User login
Concizumab looks feasible in hemophilia A and B treatment
MELBOURNE – A once-daily subcutaneous treatment that inhibits the tissue factor 4 pathway inhibitor has shown significant reductions in bleeding rates in patients with hemophilia A and B, according to findings presented at the International Society on Thrombosis and Haemostasis congress.

Jan Astermark, MD, PhD, of the Centre for Thrombosis and Haemostasis at Lund University in Sweden, presented data from two phase 2, dose-escalation trials of the monoclonal antibody concizumab.
The explorer 5 trial involved 36 adults with severe hemophilia A without inhibitors who were started on 0.15 mg/kg of concizumab for 24 weeks. If they experienced three or more bleeds during that time, they were escalated to 0.20 mg/kg, and then to 0.25mg/kg if they experienced an additional three bleeds. The initial 24-week treatment period was then extended by more than 52 weeks.
In the explorer 4 trial, 16 adults with hemophilia A and 10 adults with hemophilia B – all with inhibitors – were initially randomized 2:1 to either 24 weeks of 0.15mg/kg of concizumab, including a loading dose, or placebo, with similar dose escalation in response to breakthrough bleeds. After 24 weeks, the study continued with a 52-week extension, during which all patients were treated with concizumab.
Both studies saw reductions in bleeding rates associated with concizumab treatment.
In patients with hemophilia A and B with inhibitors, the mean annualized bleeding rate for all bleeds declined from 20.4 to 4.5 bleeds, spontaneous bleeds declined from 18.5 to 2.5, and joint bleeds declined from 15 to 3.2. All three of the reductions were statistically significant.
Almost all patients achieved a concizumab concentration of 100 ng/mL, which was the expected level based on data from the phase 1 trial. Some patients showed anticoncizumab antibodies, but these were transient and did not appear to have any effect on clinical outcomes, according to Dr. Astermark.
Most patients also reached a normal level of thrombin generation, although Dr. Astermark noted that there were some patients with hemophilia B with inhibitors who produced a lower amount of thrombin than normal.
Despite the increase in thrombin generation, there were no thromboembolic events, and no significant safety concerns emerged during the study, he reported.
“Importantly and interestingly, all patients completing the main phase went into the extension phase of this trial, indicating that it was something they think was a contribution to their treatment,” Dr. Astermark said.
Earlier in 2019, concizumab was granted breakthrough designation by the Food and Drug Administration for the treatment of patients with hemophilia B and inhibitors, allowing it to receive an accelerated review by the agency.
“What the FDA based their decision on was the B patients with inhibitors, because this is truly a group where we do not have so many options,” Dr. Astermark said in an interview. He also noted that the subcutaneous treatment, delivered via a pen-like device, was much more convenient for patients who, until now, had required repeated intravenous infusions.
Two phase 3 trials are now scheduled, in which patients will receive a higher loading dose than what was used in the phase 2 trials.
Novo Nordisk sponsored both studies. Dr. Astermark reported consultancies and research funding unrelated to the study.
SOURCE: Astermark J et al. 2019 ISTH Congress, Abstract LB 01.1.
MELBOURNE – A once-daily subcutaneous treatment that inhibits the tissue factor 4 pathway inhibitor has shown significant reductions in bleeding rates in patients with hemophilia A and B, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Jan Astermark, MD, PhD, of the Centre for Thrombosis and Haemostasis at Lund University in Sweden, presented data from two phase 2, dose-escalation trials of the monoclonal antibody concizumab.
The explorer 5 trial involved 36 adults with severe hemophilia A without inhibitors who were started on 0.15 mg/kg of concizumab for 24 weeks. If they experienced three or more bleeds during that time, they were escalated to 0.20 mg/kg, and then to 0.25mg/kg if they experienced an additional three bleeds. The initial 24-week treatment period was then extended by more than 52 weeks.
In the explorer 4 trial, 16 adults with hemophilia A and 10 adults with hemophilia B – all with inhibitors – were initially randomized 2:1 to either 24 weeks of 0.15mg/kg of concizumab, including a loading dose, or placebo, with similar dose escalation in response to breakthrough bleeds. After 24 weeks, the study continued with a 52-week extension, during which all patients were treated with concizumab.
Both studies saw reductions in bleeding rates associated with concizumab treatment.
In patients with hemophilia A and B with inhibitors, the mean annualized bleeding rate for all bleeds declined from 20.4 to 4.5 bleeds, spontaneous bleeds declined from 18.5 to 2.5, and joint bleeds declined from 15 to 3.2. All three of the reductions were statistically significant.
Almost all patients achieved a concizumab concentration of 100 ng/mL, which was the expected level based on data from the phase 1 trial. Some patients showed anticoncizumab antibodies, but these were transient and did not appear to have any effect on clinical outcomes, according to Dr. Astermark.
Most patients also reached a normal level of thrombin generation, although Dr. Astermark noted that there were some patients with hemophilia B with inhibitors who produced a lower amount of thrombin than normal.
Despite the increase in thrombin generation, there were no thromboembolic events, and no significant safety concerns emerged during the study, he reported.
“Importantly and interestingly, all patients completing the main phase went into the extension phase of this trial, indicating that it was something they think was a contribution to their treatment,” Dr. Astermark said.
Earlier in 2019, concizumab was granted breakthrough designation by the Food and Drug Administration for the treatment of patients with hemophilia B and inhibitors, allowing it to receive an accelerated review by the agency.
“What the FDA based their decision on was the B patients with inhibitors, because this is truly a group where we do not have so many options,” Dr. Astermark said in an interview. He also noted that the subcutaneous treatment, delivered via a pen-like device, was much more convenient for patients who, until now, had required repeated intravenous infusions.
Two phase 3 trials are now scheduled, in which patients will receive a higher loading dose than what was used in the phase 2 trials.
Novo Nordisk sponsored both studies. Dr. Astermark reported consultancies and research funding unrelated to the study.
SOURCE: Astermark J et al. 2019 ISTH Congress, Abstract LB 01.1.
MELBOURNE – A once-daily subcutaneous treatment that inhibits the tissue factor 4 pathway inhibitor has shown significant reductions in bleeding rates in patients with hemophilia A and B, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Jan Astermark, MD, PhD, of the Centre for Thrombosis and Haemostasis at Lund University in Sweden, presented data from two phase 2, dose-escalation trials of the monoclonal antibody concizumab.
The explorer 5 trial involved 36 adults with severe hemophilia A without inhibitors who were started on 0.15 mg/kg of concizumab for 24 weeks. If they experienced three or more bleeds during that time, they were escalated to 0.20 mg/kg, and then to 0.25mg/kg if they experienced an additional three bleeds. The initial 24-week treatment period was then extended by more than 52 weeks.
In the explorer 4 trial, 16 adults with hemophilia A and 10 adults with hemophilia B – all with inhibitors – were initially randomized 2:1 to either 24 weeks of 0.15mg/kg of concizumab, including a loading dose, or placebo, with similar dose escalation in response to breakthrough bleeds. After 24 weeks, the study continued with a 52-week extension, during which all patients were treated with concizumab.
Both studies saw reductions in bleeding rates associated with concizumab treatment.
In patients with hemophilia A and B with inhibitors, the mean annualized bleeding rate for all bleeds declined from 20.4 to 4.5 bleeds, spontaneous bleeds declined from 18.5 to 2.5, and joint bleeds declined from 15 to 3.2. All three of the reductions were statistically significant.
Almost all patients achieved a concizumab concentration of 100 ng/mL, which was the expected level based on data from the phase 1 trial. Some patients showed anticoncizumab antibodies, but these were transient and did not appear to have any effect on clinical outcomes, according to Dr. Astermark.
Most patients also reached a normal level of thrombin generation, although Dr. Astermark noted that there were some patients with hemophilia B with inhibitors who produced a lower amount of thrombin than normal.
Despite the increase in thrombin generation, there were no thromboembolic events, and no significant safety concerns emerged during the study, he reported.
“Importantly and interestingly, all patients completing the main phase went into the extension phase of this trial, indicating that it was something they think was a contribution to their treatment,” Dr. Astermark said.
Earlier in 2019, concizumab was granted breakthrough designation by the Food and Drug Administration for the treatment of patients with hemophilia B and inhibitors, allowing it to receive an accelerated review by the agency.
“What the FDA based their decision on was the B patients with inhibitors, because this is truly a group where we do not have so many options,” Dr. Astermark said in an interview. He also noted that the subcutaneous treatment, delivered via a pen-like device, was much more convenient for patients who, until now, had required repeated intravenous infusions.
Two phase 3 trials are now scheduled, in which patients will receive a higher loading dose than what was used in the phase 2 trials.
Novo Nordisk sponsored both studies. Dr. Astermark reported consultancies and research funding unrelated to the study.
SOURCE: Astermark J et al. 2019 ISTH Congress, Abstract LB 01.1.
REPORTING FROM 2019 ISTH CONGRESS
CMS is proposing higher payments for E/M visits
Physicians could be getting more money for evaluation and management (E/M) visits under a new proposal from the Centers for Medicare & Medicaid Services.

The increased funding is part of a proposed rule that provides the annual update to the Medicare physician fee schedule for 2020, as well as updates for the Quality Payment Program. The proposed rule was posted online July 29 and is scheduled to be published in the Federal Register on Aug. 14. Comments are due to CMS on Sept. 27.
CMS officials are seeking to increase Medicare payments to physicians starting in 2021 for E/M visits, based on recommendations from the American Medical Association’s Relative Value Scale Update Committee (AMA-RUC).
With this update, the agency will be “rewarding the time that doctors spend with patients,” CMS Administrator Seema Verma said during a July 29 teleconference with reporters.
A fact sheet highlighting changes in the proposed physician fee schedule update for 2020 notes that the agency also is looking to add a new CPT code for prolonged services time, also to commence in 2021.
“The RUC recommendations reflect a robust survey approach by the AMA, including surveying over 50 specialty types [that] demonstrate that office/outpatient E/M visits are generally more complex and require additional resources for most clinicians,” the fact sheet states.
Physicians would also get paid for care management services related to patients with a single chronic condition, rather than only patients with multiple chronic conditions, as current regulations state. There’s also a proposal to increase payments for transitional care management services provided after a Medicare patient is discharged from an inpatient stay or certain outpatient stays.
The proposed update to the physician fee schedule also puts into regulation a new benefit for opioid use disorder treatment that was authorized under the SUPPORT (Substance Use-Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities) Act. To meet the requirements of the law, CMS has included in the proposal definitions for opioid treatment programs and opioid use disorder treatment services; enrollment policies for programs; bundled payment rates for treatment programs, with adjusters for geography and annual updates; flexibility for telehealth services; and zero beneficiary copays for a time-limited duration.
The American Medical Association praised proposed changes to documentation requirements that are included in the rule.
Patrice Harris, MD, the AMA president, said in a statement that the proposed rule “will streamline reporting requirements, reduce note bloat, improve workflow, and contribute to a better environment for health care professionals and their Medicare patients.”
The proposal also includes a modification to the physician supervision requirements for physician assistants that would give PAs “greater flexibility to practice more broadly in the current health system in accordance with state law and state scope of practice,” the fact sheet notes.
Overall, Dr. Harris appeared to have a good first impression of the proposed update, noting that the AMA is “pleased to see important policy revisions that will bring us closer to a more patient-centered health care system that promotes the key principles of affordability, accessibility, quality, and innovation.”
Physicians could be getting more money for evaluation and management (E/M) visits under a new proposal from the Centers for Medicare & Medicaid Services.
The increased funding is part of a proposed rule that provides the annual update to the Medicare physician fee schedule for 2020, as well as updates for the Quality Payment Program. The proposed rule was posted online July 29 and is scheduled to be published in the Federal Register on Aug. 14. Comments are due to CMS on Sept. 27.
CMS officials are seeking to increase Medicare payments to physicians starting in 2021 for E/M visits, based on recommendations from the American Medical Association’s Relative Value Scale Update Committee (AMA-RUC).
With this update, the agency will be “rewarding the time that doctors spend with patients,” CMS Administrator Seema Verma said during a July 29 teleconference with reporters.
A fact sheet highlighting changes in the proposed physician fee schedule update for 2020 notes that the agency also is looking to add a new CPT code for prolonged services time, also to commence in 2021.
“The RUC recommendations reflect a robust survey approach by the AMA, including surveying over 50 specialty types [that] demonstrate that office/outpatient E/M visits are generally more complex and require additional resources for most clinicians,” the fact sheet states.
Physicians would also get paid for care management services related to patients with a single chronic condition, rather than only patients with multiple chronic conditions, as current regulations state. There’s also a proposal to increase payments for transitional care management services provided after a Medicare patient is discharged from an inpatient stay or certain outpatient stays.
The proposed update to the physician fee schedule also puts into regulation a new benefit for opioid use disorder treatment that was authorized under the SUPPORT (Substance Use-Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities) Act. To meet the requirements of the law, CMS has included in the proposal definitions for opioid treatment programs and opioid use disorder treatment services; enrollment policies for programs; bundled payment rates for treatment programs, with adjusters for geography and annual updates; flexibility for telehealth services; and zero beneficiary copays for a time-limited duration.
The American Medical Association praised proposed changes to documentation requirements that are included in the rule.
Patrice Harris, MD, the AMA president, said in a statement that the proposed rule “will streamline reporting requirements, reduce note bloat, improve workflow, and contribute to a better environment for health care professionals and their Medicare patients.”
The proposal also includes a modification to the physician supervision requirements for physician assistants that would give PAs “greater flexibility to practice more broadly in the current health system in accordance with state law and state scope of practice,” the fact sheet notes.
Overall, Dr. Harris appeared to have a good first impression of the proposed update, noting that the AMA is “pleased to see important policy revisions that will bring us closer to a more patient-centered health care system that promotes the key principles of affordability, accessibility, quality, and innovation.”
Physicians could be getting more money for evaluation and management (E/M) visits under a new proposal from the Centers for Medicare & Medicaid Services.
The increased funding is part of a proposed rule that provides the annual update to the Medicare physician fee schedule for 2020, as well as updates for the Quality Payment Program. The proposed rule was posted online July 29 and is scheduled to be published in the Federal Register on Aug. 14. Comments are due to CMS on Sept. 27.
CMS officials are seeking to increase Medicare payments to physicians starting in 2021 for E/M visits, based on recommendations from the American Medical Association’s Relative Value Scale Update Committee (AMA-RUC).
With this update, the agency will be “rewarding the time that doctors spend with patients,” CMS Administrator Seema Verma said during a July 29 teleconference with reporters.
A fact sheet highlighting changes in the proposed physician fee schedule update for 2020 notes that the agency also is looking to add a new CPT code for prolonged services time, also to commence in 2021.
“The RUC recommendations reflect a robust survey approach by the AMA, including surveying over 50 specialty types [that] demonstrate that office/outpatient E/M visits are generally more complex and require additional resources for most clinicians,” the fact sheet states.
Physicians would also get paid for care management services related to patients with a single chronic condition, rather than only patients with multiple chronic conditions, as current regulations state. There’s also a proposal to increase payments for transitional care management services provided after a Medicare patient is discharged from an inpatient stay or certain outpatient stays.
The proposed update to the physician fee schedule also puts into regulation a new benefit for opioid use disorder treatment that was authorized under the SUPPORT (Substance Use-Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities) Act. To meet the requirements of the law, CMS has included in the proposal definitions for opioid treatment programs and opioid use disorder treatment services; enrollment policies for programs; bundled payment rates for treatment programs, with adjusters for geography and annual updates; flexibility for telehealth services; and zero beneficiary copays for a time-limited duration.
The American Medical Association praised proposed changes to documentation requirements that are included in the rule.
Patrice Harris, MD, the AMA president, said in a statement that the proposed rule “will streamline reporting requirements, reduce note bloat, improve workflow, and contribute to a better environment for health care professionals and their Medicare patients.”
The proposal also includes a modification to the physician supervision requirements for physician assistants that would give PAs “greater flexibility to practice more broadly in the current health system in accordance with state law and state scope of practice,” the fact sheet notes.
Overall, Dr. Harris appeared to have a good first impression of the proposed update, noting that the AMA is “pleased to see important policy revisions that will bring us closer to a more patient-centered health care system that promotes the key principles of affordability, accessibility, quality, and innovation.”
AAD, NPF update use of phototherapy for psoriasis
, according to updated guidelines issued jointly by the American Academy of Dermatology and the National Psoriasis Foundation.

“Phototherapy serves as a reasonable and effective treatment option for patients requiring more than topical medications and/or those wishing to avoid systemic medications or simply seeking an adjunct to a failing regimen,” wrote working group cochair Craig A. Elmets, MD, professor of dermatology at the University of Alabama at Birmingham, and coauthors.
The guidelines, which focus on phototherapy for adults with psoriasis, join a multipart series on psoriasis being published this year in the Journal of the American Academy of Dermatology.
The working group used an evidence-based model to examine efficacy, effectiveness, and adverse effects of the following modalities: narrow-band ultraviolet B (NB-UVB); broadband ultraviolet B (BB-UVB); targeted phototherapy using excimer laser and excimer lamp; psoralen plus ultraviolet A (PUVA) therapy, including topical, oral, and bath PUVA; photodynamic therapy (PDT), grenz ray therapy, climatotherapy; visible light therapy; Goeckerman therapy; and pulsed dye laser/intense pulsed light.
NB-UVB, which can be used to treat generalized plaque psoriasis, refers to wavelengths of 311-313 nm. The recommended treatment is two or three times a week, with a starting dose based on skin phenotype or minimal erythema dose. Although oral PUVA has shown higher clearance rates, compared with NB-UVB, NB-UVB has demonstrated fewer side effects. NB-UVB also has shown effectiveness for psoriasis in combination with medications including oral retinoids, “particularly useful in patients at increased risk for skin cancer,” the working group wrote. Genital shielding and eye protection are recommended during all phototherapy sessions to reduce the risk of cancer and cataracts, they emphasized.
BB-UVB, an older version of NB-UVB, is still effective for generalized plaque psoriasis as monotherapy, but evidence does not support additional benefit in combination with other treatments, and overall BB-UVB is less effective than either NB-UVB or oral PUVA, the working group said.
For treatment of localized psoriatic lesions, some evidence supports the ability of targeted UVB therapy to improve lesions in fewer treatments and at a lower cumulative dose, compared with nontargeted phototherapy, for palmoplantar plaque psoriasis and palmoplantar pustulosis. Excimer lasers also have shown effectiveness against scalp psoriasis, the working group noted. However, “there is insufficient evidence to recommend the excimer laser rather than topical PUVA for treatment of localized plaque psoriasis,” they said.
PUVA treatments are available as topical creams, or they can be taken orally, or mixed with bath water. All forms of PUVA include psoralens, photosensitizing agents that prepare target cells for the effects of UVA light. Topical PUVA has demonstrated particular effectiveness for palmoplantar psoriasis, the working group noted, but there is a risk of phototoxicity, so it has become less popular, they added. Similarly, evidence supports effectiveness of oral and bath PUVA, but all forms are used less frequently because of the increased availability of NB-UVB phototherapy, they said.
PDT is primarily used to destroy premalignant or malignant cells, and in theory “PDT-induced apoptosis of T lymphocytes could lead to reductions in inflammatory cytokines and, in turn, to improvement of psoriasis,” the working group noted. However, “clinical studies have failed to find significant benefit” of PDT using either 5-aminolevulinic acid (ALA) or methyl aminolevulinic acid (MAL) for psoriasis, or any significant benefits of MAL-PDT for nail psoriasis.
The grenz ray is an effective, but rarely used treatment in which 75% of long-wavelength ionizing radiation is absorbed by the first 1 mm of skin and 95% is absorbed within the first 3 mm of skin to protect the deeper tissues from radiation. Although more alternatives are available, grenz rays can be used for psoriasis patients unable to tolerate UV therapy, according to the working group.
Climatotherapy involves temporary or permanent relocation of the patient to a part of the world with a climate that could be favorable for psoriasis because of the unique effects of environmental factors in those areas. The evidence to support climatotherapy is both subjective and objective, but considered safe.
Visible light has been associated with improvement in erythema in psoriasis, with hyperpigmentation as the only notable side effect based on the evidence reviewed. However, the working group found the current evidence insufficient to recommend the use of intense pulsed light for treating psoriasis.

Goeckerman therapy, a method that combines coal tar and UVB phototherapy, has shown safety and effectiveness for patients with recalcitrant or severe psoriasis, and remains a recommended treatment, according to the working group research. However, this method is underused, especially in the United States, because of the messiness of the application, challenge of insurance reimbursement, and investment of time for outpatient care, the work group noted.
Pulsed dye laser treatment is effective for nail psoriasis, and reported adverse effects have been mild, according to the working group.
Overall, the guidelines emphasize that quality of life and disease severity should be considered and discussed with patients along with efficacy and safety information so they can make informed decisions about adding phototherapy to a current regimen or switching among modalities.
The guidelines have no funding sources. Several coauthors disclosed relationships with multiple companies, including manufacturers of psoriasis products; however, a minimum of 51% of the work group had no relevant financial conflicts to disclose, in accordance with AAD policy. Work group members with potential conflicts recused themselves from discussion and drafting of recommendations in the relevant topic areas. Alan Menter, MD, chairman of the division of dermatology, Baylor University Medical Center, Dallas, is the other cochair of the work group.
SOURCE: Elmets CA et al. J Am Acad Dermatol. 2019 Jul 18. doi: 10.1016/j.jaad.2019.04.042.
, according to updated guidelines issued jointly by the American Academy of Dermatology and the National Psoriasis Foundation.
“Phototherapy serves as a reasonable and effective treatment option for patients requiring more than topical medications and/or those wishing to avoid systemic medications or simply seeking an adjunct to a failing regimen,” wrote working group cochair Craig A. Elmets, MD, professor of dermatology at the University of Alabama at Birmingham, and coauthors.
The guidelines, which focus on phototherapy for adults with psoriasis, join a multipart series on psoriasis being published this year in the Journal of the American Academy of Dermatology.
The working group used an evidence-based model to examine efficacy, effectiveness, and adverse effects of the following modalities: narrow-band ultraviolet B (NB-UVB); broadband ultraviolet B (BB-UVB); targeted phototherapy using excimer laser and excimer lamp; psoralen plus ultraviolet A (PUVA) therapy, including topical, oral, and bath PUVA; photodynamic therapy (PDT), grenz ray therapy, climatotherapy; visible light therapy; Goeckerman therapy; and pulsed dye laser/intense pulsed light.
NB-UVB, which can be used to treat generalized plaque psoriasis, refers to wavelengths of 311-313 nm. The recommended treatment is two or three times a week, with a starting dose based on skin phenotype or minimal erythema dose. Although oral PUVA has shown higher clearance rates, compared with NB-UVB, NB-UVB has demonstrated fewer side effects. NB-UVB also has shown effectiveness for psoriasis in combination with medications including oral retinoids, “particularly useful in patients at increased risk for skin cancer,” the working group wrote. Genital shielding and eye protection are recommended during all phototherapy sessions to reduce the risk of cancer and cataracts, they emphasized.
BB-UVB, an older version of NB-UVB, is still effective for generalized plaque psoriasis as monotherapy, but evidence does not support additional benefit in combination with other treatments, and overall BB-UVB is less effective than either NB-UVB or oral PUVA, the working group said.
For treatment of localized psoriatic lesions, some evidence supports the ability of targeted UVB therapy to improve lesions in fewer treatments and at a lower cumulative dose, compared with nontargeted phototherapy, for palmoplantar plaque psoriasis and palmoplantar pustulosis. Excimer lasers also have shown effectiveness against scalp psoriasis, the working group noted. However, “there is insufficient evidence to recommend the excimer laser rather than topical PUVA for treatment of localized plaque psoriasis,” they said.
PUVA treatments are available as topical creams, or they can be taken orally, or mixed with bath water. All forms of PUVA include psoralens, photosensitizing agents that prepare target cells for the effects of UVA light. Topical PUVA has demonstrated particular effectiveness for palmoplantar psoriasis, the working group noted, but there is a risk of phototoxicity, so it has become less popular, they added. Similarly, evidence supports effectiveness of oral and bath PUVA, but all forms are used less frequently because of the increased availability of NB-UVB phototherapy, they said.
PDT is primarily used to destroy premalignant or malignant cells, and in theory “PDT-induced apoptosis of T lymphocytes could lead to reductions in inflammatory cytokines and, in turn, to improvement of psoriasis,” the working group noted. However, “clinical studies have failed to find significant benefit” of PDT using either 5-aminolevulinic acid (ALA) or methyl aminolevulinic acid (MAL) for psoriasis, or any significant benefits of MAL-PDT for nail psoriasis.
The grenz ray is an effective, but rarely used treatment in which 75% of long-wavelength ionizing radiation is absorbed by the first 1 mm of skin and 95% is absorbed within the first 3 mm of skin to protect the deeper tissues from radiation. Although more alternatives are available, grenz rays can be used for psoriasis patients unable to tolerate UV therapy, according to the working group.
Climatotherapy involves temporary or permanent relocation of the patient to a part of the world with a climate that could be favorable for psoriasis because of the unique effects of environmental factors in those areas. The evidence to support climatotherapy is both subjective and objective, but considered safe.
Visible light has been associated with improvement in erythema in psoriasis, with hyperpigmentation as the only notable side effect based on the evidence reviewed. However, the working group found the current evidence insufficient to recommend the use of intense pulsed light for treating psoriasis.
Goeckerman therapy, a method that combines coal tar and UVB phototherapy, has shown safety and effectiveness for patients with recalcitrant or severe psoriasis, and remains a recommended treatment, according to the working group research. However, this method is underused, especially in the United States, because of the messiness of the application, challenge of insurance reimbursement, and investment of time for outpatient care, the work group noted.
Pulsed dye laser treatment is effective for nail psoriasis, and reported adverse effects have been mild, according to the working group.
Overall, the guidelines emphasize that quality of life and disease severity should be considered and discussed with patients along with efficacy and safety information so they can make informed decisions about adding phototherapy to a current regimen or switching among modalities.
The guidelines have no funding sources. Several coauthors disclosed relationships with multiple companies, including manufacturers of psoriasis products; however, a minimum of 51% of the work group had no relevant financial conflicts to disclose, in accordance with AAD policy. Work group members with potential conflicts recused themselves from discussion and drafting of recommendations in the relevant topic areas. Alan Menter, MD, chairman of the division of dermatology, Baylor University Medical Center, Dallas, is the other cochair of the work group.
SOURCE: Elmets CA et al. J Am Acad Dermatol. 2019 Jul 18. doi: 10.1016/j.jaad.2019.04.042.
, according to updated guidelines issued jointly by the American Academy of Dermatology and the National Psoriasis Foundation.
“Phototherapy serves as a reasonable and effective treatment option for patients requiring more than topical medications and/or those wishing to avoid systemic medications or simply seeking an adjunct to a failing regimen,” wrote working group cochair Craig A. Elmets, MD, professor of dermatology at the University of Alabama at Birmingham, and coauthors.
The guidelines, which focus on phototherapy for adults with psoriasis, join a multipart series on psoriasis being published this year in the Journal of the American Academy of Dermatology.
The working group used an evidence-based model to examine efficacy, effectiveness, and adverse effects of the following modalities: narrow-band ultraviolet B (NB-UVB); broadband ultraviolet B (BB-UVB); targeted phototherapy using excimer laser and excimer lamp; psoralen plus ultraviolet A (PUVA) therapy, including topical, oral, and bath PUVA; photodynamic therapy (PDT), grenz ray therapy, climatotherapy; visible light therapy; Goeckerman therapy; and pulsed dye laser/intense pulsed light.
NB-UVB, which can be used to treat generalized plaque psoriasis, refers to wavelengths of 311-313 nm. The recommended treatment is two or three times a week, with a starting dose based on skin phenotype or minimal erythema dose. Although oral PUVA has shown higher clearance rates, compared with NB-UVB, NB-UVB has demonstrated fewer side effects. NB-UVB also has shown effectiveness for psoriasis in combination with medications including oral retinoids, “particularly useful in patients at increased risk for skin cancer,” the working group wrote. Genital shielding and eye protection are recommended during all phototherapy sessions to reduce the risk of cancer and cataracts, they emphasized.
BB-UVB, an older version of NB-UVB, is still effective for generalized plaque psoriasis as monotherapy, but evidence does not support additional benefit in combination with other treatments, and overall BB-UVB is less effective than either NB-UVB or oral PUVA, the working group said.
For treatment of localized psoriatic lesions, some evidence supports the ability of targeted UVB therapy to improve lesions in fewer treatments and at a lower cumulative dose, compared with nontargeted phototherapy, for palmoplantar plaque psoriasis and palmoplantar pustulosis. Excimer lasers also have shown effectiveness against scalp psoriasis, the working group noted. However, “there is insufficient evidence to recommend the excimer laser rather than topical PUVA for treatment of localized plaque psoriasis,” they said.
PUVA treatments are available as topical creams, or they can be taken orally, or mixed with bath water. All forms of PUVA include psoralens, photosensitizing agents that prepare target cells for the effects of UVA light. Topical PUVA has demonstrated particular effectiveness for palmoplantar psoriasis, the working group noted, but there is a risk of phototoxicity, so it has become less popular, they added. Similarly, evidence supports effectiveness of oral and bath PUVA, but all forms are used less frequently because of the increased availability of NB-UVB phototherapy, they said.
PDT is primarily used to destroy premalignant or malignant cells, and in theory “PDT-induced apoptosis of T lymphocytes could lead to reductions in inflammatory cytokines and, in turn, to improvement of psoriasis,” the working group noted. However, “clinical studies have failed to find significant benefit” of PDT using either 5-aminolevulinic acid (ALA) or methyl aminolevulinic acid (MAL) for psoriasis, or any significant benefits of MAL-PDT for nail psoriasis.
The grenz ray is an effective, but rarely used treatment in which 75% of long-wavelength ionizing radiation is absorbed by the first 1 mm of skin and 95% is absorbed within the first 3 mm of skin to protect the deeper tissues from radiation. Although more alternatives are available, grenz rays can be used for psoriasis patients unable to tolerate UV therapy, according to the working group.
Climatotherapy involves temporary or permanent relocation of the patient to a part of the world with a climate that could be favorable for psoriasis because of the unique effects of environmental factors in those areas. The evidence to support climatotherapy is both subjective and objective, but considered safe.
Visible light has been associated with improvement in erythema in psoriasis, with hyperpigmentation as the only notable side effect based on the evidence reviewed. However, the working group found the current evidence insufficient to recommend the use of intense pulsed light for treating psoriasis.
Goeckerman therapy, a method that combines coal tar and UVB phototherapy, has shown safety and effectiveness for patients with recalcitrant or severe psoriasis, and remains a recommended treatment, according to the working group research. However, this method is underused, especially in the United States, because of the messiness of the application, challenge of insurance reimbursement, and investment of time for outpatient care, the work group noted.
Pulsed dye laser treatment is effective for nail psoriasis, and reported adverse effects have been mild, according to the working group.
Overall, the guidelines emphasize that quality of life and disease severity should be considered and discussed with patients along with efficacy and safety information so they can make informed decisions about adding phototherapy to a current regimen or switching among modalities.
The guidelines have no funding sources. Several coauthors disclosed relationships with multiple companies, including manufacturers of psoriasis products; however, a minimum of 51% of the work group had no relevant financial conflicts to disclose, in accordance with AAD policy. Work group members with potential conflicts recused themselves from discussion and drafting of recommendations in the relevant topic areas. Alan Menter, MD, chairman of the division of dermatology, Baylor University Medical Center, Dallas, is the other cochair of the work group.
SOURCE: Elmets CA et al. J Am Acad Dermatol. 2019 Jul 18. doi: 10.1016/j.jaad.2019.04.042.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Darkening and Eruptive Nevi During Treatment With Erlotinib
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
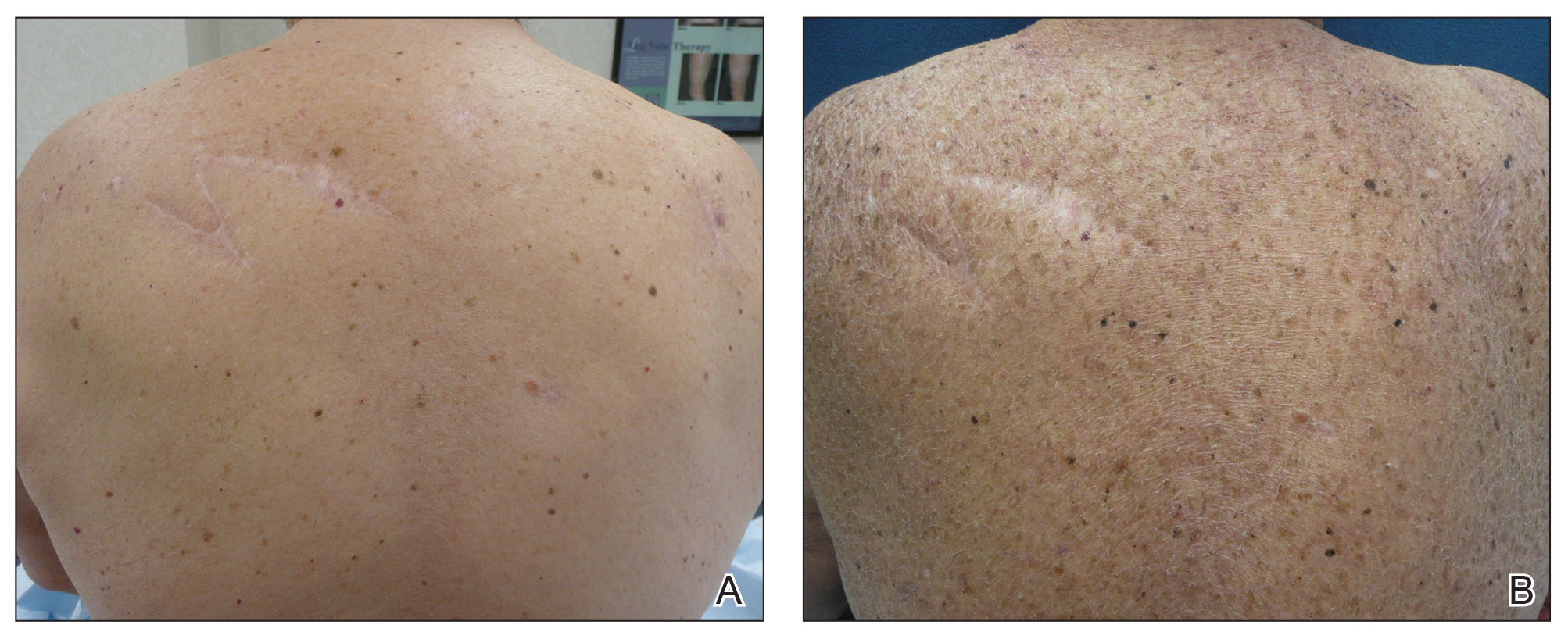
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
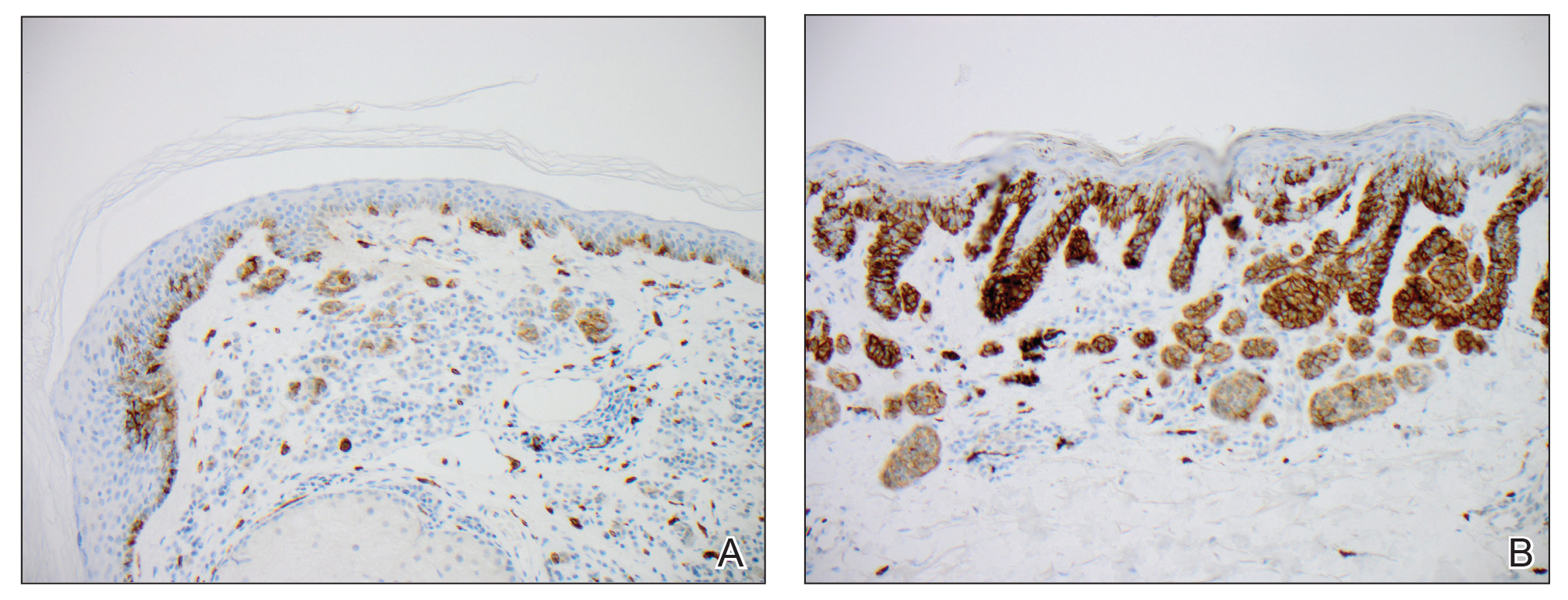
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
Practice Points
- Cutaneous side effects of erlotinib include acneform eruption, xerosis, paronychia, and pruritus.
- Clinicians should monitor patients for darkening and/or eruptive nevi as well as melanoma during treatment with erlotinib.
Hypothyroidism may have more impact on cardiac health than hyperthyroidism
ORLANDO – Thyroid disorders can have significant effects on the heart and the cardiovascular system, Christine Kessler, MN, ANP-C, CNS, BC-ADM, FAANP, said at the Cardiovascular & Respiratory Summit by Global Academy for Medical Education.

Even subclinical hypothyroidism “can be really impactful,” said Ms. Kessler, the founder of Metabolic Medicine Associates in King George, Va.
Thyroid function should be evaluated in patients with a fast resting heart fate, new-onset atrial fibrillation (AFib), idiopathic heart failure, bradycardia, using amiodarone, resistant hypertension, pericardial effusion, and statin-resistant hyperlipidemia, said Ms. Kessler, who also is a nurse practitioner and researcher. Other patients who should be evaluated: those older than 60 years, with a family history of autoimmune disease, fertility issues, new-onset anxiety/depression, and patients with high-risk pregnancy. Hypothyroidism may have more impact on cardiac health than does hyperthyroidism.
Levels of thyroid-stimulating hormone (TSH), triiodothyronine (T3), and free thyroxine (FT4) are typically used to evaluate thyroid function. High TSH levels are usually indicative of hypothyroidism if FT4 and T3 are low; hyperthyroidism is likely the diagnosis if TSH is low while FT4 and T3 are high. Subclinical hypothyroidism is characterized by high TSH and normal FT4 and T3 levels; subclinical hyperthyroidism is associated with low TSH with normal FT4 and T3 levels.*
Hypothyroidism can cause increased diastolic hypertension and systemic vascular resistance, elevated levels of C-reactive protein (CRP) and homocysteine, decreased myocardial contractility, decreased cardiac output, reduced heart rate, and liver function abnormalities. Most commonly, it is caused by the autoimmune disease Hashimoto’s hypothyroidism but also can result from radiation, thyroidectomy, nontoxic multinodular goiter, and drugs with antithyroid activity such as birth control medications that contain estrogen. Hypothyroidism also raises the risk of coronary artery disease through lipid aberrations such as increased LDL level and decreased number of LDL cholesterol receptors. Myocardial contractility from hypothyroidism also increases the risk of mortality in patients with heart disease and heart failure. Clinicians also should take special precautions with narcotics and anesthesia when caring for patients with hypothyroidism because of the risk of bradycardia, Ms. Kessler said.
In subclinical hypothyroidism, clinicians should treat with half the recommended dose of levothyroxine in patients aged 45-65 years and who have high levels of lipids and CRP. “At the age of 70, I don’t worry if you’re subclinical unless [the patient is] profoundly, profoundly symptomatic,” she said. In overt hypotension, the main concern is an increased risk of ischemic heart disease that can result from overtreatment, and these patients usually are started on a low dose of levothyroxine with escalated doses until the patient achieves a euthyroid state.
Hyperthyroidism is most commonly caused by Grave’s disease, with 85% of those affected younger than age 40 years. Other causes include toxic multinodular goiter, toxic adenoma, TSH-producing pituitary adenomas, resistance to thyroid hormone, thyroiditis, excessive ingestion of iodine, shrinking of the thyroid gland, and a human chorionic gonadotropin–producing tumor such as struma ovarii. Common cardiac complications of hyperthyroidism are increased heart rate and blood pressure, reduced systemic vascular resistance, and increased cardiovascular disease and mortality in addition to increased cardiac hypertrophy, pulmonary hypertension, and heart failure. Heart rhythm, atrial fibrillation, atrial flutter, increased sinus tachycardia, and increased angina are all possible complications of hyperthyroidism.
Treatment priorities for hyperthyroidism are the immediately relieving any symptoms, reducing thyroid hormone production, and blocking the conversion of T4 to T3. For symptomatic patients, beta-blockers will help relieve symptoms and block the T4/T3 conversion. Follow-up treatment should include antithyroid drugs such as methimazole or propylthiouracil.
Patients who have subclinical hyperthyroidism are usually asymptomatic and may not always require treatment.
“In subclinical hypothyroidism, keep your mitts off the older patients. They’re usually going to do better, and you don’t want to throw them into hyperthyroidism,” she said. “If they’re [experiencing] subclinical hyperthyroidism, you’re going to treat them, because if not, they’re going to go into AFib, cardiovascular death, [and] they’re going to have osteoporosis. It’s not a good thing.”
Ms. Kessler reports being on the speakers bureau and an adviser for Novo Nordisk on obesity, and an adviser for them on type 2 diabetes, as well. She also reports being the study chairperson for the Florajen Patient Trial Program. The Cardiovascular & Respiratory Summit is part of Global Academy for Medical Education. Global Academy for Medical Education and this news organization are owned by the same parent company.
Correction, 7/31/19: An earlier version of this article misstated the definition of subclinical hypothyroidism.
ORLANDO – Thyroid disorders can have significant effects on the heart and the cardiovascular system, Christine Kessler, MN, ANP-C, CNS, BC-ADM, FAANP, said at the Cardiovascular & Respiratory Summit by Global Academy for Medical Education.
Even subclinical hypothyroidism “can be really impactful,” said Ms. Kessler, the founder of Metabolic Medicine Associates in King George, Va.
Thyroid function should be evaluated in patients with a fast resting heart fate, new-onset atrial fibrillation (AFib), idiopathic heart failure, bradycardia, using amiodarone, resistant hypertension, pericardial effusion, and statin-resistant hyperlipidemia, said Ms. Kessler, who also is a nurse practitioner and researcher. Other patients who should be evaluated: those older than 60 years, with a family history of autoimmune disease, fertility issues, new-onset anxiety/depression, and patients with high-risk pregnancy. Hypothyroidism may have more impact on cardiac health than does hyperthyroidism.
Levels of thyroid-stimulating hormone (TSH), triiodothyronine (T3), and free thyroxine (FT4) are typically used to evaluate thyroid function. High TSH levels are usually indicative of hypothyroidism if FT4 and T3 are low; hyperthyroidism is likely the diagnosis if TSH is low while FT4 and T3 are high. Subclinical hypothyroidism is characterized by high TSH and normal FT4 and T3 levels; subclinical hyperthyroidism is associated with low TSH with normal FT4 and T3 levels.*
Hypothyroidism can cause increased diastolic hypertension and systemic vascular resistance, elevated levels of C-reactive protein (CRP) and homocysteine, decreased myocardial contractility, decreased cardiac output, reduced heart rate, and liver function abnormalities. Most commonly, it is caused by the autoimmune disease Hashimoto’s hypothyroidism but also can result from radiation, thyroidectomy, nontoxic multinodular goiter, and drugs with antithyroid activity such as birth control medications that contain estrogen. Hypothyroidism also raises the risk of coronary artery disease through lipid aberrations such as increased LDL level and decreased number of LDL cholesterol receptors. Myocardial contractility from hypothyroidism also increases the risk of mortality in patients with heart disease and heart failure. Clinicians also should take special precautions with narcotics and anesthesia when caring for patients with hypothyroidism because of the risk of bradycardia, Ms. Kessler said.
In subclinical hypothyroidism, clinicians should treat with half the recommended dose of levothyroxine in patients aged 45-65 years and who have high levels of lipids and CRP. “At the age of 70, I don’t worry if you’re subclinical unless [the patient is] profoundly, profoundly symptomatic,” she said. In overt hypotension, the main concern is an increased risk of ischemic heart disease that can result from overtreatment, and these patients usually are started on a low dose of levothyroxine with escalated doses until the patient achieves a euthyroid state.
Hyperthyroidism is most commonly caused by Grave’s disease, with 85% of those affected younger than age 40 years. Other causes include toxic multinodular goiter, toxic adenoma, TSH-producing pituitary adenomas, resistance to thyroid hormone, thyroiditis, excessive ingestion of iodine, shrinking of the thyroid gland, and a human chorionic gonadotropin–producing tumor such as struma ovarii. Common cardiac complications of hyperthyroidism are increased heart rate and blood pressure, reduced systemic vascular resistance, and increased cardiovascular disease and mortality in addition to increased cardiac hypertrophy, pulmonary hypertension, and heart failure. Heart rhythm, atrial fibrillation, atrial flutter, increased sinus tachycardia, and increased angina are all possible complications of hyperthyroidism.
Treatment priorities for hyperthyroidism are the immediately relieving any symptoms, reducing thyroid hormone production, and blocking the conversion of T4 to T3. For symptomatic patients, beta-blockers will help relieve symptoms and block the T4/T3 conversion. Follow-up treatment should include antithyroid drugs such as methimazole or propylthiouracil.
Patients who have subclinical hyperthyroidism are usually asymptomatic and may not always require treatment.
“In subclinical hypothyroidism, keep your mitts off the older patients. They’re usually going to do better, and you don’t want to throw them into hyperthyroidism,” she said. “If they’re [experiencing] subclinical hyperthyroidism, you’re going to treat them, because if not, they’re going to go into AFib, cardiovascular death, [and] they’re going to have osteoporosis. It’s not a good thing.”
Ms. Kessler reports being on the speakers bureau and an adviser for Novo Nordisk on obesity, and an adviser for them on type 2 diabetes, as well. She also reports being the study chairperson for the Florajen Patient Trial Program. The Cardiovascular & Respiratory Summit is part of Global Academy for Medical Education. Global Academy for Medical Education and this news organization are owned by the same parent company.
Correction, 7/31/19: An earlier version of this article misstated the definition of subclinical hypothyroidism.
ORLANDO – Thyroid disorders can have significant effects on the heart and the cardiovascular system, Christine Kessler, MN, ANP-C, CNS, BC-ADM, FAANP, said at the Cardiovascular & Respiratory Summit by Global Academy for Medical Education.
Even subclinical hypothyroidism “can be really impactful,” said Ms. Kessler, the founder of Metabolic Medicine Associates in King George, Va.
Thyroid function should be evaluated in patients with a fast resting heart fate, new-onset atrial fibrillation (AFib), idiopathic heart failure, bradycardia, using amiodarone, resistant hypertension, pericardial effusion, and statin-resistant hyperlipidemia, said Ms. Kessler, who also is a nurse practitioner and researcher. Other patients who should be evaluated: those older than 60 years, with a family history of autoimmune disease, fertility issues, new-onset anxiety/depression, and patients with high-risk pregnancy. Hypothyroidism may have more impact on cardiac health than does hyperthyroidism.
Levels of thyroid-stimulating hormone (TSH), triiodothyronine (T3), and free thyroxine (FT4) are typically used to evaluate thyroid function. High TSH levels are usually indicative of hypothyroidism if FT4 and T3 are low; hyperthyroidism is likely the diagnosis if TSH is low while FT4 and T3 are high. Subclinical hypothyroidism is characterized by high TSH and normal FT4 and T3 levels; subclinical hyperthyroidism is associated with low TSH with normal FT4 and T3 levels.*
Hypothyroidism can cause increased diastolic hypertension and systemic vascular resistance, elevated levels of C-reactive protein (CRP) and homocysteine, decreased myocardial contractility, decreased cardiac output, reduced heart rate, and liver function abnormalities. Most commonly, it is caused by the autoimmune disease Hashimoto’s hypothyroidism but also can result from radiation, thyroidectomy, nontoxic multinodular goiter, and drugs with antithyroid activity such as birth control medications that contain estrogen. Hypothyroidism also raises the risk of coronary artery disease through lipid aberrations such as increased LDL level and decreased number of LDL cholesterol receptors. Myocardial contractility from hypothyroidism also increases the risk of mortality in patients with heart disease and heart failure. Clinicians also should take special precautions with narcotics and anesthesia when caring for patients with hypothyroidism because of the risk of bradycardia, Ms. Kessler said.
In subclinical hypothyroidism, clinicians should treat with half the recommended dose of levothyroxine in patients aged 45-65 years and who have high levels of lipids and CRP. “At the age of 70, I don’t worry if you’re subclinical unless [the patient is] profoundly, profoundly symptomatic,” she said. In overt hypotension, the main concern is an increased risk of ischemic heart disease that can result from overtreatment, and these patients usually are started on a low dose of levothyroxine with escalated doses until the patient achieves a euthyroid state.
Hyperthyroidism is most commonly caused by Grave’s disease, with 85% of those affected younger than age 40 years. Other causes include toxic multinodular goiter, toxic adenoma, TSH-producing pituitary adenomas, resistance to thyroid hormone, thyroiditis, excessive ingestion of iodine, shrinking of the thyroid gland, and a human chorionic gonadotropin–producing tumor such as struma ovarii. Common cardiac complications of hyperthyroidism are increased heart rate and blood pressure, reduced systemic vascular resistance, and increased cardiovascular disease and mortality in addition to increased cardiac hypertrophy, pulmonary hypertension, and heart failure. Heart rhythm, atrial fibrillation, atrial flutter, increased sinus tachycardia, and increased angina are all possible complications of hyperthyroidism.
Treatment priorities for hyperthyroidism are the immediately relieving any symptoms, reducing thyroid hormone production, and blocking the conversion of T4 to T3. For symptomatic patients, beta-blockers will help relieve symptoms and block the T4/T3 conversion. Follow-up treatment should include antithyroid drugs such as methimazole or propylthiouracil.
Patients who have subclinical hyperthyroidism are usually asymptomatic and may not always require treatment.
“In subclinical hypothyroidism, keep your mitts off the older patients. They’re usually going to do better, and you don’t want to throw them into hyperthyroidism,” she said. “If they’re [experiencing] subclinical hyperthyroidism, you’re going to treat them, because if not, they’re going to go into AFib, cardiovascular death, [and] they’re going to have osteoporosis. It’s not a good thing.”
Ms. Kessler reports being on the speakers bureau and an adviser for Novo Nordisk on obesity, and an adviser for them on type 2 diabetes, as well. She also reports being the study chairperson for the Florajen Patient Trial Program. The Cardiovascular & Respiratory Summit is part of Global Academy for Medical Education. Global Academy for Medical Education and this news organization are owned by the same parent company.
Correction, 7/31/19: An earlier version of this article misstated the definition of subclinical hypothyroidism.
EXPERT ANALYSIS FROM CARPS 2019
Metastatic Adamantinoma Presenting as a Cutaneous Papule
To the Editor:
A 34-year-old woman with a history of adamantinoma of the right tibia that had been surgically resected with tibial reconstruction 5 years prior presented with a mildly tender, enlarging lesion on the right distal shin of 6 months’ duration that had started to change color. Review of systems was otherwise negative. Physical examination revealed an 8-mm, slightly tender, rubbery, pink papule adjacent to the surgical scar over the right tibia (Figure 1). Given the rapid growth of the lesion and its proximity to the surgical site, a punch biopsy was performed.
Histopathologic examination demonstrated a densely cellular dermal tumor composed of spindle cells with large hyperchromatic nuclei, numerous mitotic figures, and minimal eosinophilic cytoplasm (Figure 2A). Immunohistochemical studies revealed that approximately 40% of the tumor nuclei were immunoreactive to Ki-67 (Figure 2B), and total cytokeratin was focally positive (Figure 2C). A diagnosis of metastatic adamantinoma was made. Positron emission tomography and magnetic resonance imaging revealed new lytic lesions involving the T10 and L2 vertebrae (without frank spinal cord compression) and the right superior sacrum. Additionally, a small pulmonary nodule on the left upper lobe was noted on positron emission tomography, but it was below the size threshold for reliable detection. A computed tomography–guided biopsy of the T10 lesion demonstrated metastatic adamantinoma. The patient underwent a spinal stabilization procedure and discussed options regarding further oncologic and palliative management.
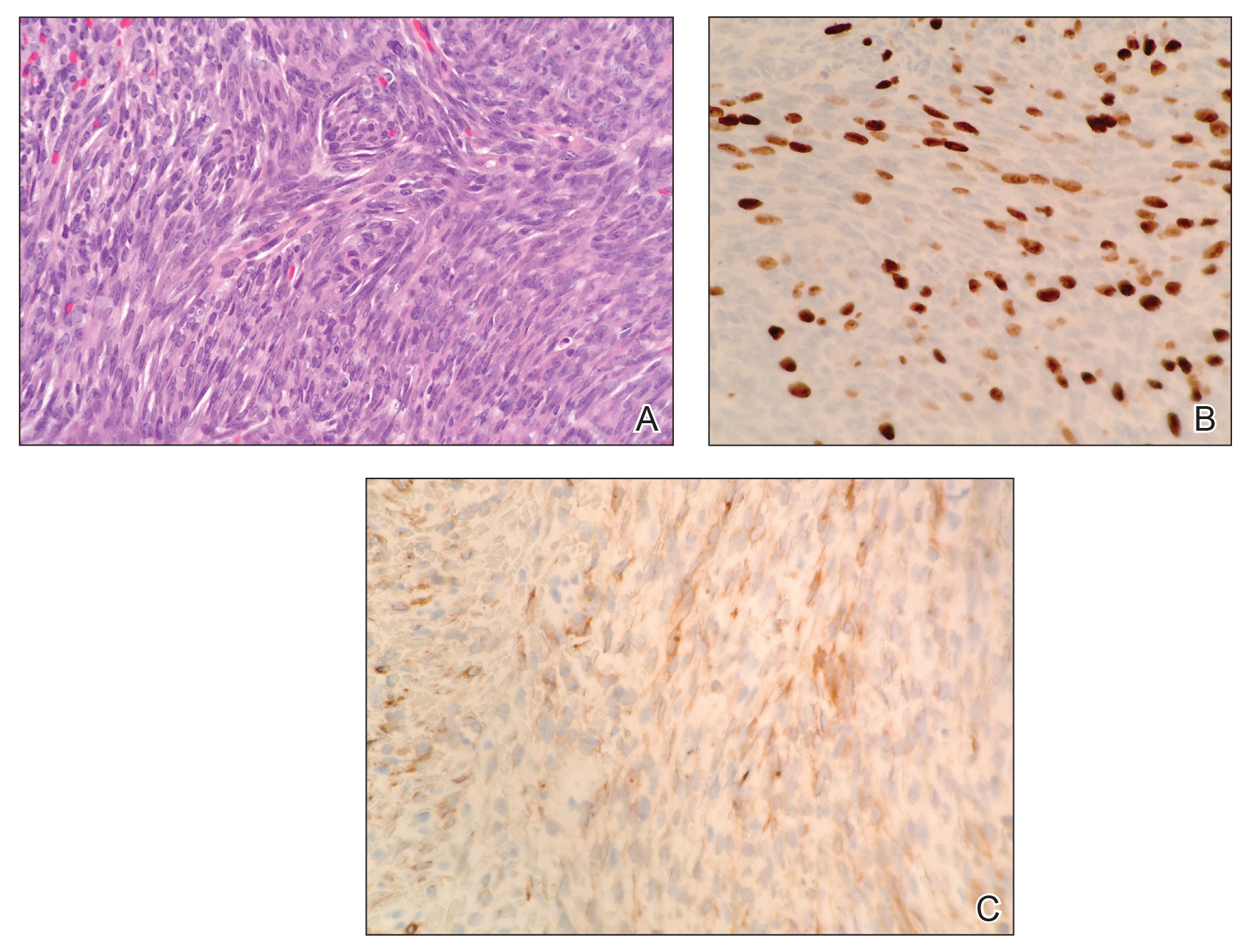
Adamantinoma is an extremely rare primary malignant bone tumor that typically involves the anterior portion of the tibial metaphysis or diaphysis in approximately 90% of cases. Young adults most commonly are affected in the third or fourth decades of life.1 Although the histogenesis is not clearly understood, experts have theorized that fetal implantation during embryogenesis or traumatic implantation of epithelial cells may be causes of this tumor and may explain the close pathologic similarity to basal cell carcinoma.2
Adamantinomas are slow growing, and as a result, patients often present with gradual onset of pain and swelling that persists for years.3,4 Metastasis occurs in 10% to 30% of patients, typically located in regional lymph nodes, the lungs, and distant bone.1,4 Our case represents a rare instance of adamantinoma metastasis to the skin. Although primary adamantinomas consist of both epithelial and stromal components, the typical metastatic lesions of adamantinomas are solely epithelial (often in a spindle-cell pattern),1 as was seen in our patient.
Operative removal via amputation or en bloc resection with limb salvage is the current treatment of choice. Adamantinomas are highly radioresistant, and chemotherapy has shown minimal efficacy.3,5
In conclusion, the presence of cutaneous metastasis from an adamantinoma is rare. Our case emphasizes this tumor’s potential for late metastasis as well as late recurrence.3,6 Most importantly, dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
- Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95-97.
- Jain D, Jain VK, Vasishta RK, et al. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
- Qureshi AA, Shott S, Mallin BA, et al. Current trends in the management of adamantinoma of long bones. an international study. J Bone Joint Surg Am. 2000;82-A:1122-1131.
- Desai SS, Jambhekar N, Agarwal M, et al. Adamantinoma of tibia: a study of 12 cases. J Surg Oncol. 2006;93:429-433.
- Weiss SW, Dorfman HD. Adamantinoma of long bone. an analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141-153.
- Szendroi M, Antal I, Arató G. Adamantinoma of long bones: a long-term follow-up study of 11 cases. Pathol Oncol Res. 2009;15:209-216.
To the Editor:
A 34-year-old woman with a history of adamantinoma of the right tibia that had been surgically resected with tibial reconstruction 5 years prior presented with a mildly tender, enlarging lesion on the right distal shin of 6 months’ duration that had started to change color. Review of systems was otherwise negative. Physical examination revealed an 8-mm, slightly tender, rubbery, pink papule adjacent to the surgical scar over the right tibia (Figure 1). Given the rapid growth of the lesion and its proximity to the surgical site, a punch biopsy was performed.
Histopathologic examination demonstrated a densely cellular dermal tumor composed of spindle cells with large hyperchromatic nuclei, numerous mitotic figures, and minimal eosinophilic cytoplasm (Figure 2A). Immunohistochemical studies revealed that approximately 40% of the tumor nuclei were immunoreactive to Ki-67 (Figure 2B), and total cytokeratin was focally positive (Figure 2C). A diagnosis of metastatic adamantinoma was made. Positron emission tomography and magnetic resonance imaging revealed new lytic lesions involving the T10 and L2 vertebrae (without frank spinal cord compression) and the right superior sacrum. Additionally, a small pulmonary nodule on the left upper lobe was noted on positron emission tomography, but it was below the size threshold for reliable detection. A computed tomography–guided biopsy of the T10 lesion demonstrated metastatic adamantinoma. The patient underwent a spinal stabilization procedure and discussed options regarding further oncologic and palliative management.
Adamantinoma is an extremely rare primary malignant bone tumor that typically involves the anterior portion of the tibial metaphysis or diaphysis in approximately 90% of cases. Young adults most commonly are affected in the third or fourth decades of life.1 Although the histogenesis is not clearly understood, experts have theorized that fetal implantation during embryogenesis or traumatic implantation of epithelial cells may be causes of this tumor and may explain the close pathologic similarity to basal cell carcinoma.2
Adamantinomas are slow growing, and as a result, patients often present with gradual onset of pain and swelling that persists for years.3,4 Metastasis occurs in 10% to 30% of patients, typically located in regional lymph nodes, the lungs, and distant bone.1,4 Our case represents a rare instance of adamantinoma metastasis to the skin. Although primary adamantinomas consist of both epithelial and stromal components, the typical metastatic lesions of adamantinomas are solely epithelial (often in a spindle-cell pattern),1 as was seen in our patient.
Operative removal via amputation or en bloc resection with limb salvage is the current treatment of choice. Adamantinomas are highly radioresistant, and chemotherapy has shown minimal efficacy.3,5
In conclusion, the presence of cutaneous metastasis from an adamantinoma is rare. Our case emphasizes this tumor’s potential for late metastasis as well as late recurrence.3,6 Most importantly, dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
To the Editor:
A 34-year-old woman with a history of adamantinoma of the right tibia that had been surgically resected with tibial reconstruction 5 years prior presented with a mildly tender, enlarging lesion on the right distal shin of 6 months’ duration that had started to change color. Review of systems was otherwise negative. Physical examination revealed an 8-mm, slightly tender, rubbery, pink papule adjacent to the surgical scar over the right tibia (Figure 1). Given the rapid growth of the lesion and its proximity to the surgical site, a punch biopsy was performed.
Histopathologic examination demonstrated a densely cellular dermal tumor composed of spindle cells with large hyperchromatic nuclei, numerous mitotic figures, and minimal eosinophilic cytoplasm (Figure 2A). Immunohistochemical studies revealed that approximately 40% of the tumor nuclei were immunoreactive to Ki-67 (Figure 2B), and total cytokeratin was focally positive (Figure 2C). A diagnosis of metastatic adamantinoma was made. Positron emission tomography and magnetic resonance imaging revealed new lytic lesions involving the T10 and L2 vertebrae (without frank spinal cord compression) and the right superior sacrum. Additionally, a small pulmonary nodule on the left upper lobe was noted on positron emission tomography, but it was below the size threshold for reliable detection. A computed tomography–guided biopsy of the T10 lesion demonstrated metastatic adamantinoma. The patient underwent a spinal stabilization procedure and discussed options regarding further oncologic and palliative management.
Adamantinoma is an extremely rare primary malignant bone tumor that typically involves the anterior portion of the tibial metaphysis or diaphysis in approximately 90% of cases. Young adults most commonly are affected in the third or fourth decades of life.1 Although the histogenesis is not clearly understood, experts have theorized that fetal implantation during embryogenesis or traumatic implantation of epithelial cells may be causes of this tumor and may explain the close pathologic similarity to basal cell carcinoma.2
Adamantinomas are slow growing, and as a result, patients often present with gradual onset of pain and swelling that persists for years.3,4 Metastasis occurs in 10% to 30% of patients, typically located in regional lymph nodes, the lungs, and distant bone.1,4 Our case represents a rare instance of adamantinoma metastasis to the skin. Although primary adamantinomas consist of both epithelial and stromal components, the typical metastatic lesions of adamantinomas are solely epithelial (often in a spindle-cell pattern),1 as was seen in our patient.
Operative removal via amputation or en bloc resection with limb salvage is the current treatment of choice. Adamantinomas are highly radioresistant, and chemotherapy has shown minimal efficacy.3,5
In conclusion, the presence of cutaneous metastasis from an adamantinoma is rare. Our case emphasizes this tumor’s potential for late metastasis as well as late recurrence.3,6 Most importantly, dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
- Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95-97.
- Jain D, Jain VK, Vasishta RK, et al. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
- Qureshi AA, Shott S, Mallin BA, et al. Current trends in the management of adamantinoma of long bones. an international study. J Bone Joint Surg Am. 2000;82-A:1122-1131.
- Desai SS, Jambhekar N, Agarwal M, et al. Adamantinoma of tibia: a study of 12 cases. J Surg Oncol. 2006;93:429-433.
- Weiss SW, Dorfman HD. Adamantinoma of long bone. an analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141-153.
- Szendroi M, Antal I, Arató G. Adamantinoma of long bones: a long-term follow-up study of 11 cases. Pathol Oncol Res. 2009;15:209-216.
- Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95-97.
- Jain D, Jain VK, Vasishta RK, et al. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
- Qureshi AA, Shott S, Mallin BA, et al. Current trends in the management of adamantinoma of long bones. an international study. J Bone Joint Surg Am. 2000;82-A:1122-1131.
- Desai SS, Jambhekar N, Agarwal M, et al. Adamantinoma of tibia: a study of 12 cases. J Surg Oncol. 2006;93:429-433.
- Weiss SW, Dorfman HD. Adamantinoma of long bone. an analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141-153.
- Szendroi M, Antal I, Arató G. Adamantinoma of long bones: a long-term follow-up study of 11 cases. Pathol Oncol Res. 2009;15:209-216.
Practice Points
- Metastatic adamantinoma of the skin is a rare clinical scenario.
- Dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
Mayo Clinic takes honors as top hospital
The Rochester, Minn., hospital has now been No. 1 on the U.S. News Honor Roll for 4 consecutive years. The last institution to head the list before that streak, Massachusetts General Hospital in Boston, was second this year, followed by Johns Hopkins Hospital in Baltimore, the Cleveland Clinic, and New York-Presbyterian Hospital–Columbia and Cornell in New York, U.S. News reported July 30.
The Mayo Clinic finished first in five of the 16 specialties used in the evaluation process – diabetes and endocrinology; ear, nose, and throat; gastroenterology and GI surgery; urology; and nephrology – and had a total of 13 top-five rankings. Massachusetts General had one first place in psychiatry and six total top fives, while Johns Hopkins, despite its lower overall ranking, had three top rankings – geriatrics, neurology and neurosurgery, and rheumatology – and nine top fives, the U.S. News data show.
This year’s 30th edition of the rankings involved 4,653 community inpatient hospitals, of which 1,447 received a “High Performing” rating in at least one of the nine procedures and conditions assessed and 165 were nationally ranked (top 50) in at least one of the 16 specialties. Twelve specialties are ranked largely on objective data, with the three most recent years of an annual expert opinion survey of specialized physicians also factored in. The other four specialties are ranked based entirely on the survey of expert opinion.
The research organization RTI International conducted the physician survey and produced the Best Hospitals methodology and national rankings under contract with U.S. News.
The Rochester, Minn., hospital has now been No. 1 on the U.S. News Honor Roll for 4 consecutive years. The last institution to head the list before that streak, Massachusetts General Hospital in Boston, was second this year, followed by Johns Hopkins Hospital in Baltimore, the Cleveland Clinic, and New York-Presbyterian Hospital–Columbia and Cornell in New York, U.S. News reported July 30.
The Mayo Clinic finished first in five of the 16 specialties used in the evaluation process – diabetes and endocrinology; ear, nose, and throat; gastroenterology and GI surgery; urology; and nephrology – and had a total of 13 top-five rankings. Massachusetts General had one first place in psychiatry and six total top fives, while Johns Hopkins, despite its lower overall ranking, had three top rankings – geriatrics, neurology and neurosurgery, and rheumatology – and nine top fives, the U.S. News data show.
This year’s 30th edition of the rankings involved 4,653 community inpatient hospitals, of which 1,447 received a “High Performing” rating in at least one of the nine procedures and conditions assessed and 165 were nationally ranked (top 50) in at least one of the 16 specialties. Twelve specialties are ranked largely on objective data, with the three most recent years of an annual expert opinion survey of specialized physicians also factored in. The other four specialties are ranked based entirely on the survey of expert opinion.
The research organization RTI International conducted the physician survey and produced the Best Hospitals methodology and national rankings under contract with U.S. News.
The Rochester, Minn., hospital has now been No. 1 on the U.S. News Honor Roll for 4 consecutive years. The last institution to head the list before that streak, Massachusetts General Hospital in Boston, was second this year, followed by Johns Hopkins Hospital in Baltimore, the Cleveland Clinic, and New York-Presbyterian Hospital–Columbia and Cornell in New York, U.S. News reported July 30.
The Mayo Clinic finished first in five of the 16 specialties used in the evaluation process – diabetes and endocrinology; ear, nose, and throat; gastroenterology and GI surgery; urology; and nephrology – and had a total of 13 top-five rankings. Massachusetts General had one first place in psychiatry and six total top fives, while Johns Hopkins, despite its lower overall ranking, had three top rankings – geriatrics, neurology and neurosurgery, and rheumatology – and nine top fives, the U.S. News data show.
This year’s 30th edition of the rankings involved 4,653 community inpatient hospitals, of which 1,447 received a “High Performing” rating in at least one of the nine procedures and conditions assessed and 165 were nationally ranked (top 50) in at least one of the 16 specialties. Twelve specialties are ranked largely on objective data, with the three most recent years of an annual expert opinion survey of specialized physicians also factored in. The other four specialties are ranked based entirely on the survey of expert opinion.
The research organization RTI International conducted the physician survey and produced the Best Hospitals methodology and national rankings under contract with U.S. News.
FDA accepts dasotraline NDA for binge-eating disorder
The Food and Drug Administration has accepted the new drug application for dasotraline for the treatment of moderate to severe binge-eating disorder, the drug’s developer, Sunovion, announced July 30.

Dasotraline, a dopamine and norepinephrine reuptake inhibitor, demonstrated significant efficacy in a pair of 12-week, randomized, placebo-controlled studies (SEP360-221 and SEP360-321). The drug also was found to be well tolerated by patients with binge-eating disorder (BED), both in those studies and in a long-term safety study that followed patients for up to a year (SEP360-322).
The medication – characterized by an extended half-life – is to be taken once a day. The most common adverse events reported by patients who took dasotraline include insomnia, dry mouth, decreased appetite, anxiety, nausea, and decreased weight.
BED is more common than any other eating disorder, with an estimated lifetime prevalence among U.S. adults of 1.25% for women and 0.42% for men (CNS Spectr. 2019 Jun 14. doi: 10.1017/S109285291900103). The condition also might run in families. BED often is comorbid with other psychiatric and behavioral disorders, such as depression, substance use, and PTSD, noted Antony Loebel, MD, president and CEO of Sunovion, in a press release. He also said BED often is underrecognized and undertreated.
Meta-analytic reviews show that cognitive-behavioral therapy is considered first-line treatment for BED. However, limited access to such psychological treatments makes the development of medication options such as dasotraline important.
Last year, the agency rejected a new drug application for dasotraline for the treatment of ADHD, citing a need for additional data.
The Food and Drug Administration has accepted the new drug application for dasotraline for the treatment of moderate to severe binge-eating disorder, the drug’s developer, Sunovion, announced July 30.
Dasotraline, a dopamine and norepinephrine reuptake inhibitor, demonstrated significant efficacy in a pair of 12-week, randomized, placebo-controlled studies (SEP360-221 and SEP360-321). The drug also was found to be well tolerated by patients with binge-eating disorder (BED), both in those studies and in a long-term safety study that followed patients for up to a year (SEP360-322).
The medication – characterized by an extended half-life – is to be taken once a day. The most common adverse events reported by patients who took dasotraline include insomnia, dry mouth, decreased appetite, anxiety, nausea, and decreased weight.
BED is more common than any other eating disorder, with an estimated lifetime prevalence among U.S. adults of 1.25% for women and 0.42% for men (CNS Spectr. 2019 Jun 14. doi: 10.1017/S109285291900103). The condition also might run in families. BED often is comorbid with other psychiatric and behavioral disorders, such as depression, substance use, and PTSD, noted Antony Loebel, MD, president and CEO of Sunovion, in a press release. He also said BED often is underrecognized and undertreated.
Meta-analytic reviews show that cognitive-behavioral therapy is considered first-line treatment for BED. However, limited access to such psychological treatments makes the development of medication options such as dasotraline important.
Last year, the agency rejected a new drug application for dasotraline for the treatment of ADHD, citing a need for additional data.
The Food and Drug Administration has accepted the new drug application for dasotraline for the treatment of moderate to severe binge-eating disorder, the drug’s developer, Sunovion, announced July 30.
Dasotraline, a dopamine and norepinephrine reuptake inhibitor, demonstrated significant efficacy in a pair of 12-week, randomized, placebo-controlled studies (SEP360-221 and SEP360-321). The drug also was found to be well tolerated by patients with binge-eating disorder (BED), both in those studies and in a long-term safety study that followed patients for up to a year (SEP360-322).
The medication – characterized by an extended half-life – is to be taken once a day. The most common adverse events reported by patients who took dasotraline include insomnia, dry mouth, decreased appetite, anxiety, nausea, and decreased weight.
BED is more common than any other eating disorder, with an estimated lifetime prevalence among U.S. adults of 1.25% for women and 0.42% for men (CNS Spectr. 2019 Jun 14. doi: 10.1017/S109285291900103). The condition also might run in families. BED often is comorbid with other psychiatric and behavioral disorders, such as depression, substance use, and PTSD, noted Antony Loebel, MD, president and CEO of Sunovion, in a press release. He also said BED often is underrecognized and undertreated.
Meta-analytic reviews show that cognitive-behavioral therapy is considered first-line treatment for BED. However, limited access to such psychological treatments makes the development of medication options such as dasotraline important.
Last year, the agency rejected a new drug application for dasotraline for the treatment of ADHD, citing a need for additional data.
Clues to eczematous cheilitis may lie in the history
NEW YORK – , but patients may be slow to seek help, Bethanee Schlosser, MD, PhD, said at the American Academy of Dermatology summer meeting.

One of the challenges in helping patients with lip problems is that lips are constantly in motion and constantly interacting with the outside world, said Dr. Schlosser, of the department of dermatology, Northwestern University, Chicago. There’s ongoing low-level trauma with phonation, eating, drinking, and general environmental exposure, she said. Eczematous cheilitis will present with scaling and erythema of the vermilion lips, with lower lip involvement often more pronounced than symptoms on the upper lip. Fissuring and erosion are sometimes, but not always, present as well.
In addition to flaking and redness, Dr. Schlosser noted that patients will complain of dry lips, irritation, itching, and sometimes tingling.
Sorting out the etiology of eczematous cheilitis requires a thorough history. “Ask about habits, such as lip licking, picking, or biting,” she said. Recent dental work, braces, or other appliances for alignment or temporomandibular joint problems can introduce both mechanical irritation and potential allergens, and even musical instruments can be culprits, such as when an oboe reed causes an allergic reaction.
Personal hygiene products, cosmetics, gum chewing, and candy consumption can be the irritant culprits, noted Dr. Schlosser. Careful questioning of patients and examination of the products used can provide clues, since dyes and pigments in cosmetics and gum may provoke reactions.
History taking should also include questions about tobacco in all forms, marijuana, and prescription medication, which can cause lip problems. And it’s important to ask about skin disease in general, to determine if symptoms are present in other anatomic locations, and to ask about any family history of skin disease, she said.
Endogenous contributors can include true atopic dermatitis, psoriasis, and nutritional deficiencies. Psoriatic cheilitis can have prominent crusting and exfoliation. In a Brazilian study that evaluated patents with cutaneous psoriasis and age-, race-, and sex-matched controls with no history of skin disease, psoriasis was associated with geographic tongue, with an odds ratio of 5.0 (95% CI 1.5-16.8). Geographic stomatitis can also be seen, said Dr. Schlosser. Tongue fissures were also more common among those with psoriasis cheilitis (OR 2.7, 95% confidence interval, 1.3-5.6) in the same study (Med Oral Patol Oral Cir Bucal. 2009 Aug 1;14[8]:e371-5).
For psoriatic cheilitis, looking beyond the lips can help refine the diagnosis, she noted. There may be intra-oral signs or signs of extra-oral involvement, especially on the scalp, ears, and genitalia. Koebnerization may be difficult to detect on the lips, but may be present elsewhere. A family history of psoriasis may also tip the scales toward this diagnosis.
Exogenous causes of eczematous cheilitis are much more common and can include contact with irritants and allergens, factitial cheilitis, and cheilitis medicamentosa, Dr Schlosser pointed out.
Allergic contact dermatitis can come from local exposure (to cosmetics and other personal care items, for example) or from incidental exposures. Components of saliva can become concentrated when saliva dries outside the oral cavity, so for chronic lip lickers, saliva alone can be sufficiently irritating to provoke a cheilitis, Dr. Schlosser said.
Transfer of an irritant or allergen is also possible from other body sites, as when a nail-chewer develops allergic cheilitis from an ingredient in nail polish. Transfer from products used on other facial areas and the hair is also possible, as is “connubial transfer,” when an allergen is transferred from an intimate partner.
Cutaneous patch tests can be helpful in pinpointing the offending agent, or agents, according to Dr. Schlosser. She cited a study of 91 patients (77% of whom were female) who underwent patch testing for eczematous cheilitis. The researchers determined that 45% of patients had allergic contact cheilitis (Int J Dermatol. 2016 Jul;55[7]:e386-91).
The patch testing revealed that fragrances, balsam of Peru (Myroxylon pereirae resin), preservatives, and even metals such as nickel and gold were common allergens. The findings echo those in another database review that showed fragrances, M. pereirae, and nickel as the top three allergens on patch testing for lip cheilitis.
Dr. Schlosser said that the most common offending sources are lipsticks, makeup, other cosmetic products, and moisturizer, which are responsible for 10% or more of reactions.
Whatever the etiology, the treatment of eczematous cheilitis can be divided conceptually into two phases. During the induction phase, use of a low- to mid-potency topical corticosteroid ointment quiets inflammation. Examples include alclometasone 0.05%, desonide 0.05%, fluticasone 0.005%, or triamcinolone 0.1%. “Ointment formulations are preferred,” said Dr. Schlosser, since they won’t dissolve so easily with lip licking and will adhere well to the surface of the vermilion lip.
Next, a topical calcineurin inhibitor such as tacrolimus 0.1% can be used for maintenance. Other topical medications, especially topical anesthetics, should be used with caution, she said.
For psoriatic cheilitis, induction with 5% salicylic acid ointment can be followed by the topical calcineurin inhibitor phase, said Dr. Schlosser.
Dr. Schlosser disclosed financial relationships with Beiersdorf, Decision Support in Medicine, and UpToDate.
[email protected]
NEW YORK – , but patients may be slow to seek help, Bethanee Schlosser, MD, PhD, said at the American Academy of Dermatology summer meeting.
One of the challenges in helping patients with lip problems is that lips are constantly in motion and constantly interacting with the outside world, said Dr. Schlosser, of the department of dermatology, Northwestern University, Chicago. There’s ongoing low-level trauma with phonation, eating, drinking, and general environmental exposure, she said. Eczematous cheilitis will present with scaling and erythema of the vermilion lips, with lower lip involvement often more pronounced than symptoms on the upper lip. Fissuring and erosion are sometimes, but not always, present as well.
In addition to flaking and redness, Dr. Schlosser noted that patients will complain of dry lips, irritation, itching, and sometimes tingling.
Sorting out the etiology of eczematous cheilitis requires a thorough history. “Ask about habits, such as lip licking, picking, or biting,” she said. Recent dental work, braces, or other appliances for alignment or temporomandibular joint problems can introduce both mechanical irritation and potential allergens, and even musical instruments can be culprits, such as when an oboe reed causes an allergic reaction.
Personal hygiene products, cosmetics, gum chewing, and candy consumption can be the irritant culprits, noted Dr. Schlosser. Careful questioning of patients and examination of the products used can provide clues, since dyes and pigments in cosmetics and gum may provoke reactions.
History taking should also include questions about tobacco in all forms, marijuana, and prescription medication, which can cause lip problems. And it’s important to ask about skin disease in general, to determine if symptoms are present in other anatomic locations, and to ask about any family history of skin disease, she said.
Endogenous contributors can include true atopic dermatitis, psoriasis, and nutritional deficiencies. Psoriatic cheilitis can have prominent crusting and exfoliation. In a Brazilian study that evaluated patents with cutaneous psoriasis and age-, race-, and sex-matched controls with no history of skin disease, psoriasis was associated with geographic tongue, with an odds ratio of 5.0 (95% CI 1.5-16.8). Geographic stomatitis can also be seen, said Dr. Schlosser. Tongue fissures were also more common among those with psoriasis cheilitis (OR 2.7, 95% confidence interval, 1.3-5.6) in the same study (Med Oral Patol Oral Cir Bucal. 2009 Aug 1;14[8]:e371-5).
For psoriatic cheilitis, looking beyond the lips can help refine the diagnosis, she noted. There may be intra-oral signs or signs of extra-oral involvement, especially on the scalp, ears, and genitalia. Koebnerization may be difficult to detect on the lips, but may be present elsewhere. A family history of psoriasis may also tip the scales toward this diagnosis.
Exogenous causes of eczematous cheilitis are much more common and can include contact with irritants and allergens, factitial cheilitis, and cheilitis medicamentosa, Dr Schlosser pointed out.
Allergic contact dermatitis can come from local exposure (to cosmetics and other personal care items, for example) or from incidental exposures. Components of saliva can become concentrated when saliva dries outside the oral cavity, so for chronic lip lickers, saliva alone can be sufficiently irritating to provoke a cheilitis, Dr. Schlosser said.
Transfer of an irritant or allergen is also possible from other body sites, as when a nail-chewer develops allergic cheilitis from an ingredient in nail polish. Transfer from products used on other facial areas and the hair is also possible, as is “connubial transfer,” when an allergen is transferred from an intimate partner.
Cutaneous patch tests can be helpful in pinpointing the offending agent, or agents, according to Dr. Schlosser. She cited a study of 91 patients (77% of whom were female) who underwent patch testing for eczematous cheilitis. The researchers determined that 45% of patients had allergic contact cheilitis (Int J Dermatol. 2016 Jul;55[7]:e386-91).
The patch testing revealed that fragrances, balsam of Peru (Myroxylon pereirae resin), preservatives, and even metals such as nickel and gold were common allergens. The findings echo those in another database review that showed fragrances, M. pereirae, and nickel as the top three allergens on patch testing for lip cheilitis.
Dr. Schlosser said that the most common offending sources are lipsticks, makeup, other cosmetic products, and moisturizer, which are responsible for 10% or more of reactions.
Whatever the etiology, the treatment of eczematous cheilitis can be divided conceptually into two phases. During the induction phase, use of a low- to mid-potency topical corticosteroid ointment quiets inflammation. Examples include alclometasone 0.05%, desonide 0.05%, fluticasone 0.005%, or triamcinolone 0.1%. “Ointment formulations are preferred,” said Dr. Schlosser, since they won’t dissolve so easily with lip licking and will adhere well to the surface of the vermilion lip.
Next, a topical calcineurin inhibitor such as tacrolimus 0.1% can be used for maintenance. Other topical medications, especially topical anesthetics, should be used with caution, she said.
For psoriatic cheilitis, induction with 5% salicylic acid ointment can be followed by the topical calcineurin inhibitor phase, said Dr. Schlosser.
Dr. Schlosser disclosed financial relationships with Beiersdorf, Decision Support in Medicine, and UpToDate.
[email protected]
NEW YORK – , but patients may be slow to seek help, Bethanee Schlosser, MD, PhD, said at the American Academy of Dermatology summer meeting.
One of the challenges in helping patients with lip problems is that lips are constantly in motion and constantly interacting with the outside world, said Dr. Schlosser, of the department of dermatology, Northwestern University, Chicago. There’s ongoing low-level trauma with phonation, eating, drinking, and general environmental exposure, she said. Eczematous cheilitis will present with scaling and erythema of the vermilion lips, with lower lip involvement often more pronounced than symptoms on the upper lip. Fissuring and erosion are sometimes, but not always, present as well.
In addition to flaking and redness, Dr. Schlosser noted that patients will complain of dry lips, irritation, itching, and sometimes tingling.
Sorting out the etiology of eczematous cheilitis requires a thorough history. “Ask about habits, such as lip licking, picking, or biting,” she said. Recent dental work, braces, or other appliances for alignment or temporomandibular joint problems can introduce both mechanical irritation and potential allergens, and even musical instruments can be culprits, such as when an oboe reed causes an allergic reaction.
Personal hygiene products, cosmetics, gum chewing, and candy consumption can be the irritant culprits, noted Dr. Schlosser. Careful questioning of patients and examination of the products used can provide clues, since dyes and pigments in cosmetics and gum may provoke reactions.
History taking should also include questions about tobacco in all forms, marijuana, and prescription medication, which can cause lip problems. And it’s important to ask about skin disease in general, to determine if symptoms are present in other anatomic locations, and to ask about any family history of skin disease, she said.
Endogenous contributors can include true atopic dermatitis, psoriasis, and nutritional deficiencies. Psoriatic cheilitis can have prominent crusting and exfoliation. In a Brazilian study that evaluated patents with cutaneous psoriasis and age-, race-, and sex-matched controls with no history of skin disease, psoriasis was associated with geographic tongue, with an odds ratio of 5.0 (95% CI 1.5-16.8). Geographic stomatitis can also be seen, said Dr. Schlosser. Tongue fissures were also more common among those with psoriasis cheilitis (OR 2.7, 95% confidence interval, 1.3-5.6) in the same study (Med Oral Patol Oral Cir Bucal. 2009 Aug 1;14[8]:e371-5).
For psoriatic cheilitis, looking beyond the lips can help refine the diagnosis, she noted. There may be intra-oral signs or signs of extra-oral involvement, especially on the scalp, ears, and genitalia. Koebnerization may be difficult to detect on the lips, but may be present elsewhere. A family history of psoriasis may also tip the scales toward this diagnosis.
Exogenous causes of eczematous cheilitis are much more common and can include contact with irritants and allergens, factitial cheilitis, and cheilitis medicamentosa, Dr Schlosser pointed out.
Allergic contact dermatitis can come from local exposure (to cosmetics and other personal care items, for example) or from incidental exposures. Components of saliva can become concentrated when saliva dries outside the oral cavity, so for chronic lip lickers, saliva alone can be sufficiently irritating to provoke a cheilitis, Dr. Schlosser said.
Transfer of an irritant or allergen is also possible from other body sites, as when a nail-chewer develops allergic cheilitis from an ingredient in nail polish. Transfer from products used on other facial areas and the hair is also possible, as is “connubial transfer,” when an allergen is transferred from an intimate partner.
Cutaneous patch tests can be helpful in pinpointing the offending agent, or agents, according to Dr. Schlosser. She cited a study of 91 patients (77% of whom were female) who underwent patch testing for eczematous cheilitis. The researchers determined that 45% of patients had allergic contact cheilitis (Int J Dermatol. 2016 Jul;55[7]:e386-91).
The patch testing revealed that fragrances, balsam of Peru (Myroxylon pereirae resin), preservatives, and even metals such as nickel and gold were common allergens. The findings echo those in another database review that showed fragrances, M. pereirae, and nickel as the top three allergens on patch testing for lip cheilitis.
Dr. Schlosser said that the most common offending sources are lipsticks, makeup, other cosmetic products, and moisturizer, which are responsible for 10% or more of reactions.
Whatever the etiology, the treatment of eczematous cheilitis can be divided conceptually into two phases. During the induction phase, use of a low- to mid-potency topical corticosteroid ointment quiets inflammation. Examples include alclometasone 0.05%, desonide 0.05%, fluticasone 0.005%, or triamcinolone 0.1%. “Ointment formulations are preferred,” said Dr. Schlosser, since they won’t dissolve so easily with lip licking and will adhere well to the surface of the vermilion lip.
Next, a topical calcineurin inhibitor such as tacrolimus 0.1% can be used for maintenance. Other topical medications, especially topical anesthetics, should be used with caution, she said.
For psoriatic cheilitis, induction with 5% salicylic acid ointment can be followed by the topical calcineurin inhibitor phase, said Dr. Schlosser.
Dr. Schlosser disclosed financial relationships with Beiersdorf, Decision Support in Medicine, and UpToDate.
[email protected]
EXPERT ANALYSIS FROM SUMMER AAD 2019
What’s the best way to sort out red nail discoloration?
NEW YORK – Nail discoloration, in all its variety, has a wide differential. And while that differential narrows when a patient presents with concerns about nails with red discoloration, there’s still a long list of diagnoses to consider.
During a nail-focused session at the American Academy of Dermatology summer meeting, Shari Lipner, MD, PhD, took attendees through a presentation-based approach that gets to the root etiology of erythronychia and guides diagnosis and treatment options.
“However, regardless of the etiology, erythronychia shares a common pathogenesis,” said Dr. Lipner, a dermatologist at New York–Presbyterian Hospital and Weil Cornell Medicine. The process begins in the distal nail matrix, resulting in a thin long strip of ventral nail becoming discolored, with the nail bed filling in the concavity. The engorged nail bed also makes the affected nail unit prone to splinter hemorrhages, and the thinned, transparent nail makes the erythema more visible, she explained.
Polydactylous longitudinal erythronychia
For erythronychia affecting several nails, onychotillomania is among the possible causes. This condition “often goes hand in hand with onychophagia,” and trichotillomania, skin-picking, or other self-mutilating disorders may also be present, she said. In both the adult and pediatric population, onychotillomania can accompany psychiatric disorders, including depression and obsessive-compulsive disorder, and may be associated with suicidal ideation, she added.
When onychotillomania is the cause, erythronychia may be accompanied by paronychia, and patients will often have a shortened nail bed and an atrophic nail plate. Dorsal pterygium may also be present.
Dermoscopy can provide some clues that onychotillomania is the culprit, said Dr. Lipner, citing a study that looked at dermoscopic images of 36 cases, which found scales in 94%, absence of the nail plate in 83%, and characteristic wavy lines in 69% (J Am Acad Dermatol. 2018 Oct;79[4]:702-5). Other frequent dermoscopy findings included hemorrhages (64%), crusts (61%), nail bed pigmentation (47%), and speckled dots (39%).
Lichen planus can also affect the nails, with erythronychia among its manifestations, she noted. Though lichen planus is thought of as a disease of middle or older age, usually affecting those aged 50-70 years, “15% of those affected are less than 20 years old,” she said.
The erythronychia of lichen planus is often accompanied by longitudinal riding, splitting, and atrophy of the nail plate, she said. Pterygium can also be present, representing a scar in the nail matrix. Dermatopathology will reveal a patchy, bandlike lichenoid infiltrate, with variable sawtooth hypergranulosis and hyperplasia.
“There’s not much evidence about how to treat lichen planus of the nails,” noted Dr. Lipner. Options can include intralesional corticosteroid injections at the nail matrix, topical corticosteroids, oral methotrexate, and retinoids.
Darier disease, an inherited condition caused by mutations in ATP2A2, has both skin and nail manifestations. Characteristic skin signs include hyperkeratotic papules, cobblestone papules, and palmar pits, she said. When nails are affected – as they are in up to 95% of Darier disease patients – they can have a characteristic “candy cane” appearance, with bands of longitudinal erythronychia alternating with normal-colored nail. The nails can also have V-shaped notching, she added.
Patients with systemic lupus erythematosus can also have longitudinal erythronychia; here, dermoscopy will show the characteristic prominent capillary loops in the proximal nail folds, she said.
Foreign substances such as nail polish and dyes, when they’re the source of erythronychia in one or several nails, can usually be wiped off with alcohol or acetone; also, “the proximal margin of discoloration will follow the same pattern as the nail fold,” said Dr. Lipner.
Localized longitudinal erythronychia
When the red discoloration is limited to a single nail, the differential shifts, said Dr. Lipner. With a hematoma, there’s often a history of trauma, and dermoscopy will show characteristic globules and streaks.
The first clue that erythronychia caused by onychopapilloma can be seen at the lunula: There will often be a comet- or pencil-shaped point to the discoloration in that region. But, she said, “the key is to do dermoscopy at the free edge of the nail plate. In 100 percent of cases, there will be a subungual hyperkeratotic papule” that will solidify the diagnosis.
Histopathology of onychopapillomas – a relatively recently recognized entity – will show nail bed and distal matrix acanthosis, with the distal nail bed showing matrix metaplasia, Dr. Lipner said.
Glomus tumors arise from cells of the glomus body, a specialized vascular apparatus that is involved in temperature regulation. “These structures are abundant in the digits and the subungual region,” said Dr. Lipner, which means that glomus tumors – a benign lesion – may develop subungually. Glomus tumors can be idiopathic, but she added, “think neurofibromatosis type 1 if you see multiple glomus tumors.”
Glomus tumors will present with longitudinal erythronychia, with a distal split of the nail sometimes accompanying the discoloration. A round bluish to gray nodule can also be seen. A thorough history and exam can help solidify the diagnosis, said Dr. Lipner; there’s a characteristic triad of point tenderness with the application of pressure to the nail, pain, and cold hypersensitivity.
Classically, the point tenderness is assessed by Love’s test, which elicits severe pain when the subungual tumor is pressed with a small object like the end of paper clip or a ballpoint pen. Application of a tourniquet to the arm after elevation, termed Hildreth’s test, should alleviate subungual glomus tumor pain, and release of the tourniquet causes an abrupt and marked recurrence of pain. Immersing the patient’s affected hand in cold water also increases glomus tumor pain.
“Imaging can be quite helpful” to confirm a glomus tumor diagnosis, said Dr. Lipner. “X-ray is cheap, and you can see erosions in 50% of patients.” However, she added, doppler ultrasound and magnetic resonance imaging are more sensitive, detecting tumors as small as 2 mm.
“Biopsy with histopathology is the gold standard in making the diagnosis” of glomus tumor, though, she noted. Pathologic examination will show monomorphic cells with small caliber vascular channels.
Malignancy is actually uncommon with erythronychia, though it’s always in the differential diagnosis, she said.
In one case series examining subungual squamous cell carcinomas, common findings included onycholysis and localized hyperkeratosis, along with longitudinal erythronychia. “This may be subtle; splinter hemorrhages can also be present,” noted Dr. Lipner, adding, “If there are any symptoms, or an evolving band, a biopsy is indicated.”
“Even with erythronychia, the presentation can be quite variable,” she said. Nail unit melanoma can count erythronychia among the presenting signs. Clues that should raise suspicion for melanoma can include a wide band and prominent onycholysis, especially distal onycholysis and splintering. Again, she said, “biopsy with histopathology is the gold standard in making the diagnosis”; any symptoms or evidence of an evolving band should trigger a biopsy.
Dr. Lipner had no conflicts of interest relevant to her presentation.
SOURCE: Lipner S. Summer AAD 2019, Presentation F035.
NEW YORK – Nail discoloration, in all its variety, has a wide differential. And while that differential narrows when a patient presents with concerns about nails with red discoloration, there’s still a long list of diagnoses to consider.
During a nail-focused session at the American Academy of Dermatology summer meeting, Shari Lipner, MD, PhD, took attendees through a presentation-based approach that gets to the root etiology of erythronychia and guides diagnosis and treatment options.
“However, regardless of the etiology, erythronychia shares a common pathogenesis,” said Dr. Lipner, a dermatologist at New York–Presbyterian Hospital and Weil Cornell Medicine. The process begins in the distal nail matrix, resulting in a thin long strip of ventral nail becoming discolored, with the nail bed filling in the concavity. The engorged nail bed also makes the affected nail unit prone to splinter hemorrhages, and the thinned, transparent nail makes the erythema more visible, she explained.
Polydactylous longitudinal erythronychia
For erythronychia affecting several nails, onychotillomania is among the possible causes. This condition “often goes hand in hand with onychophagia,” and trichotillomania, skin-picking, or other self-mutilating disorders may also be present, she said. In both the adult and pediatric population, onychotillomania can accompany psychiatric disorders, including depression and obsessive-compulsive disorder, and may be associated with suicidal ideation, she added.
When onychotillomania is the cause, erythronychia may be accompanied by paronychia, and patients will often have a shortened nail bed and an atrophic nail plate. Dorsal pterygium may also be present.
Dermoscopy can provide some clues that onychotillomania is the culprit, said Dr. Lipner, citing a study that looked at dermoscopic images of 36 cases, which found scales in 94%, absence of the nail plate in 83%, and characteristic wavy lines in 69% (J Am Acad Dermatol. 2018 Oct;79[4]:702-5). Other frequent dermoscopy findings included hemorrhages (64%), crusts (61%), nail bed pigmentation (47%), and speckled dots (39%).
Lichen planus can also affect the nails, with erythronychia among its manifestations, she noted. Though lichen planus is thought of as a disease of middle or older age, usually affecting those aged 50-70 years, “15% of those affected are less than 20 years old,” she said.
The erythronychia of lichen planus is often accompanied by longitudinal riding, splitting, and atrophy of the nail plate, she said. Pterygium can also be present, representing a scar in the nail matrix. Dermatopathology will reveal a patchy, bandlike lichenoid infiltrate, with variable sawtooth hypergranulosis and hyperplasia.
“There’s not much evidence about how to treat lichen planus of the nails,” noted Dr. Lipner. Options can include intralesional corticosteroid injections at the nail matrix, topical corticosteroids, oral methotrexate, and retinoids.
Darier disease, an inherited condition caused by mutations in ATP2A2, has both skin and nail manifestations. Characteristic skin signs include hyperkeratotic papules, cobblestone papules, and palmar pits, she said. When nails are affected – as they are in up to 95% of Darier disease patients – they can have a characteristic “candy cane” appearance, with bands of longitudinal erythronychia alternating with normal-colored nail. The nails can also have V-shaped notching, she added.
Patients with systemic lupus erythematosus can also have longitudinal erythronychia; here, dermoscopy will show the characteristic prominent capillary loops in the proximal nail folds, she said.
Foreign substances such as nail polish and dyes, when they’re the source of erythronychia in one or several nails, can usually be wiped off with alcohol or acetone; also, “the proximal margin of discoloration will follow the same pattern as the nail fold,” said Dr. Lipner.
Localized longitudinal erythronychia
When the red discoloration is limited to a single nail, the differential shifts, said Dr. Lipner. With a hematoma, there’s often a history of trauma, and dermoscopy will show characteristic globules and streaks.
The first clue that erythronychia caused by onychopapilloma can be seen at the lunula: There will often be a comet- or pencil-shaped point to the discoloration in that region. But, she said, “the key is to do dermoscopy at the free edge of the nail plate. In 100 percent of cases, there will be a subungual hyperkeratotic papule” that will solidify the diagnosis.
Histopathology of onychopapillomas – a relatively recently recognized entity – will show nail bed and distal matrix acanthosis, with the distal nail bed showing matrix metaplasia, Dr. Lipner said.
Glomus tumors arise from cells of the glomus body, a specialized vascular apparatus that is involved in temperature regulation. “These structures are abundant in the digits and the subungual region,” said Dr. Lipner, which means that glomus tumors – a benign lesion – may develop subungually. Glomus tumors can be idiopathic, but she added, “think neurofibromatosis type 1 if you see multiple glomus tumors.”
Glomus tumors will present with longitudinal erythronychia, with a distal split of the nail sometimes accompanying the discoloration. A round bluish to gray nodule can also be seen. A thorough history and exam can help solidify the diagnosis, said Dr. Lipner; there’s a characteristic triad of point tenderness with the application of pressure to the nail, pain, and cold hypersensitivity.
Classically, the point tenderness is assessed by Love’s test, which elicits severe pain when the subungual tumor is pressed with a small object like the end of paper clip or a ballpoint pen. Application of a tourniquet to the arm after elevation, termed Hildreth’s test, should alleviate subungual glomus tumor pain, and release of the tourniquet causes an abrupt and marked recurrence of pain. Immersing the patient’s affected hand in cold water also increases glomus tumor pain.
“Imaging can be quite helpful” to confirm a glomus tumor diagnosis, said Dr. Lipner. “X-ray is cheap, and you can see erosions in 50% of patients.” However, she added, doppler ultrasound and magnetic resonance imaging are more sensitive, detecting tumors as small as 2 mm.
“Biopsy with histopathology is the gold standard in making the diagnosis” of glomus tumor, though, she noted. Pathologic examination will show monomorphic cells with small caliber vascular channels.
Malignancy is actually uncommon with erythronychia, though it’s always in the differential diagnosis, she said.
In one case series examining subungual squamous cell carcinomas, common findings included onycholysis and localized hyperkeratosis, along with longitudinal erythronychia. “This may be subtle; splinter hemorrhages can also be present,” noted Dr. Lipner, adding, “If there are any symptoms, or an evolving band, a biopsy is indicated.”
“Even with erythronychia, the presentation can be quite variable,” she said. Nail unit melanoma can count erythronychia among the presenting signs. Clues that should raise suspicion for melanoma can include a wide band and prominent onycholysis, especially distal onycholysis and splintering. Again, she said, “biopsy with histopathology is the gold standard in making the diagnosis”; any symptoms or evidence of an evolving band should trigger a biopsy.
Dr. Lipner had no conflicts of interest relevant to her presentation.
SOURCE: Lipner S. Summer AAD 2019, Presentation F035.
NEW YORK – Nail discoloration, in all its variety, has a wide differential. And while that differential narrows when a patient presents with concerns about nails with red discoloration, there’s still a long list of diagnoses to consider.
During a nail-focused session at the American Academy of Dermatology summer meeting, Shari Lipner, MD, PhD, took attendees through a presentation-based approach that gets to the root etiology of erythronychia and guides diagnosis and treatment options.
“However, regardless of the etiology, erythronychia shares a common pathogenesis,” said Dr. Lipner, a dermatologist at New York–Presbyterian Hospital and Weil Cornell Medicine. The process begins in the distal nail matrix, resulting in a thin long strip of ventral nail becoming discolored, with the nail bed filling in the concavity. The engorged nail bed also makes the affected nail unit prone to splinter hemorrhages, and the thinned, transparent nail makes the erythema more visible, she explained.
Polydactylous longitudinal erythronychia
For erythronychia affecting several nails, onychotillomania is among the possible causes. This condition “often goes hand in hand with onychophagia,” and trichotillomania, skin-picking, or other self-mutilating disorders may also be present, she said. In both the adult and pediatric population, onychotillomania can accompany psychiatric disorders, including depression and obsessive-compulsive disorder, and may be associated with suicidal ideation, she added.
When onychotillomania is the cause, erythronychia may be accompanied by paronychia, and patients will often have a shortened nail bed and an atrophic nail plate. Dorsal pterygium may also be present.
Dermoscopy can provide some clues that onychotillomania is the culprit, said Dr. Lipner, citing a study that looked at dermoscopic images of 36 cases, which found scales in 94%, absence of the nail plate in 83%, and characteristic wavy lines in 69% (J Am Acad Dermatol. 2018 Oct;79[4]:702-5). Other frequent dermoscopy findings included hemorrhages (64%), crusts (61%), nail bed pigmentation (47%), and speckled dots (39%).
Lichen planus can also affect the nails, with erythronychia among its manifestations, she noted. Though lichen planus is thought of as a disease of middle or older age, usually affecting those aged 50-70 years, “15% of those affected are less than 20 years old,” she said.
The erythronychia of lichen planus is often accompanied by longitudinal riding, splitting, and atrophy of the nail plate, she said. Pterygium can also be present, representing a scar in the nail matrix. Dermatopathology will reveal a patchy, bandlike lichenoid infiltrate, with variable sawtooth hypergranulosis and hyperplasia.
“There’s not much evidence about how to treat lichen planus of the nails,” noted Dr. Lipner. Options can include intralesional corticosteroid injections at the nail matrix, topical corticosteroids, oral methotrexate, and retinoids.
Darier disease, an inherited condition caused by mutations in ATP2A2, has both skin and nail manifestations. Characteristic skin signs include hyperkeratotic papules, cobblestone papules, and palmar pits, she said. When nails are affected – as they are in up to 95% of Darier disease patients – they can have a characteristic “candy cane” appearance, with bands of longitudinal erythronychia alternating with normal-colored nail. The nails can also have V-shaped notching, she added.
Patients with systemic lupus erythematosus can also have longitudinal erythronychia; here, dermoscopy will show the characteristic prominent capillary loops in the proximal nail folds, she said.
Foreign substances such as nail polish and dyes, when they’re the source of erythronychia in one or several nails, can usually be wiped off with alcohol or acetone; also, “the proximal margin of discoloration will follow the same pattern as the nail fold,” said Dr. Lipner.
Localized longitudinal erythronychia
When the red discoloration is limited to a single nail, the differential shifts, said Dr. Lipner. With a hematoma, there’s often a history of trauma, and dermoscopy will show characteristic globules and streaks.
The first clue that erythronychia caused by onychopapilloma can be seen at the lunula: There will often be a comet- or pencil-shaped point to the discoloration in that region. But, she said, “the key is to do dermoscopy at the free edge of the nail plate. In 100 percent of cases, there will be a subungual hyperkeratotic papule” that will solidify the diagnosis.
Histopathology of onychopapillomas – a relatively recently recognized entity – will show nail bed and distal matrix acanthosis, with the distal nail bed showing matrix metaplasia, Dr. Lipner said.
Glomus tumors arise from cells of the glomus body, a specialized vascular apparatus that is involved in temperature regulation. “These structures are abundant in the digits and the subungual region,” said Dr. Lipner, which means that glomus tumors – a benign lesion – may develop subungually. Glomus tumors can be idiopathic, but she added, “think neurofibromatosis type 1 if you see multiple glomus tumors.”
Glomus tumors will present with longitudinal erythronychia, with a distal split of the nail sometimes accompanying the discoloration. A round bluish to gray nodule can also be seen. A thorough history and exam can help solidify the diagnosis, said Dr. Lipner; there’s a characteristic triad of point tenderness with the application of pressure to the nail, pain, and cold hypersensitivity.
Classically, the point tenderness is assessed by Love’s test, which elicits severe pain when the subungual tumor is pressed with a small object like the end of paper clip or a ballpoint pen. Application of a tourniquet to the arm after elevation, termed Hildreth’s test, should alleviate subungual glomus tumor pain, and release of the tourniquet causes an abrupt and marked recurrence of pain. Immersing the patient’s affected hand in cold water also increases glomus tumor pain.
“Imaging can be quite helpful” to confirm a glomus tumor diagnosis, said Dr. Lipner. “X-ray is cheap, and you can see erosions in 50% of patients.” However, she added, doppler ultrasound and magnetic resonance imaging are more sensitive, detecting tumors as small as 2 mm.
“Biopsy with histopathology is the gold standard in making the diagnosis” of glomus tumor, though, she noted. Pathologic examination will show monomorphic cells with small caliber vascular channels.
Malignancy is actually uncommon with erythronychia, though it’s always in the differential diagnosis, she said.
In one case series examining subungual squamous cell carcinomas, common findings included onycholysis and localized hyperkeratosis, along with longitudinal erythronychia. “This may be subtle; splinter hemorrhages can also be present,” noted Dr. Lipner, adding, “If there are any symptoms, or an evolving band, a biopsy is indicated.”
“Even with erythronychia, the presentation can be quite variable,” she said. Nail unit melanoma can count erythronychia among the presenting signs. Clues that should raise suspicion for melanoma can include a wide band and prominent onycholysis, especially distal onycholysis and splintering. Again, she said, “biopsy with histopathology is the gold standard in making the diagnosis”; any symptoms or evidence of an evolving band should trigger a biopsy.
Dr. Lipner had no conflicts of interest relevant to her presentation.
SOURCE: Lipner S. Summer AAD 2019, Presentation F035.
EXPERT ANALYSIS FROM SUMMER AAD 2019