User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
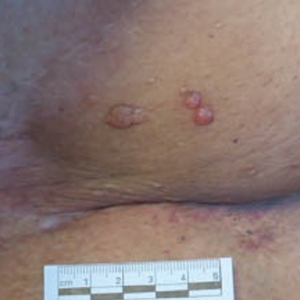
Pruritic Nodules on the Breast
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
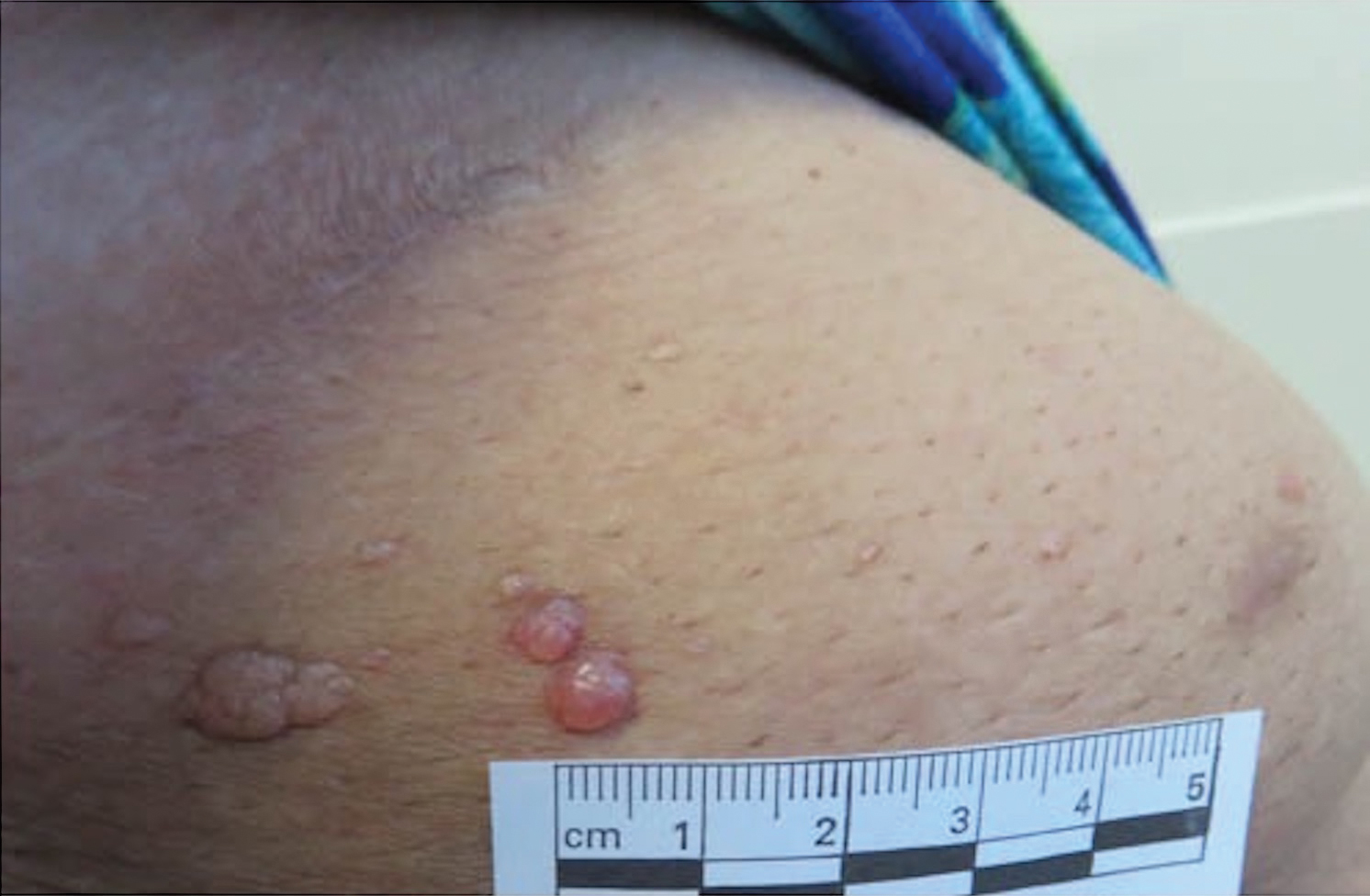
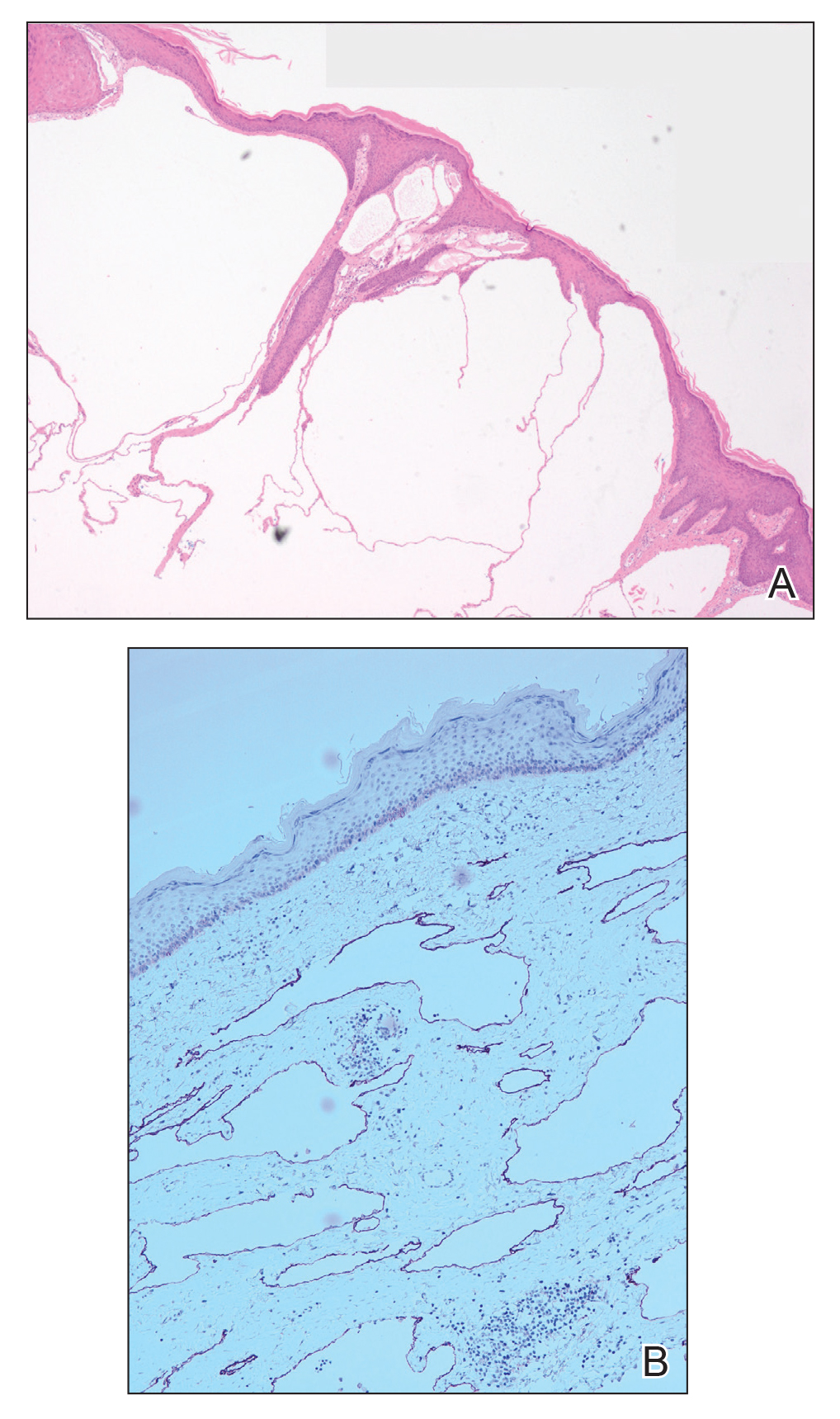
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
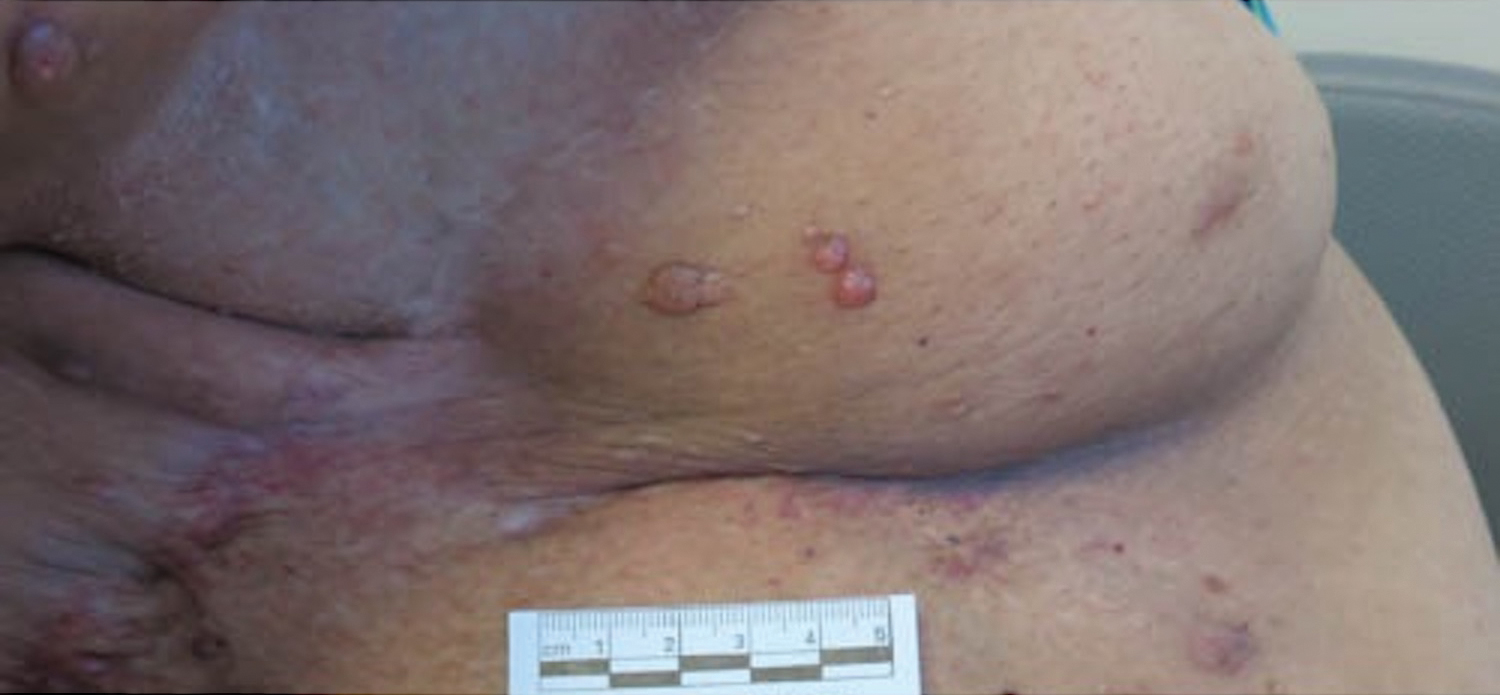
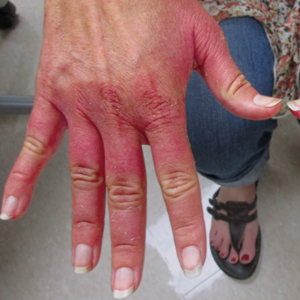
A 51-year-old woman with a history of bilateral breast cancer presented for evaluation of lesions on the underside of the right breast. She was first diagnosed with stage II cancer of the right breast that was subsequently treated with a mastectomy and adjuvant chemotherapy 7 years prior to presentation. One year later, she developed stage IIIC adenocarcinoma of the left breast and was treated with a modified radical mastectomy, adjuvant chemotherapy, and radiation. She had been followed closely by her oncologist with regular surveillance imaging (last at 7 months prior to presentation) that had all been negative for recurrent breast cancer. She presented to our dermatology clinic for evaluation of lesions on the underside of the right breast that were pruritic and occasionally painful with a burning quality. These lesions had recently begun to bleed when scratched but were not otherwise growing or spreading. On physical examination she was afebrile with stable vital signs. Skin examination was notable for numerous violaceous and translucent papules and nodules underneath the right breast and axilla overlying a well-healed mastectomy scar. No lymphadenopathy was present. Shave biopsies were performed and showed well-circumscribed nodular lesions with ectatic vascular channels separated by thin fibrous walls and filled with eosinophilic proteinaceous material and scattered red blood cells. Immunohistochemical staining also showed positivity for D2-40.
Supercharge Your On-Call Bag: 4 Must-Have Items for Dermatology Residents
It is no secret that a well-stocked on-call bag is one of the keys to providing inpatient care as a dermatology resident. Beyond the basic items that should never be left at home, there are some lesser-known tools that I have learned about from my book- and street-smart attendings and co-residents in the Department of Dermatology at the State University of New York Downstate Medical Center (referred to here as Downstate). Here are our top 4 items to pack the next time you are on call. (Bonus: you will find them helpful in clinic, too.)
Item 1: WoundSeal Powder
The most valuable player in my on-call bag, WoundSeal Powder (Biolife) is an over-the-counter hemostatic agent that I learned about from Daniel M. Siegel, MD, MS, a Mohs surgeon at Downstate and former president of the American Academy of Dermatology. The powder consists of a hydrophilic polymer and potassium ferrate.1 When poured over a bleeding wound and pressed in place (eg, with a sterile cotton-tipped swab), the hydrophilic polymer absorbs plasma while the iron in potassium ferrate agglomerates blood solids. The result is a scablike seal that is safe to leave in place until the wound has healed.1
Since Dr. Siegel introduced WoundSeal to Downstate about a decade ago, it has become our department’s go-to hemostatic agent for most punch biopsies performed in the inpatient setting. In our experience, achieving hemostasis in the hospital usually is easier, safer, and faster with WoundSeal than suture. Furthermore, using WoundSeal eliminates the need for patients to follow up for suture removal. From a practical perspective, WoundSeal works best when the biopsy defect is positioned parallel to the ground so the powder can be poured directly over and into the defect. From a cosmetic perspective, we have found that WoundSeal and suture have similar outcomes when used for punch biopsies up to 4 mm in size on the trunk and extremities in both adult and pediatric patients. Working with other dermatology attendings such as Sharon A. Glick, MD; Eve Lowenstein, MD, PhD; and Jeannette Jakus, MD, MBA, I also have found WoundSeal helpful when taking care of suture-phobic children or patients with lesions that are less amenable to suture, such as an ulcer or indurated plaque.
Item 2: Purple Surgical Marker
Another tip I have learned from Drs. Siegel and Jakus: If you are ever in a bind for a topical antibacterial or antifungal agent, look no further than a sterile purple surgical marker. These markers are a surprising source of gentian violet, the same purple dye that is the basis of Gram staining and sold as an over-the-counter antiseptic in 1% to 2% concentrations. Purple surgical markers, on the other hand, are 2.5% to 10% gentian violet.2
Gentian violet has been shown to have antibacterial, antifungal, antiviral, antihelminthic, and antitrypanosomal properties, but its efficacy has been mostly demonstrated against Streptococcus, methicillin-sensitive and methicillin-resistant Staphylococcus aureus, and Candida.3 Given the dermatologic relevance of these organisms, gentian violet is a favorite among attendings at my residency program; it is not uncommon to remove a patient’s dressing and uncover an iatrogenically purple wound. Best of all, pediatric patients are invariably amused when they see someone drawing on their skin with a purple marker.
When using a sterile surgical marker to apply gentian violet to the skin, we use either the marker tip or the ink core, which Dr. Siegel taught me can be easily accessed by snapping most plastic markers in half.
Item 3: Handheld Blacklight
The Wood lamp is a useful tool in the diagnosis of various infectious diseases and pigmentary disorders,4 but it is not always practical to use when on call, as standard ones are relatively large and corded, so they must be plugged into an electric outlet to work. You can therefore imagine the gratitude I have for my co-residents Miriam Lieberman, MD; Jaime Alexander, MD; Nicole Weiler, MD; and Alessandra Haskin, MD, for introducing me to the most convenient Wood lamp: the handheld blacklight. For less than $10, this gadget combines the diagnostic power of UV light with the portability of a pocket-sized, battery-powered flashlight. You will never want to use another Wood lamp again.
Item 4: Normal Saline Flush
Normal saline can be used for more than storing specimens for frozen section or tissue culture; it also can substitute for Michel solution when storing specimens for direct immunofluorescence (DIF) studies. I learned this tip from Edward Heilman, MD, a dermatopathologist at Downstate. For the last 20 years, Dr. Heilman has been successfully storing DIF specimens in refrigerated normal saline for up to 24 hours when Michel solution is unavailable, after which the specimen is processed or transferred to Michel solution for further storage while being transported to an immunofluorescence laboratory.
In 2004, Vodegel et al5 formally studied this technique in 25 patients with autoimmune skin diseases such as pemphigus and pemphigoid. (Thanks to Dr. Lieberman for telling me about this study.) The experiment involved taking 4 punch biopsies from each patient and placing them in either normal saline at −80°C for 24 or 48 hours, room temperature Michel solution for 48 hours, or liquid nitrogen for up to 2 weeks before being processed for DIF and analyzed by a blinded interpreter. Interestingly, specimens stored in normal saline for 24 hours were the most diagnostic, with a conclusive diagnosis reached in 21 of 25 specimens (84%). This result was attributed to the statistically significant reduction (P<.01) in background fluorescence with normal saline compared to Michel solution and liquid nitrogen, which in turn allowed for easier detection of diagnostic immunoreactants. Similar to Dr. Heilman, the authors cautioned against placing DIF specimens in normal saline for more than 24 hours; in their experience, the risk for an artefactual split developing at the dermoepidermal junction increases with this practice.5
- Biolife. How WoundSeal works. WoundSeal website. http://woundseal.com/how-it-works. Accessed March 7, 2019.
- Viscot Medical, LLC. Safety data sheet. http://www.viscot.com/download/MSDS%20Gentian%20Violet%20Ink.pdf. Published September 11, 2014. Accessed March 7, 2019.
- Maley AM, Arbiser JL. Gentian violet: a 19th century drug re-emerges in the 21st century. Exp Dermatol. 2013;22:775-780.
- Klatte JL, van der Beek N, Kemperman PM. 100 years of Wood’s lamp revised. J Eur Acad Dermatol Venereol. 2015;29:842-847.
- Vodegel RM, de Jong MC, Meijer HJ, et al. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
It is no secret that a well-stocked on-call bag is one of the keys to providing inpatient care as a dermatology resident. Beyond the basic items that should never be left at home, there are some lesser-known tools that I have learned about from my book- and street-smart attendings and co-residents in the Department of Dermatology at the State University of New York Downstate Medical Center (referred to here as Downstate). Here are our top 4 items to pack the next time you are on call. (Bonus: you will find them helpful in clinic, too.)
Item 1: WoundSeal Powder
The most valuable player in my on-call bag, WoundSeal Powder (Biolife) is an over-the-counter hemostatic agent that I learned about from Daniel M. Siegel, MD, MS, a Mohs surgeon at Downstate and former president of the American Academy of Dermatology. The powder consists of a hydrophilic polymer and potassium ferrate.1 When poured over a bleeding wound and pressed in place (eg, with a sterile cotton-tipped swab), the hydrophilic polymer absorbs plasma while the iron in potassium ferrate agglomerates blood solids. The result is a scablike seal that is safe to leave in place until the wound has healed.1
Since Dr. Siegel introduced WoundSeal to Downstate about a decade ago, it has become our department’s go-to hemostatic agent for most punch biopsies performed in the inpatient setting. In our experience, achieving hemostasis in the hospital usually is easier, safer, and faster with WoundSeal than suture. Furthermore, using WoundSeal eliminates the need for patients to follow up for suture removal. From a practical perspective, WoundSeal works best when the biopsy defect is positioned parallel to the ground so the powder can be poured directly over and into the defect. From a cosmetic perspective, we have found that WoundSeal and suture have similar outcomes when used for punch biopsies up to 4 mm in size on the trunk and extremities in both adult and pediatric patients. Working with other dermatology attendings such as Sharon A. Glick, MD; Eve Lowenstein, MD, PhD; and Jeannette Jakus, MD, MBA, I also have found WoundSeal helpful when taking care of suture-phobic children or patients with lesions that are less amenable to suture, such as an ulcer or indurated plaque.
Item 2: Purple Surgical Marker
Another tip I have learned from Drs. Siegel and Jakus: If you are ever in a bind for a topical antibacterial or antifungal agent, look no further than a sterile purple surgical marker. These markers are a surprising source of gentian violet, the same purple dye that is the basis of Gram staining and sold as an over-the-counter antiseptic in 1% to 2% concentrations. Purple surgical markers, on the other hand, are 2.5% to 10% gentian violet.2
Gentian violet has been shown to have antibacterial, antifungal, antiviral, antihelminthic, and antitrypanosomal properties, but its efficacy has been mostly demonstrated against Streptococcus, methicillin-sensitive and methicillin-resistant Staphylococcus aureus, and Candida.3 Given the dermatologic relevance of these organisms, gentian violet is a favorite among attendings at my residency program; it is not uncommon to remove a patient’s dressing and uncover an iatrogenically purple wound. Best of all, pediatric patients are invariably amused when they see someone drawing on their skin with a purple marker.
When using a sterile surgical marker to apply gentian violet to the skin, we use either the marker tip or the ink core, which Dr. Siegel taught me can be easily accessed by snapping most plastic markers in half.
Item 3: Handheld Blacklight
The Wood lamp is a useful tool in the diagnosis of various infectious diseases and pigmentary disorders,4 but it is not always practical to use when on call, as standard ones are relatively large and corded, so they must be plugged into an electric outlet to work. You can therefore imagine the gratitude I have for my co-residents Miriam Lieberman, MD; Jaime Alexander, MD; Nicole Weiler, MD; and Alessandra Haskin, MD, for introducing me to the most convenient Wood lamp: the handheld blacklight. For less than $10, this gadget combines the diagnostic power of UV light with the portability of a pocket-sized, battery-powered flashlight. You will never want to use another Wood lamp again.
Item 4: Normal Saline Flush
Normal saline can be used for more than storing specimens for frozen section or tissue culture; it also can substitute for Michel solution when storing specimens for direct immunofluorescence (DIF) studies. I learned this tip from Edward Heilman, MD, a dermatopathologist at Downstate. For the last 20 years, Dr. Heilman has been successfully storing DIF specimens in refrigerated normal saline for up to 24 hours when Michel solution is unavailable, after which the specimen is processed or transferred to Michel solution for further storage while being transported to an immunofluorescence laboratory.
In 2004, Vodegel et al5 formally studied this technique in 25 patients with autoimmune skin diseases such as pemphigus and pemphigoid. (Thanks to Dr. Lieberman for telling me about this study.) The experiment involved taking 4 punch biopsies from each patient and placing them in either normal saline at −80°C for 24 or 48 hours, room temperature Michel solution for 48 hours, or liquid nitrogen for up to 2 weeks before being processed for DIF and analyzed by a blinded interpreter. Interestingly, specimens stored in normal saline for 24 hours were the most diagnostic, with a conclusive diagnosis reached in 21 of 25 specimens (84%). This result was attributed to the statistically significant reduction (P<.01) in background fluorescence with normal saline compared to Michel solution and liquid nitrogen, which in turn allowed for easier detection of diagnostic immunoreactants. Similar to Dr. Heilman, the authors cautioned against placing DIF specimens in normal saline for more than 24 hours; in their experience, the risk for an artefactual split developing at the dermoepidermal junction increases with this practice.5
It is no secret that a well-stocked on-call bag is one of the keys to providing inpatient care as a dermatology resident. Beyond the basic items that should never be left at home, there are some lesser-known tools that I have learned about from my book- and street-smart attendings and co-residents in the Department of Dermatology at the State University of New York Downstate Medical Center (referred to here as Downstate). Here are our top 4 items to pack the next time you are on call. (Bonus: you will find them helpful in clinic, too.)
Item 1: WoundSeal Powder
The most valuable player in my on-call bag, WoundSeal Powder (Biolife) is an over-the-counter hemostatic agent that I learned about from Daniel M. Siegel, MD, MS, a Mohs surgeon at Downstate and former president of the American Academy of Dermatology. The powder consists of a hydrophilic polymer and potassium ferrate.1 When poured over a bleeding wound and pressed in place (eg, with a sterile cotton-tipped swab), the hydrophilic polymer absorbs plasma while the iron in potassium ferrate agglomerates blood solids. The result is a scablike seal that is safe to leave in place until the wound has healed.1
Since Dr. Siegel introduced WoundSeal to Downstate about a decade ago, it has become our department’s go-to hemostatic agent for most punch biopsies performed in the inpatient setting. In our experience, achieving hemostasis in the hospital usually is easier, safer, and faster with WoundSeal than suture. Furthermore, using WoundSeal eliminates the need for patients to follow up for suture removal. From a practical perspective, WoundSeal works best when the biopsy defect is positioned parallel to the ground so the powder can be poured directly over and into the defect. From a cosmetic perspective, we have found that WoundSeal and suture have similar outcomes when used for punch biopsies up to 4 mm in size on the trunk and extremities in both adult and pediatric patients. Working with other dermatology attendings such as Sharon A. Glick, MD; Eve Lowenstein, MD, PhD; and Jeannette Jakus, MD, MBA, I also have found WoundSeal helpful when taking care of suture-phobic children or patients with lesions that are less amenable to suture, such as an ulcer or indurated plaque.
Item 2: Purple Surgical Marker
Another tip I have learned from Drs. Siegel and Jakus: If you are ever in a bind for a topical antibacterial or antifungal agent, look no further than a sterile purple surgical marker. These markers are a surprising source of gentian violet, the same purple dye that is the basis of Gram staining and sold as an over-the-counter antiseptic in 1% to 2% concentrations. Purple surgical markers, on the other hand, are 2.5% to 10% gentian violet.2
Gentian violet has been shown to have antibacterial, antifungal, antiviral, antihelminthic, and antitrypanosomal properties, but its efficacy has been mostly demonstrated against Streptococcus, methicillin-sensitive and methicillin-resistant Staphylococcus aureus, and Candida.3 Given the dermatologic relevance of these organisms, gentian violet is a favorite among attendings at my residency program; it is not uncommon to remove a patient’s dressing and uncover an iatrogenically purple wound. Best of all, pediatric patients are invariably amused when they see someone drawing on their skin with a purple marker.
When using a sterile surgical marker to apply gentian violet to the skin, we use either the marker tip or the ink core, which Dr. Siegel taught me can be easily accessed by snapping most plastic markers in half.
Item 3: Handheld Blacklight
The Wood lamp is a useful tool in the diagnosis of various infectious diseases and pigmentary disorders,4 but it is not always practical to use when on call, as standard ones are relatively large and corded, so they must be plugged into an electric outlet to work. You can therefore imagine the gratitude I have for my co-residents Miriam Lieberman, MD; Jaime Alexander, MD; Nicole Weiler, MD; and Alessandra Haskin, MD, for introducing me to the most convenient Wood lamp: the handheld blacklight. For less than $10, this gadget combines the diagnostic power of UV light with the portability of a pocket-sized, battery-powered flashlight. You will never want to use another Wood lamp again.
Item 4: Normal Saline Flush
Normal saline can be used for more than storing specimens for frozen section or tissue culture; it also can substitute for Michel solution when storing specimens for direct immunofluorescence (DIF) studies. I learned this tip from Edward Heilman, MD, a dermatopathologist at Downstate. For the last 20 years, Dr. Heilman has been successfully storing DIF specimens in refrigerated normal saline for up to 24 hours when Michel solution is unavailable, after which the specimen is processed or transferred to Michel solution for further storage while being transported to an immunofluorescence laboratory.
In 2004, Vodegel et al5 formally studied this technique in 25 patients with autoimmune skin diseases such as pemphigus and pemphigoid. (Thanks to Dr. Lieberman for telling me about this study.) The experiment involved taking 4 punch biopsies from each patient and placing them in either normal saline at −80°C for 24 or 48 hours, room temperature Michel solution for 48 hours, or liquid nitrogen for up to 2 weeks before being processed for DIF and analyzed by a blinded interpreter. Interestingly, specimens stored in normal saline for 24 hours were the most diagnostic, with a conclusive diagnosis reached in 21 of 25 specimens (84%). This result was attributed to the statistically significant reduction (P<.01) in background fluorescence with normal saline compared to Michel solution and liquid nitrogen, which in turn allowed for easier detection of diagnostic immunoreactants. Similar to Dr. Heilman, the authors cautioned against placing DIF specimens in normal saline for more than 24 hours; in their experience, the risk for an artefactual split developing at the dermoepidermal junction increases with this practice.5
- Biolife. How WoundSeal works. WoundSeal website. http://woundseal.com/how-it-works. Accessed March 7, 2019.
- Viscot Medical, LLC. Safety data sheet. http://www.viscot.com/download/MSDS%20Gentian%20Violet%20Ink.pdf. Published September 11, 2014. Accessed March 7, 2019.
- Maley AM, Arbiser JL. Gentian violet: a 19th century drug re-emerges in the 21st century. Exp Dermatol. 2013;22:775-780.
- Klatte JL, van der Beek N, Kemperman PM. 100 years of Wood’s lamp revised. J Eur Acad Dermatol Venereol. 2015;29:842-847.
- Vodegel RM, de Jong MC, Meijer HJ, et al. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
- Biolife. How WoundSeal works. WoundSeal website. http://woundseal.com/how-it-works. Accessed March 7, 2019.
- Viscot Medical, LLC. Safety data sheet. http://www.viscot.com/download/MSDS%20Gentian%20Violet%20Ink.pdf. Published September 11, 2014. Accessed March 7, 2019.
- Maley AM, Arbiser JL. Gentian violet: a 19th century drug re-emerges in the 21st century. Exp Dermatol. 2013;22:775-780.
- Klatte JL, van der Beek N, Kemperman PM. 100 years of Wood’s lamp revised. J Eur Acad Dermatol Venereol. 2015;29:842-847.
- Vodegel RM, de Jong MC, Meijer HJ, et al. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
Resident Pearl
- The following unconventional items will come in handy the next time you are on call (or in clinic) and need an alternative to a suture, topical antimicrobial, Wood lamp, or Michel solution.

Concurrent Keratoacanthomas and Nonsarcoidal Granulomatous Reactions in New and Preexisting Tattoos
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
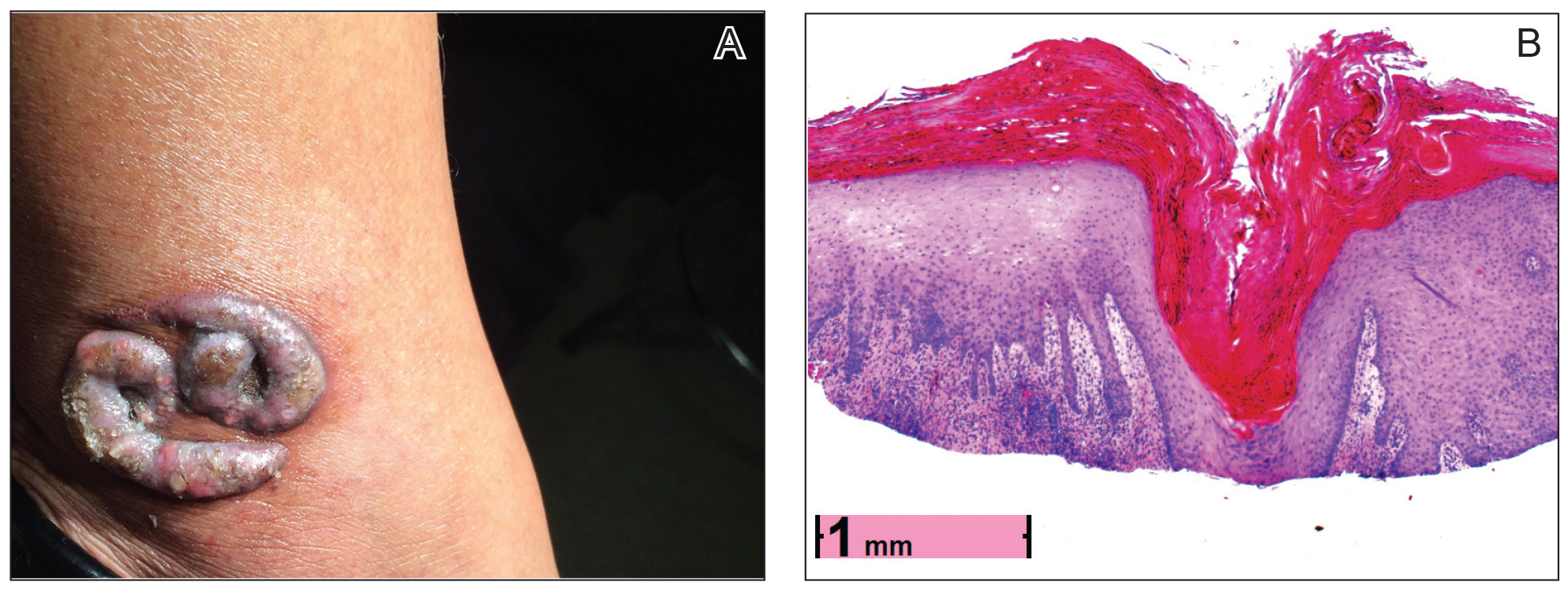
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.

contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
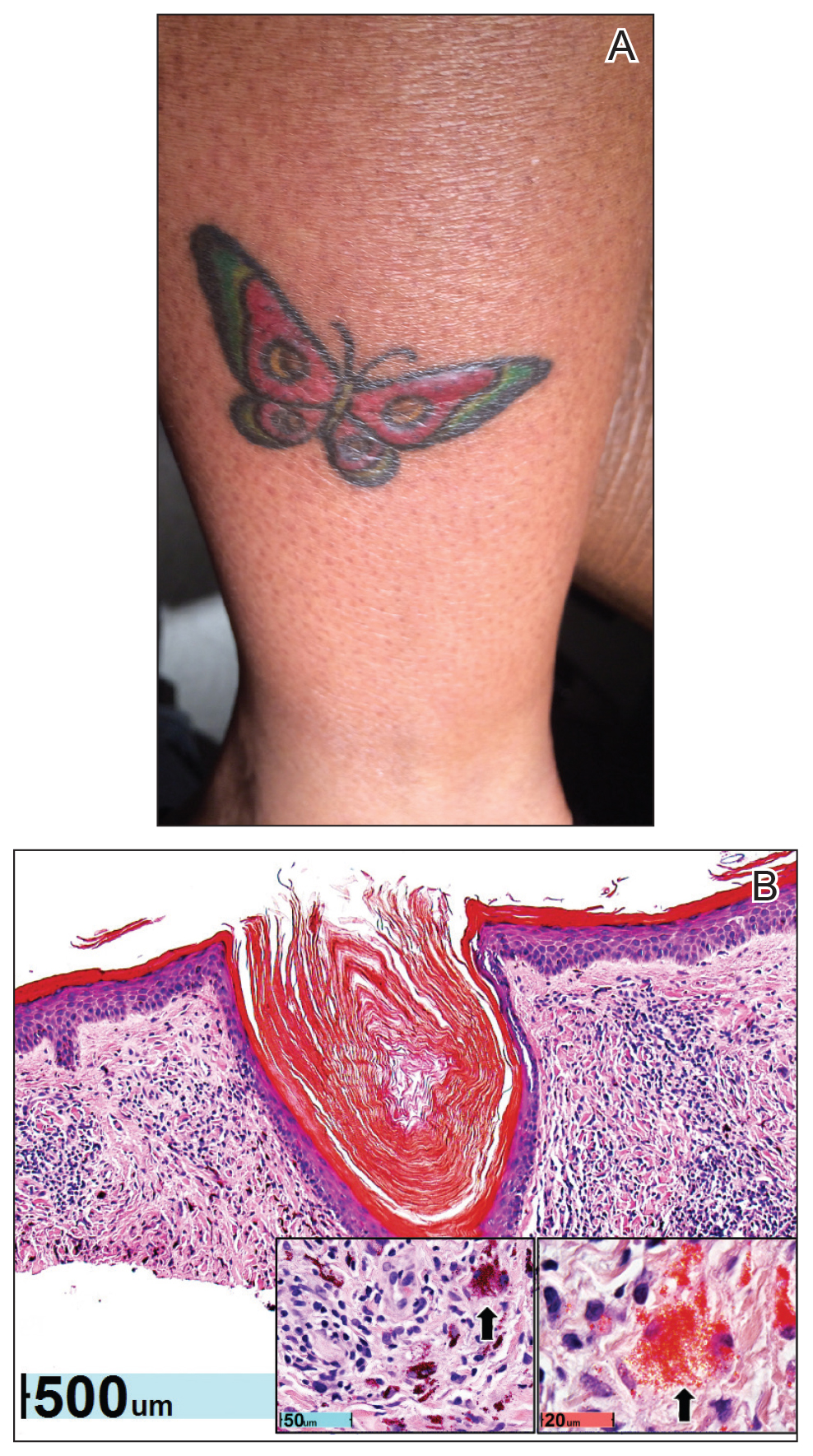
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
Practice Points
- Keratoacanthomas (KAs) are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution.
- The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, scarring disorders, and trauma (including tattoos).
- Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Our case adds additional weight to the idea that some KAs are primarily reactive phenomena sharing features of other reactive cutaneous proliferations such as foreign body granulomas.

Indurated Plaque on the Shoulder
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
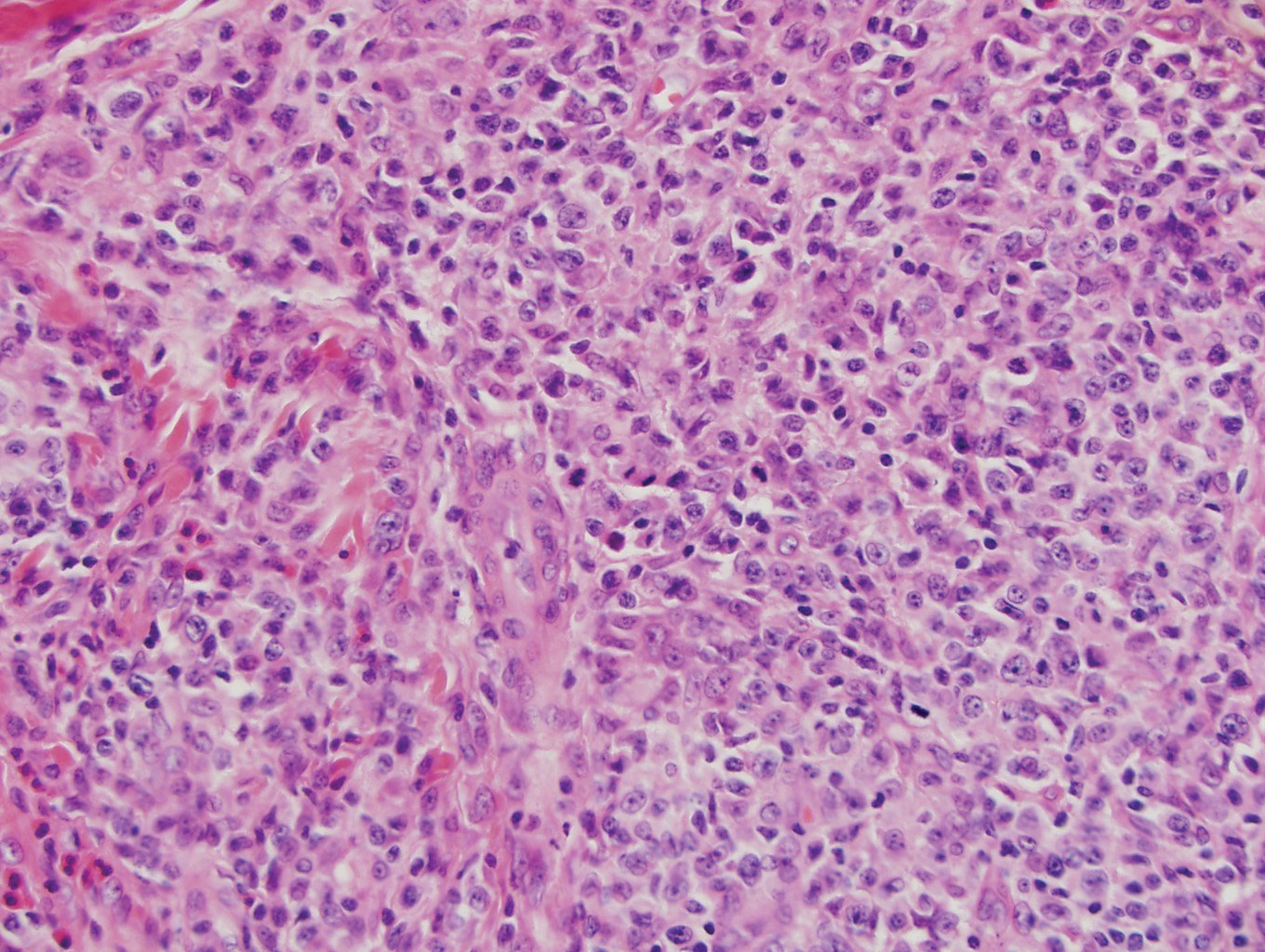
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
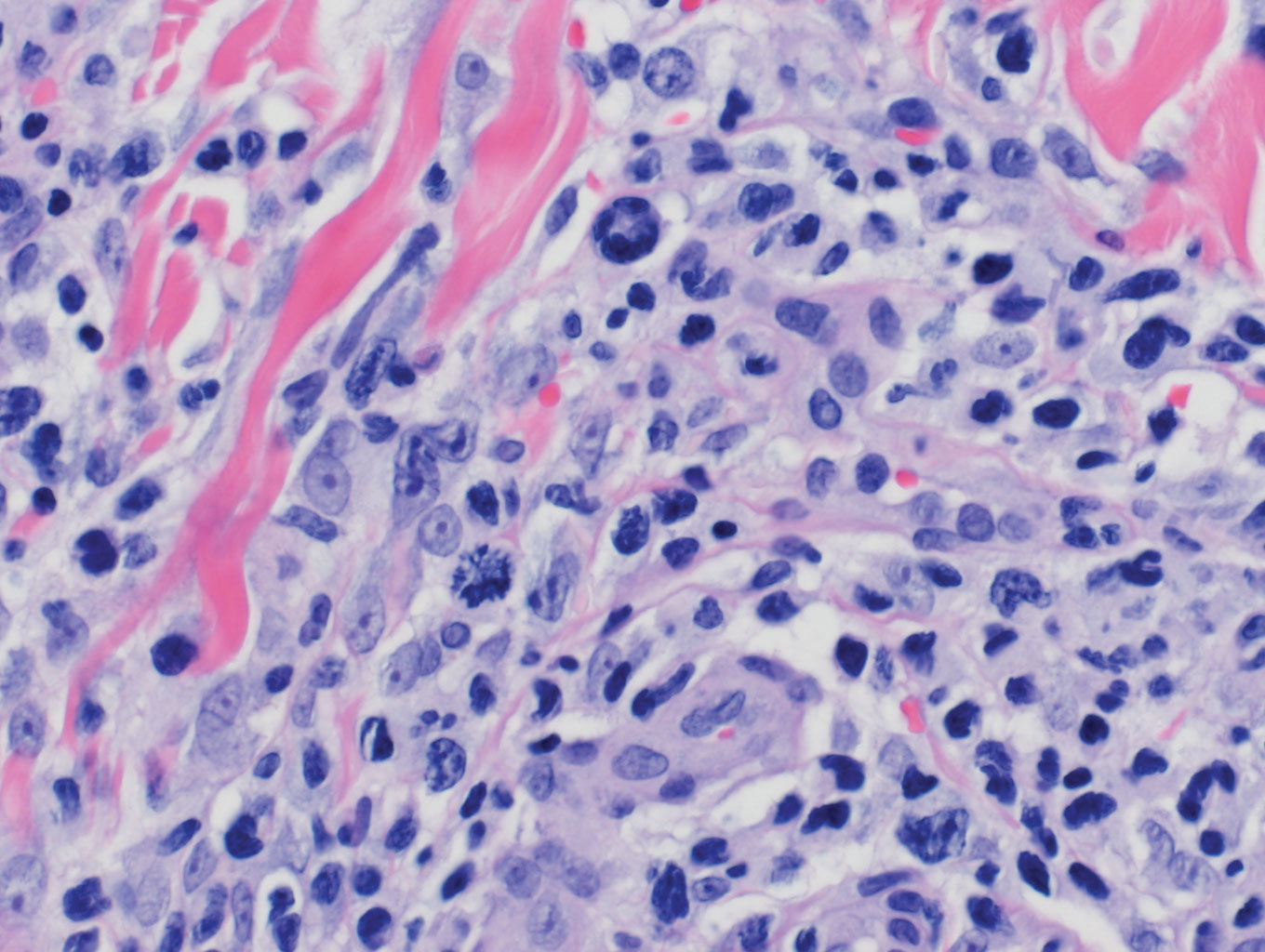
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
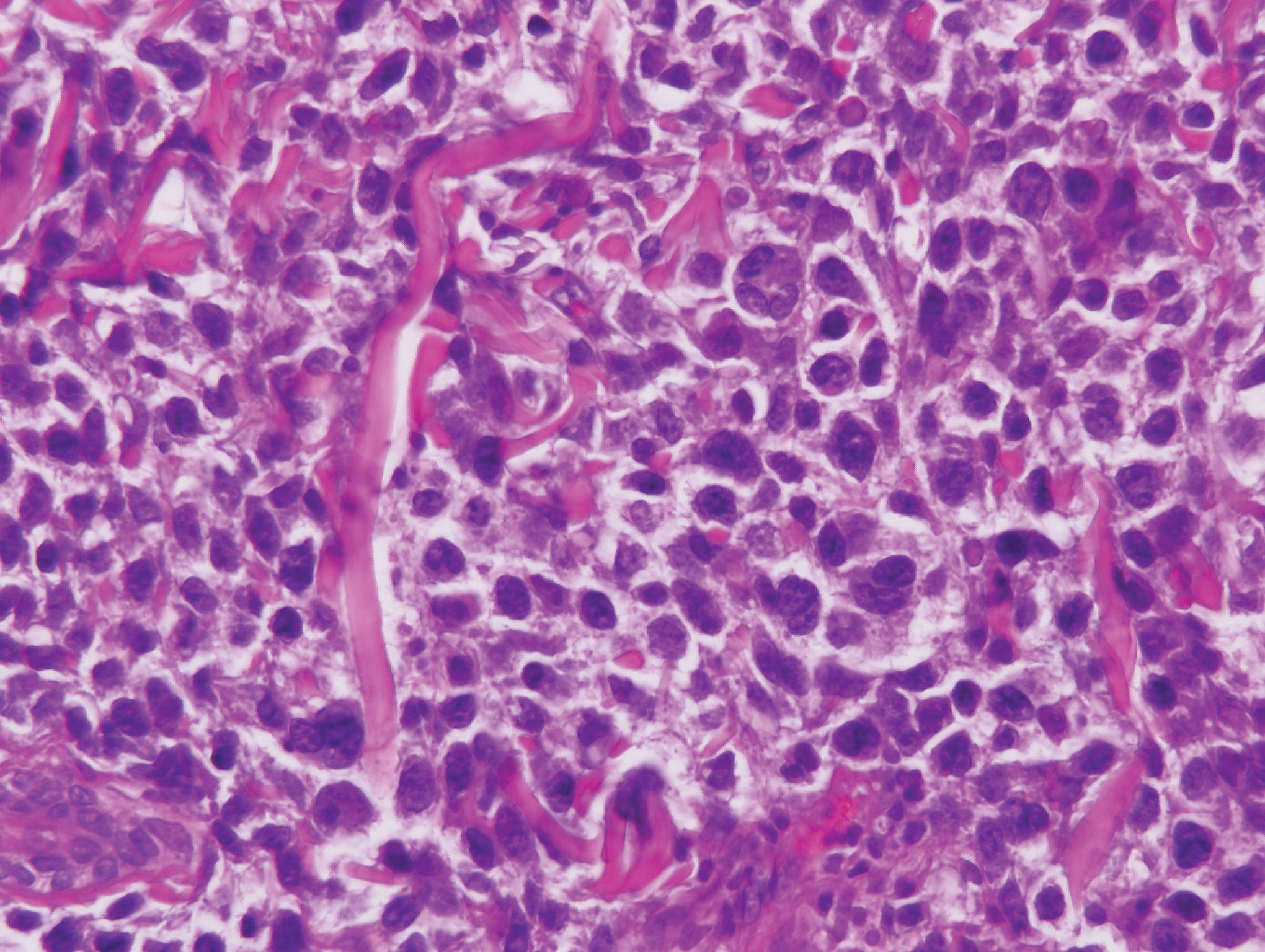
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
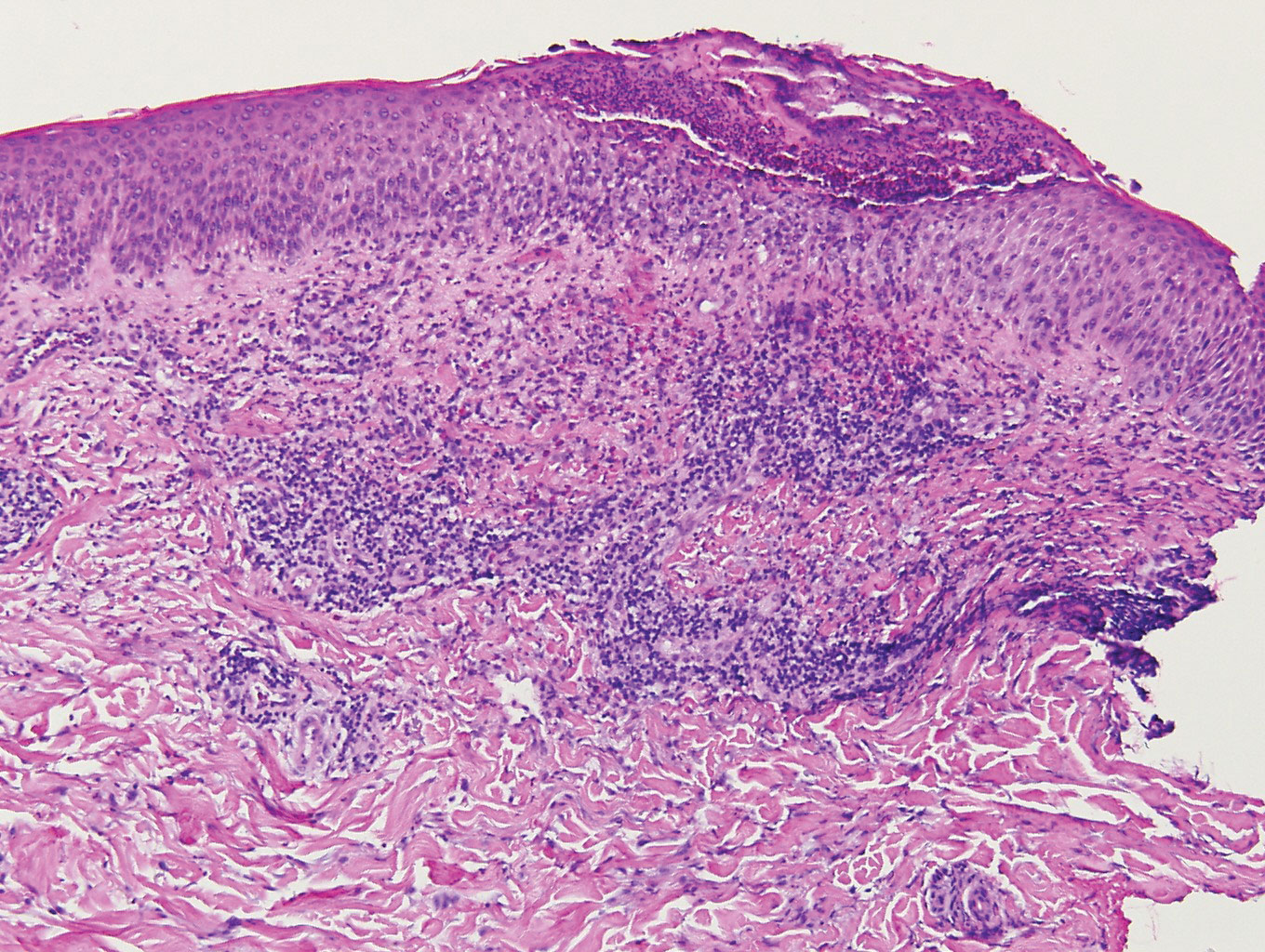
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
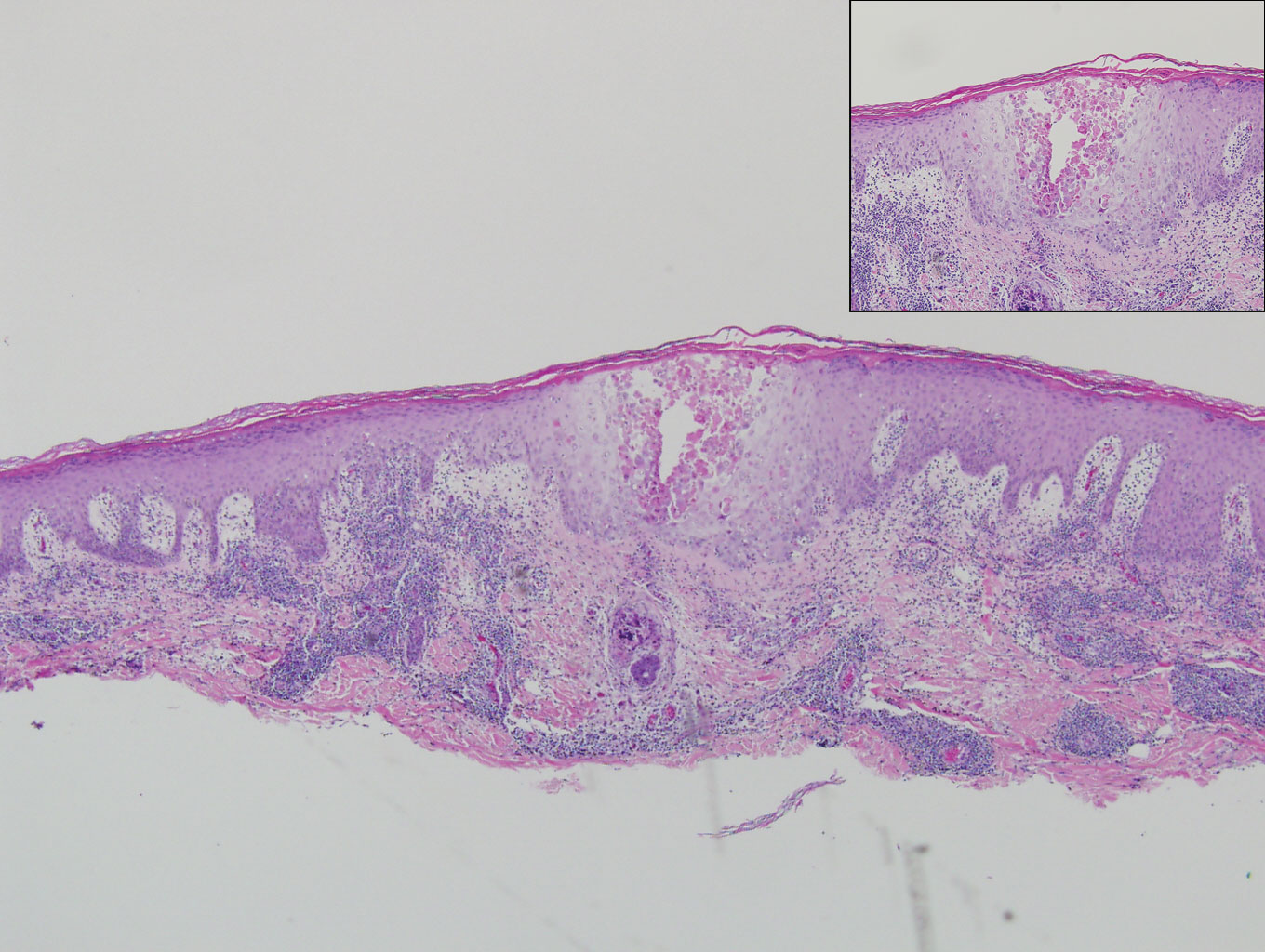
A 66-year-old man with mycosis fungoides presented with a new indurated plaque on the left shoulder. Biopsies of the left shoulder and back lesions were obtained.
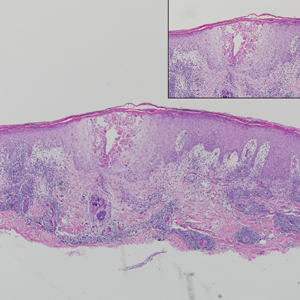
Infographic: Laser Hair Removal
Dermatologists are best equipped to treat patients who are interested in removing unwanted hair safely and effectively. Unfortunately, many patients often undergo laser hair removal treatments at spas by practitioners with limited training. Dermatologists must encourage patients to seek treatment from a board-certified dermatologist.
Full survey results and commentary from Dr. Shari Lipner are available at bit.ly/2tzNbSg.
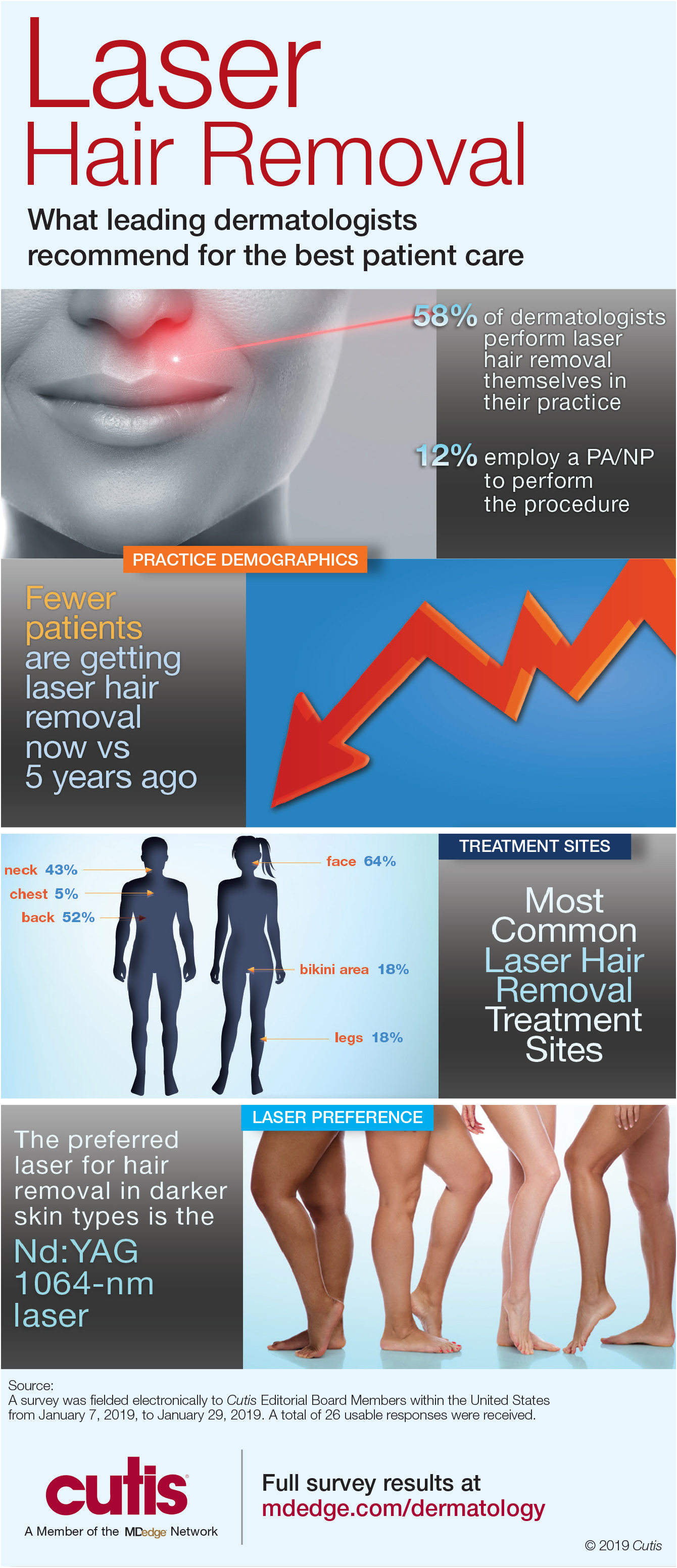
Dermatologists are best equipped to treat patients who are interested in removing unwanted hair safely and effectively. Unfortunately, many patients often undergo laser hair removal treatments at spas by practitioners with limited training. Dermatologists must encourage patients to seek treatment from a board-certified dermatologist.
Full survey results and commentary from Dr. Shari Lipner are available at bit.ly/2tzNbSg.
Dermatologists are best equipped to treat patients who are interested in removing unwanted hair safely and effectively. Unfortunately, many patients often undergo laser hair removal treatments at spas by practitioners with limited training. Dermatologists must encourage patients to seek treatment from a board-certified dermatologist.
Full survey results and commentary from Dr. Shari Lipner are available at bit.ly/2tzNbSg.

Cutaneous Gummatous Tuberculosis in a Kidney Transplant Patient
Case Report
A 60-year-old Cambodian woman presented with recurrent fever (temperature, up to 38.8°C) 7 months after receiving a kidney transplant secondary to polycystic kidney disease. Fever was attributed to recurrent pyelonephritis of the native kidneys while on mycophenolate mofetil, tacrolimus, and prednisone. As a result, she underwent a bilateral native nephrectomy and was found to have peritoneal nodules. Pathology of both native kidneys and peritoneal tissue revealed caseating granulomas and acid-fast bacilli (AFB) diagnostic for kidney and peritoneal tuberculosis (TB). She had no history of TB, and a TB skin test (purified protein derivative [PPD]) upon entering the United States from Cambodia a decade earlier was negative. Additionally, her pretransplantation PPD was negative.
Treatment with isoniazid, ethambutol, pyrazinamide, and levofloxacin was initiated immediately upon diagnosis, and all of her immunosuppressive medications—mycophenolate mofetil, tacrolimus, and prednisone—were discontinued. Her symptoms subsided within 1 week, and she was discharged from the hospital. Over the next 2 months, her immunosuppressive medications were restarted, and her TB medications were periodically discontinued by the Tuberculosis Control Program at the Department of Health (Philadelphia, Pennsylvania) due to severe thrombocytopenia. During this time, she was closely monitored twice weekly in the clinic with blood draws performed weekly.
Approximately 10 weeks after initiation of treatment, she noted recurrent subjective fever (temperature, up to 38.8°C) and painful lesions on the right side of the flank, left breast, and left arm of 3 days’ duration. Physical examination revealed a warm, dull red, tender nodule on the right side of the flank (Figure 1) and subcutaneous nodules with no overlying skin changes on the left breast and left arm. A biopsy of the lesion on the right side of the flank was performed, which resulted in substantial purulent drainage. Histologic analysis showed an inflammatory infiltrate within the deep dermis composed of neutrophils, macrophages, and giant cells, indicative of suppurative granulomatous dermatitis (Figure 2). Ziehl-Neelsen stain demonstrated rare AFB within the cytoplasm of macrophages, suggestive of Mycobacterium tuberculosis infection (Figure 3). A repeat chest radiograph was normal.
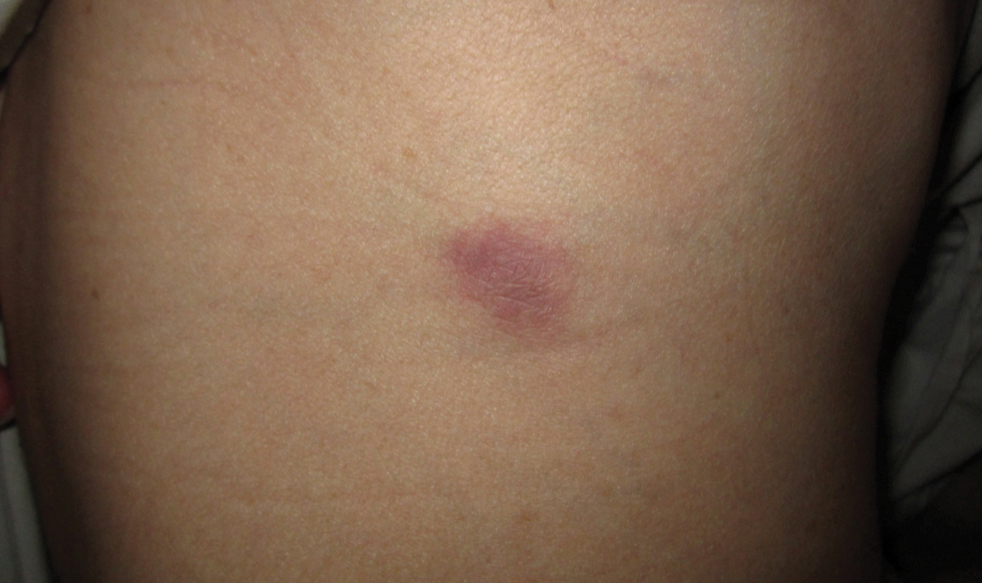
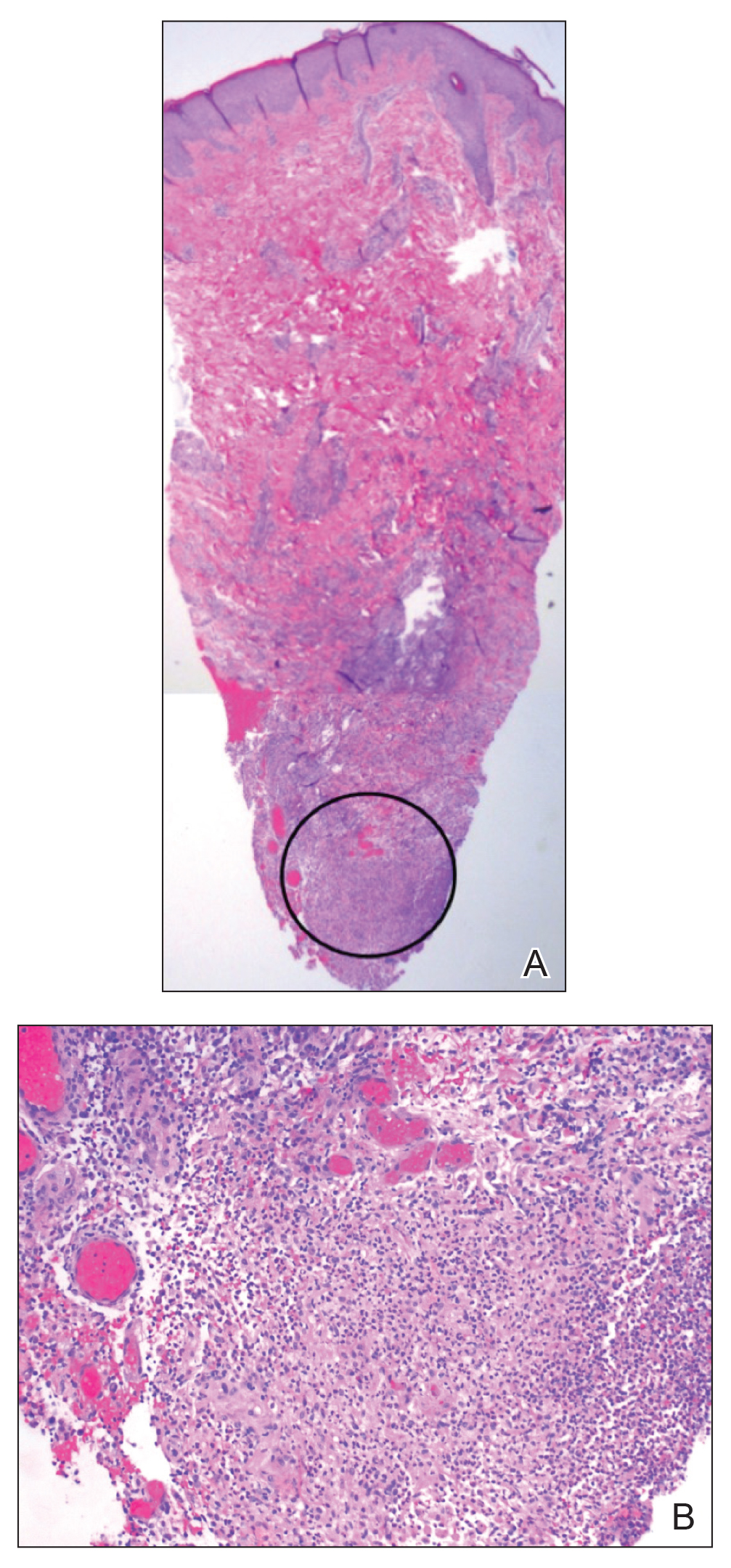
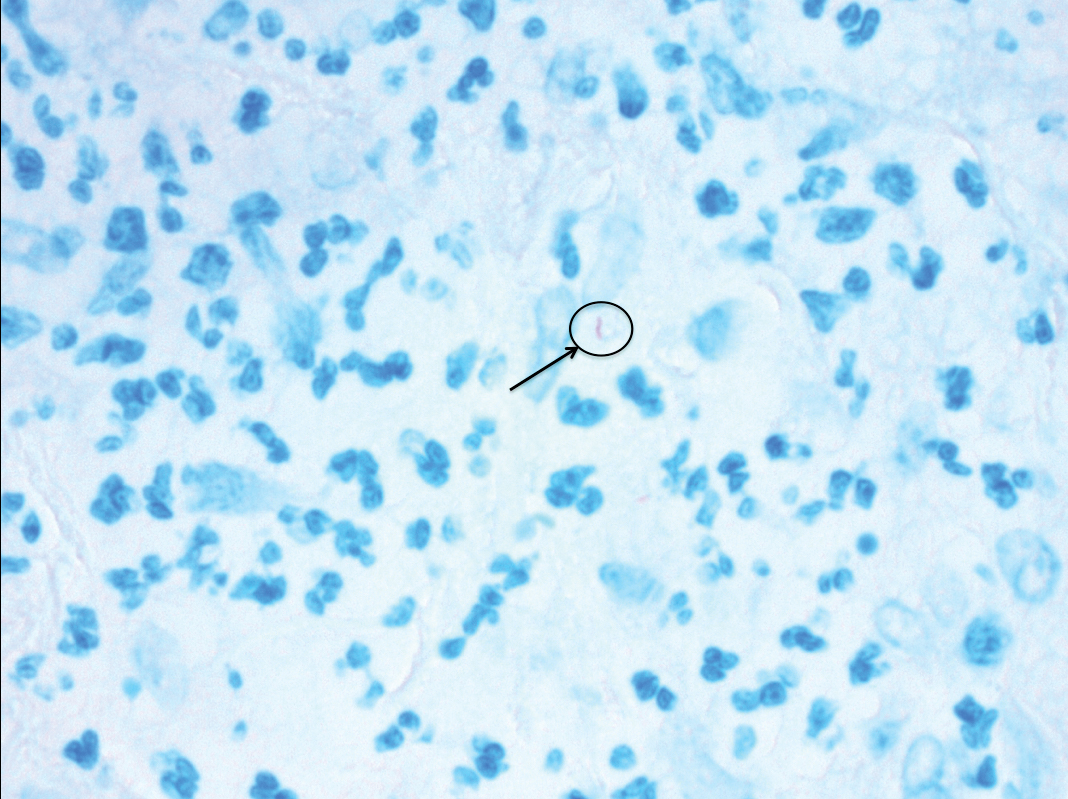
Based on the patient’s history and clinical presentation, she was continued on isoniazid, ethambutol, and levofloxacin, with complete resolution of symptoms and cutaneous lesions. Over the subsequent 2 months, the therapy was modified to rifabutin, pyrazinamide, and levofloxacin, and subsequently pyrazinamide was stopped. A subsequent biopsy of the left breast and histologic analysis indicated that the specimen was benign; stains for AFB were negative. Currently, both the fever and skin lesions have completely resolved, and she remains on anti-TB therapy.
Comment
Clinical Presentation
Cutaneous TB is an uncommon manifestation of TB that can occur either exogenously or endogenously.1 It tends to occur primarily in previously infected TB patients through hematogenous, lymphatic, or contiguous spread.2 Due to their immunocompromised state, solid organ transplant recipients have an increased incidence of primary and reactivated latent TB reported to be 20 to 74 times greater than the general population.3,4 One report stated the total incidence of posttransplant TB as 0.48% in the West and 11.8% in endemic regions such as India.5 The occurrence of cutaneous TB is rare among solid organ transplant recipients.1 On average, a diagnosis of latent TB is made 9 months after transplantation because of the opportunistic nature of M tuberculosis in an immunosuppressed environment.6
TB Subtypes
Cutaneous TB can be in the form of localized disease (eg, primary tuberculous chancre, TB verrucosa cutis, lupus vulgaris, smear-negative scrofuloderma), disseminated disease (eg, disseminated TB, TB gumma, orificial TB, miliary cutaneous TB), or tuberculids (eg, papulonecrotic tuberculid, lichen scrofulosorum, erythema induratum).7 Due to the pustular epithelioid cell granulomas and AFB positivity of the involved cutaneous lesions, our patient’s TB can be classified as a metastatic TB abscess or gummatous TB.7
Metastatic TB abscess, an uncommon subtype of cutaneous TB, generally is only seen in malnourished children and notably immunocompromised individuals.2,8,9 In these individuals, systemic failure of cell-mediated immunity enables M tuberculosis to hematogenously infect various organs of the body, resulting in alternative forms of TB, such as gummatous-type TB.10 One study reported that of the 0.1% of dermatology patients presenting with cutaneous TB, only 5.4% of these individuals had the rarer gummatous form.7 These metastatic TB abscesses begin as a single or multiple nontender subcutaneous nodule(s), which breaks down and softens to form a draining sinus abscess.2,8,9 Abscesses are most commonly seen on the trunk and extremities; however, they can be found nearly anywhere on the body.8 The pathology of cutaneous TB lesions demonstrates caseating necrosis with epithelioid and giant cells forming a surrounding rim.9
Diagnosis
Diagnosis may be difficult because of the vast number of dermatologic conditions that resemble cutaneous TB, including mycoses, sarcoidosis, leishmaniasis, leprosy, syphilis, other non-TB mycobacteria, and Wegener granulomatosis.9 Thus, confirmatory diagnosis is made via clinical presentation, detailed history and physical examination, and laboratory tests.11 These tests include the Mantoux tuberculin skin test (PPD or TST) or IFN-γ release assays (QuantiFERON-TB Gold test), identification of AFB on skin biopsy, and isolation of M tuberculosis from tissue culture or polymerase chain reaction.11
At-Risk Populations
The recommendation for the identification of at-risk populations for latent TB testing and treatment have been clearly defined by the World Health Organization (Table).12 Our patient met 2 of these criteria: she had been preparing for organ transplantation and was from a country with high TB burden. Such at-risk patients should be tested for a latent TB infection with either IFN-γ release assays or PPD.12

Treatment
The recommended treatment of active TB in transplant recipients is based on randomized trials in immunocompetent hosts, and thus the same as that used by the general population.16 This anti-TB regimen includes the use of 4 drugs—typically rifampicin, isoniazid, ethambutol, and pyrazinamide—for a 6-month duration.11 Unfortunately, the management of TB in an immunocompromised patient is more challenging due to the potential side effects and drug interactions.
Finally, thrombocytopenia is an infrequent, life-threatening complication that can be acquired by immunocompromised patients on anti-TB therapy.17 Drug-induced thrombocytopenia can be caused by a variety of medications, including rifampicin, isoniazid, ethambutol, and pyrazinamide. Diagnosis of drug-induced thrombocytopenia can be confirmed only after discontinuation of the suspected drug and subsequent resolution of the thrombocytopenia.17 Our patient initially became thrombocytopenic while taking isoniazid, ethambutol, pyrazinamide, and levofloxacin. However, her platelet levels improved once the pyrazinamide was discontinued, thereby suggesting pyrazinamide-induced thrombocytopenia.
Conclusion
The risk for infectious disease reactivation in an immunocompromised patient undergoing transplant surgery is notable. Our findings emphasize the value of a comprehensive pretransplant evaluation, vigilance even when test results appear negative, and treatment of latent TB within this population.16,18,19 Furthermore, this case illustrates a noteworthy example of a rare form of cutaneous TB, which should be considered and included in the differential for cutaneous lesions in an immunosuppressed patient.
- Sakhuja V, Jha V, Varma PP, et al. The high incidence of tuberculosis among renal transplant recipients in India. Transplantation. 1996;61:211-215.
- Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
- Schultz V, Marroni CA, Amorim CS, et al. Risk factors for hepatotoxicity in solid organ transplants recipients being treated for tuberculosis. Transplant Proc. 2014;46:3606-3610.
- Tabarsi P, Farshidpour M, Marjani M, et al. Mycobacterial infection and the impact of rifabutin treatment in organ transplant recipients: a single-center study. Saudi J Kidney Dis Transpl. 2015;26:6-11.
- Rathi M, Gundlapalli S, Ramachandran R, et al. A rare case of cytomegalovirus, scedosporium apiospermum and mycobacterium tuberculosis in a renal transplant recipient. BMC Infect Dis. 2014;14:259.
- Hickey MD, Quan DJ, Chin-Hong PV, et al. Use of rifabutin for the treatment of a latent tuberculosis infection in a patient after solid organ transplantation. Liver Transpl. 2013;19:457-461.
- Kumar B, Muralidhar S. Cutaneous tuberculosis: a twenty-year prospective study. Int J Tuberc Lung Dis. 1999;3:494-500.
- Dekeyzer S, Moerman F, Callens S, et al. Cutaneous metastatic tuberculous abscess in patient with cervico-mediastinal lymphatic tuberculosis. Acta Clin Belg. 2013;68:34-36.
- Ko M, Wu C, Chiu H. Tuberculous gumma (cutaneous metastatic tuberculous abscess). Dermatol Sinica. 2005;23:27-31.
- Steger JW, Barrett TL. Cutaneous tuberculosis. In: James WD, ed. Textbook of Military Medicine: Military Dermatology. Washington, DC: Borden Institute; 1994:355-389.
- Santos JB, Figueiredo AR, Ferraz CE, et al. Cutaneous tuberculosis: diagnosis, histopathology and treatment - part II. An Bras Dermatol. 2014;89:545-555.
- Guidelines on the Management of Latent Tuberculosis Infection. Geneva, Switzerland: World Health Organization; 2015.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 pt 2):S221-S247.
- Mycobacterium tuberculosis. Am J Transplant. 2004;4(suppl 10):37-41.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis. 2009;48:1276-1284.
- Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
- Kant S, Verma SK, Gupta V, et al. Pyrazinamide induced thrombocytopenia. Indian J Pharmacol. 2010;42:108-109.
- Screening for tuberculosis and tuberculosis infection in high-risk populations. recommendations of the Advisory Council for the Elimination of Tuberculosis. MMWR Recomm Rep. 1995;44:19-34.
- Fischer SA, Avery RK; AST Infectious Disease Community of Practice. Screening of donor and recipient prior to solid organ transplantation. Am J Transplant. 2009;9(suppl 4):S7-S18.
Case Report
A 60-year-old Cambodian woman presented with recurrent fever (temperature, up to 38.8°C) 7 months after receiving a kidney transplant secondary to polycystic kidney disease. Fever was attributed to recurrent pyelonephritis of the native kidneys while on mycophenolate mofetil, tacrolimus, and prednisone. As a result, she underwent a bilateral native nephrectomy and was found to have peritoneal nodules. Pathology of both native kidneys and peritoneal tissue revealed caseating granulomas and acid-fast bacilli (AFB) diagnostic for kidney and peritoneal tuberculosis (TB). She had no history of TB, and a TB skin test (purified protein derivative [PPD]) upon entering the United States from Cambodia a decade earlier was negative. Additionally, her pretransplantation PPD was negative.
Treatment with isoniazid, ethambutol, pyrazinamide, and levofloxacin was initiated immediately upon diagnosis, and all of her immunosuppressive medications—mycophenolate mofetil, tacrolimus, and prednisone—were discontinued. Her symptoms subsided within 1 week, and she was discharged from the hospital. Over the next 2 months, her immunosuppressive medications were restarted, and her TB medications were periodically discontinued by the Tuberculosis Control Program at the Department of Health (Philadelphia, Pennsylvania) due to severe thrombocytopenia. During this time, she was closely monitored twice weekly in the clinic with blood draws performed weekly.
Approximately 10 weeks after initiation of treatment, she noted recurrent subjective fever (temperature, up to 38.8°C) and painful lesions on the right side of the flank, left breast, and left arm of 3 days’ duration. Physical examination revealed a warm, dull red, tender nodule on the right side of the flank (Figure 1) and subcutaneous nodules with no overlying skin changes on the left breast and left arm. A biopsy of the lesion on the right side of the flank was performed, which resulted in substantial purulent drainage. Histologic analysis showed an inflammatory infiltrate within the deep dermis composed of neutrophils, macrophages, and giant cells, indicative of suppurative granulomatous dermatitis (Figure 2). Ziehl-Neelsen stain demonstrated rare AFB within the cytoplasm of macrophages, suggestive of Mycobacterium tuberculosis infection (Figure 3). A repeat chest radiograph was normal.
Based on the patient’s history and clinical presentation, she was continued on isoniazid, ethambutol, and levofloxacin, with complete resolution of symptoms and cutaneous lesions. Over the subsequent 2 months, the therapy was modified to rifabutin, pyrazinamide, and levofloxacin, and subsequently pyrazinamide was stopped. A subsequent biopsy of the left breast and histologic analysis indicated that the specimen was benign; stains for AFB were negative. Currently, both the fever and skin lesions have completely resolved, and she remains on anti-TB therapy.
Comment
Clinical Presentation
Cutaneous TB is an uncommon manifestation of TB that can occur either exogenously or endogenously.1 It tends to occur primarily in previously infected TB patients through hematogenous, lymphatic, or contiguous spread.2 Due to their immunocompromised state, solid organ transplant recipients have an increased incidence of primary and reactivated latent TB reported to be 20 to 74 times greater than the general population.3,4 One report stated the total incidence of posttransplant TB as 0.48% in the West and 11.8% in endemic regions such as India.5 The occurrence of cutaneous TB is rare among solid organ transplant recipients.1 On average, a diagnosis of latent TB is made 9 months after transplantation because of the opportunistic nature of M tuberculosis in an immunosuppressed environment.6
TB Subtypes
Cutaneous TB can be in the form of localized disease (eg, primary tuberculous chancre, TB verrucosa cutis, lupus vulgaris, smear-negative scrofuloderma), disseminated disease (eg, disseminated TB, TB gumma, orificial TB, miliary cutaneous TB), or tuberculids (eg, papulonecrotic tuberculid, lichen scrofulosorum, erythema induratum).7 Due to the pustular epithelioid cell granulomas and AFB positivity of the involved cutaneous lesions, our patient’s TB can be classified as a metastatic TB abscess or gummatous TB.7
Metastatic TB abscess, an uncommon subtype of cutaneous TB, generally is only seen in malnourished children and notably immunocompromised individuals.2,8,9 In these individuals, systemic failure of cell-mediated immunity enables M tuberculosis to hematogenously infect various organs of the body, resulting in alternative forms of TB, such as gummatous-type TB.10 One study reported that of the 0.1% of dermatology patients presenting with cutaneous TB, only 5.4% of these individuals had the rarer gummatous form.7 These metastatic TB abscesses begin as a single or multiple nontender subcutaneous nodule(s), which breaks down and softens to form a draining sinus abscess.2,8,9 Abscesses are most commonly seen on the trunk and extremities; however, they can be found nearly anywhere on the body.8 The pathology of cutaneous TB lesions demonstrates caseating necrosis with epithelioid and giant cells forming a surrounding rim.9
Diagnosis
Diagnosis may be difficult because of the vast number of dermatologic conditions that resemble cutaneous TB, including mycoses, sarcoidosis, leishmaniasis, leprosy, syphilis, other non-TB mycobacteria, and Wegener granulomatosis.9 Thus, confirmatory diagnosis is made via clinical presentation, detailed history and physical examination, and laboratory tests.11 These tests include the Mantoux tuberculin skin test (PPD or TST) or IFN-γ release assays (QuantiFERON-TB Gold test), identification of AFB on skin biopsy, and isolation of M tuberculosis from tissue culture or polymerase chain reaction.11
At-Risk Populations
The recommendation for the identification of at-risk populations for latent TB testing and treatment have been clearly defined by the World Health Organization (Table).12 Our patient met 2 of these criteria: she had been preparing for organ transplantation and was from a country with high TB burden. Such at-risk patients should be tested for a latent TB infection with either IFN-γ release assays or PPD.12
Treatment
The recommended treatment of active TB in transplant recipients is based on randomized trials in immunocompetent hosts, and thus the same as that used by the general population.16 This anti-TB regimen includes the use of 4 drugs—typically rifampicin, isoniazid, ethambutol, and pyrazinamide—for a 6-month duration.11 Unfortunately, the management of TB in an immunocompromised patient is more challenging due to the potential side effects and drug interactions.
Finally, thrombocytopenia is an infrequent, life-threatening complication that can be acquired by immunocompromised patients on anti-TB therapy.17 Drug-induced thrombocytopenia can be caused by a variety of medications, including rifampicin, isoniazid, ethambutol, and pyrazinamide. Diagnosis of drug-induced thrombocytopenia can be confirmed only after discontinuation of the suspected drug and subsequent resolution of the thrombocytopenia.17 Our patient initially became thrombocytopenic while taking isoniazid, ethambutol, pyrazinamide, and levofloxacin. However, her platelet levels improved once the pyrazinamide was discontinued, thereby suggesting pyrazinamide-induced thrombocytopenia.
Conclusion
The risk for infectious disease reactivation in an immunocompromised patient undergoing transplant surgery is notable. Our findings emphasize the value of a comprehensive pretransplant evaluation, vigilance even when test results appear negative, and treatment of latent TB within this population.16,18,19 Furthermore, this case illustrates a noteworthy example of a rare form of cutaneous TB, which should be considered and included in the differential for cutaneous lesions in an immunosuppressed patient.
Case Report
A 60-year-old Cambodian woman presented with recurrent fever (temperature, up to 38.8°C) 7 months after receiving a kidney transplant secondary to polycystic kidney disease. Fever was attributed to recurrent pyelonephritis of the native kidneys while on mycophenolate mofetil, tacrolimus, and prednisone. As a result, she underwent a bilateral native nephrectomy and was found to have peritoneal nodules. Pathology of both native kidneys and peritoneal tissue revealed caseating granulomas and acid-fast bacilli (AFB) diagnostic for kidney and peritoneal tuberculosis (TB). She had no history of TB, and a TB skin test (purified protein derivative [PPD]) upon entering the United States from Cambodia a decade earlier was negative. Additionally, her pretransplantation PPD was negative.
Treatment with isoniazid, ethambutol, pyrazinamide, and levofloxacin was initiated immediately upon diagnosis, and all of her immunosuppressive medications—mycophenolate mofetil, tacrolimus, and prednisone—were discontinued. Her symptoms subsided within 1 week, and she was discharged from the hospital. Over the next 2 months, her immunosuppressive medications were restarted, and her TB medications were periodically discontinued by the Tuberculosis Control Program at the Department of Health (Philadelphia, Pennsylvania) due to severe thrombocytopenia. During this time, she was closely monitored twice weekly in the clinic with blood draws performed weekly.
Approximately 10 weeks after initiation of treatment, she noted recurrent subjective fever (temperature, up to 38.8°C) and painful lesions on the right side of the flank, left breast, and left arm of 3 days’ duration. Physical examination revealed a warm, dull red, tender nodule on the right side of the flank (Figure 1) and subcutaneous nodules with no overlying skin changes on the left breast and left arm. A biopsy of the lesion on the right side of the flank was performed, which resulted in substantial purulent drainage. Histologic analysis showed an inflammatory infiltrate within the deep dermis composed of neutrophils, macrophages, and giant cells, indicative of suppurative granulomatous dermatitis (Figure 2). Ziehl-Neelsen stain demonstrated rare AFB within the cytoplasm of macrophages, suggestive of Mycobacterium tuberculosis infection (Figure 3). A repeat chest radiograph was normal.
Based on the patient’s history and clinical presentation, she was continued on isoniazid, ethambutol, and levofloxacin, with complete resolution of symptoms and cutaneous lesions. Over the subsequent 2 months, the therapy was modified to rifabutin, pyrazinamide, and levofloxacin, and subsequently pyrazinamide was stopped. A subsequent biopsy of the left breast and histologic analysis indicated that the specimen was benign; stains for AFB were negative. Currently, both the fever and skin lesions have completely resolved, and she remains on anti-TB therapy.
Comment
Clinical Presentation
Cutaneous TB is an uncommon manifestation of TB that can occur either exogenously or endogenously.1 It tends to occur primarily in previously infected TB patients through hematogenous, lymphatic, or contiguous spread.2 Due to their immunocompromised state, solid organ transplant recipients have an increased incidence of primary and reactivated latent TB reported to be 20 to 74 times greater than the general population.3,4 One report stated the total incidence of posttransplant TB as 0.48% in the West and 11.8% in endemic regions such as India.5 The occurrence of cutaneous TB is rare among solid organ transplant recipients.1 On average, a diagnosis of latent TB is made 9 months after transplantation because of the opportunistic nature of M tuberculosis in an immunosuppressed environment.6
TB Subtypes
Cutaneous TB can be in the form of localized disease (eg, primary tuberculous chancre, TB verrucosa cutis, lupus vulgaris, smear-negative scrofuloderma), disseminated disease (eg, disseminated TB, TB gumma, orificial TB, miliary cutaneous TB), or tuberculids (eg, papulonecrotic tuberculid, lichen scrofulosorum, erythema induratum).7 Due to the pustular epithelioid cell granulomas and AFB positivity of the involved cutaneous lesions, our patient’s TB can be classified as a metastatic TB abscess or gummatous TB.7
Metastatic TB abscess, an uncommon subtype of cutaneous TB, generally is only seen in malnourished children and notably immunocompromised individuals.2,8,9 In these individuals, systemic failure of cell-mediated immunity enables M tuberculosis to hematogenously infect various organs of the body, resulting in alternative forms of TB, such as gummatous-type TB.10 One study reported that of the 0.1% of dermatology patients presenting with cutaneous TB, only 5.4% of these individuals had the rarer gummatous form.7 These metastatic TB abscesses begin as a single or multiple nontender subcutaneous nodule(s), which breaks down and softens to form a draining sinus abscess.2,8,9 Abscesses are most commonly seen on the trunk and extremities; however, they can be found nearly anywhere on the body.8 The pathology of cutaneous TB lesions demonstrates caseating necrosis with epithelioid and giant cells forming a surrounding rim.9
Diagnosis
Diagnosis may be difficult because of the vast number of dermatologic conditions that resemble cutaneous TB, including mycoses, sarcoidosis, leishmaniasis, leprosy, syphilis, other non-TB mycobacteria, and Wegener granulomatosis.9 Thus, confirmatory diagnosis is made via clinical presentation, detailed history and physical examination, and laboratory tests.11 These tests include the Mantoux tuberculin skin test (PPD or TST) or IFN-γ release assays (QuantiFERON-TB Gold test), identification of AFB on skin biopsy, and isolation of M tuberculosis from tissue culture or polymerase chain reaction.11
At-Risk Populations
The recommendation for the identification of at-risk populations for latent TB testing and treatment have been clearly defined by the World Health Organization (Table).12 Our patient met 2 of these criteria: she had been preparing for organ transplantation and was from a country with high TB burden. Such at-risk patients should be tested for a latent TB infection with either IFN-γ release assays or PPD.12
Treatment
The recommended treatment of active TB in transplant recipients is based on randomized trials in immunocompetent hosts, and thus the same as that used by the general population.16 This anti-TB regimen includes the use of 4 drugs—typically rifampicin, isoniazid, ethambutol, and pyrazinamide—for a 6-month duration.11 Unfortunately, the management of TB in an immunocompromised patient is more challenging due to the potential side effects and drug interactions.
Finally, thrombocytopenia is an infrequent, life-threatening complication that can be acquired by immunocompromised patients on anti-TB therapy.17 Drug-induced thrombocytopenia can be caused by a variety of medications, including rifampicin, isoniazid, ethambutol, and pyrazinamide. Diagnosis of drug-induced thrombocytopenia can be confirmed only after discontinuation of the suspected drug and subsequent resolution of the thrombocytopenia.17 Our patient initially became thrombocytopenic while taking isoniazid, ethambutol, pyrazinamide, and levofloxacin. However, her platelet levels improved once the pyrazinamide was discontinued, thereby suggesting pyrazinamide-induced thrombocytopenia.
Conclusion
The risk for infectious disease reactivation in an immunocompromised patient undergoing transplant surgery is notable. Our findings emphasize the value of a comprehensive pretransplant evaluation, vigilance even when test results appear negative, and treatment of latent TB within this population.16,18,19 Furthermore, this case illustrates a noteworthy example of a rare form of cutaneous TB, which should be considered and included in the differential for cutaneous lesions in an immunosuppressed patient.
- Sakhuja V, Jha V, Varma PP, et al. The high incidence of tuberculosis among renal transplant recipients in India. Transplantation. 1996;61:211-215.
- Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
- Schultz V, Marroni CA, Amorim CS, et al. Risk factors for hepatotoxicity in solid organ transplants recipients being treated for tuberculosis. Transplant Proc. 2014;46:3606-3610.
- Tabarsi P, Farshidpour M, Marjani M, et al. Mycobacterial infection and the impact of rifabutin treatment in organ transplant recipients: a single-center study. Saudi J Kidney Dis Transpl. 2015;26:6-11.
- Rathi M, Gundlapalli S, Ramachandran R, et al. A rare case of cytomegalovirus, scedosporium apiospermum and mycobacterium tuberculosis in a renal transplant recipient. BMC Infect Dis. 2014;14:259.
- Hickey MD, Quan DJ, Chin-Hong PV, et al. Use of rifabutin for the treatment of a latent tuberculosis infection in a patient after solid organ transplantation. Liver Transpl. 2013;19:457-461.
- Kumar B, Muralidhar S. Cutaneous tuberculosis: a twenty-year prospective study. Int J Tuberc Lung Dis. 1999;3:494-500.
- Dekeyzer S, Moerman F, Callens S, et al. Cutaneous metastatic tuberculous abscess in patient with cervico-mediastinal lymphatic tuberculosis. Acta Clin Belg. 2013;68:34-36.
- Ko M, Wu C, Chiu H. Tuberculous gumma (cutaneous metastatic tuberculous abscess). Dermatol Sinica. 2005;23:27-31.
- Steger JW, Barrett TL. Cutaneous tuberculosis. In: James WD, ed. Textbook of Military Medicine: Military Dermatology. Washington, DC: Borden Institute; 1994:355-389.
- Santos JB, Figueiredo AR, Ferraz CE, et al. Cutaneous tuberculosis: diagnosis, histopathology and treatment - part II. An Bras Dermatol. 2014;89:545-555.
- Guidelines on the Management of Latent Tuberculosis Infection. Geneva, Switzerland: World Health Organization; 2015.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 pt 2):S221-S247.
- Mycobacterium tuberculosis. Am J Transplant. 2004;4(suppl 10):37-41.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis. 2009;48:1276-1284.
- Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
- Kant S, Verma SK, Gupta V, et al. Pyrazinamide induced thrombocytopenia. Indian J Pharmacol. 2010;42:108-109.
- Screening for tuberculosis and tuberculosis infection in high-risk populations. recommendations of the Advisory Council for the Elimination of Tuberculosis. MMWR Recomm Rep. 1995;44:19-34.
- Fischer SA, Avery RK; AST Infectious Disease Community of Practice. Screening of donor and recipient prior to solid organ transplantation. Am J Transplant. 2009;9(suppl 4):S7-S18.
- Sakhuja V, Jha V, Varma PP, et al. The high incidence of tuberculosis among renal transplant recipients in India. Transplantation. 1996;61:211-215.
- Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
- Schultz V, Marroni CA, Amorim CS, et al. Risk factors for hepatotoxicity in solid organ transplants recipients being treated for tuberculosis. Transplant Proc. 2014;46:3606-3610.
- Tabarsi P, Farshidpour M, Marjani M, et al. Mycobacterial infection and the impact of rifabutin treatment in organ transplant recipients: a single-center study. Saudi J Kidney Dis Transpl. 2015;26:6-11.
- Rathi M, Gundlapalli S, Ramachandran R, et al. A rare case of cytomegalovirus, scedosporium apiospermum and mycobacterium tuberculosis in a renal transplant recipient. BMC Infect Dis. 2014;14:259.
- Hickey MD, Quan DJ, Chin-Hong PV, et al. Use of rifabutin for the treatment of a latent tuberculosis infection in a patient after solid organ transplantation. Liver Transpl. 2013;19:457-461.
- Kumar B, Muralidhar S. Cutaneous tuberculosis: a twenty-year prospective study. Int J Tuberc Lung Dis. 1999;3:494-500.
- Dekeyzer S, Moerman F, Callens S, et al. Cutaneous metastatic tuberculous abscess in patient with cervico-mediastinal lymphatic tuberculosis. Acta Clin Belg. 2013;68:34-36.
- Ko M, Wu C, Chiu H. Tuberculous gumma (cutaneous metastatic tuberculous abscess). Dermatol Sinica. 2005;23:27-31.
- Steger JW, Barrett TL. Cutaneous tuberculosis. In: James WD, ed. Textbook of Military Medicine: Military Dermatology. Washington, DC: Borden Institute; 1994:355-389.
- Santos JB, Figueiredo AR, Ferraz CE, et al. Cutaneous tuberculosis: diagnosis, histopathology and treatment - part II. An Bras Dermatol. 2014;89:545-555.
- Guidelines on the Management of Latent Tuberculosis Infection. Geneva, Switzerland: World Health Organization; 2015.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 pt 2):S221-S247.
- Mycobacterium tuberculosis. Am J Transplant. 2004;4(suppl 10):37-41.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis. 2009;48:1276-1284.
- Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
- Kant S, Verma SK, Gupta V, et al. Pyrazinamide induced thrombocytopenia. Indian J Pharmacol. 2010;42:108-109.
- Screening for tuberculosis and tuberculosis infection in high-risk populations. recommendations of the Advisory Council for the Elimination of Tuberculosis. MMWR Recomm Rep. 1995;44:19-34.
- Fischer SA, Avery RK; AST Infectious Disease Community of Practice. Screening of donor and recipient prior to solid organ transplantation. Am J Transplant. 2009;9(suppl 4):S7-S18.
Practice Points
- Transplant patients are at increased risk for infection given their immunosuppressed state.
- Although rare, cutaneous tuberculosis should be considered in the differential for cutaneous lesions in an immunosuppressed patient.
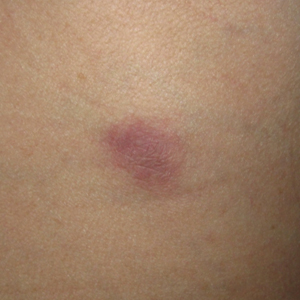
Annular Atrophic Plaques on the Forearm
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
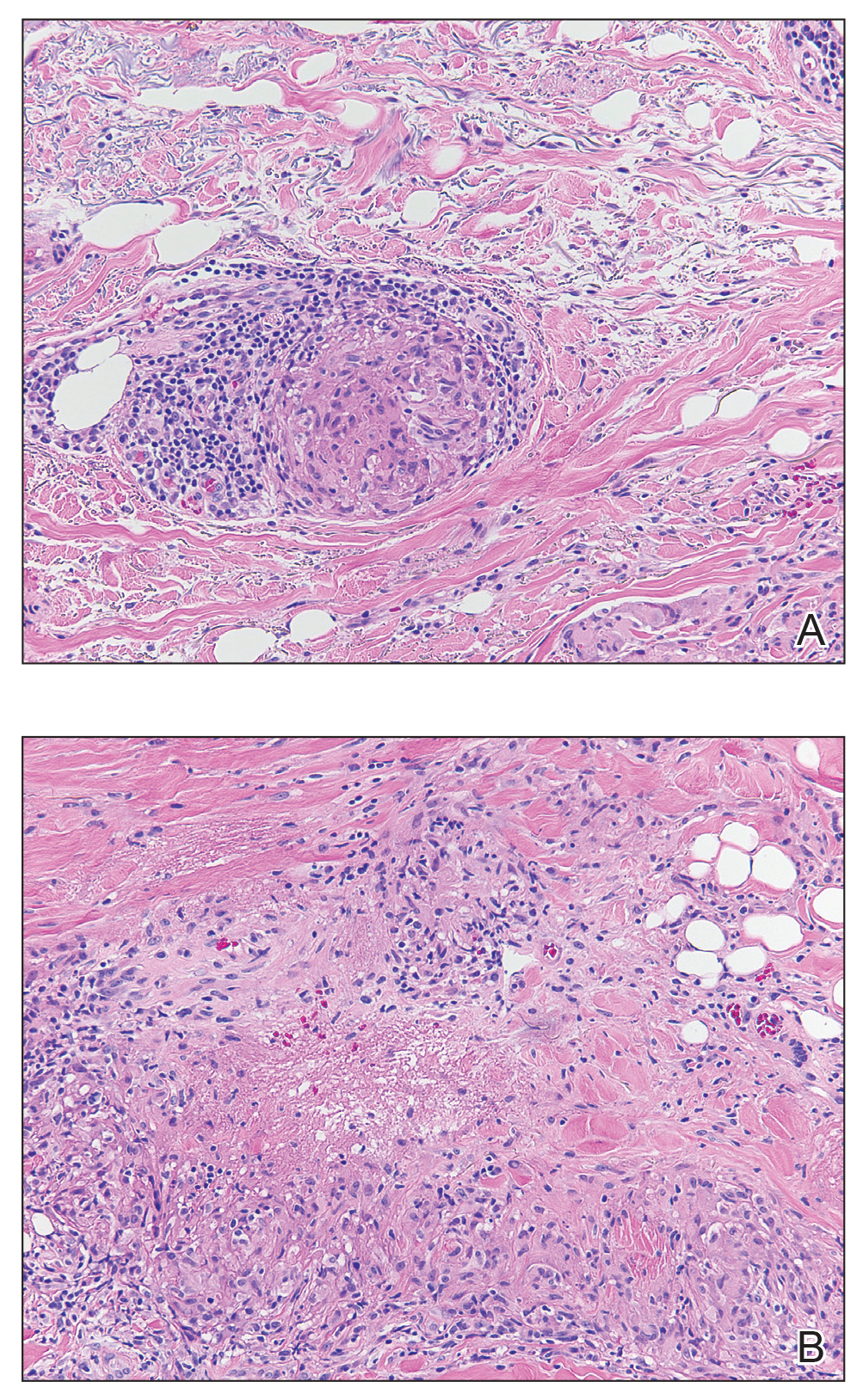
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
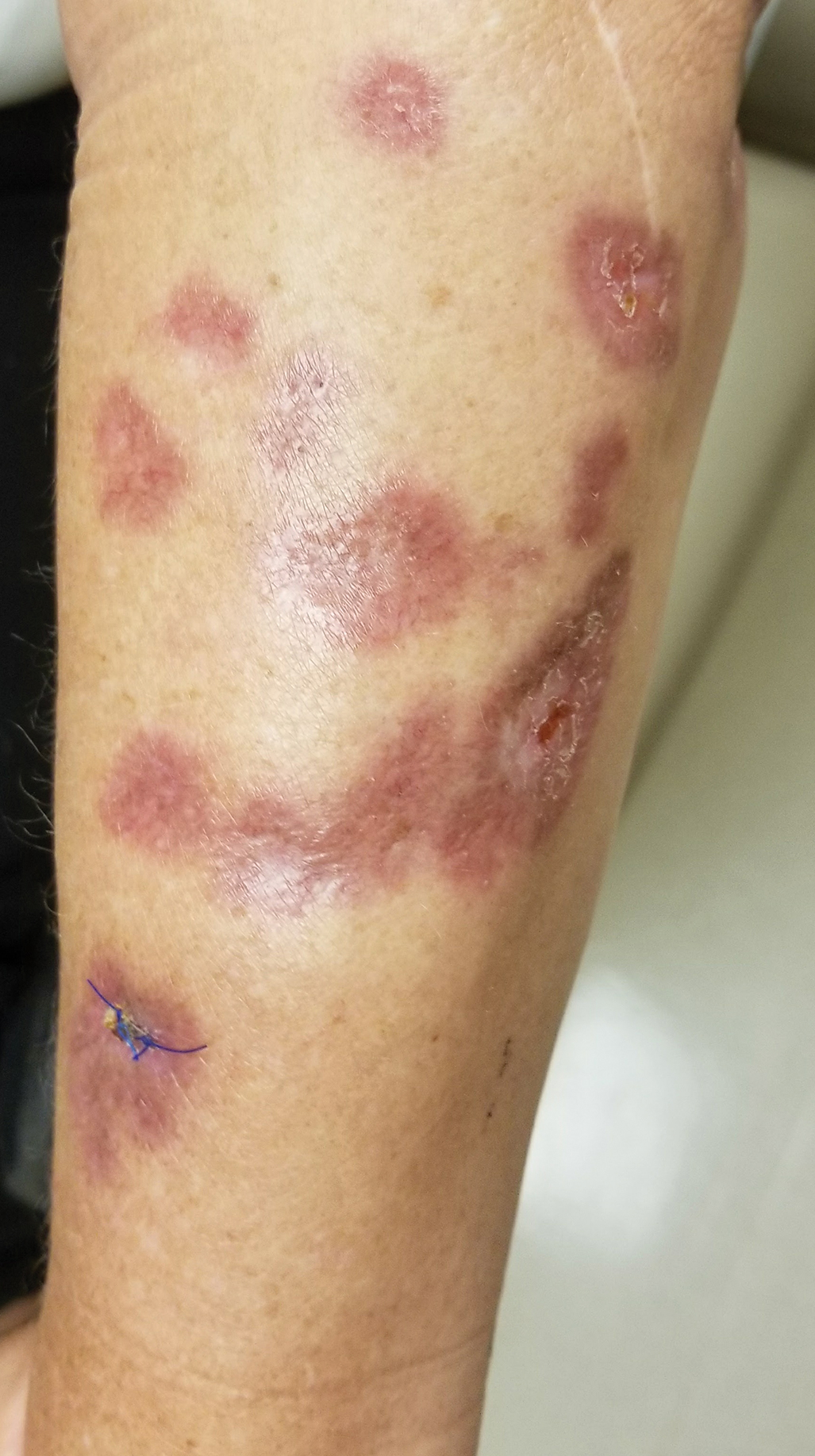
A 57-year-old woman presented with several lesions on the left extensor forearm of 10 years’ duration. A single annular indurated lesion with central atrophy initially developed near a prior surgical site. The lesions were pruritic with no associated pain or bleeding. Over 5 years, similar lesions had developed extending up the arm. No benefit was seen with low-potency topical steroid application. Biopsy for histopathologic examination was performed to confirm the diagnosis.
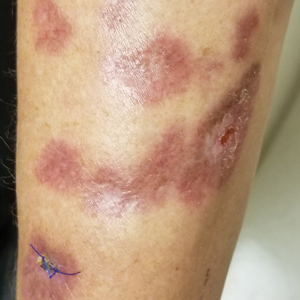
March 2019 Highlights


Erythematous Edematous Plaques on the Dorsal Aspects of the Hands
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
A 48-year-old woman presented with erythematous swelling of the dorsal aspects of the bilateral hands followed by desquamation and pruritus of 2 weeks’ duration. She denied any recent contact with plants, chemicals, or topical products or use of over-the-counter medications. A 6-day course of prednisone provided by her primary care physician relieved the swelling and pruritus; however, the erythema persisted. Physical examination revealed clearly demarcated, erythematous to violaceous, edematous plaques with peripheral scaling that involved all digits. There was notable sparing of the proximal interphalangeal joints and volar aspects of the hands extending proximally to the metacarpophalangeal joints.