User login
Meet the CHEST President-Designate
Steven Q. Simpson, MD, FCCP, is a pulmonologist and intensivist with an extensive background in sepsis and in critical care quality improvement. Dr. Simpson acts as a CHEST Regent-at-Large of the Board of Regents, board liaison for the Guidelines Oversight Committee, sits on numerous board task forces and subcommittees and is a member of the CHEST SEEK Critical Care Medicine Editorial Board. He will serve as CHEST President for the 2020-2021 term.

Dr. Simpson is Professor of Medicine in the Division of Pulmonary and Critical Care Medicine at the University of Kansas. He is also senior advisor to the Solving Sepsis initiative of the Biomedical Advanced Research and Development Authority (BARDA) of the US Department of Health and Human Services. He has conducted research in all areas of severe sepsis, including molecular and cellular mechanisms, translational, quality improvement, and computer modeling studies. He was a founder in 2005 of the Midwest Critical Care Collaborative, a multidisciplinary and interprofessional collaborative effort to improve the quality of critical care services throughout the Midwest. In 2007, he initiated the Kansas Sepsis Project, a statewide program to improve severe sepsis care and outcomes via continuing education both in sepsis and in quality improvement principles and via interprofessional collaborations. Dr. Simpson is an author of the 2016 and 2020 updates of the Surviving Sepsis Campaign Guidelines. He is a member of the board of directors and Chief Medical Officer of Sepsis Alliance, a nationwide patient information and advocacy organization.
During his tenure at the University of New Mexico, he contributed to the discovery of a particular form of sepsis, the hantavirus pulmonary syndrome, and published numerous papers on the clinical description, the hemodynamic description, and the approach to supportive care for patients with the syndrome, including extracorporeal hemodynamic and oxygenation support. Dr. Simpson has authored over 180 scientific articles, book chapters, editorials, abstracts and electronic media publications. He was awarded the 2009 Eli Lilly Distinguished Scholar in Critical Care Medicine Award of the American College of Chest Physicians and the 2013 Roger C. Bone Memorial Lecture in Critical Care Medicine, which recognizes career contributions to the field. He has also been recognized as a Distinguished CHEST Educator in 2017 and 2018.
Steven Q. Simpson, MD, FCCP, is a pulmonologist and intensivist with an extensive background in sepsis and in critical care quality improvement. Dr. Simpson acts as a CHEST Regent-at-Large of the Board of Regents, board liaison for the Guidelines Oversight Committee, sits on numerous board task forces and subcommittees and is a member of the CHEST SEEK Critical Care Medicine Editorial Board. He will serve as CHEST President for the 2020-2021 term.
Dr. Simpson is Professor of Medicine in the Division of Pulmonary and Critical Care Medicine at the University of Kansas. He is also senior advisor to the Solving Sepsis initiative of the Biomedical Advanced Research and Development Authority (BARDA) of the US Department of Health and Human Services. He has conducted research in all areas of severe sepsis, including molecular and cellular mechanisms, translational, quality improvement, and computer modeling studies. He was a founder in 2005 of the Midwest Critical Care Collaborative, a multidisciplinary and interprofessional collaborative effort to improve the quality of critical care services throughout the Midwest. In 2007, he initiated the Kansas Sepsis Project, a statewide program to improve severe sepsis care and outcomes via continuing education both in sepsis and in quality improvement principles and via interprofessional collaborations. Dr. Simpson is an author of the 2016 and 2020 updates of the Surviving Sepsis Campaign Guidelines. He is a member of the board of directors and Chief Medical Officer of Sepsis Alliance, a nationwide patient information and advocacy organization.
During his tenure at the University of New Mexico, he contributed to the discovery of a particular form of sepsis, the hantavirus pulmonary syndrome, and published numerous papers on the clinical description, the hemodynamic description, and the approach to supportive care for patients with the syndrome, including extracorporeal hemodynamic and oxygenation support. Dr. Simpson has authored over 180 scientific articles, book chapters, editorials, abstracts and electronic media publications. He was awarded the 2009 Eli Lilly Distinguished Scholar in Critical Care Medicine Award of the American College of Chest Physicians and the 2013 Roger C. Bone Memorial Lecture in Critical Care Medicine, which recognizes career contributions to the field. He has also been recognized as a Distinguished CHEST Educator in 2017 and 2018.
Steven Q. Simpson, MD, FCCP, is a pulmonologist and intensivist with an extensive background in sepsis and in critical care quality improvement. Dr. Simpson acts as a CHEST Regent-at-Large of the Board of Regents, board liaison for the Guidelines Oversight Committee, sits on numerous board task forces and subcommittees and is a member of the CHEST SEEK Critical Care Medicine Editorial Board. He will serve as CHEST President for the 2020-2021 term.
Dr. Simpson is Professor of Medicine in the Division of Pulmonary and Critical Care Medicine at the University of Kansas. He is also senior advisor to the Solving Sepsis initiative of the Biomedical Advanced Research and Development Authority (BARDA) of the US Department of Health and Human Services. He has conducted research in all areas of severe sepsis, including molecular and cellular mechanisms, translational, quality improvement, and computer modeling studies. He was a founder in 2005 of the Midwest Critical Care Collaborative, a multidisciplinary and interprofessional collaborative effort to improve the quality of critical care services throughout the Midwest. In 2007, he initiated the Kansas Sepsis Project, a statewide program to improve severe sepsis care and outcomes via continuing education both in sepsis and in quality improvement principles and via interprofessional collaborations. Dr. Simpson is an author of the 2016 and 2020 updates of the Surviving Sepsis Campaign Guidelines. He is a member of the board of directors and Chief Medical Officer of Sepsis Alliance, a nationwide patient information and advocacy organization.
During his tenure at the University of New Mexico, he contributed to the discovery of a particular form of sepsis, the hantavirus pulmonary syndrome, and published numerous papers on the clinical description, the hemodynamic description, and the approach to supportive care for patients with the syndrome, including extracorporeal hemodynamic and oxygenation support. Dr. Simpson has authored over 180 scientific articles, book chapters, editorials, abstracts and electronic media publications. He was awarded the 2009 Eli Lilly Distinguished Scholar in Critical Care Medicine Award of the American College of Chest Physicians and the 2013 Roger C. Bone Memorial Lecture in Critical Care Medicine, which recognizes career contributions to the field. He has also been recognized as a Distinguished CHEST Educator in 2017 and 2018.
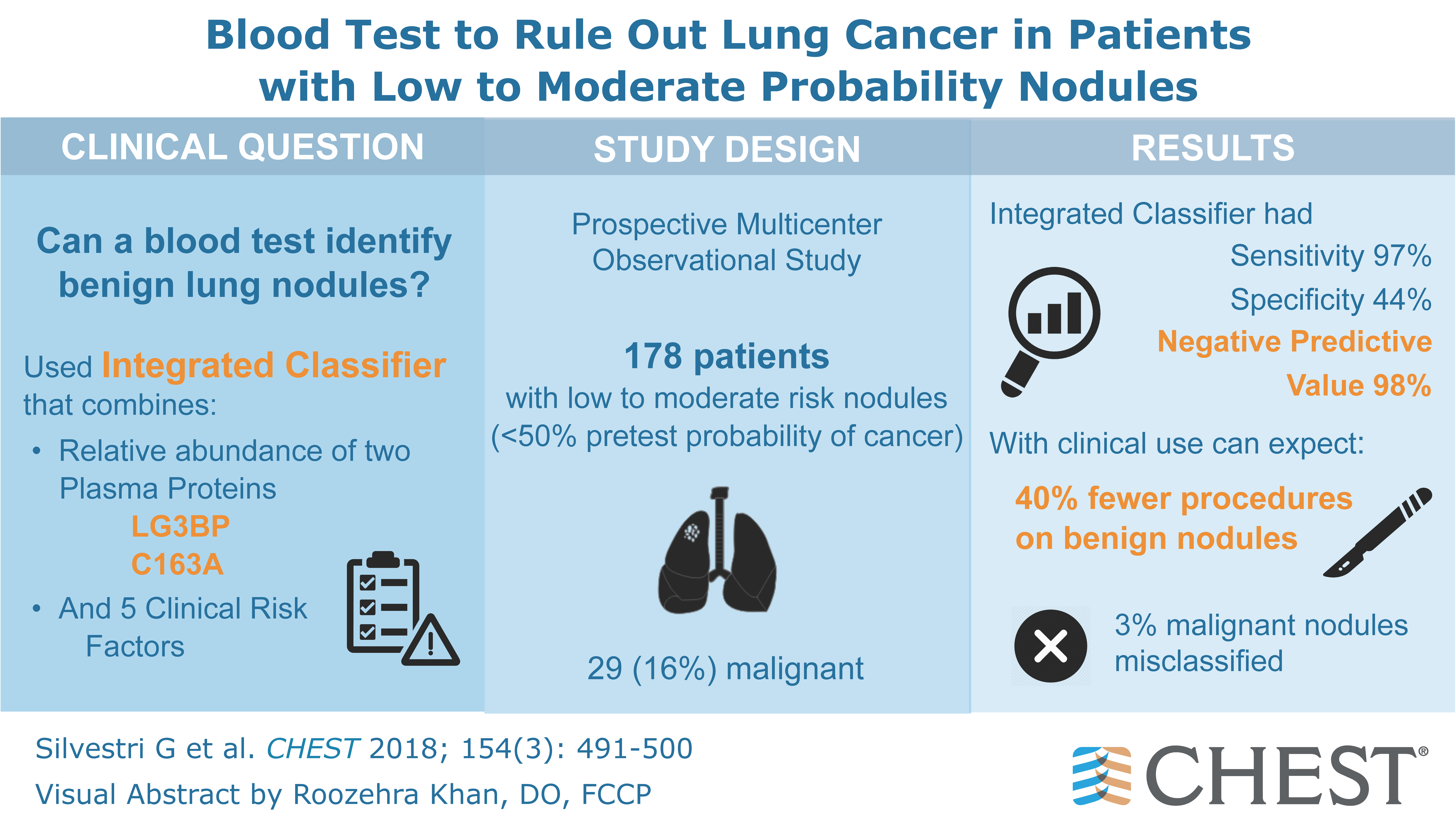
Visual abstracts enhance journal readers’ experience
Physicians’ time is decreasingly their own, and, yet, keeping abreast of clinical literature is increasingly more important. The journal CHEST has introduced a new feature aimed at easing that task and broadening the reach of journal content: visual abstracts.
At the direction of CHEST Editor in Chief Richard Irwin, MD, Master FCCP, Dr. Carroll assembled a team to help carry out an ambitious multimedia strategy (see box). Dr. Irwin charged the Web and Multimedia editorial team with not only extending the reach of journal content but also enhancing readers’ engagement with and understanding of it.
“Our first project was the development of visual abstracts, a type of infographics used to distill the key points of a research abstract into an easily digested graphic form,” says Dr. Carroll, who also is research director of pediatric critical care at Connecticut Children’s Medical Center, Hartford, and a professor of pediatrics at the University of Connecticut School of Medicine, Farmington.
The first visual abstracts were posted to accompany two articles in the July 2018 issue of CHEST. With the exception of August 2018, every issue since has been enhanced with infographics. Insert infographic here (A full gallery of all the visual abstracts so far is available at https://journal.chestnet.org/infographics.) The visual abstracts are available through a number of vehicles: the journal’s website (https://journal.chestnet.org/), the journal’s mobile app (https://journal.chestnet.org/content/mobileaccessinstructions), and social media platforms such as Facebook (https://www.facebook.com/accpchest/) and Twitter (https://twitter.com/accpchest).
“Our goal with the infographics is to promote the exciting research CHEST publishes and to get readers to click through and read the entire article,” says Dr. Carroll. “So far, we’re happy with our results—and we’re looking forward to even greater reach in 2019.”
CHEST Web and Multimedia Section
Editor
Christopher Carroll, MD, MS
Assistant Editors
Yonatan Y. Greenstein, MD, FCCP, Newark, NJ
Roozehra Khan, DO, FCCP, Los Angeles, CA
Dominique J. Pepper, MD, MBChB, MHSc, Bethesda, MD.
###
Physicians’ time is decreasingly their own, and, yet, keeping abreast of clinical literature is increasingly more important. The journal CHEST has introduced a new feature aimed at easing that task and broadening the reach of journal content: visual abstracts.
At the direction of CHEST Editor in Chief Richard Irwin, MD, Master FCCP, Dr. Carroll assembled a team to help carry out an ambitious multimedia strategy (see box). Dr. Irwin charged the Web and Multimedia editorial team with not only extending the reach of journal content but also enhancing readers’ engagement with and understanding of it.
“Our first project was the development of visual abstracts, a type of infographics used to distill the key points of a research abstract into an easily digested graphic form,” says Dr. Carroll, who also is research director of pediatric critical care at Connecticut Children’s Medical Center, Hartford, and a professor of pediatrics at the University of Connecticut School of Medicine, Farmington.
The first visual abstracts were posted to accompany two articles in the July 2018 issue of CHEST. With the exception of August 2018, every issue since has been enhanced with infographics. Insert infographic here (A full gallery of all the visual abstracts so far is available at https://journal.chestnet.org/infographics.) The visual abstracts are available through a number of vehicles: the journal’s website (https://journal.chestnet.org/), the journal’s mobile app (https://journal.chestnet.org/content/mobileaccessinstructions), and social media platforms such as Facebook (https://www.facebook.com/accpchest/) and Twitter (https://twitter.com/accpchest).
“Our goal with the infographics is to promote the exciting research CHEST publishes and to get readers to click through and read the entire article,” says Dr. Carroll. “So far, we’re happy with our results—and we’re looking forward to even greater reach in 2019.”
CHEST Web and Multimedia Section
Editor
Christopher Carroll, MD, MS
Assistant Editors
Yonatan Y. Greenstein, MD, FCCP, Newark, NJ
Roozehra Khan, DO, FCCP, Los Angeles, CA
Dominique J. Pepper, MD, MBChB, MHSc, Bethesda, MD.
###
Physicians’ time is decreasingly their own, and, yet, keeping abreast of clinical literature is increasingly more important. The journal CHEST has introduced a new feature aimed at easing that task and broadening the reach of journal content: visual abstracts.
At the direction of CHEST Editor in Chief Richard Irwin, MD, Master FCCP, Dr. Carroll assembled a team to help carry out an ambitious multimedia strategy (see box). Dr. Irwin charged the Web and Multimedia editorial team with not only extending the reach of journal content but also enhancing readers’ engagement with and understanding of it.
“Our first project was the development of visual abstracts, a type of infographics used to distill the key points of a research abstract into an easily digested graphic form,” says Dr. Carroll, who also is research director of pediatric critical care at Connecticut Children’s Medical Center, Hartford, and a professor of pediatrics at the University of Connecticut School of Medicine, Farmington.
The first visual abstracts were posted to accompany two articles in the July 2018 issue of CHEST. With the exception of August 2018, every issue since has been enhanced with infographics. Insert infographic here (A full gallery of all the visual abstracts so far is available at https://journal.chestnet.org/infographics.) The visual abstracts are available through a number of vehicles: the journal’s website (https://journal.chestnet.org/), the journal’s mobile app (https://journal.chestnet.org/content/mobileaccessinstructions), and social media platforms such as Facebook (https://www.facebook.com/accpchest/) and Twitter (https://twitter.com/accpchest).
“Our goal with the infographics is to promote the exciting research CHEST publishes and to get readers to click through and read the entire article,” says Dr. Carroll. “So far, we’re happy with our results—and we’re looking forward to even greater reach in 2019.”
CHEST Web and Multimedia Section
Editor
Christopher Carroll, MD, MS
Assistant Editors
Yonatan Y. Greenstein, MD, FCCP, Newark, NJ
Roozehra Khan, DO, FCCP, Los Angeles, CA
Dominique J. Pepper, MD, MBChB, MHSc, Bethesda, MD.
###
Five steering committees examine the literature
Airways Disorders
Defining and treating early COPD: Can we make a difference?
There is growing evidence that early COPD—before currently accepted spirometric or symptomatic criteria are present—may be an important clinical entity. The primary pathobiologic mechanisms in early COPD development include both abnormal lung development and accelerated lung aging (Augustí et al. Am J Respir Crit Care Med. 2018 Oct 15;198:8:978).

Martinez and colleagues recently proposed defining early COPD as age <50 with 10+ pack-year smoking history and at least one of the following: (1) early airflow limitation (postbronchodilator FEV1/FVC < lower limit of normal), (2) compatible CT scan abnormalities, (3) rapid decline in FEV1 (≥60 mL/yr) that is accelerated relative to FVC (Martinez et al. Am J Respir Crit Care Med. 2018 Jun 15;197[12]:1540).
A novel multiresolution CT scan imaging protocol described by Koo and coworkers found that substantial loss of small airways— specifically the terminal and transitional bronchioles—occurs in patients with mild-to-moderate COPD even prior to the development of emphysema on CT scan. These findings show that significant destruction of the small airways has occurred prior to the development of mild COPD (Koo et al. Lancet Respir Med. 2018 Aug;6:591).

Pharmacologic treatment for COPD is targeted at the reduction of symptoms and risk of exacerbation, as there remains no conclusive evidence that existing therapies modify long-term decline in lung function. It is unknown if pharmacotherapy for “early COPD” will alter the disease course. While not directly addressing this subset, information may be gleaned from trials on younger, more mild GOLD Stage 1 or Stage 2 patients. The Tie-COPD trial, the largest powered study to date of mild-to-moderate COPD, found that among patients with GOLD stage 1 or 2 COPD treatment with tiotropium compared with placebo for 2 years resulted in significantly higher FEV1 before bronchodilator use (between group difference of 157 mL) and slowed annual decline in FEV1 after bronchodilator use (Zhou et al. N Engl J Med. 2017 Sep 7;377[10]:923).
As our understanding of heterogeneity within COPD increases, striving for improved outcomes from our therapies—an impact on lung function in addition to symptom and exacerbation risk—may need to begin with the study of earlier treatment.
Megan Conroy, MD
Steering Committee Fellow-in-Training
Allen J. Blaivas, DO, FCCP
Steering Committee Vice-Chair
Clinical Pulmonary Medicine
Asthma-COPD overlap: An underappreciated phenotype of obstructive airway disease (OAD)
Asthma-COPD overlap (ACO) is a common yet underappreciated clinical entity within the complex OAD spectrum. Currently, there is no consensus criteria to define ACO; however, a roundtable consensus from an international group (Sin et al. Eur Respir J. 2016 Sep;48:664) suggests using major and minor criteria, with key features being airflow limitation, asthma history, and cigarette or biomass exposure. Several studies have shown that patients with ACO have severe disease, faster lung function decline, greater morbidity and mortality, and lower QoL (Alshabanat et al. PLoS One. 2015 Sep 3;10:e0136065).

There is paucity of data on the pathophysiology, risk factors, and clinical management given exclusion of these patients from clinical trials of asthma and COPD. Indeed, clinicians and researchers now realize that ACO is an umbrella term for multiple subphenotypes, including patients who have predominant asthma with some COPD features and others with predominant COPD with some asthma features. Overall, IgE level, FeNO, sputum, and blood eosinophils are usually higher in ACO than in COPD and relatively similar compared with asthma (Kobayashi et al. Int J Chron Obs Pulmon Dis. 2016 May 26;11:2117).

Most recently, a longitudinal study looked at predictors of ACO among NY firefighters exposed to WTC dust (Singh et al. CHEST. 2018 Dec;154[6]:1301). Pre-exposure low lung function and elevated blood eosinophils and IL4 (T2 inflammatory cytokine) increased risk of developing ACO among those exposed to WTC dust. Further research is required to better understand the interaction of environmental exposure and risk factors in the pathophysiology of ACO. It may be more pragmatic to use the unifying term OAD, as originally proposed in the Dutch hypothesis, and further delineate how several phenotypes of airway disease can be classified by combining traditional approaches with molecular and genomic analysis.
Munish Luthra, MD, FCCP
Steering Committee Member
Samantha D’Annunzio, MD
Steering Committee Member
Critical Care
Mechanical ventilation: One size fits all?
Mechanical ventilation (MV) is a lifesaving intervention in the ICU, but it has been associated with numerous complications ranging from overuse of sedation, atelectasis, and baro or volutrauma.

After 2000, it became well known that using a low tidal volume (VT) strategy (6 mL/kg predicted body weight, PBW) in patients with ARDS produced lower mortality and more ventilator-free days (N Engl J Med. 2000 May 4;342[18]:1301). In addition, a meta-analysis in 2012 demonstrated a lower relative risk of new lung injury, mortality, and pulmonary infections with low VT in non-ARDS patients (Serpa et al. JAMA. 2012 Oct 24/31;308[16]:1651). However, the included studies varied widely in their use of VT (9-12 mL/kg), duration of MV, and in mixed settings (ICU or operating room).

Recently, a large randomized clinical trial compared the effect of low (4-6 mL/kg, PBW) vs intermediate (8-10 mL/kg, PBW) VT ventilation strategy in non-ARDS ICU patients. Interestingly, the study concluded that there is no significant difference in ventilator-free days (21 days in each group), median length ICU and hospital stay, ICU mortality rates, and 28- and 90-day mortality. Also, there was no difference in new-onset ARDS, severe atelectasis, sedation use, and delirium (JAMA. 2018; 320[18]:1872). This study suggests that in non-ARDS patients, MV should be individualized according to each patient’s clinical situation, the nature of the disease, and its effect on lung mechanics, especially in patients who cannot tolerate low tidal volumes.
Margaret A. Disselkamp, MD
Steering Committee Member
Mohammed A. Megri, MD
Fellow-in-Training Steering Committee Member
Home-Based Mechanical Ventilation and Neuromuscular Disease
Improving access to sleep medicine care for patients with NMD
Sleep-disordered breathing (SDB) occurs in up to 5% of children, with adverse implications for growth and development. Children with neuromuscular disease are at significantly higher risk than unaffected children (Chiang et al. Children. 2018;5:e78). Respiratory dysfunction that may present as SDB before daytime impairment in gas exchange is evident. Diagnosing and treating SDB (to include OSA, CSA, and hypoventilation syndromes) early can significantly improve morbidity and mortality.

Unfortunately, diagnostic sleep medicine resources are limited. Children may wait up to a year or more for definitive testing with in-laboratory, attended polysomnography (PSG). Among children with neuromuscular disease, fewer than 10% may undergo a sleep clinic evaluation, and, of those that do, they may have only one visit over a 3-year period of care (Rose et al. Pediatr Pulmonol. 2018 Oct;53:1378). Home sleep testing (HST) has been evaluated as an alternative to PSG given lower cost, availability, and advantage of the child sleeping in his/her own bed. Although HST is indicated in adults with a high pretest probability for moderate to severe OSA, it is not indicated in children, given the potential to underestimate disease severity or to miss the diagnosis entirely (Kirk et al. J Clin Sleep Med. 2017 Oct 15;13[10]:1199). HST lacks electroencephalogram (EEG) and capnography. Technical recording mishaps are more common in children, but in-lab PSG has the advantage of on-site troubleshooting by a technologist.
A recently published study by Fishman and colleagues attempted to compare gold standard in-lab PSG to HST with capnography (Fishman et al. J Clin Sleep Med. 2018 Dec 15;14[12]:2013). Despite a well-designed study with a carefully selected population, HST failed to reliably diagnose SDB. HST underestimated disease severity and, in some cases, missed the diagnosis of SDB entirely. The addition of end tidal CO2 monitoring failed to improve diagnostic accuracy, and HST and PSG-ETco2 values were poorly correlated.
Although children with neuromuscular disease face long wait times for sleep evaluations, HST is clearly not the solution for now. It remains to be seen if innovations in HST with extended monitoring (and transcutaneous CO2) become viable. In the meantime, finding ways to improve access to sleep medicine care for children with neuromuscular disease is a must.
Jacob Collen, MD, FCCP
Steering Committee Member
Interstitial and Diffuse Lung Disease
Idiopathic pneumonias that are not all that idiopathic
Despite being defined as an individual entity for research purposes in 2015 (Fisher et al. Eur Respir J. 2015;46:976), interstitial pneumonias with autoimmune features (IPAF) remain a heterogeneous group of interstitial lung diseases that puzzle the clinician. Since the introduction of the IPAF definition, there have been attempts to validate the diagnostic criteria and study their prognostic implications. Some of these studies showed differential prognosis in patients who met the IPAF criteria (Oldham et al. Eur Respir J. 2016;47:1767).
Although the implications of the presence of autoimmune antibodies in idiopathic interstitial pneumonias (IIPs) is not fully understood, the treatment often entails immunosuppression, especially in those with non-UIP patterns of disease and/or clinical features of autoimmune disease. The stakes are high when IIPs are associated with antibodies correlated with rapidly progressive disease, such as MDA-5 antibody or antisynthetase antibodies. Pulmonologists often lack the clinical expertise to detect occult autoimmune disorders, though the role of the rheumatologist in facilitating the diagnosis and treatment of IPAF is not well delineated. Most health-care systems are not equipped with collaborative ILD-rheumatology clinics or even easy access to a rheumatologist. There is a need for real-world pragmatic studies to establish the optimal way to evaluate patients with ILD for autoimmune features and identify patients who would benefit most from an early referral to rheumatology to aid with diagnosis, treatment, and sometimes monitoring for extrapulmonary manifestations of autoimmune disorders.
Avanthika Thanushi Wynn, MD
Steering Committee Fellow-in-Training
Airways Disorders
Defining and treating early COPD: Can we make a difference?
There is growing evidence that early COPD—before currently accepted spirometric or symptomatic criteria are present—may be an important clinical entity. The primary pathobiologic mechanisms in early COPD development include both abnormal lung development and accelerated lung aging (Augustí et al. Am J Respir Crit Care Med. 2018 Oct 15;198:8:978).
Martinez and colleagues recently proposed defining early COPD as age <50 with 10+ pack-year smoking history and at least one of the following: (1) early airflow limitation (postbronchodilator FEV1/FVC < lower limit of normal), (2) compatible CT scan abnormalities, (3) rapid decline in FEV1 (≥60 mL/yr) that is accelerated relative to FVC (Martinez et al. Am J Respir Crit Care Med. 2018 Jun 15;197[12]:1540).
A novel multiresolution CT scan imaging protocol described by Koo and coworkers found that substantial loss of small airways— specifically the terminal and transitional bronchioles—occurs in patients with mild-to-moderate COPD even prior to the development of emphysema on CT scan. These findings show that significant destruction of the small airways has occurred prior to the development of mild COPD (Koo et al. Lancet Respir Med. 2018 Aug;6:591).
Pharmacologic treatment for COPD is targeted at the reduction of symptoms and risk of exacerbation, as there remains no conclusive evidence that existing therapies modify long-term decline in lung function. It is unknown if pharmacotherapy for “early COPD” will alter the disease course. While not directly addressing this subset, information may be gleaned from trials on younger, more mild GOLD Stage 1 or Stage 2 patients. The Tie-COPD trial, the largest powered study to date of mild-to-moderate COPD, found that among patients with GOLD stage 1 or 2 COPD treatment with tiotropium compared with placebo for 2 years resulted in significantly higher FEV1 before bronchodilator use (between group difference of 157 mL) and slowed annual decline in FEV1 after bronchodilator use (Zhou et al. N Engl J Med. 2017 Sep 7;377[10]:923).
As our understanding of heterogeneity within COPD increases, striving for improved outcomes from our therapies—an impact on lung function in addition to symptom and exacerbation risk—may need to begin with the study of earlier treatment.
Megan Conroy, MD
Steering Committee Fellow-in-Training
Allen J. Blaivas, DO, FCCP
Steering Committee Vice-Chair
Clinical Pulmonary Medicine
Asthma-COPD overlap: An underappreciated phenotype of obstructive airway disease (OAD)
Asthma-COPD overlap (ACO) is a common yet underappreciated clinical entity within the complex OAD spectrum. Currently, there is no consensus criteria to define ACO; however, a roundtable consensus from an international group (Sin et al. Eur Respir J. 2016 Sep;48:664) suggests using major and minor criteria, with key features being airflow limitation, asthma history, and cigarette or biomass exposure. Several studies have shown that patients with ACO have severe disease, faster lung function decline, greater morbidity and mortality, and lower QoL (Alshabanat et al. PLoS One. 2015 Sep 3;10:e0136065).
There is paucity of data on the pathophysiology, risk factors, and clinical management given exclusion of these patients from clinical trials of asthma and COPD. Indeed, clinicians and researchers now realize that ACO is an umbrella term for multiple subphenotypes, including patients who have predominant asthma with some COPD features and others with predominant COPD with some asthma features. Overall, IgE level, FeNO, sputum, and blood eosinophils are usually higher in ACO than in COPD and relatively similar compared with asthma (Kobayashi et al. Int J Chron Obs Pulmon Dis. 2016 May 26;11:2117).
Most recently, a longitudinal study looked at predictors of ACO among NY firefighters exposed to WTC dust (Singh et al. CHEST. 2018 Dec;154[6]:1301). Pre-exposure low lung function and elevated blood eosinophils and IL4 (T2 inflammatory cytokine) increased risk of developing ACO among those exposed to WTC dust. Further research is required to better understand the interaction of environmental exposure and risk factors in the pathophysiology of ACO. It may be more pragmatic to use the unifying term OAD, as originally proposed in the Dutch hypothesis, and further delineate how several phenotypes of airway disease can be classified by combining traditional approaches with molecular and genomic analysis.
Munish Luthra, MD, FCCP
Steering Committee Member
Samantha D’Annunzio, MD
Steering Committee Member
Critical Care
Mechanical ventilation: One size fits all?
Mechanical ventilation (MV) is a lifesaving intervention in the ICU, but it has been associated with numerous complications ranging from overuse of sedation, atelectasis, and baro or volutrauma.
After 2000, it became well known that using a low tidal volume (VT) strategy (6 mL/kg predicted body weight, PBW) in patients with ARDS produced lower mortality and more ventilator-free days (N Engl J Med. 2000 May 4;342[18]:1301). In addition, a meta-analysis in 2012 demonstrated a lower relative risk of new lung injury, mortality, and pulmonary infections with low VT in non-ARDS patients (Serpa et al. JAMA. 2012 Oct 24/31;308[16]:1651). However, the included studies varied widely in their use of VT (9-12 mL/kg), duration of MV, and in mixed settings (ICU or operating room).
Recently, a large randomized clinical trial compared the effect of low (4-6 mL/kg, PBW) vs intermediate (8-10 mL/kg, PBW) VT ventilation strategy in non-ARDS ICU patients. Interestingly, the study concluded that there is no significant difference in ventilator-free days (21 days in each group), median length ICU and hospital stay, ICU mortality rates, and 28- and 90-day mortality. Also, there was no difference in new-onset ARDS, severe atelectasis, sedation use, and delirium (JAMA. 2018; 320[18]:1872). This study suggests that in non-ARDS patients, MV should be individualized according to each patient’s clinical situation, the nature of the disease, and its effect on lung mechanics, especially in patients who cannot tolerate low tidal volumes.
Margaret A. Disselkamp, MD
Steering Committee Member
Mohammed A. Megri, MD
Fellow-in-Training Steering Committee Member
Home-Based Mechanical Ventilation and Neuromuscular Disease
Improving access to sleep medicine care for patients with NMD
Sleep-disordered breathing (SDB) occurs in up to 5% of children, with adverse implications for growth and development. Children with neuromuscular disease are at significantly higher risk than unaffected children (Chiang et al. Children. 2018;5:e78). Respiratory dysfunction that may present as SDB before daytime impairment in gas exchange is evident. Diagnosing and treating SDB (to include OSA, CSA, and hypoventilation syndromes) early can significantly improve morbidity and mortality.
Unfortunately, diagnostic sleep medicine resources are limited. Children may wait up to a year or more for definitive testing with in-laboratory, attended polysomnography (PSG). Among children with neuromuscular disease, fewer than 10% may undergo a sleep clinic evaluation, and, of those that do, they may have only one visit over a 3-year period of care (Rose et al. Pediatr Pulmonol. 2018 Oct;53:1378). Home sleep testing (HST) has been evaluated as an alternative to PSG given lower cost, availability, and advantage of the child sleeping in his/her own bed. Although HST is indicated in adults with a high pretest probability for moderate to severe OSA, it is not indicated in children, given the potential to underestimate disease severity or to miss the diagnosis entirely (Kirk et al. J Clin Sleep Med. 2017 Oct 15;13[10]:1199). HST lacks electroencephalogram (EEG) and capnography. Technical recording mishaps are more common in children, but in-lab PSG has the advantage of on-site troubleshooting by a technologist.
A recently published study by Fishman and colleagues attempted to compare gold standard in-lab PSG to HST with capnography (Fishman et al. J Clin Sleep Med. 2018 Dec 15;14[12]:2013). Despite a well-designed study with a carefully selected population, HST failed to reliably diagnose SDB. HST underestimated disease severity and, in some cases, missed the diagnosis of SDB entirely. The addition of end tidal CO2 monitoring failed to improve diagnostic accuracy, and HST and PSG-ETco2 values were poorly correlated.
Although children with neuromuscular disease face long wait times for sleep evaluations, HST is clearly not the solution for now. It remains to be seen if innovations in HST with extended monitoring (and transcutaneous CO2) become viable. In the meantime, finding ways to improve access to sleep medicine care for children with neuromuscular disease is a must.
Jacob Collen, MD, FCCP
Steering Committee Member
Interstitial and Diffuse Lung Disease
Idiopathic pneumonias that are not all that idiopathic
Despite being defined as an individual entity for research purposes in 2015 (Fisher et al. Eur Respir J. 2015;46:976), interstitial pneumonias with autoimmune features (IPAF) remain a heterogeneous group of interstitial lung diseases that puzzle the clinician. Since the introduction of the IPAF definition, there have been attempts to validate the diagnostic criteria and study their prognostic implications. Some of these studies showed differential prognosis in patients who met the IPAF criteria (Oldham et al. Eur Respir J. 2016;47:1767).
Although the implications of the presence of autoimmune antibodies in idiopathic interstitial pneumonias (IIPs) is not fully understood, the treatment often entails immunosuppression, especially in those with non-UIP patterns of disease and/or clinical features of autoimmune disease. The stakes are high when IIPs are associated with antibodies correlated with rapidly progressive disease, such as MDA-5 antibody or antisynthetase antibodies. Pulmonologists often lack the clinical expertise to detect occult autoimmune disorders, though the role of the rheumatologist in facilitating the diagnosis and treatment of IPAF is not well delineated. Most health-care systems are not equipped with collaborative ILD-rheumatology clinics or even easy access to a rheumatologist. There is a need for real-world pragmatic studies to establish the optimal way to evaluate patients with ILD for autoimmune features and identify patients who would benefit most from an early referral to rheumatology to aid with diagnosis, treatment, and sometimes monitoring for extrapulmonary manifestations of autoimmune disorders.
Avanthika Thanushi Wynn, MD
Steering Committee Fellow-in-Training
Airways Disorders
Defining and treating early COPD: Can we make a difference?
There is growing evidence that early COPD—before currently accepted spirometric or symptomatic criteria are present—may be an important clinical entity. The primary pathobiologic mechanisms in early COPD development include both abnormal lung development and accelerated lung aging (Augustí et al. Am J Respir Crit Care Med. 2018 Oct 15;198:8:978).
Martinez and colleagues recently proposed defining early COPD as age <50 with 10+ pack-year smoking history and at least one of the following: (1) early airflow limitation (postbronchodilator FEV1/FVC < lower limit of normal), (2) compatible CT scan abnormalities, (3) rapid decline in FEV1 (≥60 mL/yr) that is accelerated relative to FVC (Martinez et al. Am J Respir Crit Care Med. 2018 Jun 15;197[12]:1540).
A novel multiresolution CT scan imaging protocol described by Koo and coworkers found that substantial loss of small airways— specifically the terminal and transitional bronchioles—occurs in patients with mild-to-moderate COPD even prior to the development of emphysema on CT scan. These findings show that significant destruction of the small airways has occurred prior to the development of mild COPD (Koo et al. Lancet Respir Med. 2018 Aug;6:591).
Pharmacologic treatment for COPD is targeted at the reduction of symptoms and risk of exacerbation, as there remains no conclusive evidence that existing therapies modify long-term decline in lung function. It is unknown if pharmacotherapy for “early COPD” will alter the disease course. While not directly addressing this subset, information may be gleaned from trials on younger, more mild GOLD Stage 1 or Stage 2 patients. The Tie-COPD trial, the largest powered study to date of mild-to-moderate COPD, found that among patients with GOLD stage 1 or 2 COPD treatment with tiotropium compared with placebo for 2 years resulted in significantly higher FEV1 before bronchodilator use (between group difference of 157 mL) and slowed annual decline in FEV1 after bronchodilator use (Zhou et al. N Engl J Med. 2017 Sep 7;377[10]:923).
As our understanding of heterogeneity within COPD increases, striving for improved outcomes from our therapies—an impact on lung function in addition to symptom and exacerbation risk—may need to begin with the study of earlier treatment.
Megan Conroy, MD
Steering Committee Fellow-in-Training
Allen J. Blaivas, DO, FCCP
Steering Committee Vice-Chair
Clinical Pulmonary Medicine
Asthma-COPD overlap: An underappreciated phenotype of obstructive airway disease (OAD)
Asthma-COPD overlap (ACO) is a common yet underappreciated clinical entity within the complex OAD spectrum. Currently, there is no consensus criteria to define ACO; however, a roundtable consensus from an international group (Sin et al. Eur Respir J. 2016 Sep;48:664) suggests using major and minor criteria, with key features being airflow limitation, asthma history, and cigarette or biomass exposure. Several studies have shown that patients with ACO have severe disease, faster lung function decline, greater morbidity and mortality, and lower QoL (Alshabanat et al. PLoS One. 2015 Sep 3;10:e0136065).
There is paucity of data on the pathophysiology, risk factors, and clinical management given exclusion of these patients from clinical trials of asthma and COPD. Indeed, clinicians and researchers now realize that ACO is an umbrella term for multiple subphenotypes, including patients who have predominant asthma with some COPD features and others with predominant COPD with some asthma features. Overall, IgE level, FeNO, sputum, and blood eosinophils are usually higher in ACO than in COPD and relatively similar compared with asthma (Kobayashi et al. Int J Chron Obs Pulmon Dis. 2016 May 26;11:2117).
Most recently, a longitudinal study looked at predictors of ACO among NY firefighters exposed to WTC dust (Singh et al. CHEST. 2018 Dec;154[6]:1301). Pre-exposure low lung function and elevated blood eosinophils and IL4 (T2 inflammatory cytokine) increased risk of developing ACO among those exposed to WTC dust. Further research is required to better understand the interaction of environmental exposure and risk factors in the pathophysiology of ACO. It may be more pragmatic to use the unifying term OAD, as originally proposed in the Dutch hypothesis, and further delineate how several phenotypes of airway disease can be classified by combining traditional approaches with molecular and genomic analysis.
Munish Luthra, MD, FCCP
Steering Committee Member
Samantha D’Annunzio, MD
Steering Committee Member
Critical Care
Mechanical ventilation: One size fits all?
Mechanical ventilation (MV) is a lifesaving intervention in the ICU, but it has been associated with numerous complications ranging from overuse of sedation, atelectasis, and baro or volutrauma.
After 2000, it became well known that using a low tidal volume (VT) strategy (6 mL/kg predicted body weight, PBW) in patients with ARDS produced lower mortality and more ventilator-free days (N Engl J Med. 2000 May 4;342[18]:1301). In addition, a meta-analysis in 2012 demonstrated a lower relative risk of new lung injury, mortality, and pulmonary infections with low VT in non-ARDS patients (Serpa et al. JAMA. 2012 Oct 24/31;308[16]:1651). However, the included studies varied widely in their use of VT (9-12 mL/kg), duration of MV, and in mixed settings (ICU or operating room).
Recently, a large randomized clinical trial compared the effect of low (4-6 mL/kg, PBW) vs intermediate (8-10 mL/kg, PBW) VT ventilation strategy in non-ARDS ICU patients. Interestingly, the study concluded that there is no significant difference in ventilator-free days (21 days in each group), median length ICU and hospital stay, ICU mortality rates, and 28- and 90-day mortality. Also, there was no difference in new-onset ARDS, severe atelectasis, sedation use, and delirium (JAMA. 2018; 320[18]:1872). This study suggests that in non-ARDS patients, MV should be individualized according to each patient’s clinical situation, the nature of the disease, and its effect on lung mechanics, especially in patients who cannot tolerate low tidal volumes.
Margaret A. Disselkamp, MD
Steering Committee Member
Mohammed A. Megri, MD
Fellow-in-Training Steering Committee Member
Home-Based Mechanical Ventilation and Neuromuscular Disease
Improving access to sleep medicine care for patients with NMD
Sleep-disordered breathing (SDB) occurs in up to 5% of children, with adverse implications for growth and development. Children with neuromuscular disease are at significantly higher risk than unaffected children (Chiang et al. Children. 2018;5:e78). Respiratory dysfunction that may present as SDB before daytime impairment in gas exchange is evident. Diagnosing and treating SDB (to include OSA, CSA, and hypoventilation syndromes) early can significantly improve morbidity and mortality.
Unfortunately, diagnostic sleep medicine resources are limited. Children may wait up to a year or more for definitive testing with in-laboratory, attended polysomnography (PSG). Among children with neuromuscular disease, fewer than 10% may undergo a sleep clinic evaluation, and, of those that do, they may have only one visit over a 3-year period of care (Rose et al. Pediatr Pulmonol. 2018 Oct;53:1378). Home sleep testing (HST) has been evaluated as an alternative to PSG given lower cost, availability, and advantage of the child sleeping in his/her own bed. Although HST is indicated in adults with a high pretest probability for moderate to severe OSA, it is not indicated in children, given the potential to underestimate disease severity or to miss the diagnosis entirely (Kirk et al. J Clin Sleep Med. 2017 Oct 15;13[10]:1199). HST lacks electroencephalogram (EEG) and capnography. Technical recording mishaps are more common in children, but in-lab PSG has the advantage of on-site troubleshooting by a technologist.
A recently published study by Fishman and colleagues attempted to compare gold standard in-lab PSG to HST with capnography (Fishman et al. J Clin Sleep Med. 2018 Dec 15;14[12]:2013). Despite a well-designed study with a carefully selected population, HST failed to reliably diagnose SDB. HST underestimated disease severity and, in some cases, missed the diagnosis of SDB entirely. The addition of end tidal CO2 monitoring failed to improve diagnostic accuracy, and HST and PSG-ETco2 values were poorly correlated.
Although children with neuromuscular disease face long wait times for sleep evaluations, HST is clearly not the solution for now. It remains to be seen if innovations in HST with extended monitoring (and transcutaneous CO2) become viable. In the meantime, finding ways to improve access to sleep medicine care for children with neuromuscular disease is a must.
Jacob Collen, MD, FCCP
Steering Committee Member
Interstitial and Diffuse Lung Disease
Idiopathic pneumonias that are not all that idiopathic
Despite being defined as an individual entity for research purposes in 2015 (Fisher et al. Eur Respir J. 2015;46:976), interstitial pneumonias with autoimmune features (IPAF) remain a heterogeneous group of interstitial lung diseases that puzzle the clinician. Since the introduction of the IPAF definition, there have been attempts to validate the diagnostic criteria and study their prognostic implications. Some of these studies showed differential prognosis in patients who met the IPAF criteria (Oldham et al. Eur Respir J. 2016;47:1767).
Although the implications of the presence of autoimmune antibodies in idiopathic interstitial pneumonias (IIPs) is not fully understood, the treatment often entails immunosuppression, especially in those with non-UIP patterns of disease and/or clinical features of autoimmune disease. The stakes are high when IIPs are associated with antibodies correlated with rapidly progressive disease, such as MDA-5 antibody or antisynthetase antibodies. Pulmonologists often lack the clinical expertise to detect occult autoimmune disorders, though the role of the rheumatologist in facilitating the diagnosis and treatment of IPAF is not well delineated. Most health-care systems are not equipped with collaborative ILD-rheumatology clinics or even easy access to a rheumatologist. There is a need for real-world pragmatic studies to establish the optimal way to evaluate patients with ILD for autoimmune features and identify patients who would benefit most from an early referral to rheumatology to aid with diagnosis, treatment, and sometimes monitoring for extrapulmonary manifestations of autoimmune disorders.
Avanthika Thanushi Wynn, MD
Steering Committee Fellow-in-Training
Renal replacement therapy in the ICU: Vexed questions and team dynamics
More than 5 million patients are admitted to ICUs each year in the United States, and approximately 2% to 10% of these patients develop acute kidney injury requiring renal replacement therapy (AKI-RRT). AKI-RRT carries high morbidity and mortality (Hoste EA, et al. Intensive Care Med. 2015;41:1411) and is associated with renal and systemic complications, such as cardiovascular disease. RRT, frequently provided by nephrologists and/or intensivists, is a supportive therapy that can be life-saving when provided to the right patient at the right time. However, several questions related to the provision of RRT still remain, including the optimal timing of RRT initiation, the development of quality metrics for optimal RRT deliverables and monitoring, and the optimal strategy of RRT de-escalation and risk-stratification of renal recovery. Overall, there is paucity of randomized trials and standardized risk-stratification tools that can guide RRT in the ICU.
Current vexed questions of RRT deliverables in the ICU
There is ongoing research aiming to answer critical questions that can potentially improve current standards of RRT.
What is the optimal time of RRT initiation for critically ill patients with AKI?
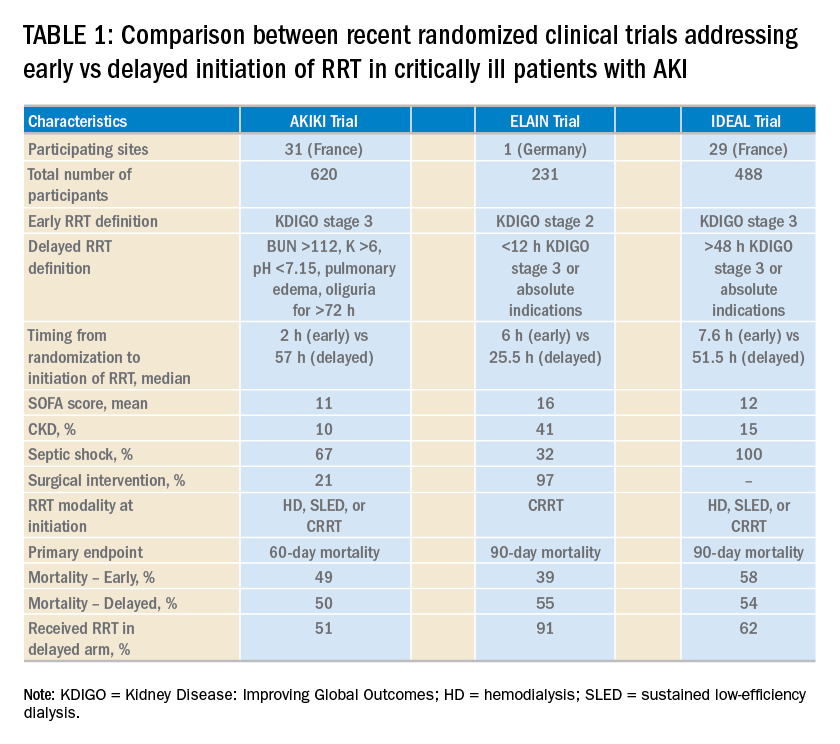
Over the last 2 years, three randomized clinical trials have attempted to address this important question involving heterogeneous ICU populations and distinct research hypotheses and study designs. Two of these studies, AKIKI (Gaudry S, et al. N Engl J Med. 2016;375:122) and IDEAL-ICU (Barbar SD, et al. N Engl J Med. 2018;379:1431) yielded no significant difference in the primary outcome of 60-day and 90-day all-cause mortality between the early vs delayed RRT initiation strategies, respectively (Table 1). Further, AKIKI showed no difference in RRT dependence at 60 days and higher catheter-related infections and hypophosphatemia in the early initiation arm. It is important to note that IDEAL-ICU was stopped early for futility after the second planned interim analysis with only 56% of patients enrolled (
How can RRT deliverables in the ICU be effectively and systematically monitored?

The provision of RRT to ICU patients with AKI requires an iterative adjustment of the RRT prescription and goals of therapy to accommodate changes in the clinical status with emphasis in hemodynamics, multiorgan failure, and fluid overload (Neyra JA. Clin Nephrol. 2018;90:1). The utilization of static and functional tests or point-of-care ultrasonography to assess hemodynamic variables can be useful. Furthermore, the implementation of customized and automated flowsheets in the electronic health record can facilitate remote monitoring. It is, therefore, essential that the multidisciplinary ICU team develops a process to monitor and ensure RRT deliverables. In this context, the standardization and monitoring of quality metrics (dose, modality, anticoagulation, filter life, downtime, etc) and the development of effective quality management systems are critically important. However, big multicenter data are direly needed to provide insight in this arena.
How can renal recovery be assessed and RRT effectively de-escalated?

The continuous examination of renal recovery in ICU patients with AKI-RRT is mostly based on urine output trend and, if feasible, interdialytic solute control. Sometimes, the transition from continuous RRT to intermittent modalities is necessary in the context of multiorgan recovery and de-escalation of care. However, clinical risk-prediction tools that identify patients who can potentially recover or already exhibit early signs of renal function recovery are needed. Current advances in clinical informatics can help to incorporate time-varying clinical parameters that may be informative for risk-prediction models. In addition, incorporating novel biomarkers of AKI repair and functional tests (eg, furosemide stress test, functional MRI) into these models may further inform these tools and aid the development of clinical decision support systems that enhance interventions to promote AKI recovery (Neyra JA, et al. Nephron. 2018;140: 99).
Is post-AKI outpatient care beneficial for ICU survivors who suffered from AKI-RRT?
Specialized AKI survivor clinics have been implemented in some centers. In general, this outpatient follow-up model includes survivors who suffered from AKI stage 2 or 3, some of them requiring RRT, and tailors individualized interventions for post-AKI complications (preventing recurrent AKI, attenuating incident or progressive CKD). However, the value of this outpatient model needs to be further evaluated with emphasis on clinical outcomes (eg, recurrent AKI, CKD, readmissions, or death) and elements that impact quality of life. This is an area of evolving research and a great opportunity for the nephrology and critical care communities to integrate and enhance post-ICU outpatient care and research collaboration.
Interdisciplinary communication among acute care team members
Two essential elements to provide effective RRT to ICU patients with AKI are: (1) the dynamics of the ICU team (intensivists, nephrologists, pharmacists, nurses, nutritionists, physical therapists, etc) to enhance the delivery of personalized therapy (RRT candidacy, timing of initiation, goals for solute control and fluid removal/regulation, renal recovery evaluation, RRT de-escalation, etc.) and (2) the frequent assessment and adjustment of RRT goals according to the clinical status of the patient. Therefore, effective RRT provision in the ICU requires the development of optimal channels of communication among all members of the acute care team and the systematic monitoring of the clinical status of the patient and RRT-specific goals and deliverables.
Perspective from a nurse and quality improvement officer for the provision of RRT in the ICU
The provision of continuous RRT (CRRT) to critically ill patients requires close communication between the bedside nurse and the rest of the ICU team. The physician typically prescribes CRRT and determines the specific goals of therapy. The pharmacist works closely with the nephrologist/intensivist and bedside nurse, especially in regards to customized CRRT solutions (when indicated) and medication dosing. Because CRRT can alter drug pharmacokinetics, the pharmacist closely and constantly monitors the patient’s clinical status, CRRT prescription, and all active medications. CRRT can also affect the nutritional and metabolic status of critically ill patients; therefore, the input of the nutritionist is necessary. The syndrome of ICU-acquired weakness is commonly encountered in ICU patients and is related to physical immobility. While ICU patients with AKI are already at risk for decreased mobility, the continuous connection to an immobile extracorporeal machine for the provision of CRRT may further contribute to immobilization and can also preclude the provision of optimal physical therapy. Therefore, the bedside nurse should assist the physical therapist for the timely and effective delivery of physical therapy according to the clinical status of the patient.
The clinical scenarios discussed above provide a small glimpse into the importance of developing an interdisciplinary ICU team caring for critically ill patients receiving CRRT. In the context of how integral the specific role of each team member is, it becomes clear that the bedside nurse’s role is not only to deliver hands-on patient care but also the orchestration of collaborative communication among all health-care providers for the effective provision of CRRT to critically ill patients in the ICU.
Dr. Neyra and Ms. Hauschild are with the Department of Internal Medicine; Division of Nephrology; Bone and Mineral Metabolism; University of Kentucky; Lexington, Kentucky.
More than 5 million patients are admitted to ICUs each year in the United States, and approximately 2% to 10% of these patients develop acute kidney injury requiring renal replacement therapy (AKI-RRT). AKI-RRT carries high morbidity and mortality (Hoste EA, et al. Intensive Care Med. 2015;41:1411) and is associated with renal and systemic complications, such as cardiovascular disease. RRT, frequently provided by nephrologists and/or intensivists, is a supportive therapy that can be life-saving when provided to the right patient at the right time. However, several questions related to the provision of RRT still remain, including the optimal timing of RRT initiation, the development of quality metrics for optimal RRT deliverables and monitoring, and the optimal strategy of RRT de-escalation and risk-stratification of renal recovery. Overall, there is paucity of randomized trials and standardized risk-stratification tools that can guide RRT in the ICU.
Current vexed questions of RRT deliverables in the ICU
There is ongoing research aiming to answer critical questions that can potentially improve current standards of RRT.
What is the optimal time of RRT initiation for critically ill patients with AKI?
Over the last 2 years, three randomized clinical trials have attempted to address this important question involving heterogeneous ICU populations and distinct research hypotheses and study designs. Two of these studies, AKIKI (Gaudry S, et al. N Engl J Med. 2016;375:122) and IDEAL-ICU (Barbar SD, et al. N Engl J Med. 2018;379:1431) yielded no significant difference in the primary outcome of 60-day and 90-day all-cause mortality between the early vs delayed RRT initiation strategies, respectively (Table 1). Further, AKIKI showed no difference in RRT dependence at 60 days and higher catheter-related infections and hypophosphatemia in the early initiation arm. It is important to note that IDEAL-ICU was stopped early for futility after the second planned interim analysis with only 56% of patients enrolled (
How can RRT deliverables in the ICU be effectively and systematically monitored?
The provision of RRT to ICU patients with AKI requires an iterative adjustment of the RRT prescription and goals of therapy to accommodate changes in the clinical status with emphasis in hemodynamics, multiorgan failure, and fluid overload (Neyra JA. Clin Nephrol. 2018;90:1). The utilization of static and functional tests or point-of-care ultrasonography to assess hemodynamic variables can be useful. Furthermore, the implementation of customized and automated flowsheets in the electronic health record can facilitate remote monitoring. It is, therefore, essential that the multidisciplinary ICU team develops a process to monitor and ensure RRT deliverables. In this context, the standardization and monitoring of quality metrics (dose, modality, anticoagulation, filter life, downtime, etc) and the development of effective quality management systems are critically important. However, big multicenter data are direly needed to provide insight in this arena.
How can renal recovery be assessed and RRT effectively de-escalated?
The continuous examination of renal recovery in ICU patients with AKI-RRT is mostly based on urine output trend and, if feasible, interdialytic solute control. Sometimes, the transition from continuous RRT to intermittent modalities is necessary in the context of multiorgan recovery and de-escalation of care. However, clinical risk-prediction tools that identify patients who can potentially recover or already exhibit early signs of renal function recovery are needed. Current advances in clinical informatics can help to incorporate time-varying clinical parameters that may be informative for risk-prediction models. In addition, incorporating novel biomarkers of AKI repair and functional tests (eg, furosemide stress test, functional MRI) into these models may further inform these tools and aid the development of clinical decision support systems that enhance interventions to promote AKI recovery (Neyra JA, et al. Nephron. 2018;140: 99).
Is post-AKI outpatient care beneficial for ICU survivors who suffered from AKI-RRT?
Specialized AKI survivor clinics have been implemented in some centers. In general, this outpatient follow-up model includes survivors who suffered from AKI stage 2 or 3, some of them requiring RRT, and tailors individualized interventions for post-AKI complications (preventing recurrent AKI, attenuating incident or progressive CKD). However, the value of this outpatient model needs to be further evaluated with emphasis on clinical outcomes (eg, recurrent AKI, CKD, readmissions, or death) and elements that impact quality of life. This is an area of evolving research and a great opportunity for the nephrology and critical care communities to integrate and enhance post-ICU outpatient care and research collaboration.
Interdisciplinary communication among acute care team members
Two essential elements to provide effective RRT to ICU patients with AKI are: (1) the dynamics of the ICU team (intensivists, nephrologists, pharmacists, nurses, nutritionists, physical therapists, etc) to enhance the delivery of personalized therapy (RRT candidacy, timing of initiation, goals for solute control and fluid removal/regulation, renal recovery evaluation, RRT de-escalation, etc.) and (2) the frequent assessment and adjustment of RRT goals according to the clinical status of the patient. Therefore, effective RRT provision in the ICU requires the development of optimal channels of communication among all members of the acute care team and the systematic monitoring of the clinical status of the patient and RRT-specific goals and deliverables.
Perspective from a nurse and quality improvement officer for the provision of RRT in the ICU
The provision of continuous RRT (CRRT) to critically ill patients requires close communication between the bedside nurse and the rest of the ICU team. The physician typically prescribes CRRT and determines the specific goals of therapy. The pharmacist works closely with the nephrologist/intensivist and bedside nurse, especially in regards to customized CRRT solutions (when indicated) and medication dosing. Because CRRT can alter drug pharmacokinetics, the pharmacist closely and constantly monitors the patient’s clinical status, CRRT prescription, and all active medications. CRRT can also affect the nutritional and metabolic status of critically ill patients; therefore, the input of the nutritionist is necessary. The syndrome of ICU-acquired weakness is commonly encountered in ICU patients and is related to physical immobility. While ICU patients with AKI are already at risk for decreased mobility, the continuous connection to an immobile extracorporeal machine for the provision of CRRT may further contribute to immobilization and can also preclude the provision of optimal physical therapy. Therefore, the bedside nurse should assist the physical therapist for the timely and effective delivery of physical therapy according to the clinical status of the patient.
The clinical scenarios discussed above provide a small glimpse into the importance of developing an interdisciplinary ICU team caring for critically ill patients receiving CRRT. In the context of how integral the specific role of each team member is, it becomes clear that the bedside nurse’s role is not only to deliver hands-on patient care but also the orchestration of collaborative communication among all health-care providers for the effective provision of CRRT to critically ill patients in the ICU.
Dr. Neyra and Ms. Hauschild are with the Department of Internal Medicine; Division of Nephrology; Bone and Mineral Metabolism; University of Kentucky; Lexington, Kentucky.
More than 5 million patients are admitted to ICUs each year in the United States, and approximately 2% to 10% of these patients develop acute kidney injury requiring renal replacement therapy (AKI-RRT). AKI-RRT carries high morbidity and mortality (Hoste EA, et al. Intensive Care Med. 2015;41:1411) and is associated with renal and systemic complications, such as cardiovascular disease. RRT, frequently provided by nephrologists and/or intensivists, is a supportive therapy that can be life-saving when provided to the right patient at the right time. However, several questions related to the provision of RRT still remain, including the optimal timing of RRT initiation, the development of quality metrics for optimal RRT deliverables and monitoring, and the optimal strategy of RRT de-escalation and risk-stratification of renal recovery. Overall, there is paucity of randomized trials and standardized risk-stratification tools that can guide RRT in the ICU.
Current vexed questions of RRT deliverables in the ICU
There is ongoing research aiming to answer critical questions that can potentially improve current standards of RRT.
What is the optimal time of RRT initiation for critically ill patients with AKI?
Over the last 2 years, three randomized clinical trials have attempted to address this important question involving heterogeneous ICU populations and distinct research hypotheses and study designs. Two of these studies, AKIKI (Gaudry S, et al. N Engl J Med. 2016;375:122) and IDEAL-ICU (Barbar SD, et al. N Engl J Med. 2018;379:1431) yielded no significant difference in the primary outcome of 60-day and 90-day all-cause mortality between the early vs delayed RRT initiation strategies, respectively (Table 1). Further, AKIKI showed no difference in RRT dependence at 60 days and higher catheter-related infections and hypophosphatemia in the early initiation arm. It is important to note that IDEAL-ICU was stopped early for futility after the second planned interim analysis with only 56% of patients enrolled (
How can RRT deliverables in the ICU be effectively and systematically monitored?
The provision of RRT to ICU patients with AKI requires an iterative adjustment of the RRT prescription and goals of therapy to accommodate changes in the clinical status with emphasis in hemodynamics, multiorgan failure, and fluid overload (Neyra JA. Clin Nephrol. 2018;90:1). The utilization of static and functional tests or point-of-care ultrasonography to assess hemodynamic variables can be useful. Furthermore, the implementation of customized and automated flowsheets in the electronic health record can facilitate remote monitoring. It is, therefore, essential that the multidisciplinary ICU team develops a process to monitor and ensure RRT deliverables. In this context, the standardization and monitoring of quality metrics (dose, modality, anticoagulation, filter life, downtime, etc) and the development of effective quality management systems are critically important. However, big multicenter data are direly needed to provide insight in this arena.
How can renal recovery be assessed and RRT effectively de-escalated?
The continuous examination of renal recovery in ICU patients with AKI-RRT is mostly based on urine output trend and, if feasible, interdialytic solute control. Sometimes, the transition from continuous RRT to intermittent modalities is necessary in the context of multiorgan recovery and de-escalation of care. However, clinical risk-prediction tools that identify patients who can potentially recover or already exhibit early signs of renal function recovery are needed. Current advances in clinical informatics can help to incorporate time-varying clinical parameters that may be informative for risk-prediction models. In addition, incorporating novel biomarkers of AKI repair and functional tests (eg, furosemide stress test, functional MRI) into these models may further inform these tools and aid the development of clinical decision support systems that enhance interventions to promote AKI recovery (Neyra JA, et al. Nephron. 2018;140: 99).
Is post-AKI outpatient care beneficial for ICU survivors who suffered from AKI-RRT?
Specialized AKI survivor clinics have been implemented in some centers. In general, this outpatient follow-up model includes survivors who suffered from AKI stage 2 or 3, some of them requiring RRT, and tailors individualized interventions for post-AKI complications (preventing recurrent AKI, attenuating incident or progressive CKD). However, the value of this outpatient model needs to be further evaluated with emphasis on clinical outcomes (eg, recurrent AKI, CKD, readmissions, or death) and elements that impact quality of life. This is an area of evolving research and a great opportunity for the nephrology and critical care communities to integrate and enhance post-ICU outpatient care and research collaboration.
Interdisciplinary communication among acute care team members
Two essential elements to provide effective RRT to ICU patients with AKI are: (1) the dynamics of the ICU team (intensivists, nephrologists, pharmacists, nurses, nutritionists, physical therapists, etc) to enhance the delivery of personalized therapy (RRT candidacy, timing of initiation, goals for solute control and fluid removal/regulation, renal recovery evaluation, RRT de-escalation, etc.) and (2) the frequent assessment and adjustment of RRT goals according to the clinical status of the patient. Therefore, effective RRT provision in the ICU requires the development of optimal channels of communication among all members of the acute care team and the systematic monitoring of the clinical status of the patient and RRT-specific goals and deliverables.
Perspective from a nurse and quality improvement officer for the provision of RRT in the ICU
The provision of continuous RRT (CRRT) to critically ill patients requires close communication between the bedside nurse and the rest of the ICU team. The physician typically prescribes CRRT and determines the specific goals of therapy. The pharmacist works closely with the nephrologist/intensivist and bedside nurse, especially in regards to customized CRRT solutions (when indicated) and medication dosing. Because CRRT can alter drug pharmacokinetics, the pharmacist closely and constantly monitors the patient’s clinical status, CRRT prescription, and all active medications. CRRT can also affect the nutritional and metabolic status of critically ill patients; therefore, the input of the nutritionist is necessary. The syndrome of ICU-acquired weakness is commonly encountered in ICU patients and is related to physical immobility. While ICU patients with AKI are already at risk for decreased mobility, the continuous connection to an immobile extracorporeal machine for the provision of CRRT may further contribute to immobilization and can also preclude the provision of optimal physical therapy. Therefore, the bedside nurse should assist the physical therapist for the timely and effective delivery of physical therapy according to the clinical status of the patient.
The clinical scenarios discussed above provide a small glimpse into the importance of developing an interdisciplinary ICU team caring for critically ill patients receiving CRRT. In the context of how integral the specific role of each team member is, it becomes clear that the bedside nurse’s role is not only to deliver hands-on patient care but also the orchestration of collaborative communication among all health-care providers for the effective provision of CRRT to critically ill patients in the ICU.
Dr. Neyra and Ms. Hauschild are with the Department of Internal Medicine; Division of Nephrology; Bone and Mineral Metabolism; University of Kentucky; Lexington, Kentucky.
An update on chronic thromboembolic pulmonary hypertension
The “fixable” form of PH that you don’t want to miss
Chronic thromboembolic pulmonary hypertension (CTEPH) is an elevation in pulmonary vascular resistance (PVR) resulting from chronic, “scarred-in” thromboembolic material partially occluding the pulmonary arteries. This vascular obstruction, over time, results in failure of the right ventricle and early mortality.

CTEPH was first characterized in an autopsy series from the Massachusetts General Hospital in 1931. On these postmortem examinations, it was noted that the affected patients had large pulmonary artery vascular obstruction, but also normal pulmonary parenchyma distal to this vascular obstruction and extensive bronchial collateral blood flow (Means J. Ann Intern Med. 1931;5:417). Although this observation set the groundwork for the theory that surgically removing the vascular obstruction to this preserved lung tissue could improve the condition of these patients, it would take until the mid-20th century until imaging and cardiac catheterization techniques allowed the recognition of the disease in real time.
CTEPH is thought to begin with an acute pulmonary embolus, but in approximately 3.4% of patients, rather than resolving over time, the thrombus will organize and incorporate into the pulmonary artery intimal layer (Simonneau G, et al. Eur Respir Rev. 2017;26:160112) A history of venous thromboembolism in a patient with persistent dyspnea should spur a screening evaluation for CTEPH; 75% of patients with CTEPH have a history of prior known acute pulmonary embolus and 56% of patients report a prior diagnosis of deep venous thrombosis. An acute pulmonary embolus will fibrinolyse early with the vast majority of the vascular obstruction resolving by the third month. Therefore, if the patient continues to report a significant exercise limitation after 3 months of therapeutic anticoagulation therapy, or has concerning physical exam signs, a workup should be pursued. The initial evaluation for CTEPH begins with a transthoracic echocardiogram (TTE) and ventilation/perfusion (V/Q) scintigraphy. A retrospective study comparing V/Q scan and multidetector CT scan revealed that V/Q scanning had a sensitivity and specificity of 97% and 95% for CTEPH, while CTPA had good specificity at 99% but only 51% sensitivity (Tunariu N, et al. J Nuc Med. 2007;48(5):680). If these are abnormal, then right-sided heart catheterization and invasive biplane digital subtraction pulmonary angiography are recommended. These studies confirm the diagnosis, grade its severity, and allow an evaluation for surgically accessible vs distal disease. Some CTEPH centers utilize additional imaging techniques, such as magnetic resonance angiography, optical resonance imaging, spectral CT scanning with iodine perfusion images, and intravascular ultrasound. These modalities and their place in the diagnostic algorithm are under investigation.
The goal of the initial evaluation process is to determine if the patient can undergo surgical pulmonary thromboendarterectomy (PTE), because in experienced hands, this procedure ensures the best long-term outcome for the patient. The first pulmonary thromboendarterectomy was performed at the University of California San Diego in 1970. Because the disease involves the intimal layer of the pulmonary artery, the surgery had to involve not just removal of the intravascular obstruction but also a pulmonary artery intimectomy. Surgical mortality rates were high in the initial experience. In 1984, a review of 85 worldwide cases reported an average mortality rate of 22%, and as high as 40% in some centers (Chitwood WR, Jr, et al. Clin Chest Med. 1984;5(3):507).
Over the ensuing years, refinements in surgical technique, the utilization of deep hypothermia and cardiac arrest during the procedure, development of new surgical instruments, and standardization of surgical selection and postoperative care have improved surgical mortality to <5% in experienced centers. Long-term outcomes of successful PTE surgery remain good, with 90% 3-year survival vs 70% for those who do not undergo surgery and are medically treated. Importantly, 90% of postoperative patients report functional class I or II symptoms at 1 year (Condliffe R, et al. Am J Reslpir Crit Care Med. 2008:177(10);1122). Because of this difference in early mortality and symptoms, PTE surgery remains the treatment of choice for CTEPH.
Despite the advances in PTE surgery, some patients are not operative candidates either due to surgically inaccessible disease or due to comorbidities. In 2001, Feinstein and colleagues described a series of 18 CTEPH cases treated with balloon pulmonary angioplasty (BPA). Promising hemodynamics effects were reported; however, the procedure had an unacceptable complication rate in which 11 patients developed reperfusion lung injury, 3 patients required mechanical ventilation, and 1 patient died. In the ensuing years, Japanese and Norwegian groups have independently developed and improved techniques for BPA. The procedure is done in a series of sessions (average four to six), 1 to 4 weeks apart, where small (2-3 mm) balloons are directed toward distal, diseased pulmonary vessels. Common complications include reperfusion injury, vessel injury, hemoptysis, and, more rarely, respiratory failure. Still, early experience suggests this procedure decreases pulmonary vascular resistance over time, improves right ventricular function, and improves patients’ symptoms (Andreassen A, et al. Heart. 2013;99(19):1415). The experience with this procedure is limited but growing in the United States, with only a handful of centers currently performing BPAs and collecting data.
Lifelong anticoagulation, oxygen, and diuretics for right-sided heart failure are recommended for patients with CTEPH. The first successful large phase III medication study for CTEPH was the CHEST-1 trial published in 2013. This was a multicenter, randomized, placebo-controlled trial of the soluble guanylate cyclase stimulator riociguat. The study enrolled 261 patients with inoperable CTEPH or persistent pulmonary hypertension after surgery. The primary end point was 6-minute walk distance at 12 weeks. The treatment group showed a 46 m improvement (P<.001). Secondary end points of pulmonary vascular resistance, NT-proBNP level, and functional class also improved. This pivotal trial led to the FDA approval of riociguat for inoperable or persistent postoperative CTEPH.
MERIT-1, a phase II, randomized placebo-controlled double trial of macitentan (an oral endothelin receptor antagonist) was recently completed. It enrolled 80 patients with inoperable CTEPH. The primary endpoint was pulmonary vascular resistance at week 16, expressed as a percentage of baseline. At week 16, the patients in the treatment arm had a PVR 73% of baseline vs 87.2% in the treatment group. This medication is not yet FDA-approved for the treatment of inoperable CTEPH (Ghofrani H, et al. Lancet Respir Med. 2017;5(10):785-794).
Pulmonary hypertension medication has been postulated as a possible way to “pretreat” patients before pulmonary thromboendarterectomy surgery, perhaps lowering preoperative pulmonary vascular resistance and surgical risk. However, there are currently no convincing data to support this practice, and medical treatment has been associated with a possible counterproductive delay in surgery. A phase II study including CTEPH patients with high PVR for preoperative treatment with riociguat vs placebo is currently enrolling to determine if “induction” treatment with medication prior to surgery reduces risk or delays definitive surgery. Occasionally, patients are found who have persistent thrombus but not pulmonary hypertension. Chronic thromboembolic disease (CTED) is a recently coined term describing patients who have chronic thromboembolism on imaging but have normal resting hemodynamics. Whether CTED represents simply unresolved clot that will never progress to CTEPH or is an early point on the continuum of disease not well-defined and a controversial topic among experts. At many centers, patients with CTED and symptoms will undergo exercise testing to look for exercise -induced pulmonary hypertension or an increase in dead space ventilation as a cause of their symptoms. A retrospective series of carefully chosen CTED patients who underwent PTE surgery reported improvements in symptoms and overall quality of life, without increased complications (Taboada D, et al. Eur Respir J. 2014 44(6):1635). The operation carries risk, however, and further work into the epidemiology and prognosis of CTED is required before operative intervention can be recommended.
In conclusion, CTEPH is a disease that rarely occurs after an acute PE but when undiagnosed and untreated portends a poor prognosis. The definitive treatment for this disease is surgical PTE, but to achieve the best outcomes, this procedure needs to be performed at expert centers with multidisciplinary team experience. Patients who are poor operative candidates or with surgically inaccessible disease may be considered for balloon pulmonary angioplasty. For patients without more curative options, medication improves exercise tolerance. The field of CTEPH has been rapidly expanding over the last decade, leading to better patient outcomes and more treatment options.
Dr. Bartolome is Associate Professor, Pulmonary and Critical Care Medicine; Director, CTEPH Program; and Associate Director, PH Program; UT Southwestern Medical Center, Dallas, Texas.
The “fixable” form of PH that you don’t want to miss
The “fixable” form of PH that you don’t want to miss
Chronic thromboembolic pulmonary hypertension (CTEPH) is an elevation in pulmonary vascular resistance (PVR) resulting from chronic, “scarred-in” thromboembolic material partially occluding the pulmonary arteries. This vascular obstruction, over time, results in failure of the right ventricle and early mortality.
CTEPH was first characterized in an autopsy series from the Massachusetts General Hospital in 1931. On these postmortem examinations, it was noted that the affected patients had large pulmonary artery vascular obstruction, but also normal pulmonary parenchyma distal to this vascular obstruction and extensive bronchial collateral blood flow (Means J. Ann Intern Med. 1931;5:417). Although this observation set the groundwork for the theory that surgically removing the vascular obstruction to this preserved lung tissue could improve the condition of these patients, it would take until the mid-20th century until imaging and cardiac catheterization techniques allowed the recognition of the disease in real time.
CTEPH is thought to begin with an acute pulmonary embolus, but in approximately 3.4% of patients, rather than resolving over time, the thrombus will organize and incorporate into the pulmonary artery intimal layer (Simonneau G, et al. Eur Respir Rev. 2017;26:160112) A history of venous thromboembolism in a patient with persistent dyspnea should spur a screening evaluation for CTEPH; 75% of patients with CTEPH have a history of prior known acute pulmonary embolus and 56% of patients report a prior diagnosis of deep venous thrombosis. An acute pulmonary embolus will fibrinolyse early with the vast majority of the vascular obstruction resolving by the third month. Therefore, if the patient continues to report a significant exercise limitation after 3 months of therapeutic anticoagulation therapy, or has concerning physical exam signs, a workup should be pursued. The initial evaluation for CTEPH begins with a transthoracic echocardiogram (TTE) and ventilation/perfusion (V/Q) scintigraphy. A retrospective study comparing V/Q scan and multidetector CT scan revealed that V/Q scanning had a sensitivity and specificity of 97% and 95% for CTEPH, while CTPA had good specificity at 99% but only 51% sensitivity (Tunariu N, et al. J Nuc Med. 2007;48(5):680). If these are abnormal, then right-sided heart catheterization and invasive biplane digital subtraction pulmonary angiography are recommended. These studies confirm the diagnosis, grade its severity, and allow an evaluation for surgically accessible vs distal disease. Some CTEPH centers utilize additional imaging techniques, such as magnetic resonance angiography, optical resonance imaging, spectral CT scanning with iodine perfusion images, and intravascular ultrasound. These modalities and their place in the diagnostic algorithm are under investigation.
The goal of the initial evaluation process is to determine if the patient can undergo surgical pulmonary thromboendarterectomy (PTE), because in experienced hands, this procedure ensures the best long-term outcome for the patient. The first pulmonary thromboendarterectomy was performed at the University of California San Diego in 1970. Because the disease involves the intimal layer of the pulmonary artery, the surgery had to involve not just removal of the intravascular obstruction but also a pulmonary artery intimectomy. Surgical mortality rates were high in the initial experience. In 1984, a review of 85 worldwide cases reported an average mortality rate of 22%, and as high as 40% in some centers (Chitwood WR, Jr, et al. Clin Chest Med. 1984;5(3):507).
Over the ensuing years, refinements in surgical technique, the utilization of deep hypothermia and cardiac arrest during the procedure, development of new surgical instruments, and standardization of surgical selection and postoperative care have improved surgical mortality to <5% in experienced centers. Long-term outcomes of successful PTE surgery remain good, with 90% 3-year survival vs 70% for those who do not undergo surgery and are medically treated. Importantly, 90% of postoperative patients report functional class I or II symptoms at 1 year (Condliffe R, et al. Am J Reslpir Crit Care Med. 2008:177(10);1122). Because of this difference in early mortality and symptoms, PTE surgery remains the treatment of choice for CTEPH.
Despite the advances in PTE surgery, some patients are not operative candidates either due to surgically inaccessible disease or due to comorbidities. In 2001, Feinstein and colleagues described a series of 18 CTEPH cases treated with balloon pulmonary angioplasty (BPA). Promising hemodynamics effects were reported; however, the procedure had an unacceptable complication rate in which 11 patients developed reperfusion lung injury, 3 patients required mechanical ventilation, and 1 patient died. In the ensuing years, Japanese and Norwegian groups have independently developed and improved techniques for BPA. The procedure is done in a series of sessions (average four to six), 1 to 4 weeks apart, where small (2-3 mm) balloons are directed toward distal, diseased pulmonary vessels. Common complications include reperfusion injury, vessel injury, hemoptysis, and, more rarely, respiratory failure. Still, early experience suggests this procedure decreases pulmonary vascular resistance over time, improves right ventricular function, and improves patients’ symptoms (Andreassen A, et al. Heart. 2013;99(19):1415). The experience with this procedure is limited but growing in the United States, with only a handful of centers currently performing BPAs and collecting data.
Lifelong anticoagulation, oxygen, and diuretics for right-sided heart failure are recommended for patients with CTEPH. The first successful large phase III medication study for CTEPH was the CHEST-1 trial published in 2013. This was a multicenter, randomized, placebo-controlled trial of the soluble guanylate cyclase stimulator riociguat. The study enrolled 261 patients with inoperable CTEPH or persistent pulmonary hypertension after surgery. The primary end point was 6-minute walk distance at 12 weeks. The treatment group showed a 46 m improvement (P<.001). Secondary end points of pulmonary vascular resistance, NT-proBNP level, and functional class also improved. This pivotal trial led to the FDA approval of riociguat for inoperable or persistent postoperative CTEPH.
MERIT-1, a phase II, randomized placebo-controlled double trial of macitentan (an oral endothelin receptor antagonist) was recently completed. It enrolled 80 patients with inoperable CTEPH. The primary endpoint was pulmonary vascular resistance at week 16, expressed as a percentage of baseline. At week 16, the patients in the treatment arm had a PVR 73% of baseline vs 87.2% in the treatment group. This medication is not yet FDA-approved for the treatment of inoperable CTEPH (Ghofrani H, et al. Lancet Respir Med. 2017;5(10):785-794).
Pulmonary hypertension medication has been postulated as a possible way to “pretreat” patients before pulmonary thromboendarterectomy surgery, perhaps lowering preoperative pulmonary vascular resistance and surgical risk. However, there are currently no convincing data to support this practice, and medical treatment has been associated with a possible counterproductive delay in surgery. A phase II study including CTEPH patients with high PVR for preoperative treatment with riociguat vs placebo is currently enrolling to determine if “induction” treatment with medication prior to surgery reduces risk or delays definitive surgery. Occasionally, patients are found who have persistent thrombus but not pulmonary hypertension. Chronic thromboembolic disease (CTED) is a recently coined term describing patients who have chronic thromboembolism on imaging but have normal resting hemodynamics. Whether CTED represents simply unresolved clot that will never progress to CTEPH or is an early point on the continuum of disease not well-defined and a controversial topic among experts. At many centers, patients with CTED and symptoms will undergo exercise testing to look for exercise -induced pulmonary hypertension or an increase in dead space ventilation as a cause of their symptoms. A retrospective series of carefully chosen CTED patients who underwent PTE surgery reported improvements in symptoms and overall quality of life, without increased complications (Taboada D, et al. Eur Respir J. 2014 44(6):1635). The operation carries risk, however, and further work into the epidemiology and prognosis of CTED is required before operative intervention can be recommended.
In conclusion, CTEPH is a disease that rarely occurs after an acute PE but when undiagnosed and untreated portends a poor prognosis. The definitive treatment for this disease is surgical PTE, but to achieve the best outcomes, this procedure needs to be performed at expert centers with multidisciplinary team experience. Patients who are poor operative candidates or with surgically inaccessible disease may be considered for balloon pulmonary angioplasty. For patients without more curative options, medication improves exercise tolerance. The field of CTEPH has been rapidly expanding over the last decade, leading to better patient outcomes and more treatment options.
Dr. Bartolome is Associate Professor, Pulmonary and Critical Care Medicine; Director, CTEPH Program; and Associate Director, PH Program; UT Southwestern Medical Center, Dallas, Texas.
Chronic thromboembolic pulmonary hypertension (CTEPH) is an elevation in pulmonary vascular resistance (PVR) resulting from chronic, “scarred-in” thromboembolic material partially occluding the pulmonary arteries. This vascular obstruction, over time, results in failure of the right ventricle and early mortality.
CTEPH was first characterized in an autopsy series from the Massachusetts General Hospital in 1931. On these postmortem examinations, it was noted that the affected patients had large pulmonary artery vascular obstruction, but also normal pulmonary parenchyma distal to this vascular obstruction and extensive bronchial collateral blood flow (Means J. Ann Intern Med. 1931;5:417). Although this observation set the groundwork for the theory that surgically removing the vascular obstruction to this preserved lung tissue could improve the condition of these patients, it would take until the mid-20th century until imaging and cardiac catheterization techniques allowed the recognition of the disease in real time.
CTEPH is thought to begin with an acute pulmonary embolus, but in approximately 3.4% of patients, rather than resolving over time, the thrombus will organize and incorporate into the pulmonary artery intimal layer (Simonneau G, et al. Eur Respir Rev. 2017;26:160112) A history of venous thromboembolism in a patient with persistent dyspnea should spur a screening evaluation for CTEPH; 75% of patients with CTEPH have a history of prior known acute pulmonary embolus and 56% of patients report a prior diagnosis of deep venous thrombosis. An acute pulmonary embolus will fibrinolyse early with the vast majority of the vascular obstruction resolving by the third month. Therefore, if the patient continues to report a significant exercise limitation after 3 months of therapeutic anticoagulation therapy, or has concerning physical exam signs, a workup should be pursued. The initial evaluation for CTEPH begins with a transthoracic echocardiogram (TTE) and ventilation/perfusion (V/Q) scintigraphy. A retrospective study comparing V/Q scan and multidetector CT scan revealed that V/Q scanning had a sensitivity and specificity of 97% and 95% for CTEPH, while CTPA had good specificity at 99% but only 51% sensitivity (Tunariu N, et al. J Nuc Med. 2007;48(5):680). If these are abnormal, then right-sided heart catheterization and invasive biplane digital subtraction pulmonary angiography are recommended. These studies confirm the diagnosis, grade its severity, and allow an evaluation for surgically accessible vs distal disease. Some CTEPH centers utilize additional imaging techniques, such as magnetic resonance angiography, optical resonance imaging, spectral CT scanning with iodine perfusion images, and intravascular ultrasound. These modalities and their place in the diagnostic algorithm are under investigation.
The goal of the initial evaluation process is to determine if the patient can undergo surgical pulmonary thromboendarterectomy (PTE), because in experienced hands, this procedure ensures the best long-term outcome for the patient. The first pulmonary thromboendarterectomy was performed at the University of California San Diego in 1970. Because the disease involves the intimal layer of the pulmonary artery, the surgery had to involve not just removal of the intravascular obstruction but also a pulmonary artery intimectomy. Surgical mortality rates were high in the initial experience. In 1984, a review of 85 worldwide cases reported an average mortality rate of 22%, and as high as 40% in some centers (Chitwood WR, Jr, et al. Clin Chest Med. 1984;5(3):507).
Over the ensuing years, refinements in surgical technique, the utilization of deep hypothermia and cardiac arrest during the procedure, development of new surgical instruments, and standardization of surgical selection and postoperative care have improved surgical mortality to <5% in experienced centers. Long-term outcomes of successful PTE surgery remain good, with 90% 3-year survival vs 70% for those who do not undergo surgery and are medically treated. Importantly, 90% of postoperative patients report functional class I or II symptoms at 1 year (Condliffe R, et al. Am J Reslpir Crit Care Med. 2008:177(10);1122). Because of this difference in early mortality and symptoms, PTE surgery remains the treatment of choice for CTEPH.
Despite the advances in PTE surgery, some patients are not operative candidates either due to surgically inaccessible disease or due to comorbidities. In 2001, Feinstein and colleagues described a series of 18 CTEPH cases treated with balloon pulmonary angioplasty (BPA). Promising hemodynamics effects were reported; however, the procedure had an unacceptable complication rate in which 11 patients developed reperfusion lung injury, 3 patients required mechanical ventilation, and 1 patient died. In the ensuing years, Japanese and Norwegian groups have independently developed and improved techniques for BPA. The procedure is done in a series of sessions (average four to six), 1 to 4 weeks apart, where small (2-3 mm) balloons are directed toward distal, diseased pulmonary vessels. Common complications include reperfusion injury, vessel injury, hemoptysis, and, more rarely, respiratory failure. Still, early experience suggests this procedure decreases pulmonary vascular resistance over time, improves right ventricular function, and improves patients’ symptoms (Andreassen A, et al. Heart. 2013;99(19):1415). The experience with this procedure is limited but growing in the United States, with only a handful of centers currently performing BPAs and collecting data.
Lifelong anticoagulation, oxygen, and diuretics for right-sided heart failure are recommended for patients with CTEPH. The first successful large phase III medication study for CTEPH was the CHEST-1 trial published in 2013. This was a multicenter, randomized, placebo-controlled trial of the soluble guanylate cyclase stimulator riociguat. The study enrolled 261 patients with inoperable CTEPH or persistent pulmonary hypertension after surgery. The primary end point was 6-minute walk distance at 12 weeks. The treatment group showed a 46 m improvement (P<.001). Secondary end points of pulmonary vascular resistance, NT-proBNP level, and functional class also improved. This pivotal trial led to the FDA approval of riociguat for inoperable or persistent postoperative CTEPH.
MERIT-1, a phase II, randomized placebo-controlled double trial of macitentan (an oral endothelin receptor antagonist) was recently completed. It enrolled 80 patients with inoperable CTEPH. The primary endpoint was pulmonary vascular resistance at week 16, expressed as a percentage of baseline. At week 16, the patients in the treatment arm had a PVR 73% of baseline vs 87.2% in the treatment group. This medication is not yet FDA-approved for the treatment of inoperable CTEPH (Ghofrani H, et al. Lancet Respir Med. 2017;5(10):785-794).
Pulmonary hypertension medication has been postulated as a possible way to “pretreat” patients before pulmonary thromboendarterectomy surgery, perhaps lowering preoperative pulmonary vascular resistance and surgical risk. However, there are currently no convincing data to support this practice, and medical treatment has been associated with a possible counterproductive delay in surgery. A phase II study including CTEPH patients with high PVR for preoperative treatment with riociguat vs placebo is currently enrolling to determine if “induction” treatment with medication prior to surgery reduces risk or delays definitive surgery. Occasionally, patients are found who have persistent thrombus but not pulmonary hypertension. Chronic thromboembolic disease (CTED) is a recently coined term describing patients who have chronic thromboembolism on imaging but have normal resting hemodynamics. Whether CTED represents simply unresolved clot that will never progress to CTEPH or is an early point on the continuum of disease not well-defined and a controversial topic among experts. At many centers, patients with CTED and symptoms will undergo exercise testing to look for exercise -induced pulmonary hypertension or an increase in dead space ventilation as a cause of their symptoms. A retrospective series of carefully chosen CTED patients who underwent PTE surgery reported improvements in symptoms and overall quality of life, without increased complications (Taboada D, et al. Eur Respir J. 2014 44(6):1635). The operation carries risk, however, and further work into the epidemiology and prognosis of CTED is required before operative intervention can be recommended.
In conclusion, CTEPH is a disease that rarely occurs after an acute PE but when undiagnosed and untreated portends a poor prognosis. The definitive treatment for this disease is surgical PTE, but to achieve the best outcomes, this procedure needs to be performed at expert centers with multidisciplinary team experience. Patients who are poor operative candidates or with surgically inaccessible disease may be considered for balloon pulmonary angioplasty. For patients without more curative options, medication improves exercise tolerance. The field of CTEPH has been rapidly expanding over the last decade, leading to better patient outcomes and more treatment options.
Dr. Bartolome is Associate Professor, Pulmonary and Critical Care Medicine; Director, CTEPH Program; and Associate Director, PH Program; UT Southwestern Medical Center, Dallas, Texas.
Secure a CHEST Foundation Research Award
In anticipation of the 2019 CHEST Foundation grants cycle, opening in late February, CHEST Foundation staff sat down with 2017 CHEST Foundation Community Service grant winner, Sharon Armstead, RRT, Director of Clinical Education & Clinical Assistant Professor for the Department of Respiratory Care at Texas State University, to learn more about her project supporting respiratory asthma clinics in Guyana.

Ms. Armstead’s program takes respiratory care students from her institution on a study abroad trip to Guyana with aims to educate Guyanese student populations about asthma and teach them self-management skills. Additionally, she and her students work alongside clinicians at Georgetown Public Hospital to host a mobile asthma clinic that provides asthma screenings and education for Guyanese students, the first of its kind at Texas State University.
This passion for supporting clinics in Guyana stems from a deeply personal place. “Guyana is my country of birth. I left when I was 14. I came back many years later realizing that I can give back to the county that gave me so much.” Ms. Armstead shared.
“The CHEST Foundation grant opened doors for me that had never been opened before. Members of the community were very open to hearing what we had to say and receptive to the changes we suggested they make in their daily lives. The financial portion of the award allowed me to purchase additional spirometers for the asthma clinic, allowing for a whole new level of outpatient testing and outreach in the community.”
In addition to the impact she and her students have in Georgetown, Ms. Armstead says opportunity provided to her students was life-changing for them. “To watch my students communicate with people in a different country really helps build their confidence as future clinicians.” Her study program received a significant growth in attendance over the past few years. “When we first started doing this study abroad in Guyana, I only had 2 students interested… We took 14 respiratory care students to Guyana in 2017. It’s really elevated this study abroad program at my institution.”
The CHEST Foundation’s grants cycle opens in late February. Visit our grants page to view the RFPs for our 2019 offerings and see a step-by-step walkthrough of how simple it is to apply for funding! Be a champion of lung health, and secure your research award today!
In anticipation of the 2019 CHEST Foundation grants cycle, opening in late February, CHEST Foundation staff sat down with 2017 CHEST Foundation Community Service grant winner, Sharon Armstead, RRT, Director of Clinical Education & Clinical Assistant Professor for the Department of Respiratory Care at Texas State University, to learn more about her project supporting respiratory asthma clinics in Guyana.
Ms. Armstead’s program takes respiratory care students from her institution on a study abroad trip to Guyana with aims to educate Guyanese student populations about asthma and teach them self-management skills. Additionally, she and her students work alongside clinicians at Georgetown Public Hospital to host a mobile asthma clinic that provides asthma screenings and education for Guyanese students, the first of its kind at Texas State University.
This passion for supporting clinics in Guyana stems from a deeply personal place. “Guyana is my country of birth. I left when I was 14. I came back many years later realizing that I can give back to the county that gave me so much.” Ms. Armstead shared.
“The CHEST Foundation grant opened doors for me that had never been opened before. Members of the community were very open to hearing what we had to say and receptive to the changes we suggested they make in their daily lives. The financial portion of the award allowed me to purchase additional spirometers for the asthma clinic, allowing for a whole new level of outpatient testing and outreach in the community.”
In addition to the impact she and her students have in Georgetown, Ms. Armstead says opportunity provided to her students was life-changing for them. “To watch my students communicate with people in a different country really helps build their confidence as future clinicians.” Her study program received a significant growth in attendance over the past few years. “When we first started doing this study abroad in Guyana, I only had 2 students interested… We took 14 respiratory care students to Guyana in 2017. It’s really elevated this study abroad program at my institution.”
The CHEST Foundation’s grants cycle opens in late February. Visit our grants page to view the RFPs for our 2019 offerings and see a step-by-step walkthrough of how simple it is to apply for funding! Be a champion of lung health, and secure your research award today!
In anticipation of the 2019 CHEST Foundation grants cycle, opening in late February, CHEST Foundation staff sat down with 2017 CHEST Foundation Community Service grant winner, Sharon Armstead, RRT, Director of Clinical Education & Clinical Assistant Professor for the Department of Respiratory Care at Texas State University, to learn more about her project supporting respiratory asthma clinics in Guyana.
Ms. Armstead’s program takes respiratory care students from her institution on a study abroad trip to Guyana with aims to educate Guyanese student populations about asthma and teach them self-management skills. Additionally, she and her students work alongside clinicians at Georgetown Public Hospital to host a mobile asthma clinic that provides asthma screenings and education for Guyanese students, the first of its kind at Texas State University.
This passion for supporting clinics in Guyana stems from a deeply personal place. “Guyana is my country of birth. I left when I was 14. I came back many years later realizing that I can give back to the county that gave me so much.” Ms. Armstead shared.
“The CHEST Foundation grant opened doors for me that had never been opened before. Members of the community were very open to hearing what we had to say and receptive to the changes we suggested they make in their daily lives. The financial portion of the award allowed me to purchase additional spirometers for the asthma clinic, allowing for a whole new level of outpatient testing and outreach in the community.”
In addition to the impact she and her students have in Georgetown, Ms. Armstead says opportunity provided to her students was life-changing for them. “To watch my students communicate with people in a different country really helps build their confidence as future clinicians.” Her study program received a significant growth in attendance over the past few years. “When we first started doing this study abroad in Guyana, I only had 2 students interested… We took 14 respiratory care students to Guyana in 2017. It’s really elevated this study abroad program at my institution.”
The CHEST Foundation’s grants cycle opens in late February. Visit our grants page to view the RFPs for our 2019 offerings and see a step-by-step walkthrough of how simple it is to apply for funding! Be a champion of lung health, and secure your research award today!
Greetings, readers!
One year ago, I wrote in these pages with regard to my two main goals for CHEST Physician for 2018, namely allowing more space in our pages for leaders and members to express their views, and improving interactivity between the staff here and our readership to help us better craft a publication that met your needs.
While I think we’ve met the first goal quite well, with a greater number of educational write-ups from our NetWork leadership and high-quality editorials and commentaries from other CHEST dignitaries, we have not yet heard much from the most important resource we have, our readers. So for the coming year, I would welcome you to drop us a line every now and then. See something in our pages that you like, or with which you disagree? Is there something in the news relevant to pulmonary, critical care, or sleep medicine that you think we should have covered but did not? Send us an email at [email protected]
I look forward to closer contact with you over the coming year. Let’s make CHEST Physician even better together!
David
One year ago, I wrote in these pages with regard to my two main goals for CHEST Physician for 2018, namely allowing more space in our pages for leaders and members to express their views, and improving interactivity between the staff here and our readership to help us better craft a publication that met your needs.
While I think we’ve met the first goal quite well, with a greater number of educational write-ups from our NetWork leadership and high-quality editorials and commentaries from other CHEST dignitaries, we have not yet heard much from the most important resource we have, our readers. So for the coming year, I would welcome you to drop us a line every now and then. See something in our pages that you like, or with which you disagree? Is there something in the news relevant to pulmonary, critical care, or sleep medicine that you think we should have covered but did not? Send us an email at [email protected]
I look forward to closer contact with you over the coming year. Let’s make CHEST Physician even better together!
David
One year ago, I wrote in these pages with regard to my two main goals for CHEST Physician for 2018, namely allowing more space in our pages for leaders and members to express their views, and improving interactivity between the staff here and our readership to help us better craft a publication that met your needs.
While I think we’ve met the first goal quite well, with a greater number of educational write-ups from our NetWork leadership and high-quality editorials and commentaries from other CHEST dignitaries, we have not yet heard much from the most important resource we have, our readers. So for the coming year, I would welcome you to drop us a line every now and then. See something in our pages that you like, or with which you disagree? Is there something in the news relevant to pulmonary, critical care, or sleep medicine that you think we should have covered but did not? Send us an email at [email protected]
I look forward to closer contact with you over the coming year. Let’s make CHEST Physician even better together!
David
BCL expression intensity key in distinguishing FL lesions
Intensity of BCL2 expression, and to a lesser extent expression of t(14;18), may help distinguish common and indolent cutaneous lymphomas from poorer-prognosis cutaneous lesions secondary to systemic follicular lymphomas, results of a recent investigation show.
Strong expression of BCL2 was almost always associated with secondary cutaneous follicular lymphoma (SCFL), and infrequently associated with primary cutaneous follicular center-cell lymphoma (PCFCL), according to the study results.
The translocation t(14;18) was likewise linked to secondary lesions, occurring less frequently in PCFCL in the study, reported recently in the Journal of Cutaneous Pathology.
“BCL2 expression intensity is the single most valuable clue in differentiating PCFCL from SCFL cases on histopathological grounds,” said Ramon M. Pujol, MD, PhD, of Hospital del Mar, Barcelona, Spain, and colleagues.
One of the main cutaneous B-cell lymphoma subtypes, PCFCL is marked by frequent relapses, but little incidence of systemic spread, meaning that conservative, skin-based therapies are usually warranted. By contrast, patients with SCFLs have a poor prognosis and may require systemic therapy, the investigators noted in their report.
Previous investigations have yielded conflicting results on the role of BCL2 expression, CD10 expression, and presence of t(14;18) translocation in distinguishing PCFCL from SCFL.
While early studies suggested most PCFCLs were negative for these markers, some recent reports suggested BCL positivity in PCFCLs is as high as 86%, the investigators said.
Accordingly, Dr. Pujol and colleagues evaluated clinicopathologic and genetic features in a large series of patients, including 59 with PCFCL and 22 with SCFL.
Significant BCL2 expression was seen in 69% of PCFCLs and in 100% of SCFLs (P = .003) in this patient series; however, when looking at BCL2 intensity, investigators found strong expression almost exclusively in SCFL. Strong expression was seen in 46% of those patients with secondary lymphomas, versus just 4%, or two cases, in the PCFCL group (P = .001).
The t(14;18) translocation was seen in 64% of SCFLs and only 9.1% of PCFCLs (P = .001).
Similar to what was seen for BCL2, expression of CD10 was observed in 66% of PCFCLs and 91% of SCFLs, and again, intensity differences mattered. Strong CD10 expression was seen in 62% of secondary lymphomas and 16% of PCFCLs (P = .01). But the high number of positive PCFCLs made this marker less useful than BCL2, the investigators said.
“We believe that differences in BCL2 and CD10 expression between our results and older previous studies could reflect the improvement of antigen retrieval laboratory techniques,” they said.
The investigators did not report disclosures related to the research.
SOURCE: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
Intensity of BCL2 expression, and to a lesser extent expression of t(14;18), may help distinguish common and indolent cutaneous lymphomas from poorer-prognosis cutaneous lesions secondary to systemic follicular lymphomas, results of a recent investigation show.
Strong expression of BCL2 was almost always associated with secondary cutaneous follicular lymphoma (SCFL), and infrequently associated with primary cutaneous follicular center-cell lymphoma (PCFCL), according to the study results.
The translocation t(14;18) was likewise linked to secondary lesions, occurring less frequently in PCFCL in the study, reported recently in the Journal of Cutaneous Pathology.
“BCL2 expression intensity is the single most valuable clue in differentiating PCFCL from SCFL cases on histopathological grounds,” said Ramon M. Pujol, MD, PhD, of Hospital del Mar, Barcelona, Spain, and colleagues.
One of the main cutaneous B-cell lymphoma subtypes, PCFCL is marked by frequent relapses, but little incidence of systemic spread, meaning that conservative, skin-based therapies are usually warranted. By contrast, patients with SCFLs have a poor prognosis and may require systemic therapy, the investigators noted in their report.
Previous investigations have yielded conflicting results on the role of BCL2 expression, CD10 expression, and presence of t(14;18) translocation in distinguishing PCFCL from SCFL.
While early studies suggested most PCFCLs were negative for these markers, some recent reports suggested BCL positivity in PCFCLs is as high as 86%, the investigators said.
Accordingly, Dr. Pujol and colleagues evaluated clinicopathologic and genetic features in a large series of patients, including 59 with PCFCL and 22 with SCFL.
Significant BCL2 expression was seen in 69% of PCFCLs and in 100% of SCFLs (P = .003) in this patient series; however, when looking at BCL2 intensity, investigators found strong expression almost exclusively in SCFL. Strong expression was seen in 46% of those patients with secondary lymphomas, versus just 4%, or two cases, in the PCFCL group (P = .001).
The t(14;18) translocation was seen in 64% of SCFLs and only 9.1% of PCFCLs (P = .001).
Similar to what was seen for BCL2, expression of CD10 was observed in 66% of PCFCLs and 91% of SCFLs, and again, intensity differences mattered. Strong CD10 expression was seen in 62% of secondary lymphomas and 16% of PCFCLs (P = .01). But the high number of positive PCFCLs made this marker less useful than BCL2, the investigators said.
“We believe that differences in BCL2 and CD10 expression between our results and older previous studies could reflect the improvement of antigen retrieval laboratory techniques,” they said.
The investigators did not report disclosures related to the research.
SOURCE: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
Intensity of BCL2 expression, and to a lesser extent expression of t(14;18), may help distinguish common and indolent cutaneous lymphomas from poorer-prognosis cutaneous lesions secondary to systemic follicular lymphomas, results of a recent investigation show.
Strong expression of BCL2 was almost always associated with secondary cutaneous follicular lymphoma (SCFL), and infrequently associated with primary cutaneous follicular center-cell lymphoma (PCFCL), according to the study results.
The translocation t(14;18) was likewise linked to secondary lesions, occurring less frequently in PCFCL in the study, reported recently in the Journal of Cutaneous Pathology.
“BCL2 expression intensity is the single most valuable clue in differentiating PCFCL from SCFL cases on histopathological grounds,” said Ramon M. Pujol, MD, PhD, of Hospital del Mar, Barcelona, Spain, and colleagues.
One of the main cutaneous B-cell lymphoma subtypes, PCFCL is marked by frequent relapses, but little incidence of systemic spread, meaning that conservative, skin-based therapies are usually warranted. By contrast, patients with SCFLs have a poor prognosis and may require systemic therapy, the investigators noted in their report.
Previous investigations have yielded conflicting results on the role of BCL2 expression, CD10 expression, and presence of t(14;18) translocation in distinguishing PCFCL from SCFL.
While early studies suggested most PCFCLs were negative for these markers, some recent reports suggested BCL positivity in PCFCLs is as high as 86%, the investigators said.
Accordingly, Dr. Pujol and colleagues evaluated clinicopathologic and genetic features in a large series of patients, including 59 with PCFCL and 22 with SCFL.
Significant BCL2 expression was seen in 69% of PCFCLs and in 100% of SCFLs (P = .003) in this patient series; however, when looking at BCL2 intensity, investigators found strong expression almost exclusively in SCFL. Strong expression was seen in 46% of those patients with secondary lymphomas, versus just 4%, or two cases, in the PCFCL group (P = .001).
The t(14;18) translocation was seen in 64% of SCFLs and only 9.1% of PCFCLs (P = .001).
Similar to what was seen for BCL2, expression of CD10 was observed in 66% of PCFCLs and 91% of SCFLs, and again, intensity differences mattered. Strong CD10 expression was seen in 62% of secondary lymphomas and 16% of PCFCLs (P = .01). But the high number of positive PCFCLs made this marker less useful than BCL2, the investigators said.
“We believe that differences in BCL2 and CD10 expression between our results and older previous studies could reflect the improvement of antigen retrieval laboratory techniques,” they said.
The investigators did not report disclosures related to the research.
SOURCE: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
FROM THE JOURNAL OF CUTANEOUS PATHOLOGY
Key clinical point:
Major finding: Strong BCL2 expression was seen in 46% of secondary lymphomas, versus just 4% of primary cutaneous follicular center-cell lymphomas (P = .001).
Study details: A comparative study evaluating clinicopathologic and genetic features in a series of patients, including 59 with PCFCL and 22 with SCFL.
Disclosures: Investigators did not report disclosures related to the research.
Source: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
Laparoscopic distal gastrectomy safe alternative to open surgery
When experienced surgeons are involved, laparoscopic distal gastrectomy is a safe alternative to open surgery in patients with early-stage gastric cancer, results of a randomized trial suggest.
Five-year overall survival exceeded 93% for both laparoscopic and open surgery groups in the multicenter trial, which included 1,416 patients with stage I gastric cancer treated by 15 surgeons who each had performed at least 100 gastrectomies.
Cancer-specific survival and recurrence were not significantly different between groups, while an intent-to-treat analysis confirmed the noninferiority of laparoscopic gastrectomy versus the open procedure, said investigators, led by Hyung-Ho Kim MD, PhD, of the Korean Laparoendoscopic Gastrointestinal Surgery Study (KLASS) group.
“Our trial supports the use of laparoscopic distal gastrectomy as a standard treatment option for clinical stage I distal gastric cancer when it can be performed by surgeons with sufficient experience,” Dr. Kim and his coauthors wrote in JAMA Oncology.
Some had doubted the oncologic safety of the laparoscopic approach because of the potential for inadequate lymphadenectomy leading to an increased risk of locoregional recurrence, said Dr. Kim and his coauthors in the KLASS group, “which includes 15 surgeons from 13 institutes.” However, among patients in this phase 3 randomized trial, known as KLASS-01, the mean number of retrieved lymph nodes was similar for the laparoscopic and open surgery groups, and there was no surgical margin involvement in any patient, investigators reported.
“We thus anticipated comparable long-term oncologic outcomes for overall and cancer-specific survival because these early outcomes indicated the oncologic safety of the laparoscopic procedure,” they said. Moreover, they said, earlier publications on the KLASS-01 study demonstrated that the laparoscopic approach was associated with less blood loss, fewer wound complications, and shorter hospital stays, compared with open distal gastrectomy.
In the current study, Dr. Kim and his coauthors reported that, with a median follow-up of about 100 months, the 5-year overall survival rate was 94.2% for the laparoscopic group and 93.3% for the open group (P = .64), while Similarly, the 5-year cancer-specific survival rates were 97.1% and 97.2% for the laparoscopic and open approach, respectively (P = .91), while recurrence was not significantly different at 5.6% and 4.8% (P = .49).
The investigators cited several limitations. One is that the investigators looked only at patients with stage I cancer “suitable for distal subtotal gastrectomy. Applying laparoscopic surgery for more advanced cancers and different operations, such as total gastrectomy, needs to be verified through other clinical trials,” they said.
Nevertheless, they wrote, “These long-term oncologic outcomes of [laparoscopy-assisted distal gastrectomy] support the adoption of this procedure as a standard treatment for clinical stage I gastric cancer.”
Dr. Kim and his coauthors reported no conflicts of interest related to the study, which was supported by a grant from the Ministry of Health & Welfare, Republic of Korea.
SOURCE: Kim HH et al. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6727.
When experienced surgeons are involved, laparoscopic distal gastrectomy is a safe alternative to open surgery in patients with early-stage gastric cancer, results of a randomized trial suggest.
Five-year overall survival exceeded 93% for both laparoscopic and open surgery groups in the multicenter trial, which included 1,416 patients with stage I gastric cancer treated by 15 surgeons who each had performed at least 100 gastrectomies.
Cancer-specific survival and recurrence were not significantly different between groups, while an intent-to-treat analysis confirmed the noninferiority of laparoscopic gastrectomy versus the open procedure, said investigators, led by Hyung-Ho Kim MD, PhD, of the Korean Laparoendoscopic Gastrointestinal Surgery Study (KLASS) group.
“Our trial supports the use of laparoscopic distal gastrectomy as a standard treatment option for clinical stage I distal gastric cancer when it can be performed by surgeons with sufficient experience,” Dr. Kim and his coauthors wrote in JAMA Oncology.
Some had doubted the oncologic safety of the laparoscopic approach because of the potential for inadequate lymphadenectomy leading to an increased risk of locoregional recurrence, said Dr. Kim and his coauthors in the KLASS group, “which includes 15 surgeons from 13 institutes.” However, among patients in this phase 3 randomized trial, known as KLASS-01, the mean number of retrieved lymph nodes was similar for the laparoscopic and open surgery groups, and there was no surgical margin involvement in any patient, investigators reported.
“We thus anticipated comparable long-term oncologic outcomes for overall and cancer-specific survival because these early outcomes indicated the oncologic safety of the laparoscopic procedure,” they said. Moreover, they said, earlier publications on the KLASS-01 study demonstrated that the laparoscopic approach was associated with less blood loss, fewer wound complications, and shorter hospital stays, compared with open distal gastrectomy.
In the current study, Dr. Kim and his coauthors reported that, with a median follow-up of about 100 months, the 5-year overall survival rate was 94.2% for the laparoscopic group and 93.3% for the open group (P = .64), while Similarly, the 5-year cancer-specific survival rates were 97.1% and 97.2% for the laparoscopic and open approach, respectively (P = .91), while recurrence was not significantly different at 5.6% and 4.8% (P = .49).
The investigators cited several limitations. One is that the investigators looked only at patients with stage I cancer “suitable for distal subtotal gastrectomy. Applying laparoscopic surgery for more advanced cancers and different operations, such as total gastrectomy, needs to be verified through other clinical trials,” they said.
Nevertheless, they wrote, “These long-term oncologic outcomes of [laparoscopy-assisted distal gastrectomy] support the adoption of this procedure as a standard treatment for clinical stage I gastric cancer.”
Dr. Kim and his coauthors reported no conflicts of interest related to the study, which was supported by a grant from the Ministry of Health & Welfare, Republic of Korea.
SOURCE: Kim HH et al. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6727.
When experienced surgeons are involved, laparoscopic distal gastrectomy is a safe alternative to open surgery in patients with early-stage gastric cancer, results of a randomized trial suggest.
Five-year overall survival exceeded 93% for both laparoscopic and open surgery groups in the multicenter trial, which included 1,416 patients with stage I gastric cancer treated by 15 surgeons who each had performed at least 100 gastrectomies.
Cancer-specific survival and recurrence were not significantly different between groups, while an intent-to-treat analysis confirmed the noninferiority of laparoscopic gastrectomy versus the open procedure, said investigators, led by Hyung-Ho Kim MD, PhD, of the Korean Laparoendoscopic Gastrointestinal Surgery Study (KLASS) group.
“Our trial supports the use of laparoscopic distal gastrectomy as a standard treatment option for clinical stage I distal gastric cancer when it can be performed by surgeons with sufficient experience,” Dr. Kim and his coauthors wrote in JAMA Oncology.
Some had doubted the oncologic safety of the laparoscopic approach because of the potential for inadequate lymphadenectomy leading to an increased risk of locoregional recurrence, said Dr. Kim and his coauthors in the KLASS group, “which includes 15 surgeons from 13 institutes.” However, among patients in this phase 3 randomized trial, known as KLASS-01, the mean number of retrieved lymph nodes was similar for the laparoscopic and open surgery groups, and there was no surgical margin involvement in any patient, investigators reported.
“We thus anticipated comparable long-term oncologic outcomes for overall and cancer-specific survival because these early outcomes indicated the oncologic safety of the laparoscopic procedure,” they said. Moreover, they said, earlier publications on the KLASS-01 study demonstrated that the laparoscopic approach was associated with less blood loss, fewer wound complications, and shorter hospital stays, compared with open distal gastrectomy.
In the current study, Dr. Kim and his coauthors reported that, with a median follow-up of about 100 months, the 5-year overall survival rate was 94.2% for the laparoscopic group and 93.3% for the open group (P = .64), while Similarly, the 5-year cancer-specific survival rates were 97.1% and 97.2% for the laparoscopic and open approach, respectively (P = .91), while recurrence was not significantly different at 5.6% and 4.8% (P = .49).
The investigators cited several limitations. One is that the investigators looked only at patients with stage I cancer “suitable for distal subtotal gastrectomy. Applying laparoscopic surgery for more advanced cancers and different operations, such as total gastrectomy, needs to be verified through other clinical trials,” they said.
Nevertheless, they wrote, “These long-term oncologic outcomes of [laparoscopy-assisted distal gastrectomy] support the adoption of this procedure as a standard treatment for clinical stage I gastric cancer.”
Dr. Kim and his coauthors reported no conflicts of interest related to the study, which was supported by a grant from the Ministry of Health & Welfare, Republic of Korea.
SOURCE: Kim HH et al. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6727.
FROM JAMA ONCOLOGY
Key clinical point: Laparoscopic distal gastrectomy performed by experienced surgeons is a safe alternative to open surgery in patient with early-stage gastric cancer.
Major finding: The 5-year overall survival rate was 94.2% for the laparoscopic group and 93.3% for the open group (P = .64).
Study details: The KLASS-01 trial, which included 1,416 patients with stage I gastric cancer treated by 15 experienced surgeons.
Disclosures: The coauthors reported no conflicts of interest related to the study, which was supported by a grant from the Ministry of Health &Welfare, Republic of Korea.
Source: Kim HH et al. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6727.
Consider adopting the MESA 10-year CHD risk calculator
SNOWMASS, COLO. – The Multi-Ethnic Study of Atherosclerosis–based 10-year coronary heart disease risk calculator offers significant advantages over the far more widely used Atherosclerotic Cardiovascular Disease risk estimator based upon the pooled cohort equations recommended in the American College of Cardiology/American Heart Association guidelines, Robert A. Vogel, MD, said at the Annual Cardiovascular Conference at Snowmass.

“I don’t use the PCE very much. I use the MESA [Multi-Ethnic Study of Atherosclerosis]. I like it because it gives me options I don’t have with the PCE [pooled cohort equations],” Dr. Vogel, a preventive cardiology expert at the University of Colorado at Denver, Aurora, said at the meeting sponsored by the American College of Cardiology.
Those added options include, importantly, the ability to plug in a patient’s coronary artery calcium score. MESA, a landmark longitudinal National Institutes of Health–sponsored study, generated a great deal of the data that established the prognostic value of measuring coronary artery calcium.
“You can do the MESA worksheet with or without coronary artery calcium, either way,” Dr. Vogel noted.
Another big advantage for the MESA risk calculator: It asks a yes/no question about family history of heart attack in first-degree relatives. That’s truly risk-altering information, and yet it’s absent from the ACC/AHA ASCVD (Atherosclerotic Cardiovascular Disease) risk calculator, the cardiologist continued.
Plus, the MESA risk calculator includes four ethnic options: Caucasian, African American, Chinese, or Hispanic, while the ACC/AHA’s PCE-based tool includes only the categories of white, African American, or other.
Dr. Vogel offered a case example for purposes of comparison and contrast of the two risk calculators: a 48-year-old white man with no history or symptoms of cardiovascular disease whose father had an MI at age 52. He wants to know whether he should start taking a statin. The patient is a former smoker who quit 3 years ago. His physical exam is normal. He has a body mass index of 29 kg/m2, an LDL cholesterol of 134 mg/dL, a total cholesterol of 194 mg/dL, a triglyceride level of 92 mg/dL, blood pressure of 128/78 mm Hg, an HDL cholesterol of 42 mg/dL, and a hemoglobin A1c of 5.9%.
Using the PCE-based risk calculator, the man’s 10-year risk of cardiovascular disease is only 3.3%, which falls below the ACC/AHA guideline-recommended threshold for statin therapy for primary prevention. But with the MESA score, as a result of the additional information regarding the family history of premature cardiovascular disease, the patient’s risk jumps to 4.9%, even without including a coronary artery calcium score.
“This shows you how big a factor the family history is. I think it’s unfortunate that the PCE doesn’t put it in,” Dr. Vogel said.
Indeed, in an analysis from MESA, a 60-year-old man with a lipid and blood pressure profile similar to that of Dr. Vogel’s patient would have a 10-year cardiovascular disease risk of 6% with a negative family history and a 9% risk with a positive history, he noted.
In the 48-year-old patient used as an example by Dr. Vogel, plugging into the MESA risk calculator a coronary artery calcium score of, say, 50, 100, or 150 Agatston units would boost the 10-year cardiovascular disease risk to 7.5%, 8.9%, and 9.9%, respectively.
Both the MESA and the ACC/AHA risk calculators incorporate diabetes as a simple yes/no item. It’s either present or absent. This is too crude a dichotomy for Dr. Vogel’s liking in light of data showing that individuals with impaired glucose tolerance have a cumulative risk of cardiovascular mortality that’s intermediate between diabetic and normoglycemic individuals. And of course, the patient in his case example had a HbA1c of 5.9%. So what is a physician to do?
“I fudge the numbers,” he said. “
He also adjusts a former smoker’s estimated 10-year cardiovascular risk based upon how long ago the smoker quit. An ex-smoker’s hazard ratio for coronary heart disease doesn’t drop to that of a never-smoker until 8 years after quitting. And since the patient in this example has been tobacco-free for only 3 years, Dr. Vogel bumps up that individual’s risk level once again. So at this point, even without the additional information that would be provided by a coronary calcium score, the patient’s adjusted MESA 10-year risk is in the 10% range, compared with the 3.3% figure derived using the PCE-based risk calculator.
He reported serving as a paid consultant to the National Football League and the Pritikin Longevity Center, receiving research grants from Sanofi, and serving on speakers bureaus for Regeneron and Sanofi.
SNOWMASS, COLO. – The Multi-Ethnic Study of Atherosclerosis–based 10-year coronary heart disease risk calculator offers significant advantages over the far more widely used Atherosclerotic Cardiovascular Disease risk estimator based upon the pooled cohort equations recommended in the American College of Cardiology/American Heart Association guidelines, Robert A. Vogel, MD, said at the Annual Cardiovascular Conference at Snowmass.
“I don’t use the PCE very much. I use the MESA [Multi-Ethnic Study of Atherosclerosis]. I like it because it gives me options I don’t have with the PCE [pooled cohort equations],” Dr. Vogel, a preventive cardiology expert at the University of Colorado at Denver, Aurora, said at the meeting sponsored by the American College of Cardiology.
Those added options include, importantly, the ability to plug in a patient’s coronary artery calcium score. MESA, a landmark longitudinal National Institutes of Health–sponsored study, generated a great deal of the data that established the prognostic value of measuring coronary artery calcium.
“You can do the MESA worksheet with or without coronary artery calcium, either way,” Dr. Vogel noted.
Another big advantage for the MESA risk calculator: It asks a yes/no question about family history of heart attack in first-degree relatives. That’s truly risk-altering information, and yet it’s absent from the ACC/AHA ASCVD (Atherosclerotic Cardiovascular Disease) risk calculator, the cardiologist continued.
Plus, the MESA risk calculator includes four ethnic options: Caucasian, African American, Chinese, or Hispanic, while the ACC/AHA’s PCE-based tool includes only the categories of white, African American, or other.
Dr. Vogel offered a case example for purposes of comparison and contrast of the two risk calculators: a 48-year-old white man with no history or symptoms of cardiovascular disease whose father had an MI at age 52. He wants to know whether he should start taking a statin. The patient is a former smoker who quit 3 years ago. His physical exam is normal. He has a body mass index of 29 kg/m2, an LDL cholesterol of 134 mg/dL, a total cholesterol of 194 mg/dL, a triglyceride level of 92 mg/dL, blood pressure of 128/78 mm Hg, an HDL cholesterol of 42 mg/dL, and a hemoglobin A1c of 5.9%.
Using the PCE-based risk calculator, the man’s 10-year risk of cardiovascular disease is only 3.3%, which falls below the ACC/AHA guideline-recommended threshold for statin therapy for primary prevention. But with the MESA score, as a result of the additional information regarding the family history of premature cardiovascular disease, the patient’s risk jumps to 4.9%, even without including a coronary artery calcium score.
“This shows you how big a factor the family history is. I think it’s unfortunate that the PCE doesn’t put it in,” Dr. Vogel said.
Indeed, in an analysis from MESA, a 60-year-old man with a lipid and blood pressure profile similar to that of Dr. Vogel’s patient would have a 10-year cardiovascular disease risk of 6% with a negative family history and a 9% risk with a positive history, he noted.
In the 48-year-old patient used as an example by Dr. Vogel, plugging into the MESA risk calculator a coronary artery calcium score of, say, 50, 100, or 150 Agatston units would boost the 10-year cardiovascular disease risk to 7.5%, 8.9%, and 9.9%, respectively.
Both the MESA and the ACC/AHA risk calculators incorporate diabetes as a simple yes/no item. It’s either present or absent. This is too crude a dichotomy for Dr. Vogel’s liking in light of data showing that individuals with impaired glucose tolerance have a cumulative risk of cardiovascular mortality that’s intermediate between diabetic and normoglycemic individuals. And of course, the patient in his case example had a HbA1c of 5.9%. So what is a physician to do?
“I fudge the numbers,” he said. “
He also adjusts a former smoker’s estimated 10-year cardiovascular risk based upon how long ago the smoker quit. An ex-smoker’s hazard ratio for coronary heart disease doesn’t drop to that of a never-smoker until 8 years after quitting. And since the patient in this example has been tobacco-free for only 3 years, Dr. Vogel bumps up that individual’s risk level once again. So at this point, even without the additional information that would be provided by a coronary calcium score, the patient’s adjusted MESA 10-year risk is in the 10% range, compared with the 3.3% figure derived using the PCE-based risk calculator.
He reported serving as a paid consultant to the National Football League and the Pritikin Longevity Center, receiving research grants from Sanofi, and serving on speakers bureaus for Regeneron and Sanofi.
SNOWMASS, COLO. – The Multi-Ethnic Study of Atherosclerosis–based 10-year coronary heart disease risk calculator offers significant advantages over the far more widely used Atherosclerotic Cardiovascular Disease risk estimator based upon the pooled cohort equations recommended in the American College of Cardiology/American Heart Association guidelines, Robert A. Vogel, MD, said at the Annual Cardiovascular Conference at Snowmass.
“I don’t use the PCE very much. I use the MESA [Multi-Ethnic Study of Atherosclerosis]. I like it because it gives me options I don’t have with the PCE [pooled cohort equations],” Dr. Vogel, a preventive cardiology expert at the University of Colorado at Denver, Aurora, said at the meeting sponsored by the American College of Cardiology.
Those added options include, importantly, the ability to plug in a patient’s coronary artery calcium score. MESA, a landmark longitudinal National Institutes of Health–sponsored study, generated a great deal of the data that established the prognostic value of measuring coronary artery calcium.
“You can do the MESA worksheet with or without coronary artery calcium, either way,” Dr. Vogel noted.
Another big advantage for the MESA risk calculator: It asks a yes/no question about family history of heart attack in first-degree relatives. That’s truly risk-altering information, and yet it’s absent from the ACC/AHA ASCVD (Atherosclerotic Cardiovascular Disease) risk calculator, the cardiologist continued.
Plus, the MESA risk calculator includes four ethnic options: Caucasian, African American, Chinese, or Hispanic, while the ACC/AHA’s PCE-based tool includes only the categories of white, African American, or other.
Dr. Vogel offered a case example for purposes of comparison and contrast of the two risk calculators: a 48-year-old white man with no history or symptoms of cardiovascular disease whose father had an MI at age 52. He wants to know whether he should start taking a statin. The patient is a former smoker who quit 3 years ago. His physical exam is normal. He has a body mass index of 29 kg/m2, an LDL cholesterol of 134 mg/dL, a total cholesterol of 194 mg/dL, a triglyceride level of 92 mg/dL, blood pressure of 128/78 mm Hg, an HDL cholesterol of 42 mg/dL, and a hemoglobin A1c of 5.9%.
Using the PCE-based risk calculator, the man’s 10-year risk of cardiovascular disease is only 3.3%, which falls below the ACC/AHA guideline-recommended threshold for statin therapy for primary prevention. But with the MESA score, as a result of the additional information regarding the family history of premature cardiovascular disease, the patient’s risk jumps to 4.9%, even without including a coronary artery calcium score.
“This shows you how big a factor the family history is. I think it’s unfortunate that the PCE doesn’t put it in,” Dr. Vogel said.
Indeed, in an analysis from MESA, a 60-year-old man with a lipid and blood pressure profile similar to that of Dr. Vogel’s patient would have a 10-year cardiovascular disease risk of 6% with a negative family history and a 9% risk with a positive history, he noted.
In the 48-year-old patient used as an example by Dr. Vogel, plugging into the MESA risk calculator a coronary artery calcium score of, say, 50, 100, or 150 Agatston units would boost the 10-year cardiovascular disease risk to 7.5%, 8.9%, and 9.9%, respectively.
Both the MESA and the ACC/AHA risk calculators incorporate diabetes as a simple yes/no item. It’s either present or absent. This is too crude a dichotomy for Dr. Vogel’s liking in light of data showing that individuals with impaired glucose tolerance have a cumulative risk of cardiovascular mortality that’s intermediate between diabetic and normoglycemic individuals. And of course, the patient in his case example had a HbA1c of 5.9%. So what is a physician to do?
“I fudge the numbers,” he said. “
He also adjusts a former smoker’s estimated 10-year cardiovascular risk based upon how long ago the smoker quit. An ex-smoker’s hazard ratio for coronary heart disease doesn’t drop to that of a never-smoker until 8 years after quitting. And since the patient in this example has been tobacco-free for only 3 years, Dr. Vogel bumps up that individual’s risk level once again. So at this point, even without the additional information that would be provided by a coronary calcium score, the patient’s adjusted MESA 10-year risk is in the 10% range, compared with the 3.3% figure derived using the PCE-based risk calculator.
He reported serving as a paid consultant to the National Football League and the Pritikin Longevity Center, receiving research grants from Sanofi, and serving on speakers bureaus for Regeneron and Sanofi.
REPORTING FROM ACC SNOWMASS 2019