User login
Lucas Franki is an associate editor for MDedge News, and has been with the company since 2014. He has a BA in English from Penn State University and is an Eagle Scout.
FDA approves mAb combo for hepatocellular carcinoma
The approval was supported by results from the IMbrave150 trial (N Engl J Med. 2020;382:1894-1905). This phase 3 trial enrolled 501 patients with hepatocellular carcinoma who were randomized to receive either sorafenib or atezolizumab plus bevacizumab.
The median overall survival was not reached in patients who received atezolizumab plus bevacizumab, but it was 13.2 months in patients who received sorafenib (hazard ratio, 0.58; 95% confidence interval, 0.42-0.79; P = .0006). The median progression-free survival was 6.8 months in patients who received atezolizumab plus bevacizumab and 4.3 months for those who received sorafenib.
The most common adverse events seen in the atezolizumab-bevacizumab arm were hypertension, fatigue, and proteinuria.
The recommended atezolizumab dose is 1,200 mg, followed by 15 mg/kg bevacizumab on the same day every 3 weeks.
The FDA collaborated with regulatory agencies from Canada, Australia, and Singapore on the review of the atezolizumab application, as part of Project Orbis. The FDA approved the application ahead of schedule. It is still under review for the other agencies.
The approval was supported by results from the IMbrave150 trial (N Engl J Med. 2020;382:1894-1905). This phase 3 trial enrolled 501 patients with hepatocellular carcinoma who were randomized to receive either sorafenib or atezolizumab plus bevacizumab.
The median overall survival was not reached in patients who received atezolizumab plus bevacizumab, but it was 13.2 months in patients who received sorafenib (hazard ratio, 0.58; 95% confidence interval, 0.42-0.79; P = .0006). The median progression-free survival was 6.8 months in patients who received atezolizumab plus bevacizumab and 4.3 months for those who received sorafenib.
The most common adverse events seen in the atezolizumab-bevacizumab arm were hypertension, fatigue, and proteinuria.
The recommended atezolizumab dose is 1,200 mg, followed by 15 mg/kg bevacizumab on the same day every 3 weeks.
The FDA collaborated with regulatory agencies from Canada, Australia, and Singapore on the review of the atezolizumab application, as part of Project Orbis. The FDA approved the application ahead of schedule. It is still under review for the other agencies.
The approval was supported by results from the IMbrave150 trial (N Engl J Med. 2020;382:1894-1905). This phase 3 trial enrolled 501 patients with hepatocellular carcinoma who were randomized to receive either sorafenib or atezolizumab plus bevacizumab.
The median overall survival was not reached in patients who received atezolizumab plus bevacizumab, but it was 13.2 months in patients who received sorafenib (hazard ratio, 0.58; 95% confidence interval, 0.42-0.79; P = .0006). The median progression-free survival was 6.8 months in patients who received atezolizumab plus bevacizumab and 4.3 months for those who received sorafenib.
The most common adverse events seen in the atezolizumab-bevacizumab arm were hypertension, fatigue, and proteinuria.
The recommended atezolizumab dose is 1,200 mg, followed by 15 mg/kg bevacizumab on the same day every 3 weeks.
The FDA collaborated with regulatory agencies from Canada, Australia, and Singapore on the review of the atezolizumab application, as part of Project Orbis. The FDA approved the application ahead of schedule. It is still under review for the other agencies.
Aripiprazole lauroxil rivals paliperidone palmitate in schizophrenia
PANSS scores prove comparable for newly hospitalized patients
Aripiprazole lauroxil (AL) and paliperidone palmitate (PP) are effective and well tolerated for initiating treatment of schizophrenia in the hospital and for continuing outpatient treatment, results of a phase 3, double-blind trial show.
A total of 200 patients completed the study, 99 of whom received aripiprazole lauroxil (Aristada Initio) and 101 of whom received paliperidone palmitate (Invega Sustenna). The baseline Positive and Negative Syndrome scale (PANSS) total score for those who received aripiprazole lauroxil was 94.1 and 94.6 for those who received paliperidone palmitate, Peter J. Weiden, MD, of Alkermes, and his associates reported in the Journal of Clinical Psychiatry.
After 4 weeks of treatment, patients who received aripiprazole lauroxil saw a mean PANSS score reduction of 17.4 points; after 9 weeks the reduction was 19.8 points and was 23.3 points after 25 weeks. For patients who received paliperidone palmitate the reduction in PANSS score was 20.1 points at week 4, 22.5 points at week 9, and 21.7 points at week 25. The most common adverse events in both groups were injection-site pain (17.2% and 24.8%, respectively), increased weight (9.1% and 16.8%, respectively), and akathisia (9.1% and 10.9%, respectively).
“Acutely symptomatic patients with schizophrenia on a 2-month dose regimen of AL with the 1-day initiation regimen demonstrated safety and efficacy consistent with that seen in prior AL studies. ... The inclusion of PP provided an active control with known safety and efficacy that can also be rapidly initiated. the investigators concluded.
The study was sponsored by Alkermes, manufacturer of Aristada Initio. Invega Sustenna is produced by Janssen Pharmaceuticals. The investigators reported being employed by, owning stock/options in, or receiving research support from Alkermes; two investigators reported receiving research support from numerous other companies.
SOURCE: Weiden PJ et al. J Clin Psychiatry. 2020 May 19. doi: 10.4088/JCP.19m13207.
PANSS scores prove comparable for newly hospitalized patients
PANSS scores prove comparable for newly hospitalized patients
Aripiprazole lauroxil (AL) and paliperidone palmitate (PP) are effective and well tolerated for initiating treatment of schizophrenia in the hospital and for continuing outpatient treatment, results of a phase 3, double-blind trial show.
A total of 200 patients completed the study, 99 of whom received aripiprazole lauroxil (Aristada Initio) and 101 of whom received paliperidone palmitate (Invega Sustenna). The baseline Positive and Negative Syndrome scale (PANSS) total score for those who received aripiprazole lauroxil was 94.1 and 94.6 for those who received paliperidone palmitate, Peter J. Weiden, MD, of Alkermes, and his associates reported in the Journal of Clinical Psychiatry.
After 4 weeks of treatment, patients who received aripiprazole lauroxil saw a mean PANSS score reduction of 17.4 points; after 9 weeks the reduction was 19.8 points and was 23.3 points after 25 weeks. For patients who received paliperidone palmitate the reduction in PANSS score was 20.1 points at week 4, 22.5 points at week 9, and 21.7 points at week 25. The most common adverse events in both groups were injection-site pain (17.2% and 24.8%, respectively), increased weight (9.1% and 16.8%, respectively), and akathisia (9.1% and 10.9%, respectively).
“Acutely symptomatic patients with schizophrenia on a 2-month dose regimen of AL with the 1-day initiation regimen demonstrated safety and efficacy consistent with that seen in prior AL studies. ... The inclusion of PP provided an active control with known safety and efficacy that can also be rapidly initiated. the investigators concluded.
The study was sponsored by Alkermes, manufacturer of Aristada Initio. Invega Sustenna is produced by Janssen Pharmaceuticals. The investigators reported being employed by, owning stock/options in, or receiving research support from Alkermes; two investigators reported receiving research support from numerous other companies.
SOURCE: Weiden PJ et al. J Clin Psychiatry. 2020 May 19. doi: 10.4088/JCP.19m13207.
Aripiprazole lauroxil (AL) and paliperidone palmitate (PP) are effective and well tolerated for initiating treatment of schizophrenia in the hospital and for continuing outpatient treatment, results of a phase 3, double-blind trial show.
A total of 200 patients completed the study, 99 of whom received aripiprazole lauroxil (Aristada Initio) and 101 of whom received paliperidone palmitate (Invega Sustenna). The baseline Positive and Negative Syndrome scale (PANSS) total score for those who received aripiprazole lauroxil was 94.1 and 94.6 for those who received paliperidone palmitate, Peter J. Weiden, MD, of Alkermes, and his associates reported in the Journal of Clinical Psychiatry.
After 4 weeks of treatment, patients who received aripiprazole lauroxil saw a mean PANSS score reduction of 17.4 points; after 9 weeks the reduction was 19.8 points and was 23.3 points after 25 weeks. For patients who received paliperidone palmitate the reduction in PANSS score was 20.1 points at week 4, 22.5 points at week 9, and 21.7 points at week 25. The most common adverse events in both groups were injection-site pain (17.2% and 24.8%, respectively), increased weight (9.1% and 16.8%, respectively), and akathisia (9.1% and 10.9%, respectively).
“Acutely symptomatic patients with schizophrenia on a 2-month dose regimen of AL with the 1-day initiation regimen demonstrated safety and efficacy consistent with that seen in prior AL studies. ... The inclusion of PP provided an active control with known safety and efficacy that can also be rapidly initiated. the investigators concluded.
The study was sponsored by Alkermes, manufacturer of Aristada Initio. Invega Sustenna is produced by Janssen Pharmaceuticals. The investigators reported being employed by, owning stock/options in, or receiving research support from Alkermes; two investigators reported receiving research support from numerous other companies.
SOURCE: Weiden PJ et al. J Clin Psychiatry. 2020 May 19. doi: 10.4088/JCP.19m13207.
FROM THE JOURNAL OF CLINICAL PSYCHIATRY
Hyperkalemia most common adverse event in women taking spironolactone
, according to new research.

Spironolactone, which is approved to treat heart failure, hypertension, edema, and primary hyperaldosteronism, has antagonistic effects on progesterone and androgen receptors and has been used as an off-label treatment for acne in women. “Numerous guidelines have recommended its off-label use for acne therapy to avoid antibiotic resistance and potential side effects,” wrote Yu Wang of Stony Brook (N.Y.) University and Shari R. Lipner MD, PhD, of Weill Cornell Medicine, New York. Their report is in the International Journal of Women’s Dermatology.
In a retrospective study, the investigators analyzed 7,920 adverse events with spironolactone reported by women of all ages between Jan. 1, 1969, and Dec. 30, 2018, to the Food and Drug Administration’s Adverse Event Reporting System database, for all indications. The most common adverse event was hyperkalemia, reported in 16.1%, followed by kidney injury (15.2%) and drug interactions (9%). Of the 1,272 cases of hyperkalemia reported, 25 occurred in women aged 45 years or younger; 59.3% occurred in women aged 65-85 years.
While spironolactone prescribing information was not available, the investigators compared yearly reports of adverse events with annual public interest in spironolactone using the Google Trends search term spironolactone and annual scholarly mentions of spironolactone in the Altmetric database. There was a strong correlation between the number of cases reported to the FDA and the Google Trends search (Spearman coefficient, 0.94; P less than .001) and to the Altmetric database (Spearman coefficient, 0.64; P less than .01).
Noting that hyperkalemia is “exceptionally uncommon” in women aged 45 years and younger, the investigators concluded that “in the absence of risk factors for hyperkalemia or reduced renal function, potassium laboratory monitoring is unnecessary in younger females taking spironolactone.” Because the incidence increases with age, “interval laboratory monitoring is recommended for females older than 45 years old,” they noted.
Limitations of the study, they noted, include the retrospective design and no available data before 1969. “In addition, since the [FDA Adverse Event Reporting System] data does not differentiate whether spironolactone was prescribed for heart failure, hypertension, edema, primary hyperaldosteronism, or for acne,” the study could not control for these or other confounding comorbidities or associated therapies.
“For future studies, it is important to analyze drug interactions more carefully to determine which other medications may potentiate the risk for hyperkalemia in patients taking spironolactone. It is also important to quantitate overall U.S. prescription data to better understand the relative frequency of these adverse effects reported to the FDA,” they wrote.
The investigators reported that they had no conflicts of interest; the study had no funding.
SOURCE: Wang Y, Lipner SR. Int J Womens Dermatol. 2020 May 18. doi: 10.1016/j.ijwd.2020.05.002.
, according to new research.
Spironolactone, which is approved to treat heart failure, hypertension, edema, and primary hyperaldosteronism, has antagonistic effects on progesterone and androgen receptors and has been used as an off-label treatment for acne in women. “Numerous guidelines have recommended its off-label use for acne therapy to avoid antibiotic resistance and potential side effects,” wrote Yu Wang of Stony Brook (N.Y.) University and Shari R. Lipner MD, PhD, of Weill Cornell Medicine, New York. Their report is in the International Journal of Women’s Dermatology.
In a retrospective study, the investigators analyzed 7,920 adverse events with spironolactone reported by women of all ages between Jan. 1, 1969, and Dec. 30, 2018, to the Food and Drug Administration’s Adverse Event Reporting System database, for all indications. The most common adverse event was hyperkalemia, reported in 16.1%, followed by kidney injury (15.2%) and drug interactions (9%). Of the 1,272 cases of hyperkalemia reported, 25 occurred in women aged 45 years or younger; 59.3% occurred in women aged 65-85 years.
While spironolactone prescribing information was not available, the investigators compared yearly reports of adverse events with annual public interest in spironolactone using the Google Trends search term spironolactone and annual scholarly mentions of spironolactone in the Altmetric database. There was a strong correlation between the number of cases reported to the FDA and the Google Trends search (Spearman coefficient, 0.94; P less than .001) and to the Altmetric database (Spearman coefficient, 0.64; P less than .01).
Noting that hyperkalemia is “exceptionally uncommon” in women aged 45 years and younger, the investigators concluded that “in the absence of risk factors for hyperkalemia or reduced renal function, potassium laboratory monitoring is unnecessary in younger females taking spironolactone.” Because the incidence increases with age, “interval laboratory monitoring is recommended for females older than 45 years old,” they noted.
Limitations of the study, they noted, include the retrospective design and no available data before 1969. “In addition, since the [FDA Adverse Event Reporting System] data does not differentiate whether spironolactone was prescribed for heart failure, hypertension, edema, primary hyperaldosteronism, or for acne,” the study could not control for these or other confounding comorbidities or associated therapies.
“For future studies, it is important to analyze drug interactions more carefully to determine which other medications may potentiate the risk for hyperkalemia in patients taking spironolactone. It is also important to quantitate overall U.S. prescription data to better understand the relative frequency of these adverse effects reported to the FDA,” they wrote.
The investigators reported that they had no conflicts of interest; the study had no funding.
SOURCE: Wang Y, Lipner SR. Int J Womens Dermatol. 2020 May 18. doi: 10.1016/j.ijwd.2020.05.002.
, according to new research.
Spironolactone, which is approved to treat heart failure, hypertension, edema, and primary hyperaldosteronism, has antagonistic effects on progesterone and androgen receptors and has been used as an off-label treatment for acne in women. “Numerous guidelines have recommended its off-label use for acne therapy to avoid antibiotic resistance and potential side effects,” wrote Yu Wang of Stony Brook (N.Y.) University and Shari R. Lipner MD, PhD, of Weill Cornell Medicine, New York. Their report is in the International Journal of Women’s Dermatology.
In a retrospective study, the investigators analyzed 7,920 adverse events with spironolactone reported by women of all ages between Jan. 1, 1969, and Dec. 30, 2018, to the Food and Drug Administration’s Adverse Event Reporting System database, for all indications. The most common adverse event was hyperkalemia, reported in 16.1%, followed by kidney injury (15.2%) and drug interactions (9%). Of the 1,272 cases of hyperkalemia reported, 25 occurred in women aged 45 years or younger; 59.3% occurred in women aged 65-85 years.
While spironolactone prescribing information was not available, the investigators compared yearly reports of adverse events with annual public interest in spironolactone using the Google Trends search term spironolactone and annual scholarly mentions of spironolactone in the Altmetric database. There was a strong correlation between the number of cases reported to the FDA and the Google Trends search (Spearman coefficient, 0.94; P less than .001) and to the Altmetric database (Spearman coefficient, 0.64; P less than .01).
Noting that hyperkalemia is “exceptionally uncommon” in women aged 45 years and younger, the investigators concluded that “in the absence of risk factors for hyperkalemia or reduced renal function, potassium laboratory monitoring is unnecessary in younger females taking spironolactone.” Because the incidence increases with age, “interval laboratory monitoring is recommended for females older than 45 years old,” they noted.
Limitations of the study, they noted, include the retrospective design and no available data before 1969. “In addition, since the [FDA Adverse Event Reporting System] data does not differentiate whether spironolactone was prescribed for heart failure, hypertension, edema, primary hyperaldosteronism, or for acne,” the study could not control for these or other confounding comorbidities or associated therapies.
“For future studies, it is important to analyze drug interactions more carefully to determine which other medications may potentiate the risk for hyperkalemia in patients taking spironolactone. It is also important to quantitate overall U.S. prescription data to better understand the relative frequency of these adverse effects reported to the FDA,” they wrote.
The investigators reported that they had no conflicts of interest; the study had no funding.
SOURCE: Wang Y, Lipner SR. Int J Womens Dermatol. 2020 May 18. doi: 10.1016/j.ijwd.2020.05.002.
FROM THE INTERNATIONAL JOURNAL OF WOMEN’S DERMATOLOGY
FDA approves Phexxi for use as an on-demand contraceptive

Evofem Biosciences expects to release Phexxi – the first nonhormonal, on-demand, vaginal pH regulator contraceptive designed to maintain vaginal pH within the range of 3.5-4.5 – in September 2020 alongside the Phexxi Concierge Experience, a comprehensive patient and health care provider telemedicine support system, according to the company’s press release. The service is designed to provide physicians with on-demand educational support, and to speed and simplify women’s access to Phexxi.
In an open-label multicenter trial, women aged 18-35 with regular menstrual cycles intravaginally administered a 5-gram dose of Phexxi vaginal gel up to 1 hour prior to intercourse; they did so for up to seven cycles. There were 101 pregnancies in 1,183 subjects during 4,769 cycles. The 7-cycle cumulative pregnancy rate was 14% (95% confidence interval: 10.0%, 17.5%).
The most common adverse events associated with Phexxi were vulvovaginal burning sensation, vulvovaginal pruritus, vulvovaginal mycotic infection, urinary tract infection, bacterial vaginosis, vaginal discharge, dysuria, and vulvovaginal pain.
Evofem Biosciences expects to release Phexxi – the first nonhormonal, on-demand, vaginal pH regulator contraceptive designed to maintain vaginal pH within the range of 3.5-4.5 – in September 2020 alongside the Phexxi Concierge Experience, a comprehensive patient and health care provider telemedicine support system, according to the company’s press release. The service is designed to provide physicians with on-demand educational support, and to speed and simplify women’s access to Phexxi.
In an open-label multicenter trial, women aged 18-35 with regular menstrual cycles intravaginally administered a 5-gram dose of Phexxi vaginal gel up to 1 hour prior to intercourse; they did so for up to seven cycles. There were 101 pregnancies in 1,183 subjects during 4,769 cycles. The 7-cycle cumulative pregnancy rate was 14% (95% confidence interval: 10.0%, 17.5%).
The most common adverse events associated with Phexxi were vulvovaginal burning sensation, vulvovaginal pruritus, vulvovaginal mycotic infection, urinary tract infection, bacterial vaginosis, vaginal discharge, dysuria, and vulvovaginal pain.
Evofem Biosciences expects to release Phexxi – the first nonhormonal, on-demand, vaginal pH regulator contraceptive designed to maintain vaginal pH within the range of 3.5-4.5 – in September 2020 alongside the Phexxi Concierge Experience, a comprehensive patient and health care provider telemedicine support system, according to the company’s press release. The service is designed to provide physicians with on-demand educational support, and to speed and simplify women’s access to Phexxi.
In an open-label multicenter trial, women aged 18-35 with regular menstrual cycles intravaginally administered a 5-gram dose of Phexxi vaginal gel up to 1 hour prior to intercourse; they did so for up to seven cycles. There were 101 pregnancies in 1,183 subjects during 4,769 cycles. The 7-cycle cumulative pregnancy rate was 14% (95% confidence interval: 10.0%, 17.5%).
The most common adverse events associated with Phexxi were vulvovaginal burning sensation, vulvovaginal pruritus, vulvovaginal mycotic infection, urinary tract infection, bacterial vaginosis, vaginal discharge, dysuria, and vulvovaginal pain.
Before pandemic, gastroenterologist earnings were holding steady
COVID-19 has changed many things in the medical landscape as practices have closed, many physicians are transitioning to telemedicine, and emergency departments struggle to provide safe environments for their employees.
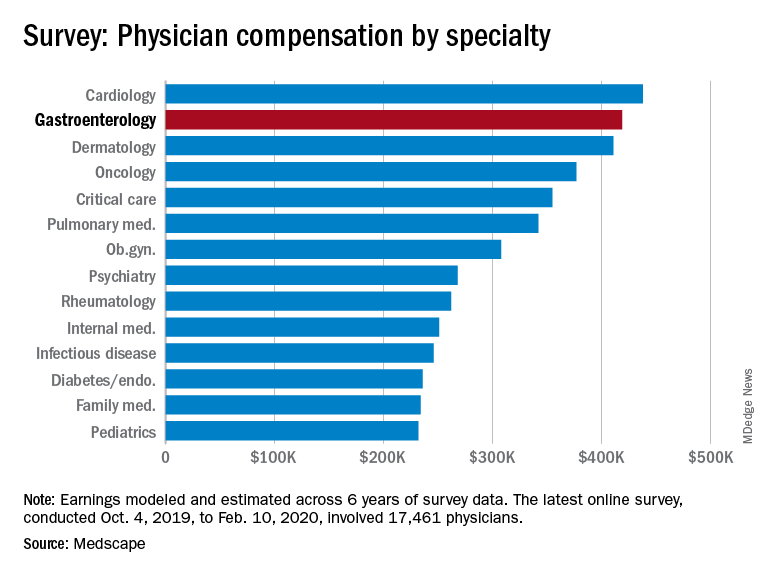
Medscape’s latest physician survey, conducted from Oct. 4, 2019, to Feb. 10, 2020, illustrates what gastroenterology looked like just before the coronavirus arrived.
While the average gastroenterologist salary did rise from 2019, the increase was minimal, going from $417,000 in 2019 to $419,000 in 2020, a 0.48% increase. In comparison, average income for all specialists was $346,000 in this year’s survey, up by 1.5% from the $341,000 earned in 2019, Medscape reported.
Prospects for next year, however, are grim. “We found out that we have a 10% salary decrease effective May 2 to Dec. 25. Our bonus will be based on clinical productivity, and since our numbers are down, that is likely to go away,” a pediatric emergency physician told Medscape.
Many gastroenterologists felt unfairly compensated in 2020, with only 52% reporting that they were satisfied with their salary. This was on the lower end of the 29 specialties included in the survey, which ranged from nephrology at 44% to oncology, emergency medicine, and radiology at 67%.
There was a notable disparity in the number of hours men spent seeing patients in comparison with women – while female GIs worked 38.3 hours a week, male GIs worked 42.5 hours. The average specialist saw patients 38 hours a week. This disparity in hours also translated into a notably higher salary for men: $430,000 versus $375,000. The number of hours gastroenterologists spent on paperwork and administration was roughly middle of the pack at 14.3 hours a week.
In the end, 80% of gastroenterologists said that they would choose to practice medicine again, compared with 77% for all physicians, and 91% said that they would choose gastroenterology again.
The respondents were Medscape members who had been invited to participate. The sample size was 17,461 physicians, and compensation was modeled and estimated based on a range of variables across 6 years of survey data. The sampling error was ±0.74%.
COVID-19 has changed many things in the medical landscape as practices have closed, many physicians are transitioning to telemedicine, and emergency departments struggle to provide safe environments for their employees.
Medscape’s latest physician survey, conducted from Oct. 4, 2019, to Feb. 10, 2020, illustrates what gastroenterology looked like just before the coronavirus arrived.
While the average gastroenterologist salary did rise from 2019, the increase was minimal, going from $417,000 in 2019 to $419,000 in 2020, a 0.48% increase. In comparison, average income for all specialists was $346,000 in this year’s survey, up by 1.5% from the $341,000 earned in 2019, Medscape reported.
Prospects for next year, however, are grim. “We found out that we have a 10% salary decrease effective May 2 to Dec. 25. Our bonus will be based on clinical productivity, and since our numbers are down, that is likely to go away,” a pediatric emergency physician told Medscape.
Many gastroenterologists felt unfairly compensated in 2020, with only 52% reporting that they were satisfied with their salary. This was on the lower end of the 29 specialties included in the survey, which ranged from nephrology at 44% to oncology, emergency medicine, and radiology at 67%.
There was a notable disparity in the number of hours men spent seeing patients in comparison with women – while female GIs worked 38.3 hours a week, male GIs worked 42.5 hours. The average specialist saw patients 38 hours a week. This disparity in hours also translated into a notably higher salary for men: $430,000 versus $375,000. The number of hours gastroenterologists spent on paperwork and administration was roughly middle of the pack at 14.3 hours a week.
In the end, 80% of gastroenterologists said that they would choose to practice medicine again, compared with 77% for all physicians, and 91% said that they would choose gastroenterology again.
The respondents were Medscape members who had been invited to participate. The sample size was 17,461 physicians, and compensation was modeled and estimated based on a range of variables across 6 years of survey data. The sampling error was ±0.74%.
COVID-19 has changed many things in the medical landscape as practices have closed, many physicians are transitioning to telemedicine, and emergency departments struggle to provide safe environments for their employees.
Medscape’s latest physician survey, conducted from Oct. 4, 2019, to Feb. 10, 2020, illustrates what gastroenterology looked like just before the coronavirus arrived.
While the average gastroenterologist salary did rise from 2019, the increase was minimal, going from $417,000 in 2019 to $419,000 in 2020, a 0.48% increase. In comparison, average income for all specialists was $346,000 in this year’s survey, up by 1.5% from the $341,000 earned in 2019, Medscape reported.
Prospects for next year, however, are grim. “We found out that we have a 10% salary decrease effective May 2 to Dec. 25. Our bonus will be based on clinical productivity, and since our numbers are down, that is likely to go away,” a pediatric emergency physician told Medscape.
Many gastroenterologists felt unfairly compensated in 2020, with only 52% reporting that they were satisfied with their salary. This was on the lower end of the 29 specialties included in the survey, which ranged from nephrology at 44% to oncology, emergency medicine, and radiology at 67%.
There was a notable disparity in the number of hours men spent seeing patients in comparison with women – while female GIs worked 38.3 hours a week, male GIs worked 42.5 hours. The average specialist saw patients 38 hours a week. This disparity in hours also translated into a notably higher salary for men: $430,000 versus $375,000. The number of hours gastroenterologists spent on paperwork and administration was roughly middle of the pack at 14.3 hours a week.
In the end, 80% of gastroenterologists said that they would choose to practice medicine again, compared with 77% for all physicians, and 91% said that they would choose gastroenterology again.
The respondents were Medscape members who had been invited to participate. The sample size was 17,461 physicians, and compensation was modeled and estimated based on a range of variables across 6 years of survey data. The sampling error was ±0.74%.
Before pandemic, rheumatologists saw small salary increase
COVID-19 has changed many things in the medical landscape as practices have closed, many physicians are transitioning to telemedicine, and EDs struggle to provide safe environments for their employees.
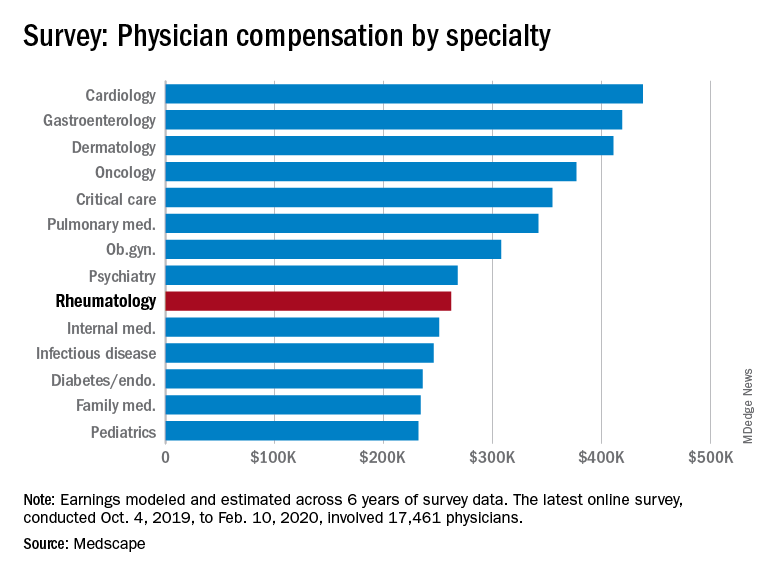
Medscape’s latest physician survey, conducted from Oct. 4, 2019, to Feb. 10, 2020, illustrates what rheumatology looked like just before the coronavirus arrived.
Rheumatologists saw a small increase in average salary in 2020, rising from $259,000 in 2019 to $262,000, an increase of 1.16%. In comparison, average income for all specialists was $346,000 in this year’s survey, up by 1.5% from the $341,000 earned in 2019, Medscape reported. Male rheumatologists earned significantly more than women at $288,000 versus $240,000.
Prospects for next year, however, are grim. “We found out that we have a 10% salary decrease effective May 2 to Dec. 25. Our bonus will be based on clinical productivity, and since our numbers are down, that is likely to go away,” a pediatric emergency physician told Medscape.
Before the pandemic, 55% of rheumatologists felt they were fairly compensated. This was about average among the 29 specialties included in the survey, which ranged from nephrology at 44% to oncology, emergency medicine, and radiology at 67%.
Rheumatologists were more likely than the average physician to report that “having so many rules and regulations” was the most challenging part of their job, according to the survey. A similar number of rheumatologists said that “gratitude/relationships with patients” was the most rewarding part of the job at 27%.
When asked if they would choose medicine again, 79% of rheumatologists said yes, slightly more than the 77% for all physicians; 81% said that they’d choose rheumatology again.
The respondents were Medscape members who had been invited to participate. The sample size was 17,461 physicians, and compensation was modeled and estimated based on a range of variables across 6 years of survey data. The sampling error was ±0.74%.
COVID-19 has changed many things in the medical landscape as practices have closed, many physicians are transitioning to telemedicine, and EDs struggle to provide safe environments for their employees.
Medscape’s latest physician survey, conducted from Oct. 4, 2019, to Feb. 10, 2020, illustrates what rheumatology looked like just before the coronavirus arrived.
Rheumatologists saw a small increase in average salary in 2020, rising from $259,000 in 2019 to $262,000, an increase of 1.16%. In comparison, average income for all specialists was $346,000 in this year’s survey, up by 1.5% from the $341,000 earned in 2019, Medscape reported. Male rheumatologists earned significantly more than women at $288,000 versus $240,000.
Prospects for next year, however, are grim. “We found out that we have a 10% salary decrease effective May 2 to Dec. 25. Our bonus will be based on clinical productivity, and since our numbers are down, that is likely to go away,” a pediatric emergency physician told Medscape.
Before the pandemic, 55% of rheumatologists felt they were fairly compensated. This was about average among the 29 specialties included in the survey, which ranged from nephrology at 44% to oncology, emergency medicine, and radiology at 67%.
Rheumatologists were more likely than the average physician to report that “having so many rules and regulations” was the most challenging part of their job, according to the survey. A similar number of rheumatologists said that “gratitude/relationships with patients” was the most rewarding part of the job at 27%.
When asked if they would choose medicine again, 79% of rheumatologists said yes, slightly more than the 77% for all physicians; 81% said that they’d choose rheumatology again.
The respondents were Medscape members who had been invited to participate. The sample size was 17,461 physicians, and compensation was modeled and estimated based on a range of variables across 6 years of survey data. The sampling error was ±0.74%.
COVID-19 has changed many things in the medical landscape as practices have closed, many physicians are transitioning to telemedicine, and EDs struggle to provide safe environments for their employees.
Medscape’s latest physician survey, conducted from Oct. 4, 2019, to Feb. 10, 2020, illustrates what rheumatology looked like just before the coronavirus arrived.
Rheumatologists saw a small increase in average salary in 2020, rising from $259,000 in 2019 to $262,000, an increase of 1.16%. In comparison, average income for all specialists was $346,000 in this year’s survey, up by 1.5% from the $341,000 earned in 2019, Medscape reported. Male rheumatologists earned significantly more than women at $288,000 versus $240,000.
Prospects for next year, however, are grim. “We found out that we have a 10% salary decrease effective May 2 to Dec. 25. Our bonus will be based on clinical productivity, and since our numbers are down, that is likely to go away,” a pediatric emergency physician told Medscape.
Before the pandemic, 55% of rheumatologists felt they were fairly compensated. This was about average among the 29 specialties included in the survey, which ranged from nephrology at 44% to oncology, emergency medicine, and radiology at 67%.
Rheumatologists were more likely than the average physician to report that “having so many rules and regulations” was the most challenging part of their job, according to the survey. A similar number of rheumatologists said that “gratitude/relationships with patients” was the most rewarding part of the job at 27%.
When asked if they would choose medicine again, 79% of rheumatologists said yes, slightly more than the 77% for all physicians; 81% said that they’d choose rheumatology again.
The respondents were Medscape members who had been invited to participate. The sample size was 17,461 physicians, and compensation was modeled and estimated based on a range of variables across 6 years of survey data. The sampling error was ±0.74%.
FDA approves Fensolvi for central precocious puberty treatment

Approval was based on results from a multicenter, open-label, single-arm, phase 3 study of 64 children with central precocious puberty, a rare disease described as onset of puberty before age 8 years in girls and before age 9 in boys. The primary study endpoint was achieved, with 87% of children achieving a serum luteinizing-hormone concentration of less than 4 IU/L within 6 months post injection. Sex hormones were suppressed to prepubertal levels, and clinical signs of puberty were halted or reversed.
Adverse events during the study were mostly mild or moderate; none led to withdrawal from the study. The most common adverse events reported were injection-site pain (31%), nasopharyngitis (22%), and fever (17%).
“Children with CPP require treatment for several years and missing treatment or stopping treatment too soon may lead to significant short stature and misalignment between chronological age and physical and emotional development. Fensolvi offers treating physicians and their patients with CPP a safe and effective treatment option that is administered twice a year with a small injection volume that has the potential to improve compliance,” Karen Klein, MD, of Rady Children’s Hospital in San Diego, said in the press release.
Approval was based on results from a multicenter, open-label, single-arm, phase 3 study of 64 children with central precocious puberty, a rare disease described as onset of puberty before age 8 years in girls and before age 9 in boys. The primary study endpoint was achieved, with 87% of children achieving a serum luteinizing-hormone concentration of less than 4 IU/L within 6 months post injection. Sex hormones were suppressed to prepubertal levels, and clinical signs of puberty were halted or reversed.
Adverse events during the study were mostly mild or moderate; none led to withdrawal from the study. The most common adverse events reported were injection-site pain (31%), nasopharyngitis (22%), and fever (17%).
“Children with CPP require treatment for several years and missing treatment or stopping treatment too soon may lead to significant short stature and misalignment between chronological age and physical and emotional development. Fensolvi offers treating physicians and their patients with CPP a safe and effective treatment option that is administered twice a year with a small injection volume that has the potential to improve compliance,” Karen Klein, MD, of Rady Children’s Hospital in San Diego, said in the press release.
Approval was based on results from a multicenter, open-label, single-arm, phase 3 study of 64 children with central precocious puberty, a rare disease described as onset of puberty before age 8 years in girls and before age 9 in boys. The primary study endpoint was achieved, with 87% of children achieving a serum luteinizing-hormone concentration of less than 4 IU/L within 6 months post injection. Sex hormones were suppressed to prepubertal levels, and clinical signs of puberty were halted or reversed.
Adverse events during the study were mostly mild or moderate; none led to withdrawal from the study. The most common adverse events reported were injection-site pain (31%), nasopharyngitis (22%), and fever (17%).
“Children with CPP require treatment for several years and missing treatment or stopping treatment too soon may lead to significant short stature and misalignment between chronological age and physical and emotional development. Fensolvi offers treating physicians and their patients with CPP a safe and effective treatment option that is administered twice a year with a small injection volume that has the potential to improve compliance,” Karen Klein, MD, of Rady Children’s Hospital in San Diego, said in the press release.
FDA approves hyaluronic acid filler for lip augmentation, perioral rhytids
, the manufacturer has announced.
Approval was supported by results of a phase 3 clinical trial in which a lower amount of Restylane Kysse was needed to see an improvement in lip fullness (1.82 mL) vs. a comparator (2.24 mL), according to the press release issued by Galderma. After 1 year, 78% of those who received the Restylane product were satisfied, and it was also shown to be safe and well tolerated, the release said.
In the statement, the company said that it is “working to determine the appropriate launch timing and availability” of this new product.
[email protected]
, the manufacturer has announced.
Approval was supported by results of a phase 3 clinical trial in which a lower amount of Restylane Kysse was needed to see an improvement in lip fullness (1.82 mL) vs. a comparator (2.24 mL), according to the press release issued by Galderma. After 1 year, 78% of those who received the Restylane product were satisfied, and it was also shown to be safe and well tolerated, the release said.
In the statement, the company said that it is “working to determine the appropriate launch timing and availability” of this new product.
[email protected]
, the manufacturer has announced.
Approval was supported by results of a phase 3 clinical trial in which a lower amount of Restylane Kysse was needed to see an improvement in lip fullness (1.82 mL) vs. a comparator (2.24 mL), according to the press release issued by Galderma. After 1 year, 78% of those who received the Restylane product were satisfied, and it was also shown to be safe and well tolerated, the release said.
In the statement, the company said that it is “working to determine the appropriate launch timing and availability” of this new product.
[email protected]
FDA grants EUA to muscle stimulator to reduce mechanical ventilator usage
The Food and Drug Administration has issued an Emergency Use Authorization (EUA) for the VentFree Respiratory Muscle Stimulator in order to potentially reduce the number of days adult patients, including those with COVID-19, require mechanical ventilation, according to a press release from Liberate Medical.
In comparison with mechanical ventilation, which is invasive and commonly weakens the breathing muscles, the VentFree system uses noninvasive neuromuscular electrical stimulation to contract the abdominal wall muscles in synchrony with exhalation during mechanical ventilation, according to the press release. This allows patients to begin treatment during the early stages of ventilation while they are sedated and to continue until they are weaned off of ventilation.
A pair of pilot randomized, controlled studies, completed in Europe and Australia, showed that VentFree helped to reduce ventilation duration and ICU length of stay, compared with placebo stimulation. The FDA granted VentFree Breakthrough Device status in 2019.
“We are grateful to the FDA for recognizing the potential of VentFree and feel privileged to have the opportunity to help patients on mechanical ventilation during the COVID-19 pandemic,” Angus McLachlan PhD, cofounder and CEO of Liberate Medical, said in the press release.
VentFree has been authorized for use only for the duration of the current COVID-19 emergency, as it has not yet been approved or cleared for usage by primary care providers.
The Food and Drug Administration has issued an Emergency Use Authorization (EUA) for the VentFree Respiratory Muscle Stimulator in order to potentially reduce the number of days adult patients, including those with COVID-19, require mechanical ventilation, according to a press release from Liberate Medical.
In comparison with mechanical ventilation, which is invasive and commonly weakens the breathing muscles, the VentFree system uses noninvasive neuromuscular electrical stimulation to contract the abdominal wall muscles in synchrony with exhalation during mechanical ventilation, according to the press release. This allows patients to begin treatment during the early stages of ventilation while they are sedated and to continue until they are weaned off of ventilation.
A pair of pilot randomized, controlled studies, completed in Europe and Australia, showed that VentFree helped to reduce ventilation duration and ICU length of stay, compared with placebo stimulation. The FDA granted VentFree Breakthrough Device status in 2019.
“We are grateful to the FDA for recognizing the potential of VentFree and feel privileged to have the opportunity to help patients on mechanical ventilation during the COVID-19 pandemic,” Angus McLachlan PhD, cofounder and CEO of Liberate Medical, said in the press release.
VentFree has been authorized for use only for the duration of the current COVID-19 emergency, as it has not yet been approved or cleared for usage by primary care providers.
The Food and Drug Administration has issued an Emergency Use Authorization (EUA) for the VentFree Respiratory Muscle Stimulator in order to potentially reduce the number of days adult patients, including those with COVID-19, require mechanical ventilation, according to a press release from Liberate Medical.
In comparison with mechanical ventilation, which is invasive and commonly weakens the breathing muscles, the VentFree system uses noninvasive neuromuscular electrical stimulation to contract the abdominal wall muscles in synchrony with exhalation during mechanical ventilation, according to the press release. This allows patients to begin treatment during the early stages of ventilation while they are sedated and to continue until they are weaned off of ventilation.
A pair of pilot randomized, controlled studies, completed in Europe and Australia, showed that VentFree helped to reduce ventilation duration and ICU length of stay, compared with placebo stimulation. The FDA granted VentFree Breakthrough Device status in 2019.
“We are grateful to the FDA for recognizing the potential of VentFree and feel privileged to have the opportunity to help patients on mechanical ventilation during the COVID-19 pandemic,” Angus McLachlan PhD, cofounder and CEO of Liberate Medical, said in the press release.
VentFree has been authorized for use only for the duration of the current COVID-19 emergency, as it has not yet been approved or cleared for usage by primary care providers.
FDA grants Breakthrough Therapy status to sotatercept for PAH treatment
Approval for sotatercept, “a selective ligand trap for members of the TGF-beta [transforming growth factor-beta] superfamily which rebalances BMPR-II [bone morphogenetic protein receptor type II] signaling,” was based on two types of research. It was based on results of preclinical research indicating “reversed pulmonary vessel muscularization and improved indicators of right heart failure,” as well as results of the phase 2, placebo-controlled PULSAR study, in which sotatercept showed positive results, meeting primary and secondary endpoints.
Adverse events during PULSAR “were consistent with previously published data on sotatercept” in other diseases. The drug is also under investigation in the phase 2 SPECTRA trial, which includes patients with PAH.
“We believe that sotatercept has the potential to shift the current treatment paradigm and provide significant benefit to patients with PAH on top of currently available therapies. Thus, we’re thrilled that the FDA has granted this Breakthrough Therapy designation – a first for an Acceleron-discovered medicine and for a therapeutic candidate in PAH – as it supports and aligns with our mission to deliver novel therapeutic options to patients in need as quickly as possible,” Habib Dable, president and CEO of Acceleron Pharma, said in the press release.
Approval for sotatercept, “a selective ligand trap for members of the TGF-beta [transforming growth factor-beta] superfamily which rebalances BMPR-II [bone morphogenetic protein receptor type II] signaling,” was based on two types of research. It was based on results of preclinical research indicating “reversed pulmonary vessel muscularization and improved indicators of right heart failure,” as well as results of the phase 2, placebo-controlled PULSAR study, in which sotatercept showed positive results, meeting primary and secondary endpoints.
Adverse events during PULSAR “were consistent with previously published data on sotatercept” in other diseases. The drug is also under investigation in the phase 2 SPECTRA trial, which includes patients with PAH.
“We believe that sotatercept has the potential to shift the current treatment paradigm and provide significant benefit to patients with PAH on top of currently available therapies. Thus, we’re thrilled that the FDA has granted this Breakthrough Therapy designation – a first for an Acceleron-discovered medicine and for a therapeutic candidate in PAH – as it supports and aligns with our mission to deliver novel therapeutic options to patients in need as quickly as possible,” Habib Dable, president and CEO of Acceleron Pharma, said in the press release.
Approval for sotatercept, “a selective ligand trap for members of the TGF-beta [transforming growth factor-beta] superfamily which rebalances BMPR-II [bone morphogenetic protein receptor type II] signaling,” was based on two types of research. It was based on results of preclinical research indicating “reversed pulmonary vessel muscularization and improved indicators of right heart failure,” as well as results of the phase 2, placebo-controlled PULSAR study, in which sotatercept showed positive results, meeting primary and secondary endpoints.
Adverse events during PULSAR “were consistent with previously published data on sotatercept” in other diseases. The drug is also under investigation in the phase 2 SPECTRA trial, which includes patients with PAH.
“We believe that sotatercept has the potential to shift the current treatment paradigm and provide significant benefit to patients with PAH on top of currently available therapies. Thus, we’re thrilled that the FDA has granted this Breakthrough Therapy designation – a first for an Acceleron-discovered medicine and for a therapeutic candidate in PAH – as it supports and aligns with our mission to deliver novel therapeutic options to patients in need as quickly as possible,” Habib Dable, president and CEO of Acceleron Pharma, said in the press release.