User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Plaquelike Syringoma Mimicking Microcystic Adnexal Carcinoma: A Potential Histologic Pitfall
To the Editor:
Plaquelike or plaque-type syringoma is a lesser-known variant of syringoma that can appear histologically indistinguishable from the superficial portion of microcystic adnexal carcinoma (MAC). The plaquelike variant of syringoma holds a benign clinical course, and no treatment is necessary. Microcystic adnexal carcinoma is distinguished from plaquelike syringoma by an aggressive growth pattern with a high risk for local invasion and recurrence if inadequately treated. Thus, treatment with Mohs micrographic surgery (MMS) has been recommended as the mainstay for MAC. If superficial biopsy specimens reveal suspicion for MAC and patients are referred for MMS, careful consideration should be made to differentiate MAC and plaquelike syringoma early to prevent unnecessary morbidity.
.")
A 78-year-old woman was referred for MMS for a left forehead lesion that was diagnosed via shave biopsy as a desmoplastic and cystic adnexal neoplasm with suspicion for desmoplastic trichoepithelioma or MAC (Figure 1). Upon presentation for MMS, a well-healed, 1.0×0.9-cm scar at the biopsy site on the left forehead was observed (Figure 2A). One stage was obtained by standard MMS technique and sent for intraoperative processing (Figure 2B). Frozen section examination of the first stage demonstrated peripheral margin involvement with syringomatous change confined to the superficial and mid dermis (Figure 3). Before proceeding further, these findings were reviewed with an in-house dermatopathologist, and it was determined that no infiltrative tumor, perineural involvement, or other features to indicate malignancy were noted. A decision was made to refrain from obtaining any additional layers and to send excised Burow triangles for permanent section analysis. A primary linear closure was performed without complication, and the patient was discharged from the ambulatory surgery suite. Histopathologic examination of the Burow triangles later confirmed findings consistent with plaquelike syringoma with no evidence of malignancy (Figure 4).

Syringomas present as small flesh-colored papules in the periorbital areas. These benign neoplasms previously have been classified into 4 major clinical variants: localized, generalized, Down syndrome associated, and familial.1 The lesser-known plaquelike variant of syringoma was first described by Kikuchi et al2 in 1979. Aside from our report, a PubMed search of articles indexed for MEDLINE using the terms plaquelike or plaque-type syringoma yielded 16 cases in the literature.2-14 Of these, 6 were referred to or encountered in the MMS setting.8,9,11,12,14 Plaquelike syringoma can be solitary or multiple in presentation.6 It most commonly involves the head and neck but also can present on the trunk, arms, legs, and groin areas. The clinical size of plaquelike syringoma is variable, with the largest reported cases extending several centimeters in diameter.2,6 Similar to reported associations with conventional syringoma, the plaquelike subtype of syringoma has been reported in association with Down syndrome.13
.")
Histopathologically, plaquelike syringoma shares features with MAC as well as desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma. Plaquelike syringoma demonstrates broad proliferations of small tubules morphologically reminiscent of tadpoles confined within the dermis. Ducts typically are lined with 2 or 3 layers of small cuboidal cells. Microcystic adnexal carcinoma typically features asymmetric ductal structures lined with single cells extending from the dermis into the subcutis and even underlying muscle, cartilage, or bone.8 There are no reliable immunohistochemical stains to differentiate between these 2 entities; thus, the primary distinction lies in the depth of involvement. Desmoplastic trichoepithelioma is composed of narrow cords and nests of basaloid cells of follicular origin commonly admixed with small cornifying cysts appearing in the dermis.8 Colonizing Merkel cells positive for cytokeratin 20 often are present in desmoplastic trichoepithelioma and not in syringoma or MAC.15 Desmoplastic basal cell carcinoma demonstrates narrow strands of basaloid cells of follicular origin appearing in the dermis. Desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma are each fundamentally differentiated from plaquelike syringoma in that proliferations of cords and nests are not of eccrine or apocrine origin.
.")
Several cases of plaquelike syringoma have been challenging to distinguish from MAC in performing MMS.8,9,11 Underlying extension of this syringoma variant can be far-reaching, extending to several centimeters in size and involving multiple cosmetic subunits.6,11,14 Inadvertent overtreatment with multiple MMS stages can be avoided with careful recognition of the differentiating histopathologic features. Syringomatous lesions commonly are encountered in MMS and may even be present at the edge of other tumor types. Plaquelike syringoma has been reported as a coexistent entity with nodular basal cell carcinoma.12 Boos et al16 similarly reported the presence of deceptive ductal proliferations along the immediate peripheral margin of MAC, which prompted multiple re-excisions. Pursuit of permanent section analysis in these cases revealed the appearance of small syringomas, and a diagnosis of benign subclinical syringomatous proliferations was made, averting further intervention.16
Our case sheds light on the threat of commission bias in dermatologic surgery, which is the tendency for action rather than inaction.17 In this context, it is important to avoid the perspective that harm to the patient can only be prevented by active intervention. Cognitive bias has been increasingly recognized as a source of medical error, and methods to mitigate bias in medical practice have been well described.17 Microcystic adnexal carcinoma and plaquelike syringoma can be hard to differentiate especially initially, as demonstrated in our case, which particularly illustrates the importance of slowing down a surgical case at the appropriate time, considering and revisiting alternative diagnoses, implementing checklists, and seeking histopathologic collaboration with colleagues when necessary. Our attempted implementation of these principles, especially early collaboration with colleagues, led to intraoperative recognition of plaquelike syringoma within the first stage of MMS.
We seek to raise the index of suspicion for plaquelike syringoma among dermatologists and dermatologic surgeons, especially when syringomatous structures are limited to the superficial dermis. We encourage familiarity with the plaquelike syringoma entity as well as careful consideration of further investigation via scouting biopsies or permanent section analysis when other characteristic features of MAC are unclear or lacking. Adequate sampling as well as collaboration with a dermatopathologist in cases of suspected syringoma can help to reduce the susceptibility to commission bias and prevent histopathologic pitfalls and unwarranted surgical morbidity.
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Dekio S, Jidoi J. Submammary syringoma—report of a case. J Dermatol. 1988;15:351-352.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Nguyen DB, Patterson JW, Wilson BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49:337-339.
- Rongioletti F, Semino MT, Rebora A. Unilateral multiple plaque-like syringomas. Br J Dermatol. 1996;135:623-625.
- Chi HI. A case of unusual syringoma: unilateral linear distribution and plaque formation. J Dermatol. 1996;23:505-506.
- Suwatee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:E206-E207.
- Schleich C, Ferringer T, Petrick M. Plaque type syringoma mimicking a microcystic adnexal carcinoma. J Am Acad Dermatol. 2016;74(suppl 1):AB287.
- Yang Y, Srivastava D. Plaque-type syringoma coexisting with basal cell carcinoma. Dermatol Surg. 2018;44:1464-1466.
- Motegi SI, Sekiguchi A, Fujiwara C, et al. Milia-like idiopathic calcinosis cutis and plaque-type syringoma in a girl with Down syndrome. J Dermatol. 2019;46:E136-E137.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Boos MD, Elenitsas R, Seykora J, et al. Benign subclinical syringomatous proliferations adjacent to a microcystic adnexal carcinoma: a tumor mimic with significant patient implications. Am J Dermatopathol. 2014;36:174-178.
- O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48:225-232.
To the Editor:
Plaquelike or plaque-type syringoma is a lesser-known variant of syringoma that can appear histologically indistinguishable from the superficial portion of microcystic adnexal carcinoma (MAC). The plaquelike variant of syringoma holds a benign clinical course, and no treatment is necessary. Microcystic adnexal carcinoma is distinguished from plaquelike syringoma by an aggressive growth pattern with a high risk for local invasion and recurrence if inadequately treated. Thus, treatment with Mohs micrographic surgery (MMS) has been recommended as the mainstay for MAC. If superficial biopsy specimens reveal suspicion for MAC and patients are referred for MMS, careful consideration should be made to differentiate MAC and plaquelike syringoma early to prevent unnecessary morbidity.
A 78-year-old woman was referred for MMS for a left forehead lesion that was diagnosed via shave biopsy as a desmoplastic and cystic adnexal neoplasm with suspicion for desmoplastic trichoepithelioma or MAC (Figure 1). Upon presentation for MMS, a well-healed, 1.0×0.9-cm scar at the biopsy site on the left forehead was observed (Figure 2A). One stage was obtained by standard MMS technique and sent for intraoperative processing (Figure 2B). Frozen section examination of the first stage demonstrated peripheral margin involvement with syringomatous change confined to the superficial and mid dermis (Figure 3). Before proceeding further, these findings were reviewed with an in-house dermatopathologist, and it was determined that no infiltrative tumor, perineural involvement, or other features to indicate malignancy were noted. A decision was made to refrain from obtaining any additional layers and to send excised Burow triangles for permanent section analysis. A primary linear closure was performed without complication, and the patient was discharged from the ambulatory surgery suite. Histopathologic examination of the Burow triangles later confirmed findings consistent with plaquelike syringoma with no evidence of malignancy (Figure 4).
Syringomas present as small flesh-colored papules in the periorbital areas. These benign neoplasms previously have been classified into 4 major clinical variants: localized, generalized, Down syndrome associated, and familial.1 The lesser-known plaquelike variant of syringoma was first described by Kikuchi et al2 in 1979. Aside from our report, a PubMed search of articles indexed for MEDLINE using the terms plaquelike or plaque-type syringoma yielded 16 cases in the literature.2-14 Of these, 6 were referred to or encountered in the MMS setting.8,9,11,12,14 Plaquelike syringoma can be solitary or multiple in presentation.6 It most commonly involves the head and neck but also can present on the trunk, arms, legs, and groin areas. The clinical size of plaquelike syringoma is variable, with the largest reported cases extending several centimeters in diameter.2,6 Similar to reported associations with conventional syringoma, the plaquelike subtype of syringoma has been reported in association with Down syndrome.13
Histopathologically, plaquelike syringoma shares features with MAC as well as desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma. Plaquelike syringoma demonstrates broad proliferations of small tubules morphologically reminiscent of tadpoles confined within the dermis. Ducts typically are lined with 2 or 3 layers of small cuboidal cells. Microcystic adnexal carcinoma typically features asymmetric ductal structures lined with single cells extending from the dermis into the subcutis and even underlying muscle, cartilage, or bone.8 There are no reliable immunohistochemical stains to differentiate between these 2 entities; thus, the primary distinction lies in the depth of involvement. Desmoplastic trichoepithelioma is composed of narrow cords and nests of basaloid cells of follicular origin commonly admixed with small cornifying cysts appearing in the dermis.8 Colonizing Merkel cells positive for cytokeratin 20 often are present in desmoplastic trichoepithelioma and not in syringoma or MAC.15 Desmoplastic basal cell carcinoma demonstrates narrow strands of basaloid cells of follicular origin appearing in the dermis. Desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma are each fundamentally differentiated from plaquelike syringoma in that proliferations of cords and nests are not of eccrine or apocrine origin.
Several cases of plaquelike syringoma have been challenging to distinguish from MAC in performing MMS.8,9,11 Underlying extension of this syringoma variant can be far-reaching, extending to several centimeters in size and involving multiple cosmetic subunits.6,11,14 Inadvertent overtreatment with multiple MMS stages can be avoided with careful recognition of the differentiating histopathologic features. Syringomatous lesions commonly are encountered in MMS and may even be present at the edge of other tumor types. Plaquelike syringoma has been reported as a coexistent entity with nodular basal cell carcinoma.12 Boos et al16 similarly reported the presence of deceptive ductal proliferations along the immediate peripheral margin of MAC, which prompted multiple re-excisions. Pursuit of permanent section analysis in these cases revealed the appearance of small syringomas, and a diagnosis of benign subclinical syringomatous proliferations was made, averting further intervention.16
Our case sheds light on the threat of commission bias in dermatologic surgery, which is the tendency for action rather than inaction.17 In this context, it is important to avoid the perspective that harm to the patient can only be prevented by active intervention. Cognitive bias has been increasingly recognized as a source of medical error, and methods to mitigate bias in medical practice have been well described.17 Microcystic adnexal carcinoma and plaquelike syringoma can be hard to differentiate especially initially, as demonstrated in our case, which particularly illustrates the importance of slowing down a surgical case at the appropriate time, considering and revisiting alternative diagnoses, implementing checklists, and seeking histopathologic collaboration with colleagues when necessary. Our attempted implementation of these principles, especially early collaboration with colleagues, led to intraoperative recognition of plaquelike syringoma within the first stage of MMS.
We seek to raise the index of suspicion for plaquelike syringoma among dermatologists and dermatologic surgeons, especially when syringomatous structures are limited to the superficial dermis. We encourage familiarity with the plaquelike syringoma entity as well as careful consideration of further investigation via scouting biopsies or permanent section analysis when other characteristic features of MAC are unclear or lacking. Adequate sampling as well as collaboration with a dermatopathologist in cases of suspected syringoma can help to reduce the susceptibility to commission bias and prevent histopathologic pitfalls and unwarranted surgical morbidity.
To the Editor:
Plaquelike or plaque-type syringoma is a lesser-known variant of syringoma that can appear histologically indistinguishable from the superficial portion of microcystic adnexal carcinoma (MAC). The plaquelike variant of syringoma holds a benign clinical course, and no treatment is necessary. Microcystic adnexal carcinoma is distinguished from plaquelike syringoma by an aggressive growth pattern with a high risk for local invasion and recurrence if inadequately treated. Thus, treatment with Mohs micrographic surgery (MMS) has been recommended as the mainstay for MAC. If superficial biopsy specimens reveal suspicion for MAC and patients are referred for MMS, careful consideration should be made to differentiate MAC and plaquelike syringoma early to prevent unnecessary morbidity.
A 78-year-old woman was referred for MMS for a left forehead lesion that was diagnosed via shave biopsy as a desmoplastic and cystic adnexal neoplasm with suspicion for desmoplastic trichoepithelioma or MAC (Figure 1). Upon presentation for MMS, a well-healed, 1.0×0.9-cm scar at the biopsy site on the left forehead was observed (Figure 2A). One stage was obtained by standard MMS technique and sent for intraoperative processing (Figure 2B). Frozen section examination of the first stage demonstrated peripheral margin involvement with syringomatous change confined to the superficial and mid dermis (Figure 3). Before proceeding further, these findings were reviewed with an in-house dermatopathologist, and it was determined that no infiltrative tumor, perineural involvement, or other features to indicate malignancy were noted. A decision was made to refrain from obtaining any additional layers and to send excised Burow triangles for permanent section analysis. A primary linear closure was performed without complication, and the patient was discharged from the ambulatory surgery suite. Histopathologic examination of the Burow triangles later confirmed findings consistent with plaquelike syringoma with no evidence of malignancy (Figure 4).
Syringomas present as small flesh-colored papules in the periorbital areas. These benign neoplasms previously have been classified into 4 major clinical variants: localized, generalized, Down syndrome associated, and familial.1 The lesser-known plaquelike variant of syringoma was first described by Kikuchi et al2 in 1979. Aside from our report, a PubMed search of articles indexed for MEDLINE using the terms plaquelike or plaque-type syringoma yielded 16 cases in the literature.2-14 Of these, 6 were referred to or encountered in the MMS setting.8,9,11,12,14 Plaquelike syringoma can be solitary or multiple in presentation.6 It most commonly involves the head and neck but also can present on the trunk, arms, legs, and groin areas. The clinical size of plaquelike syringoma is variable, with the largest reported cases extending several centimeters in diameter.2,6 Similar to reported associations with conventional syringoma, the plaquelike subtype of syringoma has been reported in association with Down syndrome.13
Histopathologically, plaquelike syringoma shares features with MAC as well as desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma. Plaquelike syringoma demonstrates broad proliferations of small tubules morphologically reminiscent of tadpoles confined within the dermis. Ducts typically are lined with 2 or 3 layers of small cuboidal cells. Microcystic adnexal carcinoma typically features asymmetric ductal structures lined with single cells extending from the dermis into the subcutis and even underlying muscle, cartilage, or bone.8 There are no reliable immunohistochemical stains to differentiate between these 2 entities; thus, the primary distinction lies in the depth of involvement. Desmoplastic trichoepithelioma is composed of narrow cords and nests of basaloid cells of follicular origin commonly admixed with small cornifying cysts appearing in the dermis.8 Colonizing Merkel cells positive for cytokeratin 20 often are present in desmoplastic trichoepithelioma and not in syringoma or MAC.15 Desmoplastic basal cell carcinoma demonstrates narrow strands of basaloid cells of follicular origin appearing in the dermis. Desmoplastic trichoepithelioma and desmoplastic basal cell carcinoma are each fundamentally differentiated from plaquelike syringoma in that proliferations of cords and nests are not of eccrine or apocrine origin.
Several cases of plaquelike syringoma have been challenging to distinguish from MAC in performing MMS.8,9,11 Underlying extension of this syringoma variant can be far-reaching, extending to several centimeters in size and involving multiple cosmetic subunits.6,11,14 Inadvertent overtreatment with multiple MMS stages can be avoided with careful recognition of the differentiating histopathologic features. Syringomatous lesions commonly are encountered in MMS and may even be present at the edge of other tumor types. Plaquelike syringoma has been reported as a coexistent entity with nodular basal cell carcinoma.12 Boos et al16 similarly reported the presence of deceptive ductal proliferations along the immediate peripheral margin of MAC, which prompted multiple re-excisions. Pursuit of permanent section analysis in these cases revealed the appearance of small syringomas, and a diagnosis of benign subclinical syringomatous proliferations was made, averting further intervention.16
Our case sheds light on the threat of commission bias in dermatologic surgery, which is the tendency for action rather than inaction.17 In this context, it is important to avoid the perspective that harm to the patient can only be prevented by active intervention. Cognitive bias has been increasingly recognized as a source of medical error, and methods to mitigate bias in medical practice have been well described.17 Microcystic adnexal carcinoma and plaquelike syringoma can be hard to differentiate especially initially, as demonstrated in our case, which particularly illustrates the importance of slowing down a surgical case at the appropriate time, considering and revisiting alternative diagnoses, implementing checklists, and seeking histopathologic collaboration with colleagues when necessary. Our attempted implementation of these principles, especially early collaboration with colleagues, led to intraoperative recognition of plaquelike syringoma within the first stage of MMS.
We seek to raise the index of suspicion for plaquelike syringoma among dermatologists and dermatologic surgeons, especially when syringomatous structures are limited to the superficial dermis. We encourage familiarity with the plaquelike syringoma entity as well as careful consideration of further investigation via scouting biopsies or permanent section analysis when other characteristic features of MAC are unclear or lacking. Adequate sampling as well as collaboration with a dermatopathologist in cases of suspected syringoma can help to reduce the susceptibility to commission bias and prevent histopathologic pitfalls and unwarranted surgical morbidity.
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Dekio S, Jidoi J. Submammary syringoma—report of a case. J Dermatol. 1988;15:351-352.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Nguyen DB, Patterson JW, Wilson BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49:337-339.
- Rongioletti F, Semino MT, Rebora A. Unilateral multiple plaque-like syringomas. Br J Dermatol. 1996;135:623-625.
- Chi HI. A case of unusual syringoma: unilateral linear distribution and plaque formation. J Dermatol. 1996;23:505-506.
- Suwatee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:E206-E207.
- Schleich C, Ferringer T, Petrick M. Plaque type syringoma mimicking a microcystic adnexal carcinoma. J Am Acad Dermatol. 2016;74(suppl 1):AB287.
- Yang Y, Srivastava D. Plaque-type syringoma coexisting with basal cell carcinoma. Dermatol Surg. 2018;44:1464-1466.
- Motegi SI, Sekiguchi A, Fujiwara C, et al. Milia-like idiopathic calcinosis cutis and plaque-type syringoma in a girl with Down syndrome. J Dermatol. 2019;46:E136-E137.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Boos MD, Elenitsas R, Seykora J, et al. Benign subclinical syringomatous proliferations adjacent to a microcystic adnexal carcinoma: a tumor mimic with significant patient implications. Am J Dermatopathol. 2014;36:174-178.
- O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48:225-232.
- Friedman SJ, Butler DF. Syringoma presenting as milia. J Am Acad Dermatol. 1987;16:310-314.
- Kikuchi I, Idemori M, Okazaki M. Plaque type syringoma. J Dermatol. 1979;6:329-331.
- Dekio S, Jidoi J. Submammary syringoma—report of a case. J Dermatol. 1988;15:351-352.
- Patrizi A, Neri I, Marzaduri S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78:460-462.
- Nguyen DB, Patterson JW, Wilson BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49:337-339.
- Rongioletti F, Semino MT, Rebora A. Unilateral multiple plaque-like syringomas. Br J Dermatol. 1996;135:623-625.
- Chi HI. A case of unusual syringoma: unilateral linear distribution and plaque formation. J Dermatol. 1996;23:505-506.
- Suwatee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Wallace JS, Bond JS, Seidel GD, et al. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg. 2014;40:810-812.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:E206-E207.
- Schleich C, Ferringer T, Petrick M. Plaque type syringoma mimicking a microcystic adnexal carcinoma. J Am Acad Dermatol. 2016;74(suppl 1):AB287.
- Yang Y, Srivastava D. Plaque-type syringoma coexisting with basal cell carcinoma. Dermatol Surg. 2018;44:1464-1466.
- Motegi SI, Sekiguchi A, Fujiwara C, et al. Milia-like idiopathic calcinosis cutis and plaque-type syringoma in a girl with Down syndrome. J Dermatol. 2019;46:E136-E137.
- Clark M, Duprey C, Sutton A, et al. Plaque-type syringoma masquerading as microcystic adnexal carcinoma: review of the literature and description of a novel technique that emphasizes lesion architecture to help make the diagnosis. Am J Dermatopathol. 2019;41:E98-E101.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Boos MD, Elenitsas R, Seykora J, et al. Benign subclinical syringomatous proliferations adjacent to a microcystic adnexal carcinoma: a tumor mimic with significant patient implications. Am J Dermatopathol. 2014;36:174-178.
- O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48:225-232.
Practice Points
- Dermatologists should familiarize themselves with the plaquelike subtype of syringoma, which can histologically mimic the superficial portion of microcystic adnexal carcinoma (MAC).
- Careful recognition of plaquelike syringoma in the Mohs micrographic surgery setting may prevent unnecessary surgical morbidity.
- Further diagnostic investigation is warranted for superficial biopsies suggestive of MAC or when other characteristic features are lacking.

Blue Nodules on the Forearms in an Active-Duty Military Servicemember
The Diagnosis: Glomangiomyoma
A punch biopsy of the right forearm revealed a collection of vascular and smooth muscle components with small and spindled bland cells containing minimal eosinophilic cytoplasm (Figure 1), confirming the diagnosis of glomangiomyoma. Immunohistochemical stains also supported the diagnosis and were positive for smooth muscle actin, desmin, and CD34 (Figure 2). Magnetic resonance imaging from a prior attempt at treatment with sclerotherapy demonstrated scattered vascular malformations with no notable internal derangement. There was no improvement with sclerotherapy. Given the number and vascular nature of the lesions, a trial of pulsed dye laser (PDL) therapy was administered and tolerated by the patient. He subsequently moved to a new military duty station. On follow-up, he reported no noticeable clinical improvement in the lesions after PDL and opted not to continue with laser treatment.

Glomangiomyoma is a rare and benign glomus tumor variant that demonstrates differentiation into the smooth muscle and potentially can result in substantial complications.1 Glomus tumors generally are benign neoplasms of the glomus apparatus, and glomus cells function as thermoregulators in the reticular dermis.2 Glomus tumors comprise less than 2% of soft tissue neoplasms and generally are solitary nodules; only 10% of glomus tumors occur with multiple lesions, and among them, glomangiomyoma is the rarest subtype, presenting in only 15% of cases.2,3 The 3 main subtypes of glomus tumors are solid, glomangioma, and glomangiomyoma.4 Clinically, the lesions may present as small blue nodules with associated pain and cold or pressure sensitivity. Although there appears to be variation of the nomenclature depending on the source in the literature, glomangiomas are characterized by their predominant vascular malformations on biopsy. Glomangiomyomas are a subset of glomus tumors with distinct smooth muscle differentiation.4 Given their pathologic presentation, our patient’s lesions were most consistent with the diagnosis of glomangiomyoma.

The small size of the lesions may result in difficulty establishing a clinical diagnosis, particularly if there is no hand involvement, where lesions most commonly occur.2 Therefore, histopathologic evaluation is essential and is the best initial step in evaluating glomangiomyomas.4 Biopsy is the most reliable means of confirming a diagnosis2,4,5; however, diagnostic imaging such as a computed tomography also should be performed if considering blue rubber bleb nevus syndrome due to the primary site of involvement. Surgical excision is the treatment of choice after confirming the diagnosis in most cases of symptomatic glomangiomyomas, particularly with painful lesions.6
Neurilemmomas (also known as schwannomas) are benign lesions that generally present as asymptomatic, soft, smooth nodules most often on the neck; however, they also may present on the flexor extremities or in internal organs. Although primarily asymptomatic, the tumors may be associated with pain and paresthesia as they enlarge and affect surrounding structures. Neurilemmomas may occur spontaneously or as part of a syndrome, such as neurofibromatosis type 2 or Carney complex.7
Hereditary hemorrhagic telangiectasia (formerly known as Osler-Weber-Rendu syndrome) is an autosomal-dominant disease that presents with arteriovenous malformations and telangiectases. Patients generally present in the third decade of life, with the main concern generally being epistaxis.8
Kaposi sarcoma is a viral infection secondary to human herpesvirus 8 that results in red-purple lesions commonly on mucocutaneous sites. Kaposi sarcoma can be AIDS associated and non-HIV associated. Although clinically indistinguishable, a few subtle histologic features can assist in differentiating the 2 etiologies. In addition to a potential history of immunodeficiency, evaluating for involvement of the lymphatic system, respiratory tract, or gastrointestinal tract can aid in differentiating this entity from glomus tumors.9
Leiomyomas are smooth muscle lesions divided into 3 subcategories: angioleiomyoma, piloleiomyoma, and genital leiomyoma. The clinical presentation and histopathology will vary depending on the subcategory. Although cutaneous leiomyomas are benign, further workup for piloleiomyoma may be required given the reported association with hereditary leiomyomatosis and renal cell cancer (Reed syndrome).10
Imaging can be helpful when the clinical diagnosis of a glomus tumor vs other painful neoplasms of the skin is unclear, such as in blue rubber bleb nevus syndrome, angioleiomyomas, neuromas, glomus tumors, leiomyomas, eccrine spiradenomas, congenital vascular malformations, schwannomas, or hemangiomas.4 Radiologic findings for glomus tumors may demonstrate cortical or cystic osseous defects. Magnetic resonance imaging and ultrasonography can help provide additional information on the lesion size and depth of involvement.1 Additionally, deeper glomangiomyomas have been associated with malignancy,2 potentially highlighting the benefit of early incorporation of imaging in the workup for this condition. Malignant transformation is rare and has been reported in less than 1% of cases.6
Treatment of glomus tumors predominantly is directed to the patient’s symptoms; asymptomatic lesions may be monitored.4 For symptomatic lesions, therapeutic options include wide local excision; sclerotherapy; and incorporation of various lasers, including Nd:YAG, CO2, and flashlamp tunable dye laser.4,5 One case report documented use of a PDL that successfully eliminated the pain associated with glomangiomyoma; however, the lesion in that report was not biopsy proven.11
Our case highlights the need to consider glomus tumors in patients presenting with multiple small nodules given the potential for misdiagnosis, impact on quality of life with associated psychological distress, and potential utility of incorporating PDL in treatment. Although our patient did not report clinical improvement in the appearance of the lesions with PDL therapy, additional treatment sessions may have helped,11 but he opted to discontinue. Follow-up for persistently symptomatic or changing lesions is necessary, given the minimal risk for malignant transformation.6
- Lee DY, Hwang SC, Jeong ST, et al. The value of diagnostic ultrasonography in the assessment of a glomus tumor of the subcutaneous layer of the forearm mimicking a hemangioma: a case report. J Med Case Rep. 2015;9:191. doi:10.1186/s13256-015-0672-y
- Li L, Bardsley V, Grainger A, et al. Extradigital glomangiomyoma of the forearm mimicking peripheral nerve sheath tumour and thrombosed varicose vein. BMJ Case Rep. 2021;14:E241221. doi: 10.1136 /bcr-2020-241221
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mohammadi O, Suarez M. Glomus cancer. StatPearls [Internet]. StatPearls Publishing; 2021.
- Maxey ML, Houghton CC, Mastriani KS, et al. Large prepatellar glomangioma: a case report [published online July 10, 2015]. Int J Surg Case Rep. 2015;14:80-84. doi:10.1016/j.ijscr.2015.07.002
- Brathwaite CD, Poppiti RJ Jr. Malignant glomus tumor. a case report of widespread metastases in a patient with multiple glomus body hamartomas. Am J Surg Pathol. 1996;20:233-238. doi:10.1097/00000478-199602000-00012
- Davis DD, Kane SM. Neurilemmoma. StatPearls [Internet]. StatPearls Publishing; 2022.
- Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary hemorrhagic telangiectasia. Otolaryngol Clin North Am. 2018;51:237-254. doi:10.1016/j.otc.2017.09.017
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. StatPearls [Internet]. StatPearls Publishing; 2022.
- Antony FC, Cliff S, Cowley N. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol. 2003;28:617-619. doi:10.1046/j.1365-2230.2003.01403.x
The Diagnosis: Glomangiomyoma
A punch biopsy of the right forearm revealed a collection of vascular and smooth muscle components with small and spindled bland cells containing minimal eosinophilic cytoplasm (Figure 1), confirming the diagnosis of glomangiomyoma. Immunohistochemical stains also supported the diagnosis and were positive for smooth muscle actin, desmin, and CD34 (Figure 2). Magnetic resonance imaging from a prior attempt at treatment with sclerotherapy demonstrated scattered vascular malformations with no notable internal derangement. There was no improvement with sclerotherapy. Given the number and vascular nature of the lesions, a trial of pulsed dye laser (PDL) therapy was administered and tolerated by the patient. He subsequently moved to a new military duty station. On follow-up, he reported no noticeable clinical improvement in the lesions after PDL and opted not to continue with laser treatment.
Glomangiomyoma is a rare and benign glomus tumor variant that demonstrates differentiation into the smooth muscle and potentially can result in substantial complications.1 Glomus tumors generally are benign neoplasms of the glomus apparatus, and glomus cells function as thermoregulators in the reticular dermis.2 Glomus tumors comprise less than 2% of soft tissue neoplasms and generally are solitary nodules; only 10% of glomus tumors occur with multiple lesions, and among them, glomangiomyoma is the rarest subtype, presenting in only 15% of cases.2,3 The 3 main subtypes of glomus tumors are solid, glomangioma, and glomangiomyoma.4 Clinically, the lesions may present as small blue nodules with associated pain and cold or pressure sensitivity. Although there appears to be variation of the nomenclature depending on the source in the literature, glomangiomas are characterized by their predominant vascular malformations on biopsy. Glomangiomyomas are a subset of glomus tumors with distinct smooth muscle differentiation.4 Given their pathologic presentation, our patient’s lesions were most consistent with the diagnosis of glomangiomyoma.
The small size of the lesions may result in difficulty establishing a clinical diagnosis, particularly if there is no hand involvement, where lesions most commonly occur.2 Therefore, histopathologic evaluation is essential and is the best initial step in evaluating glomangiomyomas.4 Biopsy is the most reliable means of confirming a diagnosis2,4,5; however, diagnostic imaging such as a computed tomography also should be performed if considering blue rubber bleb nevus syndrome due to the primary site of involvement. Surgical excision is the treatment of choice after confirming the diagnosis in most cases of symptomatic glomangiomyomas, particularly with painful lesions.6
Neurilemmomas (also known as schwannomas) are benign lesions that generally present as asymptomatic, soft, smooth nodules most often on the neck; however, they also may present on the flexor extremities or in internal organs. Although primarily asymptomatic, the tumors may be associated with pain and paresthesia as they enlarge and affect surrounding structures. Neurilemmomas may occur spontaneously or as part of a syndrome, such as neurofibromatosis type 2 or Carney complex.7
Hereditary hemorrhagic telangiectasia (formerly known as Osler-Weber-Rendu syndrome) is an autosomal-dominant disease that presents with arteriovenous malformations and telangiectases. Patients generally present in the third decade of life, with the main concern generally being epistaxis.8
Kaposi sarcoma is a viral infection secondary to human herpesvirus 8 that results in red-purple lesions commonly on mucocutaneous sites. Kaposi sarcoma can be AIDS associated and non-HIV associated. Although clinically indistinguishable, a few subtle histologic features can assist in differentiating the 2 etiologies. In addition to a potential history of immunodeficiency, evaluating for involvement of the lymphatic system, respiratory tract, or gastrointestinal tract can aid in differentiating this entity from glomus tumors.9
Leiomyomas are smooth muscle lesions divided into 3 subcategories: angioleiomyoma, piloleiomyoma, and genital leiomyoma. The clinical presentation and histopathology will vary depending on the subcategory. Although cutaneous leiomyomas are benign, further workup for piloleiomyoma may be required given the reported association with hereditary leiomyomatosis and renal cell cancer (Reed syndrome).10
Imaging can be helpful when the clinical diagnosis of a glomus tumor vs other painful neoplasms of the skin is unclear, such as in blue rubber bleb nevus syndrome, angioleiomyomas, neuromas, glomus tumors, leiomyomas, eccrine spiradenomas, congenital vascular malformations, schwannomas, or hemangiomas.4 Radiologic findings for glomus tumors may demonstrate cortical or cystic osseous defects. Magnetic resonance imaging and ultrasonography can help provide additional information on the lesion size and depth of involvement.1 Additionally, deeper glomangiomyomas have been associated with malignancy,2 potentially highlighting the benefit of early incorporation of imaging in the workup for this condition. Malignant transformation is rare and has been reported in less than 1% of cases.6
Treatment of glomus tumors predominantly is directed to the patient’s symptoms; asymptomatic lesions may be monitored.4 For symptomatic lesions, therapeutic options include wide local excision; sclerotherapy; and incorporation of various lasers, including Nd:YAG, CO2, and flashlamp tunable dye laser.4,5 One case report documented use of a PDL that successfully eliminated the pain associated with glomangiomyoma; however, the lesion in that report was not biopsy proven.11
Our case highlights the need to consider glomus tumors in patients presenting with multiple small nodules given the potential for misdiagnosis, impact on quality of life with associated psychological distress, and potential utility of incorporating PDL in treatment. Although our patient did not report clinical improvement in the appearance of the lesions with PDL therapy, additional treatment sessions may have helped,11 but he opted to discontinue. Follow-up for persistently symptomatic or changing lesions is necessary, given the minimal risk for malignant transformation.6
The Diagnosis: Glomangiomyoma
A punch biopsy of the right forearm revealed a collection of vascular and smooth muscle components with small and spindled bland cells containing minimal eosinophilic cytoplasm (Figure 1), confirming the diagnosis of glomangiomyoma. Immunohistochemical stains also supported the diagnosis and were positive for smooth muscle actin, desmin, and CD34 (Figure 2). Magnetic resonance imaging from a prior attempt at treatment with sclerotherapy demonstrated scattered vascular malformations with no notable internal derangement. There was no improvement with sclerotherapy. Given the number and vascular nature of the lesions, a trial of pulsed dye laser (PDL) therapy was administered and tolerated by the patient. He subsequently moved to a new military duty station. On follow-up, he reported no noticeable clinical improvement in the lesions after PDL and opted not to continue with laser treatment.
Glomangiomyoma is a rare and benign glomus tumor variant that demonstrates differentiation into the smooth muscle and potentially can result in substantial complications.1 Glomus tumors generally are benign neoplasms of the glomus apparatus, and glomus cells function as thermoregulators in the reticular dermis.2 Glomus tumors comprise less than 2% of soft tissue neoplasms and generally are solitary nodules; only 10% of glomus tumors occur with multiple lesions, and among them, glomangiomyoma is the rarest subtype, presenting in only 15% of cases.2,3 The 3 main subtypes of glomus tumors are solid, glomangioma, and glomangiomyoma.4 Clinically, the lesions may present as small blue nodules with associated pain and cold or pressure sensitivity. Although there appears to be variation of the nomenclature depending on the source in the literature, glomangiomas are characterized by their predominant vascular malformations on biopsy. Glomangiomyomas are a subset of glomus tumors with distinct smooth muscle differentiation.4 Given their pathologic presentation, our patient’s lesions were most consistent with the diagnosis of glomangiomyoma.
The small size of the lesions may result in difficulty establishing a clinical diagnosis, particularly if there is no hand involvement, where lesions most commonly occur.2 Therefore, histopathologic evaluation is essential and is the best initial step in evaluating glomangiomyomas.4 Biopsy is the most reliable means of confirming a diagnosis2,4,5; however, diagnostic imaging such as a computed tomography also should be performed if considering blue rubber bleb nevus syndrome due to the primary site of involvement. Surgical excision is the treatment of choice after confirming the diagnosis in most cases of symptomatic glomangiomyomas, particularly with painful lesions.6
Neurilemmomas (also known as schwannomas) are benign lesions that generally present as asymptomatic, soft, smooth nodules most often on the neck; however, they also may present on the flexor extremities or in internal organs. Although primarily asymptomatic, the tumors may be associated with pain and paresthesia as they enlarge and affect surrounding structures. Neurilemmomas may occur spontaneously or as part of a syndrome, such as neurofibromatosis type 2 or Carney complex.7
Hereditary hemorrhagic telangiectasia (formerly known as Osler-Weber-Rendu syndrome) is an autosomal-dominant disease that presents with arteriovenous malformations and telangiectases. Patients generally present in the third decade of life, with the main concern generally being epistaxis.8
Kaposi sarcoma is a viral infection secondary to human herpesvirus 8 that results in red-purple lesions commonly on mucocutaneous sites. Kaposi sarcoma can be AIDS associated and non-HIV associated. Although clinically indistinguishable, a few subtle histologic features can assist in differentiating the 2 etiologies. In addition to a potential history of immunodeficiency, evaluating for involvement of the lymphatic system, respiratory tract, or gastrointestinal tract can aid in differentiating this entity from glomus tumors.9
Leiomyomas are smooth muscle lesions divided into 3 subcategories: angioleiomyoma, piloleiomyoma, and genital leiomyoma. The clinical presentation and histopathology will vary depending on the subcategory. Although cutaneous leiomyomas are benign, further workup for piloleiomyoma may be required given the reported association with hereditary leiomyomatosis and renal cell cancer (Reed syndrome).10
Imaging can be helpful when the clinical diagnosis of a glomus tumor vs other painful neoplasms of the skin is unclear, such as in blue rubber bleb nevus syndrome, angioleiomyomas, neuromas, glomus tumors, leiomyomas, eccrine spiradenomas, congenital vascular malformations, schwannomas, or hemangiomas.4 Radiologic findings for glomus tumors may demonstrate cortical or cystic osseous defects. Magnetic resonance imaging and ultrasonography can help provide additional information on the lesion size and depth of involvement.1 Additionally, deeper glomangiomyomas have been associated with malignancy,2 potentially highlighting the benefit of early incorporation of imaging in the workup for this condition. Malignant transformation is rare and has been reported in less than 1% of cases.6
Treatment of glomus tumors predominantly is directed to the patient’s symptoms; asymptomatic lesions may be monitored.4 For symptomatic lesions, therapeutic options include wide local excision; sclerotherapy; and incorporation of various lasers, including Nd:YAG, CO2, and flashlamp tunable dye laser.4,5 One case report documented use of a PDL that successfully eliminated the pain associated with glomangiomyoma; however, the lesion in that report was not biopsy proven.11
Our case highlights the need to consider glomus tumors in patients presenting with multiple small nodules given the potential for misdiagnosis, impact on quality of life with associated psychological distress, and potential utility of incorporating PDL in treatment. Although our patient did not report clinical improvement in the appearance of the lesions with PDL therapy, additional treatment sessions may have helped,11 but he opted to discontinue. Follow-up for persistently symptomatic or changing lesions is necessary, given the minimal risk for malignant transformation.6
- Lee DY, Hwang SC, Jeong ST, et al. The value of diagnostic ultrasonography in the assessment of a glomus tumor of the subcutaneous layer of the forearm mimicking a hemangioma: a case report. J Med Case Rep. 2015;9:191. doi:10.1186/s13256-015-0672-y
- Li L, Bardsley V, Grainger A, et al. Extradigital glomangiomyoma of the forearm mimicking peripheral nerve sheath tumour and thrombosed varicose vein. BMJ Case Rep. 2021;14:E241221. doi: 10.1136 /bcr-2020-241221
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mohammadi O, Suarez M. Glomus cancer. StatPearls [Internet]. StatPearls Publishing; 2021.
- Maxey ML, Houghton CC, Mastriani KS, et al. Large prepatellar glomangioma: a case report [published online July 10, 2015]. Int J Surg Case Rep. 2015;14:80-84. doi:10.1016/j.ijscr.2015.07.002
- Brathwaite CD, Poppiti RJ Jr. Malignant glomus tumor. a case report of widespread metastases in a patient with multiple glomus body hamartomas. Am J Surg Pathol. 1996;20:233-238. doi:10.1097/00000478-199602000-00012
- Davis DD, Kane SM. Neurilemmoma. StatPearls [Internet]. StatPearls Publishing; 2022.
- Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary hemorrhagic telangiectasia. Otolaryngol Clin North Am. 2018;51:237-254. doi:10.1016/j.otc.2017.09.017
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. StatPearls [Internet]. StatPearls Publishing; 2022.
- Antony FC, Cliff S, Cowley N. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol. 2003;28:617-619. doi:10.1046/j.1365-2230.2003.01403.x
- Lee DY, Hwang SC, Jeong ST, et al. The value of diagnostic ultrasonography in the assessment of a glomus tumor of the subcutaneous layer of the forearm mimicking a hemangioma: a case report. J Med Case Rep. 2015;9:191. doi:10.1186/s13256-015-0672-y
- Li L, Bardsley V, Grainger A, et al. Extradigital glomangiomyoma of the forearm mimicking peripheral nerve sheath tumour and thrombosed varicose vein. BMJ Case Rep. 2021;14:E241221. doi: 10.1136 /bcr-2020-241221
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mohammadi O, Suarez M. Glomus cancer. StatPearls [Internet]. StatPearls Publishing; 2021.
- Maxey ML, Houghton CC, Mastriani KS, et al. Large prepatellar glomangioma: a case report [published online July 10, 2015]. Int J Surg Case Rep. 2015;14:80-84. doi:10.1016/j.ijscr.2015.07.002
- Brathwaite CD, Poppiti RJ Jr. Malignant glomus tumor. a case report of widespread metastases in a patient with multiple glomus body hamartomas. Am J Surg Pathol. 1996;20:233-238. doi:10.1097/00000478-199602000-00012
- Davis DD, Kane SM. Neurilemmoma. StatPearls [Internet]. StatPearls Publishing; 2022.
- Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary hemorrhagic telangiectasia. Otolaryngol Clin North Am. 2018;51:237-254. doi:10.1016/j.otc.2017.09.017
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. StatPearls [Internet]. StatPearls Publishing; 2022.
- Antony FC, Cliff S, Cowley N. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol. 2003;28:617-619. doi:10.1046/j.1365-2230.2003.01403.x
A 31-year-old active-duty military servicemember presented to the dermatology clinic for evaluation of 0.3- to 2-cm, tender, blue nodules on the wrists and forearms. The lesions first appeared on the right volar wrist secondary to a presumed injury sustained approximately 10 years prior to presentation and spread to the proximal forearm as well as the left wrist and forearm. He denied fevers, chills, chest pain, hematochezia, hematuria, or other skin findings. Physical examination revealed blue-violaceous, firm nodules on the right volar wrist and forearm that were tender to palpation. Blue-violaceous, papulonodular lesions on the left volar wrist and dorsal hand were not tender to palpation. A punch biopsy was performed.


What’s Eating You? Phlebotomine Sandflies and Leishmania Parasites
The genus Leishmania comprises protozoan parasites that cause approximately 2 million new cases of leishmaniasis each year across 98 countries.1 These protozoa are obligate intracellular parasites of phlebotomine sandfly species that transmit leishmaniasis and result in a considerable parasitic cause of fatalities globally, second only to malaria.2,3
Phlebotomine sandflies primarily live in tropical and subtropical regions and function as vectors for many pathogens in addition to Leishmania species, such as Bartonella species and arboviruses.3 In 2004, it was noted that the majority of leishmaniasis cases affected developing countries: 90% of visceral leishmaniasis cases occurred in Bangladesh, India, Nepal, Sudan, and Brazil, and 90% of cutaneous leishmaniasis cases occurred in Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia, and Syria.4 Of note, with recent environmental changes, phlebotomine sandflies have gradually migrated to more northerly latitudes, extending into Europe.5
Twenty Leishmania species and 30 sandfly species have been identified as causes of leishmaniasis.4 Leishmania infection occurs when an infected sandfly bites a mammalian host and transmits the parasite’s flagellated form, known as a promastigote. Host inflammatory cells, such as monocytes and dendritic cells, phagocytize parasites that enter the skin. The interaction between parasites and dendritic cells become an important factor in the outcome of Leishmania infection in the host because dendritic cells promote development of CD4 and CD8 T lymphocytes with specificity to target Leishmania parasites and protect the host.1
The number of cases of leishmaniasis has increased worldwide, most likely due to changes in the environment and human behaviors such as urbanization, the creation of new settlements, and migration from rural to urban areas.3,5 Important risk factors in individual patients include malnutrition; low-quality housing and sanitation; a history of migration or travel; and immunosuppression, such as that caused by HIV co-infection.2,5
Case Report
An otherwise healthy 25-year-old Bangladeshi man presented to our community hospital for evaluation of a painful leg ulcer of 1 month’s duration. The patient had migrated from Bangladesh to Panama, then to Costa Rica, followed by Guatemala, Honduras, Mexico, and, last, Texas. In Texas, he was identified by the US Immigration and Customs Enforcement, transported to a detention facility, and transferred to this hospital shortly afterward.
The patient reported that, during his extensive migration, he had lived in the jungle and reported what he described as mosquito bites on the legs. He subsequently developed a 3-cm ulcerated and crusted plaque with rolled borders on the right medial ankle (Figure 1). In addition, he had a palpable nodular cord on the medial leg from the ankle lesion to the mid thigh that was consistent with lymphocutaneous spread. Ultrasonography was negative for deep-vein thrombosis.

Because the patient’s recent migration from Central America was highly concerning for microbial infection, vancomycin and piperacillin-tazobactam were started empirically on admission. A punch biopsy from the right medial ankle was nondiagnostic, showing acute and chronic necrotizing inflammation along with numerous epithelioid histiocytes with a vaguely granulomatous appearance (Figure 2). A specimen from the right medial ankle that had already been taken by an astute border patrol medical provider was sent to the Centers for Disease Control and Prevention (CDC) for polymerase chain reaction analysis following admission and was found to be positive for Leishmania panamensis.
.")
Given the concern for mucocutaneous leishmaniasis with this particular species, otolaryngology was consulted; however, the patient did not demonstrate mucocutaneous disease. Because of the elevated risk for persistent disease with L panamensis, systemic therapy was indicated and administered: IV amphotericin B 200 mg on days 1 through 5 and again on day 10. Improvement in the ulcer was seen after the 10-day regimen was completed.
Comment
Leishmaniasis can be broadly classified by geographic region or clinical presentation. Under the geographic region system, leishmaniasis can be categorized as Old World or New World. Old World leishmaniasis primarily is transmitted by Phlebotomus sandflies and carries the parasites Leishmania major and Leishmania tropica, among others. New World leishmaniasis is caused by Lutzomyia sandflies, which carry Leishmania mexicana, Leishmania braziliensis, Leishmania amazonensis, and others.6
Our patient presented with cutaneous leishmaniasis, one of 4 primary clinical disease forms of leishmaniasis; the other 3 forms under this classification system are diffuse cutaneous, mucocutaneous, and visceral leishmaniasis, also known as kala-azar.3,6 Cutaneous leishmaniasis is limited to the skin, particularly the face and extremities. This form is more common with Old World vectors, with most cases occurring in Peru, Brazil, and the Middle East. In Old World cutaneous leishmaniasis, the disease begins with a solitary nodule at the site of the bite that ulcerates and can continue to spread in a sporotrichoid pattern. This cutaneous form tends to heal slowly over months to years with residual scarring. New World cutaneous leishmaniasis can present with a variety of clinical manifestations, including ulcerative, sarcoidlike, miliary, and nodular lesions.6,7
The diffuse form of cutaneous leishmaniasis begins in a similar manner to the Old World cutaneous form: a single nodule spreads widely over the body, especially the nose, and covers the patient’s skin with keloidal or verrucous lesions that do not ulcerate. These nodules contain large groupings of Leishmania-filled foamy macrophages. Often, patients with diffuse cutaneous leishmaniasis are immunosuppressed and are unable to develop an immune response to leishmanin and other skin antigens.6,7
Mucocutaneous leishmaniasis predominantly is caused by the New World species L braziliensis but also has been attributed to L amazonensis, L panamensis, and L guyanensis. This form manifests as mucosal lesions that can develop simultaneously with cutaneous lesions but more commonly appear months to years after resolution of the skin infection. Patients often present with ulceration of the lip, nose, and oropharynx, and destruction of the nasopharynx can result in severe consequences such as obstruction of the airway and perforation of the nasal septum (also known as espundia).6,7
The most severe presentation of leishmaniasis is the visceral form (kala-azar), which presents with parasitic infection of the liver, spleen, and bone marrow. Most commonly caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi, this form has a long incubation period spanning months to years before presenting with diarrhea, hepatomegaly, splenomegaly, darkening of the skin (in Hindi, kala-azar means “black fever”), pancytopenia, lymphadenopathy, nephritis, and intestinal hemorrhage, among other severe manifestations. Visceral leishmaniasis has a poor prognosis: patients succumb to disease within 2 years if not treated.6,7
Diagnosis—Diagnosing leishmaniasis starts with a complete personal and medical history, paying close attention to travel and exposures. Diagnosis is most successfully performed by polymerase chain reaction analysis, which is both highly sensitive and specific but also can be determined by culture using Novy-McNeal-Nicolle medium or by light microscopy. Histologic findings include the marquee sign, which describes an array of amastigotes (promastigotes that have developed into the intracellular tissue-stage form) with kinetoplasts surrounding the periphery of parasitized histiocytes. Giemsa staining can be helpful in identifying organisms.2,6,7
The diagnosis in our case was challenging, as none of the above findings were seen in our patient. The specimen taken by the border patrol medical provider was negative on Gram, Giemsa, and Grocott-Gömöri methenamine silver staining; no amastigotes were identified. Another diagnostic modality (not performed in our patient) is the Montenegro delayed skin-reaction test, which often is positive in patients with cutaneous leishmaniasis but also yields a positive result in patients who have been cured of Leishmania infection.6
An important consideration in the diagnostic workup of leishmaniasis is that collaboration with the CDC can be helpful, such as in our case, as they provide clear guidance for specimen collection and processing.2
Treatment—Treating leishmaniasis is challenging and complex. Even the initial decision to treat depends on several factors, including the form of infection. Most visceral and mucocutaneous infections should be treated due to both the lack of self-resolution of these forms and the higher risk for a potentially life-threatening disease course; in contrast, cutaneous forms require further consideration before initiating treatment. Some indicators for treating cutaneous leishmaniasis include widespread infection, intention to decrease scarring, and lesions with the potential to cause further complications (eg, on the face or ears or close to joints).6-8
The treatment of choice for cutaneous and mucocutaneous leishmaniasis is pentavalent antimony; however, this drug can only be obtained in the United States for investigational use, requiring approval by the CDC. A 20-day intravenous or intramuscular course of 20 mg/kg per day typically is used for cutaneous cases; a 28-day course typically is used for mucosal forms.
Amphotericin B is not only the treatment of choice for visceral leishmaniasis but also is an important alternative therapy for patients with mucosal leishmaniasis or who are co-infected with HIV. Patients with visceral infection also should receive supportive care for any concomitant afflictions, such as malnutrition or other infections. Although different regimens have been described, the US Food and Drug Administration has created outlines of specific intravenous infusion schedules for liposomal amphotericin B in immunocompetent and immunosuppressed patients.8 Liposomal amphotericin B also has a more favorable toxicity profile than conventional amphotericin B deoxycholate, which is otherwise effective in combating visceral leishmaniasis.6-8
Other treatments that have been attempted include pentamidine, miltefosine, thermotherapy, oral itraconazole and fluconazole, rifampicin, metronidazole and cotrimoxazole, dapsone, photodynamic therapy, thermotherapy, topical paromomycin formulations, intralesional pentavalent antimony, and laser cryotherapy. Notable among these other agents is miltefosine, a US Food and Drug Administration–approved oral medication for adults and adolescents (used off-label for patients younger than 12 years) with cutaneous leishmaniasis caused by L braziliensis, L panamensis, or L guyanensis. Other oral options mentioned include the so-called azole antifungal medications, which historically have produced variable results. From the CDC’s reports, ketoconazole was moderately effective in Guatemala and Panama,8 whereas itraconazole did not demonstrate efficacy in Colombia, and the efficacy of fluconazole was inconsistent in different countries.8 When considering one of the local (as opposed to oral and parenteral) therapies mentioned, the extent of cutaneous findings as well as the risk of mucosal spread should be factored in.6-8
Understandably, a number of considerations can come into play in determining the appropriate treatment modality, including body region affected, clinical form, severity, and Leishmania species.6-8 Our case is of particular interest because it demonstrates the complexities behind the diagnosis and treatment of cutaneous leishmaniasis, with careful consideration geared toward the species; for example, because our patient was infected with L panamensis, which is known to cause mucocutaneous disease, the infectious disease service decided to pursue systemic therapy with amphotericin B rather than topical treatment.
Prevention—Vector control is the primary means of preventing leishmaniasis under 2 umbrellas: environmental management and synthetic insecticides. The goal of environmental management is to eliminate the phlebotomine sandfly habitat; this was the primary method of vector control until 1940. Until that time, tree stumps were removed, indoor cracks and crevices were filled to prevent sandfly emergence, and areas around animal shelters were cleaned. These methods were highly dependent on community awareness and involvement; today, they can be combined with synthetic insecticides to offer maximum protection.
Synthetic insecticides include indoor sprays, treated nets, repellents, and impregnated dog collars, all of which control sandflies. However, the use of these insecticides in endemic areas, such as India, has driven development of insecticide resistance in many sandfly vector species.3
As of 2020, 5 vaccines against Leishmania have been created. Two are approved–one in Brazil and one in Uzbekistan–for human use as immunotherapy, while the other 3 have been developed to immunize dogs in Brazil. However, the effectiveness of these vaccines is under debate. First, one of the vaccines used as immunotherapy for cutaneous leishmaniasis must be used in combination with conventional chemotherapy; second, long-term effects of the canine vaccine are unknown.1 A preventive vaccine for humans is under development.1,3
Final Thoughts
Leishmaniasis remains a notable parasitic disease that is increasing in prevalence worldwide. Clinicians should be aware of this disease because early detection and treatment are essential to control infection.3 Health care providers in the United States should be especially aware of this condition among patients who have a history of travel or migration; those in Texas should recognize the current endemic status of leishmaniasis there.4,6
- Coutinho De Oliveira B, Duthie MS, Alves Pereira VR. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum Vaccin Immunother. 2020;16:919-930. doi:10.1080/21645515.2019.1678998
- Chan CX, Simmons BJ, Call JE, et al. Cutaneous leishmaniasis successfully treated with miltefosine. Cutis. 2020;106:206-209. doi:10.12788/cutis.0086
- Balaska S, Fotakis EA, Chaskopoulou A, et al. Chemical control and insecticide resistance status of sand fly vectors worldwide. PLoS Negl Trop Dis. 2021;15:E0009586. doi:10.1371/journal.pntd.0009586
- Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692. doi:10.1038/nrmicro981
- Michelutti A, Toniolo F, Bertola M, et al. Occurrence of Phlebotomine sand flies (Diptera: Psychodidae) in the northeastern plain of Italy. Parasit Vectors. 2021;14:164. doi:10.1186/s13071-021-04652-2
- Alkihan A, Hocker TLH. Infectious diseases: parasites and other creatures: protozoa. In: Alikhan A, Hocker TLH, eds. Review of Dermatology. Elsevier; 2024:329-331.
- Dinulos JGH. Infestations and bites. In: Habif TP, ed. Clinical Dermatology. Elsevier; 2016:630-634.
- Centers for Disease Control and Prevention. Leishmaniasis: resources for health professionals. US Department of Health and Human Services. March 20, 2023. Accessed October 5, 2023. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#:~:text=Liposomal%20amphotericin%20B%20is%20FDA,treatment%20of%20choice%20for%20U.S
The genus Leishmania comprises protozoan parasites that cause approximately 2 million new cases of leishmaniasis each year across 98 countries.1 These protozoa are obligate intracellular parasites of phlebotomine sandfly species that transmit leishmaniasis and result in a considerable parasitic cause of fatalities globally, second only to malaria.2,3
Phlebotomine sandflies primarily live in tropical and subtropical regions and function as vectors for many pathogens in addition to Leishmania species, such as Bartonella species and arboviruses.3 In 2004, it was noted that the majority of leishmaniasis cases affected developing countries: 90% of visceral leishmaniasis cases occurred in Bangladesh, India, Nepal, Sudan, and Brazil, and 90% of cutaneous leishmaniasis cases occurred in Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia, and Syria.4 Of note, with recent environmental changes, phlebotomine sandflies have gradually migrated to more northerly latitudes, extending into Europe.5
Twenty Leishmania species and 30 sandfly species have been identified as causes of leishmaniasis.4 Leishmania infection occurs when an infected sandfly bites a mammalian host and transmits the parasite’s flagellated form, known as a promastigote. Host inflammatory cells, such as monocytes and dendritic cells, phagocytize parasites that enter the skin. The interaction between parasites and dendritic cells become an important factor in the outcome of Leishmania infection in the host because dendritic cells promote development of CD4 and CD8 T lymphocytes with specificity to target Leishmania parasites and protect the host.1
The number of cases of leishmaniasis has increased worldwide, most likely due to changes in the environment and human behaviors such as urbanization, the creation of new settlements, and migration from rural to urban areas.3,5 Important risk factors in individual patients include malnutrition; low-quality housing and sanitation; a history of migration or travel; and immunosuppression, such as that caused by HIV co-infection.2,5
Case Report
An otherwise healthy 25-year-old Bangladeshi man presented to our community hospital for evaluation of a painful leg ulcer of 1 month’s duration. The patient had migrated from Bangladesh to Panama, then to Costa Rica, followed by Guatemala, Honduras, Mexico, and, last, Texas. In Texas, he was identified by the US Immigration and Customs Enforcement, transported to a detention facility, and transferred to this hospital shortly afterward.
The patient reported that, during his extensive migration, he had lived in the jungle and reported what he described as mosquito bites on the legs. He subsequently developed a 3-cm ulcerated and crusted plaque with rolled borders on the right medial ankle (Figure 1). In addition, he had a palpable nodular cord on the medial leg from the ankle lesion to the mid thigh that was consistent with lymphocutaneous spread. Ultrasonography was negative for deep-vein thrombosis.
Because the patient’s recent migration from Central America was highly concerning for microbial infection, vancomycin and piperacillin-tazobactam were started empirically on admission. A punch biopsy from the right medial ankle was nondiagnostic, showing acute and chronic necrotizing inflammation along with numerous epithelioid histiocytes with a vaguely granulomatous appearance (Figure 2). A specimen from the right medial ankle that had already been taken by an astute border patrol medical provider was sent to the Centers for Disease Control and Prevention (CDC) for polymerase chain reaction analysis following admission and was found to be positive for Leishmania panamensis.
Given the concern for mucocutaneous leishmaniasis with this particular species, otolaryngology was consulted; however, the patient did not demonstrate mucocutaneous disease. Because of the elevated risk for persistent disease with L panamensis, systemic therapy was indicated and administered: IV amphotericin B 200 mg on days 1 through 5 and again on day 10. Improvement in the ulcer was seen after the 10-day regimen was completed.
Comment
Leishmaniasis can be broadly classified by geographic region or clinical presentation. Under the geographic region system, leishmaniasis can be categorized as Old World or New World. Old World leishmaniasis primarily is transmitted by Phlebotomus sandflies and carries the parasites Leishmania major and Leishmania tropica, among others. New World leishmaniasis is caused by Lutzomyia sandflies, which carry Leishmania mexicana, Leishmania braziliensis, Leishmania amazonensis, and others.6
Our patient presented with cutaneous leishmaniasis, one of 4 primary clinical disease forms of leishmaniasis; the other 3 forms under this classification system are diffuse cutaneous, mucocutaneous, and visceral leishmaniasis, also known as kala-azar.3,6 Cutaneous leishmaniasis is limited to the skin, particularly the face and extremities. This form is more common with Old World vectors, with most cases occurring in Peru, Brazil, and the Middle East. In Old World cutaneous leishmaniasis, the disease begins with a solitary nodule at the site of the bite that ulcerates and can continue to spread in a sporotrichoid pattern. This cutaneous form tends to heal slowly over months to years with residual scarring. New World cutaneous leishmaniasis can present with a variety of clinical manifestations, including ulcerative, sarcoidlike, miliary, and nodular lesions.6,7
The diffuse form of cutaneous leishmaniasis begins in a similar manner to the Old World cutaneous form: a single nodule spreads widely over the body, especially the nose, and covers the patient’s skin with keloidal or verrucous lesions that do not ulcerate. These nodules contain large groupings of Leishmania-filled foamy macrophages. Often, patients with diffuse cutaneous leishmaniasis are immunosuppressed and are unable to develop an immune response to leishmanin and other skin antigens.6,7
Mucocutaneous leishmaniasis predominantly is caused by the New World species L braziliensis but also has been attributed to L amazonensis, L panamensis, and L guyanensis. This form manifests as mucosal lesions that can develop simultaneously with cutaneous lesions but more commonly appear months to years after resolution of the skin infection. Patients often present with ulceration of the lip, nose, and oropharynx, and destruction of the nasopharynx can result in severe consequences such as obstruction of the airway and perforation of the nasal septum (also known as espundia).6,7
The most severe presentation of leishmaniasis is the visceral form (kala-azar), which presents with parasitic infection of the liver, spleen, and bone marrow. Most commonly caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi, this form has a long incubation period spanning months to years before presenting with diarrhea, hepatomegaly, splenomegaly, darkening of the skin (in Hindi, kala-azar means “black fever”), pancytopenia, lymphadenopathy, nephritis, and intestinal hemorrhage, among other severe manifestations. Visceral leishmaniasis has a poor prognosis: patients succumb to disease within 2 years if not treated.6,7
Diagnosis—Diagnosing leishmaniasis starts with a complete personal and medical history, paying close attention to travel and exposures. Diagnosis is most successfully performed by polymerase chain reaction analysis, which is both highly sensitive and specific but also can be determined by culture using Novy-McNeal-Nicolle medium or by light microscopy. Histologic findings include the marquee sign, which describes an array of amastigotes (promastigotes that have developed into the intracellular tissue-stage form) with kinetoplasts surrounding the periphery of parasitized histiocytes. Giemsa staining can be helpful in identifying organisms.2,6,7
The diagnosis in our case was challenging, as none of the above findings were seen in our patient. The specimen taken by the border patrol medical provider was negative on Gram, Giemsa, and Grocott-Gömöri methenamine silver staining; no amastigotes were identified. Another diagnostic modality (not performed in our patient) is the Montenegro delayed skin-reaction test, which often is positive in patients with cutaneous leishmaniasis but also yields a positive result in patients who have been cured of Leishmania infection.6
An important consideration in the diagnostic workup of leishmaniasis is that collaboration with the CDC can be helpful, such as in our case, as they provide clear guidance for specimen collection and processing.2
Treatment—Treating leishmaniasis is challenging and complex. Even the initial decision to treat depends on several factors, including the form of infection. Most visceral and mucocutaneous infections should be treated due to both the lack of self-resolution of these forms and the higher risk for a potentially life-threatening disease course; in contrast, cutaneous forms require further consideration before initiating treatment. Some indicators for treating cutaneous leishmaniasis include widespread infection, intention to decrease scarring, and lesions with the potential to cause further complications (eg, on the face or ears or close to joints).6-8
The treatment of choice for cutaneous and mucocutaneous leishmaniasis is pentavalent antimony; however, this drug can only be obtained in the United States for investigational use, requiring approval by the CDC. A 20-day intravenous or intramuscular course of 20 mg/kg per day typically is used for cutaneous cases; a 28-day course typically is used for mucosal forms.
Amphotericin B is not only the treatment of choice for visceral leishmaniasis but also is an important alternative therapy for patients with mucosal leishmaniasis or who are co-infected with HIV. Patients with visceral infection also should receive supportive care for any concomitant afflictions, such as malnutrition or other infections. Although different regimens have been described, the US Food and Drug Administration has created outlines of specific intravenous infusion schedules for liposomal amphotericin B in immunocompetent and immunosuppressed patients.8 Liposomal amphotericin B also has a more favorable toxicity profile than conventional amphotericin B deoxycholate, which is otherwise effective in combating visceral leishmaniasis.6-8
Other treatments that have been attempted include pentamidine, miltefosine, thermotherapy, oral itraconazole and fluconazole, rifampicin, metronidazole and cotrimoxazole, dapsone, photodynamic therapy, thermotherapy, topical paromomycin formulations, intralesional pentavalent antimony, and laser cryotherapy. Notable among these other agents is miltefosine, a US Food and Drug Administration–approved oral medication for adults and adolescents (used off-label for patients younger than 12 years) with cutaneous leishmaniasis caused by L braziliensis, L panamensis, or L guyanensis. Other oral options mentioned include the so-called azole antifungal medications, which historically have produced variable results. From the CDC’s reports, ketoconazole was moderately effective in Guatemala and Panama,8 whereas itraconazole did not demonstrate efficacy in Colombia, and the efficacy of fluconazole was inconsistent in different countries.8 When considering one of the local (as opposed to oral and parenteral) therapies mentioned, the extent of cutaneous findings as well as the risk of mucosal spread should be factored in.6-8
Understandably, a number of considerations can come into play in determining the appropriate treatment modality, including body region affected, clinical form, severity, and Leishmania species.6-8 Our case is of particular interest because it demonstrates the complexities behind the diagnosis and treatment of cutaneous leishmaniasis, with careful consideration geared toward the species; for example, because our patient was infected with L panamensis, which is known to cause mucocutaneous disease, the infectious disease service decided to pursue systemic therapy with amphotericin B rather than topical treatment.
Prevention—Vector control is the primary means of preventing leishmaniasis under 2 umbrellas: environmental management and synthetic insecticides. The goal of environmental management is to eliminate the phlebotomine sandfly habitat; this was the primary method of vector control until 1940. Until that time, tree stumps were removed, indoor cracks and crevices were filled to prevent sandfly emergence, and areas around animal shelters were cleaned. These methods were highly dependent on community awareness and involvement; today, they can be combined with synthetic insecticides to offer maximum protection.
Synthetic insecticides include indoor sprays, treated nets, repellents, and impregnated dog collars, all of which control sandflies. However, the use of these insecticides in endemic areas, such as India, has driven development of insecticide resistance in many sandfly vector species.3
As of 2020, 5 vaccines against Leishmania have been created. Two are approved–one in Brazil and one in Uzbekistan–for human use as immunotherapy, while the other 3 have been developed to immunize dogs in Brazil. However, the effectiveness of these vaccines is under debate. First, one of the vaccines used as immunotherapy for cutaneous leishmaniasis must be used in combination with conventional chemotherapy; second, long-term effects of the canine vaccine are unknown.1 A preventive vaccine for humans is under development.1,3
Final Thoughts
Leishmaniasis remains a notable parasitic disease that is increasing in prevalence worldwide. Clinicians should be aware of this disease because early detection and treatment are essential to control infection.3 Health care providers in the United States should be especially aware of this condition among patients who have a history of travel or migration; those in Texas should recognize the current endemic status of leishmaniasis there.4,6
The genus Leishmania comprises protozoan parasites that cause approximately 2 million new cases of leishmaniasis each year across 98 countries.1 These protozoa are obligate intracellular parasites of phlebotomine sandfly species that transmit leishmaniasis and result in a considerable parasitic cause of fatalities globally, second only to malaria.2,3
Phlebotomine sandflies primarily live in tropical and subtropical regions and function as vectors for many pathogens in addition to Leishmania species, such as Bartonella species and arboviruses.3 In 2004, it was noted that the majority of leishmaniasis cases affected developing countries: 90% of visceral leishmaniasis cases occurred in Bangladesh, India, Nepal, Sudan, and Brazil, and 90% of cutaneous leishmaniasis cases occurred in Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia, and Syria.4 Of note, with recent environmental changes, phlebotomine sandflies have gradually migrated to more northerly latitudes, extending into Europe.5
Twenty Leishmania species and 30 sandfly species have been identified as causes of leishmaniasis.4 Leishmania infection occurs when an infected sandfly bites a mammalian host and transmits the parasite’s flagellated form, known as a promastigote. Host inflammatory cells, such as monocytes and dendritic cells, phagocytize parasites that enter the skin. The interaction between parasites and dendritic cells become an important factor in the outcome of Leishmania infection in the host because dendritic cells promote development of CD4 and CD8 T lymphocytes with specificity to target Leishmania parasites and protect the host.1
The number of cases of leishmaniasis has increased worldwide, most likely due to changes in the environment and human behaviors such as urbanization, the creation of new settlements, and migration from rural to urban areas.3,5 Important risk factors in individual patients include malnutrition; low-quality housing and sanitation; a history of migration or travel; and immunosuppression, such as that caused by HIV co-infection.2,5
Case Report
An otherwise healthy 25-year-old Bangladeshi man presented to our community hospital for evaluation of a painful leg ulcer of 1 month’s duration. The patient had migrated from Bangladesh to Panama, then to Costa Rica, followed by Guatemala, Honduras, Mexico, and, last, Texas. In Texas, he was identified by the US Immigration and Customs Enforcement, transported to a detention facility, and transferred to this hospital shortly afterward.
The patient reported that, during his extensive migration, he had lived in the jungle and reported what he described as mosquito bites on the legs. He subsequently developed a 3-cm ulcerated and crusted plaque with rolled borders on the right medial ankle (Figure 1). In addition, he had a palpable nodular cord on the medial leg from the ankle lesion to the mid thigh that was consistent with lymphocutaneous spread. Ultrasonography was negative for deep-vein thrombosis.
Because the patient’s recent migration from Central America was highly concerning for microbial infection, vancomycin and piperacillin-tazobactam were started empirically on admission. A punch biopsy from the right medial ankle was nondiagnostic, showing acute and chronic necrotizing inflammation along with numerous epithelioid histiocytes with a vaguely granulomatous appearance (Figure 2). A specimen from the right medial ankle that had already been taken by an astute border patrol medical provider was sent to the Centers for Disease Control and Prevention (CDC) for polymerase chain reaction analysis following admission and was found to be positive for Leishmania panamensis.
Given the concern for mucocutaneous leishmaniasis with this particular species, otolaryngology was consulted; however, the patient did not demonstrate mucocutaneous disease. Because of the elevated risk for persistent disease with L panamensis, systemic therapy was indicated and administered: IV amphotericin B 200 mg on days 1 through 5 and again on day 10. Improvement in the ulcer was seen after the 10-day regimen was completed.
Comment
Leishmaniasis can be broadly classified by geographic region or clinical presentation. Under the geographic region system, leishmaniasis can be categorized as Old World or New World. Old World leishmaniasis primarily is transmitted by Phlebotomus sandflies and carries the parasites Leishmania major and Leishmania tropica, among others. New World leishmaniasis is caused by Lutzomyia sandflies, which carry Leishmania mexicana, Leishmania braziliensis, Leishmania amazonensis, and others.6
Our patient presented with cutaneous leishmaniasis, one of 4 primary clinical disease forms of leishmaniasis; the other 3 forms under this classification system are diffuse cutaneous, mucocutaneous, and visceral leishmaniasis, also known as kala-azar.3,6 Cutaneous leishmaniasis is limited to the skin, particularly the face and extremities. This form is more common with Old World vectors, with most cases occurring in Peru, Brazil, and the Middle East. In Old World cutaneous leishmaniasis, the disease begins with a solitary nodule at the site of the bite that ulcerates and can continue to spread in a sporotrichoid pattern. This cutaneous form tends to heal slowly over months to years with residual scarring. New World cutaneous leishmaniasis can present with a variety of clinical manifestations, including ulcerative, sarcoidlike, miliary, and nodular lesions.6,7
The diffuse form of cutaneous leishmaniasis begins in a similar manner to the Old World cutaneous form: a single nodule spreads widely over the body, especially the nose, and covers the patient’s skin with keloidal or verrucous lesions that do not ulcerate. These nodules contain large groupings of Leishmania-filled foamy macrophages. Often, patients with diffuse cutaneous leishmaniasis are immunosuppressed and are unable to develop an immune response to leishmanin and other skin antigens.6,7
Mucocutaneous leishmaniasis predominantly is caused by the New World species L braziliensis but also has been attributed to L amazonensis, L panamensis, and L guyanensis. This form manifests as mucosal lesions that can develop simultaneously with cutaneous lesions but more commonly appear months to years after resolution of the skin infection. Patients often present with ulceration of the lip, nose, and oropharynx, and destruction of the nasopharynx can result in severe consequences such as obstruction of the airway and perforation of the nasal septum (also known as espundia).6,7
The most severe presentation of leishmaniasis is the visceral form (kala-azar), which presents with parasitic infection of the liver, spleen, and bone marrow. Most commonly caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi, this form has a long incubation period spanning months to years before presenting with diarrhea, hepatomegaly, splenomegaly, darkening of the skin (in Hindi, kala-azar means “black fever”), pancytopenia, lymphadenopathy, nephritis, and intestinal hemorrhage, among other severe manifestations. Visceral leishmaniasis has a poor prognosis: patients succumb to disease within 2 years if not treated.6,7
Diagnosis—Diagnosing leishmaniasis starts with a complete personal and medical history, paying close attention to travel and exposures. Diagnosis is most successfully performed by polymerase chain reaction analysis, which is both highly sensitive and specific but also can be determined by culture using Novy-McNeal-Nicolle medium or by light microscopy. Histologic findings include the marquee sign, which describes an array of amastigotes (promastigotes that have developed into the intracellular tissue-stage form) with kinetoplasts surrounding the periphery of parasitized histiocytes. Giemsa staining can be helpful in identifying organisms.2,6,7
The diagnosis in our case was challenging, as none of the above findings were seen in our patient. The specimen taken by the border patrol medical provider was negative on Gram, Giemsa, and Grocott-Gömöri methenamine silver staining; no amastigotes were identified. Another diagnostic modality (not performed in our patient) is the Montenegro delayed skin-reaction test, which often is positive in patients with cutaneous leishmaniasis but also yields a positive result in patients who have been cured of Leishmania infection.6
An important consideration in the diagnostic workup of leishmaniasis is that collaboration with the CDC can be helpful, such as in our case, as they provide clear guidance for specimen collection and processing.2
Treatment—Treating leishmaniasis is challenging and complex. Even the initial decision to treat depends on several factors, including the form of infection. Most visceral and mucocutaneous infections should be treated due to both the lack of self-resolution of these forms and the higher risk for a potentially life-threatening disease course; in contrast, cutaneous forms require further consideration before initiating treatment. Some indicators for treating cutaneous leishmaniasis include widespread infection, intention to decrease scarring, and lesions with the potential to cause further complications (eg, on the face or ears or close to joints).6-8
The treatment of choice for cutaneous and mucocutaneous leishmaniasis is pentavalent antimony; however, this drug can only be obtained in the United States for investigational use, requiring approval by the CDC. A 20-day intravenous or intramuscular course of 20 mg/kg per day typically is used for cutaneous cases; a 28-day course typically is used for mucosal forms.
Amphotericin B is not only the treatment of choice for visceral leishmaniasis but also is an important alternative therapy for patients with mucosal leishmaniasis or who are co-infected with HIV. Patients with visceral infection also should receive supportive care for any concomitant afflictions, such as malnutrition or other infections. Although different regimens have been described, the US Food and Drug Administration has created outlines of specific intravenous infusion schedules for liposomal amphotericin B in immunocompetent and immunosuppressed patients.8 Liposomal amphotericin B also has a more favorable toxicity profile than conventional amphotericin B deoxycholate, which is otherwise effective in combating visceral leishmaniasis.6-8
Other treatments that have been attempted include pentamidine, miltefosine, thermotherapy, oral itraconazole and fluconazole, rifampicin, metronidazole and cotrimoxazole, dapsone, photodynamic therapy, thermotherapy, topical paromomycin formulations, intralesional pentavalent antimony, and laser cryotherapy. Notable among these other agents is miltefosine, a US Food and Drug Administration–approved oral medication for adults and adolescents (used off-label for patients younger than 12 years) with cutaneous leishmaniasis caused by L braziliensis, L panamensis, or L guyanensis. Other oral options mentioned include the so-called azole antifungal medications, which historically have produced variable results. From the CDC’s reports, ketoconazole was moderately effective in Guatemala and Panama,8 whereas itraconazole did not demonstrate efficacy in Colombia, and the efficacy of fluconazole was inconsistent in different countries.8 When considering one of the local (as opposed to oral and parenteral) therapies mentioned, the extent of cutaneous findings as well as the risk of mucosal spread should be factored in.6-8
Understandably, a number of considerations can come into play in determining the appropriate treatment modality, including body region affected, clinical form, severity, and Leishmania species.6-8 Our case is of particular interest because it demonstrates the complexities behind the diagnosis and treatment of cutaneous leishmaniasis, with careful consideration geared toward the species; for example, because our patient was infected with L panamensis, which is known to cause mucocutaneous disease, the infectious disease service decided to pursue systemic therapy with amphotericin B rather than topical treatment.
Prevention—Vector control is the primary means of preventing leishmaniasis under 2 umbrellas: environmental management and synthetic insecticides. The goal of environmental management is to eliminate the phlebotomine sandfly habitat; this was the primary method of vector control until 1940. Until that time, tree stumps were removed, indoor cracks and crevices were filled to prevent sandfly emergence, and areas around animal shelters were cleaned. These methods were highly dependent on community awareness and involvement; today, they can be combined with synthetic insecticides to offer maximum protection.
Synthetic insecticides include indoor sprays, treated nets, repellents, and impregnated dog collars, all of which control sandflies. However, the use of these insecticides in endemic areas, such as India, has driven development of insecticide resistance in many sandfly vector species.3
As of 2020, 5 vaccines against Leishmania have been created. Two are approved–one in Brazil and one in Uzbekistan–for human use as immunotherapy, while the other 3 have been developed to immunize dogs in Brazil. However, the effectiveness of these vaccines is under debate. First, one of the vaccines used as immunotherapy for cutaneous leishmaniasis must be used in combination with conventional chemotherapy; second, long-term effects of the canine vaccine are unknown.1 A preventive vaccine for humans is under development.1,3
Final Thoughts
Leishmaniasis remains a notable parasitic disease that is increasing in prevalence worldwide. Clinicians should be aware of this disease because early detection and treatment are essential to control infection.3 Health care providers in the United States should be especially aware of this condition among patients who have a history of travel or migration; those in Texas should recognize the current endemic status of leishmaniasis there.4,6
- Coutinho De Oliveira B, Duthie MS, Alves Pereira VR. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum Vaccin Immunother. 2020;16:919-930. doi:10.1080/21645515.2019.1678998
- Chan CX, Simmons BJ, Call JE, et al. Cutaneous leishmaniasis successfully treated with miltefosine. Cutis. 2020;106:206-209. doi:10.12788/cutis.0086
- Balaska S, Fotakis EA, Chaskopoulou A, et al. Chemical control and insecticide resistance status of sand fly vectors worldwide. PLoS Negl Trop Dis. 2021;15:E0009586. doi:10.1371/journal.pntd.0009586
- Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692. doi:10.1038/nrmicro981
- Michelutti A, Toniolo F, Bertola M, et al. Occurrence of Phlebotomine sand flies (Diptera: Psychodidae) in the northeastern plain of Italy. Parasit Vectors. 2021;14:164. doi:10.1186/s13071-021-04652-2
- Alkihan A, Hocker TLH. Infectious diseases: parasites and other creatures: protozoa. In: Alikhan A, Hocker TLH, eds. Review of Dermatology. Elsevier; 2024:329-331.
- Dinulos JGH. Infestations and bites. In: Habif TP, ed. Clinical Dermatology. Elsevier; 2016:630-634.
- Centers for Disease Control and Prevention. Leishmaniasis: resources for health professionals. US Department of Health and Human Services. March 20, 2023. Accessed October 5, 2023. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#:~:text=Liposomal%20amphotericin%20B%20is%20FDA,treatment%20of%20choice%20for%20U.S
- Coutinho De Oliveira B, Duthie MS, Alves Pereira VR. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum Vaccin Immunother. 2020;16:919-930. doi:10.1080/21645515.2019.1678998
- Chan CX, Simmons BJ, Call JE, et al. Cutaneous leishmaniasis successfully treated with miltefosine. Cutis. 2020;106:206-209. doi:10.12788/cutis.0086
- Balaska S, Fotakis EA, Chaskopoulou A, et al. Chemical control and insecticide resistance status of sand fly vectors worldwide. PLoS Negl Trop Dis. 2021;15:E0009586. doi:10.1371/journal.pntd.0009586
- Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692. doi:10.1038/nrmicro981
- Michelutti A, Toniolo F, Bertola M, et al. Occurrence of Phlebotomine sand flies (Diptera: Psychodidae) in the northeastern plain of Italy. Parasit Vectors. 2021;14:164. doi:10.1186/s13071-021-04652-2
- Alkihan A, Hocker TLH. Infectious diseases: parasites and other creatures: protozoa. In: Alikhan A, Hocker TLH, eds. Review of Dermatology. Elsevier; 2024:329-331.
- Dinulos JGH. Infestations and bites. In: Habif TP, ed. Clinical Dermatology. Elsevier; 2016:630-634.
- Centers for Disease Control and Prevention. Leishmaniasis: resources for health professionals. US Department of Health and Human Services. March 20, 2023. Accessed October 5, 2023. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#:~:text=Liposomal%20amphotericin%20B%20is%20FDA,treatment%20of%20choice%20for%20U.S
Practice Points
- The Phlebotomus and Lutzomyia genera of sandflies are vectors of Leishmania parasites, which can result in an array of clinical findings associated with leishmaniasis.
- Treatment options for leishmaniasis differ based on whether the infection is considered uncomplicated or complicated, which depends on the species of Leishmania; the number, size, and location of the lesion(s); and host immune status.
- All US practitioners should be aware of this pathogen, especially with regard to patients who have a history of travel to other countries. Health care professionals in states such as Texas and Oklahoma should be especially cognizant because these constitute one of the few areas in the United States where locally acquired cases of leishmaniasis have been reported.

Atopic Dermatitis Triggered by Omalizumab and Treated With Dupilumab
To the Editor:
A 16-year-old adolescent boy presented to our pediatric dermatology clinic for evaluation of long-standing mild atopic dermatitis (AD) that had become severe over the last year after omalizumab was initiated for severe asthma. The patient had a history of multiple hospitalizations for severe asthma. Despite excellent control of asthma with omalizumab given every 2 weeks, he developed widespread eczematous plaques on the neck, trunk, and extremities over the course of a year. The AD often was complicated by superimposed folliculitis due to scratching from severe pruritus. Treatment with topical corticosteroids including triamcinolone ointment 0.1% to AD on the body, plus clobetasol ointment 0.05% for prurigolike lesions on the legs resulted in modest improvement; however, the AD consistently recurred within a few days after the biweekly omalizumab injection (Figure 1). When the omalizumab injections were delayed, the flares temporarily improved, and when injections were decreased to once monthly, the exacerbations subsided partially but not fully.

Because omalizumab resulted in dramatic improvement in the patient’s asthma, there was hesitation to discontinue it initially; however, the patient and his parents in conjunction with the dermatology and pulmonary teams decided to transition to dupilumab. The patient reported vast improvement of AD 1 month after initiation of dupilumab (Figure 2), which remained well controlled more than 1 year later. Mid-potency topical corticosteroids for the treatment of occasional mild eczematous flares on the extremities were used. The patient’s asthma has remained well controlled on dupilumab without any exacerbations.

Omalizumab is a recombinant DNA-derived humanized monoclonal antibody that binds both circulating and membrane-bound IgE. It has been proposed as a possible treatment for severe and/or recalcitrant AD, with mixed treatment results.1 A case series and review of 174 patients demonstrated a moderate to complete AD response to treatment with omalizumab in 74.1% of patients.2 The Atopic Dermatitis Anti-IgE Pediatric Trial (ADAPT) showed a statistically significant reduction in the Scoring Atopic Dermatitis (SCORAD) index (P=.01), along with improved quality of life in children treated with omalizumab vs those treated with placebo.3 However, a prior randomized, placebo-controlled, double-blind study did not show a significant difference in clinical disease parameters in patients treated with omalizumab.4
The humanized monoclonal antibody dupilumab, an anti–IL-4/IL-13 agent, has demonstrated more consistent efficacy for the treatment of AD in children and adults.1 Dupilumab is effective for both intrinsic and extrinsic AD1 because its clinical efficacy is unrelated to circulating levels of IgE in the bloodstream. Although IgE may have a role in childhood AD, our case demonstrated a different pathophysiologic mechanism independent of IgE. Our patient’s AD flares occurred within a few days of omalizumab injection, which may have resulted in a paradoxical increase in basophil sensitivity to other cytokines such as IL-335 and led to an increase in IL-4/IL-13 production within the skin. In our patient, this increase was successfully blocked by dupilumab. Furthermore, omalizumab has been shown to modulate helper T cell (TH2) cytokine response such as thymic stromal lymphopoietin.6 A cytokine imbalance could have exacerbated AD in our case.
Although additional work to clarify the pathogenesis of AD is needed, it is important to recognize the potential for the occurrence of paradoxical AD flares in patients treated with omalizumab, which is analogous to the well-documented entity of tumor necrosis factor α inhibitor–induced psoriasis. It is equally important to recognize the potential benefit for patients treated with dupilumab.
- Nygaard U, Vestergaard C, Deleuran M. Emerging treatment options in atopic dermatitis: systemic therapies. Dermatology. 2017;233:344-357.
- Holm JG, Agner T, Sand C, et al. Omalizumab for atopic dermatitis: case series and a systematic review of the literature. Int J Dermatol. 2017;56:18-26.
- Chan S, Cornelius V, Cro S, et al. Treatment effect of omalizumab on severe pediatric atopic dermatitis: the ADAPT randomized clinical trial. JAMA Pediatr. 2020;174:29-37.
- Heil PM, Maurer D, Klein B, et al. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course – a randomized placebo-controlled and double blind pilot study. J Dtsch Dermatol Ges. 2010;8:990-998.
- Imai Y. Interleukin-33 in atopic dermatitis. J Dermatol Sci. 2019;96:2-7.
- Iyengar SR, Hoyte EG, Loza A, et al. Immunologic effects of omalizumab in children with severe refractory atopic dermatitis: a randomized, placebo-controlled clinical trial. Int Arch Allergy Immunol. 2013;162:89-93.
To the Editor:
A 16-year-old adolescent boy presented to our pediatric dermatology clinic for evaluation of long-standing mild atopic dermatitis (AD) that had become severe over the last year after omalizumab was initiated for severe asthma. The patient had a history of multiple hospitalizations for severe asthma. Despite excellent control of asthma with omalizumab given every 2 weeks, he developed widespread eczematous plaques on the neck, trunk, and extremities over the course of a year. The AD often was complicated by superimposed folliculitis due to scratching from severe pruritus. Treatment with topical corticosteroids including triamcinolone ointment 0.1% to AD on the body, plus clobetasol ointment 0.05% for prurigolike lesions on the legs resulted in modest improvement; however, the AD consistently recurred within a few days after the biweekly omalizumab injection (Figure 1). When the omalizumab injections were delayed, the flares temporarily improved, and when injections were decreased to once monthly, the exacerbations subsided partially but not fully.
Because omalizumab resulted in dramatic improvement in the patient’s asthma, there was hesitation to discontinue it initially; however, the patient and his parents in conjunction with the dermatology and pulmonary teams decided to transition to dupilumab. The patient reported vast improvement of AD 1 month after initiation of dupilumab (Figure 2), which remained well controlled more than 1 year later. Mid-potency topical corticosteroids for the treatment of occasional mild eczematous flares on the extremities were used. The patient’s asthma has remained well controlled on dupilumab without any exacerbations.
Omalizumab is a recombinant DNA-derived humanized monoclonal antibody that binds both circulating and membrane-bound IgE. It has been proposed as a possible treatment for severe and/or recalcitrant AD, with mixed treatment results.1 A case series and review of 174 patients demonstrated a moderate to complete AD response to treatment with omalizumab in 74.1% of patients.2 The Atopic Dermatitis Anti-IgE Pediatric Trial (ADAPT) showed a statistically significant reduction in the Scoring Atopic Dermatitis (SCORAD) index (P=.01), along with improved quality of life in children treated with omalizumab vs those treated with placebo.3 However, a prior randomized, placebo-controlled, double-blind study did not show a significant difference in clinical disease parameters in patients treated with omalizumab.4
The humanized monoclonal antibody dupilumab, an anti–IL-4/IL-13 agent, has demonstrated more consistent efficacy for the treatment of AD in children and adults.1 Dupilumab is effective for both intrinsic and extrinsic AD1 because its clinical efficacy is unrelated to circulating levels of IgE in the bloodstream. Although IgE may have a role in childhood AD, our case demonstrated a different pathophysiologic mechanism independent of IgE. Our patient’s AD flares occurred within a few days of omalizumab injection, which may have resulted in a paradoxical increase in basophil sensitivity to other cytokines such as IL-335 and led to an increase in IL-4/IL-13 production within the skin. In our patient, this increase was successfully blocked by dupilumab. Furthermore, omalizumab has been shown to modulate helper T cell (TH2) cytokine response such as thymic stromal lymphopoietin.6 A cytokine imbalance could have exacerbated AD in our case.
Although additional work to clarify the pathogenesis of AD is needed, it is important to recognize the potential for the occurrence of paradoxical AD flares in patients treated with omalizumab, which is analogous to the well-documented entity of tumor necrosis factor α inhibitor–induced psoriasis. It is equally important to recognize the potential benefit for patients treated with dupilumab.
To the Editor:
A 16-year-old adolescent boy presented to our pediatric dermatology clinic for evaluation of long-standing mild atopic dermatitis (AD) that had become severe over the last year after omalizumab was initiated for severe asthma. The patient had a history of multiple hospitalizations for severe asthma. Despite excellent control of asthma with omalizumab given every 2 weeks, he developed widespread eczematous plaques on the neck, trunk, and extremities over the course of a year. The AD often was complicated by superimposed folliculitis due to scratching from severe pruritus. Treatment with topical corticosteroids including triamcinolone ointment 0.1% to AD on the body, plus clobetasol ointment 0.05% for prurigolike lesions on the legs resulted in modest improvement; however, the AD consistently recurred within a few days after the biweekly omalizumab injection (Figure 1). When the omalizumab injections were delayed, the flares temporarily improved, and when injections were decreased to once monthly, the exacerbations subsided partially but not fully.
Because omalizumab resulted in dramatic improvement in the patient’s asthma, there was hesitation to discontinue it initially; however, the patient and his parents in conjunction with the dermatology and pulmonary teams decided to transition to dupilumab. The patient reported vast improvement of AD 1 month after initiation of dupilumab (Figure 2), which remained well controlled more than 1 year later. Mid-potency topical corticosteroids for the treatment of occasional mild eczematous flares on the extremities were used. The patient’s asthma has remained well controlled on dupilumab without any exacerbations.
Omalizumab is a recombinant DNA-derived humanized monoclonal antibody that binds both circulating and membrane-bound IgE. It has been proposed as a possible treatment for severe and/or recalcitrant AD, with mixed treatment results.1 A case series and review of 174 patients demonstrated a moderate to complete AD response to treatment with omalizumab in 74.1% of patients.2 The Atopic Dermatitis Anti-IgE Pediatric Trial (ADAPT) showed a statistically significant reduction in the Scoring Atopic Dermatitis (SCORAD) index (P=.01), along with improved quality of life in children treated with omalizumab vs those treated with placebo.3 However, a prior randomized, placebo-controlled, double-blind study did not show a significant difference in clinical disease parameters in patients treated with omalizumab.4
The humanized monoclonal antibody dupilumab, an anti–IL-4/IL-13 agent, has demonstrated more consistent efficacy for the treatment of AD in children and adults.1 Dupilumab is effective for both intrinsic and extrinsic AD1 because its clinical efficacy is unrelated to circulating levels of IgE in the bloodstream. Although IgE may have a role in childhood AD, our case demonstrated a different pathophysiologic mechanism independent of IgE. Our patient’s AD flares occurred within a few days of omalizumab injection, which may have resulted in a paradoxical increase in basophil sensitivity to other cytokines such as IL-335 and led to an increase in IL-4/IL-13 production within the skin. In our patient, this increase was successfully blocked by dupilumab. Furthermore, omalizumab has been shown to modulate helper T cell (TH2) cytokine response such as thymic stromal lymphopoietin.6 A cytokine imbalance could have exacerbated AD in our case.
Although additional work to clarify the pathogenesis of AD is needed, it is important to recognize the potential for the occurrence of paradoxical AD flares in patients treated with omalizumab, which is analogous to the well-documented entity of tumor necrosis factor α inhibitor–induced psoriasis. It is equally important to recognize the potential benefit for patients treated with dupilumab.
- Nygaard U, Vestergaard C, Deleuran M. Emerging treatment options in atopic dermatitis: systemic therapies. Dermatology. 2017;233:344-357.
- Holm JG, Agner T, Sand C, et al. Omalizumab for atopic dermatitis: case series and a systematic review of the literature. Int J Dermatol. 2017;56:18-26.
- Chan S, Cornelius V, Cro S, et al. Treatment effect of omalizumab on severe pediatric atopic dermatitis: the ADAPT randomized clinical trial. JAMA Pediatr. 2020;174:29-37.
- Heil PM, Maurer D, Klein B, et al. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course – a randomized placebo-controlled and double blind pilot study. J Dtsch Dermatol Ges. 2010;8:990-998.
- Imai Y. Interleukin-33 in atopic dermatitis. J Dermatol Sci. 2019;96:2-7.
- Iyengar SR, Hoyte EG, Loza A, et al. Immunologic effects of omalizumab in children with severe refractory atopic dermatitis: a randomized, placebo-controlled clinical trial. Int Arch Allergy Immunol. 2013;162:89-93.
- Nygaard U, Vestergaard C, Deleuran M. Emerging treatment options in atopic dermatitis: systemic therapies. Dermatology. 2017;233:344-357.
- Holm JG, Agner T, Sand C, et al. Omalizumab for atopic dermatitis: case series and a systematic review of the literature. Int J Dermatol. 2017;56:18-26.
- Chan S, Cornelius V, Cro S, et al. Treatment effect of omalizumab on severe pediatric atopic dermatitis: the ADAPT randomized clinical trial. JAMA Pediatr. 2020;174:29-37.
- Heil PM, Maurer D, Klein B, et al. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course – a randomized placebo-controlled and double blind pilot study. J Dtsch Dermatol Ges. 2010;8:990-998.
- Imai Y. Interleukin-33 in atopic dermatitis. J Dermatol Sci. 2019;96:2-7.
- Iyengar SR, Hoyte EG, Loza A, et al. Immunologic effects of omalizumab in children with severe refractory atopic dermatitis: a randomized, placebo-controlled clinical trial. Int Arch Allergy Immunol. 2013;162:89-93.
Practice Points
- Monoclonal antibodies are promising therapies for atopic conditions, although its efficacy for atopic dermatitis (AD) is debated and the side-effect profile is not entirely known.
- Omalizumab may cause a paradoxical exacerbation of AD in select patients analogous to tumor necrosis factor α inhibitor–induced psoriasis.

Tender Nodular Lesions in the Axilla and Vulva
The Diagnosis: Cutaneous Langerhans Cell Histiocytosis
Histopathologic findings of the left axillary lesion included a diffuse infiltrate of irregular hematolymphoid cells with reniform nuclei that strongly and diffusely stained positively with CD1a and S-100 but were negative for CD138 and CD163 (Figure). Numerous eosinophils also were present. The surrounding lymphocytic infiltrate stained positively with CD45. Polymerase chain reaction of the vaginal lesion was negative for herpes simplex virus types 1 and 2. Biopsy of the vaginal lesion revealed a mildly acanthotic epidermis and an aggregation of epithelioid cells with reniform nuclei in the papillary dermis. Positron emission tomography revealed widely disseminated disease. Sequencing of the mitogen-activated protein kinase/extracellular signalregulated kinase pathway showed amplified expression of these genes but found no mutations. These results led to a diagnosis of cutaneous Langerhans cell histiocytosis (LCH) with a background of hidradenitis suppurativa (HS). Our patient has since initiated therapy with trametinib leading to disease improvement without known recurrence.

Langerhans cell histiocytosis is a rare disease of clonal dendritic cells (Langerhans cells) that can present in any organ.1 Most LCH diagnoses are made in pediatric patients, most often presenting in the bones, with other presentations in the skin, hypophysis, liver, lymph nodes, lungs, and spleen occurring less commonly.2 Proto-oncogene BRAF V600E mutations are a common determinant of LCH, with half of cases linked with this mutation that leads to enhanced activation of the mitogen-activated protein kinase pathway, though other mutations have been reported.3,4 These genetic alterations suggest LCH is neoplastic in nature; however, this is controversial, as spontaneous regression among pulmonary LCH has been observed, pointing to a reactive inflammatory process.5 Cutaneous LCH can present as a distinct papular or nodular lesion or multiple lesions with possible ulceration, but it is rare that LCH first presents on the skin.2,6 There is a substantial association of cutaneous LCH with the development of systemically disseminated LCH as well as other blood tumors, such as myelomonocytic leukemia, histiocytic sarcoma, and multiple lymphomas; this association is thought to be due to the common origin of LCH and other blood diseases in the bone marrow.6
Histopathology of LCH shows a diffuse papillary dermal infiltrate of clonal proliferation of reniform or cleaved histiocytes.5 Epidermal ulceration and epidermotropism also are common. Neoplastic cells are found admixed with variable levels of eosinophils, lymphocytes, plasma cells, and neutrophils, though eosinophils typically are elevated. Immunohistochemistry characteristically shows the expression of CD1a, S-100, and/or CD207, and the absence of CD163 expression.
Treatment of LCH is primarily dependent on disease dissemination status, with splenic and hepatic involvement, genetic panel results, and central nervous system risk considered in the treatment plan.5 Langerhans cell histiocytosis localized to the skin may require follow-up and monitoring, as spontaneous regression of cutaneous LCH is common. However, topical steroids or psoralen and long-wave UV radiation are potential treatments. Physicians who diagnose unifocal cutaneous LCH should have high clinical suspicion of disseminated LCH, and laboratory and radiographic evaluation may be necessary to rule out systemic disease, as more than 40% of patients with cutaneous LCH have systemic disease upon full evaluation.7 With systemic involvement, systemic chemotherapy may reduce morbidity and mortality, but clinical response should be monitored after 6 weeks of treatment, as results are variably effective. Vinblastine is the most common chemotherapy regimen, with an 84% survival rate and 51.5% event-free survival rate after 8 years.8 Targeted therapy for common genetic mutations also is possible, as vemurafenib has been used to treat patients with the BRAF V600E mutation.
Due to the variable clinical presentation of cutaneous LCH, the lesions can mimic other common skin diseases such as eczema or seborrheic dermatitis.7 However, there are limited data on LCH presenting in infiltrative skin disease. Langerhans cell histiocytosis that was misdiagnosed as HS has been reported,9-11 but LCH presenting alongside long-standing HS is rare. Although LCH often mimics infiltrative skin diseases, its simultaneous presentation with a previously confirmed diagnosis of HS was notable in our patient.
In our patient, the differential diagnosis included HS, Actinomyces infection, lymphomatoid papulosis, and dermatofibrosarcoma protuberans. Cutaneous findings in HS include chronic acneform nodules with follicular plugging, ruptured ducts leading to epithelized sinuses, inflammation, and abscesses in the axillae or inguinal and perineal areas.11 Histopathology reveals follicular occlusion and hyperkeratinization, which cause destruction of the pilosebaceous glands. Hidradenitis suppurativa features on immunohistochemistry often are conflicting, but there consistently is co-localization of keratinocyte hyperplasia with CD3-, CD4-, CD8-, and CD68-positive staining of cells that produce tumor necrosis factor α, IL-12, IL-23, and IL-32, with CD1a staining variable.12 An infection with Actinomyces, a slow-progressing anaerobic or microaerophilic bacteria, may present in the skin with chronic suppurative inflammation on the neck, trunk, and abdomen. The classic presentation is subcutaneous nodules with localized infiltration of abscesses, fistulas, and draining sinuses.13 Morphologically, Actinomyces causes chronic granulomatous infection with 0.1- to 1-mm sulfur granules, which are seen as basophilic masses with eosinophilic terminal clubs on hematoxylin and eosin staining.14 Histopathology reveals grampositive filamentous Actinomyces bacteria that branch at the edge of the granules. Lymphomatoid papulosis, a nonaggressive T-cell lymphoma, presents as papulonodular and sometimes necrotic disseminated lesions that spontaneously can regress or can cause a higher risk for the development of more aggressive lymphomas.15 Histopathology shows consistently dense, dermal, lymphocytic infiltration. Immunohistochemistry is characterized by lymphocytes expressing CD30 of varying degrees: type A with many CD30 staining cells, type B presenting similar to mycosis fungoides with little CD30 staining, and type C with lymphocytic CD30-staining plaques. Dermatofibrosarcoma protuberans is a low-grade soft-tissue malignant tumor with extensive local infiltration characterized by asymptomatic plaques on the trunk and proximal extremities that are indurated and adhered to the skin.16 Histopathology shows extensive invasion into the adjacent tissue far from the original focus of the tumor.
- Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;8:72. doi:10.1186/1750-1172-8-72
- Flores-Terry MA, Sanz-Trenado JL, García-Arpa M, et al. Cutaneous Langerhans cell histiocytosis presenting in adulthood. Actas Dermosifiliogr (Engl Ed). 2019;110:167-169. doi:10.1016/j .adengl.2018.12.005
- Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
- Bohn OL, Teruya-Feldstein J, Sanchez-Sosa S. Skin biopsy diagnosis of Langerhans cell neoplasms. In: Fernando S, ed. Skin Biopsy: Diagnosis and Treatment [Internet]. InTechOpen; 2013. http://dx.doi .org/10.5772/55893
- Edelbroek JR, Vermeer MH, Jansen PM, et al. Langerhans cell histiocytosis first presenting in the skin in adults: frequent association with a second haematological malignancy. Br J Dermatol. 2012;167:1287-1294. doi:10.1111/j.1365-2133.2012.11169.x
- Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165: 990-996. doi:10.1016/j.jpeds.2014.07.063
- Yag˘ ci B, Varan A, Cag˘ lar M, et al. Langerhans cell histiocytosis: retrospective analysis of 217 cases in a single center. Pediatr Hematol Oncol. 2008;25:399-408. doi:10.1080/08880010802107356
- Kalen JE, Shokeen D, Mislankar M, et al. Langerhans cell histiocytosis with clinical and histologic features of hidradenitis suppurativa: brief report and review. Am J Dermatopathol. 2018;40:502-505. doi:10.1097/dad.0000000000001005
- Chertoff J, Chung J, Ataya A. Adult Langerhans cell histiocytosis masquerading as hidradenitis suppurativa. Am J Respir Crit Care Med. 2017;195:E34-E36. doi:10.1164/rccm.201610-2082IM
- St. Claire K, Bunney R, Ashack KA, et al. Langerhans cell histiocytosis: a great imitator. Clin Dermatol. 2020;38:223-234. doi:10.1016/j.clindermatol.2019.10.007
- Frew JW, Hawkes JE, Krueger JG. A systematic review and critical evaluation of immunohistochemical associations in hidradenitis suppurativa. F1000Research. 2019;7:1923. doi:10.12688/f1000research.17268.2
- Robati RM, Niknezhad N, Bidari-Zerehpoush F, et al. Primary cutaneous actinomycosis along with the surgical scar on the hand [published online November 9, 2016]. Case Rep Infect Dis. doi:10.1155/2016/5943932
- Ferry T, Valour F, Karsenty J, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Res. 2014;2014:183-197. doi:10.2147/idr.s39601
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182 /blood-2004-09-3502
- Tsai Y, Lin P, Chew K, et al. Dermatofibrosarcoma protuberans in children and adolescents: clinical presentation, histology, treatment, and review of the literature. J Plast Reconstr Aesthet Surg. 2014;67:1222-1229. doi:10.1016/j.bjps.2014.05.03
The Diagnosis: Cutaneous Langerhans Cell Histiocytosis
Histopathologic findings of the left axillary lesion included a diffuse infiltrate of irregular hematolymphoid cells with reniform nuclei that strongly and diffusely stained positively with CD1a and S-100 but were negative for CD138 and CD163 (Figure). Numerous eosinophils also were present. The surrounding lymphocytic infiltrate stained positively with CD45. Polymerase chain reaction of the vaginal lesion was negative for herpes simplex virus types 1 and 2. Biopsy of the vaginal lesion revealed a mildly acanthotic epidermis and an aggregation of epithelioid cells with reniform nuclei in the papillary dermis. Positron emission tomography revealed widely disseminated disease. Sequencing of the mitogen-activated protein kinase/extracellular signalregulated kinase pathway showed amplified expression of these genes but found no mutations. These results led to a diagnosis of cutaneous Langerhans cell histiocytosis (LCH) with a background of hidradenitis suppurativa (HS). Our patient has since initiated therapy with trametinib leading to disease improvement without known recurrence.
Langerhans cell histiocytosis is a rare disease of clonal dendritic cells (Langerhans cells) that can present in any organ.1 Most LCH diagnoses are made in pediatric patients, most often presenting in the bones, with other presentations in the skin, hypophysis, liver, lymph nodes, lungs, and spleen occurring less commonly.2 Proto-oncogene BRAF V600E mutations are a common determinant of LCH, with half of cases linked with this mutation that leads to enhanced activation of the mitogen-activated protein kinase pathway, though other mutations have been reported.3,4 These genetic alterations suggest LCH is neoplastic in nature; however, this is controversial, as spontaneous regression among pulmonary LCH has been observed, pointing to a reactive inflammatory process.5 Cutaneous LCH can present as a distinct papular or nodular lesion or multiple lesions with possible ulceration, but it is rare that LCH first presents on the skin.2,6 There is a substantial association of cutaneous LCH with the development of systemically disseminated LCH as well as other blood tumors, such as myelomonocytic leukemia, histiocytic sarcoma, and multiple lymphomas; this association is thought to be due to the common origin of LCH and other blood diseases in the bone marrow.6
Histopathology of LCH shows a diffuse papillary dermal infiltrate of clonal proliferation of reniform or cleaved histiocytes.5 Epidermal ulceration and epidermotropism also are common. Neoplastic cells are found admixed with variable levels of eosinophils, lymphocytes, plasma cells, and neutrophils, though eosinophils typically are elevated. Immunohistochemistry characteristically shows the expression of CD1a, S-100, and/or CD207, and the absence of CD163 expression.
Treatment of LCH is primarily dependent on disease dissemination status, with splenic and hepatic involvement, genetic panel results, and central nervous system risk considered in the treatment plan.5 Langerhans cell histiocytosis localized to the skin may require follow-up and monitoring, as spontaneous regression of cutaneous LCH is common. However, topical steroids or psoralen and long-wave UV radiation are potential treatments. Physicians who diagnose unifocal cutaneous LCH should have high clinical suspicion of disseminated LCH, and laboratory and radiographic evaluation may be necessary to rule out systemic disease, as more than 40% of patients with cutaneous LCH have systemic disease upon full evaluation.7 With systemic involvement, systemic chemotherapy may reduce morbidity and mortality, but clinical response should be monitored after 6 weeks of treatment, as results are variably effective. Vinblastine is the most common chemotherapy regimen, with an 84% survival rate and 51.5% event-free survival rate after 8 years.8 Targeted therapy for common genetic mutations also is possible, as vemurafenib has been used to treat patients with the BRAF V600E mutation.
Due to the variable clinical presentation of cutaneous LCH, the lesions can mimic other common skin diseases such as eczema or seborrheic dermatitis.7 However, there are limited data on LCH presenting in infiltrative skin disease. Langerhans cell histiocytosis that was misdiagnosed as HS has been reported,9-11 but LCH presenting alongside long-standing HS is rare. Although LCH often mimics infiltrative skin diseases, its simultaneous presentation with a previously confirmed diagnosis of HS was notable in our patient.
In our patient, the differential diagnosis included HS, Actinomyces infection, lymphomatoid papulosis, and dermatofibrosarcoma protuberans. Cutaneous findings in HS include chronic acneform nodules with follicular plugging, ruptured ducts leading to epithelized sinuses, inflammation, and abscesses in the axillae or inguinal and perineal areas.11 Histopathology reveals follicular occlusion and hyperkeratinization, which cause destruction of the pilosebaceous glands. Hidradenitis suppurativa features on immunohistochemistry often are conflicting, but there consistently is co-localization of keratinocyte hyperplasia with CD3-, CD4-, CD8-, and CD68-positive staining of cells that produce tumor necrosis factor α, IL-12, IL-23, and IL-32, with CD1a staining variable.12 An infection with Actinomyces, a slow-progressing anaerobic or microaerophilic bacteria, may present in the skin with chronic suppurative inflammation on the neck, trunk, and abdomen. The classic presentation is subcutaneous nodules with localized infiltration of abscesses, fistulas, and draining sinuses.13 Morphologically, Actinomyces causes chronic granulomatous infection with 0.1- to 1-mm sulfur granules, which are seen as basophilic masses with eosinophilic terminal clubs on hematoxylin and eosin staining.14 Histopathology reveals grampositive filamentous Actinomyces bacteria that branch at the edge of the granules. Lymphomatoid papulosis, a nonaggressive T-cell lymphoma, presents as papulonodular and sometimes necrotic disseminated lesions that spontaneously can regress or can cause a higher risk for the development of more aggressive lymphomas.15 Histopathology shows consistently dense, dermal, lymphocytic infiltration. Immunohistochemistry is characterized by lymphocytes expressing CD30 of varying degrees: type A with many CD30 staining cells, type B presenting similar to mycosis fungoides with little CD30 staining, and type C with lymphocytic CD30-staining plaques. Dermatofibrosarcoma protuberans is a low-grade soft-tissue malignant tumor with extensive local infiltration characterized by asymptomatic plaques on the trunk and proximal extremities that are indurated and adhered to the skin.16 Histopathology shows extensive invasion into the adjacent tissue far from the original focus of the tumor.
The Diagnosis: Cutaneous Langerhans Cell Histiocytosis
Histopathologic findings of the left axillary lesion included a diffuse infiltrate of irregular hematolymphoid cells with reniform nuclei that strongly and diffusely stained positively with CD1a and S-100 but were negative for CD138 and CD163 (Figure). Numerous eosinophils also were present. The surrounding lymphocytic infiltrate stained positively with CD45. Polymerase chain reaction of the vaginal lesion was negative for herpes simplex virus types 1 and 2. Biopsy of the vaginal lesion revealed a mildly acanthotic epidermis and an aggregation of epithelioid cells with reniform nuclei in the papillary dermis. Positron emission tomography revealed widely disseminated disease. Sequencing of the mitogen-activated protein kinase/extracellular signalregulated kinase pathway showed amplified expression of these genes but found no mutations. These results led to a diagnosis of cutaneous Langerhans cell histiocytosis (LCH) with a background of hidradenitis suppurativa (HS). Our patient has since initiated therapy with trametinib leading to disease improvement without known recurrence.
Langerhans cell histiocytosis is a rare disease of clonal dendritic cells (Langerhans cells) that can present in any organ.1 Most LCH diagnoses are made in pediatric patients, most often presenting in the bones, with other presentations in the skin, hypophysis, liver, lymph nodes, lungs, and spleen occurring less commonly.2 Proto-oncogene BRAF V600E mutations are a common determinant of LCH, with half of cases linked with this mutation that leads to enhanced activation of the mitogen-activated protein kinase pathway, though other mutations have been reported.3,4 These genetic alterations suggest LCH is neoplastic in nature; however, this is controversial, as spontaneous regression among pulmonary LCH has been observed, pointing to a reactive inflammatory process.5 Cutaneous LCH can present as a distinct papular or nodular lesion or multiple lesions with possible ulceration, but it is rare that LCH first presents on the skin.2,6 There is a substantial association of cutaneous LCH with the development of systemically disseminated LCH as well as other blood tumors, such as myelomonocytic leukemia, histiocytic sarcoma, and multiple lymphomas; this association is thought to be due to the common origin of LCH and other blood diseases in the bone marrow.6
Histopathology of LCH shows a diffuse papillary dermal infiltrate of clonal proliferation of reniform or cleaved histiocytes.5 Epidermal ulceration and epidermotropism also are common. Neoplastic cells are found admixed with variable levels of eosinophils, lymphocytes, plasma cells, and neutrophils, though eosinophils typically are elevated. Immunohistochemistry characteristically shows the expression of CD1a, S-100, and/or CD207, and the absence of CD163 expression.
Treatment of LCH is primarily dependent on disease dissemination status, with splenic and hepatic involvement, genetic panel results, and central nervous system risk considered in the treatment plan.5 Langerhans cell histiocytosis localized to the skin may require follow-up and monitoring, as spontaneous regression of cutaneous LCH is common. However, topical steroids or psoralen and long-wave UV radiation are potential treatments. Physicians who diagnose unifocal cutaneous LCH should have high clinical suspicion of disseminated LCH, and laboratory and radiographic evaluation may be necessary to rule out systemic disease, as more than 40% of patients with cutaneous LCH have systemic disease upon full evaluation.7 With systemic involvement, systemic chemotherapy may reduce morbidity and mortality, but clinical response should be monitored after 6 weeks of treatment, as results are variably effective. Vinblastine is the most common chemotherapy regimen, with an 84% survival rate and 51.5% event-free survival rate after 8 years.8 Targeted therapy for common genetic mutations also is possible, as vemurafenib has been used to treat patients with the BRAF V600E mutation.
Due to the variable clinical presentation of cutaneous LCH, the lesions can mimic other common skin diseases such as eczema or seborrheic dermatitis.7 However, there are limited data on LCH presenting in infiltrative skin disease. Langerhans cell histiocytosis that was misdiagnosed as HS has been reported,9-11 but LCH presenting alongside long-standing HS is rare. Although LCH often mimics infiltrative skin diseases, its simultaneous presentation with a previously confirmed diagnosis of HS was notable in our patient.
In our patient, the differential diagnosis included HS, Actinomyces infection, lymphomatoid papulosis, and dermatofibrosarcoma protuberans. Cutaneous findings in HS include chronic acneform nodules with follicular plugging, ruptured ducts leading to epithelized sinuses, inflammation, and abscesses in the axillae or inguinal and perineal areas.11 Histopathology reveals follicular occlusion and hyperkeratinization, which cause destruction of the pilosebaceous glands. Hidradenitis suppurativa features on immunohistochemistry often are conflicting, but there consistently is co-localization of keratinocyte hyperplasia with CD3-, CD4-, CD8-, and CD68-positive staining of cells that produce tumor necrosis factor α, IL-12, IL-23, and IL-32, with CD1a staining variable.12 An infection with Actinomyces, a slow-progressing anaerobic or microaerophilic bacteria, may present in the skin with chronic suppurative inflammation on the neck, trunk, and abdomen. The classic presentation is subcutaneous nodules with localized infiltration of abscesses, fistulas, and draining sinuses.13 Morphologically, Actinomyces causes chronic granulomatous infection with 0.1- to 1-mm sulfur granules, which are seen as basophilic masses with eosinophilic terminal clubs on hematoxylin and eosin staining.14 Histopathology reveals grampositive filamentous Actinomyces bacteria that branch at the edge of the granules. Lymphomatoid papulosis, a nonaggressive T-cell lymphoma, presents as papulonodular and sometimes necrotic disseminated lesions that spontaneously can regress or can cause a higher risk for the development of more aggressive lymphomas.15 Histopathology shows consistently dense, dermal, lymphocytic infiltration. Immunohistochemistry is characterized by lymphocytes expressing CD30 of varying degrees: type A with many CD30 staining cells, type B presenting similar to mycosis fungoides with little CD30 staining, and type C with lymphocytic CD30-staining plaques. Dermatofibrosarcoma protuberans is a low-grade soft-tissue malignant tumor with extensive local infiltration characterized by asymptomatic plaques on the trunk and proximal extremities that are indurated and adhered to the skin.16 Histopathology shows extensive invasion into the adjacent tissue far from the original focus of the tumor.
- Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;8:72. doi:10.1186/1750-1172-8-72
- Flores-Terry MA, Sanz-Trenado JL, García-Arpa M, et al. Cutaneous Langerhans cell histiocytosis presenting in adulthood. Actas Dermosifiliogr (Engl Ed). 2019;110:167-169. doi:10.1016/j .adengl.2018.12.005
- Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
- Bohn OL, Teruya-Feldstein J, Sanchez-Sosa S. Skin biopsy diagnosis of Langerhans cell neoplasms. In: Fernando S, ed. Skin Biopsy: Diagnosis and Treatment [Internet]. InTechOpen; 2013. http://dx.doi .org/10.5772/55893
- Edelbroek JR, Vermeer MH, Jansen PM, et al. Langerhans cell histiocytosis first presenting in the skin in adults: frequent association with a second haematological malignancy. Br J Dermatol. 2012;167:1287-1294. doi:10.1111/j.1365-2133.2012.11169.x
- Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165: 990-996. doi:10.1016/j.jpeds.2014.07.063
- Yag˘ ci B, Varan A, Cag˘ lar M, et al. Langerhans cell histiocytosis: retrospective analysis of 217 cases in a single center. Pediatr Hematol Oncol. 2008;25:399-408. doi:10.1080/08880010802107356
- Kalen JE, Shokeen D, Mislankar M, et al. Langerhans cell histiocytosis with clinical and histologic features of hidradenitis suppurativa: brief report and review. Am J Dermatopathol. 2018;40:502-505. doi:10.1097/dad.0000000000001005
- Chertoff J, Chung J, Ataya A. Adult Langerhans cell histiocytosis masquerading as hidradenitis suppurativa. Am J Respir Crit Care Med. 2017;195:E34-E36. doi:10.1164/rccm.201610-2082IM
- St. Claire K, Bunney R, Ashack KA, et al. Langerhans cell histiocytosis: a great imitator. Clin Dermatol. 2020;38:223-234. doi:10.1016/j.clindermatol.2019.10.007
- Frew JW, Hawkes JE, Krueger JG. A systematic review and critical evaluation of immunohistochemical associations in hidradenitis suppurativa. F1000Research. 2019;7:1923. doi:10.12688/f1000research.17268.2
- Robati RM, Niknezhad N, Bidari-Zerehpoush F, et al. Primary cutaneous actinomycosis along with the surgical scar on the hand [published online November 9, 2016]. Case Rep Infect Dis. doi:10.1155/2016/5943932
- Ferry T, Valour F, Karsenty J, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Res. 2014;2014:183-197. doi:10.2147/idr.s39601
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182 /blood-2004-09-3502
- Tsai Y, Lin P, Chew K, et al. Dermatofibrosarcoma protuberans in children and adolescents: clinical presentation, histology, treatment, and review of the literature. J Plast Reconstr Aesthet Surg. 2014;67:1222-1229. doi:10.1016/j.bjps.2014.05.03
- Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;8:72. doi:10.1186/1750-1172-8-72
- Flores-Terry MA, Sanz-Trenado JL, García-Arpa M, et al. Cutaneous Langerhans cell histiocytosis presenting in adulthood. Actas Dermosifiliogr (Engl Ed). 2019;110:167-169. doi:10.1016/j .adengl.2018.12.005
- Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
- Bohn OL, Teruya-Feldstein J, Sanchez-Sosa S. Skin biopsy diagnosis of Langerhans cell neoplasms. In: Fernando S, ed. Skin Biopsy: Diagnosis and Treatment [Internet]. InTechOpen; 2013. http://dx.doi .org/10.5772/55893
- Edelbroek JR, Vermeer MH, Jansen PM, et al. Langerhans cell histiocytosis first presenting in the skin in adults: frequent association with a second haematological malignancy. Br J Dermatol. 2012;167:1287-1294. doi:10.1111/j.1365-2133.2012.11169.x
- Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165: 990-996. doi:10.1016/j.jpeds.2014.07.063
- Yag˘ ci B, Varan A, Cag˘ lar M, et al. Langerhans cell histiocytosis: retrospective analysis of 217 cases in a single center. Pediatr Hematol Oncol. 2008;25:399-408. doi:10.1080/08880010802107356
- Kalen JE, Shokeen D, Mislankar M, et al. Langerhans cell histiocytosis with clinical and histologic features of hidradenitis suppurativa: brief report and review. Am J Dermatopathol. 2018;40:502-505. doi:10.1097/dad.0000000000001005
- Chertoff J, Chung J, Ataya A. Adult Langerhans cell histiocytosis masquerading as hidradenitis suppurativa. Am J Respir Crit Care Med. 2017;195:E34-E36. doi:10.1164/rccm.201610-2082IM
- St. Claire K, Bunney R, Ashack KA, et al. Langerhans cell histiocytosis: a great imitator. Clin Dermatol. 2020;38:223-234. doi:10.1016/j.clindermatol.2019.10.007
- Frew JW, Hawkes JE, Krueger JG. A systematic review and critical evaluation of immunohistochemical associations in hidradenitis suppurativa. F1000Research. 2019;7:1923. doi:10.12688/f1000research.17268.2
- Robati RM, Niknezhad N, Bidari-Zerehpoush F, et al. Primary cutaneous actinomycosis along with the surgical scar on the hand [published online November 9, 2016]. Case Rep Infect Dis. doi:10.1155/2016/5943932
- Ferry T, Valour F, Karsenty J, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Res. 2014;2014:183-197. doi:10.2147/idr.s39601
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182 /blood-2004-09-3502
- Tsai Y, Lin P, Chew K, et al. Dermatofibrosarcoma protuberans in children and adolescents: clinical presentation, histology, treatment, and review of the literature. J Plast Reconstr Aesthet Surg. 2014;67:1222-1229. doi:10.1016/j.bjps.2014.05.03
A 28-year-old woman presented with tender burning lesions of the left axillary and vaginal skin that had worsened over the last year. Her medical history was notable for hidradenitis suppurativa, which had been present since adolescence, as well as pulmonary Langerhans cell histiocytosis diagnosed 7 years prior to the current presentation after a spontaneous pneumothorax that eventually led to a pulmonary transplantation 3 years prior. The patient’s Langerhans cell histiocytosis was believed to have resolved without treatment after smoking cessation. Physical examination revealed nodular inflammation and scarring with deep undermining along the left axilla as well as swelling of the mons pubis with erosive skin lesions in the surrounding vaginal area. Bilateral cervical, axillary, inguinal, supraclavicular, and femoral lymph node chains were negative for adenopathy. A shave biopsy was performed on the axillary nodule.


Cutaneous Presentation of Metastatic Salivary Duct Carcinoma
To the Editor:
Metastatic spread of salivary duct carcinoma (SDC) to the skin is rare. Diagnosing SDC can be challenging because the cutaneous manifestations of this disease are variable and include nodules, papules, and erysipelaslike inflammation (also known as shield sign) with purpuric papules and pseudovesicles. We describe a case of cutaneous metastatic SDC that originated from the parotid gland and presented with 2 distinct cutaneous findings: sharply demarcated erythematous plaques and focally hemorrhagic angiomatous papules.
A 60-year-old man presented with a persistent polymorphous pruritic eruption of several months’ duration involving the entire face, ears, neck, and upper chest. He had a history of unspecified adenocarcinoma of the parotid gland diagnosed 2 years prior and underwent multiple treatment cycles with several chemotherapeutic agents over the course of 18 months. Physical examination showed erythematous papules and nodules on the face and neck with slight overlying scale. Sharply demarcated, erythematous plaques studded with focally hemorrhagic, angiomatous papules were noted on the neck and chest (Figure 1). Two 4-mm punch biopsies were sampled from representative nodular areas. Histopathology showed multiple round solid-tumor nodules with central necrosis in the superficial and deep dermis that were not associated with the overlying epidermis (Figures 2A and 2B). The tumor cells appeared polygonal and contained ample eosinophilic cytoplasm. Tumor nuclei showed marked pleomorphism, and numerous atypical mitotic figures were readily identifiable (Figure 2C). There was diffuse cytoplasmic staining with cytokeratin 7 and nuclear staining with androgen receptor (Figure 2D). These findings were consistent with a diagnosis of SDC metastatic to the skin.

The patient underwent 8 cycles of docetaxel chemotherapy. With disease progression, the chemotherapy regimen was changed to gemcitabine and methotrexate. The patient continued to experience disease progression and died 9 months after diagnosis of skin metastases.
. B, A tumor nodule with central zone of cellular necrosis (H&E, original magnification ×20).")
Salivary duct carcinoma is rare and is estimated to represent 1% to 3% of all salivary malignancies.1 It is a highly aggressive form of salivary gland carcinoma and is associated with a poor clinical outcome. The 3-year overall survival rate for stage I disease is 42% and only 23% for stage IV disease.2 Salivary duct carcinoma has a high rate of distant metastasis,3 but cases of cutaneous metastases are rare.3-8 Previously reported cases of SDC that metastasized to the skin originated from the parotid gland (n=6) and submandibular gland (n=1).3
The diagnosis of cutaneous metastases is challenging due to the variability of the skin manifestations. Three cases described small firm nodules in patients,3-5 while others presented with purpuric papules and pseudovesicles.6-8 Our patient presented with sharply demarcated, erythematous plaques studded with focally hemorrhagic, angiomatous papules, which further emphasizes the capricious nature of skin findings.
The morphology of SDC is strikingly similar to ductal adenocarcinoma of the breast, which can lead to diagnostic confusion. Both carcinomas may show oncocytic cells, ductal formations, and cribriform structures with central comedo necrosis. Moreover, immunohistochemical features overlap, including positive staining for cytokeratin 7 and gross cystic disease fluid protein 15. Positive immunohistochemistry with androgen receptor is consistent with SDC but also can be expressed in some cases of breast carcinoma.9,10 Therefore, the diagnosis of cutaneous involvement from metastatic SDC requires not just an evaluation of the pathologic features but careful attention to the clinical history and a thorough staging evaluation.
- D’heygere E, Meulemans J, Vander Poorten V. Salivary duct carcinoma. Curr Opin Otolaryngol Head Neck Surg. 2018;26:142-151.
- Gilbert MR, Sharma A, Schmitt NC, et al. A 20-year review of 75 cases of salivary duct carcinoma. JAMA Otolaryngol Head Neck Surg. 2016;142:489-495.
- Chakari W, Andersen L, Andersen JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Tok J, Kao GF, Berberian BJ, et al. Cutaneous metastasis from a parotid adenocarcinoma. Report of a case with immunohistochemical findings and review of the literature. Am J Dermatopathol. 1995;17:303-306.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Hafiji J, Rytina E, Jani P, et al. A rare cutaneous presentation of metastatic parotid adenocarcinoma. Australas J Dermatol. 2013;54:E40-E42.
- Zanca A, Ferracini U, Bertazzoni MG. Telangiectatic metastasis from ductal carcinoma of the parotid gland. J Am Acad Dermatol. 1993;28:113-114.
- Brys´ M, Wójcik M, Romanowicz-Makowska H, et al. Androgen receptor status in female breast cancer: RT-PCR and Western blot studies. J Cancer Res Clin Oncol. 2002;128:85-90.
- Udager AM, Chiosea SI. Salivary duct carcinoma: an update on morphologic mimics and diagnostic use of androgen receptor immunohistochemistry. Head Neck Pathol. 2017;11:288-294.
To the Editor:
Metastatic spread of salivary duct carcinoma (SDC) to the skin is rare. Diagnosing SDC can be challenging because the cutaneous manifestations of this disease are variable and include nodules, papules, and erysipelaslike inflammation (also known as shield sign) with purpuric papules and pseudovesicles. We describe a case of cutaneous metastatic SDC that originated from the parotid gland and presented with 2 distinct cutaneous findings: sharply demarcated erythematous plaques and focally hemorrhagic angiomatous papules.
A 60-year-old man presented with a persistent polymorphous pruritic eruption of several months’ duration involving the entire face, ears, neck, and upper chest. He had a history of unspecified adenocarcinoma of the parotid gland diagnosed 2 years prior and underwent multiple treatment cycles with several chemotherapeutic agents over the course of 18 months. Physical examination showed erythematous papules and nodules on the face and neck with slight overlying scale. Sharply demarcated, erythematous plaques studded with focally hemorrhagic, angiomatous papules were noted on the neck and chest (Figure 1). Two 4-mm punch biopsies were sampled from representative nodular areas. Histopathology showed multiple round solid-tumor nodules with central necrosis in the superficial and deep dermis that were not associated with the overlying epidermis (Figures 2A and 2B). The tumor cells appeared polygonal and contained ample eosinophilic cytoplasm. Tumor nuclei showed marked pleomorphism, and numerous atypical mitotic figures were readily identifiable (Figure 2C). There was diffuse cytoplasmic staining with cytokeratin 7 and nuclear staining with androgen receptor (Figure 2D). These findings were consistent with a diagnosis of SDC metastatic to the skin.
The patient underwent 8 cycles of docetaxel chemotherapy. With disease progression, the chemotherapy regimen was changed to gemcitabine and methotrexate. The patient continued to experience disease progression and died 9 months after diagnosis of skin metastases.
Salivary duct carcinoma is rare and is estimated to represent 1% to 3% of all salivary malignancies.1 It is a highly aggressive form of salivary gland carcinoma and is associated with a poor clinical outcome. The 3-year overall survival rate for stage I disease is 42% and only 23% for stage IV disease.2 Salivary duct carcinoma has a high rate of distant metastasis,3 but cases of cutaneous metastases are rare.3-8 Previously reported cases of SDC that metastasized to the skin originated from the parotid gland (n=6) and submandibular gland (n=1).3
The diagnosis of cutaneous metastases is challenging due to the variability of the skin manifestations. Three cases described small firm nodules in patients,3-5 while others presented with purpuric papules and pseudovesicles.6-8 Our patient presented with sharply demarcated, erythematous plaques studded with focally hemorrhagic, angiomatous papules, which further emphasizes the capricious nature of skin findings.
The morphology of SDC is strikingly similar to ductal adenocarcinoma of the breast, which can lead to diagnostic confusion. Both carcinomas may show oncocytic cells, ductal formations, and cribriform structures with central comedo necrosis. Moreover, immunohistochemical features overlap, including positive staining for cytokeratin 7 and gross cystic disease fluid protein 15. Positive immunohistochemistry with androgen receptor is consistent with SDC but also can be expressed in some cases of breast carcinoma.9,10 Therefore, the diagnosis of cutaneous involvement from metastatic SDC requires not just an evaluation of the pathologic features but careful attention to the clinical history and a thorough staging evaluation.
To the Editor:
Metastatic spread of salivary duct carcinoma (SDC) to the skin is rare. Diagnosing SDC can be challenging because the cutaneous manifestations of this disease are variable and include nodules, papules, and erysipelaslike inflammation (also known as shield sign) with purpuric papules and pseudovesicles. We describe a case of cutaneous metastatic SDC that originated from the parotid gland and presented with 2 distinct cutaneous findings: sharply demarcated erythematous plaques and focally hemorrhagic angiomatous papules.
A 60-year-old man presented with a persistent polymorphous pruritic eruption of several months’ duration involving the entire face, ears, neck, and upper chest. He had a history of unspecified adenocarcinoma of the parotid gland diagnosed 2 years prior and underwent multiple treatment cycles with several chemotherapeutic agents over the course of 18 months. Physical examination showed erythematous papules and nodules on the face and neck with slight overlying scale. Sharply demarcated, erythematous plaques studded with focally hemorrhagic, angiomatous papules were noted on the neck and chest (Figure 1). Two 4-mm punch biopsies were sampled from representative nodular areas. Histopathology showed multiple round solid-tumor nodules with central necrosis in the superficial and deep dermis that were not associated with the overlying epidermis (Figures 2A and 2B). The tumor cells appeared polygonal and contained ample eosinophilic cytoplasm. Tumor nuclei showed marked pleomorphism, and numerous atypical mitotic figures were readily identifiable (Figure 2C). There was diffuse cytoplasmic staining with cytokeratin 7 and nuclear staining with androgen receptor (Figure 2D). These findings were consistent with a diagnosis of SDC metastatic to the skin.
The patient underwent 8 cycles of docetaxel chemotherapy. With disease progression, the chemotherapy regimen was changed to gemcitabine and methotrexate. The patient continued to experience disease progression and died 9 months after diagnosis of skin metastases.
Salivary duct carcinoma is rare and is estimated to represent 1% to 3% of all salivary malignancies.1 It is a highly aggressive form of salivary gland carcinoma and is associated with a poor clinical outcome. The 3-year overall survival rate for stage I disease is 42% and only 23% for stage IV disease.2 Salivary duct carcinoma has a high rate of distant metastasis,3 but cases of cutaneous metastases are rare.3-8 Previously reported cases of SDC that metastasized to the skin originated from the parotid gland (n=6) and submandibular gland (n=1).3
The diagnosis of cutaneous metastases is challenging due to the variability of the skin manifestations. Three cases described small firm nodules in patients,3-5 while others presented with purpuric papules and pseudovesicles.6-8 Our patient presented with sharply demarcated, erythematous plaques studded with focally hemorrhagic, angiomatous papules, which further emphasizes the capricious nature of skin findings.
The morphology of SDC is strikingly similar to ductal adenocarcinoma of the breast, which can lead to diagnostic confusion. Both carcinomas may show oncocytic cells, ductal formations, and cribriform structures with central comedo necrosis. Moreover, immunohistochemical features overlap, including positive staining for cytokeratin 7 and gross cystic disease fluid protein 15. Positive immunohistochemistry with androgen receptor is consistent with SDC but also can be expressed in some cases of breast carcinoma.9,10 Therefore, the diagnosis of cutaneous involvement from metastatic SDC requires not just an evaluation of the pathologic features but careful attention to the clinical history and a thorough staging evaluation.
- D’heygere E, Meulemans J, Vander Poorten V. Salivary duct carcinoma. Curr Opin Otolaryngol Head Neck Surg. 2018;26:142-151.
- Gilbert MR, Sharma A, Schmitt NC, et al. A 20-year review of 75 cases of salivary duct carcinoma. JAMA Otolaryngol Head Neck Surg. 2016;142:489-495.
- Chakari W, Andersen L, Andersen JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Tok J, Kao GF, Berberian BJ, et al. Cutaneous metastasis from a parotid adenocarcinoma. Report of a case with immunohistochemical findings and review of the literature. Am J Dermatopathol. 1995;17:303-306.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Hafiji J, Rytina E, Jani P, et al. A rare cutaneous presentation of metastatic parotid adenocarcinoma. Australas J Dermatol. 2013;54:E40-E42.
- Zanca A, Ferracini U, Bertazzoni MG. Telangiectatic metastasis from ductal carcinoma of the parotid gland. J Am Acad Dermatol. 1993;28:113-114.
- Brys´ M, Wójcik M, Romanowicz-Makowska H, et al. Androgen receptor status in female breast cancer: RT-PCR and Western blot studies. J Cancer Res Clin Oncol. 2002;128:85-90.
- Udager AM, Chiosea SI. Salivary duct carcinoma: an update on morphologic mimics and diagnostic use of androgen receptor immunohistochemistry. Head Neck Pathol. 2017;11:288-294.
- D’heygere E, Meulemans J, Vander Poorten V. Salivary duct carcinoma. Curr Opin Otolaryngol Head Neck Surg. 2018;26:142-151.
- Gilbert MR, Sharma A, Schmitt NC, et al. A 20-year review of 75 cases of salivary duct carcinoma. JAMA Otolaryngol Head Neck Surg. 2016;142:489-495.
- Chakari W, Andersen L, Andersen JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Tok J, Kao GF, Berberian BJ, et al. Cutaneous metastasis from a parotid adenocarcinoma. Report of a case with immunohistochemical findings and review of the literature. Am J Dermatopathol. 1995;17:303-306.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Hafiji J, Rytina E, Jani P, et al. A rare cutaneous presentation of metastatic parotid adenocarcinoma. Australas J Dermatol. 2013;54:E40-E42.
- Zanca A, Ferracini U, Bertazzoni MG. Telangiectatic metastasis from ductal carcinoma of the parotid gland. J Am Acad Dermatol. 1993;28:113-114.
- Brys´ M, Wójcik M, Romanowicz-Makowska H, et al. Androgen receptor status in female breast cancer: RT-PCR and Western blot studies. J Cancer Res Clin Oncol. 2002;128:85-90.
- Udager AM, Chiosea SI. Salivary duct carcinoma: an update on morphologic mimics and diagnostic use of androgen receptor immunohistochemistry. Head Neck Pathol. 2017;11:288-294.
Practice Points
- Skin manifestations of metastatic salivary duct carcinoma can be variable, ranging from nodules to erysipelaslike inflammation (also known as shield sign) with purpuric papules and pseudovesicles.
- The specific clinical findings as well as histologic and immunohistochemical characteristics can aid in the diagnosis of this rare disease.

Iododerma Simulating Cryptococcal Infection
To the Editor:
A woman in her 40s presented with acute onset of rapidly spreading lesions on the face, trunk, and extremities. She reported high fever and endorsed malaise. She had a history of end-stage renal disease and was on renal dialysis. She recently underwent revision of an arteriovenous fistula.
Physical examination revealed diffuse, erythematous, firm papules and plaques with central hemorrhage and umbilication on the dorsal aspect of the nose, forehead, temples, and cheeks. There also were purpuric papules and plaques with a peripheral rim of vesiculation (Figure 1) on the medial and posterior thighs and buttocks. Histopathology of a biopsy specimen revealed an interstitial neutrophilic infiltrate in the superficial dermis and mid dermis with scattered, haloed, acellular structures simulating cryptococcal organisms (Figure 2). Periodic acid–Schiff (PAS), Grocott methenamine-silver, and mucicarmine staining was negative. Repeat biopsy showed similar findings. A (1-3)-β-

The findings compatible with a diagnosis of iododerma included umbilicated hemorrhagic papules and plaques, cryptococcal-like structures with negative staining on histopathology, and elevated iodine levels with a negative infectious workup. The patient was treated with topical corticosteroids. At 1-month follow-up, the lesions had resolved.
. Reference bar indicates 200 μm.")
Iododerma is a halogenoderma, a skin eruption that occurs after ingestion of or exposure to a halogen-containing substance (eg, iodine, bromine, fluorine) or medication (eg, lithium).1 Common sources of iodine include iodinated contrast media, potassium iodide ingestion, topical application of povidone–iodine, radioactive iodine administration, and the antiarrhythmic amiodarone. Excess exposure to iodine-containing compounds typically occurs in the setting of kidney disease or failure as well as due to reduced iodine clearance.1 Although the pathogenesis of iododerma is unknown, the most common hypothesis is that lesions are delayed hypersensitivity reactions secondary to formation of a protein-halogen complex.2
The presentation of iododerma is polymorphous and includes acneform, vegetative, or pustular eruptions; umbilicated papules and plaques can be present.2,3 Lesions can be either asymptomatic or painful and pruritic. Timing between iodine exposure and onset of lesions varies from hours to days to years.2,4
Systemic symptoms of iododerma can occur, including salivary gland swelling, hypotension and bradycardia, kidney injury, or thyroid and liver abnormalities. Histopathologic analysis demonstrates a dense neutrophilic dermatitis with negative staining for infectious causes.4,5 Cryptococcal-like structures have been described in iododerma3; neutrophilic dermatoses of various causes that mimic cryptococcal infection have been reported.6 Ultimately, iododerma remains a diagnosis of exclusion.
Withdrawal of an offending compound is remedial. Dialysis is beneficial in end-stage renal disease. Topical, intralesional, and systemic corticosteroids, as well as antibiotics, provide variable benefit.4,7 Lesions can take 4 to 6 weeks to clear after withdrawal of the offending agent. It is unclear whether recurrences happen; iodine-containing compounds need to be avoided after a patient has been affected.
Iododerma has a broad differential diagnosis due to the polymorphous presentation of the disorder, including acute febrile neutrophilic dermatosis (also known as Sweet syndrome), cutaneous cryptococcosis, and cutaneous histoplasmosis. Sweet syndrome presents as abrupt onset of edematous erythematous plaques with fever and leukocytosis. It is associated with infection, inflammatory disorders, medication, and malignancy.8 Histopathologic analysis reveals papillary dermal edema and a neutrophilic dermatosis. Cytoplasmic vacuolization resembling C neoformans has been reported.9 The diagnosis is less favored in the presence of renal disease, temporal association of the eruption with iodine exposure, and elevated blood and urine iodine levels, as in our patient.
Cutaneous cryptococcosis, an infection caused by C neoformans, typically occurs secondary to dissemination from the lungs; rarely, the disease is primary. Acneform plaques, vegetative plaques, and umbilicated lesions are seen.10 Histopathologic analysis shows characteristic yeast forms of cryptococcosis surrounded by gelatinous edema, which create a haloed effect, typically throughout the dermis. Capsules are positive for PAS or mucicarmine staining. Although C neoformans can closely mimic iododerma both clinically and histopathologically, negative infectious staining, localization of haloed structures to the upper dermis, a negative test for cryptococcal antigen, and elevated blood and urine iodine levels in this case all favored iododerma.
Cutaneous histoplasmosis is an infection caused by Histoplasma capsulatum, most commonly as secondary dissemination from pulmonary infection but rarely from direct inoculation of the skin.11 Presentation includes erythematous to hemorrhagic, umbilicated papules and plaques. Histopathologic findings are round to oval, narrow-based, budding yeasts that stain positive for PAS or mucicarmine. Although histoplasmosis can clinically mimic iododerma, the disease is distinguished histologically by the presence of fungal microorganisms that lack the gelatinous edema and haloed effect of iododerma.
We presented a unique case of iododerma simulating cryptococcal infection both clinically and histopathologically. Prompt recognition of histologic mimickers of true infectious microorganisms is essential to prevent unnecessary delay of withdrawal of the offending substance and to initiate appropriate therapy.
- Alagheband M, Engineer L. Lithium and halogenoderma. Arch Dermatol. 2000;136:126-127. doi:10.1001/archderm.136.1.126
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379. doi:10.1111/bjd.12852
- Runge M, Williams K, Scharnitz T, et al. Iodine toxicity after iodinated contrast: new observations in iododerma. JAAD Case Rep. 2020;6:319-322. doi:10.1016/j.jdcr.2020.02.006
- Chalela JG, Aguilar L. Iododerma from contrast material. N Engl J Med. 2016;374:2477. doi:10.1056/NEJMicm1512512
- Chang MW, Miner JE, Moiin A, et al. Iododerma after computed tomographic scan with intravenous radiopaque contrast media. J Am Acad Dermatol. 1997;36:1014-1016. doi:10.1016/s0190-9622(97)80291-5
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi:10.1111/cup.12019
- Pranteda G, Grimaldi M, Salzetta M, et al. Vegetating iododerma and pulmonary eosinophilic infiltration. a simple co-occurrence? Acta Derm Venereol. 2004;84:480-481.
- Nelson CA, Stephen S, Ashchyan HJ, et al. M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.064
- Wilson J, Gleghorn K, Kelly B. Cryptococcoid Sweet’s syndrome: two reports of Sweet’s syndrome mimicking cutaneous cryptococcosis. J Cutan Pathol. 2017;44:413-419. doi:10.1111/cup.12921
- Beatson M, Harwood M, Reese V, et al. Primary cutaneous cryptococcosis in an elderly pigeon breeder. JAAD Case Rep. 2019;5:433-435. doi:10.1016/j.jdcr.2019.03.006
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348. doi:10.1177/0145561318097010-1108
To the Editor:
A woman in her 40s presented with acute onset of rapidly spreading lesions on the face, trunk, and extremities. She reported high fever and endorsed malaise. She had a history of end-stage renal disease and was on renal dialysis. She recently underwent revision of an arteriovenous fistula.
Physical examination revealed diffuse, erythematous, firm papules and plaques with central hemorrhage and umbilication on the dorsal aspect of the nose, forehead, temples, and cheeks. There also were purpuric papules and plaques with a peripheral rim of vesiculation (Figure 1) on the medial and posterior thighs and buttocks. Histopathology of a biopsy specimen revealed an interstitial neutrophilic infiltrate in the superficial dermis and mid dermis with scattered, haloed, acellular structures simulating cryptococcal organisms (Figure 2). Periodic acid–Schiff (PAS), Grocott methenamine-silver, and mucicarmine staining was negative. Repeat biopsy showed similar findings. A (1-3)-β-
The findings compatible with a diagnosis of iododerma included umbilicated hemorrhagic papules and plaques, cryptococcal-like structures with negative staining on histopathology, and elevated iodine levels with a negative infectious workup. The patient was treated with topical corticosteroids. At 1-month follow-up, the lesions had resolved.
Iododerma is a halogenoderma, a skin eruption that occurs after ingestion of or exposure to a halogen-containing substance (eg, iodine, bromine, fluorine) or medication (eg, lithium).1 Common sources of iodine include iodinated contrast media, potassium iodide ingestion, topical application of povidone–iodine, radioactive iodine administration, and the antiarrhythmic amiodarone. Excess exposure to iodine-containing compounds typically occurs in the setting of kidney disease or failure as well as due to reduced iodine clearance.1 Although the pathogenesis of iododerma is unknown, the most common hypothesis is that lesions are delayed hypersensitivity reactions secondary to formation of a protein-halogen complex.2
The presentation of iododerma is polymorphous and includes acneform, vegetative, or pustular eruptions; umbilicated papules and plaques can be present.2,3 Lesions can be either asymptomatic or painful and pruritic. Timing between iodine exposure and onset of lesions varies from hours to days to years.2,4
Systemic symptoms of iododerma can occur, including salivary gland swelling, hypotension and bradycardia, kidney injury, or thyroid and liver abnormalities. Histopathologic analysis demonstrates a dense neutrophilic dermatitis with negative staining for infectious causes.4,5 Cryptococcal-like structures have been described in iododerma3; neutrophilic dermatoses of various causes that mimic cryptococcal infection have been reported.6 Ultimately, iododerma remains a diagnosis of exclusion.
Withdrawal of an offending compound is remedial. Dialysis is beneficial in end-stage renal disease. Topical, intralesional, and systemic corticosteroids, as well as antibiotics, provide variable benefit.4,7 Lesions can take 4 to 6 weeks to clear after withdrawal of the offending agent. It is unclear whether recurrences happen; iodine-containing compounds need to be avoided after a patient has been affected.
Iododerma has a broad differential diagnosis due to the polymorphous presentation of the disorder, including acute febrile neutrophilic dermatosis (also known as Sweet syndrome), cutaneous cryptococcosis, and cutaneous histoplasmosis. Sweet syndrome presents as abrupt onset of edematous erythematous plaques with fever and leukocytosis. It is associated with infection, inflammatory disorders, medication, and malignancy.8 Histopathologic analysis reveals papillary dermal edema and a neutrophilic dermatosis. Cytoplasmic vacuolization resembling C neoformans has been reported.9 The diagnosis is less favored in the presence of renal disease, temporal association of the eruption with iodine exposure, and elevated blood and urine iodine levels, as in our patient.
Cutaneous cryptococcosis, an infection caused by C neoformans, typically occurs secondary to dissemination from the lungs; rarely, the disease is primary. Acneform plaques, vegetative plaques, and umbilicated lesions are seen.10 Histopathologic analysis shows characteristic yeast forms of cryptococcosis surrounded by gelatinous edema, which create a haloed effect, typically throughout the dermis. Capsules are positive for PAS or mucicarmine staining. Although C neoformans can closely mimic iododerma both clinically and histopathologically, negative infectious staining, localization of haloed structures to the upper dermis, a negative test for cryptococcal antigen, and elevated blood and urine iodine levels in this case all favored iododerma.
Cutaneous histoplasmosis is an infection caused by Histoplasma capsulatum, most commonly as secondary dissemination from pulmonary infection but rarely from direct inoculation of the skin.11 Presentation includes erythematous to hemorrhagic, umbilicated papules and plaques. Histopathologic findings are round to oval, narrow-based, budding yeasts that stain positive for PAS or mucicarmine. Although histoplasmosis can clinically mimic iododerma, the disease is distinguished histologically by the presence of fungal microorganisms that lack the gelatinous edema and haloed effect of iododerma.
We presented a unique case of iododerma simulating cryptococcal infection both clinically and histopathologically. Prompt recognition of histologic mimickers of true infectious microorganisms is essential to prevent unnecessary delay of withdrawal of the offending substance and to initiate appropriate therapy.
To the Editor:
A woman in her 40s presented with acute onset of rapidly spreading lesions on the face, trunk, and extremities. She reported high fever and endorsed malaise. She had a history of end-stage renal disease and was on renal dialysis. She recently underwent revision of an arteriovenous fistula.
Physical examination revealed diffuse, erythematous, firm papules and plaques with central hemorrhage and umbilication on the dorsal aspect of the nose, forehead, temples, and cheeks. There also were purpuric papules and plaques with a peripheral rim of vesiculation (Figure 1) on the medial and posterior thighs and buttocks. Histopathology of a biopsy specimen revealed an interstitial neutrophilic infiltrate in the superficial dermis and mid dermis with scattered, haloed, acellular structures simulating cryptococcal organisms (Figure 2). Periodic acid–Schiff (PAS), Grocott methenamine-silver, and mucicarmine staining was negative. Repeat biopsy showed similar findings. A (1-3)-β-
The findings compatible with a diagnosis of iododerma included umbilicated hemorrhagic papules and plaques, cryptococcal-like structures with negative staining on histopathology, and elevated iodine levels with a negative infectious workup. The patient was treated with topical corticosteroids. At 1-month follow-up, the lesions had resolved.
Iododerma is a halogenoderma, a skin eruption that occurs after ingestion of or exposure to a halogen-containing substance (eg, iodine, bromine, fluorine) or medication (eg, lithium).1 Common sources of iodine include iodinated contrast media, potassium iodide ingestion, topical application of povidone–iodine, radioactive iodine administration, and the antiarrhythmic amiodarone. Excess exposure to iodine-containing compounds typically occurs in the setting of kidney disease or failure as well as due to reduced iodine clearance.1 Although the pathogenesis of iododerma is unknown, the most common hypothesis is that lesions are delayed hypersensitivity reactions secondary to formation of a protein-halogen complex.2
The presentation of iododerma is polymorphous and includes acneform, vegetative, or pustular eruptions; umbilicated papules and plaques can be present.2,3 Lesions can be either asymptomatic or painful and pruritic. Timing between iodine exposure and onset of lesions varies from hours to days to years.2,4
Systemic symptoms of iododerma can occur, including salivary gland swelling, hypotension and bradycardia, kidney injury, or thyroid and liver abnormalities. Histopathologic analysis demonstrates a dense neutrophilic dermatitis with negative staining for infectious causes.4,5 Cryptococcal-like structures have been described in iododerma3; neutrophilic dermatoses of various causes that mimic cryptococcal infection have been reported.6 Ultimately, iododerma remains a diagnosis of exclusion.
Withdrawal of an offending compound is remedial. Dialysis is beneficial in end-stage renal disease. Topical, intralesional, and systemic corticosteroids, as well as antibiotics, provide variable benefit.4,7 Lesions can take 4 to 6 weeks to clear after withdrawal of the offending agent. It is unclear whether recurrences happen; iodine-containing compounds need to be avoided after a patient has been affected.
Iododerma has a broad differential diagnosis due to the polymorphous presentation of the disorder, including acute febrile neutrophilic dermatosis (also known as Sweet syndrome), cutaneous cryptococcosis, and cutaneous histoplasmosis. Sweet syndrome presents as abrupt onset of edematous erythematous plaques with fever and leukocytosis. It is associated with infection, inflammatory disorders, medication, and malignancy.8 Histopathologic analysis reveals papillary dermal edema and a neutrophilic dermatosis. Cytoplasmic vacuolization resembling C neoformans has been reported.9 The diagnosis is less favored in the presence of renal disease, temporal association of the eruption with iodine exposure, and elevated blood and urine iodine levels, as in our patient.
Cutaneous cryptococcosis, an infection caused by C neoformans, typically occurs secondary to dissemination from the lungs; rarely, the disease is primary. Acneform plaques, vegetative plaques, and umbilicated lesions are seen.10 Histopathologic analysis shows characteristic yeast forms of cryptococcosis surrounded by gelatinous edema, which create a haloed effect, typically throughout the dermis. Capsules are positive for PAS or mucicarmine staining. Although C neoformans can closely mimic iododerma both clinically and histopathologically, negative infectious staining, localization of haloed structures to the upper dermis, a negative test for cryptococcal antigen, and elevated blood and urine iodine levels in this case all favored iododerma.
Cutaneous histoplasmosis is an infection caused by Histoplasma capsulatum, most commonly as secondary dissemination from pulmonary infection but rarely from direct inoculation of the skin.11 Presentation includes erythematous to hemorrhagic, umbilicated papules and plaques. Histopathologic findings are round to oval, narrow-based, budding yeasts that stain positive for PAS or mucicarmine. Although histoplasmosis can clinically mimic iododerma, the disease is distinguished histologically by the presence of fungal microorganisms that lack the gelatinous edema and haloed effect of iododerma.
We presented a unique case of iododerma simulating cryptococcal infection both clinically and histopathologically. Prompt recognition of histologic mimickers of true infectious microorganisms is essential to prevent unnecessary delay of withdrawal of the offending substance and to initiate appropriate therapy.
- Alagheband M, Engineer L. Lithium and halogenoderma. Arch Dermatol. 2000;136:126-127. doi:10.1001/archderm.136.1.126
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379. doi:10.1111/bjd.12852
- Runge M, Williams K, Scharnitz T, et al. Iodine toxicity after iodinated contrast: new observations in iododerma. JAAD Case Rep. 2020;6:319-322. doi:10.1016/j.jdcr.2020.02.006
- Chalela JG, Aguilar L. Iododerma from contrast material. N Engl J Med. 2016;374:2477. doi:10.1056/NEJMicm1512512
- Chang MW, Miner JE, Moiin A, et al. Iododerma after computed tomographic scan with intravenous radiopaque contrast media. J Am Acad Dermatol. 1997;36:1014-1016. doi:10.1016/s0190-9622(97)80291-5
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi:10.1111/cup.12019
- Pranteda G, Grimaldi M, Salzetta M, et al. Vegetating iododerma and pulmonary eosinophilic infiltration. a simple co-occurrence? Acta Derm Venereol. 2004;84:480-481.
- Nelson CA, Stephen S, Ashchyan HJ, et al. M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.064
- Wilson J, Gleghorn K, Kelly B. Cryptococcoid Sweet’s syndrome: two reports of Sweet’s syndrome mimicking cutaneous cryptococcosis. J Cutan Pathol. 2017;44:413-419. doi:10.1111/cup.12921
- Beatson M, Harwood M, Reese V, et al. Primary cutaneous cryptococcosis in an elderly pigeon breeder. JAAD Case Rep. 2019;5:433-435. doi:10.1016/j.jdcr.2019.03.006
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348. doi:10.1177/0145561318097010-1108
- Alagheband M, Engineer L. Lithium and halogenoderma. Arch Dermatol. 2000;136:126-127. doi:10.1001/archderm.136.1.126
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379. doi:10.1111/bjd.12852
- Runge M, Williams K, Scharnitz T, et al. Iodine toxicity after iodinated contrast: new observations in iododerma. JAAD Case Rep. 2020;6:319-322. doi:10.1016/j.jdcr.2020.02.006
- Chalela JG, Aguilar L. Iododerma from contrast material. N Engl J Med. 2016;374:2477. doi:10.1056/NEJMicm1512512
- Chang MW, Miner JE, Moiin A, et al. Iododerma after computed tomographic scan with intravenous radiopaque contrast media. J Am Acad Dermatol. 1997;36:1014-1016. doi:10.1016/s0190-9622(97)80291-5
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi:10.1111/cup.12019
- Pranteda G, Grimaldi M, Salzetta M, et al. Vegetating iododerma and pulmonary eosinophilic infiltration. a simple co-occurrence? Acta Derm Venereol. 2004;84:480-481.
- Nelson CA, Stephen S, Ashchyan HJ, et al. M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.064
- Wilson J, Gleghorn K, Kelly B. Cryptococcoid Sweet’s syndrome: two reports of Sweet’s syndrome mimicking cutaneous cryptococcosis. J Cutan Pathol. 2017;44:413-419. doi:10.1111/cup.12921
- Beatson M, Harwood M, Reese V, et al. Primary cutaneous cryptococcosis in an elderly pigeon breeder. JAAD Case Rep. 2019;5:433-435. doi:10.1016/j.jdcr.2019.03.006
- Raggio B. Primary cutaneous histoplasmosis. Ear Nose Throat J. 2018;97:346-348. doi:10.1177/0145561318097010-1108
Practice Points
- Halogenodermas are rare cutaneous reactions to excess exposure to or ingestion of halogen-containing drugs or substances such as bromine, iodine (iododerma), fluorine, and rarely lithium.
- The clinical presentation of a halogenoderma varies; the most characteristic manifestation is a vegetative or exudative plaque with a peripheral rim of pustules.
- Histologically, lesions of a halogenoderma are characterized by pseudoepitheliomatous hyperplasia associated with numerous intraepidermal microabscesses overlying a dense mixed inflammatory infiltrate of neutrophils, plasma cells, eosinophils, histiocytes, and scattered multinucleated giant cells.
- Rarely, the dermal infiltrate of a halogenoderma contains abundant acellular bodies surrounded by capsulelike vacuolated spaces mimicking Cryptococcus neoformans.

Nonhealing Ulcer in a Patient With Crohn Disease
The Diagnosis: Mycobacterium abscessus Infection
Upon further testing, cultures were positive for Mycobacterium abscessus. Our patient was referred to infectious disease for co-management, and his treatment plan consisted of intravenous amikacin 885 mg 3 times weekly, intravenous imipenem 1 g twice daily, azithromycin 500 mg/d, and omadacycline 150 mg/d for at least 3 months. Magnetic resonance imaging findings were consistent with a combination of cellulitis and osteomyelitis, and our patient was referred to plastic surgery for debridement. He subsequently was lost to follow-up.
Mycobacterium abscessus is classified as both a nontuberculous and rapidly growing mycobacterium. Mycobacterium abscessus recently has emerged as a pathogen of increasing public health concern, especially due to its high rate of antibiotic resistance.1-5 It is highly prevalent in the environment, and infection has been reported from a wide variety of environmental sources.6-8 Immunocompromised individuals, such as our patient, undergoing anti–tumor necrosis factor therapy are at increased risk for infection from all Mycobacterium species.9-11 Recognizing these infections quickly is a priority for patient care, as M abscessus can lead to disseminated infection and high mortality rates.1
Histopathology of M abscessus consists of granulomatous inflammation with mixed granulomas12; however, these findings are not always appreciable, and staining does not always reveal visible organisms. In our patient, histopathology revealed patchy plasmalymphocytic infiltrates of the dermis and subcutaneous tissue, which are signs of generalized inflammation (Figure). Therefore, cultures positive for M abscessus are the gold standard for diagnosis and established the diagnosis in this case.
.")
The differential diagnoses for our patient’s ulceration included squamous cell carcinoma, pyoderma gangrenosum, aseptic abscess ulcer, and pyodermatitispyostomatitis vegetans. Immunosuppressive therapy is a risk factor for squamous cell carcinoma13,14; however, ulcerated squamous cell carcinoma typically presents with prominent everted edges with a necrotic tumor base.15 Biopsy reveals cells with abundant eosinophilic cytoplasm, large nuclei, and variable keratin pearls.16 Pyoderma gangrenosum is an inflammatory skin condition associated with Crohn disease and often is a diagnosis of exclusion characterized by neutrophilic infiltrates on biopsy.17-19 Aseptic abscess ulcers are characterized by neutrophil-filled lesions that respond to corticosteroids but not antibiotics.20 Pyodermatitis-pyostomatitis vegetans is a rare skin manifestation of inflammatory bowel disease associated with a pustular eruption of the skin and/or mouth. Histopathology reveals pustules within or below the epidermis with many eosinophils or neutrophils. Granulomas do not occur as in M abscessus.21
Treatment of M abscessus infection requires the coadministration of several antibiotics across multiple classes to ensure complete disease resolution. High rates of antibiotic resistance are characterized by at least partial resistance to almost every antibiotic; clarithromycin has near-complete efficacy, but resistant strains have started to emerge. Amikacin and cefoxitin are other antibiotics that have reported a resistance rate of less than 50%, but they are only effective 90% and 70% of the time, respectively.1,22 The antibiotic omadacycline, which is approved by the US Food and Drug Administration to treat acute bacterial skin and soft-tissue infections, also may have utility in treating M abscessus infections.23,24 Finally, phage therapy may offer a potential mode of treatment for this bacterium and was used to treat pulmonary infection in a patient with cystic fibrosis.25 Despite these newer innovations, the current standard of care involves clarithromycin or azithromycin in combination with a parenteral antibiotic such as cefoxitin, amikacin, or imipenem for at least 4 months.1
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus complex. Semin Respir Crit Care Med. 2018;39:362-376.
- Cardenas DD, Yasmin T, Ahmed S. A rare insidious case of skin and soft tissue infection due to Mycobacterium abscessus: a case report. Cureus. 2022;14:E25725.
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-577.
- Dickison P, Howard V, O’Kane G, et al. Mycobacterium abscessus infection following penetrations through wetsuits. Australas J Dermatol. 2019;60:57-59.
- Choi H, Kim YI, Na CH, et al. Mycobacterium abscessus skin infection associated with shaving activity in a 75-year-old man. Ann Geriatr Med Res. 2018;22:204.
- Costa-Silva M, Cesar A, Gomes NP, et al. Mycobacterium abscessus infection in a spa worker. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:159-161.
- Besada E. Rapid growing mycobacteria and TNF-α blockers: case report of a fatal lung infection with Mycobacterium abscessus. Clin Exp Rheumatol. 2011;29:705-707.
- Mufti AH, Toye BW, Mckendry RR, et al. Mycobacterium abscessus infection after use of tumor necrosis factor α inhibitor therapy: case report and review of infectious complications associated with tumor necrosis factor α inhibitor use. Diagn Microbiol Infect Dis. 2005;53:233-238.
- Lee SK, Kim SY, Kim EY, et al. Mycobacterial infections in patients treated with tumor necrosis factor antagonists in South Korea. Lung. 2013;191:565-571.
- Rodríguez G, Ortegón M, Camargo D, et al. Iatrogenic Mycobacterium abscessus infection: histopathology of 71 patients. Br J Dermatol. 1997;137:214-218.
- Firnhaber JM. Diagnosis and treatment of basal cell and squamous cell carcinoma. Am Fam Physician. 2012;86:161-168.
- Walker HS, Hardwicke J. Non-melanoma skin cancer. Surgery (Oxford). 2022;40:39-45.
- Browse NL. The skin. In: Browse NL, ed. An Introduction to the Symptoms and Signs of Surgical Disease. 3rd ed. London Arnold Publications; 2001:66-69.
- Weedon D. Squamous cell carcinoma. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010;691-700.
- Powell F, Schroeter A, Su W, et al. Pyoderma gangrenosum: a review of 86 patients. QJM Int J Med. 1985;55:173-186.
- Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118:743-768.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- André MFJ, Piette JC, Kémény JL, et al. Aseptic abscesses: a study of 30 patients with or without inflammatory bowel disease and review of the literature. Medicine (Baltimore). 2007;86:145. doi:10.1097/md.0b013e18064f9f3
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015;36:67-78.
- Duah M, Beshay M. Omadacycline in first-line combination therapy for pulmonary Mycobacterium abscessus infection: a case series. Int J Infect Dis. 2022;122:953-956.
- Minhas R, Sharma S, Kundu S. Utilizing the promise of omadacycline in a resistant, non-tubercular mycobacterial pulmonary infection. Cureus. 2019;11:E5112.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-733.
The Diagnosis: Mycobacterium abscessus Infection
Upon further testing, cultures were positive for Mycobacterium abscessus. Our patient was referred to infectious disease for co-management, and his treatment plan consisted of intravenous amikacin 885 mg 3 times weekly, intravenous imipenem 1 g twice daily, azithromycin 500 mg/d, and omadacycline 150 mg/d for at least 3 months. Magnetic resonance imaging findings were consistent with a combination of cellulitis and osteomyelitis, and our patient was referred to plastic surgery for debridement. He subsequently was lost to follow-up.
Mycobacterium abscessus is classified as both a nontuberculous and rapidly growing mycobacterium. Mycobacterium abscessus recently has emerged as a pathogen of increasing public health concern, especially due to its high rate of antibiotic resistance.1-5 It is highly prevalent in the environment, and infection has been reported from a wide variety of environmental sources.6-8 Immunocompromised individuals, such as our patient, undergoing anti–tumor necrosis factor therapy are at increased risk for infection from all Mycobacterium species.9-11 Recognizing these infections quickly is a priority for patient care, as M abscessus can lead to disseminated infection and high mortality rates.1
Histopathology of M abscessus consists of granulomatous inflammation with mixed granulomas12; however, these findings are not always appreciable, and staining does not always reveal visible organisms. In our patient, histopathology revealed patchy plasmalymphocytic infiltrates of the dermis and subcutaneous tissue, which are signs of generalized inflammation (Figure). Therefore, cultures positive for M abscessus are the gold standard for diagnosis and established the diagnosis in this case.
The differential diagnoses for our patient’s ulceration included squamous cell carcinoma, pyoderma gangrenosum, aseptic abscess ulcer, and pyodermatitispyostomatitis vegetans. Immunosuppressive therapy is a risk factor for squamous cell carcinoma13,14; however, ulcerated squamous cell carcinoma typically presents with prominent everted edges with a necrotic tumor base.15 Biopsy reveals cells with abundant eosinophilic cytoplasm, large nuclei, and variable keratin pearls.16 Pyoderma gangrenosum is an inflammatory skin condition associated with Crohn disease and often is a diagnosis of exclusion characterized by neutrophilic infiltrates on biopsy.17-19 Aseptic abscess ulcers are characterized by neutrophil-filled lesions that respond to corticosteroids but not antibiotics.20 Pyodermatitis-pyostomatitis vegetans is a rare skin manifestation of inflammatory bowel disease associated with a pustular eruption of the skin and/or mouth. Histopathology reveals pustules within or below the epidermis with many eosinophils or neutrophils. Granulomas do not occur as in M abscessus.21
Treatment of M abscessus infection requires the coadministration of several antibiotics across multiple classes to ensure complete disease resolution. High rates of antibiotic resistance are characterized by at least partial resistance to almost every antibiotic; clarithromycin has near-complete efficacy, but resistant strains have started to emerge. Amikacin and cefoxitin are other antibiotics that have reported a resistance rate of less than 50%, but they are only effective 90% and 70% of the time, respectively.1,22 The antibiotic omadacycline, which is approved by the US Food and Drug Administration to treat acute bacterial skin and soft-tissue infections, also may have utility in treating M abscessus infections.23,24 Finally, phage therapy may offer a potential mode of treatment for this bacterium and was used to treat pulmonary infection in a patient with cystic fibrosis.25 Despite these newer innovations, the current standard of care involves clarithromycin or azithromycin in combination with a parenteral antibiotic such as cefoxitin, amikacin, or imipenem for at least 4 months.1
The Diagnosis: Mycobacterium abscessus Infection
Upon further testing, cultures were positive for Mycobacterium abscessus. Our patient was referred to infectious disease for co-management, and his treatment plan consisted of intravenous amikacin 885 mg 3 times weekly, intravenous imipenem 1 g twice daily, azithromycin 500 mg/d, and omadacycline 150 mg/d for at least 3 months. Magnetic resonance imaging findings were consistent with a combination of cellulitis and osteomyelitis, and our patient was referred to plastic surgery for debridement. He subsequently was lost to follow-up.
Mycobacterium abscessus is classified as both a nontuberculous and rapidly growing mycobacterium. Mycobacterium abscessus recently has emerged as a pathogen of increasing public health concern, especially due to its high rate of antibiotic resistance.1-5 It is highly prevalent in the environment, and infection has been reported from a wide variety of environmental sources.6-8 Immunocompromised individuals, such as our patient, undergoing anti–tumor necrosis factor therapy are at increased risk for infection from all Mycobacterium species.9-11 Recognizing these infections quickly is a priority for patient care, as M abscessus can lead to disseminated infection and high mortality rates.1
Histopathology of M abscessus consists of granulomatous inflammation with mixed granulomas12; however, these findings are not always appreciable, and staining does not always reveal visible organisms. In our patient, histopathology revealed patchy plasmalymphocytic infiltrates of the dermis and subcutaneous tissue, which are signs of generalized inflammation (Figure). Therefore, cultures positive for M abscessus are the gold standard for diagnosis and established the diagnosis in this case.
The differential diagnoses for our patient’s ulceration included squamous cell carcinoma, pyoderma gangrenosum, aseptic abscess ulcer, and pyodermatitispyostomatitis vegetans. Immunosuppressive therapy is a risk factor for squamous cell carcinoma13,14; however, ulcerated squamous cell carcinoma typically presents with prominent everted edges with a necrotic tumor base.15 Biopsy reveals cells with abundant eosinophilic cytoplasm, large nuclei, and variable keratin pearls.16 Pyoderma gangrenosum is an inflammatory skin condition associated with Crohn disease and often is a diagnosis of exclusion characterized by neutrophilic infiltrates on biopsy.17-19 Aseptic abscess ulcers are characterized by neutrophil-filled lesions that respond to corticosteroids but not antibiotics.20 Pyodermatitis-pyostomatitis vegetans is a rare skin manifestation of inflammatory bowel disease associated with a pustular eruption of the skin and/or mouth. Histopathology reveals pustules within or below the epidermis with many eosinophils or neutrophils. Granulomas do not occur as in M abscessus.21
Treatment of M abscessus infection requires the coadministration of several antibiotics across multiple classes to ensure complete disease resolution. High rates of antibiotic resistance are characterized by at least partial resistance to almost every antibiotic; clarithromycin has near-complete efficacy, but resistant strains have started to emerge. Amikacin and cefoxitin are other antibiotics that have reported a resistance rate of less than 50%, but they are only effective 90% and 70% of the time, respectively.1,22 The antibiotic omadacycline, which is approved by the US Food and Drug Administration to treat acute bacterial skin and soft-tissue infections, also may have utility in treating M abscessus infections.23,24 Finally, phage therapy may offer a potential mode of treatment for this bacterium and was used to treat pulmonary infection in a patient with cystic fibrosis.25 Despite these newer innovations, the current standard of care involves clarithromycin or azithromycin in combination with a parenteral antibiotic such as cefoxitin, amikacin, or imipenem for at least 4 months.1
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus complex. Semin Respir Crit Care Med. 2018;39:362-376.
- Cardenas DD, Yasmin T, Ahmed S. A rare insidious case of skin and soft tissue infection due to Mycobacterium abscessus: a case report. Cureus. 2022;14:E25725.
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-577.
- Dickison P, Howard V, O’Kane G, et al. Mycobacterium abscessus infection following penetrations through wetsuits. Australas J Dermatol. 2019;60:57-59.
- Choi H, Kim YI, Na CH, et al. Mycobacterium abscessus skin infection associated with shaving activity in a 75-year-old man. Ann Geriatr Med Res. 2018;22:204.
- Costa-Silva M, Cesar A, Gomes NP, et al. Mycobacterium abscessus infection in a spa worker. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:159-161.
- Besada E. Rapid growing mycobacteria and TNF-α blockers: case report of a fatal lung infection with Mycobacterium abscessus. Clin Exp Rheumatol. 2011;29:705-707.
- Mufti AH, Toye BW, Mckendry RR, et al. Mycobacterium abscessus infection after use of tumor necrosis factor α inhibitor therapy: case report and review of infectious complications associated with tumor necrosis factor α inhibitor use. Diagn Microbiol Infect Dis. 2005;53:233-238.
- Lee SK, Kim SY, Kim EY, et al. Mycobacterial infections in patients treated with tumor necrosis factor antagonists in South Korea. Lung. 2013;191:565-571.
- Rodríguez G, Ortegón M, Camargo D, et al. Iatrogenic Mycobacterium abscessus infection: histopathology of 71 patients. Br J Dermatol. 1997;137:214-218.
- Firnhaber JM. Diagnosis and treatment of basal cell and squamous cell carcinoma. Am Fam Physician. 2012;86:161-168.
- Walker HS, Hardwicke J. Non-melanoma skin cancer. Surgery (Oxford). 2022;40:39-45.
- Browse NL. The skin. In: Browse NL, ed. An Introduction to the Symptoms and Signs of Surgical Disease. 3rd ed. London Arnold Publications; 2001:66-69.
- Weedon D. Squamous cell carcinoma. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010;691-700.
- Powell F, Schroeter A, Su W, et al. Pyoderma gangrenosum: a review of 86 patients. QJM Int J Med. 1985;55:173-186.
- Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118:743-768.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- André MFJ, Piette JC, Kémény JL, et al. Aseptic abscesses: a study of 30 patients with or without inflammatory bowel disease and review of the literature. Medicine (Baltimore). 2007;86:145. doi:10.1097/md.0b013e18064f9f3
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015;36:67-78.
- Duah M, Beshay M. Omadacycline in first-line combination therapy for pulmonary Mycobacterium abscessus infection: a case series. Int J Infect Dis. 2022;122:953-956.
- Minhas R, Sharma S, Kundu S. Utilizing the promise of omadacycline in a resistant, non-tubercular mycobacterial pulmonary infection. Cureus. 2019;11:E5112.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-733.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus complex. Semin Respir Crit Care Med. 2018;39:362-376.
- Cardenas DD, Yasmin T, Ahmed S. A rare insidious case of skin and soft tissue infection due to Mycobacterium abscessus: a case report. Cureus. 2022;14:E25725.
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-577.
- Dickison P, Howard V, O’Kane G, et al. Mycobacterium abscessus infection following penetrations through wetsuits. Australas J Dermatol. 2019;60:57-59.
- Choi H, Kim YI, Na CH, et al. Mycobacterium abscessus skin infection associated with shaving activity in a 75-year-old man. Ann Geriatr Med Res. 2018;22:204.
- Costa-Silva M, Cesar A, Gomes NP, et al. Mycobacterium abscessus infection in a spa worker. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:159-161.
- Besada E. Rapid growing mycobacteria and TNF-α blockers: case report of a fatal lung infection with Mycobacterium abscessus. Clin Exp Rheumatol. 2011;29:705-707.
- Mufti AH, Toye BW, Mckendry RR, et al. Mycobacterium abscessus infection after use of tumor necrosis factor α inhibitor therapy: case report and review of infectious complications associated with tumor necrosis factor α inhibitor use. Diagn Microbiol Infect Dis. 2005;53:233-238.
- Lee SK, Kim SY, Kim EY, et al. Mycobacterial infections in patients treated with tumor necrosis factor antagonists in South Korea. Lung. 2013;191:565-571.
- Rodríguez G, Ortegón M, Camargo D, et al. Iatrogenic Mycobacterium abscessus infection: histopathology of 71 patients. Br J Dermatol. 1997;137:214-218.
- Firnhaber JM. Diagnosis and treatment of basal cell and squamous cell carcinoma. Am Fam Physician. 2012;86:161-168.
- Walker HS, Hardwicke J. Non-melanoma skin cancer. Surgery (Oxford). 2022;40:39-45.
- Browse NL. The skin. In: Browse NL, ed. An Introduction to the Symptoms and Signs of Surgical Disease. 3rd ed. London Arnold Publications; 2001:66-69.
- Weedon D. Squamous cell carcinoma. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010;691-700.
- Powell F, Schroeter A, Su W, et al. Pyoderma gangrenosum: a review of 86 patients. QJM Int J Med. 1985;55:173-186.
- Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (ecthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118:743-768.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- André MFJ, Piette JC, Kémény JL, et al. Aseptic abscesses: a study of 30 patients with or without inflammatory bowel disease and review of the literature. Medicine (Baltimore). 2007;86:145. doi:10.1097/md.0b013e18064f9f3
- Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal. 2009;14:E114-E117.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015;36:67-78.
- Duah M, Beshay M. Omadacycline in first-line combination therapy for pulmonary Mycobacterium abscessus infection: a case series. Int J Infect Dis. 2022;122:953-956.
- Minhas R, Sharma S, Kundu S. Utilizing the promise of omadacycline in a resistant, non-tubercular mycobacterial pulmonary infection. Cureus. 2019;11:E5112.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-733.
A 24-year-old man presented to our dermatology clinic with a painful lesion on the right buccal cheek of 4 months’ duration that had not changed in size or appearance. He had a history of Crohn disease that was being treated with 6-mercaptopurine and infliximab. He underwent jaw surgery 7 years prior for correction of an underbite, followed by subsequent surgery to remove the hardware 1 year after the initial procedure. He experienced recurring skin abscesses following the initial jaw surgery roughly once a year that were treated with bedside incision and drainage procedures in the emergency department followed by trimethoprim-sulfamethoxazole with complete resolution; however, treatment with mupirocin ointment 2%, trimethoprim-sulfamethoxazole, and azithromycin did not provide symptomatic relief or resolution for the current lesion. Physical examination revealed a 4-cm ulceration with actively draining serosanguineous discharge. Two punch biopsies were performed; 48-hour bacterial and fungal cultures, as well as Giemsa, acid-fast bacilli, and periodic acid–Schiff staining were negative.


Allergic Contact Dermatitis
THE COMPARISON
A An 11-year-old Hispanic boy with allergic contact dermatitis (ACD) on the abdomen. The geometric nature of the eruption and proximity to the belt buckle were highly suggestive of ACD to nickel; patch testing was not needed.
B A Black woman with ACD on the neck. A punch biopsy demonstrated spongiotic dermatitis that was typical of ACD. The diagnosis was supported by the patient’s history of dermatitis that developed after new products were applied to the hair. The patient declined patch testing.
C A Hispanic man with ACD on hair-bearing areas on the face where hair dye was used. The patient’s history of dermatitis following the application of hair dye was highly suggestive of ACD; patch testing confirmed the allergen was paraphenylenediamine (PPD).
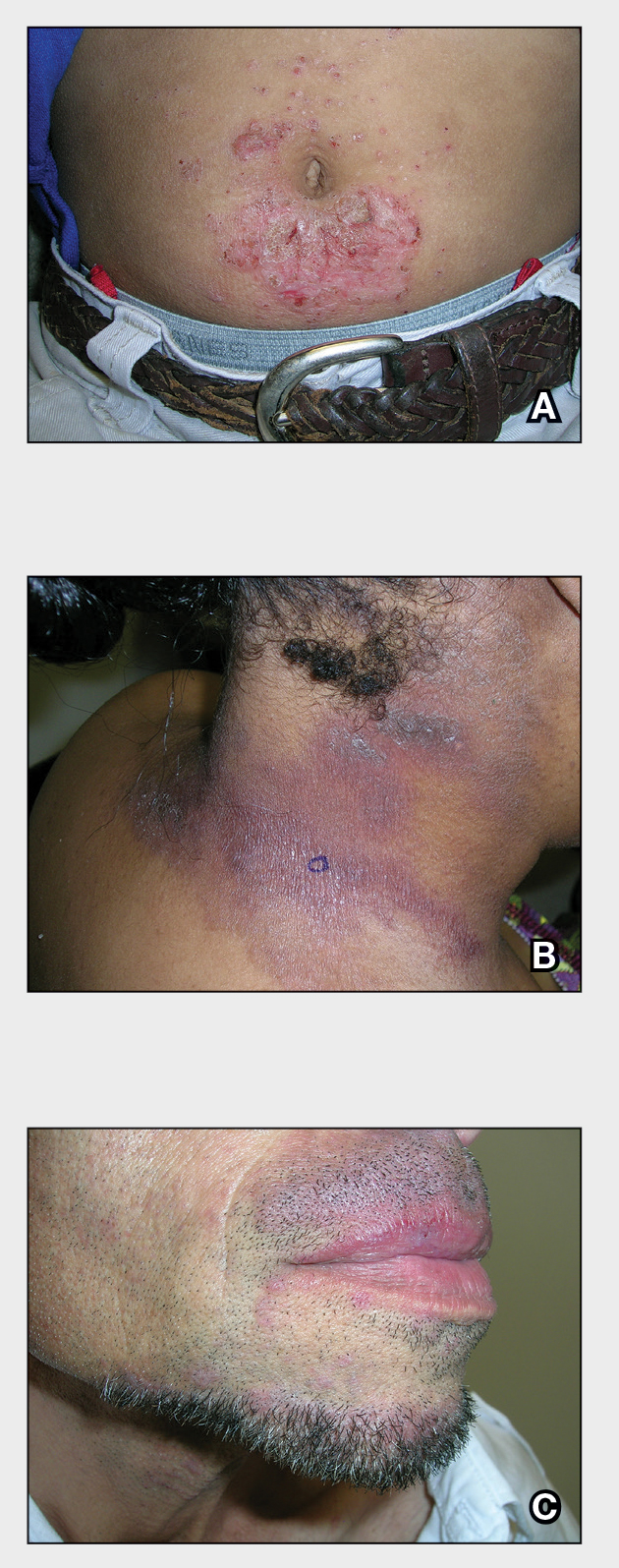
Allergic contact dermatitis (ACD) is an inflammatory condition of the skin caused by an immunologic response to one or more identifiable allergens. A delayed-type immune response (type IV hypersensitivity reaction) occurs after the skin is reexposed to an offending allergen.1 Severe pruritus is the main symptom of ACD in the early stages, accompanied by erythema, vesicles, and scaling in a distinct pattern corresponding to the allergen’s contact with the skin.2 Delayed widespread dermatitis after exposure to an allergen—a phenomenon known as autoeczematization (id reaction)—also may occur.3
The gold-standard diagnostic tool for ACD is patch testing, in which the patient is re-exposed to the suspected contact allergen(s) and observed for the development of dermatitis.4 However, ACD can be diagnosed with a detailed patient history including occupation, hobbies, personal care practices, and possible triggers with subsequent rashes. Thorough clinical examination of the skin is paramount. Indicators of possible ACD include dermatitis that persists despite use of appropriate treatment, an unexplained flare of previously quiescent dermatitis, and a diagnosis of dermatitis without a clear cause.1
Hairdressers, health care workers, and metal workers are at higher risk for ACD.5 Occupational ACD has notable socioeconomic implications, as it can result in frequent sick days, inability to perform tasks at work, and in some cases job loss.6
Patients with atopic dermatitis have impaired barrier function of the skin, permitting the entrance of allergens and subsequent sensitization.7 Allergic contact dermatitis is a challenge to manage, as complete avoidance of the allergen may not be possible.8
The underrepresentation of patients with skin of color (SOC) in educational materials as well as socioeconomic health disparities may contribute to the lower rates of diagnosis, patch testing, and treatment of ACD in this patient population.
Epidemiology
An ACD prevalence of 15.2% was reported in a study of 793 Danish patients who underwent skin prick and patch testing.9 Alinaghi et al10 conducted a meta-analysis of 20,107 patients across 28 studies who were patch tested to determine the prevalence of ACD in the general population. The researchers concluded that 20.1% (95% CI, 16.8%- 23.7%) of the general population experienced ACD. They analyzed 22 studies to determine the prevalence of ACD based on specific geographic area including 18,709 individuals from Europe with a prevalence of 19.5% (95% CI, 15.8%-23.4%), 1639 individuals from North America with a prevalence of 20.6% (95% CI, 9.2%-35.2%), and 2 studies from China (no other studies from Asia found) with a prevalence of 20.6% (95% CI, 17.4%-23.9%). Researchers did not find data from studies conducted in Africa or South America.10
The current available epidemiologic data on ACD are not representative of SOC populations. DeLeo et al11 looked at patch test reaction patterns in association with race and ethnicity in a large sample size (N=19,457); 17,803 (92.9%) of these patients were White and only 1360 (7.1%) were Black. Large-scale, inclusive studies are needed, which can only be achieved with increased suspicion for ACD and increased access to patch testing.
Allergic contact dermatitis is more common in women, with nickel being the most frequently identified allergen (Figure, A).10 Personal care products often are linked to ACD (Figure, B). An analysis of data from the North American Contact Dermatitis Group revealed that the top 5 personal care product allergens were methylisothiazolinone (a preservative), fragrance mix I, balsam of Peru, quaternium-15 (a preservative), and paraphenylenediamine (PPD)(a common component of hair dye) (Figure, C).12
There is a paucity of epidemiologic data among various ethnic groups; however, a few studies have suggested that there is no difference in the frequency rates of positive patch test results in Black vs White populations.11,13,14 One study of patch test results from 114 Black patients and 877 White patients at the Cleveland Clinic Foundation in Ohio demonstrated a similar allergy frequency of 43.0% and 43.6%, respectively.13 However, there were differences in the types of allergen sensitization. Black patients had higher positive patch test rates for PPD than White patients (10.6% vs 4.5%). Black men had a higher frequency of sensitivity to PPD (21.2% vs 4.2%) and imidazolidinyl urea (a formaldehyde-releasing preservative) (9.1% vs 2.6%) compared to White men.13
Ethnicity and cultural practices influence epidemiologic patterns of ACD. Darker hair dyes used in Black patients14 and deeply pigmented PPD dye found in henna tattoos used in Indian and Black patients15 may lead to increased sensitization to PPD. Allergic contact dermatitis due to formaldehyde is more common in White patients, possibly due to more frequent use of formaldehyde-containing moisturizers, shampoos, and creams.15
Key clinical features in people with darker skin tones
In patients with SOC, the clinical features of ACD vary, posing a diagnostic challenge. Hyperpigmentation, lichenification, and induration are more likely to be seen than the papules, vesicles, and erythematous dermatitis often described in lighter skin tones or acute ACD. Erythema can be difficult to assess on darker skin and may appear violaceous or very faint pink.16
Worth noting
A high index of suspicion is necessary when interpreting patch tests in patients with SOC, as patch test kits use a reading plate with graduated intensities of erythema, papulation, and vesicular reactions to determine the likelihood of ACD. The potential contact allergens are placed on the skin on day 1 and covered. Then, on day 3 the allergens are removed. The skin is clinically evaluated using visual assessment and skin palpation. The reactions are graded as negative, irritant reaction, equivocal, weak positive, strong positive, or extreme reaction at around days 3 and 5 to capture both early and delayed reactions.17 A patch test may be positive even if obvious signs of erythema are not appreciated as expected.
Adjusting the lighting in the examination room, including side lighting, or using a blue background can be helpful in identifying erythema in darker skin tones.15,16,18 Palpation of the skin also is useful, as even slight texture changes and induration are indicators of a possible skin reaction to the test allergen.15
Health disparity highlight
Clinical photographs of ACD and patch test results in patients with SOC are not commonplace in the literature. Positive patch test results in patients with darker skin tones vary from those of patients with lighter skin tones, and if the clinician reading the patch test result is not familiar with the findings in darker skin tones, the diagnosis may be delayed or missed.15
Furthermore, Scott et al15 highlighted that many dermatology residency training programs have a paucity of SOC education in their curriculum. This lack of representation may contribute to the diagnostic challenges encountered by health care providers.
Timely access to health care and education as well as economic stability are essential for the successful management of patients with ACD. Some individuals with SOC have been disproportionately affected by social determinants of health. Rodriguez-Homs et al19 demonstrated that the distance needed to travel to a clinic and the poverty rate of the county the patient lives in play a role in referral to a clinician specializing in contact dermatitis.
A retrospective registry review of 2310 patients undergoing patch testing at the Massachusetts General Hospital in Boston revealed that 2.5% were Black, 5.5% were Latinx, 8.3% were Asian, and the remaining 83.7% were White.20 Qian et al21 also looked at patch testing patterns among various sociodemographic groups (N=1,107,530) and found that 69% of patients were White and 59% were female. Rates of patch testing among patients who were Black, lesser educated, male, lower income, and younger (children aged 0–12 years) were significantly lower than for other groups when ACD was suspected (P<.0001).21 The lower rates of patch testing in patients with SOC may be due to low suspicion of diagnosis, low referral rates due to limited medical insurance, and financial instability, as well as other socioeconomic factors.20
Tamazian et al16 reviewed pediatric populations at 13 US centers and found that Black children received patch testing less frequently than White and Hispanic children. Another review of pediatric patch testing in patients with SOC found that a less comprehensive panel of allergens was used in this population.22
The key to resolution of ACD is removal of the offending antigen, and if patients are not being tested, then they risk having a prolonged and complicated course of ACD with a poor prognosis. Patients with SOC also experience greater negative psychosocial impact due to ACD disease burden.21,23
The lower rates of patch testing in Black patients cannot solely be attributed to difficulty diagnosing ACD in darker skin tones; it is likely due to the impact of social determinants of health. Alleviating health disparities will improve patient outcomes and quality of life.
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74: 1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Bertoli MJ, Schwartz RA, Janniger CK. Autoeczematization: a strange id reaction of the skin. Cutis. 2021;108:163-166. doi:10.12788/cutis.0342
- Johansen JD, Bonefeld CM, Schwensen JFB, et al. Novel insights into contact dermatitis. J Allergy Clin Immunol. 2022;149:1162-1171. doi:10.1016/j.jaci.2022.02.002
- Karagounis TK, Cohen DE. Occupational hand dermatitis. Curr Allergy Asthma Rep. 2023;23:201-212. doi:10.1007/s11882-023-01070-5
- Cvetkovski RS, Rothman KJ, Olsen J, et al. Relation between diagnoses on severity, sick leave and loss of job among patients with occupational hand eczema. Br J Dermatol. 2005;152:93-98. doi:10.1111/j .1365-2133.2005.06415.x
- Owen JL, Vakharia PP, Silverberg JI. The role and diagnosis of allergic contact dermatitis in patients with atopic dermatitis. Am J Clin Dermatol. 2018;19:293-302. doi:10.1007/s40257-017-0340-7
- Brites GS, Ferreira I, Sebastião AI, et al. Allergic contact dermatitis: from pathophysiology to development of new preventive strategies. Pharmacol Res. 2020;162:105282. doi:10.1016/j.phrs.2020.105282
- Nielsen NH, Menne T. The relationship between IgE‐mediated and cell‐mediated hypersensitivities in an unselected Danish population: the Glostrup Allergy Study, Denmark. Br J Dermatol. 1996;134:669-672. doi:10.1111/j.1365-2133.1996.tb06967.x
- Alinaghi F, Bennike NH, Egeberg A, et al. Prevalence of contact allergy in the general population: a systematic review and meta‐analysis. Contact Dermatitis. 2019;80:77-85. doi:10.1111/cod.13119
- DeLeo VA, Alexis A, Warshaw EM, et al. The association of race/ethnicity and patch test results: North American Contact Dermatitis Group, 1998- 2006. Dermatitis. 2016;27:288-292. doi:10.1097/DER.0000000000000220
- Warshaw EM, Schlarbaum JP, Silverberg JI, et al. Contact dermatitis to personal care products is increasing (but different!) in males and females: North American Contact Dermatitis Group data, 1996-2016. J Am Acad Dermatol. 2021;85:1446-1455. doi:10.1016/j.jaad.2020.10.003
- Dickel H, Taylor JS, Evey P, et al. Comparison of patch test results with a standard series among white and black racial groups. Am J Contact Dermatol. 2001;12:77-82. doi:10.1053/ajcd.2001.20110
- DeLeo VA, Taylor SC, Belsito DV, et al. The effect of race and ethnicity on patch test results. J Am Acad Dermatol. 2002;46(2 suppl):S107-S112. doi:10.1067/mjd.2002.120792
- Scott I, Atwater AR, Reeder M. Update on contact dermatitis and patch testing in patients with skin of color. Cutis. 2021;108:10-12. doi:10.12788/cutis.0292
- Tamazian S, Oboite M, Treat JR. Patch testing in skin of color: a brief report. Pediatr Dermatol. 2021;38:952-953. doi:10.1111/pde.14578
- Litchman G, Nair PA, Atwater AR, et al. Contact dermatitis. StatPearls [Internet]. Updated February 9, 2023. Accessed September 25, 2023. https://www.ncbi.nlm.nih.gov/books/NBK459230/
- Alexis AF, Callender VD, Baldwin HE, et al. Global epidemiology and clinical spectrum of rosacea, highlighting skin of color: review and clinical practice experience. J Am Acad Dermatol. 2019;80:1722-1729. doi:10.1016/j.jaad.2018.08.049
- Rodriguez-Homs LG, Liu B, Green CL, et al. Duration of dermatitis before patch test appointment is associated with distance to clinic and county poverty rate. Dermatitis. 2020;31:259-264. doi:10.1097 /DER.0000000000000581
- Foschi CM, Tam I, Schalock PC, et al. Patch testing results in skin of color: a retrospective review from the Massachusetts General Hospital contact dermatitis clinic. J Am Acad Dermatol. 2022;87:452-454. doi:10.1016/j.jaad.2021.09.022
- Qian MF, Li S, Honari G, et al. Sociodemographic disparities in patch testing for commercially insured patients with dermatitis: a retrospective analysis of administrative claims data. J Am Acad Dermatol. 2022;87:1411-1413. doi:10.1016/j.jaad.2022.08.041
- Young K, Collis RW, Sheinbein D, et al. Retrospective review of pediatric patch testing results in skin of color. J Am Acad Dermatol. 2023;88:953-954. doi:10.1016/j.jaad.2022.11.031
- Kadyk DL, Hall S, Belsito DV. Quality of life of patients with allergic contact dermatitis: an exploratory analysis by gender, ethnicity, age, and occupation. Dermatitis. 2004;15:117-124.
THE COMPARISON
A An 11-year-old Hispanic boy with allergic contact dermatitis (ACD) on the abdomen. The geometric nature of the eruption and proximity to the belt buckle were highly suggestive of ACD to nickel; patch testing was not needed.
B A Black woman with ACD on the neck. A punch biopsy demonstrated spongiotic dermatitis that was typical of ACD. The diagnosis was supported by the patient’s history of dermatitis that developed after new products were applied to the hair. The patient declined patch testing.
C A Hispanic man with ACD on hair-bearing areas on the face where hair dye was used. The patient’s history of dermatitis following the application of hair dye was highly suggestive of ACD; patch testing confirmed the allergen was paraphenylenediamine (PPD).
Allergic contact dermatitis (ACD) is an inflammatory condition of the skin caused by an immunologic response to one or more identifiable allergens. A delayed-type immune response (type IV hypersensitivity reaction) occurs after the skin is reexposed to an offending allergen.1 Severe pruritus is the main symptom of ACD in the early stages, accompanied by erythema, vesicles, and scaling in a distinct pattern corresponding to the allergen’s contact with the skin.2 Delayed widespread dermatitis after exposure to an allergen—a phenomenon known as autoeczematization (id reaction)—also may occur.3
The gold-standard diagnostic tool for ACD is patch testing, in which the patient is re-exposed to the suspected contact allergen(s) and observed for the development of dermatitis.4 However, ACD can be diagnosed with a detailed patient history including occupation, hobbies, personal care practices, and possible triggers with subsequent rashes. Thorough clinical examination of the skin is paramount. Indicators of possible ACD include dermatitis that persists despite use of appropriate treatment, an unexplained flare of previously quiescent dermatitis, and a diagnosis of dermatitis without a clear cause.1
Hairdressers, health care workers, and metal workers are at higher risk for ACD.5 Occupational ACD has notable socioeconomic implications, as it can result in frequent sick days, inability to perform tasks at work, and in some cases job loss.6
Patients with atopic dermatitis have impaired barrier function of the skin, permitting the entrance of allergens and subsequent sensitization.7 Allergic contact dermatitis is a challenge to manage, as complete avoidance of the allergen may not be possible.8
The underrepresentation of patients with skin of color (SOC) in educational materials as well as socioeconomic health disparities may contribute to the lower rates of diagnosis, patch testing, and treatment of ACD in this patient population.
Epidemiology
An ACD prevalence of 15.2% was reported in a study of 793 Danish patients who underwent skin prick and patch testing.9 Alinaghi et al10 conducted a meta-analysis of 20,107 patients across 28 studies who were patch tested to determine the prevalence of ACD in the general population. The researchers concluded that 20.1% (95% CI, 16.8%- 23.7%) of the general population experienced ACD. They analyzed 22 studies to determine the prevalence of ACD based on specific geographic area including 18,709 individuals from Europe with a prevalence of 19.5% (95% CI, 15.8%-23.4%), 1639 individuals from North America with a prevalence of 20.6% (95% CI, 9.2%-35.2%), and 2 studies from China (no other studies from Asia found) with a prevalence of 20.6% (95% CI, 17.4%-23.9%). Researchers did not find data from studies conducted in Africa or South America.10
The current available epidemiologic data on ACD are not representative of SOC populations. DeLeo et al11 looked at patch test reaction patterns in association with race and ethnicity in a large sample size (N=19,457); 17,803 (92.9%) of these patients were White and only 1360 (7.1%) were Black. Large-scale, inclusive studies are needed, which can only be achieved with increased suspicion for ACD and increased access to patch testing.
Allergic contact dermatitis is more common in women, with nickel being the most frequently identified allergen (Figure, A).10 Personal care products often are linked to ACD (Figure, B). An analysis of data from the North American Contact Dermatitis Group revealed that the top 5 personal care product allergens were methylisothiazolinone (a preservative), fragrance mix I, balsam of Peru, quaternium-15 (a preservative), and paraphenylenediamine (PPD)(a common component of hair dye) (Figure, C).12
There is a paucity of epidemiologic data among various ethnic groups; however, a few studies have suggested that there is no difference in the frequency rates of positive patch test results in Black vs White populations.11,13,14 One study of patch test results from 114 Black patients and 877 White patients at the Cleveland Clinic Foundation in Ohio demonstrated a similar allergy frequency of 43.0% and 43.6%, respectively.13 However, there were differences in the types of allergen sensitization. Black patients had higher positive patch test rates for PPD than White patients (10.6% vs 4.5%). Black men had a higher frequency of sensitivity to PPD (21.2% vs 4.2%) and imidazolidinyl urea (a formaldehyde-releasing preservative) (9.1% vs 2.6%) compared to White men.13
Ethnicity and cultural practices influence epidemiologic patterns of ACD. Darker hair dyes used in Black patients14 and deeply pigmented PPD dye found in henna tattoos used in Indian and Black patients15 may lead to increased sensitization to PPD. Allergic contact dermatitis due to formaldehyde is more common in White patients, possibly due to more frequent use of formaldehyde-containing moisturizers, shampoos, and creams.15
Key clinical features in people with darker skin tones
In patients with SOC, the clinical features of ACD vary, posing a diagnostic challenge. Hyperpigmentation, lichenification, and induration are more likely to be seen than the papules, vesicles, and erythematous dermatitis often described in lighter skin tones or acute ACD. Erythema can be difficult to assess on darker skin and may appear violaceous or very faint pink.16
Worth noting
A high index of suspicion is necessary when interpreting patch tests in patients with SOC, as patch test kits use a reading plate with graduated intensities of erythema, papulation, and vesicular reactions to determine the likelihood of ACD. The potential contact allergens are placed on the skin on day 1 and covered. Then, on day 3 the allergens are removed. The skin is clinically evaluated using visual assessment and skin palpation. The reactions are graded as negative, irritant reaction, equivocal, weak positive, strong positive, or extreme reaction at around days 3 and 5 to capture both early and delayed reactions.17 A patch test may be positive even if obvious signs of erythema are not appreciated as expected.
Adjusting the lighting in the examination room, including side lighting, or using a blue background can be helpful in identifying erythema in darker skin tones.15,16,18 Palpation of the skin also is useful, as even slight texture changes and induration are indicators of a possible skin reaction to the test allergen.15
Health disparity highlight
Clinical photographs of ACD and patch test results in patients with SOC are not commonplace in the literature. Positive patch test results in patients with darker skin tones vary from those of patients with lighter skin tones, and if the clinician reading the patch test result is not familiar with the findings in darker skin tones, the diagnosis may be delayed or missed.15
Furthermore, Scott et al15 highlighted that many dermatology residency training programs have a paucity of SOC education in their curriculum. This lack of representation may contribute to the diagnostic challenges encountered by health care providers.
Timely access to health care and education as well as economic stability are essential for the successful management of patients with ACD. Some individuals with SOC have been disproportionately affected by social determinants of health. Rodriguez-Homs et al19 demonstrated that the distance needed to travel to a clinic and the poverty rate of the county the patient lives in play a role in referral to a clinician specializing in contact dermatitis.
A retrospective registry review of 2310 patients undergoing patch testing at the Massachusetts General Hospital in Boston revealed that 2.5% were Black, 5.5% were Latinx, 8.3% were Asian, and the remaining 83.7% were White.20 Qian et al21 also looked at patch testing patterns among various sociodemographic groups (N=1,107,530) and found that 69% of patients were White and 59% were female. Rates of patch testing among patients who were Black, lesser educated, male, lower income, and younger (children aged 0–12 years) were significantly lower than for other groups when ACD was suspected (P<.0001).21 The lower rates of patch testing in patients with SOC may be due to low suspicion of diagnosis, low referral rates due to limited medical insurance, and financial instability, as well as other socioeconomic factors.20
Tamazian et al16 reviewed pediatric populations at 13 US centers and found that Black children received patch testing less frequently than White and Hispanic children. Another review of pediatric patch testing in patients with SOC found that a less comprehensive panel of allergens was used in this population.22
The key to resolution of ACD is removal of the offending antigen, and if patients are not being tested, then they risk having a prolonged and complicated course of ACD with a poor prognosis. Patients with SOC also experience greater negative psychosocial impact due to ACD disease burden.21,23
The lower rates of patch testing in Black patients cannot solely be attributed to difficulty diagnosing ACD in darker skin tones; it is likely due to the impact of social determinants of health. Alleviating health disparities will improve patient outcomes and quality of life.
THE COMPARISON
A An 11-year-old Hispanic boy with allergic contact dermatitis (ACD) on the abdomen. The geometric nature of the eruption and proximity to the belt buckle were highly suggestive of ACD to nickel; patch testing was not needed.
B A Black woman with ACD on the neck. A punch biopsy demonstrated spongiotic dermatitis that was typical of ACD. The diagnosis was supported by the patient’s history of dermatitis that developed after new products were applied to the hair. The patient declined patch testing.
C A Hispanic man with ACD on hair-bearing areas on the face where hair dye was used. The patient’s history of dermatitis following the application of hair dye was highly suggestive of ACD; patch testing confirmed the allergen was paraphenylenediamine (PPD).
Allergic contact dermatitis (ACD) is an inflammatory condition of the skin caused by an immunologic response to one or more identifiable allergens. A delayed-type immune response (type IV hypersensitivity reaction) occurs after the skin is reexposed to an offending allergen.1 Severe pruritus is the main symptom of ACD in the early stages, accompanied by erythema, vesicles, and scaling in a distinct pattern corresponding to the allergen’s contact with the skin.2 Delayed widespread dermatitis after exposure to an allergen—a phenomenon known as autoeczematization (id reaction)—also may occur.3
The gold-standard diagnostic tool for ACD is patch testing, in which the patient is re-exposed to the suspected contact allergen(s) and observed for the development of dermatitis.4 However, ACD can be diagnosed with a detailed patient history including occupation, hobbies, personal care practices, and possible triggers with subsequent rashes. Thorough clinical examination of the skin is paramount. Indicators of possible ACD include dermatitis that persists despite use of appropriate treatment, an unexplained flare of previously quiescent dermatitis, and a diagnosis of dermatitis without a clear cause.1
Hairdressers, health care workers, and metal workers are at higher risk for ACD.5 Occupational ACD has notable socioeconomic implications, as it can result in frequent sick days, inability to perform tasks at work, and in some cases job loss.6
Patients with atopic dermatitis have impaired barrier function of the skin, permitting the entrance of allergens and subsequent sensitization.7 Allergic contact dermatitis is a challenge to manage, as complete avoidance of the allergen may not be possible.8
The underrepresentation of patients with skin of color (SOC) in educational materials as well as socioeconomic health disparities may contribute to the lower rates of diagnosis, patch testing, and treatment of ACD in this patient population.
Epidemiology
An ACD prevalence of 15.2% was reported in a study of 793 Danish patients who underwent skin prick and patch testing.9 Alinaghi et al10 conducted a meta-analysis of 20,107 patients across 28 studies who were patch tested to determine the prevalence of ACD in the general population. The researchers concluded that 20.1% (95% CI, 16.8%- 23.7%) of the general population experienced ACD. They analyzed 22 studies to determine the prevalence of ACD based on specific geographic area including 18,709 individuals from Europe with a prevalence of 19.5% (95% CI, 15.8%-23.4%), 1639 individuals from North America with a prevalence of 20.6% (95% CI, 9.2%-35.2%), and 2 studies from China (no other studies from Asia found) with a prevalence of 20.6% (95% CI, 17.4%-23.9%). Researchers did not find data from studies conducted in Africa or South America.10
The current available epidemiologic data on ACD are not representative of SOC populations. DeLeo et al11 looked at patch test reaction patterns in association with race and ethnicity in a large sample size (N=19,457); 17,803 (92.9%) of these patients were White and only 1360 (7.1%) were Black. Large-scale, inclusive studies are needed, which can only be achieved with increased suspicion for ACD and increased access to patch testing.
Allergic contact dermatitis is more common in women, with nickel being the most frequently identified allergen (Figure, A).10 Personal care products often are linked to ACD (Figure, B). An analysis of data from the North American Contact Dermatitis Group revealed that the top 5 personal care product allergens were methylisothiazolinone (a preservative), fragrance mix I, balsam of Peru, quaternium-15 (a preservative), and paraphenylenediamine (PPD)(a common component of hair dye) (Figure, C).12
There is a paucity of epidemiologic data among various ethnic groups; however, a few studies have suggested that there is no difference in the frequency rates of positive patch test results in Black vs White populations.11,13,14 One study of patch test results from 114 Black patients and 877 White patients at the Cleveland Clinic Foundation in Ohio demonstrated a similar allergy frequency of 43.0% and 43.6%, respectively.13 However, there were differences in the types of allergen sensitization. Black patients had higher positive patch test rates for PPD than White patients (10.6% vs 4.5%). Black men had a higher frequency of sensitivity to PPD (21.2% vs 4.2%) and imidazolidinyl urea (a formaldehyde-releasing preservative) (9.1% vs 2.6%) compared to White men.13
Ethnicity and cultural practices influence epidemiologic patterns of ACD. Darker hair dyes used in Black patients14 and deeply pigmented PPD dye found in henna tattoos used in Indian and Black patients15 may lead to increased sensitization to PPD. Allergic contact dermatitis due to formaldehyde is more common in White patients, possibly due to more frequent use of formaldehyde-containing moisturizers, shampoos, and creams.15
Key clinical features in people with darker skin tones
In patients with SOC, the clinical features of ACD vary, posing a diagnostic challenge. Hyperpigmentation, lichenification, and induration are more likely to be seen than the papules, vesicles, and erythematous dermatitis often described in lighter skin tones or acute ACD. Erythema can be difficult to assess on darker skin and may appear violaceous or very faint pink.16
Worth noting
A high index of suspicion is necessary when interpreting patch tests in patients with SOC, as patch test kits use a reading plate with graduated intensities of erythema, papulation, and vesicular reactions to determine the likelihood of ACD. The potential contact allergens are placed on the skin on day 1 and covered. Then, on day 3 the allergens are removed. The skin is clinically evaluated using visual assessment and skin palpation. The reactions are graded as negative, irritant reaction, equivocal, weak positive, strong positive, or extreme reaction at around days 3 and 5 to capture both early and delayed reactions.17 A patch test may be positive even if obvious signs of erythema are not appreciated as expected.
Adjusting the lighting in the examination room, including side lighting, or using a blue background can be helpful in identifying erythema in darker skin tones.15,16,18 Palpation of the skin also is useful, as even slight texture changes and induration are indicators of a possible skin reaction to the test allergen.15
Health disparity highlight
Clinical photographs of ACD and patch test results in patients with SOC are not commonplace in the literature. Positive patch test results in patients with darker skin tones vary from those of patients with lighter skin tones, and if the clinician reading the patch test result is not familiar with the findings in darker skin tones, the diagnosis may be delayed or missed.15
Furthermore, Scott et al15 highlighted that many dermatology residency training programs have a paucity of SOC education in their curriculum. This lack of representation may contribute to the diagnostic challenges encountered by health care providers.
Timely access to health care and education as well as economic stability are essential for the successful management of patients with ACD. Some individuals with SOC have been disproportionately affected by social determinants of health. Rodriguez-Homs et al19 demonstrated that the distance needed to travel to a clinic and the poverty rate of the county the patient lives in play a role in referral to a clinician specializing in contact dermatitis.
A retrospective registry review of 2310 patients undergoing patch testing at the Massachusetts General Hospital in Boston revealed that 2.5% were Black, 5.5% were Latinx, 8.3% were Asian, and the remaining 83.7% were White.20 Qian et al21 also looked at patch testing patterns among various sociodemographic groups (N=1,107,530) and found that 69% of patients were White and 59% were female. Rates of patch testing among patients who were Black, lesser educated, male, lower income, and younger (children aged 0–12 years) were significantly lower than for other groups when ACD was suspected (P<.0001).21 The lower rates of patch testing in patients with SOC may be due to low suspicion of diagnosis, low referral rates due to limited medical insurance, and financial instability, as well as other socioeconomic factors.20
Tamazian et al16 reviewed pediatric populations at 13 US centers and found that Black children received patch testing less frequently than White and Hispanic children. Another review of pediatric patch testing in patients with SOC found that a less comprehensive panel of allergens was used in this population.22
The key to resolution of ACD is removal of the offending antigen, and if patients are not being tested, then they risk having a prolonged and complicated course of ACD with a poor prognosis. Patients with SOC also experience greater negative psychosocial impact due to ACD disease burden.21,23
The lower rates of patch testing in Black patients cannot solely be attributed to difficulty diagnosing ACD in darker skin tones; it is likely due to the impact of social determinants of health. Alleviating health disparities will improve patient outcomes and quality of life.
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74: 1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Bertoli MJ, Schwartz RA, Janniger CK. Autoeczematization: a strange id reaction of the skin. Cutis. 2021;108:163-166. doi:10.12788/cutis.0342
- Johansen JD, Bonefeld CM, Schwensen JFB, et al. Novel insights into contact dermatitis. J Allergy Clin Immunol. 2022;149:1162-1171. doi:10.1016/j.jaci.2022.02.002
- Karagounis TK, Cohen DE. Occupational hand dermatitis. Curr Allergy Asthma Rep. 2023;23:201-212. doi:10.1007/s11882-023-01070-5
- Cvetkovski RS, Rothman KJ, Olsen J, et al. Relation between diagnoses on severity, sick leave and loss of job among patients with occupational hand eczema. Br J Dermatol. 2005;152:93-98. doi:10.1111/j .1365-2133.2005.06415.x
- Owen JL, Vakharia PP, Silverberg JI. The role and diagnosis of allergic contact dermatitis in patients with atopic dermatitis. Am J Clin Dermatol. 2018;19:293-302. doi:10.1007/s40257-017-0340-7
- Brites GS, Ferreira I, Sebastião AI, et al. Allergic contact dermatitis: from pathophysiology to development of new preventive strategies. Pharmacol Res. 2020;162:105282. doi:10.1016/j.phrs.2020.105282
- Nielsen NH, Menne T. The relationship between IgE‐mediated and cell‐mediated hypersensitivities in an unselected Danish population: the Glostrup Allergy Study, Denmark. Br J Dermatol. 1996;134:669-672. doi:10.1111/j.1365-2133.1996.tb06967.x
- Alinaghi F, Bennike NH, Egeberg A, et al. Prevalence of contact allergy in the general population: a systematic review and meta‐analysis. Contact Dermatitis. 2019;80:77-85. doi:10.1111/cod.13119
- DeLeo VA, Alexis A, Warshaw EM, et al. The association of race/ethnicity and patch test results: North American Contact Dermatitis Group, 1998- 2006. Dermatitis. 2016;27:288-292. doi:10.1097/DER.0000000000000220
- Warshaw EM, Schlarbaum JP, Silverberg JI, et al. Contact dermatitis to personal care products is increasing (but different!) in males and females: North American Contact Dermatitis Group data, 1996-2016. J Am Acad Dermatol. 2021;85:1446-1455. doi:10.1016/j.jaad.2020.10.003
- Dickel H, Taylor JS, Evey P, et al. Comparison of patch test results with a standard series among white and black racial groups. Am J Contact Dermatol. 2001;12:77-82. doi:10.1053/ajcd.2001.20110
- DeLeo VA, Taylor SC, Belsito DV, et al. The effect of race and ethnicity on patch test results. J Am Acad Dermatol. 2002;46(2 suppl):S107-S112. doi:10.1067/mjd.2002.120792
- Scott I, Atwater AR, Reeder M. Update on contact dermatitis and patch testing in patients with skin of color. Cutis. 2021;108:10-12. doi:10.12788/cutis.0292
- Tamazian S, Oboite M, Treat JR. Patch testing in skin of color: a brief report. Pediatr Dermatol. 2021;38:952-953. doi:10.1111/pde.14578
- Litchman G, Nair PA, Atwater AR, et al. Contact dermatitis. StatPearls [Internet]. Updated February 9, 2023. Accessed September 25, 2023. https://www.ncbi.nlm.nih.gov/books/NBK459230/
- Alexis AF, Callender VD, Baldwin HE, et al. Global epidemiology and clinical spectrum of rosacea, highlighting skin of color: review and clinical practice experience. J Am Acad Dermatol. 2019;80:1722-1729. doi:10.1016/j.jaad.2018.08.049
- Rodriguez-Homs LG, Liu B, Green CL, et al. Duration of dermatitis before patch test appointment is associated with distance to clinic and county poverty rate. Dermatitis. 2020;31:259-264. doi:10.1097 /DER.0000000000000581
- Foschi CM, Tam I, Schalock PC, et al. Patch testing results in skin of color: a retrospective review from the Massachusetts General Hospital contact dermatitis clinic. J Am Acad Dermatol. 2022;87:452-454. doi:10.1016/j.jaad.2021.09.022
- Qian MF, Li S, Honari G, et al. Sociodemographic disparities in patch testing for commercially insured patients with dermatitis: a retrospective analysis of administrative claims data. J Am Acad Dermatol. 2022;87:1411-1413. doi:10.1016/j.jaad.2022.08.041
- Young K, Collis RW, Sheinbein D, et al. Retrospective review of pediatric patch testing results in skin of color. J Am Acad Dermatol. 2023;88:953-954. doi:10.1016/j.jaad.2022.11.031
- Kadyk DL, Hall S, Belsito DV. Quality of life of patients with allergic contact dermatitis: an exploratory analysis by gender, ethnicity, age, and occupation. Dermatitis. 2004;15:117-124.
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74: 1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Bertoli MJ, Schwartz RA, Janniger CK. Autoeczematization: a strange id reaction of the skin. Cutis. 2021;108:163-166. doi:10.12788/cutis.0342
- Johansen JD, Bonefeld CM, Schwensen JFB, et al. Novel insights into contact dermatitis. J Allergy Clin Immunol. 2022;149:1162-1171. doi:10.1016/j.jaci.2022.02.002
- Karagounis TK, Cohen DE. Occupational hand dermatitis. Curr Allergy Asthma Rep. 2023;23:201-212. doi:10.1007/s11882-023-01070-5
- Cvetkovski RS, Rothman KJ, Olsen J, et al. Relation between diagnoses on severity, sick leave and loss of job among patients with occupational hand eczema. Br J Dermatol. 2005;152:93-98. doi:10.1111/j .1365-2133.2005.06415.x
- Owen JL, Vakharia PP, Silverberg JI. The role and diagnosis of allergic contact dermatitis in patients with atopic dermatitis. Am J Clin Dermatol. 2018;19:293-302. doi:10.1007/s40257-017-0340-7
- Brites GS, Ferreira I, Sebastião AI, et al. Allergic contact dermatitis: from pathophysiology to development of new preventive strategies. Pharmacol Res. 2020;162:105282. doi:10.1016/j.phrs.2020.105282
- Nielsen NH, Menne T. The relationship between IgE‐mediated and cell‐mediated hypersensitivities in an unselected Danish population: the Glostrup Allergy Study, Denmark. Br J Dermatol. 1996;134:669-672. doi:10.1111/j.1365-2133.1996.tb06967.x
- Alinaghi F, Bennike NH, Egeberg A, et al. Prevalence of contact allergy in the general population: a systematic review and meta‐analysis. Contact Dermatitis. 2019;80:77-85. doi:10.1111/cod.13119
- DeLeo VA, Alexis A, Warshaw EM, et al. The association of race/ethnicity and patch test results: North American Contact Dermatitis Group, 1998- 2006. Dermatitis. 2016;27:288-292. doi:10.1097/DER.0000000000000220
- Warshaw EM, Schlarbaum JP, Silverberg JI, et al. Contact dermatitis to personal care products is increasing (but different!) in males and females: North American Contact Dermatitis Group data, 1996-2016. J Am Acad Dermatol. 2021;85:1446-1455. doi:10.1016/j.jaad.2020.10.003
- Dickel H, Taylor JS, Evey P, et al. Comparison of patch test results with a standard series among white and black racial groups. Am J Contact Dermatol. 2001;12:77-82. doi:10.1053/ajcd.2001.20110
- DeLeo VA, Taylor SC, Belsito DV, et al. The effect of race and ethnicity on patch test results. J Am Acad Dermatol. 2002;46(2 suppl):S107-S112. doi:10.1067/mjd.2002.120792
- Scott I, Atwater AR, Reeder M. Update on contact dermatitis and patch testing in patients with skin of color. Cutis. 2021;108:10-12. doi:10.12788/cutis.0292
- Tamazian S, Oboite M, Treat JR. Patch testing in skin of color: a brief report. Pediatr Dermatol. 2021;38:952-953. doi:10.1111/pde.14578
- Litchman G, Nair PA, Atwater AR, et al. Contact dermatitis. StatPearls [Internet]. Updated February 9, 2023. Accessed September 25, 2023. https://www.ncbi.nlm.nih.gov/books/NBK459230/
- Alexis AF, Callender VD, Baldwin HE, et al. Global epidemiology and clinical spectrum of rosacea, highlighting skin of color: review and clinical practice experience. J Am Acad Dermatol. 2019;80:1722-1729. doi:10.1016/j.jaad.2018.08.049
- Rodriguez-Homs LG, Liu B, Green CL, et al. Duration of dermatitis before patch test appointment is associated with distance to clinic and county poverty rate. Dermatitis. 2020;31:259-264. doi:10.1097 /DER.0000000000000581
- Foschi CM, Tam I, Schalock PC, et al. Patch testing results in skin of color: a retrospective review from the Massachusetts General Hospital contact dermatitis clinic. J Am Acad Dermatol. 2022;87:452-454. doi:10.1016/j.jaad.2021.09.022
- Qian MF, Li S, Honari G, et al. Sociodemographic disparities in patch testing for commercially insured patients with dermatitis: a retrospective analysis of administrative claims data. J Am Acad Dermatol. 2022;87:1411-1413. doi:10.1016/j.jaad.2022.08.041
- Young K, Collis RW, Sheinbein D, et al. Retrospective review of pediatric patch testing results in skin of color. J Am Acad Dermatol. 2023;88:953-954. doi:10.1016/j.jaad.2022.11.031
- Kadyk DL, Hall S, Belsito DV. Quality of life of patients with allergic contact dermatitis: an exploratory analysis by gender, ethnicity, age, and occupation. Dermatitis. 2004;15:117-124.

Reticular Hyperpigmentation With Keratotic Papules in the Axillae and Groin
The Diagnosis: Galli-Galli Disease
Several cutaneous conditions can present as reticulated hyperpigmentation or keratotic papules. Although genetic testing can help identify some of these dermatoses, biopsy typically is sufficient for diagnosis, and genetic testing can be considered for more clinically challenging cases. In our case, the clinical evidence and histopathologic findings were diagnostic of Galli-Galli disease (GGD), an autosomal-dominant genodermatosis with incomplete penetrance. Our patient was unaware of any family members with a diagnosis of GGD; however, she reported a great uncle with similar clinical findings.
Galli-Galli disease is a rare allelic variant of Dowling- Degos disease (DDD), both caused by a loss-of-function mutation in the keratin 5 gene, KRT5. Both conditions present as reticulated papules distributed symmetrically in the flexural regions, most commonly the axillae and groin, but also as comedolike papules, typically in patients aged 30 to 50 years.1 Cutaneous lesions primarily are of cosmetic concern but can be extremely pruritic, especially for patients with GGD. Gene mutations in protein O-fucosyltransferase 1, POFUT1; protein O-glucosyltransferase 1, POGLUT1; and presenilin enhancer 2, PSENEN, also have been discovered in cases of DDD and GGD.2,3
Galli-Galli disease and DDD are distinguishable by their histologic appearance. Both diseases show elongated fingerlike rete ridges and a thin suprapapillary epidermis. The basal projections often are described as bulbous or resembling antler horns.4 Galli-Galli disease can be differentiated from DDD by focal suprabasal acantholysis with minimal dyskeratosis (quiz images).5 Due to the genetic and clinical similarities, many consider GGD an acantholytic variant of DDD rather than its own entity. Indeed, some patients have shown acantholysis in one area of biopsy but not others.6
Hailey-Hailey disease (HHD)(also known as benign familial or benign chronic pemphigus) is an autosomaldominant disorder caused by mutation of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1. Clinically, patients tend to present at a wide age range with fragile flaccid vesicles that commonly develop on the neck, axillae, and groin. Histologically, the epidermis is acanthotic with a dilapidated brick wall– like appearance from a few persistent intercellular connections amid widespread acantholysis (Figure 1).7 Unlike in autoimmune pemphigus, direct immunofluorescence is negative, and acantholysis spares the adnexal structures. Hailey-Hailey disease does not involve reticulated hyperpigmentation or the elongated bulbous rete seen in GGD. Confluent and reticulated papillomatosis is a rare, typically asymptomatic, hyperpigmented dermatosis. It presents as a conglomeration of scaly hyperpigmented macules or papillomatous papules that coalesce centrally and are reticulated toward the periphery.

Confluent and reticulated papillomatosis most commonly is seen on the trunk, initially presenting in adolescents and young adults. Confluent and reticulated papillomatosis is histologically similar to acanthosis nigricans. Histopathology will show hyperkeratosis, papillomatosis, and minimal to no inflammatory infiltrate, with no elongated rete ridges or acantholysis (Figure 2).8

Pemphigus vulgaris is a blistering disease resulting from the development of autoantibodies against desmogleins 1 and 3. Similar to GGD, there is suprabasal acantholysis, which often results in a tombstonelike appearance consisting of separation between the basal layer cells of the epidermis but with maintained attachment to the underlying basement membrane zone. Unlike HHD, the acantholysis tends to involve the follicular epithelium in pemphigus vulgaris (Figure 3). Clinically, the blisters are positive for Nikolsky sign and can be both cutaneous or mucosal, commonly arising initially in the mouth during the fourth or fifth decades of life. Ruptured blisters can result in painful and hemorrhagic erosions.9 Direct immunofluorescence exhibits a classic chicken wire–like deposition of IgG and C3 between keratinocytes of the epidermis. Although sometimes difficult to appreciate, the deposition can be more prominent in the lower epidermis, in contrast to pemphigus foliaceus, which can have more prominent deposition in the upper epidermis.

Darier disease (or dyskeratosis follicularis) is an autosomal-dominant genodermatosis caused by mutation of the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2. Clinically, this disorder arises in adolescents as red-brown, greasy, crusted papules in seborrheic areas that may coalesce into papillomatous clusters. Palmar punctate keratoses and pits also are common. Histologically, Darier disease can appear similar to GGD, as both can show acantholysis and dyskeratosis. Darier disease will tend to show more prominent dyskeratosis with corps ronds and grains, as well as thicker villilike projections of keratinocytes into the papillary dermis, in contrast to the thinner, fingerlike or bulbous projections that hang down from the epidermis in GGD (Figure 4).10

- Hanneken S, Rütten A, Eigelshoven S, et al. Morbus Galli-Galli. Hautarzt. 2013;64:282.
- Wilson NJ, Cole C, Kroboth K, et al. Mutations in POGLUT1 in Galli- Galli/Dowling-Degos disease. Br J Dermatol. 2017;176:270-274.
- Ralser DJ, Basmanav FB, Tafazzoli A, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-1490.
- Desai CA, Virmani N, Sakhiya J, et al. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol. 2016; 82:720-723.
- Joshi TP, Shaver S, Tschen J. Exacerbation of Galli-Galli disease following dialysis treatment: a case report and review of aggravating factors. Cureus. 2021;13:E15401.
- Muller CS, Pfohler C, Tilgen W. Changing a concept—controversy on the confusion spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol. 2008;36:44-48.
- Dai Y, Yu L, Wang Y, et al. Case report: a case of Hailey-Hailey disease mimicking condyloma acuminatum and a novel splice-site mutation of ATP2C1 gene. Front Genet. 2021;12:777630.
- Banjar TA, Abdulwahab RA, Al Hawsawi KA. Confluent and reticulated papillomatosis of Gougerot and Carteaud: a case report and review of the literature. Cureus. 2022;14:E24557.
- Porro AM, Seque CA, Ferreira MCC, et al. Pemphigus vulgaris. An Bras Dermatol. 2019;94:264-278.
- Bachar-Wikström E, Wikström JD. Darier disease—a multi-organ condition? Acta Derm Venereol. 2021;101:adv00430.
The Diagnosis: Galli-Galli Disease
Several cutaneous conditions can present as reticulated hyperpigmentation or keratotic papules. Although genetic testing can help identify some of these dermatoses, biopsy typically is sufficient for diagnosis, and genetic testing can be considered for more clinically challenging cases. In our case, the clinical evidence and histopathologic findings were diagnostic of Galli-Galli disease (GGD), an autosomal-dominant genodermatosis with incomplete penetrance. Our patient was unaware of any family members with a diagnosis of GGD; however, she reported a great uncle with similar clinical findings.
Galli-Galli disease is a rare allelic variant of Dowling- Degos disease (DDD), both caused by a loss-of-function mutation in the keratin 5 gene, KRT5. Both conditions present as reticulated papules distributed symmetrically in the flexural regions, most commonly the axillae and groin, but also as comedolike papules, typically in patients aged 30 to 50 years.1 Cutaneous lesions primarily are of cosmetic concern but can be extremely pruritic, especially for patients with GGD. Gene mutations in protein O-fucosyltransferase 1, POFUT1; protein O-glucosyltransferase 1, POGLUT1; and presenilin enhancer 2, PSENEN, also have been discovered in cases of DDD and GGD.2,3
Galli-Galli disease and DDD are distinguishable by their histologic appearance. Both diseases show elongated fingerlike rete ridges and a thin suprapapillary epidermis. The basal projections often are described as bulbous or resembling antler horns.4 Galli-Galli disease can be differentiated from DDD by focal suprabasal acantholysis with minimal dyskeratosis (quiz images).5 Due to the genetic and clinical similarities, many consider GGD an acantholytic variant of DDD rather than its own entity. Indeed, some patients have shown acantholysis in one area of biopsy but not others.6
Hailey-Hailey disease (HHD)(also known as benign familial or benign chronic pemphigus) is an autosomaldominant disorder caused by mutation of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1. Clinically, patients tend to present at a wide age range with fragile flaccid vesicles that commonly develop on the neck, axillae, and groin. Histologically, the epidermis is acanthotic with a dilapidated brick wall– like appearance from a few persistent intercellular connections amid widespread acantholysis (Figure 1).7 Unlike in autoimmune pemphigus, direct immunofluorescence is negative, and acantholysis spares the adnexal structures. Hailey-Hailey disease does not involve reticulated hyperpigmentation or the elongated bulbous rete seen in GGD. Confluent and reticulated papillomatosis is a rare, typically asymptomatic, hyperpigmented dermatosis. It presents as a conglomeration of scaly hyperpigmented macules or papillomatous papules that coalesce centrally and are reticulated toward the periphery.
Confluent and reticulated papillomatosis most commonly is seen on the trunk, initially presenting in adolescents and young adults. Confluent and reticulated papillomatosis is histologically similar to acanthosis nigricans. Histopathology will show hyperkeratosis, papillomatosis, and minimal to no inflammatory infiltrate, with no elongated rete ridges or acantholysis (Figure 2).8
Pemphigus vulgaris is a blistering disease resulting from the development of autoantibodies against desmogleins 1 and 3. Similar to GGD, there is suprabasal acantholysis, which often results in a tombstonelike appearance consisting of separation between the basal layer cells of the epidermis but with maintained attachment to the underlying basement membrane zone. Unlike HHD, the acantholysis tends to involve the follicular epithelium in pemphigus vulgaris (Figure 3). Clinically, the blisters are positive for Nikolsky sign and can be both cutaneous or mucosal, commonly arising initially in the mouth during the fourth or fifth decades of life. Ruptured blisters can result in painful and hemorrhagic erosions.9 Direct immunofluorescence exhibits a classic chicken wire–like deposition of IgG and C3 between keratinocytes of the epidermis. Although sometimes difficult to appreciate, the deposition can be more prominent in the lower epidermis, in contrast to pemphigus foliaceus, which can have more prominent deposition in the upper epidermis.
Darier disease (or dyskeratosis follicularis) is an autosomal-dominant genodermatosis caused by mutation of the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2. Clinically, this disorder arises in adolescents as red-brown, greasy, crusted papules in seborrheic areas that may coalesce into papillomatous clusters. Palmar punctate keratoses and pits also are common. Histologically, Darier disease can appear similar to GGD, as both can show acantholysis and dyskeratosis. Darier disease will tend to show more prominent dyskeratosis with corps ronds and grains, as well as thicker villilike projections of keratinocytes into the papillary dermis, in contrast to the thinner, fingerlike or bulbous projections that hang down from the epidermis in GGD (Figure 4).10
The Diagnosis: Galli-Galli Disease
Several cutaneous conditions can present as reticulated hyperpigmentation or keratotic papules. Although genetic testing can help identify some of these dermatoses, biopsy typically is sufficient for diagnosis, and genetic testing can be considered for more clinically challenging cases. In our case, the clinical evidence and histopathologic findings were diagnostic of Galli-Galli disease (GGD), an autosomal-dominant genodermatosis with incomplete penetrance. Our patient was unaware of any family members with a diagnosis of GGD; however, she reported a great uncle with similar clinical findings.
Galli-Galli disease is a rare allelic variant of Dowling- Degos disease (DDD), both caused by a loss-of-function mutation in the keratin 5 gene, KRT5. Both conditions present as reticulated papules distributed symmetrically in the flexural regions, most commonly the axillae and groin, but also as comedolike papules, typically in patients aged 30 to 50 years.1 Cutaneous lesions primarily are of cosmetic concern but can be extremely pruritic, especially for patients with GGD. Gene mutations in protein O-fucosyltransferase 1, POFUT1; protein O-glucosyltransferase 1, POGLUT1; and presenilin enhancer 2, PSENEN, also have been discovered in cases of DDD and GGD.2,3
Galli-Galli disease and DDD are distinguishable by their histologic appearance. Both diseases show elongated fingerlike rete ridges and a thin suprapapillary epidermis. The basal projections often are described as bulbous or resembling antler horns.4 Galli-Galli disease can be differentiated from DDD by focal suprabasal acantholysis with minimal dyskeratosis (quiz images).5 Due to the genetic and clinical similarities, many consider GGD an acantholytic variant of DDD rather than its own entity. Indeed, some patients have shown acantholysis in one area of biopsy but not others.6
Hailey-Hailey disease (HHD)(also known as benign familial or benign chronic pemphigus) is an autosomaldominant disorder caused by mutation of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1. Clinically, patients tend to present at a wide age range with fragile flaccid vesicles that commonly develop on the neck, axillae, and groin. Histologically, the epidermis is acanthotic with a dilapidated brick wall– like appearance from a few persistent intercellular connections amid widespread acantholysis (Figure 1).7 Unlike in autoimmune pemphigus, direct immunofluorescence is negative, and acantholysis spares the adnexal structures. Hailey-Hailey disease does not involve reticulated hyperpigmentation or the elongated bulbous rete seen in GGD. Confluent and reticulated papillomatosis is a rare, typically asymptomatic, hyperpigmented dermatosis. It presents as a conglomeration of scaly hyperpigmented macules or papillomatous papules that coalesce centrally and are reticulated toward the periphery.
Confluent and reticulated papillomatosis most commonly is seen on the trunk, initially presenting in adolescents and young adults. Confluent and reticulated papillomatosis is histologically similar to acanthosis nigricans. Histopathology will show hyperkeratosis, papillomatosis, and minimal to no inflammatory infiltrate, with no elongated rete ridges or acantholysis (Figure 2).8
Pemphigus vulgaris is a blistering disease resulting from the development of autoantibodies against desmogleins 1 and 3. Similar to GGD, there is suprabasal acantholysis, which often results in a tombstonelike appearance consisting of separation between the basal layer cells of the epidermis but with maintained attachment to the underlying basement membrane zone. Unlike HHD, the acantholysis tends to involve the follicular epithelium in pemphigus vulgaris (Figure 3). Clinically, the blisters are positive for Nikolsky sign and can be both cutaneous or mucosal, commonly arising initially in the mouth during the fourth or fifth decades of life. Ruptured blisters can result in painful and hemorrhagic erosions.9 Direct immunofluorescence exhibits a classic chicken wire–like deposition of IgG and C3 between keratinocytes of the epidermis. Although sometimes difficult to appreciate, the deposition can be more prominent in the lower epidermis, in contrast to pemphigus foliaceus, which can have more prominent deposition in the upper epidermis.
Darier disease (or dyskeratosis follicularis) is an autosomal-dominant genodermatosis caused by mutation of the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2. Clinically, this disorder arises in adolescents as red-brown, greasy, crusted papules in seborrheic areas that may coalesce into papillomatous clusters. Palmar punctate keratoses and pits also are common. Histologically, Darier disease can appear similar to GGD, as both can show acantholysis and dyskeratosis. Darier disease will tend to show more prominent dyskeratosis with corps ronds and grains, as well as thicker villilike projections of keratinocytes into the papillary dermis, in contrast to the thinner, fingerlike or bulbous projections that hang down from the epidermis in GGD (Figure 4).10
- Hanneken S, Rütten A, Eigelshoven S, et al. Morbus Galli-Galli. Hautarzt. 2013;64:282.
- Wilson NJ, Cole C, Kroboth K, et al. Mutations in POGLUT1 in Galli- Galli/Dowling-Degos disease. Br J Dermatol. 2017;176:270-274.
- Ralser DJ, Basmanav FB, Tafazzoli A, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-1490.
- Desai CA, Virmani N, Sakhiya J, et al. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol. 2016; 82:720-723.
- Joshi TP, Shaver S, Tschen J. Exacerbation of Galli-Galli disease following dialysis treatment: a case report and review of aggravating factors. Cureus. 2021;13:E15401.
- Muller CS, Pfohler C, Tilgen W. Changing a concept—controversy on the confusion spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol. 2008;36:44-48.
- Dai Y, Yu L, Wang Y, et al. Case report: a case of Hailey-Hailey disease mimicking condyloma acuminatum and a novel splice-site mutation of ATP2C1 gene. Front Genet. 2021;12:777630.
- Banjar TA, Abdulwahab RA, Al Hawsawi KA. Confluent and reticulated papillomatosis of Gougerot and Carteaud: a case report and review of the literature. Cureus. 2022;14:E24557.
- Porro AM, Seque CA, Ferreira MCC, et al. Pemphigus vulgaris. An Bras Dermatol. 2019;94:264-278.
- Bachar-Wikström E, Wikström JD. Darier disease—a multi-organ condition? Acta Derm Venereol. 2021;101:adv00430.
- Hanneken S, Rütten A, Eigelshoven S, et al. Morbus Galli-Galli. Hautarzt. 2013;64:282.
- Wilson NJ, Cole C, Kroboth K, et al. Mutations in POGLUT1 in Galli- Galli/Dowling-Degos disease. Br J Dermatol. 2017;176:270-274.
- Ralser DJ, Basmanav FB, Tafazzoli A, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-1490.
- Desai CA, Virmani N, Sakhiya J, et al. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol. 2016; 82:720-723.
- Joshi TP, Shaver S, Tschen J. Exacerbation of Galli-Galli disease following dialysis treatment: a case report and review of aggravating factors. Cureus. 2021;13:E15401.
- Muller CS, Pfohler C, Tilgen W. Changing a concept—controversy on the confusion spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol. 2008;36:44-48.
- Dai Y, Yu L, Wang Y, et al. Case report: a case of Hailey-Hailey disease mimicking condyloma acuminatum and a novel splice-site mutation of ATP2C1 gene. Front Genet. 2021;12:777630.
- Banjar TA, Abdulwahab RA, Al Hawsawi KA. Confluent and reticulated papillomatosis of Gougerot and Carteaud: a case report and review of the literature. Cureus. 2022;14:E24557.
- Porro AM, Seque CA, Ferreira MCC, et al. Pemphigus vulgaris. An Bras Dermatol. 2019;94:264-278.
- Bachar-Wikström E, Wikström JD. Darier disease—a multi-organ condition? Acta Derm Venereol. 2021;101:adv00430.
A 37-year-old woman presented with multiple hyperkeratotic small papules in the axillae and groin of 1 year’s duration. She reported pruritus and occasional sleep disruption. Subtle background reticulated hyperpigmentation was present. The patient reported that she had a great uncle with similar findings.


