User login
Compounding rules challenge practice norms
As new rules about drug compounding get shaped, rheumatologists seek to protect their ability to combine injectable drugs – most commonly a steroid and a local anesthetic – in their own offices.
In a position statement sent to government agencies and members of Congress in February, the American College of Rheumatology voiced concerns that the practice, which it called “critical,” could become a casualty of drug-compounding regulations under revision by the United States Pharmacopeial Convention (USP), a nonprofit group whose standards are enforceable by state and federal regulators.

In the same position statement on compounding, the ACR said it also seeks a change to a Food and Drug Administration rule limiting practitioners’ access to quinacrine, a drug only available through compounding pharmacies that is sometimes used to treat lupus patients. Quinacrine is not on the FDA’s current list of bulk substances approved for compounding, except by special permission. The ACR has asked the agency to add quinacrine to the list, but no one knows when this will happen.
Rheumatologists may also be more restricted than before in terms of which compounding pharmacies they can turn to, as new federal standards divide them into two types – those that can provide medicines in larger quantities and those that can’t.
Steroid fiasco sparked rule revisions
The ACR’s concerns follow a tighter focus by state and federal agencies on drug compounding after a fungal meningitis outbreak in 2012 was traced to contaminated steroids produced in bulk by a compounding pharmacy.
More than 800 infections, 64 of them fatal, occurred after the New England Compounding Center in Framingham, Mass., sold contaminated methylprednisolone acetate that was used in epidural and intra-articular joint injections.
The following year Congress passed the Drug Quality and Security Act, which aims, in part, to prevent compounding pharmacies from engaging in what amounts to unregulated manufacturing.
As part of the law, the FDA created a list of drugs appropriate for compounding and a process by which larger compounding pharmacies must register with the FDA, and agree to inspections. The USP standards, meanwhile, address detailed technical and safety aspects of compounding and are enforceable by the FDA and state agencies.
“USP and FDA have had the ability to regulate compounding for over a decade, but only recently have the rules become actively enforced,” said Donald Miller, PharmD, of North Dakota State University, Fargo, who helped shape the ACR’s position statement on compounding with the help of rheumatologists in private practice.
“When you make guidelines for safety, they make sense, but then you can’t anticipate the way it’s going to affect individuals’ practice. And that’s where rheumatology got caught up,” said Dr. Miller, who was a member of the FDA Arthritis Advisory Committee in 2014-2016.
In-office mixing a top concern
Other specialties, including dermatology and immunology, also stand to be affected by various changes to compounding law and practice – and their societies have been active in voicing concerns.
Though the latest revisions of USP chapter 797, which impacts in-office mixing, are still being sorted out, it’s the No. 1 compounding-related concern for rheumatologists, Dr. Miller said.
Rheumatologists routinely mix an analgesic and a steroid for injection. The analgesic makes the steroids less viscous, and offers patients hours of immediate relief. They also add analgesics to hyaluronic acid injected for viscosupplementation. The mixing is usually conducted bedside, and the injections are administered right away.
Technically, combining these products amounts to sterile compounding, Dr. Miller explained. “And theoretically, under these rules, a physician could still do this, but they’d have to do it under a sterile hood like you find in a pharmacy, and that’s just not practical. It also becomes a matter of interpretation.”
USP chapter 797 sanctions in-office mixing for “immediate use” with individual patients – which is nearly always the case for the steroid injections used in rheumatology. But it’s unclear whether “immediate use” means emergency use only, or allows for routine use, as rheumatologists hope.
“One reason this came to rheumatology’s attention is that some state boards of medicine were inspecting and saying ‘Hey, you can’t do that,’ ” Dr. Miller said.

“There’s that law of unintended consequences where you snare things in a net that you really don’t want to,” Dr. Huffstutter said.
Marcus Snow, MD, a rheumatologist at the University of Nebraska, Omaha, who also worked on the statement, said that most rheumatologists are likely unaware that their ability to mix drugs in-office has been called into question.
“I brought it up at our division meeting with a group of 10 rheumatologists, and no one was aware that this was coming down the pike,” Dr. Snow said in an interview.

Pediatric issues
Pediatric rheumatologists, and adult rheumatologists who see children occasionally, use compounding pharmacies to create palatable oral medicines and adjusted doses of adult treatments.
They also use injections combining steroids with analgesics, and consider the addition of the analgesic a key aid to compliance.
“The biggest barrier we have is patient and parent anxiety about doing the procedure and the associated pain. We always administer our steroids mixed with lidocaine to help with the postprocedural discomfort,” said Adam Reinhardt, MD, chief of pediatric rheumatology at the University of Nebraska and Children’s Hospital and Medical Center in Omaha.

Steroid injections can mean avoiding or delaying systemic treatment in children with oligoarticular arthritis, he said. “Most of us consider them a first-line therapy. The hope is that you can get by without having to use meds like methotrexate if you can get a prolonged response in the one or two joints that are active in that patient.”
But Dr. Reinhardt said that, while he mixed his own injections during his fellowship training, Children’s of Omaha now insists that they be prepared by in-house pharmacists, working under sterile hoods. The delay to receiving them in the clinic or procedure room is 40 minutes to an hour, he said, which the clinicians accommodate through careful scheduling.
The change from mixing in-clinic to relying on the central pharmacy came about in recent years, Dr. Reinhardt said, because of broader concerns related to medication storage in the clinics. While ordering from the central pharmacy works for his practice, he said, “I probably only inject maybe 50-70 joints a year, while adult rheumatologists are injecting far more than that. For a busy private practice, I can see that being a huge time constraint,” he said.
Relevance of rules
None of the rheumatologists interviewed questioned the need for tightened state and federal oversight of compounding practices overall – just the applicability of certain rules to their own practice.
Dr. Snow and Dr. Huffstutter noted that reports of infected joints – a potential result of a contaminated injection – are sporadic and rare. “There’s very little research in this, but [these types of injections] have been standard practice for decades,” Dr. Snow said.
Srikanth Mukkera, MD, a rheumatologist in Tupelo, Miss., agreed that “sporadic cases of joint infection do happen following injection, but it can be hard to show if an injection was the cause.”
Assuring that medicines are mixed only immediately prior to injection, and not stored, reduces the likelihood of contamination, Dr. Mukkera said. Moreover, he noted, epidural injections such as those that resulted in the 2012 meningitis outbreak carry different risks than those seen in intra-articular injections.
Dr. Miller, the lead author of the ACR statement, said that the rheumatologists on our committee “don’t know of anyone that’s had a knee or other joint infection from a contaminated injection. They feel that unless somebody finds some evidence of that, they should be allowed to continue” with their usual practice.
He said that he feels that the USP will ultimately heed the concerns of rheumatologists and hopefully provide a more relaxed interpretation of in-office compounding. “We’re hoping they’ll make some exceptions when they revise 797 standards or at least maybe leave room for organizations to create a best practice statement. We’ll see,” Dr. Miller said.
But this is in no way guaranteed. Dr. Huffstutter said he fears that, if the rules come to be interpreted more narrowly, even standard practices like reconstituting biologic drugs for infusion – something that’s also a routine part of in-office practice – could fall under the rubric of sterile compounding and come into question.
The quinacrine problem
A separate compounding-related issue in rheumatology is clinicians’ access to quinacrine, an antimalarial rheumatology drug that, while infrequently used, represents the only alternative to hydroxychloroquine for some lupus patients.
“There are no alternatives out there for hydroxychloroquine, so we need it as a backup,” Dr. Snow said. “If hydroxychloroquine isn’t an option, there’s nothing out there that we can use. There’s no easy replacement.”
Dr. Huffstutter said he currently had no patients on quinacrine. “It’s not very often that we use it, but in those patients that really need it, it can make a huge difference in how they do.”
Quinacrine is no longer manufactured commercially as a finished drug product but is available in a powder that compounding physicians put into 100-mg capsules. It is not on the FDA’s current list of drugs available for compounding except with special permission.
While the ACR has requested that the FDA add it the list of bulk drug substances that can be used in compounding, quinacrine remains off the list for now – and, providers say, hard to find.
Moreover, while rheumatologists may have previously been able to order and store quantities of quinacrine and other compounded nonsterile medications to dispense to their patients, they can no longer easily do so, as only the FDA-approved compounding “outsourcing facilities” are allowed to process larger orders; the rest can only respond to prescriptions for individual patients.
Dr. Miller said it’s likely that quinacrine will make it onto the FDA’s next list of bulk drugs available for compounding. “The FDA has kind of said, ‘Don’t worry about it,’ ” he said.
As new rules about drug compounding get shaped, rheumatologists seek to protect their ability to combine injectable drugs – most commonly a steroid and a local anesthetic – in their own offices.
In a position statement sent to government agencies and members of Congress in February, the American College of Rheumatology voiced concerns that the practice, which it called “critical,” could become a casualty of drug-compounding regulations under revision by the United States Pharmacopeial Convention (USP), a nonprofit group whose standards are enforceable by state and federal regulators.
In the same position statement on compounding, the ACR said it also seeks a change to a Food and Drug Administration rule limiting practitioners’ access to quinacrine, a drug only available through compounding pharmacies that is sometimes used to treat lupus patients. Quinacrine is not on the FDA’s current list of bulk substances approved for compounding, except by special permission. The ACR has asked the agency to add quinacrine to the list, but no one knows when this will happen.
Rheumatologists may also be more restricted than before in terms of which compounding pharmacies they can turn to, as new federal standards divide them into two types – those that can provide medicines in larger quantities and those that can’t.
Steroid fiasco sparked rule revisions
The ACR’s concerns follow a tighter focus by state and federal agencies on drug compounding after a fungal meningitis outbreak in 2012 was traced to contaminated steroids produced in bulk by a compounding pharmacy.
More than 800 infections, 64 of them fatal, occurred after the New England Compounding Center in Framingham, Mass., sold contaminated methylprednisolone acetate that was used in epidural and intra-articular joint injections.
The following year Congress passed the Drug Quality and Security Act, which aims, in part, to prevent compounding pharmacies from engaging in what amounts to unregulated manufacturing.
As part of the law, the FDA created a list of drugs appropriate for compounding and a process by which larger compounding pharmacies must register with the FDA, and agree to inspections. The USP standards, meanwhile, address detailed technical and safety aspects of compounding and are enforceable by the FDA and state agencies.
“USP and FDA have had the ability to regulate compounding for over a decade, but only recently have the rules become actively enforced,” said Donald Miller, PharmD, of North Dakota State University, Fargo, who helped shape the ACR’s position statement on compounding with the help of rheumatologists in private practice.
“When you make guidelines for safety, they make sense, but then you can’t anticipate the way it’s going to affect individuals’ practice. And that’s where rheumatology got caught up,” said Dr. Miller, who was a member of the FDA Arthritis Advisory Committee in 2014-2016.
In-office mixing a top concern
Other specialties, including dermatology and immunology, also stand to be affected by various changes to compounding law and practice – and their societies have been active in voicing concerns.
Though the latest revisions of USP chapter 797, which impacts in-office mixing, are still being sorted out, it’s the No. 1 compounding-related concern for rheumatologists, Dr. Miller said.
Rheumatologists routinely mix an analgesic and a steroid for injection. The analgesic makes the steroids less viscous, and offers patients hours of immediate relief. They also add analgesics to hyaluronic acid injected for viscosupplementation. The mixing is usually conducted bedside, and the injections are administered right away.
Technically, combining these products amounts to sterile compounding, Dr. Miller explained. “And theoretically, under these rules, a physician could still do this, but they’d have to do it under a sterile hood like you find in a pharmacy, and that’s just not practical. It also becomes a matter of interpretation.”
USP chapter 797 sanctions in-office mixing for “immediate use” with individual patients – which is nearly always the case for the steroid injections used in rheumatology. But it’s unclear whether “immediate use” means emergency use only, or allows for routine use, as rheumatologists hope.
“One reason this came to rheumatology’s attention is that some state boards of medicine were inspecting and saying ‘Hey, you can’t do that,’ ” Dr. Miller said.
“There’s that law of unintended consequences where you snare things in a net that you really don’t want to,” Dr. Huffstutter said.
Marcus Snow, MD, a rheumatologist at the University of Nebraska, Omaha, who also worked on the statement, said that most rheumatologists are likely unaware that their ability to mix drugs in-office has been called into question.
“I brought it up at our division meeting with a group of 10 rheumatologists, and no one was aware that this was coming down the pike,” Dr. Snow said in an interview.
Pediatric issues
Pediatric rheumatologists, and adult rheumatologists who see children occasionally, use compounding pharmacies to create palatable oral medicines and adjusted doses of adult treatments.
They also use injections combining steroids with analgesics, and consider the addition of the analgesic a key aid to compliance.
“The biggest barrier we have is patient and parent anxiety about doing the procedure and the associated pain. We always administer our steroids mixed with lidocaine to help with the postprocedural discomfort,” said Adam Reinhardt, MD, chief of pediatric rheumatology at the University of Nebraska and Children’s Hospital and Medical Center in Omaha.
Steroid injections can mean avoiding or delaying systemic treatment in children with oligoarticular arthritis, he said. “Most of us consider them a first-line therapy. The hope is that you can get by without having to use meds like methotrexate if you can get a prolonged response in the one or two joints that are active in that patient.”
But Dr. Reinhardt said that, while he mixed his own injections during his fellowship training, Children’s of Omaha now insists that they be prepared by in-house pharmacists, working under sterile hoods. The delay to receiving them in the clinic or procedure room is 40 minutes to an hour, he said, which the clinicians accommodate through careful scheduling.
The change from mixing in-clinic to relying on the central pharmacy came about in recent years, Dr. Reinhardt said, because of broader concerns related to medication storage in the clinics. While ordering from the central pharmacy works for his practice, he said, “I probably only inject maybe 50-70 joints a year, while adult rheumatologists are injecting far more than that. For a busy private practice, I can see that being a huge time constraint,” he said.
Relevance of rules
None of the rheumatologists interviewed questioned the need for tightened state and federal oversight of compounding practices overall – just the applicability of certain rules to their own practice.
Dr. Snow and Dr. Huffstutter noted that reports of infected joints – a potential result of a contaminated injection – are sporadic and rare. “There’s very little research in this, but [these types of injections] have been standard practice for decades,” Dr. Snow said.
Srikanth Mukkera, MD, a rheumatologist in Tupelo, Miss., agreed that “sporadic cases of joint infection do happen following injection, but it can be hard to show if an injection was the cause.”
Assuring that medicines are mixed only immediately prior to injection, and not stored, reduces the likelihood of contamination, Dr. Mukkera said. Moreover, he noted, epidural injections such as those that resulted in the 2012 meningitis outbreak carry different risks than those seen in intra-articular injections.
Dr. Miller, the lead author of the ACR statement, said that the rheumatologists on our committee “don’t know of anyone that’s had a knee or other joint infection from a contaminated injection. They feel that unless somebody finds some evidence of that, they should be allowed to continue” with their usual practice.
He said that he feels that the USP will ultimately heed the concerns of rheumatologists and hopefully provide a more relaxed interpretation of in-office compounding. “We’re hoping they’ll make some exceptions when they revise 797 standards or at least maybe leave room for organizations to create a best practice statement. We’ll see,” Dr. Miller said.
But this is in no way guaranteed. Dr. Huffstutter said he fears that, if the rules come to be interpreted more narrowly, even standard practices like reconstituting biologic drugs for infusion – something that’s also a routine part of in-office practice – could fall under the rubric of sterile compounding and come into question.
The quinacrine problem
A separate compounding-related issue in rheumatology is clinicians’ access to quinacrine, an antimalarial rheumatology drug that, while infrequently used, represents the only alternative to hydroxychloroquine for some lupus patients.
“There are no alternatives out there for hydroxychloroquine, so we need it as a backup,” Dr. Snow said. “If hydroxychloroquine isn’t an option, there’s nothing out there that we can use. There’s no easy replacement.”
Dr. Huffstutter said he currently had no patients on quinacrine. “It’s not very often that we use it, but in those patients that really need it, it can make a huge difference in how they do.”
Quinacrine is no longer manufactured commercially as a finished drug product but is available in a powder that compounding physicians put into 100-mg capsules. It is not on the FDA’s current list of drugs available for compounding except with special permission.
While the ACR has requested that the FDA add it the list of bulk drug substances that can be used in compounding, quinacrine remains off the list for now – and, providers say, hard to find.
Moreover, while rheumatologists may have previously been able to order and store quantities of quinacrine and other compounded nonsterile medications to dispense to their patients, they can no longer easily do so, as only the FDA-approved compounding “outsourcing facilities” are allowed to process larger orders; the rest can only respond to prescriptions for individual patients.
Dr. Miller said it’s likely that quinacrine will make it onto the FDA’s next list of bulk drugs available for compounding. “The FDA has kind of said, ‘Don’t worry about it,’ ” he said.
As new rules about drug compounding get shaped, rheumatologists seek to protect their ability to combine injectable drugs – most commonly a steroid and a local anesthetic – in their own offices.
In a position statement sent to government agencies and members of Congress in February, the American College of Rheumatology voiced concerns that the practice, which it called “critical,” could become a casualty of drug-compounding regulations under revision by the United States Pharmacopeial Convention (USP), a nonprofit group whose standards are enforceable by state and federal regulators.
In the same position statement on compounding, the ACR said it also seeks a change to a Food and Drug Administration rule limiting practitioners’ access to quinacrine, a drug only available through compounding pharmacies that is sometimes used to treat lupus patients. Quinacrine is not on the FDA’s current list of bulk substances approved for compounding, except by special permission. The ACR has asked the agency to add quinacrine to the list, but no one knows when this will happen.
Rheumatologists may also be more restricted than before in terms of which compounding pharmacies they can turn to, as new federal standards divide them into two types – those that can provide medicines in larger quantities and those that can’t.
Steroid fiasco sparked rule revisions
The ACR’s concerns follow a tighter focus by state and federal agencies on drug compounding after a fungal meningitis outbreak in 2012 was traced to contaminated steroids produced in bulk by a compounding pharmacy.
More than 800 infections, 64 of them fatal, occurred after the New England Compounding Center in Framingham, Mass., sold contaminated methylprednisolone acetate that was used in epidural and intra-articular joint injections.
The following year Congress passed the Drug Quality and Security Act, which aims, in part, to prevent compounding pharmacies from engaging in what amounts to unregulated manufacturing.
As part of the law, the FDA created a list of drugs appropriate for compounding and a process by which larger compounding pharmacies must register with the FDA, and agree to inspections. The USP standards, meanwhile, address detailed technical and safety aspects of compounding and are enforceable by the FDA and state agencies.
“USP and FDA have had the ability to regulate compounding for over a decade, but only recently have the rules become actively enforced,” said Donald Miller, PharmD, of North Dakota State University, Fargo, who helped shape the ACR’s position statement on compounding with the help of rheumatologists in private practice.
“When you make guidelines for safety, they make sense, but then you can’t anticipate the way it’s going to affect individuals’ practice. And that’s where rheumatology got caught up,” said Dr. Miller, who was a member of the FDA Arthritis Advisory Committee in 2014-2016.
In-office mixing a top concern
Other specialties, including dermatology and immunology, also stand to be affected by various changes to compounding law and practice – and their societies have been active in voicing concerns.
Though the latest revisions of USP chapter 797, which impacts in-office mixing, are still being sorted out, it’s the No. 1 compounding-related concern for rheumatologists, Dr. Miller said.
Rheumatologists routinely mix an analgesic and a steroid for injection. The analgesic makes the steroids less viscous, and offers patients hours of immediate relief. They also add analgesics to hyaluronic acid injected for viscosupplementation. The mixing is usually conducted bedside, and the injections are administered right away.
Technically, combining these products amounts to sterile compounding, Dr. Miller explained. “And theoretically, under these rules, a physician could still do this, but they’d have to do it under a sterile hood like you find in a pharmacy, and that’s just not practical. It also becomes a matter of interpretation.”
USP chapter 797 sanctions in-office mixing for “immediate use” with individual patients – which is nearly always the case for the steroid injections used in rheumatology. But it’s unclear whether “immediate use” means emergency use only, or allows for routine use, as rheumatologists hope.
“One reason this came to rheumatology’s attention is that some state boards of medicine were inspecting and saying ‘Hey, you can’t do that,’ ” Dr. Miller said.
“There’s that law of unintended consequences where you snare things in a net that you really don’t want to,” Dr. Huffstutter said.
Marcus Snow, MD, a rheumatologist at the University of Nebraska, Omaha, who also worked on the statement, said that most rheumatologists are likely unaware that their ability to mix drugs in-office has been called into question.
“I brought it up at our division meeting with a group of 10 rheumatologists, and no one was aware that this was coming down the pike,” Dr. Snow said in an interview.
Pediatric issues
Pediatric rheumatologists, and adult rheumatologists who see children occasionally, use compounding pharmacies to create palatable oral medicines and adjusted doses of adult treatments.
They also use injections combining steroids with analgesics, and consider the addition of the analgesic a key aid to compliance.
“The biggest barrier we have is patient and parent anxiety about doing the procedure and the associated pain. We always administer our steroids mixed with lidocaine to help with the postprocedural discomfort,” said Adam Reinhardt, MD, chief of pediatric rheumatology at the University of Nebraska and Children’s Hospital and Medical Center in Omaha.
Steroid injections can mean avoiding or delaying systemic treatment in children with oligoarticular arthritis, he said. “Most of us consider them a first-line therapy. The hope is that you can get by without having to use meds like methotrexate if you can get a prolonged response in the one or two joints that are active in that patient.”
But Dr. Reinhardt said that, while he mixed his own injections during his fellowship training, Children’s of Omaha now insists that they be prepared by in-house pharmacists, working under sterile hoods. The delay to receiving them in the clinic or procedure room is 40 minutes to an hour, he said, which the clinicians accommodate through careful scheduling.
The change from mixing in-clinic to relying on the central pharmacy came about in recent years, Dr. Reinhardt said, because of broader concerns related to medication storage in the clinics. While ordering from the central pharmacy works for his practice, he said, “I probably only inject maybe 50-70 joints a year, while adult rheumatologists are injecting far more than that. For a busy private practice, I can see that being a huge time constraint,” he said.
Relevance of rules
None of the rheumatologists interviewed questioned the need for tightened state and federal oversight of compounding practices overall – just the applicability of certain rules to their own practice.
Dr. Snow and Dr. Huffstutter noted that reports of infected joints – a potential result of a contaminated injection – are sporadic and rare. “There’s very little research in this, but [these types of injections] have been standard practice for decades,” Dr. Snow said.
Srikanth Mukkera, MD, a rheumatologist in Tupelo, Miss., agreed that “sporadic cases of joint infection do happen following injection, but it can be hard to show if an injection was the cause.”
Assuring that medicines are mixed only immediately prior to injection, and not stored, reduces the likelihood of contamination, Dr. Mukkera said. Moreover, he noted, epidural injections such as those that resulted in the 2012 meningitis outbreak carry different risks than those seen in intra-articular injections.
Dr. Miller, the lead author of the ACR statement, said that the rheumatologists on our committee “don’t know of anyone that’s had a knee or other joint infection from a contaminated injection. They feel that unless somebody finds some evidence of that, they should be allowed to continue” with their usual practice.
He said that he feels that the USP will ultimately heed the concerns of rheumatologists and hopefully provide a more relaxed interpretation of in-office compounding. “We’re hoping they’ll make some exceptions when they revise 797 standards or at least maybe leave room for organizations to create a best practice statement. We’ll see,” Dr. Miller said.
But this is in no way guaranteed. Dr. Huffstutter said he fears that, if the rules come to be interpreted more narrowly, even standard practices like reconstituting biologic drugs for infusion – something that’s also a routine part of in-office practice – could fall under the rubric of sterile compounding and come into question.
The quinacrine problem
A separate compounding-related issue in rheumatology is clinicians’ access to quinacrine, an antimalarial rheumatology drug that, while infrequently used, represents the only alternative to hydroxychloroquine for some lupus patients.
“There are no alternatives out there for hydroxychloroquine, so we need it as a backup,” Dr. Snow said. “If hydroxychloroquine isn’t an option, there’s nothing out there that we can use. There’s no easy replacement.”
Dr. Huffstutter said he currently had no patients on quinacrine. “It’s not very often that we use it, but in those patients that really need it, it can make a huge difference in how they do.”
Quinacrine is no longer manufactured commercially as a finished drug product but is available in a powder that compounding physicians put into 100-mg capsules. It is not on the FDA’s current list of drugs available for compounding except with special permission.
While the ACR has requested that the FDA add it the list of bulk drug substances that can be used in compounding, quinacrine remains off the list for now – and, providers say, hard to find.
Moreover, while rheumatologists may have previously been able to order and store quantities of quinacrine and other compounded nonsterile medications to dispense to their patients, they can no longer easily do so, as only the FDA-approved compounding “outsourcing facilities” are allowed to process larger orders; the rest can only respond to prescriptions for individual patients.
Dr. Miller said it’s likely that quinacrine will make it onto the FDA’s next list of bulk drugs available for compounding. “The FDA has kind of said, ‘Don’t worry about it,’ ” he said.
Stopping TNF inhibitors for pregnancy may invite flares
Women with rheumatoid arthritis or axial spondyloarthritis who stop treatment with tumor necrosis factor inhibitors when they become pregnant may be inviting disease flares during the pregnancy, according to a report published in Arthritis Research & Therapy.
To examine the frequency of rheumatoid arthritis (RA) and axial spondyloarthritis (axSpA) flares during pregnancy, researchers prospectively followed 136 women treated at the Center for Pregnancy in Rheumatic Diseases at Inselspital Bern (Switzerland) during a 5-year period. These patients – 75 with RA and 61 with axSpA – were assessed before conception, during each trimester, and 6-8 weeks postpartum for disease activity and medication use, said Stephanie van den Brandt, MD, of the department of rheumatology, immunology, and allergology at the University of Bern, and her associates.
The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at the time of a positive pregnancy test. In comparison, rheumatic disease remained stable throughout pregnancy in most women who were not taking TNF inhibitors before pregnancy, the investigators said (Arthritis Res Ther. 2017 Mar 20. doi: 10.1186/s13075-017-1269-1).
Most disease flares occurred in the first trimester among women with RA and in the second half of pregnancy among women with axSpA. Most women with RA who resumed taking TNF inhibitors when their disease flared responded well to the treatment, with CRP levels dropping by 70% and remission being achieved rapidly. In contrast, most women with axSpA who resumed taking TNF inhibitors did not respond as well, with CRP levels dropping by only 35%. Their disease was ameliorated but not controlled by restarting the therapy.
No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Women with rheumatoid arthritis or axial spondyloarthritis who stop treatment with tumor necrosis factor inhibitors when they become pregnant may be inviting disease flares during the pregnancy, according to a report published in Arthritis Research & Therapy.
To examine the frequency of rheumatoid arthritis (RA) and axial spondyloarthritis (axSpA) flares during pregnancy, researchers prospectively followed 136 women treated at the Center for Pregnancy in Rheumatic Diseases at Inselspital Bern (Switzerland) during a 5-year period. These patients – 75 with RA and 61 with axSpA – were assessed before conception, during each trimester, and 6-8 weeks postpartum for disease activity and medication use, said Stephanie van den Brandt, MD, of the department of rheumatology, immunology, and allergology at the University of Bern, and her associates.
The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at the time of a positive pregnancy test. In comparison, rheumatic disease remained stable throughout pregnancy in most women who were not taking TNF inhibitors before pregnancy, the investigators said (Arthritis Res Ther. 2017 Mar 20. doi: 10.1186/s13075-017-1269-1).
Most disease flares occurred in the first trimester among women with RA and in the second half of pregnancy among women with axSpA. Most women with RA who resumed taking TNF inhibitors when their disease flared responded well to the treatment, with CRP levels dropping by 70% and remission being achieved rapidly. In contrast, most women with axSpA who resumed taking TNF inhibitors did not respond as well, with CRP levels dropping by only 35%. Their disease was ameliorated but not controlled by restarting the therapy.
No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Women with rheumatoid arthritis or axial spondyloarthritis who stop treatment with tumor necrosis factor inhibitors when they become pregnant may be inviting disease flares during the pregnancy, according to a report published in Arthritis Research & Therapy.
To examine the frequency of rheumatoid arthritis (RA) and axial spondyloarthritis (axSpA) flares during pregnancy, researchers prospectively followed 136 women treated at the Center for Pregnancy in Rheumatic Diseases at Inselspital Bern (Switzerland) during a 5-year period. These patients – 75 with RA and 61 with axSpA – were assessed before conception, during each trimester, and 6-8 weeks postpartum for disease activity and medication use, said Stephanie van den Brandt, MD, of the department of rheumatology, immunology, and allergology at the University of Bern, and her associates.
The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at the time of a positive pregnancy test. In comparison, rheumatic disease remained stable throughout pregnancy in most women who were not taking TNF inhibitors before pregnancy, the investigators said (Arthritis Res Ther. 2017 Mar 20. doi: 10.1186/s13075-017-1269-1).
Most disease flares occurred in the first trimester among women with RA and in the second half of pregnancy among women with axSpA. Most women with RA who resumed taking TNF inhibitors when their disease flared responded well to the treatment, with CRP levels dropping by 70% and remission being achieved rapidly. In contrast, most women with axSpA who resumed taking TNF inhibitors did not respond as well, with CRP levels dropping by only 35%. Their disease was ameliorated but not controlled by restarting the therapy.
No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Key clinical point:
Major finding: The relative risk of a disease flare was 3.33 among RA patients and 3.08 among axSpA patients who discontinued TNF inhibitors at conception.
Data source: A prospective cohort study involving 75 pregnant women with RA and 61 with axial spondyloarthritis treated at one Swiss specialty center in 2000-2015.
Disclosures: No sponsor was cited for this study. Dr. van den Brandt and her associates reported having no relevant financial disclosures.
Etanercept found not optimal for reducing anterior uveitis in ankylosing spondylitis
Two anti–tumor necrosis factor monoclonal antibodies, adalimumab and infliximab, showed evidence of being markedly more effective than the anti-TNF–receptor inhibitor etanercept at reducing the rate of anterior uveitis in patients with ankylosing spondylitis in a retrospective Swedish cohort study.
To compare the efficacy of the three TNF inhibitors, researchers analyzed data in nationwide Swedish population-based registries for 1,365 ankylosing spondylitis (AS) patients who initiated treatment during a 7-year period. Treatment began with adalimumab in 406 patients, infliximab in 605, and etanercept in 354, said Elisabeth Lie, MD, of the department of rheumatology and inflammation research at the University of Gothenburg (Sweden), and her associates.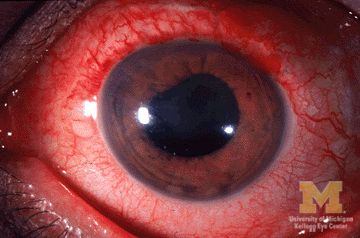
“Compared with the rates [of anterior uveitis] pretreatment, the rates increased when initiating treatment with etanercept, but decreased when starting adalimumab or infliximab,” the investigators wrote (Ann Rheum Dis. 2017 Mar 2. doi: 10.1136/annrheumdis-2016-210931).
The biological explanation for this discrepancy is unclear. It is possible that etanercept simply isn’t as protective as the other two agents, but it also appears possible that etanercept may act paradoxically to induce anterior uveitis in some patients. However, it should be noted that “previous studies have indicated that etanercept still reduces the number of uveitis flares more effectively than placebo,” Dr. Lie and her associates noted.
Regardless of the underlying reason, these findings, taken together with those of previous studies, “support the choice of another TNF inhibitor than etanercept in patients with AS with a history of anterior uveitis,” they said.
Dr. Lie also reported the results at the 2015 American College of Rheumatology annual meeting.
This study was supported by the Swedish Research Council, Gothenburg University, the Stockholm County Council, the Swedish National Rheumatism Association, the Swedish COMBINE public-private research program, the Swedish Cancer Society, the EU-IMI BT Cure project, and the Swedish Foundation for Strategic Research. Dr. Lie reported receiving personal fees from AbbVie, Bristol-Myers Squibb, Hospira, Pfizer, and UCB; her associates reported ties to numerous industry sources.
Two anti–tumor necrosis factor monoclonal antibodies, adalimumab and infliximab, showed evidence of being markedly more effective than the anti-TNF–receptor inhibitor etanercept at reducing the rate of anterior uveitis in patients with ankylosing spondylitis in a retrospective Swedish cohort study.
To compare the efficacy of the three TNF inhibitors, researchers analyzed data in nationwide Swedish population-based registries for 1,365 ankylosing spondylitis (AS) patients who initiated treatment during a 7-year period. Treatment began with adalimumab in 406 patients, infliximab in 605, and etanercept in 354, said Elisabeth Lie, MD, of the department of rheumatology and inflammation research at the University of Gothenburg (Sweden), and her associates.
“Compared with the rates [of anterior uveitis] pretreatment, the rates increased when initiating treatment with etanercept, but decreased when starting adalimumab or infliximab,” the investigators wrote (Ann Rheum Dis. 2017 Mar 2. doi: 10.1136/annrheumdis-2016-210931).
The biological explanation for this discrepancy is unclear. It is possible that etanercept simply isn’t as protective as the other two agents, but it also appears possible that etanercept may act paradoxically to induce anterior uveitis in some patients. However, it should be noted that “previous studies have indicated that etanercept still reduces the number of uveitis flares more effectively than placebo,” Dr. Lie and her associates noted.
Regardless of the underlying reason, these findings, taken together with those of previous studies, “support the choice of another TNF inhibitor than etanercept in patients with AS with a history of anterior uveitis,” they said.
Dr. Lie also reported the results at the 2015 American College of Rheumatology annual meeting.
This study was supported by the Swedish Research Council, Gothenburg University, the Stockholm County Council, the Swedish National Rheumatism Association, the Swedish COMBINE public-private research program, the Swedish Cancer Society, the EU-IMI BT Cure project, and the Swedish Foundation for Strategic Research. Dr. Lie reported receiving personal fees from AbbVie, Bristol-Myers Squibb, Hospira, Pfizer, and UCB; her associates reported ties to numerous industry sources.
Two anti–tumor necrosis factor monoclonal antibodies, adalimumab and infliximab, showed evidence of being markedly more effective than the anti-TNF–receptor inhibitor etanercept at reducing the rate of anterior uveitis in patients with ankylosing spondylitis in a retrospective Swedish cohort study.
To compare the efficacy of the three TNF inhibitors, researchers analyzed data in nationwide Swedish population-based registries for 1,365 ankylosing spondylitis (AS) patients who initiated treatment during a 7-year period. Treatment began with adalimumab in 406 patients, infliximab in 605, and etanercept in 354, said Elisabeth Lie, MD, of the department of rheumatology and inflammation research at the University of Gothenburg (Sweden), and her associates.
“Compared with the rates [of anterior uveitis] pretreatment, the rates increased when initiating treatment with etanercept, but decreased when starting adalimumab or infliximab,” the investigators wrote (Ann Rheum Dis. 2017 Mar 2. doi: 10.1136/annrheumdis-2016-210931).
The biological explanation for this discrepancy is unclear. It is possible that etanercept simply isn’t as protective as the other two agents, but it also appears possible that etanercept may act paradoxically to induce anterior uveitis in some patients. However, it should be noted that “previous studies have indicated that etanercept still reduces the number of uveitis flares more effectively than placebo,” Dr. Lie and her associates noted.
Regardless of the underlying reason, these findings, taken together with those of previous studies, “support the choice of another TNF inhibitor than etanercept in patients with AS with a history of anterior uveitis,” they said.
Dr. Lie also reported the results at the 2015 American College of Rheumatology annual meeting.
This study was supported by the Swedish Research Council, Gothenburg University, the Stockholm County Council, the Swedish National Rheumatism Association, the Swedish COMBINE public-private research program, the Swedish Cancer Society, the EU-IMI BT Cure project, and the Swedish Foundation for Strategic Research. Dr. Lie reported receiving personal fees from AbbVie, Bristol-Myers Squibb, Hospira, Pfizer, and UCB; her associates reported ties to numerous industry sources.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: Etanercept was associated with nearly a fourfold higher risk of developing uveitis than was adalimumab (HR, 3.86) and a twofold higher risk than was infliximab (HR, 1.99), but there was no difference in risk between adalimumab and infliximab.
Data source: A retrospective cohort study involving 1,365 AS patients enrolled in nationwide Swedish registries during a 7-year period.
Disclosures: This study was supported by the Swedish Research Council, Gothenburg University, the Stockholm County Council, the Swedish National Rheumatism Association, the Swedish COMBINE public-private research program, the Swedish Cancer Society, the EU-IMI BT Cure project, and the Swedish Foundation for Strategic Research. Dr. Lie reported receiving personal fees from AbbVie, Bristol-Myers Squibb, Hospira, Pfizer, and UCB; her associates reported ties to numerous industry sources.
Shingles vaccine deemed effective in people with autoimmune disease
The herpes zoster vaccine reduces the risk of shingles in older adults with autoimmune disease, even if they are taking immunosuppressants for their condition, but the protection begins to wane after about 5 years, a recent retrospective study found.
“There has been some concern that patients with autoimmune conditions might have a lower immunogenic response to herpes zoster vaccination, especially when treated with immunosuppressive medications such as glucocorticoids,” wrote Huifeng Yun, PhD, of the University of Alabama at Birmingham, and her colleagues.
The researchers used 2006-2013 Medicare data to calculate the risk of shingles among Medicare recipients who had an autoimmune disease and either did or did not receive the herpes zoster vaccine. All the patients had been enrolled in Medicare for at least 12 continuous months and had a diagnosis of ankylosing spondylitis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or rheumatoid arthritis.
The researchers matched 59,627 patients who received the herpes zoster vaccine with 119,254 unvaccinated patients, based on age, sex, race, calendar year, autoimmune disease type, and use of autoimmune drugs (biologics, disease-modifying antirheumatic drugs, and glucocorticoids). During a follow-up of up to 7 years, the researchers additionally accounted for comorbid medical conditions and concurrent medications each year.
The cohort, with an average age of 73.5 years in both groups, included 53.1% of adults with rheumatoid arthritis, 31.6% with psoriasis, 20.9% with inflammatory bowel disease, 4.7% with psoriatic arthritis, and 1.4% with ankylosing spondylitis.
Those who received the vaccine had a rate of 0.75 herpes zoster cases per 100 people during the first year, which rose to 1.25 cases per 100 people per year at the seventh year after vaccination. The rate among unvaccinated individuals stayed steady at approximately 1.3-1.7 cases per 100 people per year throughout the study period. These rates, as expected, were approximately 50% higher than in the general population over age 70 without autoimmune disease.
Compared with unvaccinated individuals, vaccinated individuals had a reduced relative risk for shingles of 0.74-0.77 after adjustment for confounders, but the risk reduction only remained statistically significant for the first 5 years after vaccination.
The waning seen with the vaccine’s effectiveness “raises the possibility that patients might benefit from a booster vaccine at some point after initial vaccination, although no recommendation currently exists that would support such a practice,” the authors wrote.
Dr. Yun has received research funding from Amgen. Other authors disclosed ties to Amgen, AstraZeneca, Bristol-Myers Squibb, Crescendo Bioscience, Janssen, and Pfizer. One author has received research support and consulting fees from Corrona. The study did not note an external source of funding.
The herpes zoster vaccine reduces the risk of shingles in older adults with autoimmune disease, even if they are taking immunosuppressants for their condition, but the protection begins to wane after about 5 years, a recent retrospective study found.
“There has been some concern that patients with autoimmune conditions might have a lower immunogenic response to herpes zoster vaccination, especially when treated with immunosuppressive medications such as glucocorticoids,” wrote Huifeng Yun, PhD, of the University of Alabama at Birmingham, and her colleagues.
The researchers used 2006-2013 Medicare data to calculate the risk of shingles among Medicare recipients who had an autoimmune disease and either did or did not receive the herpes zoster vaccine. All the patients had been enrolled in Medicare for at least 12 continuous months and had a diagnosis of ankylosing spondylitis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or rheumatoid arthritis.
The researchers matched 59,627 patients who received the herpes zoster vaccine with 119,254 unvaccinated patients, based on age, sex, race, calendar year, autoimmune disease type, and use of autoimmune drugs (biologics, disease-modifying antirheumatic drugs, and glucocorticoids). During a follow-up of up to 7 years, the researchers additionally accounted for comorbid medical conditions and concurrent medications each year.
The cohort, with an average age of 73.5 years in both groups, included 53.1% of adults with rheumatoid arthritis, 31.6% with psoriasis, 20.9% with inflammatory bowel disease, 4.7% with psoriatic arthritis, and 1.4% with ankylosing spondylitis.
Those who received the vaccine had a rate of 0.75 herpes zoster cases per 100 people during the first year, which rose to 1.25 cases per 100 people per year at the seventh year after vaccination. The rate among unvaccinated individuals stayed steady at approximately 1.3-1.7 cases per 100 people per year throughout the study period. These rates, as expected, were approximately 50% higher than in the general population over age 70 without autoimmune disease.
Compared with unvaccinated individuals, vaccinated individuals had a reduced relative risk for shingles of 0.74-0.77 after adjustment for confounders, but the risk reduction only remained statistically significant for the first 5 years after vaccination.
The waning seen with the vaccine’s effectiveness “raises the possibility that patients might benefit from a booster vaccine at some point after initial vaccination, although no recommendation currently exists that would support such a practice,” the authors wrote.
Dr. Yun has received research funding from Amgen. Other authors disclosed ties to Amgen, AstraZeneca, Bristol-Myers Squibb, Crescendo Bioscience, Janssen, and Pfizer. One author has received research support and consulting fees from Corrona. The study did not note an external source of funding.
The herpes zoster vaccine reduces the risk of shingles in older adults with autoimmune disease, even if they are taking immunosuppressants for their condition, but the protection begins to wane after about 5 years, a recent retrospective study found.
“There has been some concern that patients with autoimmune conditions might have a lower immunogenic response to herpes zoster vaccination, especially when treated with immunosuppressive medications such as glucocorticoids,” wrote Huifeng Yun, PhD, of the University of Alabama at Birmingham, and her colleagues.
The researchers used 2006-2013 Medicare data to calculate the risk of shingles among Medicare recipients who had an autoimmune disease and either did or did not receive the herpes zoster vaccine. All the patients had been enrolled in Medicare for at least 12 continuous months and had a diagnosis of ankylosing spondylitis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or rheumatoid arthritis.
The researchers matched 59,627 patients who received the herpes zoster vaccine with 119,254 unvaccinated patients, based on age, sex, race, calendar year, autoimmune disease type, and use of autoimmune drugs (biologics, disease-modifying antirheumatic drugs, and glucocorticoids). During a follow-up of up to 7 years, the researchers additionally accounted for comorbid medical conditions and concurrent medications each year.
The cohort, with an average age of 73.5 years in both groups, included 53.1% of adults with rheumatoid arthritis, 31.6% with psoriasis, 20.9% with inflammatory bowel disease, 4.7% with psoriatic arthritis, and 1.4% with ankylosing spondylitis.
Those who received the vaccine had a rate of 0.75 herpes zoster cases per 100 people during the first year, which rose to 1.25 cases per 100 people per year at the seventh year after vaccination. The rate among unvaccinated individuals stayed steady at approximately 1.3-1.7 cases per 100 people per year throughout the study period. These rates, as expected, were approximately 50% higher than in the general population over age 70 without autoimmune disease.
Compared with unvaccinated individuals, vaccinated individuals had a reduced relative risk for shingles of 0.74-0.77 after adjustment for confounders, but the risk reduction only remained statistically significant for the first 5 years after vaccination.
The waning seen with the vaccine’s effectiveness “raises the possibility that patients might benefit from a booster vaccine at some point after initial vaccination, although no recommendation currently exists that would support such a practice,” the authors wrote.
Dr. Yun has received research funding from Amgen. Other authors disclosed ties to Amgen, AstraZeneca, Bristol-Myers Squibb, Crescendo Bioscience, Janssen, and Pfizer. One author has received research support and consulting fees from Corrona. The study did not note an external source of funding.
Key clinical point:
Major finding: Medicare patients with autoimmune disease had a 23%-26% reduced risk of shingles for 5 years after receiving the herpes zoster vaccine.
Data source: The findings are based on analysis of 2006-2013 Medicare data on 59,627 patients who received the herpes zoster vaccine and 119,254 patients who didn’t.
Disclosures: Dr. Yun has received research funding from Amgen. Other authors disclosed ties to Amgen, AstraZeneca, Bristol-Myers Squibb, Crescendo Bioscience, Janssen, and Pfizer. One author has received research support and consulting fees from Corrona. The study did not note an external source of funding.
Biosimilars: No big dollar savings, but are clinically ‘dead on’
SNOWMASS, COLO. – If you thought biosimilars would bring sharply reduced pricing compared with their parent agents, with resultant greater patient access to highly effective therapies for rheumatic diseases ... think again.
“The promise to our patients of biosimilars – greater access to treatments – is something I think we’re just not going to see, at least not here in the U.S.,” Michael E. Weinblatt, MD, declared at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.

In contrast, the safety and efficacy of the biosimilars, as well as their interchangeability with their reference products, appear to be as hoped for. At the 2016 annual meeting of the American College of Rheumatology, Dr. Weinblatt presented the week 24 results of a phase III, randomized trial involving rheumatoid arthritis patients on background methotrexate plus either adalimumab (Humira) or its biosimilar SB5.
“Essentially, they’re dead on in clinical response, they’re dead on in antibody levels, and they’re dead on in toxicity. And, you can put any of the biosimilars up there and the results are the same. If they get approved, this is what you’re going to see,” the rheumatologist said.
Also at the 2016 ACR annual meeting, he noted, Danish investigators presented reassuring 1-year follow-up data on 802 Danes with inflammatory rheumatic diseases who switched from infliximab (Remicade) to its biosimilar Remsima. Disease activity and flare rates in the year following the switch were similar to those in the year before. The 1-year rate of adherence to Remsima was 84%, similar to the historical 86% 1-year rate with infliximab.
“So, I’m pretty comfortable with the biosimilars,” Dr. Weinblatt continued.
He observed that, of all the systemic rheumatic diseases, the greatest progress has occurred in the treatment of rheumatoid arthritis.
“We have made great advances in the treatment of this disease, unlike many of our other diseases. Methotrexate and combination therapies with small molecules and biologics has dramatically changed the course of the disease,” he noted. “The greatest challenge we have now as rheumatologists is access barriers for our patients.”
Dr. Weinblatt reported receiving research grants from half a dozen companies and serving as a consultant to more than two dozen.
SNOWMASS, COLO. – If you thought biosimilars would bring sharply reduced pricing compared with their parent agents, with resultant greater patient access to highly effective therapies for rheumatic diseases ... think again.
“The promise to our patients of biosimilars – greater access to treatments – is something I think we’re just not going to see, at least not here in the U.S.,” Michael E. Weinblatt, MD, declared at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
In contrast, the safety and efficacy of the biosimilars, as well as their interchangeability with their reference products, appear to be as hoped for. At the 2016 annual meeting of the American College of Rheumatology, Dr. Weinblatt presented the week 24 results of a phase III, randomized trial involving rheumatoid arthritis patients on background methotrexate plus either adalimumab (Humira) or its biosimilar SB5.
“Essentially, they’re dead on in clinical response, they’re dead on in antibody levels, and they’re dead on in toxicity. And, you can put any of the biosimilars up there and the results are the same. If they get approved, this is what you’re going to see,” the rheumatologist said.
Also at the 2016 ACR annual meeting, he noted, Danish investigators presented reassuring 1-year follow-up data on 802 Danes with inflammatory rheumatic diseases who switched from infliximab (Remicade) to its biosimilar Remsima. Disease activity and flare rates in the year following the switch were similar to those in the year before. The 1-year rate of adherence to Remsima was 84%, similar to the historical 86% 1-year rate with infliximab.
“So, I’m pretty comfortable with the biosimilars,” Dr. Weinblatt continued.
He observed that, of all the systemic rheumatic diseases, the greatest progress has occurred in the treatment of rheumatoid arthritis.
“We have made great advances in the treatment of this disease, unlike many of our other diseases. Methotrexate and combination therapies with small molecules and biologics has dramatically changed the course of the disease,” he noted. “The greatest challenge we have now as rheumatologists is access barriers for our patients.”
Dr. Weinblatt reported receiving research grants from half a dozen companies and serving as a consultant to more than two dozen.
SNOWMASS, COLO. – If you thought biosimilars would bring sharply reduced pricing compared with their parent agents, with resultant greater patient access to highly effective therapies for rheumatic diseases ... think again.
“The promise to our patients of biosimilars – greater access to treatments – is something I think we’re just not going to see, at least not here in the U.S.,” Michael E. Weinblatt, MD, declared at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
In contrast, the safety and efficacy of the biosimilars, as well as their interchangeability with their reference products, appear to be as hoped for. At the 2016 annual meeting of the American College of Rheumatology, Dr. Weinblatt presented the week 24 results of a phase III, randomized trial involving rheumatoid arthritis patients on background methotrexate plus either adalimumab (Humira) or its biosimilar SB5.
“Essentially, they’re dead on in clinical response, they’re dead on in antibody levels, and they’re dead on in toxicity. And, you can put any of the biosimilars up there and the results are the same. If they get approved, this is what you’re going to see,” the rheumatologist said.
Also at the 2016 ACR annual meeting, he noted, Danish investigators presented reassuring 1-year follow-up data on 802 Danes with inflammatory rheumatic diseases who switched from infliximab (Remicade) to its biosimilar Remsima. Disease activity and flare rates in the year following the switch were similar to those in the year before. The 1-year rate of adherence to Remsima was 84%, similar to the historical 86% 1-year rate with infliximab.
“So, I’m pretty comfortable with the biosimilars,” Dr. Weinblatt continued.
He observed that, of all the systemic rheumatic diseases, the greatest progress has occurred in the treatment of rheumatoid arthritis.
“We have made great advances in the treatment of this disease, unlike many of our other diseases. Methotrexate and combination therapies with small molecules and biologics has dramatically changed the course of the disease,” he noted. “The greatest challenge we have now as rheumatologists is access barriers for our patients.”
Dr. Weinblatt reported receiving research grants from half a dozen companies and serving as a consultant to more than two dozen.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM
Familial and sporadic ankylosing spondylitis differ in small ways
While differences do exist between familial and sporadic ankylosing spondylitis, key similarities suggest that the two conditions can be treated the same way, according to a new study presented at the annual meeting of the Canadian Rheumatology Association.
“AS [ankylosing spondylitis] patients with a family history of AS are not very different from patients without any family history,” Nigil Haroon, MD, of the University Health Network in Toronto, explained in an interview. “They have similar disease activity as measured by markers of inflammation [and] similar disease severity as assessed by radiographic scoring for spinal damage.”
Dr. Haroon, along with his coinvestigators – including Bruce Sheng, MD,of the same institution, who presented the study at the meeting – prospectively followed AS patients satisfying the New York criteria for a period of 15 years, collecting data on 888 eligible subjects who were eventually included in the study. Of the subjects included, 74% were male, the average age was 45.6 years (standard deviation, 13.7 years), and average disease duration was 15 years (SD, 11.5 years).

The investigators found some similarities between the 177 (20%) patients with familial AS who had at least one first- or second-generation relative with the disease and the 711 with sporadic AS. Anti–tumor necrosis factor (anti-TNF) treatment failed in 23.1% of familial AS patients and 23.6% of sporadic disease patients based on the lack of a “sustained clinical effect” for more than 1 year. There were also no differences found between the groups in clinical and radiographic severity of disease.
However, patients with familial AS did record earlier onset of disease (22.5 years vs. 24.3 years; P = .016), longer disease duration (17.4 years vs. 14.3 years; P = .003), and higher HLA-B27 positivity (90% vs. 65%; P less than .001), along with higher rates of uveitis, psoriatic arthritis, and inflammatory bowel disease.
“Some of the findings are expected, including the higher prevalence of HLA-B27 due to gene sharing in the family. ... The higher B27 sharing may also affect the uveitis prevalence as well in familial AS,” Dr. Haroon explained. “The similar radiographic progression rates and treatment responses are interesting findings.”
In terms of the ramifications of these findings, Dr. Haroon stated that clinicians should reevaluate how they prescribe drugs to their AS patients.
“The high likelihood of uveitis in familial AS patients – 43% versus 29% – may affect the choice of treatment as all drugs are not equally effective in uveitis,” he said. “As the family history of extra-articular manifestations is high in familial AS, it remains to be seen if a lower threshold for investigating symptoms suggestive of IBD/uveitis will decrease delays in diagnosis of these conditions in individuals with a family history of AS.”
Moving forward from here, Dr. Haroon called for family studies, especially those including families with multiple individuals affected with AS, as these can help identify genetic risk factors that may be contribute to the development of AS.
“There is paucity of data on familial AS,” Dr. Haroon said. “The strength of this study is the large dataset.”
The study was funded by the Canadian Rheumatology Association’s Summer Research Program, which supported Dr. Sheng. Dr. Sheng and Dr. Haroon did not report any other relevant financial disclosures.
While differences do exist between familial and sporadic ankylosing spondylitis, key similarities suggest that the two conditions can be treated the same way, according to a new study presented at the annual meeting of the Canadian Rheumatology Association.
“AS [ankylosing spondylitis] patients with a family history of AS are not very different from patients without any family history,” Nigil Haroon, MD, of the University Health Network in Toronto, explained in an interview. “They have similar disease activity as measured by markers of inflammation [and] similar disease severity as assessed by radiographic scoring for spinal damage.”
Dr. Haroon, along with his coinvestigators – including Bruce Sheng, MD,of the same institution, who presented the study at the meeting – prospectively followed AS patients satisfying the New York criteria for a period of 15 years, collecting data on 888 eligible subjects who were eventually included in the study. Of the subjects included, 74% were male, the average age was 45.6 years (standard deviation, 13.7 years), and average disease duration was 15 years (SD, 11.5 years).
The investigators found some similarities between the 177 (20%) patients with familial AS who had at least one first- or second-generation relative with the disease and the 711 with sporadic AS. Anti–tumor necrosis factor (anti-TNF) treatment failed in 23.1% of familial AS patients and 23.6% of sporadic disease patients based on the lack of a “sustained clinical effect” for more than 1 year. There were also no differences found between the groups in clinical and radiographic severity of disease.
However, patients with familial AS did record earlier onset of disease (22.5 years vs. 24.3 years; P = .016), longer disease duration (17.4 years vs. 14.3 years; P = .003), and higher HLA-B27 positivity (90% vs. 65%; P less than .001), along with higher rates of uveitis, psoriatic arthritis, and inflammatory bowel disease.
“Some of the findings are expected, including the higher prevalence of HLA-B27 due to gene sharing in the family. ... The higher B27 sharing may also affect the uveitis prevalence as well in familial AS,” Dr. Haroon explained. “The similar radiographic progression rates and treatment responses are interesting findings.”
In terms of the ramifications of these findings, Dr. Haroon stated that clinicians should reevaluate how they prescribe drugs to their AS patients.
“The high likelihood of uveitis in familial AS patients – 43% versus 29% – may affect the choice of treatment as all drugs are not equally effective in uveitis,” he said. “As the family history of extra-articular manifestations is high in familial AS, it remains to be seen if a lower threshold for investigating symptoms suggestive of IBD/uveitis will decrease delays in diagnosis of these conditions in individuals with a family history of AS.”
Moving forward from here, Dr. Haroon called for family studies, especially those including families with multiple individuals affected with AS, as these can help identify genetic risk factors that may be contribute to the development of AS.
“There is paucity of data on familial AS,” Dr. Haroon said. “The strength of this study is the large dataset.”
The study was funded by the Canadian Rheumatology Association’s Summer Research Program, which supported Dr. Sheng. Dr. Sheng and Dr. Haroon did not report any other relevant financial disclosures.
While differences do exist between familial and sporadic ankylosing spondylitis, key similarities suggest that the two conditions can be treated the same way, according to a new study presented at the annual meeting of the Canadian Rheumatology Association.
“AS [ankylosing spondylitis] patients with a family history of AS are not very different from patients without any family history,” Nigil Haroon, MD, of the University Health Network in Toronto, explained in an interview. “They have similar disease activity as measured by markers of inflammation [and] similar disease severity as assessed by radiographic scoring for spinal damage.”
Dr. Haroon, along with his coinvestigators – including Bruce Sheng, MD,of the same institution, who presented the study at the meeting – prospectively followed AS patients satisfying the New York criteria for a period of 15 years, collecting data on 888 eligible subjects who were eventually included in the study. Of the subjects included, 74% were male, the average age was 45.6 years (standard deviation, 13.7 years), and average disease duration was 15 years (SD, 11.5 years).
The investigators found some similarities between the 177 (20%) patients with familial AS who had at least one first- or second-generation relative with the disease and the 711 with sporadic AS. Anti–tumor necrosis factor (anti-TNF) treatment failed in 23.1% of familial AS patients and 23.6% of sporadic disease patients based on the lack of a “sustained clinical effect” for more than 1 year. There were also no differences found between the groups in clinical and radiographic severity of disease.
However, patients with familial AS did record earlier onset of disease (22.5 years vs. 24.3 years; P = .016), longer disease duration (17.4 years vs. 14.3 years; P = .003), and higher HLA-B27 positivity (90% vs. 65%; P less than .001), along with higher rates of uveitis, psoriatic arthritis, and inflammatory bowel disease.
“Some of the findings are expected, including the higher prevalence of HLA-B27 due to gene sharing in the family. ... The higher B27 sharing may also affect the uveitis prevalence as well in familial AS,” Dr. Haroon explained. “The similar radiographic progression rates and treatment responses are interesting findings.”
In terms of the ramifications of these findings, Dr. Haroon stated that clinicians should reevaluate how they prescribe drugs to their AS patients.
“The high likelihood of uveitis in familial AS patients – 43% versus 29% – may affect the choice of treatment as all drugs are not equally effective in uveitis,” he said. “As the family history of extra-articular manifestations is high in familial AS, it remains to be seen if a lower threshold for investigating symptoms suggestive of IBD/uveitis will decrease delays in diagnosis of these conditions in individuals with a family history of AS.”
Moving forward from here, Dr. Haroon called for family studies, especially those including families with multiple individuals affected with AS, as these can help identify genetic risk factors that may be contribute to the development of AS.
“There is paucity of data on familial AS,” Dr. Haroon said. “The strength of this study is the large dataset.”
The study was funded by the Canadian Rheumatology Association’s Summer Research Program, which supported Dr. Sheng. Dr. Sheng and Dr. Haroon did not report any other relevant financial disclosures.
FROM THE CRA SCIENTIFIC CONFERENCE
Key clinical point:
Major finding: Anti-TNF treatment failed in 23.1% of familial AS patients and 23.6% of sporadic disease patients based on the lack of a “sustained clinical effect” for more than 1 year.
Data source: Prospective cohort study of 888 patients with AS over 15 years.
Disclosures: Funded by the Canadian Rheumatology Association’s Summer Research Program. No other relevant disclosures were reported.
Perioperative infliximab does not increase serious infection risk
Administration of infliximab within 4 weeks of elective knee or hip arthroplasty did not have any significant effect on patients’ risk of serious infection after surgery, whereas the use of glucocorticoids increased that risk, in an analysis of a Medicare claims database.
“This increased risk with glucocorticoids has been suggested by previous studies [and] although this risk may be related in part to increased disease severity among glucocorticoid treated patients, a direct medication effect is likely. [These data suggest] that prolonged interruptions in infliximab therapy prior to surgery may be counterproductive if higher dose glucocorticoid therapy is used in substitution,” wrote the authors of the new study, led by Michael D. George, MD, of the University of Pennsylvania in Philadelphia.
Dr. George and his colleagues examined data from the U.S. Medicare claims system on 4,288 elective knee or hip arthroplasties in individuals with rheumatoid arthritis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or ankylosing spondylitis who received infliximab within 6 months prior to the operation during 2007-2013 (Arthritis Care Res. 2017 Jan 27. doi: 10.1002/acr.23209).
The patients had to have received infliximab at least three times within a year of their procedure to establish that they were receiving stable therapy over a long-term period. The investigators also looked at oral prednisone, prednisolone, and methylprednisolone prescriptions and used data on average dosing to determine how much was administered to each subject.
“Although previous studies have treated TNF stopping vs. not stopping as a dichotomous exposure based on an arbitrary (and variable) stopping definition, in this study the primary analysis evaluated stop timing as a more general categorical exposure using 4-week intervals (half the standard rheumatoid arthritis dosing interval) to allow better assessment of the optimal stop timing,” the authors explained.
Stopping infliximab within 4 weeks of the operation did not significantly influence the rate of serious infection within 30 days (adjusted odds ratio, 0.90; 95% CI, 0.60-1.34) and neither did stopping within 4-8 weeks (OR, 0.95; 95% CI, 0.62-1.36) when compared against stopping 8-12 weeks before surgery. Of the 4,288 arthroplasties, 270 serious infections (6.3%) occurred within 30 days of the operation.
There also was no significant difference between stopping within 4 weeks and 8-12 weeks in the rate of prosthetic joint infection within 1 year of the operation (hazard ratio, 0.98; 95% CI, 0.52-1.87). Overall, prosthetic joint infection occurred 2.9 times per 100 person-years.
However, glucocorticoid doses of more than 10 mg per day were risky. The odds for a serious infection within 30 days after surgery more than doubled with that level of use (OR, 2.11; 95% CI, 1.30-3.40), while the risk for a prosthetic joint infection within 1 year of the surgery also rose significantly (HR, 2.70; 95% CI, 1.30-5.60).

“This is a very well done paper that adds important observational data to our understanding of perioperative medication risk,” Dr. Goodman said.
But the study results will not, at least initially, bring about any changes to the proposed guidelines for perioperative management of patients taking antirheumatic drugs that were described at the 2016 annual meeting of the American College of Rheumatology, she said.
“We were aware of the abstract, which was also presented at the ACR last fall at the time the current perioperative medication management guidelines were presented, and it won’t change guidelines at this point,” said Dr. Goodman, who is one of the lead authors of the proposed guidelines. “[But] I think [the study] could provide important background information to use in a randomized clinical trial to compare infection on [and] not on TNF inhibitors.”
The proposed guidelines conditionally recommend that all biologics should be withheld prior to surgery in patients with inflammatory arthritis, that surgery should be planned for the end of the dosing cycle, and that current daily doses of glucocorticoids, rather than supraphysiologic doses, should be continued in adults with rheumatoid arthritis, lupus, or inflammatory arthritis.
The National Institutes of Health, the Rheumatology Research Foundation, and the Department of Veterans Affairs funded the study. Dr. George did not report any relevant financial disclosures. Two coauthors disclosed receiving research grants or consulting fees from pharmaceutical companies for unrelated work.
Administration of infliximab within 4 weeks of elective knee or hip arthroplasty did not have any significant effect on patients’ risk of serious infection after surgery, whereas the use of glucocorticoids increased that risk, in an analysis of a Medicare claims database.
“This increased risk with glucocorticoids has been suggested by previous studies [and] although this risk may be related in part to increased disease severity among glucocorticoid treated patients, a direct medication effect is likely. [These data suggest] that prolonged interruptions in infliximab therapy prior to surgery may be counterproductive if higher dose glucocorticoid therapy is used in substitution,” wrote the authors of the new study, led by Michael D. George, MD, of the University of Pennsylvania in Philadelphia.
Dr. George and his colleagues examined data from the U.S. Medicare claims system on 4,288 elective knee or hip arthroplasties in individuals with rheumatoid arthritis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or ankylosing spondylitis who received infliximab within 6 months prior to the operation during 2007-2013 (Arthritis Care Res. 2017 Jan 27. doi: 10.1002/acr.23209).
The patients had to have received infliximab at least three times within a year of their procedure to establish that they were receiving stable therapy over a long-term period. The investigators also looked at oral prednisone, prednisolone, and methylprednisolone prescriptions and used data on average dosing to determine how much was administered to each subject.
“Although previous studies have treated TNF stopping vs. not stopping as a dichotomous exposure based on an arbitrary (and variable) stopping definition, in this study the primary analysis evaluated stop timing as a more general categorical exposure using 4-week intervals (half the standard rheumatoid arthritis dosing interval) to allow better assessment of the optimal stop timing,” the authors explained.
Stopping infliximab within 4 weeks of the operation did not significantly influence the rate of serious infection within 30 days (adjusted odds ratio, 0.90; 95% CI, 0.60-1.34) and neither did stopping within 4-8 weeks (OR, 0.95; 95% CI, 0.62-1.36) when compared against stopping 8-12 weeks before surgery. Of the 4,288 arthroplasties, 270 serious infections (6.3%) occurred within 30 days of the operation.
There also was no significant difference between stopping within 4 weeks and 8-12 weeks in the rate of prosthetic joint infection within 1 year of the operation (hazard ratio, 0.98; 95% CI, 0.52-1.87). Overall, prosthetic joint infection occurred 2.9 times per 100 person-years.
However, glucocorticoid doses of more than 10 mg per day were risky. The odds for a serious infection within 30 days after surgery more than doubled with that level of use (OR, 2.11; 95% CI, 1.30-3.40), while the risk for a prosthetic joint infection within 1 year of the surgery also rose significantly (HR, 2.70; 95% CI, 1.30-5.60).
“This is a very well done paper that adds important observational data to our understanding of perioperative medication risk,” Dr. Goodman said.
But the study results will not, at least initially, bring about any changes to the proposed guidelines for perioperative management of patients taking antirheumatic drugs that were described at the 2016 annual meeting of the American College of Rheumatology, she said.
“We were aware of the abstract, which was also presented at the ACR last fall at the time the current perioperative medication management guidelines were presented, and it won’t change guidelines at this point,” said Dr. Goodman, who is one of the lead authors of the proposed guidelines. “[But] I think [the study] could provide important background information to use in a randomized clinical trial to compare infection on [and] not on TNF inhibitors.”
The proposed guidelines conditionally recommend that all biologics should be withheld prior to surgery in patients with inflammatory arthritis, that surgery should be planned for the end of the dosing cycle, and that current daily doses of glucocorticoids, rather than supraphysiologic doses, should be continued in adults with rheumatoid arthritis, lupus, or inflammatory arthritis.
The National Institutes of Health, the Rheumatology Research Foundation, and the Department of Veterans Affairs funded the study. Dr. George did not report any relevant financial disclosures. Two coauthors disclosed receiving research grants or consulting fees from pharmaceutical companies for unrelated work.
Administration of infliximab within 4 weeks of elective knee or hip arthroplasty did not have any significant effect on patients’ risk of serious infection after surgery, whereas the use of glucocorticoids increased that risk, in an analysis of a Medicare claims database.
“This increased risk with glucocorticoids has been suggested by previous studies [and] although this risk may be related in part to increased disease severity among glucocorticoid treated patients, a direct medication effect is likely. [These data suggest] that prolonged interruptions in infliximab therapy prior to surgery may be counterproductive if higher dose glucocorticoid therapy is used in substitution,” wrote the authors of the new study, led by Michael D. George, MD, of the University of Pennsylvania in Philadelphia.
Dr. George and his colleagues examined data from the U.S. Medicare claims system on 4,288 elective knee or hip arthroplasties in individuals with rheumatoid arthritis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or ankylosing spondylitis who received infliximab within 6 months prior to the operation during 2007-2013 (Arthritis Care Res. 2017 Jan 27. doi: 10.1002/acr.23209).
The patients had to have received infliximab at least three times within a year of their procedure to establish that they were receiving stable therapy over a long-term period. The investigators also looked at oral prednisone, prednisolone, and methylprednisolone prescriptions and used data on average dosing to determine how much was administered to each subject.
“Although previous studies have treated TNF stopping vs. not stopping as a dichotomous exposure based on an arbitrary (and variable) stopping definition, in this study the primary analysis evaluated stop timing as a more general categorical exposure using 4-week intervals (half the standard rheumatoid arthritis dosing interval) to allow better assessment of the optimal stop timing,” the authors explained.
Stopping infliximab within 4 weeks of the operation did not significantly influence the rate of serious infection within 30 days (adjusted odds ratio, 0.90; 95% CI, 0.60-1.34) and neither did stopping within 4-8 weeks (OR, 0.95; 95% CI, 0.62-1.36) when compared against stopping 8-12 weeks before surgery. Of the 4,288 arthroplasties, 270 serious infections (6.3%) occurred within 30 days of the operation.
There also was no significant difference between stopping within 4 weeks and 8-12 weeks in the rate of prosthetic joint infection within 1 year of the operation (hazard ratio, 0.98; 95% CI, 0.52-1.87). Overall, prosthetic joint infection occurred 2.9 times per 100 person-years.
However, glucocorticoid doses of more than 10 mg per day were risky. The odds for a serious infection within 30 days after surgery more than doubled with that level of use (OR, 2.11; 95% CI, 1.30-3.40), while the risk for a prosthetic joint infection within 1 year of the surgery also rose significantly (HR, 2.70; 95% CI, 1.30-5.60).
“This is a very well done paper that adds important observational data to our understanding of perioperative medication risk,” Dr. Goodman said.
But the study results will not, at least initially, bring about any changes to the proposed guidelines for perioperative management of patients taking antirheumatic drugs that were described at the 2016 annual meeting of the American College of Rheumatology, she said.
“We were aware of the abstract, which was also presented at the ACR last fall at the time the current perioperative medication management guidelines were presented, and it won’t change guidelines at this point,” said Dr. Goodman, who is one of the lead authors of the proposed guidelines. “[But] I think [the study] could provide important background information to use in a randomized clinical trial to compare infection on [and] not on TNF inhibitors.”
The proposed guidelines conditionally recommend that all biologics should be withheld prior to surgery in patients with inflammatory arthritis, that surgery should be planned for the end of the dosing cycle, and that current daily doses of glucocorticoids, rather than supraphysiologic doses, should be continued in adults with rheumatoid arthritis, lupus, or inflammatory arthritis.
The National Institutes of Health, the Rheumatology Research Foundation, and the Department of Veterans Affairs funded the study. Dr. George did not report any relevant financial disclosures. Two coauthors disclosed receiving research grants or consulting fees from pharmaceutical companies for unrelated work.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: Subjects on glucocorticoids had an OR of 2.11 (95% CI 1.30-3.40) for serious infection within 30 days and an HR of 2.70 (95% CI 1.30-5.60) for prosthetic joint infection within 1 year.
Data source: Retrospective cohort study of 4,288 elective knee and hip arthroplasties in Medicare patients with rheumatoid arthritis, inflammatory bowel disease, psoriasis, psoriatic arthritis, or ankylosing spondylitis during 2007-2013.
Disclosures: The National Institutes of Health, the Rheumatology Research Foundation, and the Department of Veterans Affairs funded the study. Dr. George did not report any relevant financial disclosures. Two coauthors disclosed receiving research grants or consulting fees from pharmaceutical companies for unrelated work.
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.

“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).

“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
[email protected]
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
[email protected]
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
[email protected]
On Twitter @alz_gal
Reports of new-onset joint pain differ after starting vedolizumab
Two recent reports that provide opposing evidence about the potential for inflammatory bowel disease patients to develop articular manifestations after starting vedolizumab raise questions for future studies to answer in regard to a plausible mechanism for the adverse event and its relative importance.
The two reports, one a case series of 5 patients with inflammatory bowel disease (IBD) who developed articular manifestations after beginning vedolizumab (Entyvio) and the other a prospective cohort study of 53 patients with IBD who started vedolizumab without any subsequent cases of induction or flare of arthritis and/or sacroiliitis, came to somewhat different conclusions about the beneficial or paradoxical effects of vedolizumab’s blockade of the alpha4beta7 receptor on articular manifestations of IBD.
The five-patient cases series reported by Gaëlle Varkas, MD, a doctoral student at the University of Ghent, Belgium, and her colleagues consisted of five IBD patients, aged 26-50 years, who developed either new onset or an exacerbation of sacroiliitis or arthritis soon after starting vedolizumab. All but one of the patients was female (Ann Rheum Dis. 2016 Nov 29. doi: 10.1136/annrheumdis-2016-210233). In these patients, the investigators said, vedolizumab did not “seem to show any efficacy in and might even induce arthritis and/or sacroiliitis.”
The first was a 50-year-old woman who had progressive back pain with MRI-confirmed bilateral sacroiliitis about 2 months after beginning vedolizumab. The second patient, a 28-year-old woman with no previous history of spondyloarthropathy, had lower limb pain, a painful left shoulder, and arthritis of one wrist. Ultrasound examination confirmed intercarpal effusions and synovial hyperproliferation.
The third patient was 30 years old and male. He had both ankylosing spondylitis and Crohn’s disease, and experienced arthralgias, elevated C-reactive protein, and MRI-confirmed axial skeletal inflammation 4 weeks after starting vedolizumab.
The fourth patient was a 47-year-old woman with no previous history of spondyloarthropathy who developed MRI-confirmed sacroiliitis after beginning vedolizumab. The fifth patient, a 26-year-old woman, developed polyarticular joint pain after starting vedolizumab. Examination of this patient showed synovitis and enthesitis of multiple joints of the appendicular skeleton.
In discussion, Dr. Varkas and her colleagues noted that “one of the many hypotheses is that integrins and adhesion molecules play a role in the interception of recirculating activated lymphocytes between the gut and the synovial membrane due to the inhibition of the alpha4beta7 integrin homing at the level of the gut.” However, the investigators also acknowledged that other hypotheses may also account for their findings. “Alternatively, in the presence of vedolizumab, cellular recruitment may be mediated by yet to be determined adhesion molecules. This recirculation theory might explain the short mean interval of 64 days between vedolizumab initiation and the expression of symptoms.”
Another group, publishing its prospective cohort study in a letter, had different findings (Ann Rheum Dis. 2017 Jan 17. doi: 10.1136/annrheumdis-2016-211011).
“Although the hypotheses proposed by the authors to explain such events sounds reasonable,” wrote Ambrogio Orlando, MD, and his coauthors, their experience of the effect of vedolizumab on spondyloarthritis differed.
In the report on 53 patients who began treatment with vedolizumab at Villa Sofia-Cervello Hospital, Palermo, Italy, where Dr. Orlando and his associates work, almost all (96%) had been steroid dependent and 81% had been treated with at least one tumor necrosis factor inhibitor. About two-thirds had completed the induction phase of vedolizumab treatment during follow-up, which lasted a mean of 2.6 months. Of the 14 patients (26%) who had active IBD-associated spondyloarthropathy when starting vedolizumab, 6 (46.2%) saw “a sharp clinical benefit after the initiation of vedolizumab,” wrote Dr. Orlando and his colleagues. Five of these six patients experienced clinical remission of gut symptoms by 12 weeks of therapy.
Dr. Orlando and his colleagues wrote that “our preliminary prospective data indicate a potential benefit of vedolizumab on IBD-associated spondyloarthropathy.”
Looking for mechanistic reasons for this apparent benefit, Dr. Orlando and his collaborators wrote that “the previous demonstration of alpha4beta7 in the joint and the recent evidence of the upregulation of mucosal vascular address in cell adhesion molecule (MadCAM-1) in the high endothelial venules of bone marrow in patients with active axial SpA seem to strengthen the hypothesis of a beneficial rather than a paradoxical effect of alpha4beta7 blockade on articular manifestations of IBD.”
Two authors of the case series reported relationships with multiple pharmaceutical companies, as did Dr. Orlando and two other authors of the letter describing the prospective study.
[email protected]
On Twitter @karioakes
Vedolizumab is gut-selective, and therefore a question that’s been raised is whether it would uncover extra-intestinal manifestations of inflammatory bowel disease (IBD).
When extra-intestinal manifestations of IBD occur with new treatments, we try to discern whether there is still active disease in the bowel. If the bowel is responding, we try to determine if the extra-intestinal symptoms are occurring in parallel to the bowel disease or if they represent a unique side effect of the medication.

In February 2017, at ECCO [the annual meeting of the European Crohn’s and Colitis Organisation], we will be presenting a post hoc analysis of the data from the vedolizumab pivotal clinical trial that examines whether joint pain was independently associated with administration of vedolizumab.
The individual case reports of joint pain with vedolizumab have not limited our using the drug for the patients who need it. It’s a matter of weighing risks and benefits, and the safety profile of this medication is overall so good that we don’t hesitate to use it. In our clinic, we have treated more than 400 IBD patients with vedolizumab, and I can only recall one patient who had to stop using it due to joint pain.
David Rubin, MD, is professor of medicine and chief of the gastroenterology, hepatology, and nutrition section of the University of Chicago. He reported that he is a consultant for and has received grant support from Takeda Pharmaceuticals. These remarks were drawn from an interview.
Vedolizumab is gut-selective, and therefore a question that’s been raised is whether it would uncover extra-intestinal manifestations of inflammatory bowel disease (IBD).
When extra-intestinal manifestations of IBD occur with new treatments, we try to discern whether there is still active disease in the bowel. If the bowel is responding, we try to determine if the extra-intestinal symptoms are occurring in parallel to the bowel disease or if they represent a unique side effect of the medication.
In February 2017, at ECCO [the annual meeting of the European Crohn’s and Colitis Organisation], we will be presenting a post hoc analysis of the data from the vedolizumab pivotal clinical trial that examines whether joint pain was independently associated with administration of vedolizumab.
The individual case reports of joint pain with vedolizumab have not limited our using the drug for the patients who need it. It’s a matter of weighing risks and benefits, and the safety profile of this medication is overall so good that we don’t hesitate to use it. In our clinic, we have treated more than 400 IBD patients with vedolizumab, and I can only recall one patient who had to stop using it due to joint pain.
David Rubin, MD, is professor of medicine and chief of the gastroenterology, hepatology, and nutrition section of the University of Chicago. He reported that he is a consultant for and has received grant support from Takeda Pharmaceuticals. These remarks were drawn from an interview.
Vedolizumab is gut-selective, and therefore a question that’s been raised is whether it would uncover extra-intestinal manifestations of inflammatory bowel disease (IBD).
When extra-intestinal manifestations of IBD occur with new treatments, we try to discern whether there is still active disease in the bowel. If the bowel is responding, we try to determine if the extra-intestinal symptoms are occurring in parallel to the bowel disease or if they represent a unique side effect of the medication.
In February 2017, at ECCO [the annual meeting of the European Crohn’s and Colitis Organisation], we will be presenting a post hoc analysis of the data from the vedolizumab pivotal clinical trial that examines whether joint pain was independently associated with administration of vedolizumab.
The individual case reports of joint pain with vedolizumab have not limited our using the drug for the patients who need it. It’s a matter of weighing risks and benefits, and the safety profile of this medication is overall so good that we don’t hesitate to use it. In our clinic, we have treated more than 400 IBD patients with vedolizumab, and I can only recall one patient who had to stop using it due to joint pain.
David Rubin, MD, is professor of medicine and chief of the gastroenterology, hepatology, and nutrition section of the University of Chicago. He reported that he is a consultant for and has received grant support from Takeda Pharmaceuticals. These remarks were drawn from an interview.
Two recent reports that provide opposing evidence about the potential for inflammatory bowel disease patients to develop articular manifestations after starting vedolizumab raise questions for future studies to answer in regard to a plausible mechanism for the adverse event and its relative importance.
The two reports, one a case series of 5 patients with inflammatory bowel disease (IBD) who developed articular manifestations after beginning vedolizumab (Entyvio) and the other a prospective cohort study of 53 patients with IBD who started vedolizumab without any subsequent cases of induction or flare of arthritis and/or sacroiliitis, came to somewhat different conclusions about the beneficial or paradoxical effects of vedolizumab’s blockade of the alpha4beta7 receptor on articular manifestations of IBD.
The five-patient cases series reported by Gaëlle Varkas, MD, a doctoral student at the University of Ghent, Belgium, and her colleagues consisted of five IBD patients, aged 26-50 years, who developed either new onset or an exacerbation of sacroiliitis or arthritis soon after starting vedolizumab. All but one of the patients was female (Ann Rheum Dis. 2016 Nov 29. doi: 10.1136/annrheumdis-2016-210233). In these patients, the investigators said, vedolizumab did not “seem to show any efficacy in and might even induce arthritis and/or sacroiliitis.”
The first was a 50-year-old woman who had progressive back pain with MRI-confirmed bilateral sacroiliitis about 2 months after beginning vedolizumab. The second patient, a 28-year-old woman with no previous history of spondyloarthropathy, had lower limb pain, a painful left shoulder, and arthritis of one wrist. Ultrasound examination confirmed intercarpal effusions and synovial hyperproliferation.
The third patient was 30 years old and male. He had both ankylosing spondylitis and Crohn’s disease, and experienced arthralgias, elevated C-reactive protein, and MRI-confirmed axial skeletal inflammation 4 weeks after starting vedolizumab.
The fourth patient was a 47-year-old woman with no previous history of spondyloarthropathy who developed MRI-confirmed sacroiliitis after beginning vedolizumab. The fifth patient, a 26-year-old woman, developed polyarticular joint pain after starting vedolizumab. Examination of this patient showed synovitis and enthesitis of multiple joints of the appendicular skeleton.
In discussion, Dr. Varkas and her colleagues noted that “one of the many hypotheses is that integrins and adhesion molecules play a role in the interception of recirculating activated lymphocytes between the gut and the synovial membrane due to the inhibition of the alpha4beta7 integrin homing at the level of the gut.” However, the investigators also acknowledged that other hypotheses may also account for their findings. “Alternatively, in the presence of vedolizumab, cellular recruitment may be mediated by yet to be determined adhesion molecules. This recirculation theory might explain the short mean interval of 64 days between vedolizumab initiation and the expression of symptoms.”
Another group, publishing its prospective cohort study in a letter, had different findings (Ann Rheum Dis. 2017 Jan 17. doi: 10.1136/annrheumdis-2016-211011).
“Although the hypotheses proposed by the authors to explain such events sounds reasonable,” wrote Ambrogio Orlando, MD, and his coauthors, their experience of the effect of vedolizumab on spondyloarthritis differed.
In the report on 53 patients who began treatment with vedolizumab at Villa Sofia-Cervello Hospital, Palermo, Italy, where Dr. Orlando and his associates work, almost all (96%) had been steroid dependent and 81% had been treated with at least one tumor necrosis factor inhibitor. About two-thirds had completed the induction phase of vedolizumab treatment during follow-up, which lasted a mean of 2.6 months. Of the 14 patients (26%) who had active IBD-associated spondyloarthropathy when starting vedolizumab, 6 (46.2%) saw “a sharp clinical benefit after the initiation of vedolizumab,” wrote Dr. Orlando and his colleagues. Five of these six patients experienced clinical remission of gut symptoms by 12 weeks of therapy.
Dr. Orlando and his colleagues wrote that “our preliminary prospective data indicate a potential benefit of vedolizumab on IBD-associated spondyloarthropathy.”
Looking for mechanistic reasons for this apparent benefit, Dr. Orlando and his collaborators wrote that “the previous demonstration of alpha4beta7 in the joint and the recent evidence of the upregulation of mucosal vascular address in cell adhesion molecule (MadCAM-1) in the high endothelial venules of bone marrow in patients with active axial SpA seem to strengthen the hypothesis of a beneficial rather than a paradoxical effect of alpha4beta7 blockade on articular manifestations of IBD.”
Two authors of the case series reported relationships with multiple pharmaceutical companies, as did Dr. Orlando and two other authors of the letter describing the prospective study.
[email protected]
On Twitter @karioakes
Two recent reports that provide opposing evidence about the potential for inflammatory bowel disease patients to develop articular manifestations after starting vedolizumab raise questions for future studies to answer in regard to a plausible mechanism for the adverse event and its relative importance.
The two reports, one a case series of 5 patients with inflammatory bowel disease (IBD) who developed articular manifestations after beginning vedolizumab (Entyvio) and the other a prospective cohort study of 53 patients with IBD who started vedolizumab without any subsequent cases of induction or flare of arthritis and/or sacroiliitis, came to somewhat different conclusions about the beneficial or paradoxical effects of vedolizumab’s blockade of the alpha4beta7 receptor on articular manifestations of IBD.
The five-patient cases series reported by Gaëlle Varkas, MD, a doctoral student at the University of Ghent, Belgium, and her colleagues consisted of five IBD patients, aged 26-50 years, who developed either new onset or an exacerbation of sacroiliitis or arthritis soon after starting vedolizumab. All but one of the patients was female (Ann Rheum Dis. 2016 Nov 29. doi: 10.1136/annrheumdis-2016-210233). In these patients, the investigators said, vedolizumab did not “seem to show any efficacy in and might even induce arthritis and/or sacroiliitis.”
The first was a 50-year-old woman who had progressive back pain with MRI-confirmed bilateral sacroiliitis about 2 months after beginning vedolizumab. The second patient, a 28-year-old woman with no previous history of spondyloarthropathy, had lower limb pain, a painful left shoulder, and arthritis of one wrist. Ultrasound examination confirmed intercarpal effusions and synovial hyperproliferation.
The third patient was 30 years old and male. He had both ankylosing spondylitis and Crohn’s disease, and experienced arthralgias, elevated C-reactive protein, and MRI-confirmed axial skeletal inflammation 4 weeks after starting vedolizumab.
The fourth patient was a 47-year-old woman with no previous history of spondyloarthropathy who developed MRI-confirmed sacroiliitis after beginning vedolizumab. The fifth patient, a 26-year-old woman, developed polyarticular joint pain after starting vedolizumab. Examination of this patient showed synovitis and enthesitis of multiple joints of the appendicular skeleton.
In discussion, Dr. Varkas and her colleagues noted that “one of the many hypotheses is that integrins and adhesion molecules play a role in the interception of recirculating activated lymphocytes between the gut and the synovial membrane due to the inhibition of the alpha4beta7 integrin homing at the level of the gut.” However, the investigators also acknowledged that other hypotheses may also account for their findings. “Alternatively, in the presence of vedolizumab, cellular recruitment may be mediated by yet to be determined adhesion molecules. This recirculation theory might explain the short mean interval of 64 days between vedolizumab initiation and the expression of symptoms.”
Another group, publishing its prospective cohort study in a letter, had different findings (Ann Rheum Dis. 2017 Jan 17. doi: 10.1136/annrheumdis-2016-211011).
“Although the hypotheses proposed by the authors to explain such events sounds reasonable,” wrote Ambrogio Orlando, MD, and his coauthors, their experience of the effect of vedolizumab on spondyloarthritis differed.
In the report on 53 patients who began treatment with vedolizumab at Villa Sofia-Cervello Hospital, Palermo, Italy, where Dr. Orlando and his associates work, almost all (96%) had been steroid dependent and 81% had been treated with at least one tumor necrosis factor inhibitor. About two-thirds had completed the induction phase of vedolizumab treatment during follow-up, which lasted a mean of 2.6 months. Of the 14 patients (26%) who had active IBD-associated spondyloarthropathy when starting vedolizumab, 6 (46.2%) saw “a sharp clinical benefit after the initiation of vedolizumab,” wrote Dr. Orlando and his colleagues. Five of these six patients experienced clinical remission of gut symptoms by 12 weeks of therapy.
Dr. Orlando and his colleagues wrote that “our preliminary prospective data indicate a potential benefit of vedolizumab on IBD-associated spondyloarthropathy.”
Looking for mechanistic reasons for this apparent benefit, Dr. Orlando and his collaborators wrote that “the previous demonstration of alpha4beta7 in the joint and the recent evidence of the upregulation of mucosal vascular address in cell adhesion molecule (MadCAM-1) in the high endothelial venules of bone marrow in patients with active axial SpA seem to strengthen the hypothesis of a beneficial rather than a paradoxical effect of alpha4beta7 blockade on articular manifestations of IBD.”
Two authors of the case series reported relationships with multiple pharmaceutical companies, as did Dr. Orlando and two other authors of the letter describing the prospective study.
[email protected]
On Twitter @karioakes
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: Imaging-confirmed arthritis or sacroiliitis after starting vedolizumab was seen in a case series of 5 patients; a prospective study of 53 patients saw zero new-onset cases of joint pain.
Data source: Case series of 5 IBD patients starting vedolizumab, and prospective surveillance at another facility of 53 IBD patients receiving vedolizumab.
Disclosures: Two authors of the case series reported multiple relationships with pharmaceutical companies, as did three authors of a letter describing a prospective study.
When primary anti-TNF fails in axial spondyloarthritis, consider comorbidities or second anti-TNF
Most of the minority of axial spondyloarthritis patients who do not respond to their first tumor necrosis factor inhibitor still meet classification criteria for the condition 5-10 years later, but more than half respond to a second TNFi and most also have comorbidities that could affect the spondyloarthritis evaluation, according to findings from a single-center cohort study.
The study’s finding of TNFi primary inefficacy in 27 (12%) of 222 patients with axial spondyloarthritis (axSpA) who were given their first TNFi during 2004-2009 is in line with previous results of about 5%-15% primary inefficacy for a first TNFi in axSpA. However, the French investigators, led by Sandra Kossi of Cochin Hospital in Paris, noted that the difficulty of making an axSpA diagnosis and the presence of certain comorbidities “could interfere either with the activity of SpA (falsely heightened disease activity) or with the response to TNFi (falsely heightened inefficacy).” They sought to report the characteristics of axSpA patients with primary inefficacy after their first TNFi in the 5- to 10-year period after their prescription (Rheumatology [Oxford]. 2017 Jan 9. doi: 10.1093/rheumatology/kew456).
A total of 25 of the patients with primary inefficacy underwent re-evaluation 5-10 years (mean of 6 years) later. The investigators defined primary inefficacy as “treatment interruption 3-4 months after treatment onset, with a rheumatologist assessment in the medical file citing lack of efficacy as primary reason for drug interruption.”
The patients with primary TNFi inefficacy had a mean age of 53 years at the time of follow-up, and about half were female. All the patients still had symptoms and back pain, but symptoms were moderate based on a mean Bath Ankylosing Spondylitis Disease Activity Index score of 42. Overall, nine were taking a TNFi and nine were taking nonsteroidal anti-inflammatory drugs, while others used analgesics or nonpharmacologic measures.
Primary TNFi inefficacy occurred significantly more often occurred among females (48% vs. 27%; P = .04), older aged patients at first TNFi use (45 vs. 39 years; P = .04), patients with higher mean Bath Ankylosing Spondylitis Functional Index scores (68 vs. 42; P = .03), and in those who did not have an abnormally elevated C-reactive protein level (33% vs. 63%; P = .02). Patients with primary TNFi inefficacy had lower rates of HLAB27 positivity (56% vs. 72%) or radiographic sacroiliitis (67% vs. 81%), but the differences were not statistically significant.
At follow-up, 21 (84%) patients met Assessment of Spondyloarthritis International Society classification criteria for axSpA, and 20 (80%) met the axSpA diagnosis according to the rheumatologists’ opinion, with 17 (68%) fulfilling both.
A total of 18 (72%) had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression. Five patients – all females – had widespread pain syndrome according to the Fibromyalgia Rapid Screening Tool questionnaire. Another 10 patients had osteoarthritis of lower-limb peripheral joints or of the spine, while an additional 8 had self-declared depression, for which 3 were taking antidepressants.
By the time of follow-up, 16 (64%) had switched to another TNFi, including 9 who had received two TNFi drugs and 7 who had received three or more. The second TNFi was considered efficacious in nine patients. The retention rate of the second TNFi at 1 year among those with primary inefficacy was 50%, and overall, nine patients were still prescribed a TNFi at the time of follow-up.
The fact that most of the patients with primary inefficacy to their first TNFi had confirmed axSpA but also had comorbidities that could affect axSpA evaluation “is important because practitioners might consider that primary inefficacy to TNFi leads to reconsidering the diagnosis of SpA (i.e. the notion of a TNFi prescription being used as the diagnostic test). We suggest here that primary inefficacy should not be considered as equivalent to a diagnostic error, and that a second prescription of TNFi may be of use in such patients, although painful comorbidities should certainly be screened for and taken into account.”
The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
Most of the minority of axial spondyloarthritis patients who do not respond to their first tumor necrosis factor inhibitor still meet classification criteria for the condition 5-10 years later, but more than half respond to a second TNFi and most also have comorbidities that could affect the spondyloarthritis evaluation, according to findings from a single-center cohort study.
The study’s finding of TNFi primary inefficacy in 27 (12%) of 222 patients with axial spondyloarthritis (axSpA) who were given their first TNFi during 2004-2009 is in line with previous results of about 5%-15% primary inefficacy for a first TNFi in axSpA. However, the French investigators, led by Sandra Kossi of Cochin Hospital in Paris, noted that the difficulty of making an axSpA diagnosis and the presence of certain comorbidities “could interfere either with the activity of SpA (falsely heightened disease activity) or with the response to TNFi (falsely heightened inefficacy).” They sought to report the characteristics of axSpA patients with primary inefficacy after their first TNFi in the 5- to 10-year period after their prescription (Rheumatology [Oxford]. 2017 Jan 9. doi: 10.1093/rheumatology/kew456).
A total of 25 of the patients with primary inefficacy underwent re-evaluation 5-10 years (mean of 6 years) later. The investigators defined primary inefficacy as “treatment interruption 3-4 months after treatment onset, with a rheumatologist assessment in the medical file citing lack of efficacy as primary reason for drug interruption.”
The patients with primary TNFi inefficacy had a mean age of 53 years at the time of follow-up, and about half were female. All the patients still had symptoms and back pain, but symptoms were moderate based on a mean Bath Ankylosing Spondylitis Disease Activity Index score of 42. Overall, nine were taking a TNFi and nine were taking nonsteroidal anti-inflammatory drugs, while others used analgesics or nonpharmacologic measures.
Primary TNFi inefficacy occurred significantly more often occurred among females (48% vs. 27%; P = .04), older aged patients at first TNFi use (45 vs. 39 years; P = .04), patients with higher mean Bath Ankylosing Spondylitis Functional Index scores (68 vs. 42; P = .03), and in those who did not have an abnormally elevated C-reactive protein level (33% vs. 63%; P = .02). Patients with primary TNFi inefficacy had lower rates of HLAB27 positivity (56% vs. 72%) or radiographic sacroiliitis (67% vs. 81%), but the differences were not statistically significant.
At follow-up, 21 (84%) patients met Assessment of Spondyloarthritis International Society classification criteria for axSpA, and 20 (80%) met the axSpA diagnosis according to the rheumatologists’ opinion, with 17 (68%) fulfilling both.
A total of 18 (72%) had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression. Five patients – all females – had widespread pain syndrome according to the Fibromyalgia Rapid Screening Tool questionnaire. Another 10 patients had osteoarthritis of lower-limb peripheral joints or of the spine, while an additional 8 had self-declared depression, for which 3 were taking antidepressants.
By the time of follow-up, 16 (64%) had switched to another TNFi, including 9 who had received two TNFi drugs and 7 who had received three or more. The second TNFi was considered efficacious in nine patients. The retention rate of the second TNFi at 1 year among those with primary inefficacy was 50%, and overall, nine patients were still prescribed a TNFi at the time of follow-up.
The fact that most of the patients with primary inefficacy to their first TNFi had confirmed axSpA but also had comorbidities that could affect axSpA evaluation “is important because practitioners might consider that primary inefficacy to TNFi leads to reconsidering the diagnosis of SpA (i.e. the notion of a TNFi prescription being used as the diagnostic test). We suggest here that primary inefficacy should not be considered as equivalent to a diagnostic error, and that a second prescription of TNFi may be of use in such patients, although painful comorbidities should certainly be screened for and taken into account.”
The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
Most of the minority of axial spondyloarthritis patients who do not respond to their first tumor necrosis factor inhibitor still meet classification criteria for the condition 5-10 years later, but more than half respond to a second TNFi and most also have comorbidities that could affect the spondyloarthritis evaluation, according to findings from a single-center cohort study.
The study’s finding of TNFi primary inefficacy in 27 (12%) of 222 patients with axial spondyloarthritis (axSpA) who were given their first TNFi during 2004-2009 is in line with previous results of about 5%-15% primary inefficacy for a first TNFi in axSpA. However, the French investigators, led by Sandra Kossi of Cochin Hospital in Paris, noted that the difficulty of making an axSpA diagnosis and the presence of certain comorbidities “could interfere either with the activity of SpA (falsely heightened disease activity) or with the response to TNFi (falsely heightened inefficacy).” They sought to report the characteristics of axSpA patients with primary inefficacy after their first TNFi in the 5- to 10-year period after their prescription (Rheumatology [Oxford]. 2017 Jan 9. doi: 10.1093/rheumatology/kew456).
A total of 25 of the patients with primary inefficacy underwent re-evaluation 5-10 years (mean of 6 years) later. The investigators defined primary inefficacy as “treatment interruption 3-4 months after treatment onset, with a rheumatologist assessment in the medical file citing lack of efficacy as primary reason for drug interruption.”
The patients with primary TNFi inefficacy had a mean age of 53 years at the time of follow-up, and about half were female. All the patients still had symptoms and back pain, but symptoms were moderate based on a mean Bath Ankylosing Spondylitis Disease Activity Index score of 42. Overall, nine were taking a TNFi and nine were taking nonsteroidal anti-inflammatory drugs, while others used analgesics or nonpharmacologic measures.
Primary TNFi inefficacy occurred significantly more often occurred among females (48% vs. 27%; P = .04), older aged patients at first TNFi use (45 vs. 39 years; P = .04), patients with higher mean Bath Ankylosing Spondylitis Functional Index scores (68 vs. 42; P = .03), and in those who did not have an abnormally elevated C-reactive protein level (33% vs. 63%; P = .02). Patients with primary TNFi inefficacy had lower rates of HLAB27 positivity (56% vs. 72%) or radiographic sacroiliitis (67% vs. 81%), but the differences were not statistically significant.
At follow-up, 21 (84%) patients met Assessment of Spondyloarthritis International Society classification criteria for axSpA, and 20 (80%) met the axSpA diagnosis according to the rheumatologists’ opinion, with 17 (68%) fulfilling both.
A total of 18 (72%) had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression. Five patients – all females – had widespread pain syndrome according to the Fibromyalgia Rapid Screening Tool questionnaire. Another 10 patients had osteoarthritis of lower-limb peripheral joints or of the spine, while an additional 8 had self-declared depression, for which 3 were taking antidepressants.
By the time of follow-up, 16 (64%) had switched to another TNFi, including 9 who had received two TNFi drugs and 7 who had received three or more. The second TNFi was considered efficacious in nine patients. The retention rate of the second TNFi at 1 year among those with primary inefficacy was 50%, and overall, nine patients were still prescribed a TNFi at the time of follow-up.
The fact that most of the patients with primary inefficacy to their first TNFi had confirmed axSpA but also had comorbidities that could affect axSpA evaluation “is important because practitioners might consider that primary inefficacy to TNFi leads to reconsidering the diagnosis of SpA (i.e. the notion of a TNFi prescription being used as the diagnostic test). We suggest here that primary inefficacy should not be considered as equivalent to a diagnostic error, and that a second prescription of TNFi may be of use in such patients, although painful comorbidities should certainly be screened for and taken into account.”
The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
Key clinical point:
Major finding: A total of 18 (72%) of 25 patients with primary TNFi inefficacy had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression.
Data source: A retrospective and prospective cohort study of 222 patients who received a first TNFi after being diagnosed with axial spondyloarthritis during 2004-2009.
Disclosures: The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.