User login
FDA approves avapritinib for adults with GIST with PDGFRA mutation
The Food and Drug Administration has approved avapritinib (Ayvakit) for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GIST) with a platelet-derived growth factor receptor–alpha (PDGFRA) exon 18 mutation.

Approval was based on results from a clinical trial of 43 patients with PDGFRA exon 18 mutations, including 38 patients with a PDGFRA D842V mutation, who received 300 mg avapritinib once daily, the FDA said in a statement.
The overall response rate was 84% (7% with complete response, 77% with partial response); the response rate in patients with a D842V mutation was 89% (8% with complete response, 82% with partial response). Median response duration was not reached, but 61% of patients had a response lasting longer than 6 months.
The most common adverse events associated with avapritinib include edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. The drug also can cause intracranial hemorrhage and have effects on the central nervous system.
“GIST harboring a PDGFRA exon 18 mutation do not respond to standard therapies for GIST. However, today’s approval provides patients with the first drug specifically approved for GIST harboring this mutation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research, said in the statement.
The Food and Drug Administration has approved avapritinib (Ayvakit) for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GIST) with a platelet-derived growth factor receptor–alpha (PDGFRA) exon 18 mutation.
Approval was based on results from a clinical trial of 43 patients with PDGFRA exon 18 mutations, including 38 patients with a PDGFRA D842V mutation, who received 300 mg avapritinib once daily, the FDA said in a statement.
The overall response rate was 84% (7% with complete response, 77% with partial response); the response rate in patients with a D842V mutation was 89% (8% with complete response, 82% with partial response). Median response duration was not reached, but 61% of patients had a response lasting longer than 6 months.
The most common adverse events associated with avapritinib include edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. The drug also can cause intracranial hemorrhage and have effects on the central nervous system.
“GIST harboring a PDGFRA exon 18 mutation do not respond to standard therapies for GIST. However, today’s approval provides patients with the first drug specifically approved for GIST harboring this mutation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research, said in the statement.
The Food and Drug Administration has approved avapritinib (Ayvakit) for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GIST) with a platelet-derived growth factor receptor–alpha (PDGFRA) exon 18 mutation.
Approval was based on results from a clinical trial of 43 patients with PDGFRA exon 18 mutations, including 38 patients with a PDGFRA D842V mutation, who received 300 mg avapritinib once daily, the FDA said in a statement.
The overall response rate was 84% (7% with complete response, 77% with partial response); the response rate in patients with a D842V mutation was 89% (8% with complete response, 82% with partial response). Median response duration was not reached, but 61% of patients had a response lasting longer than 6 months.
The most common adverse events associated with avapritinib include edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. The drug also can cause intracranial hemorrhage and have effects on the central nervous system.
“GIST harboring a PDGFRA exon 18 mutation do not respond to standard therapies for GIST. However, today’s approval provides patients with the first drug specifically approved for GIST harboring this mutation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research, said in the statement.
FUEL trial: Post-Fontan udenafil shows mixed results
PHILADELPHIA – In adolescents who have had a Fontan procedure for congenital heart disease, a randomized trial of the phosphodiesterase type 5 inhibitor udenafil showed that it achieved improved exercise performance but did not lead to significant improvement in oxygen levels or myocardial performance.
That’s according to results of the Pediatric Heart Network’s Fontan Udenafil Exercise Longitudinal Trial (FUEL) presented at the American Heart Association scientific sessions. “Treatment with udenafil was not associated with a statistically significant improvement in oxygen consumption at peak exercise, but it was associated with statistically significant improvements in exercise performance at the ventilatory anaerobic threshold,” said David J. Goldberg, MD, of Children’s Hospital of Philadelphia in reporting the FUEL results. The results were published simultaneously in Circulation (2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044352).
“This is the first large clinical trial to show improvement in measures of clinically relevant exercise performance in those with single-ventricle heart disease after Fontan palliation,” he said.
FUEL enrolled 400 male and female adolescents with a single functional ventricle after Fontan surgical palliation. In these patients, pulmonary vascular resistance (PVR) is critical for the efficient flow of blood through the lungs without the benefit of a ventricular pump. “While this circulation is typically stable through childhood, cardiovascular efficiency deteriorates over time, associated with a decline in exercise performance and the accrual of Fontan-associated morbidities,” Dr. Goldberg said. “Given the importance of pulmonary vascular resistance, modulators of PVR make sense as potential therapies.”
FUEL evaluated the effect of udenafil 87.5 mg twice daily versus placebo in post-Fontan patients who’d been on anticoagulation or antiplatelet therapy. The treatment group had a higher percentage of female patients (44% vs. 36% on placebo), but all other baseline characteristics were similar between the two groups.
While the trial found the drug was well tolerated and safe, with side effects typical of PDE5 inhibitors, it did not lead to changes in myocardial performance index, reactive hyperemia index, or log brain natriuretic peptide, Dr. Goldberg said.
At 6 months, both groups showed a decline in exercise data, “as expected,” Dr. Goldberg said. “But that decline was attenuated in the group receiving udenafil,” he said, with peak oxygen consumption declining an average of 0.23 and 0.89 mL/kg per minute in the treatment and placebo groups, respectively (P = 0.092).
Total oxygen consumption, however, actually improved in the udenafil group and declined in the placebo group, 44 mL/min on average versus –3.7 mL/min (P = 0.071).
“There was no significant difference in the change in peak heart rate or the change in peak oxygen saturation between the groups,” Dr. Goldberg said. But three measures at the ventilatory aerobic threshold (VAT) – oxygen consumption, work rate, and ventilation/carbon dioxide output – all showed statistically significant improvement in exercise performance.
“This has important clinical implications,” Dr. Goldberg said of the study findings. “Our study extends recent findings in highlighting the importance of submaximal exercise in the understanding of Fontan physiology. And unlike peak oxygen consumption, submaximal exercise is not constrained by the physiologic ceiling of central venous pressure inherent in exercise physiology after Fontan palliation.”
Maximum oxygen consumption at VAT is likely a more relevant measure after Fontan palliation than is central venous pressure, discussant Craig A. Sable, MD, a pediatric cardiologist in Potomac, Md., noted in his comments. “This is because VAT occurs at about 70% of maximum VO2 [oxygen consumption] in Fontan as opposed to 55% in two-ventricle physiology,” Dr. Sable said.
In adults with congenital heart disease, maximal VO2 of 45%-50% of predicted levels portends increased risk of heart failure and death. “Therefore, a medication that addresses the central deficiencies of Fontan physiology and results in improved exercise performance may allow for a longer period of symptom-free survival,” he said.
In an invited commentary in Circulation (2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044512), Marc Gewillig, MD, and Alexander van de Bruaene, MD, of University Hospitals Leuven (Belgium) said that the findings of FUEL and other trials of pulmonary vasodilators after Fontan leave “open for debate” whether the treatment effects of a 3%-5% improvement in oxygen consumption is clinically meaningful for adolescents. “For failing Fontan patients (not studied in FUEL), these improvements are minimal but maybe relevant,” the commentators wrote. But the studies do not resolve whether that’s enough to prevent further decline.
Dr. Goldberg disclosed receiving research grants from trial sponsor Mezzion Pharmaceuticals and the National Heart Lung and Blood Institute. Dr. Sable, Dr. Gewillig, and Dr. van de Bruaene have no financial relationships to disclose.
SOURCE: Goldberg D. AHA 2019, Late Breaking Science Session 5.
PHILADELPHIA – In adolescents who have had a Fontan procedure for congenital heart disease, a randomized trial of the phosphodiesterase type 5 inhibitor udenafil showed that it achieved improved exercise performance but did not lead to significant improvement in oxygen levels or myocardial performance.
That’s according to results of the Pediatric Heart Network’s Fontan Udenafil Exercise Longitudinal Trial (FUEL) presented at the American Heart Association scientific sessions. “Treatment with udenafil was not associated with a statistically significant improvement in oxygen consumption at peak exercise, but it was associated with statistically significant improvements in exercise performance at the ventilatory anaerobic threshold,” said David J. Goldberg, MD, of Children’s Hospital of Philadelphia in reporting the FUEL results. The results were published simultaneously in Circulation (2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044352).
“This is the first large clinical trial to show improvement in measures of clinically relevant exercise performance in those with single-ventricle heart disease after Fontan palliation,” he said.
FUEL enrolled 400 male and female adolescents with a single functional ventricle after Fontan surgical palliation. In these patients, pulmonary vascular resistance (PVR) is critical for the efficient flow of blood through the lungs without the benefit of a ventricular pump. “While this circulation is typically stable through childhood, cardiovascular efficiency deteriorates over time, associated with a decline in exercise performance and the accrual of Fontan-associated morbidities,” Dr. Goldberg said. “Given the importance of pulmonary vascular resistance, modulators of PVR make sense as potential therapies.”
FUEL evaluated the effect of udenafil 87.5 mg twice daily versus placebo in post-Fontan patients who’d been on anticoagulation or antiplatelet therapy. The treatment group had a higher percentage of female patients (44% vs. 36% on placebo), but all other baseline characteristics were similar between the two groups.
While the trial found the drug was well tolerated and safe, with side effects typical of PDE5 inhibitors, it did not lead to changes in myocardial performance index, reactive hyperemia index, or log brain natriuretic peptide, Dr. Goldberg said.
At 6 months, both groups showed a decline in exercise data, “as expected,” Dr. Goldberg said. “But that decline was attenuated in the group receiving udenafil,” he said, with peak oxygen consumption declining an average of 0.23 and 0.89 mL/kg per minute in the treatment and placebo groups, respectively (P = 0.092).
Total oxygen consumption, however, actually improved in the udenafil group and declined in the placebo group, 44 mL/min on average versus –3.7 mL/min (P = 0.071).
“There was no significant difference in the change in peak heart rate or the change in peak oxygen saturation between the groups,” Dr. Goldberg said. But three measures at the ventilatory aerobic threshold (VAT) – oxygen consumption, work rate, and ventilation/carbon dioxide output – all showed statistically significant improvement in exercise performance.
“This has important clinical implications,” Dr. Goldberg said of the study findings. “Our study extends recent findings in highlighting the importance of submaximal exercise in the understanding of Fontan physiology. And unlike peak oxygen consumption, submaximal exercise is not constrained by the physiologic ceiling of central venous pressure inherent in exercise physiology after Fontan palliation.”
Maximum oxygen consumption at VAT is likely a more relevant measure after Fontan palliation than is central venous pressure, discussant Craig A. Sable, MD, a pediatric cardiologist in Potomac, Md., noted in his comments. “This is because VAT occurs at about 70% of maximum VO2 [oxygen consumption] in Fontan as opposed to 55% in two-ventricle physiology,” Dr. Sable said.
In adults with congenital heart disease, maximal VO2 of 45%-50% of predicted levels portends increased risk of heart failure and death. “Therefore, a medication that addresses the central deficiencies of Fontan physiology and results in improved exercise performance may allow for a longer period of symptom-free survival,” he said.
In an invited commentary in Circulation (2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044512), Marc Gewillig, MD, and Alexander van de Bruaene, MD, of University Hospitals Leuven (Belgium) said that the findings of FUEL and other trials of pulmonary vasodilators after Fontan leave “open for debate” whether the treatment effects of a 3%-5% improvement in oxygen consumption is clinically meaningful for adolescents. “For failing Fontan patients (not studied in FUEL), these improvements are minimal but maybe relevant,” the commentators wrote. But the studies do not resolve whether that’s enough to prevent further decline.
Dr. Goldberg disclosed receiving research grants from trial sponsor Mezzion Pharmaceuticals and the National Heart Lung and Blood Institute. Dr. Sable, Dr. Gewillig, and Dr. van de Bruaene have no financial relationships to disclose.
SOURCE: Goldberg D. AHA 2019, Late Breaking Science Session 5.
PHILADELPHIA – In adolescents who have had a Fontan procedure for congenital heart disease, a randomized trial of the phosphodiesterase type 5 inhibitor udenafil showed that it achieved improved exercise performance but did not lead to significant improvement in oxygen levels or myocardial performance.
That’s according to results of the Pediatric Heart Network’s Fontan Udenafil Exercise Longitudinal Trial (FUEL) presented at the American Heart Association scientific sessions. “Treatment with udenafil was not associated with a statistically significant improvement in oxygen consumption at peak exercise, but it was associated with statistically significant improvements in exercise performance at the ventilatory anaerobic threshold,” said David J. Goldberg, MD, of Children’s Hospital of Philadelphia in reporting the FUEL results. The results were published simultaneously in Circulation (2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044352).
“This is the first large clinical trial to show improvement in measures of clinically relevant exercise performance in those with single-ventricle heart disease after Fontan palliation,” he said.
FUEL enrolled 400 male and female adolescents with a single functional ventricle after Fontan surgical palliation. In these patients, pulmonary vascular resistance (PVR) is critical for the efficient flow of blood through the lungs without the benefit of a ventricular pump. “While this circulation is typically stable through childhood, cardiovascular efficiency deteriorates over time, associated with a decline in exercise performance and the accrual of Fontan-associated morbidities,” Dr. Goldberg said. “Given the importance of pulmonary vascular resistance, modulators of PVR make sense as potential therapies.”
FUEL evaluated the effect of udenafil 87.5 mg twice daily versus placebo in post-Fontan patients who’d been on anticoagulation or antiplatelet therapy. The treatment group had a higher percentage of female patients (44% vs. 36% on placebo), but all other baseline characteristics were similar between the two groups.
While the trial found the drug was well tolerated and safe, with side effects typical of PDE5 inhibitors, it did not lead to changes in myocardial performance index, reactive hyperemia index, or log brain natriuretic peptide, Dr. Goldberg said.
At 6 months, both groups showed a decline in exercise data, “as expected,” Dr. Goldberg said. “But that decline was attenuated in the group receiving udenafil,” he said, with peak oxygen consumption declining an average of 0.23 and 0.89 mL/kg per minute in the treatment and placebo groups, respectively (P = 0.092).
Total oxygen consumption, however, actually improved in the udenafil group and declined in the placebo group, 44 mL/min on average versus –3.7 mL/min (P = 0.071).
“There was no significant difference in the change in peak heart rate or the change in peak oxygen saturation between the groups,” Dr. Goldberg said. But three measures at the ventilatory aerobic threshold (VAT) – oxygen consumption, work rate, and ventilation/carbon dioxide output – all showed statistically significant improvement in exercise performance.
“This has important clinical implications,” Dr. Goldberg said of the study findings. “Our study extends recent findings in highlighting the importance of submaximal exercise in the understanding of Fontan physiology. And unlike peak oxygen consumption, submaximal exercise is not constrained by the physiologic ceiling of central venous pressure inherent in exercise physiology after Fontan palliation.”
Maximum oxygen consumption at VAT is likely a more relevant measure after Fontan palliation than is central venous pressure, discussant Craig A. Sable, MD, a pediatric cardiologist in Potomac, Md., noted in his comments. “This is because VAT occurs at about 70% of maximum VO2 [oxygen consumption] in Fontan as opposed to 55% in two-ventricle physiology,” Dr. Sable said.
In adults with congenital heart disease, maximal VO2 of 45%-50% of predicted levels portends increased risk of heart failure and death. “Therefore, a medication that addresses the central deficiencies of Fontan physiology and results in improved exercise performance may allow for a longer period of symptom-free survival,” he said.
In an invited commentary in Circulation (2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044512), Marc Gewillig, MD, and Alexander van de Bruaene, MD, of University Hospitals Leuven (Belgium) said that the findings of FUEL and other trials of pulmonary vasodilators after Fontan leave “open for debate” whether the treatment effects of a 3%-5% improvement in oxygen consumption is clinically meaningful for adolescents. “For failing Fontan patients (not studied in FUEL), these improvements are minimal but maybe relevant,” the commentators wrote. But the studies do not resolve whether that’s enough to prevent further decline.
Dr. Goldberg disclosed receiving research grants from trial sponsor Mezzion Pharmaceuticals and the National Heart Lung and Blood Institute. Dr. Sable, Dr. Gewillig, and Dr. van de Bruaene have no financial relationships to disclose.
SOURCE: Goldberg D. AHA 2019, Late Breaking Science Session 5.
REPORTING FROM AHA 2019
Cognitive problems after extremely preterm birth persist
Cognitive and neuropsychological impairment associated with extremely preterm (EP) birth persists into young adulthood, according to findings from the 1995 EPICure cohort.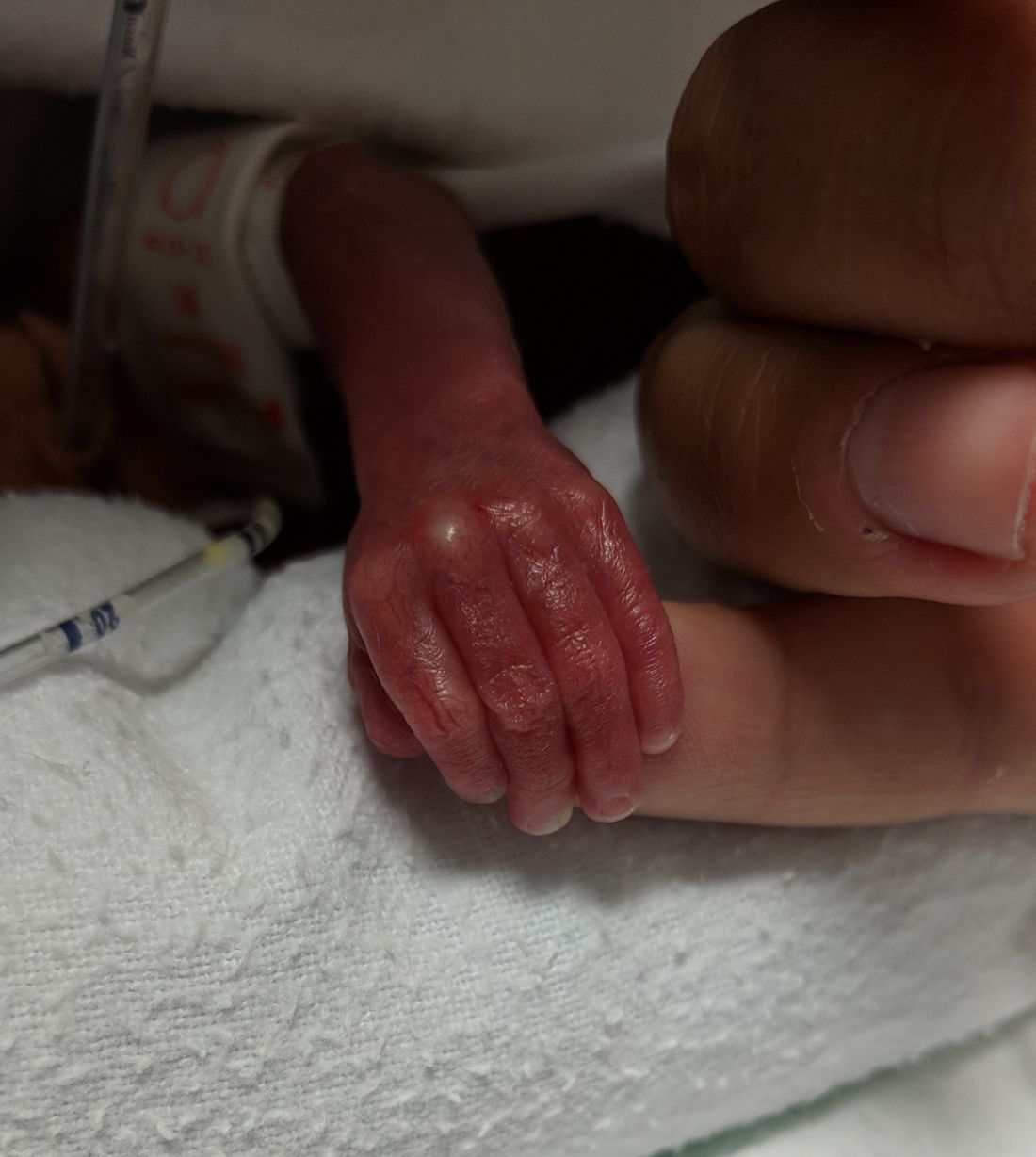
Of note, intellectual impairment increased significantly after the age of 11 years among 19-year-olds in the cohort of individuals born EP, Helen O’Reilly, PhD, of the Institute for Women’s Health at University College London and colleagues reported in Pediatrics.
Neuropsychological assessment to examine general cognitive abilities, visuomotor abilities, prospective memory, and certain aspects of executive functioning and language in 127 cases and 64 term-born controls showed significantly lower scores across all tests in those born EP.
Impairment in at least one neuropsychological domain was present in 60% of EP birth cases (compared with 21% of controls), with 35% having impairment in at least four domains. Most deficits occurred in general cognitive function and/or visuomotor abilities.
Further, and those with cognitive impairment at 11 years were at increased risk of deficit at 19 years (RR, 3.56), even after adjustment for sex and socioeconomic status, the authors wrote.
None of the term-born controls had a cognitive impairment at 11 years, and two (3%) had impairment at 19 years.
Studies of adults born very preterm have revealed that these individuals are at risk for neuropsychological impairment, but the extent of such impairment in individuals with EP birth, defined as birth before 26 weeks’ gestation, had not previously been studied in the long term.
Assessments in the EPICure cohort of individuals born EP in 1995 previously showed scores at 1.1-1.6 standard deviations lower on measures of general cognitive function, compared with standardized norms and/or term-born controls, at age 2.5, 6, and 11 years, Dr. O’Reilly and colleagues explained.
The current findings indicate that general cognitive and neuropsychological functioning problems associated with EP birth persist and can increase into early adulthood, and they “highlight the need for early and ongoing neuropsychological and educational assessment in EP children to ensure these children receive appropriate support in school and for planned educational pathways,” the investigators concluded.
In an accompanying editorial, Louis A. Schmidt, PhD, and Saroj Saigal, MD, of McMaster University, Hamilton, Ont., wrote that these findings “provide compelling evidence for persistent effects of cognitive impairments” in individuals born EP.
They highlighted three lessons from the study:
- It is important to control for anxiety in future studies like this “to eliminate potential confounding influences of anxiety when examining performance-based measures in the laboratory setting,” as individuals born EP are known to exhibit anxiety.
- Group heterogeneity also should be considered, as all survivors of prematurity are not alike.
- Measurement equivalency should be established between groups.
With respect to the latter, “although many of the measures used by O’Reilly et al. have been normed, issues of measurement invariance have not been established between EP and control groups on some of the measures reported,” Dr. Schmidt and Dr. Saigal wrote, noting that “many other studies [also] fail to consider this fundamental measurement property.”
“Considering issues of measurement equivalency is of critical importance to ensuring unbiased interpretations of findings,” they added, concluding that the findings by O’Reilly et al. represent an important contribution and confirm findings from many prior studies of extreme prematurity, which “informs how we effectively manage these problems.”
“As the percentage of preterm birth continues to rise worldwide, coupled with reduced morbidity and mortality, and with more EP infants reaching adulthood, there is a need for prospective, long-term outcome studies of extreme prematurity,” Dr. Schmidt and Dr. Saigal added.
The study was funded by the Medical Research Council United Kingdom. The authors reported having no relevant financial disclosures. The editorial by Dr. Schmidt and Dr. Saigal, who also reported having no relevant financial disclosures, was supported by the Canadian Institutes of Health Research.
SOURCES: O’Reilly H et al. Pediatrics. 2020;145(2):e20192087; Schmidt LA, Saigal S. Pediatrics. 2020;145(2):e20193359.
Cognitive and neuropsychological impairment associated with extremely preterm (EP) birth persists into young adulthood, according to findings from the 1995 EPICure cohort.
Of note, intellectual impairment increased significantly after the age of 11 years among 19-year-olds in the cohort of individuals born EP, Helen O’Reilly, PhD, of the Institute for Women’s Health at University College London and colleagues reported in Pediatrics.
Neuropsychological assessment to examine general cognitive abilities, visuomotor abilities, prospective memory, and certain aspects of executive functioning and language in 127 cases and 64 term-born controls showed significantly lower scores across all tests in those born EP.
Impairment in at least one neuropsychological domain was present in 60% of EP birth cases (compared with 21% of controls), with 35% having impairment in at least four domains. Most deficits occurred in general cognitive function and/or visuomotor abilities.
Further, and those with cognitive impairment at 11 years were at increased risk of deficit at 19 years (RR, 3.56), even after adjustment for sex and socioeconomic status, the authors wrote.
None of the term-born controls had a cognitive impairment at 11 years, and two (3%) had impairment at 19 years.
Studies of adults born very preterm have revealed that these individuals are at risk for neuropsychological impairment, but the extent of such impairment in individuals with EP birth, defined as birth before 26 weeks’ gestation, had not previously been studied in the long term.
Assessments in the EPICure cohort of individuals born EP in 1995 previously showed scores at 1.1-1.6 standard deviations lower on measures of general cognitive function, compared with standardized norms and/or term-born controls, at age 2.5, 6, and 11 years, Dr. O’Reilly and colleagues explained.
The current findings indicate that general cognitive and neuropsychological functioning problems associated with EP birth persist and can increase into early adulthood, and they “highlight the need for early and ongoing neuropsychological and educational assessment in EP children to ensure these children receive appropriate support in school and for planned educational pathways,” the investigators concluded.
In an accompanying editorial, Louis A. Schmidt, PhD, and Saroj Saigal, MD, of McMaster University, Hamilton, Ont., wrote that these findings “provide compelling evidence for persistent effects of cognitive impairments” in individuals born EP.
They highlighted three lessons from the study:
- It is important to control for anxiety in future studies like this “to eliminate potential confounding influences of anxiety when examining performance-based measures in the laboratory setting,” as individuals born EP are known to exhibit anxiety.
- Group heterogeneity also should be considered, as all survivors of prematurity are not alike.
- Measurement equivalency should be established between groups.
With respect to the latter, “although many of the measures used by O’Reilly et al. have been normed, issues of measurement invariance have not been established between EP and control groups on some of the measures reported,” Dr. Schmidt and Dr. Saigal wrote, noting that “many other studies [also] fail to consider this fundamental measurement property.”
“Considering issues of measurement equivalency is of critical importance to ensuring unbiased interpretations of findings,” they added, concluding that the findings by O’Reilly et al. represent an important contribution and confirm findings from many prior studies of extreme prematurity, which “informs how we effectively manage these problems.”
“As the percentage of preterm birth continues to rise worldwide, coupled with reduced morbidity and mortality, and with more EP infants reaching adulthood, there is a need for prospective, long-term outcome studies of extreme prematurity,” Dr. Schmidt and Dr. Saigal added.
The study was funded by the Medical Research Council United Kingdom. The authors reported having no relevant financial disclosures. The editorial by Dr. Schmidt and Dr. Saigal, who also reported having no relevant financial disclosures, was supported by the Canadian Institutes of Health Research.
SOURCES: O’Reilly H et al. Pediatrics. 2020;145(2):e20192087; Schmidt LA, Saigal S. Pediatrics. 2020;145(2):e20193359.
Cognitive and neuropsychological impairment associated with extremely preterm (EP) birth persists into young adulthood, according to findings from the 1995 EPICure cohort.
Of note, intellectual impairment increased significantly after the age of 11 years among 19-year-olds in the cohort of individuals born EP, Helen O’Reilly, PhD, of the Institute for Women’s Health at University College London and colleagues reported in Pediatrics.
Neuropsychological assessment to examine general cognitive abilities, visuomotor abilities, prospective memory, and certain aspects of executive functioning and language in 127 cases and 64 term-born controls showed significantly lower scores across all tests in those born EP.
Impairment in at least one neuropsychological domain was present in 60% of EP birth cases (compared with 21% of controls), with 35% having impairment in at least four domains. Most deficits occurred in general cognitive function and/or visuomotor abilities.
Further, and those with cognitive impairment at 11 years were at increased risk of deficit at 19 years (RR, 3.56), even after adjustment for sex and socioeconomic status, the authors wrote.
None of the term-born controls had a cognitive impairment at 11 years, and two (3%) had impairment at 19 years.
Studies of adults born very preterm have revealed that these individuals are at risk for neuropsychological impairment, but the extent of such impairment in individuals with EP birth, defined as birth before 26 weeks’ gestation, had not previously been studied in the long term.
Assessments in the EPICure cohort of individuals born EP in 1995 previously showed scores at 1.1-1.6 standard deviations lower on measures of general cognitive function, compared with standardized norms and/or term-born controls, at age 2.5, 6, and 11 years, Dr. O’Reilly and colleagues explained.
The current findings indicate that general cognitive and neuropsychological functioning problems associated with EP birth persist and can increase into early adulthood, and they “highlight the need for early and ongoing neuropsychological and educational assessment in EP children to ensure these children receive appropriate support in school and for planned educational pathways,” the investigators concluded.
In an accompanying editorial, Louis A. Schmidt, PhD, and Saroj Saigal, MD, of McMaster University, Hamilton, Ont., wrote that these findings “provide compelling evidence for persistent effects of cognitive impairments” in individuals born EP.
They highlighted three lessons from the study:
- It is important to control for anxiety in future studies like this “to eliminate potential confounding influences of anxiety when examining performance-based measures in the laboratory setting,” as individuals born EP are known to exhibit anxiety.
- Group heterogeneity also should be considered, as all survivors of prematurity are not alike.
- Measurement equivalency should be established between groups.
With respect to the latter, “although many of the measures used by O’Reilly et al. have been normed, issues of measurement invariance have not been established between EP and control groups on some of the measures reported,” Dr. Schmidt and Dr. Saigal wrote, noting that “many other studies [also] fail to consider this fundamental measurement property.”
“Considering issues of measurement equivalency is of critical importance to ensuring unbiased interpretations of findings,” they added, concluding that the findings by O’Reilly et al. represent an important contribution and confirm findings from many prior studies of extreme prematurity, which “informs how we effectively manage these problems.”
“As the percentage of preterm birth continues to rise worldwide, coupled with reduced morbidity and mortality, and with more EP infants reaching adulthood, there is a need for prospective, long-term outcome studies of extreme prematurity,” Dr. Schmidt and Dr. Saigal added.
The study was funded by the Medical Research Council United Kingdom. The authors reported having no relevant financial disclosures. The editorial by Dr. Schmidt and Dr. Saigal, who also reported having no relevant financial disclosures, was supported by the Canadian Institutes of Health Research.
SOURCES: O’Reilly H et al. Pediatrics. 2020;145(2):e20192087; Schmidt LA, Saigal S. Pediatrics. 2020;145(2):e20193359.
FROM PEDIATRICS
Oral lichen planus prevalence estimates go global
for the general population and 0.98% among clinical patients.
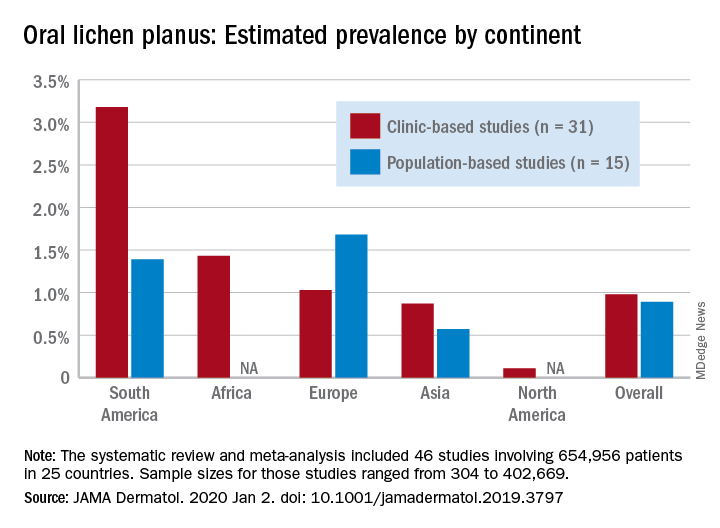
Globally, oral lichen planus (OLP) appears to be more prevalent in women than men (1.55% vs. 1.11% in population-based studies; 1.69% vs. 1.09% in clinic-based), in those aged 40 years and older (1.90% vs. 0.62% in clinic-based studies), and in non-Asian countries (see graph), Changchang Li, MD, and associates reported in JAMA Dermatology.
Of the 25 countries represented among the 46 included studies, Brazil had the highest OLP prevalence at 6.04% and India had the lowest at 0.02%. “Smokers and patients who abuse alcohol have a higher prevalence of OLP. This factor may explain why the highest prevalence … was found in Brazil, where 18.18% of residents report being smokers and 29.09% report consumption of alcoholic beverages,” wrote Dr. Li of the department of dermatology at Zhejiang University of Traditional Chinese Medicine, Wenzhou, China, and associates.
The difference in OLP prevalence by sex may be related to fluctuating female hormone levels, “especially during menstruation or menopause, and that different social roles may lead to the body being in a state of stress,” the investigators suggested.
The age-related difference in OLP could be the result of “longstanding oral habits” or changes to the oral mucosa over time, such as mucosal thinning, decreased elasticity, less saliva secretion, and greater tissue permeability. The higher prevalence among those aged 40 years and older also may be “associated with metabolic changes during aging or with decreased immunity, nutritional deficiencies, medication use, or denture wear,” they wrote.
The review and meta-analysis involved 15 studies (n = 462,993) that included general population data and 31 (n = 191,963) that used information from clinical patients. Sample sizes for those studies ranged from 308 to 402,669.
Statistically significant publication bias was seen among the clinic-based studies but not those that were population based, Dr. Li and associates wrote, adding that “our findings should be considered with caution because of the high heterogeneity of the included studies.”
The study was funded by the First-Class Discipline Construction Foundation of Guangzhou University of Chinese Medicine, the Young Top Talent Project of Scientific and Technological Innovation in Special Support Plan for Training High-level Talents, and the Youth Research and Cultivation Project of Guangzhou University of Chinese Medicine. The investigators did not report any conflicts of interest.
SOURCE: C Li et al. JAMA Dermatol. 2020 Jan 2. doi: 10.1001/jamadermatol.2019.3797.
for the general population and 0.98% among clinical patients.
Globally, oral lichen planus (OLP) appears to be more prevalent in women than men (1.55% vs. 1.11% in population-based studies; 1.69% vs. 1.09% in clinic-based), in those aged 40 years and older (1.90% vs. 0.62% in clinic-based studies), and in non-Asian countries (see graph), Changchang Li, MD, and associates reported in JAMA Dermatology.
Of the 25 countries represented among the 46 included studies, Brazil had the highest OLP prevalence at 6.04% and India had the lowest at 0.02%. “Smokers and patients who abuse alcohol have a higher prevalence of OLP. This factor may explain why the highest prevalence … was found in Brazil, where 18.18% of residents report being smokers and 29.09% report consumption of alcoholic beverages,” wrote Dr. Li of the department of dermatology at Zhejiang University of Traditional Chinese Medicine, Wenzhou, China, and associates.
The difference in OLP prevalence by sex may be related to fluctuating female hormone levels, “especially during menstruation or menopause, and that different social roles may lead to the body being in a state of stress,” the investigators suggested.
The age-related difference in OLP could be the result of “longstanding oral habits” or changes to the oral mucosa over time, such as mucosal thinning, decreased elasticity, less saliva secretion, and greater tissue permeability. The higher prevalence among those aged 40 years and older also may be “associated with metabolic changes during aging or with decreased immunity, nutritional deficiencies, medication use, or denture wear,” they wrote.
The review and meta-analysis involved 15 studies (n = 462,993) that included general population data and 31 (n = 191,963) that used information from clinical patients. Sample sizes for those studies ranged from 308 to 402,669.
Statistically significant publication bias was seen among the clinic-based studies but not those that were population based, Dr. Li and associates wrote, adding that “our findings should be considered with caution because of the high heterogeneity of the included studies.”
The study was funded by the First-Class Discipline Construction Foundation of Guangzhou University of Chinese Medicine, the Young Top Talent Project of Scientific and Technological Innovation in Special Support Plan for Training High-level Talents, and the Youth Research and Cultivation Project of Guangzhou University of Chinese Medicine. The investigators did not report any conflicts of interest.
SOURCE: C Li et al. JAMA Dermatol. 2020 Jan 2. doi: 10.1001/jamadermatol.2019.3797.
for the general population and 0.98% among clinical patients.
Globally, oral lichen planus (OLP) appears to be more prevalent in women than men (1.55% vs. 1.11% in population-based studies; 1.69% vs. 1.09% in clinic-based), in those aged 40 years and older (1.90% vs. 0.62% in clinic-based studies), and in non-Asian countries (see graph), Changchang Li, MD, and associates reported in JAMA Dermatology.
Of the 25 countries represented among the 46 included studies, Brazil had the highest OLP prevalence at 6.04% and India had the lowest at 0.02%. “Smokers and patients who abuse alcohol have a higher prevalence of OLP. This factor may explain why the highest prevalence … was found in Brazil, where 18.18% of residents report being smokers and 29.09% report consumption of alcoholic beverages,” wrote Dr. Li of the department of dermatology at Zhejiang University of Traditional Chinese Medicine, Wenzhou, China, and associates.
The difference in OLP prevalence by sex may be related to fluctuating female hormone levels, “especially during menstruation or menopause, and that different social roles may lead to the body being in a state of stress,” the investigators suggested.
The age-related difference in OLP could be the result of “longstanding oral habits” or changes to the oral mucosa over time, such as mucosal thinning, decreased elasticity, less saliva secretion, and greater tissue permeability. The higher prevalence among those aged 40 years and older also may be “associated with metabolic changes during aging or with decreased immunity, nutritional deficiencies, medication use, or denture wear,” they wrote.
The review and meta-analysis involved 15 studies (n = 462,993) that included general population data and 31 (n = 191,963) that used information from clinical patients. Sample sizes for those studies ranged from 308 to 402,669.
Statistically significant publication bias was seen among the clinic-based studies but not those that were population based, Dr. Li and associates wrote, adding that “our findings should be considered with caution because of the high heterogeneity of the included studies.”
The study was funded by the First-Class Discipline Construction Foundation of Guangzhou University of Chinese Medicine, the Young Top Talent Project of Scientific and Technological Innovation in Special Support Plan for Training High-level Talents, and the Youth Research and Cultivation Project of Guangzhou University of Chinese Medicine. The investigators did not report any conflicts of interest.
SOURCE: C Li et al. JAMA Dermatol. 2020 Jan 2. doi: 10.1001/jamadermatol.2019.3797.
FROM JAMA DERMATOLOGY
Long-term entecavir looks safe, effective in HBV
For patients with chronic hepatitis B virus (HBV) infection, up to 10 years of treatment with entecavir was safe and produced a superior rate of sustained virologic response, compared with other HBV nucleoside or nucleotide analogues in a global randomized clinical trial.
Virologic responses were confirmed and maintained in 80% of entecavir patients and 61% of patients who received other therapies, said Jin-Lin Hou, MD, of Southern Medical University in Guangzhou, China, and associates. Regardless of which treatment patients received, a sustained virologic response was associated with a significantly lower rate of liver-related hepatitis B virus (HBV) disease progression and hepatocellular carcinoma. Rates of serious treatment-related adverse events were 0.2% in the entecavir arm and 0.8% in the nonentecavir arm. Moreover, the primary outcome of time-to-adjudicated clinical outcome events “showed that entecavir treatment, compared with nonentecavir, was not associated with an increased risk of malignant neoplasms, including hepatocellular carcinoma, nonhepatocellular carcinoma malignancies, and overall malignancies,” they wrote in Clinical Gastroenterology and Hepatology.
Entecavir is approved for the treatment of adults with chronic HBV infection, and its long-term use has been linked to the regression of hepatic fibrosis and cirrhosis. In treatment-naive patients, genotypic resistance and virologic breakthrough are rare even after up to 5 years of entecavir therapy. Although human studies have not linked this treatment duration with an increased risk of adverse events, murine studies have identified benign and malignant tumors of the brain, lung, and liver in entecavir-treated mice and rats. “With the exception of lung tumors, which were limited to male mice, rodent tumors occurred only at entecavir exposures [that were] significantly higher than those achieved in human beings with standard approved doses,” the researchers wrote.
For the trial, they assigned more than 12,000 patients with chronic HBV infection to receive long-term treatment with entecavir or investigators’ choice of another HBV nucleoside or nucleotide analogue. Patients were from 229 centers in Asia, Europe, and North and South America, and a total of 6,216 received entecavir, while 6,162 received another therapy.
Compared with other HBV nucleoside and nucleotide analogues, long-term treatment with entecavir “provided a high margin of safety” and was not tied to higher rates of liver or nonliver malignancies, the researchers found. The carcinogenicity of entecavir in rodents did not appear to extend to humans. Furthermore, among 5,305 trial participants in China, a sustained virologic response was associated with a clinically and statistically significant reduction in the risk of liver-related HBV disease progression (hazard ratio, 0.09; 95% CI, 0.04-0.22) and hepatocellular carcinoma (HR, 0.03; 95% CI, 0.009-0.113).
The results confirm the appropriateness of long-term entecavir therapy for chronic HBV infection, as recommended by current guidelines, Dr. Hou and associates concluded. However, patients in this trial were relatively young, with a median age of only 39 years. Therefore, the risk of entecavir-associated malignancies in older age cohorts could not be evaluated.
Bristol-Myers Squibb designed the study, performed statistical analyses, and funded the study and manuscript preparation. The Ministry of Science and Technology of China and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program provided partial support. Dr. Hou disclosed grants and personal fees from Bristol-Myers Squibb, GlaxoSmithKline, and Novartis. Several coinvestigators also disclosed ties to Bristol-Myers Squibb and to several other pharmaceutical companies.
SOURCE: Hou J-L et al. Clin Gastroenterol Hepatol. 2019 Jul 12. doi: 10.1016/j.cgh.2019.07.010.
For patients with chronic hepatitis B virus (HBV) infection, up to 10 years of treatment with entecavir was safe and produced a superior rate of sustained virologic response, compared with other HBV nucleoside or nucleotide analogues in a global randomized clinical trial.
Virologic responses were confirmed and maintained in 80% of entecavir patients and 61% of patients who received other therapies, said Jin-Lin Hou, MD, of Southern Medical University in Guangzhou, China, and associates. Regardless of which treatment patients received, a sustained virologic response was associated with a significantly lower rate of liver-related hepatitis B virus (HBV) disease progression and hepatocellular carcinoma. Rates of serious treatment-related adverse events were 0.2% in the entecavir arm and 0.8% in the nonentecavir arm. Moreover, the primary outcome of time-to-adjudicated clinical outcome events “showed that entecavir treatment, compared with nonentecavir, was not associated with an increased risk of malignant neoplasms, including hepatocellular carcinoma, nonhepatocellular carcinoma malignancies, and overall malignancies,” they wrote in Clinical Gastroenterology and Hepatology.
Entecavir is approved for the treatment of adults with chronic HBV infection, and its long-term use has been linked to the regression of hepatic fibrosis and cirrhosis. In treatment-naive patients, genotypic resistance and virologic breakthrough are rare even after up to 5 years of entecavir therapy. Although human studies have not linked this treatment duration with an increased risk of adverse events, murine studies have identified benign and malignant tumors of the brain, lung, and liver in entecavir-treated mice and rats. “With the exception of lung tumors, which were limited to male mice, rodent tumors occurred only at entecavir exposures [that were] significantly higher than those achieved in human beings with standard approved doses,” the researchers wrote.
For the trial, they assigned more than 12,000 patients with chronic HBV infection to receive long-term treatment with entecavir or investigators’ choice of another HBV nucleoside or nucleotide analogue. Patients were from 229 centers in Asia, Europe, and North and South America, and a total of 6,216 received entecavir, while 6,162 received another therapy.
Compared with other HBV nucleoside and nucleotide analogues, long-term treatment with entecavir “provided a high margin of safety” and was not tied to higher rates of liver or nonliver malignancies, the researchers found. The carcinogenicity of entecavir in rodents did not appear to extend to humans. Furthermore, among 5,305 trial participants in China, a sustained virologic response was associated with a clinically and statistically significant reduction in the risk of liver-related HBV disease progression (hazard ratio, 0.09; 95% CI, 0.04-0.22) and hepatocellular carcinoma (HR, 0.03; 95% CI, 0.009-0.113).
The results confirm the appropriateness of long-term entecavir therapy for chronic HBV infection, as recommended by current guidelines, Dr. Hou and associates concluded. However, patients in this trial were relatively young, with a median age of only 39 years. Therefore, the risk of entecavir-associated malignancies in older age cohorts could not be evaluated.
Bristol-Myers Squibb designed the study, performed statistical analyses, and funded the study and manuscript preparation. The Ministry of Science and Technology of China and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program provided partial support. Dr. Hou disclosed grants and personal fees from Bristol-Myers Squibb, GlaxoSmithKline, and Novartis. Several coinvestigators also disclosed ties to Bristol-Myers Squibb and to several other pharmaceutical companies.
SOURCE: Hou J-L et al. Clin Gastroenterol Hepatol. 2019 Jul 12. doi: 10.1016/j.cgh.2019.07.010.
For patients with chronic hepatitis B virus (HBV) infection, up to 10 years of treatment with entecavir was safe and produced a superior rate of sustained virologic response, compared with other HBV nucleoside or nucleotide analogues in a global randomized clinical trial.
Virologic responses were confirmed and maintained in 80% of entecavir patients and 61% of patients who received other therapies, said Jin-Lin Hou, MD, of Southern Medical University in Guangzhou, China, and associates. Regardless of which treatment patients received, a sustained virologic response was associated with a significantly lower rate of liver-related hepatitis B virus (HBV) disease progression and hepatocellular carcinoma. Rates of serious treatment-related adverse events were 0.2% in the entecavir arm and 0.8% in the nonentecavir arm. Moreover, the primary outcome of time-to-adjudicated clinical outcome events “showed that entecavir treatment, compared with nonentecavir, was not associated with an increased risk of malignant neoplasms, including hepatocellular carcinoma, nonhepatocellular carcinoma malignancies, and overall malignancies,” they wrote in Clinical Gastroenterology and Hepatology.
Entecavir is approved for the treatment of adults with chronic HBV infection, and its long-term use has been linked to the regression of hepatic fibrosis and cirrhosis. In treatment-naive patients, genotypic resistance and virologic breakthrough are rare even after up to 5 years of entecavir therapy. Although human studies have not linked this treatment duration with an increased risk of adverse events, murine studies have identified benign and malignant tumors of the brain, lung, and liver in entecavir-treated mice and rats. “With the exception of lung tumors, which were limited to male mice, rodent tumors occurred only at entecavir exposures [that were] significantly higher than those achieved in human beings with standard approved doses,” the researchers wrote.
For the trial, they assigned more than 12,000 patients with chronic HBV infection to receive long-term treatment with entecavir or investigators’ choice of another HBV nucleoside or nucleotide analogue. Patients were from 229 centers in Asia, Europe, and North and South America, and a total of 6,216 received entecavir, while 6,162 received another therapy.
Compared with other HBV nucleoside and nucleotide analogues, long-term treatment with entecavir “provided a high margin of safety” and was not tied to higher rates of liver or nonliver malignancies, the researchers found. The carcinogenicity of entecavir in rodents did not appear to extend to humans. Furthermore, among 5,305 trial participants in China, a sustained virologic response was associated with a clinically and statistically significant reduction in the risk of liver-related HBV disease progression (hazard ratio, 0.09; 95% CI, 0.04-0.22) and hepatocellular carcinoma (HR, 0.03; 95% CI, 0.009-0.113).
The results confirm the appropriateness of long-term entecavir therapy for chronic HBV infection, as recommended by current guidelines, Dr. Hou and associates concluded. However, patients in this trial were relatively young, with a median age of only 39 years. Therefore, the risk of entecavir-associated malignancies in older age cohorts could not be evaluated.
Bristol-Myers Squibb designed the study, performed statistical analyses, and funded the study and manuscript preparation. The Ministry of Science and Technology of China and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program provided partial support. Dr. Hou disclosed grants and personal fees from Bristol-Myers Squibb, GlaxoSmithKline, and Novartis. Several coinvestigators also disclosed ties to Bristol-Myers Squibb and to several other pharmaceutical companies.
SOURCE: Hou J-L et al. Clin Gastroenterol Hepatol. 2019 Jul 12. doi: 10.1016/j.cgh.2019.07.010.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
PACT-HF: Transitional care derives no overall benefit
Women respond more to intervention
PHILADELPHIA – A clinical trial of a program that transitions heart failure patients after they’re discharged from the hospital didn’t result in any appreciable improvement in all-cause death, readmissions or emergency department visits after 6 months overall, but it did show that women responded more favorably than men.

Harriette G.C. Van Spall, MD, MPH, reported 6-month results of the Patient-Centered Transitional Care Services in Heart Failure (PACT-HF) trial of 2,494 HF patients at 10 hospitals in Ontario during February 2015 to March 2016. They were randomized to the care-transition program or usual care. The findings, she said at the American Heart Association scientific sessions, “highlight the gap between efficacy that’s often demonstrated in mechanistic clinical trials and effectiveness when we aim to implement these results in real-world settings.” Three-month PACT-HF results were reported previously (JAMA. 2019 Feb 26;321:753-61).
The transitional-care model consisted of a comprehensive needs assessment by a nurse who also provided self-care education, a patient-centered discharge summary, and follow-up with a family physician within 7 days of discharge, which Dr. Van Spall noted “is not current practice in our health care system.”
Patients deemed high risk for readmission or death also received nurse home visits and scheduled visits to a multidisciplinary heart function clinic within 2-4 weeks of discharge and continuing as long as clinically suitable, said Dr. Van Spall, a principal investigator at the Population Health Research Institute, Hamilton, Ont., and assistant professor in cardiology at McMaster University in Hamilton.
The trial found no difference between the intervention and usual-care groups in the two composite endpoints at 6 months, Dr. Van Spall said: all-cause death, readmissions, or ED visits (63.1% and 64.5%, respectively; P = .50); or all-cause readmissions or ED visits (60.8% and 62.4%; P = .36).
“Despite the mutual overall clinical outcomes, we noted specific differences in response to treatment,” she said. With regard to the composite endpoint that included all-cause death, “Men had an attenuated response to the treatment with a hazard ratio of 1.05 (95% confidence interval, 0.87-1.26), whereas women had a hazard ratio of 0.85 (95% CI, 0.71-1.03), demonstrating that women have more of a treatment response to this health care service,” she said.
In men, rates for the first primary composite outcome were 66.3% and 64.1% in the intervention and usual-care groups, whereas in women those rates were 59.9% and 64.8% (P = .04 for sex interaction).
In the second composite endpoint, all-cause readmission or ED visit, “again, men had an attenuated response” with a HR of 1.03, whereas women had a HR of 0.83. Results were similar to those for the first primary composite outcome: 63.4% and 61.7% for intervention and usual care in men and 57.7% and 63% in women (P = .03 for sex interaction).
In putting the findings into context, Dr. Van Spall said tailoring services to risk in HF patients may be fraught with pitfalls. “We delivered intensive services to those patients at high risk of readmission or death, but it is quite possible they are the least likely to derive benefit by virtue of their advanced heart failure,” she said. “It may be that more benefit would have been derived had we chosen low- or moderate-risk patients to receive the intervention.”
She also said the sex-specific outcomes must be interpreted with caution. “But they do give us pause to consider that services could be titrated more effectively if delivered to patients who are more likely to derive benefit,” Dr. Van Spall said. The finding that women derived more of a benefit is in line with other prospective and observational studies that have found that women have a higher sense of self-care, self-efficacy, and confidence in managing their own health care needs than men.
Dr. Van Spall has no financial relationships to disclose.
Women respond more to intervention
Women respond more to intervention
PHILADELPHIA – A clinical trial of a program that transitions heart failure patients after they’re discharged from the hospital didn’t result in any appreciable improvement in all-cause death, readmissions or emergency department visits after 6 months overall, but it did show that women responded more favorably than men.
Harriette G.C. Van Spall, MD, MPH, reported 6-month results of the Patient-Centered Transitional Care Services in Heart Failure (PACT-HF) trial of 2,494 HF patients at 10 hospitals in Ontario during February 2015 to March 2016. They were randomized to the care-transition program or usual care. The findings, she said at the American Heart Association scientific sessions, “highlight the gap between efficacy that’s often demonstrated in mechanistic clinical trials and effectiveness when we aim to implement these results in real-world settings.” Three-month PACT-HF results were reported previously (JAMA. 2019 Feb 26;321:753-61).
The transitional-care model consisted of a comprehensive needs assessment by a nurse who also provided self-care education, a patient-centered discharge summary, and follow-up with a family physician within 7 days of discharge, which Dr. Van Spall noted “is not current practice in our health care system.”
Patients deemed high risk for readmission or death also received nurse home visits and scheduled visits to a multidisciplinary heart function clinic within 2-4 weeks of discharge and continuing as long as clinically suitable, said Dr. Van Spall, a principal investigator at the Population Health Research Institute, Hamilton, Ont., and assistant professor in cardiology at McMaster University in Hamilton.
The trial found no difference between the intervention and usual-care groups in the two composite endpoints at 6 months, Dr. Van Spall said: all-cause death, readmissions, or ED visits (63.1% and 64.5%, respectively; P = .50); or all-cause readmissions or ED visits (60.8% and 62.4%; P = .36).
“Despite the mutual overall clinical outcomes, we noted specific differences in response to treatment,” she said. With regard to the composite endpoint that included all-cause death, “Men had an attenuated response to the treatment with a hazard ratio of 1.05 (95% confidence interval, 0.87-1.26), whereas women had a hazard ratio of 0.85 (95% CI, 0.71-1.03), demonstrating that women have more of a treatment response to this health care service,” she said.
In men, rates for the first primary composite outcome were 66.3% and 64.1% in the intervention and usual-care groups, whereas in women those rates were 59.9% and 64.8% (P = .04 for sex interaction).
In the second composite endpoint, all-cause readmission or ED visit, “again, men had an attenuated response” with a HR of 1.03, whereas women had a HR of 0.83. Results were similar to those for the first primary composite outcome: 63.4% and 61.7% for intervention and usual care in men and 57.7% and 63% in women (P = .03 for sex interaction).
In putting the findings into context, Dr. Van Spall said tailoring services to risk in HF patients may be fraught with pitfalls. “We delivered intensive services to those patients at high risk of readmission or death, but it is quite possible they are the least likely to derive benefit by virtue of their advanced heart failure,” she said. “It may be that more benefit would have been derived had we chosen low- or moderate-risk patients to receive the intervention.”
She also said the sex-specific outcomes must be interpreted with caution. “But they do give us pause to consider that services could be titrated more effectively if delivered to patients who are more likely to derive benefit,” Dr. Van Spall said. The finding that women derived more of a benefit is in line with other prospective and observational studies that have found that women have a higher sense of self-care, self-efficacy, and confidence in managing their own health care needs than men.
Dr. Van Spall has no financial relationships to disclose.
PHILADELPHIA – A clinical trial of a program that transitions heart failure patients after they’re discharged from the hospital didn’t result in any appreciable improvement in all-cause death, readmissions or emergency department visits after 6 months overall, but it did show that women responded more favorably than men.
Harriette G.C. Van Spall, MD, MPH, reported 6-month results of the Patient-Centered Transitional Care Services in Heart Failure (PACT-HF) trial of 2,494 HF patients at 10 hospitals in Ontario during February 2015 to March 2016. They were randomized to the care-transition program or usual care. The findings, she said at the American Heart Association scientific sessions, “highlight the gap between efficacy that’s often demonstrated in mechanistic clinical trials and effectiveness when we aim to implement these results in real-world settings.” Three-month PACT-HF results were reported previously (JAMA. 2019 Feb 26;321:753-61).
The transitional-care model consisted of a comprehensive needs assessment by a nurse who also provided self-care education, a patient-centered discharge summary, and follow-up with a family physician within 7 days of discharge, which Dr. Van Spall noted “is not current practice in our health care system.”
Patients deemed high risk for readmission or death also received nurse home visits and scheduled visits to a multidisciplinary heart function clinic within 2-4 weeks of discharge and continuing as long as clinically suitable, said Dr. Van Spall, a principal investigator at the Population Health Research Institute, Hamilton, Ont., and assistant professor in cardiology at McMaster University in Hamilton.
The trial found no difference between the intervention and usual-care groups in the two composite endpoints at 6 months, Dr. Van Spall said: all-cause death, readmissions, or ED visits (63.1% and 64.5%, respectively; P = .50); or all-cause readmissions or ED visits (60.8% and 62.4%; P = .36).
“Despite the mutual overall clinical outcomes, we noted specific differences in response to treatment,” she said. With regard to the composite endpoint that included all-cause death, “Men had an attenuated response to the treatment with a hazard ratio of 1.05 (95% confidence interval, 0.87-1.26), whereas women had a hazard ratio of 0.85 (95% CI, 0.71-1.03), demonstrating that women have more of a treatment response to this health care service,” she said.
In men, rates for the first primary composite outcome were 66.3% and 64.1% in the intervention and usual-care groups, whereas in women those rates were 59.9% and 64.8% (P = .04 for sex interaction).
In the second composite endpoint, all-cause readmission or ED visit, “again, men had an attenuated response” with a HR of 1.03, whereas women had a HR of 0.83. Results were similar to those for the first primary composite outcome: 63.4% and 61.7% for intervention and usual care in men and 57.7% and 63% in women (P = .03 for sex interaction).
In putting the findings into context, Dr. Van Spall said tailoring services to risk in HF patients may be fraught with pitfalls. “We delivered intensive services to those patients at high risk of readmission or death, but it is quite possible they are the least likely to derive benefit by virtue of their advanced heart failure,” she said. “It may be that more benefit would have been derived had we chosen low- or moderate-risk patients to receive the intervention.”
She also said the sex-specific outcomes must be interpreted with caution. “But they do give us pause to consider that services could be titrated more effectively if delivered to patients who are more likely to derive benefit,” Dr. Van Spall said. The finding that women derived more of a benefit is in line with other prospective and observational studies that have found that women have a higher sense of self-care, self-efficacy, and confidence in managing their own health care needs than men.
Dr. Van Spall has no financial relationships to disclose.
REPORTING FROM AHA 2019
Adolescent alcohol, opioid misuse linked to risky behaviors
Binge drinking and misuse of opioids led to risky behavior during adolescence, two studies from the journal Pediatrics highlighted. And the binge drinking in high school may predict risky driving behaviors up to 4 years after high school.

Federico E. Vaca, MD, of the developmental neurocognitive driving simulation research center at Yale University, New Haven, Conn., and colleagues examined the associations between risky driving behaviors and binge drinking of 2,785 adolescents in the nationally representative, longitudinal NEXT Generation Health Study. The researchers studied the effects of binge drinking on driving while impaired (DWI), riding with an impaired driver (RWI), blackouts, extreme binge drinking, and risky driving.
The adolescents were studied across seven waves, with Wave 1 beginning in the 2009-2010 school year (10th grade; mean age, 16 years), and data extended up to 4 years after high school. Of all adolescents enrolled, 91% completed Wave 1, 88% completed Wave 2, 86% completed Wave 3 (12th grade), 78% completed Wave 4, 79% completed Wave 5, 84% completed Wave 6, and 83% completed Wave 7 (4 years after leaving high school) of the study.
High school binge drinking predicts later risky behavior
About one-quarter of adolescents reported binge drinking in Waves 1-3, with an incidence of 27% in Wave 1, 24% in Wave 2, and 27% in Wave 3. Adolescents who reported binge drinking in Wave 3 had a higher likelihood of DWI in subsequent waves, with nearly six times higher odds in Wave 5 and more than twice as likely in Wave 7, researchers said. Binge drinking in Wave 3 also was associated with greater than four times higher odds of RWI in Wave 4, and more than two and a half times higher odds of RWI in Wave 7. Among adolescents who reported binge drinking across 3 years in high school, there was a higher likelihood of extreme binge drinking in Wave 7, and higher likelihood of risky driving after graduating.
Impact of parental knowledge of drinking
in some waves. Father monitoring knowledge of drinking in Waves 1-3 lowered the odds of DWI by 30% in Wave 5 and 20% in Wave 6, while also lowering the odds of RWI in Wave 4 and Wave 7 by 20%.
Mother knowledge of drinking in Waves 1-3 was associated with 60% lower odds of DWI in Wave 4, but did not lower odds in any wave for RWI.
Overall, parental support for not drinking lowered odds for DWI by 40% in Waves 4 and 5, and by 30% in Wave 7 while also lowering odds of RWI in Wave 4 by 20%.
The results are consistent with other studies examining risky driving behavior and binge drinking in adolescent populations, but researchers noted that “to an important but limited extent, parental practices while the teenager is in high school may protect against DWI, RWI, and blackouts as adolescents move into early adulthood.”
“Our findings are relevant to prevention programs that seek to incorporate alcohol screening with intentional inquiry about binge drinking. Moreover, our results may be instructive to programs that seek to leverage facets of parental practices to reduce health-risk contexts for youth,” Dr. Vaca and colleagues concluded. “Such prevention activities coupled with strengthening of policies and practices reducing adolescents’ access to alcohol could reduce later major alcohol-related health-risk behaviors and their consequences.”
Opioid misuse and risky behavior
In a second study, Devika Bhatia, MD, of the University of Colorado at Denver, Aurora, and colleagues examined opioid misuse in a nationally-representative sample of 14,765 adolescents from the Centers for Disease Control and Prevention’s 2017 Youth Risk Behavior Surveillance Survey. The researchers measured opioid misuse by categorizing adolescents into groups based on whether they had ever misused prescription opioids and whether they had engaged in risky driving behavior, violent behavior, risky sexual behavior, had a history of substance abuse, or attempted suicide.
Dr. Bhatia and colleagues found 14% of adolescents in the study reported misusing opioids, with an overrepresentation of 17-year-old and 18-year-old participants reporting opioid misuse (P less than .0001). there were no statistically significant difference between those who misused opioids and those who did not in terms of race, ethnicity, or sex.
Those adolescents who reported misusing opioids were 2.8 times more likely to not use a seatbelt; were 2.8 times more likely to have RWI; were 5.8 times more likely to have DWI; or 2.3 times more likely to have texted or emailed while driving. In each of these cases, P was less than .0001.
Adolescents who misused opioids also had significantly increased odds of engaging in risky sexual behaviors such as having sex before 13 years (3.9 times); having sex with four or more partners (4.8 times); using substances before sex (3.6 times); and not using a condom before sex (2.0 times). In each of these cases, P was less than .0001.
Additionally, adolescents in this category were between 5.4 times and 22.3 times more likely to use other substances (P less than .0001 for 10 variables); 4.9 times more likely to have attempted suicide (P less than .0001); or more likely to have engaged in violent behavior such as getting into physical fights (4.0 times), carrying a weapon (3.4 times) or a gun (5.1 times) within the last 30 days. In the four latter cases, P was less than .0001.
“With the ongoing opioid epidemic, pediatricians and child psychiatrists are likely to be more attuned to opioid misuse in their patients,” Dr. Bhatia and colleagues concluded. “If youth are screening positive for opioid misuse, pediatricians, nurses, social workers, child psychiatrists, and other providers assessing adolescents may have a new, broad range of other risky behaviors for which to screen regardless of the direction of the association.”
Substance use screening for treating substance use disorder traditionally has been is provided by a specialist, Jessica A. Kulak, PhD, MPH, said in an interview. “However, integration of care services may help to change societal norms around problematic substance use – both by decreasing stigma associated with substance use, as well as increasing clinicians’ preparedness, knowledge, and confidence in preventing and intervening on adolescents’ substance experimentation and use.” She recommended that clinicians in primary care improve their training by using the Substance Abuse and Mental Health Services Administration’s Screening, Brief Intervention, and Referral to Treatment program, which is available as a free online course.
Confidentiality is important in adolescent health, said Dr. Kulak, who is an assistant professor in the department of health, nutrition, and dietetics at State University of New York at Buffalo. “When discussing sensitive topics, such as binge drinking and opioid misuse, adolescents may fear that these or other risky activities may be disclosed to parents or law enforcement officials. Therefore, adolescent health providers should be aware of local, state, and federal laws pertaining to the confidentiality of minors.”
She added, “adolescents are often susceptible to others’ influences, so having open communication and support from a trusted adult – be it a parent or clinician – may also be protective against risky behaviors.”
The study by Vaca et al. was funded by the National Institutes of Health with support from the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development; the National Heart, Lung, and Blood Institute; the National Institute on Alcohol Abuse and Alcoholism; the National Institute on Drug Abuse; and the Maternal and Child Health Bureau of the Health Resources and Services Administration. The study by Bhatia et al. had no external funding. The authors from both studies reported no relevant financial disclosures. Dr. Kulak said she had no financial disclosures or other conflicts of interest.
SOURCE: Vaca FE et al. Pediatrics. 2020; doi: 10.1542/peds.2018-4095. Bhatia D et al. Pediatrics. 2020; doi: 10.1542/peds.2019-2470.
These newly published reports indicate the high prevalence of risky behaviors and their associations – cross-sectionally and longitudinally – with major threats to adolescent health – so asking about alcohol use, opioid misuse, and associated health risks is truly “in the lane” of clinicians, school professionals, and parents who see and care about adolescents.
At this point, I think it’s incontrovertible that clinicians should screen adolescents to learn about their physical, emotional, and behavioral health. And they should seek opportunities for professional training, skills development, and expansion of their professional networks so they are able to address – individually or collaboratively via referrals – the behavioral and psychosocial health risks of their patients.
The good news is that there is growing awareness of the importance of using validated screening tools to identify patient behavioral health risks – including those pertaining to adolescent and young adult alcohol use and opioid misuse. “Best practice” dictates that screening approaches rely on asking questions using structured tools; intuition and “just winging it” are not effective or reliable for identifying patient behavior. Forward-looking clinics and practices could be asking patients to report about health behaviors in the waiting room (on a computer tablet, for example), or even remotely (using a secure app or data collection tool) in advance of a visit. Asking should be periodic – since behaviors can change fairly rapidly among young people. The benefit is that patient-reported information can be processed in advance to cue clinician follow-up and intervention. And youth tend to share more about their behaviors when they are asked electronically, rather than face to face. Intelligent screens can provide near real-time estimation of risk – to support in-office brief intervention tailored to the risk level of a young person or to trigger follow-up.
These studies indicate that binge alcohol use and misuse of prescription opioids among adolescents are real, pervasive, and deserving of our considered attention. There is no magic bullet. However busy clinicians may have a significant role to play in identifying and addressing these problems.
Elissa Weitzman, ScD, MSc, is an associate professor of pediatrics at Harvard Medical School, Boston, and an associate scientist based in adolescent/young adult medicine and the computational health informatics program at Boston Children’s Hospital. She was asked to comment on the articles by Vaca et al. and Bhatia et al. Dr. Weitzman said she had no relevant financial disclosures.
These newly published reports indicate the high prevalence of risky behaviors and their associations – cross-sectionally and longitudinally – with major threats to adolescent health – so asking about alcohol use, opioid misuse, and associated health risks is truly “in the lane” of clinicians, school professionals, and parents who see and care about adolescents.
At this point, I think it’s incontrovertible that clinicians should screen adolescents to learn about their physical, emotional, and behavioral health. And they should seek opportunities for professional training, skills development, and expansion of their professional networks so they are able to address – individually or collaboratively via referrals – the behavioral and psychosocial health risks of their patients.
The good news is that there is growing awareness of the importance of using validated screening tools to identify patient behavioral health risks – including those pertaining to adolescent and young adult alcohol use and opioid misuse. “Best practice” dictates that screening approaches rely on asking questions using structured tools; intuition and “just winging it” are not effective or reliable for identifying patient behavior. Forward-looking clinics and practices could be asking patients to report about health behaviors in the waiting room (on a computer tablet, for example), or even remotely (using a secure app or data collection tool) in advance of a visit. Asking should be periodic – since behaviors can change fairly rapidly among young people. The benefit is that patient-reported information can be processed in advance to cue clinician follow-up and intervention. And youth tend to share more about their behaviors when they are asked electronically, rather than face to face. Intelligent screens can provide near real-time estimation of risk – to support in-office brief intervention tailored to the risk level of a young person or to trigger follow-up.
These studies indicate that binge alcohol use and misuse of prescription opioids among adolescents are real, pervasive, and deserving of our considered attention. There is no magic bullet. However busy clinicians may have a significant role to play in identifying and addressing these problems.
Elissa Weitzman, ScD, MSc, is an associate professor of pediatrics at Harvard Medical School, Boston, and an associate scientist based in adolescent/young adult medicine and the computational health informatics program at Boston Children’s Hospital. She was asked to comment on the articles by Vaca et al. and Bhatia et al. Dr. Weitzman said she had no relevant financial disclosures.
These newly published reports indicate the high prevalence of risky behaviors and their associations – cross-sectionally and longitudinally – with major threats to adolescent health – so asking about alcohol use, opioid misuse, and associated health risks is truly “in the lane” of clinicians, school professionals, and parents who see and care about adolescents.
At this point, I think it’s incontrovertible that clinicians should screen adolescents to learn about their physical, emotional, and behavioral health. And they should seek opportunities for professional training, skills development, and expansion of their professional networks so they are able to address – individually or collaboratively via referrals – the behavioral and psychosocial health risks of their patients.
The good news is that there is growing awareness of the importance of using validated screening tools to identify patient behavioral health risks – including those pertaining to adolescent and young adult alcohol use and opioid misuse. “Best practice” dictates that screening approaches rely on asking questions using structured tools; intuition and “just winging it” are not effective or reliable for identifying patient behavior. Forward-looking clinics and practices could be asking patients to report about health behaviors in the waiting room (on a computer tablet, for example), or even remotely (using a secure app or data collection tool) in advance of a visit. Asking should be periodic – since behaviors can change fairly rapidly among young people. The benefit is that patient-reported information can be processed in advance to cue clinician follow-up and intervention. And youth tend to share more about their behaviors when they are asked electronically, rather than face to face. Intelligent screens can provide near real-time estimation of risk – to support in-office brief intervention tailored to the risk level of a young person or to trigger follow-up.
These studies indicate that binge alcohol use and misuse of prescription opioids among adolescents are real, pervasive, and deserving of our considered attention. There is no magic bullet. However busy clinicians may have a significant role to play in identifying and addressing these problems.
Elissa Weitzman, ScD, MSc, is an associate professor of pediatrics at Harvard Medical School, Boston, and an associate scientist based in adolescent/young adult medicine and the computational health informatics program at Boston Children’s Hospital. She was asked to comment on the articles by Vaca et al. and Bhatia et al. Dr. Weitzman said she had no relevant financial disclosures.
Binge drinking and misuse of opioids led to risky behavior during adolescence, two studies from the journal Pediatrics highlighted. And the binge drinking in high school may predict risky driving behaviors up to 4 years after high school.
Federico E. Vaca, MD, of the developmental neurocognitive driving simulation research center at Yale University, New Haven, Conn., and colleagues examined the associations between risky driving behaviors and binge drinking of 2,785 adolescents in the nationally representative, longitudinal NEXT Generation Health Study. The researchers studied the effects of binge drinking on driving while impaired (DWI), riding with an impaired driver (RWI), blackouts, extreme binge drinking, and risky driving.
The adolescents were studied across seven waves, with Wave 1 beginning in the 2009-2010 school year (10th grade; mean age, 16 years), and data extended up to 4 years after high school. Of all adolescents enrolled, 91% completed Wave 1, 88% completed Wave 2, 86% completed Wave 3 (12th grade), 78% completed Wave 4, 79% completed Wave 5, 84% completed Wave 6, and 83% completed Wave 7 (4 years after leaving high school) of the study.
High school binge drinking predicts later risky behavior
About one-quarter of adolescents reported binge drinking in Waves 1-3, with an incidence of 27% in Wave 1, 24% in Wave 2, and 27% in Wave 3. Adolescents who reported binge drinking in Wave 3 had a higher likelihood of DWI in subsequent waves, with nearly six times higher odds in Wave 5 and more than twice as likely in Wave 7, researchers said. Binge drinking in Wave 3 also was associated with greater than four times higher odds of RWI in Wave 4, and more than two and a half times higher odds of RWI in Wave 7. Among adolescents who reported binge drinking across 3 years in high school, there was a higher likelihood of extreme binge drinking in Wave 7, and higher likelihood of risky driving after graduating.
Impact of parental knowledge of drinking
in some waves. Father monitoring knowledge of drinking in Waves 1-3 lowered the odds of DWI by 30% in Wave 5 and 20% in Wave 6, while also lowering the odds of RWI in Wave 4 and Wave 7 by 20%.
Mother knowledge of drinking in Waves 1-3 was associated with 60% lower odds of DWI in Wave 4, but did not lower odds in any wave for RWI.
Overall, parental support for not drinking lowered odds for DWI by 40% in Waves 4 and 5, and by 30% in Wave 7 while also lowering odds of RWI in Wave 4 by 20%.
The results are consistent with other studies examining risky driving behavior and binge drinking in adolescent populations, but researchers noted that “to an important but limited extent, parental practices while the teenager is in high school may protect against DWI, RWI, and blackouts as adolescents move into early adulthood.”
“Our findings are relevant to prevention programs that seek to incorporate alcohol screening with intentional inquiry about binge drinking. Moreover, our results may be instructive to programs that seek to leverage facets of parental practices to reduce health-risk contexts for youth,” Dr. Vaca and colleagues concluded. “Such prevention activities coupled with strengthening of policies and practices reducing adolescents’ access to alcohol could reduce later major alcohol-related health-risk behaviors and their consequences.”
Opioid misuse and risky behavior
In a second study, Devika Bhatia, MD, of the University of Colorado at Denver, Aurora, and colleagues examined opioid misuse in a nationally-representative sample of 14,765 adolescents from the Centers for Disease Control and Prevention’s 2017 Youth Risk Behavior Surveillance Survey. The researchers measured opioid misuse by categorizing adolescents into groups based on whether they had ever misused prescription opioids and whether they had engaged in risky driving behavior, violent behavior, risky sexual behavior, had a history of substance abuse, or attempted suicide.
Dr. Bhatia and colleagues found 14% of adolescents in the study reported misusing opioids, with an overrepresentation of 17-year-old and 18-year-old participants reporting opioid misuse (P less than .0001). there were no statistically significant difference between those who misused opioids and those who did not in terms of race, ethnicity, or sex.
Those adolescents who reported misusing opioids were 2.8 times more likely to not use a seatbelt; were 2.8 times more likely to have RWI; were 5.8 times more likely to have DWI; or 2.3 times more likely to have texted or emailed while driving. In each of these cases, P was less than .0001.
Adolescents who misused opioids also had significantly increased odds of engaging in risky sexual behaviors such as having sex before 13 years (3.9 times); having sex with four or more partners (4.8 times); using substances before sex (3.6 times); and not using a condom before sex (2.0 times). In each of these cases, P was less than .0001.
Additionally, adolescents in this category were between 5.4 times and 22.3 times more likely to use other substances (P less than .0001 for 10 variables); 4.9 times more likely to have attempted suicide (P less than .0001); or more likely to have engaged in violent behavior such as getting into physical fights (4.0 times), carrying a weapon (3.4 times) or a gun (5.1 times) within the last 30 days. In the four latter cases, P was less than .0001.
“With the ongoing opioid epidemic, pediatricians and child psychiatrists are likely to be more attuned to opioid misuse in their patients,” Dr. Bhatia and colleagues concluded. “If youth are screening positive for opioid misuse, pediatricians, nurses, social workers, child psychiatrists, and other providers assessing adolescents may have a new, broad range of other risky behaviors for which to screen regardless of the direction of the association.”
Substance use screening for treating substance use disorder traditionally has been is provided by a specialist, Jessica A. Kulak, PhD, MPH, said in an interview. “However, integration of care services may help to change societal norms around problematic substance use – both by decreasing stigma associated with substance use, as well as increasing clinicians’ preparedness, knowledge, and confidence in preventing and intervening on adolescents’ substance experimentation and use.” She recommended that clinicians in primary care improve their training by using the Substance Abuse and Mental Health Services Administration’s Screening, Brief Intervention, and Referral to Treatment program, which is available as a free online course.
Confidentiality is important in adolescent health, said Dr. Kulak, who is an assistant professor in the department of health, nutrition, and dietetics at State University of New York at Buffalo. “When discussing sensitive topics, such as binge drinking and opioid misuse, adolescents may fear that these or other risky activities may be disclosed to parents or law enforcement officials. Therefore, adolescent health providers should be aware of local, state, and federal laws pertaining to the confidentiality of minors.”
She added, “adolescents are often susceptible to others’ influences, so having open communication and support from a trusted adult – be it a parent or clinician – may also be protective against risky behaviors.”
The study by Vaca et al. was funded by the National Institutes of Health with support from the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development; the National Heart, Lung, and Blood Institute; the National Institute on Alcohol Abuse and Alcoholism; the National Institute on Drug Abuse; and the Maternal and Child Health Bureau of the Health Resources and Services Administration. The study by Bhatia et al. had no external funding. The authors from both studies reported no relevant financial disclosures. Dr. Kulak said she had no financial disclosures or other conflicts of interest.
SOURCE: Vaca FE et al. Pediatrics. 2020; doi: 10.1542/peds.2018-4095. Bhatia D et al. Pediatrics. 2020; doi: 10.1542/peds.2019-2470.
Binge drinking and misuse of opioids led to risky behavior during adolescence, two studies from the journal Pediatrics highlighted. And the binge drinking in high school may predict risky driving behaviors up to 4 years after high school.
Federico E. Vaca, MD, of the developmental neurocognitive driving simulation research center at Yale University, New Haven, Conn., and colleagues examined the associations between risky driving behaviors and binge drinking of 2,785 adolescents in the nationally representative, longitudinal NEXT Generation Health Study. The researchers studied the effects of binge drinking on driving while impaired (DWI), riding with an impaired driver (RWI), blackouts, extreme binge drinking, and risky driving.
The adolescents were studied across seven waves, with Wave 1 beginning in the 2009-2010 school year (10th grade; mean age, 16 years), and data extended up to 4 years after high school. Of all adolescents enrolled, 91% completed Wave 1, 88% completed Wave 2, 86% completed Wave 3 (12th grade), 78% completed Wave 4, 79% completed Wave 5, 84% completed Wave 6, and 83% completed Wave 7 (4 years after leaving high school) of the study.
High school binge drinking predicts later risky behavior
About one-quarter of adolescents reported binge drinking in Waves 1-3, with an incidence of 27% in Wave 1, 24% in Wave 2, and 27% in Wave 3. Adolescents who reported binge drinking in Wave 3 had a higher likelihood of DWI in subsequent waves, with nearly six times higher odds in Wave 5 and more than twice as likely in Wave 7, researchers said. Binge drinking in Wave 3 also was associated with greater than four times higher odds of RWI in Wave 4, and more than two and a half times higher odds of RWI in Wave 7. Among adolescents who reported binge drinking across 3 years in high school, there was a higher likelihood of extreme binge drinking in Wave 7, and higher likelihood of risky driving after graduating.
Impact of parental knowledge of drinking
in some waves. Father monitoring knowledge of drinking in Waves 1-3 lowered the odds of DWI by 30% in Wave 5 and 20% in Wave 6, while also lowering the odds of RWI in Wave 4 and Wave 7 by 20%.
Mother knowledge of drinking in Waves 1-3 was associated with 60% lower odds of DWI in Wave 4, but did not lower odds in any wave for RWI.
Overall, parental support for not drinking lowered odds for DWI by 40% in Waves 4 and 5, and by 30% in Wave 7 while also lowering odds of RWI in Wave 4 by 20%.
The results are consistent with other studies examining risky driving behavior and binge drinking in adolescent populations, but researchers noted that “to an important but limited extent, parental practices while the teenager is in high school may protect against DWI, RWI, and blackouts as adolescents move into early adulthood.”
“Our findings are relevant to prevention programs that seek to incorporate alcohol screening with intentional inquiry about binge drinking. Moreover, our results may be instructive to programs that seek to leverage facets of parental practices to reduce health-risk contexts for youth,” Dr. Vaca and colleagues concluded. “Such prevention activities coupled with strengthening of policies and practices reducing adolescents’ access to alcohol could reduce later major alcohol-related health-risk behaviors and their consequences.”
Opioid misuse and risky behavior
In a second study, Devika Bhatia, MD, of the University of Colorado at Denver, Aurora, and colleagues examined opioid misuse in a nationally-representative sample of 14,765 adolescents from the Centers for Disease Control and Prevention’s 2017 Youth Risk Behavior Surveillance Survey. The researchers measured opioid misuse by categorizing adolescents into groups based on whether they had ever misused prescription opioids and whether they had engaged in risky driving behavior, violent behavior, risky sexual behavior, had a history of substance abuse, or attempted suicide.
Dr. Bhatia and colleagues found 14% of adolescents in the study reported misusing opioids, with an overrepresentation of 17-year-old and 18-year-old participants reporting opioid misuse (P less than .0001). there were no statistically significant difference between those who misused opioids and those who did not in terms of race, ethnicity, or sex.
Those adolescents who reported misusing opioids were 2.8 times more likely to not use a seatbelt; were 2.8 times more likely to have RWI; were 5.8 times more likely to have DWI; or 2.3 times more likely to have texted or emailed while driving. In each of these cases, P was less than .0001.
Adolescents who misused opioids also had significantly increased odds of engaging in risky sexual behaviors such as having sex before 13 years (3.9 times); having sex with four or more partners (4.8 times); using substances before sex (3.6 times); and not using a condom before sex (2.0 times). In each of these cases, P was less than .0001.
Additionally, adolescents in this category were between 5.4 times and 22.3 times more likely to use other substances (P less than .0001 for 10 variables); 4.9 times more likely to have attempted suicide (P less than .0001); or more likely to have engaged in violent behavior such as getting into physical fights (4.0 times), carrying a weapon (3.4 times) or a gun (5.1 times) within the last 30 days. In the four latter cases, P was less than .0001.
“With the ongoing opioid epidemic, pediatricians and child psychiatrists are likely to be more attuned to opioid misuse in their patients,” Dr. Bhatia and colleagues concluded. “If youth are screening positive for opioid misuse, pediatricians, nurses, social workers, child psychiatrists, and other providers assessing adolescents may have a new, broad range of other risky behaviors for which to screen regardless of the direction of the association.”
Substance use screening for treating substance use disorder traditionally has been is provided by a specialist, Jessica A. Kulak, PhD, MPH, said in an interview. “However, integration of care services may help to change societal norms around problematic substance use – both by decreasing stigma associated with substance use, as well as increasing clinicians’ preparedness, knowledge, and confidence in preventing and intervening on adolescents’ substance experimentation and use.” She recommended that clinicians in primary care improve their training by using the Substance Abuse and Mental Health Services Administration’s Screening, Brief Intervention, and Referral to Treatment program, which is available as a free online course.
Confidentiality is important in adolescent health, said Dr. Kulak, who is an assistant professor in the department of health, nutrition, and dietetics at State University of New York at Buffalo. “When discussing sensitive topics, such as binge drinking and opioid misuse, adolescents may fear that these or other risky activities may be disclosed to parents or law enforcement officials. Therefore, adolescent health providers should be aware of local, state, and federal laws pertaining to the confidentiality of minors.”
She added, “adolescents are often susceptible to others’ influences, so having open communication and support from a trusted adult – be it a parent or clinician – may also be protective against risky behaviors.”
The study by Vaca et al. was funded by the National Institutes of Health with support from the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development; the National Heart, Lung, and Blood Institute; the National Institute on Alcohol Abuse and Alcoholism; the National Institute on Drug Abuse; and the Maternal and Child Health Bureau of the Health Resources and Services Administration. The study by Bhatia et al. had no external funding. The authors from both studies reported no relevant financial disclosures. Dr. Kulak said she had no financial disclosures or other conflicts of interest.
SOURCE: Vaca FE et al. Pediatrics. 2020; doi: 10.1542/peds.2018-4095. Bhatia D et al. Pediatrics. 2020; doi: 10.1542/peds.2019-2470.
FROM PEDIATRICS
Adenotonsillectomy doesn’t improve cognitive function in preschoolers with OSA
according to a prospective study.
The study showed no significant difference in global IQ at 12 months between children who underwent adenotonsillectomy and those who did not. However, as expected, the adenotonsillectomy group did experience improvements in sleep.

Karen A. Waters, MBBS, PhD, of the Children’s Hospital at Westmead and the University of Sydney, and her colleagues reported these results in Pediatrics. There also was a related commentary.
The study enrolled 190 children (ages 3-5 years) with mild obstructive sleep apnea. Roughly half of patients (n = 99) were randomized to early adenotonsillectomy (within 2 months), and the other half (n = 91) were randomized to no adenotonsillectomy (12-month routine wait). There were 121 patients who had global IQ assessments at 12 months, as measured by the Woodcock Johnson III Brief Intellectual Ability (BIA) test. Of these patients, 61 were in the adenotonsillectomy group, and 60 were in the control group.
Both groups had improvements in BIA scores from baseline to 12 months, and the 12-month BIA score was not significantly different between the groups.
At baseline, the mean W score (task proficiency) for BIA was 448.36 in the adenotonsillectomy group and 451.3 in the control group. At 12 months, the scores were 465.46 and 463.12, respectively (P = .29).
“Intellectual ability scores improved in both groups over time with no effect attributable to the intervention [adenotonsillectomy],” Dr. Waters and her colleagues wrote.
However, patients in the adenotonsillectomy group did have greater improvements in sleep than patients in the control group, as assessed by polysomnogram and parent reports.
In the adenotonsillectomy group, the mean total sleep time was 469.2 minutes at baseline and 481.8 minutes at 12 months. In the control group, the mean total sleep time was 463.8 minutes at baseline and 475.3 minutes at 12 months. The adjusted mean difference was –2.12 (P less than .001).
According to parent reports, children in the adenotonsillectomy group were significantly less likely than those in the control group to have trouble sleeping at night at 12 months: 8% and 74%, respectively (P less than .001).
“Children randomly assigned to adenotonsillectomy did show greater improvement in polysomnography obstructive indices and parent-reported behavior but did not demonstrate a treatment-attributable improvement in cognitive function,” David O. Francis, MD, of University of Wisconsin–Madison, and Derek J. Lam, MD, of Oregon Health & Science University in Portland, wrote in a related commentary.
The commentators noted that these results are similar to those of the CHAT study, which showed no significant differences in Developmental Neuropsychological Assessment results between children (ages 5-9 years) who underwent adenotonsillectomy and those who did not (N Engl J Med. 2013 Jun 20;368[25]:2366-76).
The current study was funded by the National Health and Medical Research Council, Sydney University, The Garnett Passe and Rodney Williams Memorial Foundation, and The Golden Casket, Brisbane. Dr. Waters, her coauthors, and the commentary authors said they have no relevant conflicts of interest. The commentators received no external funding.
SOURCE: Waters KA et al. Pediatrics. 2020;145(2):e20191450; Francis DO and Lam DJ. Pediatrics. 2020;145(2):e20192479.
according to a prospective study.
The study showed no significant difference in global IQ at 12 months between children who underwent adenotonsillectomy and those who did not. However, as expected, the adenotonsillectomy group did experience improvements in sleep.
Karen A. Waters, MBBS, PhD, of the Children’s Hospital at Westmead and the University of Sydney, and her colleagues reported these results in Pediatrics. There also was a related commentary.
The study enrolled 190 children (ages 3-5 years) with mild obstructive sleep apnea. Roughly half of patients (n = 99) were randomized to early adenotonsillectomy (within 2 months), and the other half (n = 91) were randomized to no adenotonsillectomy (12-month routine wait). There were 121 patients who had global IQ assessments at 12 months, as measured by the Woodcock Johnson III Brief Intellectual Ability (BIA) test. Of these patients, 61 were in the adenotonsillectomy group, and 60 were in the control group.
Both groups had improvements in BIA scores from baseline to 12 months, and the 12-month BIA score was not significantly different between the groups.
At baseline, the mean W score (task proficiency) for BIA was 448.36 in the adenotonsillectomy group and 451.3 in the control group. At 12 months, the scores were 465.46 and 463.12, respectively (P = .29).
“Intellectual ability scores improved in both groups over time with no effect attributable to the intervention [adenotonsillectomy],” Dr. Waters and her colleagues wrote.
However, patients in the adenotonsillectomy group did have greater improvements in sleep than patients in the control group, as assessed by polysomnogram and parent reports.
In the adenotonsillectomy group, the mean total sleep time was 469.2 minutes at baseline and 481.8 minutes at 12 months. In the control group, the mean total sleep time was 463.8 minutes at baseline and 475.3 minutes at 12 months. The adjusted mean difference was –2.12 (P less than .001).
According to parent reports, children in the adenotonsillectomy group were significantly less likely than those in the control group to have trouble sleeping at night at 12 months: 8% and 74%, respectively (P less than .001).
“Children randomly assigned to adenotonsillectomy did show greater improvement in polysomnography obstructive indices and parent-reported behavior but did not demonstrate a treatment-attributable improvement in cognitive function,” David O. Francis, MD, of University of Wisconsin–Madison, and Derek J. Lam, MD, of Oregon Health & Science University in Portland, wrote in a related commentary.
The commentators noted that these results are similar to those of the CHAT study, which showed no significant differences in Developmental Neuropsychological Assessment results between children (ages 5-9 years) who underwent adenotonsillectomy and those who did not (N Engl J Med. 2013 Jun 20;368[25]:2366-76).
The current study was funded by the National Health and Medical Research Council, Sydney University, The Garnett Passe and Rodney Williams Memorial Foundation, and The Golden Casket, Brisbane. Dr. Waters, her coauthors, and the commentary authors said they have no relevant conflicts of interest. The commentators received no external funding.
SOURCE: Waters KA et al. Pediatrics. 2020;145(2):e20191450; Francis DO and Lam DJ. Pediatrics. 2020;145(2):e20192479.
according to a prospective study.
The study showed no significant difference in global IQ at 12 months between children who underwent adenotonsillectomy and those who did not. However, as expected, the adenotonsillectomy group did experience improvements in sleep.
Karen A. Waters, MBBS, PhD, of the Children’s Hospital at Westmead and the University of Sydney, and her colleagues reported these results in Pediatrics. There also was a related commentary.
The study enrolled 190 children (ages 3-5 years) with mild obstructive sleep apnea. Roughly half of patients (n = 99) were randomized to early adenotonsillectomy (within 2 months), and the other half (n = 91) were randomized to no adenotonsillectomy (12-month routine wait). There were 121 patients who had global IQ assessments at 12 months, as measured by the Woodcock Johnson III Brief Intellectual Ability (BIA) test. Of these patients, 61 were in the adenotonsillectomy group, and 60 were in the control group.
Both groups had improvements in BIA scores from baseline to 12 months, and the 12-month BIA score was not significantly different between the groups.
At baseline, the mean W score (task proficiency) for BIA was 448.36 in the adenotonsillectomy group and 451.3 in the control group. At 12 months, the scores were 465.46 and 463.12, respectively (P = .29).
“Intellectual ability scores improved in both groups over time with no effect attributable to the intervention [adenotonsillectomy],” Dr. Waters and her colleagues wrote.
However, patients in the adenotonsillectomy group did have greater improvements in sleep than patients in the control group, as assessed by polysomnogram and parent reports.
In the adenotonsillectomy group, the mean total sleep time was 469.2 minutes at baseline and 481.8 minutes at 12 months. In the control group, the mean total sleep time was 463.8 minutes at baseline and 475.3 minutes at 12 months. The adjusted mean difference was –2.12 (P less than .001).
According to parent reports, children in the adenotonsillectomy group were significantly less likely than those in the control group to have trouble sleeping at night at 12 months: 8% and 74%, respectively (P less than .001).
“Children randomly assigned to adenotonsillectomy did show greater improvement in polysomnography obstructive indices and parent-reported behavior but did not demonstrate a treatment-attributable improvement in cognitive function,” David O. Francis, MD, of University of Wisconsin–Madison, and Derek J. Lam, MD, of Oregon Health & Science University in Portland, wrote in a related commentary.
The commentators noted that these results are similar to those of the CHAT study, which showed no significant differences in Developmental Neuropsychological Assessment results between children (ages 5-9 years) who underwent adenotonsillectomy and those who did not (N Engl J Med. 2013 Jun 20;368[25]:2366-76).
The current study was funded by the National Health and Medical Research Council, Sydney University, The Garnett Passe and Rodney Williams Memorial Foundation, and The Golden Casket, Brisbane. Dr. Waters, her coauthors, and the commentary authors said they have no relevant conflicts of interest. The commentators received no external funding.
SOURCE: Waters KA et al. Pediatrics. 2020;145(2):e20191450; Francis DO and Lam DJ. Pediatrics. 2020;145(2):e20192479.
FROM PEDIATRICS
Navitoclax may overcome ruxolitinib resistance in MF
ORLANDO – Adding navitoclax to ruxolitinib improved responses in a phase 2 trial of patients with uncontrolled myelofibrosis (MF) and prolonged exposure to ruxolitinib.

Navitoclax and ruxolitinib yielded “clinically meaningful” spleen responses, improved total symptom scores, and produced “encouraging” reductions in bone marrow fibrosis among patients with primary or secondary MF, according to Jacqueline S. Garcia, MD, of Dana-Farber Cancer Institute in Boston.
Dr. Garcia presented these results at the annual meeting of the American Society of Hematology.
Navitoclax binds with high affinity to BCL-XL, BCL-2, and BCL-W, Dr. Garcia noted. Preclinical research has shown that combining Janus kinase 2 (JAK2) inhibition with BCL-XL/BCL-2 inhibition has a synergistic cytotoxic effect on JAK2-mutated cells (Blood. 2009 Feb 12;113[7]:1522-5), and BCL-XL inhibition can overcome resistance to JAK2 inhibition (Cell Rep. 2013 Nov 27;5[4]:1047-59).
These findings led to the theory that combining navitoclax and ruxolitinib could overcome resistance to JAK2 inhibition in MF. The researchers tested this theory in a phase 2 trial (NCT03222609) of 34 MF patients.
The patients had primary MF (n = 16), post–polycythemia vera MF (n = 13), and post–essential thrombocythemia MF (n = 5). At baseline, their median age was 68 years (range 42-86 years), and 68% of them were men.
There were 33 patients with genetic testing results available. None of them were triple negative, 27 had JAK2 mutations, and 7 had CALR mutations. Roughly half of patients (n = 17) were classified as high molecular risk, with mutations in ASXL1, EZH2, IDH1/2, SRSF2, or U2AF1.
Treatment
All patients had received ruxolitinib for at least 12 weeks prior to their first dose of navitoclax. They had been receiving a stable dose of 10 mg or greater, twice daily, for at least 8 weeks. The median duration of prior ruxolitinib exposure was 21 months (range, 4-71 months).
On study, patients received navitoclax once daily plus the current stable dose of ruxolitinib (10 mg or greater twice daily). Navitoclax dosing started at 50 mg, but weekly dose escalation was allowed to a maximum daily dose of 300 mg. Treatment could continue until the loss of clinical benefit, unacceptable toxicity, or discontinuation.
There were 23 patients who received the maximum dose of navitoclax, and the median duration of navitoclax treatment was 330 days (range, 29-588 days). Of the 25 patients who started the study on a ruxolitinib dose higher than 10 mg twice daily, 22 had their dose reduced to 10 mg twice daily.
Nine patients discontinued study treatment – three due to adverse events, two due to progressive disease, and four for other reasons.
Efficacy
Navitoclax appears to overcome ruxolitinib resistance, Dr. Garcia said, citing improvements in spleen size, symptom scores, bone marrow fibrosis, white blood cell counts, and transfusion needs.
Thirty patients were evaluable for spleen response. At week 24, 30% had a spleen volume reduction of 35% or greater from baseline. At any time on study, 43% of patients had a spleen volume reduction of 35% or greater from baseline. More than half of patients (53%) had a resolution of palpable splenomegaly.
Eight of 32 patients (25%) had a reduction in bone marrow fibrosis, four with a one-grade reduction and four with a two-grade reduction.
Seventeen patients were evaluable for change in total symptom score. Eleven patients (65%) experienced a reduction in symptoms, and six (35%) had a 50% or greater reduction in symptoms. The median total symptom score was 12 (range, 0-30) at baseline and 7 (range, 0-23) at 24 weeks.
Patients had a significant reduction in white blood cells on study. The mean white blood cell reduction at week 24 was 25.8 x 109/L.
Patients’ hemoglobin levels remained stable over time, but a few patients had a decreased need for transfusions on study. Seven patients entered the study having received at least one unit of packed red blood cells in the prior 12 weeks. Four of them (57%) have had a transfusion-free period of at least 12 weeks on study.
Safety
“Navitoclax in combination with ruxolitinib appears to be well tolerated,” Dr. Garcia said.
She noted that treatment resulted in reduced platelet counts, but counts stabilized after 6-8 weeks. The mean platelet count was 232 x 109/L at baseline and 95 x 109/L at week 8.
In fact, the most common adverse event was thrombocytopenia, with any-grade thrombocytopenia occurring in 85% of patients and grade 3/4 occurring in 44%. One patient had grade 4 thrombocytopenia, but it was reversed by withholding treatment and subsequent dose modification.
Other common treatment-emergent adverse events were diarrhea (68%), fatigue (53%), nausea (35%), anemia (29%), dizziness (27%), confusion (27%), and vomiting (24%).
All 34 patients experienced at least one adverse event. Eight patients (24%) had serious adverse events, including anemia, pancytopenia, splenic infarction, upper abdominal pain, vomiting, chest pain, pneumonia, and abnormal liver function test.
One patient had a grade 5 adverse event – pneumonia – that was deemed unrelated to navitoclax.
This trial is sponsored by AbbVie. Dr. Garcia reported relationships with AbbVie, Genentech, and Pfizer.
SOURCE: Garcia JS et al. ASH 2019, Abstract 671.
ORLANDO – Adding navitoclax to ruxolitinib improved responses in a phase 2 trial of patients with uncontrolled myelofibrosis (MF) and prolonged exposure to ruxolitinib.
Navitoclax and ruxolitinib yielded “clinically meaningful” spleen responses, improved total symptom scores, and produced “encouraging” reductions in bone marrow fibrosis among patients with primary or secondary MF, according to Jacqueline S. Garcia, MD, of Dana-Farber Cancer Institute in Boston.
Dr. Garcia presented these results at the annual meeting of the American Society of Hematology.
Navitoclax binds with high affinity to BCL-XL, BCL-2, and BCL-W, Dr. Garcia noted. Preclinical research has shown that combining Janus kinase 2 (JAK2) inhibition with BCL-XL/BCL-2 inhibition has a synergistic cytotoxic effect on JAK2-mutated cells (Blood. 2009 Feb 12;113[7]:1522-5), and BCL-XL inhibition can overcome resistance to JAK2 inhibition (Cell Rep. 2013 Nov 27;5[4]:1047-59).
These findings led to the theory that combining navitoclax and ruxolitinib could overcome resistance to JAK2 inhibition in MF. The researchers tested this theory in a phase 2 trial (NCT03222609) of 34 MF patients.
The patients had primary MF (n = 16), post–polycythemia vera MF (n = 13), and post–essential thrombocythemia MF (n = 5). At baseline, their median age was 68 years (range 42-86 years), and 68% of them were men.
There were 33 patients with genetic testing results available. None of them were triple negative, 27 had JAK2 mutations, and 7 had CALR mutations. Roughly half of patients (n = 17) were classified as high molecular risk, with mutations in ASXL1, EZH2, IDH1/2, SRSF2, or U2AF1.
Treatment
All patients had received ruxolitinib for at least 12 weeks prior to their first dose of navitoclax. They had been receiving a stable dose of 10 mg or greater, twice daily, for at least 8 weeks. The median duration of prior ruxolitinib exposure was 21 months (range, 4-71 months).
On study, patients received navitoclax once daily plus the current stable dose of ruxolitinib (10 mg or greater twice daily). Navitoclax dosing started at 50 mg, but weekly dose escalation was allowed to a maximum daily dose of 300 mg. Treatment could continue until the loss of clinical benefit, unacceptable toxicity, or discontinuation.
There were 23 patients who received the maximum dose of navitoclax, and the median duration of navitoclax treatment was 330 days (range, 29-588 days). Of the 25 patients who started the study on a ruxolitinib dose higher than 10 mg twice daily, 22 had their dose reduced to 10 mg twice daily.
Nine patients discontinued study treatment – three due to adverse events, two due to progressive disease, and four for other reasons.
Efficacy
Navitoclax appears to overcome ruxolitinib resistance, Dr. Garcia said, citing improvements in spleen size, symptom scores, bone marrow fibrosis, white blood cell counts, and transfusion needs.
Thirty patients were evaluable for spleen response. At week 24, 30% had a spleen volume reduction of 35% or greater from baseline. At any time on study, 43% of patients had a spleen volume reduction of 35% or greater from baseline. More than half of patients (53%) had a resolution of palpable splenomegaly.
Eight of 32 patients (25%) had a reduction in bone marrow fibrosis, four with a one-grade reduction and four with a two-grade reduction.
Seventeen patients were evaluable for change in total symptom score. Eleven patients (65%) experienced a reduction in symptoms, and six (35%) had a 50% or greater reduction in symptoms. The median total symptom score was 12 (range, 0-30) at baseline and 7 (range, 0-23) at 24 weeks.
Patients had a significant reduction in white blood cells on study. The mean white blood cell reduction at week 24 was 25.8 x 109/L.
Patients’ hemoglobin levels remained stable over time, but a few patients had a decreased need for transfusions on study. Seven patients entered the study having received at least one unit of packed red blood cells in the prior 12 weeks. Four of them (57%) have had a transfusion-free period of at least 12 weeks on study.
Safety
“Navitoclax in combination with ruxolitinib appears to be well tolerated,” Dr. Garcia said.
She noted that treatment resulted in reduced platelet counts, but counts stabilized after 6-8 weeks. The mean platelet count was 232 x 109/L at baseline and 95 x 109/L at week 8.
In fact, the most common adverse event was thrombocytopenia, with any-grade thrombocytopenia occurring in 85% of patients and grade 3/4 occurring in 44%. One patient had grade 4 thrombocytopenia, but it was reversed by withholding treatment and subsequent dose modification.
Other common treatment-emergent adverse events were diarrhea (68%), fatigue (53%), nausea (35%), anemia (29%), dizziness (27%), confusion (27%), and vomiting (24%).
All 34 patients experienced at least one adverse event. Eight patients (24%) had serious adverse events, including anemia, pancytopenia, splenic infarction, upper abdominal pain, vomiting, chest pain, pneumonia, and abnormal liver function test.
One patient had a grade 5 adverse event – pneumonia – that was deemed unrelated to navitoclax.
This trial is sponsored by AbbVie. Dr. Garcia reported relationships with AbbVie, Genentech, and Pfizer.
SOURCE: Garcia JS et al. ASH 2019, Abstract 671.
ORLANDO – Adding navitoclax to ruxolitinib improved responses in a phase 2 trial of patients with uncontrolled myelofibrosis (MF) and prolonged exposure to ruxolitinib.
Navitoclax and ruxolitinib yielded “clinically meaningful” spleen responses, improved total symptom scores, and produced “encouraging” reductions in bone marrow fibrosis among patients with primary or secondary MF, according to Jacqueline S. Garcia, MD, of Dana-Farber Cancer Institute in Boston.
Dr. Garcia presented these results at the annual meeting of the American Society of Hematology.
Navitoclax binds with high affinity to BCL-XL, BCL-2, and BCL-W, Dr. Garcia noted. Preclinical research has shown that combining Janus kinase 2 (JAK2) inhibition with BCL-XL/BCL-2 inhibition has a synergistic cytotoxic effect on JAK2-mutated cells (Blood. 2009 Feb 12;113[7]:1522-5), and BCL-XL inhibition can overcome resistance to JAK2 inhibition (Cell Rep. 2013 Nov 27;5[4]:1047-59).
These findings led to the theory that combining navitoclax and ruxolitinib could overcome resistance to JAK2 inhibition in MF. The researchers tested this theory in a phase 2 trial (NCT03222609) of 34 MF patients.
The patients had primary MF (n = 16), post–polycythemia vera MF (n = 13), and post–essential thrombocythemia MF (n = 5). At baseline, their median age was 68 years (range 42-86 years), and 68% of them were men.
There were 33 patients with genetic testing results available. None of them were triple negative, 27 had JAK2 mutations, and 7 had CALR mutations. Roughly half of patients (n = 17) were classified as high molecular risk, with mutations in ASXL1, EZH2, IDH1/2, SRSF2, or U2AF1.
Treatment
All patients had received ruxolitinib for at least 12 weeks prior to their first dose of navitoclax. They had been receiving a stable dose of 10 mg or greater, twice daily, for at least 8 weeks. The median duration of prior ruxolitinib exposure was 21 months (range, 4-71 months).
On study, patients received navitoclax once daily plus the current stable dose of ruxolitinib (10 mg or greater twice daily). Navitoclax dosing started at 50 mg, but weekly dose escalation was allowed to a maximum daily dose of 300 mg. Treatment could continue until the loss of clinical benefit, unacceptable toxicity, or discontinuation.
There were 23 patients who received the maximum dose of navitoclax, and the median duration of navitoclax treatment was 330 days (range, 29-588 days). Of the 25 patients who started the study on a ruxolitinib dose higher than 10 mg twice daily, 22 had their dose reduced to 10 mg twice daily.
Nine patients discontinued study treatment – three due to adverse events, two due to progressive disease, and four for other reasons.
Efficacy
Navitoclax appears to overcome ruxolitinib resistance, Dr. Garcia said, citing improvements in spleen size, symptom scores, bone marrow fibrosis, white blood cell counts, and transfusion needs.
Thirty patients were evaluable for spleen response. At week 24, 30% had a spleen volume reduction of 35% or greater from baseline. At any time on study, 43% of patients had a spleen volume reduction of 35% or greater from baseline. More than half of patients (53%) had a resolution of palpable splenomegaly.
Eight of 32 patients (25%) had a reduction in bone marrow fibrosis, four with a one-grade reduction and four with a two-grade reduction.
Seventeen patients were evaluable for change in total symptom score. Eleven patients (65%) experienced a reduction in symptoms, and six (35%) had a 50% or greater reduction in symptoms. The median total symptom score was 12 (range, 0-30) at baseline and 7 (range, 0-23) at 24 weeks.
Patients had a significant reduction in white blood cells on study. The mean white blood cell reduction at week 24 was 25.8 x 109/L.
Patients’ hemoglobin levels remained stable over time, but a few patients had a decreased need for transfusions on study. Seven patients entered the study having received at least one unit of packed red blood cells in the prior 12 weeks. Four of them (57%) have had a transfusion-free period of at least 12 weeks on study.
Safety
“Navitoclax in combination with ruxolitinib appears to be well tolerated,” Dr. Garcia said.
She noted that treatment resulted in reduced platelet counts, but counts stabilized after 6-8 weeks. The mean platelet count was 232 x 109/L at baseline and 95 x 109/L at week 8.
In fact, the most common adverse event was thrombocytopenia, with any-grade thrombocytopenia occurring in 85% of patients and grade 3/4 occurring in 44%. One patient had grade 4 thrombocytopenia, but it was reversed by withholding treatment and subsequent dose modification.
Other common treatment-emergent adverse events were diarrhea (68%), fatigue (53%), nausea (35%), anemia (29%), dizziness (27%), confusion (27%), and vomiting (24%).
All 34 patients experienced at least one adverse event. Eight patients (24%) had serious adverse events, including anemia, pancytopenia, splenic infarction, upper abdominal pain, vomiting, chest pain, pneumonia, and abnormal liver function test.
One patient had a grade 5 adverse event – pneumonia – that was deemed unrelated to navitoclax.
This trial is sponsored by AbbVie. Dr. Garcia reported relationships with AbbVie, Genentech, and Pfizer.
SOURCE: Garcia JS et al. ASH 2019, Abstract 671.
REPORTING FROM ASH 2019
FDA Awards Grant To Study Temozolomide in Gist
A phase 2 study of temozolomide in gastrointestinal stromal tumors (GIST) received one of the 12 grants awarded in October by the US Food and Drug Administration (FDA) to enhance the development of medical products for patients with rare diseases. Jason Sicklick, MD, and the University of California San Diego in La Jolla will receive $1.5 million over 3 years to conduct the phase 2 study (NCT03556384). The objective of the study is to determine the efficacy at 6 months of temozolomide therapy in patients with the SDH-mutant/deficient subtype. Temozolomide is approved by the FDA to treat newly diagnosed glioblastoma multiforme and refractory anaplastic astrocytomas. It is not approved for the treatment of SDH-mutant/deficient GIST.
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. Other orphan diseases receiving grants included glomerulopathy, gliomas, Fanconi anemia, sickle cell respiratory complications, Duchenne muscular dystrophy, HPV associated respiratory papillomatosis, refractory viral infections and T-cell immunodeficiency, oral cancer, retinoblastoma, cerebellar brain tumors, and acute myeloid leukemia.
A phase 2 study of temozolomide in gastrointestinal stromal tumors (GIST) received one of the 12 grants awarded in October by the US Food and Drug Administration (FDA) to enhance the development of medical products for patients with rare diseases. Jason Sicklick, MD, and the University of California San Diego in La Jolla will receive $1.5 million over 3 years to conduct the phase 2 study (NCT03556384). The objective of the study is to determine the efficacy at 6 months of temozolomide therapy in patients with the SDH-mutant/deficient subtype. Temozolomide is approved by the FDA to treat newly diagnosed glioblastoma multiforme and refractory anaplastic astrocytomas. It is not approved for the treatment of SDH-mutant/deficient GIST.
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. Other orphan diseases receiving grants included glomerulopathy, gliomas, Fanconi anemia, sickle cell respiratory complications, Duchenne muscular dystrophy, HPV associated respiratory papillomatosis, refractory viral infections and T-cell immunodeficiency, oral cancer, retinoblastoma, cerebellar brain tumors, and acute myeloid leukemia.
A phase 2 study of temozolomide in gastrointestinal stromal tumors (GIST) received one of the 12 grants awarded in October by the US Food and Drug Administration (FDA) to enhance the development of medical products for patients with rare diseases. Jason Sicklick, MD, and the University of California San Diego in La Jolla will receive $1.5 million over 3 years to conduct the phase 2 study (NCT03556384). The objective of the study is to determine the efficacy at 6 months of temozolomide therapy in patients with the SDH-mutant/deficient subtype. Temozolomide is approved by the FDA to treat newly diagnosed glioblastoma multiforme and refractory anaplastic astrocytomas. It is not approved for the treatment of SDH-mutant/deficient GIST.
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. Other orphan diseases receiving grants included glomerulopathy, gliomas, Fanconi anemia, sickle cell respiratory complications, Duchenne muscular dystrophy, HPV associated respiratory papillomatosis, refractory viral infections and T-cell immunodeficiency, oral cancer, retinoblastoma, cerebellar brain tumors, and acute myeloid leukemia.