User login
State laws, regulatory concerns complicate biosimilars landscape
SAN FRANCISCO – A growing number of states are passing laws that set the parameters for how originator biologics may be replaced with biosimilars, and not all of them are helpful to physicians trying to maintain some knowledge and control over how prescriptions for biologics are dispensed, according to speakers at the annual meeting of the American College of Rheumatology.
These laws – now passed by a total of 19 states and Puerto Rico, including 12 in 2015 – address a requirement built into the Biologics Price Competition and Innovation Act of 2009 (part of the Affordable Care Act) that introduced the term “interchangeability” into the biosimilars approval pathway, stating that an approved biosimilar “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” The law also gives 1 year of exclusive marketing rights to the first biosimilar approved as being interchangeable with the reference product.

Although regulations on how the Food and Drug Administration will deem a biosimilar to be interchangeable have yet to be drafted, it’s important for physicians to play an active role in shaping the laws that address interchangeability, said Dr. J. Eugene Huffstutter, a rheumatologist in private practice in Hixson, Tenn., and clinical assistant professor of medicine at the University of Tennessee, Chattanooga.
Dr. Huffstutter encouraged rheumatologists and other physicians to work with their local state medical associations to contact their state legislators to explain to them the need for strict laws on how biosimilars can be dispensed. He suggested that good state legislation on biosimilars should contain provisions stating that:
• Only FDA-approved interchangeable biosimilars can be substituted.
• No substitution can be made with “dispense as written” prescription.
• The prescribing physician must be notified of substitution within 1-5 days by electronic record or fax.
• The pharmacy must maintain a record of the dispensed drug.
Organizations including the ACR, the Coalition of State Rheumatology Organizations, patient groups, electronic prescribers, state medical societies, and pharmaceutical companies (for the most part) support strong state laws on biosimilars, while the pharmacy lobby and insurance companies have opposed them, Dr. Huffstutter said.
The 19 states with biosimilars laws include California, Colorado, Delaware, Florida, Georgia, Idaho, Illinois, Indiana, Louisiana, Massachusetts, New Jersey, North Carolina, North Dakota, Oregon, Tennessee, Texas, Utah, Virginia, and Washington, along with Puerto Rico. However, the laws in Oregon and Virginia will sunset in 2016. Another seven states have 2015 or current session legislation on biosimilar substitution: Hawaii, Maryland, Michigan, Mississippi, Oklahoma, Pennsylvania, and Vermont. States with bills that failed or were not acted upon at adjournment include Arizona, Arkansas, Nevada, and Rhode Island, according to information provided to Dr. Huffstutter by the Coalition.
State laws may differ on substitution notification, he noted. In Tennessee, the law (H.B. 572) states that notification should occur in “a reasonable period of time” instead of a defined period. “What may be reasonable for one person may not be reasonable for another. I really think 5 days is the outside time, or 5 business days, because if you’re thinking about some of the biosimilars that are coming down the pike, they’ll be given on a weekly basis, so you’ll at least know before [patients] get their second shot what they’re receiving,” Dr. Huffstutter noted.
Idaho is unique in that it impaneled a board of pharmacy regulation on biosimilar substitution rather than a separate law. It states that “A pharmacist may substitute an interchangeable biosimilar product for a prescribed biological product if the biosimilar has been determined by the FDA to be interchangeable and published in the Purple Book; the prescriber does not indicate by any means that the prescribed biological product must be dispensed; and the name of the drug and the manufacturer or the NDC [National Drug Code] number is documented in the patient medical record.”
The situation may become trickier as more biosimilars are approved, noted Dr. Huffstutter, who is a member of the ACR’s Government Affairs Committee and is a liaison to the Committee on Rheumatologic Care. It could potentially be a problem when a patient is taking a biosimilar for a particular originator biologic and then other biosimilars for that originator join the marketplace. A patient could potentially be switched from one biosimilar to another when a prescription is filled if the company marketing the second biosimilar happens to win a competitive bid with an insurance company, he said.

The filgrastim biosimilar called Zarxio is the only biosimilar currently approved in the United States, but the FDA has received two applications for biosimilars for inflammatory diseases: one from Celltrion for infliximab biosimilar Remsima (August 2014) and one from Sandoz for an etanercept biosimilar (October 2015). In addition to two infliximab biosimilars that have been approved in other countries, one etanercept biosimilar that has been approved in South Korea, and one adalimumab biosimilar approved in India, there are many others in development to treat inflammatory diseases as of July 2015, including 12 for adalimumab, 9 for etanercept, 5 for infliximab, 2 for tocilizumab, and 7 for rituximab, according to Dr. Jonathan Kay, professor of medicine and director of clinical research in the division of rheumatology at the University of Massachusetts, Worcester.
Unresolved regulatory concerns
In separate presentations at the meeting, Dr. Kay described many of the nuances that define biosimilars, which go by different terminology in other countries, such as “follow-on biologic” in Japan, “subsequent-entry biological” in Canada, and “similar biological medicinal product” in the European Union. They are defined as a legitimate copy of an off-patent biopharmaceutical that has undergone rigorous analytical comparison and clinical assessment, in comparison to its reference product, and are approved by a regulatory agency according to a specific pathway for biosimilar evaluation. These differ from biomimetics, which are developed as a replica of a biopharmaceutical but have not been developed, assessed, or approved according to regulatory guidelines for biosimilars.
The FDA takes a “totality of the evidence” approach to establish biosimilarity. The biosimilar pathway for approval is different from the originator pathway by relying more on analytical and preclinical studies and less on the clinical pharmacology and clinical trial data to support biosimilarity.
However, the agency has not yet published final guidance for all steps in demonstrating biosimilarity, particularly for clinical pharmacology data. Draft guidance says the biosimilar must be analyzed and assigned to an assessment grade: not similar, similar, highly similar, and highly similar with fingerprintlike similarity.
During production, both originator and biosimilar biopharmaceuticals can have protein-folding variants, misfolding, aggregation, enzymatic cleavage, and degradation that can lead to inactivation of the protein or increased immunogenicity. Even so, most biosimilars are not identical to the originator because of post-translational modifications, but they do not matter as long as they are not clinically meaningful. Even originator products can drift over time because small changes in manufacturing processes can lead to gradual changes in the molecule, Dr. Kay said.
Extrapolation of indications from the originator to a biosimilar is another area of concern, Dr. Kay said. The FDA will consider the extrapolation of data from a clinical trial of a biosimilar conducted in one disease to support approval for additional indications for which the originator product is already licensed, but not across indications with different mechanisms of action, such as between rheumatoid arthritis and non-Hodgkin’s lymphoma for rituximab. But for which inflammatory diseases should a biosimilar be studied to provide adequate information for extrapolation of indications? Dr. Kay asked. This situation brings about questions of whether dermatologists would be comfortable using a biosimilar that has been studied for rheumatoid arthritis to treat psoriasis or gastroenterologists using a drug with data only from psoriatic arthritis to treat Crohn’s disease or ulcerative colitis.
Both the Coalition of State Rheumatology Organizations and the ACR oppose extrapolation of indications from the clinical trial data of a biosimilar in one disease to support approval of additional indications for which the originator product is already licensed, but Dr. Kay thought that “extrapolation of indications makes perfect sense” because it won’t be possible to review a biosimilar for indications not already approved for the originator and it’s natural to extrapolate to already approved indications once the protein is shown to function nearly identically to the originator.
Ideally, the nomenclature system for biosimilars should clearly identify each product to improve pharmacovigilance and to differentiate between products that have not been determined to be interchangeable, Dr. Kay said. In August, the FDA announced draft guidance proposing that all biopharmaceutical products (originator and biosimilars) would have a nonproprietary name that includes a suffix of four lowercase letters that is devoid of meaning. For example, all products that share a core name such as replicamab would be named replicamab-cznm, replicamab-hixf, and so on, Dr. Kay said. Some people have voiced concern that it would be better for names to have some meaning, such as using an abbreviated name for the developing company along with the nonproprietary name of the originator product, but others noted that method could be problematic when companies merge or are acquired by others.
Dr. Huffstutter disclosed relationships with Janssen, UCB, Lilly, Pfizer, Genentech, Bristol-Myers Squibb, and Celgene. Dr. Kay has received grant and research support from many pharmaceutical companies and has served as a consultant or on an advisory board for many pharmaceutical companies, including those developing biosimilars.
SAN FRANCISCO – A growing number of states are passing laws that set the parameters for how originator biologics may be replaced with biosimilars, and not all of them are helpful to physicians trying to maintain some knowledge and control over how prescriptions for biologics are dispensed, according to speakers at the annual meeting of the American College of Rheumatology.
These laws – now passed by a total of 19 states and Puerto Rico, including 12 in 2015 – address a requirement built into the Biologics Price Competition and Innovation Act of 2009 (part of the Affordable Care Act) that introduced the term “interchangeability” into the biosimilars approval pathway, stating that an approved biosimilar “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” The law also gives 1 year of exclusive marketing rights to the first biosimilar approved as being interchangeable with the reference product.
Although regulations on how the Food and Drug Administration will deem a biosimilar to be interchangeable have yet to be drafted, it’s important for physicians to play an active role in shaping the laws that address interchangeability, said Dr. J. Eugene Huffstutter, a rheumatologist in private practice in Hixson, Tenn., and clinical assistant professor of medicine at the University of Tennessee, Chattanooga.
Dr. Huffstutter encouraged rheumatologists and other physicians to work with their local state medical associations to contact their state legislators to explain to them the need for strict laws on how biosimilars can be dispensed. He suggested that good state legislation on biosimilars should contain provisions stating that:
• Only FDA-approved interchangeable biosimilars can be substituted.
• No substitution can be made with “dispense as written” prescription.
• The prescribing physician must be notified of substitution within 1-5 days by electronic record or fax.
• The pharmacy must maintain a record of the dispensed drug.
Organizations including the ACR, the Coalition of State Rheumatology Organizations, patient groups, electronic prescribers, state medical societies, and pharmaceutical companies (for the most part) support strong state laws on biosimilars, while the pharmacy lobby and insurance companies have opposed them, Dr. Huffstutter said.
The 19 states with biosimilars laws include California, Colorado, Delaware, Florida, Georgia, Idaho, Illinois, Indiana, Louisiana, Massachusetts, New Jersey, North Carolina, North Dakota, Oregon, Tennessee, Texas, Utah, Virginia, and Washington, along with Puerto Rico. However, the laws in Oregon and Virginia will sunset in 2016. Another seven states have 2015 or current session legislation on biosimilar substitution: Hawaii, Maryland, Michigan, Mississippi, Oklahoma, Pennsylvania, and Vermont. States with bills that failed or were not acted upon at adjournment include Arizona, Arkansas, Nevada, and Rhode Island, according to information provided to Dr. Huffstutter by the Coalition.
State laws may differ on substitution notification, he noted. In Tennessee, the law (H.B. 572) states that notification should occur in “a reasonable period of time” instead of a defined period. “What may be reasonable for one person may not be reasonable for another. I really think 5 days is the outside time, or 5 business days, because if you’re thinking about some of the biosimilars that are coming down the pike, they’ll be given on a weekly basis, so you’ll at least know before [patients] get their second shot what they’re receiving,” Dr. Huffstutter noted.
Idaho is unique in that it impaneled a board of pharmacy regulation on biosimilar substitution rather than a separate law. It states that “A pharmacist may substitute an interchangeable biosimilar product for a prescribed biological product if the biosimilar has been determined by the FDA to be interchangeable and published in the Purple Book; the prescriber does not indicate by any means that the prescribed biological product must be dispensed; and the name of the drug and the manufacturer or the NDC [National Drug Code] number is documented in the patient medical record.”
The situation may become trickier as more biosimilars are approved, noted Dr. Huffstutter, who is a member of the ACR’s Government Affairs Committee and is a liaison to the Committee on Rheumatologic Care. It could potentially be a problem when a patient is taking a biosimilar for a particular originator biologic and then other biosimilars for that originator join the marketplace. A patient could potentially be switched from one biosimilar to another when a prescription is filled if the company marketing the second biosimilar happens to win a competitive bid with an insurance company, he said.
The filgrastim biosimilar called Zarxio is the only biosimilar currently approved in the United States, but the FDA has received two applications for biosimilars for inflammatory diseases: one from Celltrion for infliximab biosimilar Remsima (August 2014) and one from Sandoz for an etanercept biosimilar (October 2015). In addition to two infliximab biosimilars that have been approved in other countries, one etanercept biosimilar that has been approved in South Korea, and one adalimumab biosimilar approved in India, there are many others in development to treat inflammatory diseases as of July 2015, including 12 for adalimumab, 9 for etanercept, 5 for infliximab, 2 for tocilizumab, and 7 for rituximab, according to Dr. Jonathan Kay, professor of medicine and director of clinical research in the division of rheumatology at the University of Massachusetts, Worcester.
Unresolved regulatory concerns
In separate presentations at the meeting, Dr. Kay described many of the nuances that define biosimilars, which go by different terminology in other countries, such as “follow-on biologic” in Japan, “subsequent-entry biological” in Canada, and “similar biological medicinal product” in the European Union. They are defined as a legitimate copy of an off-patent biopharmaceutical that has undergone rigorous analytical comparison and clinical assessment, in comparison to its reference product, and are approved by a regulatory agency according to a specific pathway for biosimilar evaluation. These differ from biomimetics, which are developed as a replica of a biopharmaceutical but have not been developed, assessed, or approved according to regulatory guidelines for biosimilars.
The FDA takes a “totality of the evidence” approach to establish biosimilarity. The biosimilar pathway for approval is different from the originator pathway by relying more on analytical and preclinical studies and less on the clinical pharmacology and clinical trial data to support biosimilarity.
However, the agency has not yet published final guidance for all steps in demonstrating biosimilarity, particularly for clinical pharmacology data. Draft guidance says the biosimilar must be analyzed and assigned to an assessment grade: not similar, similar, highly similar, and highly similar with fingerprintlike similarity.
During production, both originator and biosimilar biopharmaceuticals can have protein-folding variants, misfolding, aggregation, enzymatic cleavage, and degradation that can lead to inactivation of the protein or increased immunogenicity. Even so, most biosimilars are not identical to the originator because of post-translational modifications, but they do not matter as long as they are not clinically meaningful. Even originator products can drift over time because small changes in manufacturing processes can lead to gradual changes in the molecule, Dr. Kay said.
Extrapolation of indications from the originator to a biosimilar is another area of concern, Dr. Kay said. The FDA will consider the extrapolation of data from a clinical trial of a biosimilar conducted in one disease to support approval for additional indications for which the originator product is already licensed, but not across indications with different mechanisms of action, such as between rheumatoid arthritis and non-Hodgkin’s lymphoma for rituximab. But for which inflammatory diseases should a biosimilar be studied to provide adequate information for extrapolation of indications? Dr. Kay asked. This situation brings about questions of whether dermatologists would be comfortable using a biosimilar that has been studied for rheumatoid arthritis to treat psoriasis or gastroenterologists using a drug with data only from psoriatic arthritis to treat Crohn’s disease or ulcerative colitis.
Both the Coalition of State Rheumatology Organizations and the ACR oppose extrapolation of indications from the clinical trial data of a biosimilar in one disease to support approval of additional indications for which the originator product is already licensed, but Dr. Kay thought that “extrapolation of indications makes perfect sense” because it won’t be possible to review a biosimilar for indications not already approved for the originator and it’s natural to extrapolate to already approved indications once the protein is shown to function nearly identically to the originator.
Ideally, the nomenclature system for biosimilars should clearly identify each product to improve pharmacovigilance and to differentiate between products that have not been determined to be interchangeable, Dr. Kay said. In August, the FDA announced draft guidance proposing that all biopharmaceutical products (originator and biosimilars) would have a nonproprietary name that includes a suffix of four lowercase letters that is devoid of meaning. For example, all products that share a core name such as replicamab would be named replicamab-cznm, replicamab-hixf, and so on, Dr. Kay said. Some people have voiced concern that it would be better for names to have some meaning, such as using an abbreviated name for the developing company along with the nonproprietary name of the originator product, but others noted that method could be problematic when companies merge or are acquired by others.
Dr. Huffstutter disclosed relationships with Janssen, UCB, Lilly, Pfizer, Genentech, Bristol-Myers Squibb, and Celgene. Dr. Kay has received grant and research support from many pharmaceutical companies and has served as a consultant or on an advisory board for many pharmaceutical companies, including those developing biosimilars.
SAN FRANCISCO – A growing number of states are passing laws that set the parameters for how originator biologics may be replaced with biosimilars, and not all of them are helpful to physicians trying to maintain some knowledge and control over how prescriptions for biologics are dispensed, according to speakers at the annual meeting of the American College of Rheumatology.
These laws – now passed by a total of 19 states and Puerto Rico, including 12 in 2015 – address a requirement built into the Biologics Price Competition and Innovation Act of 2009 (part of the Affordable Care Act) that introduced the term “interchangeability” into the biosimilars approval pathway, stating that an approved biosimilar “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” The law also gives 1 year of exclusive marketing rights to the first biosimilar approved as being interchangeable with the reference product.
Although regulations on how the Food and Drug Administration will deem a biosimilar to be interchangeable have yet to be drafted, it’s important for physicians to play an active role in shaping the laws that address interchangeability, said Dr. J. Eugene Huffstutter, a rheumatologist in private practice in Hixson, Tenn., and clinical assistant professor of medicine at the University of Tennessee, Chattanooga.
Dr. Huffstutter encouraged rheumatologists and other physicians to work with their local state medical associations to contact their state legislators to explain to them the need for strict laws on how biosimilars can be dispensed. He suggested that good state legislation on biosimilars should contain provisions stating that:
• Only FDA-approved interchangeable biosimilars can be substituted.
• No substitution can be made with “dispense as written” prescription.
• The prescribing physician must be notified of substitution within 1-5 days by electronic record or fax.
• The pharmacy must maintain a record of the dispensed drug.
Organizations including the ACR, the Coalition of State Rheumatology Organizations, patient groups, electronic prescribers, state medical societies, and pharmaceutical companies (for the most part) support strong state laws on biosimilars, while the pharmacy lobby and insurance companies have opposed them, Dr. Huffstutter said.
The 19 states with biosimilars laws include California, Colorado, Delaware, Florida, Georgia, Idaho, Illinois, Indiana, Louisiana, Massachusetts, New Jersey, North Carolina, North Dakota, Oregon, Tennessee, Texas, Utah, Virginia, and Washington, along with Puerto Rico. However, the laws in Oregon and Virginia will sunset in 2016. Another seven states have 2015 or current session legislation on biosimilar substitution: Hawaii, Maryland, Michigan, Mississippi, Oklahoma, Pennsylvania, and Vermont. States with bills that failed or were not acted upon at adjournment include Arizona, Arkansas, Nevada, and Rhode Island, according to information provided to Dr. Huffstutter by the Coalition.
State laws may differ on substitution notification, he noted. In Tennessee, the law (H.B. 572) states that notification should occur in “a reasonable period of time” instead of a defined period. “What may be reasonable for one person may not be reasonable for another. I really think 5 days is the outside time, or 5 business days, because if you’re thinking about some of the biosimilars that are coming down the pike, they’ll be given on a weekly basis, so you’ll at least know before [patients] get their second shot what they’re receiving,” Dr. Huffstutter noted.
Idaho is unique in that it impaneled a board of pharmacy regulation on biosimilar substitution rather than a separate law. It states that “A pharmacist may substitute an interchangeable biosimilar product for a prescribed biological product if the biosimilar has been determined by the FDA to be interchangeable and published in the Purple Book; the prescriber does not indicate by any means that the prescribed biological product must be dispensed; and the name of the drug and the manufacturer or the NDC [National Drug Code] number is documented in the patient medical record.”
The situation may become trickier as more biosimilars are approved, noted Dr. Huffstutter, who is a member of the ACR’s Government Affairs Committee and is a liaison to the Committee on Rheumatologic Care. It could potentially be a problem when a patient is taking a biosimilar for a particular originator biologic and then other biosimilars for that originator join the marketplace. A patient could potentially be switched from one biosimilar to another when a prescription is filled if the company marketing the second biosimilar happens to win a competitive bid with an insurance company, he said.
The filgrastim biosimilar called Zarxio is the only biosimilar currently approved in the United States, but the FDA has received two applications for biosimilars for inflammatory diseases: one from Celltrion for infliximab biosimilar Remsima (August 2014) and one from Sandoz for an etanercept biosimilar (October 2015). In addition to two infliximab biosimilars that have been approved in other countries, one etanercept biosimilar that has been approved in South Korea, and one adalimumab biosimilar approved in India, there are many others in development to treat inflammatory diseases as of July 2015, including 12 for adalimumab, 9 for etanercept, 5 for infliximab, 2 for tocilizumab, and 7 for rituximab, according to Dr. Jonathan Kay, professor of medicine and director of clinical research in the division of rheumatology at the University of Massachusetts, Worcester.
Unresolved regulatory concerns
In separate presentations at the meeting, Dr. Kay described many of the nuances that define biosimilars, which go by different terminology in other countries, such as “follow-on biologic” in Japan, “subsequent-entry biological” in Canada, and “similar biological medicinal product” in the European Union. They are defined as a legitimate copy of an off-patent biopharmaceutical that has undergone rigorous analytical comparison and clinical assessment, in comparison to its reference product, and are approved by a regulatory agency according to a specific pathway for biosimilar evaluation. These differ from biomimetics, which are developed as a replica of a biopharmaceutical but have not been developed, assessed, or approved according to regulatory guidelines for biosimilars.
The FDA takes a “totality of the evidence” approach to establish biosimilarity. The biosimilar pathway for approval is different from the originator pathway by relying more on analytical and preclinical studies and less on the clinical pharmacology and clinical trial data to support biosimilarity.
However, the agency has not yet published final guidance for all steps in demonstrating biosimilarity, particularly for clinical pharmacology data. Draft guidance says the biosimilar must be analyzed and assigned to an assessment grade: not similar, similar, highly similar, and highly similar with fingerprintlike similarity.
During production, both originator and biosimilar biopharmaceuticals can have protein-folding variants, misfolding, aggregation, enzymatic cleavage, and degradation that can lead to inactivation of the protein or increased immunogenicity. Even so, most biosimilars are not identical to the originator because of post-translational modifications, but they do not matter as long as they are not clinically meaningful. Even originator products can drift over time because small changes in manufacturing processes can lead to gradual changes in the molecule, Dr. Kay said.
Extrapolation of indications from the originator to a biosimilar is another area of concern, Dr. Kay said. The FDA will consider the extrapolation of data from a clinical trial of a biosimilar conducted in one disease to support approval for additional indications for which the originator product is already licensed, but not across indications with different mechanisms of action, such as between rheumatoid arthritis and non-Hodgkin’s lymphoma for rituximab. But for which inflammatory diseases should a biosimilar be studied to provide adequate information for extrapolation of indications? Dr. Kay asked. This situation brings about questions of whether dermatologists would be comfortable using a biosimilar that has been studied for rheumatoid arthritis to treat psoriasis or gastroenterologists using a drug with data only from psoriatic arthritis to treat Crohn’s disease or ulcerative colitis.
Both the Coalition of State Rheumatology Organizations and the ACR oppose extrapolation of indications from the clinical trial data of a biosimilar in one disease to support approval of additional indications for which the originator product is already licensed, but Dr. Kay thought that “extrapolation of indications makes perfect sense” because it won’t be possible to review a biosimilar for indications not already approved for the originator and it’s natural to extrapolate to already approved indications once the protein is shown to function nearly identically to the originator.
Ideally, the nomenclature system for biosimilars should clearly identify each product to improve pharmacovigilance and to differentiate between products that have not been determined to be interchangeable, Dr. Kay said. In August, the FDA announced draft guidance proposing that all biopharmaceutical products (originator and biosimilars) would have a nonproprietary name that includes a suffix of four lowercase letters that is devoid of meaning. For example, all products that share a core name such as replicamab would be named replicamab-cznm, replicamab-hixf, and so on, Dr. Kay said. Some people have voiced concern that it would be better for names to have some meaning, such as using an abbreviated name for the developing company along with the nonproprietary name of the originator product, but others noted that method could be problematic when companies merge or are acquired by others.
Dr. Huffstutter disclosed relationships with Janssen, UCB, Lilly, Pfizer, Genentech, Bristol-Myers Squibb, and Celgene. Dr. Kay has received grant and research support from many pharmaceutical companies and has served as a consultant or on an advisory board for many pharmaceutical companies, including those developing biosimilars.
EXPERT ANALYSIS FROM THE ACR ANNUAL MEETING

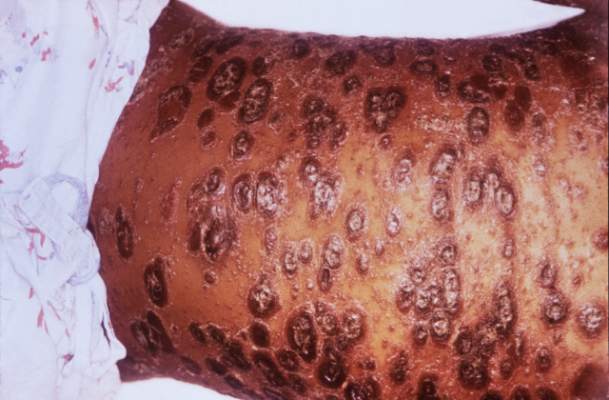
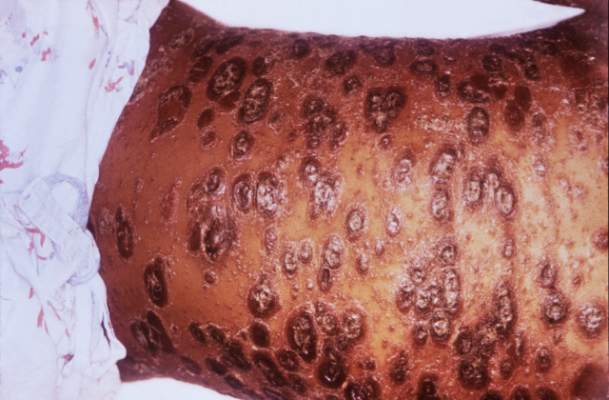
Hope for Hidradenitis Suppurativa
In September 2015, the US Food and Drug Administration approved adalimumab, the well-known injectable tumor necrosis factor (TNF)–α inhibitor indicated for psoriasis and other inflammatory conditions, for treatment of moderate to severe hidradenitis suppurativa (HS), classifying it as the first and only US Food and Drug Administration–approved therapy for adults with HS.
Pivotal studies (PIONEER/HS-I and -II, phase 3, double-blind) evaluated 633 patients (307 in HS-I and 326 in HS-II) with moderate to severe HS who were randomized to adalimumab versus placebo for 12 weeks. There was significant clinical response (at least 50% reduction in abscess and inflammatory nodule count, defined as hidradenitis suppurativa clinical response, HiSCR) in the adalimumab group (42% vs 26% in HS-I, 59% vs. 28% in HS-II, P<.001), reduction in pain (significant in the HS-II trial, 45.7% vs 20.7%, P<.001; HS-I 27.9% vs 24.8%, P>.05), and no new safety concerns when compared to other adalimumab dosages and indications. Some HS-II study patients (19.3%) were permitted to use oral antibiotics during the study.
For the indication of adult HS (moderate to severe disease), adalimumab should be administered subcutaneously at a dosing regimen of 160 mg/4 syringes on day 1 (or 80 mg/2 syringes on days 1 and 2), followed by 80 mg/2 syringes on day 15, then 40 mg/1 syringe on day 29, and every 7 days thereafter for an indefinite treatment period.
What’s the Issue?
It sits well with dermatologists when the indications for a medication with which we have great familiarity are broadened to include new disease entities; it is even better when the new entity is a condition for which every dermatologist pines for efficacious treatment options. Despite the disease burden of HS, which can include pain, scarring, disfigurement, social exclusion, and/or embarrassment, as well as the wasteful and burdensome effect that HS has on health care resources, such as injudicious use of antibiotics and unnecessary emergency department visits and inpatient hospital stays,1 there is nonetheless a wide-open and inviting playing field for effective therapies.
Although a much higher dosage of adalimumab is required in the treatment of HS compared to what is indicated for psoriasis patients, its safety concerns and side effect profile were unchanged in HS, and therefore its monitoring guidelines remain the same. Not all patients in these studies showed a notable reduction in lesion count, but given that HS classically is unresponsive to most medication regimens, will you embrace this therapy option for your patients with HS?
Reference
1. Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
In September 2015, the US Food and Drug Administration approved adalimumab, the well-known injectable tumor necrosis factor (TNF)–α inhibitor indicated for psoriasis and other inflammatory conditions, for treatment of moderate to severe hidradenitis suppurativa (HS), classifying it as the first and only US Food and Drug Administration–approved therapy for adults with HS.
Pivotal studies (PIONEER/HS-I and -II, phase 3, double-blind) evaluated 633 patients (307 in HS-I and 326 in HS-II) with moderate to severe HS who were randomized to adalimumab versus placebo for 12 weeks. There was significant clinical response (at least 50% reduction in abscess and inflammatory nodule count, defined as hidradenitis suppurativa clinical response, HiSCR) in the adalimumab group (42% vs 26% in HS-I, 59% vs. 28% in HS-II, P<.001), reduction in pain (significant in the HS-II trial, 45.7% vs 20.7%, P<.001; HS-I 27.9% vs 24.8%, P>.05), and no new safety concerns when compared to other adalimumab dosages and indications. Some HS-II study patients (19.3%) were permitted to use oral antibiotics during the study.
For the indication of adult HS (moderate to severe disease), adalimumab should be administered subcutaneously at a dosing regimen of 160 mg/4 syringes on day 1 (or 80 mg/2 syringes on days 1 and 2), followed by 80 mg/2 syringes on day 15, then 40 mg/1 syringe on day 29, and every 7 days thereafter for an indefinite treatment period.
What’s the Issue?
It sits well with dermatologists when the indications for a medication with which we have great familiarity are broadened to include new disease entities; it is even better when the new entity is a condition for which every dermatologist pines for efficacious treatment options. Despite the disease burden of HS, which can include pain, scarring, disfigurement, social exclusion, and/or embarrassment, as well as the wasteful and burdensome effect that HS has on health care resources, such as injudicious use of antibiotics and unnecessary emergency department visits and inpatient hospital stays,1 there is nonetheless a wide-open and inviting playing field for effective therapies.
Although a much higher dosage of adalimumab is required in the treatment of HS compared to what is indicated for psoriasis patients, its safety concerns and side effect profile were unchanged in HS, and therefore its monitoring guidelines remain the same. Not all patients in these studies showed a notable reduction in lesion count, but given that HS classically is unresponsive to most medication regimens, will you embrace this therapy option for your patients with HS?
In September 2015, the US Food and Drug Administration approved adalimumab, the well-known injectable tumor necrosis factor (TNF)–α inhibitor indicated for psoriasis and other inflammatory conditions, for treatment of moderate to severe hidradenitis suppurativa (HS), classifying it as the first and only US Food and Drug Administration–approved therapy for adults with HS.
Pivotal studies (PIONEER/HS-I and -II, phase 3, double-blind) evaluated 633 patients (307 in HS-I and 326 in HS-II) with moderate to severe HS who were randomized to adalimumab versus placebo for 12 weeks. There was significant clinical response (at least 50% reduction in abscess and inflammatory nodule count, defined as hidradenitis suppurativa clinical response, HiSCR) in the adalimumab group (42% vs 26% in HS-I, 59% vs. 28% in HS-II, P<.001), reduction in pain (significant in the HS-II trial, 45.7% vs 20.7%, P<.001; HS-I 27.9% vs 24.8%, P>.05), and no new safety concerns when compared to other adalimumab dosages and indications. Some HS-II study patients (19.3%) were permitted to use oral antibiotics during the study.
For the indication of adult HS (moderate to severe disease), adalimumab should be administered subcutaneously at a dosing regimen of 160 mg/4 syringes on day 1 (or 80 mg/2 syringes on days 1 and 2), followed by 80 mg/2 syringes on day 15, then 40 mg/1 syringe on day 29, and every 7 days thereafter for an indefinite treatment period.
What’s the Issue?
It sits well with dermatologists when the indications for a medication with which we have great familiarity are broadened to include new disease entities; it is even better when the new entity is a condition for which every dermatologist pines for efficacious treatment options. Despite the disease burden of HS, which can include pain, scarring, disfigurement, social exclusion, and/or embarrassment, as well as the wasteful and burdensome effect that HS has on health care resources, such as injudicious use of antibiotics and unnecessary emergency department visits and inpatient hospital stays,1 there is nonetheless a wide-open and inviting playing field for effective therapies.
Although a much higher dosage of adalimumab is required in the treatment of HS compared to what is indicated for psoriasis patients, its safety concerns and side effect profile were unchanged in HS, and therefore its monitoring guidelines remain the same. Not all patients in these studies showed a notable reduction in lesion count, but given that HS classically is unresponsive to most medication regimens, will you embrace this therapy option for your patients with HS?
Reference
1. Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
Reference
1. Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.

Common sense drives risk factor screening for systemic psoriasis therapy
LAS VEGAS – For a physician starting a patient on a systemic agent to treat psoriasis, deciding on risk factor screening and management strategies can be confusing. Who gets checked for what? Why? How often?
With a dearth of evidence to guide them, physicians should use what they know about psoriasis itself, as well as patient risk factors and the inherent risks of a given systemic therapy to guide screening, according to Dr. Kristina Callis Duffin of the department of dermatology, University of Utah, Salt Lake City.
Speaking at the Skin Disease Education Foundation’s annual Las Vegas dermatology seminar, Dr. Duffin said that in her practice, this means that all patients being considered for systemic therapy get baseline labs as recommended by the drug manufacturer, and risk-focused monitoring for cardiovascular disease, infection, and cancer. Basically, she noted, the strategy is, “How can we be effective at monitoring and surveying for those ‘bad guys’– those complications of therapy?”
“Some of this is really about using common sense and being a good physician,” she added.
To obtain a pertinent history, it’s worthwhile to formulate a directed intake questionnaire, Dr. Duffin said. Screening items should include history of recurrent infections; hepatitis, HIV, and TB status and risk; travel history; cardiovascular history, including lipid status; history of any liver or kidney problems; history and risk of diabetes mellitus; depression; personal or family history of multiple sclerosis; and history of cancer, including skin cancer, and blood disorders.
The social history should include pregnancy status and birth control method for women (as appropriate); and alcohol use for all patients. Patients should be current with age-appropriate recommended immunizations and cancer screenings.
Obtaining baseline lab tests for all patients before they start treatment with a systemic agent affords the opportunity to detect common metabolic abnormalities, such as diabetes, liver or kidney disease, and dyslipidemia, as well as rare but serious blood abnormalities, Dr. Duffin said. This approach respects the fact that “there is no question that cardiovascular risk factors are overrepresented in the moderate to severe psoriasis population.”
Published TB screening guidelines have universally recommended screening before initiating a biologic agent with a tuberculin skin test, unless the patient has had the BCG vaccine or is immunosuppressed. From there, interpretation of the tine test and further screening requires knowing the patient: “It’s all about pretest probability,” said Dr. Duffin, adding that physicians should not be afraid to use infectious disease consultants to help them sort out tricky cases.
In terms of ongoing management, communication and coordination are key. Continue to ask patients about symptoms of psoriatic arthritis, and make sure that you work with the patient’s primary care provider so that cardiovascular risk management, malignancy screening, and immunizations don’t fall through the cracks, she advised.
“Have a high index of suspicion for infection,” including deep fungal infections, she added. Though the literature does not report frequent cases of serious fungal infections, “there’s no question cases are unreported or numbers can be unclear in the registries.” Patients should know early signs of infection and know to seek care right away.
Patients should know to defer live vaccines, and the physician managing the care of psoriasis should be included in the planning and management process for any type of surgery, to oversee biologic administration. While probably slight, the risk of infection should be weighed against the risk of loss of efficacy in psoriasis or psoriatic arthritis treatment, Dr. Duffin said.
The baseline risk of solid tumors is elevated in patients with psoriasis, but “is not definitively elevated in patients on biologics,” she commented. The risk of nonmelanoma skin cancer, however, is definitely elevated for patients on biologic therapies, so an annual skin survey is a must for these patients, she said.
“Should we be discussing immunizations with our patients? Absolutely,”she added. All patients need the influenza vaccine and the pneumococcal vaccine, and for live virus immunizations, the dermatologist should coordinate administration with the primary care physician.
Managing these risks effectively can make a big difference in quality of life for patients who are able to start and stay on systemic therapy, said Dr. Duffin. The many risks of not treating psoriasis appropriately include not only the significant psychosocial impact, but also progression of psoriatic arthritis, progression of skin disease, and increased risk of heart disease and other cardiovascular comorbidities. “Our job is to treat to patient satisfaction and the best possible quality of life. A lot of this is about anticipation and communication,” she said.
Dr. Duffin reported receiving research support from and consulting for Amgen, Eli Lilly, Janssen, Stiefel, AbbVie, Bristol-Myers Squibb, Celgene, Novartis, and XenoPort; consulting and being on the scientific advisory board for Pfizer; and being on the scientific advisory board for Novartis, Eli Lilly, Janssen, Celgene, and XenoPort.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
LAS VEGAS – For a physician starting a patient on a systemic agent to treat psoriasis, deciding on risk factor screening and management strategies can be confusing. Who gets checked for what? Why? How often?
With a dearth of evidence to guide them, physicians should use what they know about psoriasis itself, as well as patient risk factors and the inherent risks of a given systemic therapy to guide screening, according to Dr. Kristina Callis Duffin of the department of dermatology, University of Utah, Salt Lake City.
Speaking at the Skin Disease Education Foundation’s annual Las Vegas dermatology seminar, Dr. Duffin said that in her practice, this means that all patients being considered for systemic therapy get baseline labs as recommended by the drug manufacturer, and risk-focused monitoring for cardiovascular disease, infection, and cancer. Basically, she noted, the strategy is, “How can we be effective at monitoring and surveying for those ‘bad guys’– those complications of therapy?”
“Some of this is really about using common sense and being a good physician,” she added.
To obtain a pertinent history, it’s worthwhile to formulate a directed intake questionnaire, Dr. Duffin said. Screening items should include history of recurrent infections; hepatitis, HIV, and TB status and risk; travel history; cardiovascular history, including lipid status; history of any liver or kidney problems; history and risk of diabetes mellitus; depression; personal or family history of multiple sclerosis; and history of cancer, including skin cancer, and blood disorders.
The social history should include pregnancy status and birth control method for women (as appropriate); and alcohol use for all patients. Patients should be current with age-appropriate recommended immunizations and cancer screenings.
Obtaining baseline lab tests for all patients before they start treatment with a systemic agent affords the opportunity to detect common metabolic abnormalities, such as diabetes, liver or kidney disease, and dyslipidemia, as well as rare but serious blood abnormalities, Dr. Duffin said. This approach respects the fact that “there is no question that cardiovascular risk factors are overrepresented in the moderate to severe psoriasis population.”
Published TB screening guidelines have universally recommended screening before initiating a biologic agent with a tuberculin skin test, unless the patient has had the BCG vaccine or is immunosuppressed. From there, interpretation of the tine test and further screening requires knowing the patient: “It’s all about pretest probability,” said Dr. Duffin, adding that physicians should not be afraid to use infectious disease consultants to help them sort out tricky cases.
In terms of ongoing management, communication and coordination are key. Continue to ask patients about symptoms of psoriatic arthritis, and make sure that you work with the patient’s primary care provider so that cardiovascular risk management, malignancy screening, and immunizations don’t fall through the cracks, she advised.
“Have a high index of suspicion for infection,” including deep fungal infections, she added. Though the literature does not report frequent cases of serious fungal infections, “there’s no question cases are unreported or numbers can be unclear in the registries.” Patients should know early signs of infection and know to seek care right away.
Patients should know to defer live vaccines, and the physician managing the care of psoriasis should be included in the planning and management process for any type of surgery, to oversee biologic administration. While probably slight, the risk of infection should be weighed against the risk of loss of efficacy in psoriasis or psoriatic arthritis treatment, Dr. Duffin said.
The baseline risk of solid tumors is elevated in patients with psoriasis, but “is not definitively elevated in patients on biologics,” she commented. The risk of nonmelanoma skin cancer, however, is definitely elevated for patients on biologic therapies, so an annual skin survey is a must for these patients, she said.
“Should we be discussing immunizations with our patients? Absolutely,”she added. All patients need the influenza vaccine and the pneumococcal vaccine, and for live virus immunizations, the dermatologist should coordinate administration with the primary care physician.
Managing these risks effectively can make a big difference in quality of life for patients who are able to start and stay on systemic therapy, said Dr. Duffin. The many risks of not treating psoriasis appropriately include not only the significant psychosocial impact, but also progression of psoriatic arthritis, progression of skin disease, and increased risk of heart disease and other cardiovascular comorbidities. “Our job is to treat to patient satisfaction and the best possible quality of life. A lot of this is about anticipation and communication,” she said.
Dr. Duffin reported receiving research support from and consulting for Amgen, Eli Lilly, Janssen, Stiefel, AbbVie, Bristol-Myers Squibb, Celgene, Novartis, and XenoPort; consulting and being on the scientific advisory board for Pfizer; and being on the scientific advisory board for Novartis, Eli Lilly, Janssen, Celgene, and XenoPort.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
LAS VEGAS – For a physician starting a patient on a systemic agent to treat psoriasis, deciding on risk factor screening and management strategies can be confusing. Who gets checked for what? Why? How often?
With a dearth of evidence to guide them, physicians should use what they know about psoriasis itself, as well as patient risk factors and the inherent risks of a given systemic therapy to guide screening, according to Dr. Kristina Callis Duffin of the department of dermatology, University of Utah, Salt Lake City.
Speaking at the Skin Disease Education Foundation’s annual Las Vegas dermatology seminar, Dr. Duffin said that in her practice, this means that all patients being considered for systemic therapy get baseline labs as recommended by the drug manufacturer, and risk-focused monitoring for cardiovascular disease, infection, and cancer. Basically, she noted, the strategy is, “How can we be effective at monitoring and surveying for those ‘bad guys’– those complications of therapy?”
“Some of this is really about using common sense and being a good physician,” she added.
To obtain a pertinent history, it’s worthwhile to formulate a directed intake questionnaire, Dr. Duffin said. Screening items should include history of recurrent infections; hepatitis, HIV, and TB status and risk; travel history; cardiovascular history, including lipid status; history of any liver or kidney problems; history and risk of diabetes mellitus; depression; personal or family history of multiple sclerosis; and history of cancer, including skin cancer, and blood disorders.
The social history should include pregnancy status and birth control method for women (as appropriate); and alcohol use for all patients. Patients should be current with age-appropriate recommended immunizations and cancer screenings.
Obtaining baseline lab tests for all patients before they start treatment with a systemic agent affords the opportunity to detect common metabolic abnormalities, such as diabetes, liver or kidney disease, and dyslipidemia, as well as rare but serious blood abnormalities, Dr. Duffin said. This approach respects the fact that “there is no question that cardiovascular risk factors are overrepresented in the moderate to severe psoriasis population.”
Published TB screening guidelines have universally recommended screening before initiating a biologic agent with a tuberculin skin test, unless the patient has had the BCG vaccine or is immunosuppressed. From there, interpretation of the tine test and further screening requires knowing the patient: “It’s all about pretest probability,” said Dr. Duffin, adding that physicians should not be afraid to use infectious disease consultants to help them sort out tricky cases.
In terms of ongoing management, communication and coordination are key. Continue to ask patients about symptoms of psoriatic arthritis, and make sure that you work with the patient’s primary care provider so that cardiovascular risk management, malignancy screening, and immunizations don’t fall through the cracks, she advised.
“Have a high index of suspicion for infection,” including deep fungal infections, she added. Though the literature does not report frequent cases of serious fungal infections, “there’s no question cases are unreported or numbers can be unclear in the registries.” Patients should know early signs of infection and know to seek care right away.
Patients should know to defer live vaccines, and the physician managing the care of psoriasis should be included in the planning and management process for any type of surgery, to oversee biologic administration. While probably slight, the risk of infection should be weighed against the risk of loss of efficacy in psoriasis or psoriatic arthritis treatment, Dr. Duffin said.
The baseline risk of solid tumors is elevated in patients with psoriasis, but “is not definitively elevated in patients on biologics,” she commented. The risk of nonmelanoma skin cancer, however, is definitely elevated for patients on biologic therapies, so an annual skin survey is a must for these patients, she said.
“Should we be discussing immunizations with our patients? Absolutely,”she added. All patients need the influenza vaccine and the pneumococcal vaccine, and for live virus immunizations, the dermatologist should coordinate administration with the primary care physician.
Managing these risks effectively can make a big difference in quality of life for patients who are able to start and stay on systemic therapy, said Dr. Duffin. The many risks of not treating psoriasis appropriately include not only the significant psychosocial impact, but also progression of psoriatic arthritis, progression of skin disease, and increased risk of heart disease and other cardiovascular comorbidities. “Our job is to treat to patient satisfaction and the best possible quality of life. A lot of this is about anticipation and communication,” she said.
Dr. Duffin reported receiving research support from and consulting for Amgen, Eli Lilly, Janssen, Stiefel, AbbVie, Bristol-Myers Squibb, Celgene, Novartis, and XenoPort; consulting and being on the scientific advisory board for Pfizer; and being on the scientific advisory board for Novartis, Eli Lilly, Janssen, Celgene, and XenoPort.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
VIDEO: Psoriasis, psoriatic arthritis improve with bariatric surgery
SAN FRANCISCO – It might be time to add psoriasis to the list of comorbidities bariatric surgery is likely to help.
New York University investigators have found a marked improvement in psoriasis and psoriatic arthritis following bariatric surgery, especially with severe disease. The more weight people lose, the better they do.
In an interview at the annual meeting of the American College of Rheumatology, investigator Dr. Soumya Reddy, codirector of NYU’s Psoriatic Arthritis Center in Manhattan, explained how the findings can be used in the clinic and their potential impact on bariatric surgery authorization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – It might be time to add psoriasis to the list of comorbidities bariatric surgery is likely to help.
New York University investigators have found a marked improvement in psoriasis and psoriatic arthritis following bariatric surgery, especially with severe disease. The more weight people lose, the better they do.
In an interview at the annual meeting of the American College of Rheumatology, investigator Dr. Soumya Reddy, codirector of NYU’s Psoriatic Arthritis Center in Manhattan, explained how the findings can be used in the clinic and their potential impact on bariatric surgery authorization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – It might be time to add psoriasis to the list of comorbidities bariatric surgery is likely to help.
New York University investigators have found a marked improvement in psoriasis and psoriatic arthritis following bariatric surgery, especially with severe disease. The more weight people lose, the better they do.
In an interview at the annual meeting of the American College of Rheumatology, investigator Dr. Soumya Reddy, codirector of NYU’s Psoriatic Arthritis Center in Manhattan, explained how the findings can be used in the clinic and their potential impact on bariatric surgery authorization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE ACR ANNUAL MEETING

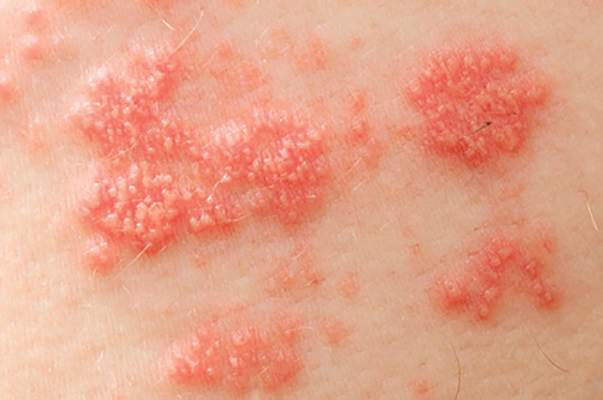
Stroke risk rose in autoimmune disease patients after herpes zoster
Stroke risk was 50% higher in the month after patients with autoimmune diseases developed herpes zoster, compared with the next 2-6 years, according to Dr. Leonard H. Calabrese.
“These data provide urgency for developing strategies to reduce the risk of varicella zoster virus in vulnerable immunosuppressed patients,” said Dr. Calabrese of the department of rheumatic and immunologic diseases at the Cleveland Clinic.

Immunosuppressive therapies increase the frequency and complexity of herpes zoster, which is a known risk factor for stroke. To examine the temporal relationship between herpes zoster and stroke among immunosuppressed patients, Dr. Calabrese and his associates studied Medicare data for almost 51,000 patients with new-onset herpes zoster who also had physician-diagnosed ankylosing spondylitis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or rheumatoid arthritis. The researchers excluded patients with a history of stroke.
In the multivariable analysis, stroke was 1.5 times more likely during the 6 months immediately after herpes zoster than in the 2-6 years after herpes zoster (95% confidence interval, 1.06-2.12). During this 6-month window, there were 9.8 strokes per 1,000 person-years, compared with 8.7 per 1,000 person-years in the 2-6 years after herpes zoster. Stroke risk also remained somewhat elevated during the entire year after herpes zoster (incidence rate ratio, 1.3; 95% CI, 1.05-1.61).
In general, stroke was more likely to occur among patients who were older, were receiving high-dose glucocorticoids, or had diabetes, hypertension, atrial fibrillation, or a history of transient ischemic attack, he said at the annual meeting of the American College of Rheumatology in San Francisco.
Dr. Calabrese disclosed relationships with Bristol-Myers Squibb, Crescendo, AbbVie, Genentech, Biogen, Pfizer, Sanofi-Aventis Pharmaceutical, and Johnson & Johnson. Two coauthors also disclosed relationships with several pharmaceutical companies. The other four coinvestigators had no disclosures.
Stroke risk was 50% higher in the month after patients with autoimmune diseases developed herpes zoster, compared with the next 2-6 years, according to Dr. Leonard H. Calabrese.
“These data provide urgency for developing strategies to reduce the risk of varicella zoster virus in vulnerable immunosuppressed patients,” said Dr. Calabrese of the department of rheumatic and immunologic diseases at the Cleveland Clinic.
Immunosuppressive therapies increase the frequency and complexity of herpes zoster, which is a known risk factor for stroke. To examine the temporal relationship between herpes zoster and stroke among immunosuppressed patients, Dr. Calabrese and his associates studied Medicare data for almost 51,000 patients with new-onset herpes zoster who also had physician-diagnosed ankylosing spondylitis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or rheumatoid arthritis. The researchers excluded patients with a history of stroke.
In the multivariable analysis, stroke was 1.5 times more likely during the 6 months immediately after herpes zoster than in the 2-6 years after herpes zoster (95% confidence interval, 1.06-2.12). During this 6-month window, there were 9.8 strokes per 1,000 person-years, compared with 8.7 per 1,000 person-years in the 2-6 years after herpes zoster. Stroke risk also remained somewhat elevated during the entire year after herpes zoster (incidence rate ratio, 1.3; 95% CI, 1.05-1.61).
In general, stroke was more likely to occur among patients who were older, were receiving high-dose glucocorticoids, or had diabetes, hypertension, atrial fibrillation, or a history of transient ischemic attack, he said at the annual meeting of the American College of Rheumatology in San Francisco.
Dr. Calabrese disclosed relationships with Bristol-Myers Squibb, Crescendo, AbbVie, Genentech, Biogen, Pfizer, Sanofi-Aventis Pharmaceutical, and Johnson & Johnson. Two coauthors also disclosed relationships with several pharmaceutical companies. The other four coinvestigators had no disclosures.
Stroke risk was 50% higher in the month after patients with autoimmune diseases developed herpes zoster, compared with the next 2-6 years, according to Dr. Leonard H. Calabrese.
“These data provide urgency for developing strategies to reduce the risk of varicella zoster virus in vulnerable immunosuppressed patients,” said Dr. Calabrese of the department of rheumatic and immunologic diseases at the Cleveland Clinic.
Immunosuppressive therapies increase the frequency and complexity of herpes zoster, which is a known risk factor for stroke. To examine the temporal relationship between herpes zoster and stroke among immunosuppressed patients, Dr. Calabrese and his associates studied Medicare data for almost 51,000 patients with new-onset herpes zoster who also had physician-diagnosed ankylosing spondylitis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or rheumatoid arthritis. The researchers excluded patients with a history of stroke.
In the multivariable analysis, stroke was 1.5 times more likely during the 6 months immediately after herpes zoster than in the 2-6 years after herpes zoster (95% confidence interval, 1.06-2.12). During this 6-month window, there were 9.8 strokes per 1,000 person-years, compared with 8.7 per 1,000 person-years in the 2-6 years after herpes zoster. Stroke risk also remained somewhat elevated during the entire year after herpes zoster (incidence rate ratio, 1.3; 95% CI, 1.05-1.61).
In general, stroke was more likely to occur among patients who were older, were receiving high-dose glucocorticoids, or had diabetes, hypertension, atrial fibrillation, or a history of transient ischemic attack, he said at the annual meeting of the American College of Rheumatology in San Francisco.
Dr. Calabrese disclosed relationships with Bristol-Myers Squibb, Crescendo, AbbVie, Genentech, Biogen, Pfizer, Sanofi-Aventis Pharmaceutical, and Johnson & Johnson. Two coauthors also disclosed relationships with several pharmaceutical companies. The other four coinvestigators had no disclosures.
FROM THE ACR ANNUAL MEETING
Key clinical point: Stroke risk increased by 50% in the month after patients with autoimmune diseases had an episode of herpes zoster.
Major finding: The risk of stroke was 50% higher in the 6 months immediately after incident herpes zoster than 2-6 years after herpes zoster (95% CI, 1.06- 2.12).
Data source: An analysis of Medicare data for 50,929 patients with autoimmune disease and incident herpes zoster between 2006 and 2012.
Disclosures: Dr. Calabrese disclosed relationships with Bristol-Myers Squibb, Crescendo, AbbVie, Genentech, Biogen, Pfizer, Sanofi-Aventis Pharmaceutical, and Johnson & Johnson. Two coauthors also disclosed relationships with several pharmaceutical companies. The other four coinvestigators had no disclosures.

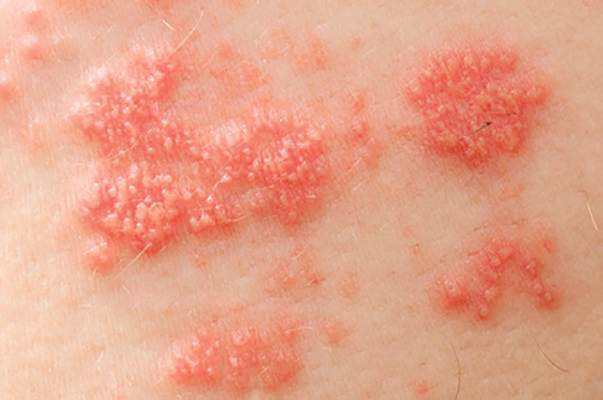
Shingles vaccine protection lasted 5-6 years in autoimmune disease patients
Protection against shingles appeared to wane between the fifth and sixth years after patients with autoimmune diseases received the live herpes zoster vaccine, according to a large retrospective cohort study presented at the annual meeting of the American College of Rheumatology.
In contrast, shingles risk remained fairly constant among unvaccinated patients, reported Dr. Huifeng Yun, assistant professor of epidemiology at the University of Alabama, Birmingham. Based on the findings, clinicians could consider revaccinating patients with autoimmune diseases about 5 years after their first live herpes zoster vaccine, she said.*
The long-term Shingles Prevention Study recently showed that the live herpes zoster vaccine is effective for about a decade among healthy older individuals, but the duration of protection for patients with autoimmune diseases was unclear, the investigators said. Therefore, they retrospectively studied Medicare data for more than 130,000 such patients between 2006 and 2012. About one-third of patients were vaccinated against herpes zoster, and 47% had rheumatoid arthritis, 32% had psoriasis, 21% had inflammatory bowel disease, 5% had psoriatic arthritis, and 1% had ankylosing spondylitis.
Rates of herpes zoster among vaccinated patients rose from 0.75 per 100 person-years during the first year after vaccination to 1.36 during the sixth year. In the adjusted analysis, vaccinated patients had about half the risk of herpes zoster, compared with unvaccinated patients, during year 1 (relative risk, 0.52; 95% confidence interval, 0.45-0.61) and remained substantially less likely to develop shingles until year 6, when the gap in risk between vaccinated and unvaccinated patients essentially closed (RR, 0.92; 95% CI, 0.45-1.86). There was no overall change in risk among unvaccinated patients during the study period, the investigators noted.
Dr. Yun disclosed a financial relationship with Amgen. The senior author and two coauthors also disclosed relationships with several pharmaceutical companies. Two investigators had no disclosures.
* Correction, 11/13/2015: The article previously misstated Dr. Yun's gender.
Protection against shingles appeared to wane between the fifth and sixth years after patients with autoimmune diseases received the live herpes zoster vaccine, according to a large retrospective cohort study presented at the annual meeting of the American College of Rheumatology.
In contrast, shingles risk remained fairly constant among unvaccinated patients, reported Dr. Huifeng Yun, assistant professor of epidemiology at the University of Alabama, Birmingham. Based on the findings, clinicians could consider revaccinating patients with autoimmune diseases about 5 years after their first live herpes zoster vaccine, she said.*
The long-term Shingles Prevention Study recently showed that the live herpes zoster vaccine is effective for about a decade among healthy older individuals, but the duration of protection for patients with autoimmune diseases was unclear, the investigators said. Therefore, they retrospectively studied Medicare data for more than 130,000 such patients between 2006 and 2012. About one-third of patients were vaccinated against herpes zoster, and 47% had rheumatoid arthritis, 32% had psoriasis, 21% had inflammatory bowel disease, 5% had psoriatic arthritis, and 1% had ankylosing spondylitis.
Rates of herpes zoster among vaccinated patients rose from 0.75 per 100 person-years during the first year after vaccination to 1.36 during the sixth year. In the adjusted analysis, vaccinated patients had about half the risk of herpes zoster, compared with unvaccinated patients, during year 1 (relative risk, 0.52; 95% confidence interval, 0.45-0.61) and remained substantially less likely to develop shingles until year 6, when the gap in risk between vaccinated and unvaccinated patients essentially closed (RR, 0.92; 95% CI, 0.45-1.86). There was no overall change in risk among unvaccinated patients during the study period, the investigators noted.
Dr. Yun disclosed a financial relationship with Amgen. The senior author and two coauthors also disclosed relationships with several pharmaceutical companies. Two investigators had no disclosures.
* Correction, 11/13/2015: The article previously misstated Dr. Yun's gender.
Protection against shingles appeared to wane between the fifth and sixth years after patients with autoimmune diseases received the live herpes zoster vaccine, according to a large retrospective cohort study presented at the annual meeting of the American College of Rheumatology.
In contrast, shingles risk remained fairly constant among unvaccinated patients, reported Dr. Huifeng Yun, assistant professor of epidemiology at the University of Alabama, Birmingham. Based on the findings, clinicians could consider revaccinating patients with autoimmune diseases about 5 years after their first live herpes zoster vaccine, she said.*
The long-term Shingles Prevention Study recently showed that the live herpes zoster vaccine is effective for about a decade among healthy older individuals, but the duration of protection for patients with autoimmune diseases was unclear, the investigators said. Therefore, they retrospectively studied Medicare data for more than 130,000 such patients between 2006 and 2012. About one-third of patients were vaccinated against herpes zoster, and 47% had rheumatoid arthritis, 32% had psoriasis, 21% had inflammatory bowel disease, 5% had psoriatic arthritis, and 1% had ankylosing spondylitis.
Rates of herpes zoster among vaccinated patients rose from 0.75 per 100 person-years during the first year after vaccination to 1.36 during the sixth year. In the adjusted analysis, vaccinated patients had about half the risk of herpes zoster, compared with unvaccinated patients, during year 1 (relative risk, 0.52; 95% confidence interval, 0.45-0.61) and remained substantially less likely to develop shingles until year 6, when the gap in risk between vaccinated and unvaccinated patients essentially closed (RR, 0.92; 95% CI, 0.45-1.86). There was no overall change in risk among unvaccinated patients during the study period, the investigators noted.
Dr. Yun disclosed a financial relationship with Amgen. The senior author and two coauthors also disclosed relationships with several pharmaceutical companies. Two investigators had no disclosures.
* Correction, 11/13/2015: The article previously misstated Dr. Yun's gender.
FROM THE ACR ANNUAL MEETING
Key clinical point: Protection against shingles appeared to wane between the fifth and sixth years after patients with autoimmune diseases received the live herpes zoster vaccine.
Major finding: The rate of herpes zoster rose from 0.75 per 100 person-years in the first year after vaccination to 1.36 in the sixth year, when it approached the rate among unvaccinated patients.
Data source: Retrospective analysis of 130,107 Medicare patients with autoimmune diseases between 2006 and 2012.
Disclosures: Dr. Yun disclosed a financial relationship with Amgen. The senior author and two coauthors also disclosed relationships with several pharmaceutical companies. Two investigators had no disclosures.
Can bariatric surgery ease psoriasis too?
LOS ANGELES – A provocative study opens the door for psoriasis and psoriatic arthritis to be added to the growing list of benefits following bariatric surgery.
“Although the natural history of psoriasis and psoriatic arthritis is typically chronic, the majority of patients experience improvement after bariatric surgery, Dr. Monica Sethi of New York University said at Obesity Week 2015.

At an average of 6 years after bariatric surgery, 55% of patients with psoriasis and 62% of those with psoriatic arthritis (PsA) reported subjective lessening of their disease.
Preoperative disease severity ratings on a 10-point scale significantly decreased at the most recent follow-up for psoriasis (5.6 vs. 3.3; P less than .01) and PsA (6.4 vs. 3.9; P = .02).
“Our results indicate an association between excess weight loss and symptomatic improvement in severe cases of psoriasis, and a possible improvement in psoriatic arthritis,” she said.
Although the effects of surgical weight loss on psoriasis and PsA are unknown, obesity is known to be more prevalent among patients with psoriasis and PsA. This correlation appears to be related to fat tissue–driven systemic inflammation, Dr. Sethi said.
The investigators surveyed 128 patients with a preoperative diagnosis of psoriasis identified from a single-center database of 9,073 bariatric surgeries performed between 2002 and 2013. A total of 86 patients completed the study, with 21 patients also having a preoperative diagnosis of PsA. Their mean preoperative weight was 288 pounds and preoperative body mass index 45.8 kg/m2. The average duration of psoriasis and PsA at the time of surgery was significant at 18.7 years, Dr. Sethi noted.
The mean time from surgery was 6.1 years, with a mean excess weight loss of 46.2% and total weight loss of 23.8%. Laparoscopic adjustable gastric banding was the most common surgery (91%), followed by Roux-en-Y bypass (7%).
In secondary analyses, a higher percent of excess weight loss at recent follow-up was significantly associated with an easing of psoriasis severity (59.5% vs. 43.5%; P = .046), while higher percent of excess weight loss at 1 year was associated with a trend in PsA improvement (55.4% vs. 43.8%; P = .47), Dr. Sethi said.
Easing of disease severity after surgery was associated with a higher rating of disease at the time of surgery (8.9 vs. 7.4; P less than .01) and older age at diagnosis (36.9 years vs. 25.9 years; P = .02), suggesting that these factors may be used to identify patients with a greater likelihood of improvement.
“Larger prospective studies are needed to further define the true effect of surgical weight loss on psoriasis and psoriatic arthritis,” she said at the meeting, presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery (ASMBS).
Session comoderator Dr. Peter T. Hallowell of the University of Virginia, Charlottesville, agreed that further studies are needed, but said the interesting thing is how bariatric surgery improves multiple problems.
“The results are pretty impressive with the moderate weight loss they got with laparoscopic gastric banding,” he said in an interview. “With gastric bypass or a sleeve, where we would expect greater weight loss and greater decrease in the inflammatory mediators, we may see an even greater improved outcome.”
A larger prospective study is being planned between New York University’s psoriasis and bariatric centers in about 60 patients with psoriasis and PsA undergoing bariatric surgery, mostly sleeve gastrectomy and gastric bypass, Dr. Sethi said in an interview.
LOS ANGELES – A provocative study opens the door for psoriasis and psoriatic arthritis to be added to the growing list of benefits following bariatric surgery.
“Although the natural history of psoriasis and psoriatic arthritis is typically chronic, the majority of patients experience improvement after bariatric surgery, Dr. Monica Sethi of New York University said at Obesity Week 2015.
At an average of 6 years after bariatric surgery, 55% of patients with psoriasis and 62% of those with psoriatic arthritis (PsA) reported subjective lessening of their disease.
Preoperative disease severity ratings on a 10-point scale significantly decreased at the most recent follow-up for psoriasis (5.6 vs. 3.3; P less than .01) and PsA (6.4 vs. 3.9; P = .02).
“Our results indicate an association between excess weight loss and symptomatic improvement in severe cases of psoriasis, and a possible improvement in psoriatic arthritis,” she said.
Although the effects of surgical weight loss on psoriasis and PsA are unknown, obesity is known to be more prevalent among patients with psoriasis and PsA. This correlation appears to be related to fat tissue–driven systemic inflammation, Dr. Sethi said.
The investigators surveyed 128 patients with a preoperative diagnosis of psoriasis identified from a single-center database of 9,073 bariatric surgeries performed between 2002 and 2013. A total of 86 patients completed the study, with 21 patients also having a preoperative diagnosis of PsA. Their mean preoperative weight was 288 pounds and preoperative body mass index 45.8 kg/m2. The average duration of psoriasis and PsA at the time of surgery was significant at 18.7 years, Dr. Sethi noted.
The mean time from surgery was 6.1 years, with a mean excess weight loss of 46.2% and total weight loss of 23.8%. Laparoscopic adjustable gastric banding was the most common surgery (91%), followed by Roux-en-Y bypass (7%).
In secondary analyses, a higher percent of excess weight loss at recent follow-up was significantly associated with an easing of psoriasis severity (59.5% vs. 43.5%; P = .046), while higher percent of excess weight loss at 1 year was associated with a trend in PsA improvement (55.4% vs. 43.8%; P = .47), Dr. Sethi said.
Easing of disease severity after surgery was associated with a higher rating of disease at the time of surgery (8.9 vs. 7.4; P less than .01) and older age at diagnosis (36.9 years vs. 25.9 years; P = .02), suggesting that these factors may be used to identify patients with a greater likelihood of improvement.
“Larger prospective studies are needed to further define the true effect of surgical weight loss on psoriasis and psoriatic arthritis,” she said at the meeting, presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery (ASMBS).
Session comoderator Dr. Peter T. Hallowell of the University of Virginia, Charlottesville, agreed that further studies are needed, but said the interesting thing is how bariatric surgery improves multiple problems.
“The results are pretty impressive with the moderate weight loss they got with laparoscopic gastric banding,” he said in an interview. “With gastric bypass or a sleeve, where we would expect greater weight loss and greater decrease in the inflammatory mediators, we may see an even greater improved outcome.”
A larger prospective study is being planned between New York University’s psoriasis and bariatric centers in about 60 patients with psoriasis and PsA undergoing bariatric surgery, mostly sleeve gastrectomy and gastric bypass, Dr. Sethi said in an interview.
LOS ANGELES – A provocative study opens the door for psoriasis and psoriatic arthritis to be added to the growing list of benefits following bariatric surgery.
“Although the natural history of psoriasis and psoriatic arthritis is typically chronic, the majority of patients experience improvement after bariatric surgery, Dr. Monica Sethi of New York University said at Obesity Week 2015.
At an average of 6 years after bariatric surgery, 55% of patients with psoriasis and 62% of those with psoriatic arthritis (PsA) reported subjective lessening of their disease.
Preoperative disease severity ratings on a 10-point scale significantly decreased at the most recent follow-up for psoriasis (5.6 vs. 3.3; P less than .01) and PsA (6.4 vs. 3.9; P = .02).
“Our results indicate an association between excess weight loss and symptomatic improvement in severe cases of psoriasis, and a possible improvement in psoriatic arthritis,” she said.
Although the effects of surgical weight loss on psoriasis and PsA are unknown, obesity is known to be more prevalent among patients with psoriasis and PsA. This correlation appears to be related to fat tissue–driven systemic inflammation, Dr. Sethi said.
The investigators surveyed 128 patients with a preoperative diagnosis of psoriasis identified from a single-center database of 9,073 bariatric surgeries performed between 2002 and 2013. A total of 86 patients completed the study, with 21 patients also having a preoperative diagnosis of PsA. Their mean preoperative weight was 288 pounds and preoperative body mass index 45.8 kg/m2. The average duration of psoriasis and PsA at the time of surgery was significant at 18.7 years, Dr. Sethi noted.
The mean time from surgery was 6.1 years, with a mean excess weight loss of 46.2% and total weight loss of 23.8%. Laparoscopic adjustable gastric banding was the most common surgery (91%), followed by Roux-en-Y bypass (7%).
In secondary analyses, a higher percent of excess weight loss at recent follow-up was significantly associated with an easing of psoriasis severity (59.5% vs. 43.5%; P = .046), while higher percent of excess weight loss at 1 year was associated with a trend in PsA improvement (55.4% vs. 43.8%; P = .47), Dr. Sethi said.
Easing of disease severity after surgery was associated with a higher rating of disease at the time of surgery (8.9 vs. 7.4; P less than .01) and older age at diagnosis (36.9 years vs. 25.9 years; P = .02), suggesting that these factors may be used to identify patients with a greater likelihood of improvement.
“Larger prospective studies are needed to further define the true effect of surgical weight loss on psoriasis and psoriatic arthritis,” she said at the meeting, presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery (ASMBS).
Session comoderator Dr. Peter T. Hallowell of the University of Virginia, Charlottesville, agreed that further studies are needed, but said the interesting thing is how bariatric surgery improves multiple problems.
“The results are pretty impressive with the moderate weight loss they got with laparoscopic gastric banding,” he said in an interview. “With gastric bypass or a sleeve, where we would expect greater weight loss and greater decrease in the inflammatory mediators, we may see an even greater improved outcome.”
A larger prospective study is being planned between New York University’s psoriasis and bariatric centers in about 60 patients with psoriasis and PsA undergoing bariatric surgery, mostly sleeve gastrectomy and gastric bypass, Dr. Sethi said in an interview.
AT OBESITY WEEK 2015
Key clinical point: Bariatric surgery in obese patients may ease the symptoms of psoriasis and psoriatic arthritis.
Major finding: Preoperative disease severity ratings declined at most recent follow-up for psoriasis (5.6 vs. 3.3; P less than .01) and psoriatic arthritis (6.4 vs. 3.9; P = .02).
Data source: Survey of 86 bariatric surgery patients with preoperative psoriasis alone or in combination with psoriatic arthritis.
Disclosures: Dr. Sethi reported having no disclosures. Three coauthors reported financial ties with Allergan Medical, one of whom is also a speaker for Apollo Endosurgery and one of whom is on the faculty for Gore.

Depression and Psoriasis
While psoriasis is a known risk factor for depression, depression can also exacerbate or trigger psoriasis. This relationship between depression and psoriasis, however, remains to be fully explored.
In an article published online on September 30 in JAMA Dermatology , Cohen et al examined the association between psoriasis and major depression in the US population. The authors conducted a population-based study that utilized individuals who were participating in the National Health and Nutrition Examination Survey from 2009 through 2012.
The authors identified 351 (2.8%) cases of psoriasis and 968 (7.8%) cases of major depression in the 12,382 participants included in the study. Of the patients with psoriasis, 58 (16.5%) met criteria for major depression. The mean (standard deviation) Patient Health Questionnaire-9 score was significantly higher among patients with a history of psoriasis than those without psoriasis (4.54 [5.7] vs 3.22 [4.3], P<.001). After adjustment for sex, age, race, body mass index, physical activity level, smoking history, alcohol use, history of myocardial infarction, history of stroke, and history of diabetes mellitus (odds ratio, 2.09 [95% confidence interval, 1.41–3.11], P<.001), psoriasis was significantly associated with major depression. Having a history of cardiovascular events did not modify the risk of major depression for patients with psoriasis. The investigators also found that the risk of major depression was not significantly different between patients with limited vs extensive psoriasis (odds ratio, 0.66 [95% confidence interval, 0.18–2.44], P=.53).
What’s the Issue?
We know that psoriasis is associated with depression. This study, however, has some surprising findings. The severity of psoriasis was unrelated to the risk of major depression. Additionally, cardiovascular events did not seem to impact major depression in participants with psoriasis. Therefore, all patients with psoriasis, regardless of severity, may be at risk for major depression. Will these findings impact your evaluation of psychological issues in individuals with psoriasis?
While psoriasis is a known risk factor for depression, depression can also exacerbate or trigger psoriasis. This relationship between depression and psoriasis, however, remains to be fully explored.
In an article published online on September 30 in JAMA Dermatology , Cohen et al examined the association between psoriasis and major depression in the US population. The authors conducted a population-based study that utilized individuals who were participating in the National Health and Nutrition Examination Survey from 2009 through 2012.
The authors identified 351 (2.8%) cases of psoriasis and 968 (7.8%) cases of major depression in the 12,382 participants included in the study. Of the patients with psoriasis, 58 (16.5%) met criteria for major depression. The mean (standard deviation) Patient Health Questionnaire-9 score was significantly higher among patients with a history of psoriasis than those without psoriasis (4.54 [5.7] vs 3.22 [4.3], P<.001). After adjustment for sex, age, race, body mass index, physical activity level, smoking history, alcohol use, history of myocardial infarction, history of stroke, and history of diabetes mellitus (odds ratio, 2.09 [95% confidence interval, 1.41–3.11], P<.001), psoriasis was significantly associated with major depression. Having a history of cardiovascular events did not modify the risk of major depression for patients with psoriasis. The investigators also found that the risk of major depression was not significantly different between patients with limited vs extensive psoriasis (odds ratio, 0.66 [95% confidence interval, 0.18–2.44], P=.53).
What’s the Issue?
We know that psoriasis is associated with depression. This study, however, has some surprising findings. The severity of psoriasis was unrelated to the risk of major depression. Additionally, cardiovascular events did not seem to impact major depression in participants with psoriasis. Therefore, all patients with psoriasis, regardless of severity, may be at risk for major depression. Will these findings impact your evaluation of psychological issues in individuals with psoriasis?
While psoriasis is a known risk factor for depression, depression can also exacerbate or trigger psoriasis. This relationship between depression and psoriasis, however, remains to be fully explored.
In an article published online on September 30 in JAMA Dermatology , Cohen et al examined the association between psoriasis and major depression in the US population. The authors conducted a population-based study that utilized individuals who were participating in the National Health and Nutrition Examination Survey from 2009 through 2012.
The authors identified 351 (2.8%) cases of psoriasis and 968 (7.8%) cases of major depression in the 12,382 participants included in the study. Of the patients with psoriasis, 58 (16.5%) met criteria for major depression. The mean (standard deviation) Patient Health Questionnaire-9 score was significantly higher among patients with a history of psoriasis than those without psoriasis (4.54 [5.7] vs 3.22 [4.3], P<.001). After adjustment for sex, age, race, body mass index, physical activity level, smoking history, alcohol use, history of myocardial infarction, history of stroke, and history of diabetes mellitus (odds ratio, 2.09 [95% confidence interval, 1.41–3.11], P<.001), psoriasis was significantly associated with major depression. Having a history of cardiovascular events did not modify the risk of major depression for patients with psoriasis. The investigators also found that the risk of major depression was not significantly different between patients with limited vs extensive psoriasis (odds ratio, 0.66 [95% confidence interval, 0.18–2.44], P=.53).
What’s the Issue?
We know that psoriasis is associated with depression. This study, however, has some surprising findings. The severity of psoriasis was unrelated to the risk of major depression. Additionally, cardiovascular events did not seem to impact major depression in participants with psoriasis. Therefore, all patients with psoriasis, regardless of severity, may be at risk for major depression. Will these findings impact your evaluation of psychological issues in individuals with psoriasis?

EADV: No cancer signal with ixekizumab
COPENHAGEN – Ixekizumab showed no hint of an increased malignancy risk of any sort in 4,208 patients with moderate to severe psoriasis who had 6,480 patient-years of exposure to the investigational interleukin-17A inhibitor in seven clinical trials.
“Patients were studied for an average of 18 months on ixekizumab. You may say that’s not enough time to assess the true risk for malignancy, and I would say you’re probably right. But 18 months isn’t shabby. From such an exposure, we can get some sense of the malignancy risk with this drug,” Dr. Bruce E. Strober asserted at the annual congress of the European Academy of Dermatology and Venereology.

That’s a key issue whenever a dermatologist prescribes a potent immune-altering anti-inflammatory medication for an indefinite period of time, he added.
In the randomized trials in which more than 2,300 patients who received 80 mg of ixekizumab every 2 or 4 weeks were compared with 739 patients assigned to etanercept (Enbrel) and 791 on placebo, malignancy rates were similarly low, at 0.1-0.3 cases per 100 patient-years across all four treatment arms, reported Dr. Strober, chair of the department of dermatology at the University of Connecticut, Farmington.
Moreover, in the long-term maintenance studies featuring 60 months of follow-up, the risks of both nonmelanoma skin cancer and other types of cancer were similarly low in patients randomized to ixekizumab at 80 mg every 2 weeks and those dosed every 4 weeks. That’s reassuring, because if a drug causes cancer one would expect to see that the more of the drug given, the higher the malignancy rate.
“You really don’t see any kind of a pattern or any difference between the various compared groups, including patients on etanercept, a drug that all of us feel very comfortable using in patients with psoriasis,” the dermatologist observed.
It’s thought that psoriasis patients have a higher background risk of malignancy than the general population due to the nature of the chronic disease, which involves immune dysregulation and chronic inflammation. In the studies to date, however, the malignancy risk in ixekizumab-treated patients is similar to those on placebo or etanercept.
“In my opinion, it’s comforting that the malignancy rates of the currently approved biologic agents for psoriasis are low and – one could argue – not above that which you would expect for the population of patients we’re treating. So I’ve always felt that monotherapy – and that’s an important point, not combination therapy with other immunosuppressive drugs – offers very little risk of de novo malignancy with the currently approved biologics,” Dr. Strober said.
Lumping together the results of the seven studies in his analysis, the incidence of squamous cell carcinoma was 0.1 cases per 100 patient-years of ixekizumab exposure, the risk of basal cell carcinoma was 0.3 cases per 100 patient-years, and the risk of malignancies other than nonmelanoma skin cancer was 0.5 per 100 patient-years of exposure to ixekizumab.
“That’s basically in line with every other biologic that I’ve examined with regard to malignancy rates: about 1 in 200 patients treated for 1 year gets a nonmelanoma skin cancer of some nature if you look at all data sources, including long-term registries and clinical trials. So it appears that this drug falls right in line with the other biologics in terms of rates of the more serious cancers, at least over the time period studied,” Dr. Strober commented.
He offered a caveat, however: “What will happen in patients given combination therapy with other immunosuppressive drugs? Or special populations, such as patients at higher risk for malignancy, be it skin cancer or other types of cancer? Or – and here’s the question we often have the most difficulty answering – what about people with a prior history of malignancy? What would be their risk of malignancy in being placed on a drug of this nature? Only time and really good registry data will allow us to answer these questions.”
Dr. Strober is a consultant to Eli Lilly, which sponsored the seven ixekizumab studies, as well as to numerous other pharmaceutical companies.
COPENHAGEN – Ixekizumab showed no hint of an increased malignancy risk of any sort in 4,208 patients with moderate to severe psoriasis who had 6,480 patient-years of exposure to the investigational interleukin-17A inhibitor in seven clinical trials.
“Patients were studied for an average of 18 months on ixekizumab. You may say that’s not enough time to assess the true risk for malignancy, and I would say you’re probably right. But 18 months isn’t shabby. From such an exposure, we can get some sense of the malignancy risk with this drug,” Dr. Bruce E. Strober asserted at the annual congress of the European Academy of Dermatology and Venereology.
That’s a key issue whenever a dermatologist prescribes a potent immune-altering anti-inflammatory medication for an indefinite period of time, he added.
In the randomized trials in which more than 2,300 patients who received 80 mg of ixekizumab every 2 or 4 weeks were compared with 739 patients assigned to etanercept (Enbrel) and 791 on placebo, malignancy rates were similarly low, at 0.1-0.3 cases per 100 patient-years across all four treatment arms, reported Dr. Strober, chair of the department of dermatology at the University of Connecticut, Farmington.
Moreover, in the long-term maintenance studies featuring 60 months of follow-up, the risks of both nonmelanoma skin cancer and other types of cancer were similarly low in patients randomized to ixekizumab at 80 mg every 2 weeks and those dosed every 4 weeks. That’s reassuring, because if a drug causes cancer one would expect to see that the more of the drug given, the higher the malignancy rate.
“You really don’t see any kind of a pattern or any difference between the various compared groups, including patients on etanercept, a drug that all of us feel very comfortable using in patients with psoriasis,” the dermatologist observed.
It’s thought that psoriasis patients have a higher background risk of malignancy than the general population due to the nature of the chronic disease, which involves immune dysregulation and chronic inflammation. In the studies to date, however, the malignancy risk in ixekizumab-treated patients is similar to those on placebo or etanercept.
“In my opinion, it’s comforting that the malignancy rates of the currently approved biologic agents for psoriasis are low and – one could argue – not above that which you would expect for the population of patients we’re treating. So I’ve always felt that monotherapy – and that’s an important point, not combination therapy with other immunosuppressive drugs – offers very little risk of de novo malignancy with the currently approved biologics,” Dr. Strober said.
Lumping together the results of the seven studies in his analysis, the incidence of squamous cell carcinoma was 0.1 cases per 100 patient-years of ixekizumab exposure, the risk of basal cell carcinoma was 0.3 cases per 100 patient-years, and the risk of malignancies other than nonmelanoma skin cancer was 0.5 per 100 patient-years of exposure to ixekizumab.
“That’s basically in line with every other biologic that I’ve examined with regard to malignancy rates: about 1 in 200 patients treated for 1 year gets a nonmelanoma skin cancer of some nature if you look at all data sources, including long-term registries and clinical trials. So it appears that this drug falls right in line with the other biologics in terms of rates of the more serious cancers, at least over the time period studied,” Dr. Strober commented.
He offered a caveat, however: “What will happen in patients given combination therapy with other immunosuppressive drugs? Or special populations, such as patients at higher risk for malignancy, be it skin cancer or other types of cancer? Or – and here’s the question we often have the most difficulty answering – what about people with a prior history of malignancy? What would be their risk of malignancy in being placed on a drug of this nature? Only time and really good registry data will allow us to answer these questions.”
Dr. Strober is a consultant to Eli Lilly, which sponsored the seven ixekizumab studies, as well as to numerous other pharmaceutical companies.
COPENHAGEN – Ixekizumab showed no hint of an increased malignancy risk of any sort in 4,208 patients with moderate to severe psoriasis who had 6,480 patient-years of exposure to the investigational interleukin-17A inhibitor in seven clinical trials.
“Patients were studied for an average of 18 months on ixekizumab. You may say that’s not enough time to assess the true risk for malignancy, and I would say you’re probably right. But 18 months isn’t shabby. From such an exposure, we can get some sense of the malignancy risk with this drug,” Dr. Bruce E. Strober asserted at the annual congress of the European Academy of Dermatology and Venereology.
That’s a key issue whenever a dermatologist prescribes a potent immune-altering anti-inflammatory medication for an indefinite period of time, he added.
In the randomized trials in which more than 2,300 patients who received 80 mg of ixekizumab every 2 or 4 weeks were compared with 739 patients assigned to etanercept (Enbrel) and 791 on placebo, malignancy rates were similarly low, at 0.1-0.3 cases per 100 patient-years across all four treatment arms, reported Dr. Strober, chair of the department of dermatology at the University of Connecticut, Farmington.
Moreover, in the long-term maintenance studies featuring 60 months of follow-up, the risks of both nonmelanoma skin cancer and other types of cancer were similarly low in patients randomized to ixekizumab at 80 mg every 2 weeks and those dosed every 4 weeks. That’s reassuring, because if a drug causes cancer one would expect to see that the more of the drug given, the higher the malignancy rate.
“You really don’t see any kind of a pattern or any difference between the various compared groups, including patients on etanercept, a drug that all of us feel very comfortable using in patients with psoriasis,” the dermatologist observed.
It’s thought that psoriasis patients have a higher background risk of malignancy than the general population due to the nature of the chronic disease, which involves immune dysregulation and chronic inflammation. In the studies to date, however, the malignancy risk in ixekizumab-treated patients is similar to those on placebo or etanercept.
“In my opinion, it’s comforting that the malignancy rates of the currently approved biologic agents for psoriasis are low and – one could argue – not above that which you would expect for the population of patients we’re treating. So I’ve always felt that monotherapy – and that’s an important point, not combination therapy with other immunosuppressive drugs – offers very little risk of de novo malignancy with the currently approved biologics,” Dr. Strober said.
Lumping together the results of the seven studies in his analysis, the incidence of squamous cell carcinoma was 0.1 cases per 100 patient-years of ixekizumab exposure, the risk of basal cell carcinoma was 0.3 cases per 100 patient-years, and the risk of malignancies other than nonmelanoma skin cancer was 0.5 per 100 patient-years of exposure to ixekizumab.
“That’s basically in line with every other biologic that I’ve examined with regard to malignancy rates: about 1 in 200 patients treated for 1 year gets a nonmelanoma skin cancer of some nature if you look at all data sources, including long-term registries and clinical trials. So it appears that this drug falls right in line with the other biologics in terms of rates of the more serious cancers, at least over the time period studied,” Dr. Strober commented.
He offered a caveat, however: “What will happen in patients given combination therapy with other immunosuppressive drugs? Or special populations, such as patients at higher risk for malignancy, be it skin cancer or other types of cancer? Or – and here’s the question we often have the most difficulty answering – what about people with a prior history of malignancy? What would be their risk of malignancy in being placed on a drug of this nature? Only time and really good registry data will allow us to answer these questions.”
Dr. Strober is a consultant to Eli Lilly, which sponsored the seven ixekizumab studies, as well as to numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: In studies conducted in psoriasis patients to date, ixekizumab doesn’t raise any malignancy concerns.
Major finding: The risk of malignancies of any type was 0.9 cases per 100 patient-years of exposure to the investigational interleukin-17A inhibitor ixekizumab, which is in line with rates associated with the currently approved biologic agents.
Data source: This was an analysis of treatment-emergent malignancies among participants in seven clinical psoriasis trials representing a total of 6,480 patient-years of exposure to ixekizumab.
Disclosures: The presenter of this analysis is a consultant to Eli Lilly, which sponsored the seven ixekizumab studies, as well as to numerous other pharmaceutical companies.

EADV: Secukinumab shows sustained high efficacy in SCULPTURE through 3 years
COPENHAGEN – Secukinumab maintained consistently high and previously unheard of long-term Psoriasis Area and Severity Index 90 and PASI 100 response rates in psoriasis patients through 3 years of prospective follow-up in an extension of the phase III SCULPTURE trial.
The key to maintaining these high response rates was to give secukinumab (Cosentyx) on a fixed schedule every 4 weeks. SCULPTURE tested the hypothesis that retreatment of initial responders only as needed based upon early evidence of relapse off treatment might be a reasonable alternative strategy. The answer turns out to be emphatically no.

“We can see from the 3-year data that retreatment as needed is probably not a good strategy over the long term. We see four times fewer patients with PASI 90s and 10 times fewer patients with PASI 100s if they are treated as needed. From the patient point of view, quality of life and satisfaction are not optimal with retreatment as needed,” Dr. Diamant Thaci said at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year extension of SCULPTURE also provided reassuring safety data, he noted.
“It’s interesting that with long-term treatment out to 3 years, we don’t see any new safety signals. In fact, the signals we saw in the preapproval studies are slightly diluted here, and now that secukinumab is approved, we’re not seeing them as problems in real life clinical practice,” said Dr. Thaci of University Hospital in Lübeck, Germany.
The 52-week SCULPTURE results were published earlier this year (J Am Acad Dermatol. 2015 Jul;73[1]:27-36). Dr. Thaci’s update extended the findings from 52 weeks through 3 years.
The double-blind phase of the core SCULPTURE study began with 966 patients with moderate to severe plaque psoriasis and a baseline PASI score of about 23. After 12 weeks on subcutaneous secukinumab, the 842 subjects who had achieved 75% or more improvement from their baseline PASI score were randomized to one of four study arms: secukinumab at either 300 mg or 150 mg, administered either every 4 weeks or by retreatment as needed when an individual’s response dropped below PASI 75.
Picking up where the published 52-week results left off, Dr. Thaci focused special attention on the PASI 90 outcomes out to 3 years because he said he believes PASI 90 will become generally accepted as the new treatment target, now that secukinumab is on the market and other supereffective biologic agents are advancing through the developmental pipeline.
At 52 weeks, 68.5% of patients on fixed-dose secukinumab at 300 mg every 4 weeks had a PASI 90 response. At 3 years, after an additional 2 years on this schedule, the PASI 90 rate was still hovering around the two-thirds mark at 63.8%. In contrast, patients on retreatment with 300 mg as needed had a PASI 90 rate of 14.8% at 52 weeks and 13% at 3 years.
Although the Food and Drug Administration recommended the 300-mg dose when it approved secukinumab as a first-in-class biologic in January, the agency added that 150 mg may be acceptable in some patients. But the 150-mg dose was clearly inferior long term in the SCULPTURE extension. At 52 weeks, 50.3% of patients on secukinumab at 150 mg every 4 weeks had a PASI 90 response; at 3 years the rate was 36.8%. With retreatment as needed using 150 mg, the 52-week and 3-year PASI 90 rates were 12% and 11.8%, Dr. Thaci reported.
The PASI 100 (clear skin) response rate at 52 weeks with fixed-dose secukinumab at 300 mg was 43.8%. At 3 years it was 42.6%.
“These excellent responders kept their excellent response by continuing treatment for 3 years,” the dermatologist noted.
The PASI 100 rates with retreatment as needed were 5.3% and 3.8%.
In terms of safety through 3 years of treatment, rates of the adverse events of greatest interest with the use of immunomodulatory therapies – Candida infections, malignancies, serious infections, and major adverse cardiovascular events – were similarly low across all four treatment arms, ranging from 0 to 1.6 events per 100 subject-years. Thus, there was no safety advantage in using the retreatment as needed strategy.
“In the past we were all talking about the risk of Candida infection caused by inhibiting [interleukin]-17. But if you treat patients in real life clinical practice, you may see a signal of Candida infection in the beginning, yet in the long term you don’t see Candida as a significant side effect in our daily practice – and in this trial the incidence decreases,” Dr. Thaci said.
One audience member rose to criticize the way the analysis of the long-term extension of SCULPTURE was structured – namely, that only patients still on secukinumab at the 3-year mark were included in the results. This “as-observed” method inflates the efficacy results, he said. How many patients dropped out over time and weren’t counted? he asked.
Dr. Thaci replied that roughly 90% of patients in the trial at 52 weeks continued on secukinumab out to 3 years.
“It’s not a massive dropout. I wish it were a nonresponder imputation analysis, but in long-term studies, it’s very difficult to show that analysis because the vast majority of the trials that have been presented in the past have been ‘as observed.’ But I don’t think you’ll see a big difference between this as-observed analysis and a nonresponder imputation analysis. This is a very fair and convincing analysis,” he said.
Secukinumab is marketed by Novartis, which sponsored the study. Dr. Thaci serves as an investigator and consultant to Novartis and other pharmaceutical companies.
COPENHAGEN – Secukinumab maintained consistently high and previously unheard of long-term Psoriasis Area and Severity Index 90 and PASI 100 response rates in psoriasis patients through 3 years of prospective follow-up in an extension of the phase III SCULPTURE trial.
The key to maintaining these high response rates was to give secukinumab (Cosentyx) on a fixed schedule every 4 weeks. SCULPTURE tested the hypothesis that retreatment of initial responders only as needed based upon early evidence of relapse off treatment might be a reasonable alternative strategy. The answer turns out to be emphatically no.
“We can see from the 3-year data that retreatment as needed is probably not a good strategy over the long term. We see four times fewer patients with PASI 90s and 10 times fewer patients with PASI 100s if they are treated as needed. From the patient point of view, quality of life and satisfaction are not optimal with retreatment as needed,” Dr. Diamant Thaci said at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year extension of SCULPTURE also provided reassuring safety data, he noted.
“It’s interesting that with long-term treatment out to 3 years, we don’t see any new safety signals. In fact, the signals we saw in the preapproval studies are slightly diluted here, and now that secukinumab is approved, we’re not seeing them as problems in real life clinical practice,” said Dr. Thaci of University Hospital in Lübeck, Germany.
The 52-week SCULPTURE results were published earlier this year (J Am Acad Dermatol. 2015 Jul;73[1]:27-36). Dr. Thaci’s update extended the findings from 52 weeks through 3 years.
The double-blind phase of the core SCULPTURE study began with 966 patients with moderate to severe plaque psoriasis and a baseline PASI score of about 23. After 12 weeks on subcutaneous secukinumab, the 842 subjects who had achieved 75% or more improvement from their baseline PASI score were randomized to one of four study arms: secukinumab at either 300 mg or 150 mg, administered either every 4 weeks or by retreatment as needed when an individual’s response dropped below PASI 75.
Picking up where the published 52-week results left off, Dr. Thaci focused special attention on the PASI 90 outcomes out to 3 years because he said he believes PASI 90 will become generally accepted as the new treatment target, now that secukinumab is on the market and other supereffective biologic agents are advancing through the developmental pipeline.
At 52 weeks, 68.5% of patients on fixed-dose secukinumab at 300 mg every 4 weeks had a PASI 90 response. At 3 years, after an additional 2 years on this schedule, the PASI 90 rate was still hovering around the two-thirds mark at 63.8%. In contrast, patients on retreatment with 300 mg as needed had a PASI 90 rate of 14.8% at 52 weeks and 13% at 3 years.
Although the Food and Drug Administration recommended the 300-mg dose when it approved secukinumab as a first-in-class biologic in January, the agency added that 150 mg may be acceptable in some patients. But the 150-mg dose was clearly inferior long term in the SCULPTURE extension. At 52 weeks, 50.3% of patients on secukinumab at 150 mg every 4 weeks had a PASI 90 response; at 3 years the rate was 36.8%. With retreatment as needed using 150 mg, the 52-week and 3-year PASI 90 rates were 12% and 11.8%, Dr. Thaci reported.
The PASI 100 (clear skin) response rate at 52 weeks with fixed-dose secukinumab at 300 mg was 43.8%. At 3 years it was 42.6%.
“These excellent responders kept their excellent response by continuing treatment for 3 years,” the dermatologist noted.
The PASI 100 rates with retreatment as needed were 5.3% and 3.8%.
In terms of safety through 3 years of treatment, rates of the adverse events of greatest interest with the use of immunomodulatory therapies – Candida infections, malignancies, serious infections, and major adverse cardiovascular events – were similarly low across all four treatment arms, ranging from 0 to 1.6 events per 100 subject-years. Thus, there was no safety advantage in using the retreatment as needed strategy.
“In the past we were all talking about the risk of Candida infection caused by inhibiting [interleukin]-17. But if you treat patients in real life clinical practice, you may see a signal of Candida infection in the beginning, yet in the long term you don’t see Candida as a significant side effect in our daily practice – and in this trial the incidence decreases,” Dr. Thaci said.
One audience member rose to criticize the way the analysis of the long-term extension of SCULPTURE was structured – namely, that only patients still on secukinumab at the 3-year mark were included in the results. This “as-observed” method inflates the efficacy results, he said. How many patients dropped out over time and weren’t counted? he asked.
Dr. Thaci replied that roughly 90% of patients in the trial at 52 weeks continued on secukinumab out to 3 years.
“It’s not a massive dropout. I wish it were a nonresponder imputation analysis, but in long-term studies, it’s very difficult to show that analysis because the vast majority of the trials that have been presented in the past have been ‘as observed.’ But I don’t think you’ll see a big difference between this as-observed analysis and a nonresponder imputation analysis. This is a very fair and convincing analysis,” he said.
Secukinumab is marketed by Novartis, which sponsored the study. Dr. Thaci serves as an investigator and consultant to Novartis and other pharmaceutical companies.
COPENHAGEN – Secukinumab maintained consistently high and previously unheard of long-term Psoriasis Area and Severity Index 90 and PASI 100 response rates in psoriasis patients through 3 years of prospective follow-up in an extension of the phase III SCULPTURE trial.
The key to maintaining these high response rates was to give secukinumab (Cosentyx) on a fixed schedule every 4 weeks. SCULPTURE tested the hypothesis that retreatment of initial responders only as needed based upon early evidence of relapse off treatment might be a reasonable alternative strategy. The answer turns out to be emphatically no.
“We can see from the 3-year data that retreatment as needed is probably not a good strategy over the long term. We see four times fewer patients with PASI 90s and 10 times fewer patients with PASI 100s if they are treated as needed. From the patient point of view, quality of life and satisfaction are not optimal with retreatment as needed,” Dr. Diamant Thaci said at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year extension of SCULPTURE also provided reassuring safety data, he noted.
“It’s interesting that with long-term treatment out to 3 years, we don’t see any new safety signals. In fact, the signals we saw in the preapproval studies are slightly diluted here, and now that secukinumab is approved, we’re not seeing them as problems in real life clinical practice,” said Dr. Thaci of University Hospital in Lübeck, Germany.
The 52-week SCULPTURE results were published earlier this year (J Am Acad Dermatol. 2015 Jul;73[1]:27-36). Dr. Thaci’s update extended the findings from 52 weeks through 3 years.
The double-blind phase of the core SCULPTURE study began with 966 patients with moderate to severe plaque psoriasis and a baseline PASI score of about 23. After 12 weeks on subcutaneous secukinumab, the 842 subjects who had achieved 75% or more improvement from their baseline PASI score were randomized to one of four study arms: secukinumab at either 300 mg or 150 mg, administered either every 4 weeks or by retreatment as needed when an individual’s response dropped below PASI 75.
Picking up where the published 52-week results left off, Dr. Thaci focused special attention on the PASI 90 outcomes out to 3 years because he said he believes PASI 90 will become generally accepted as the new treatment target, now that secukinumab is on the market and other supereffective biologic agents are advancing through the developmental pipeline.
At 52 weeks, 68.5% of patients on fixed-dose secukinumab at 300 mg every 4 weeks had a PASI 90 response. At 3 years, after an additional 2 years on this schedule, the PASI 90 rate was still hovering around the two-thirds mark at 63.8%. In contrast, patients on retreatment with 300 mg as needed had a PASI 90 rate of 14.8% at 52 weeks and 13% at 3 years.
Although the Food and Drug Administration recommended the 300-mg dose when it approved secukinumab as a first-in-class biologic in January, the agency added that 150 mg may be acceptable in some patients. But the 150-mg dose was clearly inferior long term in the SCULPTURE extension. At 52 weeks, 50.3% of patients on secukinumab at 150 mg every 4 weeks had a PASI 90 response; at 3 years the rate was 36.8%. With retreatment as needed using 150 mg, the 52-week and 3-year PASI 90 rates were 12% and 11.8%, Dr. Thaci reported.
The PASI 100 (clear skin) response rate at 52 weeks with fixed-dose secukinumab at 300 mg was 43.8%. At 3 years it was 42.6%.
“These excellent responders kept their excellent response by continuing treatment for 3 years,” the dermatologist noted.
The PASI 100 rates with retreatment as needed were 5.3% and 3.8%.
In terms of safety through 3 years of treatment, rates of the adverse events of greatest interest with the use of immunomodulatory therapies – Candida infections, malignancies, serious infections, and major adverse cardiovascular events – were similarly low across all four treatment arms, ranging from 0 to 1.6 events per 100 subject-years. Thus, there was no safety advantage in using the retreatment as needed strategy.
“In the past we were all talking about the risk of Candida infection caused by inhibiting [interleukin]-17. But if you treat patients in real life clinical practice, you may see a signal of Candida infection in the beginning, yet in the long term you don’t see Candida as a significant side effect in our daily practice – and in this trial the incidence decreases,” Dr. Thaci said.
One audience member rose to criticize the way the analysis of the long-term extension of SCULPTURE was structured – namely, that only patients still on secukinumab at the 3-year mark were included in the results. This “as-observed” method inflates the efficacy results, he said. How many patients dropped out over time and weren’t counted? he asked.
Dr. Thaci replied that roughly 90% of patients in the trial at 52 weeks continued on secukinumab out to 3 years.
“It’s not a massive dropout. I wish it were a nonresponder imputation analysis, but in long-term studies, it’s very difficult to show that analysis because the vast majority of the trials that have been presented in the past have been ‘as observed.’ But I don’t think you’ll see a big difference between this as-observed analysis and a nonresponder imputation analysis. This is a very fair and convincing analysis,” he said.
Secukinumab is marketed by Novartis, which sponsored the study. Dr. Thaci serves as an investigator and consultant to Novartis and other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: Three-year data from a phase III trial provide reassurance regarding the long-term safety and efficacy of secukinumab for psoriasis.
Major finding: The high level of improvement seen in the 52-week core study of secukinumab, at the dose of 300 mg every 4 weeks, in patients with psoriasis was largely maintained with 2 more years of therapy, with a 63.8% PASI-90 rate at 3 years.
Data source: A prospective 2-year extension of a 1-year, randomized double-blind trial of 842 psoriasis patients, who initially achieved at least a PASI-75 response to secukinumab.
Disclosures: The study was sponsored by Novartis. The presenter serves as a paid investigator and consultant for Novartis and other pharmaceutical companies.
