User login
Adjuvant-boosted shingles vaccine earns FDA panel’s unanimous nod
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
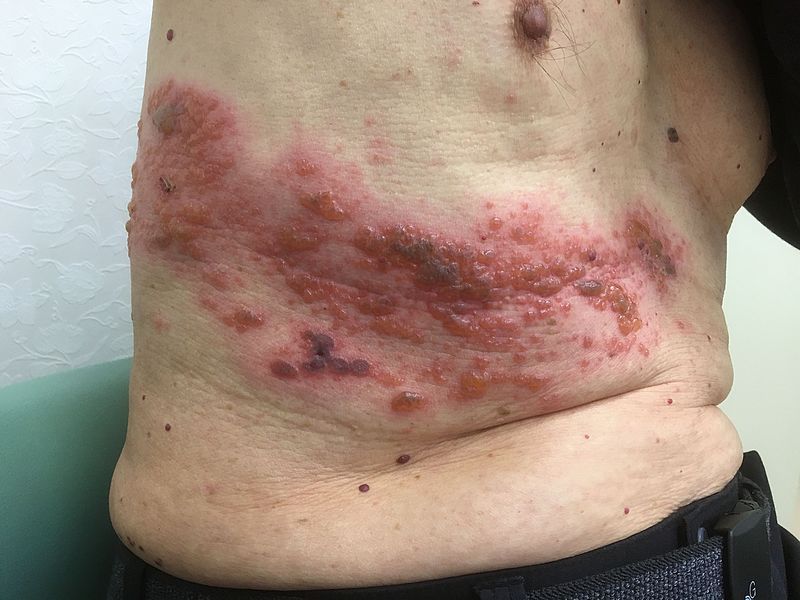
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
[email protected]
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
[email protected]
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
[email protected]
On Twitter @karioakes
Postpartum NSAIDs didn’t up hypertension risk in preeclampsia
Women with severe preeclampsia who received nonsteroidal anti-inflammatory drugs during the postpartum period had no greater risk of persistent postpartum hypertension than women who didn’t take them, a study showed.
Additionally, though the numbers of women affected were small, there was no increased risk of severe maternal morbidity. Rates of pulmonary hypertension, renal failure, eclampsia, or intensive care unit admission were similar between women who received NSAIDs during the postpartum period and women who did not.
The single-center retrospective cohort study examined the records of 399 women with severe preeclampsia, 324 of whom (81%) were still hypertensive 24 hours post delivery (Obstet Gynecol. 2017;130:830-5. doi: 10.1097/AOG.0000000000002247). Of this group, three-quarters (n = 243) received NSAIDs, while one-quarter (n = 81) did not.
After multivariable analysis, first author Oscar Viteri, MD, and his colleagues reported that 70% of patients who received NSAIDs had persistent postpartum hypertension, defined as a blood pressure of at least 150 mm Hg or a diastolic BP of at least 100 mm Hg obtained on two occasions at least 4 hours apart. This compared with a rate of 73% for the women who did not receive NSAIDs (adjusted odds ratio, 1.1; 95% confidence interval [CI], 0.6-2.0; P = .57).
Relatively small numbers of women in each group experienced severe morbidity, limiting statistical analysis of these secondary outcome measures. Just six women who received NSAIDs and eight who did not (3% and 10%) developed pulmonary edema (OR, 4.4; 95% CI, 1.5-13.1).
Renal dysfunction occurred in 5% of the NSAIDs users vs. 8% of the nonusers (OR, 1.7; 95% CI, 0.6-4.8), and eclampsia occurred in two patients who took NSAIDs and none of the nonusers. Of those who took NSAIDs, 3% had an intensive care unit admission, compared with 8% of those who did not take these drugs (OR 2.4; 95% CI, 0.8-7.1).
Dr. Viteri and his coauthors at the University of Texas Health Science Center, Houston, noted that a high proportion of women with severe preeclampsia received ibuprofen (40%), ketorolac (6%), or both (54%) during their postpartum hospital stay. This occurred despite a 2013 recommendation from the American College of Obstetricians and Gynecologists Task Force for Hypertension in Pregnancy urging clinicians to avoid NSAIDs in women with hypertension persisting for 24 hours post partum.
In nonpregnant women with hypertension who are taking beta-blockers or angiotensin-converting enzyme inhibitors, NSAIDs use has been associated with increased systolic and diastolic blood pressure, said Dr. Viteri and his colleagues. There are several plausible physiologic mechanisms for this effect, including increased renal sodium retention from inhibition of prostaglandin E2. This potential effect, in particular, may have implications for women in the puerperum, since 6-8 L of fluid are returned to the maternal intravascular space during the early postpartum period.
However, “evidence on the effects of NSAIDs in otherwise healthy puerperal women with preeclampsia before delivery remains conflicting,” the investigators wrote. This study helps to fill the knowledge gap, though there are some limitations, including the fact that the non-NSAIDs arm was small, leaving an unbalanced study that was underpowered to detect differences in “rare but clinically significant” severe maternal morbidity. Also, the study captured only the inpatient period; because the mean duration of hospital stay was 4.5 days, the study missed a portion of the window of fluid volume redistribution, which occurs mostly during postpartum days 3-6.
Still, the findings from this large retrospective study warrant an adequately powered clinical trial to settle the question of the safety of NSAIDs for women with preeclampsia, the investigators said.
Dr. Viteri and his colleagues reported having no relevant conflicts of interest.
Women with severe preeclampsia who received nonsteroidal anti-inflammatory drugs during the postpartum period had no greater risk of persistent postpartum hypertension than women who didn’t take them, a study showed.
Additionally, though the numbers of women affected were small, there was no increased risk of severe maternal morbidity. Rates of pulmonary hypertension, renal failure, eclampsia, or intensive care unit admission were similar between women who received NSAIDs during the postpartum period and women who did not.
The single-center retrospective cohort study examined the records of 399 women with severe preeclampsia, 324 of whom (81%) were still hypertensive 24 hours post delivery (Obstet Gynecol. 2017;130:830-5. doi: 10.1097/AOG.0000000000002247). Of this group, three-quarters (n = 243) received NSAIDs, while one-quarter (n = 81) did not.
After multivariable analysis, first author Oscar Viteri, MD, and his colleagues reported that 70% of patients who received NSAIDs had persistent postpartum hypertension, defined as a blood pressure of at least 150 mm Hg or a diastolic BP of at least 100 mm Hg obtained on two occasions at least 4 hours apart. This compared with a rate of 73% for the women who did not receive NSAIDs (adjusted odds ratio, 1.1; 95% confidence interval [CI], 0.6-2.0; P = .57).
Relatively small numbers of women in each group experienced severe morbidity, limiting statistical analysis of these secondary outcome measures. Just six women who received NSAIDs and eight who did not (3% and 10%) developed pulmonary edema (OR, 4.4; 95% CI, 1.5-13.1).
Renal dysfunction occurred in 5% of the NSAIDs users vs. 8% of the nonusers (OR, 1.7; 95% CI, 0.6-4.8), and eclampsia occurred in two patients who took NSAIDs and none of the nonusers. Of those who took NSAIDs, 3% had an intensive care unit admission, compared with 8% of those who did not take these drugs (OR 2.4; 95% CI, 0.8-7.1).
Dr. Viteri and his coauthors at the University of Texas Health Science Center, Houston, noted that a high proportion of women with severe preeclampsia received ibuprofen (40%), ketorolac (6%), or both (54%) during their postpartum hospital stay. This occurred despite a 2013 recommendation from the American College of Obstetricians and Gynecologists Task Force for Hypertension in Pregnancy urging clinicians to avoid NSAIDs in women with hypertension persisting for 24 hours post partum.
In nonpregnant women with hypertension who are taking beta-blockers or angiotensin-converting enzyme inhibitors, NSAIDs use has been associated with increased systolic and diastolic blood pressure, said Dr. Viteri and his colleagues. There are several plausible physiologic mechanisms for this effect, including increased renal sodium retention from inhibition of prostaglandin E2. This potential effect, in particular, may have implications for women in the puerperum, since 6-8 L of fluid are returned to the maternal intravascular space during the early postpartum period.
However, “evidence on the effects of NSAIDs in otherwise healthy puerperal women with preeclampsia before delivery remains conflicting,” the investigators wrote. This study helps to fill the knowledge gap, though there are some limitations, including the fact that the non-NSAIDs arm was small, leaving an unbalanced study that was underpowered to detect differences in “rare but clinically significant” severe maternal morbidity. Also, the study captured only the inpatient period; because the mean duration of hospital stay was 4.5 days, the study missed a portion of the window of fluid volume redistribution, which occurs mostly during postpartum days 3-6.
Still, the findings from this large retrospective study warrant an adequately powered clinical trial to settle the question of the safety of NSAIDs for women with preeclampsia, the investigators said.
Dr. Viteri and his colleagues reported having no relevant conflicts of interest.
Women with severe preeclampsia who received nonsteroidal anti-inflammatory drugs during the postpartum period had no greater risk of persistent postpartum hypertension than women who didn’t take them, a study showed.
Additionally, though the numbers of women affected were small, there was no increased risk of severe maternal morbidity. Rates of pulmonary hypertension, renal failure, eclampsia, or intensive care unit admission were similar between women who received NSAIDs during the postpartum period and women who did not.
The single-center retrospective cohort study examined the records of 399 women with severe preeclampsia, 324 of whom (81%) were still hypertensive 24 hours post delivery (Obstet Gynecol. 2017;130:830-5. doi: 10.1097/AOG.0000000000002247). Of this group, three-quarters (n = 243) received NSAIDs, while one-quarter (n = 81) did not.
After multivariable analysis, first author Oscar Viteri, MD, and his colleagues reported that 70% of patients who received NSAIDs had persistent postpartum hypertension, defined as a blood pressure of at least 150 mm Hg or a diastolic BP of at least 100 mm Hg obtained on two occasions at least 4 hours apart. This compared with a rate of 73% for the women who did not receive NSAIDs (adjusted odds ratio, 1.1; 95% confidence interval [CI], 0.6-2.0; P = .57).
Relatively small numbers of women in each group experienced severe morbidity, limiting statistical analysis of these secondary outcome measures. Just six women who received NSAIDs and eight who did not (3% and 10%) developed pulmonary edema (OR, 4.4; 95% CI, 1.5-13.1).
Renal dysfunction occurred in 5% of the NSAIDs users vs. 8% of the nonusers (OR, 1.7; 95% CI, 0.6-4.8), and eclampsia occurred in two patients who took NSAIDs and none of the nonusers. Of those who took NSAIDs, 3% had an intensive care unit admission, compared with 8% of those who did not take these drugs (OR 2.4; 95% CI, 0.8-7.1).
Dr. Viteri and his coauthors at the University of Texas Health Science Center, Houston, noted that a high proportion of women with severe preeclampsia received ibuprofen (40%), ketorolac (6%), or both (54%) during their postpartum hospital stay. This occurred despite a 2013 recommendation from the American College of Obstetricians and Gynecologists Task Force for Hypertension in Pregnancy urging clinicians to avoid NSAIDs in women with hypertension persisting for 24 hours post partum.
In nonpregnant women with hypertension who are taking beta-blockers or angiotensin-converting enzyme inhibitors, NSAIDs use has been associated with increased systolic and diastolic blood pressure, said Dr. Viteri and his colleagues. There are several plausible physiologic mechanisms for this effect, including increased renal sodium retention from inhibition of prostaglandin E2. This potential effect, in particular, may have implications for women in the puerperum, since 6-8 L of fluid are returned to the maternal intravascular space during the early postpartum period.
However, “evidence on the effects of NSAIDs in otherwise healthy puerperal women with preeclampsia before delivery remains conflicting,” the investigators wrote. This study helps to fill the knowledge gap, though there are some limitations, including the fact that the non-NSAIDs arm was small, leaving an unbalanced study that was underpowered to detect differences in “rare but clinically significant” severe maternal morbidity. Also, the study captured only the inpatient period; because the mean duration of hospital stay was 4.5 days, the study missed a portion of the window of fluid volume redistribution, which occurs mostly during postpartum days 3-6.
Still, the findings from this large retrospective study warrant an adequately powered clinical trial to settle the question of the safety of NSAIDs for women with preeclampsia, the investigators said.
Dr. Viteri and his colleagues reported having no relevant conflicts of interest.
FROM OBSTETRICS AND GYNECOLOGY
Key clinical point:
Major finding: In total, 70% of women with severe preeclampsia taking NSAIDs, and 73% of those who did not, had persistent postpartum hypertension.
Study details: A retrospective cohort study of 324 women with severe preeclampsia who remained hypertensive for more than 24 hours after delivery.
Disclosures: None of the study authors reported having relevant conflicts of interest.
Patient and physician outreach boost CRC screening rates
Can outreach improve the globally low rates of adherence to colorectal cancer screening? Yes, according to two recent studies in JAMA; the studies found that both patient-focused and physician-focused outreach approaches can result in significantly better patient participation in colorectal cancer (CRC) screening.
The first study (JAMA. 2017;318[9]:806-15) compared a colonoscopy outreach program and a fecal immunochemical test (FIT) outreach program both with each other and with usual care. The results of the pragmatic, single-site, randomized, clinical trial showed that completed screenings were higher for both outreach groups, compared with the usual care group.
The primary outcome measure of the study was completion of the screening process, wrote Amit Singal, MD, and his coauthors. This was defined as any adherence to colonoscopy completion, the completion of annual testing for patients who had a normal FIT test, or treatment evaluation if CRC was detected during the screening process. Screenings were considered complete even if, for example, a patient in the colonoscopy arm eventually went on to have three consecutive annual FIT tests rather than a colonoscopy.
A total of 5,999 patients eligible for screening were initially randomized to one of the three study arms. Across all study arms, approximately half were lost to follow-up. These patients were excluded from the primary analysis but were included in an additional intention-to-screen analysis. A total of 2,400 patients received a colonoscopy outreach mailing; 2,400 received FIT outreach, including a letter, the home FIT testing kit and instructions; 1,199 received usual care. Patients in both intervention arms also received up to two phone calls if they didn’t respond to the initial mailing within 2 weeks. Mailings and phone calls were conducted in English or Spanish, according to the patients’ stated language preferences (those whose spoke neither language were excluded from the study).
Of the patients in the colonoscopy outreach group, 922 (38.4%) completed the screening process, compared with 671 (28.0%) in the FIT outreach group and 128 (10.7%) in the usual care group.
Compared with the group receiving usual care, completion of the screening process was 27.7% higher in the colonoscopy outreach group and 17.3% higher in the FIT outreach group. Screening process completion was 10.4% higher for the colonoscopy outreach group, compared with the FIT outreach group (P less than .001 for all).
Dr. Singal, who is with the department of internal medicine at UT Southwestern Medical Center, Dallas, and his colleagues also performed several post-hoc secondary analyses. In one, they used a less-stringent definition of screening process completion in which biennial FIT testing was considered satisfactory. When this definition was applied, the colonoscopy outreach group had 0.5% lower screening process completion than the FIT outreach group. The chances of a patient receiving any screening during the study period was highest in the FIT group (65%), with 51.7% of those in the colonoscopy outreach group and 39% of those in the usual care group receiving any screening.
“FIT has lower barriers to one-time participation but requires annual screening and diagnostic evaluation of abnormal results,” wrote Dr. Singal and his colleagues.
Strengths of the study, said Dr. Singal and his colleagues, included the fact that the study took place at a “safety net” institution with a racially and socioeconomically diverse population. Also, the study design avoided volunteer bias, and offered a pragmatic head-to-head comparison of colonoscopy and FIT.
The second study took place in western France, and targeted outreach to physicians rather than patients (JAMA. 2017;318[9];816-84). When physicians were given a list of their own patients who were not up to date on CRC screening, investigators saw a small, but significant, uptick in patient participation in FIT screening.
One year after the reminders went out, FIT screening had been initiated in 24.8% of patients whose physicians had received the list, compared with 21.7% of patients of physicians who had received a more generic notice and 20.6% of patients whose physicians received no notification, according to first author Cedric Rat, MD, and his colleagues.
The study examined which notification approach was most effective in increasing FIT screening among the physicians’ patient panels: sending general practitioners (GPs) letters that included a list of their own patients who had not undergone CRC screening, or sending them generic letters describing CRC screening adherence rates specific to their region. A usual care group of practices received no notifications in this 3-group randomized cluster design.
Patients in the patient-specific reminders group had an odds ratio of 1.27 for participation in FIT screening (P less than .001) compared to the usual care group. The odds ratio for the generic reminders group was 1.09, a nonsignificant difference.
Between-group comparison showed statistical significance for both the 3.1% difference between the patient-specific and generic reminders groups, and for the 4.2% difference between the patient-specific and usual care groups (P less than .001 for both). There was no significant difference between the generic reminders group and the usual care group.
Dr. Rat, professor of medicine at the Faculty of Medicine, Nantes, France, and his colleagues enrolled GPs in a total of 801 practices that included patients aged 50participating GPs caring for 33,044 patients who met study criteria.
Physician characteristics that were associated with higher FIT participation included younger age and an initially smaller number of unscreened patients. Patients with low socioeconomic status and those with a higher chronic disease burden were less likely to participate in FIT screening.
Also, Dr. Rat and his colleagues noted that the busiest practices actually had higher CRC screening rates. The investigators hypothesized that a recent physician pay-for-performance grant for CRC completion might be more appealing for some busy physicians.
This was the largest study of CRC screening participation to date, according to Dr. Rat and his coauthors, and showed the small but detectable efficacy of an inexpensive intervention that, given complete patient records, is relatively easy to effect. Though the effect size was smaller than the 12% difference the investigators had anticipated seeing for the patient-specific reminders group, the study still showed that targeting physicians can be an effective public health intervention to increase CRC screening rates, said Dr. Rat and his colleagues.
None of the investigators in either study reported conflicts of interest.
Both studies, though they used different outreach interventions, highlight the same problem: the need to identify and execute effective colorectal cancer (CRC) screening programs. Effective screening has great lifesaving potential; if screening rates were elevated to greater than 80% in the United States, an estimated 200,000 deaths would be prevented within the next 2 decades.
The nature of CRC screening options means that a home fecal sample collection is inexpensive, and will result in an initial higher screening rate; however, complete screening via fecal occult blood testing requires annual repeats of negative tests, and patients with positive fecal occult blood tests still need colonoscopy.
Colonoscopy, although it’s burdensome for patients and perhaps cost prohibitive for those without health insurance, offers a one-time test that, if negative, provides patients with a 10-year window of screening coverage.
Any effective programs to increase CRC screening rates will need to use a systems change approach, with creative interventions that take patient education, and even delivery of preventive health services, out of the context of the already too-full office visit.
Staff supports, such as the follow-up telephone calls used in the patient-targeted intervention, are key to effective interventions, especially for vulnerable populations. Additionally, institutions must ensure that they have adequate physical and staff resources to support the increased screening they are seeking to achieve.
Dr. Michael Pignone is a professor of medicine at the University of Texas at Austin. Dr. David Miller is a professor of internal medicine, Wake Forest University, Winston-Salem, N.C. Dr. Pignone is a medical director for Healthwise; Dr. Miller reported no relevant conflicts of interest These remarks were drawn from an editorial accompanying the two clinical trials.
Both studies, though they used different outreach interventions, highlight the same problem: the need to identify and execute effective colorectal cancer (CRC) screening programs. Effective screening has great lifesaving potential; if screening rates were elevated to greater than 80% in the United States, an estimated 200,000 deaths would be prevented within the next 2 decades.
The nature of CRC screening options means that a home fecal sample collection is inexpensive, and will result in an initial higher screening rate; however, complete screening via fecal occult blood testing requires annual repeats of negative tests, and patients with positive fecal occult blood tests still need colonoscopy.
Colonoscopy, although it’s burdensome for patients and perhaps cost prohibitive for those without health insurance, offers a one-time test that, if negative, provides patients with a 10-year window of screening coverage.
Any effective programs to increase CRC screening rates will need to use a systems change approach, with creative interventions that take patient education, and even delivery of preventive health services, out of the context of the already too-full office visit.
Staff supports, such as the follow-up telephone calls used in the patient-targeted intervention, are key to effective interventions, especially for vulnerable populations. Additionally, institutions must ensure that they have adequate physical and staff resources to support the increased screening they are seeking to achieve.
Dr. Michael Pignone is a professor of medicine at the University of Texas at Austin. Dr. David Miller is a professor of internal medicine, Wake Forest University, Winston-Salem, N.C. Dr. Pignone is a medical director for Healthwise; Dr. Miller reported no relevant conflicts of interest These remarks were drawn from an editorial accompanying the two clinical trials.
Both studies, though they used different outreach interventions, highlight the same problem: the need to identify and execute effective colorectal cancer (CRC) screening programs. Effective screening has great lifesaving potential; if screening rates were elevated to greater than 80% in the United States, an estimated 200,000 deaths would be prevented within the next 2 decades.
The nature of CRC screening options means that a home fecal sample collection is inexpensive, and will result in an initial higher screening rate; however, complete screening via fecal occult blood testing requires annual repeats of negative tests, and patients with positive fecal occult blood tests still need colonoscopy.
Colonoscopy, although it’s burdensome for patients and perhaps cost prohibitive for those without health insurance, offers a one-time test that, if negative, provides patients with a 10-year window of screening coverage.
Any effective programs to increase CRC screening rates will need to use a systems change approach, with creative interventions that take patient education, and even delivery of preventive health services, out of the context of the already too-full office visit.
Staff supports, such as the follow-up telephone calls used in the patient-targeted intervention, are key to effective interventions, especially for vulnerable populations. Additionally, institutions must ensure that they have adequate physical and staff resources to support the increased screening they are seeking to achieve.
Dr. Michael Pignone is a professor of medicine at the University of Texas at Austin. Dr. David Miller is a professor of internal medicine, Wake Forest University, Winston-Salem, N.C. Dr. Pignone is a medical director for Healthwise; Dr. Miller reported no relevant conflicts of interest These remarks were drawn from an editorial accompanying the two clinical trials.
Can outreach improve the globally low rates of adherence to colorectal cancer screening? Yes, according to two recent studies in JAMA; the studies found that both patient-focused and physician-focused outreach approaches can result in significantly better patient participation in colorectal cancer (CRC) screening.
The first study (JAMA. 2017;318[9]:806-15) compared a colonoscopy outreach program and a fecal immunochemical test (FIT) outreach program both with each other and with usual care. The results of the pragmatic, single-site, randomized, clinical trial showed that completed screenings were higher for both outreach groups, compared with the usual care group.
The primary outcome measure of the study was completion of the screening process, wrote Amit Singal, MD, and his coauthors. This was defined as any adherence to colonoscopy completion, the completion of annual testing for patients who had a normal FIT test, or treatment evaluation if CRC was detected during the screening process. Screenings were considered complete even if, for example, a patient in the colonoscopy arm eventually went on to have three consecutive annual FIT tests rather than a colonoscopy.
A total of 5,999 patients eligible for screening were initially randomized to one of the three study arms. Across all study arms, approximately half were lost to follow-up. These patients were excluded from the primary analysis but were included in an additional intention-to-screen analysis. A total of 2,400 patients received a colonoscopy outreach mailing; 2,400 received FIT outreach, including a letter, the home FIT testing kit and instructions; 1,199 received usual care. Patients in both intervention arms also received up to two phone calls if they didn’t respond to the initial mailing within 2 weeks. Mailings and phone calls were conducted in English or Spanish, according to the patients’ stated language preferences (those whose spoke neither language were excluded from the study).
Of the patients in the colonoscopy outreach group, 922 (38.4%) completed the screening process, compared with 671 (28.0%) in the FIT outreach group and 128 (10.7%) in the usual care group.
Compared with the group receiving usual care, completion of the screening process was 27.7% higher in the colonoscopy outreach group and 17.3% higher in the FIT outreach group. Screening process completion was 10.4% higher for the colonoscopy outreach group, compared with the FIT outreach group (P less than .001 for all).
Dr. Singal, who is with the department of internal medicine at UT Southwestern Medical Center, Dallas, and his colleagues also performed several post-hoc secondary analyses. In one, they used a less-stringent definition of screening process completion in which biennial FIT testing was considered satisfactory. When this definition was applied, the colonoscopy outreach group had 0.5% lower screening process completion than the FIT outreach group. The chances of a patient receiving any screening during the study period was highest in the FIT group (65%), with 51.7% of those in the colonoscopy outreach group and 39% of those in the usual care group receiving any screening.
“FIT has lower barriers to one-time participation but requires annual screening and diagnostic evaluation of abnormal results,” wrote Dr. Singal and his colleagues.
Strengths of the study, said Dr. Singal and his colleagues, included the fact that the study took place at a “safety net” institution with a racially and socioeconomically diverse population. Also, the study design avoided volunteer bias, and offered a pragmatic head-to-head comparison of colonoscopy and FIT.
The second study took place in western France, and targeted outreach to physicians rather than patients (JAMA. 2017;318[9];816-84). When physicians were given a list of their own patients who were not up to date on CRC screening, investigators saw a small, but significant, uptick in patient participation in FIT screening.
One year after the reminders went out, FIT screening had been initiated in 24.8% of patients whose physicians had received the list, compared with 21.7% of patients of physicians who had received a more generic notice and 20.6% of patients whose physicians received no notification, according to first author Cedric Rat, MD, and his colleagues.
The study examined which notification approach was most effective in increasing FIT screening among the physicians’ patient panels: sending general practitioners (GPs) letters that included a list of their own patients who had not undergone CRC screening, or sending them generic letters describing CRC screening adherence rates specific to their region. A usual care group of practices received no notifications in this 3-group randomized cluster design.
Patients in the patient-specific reminders group had an odds ratio of 1.27 for participation in FIT screening (P less than .001) compared to the usual care group. The odds ratio for the generic reminders group was 1.09, a nonsignificant difference.
Between-group comparison showed statistical significance for both the 3.1% difference between the patient-specific and generic reminders groups, and for the 4.2% difference between the patient-specific and usual care groups (P less than .001 for both). There was no significant difference between the generic reminders group and the usual care group.
Dr. Rat, professor of medicine at the Faculty of Medicine, Nantes, France, and his colleagues enrolled GPs in a total of 801 practices that included patients aged 50participating GPs caring for 33,044 patients who met study criteria.
Physician characteristics that were associated with higher FIT participation included younger age and an initially smaller number of unscreened patients. Patients with low socioeconomic status and those with a higher chronic disease burden were less likely to participate in FIT screening.
Also, Dr. Rat and his colleagues noted that the busiest practices actually had higher CRC screening rates. The investigators hypothesized that a recent physician pay-for-performance grant for CRC completion might be more appealing for some busy physicians.
This was the largest study of CRC screening participation to date, according to Dr. Rat and his coauthors, and showed the small but detectable efficacy of an inexpensive intervention that, given complete patient records, is relatively easy to effect. Though the effect size was smaller than the 12% difference the investigators had anticipated seeing for the patient-specific reminders group, the study still showed that targeting physicians can be an effective public health intervention to increase CRC screening rates, said Dr. Rat and his colleagues.
None of the investigators in either study reported conflicts of interest.
Can outreach improve the globally low rates of adherence to colorectal cancer screening? Yes, according to two recent studies in JAMA; the studies found that both patient-focused and physician-focused outreach approaches can result in significantly better patient participation in colorectal cancer (CRC) screening.
The first study (JAMA. 2017;318[9]:806-15) compared a colonoscopy outreach program and a fecal immunochemical test (FIT) outreach program both with each other and with usual care. The results of the pragmatic, single-site, randomized, clinical trial showed that completed screenings were higher for both outreach groups, compared with the usual care group.
The primary outcome measure of the study was completion of the screening process, wrote Amit Singal, MD, and his coauthors. This was defined as any adherence to colonoscopy completion, the completion of annual testing for patients who had a normal FIT test, or treatment evaluation if CRC was detected during the screening process. Screenings were considered complete even if, for example, a patient in the colonoscopy arm eventually went on to have three consecutive annual FIT tests rather than a colonoscopy.
A total of 5,999 patients eligible for screening were initially randomized to one of the three study arms. Across all study arms, approximately half were lost to follow-up. These patients were excluded from the primary analysis but were included in an additional intention-to-screen analysis. A total of 2,400 patients received a colonoscopy outreach mailing; 2,400 received FIT outreach, including a letter, the home FIT testing kit and instructions; 1,199 received usual care. Patients in both intervention arms also received up to two phone calls if they didn’t respond to the initial mailing within 2 weeks. Mailings and phone calls were conducted in English or Spanish, according to the patients’ stated language preferences (those whose spoke neither language were excluded from the study).
Of the patients in the colonoscopy outreach group, 922 (38.4%) completed the screening process, compared with 671 (28.0%) in the FIT outreach group and 128 (10.7%) in the usual care group.
Compared with the group receiving usual care, completion of the screening process was 27.7% higher in the colonoscopy outreach group and 17.3% higher in the FIT outreach group. Screening process completion was 10.4% higher for the colonoscopy outreach group, compared with the FIT outreach group (P less than .001 for all).
Dr. Singal, who is with the department of internal medicine at UT Southwestern Medical Center, Dallas, and his colleagues also performed several post-hoc secondary analyses. In one, they used a less-stringent definition of screening process completion in which biennial FIT testing was considered satisfactory. When this definition was applied, the colonoscopy outreach group had 0.5% lower screening process completion than the FIT outreach group. The chances of a patient receiving any screening during the study period was highest in the FIT group (65%), with 51.7% of those in the colonoscopy outreach group and 39% of those in the usual care group receiving any screening.
“FIT has lower barriers to one-time participation but requires annual screening and diagnostic evaluation of abnormal results,” wrote Dr. Singal and his colleagues.
Strengths of the study, said Dr. Singal and his colleagues, included the fact that the study took place at a “safety net” institution with a racially and socioeconomically diverse population. Also, the study design avoided volunteer bias, and offered a pragmatic head-to-head comparison of colonoscopy and FIT.
The second study took place in western France, and targeted outreach to physicians rather than patients (JAMA. 2017;318[9];816-84). When physicians were given a list of their own patients who were not up to date on CRC screening, investigators saw a small, but significant, uptick in patient participation in FIT screening.
One year after the reminders went out, FIT screening had been initiated in 24.8% of patients whose physicians had received the list, compared with 21.7% of patients of physicians who had received a more generic notice and 20.6% of patients whose physicians received no notification, according to first author Cedric Rat, MD, and his colleagues.
The study examined which notification approach was most effective in increasing FIT screening among the physicians’ patient panels: sending general practitioners (GPs) letters that included a list of their own patients who had not undergone CRC screening, or sending them generic letters describing CRC screening adherence rates specific to their region. A usual care group of practices received no notifications in this 3-group randomized cluster design.
Patients in the patient-specific reminders group had an odds ratio of 1.27 for participation in FIT screening (P less than .001) compared to the usual care group. The odds ratio for the generic reminders group was 1.09, a nonsignificant difference.
Between-group comparison showed statistical significance for both the 3.1% difference between the patient-specific and generic reminders groups, and for the 4.2% difference between the patient-specific and usual care groups (P less than .001 for both). There was no significant difference between the generic reminders group and the usual care group.
Dr. Rat, professor of medicine at the Faculty of Medicine, Nantes, France, and his colleagues enrolled GPs in a total of 801 practices that included patients aged 50participating GPs caring for 33,044 patients who met study criteria.
Physician characteristics that were associated with higher FIT participation included younger age and an initially smaller number of unscreened patients. Patients with low socioeconomic status and those with a higher chronic disease burden were less likely to participate in FIT screening.
Also, Dr. Rat and his colleagues noted that the busiest practices actually had higher CRC screening rates. The investigators hypothesized that a recent physician pay-for-performance grant for CRC completion might be more appealing for some busy physicians.
This was the largest study of CRC screening participation to date, according to Dr. Rat and his coauthors, and showed the small but detectable efficacy of an inexpensive intervention that, given complete patient records, is relatively easy to effect. Though the effect size was smaller than the 12% difference the investigators had anticipated seeing for the patient-specific reminders group, the study still showed that targeting physicians can be an effective public health intervention to increase CRC screening rates, said Dr. Rat and his colleagues.
None of the investigators in either study reported conflicts of interest.
FROM JAMA
Key clinical point: Both a physician-directed outreach effort and one that targeted patients increased CRC screening rates.
Major finding: The patient-directed effort increased CRC screening completion by 17%-28%, compared with usual care; a single physician-directed outreach intervention increased screening by 4.2% compared to usual care.
Study details: A single-center pragmatic study of 5,999 patients eligible for CRC screening, and a three-group randomized cluster study of 1,446 general practitioners caring for 33,044 patients eligible for CRC screening.
Disclosures: No study authors reported conflicts of interest.
Plerixafor doesn’t overcome HPC failure in R-hyperCVAD for mantle cell lymphoma
A commonly-used intensive induction regimen was associated with higher rates of hematopoietic progenitor cell mobilization failure in patients with mantle cell lymphoma, even when plerixafor rescue was attempted, based on a study by Amandeep Salhotra, MD, and his colleagues at City of Hope, Duarte, Calif.
Patients who received rituximab and hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (R-hyperCVAD) in the era after plerixafor came into use experienced significantly higher rates of peripheral blood stem cell (PBSC) collection failure than did patients receiving other induction regimens (17% vs. 4% failure rate, P = .04).
“Plerixafor does not overcome the negative impact of R-hyperCVAD on PBSC mobilization, and caution is warranted in using R-hyperCVAD in patients with newly diagnosed MCL who are candidates for ASCT (autologous stem cell transplant),” wrote Dr. Salhotra and his colleagues.
The higher rate of hematopoietic progenitor cell collection failure for R-hyperCVAD patients could not be attributed to their age at time of mantle cell lymphoma diagnosis or to the amount of time between diagnosis and collection.
Treatment records for 181 consecutive mantle cell lymphoma patients were examined for a 10 year period in the retrospective single-site study. Plerixafor, a C-X-C chemokine receptor agonist that reduces hematopoietic progenitor cells’ ability to bind to bone marrow stroma, was introduced on August 16, 2009; a total of 71 patients were treated before this point, and 110 were treated afterward.
The R-hyperCVAD regimen was received by 34 pre-plerixafor patients (45%) and by 42 of the post-plerixafor era patients (55%). Other regimens were received by 37 (35%) and 68 (65%) of the pre- and post-plerixafor era patients, respectively.
Before plerixafor came into use, Dr. Salhotra, of City of Hope’s department of hematology and hematopoietic cell transplantation, and his coinvestigators saw no significant difference among their study population in the rates of PBSC collection failure between those receiving R-hyperCVAD (11%) and those receiving other regimens (12%). The findings were reported in Biology of Blood and Marrow Transplantation.
The study was conducted in the context of other recent work that showed higher rates of PBSC collection failure and fewer CD34+ cells collected with the use of an R-hyperCVAD conditioning regimen. The fact that PBSC mobilization rates were significantly lower in R-hyperCVAD patients post-plerixafor surprised the investigators, who had hypothesized that the use of plerixafor would overcome PBSC mobilization failures without regard to the conditioning regimen used.
“It may be worthwhile to consider using a more aggressive strategy for [hematopoetic progenitor cell] mobilization in patients who have received R-hyperCVAD chemotherapy upfront or as salvage for aggressive lymphomas,” the researchers wrote. This might include the use of plerixafor upfront when patients have low CD34 counts before apheresis.
The researchers plan to examine their data to see how the choice of induction regimen and plerixafor usage impact patient survival.
The study authors reported no conflicts of interest.
Source: Amandeep Salhotra, et al. Hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone chemotherapy in mantle cell lymphoma patients is associated with higher rates of hematopoietic progenitor cell mobilization failure despite plerixafor rescue. Biol Blood Marrow Transplant 2017; 23:1264-1268.
SOURCE: Biol Blood Marrow Transplant 2017; 23:1264-1268. http://dx.doi.org/10.1016/j.bbmt.2017.04.011
A commonly-used intensive induction regimen was associated with higher rates of hematopoietic progenitor cell mobilization failure in patients with mantle cell lymphoma, even when plerixafor rescue was attempted, based on a study by Amandeep Salhotra, MD, and his colleagues at City of Hope, Duarte, Calif.
Patients who received rituximab and hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (R-hyperCVAD) in the era after plerixafor came into use experienced significantly higher rates of peripheral blood stem cell (PBSC) collection failure than did patients receiving other induction regimens (17% vs. 4% failure rate, P = .04).
“Plerixafor does not overcome the negative impact of R-hyperCVAD on PBSC mobilization, and caution is warranted in using R-hyperCVAD in patients with newly diagnosed MCL who are candidates for ASCT (autologous stem cell transplant),” wrote Dr. Salhotra and his colleagues.
The higher rate of hematopoietic progenitor cell collection failure for R-hyperCVAD patients could not be attributed to their age at time of mantle cell lymphoma diagnosis or to the amount of time between diagnosis and collection.
Treatment records for 181 consecutive mantle cell lymphoma patients were examined for a 10 year period in the retrospective single-site study. Plerixafor, a C-X-C chemokine receptor agonist that reduces hematopoietic progenitor cells’ ability to bind to bone marrow stroma, was introduced on August 16, 2009; a total of 71 patients were treated before this point, and 110 were treated afterward.
The R-hyperCVAD regimen was received by 34 pre-plerixafor patients (45%) and by 42 of the post-plerixafor era patients (55%). Other regimens were received by 37 (35%) and 68 (65%) of the pre- and post-plerixafor era patients, respectively.
Before plerixafor came into use, Dr. Salhotra, of City of Hope’s department of hematology and hematopoietic cell transplantation, and his coinvestigators saw no significant difference among their study population in the rates of PBSC collection failure between those receiving R-hyperCVAD (11%) and those receiving other regimens (12%). The findings were reported in Biology of Blood and Marrow Transplantation.
The study was conducted in the context of other recent work that showed higher rates of PBSC collection failure and fewer CD34+ cells collected with the use of an R-hyperCVAD conditioning regimen. The fact that PBSC mobilization rates were significantly lower in R-hyperCVAD patients post-plerixafor surprised the investigators, who had hypothesized that the use of plerixafor would overcome PBSC mobilization failures without regard to the conditioning regimen used.
“It may be worthwhile to consider using a more aggressive strategy for [hematopoetic progenitor cell] mobilization in patients who have received R-hyperCVAD chemotherapy upfront or as salvage for aggressive lymphomas,” the researchers wrote. This might include the use of plerixafor upfront when patients have low CD34 counts before apheresis.
The researchers plan to examine their data to see how the choice of induction regimen and plerixafor usage impact patient survival.
The study authors reported no conflicts of interest.
Source: Amandeep Salhotra, et al. Hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone chemotherapy in mantle cell lymphoma patients is associated with higher rates of hematopoietic progenitor cell mobilization failure despite plerixafor rescue. Biol Blood Marrow Transplant 2017; 23:1264-1268.
SOURCE: Biol Blood Marrow Transplant 2017; 23:1264-1268. http://dx.doi.org/10.1016/j.bbmt.2017.04.011
A commonly-used intensive induction regimen was associated with higher rates of hematopoietic progenitor cell mobilization failure in patients with mantle cell lymphoma, even when plerixafor rescue was attempted, based on a study by Amandeep Salhotra, MD, and his colleagues at City of Hope, Duarte, Calif.
Patients who received rituximab and hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (R-hyperCVAD) in the era after plerixafor came into use experienced significantly higher rates of peripheral blood stem cell (PBSC) collection failure than did patients receiving other induction regimens (17% vs. 4% failure rate, P = .04).
“Plerixafor does not overcome the negative impact of R-hyperCVAD on PBSC mobilization, and caution is warranted in using R-hyperCVAD in patients with newly diagnosed MCL who are candidates for ASCT (autologous stem cell transplant),” wrote Dr. Salhotra and his colleagues.
The higher rate of hematopoietic progenitor cell collection failure for R-hyperCVAD patients could not be attributed to their age at time of mantle cell lymphoma diagnosis or to the amount of time between diagnosis and collection.
Treatment records for 181 consecutive mantle cell lymphoma patients were examined for a 10 year period in the retrospective single-site study. Plerixafor, a C-X-C chemokine receptor agonist that reduces hematopoietic progenitor cells’ ability to bind to bone marrow stroma, was introduced on August 16, 2009; a total of 71 patients were treated before this point, and 110 were treated afterward.
The R-hyperCVAD regimen was received by 34 pre-plerixafor patients (45%) and by 42 of the post-plerixafor era patients (55%). Other regimens were received by 37 (35%) and 68 (65%) of the pre- and post-plerixafor era patients, respectively.
Before plerixafor came into use, Dr. Salhotra, of City of Hope’s department of hematology and hematopoietic cell transplantation, and his coinvestigators saw no significant difference among their study population in the rates of PBSC collection failure between those receiving R-hyperCVAD (11%) and those receiving other regimens (12%). The findings were reported in Biology of Blood and Marrow Transplantation.
The study was conducted in the context of other recent work that showed higher rates of PBSC collection failure and fewer CD34+ cells collected with the use of an R-hyperCVAD conditioning regimen. The fact that PBSC mobilization rates were significantly lower in R-hyperCVAD patients post-plerixafor surprised the investigators, who had hypothesized that the use of plerixafor would overcome PBSC mobilization failures without regard to the conditioning regimen used.
“It may be worthwhile to consider using a more aggressive strategy for [hematopoetic progenitor cell] mobilization in patients who have received R-hyperCVAD chemotherapy upfront or as salvage for aggressive lymphomas,” the researchers wrote. This might include the use of plerixafor upfront when patients have low CD34 counts before apheresis.
The researchers plan to examine their data to see how the choice of induction regimen and plerixafor usage impact patient survival.
The study authors reported no conflicts of interest.
Source: Amandeep Salhotra, et al. Hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone chemotherapy in mantle cell lymphoma patients is associated with higher rates of hematopoietic progenitor cell mobilization failure despite plerixafor rescue. Biol Blood Marrow Transplant 2017; 23:1264-1268.
SOURCE: Biol Blood Marrow Transplant 2017; 23:1264-1268. http://dx.doi.org/10.1016/j.bbmt.2017.04.011
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
Key clinical point: R-hyperCVAD was associated with increased peripheral blood stem cell (PBSC) collection failure in the post-plerixafor era.
Major finding: Patients receiving R-hyperCVAD in the post-plerixafor era had a 17% PBSC collection failure rate, compared to a 4% rate for those receiving other chemotherapy (P = 0.04).
Study details: Single-center retrospective study of 181 consecutive patients with mantle cell lymphoma over a 10-year period spanning the introduction of plerixafor.
Disclosures: The study was sponsored by City of Hope and the National Cancer Institute; the authors reported no conflicts of interest.
Source: Amandeep Salhotra, et al. Hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone chemotherapy in mantle cell lymphoma patients is associated with higher rates of hematopoietic progenitor cell mobilization failure despite plerixafor rescue. Biol Blood Marrow Transplant 2017; 23:1264-1268.
‘Making a difference in cancer care’
Survival improves when patients with cancer self-report symptoms
Key clinical point Patients with metastatic cancer who self-reported symptoms experienced significant improvement in overall survival. Major finding Median overall survival with self-reporting of symptoms compared with usual care was 31.2 and 26 months, respectively. Data source A randomized controlled clinical trial of 766 patients. Funding and disclosures This study was supported by the National Institutes of Health and the Conquer Cancer Foundation of the American Society of Clinical Oncology. Dr Basch and Dr Burstein each reported having no disclosures.
Patients with metastatic cancer who self-reported symptoms during routine cancer treatment experienced a number of benefits, including a statistically significant improvement in overall survival, according to findings from a randomized, controlled clinical trial (see p. e184). The median overall survival in 441 patients receiving treatment for metastatic breast, lung, genitourinary, or gynecologic cancer who were randomized to the self-reporting intervention arm was more than 5 months longer (a nearly 20% increase) than in 325 patients receiving standard care (31.2 vs. 26 months), Ethan Basch, MD, of the Lineberger Comprehensive Cancer Center at the University of North Carolina, Chapel Hill, said at the meeting. “Another way to think of this is [in terms of] 5-year survival. At 5 years, 8% more patients were alive in the self-reporting group,” he said.

In addition, 31% of patients in the intervention arm had better quality of life/physical functioning, compared with those in the control arm, and 7% fewer patients in the intervention arm visited an emergency room during the study. The duration of potentially life-prolonging chemotherapy was increased by an average of 2 months in the intervention arm, he said (JAMA. 2017 Jun 4. doi: 10.1001/jama.2017.7156).
Symptoms such as nausea, pain, and fatigue are common among patients with metastatic cancer and can often go undetected by doctors and nurses until they become severe and physically debilitating, Dr Basch added, noting that patients are often hesitant to call the office between visits to report symptoms.
Dr Basch and his colleagues hypothesized that self-reporting of patient symptoms between visits or before a visit while the patient was in the clinic waiting area would prompt earlier intervention and improve symptom control and outcomes.
Study participants were patients at Memorial Sloan Kettering Cancer Center who had advanced solid genitourinary, gynecologic, breast, or lung tumors and who were receiving outpatient chemotherapy. Those assigned to the intervention group used tablet computers and an online web survey system to report on 12 symptoms commonly experienced during chemotherapy. The system triggers an alert to a nurse when a severe or worsening symptom is reported. Patients in the usual care group discussed symptoms during office visits and were encouraged to call the office between visits if they were concerned about symptoms.
Patients remained on the study until discontinuation of all cancer treatment, hospice, or death.
One possible explanation for the findings is that this self-reporting approach prompts clinicians to manage symptoms before they cause serious downstream complications, Dr Basch said.
The approach may also keep patients more physically functional, which is known from previous studies to have a strong association with better survival, and it may also improve management of chemotherapy side effects, enabling longer duration of beneficial cancer treatment. “In oncology, we often are limited in our ability to give life-prolonging treatment because people don’t tolerate it well,” Dr Basch explained.
“This approach should be considered for inclusion in standard symptoms management as a component of high-quality cancer care,” he concluded, noting that efforts are underway to test the next generation of systems to improve communication between patients and care teams and to figure out how best to integrate these tools into oncology practice.
The system used in the this study was designed for research, but a number of companies have tools currently available for patient-reported outcomes, and others are being developed, Dr Basch said. A National Cancer Institute questionnaire, the PRO-CTCAE, is publicly available and can be loaded into patients’ electronic health records for this purpose as well.
Harold J Burstein, MD, of Dana-Farber Cancer Institute, Boston, said the study findings validate the feeling among many clinicians that patient-focused, team-based care can improve outcomes in a meaningful way for patients. If this were a drug … it would be worth tens, if not hundreds of thousands, of dollars per year … We don’t have those same kinds of dollars to help implement these into our electronic health records or our systems. We need to find ways to support that and make it happen,” he said.
— Sharon Worcester
TRK inhibitor shows ‘striking’ activity and durability across diverse cancers
Key clinical point Larotrectinib has good, durable efficacy when used to treat advanced cancers harboring TRK fusions. Major finding The overall response rate was 76%, and 79% of responses were still ongoing at 12 months. Data source An integrated analysis of phase 1 and 2 trials among 55 children and adults having 17 discrete types of advanced cancer with TRK fusions. Funding and disclosures Loxo Oncology funded the study. Dr Hyman disclosed that he has a consulting or advisory role with Atara Biotherapeutics, Chugai Pharma, and CytomX Therapeutics, and that he receives research funding from AstraZeneca and Puma Biotechnology.
Larotrectinib, an oral inhibitor of tropomyosin receptor kinase (TRK), has durable efficacy across diverse adult and pediatric cancers that harbor a genetic aberration known as TRK fusion, according to findings from an analysis of 3 trials reported at the meeting (see p. e184). Fusion of a TRK gene with an unrelated gene leads to uncontrolled signaling in the TRK pathway, potentially causing tumor growth and addiction to this input, David Hyman, MD, chief of early drug development at Memorial Sloan Kettering Cancer Center in New York, explained in a press briefing.
“One of the defining features of TRK fusions is that they are not just found in one cancer type, but in dozens of different cancer types, and not just in adults, but children as well, spanning the entire lifetime of the person,” he noted. They are rare in common cancers and nearly universal in certain uncommon cancers; collectively, they are present in possibly 5,000 cancers diagnosed each year in the United States.
Dr Hyman and his colleagues analyzed data from 55 patients having 17 discrete types of advanced cancer harboring TRK fusions who were treated with larotrectinib in phase 1 and 2 trials. Results showed an overall response rate of 76%, and the majority of responses were still ongoing at 12 months. “I believe these data support larotrectinib as a potential new standard of care for these patients,” he said. “However, I want to emphasize that really recognizing this benefit in the community will require that we test patients more universally for the presence of TRK fusions or other tumor-agnostic biomarkers, such as microsatellite instability.”
Study details
The investigators analyzed data from 3 trials in which patients with advanced TRK fusion-positive solid cancers received larotrectinib (LOXO-101): a phase 1 trial among 8 adult patients, a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 “basket” trial among 35 adult and adolescent patients (NAVIGATE).
“These patients were identified by local testing,” Dr Hyman noted. “We did not perform central screening to find the TRK fusions, and in fact, 50 different laboratories identified the 55 patients. So in a sense, this really represents the real-world identification of these patients.”
In an integrated analysis, the overall rate of confirmed response as assessed by investigators was 76%, with complete response in 12% of patients and partial response in 64%. Two patients had such deep tumor regression that they experienced downstaging enabling them to undergo potentially curative surgery. Efficacy was consistent regardless of tumor type, which TRK gene was affected, and the fusion partner gene.
Median time to response was 1.8 months. “This is just a reflection of when the first scan was obtained. But in the clinic, patients reported dramatic improvement of their symptoms within days of starting therapy,” Dr Hyman said.
With a median follow-up of 5.8 months, the median duration of response was not yet reached. In all, 79% of responses were still ongoing at 12 months. Median progression-free survival (PFS) was likewise not reached; the 12-month rate was 63%.
The leading treatment-emergent adverse events were fatigue (38%), dizziness (27%), nausea (26%), and anemia (26%). “This is an extremely well tolerated therapy with only 13% of patients requiring any form of dose modification and not a single patient discontinuing due to adverse events,” he said.
It is not clear why some patients had apparent primary resistance to larotrectinib, but their TRK fusion test results may have been incorrect, Dr Hyman speculated. In all, 6 patients developed acquired resistance to larotrectinib; 5 of them were found to have an identical resistance mutation, and 2 went on to receive and have a response to LOXO-195, a next-generation TRK inhibitor that seems to retain activity in the presence of this mutation (Cancer Discov. 2017 June 3. doi: 10.1158/2159-8290.CD-17-0507).
TRK testing
Several next-generation sequencing-based tests already available clinically can pick up TRK fusions, Dr Hyman pointed out. “But it is important for the ordering physician to understand whether the tests they are ordering include fusion detection and, if it’s an option, to select it. Otherwise, they will not find TRK fusions. “The list price for these tests is in the low thousands of dollars,” he noted. In cancers in which sequential single-gene testing is already being done as standard of care, there is “minimal” incremental cost of instead using comprehensive testing that would detect TRK fusions.
Oncologists should be aware that obtaining test results can take weeks, Dr Hyman stressed. “This [testing] should be more broadly adopted and should be adopted at a point in the patient’s treatment [so that they] don’t become too sick, then don’t have an opportunity to be treated even when the test results come back positive.”
— Susan London
QoL preserved with ribociclib-letrozole for advanced breast cancer
Key clinical point Patients who took ribociclib plus letrozole had less pain and no drop in QoL compared with letrozole alone. Major finding QoL was sustained and pain scores decreased when ribociclib was added to letrozole for patients with advanced breast cancer. Data source Double-blind, placebo-controlled phase 3 trial of letrozole plus ribociclib compared with letrozole plus placebo in 668 patients with advanced hormone receptor–positive, HER2-negative breast cancer. Disclosures Dr Verma reported financial relationships with Novartis, which markets ribociclib, and other firms.
Patients with advanced breast cancer whose aromatase inhibitor therapy was supplemented with a cycline-dependent kinase inhibitor had better PFS with no drop in quality of life (QoL). Health-related QoL for patients on the combination therapy was equivalent to that of patients on monotherapy in most aspects, but patients receiving both therapies had a sustained and clinically meaningful decrease in pain.
In addition, the time to definitive deterioration by 10% or more of the global health status/QoL scale score was similar between treatment arms (hazard ratio [HR], 0.944; 95% confidence interval, 0.720-1.237).
The MONALEESA-2 trial had previously shown that the CDK4/6 inhibitor ribociclib, when added to the aromatase inhibitor letrozole, significantly improved PFS for postmenopausal patients with hormone receptor–positive, HER2-negative advanced breast cancer, when compared with letrozole in combination with placebo.
Sunil Verma, MD, reported on health-related QoL and symptoms in the 2 arms of MONALEESA-2, showing change from baseline, time to a definitive 10% deterioration, and the mean scores for on-treatment time compared with end of treatment on the global health-status QoL subscale of the European Organization for Research and Treatment of Cancer's (EORTC's) 30-item core QoL questionnaire.
“During treatment, overall health-related quality of life was maintained from baseline and was similar in both arms,” said Dr Verma, the study’s first author. Changes during treatment were not statistically significant, and did not reach the predetermined threshold for a clinically meaningful difference. The effect of key symptoms such as fatigue, nausea, and vomiting on QoL was similar for patients receiving ribociclib or placebo, he said. Although symptom scores were slightly higher for patients in the active arm of the study, the results were not clinically significant.
Reporting validated, cancer-specific patient-reported outcomes from the trial, Dr Verma, professor and head of the department of oncology at the University of Calgary in Alberta, Canada, sought to “highlight the patient experience with a focus on health-related quality of life and symptoms,” he said during his presentation at the meeting. “A clinically meaningful – more than 5 points – improvement from baseline in pain score was maintained up to and including cycle 15 in the ribociclib plus letrozole arm.” The placebo arm had a mild, clinically insignificant improvement at most assessment points. For both treatment arms, pain scores increased a bit above baseline levels at the time of disease progression or end of therapy, he said.
Patients completed the EORTC 30-item core QoL questionnaire at their screening visit, and then every 8 weeks for the first 18 months. Then, they completed the questionnaires every 12 weeks until they experienced disease progression, died, were lost to follow-up or withdrew from the study, or stopped treatment.
Statistical analysis of the questionnaire results took into account the patients’ baseline scores, treatments received, and how they were stratified. Investigators assessed both statistical significance and the clinical meaningfulness of changes, defined as a change of 5-10 points.
In MONALEESA-2, 334 patients each were allocated to the ribociclib-letrozole arm and the placebo-letrozole arm. Patients in both arms, said Dr Verma, were very compliant with questionnaire completion. More than 90% of patients who were eligible completed questionnaire through cycle 19 of ribociclib or placebo. He explained that the overall numbers completing the questionnaire declined with time, as more patients had disease progression. Dr Verma said it is important to include those measures, because patients and their families care about “the quality of the time gained,” so patient-reported outcomes should be a part of risk-benefit assessments for new cancer therapies. “While delaying disease progression may help maintain patient quality of life, the addition of novel treatments to existing therapies can add toxicities, which may diminish quality of life,” he said.
— Kari Oakes
Ibrutinib and beyond: optimizing therapy in relapsed CLL
Key clinical point Ibrutinib had durable efficacy in relapsed CLL, and combinations with other targeted agents or with CAR-T cells are promising. Major finding Long-term PFS was better with ibrutinib than with ofatumumab (HR, 0.133). The overall response rate with the triplet of ibrutinib, ublituximab, and umbralisib was 100%. In all, 89% of patients achieved no evidence of disease in marrow when anti-CD19 CAR-T cells were added to ibrutinib. Data source An update of a phase 3 randomized trial among 391 patients with previously treated CLL or SLL (RESONATE). A phase 1/1b trial including 19 patients with mainly relapsed or refractory CLL or SLL. A pilot trial among 10 patients with previously treated, mainly higher-risk, CLL or SLL. Disclosures See article text.
Ibrutinib monotherapy
In the phase 3 randomized RESONATE trial, investigators compared ibrutinib with ofatumumab, an anti-CD20 antibody, among 391 patients with CLL or small lymphocytic lymphoma (SLL), with crossover allowed. Initial results favored ibrutinib.
Investigators led by John C Byrd, MD, director of the division of hematology at the Ohio State University Comprehensive Cancer Center in Columbus, reported updated data in a poster session at the meeting, now with a median 44 months of follow-up in the ibrutinib arm.
Median PFS was not reached with ibrutinib, compared with 8.1 months with ofatumumab (HR, 0.133). The 3-year rate of PFS for ibrutinib and ofatumumab was 59% and 3%, respectively.
The pattern was generally similar across patients stratified by cytogenetics (deletion of 17p, deletion of 11q, or neither), IGHV and TP53 mutation status, and previous lines of therapy, reported Dr Byrd. The 3-year overall survival rate for ibrutinib was 74%. In analyses adjusted for crossover, patients given the inhibitor had a markedly lower risk of death than did peers given the antibody (HR, 0.37). The overall response rate with ibrutinib was 91%. Although the rate of complete response increased with follow-up, it was still just 9%.
The most common grade 3 or worse adverse events were neutropenia (23%), pneumonia (17%), and anemia, thrombocytopenia, and hypertension (8% each). Major hemorrhage was reported in 2 patients (1%) in the ibrutinib group, and 3 (2%) in the ofatumumab group.
The investigators emphasized that these long-term results “show that extended treatment with ibrutinib is tolerable and continues to show sustained PFS in previously treated patients with CLL regardless of high-risk cytogenetics, adding that traditional poor prognostic factors for survival with chemoimmunotherapy, including del(17p) and del(11q), were not significant factors predictive of PFS outcomes with ibrutinib.
Funding and disclosures: Pharmacyclics funded the trial. Dr Byrd disclosed that he receives research funding from Pharmacyclics and other companies.
Ibrutinib plus ublituximab and umbralisib
Investigators led by Loretta J Nastoupil, MD, tested a triplet consisting of ibrutinib with ublituximab (another anti-CD20 antibody) and umbralisib (TGR-1202), an oral PI3 kinase-delta inhibitor, in a phase 1/1b trial of 38 patients with generally heavily pretreated leukemias and lymphomas, including 20 with CLL or SLL. Notably, 8 patients with CLL (50%) had a 17p or 11q deletion.
The median time on study was 11.1 months, reported Dr Nastoupil, of the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston. The overall response rate for the 19 evaluable patients with CLL or SLL was 100% (complete response in 32%, partial response in 68%). The main grade 3 or 4 adverse events in the entire trial population were neutropenia (18%) and pyrexia (8%). Only two patients discontinued treatment because of adverse events.
“An expansion cohort is ongoing at the highest dose, and we will clearly need more patients treated and longer follow-up to really know the efficacy,” commented invited discussant Jennifer R Brown, MD, PhD, director of the Chronic Lymphocytic Leukemia Center at the Dana-Farber Cancer Institute in Boston.
Funding and disclosures: TG Therapeutics funded the trial. Dr Nastoupil has received honoraria and research funding from TG Therapeutics, and honoraria from Pharmacyclics.
Ibrutinib plus CAR-T cells
Investigators in a pilot trial led by Saar Gill, MD, PhD, of the hospital of the University of Pennsylvania in Philadelphia, tested the combination of ibrutinib with chimeric antigen receptor-T cells (CAR-T cells) against CD19, on the basis of preclinical evidence of synergy. The trial participants were 10 patients with CLL or SLL who had not achieved complete response with ibrutinib. All had a 17p deletion or a p53 mutation, or a complex karyotype, and some had a known ibrutinib resistance mutation.
At 3 months, 8 of 9 evaluable patients (89%) had no evidence of disease in bone marrow, reported Dr Gill. Seven patients (78%) achieved a complete or partial radiographic response in the spleen and lymph nodes. Overall, the treatment was well tolerated. One patient developed grade 4 tumor lysis syndrome, and two developed grade 3 cytokine release syndrome. But, none required anticytokine therapy.
Most patients remain on ibrutinib and are being monitored. In addition, the researchers plan to treat 25 more patients with the same combination.
Funding and disclosures: Novartis funded the trial. Dr Gill disclosed that he receives research funding from Novartis (institutional) and has patents for CAR-T cells for AML.
— Susan London
Pazopanib falls short as adjuvant therapy for high-risk RCC
Key clinical point Pazopanib is not efficacious for treating resected high-risk locally advanced RCC. Major finding Compared with placebo, pazopanib started at 600 mg daily did not significantly reduce the risk of DFS events (HR, 0.86; P = .16). Data source A phase 3 randomized controlled trial among 1,538 patients who had undergone nephrectomy for high-risk locally advanced RCC (PROTECT trial). Disclosures Novartis Oncology funded the trial. Dr Motzer disclosed that he is a consultant to Novartis and receives research funding from Novartis (institutional).
The antiangiogenic agent pazopanib is not efficacious when used as adjuvant therapy for resected renal cell carcinoma (RCC) with features that confer a high risk of recurrence, investigators in the PROTECT investigators reported at the meeting. “The trial did not meet its primary endpoint,” concluded lead investigator Robert J Motzer, MD, of Memorial Sloan-Kettering Cancer Center in New York. “Pazopanib is not recommended for adjuvant therapy following resection of locally advanced RCC.”

Adjuvant use of other agents in this class has yielded mixed results, Dr Motzer noted, citing the earlier ASSURE trial found that neither sunitinib nor sorafenib improved disease-free survival (DFS) or overall survival (Lancet 2016;387:2008-2016), and the S-TRAC trial that found that sunitinib improved DFS (N Engl J Med. 2016;375:2246-2254).
Pazopanib is an oral multitargeted tyrosine kinase inhibitor active against the vascular endothelial growth factor receptor (VEGFR). The PROTECT trial included 1,538 patients with resected pT2 (high grade), pT3, or greater nonmetastatic clear cell RCC, a highly vascular tumor typically reliant on aberrant signaling in the VEGF pathway. The patients were assigned evenly to receive pazopanib or placebo for 1 year. The starting dose was lowered from 800 mg daily to 600 mg daily (with escalation permitted) after 403 patients had been treated.
In intention-to-treat analysis among patients started on the 600-mg dose and having a median follow-up of about 31 months, DFS was better with pazopanib but not significantly so (HR, 0.86; P = .16). In secondary analyses, pazopanib had a significant DFS benefit in patients started on the 800-mg dose (HR, 0.69; P = .02) and among the entire trial population started on either dose (HR, 0.80; P = .01).
One possible explanation for the differing results seen with the 2 doses was the difference in follow-up, because the 800-mg group was treated earlier in the trial, said Dr Motzer. But with an additional year of blinded follow-up, the benefit in the 600-mg group diminished, but not in the 800-mg group.
Another possibility was the somewhat better performance of the placebo group used for comparison with the lower dose: the 3-year DFS rate with placebo was 64% for the 600-mg comparison but 56% for the 800-mg comparison. “One factor that could explain differences in the outcomes of the placebo groups includes unidentified patient demographic characteristics,” Dr Motzer added.
Overall survival was statistically indistinguishable between the pazopanib and placebo groups, regardless of dose. However, “the results are inconclusive as the data are not yet mature,” he said, with a definitive analysis planned for 2019.
Patients started on the 600-mg dose of pazopanib had a higher rate of grade 3 or 4 adverse events overall compared with those given placebo (60% vs 21%, respectively), driven in part by higher rates of hypertension and increased alanine aminotransferase levels. “Although the intent of modifying the protocol dose of pazopanib from 800 mg to 600 mg was to reduce the rate of discontinuation and improve the safety profile ... both cohorts had similar discontinuation rates and safety profiles,” Dr Motzer noted.
A QoL analysis for the 600-mg group using the 19-item Functional Assessment of Cancer Therapy Kidney Symptom Index showed values were consistently lower with the drug than with placebo during treatment, with a crossing of the threshold for a minimally important difference at week 8. Pharmacokinetic analyses from the trial, reported in a poster at the meeting, showed that in the group starting pazopanib at 600 mg, DFS was longer in patients who achieved higher drug trough concentrations at week 3 or 5.
— Susan London
Survival improves when patients with cancer self-report symptoms
Key clinical point Patients with metastatic cancer who self-reported symptoms experienced significant improvement in overall survival. Major finding Median overall survival with self-reporting of symptoms compared with usual care was 31.2 and 26 months, respectively. Data source A randomized controlled clinical trial of 766 patients. Funding and disclosures This study was supported by the National Institutes of Health and the Conquer Cancer Foundation of the American Society of Clinical Oncology. Dr Basch and Dr Burstein each reported having no disclosures.
Patients with metastatic cancer who self-reported symptoms during routine cancer treatment experienced a number of benefits, including a statistically significant improvement in overall survival, according to findings from a randomized, controlled clinical trial (see p. e184). The median overall survival in 441 patients receiving treatment for metastatic breast, lung, genitourinary, or gynecologic cancer who were randomized to the self-reporting intervention arm was more than 5 months longer (a nearly 20% increase) than in 325 patients receiving standard care (31.2 vs. 26 months), Ethan Basch, MD, of the Lineberger Comprehensive Cancer Center at the University of North Carolina, Chapel Hill, said at the meeting. “Another way to think of this is [in terms of] 5-year survival. At 5 years, 8% more patients were alive in the self-reporting group,” he said.
In addition, 31% of patients in the intervention arm had better quality of life/physical functioning, compared with those in the control arm, and 7% fewer patients in the intervention arm visited an emergency room during the study. The duration of potentially life-prolonging chemotherapy was increased by an average of 2 months in the intervention arm, he said (JAMA. 2017 Jun 4. doi: 10.1001/jama.2017.7156).
Symptoms such as nausea, pain, and fatigue are common among patients with metastatic cancer and can often go undetected by doctors and nurses until they become severe and physically debilitating, Dr Basch added, noting that patients are often hesitant to call the office between visits to report symptoms.
Dr Basch and his colleagues hypothesized that self-reporting of patient symptoms between visits or before a visit while the patient was in the clinic waiting area would prompt earlier intervention and improve symptom control and outcomes.
Study participants were patients at Memorial Sloan Kettering Cancer Center who had advanced solid genitourinary, gynecologic, breast, or lung tumors and who were receiving outpatient chemotherapy. Those assigned to the intervention group used tablet computers and an online web survey system to report on 12 symptoms commonly experienced during chemotherapy. The system triggers an alert to a nurse when a severe or worsening symptom is reported. Patients in the usual care group discussed symptoms during office visits and were encouraged to call the office between visits if they were concerned about symptoms.
Patients remained on the study until discontinuation of all cancer treatment, hospice, or death.
One possible explanation for the findings is that this self-reporting approach prompts clinicians to manage symptoms before they cause serious downstream complications, Dr Basch said.
The approach may also keep patients more physically functional, which is known from previous studies to have a strong association with better survival, and it may also improve management of chemotherapy side effects, enabling longer duration of beneficial cancer treatment. “In oncology, we often are limited in our ability to give life-prolonging treatment because people don’t tolerate it well,” Dr Basch explained.
“This approach should be considered for inclusion in standard symptoms management as a component of high-quality cancer care,” he concluded, noting that efforts are underway to test the next generation of systems to improve communication between patients and care teams and to figure out how best to integrate these tools into oncology practice.
The system used in the this study was designed for research, but a number of companies have tools currently available for patient-reported outcomes, and others are being developed, Dr Basch said. A National Cancer Institute questionnaire, the PRO-CTCAE, is publicly available and can be loaded into patients’ electronic health records for this purpose as well.
Harold J Burstein, MD, of Dana-Farber Cancer Institute, Boston, said the study findings validate the feeling among many clinicians that patient-focused, team-based care can improve outcomes in a meaningful way for patients. If this were a drug … it would be worth tens, if not hundreds of thousands, of dollars per year … We don’t have those same kinds of dollars to help implement these into our electronic health records or our systems. We need to find ways to support that and make it happen,” he said.
— Sharon Worcester
TRK inhibitor shows ‘striking’ activity and durability across diverse cancers
Key clinical point Larotrectinib has good, durable efficacy when used to treat advanced cancers harboring TRK fusions. Major finding The overall response rate was 76%, and 79% of responses were still ongoing at 12 months. Data source An integrated analysis of phase 1 and 2 trials among 55 children and adults having 17 discrete types of advanced cancer with TRK fusions. Funding and disclosures Loxo Oncology funded the study. Dr Hyman disclosed that he has a consulting or advisory role with Atara Biotherapeutics, Chugai Pharma, and CytomX Therapeutics, and that he receives research funding from AstraZeneca and Puma Biotechnology.
Larotrectinib, an oral inhibitor of tropomyosin receptor kinase (TRK), has durable efficacy across diverse adult and pediatric cancers that harbor a genetic aberration known as TRK fusion, according to findings from an analysis of 3 trials reported at the meeting (see p. e184). Fusion of a TRK gene with an unrelated gene leads to uncontrolled signaling in the TRK pathway, potentially causing tumor growth and addiction to this input, David Hyman, MD, chief of early drug development at Memorial Sloan Kettering Cancer Center in New York, explained in a press briefing.
“One of the defining features of TRK fusions is that they are not just found in one cancer type, but in dozens of different cancer types, and not just in adults, but children as well, spanning the entire lifetime of the person,” he noted. They are rare in common cancers and nearly universal in certain uncommon cancers; collectively, they are present in possibly 5,000 cancers diagnosed each year in the United States.
Dr Hyman and his colleagues analyzed data from 55 patients having 17 discrete types of advanced cancer harboring TRK fusions who were treated with larotrectinib in phase 1 and 2 trials. Results showed an overall response rate of 76%, and the majority of responses were still ongoing at 12 months. “I believe these data support larotrectinib as a potential new standard of care for these patients,” he said. “However, I want to emphasize that really recognizing this benefit in the community will require that we test patients more universally for the presence of TRK fusions or other tumor-agnostic biomarkers, such as microsatellite instability.”
Study details
The investigators analyzed data from 3 trials in which patients with advanced TRK fusion-positive solid cancers received larotrectinib (LOXO-101): a phase 1 trial among 8 adult patients, a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 “basket” trial among 35 adult and adolescent patients (NAVIGATE).
“These patients were identified by local testing,” Dr Hyman noted. “We did not perform central screening to find the TRK fusions, and in fact, 50 different laboratories identified the 55 patients. So in a sense, this really represents the real-world identification of these patients.”
In an integrated analysis, the overall rate of confirmed response as assessed by investigators was 76%, with complete response in 12% of patients and partial response in 64%. Two patients had such deep tumor regression that they experienced downstaging enabling them to undergo potentially curative surgery. Efficacy was consistent regardless of tumor type, which TRK gene was affected, and the fusion partner gene.
Median time to response was 1.8 months. “This is just a reflection of when the first scan was obtained. But in the clinic, patients reported dramatic improvement of their symptoms within days of starting therapy,” Dr Hyman said.
With a median follow-up of 5.8 months, the median duration of response was not yet reached. In all, 79% of responses were still ongoing at 12 months. Median progression-free survival (PFS) was likewise not reached; the 12-month rate was 63%.
The leading treatment-emergent adverse events were fatigue (38%), dizziness (27%), nausea (26%), and anemia (26%). “This is an extremely well tolerated therapy with only 13% of patients requiring any form of dose modification and not a single patient discontinuing due to adverse events,” he said.
It is not clear why some patients had apparent primary resistance to larotrectinib, but their TRK fusion test results may have been incorrect, Dr Hyman speculated. In all, 6 patients developed acquired resistance to larotrectinib; 5 of them were found to have an identical resistance mutation, and 2 went on to receive and have a response to LOXO-195, a next-generation TRK inhibitor that seems to retain activity in the presence of this mutation (Cancer Discov. 2017 June 3. doi: 10.1158/2159-8290.CD-17-0507).
TRK testing
Several next-generation sequencing-based tests already available clinically can pick up TRK fusions, Dr Hyman pointed out. “But it is important for the ordering physician to understand whether the tests they are ordering include fusion detection and, if it’s an option, to select it. Otherwise, they will not find TRK fusions. “The list price for these tests is in the low thousands of dollars,” he noted. In cancers in which sequential single-gene testing is already being done as standard of care, there is “minimal” incremental cost of instead using comprehensive testing that would detect TRK fusions.
Oncologists should be aware that obtaining test results can take weeks, Dr Hyman stressed. “This [testing] should be more broadly adopted and should be adopted at a point in the patient’s treatment [so that they] don’t become too sick, then don’t have an opportunity to be treated even when the test results come back positive.”
— Susan London
QoL preserved with ribociclib-letrozole for advanced breast cancer
Key clinical point Patients who took ribociclib plus letrozole had less pain and no drop in QoL compared with letrozole alone. Major finding QoL was sustained and pain scores decreased when ribociclib was added to letrozole for patients with advanced breast cancer. Data source Double-blind, placebo-controlled phase 3 trial of letrozole plus ribociclib compared with letrozole plus placebo in 668 patients with advanced hormone receptor–positive, HER2-negative breast cancer. Disclosures Dr Verma reported financial relationships with Novartis, which markets ribociclib, and other firms.
Patients with advanced breast cancer whose aromatase inhibitor therapy was supplemented with a cycline-dependent kinase inhibitor had better PFS with no drop in quality of life (QoL). Health-related QoL for patients on the combination therapy was equivalent to that of patients on monotherapy in most aspects, but patients receiving both therapies had a sustained and clinically meaningful decrease in pain.
In addition, the time to definitive deterioration by 10% or more of the global health status/QoL scale score was similar between treatment arms (hazard ratio [HR], 0.944; 95% confidence interval, 0.720-1.237).
The MONALEESA-2 trial had previously shown that the CDK4/6 inhibitor ribociclib, when added to the aromatase inhibitor letrozole, significantly improved PFS for postmenopausal patients with hormone receptor–positive, HER2-negative advanced breast cancer, when compared with letrozole in combination with placebo.
Sunil Verma, MD, reported on health-related QoL and symptoms in the 2 arms of MONALEESA-2, showing change from baseline, time to a definitive 10% deterioration, and the mean scores for on-treatment time compared with end of treatment on the global health-status QoL subscale of the European Organization for Research and Treatment of Cancer's (EORTC's) 30-item core QoL questionnaire.
“During treatment, overall health-related quality of life was maintained from baseline and was similar in both arms,” said Dr Verma, the study’s first author. Changes during treatment were not statistically significant, and did not reach the predetermined threshold for a clinically meaningful difference. The effect of key symptoms such as fatigue, nausea, and vomiting on QoL was similar for patients receiving ribociclib or placebo, he said. Although symptom scores were slightly higher for patients in the active arm of the study, the results were not clinically significant.
Reporting validated, cancer-specific patient-reported outcomes from the trial, Dr Verma, professor and head of the department of oncology at the University of Calgary in Alberta, Canada, sought to “highlight the patient experience with a focus on health-related quality of life and symptoms,” he said during his presentation at the meeting. “A clinically meaningful – more than 5 points – improvement from baseline in pain score was maintained up to and including cycle 15 in the ribociclib plus letrozole arm.” The placebo arm had a mild, clinically insignificant improvement at most assessment points. For both treatment arms, pain scores increased a bit above baseline levels at the time of disease progression or end of therapy, he said.
Patients completed the EORTC 30-item core QoL questionnaire at their screening visit, and then every 8 weeks for the first 18 months. Then, they completed the questionnaires every 12 weeks until they experienced disease progression, died, were lost to follow-up or withdrew from the study, or stopped treatment.
Statistical analysis of the questionnaire results took into account the patients’ baseline scores, treatments received, and how they were stratified. Investigators assessed both statistical significance and the clinical meaningfulness of changes, defined as a change of 5-10 points.
In MONALEESA-2, 334 patients each were allocated to the ribociclib-letrozole arm and the placebo-letrozole arm. Patients in both arms, said Dr Verma, were very compliant with questionnaire completion. More than 90% of patients who were eligible completed questionnaire through cycle 19 of ribociclib or placebo. He explained that the overall numbers completing the questionnaire declined with time, as more patients had disease progression. Dr Verma said it is important to include those measures, because patients and their families care about “the quality of the time gained,” so patient-reported outcomes should be a part of risk-benefit assessments for new cancer therapies. “While delaying disease progression may help maintain patient quality of life, the addition of novel treatments to existing therapies can add toxicities, which may diminish quality of life,” he said.
— Kari Oakes
Ibrutinib and beyond: optimizing therapy in relapsed CLL
Key clinical point Ibrutinib had durable efficacy in relapsed CLL, and combinations with other targeted agents or with CAR-T cells are promising. Major finding Long-term PFS was better with ibrutinib than with ofatumumab (HR, 0.133). The overall response rate with the triplet of ibrutinib, ublituximab, and umbralisib was 100%. In all, 89% of patients achieved no evidence of disease in marrow when anti-CD19 CAR-T cells were added to ibrutinib. Data source An update of a phase 3 randomized trial among 391 patients with previously treated CLL or SLL (RESONATE). A phase 1/1b trial including 19 patients with mainly relapsed or refractory CLL or SLL. A pilot trial among 10 patients with previously treated, mainly higher-risk, CLL or SLL. Disclosures See article text.
Ibrutinib monotherapy
In the phase 3 randomized RESONATE trial, investigators compared ibrutinib with ofatumumab, an anti-CD20 antibody, among 391 patients with CLL or small lymphocytic lymphoma (SLL), with crossover allowed. Initial results favored ibrutinib.
Investigators led by John C Byrd, MD, director of the division of hematology at the Ohio State University Comprehensive Cancer Center in Columbus, reported updated data in a poster session at the meeting, now with a median 44 months of follow-up in the ibrutinib arm.
Median PFS was not reached with ibrutinib, compared with 8.1 months with ofatumumab (HR, 0.133). The 3-year rate of PFS for ibrutinib and ofatumumab was 59% and 3%, respectively.
The pattern was generally similar across patients stratified by cytogenetics (deletion of 17p, deletion of 11q, or neither), IGHV and TP53 mutation status, and previous lines of therapy, reported Dr Byrd. The 3-year overall survival rate for ibrutinib was 74%. In analyses adjusted for crossover, patients given the inhibitor had a markedly lower risk of death than did peers given the antibody (HR, 0.37). The overall response rate with ibrutinib was 91%. Although the rate of complete response increased with follow-up, it was still just 9%.
The most common grade 3 or worse adverse events were neutropenia (23%), pneumonia (17%), and anemia, thrombocytopenia, and hypertension (8% each). Major hemorrhage was reported in 2 patients (1%) in the ibrutinib group, and 3 (2%) in the ofatumumab group.
The investigators emphasized that these long-term results “show that extended treatment with ibrutinib is tolerable and continues to show sustained PFS in previously treated patients with CLL regardless of high-risk cytogenetics, adding that traditional poor prognostic factors for survival with chemoimmunotherapy, including del(17p) and del(11q), were not significant factors predictive of PFS outcomes with ibrutinib.
Funding and disclosures: Pharmacyclics funded the trial. Dr Byrd disclosed that he receives research funding from Pharmacyclics and other companies.
Ibrutinib plus ublituximab and umbralisib
Investigators led by Loretta J Nastoupil, MD, tested a triplet consisting of ibrutinib with ublituximab (another anti-CD20 antibody) and umbralisib (TGR-1202), an oral PI3 kinase-delta inhibitor, in a phase 1/1b trial of 38 patients with generally heavily pretreated leukemias and lymphomas, including 20 with CLL or SLL. Notably, 8 patients with CLL (50%) had a 17p or 11q deletion.
The median time on study was 11.1 months, reported Dr Nastoupil, of the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston. The overall response rate for the 19 evaluable patients with CLL or SLL was 100% (complete response in 32%, partial response in 68%). The main grade 3 or 4 adverse events in the entire trial population were neutropenia (18%) and pyrexia (8%). Only two patients discontinued treatment because of adverse events.
“An expansion cohort is ongoing at the highest dose, and we will clearly need more patients treated and longer follow-up to really know the efficacy,” commented invited discussant Jennifer R Brown, MD, PhD, director of the Chronic Lymphocytic Leukemia Center at the Dana-Farber Cancer Institute in Boston.
Funding and disclosures: TG Therapeutics funded the trial. Dr Nastoupil has received honoraria and research funding from TG Therapeutics, and honoraria from Pharmacyclics.
Ibrutinib plus CAR-T cells
Investigators in a pilot trial led by Saar Gill, MD, PhD, of the hospital of the University of Pennsylvania in Philadelphia, tested the combination of ibrutinib with chimeric antigen receptor-T cells (CAR-T cells) against CD19, on the basis of preclinical evidence of synergy. The trial participants were 10 patients with CLL or SLL who had not achieved complete response with ibrutinib. All had a 17p deletion or a p53 mutation, or a complex karyotype, and some had a known ibrutinib resistance mutation.
At 3 months, 8 of 9 evaluable patients (89%) had no evidence of disease in bone marrow, reported Dr Gill. Seven patients (78%) achieved a complete or partial radiographic response in the spleen and lymph nodes. Overall, the treatment was well tolerated. One patient developed grade 4 tumor lysis syndrome, and two developed grade 3 cytokine release syndrome. But, none required anticytokine therapy.
Most patients remain on ibrutinib and are being monitored. In addition, the researchers plan to treat 25 more patients with the same combination.
Funding and disclosures: Novartis funded the trial. Dr Gill disclosed that he receives research funding from Novartis (institutional) and has patents for CAR-T cells for AML.
— Susan London
Pazopanib falls short as adjuvant therapy for high-risk RCC
Key clinical point Pazopanib is not efficacious for treating resected high-risk locally advanced RCC. Major finding Compared with placebo, pazopanib started at 600 mg daily did not significantly reduce the risk of DFS events (HR, 0.86; P = .16). Data source A phase 3 randomized controlled trial among 1,538 patients who had undergone nephrectomy for high-risk locally advanced RCC (PROTECT trial). Disclosures Novartis Oncology funded the trial. Dr Motzer disclosed that he is a consultant to Novartis and receives research funding from Novartis (institutional).
The antiangiogenic agent pazopanib is not efficacious when used as adjuvant therapy for resected renal cell carcinoma (RCC) with features that confer a high risk of recurrence, investigators in the PROTECT investigators reported at the meeting. “The trial did not meet its primary endpoint,” concluded lead investigator Robert J Motzer, MD, of Memorial Sloan-Kettering Cancer Center in New York. “Pazopanib is not recommended for adjuvant therapy following resection of locally advanced RCC.”
Adjuvant use of other agents in this class has yielded mixed results, Dr Motzer noted, citing the earlier ASSURE trial found that neither sunitinib nor sorafenib improved disease-free survival (DFS) or overall survival (Lancet 2016;387:2008-2016), and the S-TRAC trial that found that sunitinib improved DFS (N Engl J Med. 2016;375:2246-2254).
Pazopanib is an oral multitargeted tyrosine kinase inhibitor active against the vascular endothelial growth factor receptor (VEGFR). The PROTECT trial included 1,538 patients with resected pT2 (high grade), pT3, or greater nonmetastatic clear cell RCC, a highly vascular tumor typically reliant on aberrant signaling in the VEGF pathway. The patients were assigned evenly to receive pazopanib or placebo for 1 year. The starting dose was lowered from 800 mg daily to 600 mg daily (with escalation permitted) after 403 patients had been treated.
In intention-to-treat analysis among patients started on the 600-mg dose and having a median follow-up of about 31 months, DFS was better with pazopanib but not significantly so (HR, 0.86; P = .16). In secondary analyses, pazopanib had a significant DFS benefit in patients started on the 800-mg dose (HR, 0.69; P = .02) and among the entire trial population started on either dose (HR, 0.80; P = .01).
One possible explanation for the differing results seen with the 2 doses was the difference in follow-up, because the 800-mg group was treated earlier in the trial, said Dr Motzer. But with an additional year of blinded follow-up, the benefit in the 600-mg group diminished, but not in the 800-mg group.
Another possibility was the somewhat better performance of the placebo group used for comparison with the lower dose: the 3-year DFS rate with placebo was 64% for the 600-mg comparison but 56% for the 800-mg comparison. “One factor that could explain differences in the outcomes of the placebo groups includes unidentified patient demographic characteristics,” Dr Motzer added.
Overall survival was statistically indistinguishable between the pazopanib and placebo groups, regardless of dose. However, “the results are inconclusive as the data are not yet mature,” he said, with a definitive analysis planned for 2019.
Patients started on the 600-mg dose of pazopanib had a higher rate of grade 3 or 4 adverse events overall compared with those given placebo (60% vs 21%, respectively), driven in part by higher rates of hypertension and increased alanine aminotransferase levels. “Although the intent of modifying the protocol dose of pazopanib from 800 mg to 600 mg was to reduce the rate of discontinuation and improve the safety profile ... both cohorts had similar discontinuation rates and safety profiles,” Dr Motzer noted.
A QoL analysis for the 600-mg group using the 19-item Functional Assessment of Cancer Therapy Kidney Symptom Index showed values were consistently lower with the drug than with placebo during treatment, with a crossing of the threshold for a minimally important difference at week 8. Pharmacokinetic analyses from the trial, reported in a poster at the meeting, showed that in the group starting pazopanib at 600 mg, DFS was longer in patients who achieved higher drug trough concentrations at week 3 or 5.
— Susan London
Survival improves when patients with cancer self-report symptoms
Key clinical point Patients with metastatic cancer who self-reported symptoms experienced significant improvement in overall survival. Major finding Median overall survival with self-reporting of symptoms compared with usual care was 31.2 and 26 months, respectively. Data source A randomized controlled clinical trial of 766 patients. Funding and disclosures This study was supported by the National Institutes of Health and the Conquer Cancer Foundation of the American Society of Clinical Oncology. Dr Basch and Dr Burstein each reported having no disclosures.
Patients with metastatic cancer who self-reported symptoms during routine cancer treatment experienced a number of benefits, including a statistically significant improvement in overall survival, according to findings from a randomized, controlled clinical trial (see p. e184). The median overall survival in 441 patients receiving treatment for metastatic breast, lung, genitourinary, or gynecologic cancer who were randomized to the self-reporting intervention arm was more than 5 months longer (a nearly 20% increase) than in 325 patients receiving standard care (31.2 vs. 26 months), Ethan Basch, MD, of the Lineberger Comprehensive Cancer Center at the University of North Carolina, Chapel Hill, said at the meeting. “Another way to think of this is [in terms of] 5-year survival. At 5 years, 8% more patients were alive in the self-reporting group,” he said.
In addition, 31% of patients in the intervention arm had better quality of life/physical functioning, compared with those in the control arm, and 7% fewer patients in the intervention arm visited an emergency room during the study. The duration of potentially life-prolonging chemotherapy was increased by an average of 2 months in the intervention arm, he said (JAMA. 2017 Jun 4. doi: 10.1001/jama.2017.7156).
Symptoms such as nausea, pain, and fatigue are common among patients with metastatic cancer and can often go undetected by doctors and nurses until they become severe and physically debilitating, Dr Basch added, noting that patients are often hesitant to call the office between visits to report symptoms.
Dr Basch and his colleagues hypothesized that self-reporting of patient symptoms between visits or before a visit while the patient was in the clinic waiting area would prompt earlier intervention and improve symptom control and outcomes.
Study participants were patients at Memorial Sloan Kettering Cancer Center who had advanced solid genitourinary, gynecologic, breast, or lung tumors and who were receiving outpatient chemotherapy. Those assigned to the intervention group used tablet computers and an online web survey system to report on 12 symptoms commonly experienced during chemotherapy. The system triggers an alert to a nurse when a severe or worsening symptom is reported. Patients in the usual care group discussed symptoms during office visits and were encouraged to call the office between visits if they were concerned about symptoms.
Patients remained on the study until discontinuation of all cancer treatment, hospice, or death.
One possible explanation for the findings is that this self-reporting approach prompts clinicians to manage symptoms before they cause serious downstream complications, Dr Basch said.
The approach may also keep patients more physically functional, which is known from previous studies to have a strong association with better survival, and it may also improve management of chemotherapy side effects, enabling longer duration of beneficial cancer treatment. “In oncology, we often are limited in our ability to give life-prolonging treatment because people don’t tolerate it well,” Dr Basch explained.
“This approach should be considered for inclusion in standard symptoms management as a component of high-quality cancer care,” he concluded, noting that efforts are underway to test the next generation of systems to improve communication between patients and care teams and to figure out how best to integrate these tools into oncology practice.
The system used in the this study was designed for research, but a number of companies have tools currently available for patient-reported outcomes, and others are being developed, Dr Basch said. A National Cancer Institute questionnaire, the PRO-CTCAE, is publicly available and can be loaded into patients’ electronic health records for this purpose as well.
Harold J Burstein, MD, of Dana-Farber Cancer Institute, Boston, said the study findings validate the feeling among many clinicians that patient-focused, team-based care can improve outcomes in a meaningful way for patients. If this were a drug … it would be worth tens, if not hundreds of thousands, of dollars per year … We don’t have those same kinds of dollars to help implement these into our electronic health records or our systems. We need to find ways to support that and make it happen,” he said.
— Sharon Worcester
TRK inhibitor shows ‘striking’ activity and durability across diverse cancers
Key clinical point Larotrectinib has good, durable efficacy when used to treat advanced cancers harboring TRK fusions. Major finding The overall response rate was 76%, and 79% of responses were still ongoing at 12 months. Data source An integrated analysis of phase 1 and 2 trials among 55 children and adults having 17 discrete types of advanced cancer with TRK fusions. Funding and disclosures Loxo Oncology funded the study. Dr Hyman disclosed that he has a consulting or advisory role with Atara Biotherapeutics, Chugai Pharma, and CytomX Therapeutics, and that he receives research funding from AstraZeneca and Puma Biotechnology.
Larotrectinib, an oral inhibitor of tropomyosin receptor kinase (TRK), has durable efficacy across diverse adult and pediatric cancers that harbor a genetic aberration known as TRK fusion, according to findings from an analysis of 3 trials reported at the meeting (see p. e184). Fusion of a TRK gene with an unrelated gene leads to uncontrolled signaling in the TRK pathway, potentially causing tumor growth and addiction to this input, David Hyman, MD, chief of early drug development at Memorial Sloan Kettering Cancer Center in New York, explained in a press briefing.
“One of the defining features of TRK fusions is that they are not just found in one cancer type, but in dozens of different cancer types, and not just in adults, but children as well, spanning the entire lifetime of the person,” he noted. They are rare in common cancers and nearly universal in certain uncommon cancers; collectively, they are present in possibly 5,000 cancers diagnosed each year in the United States.
Dr Hyman and his colleagues analyzed data from 55 patients having 17 discrete types of advanced cancer harboring TRK fusions who were treated with larotrectinib in phase 1 and 2 trials. Results showed an overall response rate of 76%, and the majority of responses were still ongoing at 12 months. “I believe these data support larotrectinib as a potential new standard of care for these patients,” he said. “However, I want to emphasize that really recognizing this benefit in the community will require that we test patients more universally for the presence of TRK fusions or other tumor-agnostic biomarkers, such as microsatellite instability.”
Study details
The investigators analyzed data from 3 trials in which patients with advanced TRK fusion-positive solid cancers received larotrectinib (LOXO-101): a phase 1 trial among 8 adult patients, a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 “basket” trial among 35 adult and adolescent patients (NAVIGATE).
“These patients were identified by local testing,” Dr Hyman noted. “We did not perform central screening to find the TRK fusions, and in fact, 50 different laboratories identified the 55 patients. So in a sense, this really represents the real-world identification of these patients.”
In an integrated analysis, the overall rate of confirmed response as assessed by investigators was 76%, with complete response in 12% of patients and partial response in 64%. Two patients had such deep tumor regression that they experienced downstaging enabling them to undergo potentially curative surgery. Efficacy was consistent regardless of tumor type, which TRK gene was affected, and the fusion partner gene.
Median time to response was 1.8 months. “This is just a reflection of when the first scan was obtained. But in the clinic, patients reported dramatic improvement of their symptoms within days of starting therapy,” Dr Hyman said.
With a median follow-up of 5.8 months, the median duration of response was not yet reached. In all, 79% of responses were still ongoing at 12 months. Median progression-free survival (PFS) was likewise not reached; the 12-month rate was 63%.
The leading treatment-emergent adverse events were fatigue (38%), dizziness (27%), nausea (26%), and anemia (26%). “This is an extremely well tolerated therapy with only 13% of patients requiring any form of dose modification and not a single patient discontinuing due to adverse events,” he said.
It is not clear why some patients had apparent primary resistance to larotrectinib, but their TRK fusion test results may have been incorrect, Dr Hyman speculated. In all, 6 patients developed acquired resistance to larotrectinib; 5 of them were found to have an identical resistance mutation, and 2 went on to receive and have a response to LOXO-195, a next-generation TRK inhibitor that seems to retain activity in the presence of this mutation (Cancer Discov. 2017 June 3. doi: 10.1158/2159-8290.CD-17-0507).
TRK testing
Several next-generation sequencing-based tests already available clinically can pick up TRK fusions, Dr Hyman pointed out. “But it is important for the ordering physician to understand whether the tests they are ordering include fusion detection and, if it’s an option, to select it. Otherwise, they will not find TRK fusions. “The list price for these tests is in the low thousands of dollars,” he noted. In cancers in which sequential single-gene testing is already being done as standard of care, there is “minimal” incremental cost of instead using comprehensive testing that would detect TRK fusions.
Oncologists should be aware that obtaining test results can take weeks, Dr Hyman stressed. “This [testing] should be more broadly adopted and should be adopted at a point in the patient’s treatment [so that they] don’t become too sick, then don’t have an opportunity to be treated even when the test results come back positive.”
— Susan London
QoL preserved with ribociclib-letrozole for advanced breast cancer
Key clinical point Patients who took ribociclib plus letrozole had less pain and no drop in QoL compared with letrozole alone. Major finding QoL was sustained and pain scores decreased when ribociclib was added to letrozole for patients with advanced breast cancer. Data source Double-blind, placebo-controlled phase 3 trial of letrozole plus ribociclib compared with letrozole plus placebo in 668 patients with advanced hormone receptor–positive, HER2-negative breast cancer. Disclosures Dr Verma reported financial relationships with Novartis, which markets ribociclib, and other firms.
Patients with advanced breast cancer whose aromatase inhibitor therapy was supplemented with a cycline-dependent kinase inhibitor had better PFS with no drop in quality of life (QoL). Health-related QoL for patients on the combination therapy was equivalent to that of patients on monotherapy in most aspects, but patients receiving both therapies had a sustained and clinically meaningful decrease in pain.
In addition, the time to definitive deterioration by 10% or more of the global health status/QoL scale score was similar between treatment arms (hazard ratio [HR], 0.944; 95% confidence interval, 0.720-1.237).
The MONALEESA-2 trial had previously shown that the CDK4/6 inhibitor ribociclib, when added to the aromatase inhibitor letrozole, significantly improved PFS for postmenopausal patients with hormone receptor–positive, HER2-negative advanced breast cancer, when compared with letrozole in combination with placebo.
Sunil Verma, MD, reported on health-related QoL and symptoms in the 2 arms of MONALEESA-2, showing change from baseline, time to a definitive 10% deterioration, and the mean scores for on-treatment time compared with end of treatment on the global health-status QoL subscale of the European Organization for Research and Treatment of Cancer's (EORTC's) 30-item core QoL questionnaire.
“During treatment, overall health-related quality of life was maintained from baseline and was similar in both arms,” said Dr Verma, the study’s first author. Changes during treatment were not statistically significant, and did not reach the predetermined threshold for a clinically meaningful difference. The effect of key symptoms such as fatigue, nausea, and vomiting on QoL was similar for patients receiving ribociclib or placebo, he said. Although symptom scores were slightly higher for patients in the active arm of the study, the results were not clinically significant.
Reporting validated, cancer-specific patient-reported outcomes from the trial, Dr Verma, professor and head of the department of oncology at the University of Calgary in Alberta, Canada, sought to “highlight the patient experience with a focus on health-related quality of life and symptoms,” he said during his presentation at the meeting. “A clinically meaningful – more than 5 points – improvement from baseline in pain score was maintained up to and including cycle 15 in the ribociclib plus letrozole arm.” The placebo arm had a mild, clinically insignificant improvement at most assessment points. For both treatment arms, pain scores increased a bit above baseline levels at the time of disease progression or end of therapy, he said.
Patients completed the EORTC 30-item core QoL questionnaire at their screening visit, and then every 8 weeks for the first 18 months. Then, they completed the questionnaires every 12 weeks until they experienced disease progression, died, were lost to follow-up or withdrew from the study, or stopped treatment.
Statistical analysis of the questionnaire results took into account the patients’ baseline scores, treatments received, and how they were stratified. Investigators assessed both statistical significance and the clinical meaningfulness of changes, defined as a change of 5-10 points.
In MONALEESA-2, 334 patients each were allocated to the ribociclib-letrozole arm and the placebo-letrozole arm. Patients in both arms, said Dr Verma, were very compliant with questionnaire completion. More than 90% of patients who were eligible completed questionnaire through cycle 19 of ribociclib or placebo. He explained that the overall numbers completing the questionnaire declined with time, as more patients had disease progression. Dr Verma said it is important to include those measures, because patients and their families care about “the quality of the time gained,” so patient-reported outcomes should be a part of risk-benefit assessments for new cancer therapies. “While delaying disease progression may help maintain patient quality of life, the addition of novel treatments to existing therapies can add toxicities, which may diminish quality of life,” he said.
— Kari Oakes
Ibrutinib and beyond: optimizing therapy in relapsed CLL
Key clinical point Ibrutinib had durable efficacy in relapsed CLL, and combinations with other targeted agents or with CAR-T cells are promising. Major finding Long-term PFS was better with ibrutinib than with ofatumumab (HR, 0.133). The overall response rate with the triplet of ibrutinib, ublituximab, and umbralisib was 100%. In all, 89% of patients achieved no evidence of disease in marrow when anti-CD19 CAR-T cells were added to ibrutinib. Data source An update of a phase 3 randomized trial among 391 patients with previously treated CLL or SLL (RESONATE). A phase 1/1b trial including 19 patients with mainly relapsed or refractory CLL or SLL. A pilot trial among 10 patients with previously treated, mainly higher-risk, CLL or SLL. Disclosures See article text.
Ibrutinib monotherapy
In the phase 3 randomized RESONATE trial, investigators compared ibrutinib with ofatumumab, an anti-CD20 antibody, among 391 patients with CLL or small lymphocytic lymphoma (SLL), with crossover allowed. Initial results favored ibrutinib.
Investigators led by John C Byrd, MD, director of the division of hematology at the Ohio State University Comprehensive Cancer Center in Columbus, reported updated data in a poster session at the meeting, now with a median 44 months of follow-up in the ibrutinib arm.
Median PFS was not reached with ibrutinib, compared with 8.1 months with ofatumumab (HR, 0.133). The 3-year rate of PFS for ibrutinib and ofatumumab was 59% and 3%, respectively.
The pattern was generally similar across patients stratified by cytogenetics (deletion of 17p, deletion of 11q, or neither), IGHV and TP53 mutation status, and previous lines of therapy, reported Dr Byrd. The 3-year overall survival rate for ibrutinib was 74%. In analyses adjusted for crossover, patients given the inhibitor had a markedly lower risk of death than did peers given the antibody (HR, 0.37). The overall response rate with ibrutinib was 91%. Although the rate of complete response increased with follow-up, it was still just 9%.
The most common grade 3 or worse adverse events were neutropenia (23%), pneumonia (17%), and anemia, thrombocytopenia, and hypertension (8% each). Major hemorrhage was reported in 2 patients (1%) in the ibrutinib group, and 3 (2%) in the ofatumumab group.
The investigators emphasized that these long-term results “show that extended treatment with ibrutinib is tolerable and continues to show sustained PFS in previously treated patients with CLL regardless of high-risk cytogenetics, adding that traditional poor prognostic factors for survival with chemoimmunotherapy, including del(17p) and del(11q), were not significant factors predictive of PFS outcomes with ibrutinib.
Funding and disclosures: Pharmacyclics funded the trial. Dr Byrd disclosed that he receives research funding from Pharmacyclics and other companies.
Ibrutinib plus ublituximab and umbralisib
Investigators led by Loretta J Nastoupil, MD, tested a triplet consisting of ibrutinib with ublituximab (another anti-CD20 antibody) and umbralisib (TGR-1202), an oral PI3 kinase-delta inhibitor, in a phase 1/1b trial of 38 patients with generally heavily pretreated leukemias and lymphomas, including 20 with CLL or SLL. Notably, 8 patients with CLL (50%) had a 17p or 11q deletion.
The median time on study was 11.1 months, reported Dr Nastoupil, of the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston. The overall response rate for the 19 evaluable patients with CLL or SLL was 100% (complete response in 32%, partial response in 68%). The main grade 3 or 4 adverse events in the entire trial population were neutropenia (18%) and pyrexia (8%). Only two patients discontinued treatment because of adverse events.
“An expansion cohort is ongoing at the highest dose, and we will clearly need more patients treated and longer follow-up to really know the efficacy,” commented invited discussant Jennifer R Brown, MD, PhD, director of the Chronic Lymphocytic Leukemia Center at the Dana-Farber Cancer Institute in Boston.
Funding and disclosures: TG Therapeutics funded the trial. Dr Nastoupil has received honoraria and research funding from TG Therapeutics, and honoraria from Pharmacyclics.
Ibrutinib plus CAR-T cells
Investigators in a pilot trial led by Saar Gill, MD, PhD, of the hospital of the University of Pennsylvania in Philadelphia, tested the combination of ibrutinib with chimeric antigen receptor-T cells (CAR-T cells) against CD19, on the basis of preclinical evidence of synergy. The trial participants were 10 patients with CLL or SLL who had not achieved complete response with ibrutinib. All had a 17p deletion or a p53 mutation, or a complex karyotype, and some had a known ibrutinib resistance mutation.
At 3 months, 8 of 9 evaluable patients (89%) had no evidence of disease in bone marrow, reported Dr Gill. Seven patients (78%) achieved a complete or partial radiographic response in the spleen and lymph nodes. Overall, the treatment was well tolerated. One patient developed grade 4 tumor lysis syndrome, and two developed grade 3 cytokine release syndrome. But, none required anticytokine therapy.
Most patients remain on ibrutinib and are being monitored. In addition, the researchers plan to treat 25 more patients with the same combination.
Funding and disclosures: Novartis funded the trial. Dr Gill disclosed that he receives research funding from Novartis (institutional) and has patents for CAR-T cells for AML.
— Susan London
Pazopanib falls short as adjuvant therapy for high-risk RCC
Key clinical point Pazopanib is not efficacious for treating resected high-risk locally advanced RCC. Major finding Compared with placebo, pazopanib started at 600 mg daily did not significantly reduce the risk of DFS events (HR, 0.86; P = .16). Data source A phase 3 randomized controlled trial among 1,538 patients who had undergone nephrectomy for high-risk locally advanced RCC (PROTECT trial). Disclosures Novartis Oncology funded the trial. Dr Motzer disclosed that he is a consultant to Novartis and receives research funding from Novartis (institutional).
The antiangiogenic agent pazopanib is not efficacious when used as adjuvant therapy for resected renal cell carcinoma (RCC) with features that confer a high risk of recurrence, investigators in the PROTECT investigators reported at the meeting. “The trial did not meet its primary endpoint,” concluded lead investigator Robert J Motzer, MD, of Memorial Sloan-Kettering Cancer Center in New York. “Pazopanib is not recommended for adjuvant therapy following resection of locally advanced RCC.”
Adjuvant use of other agents in this class has yielded mixed results, Dr Motzer noted, citing the earlier ASSURE trial found that neither sunitinib nor sorafenib improved disease-free survival (DFS) or overall survival (Lancet 2016;387:2008-2016), and the S-TRAC trial that found that sunitinib improved DFS (N Engl J Med. 2016;375:2246-2254).
Pazopanib is an oral multitargeted tyrosine kinase inhibitor active against the vascular endothelial growth factor receptor (VEGFR). The PROTECT trial included 1,538 patients with resected pT2 (high grade), pT3, or greater nonmetastatic clear cell RCC, a highly vascular tumor typically reliant on aberrant signaling in the VEGF pathway. The patients were assigned evenly to receive pazopanib or placebo for 1 year. The starting dose was lowered from 800 mg daily to 600 mg daily (with escalation permitted) after 403 patients had been treated.
In intention-to-treat analysis among patients started on the 600-mg dose and having a median follow-up of about 31 months, DFS was better with pazopanib but not significantly so (HR, 0.86; P = .16). In secondary analyses, pazopanib had a significant DFS benefit in patients started on the 800-mg dose (HR, 0.69; P = .02) and among the entire trial population started on either dose (HR, 0.80; P = .01).
One possible explanation for the differing results seen with the 2 doses was the difference in follow-up, because the 800-mg group was treated earlier in the trial, said Dr Motzer. But with an additional year of blinded follow-up, the benefit in the 600-mg group diminished, but not in the 800-mg group.
Another possibility was the somewhat better performance of the placebo group used for comparison with the lower dose: the 3-year DFS rate with placebo was 64% for the 600-mg comparison but 56% for the 800-mg comparison. “One factor that could explain differences in the outcomes of the placebo groups includes unidentified patient demographic characteristics,” Dr Motzer added.
Overall survival was statistically indistinguishable between the pazopanib and placebo groups, regardless of dose. However, “the results are inconclusive as the data are not yet mature,” he said, with a definitive analysis planned for 2019.
Patients started on the 600-mg dose of pazopanib had a higher rate of grade 3 or 4 adverse events overall compared with those given placebo (60% vs 21%, respectively), driven in part by higher rates of hypertension and increased alanine aminotransferase levels. “Although the intent of modifying the protocol dose of pazopanib from 800 mg to 600 mg was to reduce the rate of discontinuation and improve the safety profile ... both cohorts had similar discontinuation rates and safety profiles,” Dr Motzer noted.
A QoL analysis for the 600-mg group using the 19-item Functional Assessment of Cancer Therapy Kidney Symptom Index showed values were consistently lower with the drug than with placebo during treatment, with a crossing of the threshold for a minimally important difference at week 8. Pharmacokinetic analyses from the trial, reported in a poster at the meeting, showed that in the group starting pazopanib at 600 mg, DFS was longer in patients who achieved higher drug trough concentrations at week 3 or 5.
— Susan London
Do staphylococci play a role in acne?
Clues are emerging that Staphylococcus species may be contributors to acne vulgaris, according to a new study that examined changes in the skin microbiota of acne patients undergoing topical treatment.
Comparing topical antibiotic treatment for acne with a novel cosmetic formulation, Brigitte Dreno, MD, PhD, and her colleagues found that normal skin had fewer surface staphylococci than did skin with comedones or papulopustular eruptions (P = .004 for comedones; P = .003 for papules and pustules). Further, the number of staphylococci increased as acne severity increased (P less than .05 for the increase seen between scores of GEA-2 and GEA-3 on the Global Acne Severity Scale).
These results were seen in a split-face study of 26 adults with mild to moderate acne (GEA-2 and GEA-3) that compared a topical 4% erythromycin gel to a “dermocosmetic” containing lipohydroxy, linoleic, and salicylic acids, niacinamide, piroctone olamine, a ceramide, and water from a thermal spring. Each patient used each product on half of his or her face for 28 consecutive days while avoiding use of other skin care products or topical medications during the study period (Exp Dermatol. 2017;26[9]:798-803; doi: 10.1111/exd.13296).
Patients’ acne severity was assessed at days 0, 14, and 28 using the GEA scale, together with a count of inflammatory and noninflammatory lesions. The mean GEA grade for patients at enrollment was 2.4. For each patient, skin microbiota samples were obtained from an area with comedones, an area with papulopustular lesions, and an unaffected area.
Dr. Dreno, professor of dermatology at University Hospital, Nantes, France, and her coauthors noted that the range of bacterial diversity was similar on all the facial areas sampled.
After 28 days of treatment, the antibiotic-treated facial skin had significantly less Actinobacteria species, including corynebacteria and propionibacteria. However, the effect on Staphylococci was limited, “potentially confirming the increased resistance of the bacterium to macrolides,” they wrote.
Facial skin treated with the cosmetic, by contrast, had fewer Actinobacteria and fewer Staphylococcus species by the end of the study period, though staphylococci remained the predominant organisms after treatment. Patients saw a significant reduction in both inflammatory and noninflammatory lesions during the study period (P less than .05), with no significant difference between antibiotic- and cosmetic-treated skin.
The authors said that previous studies have shown that propionibacteria are common commensal organisms, representing up to 30% of the bacteria on healthy skin. For the individuals enrolled in the study, though, propionibacteria made up just 2% of the bacteria on the surface of both healthy skin and skin with acne.
Staphylococci, and S. epidermidis in particular, may use fermentation as a defense against other organisms in the skin microbiota. Since staphylococci are aerobic and facultative anaerobic species, they flourish on the skin surface. Propionibacterium acnes, by contrast, is anaerobic, and primarily inhabits the sebaceous follicle.
The complex interplay of skin microbiota contribute to acne and other dermatologic pathology in ways that are just beginning to be understood. “Even though the association between P. acnes and acne vulgaris is well established, very few studies have investigated the entire facial skin microbiota of patients with acne,” wrote Dr. Dreno and her coauthors.
One of the six study authors are employees of L’Oreal; two are employees of La Roche–Posay Dermatological Laboratory, which funded the study and produced the cosmetic product used by the researchers.
[email protected]
On Twitter @karioakes
Clues are emerging that Staphylococcus species may be contributors to acne vulgaris, according to a new study that examined changes in the skin microbiota of acne patients undergoing topical treatment.
Comparing topical antibiotic treatment for acne with a novel cosmetic formulation, Brigitte Dreno, MD, PhD, and her colleagues found that normal skin had fewer surface staphylococci than did skin with comedones or papulopustular eruptions (P = .004 for comedones; P = .003 for papules and pustules). Further, the number of staphylococci increased as acne severity increased (P less than .05 for the increase seen between scores of GEA-2 and GEA-3 on the Global Acne Severity Scale).
These results were seen in a split-face study of 26 adults with mild to moderate acne (GEA-2 and GEA-3) that compared a topical 4% erythromycin gel to a “dermocosmetic” containing lipohydroxy, linoleic, and salicylic acids, niacinamide, piroctone olamine, a ceramide, and water from a thermal spring. Each patient used each product on half of his or her face for 28 consecutive days while avoiding use of other skin care products or topical medications during the study period (Exp Dermatol. 2017;26[9]:798-803; doi: 10.1111/exd.13296).
Patients’ acne severity was assessed at days 0, 14, and 28 using the GEA scale, together with a count of inflammatory and noninflammatory lesions. The mean GEA grade for patients at enrollment was 2.4. For each patient, skin microbiota samples were obtained from an area with comedones, an area with papulopustular lesions, and an unaffected area.
Dr. Dreno, professor of dermatology at University Hospital, Nantes, France, and her coauthors noted that the range of bacterial diversity was similar on all the facial areas sampled.
After 28 days of treatment, the antibiotic-treated facial skin had significantly less Actinobacteria species, including corynebacteria and propionibacteria. However, the effect on Staphylococci was limited, “potentially confirming the increased resistance of the bacterium to macrolides,” they wrote.
Facial skin treated with the cosmetic, by contrast, had fewer Actinobacteria and fewer Staphylococcus species by the end of the study period, though staphylococci remained the predominant organisms after treatment. Patients saw a significant reduction in both inflammatory and noninflammatory lesions during the study period (P less than .05), with no significant difference between antibiotic- and cosmetic-treated skin.
The authors said that previous studies have shown that propionibacteria are common commensal organisms, representing up to 30% of the bacteria on healthy skin. For the individuals enrolled in the study, though, propionibacteria made up just 2% of the bacteria on the surface of both healthy skin and skin with acne.
Staphylococci, and S. epidermidis in particular, may use fermentation as a defense against other organisms in the skin microbiota. Since staphylococci are aerobic and facultative anaerobic species, they flourish on the skin surface. Propionibacterium acnes, by contrast, is anaerobic, and primarily inhabits the sebaceous follicle.
The complex interplay of skin microbiota contribute to acne and other dermatologic pathology in ways that are just beginning to be understood. “Even though the association between P. acnes and acne vulgaris is well established, very few studies have investigated the entire facial skin microbiota of patients with acne,” wrote Dr. Dreno and her coauthors.
One of the six study authors are employees of L’Oreal; two are employees of La Roche–Posay Dermatological Laboratory, which funded the study and produced the cosmetic product used by the researchers.
[email protected]
On Twitter @karioakes
Clues are emerging that Staphylococcus species may be contributors to acne vulgaris, according to a new study that examined changes in the skin microbiota of acne patients undergoing topical treatment.
Comparing topical antibiotic treatment for acne with a novel cosmetic formulation, Brigitte Dreno, MD, PhD, and her colleagues found that normal skin had fewer surface staphylococci than did skin with comedones or papulopustular eruptions (P = .004 for comedones; P = .003 for papules and pustules). Further, the number of staphylococci increased as acne severity increased (P less than .05 for the increase seen between scores of GEA-2 and GEA-3 on the Global Acne Severity Scale).
These results were seen in a split-face study of 26 adults with mild to moderate acne (GEA-2 and GEA-3) that compared a topical 4% erythromycin gel to a “dermocosmetic” containing lipohydroxy, linoleic, and salicylic acids, niacinamide, piroctone olamine, a ceramide, and water from a thermal spring. Each patient used each product on half of his or her face for 28 consecutive days while avoiding use of other skin care products or topical medications during the study period (Exp Dermatol. 2017;26[9]:798-803; doi: 10.1111/exd.13296).
Patients’ acne severity was assessed at days 0, 14, and 28 using the GEA scale, together with a count of inflammatory and noninflammatory lesions. The mean GEA grade for patients at enrollment was 2.4. For each patient, skin microbiota samples were obtained from an area with comedones, an area with papulopustular lesions, and an unaffected area.
Dr. Dreno, professor of dermatology at University Hospital, Nantes, France, and her coauthors noted that the range of bacterial diversity was similar on all the facial areas sampled.
After 28 days of treatment, the antibiotic-treated facial skin had significantly less Actinobacteria species, including corynebacteria and propionibacteria. However, the effect on Staphylococci was limited, “potentially confirming the increased resistance of the bacterium to macrolides,” they wrote.
Facial skin treated with the cosmetic, by contrast, had fewer Actinobacteria and fewer Staphylococcus species by the end of the study period, though staphylococci remained the predominant organisms after treatment. Patients saw a significant reduction in both inflammatory and noninflammatory lesions during the study period (P less than .05), with no significant difference between antibiotic- and cosmetic-treated skin.
The authors said that previous studies have shown that propionibacteria are common commensal organisms, representing up to 30% of the bacteria on healthy skin. For the individuals enrolled in the study, though, propionibacteria made up just 2% of the bacteria on the surface of both healthy skin and skin with acne.
Staphylococci, and S. epidermidis in particular, may use fermentation as a defense against other organisms in the skin microbiota. Since staphylococci are aerobic and facultative anaerobic species, they flourish on the skin surface. Propionibacterium acnes, by contrast, is anaerobic, and primarily inhabits the sebaceous follicle.
The complex interplay of skin microbiota contribute to acne and other dermatologic pathology in ways that are just beginning to be understood. “Even though the association between P. acnes and acne vulgaris is well established, very few studies have investigated the entire facial skin microbiota of patients with acne,” wrote Dr. Dreno and her coauthors.
One of the six study authors are employees of L’Oreal; two are employees of La Roche–Posay Dermatological Laboratory, which funded the study and produced the cosmetic product used by the researchers.
[email protected]
On Twitter @karioakes
FROM EXPERIMENTAL DERMATOLOGY
Key clinical point:
Major finding: Staphylococci increased as acne severity increased (P less than .05 for increase from GEA-2 to GEA-3 on the Global Acne Severity Scale).
Data source: A split-face study of erythromycin gel and a novel cosmetic product in 26 adult patients with acne.
Disclosures: One of the six study authors are employees of L’Oreal; two are employees of La Roche–Posay Dermatological Laboratory, which funded the study and produced the cosmetic product used by the researchers.
Is pain or dependency driving elevated opioid use among long-term cancer survivors?
Rates of opioid prescribing were about 1.2 times higher overall among cancer survivors up to 10 years after diagnosis, compared with matched controls, with more than threefold higher rates of opioid prescriptions for survivors of some cancers, according to a Canadian population-based cohort study.
Searching for the cause of these elevated rates reveals the complexity of the survivorship experience, and may also point the way to areas where there’s work to be done, according to physicians whose practices touch the lives of cancer survivors.
In a retrospective matched cohort study of participants in the Ontario Health Insurance Plan and the Ontario Drug Benefits Program, Rinku Sutradhar, PhD, of the University of Toronto, and her colleagues, identified patients aged 18-64 who had a cancer diagnosis at least 5 years previously. Patients were included only if they had not had a cancer recurrence or another malignancy. When compared 1:1 to age- and sex-matched controls, the 8,601 cancer survivors had a relative rate of opioid prescribing of 1.220 (95% confidence interval [CI], 1.209-1.232), the investigators reported (Cancer 2017 Aug 17. doi: 10.1002/cncr.30839).
Opioid prescribing rates varied according to the type of cancer the survivor had had, with a relative rate of 3.119 for noncolorectal gastrointestinal cancer survivors and a rate of 2.066 for lung cancer survivors. Individuals with nonprostate genitourinary cancers had a prescribing rate of 1.619. All of these differences were statistically significant.
Elevated prescribing rates were not seen in patients with brain, breast, colorectal, head and neck, or prostate cancers. The relative rate of prescribing for hematologic cancers was 1.383, a difference that approached, but did not quite reach, statistical significance (P = .0512).
When multivariable analysis was used to stratify individuals by length of time since cancer diagnosis, a significantly elevated relative rate of opioid prescribing persisted; for those 5-10 years from diagnosis, the relative rate was 1.190 (95% CI, 1.040-1.362, P = .011), while for those diagnosed at least 10 years ago, the relative rate was 1.244 (95% CI, 1.090-1.420; P = .00118).
Multivariable analysis was also used to control for income, rural residence, and comorbidities. However, the study could capture only opioids that were obtained with a prescription, and could not track whether medications were taken by the person for whom they were prescribed, Dr. Sutradhar and her colleagues said.
Cancer survivors may have a higher prevalence of chronic pain than the general population for reasons related both to their initial diagnosis and the sequelae of treatments such as surgery, chemotherapy, and radiation therapy, the investigators noted, adding, “it is also possible that a higher rate of opioid prescribing among survivors is due to a dependency that originated from opioid use earlier in the disease trajectory.”
Because of the potential for opioid use disorder and the many adverse effects that can be associated with long-term opioid use, they said, “primary care providers who treat cancer survivors should be encouraged to critically examine reasons for lingering opioid use among their patients.”
The oncologist’s perspective
Walter M. Stadler, MD, an oncologist who treats genitourinary and hematologic cancers, also wonders whether it’s pain or dependency that’s driving the increased prescribing rates in cancer survivors.

One opportunity to reassess which medications and treatment modalities are appropriate for the cancer survivor, said Dr. Stadler, is at the point of discharge from oncology care, which usually happens at about the 5-year mark for patients with no evidence of disease. Survivorship has received more attention since the 2005 Institute of Medicine report calling for increased attention to cancer survivors’ ongoing care. However, he said, “it’s not clear that we do a very good job in terms of educating either the patient or their primary care physician in regards to the kinds of things that we expect, the kinds of things that need to be done, or even a good summary of the therapy that was provided.”
There are resources that can help, he said. “That’s why organizations like [the American Society for Clinical Oncology] have put together some more formal survivorship plans that should be provided when patients are transitioned.”
The realities of clinical life can get in the way of implementation, though. Oncologists are already stretched thin, and most electronic health record systems don’t integrate well with survivorship documentation. Finding staff who can spend the time to gather and package all the necessary information can also be a problem: “People are expensive, and none of us have extra cash lying around,” said Dr. Stadler.
Still, he said, “like a lot of good papers, this raises some issues and areas for further investigation.” First, he said, physicians must assess whether cancer survivors are having chronic pain, and then sort things out from there. “What are the pain syndromes – and what are we doing about them? – because it’s not something that’s been well addressed.”
What can primary care offer?
Larissa Nekhlyudov, MD, is an internal medicine physician whose clinical practice straddles two domains. She sees patients, including some cancer survivors, as a primary care provider; she also provides care in a survivorship clinic to adult survivors of childhood cancers. There, she is able to focus more on survivorship care, developing a care plan and communicating with primary care providers about care elements her patients need.

It’s reasonable to think that there might be an increased risk for chronic pain syndromes in some of the types of cancer in which elevated opioid prescribing rates were seen, said Dr. Nekhlyudov. “Maybe this is okay.
“Pain in cancer survivors is so multidimensional that it’s quite possible that some of these cancer survivors – gynecologic, lung, other gastrointestinal, genitourinary – might have peripheral neuropathy, adhesions, and so many potential late effects,” said Dr. Nekhlyudov. “However, narcotics are not necessarily the preferred and the only method to treat this pain,” she said, noting that optimal survivorship care might seek to transition these patients to nonopioid therapies or, at least, a multimodal approach.
When she’s wearing her survivorship care hat, said Dr. Nekhlyudov, managing pain medication isn’t always at the top of the to-do list in an office visit. “It’s certainly not uncommon that patients will have a variety of pain issues. But in the survivorship domain, I think that we don’t take the role of managing their pain medications; that piece belongs, really, to their primary care provider,” she said.
“In many ways, it’s difficult to distinguish how much of their pain is related to their cancer, versus not, and figuring out alternatives,” said Dr. Nekhlyudov, applauding the authors’ recognition of the need for a multimodal approach in cancer survivors with pain. However, she said, “that sounds really great on paper, but it’s really not readily available.”
Even in the resource-rich greater Boston area where she practices, said Dr. Nekhlyudov, “it’s very difficult for cancer patients – and noncancer patients – to get hooked into a multidisciplinary, holistic program for pain.”
Although the long-term perspective is helpful, Dr. Nekhlyudov hopes for research that can help identify at what point, and by whom, the opioids were initiated in the cancer survivor population. “What is their trajectory from the time of diagnosis? Are these patients who are started on narcotics during their cancer treatment, and then continue on forever, or are some of these patients being started later, because of late effects?”
In any case, she said, “one of the key pieces is the ownership for this really belongs with both oncologists and the primary care providers.”
Mental health implications of survivorship
Viewing the issue through the lens of mental health offers a slightly different perspective. Thomas B. Strouse, MD, is a psychiatrist who holds the Maddie Katz Chair in palliative care research and education at the University of California, Los Angeles. He said he laments the current “opioidophobia” that calls into question any long-term opioid prescribing.
Acknowledging that there’s certainly a serious nationwide problem with both prescription and nonprescription opioid abuse, Dr. Strouse said he still finds it unfortunate that the current situation has “reactivated for many people a certain set of reflexes that say that any chronic opioid use is always a bad thing. That’s simply not true,” he said.
“Whether opioids are the right treatment for all of those patients, of course, is an entirely fair question. But it’s unfortunate, or wrong, for everybody to approach this article and to say that we know that for all of these patients, chronic opioid therapy is not appropriate,” he added.
Chronic pain that lingers after cancer treatments affects “a very significant minority of cancer survivors,” he said. It’s also true that the meaning of pain can be different for cancer survivors, said Dr. Strouse. For a cancer survivor, “any new pain is cancer pain until proven otherwise,” he said.
Further, pivoting from the attentive, multidisciplinary, wraparound care often received during cancer treatment to the relatively unsupported survivorship experience can be a rough transition for some. Despite the grim reason for the connection, “frequently, it’s the best experience of patients’ lives from a human relations perspective. … We don’t think enough about the loss that the end of cancer treatment may mean for people who may have otherwise unsatisfactory relationships in their lives,” he said.
Dr. Strouse, who works extensively with cancer survivors, said that the elevated rate of opioid prescribing seen in this study “opens the door to a bigger discussion about the challenges in the relatively empty domain of survivorship.” After discharge from cancer care, patients are all too often left without a navigator to help them through the years when, though their treatment is complete, anxiety, financial and social strain, and pain may linger.
The study, he said, should be a call to physicians for “a more meaningful commitment to understanding the burdens of survivorship, and actually offering meaningful clinical services to those people in an integrated and appropriate way.” This might include determining a patient’s absolute minimum opioid requirement, with a goal of getting the patient off opioids, but also making sure the patient has knowledge of and access to alternative pharmacologic and nonpharmacologic treatments for pain. “That seems like a reasonable approach,” said Dr. Strouse.
None of the study’s authors or the physicians interviewed for commentary had relevant conflicts of interest.
[email protected]
On Twitter @karioakes
Rates of opioid prescribing were about 1.2 times higher overall among cancer survivors up to 10 years after diagnosis, compared with matched controls, with more than threefold higher rates of opioid prescriptions for survivors of some cancers, according to a Canadian population-based cohort study.
Searching for the cause of these elevated rates reveals the complexity of the survivorship experience, and may also point the way to areas where there’s work to be done, according to physicians whose practices touch the lives of cancer survivors.
In a retrospective matched cohort study of participants in the Ontario Health Insurance Plan and the Ontario Drug Benefits Program, Rinku Sutradhar, PhD, of the University of Toronto, and her colleagues, identified patients aged 18-64 who had a cancer diagnosis at least 5 years previously. Patients were included only if they had not had a cancer recurrence or another malignancy. When compared 1:1 to age- and sex-matched controls, the 8,601 cancer survivors had a relative rate of opioid prescribing of 1.220 (95% confidence interval [CI], 1.209-1.232), the investigators reported (Cancer 2017 Aug 17. doi: 10.1002/cncr.30839).
Opioid prescribing rates varied according to the type of cancer the survivor had had, with a relative rate of 3.119 for noncolorectal gastrointestinal cancer survivors and a rate of 2.066 for lung cancer survivors. Individuals with nonprostate genitourinary cancers had a prescribing rate of 1.619. All of these differences were statistically significant.
Elevated prescribing rates were not seen in patients with brain, breast, colorectal, head and neck, or prostate cancers. The relative rate of prescribing for hematologic cancers was 1.383, a difference that approached, but did not quite reach, statistical significance (P = .0512).
When multivariable analysis was used to stratify individuals by length of time since cancer diagnosis, a significantly elevated relative rate of opioid prescribing persisted; for those 5-10 years from diagnosis, the relative rate was 1.190 (95% CI, 1.040-1.362, P = .011), while for those diagnosed at least 10 years ago, the relative rate was 1.244 (95% CI, 1.090-1.420; P = .00118).
Multivariable analysis was also used to control for income, rural residence, and comorbidities. However, the study could capture only opioids that were obtained with a prescription, and could not track whether medications were taken by the person for whom they were prescribed, Dr. Sutradhar and her colleagues said.
Cancer survivors may have a higher prevalence of chronic pain than the general population for reasons related both to their initial diagnosis and the sequelae of treatments such as surgery, chemotherapy, and radiation therapy, the investigators noted, adding, “it is also possible that a higher rate of opioid prescribing among survivors is due to a dependency that originated from opioid use earlier in the disease trajectory.”
Because of the potential for opioid use disorder and the many adverse effects that can be associated with long-term opioid use, they said, “primary care providers who treat cancer survivors should be encouraged to critically examine reasons for lingering opioid use among their patients.”
The oncologist’s perspective
Walter M. Stadler, MD, an oncologist who treats genitourinary and hematologic cancers, also wonders whether it’s pain or dependency that’s driving the increased prescribing rates in cancer survivors.
One opportunity to reassess which medications and treatment modalities are appropriate for the cancer survivor, said Dr. Stadler, is at the point of discharge from oncology care, which usually happens at about the 5-year mark for patients with no evidence of disease. Survivorship has received more attention since the 2005 Institute of Medicine report calling for increased attention to cancer survivors’ ongoing care. However, he said, “it’s not clear that we do a very good job in terms of educating either the patient or their primary care physician in regards to the kinds of things that we expect, the kinds of things that need to be done, or even a good summary of the therapy that was provided.”
There are resources that can help, he said. “That’s why organizations like [the American Society for Clinical Oncology] have put together some more formal survivorship plans that should be provided when patients are transitioned.”
The realities of clinical life can get in the way of implementation, though. Oncologists are already stretched thin, and most electronic health record systems don’t integrate well with survivorship documentation. Finding staff who can spend the time to gather and package all the necessary information can also be a problem: “People are expensive, and none of us have extra cash lying around,” said Dr. Stadler.
Still, he said, “like a lot of good papers, this raises some issues and areas for further investigation.” First, he said, physicians must assess whether cancer survivors are having chronic pain, and then sort things out from there. “What are the pain syndromes – and what are we doing about them? – because it’s not something that’s been well addressed.”
What can primary care offer?
Larissa Nekhlyudov, MD, is an internal medicine physician whose clinical practice straddles two domains. She sees patients, including some cancer survivors, as a primary care provider; she also provides care in a survivorship clinic to adult survivors of childhood cancers. There, she is able to focus more on survivorship care, developing a care plan and communicating with primary care providers about care elements her patients need.
It’s reasonable to think that there might be an increased risk for chronic pain syndromes in some of the types of cancer in which elevated opioid prescribing rates were seen, said Dr. Nekhlyudov. “Maybe this is okay.
“Pain in cancer survivors is so multidimensional that it’s quite possible that some of these cancer survivors – gynecologic, lung, other gastrointestinal, genitourinary – might have peripheral neuropathy, adhesions, and so many potential late effects,” said Dr. Nekhlyudov. “However, narcotics are not necessarily the preferred and the only method to treat this pain,” she said, noting that optimal survivorship care might seek to transition these patients to nonopioid therapies or, at least, a multimodal approach.
When she’s wearing her survivorship care hat, said Dr. Nekhlyudov, managing pain medication isn’t always at the top of the to-do list in an office visit. “It’s certainly not uncommon that patients will have a variety of pain issues. But in the survivorship domain, I think that we don’t take the role of managing their pain medications; that piece belongs, really, to their primary care provider,” she said.
“In many ways, it’s difficult to distinguish how much of their pain is related to their cancer, versus not, and figuring out alternatives,” said Dr. Nekhlyudov, applauding the authors’ recognition of the need for a multimodal approach in cancer survivors with pain. However, she said, “that sounds really great on paper, but it’s really not readily available.”
Even in the resource-rich greater Boston area where she practices, said Dr. Nekhlyudov, “it’s very difficult for cancer patients – and noncancer patients – to get hooked into a multidisciplinary, holistic program for pain.”
Although the long-term perspective is helpful, Dr. Nekhlyudov hopes for research that can help identify at what point, and by whom, the opioids were initiated in the cancer survivor population. “What is their trajectory from the time of diagnosis? Are these patients who are started on narcotics during their cancer treatment, and then continue on forever, or are some of these patients being started later, because of late effects?”
In any case, she said, “one of the key pieces is the ownership for this really belongs with both oncologists and the primary care providers.”
Mental health implications of survivorship
Viewing the issue through the lens of mental health offers a slightly different perspective. Thomas B. Strouse, MD, is a psychiatrist who holds the Maddie Katz Chair in palliative care research and education at the University of California, Los Angeles. He said he laments the current “opioidophobia” that calls into question any long-term opioid prescribing.
Acknowledging that there’s certainly a serious nationwide problem with both prescription and nonprescription opioid abuse, Dr. Strouse said he still finds it unfortunate that the current situation has “reactivated for many people a certain set of reflexes that say that any chronic opioid use is always a bad thing. That’s simply not true,” he said.
“Whether opioids are the right treatment for all of those patients, of course, is an entirely fair question. But it’s unfortunate, or wrong, for everybody to approach this article and to say that we know that for all of these patients, chronic opioid therapy is not appropriate,” he added.
Chronic pain that lingers after cancer treatments affects “a very significant minority of cancer survivors,” he said. It’s also true that the meaning of pain can be different for cancer survivors, said Dr. Strouse. For a cancer survivor, “any new pain is cancer pain until proven otherwise,” he said.
Further, pivoting from the attentive, multidisciplinary, wraparound care often received during cancer treatment to the relatively unsupported survivorship experience can be a rough transition for some. Despite the grim reason for the connection, “frequently, it’s the best experience of patients’ lives from a human relations perspective. … We don’t think enough about the loss that the end of cancer treatment may mean for people who may have otherwise unsatisfactory relationships in their lives,” he said.
Dr. Strouse, who works extensively with cancer survivors, said that the elevated rate of opioid prescribing seen in this study “opens the door to a bigger discussion about the challenges in the relatively empty domain of survivorship.” After discharge from cancer care, patients are all too often left without a navigator to help them through the years when, though their treatment is complete, anxiety, financial and social strain, and pain may linger.
The study, he said, should be a call to physicians for “a more meaningful commitment to understanding the burdens of survivorship, and actually offering meaningful clinical services to those people in an integrated and appropriate way.” This might include determining a patient’s absolute minimum opioid requirement, with a goal of getting the patient off opioids, but also making sure the patient has knowledge of and access to alternative pharmacologic and nonpharmacologic treatments for pain. “That seems like a reasonable approach,” said Dr. Strouse.
None of the study’s authors or the physicians interviewed for commentary had relevant conflicts of interest.
[email protected]
On Twitter @karioakes
Rates of opioid prescribing were about 1.2 times higher overall among cancer survivors up to 10 years after diagnosis, compared with matched controls, with more than threefold higher rates of opioid prescriptions for survivors of some cancers, according to a Canadian population-based cohort study.
Searching for the cause of these elevated rates reveals the complexity of the survivorship experience, and may also point the way to areas where there’s work to be done, according to physicians whose practices touch the lives of cancer survivors.
In a retrospective matched cohort study of participants in the Ontario Health Insurance Plan and the Ontario Drug Benefits Program, Rinku Sutradhar, PhD, of the University of Toronto, and her colleagues, identified patients aged 18-64 who had a cancer diagnosis at least 5 years previously. Patients were included only if they had not had a cancer recurrence or another malignancy. When compared 1:1 to age- and sex-matched controls, the 8,601 cancer survivors had a relative rate of opioid prescribing of 1.220 (95% confidence interval [CI], 1.209-1.232), the investigators reported (Cancer 2017 Aug 17. doi: 10.1002/cncr.30839).
Opioid prescribing rates varied according to the type of cancer the survivor had had, with a relative rate of 3.119 for noncolorectal gastrointestinal cancer survivors and a rate of 2.066 for lung cancer survivors. Individuals with nonprostate genitourinary cancers had a prescribing rate of 1.619. All of these differences were statistically significant.
Elevated prescribing rates were not seen in patients with brain, breast, colorectal, head and neck, or prostate cancers. The relative rate of prescribing for hematologic cancers was 1.383, a difference that approached, but did not quite reach, statistical significance (P = .0512).
When multivariable analysis was used to stratify individuals by length of time since cancer diagnosis, a significantly elevated relative rate of opioid prescribing persisted; for those 5-10 years from diagnosis, the relative rate was 1.190 (95% CI, 1.040-1.362, P = .011), while for those diagnosed at least 10 years ago, the relative rate was 1.244 (95% CI, 1.090-1.420; P = .00118).
Multivariable analysis was also used to control for income, rural residence, and comorbidities. However, the study could capture only opioids that were obtained with a prescription, and could not track whether medications were taken by the person for whom they were prescribed, Dr. Sutradhar and her colleagues said.
Cancer survivors may have a higher prevalence of chronic pain than the general population for reasons related both to their initial diagnosis and the sequelae of treatments such as surgery, chemotherapy, and radiation therapy, the investigators noted, adding, “it is also possible that a higher rate of opioid prescribing among survivors is due to a dependency that originated from opioid use earlier in the disease trajectory.”
Because of the potential for opioid use disorder and the many adverse effects that can be associated with long-term opioid use, they said, “primary care providers who treat cancer survivors should be encouraged to critically examine reasons for lingering opioid use among their patients.”
The oncologist’s perspective
Walter M. Stadler, MD, an oncologist who treats genitourinary and hematologic cancers, also wonders whether it’s pain or dependency that’s driving the increased prescribing rates in cancer survivors.
One opportunity to reassess which medications and treatment modalities are appropriate for the cancer survivor, said Dr. Stadler, is at the point of discharge from oncology care, which usually happens at about the 5-year mark for patients with no evidence of disease. Survivorship has received more attention since the 2005 Institute of Medicine report calling for increased attention to cancer survivors’ ongoing care. However, he said, “it’s not clear that we do a very good job in terms of educating either the patient or their primary care physician in regards to the kinds of things that we expect, the kinds of things that need to be done, or even a good summary of the therapy that was provided.”
There are resources that can help, he said. “That’s why organizations like [the American Society for Clinical Oncology] have put together some more formal survivorship plans that should be provided when patients are transitioned.”
The realities of clinical life can get in the way of implementation, though. Oncologists are already stretched thin, and most electronic health record systems don’t integrate well with survivorship documentation. Finding staff who can spend the time to gather and package all the necessary information can also be a problem: “People are expensive, and none of us have extra cash lying around,” said Dr. Stadler.
Still, he said, “like a lot of good papers, this raises some issues and areas for further investigation.” First, he said, physicians must assess whether cancer survivors are having chronic pain, and then sort things out from there. “What are the pain syndromes – and what are we doing about them? – because it’s not something that’s been well addressed.”
What can primary care offer?
Larissa Nekhlyudov, MD, is an internal medicine physician whose clinical practice straddles two domains. She sees patients, including some cancer survivors, as a primary care provider; she also provides care in a survivorship clinic to adult survivors of childhood cancers. There, she is able to focus more on survivorship care, developing a care plan and communicating with primary care providers about care elements her patients need.
It’s reasonable to think that there might be an increased risk for chronic pain syndromes in some of the types of cancer in which elevated opioid prescribing rates were seen, said Dr. Nekhlyudov. “Maybe this is okay.
“Pain in cancer survivors is so multidimensional that it’s quite possible that some of these cancer survivors – gynecologic, lung, other gastrointestinal, genitourinary – might have peripheral neuropathy, adhesions, and so many potential late effects,” said Dr. Nekhlyudov. “However, narcotics are not necessarily the preferred and the only method to treat this pain,” she said, noting that optimal survivorship care might seek to transition these patients to nonopioid therapies or, at least, a multimodal approach.
When she’s wearing her survivorship care hat, said Dr. Nekhlyudov, managing pain medication isn’t always at the top of the to-do list in an office visit. “It’s certainly not uncommon that patients will have a variety of pain issues. But in the survivorship domain, I think that we don’t take the role of managing their pain medications; that piece belongs, really, to their primary care provider,” she said.
“In many ways, it’s difficult to distinguish how much of their pain is related to their cancer, versus not, and figuring out alternatives,” said Dr. Nekhlyudov, applauding the authors’ recognition of the need for a multimodal approach in cancer survivors with pain. However, she said, “that sounds really great on paper, but it’s really not readily available.”
Even in the resource-rich greater Boston area where she practices, said Dr. Nekhlyudov, “it’s very difficult for cancer patients – and noncancer patients – to get hooked into a multidisciplinary, holistic program for pain.”
Although the long-term perspective is helpful, Dr. Nekhlyudov hopes for research that can help identify at what point, and by whom, the opioids were initiated in the cancer survivor population. “What is their trajectory from the time of diagnosis? Are these patients who are started on narcotics during their cancer treatment, and then continue on forever, or are some of these patients being started later, because of late effects?”
In any case, she said, “one of the key pieces is the ownership for this really belongs with both oncologists and the primary care providers.”
Mental health implications of survivorship
Viewing the issue through the lens of mental health offers a slightly different perspective. Thomas B. Strouse, MD, is a psychiatrist who holds the Maddie Katz Chair in palliative care research and education at the University of California, Los Angeles. He said he laments the current “opioidophobia” that calls into question any long-term opioid prescribing.
Acknowledging that there’s certainly a serious nationwide problem with both prescription and nonprescription opioid abuse, Dr. Strouse said he still finds it unfortunate that the current situation has “reactivated for many people a certain set of reflexes that say that any chronic opioid use is always a bad thing. That’s simply not true,” he said.
“Whether opioids are the right treatment for all of those patients, of course, is an entirely fair question. But it’s unfortunate, or wrong, for everybody to approach this article and to say that we know that for all of these patients, chronic opioid therapy is not appropriate,” he added.
Chronic pain that lingers after cancer treatments affects “a very significant minority of cancer survivors,” he said. It’s also true that the meaning of pain can be different for cancer survivors, said Dr. Strouse. For a cancer survivor, “any new pain is cancer pain until proven otherwise,” he said.
Further, pivoting from the attentive, multidisciplinary, wraparound care often received during cancer treatment to the relatively unsupported survivorship experience can be a rough transition for some. Despite the grim reason for the connection, “frequently, it’s the best experience of patients’ lives from a human relations perspective. … We don’t think enough about the loss that the end of cancer treatment may mean for people who may have otherwise unsatisfactory relationships in their lives,” he said.
Dr. Strouse, who works extensively with cancer survivors, said that the elevated rate of opioid prescribing seen in this study “opens the door to a bigger discussion about the challenges in the relatively empty domain of survivorship.” After discharge from cancer care, patients are all too often left without a navigator to help them through the years when, though their treatment is complete, anxiety, financial and social strain, and pain may linger.
The study, he said, should be a call to physicians for “a more meaningful commitment to understanding the burdens of survivorship, and actually offering meaningful clinical services to those people in an integrated and appropriate way.” This might include determining a patient’s absolute minimum opioid requirement, with a goal of getting the patient off opioids, but also making sure the patient has knowledge of and access to alternative pharmacologic and nonpharmacologic treatments for pain. “That seems like a reasonable approach,” said Dr. Strouse.
None of the study’s authors or the physicians interviewed for commentary had relevant conflicts of interest.
[email protected]
On Twitter @karioakes
Extended-release amantadine approved for treatment of dyskinesia in Parkinson’s
An extended-release formulation of amantadine received approval from the Food and Drug Administration on Aug. 24 for the treatment of dyskinesia in patients with Parkinson’s disease. The significant increase in functional time for Parkinson’s disease patients with dyskinesia who took extended-release amantadine was attributable both to a reduction in off-time and to a decrease in troublesome dyskinesia during on-time.
This is the first FDA approval for a drug to treat levodopa therapy-related dyskinesia in patients with Parkinson’s disease, according to an announcement from its manufacturer, Adamas Pharmaceuticals.
The second study also showed clinically relevant and statistically significant results, with a 46% reduction on the UDysRS for those taking ER amantadine, compared with a 16% reduction for those taking placebo.
The oral ER amantadine formulation delivers 274 mg of amantadine once daily at bedtime, allowing sustained high levels of the drug during waking hours, with peak levels delivered during the morning and throughout the day and a trough near bedtime.
When investigators of the two studies analyzed diaries that had been kept by Parkinson’s disease patients, they found that patients in the two studies who were taking ER amantadine experienced a placebo-adjusted reduction in off-time of about 1 hour per day.
Patients in the ER amantadine arm of the first study also had an increase of 3.6 hours per day of functional time, compared with a 0.8-hour increase for patients taking placebo. In the second study, functional time went up by 4.0 hours per day for patients in the ER amantadine arm, compared with an increase of 2.1 hours per day for those on placebo. Functional time was defined as on-time without troublesome dyskinesia.
Adverse reactions to ER amantadine that occurred in more than 10% of patients in the active arms of the study, and which occurred more frequently than in those taking placebo, included hallucinations, falls, orthostatic hypotension, dizziness, peripheral edema, dry mouth, and constipation.
The medication is contraindicated in those with creatinine clearance below 15 mL/min/1.73 m2. Prescribing information advises that amantadine ER be avoided or used with caution in patients with a history of suicidality and depression, hallucinations or psychotic behavior, and orthostatic hypotension or dizziness.
Patients taking ER amantadine may also experience impulsivity and sexual, spending, or gambling urges. Abrupt withdrawal or rapid dose reduction may result in withdrawal-emergent hyperpyrexia or confusion, including delirium, hallucinations, stupor, and slurred speech.
The clinical trials upon which the approval is based were funded by Adamas Pharmaceuticals.
[email protected]
On Twitter @karioakes
An extended-release formulation of amantadine received approval from the Food and Drug Administration on Aug. 24 for the treatment of dyskinesia in patients with Parkinson’s disease. The significant increase in functional time for Parkinson’s disease patients with dyskinesia who took extended-release amantadine was attributable both to a reduction in off-time and to a decrease in troublesome dyskinesia during on-time.
This is the first FDA approval for a drug to treat levodopa therapy-related dyskinesia in patients with Parkinson’s disease, according to an announcement from its manufacturer, Adamas Pharmaceuticals.
The second study also showed clinically relevant and statistically significant results, with a 46% reduction on the UDysRS for those taking ER amantadine, compared with a 16% reduction for those taking placebo.
The oral ER amantadine formulation delivers 274 mg of amantadine once daily at bedtime, allowing sustained high levels of the drug during waking hours, with peak levels delivered during the morning and throughout the day and a trough near bedtime.
When investigators of the two studies analyzed diaries that had been kept by Parkinson’s disease patients, they found that patients in the two studies who were taking ER amantadine experienced a placebo-adjusted reduction in off-time of about 1 hour per day.
Patients in the ER amantadine arm of the first study also had an increase of 3.6 hours per day of functional time, compared with a 0.8-hour increase for patients taking placebo. In the second study, functional time went up by 4.0 hours per day for patients in the ER amantadine arm, compared with an increase of 2.1 hours per day for those on placebo. Functional time was defined as on-time without troublesome dyskinesia.
Adverse reactions to ER amantadine that occurred in more than 10% of patients in the active arms of the study, and which occurred more frequently than in those taking placebo, included hallucinations, falls, orthostatic hypotension, dizziness, peripheral edema, dry mouth, and constipation.
The medication is contraindicated in those with creatinine clearance below 15 mL/min/1.73 m2. Prescribing information advises that amantadine ER be avoided or used with caution in patients with a history of suicidality and depression, hallucinations or psychotic behavior, and orthostatic hypotension or dizziness.
Patients taking ER amantadine may also experience impulsivity and sexual, spending, or gambling urges. Abrupt withdrawal or rapid dose reduction may result in withdrawal-emergent hyperpyrexia or confusion, including delirium, hallucinations, stupor, and slurred speech.
The clinical trials upon which the approval is based were funded by Adamas Pharmaceuticals.
[email protected]
On Twitter @karioakes
An extended-release formulation of amantadine received approval from the Food and Drug Administration on Aug. 24 for the treatment of dyskinesia in patients with Parkinson’s disease. The significant increase in functional time for Parkinson’s disease patients with dyskinesia who took extended-release amantadine was attributable both to a reduction in off-time and to a decrease in troublesome dyskinesia during on-time.
This is the first FDA approval for a drug to treat levodopa therapy-related dyskinesia in patients with Parkinson’s disease, according to an announcement from its manufacturer, Adamas Pharmaceuticals.
The second study also showed clinically relevant and statistically significant results, with a 46% reduction on the UDysRS for those taking ER amantadine, compared with a 16% reduction for those taking placebo.
The oral ER amantadine formulation delivers 274 mg of amantadine once daily at bedtime, allowing sustained high levels of the drug during waking hours, with peak levels delivered during the morning and throughout the day and a trough near bedtime.
When investigators of the two studies analyzed diaries that had been kept by Parkinson’s disease patients, they found that patients in the two studies who were taking ER amantadine experienced a placebo-adjusted reduction in off-time of about 1 hour per day.
Patients in the ER amantadine arm of the first study also had an increase of 3.6 hours per day of functional time, compared with a 0.8-hour increase for patients taking placebo. In the second study, functional time went up by 4.0 hours per day for patients in the ER amantadine arm, compared with an increase of 2.1 hours per day for those on placebo. Functional time was defined as on-time without troublesome dyskinesia.
Adverse reactions to ER amantadine that occurred in more than 10% of patients in the active arms of the study, and which occurred more frequently than in those taking placebo, included hallucinations, falls, orthostatic hypotension, dizziness, peripheral edema, dry mouth, and constipation.
The medication is contraindicated in those with creatinine clearance below 15 mL/min/1.73 m2. Prescribing information advises that amantadine ER be avoided or used with caution in patients with a history of suicidality and depression, hallucinations or psychotic behavior, and orthostatic hypotension or dizziness.
Patients taking ER amantadine may also experience impulsivity and sexual, spending, or gambling urges. Abrupt withdrawal or rapid dose reduction may result in withdrawal-emergent hyperpyrexia or confusion, including delirium, hallucinations, stupor, and slurred speech.
The clinical trials upon which the approval is based were funded by Adamas Pharmaceuticals.
[email protected]
On Twitter @karioakes
Dermatologists have a role in managing GVHD
NEW YORK – Dermatologists have an important role to play in caring for patients with chronic graft versus host disease (GVHD), a condition whose cutaneous manifestations are many, stubborn, and often disabling.
Although a wide range of systemic therapies are available, topical and intralesional treatment with such agents as potent steroids and calcineurin inhibitors can also help with cutaneous manifestations of GVHD in some instances, said Kathryn Martires, MD, at the American Academy of Dermatology summer meeting. However, she noted, “there are no studies or series examining the use of topical steroids alone in these patients, partly speaking to the complexity of these patients and required other care, but partly also due to the lack of dermatologists’ involvement in the care of these patients on a wide scale.”
“The types of GVHD that are particularly amenable to high dose steroids are predominantly the epidermal types,” she said. These include ichthyotic and eczematous as well as lichen planus–like cutaneous GVHD. “We also use topical steroids frequently in the papulosquamous type, though this is a rare variant,” she added.
Topical steroids can be used for dermal skin changes of GVHD as well, including lichen sclerosus–like and focal morphea–like plaques, according to Dr. Martires of the department of dermatology at Stanford (Calif.) University. These lesions are often first seen in the skin folds of the neck.
Even for patients with more diffuse dermal sclerosis, topical steroids have a role in quieting specific areas where active flares are occurring, she noted. These flares can look like erythematous, scaly patches and are “particularly amenable” to spot treatment with topical steroids.
“Just like in vitiligo that’s not associated with GVHD, certainly, topical steroids have their role in treating vitiligo that’s associated with chronic GVHD,” Dr. Martires said. This scenario stands in contrast to the situation where a patient has postinflammatory hyperpigmentation, for example, further along in the course of epidermal GVHD. Steroids should be avoided in situations where there’s hyperpigmentation.
Topical steroids are not usually useful for chronic poikilodermatous GVHD, or, generally, when patients have little epidermal change and the GVHD-associated changes are mostly dermal or subcutaneous, she said.
“Intralesional steroids have their role” in GVHD, although this is another instance where there are no studies to back up their efficacy, and recommendations are based on consensus, Dr. Martires pointed out. Nodular sclerotic GVHD is a rare manifestation, with firm, keloid-like lesions. These can flatten with intralesional injections, said Dr. Martires.
Intralesional injections have also been described in the literature as a treatment for ulcerative oral GVHD, she noted. Other therapy options for oral mucosal GVHD are fluocinonide gel 0.05% or clobetasol gel 0.05%, with spot application to the lesions. When there’s more diffuse lichenoid GVHD of the mouth, dexamethasone or prednisolone oral rinses can also be used, but should be combined with nystatin to prevent thrush, she advised. Triamcinolone 0.1% can be used with topical benzocaine dental paste (Orabase).
Calcineurin inhibitors are another option for oral lesions. Patients generally have a good comfort level with starting topical calcineurin inhibitors, said Dr. Martires, because they’ve likely had exposure to the systemic formulation. Case series have reported improvement “primarily in lichenoid GVHD” with the adjunctive use of topical calcineurin inhibitors, she said. In the mouth, tacrolimus 0.1% can be put in dental paste for focal lesions, and cyclosporine and azathioprine oral solutions can also be used.
Dry mouth is common in GVHD. “Remember, in patients who have other skin symptoms like pruritus, to ask about oral sicca symptoms in order to avoid things that might exacerbate it, like antihistamines and [tricyclic antidepressants],” she added.
Genital mucosal GVHD can respond to topical steroids, with ointment as the preferred vehicle, said Dr. Martires, noting that clobetasol 0.05% ointment and fluocinolone 0.025% ointment are good options, and tacrolimus 0.1% ointment is a logical nonsteroidal topical choice for the genital mucosa.
“Intralesionals are also first-line therapy here,” and “may prevent progression and permanent scarring if initiated early,” she pointed out. However, these injections are quite painful, so “patients have to be quite motivated” to be on board with this line of therapy, she said, adding that numbing prior to injections can help with pain.
Genital discomfort in women may not all be GVHD-related. “Remember, in patients who have undergone several cycles of chemotherapy prior to transplant, that they often have been experiencing menopausal symptoms, sometimes for years, so estrogen cream can sometimes go a long way,” said Dr. Martires, adding, “Certainly, a reminder about lubrication during intercourse is appropriate.”
Also, she said, dermatologists can help patients understand how important it is to be vigilant in preserving skin integrity by, for example, keeping skin well moisturized, avoiding aggressive nail care, and wearing gloves for wet work.
Dr. Martires reported no relevant financial relationships.
[email protected]
On Twitter @karioakes
NEW YORK – Dermatologists have an important role to play in caring for patients with chronic graft versus host disease (GVHD), a condition whose cutaneous manifestations are many, stubborn, and often disabling.
Although a wide range of systemic therapies are available, topical and intralesional treatment with such agents as potent steroids and calcineurin inhibitors can also help with cutaneous manifestations of GVHD in some instances, said Kathryn Martires, MD, at the American Academy of Dermatology summer meeting. However, she noted, “there are no studies or series examining the use of topical steroids alone in these patients, partly speaking to the complexity of these patients and required other care, but partly also due to the lack of dermatologists’ involvement in the care of these patients on a wide scale.”
“The types of GVHD that are particularly amenable to high dose steroids are predominantly the epidermal types,” she said. These include ichthyotic and eczematous as well as lichen planus–like cutaneous GVHD. “We also use topical steroids frequently in the papulosquamous type, though this is a rare variant,” she added.
Topical steroids can be used for dermal skin changes of GVHD as well, including lichen sclerosus–like and focal morphea–like plaques, according to Dr. Martires of the department of dermatology at Stanford (Calif.) University. These lesions are often first seen in the skin folds of the neck.
Even for patients with more diffuse dermal sclerosis, topical steroids have a role in quieting specific areas where active flares are occurring, she noted. These flares can look like erythematous, scaly patches and are “particularly amenable” to spot treatment with topical steroids.
“Just like in vitiligo that’s not associated with GVHD, certainly, topical steroids have their role in treating vitiligo that’s associated with chronic GVHD,” Dr. Martires said. This scenario stands in contrast to the situation where a patient has postinflammatory hyperpigmentation, for example, further along in the course of epidermal GVHD. Steroids should be avoided in situations where there’s hyperpigmentation.
Topical steroids are not usually useful for chronic poikilodermatous GVHD, or, generally, when patients have little epidermal change and the GVHD-associated changes are mostly dermal or subcutaneous, she said.
“Intralesional steroids have their role” in GVHD, although this is another instance where there are no studies to back up their efficacy, and recommendations are based on consensus, Dr. Martires pointed out. Nodular sclerotic GVHD is a rare manifestation, with firm, keloid-like lesions. These can flatten with intralesional injections, said Dr. Martires.
Intralesional injections have also been described in the literature as a treatment for ulcerative oral GVHD, she noted. Other therapy options for oral mucosal GVHD are fluocinonide gel 0.05% or clobetasol gel 0.05%, with spot application to the lesions. When there’s more diffuse lichenoid GVHD of the mouth, dexamethasone or prednisolone oral rinses can also be used, but should be combined with nystatin to prevent thrush, she advised. Triamcinolone 0.1% can be used with topical benzocaine dental paste (Orabase).
Calcineurin inhibitors are another option for oral lesions. Patients generally have a good comfort level with starting topical calcineurin inhibitors, said Dr. Martires, because they’ve likely had exposure to the systemic formulation. Case series have reported improvement “primarily in lichenoid GVHD” with the adjunctive use of topical calcineurin inhibitors, she said. In the mouth, tacrolimus 0.1% can be put in dental paste for focal lesions, and cyclosporine and azathioprine oral solutions can also be used.
Dry mouth is common in GVHD. “Remember, in patients who have other skin symptoms like pruritus, to ask about oral sicca symptoms in order to avoid things that might exacerbate it, like antihistamines and [tricyclic antidepressants],” she added.
Genital mucosal GVHD can respond to topical steroids, with ointment as the preferred vehicle, said Dr. Martires, noting that clobetasol 0.05% ointment and fluocinolone 0.025% ointment are good options, and tacrolimus 0.1% ointment is a logical nonsteroidal topical choice for the genital mucosa.
“Intralesionals are also first-line therapy here,” and “may prevent progression and permanent scarring if initiated early,” she pointed out. However, these injections are quite painful, so “patients have to be quite motivated” to be on board with this line of therapy, she said, adding that numbing prior to injections can help with pain.
Genital discomfort in women may not all be GVHD-related. “Remember, in patients who have undergone several cycles of chemotherapy prior to transplant, that they often have been experiencing menopausal symptoms, sometimes for years, so estrogen cream can sometimes go a long way,” said Dr. Martires, adding, “Certainly, a reminder about lubrication during intercourse is appropriate.”
Also, she said, dermatologists can help patients understand how important it is to be vigilant in preserving skin integrity by, for example, keeping skin well moisturized, avoiding aggressive nail care, and wearing gloves for wet work.
Dr. Martires reported no relevant financial relationships.
[email protected]
On Twitter @karioakes
NEW YORK – Dermatologists have an important role to play in caring for patients with chronic graft versus host disease (GVHD), a condition whose cutaneous manifestations are many, stubborn, and often disabling.
Although a wide range of systemic therapies are available, topical and intralesional treatment with such agents as potent steroids and calcineurin inhibitors can also help with cutaneous manifestations of GVHD in some instances, said Kathryn Martires, MD, at the American Academy of Dermatology summer meeting. However, she noted, “there are no studies or series examining the use of topical steroids alone in these patients, partly speaking to the complexity of these patients and required other care, but partly also due to the lack of dermatologists’ involvement in the care of these patients on a wide scale.”
“The types of GVHD that are particularly amenable to high dose steroids are predominantly the epidermal types,” she said. These include ichthyotic and eczematous as well as lichen planus–like cutaneous GVHD. “We also use topical steroids frequently in the papulosquamous type, though this is a rare variant,” she added.
Topical steroids can be used for dermal skin changes of GVHD as well, including lichen sclerosus–like and focal morphea–like plaques, according to Dr. Martires of the department of dermatology at Stanford (Calif.) University. These lesions are often first seen in the skin folds of the neck.
Even for patients with more diffuse dermal sclerosis, topical steroids have a role in quieting specific areas where active flares are occurring, she noted. These flares can look like erythematous, scaly patches and are “particularly amenable” to spot treatment with topical steroids.
“Just like in vitiligo that’s not associated with GVHD, certainly, topical steroids have their role in treating vitiligo that’s associated with chronic GVHD,” Dr. Martires said. This scenario stands in contrast to the situation where a patient has postinflammatory hyperpigmentation, for example, further along in the course of epidermal GVHD. Steroids should be avoided in situations where there’s hyperpigmentation.
Topical steroids are not usually useful for chronic poikilodermatous GVHD, or, generally, when patients have little epidermal change and the GVHD-associated changes are mostly dermal or subcutaneous, she said.
“Intralesional steroids have their role” in GVHD, although this is another instance where there are no studies to back up their efficacy, and recommendations are based on consensus, Dr. Martires pointed out. Nodular sclerotic GVHD is a rare manifestation, with firm, keloid-like lesions. These can flatten with intralesional injections, said Dr. Martires.
Intralesional injections have also been described in the literature as a treatment for ulcerative oral GVHD, she noted. Other therapy options for oral mucosal GVHD are fluocinonide gel 0.05% or clobetasol gel 0.05%, with spot application to the lesions. When there’s more diffuse lichenoid GVHD of the mouth, dexamethasone or prednisolone oral rinses can also be used, but should be combined with nystatin to prevent thrush, she advised. Triamcinolone 0.1% can be used with topical benzocaine dental paste (Orabase).
Calcineurin inhibitors are another option for oral lesions. Patients generally have a good comfort level with starting topical calcineurin inhibitors, said Dr. Martires, because they’ve likely had exposure to the systemic formulation. Case series have reported improvement “primarily in lichenoid GVHD” with the adjunctive use of topical calcineurin inhibitors, she said. In the mouth, tacrolimus 0.1% can be put in dental paste for focal lesions, and cyclosporine and azathioprine oral solutions can also be used.
Dry mouth is common in GVHD. “Remember, in patients who have other skin symptoms like pruritus, to ask about oral sicca symptoms in order to avoid things that might exacerbate it, like antihistamines and [tricyclic antidepressants],” she added.
Genital mucosal GVHD can respond to topical steroids, with ointment as the preferred vehicle, said Dr. Martires, noting that clobetasol 0.05% ointment and fluocinolone 0.025% ointment are good options, and tacrolimus 0.1% ointment is a logical nonsteroidal topical choice for the genital mucosa.
“Intralesionals are also first-line therapy here,” and “may prevent progression and permanent scarring if initiated early,” she pointed out. However, these injections are quite painful, so “patients have to be quite motivated” to be on board with this line of therapy, she said, adding that numbing prior to injections can help with pain.
Genital discomfort in women may not all be GVHD-related. “Remember, in patients who have undergone several cycles of chemotherapy prior to transplant, that they often have been experiencing menopausal symptoms, sometimes for years, so estrogen cream can sometimes go a long way,” said Dr. Martires, adding, “Certainly, a reminder about lubrication during intercourse is appropriate.”
Also, she said, dermatologists can help patients understand how important it is to be vigilant in preserving skin integrity by, for example, keeping skin well moisturized, avoiding aggressive nail care, and wearing gloves for wet work.
Dr. Martires reported no relevant financial relationships.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
The microbiota matters: In acne, it’s not us versus them
NEW YORK – Just as an imbalance in the intestinal microbiota can disrupt gut function, dysbiosis of the facial skin can allow acne-causing bacteria to flourish.
In acne, said Adam Friedman, MD, “we’ve always been talking about bacteria,” but now the thinking has shifted from just controlling Propionibacterium acnes to a subtler understanding of what’s happening on the skin of individuals with acne. Individuals may have their own unique skin microbiota – the community of organisms resident on the skin – but dysbiosis characterized by a lack of diversity is increasingly understood as a common theme in many skin disorders, and acne is no exception.
As in many other areas of medicine, dermatology’s understanding has been informed by genetic work that moves beyond the human genome. “Using newer technology, we were able to identify that our genome really was overshadowed by the microbial genome that makes up the populations in our skin, in our gut, and what have you,” said Dr. Friedman, speaking at the summer meeting of the American Academy of Dermatology.

The human body is like a planet to the bacteria that live on the human skin, and like a planet, the skin provides multiple “climates” for many bacterial ecosystems, said Dr. Friedman, director of translational research and dermatology residency program director, at George Washington University, Washington, DC.
Some areas are dry, some are moist; some are more oily, and some areas of the skin produce little sebum; while some are mostly dark and some are more likely to be exposed to light.
Considering skin from this perspective, it makes sense that bacterial microbiota for these disparate areas varies widely, with a different mix of bacteria found in the groin than on the forearm, he noted. Further, “each individual has his or her own microbiota fingerprint,” said Dr. Friedman, citing a 2012 study showing that in four healthy volunteers, the microbiota from swabs at four sites (antecubital fossa, back, nare, and plantar heel) varied widely both in diversity and composition (Genome Res. 2012 May;22[5]:850-9).
Multiple factors can contribute to this variability, which can include endogenous factors, such as host genotype, sex, age, immune system, and pathobiology. Exogenous factors, such as climate, geographic location, and occupational exposures, also play a part.
Increasingly, said Dr. Friedman, lack of bacterial diversity in skin microbiota is recognized as an important factor in many disease states, including atopic dermatitis and psoriasis. And bacterial diversity has recently been shown to be reduced on the facial skin of patients with acne, even on areas of clear skin.
When acne treatments work, a healthy facial microbiota is restored. And perhaps counterintuitively, patients with acne who receive isotretinoin and antibiotics have much greater diversity in the microbiota of their facial skin after treatment than before, according to a study recently published online (Exp Dermatol. 2017 Jun 21. doi: 10.1111/exd.13397).
For now, this is still a chicken-and-egg situation, Dr. Friedman said. “Does the disease cause the lack of diversity, or does the lack of diversity cause the disease to develop? We don’t know yet.” (Nat Rev Microbiol. 2011 Apr;9[4]:244-53).
“If we’re going to think about the surface of our skin as a barrier, we must consider the microbiota as part of that barrier.”
P. acnes “is a clear instigator in eliciting a host inflammatory response,” through its recognition by toll-like receptors and the inflammasome to induce inflammation, Dr. Friedman said. However, it can also help prevent the colonization of opportunistic pathogens, including methicillin-resistant Staphylococcus aureus and Streptococcus pyogenes by helping maintain an acidic skin pH. “When and how does a commensal [organism] become a pathogen?” he asked.
The fact that P. acnes is a commensal bacterium on healthy skin seems to muddy the picture, until one also recognizes that there are different strains of P. acnes. Only some of these phylotypes cause acne, with an exaggerated host inflammatory response being one possible causative factor, noted Dr. Friedman.
A clue to how this occurs comes from a recent study that found that some types of P. acnes actually convert sebum to short-chain fatty acids that “interfere with how our bodies regulate toll-like receptors, uncoupling them and then laying them loose to create inflammation,” said Dr. Friedman (Sci Immunol. 2016 Oct 28;1[4]. pii: eaah4609).
When considering what to do with the available information, something for dermatologists to consider is the effect moisturizers have on the skin of patients with acne, Dr. Friedman said. A moisturizer contains water; it may also contain a carbon source in the form of a sugar like mannose, nitrogen in the form of amino acids, and some oligoelements such as calcium, magnesium, manganese, strontium, and selenium. All of these ingredients really serve as prebiotics for the skin microbiota, Dr. Friedman noted, adding that products that create a prebiotic environment where acnegenic P. acnes are suppressed and a healthy microbiota can flourish are being developed.
“What does all this mean? We do not know yet,” said Dr. Friedman. But, he added, “clearly, what we’re using is having an effect, and we need to figure it out.”
Dr. Friedman reported financial relationships with several pharmaceutical and skin care companies. He serves on the editorial board of Dermatology News.
[email protected]
On Twitter @karioakes
NEW YORK – Just as an imbalance in the intestinal microbiota can disrupt gut function, dysbiosis of the facial skin can allow acne-causing bacteria to flourish.
In acne, said Adam Friedman, MD, “we’ve always been talking about bacteria,” but now the thinking has shifted from just controlling Propionibacterium acnes to a subtler understanding of what’s happening on the skin of individuals with acne. Individuals may have their own unique skin microbiota – the community of organisms resident on the skin – but dysbiosis characterized by a lack of diversity is increasingly understood as a common theme in many skin disorders, and acne is no exception.
As in many other areas of medicine, dermatology’s understanding has been informed by genetic work that moves beyond the human genome. “Using newer technology, we were able to identify that our genome really was overshadowed by the microbial genome that makes up the populations in our skin, in our gut, and what have you,” said Dr. Friedman, speaking at the summer meeting of the American Academy of Dermatology.
The human body is like a planet to the bacteria that live on the human skin, and like a planet, the skin provides multiple “climates” for many bacterial ecosystems, said Dr. Friedman, director of translational research and dermatology residency program director, at George Washington University, Washington, DC.
Some areas are dry, some are moist; some are more oily, and some areas of the skin produce little sebum; while some are mostly dark and some are more likely to be exposed to light.
Considering skin from this perspective, it makes sense that bacterial microbiota for these disparate areas varies widely, with a different mix of bacteria found in the groin than on the forearm, he noted. Further, “each individual has his or her own microbiota fingerprint,” said Dr. Friedman, citing a 2012 study showing that in four healthy volunteers, the microbiota from swabs at four sites (antecubital fossa, back, nare, and plantar heel) varied widely both in diversity and composition (Genome Res. 2012 May;22[5]:850-9).
Multiple factors can contribute to this variability, which can include endogenous factors, such as host genotype, sex, age, immune system, and pathobiology. Exogenous factors, such as climate, geographic location, and occupational exposures, also play a part.
Increasingly, said Dr. Friedman, lack of bacterial diversity in skin microbiota is recognized as an important factor in many disease states, including atopic dermatitis and psoriasis. And bacterial diversity has recently been shown to be reduced on the facial skin of patients with acne, even on areas of clear skin.
When acne treatments work, a healthy facial microbiota is restored. And perhaps counterintuitively, patients with acne who receive isotretinoin and antibiotics have much greater diversity in the microbiota of their facial skin after treatment than before, according to a study recently published online (Exp Dermatol. 2017 Jun 21. doi: 10.1111/exd.13397).
For now, this is still a chicken-and-egg situation, Dr. Friedman said. “Does the disease cause the lack of diversity, or does the lack of diversity cause the disease to develop? We don’t know yet.” (Nat Rev Microbiol. 2011 Apr;9[4]:244-53).
“If we’re going to think about the surface of our skin as a barrier, we must consider the microbiota as part of that barrier.”
P. acnes “is a clear instigator in eliciting a host inflammatory response,” through its recognition by toll-like receptors and the inflammasome to induce inflammation, Dr. Friedman said. However, it can also help prevent the colonization of opportunistic pathogens, including methicillin-resistant Staphylococcus aureus and Streptococcus pyogenes by helping maintain an acidic skin pH. “When and how does a commensal [organism] become a pathogen?” he asked.
The fact that P. acnes is a commensal bacterium on healthy skin seems to muddy the picture, until one also recognizes that there are different strains of P. acnes. Only some of these phylotypes cause acne, with an exaggerated host inflammatory response being one possible causative factor, noted Dr. Friedman.
A clue to how this occurs comes from a recent study that found that some types of P. acnes actually convert sebum to short-chain fatty acids that “interfere with how our bodies regulate toll-like receptors, uncoupling them and then laying them loose to create inflammation,” said Dr. Friedman (Sci Immunol. 2016 Oct 28;1[4]. pii: eaah4609).
When considering what to do with the available information, something for dermatologists to consider is the effect moisturizers have on the skin of patients with acne, Dr. Friedman said. A moisturizer contains water; it may also contain a carbon source in the form of a sugar like mannose, nitrogen in the form of amino acids, and some oligoelements such as calcium, magnesium, manganese, strontium, and selenium. All of these ingredients really serve as prebiotics for the skin microbiota, Dr. Friedman noted, adding that products that create a prebiotic environment where acnegenic P. acnes are suppressed and a healthy microbiota can flourish are being developed.
“What does all this mean? We do not know yet,” said Dr. Friedman. But, he added, “clearly, what we’re using is having an effect, and we need to figure it out.”
Dr. Friedman reported financial relationships with several pharmaceutical and skin care companies. He serves on the editorial board of Dermatology News.
[email protected]
On Twitter @karioakes
NEW YORK – Just as an imbalance in the intestinal microbiota can disrupt gut function, dysbiosis of the facial skin can allow acne-causing bacteria to flourish.
In acne, said Adam Friedman, MD, “we’ve always been talking about bacteria,” but now the thinking has shifted from just controlling Propionibacterium acnes to a subtler understanding of what’s happening on the skin of individuals with acne. Individuals may have their own unique skin microbiota – the community of organisms resident on the skin – but dysbiosis characterized by a lack of diversity is increasingly understood as a common theme in many skin disorders, and acne is no exception.
As in many other areas of medicine, dermatology’s understanding has been informed by genetic work that moves beyond the human genome. “Using newer technology, we were able to identify that our genome really was overshadowed by the microbial genome that makes up the populations in our skin, in our gut, and what have you,” said Dr. Friedman, speaking at the summer meeting of the American Academy of Dermatology.
The human body is like a planet to the bacteria that live on the human skin, and like a planet, the skin provides multiple “climates” for many bacterial ecosystems, said Dr. Friedman, director of translational research and dermatology residency program director, at George Washington University, Washington, DC.
Some areas are dry, some are moist; some are more oily, and some areas of the skin produce little sebum; while some are mostly dark and some are more likely to be exposed to light.
Considering skin from this perspective, it makes sense that bacterial microbiota for these disparate areas varies widely, with a different mix of bacteria found in the groin than on the forearm, he noted. Further, “each individual has his or her own microbiota fingerprint,” said Dr. Friedman, citing a 2012 study showing that in four healthy volunteers, the microbiota from swabs at four sites (antecubital fossa, back, nare, and plantar heel) varied widely both in diversity and composition (Genome Res. 2012 May;22[5]:850-9).
Multiple factors can contribute to this variability, which can include endogenous factors, such as host genotype, sex, age, immune system, and pathobiology. Exogenous factors, such as climate, geographic location, and occupational exposures, also play a part.
Increasingly, said Dr. Friedman, lack of bacterial diversity in skin microbiota is recognized as an important factor in many disease states, including atopic dermatitis and psoriasis. And bacterial diversity has recently been shown to be reduced on the facial skin of patients with acne, even on areas of clear skin.
When acne treatments work, a healthy facial microbiota is restored. And perhaps counterintuitively, patients with acne who receive isotretinoin and antibiotics have much greater diversity in the microbiota of their facial skin after treatment than before, according to a study recently published online (Exp Dermatol. 2017 Jun 21. doi: 10.1111/exd.13397).
For now, this is still a chicken-and-egg situation, Dr. Friedman said. “Does the disease cause the lack of diversity, or does the lack of diversity cause the disease to develop? We don’t know yet.” (Nat Rev Microbiol. 2011 Apr;9[4]:244-53).
“If we’re going to think about the surface of our skin as a barrier, we must consider the microbiota as part of that barrier.”
P. acnes “is a clear instigator in eliciting a host inflammatory response,” through its recognition by toll-like receptors and the inflammasome to induce inflammation, Dr. Friedman said. However, it can also help prevent the colonization of opportunistic pathogens, including methicillin-resistant Staphylococcus aureus and Streptococcus pyogenes by helping maintain an acidic skin pH. “When and how does a commensal [organism] become a pathogen?” he asked.
The fact that P. acnes is a commensal bacterium on healthy skin seems to muddy the picture, until one also recognizes that there are different strains of P. acnes. Only some of these phylotypes cause acne, with an exaggerated host inflammatory response being one possible causative factor, noted Dr. Friedman.
A clue to how this occurs comes from a recent study that found that some types of P. acnes actually convert sebum to short-chain fatty acids that “interfere with how our bodies regulate toll-like receptors, uncoupling them and then laying them loose to create inflammation,” said Dr. Friedman (Sci Immunol. 2016 Oct 28;1[4]. pii: eaah4609).
When considering what to do with the available information, something for dermatologists to consider is the effect moisturizers have on the skin of patients with acne, Dr. Friedman said. A moisturizer contains water; it may also contain a carbon source in the form of a sugar like mannose, nitrogen in the form of amino acids, and some oligoelements such as calcium, magnesium, manganese, strontium, and selenium. All of these ingredients really serve as prebiotics for the skin microbiota, Dr. Friedman noted, adding that products that create a prebiotic environment where acnegenic P. acnes are suppressed and a healthy microbiota can flourish are being developed.
“What does all this mean? We do not know yet,” said Dr. Friedman. But, he added, “clearly, what we’re using is having an effect, and we need to figure it out.”
Dr. Friedman reported financial relationships with several pharmaceutical and skin care companies. He serves on the editorial board of Dermatology News.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING