User login
Motherhood can get old fast, and snubbing can become phubbing
Killer babies and their aging mommies
The joys of new parenthood are endless, like the long nights and functioning on 4 hours of sleep. But those babies sure are sweet, and deadly. That’s right, little Johnny junior is shaving years off of your life.

. But hold on, that doesn’t mean mothers need to update their driver licenses. There’s a difference between biological and chronological age.
Biological aging is measured by epigenetics, which analyzes changes in DNA over time by determining whether coding for certain proteins is turned on or off. The process acts as a sort of clock, lead author Judith E. Carroll, PhD, said in a separate statement, allowing scientists to estimate a person’s biological age.
Although loss of sleep may accelerate biological aging and increase health risks, the researchers don’t want people to think that lack of sleep during infant care is going to automatically cause permanent damage. The jury is still out on whether the effects are long lasting. Instead, they emphasized the importance of prioritizing sleep needs and getting some help from others to do it.
“With every hour of additional sleep, the mother’s biological age was younger,” Dr. Carroll said. “I, and many other sleep scientists, consider sleep health to be just as vital to overall health as diet and exercise.”
So, new moms, fix that gourmet dinner after you go for that run because you’re already up at 4 a.m. anyway. It’s all about balance.
Me and my phone-y phriends
It’s been months since you’ve seen your friends in person. You got your vaccine and so, after all this time, you can finally meet with your friends in real life. No more Zoom. It’s a strange dream come true.

The problem is that half your friends barely seem interested, spending much of your time together staring at their phones. Naturally, there’s a clever term for this: You’ve just been the victim of phubbing, specifically friend phubbing or fphubbing (we’re not sure there are enough “f” sounds at the beginning of that word), and it’s been the focus of a new study from the University of Georgia.
So who are these fphubbers? Researchers found that neurotic and depressed individuals are more likely to fphub, as were those with social anxiety, since they may actually prefer online interaction over face-to-face conversation. On the flip side, people with agreeable traits were less likely to fphub, as they felt doing so would be rude and impolite. Quite a bold stance right there, we know.
The researchers noted the complete ordinariness of people pulling their phones out while with friends, and the rapid acceptance of something many people may still consider rude. It could speak to casual smartphone addiction and the urge we all get when we hear that notification in our pocket. Maybe what we need when we see friends is the equivalent of those PSAs before movies telling you to turn off your cell phones. Then you can all go down to the lobby and get yourselves a treat.
Who needs a vaccine when there’s horse paste?
It’s not the first time, and it won’t be the last, that some people think they know best when it comes to COVID-19 safety.

What is the newest “trend” for prevention and treatment? Enter, ivermectin, a Food and Drug Administration–approved drug for treating conditions caused by parasitic worms. The prescription form is hard to find these days, so some folks have been “raiding rural tractor supply stores in search of ivermectin horse paste (packed with ‘apple flavor’!) and [weighing] the benefits of taking ivermectin ‘sheep drench’,” according to the Daily Beast.
The FDA does not condone the use of ivermectin for COVID-19 and warns that the types meant for animals can be harmful to humans if taken in large doses. Facebook has played its part, as groups are forming to share conflicting information about how the drug can be used for COVID-19. The medication often comes from sketchy sources, and it’s seemingly causing more harm than good. Pharmacies are even starting to treat ivermectin as if it’s an opioid.
“My ‘horse’ had no negative side effects, and now he tells me he feels like a million bucks and is now COVID free,” one social media poster wrote in code, according to the Daily Beast.
When the card fits, COVID-19 will take a hit
Good news! We have figured out the problem behind the whole COVID-19 vaccine-denial business.

And by “we,” of course, we mean someone else. But we’re telling you about it, and isn’t that really the important part?
Anyway, back to the problem. It’s not the vaccines themselves, it’s the vaccine cards. They’re the wrong size.
The Atlantic’s Amanda Mull explains: “When I got my first shot, in late February, I sat in the mandatory waiting area, holding my new card in one hand and my wallet in the other, trying to understand why the two objects weren’t compatible.”
She didn’t get very far with the CDC, but Chelsea Cirruzzo, a public-health reporter at U.S. News & World Report who has been tweeting about the vaccine cards, suggested that “someone just printed out a bunch of cards that are easy to write your name and vaccine brand on, without thinking about wallets.”
The evidence does fit the nobody-really-gave-it-any-thought argument. The template was available to the public on some state government websites when the vaccine was approved and can still be found on Florida’s, Ms. Mull notes. “Try to imagine governments freely distributing their templates for driver’s licenses, passports, or other documents intended to certify a particular identity or status.” The FBI, we understand, frowns upon this sort of thing.
Well, there you have it, America. When the card fits in a wallet, the vaccine problem will go away. Just remember where you read it, not where we read it.
Killer babies and their aging mommies
The joys of new parenthood are endless, like the long nights and functioning on 4 hours of sleep. But those babies sure are sweet, and deadly. That’s right, little Johnny junior is shaving years off of your life.
. But hold on, that doesn’t mean mothers need to update their driver licenses. There’s a difference between biological and chronological age.
Biological aging is measured by epigenetics, which analyzes changes in DNA over time by determining whether coding for certain proteins is turned on or off. The process acts as a sort of clock, lead author Judith E. Carroll, PhD, said in a separate statement, allowing scientists to estimate a person’s biological age.
Although loss of sleep may accelerate biological aging and increase health risks, the researchers don’t want people to think that lack of sleep during infant care is going to automatically cause permanent damage. The jury is still out on whether the effects are long lasting. Instead, they emphasized the importance of prioritizing sleep needs and getting some help from others to do it.
“With every hour of additional sleep, the mother’s biological age was younger,” Dr. Carroll said. “I, and many other sleep scientists, consider sleep health to be just as vital to overall health as diet and exercise.”
So, new moms, fix that gourmet dinner after you go for that run because you’re already up at 4 a.m. anyway. It’s all about balance.
Me and my phone-y phriends
It’s been months since you’ve seen your friends in person. You got your vaccine and so, after all this time, you can finally meet with your friends in real life. No more Zoom. It’s a strange dream come true.
The problem is that half your friends barely seem interested, spending much of your time together staring at their phones. Naturally, there’s a clever term for this: You’ve just been the victim of phubbing, specifically friend phubbing or fphubbing (we’re not sure there are enough “f” sounds at the beginning of that word), and it’s been the focus of a new study from the University of Georgia.
So who are these fphubbers? Researchers found that neurotic and depressed individuals are more likely to fphub, as were those with social anxiety, since they may actually prefer online interaction over face-to-face conversation. On the flip side, people with agreeable traits were less likely to fphub, as they felt doing so would be rude and impolite. Quite a bold stance right there, we know.
The researchers noted the complete ordinariness of people pulling their phones out while with friends, and the rapid acceptance of something many people may still consider rude. It could speak to casual smartphone addiction and the urge we all get when we hear that notification in our pocket. Maybe what we need when we see friends is the equivalent of those PSAs before movies telling you to turn off your cell phones. Then you can all go down to the lobby and get yourselves a treat.
Who needs a vaccine when there’s horse paste?
It’s not the first time, and it won’t be the last, that some people think they know best when it comes to COVID-19 safety.
What is the newest “trend” for prevention and treatment? Enter, ivermectin, a Food and Drug Administration–approved drug for treating conditions caused by parasitic worms. The prescription form is hard to find these days, so some folks have been “raiding rural tractor supply stores in search of ivermectin horse paste (packed with ‘apple flavor’!) and [weighing] the benefits of taking ivermectin ‘sheep drench’,” according to the Daily Beast.
The FDA does not condone the use of ivermectin for COVID-19 and warns that the types meant for animals can be harmful to humans if taken in large doses. Facebook has played its part, as groups are forming to share conflicting information about how the drug can be used for COVID-19. The medication often comes from sketchy sources, and it’s seemingly causing more harm than good. Pharmacies are even starting to treat ivermectin as if it’s an opioid.
“My ‘horse’ had no negative side effects, and now he tells me he feels like a million bucks and is now COVID free,” one social media poster wrote in code, according to the Daily Beast.
When the card fits, COVID-19 will take a hit
Good news! We have figured out the problem behind the whole COVID-19 vaccine-denial business.
And by “we,” of course, we mean someone else. But we’re telling you about it, and isn’t that really the important part?
Anyway, back to the problem. It’s not the vaccines themselves, it’s the vaccine cards. They’re the wrong size.
The Atlantic’s Amanda Mull explains: “When I got my first shot, in late February, I sat in the mandatory waiting area, holding my new card in one hand and my wallet in the other, trying to understand why the two objects weren’t compatible.”
She didn’t get very far with the CDC, but Chelsea Cirruzzo, a public-health reporter at U.S. News & World Report who has been tweeting about the vaccine cards, suggested that “someone just printed out a bunch of cards that are easy to write your name and vaccine brand on, without thinking about wallets.”
The evidence does fit the nobody-really-gave-it-any-thought argument. The template was available to the public on some state government websites when the vaccine was approved and can still be found on Florida’s, Ms. Mull notes. “Try to imagine governments freely distributing their templates for driver’s licenses, passports, or other documents intended to certify a particular identity or status.” The FBI, we understand, frowns upon this sort of thing.
Well, there you have it, America. When the card fits in a wallet, the vaccine problem will go away. Just remember where you read it, not where we read it.
Killer babies and their aging mommies
The joys of new parenthood are endless, like the long nights and functioning on 4 hours of sleep. But those babies sure are sweet, and deadly. That’s right, little Johnny junior is shaving years off of your life.
. But hold on, that doesn’t mean mothers need to update their driver licenses. There’s a difference between biological and chronological age.
Biological aging is measured by epigenetics, which analyzes changes in DNA over time by determining whether coding for certain proteins is turned on or off. The process acts as a sort of clock, lead author Judith E. Carroll, PhD, said in a separate statement, allowing scientists to estimate a person’s biological age.
Although loss of sleep may accelerate biological aging and increase health risks, the researchers don’t want people to think that lack of sleep during infant care is going to automatically cause permanent damage. The jury is still out on whether the effects are long lasting. Instead, they emphasized the importance of prioritizing sleep needs and getting some help from others to do it.
“With every hour of additional sleep, the mother’s biological age was younger,” Dr. Carroll said. “I, and many other sleep scientists, consider sleep health to be just as vital to overall health as diet and exercise.”
So, new moms, fix that gourmet dinner after you go for that run because you’re already up at 4 a.m. anyway. It’s all about balance.
Me and my phone-y phriends
It’s been months since you’ve seen your friends in person. You got your vaccine and so, after all this time, you can finally meet with your friends in real life. No more Zoom. It’s a strange dream come true.
The problem is that half your friends barely seem interested, spending much of your time together staring at their phones. Naturally, there’s a clever term for this: You’ve just been the victim of phubbing, specifically friend phubbing or fphubbing (we’re not sure there are enough “f” sounds at the beginning of that word), and it’s been the focus of a new study from the University of Georgia.
So who are these fphubbers? Researchers found that neurotic and depressed individuals are more likely to fphub, as were those with social anxiety, since they may actually prefer online interaction over face-to-face conversation. On the flip side, people with agreeable traits were less likely to fphub, as they felt doing so would be rude and impolite. Quite a bold stance right there, we know.
The researchers noted the complete ordinariness of people pulling their phones out while with friends, and the rapid acceptance of something many people may still consider rude. It could speak to casual smartphone addiction and the urge we all get when we hear that notification in our pocket. Maybe what we need when we see friends is the equivalent of those PSAs before movies telling you to turn off your cell phones. Then you can all go down to the lobby and get yourselves a treat.
Who needs a vaccine when there’s horse paste?
It’s not the first time, and it won’t be the last, that some people think they know best when it comes to COVID-19 safety.
What is the newest “trend” for prevention and treatment? Enter, ivermectin, a Food and Drug Administration–approved drug for treating conditions caused by parasitic worms. The prescription form is hard to find these days, so some folks have been “raiding rural tractor supply stores in search of ivermectin horse paste (packed with ‘apple flavor’!) and [weighing] the benefits of taking ivermectin ‘sheep drench’,” according to the Daily Beast.
The FDA does not condone the use of ivermectin for COVID-19 and warns that the types meant for animals can be harmful to humans if taken in large doses. Facebook has played its part, as groups are forming to share conflicting information about how the drug can be used for COVID-19. The medication often comes from sketchy sources, and it’s seemingly causing more harm than good. Pharmacies are even starting to treat ivermectin as if it’s an opioid.
“My ‘horse’ had no negative side effects, and now he tells me he feels like a million bucks and is now COVID free,” one social media poster wrote in code, according to the Daily Beast.
When the card fits, COVID-19 will take a hit
Good news! We have figured out the problem behind the whole COVID-19 vaccine-denial business.
And by “we,” of course, we mean someone else. But we’re telling you about it, and isn’t that really the important part?
Anyway, back to the problem. It’s not the vaccines themselves, it’s the vaccine cards. They’re the wrong size.
The Atlantic’s Amanda Mull explains: “When I got my first shot, in late February, I sat in the mandatory waiting area, holding my new card in one hand and my wallet in the other, trying to understand why the two objects weren’t compatible.”
She didn’t get very far with the CDC, but Chelsea Cirruzzo, a public-health reporter at U.S. News & World Report who has been tweeting about the vaccine cards, suggested that “someone just printed out a bunch of cards that are easy to write your name and vaccine brand on, without thinking about wallets.”
The evidence does fit the nobody-really-gave-it-any-thought argument. The template was available to the public on some state government websites when the vaccine was approved and can still be found on Florida’s, Ms. Mull notes. “Try to imagine governments freely distributing their templates for driver’s licenses, passports, or other documents intended to certify a particular identity or status.” The FBI, we understand, frowns upon this sort of thing.
Well, there you have it, America. When the card fits in a wallet, the vaccine problem will go away. Just remember where you read it, not where we read it.
Multiple pigmented patches

This patient was given a diagnosis of erythema dyschromicum perstans (EDP), also known as ashy dermatosis because of the hyperpigmented macules that come together into confluent patches that look like burned wood. There is often an inflammatory erythematous aspect to EDP.
The etiology of EDP is unknown. It is not related to sun exposure and occurs most commonly on the trunk. Although there are case reports implicating medications or infections, no clear connection has been found. This patient’s chemotherapy may have been an inciting factor, based on her history, but it is not likely that cancer caused the EDP.
EDP tends to be chronic and difficult to treat. Fortunately, other than the itching and skin discoloration, it is usually asymptomatic and benign. Large-scale trials are lacking, but there are case reports showing benefit from narrow beam UVB treatments and topical tacrolimus.1 Laser has not proven very helpful, and the hyperpigmentation can recur.
Based on the clinical appearance of this patient’s lesion, and the fact that a previous biopsy in the same location was consistent with her diagnosis, no further testing was performed. The patient was advised to apply topical diphenhydramine to her back 4 times daily for a 2-week trial. If the diphenhydramine failed to provide relief, the next step in her treatment would have been topical tacrolimus.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Leung N, Oliveira M, Selim MA, et al. Erythema dyschromicum perstans: a case report and systematic review of histologic presentation and treatment. Int J Womens Dermatol. 2018;4:216-222. doi: 10.1016/j.ijwd.2018.08.003
This patient was given a diagnosis of erythema dyschromicum perstans (EDP), also known as ashy dermatosis because of the hyperpigmented macules that come together into confluent patches that look like burned wood. There is often an inflammatory erythematous aspect to EDP.
The etiology of EDP is unknown. It is not related to sun exposure and occurs most commonly on the trunk. Although there are case reports implicating medications or infections, no clear connection has been found. This patient’s chemotherapy may have been an inciting factor, based on her history, but it is not likely that cancer caused the EDP.
EDP tends to be chronic and difficult to treat. Fortunately, other than the itching and skin discoloration, it is usually asymptomatic and benign. Large-scale trials are lacking, but there are case reports showing benefit from narrow beam UVB treatments and topical tacrolimus.1 Laser has not proven very helpful, and the hyperpigmentation can recur.
Based on the clinical appearance of this patient’s lesion, and the fact that a previous biopsy in the same location was consistent with her diagnosis, no further testing was performed. The patient was advised to apply topical diphenhydramine to her back 4 times daily for a 2-week trial. If the diphenhydramine failed to provide relief, the next step in her treatment would have been topical tacrolimus.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
This patient was given a diagnosis of erythema dyschromicum perstans (EDP), also known as ashy dermatosis because of the hyperpigmented macules that come together into confluent patches that look like burned wood. There is often an inflammatory erythematous aspect to EDP.
The etiology of EDP is unknown. It is not related to sun exposure and occurs most commonly on the trunk. Although there are case reports implicating medications or infections, no clear connection has been found. This patient’s chemotherapy may have been an inciting factor, based on her history, but it is not likely that cancer caused the EDP.
EDP tends to be chronic and difficult to treat. Fortunately, other than the itching and skin discoloration, it is usually asymptomatic and benign. Large-scale trials are lacking, but there are case reports showing benefit from narrow beam UVB treatments and topical tacrolimus.1 Laser has not proven very helpful, and the hyperpigmentation can recur.
Based on the clinical appearance of this patient’s lesion, and the fact that a previous biopsy in the same location was consistent with her diagnosis, no further testing was performed. The patient was advised to apply topical diphenhydramine to her back 4 times daily for a 2-week trial. If the diphenhydramine failed to provide relief, the next step in her treatment would have been topical tacrolimus.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Leung N, Oliveira M, Selim MA, et al. Erythema dyschromicum perstans: a case report and systematic review of histologic presentation and treatment. Int J Womens Dermatol. 2018;4:216-222. doi: 10.1016/j.ijwd.2018.08.003
1. Leung N, Oliveira M, Selim MA, et al. Erythema dyschromicum perstans: a case report and systematic review of histologic presentation and treatment. Int J Womens Dermatol. 2018;4:216-222. doi: 10.1016/j.ijwd.2018.08.003

Opioid prescribing laws having an impact
State laws capping initial opioid prescriptions to 7 days or less have led to a reduction in opioid prescribing, a new analysis of Medicare data shows.
While overall opioid prescribing has decreased, the reduction in states with legislation restricting opioid prescribing was “significantly greater than in states without such legislation,” study investigator Michael Brenner, MD, University of Michigan, Ann Arbor, said in an interview.
The study was published online August 9 in JAMA Internal Medicine.
Significant but limited effect
Because of rising concern around the opioid crisis, 23 states representing 43% of the U.S. population passed laws from 2016 through 2018 limiting initial opioid prescription to 7 days or less.
Using Medicare data from 2013 through 2018, Dr. Brenner and colleagues conducted a before-and-after study to assess the effect of these laws.
They found that on average, the number of days an opioid was prescribed for each Medicare beneficiary decreased by 11.6 days (from 44.2 days in 2013 to 32.7 days in 2018) in states that imposed duration limits, compared with 10.1 days in states without these laws (from 43.4 days in 2013 to 33.3 days in 2018).
Prior to the start of duration limits in 2016, days an opioid was prescribed were comparable among states.
After adjusting for state-level differences in race, urbanization, median income, tobacco and alcohol use, serious mental illness, and other factors, state laws limiting opioid prescriptions to 7 days or less were associated with a reduction in prescribing of 1.7 days per enrollee, “suggesting a significant but limited outcome” for these laws, the researchers note.
, but this was not significantly different in states with limit laws versus those without. However, state laws limiting duration led to a significant reduction in days of opioid prescribed among surgeons, dentists, pain specialists, and other specialists.
Inadequate pain control?
The researchers note the study was limited to Medicare beneficiaries; however, excess opioid prescribing is prevalent across all patient populations.
In addition, it’s not possible to tell from the data whether acute pain was adequately controlled with fewer pills.
“The question of adequacy of pain control is a crucial one that has been investigated extensively in prior work but was not possible to evaluate in this particular study,” said Dr. Brenner.
However, “ample evidence supports a role for reducing opioid prescribing and that such reduction can be achieved while ensuring that pain is adequately controlled with fewer pills,” he noted.
“A persistent misconception is that opioids are uniquely powerful and effective for controlling pain. Patients may perceive that effective analgesia is being withheld when opioids are not included in a regimen,” Dr. Brenner added.
“Yet, the evidence from meta-analyses derived from large numbers of randomized clinical trials finds that [nonsteroidal anti-inflammatory drugs] NSAIDS combined with acetaminophen provide similar or improved acute pain when compared to commonly prescribed opioid regimens, based on number-needed-to-treat analyses,” he added.
In a related editorial, Deborah Grady, MD, MPH, with University of California, San Francisco, and Mitchell H. Katz, MD, president and CEO of NYC Health + Hospitals, say the decrease in opioid prescribing with duration limits was “small but probably meaningful.”
Restricting initial prescriptions to seven or fewer days is “reasonable because patients with new onset of pain should be re-evaluated in a week if the pain continues,” they write.
However, Dr. Grady and Dr. Katz “worry” that restricting initial prescriptions to shorter periods, such as 3 or 5 days, as has occurred in six states, “may result in patients with acute pain going untreated or having to go to extraordinary effort to obtain adequate pain relief.”
In their view, the data from this study suggest that limiting initial prescriptions to seven or fewer days is “helpful, but we would not restrict any further given that we do not know how it affected patients with acute pain.”
The study had no specific funding. Dr. Brenner, Dr. Grady, and Dr. Katz have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
State laws capping initial opioid prescriptions to 7 days or less have led to a reduction in opioid prescribing, a new analysis of Medicare data shows.
While overall opioid prescribing has decreased, the reduction in states with legislation restricting opioid prescribing was “significantly greater than in states without such legislation,” study investigator Michael Brenner, MD, University of Michigan, Ann Arbor, said in an interview.
The study was published online August 9 in JAMA Internal Medicine.
Significant but limited effect
Because of rising concern around the opioid crisis, 23 states representing 43% of the U.S. population passed laws from 2016 through 2018 limiting initial opioid prescription to 7 days or less.
Using Medicare data from 2013 through 2018, Dr. Brenner and colleagues conducted a before-and-after study to assess the effect of these laws.
They found that on average, the number of days an opioid was prescribed for each Medicare beneficiary decreased by 11.6 days (from 44.2 days in 2013 to 32.7 days in 2018) in states that imposed duration limits, compared with 10.1 days in states without these laws (from 43.4 days in 2013 to 33.3 days in 2018).
Prior to the start of duration limits in 2016, days an opioid was prescribed were comparable among states.
After adjusting for state-level differences in race, urbanization, median income, tobacco and alcohol use, serious mental illness, and other factors, state laws limiting opioid prescriptions to 7 days or less were associated with a reduction in prescribing of 1.7 days per enrollee, “suggesting a significant but limited outcome” for these laws, the researchers note.
, but this was not significantly different in states with limit laws versus those without. However, state laws limiting duration led to a significant reduction in days of opioid prescribed among surgeons, dentists, pain specialists, and other specialists.
Inadequate pain control?
The researchers note the study was limited to Medicare beneficiaries; however, excess opioid prescribing is prevalent across all patient populations.
In addition, it’s not possible to tell from the data whether acute pain was adequately controlled with fewer pills.
“The question of adequacy of pain control is a crucial one that has been investigated extensively in prior work but was not possible to evaluate in this particular study,” said Dr. Brenner.
However, “ample evidence supports a role for reducing opioid prescribing and that such reduction can be achieved while ensuring that pain is adequately controlled with fewer pills,” he noted.
“A persistent misconception is that opioids are uniquely powerful and effective for controlling pain. Patients may perceive that effective analgesia is being withheld when opioids are not included in a regimen,” Dr. Brenner added.
“Yet, the evidence from meta-analyses derived from large numbers of randomized clinical trials finds that [nonsteroidal anti-inflammatory drugs] NSAIDS combined with acetaminophen provide similar or improved acute pain when compared to commonly prescribed opioid regimens, based on number-needed-to-treat analyses,” he added.
In a related editorial, Deborah Grady, MD, MPH, with University of California, San Francisco, and Mitchell H. Katz, MD, president and CEO of NYC Health + Hospitals, say the decrease in opioid prescribing with duration limits was “small but probably meaningful.”
Restricting initial prescriptions to seven or fewer days is “reasonable because patients with new onset of pain should be re-evaluated in a week if the pain continues,” they write.
However, Dr. Grady and Dr. Katz “worry” that restricting initial prescriptions to shorter periods, such as 3 or 5 days, as has occurred in six states, “may result in patients with acute pain going untreated or having to go to extraordinary effort to obtain adequate pain relief.”
In their view, the data from this study suggest that limiting initial prescriptions to seven or fewer days is “helpful, but we would not restrict any further given that we do not know how it affected patients with acute pain.”
The study had no specific funding. Dr. Brenner, Dr. Grady, and Dr. Katz have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
State laws capping initial opioid prescriptions to 7 days or less have led to a reduction in opioid prescribing, a new analysis of Medicare data shows.
While overall opioid prescribing has decreased, the reduction in states with legislation restricting opioid prescribing was “significantly greater than in states without such legislation,” study investigator Michael Brenner, MD, University of Michigan, Ann Arbor, said in an interview.
The study was published online August 9 in JAMA Internal Medicine.
Significant but limited effect
Because of rising concern around the opioid crisis, 23 states representing 43% of the U.S. population passed laws from 2016 through 2018 limiting initial opioid prescription to 7 days or less.
Using Medicare data from 2013 through 2018, Dr. Brenner and colleagues conducted a before-and-after study to assess the effect of these laws.
They found that on average, the number of days an opioid was prescribed for each Medicare beneficiary decreased by 11.6 days (from 44.2 days in 2013 to 32.7 days in 2018) in states that imposed duration limits, compared with 10.1 days in states without these laws (from 43.4 days in 2013 to 33.3 days in 2018).
Prior to the start of duration limits in 2016, days an opioid was prescribed were comparable among states.
After adjusting for state-level differences in race, urbanization, median income, tobacco and alcohol use, serious mental illness, and other factors, state laws limiting opioid prescriptions to 7 days or less were associated with a reduction in prescribing of 1.7 days per enrollee, “suggesting a significant but limited outcome” for these laws, the researchers note.
, but this was not significantly different in states with limit laws versus those without. However, state laws limiting duration led to a significant reduction in days of opioid prescribed among surgeons, dentists, pain specialists, and other specialists.
Inadequate pain control?
The researchers note the study was limited to Medicare beneficiaries; however, excess opioid prescribing is prevalent across all patient populations.
In addition, it’s not possible to tell from the data whether acute pain was adequately controlled with fewer pills.
“The question of adequacy of pain control is a crucial one that has been investigated extensively in prior work but was not possible to evaluate in this particular study,” said Dr. Brenner.
However, “ample evidence supports a role for reducing opioid prescribing and that such reduction can be achieved while ensuring that pain is adequately controlled with fewer pills,” he noted.
“A persistent misconception is that opioids are uniquely powerful and effective for controlling pain. Patients may perceive that effective analgesia is being withheld when opioids are not included in a regimen,” Dr. Brenner added.
“Yet, the evidence from meta-analyses derived from large numbers of randomized clinical trials finds that [nonsteroidal anti-inflammatory drugs] NSAIDS combined with acetaminophen provide similar or improved acute pain when compared to commonly prescribed opioid regimens, based on number-needed-to-treat analyses,” he added.
In a related editorial, Deborah Grady, MD, MPH, with University of California, San Francisco, and Mitchell H. Katz, MD, president and CEO of NYC Health + Hospitals, say the decrease in opioid prescribing with duration limits was “small but probably meaningful.”
Restricting initial prescriptions to seven or fewer days is “reasonable because patients with new onset of pain should be re-evaluated in a week if the pain continues,” they write.
However, Dr. Grady and Dr. Katz “worry” that restricting initial prescriptions to shorter periods, such as 3 or 5 days, as has occurred in six states, “may result in patients with acute pain going untreated or having to go to extraordinary effort to obtain adequate pain relief.”
In their view, the data from this study suggest that limiting initial prescriptions to seven or fewer days is “helpful, but we would not restrict any further given that we do not know how it affected patients with acute pain.”
The study had no specific funding. Dr. Brenner, Dr. Grady, and Dr. Katz have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Insurance coverage for vitiligo varies widely in the U.S., analysis finds
, which may disproportionately affect patients of color.
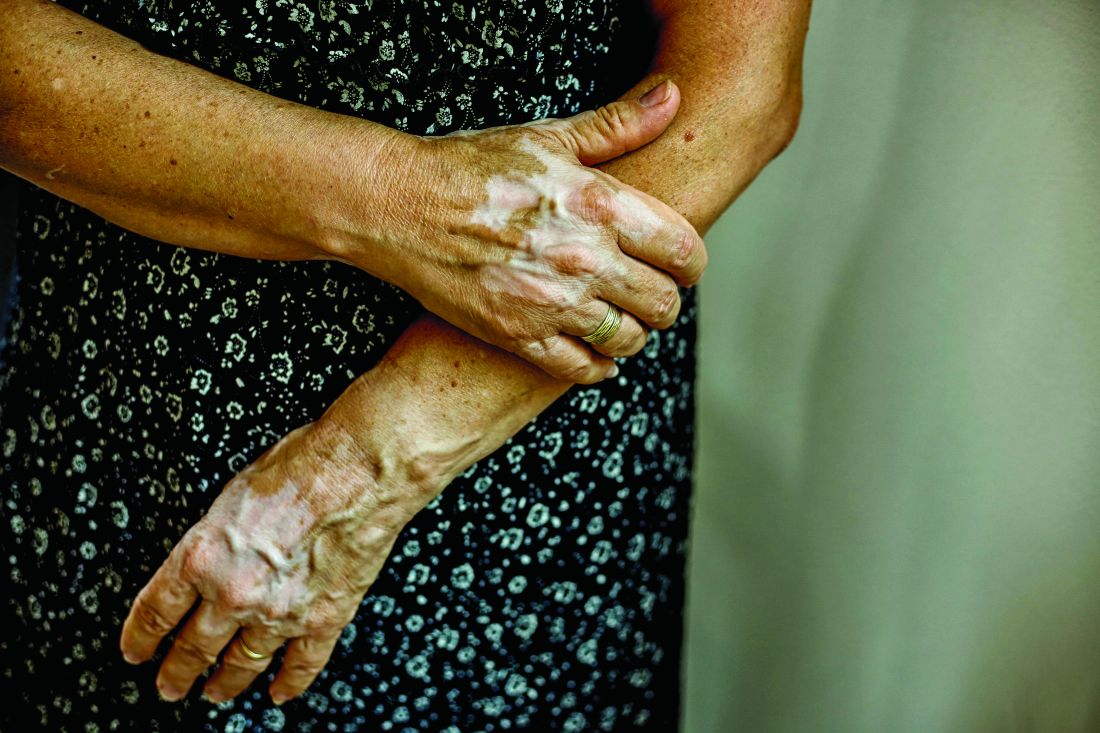
Those are the conclusions from an analysis of vitiligo treatment coverage policies across major health insurers in the United States.
“Vitiligo can be less noticeable in patients with lighter skin types, becoming apparent only when affected patches fail to tan,” first authors Andrew Blundell, MD, MSc, and Moniyka Sachar, MD, wrote in a study published online on July 16 in Pediatric Dermatology. However, they pointed out that, in patients with darker skin types, “vitiligo can be far more evident due to the stark contrast of involved versus uninvolved skin, and as such can lead to a significant impact on quality of life, as well as heightened stigmatization.”
Nevertheless, they noted many health care insurers consider vitiligo as a cosmetic condition, and do not cover treatments, and for the 1%-2% of the general population with vitiligo, “this lack of recognition from health care insurers makes treatments both less accessible and affordable, and only further marginalizes patients with this condition.”
Dr. Blundell, of San Juan Bautista School of Medicine, Caguas, P.R., and Dr. Sachar, of the department of dermatology at Brown University, Providence, R.I., and colleagues surveyed 15 commercial health care insurers, 50 BlueCross BlueShield plans, Medicare, Medicaid, and Veterans Affairs to determine the level of treatment coverage for vitiligo. They looked at office visits, medications (the topical calcineurin inhibitors [TCIs] pimecrolimus, and tacrolimus), excimer laser therapy, and phototherapy (psoralen with UVA [PUVA] and narrow-band UVB [nbUVB]). They collected information from medical policies available online or by direct contact with the plans in 2018.
The researchers reported data from 17 organizations with regional or national coverage policies for vitiligo treatment and two others – BlueCross BlueShield and Medicaid – which had policies that differed by state and plan. Of the 17 organizations, only 12% did not cover TCIs, 56% did not cover nbUVB phototherapy, 53% did not cover PUVA phototherapy, and 41% did not cover laser therapy.
As for BlueCross BlueShield, the health plan did not cover pimecrolimus and tacrolimus in 39% and 35% of states, respectively. At the same time, NbUVB and PUVA therapy were not covered in 20% and 10% of states, respectively, while excimer laser therapy was not covered in 82% of states.
Of accessible Medicaid information from 32 states, 11 did not cover topicals, 5 did not cover nbUVB, 4 did not cover PUVA, and 7 did not cover laser therapy. “The two most commonly cited reasons for denial of coverage were (a) vitiligo is considered a cosmetic condition and (b) certain therapies are not FDA-approved for vitiligo, though they may be approved for other skin conditions,” the study authors wrote.
While the analysis revealed that topical TCI therapy is more widely covered by insurance companies, compared with phototherapy, “multiple studies have shown that a combination of both topical and phototherapy is more effective in treating vitiligo than either alone,” they noted. “Vitiligo treatments can delay the progression of the disease and result in better outcomes when started early, furthering the need for insurance coverage of these treatments. If all proven and accepted vitiligo treatments were covered by their health insurers, patients would have better access, as well as timely and affordable ways by which to limit depigmentation and to repigment affected areas.”
In addition, lack of access to treatments “may increase health disparities among already-marginalized groups, such as children and adults of darker skin phototypes,” they wrote.

Seemal R. Desai, MD, who was asked to comment on the study, said that the findings resonate with him based on his clinical experience as a dermatologist at the University of Texas Southwestern Medical Center in Dallas and in clinical practice. “Vitiligo has a high psychological impact, continues to increase in its prevalence, and has been shown to be an autoimmune, chronic, inflammatory skin disease, yet we’re still having challenges with treatment,” said Dr. Desai, who is also a member of the board of directors for the American Academy of Dermatology and the Global Vitiligo Foundation (GVF).
He said that he is working with the AAD, the GVF, and other stakeholders to improve treatment coverage. For example, in Massachusetts, the Tufts Health Plan had stopped covering treatment for vitiligo. “Through a series of advocacy efforts, that was reversed a couple of years ago,” said Dr. Desai, who is also a past president of the Skin of Color Society. “We also have seen isolated reports of Medicaid and Medicare coverage where local contractors aren’t following national Centers for Medicare and Medicaid Service directive guidance. The challenge becomes, how do you get consistency in treatment coverage, and how do you make sure patients continue to get access to treatment?”
Turning the tide will require “a concerted effort” by dermatologists to engage with the payers, he added. “I’ve had to get on the phone with countless insurance companies on behalf of my patients and make them understand the comorbidities associated with vitiligo, sending them copies of studies that show it’s an autoimmune disease linked to thyroid issues,” Dr. Desai continued. “We talk a lot about the psychological burden and quality of life. There’s still a lot of work to be done in this sphere, but I think we’re making progress.”
With hopes that Janus kinase (JAK) inhibitors and other new products being investigated will soon be approved as a treatment option for vitiligo, Dr. Desai said that now is the time to standardize coverage for patients. “It’s important that we start talking about insurance coverage and denial issues now and get ahead of it, so that when we get those JAK inhibitors available, we don’t fight coverage decisions then.”
The researchers acknowledged certain limitations of the study, including the fact that it was based on insurance coverage from 2017 to 2018 and the lack of easily available state Medicaid policies.
The study coauthors were Colleen K. Gabel, MD, of the University of Massachusetts, Worcester, and Lionel G. Bercovitch, MD, of Brown University. None of the study authors reported financial disclosures.
Dr. Desai disclosed that he has conducted vitiligo research trials and has done consulting work for several pharmaceutical companies.
, which may disproportionately affect patients of color.
Those are the conclusions from an analysis of vitiligo treatment coverage policies across major health insurers in the United States.
“Vitiligo can be less noticeable in patients with lighter skin types, becoming apparent only when affected patches fail to tan,” first authors Andrew Blundell, MD, MSc, and Moniyka Sachar, MD, wrote in a study published online on July 16 in Pediatric Dermatology. However, they pointed out that, in patients with darker skin types, “vitiligo can be far more evident due to the stark contrast of involved versus uninvolved skin, and as such can lead to a significant impact on quality of life, as well as heightened stigmatization.”
Nevertheless, they noted many health care insurers consider vitiligo as a cosmetic condition, and do not cover treatments, and for the 1%-2% of the general population with vitiligo, “this lack of recognition from health care insurers makes treatments both less accessible and affordable, and only further marginalizes patients with this condition.”
Dr. Blundell, of San Juan Bautista School of Medicine, Caguas, P.R., and Dr. Sachar, of the department of dermatology at Brown University, Providence, R.I., and colleagues surveyed 15 commercial health care insurers, 50 BlueCross BlueShield plans, Medicare, Medicaid, and Veterans Affairs to determine the level of treatment coverage for vitiligo. They looked at office visits, medications (the topical calcineurin inhibitors [TCIs] pimecrolimus, and tacrolimus), excimer laser therapy, and phototherapy (psoralen with UVA [PUVA] and narrow-band UVB [nbUVB]). They collected information from medical policies available online or by direct contact with the plans in 2018.
The researchers reported data from 17 organizations with regional or national coverage policies for vitiligo treatment and two others – BlueCross BlueShield and Medicaid – which had policies that differed by state and plan. Of the 17 organizations, only 12% did not cover TCIs, 56% did not cover nbUVB phototherapy, 53% did not cover PUVA phototherapy, and 41% did not cover laser therapy.
As for BlueCross BlueShield, the health plan did not cover pimecrolimus and tacrolimus in 39% and 35% of states, respectively. At the same time, NbUVB and PUVA therapy were not covered in 20% and 10% of states, respectively, while excimer laser therapy was not covered in 82% of states.
Of accessible Medicaid information from 32 states, 11 did not cover topicals, 5 did not cover nbUVB, 4 did not cover PUVA, and 7 did not cover laser therapy. “The two most commonly cited reasons for denial of coverage were (a) vitiligo is considered a cosmetic condition and (b) certain therapies are not FDA-approved for vitiligo, though they may be approved for other skin conditions,” the study authors wrote.
While the analysis revealed that topical TCI therapy is more widely covered by insurance companies, compared with phototherapy, “multiple studies have shown that a combination of both topical and phototherapy is more effective in treating vitiligo than either alone,” they noted. “Vitiligo treatments can delay the progression of the disease and result in better outcomes when started early, furthering the need for insurance coverage of these treatments. If all proven and accepted vitiligo treatments were covered by their health insurers, patients would have better access, as well as timely and affordable ways by which to limit depigmentation and to repigment affected areas.”
In addition, lack of access to treatments “may increase health disparities among already-marginalized groups, such as children and adults of darker skin phototypes,” they wrote.
Seemal R. Desai, MD, who was asked to comment on the study, said that the findings resonate with him based on his clinical experience as a dermatologist at the University of Texas Southwestern Medical Center in Dallas and in clinical practice. “Vitiligo has a high psychological impact, continues to increase in its prevalence, and has been shown to be an autoimmune, chronic, inflammatory skin disease, yet we’re still having challenges with treatment,” said Dr. Desai, who is also a member of the board of directors for the American Academy of Dermatology and the Global Vitiligo Foundation (GVF).
He said that he is working with the AAD, the GVF, and other stakeholders to improve treatment coverage. For example, in Massachusetts, the Tufts Health Plan had stopped covering treatment for vitiligo. “Through a series of advocacy efforts, that was reversed a couple of years ago,” said Dr. Desai, who is also a past president of the Skin of Color Society. “We also have seen isolated reports of Medicaid and Medicare coverage where local contractors aren’t following national Centers for Medicare and Medicaid Service directive guidance. The challenge becomes, how do you get consistency in treatment coverage, and how do you make sure patients continue to get access to treatment?”
Turning the tide will require “a concerted effort” by dermatologists to engage with the payers, he added. “I’ve had to get on the phone with countless insurance companies on behalf of my patients and make them understand the comorbidities associated with vitiligo, sending them copies of studies that show it’s an autoimmune disease linked to thyroid issues,” Dr. Desai continued. “We talk a lot about the psychological burden and quality of life. There’s still a lot of work to be done in this sphere, but I think we’re making progress.”
With hopes that Janus kinase (JAK) inhibitors and other new products being investigated will soon be approved as a treatment option for vitiligo, Dr. Desai said that now is the time to standardize coverage for patients. “It’s important that we start talking about insurance coverage and denial issues now and get ahead of it, so that when we get those JAK inhibitors available, we don’t fight coverage decisions then.”
The researchers acknowledged certain limitations of the study, including the fact that it was based on insurance coverage from 2017 to 2018 and the lack of easily available state Medicaid policies.
The study coauthors were Colleen K. Gabel, MD, of the University of Massachusetts, Worcester, and Lionel G. Bercovitch, MD, of Brown University. None of the study authors reported financial disclosures.
Dr. Desai disclosed that he has conducted vitiligo research trials and has done consulting work for several pharmaceutical companies.
, which may disproportionately affect patients of color.
Those are the conclusions from an analysis of vitiligo treatment coverage policies across major health insurers in the United States.
“Vitiligo can be less noticeable in patients with lighter skin types, becoming apparent only when affected patches fail to tan,” first authors Andrew Blundell, MD, MSc, and Moniyka Sachar, MD, wrote in a study published online on July 16 in Pediatric Dermatology. However, they pointed out that, in patients with darker skin types, “vitiligo can be far more evident due to the stark contrast of involved versus uninvolved skin, and as such can lead to a significant impact on quality of life, as well as heightened stigmatization.”
Nevertheless, they noted many health care insurers consider vitiligo as a cosmetic condition, and do not cover treatments, and for the 1%-2% of the general population with vitiligo, “this lack of recognition from health care insurers makes treatments both less accessible and affordable, and only further marginalizes patients with this condition.”
Dr. Blundell, of San Juan Bautista School of Medicine, Caguas, P.R., and Dr. Sachar, of the department of dermatology at Brown University, Providence, R.I., and colleagues surveyed 15 commercial health care insurers, 50 BlueCross BlueShield plans, Medicare, Medicaid, and Veterans Affairs to determine the level of treatment coverage for vitiligo. They looked at office visits, medications (the topical calcineurin inhibitors [TCIs] pimecrolimus, and tacrolimus), excimer laser therapy, and phototherapy (psoralen with UVA [PUVA] and narrow-band UVB [nbUVB]). They collected information from medical policies available online or by direct contact with the plans in 2018.
The researchers reported data from 17 organizations with regional or national coverage policies for vitiligo treatment and two others – BlueCross BlueShield and Medicaid – which had policies that differed by state and plan. Of the 17 organizations, only 12% did not cover TCIs, 56% did not cover nbUVB phototherapy, 53% did not cover PUVA phototherapy, and 41% did not cover laser therapy.
As for BlueCross BlueShield, the health plan did not cover pimecrolimus and tacrolimus in 39% and 35% of states, respectively. At the same time, NbUVB and PUVA therapy were not covered in 20% and 10% of states, respectively, while excimer laser therapy was not covered in 82% of states.
Of accessible Medicaid information from 32 states, 11 did not cover topicals, 5 did not cover nbUVB, 4 did not cover PUVA, and 7 did not cover laser therapy. “The two most commonly cited reasons for denial of coverage were (a) vitiligo is considered a cosmetic condition and (b) certain therapies are not FDA-approved for vitiligo, though they may be approved for other skin conditions,” the study authors wrote.
While the analysis revealed that topical TCI therapy is more widely covered by insurance companies, compared with phototherapy, “multiple studies have shown that a combination of both topical and phototherapy is more effective in treating vitiligo than either alone,” they noted. “Vitiligo treatments can delay the progression of the disease and result in better outcomes when started early, furthering the need for insurance coverage of these treatments. If all proven and accepted vitiligo treatments were covered by their health insurers, patients would have better access, as well as timely and affordable ways by which to limit depigmentation and to repigment affected areas.”
In addition, lack of access to treatments “may increase health disparities among already-marginalized groups, such as children and adults of darker skin phototypes,” they wrote.
Seemal R. Desai, MD, who was asked to comment on the study, said that the findings resonate with him based on his clinical experience as a dermatologist at the University of Texas Southwestern Medical Center in Dallas and in clinical practice. “Vitiligo has a high psychological impact, continues to increase in its prevalence, and has been shown to be an autoimmune, chronic, inflammatory skin disease, yet we’re still having challenges with treatment,” said Dr. Desai, who is also a member of the board of directors for the American Academy of Dermatology and the Global Vitiligo Foundation (GVF).
He said that he is working with the AAD, the GVF, and other stakeholders to improve treatment coverage. For example, in Massachusetts, the Tufts Health Plan had stopped covering treatment for vitiligo. “Through a series of advocacy efforts, that was reversed a couple of years ago,” said Dr. Desai, who is also a past president of the Skin of Color Society. “We also have seen isolated reports of Medicaid and Medicare coverage where local contractors aren’t following national Centers for Medicare and Medicaid Service directive guidance. The challenge becomes, how do you get consistency in treatment coverage, and how do you make sure patients continue to get access to treatment?”
Turning the tide will require “a concerted effort” by dermatologists to engage with the payers, he added. “I’ve had to get on the phone with countless insurance companies on behalf of my patients and make them understand the comorbidities associated with vitiligo, sending them copies of studies that show it’s an autoimmune disease linked to thyroid issues,” Dr. Desai continued. “We talk a lot about the psychological burden and quality of life. There’s still a lot of work to be done in this sphere, but I think we’re making progress.”
With hopes that Janus kinase (JAK) inhibitors and other new products being investigated will soon be approved as a treatment option for vitiligo, Dr. Desai said that now is the time to standardize coverage for patients. “It’s important that we start talking about insurance coverage and denial issues now and get ahead of it, so that when we get those JAK inhibitors available, we don’t fight coverage decisions then.”
The researchers acknowledged certain limitations of the study, including the fact that it was based on insurance coverage from 2017 to 2018 and the lack of easily available state Medicaid policies.
The study coauthors were Colleen K. Gabel, MD, of the University of Massachusetts, Worcester, and Lionel G. Bercovitch, MD, of Brown University. None of the study authors reported financial disclosures.
Dr. Desai disclosed that he has conducted vitiligo research trials and has done consulting work for several pharmaceutical companies.
FROM PEDIATRIC DERMATOLOGY
Being a good neighbor
My neighbor’s house got burglarized recently.

They were on vacation, and so the thieves were able to take their time inside late at night. The neighborhood wasn’t aware anything was going on until they’d left, with a lot of jewelry and other valuables. As of this writing, they haven’t been caught.
I’m not the kind of person who needs to be close with my neighbors. Some people want a cohesive bunch that does stuff together. That’s not me. I’m fine just being collegial. I wave, I say hi, I let them know if they left a garage door open. I keep to myself and hope they do the same. If we’d been suspicious about a burglary, though, I definitely would have called 911, but all of us were asleep.
I get along with the family that lives there. We occasionally chat about nothing in particular when getting the mail or rolling out the recycling can. I’m pretty sure they don’t vote the way I do, or have the same religious beliefs, but that’s life. I mean, isn’t that the point of America, or even civilization? That we’re all supposed to get along, accept our differences, and work together for the common good? In spite of politicians trying to push the country as an us-against-them narrative, the bottom line is that .
I and the rest of the block offered them any help we could provide in the aftermath. A burglary isn’t as serious as a house fire or medical emergency, but it’s still something that you want to assist with if possible.
A crisis, minor or major, is a good time to step back from the inflammatory rhetoric that television’s talking heads and pundits push. The majority of us live in peace with our neighbors, want to help them if needed, and don’t take any joy in their predicaments – regardless of what we each might believe. After all, next time it could be me.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
My neighbor’s house got burglarized recently.
They were on vacation, and so the thieves were able to take their time inside late at night. The neighborhood wasn’t aware anything was going on until they’d left, with a lot of jewelry and other valuables. As of this writing, they haven’t been caught.
I’m not the kind of person who needs to be close with my neighbors. Some people want a cohesive bunch that does stuff together. That’s not me. I’m fine just being collegial. I wave, I say hi, I let them know if they left a garage door open. I keep to myself and hope they do the same. If we’d been suspicious about a burglary, though, I definitely would have called 911, but all of us were asleep.
I get along with the family that lives there. We occasionally chat about nothing in particular when getting the mail or rolling out the recycling can. I’m pretty sure they don’t vote the way I do, or have the same religious beliefs, but that’s life. I mean, isn’t that the point of America, or even civilization? That we’re all supposed to get along, accept our differences, and work together for the common good? In spite of politicians trying to push the country as an us-against-them narrative, the bottom line is that .
I and the rest of the block offered them any help we could provide in the aftermath. A burglary isn’t as serious as a house fire or medical emergency, but it’s still something that you want to assist with if possible.
A crisis, minor or major, is a good time to step back from the inflammatory rhetoric that television’s talking heads and pundits push. The majority of us live in peace with our neighbors, want to help them if needed, and don’t take any joy in their predicaments – regardless of what we each might believe. After all, next time it could be me.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
My neighbor’s house got burglarized recently.
They were on vacation, and so the thieves were able to take their time inside late at night. The neighborhood wasn’t aware anything was going on until they’d left, with a lot of jewelry and other valuables. As of this writing, they haven’t been caught.
I’m not the kind of person who needs to be close with my neighbors. Some people want a cohesive bunch that does stuff together. That’s not me. I’m fine just being collegial. I wave, I say hi, I let them know if they left a garage door open. I keep to myself and hope they do the same. If we’d been suspicious about a burglary, though, I definitely would have called 911, but all of us were asleep.
I get along with the family that lives there. We occasionally chat about nothing in particular when getting the mail or rolling out the recycling can. I’m pretty sure they don’t vote the way I do, or have the same religious beliefs, but that’s life. I mean, isn’t that the point of America, or even civilization? That we’re all supposed to get along, accept our differences, and work together for the common good? In spite of politicians trying to push the country as an us-against-them narrative, the bottom line is that .
I and the rest of the block offered them any help we could provide in the aftermath. A burglary isn’t as serious as a house fire or medical emergency, but it’s still something that you want to assist with if possible.
A crisis, minor or major, is a good time to step back from the inflammatory rhetoric that television’s talking heads and pundits push. The majority of us live in peace with our neighbors, want to help them if needed, and don’t take any joy in their predicaments – regardless of what we each might believe. After all, next time it could be me.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
COVID-19 mitigation measures led to shifts in typical annual respiratory virus patterns
Nonpharmaceutical interventions, such as masking, staying home, limiting travel, and social distancing, have been doing more than reducing the risk for COVID-19. They’re also having an impact on infection rates and the timing of seasonal surges of other common respiratory diseases, according to an article published July 23 in Morbidity and Mortality Weekly Report.
Typically, respiratory pathogens such as respiratory syncytial virus (RSV), common cold coronaviruses, parainfluenza viruses, and respiratory adenoviruses increase in the fall and remain high throughout winter, following the same basic patterns as influenza. Although the historically low rates of influenza remained low into spring 2021, that’s not the case for several other common respiratory viruses.
“Clinicians should be aware of increases in some respiratory virus activity and remain vigilant for off-season increases,” wrote Sonja J. Olsen, PhD, and her colleagues at the Centers for Disease Control and Prevention. She told this news organization that clinicians should use multipathogen testing to help guide treatment.
The authors also underscore the importance of fall influenza vaccination campaigns for anyone aged 6 months or older.
Timothy Brewer, MD, MPH, a professor of medicine in the Division of Infectious Diseases at the University of California, Los Angeles (UCLA), and of epidemiology at the UCLA Fielding School of Public Health, agreed that it’s important for health care professionals to consider off-season illnesses in their patients.
“Practitioners should be aware that if they see a sick child in the summer, outside of what normally might be influenza season, but they look like they have influenza, consider potentially influenza and test for it, because it might be possible that we may have disrupted that natural pattern,” Dr. Brewer told this news organization. Dr. Brewer, who was not involved in the CDC research, said it’s also “critically important” to encourage influenza vaccination as the season approaches.
The CDC researchers used the U.S. World Health Organization Collaborating Laboratories System and the CDC’s National Respiratory and Enteric Virus Surveillance System to analyze virologic data from Oct. 3, 2020, to May 22, 2021, for influenza and Jan. 4, 2020, to May 22, 2021, for other respiratory viruses. The authors compared virus circulation during these periods to circulation during the same dates from four previous years.
Data to calculate influenza and RSV hospitalization rates came from the Influenza Hospitalization Surveillance Network and RSV Hospitalization Surveillance Network.
The authors report that flu activity dropped dramatically in March 2020 to its lowest levels since 1997, the earliest season for which data are available. Only 0.2% of more than 1 million specimens tested positive for influenza; the rate of hospitalizations for lab-confirmed flu was 0.8 per 100,000 people. Flu levels remained low through the summer, fall, and on to May 2021.
A potential drawback to this low activity, however, is a more prevalent and severe upcoming flu season, the authors write. The repeated exposure to flu viruses every year often “does not lead to illness, but it does serve to boost our immune response to influenza viruses,” Dr. Olsen said in an interview. “The absence of influenza viruses in the community over the last year means that we are not getting these regular boosts to our immune system. When we finally get exposed, our body may mount a weak response, and this could mean we develop a more clinically severe illness.”
Children are most susceptible to that phenomenon because they haven’t had a lifetime of exposure to flu viruses, Dr. Olsen said.
“An immunologically naive child may be more likely to develop a severe illness than someone who has lived through several influenza seasons,” she said. “This is why it is especially important for everyone 6 months and older to get vaccinated against influenza this season.”
Rhinovirus and enterovirus infections rebounded fairly quickly after their decline in March 2020 and started increasing in May 2020 until they reached “near prepandemic seasonal levels,” the authors write.
RSV infections dropped from 15.3% of weekly positive results in January 2020 to 1.4% by April and then stayed below 1% through the end of 2020. In past years, weekly positive results climbed to 3% in October and peaked at 12.5% to 16.7% in late December. Instead, RSV weekly positive results began increasing in April 2021, rising from 1.1% to 2.8% in May.
The “unusually timed” late spring increase in RSV “is probably associated with various nonpharmaceutical measures that have been in place but are now relaxing,” Dr. Olsen stated.
The RSV hospitalization rate was 0.3 per 100,000 people from October 2020 to April 2021, compared to 27.1 and 33.4 per 100,000 people in the previous 2 years. Of all RSV hospitalizations in the past year, 76.5% occurred in April-May 2021.
Rates of illness caused by the four common human coronaviruses (OC43, NL63, 229E, and HKU1) dropped from 7.5% of weekly positive results in January 2020 to 1.3% in April 2020 and stayed below 1% through February 2021. Then they climbed to 6.6% by May 2021. Infection rates of parainfluenza viruses types 1-4 similarly dropped from 2.6% in January 2020 to 1% in March 2020 and stayed below 1% until April 2021. Since then, rates of the common coronaviruses increased to 6.6% and parainfluenza viruses to 10.9% in May 2021.
Normally, parainfluenza viruses peak in October-November and May-June, so “the current increase could represent a return to prepandemic seasonality,” the authors write.
Human pneumoviruses’ weekly positive results initially increased from 4.2% in January 2020 to 7% in March and then fell to 1.9% the second week of April and remained below 1% through May 2021. In typical years, these viruses peak from 6.2% to 7.7% in March-April. Respiratory adenovirus activity similarly dropped to historically low levels in April 2021 and then began increasing to reach 3% by May 2021, the usual level for that month.
“The different circulation patterns observed across respiratory viruses probably also reflect differences in the virus transmission routes and how effective various nonpharmaceutical measures are at stopping transmission,” Dr. Olsen said in an interview. “As pandemic mitigation measures continue to be adjusted, we expect to see more changes in the circulation of these viruses, including a return to prepandemic circulation, as seen for rhinoviruses and enteroviruses.”
Rhinovirus and enterovirus rates dropped from 14.9% in March 2020 to 3.2% in May – lower than typical – and then climbed to a peak in October 2020. The peak (21.7% weekly positive results) was, however, still lower than the usual median of 32.8%. After dropping to 9.9% in January 2021, it then rose 19.1% in May, potentially reflecting “the usual spring peak that has occurred in previous years,” the authors write.
The authors note that it’s not yet clear how the COVID-19 pandemic and related mitigation measures will continue to affect respiratory virus circulation.
The authors hypothesize that the reasons for a seeming return to seasonal activity of respiratory adenoviruses, rhinoviruses, and enteroviruses could involve “different transmission mechanisms, the role of asymptomatic transmission, and prolonged survival of these nonenveloped viruses on surfaces, all of which might make these viruses less susceptible to nonpharmaceutical interventions.”
Dr. Brewer, of UCLA, agreed.
All the viruses basically “flatline except for adenoviruses and enteroviruses, and they behave a little differently in terms of how they spread,” he said. “Enteroviruses are much more likely to be fecal-oral spread than the other viruses [in the study].”
The delayed circulation of parainfluenza and human coronaviruses may have resulted from suspension of in-person classes through late winter 2020, they write, but that doesn’t explain the relative absence of pneumovirus activity, which usually affects the same young pediatric populations as RSV.
Dr. Brewer said California is seeing a surge of RSV right now, as are many states, especially throughout in the South. He’s not surprised by RSV’s deferred season, because those most affected – children younger than 2 years – are less likely to wear masks now and were “not going to daycare, not being out in public” in 2020. “As people are doing more activities, that’s probably why RSV has been starting to go up since April,” he said.
Despite the fact that, unlike many East Asian cultures, the United States has not traditionally been a mask-wearing culture, Dr. Brewer wouldn’t be surprised if more Americans begin wearing masks during flu season. “Hopefully another thing that will come out of this is better hand hygiene, with people just getting used to washing their hands more, particularly after they come home from being out,” he added.
Dr. Brewer similarly emphasized the importance of flu vaccination for the upcoming season, especially for younger children who may have poorer natural immunity to influenza, owing to its low circulation rates in 2020-2021.
The study was funded by the CDC. Dr. Brewer and Dr. Olsen have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Nonpharmaceutical interventions, such as masking, staying home, limiting travel, and social distancing, have been doing more than reducing the risk for COVID-19. They’re also having an impact on infection rates and the timing of seasonal surges of other common respiratory diseases, according to an article published July 23 in Morbidity and Mortality Weekly Report.
Typically, respiratory pathogens such as respiratory syncytial virus (RSV), common cold coronaviruses, parainfluenza viruses, and respiratory adenoviruses increase in the fall and remain high throughout winter, following the same basic patterns as influenza. Although the historically low rates of influenza remained low into spring 2021, that’s not the case for several other common respiratory viruses.
“Clinicians should be aware of increases in some respiratory virus activity and remain vigilant for off-season increases,” wrote Sonja J. Olsen, PhD, and her colleagues at the Centers for Disease Control and Prevention. She told this news organization that clinicians should use multipathogen testing to help guide treatment.
The authors also underscore the importance of fall influenza vaccination campaigns for anyone aged 6 months or older.
Timothy Brewer, MD, MPH, a professor of medicine in the Division of Infectious Diseases at the University of California, Los Angeles (UCLA), and of epidemiology at the UCLA Fielding School of Public Health, agreed that it’s important for health care professionals to consider off-season illnesses in their patients.
“Practitioners should be aware that if they see a sick child in the summer, outside of what normally might be influenza season, but they look like they have influenza, consider potentially influenza and test for it, because it might be possible that we may have disrupted that natural pattern,” Dr. Brewer told this news organization. Dr. Brewer, who was not involved in the CDC research, said it’s also “critically important” to encourage influenza vaccination as the season approaches.
The CDC researchers used the U.S. World Health Organization Collaborating Laboratories System and the CDC’s National Respiratory and Enteric Virus Surveillance System to analyze virologic data from Oct. 3, 2020, to May 22, 2021, for influenza and Jan. 4, 2020, to May 22, 2021, for other respiratory viruses. The authors compared virus circulation during these periods to circulation during the same dates from four previous years.
Data to calculate influenza and RSV hospitalization rates came from the Influenza Hospitalization Surveillance Network and RSV Hospitalization Surveillance Network.
The authors report that flu activity dropped dramatically in March 2020 to its lowest levels since 1997, the earliest season for which data are available. Only 0.2% of more than 1 million specimens tested positive for influenza; the rate of hospitalizations for lab-confirmed flu was 0.8 per 100,000 people. Flu levels remained low through the summer, fall, and on to May 2021.
A potential drawback to this low activity, however, is a more prevalent and severe upcoming flu season, the authors write. The repeated exposure to flu viruses every year often “does not lead to illness, but it does serve to boost our immune response to influenza viruses,” Dr. Olsen said in an interview. “The absence of influenza viruses in the community over the last year means that we are not getting these regular boosts to our immune system. When we finally get exposed, our body may mount a weak response, and this could mean we develop a more clinically severe illness.”
Children are most susceptible to that phenomenon because they haven’t had a lifetime of exposure to flu viruses, Dr. Olsen said.
“An immunologically naive child may be more likely to develop a severe illness than someone who has lived through several influenza seasons,” she said. “This is why it is especially important for everyone 6 months and older to get vaccinated against influenza this season.”
Rhinovirus and enterovirus infections rebounded fairly quickly after their decline in March 2020 and started increasing in May 2020 until they reached “near prepandemic seasonal levels,” the authors write.
RSV infections dropped from 15.3% of weekly positive results in January 2020 to 1.4% by April and then stayed below 1% through the end of 2020. In past years, weekly positive results climbed to 3% in October and peaked at 12.5% to 16.7% in late December. Instead, RSV weekly positive results began increasing in April 2021, rising from 1.1% to 2.8% in May.
The “unusually timed” late spring increase in RSV “is probably associated with various nonpharmaceutical measures that have been in place but are now relaxing,” Dr. Olsen stated.
The RSV hospitalization rate was 0.3 per 100,000 people from October 2020 to April 2021, compared to 27.1 and 33.4 per 100,000 people in the previous 2 years. Of all RSV hospitalizations in the past year, 76.5% occurred in April-May 2021.
Rates of illness caused by the four common human coronaviruses (OC43, NL63, 229E, and HKU1) dropped from 7.5% of weekly positive results in January 2020 to 1.3% in April 2020 and stayed below 1% through February 2021. Then they climbed to 6.6% by May 2021. Infection rates of parainfluenza viruses types 1-4 similarly dropped from 2.6% in January 2020 to 1% in March 2020 and stayed below 1% until April 2021. Since then, rates of the common coronaviruses increased to 6.6% and parainfluenza viruses to 10.9% in May 2021.
Normally, parainfluenza viruses peak in October-November and May-June, so “the current increase could represent a return to prepandemic seasonality,” the authors write.
Human pneumoviruses’ weekly positive results initially increased from 4.2% in January 2020 to 7% in March and then fell to 1.9% the second week of April and remained below 1% through May 2021. In typical years, these viruses peak from 6.2% to 7.7% in March-April. Respiratory adenovirus activity similarly dropped to historically low levels in April 2021 and then began increasing to reach 3% by May 2021, the usual level for that month.
“The different circulation patterns observed across respiratory viruses probably also reflect differences in the virus transmission routes and how effective various nonpharmaceutical measures are at stopping transmission,” Dr. Olsen said in an interview. “As pandemic mitigation measures continue to be adjusted, we expect to see more changes in the circulation of these viruses, including a return to prepandemic circulation, as seen for rhinoviruses and enteroviruses.”
Rhinovirus and enterovirus rates dropped from 14.9% in March 2020 to 3.2% in May – lower than typical – and then climbed to a peak in October 2020. The peak (21.7% weekly positive results) was, however, still lower than the usual median of 32.8%. After dropping to 9.9% in January 2021, it then rose 19.1% in May, potentially reflecting “the usual spring peak that has occurred in previous years,” the authors write.
The authors note that it’s not yet clear how the COVID-19 pandemic and related mitigation measures will continue to affect respiratory virus circulation.
The authors hypothesize that the reasons for a seeming return to seasonal activity of respiratory adenoviruses, rhinoviruses, and enteroviruses could involve “different transmission mechanisms, the role of asymptomatic transmission, and prolonged survival of these nonenveloped viruses on surfaces, all of which might make these viruses less susceptible to nonpharmaceutical interventions.”
Dr. Brewer, of UCLA, agreed.
All the viruses basically “flatline except for adenoviruses and enteroviruses, and they behave a little differently in terms of how they spread,” he said. “Enteroviruses are much more likely to be fecal-oral spread than the other viruses [in the study].”
The delayed circulation of parainfluenza and human coronaviruses may have resulted from suspension of in-person classes through late winter 2020, they write, but that doesn’t explain the relative absence of pneumovirus activity, which usually affects the same young pediatric populations as RSV.
Dr. Brewer said California is seeing a surge of RSV right now, as are many states, especially throughout in the South. He’s not surprised by RSV’s deferred season, because those most affected – children younger than 2 years – are less likely to wear masks now and were “not going to daycare, not being out in public” in 2020. “As people are doing more activities, that’s probably why RSV has been starting to go up since April,” he said.
Despite the fact that, unlike many East Asian cultures, the United States has not traditionally been a mask-wearing culture, Dr. Brewer wouldn’t be surprised if more Americans begin wearing masks during flu season. “Hopefully another thing that will come out of this is better hand hygiene, with people just getting used to washing their hands more, particularly after they come home from being out,” he added.
Dr. Brewer similarly emphasized the importance of flu vaccination for the upcoming season, especially for younger children who may have poorer natural immunity to influenza, owing to its low circulation rates in 2020-2021.
The study was funded by the CDC. Dr. Brewer and Dr. Olsen have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Nonpharmaceutical interventions, such as masking, staying home, limiting travel, and social distancing, have been doing more than reducing the risk for COVID-19. They’re also having an impact on infection rates and the timing of seasonal surges of other common respiratory diseases, according to an article published July 23 in Morbidity and Mortality Weekly Report.
Typically, respiratory pathogens such as respiratory syncytial virus (RSV), common cold coronaviruses, parainfluenza viruses, and respiratory adenoviruses increase in the fall and remain high throughout winter, following the same basic patterns as influenza. Although the historically low rates of influenza remained low into spring 2021, that’s not the case for several other common respiratory viruses.
“Clinicians should be aware of increases in some respiratory virus activity and remain vigilant for off-season increases,” wrote Sonja J. Olsen, PhD, and her colleagues at the Centers for Disease Control and Prevention. She told this news organization that clinicians should use multipathogen testing to help guide treatment.
The authors also underscore the importance of fall influenza vaccination campaigns for anyone aged 6 months or older.
Timothy Brewer, MD, MPH, a professor of medicine in the Division of Infectious Diseases at the University of California, Los Angeles (UCLA), and of epidemiology at the UCLA Fielding School of Public Health, agreed that it’s important for health care professionals to consider off-season illnesses in their patients.
“Practitioners should be aware that if they see a sick child in the summer, outside of what normally might be influenza season, but they look like they have influenza, consider potentially influenza and test for it, because it might be possible that we may have disrupted that natural pattern,” Dr. Brewer told this news organization. Dr. Brewer, who was not involved in the CDC research, said it’s also “critically important” to encourage influenza vaccination as the season approaches.
The CDC researchers used the U.S. World Health Organization Collaborating Laboratories System and the CDC’s National Respiratory and Enteric Virus Surveillance System to analyze virologic data from Oct. 3, 2020, to May 22, 2021, for influenza and Jan. 4, 2020, to May 22, 2021, for other respiratory viruses. The authors compared virus circulation during these periods to circulation during the same dates from four previous years.
Data to calculate influenza and RSV hospitalization rates came from the Influenza Hospitalization Surveillance Network and RSV Hospitalization Surveillance Network.
The authors report that flu activity dropped dramatically in March 2020 to its lowest levels since 1997, the earliest season for which data are available. Only 0.2% of more than 1 million specimens tested positive for influenza; the rate of hospitalizations for lab-confirmed flu was 0.8 per 100,000 people. Flu levels remained low through the summer, fall, and on to May 2021.
A potential drawback to this low activity, however, is a more prevalent and severe upcoming flu season, the authors write. The repeated exposure to flu viruses every year often “does not lead to illness, but it does serve to boost our immune response to influenza viruses,” Dr. Olsen said in an interview. “The absence of influenza viruses in the community over the last year means that we are not getting these regular boosts to our immune system. When we finally get exposed, our body may mount a weak response, and this could mean we develop a more clinically severe illness.”
Children are most susceptible to that phenomenon because they haven’t had a lifetime of exposure to flu viruses, Dr. Olsen said.
“An immunologically naive child may be more likely to develop a severe illness than someone who has lived through several influenza seasons,” she said. “This is why it is especially important for everyone 6 months and older to get vaccinated against influenza this season.”
Rhinovirus and enterovirus infections rebounded fairly quickly after their decline in March 2020 and started increasing in May 2020 until they reached “near prepandemic seasonal levels,” the authors write.
RSV infections dropped from 15.3% of weekly positive results in January 2020 to 1.4% by April and then stayed below 1% through the end of 2020. In past years, weekly positive results climbed to 3% in October and peaked at 12.5% to 16.7% in late December. Instead, RSV weekly positive results began increasing in April 2021, rising from 1.1% to 2.8% in May.
The “unusually timed” late spring increase in RSV “is probably associated with various nonpharmaceutical measures that have been in place but are now relaxing,” Dr. Olsen stated.
The RSV hospitalization rate was 0.3 per 100,000 people from October 2020 to April 2021, compared to 27.1 and 33.4 per 100,000 people in the previous 2 years. Of all RSV hospitalizations in the past year, 76.5% occurred in April-May 2021.
Rates of illness caused by the four common human coronaviruses (OC43, NL63, 229E, and HKU1) dropped from 7.5% of weekly positive results in January 2020 to 1.3% in April 2020 and stayed below 1% through February 2021. Then they climbed to 6.6% by May 2021. Infection rates of parainfluenza viruses types 1-4 similarly dropped from 2.6% in January 2020 to 1% in March 2020 and stayed below 1% until April 2021. Since then, rates of the common coronaviruses increased to 6.6% and parainfluenza viruses to 10.9% in May 2021.
Normally, parainfluenza viruses peak in October-November and May-June, so “the current increase could represent a return to prepandemic seasonality,” the authors write.
Human pneumoviruses’ weekly positive results initially increased from 4.2% in January 2020 to 7% in March and then fell to 1.9% the second week of April and remained below 1% through May 2021. In typical years, these viruses peak from 6.2% to 7.7% in March-April. Respiratory adenovirus activity similarly dropped to historically low levels in April 2021 and then began increasing to reach 3% by May 2021, the usual level for that month.
“The different circulation patterns observed across respiratory viruses probably also reflect differences in the virus transmission routes and how effective various nonpharmaceutical measures are at stopping transmission,” Dr. Olsen said in an interview. “As pandemic mitigation measures continue to be adjusted, we expect to see more changes in the circulation of these viruses, including a return to prepandemic circulation, as seen for rhinoviruses and enteroviruses.”
Rhinovirus and enterovirus rates dropped from 14.9% in March 2020 to 3.2% in May – lower than typical – and then climbed to a peak in October 2020. The peak (21.7% weekly positive results) was, however, still lower than the usual median of 32.8%. After dropping to 9.9% in January 2021, it then rose 19.1% in May, potentially reflecting “the usual spring peak that has occurred in previous years,” the authors write.
The authors note that it’s not yet clear how the COVID-19 pandemic and related mitigation measures will continue to affect respiratory virus circulation.
The authors hypothesize that the reasons for a seeming return to seasonal activity of respiratory adenoviruses, rhinoviruses, and enteroviruses could involve “different transmission mechanisms, the role of asymptomatic transmission, and prolonged survival of these nonenveloped viruses on surfaces, all of which might make these viruses less susceptible to nonpharmaceutical interventions.”
Dr. Brewer, of UCLA, agreed.
All the viruses basically “flatline except for adenoviruses and enteroviruses, and they behave a little differently in terms of how they spread,” he said. “Enteroviruses are much more likely to be fecal-oral spread than the other viruses [in the study].”
The delayed circulation of parainfluenza and human coronaviruses may have resulted from suspension of in-person classes through late winter 2020, they write, but that doesn’t explain the relative absence of pneumovirus activity, which usually affects the same young pediatric populations as RSV.
Dr. Brewer said California is seeing a surge of RSV right now, as are many states, especially throughout in the South. He’s not surprised by RSV’s deferred season, because those most affected – children younger than 2 years – are less likely to wear masks now and were “not going to daycare, not being out in public” in 2020. “As people are doing more activities, that’s probably why RSV has been starting to go up since April,” he said.
Despite the fact that, unlike many East Asian cultures, the United States has not traditionally been a mask-wearing culture, Dr. Brewer wouldn’t be surprised if more Americans begin wearing masks during flu season. “Hopefully another thing that will come out of this is better hand hygiene, with people just getting used to washing their hands more, particularly after they come home from being out,” he added.
Dr. Brewer similarly emphasized the importance of flu vaccination for the upcoming season, especially for younger children who may have poorer natural immunity to influenza, owing to its low circulation rates in 2020-2021.
The study was funded by the CDC. Dr. Brewer and Dr. Olsen have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Why aren’t more women doctors in the top-paying specialties?
Women compose only 6% of orthopedic surgeons, 8% of interventional cardiologists, 10% of urologists, 17% of plastic surgeons, and 18% of otolaryngologists, according to the 2020 Association of American Medical Colleges Physician Specialty Data Report.
Plastic surgeons earn an average of $526,000 annually, which is the highest-paying specialty. Otolaryngologists earn an average of $417,000 annually, and urologists earn $427,000, according to the Medscape Physician Compensation Report 2021: The Recovery Begins.
Yet, far more women are practicing in specialties that pay less. Women are the majority in pediatrics (64%), ob.gyn. (59%), internal medicine (53%), and endocrinology (51%), the AAMC data show. The exception is dermatology, which pays well and in which 51% are women. The annual average pay is $394,000.
Why are so many women avoiding the top-paying specialties?
Several physician researchers and leaders in the top-paying specialties point to four main factors: Women are attracted to specialties that have more women in faculty and leadership positions, women prioritize work-life balance over pay, women residents may be deterred from the high-paying specialties because of gender discrimination and sexual harassment, and the longer training periods for surgical specialties may be a deterrent for women who want to have children.
Lack of women leaders
The specialties with the most women tend to have the highest proportion of women in leadership positions. For example, obstetrics and gynecology had the highest proportion of women department chairs (24.1%) and vice chairs (38.8). Pediatrics had the highest proportion of women division directors (31.5%) and residency program directors (64.6%), a study shows.
Surgical specialties, on the other hand, may have a harder time attracting female residents, possibly because of a lack of women in leadership positions. A recent study that examined gender differences in attitudes toward surgery training found that women would be more likely to go into surgery if there were more surgical faculty and residents of their same gender.
An analysis of orthopedic residency programs shows that more trainees were drawn to programs that had more female faculty members, including associate professors and women in leadership positions.

Terri Malcolm, MD, a board-certified ob.gyn. and CEO/founder of Master Physician Leaders, said women need to consider whether they want to be a trailblazer in a specialty that has fewer women. “What support systems are in place to accommodate your goals, whether it’s career advancement, having a family, or mentorship? Where can you show up as your whole self and be supported in that?”
Being the only woman in a residency program can be a challenge, said Dr. Malcolm. If the residents and attendings are predominantly men, for example, they may not think about creating a call schedule that takes into account maternity leave or the fact that women tend to be caretakers for their children and parents.
The study of gender differences toward surgery training shows that 75% of women, in comparison with 46% of men, would be more willing to enter surgery if maternity leave and childcare were made available to female residents and attending physicians.
Women want work-life balance
Although both men and women want families, women still shoulder more family and childcare responsibilities. That may explain why women physicians ranked work-life balance first and compensation second in the Medscape Women Physicians 2020 Report: The Issues They Care About.
“My physician colleagues have been and are supportive of intellectual abilities, but I feel they don’t fully understand the uneven distribution of childcare issues on women,” a woman dermatologist commented.

Women may want to work fewer hours or have a more flexible schedule to take care of children. “I can count on one hand the number of women who have a part-time job in orthopedics. It’s very rare, and working part time absolutely is a barrier for someone who wants to be a surgeon,” said Julie Samora, MD, PhD, a researcher and pediatric hand surgeon at Nationwide Children’s Hospital, in Columbus, Ohio. She is also a spokesperson for the American Association of Orthopedic Surgeons.
Preeti Malani, MD, a professor of medicine who specializes in infectious diseases at the University of Michigan, chose to work full-time in academia while raising two children with her husband. In a decade, she rose through the ranks to full professor. “I took the advice of a woman who wanted to recruit me to have a full-time position with maximum flexibility rather than work part time, often for more hours and less pay. I also have tried to build my career so I was not doing all clinical work.”
Her husband is a surgeon at the University of Michigan. His schedule was not flexible, and he was unable to take on family responsibilities, said Dr. Malani. “I knew someone had to be able to grab the kids from daycare or pick them up at school if they were sick.” She also took work home and worked weekends.
Young women physicians in particular are thinking about combining parenting with work – in the Medscape report, that issue ranked third among the issues women care about. Seeing other women doctors navigate that in their particular specialty can have a positive impact.
“When I chose adolescent medicine, I remember working with a doctor in this field who talked about how much she enjoyed raising her kids even as teenagers and how much she was enjoying them as young adults. She seemed so balanced and happy in her family, and it gave me a nice feeling about the field,” said Nancy Dodson, MD, MPH, a pediatrician specializing in adolescent medicine at Pediatrics on Hudson in New York.
Rachel Zhuk, MD, a reproductive psychiatrist in New York, took a break after medical school to spend time with her newborn son. She met a woman who was also a young parent and a psychiatrist. “We were both figuring out parenting together – it was like looking into my future.” That friendship and her desire to have more time with patients influenced her decision to pursue psychiatry instead of internal medicine.
Discrimination and harassment influence specialty choice
Women doctors in the top-paying surgical and other specialties have reported experiencing more discrimination and harassment than men.
Of 927 orthopedic surgeons who responded to an AAOS survey, 66% said they experienced gender discrimination, bullying, sexual harassment, or harassment in the health care workplace. More than twice as many women (81%) experienced these behaviors as men (35%).
“This study shows that women in orthopedic surgery disproportionately experience these negative behaviors, and only a handful of institutions in the United States provide any type of training to prevent them,” said Dr. Samora, the lead author of the AAOS report.
Radiology is another male-dominated field – women represent 26% of all radiologists, the 2020 AAMC specialty report shows. A systematic review shows that 40% of women radiologists experienced gender discrimination at work, compared with 1% of men, and that 47% of women experienced sexual harassment.
Female trainees in surgery have also reported disproportionate rates of discrimination and harassment. Female general surgical residents have experienced more gender discrimination than male residents (65.1% vs. 10.0%) and more sexual harassment than male residents (19.9% vs. 3.9), a national survey indicates.
When medical students are exposed to these behaviors through personal experience, witnessing, or hearing about them, it can affect which specialty they choose. A survey of fourth-year medical students shows that far more women than men reported that exposure to gender discrimination and sexual harassment influenced their specialty choices (45.3% vs. 16.4%) and residency rankings (25.3% vs. 10.9%). Women who chose general surgery were the most likely to experience gender discrimination and sexual harassment during residency selection; women who chose psychiatry were the least likely to experience such behaviors, the report shows.
“If young trainees witness such behaviors in a specific field, they would naturally migrate toward a different specialty,” said Dr. Samora.
Trainees can also be put off by residency directors asking them inappropriate questions. Of nearly 500 female orthopedic surgeons surveyed, 62% reported that they were asked inappropriate questions during their residency interviews. “Inappropriate questions and comments directed toward women during residency interviews are clearly not conducive to women entering the field,” the authors stated. They found that little changed during the study period from 1971 to 2015.
The most frequent inappropriate questions concerned whether the prospective residents would be getting pregnant or raising children during residency and their marital status. One female orthopedic surgeon reported: “I was asked if I have children and was told that it would be too difficult to complete an orthopedic residency with children.”
The interviewers also made frequent comments about the inferiority of women to men. For example, “I was told by one program interviewer that ‘I don’t have a bias about women in medicine, I have a bias about women in orthopedic surgery,’ ” another female orthopedic surgeon commented.
Longer training
Residency training for the top-paying surgical specialties, including orthopedic surgery, plastic surgery, and otolaryngology, lasts 5-6 years. This compares with 3-4 years for the lower-paying specialties, such as pediatrics, internal medicine, and ob.gyn., according to data from the American Medical Association.
Women doctors are in their prime childbearing years during residency. Women who want to start a family will consider whether they want to get pregnant during residency or wait until they finish their training, said Dr. Malcolm.
The vast majority (84%) of 190 female orthopedic surgery trainees who responded to a survey indicated that they did not have children or were pregnant during residency. Nearly half (48%) reported that they had postponed having children because they were in training.
“The longer training is definitely a concerning issue for women of childbearing age. Many professional women are waiting to have children, for multiple reasons, but one major fear is the stigma due to taking time off from work obligations. There is a risk of irritating your peers because they may have to take on more work and cover more calls for you during your absence,” said Dr. Samora.
That fear is not unfounded. At least half of the 190 female orthopedic residents reported that they encountered bias against becoming pregnant during training from both coresidents (60%) and attendings (50%), according to the study.
Another recent survey suggests that pregnant surgical residents face several barriers during their training, including a lack of salary for extended family leave, resentment from fellow residents who need to cover for them during maternity leave, and a lack of formal lactation policies.
A few policy changes by national board organizations, including those in the surgical specialties, may make life a little easier for female trainees to have children, suggested Dr. Samora.
Residents and fellows are now allowed a minimum of 6 weeks away for medical leave or caregiving once during training, without having to use vacation or sick leave and without having to extend their training, the American Board of Medical Specialties has announced.
In addition, the American Board of Orthopaedic Surgery and the American Board of Surgery have enacted policies that allow lactating women to take a break to pump during their board exams.
A version of this article first appeared on Medscape.com.
Women compose only 6% of orthopedic surgeons, 8% of interventional cardiologists, 10% of urologists, 17% of plastic surgeons, and 18% of otolaryngologists, according to the 2020 Association of American Medical Colleges Physician Specialty Data Report.
Plastic surgeons earn an average of $526,000 annually, which is the highest-paying specialty. Otolaryngologists earn an average of $417,000 annually, and urologists earn $427,000, according to the Medscape Physician Compensation Report 2021: The Recovery Begins.
Yet, far more women are practicing in specialties that pay less. Women are the majority in pediatrics (64%), ob.gyn. (59%), internal medicine (53%), and endocrinology (51%), the AAMC data show. The exception is dermatology, which pays well and in which 51% are women. The annual average pay is $394,000.
Why are so many women avoiding the top-paying specialties?
Several physician researchers and leaders in the top-paying specialties point to four main factors: Women are attracted to specialties that have more women in faculty and leadership positions, women prioritize work-life balance over pay, women residents may be deterred from the high-paying specialties because of gender discrimination and sexual harassment, and the longer training periods for surgical specialties may be a deterrent for women who want to have children.
Lack of women leaders
The specialties with the most women tend to have the highest proportion of women in leadership positions. For example, obstetrics and gynecology had the highest proportion of women department chairs (24.1%) and vice chairs (38.8). Pediatrics had the highest proportion of women division directors (31.5%) and residency program directors (64.6%), a study shows.
Surgical specialties, on the other hand, may have a harder time attracting female residents, possibly because of a lack of women in leadership positions. A recent study that examined gender differences in attitudes toward surgery training found that women would be more likely to go into surgery if there were more surgical faculty and residents of their same gender.
An analysis of orthopedic residency programs shows that more trainees were drawn to programs that had more female faculty members, including associate professors and women in leadership positions.
Terri Malcolm, MD, a board-certified ob.gyn. and CEO/founder of Master Physician Leaders, said women need to consider whether they want to be a trailblazer in a specialty that has fewer women. “What support systems are in place to accommodate your goals, whether it’s career advancement, having a family, or mentorship? Where can you show up as your whole self and be supported in that?”
Being the only woman in a residency program can be a challenge, said Dr. Malcolm. If the residents and attendings are predominantly men, for example, they may not think about creating a call schedule that takes into account maternity leave or the fact that women tend to be caretakers for their children and parents.
The study of gender differences toward surgery training shows that 75% of women, in comparison with 46% of men, would be more willing to enter surgery if maternity leave and childcare were made available to female residents and attending physicians.
Women want work-life balance
Although both men and women want families, women still shoulder more family and childcare responsibilities. That may explain why women physicians ranked work-life balance first and compensation second in the Medscape Women Physicians 2020 Report: The Issues They Care About.
“My physician colleagues have been and are supportive of intellectual abilities, but I feel they don’t fully understand the uneven distribution of childcare issues on women,” a woman dermatologist commented.
Women may want to work fewer hours or have a more flexible schedule to take care of children. “I can count on one hand the number of women who have a part-time job in orthopedics. It’s very rare, and working part time absolutely is a barrier for someone who wants to be a surgeon,” said Julie Samora, MD, PhD, a researcher and pediatric hand surgeon at Nationwide Children’s Hospital, in Columbus, Ohio. She is also a spokesperson for the American Association of Orthopedic Surgeons.
Preeti Malani, MD, a professor of medicine who specializes in infectious diseases at the University of Michigan, chose to work full-time in academia while raising two children with her husband. In a decade, she rose through the ranks to full professor. “I took the advice of a woman who wanted to recruit me to have a full-time position with maximum flexibility rather than work part time, often for more hours and less pay. I also have tried to build my career so I was not doing all clinical work.”
Her husband is a surgeon at the University of Michigan. His schedule was not flexible, and he was unable to take on family responsibilities, said Dr. Malani. “I knew someone had to be able to grab the kids from daycare or pick them up at school if they were sick.” She also took work home and worked weekends.
Young women physicians in particular are thinking about combining parenting with work – in the Medscape report, that issue ranked third among the issues women care about. Seeing other women doctors navigate that in their particular specialty can have a positive impact.
“When I chose adolescent medicine, I remember working with a doctor in this field who talked about how much she enjoyed raising her kids even as teenagers and how much she was enjoying them as young adults. She seemed so balanced and happy in her family, and it gave me a nice feeling about the field,” said Nancy Dodson, MD, MPH, a pediatrician specializing in adolescent medicine at Pediatrics on Hudson in New York.
Rachel Zhuk, MD, a reproductive psychiatrist in New York, took a break after medical school to spend time with her newborn son. She met a woman who was also a young parent and a psychiatrist. “We were both figuring out parenting together – it was like looking into my future.” That friendship and her desire to have more time with patients influenced her decision to pursue psychiatry instead of internal medicine.
Discrimination and harassment influence specialty choice
Women doctors in the top-paying surgical and other specialties have reported experiencing more discrimination and harassment than men.
Of 927 orthopedic surgeons who responded to an AAOS survey, 66% said they experienced gender discrimination, bullying, sexual harassment, or harassment in the health care workplace. More than twice as many women (81%) experienced these behaviors as men (35%).
“This study shows that women in orthopedic surgery disproportionately experience these negative behaviors, and only a handful of institutions in the United States provide any type of training to prevent them,” said Dr. Samora, the lead author of the AAOS report.
Radiology is another male-dominated field – women represent 26% of all radiologists, the 2020 AAMC specialty report shows. A systematic review shows that 40% of women radiologists experienced gender discrimination at work, compared with 1% of men, and that 47% of women experienced sexual harassment.
Female trainees in surgery have also reported disproportionate rates of discrimination and harassment. Female general surgical residents have experienced more gender discrimination than male residents (65.1% vs. 10.0%) and more sexual harassment than male residents (19.9% vs. 3.9), a national survey indicates.
When medical students are exposed to these behaviors through personal experience, witnessing, or hearing about them, it can affect which specialty they choose. A survey of fourth-year medical students shows that far more women than men reported that exposure to gender discrimination and sexual harassment influenced their specialty choices (45.3% vs. 16.4%) and residency rankings (25.3% vs. 10.9%). Women who chose general surgery were the most likely to experience gender discrimination and sexual harassment during residency selection; women who chose psychiatry were the least likely to experience such behaviors, the report shows.
“If young trainees witness such behaviors in a specific field, they would naturally migrate toward a different specialty,” said Dr. Samora.
Trainees can also be put off by residency directors asking them inappropriate questions. Of nearly 500 female orthopedic surgeons surveyed, 62% reported that they were asked inappropriate questions during their residency interviews. “Inappropriate questions and comments directed toward women during residency interviews are clearly not conducive to women entering the field,” the authors stated. They found that little changed during the study period from 1971 to 2015.
The most frequent inappropriate questions concerned whether the prospective residents would be getting pregnant or raising children during residency and their marital status. One female orthopedic surgeon reported: “I was asked if I have children and was told that it would be too difficult to complete an orthopedic residency with children.”
The interviewers also made frequent comments about the inferiority of women to men. For example, “I was told by one program interviewer that ‘I don’t have a bias about women in medicine, I have a bias about women in orthopedic surgery,’ ” another female orthopedic surgeon commented.
Longer training
Residency training for the top-paying surgical specialties, including orthopedic surgery, plastic surgery, and otolaryngology, lasts 5-6 years. This compares with 3-4 years for the lower-paying specialties, such as pediatrics, internal medicine, and ob.gyn., according to data from the American Medical Association.
Women doctors are in their prime childbearing years during residency. Women who want to start a family will consider whether they want to get pregnant during residency or wait until they finish their training, said Dr. Malcolm.
The vast majority (84%) of 190 female orthopedic surgery trainees who responded to a survey indicated that they did not have children or were pregnant during residency. Nearly half (48%) reported that they had postponed having children because they were in training.
“The longer training is definitely a concerning issue for women of childbearing age. Many professional women are waiting to have children, for multiple reasons, but one major fear is the stigma due to taking time off from work obligations. There is a risk of irritating your peers because they may have to take on more work and cover more calls for you during your absence,” said Dr. Samora.
That fear is not unfounded. At least half of the 190 female orthopedic residents reported that they encountered bias against becoming pregnant during training from both coresidents (60%) and attendings (50%), according to the study.
Another recent survey suggests that pregnant surgical residents face several barriers during their training, including a lack of salary for extended family leave, resentment from fellow residents who need to cover for them during maternity leave, and a lack of formal lactation policies.
A few policy changes by national board organizations, including those in the surgical specialties, may make life a little easier for female trainees to have children, suggested Dr. Samora.
Residents and fellows are now allowed a minimum of 6 weeks away for medical leave or caregiving once during training, without having to use vacation or sick leave and without having to extend their training, the American Board of Medical Specialties has announced.
In addition, the American Board of Orthopaedic Surgery and the American Board of Surgery have enacted policies that allow lactating women to take a break to pump during their board exams.
A version of this article first appeared on Medscape.com.
Women compose only 6% of orthopedic surgeons, 8% of interventional cardiologists, 10% of urologists, 17% of plastic surgeons, and 18% of otolaryngologists, according to the 2020 Association of American Medical Colleges Physician Specialty Data Report.
Plastic surgeons earn an average of $526,000 annually, which is the highest-paying specialty. Otolaryngologists earn an average of $417,000 annually, and urologists earn $427,000, according to the Medscape Physician Compensation Report 2021: The Recovery Begins.
Yet, far more women are practicing in specialties that pay less. Women are the majority in pediatrics (64%), ob.gyn. (59%), internal medicine (53%), and endocrinology (51%), the AAMC data show. The exception is dermatology, which pays well and in which 51% are women. The annual average pay is $394,000.
Why are so many women avoiding the top-paying specialties?
Several physician researchers and leaders in the top-paying specialties point to four main factors: Women are attracted to specialties that have more women in faculty and leadership positions, women prioritize work-life balance over pay, women residents may be deterred from the high-paying specialties because of gender discrimination and sexual harassment, and the longer training periods for surgical specialties may be a deterrent for women who want to have children.
Lack of women leaders
The specialties with the most women tend to have the highest proportion of women in leadership positions. For example, obstetrics and gynecology had the highest proportion of women department chairs (24.1%) and vice chairs (38.8). Pediatrics had the highest proportion of women division directors (31.5%) and residency program directors (64.6%), a study shows.
Surgical specialties, on the other hand, may have a harder time attracting female residents, possibly because of a lack of women in leadership positions. A recent study that examined gender differences in attitudes toward surgery training found that women would be more likely to go into surgery if there were more surgical faculty and residents of their same gender.
An analysis of orthopedic residency programs shows that more trainees were drawn to programs that had more female faculty members, including associate professors and women in leadership positions.
Terri Malcolm, MD, a board-certified ob.gyn. and CEO/founder of Master Physician Leaders, said women need to consider whether they want to be a trailblazer in a specialty that has fewer women. “What support systems are in place to accommodate your goals, whether it’s career advancement, having a family, or mentorship? Where can you show up as your whole self and be supported in that?”
Being the only woman in a residency program can be a challenge, said Dr. Malcolm. If the residents and attendings are predominantly men, for example, they may not think about creating a call schedule that takes into account maternity leave or the fact that women tend to be caretakers for their children and parents.
The study of gender differences toward surgery training shows that 75% of women, in comparison with 46% of men, would be more willing to enter surgery if maternity leave and childcare were made available to female residents and attending physicians.
Women want work-life balance
Although both men and women want families, women still shoulder more family and childcare responsibilities. That may explain why women physicians ranked work-life balance first and compensation second in the Medscape Women Physicians 2020 Report: The Issues They Care About.
“My physician colleagues have been and are supportive of intellectual abilities, but I feel they don’t fully understand the uneven distribution of childcare issues on women,” a woman dermatologist commented.
Women may want to work fewer hours or have a more flexible schedule to take care of children. “I can count on one hand the number of women who have a part-time job in orthopedics. It’s very rare, and working part time absolutely is a barrier for someone who wants to be a surgeon,” said Julie Samora, MD, PhD, a researcher and pediatric hand surgeon at Nationwide Children’s Hospital, in Columbus, Ohio. She is also a spokesperson for the American Association of Orthopedic Surgeons.
Preeti Malani, MD, a professor of medicine who specializes in infectious diseases at the University of Michigan, chose to work full-time in academia while raising two children with her husband. In a decade, she rose through the ranks to full professor. “I took the advice of a woman who wanted to recruit me to have a full-time position with maximum flexibility rather than work part time, often for more hours and less pay. I also have tried to build my career so I was not doing all clinical work.”
Her husband is a surgeon at the University of Michigan. His schedule was not flexible, and he was unable to take on family responsibilities, said Dr. Malani. “I knew someone had to be able to grab the kids from daycare or pick them up at school if they were sick.” She also took work home and worked weekends.
Young women physicians in particular are thinking about combining parenting with work – in the Medscape report, that issue ranked third among the issues women care about. Seeing other women doctors navigate that in their particular specialty can have a positive impact.
“When I chose adolescent medicine, I remember working with a doctor in this field who talked about how much she enjoyed raising her kids even as teenagers and how much she was enjoying them as young adults. She seemed so balanced and happy in her family, and it gave me a nice feeling about the field,” said Nancy Dodson, MD, MPH, a pediatrician specializing in adolescent medicine at Pediatrics on Hudson in New York.
Rachel Zhuk, MD, a reproductive psychiatrist in New York, took a break after medical school to spend time with her newborn son. She met a woman who was also a young parent and a psychiatrist. “We were both figuring out parenting together – it was like looking into my future.” That friendship and her desire to have more time with patients influenced her decision to pursue psychiatry instead of internal medicine.
Discrimination and harassment influence specialty choice
Women doctors in the top-paying surgical and other specialties have reported experiencing more discrimination and harassment than men.
Of 927 orthopedic surgeons who responded to an AAOS survey, 66% said they experienced gender discrimination, bullying, sexual harassment, or harassment in the health care workplace. More than twice as many women (81%) experienced these behaviors as men (35%).
“This study shows that women in orthopedic surgery disproportionately experience these negative behaviors, and only a handful of institutions in the United States provide any type of training to prevent them,” said Dr. Samora, the lead author of the AAOS report.
Radiology is another male-dominated field – women represent 26% of all radiologists, the 2020 AAMC specialty report shows. A systematic review shows that 40% of women radiologists experienced gender discrimination at work, compared with 1% of men, and that 47% of women experienced sexual harassment.
Female trainees in surgery have also reported disproportionate rates of discrimination and harassment. Female general surgical residents have experienced more gender discrimination than male residents (65.1% vs. 10.0%) and more sexual harassment than male residents (19.9% vs. 3.9), a national survey indicates.
When medical students are exposed to these behaviors through personal experience, witnessing, or hearing about them, it can affect which specialty they choose. A survey of fourth-year medical students shows that far more women than men reported that exposure to gender discrimination and sexual harassment influenced their specialty choices (45.3% vs. 16.4%) and residency rankings (25.3% vs. 10.9%). Women who chose general surgery were the most likely to experience gender discrimination and sexual harassment during residency selection; women who chose psychiatry were the least likely to experience such behaviors, the report shows.
“If young trainees witness such behaviors in a specific field, they would naturally migrate toward a different specialty,” said Dr. Samora.
Trainees can also be put off by residency directors asking them inappropriate questions. Of nearly 500 female orthopedic surgeons surveyed, 62% reported that they were asked inappropriate questions during their residency interviews. “Inappropriate questions and comments directed toward women during residency interviews are clearly not conducive to women entering the field,” the authors stated. They found that little changed during the study period from 1971 to 2015.
The most frequent inappropriate questions concerned whether the prospective residents would be getting pregnant or raising children during residency and their marital status. One female orthopedic surgeon reported: “I was asked if I have children and was told that it would be too difficult to complete an orthopedic residency with children.”
The interviewers also made frequent comments about the inferiority of women to men. For example, “I was told by one program interviewer that ‘I don’t have a bias about women in medicine, I have a bias about women in orthopedic surgery,’ ” another female orthopedic surgeon commented.
Longer training
Residency training for the top-paying surgical specialties, including orthopedic surgery, plastic surgery, and otolaryngology, lasts 5-6 years. This compares with 3-4 years for the lower-paying specialties, such as pediatrics, internal medicine, and ob.gyn., according to data from the American Medical Association.
Women doctors are in their prime childbearing years during residency. Women who want to start a family will consider whether they want to get pregnant during residency or wait until they finish their training, said Dr. Malcolm.
The vast majority (84%) of 190 female orthopedic surgery trainees who responded to a survey indicated that they did not have children or were pregnant during residency. Nearly half (48%) reported that they had postponed having children because they were in training.
“The longer training is definitely a concerning issue for women of childbearing age. Many professional women are waiting to have children, for multiple reasons, but one major fear is the stigma due to taking time off from work obligations. There is a risk of irritating your peers because they may have to take on more work and cover more calls for you during your absence,” said Dr. Samora.
That fear is not unfounded. At least half of the 190 female orthopedic residents reported that they encountered bias against becoming pregnant during training from both coresidents (60%) and attendings (50%), according to the study.
Another recent survey suggests that pregnant surgical residents face several barriers during their training, including a lack of salary for extended family leave, resentment from fellow residents who need to cover for them during maternity leave, and a lack of formal lactation policies.
A few policy changes by national board organizations, including those in the surgical specialties, may make life a little easier for female trainees to have children, suggested Dr. Samora.
Residents and fellows are now allowed a minimum of 6 weeks away for medical leave or caregiving once during training, without having to use vacation or sick leave and without having to extend their training, the American Board of Medical Specialties has announced.
In addition, the American Board of Orthopaedic Surgery and the American Board of Surgery have enacted policies that allow lactating women to take a break to pump during their board exams.
A version of this article first appeared on Medscape.com.
Bullous Retiform Purpura on the Ears and Legs
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
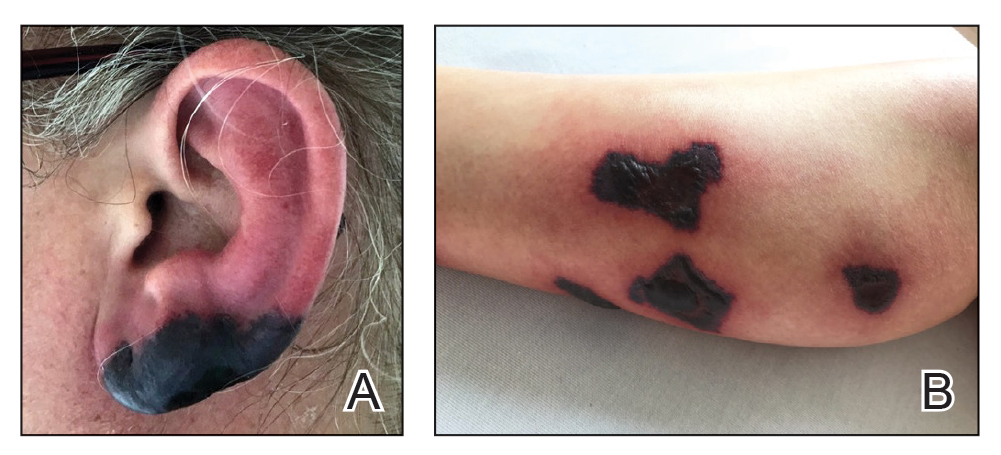
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
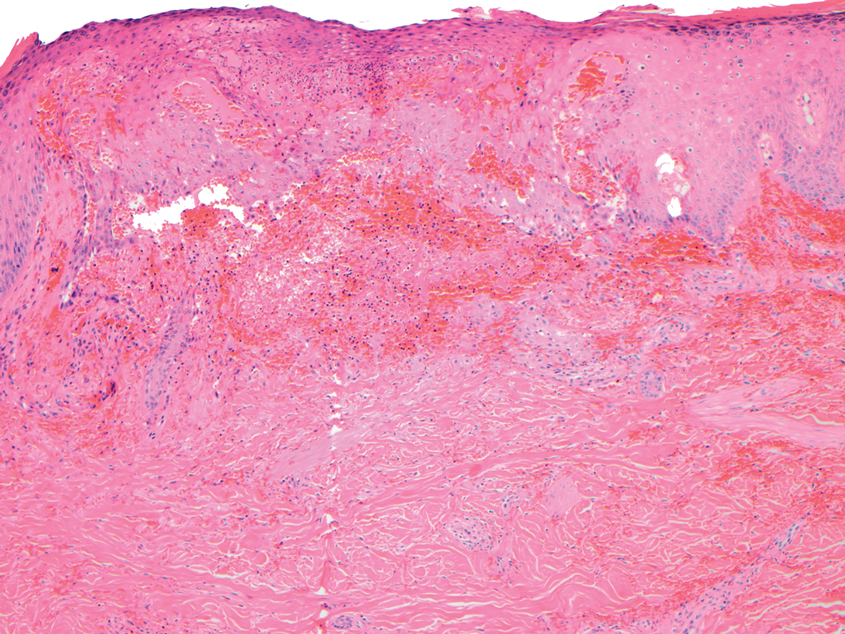
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
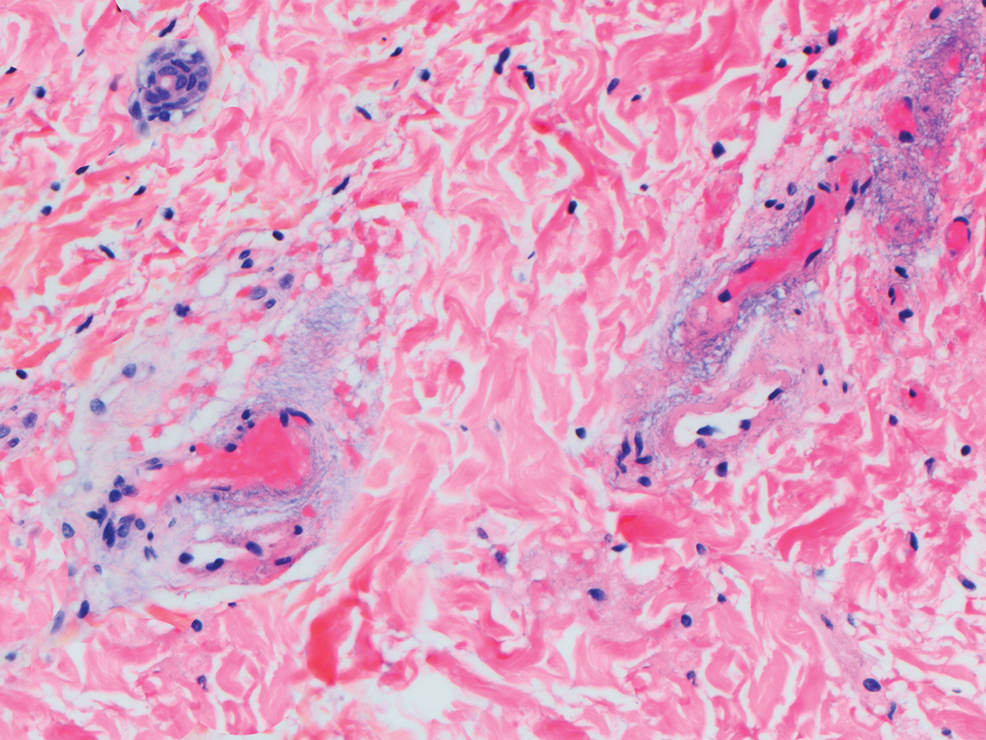
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
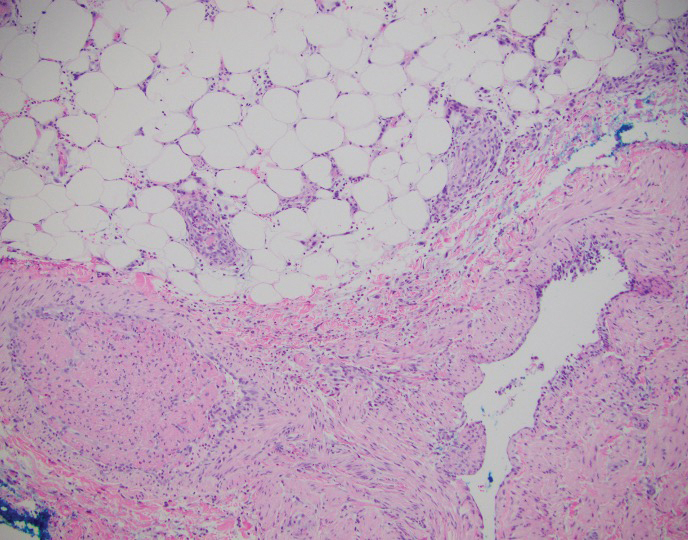
A 40-year-old woman presented with a progressive painful rash on the ears and legs of 2 weeks’ duration. She described the rash as initially red and nonpainful; it started on the right leg and progressed to the left leg, eventually involving the earlobes 4 days prior to presentation. Physical examination revealed edematous purpura of the earlobes and bullous retiform purpura on the lower extremities. Laboratory studies revealed leukopenia (3.6×103 /cm2 [reference range, 4.0–10.5×103 /cm2 ]) and elevated antineutrophil cytoplasmic antibodies (1:320 titer [reference range, <1:40]) in a perinuclear pattern (perinuclear antineutrophil cytoplasmic antibodies). Urine toxicology screening was positive for cocaine and opiates. A punch biopsy of a bullous retiform purpura on the right thigh was obtained for standard hematoxylin and eosin staining.
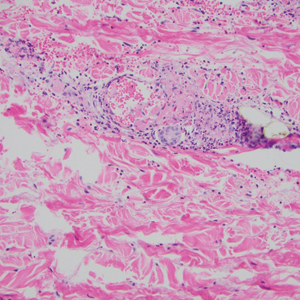
Myocarditis in adolescents after COVID-19 vaccine typically mild
Adolescents can develop mild myocarditis as a rare complication after COVID-19 vaccination, as has been reported in adults, an early case series from Boston confirms.
The adolescents who developed heart inflammation after vaccination typically had a benign course, with symptoms resolving without treatment, although one patient had persistent borderline low left ventricular (LV) function, report Audrey Dionne, MD, and colleagues at Boston Children’s Hospital.
“Despite the risks of myocarditis associated with vaccination, the benefits of vaccination likely outweigh risks in children and adolescents,” they say.
They estimate that for males 12-29 years of age COVID-19 vaccination prevents 11,000 COVID-19 cases, 560 hospitalizations, 138 intensive care unit admissions, and six deaths, compared with 39-47 expected myocarditis cases.
The case series was published online Aug. 10 in JAMA Cardiology.
Long-term risks unknown
Dr. Dionne and colleagues reviewed the results of comprehensive cardiac imaging in 14 boys and 1 girl, 12-18 years of age (median, 15 years), who were hospitalized with myocarditis after receiving the Pfizer-BioNTech messenger RNA COVID-19 vaccine.
Symptoms started 1-6 days after vaccine administration (most after the second dose) and included chest pain in all 15 patients, fever in 10 (67%), myalgia in eight (53%), and headache in six (40%).
On admission, all patients had elevated troponin levels (median, 0.25 ng/mL; range, 0.08-3.15 ng/mL). Troponin levels peaked 0.1-2.3 days after admission.
Echocardiography revealed decreased LV ejection fraction (EF) in three patients (20%) and abnormal global longitudinal or circumferential strain in five patients (33%). No patient had a pericardial effusion.
Cardiac MRI findings were consistent with myocarditis in 13 patients (87%), including late gadolinium enhancement in 12 (80%), regional hyperintensity on T2-weighted imaging in two (13%), elevated extracellular volume fraction in three (20%), and elevated LV global native T1 in two (20%).
The patients remained in the hospital for 1-5 days (median, 2 days) and were discharged. No patient required admission to the intensive care unit.
In follow-up assessments performed 1-13 days after hospital discharge, symptoms of myocarditis had resolved in 11 patients (73%).
One patient (7%) had persistent borderline low LV systolic function on echocardiogram (LVEF, 54%).
Troponin levels remained mildly elevated in three patients (20%). One patient (7%) had nonsustained ventricular tachycardia on ambulatory monitor.
The authors say longitudinal studies of patients with myocarditis after COVID-19 vaccine “will be important to better understand long-term risks.”
In a statement from the UK nonprofit Science Media Centre, Peter Openshaw, FMedSci, Imperial College London, says: “The problem with case series of this type is the lack of comparison groups. How many cases of myocarditis might be seen in normal children, or those given other vaccines (including those that are not for COVID), or in teenagers infected with SARS-CoV-2?”
“As the authors note, myocarditis does happen after other vaccines. The estimated rate (62.8 cases per million) makes this a rare event,” Dr. Openshaw says.
“My view that teenagers should be considered for vaccination is not changed by this new publication,” he adds.
This study was funded by the McCance Foundation. The authors have declared no relevant conflicts of interest. Dr. Openshaw has served on scientific advisory boards for Janssen/J&J, Oxford Immunotech, GSK, Nestle, and Pfizer in relation to immunity to viruses (fees paid to Imperial College London).
A version of this article first appeared on Medscape.com.
Adolescents can develop mild myocarditis as a rare complication after COVID-19 vaccination, as has been reported in adults, an early case series from Boston confirms.
The adolescents who developed heart inflammation after vaccination typically had a benign course, with symptoms resolving without treatment, although one patient had persistent borderline low left ventricular (LV) function, report Audrey Dionne, MD, and colleagues at Boston Children’s Hospital.
“Despite the risks of myocarditis associated with vaccination, the benefits of vaccination likely outweigh risks in children and adolescents,” they say.
They estimate that for males 12-29 years of age COVID-19 vaccination prevents 11,000 COVID-19 cases, 560 hospitalizations, 138 intensive care unit admissions, and six deaths, compared with 39-47 expected myocarditis cases.
The case series was published online Aug. 10 in JAMA Cardiology.
Long-term risks unknown
Dr. Dionne and colleagues reviewed the results of comprehensive cardiac imaging in 14 boys and 1 girl, 12-18 years of age (median, 15 years), who were hospitalized with myocarditis after receiving the Pfizer-BioNTech messenger RNA COVID-19 vaccine.
Symptoms started 1-6 days after vaccine administration (most after the second dose) and included chest pain in all 15 patients, fever in 10 (67%), myalgia in eight (53%), and headache in six (40%).
On admission, all patients had elevated troponin levels (median, 0.25 ng/mL; range, 0.08-3.15 ng/mL). Troponin levels peaked 0.1-2.3 days after admission.
Echocardiography revealed decreased LV ejection fraction (EF) in three patients (20%) and abnormal global longitudinal or circumferential strain in five patients (33%). No patient had a pericardial effusion.
Cardiac MRI findings were consistent with myocarditis in 13 patients (87%), including late gadolinium enhancement in 12 (80%), regional hyperintensity on T2-weighted imaging in two (13%), elevated extracellular volume fraction in three (20%), and elevated LV global native T1 in two (20%).
The patients remained in the hospital for 1-5 days (median, 2 days) and were discharged. No patient required admission to the intensive care unit.
In follow-up assessments performed 1-13 days after hospital discharge, symptoms of myocarditis had resolved in 11 patients (73%).
One patient (7%) had persistent borderline low LV systolic function on echocardiogram (LVEF, 54%).
Troponin levels remained mildly elevated in three patients (20%). One patient (7%) had nonsustained ventricular tachycardia on ambulatory monitor.
The authors say longitudinal studies of patients with myocarditis after COVID-19 vaccine “will be important to better understand long-term risks.”
In a statement from the UK nonprofit Science Media Centre, Peter Openshaw, FMedSci, Imperial College London, says: “The problem with case series of this type is the lack of comparison groups. How many cases of myocarditis might be seen in normal children, or those given other vaccines (including those that are not for COVID), or in teenagers infected with SARS-CoV-2?”
“As the authors note, myocarditis does happen after other vaccines. The estimated rate (62.8 cases per million) makes this a rare event,” Dr. Openshaw says.
“My view that teenagers should be considered for vaccination is not changed by this new publication,” he adds.
This study was funded by the McCance Foundation. The authors have declared no relevant conflicts of interest. Dr. Openshaw has served on scientific advisory boards for Janssen/J&J, Oxford Immunotech, GSK, Nestle, and Pfizer in relation to immunity to viruses (fees paid to Imperial College London).
A version of this article first appeared on Medscape.com.
Adolescents can develop mild myocarditis as a rare complication after COVID-19 vaccination, as has been reported in adults, an early case series from Boston confirms.
The adolescents who developed heart inflammation after vaccination typically had a benign course, with symptoms resolving without treatment, although one patient had persistent borderline low left ventricular (LV) function, report Audrey Dionne, MD, and colleagues at Boston Children’s Hospital.
“Despite the risks of myocarditis associated with vaccination, the benefits of vaccination likely outweigh risks in children and adolescents,” they say.
They estimate that for males 12-29 years of age COVID-19 vaccination prevents 11,000 COVID-19 cases, 560 hospitalizations, 138 intensive care unit admissions, and six deaths, compared with 39-47 expected myocarditis cases.
The case series was published online Aug. 10 in JAMA Cardiology.
Long-term risks unknown
Dr. Dionne and colleagues reviewed the results of comprehensive cardiac imaging in 14 boys and 1 girl, 12-18 years of age (median, 15 years), who were hospitalized with myocarditis after receiving the Pfizer-BioNTech messenger RNA COVID-19 vaccine.
Symptoms started 1-6 days after vaccine administration (most after the second dose) and included chest pain in all 15 patients, fever in 10 (67%), myalgia in eight (53%), and headache in six (40%).
On admission, all patients had elevated troponin levels (median, 0.25 ng/mL; range, 0.08-3.15 ng/mL). Troponin levels peaked 0.1-2.3 days after admission.
Echocardiography revealed decreased LV ejection fraction (EF) in three patients (20%) and abnormal global longitudinal or circumferential strain in five patients (33%). No patient had a pericardial effusion.
Cardiac MRI findings were consistent with myocarditis in 13 patients (87%), including late gadolinium enhancement in 12 (80%), regional hyperintensity on T2-weighted imaging in two (13%), elevated extracellular volume fraction in three (20%), and elevated LV global native T1 in two (20%).
The patients remained in the hospital for 1-5 days (median, 2 days) and were discharged. No patient required admission to the intensive care unit.
In follow-up assessments performed 1-13 days after hospital discharge, symptoms of myocarditis had resolved in 11 patients (73%).
One patient (7%) had persistent borderline low LV systolic function on echocardiogram (LVEF, 54%).
Troponin levels remained mildly elevated in three patients (20%). One patient (7%) had nonsustained ventricular tachycardia on ambulatory monitor.
The authors say longitudinal studies of patients with myocarditis after COVID-19 vaccine “will be important to better understand long-term risks.”
In a statement from the UK nonprofit Science Media Centre, Peter Openshaw, FMedSci, Imperial College London, says: “The problem with case series of this type is the lack of comparison groups. How many cases of myocarditis might be seen in normal children, or those given other vaccines (including those that are not for COVID), or in teenagers infected with SARS-CoV-2?”
“As the authors note, myocarditis does happen after other vaccines. The estimated rate (62.8 cases per million) makes this a rare event,” Dr. Openshaw says.
“My view that teenagers should be considered for vaccination is not changed by this new publication,” he adds.
This study was funded by the McCance Foundation. The authors have declared no relevant conflicts of interest. Dr. Openshaw has served on scientific advisory boards for Janssen/J&J, Oxford Immunotech, GSK, Nestle, and Pfizer in relation to immunity to viruses (fees paid to Imperial College London).
A version of this article first appeared on Medscape.com.
Real-world COVID-19 vaccine protection high in transplant patients
Real-world protection from COVID-19 vaccination is better than expected in transplant recipients, reducing the risk of symptomatic infection by almost 80% in those who have had both doses compared with unvaccinated controls, a new transplant registry analysis shows.
“Persons who have received an organ transplant are considered to be at increased risk for COVID-19 and for a severe outcome because their immune systems are necessarily suppressed to ensure their transplants are successful and lasting,” lead author Saima Aslam, MD, professor of medicine, University of California, San Diego, said in a statement.
Because numerous studies have demonstrated reduced antibody responses to SARS-CoV-2 in solid organ transplant recipients and variable effect on T-cell responses, there has been a need to study clinical effectiveness and breakthrough infections in those who are vaccinated, they explained.
“These findings offer strong evidence that getting vaccinated provides significant protection,” Dr. Aslam noted.
The investigators say that recent data from France, as well as other studies, show an increased rate of detectable antibodies following a third dose of the Pfizer-BioNTech COVID-19 vaccine in organ transplant recipients, “but based on our data it is unclear if a third dose is clinically warranted.”
The researchers stressed that almost half of the solid organ transplant recipients analyzed in the study had not been vaccinated at all, even by the beginning of 2021 when the United States was well into a third wave of COVID-19 infections.
So there is still a significant need, the authors said, to continue to improve outreach efforts to those in the transplant community and promote the benefits of being fully vaccinated.
The study was published online recently in Transplant Infectious Disease.
Transplant registry
The researchers analyzed clinical data from the UC San Diego transplant registry from Jan. 1 through June 2 of this year, with 2,151 solid organ transplant recipients identified. The patients had received a variety of solid organ transplants including kidney, liver, lung, and heart; the largest percentage received a donor kidney.
Among all patients, 912 were fully vaccinated and 1,239 were not (1,151 of those 1,239 received no vaccine at all and 88 had been partially vaccinated; these 1,239 served as the control group).
Fully vaccinated patients had received two shots of either the Pfizer-BioNTech COVID-19 or the Moderna vaccine, or a single dose of the Johnson & Johnson vaccine. The majority, at nearly 70%, had received the Moderna vaccine. The mean age of the cohort was 57 years and the median time since patients had undergone transplantation was almost 5 years (57.5 months).
During the 6-month study interval, 65 cases of COVID-19 were documented in the group overall. Only 4 cases occurred among fully vaccinated individuals whereas 61 cases occurred among the unvaccinated, including in 2 patients who had been partially vaccinated.
Among the four cases that occurred among the fully vaccinated, two were considered mild and were treated on an outpatient basis, and the other two were moderate, requiring hospitalization and treatment with remdesivir.
There were no COVID-19–related deaths among the 4 patients who experienced breakthrough infections, whereas 2 (3.3%) of 61 of control patients died of COVID-19–related causes.
The authors noted that the incidence rate for COVID-19 was 0.065 per 1,000-person days among the fully vaccinated compared with an incidence rate of 0.34 per 1,000-person days in the control group.
Booster doses for especially vulnerable transplant recipients?
“These findings are encouraging for a couple of reasons,” said coauthor Kristin Mekeel, MD, chief of transplant and hepatobiliary surgery at UCSD.
“First, they demonstrate real-world clinical effectiveness of COVID-19 vaccination in a vulnerable population,” she noted.
“Second, the effectiveness is better than expected,” she added, “given that studies have found that only about half of solid organ transplant recipients develop detectable antibodies after vaccination.”
Although calls for patients who are immunosuppressed to receive a third booster dose of a COVID-19 vaccine may not be necessary, “prioritizing at-risk subsets of transplant recipients based on immunological profiles and clinical characteristics for a third vaccine dose could be considered,” they said, adding it’s still vitally important for transplant patients to continue to mask and practice social distancing.
And it is especially important for transplant recipients to encourage household members to get vaccinated, too, especially given the current COVID-19 surge in San Diego.
The study was funded by the Cystic Fibrosis Foundation.
Dr. Aslam reports receiving grants from the Cystic Fibrosis Foundation as well as honoraria from Gilead and Merck. Study author Susan J. Little, MD, received grant funding from Gilead Sciences. Dr. Mekeel has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Real-world protection from COVID-19 vaccination is better than expected in transplant recipients, reducing the risk of symptomatic infection by almost 80% in those who have had both doses compared with unvaccinated controls, a new transplant registry analysis shows.
“Persons who have received an organ transplant are considered to be at increased risk for COVID-19 and for a severe outcome because their immune systems are necessarily suppressed to ensure their transplants are successful and lasting,” lead author Saima Aslam, MD, professor of medicine, University of California, San Diego, said in a statement.
Because numerous studies have demonstrated reduced antibody responses to SARS-CoV-2 in solid organ transplant recipients and variable effect on T-cell responses, there has been a need to study clinical effectiveness and breakthrough infections in those who are vaccinated, they explained.
“These findings offer strong evidence that getting vaccinated provides significant protection,” Dr. Aslam noted.
The investigators say that recent data from France, as well as other studies, show an increased rate of detectable antibodies following a third dose of the Pfizer-BioNTech COVID-19 vaccine in organ transplant recipients, “but based on our data it is unclear if a third dose is clinically warranted.”
The researchers stressed that almost half of the solid organ transplant recipients analyzed in the study had not been vaccinated at all, even by the beginning of 2021 when the United States was well into a third wave of COVID-19 infections.
So there is still a significant need, the authors said, to continue to improve outreach efforts to those in the transplant community and promote the benefits of being fully vaccinated.
The study was published online recently in Transplant Infectious Disease.
Transplant registry
The researchers analyzed clinical data from the UC San Diego transplant registry from Jan. 1 through June 2 of this year, with 2,151 solid organ transplant recipients identified. The patients had received a variety of solid organ transplants including kidney, liver, lung, and heart; the largest percentage received a donor kidney.
Among all patients, 912 were fully vaccinated and 1,239 were not (1,151 of those 1,239 received no vaccine at all and 88 had been partially vaccinated; these 1,239 served as the control group).
Fully vaccinated patients had received two shots of either the Pfizer-BioNTech COVID-19 or the Moderna vaccine, or a single dose of the Johnson & Johnson vaccine. The majority, at nearly 70%, had received the Moderna vaccine. The mean age of the cohort was 57 years and the median time since patients had undergone transplantation was almost 5 years (57.5 months).
During the 6-month study interval, 65 cases of COVID-19 were documented in the group overall. Only 4 cases occurred among fully vaccinated individuals whereas 61 cases occurred among the unvaccinated, including in 2 patients who had been partially vaccinated.
Among the four cases that occurred among the fully vaccinated, two were considered mild and were treated on an outpatient basis, and the other two were moderate, requiring hospitalization and treatment with remdesivir.
There were no COVID-19–related deaths among the 4 patients who experienced breakthrough infections, whereas 2 (3.3%) of 61 of control patients died of COVID-19–related causes.
The authors noted that the incidence rate for COVID-19 was 0.065 per 1,000-person days among the fully vaccinated compared with an incidence rate of 0.34 per 1,000-person days in the control group.
Booster doses for especially vulnerable transplant recipients?
“These findings are encouraging for a couple of reasons,” said coauthor Kristin Mekeel, MD, chief of transplant and hepatobiliary surgery at UCSD.
“First, they demonstrate real-world clinical effectiveness of COVID-19 vaccination in a vulnerable population,” she noted.
“Second, the effectiveness is better than expected,” she added, “given that studies have found that only about half of solid organ transplant recipients develop detectable antibodies after vaccination.”
Although calls for patients who are immunosuppressed to receive a third booster dose of a COVID-19 vaccine may not be necessary, “prioritizing at-risk subsets of transplant recipients based on immunological profiles and clinical characteristics for a third vaccine dose could be considered,” they said, adding it’s still vitally important for transplant patients to continue to mask and practice social distancing.
And it is especially important for transplant recipients to encourage household members to get vaccinated, too, especially given the current COVID-19 surge in San Diego.
The study was funded by the Cystic Fibrosis Foundation.
Dr. Aslam reports receiving grants from the Cystic Fibrosis Foundation as well as honoraria from Gilead and Merck. Study author Susan J. Little, MD, received grant funding from Gilead Sciences. Dr. Mekeel has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Real-world protection from COVID-19 vaccination is better than expected in transplant recipients, reducing the risk of symptomatic infection by almost 80% in those who have had both doses compared with unvaccinated controls, a new transplant registry analysis shows.
“Persons who have received an organ transplant are considered to be at increased risk for COVID-19 and for a severe outcome because their immune systems are necessarily suppressed to ensure their transplants are successful and lasting,” lead author Saima Aslam, MD, professor of medicine, University of California, San Diego, said in a statement.
Because numerous studies have demonstrated reduced antibody responses to SARS-CoV-2 in solid organ transplant recipients and variable effect on T-cell responses, there has been a need to study clinical effectiveness and breakthrough infections in those who are vaccinated, they explained.
“These findings offer strong evidence that getting vaccinated provides significant protection,” Dr. Aslam noted.
The investigators say that recent data from France, as well as other studies, show an increased rate of detectable antibodies following a third dose of the Pfizer-BioNTech COVID-19 vaccine in organ transplant recipients, “but based on our data it is unclear if a third dose is clinically warranted.”
The researchers stressed that almost half of the solid organ transplant recipients analyzed in the study had not been vaccinated at all, even by the beginning of 2021 when the United States was well into a third wave of COVID-19 infections.
So there is still a significant need, the authors said, to continue to improve outreach efforts to those in the transplant community and promote the benefits of being fully vaccinated.
The study was published online recently in Transplant Infectious Disease.
Transplant registry
The researchers analyzed clinical data from the UC San Diego transplant registry from Jan. 1 through June 2 of this year, with 2,151 solid organ transplant recipients identified. The patients had received a variety of solid organ transplants including kidney, liver, lung, and heart; the largest percentage received a donor kidney.
Among all patients, 912 were fully vaccinated and 1,239 were not (1,151 of those 1,239 received no vaccine at all and 88 had been partially vaccinated; these 1,239 served as the control group).
Fully vaccinated patients had received two shots of either the Pfizer-BioNTech COVID-19 or the Moderna vaccine, or a single dose of the Johnson & Johnson vaccine. The majority, at nearly 70%, had received the Moderna vaccine. The mean age of the cohort was 57 years and the median time since patients had undergone transplantation was almost 5 years (57.5 months).
During the 6-month study interval, 65 cases of COVID-19 were documented in the group overall. Only 4 cases occurred among fully vaccinated individuals whereas 61 cases occurred among the unvaccinated, including in 2 patients who had been partially vaccinated.
Among the four cases that occurred among the fully vaccinated, two were considered mild and were treated on an outpatient basis, and the other two were moderate, requiring hospitalization and treatment with remdesivir.
There were no COVID-19–related deaths among the 4 patients who experienced breakthrough infections, whereas 2 (3.3%) of 61 of control patients died of COVID-19–related causes.
The authors noted that the incidence rate for COVID-19 was 0.065 per 1,000-person days among the fully vaccinated compared with an incidence rate of 0.34 per 1,000-person days in the control group.
Booster doses for especially vulnerable transplant recipients?
“These findings are encouraging for a couple of reasons,” said coauthor Kristin Mekeel, MD, chief of transplant and hepatobiliary surgery at UCSD.
“First, they demonstrate real-world clinical effectiveness of COVID-19 vaccination in a vulnerable population,” she noted.
“Second, the effectiveness is better than expected,” she added, “given that studies have found that only about half of solid organ transplant recipients develop detectable antibodies after vaccination.”
Although calls for patients who are immunosuppressed to receive a third booster dose of a COVID-19 vaccine may not be necessary, “prioritizing at-risk subsets of transplant recipients based on immunological profiles and clinical characteristics for a third vaccine dose could be considered,” they said, adding it’s still vitally important for transplant patients to continue to mask and practice social distancing.
And it is especially important for transplant recipients to encourage household members to get vaccinated, too, especially given the current COVID-19 surge in San Diego.
The study was funded by the Cystic Fibrosis Foundation.
Dr. Aslam reports receiving grants from the Cystic Fibrosis Foundation as well as honoraria from Gilead and Merck. Study author Susan J. Little, MD, received grant funding from Gilead Sciences. Dr. Mekeel has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.