enactment.
The plaintiffs’ petition to the Supreme Court is due in April. If the case is accepted, oral arguments would likely be scheduled for fall 2019 or winter 2020, according to court analysts.
The U.S. Supreme Court has temporarily barred a Louisiana law that would require stricter requirements for physicians who provide abortion care, the first abortion-related decision for the current conservative-leaning high court.
In a Feb. 7, 2019, order, Supreme Court justices stopped the law from moving forward until they can decide whether to accept the case for oral argument. The law in question would require Louisiana physicians who perform abortions to have admitting privileges at a hospital within 30 miles of the clinic where they offer abortion care.
A group of health professionals sued over the Louisiana statute after it was enacted in 2014, arguing that the requirement was unconstitutional because it placed an undue burden on women seeking abortions. A federal court agreed, concluding that the law would leave a significant number of Louisiana women unable to get an abortion. The state appealed to the 5th U.S. Circuit Court of Appeals, which reversed the decision in January 2019. The physician plaintiffs then urged the Supreme Court to stop the law, scheduled to take effect on Feb. 4 while the case continued through the courts. The health professionals argue that no physicians in Louisiana would be available to perform abortions after 17 weeks of pregnancy if the law proceeds and that only one physician in the state would be available to provide an abortion in the earlier stages of pregnancy. Attorneys for the state countered that health providers are overestimating the law’s effect and requested that the measure be allowed to go forward.
In a Feb. 1 order, the Supreme Court provided the plaintiffs a short stay while they reviewed briefs in the case. Then, in a 5-4 decision on Feb. 7, the majority court halted the legal challenge indefinitely until they can decide whether to take up the case.
Four justices – Clarence Thomas, Samuel Alito, Neil Gorsuch, and Brett Kavanaugh – dissented from the majority, writing that they would have allowed Louisiana to enforce the law. Chief Justice John Roberts joined the high court’s four more liberal justices in stopping the law’s enactment.
The plaintiffs’ petition to the Supreme Court is due in April. If the case is accepted, oral arguments would likely be scheduled for fall 2019 or winter 2020, according to court analysts.
The U.S. Supreme Court has temporarily barred a Louisiana law that would require stricter requirements for physicians who provide abortion care, the first abortion-related decision for the current conservative-leaning high court.
In a Feb. 7, 2019, order, Supreme Court justices stopped the law from moving forward until they can decide whether to accept the case for oral argument. The law in question would require Louisiana physicians who perform abortions to have admitting privileges at a hospital within 30 miles of the clinic where they offer abortion care.
A group of health professionals sued over the Louisiana statute after it was enacted in 2014, arguing that the requirement was unconstitutional because it placed an undue burden on women seeking abortions. A federal court agreed, concluding that the law would leave a significant number of Louisiana women unable to get an abortion. The state appealed to the 5th U.S. Circuit Court of Appeals, which reversed the decision in January 2019. The physician plaintiffs then urged the Supreme Court to stop the law, scheduled to take effect on Feb. 4 while the case continued through the courts. The health professionals argue that no physicians in Louisiana would be available to perform abortions after 17 weeks of pregnancy if the law proceeds and that only one physician in the state would be available to provide an abortion in the earlier stages of pregnancy. Attorneys for the state countered that health providers are overestimating the law’s effect and requested that the measure be allowed to go forward.
In a Feb. 1 order, the Supreme Court provided the plaintiffs a short stay while they reviewed briefs in the case. Then, in a 5-4 decision on Feb. 7, the majority court halted the legal challenge indefinitely until they can decide whether to take up the case.
Four justices – Clarence Thomas, Samuel Alito, Neil Gorsuch, and Brett Kavanaugh – dissented from the majority, writing that they would have allowed Louisiana to enforce the law. Chief Justice John Roberts joined the high court’s four more liberal justices in stopping the law’s enactment.
The plaintiffs’ petition to the Supreme Court is due in April. If the case is accepted, oral arguments would likely be scheduled for fall 2019 or winter 2020, according to court analysts.
What is your diagnosis?
It most commonly affects young girls. The pathogenesis of LAHS is thought to involve a sporadic, autosomal dominant mutation that leads to a defect between the hair cuticle and the inner root sheath.1 This defect results in the hair being poorly anchored to the scalp, and therefore easily and painlessly plucked or lost during normal hair care.

The classic presentation of LAHS is that of hair thinning and hair that may be unruly and/or lackluster; the hair rarely, if ever, requires cutting.2 The key feature is the ability to easily and painlessly pluck hairs from the patient’s scalp. The affected area is limited to the scalp, and loss of eyebrows, eyelashes, and body hair should not be seen.
Diagnosis and consideration of the differential
The diagnosis of LAHS can in some cases be made on history and physical exam alone. Patients with LAHS typically will show hair thinning with or without dullness or unruliness. They lack evidence of scalp inflammation, such as erythema, scale, pruritus, and pain. Areas of hair thinning or aberration are typically not well demarcated, and there are typically not areas of complete hair loss. There is no scarring or atrophy of the scalp itself.

Diagnostic tests include the “hair pull test,” as well as trichogram testing. In the “hair pull test” a provider grasps a set of hair at the proximal shaft near the scalp. The traction applied should result in the painless and easy extraction of more than 10% of grasped hairs in a patient with LAHS. Removal of less than 10% of hair is a normal finding, as patients without LAHS typically have about 10% of their scalp hair in the telogen phase at any given time, which would result in removal during the hair pull test.3 In trichography, plucked hairs are examined under magnification, with or without the use of selective dyes. Cinnamaldehyde is a dye that stains citrulline, which is abundant in the inner root sheath, and can be a tool in identifying its presence and/or aberrations.4 A trichogram of the pulled hairs in a patient with LAHS may classically show ruffled appearance of the cuticle, misshapen anagen hair bulbs, and absence of the inner root sheath.5 Examination under magnification also allows providers to better identify telogen versus anagen hairs, which aids in the diagnosis. By carefully considering the patient history, physical exam, and results of additional hair tests, providers can make the diagnosis of LAHS and avoid unnecessary blood work and invasive procedures like scalp biopsies.
The differential diagnosis of hair loss frequently includes alopecia areata. However, in alopecia areata, patients typically have sharply demarcated areas of hair loss, which may involve the eyebrows, eyelids, and body hairs. In alopecia areata, providers may be able to identify the “exclamation point sign” in which the hair shaft thins proximally, leading to the appearance of more pigmented, thicker hairs floating above the scalp.
Telogen effluvium is a condition in which a medical illness or stress, such as systemic illness, surgery, severe emotional distress, childbirth, dietary changes, or another traumatic event, causes a disruption in the natural cycle of hair growth such that the percentage of hairs in the telogen phase increases from about 10% to up to 70%.6 Unlike in LAHS, in which shed hairs are in the anagen phase, the hair that is shed in telogen effluvium is in the telogen phase and will have a different appearance when magnified.
Anagen effluvium, loss of hairs in their growing phase, is typically associated with chemotherapy. The hairs become broken and fractured at the shaft leading to breakage at different points throughout the scalp. Affected areas can include the eyebrows, eyelashes, and body hair. In the absence of a history of administration of a chemotherapy agent (or other drug known to trigger hair loss), the diagnosis of anagen effluvium should not be made.
Patients with trichotillosis (also known as trichotillomania) present with areas of hair loss caused by intentional or subconscious hair pulling. It is considered a psychological condition that can be associated with obsessive compulsive disorder, although the presence of a secondary psychological diagnosis is not required. Providers may see irregular geometric shapes of hair loss, and on close inspection see broken hair shafts of different lengths. Patients most often pull hair from their scalps (over 70% of patients), but also can pull eyelashes, eyebrow hairs, and pubic hairs.7
Treatment
LAHS is self-limited and does not necessitate treatment. However, if patients or parents feel there is significant disease burden, perhaps with poor effects on quality of life or with psychosocial impairment, treatment with minoxidil 5% solution has been studied with some success reported in the literature.1,8,9
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Natsis and Dr. Eichenfield had no relevant financial disclosures. Email them at [email protected].
References
1. Arch Dermatol. 2002;138(4):501-6.
2. Int J Trichology. 2010;2(2):96-100.
3. Pediatric Dermatol. 2016:33(50):507-10.
4. Dermatol Clin. 1986;14:745-51.
5. Arch Dermatol. 2009;145(10):1123-8.
6. J Clin Diagn Res. 2015;9(9):WE01-3.
7. Am J Psychiatry. 2016;173(9):868-74.
8. Australas J Dermatol. 2018;59:e286-e287.
9. Pediatr Dermatol. 2014;31:389-90.
It most commonly affects young girls. The pathogenesis of LAHS is thought to involve a sporadic, autosomal dominant mutation that leads to a defect between the hair cuticle and the inner root sheath.1 This defect results in the hair being poorly anchored to the scalp, and therefore easily and painlessly plucked or lost during normal hair care.
The classic presentation of LAHS is that of hair thinning and hair that may be unruly and/or lackluster; the hair rarely, if ever, requires cutting.2 The key feature is the ability to easily and painlessly pluck hairs from the patient’s scalp. The affected area is limited to the scalp, and loss of eyebrows, eyelashes, and body hair should not be seen.
Diagnosis and consideration of the differential
The diagnosis of LAHS can in some cases be made on history and physical exam alone. Patients with LAHS typically will show hair thinning with or without dullness or unruliness. They lack evidence of scalp inflammation, such as erythema, scale, pruritus, and pain. Areas of hair thinning or aberration are typically not well demarcated, and there are typically not areas of complete hair loss. There is no scarring or atrophy of the scalp itself.
Diagnostic tests include the “hair pull test,” as well as trichogram testing. In the “hair pull test” a provider grasps a set of hair at the proximal shaft near the scalp. The traction applied should result in the painless and easy extraction of more than 10% of grasped hairs in a patient with LAHS. Removal of less than 10% of hair is a normal finding, as patients without LAHS typically have about 10% of their scalp hair in the telogen phase at any given time, which would result in removal during the hair pull test.3 In trichography, plucked hairs are examined under magnification, with or without the use of selective dyes. Cinnamaldehyde is a dye that stains citrulline, which is abundant in the inner root sheath, and can be a tool in identifying its presence and/or aberrations.4 A trichogram of the pulled hairs in a patient with LAHS may classically show ruffled appearance of the cuticle, misshapen anagen hair bulbs, and absence of the inner root sheath.5 Examination under magnification also allows providers to better identify telogen versus anagen hairs, which aids in the diagnosis. By carefully considering the patient history, physical exam, and results of additional hair tests, providers can make the diagnosis of LAHS and avoid unnecessary blood work and invasive procedures like scalp biopsies.
The differential diagnosis of hair loss frequently includes alopecia areata. However, in alopecia areata, patients typically have sharply demarcated areas of hair loss, which may involve the eyebrows, eyelids, and body hairs. In alopecia areata, providers may be able to identify the “exclamation point sign” in which the hair shaft thins proximally, leading to the appearance of more pigmented, thicker hairs floating above the scalp.
Telogen effluvium is a condition in which a medical illness or stress, such as systemic illness, surgery, severe emotional distress, childbirth, dietary changes, or another traumatic event, causes a disruption in the natural cycle of hair growth such that the percentage of hairs in the telogen phase increases from about 10% to up to 70%.6 Unlike in LAHS, in which shed hairs are in the anagen phase, the hair that is shed in telogen effluvium is in the telogen phase and will have a different appearance when magnified.
Anagen effluvium, loss of hairs in their growing phase, is typically associated with chemotherapy. The hairs become broken and fractured at the shaft leading to breakage at different points throughout the scalp. Affected areas can include the eyebrows, eyelashes, and body hair. In the absence of a history of administration of a chemotherapy agent (or other drug known to trigger hair loss), the diagnosis of anagen effluvium should not be made.
Patients with trichotillosis (also known as trichotillomania) present with areas of hair loss caused by intentional or subconscious hair pulling. It is considered a psychological condition that can be associated with obsessive compulsive disorder, although the presence of a secondary psychological diagnosis is not required. Providers may see irregular geometric shapes of hair loss, and on close inspection see broken hair shafts of different lengths. Patients most often pull hair from their scalps (over 70% of patients), but also can pull eyelashes, eyebrow hairs, and pubic hairs.7
Treatment
LAHS is self-limited and does not necessitate treatment. However, if patients or parents feel there is significant disease burden, perhaps with poor effects on quality of life or with psychosocial impairment, treatment with minoxidil 5% solution has been studied with some success reported in the literature.1,8,9
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Natsis and Dr. Eichenfield had no relevant financial disclosures. Email them at [email protected].
References
1. Arch Dermatol. 2002;138(4):501-6.
2. Int J Trichology. 2010;2(2):96-100.
3. Pediatric Dermatol. 2016:33(50):507-10.
4. Dermatol Clin. 1986;14:745-51.
5. Arch Dermatol. 2009;145(10):1123-8.
6. J Clin Diagn Res. 2015;9(9):WE01-3.
7. Am J Psychiatry. 2016;173(9):868-74.
8. Australas J Dermatol. 2018;59:e286-e287.
9. Pediatr Dermatol. 2014;31:389-90.
It most commonly affects young girls. The pathogenesis of LAHS is thought to involve a sporadic, autosomal dominant mutation that leads to a defect between the hair cuticle and the inner root sheath.1 This defect results in the hair being poorly anchored to the scalp, and therefore easily and painlessly plucked or lost during normal hair care.
The classic presentation of LAHS is that of hair thinning and hair that may be unruly and/or lackluster; the hair rarely, if ever, requires cutting.2 The key feature is the ability to easily and painlessly pluck hairs from the patient’s scalp. The affected area is limited to the scalp, and loss of eyebrows, eyelashes, and body hair should not be seen.
Diagnosis and consideration of the differential
The diagnosis of LAHS can in some cases be made on history and physical exam alone. Patients with LAHS typically will show hair thinning with or without dullness or unruliness. They lack evidence of scalp inflammation, such as erythema, scale, pruritus, and pain. Areas of hair thinning or aberration are typically not well demarcated, and there are typically not areas of complete hair loss. There is no scarring or atrophy of the scalp itself.
Diagnostic tests include the “hair pull test,” as well as trichogram testing. In the “hair pull test” a provider grasps a set of hair at the proximal shaft near the scalp. The traction applied should result in the painless and easy extraction of more than 10% of grasped hairs in a patient with LAHS. Removal of less than 10% of hair is a normal finding, as patients without LAHS typically have about 10% of their scalp hair in the telogen phase at any given time, which would result in removal during the hair pull test.3 In trichography, plucked hairs are examined under magnification, with or without the use of selective dyes. Cinnamaldehyde is a dye that stains citrulline, which is abundant in the inner root sheath, and can be a tool in identifying its presence and/or aberrations.4 A trichogram of the pulled hairs in a patient with LAHS may classically show ruffled appearance of the cuticle, misshapen anagen hair bulbs, and absence of the inner root sheath.5 Examination under magnification also allows providers to better identify telogen versus anagen hairs, which aids in the diagnosis. By carefully considering the patient history, physical exam, and results of additional hair tests, providers can make the diagnosis of LAHS and avoid unnecessary blood work and invasive procedures like scalp biopsies.
The differential diagnosis of hair loss frequently includes alopecia areata. However, in alopecia areata, patients typically have sharply demarcated areas of hair loss, which may involve the eyebrows, eyelids, and body hairs. In alopecia areata, providers may be able to identify the “exclamation point sign” in which the hair shaft thins proximally, leading to the appearance of more pigmented, thicker hairs floating above the scalp.
Telogen effluvium is a condition in which a medical illness or stress, such as systemic illness, surgery, severe emotional distress, childbirth, dietary changes, or another traumatic event, causes a disruption in the natural cycle of hair growth such that the percentage of hairs in the telogen phase increases from about 10% to up to 70%.6 Unlike in LAHS, in which shed hairs are in the anagen phase, the hair that is shed in telogen effluvium is in the telogen phase and will have a different appearance when magnified.
Anagen effluvium, loss of hairs in their growing phase, is typically associated with chemotherapy. The hairs become broken and fractured at the shaft leading to breakage at different points throughout the scalp. Affected areas can include the eyebrows, eyelashes, and body hair. In the absence of a history of administration of a chemotherapy agent (or other drug known to trigger hair loss), the diagnosis of anagen effluvium should not be made.
Patients with trichotillosis (also known as trichotillomania) present with areas of hair loss caused by intentional or subconscious hair pulling. It is considered a psychological condition that can be associated with obsessive compulsive disorder, although the presence of a secondary psychological diagnosis is not required. Providers may see irregular geometric shapes of hair loss, and on close inspection see broken hair shafts of different lengths. Patients most often pull hair from their scalps (over 70% of patients), but also can pull eyelashes, eyebrow hairs, and pubic hairs.7
Treatment
LAHS is self-limited and does not necessitate treatment. However, if patients or parents feel there is significant disease burden, perhaps with poor effects on quality of life or with psychosocial impairment, treatment with minoxidil 5% solution has been studied with some success reported in the literature.1,8,9
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Natsis and Dr. Eichenfield had no relevant financial disclosures. Email them at [email protected].
References
1. Arch Dermatol. 2002;138(4):501-6.
2. Int J Trichology. 2010;2(2):96-100.
3. Pediatric Dermatol. 2016:33(50):507-10.
4. Dermatol Clin. 1986;14:745-51.
5. Arch Dermatol. 2009;145(10):1123-8.
6. J Clin Diagn Res. 2015;9(9):WE01-3.
7. Am J Psychiatry. 2016;173(9):868-74.
8. Australas J Dermatol. 2018;59:e286-e287.
9. Pediatr Dermatol. 2014;31:389-90.
A 5-year-old female is brought to clinic for hair loss. The mother reports that when styling her daughter's hair, she has noticed areas of hair thinning, especially at the temples and at the occiput. The mother denies scale, pruritus, and erythema. The patient reports that her scalp does not hurt. She has never had a haircut because her hair hasn't grown long enough to cut. There is no history of specific bald spots. The patient has no personal history of psoriasis, seborrheic dermatitis, or autoimmune disease. No picking has been noted, and there is no history of compulsive behaviors or anxiety. The patient's mother has a history of Graves disease. The mother reports that the patient's older sister may have had hair thinning when she was younger as well, but no longer has thin hair.
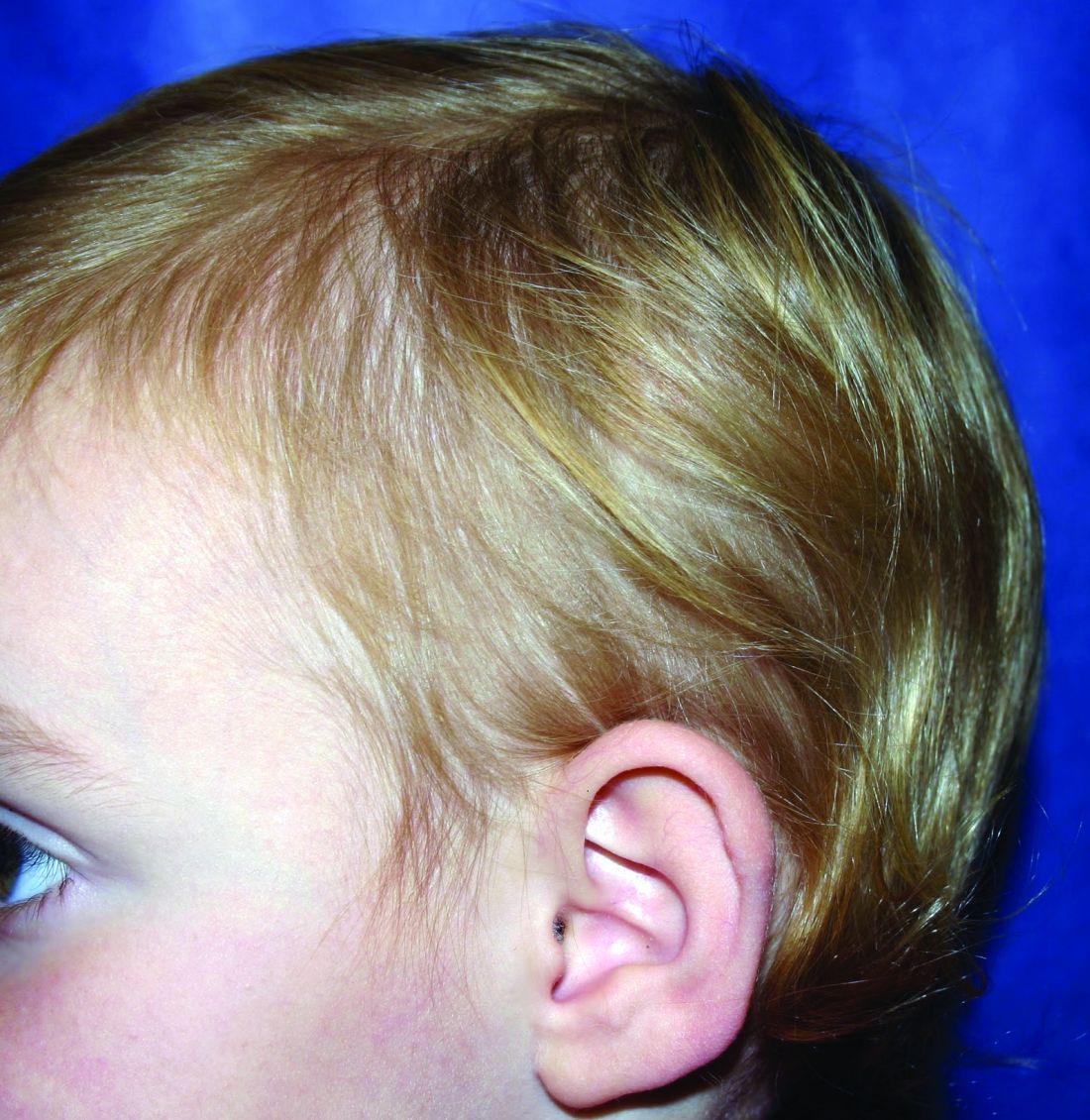
The patient was previously seen by another provider who prescribed hydrocortisone 2.5% ointment, which the mother has been applying nightly without improvement.
The child is otherwise medically well and thriving, with no recent change in activity level and with growth parameters consistently around the 75th percentile for height and weight. On physical exam, the patient has blondish, fine hair with areas of poorly demarcated hair thinning at the left temple and at the occiput. The hair remaining at the occiput is normal in texture. There are no areas of complete hair loss. There is no scale, erythema, or abnormal pigmentation, and no cervical or occipital adenopathy. The patient has intact eyebrows and eyelashes.
Trial supports less aggressive myeloma treatment
For patients with multiple myeloma that remains symptomatic within a year of starting therapy, neither a second autologous stem cell transplant nor more intensive consolidation therapy offered survival benefits superior to those seen with a single first autologous transplant and lenalidomide maintenance, reported investigators in a multicenter U.S. trial.
Among 758 patients with multiple myeloma (MM) who underwent standard induction therapy, followed by melphalan conditioning and autologous hematopoietic cell transplant (AHCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) between the three treatment arms, reported Edward A. Stadtmauer, MD, from the University of Pennsylvania, Philadelphia, and his colleagues.
Patients were randomized to either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide, bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant followed by lenalidomide maintenance.
“Single AHCT followed by len[alidomide] remains the standard of care. Greater than 80% of patients were alive at 38 months, which highlights excellent contemporary outcomes of patients with MM when treated with a standard approach of a multidrug induction followed by AHCT consolidation and maintenance,” they wrote in the Journal of Clinical Oncology.
The investigators hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and AHCT would improve survival, compared with a second AHCT.
To test this idea, they enrolled 758 patients from 54 U.S. centers and randomized them to one of three post-transplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and AHCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta-2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
The patients, who were a median age of 56 years old, had symptomatic multiple myeloma 12 months from the start of therapy without disease progression. They were randomly assigned to either AHCT followed by a second transplant and lenalidomide maintenance (247 patients), single transplant followed by RVD and lenalidomide maintenance (254), or single AHCT plus lenalidomide maintenance (257).
There were no significant differences between the groups in the primary endpoint of PFS at 38 months, with rates of 58.5% for the dual AHCT plus lenalidomide group, 57.8% for AHCT/RVD/lenalidomide, and 53.9% for AHCT/lenalidomide. Respective OS rates also did not differ significantly, at 81.8%, 85.4%, and 83.7%.
Complete response rates at 1 year were 50.5%, 58.4%, and 47.1%, respectively.
The three regimens also were similar in their toxicity profiles and in the risk of second malignancies.
The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
SOURCE: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
For patients with multiple myeloma that remains symptomatic within a year of starting therapy, neither a second autologous stem cell transplant nor more intensive consolidation therapy offered survival benefits superior to those seen with a single first autologous transplant and lenalidomide maintenance, reported investigators in a multicenter U.S. trial.
Among 758 patients with multiple myeloma (MM) who underwent standard induction therapy, followed by melphalan conditioning and autologous hematopoietic cell transplant (AHCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) between the three treatment arms, reported Edward A. Stadtmauer, MD, from the University of Pennsylvania, Philadelphia, and his colleagues.
Patients were randomized to either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide, bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant followed by lenalidomide maintenance.
“Single AHCT followed by len[alidomide] remains the standard of care. Greater than 80% of patients were alive at 38 months, which highlights excellent contemporary outcomes of patients with MM when treated with a standard approach of a multidrug induction followed by AHCT consolidation and maintenance,” they wrote in the Journal of Clinical Oncology.
The investigators hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and AHCT would improve survival, compared with a second AHCT.
To test this idea, they enrolled 758 patients from 54 U.S. centers and randomized them to one of three post-transplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and AHCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta-2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
The patients, who were a median age of 56 years old, had symptomatic multiple myeloma 12 months from the start of therapy without disease progression. They were randomly assigned to either AHCT followed by a second transplant and lenalidomide maintenance (247 patients), single transplant followed by RVD and lenalidomide maintenance (254), or single AHCT plus lenalidomide maintenance (257).
There were no significant differences between the groups in the primary endpoint of PFS at 38 months, with rates of 58.5% for the dual AHCT plus lenalidomide group, 57.8% for AHCT/RVD/lenalidomide, and 53.9% for AHCT/lenalidomide. Respective OS rates also did not differ significantly, at 81.8%, 85.4%, and 83.7%.
Complete response rates at 1 year were 50.5%, 58.4%, and 47.1%, respectively.
The three regimens also were similar in their toxicity profiles and in the risk of second malignancies.
The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
SOURCE: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
For patients with multiple myeloma that remains symptomatic within a year of starting therapy, neither a second autologous stem cell transplant nor more intensive consolidation therapy offered survival benefits superior to those seen with a single first autologous transplant and lenalidomide maintenance, reported investigators in a multicenter U.S. trial.
Among 758 patients with multiple myeloma (MM) who underwent standard induction therapy, followed by melphalan conditioning and autologous hematopoietic cell transplant (AHCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) between the three treatment arms, reported Edward A. Stadtmauer, MD, from the University of Pennsylvania, Philadelphia, and his colleagues.
Patients were randomized to either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide, bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant followed by lenalidomide maintenance.
“Single AHCT followed by len[alidomide] remains the standard of care. Greater than 80% of patients were alive at 38 months, which highlights excellent contemporary outcomes of patients with MM when treated with a standard approach of a multidrug induction followed by AHCT consolidation and maintenance,” they wrote in the Journal of Clinical Oncology.
The investigators hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and AHCT would improve survival, compared with a second AHCT.
To test this idea, they enrolled 758 patients from 54 U.S. centers and randomized them to one of three post-transplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and AHCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta-2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
The patients, who were a median age of 56 years old, had symptomatic multiple myeloma 12 months from the start of therapy without disease progression. They were randomly assigned to either AHCT followed by a second transplant and lenalidomide maintenance (247 patients), single transplant followed by RVD and lenalidomide maintenance (254), or single AHCT plus lenalidomide maintenance (257).
There were no significant differences between the groups in the primary endpoint of PFS at 38 months, with rates of 58.5% for the dual AHCT plus lenalidomide group, 57.8% for AHCT/RVD/lenalidomide, and 53.9% for AHCT/lenalidomide. Respective OS rates also did not differ significantly, at 81.8%, 85.4%, and 83.7%.
Complete response rates at 1 year were 50.5%, 58.4%, and 47.1%, respectively.
The three regimens also were similar in their toxicity profiles and in the risk of second malignancies.
The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
SOURCE: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: There were no differences in progression-free survival or overall survival among the three trial arms.
Study details: Randomized clinical trial with 758 patients with multiple myeloma.
Disclosures: The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
Source: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
Consider hysterectomy in patients with post-irradiated residual cervical cancer
according to a study of Bangladeshi patients with cervical cancer.
“Surgical intervention by either extrafascial or radical hysterectomy should be strongly considered for women with biopsy-confirmed residual disease after chemoradiation,” wrote lead authors Shahana Pervin, MBBS, and Farzana Islam Ruma, MBBS, of the National Institute of Cancer Research and Hospital and of the Railway General Hospital, respectively, in Dhaka, Bangladesh, and their associates. The study was published in the Journal of Global Oncology.
From 2009 to June 2013, this prospective longitudinal study collected data from 40 patients with biopsy-confirmed persistence of cervical cancer. The patients, who were being treated at one of two hospitals in Dhaka, Bangladesh, underwent either radical or extrafascial hysterectomy at least 12 weeks after initial radiation therapy.
At 5 years of follow-up, 36 (90%) had no evidence of disease. Of the 29 women who underwent extrafascial hysterectomy, 4 (14%) developed recurrent disease and 1 died. None of the 11 women who underwent radical hysterectomy had recurrences during the study period; that group, however, did suffer from “intraoperative, postoperative, and long-term complications.”
The investigators acknowledged the study’s several limitations, including a lack of standardized preoperative therapy, incomplete records of radiation dosing, and a limited number of patients. Along the same lines, another larger prospective trial to evaluate the two types of hysterectomy would “help guide what should be the standard of care for salvage therapy,” they wrote.
However, their findings emphasized the need for physicians in limited resource areas to have “a strong index of suspicion” when evaluating patients with locally advanced cervical cancer for residual disease. “Close clinical follow-up is crucial to identify these women in a timely manner,” the investigators added.
The Massachusetts General Hospital Gynecologic Oncology Global Health Fund supported the study. The authors reported no conflicts of interest.
SOURCE: Pervin S et al. J Glob Oncol. 2019 Feb 1. doi: 10.1200/JGO.18.00157.
according to a study of Bangladeshi patients with cervical cancer.
“Surgical intervention by either extrafascial or radical hysterectomy should be strongly considered for women with biopsy-confirmed residual disease after chemoradiation,” wrote lead authors Shahana Pervin, MBBS, and Farzana Islam Ruma, MBBS, of the National Institute of Cancer Research and Hospital and of the Railway General Hospital, respectively, in Dhaka, Bangladesh, and their associates. The study was published in the Journal of Global Oncology.
From 2009 to June 2013, this prospective longitudinal study collected data from 40 patients with biopsy-confirmed persistence of cervical cancer. The patients, who were being treated at one of two hospitals in Dhaka, Bangladesh, underwent either radical or extrafascial hysterectomy at least 12 weeks after initial radiation therapy.
At 5 years of follow-up, 36 (90%) had no evidence of disease. Of the 29 women who underwent extrafascial hysterectomy, 4 (14%) developed recurrent disease and 1 died. None of the 11 women who underwent radical hysterectomy had recurrences during the study period; that group, however, did suffer from “intraoperative, postoperative, and long-term complications.”
The investigators acknowledged the study’s several limitations, including a lack of standardized preoperative therapy, incomplete records of radiation dosing, and a limited number of patients. Along the same lines, another larger prospective trial to evaluate the two types of hysterectomy would “help guide what should be the standard of care for salvage therapy,” they wrote.
However, their findings emphasized the need for physicians in limited resource areas to have “a strong index of suspicion” when evaluating patients with locally advanced cervical cancer for residual disease. “Close clinical follow-up is crucial to identify these women in a timely manner,” the investigators added.
The Massachusetts General Hospital Gynecologic Oncology Global Health Fund supported the study. The authors reported no conflicts of interest.
SOURCE: Pervin S et al. J Glob Oncol. 2019 Feb 1. doi: 10.1200/JGO.18.00157.
according to a study of Bangladeshi patients with cervical cancer.
“Surgical intervention by either extrafascial or radical hysterectomy should be strongly considered for women with biopsy-confirmed residual disease after chemoradiation,” wrote lead authors Shahana Pervin, MBBS, and Farzana Islam Ruma, MBBS, of the National Institute of Cancer Research and Hospital and of the Railway General Hospital, respectively, in Dhaka, Bangladesh, and their associates. The study was published in the Journal of Global Oncology.
From 2009 to June 2013, this prospective longitudinal study collected data from 40 patients with biopsy-confirmed persistence of cervical cancer. The patients, who were being treated at one of two hospitals in Dhaka, Bangladesh, underwent either radical or extrafascial hysterectomy at least 12 weeks after initial radiation therapy.
At 5 years of follow-up, 36 (90%) had no evidence of disease. Of the 29 women who underwent extrafascial hysterectomy, 4 (14%) developed recurrent disease and 1 died. None of the 11 women who underwent radical hysterectomy had recurrences during the study period; that group, however, did suffer from “intraoperative, postoperative, and long-term complications.”
The investigators acknowledged the study’s several limitations, including a lack of standardized preoperative therapy, incomplete records of radiation dosing, and a limited number of patients. Along the same lines, another larger prospective trial to evaluate the two types of hysterectomy would “help guide what should be the standard of care for salvage therapy,” they wrote.
However, their findings emphasized the need for physicians in limited resource areas to have “a strong index of suspicion” when evaluating patients with locally advanced cervical cancer for residual disease. “Close clinical follow-up is crucial to identify these women in a timely manner,” the investigators added.
The Massachusetts General Hospital Gynecologic Oncology Global Health Fund supported the study. The authors reported no conflicts of interest.
SOURCE: Pervin S et al. J Glob Oncol. 2019 Feb 1. doi: 10.1200/JGO.18.00157.
FROM THE JOURNAL OF GLOBAL ONCOLOGY
Key clinical point: Hysterectomy should be strongly considered for women with biopsy-confirmed residual cervical cancer after radiation.
Major finding: At 5 years of follow-up, 90% of the patients who underwent hysterectomy had no evidence of disease.
Study details: A prospective longitudinal study of 40 patients with locally advanced cervical cancer who underwent hysterectomy after radiation at one of two hospitals in Dhaka, Bangladesh.
Disclosures: The Massachusetts General Hospital Gynecologic Oncology Global Health Fund supported the study. The authors reported no conflicts of interest.
Source: Pervin S et al. J Glob Oncol. 2019 Feb 1. doi: 10.1200/JGO.18.00157.