User login
HR+/HER2-negative breast cancer: Adding palbociclib to fulvestrant prolongs PFS in phase 2
Key clinical point: Palbociclib+fulvestrant prolonged progression-free survival (PFS) compared with placebo+fulvestrant in patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, endocrine-sensitive advanced breast cancer (BC).
Major finding: Palbociclib+fulvestrant vs placebo+fulvestrant improved 1-year PFS (83.5% vs 71.9%) and reduced the risk for progressive disease by 45% (hazard ratio [HR], 0.55; 80% CI, 0.36-0.83). Grade 3 or higher adverse events were reported by 80.9% vs 37.9% of patients in palbociclib/fulvestrant vs placebo/fulvestrant arms.
Study details: This was a phase 2 FLIPPER study of 189 postmenopausal women with HR-positive, HER2-negative, endocrine-sensitive advanced BC who were randomly assigned to palbociclib+fulvestrant or placebo+fulvestrant.
Disclosures: This study was funded by GEICAM Spanish Breast Cancer Group, AstraZeneca, and Pfizer. The authors reported receiving research grants, advisory board fees, consulting fees, honoraria, and travel and accommodation support from several sources.
Source: Albanell J et al. Eur J Cancer. 2021 Dec 11. doi: 10.1016/j.ejca.2021.11.010.
Key clinical point: Palbociclib+fulvestrant prolonged progression-free survival (PFS) compared with placebo+fulvestrant in patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, endocrine-sensitive advanced breast cancer (BC).
Major finding: Palbociclib+fulvestrant vs placebo+fulvestrant improved 1-year PFS (83.5% vs 71.9%) and reduced the risk for progressive disease by 45% (hazard ratio [HR], 0.55; 80% CI, 0.36-0.83). Grade 3 or higher adverse events were reported by 80.9% vs 37.9% of patients in palbociclib/fulvestrant vs placebo/fulvestrant arms.
Study details: This was a phase 2 FLIPPER study of 189 postmenopausal women with HR-positive, HER2-negative, endocrine-sensitive advanced BC who were randomly assigned to palbociclib+fulvestrant or placebo+fulvestrant.
Disclosures: This study was funded by GEICAM Spanish Breast Cancer Group, AstraZeneca, and Pfizer. The authors reported receiving research grants, advisory board fees, consulting fees, honoraria, and travel and accommodation support from several sources.
Source: Albanell J et al. Eur J Cancer. 2021 Dec 11. doi: 10.1016/j.ejca.2021.11.010.
Key clinical point: Palbociclib+fulvestrant prolonged progression-free survival (PFS) compared with placebo+fulvestrant in patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, endocrine-sensitive advanced breast cancer (BC).
Major finding: Palbociclib+fulvestrant vs placebo+fulvestrant improved 1-year PFS (83.5% vs 71.9%) and reduced the risk for progressive disease by 45% (hazard ratio [HR], 0.55; 80% CI, 0.36-0.83). Grade 3 or higher adverse events were reported by 80.9% vs 37.9% of patients in palbociclib/fulvestrant vs placebo/fulvestrant arms.
Study details: This was a phase 2 FLIPPER study of 189 postmenopausal women with HR-positive, HER2-negative, endocrine-sensitive advanced BC who were randomly assigned to palbociclib+fulvestrant or placebo+fulvestrant.
Disclosures: This study was funded by GEICAM Spanish Breast Cancer Group, AstraZeneca, and Pfizer. The authors reported receiving research grants, advisory board fees, consulting fees, honoraria, and travel and accommodation support from several sources.
Source: Albanell J et al. Eur J Cancer. 2021 Dec 11. doi: 10.1016/j.ejca.2021.11.010.
Metastatic BC: Improved survival in long-term responders with no evidence of disease vs residual disease
Key clinical point: Women with metastatic breast cancer (BC) with long-term response to first-line human epidermal growth factor receptor 2 (HER2)-targeted therapy who achieved no evidence of disease (NED) showed improved survival than those with residual disease (RES).
Major finding: Women with NED vs RES achieved longer median progression-free survival (not reached vs 3.08 years; P < .001) and superior overall survival (not reached vs 5.38 years; P < .001) with premenopausal status (P = .006) and de novo metastases (P = .002) associated with higher chances of achieving NED.
Study details: Findings are from a retrospective study including 103 women with HER2-positive metastatic BC who received first-line chemotherapy+trastuzumab or taxane+trastuzumab+pertuzumab and showed a response duration ≥2-fold higher than those observed in pivotal trials.
Disclosures: This study did not report any source of funding. The authors declared serving as a member of a trial steering committee and/or receiving honoraria, funding, consultancy, and advisory fees from several sources.
Source: Veitch Z et al. Br J Cancer. 2021 Dec 20. doi: 10.1038/s41416-021-01676-4.
Key clinical point: Women with metastatic breast cancer (BC) with long-term response to first-line human epidermal growth factor receptor 2 (HER2)-targeted therapy who achieved no evidence of disease (NED) showed improved survival than those with residual disease (RES).
Major finding: Women with NED vs RES achieved longer median progression-free survival (not reached vs 3.08 years; P < .001) and superior overall survival (not reached vs 5.38 years; P < .001) with premenopausal status (P = .006) and de novo metastases (P = .002) associated with higher chances of achieving NED.
Study details: Findings are from a retrospective study including 103 women with HER2-positive metastatic BC who received first-line chemotherapy+trastuzumab or taxane+trastuzumab+pertuzumab and showed a response duration ≥2-fold higher than those observed in pivotal trials.
Disclosures: This study did not report any source of funding. The authors declared serving as a member of a trial steering committee and/or receiving honoraria, funding, consultancy, and advisory fees from several sources.
Source: Veitch Z et al. Br J Cancer. 2021 Dec 20. doi: 10.1038/s41416-021-01676-4.
Key clinical point: Women with metastatic breast cancer (BC) with long-term response to first-line human epidermal growth factor receptor 2 (HER2)-targeted therapy who achieved no evidence of disease (NED) showed improved survival than those with residual disease (RES).
Major finding: Women with NED vs RES achieved longer median progression-free survival (not reached vs 3.08 years; P < .001) and superior overall survival (not reached vs 5.38 years; P < .001) with premenopausal status (P = .006) and de novo metastases (P = .002) associated with higher chances of achieving NED.
Study details: Findings are from a retrospective study including 103 women with HER2-positive metastatic BC who received first-line chemotherapy+trastuzumab or taxane+trastuzumab+pertuzumab and showed a response duration ≥2-fold higher than those observed in pivotal trials.
Disclosures: This study did not report any source of funding. The authors declared serving as a member of a trial steering committee and/or receiving honoraria, funding, consultancy, and advisory fees from several sources.
Source: Veitch Z et al. Br J Cancer. 2021 Dec 20. doi: 10.1038/s41416-021-01676-4.
Tucatinib and trastuzumab+capecitabine combo offers survival benefit in HER2+ metastatic breast cancer
Key clinical point: Addition of tucatinib to trastuzumab and capecitabine continued to improve survival along with good tolerability in patients with human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer (BC) who progressed on HER2-targeted therapies.
Major finding: Addition of tucatinib to trastuzumab and capecitabine significantly improved overall survival (hazard ratio [HR] for death, 0.73; P = .004) and progression-free survival (HR for disease-progression or death, 0.57; P < .00001) compared with placebo. Rates of grade 3 or higher adverse events (AEs) were similar between treatment arms, with only 5.9% of patients discontinuing treatment because of AEs.
Study details: These are the final outcomes from the phase 2 HER2CLIMB study including 612 patients with HER2-positive metastatic BC who progressed on trastuzumab, pertuzumab, and trastuzumab emtansine and were randomly assigned to tucatinib or placebo, each in combination with trastuzumab and capecitabine.
Disclosures: This work was supported by Seagen Inc. and Merck Sharp & Dohme Corp. Four authors declared being employees of Seagen and other sources, and other authors reported ties with various sources.
Source: Curigliano G et al. Ann Oncol. 2021 Dec 22. doi: 10.1016/j.annonc.2021.12.005.
Key clinical point: Addition of tucatinib to trastuzumab and capecitabine continued to improve survival along with good tolerability in patients with human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer (BC) who progressed on HER2-targeted therapies.
Major finding: Addition of tucatinib to trastuzumab and capecitabine significantly improved overall survival (hazard ratio [HR] for death, 0.73; P = .004) and progression-free survival (HR for disease-progression or death, 0.57; P < .00001) compared with placebo. Rates of grade 3 or higher adverse events (AEs) were similar between treatment arms, with only 5.9% of patients discontinuing treatment because of AEs.
Study details: These are the final outcomes from the phase 2 HER2CLIMB study including 612 patients with HER2-positive metastatic BC who progressed on trastuzumab, pertuzumab, and trastuzumab emtansine and were randomly assigned to tucatinib or placebo, each in combination with trastuzumab and capecitabine.
Disclosures: This work was supported by Seagen Inc. and Merck Sharp & Dohme Corp. Four authors declared being employees of Seagen and other sources, and other authors reported ties with various sources.
Source: Curigliano G et al. Ann Oncol. 2021 Dec 22. doi: 10.1016/j.annonc.2021.12.005.
Key clinical point: Addition of tucatinib to trastuzumab and capecitabine continued to improve survival along with good tolerability in patients with human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer (BC) who progressed on HER2-targeted therapies.
Major finding: Addition of tucatinib to trastuzumab and capecitabine significantly improved overall survival (hazard ratio [HR] for death, 0.73; P = .004) and progression-free survival (HR for disease-progression or death, 0.57; P < .00001) compared with placebo. Rates of grade 3 or higher adverse events (AEs) were similar between treatment arms, with only 5.9% of patients discontinuing treatment because of AEs.
Study details: These are the final outcomes from the phase 2 HER2CLIMB study including 612 patients with HER2-positive metastatic BC who progressed on trastuzumab, pertuzumab, and trastuzumab emtansine and were randomly assigned to tucatinib or placebo, each in combination with trastuzumab and capecitabine.
Disclosures: This work was supported by Seagen Inc. and Merck Sharp & Dohme Corp. Four authors declared being employees of Seagen and other sources, and other authors reported ties with various sources.
Source: Curigliano G et al. Ann Oncol. 2021 Dec 22. doi: 10.1016/j.annonc.2021.12.005.
GnRHa protects ovarian function in premenopausal women receiving chemotherapy for breast cancer
Key clinical point: Administration of gonadotropin-releasing hormone analogs (GnRHa) with chemotherapy reduced the risk for premature ovarian insufficiency (POI) in premenopausal women with breast cancer.
Major finding: At 12 months after chemotherapy, POI was reported by 10.3% vs 44.5% of patients in the GnRHa + chemotherapy vs chemotherapy-only group (odds ratio, 0.23; P < .001). No serious adverse events were reported.
Study details: Findings are from a phase 3 superiority trial including 330 premenopausal women with stages I-III operable breast cancer who were randomly assigned to GnRHa (3.6 mg of goserelin or 3.75 mg of leuprorelin) + chemotherapy or chemotherapy alone.
Disclosures: This study was funded by the Science and Technology Commission, Shanghai, and Zhejiang Medical Association. Dr. Zong reported receiving grants from the Science and Technology Commission of Shanghai Municipality.
Source: Zong X et al. JAMA Oncol. 2021 Dec 30. doi: 10.1001/jamaoncol.2021.6214.
Key clinical point: Administration of gonadotropin-releasing hormone analogs (GnRHa) with chemotherapy reduced the risk for premature ovarian insufficiency (POI) in premenopausal women with breast cancer.
Major finding: At 12 months after chemotherapy, POI was reported by 10.3% vs 44.5% of patients in the GnRHa + chemotherapy vs chemotherapy-only group (odds ratio, 0.23; P < .001). No serious adverse events were reported.
Study details: Findings are from a phase 3 superiority trial including 330 premenopausal women with stages I-III operable breast cancer who were randomly assigned to GnRHa (3.6 mg of goserelin or 3.75 mg of leuprorelin) + chemotherapy or chemotherapy alone.
Disclosures: This study was funded by the Science and Technology Commission, Shanghai, and Zhejiang Medical Association. Dr. Zong reported receiving grants from the Science and Technology Commission of Shanghai Municipality.
Source: Zong X et al. JAMA Oncol. 2021 Dec 30. doi: 10.1001/jamaoncol.2021.6214.
Key clinical point: Administration of gonadotropin-releasing hormone analogs (GnRHa) with chemotherapy reduced the risk for premature ovarian insufficiency (POI) in premenopausal women with breast cancer.
Major finding: At 12 months after chemotherapy, POI was reported by 10.3% vs 44.5% of patients in the GnRHa + chemotherapy vs chemotherapy-only group (odds ratio, 0.23; P < .001). No serious adverse events were reported.
Study details: Findings are from a phase 3 superiority trial including 330 premenopausal women with stages I-III operable breast cancer who were randomly assigned to GnRHa (3.6 mg of goserelin or 3.75 mg of leuprorelin) + chemotherapy or chemotherapy alone.
Disclosures: This study was funded by the Science and Technology Commission, Shanghai, and Zhejiang Medical Association. Dr. Zong reported receiving grants from the Science and Technology Commission of Shanghai Municipality.
Source: Zong X et al. JAMA Oncol. 2021 Dec 30. doi: 10.1001/jamaoncol.2021.6214.
Residual cancer burden prognostic across all breast cancer subtypes
Key clinical point: Residual cancer burden (RCB) after neoadjuvant chemotherapy was prognostic for event-free survival (EFS) in each hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) subtype of breast cancer.
Major finding: RCB was prognostic for EFS with the hazard ratio associated with each unit increase in RCB being 1.69 (P < .0001) for the overall population and ranging from 1.52 in HR-positive/HER2-negative group to 2.09 in HR-negative/HER2-positive group (P < .0001 for all subtypes).
Study details: Findings are pooled analysis of 4 trials and 8 clinical cohorts, including 5,161 adult patients with primary stage I-III breast cancer treated with neoadjuvant chemotherapy followed by surgery.
Disclosures: This study was funded by the National Cancer Institute, USA. Some of the authors declared serving as a consultant, data and safety monitoring advisor, and/or receiving grants, funding, personal fees, travel support, and honoraria from several sources.
Source: Yau C et al. Lancet Oncol. 2021 Dec 10. doi: 10.1016/S1470-2045(21)00589-1.
Key clinical point: Residual cancer burden (RCB) after neoadjuvant chemotherapy was prognostic for event-free survival (EFS) in each hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) subtype of breast cancer.
Major finding: RCB was prognostic for EFS with the hazard ratio associated with each unit increase in RCB being 1.69 (P < .0001) for the overall population and ranging from 1.52 in HR-positive/HER2-negative group to 2.09 in HR-negative/HER2-positive group (P < .0001 for all subtypes).
Study details: Findings are pooled analysis of 4 trials and 8 clinical cohorts, including 5,161 adult patients with primary stage I-III breast cancer treated with neoadjuvant chemotherapy followed by surgery.
Disclosures: This study was funded by the National Cancer Institute, USA. Some of the authors declared serving as a consultant, data and safety monitoring advisor, and/or receiving grants, funding, personal fees, travel support, and honoraria from several sources.
Source: Yau C et al. Lancet Oncol. 2021 Dec 10. doi: 10.1016/S1470-2045(21)00589-1.
Key clinical point: Residual cancer burden (RCB) after neoadjuvant chemotherapy was prognostic for event-free survival (EFS) in each hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) subtype of breast cancer.
Major finding: RCB was prognostic for EFS with the hazard ratio associated with each unit increase in RCB being 1.69 (P < .0001) for the overall population and ranging from 1.52 in HR-positive/HER2-negative group to 2.09 in HR-negative/HER2-positive group (P < .0001 for all subtypes).
Study details: Findings are pooled analysis of 4 trials and 8 clinical cohorts, including 5,161 adult patients with primary stage I-III breast cancer treated with neoadjuvant chemotherapy followed by surgery.
Disclosures: This study was funded by the National Cancer Institute, USA. Some of the authors declared serving as a consultant, data and safety monitoring advisor, and/or receiving grants, funding, personal fees, travel support, and honoraria from several sources.
Source: Yau C et al. Lancet Oncol. 2021 Dec 10. doi: 10.1016/S1470-2045(21)00589-1.
Study finds genetic factor for COVID smell and taste loss
, according to a new study published in the journal Nature Genetics
The finding could eventually help the 1.6 million people in the United States who still can’t smell or have had a change in their ability to smell more than 6 months after getting the coronavirus. The exact cause related to COVID-19 is still unknown, but researchers believe it could be because of damage in a part of the nose called the olfactory epithelium.
“How we get from infection to smell loss remains unclear,” Justin Turner, MD, an associate professor of otolaryngology at Vanderbilt University, Nashville, Tenn., told NBC News. Dr. Turner was not part of the research team.
“Early data suggest that supporting cells of the olfactory epithelium are the ones mostly being infected by the virus, and presumably this leads to the death of the neurons themselves,” he said. “But we don’t really, really know why and when that happens, and why it seems to preferentially happen in certain individuals.”
Researchers at 23andMe, a genomics and biotechnology company, did the study as part of a larger COVID-19 project, which includes people in the United States and the United Kingdom. They analyzed data from nearly 70,000 people who took online surveys after receiving a positive coronavirus test. Among those, 68% reported a loss of smell or taste as a symptom.
The study team compared the genetic differences between those who lost their sense of smell and taste and those who didn’t. They found that a location near two olfactory genes – UGT2A1 and UGT2A2 – is associated with COVID-19 loss of smell and taste. The genetic risk factor makes it 11% more likely for a person with COVID-19 to lose their sense of smell or taste.
The research team also found that women were 11% more likely than men to report a loss of smell and taste. About 73% of those who reported a loss of smell and taste were ages 26-35.
The researchers aren’t sure how the genes are involved, though they suspect that infected cells could lead to smell loss. Typically, the genes are expressed in tissue inside the nose involved with smell and play a role in processing things that have an odor. To use the findings, researchers need to learn more about the genes, how they are expressed, and what their functions are, NBC News reported.
The findings could help lead to treatments. Other research has shown that the loss of taste and smell is related to a “failure to protect the sensory cells of the nose and tongue from viral infection,” Danielle Reed, PhD, associate director of the Monell Chemical Senses Center in Philadelphia, told NBC News. She was not part of the research team but studies person-to-person differences in the loss of these senses because of COVID-19.
“This study suggests a different direction,” she said. “The pathways that break down the chemicals that cause taste and smell in the first place might be over or underactive, reducing or distorting the ability to taste and smell.”
A version of this article first appeared on WebMD.com.
, according to a new study published in the journal Nature Genetics
The finding could eventually help the 1.6 million people in the United States who still can’t smell or have had a change in their ability to smell more than 6 months after getting the coronavirus. The exact cause related to COVID-19 is still unknown, but researchers believe it could be because of damage in a part of the nose called the olfactory epithelium.
“How we get from infection to smell loss remains unclear,” Justin Turner, MD, an associate professor of otolaryngology at Vanderbilt University, Nashville, Tenn., told NBC News. Dr. Turner was not part of the research team.
“Early data suggest that supporting cells of the olfactory epithelium are the ones mostly being infected by the virus, and presumably this leads to the death of the neurons themselves,” he said. “But we don’t really, really know why and when that happens, and why it seems to preferentially happen in certain individuals.”
Researchers at 23andMe, a genomics and biotechnology company, did the study as part of a larger COVID-19 project, which includes people in the United States and the United Kingdom. They analyzed data from nearly 70,000 people who took online surveys after receiving a positive coronavirus test. Among those, 68% reported a loss of smell or taste as a symptom.
The study team compared the genetic differences between those who lost their sense of smell and taste and those who didn’t. They found that a location near two olfactory genes – UGT2A1 and UGT2A2 – is associated with COVID-19 loss of smell and taste. The genetic risk factor makes it 11% more likely for a person with COVID-19 to lose their sense of smell or taste.
The research team also found that women were 11% more likely than men to report a loss of smell and taste. About 73% of those who reported a loss of smell and taste were ages 26-35.
The researchers aren’t sure how the genes are involved, though they suspect that infected cells could lead to smell loss. Typically, the genes are expressed in tissue inside the nose involved with smell and play a role in processing things that have an odor. To use the findings, researchers need to learn more about the genes, how they are expressed, and what their functions are, NBC News reported.
The findings could help lead to treatments. Other research has shown that the loss of taste and smell is related to a “failure to protect the sensory cells of the nose and tongue from viral infection,” Danielle Reed, PhD, associate director of the Monell Chemical Senses Center in Philadelphia, told NBC News. She was not part of the research team but studies person-to-person differences in the loss of these senses because of COVID-19.
“This study suggests a different direction,” she said. “The pathways that break down the chemicals that cause taste and smell in the first place might be over or underactive, reducing or distorting the ability to taste and smell.”
A version of this article first appeared on WebMD.com.
, according to a new study published in the journal Nature Genetics
The finding could eventually help the 1.6 million people in the United States who still can’t smell or have had a change in their ability to smell more than 6 months after getting the coronavirus. The exact cause related to COVID-19 is still unknown, but researchers believe it could be because of damage in a part of the nose called the olfactory epithelium.
“How we get from infection to smell loss remains unclear,” Justin Turner, MD, an associate professor of otolaryngology at Vanderbilt University, Nashville, Tenn., told NBC News. Dr. Turner was not part of the research team.
“Early data suggest that supporting cells of the olfactory epithelium are the ones mostly being infected by the virus, and presumably this leads to the death of the neurons themselves,” he said. “But we don’t really, really know why and when that happens, and why it seems to preferentially happen in certain individuals.”
Researchers at 23andMe, a genomics and biotechnology company, did the study as part of a larger COVID-19 project, which includes people in the United States and the United Kingdom. They analyzed data from nearly 70,000 people who took online surveys after receiving a positive coronavirus test. Among those, 68% reported a loss of smell or taste as a symptom.
The study team compared the genetic differences between those who lost their sense of smell and taste and those who didn’t. They found that a location near two olfactory genes – UGT2A1 and UGT2A2 – is associated with COVID-19 loss of smell and taste. The genetic risk factor makes it 11% more likely for a person with COVID-19 to lose their sense of smell or taste.
The research team also found that women were 11% more likely than men to report a loss of smell and taste. About 73% of those who reported a loss of smell and taste were ages 26-35.
The researchers aren’t sure how the genes are involved, though they suspect that infected cells could lead to smell loss. Typically, the genes are expressed in tissue inside the nose involved with smell and play a role in processing things that have an odor. To use the findings, researchers need to learn more about the genes, how they are expressed, and what their functions are, NBC News reported.
The findings could help lead to treatments. Other research has shown that the loss of taste and smell is related to a “failure to protect the sensory cells of the nose and tongue from viral infection,” Danielle Reed, PhD, associate director of the Monell Chemical Senses Center in Philadelphia, told NBC News. She was not part of the research team but studies person-to-person differences in the loss of these senses because of COVID-19.
“This study suggests a different direction,” she said. “The pathways that break down the chemicals that cause taste and smell in the first place might be over or underactive, reducing or distorting the ability to taste and smell.”
A version of this article first appeared on WebMD.com.
FROM NATURE GENETICS
Fourth vaccine shot less effective against Omicron, Israeli study says
, according to new research at an Israeli hospital.
The preliminary results, released on Jan. 17, challenge the idea of giving a second booster dose to slow the spread of the coronavirus, according to USA Today.
“Despite increased antibody levels, the fourth vaccine only offers a partial defense against the virus,” Gili Regev-Yochay, MD, director of the hospital’s infection prevention and control units, told reporters.
“The vaccines, which were more effective against previous variants, offer less protection versus Omicron,” she said.
In a clinical trial, 274 medical workers at Sheba Medical Center near Tel Aviv received a fourth vaccine dose in December – 154 got the Pfizer vaccine and 120 got the Moderna vaccine – after previously getting three Pfizer shots.
Both groups received a boost in antibodies that was “slightly higher” than after the third shot, Dr. Regev-Yochay said. But when compared with a control group that didn’t receive the fourth dose, the extra boost didn’t prevent the spread of Omicron.
“We see many infected with Omicron who received the fourth dose,” Dr. Regev-Yochay said. “Granted, a bit less than in the control group, but still a lot of infections.”
Some public health officials in Israel say the campaign for fourth doses is still worthwhile, according to The Times of Israel. The vaccine still works well against the Alpha and Delta variants, Dr. Regev-Yochay said, and a fourth shot should go to older adults and those who face higher risks for severe COVID-19.
Hours after releasing the preliminary results, Sheba Medical Center published a statement calling for “continuing the vaccination drive for risk groups at this time, even though the vaccine doesn’t provide optimal protection against getting infected with the variant.” News outlets reported that the hospital was pressured into issuing the statement after Israel’s Health Ministry didn’t like the release of the early study results, The Times of Israel reported.
The second booster “returns the level of antibodies to what it was at the beginning of the third booster,” Nachman Ash, MD, director of Israel’s Health Ministry, told Channel 13 TV in Israel, according to The Associated Press.
“That has great importance, especially among the older population,” he said.
As of Sunday, more than 500,000 people in Israel had received fourth doses since the country began offering them last month to medical workers, immunocompromised patients, and people ages 60 years and older, the AP reported. At the same time, the country has faced a recent coronavirus surge that has led to record-breaking numbers of cases and rising hospitalizations.
On Tuesday, the Israeli government said it would shorten the mandatory quarantine period from 7 days to 5 days, the AP reported.
“This decision will enable us to continue safeguarding public health on the one hand and to keep the economy going at this time on the other, even though it is difficult, so that we can get through this wave safely,” Prime Minister Naftali Bennett said.
A version of this article first appeared on WebMD.com.
, according to new research at an Israeli hospital.
The preliminary results, released on Jan. 17, challenge the idea of giving a second booster dose to slow the spread of the coronavirus, according to USA Today.
“Despite increased antibody levels, the fourth vaccine only offers a partial defense against the virus,” Gili Regev-Yochay, MD, director of the hospital’s infection prevention and control units, told reporters.
“The vaccines, which were more effective against previous variants, offer less protection versus Omicron,” she said.
In a clinical trial, 274 medical workers at Sheba Medical Center near Tel Aviv received a fourth vaccine dose in December – 154 got the Pfizer vaccine and 120 got the Moderna vaccine – after previously getting three Pfizer shots.
Both groups received a boost in antibodies that was “slightly higher” than after the third shot, Dr. Regev-Yochay said. But when compared with a control group that didn’t receive the fourth dose, the extra boost didn’t prevent the spread of Omicron.
“We see many infected with Omicron who received the fourth dose,” Dr. Regev-Yochay said. “Granted, a bit less than in the control group, but still a lot of infections.”
Some public health officials in Israel say the campaign for fourth doses is still worthwhile, according to The Times of Israel. The vaccine still works well against the Alpha and Delta variants, Dr. Regev-Yochay said, and a fourth shot should go to older adults and those who face higher risks for severe COVID-19.
Hours after releasing the preliminary results, Sheba Medical Center published a statement calling for “continuing the vaccination drive for risk groups at this time, even though the vaccine doesn’t provide optimal protection against getting infected with the variant.” News outlets reported that the hospital was pressured into issuing the statement after Israel’s Health Ministry didn’t like the release of the early study results, The Times of Israel reported.
The second booster “returns the level of antibodies to what it was at the beginning of the third booster,” Nachman Ash, MD, director of Israel’s Health Ministry, told Channel 13 TV in Israel, according to The Associated Press.
“That has great importance, especially among the older population,” he said.
As of Sunday, more than 500,000 people in Israel had received fourth doses since the country began offering them last month to medical workers, immunocompromised patients, and people ages 60 years and older, the AP reported. At the same time, the country has faced a recent coronavirus surge that has led to record-breaking numbers of cases and rising hospitalizations.
On Tuesday, the Israeli government said it would shorten the mandatory quarantine period from 7 days to 5 days, the AP reported.
“This decision will enable us to continue safeguarding public health on the one hand and to keep the economy going at this time on the other, even though it is difficult, so that we can get through this wave safely,” Prime Minister Naftali Bennett said.
A version of this article first appeared on WebMD.com.
, according to new research at an Israeli hospital.
The preliminary results, released on Jan. 17, challenge the idea of giving a second booster dose to slow the spread of the coronavirus, according to USA Today.
“Despite increased antibody levels, the fourth vaccine only offers a partial defense against the virus,” Gili Regev-Yochay, MD, director of the hospital’s infection prevention and control units, told reporters.
“The vaccines, which were more effective against previous variants, offer less protection versus Omicron,” she said.
In a clinical trial, 274 medical workers at Sheba Medical Center near Tel Aviv received a fourth vaccine dose in December – 154 got the Pfizer vaccine and 120 got the Moderna vaccine – after previously getting three Pfizer shots.
Both groups received a boost in antibodies that was “slightly higher” than after the third shot, Dr. Regev-Yochay said. But when compared with a control group that didn’t receive the fourth dose, the extra boost didn’t prevent the spread of Omicron.
“We see many infected with Omicron who received the fourth dose,” Dr. Regev-Yochay said. “Granted, a bit less than in the control group, but still a lot of infections.”
Some public health officials in Israel say the campaign for fourth doses is still worthwhile, according to The Times of Israel. The vaccine still works well against the Alpha and Delta variants, Dr. Regev-Yochay said, and a fourth shot should go to older adults and those who face higher risks for severe COVID-19.
Hours after releasing the preliminary results, Sheba Medical Center published a statement calling for “continuing the vaccination drive for risk groups at this time, even though the vaccine doesn’t provide optimal protection against getting infected with the variant.” News outlets reported that the hospital was pressured into issuing the statement after Israel’s Health Ministry didn’t like the release of the early study results, The Times of Israel reported.
The second booster “returns the level of antibodies to what it was at the beginning of the third booster,” Nachman Ash, MD, director of Israel’s Health Ministry, told Channel 13 TV in Israel, according to The Associated Press.
“That has great importance, especially among the older population,” he said.
As of Sunday, more than 500,000 people in Israel had received fourth doses since the country began offering them last month to medical workers, immunocompromised patients, and people ages 60 years and older, the AP reported. At the same time, the country has faced a recent coronavirus surge that has led to record-breaking numbers of cases and rising hospitalizations.
On Tuesday, the Israeli government said it would shorten the mandatory quarantine period from 7 days to 5 days, the AP reported.
“This decision will enable us to continue safeguarding public health on the one hand and to keep the economy going at this time on the other, even though it is difficult, so that we can get through this wave safely,” Prime Minister Naftali Bennett said.
A version of this article first appeared on WebMD.com.
Blisters in a Comatose Elderly Woman
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
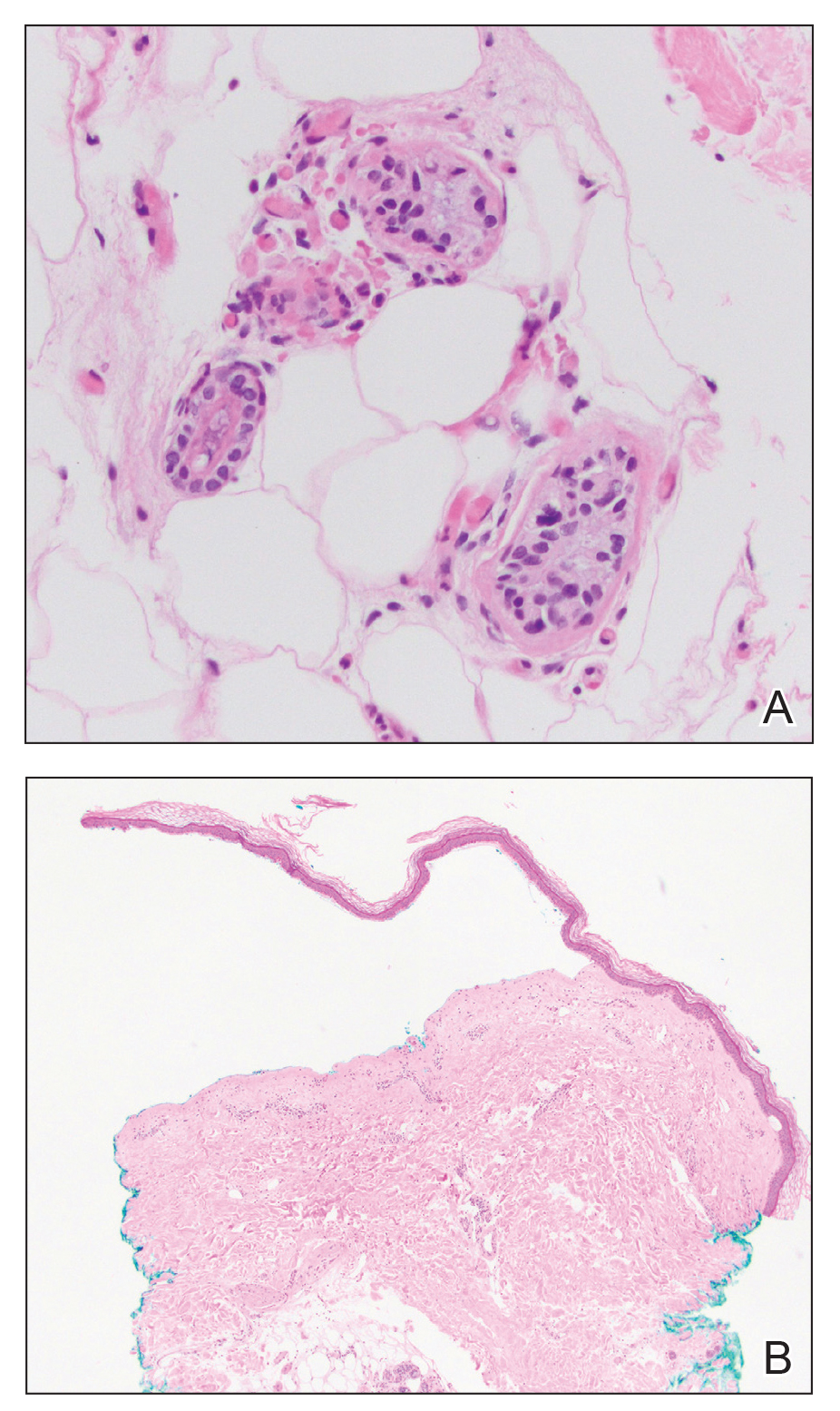
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
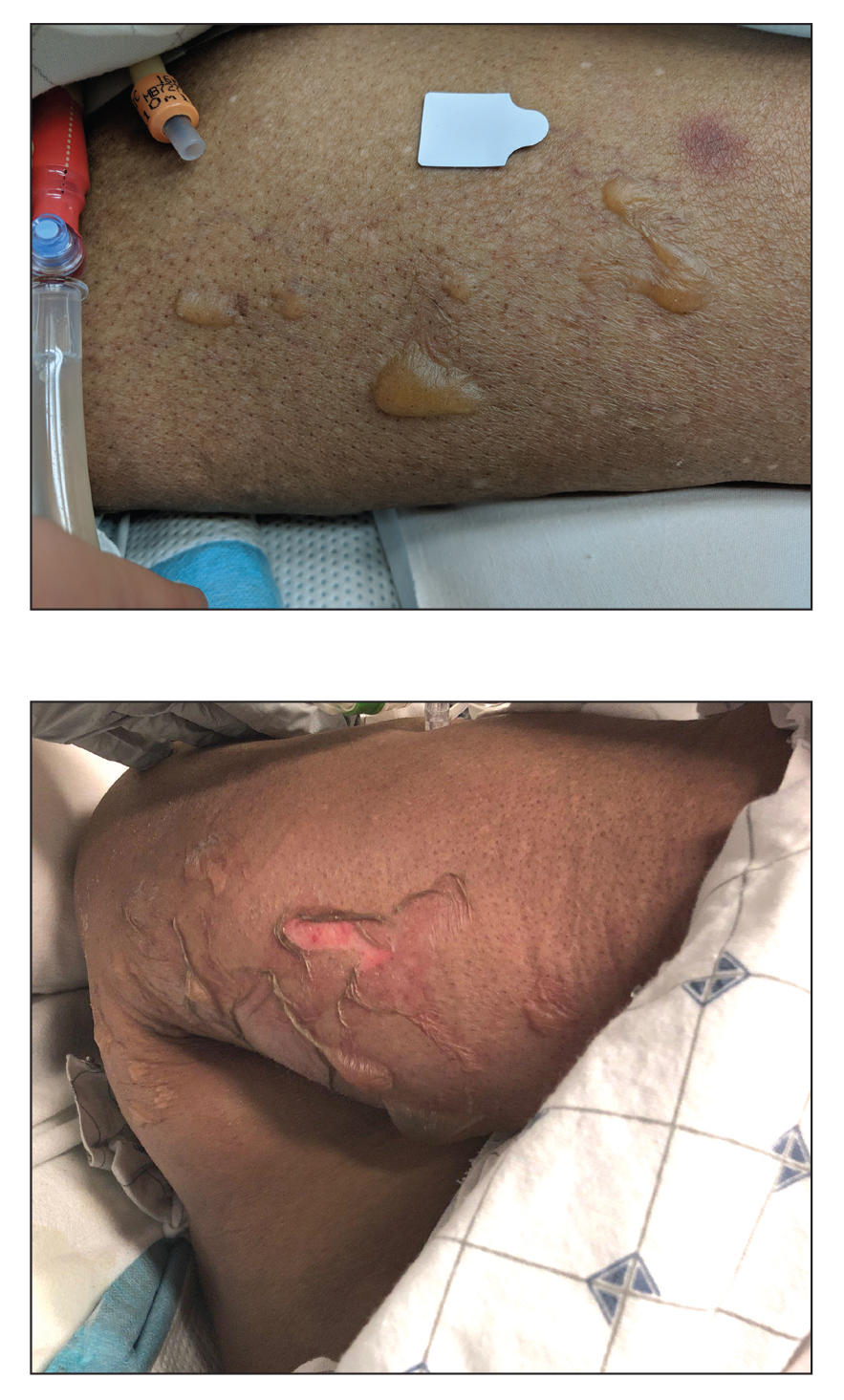
An 82-year-old woman presented to the emergency department after her daughter found her unconscious in the bathroom laying on her right side. Her medical history was notable for hypertension and asthma for which she was on losartan, furosemide, diltiazem, and albuterol. She recently had been prescribed lorazepam for insomnia and had started taking the medication 2 days prior. She underwent intubation and was noted to have flaccid, fluid-filled bullae on the right thigh (top) along with large areas of desquamation on the right lateral arm (bottom) with minimal surrounding erythema. There was no mucous membrane involvement. Urine toxicology was positive for benzodiazepines and negative for all other drugs, including barbiturates.
Shortness of breath and abdominal pain
On the basis of the patient's history and clinical presentation, the likely diagnosis is ketosis-prone diabetes (KPD) type 2. KPD is widely thought of as an atypical diabetes syndrome, though some groups consider it to be a common clinical presentation in newly diagnosed patients with type 2 diabetes (T2D) rather than a subtype of disease. The condition is more prevalent in males and among Black and Hispanic populations.
Definitive diagnosis of the type of diabetes can present a clinical challenge during acute presentation. Although the majority of diabetic ketoacidosis (DKA) episodes occur in patients previously diagnosed with type 1 diabetes, an estimated 34% occur in patients with T2D. For patients who are obese or have a family history of diabetes, there should be a high index of suspicion for new-onset DKA in type 2 diabetes.
Patients with KPD typically present in a state of ketoacidosis with a brief but acute history of hyperglycemic symptoms. Hyperglycemia, elevated anion gap acidosis, and ketonemia form the expected constellation of DKA symptoms. A key consideration in the differential diagnosis is hyperosmolar hyperglycemic state (HHS). Patients with HHS are much more likely to have altered mental status than are patients with DKA. Metabolic acidosis and ketonemia are absent or mild, and anion gap is either normal or slightly elevated. In HHS, extreme elevations of glucose are seen, with a lack of significant ketoacidosis. Glucose levels tend to be higher in HHS than in DKA; they are almost always > 600 mg/dL, and levels > 1000 mg/dL are not uncommon. In DKA, glucose levels are still markedly high — generally 500-800 mg/dL but rarely exceeding 900 mg/dL. Patients with DKA also usually present with an A1c > 10% and a blood pH < 7.30.
Treatment for KPD is initially acute, beginning with aggressive intravenous fluid and insulin therapy A. Once this state resolves, insulin requirements typically decrease for patients with T2D, and they are able to maintain adequate glycemic control with an oral therapy regimen.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia.
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
On the basis of the patient's history and clinical presentation, the likely diagnosis is ketosis-prone diabetes (KPD) type 2. KPD is widely thought of as an atypical diabetes syndrome, though some groups consider it to be a common clinical presentation in newly diagnosed patients with type 2 diabetes (T2D) rather than a subtype of disease. The condition is more prevalent in males and among Black and Hispanic populations.
Definitive diagnosis of the type of diabetes can present a clinical challenge during acute presentation. Although the majority of diabetic ketoacidosis (DKA) episodes occur in patients previously diagnosed with type 1 diabetes, an estimated 34% occur in patients with T2D. For patients who are obese or have a family history of diabetes, there should be a high index of suspicion for new-onset DKA in type 2 diabetes.
Patients with KPD typically present in a state of ketoacidosis with a brief but acute history of hyperglycemic symptoms. Hyperglycemia, elevated anion gap acidosis, and ketonemia form the expected constellation of DKA symptoms. A key consideration in the differential diagnosis is hyperosmolar hyperglycemic state (HHS). Patients with HHS are much more likely to have altered mental status than are patients with DKA. Metabolic acidosis and ketonemia are absent or mild, and anion gap is either normal or slightly elevated. In HHS, extreme elevations of glucose are seen, with a lack of significant ketoacidosis. Glucose levels tend to be higher in HHS than in DKA; they are almost always > 600 mg/dL, and levels > 1000 mg/dL are not uncommon. In DKA, glucose levels are still markedly high — generally 500-800 mg/dL but rarely exceeding 900 mg/dL. Patients with DKA also usually present with an A1c > 10% and a blood pH < 7.30.
Treatment for KPD is initially acute, beginning with aggressive intravenous fluid and insulin therapy A. Once this state resolves, insulin requirements typically decrease for patients with T2D, and they are able to maintain adequate glycemic control with an oral therapy regimen.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia.
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
On the basis of the patient's history and clinical presentation, the likely diagnosis is ketosis-prone diabetes (KPD) type 2. KPD is widely thought of as an atypical diabetes syndrome, though some groups consider it to be a common clinical presentation in newly diagnosed patients with type 2 diabetes (T2D) rather than a subtype of disease. The condition is more prevalent in males and among Black and Hispanic populations.
Definitive diagnosis of the type of diabetes can present a clinical challenge during acute presentation. Although the majority of diabetic ketoacidosis (DKA) episodes occur in patients previously diagnosed with type 1 diabetes, an estimated 34% occur in patients with T2D. For patients who are obese or have a family history of diabetes, there should be a high index of suspicion for new-onset DKA in type 2 diabetes.
Patients with KPD typically present in a state of ketoacidosis with a brief but acute history of hyperglycemic symptoms. Hyperglycemia, elevated anion gap acidosis, and ketonemia form the expected constellation of DKA symptoms. A key consideration in the differential diagnosis is hyperosmolar hyperglycemic state (HHS). Patients with HHS are much more likely to have altered mental status than are patients with DKA. Metabolic acidosis and ketonemia are absent or mild, and anion gap is either normal or slightly elevated. In HHS, extreme elevations of glucose are seen, with a lack of significant ketoacidosis. Glucose levels tend to be higher in HHS than in DKA; they are almost always > 600 mg/dL, and levels > 1000 mg/dL are not uncommon. In DKA, glucose levels are still markedly high — generally 500-800 mg/dL but rarely exceeding 900 mg/dL. Patients with DKA also usually present with an A1c > 10% and a blood pH < 7.30.
Treatment for KPD is initially acute, beginning with aggressive intravenous fluid and insulin therapy A. Once this state resolves, insulin requirements typically decrease for patients with T2D, and they are able to maintain adequate glycemic control with an oral therapy regimen.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia.
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
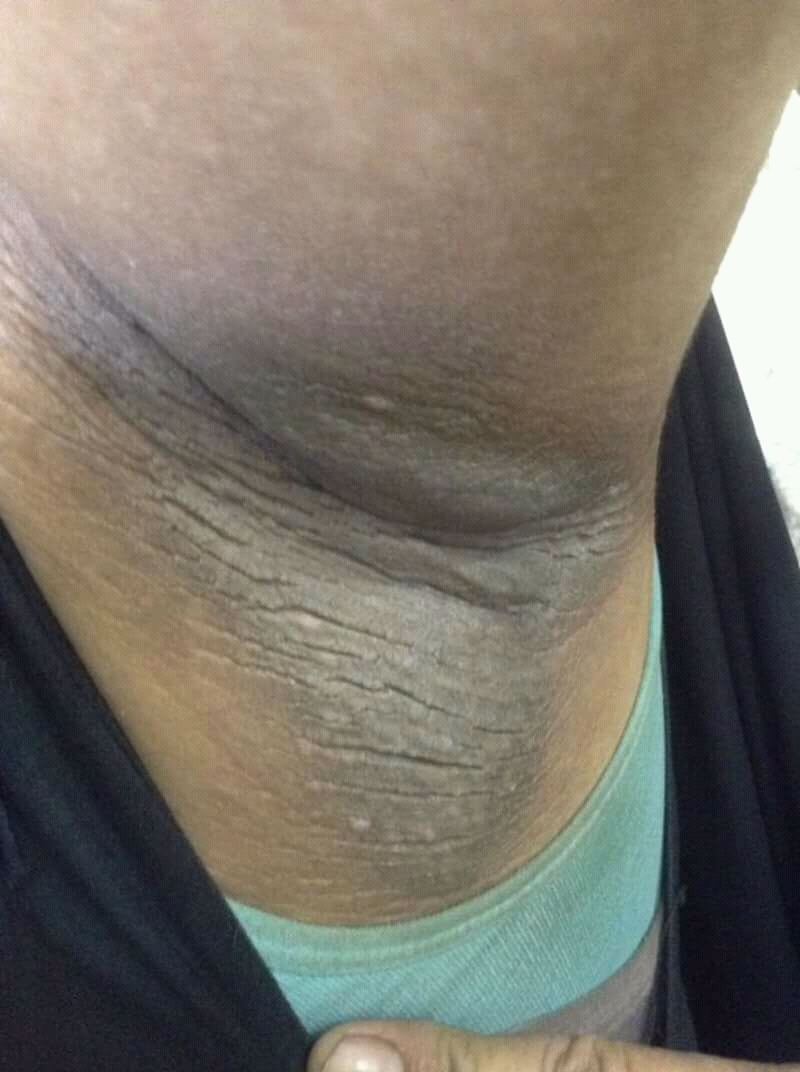
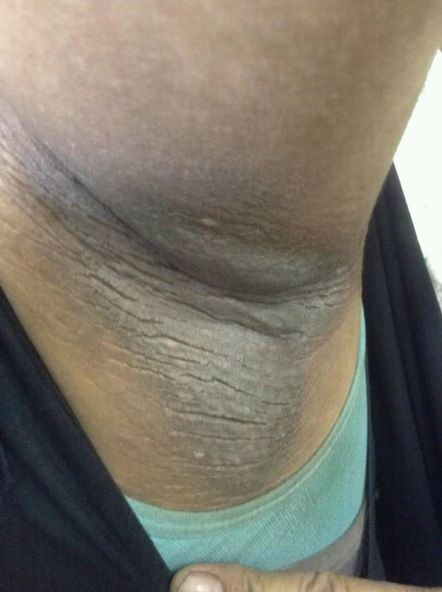
A 21-year-old man presents with shortness of breath and abdominal pain. He has a BMI of 34.6 and explains that he has had asthma for several years, using an inhaler when needed. He reports a few weeks of polydipsia and polyuria. The patient notes that his father has kidney disease. He believes other close relatives are managing chronic metabolic conditions but does not know any further detail regarding their diagnoses. On laboratory testing, blood pH is 6.30 and bicarbonate level is 11.1 mmol/L. A1c is 12.0%. Acanthosis nigricans are noted on the neck and in the axilla bilaterally.
2022 Update on obstetrics
Obstetrical practice saw updates in 2021 to 3 major areas of pregnancy management: preterm birth prevention, antepartum fetal surveillance, and the use of antenatal corticosteroids.
Updated guidance on predicting and preventing spontaneous PTB
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins–Obstetrics. Prediction and prevention of spontaneous preterm birth: ACOG practice bulletin, number 234. Obstet Gynecol. 2021;138:e65-e90.
Preterm birth (PTB) continues to pose a challenge in clinical obstetrics, with the most recently reported rate of 10.2% in the United States.1 This accounts for almost 75% of perinatal mortality and more than half of neonatal morbidity, in which effects last well past the neonatal period. PTB is classified as spontaneous (following preterm labor, preterm prelabor rupture of membranes, or cervical insufficiency) or iatrogenic (indicated due to maternal and/or fetal complications).
Assessing risk for PTB
The single strongest predictor of subsequent PTB is a history of spontaneous PTB. Recurrence risk is further increased by the number of prior PTBs and the gestational age at prior PTB. Identification of and intervention for a short cervix has been shown to prolong gestation. Transvaginal ultrasonography of the cervix is the most accurate method for evaluating cervical length (CL). Specific examination criteria exist to ensure that CL measurements are reproducible and reliable.2 A short CL is generally defined as a measurement of less than 25 mm between 16 and 24 weeks’ gestation.
Screening strategies
The American College of Obstetricians and Gynecologists (ACOG), with an endorsement from the Society for Maternal-Fetal Medicine (SMFM), recommends cervical evaluation during the anatomy ultrasound exam between 18 0/7 and 22 6/7 weeks’ gestation in all pregnant patients regardless of prior PTB.3 If transabdominal imaging is concerning for a shortened cervix, transvaginal ultrasonography should be performed to assess the CL.
Serial transvaginal CL measurements are recommended between 16 0/7 and 24 0/7 weeks’ gestation for patients with a current singleton pregnancy and history of a spontaneous PTB, but not for patients with a history of iatrogenic or indicated PTB.
Interventions: Mind your p’s and c’s
Interventions to reduce the risk of spontaneous PTB depend on whether the current pregnancy is a singleton, twins, or higher-order multiples; CL measurement; and history of spontaneous PTB. Preconception optimization of underlying medical conditions also is important to reduce the risk of recurrent indicated PTB.
Continue to: Progesterone...
Progesterone
Vaginal administration. Several trials have shown that vaginal progesterone can be used to reduce the risk of spontaneous PTB in asymptomatic patients with a singleton pregnancy, incidental finding of a short cervix (<25 mm), and no history of spontaneous PTB. This is a change from the prior recommendation of CL of less than 20 mm. In the setting of a twin pregnancy, regardless of CL, data do not definitively support the use of vaginal progesterone.
Intramuscular administration.4,5 The popularity of intramuscular progesterone has waxed and waned. At present, ACOG recommends that all patients with a singleton pregnancy and history of spontaneous PTB be offered progesterone beginning at 16 0/7 weeks’ gestation following a shared decision-making process that includes the limited data of efficacy noted in existing studies.
In a twin pregnancy with no history of spontaneous PTB, the use of intramuscular progesterone has been shown to potentially increase the risk of PTB and admission to the neonatal intensive care unit. As such, intramuscular progesterone in the setting of a twin gestation without a history of spontaneous PTB is not recommended. When a prior spontaneous PTB has occurred, there may be some benefit to intramuscular progesterone in twin gestations.
Cerclage
Ultrasound indicated. In a singleton pregnancy with an incidental finding of short cervix (<25 mm) and no history of PTB, the use of cerclage is of uncertain benefit. Effectiveness may be seen if the cervix is less than 10 mm. Ultrasound-indicated cerclage should be considered in a singleton pregnancy with a CL less than 25 mm and a history of spontaneous PTB.
Possibly one of the most controversial topics is ultrasound-indicated cerclage placement in twin gestation. As with many situations in obstetrics, data regarding ultrasound-indicated cerclage in twin gestation is based on small retrospective studies fraught with bias. Results from these studies range from no benefit, to potential benefit, to even possible increased risk of PTB. Since data are limited, as we await more evidence, it is recommended that the clinician and patient use shared decision making to decide on cerclage placement in a twin gestation.
Exam indicated. In a singleton pregnancy with a dilated cervix on digital or speculum exam between 16 0/7 to 23 6/7 weeks’ gestation, a physical exam–indicated cerclage should be offered. Exam-indicated cerclage also may reduce the incidence of PTB in twin gestations with cervical dilation between 16 0/7 and 23 6/7 weeks’ gestation. Indomethacin tocolysis and perioperative antibiotics should be considered when an exam-indicated cerclage is placed.
As the limits of viability are continually pushed earlier, more in-depth conversation is needed with patients who are considering an exam-indicated cerclage. The nuances of periviability and the likelihood that an exam-indicated cerclage will commit a pregnancy to a periviable or extremely preterm birth should be discussed in detail using a shared decision making model.
Regardless of whether the cerclage is ultrasound or exam indicated, once it is placed there is no utility in additional CL ultrasound monitoring.
Pessary
Vaginal pessaries for prevention of PTB have not gained popularity in the United States as they have in other countries. Trials are being conducted to determine the utility of vaginal pessary, but current data have not proven its effectiveness in preventing PTB in the setting of singleton pregnancy, short cervix, and no history of spontaneous PTB. So for now, pessary is not recommended. The same can be said for use in the twin gestation.
- All patients should have cervical evaluation during pregnancy. Serial imaging is reserved for those with a history of spontaneous PTB.
- Progesterone supplementation should be offered to patients with a singleton pregnancy and a history of spontaneous PTB or to patients with a singleton pregnancy and no history of spontaneous PTB who have cervical shortening at less than 24 weeks.
- Cerclage may be offered between 16 and 24 weeks for a cervical length less than 25 mm in a patient with a singleton gestation who has a history of spontaneous PTB (<10 mm if no history of spontaneous PTB) or for a dilated cervix on exam regardless of history.
- Women who have a twin gestation with cervical dilation may be offered physical exam–indicated cerclage.
Which patients may benefit from antepartum fetal surveillance and when to initiate it
American College of Obstetricians and Gynecologists’ Committee on Obstetrics Practice, Society for Maternal-Fetal Medicine. Indications for outpatient antenatal surveillance: ACOG committee opinion, number 828. Obstet Gynecol. 2021;137:e177-e197.
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics. Antepartum fetal surveillance: ACOG practice bulletin, number 229. Obstet Gynecol. 2021;137:e116-e127.
The ultimate purpose of antenatal fetal surveillance is to prevent stillbirth. However, stillbirth has multiple etiologies, not all of which are preventable with testing. In June 2021, ACOG released a new Committee Opinion containing guidelines for fetal surveillance, including suggested gestational age at initiation and frequency of testing, for the most common high-risk conditions. ACOG also released an update to the Practice Bulletin on antepartum fetal surveillance; additions include randomized controlled trial level data on the utility of fetal kick counts (FKCs) and recommendations that align with the new Committee Opinion.
Data for the efficacy of antepartum fetal surveillance are lacking, mainly due to the difficulty of performing prospective studies in stillbirth. The existing evidence is subject to intervention bias, as deliveries increase in tested patients, and recommendations rely heavily on expert consensus and nonrandomized studies. Antenatal testing is also time, cost, and labor intensive, with the risk of intervention for a false-positive result. Despite these limitations, obstetrical practices routinely perform antenatal fetal surveillance.
The new guidelines: The why, when, and how often
Why. Antepartum fetal surveillance is suggested for conditions that have a risk of stillbirth greater than 0.8 per 1,000 (that is, the false-negative rate of a biophysical profile or a modified biophysical profile) and the relative risk or odds ratio is greater than 2.0 for stillbirth compared with unaffected pregnancies.
When. For most conditions, ACOG recommends initiation of testing at 32 weeks or later, with notable earlier exceptions for some of the highest-risk patients. For certain conditions, such as fetal growth restriction and hypertensive disorders of pregnancy, the recommendation is to start “at diagnosis,” with the corollary “or at a gestational age when delivery would be considered because of abnormal results.” Shared decision making with the patient about pregnancy goals therefore is required, particularly in cases of fetal anomalies, genetic conditions, or at very early gestational ages.
How often. The recommended frequency of testing is at least weekly. Testing frequency should be increased to twice-weekly outpatient or daily inpatient for the most complicated pregnancies (for example, fetal growth restriction with abnormal umbilical artery Doppler studies, preeclampsia with severe features).
Once or twice weekly is an option for many conditions, which gives the clinician the opportunity to assess clinical stability as well as the patient’s input in terms of logistics and anxiety.
Patients with multiple conditions may fall into the “individualized” category, as do patients with suboptimal control of conditions (for example, diabetes, hypertension) that may affect the fetus as the pregnancy progresses.
New diagnoses included for surveillance
Several diagnoses not previously included now qualify for antepartum fetal surveillance under the new guidelines, most notably:
- history of obstetrical complications in the immediate preceding pregnancy
—history of prior fetal growth restriction requiring preterm delivery
—history of prior preeclampsia requiring delivery
- alcohol use of 5 or more drinks per day
- in vitro fertilization
- abnormal serum markers
—pregnancy-associated plasma protein A (PAPP-A) in the fifth or lower percentile or 0.4 multiples of the median (MoM)
—second trimester inhibin A of 2 or greater MoM
- prepregnancy body mass index (BMI)
—this is divided into 2 categories for timing of initiation of testing:
- 37 weeks for BMI of 35 to 39.9 kg/m2
- 34 weeks for BMI of or greater than 40 kg/m2.
Fetal kick counts
The major change to the updated Practice Bulletin on antenatal surveillance is the inclusion of data on FKCs, a simple modality of fetal surveillance that does not require a clinical visit. For FKCs, a meta-analysis of more than 450,000 patients did not demonstrate a difference in perinatal death between the FKC intervention group (0.54%) and the control group (0.59%). Of note, there were small but statistically significant increases in the rates of induction of labor, cesarean delivery, and preterm delivery in the FKC intervention group. Therefore, this update does not recommend a formal program of FKCs for all patients.
- The antenatal fetal surveillance guidelines are just that—guidelines, not mandates. Their use will need to be adapted for specific patient populations and practice management patterns.
- Many conditions qualify for “individualized” surveillance, which offers the opportunity for detailed discussions on the patient’s care. This includes shared decision making with patients to meet their goals for the pregnancy.
- Although patient-perceived decreased fetal movement always warrants clinical evaluation, a regular program of fetal kick count monitoring is not recommended for all patients due to lack of data supporting its benefit in reducing perinatal death.
- As with any change, new guidelines potentially are a source of frustration, so a concerted effort by obstetrical clinicians to agree on adoption of the guidelines is needed. Additional clinical resources and both clinician and patient education may be required depending on current practice style, as the new strategy may increase the number of appointments and ultrasound exams required.
Continue to: Use of antenatal corticosteroids now may be considered at 22 weeks’ gestation...
Use of antenatal corticosteroids now may be considered at 22 weeks’ gestation
American College of Obstetricians and Gynecologists. Use of antenatal corticosteroids at 22 weeks of gestation: ACOG practice advisory. September 2021. https://www .acog.org/clinical/clinical-guidance/practice-advisory /articles/2021/09/use-of-antenatal-corticosteroids-at -22-weeks-of-gestation. Accessed December 11, 2021.
In September 2021, ACOG and SMFM released a Practice Advisory updating the current recommendations for the administration of antenatal corticosteroids in the periviable period (22 to 25 6/7 weeks’ gestation). Whereas the prior lower limit of gestational age for consideration of steroids was 23 weeks, the new recommendation now extends this consideration down to 22 weeks.
The cited data include a meta-analysis of more than 2,200 patients in which the survival rate of infants born between 22 and 22 6/7 weeks who were exposed to antenatal steroids was 39% compared with 19.5% in the unexposed group. Another study of more than 1,000 patients demonstrated a statistically significant increase in overall survival in patients treated with antenatal steroids plus life support compared with life support only (38.5% vs 17.7%). Survival without major morbidity in this study, although increased from 1% to 4.4%, was still low.
Recommendation carries caveats
Given this information, the Practice Advisory offers a 2C level recommendation (weak recommendation, low quality of evidence) for antenatal steroids at 22 to 22 6/7 weeks’ gestation if neonatal resuscitation is planned, acknowledging the limitations and potential bias of the available data.
The Practice Advisory emphasizes the importance of counseling and patient involvement in the decision making. This requires a multidisciplinary collaboration among the neonatology and obstetrical teams, flexibility in the plan after birth depending on the infant’s condition, and redirection of care if appropriate. Estimated fetal weight, the presence of multiple gestations, fetal biologic sex, and any anomalies are also important in helping families make an informed decision for their particular pregnancy. As described in the Obstetric Care Consensus on periviable birth,6 it is important to remember that considerations and recommendations are not the same as requirements, and redirection of care to comfort and family memory making is not the same as withholding care.
The rest of the recommendations for the administration of antenatal steroids remain the same: Antenatal steroids are not recommended at less than 22 weeks due to lack of evidence of benefit, and they continue to be recommended at 24 weeks and beyond. ●
- Antenatal corticosteroids may be considered at 22 to 22 6/7 weeks’ gestation if, after thorough patient counseling, neonatal resuscitation is desired and planned by the family.
- The overall likelihood of survival and survival without major morbidities continues to be very low in the periviable period, especially at 22 weeks. Gestational age is only one of the many factors that must be considered in the shared decision making for this very difficult decision.
- Palliative care is a valid and appropriate option for patients facing a periviable delivery after appropriate counseling or after evaluation of the infant has occurred after birth.
- Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2019. NCHS Data Brief, no 387. Hyattsville, MD: National Center for Health Statistics. October 2020. www.cdc.gov/nchs /data/databriefs/db387-H.pdf. Accessed December 20, 2021.
- To MS, Skentou C, Chan C, et al. Cervical assessment at the routine 23-week scan: standardizing techniques. Ultrasound Obstet Gynecol. 2001;17:217-219.
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins–Obstetrics. Prediction and prevention of spontaneous preterm birth: ACOG practice bulletin, number 234. Obstet Gynecol. 2021;138:e65-e90.
- Society for Maternal-Fetal Medicine (SMFM) Publications Committee. SMFM statement: use of 17-alpha hydroxyprogesterone caproate for prevention of recurrent preterm birth. Am J Obstet Gynecol. 2020;223:B16-B18.
- Blackwell SC, Gyamfi-Bannerman C, Biggio JR Jr, et al. 17-OHPC to prevent recurrent preterm birth in singleton gestations (PROLONG study): a multicenter, international, randomized double-blind trial. Am J Perinatol. 2020;37:127-136.
- American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric care consensus no. 6: Periviable birth. Obstet Gynecol. 2017;130:e187-e199.

Jaimie L. Maines, MD
Dr. Maines is Attending Physician in Maternal-Fetal Medicine and Assistant Professor, Department of Obstetrics and Gynecology, Pennsylvania State College of Medicine, Milton S. Hershey Medical Center, Hershey, Pennsylvania.

Jaimey M. Pauli, MD
Dr. Pauli is Associate Professor of Obstetrics and Gynecology and Chief, Division of Maternal-Fetal Medicine, Pennsylvania State College of Medicine, Milton S. Hershey Medical Center, Hershey, Pennsylvania. She serves on the OBG Management Board of Editors.
Dr. Pauli reports receiving grant or research support from Pfizer. Dr. Maines reports no financial relationships relevant to this article.
Jaimie L. Maines, MD
Dr. Maines is Attending Physician in Maternal-Fetal Medicine and Assistant Professor, Department of Obstetrics and Gynecology, Pennsylvania State College of Medicine, Milton S. Hershey Medical Center, Hershey, Pennsylvania.
Jaimey M. Pauli, MD
Dr. Pauli is Associate Professor of Obstetrics and Gynecology and Chief, Division of Maternal-Fetal Medicine, Pennsylvania State College of Medicine, Milton S. Hershey Medical Center, Hershey, Pennsylvania. She serves on the OBG Management Board of Editors.
Dr. Pauli reports receiving grant or research support from Pfizer. Dr. Maines reports no financial relationships relevant to this article.
Jaimie L. Maines, MD
Dr. Maines is Attending Physician in Maternal-Fetal Medicine and Assistant Professor, Department of Obstetrics and Gynecology, Pennsylvania State College of Medicine, Milton S. Hershey Medical Center, Hershey, Pennsylvania.
Jaimey M. Pauli, MD
Dr. Pauli is Associate Professor of Obstetrics and Gynecology and Chief, Division of Maternal-Fetal Medicine, Pennsylvania State College of Medicine, Milton S. Hershey Medical Center, Hershey, Pennsylvania. She serves on the OBG Management Board of Editors.
Dr. Pauli reports receiving grant or research support from Pfizer. Dr. Maines reports no financial relationships relevant to this article.
Obstetrical practice saw updates in 2021 to 3 major areas of pregnancy management: preterm birth prevention, antepartum fetal surveillance, and the use of antenatal corticosteroids.
Updated guidance on predicting and preventing spontaneous PTB
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins–Obstetrics. Prediction and prevention of spontaneous preterm birth: ACOG practice bulletin, number 234. Obstet Gynecol. 2021;138:e65-e90.
Preterm birth (PTB) continues to pose a challenge in clinical obstetrics, with the most recently reported rate of 10.2% in the United States.1 This accounts for almost 75% of perinatal mortality and more than half of neonatal morbidity, in which effects last well past the neonatal period. PTB is classified as spontaneous (following preterm labor, preterm prelabor rupture of membranes, or cervical insufficiency) or iatrogenic (indicated due to maternal and/or fetal complications).
Assessing risk for PTB
The single strongest predictor of subsequent PTB is a history of spontaneous PTB. Recurrence risk is further increased by the number of prior PTBs and the gestational age at prior PTB. Identification of and intervention for a short cervix has been shown to prolong gestation. Transvaginal ultrasonography of the cervix is the most accurate method for evaluating cervical length (CL). Specific examination criteria exist to ensure that CL measurements are reproducible and reliable.2 A short CL is generally defined as a measurement of less than 25 mm between 16 and 24 weeks’ gestation.
Screening strategies
The American College of Obstetricians and Gynecologists (ACOG), with an endorsement from the Society for Maternal-Fetal Medicine (SMFM), recommends cervical evaluation during the anatomy ultrasound exam between 18 0/7 and 22 6/7 weeks’ gestation in all pregnant patients regardless of prior PTB.3 If transabdominal imaging is concerning for a shortened cervix, transvaginal ultrasonography should be performed to assess the CL.
Serial transvaginal CL measurements are recommended between 16 0/7 and 24 0/7 weeks’ gestation for patients with a current singleton pregnancy and history of a spontaneous PTB, but not for patients with a history of iatrogenic or indicated PTB.
Interventions: Mind your p’s and c’s
Interventions to reduce the risk of spontaneous PTB depend on whether the current pregnancy is a singleton, twins, or higher-order multiples; CL measurement; and history of spontaneous PTB. Preconception optimization of underlying medical conditions also is important to reduce the risk of recurrent indicated PTB.
Continue to: Progesterone...
Progesterone
Vaginal administration. Several trials have shown that vaginal progesterone can be used to reduce the risk of spontaneous PTB in asymptomatic patients with a singleton pregnancy, incidental finding of a short cervix (<25 mm), and no history of spontaneous PTB. This is a change from the prior recommendation of CL of less than 20 mm. In the setting of a twin pregnancy, regardless of CL, data do not definitively support the use of vaginal progesterone.
Intramuscular administration.4,5 The popularity of intramuscular progesterone has waxed and waned. At present, ACOG recommends that all patients with a singleton pregnancy and history of spontaneous PTB be offered progesterone beginning at 16 0/7 weeks’ gestation following a shared decision-making process that includes the limited data of efficacy noted in existing studies.
In a twin pregnancy with no history of spontaneous PTB, the use of intramuscular progesterone has been shown to potentially increase the risk of PTB and admission to the neonatal intensive care unit. As such, intramuscular progesterone in the setting of a twin gestation without a history of spontaneous PTB is not recommended. When a prior spontaneous PTB has occurred, there may be some benefit to intramuscular progesterone in twin gestations.
Cerclage
Ultrasound indicated. In a singleton pregnancy with an incidental finding of short cervix (<25 mm) and no history of PTB, the use of cerclage is of uncertain benefit. Effectiveness may be seen if the cervix is less than 10 mm. Ultrasound-indicated cerclage should be considered in a singleton pregnancy with a CL less than 25 mm and a history of spontaneous PTB.
Possibly one of the most controversial topics is ultrasound-indicated cerclage placement in twin gestation. As with many situations in obstetrics, data regarding ultrasound-indicated cerclage in twin gestation is based on small retrospective studies fraught with bias. Results from these studies range from no benefit, to potential benefit, to even possible increased risk of PTB. Since data are limited, as we await more evidence, it is recommended that the clinician and patient use shared decision making to decide on cerclage placement in a twin gestation.
Exam indicated. In a singleton pregnancy with a dilated cervix on digital or speculum exam between 16 0/7 to 23 6/7 weeks’ gestation, a physical exam–indicated cerclage should be offered. Exam-indicated cerclage also may reduce the incidence of PTB in twin gestations with cervical dilation between 16 0/7 and 23 6/7 weeks’ gestation. Indomethacin tocolysis and perioperative antibiotics should be considered when an exam-indicated cerclage is placed.
As the limits of viability are continually pushed earlier, more in-depth conversation is needed with patients who are considering an exam-indicated cerclage. The nuances of periviability and the likelihood that an exam-indicated cerclage will commit a pregnancy to a periviable or extremely preterm birth should be discussed in detail using a shared decision making model.
Regardless of whether the cerclage is ultrasound or exam indicated, once it is placed there is no utility in additional CL ultrasound monitoring.
Pessary
Vaginal pessaries for prevention of PTB have not gained popularity in the United States as they have in other countries. Trials are being conducted to determine the utility of vaginal pessary, but current data have not proven its effectiveness in preventing PTB in the setting of singleton pregnancy, short cervix, and no history of spontaneous PTB. So for now, pessary is not recommended. The same can be said for use in the twin gestation.
- All patients should have cervical evaluation during pregnancy. Serial imaging is reserved for those with a history of spontaneous PTB.
- Progesterone supplementation should be offered to patients with a singleton pregnancy and a history of spontaneous PTB or to patients with a singleton pregnancy and no history of spontaneous PTB who have cervical shortening at less than 24 weeks.
- Cerclage may be offered between 16 and 24 weeks for a cervical length less than 25 mm in a patient with a singleton gestation who has a history of spontaneous PTB (<10 mm if no history of spontaneous PTB) or for a dilated cervix on exam regardless of history.
- Women who have a twin gestation with cervical dilation may be offered physical exam–indicated cerclage.
Which patients may benefit from antepartum fetal surveillance and when to initiate it
American College of Obstetricians and Gynecologists’ Committee on Obstetrics Practice, Society for Maternal-Fetal Medicine. Indications for outpatient antenatal surveillance: ACOG committee opinion, number 828. Obstet Gynecol. 2021;137:e177-e197.
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics. Antepartum fetal surveillance: ACOG practice bulletin, number 229. Obstet Gynecol. 2021;137:e116-e127.
The ultimate purpose of antenatal fetal surveillance is to prevent stillbirth. However, stillbirth has multiple etiologies, not all of which are preventable with testing. In June 2021, ACOG released a new Committee Opinion containing guidelines for fetal surveillance, including suggested gestational age at initiation and frequency of testing, for the most common high-risk conditions. ACOG also released an update to the Practice Bulletin on antepartum fetal surveillance; additions include randomized controlled trial level data on the utility of fetal kick counts (FKCs) and recommendations that align with the new Committee Opinion.
Data for the efficacy of antepartum fetal surveillance are lacking, mainly due to the difficulty of performing prospective studies in stillbirth. The existing evidence is subject to intervention bias, as deliveries increase in tested patients, and recommendations rely heavily on expert consensus and nonrandomized studies. Antenatal testing is also time, cost, and labor intensive, with the risk of intervention for a false-positive result. Despite these limitations, obstetrical practices routinely perform antenatal fetal surveillance.
The new guidelines: The why, when, and how often
Why. Antepartum fetal surveillance is suggested for conditions that have a risk of stillbirth greater than 0.8 per 1,000 (that is, the false-negative rate of a biophysical profile or a modified biophysical profile) and the relative risk or odds ratio is greater than 2.0 for stillbirth compared with unaffected pregnancies.
When. For most conditions, ACOG recommends initiation of testing at 32 weeks or later, with notable earlier exceptions for some of the highest-risk patients. For certain conditions, such as fetal growth restriction and hypertensive disorders of pregnancy, the recommendation is to start “at diagnosis,” with the corollary “or at a gestational age when delivery would be considered because of abnormal results.” Shared decision making with the patient about pregnancy goals therefore is required, particularly in cases of fetal anomalies, genetic conditions, or at very early gestational ages.
How often. The recommended frequency of testing is at least weekly. Testing frequency should be increased to twice-weekly outpatient or daily inpatient for the most complicated pregnancies (for example, fetal growth restriction with abnormal umbilical artery Doppler studies, preeclampsia with severe features).
Once or twice weekly is an option for many conditions, which gives the clinician the opportunity to assess clinical stability as well as the patient’s input in terms of logistics and anxiety.
Patients with multiple conditions may fall into the “individualized” category, as do patients with suboptimal control of conditions (for example, diabetes, hypertension) that may affect the fetus as the pregnancy progresses.
New diagnoses included for surveillance
Several diagnoses not previously included now qualify for antepartum fetal surveillance under the new guidelines, most notably:
- history of obstetrical complications in the immediate preceding pregnancy
—history of prior fetal growth restriction requiring preterm delivery
—history of prior preeclampsia requiring delivery
- alcohol use of 5 or more drinks per day
- in vitro fertilization
- abnormal serum markers
—pregnancy-associated plasma protein A (PAPP-A) in the fifth or lower percentile or 0.4 multiples of the median (MoM)
—second trimester inhibin A of 2 or greater MoM
- prepregnancy body mass index (BMI)
—this is divided into 2 categories for timing of initiation of testing:
- 37 weeks for BMI of 35 to 39.9 kg/m2
- 34 weeks for BMI of or greater than 40 kg/m2.
Fetal kick counts
The major change to the updated Practice Bulletin on antenatal surveillance is the inclusion of data on FKCs, a simple modality of fetal surveillance that does not require a clinical visit. For FKCs, a meta-analysis of more than 450,000 patients did not demonstrate a difference in perinatal death between the FKC intervention group (0.54%) and the control group (0.59%). Of note, there were small but statistically significant increases in the rates of induction of labor, cesarean delivery, and preterm delivery in the FKC intervention group. Therefore, this update does not recommend a formal program of FKCs for all patients.
- The antenatal fetal surveillance guidelines are just that—guidelines, not mandates. Their use will need to be adapted for specific patient populations and practice management patterns.
- Many conditions qualify for “individualized” surveillance, which offers the opportunity for detailed discussions on the patient’s care. This includes shared decision making with patients to meet their goals for the pregnancy.
- Although patient-perceived decreased fetal movement always warrants clinical evaluation, a regular program of fetal kick count monitoring is not recommended for all patients due to lack of data supporting its benefit in reducing perinatal death.
- As with any change, new guidelines potentially are a source of frustration, so a concerted effort by obstetrical clinicians to agree on adoption of the guidelines is needed. Additional clinical resources and both clinician and patient education may be required depending on current practice style, as the new strategy may increase the number of appointments and ultrasound exams required.
Continue to: Use of antenatal corticosteroids now may be considered at 22 weeks’ gestation...
Use of antenatal corticosteroids now may be considered at 22 weeks’ gestation
American College of Obstetricians and Gynecologists. Use of antenatal corticosteroids at 22 weeks of gestation: ACOG practice advisory. September 2021. https://www .acog.org/clinical/clinical-guidance/practice-advisory /articles/2021/09/use-of-antenatal-corticosteroids-at -22-weeks-of-gestation. Accessed December 11, 2021.
In September 2021, ACOG and SMFM released a Practice Advisory updating the current recommendations for the administration of antenatal corticosteroids in the periviable period (22 to 25 6/7 weeks’ gestation). Whereas the prior lower limit of gestational age for consideration of steroids was 23 weeks, the new recommendation now extends this consideration down to 22 weeks.
The cited data include a meta-analysis of more than 2,200 patients in which the survival rate of infants born between 22 and 22 6/7 weeks who were exposed to antenatal steroids was 39% compared with 19.5% in the unexposed group. Another study of more than 1,000 patients demonstrated a statistically significant increase in overall survival in patients treated with antenatal steroids plus life support compared with life support only (38.5% vs 17.7%). Survival without major morbidity in this study, although increased from 1% to 4.4%, was still low.
Recommendation carries caveats
Given this information, the Practice Advisory offers a 2C level recommendation (weak recommendation, low quality of evidence) for antenatal steroids at 22 to 22 6/7 weeks’ gestation if neonatal resuscitation is planned, acknowledging the limitations and potential bias of the available data.
The Practice Advisory emphasizes the importance of counseling and patient involvement in the decision making. This requires a multidisciplinary collaboration among the neonatology and obstetrical teams, flexibility in the plan after birth depending on the infant’s condition, and redirection of care if appropriate. Estimated fetal weight, the presence of multiple gestations, fetal biologic sex, and any anomalies are also important in helping families make an informed decision for their particular pregnancy. As described in the Obstetric Care Consensus on periviable birth,6 it is important to remember that considerations and recommendations are not the same as requirements, and redirection of care to comfort and family memory making is not the same as withholding care.
The rest of the recommendations for the administration of antenatal steroids remain the same: Antenatal steroids are not recommended at less than 22 weeks due to lack of evidence of benefit, and they continue to be recommended at 24 weeks and beyond. ●
- Antenatal corticosteroids may be considered at 22 to 22 6/7 weeks’ gestation if, after thorough patient counseling, neonatal resuscitation is desired and planned by the family.
- The overall likelihood of survival and survival without major morbidities continues to be very low in the periviable period, especially at 22 weeks. Gestational age is only one of the many factors that must be considered in the shared decision making for this very difficult decision.
- Palliative care is a valid and appropriate option for patients facing a periviable delivery after appropriate counseling or after evaluation of the infant has occurred after birth.
Obstetrical practice saw updates in 2021 to 3 major areas of pregnancy management: preterm birth prevention, antepartum fetal surveillance, and the use of antenatal corticosteroids.
Updated guidance on predicting and preventing spontaneous PTB
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins–Obstetrics. Prediction and prevention of spontaneous preterm birth: ACOG practice bulletin, number 234. Obstet Gynecol. 2021;138:e65-e90.
Preterm birth (PTB) continues to pose a challenge in clinical obstetrics, with the most recently reported rate of 10.2% in the United States.1 This accounts for almost 75% of perinatal mortality and more than half of neonatal morbidity, in which effects last well past the neonatal period. PTB is classified as spontaneous (following preterm labor, preterm prelabor rupture of membranes, or cervical insufficiency) or iatrogenic (indicated due to maternal and/or fetal complications).
Assessing risk for PTB
The single strongest predictor of subsequent PTB is a history of spontaneous PTB. Recurrence risk is further increased by the number of prior PTBs and the gestational age at prior PTB. Identification of and intervention for a short cervix has been shown to prolong gestation. Transvaginal ultrasonography of the cervix is the most accurate method for evaluating cervical length (CL). Specific examination criteria exist to ensure that CL measurements are reproducible and reliable.2 A short CL is generally defined as a measurement of less than 25 mm between 16 and 24 weeks’ gestation.
Screening strategies
The American College of Obstetricians and Gynecologists (ACOG), with an endorsement from the Society for Maternal-Fetal Medicine (SMFM), recommends cervical evaluation during the anatomy ultrasound exam between 18 0/7 and 22 6/7 weeks’ gestation in all pregnant patients regardless of prior PTB.3 If transabdominal imaging is concerning for a shortened cervix, transvaginal ultrasonography should be performed to assess the CL.
Serial transvaginal CL measurements are recommended between 16 0/7 and 24 0/7 weeks’ gestation for patients with a current singleton pregnancy and history of a spontaneous PTB, but not for patients with a history of iatrogenic or indicated PTB.
Interventions: Mind your p’s and c’s
Interventions to reduce the risk of spontaneous PTB depend on whether the current pregnancy is a singleton, twins, or higher-order multiples; CL measurement; and history of spontaneous PTB. Preconception optimization of underlying medical conditions also is important to reduce the risk of recurrent indicated PTB.
Continue to: Progesterone...
Progesterone
Vaginal administration. Several trials have shown that vaginal progesterone can be used to reduce the risk of spontaneous PTB in asymptomatic patients with a singleton pregnancy, incidental finding of a short cervix (<25 mm), and no history of spontaneous PTB. This is a change from the prior recommendation of CL of less than 20 mm. In the setting of a twin pregnancy, regardless of CL, data do not definitively support the use of vaginal progesterone.
Intramuscular administration.4,5 The popularity of intramuscular progesterone has waxed and waned. At present, ACOG recommends that all patients with a singleton pregnancy and history of spontaneous PTB be offered progesterone beginning at 16 0/7 weeks’ gestation following a shared decision-making process that includes the limited data of efficacy noted in existing studies.
In a twin pregnancy with no history of spontaneous PTB, the use of intramuscular progesterone has been shown to potentially increase the risk of PTB and admission to the neonatal intensive care unit. As such, intramuscular progesterone in the setting of a twin gestation without a history of spontaneous PTB is not recommended. When a prior spontaneous PTB has occurred, there may be some benefit to intramuscular progesterone in twin gestations.
Cerclage
Ultrasound indicated. In a singleton pregnancy with an incidental finding of short cervix (<25 mm) and no history of PTB, the use of cerclage is of uncertain benefit. Effectiveness may be seen if the cervix is less than 10 mm. Ultrasound-indicated cerclage should be considered in a singleton pregnancy with a CL less than 25 mm and a history of spontaneous PTB.
Possibly one of the most controversial topics is ultrasound-indicated cerclage placement in twin gestation. As with many situations in obstetrics, data regarding ultrasound-indicated cerclage in twin gestation is based on small retrospective studies fraught with bias. Results from these studies range from no benefit, to potential benefit, to even possible increased risk of PTB. Since data are limited, as we await more evidence, it is recommended that the clinician and patient use shared decision making to decide on cerclage placement in a twin gestation.
Exam indicated. In a singleton pregnancy with a dilated cervix on digital or speculum exam between 16 0/7 to 23 6/7 weeks’ gestation, a physical exam–indicated cerclage should be offered. Exam-indicated cerclage also may reduce the incidence of PTB in twin gestations with cervical dilation between 16 0/7 and 23 6/7 weeks’ gestation. Indomethacin tocolysis and perioperative antibiotics should be considered when an exam-indicated cerclage is placed.
As the limits of viability are continually pushed earlier, more in-depth conversation is needed with patients who are considering an exam-indicated cerclage. The nuances of periviability and the likelihood that an exam-indicated cerclage will commit a pregnancy to a periviable or extremely preterm birth should be discussed in detail using a shared decision making model.
Regardless of whether the cerclage is ultrasound or exam indicated, once it is placed there is no utility in additional CL ultrasound monitoring.
Pessary
Vaginal pessaries for prevention of PTB have not gained popularity in the United States as they have in other countries. Trials are being conducted to determine the utility of vaginal pessary, but current data have not proven its effectiveness in preventing PTB in the setting of singleton pregnancy, short cervix, and no history of spontaneous PTB. So for now, pessary is not recommended. The same can be said for use in the twin gestation.
- All patients should have cervical evaluation during pregnancy. Serial imaging is reserved for those with a history of spontaneous PTB.
- Progesterone supplementation should be offered to patients with a singleton pregnancy and a history of spontaneous PTB or to patients with a singleton pregnancy and no history of spontaneous PTB who have cervical shortening at less than 24 weeks.
- Cerclage may be offered between 16 and 24 weeks for a cervical length less than 25 mm in a patient with a singleton gestation who has a history of spontaneous PTB (<10 mm if no history of spontaneous PTB) or for a dilated cervix on exam regardless of history.
- Women who have a twin gestation with cervical dilation may be offered physical exam–indicated cerclage.
Which patients may benefit from antepartum fetal surveillance and when to initiate it
American College of Obstetricians and Gynecologists’ Committee on Obstetrics Practice, Society for Maternal-Fetal Medicine. Indications for outpatient antenatal surveillance: ACOG committee opinion, number 828. Obstet Gynecol. 2021;137:e177-e197.
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics. Antepartum fetal surveillance: ACOG practice bulletin, number 229. Obstet Gynecol. 2021;137:e116-e127.
The ultimate purpose of antenatal fetal surveillance is to prevent stillbirth. However, stillbirth has multiple etiologies, not all of which are preventable with testing. In June 2021, ACOG released a new Committee Opinion containing guidelines for fetal surveillance, including suggested gestational age at initiation and frequency of testing, for the most common high-risk conditions. ACOG also released an update to the Practice Bulletin on antepartum fetal surveillance; additions include randomized controlled trial level data on the utility of fetal kick counts (FKCs) and recommendations that align with the new Committee Opinion.
Data for the efficacy of antepartum fetal surveillance are lacking, mainly due to the difficulty of performing prospective studies in stillbirth. The existing evidence is subject to intervention bias, as deliveries increase in tested patients, and recommendations rely heavily on expert consensus and nonrandomized studies. Antenatal testing is also time, cost, and labor intensive, with the risk of intervention for a false-positive result. Despite these limitations, obstetrical practices routinely perform antenatal fetal surveillance.
The new guidelines: The why, when, and how often
Why. Antepartum fetal surveillance is suggested for conditions that have a risk of stillbirth greater than 0.8 per 1,000 (that is, the false-negative rate of a biophysical profile or a modified biophysical profile) and the relative risk or odds ratio is greater than 2.0 for stillbirth compared with unaffected pregnancies.
When. For most conditions, ACOG recommends initiation of testing at 32 weeks or later, with notable earlier exceptions for some of the highest-risk patients. For certain conditions, such as fetal growth restriction and hypertensive disorders of pregnancy, the recommendation is to start “at diagnosis,” with the corollary “or at a gestational age when delivery would be considered because of abnormal results.” Shared decision making with the patient about pregnancy goals therefore is required, particularly in cases of fetal anomalies, genetic conditions, or at very early gestational ages.
How often. The recommended frequency of testing is at least weekly. Testing frequency should be increased to twice-weekly outpatient or daily inpatient for the most complicated pregnancies (for example, fetal growth restriction with abnormal umbilical artery Doppler studies, preeclampsia with severe features).
Once or twice weekly is an option for many conditions, which gives the clinician the opportunity to assess clinical stability as well as the patient’s input in terms of logistics and anxiety.
Patients with multiple conditions may fall into the “individualized” category, as do patients with suboptimal control of conditions (for example, diabetes, hypertension) that may affect the fetus as the pregnancy progresses.
New diagnoses included for surveillance
Several diagnoses not previously included now qualify for antepartum fetal surveillance under the new guidelines, most notably:
- history of obstetrical complications in the immediate preceding pregnancy
—history of prior fetal growth restriction requiring preterm delivery
—history of prior preeclampsia requiring delivery
- alcohol use of 5 or more drinks per day
- in vitro fertilization
- abnormal serum markers
—pregnancy-associated plasma protein A (PAPP-A) in the fifth or lower percentile or 0.4 multiples of the median (MoM)
—second trimester inhibin A of 2 or greater MoM
- prepregnancy body mass index (BMI)
—this is divided into 2 categories for timing of initiation of testing:
- 37 weeks for BMI of 35 to 39.9 kg/m2
- 34 weeks for BMI of or greater than 40 kg/m2.
Fetal kick counts
The major change to the updated Practice Bulletin on antenatal surveillance is the inclusion of data on FKCs, a simple modality of fetal surveillance that does not require a clinical visit. For FKCs, a meta-analysis of more than 450,000 patients did not demonstrate a difference in perinatal death between the FKC intervention group (0.54%) and the control group (0.59%). Of note, there were small but statistically significant increases in the rates of induction of labor, cesarean delivery, and preterm delivery in the FKC intervention group. Therefore, this update does not recommend a formal program of FKCs for all patients.
- The antenatal fetal surveillance guidelines are just that—guidelines, not mandates. Their use will need to be adapted for specific patient populations and practice management patterns.
- Many conditions qualify for “individualized” surveillance, which offers the opportunity for detailed discussions on the patient’s care. This includes shared decision making with patients to meet their goals for the pregnancy.
- Although patient-perceived decreased fetal movement always warrants clinical evaluation, a regular program of fetal kick count monitoring is not recommended for all patients due to lack of data supporting its benefit in reducing perinatal death.
- As with any change, new guidelines potentially are a source of frustration, so a concerted effort by obstetrical clinicians to agree on adoption of the guidelines is needed. Additional clinical resources and both clinician and patient education may be required depending on current practice style, as the new strategy may increase the number of appointments and ultrasound exams required.
Continue to: Use of antenatal corticosteroids now may be considered at 22 weeks’ gestation...
Use of antenatal corticosteroids now may be considered at 22 weeks’ gestation
American College of Obstetricians and Gynecologists. Use of antenatal corticosteroids at 22 weeks of gestation: ACOG practice advisory. September 2021. https://www .acog.org/clinical/clinical-guidance/practice-advisory /articles/2021/09/use-of-antenatal-corticosteroids-at -22-weeks-of-gestation. Accessed December 11, 2021.
In September 2021, ACOG and SMFM released a Practice Advisory updating the current recommendations for the administration of antenatal corticosteroids in the periviable period (22 to 25 6/7 weeks’ gestation). Whereas the prior lower limit of gestational age for consideration of steroids was 23 weeks, the new recommendation now extends this consideration down to 22 weeks.
The cited data include a meta-analysis of more than 2,200 patients in which the survival rate of infants born between 22 and 22 6/7 weeks who were exposed to antenatal steroids was 39% compared with 19.5% in the unexposed group. Another study of more than 1,000 patients demonstrated a statistically significant increase in overall survival in patients treated with antenatal steroids plus life support compared with life support only (38.5% vs 17.7%). Survival without major morbidity in this study, although increased from 1% to 4.4%, was still low.
Recommendation carries caveats
Given this information, the Practice Advisory offers a 2C level recommendation (weak recommendation, low quality of evidence) for antenatal steroids at 22 to 22 6/7 weeks’ gestation if neonatal resuscitation is planned, acknowledging the limitations and potential bias of the available data.
The Practice Advisory emphasizes the importance of counseling and patient involvement in the decision making. This requires a multidisciplinary collaboration among the neonatology and obstetrical teams, flexibility in the plan after birth depending on the infant’s condition, and redirection of care if appropriate. Estimated fetal weight, the presence of multiple gestations, fetal biologic sex, and any anomalies are also important in helping families make an informed decision for their particular pregnancy. As described in the Obstetric Care Consensus on periviable birth,6 it is important to remember that considerations and recommendations are not the same as requirements, and redirection of care to comfort and family memory making is not the same as withholding care.
The rest of the recommendations for the administration of antenatal steroids remain the same: Antenatal steroids are not recommended at less than 22 weeks due to lack of evidence of benefit, and they continue to be recommended at 24 weeks and beyond. ●
- Antenatal corticosteroids may be considered at 22 to 22 6/7 weeks’ gestation if, after thorough patient counseling, neonatal resuscitation is desired and planned by the family.
- The overall likelihood of survival and survival without major morbidities continues to be very low in the periviable period, especially at 22 weeks. Gestational age is only one of the many factors that must be considered in the shared decision making for this very difficult decision.
- Palliative care is a valid and appropriate option for patients facing a periviable delivery after appropriate counseling or after evaluation of the infant has occurred after birth.
- Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2019. NCHS Data Brief, no 387. Hyattsville, MD: National Center for Health Statistics. October 2020. www.cdc.gov/nchs /data/databriefs/db387-H.pdf. Accessed December 20, 2021.
- To MS, Skentou C, Chan C, et al. Cervical assessment at the routine 23-week scan: standardizing techniques. Ultrasound Obstet Gynecol. 2001;17:217-219.
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins–Obstetrics. Prediction and prevention of spontaneous preterm birth: ACOG practice bulletin, number 234. Obstet Gynecol. 2021;138:e65-e90.
- Society for Maternal-Fetal Medicine (SMFM) Publications Committee. SMFM statement: use of 17-alpha hydroxyprogesterone caproate for prevention of recurrent preterm birth. Am J Obstet Gynecol. 2020;223:B16-B18.
- Blackwell SC, Gyamfi-Bannerman C, Biggio JR Jr, et al. 17-OHPC to prevent recurrent preterm birth in singleton gestations (PROLONG study): a multicenter, international, randomized double-blind trial. Am J Perinatol. 2020;37:127-136.
- American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric care consensus no. 6: Periviable birth. Obstet Gynecol. 2017;130:e187-e199.
- Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2019. NCHS Data Brief, no 387. Hyattsville, MD: National Center for Health Statistics. October 2020. www.cdc.gov/nchs /data/databriefs/db387-H.pdf. Accessed December 20, 2021.
- To MS, Skentou C, Chan C, et al. Cervical assessment at the routine 23-week scan: standardizing techniques. Ultrasound Obstet Gynecol. 2001;17:217-219.
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins–Obstetrics. Prediction and prevention of spontaneous preterm birth: ACOG practice bulletin, number 234. Obstet Gynecol. 2021;138:e65-e90.
- Society for Maternal-Fetal Medicine (SMFM) Publications Committee. SMFM statement: use of 17-alpha hydroxyprogesterone caproate for prevention of recurrent preterm birth. Am J Obstet Gynecol. 2020;223:B16-B18.
- Blackwell SC, Gyamfi-Bannerman C, Biggio JR Jr, et al. 17-OHPC to prevent recurrent preterm birth in singleton gestations (PROLONG study): a multicenter, international, randomized double-blind trial. Am J Perinatol. 2020;37:127-136.
- American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric care consensus no. 6: Periviable birth. Obstet Gynecol. 2017;130:e187-e199.