User login
Intervention tied to fewer depressive symptoms, more weight loss
Adults with obesity and depression who participated in a program that addressed weight and mood saw improvement in weight loss and depressive symptoms at 12 months, results of a randomized, controlled trial of almost 350 patients show.
“To our knowledge, this study was the first and largest RTC of integrated collaborative care for coexisting obesity and depression,” wrote Jun Ma, MD, PhD, of the Institute of Health Research and Policy at the University of Illinois at Chicago, and colleagues.
Dr. Ma and colleagues enrolled 409 patients in the RAINBOW (Research Aimed at Improving Both Mood and Weight) trial between September 2014 and January 2017 from family and internal medicine departments at four medical centers in California. The RAINBOW intervention combined usual care with a weight loss treatment program used in diabetes prevention, problem-solving therapy, and prescriptions for antidepressants if indicated. About 71% of the trial participants were non-Hispanic white adults, 70% were women, and 69% had a college education.
Half the patients were randomized to receive usual care consisting of seeing personal physicians, receiving information on obesity and depression services at the clinic, and wireless activity-tracking devices. Patients were enrolled in the trial if they scored at least 10 points in the nine-item Patient Health Questionaire (PHQ-9) and had a body mass index (BMI) of 30 or higher, or a BMI of 27 or higher in Asian adults. The mean age in the cohort was 51.0 years, the mean BMI was 37.7, and the mean PHQ-9 score was 13.8.
Of the 344 patients (84.1%) who completed follow-up at 12 months, there was a decrease in mean BMI from 36.7 to 35.9 for patients who received the collaborative care intervention, compared with no change in BMI for patients who received usual care alone (between-group mean difference, −0.7; 95% confidence interval, −1.1 to −0.2; P = .01). Depressive symptoms also improved in the intervention group, with mean 20-item Depression Symptom Checklist scores decreasing from 1.5 at baseline to 1.1 at 12 months, compared with a decrease from 1.5 at baseline to 1.4 at 12 months in the usual-care group (between-group mean difference, −0.2; 95% CI, −0.4 to 0; P = .01). Overall, there were 47 adverse events or serious adverse events, with 27 events in the collaborative-care intervention group and 20 events in the usual-care group involving musculoskeletal injuries such as fracture and meniscus tear.
In addition, they cited the relative demographic homogeneity of the study sample as one of several limitations.
The study was funded in part by Palo Alto Medical Foundation Research Institute, the University of Illinois at Chicago, and an award from the National Heart, Lung, and Blood Institute. One author, Philip W. Lavori, PhD, reported receiving personal fees from Palo Alto Medical Foundation Research Institute. The other authors reported no relevant financial disclosures.
SOURCE: Ma J et al. JAMA. 2019. doi: 10.1001/jama2019.0557.
Adults with obesity and depression who participated in a program that addressed weight and mood saw improvement in weight loss and depressive symptoms at 12 months, results of a randomized, controlled trial of almost 350 patients show.
“To our knowledge, this study was the first and largest RTC of integrated collaborative care for coexisting obesity and depression,” wrote Jun Ma, MD, PhD, of the Institute of Health Research and Policy at the University of Illinois at Chicago, and colleagues.
Dr. Ma and colleagues enrolled 409 patients in the RAINBOW (Research Aimed at Improving Both Mood and Weight) trial between September 2014 and January 2017 from family and internal medicine departments at four medical centers in California. The RAINBOW intervention combined usual care with a weight loss treatment program used in diabetes prevention, problem-solving therapy, and prescriptions for antidepressants if indicated. About 71% of the trial participants were non-Hispanic white adults, 70% were women, and 69% had a college education.
Half the patients were randomized to receive usual care consisting of seeing personal physicians, receiving information on obesity and depression services at the clinic, and wireless activity-tracking devices. Patients were enrolled in the trial if they scored at least 10 points in the nine-item Patient Health Questionaire (PHQ-9) and had a body mass index (BMI) of 30 or higher, or a BMI of 27 or higher in Asian adults. The mean age in the cohort was 51.0 years, the mean BMI was 37.7, and the mean PHQ-9 score was 13.8.
Of the 344 patients (84.1%) who completed follow-up at 12 months, there was a decrease in mean BMI from 36.7 to 35.9 for patients who received the collaborative care intervention, compared with no change in BMI for patients who received usual care alone (between-group mean difference, −0.7; 95% confidence interval, −1.1 to −0.2; P = .01). Depressive symptoms also improved in the intervention group, with mean 20-item Depression Symptom Checklist scores decreasing from 1.5 at baseline to 1.1 at 12 months, compared with a decrease from 1.5 at baseline to 1.4 at 12 months in the usual-care group (between-group mean difference, −0.2; 95% CI, −0.4 to 0; P = .01). Overall, there were 47 adverse events or serious adverse events, with 27 events in the collaborative-care intervention group and 20 events in the usual-care group involving musculoskeletal injuries such as fracture and meniscus tear.
In addition, they cited the relative demographic homogeneity of the study sample as one of several limitations.
The study was funded in part by Palo Alto Medical Foundation Research Institute, the University of Illinois at Chicago, and an award from the National Heart, Lung, and Blood Institute. One author, Philip W. Lavori, PhD, reported receiving personal fees from Palo Alto Medical Foundation Research Institute. The other authors reported no relevant financial disclosures.
SOURCE: Ma J et al. JAMA. 2019. doi: 10.1001/jama2019.0557.
Adults with obesity and depression who participated in a program that addressed weight and mood saw improvement in weight loss and depressive symptoms at 12 months, results of a randomized, controlled trial of almost 350 patients show.
“To our knowledge, this study was the first and largest RTC of integrated collaborative care for coexisting obesity and depression,” wrote Jun Ma, MD, PhD, of the Institute of Health Research and Policy at the University of Illinois at Chicago, and colleagues.
Dr. Ma and colleagues enrolled 409 patients in the RAINBOW (Research Aimed at Improving Both Mood and Weight) trial between September 2014 and January 2017 from family and internal medicine departments at four medical centers in California. The RAINBOW intervention combined usual care with a weight loss treatment program used in diabetes prevention, problem-solving therapy, and prescriptions for antidepressants if indicated. About 71% of the trial participants were non-Hispanic white adults, 70% were women, and 69% had a college education.
Half the patients were randomized to receive usual care consisting of seeing personal physicians, receiving information on obesity and depression services at the clinic, and wireless activity-tracking devices. Patients were enrolled in the trial if they scored at least 10 points in the nine-item Patient Health Questionaire (PHQ-9) and had a body mass index (BMI) of 30 or higher, or a BMI of 27 or higher in Asian adults. The mean age in the cohort was 51.0 years, the mean BMI was 37.7, and the mean PHQ-9 score was 13.8.
Of the 344 patients (84.1%) who completed follow-up at 12 months, there was a decrease in mean BMI from 36.7 to 35.9 for patients who received the collaborative care intervention, compared with no change in BMI for patients who received usual care alone (between-group mean difference, −0.7; 95% confidence interval, −1.1 to −0.2; P = .01). Depressive symptoms also improved in the intervention group, with mean 20-item Depression Symptom Checklist scores decreasing from 1.5 at baseline to 1.1 at 12 months, compared with a decrease from 1.5 at baseline to 1.4 at 12 months in the usual-care group (between-group mean difference, −0.2; 95% CI, −0.4 to 0; P = .01). Overall, there were 47 adverse events or serious adverse events, with 27 events in the collaborative-care intervention group and 20 events in the usual-care group involving musculoskeletal injuries such as fracture and meniscus tear.
In addition, they cited the relative demographic homogeneity of the study sample as one of several limitations.
The study was funded in part by Palo Alto Medical Foundation Research Institute, the University of Illinois at Chicago, and an award from the National Heart, Lung, and Blood Institute. One author, Philip W. Lavori, PhD, reported receiving personal fees from Palo Alto Medical Foundation Research Institute. The other authors reported no relevant financial disclosures.
SOURCE: Ma J et al. JAMA. 2019. doi: 10.1001/jama2019.0557.
FROM JAMA
Hemostasis researcher passes away at age 72
George J. Broze Jr., MD, a former professor of medicine at Washington University School of Medicine in St. Louis, died following a heart attack on June 19, 2019, at the age of 72.

Dr. Broze was born in Seattle. He earned a bachelor’s degree from the University of Washington in Seattle and a medical degree from the University of Washington School of Medicine in St. Louis. Dr. Broze completed his internship and residency at North Carolina Memorial Hospital in Chapel Hill.
Dr. Broze became a clinical fellow in hematology at Washington University in 1976 and began teaching there in 1979. Dr. Broze practiced at the Jewish Hospital, Barnes Hospital, and Barnes-Jewish Hospital.
Dr. Broze’s research was focused on hemostasis and the relationship between coagulation and inflammation. He and his colleagues isolated and characterized tissue factor pathway inhibitor, uncovered a pathway for coagulation factor XI activation, and characterized the protein Z-dependent protease inhibitor serpinA10.
Dr. Broze is survived by his wife, two sons, and brother.
In happier news, Thomas J. Smith, MD, of the Johns Hopkins University School of Medicine in Baltimore, has won the Walther Cancer Foundation Palliative and Supportive Care in Oncology Endowed Award and Lecture from the American Society of Clinical Oncology.

The award is given to someone who “has made significant contributions to palliative care practice and research in oncology,” according to ASCO. Dr. Smith and his colleagues are known for their work showing that end-of-life palliative care can improve patient symptoms and quality of life while reducing the cost of care.
Dr. Smith will receive the award and deliver a keynote address at the 2019 Supportive Care in Oncology Symposium, which is set to take place Oct. 25-26 in San Francisco.
Meanwhile, Asya Nina Varshavsky-Yanovsky, MD, PhD, has joined Fox Chase Cancer Center in Philadelphia as an assistant professor in the hematology and bone marrow transplant programs within the department of hematology/oncology.

Dr. Varshavsky-Yanovsky earned her MD and PhD from the Technion-Israel Institute of Technology in Haifa, Israel. She joined Fox Chase Cancer Center/Temple University in 2016 for a 3-year fellowship in hematology/oncology.
Susmitha Apuri, MD, has joined Florida Cancer Specialists & Research Institute and is seeing patients in Inverness. She is board certified in medical oncology, hematology, and internal medicine.

Dr. Apuri earned her medical degree from NTR University of Health Sciences in Vijayawada, India; completed her internship and residency in internal medicine at the University of Miami Miller School of Medicine; and completed her fellowship in hematology/oncology at the University of South Florida/H. Lee Moffitt Cancer and Research Institute in Tampa. Her research and practice interests include malignant and nonmalignant hematology as well as breast, lung, and colorectal cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at [email protected], and you could be featured in Movers in Medicine.
George J. Broze Jr., MD, a former professor of medicine at Washington University School of Medicine in St. Louis, died following a heart attack on June 19, 2019, at the age of 72.
Dr. Broze was born in Seattle. He earned a bachelor’s degree from the University of Washington in Seattle and a medical degree from the University of Washington School of Medicine in St. Louis. Dr. Broze completed his internship and residency at North Carolina Memorial Hospital in Chapel Hill.
Dr. Broze became a clinical fellow in hematology at Washington University in 1976 and began teaching there in 1979. Dr. Broze practiced at the Jewish Hospital, Barnes Hospital, and Barnes-Jewish Hospital.
Dr. Broze’s research was focused on hemostasis and the relationship between coagulation and inflammation. He and his colleagues isolated and characterized tissue factor pathway inhibitor, uncovered a pathway for coagulation factor XI activation, and characterized the protein Z-dependent protease inhibitor serpinA10.
Dr. Broze is survived by his wife, two sons, and brother.
In happier news, Thomas J. Smith, MD, of the Johns Hopkins University School of Medicine in Baltimore, has won the Walther Cancer Foundation Palliative and Supportive Care in Oncology Endowed Award and Lecture from the American Society of Clinical Oncology.
The award is given to someone who “has made significant contributions to palliative care practice and research in oncology,” according to ASCO. Dr. Smith and his colleagues are known for their work showing that end-of-life palliative care can improve patient symptoms and quality of life while reducing the cost of care.
Dr. Smith will receive the award and deliver a keynote address at the 2019 Supportive Care in Oncology Symposium, which is set to take place Oct. 25-26 in San Francisco.
Meanwhile, Asya Nina Varshavsky-Yanovsky, MD, PhD, has joined Fox Chase Cancer Center in Philadelphia as an assistant professor in the hematology and bone marrow transplant programs within the department of hematology/oncology.
Dr. Varshavsky-Yanovsky earned her MD and PhD from the Technion-Israel Institute of Technology in Haifa, Israel. She joined Fox Chase Cancer Center/Temple University in 2016 for a 3-year fellowship in hematology/oncology.
Susmitha Apuri, MD, has joined Florida Cancer Specialists & Research Institute and is seeing patients in Inverness. She is board certified in medical oncology, hematology, and internal medicine.
Dr. Apuri earned her medical degree from NTR University of Health Sciences in Vijayawada, India; completed her internship and residency in internal medicine at the University of Miami Miller School of Medicine; and completed her fellowship in hematology/oncology at the University of South Florida/H. Lee Moffitt Cancer and Research Institute in Tampa. Her research and practice interests include malignant and nonmalignant hematology as well as breast, lung, and colorectal cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at [email protected], and you could be featured in Movers in Medicine.
George J. Broze Jr., MD, a former professor of medicine at Washington University School of Medicine in St. Louis, died following a heart attack on June 19, 2019, at the age of 72.
Dr. Broze was born in Seattle. He earned a bachelor’s degree from the University of Washington in Seattle and a medical degree from the University of Washington School of Medicine in St. Louis. Dr. Broze completed his internship and residency at North Carolina Memorial Hospital in Chapel Hill.
Dr. Broze became a clinical fellow in hematology at Washington University in 1976 and began teaching there in 1979. Dr. Broze practiced at the Jewish Hospital, Barnes Hospital, and Barnes-Jewish Hospital.
Dr. Broze’s research was focused on hemostasis and the relationship between coagulation and inflammation. He and his colleagues isolated and characterized tissue factor pathway inhibitor, uncovered a pathway for coagulation factor XI activation, and characterized the protein Z-dependent protease inhibitor serpinA10.
Dr. Broze is survived by his wife, two sons, and brother.
In happier news, Thomas J. Smith, MD, of the Johns Hopkins University School of Medicine in Baltimore, has won the Walther Cancer Foundation Palliative and Supportive Care in Oncology Endowed Award and Lecture from the American Society of Clinical Oncology.
The award is given to someone who “has made significant contributions to palliative care practice and research in oncology,” according to ASCO. Dr. Smith and his colleagues are known for their work showing that end-of-life palliative care can improve patient symptoms and quality of life while reducing the cost of care.
Dr. Smith will receive the award and deliver a keynote address at the 2019 Supportive Care in Oncology Symposium, which is set to take place Oct. 25-26 in San Francisco.
Meanwhile, Asya Nina Varshavsky-Yanovsky, MD, PhD, has joined Fox Chase Cancer Center in Philadelphia as an assistant professor in the hematology and bone marrow transplant programs within the department of hematology/oncology.
Dr. Varshavsky-Yanovsky earned her MD and PhD from the Technion-Israel Institute of Technology in Haifa, Israel. She joined Fox Chase Cancer Center/Temple University in 2016 for a 3-year fellowship in hematology/oncology.
Susmitha Apuri, MD, has joined Florida Cancer Specialists & Research Institute and is seeing patients in Inverness. She is board certified in medical oncology, hematology, and internal medicine.
Dr. Apuri earned her medical degree from NTR University of Health Sciences in Vijayawada, India; completed her internship and residency in internal medicine at the University of Miami Miller School of Medicine; and completed her fellowship in hematology/oncology at the University of South Florida/H. Lee Moffitt Cancer and Research Institute in Tampa. Her research and practice interests include malignant and nonmalignant hematology as well as breast, lung, and colorectal cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at [email protected], and you could be featured in Movers in Medicine.
MOVERS IN MEDICINE
Patients with focal epilepsy have progressive cortical thinning
, according to research published online July 1 in JAMA Neurology. Methods for preventing this thinning are unknown. “Our findings appear to highlight the need for longitudinal studies to develop disease-modifying treatments for epilepsy,” said Marian Galovic, MD, a doctoral student at University College London, and colleagues.
To date, neurologists have not found a definitive answer to the question of whether epilepsy is a static or progressive disease. Few longitudinal studies have examined patients with structural neuroimaging to determine whether the brain changes over time. The studies that have taken this approach have had small populations or have lacked control populations.
Comparing brain changes in patients and controls
Dr. Galovic and colleagues analyzed data for consecutive patients with focal epilepsy who underwent follow-up at the National Hospital for Neurology and Neurosurgery in London. The data were collected from Aug. 3, 2004, to Jan. 26, 2016. The researchers chose individuals who had at least 2 high-resolution T1-weighted MRI scans performed on the same scanner more than 6 months apart. They excluded patients with brain lesions other than hippocampal sclerosis, those with inadequate MRI scan quality, and those for whom clinical data were missing. To match these patients with controls, Dr. Galovic and colleagues chose three longitudinal data sets with data for healthy volunteers between ages 20 and 70 years. Each control participant had two high-resolution T1-weighted scans taken more than 6 months apart. The investigators matched patients and controls on age and sex. The automated and validated Computational Anatomy Toolbox (CAT12) estimated cortical thickness.
Dr. Galovic’s group included 190 patients with focal epilepsy, who had had 396 MRI scans, and 141 healthy controls, who had had 282 MRI scans, in their analysis. Age, sex, and image quality did not differ significantly between the two groups. Mean age was 36 years for patients and 35 years for controls. The proportion of women was 52.1% among patients and 53.9% among controls.
The rate of atrophy was doubled in patients
Approximately 77% of people with epilepsy had progressive cortical thinning that was distinct from that associated with normal aging. The mean overall annual rate of global cortical thinning was higher among patients with epilepsy (0.024) than among controls (0.011). The mean annual rate of cortical thinning increased among people with epilepsy who were older than 55 years. This rate was 0.021 in patients aged 18 to less than 35 years, compared with 0.023 in patients aged 35 to less than 55 years. Seizure frequency, number of antiepileptic drugs (AEDs) taken, and history of secondarily generalized seizures did not differ between age groups.
Compared with healthy controls, patients with focal epilepsy had widespread areas of greater progressive atrophy. Bilaterally affected areas included the lateral and posterior temporal lobes, posterior cingulate gyri, occipital lobes, pericentral gyri, and opercula. The distribution of progressive thinning in all patients with epilepsy was similar to regions connected to both hippocampi. Healthy controls had no areas of greater cortical thinning, compared with patients with epilepsy.
Progressive thinning in the left postcentral gyrus was greater in patients with left temporal lobe epilepsy (TLE) than in those with right TLE. Cortical thinning was more progressive in patients with right frontal lobe epilepsy (FLE) than in those with left FLE, particularly in right parietotemporal and right frontal areas.
Dr. Galovic and colleagues found no association between the rate of cortical thinning and seizure frequency, history of secondarily generalized seizures, or number of AEDs taken between MRIs. They found no difference in the rate of atrophy between patients with epilepsy with ongoing seizures and those without. The annual mean rate of cortical thinning was higher in people with a short duration of epilepsy (i.e., less than 5 years), compared with patients with a longer duration of epilepsy (i.e., 5 years or more).
A surrogate marker for neurodegeneration?
“The most likely cause of cortical thinning is neuronal loss, suggesting that these measurements are a surrogate marker for neurodegeneration,” said Dr. Galovic and colleagues. The finding that progressive morphologic changes were most pronounced in the first 5 years after epilepsy onset “supports the need for early diagnosis, rapid treatment, and reduction of delays of surgical referral in people with epilepsy,” they added.
One limitation of the current study is the fact that data from patients and controls were acquired using different MRI scanners. In addition, the patients included in the study had been referred to the center because their cases were more complicated, thus introducing the possibility of referral bias. The findings thus cannot be generalized readily to the overall population, said Dr. Galovic and colleagues.
Future studies should examine whether particular AEDs have differential influences on the progressive morphologic changes observed in epilepsy, said the investigators. “Future research should also address whether progressive changes in cortical morphologic characteristics correlate with deficits on serial cognitive testing or spreading of the irritative zone on EEG recordings,” they concluded.
The study and the authors received support from the Medical Research Council, the Wellcome Trust, and the University College London Hospital.
SOURCE: Galovic M et al. JAMA Neurol. 2019 Jul 1. doi: 10.1001/jamaneurol.2019.1708.
, according to research published online July 1 in JAMA Neurology. Methods for preventing this thinning are unknown. “Our findings appear to highlight the need for longitudinal studies to develop disease-modifying treatments for epilepsy,” said Marian Galovic, MD, a doctoral student at University College London, and colleagues.
To date, neurologists have not found a definitive answer to the question of whether epilepsy is a static or progressive disease. Few longitudinal studies have examined patients with structural neuroimaging to determine whether the brain changes over time. The studies that have taken this approach have had small populations or have lacked control populations.
Comparing brain changes in patients and controls
Dr. Galovic and colleagues analyzed data for consecutive patients with focal epilepsy who underwent follow-up at the National Hospital for Neurology and Neurosurgery in London. The data were collected from Aug. 3, 2004, to Jan. 26, 2016. The researchers chose individuals who had at least 2 high-resolution T1-weighted MRI scans performed on the same scanner more than 6 months apart. They excluded patients with brain lesions other than hippocampal sclerosis, those with inadequate MRI scan quality, and those for whom clinical data were missing. To match these patients with controls, Dr. Galovic and colleagues chose three longitudinal data sets with data for healthy volunteers between ages 20 and 70 years. Each control participant had two high-resolution T1-weighted scans taken more than 6 months apart. The investigators matched patients and controls on age and sex. The automated and validated Computational Anatomy Toolbox (CAT12) estimated cortical thickness.
Dr. Galovic’s group included 190 patients with focal epilepsy, who had had 396 MRI scans, and 141 healthy controls, who had had 282 MRI scans, in their analysis. Age, sex, and image quality did not differ significantly between the two groups. Mean age was 36 years for patients and 35 years for controls. The proportion of women was 52.1% among patients and 53.9% among controls.
The rate of atrophy was doubled in patients
Approximately 77% of people with epilepsy had progressive cortical thinning that was distinct from that associated with normal aging. The mean overall annual rate of global cortical thinning was higher among patients with epilepsy (0.024) than among controls (0.011). The mean annual rate of cortical thinning increased among people with epilepsy who were older than 55 years. This rate was 0.021 in patients aged 18 to less than 35 years, compared with 0.023 in patients aged 35 to less than 55 years. Seizure frequency, number of antiepileptic drugs (AEDs) taken, and history of secondarily generalized seizures did not differ between age groups.
Compared with healthy controls, patients with focal epilepsy had widespread areas of greater progressive atrophy. Bilaterally affected areas included the lateral and posterior temporal lobes, posterior cingulate gyri, occipital lobes, pericentral gyri, and opercula. The distribution of progressive thinning in all patients with epilepsy was similar to regions connected to both hippocampi. Healthy controls had no areas of greater cortical thinning, compared with patients with epilepsy.
Progressive thinning in the left postcentral gyrus was greater in patients with left temporal lobe epilepsy (TLE) than in those with right TLE. Cortical thinning was more progressive in patients with right frontal lobe epilepsy (FLE) than in those with left FLE, particularly in right parietotemporal and right frontal areas.
Dr. Galovic and colleagues found no association between the rate of cortical thinning and seizure frequency, history of secondarily generalized seizures, or number of AEDs taken between MRIs. They found no difference in the rate of atrophy between patients with epilepsy with ongoing seizures and those without. The annual mean rate of cortical thinning was higher in people with a short duration of epilepsy (i.e., less than 5 years), compared with patients with a longer duration of epilepsy (i.e., 5 years or more).
A surrogate marker for neurodegeneration?
“The most likely cause of cortical thinning is neuronal loss, suggesting that these measurements are a surrogate marker for neurodegeneration,” said Dr. Galovic and colleagues. The finding that progressive morphologic changes were most pronounced in the first 5 years after epilepsy onset “supports the need for early diagnosis, rapid treatment, and reduction of delays of surgical referral in people with epilepsy,” they added.
One limitation of the current study is the fact that data from patients and controls were acquired using different MRI scanners. In addition, the patients included in the study had been referred to the center because their cases were more complicated, thus introducing the possibility of referral bias. The findings thus cannot be generalized readily to the overall population, said Dr. Galovic and colleagues.
Future studies should examine whether particular AEDs have differential influences on the progressive morphologic changes observed in epilepsy, said the investigators. “Future research should also address whether progressive changes in cortical morphologic characteristics correlate with deficits on serial cognitive testing or spreading of the irritative zone on EEG recordings,” they concluded.
The study and the authors received support from the Medical Research Council, the Wellcome Trust, and the University College London Hospital.
SOURCE: Galovic M et al. JAMA Neurol. 2019 Jul 1. doi: 10.1001/jamaneurol.2019.1708.
, according to research published online July 1 in JAMA Neurology. Methods for preventing this thinning are unknown. “Our findings appear to highlight the need for longitudinal studies to develop disease-modifying treatments for epilepsy,” said Marian Galovic, MD, a doctoral student at University College London, and colleagues.
To date, neurologists have not found a definitive answer to the question of whether epilepsy is a static or progressive disease. Few longitudinal studies have examined patients with structural neuroimaging to determine whether the brain changes over time. The studies that have taken this approach have had small populations or have lacked control populations.
Comparing brain changes in patients and controls
Dr. Galovic and colleagues analyzed data for consecutive patients with focal epilepsy who underwent follow-up at the National Hospital for Neurology and Neurosurgery in London. The data were collected from Aug. 3, 2004, to Jan. 26, 2016. The researchers chose individuals who had at least 2 high-resolution T1-weighted MRI scans performed on the same scanner more than 6 months apart. They excluded patients with brain lesions other than hippocampal sclerosis, those with inadequate MRI scan quality, and those for whom clinical data were missing. To match these patients with controls, Dr. Galovic and colleagues chose three longitudinal data sets with data for healthy volunteers between ages 20 and 70 years. Each control participant had two high-resolution T1-weighted scans taken more than 6 months apart. The investigators matched patients and controls on age and sex. The automated and validated Computational Anatomy Toolbox (CAT12) estimated cortical thickness.
Dr. Galovic’s group included 190 patients with focal epilepsy, who had had 396 MRI scans, and 141 healthy controls, who had had 282 MRI scans, in their analysis. Age, sex, and image quality did not differ significantly between the two groups. Mean age was 36 years for patients and 35 years for controls. The proportion of women was 52.1% among patients and 53.9% among controls.
The rate of atrophy was doubled in patients
Approximately 77% of people with epilepsy had progressive cortical thinning that was distinct from that associated with normal aging. The mean overall annual rate of global cortical thinning was higher among patients with epilepsy (0.024) than among controls (0.011). The mean annual rate of cortical thinning increased among people with epilepsy who were older than 55 years. This rate was 0.021 in patients aged 18 to less than 35 years, compared with 0.023 in patients aged 35 to less than 55 years. Seizure frequency, number of antiepileptic drugs (AEDs) taken, and history of secondarily generalized seizures did not differ between age groups.
Compared with healthy controls, patients with focal epilepsy had widespread areas of greater progressive atrophy. Bilaterally affected areas included the lateral and posterior temporal lobes, posterior cingulate gyri, occipital lobes, pericentral gyri, and opercula. The distribution of progressive thinning in all patients with epilepsy was similar to regions connected to both hippocampi. Healthy controls had no areas of greater cortical thinning, compared with patients with epilepsy.
Progressive thinning in the left postcentral gyrus was greater in patients with left temporal lobe epilepsy (TLE) than in those with right TLE. Cortical thinning was more progressive in patients with right frontal lobe epilepsy (FLE) than in those with left FLE, particularly in right parietotemporal and right frontal areas.
Dr. Galovic and colleagues found no association between the rate of cortical thinning and seizure frequency, history of secondarily generalized seizures, or number of AEDs taken between MRIs. They found no difference in the rate of atrophy between patients with epilepsy with ongoing seizures and those without. The annual mean rate of cortical thinning was higher in people with a short duration of epilepsy (i.e., less than 5 years), compared with patients with a longer duration of epilepsy (i.e., 5 years or more).
A surrogate marker for neurodegeneration?
“The most likely cause of cortical thinning is neuronal loss, suggesting that these measurements are a surrogate marker for neurodegeneration,” said Dr. Galovic and colleagues. The finding that progressive morphologic changes were most pronounced in the first 5 years after epilepsy onset “supports the need for early diagnosis, rapid treatment, and reduction of delays of surgical referral in people with epilepsy,” they added.
One limitation of the current study is the fact that data from patients and controls were acquired using different MRI scanners. In addition, the patients included in the study had been referred to the center because their cases were more complicated, thus introducing the possibility of referral bias. The findings thus cannot be generalized readily to the overall population, said Dr. Galovic and colleagues.
Future studies should examine whether particular AEDs have differential influences on the progressive morphologic changes observed in epilepsy, said the investigators. “Future research should also address whether progressive changes in cortical morphologic characteristics correlate with deficits on serial cognitive testing or spreading of the irritative zone on EEG recordings,” they concluded.
The study and the authors received support from the Medical Research Council, the Wellcome Trust, and the University College London Hospital.
SOURCE: Galovic M et al. JAMA Neurol. 2019 Jul 1. doi: 10.1001/jamaneurol.2019.1708.
FROM JAMA NEUROLOGY
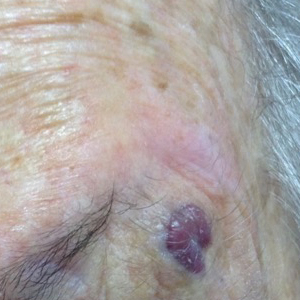
Rapidly Enlarging Neoplasm on the Face
The Diagnosis: Atypical Fibroxanthoma
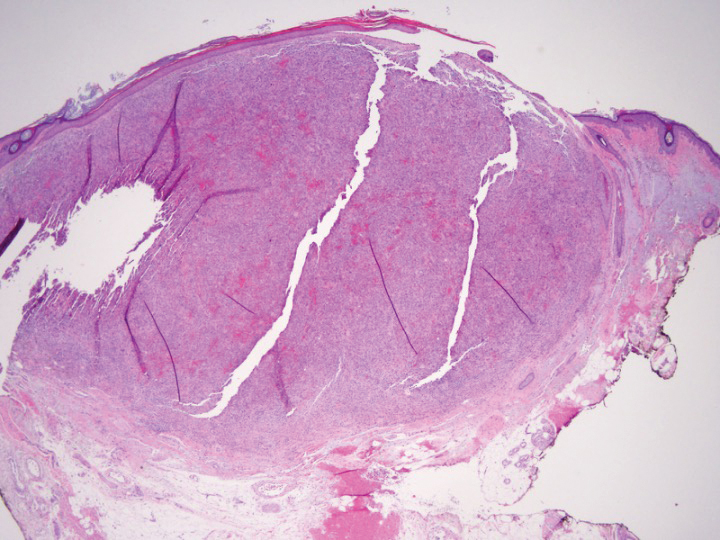
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
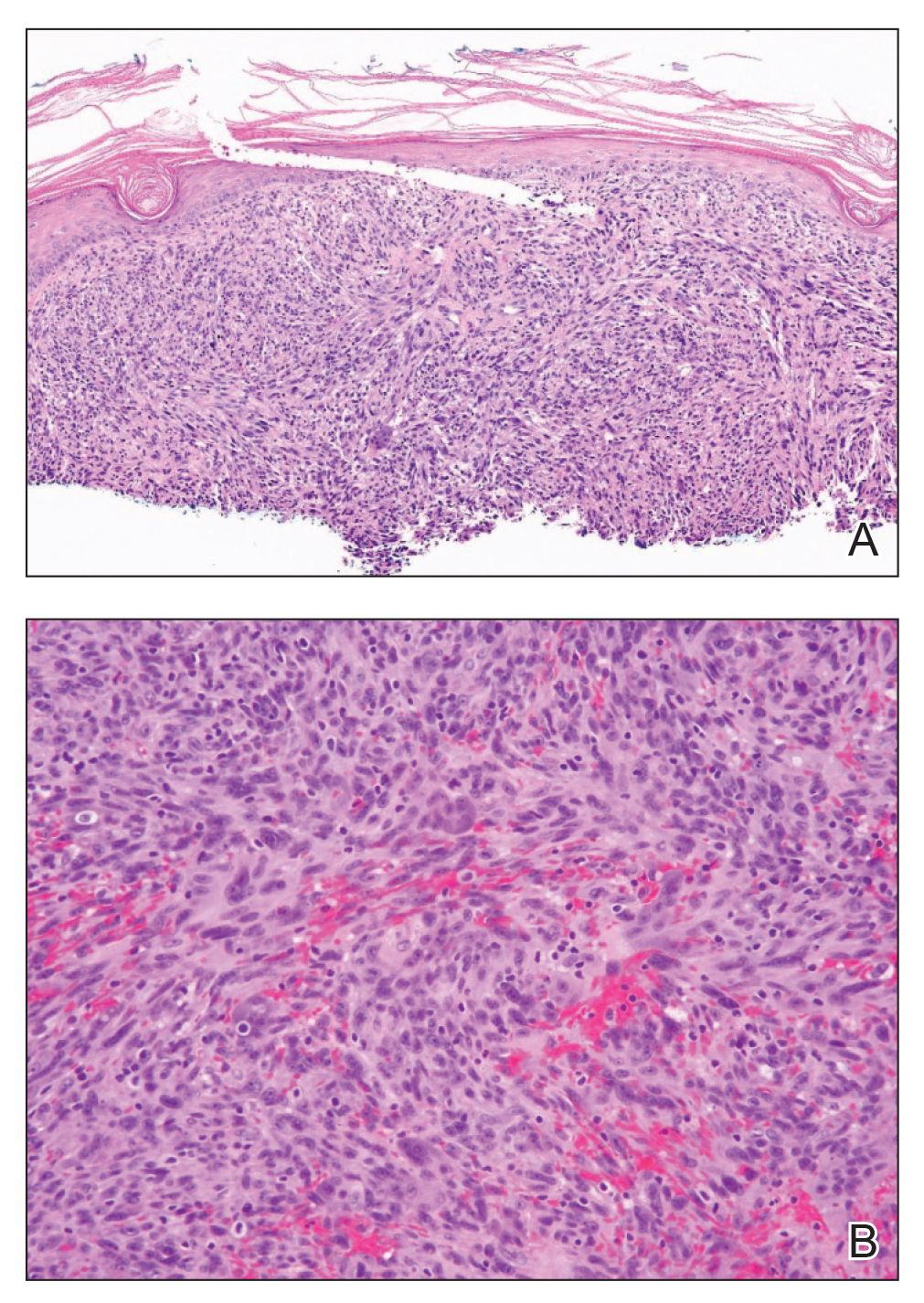
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
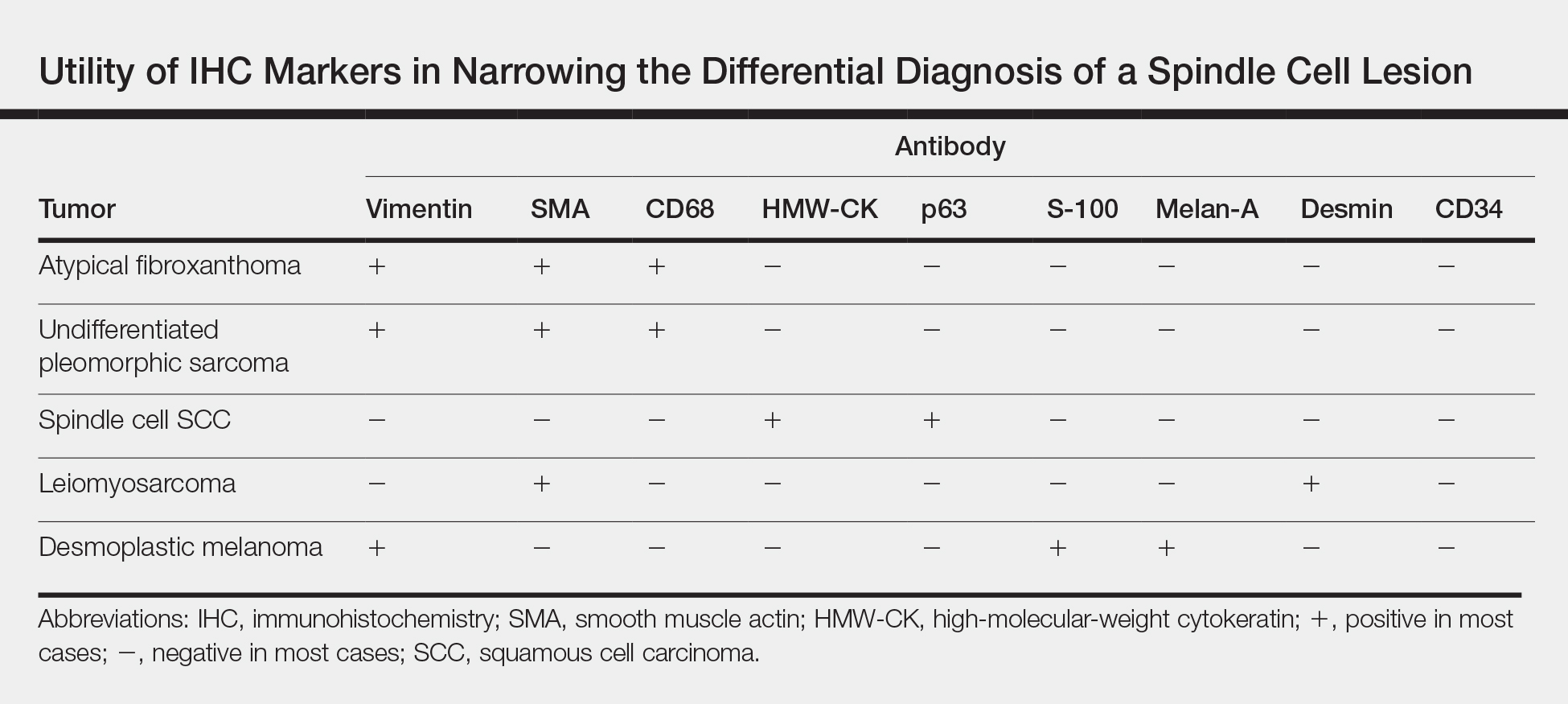
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
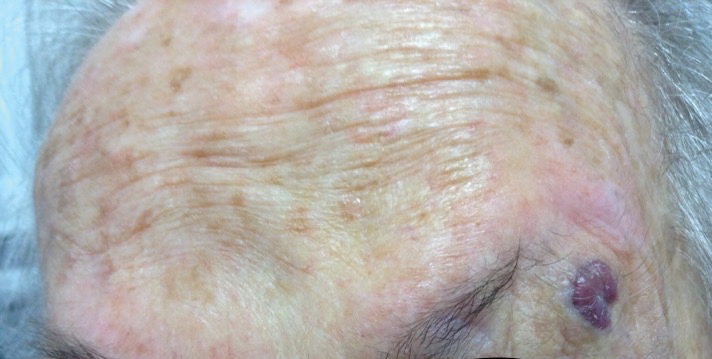
An 88-year-old woman presented for evaluation of an asymptomatic facial lesion that she first noticed 3 months prior, with rapid growth over the last month. Review of systems was negative, and she denied any history of connective tissue disease, skin cancer, or radiation to the head or neck area. Physical examination revealed a 1.5-cm, solitary, violaceous nodule on the left lateral eyebrow on a background of actinically damaged skin. The lesion was nontender and there were no similar lesions or palpable lymphadenopathy.
New findings cast more doubt on ‘fat-but-fit’ theory
SAN FRANCISCO – Can you be “fat but fit” if you’re obese but don’t suffer from metabolic syndrome? Some advocates have claimed you can, but new findings presented at the annual scientific sessions of the American Diabetes Association provide more evidence that those extra pounds translate to extra cardiac risk.

Fat-but-fit is a misnomer, Yvonne Commodore-Mensah, PhD, RN, assistant professor at Johns Hopkins School of Nursing, Baltimore, said in an interview. “The metabolically healthy obese are not so healthy. [We found] they had a higher risk of heart disease than people who were metabolically healthy and nonobese.”
Studies began supporting the fat-but-fit “paradox” in the late 1990s. They showed “that all-cause and CVD [cardiovascular] mortality risk in obese individuals, as defined by body mass index (BMI), body fat percentage, or waist circumference, who are fit (i.e., cardiorespiratory fitness level above the age-specific and sex-specific 20th percentile) is not significantly different from their normal-weight and fit counterparts” (Br J Sports Med. 2018;52[3]:151-3).
However, a 2017 study had found that “metabolically healthy obese individuals had a higher risk of coronary heart disease, cerebrovascular disease, and heart failure [compared with] normal weight, metabolically healthy individuals” (J Am Coll Cardiol. 2017;70[12]:1429-37). And a 2016 meta-analysis of 22 studies had produced similar results but also found that metabolically healthy obese individuals were better off, cardiac-health–wise, than those of normal weight who were metabolically unhealthy (Eur J Prev Cardiol. 2016;23[9]:956-66).
Dr. Commodore-Mensah and colleagues sought to establish through their study whether there was evidence of subclinical heart disease in people who are considered obese but metabolically healthy (Abstract 272-OR).
They tracked 11,884 participants in the Atherosclerosis Risk in Communities Study (ARIC) from 1990-1992 to 2016-2018. The study, which continues today, includes participants in suburban Minneapolis; Jackson, Miss.; Forsyth County, N.C.; and Washington County, Md.
None of the participants had previous cardiovascular disease at baseline (1990-1992). The researchers divided the participants into four groups at baseline: Nonobese (with metabolic syndrome, 20% of the total number of participants; or without metabolic syndrome, 51%) and obese (with metabolic syndrome, 20%; or without metabolic syndrome, 9%).
The average age range in the groups was 56-57 years. The percentage of women in the groups ranged from 53% to 58%, except for the obese and metabolically healthy group (73%). The percentage of black participants in the groups ranged from 17% (nonobese, metabolically unhealthy) to 45% (obese, metabolically healthy).
“People who were younger, women, and black were more likely to be classified as metabolically healthy obese,” Dr. Commodore-Mensah said.
According to one adjusted model with a median follow-up of 16 years and a total of 3,560 events, obese participants had a higher risk of incident cardiovascular disease, compared with their nonobese counterparts, regardless of whether they had metabolic syndrome.
When compared with the nonobese, metabolically healthy group, the risk grew in the nonobese, metabolically unhealthy group (hazard ratio, .24; 95% confidence interval, 1.12-1.36), as well as in the obese, metabolically healthy (HR, 1.33; 95% CI, 1.15-1.53) and the obese, metabolically unhealthy (HR, 2.11; 95% CI, 1.90-2.35) groups.
The researchers also focused on the cardiac biomarker known as high-sensitive cardiac troponin T (hs-cTnT), which indicates chronic myocardial damage. “This biomarker provides us with a window to the heart,” Dr. Commodore-Mensah said.
According to previous findings reported in 2014, ARIC participants who had hs-cTnT levels of 14 ng/L or higher were much more likely than were those with undetectable levels to suffer from heart failure, death from any cause, and coronary heart disease (JACC Heart Fail. 2014;2[6]:600-7).
Based on an analysis of the hs-cTnT levels in the present study, the researchers believe obese, metabolically healthy participants fell in the intermediate range of excess subclinical myocardial damage, between the nonobese and the obese participants who are also metabolically unhealthy.
“This group is not protected from heart disease,” Dr. Commodore-Mensah said. “They should be targeted, and they would benefit from behavioral changes, such as modifying their diet and increasing physical activity levels.”
The study is funded by the National Institutes of Health. Dr. Commodore-Mensah and six coauthors reported no relevant disclosures. Two coauthors reported various disclosures.
SAN FRANCISCO – Can you be “fat but fit” if you’re obese but don’t suffer from metabolic syndrome? Some advocates have claimed you can, but new findings presented at the annual scientific sessions of the American Diabetes Association provide more evidence that those extra pounds translate to extra cardiac risk.
Fat-but-fit is a misnomer, Yvonne Commodore-Mensah, PhD, RN, assistant professor at Johns Hopkins School of Nursing, Baltimore, said in an interview. “The metabolically healthy obese are not so healthy. [We found] they had a higher risk of heart disease than people who were metabolically healthy and nonobese.”
Studies began supporting the fat-but-fit “paradox” in the late 1990s. They showed “that all-cause and CVD [cardiovascular] mortality risk in obese individuals, as defined by body mass index (BMI), body fat percentage, or waist circumference, who are fit (i.e., cardiorespiratory fitness level above the age-specific and sex-specific 20th percentile) is not significantly different from their normal-weight and fit counterparts” (Br J Sports Med. 2018;52[3]:151-3).
However, a 2017 study had found that “metabolically healthy obese individuals had a higher risk of coronary heart disease, cerebrovascular disease, and heart failure [compared with] normal weight, metabolically healthy individuals” (J Am Coll Cardiol. 2017;70[12]:1429-37). And a 2016 meta-analysis of 22 studies had produced similar results but also found that metabolically healthy obese individuals were better off, cardiac-health–wise, than those of normal weight who were metabolically unhealthy (Eur J Prev Cardiol. 2016;23[9]:956-66).
Dr. Commodore-Mensah and colleagues sought to establish through their study whether there was evidence of subclinical heart disease in people who are considered obese but metabolically healthy (Abstract 272-OR).
They tracked 11,884 participants in the Atherosclerosis Risk in Communities Study (ARIC) from 1990-1992 to 2016-2018. The study, which continues today, includes participants in suburban Minneapolis; Jackson, Miss.; Forsyth County, N.C.; and Washington County, Md.
None of the participants had previous cardiovascular disease at baseline (1990-1992). The researchers divided the participants into four groups at baseline: Nonobese (with metabolic syndrome, 20% of the total number of participants; or without metabolic syndrome, 51%) and obese (with metabolic syndrome, 20%; or without metabolic syndrome, 9%).
The average age range in the groups was 56-57 years. The percentage of women in the groups ranged from 53% to 58%, except for the obese and metabolically healthy group (73%). The percentage of black participants in the groups ranged from 17% (nonobese, metabolically unhealthy) to 45% (obese, metabolically healthy).
“People who were younger, women, and black were more likely to be classified as metabolically healthy obese,” Dr. Commodore-Mensah said.
According to one adjusted model with a median follow-up of 16 years and a total of 3,560 events, obese participants had a higher risk of incident cardiovascular disease, compared with their nonobese counterparts, regardless of whether they had metabolic syndrome.
When compared with the nonobese, metabolically healthy group, the risk grew in the nonobese, metabolically unhealthy group (hazard ratio, .24; 95% confidence interval, 1.12-1.36), as well as in the obese, metabolically healthy (HR, 1.33; 95% CI, 1.15-1.53) and the obese, metabolically unhealthy (HR, 2.11; 95% CI, 1.90-2.35) groups.
The researchers also focused on the cardiac biomarker known as high-sensitive cardiac troponin T (hs-cTnT), which indicates chronic myocardial damage. “This biomarker provides us with a window to the heart,” Dr. Commodore-Mensah said.
According to previous findings reported in 2014, ARIC participants who had hs-cTnT levels of 14 ng/L or higher were much more likely than were those with undetectable levels to suffer from heart failure, death from any cause, and coronary heart disease (JACC Heart Fail. 2014;2[6]:600-7).
Based on an analysis of the hs-cTnT levels in the present study, the researchers believe obese, metabolically healthy participants fell in the intermediate range of excess subclinical myocardial damage, between the nonobese and the obese participants who are also metabolically unhealthy.
“This group is not protected from heart disease,” Dr. Commodore-Mensah said. “They should be targeted, and they would benefit from behavioral changes, such as modifying their diet and increasing physical activity levels.”
The study is funded by the National Institutes of Health. Dr. Commodore-Mensah and six coauthors reported no relevant disclosures. Two coauthors reported various disclosures.
SAN FRANCISCO – Can you be “fat but fit” if you’re obese but don’t suffer from metabolic syndrome? Some advocates have claimed you can, but new findings presented at the annual scientific sessions of the American Diabetes Association provide more evidence that those extra pounds translate to extra cardiac risk.
Fat-but-fit is a misnomer, Yvonne Commodore-Mensah, PhD, RN, assistant professor at Johns Hopkins School of Nursing, Baltimore, said in an interview. “The metabolically healthy obese are not so healthy. [We found] they had a higher risk of heart disease than people who were metabolically healthy and nonobese.”
Studies began supporting the fat-but-fit “paradox” in the late 1990s. They showed “that all-cause and CVD [cardiovascular] mortality risk in obese individuals, as defined by body mass index (BMI), body fat percentage, or waist circumference, who are fit (i.e., cardiorespiratory fitness level above the age-specific and sex-specific 20th percentile) is not significantly different from their normal-weight and fit counterparts” (Br J Sports Med. 2018;52[3]:151-3).
However, a 2017 study had found that “metabolically healthy obese individuals had a higher risk of coronary heart disease, cerebrovascular disease, and heart failure [compared with] normal weight, metabolically healthy individuals” (J Am Coll Cardiol. 2017;70[12]:1429-37). And a 2016 meta-analysis of 22 studies had produced similar results but also found that metabolically healthy obese individuals were better off, cardiac-health–wise, than those of normal weight who were metabolically unhealthy (Eur J Prev Cardiol. 2016;23[9]:956-66).
Dr. Commodore-Mensah and colleagues sought to establish through their study whether there was evidence of subclinical heart disease in people who are considered obese but metabolically healthy (Abstract 272-OR).
They tracked 11,884 participants in the Atherosclerosis Risk in Communities Study (ARIC) from 1990-1992 to 2016-2018. The study, which continues today, includes participants in suburban Minneapolis; Jackson, Miss.; Forsyth County, N.C.; and Washington County, Md.
None of the participants had previous cardiovascular disease at baseline (1990-1992). The researchers divided the participants into four groups at baseline: Nonobese (with metabolic syndrome, 20% of the total number of participants; or without metabolic syndrome, 51%) and obese (with metabolic syndrome, 20%; or without metabolic syndrome, 9%).
The average age range in the groups was 56-57 years. The percentage of women in the groups ranged from 53% to 58%, except for the obese and metabolically healthy group (73%). The percentage of black participants in the groups ranged from 17% (nonobese, metabolically unhealthy) to 45% (obese, metabolically healthy).
“People who were younger, women, and black were more likely to be classified as metabolically healthy obese,” Dr. Commodore-Mensah said.
According to one adjusted model with a median follow-up of 16 years and a total of 3,560 events, obese participants had a higher risk of incident cardiovascular disease, compared with their nonobese counterparts, regardless of whether they had metabolic syndrome.
When compared with the nonobese, metabolically healthy group, the risk grew in the nonobese, metabolically unhealthy group (hazard ratio, .24; 95% confidence interval, 1.12-1.36), as well as in the obese, metabolically healthy (HR, 1.33; 95% CI, 1.15-1.53) and the obese, metabolically unhealthy (HR, 2.11; 95% CI, 1.90-2.35) groups.
The researchers also focused on the cardiac biomarker known as high-sensitive cardiac troponin T (hs-cTnT), which indicates chronic myocardial damage. “This biomarker provides us with a window to the heart,” Dr. Commodore-Mensah said.
According to previous findings reported in 2014, ARIC participants who had hs-cTnT levels of 14 ng/L or higher were much more likely than were those with undetectable levels to suffer from heart failure, death from any cause, and coronary heart disease (JACC Heart Fail. 2014;2[6]:600-7).
Based on an analysis of the hs-cTnT levels in the present study, the researchers believe obese, metabolically healthy participants fell in the intermediate range of excess subclinical myocardial damage, between the nonobese and the obese participants who are also metabolically unhealthy.
“This group is not protected from heart disease,” Dr. Commodore-Mensah said. “They should be targeted, and they would benefit from behavioral changes, such as modifying their diet and increasing physical activity levels.”
The study is funded by the National Institutes of Health. Dr. Commodore-Mensah and six coauthors reported no relevant disclosures. Two coauthors reported various disclosures.
REPORTING FROM ADA 2019
Evidence supports accuracy of COPD diagnosis tool
The ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1:FVC) at the recommended threshold of 0.70 effectively diagnosed individuals at risk for clinically significant COPD, a longitudinal study of more than 24,000 individuals has found.
Guidelines from respiratory societies have long recommended a diagnosis of airflow obstruction when the FEV1:FVC is less than 0.70, but no rigorous, population-based studies have been conducted to support this recommendation, wrote Surya P. Bhatt, MD, of the University of Alabama at Birmingham, and colleagues.
“The selection of a threshold for defining airflow obstruction has major implications for patient care and public health as the prevalence of airflow obstruction can vary by as much as 33% depending on which threshold is selected,” they said.
In a study published in JAMA, the researchers reviewed data from 24,207 participants in the National Heart, Lung, and Blood Institute Pooled Cohorts Study to assess the accuracy of different thresholds in predicting COPD events in a large, multiethnic, U.S. population. All participants underwent spirometry; the average age at spirometry was 63 years, and 54% of the patients were women. Patients were enrolled during 1987-2000 and received follow-up longitudinally through 2016.
Overall, 3,925 participants experienced COPD-related events during an average of 15 years of follow-up (more than 340,757 person-years). These events included 3,563 hospitalizations and 447 deaths related to COPD.
The researchers compared three thresholds for FEV1:FVC ratios: a fixed optimal threshold of 0.71, a lower limit of normal (LLN) defined as 0.034, and the currently recommended 0.70.
The optimal 0.71 was not significantly different from the recommended 0.70 but was significantly more accurate than the LLN of 0.034. In addition, the 0.70 value was the optimal predictor in a subgroup analysis of ever-smokers and in multivariate analysis.
The findings were limited by several factors including the use of prebronchodilator spirometry, lack of adjustment for medication use, and limitation of outcomes to COPD mortality or clinical events mainly caused by COPD, which might exclude patients with mild to moderate disease, the researchers noted.
However, ” to help clinicians identify patients at increased risk for significant COPD, they said.
Lead author Dr. Bhatt disclosed a National Institutes of Health grant, consulting fees from Sunovion and research funds from Proterix Bio. The study was supported by grants from multiple agencies of the National Institutes of Health, including the National Heart, Lung, and Blood Institute, the National Institute of Neurological Disorders and Stroke, and the National Institute on Aging.
The ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1:FVC) at the recommended threshold of 0.70 effectively diagnosed individuals at risk for clinically significant COPD, a longitudinal study of more than 24,000 individuals has found.
Guidelines from respiratory societies have long recommended a diagnosis of airflow obstruction when the FEV1:FVC is less than 0.70, but no rigorous, population-based studies have been conducted to support this recommendation, wrote Surya P. Bhatt, MD, of the University of Alabama at Birmingham, and colleagues.
“The selection of a threshold for defining airflow obstruction has major implications for patient care and public health as the prevalence of airflow obstruction can vary by as much as 33% depending on which threshold is selected,” they said.
In a study published in JAMA, the researchers reviewed data from 24,207 participants in the National Heart, Lung, and Blood Institute Pooled Cohorts Study to assess the accuracy of different thresholds in predicting COPD events in a large, multiethnic, U.S. population. All participants underwent spirometry; the average age at spirometry was 63 years, and 54% of the patients were women. Patients were enrolled during 1987-2000 and received follow-up longitudinally through 2016.
Overall, 3,925 participants experienced COPD-related events during an average of 15 years of follow-up (more than 340,757 person-years). These events included 3,563 hospitalizations and 447 deaths related to COPD.
The researchers compared three thresholds for FEV1:FVC ratios: a fixed optimal threshold of 0.71, a lower limit of normal (LLN) defined as 0.034, and the currently recommended 0.70.
The optimal 0.71 was not significantly different from the recommended 0.70 but was significantly more accurate than the LLN of 0.034. In addition, the 0.70 value was the optimal predictor in a subgroup analysis of ever-smokers and in multivariate analysis.
The findings were limited by several factors including the use of prebronchodilator spirometry, lack of adjustment for medication use, and limitation of outcomes to COPD mortality or clinical events mainly caused by COPD, which might exclude patients with mild to moderate disease, the researchers noted.
However, ” to help clinicians identify patients at increased risk for significant COPD, they said.
Lead author Dr. Bhatt disclosed a National Institutes of Health grant, consulting fees from Sunovion and research funds from Proterix Bio. The study was supported by grants from multiple agencies of the National Institutes of Health, including the National Heart, Lung, and Blood Institute, the National Institute of Neurological Disorders and Stroke, and the National Institute on Aging.
The ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1:FVC) at the recommended threshold of 0.70 effectively diagnosed individuals at risk for clinically significant COPD, a longitudinal study of more than 24,000 individuals has found.
Guidelines from respiratory societies have long recommended a diagnosis of airflow obstruction when the FEV1:FVC is less than 0.70, but no rigorous, population-based studies have been conducted to support this recommendation, wrote Surya P. Bhatt, MD, of the University of Alabama at Birmingham, and colleagues.
“The selection of a threshold for defining airflow obstruction has major implications for patient care and public health as the prevalence of airflow obstruction can vary by as much as 33% depending on which threshold is selected,” they said.
In a study published in JAMA, the researchers reviewed data from 24,207 participants in the National Heart, Lung, and Blood Institute Pooled Cohorts Study to assess the accuracy of different thresholds in predicting COPD events in a large, multiethnic, U.S. population. All participants underwent spirometry; the average age at spirometry was 63 years, and 54% of the patients were women. Patients were enrolled during 1987-2000 and received follow-up longitudinally through 2016.
Overall, 3,925 participants experienced COPD-related events during an average of 15 years of follow-up (more than 340,757 person-years). These events included 3,563 hospitalizations and 447 deaths related to COPD.
The researchers compared three thresholds for FEV1:FVC ratios: a fixed optimal threshold of 0.71, a lower limit of normal (LLN) defined as 0.034, and the currently recommended 0.70.
The optimal 0.71 was not significantly different from the recommended 0.70 but was significantly more accurate than the LLN of 0.034. In addition, the 0.70 value was the optimal predictor in a subgroup analysis of ever-smokers and in multivariate analysis.
The findings were limited by several factors including the use of prebronchodilator spirometry, lack of adjustment for medication use, and limitation of outcomes to COPD mortality or clinical events mainly caused by COPD, which might exclude patients with mild to moderate disease, the researchers noted.
However, ” to help clinicians identify patients at increased risk for significant COPD, they said.
Lead author Dr. Bhatt disclosed a National Institutes of Health grant, consulting fees from Sunovion and research funds from Proterix Bio. The study was supported by grants from multiple agencies of the National Institutes of Health, including the National Heart, Lung, and Blood Institute, the National Institute of Neurological Disorders and Stroke, and the National Institute on Aging.
FROM JAMA
CAROLINA findings reaffirm linagliptin’s safety, free glimepiride of CV-risk stigma
SAN FRANCISCO – The sulfonylurea glimepiride (Amaryl) did not increase the risk of cardiovascular events in patients with type 2 diabetes and cardiovascular risk in a head-to-head comparison with the dipeptidyl peptidase–4 (DPP-4) inhibitor linagliptin (Tradjenta), a drug with proven cardiovascular safety, according to findings from the CAROLINA study presented at the scientific sessions of the American Diabetes Association.

Linagliptin’s cardiovascular safety, compared with placebo, was demonstrated in the CARMELINA study (JAMA. 2019;321[1]:69-79), but CAROLINA pitted the DPP-4 inhibitor against an active comparator, glimepiride, which along with other sulfonylureas, carries a warning for increased risk of cardiovascular mortality. The latest findings about the comparator were not expected, but seem to set aside the lingering doubts about cardiovascular safety in at least the modern-day sulfonylureas.
“The stigma that sulfonylureas have had for more than 50 years” has been lifted. “We take a lot of pride in this study,” said principal investigator and endocrinologist Julio Rosenstock, MD, a clinical professor of medicine at the University of Texas Southwestern Medical Center, Dallas.
In 1970, the University Group Diabetes Program trial reported 26 cardiovascular deaths in 204 patients who received with tolbutamide – a first-generation sulfonylurea no longer in common use – compared with 10 deaths in 205 patients who received placebo (Diabetes. 1970;19[Suppl]:789-830). The finding led to a warning of increased risk of cardiovascular mortality that still appears on sulfonylurea labels today.
The results were never confirmed by subsequent studies, and debate about the cardiovascular safety of sulfonylureas continued, with many physicians over the years calling for a large, rigorous trial to resolve the issue once and for all.

]And that’s what the CAROLINA findings delivered – a resolution to the “decades-long debate” about cardiovascular safety with glimepiride and likely other modern sulfonylureas, according to coinvestigator and cardiologist Nikolaus Marx, MD, a professor of medicine at Aachen (Germany) University.
The CAROLINA investigators randomized 1:1 more than 6,000 patients with type 2 diabetes at 607 sites in 43 countries to receive either linagliptin 5 mg daily or glimepiride 1-4 mg daily on a background therapy, in most cases, of metformin. The median diabetes duration was 6.3 years, and baseline hemoglobin A1c was 7.15%. In all, 42% of the patients had established cardiovascular disease, and 37% had two or more risk factors that were managed by standard care.
After a median follow-up of 6.3 years, there was no difference between linagliptin and glimepiride in the primary composite outcome of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke (11.8% and 12%, respectively; P = .76), nor were there any differences in the individual components. Likewise, there were no differences between the two drugs in hospitalization for heart failure (3.7% and 3.1%, P = .18) or all-cause mortality (10.2% and 11.2%; P = .23), and no difference in glucose control – a drop of about 0.3% in the HbA1c level at 1 year, then a slow creep back to baseline at around 4 years.
“We believe cardiovascular safety should no longer be a consideration in [deciding] between these two agents,” said Dr. Rosenstock, who cochaired the session with Dr. Marx.
However, there was a modest weight gain with glimepiride, and a marked increase in the risk of moderate to severe hypoglycemia, compared with linagliptin (30.9% and 6.5%, respectively; P less than .0001). Both weight gain and hypoglycemia are recognized side effects of sulfonylureas.
Sulfonylureas are far less expensive than the DPP-4 inhibitors are, so “other than cost consideration ... [the findings] support use of DPP-4 inhibitors before sulfonylureas if hypoglycemia and weight gain are also considerations,” Dr. Rosenstock said.
Robert H. Eckel, MD, an endocrinologist, said he has been “taking people off sulfonylureas for years because I [wasn’t] sure they were safe. This study has helped me know that I can continue [them] safely.”

However, even with the new data, “the only reason I would [use a sulfonylurea] is cost. Cost is the bottom line in the clinic,” added Dr. Eckel, a professor of medicine at the University of Colorado, Aurora.
The trial findings pose no challenge to current guidelines for type 2 diabetes and the recommendation to opt first for a second-line medication with proven cardiovascular benefit, such as a sodium-glucose transporter 2 inhibitor or glucagonlike peptide–1 receptor agonist, in at-risk patients.
Boehringer Ingelheim, the maker of linagliptin, funded the study in collaboration with comarketer, Eli Lilly. Dr. Rosenstock and Dr. Marx disclosed numerous ties to Boehringer Ingelheim and Eli Lilly, and other companies not associated with the study. Dr. Eckel disclosed ties with companies not associated with the study.
SOURCE: Rosenstock J et al. ADA 2019.
SAN FRANCISCO – The sulfonylurea glimepiride (Amaryl) did not increase the risk of cardiovascular events in patients with type 2 diabetes and cardiovascular risk in a head-to-head comparison with the dipeptidyl peptidase–4 (DPP-4) inhibitor linagliptin (Tradjenta), a drug with proven cardiovascular safety, according to findings from the CAROLINA study presented at the scientific sessions of the American Diabetes Association.
Linagliptin’s cardiovascular safety, compared with placebo, was demonstrated in the CARMELINA study (JAMA. 2019;321[1]:69-79), but CAROLINA pitted the DPP-4 inhibitor against an active comparator, glimepiride, which along with other sulfonylureas, carries a warning for increased risk of cardiovascular mortality. The latest findings about the comparator were not expected, but seem to set aside the lingering doubts about cardiovascular safety in at least the modern-day sulfonylureas.
“The stigma that sulfonylureas have had for more than 50 years” has been lifted. “We take a lot of pride in this study,” said principal investigator and endocrinologist Julio Rosenstock, MD, a clinical professor of medicine at the University of Texas Southwestern Medical Center, Dallas.
In 1970, the University Group Diabetes Program trial reported 26 cardiovascular deaths in 204 patients who received with tolbutamide – a first-generation sulfonylurea no longer in common use – compared with 10 deaths in 205 patients who received placebo (Diabetes. 1970;19[Suppl]:789-830). The finding led to a warning of increased risk of cardiovascular mortality that still appears on sulfonylurea labels today.
The results were never confirmed by subsequent studies, and debate about the cardiovascular safety of sulfonylureas continued, with many physicians over the years calling for a large, rigorous trial to resolve the issue once and for all.
]And that’s what the CAROLINA findings delivered – a resolution to the “decades-long debate” about cardiovascular safety with glimepiride and likely other modern sulfonylureas, according to coinvestigator and cardiologist Nikolaus Marx, MD, a professor of medicine at Aachen (Germany) University.
The CAROLINA investigators randomized 1:1 more than 6,000 patients with type 2 diabetes at 607 sites in 43 countries to receive either linagliptin 5 mg daily or glimepiride 1-4 mg daily on a background therapy, in most cases, of metformin. The median diabetes duration was 6.3 years, and baseline hemoglobin A1c was 7.15%. In all, 42% of the patients had established cardiovascular disease, and 37% had two or more risk factors that were managed by standard care.
After a median follow-up of 6.3 years, there was no difference between linagliptin and glimepiride in the primary composite outcome of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke (11.8% and 12%, respectively; P = .76), nor were there any differences in the individual components. Likewise, there were no differences between the two drugs in hospitalization for heart failure (3.7% and 3.1%, P = .18) or all-cause mortality (10.2% and 11.2%; P = .23), and no difference in glucose control – a drop of about 0.3% in the HbA1c level at 1 year, then a slow creep back to baseline at around 4 years.
“We believe cardiovascular safety should no longer be a consideration in [deciding] between these two agents,” said Dr. Rosenstock, who cochaired the session with Dr. Marx.
However, there was a modest weight gain with glimepiride, and a marked increase in the risk of moderate to severe hypoglycemia, compared with linagliptin (30.9% and 6.5%, respectively; P less than .0001). Both weight gain and hypoglycemia are recognized side effects of sulfonylureas.
Sulfonylureas are far less expensive than the DPP-4 inhibitors are, so “other than cost consideration ... [the findings] support use of DPP-4 inhibitors before sulfonylureas if hypoglycemia and weight gain are also considerations,” Dr. Rosenstock said.
Robert H. Eckel, MD, an endocrinologist, said he has been “taking people off sulfonylureas for years because I [wasn’t] sure they were safe. This study has helped me know that I can continue [them] safely.”
However, even with the new data, “the only reason I would [use a sulfonylurea] is cost. Cost is the bottom line in the clinic,” added Dr. Eckel, a professor of medicine at the University of Colorado, Aurora.
The trial findings pose no challenge to current guidelines for type 2 diabetes and the recommendation to opt first for a second-line medication with proven cardiovascular benefit, such as a sodium-glucose transporter 2 inhibitor or glucagonlike peptide–1 receptor agonist, in at-risk patients.
Boehringer Ingelheim, the maker of linagliptin, funded the study in collaboration with comarketer, Eli Lilly. Dr. Rosenstock and Dr. Marx disclosed numerous ties to Boehringer Ingelheim and Eli Lilly, and other companies not associated with the study. Dr. Eckel disclosed ties with companies not associated with the study.
SOURCE: Rosenstock J et al. ADA 2019.
SAN FRANCISCO – The sulfonylurea glimepiride (Amaryl) did not increase the risk of cardiovascular events in patients with type 2 diabetes and cardiovascular risk in a head-to-head comparison with the dipeptidyl peptidase–4 (DPP-4) inhibitor linagliptin (Tradjenta), a drug with proven cardiovascular safety, according to findings from the CAROLINA study presented at the scientific sessions of the American Diabetes Association.
Linagliptin’s cardiovascular safety, compared with placebo, was demonstrated in the CARMELINA study (JAMA. 2019;321[1]:69-79), but CAROLINA pitted the DPP-4 inhibitor against an active comparator, glimepiride, which along with other sulfonylureas, carries a warning for increased risk of cardiovascular mortality. The latest findings about the comparator were not expected, but seem to set aside the lingering doubts about cardiovascular safety in at least the modern-day sulfonylureas.
“The stigma that sulfonylureas have had for more than 50 years” has been lifted. “We take a lot of pride in this study,” said principal investigator and endocrinologist Julio Rosenstock, MD, a clinical professor of medicine at the University of Texas Southwestern Medical Center, Dallas.
In 1970, the University Group Diabetes Program trial reported 26 cardiovascular deaths in 204 patients who received with tolbutamide – a first-generation sulfonylurea no longer in common use – compared with 10 deaths in 205 patients who received placebo (Diabetes. 1970;19[Suppl]:789-830). The finding led to a warning of increased risk of cardiovascular mortality that still appears on sulfonylurea labels today.
The results were never confirmed by subsequent studies, and debate about the cardiovascular safety of sulfonylureas continued, with many physicians over the years calling for a large, rigorous trial to resolve the issue once and for all.
]And that’s what the CAROLINA findings delivered – a resolution to the “decades-long debate” about cardiovascular safety with glimepiride and likely other modern sulfonylureas, according to coinvestigator and cardiologist Nikolaus Marx, MD, a professor of medicine at Aachen (Germany) University.
The CAROLINA investigators randomized 1:1 more than 6,000 patients with type 2 diabetes at 607 sites in 43 countries to receive either linagliptin 5 mg daily or glimepiride 1-4 mg daily on a background therapy, in most cases, of metformin. The median diabetes duration was 6.3 years, and baseline hemoglobin A1c was 7.15%. In all, 42% of the patients had established cardiovascular disease, and 37% had two or more risk factors that were managed by standard care.
After a median follow-up of 6.3 years, there was no difference between linagliptin and glimepiride in the primary composite outcome of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke (11.8% and 12%, respectively; P = .76), nor were there any differences in the individual components. Likewise, there were no differences between the two drugs in hospitalization for heart failure (3.7% and 3.1%, P = .18) or all-cause mortality (10.2% and 11.2%; P = .23), and no difference in glucose control – a drop of about 0.3% in the HbA1c level at 1 year, then a slow creep back to baseline at around 4 years.
“We believe cardiovascular safety should no longer be a consideration in [deciding] between these two agents,” said Dr. Rosenstock, who cochaired the session with Dr. Marx.
However, there was a modest weight gain with glimepiride, and a marked increase in the risk of moderate to severe hypoglycemia, compared with linagliptin (30.9% and 6.5%, respectively; P less than .0001). Both weight gain and hypoglycemia are recognized side effects of sulfonylureas.
Sulfonylureas are far less expensive than the DPP-4 inhibitors are, so “other than cost consideration ... [the findings] support use of DPP-4 inhibitors before sulfonylureas if hypoglycemia and weight gain are also considerations,” Dr. Rosenstock said.
Robert H. Eckel, MD, an endocrinologist, said he has been “taking people off sulfonylureas for years because I [wasn’t] sure they were safe. This study has helped me know that I can continue [them] safely.”
However, even with the new data, “the only reason I would [use a sulfonylurea] is cost. Cost is the bottom line in the clinic,” added Dr. Eckel, a professor of medicine at the University of Colorado, Aurora.
The trial findings pose no challenge to current guidelines for type 2 diabetes and the recommendation to opt first for a second-line medication with proven cardiovascular benefit, such as a sodium-glucose transporter 2 inhibitor or glucagonlike peptide–1 receptor agonist, in at-risk patients.
Boehringer Ingelheim, the maker of linagliptin, funded the study in collaboration with comarketer, Eli Lilly. Dr. Rosenstock and Dr. Marx disclosed numerous ties to Boehringer Ingelheim and Eli Lilly, and other companies not associated with the study. Dr. Eckel disclosed ties with companies not associated with the study.
SOURCE: Rosenstock J et al. ADA 2019.
REPORTING FROM ADA 2019
EULAR keeps csDMARDs as top PsA drugs
MADRID – The draft revision of the European League Against Rheumatism’s recommendations for managing psoriatic arthritis sticks with the group’s already-existing conviction that psoriatic arthritis treatment best starts with an NSAID, and if that fails follow with a conventional synthetic antirheumatic drug such as methotrexate, a position in stark contrast with the 2018 recommendation from the American College of Rheumatology to first treat with a tumor necrosis factor (TNF) inhibitor.

For patients with psoriatic arthritis (PsA) manifesting with polyarthritis, conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) “should be first,” and should “start rapidly” if brief, initial treatment with an NSAID proves inadequate, Laure Gossec, MD, PhD, said while presenting a draft version of an update to the PsA management recommendations from EULAR at the European Congress of Rheumatology.
The EULAR recommendations-revision panel had about the same advice for managing PsA patients with oligoarthritis, monoarthritis, or peripheral arthritis. For oligo- and monoarthritis, “consider a csDMARD after failing NSAIDS, but also consider the patient’s prognostic factors,” like structural damage, and dactylitis. For PsA patients with peripheral arthritis, “it still makes sense to keep csDMARDs as the first line treatment,” said Dr. Gossec, professor of rheumatology at Pitie-Salpétriere Hospital and Sorbonne University, Paris. Once published, the revision will replace existing EULAR recommendations from 2015 (Ann Rheum Dis. 2016 Mar;75[3]:499-510).
The list of csDMARDs she cited included not just methotrexate, still the top csDMARD, but also sulfasalazine and leflunomide as alternatives, she noted, with methotrexate also the preferred csDMARD for patients with skin involvement. When a PsA patient fails at least one csDMARD, then switching to a biologic DMARD is recommended. For a patient with skin involvement, a drug that targets interleukin-17 or IL-12 and -23 is preferred. If skin involvement is not a major issue, then treatment with a TNF inhibitor is equally valid, she said.
The 2018 PsA management guideline from the American College of Rheumatology (ACR) proposed a strikingly different sequence, endorsing initial treatment with a TNF inhibitor first over all other options, including methotrexate and other “oral small molecules” (the ACR term for csDMARD), and also including NSAIDs (Arthritis Rheumatol. 2019 Jan;71[1]:5-32).
This schism between EULAR and the ACR could be seen as predictable, given the different constraints the two societies have set for themselves.

“EULAR recommendations take into account drug costs; the ACR guideline is supposed to be agnostic to costs,” explained Philip J. Mease, MD, a rheumatologist at Swedish Medical Center in Seattle and a member of the ACR panel that wrote the 2018 PsA guideline.
In fact it was a study Dr. Mease recently led and reported results from that provided the most recent and perhaps best assessment of a TNF inhibitor, compared with methotrexate, as initial treatment for PsA, with findings that suggest that, although the advice from the two societies may sharply differ, the viewpoints of both groups are evidence based.
The SEAM-PsA (Study of Etanercept and Methotrexate in Subjects with Psoriatic Arthritis) trial randomized 851 PsA patients receiving their first treatment to methotrexate only, the TNF inhibitor etanercept (Enbrel) only, or both drugs. The study’s two coprimary outcomes, the ACR 20 and minimal disease activity responses after 24 weeks, showed that etanercept monotherapy produced these responses in 61% and 36% of patients, respectively, while methotrexate monotherapy produced response rates of 51% and 23%, respectively. Both these differences between etanercept monotherapy and methotrexate monotherapy were statistically significant. Combining methotrexate with etanercept did not produce a significant improvement over etanercept alone.
Interpreting the meaning of this finding for clinical practice “depends on the lens you look through,” Dr. Mease said in an interview. “A lot of patients respond to methotrexate, which is good when treatment resources are challenged. But when there is no resource challenge, the data support going straight to a TNF inhibitor.”
Dr. Gossec confirmed the importance of the SEAM-PsA findings in the writing panel’s decision during discussion of the draft, replying to a question about consideration of the study’s findings. “We carefully looked at the SEAM-PsA trial results, which provide some of the only data we have on methotrexate” for PsA. “We felt that the results were in favor of methotrexate’s efficacy, and therefore did not go against our proposal to keep a graduated approach starting with a csDMARD.”
Patients who fail to receive adequate relief from a csDMARD could then try a biologic DMARD – a TNF inhibitor, IL-17 inhibitor, or IL-12/23 inhibitor, Dr. Gossec said. When skin involvement is minimal, any of these options are possible, she said. If skin involvement is significant, the panel recommended preferentially using an IL-17 or IL-12/23 inhibitor based on head-to-head trials in patients with psoriasis, she said.
When a biologic DMARD is not appropriate or fails, another option is to then try a targeted synthetic DMARD, such as a Janus kinase inhibitor. When none of these options are appropriate, or they all fail, another option for patients with mild oligo- or monoarthritis or in patients with limited skin involvement is apremilast (Otezla), a phosphodiesterase-4 inhibitor. The draft recommendations also advise clinicians to be sure to distinguish fibromyalgia pain from enthesitis involvement, and they introduce the possibility of, with “great caution,” tapering down DMARD treatment in PsA patients who show sustained remission.
Dr. Gossec and Dr. Mease have both been consultants to and received honoraria from several companies. SEAM-PsA was sponsored by Amgen, the company that markets Enbrel.
MADRID – The draft revision of the European League Against Rheumatism’s recommendations for managing psoriatic arthritis sticks with the group’s already-existing conviction that psoriatic arthritis treatment best starts with an NSAID, and if that fails follow with a conventional synthetic antirheumatic drug such as methotrexate, a position in stark contrast with the 2018 recommendation from the American College of Rheumatology to first treat with a tumor necrosis factor (TNF) inhibitor.
For patients with psoriatic arthritis (PsA) manifesting with polyarthritis, conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) “should be first,” and should “start rapidly” if brief, initial treatment with an NSAID proves inadequate, Laure Gossec, MD, PhD, said while presenting a draft version of an update to the PsA management recommendations from EULAR at the European Congress of Rheumatology.
The EULAR recommendations-revision panel had about the same advice for managing PsA patients with oligoarthritis, monoarthritis, or peripheral arthritis. For oligo- and monoarthritis, “consider a csDMARD after failing NSAIDS, but also consider the patient’s prognostic factors,” like structural damage, and dactylitis. For PsA patients with peripheral arthritis, “it still makes sense to keep csDMARDs as the first line treatment,” said Dr. Gossec, professor of rheumatology at Pitie-Salpétriere Hospital and Sorbonne University, Paris. Once published, the revision will replace existing EULAR recommendations from 2015 (Ann Rheum Dis. 2016 Mar;75[3]:499-510).
The list of csDMARDs she cited included not just methotrexate, still the top csDMARD, but also sulfasalazine and leflunomide as alternatives, she noted, with methotrexate also the preferred csDMARD for patients with skin involvement. When a PsA patient fails at least one csDMARD, then switching to a biologic DMARD is recommended. For a patient with skin involvement, a drug that targets interleukin-17 or IL-12 and -23 is preferred. If skin involvement is not a major issue, then treatment with a TNF inhibitor is equally valid, she said.
The 2018 PsA management guideline from the American College of Rheumatology (ACR) proposed a strikingly different sequence, endorsing initial treatment with a TNF inhibitor first over all other options, including methotrexate and other “oral small molecules” (the ACR term for csDMARD), and also including NSAIDs (Arthritis Rheumatol. 2019 Jan;71[1]:5-32).
This schism between EULAR and the ACR could be seen as predictable, given the different constraints the two societies have set for themselves.
“EULAR recommendations take into account drug costs; the ACR guideline is supposed to be agnostic to costs,” explained Philip J. Mease, MD, a rheumatologist at Swedish Medical Center in Seattle and a member of the ACR panel that wrote the 2018 PsA guideline.
In fact it was a study Dr. Mease recently led and reported results from that provided the most recent and perhaps best assessment of a TNF inhibitor, compared with methotrexate, as initial treatment for PsA, with findings that suggest that, although the advice from the two societies may sharply differ, the viewpoints of both groups are evidence based.
The SEAM-PsA (Study of Etanercept and Methotrexate in Subjects with Psoriatic Arthritis) trial randomized 851 PsA patients receiving their first treatment to methotrexate only, the TNF inhibitor etanercept (Enbrel) only, or both drugs. The study’s two coprimary outcomes, the ACR 20 and minimal disease activity responses after 24 weeks, showed that etanercept monotherapy produced these responses in 61% and 36% of patients, respectively, while methotrexate monotherapy produced response rates of 51% and 23%, respectively. Both these differences between etanercept monotherapy and methotrexate monotherapy were statistically significant. Combining methotrexate with etanercept did not produce a significant improvement over etanercept alone.
Interpreting the meaning of this finding for clinical practice “depends on the lens you look through,” Dr. Mease said in an interview. “A lot of patients respond to methotrexate, which is good when treatment resources are challenged. But when there is no resource challenge, the data support going straight to a TNF inhibitor.”
Dr. Gossec confirmed the importance of the SEAM-PsA findings in the writing panel’s decision during discussion of the draft, replying to a question about consideration of the study’s findings. “We carefully looked at the SEAM-PsA trial results, which provide some of the only data we have on methotrexate” for PsA. “We felt that the results were in favor of methotrexate’s efficacy, and therefore did not go against our proposal to keep a graduated approach starting with a csDMARD.”
Patients who fail to receive adequate relief from a csDMARD could then try a biologic DMARD – a TNF inhibitor, IL-17 inhibitor, or IL-12/23 inhibitor, Dr. Gossec said. When skin involvement is minimal, any of these options are possible, she said. If skin involvement is significant, the panel recommended preferentially using an IL-17 or IL-12/23 inhibitor based on head-to-head trials in patients with psoriasis, she said.
When a biologic DMARD is not appropriate or fails, another option is to then try a targeted synthetic DMARD, such as a Janus kinase inhibitor. When none of these options are appropriate, or they all fail, another option for patients with mild oligo- or monoarthritis or in patients with limited skin involvement is apremilast (Otezla), a phosphodiesterase-4 inhibitor. The draft recommendations also advise clinicians to be sure to distinguish fibromyalgia pain from enthesitis involvement, and they introduce the possibility of, with “great caution,” tapering down DMARD treatment in PsA patients who show sustained remission.
Dr. Gossec and Dr. Mease have both been consultants to and received honoraria from several companies. SEAM-PsA was sponsored by Amgen, the company that markets Enbrel.
MADRID – The draft revision of the European League Against Rheumatism’s recommendations for managing psoriatic arthritis sticks with the group’s already-existing conviction that psoriatic arthritis treatment best starts with an NSAID, and if that fails follow with a conventional synthetic antirheumatic drug such as methotrexate, a position in stark contrast with the 2018 recommendation from the American College of Rheumatology to first treat with a tumor necrosis factor (TNF) inhibitor.
For patients with psoriatic arthritis (PsA) manifesting with polyarthritis, conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) “should be first,” and should “start rapidly” if brief, initial treatment with an NSAID proves inadequate, Laure Gossec, MD, PhD, said while presenting a draft version of an update to the PsA management recommendations from EULAR at the European Congress of Rheumatology.
The EULAR recommendations-revision panel had about the same advice for managing PsA patients with oligoarthritis, monoarthritis, or peripheral arthritis. For oligo- and monoarthritis, “consider a csDMARD after failing NSAIDS, but also consider the patient’s prognostic factors,” like structural damage, and dactylitis. For PsA patients with peripheral arthritis, “it still makes sense to keep csDMARDs as the first line treatment,” said Dr. Gossec, professor of rheumatology at Pitie-Salpétriere Hospital and Sorbonne University, Paris. Once published, the revision will replace existing EULAR recommendations from 2015 (Ann Rheum Dis. 2016 Mar;75[3]:499-510).
The list of csDMARDs she cited included not just methotrexate, still the top csDMARD, but also sulfasalazine and leflunomide as alternatives, she noted, with methotrexate also the preferred csDMARD for patients with skin involvement. When a PsA patient fails at least one csDMARD, then switching to a biologic DMARD is recommended. For a patient with skin involvement, a drug that targets interleukin-17 or IL-12 and -23 is preferred. If skin involvement is not a major issue, then treatment with a TNF inhibitor is equally valid, she said.
The 2018 PsA management guideline from the American College of Rheumatology (ACR) proposed a strikingly different sequence, endorsing initial treatment with a TNF inhibitor first over all other options, including methotrexate and other “oral small molecules” (the ACR term for csDMARD), and also including NSAIDs (Arthritis Rheumatol. 2019 Jan;71[1]:5-32).
This schism between EULAR and the ACR could be seen as predictable, given the different constraints the two societies have set for themselves.
“EULAR recommendations take into account drug costs; the ACR guideline is supposed to be agnostic to costs,” explained Philip J. Mease, MD, a rheumatologist at Swedish Medical Center in Seattle and a member of the ACR panel that wrote the 2018 PsA guideline.
In fact it was a study Dr. Mease recently led and reported results from that provided the most recent and perhaps best assessment of a TNF inhibitor, compared with methotrexate, as initial treatment for PsA, with findings that suggest that, although the advice from the two societies may sharply differ, the viewpoints of both groups are evidence based.
The SEAM-PsA (Study of Etanercept and Methotrexate in Subjects with Psoriatic Arthritis) trial randomized 851 PsA patients receiving their first treatment to methotrexate only, the TNF inhibitor etanercept (Enbrel) only, or both drugs. The study’s two coprimary outcomes, the ACR 20 and minimal disease activity responses after 24 weeks, showed that etanercept monotherapy produced these responses in 61% and 36% of patients, respectively, while methotrexate monotherapy produced response rates of 51% and 23%, respectively. Both these differences between etanercept monotherapy and methotrexate monotherapy were statistically significant. Combining methotrexate with etanercept did not produce a significant improvement over etanercept alone.
Interpreting the meaning of this finding for clinical practice “depends on the lens you look through,” Dr. Mease said in an interview. “A lot of patients respond to methotrexate, which is good when treatment resources are challenged. But when there is no resource challenge, the data support going straight to a TNF inhibitor.”
Dr. Gossec confirmed the importance of the SEAM-PsA findings in the writing panel’s decision during discussion of the draft, replying to a question about consideration of the study’s findings. “We carefully looked at the SEAM-PsA trial results, which provide some of the only data we have on methotrexate” for PsA. “We felt that the results were in favor of methotrexate’s efficacy, and therefore did not go against our proposal to keep a graduated approach starting with a csDMARD.”
Patients who fail to receive adequate relief from a csDMARD could then try a biologic DMARD – a TNF inhibitor, IL-17 inhibitor, or IL-12/23 inhibitor, Dr. Gossec said. When skin involvement is minimal, any of these options are possible, she said. If skin involvement is significant, the panel recommended preferentially using an IL-17 or IL-12/23 inhibitor based on head-to-head trials in patients with psoriasis, she said.
When a biologic DMARD is not appropriate or fails, another option is to then try a targeted synthetic DMARD, such as a Janus kinase inhibitor. When none of these options are appropriate, or they all fail, another option for patients with mild oligo- or monoarthritis or in patients with limited skin involvement is apremilast (Otezla), a phosphodiesterase-4 inhibitor. The draft recommendations also advise clinicians to be sure to distinguish fibromyalgia pain from enthesitis involvement, and they introduce the possibility of, with “great caution,” tapering down DMARD treatment in PsA patients who show sustained remission.
Dr. Gossec and Dr. Mease have both been consultants to and received honoraria from several companies. SEAM-PsA was sponsored by Amgen, the company that markets Enbrel.
REPORTING FROM EULAR 2019 CONGRESS
Could some elderly skip statins based on negative risk markers?
Older individuals with certain negative risk markers have a very low risk of atherosclerotic cardiovascular disease, raising the possibility that some could forgo preventive treatment even if it’s indicated by current standards of risk assessment.
Low levels of coronary artery calcification (CAC), low galectin-3 levels, and absence of carotid plaque were all linked to a lower likelihood of disease than might be expected based on traditional risk assessment, according to the authors of analysis of a large, contemporary cohort of elderly individuals published in the Journal of the American College of Cardiology.
“Our results hold the potential to markedly improve statin allocation in elderly individuals by de-escalating or even withholding preventive therapy in elderly individuals at truly low atherosclerotic cardiovascular disease risk despite advancing age,“ wrote Martin Bødtker Mortensen, MD, PhD, of Aarhus (Denmark) University Hospital.
Most elderly individuals now qualify for lifelong preventive statin treatment, based on the broader indication for treatment in recent guidelines, and the substantial impact that age has when risk is being calculated, Dr. Mortensen and coauthors said in their report.
“Because frailty, comorbidity, and polypharmacy are increasing concerns in elderly individuals and have been proposed to increase the risk for adverse effects, the appropriateness of treating almost all elderly individuals is questionable,” they said in the report.
In their study, Dr. Mortensen and colleagues evaluated a set of 13 biomarkers or imaging tests that they though had potential to “downgrade” risk of coronary heart disease (CHD) and cardiovascular disease (CVD). They based their analysis on 5,805 patients in the BioImage Study, a prospective cohort study of elderly men and women with no atherosclerotic cardiovascular disease at the time of enrollment in 2008 and 2009. The mean age at the time of enrollment was 69 years, and the mean follow-up in this analysis was 2.7 years.
The overall rate of CHD was 6.1 per 1,000 person-years, though looking at negative risk markers, the event rate was just 0.9 for individuals with CAC of 0 and also 0.9 for those with a CAC of 10 or less, followed by 1.7 for absence of carotid plaque, and 2.6 for galectin-3 in the bottom 25th percentile, according to Dr. Mortensen and coinvestigators. Similarly, the rate of CVD was 9.2 per 1,000 person-years overall, and just 3.2 for a CAC of 0, 2.8 for a CAC of 10 or lower, 4.4 for no carotid plaque, and 4.0 for low galectin-3.
Results were less impressive for other negative risk markers, including normal ankle-brachial index (ABI) test, lack of family history, and low levels of circulating biomarkers such as high-sensitivity C-reactive protein and lipoprotein (a).
Investigators also calculated diagnostic likelihood ratios (DLR), a measure they said assesses the value of performing a diagnostic test, with values lower than 1 indicating a specific marker has value for downgrading risk.
Zero or low CAC exerted the greatest downward change in pre- to post-test risk, according to the investigators, with a multivariable-adjusted DLR of 0.20 for CHD, translating into an 80% relative risk reduction. Similarly, the adjusted DLRs for zero or low CAC for CVD were 0.48 and 0.41, respectively, translating into a 59% risk reduction.
Low galectin-3 also resulted in significant downward change in that pre- to post-test risk, investigators added.
The BioImage Study was funded by Abbott, AstraZeneca, Merck, Philips, and Takeda. Dr. Mortensen had no disclosures related to the present analysis. Coauthors provided disclosures related to G3 Pharmaceuticals, Abbott Laboratories, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Eli Lilly/DSI, Medtronic, Novartis Pharmaceuticals, OrbusNeich, and PLC/Renal Guard, among others.
SOURCE: Mortensen MB et al. J Am Coll Cardiol. 2019 Jul 1. doi: 10.1016/j.jacc.2019.04.049.
This study suggests that atherosclerosis imaging tests are the strongest negative risk factors identified to date in cardiovascular medicine. The results are clinically actionable and should shape our approach to these tests in clinical practice.
Atherosclerosis imaging tests could be important in preventing overuse of pharmacotherapy for primary prevention in older adults, nearly all of whom would be considered at elevated risk by current standards.
Results for other candidate markers in the study were generally unimpressive, though the finding that low galectin-3 levels predicted low cardiovascular risk is novel, highly interesting, and deserving of further study.
Meanwhile, guidelines are already taking notice of an emerging consensus on the value of imaging studies as a negative risk factor.
The 2018 prevention guidelines from the American College of Cardiology/American Heart Association recommend coronary artery calcium (CAC) testing to guide individualized patient decision-making in certain adults between the ages of 40 and 75 years who are at borderline to intermediate risk, and that using CAC results to reclassify risk is reasonable in those aged 76-80 years, they said.
For the first time, the ACC/AHA guidelines devoted a section to negative risk factors, specifically highlighting the value of CAC = 0 and stating that intensive statin therapy is of less value in such patients and can potentially be avoided.
Likewise, 2017 guidelines from the Society of Cardiovascular Computed Tomography say that aspirin for primary prevention can almost always be forgone in patients with a CAC of 0.
“The bar for preventive therapy has justifiably fallen, reaching a point where many patients will qualify based on their age alone,” the authors said.
In light of that development, negative risk factors might meaningfully downgrade risk and help identify individuals who can safely focus on lifestyle therapies and defer preventive medication.
Michael J. Blaha, MD, MPH, and Khurram Nasir, MD, MPH are with the Johns Hopkins Ciccarone Center for the Prevention of Cardiovascular Disease in Baltimore; Dr. Nasir also is affiliated with the Center for Outcomes Research and Evaluation, Yale University, New Haven, Conn. Ron Blankstein, MD, is with the cardiovascular division, department of medicine, Brigham and Women’s Hospital, Boston. These comments are adapted from their editorial (J Am Coll Cardiol. 2019 Jul 1. doi: 10.1016/j.jacc.2019.05.032 ). Dr. Blankstein reported research funding/grant support from Amgen and Astellas; Dr. Blaha and Dr. Nasir said they had no disclosures to report.
This study suggests that atherosclerosis imaging tests are the strongest negative risk factors identified to date in cardiovascular medicine. The results are clinically actionable and should shape our approach to these tests in clinical practice.
Atherosclerosis imaging tests could be important in preventing overuse of pharmacotherapy for primary prevention in older adults, nearly all of whom would be considered at elevated risk by current standards.
Results for other candidate markers in the study were generally unimpressive, though the finding that low galectin-3 levels predicted low cardiovascular risk is novel, highly interesting, and deserving of further study.
Meanwhile, guidelines are already taking notice of an emerging consensus on the value of imaging studies as a negative risk factor.
The 2018 prevention guidelines from the American College of Cardiology/American Heart Association recommend coronary artery calcium (CAC) testing to guide individualized patient decision-making in certain adults between the ages of 40 and 75 years who are at borderline to intermediate risk, and that using CAC results to reclassify risk is reasonable in those aged 76-80 years, they said.
For the first time, the ACC/AHA guidelines devoted a section to negative risk factors, specifically highlighting the value of CAC = 0 and stating that intensive statin therapy is of less value in such patients and can potentially be avoided.
Likewise, 2017 guidelines from the Society of Cardiovascular Computed Tomography say that aspirin for primary prevention can almost always be forgone in patients with a CAC of 0.
“The bar for preventive therapy has justifiably fallen, reaching a point where many patients will qualify based on their age alone,” the authors said.
In light of that development, negative risk factors might meaningfully downgrade risk and help identify individuals who can safely focus on lifestyle therapies and defer preventive medication.
Michael J. Blaha, MD, MPH, and Khurram Nasir, MD, MPH are with the Johns Hopkins Ciccarone Center for the Prevention of Cardiovascular Disease in Baltimore; Dr. Nasir also is affiliated with the Center for Outcomes Research and Evaluation, Yale University, New Haven, Conn. Ron Blankstein, MD, is with the cardiovascular division, department of medicine, Brigham and Women’s Hospital, Boston. These comments are adapted from their editorial (J Am Coll Cardiol. 2019 Jul 1. doi: 10.1016/j.jacc.2019.05.032 ). Dr. Blankstein reported research funding/grant support from Amgen and Astellas; Dr. Blaha and Dr. Nasir said they had no disclosures to report.
This study suggests that atherosclerosis imaging tests are the strongest negative risk factors identified to date in cardiovascular medicine. The results are clinically actionable and should shape our approach to these tests in clinical practice.
Atherosclerosis imaging tests could be important in preventing overuse of pharmacotherapy for primary prevention in older adults, nearly all of whom would be considered at elevated risk by current standards.
Results for other candidate markers in the study were generally unimpressive, though the finding that low galectin-3 levels predicted low cardiovascular risk is novel, highly interesting, and deserving of further study.
Meanwhile, guidelines are already taking notice of an emerging consensus on the value of imaging studies as a negative risk factor.
The 2018 prevention guidelines from the American College of Cardiology/American Heart Association recommend coronary artery calcium (CAC) testing to guide individualized patient decision-making in certain adults between the ages of 40 and 75 years who are at borderline to intermediate risk, and that using CAC results to reclassify risk is reasonable in those aged 76-80 years, they said.
For the first time, the ACC/AHA guidelines devoted a section to negative risk factors, specifically highlighting the value of CAC = 0 and stating that intensive statin therapy is of less value in such patients and can potentially be avoided.
Likewise, 2017 guidelines from the Society of Cardiovascular Computed Tomography say that aspirin for primary prevention can almost always be forgone in patients with a CAC of 0.
“The bar for preventive therapy has justifiably fallen, reaching a point where many patients will qualify based on their age alone,” the authors said.
In light of that development, negative risk factors might meaningfully downgrade risk and help identify individuals who can safely focus on lifestyle therapies and defer preventive medication.
Michael J. Blaha, MD, MPH, and Khurram Nasir, MD, MPH are with the Johns Hopkins Ciccarone Center for the Prevention of Cardiovascular Disease in Baltimore; Dr. Nasir also is affiliated with the Center for Outcomes Research and Evaluation, Yale University, New Haven, Conn. Ron Blankstein, MD, is with the cardiovascular division, department of medicine, Brigham and Women’s Hospital, Boston. These comments are adapted from their editorial (J Am Coll Cardiol. 2019 Jul 1. doi: 10.1016/j.jacc.2019.05.032 ). Dr. Blankstein reported research funding/grant support from Amgen and Astellas; Dr. Blaha and Dr. Nasir said they had no disclosures to report.
Older individuals with certain negative risk markers have a very low risk of atherosclerotic cardiovascular disease, raising the possibility that some could forgo preventive treatment even if it’s indicated by current standards of risk assessment.
Low levels of coronary artery calcification (CAC), low galectin-3 levels, and absence of carotid plaque were all linked to a lower likelihood of disease than might be expected based on traditional risk assessment, according to the authors of analysis of a large, contemporary cohort of elderly individuals published in the Journal of the American College of Cardiology.
“Our results hold the potential to markedly improve statin allocation in elderly individuals by de-escalating or even withholding preventive therapy in elderly individuals at truly low atherosclerotic cardiovascular disease risk despite advancing age,“ wrote Martin Bødtker Mortensen, MD, PhD, of Aarhus (Denmark) University Hospital.
Most elderly individuals now qualify for lifelong preventive statin treatment, based on the broader indication for treatment in recent guidelines, and the substantial impact that age has when risk is being calculated, Dr. Mortensen and coauthors said in their report.
“Because frailty, comorbidity, and polypharmacy are increasing concerns in elderly individuals and have been proposed to increase the risk for adverse effects, the appropriateness of treating almost all elderly individuals is questionable,” they said in the report.
In their study, Dr. Mortensen and colleagues evaluated a set of 13 biomarkers or imaging tests that they though had potential to “downgrade” risk of coronary heart disease (CHD) and cardiovascular disease (CVD). They based their analysis on 5,805 patients in the BioImage Study, a prospective cohort study of elderly men and women with no atherosclerotic cardiovascular disease at the time of enrollment in 2008 and 2009. The mean age at the time of enrollment was 69 years, and the mean follow-up in this analysis was 2.7 years.
The overall rate of CHD was 6.1 per 1,000 person-years, though looking at negative risk markers, the event rate was just 0.9 for individuals with CAC of 0 and also 0.9 for those with a CAC of 10 or less, followed by 1.7 for absence of carotid plaque, and 2.6 for galectin-3 in the bottom 25th percentile, according to Dr. Mortensen and coinvestigators. Similarly, the rate of CVD was 9.2 per 1,000 person-years overall, and just 3.2 for a CAC of 0, 2.8 for a CAC of 10 or lower, 4.4 for no carotid plaque, and 4.0 for low galectin-3.
Results were less impressive for other negative risk markers, including normal ankle-brachial index (ABI) test, lack of family history, and low levels of circulating biomarkers such as high-sensitivity C-reactive protein and lipoprotein (a).
Investigators also calculated diagnostic likelihood ratios (DLR), a measure they said assesses the value of performing a diagnostic test, with values lower than 1 indicating a specific marker has value for downgrading risk.
Zero or low CAC exerted the greatest downward change in pre- to post-test risk, according to the investigators, with a multivariable-adjusted DLR of 0.20 for CHD, translating into an 80% relative risk reduction. Similarly, the adjusted DLRs for zero or low CAC for CVD were 0.48 and 0.41, respectively, translating into a 59% risk reduction.
Low galectin-3 also resulted in significant downward change in that pre- to post-test risk, investigators added.
The BioImage Study was funded by Abbott, AstraZeneca, Merck, Philips, and Takeda. Dr. Mortensen had no disclosures related to the present analysis. Coauthors provided disclosures related to G3 Pharmaceuticals, Abbott Laboratories, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Eli Lilly/DSI, Medtronic, Novartis Pharmaceuticals, OrbusNeich, and PLC/Renal Guard, among others.
SOURCE: Mortensen MB et al. J Am Coll Cardiol. 2019 Jul 1. doi: 10.1016/j.jacc.2019.04.049.
Older individuals with certain negative risk markers have a very low risk of atherosclerotic cardiovascular disease, raising the possibility that some could forgo preventive treatment even if it’s indicated by current standards of risk assessment.
Low levels of coronary artery calcification (CAC), low galectin-3 levels, and absence of carotid plaque were all linked to a lower likelihood of disease than might be expected based on traditional risk assessment, according to the authors of analysis of a large, contemporary cohort of elderly individuals published in the Journal of the American College of Cardiology.
“Our results hold the potential to markedly improve statin allocation in elderly individuals by de-escalating or even withholding preventive therapy in elderly individuals at truly low atherosclerotic cardiovascular disease risk despite advancing age,“ wrote Martin Bødtker Mortensen, MD, PhD, of Aarhus (Denmark) University Hospital.
Most elderly individuals now qualify for lifelong preventive statin treatment, based on the broader indication for treatment in recent guidelines, and the substantial impact that age has when risk is being calculated, Dr. Mortensen and coauthors said in their report.
“Because frailty, comorbidity, and polypharmacy are increasing concerns in elderly individuals and have been proposed to increase the risk for adverse effects, the appropriateness of treating almost all elderly individuals is questionable,” they said in the report.
In their study, Dr. Mortensen and colleagues evaluated a set of 13 biomarkers or imaging tests that they though had potential to “downgrade” risk of coronary heart disease (CHD) and cardiovascular disease (CVD). They based their analysis on 5,805 patients in the BioImage Study, a prospective cohort study of elderly men and women with no atherosclerotic cardiovascular disease at the time of enrollment in 2008 and 2009. The mean age at the time of enrollment was 69 years, and the mean follow-up in this analysis was 2.7 years.
The overall rate of CHD was 6.1 per 1,000 person-years, though looking at negative risk markers, the event rate was just 0.9 for individuals with CAC of 0 and also 0.9 for those with a CAC of 10 or less, followed by 1.7 for absence of carotid plaque, and 2.6 for galectin-3 in the bottom 25th percentile, according to Dr. Mortensen and coinvestigators. Similarly, the rate of CVD was 9.2 per 1,000 person-years overall, and just 3.2 for a CAC of 0, 2.8 for a CAC of 10 or lower, 4.4 for no carotid plaque, and 4.0 for low galectin-3.
Results were less impressive for other negative risk markers, including normal ankle-brachial index (ABI) test, lack of family history, and low levels of circulating biomarkers such as high-sensitivity C-reactive protein and lipoprotein (a).
Investigators also calculated diagnostic likelihood ratios (DLR), a measure they said assesses the value of performing a diagnostic test, with values lower than 1 indicating a specific marker has value for downgrading risk.
Zero or low CAC exerted the greatest downward change in pre- to post-test risk, according to the investigators, with a multivariable-adjusted DLR of 0.20 for CHD, translating into an 80% relative risk reduction. Similarly, the adjusted DLRs for zero or low CAC for CVD were 0.48 and 0.41, respectively, translating into a 59% risk reduction.
Low galectin-3 also resulted in significant downward change in that pre- to post-test risk, investigators added.
The BioImage Study was funded by Abbott, AstraZeneca, Merck, Philips, and Takeda. Dr. Mortensen had no disclosures related to the present analysis. Coauthors provided disclosures related to G3 Pharmaceuticals, Abbott Laboratories, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Eli Lilly/DSI, Medtronic, Novartis Pharmaceuticals, OrbusNeich, and PLC/Renal Guard, among others.
SOURCE: Mortensen MB et al. J Am Coll Cardiol. 2019 Jul 1. doi: 10.1016/j.jacc.2019.04.049.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point: Low or no coronary artery calcification (CAC), low galectin-3 levels, and absence of carotid plaque were all linked to a lower likelihood of atherosclerotic cardiovascular disease in elderly patients, suggesting pharmacotherapy might be avoidable in a certain proportion of them.
Major finding: The diagnostic likelihood ratios (DLR) were low for those negative risk markers, e.g., 0.20 for no or low CAC, indicating a relative risk reduction of about 80%.
Study details: Analysis of data from the BioImage Study, a prospective cohort study of 5,805 older men and women who had no atherosclerotic cardiovascular disease at the time of enrollment.
Disclosures: The BioImage Study was funded by Abbott, AstraZeneca, Merck, Philips, and Takeda. Authors of the present analysis provided disclosures related to G3 Pharmaceuticals, Abbott Laboratories, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Eli Lilly/DSI, Medtronic, Novartis Pharmaceuticals, OrbusNeich, and PLC/Renal Guard, among others.
Source: Mortensen MB et al. J Am Coll Cardiol. 2019 Jul 1. doi: j.jacc.2019.04.049.
Lipoprotein(a) levels tied to higher ischemic stroke risk
High levels of lipoprotein(a) [Lp(a)] and LPA genotypes were linked to increased ischemic stroke risk in a recent large, contemporary general population study, investigators are reporting in the Journal of the American College of Cardiology.
Anne Langsted, MD, with Copenhagen University Hospital and the University of Copenhagen in Denmark, and her co-researchers evaluated the impact of high Lp(a) levels in a large contemporary cohort of 49,699 individuals in the Copenhagen General Population Study, and another 10,813 individuals in the Copenhagen City Heart Study.
Measurements assessed included plasma lipoprotein(a) levels and carrier or noncarrier status for LPA rs10455872. The endpoint of ischemic stroke was ascertained from Danish national health registries and confirmed by physicians.
Although risk estimates were less pronounced than what was reported before regarding the link between Lp(a) for ischemic heart disease and aortic valve stenosis, the risk of stroke was increased by a factor of 1.6 among individuals with high Lp(a) levels as compared to those with lower levels, the investigators said.
Compared with noncarriers of LPA rs1045572, the hazard ratio for ischemic stroke was 1.23 for carriers of LPA rs1045572, which was associated with high levels plasma lipoprotein(a) levels, according to the researchers.
“Our results indicate a causal association of Lp(a) with risk of ischemic stroke, and emphasize the need for randomized, controlled clinical trials on the effect of Lp(a)-lowering to prevent cardiovascular disease including ischemic stroke,” About 20% of the general population have high Lp(a) levels, and some individuals have extremely high levels, Dr. Langsted and co-authors said in their report.
Interest in Lp(a) as a risk factor for cardiovascular disease has been reignited following large studies showing that high Lp(a) levels were linked to increased risk of myocardial infarction and aortic valve stenosis, according to the investigators.
However, results of various studies are conflicting as to whether high Lp(a) levels increase risk of hemorrhagic or ischemic stroke, they said.
Both cohort studies used in the analysis were supported by sources in Denmark including the Danish Medical Research Council and Copenhagen University Hospital. Dr. Langsted had no disclosures. One co-author reported disclosures related to Akcea, Amgen, Sanofi, Regeneron, and AstraZeneca.
SOURCE: Langsted A, et al. JACC 2019;74[1]: 54-66. doi: 10.1016/j.jacc.2019.03.524
This study linking high lipoprotein(a) [Lp(a)] levels to stroke risk, taken together with previous research, provide a sound basis to routinely perform one-time screening so that individuals with inherited high levels can try to avoid adverse cardiovascular outcomes, according to Christie M. Ballantyne, MD.
“As someone in the dual role of preventive cardiologist and patient with a strong family history of cardiovascular disease, I think that we have sufficient evidence that high Lp(a) is strongly associated with an increased risk of myocardial infarction, stroke, and aortic valve stenosis,” Dr. Ballantyne wrote in an editorial comment on the study.
Evidence is now “overwhelming” that high Lp(a) is linked to myocardial infarction and stroke, and it’s known that statins and aspirin reduce risk of these outcomes, he said in the commentary.
Despite that, scientific statements do not recommend routine Lp(a) testing due to a lack of clinical trials evidence; as a result, clinical trials are not including Lp(a) as a routine measurement: “We thus have a loop of futility—lack of routine measurement leads to lack of data,” he said.
This most recent study from Langsted and colleagues demonstrates that high Lp(a) levels, and genetic variants associated with Lp(a), are associated with increased ischemic stroke risk. “The genetics strongly supported that high Lp(a) levels were in the causal pathway for ischemic stroke and coronary heart disease,” Dr. Ballantyne said.
One major strength and weakness of the study is its large and relatively homogeneous European population—that bolstered the genetic analyses, but also means the data can’t be extrapolated to other populations, such as Africans and East Asians, who have higher stroke rates compared with Europeans, Dr. Ballantyne said.
Dr. Ballantyne is with the Department of Medicine and Center for Cardiometabolic Disease Prevention, Baylor College of Medicine, Houston, Tex. His editorial comment appears in the Journal of the American College of Cardiology (2019;74[1]:67-9. doi:10.1016/j.jacc.2019.05.029 . Dr. Ballantyne reported disclosures related to Akcea, Amgen, and Novartis.
This study linking high lipoprotein(a) [Lp(a)] levels to stroke risk, taken together with previous research, provide a sound basis to routinely perform one-time screening so that individuals with inherited high levels can try to avoid adverse cardiovascular outcomes, according to Christie M. Ballantyne, MD.
“As someone in the dual role of preventive cardiologist and patient with a strong family history of cardiovascular disease, I think that we have sufficient evidence that high Lp(a) is strongly associated with an increased risk of myocardial infarction, stroke, and aortic valve stenosis,” Dr. Ballantyne wrote in an editorial comment on the study.
Evidence is now “overwhelming” that high Lp(a) is linked to myocardial infarction and stroke, and it’s known that statins and aspirin reduce risk of these outcomes, he said in the commentary.
Despite that, scientific statements do not recommend routine Lp(a) testing due to a lack of clinical trials evidence; as a result, clinical trials are not including Lp(a) as a routine measurement: “We thus have a loop of futility—lack of routine measurement leads to lack of data,” he said.
This most recent study from Langsted and colleagues demonstrates that high Lp(a) levels, and genetic variants associated with Lp(a), are associated with increased ischemic stroke risk. “The genetics strongly supported that high Lp(a) levels were in the causal pathway for ischemic stroke and coronary heart disease,” Dr. Ballantyne said.
One major strength and weakness of the study is its large and relatively homogeneous European population—that bolstered the genetic analyses, but also means the data can’t be extrapolated to other populations, such as Africans and East Asians, who have higher stroke rates compared with Europeans, Dr. Ballantyne said.
Dr. Ballantyne is with the Department of Medicine and Center for Cardiometabolic Disease Prevention, Baylor College of Medicine, Houston, Tex. His editorial comment appears in the Journal of the American College of Cardiology (2019;74[1]:67-9. doi:10.1016/j.jacc.2019.05.029 . Dr. Ballantyne reported disclosures related to Akcea, Amgen, and Novartis.
This study linking high lipoprotein(a) [Lp(a)] levels to stroke risk, taken together with previous research, provide a sound basis to routinely perform one-time screening so that individuals with inherited high levels can try to avoid adverse cardiovascular outcomes, according to Christie M. Ballantyne, MD.
“As someone in the dual role of preventive cardiologist and patient with a strong family history of cardiovascular disease, I think that we have sufficient evidence that high Lp(a) is strongly associated with an increased risk of myocardial infarction, stroke, and aortic valve stenosis,” Dr. Ballantyne wrote in an editorial comment on the study.
Evidence is now “overwhelming” that high Lp(a) is linked to myocardial infarction and stroke, and it’s known that statins and aspirin reduce risk of these outcomes, he said in the commentary.
Despite that, scientific statements do not recommend routine Lp(a) testing due to a lack of clinical trials evidence; as a result, clinical trials are not including Lp(a) as a routine measurement: “We thus have a loop of futility—lack of routine measurement leads to lack of data,” he said.
This most recent study from Langsted and colleagues demonstrates that high Lp(a) levels, and genetic variants associated with Lp(a), are associated with increased ischemic stroke risk. “The genetics strongly supported that high Lp(a) levels were in the causal pathway for ischemic stroke and coronary heart disease,” Dr. Ballantyne said.
One major strength and weakness of the study is its large and relatively homogeneous European population—that bolstered the genetic analyses, but also means the data can’t be extrapolated to other populations, such as Africans and East Asians, who have higher stroke rates compared with Europeans, Dr. Ballantyne said.
Dr. Ballantyne is with the Department of Medicine and Center for Cardiometabolic Disease Prevention, Baylor College of Medicine, Houston, Tex. His editorial comment appears in the Journal of the American College of Cardiology (2019;74[1]:67-9. doi:10.1016/j.jacc.2019.05.029 . Dr. Ballantyne reported disclosures related to Akcea, Amgen, and Novartis.
High levels of lipoprotein(a) [Lp(a)] and LPA genotypes were linked to increased ischemic stroke risk in a recent large, contemporary general population study, investigators are reporting in the Journal of the American College of Cardiology.
Anne Langsted, MD, with Copenhagen University Hospital and the University of Copenhagen in Denmark, and her co-researchers evaluated the impact of high Lp(a) levels in a large contemporary cohort of 49,699 individuals in the Copenhagen General Population Study, and another 10,813 individuals in the Copenhagen City Heart Study.
Measurements assessed included plasma lipoprotein(a) levels and carrier or noncarrier status for LPA rs10455872. The endpoint of ischemic stroke was ascertained from Danish national health registries and confirmed by physicians.
Although risk estimates were less pronounced than what was reported before regarding the link between Lp(a) for ischemic heart disease and aortic valve stenosis, the risk of stroke was increased by a factor of 1.6 among individuals with high Lp(a) levels as compared to those with lower levels, the investigators said.
Compared with noncarriers of LPA rs1045572, the hazard ratio for ischemic stroke was 1.23 for carriers of LPA rs1045572, which was associated with high levels plasma lipoprotein(a) levels, according to the researchers.
“Our results indicate a causal association of Lp(a) with risk of ischemic stroke, and emphasize the need for randomized, controlled clinical trials on the effect of Lp(a)-lowering to prevent cardiovascular disease including ischemic stroke,” About 20% of the general population have high Lp(a) levels, and some individuals have extremely high levels, Dr. Langsted and co-authors said in their report.
Interest in Lp(a) as a risk factor for cardiovascular disease has been reignited following large studies showing that high Lp(a) levels were linked to increased risk of myocardial infarction and aortic valve stenosis, according to the investigators.
However, results of various studies are conflicting as to whether high Lp(a) levels increase risk of hemorrhagic or ischemic stroke, they said.
Both cohort studies used in the analysis were supported by sources in Denmark including the Danish Medical Research Council and Copenhagen University Hospital. Dr. Langsted had no disclosures. One co-author reported disclosures related to Akcea, Amgen, Sanofi, Regeneron, and AstraZeneca.
SOURCE: Langsted A, et al. JACC 2019;74[1]: 54-66. doi: 10.1016/j.jacc.2019.03.524
High levels of lipoprotein(a) [Lp(a)] and LPA genotypes were linked to increased ischemic stroke risk in a recent large, contemporary general population study, investigators are reporting in the Journal of the American College of Cardiology.
Anne Langsted, MD, with Copenhagen University Hospital and the University of Copenhagen in Denmark, and her co-researchers evaluated the impact of high Lp(a) levels in a large contemporary cohort of 49,699 individuals in the Copenhagen General Population Study, and another 10,813 individuals in the Copenhagen City Heart Study.
Measurements assessed included plasma lipoprotein(a) levels and carrier or noncarrier status for LPA rs10455872. The endpoint of ischemic stroke was ascertained from Danish national health registries and confirmed by physicians.
Although risk estimates were less pronounced than what was reported before regarding the link between Lp(a) for ischemic heart disease and aortic valve stenosis, the risk of stroke was increased by a factor of 1.6 among individuals with high Lp(a) levels as compared to those with lower levels, the investigators said.
Compared with noncarriers of LPA rs1045572, the hazard ratio for ischemic stroke was 1.23 for carriers of LPA rs1045572, which was associated with high levels plasma lipoprotein(a) levels, according to the researchers.
“Our results indicate a causal association of Lp(a) with risk of ischemic stroke, and emphasize the need for randomized, controlled clinical trials on the effect of Lp(a)-lowering to prevent cardiovascular disease including ischemic stroke,” About 20% of the general population have high Lp(a) levels, and some individuals have extremely high levels, Dr. Langsted and co-authors said in their report.
Interest in Lp(a) as a risk factor for cardiovascular disease has been reignited following large studies showing that high Lp(a) levels were linked to increased risk of myocardial infarction and aortic valve stenosis, according to the investigators.
However, results of various studies are conflicting as to whether high Lp(a) levels increase risk of hemorrhagic or ischemic stroke, they said.
Both cohort studies used in the analysis were supported by sources in Denmark including the Danish Medical Research Council and Copenhagen University Hospital. Dr. Langsted had no disclosures. One co-author reported disclosures related to Akcea, Amgen, Sanofi, Regeneron, and AstraZeneca.
SOURCE: Langsted A, et al. JACC 2019;74[1]: 54-66. doi: 10.1016/j.jacc.2019.03.524
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point:
Major finding: Stroke risk was 1.6X higher with high Lp(a) levels.
Study details: Analysis of 49,699 individuals in the Copenhagen General Population Study, and 10,813 individuals in the Copenhagen City Heart Study.
Disclosures: Both studies were supported by the sources in Denmark including the Danish Medical Research Council and Copenhagen University Hospital. Dr. Langsted had no disclosures.
Source: Langsted A, et al. JACC 2019;74[1]: 54-66. doi: 10.1016/j.jacc.2019.03.524