User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Argyria From a Topical Home Remedy
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
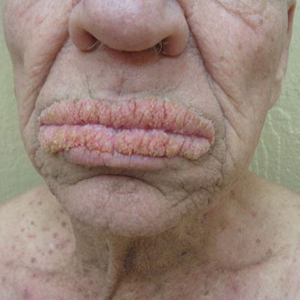
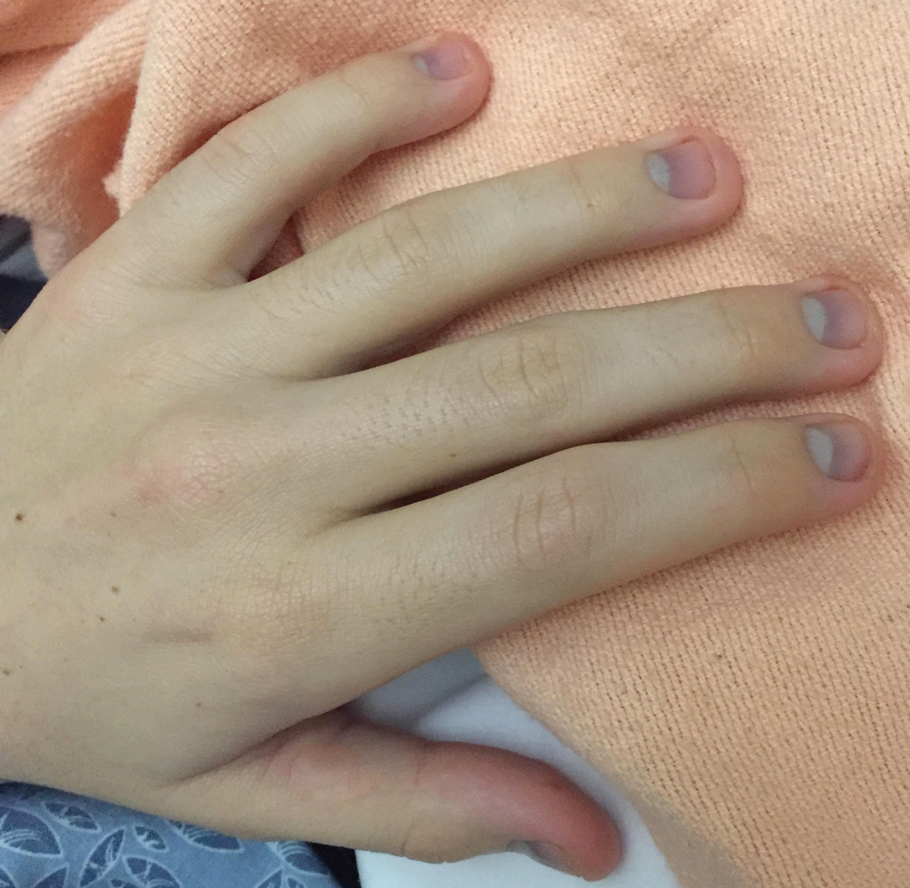
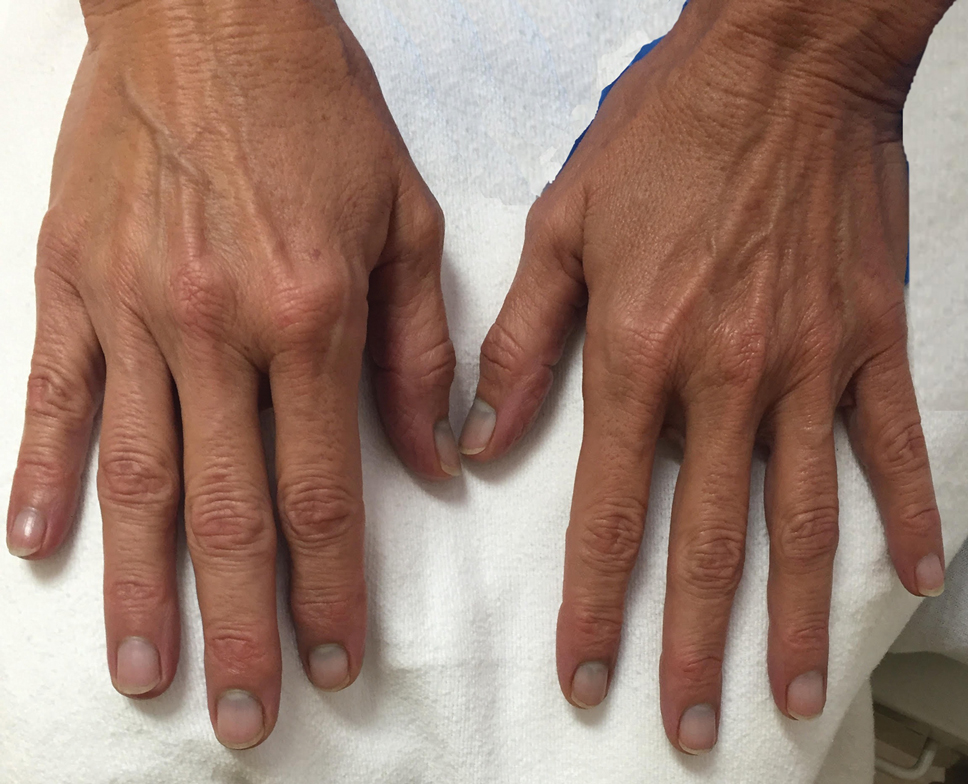
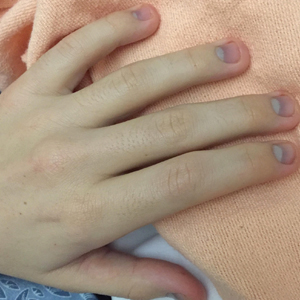
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
Practice Points
- Argyria results from chronic exposure to products with a high silver content and may result in abnormalities of the skin and internal organs.
- Examination of the fingernails can provide important clues to underlying systemic conditions or external exposures.
Squamoid Eccrine Ductal Carcinoma
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
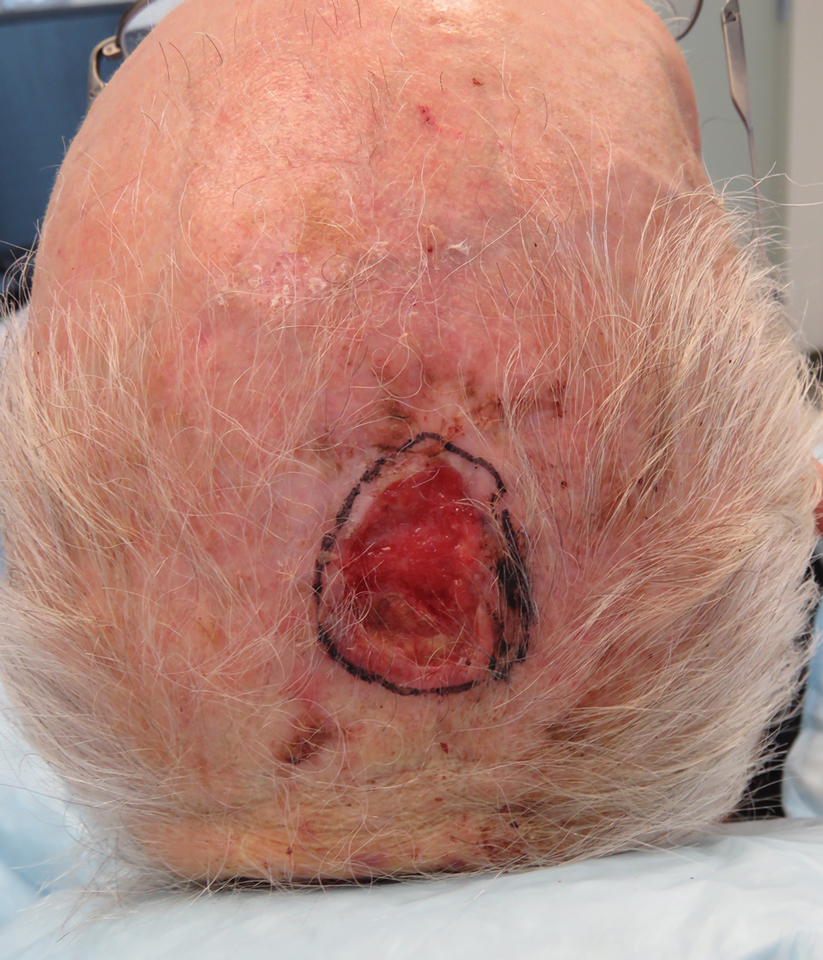
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
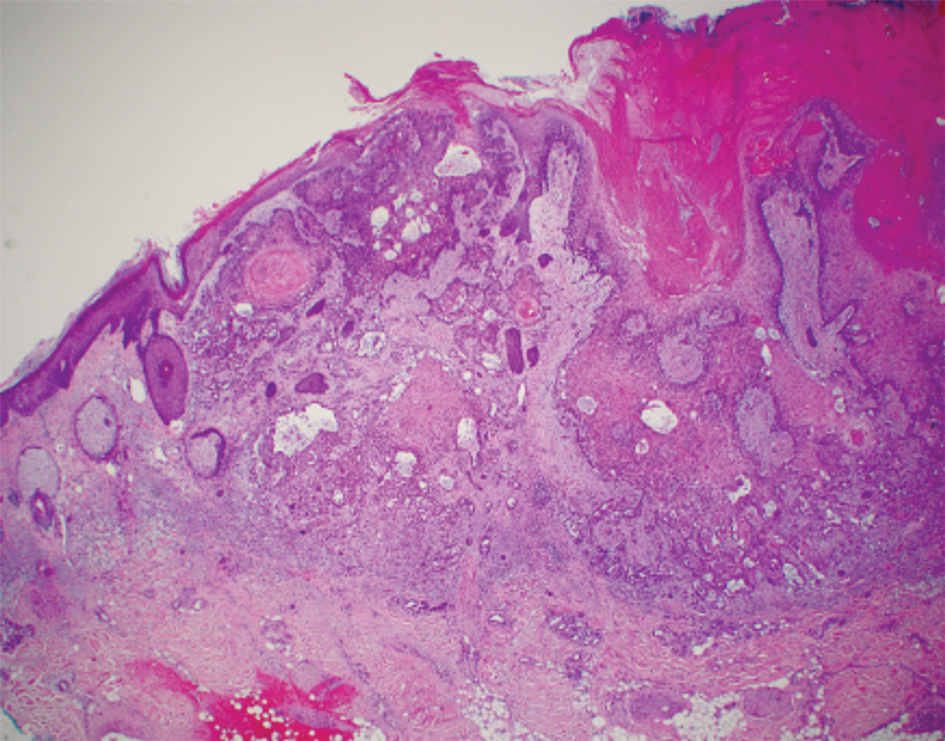
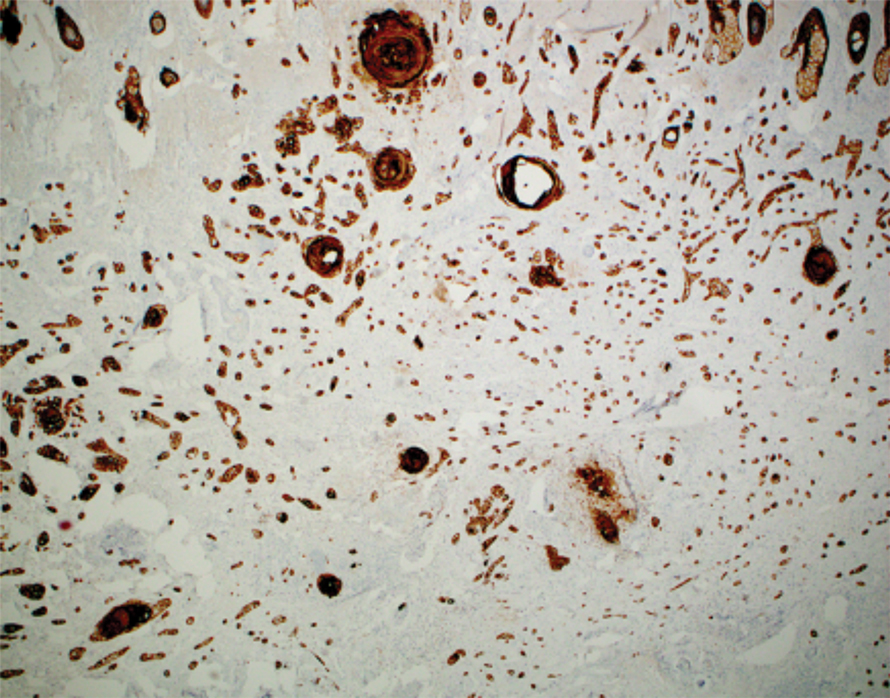
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.


The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
PRACTICE POINTS
- Squamoid eccrine ductal carcinoma is an aggressive underrecognized cutaneous malignancy that often is misdiagnosed as squamous cell carcinoma (SCC) during initial biopsy.
- Squamoid eccrine ductal carcinoma has a biphasic histologic appearance with a superficial portion resembling well-differentiated SCC and a deeply invasive portion comprised of infiltrative irregular cords with ductal differentiation.
- Excision with complete circumferential peripheral and deep margin assessment with close follow-up is recommended for these patients because of the high risk for recurrence and metastasis.
Nivolumab-Induced Granuloma Annulare
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
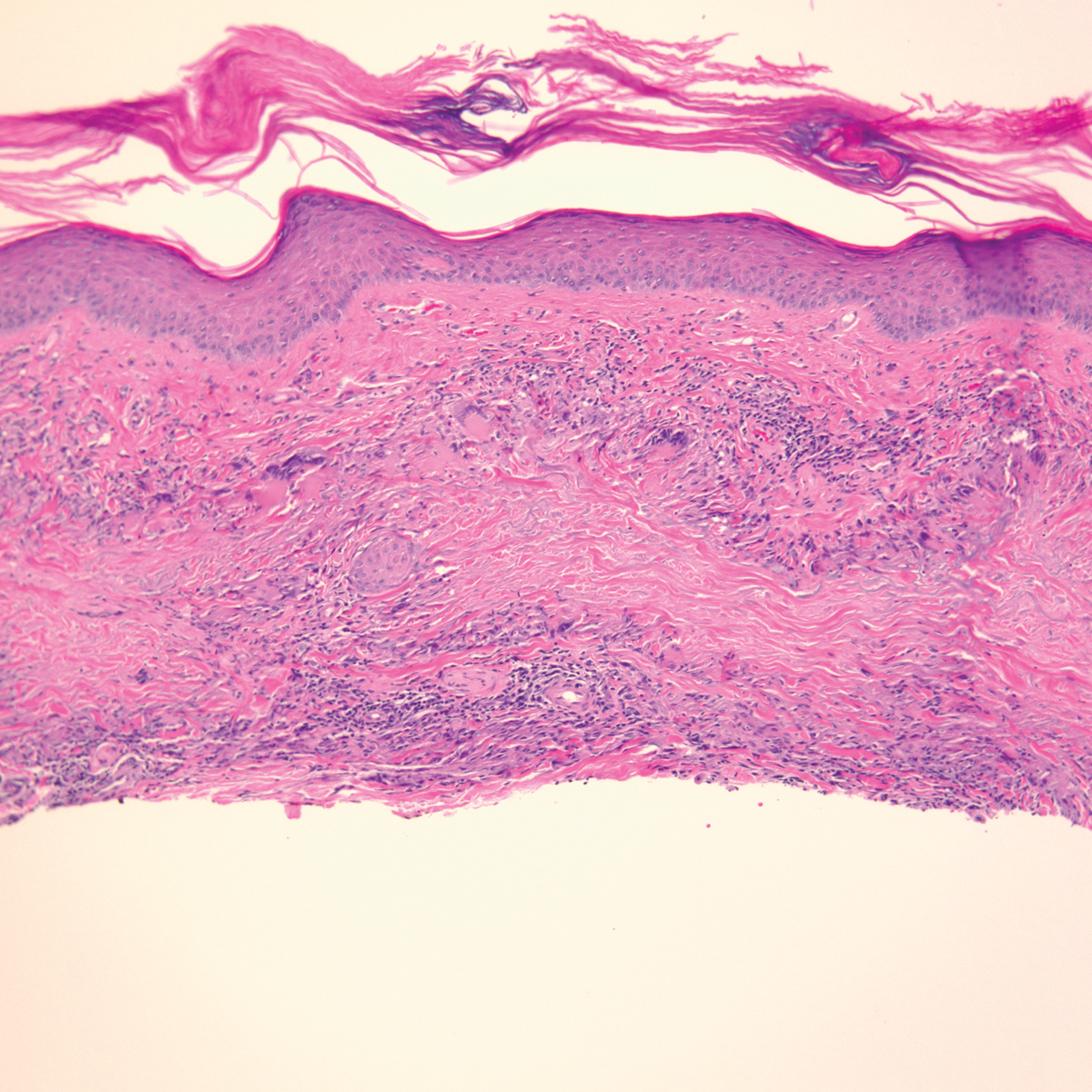
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
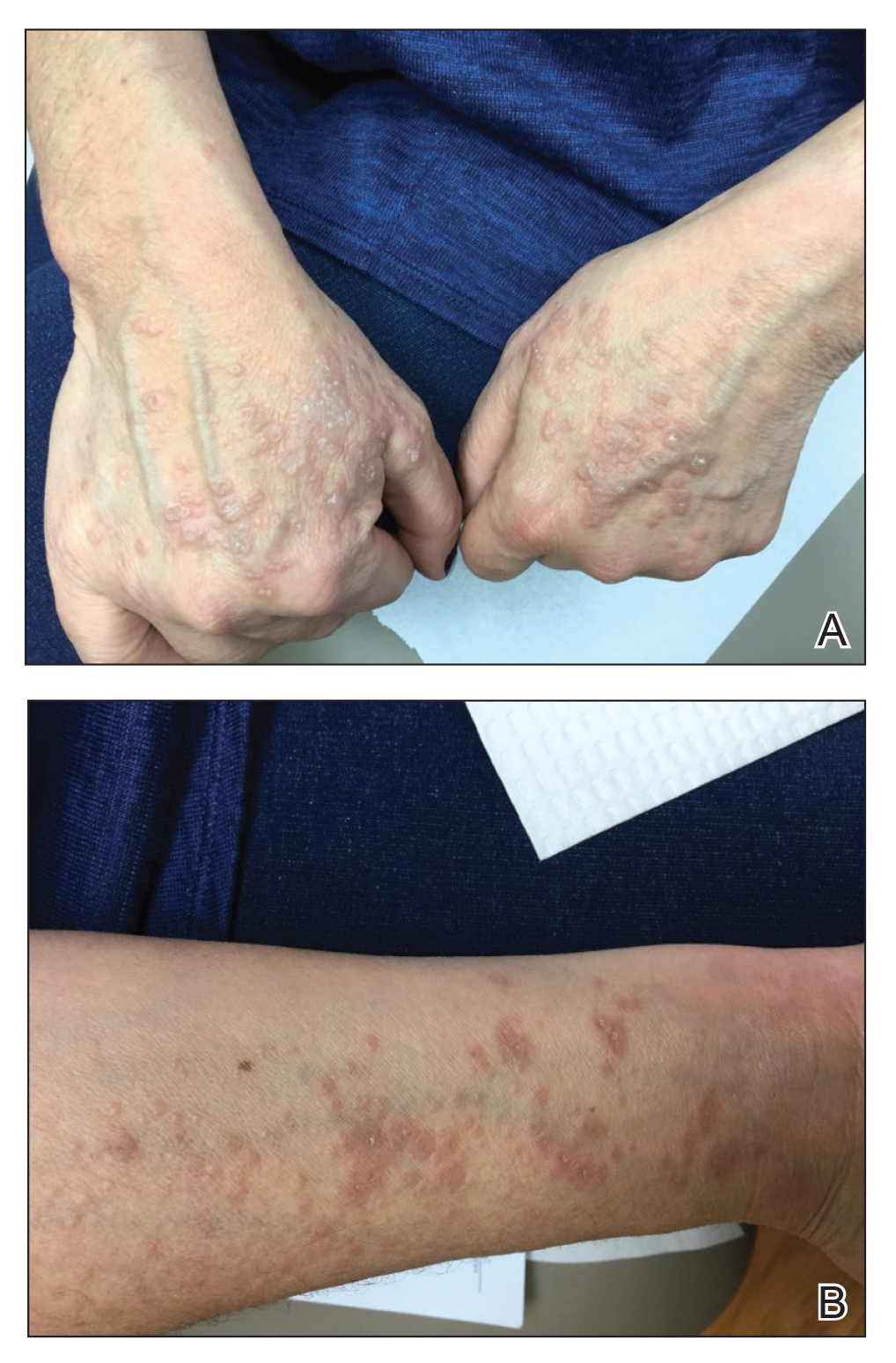
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Practice Points
- Immune-related adverse events (irAEs) frequently occur in patients on immunotherapy, with the skin representing the most common site of involvement.
- Although rare, granulomatous reactions such as granuloma annulare increasingly are recognized as potential irAEs.
- Clinicians should be aware of this novel association to accurately diagnose and effectively treat adverse reactions in patients receiving immunotherapy.
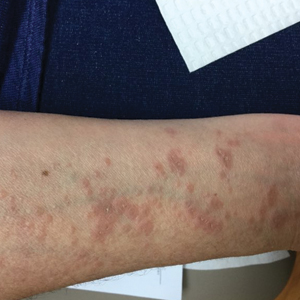
Atopic Dermatitis
The Comparison
A Pink scaling plaques and erythematous erosions in the antecubital fossae of a 6-year-old White boy.
B Violaceous, hyperpigmented, nummular plaques on the back and extensor surface of the right arm of a 16-month-old Black girl.
C Atopic dermatitis and follicular prominence/accentuation on the neck of a young Black girl.
Epidemiology
People of African descent have the highest atopic dermatitis prevalence and severity.
Key clinical features in people with darker skin tones include:
- follicular prominence
- papular morphology
- prurigo nodules
- hyperpigmented, violaceous-brown or gray plaques instead of erythematous plaques
- lichenification
- treatment resistant.1,2
Worth noting
Postinflammatory hyperpigmentation and postinflammatory hypopigmentation may be more distressing to the patient/family than the atopic dermatitis itself.
Health disparity highlight
In the United States, patients with skin of color are more likely to be hospitalized with severe atopic dermatitis, have more substantial out-ofpocket costs, be underinsured, and have an increased number of missed days of work. Limited access to outpatient health care plays a role in exacerbating this health disparity.3,4
- McKenzie C, Silverberg JI. The prevalence and persistence of atopic dermatitis in urban United States children. Ann Allergy Asthma Immunol. 2019;123:173-178.e1. doi:10.1016 /j.anai.2019.05.014
- Kim Y, Bloomberg M, Rifas-Shiman SL, et al. Racial/ethnic differences in incidence and persistence of childhood atopic dermatitis. J Invest Dermatol. 2019;139:827-834. doi:10.1016 /j.jid.2018.10.029
- Narla S, Hsu DY, Thyssen JP, et al. Predictors of hospitalization, length of stay, and costs of care among adult and pediatric inpatients with atopic dermatitis in the United States. Dermatitis. 2018;29:22-31. doi:10.1097/DER.0000000000000323
- Silverberg JI. Health care utilization, patient costs, and access to care in US adults with eczema. JAMA Dermatol. 2015;151:743-752. doi:10.1001/jamadermatol.2014.5432
The Comparison
A Pink scaling plaques and erythematous erosions in the antecubital fossae of a 6-year-old White boy.
B Violaceous, hyperpigmented, nummular plaques on the back and extensor surface of the right arm of a 16-month-old Black girl.
C Atopic dermatitis and follicular prominence/accentuation on the neck of a young Black girl.
Epidemiology
People of African descent have the highest atopic dermatitis prevalence and severity.
Key clinical features in people with darker skin tones include:
- follicular prominence
- papular morphology
- prurigo nodules
- hyperpigmented, violaceous-brown or gray plaques instead of erythematous plaques
- lichenification
- treatment resistant.1,2
Worth noting
Postinflammatory hyperpigmentation and postinflammatory hypopigmentation may be more distressing to the patient/family than the atopic dermatitis itself.
Health disparity highlight
In the United States, patients with skin of color are more likely to be hospitalized with severe atopic dermatitis, have more substantial out-ofpocket costs, be underinsured, and have an increased number of missed days of work. Limited access to outpatient health care plays a role in exacerbating this health disparity.3,4
The Comparison
A Pink scaling plaques and erythematous erosions in the antecubital fossae of a 6-year-old White boy.
B Violaceous, hyperpigmented, nummular plaques on the back and extensor surface of the right arm of a 16-month-old Black girl.
C Atopic dermatitis and follicular prominence/accentuation on the neck of a young Black girl.
Epidemiology
People of African descent have the highest atopic dermatitis prevalence and severity.
Key clinical features in people with darker skin tones include:
- follicular prominence
- papular morphology
- prurigo nodules
- hyperpigmented, violaceous-brown or gray plaques instead of erythematous plaques
- lichenification
- treatment resistant.1,2
Worth noting
Postinflammatory hyperpigmentation and postinflammatory hypopigmentation may be more distressing to the patient/family than the atopic dermatitis itself.
Health disparity highlight
In the United States, patients with skin of color are more likely to be hospitalized with severe atopic dermatitis, have more substantial out-ofpocket costs, be underinsured, and have an increased number of missed days of work. Limited access to outpatient health care plays a role in exacerbating this health disparity.3,4
- McKenzie C, Silverberg JI. The prevalence and persistence of atopic dermatitis in urban United States children. Ann Allergy Asthma Immunol. 2019;123:173-178.e1. doi:10.1016 /j.anai.2019.05.014
- Kim Y, Bloomberg M, Rifas-Shiman SL, et al. Racial/ethnic differences in incidence and persistence of childhood atopic dermatitis. J Invest Dermatol. 2019;139:827-834. doi:10.1016 /j.jid.2018.10.029
- Narla S, Hsu DY, Thyssen JP, et al. Predictors of hospitalization, length of stay, and costs of care among adult and pediatric inpatients with atopic dermatitis in the United States. Dermatitis. 2018;29:22-31. doi:10.1097/DER.0000000000000323
- Silverberg JI. Health care utilization, patient costs, and access to care in US adults with eczema. JAMA Dermatol. 2015;151:743-752. doi:10.1001/jamadermatol.2014.5432
- McKenzie C, Silverberg JI. The prevalence and persistence of atopic dermatitis in urban United States children. Ann Allergy Asthma Immunol. 2019;123:173-178.e1. doi:10.1016 /j.anai.2019.05.014
- Kim Y, Bloomberg M, Rifas-Shiman SL, et al. Racial/ethnic differences in incidence and persistence of childhood atopic dermatitis. J Invest Dermatol. 2019;139:827-834. doi:10.1016 /j.jid.2018.10.029
- Narla S, Hsu DY, Thyssen JP, et al. Predictors of hospitalization, length of stay, and costs of care among adult and pediatric inpatients with atopic dermatitis in the United States. Dermatitis. 2018;29:22-31. doi:10.1097/DER.0000000000000323
- Silverberg JI. Health care utilization, patient costs, and access to care in US adults with eczema. JAMA Dermatol. 2015;151:743-752. doi:10.1001/jamadermatol.2014.5432
Atrophic Lesions in a Pregnant Woman
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
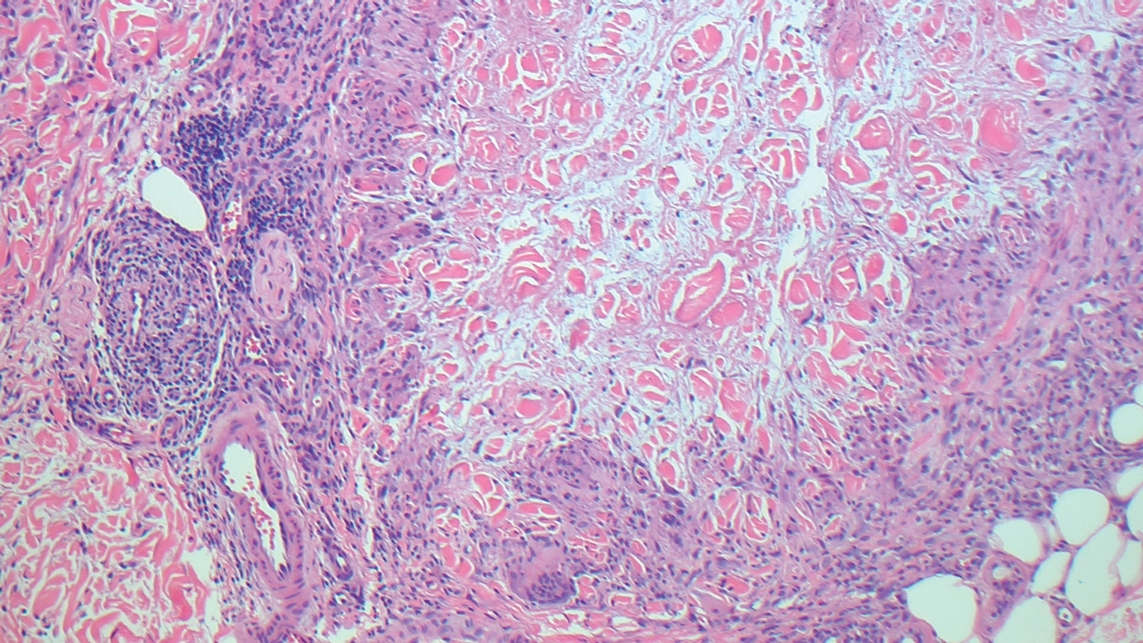
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
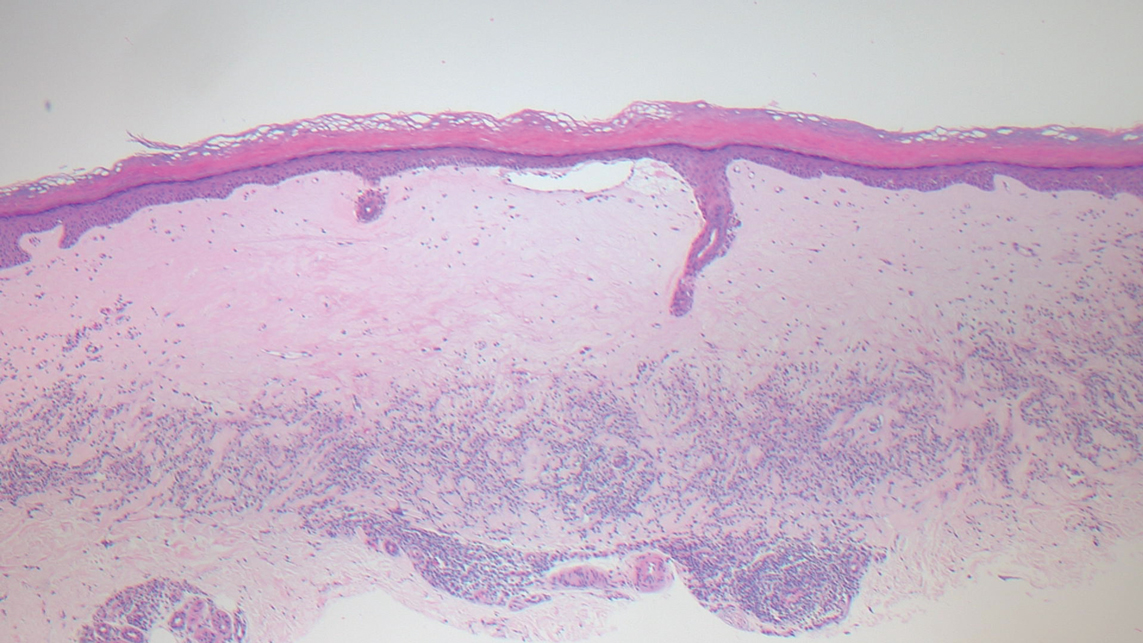
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
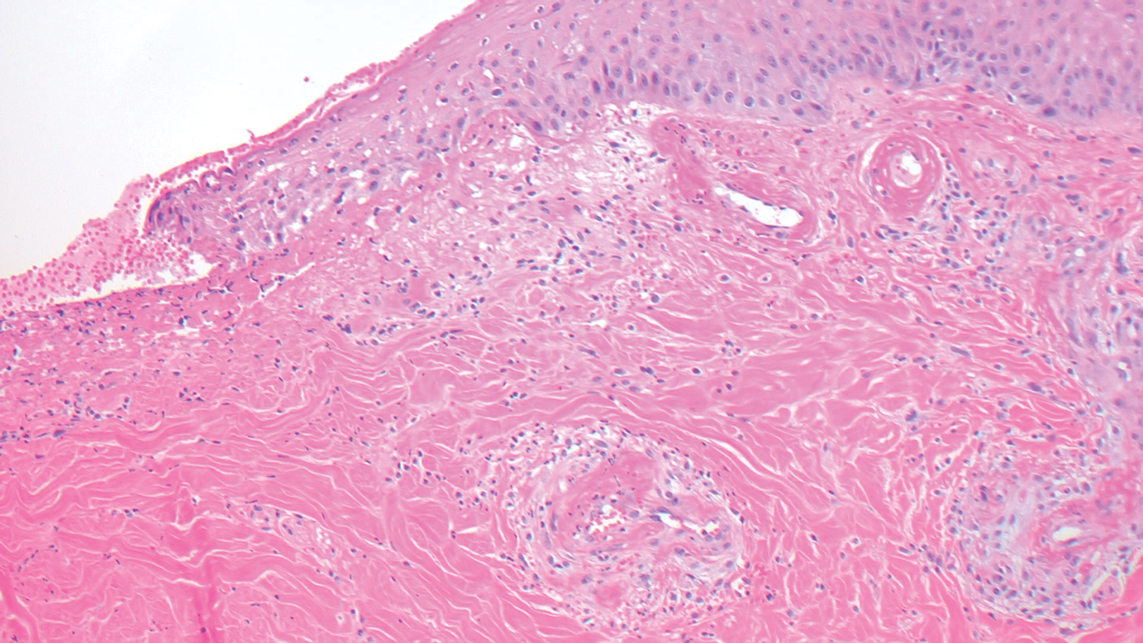
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
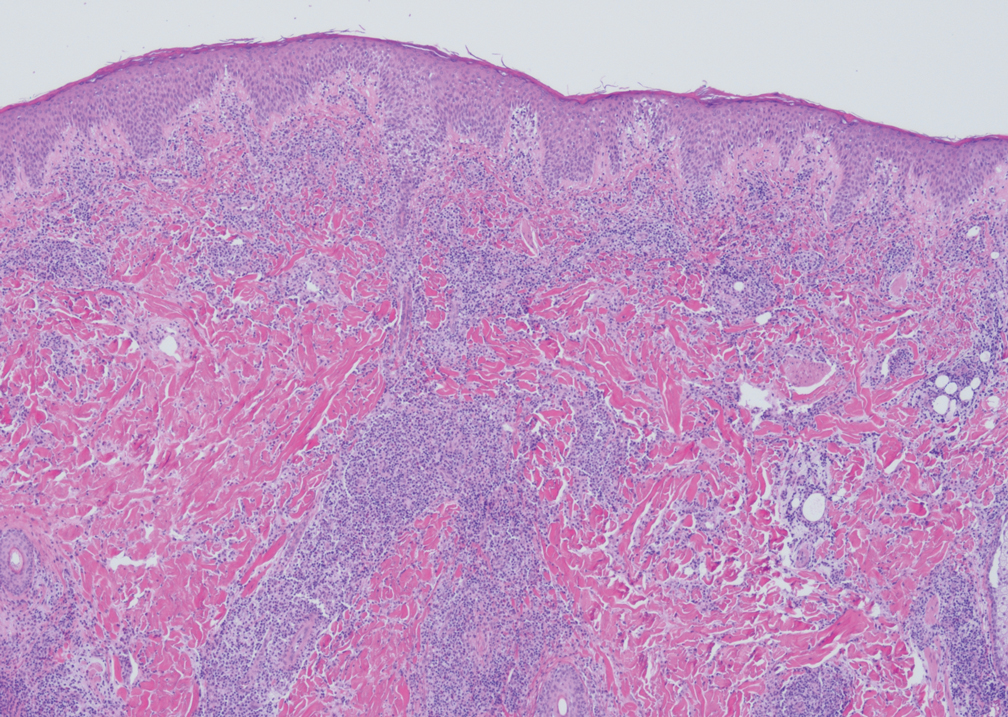
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
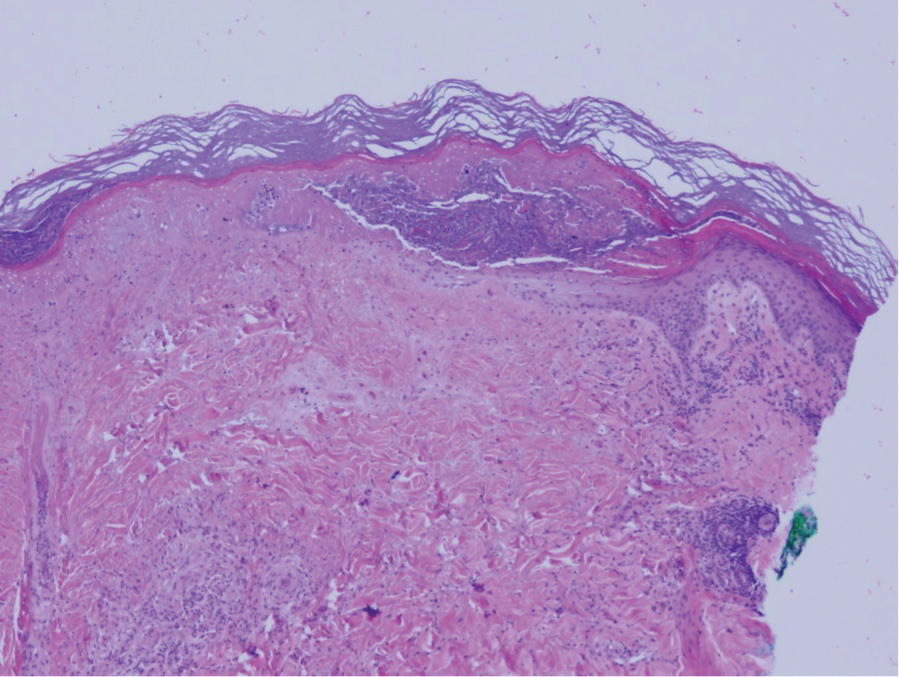
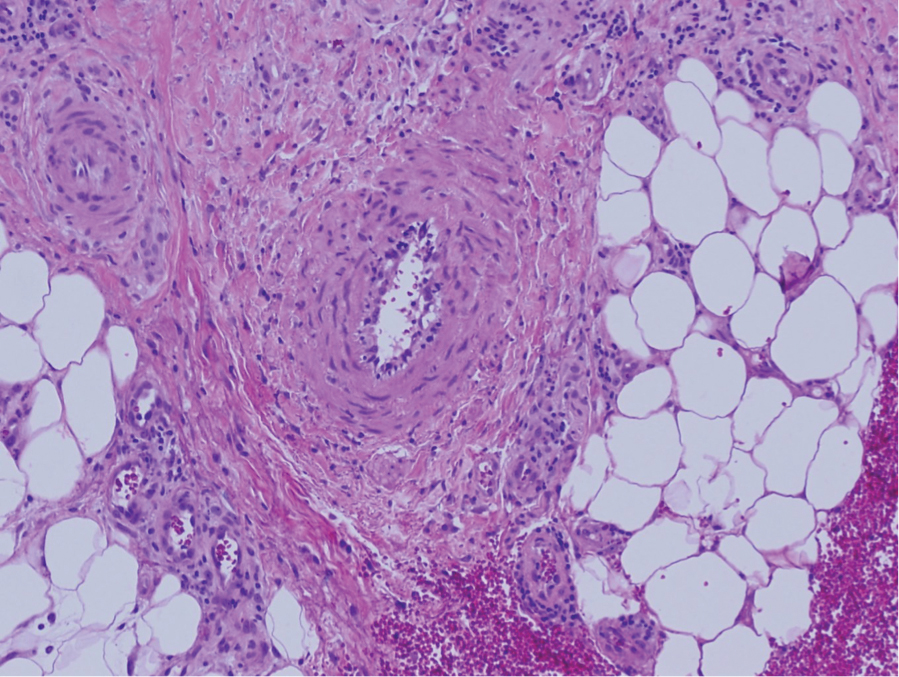
A 36-year-old pregnant woman presented with painful erythematous papules on the palms and fingers of 2 months’ duration. Similar lesions developed on the thighs and feet several weeks later. Two tender macules with central areas of porcelain white scarring rimmed by telangiectases on the right foot also were present. A punch biopsy of these lesions demonstrated a wedge-shaped area of ischemic necrosis associated with dermal mucin without associated necrobiosis. Fibrin thrombi were seen within several small dermal vessels and were associated with a perivascular lymphocytic infiltrate. Endotheliitis was observed within a deep dermal vessel. Laboratory workup including syphilis IgG, antinuclear antibodies, extractable nuclear antigen antibodies, anti–double-stranded DNA, antistreptolysin O antibodies, Russell viper venom time, cryoglobulin, hepatitis screening, perinuclear antineutrophil cytoplasmic antibodies (ANCA), and cytoplasmic ANCA was unremarkable. Hypercoagulable studies including prothrombin gene mutation, factor V Leiden, plasminogen, proteins C and S, antithrombin III, homocysteine, and antiphospholipid IgM and IgG antibodies were notable only for heterozygosity for factor V Leiden.
Reporting Biopsy Margin Status for Cutaneous Basal Cell Carcinoma: To Do or Not to Do
To the Editor:
In an interesting analysis, Brady and Hossler1 (Cutis. 2020;106:315-317) highlighted the limitations of histopathologic biopsy margin evaluation for cutaneous basal cell carcinoma (BCC). Taking into consideration the high prevalence of BCC and its medical and economic impact on the health care system, the issue raised by the authors is an important one. They proposed that pathologists may omit reporting margins or clarify the limitations in their reports. It is a valid suggestion; however, in practice, margin evaluation is not always a simple process and is influenced by a number of factors.
The subject of optimum margins for BCC has been debated over decades now; however, ambiguity and lack of definitive guidelines on certain aspects still remain, leading to a lack of standardization and variability in reporting, which opens potential for error. In anatomical pathology, the biopsies for malignancies are interpreted to confirm diagnosis and perform risk assessment, with evaluation of margins generally reserved for subsequent definitive resections. Typically, margins are not required by clinicians or reported by pathologists in common endoscopic (eg, stomach, colon) or needle core (eg, prostate, breast) biopsies. Skin holds a rather unique position in which margin evaluation is not just limited to excisions. With the exception of samples generated from electrodesiccation and curettage, it is common practice by some laboratories to report margins on most specimens of cutaneous malignancies.
In simple terms, when margins are labeled negative there should be no residual disease, and when they are deemed positive there should be disease still persisting in the patient. Margin evaluation for BCC on biopsies falls short on both fronts. In one analysis, 24% (34/143) of shave biopsies reported with negative margins displayed residual BCC in ensuing re-excisions (negative predictive value: 76%).2 Standard bread-loafing, en-face margins and inking for orientation utilized to provide a thorough margin evaluation of excisions cannot be optimally achieved on small skin biopsies. Microscopic sections for analysis are 2-dimensional representations of 3-dimensional structures. Slides prepared can miss deeply embedded outermost margins, positioned parallel to the plane of sectioning, thereby creating blind spots where margins cannot be precisely assessed and generating an inherent limitation in evaluation. Exhaustive deeper levels done routinely can address this issue to a certain degree; however, it can be an impractical solution with cost implications and delay in turnaround time.
Conversely, it also is common to encounter absence of residual BCC in re-excisions in which the original biopsy margins were labeled positive. In one analysis, 49% of BCC patients (n=100) with positive biopsy margins did not display residual neoplasm on following re-excisions.3 Localized biopsy site immune response as a cause of postbiopsy regression of residual tumor has been hypothesized to produce this phenomenon. Moreover, initial biopsies may eliminate the majority of the tumor with only minimal disease persisting. Re-excisions submitted in toto allow for a systematic examination; however, areas in between sections still remain where minute residual tumor may hide. Searching for such occult foci generally is not aggressively pursued via deeper levels unless the margins of re-excision are in question.
Superficial-type BCC (or superficial multifocal BCC) is a major factor in precluding precise biopsy margin evaluation. In a study where initial biopsies reported with negative margins displayed residual BCC in subsequent re-excisions, 91% (31/34) of residual BCCs were of superficial variety.2 Clinically, superficial BCC frequently has indistinct borders with subtle subclinical peripheral progression. It has a tendency to expand radially, with the clinical appearance deceptively smaller than its true extent. In a plane of histopathologic section, superficial BCC may exhibit skip zones within the epidermis. Even though the margin may seem uninvolved on the slide, a noncontiguous focus may still emerge beyond the “negative” margin. Because superficial pattern is not unusual as one of the components of mixed histology (composite) BCC, this issue is not just limited to tumors specifically designated as superficial type.4
The intent of a procedure is important to recognize. If a biopsy is done with the intention of diagnosis only, the pathologic assessment can be limited to tumor identification and core data elements, with margin evaluation reserved for excisions done with therapeutic intent. However, the intent is not always clear, which adds to ambiguity on when to report margins. It is not uncommon to find saucerization shaves or large punch biopsies for BCC carried out with a therapeutic intent. The status of margin is desired in such samples; however, the intent is not always clearly communicated on requisitions. To avoid any gaps in communication, some pathologists may err on the side of caution and start routinely reporting margins on biopsies.
Taking into account the inaccuracy of margin assessment in biopsies, an argument for omitting margin reporting is plausible. Although dermatologists are the major contributors of skin samples, pathology laboratories cater to a broader clientele. Other physicians from different surgical and medical specialities also perform skin biopsies, and catering to a variety of specialities adds another layer of complexity. A dermatologist may appreciate the debate regarding reliability of margins; however, a physician from another speciality who is not as familiar with the diseases of the integument may lack proper understanding. Omitting margin reporting may lead to misinterpretations or false assumptions, such as, “The margins must be uninvolved, otherwise the pathologist would have said something.” This also can generate additional phone or email inquiries and second review requests. Rather than completely omitting them, another strategy can be to report margins in more quantitative terms. One reporting approach is to have 3 categories of involved, uninvolved, and uninvolved but close for margins less than 1 mm. The cases in the third category may require greater scrutiny by deeper levels or an added caveat in the comment addressing the limitation. If the status of margins is not reported due to a certain reason, a short comment can be added to explain the reason.
In sum, clinicians should recognize that “margin negative” on skin biopsy does not always equate to “completely excised.” Margin status on biopsies is a data item that essentially provides a probability of margin clearance. Completely omitting the margin status on all biopsies may not be the most prudent approach; however, improved guidelines and modifications to enhance the reporting are definitely required.
References
- Brady MC, Hossler EW. Reliability of biopsy margin status for basal cell carcinoma: a retrospective study. Cutis. 2020;106:315-317.
- Willardson HB, Lombardo J, Raines M, et al. Predictive value of basal cell carcinoma biopsies with negative margins: a retrospective cohort study. J Am Acad Dermatol. 2018;79:42-46.
- Yuan Y, Duff ML, Sammons DL, et al. Retrospective chart review of skin cancer presence in the wide excisions. World J Clin Cases. 2014;2:52-56.
- Cohen PR, Schulze KE, Nelson BR. Basal cell carcinoma with mixed histology: a possible pathogenesis for recurrent skin cancer. Dermatol Surg. 2006;32:542-551.
Continue to: Author's Response...
Authors’ Response
We appreciate the thorough and thoughtful comments in the Letter to the Editor. We agree with the author’s assertion that negative margins on skin specimens does not equate to “completely excised” and that the intent of the clinician is not always clear, even when the pathologist has ready access to the clinician’s notes, as was the case for the majority of specimens included in our study.
There is already variability in how pathologists report margins, including the specific verbiage used, at least for melanocytic lesions.1 The choice of whether or not to report margins and the meaning of those margins is complex due to the uncertainty inherent in margin assessment. Quantifying this uncertainty was the main reason for our study. Ultimately, the pathologist’s decision on whether and how to report margins should be focused on improving patient outcomes. There are benefits and drawbacks to all approaches, and our goal is to provide more information for clinicians and pathologists so that they may better care for their patients. Understanding the limitations of margins on submitted skin specimens—whether margins are reported or not—can only serve to guide improve clinical decision-making.
We also agree that the breadth of specialties of submitting clinicians make reporting of margins difficult, and there is likely similar breadth in their understanding of the nuances of margin assessment and reports. The solution to this problem is adequate education regarding the limitations of a pathology report, and specifically what is meant when margins are (or are not) reported on skin specimens. How to best educate the myriad clinicians who submit biopsies is, of course, the ultimate challenge.
We hope that our study adds information to this ongoing debate regarding margin status reporting, and we appreciate the discussion points raised by the author.
Eric Hossler, MD; Mary Brady, MD
From the Department of Dermatology, Geisinger Health System, Danville, Pennsylvania.
The authors report no conflict of interest.
Reference
- Sellheyer K, Bergfeld WF, Stewart E, et al. Evaluation of surgical margins in melanocytic lesions: a survey among 152 dermatopathologists.J Cutan Pathol. 2005;32:293-299.
To the Editor:
In an interesting analysis, Brady and Hossler1 (Cutis. 2020;106:315-317) highlighted the limitations of histopathologic biopsy margin evaluation for cutaneous basal cell carcinoma (BCC). Taking into consideration the high prevalence of BCC and its medical and economic impact on the health care system, the issue raised by the authors is an important one. They proposed that pathologists may omit reporting margins or clarify the limitations in their reports. It is a valid suggestion; however, in practice, margin evaluation is not always a simple process and is influenced by a number of factors.
The subject of optimum margins for BCC has been debated over decades now; however, ambiguity and lack of definitive guidelines on certain aspects still remain, leading to a lack of standardization and variability in reporting, which opens potential for error. In anatomical pathology, the biopsies for malignancies are interpreted to confirm diagnosis and perform risk assessment, with evaluation of margins generally reserved for subsequent definitive resections. Typically, margins are not required by clinicians or reported by pathologists in common endoscopic (eg, stomach, colon) or needle core (eg, prostate, breast) biopsies. Skin holds a rather unique position in which margin evaluation is not just limited to excisions. With the exception of samples generated from electrodesiccation and curettage, it is common practice by some laboratories to report margins on most specimens of cutaneous malignancies.
In simple terms, when margins are labeled negative there should be no residual disease, and when they are deemed positive there should be disease still persisting in the patient. Margin evaluation for BCC on biopsies falls short on both fronts. In one analysis, 24% (34/143) of shave biopsies reported with negative margins displayed residual BCC in ensuing re-excisions (negative predictive value: 76%).2 Standard bread-loafing, en-face margins and inking for orientation utilized to provide a thorough margin evaluation of excisions cannot be optimally achieved on small skin biopsies. Microscopic sections for analysis are 2-dimensional representations of 3-dimensional structures. Slides prepared can miss deeply embedded outermost margins, positioned parallel to the plane of sectioning, thereby creating blind spots where margins cannot be precisely assessed and generating an inherent limitation in evaluation. Exhaustive deeper levels done routinely can address this issue to a certain degree; however, it can be an impractical solution with cost implications and delay in turnaround time.
Conversely, it also is common to encounter absence of residual BCC in re-excisions in which the original biopsy margins were labeled positive. In one analysis, 49% of BCC patients (n=100) with positive biopsy margins did not display residual neoplasm on following re-excisions.3 Localized biopsy site immune response as a cause of postbiopsy regression of residual tumor has been hypothesized to produce this phenomenon. Moreover, initial biopsies may eliminate the majority of the tumor with only minimal disease persisting. Re-excisions submitted in toto allow for a systematic examination; however, areas in between sections still remain where minute residual tumor may hide. Searching for such occult foci generally is not aggressively pursued via deeper levels unless the margins of re-excision are in question.
Superficial-type BCC (or superficial multifocal BCC) is a major factor in precluding precise biopsy margin evaluation. In a study where initial biopsies reported with negative margins displayed residual BCC in subsequent re-excisions, 91% (31/34) of residual BCCs were of superficial variety.2 Clinically, superficial BCC frequently has indistinct borders with subtle subclinical peripheral progression. It has a tendency to expand radially, with the clinical appearance deceptively smaller than its true extent. In a plane of histopathologic section, superficial BCC may exhibit skip zones within the epidermis. Even though the margin may seem uninvolved on the slide, a noncontiguous focus may still emerge beyond the “negative” margin. Because superficial pattern is not unusual as one of the components of mixed histology (composite) BCC, this issue is not just limited to tumors specifically designated as superficial type.4
The intent of a procedure is important to recognize. If a biopsy is done with the intention of diagnosis only, the pathologic assessment can be limited to tumor identification and core data elements, with margin evaluation reserved for excisions done with therapeutic intent. However, the intent is not always clear, which adds to ambiguity on when to report margins. It is not uncommon to find saucerization shaves or large punch biopsies for BCC carried out with a therapeutic intent. The status of margin is desired in such samples; however, the intent is not always clearly communicated on requisitions. To avoid any gaps in communication, some pathologists may err on the side of caution and start routinely reporting margins on biopsies.
Taking into account the inaccuracy of margin assessment in biopsies, an argument for omitting margin reporting is plausible. Although dermatologists are the major contributors of skin samples, pathology laboratories cater to a broader clientele. Other physicians from different surgical and medical specialities also perform skin biopsies, and catering to a variety of specialities adds another layer of complexity. A dermatologist may appreciate the debate regarding reliability of margins; however, a physician from another speciality who is not as familiar with the diseases of the integument may lack proper understanding. Omitting margin reporting may lead to misinterpretations or false assumptions, such as, “The margins must be uninvolved, otherwise the pathologist would have said something.” This also can generate additional phone or email inquiries and second review requests. Rather than completely omitting them, another strategy can be to report margins in more quantitative terms. One reporting approach is to have 3 categories of involved, uninvolved, and uninvolved but close for margins less than 1 mm. The cases in the third category may require greater scrutiny by deeper levels or an added caveat in the comment addressing the limitation. If the status of margins is not reported due to a certain reason, a short comment can be added to explain the reason.
In sum, clinicians should recognize that “margin negative” on skin biopsy does not always equate to “completely excised.” Margin status on biopsies is a data item that essentially provides a probability of margin clearance. Completely omitting the margin status on all biopsies may not be the most prudent approach; however, improved guidelines and modifications to enhance the reporting are definitely required.
References
- Brady MC, Hossler EW. Reliability of biopsy margin status for basal cell carcinoma: a retrospective study. Cutis. 2020;106:315-317.
- Willardson HB, Lombardo J, Raines M, et al. Predictive value of basal cell carcinoma biopsies with negative margins: a retrospective cohort study. J Am Acad Dermatol. 2018;79:42-46.
- Yuan Y, Duff ML, Sammons DL, et al. Retrospective chart review of skin cancer presence in the wide excisions. World J Clin Cases. 2014;2:52-56.
- Cohen PR, Schulze KE, Nelson BR. Basal cell carcinoma with mixed histology: a possible pathogenesis for recurrent skin cancer. Dermatol Surg. 2006;32:542-551.
Continue to: Author's Response...
Authors’ Response
We appreciate the thorough and thoughtful comments in the Letter to the Editor. We agree with the author’s assertion that negative margins on skin specimens does not equate to “completely excised” and that the intent of the clinician is not always clear, even when the pathologist has ready access to the clinician’s notes, as was the case for the majority of specimens included in our study.
There is already variability in how pathologists report margins, including the specific verbiage used, at least for melanocytic lesions.1 The choice of whether or not to report margins and the meaning of those margins is complex due to the uncertainty inherent in margin assessment. Quantifying this uncertainty was the main reason for our study. Ultimately, the pathologist’s decision on whether and how to report margins should be focused on improving patient outcomes. There are benefits and drawbacks to all approaches, and our goal is to provide more information for clinicians and pathologists so that they may better care for their patients. Understanding the limitations of margins on submitted skin specimens—whether margins are reported or not—can only serve to guide improve clinical decision-making.
We also agree that the breadth of specialties of submitting clinicians make reporting of margins difficult, and there is likely similar breadth in their understanding of the nuances of margin assessment and reports. The solution to this problem is adequate education regarding the limitations of a pathology report, and specifically what is meant when margins are (or are not) reported on skin specimens. How to best educate the myriad clinicians who submit biopsies is, of course, the ultimate challenge.
We hope that our study adds information to this ongoing debate regarding margin status reporting, and we appreciate the discussion points raised by the author.
Eric Hossler, MD; Mary Brady, MD
From the Department of Dermatology, Geisinger Health System, Danville, Pennsylvania.
The authors report no conflict of interest.
Reference
- Sellheyer K, Bergfeld WF, Stewart E, et al. Evaluation of surgical margins in melanocytic lesions: a survey among 152 dermatopathologists.J Cutan Pathol. 2005;32:293-299.
To the Editor:
In an interesting analysis, Brady and Hossler1 (Cutis. 2020;106:315-317) highlighted the limitations of histopathologic biopsy margin evaluation for cutaneous basal cell carcinoma (BCC). Taking into consideration the high prevalence of BCC and its medical and economic impact on the health care system, the issue raised by the authors is an important one. They proposed that pathologists may omit reporting margins or clarify the limitations in their reports. It is a valid suggestion; however, in practice, margin evaluation is not always a simple process and is influenced by a number of factors.
The subject of optimum margins for BCC has been debated over decades now; however, ambiguity and lack of definitive guidelines on certain aspects still remain, leading to a lack of standardization and variability in reporting, which opens potential for error. In anatomical pathology, the biopsies for malignancies are interpreted to confirm diagnosis and perform risk assessment, with evaluation of margins generally reserved for subsequent definitive resections. Typically, margins are not required by clinicians or reported by pathologists in common endoscopic (eg, stomach, colon) or needle core (eg, prostate, breast) biopsies. Skin holds a rather unique position in which margin evaluation is not just limited to excisions. With the exception of samples generated from electrodesiccation and curettage, it is common practice by some laboratories to report margins on most specimens of cutaneous malignancies.
In simple terms, when margins are labeled negative there should be no residual disease, and when they are deemed positive there should be disease still persisting in the patient. Margin evaluation for BCC on biopsies falls short on both fronts. In one analysis, 24% (34/143) of shave biopsies reported with negative margins displayed residual BCC in ensuing re-excisions (negative predictive value: 76%).2 Standard bread-loafing, en-face margins and inking for orientation utilized to provide a thorough margin evaluation of excisions cannot be optimally achieved on small skin biopsies. Microscopic sections for analysis are 2-dimensional representations of 3-dimensional structures. Slides prepared can miss deeply embedded outermost margins, positioned parallel to the plane of sectioning, thereby creating blind spots where margins cannot be precisely assessed and generating an inherent limitation in evaluation. Exhaustive deeper levels done routinely can address this issue to a certain degree; however, it can be an impractical solution with cost implications and delay in turnaround time.
Conversely, it also is common to encounter absence of residual BCC in re-excisions in which the original biopsy margins were labeled positive. In one analysis, 49% of BCC patients (n=100) with positive biopsy margins did not display residual neoplasm on following re-excisions.3 Localized biopsy site immune response as a cause of postbiopsy regression of residual tumor has been hypothesized to produce this phenomenon. Moreover, initial biopsies may eliminate the majority of the tumor with only minimal disease persisting. Re-excisions submitted in toto allow for a systematic examination; however, areas in between sections still remain where minute residual tumor may hide. Searching for such occult foci generally is not aggressively pursued via deeper levels unless the margins of re-excision are in question.
Superficial-type BCC (or superficial multifocal BCC) is a major factor in precluding precise biopsy margin evaluation. In a study where initial biopsies reported with negative margins displayed residual BCC in subsequent re-excisions, 91% (31/34) of residual BCCs were of superficial variety.2 Clinically, superficial BCC frequently has indistinct borders with subtle subclinical peripheral progression. It has a tendency to expand radially, with the clinical appearance deceptively smaller than its true extent. In a plane of histopathologic section, superficial BCC may exhibit skip zones within the epidermis. Even though the margin may seem uninvolved on the slide, a noncontiguous focus may still emerge beyond the “negative” margin. Because superficial pattern is not unusual as one of the components of mixed histology (composite) BCC, this issue is not just limited to tumors specifically designated as superficial type.4
The intent of a procedure is important to recognize. If a biopsy is done with the intention of diagnosis only, the pathologic assessment can be limited to tumor identification and core data elements, with margin evaluation reserved for excisions done with therapeutic intent. However, the intent is not always clear, which adds to ambiguity on when to report margins. It is not uncommon to find saucerization shaves or large punch biopsies for BCC carried out with a therapeutic intent. The status of margin is desired in such samples; however, the intent is not always clearly communicated on requisitions. To avoid any gaps in communication, some pathologists may err on the side of caution and start routinely reporting margins on biopsies.
Taking into account the inaccuracy of margin assessment in biopsies, an argument for omitting margin reporting is plausible. Although dermatologists are the major contributors of skin samples, pathology laboratories cater to a broader clientele. Other physicians from different surgical and medical specialities also perform skin biopsies, and catering to a variety of specialities adds another layer of complexity. A dermatologist may appreciate the debate regarding reliability of margins; however, a physician from another speciality who is not as familiar with the diseases of the integument may lack proper understanding. Omitting margin reporting may lead to misinterpretations or false assumptions, such as, “The margins must be uninvolved, otherwise the pathologist would have said something.” This also can generate additional phone or email inquiries and second review requests. Rather than completely omitting them, another strategy can be to report margins in more quantitative terms. One reporting approach is to have 3 categories of involved, uninvolved, and uninvolved but close for margins less than 1 mm. The cases in the third category may require greater scrutiny by deeper levels or an added caveat in the comment addressing the limitation. If the status of margins is not reported due to a certain reason, a short comment can be added to explain the reason.
In sum, clinicians should recognize that “margin negative” on skin biopsy does not always equate to “completely excised.” Margin status on biopsies is a data item that essentially provides a probability of margin clearance. Completely omitting the margin status on all biopsies may not be the most prudent approach; however, improved guidelines and modifications to enhance the reporting are definitely required.
References
- Brady MC, Hossler EW. Reliability of biopsy margin status for basal cell carcinoma: a retrospective study. Cutis. 2020;106:315-317.
- Willardson HB, Lombardo J, Raines M, et al. Predictive value of basal cell carcinoma biopsies with negative margins: a retrospective cohort study. J Am Acad Dermatol. 2018;79:42-46.
- Yuan Y, Duff ML, Sammons DL, et al. Retrospective chart review of skin cancer presence in the wide excisions. World J Clin Cases. 2014;2:52-56.
- Cohen PR, Schulze KE, Nelson BR. Basal cell carcinoma with mixed histology: a possible pathogenesis for recurrent skin cancer. Dermatol Surg. 2006;32:542-551.
Continue to: Author's Response...
Authors’ Response
We appreciate the thorough and thoughtful comments in the Letter to the Editor. We agree with the author’s assertion that negative margins on skin specimens does not equate to “completely excised” and that the intent of the clinician is not always clear, even when the pathologist has ready access to the clinician’s notes, as was the case for the majority of specimens included in our study.
There is already variability in how pathologists report margins, including the specific verbiage used, at least for melanocytic lesions.1 The choice of whether or not to report margins and the meaning of those margins is complex due to the uncertainty inherent in margin assessment. Quantifying this uncertainty was the main reason for our study. Ultimately, the pathologist’s decision on whether and how to report margins should be focused on improving patient outcomes. There are benefits and drawbacks to all approaches, and our goal is to provide more information for clinicians and pathologists so that they may better care for their patients. Understanding the limitations of margins on submitted skin specimens—whether margins are reported or not—can only serve to guide improve clinical decision-making.
We also agree that the breadth of specialties of submitting clinicians make reporting of margins difficult, and there is likely similar breadth in their understanding of the nuances of margin assessment and reports. The solution to this problem is adequate education regarding the limitations of a pathology report, and specifically what is meant when margins are (or are not) reported on skin specimens. How to best educate the myriad clinicians who submit biopsies is, of course, the ultimate challenge.
We hope that our study adds information to this ongoing debate regarding margin status reporting, and we appreciate the discussion points raised by the author.
Eric Hossler, MD; Mary Brady, MD
From the Department of Dermatology, Geisinger Health System, Danville, Pennsylvania.
The authors report no conflict of interest.
Reference
- Sellheyer K, Bergfeld WF, Stewart E, et al. Evaluation of surgical margins in melanocytic lesions: a survey among 152 dermatopathologists.J Cutan Pathol. 2005;32:293-299.
USMLE Step 1 Changes: Dermatology Program Director Perspectives and Implications
To the Editor:
With a trend toward increasing pass/fail medical school curricula, residency program directors (PDs) have relied on the US Medical Licensing Examination (USMLE) Step 1 as an objective measurement of applicant achievement, which is particularly true in competitive subspecialties such as dermatology, plastic surgery, orthopedic surgery, ophthalmology, and neurosurgery, in which reported Step 1 scores are consistently the highest among matched applicants.1 Program directors in dermatology have indicated that Step 1 scores are a priority when considering an applicant.2 However, among PDs, the general perception of plans to change Step 1 scores to pass/fail has largely been negative.3 Although the impact of this change on the dermatology residency selection process remains unknown, we undertook a study to determine dermatology PDs’ perspectives on the scoring change and discuss its potential implications among all competitive specialties.
A 19-question survey was designed that assessed PD demographics and opinions of the changes and potential implications of the Step 1 scoring change (eTable). A list of current US dermatology PDs at osteopathic and allopathic programs was obtained through the 2019-2020 Accreditation Council for Graduate Medical Education list of accredited programs. Surveys were piloted at our institution to assess for internal validity and misleading questions, and then were distributed electronically through REDCap software (https://www.project-redcap.org/). All responses were kept anonymous. Institutional review board approval was obtained. Variables were assessed with means, proportions, and CIs. Results were deemed statistically significant with nonoverlapping 99% CIs (P<.01).
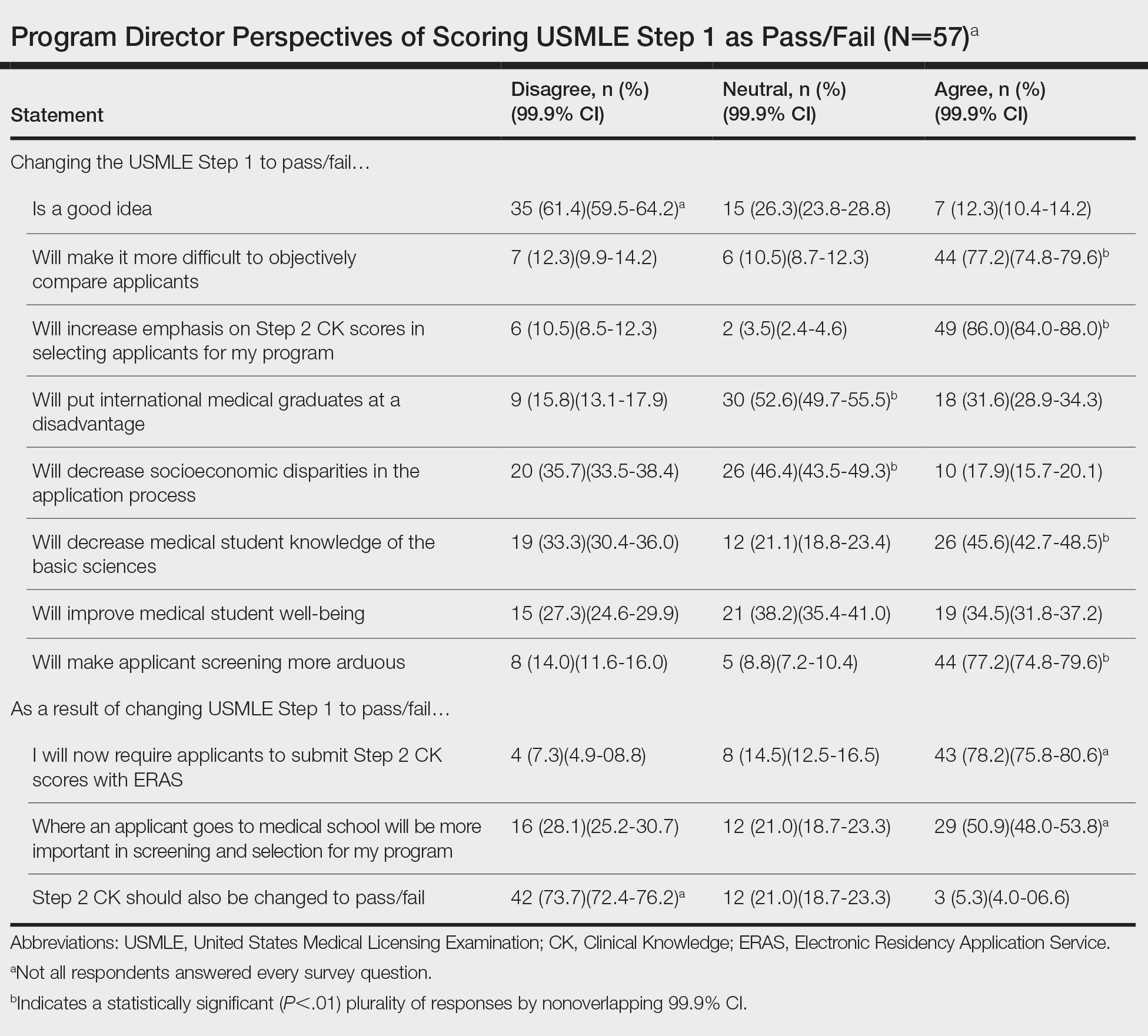
Of 139 surveys, 57 (41.0%) were completed. Most PDs (54.4% [31/57]) were women. The average years of service as a PD was 8.5 years. Most PDs (61.4% [35/57]) disagreed with the scoring change; 77.2% (44/57) of PDs noted that it would make it difficult to objectively assess candidates. Program directors indicated that this change would increase the emphasis they place on USMLE Step 2 Clinical Knowledge (CK) scores (86.0% [49/57]); 78.2% (43/55) reported that they would start requiring Step 2 CK results with submitted applications.
Meanwhile, 73.7% (42/57) of PDs disagreed that Step 2 CK should be changed to pass/fail. Most PDs (50.9% [29/57]) thought that binary Step 1 scoring would increase the importance of medical school reputation in application decisions. The percentage of PDs who were neutral (eTable) on whether pass/fail scoring would place international graduates at a disadvantage was 52.6% (30/57), decrease socioeconomic disparities in the application process was 46.4% (26/56), and improve student well-being was 38.2% (21/55).
Results of our survey indicate generally negative perceptions by dermatology PDs to pass/fail scoring of the USMLE Step 1. A primary goal of introducing binary scoring in both medical school grading and the USMLE was to improve student well-being, as traditional grading systems have been associated with a higher rate of medical student burnout.4-6 However, PDs were equivocal about such an impact on student well-being. Furthermore, PDs indicated that the importance of objective measures would merely shift to the USMLE Step 2 CK, which will still be graded with a 3-digit numeric score. Therefore, Step 2 likely will become the source of anxiety for medical students that was once synonymous with Step 1.
Another goal of the scoring change was to encourage a more holistic approach to applicant review, rather than focusing on numerical metrics. However, with most curricula adopting pass/fail models, there is already a lack of objective measures. Although removal of USMLE Step 1 scores could increase the focus on subjective measures, such as letters of recommendation and rank in medical school class (as indicated by our survey), these are susceptible to bias and may not be the best indicators of applicant suitability. This finding also is concerning for maintaining an equitable application process: PDs indicated that the USMLE Step 1 scoring change would not decrease socioeconomic disparities within the selection process.
In dermatology and other competitive specialties, in which USMLE Step 1 scores have become an important consideration, PDs and residency programs will need to identify additional metrics to compare applicants. Examples include research productivity, grades on relevant rotations, and shelf examination scores. Although more reliable subjective measures, such as interviews and performance on away rotations, are already important, they may become of greater significance.
The findings of our survey suggest that PDs are skeptical about changes to Step 1 and more diligence is necessary to maintain a fair and impartial selection process. Increased emphasis on other objective measurements, such as shelf examination scores, graded curricular components, and research productivity, could help maintain an unbiased approach. With changes to USMLE Step 1 expected to be implemented in the 2022 application cycle, programs may need to explore additional options to maintain reliable and transparent applicant review practices.
- National Resident Matching Program. Charting Outcomes in the Match: U.S Allopathic Seniors, 2018. 2nd ed. National Resident Matching Program; July 2018. Accessed May 12, 2021. https://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf
- Grading systems use by US medical schools. Association of American Medical Colleges. Accessed May 12, 2021. https://www.aamc.org/data-reports/curriculum-reports/interactive-data/grading-systems-use-us-medical-schools
- Makhoul AT, Pontell ME, Ganesh Kumar N, et al. Objective measures needed—program directors’ perspectives on a pass/fail USMLE Step 1. N Engl J Med; 2020;382:2389-2392. doi:10.1056/NEJMp2006148
- Change to pass/fail score reporting for Step 1. United States Medical Licensing Examination. Accessed May 12, 2021. https://www.usmle.org/incus/
- Reed DA, Shanafelt TD, Satele DW, et al. Relationship of pass/fail grading and curriculum structure with well-being among preclinical medical students: a multi-institutional study. Acad Med. 2011;86:1367-1373. doi:10.1097/ACM.0b013e3182305d81
- Summary report and preliminary recommendations from the Invitational Conference on USMLE Scoring (InCUS). United States Medical Licensing Examination. March 11-12, 2019. Accessed May 12, 2021. https://www.usmle.org/pdfs/incus/incus_summary_report.pdf
To the Editor:
With a trend toward increasing pass/fail medical school curricula, residency program directors (PDs) have relied on the US Medical Licensing Examination (USMLE) Step 1 as an objective measurement of applicant achievement, which is particularly true in competitive subspecialties such as dermatology, plastic surgery, orthopedic surgery, ophthalmology, and neurosurgery, in which reported Step 1 scores are consistently the highest among matched applicants.1 Program directors in dermatology have indicated that Step 1 scores are a priority when considering an applicant.2 However, among PDs, the general perception of plans to change Step 1 scores to pass/fail has largely been negative.3 Although the impact of this change on the dermatology residency selection process remains unknown, we undertook a study to determine dermatology PDs’ perspectives on the scoring change and discuss its potential implications among all competitive specialties.
A 19-question survey was designed that assessed PD demographics and opinions of the changes and potential implications of the Step 1 scoring change (eTable). A list of current US dermatology PDs at osteopathic and allopathic programs was obtained through the 2019-2020 Accreditation Council for Graduate Medical Education list of accredited programs. Surveys were piloted at our institution to assess for internal validity and misleading questions, and then were distributed electronically through REDCap software (https://www.project-redcap.org/). All responses were kept anonymous. Institutional review board approval was obtained. Variables were assessed with means, proportions, and CIs. Results were deemed statistically significant with nonoverlapping 99% CIs (P<.01).
Of 139 surveys, 57 (41.0%) were completed. Most PDs (54.4% [31/57]) were women. The average years of service as a PD was 8.5 years. Most PDs (61.4% [35/57]) disagreed with the scoring change; 77.2% (44/57) of PDs noted that it would make it difficult to objectively assess candidates. Program directors indicated that this change would increase the emphasis they place on USMLE Step 2 Clinical Knowledge (CK) scores (86.0% [49/57]); 78.2% (43/55) reported that they would start requiring Step 2 CK results with submitted applications.
Meanwhile, 73.7% (42/57) of PDs disagreed that Step 2 CK should be changed to pass/fail. Most PDs (50.9% [29/57]) thought that binary Step 1 scoring would increase the importance of medical school reputation in application decisions. The percentage of PDs who were neutral (eTable) on whether pass/fail scoring would place international graduates at a disadvantage was 52.6% (30/57), decrease socioeconomic disparities in the application process was 46.4% (26/56), and improve student well-being was 38.2% (21/55).
Results of our survey indicate generally negative perceptions by dermatology PDs to pass/fail scoring of the USMLE Step 1. A primary goal of introducing binary scoring in both medical school grading and the USMLE was to improve student well-being, as traditional grading systems have been associated with a higher rate of medical student burnout.4-6 However, PDs were equivocal about such an impact on student well-being. Furthermore, PDs indicated that the importance of objective measures would merely shift to the USMLE Step 2 CK, which will still be graded with a 3-digit numeric score. Therefore, Step 2 likely will become the source of anxiety for medical students that was once synonymous with Step 1.
Another goal of the scoring change was to encourage a more holistic approach to applicant review, rather than focusing on numerical metrics. However, with most curricula adopting pass/fail models, there is already a lack of objective measures. Although removal of USMLE Step 1 scores could increase the focus on subjective measures, such as letters of recommendation and rank in medical school class (as indicated by our survey), these are susceptible to bias and may not be the best indicators of applicant suitability. This finding also is concerning for maintaining an equitable application process: PDs indicated that the USMLE Step 1 scoring change would not decrease socioeconomic disparities within the selection process.
In dermatology and other competitive specialties, in which USMLE Step 1 scores have become an important consideration, PDs and residency programs will need to identify additional metrics to compare applicants. Examples include research productivity, grades on relevant rotations, and shelf examination scores. Although more reliable subjective measures, such as interviews and performance on away rotations, are already important, they may become of greater significance.
The findings of our survey suggest that PDs are skeptical about changes to Step 1 and more diligence is necessary to maintain a fair and impartial selection process. Increased emphasis on other objective measurements, such as shelf examination scores, graded curricular components, and research productivity, could help maintain an unbiased approach. With changes to USMLE Step 1 expected to be implemented in the 2022 application cycle, programs may need to explore additional options to maintain reliable and transparent applicant review practices.
To the Editor:
With a trend toward increasing pass/fail medical school curricula, residency program directors (PDs) have relied on the US Medical Licensing Examination (USMLE) Step 1 as an objective measurement of applicant achievement, which is particularly true in competitive subspecialties such as dermatology, plastic surgery, orthopedic surgery, ophthalmology, and neurosurgery, in which reported Step 1 scores are consistently the highest among matched applicants.1 Program directors in dermatology have indicated that Step 1 scores are a priority when considering an applicant.2 However, among PDs, the general perception of plans to change Step 1 scores to pass/fail has largely been negative.3 Although the impact of this change on the dermatology residency selection process remains unknown, we undertook a study to determine dermatology PDs’ perspectives on the scoring change and discuss its potential implications among all competitive specialties.
A 19-question survey was designed that assessed PD demographics and opinions of the changes and potential implications of the Step 1 scoring change (eTable). A list of current US dermatology PDs at osteopathic and allopathic programs was obtained through the 2019-2020 Accreditation Council for Graduate Medical Education list of accredited programs. Surveys were piloted at our institution to assess for internal validity and misleading questions, and then were distributed electronically through REDCap software (https://www.project-redcap.org/). All responses were kept anonymous. Institutional review board approval was obtained. Variables were assessed with means, proportions, and CIs. Results were deemed statistically significant with nonoverlapping 99% CIs (P<.01).
Of 139 surveys, 57 (41.0%) were completed. Most PDs (54.4% [31/57]) were women. The average years of service as a PD was 8.5 years. Most PDs (61.4% [35/57]) disagreed with the scoring change; 77.2% (44/57) of PDs noted that it would make it difficult to objectively assess candidates. Program directors indicated that this change would increase the emphasis they place on USMLE Step 2 Clinical Knowledge (CK) scores (86.0% [49/57]); 78.2% (43/55) reported that they would start requiring Step 2 CK results with submitted applications.
Meanwhile, 73.7% (42/57) of PDs disagreed that Step 2 CK should be changed to pass/fail. Most PDs (50.9% [29/57]) thought that binary Step 1 scoring would increase the importance of medical school reputation in application decisions. The percentage of PDs who were neutral (eTable) on whether pass/fail scoring would place international graduates at a disadvantage was 52.6% (30/57), decrease socioeconomic disparities in the application process was 46.4% (26/56), and improve student well-being was 38.2% (21/55).
Results of our survey indicate generally negative perceptions by dermatology PDs to pass/fail scoring of the USMLE Step 1. A primary goal of introducing binary scoring in both medical school grading and the USMLE was to improve student well-being, as traditional grading systems have been associated with a higher rate of medical student burnout.4-6 However, PDs were equivocal about such an impact on student well-being. Furthermore, PDs indicated that the importance of objective measures would merely shift to the USMLE Step 2 CK, which will still be graded with a 3-digit numeric score. Therefore, Step 2 likely will become the source of anxiety for medical students that was once synonymous with Step 1.
Another goal of the scoring change was to encourage a more holistic approach to applicant review, rather than focusing on numerical metrics. However, with most curricula adopting pass/fail models, there is already a lack of objective measures. Although removal of USMLE Step 1 scores could increase the focus on subjective measures, such as letters of recommendation and rank in medical school class (as indicated by our survey), these are susceptible to bias and may not be the best indicators of applicant suitability. This finding also is concerning for maintaining an equitable application process: PDs indicated that the USMLE Step 1 scoring change would not decrease socioeconomic disparities within the selection process.
In dermatology and other competitive specialties, in which USMLE Step 1 scores have become an important consideration, PDs and residency programs will need to identify additional metrics to compare applicants. Examples include research productivity, grades on relevant rotations, and shelf examination scores. Although more reliable subjective measures, such as interviews and performance on away rotations, are already important, they may become of greater significance.
The findings of our survey suggest that PDs are skeptical about changes to Step 1 and more diligence is necessary to maintain a fair and impartial selection process. Increased emphasis on other objective measurements, such as shelf examination scores, graded curricular components, and research productivity, could help maintain an unbiased approach. With changes to USMLE Step 1 expected to be implemented in the 2022 application cycle, programs may need to explore additional options to maintain reliable and transparent applicant review practices.
- National Resident Matching Program. Charting Outcomes in the Match: U.S Allopathic Seniors, 2018. 2nd ed. National Resident Matching Program; July 2018. Accessed May 12, 2021. https://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf
- Grading systems use by US medical schools. Association of American Medical Colleges. Accessed May 12, 2021. https://www.aamc.org/data-reports/curriculum-reports/interactive-data/grading-systems-use-us-medical-schools
- Makhoul AT, Pontell ME, Ganesh Kumar N, et al. Objective measures needed—program directors’ perspectives on a pass/fail USMLE Step 1. N Engl J Med; 2020;382:2389-2392. doi:10.1056/NEJMp2006148
- Change to pass/fail score reporting for Step 1. United States Medical Licensing Examination. Accessed May 12, 2021. https://www.usmle.org/incus/
- Reed DA, Shanafelt TD, Satele DW, et al. Relationship of pass/fail grading and curriculum structure with well-being among preclinical medical students: a multi-institutional study. Acad Med. 2011;86:1367-1373. doi:10.1097/ACM.0b013e3182305d81
- Summary report and preliminary recommendations from the Invitational Conference on USMLE Scoring (InCUS). United States Medical Licensing Examination. March 11-12, 2019. Accessed May 12, 2021. https://www.usmle.org/pdfs/incus/incus_summary_report.pdf
- National Resident Matching Program. Charting Outcomes in the Match: U.S Allopathic Seniors, 2018. 2nd ed. National Resident Matching Program; July 2018. Accessed May 12, 2021. https://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf
- Grading systems use by US medical schools. Association of American Medical Colleges. Accessed May 12, 2021. https://www.aamc.org/data-reports/curriculum-reports/interactive-data/grading-systems-use-us-medical-schools
- Makhoul AT, Pontell ME, Ganesh Kumar N, et al. Objective measures needed—program directors’ perspectives on a pass/fail USMLE Step 1. N Engl J Med; 2020;382:2389-2392. doi:10.1056/NEJMp2006148
- Change to pass/fail score reporting for Step 1. United States Medical Licensing Examination. Accessed May 12, 2021. https://www.usmle.org/incus/
- Reed DA, Shanafelt TD, Satele DW, et al. Relationship of pass/fail grading and curriculum structure with well-being among preclinical medical students: a multi-institutional study. Acad Med. 2011;86:1367-1373. doi:10.1097/ACM.0b013e3182305d81
- Summary report and preliminary recommendations from the Invitational Conference on USMLE Scoring (InCUS). United States Medical Licensing Examination. March 11-12, 2019. Accessed May 12, 2021. https://www.usmle.org/pdfs/incus/incus_summary_report.pdf
Practice Points
- The changes to US Medical Licensing Examination (USMLE) Step 1 were met with mixed reactions from dermatology program directors.
- These changes likely will increase the emphasis on USMLE Step 2 and other objective measures.
Wound Healing on the Dorsal Hands: An Intrapatient Comparison of Primary Closure, Purse-String Closure, and Secondary Intention
Practice Gap
Many cutaneous surgery wounds can be closed primarily; however, in certain cases, other repair options might be appropriate and should be evaluated on a case-by-case basis with input from the patient. Defects on the dorsal aspect of the hands—where nonmelanoma skin cancer is common and reserve tissue is limited—often heal by secondary intention with good cosmetic and functional results. Patients often express a desire to reduce the time spent in the surgical suite and restrictions on postoperative activity, making secondary intention healing more appealing. An additional advantage is obviation of the need to remove additional tissue in the form of Burow triangles, which would lead to a longer wound. The major disadvantage of secondary intention healing is longer time to wound maturity; we often minimize this disadvantage with purse-string closure to decrease the size of the wound defect, which can be done quickly and without removing additional tissue.
The Technique
An elderly man had 3 nonmelanoma skin cancers—all on the dorsal aspect of the left hand—that were treated on the same day, leaving 3 similar wound defects after Mohs micrographic surgery. The wound defects (distal to proximal) measured 12 mm, 12 mm, and 10 mm in diameter (Figure 1) and were repaired by primary closure, secondary intention, and purse-string circumferential closure, respectively. Purse-string closure1 was performed with a 4-0 polyglactin 901 suture and left to heal without external sutures (Figure 2). Figure 3 shows the 3 types of repairs immediately following closure. All wounds healed with excellent and essentially equivalent cosmetic results, with excellent patient satisfaction at 6-month follow-up (Figure 4).

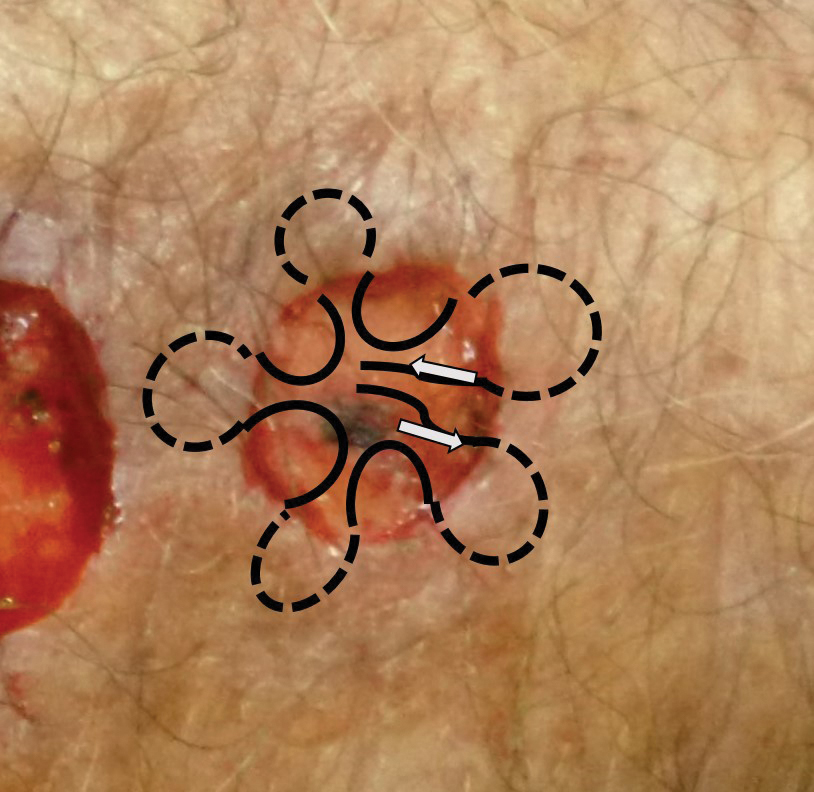


Practical Implications
Our case illustrates different modalities of wound repair during precisely the same time frame and essentially on the same location. Skin of the dorsal hand often is tight; depending on the size of the defect, large primary closure can be tedious to perform, can lead to increased wound tension and risk of dehiscence, and can be uncomfortable for the patient during healing. However, primary closure typically will lead to faster healing.
Secondary intention healing and purse-string closure require less surgery and therefore cost less; these modalities yield similar cosmesis and satisfaction. In the appropriate context, secondary intention has been highlighted as a suitable alternative to primary closure2-4; in our experience (and that of others5), patient satisfaction is not diminished with healing by secondary intention. Purse-string closure also can minimize wound size and healing time.
For small shallow wounds on the dorsal hand, dermatologic surgeons should have confidence that secondary intention healing, with or without wound reduction using purse-string repair, likely will lead to acceptable cosmetic and functional results. Of course, repair should be tailored to the circumstances and wishes of the individual patient.
- Peled IJ, Zagher U, Wexler MR. Purse-string suture for reduction and closure of skin defects. Ann Plast Surg. 1985;14:465-469. doi:10.1097/00000637-198505000-00012
- Zitelli JA. Secondary intention healing: an alternative to surgical repair. Clin Dermatol. 1984;2:92-106. doi:10.1016/0738-081x(84)90031-2
- Fazio MJ, Zitelli JA. Principles of reconstruction following excision of nonmelanoma skin cancer. Clin Dermatol. 1995;13:601-616. doi:10.1016/0738-081x(95)00099-2
- Bosley R, Leithauser L, Turner M, et al. The efficacy of second-intention healing in the management of defects on the dorsal surface of the hands and fingers after Mohs micrographic surgery. Dermatol Surg. 2012;38:647-653. doi:10.1111/j.1524-4725.2011.02258.x
- Stebbins WG, Gusev J, Higgins HW 2nd, et al. Evaluation of patient satisfaction with second intention healing versus primary surgical closure. J Am Acad Dermatol. 2015;73:865-867.e1. doi:10.1016/j.jaad.2015.07.019
Practice Gap
Many cutaneous surgery wounds can be closed primarily; however, in certain cases, other repair options might be appropriate and should be evaluated on a case-by-case basis with input from the patient. Defects on the dorsal aspect of the hands—where nonmelanoma skin cancer is common and reserve tissue is limited—often heal by secondary intention with good cosmetic and functional results. Patients often express a desire to reduce the time spent in the surgical suite and restrictions on postoperative activity, making secondary intention healing more appealing. An additional advantage is obviation of the need to remove additional tissue in the form of Burow triangles, which would lead to a longer wound. The major disadvantage of secondary intention healing is longer time to wound maturity; we often minimize this disadvantage with purse-string closure to decrease the size of the wound defect, which can be done quickly and without removing additional tissue.
The Technique
An elderly man had 3 nonmelanoma skin cancers—all on the dorsal aspect of the left hand—that were treated on the same day, leaving 3 similar wound defects after Mohs micrographic surgery. The wound defects (distal to proximal) measured 12 mm, 12 mm, and 10 mm in diameter (Figure 1) and were repaired by primary closure, secondary intention, and purse-string circumferential closure, respectively. Purse-string closure1 was performed with a 4-0 polyglactin 901 suture and left to heal without external sutures (Figure 2). Figure 3 shows the 3 types of repairs immediately following closure. All wounds healed with excellent and essentially equivalent cosmetic results, with excellent patient satisfaction at 6-month follow-up (Figure 4).
Practical Implications
Our case illustrates different modalities of wound repair during precisely the same time frame and essentially on the same location. Skin of the dorsal hand often is tight; depending on the size of the defect, large primary closure can be tedious to perform, can lead to increased wound tension and risk of dehiscence, and can be uncomfortable for the patient during healing. However, primary closure typically will lead to faster healing.
Secondary intention healing and purse-string closure require less surgery and therefore cost less; these modalities yield similar cosmesis and satisfaction. In the appropriate context, secondary intention has been highlighted as a suitable alternative to primary closure2-4; in our experience (and that of others5), patient satisfaction is not diminished with healing by secondary intention. Purse-string closure also can minimize wound size and healing time.
For small shallow wounds on the dorsal hand, dermatologic surgeons should have confidence that secondary intention healing, with or without wound reduction using purse-string repair, likely will lead to acceptable cosmetic and functional results. Of course, repair should be tailored to the circumstances and wishes of the individual patient.
Practice Gap
Many cutaneous surgery wounds can be closed primarily; however, in certain cases, other repair options might be appropriate and should be evaluated on a case-by-case basis with input from the patient. Defects on the dorsal aspect of the hands—where nonmelanoma skin cancer is common and reserve tissue is limited—often heal by secondary intention with good cosmetic and functional results. Patients often express a desire to reduce the time spent in the surgical suite and restrictions on postoperative activity, making secondary intention healing more appealing. An additional advantage is obviation of the need to remove additional tissue in the form of Burow triangles, which would lead to a longer wound. The major disadvantage of secondary intention healing is longer time to wound maturity; we often minimize this disadvantage with purse-string closure to decrease the size of the wound defect, which can be done quickly and without removing additional tissue.
The Technique
An elderly man had 3 nonmelanoma skin cancers—all on the dorsal aspect of the left hand—that were treated on the same day, leaving 3 similar wound defects after Mohs micrographic surgery. The wound defects (distal to proximal) measured 12 mm, 12 mm, and 10 mm in diameter (Figure 1) and were repaired by primary closure, secondary intention, and purse-string circumferential closure, respectively. Purse-string closure1 was performed with a 4-0 polyglactin 901 suture and left to heal without external sutures (Figure 2). Figure 3 shows the 3 types of repairs immediately following closure. All wounds healed with excellent and essentially equivalent cosmetic results, with excellent patient satisfaction at 6-month follow-up (Figure 4).
Practical Implications
Our case illustrates different modalities of wound repair during precisely the same time frame and essentially on the same location. Skin of the dorsal hand often is tight; depending on the size of the defect, large primary closure can be tedious to perform, can lead to increased wound tension and risk of dehiscence, and can be uncomfortable for the patient during healing. However, primary closure typically will lead to faster healing.
Secondary intention healing and purse-string closure require less surgery and therefore cost less; these modalities yield similar cosmesis and satisfaction. In the appropriate context, secondary intention has been highlighted as a suitable alternative to primary closure2-4; in our experience (and that of others5), patient satisfaction is not diminished with healing by secondary intention. Purse-string closure also can minimize wound size and healing time.
For small shallow wounds on the dorsal hand, dermatologic surgeons should have confidence that secondary intention healing, with or without wound reduction using purse-string repair, likely will lead to acceptable cosmetic and functional results. Of course, repair should be tailored to the circumstances and wishes of the individual patient.
- Peled IJ, Zagher U, Wexler MR. Purse-string suture for reduction and closure of skin defects. Ann Plast Surg. 1985;14:465-469. doi:10.1097/00000637-198505000-00012
- Zitelli JA. Secondary intention healing: an alternative to surgical repair. Clin Dermatol. 1984;2:92-106. doi:10.1016/0738-081x(84)90031-2
- Fazio MJ, Zitelli JA. Principles of reconstruction following excision of nonmelanoma skin cancer. Clin Dermatol. 1995;13:601-616. doi:10.1016/0738-081x(95)00099-2
- Bosley R, Leithauser L, Turner M, et al. The efficacy of second-intention healing in the management of defects on the dorsal surface of the hands and fingers after Mohs micrographic surgery. Dermatol Surg. 2012;38:647-653. doi:10.1111/j.1524-4725.2011.02258.x
- Stebbins WG, Gusev J, Higgins HW 2nd, et al. Evaluation of patient satisfaction with second intention healing versus primary surgical closure. J Am Acad Dermatol. 2015;73:865-867.e1. doi:10.1016/j.jaad.2015.07.019
- Peled IJ, Zagher U, Wexler MR. Purse-string suture for reduction and closure of skin defects. Ann Plast Surg. 1985;14:465-469. doi:10.1097/00000637-198505000-00012
- Zitelli JA. Secondary intention healing: an alternative to surgical repair. Clin Dermatol. 1984;2:92-106. doi:10.1016/0738-081x(84)90031-2
- Fazio MJ, Zitelli JA. Principles of reconstruction following excision of nonmelanoma skin cancer. Clin Dermatol. 1995;13:601-616. doi:10.1016/0738-081x(95)00099-2
- Bosley R, Leithauser L, Turner M, et al. The efficacy of second-intention healing in the management of defects on the dorsal surface of the hands and fingers after Mohs micrographic surgery. Dermatol Surg. 2012;38:647-653. doi:10.1111/j.1524-4725.2011.02258.x
- Stebbins WG, Gusev J, Higgins HW 2nd, et al. Evaluation of patient satisfaction with second intention healing versus primary surgical closure. J Am Acad Dermatol. 2015;73:865-867.e1. doi:10.1016/j.jaad.2015.07.019
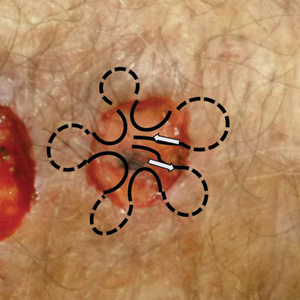
Oral Verrucous Plaques in a Patient With Urothelial Cancer
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
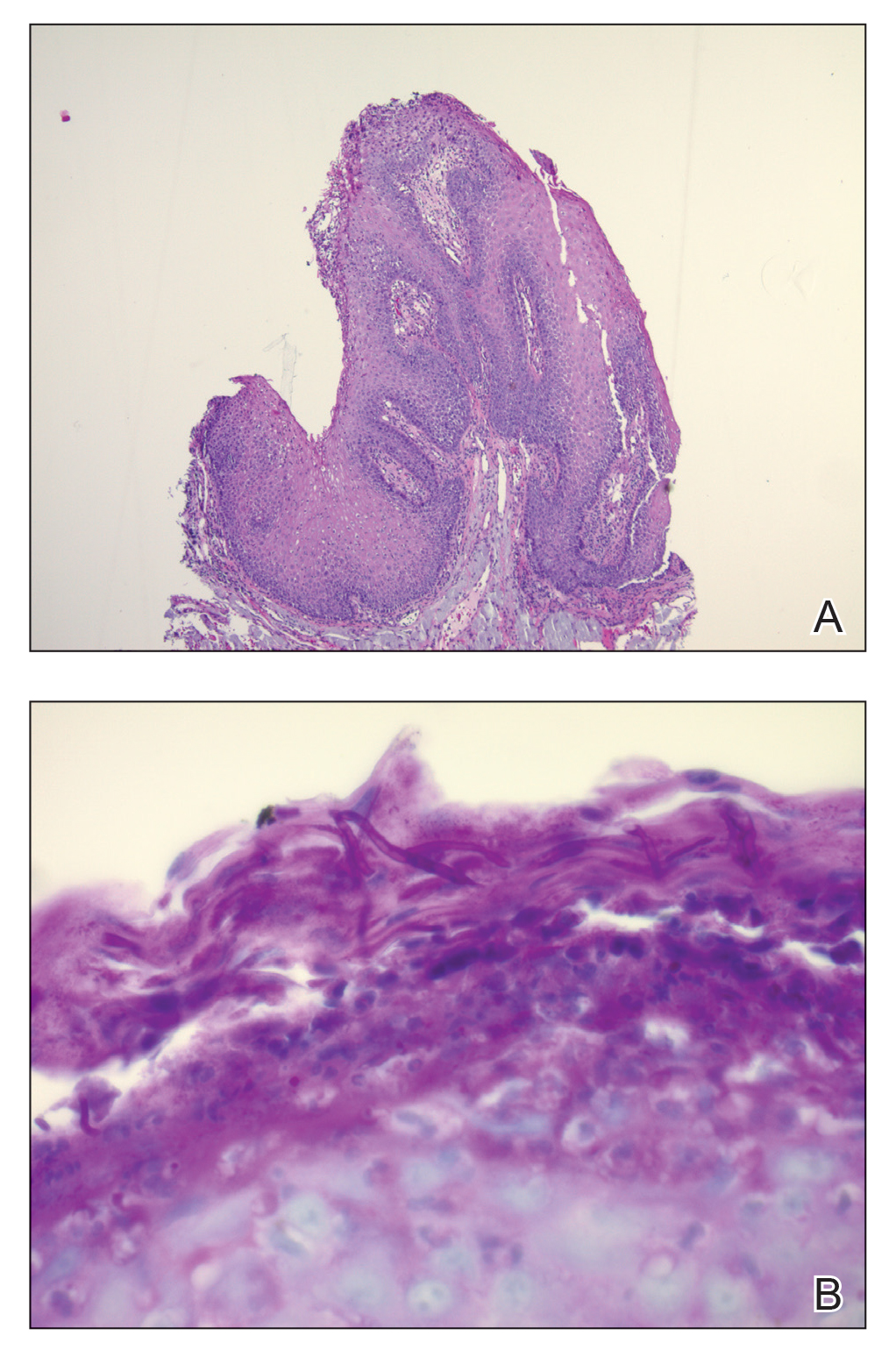
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
A 75-year-old nonobese man with metastatic urothelial carcinoma presented for evaluation and treatment of swollen lips. The patient stated that his lips began to swell and crack shortly after beginning pembrolizumab approximately 5 months prior. The swelling had progressively worsened, prompting discontinuation of the pembrolizumab by oncology about 2 months prior to presentation to our dermatology clinic. He reported slight improvement after the discontinuation of pembrolizumab, and he had since been started on carboplatin and gemcitabine. He previously was treated with oral corticosteroids without improvement. His oncologist started him on oral fluconazole for treatment of oral thrush on the day of presentation to our clinic. Physical examination revealed diffuse papillomatous and verrucous plaques of the upper and lower lips with involvement of the buccal mucosa. He also had deep fissures and white plaques on the tongue. Velvety hyperpigmented plaques were noted in the axillae, and he had confluent thickening of the palms. A 3-mm punch biopsy from the lower lip was performed. The patient subsequently was evaluated 2 weeks after the initial appointment, and minor improvement in the oral verrucous hyperplasia was noted following antifungal therapy, with resolution of the candidiasis.