User login
Antidepressants highly effective against binge-eating disorder
People with binge-eating disorder have the greatest chance of achieving normal eating habits and alleviating symptoms associated with the disorder by taking second-generation antidepressants, topiramate, and lisdexamfetamine and engaging in cognitive-behavioral therapy, an analysis of several studies showed.
The findings should be used to “address other treatments, combinations of treatments, and comparisons between treatments; treatment for postbariatric surgery patients and children; and the course of these illnesses,” according to the report, released as part of the Comparative Effectiveness Review No. 160 by the Agency for Healthcare Research and Quality.
The authors of the report examined a total of 52 randomized controlled trials and 15 observational studies collected through searches of MEDLINE, EMBASE, the Cochrane Library, Academic OneFile, and the Cumulative Index to Nursing and Allied Health Literature databases, with 48 of the included studies specifically concerning binge-eating disorder (BED). English-language studies up through Jan. 19, 2015, were included for analysis, and the investigators specifically looked for studies of individuals who met DSM-IV or DSM-5 criteria for BED and studies of postbariatric surgery patients, including children, experiencing loss-of-control (LOC) eating habits.
Each study was evaluated based on a set of 15 “key questions” to determine the effectiveness and harms of the treatments involved. The key questions used by the investigators sought to determine the evidence of effectiveness and harms of BED treatments; LOC eating among bariatric surgery patients; and the effectiveness of any LOC treatments based on age, sex, race, ethnicity, initial body mass index, duration of illness, and coexisting conditions. In addition, similar questions were used to ascertain the effectiveness of treatments on pediatric patients.
“Broadly, we included pharmacological, psychological, behavioral, and combination interventions,” the report stated. “We considered physical and psychological health outcomes in four major categories: binge behavior (binge eating or LOC eating); binge-eating–related psychopathology (e.g., weight and shape concerns, dietary restraint); physical health functioning (i.e., weight and other indexes of metabolic health, e.g., diabetes); and general psychopathology (e.g., depression, anxiety).”
Antidepressants were found to be more effective than placebos across the studies included in the survey, specifically second-generation antidepressants, and were 1.67 times more likely to help BED patients achieve abstinence than placebos used in these trials; 41% of subjects receiving antidepressants ultimately achieved abstinence, compared with 23% on placebos.
With topiramate, binge eating generally decreased to as little as one episode per week, and a higher portion of subjects (58%) achieved abstinence than those on placebo (28%). In addition, topiramate was found to decrease “obsessive thoughts and compulsions related to binge eating” by nearly 30%, versus 23% in subjects taking placebos.
Studies involving lisdexamfetamine showed abstinence achieved in 40% of subjects, far higher than the 15% on placebos, and a likelihood of achieving abstinence 2.61 times higher than for those in the placebo cohorts. Binge-eating episodes per week also decreased, and were, on average, anywhere from 1.7 to 1.3 fewer than those in subjects taking placebo. Subjects receiving cognitive-behavioral therapy – whether led by a therapist or self-led, though the former was found to have stronger evidence of effectiveness than the latter – had an average of 2.3 fewer binge-eating episodes per week, and subjects involved with therapy were 4.95 times more likely to achieve abstinence than those who were not receiving therapy.
“Findings about BED treatment interventions are likely to be applicable to all adults age 18 and older with the disorder, but chiefly to overweight or obese women,” the report stated. “We cannot comment on the applicability of treatment findings for specific subgroups of adults (even among women) or whether findings extend to BED patients diagnosed based on DSM-5 criteria.”
The authors also noted that the findings are unclear with respect to adolescents with BED or members of ethnic groups, and children with loss-of-control eating or who have undergone bariatric surgery.
“A convention for reporting and analyzing” outcomes is necessary for the findings of this study to take on real-world applications that can be beneficial to clinicians and their patients in the near future, the authors concluded. However, more multisite randomized, controlled trials are needed.
People with binge-eating disorder have the greatest chance of achieving normal eating habits and alleviating symptoms associated with the disorder by taking second-generation antidepressants, topiramate, and lisdexamfetamine and engaging in cognitive-behavioral therapy, an analysis of several studies showed.
The findings should be used to “address other treatments, combinations of treatments, and comparisons between treatments; treatment for postbariatric surgery patients and children; and the course of these illnesses,” according to the report, released as part of the Comparative Effectiveness Review No. 160 by the Agency for Healthcare Research and Quality.
The authors of the report examined a total of 52 randomized controlled trials and 15 observational studies collected through searches of MEDLINE, EMBASE, the Cochrane Library, Academic OneFile, and the Cumulative Index to Nursing and Allied Health Literature databases, with 48 of the included studies specifically concerning binge-eating disorder (BED). English-language studies up through Jan. 19, 2015, were included for analysis, and the investigators specifically looked for studies of individuals who met DSM-IV or DSM-5 criteria for BED and studies of postbariatric surgery patients, including children, experiencing loss-of-control (LOC) eating habits.
Each study was evaluated based on a set of 15 “key questions” to determine the effectiveness and harms of the treatments involved. The key questions used by the investigators sought to determine the evidence of effectiveness and harms of BED treatments; LOC eating among bariatric surgery patients; and the effectiveness of any LOC treatments based on age, sex, race, ethnicity, initial body mass index, duration of illness, and coexisting conditions. In addition, similar questions were used to ascertain the effectiveness of treatments on pediatric patients.
“Broadly, we included pharmacological, psychological, behavioral, and combination interventions,” the report stated. “We considered physical and psychological health outcomes in four major categories: binge behavior (binge eating or LOC eating); binge-eating–related psychopathology (e.g., weight and shape concerns, dietary restraint); physical health functioning (i.e., weight and other indexes of metabolic health, e.g., diabetes); and general psychopathology (e.g., depression, anxiety).”
Antidepressants were found to be more effective than placebos across the studies included in the survey, specifically second-generation antidepressants, and were 1.67 times more likely to help BED patients achieve abstinence than placebos used in these trials; 41% of subjects receiving antidepressants ultimately achieved abstinence, compared with 23% on placebos.
With topiramate, binge eating generally decreased to as little as one episode per week, and a higher portion of subjects (58%) achieved abstinence than those on placebo (28%). In addition, topiramate was found to decrease “obsessive thoughts and compulsions related to binge eating” by nearly 30%, versus 23% in subjects taking placebos.
Studies involving lisdexamfetamine showed abstinence achieved in 40% of subjects, far higher than the 15% on placebos, and a likelihood of achieving abstinence 2.61 times higher than for those in the placebo cohorts. Binge-eating episodes per week also decreased, and were, on average, anywhere from 1.7 to 1.3 fewer than those in subjects taking placebo. Subjects receiving cognitive-behavioral therapy – whether led by a therapist or self-led, though the former was found to have stronger evidence of effectiveness than the latter – had an average of 2.3 fewer binge-eating episodes per week, and subjects involved with therapy were 4.95 times more likely to achieve abstinence than those who were not receiving therapy.
“Findings about BED treatment interventions are likely to be applicable to all adults age 18 and older with the disorder, but chiefly to overweight or obese women,” the report stated. “We cannot comment on the applicability of treatment findings for specific subgroups of adults (even among women) or whether findings extend to BED patients diagnosed based on DSM-5 criteria.”
The authors also noted that the findings are unclear with respect to adolescents with BED or members of ethnic groups, and children with loss-of-control eating or who have undergone bariatric surgery.
“A convention for reporting and analyzing” outcomes is necessary for the findings of this study to take on real-world applications that can be beneficial to clinicians and their patients in the near future, the authors concluded. However, more multisite randomized, controlled trials are needed.
People with binge-eating disorder have the greatest chance of achieving normal eating habits and alleviating symptoms associated with the disorder by taking second-generation antidepressants, topiramate, and lisdexamfetamine and engaging in cognitive-behavioral therapy, an analysis of several studies showed.
The findings should be used to “address other treatments, combinations of treatments, and comparisons between treatments; treatment for postbariatric surgery patients and children; and the course of these illnesses,” according to the report, released as part of the Comparative Effectiveness Review No. 160 by the Agency for Healthcare Research and Quality.
The authors of the report examined a total of 52 randomized controlled trials and 15 observational studies collected through searches of MEDLINE, EMBASE, the Cochrane Library, Academic OneFile, and the Cumulative Index to Nursing and Allied Health Literature databases, with 48 of the included studies specifically concerning binge-eating disorder (BED). English-language studies up through Jan. 19, 2015, were included for analysis, and the investigators specifically looked for studies of individuals who met DSM-IV or DSM-5 criteria for BED and studies of postbariatric surgery patients, including children, experiencing loss-of-control (LOC) eating habits.
Each study was evaluated based on a set of 15 “key questions” to determine the effectiveness and harms of the treatments involved. The key questions used by the investigators sought to determine the evidence of effectiveness and harms of BED treatments; LOC eating among bariatric surgery patients; and the effectiveness of any LOC treatments based on age, sex, race, ethnicity, initial body mass index, duration of illness, and coexisting conditions. In addition, similar questions were used to ascertain the effectiveness of treatments on pediatric patients.
“Broadly, we included pharmacological, psychological, behavioral, and combination interventions,” the report stated. “We considered physical and psychological health outcomes in four major categories: binge behavior (binge eating or LOC eating); binge-eating–related psychopathology (e.g., weight and shape concerns, dietary restraint); physical health functioning (i.e., weight and other indexes of metabolic health, e.g., diabetes); and general psychopathology (e.g., depression, anxiety).”
Antidepressants were found to be more effective than placebos across the studies included in the survey, specifically second-generation antidepressants, and were 1.67 times more likely to help BED patients achieve abstinence than placebos used in these trials; 41% of subjects receiving antidepressants ultimately achieved abstinence, compared with 23% on placebos.
With topiramate, binge eating generally decreased to as little as one episode per week, and a higher portion of subjects (58%) achieved abstinence than those on placebo (28%). In addition, topiramate was found to decrease “obsessive thoughts and compulsions related to binge eating” by nearly 30%, versus 23% in subjects taking placebos.
Studies involving lisdexamfetamine showed abstinence achieved in 40% of subjects, far higher than the 15% on placebos, and a likelihood of achieving abstinence 2.61 times higher than for those in the placebo cohorts. Binge-eating episodes per week also decreased, and were, on average, anywhere from 1.7 to 1.3 fewer than those in subjects taking placebo. Subjects receiving cognitive-behavioral therapy – whether led by a therapist or self-led, though the former was found to have stronger evidence of effectiveness than the latter – had an average of 2.3 fewer binge-eating episodes per week, and subjects involved with therapy were 4.95 times more likely to achieve abstinence than those who were not receiving therapy.
“Findings about BED treatment interventions are likely to be applicable to all adults age 18 and older with the disorder, but chiefly to overweight or obese women,” the report stated. “We cannot comment on the applicability of treatment findings for specific subgroups of adults (even among women) or whether findings extend to BED patients diagnosed based on DSM-5 criteria.”
The authors also noted that the findings are unclear with respect to adolescents with BED or members of ethnic groups, and children with loss-of-control eating or who have undergone bariatric surgery.
“A convention for reporting and analyzing” outcomes is necessary for the findings of this study to take on real-world applications that can be beneficial to clinicians and their patients in the near future, the authors concluded. However, more multisite randomized, controlled trials are needed.
Malignant catatonia and aphasia follow multiple-drug overdose
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
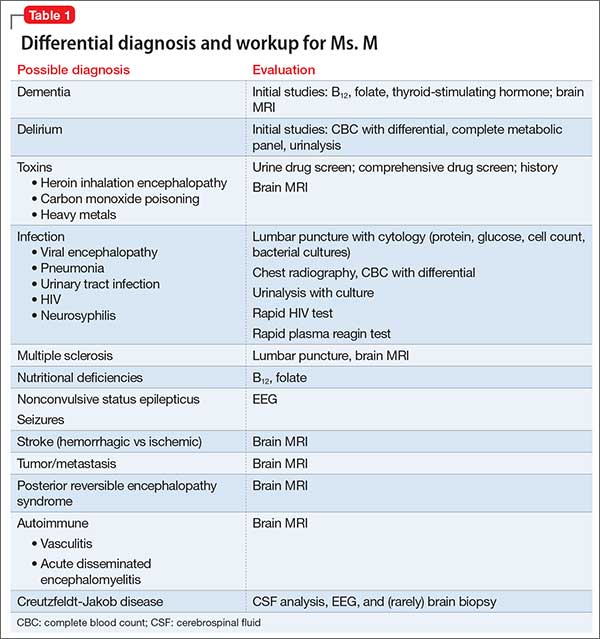
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.

Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
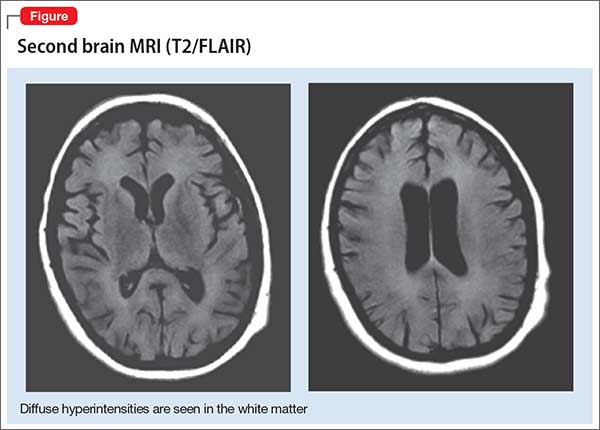
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
Antidepressants for functional dyspepsia
Functional, a.k.a. “nonulcer,” dyspepsia is a challenging diagnosis and likely afflicts many more patients than we have identified in our practices. Functional dyspepsia (FD) is defined by the presence of postprandial fullness, early satiety, epigastric pain or burning, and no evidence of structural disease. These are the patients who do not get better with proton pump inhibitors or feel better after a bowel movement.
After a negative upper endoscopy and Helicobacter pylori stool antigen test, the task turns to symptom control. But what’s the best treatment?
Dr. Nicholas J. Talley of the University of Newcastle in Callaghan, Australia, and colleagues conducted a multicenter, randomized trial evaluating the comparative efficacy of amitriptyline or escitalopram for symptom control, gastric emptying, and meal-induced satiety in patients with FD (Gastroenterology. 2015;149(2):340-9.e2).
Participants were enrolled if they met Rome II criteria for FD requiring that folks in the preceding 12 months have at least 12 weeks of dyspepsia, absence of organic disease, and no relationship to defecation. Patients were randomized to placebo, amitriptyline 50 mg (titrated), or escitalopram 10 mg. Medication was given for 10 weeks. The primary endpoint was adequate relief of symptoms for at least 5 weeks.
A total of 292 patients (most of whom [75%] were female) with an average age of 44 years were randomized. Seventy percent had dysmotility-like FD and 30% had ulcer-like FD.
Patients with ulcer-like FD receiving amitriptyline were more likely to report adequate relief (odds ratio, 3.1; 95% confidence interval, 1.1-9.0). Neither medication affected gastric emptying or meal-induced satiety. Both medications improved overall quality of life.
The data support the use of amitriptyline for ulcer-like FD. Some of these patients may have comorbid psychiatric illness that may be improved with escitalopram. Perhaps this is what is impacting the quality-of-life metric that taps into dimensions above and beyond relief of symptoms (such as sleep disturbance or work/study).
Proton pump inhibitors tend to be overused, and many of our patients take them indefinitely without trying to see how they do off of them. Some patients for whom we have not considered a diagnosis of FD may be on PPIs because we have had nothing else to offer them. Maybe they felt better because of a PPI placebo effect and we have continued them.
If we can, we should review the diagnosis of dyspepsia, consider FD as a possibility etiology for gastrointestinal distress, stop the PPIs, and try amitriptyline.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.
Functional, a.k.a. “nonulcer,” dyspepsia is a challenging diagnosis and likely afflicts many more patients than we have identified in our practices. Functional dyspepsia (FD) is defined by the presence of postprandial fullness, early satiety, epigastric pain or burning, and no evidence of structural disease. These are the patients who do not get better with proton pump inhibitors or feel better after a bowel movement.
After a negative upper endoscopy and Helicobacter pylori stool antigen test, the task turns to symptom control. But what’s the best treatment?
Dr. Nicholas J. Talley of the University of Newcastle in Callaghan, Australia, and colleagues conducted a multicenter, randomized trial evaluating the comparative efficacy of amitriptyline or escitalopram for symptom control, gastric emptying, and meal-induced satiety in patients with FD (Gastroenterology. 2015;149(2):340-9.e2).
Participants were enrolled if they met Rome II criteria for FD requiring that folks in the preceding 12 months have at least 12 weeks of dyspepsia, absence of organic disease, and no relationship to defecation. Patients were randomized to placebo, amitriptyline 50 mg (titrated), or escitalopram 10 mg. Medication was given for 10 weeks. The primary endpoint was adequate relief of symptoms for at least 5 weeks.
A total of 292 patients (most of whom [75%] were female) with an average age of 44 years were randomized. Seventy percent had dysmotility-like FD and 30% had ulcer-like FD.
Patients with ulcer-like FD receiving amitriptyline were more likely to report adequate relief (odds ratio, 3.1; 95% confidence interval, 1.1-9.0). Neither medication affected gastric emptying or meal-induced satiety. Both medications improved overall quality of life.
The data support the use of amitriptyline for ulcer-like FD. Some of these patients may have comorbid psychiatric illness that may be improved with escitalopram. Perhaps this is what is impacting the quality-of-life metric that taps into dimensions above and beyond relief of symptoms (such as sleep disturbance or work/study).
Proton pump inhibitors tend to be overused, and many of our patients take them indefinitely without trying to see how they do off of them. Some patients for whom we have not considered a diagnosis of FD may be on PPIs because we have had nothing else to offer them. Maybe they felt better because of a PPI placebo effect and we have continued them.
If we can, we should review the diagnosis of dyspepsia, consider FD as a possibility etiology for gastrointestinal distress, stop the PPIs, and try amitriptyline.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.
Functional, a.k.a. “nonulcer,” dyspepsia is a challenging diagnosis and likely afflicts many more patients than we have identified in our practices. Functional dyspepsia (FD) is defined by the presence of postprandial fullness, early satiety, epigastric pain or burning, and no evidence of structural disease. These are the patients who do not get better with proton pump inhibitors or feel better after a bowel movement.
After a negative upper endoscopy and Helicobacter pylori stool antigen test, the task turns to symptom control. But what’s the best treatment?
Dr. Nicholas J. Talley of the University of Newcastle in Callaghan, Australia, and colleagues conducted a multicenter, randomized trial evaluating the comparative efficacy of amitriptyline or escitalopram for symptom control, gastric emptying, and meal-induced satiety in patients with FD (Gastroenterology. 2015;149(2):340-9.e2).
Participants were enrolled if they met Rome II criteria for FD requiring that folks in the preceding 12 months have at least 12 weeks of dyspepsia, absence of organic disease, and no relationship to defecation. Patients were randomized to placebo, amitriptyline 50 mg (titrated), or escitalopram 10 mg. Medication was given for 10 weeks. The primary endpoint was adequate relief of symptoms for at least 5 weeks.
A total of 292 patients (most of whom [75%] were female) with an average age of 44 years were randomized. Seventy percent had dysmotility-like FD and 30% had ulcer-like FD.
Patients with ulcer-like FD receiving amitriptyline were more likely to report adequate relief (odds ratio, 3.1; 95% confidence interval, 1.1-9.0). Neither medication affected gastric emptying or meal-induced satiety. Both medications improved overall quality of life.
The data support the use of amitriptyline for ulcer-like FD. Some of these patients may have comorbid psychiatric illness that may be improved with escitalopram. Perhaps this is what is impacting the quality-of-life metric that taps into dimensions above and beyond relief of symptoms (such as sleep disturbance or work/study).
Proton pump inhibitors tend to be overused, and many of our patients take them indefinitely without trying to see how they do off of them. Some patients for whom we have not considered a diagnosis of FD may be on PPIs because we have had nothing else to offer them. Maybe they felt better because of a PPI placebo effect and we have continued them.
If we can, we should review the diagnosis of dyspepsia, consider FD as a possibility etiology for gastrointestinal distress, stop the PPIs, and try amitriptyline.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.

Coronary artery disease and mental illness

Your significant role in modifying risk factors for coronary artery disease and managing problems subsequently
The problem is enormous: Heart disease is the leading cause of death in the United States, and coronary artery disease (CAD) is the most common form of heart disease—responsible for 385,000 deaths in the United States in 2009 (http://www.cdc.gov/heartdisease/facts. htm). Patients with psychiatric illness have higher rates of morbidity and mortality from CAD than the general population, and warrant consideration as a special population. You should be familiar with routine cardiac medications; your patients’ medical problems; potential cardiac-related interactions among their psychotropic medications; and interactions among illnesses in their mental health and medical health domains (Box).
CASE Type 2 diabetes mellitus plus a long history of heavy smoking
Ms. S, age 57, is an African American woman with chronic paranoid schizophrenia who has been seeing a psychiatrist for the past 10 years. Ms. S’s psychiatric symptoms have been well controlled on risperidone, 3 mg/d.
Ms. S has a family history of diabetes, hypertension, and early CAD (a brother died of a myocardial infarction [MI] in his late 40s). She continues to smoke 2 packs of unfiltered cigarettes daily, as she has done for the past 40 years.
The psychiatrist has been following American Diabetes Association/American Psychiatric Association guidelines for monitoring; he has noticed that Ms. S’s body mass index (BMI) has increased from 27 to 31 kg/m2 over the past year. She has developed type 2 diabetes mellitus (T2DM).
At today’s visit, Ms. S arrives a few minutes late and appears flustered and out of breath. She explains that she had to climb a flight of stairs to get to office because the elevator is broken.
During the visit, the psychiatrist notes that Ms. S occasionally winces and massages her left shoulder.
Questions to ponder
• What else could the psychiatrist do to modify Ms. S’s cardiac risk factors?
• What is Ms. S’s 10-year risk of an acute coronary event?
• What should her physician do now?
Overview: Cardiac risk in patients with mental illness
Modifiable risks for CAD include hypertension, hypercholesterolemia, T2DM, obesity (all of which, taken together, constitute the metabolic syndrome), smoking, and a sedentary lifestyle. Some risk factors, including sex, age, and family history, are not modifiable. Whether or not this modification leads to better outcomes, psychiatric comorbidity is associated with higher morbidity and mortality from CAD.
Whether a common underlying pathological process manifesting in both CAD and mental illness exists, or whether the association is causal, are not well understood. Symptoms characteristic of depression (apathy, amotivation) and schizophrenia (disorganization, paranoia) could lead to poor self-care or impaired adherence to programs designed to lower CAD risk factors.1,2
People with mental illness smoke at a higher rate than those who do not have mental illness.3 This finding is of particular relevance because smoking contributes to worse outcomes with respect to CAD, even when medications are prescribed to address metabolic risks.4
Lower socioeconomic status is associated with poorer prognosis from CAD5 and is a risk factor for depression.6 Depression is a strong independent predictor of worse survival in acute coronary syndromes.5 Some experts consider depression to be a stronger risk factor for MI than traditional medical risk factors such as obesity, hypertension, and second-hand smoke.7
Interventions used to treat certain mental illnesses can exacerbate, or predispose to, metabolic syndrome (which, in turn, increases the risk of CAD). Although some studies have demonstrated metabolic derangements in medication-naïve patients who have a new diagnosis of schizophrenia,8 there is a clearly established association between second-generation antipsychotic use and obesity, hypertension, hyperlipidemia, and T2DM. This association prompted development in 2004 of consensus recommendations for cardiovascular monitoring of patients who are taking an atypical antipsychotic.9
Some studies suggest that the stress of mental illness contributes to the pathogen esis of CAD.8 Hypothesized mechanisms include:
• sympathetic activation
• vagal deactivation
• platelet activation
• hypothalamic-pituitary-adrenocortical pathways
• anticholinergic mechanisms
• inflammatory mediators, including cytokines.
Mental stress itself has the capacity to induce coronary ischemia.10 The mental stress of psychiatric illness could have an important pathophysiologic role in CAD. It can be tempting to disregard chest pain in a patient who is known to have panic disorder, but that patient might in fact be experiencing stress-induced myocardial ischemia.11
As many as 30% to 40% of patients with CAD suffer from clinically significant symptoms of depression; as many as 20% of patients with CAD meet criteria for major depressive disorder, compared with 5% to 10% of people who do not have CAD.2 Depression post-MI has been associated with a higher rate of sudden cardiac death and worse outcomes.12
Anxiety also can portend worse outcomes from CAD,13 including higher all-cause mortality.14 There is some hope, but limited evidence, that treating depression and anxiety, whether with antidepressant medication or behavioral therapy, can improve CAD outcomes.10,15
Making a diagnosis of CAD
CAD can present in a variety of ways, ranging from unrecognized or so-called silent CAD (there is an association between T2DM and unrecognized CAD and between hypertension and unrecognized CAD) to stable angina, unstable angina, acute coronary syndrome, MI, and sudden cardiac death. A variety of abnormalities on resting and exercise electrocardiogram (ECG), including ST segment depression, ST elevation, Q waves, and other morphological changes are indicative of CAD.
Other modalities, including coronary calcification score on computed tomography and coronary angiography can confirm the presence of CAD. Some clinicians recommend periodic ECG treadmill testing in patients who have:
• a total cholesterol level is >240 mg/dL
• systolic blood pressure >140 mm Hg, diastolic blood pressure >90 mm Hg, or both
• a family history of MI or sudden cardiac death in young (age <60) first-degree relatives
• a history of smoking
• diabetes.
Preventive guidelines
Risk stratification. A low (<10%), moderate (10% to 20%), or high (>20%) 10-year risk of CAD can be ascertained using a risk calculator, such as one that is available through the Framingham Heart Study (Figure) and the National Heart, Lung, and Blood Institute (http://cvdrisk.nhlbi. nih.gov). Because patients with risk factors for CAD should be offered interventions— including smoking cessation therapy, diet and exercise, aspirin, lipid-lowering therapy, and blood pressure modification strategies—whether or not they have evidence of CAD, the United States Preventive Services Task Force does not recommend for or against diagnostic screening in patients at moderate or elevated risk of CAD.16
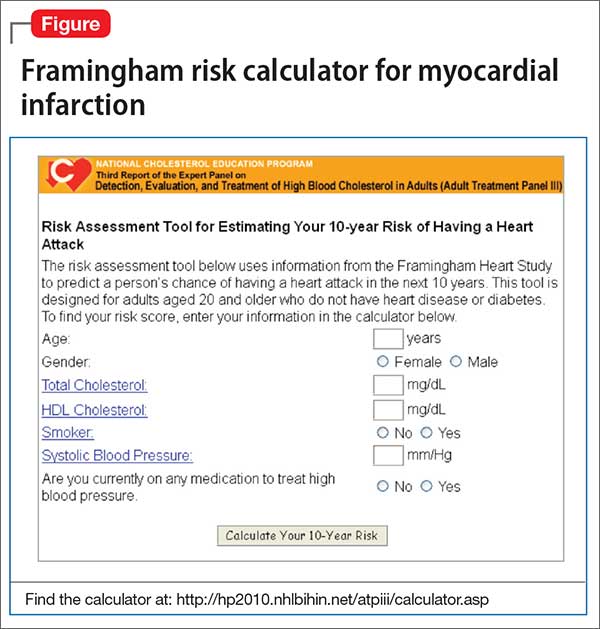
There are guidelines in the literature recommending specific screening strategies for patients with mental illness, although the vetting and update process has been ill defined. Among patients with schizophrenia, though, regardless of antipsychotic prescription status, baseline and then regular monitoring of metabolic risk parameters is recommended.17
Primary prevention. Lifestyle modification and attention to modifiable coronary risk factors are important primary prevention strategies. Dietary modifications, exercise, not smoking, and maintenance of a normal BMI (<25 kg/m2) are associated with a lower risk of CAD.18,19
Lifestyle modifications can be challenging for patients with persistent mental illness, however: For example, patients with schizophrenia smoke more, eat less healthfully, and participate less in behavioral modification that targets risk factors than patients who do not have schizophrenia.20,21
According to 2012 evidence-based practice guidelines established by a collaboration that included the American College of Physicians and several cardiology and thoracic medicine societies, persons age >50 who do not have symptomatic CAD should take low-dose (75 to 100 mg/d) aspirin; the benefit of low-dose aspirin in persons at moderate or high risk of CAD is even greater. Other medications, including statins and fixed-dose combinations of antihypertensive medications in combination with a statin are not clearly beneficial as primary prevention strategies across the board, although selected high-risk populations might benefit.
Regrettably, the high-risk population of persons with mental illness and whose primary care is suboptimal has not been studied. It stands to reason that these patients would especially benefit from more attentive monitoring and intervention.
Collaborative care? Although many psychiatrists do not practice in such a model, a comprehensive approach to the care of their patients, using a collaborative care strategy that includes attention to the mental health diagnosis along with medical health, can result in improved health in both domains.22 However, enlisting patients with paranoia or an inherent distrust of medications and health care providers to adhere to either a medication regimen or lifestyle modification can be challenging.
Common-sense strategies, such as creating a multidisciplinary team with the psychiatrist coordinating care and optimizing antipsychotic treatment, might provide benefit.1 Data demonstrate that patients with severe mental illness who experience acute coronary events undergo revascularization at a lower rate than their mentally heathy counterparts, despite the fact that patients with severe mental illness die at a higher rate from their CAD than patients who do not have mental illness. An important role for the psychiatrist, even in the absence of a collaborative care program, is to be an advocate for appropriate guideline-based care.23
Secondary prevention. Once a patient develops CAD, ongoing risk factor modification is important. Adherence to a therapeutic regimen that variously combines a platelet inhibitor, beta blocker, statin, and angiotensin-converting enzyme (ACE) inhibitor is associated with improved outcomes in patients with CAD.24 Specific antiplatelet recommendations and a recommendation for single vs combination antiplatelet therapy depends on chronicity and type of revascularization in a setting of CAD.25
Summary of guideline-based recommendations
Treatment guidelines published in the National Guidelines Clearinghouse address depression, CAD screening, and specific cardiac therapies, including ACE inhibitors, angiotensin-receptor blockers, oral anticoagulants, platelet inhibitors, beta blockers, and lifestyle modification.
Primary prevention. Recommendations for treatment to prevent CAD are listed in Table 1.

Secondary prevention. Recommendations for treatment after a diagnosis of CAD are listed in Table 2.

Special considerations for psychiatric providers
You should be comfortable with patients’ use of antihypertensive therapies and familiar with the potential these agents have to interact with psychotropics; in addition, you can take a more active role in prescribing, and monitoring patients’ responses to, these medications. Provide appropriate monitoring of ACE inhibitors, statins, and beta blockers; also, provide appropriate monitoring of psychotropics in patients who take recommended cardioprotective medications.
In situations that prompt referral (such as recent MI, new symptoms of heart failure, any history of syncope or new identification of T2DM), ideally you should collaborate with the patient’s primary care provider to help enhance adherence to recommended treatment strategies. You also should employ motivational interviewing techniques and offer strategies by which patients can engage in meaningful lifestyle modification.
There are official recommendations for depression screening strategies26 and psychosocial risk screening for patients in whom CAD has been identified.27 Official screening strategies for CAD in patients with psychiatric illness have not, however, been spelled out.
Primary CAD prevention with medication is not routinely recommended for the general population, but the increased risk of CAD associated with psychiatric diagnoses (particularly schizophrenia, as well as the medications used to treat it) might warrant consideration of aggressive primary prevention strategies.28 For example, some experts recommend starting metformin to reduce the risk of T2DM in patients who have been started on olanzapine or clozapine, regardless of the baseline fasting blood glucose level.29
You should be fully informed and aware of patients’ underlying medical conditions and the medications that are recommended to treat their conditions. Ideally, an integrated care strategy or, at the least, clear communication between you and the patient’s primary care providers should be in place to avoid foreseeable problems.
Stimulants. Systematic reviews suggest an association between prescription stimulants and at least the 2 cardiovascular risk factors of elevated heart rate and blood pressure. Stimulants are not recommended, therefore, for routine use in patients who have known hypertension or CAD.30
Second-generation antipsychotics are associated with significant weight gain and development of metabolic syndrome.
Selective serotonin reuptake inhibitors are associated with an increased risk of gastrointestinal bleeding risk related to platelet inhibition and gastric effects. Risk increases with additional platelet inhibitors, such as aspirin or clopidogrel.31
Lithium is excreted solely by the kidney. Guidelines recommend ACE inhibitors and angiotensin receptor-blockers for patients with CAD or T2DM, and many patients with symptomatic congestive heart failure are prescribed a diuretic; all of these classes of medications impair excretion of lithium. In a nested case-control study, 3% of observed cases of lithium toxicity were attributable to a newly initiated ACE inhibitor or angiotensin receptor-blocker.32 It is essential that you, and your patients taking lithium, be aware of the need to monitor the drug level frequently and be vigilant for symptoms of mild toxicity.
Beta blockers. No prospectively collected data support a association between beta blockers and depression.33 Patients with CAD should be given a trial of a beta blocker to achieve optimal medical management; because they are at increased risk of depression in the first place, all patients with CAD should undergo monitoring for depressive symptoms.
Clopidogrel is activated through the cytochrome P450 2C19 isoenzyme; medications such as fluoxetine and fluvoxamine that inhibit the function of CYP2C19 can impair the effectiveness of clopidogrel.31
Other considerations. Patients taking a second-generation antipsychotic should have baseline and periodic (monthly for the first quarter, then quarterly) assessments of BMI and, after monitoring at 3 months after baseline, annual monitoring of blood pressure, the fasting glucose level, and abdominal waist circumference. Lipid levels should be monitored every 5 years9 (Table 3).

Baseline and periodic monitoring of hepatic enzymes is recommended for patients taking a statin. You, and the patient, should be alert to the possible development of muscle weakness or pain; establish a low threshold for screening for an elevated creatine kinase level, which signals rhabdomyolysis.
Case concluded
Ms. S’s psychiatrist measures her blood pressure and finds that it is 147/92 mm Hg. He uses the Pooled Cohort Equations to determine that her lifetime risk of cardiovascular event is 50% (compared with a 8% lifetime risk among a cohort in whom risk factors are optimized) and that her 10-year risk is 41% (compared with a 2.2% risk among optimized controls).
At this point, the psychiatrist starts metformin to prevent T2DM. He also starts Ms. S on a statin to prevent CAD in a setting of diagnosed T2DM.
Ms. S’s exertional dyspnea and shoulder discomfort could be associated with angina, and the physician wisely refers her for urgent evaluation. Because he is aware of the literature demonstrating decreased revascularization among patients with mental illness, he urges her other health care providers to provide her with guideline-based strategies to treat her cardiovascular disease.
Bottom Line
Patients with psychiatric illness have higher rates of morbidity and mortality from coronary artery disease (CAD) than the general population. Symptoms characteristic of depression and schizophrenia could lead to poor self-care or impaired adherence to programs designed to lower CAD risk factors. Institute strategies for primary and secondary prevention of CAD among your patients, based on published guidelines, and be aware of, and alert for, adverse cardiac effects and an increase in risk factors for CAD from the use of psychotropics.
Related Resources
• Elderon L, Whooley MA. Depression and cardiovascular disease. Prog Cardiovasc Dis. 2013;55(6):511-523.
• Interactive cardiovascular risk calculator developed from the Framingham Heart Study. https://www.framingham heartstudy.org/risk-functions/cardiovascular-disease/ 10-year-risk.php.
• Pooled Cohort Equations calculator. To determine estimated cardiovascular risk in comparison with peers with optimized risk factors. http://clincalc.com/cardiology/ascvd/ pooledcohort.aspx.
• To learn more about traditional cardiovascular risk factors from the Framingham Heart Study. http://www.framinghamheart study.org/risk-functions/.
Drug Brand Names
Amlodipine • Norvasc
Clozapine • Clozaril
Clopidogrel • Plavix
Felodipine • Plendil
Fluoxetine • Prozac
Fluvoxamine • Luvox
Lithium • Eskalith, Lithobid
Metformin • Glucophage
Olanzapine • Zyprexa
Risperidone • Risperdal
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Heald A, Montejo AL, Millar H, et al. Management of physical health in patients with schizophrenia: practical recommendations. Eur Psychiatry. 2010;25(suppl 2):S41-S45.
2. Huffman JC, Celano CM, Beach SR, et al. Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925. doi: 10.1155/2013/695925.
3. Lawrence D, Mitrou F, Zubrick ZR. Smoking and mental illness: results from population surveys in Australia and the United States. BMC Public Health. 2009;9:285.
4. Athyros VG, Tziomalos K, Katsiki N, et al; GREACE Study Collaborative Group. The impact of smoking on cardiovascular outcomes and comorbidities in statin-treated patients with coronary artery disease: a post hoc analysis of the GREACE study. Curr Vasc Pharmacol. 2013;11(5):779-784.
5. Fihn SD, Gardin JM, Abrams J, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; Preventive Cardiovascular Nurses Association; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2012 AACF/ AHA/ACP/AATS/PCNA/SCAI/STS Guidelines for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2012;60(24):e44-e164.
6. Gilman SE, Kawachi I, Fitzmaurice GM, et al. Socioeconomic status in childhood and the lifetime risk of major depression. Int J Epidemiol. 2002;31(2):359-367.
7. Pozuelo L, Tesar G, Zhang J, et al. Depression and heart disease: what do we know, and where are we headed? Cleve Clin J Med. 2009;76(1):59-70.
8. Osborn DP, Wright CA, Levy G, et al. Relative risk of diabetes, dyslipidaemia, hypertension and the metabolic syndrome in people with severe mental illnesses: systematic review and metaanalysis. BMC Psychiatry. 2008;8:84.
9. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
10. Jiang W, Velazquez EJ, Kuchibhatla M, et al. Effect of escitalopram on mental stress-induced myocardial ischemia: results of the REMIT trial. JAMA. 2013;309(20):2139-2049.
11. Soares-Filho GL, Mesquita CT, Mesquita ET, et al. Panic attack triggering myocardial ischemia documented by myocardial perfusion imaging study. A case report. Int Arch Med. 2012;5(1):24.
12. Khawaja IS, Westermeyer JJ, Gajwani P, et al. Depression and coronary artery disease: the association, mechanisms, and therapeutic implications. Psychiatry (Edgmont). 2009;6(1):38-51.
13. Wang G, Cui J, Wang Y, et al. Anxiety and adverse coronary artery disease outcomes in Chinese patients. Psychosom Med. 2013;75(6):530-536.
14. Watkins LL, Koch GG, Sherwood A, et al. Association of anxiety and depression with all-cause mortality in individuals with coronary heart disease. J Am Heart Assoc. 2013;2(2):e000068. doi: 10.1161/JAHA.112.000068.
15. Chiavarino C, Rabellino D, Ardito RB, et al. Emotional coping is a better predictor of cardiac prognosis than depression and anxiety. J Psychosom Res. 2012;73(6):473-475.
16. Moyer VA; U.S. Preventive Services Task Force. Screening for coronary heart disease with electrocardiography: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(7):512-518.
17. De Hert M, Vancampfort D, Correll CU, et al. Guidelines for screening and monitoring of cardiometabolic risk in schizophrenia: systematic evaluation. Br J Psychiatry. 2012;199(2):99-105.
18. Hartley L, Igbinedion E, Holmes J, et al. Increased consumption of fruit and vegetables for the primary prevention of cardiovascular diseases. Cochrane Database Syst Rev. 2013;6:CD009874. doi: 10.1002/14651858.CD009874.pub2.
19. Chiuve SE, Fung TT, Rexrode KM, et al. Adherence to a low-risk, healthy lifestyle and risk of sudden cardiac death among women. JAMA. 2011;306(1):62-69.
20. Davidson M. Risk of cardiovascular disease and sudden death in schizophrenia. J Clin Psychiatry. 2002;63(suppl 9):5-11.
21. Dipasquale S, Pariante CM, Dazzan P, et al. The dietary pattern of patients with schizophrenia: a systematic review. J Psychiatr Res. 2013;47(2):197-207.
22. Katon WJ, Lin EH, Von Korff M, et al. Collaborative care for patients with depression and chronic illness. N Engl J Med. 2010;363(27):2611-2620.
23. Manderbacka K, Arffman M, Sund R, et al. How does a history of psychiatric hospital care influence access to coronary care: a cohort study. BMJ Open. 2012;2(2):e000831. doi: 10.1136/bmjopen-2012-000831.
24. Kumbhani DJ, Steg PG, Cannon CP, et al; REduction of Atherothrombosis for Continued Health Registry Investigators. Adherence to secondary prevention medications and four-year outcomes in outpatients with atherosclerosis. Am J Med. 2013;126(8):693-700.
25. Vandvik PO, Lincoff AM, Gore JM, et al. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence- Based Clinical Practice Guidelines. Chest. 2012;141(suppl 2):e637S-e668S.
26. Lichtman JH, Bigger T, Blumenthal JA, et al; American Heart Association Prevention Committee of the Council on Cardiovascular Nursing; American Heart Association Council on Clinical Cardiology; American Heart Association Council on Epidemiology and Prevention; American Heart Association Interdisciplinary Council on Quality of Care and Outcomes Research; American Psychiatric Association. Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation. 2008;118:1768-1775.
27. Albus C, Jordan J, Herrmann-Lingen C. Screening for psychosocial risk factors in patients with coronary heart disease-recommendations for clinical practice. Eur J Cardiovasc Prev Rehabil. 2004;11(1):75-79.
28. Srihari VH, Phutane VH, Ozkan B, et al. Cardiovascular mortality in schizophrenia: defining a critical period for prevention. Schizophr Res. 2013;146(1-3):64-68.
29. Brooks JO 3rd, Chang HS, Krasnykh O. Metabolic risks in older adults receiving second-generation antipsychotic medication. Curr Psychiatry Rep. 2009;11(1):33-40.
30. Martinez-Raga J, Knecht C, Szerman N, et al. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15-30.
31. Andrade C. Drug interactions in the treatment of depression in patients with ischemic heart disease. J Clin Psychiatry. 2012;73(12):e1475-e1477.
32. Juurlink DN, Mamdani MM, Kopp A, et al. Drug-induced lithium toxicity in the elderly: a population-based study. J Am Geriatr Soc. 2004;52(5):794-798.
33. Muzyk AJ, Gagliardi JP. Do beta blockers cause depression? Current Psychiatry. 2010;9(5):50,51,55.
The problem is enormous: Heart disease is the leading cause of death in the United States, and coronary artery disease (CAD) is the most common form of heart disease—responsible for 385,000 deaths in the United States in 2009 (http://www.cdc.gov/heartdisease/facts. htm). Patients with psychiatric illness have higher rates of morbidity and mortality from CAD than the general population, and warrant consideration as a special population. You should be familiar with routine cardiac medications; your patients’ medical problems; potential cardiac-related interactions among their psychotropic medications; and interactions among illnesses in their mental health and medical health domains (Box).
CASE Type 2 diabetes mellitus plus a long history of heavy smoking
Ms. S, age 57, is an African American woman with chronic paranoid schizophrenia who has been seeing a psychiatrist for the past 10 years. Ms. S’s psychiatric symptoms have been well controlled on risperidone, 3 mg/d.
Ms. S has a family history of diabetes, hypertension, and early CAD (a brother died of a myocardial infarction [MI] in his late 40s). She continues to smoke 2 packs of unfiltered cigarettes daily, as she has done for the past 40 years.
The psychiatrist has been following American Diabetes Association/American Psychiatric Association guidelines for monitoring; he has noticed that Ms. S’s body mass index (BMI) has increased from 27 to 31 kg/m2 over the past year. She has developed type 2 diabetes mellitus (T2DM).
At today’s visit, Ms. S arrives a few minutes late and appears flustered and out of breath. She explains that she had to climb a flight of stairs to get to office because the elevator is broken.
During the visit, the psychiatrist notes that Ms. S occasionally winces and massages her left shoulder.
Questions to ponder
• What else could the psychiatrist do to modify Ms. S’s cardiac risk factors?
• What is Ms. S’s 10-year risk of an acute coronary event?
• What should her physician do now?
Overview: Cardiac risk in patients with mental illness
Modifiable risks for CAD include hypertension, hypercholesterolemia, T2DM, obesity (all of which, taken together, constitute the metabolic syndrome), smoking, and a sedentary lifestyle. Some risk factors, including sex, age, and family history, are not modifiable. Whether or not this modification leads to better outcomes, psychiatric comorbidity is associated with higher morbidity and mortality from CAD.
Whether a common underlying pathological process manifesting in both CAD and mental illness exists, or whether the association is causal, are not well understood. Symptoms characteristic of depression (apathy, amotivation) and schizophrenia (disorganization, paranoia) could lead to poor self-care or impaired adherence to programs designed to lower CAD risk factors.1,2
People with mental illness smoke at a higher rate than those who do not have mental illness.3 This finding is of particular relevance because smoking contributes to worse outcomes with respect to CAD, even when medications are prescribed to address metabolic risks.4
Lower socioeconomic status is associated with poorer prognosis from CAD5 and is a risk factor for depression.6 Depression is a strong independent predictor of worse survival in acute coronary syndromes.5 Some experts consider depression to be a stronger risk factor for MI than traditional medical risk factors such as obesity, hypertension, and second-hand smoke.7
Interventions used to treat certain mental illnesses can exacerbate, or predispose to, metabolic syndrome (which, in turn, increases the risk of CAD). Although some studies have demonstrated metabolic derangements in medication-naïve patients who have a new diagnosis of schizophrenia,8 there is a clearly established association between second-generation antipsychotic use and obesity, hypertension, hyperlipidemia, and T2DM. This association prompted development in 2004 of consensus recommendations for cardiovascular monitoring of patients who are taking an atypical antipsychotic.9
Some studies suggest that the stress of mental illness contributes to the pathogen esis of CAD.8 Hypothesized mechanisms include:
• sympathetic activation
• vagal deactivation
• platelet activation
• hypothalamic-pituitary-adrenocortical pathways
• anticholinergic mechanisms
• inflammatory mediators, including cytokines.
Mental stress itself has the capacity to induce coronary ischemia.10 The mental stress of psychiatric illness could have an important pathophysiologic role in CAD. It can be tempting to disregard chest pain in a patient who is known to have panic disorder, but that patient might in fact be experiencing stress-induced myocardial ischemia.11
As many as 30% to 40% of patients with CAD suffer from clinically significant symptoms of depression; as many as 20% of patients with CAD meet criteria for major depressive disorder, compared with 5% to 10% of people who do not have CAD.2 Depression post-MI has been associated with a higher rate of sudden cardiac death and worse outcomes.12
Anxiety also can portend worse outcomes from CAD,13 including higher all-cause mortality.14 There is some hope, but limited evidence, that treating depression and anxiety, whether with antidepressant medication or behavioral therapy, can improve CAD outcomes.10,15
Making a diagnosis of CAD
CAD can present in a variety of ways, ranging from unrecognized or so-called silent CAD (there is an association between T2DM and unrecognized CAD and between hypertension and unrecognized CAD) to stable angina, unstable angina, acute coronary syndrome, MI, and sudden cardiac death. A variety of abnormalities on resting and exercise electrocardiogram (ECG), including ST segment depression, ST elevation, Q waves, and other morphological changes are indicative of CAD.
Other modalities, including coronary calcification score on computed tomography and coronary angiography can confirm the presence of CAD. Some clinicians recommend periodic ECG treadmill testing in patients who have:
• a total cholesterol level is >240 mg/dL
• systolic blood pressure >140 mm Hg, diastolic blood pressure >90 mm Hg, or both
• a family history of MI or sudden cardiac death in young (age <60) first-degree relatives
• a history of smoking
• diabetes.
Preventive guidelines
Risk stratification. A low (<10%), moderate (10% to 20%), or high (>20%) 10-year risk of CAD can be ascertained using a risk calculator, such as one that is available through the Framingham Heart Study (Figure) and the National Heart, Lung, and Blood Institute (http://cvdrisk.nhlbi. nih.gov). Because patients with risk factors for CAD should be offered interventions— including smoking cessation therapy, diet and exercise, aspirin, lipid-lowering therapy, and blood pressure modification strategies—whether or not they have evidence of CAD, the United States Preventive Services Task Force does not recommend for or against diagnostic screening in patients at moderate or elevated risk of CAD.16
There are guidelines in the literature recommending specific screening strategies for patients with mental illness, although the vetting and update process has been ill defined. Among patients with schizophrenia, though, regardless of antipsychotic prescription status, baseline and then regular monitoring of metabolic risk parameters is recommended.17
Primary prevention. Lifestyle modification and attention to modifiable coronary risk factors are important primary prevention strategies. Dietary modifications, exercise, not smoking, and maintenance of a normal BMI (<25 kg/m2) are associated with a lower risk of CAD.18,19
Lifestyle modifications can be challenging for patients with persistent mental illness, however: For example, patients with schizophrenia smoke more, eat less healthfully, and participate less in behavioral modification that targets risk factors than patients who do not have schizophrenia.20,21
According to 2012 evidence-based practice guidelines established by a collaboration that included the American College of Physicians and several cardiology and thoracic medicine societies, persons age >50 who do not have symptomatic CAD should take low-dose (75 to 100 mg/d) aspirin; the benefit of low-dose aspirin in persons at moderate or high risk of CAD is even greater. Other medications, including statins and fixed-dose combinations of antihypertensive medications in combination with a statin are not clearly beneficial as primary prevention strategies across the board, although selected high-risk populations might benefit.
Regrettably, the high-risk population of persons with mental illness and whose primary care is suboptimal has not been studied. It stands to reason that these patients would especially benefit from more attentive monitoring and intervention.
Collaborative care? Although many psychiatrists do not practice in such a model, a comprehensive approach to the care of their patients, using a collaborative care strategy that includes attention to the mental health diagnosis along with medical health, can result in improved health in both domains.22 However, enlisting patients with paranoia or an inherent distrust of medications and health care providers to adhere to either a medication regimen or lifestyle modification can be challenging.
Common-sense strategies, such as creating a multidisciplinary team with the psychiatrist coordinating care and optimizing antipsychotic treatment, might provide benefit.1 Data demonstrate that patients with severe mental illness who experience acute coronary events undergo revascularization at a lower rate than their mentally heathy counterparts, despite the fact that patients with severe mental illness die at a higher rate from their CAD than patients who do not have mental illness. An important role for the psychiatrist, even in the absence of a collaborative care program, is to be an advocate for appropriate guideline-based care.23
Secondary prevention. Once a patient develops CAD, ongoing risk factor modification is important. Adherence to a therapeutic regimen that variously combines a platelet inhibitor, beta blocker, statin, and angiotensin-converting enzyme (ACE) inhibitor is associated with improved outcomes in patients with CAD.24 Specific antiplatelet recommendations and a recommendation for single vs combination antiplatelet therapy depends on chronicity and type of revascularization in a setting of CAD.25
Summary of guideline-based recommendations
Treatment guidelines published in the National Guidelines Clearinghouse address depression, CAD screening, and specific cardiac therapies, including ACE inhibitors, angiotensin-receptor blockers, oral anticoagulants, platelet inhibitors, beta blockers, and lifestyle modification.
Primary prevention. Recommendations for treatment to prevent CAD are listed in Table 1.
Secondary prevention. Recommendations for treatment after a diagnosis of CAD are listed in Table 2.
Special considerations for psychiatric providers
You should be comfortable with patients’ use of antihypertensive therapies and familiar with the potential these agents have to interact with psychotropics; in addition, you can take a more active role in prescribing, and monitoring patients’ responses to, these medications. Provide appropriate monitoring of ACE inhibitors, statins, and beta blockers; also, provide appropriate monitoring of psychotropics in patients who take recommended cardioprotective medications.
In situations that prompt referral (such as recent MI, new symptoms of heart failure, any history of syncope or new identification of T2DM), ideally you should collaborate with the patient’s primary care provider to help enhance adherence to recommended treatment strategies. You also should employ motivational interviewing techniques and offer strategies by which patients can engage in meaningful lifestyle modification.
There are official recommendations for depression screening strategies26 and psychosocial risk screening for patients in whom CAD has been identified.27 Official screening strategies for CAD in patients with psychiatric illness have not, however, been spelled out.
Primary CAD prevention with medication is not routinely recommended for the general population, but the increased risk of CAD associated with psychiatric diagnoses (particularly schizophrenia, as well as the medications used to treat it) might warrant consideration of aggressive primary prevention strategies.28 For example, some experts recommend starting metformin to reduce the risk of T2DM in patients who have been started on olanzapine or clozapine, regardless of the baseline fasting blood glucose level.29
You should be fully informed and aware of patients’ underlying medical conditions and the medications that are recommended to treat their conditions. Ideally, an integrated care strategy or, at the least, clear communication between you and the patient’s primary care providers should be in place to avoid foreseeable problems.
Stimulants. Systematic reviews suggest an association between prescription stimulants and at least the 2 cardiovascular risk factors of elevated heart rate and blood pressure. Stimulants are not recommended, therefore, for routine use in patients who have known hypertension or CAD.30
Second-generation antipsychotics are associated with significant weight gain and development of metabolic syndrome.
Selective serotonin reuptake inhibitors are associated with an increased risk of gastrointestinal bleeding risk related to platelet inhibition and gastric effects. Risk increases with additional platelet inhibitors, such as aspirin or clopidogrel.31
Lithium is excreted solely by the kidney. Guidelines recommend ACE inhibitors and angiotensin receptor-blockers for patients with CAD or T2DM, and many patients with symptomatic congestive heart failure are prescribed a diuretic; all of these classes of medications impair excretion of lithium. In a nested case-control study, 3% of observed cases of lithium toxicity were attributable to a newly initiated ACE inhibitor or angiotensin receptor-blocker.32 It is essential that you, and your patients taking lithium, be aware of the need to monitor the drug level frequently and be vigilant for symptoms of mild toxicity.
Beta blockers. No prospectively collected data support a association between beta blockers and depression.33 Patients with CAD should be given a trial of a beta blocker to achieve optimal medical management; because they are at increased risk of depression in the first place, all patients with CAD should undergo monitoring for depressive symptoms.
Clopidogrel is activated through the cytochrome P450 2C19 isoenzyme; medications such as fluoxetine and fluvoxamine that inhibit the function of CYP2C19 can impair the effectiveness of clopidogrel.31
Other considerations. Patients taking a second-generation antipsychotic should have baseline and periodic (monthly for the first quarter, then quarterly) assessments of BMI and, after monitoring at 3 months after baseline, annual monitoring of blood pressure, the fasting glucose level, and abdominal waist circumference. Lipid levels should be monitored every 5 years9 (Table 3).
Baseline and periodic monitoring of hepatic enzymes is recommended for patients taking a statin. You, and the patient, should be alert to the possible development of muscle weakness or pain; establish a low threshold for screening for an elevated creatine kinase level, which signals rhabdomyolysis.
Case concluded
Ms. S’s psychiatrist measures her blood pressure and finds that it is 147/92 mm Hg. He uses the Pooled Cohort Equations to determine that her lifetime risk of cardiovascular event is 50% (compared with a 8% lifetime risk among a cohort in whom risk factors are optimized) and that her 10-year risk is 41% (compared with a 2.2% risk among optimized controls).
At this point, the psychiatrist starts metformin to prevent T2DM. He also starts Ms. S on a statin to prevent CAD in a setting of diagnosed T2DM.
Ms. S’s exertional dyspnea and shoulder discomfort could be associated with angina, and the physician wisely refers her for urgent evaluation. Because he is aware of the literature demonstrating decreased revascularization among patients with mental illness, he urges her other health care providers to provide her with guideline-based strategies to treat her cardiovascular disease.
Bottom Line
Patients with psychiatric illness have higher rates of morbidity and mortality from coronary artery disease (CAD) than the general population. Symptoms characteristic of depression and schizophrenia could lead to poor self-care or impaired adherence to programs designed to lower CAD risk factors. Institute strategies for primary and secondary prevention of CAD among your patients, based on published guidelines, and be aware of, and alert for, adverse cardiac effects and an increase in risk factors for CAD from the use of psychotropics.
Related Resources
• Elderon L, Whooley MA. Depression and cardiovascular disease. Prog Cardiovasc Dis. 2013;55(6):511-523.
• Interactive cardiovascular risk calculator developed from the Framingham Heart Study. https://www.framingham heartstudy.org/risk-functions/cardiovascular-disease/ 10-year-risk.php.
• Pooled Cohort Equations calculator. To determine estimated cardiovascular risk in comparison with peers with optimized risk factors. http://clincalc.com/cardiology/ascvd/ pooledcohort.aspx.
• To learn more about traditional cardiovascular risk factors from the Framingham Heart Study. http://www.framinghamheart study.org/risk-functions/.
Drug Brand Names
Amlodipine • Norvasc
Clozapine • Clozaril
Clopidogrel • Plavix
Felodipine • Plendil
Fluoxetine • Prozac
Fluvoxamine • Luvox
Lithium • Eskalith, Lithobid
Metformin • Glucophage
Olanzapine • Zyprexa
Risperidone • Risperdal
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
The problem is enormous: Heart disease is the leading cause of death in the United States, and coronary artery disease (CAD) is the most common form of heart disease—responsible for 385,000 deaths in the United States in 2009 (http://www.cdc.gov/heartdisease/facts. htm). Patients with psychiatric illness have higher rates of morbidity and mortality from CAD than the general population, and warrant consideration as a special population. You should be familiar with routine cardiac medications; your patients’ medical problems; potential cardiac-related interactions among their psychotropic medications; and interactions among illnesses in their mental health and medical health domains (Box).
CASE Type 2 diabetes mellitus plus a long history of heavy smoking
Ms. S, age 57, is an African American woman with chronic paranoid schizophrenia who has been seeing a psychiatrist for the past 10 years. Ms. S’s psychiatric symptoms have been well controlled on risperidone, 3 mg/d.
Ms. S has a family history of diabetes, hypertension, and early CAD (a brother died of a myocardial infarction [MI] in his late 40s). She continues to smoke 2 packs of unfiltered cigarettes daily, as she has done for the past 40 years.
The psychiatrist has been following American Diabetes Association/American Psychiatric Association guidelines for monitoring; he has noticed that Ms. S’s body mass index (BMI) has increased from 27 to 31 kg/m2 over the past year. She has developed type 2 diabetes mellitus (T2DM).
At today’s visit, Ms. S arrives a few minutes late and appears flustered and out of breath. She explains that she had to climb a flight of stairs to get to office because the elevator is broken.
During the visit, the psychiatrist notes that Ms. S occasionally winces and massages her left shoulder.
Questions to ponder
• What else could the psychiatrist do to modify Ms. S’s cardiac risk factors?
• What is Ms. S’s 10-year risk of an acute coronary event?
• What should her physician do now?
Overview: Cardiac risk in patients with mental illness
Modifiable risks for CAD include hypertension, hypercholesterolemia, T2DM, obesity (all of which, taken together, constitute the metabolic syndrome), smoking, and a sedentary lifestyle. Some risk factors, including sex, age, and family history, are not modifiable. Whether or not this modification leads to better outcomes, psychiatric comorbidity is associated with higher morbidity and mortality from CAD.
Whether a common underlying pathological process manifesting in both CAD and mental illness exists, or whether the association is causal, are not well understood. Symptoms characteristic of depression (apathy, amotivation) and schizophrenia (disorganization, paranoia) could lead to poor self-care or impaired adherence to programs designed to lower CAD risk factors.1,2
People with mental illness smoke at a higher rate than those who do not have mental illness.3 This finding is of particular relevance because smoking contributes to worse outcomes with respect to CAD, even when medications are prescribed to address metabolic risks.4
Lower socioeconomic status is associated with poorer prognosis from CAD5 and is a risk factor for depression.6 Depression is a strong independent predictor of worse survival in acute coronary syndromes.5 Some experts consider depression to be a stronger risk factor for MI than traditional medical risk factors such as obesity, hypertension, and second-hand smoke.7
Interventions used to treat certain mental illnesses can exacerbate, or predispose to, metabolic syndrome (which, in turn, increases the risk of CAD). Although some studies have demonstrated metabolic derangements in medication-naïve patients who have a new diagnosis of schizophrenia,8 there is a clearly established association between second-generation antipsychotic use and obesity, hypertension, hyperlipidemia, and T2DM. This association prompted development in 2004 of consensus recommendations for cardiovascular monitoring of patients who are taking an atypical antipsychotic.9
Some studies suggest that the stress of mental illness contributes to the pathogen esis of CAD.8 Hypothesized mechanisms include:
• sympathetic activation
• vagal deactivation
• platelet activation
• hypothalamic-pituitary-adrenocortical pathways
• anticholinergic mechanisms
• inflammatory mediators, including cytokines.
Mental stress itself has the capacity to induce coronary ischemia.10 The mental stress of psychiatric illness could have an important pathophysiologic role in CAD. It can be tempting to disregard chest pain in a patient who is known to have panic disorder, but that patient might in fact be experiencing stress-induced myocardial ischemia.11
As many as 30% to 40% of patients with CAD suffer from clinically significant symptoms of depression; as many as 20% of patients with CAD meet criteria for major depressive disorder, compared with 5% to 10% of people who do not have CAD.2 Depression post-MI has been associated with a higher rate of sudden cardiac death and worse outcomes.12
Anxiety also can portend worse outcomes from CAD,13 including higher all-cause mortality.14 There is some hope, but limited evidence, that treating depression and anxiety, whether with antidepressant medication or behavioral therapy, can improve CAD outcomes.10,15
Making a diagnosis of CAD
CAD can present in a variety of ways, ranging from unrecognized or so-called silent CAD (there is an association between T2DM and unrecognized CAD and between hypertension and unrecognized CAD) to stable angina, unstable angina, acute coronary syndrome, MI, and sudden cardiac death. A variety of abnormalities on resting and exercise electrocardiogram (ECG), including ST segment depression, ST elevation, Q waves, and other morphological changes are indicative of CAD.
Other modalities, including coronary calcification score on computed tomography and coronary angiography can confirm the presence of CAD. Some clinicians recommend periodic ECG treadmill testing in patients who have:
• a total cholesterol level is >240 mg/dL
• systolic blood pressure >140 mm Hg, diastolic blood pressure >90 mm Hg, or both
• a family history of MI or sudden cardiac death in young (age <60) first-degree relatives
• a history of smoking
• diabetes.
Preventive guidelines
Risk stratification. A low (<10%), moderate (10% to 20%), or high (>20%) 10-year risk of CAD can be ascertained using a risk calculator, such as one that is available through the Framingham Heart Study (Figure) and the National Heart, Lung, and Blood Institute (http://cvdrisk.nhlbi. nih.gov). Because patients with risk factors for CAD should be offered interventions— including smoking cessation therapy, diet and exercise, aspirin, lipid-lowering therapy, and blood pressure modification strategies—whether or not they have evidence of CAD, the United States Preventive Services Task Force does not recommend for or against diagnostic screening in patients at moderate or elevated risk of CAD.16
There are guidelines in the literature recommending specific screening strategies for patients with mental illness, although the vetting and update process has been ill defined. Among patients with schizophrenia, though, regardless of antipsychotic prescription status, baseline and then regular monitoring of metabolic risk parameters is recommended.17
Primary prevention. Lifestyle modification and attention to modifiable coronary risk factors are important primary prevention strategies. Dietary modifications, exercise, not smoking, and maintenance of a normal BMI (<25 kg/m2) are associated with a lower risk of CAD.18,19
Lifestyle modifications can be challenging for patients with persistent mental illness, however: For example, patients with schizophrenia smoke more, eat less healthfully, and participate less in behavioral modification that targets risk factors than patients who do not have schizophrenia.20,21
According to 2012 evidence-based practice guidelines established by a collaboration that included the American College of Physicians and several cardiology and thoracic medicine societies, persons age >50 who do not have symptomatic CAD should take low-dose (75 to 100 mg/d) aspirin; the benefit of low-dose aspirin in persons at moderate or high risk of CAD is even greater. Other medications, including statins and fixed-dose combinations of antihypertensive medications in combination with a statin are not clearly beneficial as primary prevention strategies across the board, although selected high-risk populations might benefit.
Regrettably, the high-risk population of persons with mental illness and whose primary care is suboptimal has not been studied. It stands to reason that these patients would especially benefit from more attentive monitoring and intervention.
Collaborative care? Although many psychiatrists do not practice in such a model, a comprehensive approach to the care of their patients, using a collaborative care strategy that includes attention to the mental health diagnosis along with medical health, can result in improved health in both domains.22 However, enlisting patients with paranoia or an inherent distrust of medications and health care providers to adhere to either a medication regimen or lifestyle modification can be challenging.
Common-sense strategies, such as creating a multidisciplinary team with the psychiatrist coordinating care and optimizing antipsychotic treatment, might provide benefit.1 Data demonstrate that patients with severe mental illness who experience acute coronary events undergo revascularization at a lower rate than their mentally heathy counterparts, despite the fact that patients with severe mental illness die at a higher rate from their CAD than patients who do not have mental illness. An important role for the psychiatrist, even in the absence of a collaborative care program, is to be an advocate for appropriate guideline-based care.23
Secondary prevention. Once a patient develops CAD, ongoing risk factor modification is important. Adherence to a therapeutic regimen that variously combines a platelet inhibitor, beta blocker, statin, and angiotensin-converting enzyme (ACE) inhibitor is associated with improved outcomes in patients with CAD.24 Specific antiplatelet recommendations and a recommendation for single vs combination antiplatelet therapy depends on chronicity and type of revascularization in a setting of CAD.25
Summary of guideline-based recommendations
Treatment guidelines published in the National Guidelines Clearinghouse address depression, CAD screening, and specific cardiac therapies, including ACE inhibitors, angiotensin-receptor blockers, oral anticoagulants, platelet inhibitors, beta blockers, and lifestyle modification.
Primary prevention. Recommendations for treatment to prevent CAD are listed in Table 1.
Secondary prevention. Recommendations for treatment after a diagnosis of CAD are listed in Table 2.
Special considerations for psychiatric providers
You should be comfortable with patients’ use of antihypertensive therapies and familiar with the potential these agents have to interact with psychotropics; in addition, you can take a more active role in prescribing, and monitoring patients’ responses to, these medications. Provide appropriate monitoring of ACE inhibitors, statins, and beta blockers; also, provide appropriate monitoring of psychotropics in patients who take recommended cardioprotective medications.
In situations that prompt referral (such as recent MI, new symptoms of heart failure, any history of syncope or new identification of T2DM), ideally you should collaborate with the patient’s primary care provider to help enhance adherence to recommended treatment strategies. You also should employ motivational interviewing techniques and offer strategies by which patients can engage in meaningful lifestyle modification.
There are official recommendations for depression screening strategies26 and psychosocial risk screening for patients in whom CAD has been identified.27 Official screening strategies for CAD in patients with psychiatric illness have not, however, been spelled out.
Primary CAD prevention with medication is not routinely recommended for the general population, but the increased risk of CAD associated with psychiatric diagnoses (particularly schizophrenia, as well as the medications used to treat it) might warrant consideration of aggressive primary prevention strategies.28 For example, some experts recommend starting metformin to reduce the risk of T2DM in patients who have been started on olanzapine or clozapine, regardless of the baseline fasting blood glucose level.29
You should be fully informed and aware of patients’ underlying medical conditions and the medications that are recommended to treat their conditions. Ideally, an integrated care strategy or, at the least, clear communication between you and the patient’s primary care providers should be in place to avoid foreseeable problems.
Stimulants. Systematic reviews suggest an association between prescription stimulants and at least the 2 cardiovascular risk factors of elevated heart rate and blood pressure. Stimulants are not recommended, therefore, for routine use in patients who have known hypertension or CAD.30
Second-generation antipsychotics are associated with significant weight gain and development of metabolic syndrome.
Selective serotonin reuptake inhibitors are associated with an increased risk of gastrointestinal bleeding risk related to platelet inhibition and gastric effects. Risk increases with additional platelet inhibitors, such as aspirin or clopidogrel.31
Lithium is excreted solely by the kidney. Guidelines recommend ACE inhibitors and angiotensin receptor-blockers for patients with CAD or T2DM, and many patients with symptomatic congestive heart failure are prescribed a diuretic; all of these classes of medications impair excretion of lithium. In a nested case-control study, 3% of observed cases of lithium toxicity were attributable to a newly initiated ACE inhibitor or angiotensin receptor-blocker.32 It is essential that you, and your patients taking lithium, be aware of the need to monitor the drug level frequently and be vigilant for symptoms of mild toxicity.
Beta blockers. No prospectively collected data support a association between beta blockers and depression.33 Patients with CAD should be given a trial of a beta blocker to achieve optimal medical management; because they are at increased risk of depression in the first place, all patients with CAD should undergo monitoring for depressive symptoms.
Clopidogrel is activated through the cytochrome P450 2C19 isoenzyme; medications such as fluoxetine and fluvoxamine that inhibit the function of CYP2C19 can impair the effectiveness of clopidogrel.31
Other considerations. Patients taking a second-generation antipsychotic should have baseline and periodic (monthly for the first quarter, then quarterly) assessments of BMI and, after monitoring at 3 months after baseline, annual monitoring of blood pressure, the fasting glucose level, and abdominal waist circumference. Lipid levels should be monitored every 5 years9 (Table 3).
Baseline and periodic monitoring of hepatic enzymes is recommended for patients taking a statin. You, and the patient, should be alert to the possible development of muscle weakness or pain; establish a low threshold for screening for an elevated creatine kinase level, which signals rhabdomyolysis.
Case concluded
Ms. S’s psychiatrist measures her blood pressure and finds that it is 147/92 mm Hg. He uses the Pooled Cohort Equations to determine that her lifetime risk of cardiovascular event is 50% (compared with a 8% lifetime risk among a cohort in whom risk factors are optimized) and that her 10-year risk is 41% (compared with a 2.2% risk among optimized controls).
At this point, the psychiatrist starts metformin to prevent T2DM. He also starts Ms. S on a statin to prevent CAD in a setting of diagnosed T2DM.
Ms. S’s exertional dyspnea and shoulder discomfort could be associated with angina, and the physician wisely refers her for urgent evaluation. Because he is aware of the literature demonstrating decreased revascularization among patients with mental illness, he urges her other health care providers to provide her with guideline-based strategies to treat her cardiovascular disease.
Bottom Line
Patients with psychiatric illness have higher rates of morbidity and mortality from coronary artery disease (CAD) than the general population. Symptoms characteristic of depression and schizophrenia could lead to poor self-care or impaired adherence to programs designed to lower CAD risk factors. Institute strategies for primary and secondary prevention of CAD among your patients, based on published guidelines, and be aware of, and alert for, adverse cardiac effects and an increase in risk factors for CAD from the use of psychotropics.
Related Resources
• Elderon L, Whooley MA. Depression and cardiovascular disease. Prog Cardiovasc Dis. 2013;55(6):511-523.
• Interactive cardiovascular risk calculator developed from the Framingham Heart Study. https://www.framingham heartstudy.org/risk-functions/cardiovascular-disease/ 10-year-risk.php.
• Pooled Cohort Equations calculator. To determine estimated cardiovascular risk in comparison with peers with optimized risk factors. http://clincalc.com/cardiology/ascvd/ pooledcohort.aspx.
• To learn more about traditional cardiovascular risk factors from the Framingham Heart Study. http://www.framinghamheart study.org/risk-functions/.
Drug Brand Names
Amlodipine • Norvasc
Clozapine • Clozaril
Clopidogrel • Plavix
Felodipine • Plendil
Fluoxetine • Prozac
Fluvoxamine • Luvox
Lithium • Eskalith, Lithobid
Metformin • Glucophage
Olanzapine • Zyprexa
Risperidone • Risperdal
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Heald A, Montejo AL, Millar H, et al. Management of physical health in patients with schizophrenia: practical recommendations. Eur Psychiatry. 2010;25(suppl 2):S41-S45.
2. Huffman JC, Celano CM, Beach SR, et al. Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925. doi: 10.1155/2013/695925.
3. Lawrence D, Mitrou F, Zubrick ZR. Smoking and mental illness: results from population surveys in Australia and the United States. BMC Public Health. 2009;9:285.
4. Athyros VG, Tziomalos K, Katsiki N, et al; GREACE Study Collaborative Group. The impact of smoking on cardiovascular outcomes and comorbidities in statin-treated patients with coronary artery disease: a post hoc analysis of the GREACE study. Curr Vasc Pharmacol. 2013;11(5):779-784.
5. Fihn SD, Gardin JM, Abrams J, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; Preventive Cardiovascular Nurses Association; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2012 AACF/ AHA/ACP/AATS/PCNA/SCAI/STS Guidelines for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2012;60(24):e44-e164.
6. Gilman SE, Kawachi I, Fitzmaurice GM, et al. Socioeconomic status in childhood and the lifetime risk of major depression. Int J Epidemiol. 2002;31(2):359-367.
7. Pozuelo L, Tesar G, Zhang J, et al. Depression and heart disease: what do we know, and where are we headed? Cleve Clin J Med. 2009;76(1):59-70.
8. Osborn DP, Wright CA, Levy G, et al. Relative risk of diabetes, dyslipidaemia, hypertension and the metabolic syndrome in people with severe mental illnesses: systematic review and metaanalysis. BMC Psychiatry. 2008;8:84.
9. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
10. Jiang W, Velazquez EJ, Kuchibhatla M, et al. Effect of escitalopram on mental stress-induced myocardial ischemia: results of the REMIT trial. JAMA. 2013;309(20):2139-2049.
11. Soares-Filho GL, Mesquita CT, Mesquita ET, et al. Panic attack triggering myocardial ischemia documented by myocardial perfusion imaging study. A case report. Int Arch Med. 2012;5(1):24.
12. Khawaja IS, Westermeyer JJ, Gajwani P, et al. Depression and coronary artery disease: the association, mechanisms, and therapeutic implications. Psychiatry (Edgmont). 2009;6(1):38-51.
13. Wang G, Cui J, Wang Y, et al. Anxiety and adverse coronary artery disease outcomes in Chinese patients. Psychosom Med. 2013;75(6):530-536.
14. Watkins LL, Koch GG, Sherwood A, et al. Association of anxiety and depression with all-cause mortality in individuals with coronary heart disease. J Am Heart Assoc. 2013;2(2):e000068. doi: 10.1161/JAHA.112.000068.
15. Chiavarino C, Rabellino D, Ardito RB, et al. Emotional coping is a better predictor of cardiac prognosis than depression and anxiety. J Psychosom Res. 2012;73(6):473-475.
16. Moyer VA; U.S. Preventive Services Task Force. Screening for coronary heart disease with electrocardiography: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(7):512-518.
17. De Hert M, Vancampfort D, Correll CU, et al. Guidelines for screening and monitoring of cardiometabolic risk in schizophrenia: systematic evaluation. Br J Psychiatry. 2012;199(2):99-105.
18. Hartley L, Igbinedion E, Holmes J, et al. Increased consumption of fruit and vegetables for the primary prevention of cardiovascular diseases. Cochrane Database Syst Rev. 2013;6:CD009874. doi: 10.1002/14651858.CD009874.pub2.
19. Chiuve SE, Fung TT, Rexrode KM, et al. Adherence to a low-risk, healthy lifestyle and risk of sudden cardiac death among women. JAMA. 2011;306(1):62-69.
20. Davidson M. Risk of cardiovascular disease and sudden death in schizophrenia. J Clin Psychiatry. 2002;63(suppl 9):5-11.
21. Dipasquale S, Pariante CM, Dazzan P, et al. The dietary pattern of patients with schizophrenia: a systematic review. J Psychiatr Res. 2013;47(2):197-207.
22. Katon WJ, Lin EH, Von Korff M, et al. Collaborative care for patients with depression and chronic illness. N Engl J Med. 2010;363(27):2611-2620.
23. Manderbacka K, Arffman M, Sund R, et al. How does a history of psychiatric hospital care influence access to coronary care: a cohort study. BMJ Open. 2012;2(2):e000831. doi: 10.1136/bmjopen-2012-000831.
24. Kumbhani DJ, Steg PG, Cannon CP, et al; REduction of Atherothrombosis for Continued Health Registry Investigators. Adherence to secondary prevention medications and four-year outcomes in outpatients with atherosclerosis. Am J Med. 2013;126(8):693-700.
25. Vandvik PO, Lincoff AM, Gore JM, et al. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence- Based Clinical Practice Guidelines. Chest. 2012;141(suppl 2):e637S-e668S.
26. Lichtman JH, Bigger T, Blumenthal JA, et al; American Heart Association Prevention Committee of the Council on Cardiovascular Nursing; American Heart Association Council on Clinical Cardiology; American Heart Association Council on Epidemiology and Prevention; American Heart Association Interdisciplinary Council on Quality of Care and Outcomes Research; American Psychiatric Association. Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation. 2008;118:1768-1775.
27. Albus C, Jordan J, Herrmann-Lingen C. Screening for psychosocial risk factors in patients with coronary heart disease-recommendations for clinical practice. Eur J Cardiovasc Prev Rehabil. 2004;11(1):75-79.
28. Srihari VH, Phutane VH, Ozkan B, et al. Cardiovascular mortality in schizophrenia: defining a critical period for prevention. Schizophr Res. 2013;146(1-3):64-68.
29. Brooks JO 3rd, Chang HS, Krasnykh O. Metabolic risks in older adults receiving second-generation antipsychotic medication. Curr Psychiatry Rep. 2009;11(1):33-40.
30. Martinez-Raga J, Knecht C, Szerman N, et al. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15-30.
31. Andrade C. Drug interactions in the treatment of depression in patients with ischemic heart disease. J Clin Psychiatry. 2012;73(12):e1475-e1477.
32. Juurlink DN, Mamdani MM, Kopp A, et al. Drug-induced lithium toxicity in the elderly: a population-based study. J Am Geriatr Soc. 2004;52(5):794-798.
33. Muzyk AJ, Gagliardi JP. Do beta blockers cause depression? Current Psychiatry. 2010;9(5):50,51,55.
1. Heald A, Montejo AL, Millar H, et al. Management of physical health in patients with schizophrenia: practical recommendations. Eur Psychiatry. 2010;25(suppl 2):S41-S45.
2. Huffman JC, Celano CM, Beach SR, et al. Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925. doi: 10.1155/2013/695925.
3. Lawrence D, Mitrou F, Zubrick ZR. Smoking and mental illness: results from population surveys in Australia and the United States. BMC Public Health. 2009;9:285.
4. Athyros VG, Tziomalos K, Katsiki N, et al; GREACE Study Collaborative Group. The impact of smoking on cardiovascular outcomes and comorbidities in statin-treated patients with coronary artery disease: a post hoc analysis of the GREACE study. Curr Vasc Pharmacol. 2013;11(5):779-784.
5. Fihn SD, Gardin JM, Abrams J, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; Preventive Cardiovascular Nurses Association; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2012 AACF/ AHA/ACP/AATS/PCNA/SCAI/STS Guidelines for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2012;60(24):e44-e164.
6. Gilman SE, Kawachi I, Fitzmaurice GM, et al. Socioeconomic status in childhood and the lifetime risk of major depression. Int J Epidemiol. 2002;31(2):359-367.
7. Pozuelo L, Tesar G, Zhang J, et al. Depression and heart disease: what do we know, and where are we headed? Cleve Clin J Med. 2009;76(1):59-70.
8. Osborn DP, Wright CA, Levy G, et al. Relative risk of diabetes, dyslipidaemia, hypertension and the metabolic syndrome in people with severe mental illnesses: systematic review and metaanalysis. BMC Psychiatry. 2008;8:84.
9. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
10. Jiang W, Velazquez EJ, Kuchibhatla M, et al. Effect of escitalopram on mental stress-induced myocardial ischemia: results of the REMIT trial. JAMA. 2013;309(20):2139-2049.
11. Soares-Filho GL, Mesquita CT, Mesquita ET, et al. Panic attack triggering myocardial ischemia documented by myocardial perfusion imaging study. A case report. Int Arch Med. 2012;5(1):24.
12. Khawaja IS, Westermeyer JJ, Gajwani P, et al. Depression and coronary artery disease: the association, mechanisms, and therapeutic implications. Psychiatry (Edgmont). 2009;6(1):38-51.
13. Wang G, Cui J, Wang Y, et al. Anxiety and adverse coronary artery disease outcomes in Chinese patients. Psychosom Med. 2013;75(6):530-536.
14. Watkins LL, Koch GG, Sherwood A, et al. Association of anxiety and depression with all-cause mortality in individuals with coronary heart disease. J Am Heart Assoc. 2013;2(2):e000068. doi: 10.1161/JAHA.112.000068.
15. Chiavarino C, Rabellino D, Ardito RB, et al. Emotional coping is a better predictor of cardiac prognosis than depression and anxiety. J Psychosom Res. 2012;73(6):473-475.
16. Moyer VA; U.S. Preventive Services Task Force. Screening for coronary heart disease with electrocardiography: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(7):512-518.
17. De Hert M, Vancampfort D, Correll CU, et al. Guidelines for screening and monitoring of cardiometabolic risk in schizophrenia: systematic evaluation. Br J Psychiatry. 2012;199(2):99-105.
18. Hartley L, Igbinedion E, Holmes J, et al. Increased consumption of fruit and vegetables for the primary prevention of cardiovascular diseases. Cochrane Database Syst Rev. 2013;6:CD009874. doi: 10.1002/14651858.CD009874.pub2.
19. Chiuve SE, Fung TT, Rexrode KM, et al. Adherence to a low-risk, healthy lifestyle and risk of sudden cardiac death among women. JAMA. 2011;306(1):62-69.
20. Davidson M. Risk of cardiovascular disease and sudden death in schizophrenia. J Clin Psychiatry. 2002;63(suppl 9):5-11.
21. Dipasquale S, Pariante CM, Dazzan P, et al. The dietary pattern of patients with schizophrenia: a systematic review. J Psychiatr Res. 2013;47(2):197-207.
22. Katon WJ, Lin EH, Von Korff M, et al. Collaborative care for patients with depression and chronic illness. N Engl J Med. 2010;363(27):2611-2620.
23. Manderbacka K, Arffman M, Sund R, et al. How does a history of psychiatric hospital care influence access to coronary care: a cohort study. BMJ Open. 2012;2(2):e000831. doi: 10.1136/bmjopen-2012-000831.
24. Kumbhani DJ, Steg PG, Cannon CP, et al; REduction of Atherothrombosis for Continued Health Registry Investigators. Adherence to secondary prevention medications and four-year outcomes in outpatients with atherosclerosis. Am J Med. 2013;126(8):693-700.
25. Vandvik PO, Lincoff AM, Gore JM, et al. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence- Based Clinical Practice Guidelines. Chest. 2012;141(suppl 2):e637S-e668S.
26. Lichtman JH, Bigger T, Blumenthal JA, et al; American Heart Association Prevention Committee of the Council on Cardiovascular Nursing; American Heart Association Council on Clinical Cardiology; American Heart Association Council on Epidemiology and Prevention; American Heart Association Interdisciplinary Council on Quality of Care and Outcomes Research; American Psychiatric Association. Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation. 2008;118:1768-1775.
27. Albus C, Jordan J, Herrmann-Lingen C. Screening for psychosocial risk factors in patients with coronary heart disease-recommendations for clinical practice. Eur J Cardiovasc Prev Rehabil. 2004;11(1):75-79.
28. Srihari VH, Phutane VH, Ozkan B, et al. Cardiovascular mortality in schizophrenia: defining a critical period for prevention. Schizophr Res. 2013;146(1-3):64-68.
29. Brooks JO 3rd, Chang HS, Krasnykh O. Metabolic risks in older adults receiving second-generation antipsychotic medication. Curr Psychiatry Rep. 2009;11(1):33-40.
30. Martinez-Raga J, Knecht C, Szerman N, et al. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15-30.
31. Andrade C. Drug interactions in the treatment of depression in patients with ischemic heart disease. J Clin Psychiatry. 2012;73(12):e1475-e1477.
32. Juurlink DN, Mamdani MM, Kopp A, et al. Drug-induced lithium toxicity in the elderly: a population-based study. J Am Geriatr Soc. 2004;52(5):794-798.
33. Muzyk AJ, Gagliardi JP. Do beta blockers cause depression? Current Psychiatry. 2010;9(5):50,51,55.

Malnourished and psychotic, and found incompetent to stand trial
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
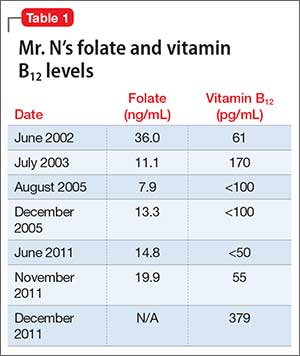
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
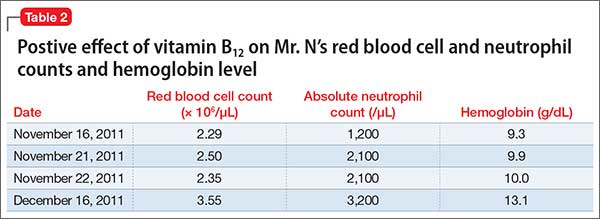
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
CBT improves depression but not self-care in heart failure patients
Cognitive behavioral therapy significantly improved major depression but did not improve self-care by heart failure patients, investigators reported online in JAMA Internal Medicine.
“The results suggest that CBT is superior to usual care for depression in patients with heart failure,” said Dr. Kenneth Freedland and his associates at Washington University in St. Louis. They called the findings “especially encouraging” in light of recent negative results from the SADHART-CHF and MOOD-HF trials of selective serotonin reuptake inhibitors in this population.Patients in heart failure often have major depression, which increases their chances of poor self-care, hospitalization, and mortality, the researchers noted. Their single-blind, randomized trial included 158 patients who were in New York Heart Association class I, II, or III heart failure and met criteria for major depression. Patients in the intervention group received standard medical care, plus up to 6 months of CBT designed for cardiac patients.Patients received CBT weekly, then biweekly, and then monthly as they reached their treatment goals, but they also received telephone follow-up to help prevent relapse. The control group received standard medical care plus consultation with a cardiac nurse, written materials on heart failure self-care, and three follow-up phone calls with the nurse (JAMA Intern Med. 2015 Sept. 28. doi:10.1001/jamainternmed.2015.5220). At 6 months, the CBT group scored significantly lower on the BDI-II than did controls (mean score, 12.8 [standard deviation, 10.6] vs. 17.3 [10.7]; P = .008), the researchers said. Remission rates with CBT were 46% based on the BDI-II and 51% based on the Hamilton Depression Scale, both of which significantly exceeded remission rates of 19-20% for controls. The CBT group also improved significantly more than did controls on standard measures for anxiety, heart failure-related quality of life, mental health–related quality of life, fatigue, and social functioning, but not on measures of physical functioning, the researchers reported.
The National Heart, Lung, and Blood Institute partially funded the study. The researchers declared no competing interests.
When depression occurs in patients with heart failure, which is often, the illness burden and management complexity increase multifold. Freedland et al. tested the hypothesis that the effective treatment of comorbid depression with cognitive behavioral therapy (CBT) would also lead to improvements in heart failure self-care and physical functioning and found that it did not. The good news is that CBT did significantly improve emotional health and overall quality of life, and the improvement in depressive symptoms associated with CBT was larger than observed in pharmacotherapy trials for depression in patients with heart disease. This supports evidence for a shift in practice away from so much pharmacotherapy and more use of psychotherapy to achieve better mental health and overall quality of life outcomes in patients with heart failure. In reframing how we think about the management of depression in patients with heart failure, we should be talking more and prescribing less.
Dr. Patrick G. O’Malley is deputy editor of JAMA Internal Medicine. He declared no competing interests. These comments were taken from his accompanying editorial (JAMA Intern Med. 2015 Sept. 28).
When depression occurs in patients with heart failure, which is often, the illness burden and management complexity increase multifold. Freedland et al. tested the hypothesis that the effective treatment of comorbid depression with cognitive behavioral therapy (CBT) would also lead to improvements in heart failure self-care and physical functioning and found that it did not. The good news is that CBT did significantly improve emotional health and overall quality of life, and the improvement in depressive symptoms associated with CBT was larger than observed in pharmacotherapy trials for depression in patients with heart disease. This supports evidence for a shift in practice away from so much pharmacotherapy and more use of psychotherapy to achieve better mental health and overall quality of life outcomes in patients with heart failure. In reframing how we think about the management of depression in patients with heart failure, we should be talking more and prescribing less.
Dr. Patrick G. O’Malley is deputy editor of JAMA Internal Medicine. He declared no competing interests. These comments were taken from his accompanying editorial (JAMA Intern Med. 2015 Sept. 28).
When depression occurs in patients with heart failure, which is often, the illness burden and management complexity increase multifold. Freedland et al. tested the hypothesis that the effective treatment of comorbid depression with cognitive behavioral therapy (CBT) would also lead to improvements in heart failure self-care and physical functioning and found that it did not. The good news is that CBT did significantly improve emotional health and overall quality of life, and the improvement in depressive symptoms associated with CBT was larger than observed in pharmacotherapy trials for depression in patients with heart disease. This supports evidence for a shift in practice away from so much pharmacotherapy and more use of psychotherapy to achieve better mental health and overall quality of life outcomes in patients with heart failure. In reframing how we think about the management of depression in patients with heart failure, we should be talking more and prescribing less.
Dr. Patrick G. O’Malley is deputy editor of JAMA Internal Medicine. He declared no competing interests. These comments were taken from his accompanying editorial (JAMA Intern Med. 2015 Sept. 28).
Cognitive behavioral therapy significantly improved major depression but did not improve self-care by heart failure patients, investigators reported online in JAMA Internal Medicine.
“The results suggest that CBT is superior to usual care for depression in patients with heart failure,” said Dr. Kenneth Freedland and his associates at Washington University in St. Louis. They called the findings “especially encouraging” in light of recent negative results from the SADHART-CHF and MOOD-HF trials of selective serotonin reuptake inhibitors in this population.Patients in heart failure often have major depression, which increases their chances of poor self-care, hospitalization, and mortality, the researchers noted. Their single-blind, randomized trial included 158 patients who were in New York Heart Association class I, II, or III heart failure and met criteria for major depression. Patients in the intervention group received standard medical care, plus up to 6 months of CBT designed for cardiac patients.Patients received CBT weekly, then biweekly, and then monthly as they reached their treatment goals, but they also received telephone follow-up to help prevent relapse. The control group received standard medical care plus consultation with a cardiac nurse, written materials on heart failure self-care, and three follow-up phone calls with the nurse (JAMA Intern Med. 2015 Sept. 28. doi:10.1001/jamainternmed.2015.5220). At 6 months, the CBT group scored significantly lower on the BDI-II than did controls (mean score, 12.8 [standard deviation, 10.6] vs. 17.3 [10.7]; P = .008), the researchers said. Remission rates with CBT were 46% based on the BDI-II and 51% based on the Hamilton Depression Scale, both of which significantly exceeded remission rates of 19-20% for controls. The CBT group also improved significantly more than did controls on standard measures for anxiety, heart failure-related quality of life, mental health–related quality of life, fatigue, and social functioning, but not on measures of physical functioning, the researchers reported.
The National Heart, Lung, and Blood Institute partially funded the study. The researchers declared no competing interests.
Cognitive behavioral therapy significantly improved major depression but did not improve self-care by heart failure patients, investigators reported online in JAMA Internal Medicine.
“The results suggest that CBT is superior to usual care for depression in patients with heart failure,” said Dr. Kenneth Freedland and his associates at Washington University in St. Louis. They called the findings “especially encouraging” in light of recent negative results from the SADHART-CHF and MOOD-HF trials of selective serotonin reuptake inhibitors in this population.Patients in heart failure often have major depression, which increases their chances of poor self-care, hospitalization, and mortality, the researchers noted. Their single-blind, randomized trial included 158 patients who were in New York Heart Association class I, II, or III heart failure and met criteria for major depression. Patients in the intervention group received standard medical care, plus up to 6 months of CBT designed for cardiac patients.Patients received CBT weekly, then biweekly, and then monthly as they reached their treatment goals, but they also received telephone follow-up to help prevent relapse. The control group received standard medical care plus consultation with a cardiac nurse, written materials on heart failure self-care, and three follow-up phone calls with the nurse (JAMA Intern Med. 2015 Sept. 28. doi:10.1001/jamainternmed.2015.5220). At 6 months, the CBT group scored significantly lower on the BDI-II than did controls (mean score, 12.8 [standard deviation, 10.6] vs. 17.3 [10.7]; P = .008), the researchers said. Remission rates with CBT were 46% based on the BDI-II and 51% based on the Hamilton Depression Scale, both of which significantly exceeded remission rates of 19-20% for controls. The CBT group also improved significantly more than did controls on standard measures for anxiety, heart failure-related quality of life, mental health–related quality of life, fatigue, and social functioning, but not on measures of physical functioning, the researchers reported.
The National Heart, Lung, and Blood Institute partially funded the study. The researchers declared no competing interests.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Cognitive behavior therapy significantly improved major depression but not self-care among patients with heart failure.
Major finding: At 6 months, mean BDI-II scores were 12.8 for CBT vs. 17.3 for enhanced usual care (P = .008).
Data source: Single-blind, randomized trial of 158 patients.
Disclosures: The National Heart, Lung, and Blood Institute partially funded the study. The researchers declared no conflicts of interest.
Assessing head pain

A medication change, then involuntary lip smacking and tongue rolling
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
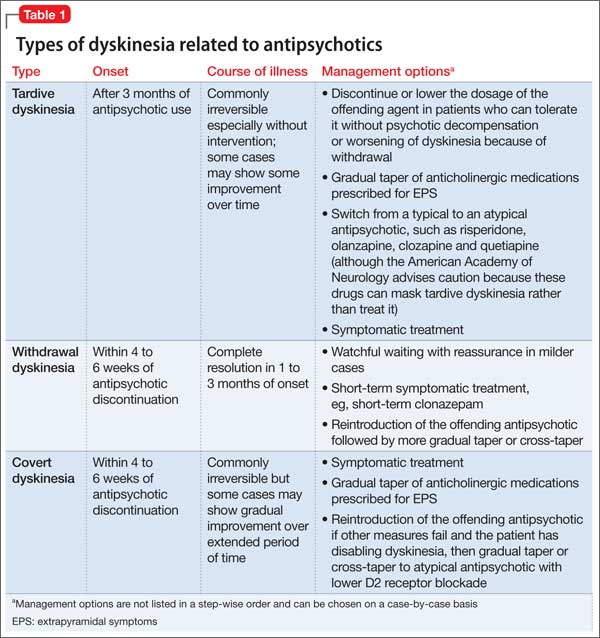
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
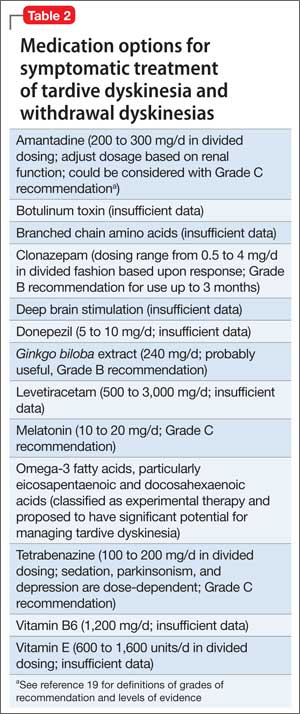
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
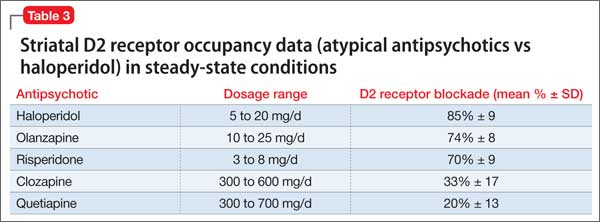
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Insurer denies drug coverage
Ms. X, age 65, has a 35-year history of bipolar I disorder (BD I) characterized by psychotic mania and severe suicidal depression. For the past year, her symptoms have been well controlled with aripiprazole, 5 mg/d; trazodone, 50 mg at bedtime; and citalopram, 20 mg/d. Because her health insurance has changed, Ms. X asks to be switched to an alternative antipsychotic because the new provider denied coverage of aripiprazole.
While taking aripiprazole, Ms. X did not report any extrapyramidal side effects, including tardive dyskinesia. Her Abnormal Involuntary Movement Scale (AIMS) score is 4. No significant abnormal movements were noted on examination during previous medication management sessions.
We decide to replace aripiprazole with quetiapine, 50 mg/d. At a 2-week follow-up visit, Ms. X is noted to have euphoric mood and reduced need to sleep, flight of ideas, increased talkativeness, and paranoia. We also notice that she has significant tongue rolling and lip smacking, which she says started 10 days after changing from aripiprazole to quetiapine. Her AIMS score is 17.
What could be causing Ms. X’s tongue rolling and lip smacking?
a) an irreversible syndrome usually starting after 1 or 2 years of continuous exposure to antipsychotics
b) a self-limited condition expected to resolve completely within 12 weeks
c) an acute manifestation of an antipsychotic that can respond to an anticholinergic agent
d) none of the above
The authors’ observations
Tardive dyskinesia (TD) refers to at least moderate abnormal involuntary movements in ≥1 areas of the body or at least mild movements in ≥2 areas of the body, developing after ≥3 months of cumulative exposure (continuous or discontinuous) to dopamine D2 receptor-blocking agents.1 AIMS is a 14-item, clinician-administered questionnaire designed to evaluate such movements and track their severity over time. The first 10 items are rated on 5-point scale (0 = none; 1 = minimal; 2 = mild; 3 = moderate; 4 = severe), with items 1 to 4 assessing orofacial movements, 5 to 7 assessing extremity and truncal movements, and 8 to 10 assessing overall severity, impairment, and subjective distress. Items 11 to 13 assess dental status because lack of teeth can result in oral movements mimicking TDs. The last item assesses whether these movements disappear during sleep.
HISTORY Poor response
Ms. X was given a diagnosis of BD I at age 30; she first started taking antipsychotics 10 years later. Previous psychotropic trials included lamotrigine, divalproex sodium, risperidone, and ziprasidone, which were ineffective or poorly tolerated. Her medical history includes obstructive sleep apnea, narcolepsy, type 2 diabetes mellitus, hypertension, dyslipidemia, fibromyalgia, gastroesophageal reflux disease, and hypothyroidism. She takes metformin, omeprazole, pravastatin, carvedilol, insulin, levothyroxine, methylphenidate (for hypersomnia), and enalapril.
What is the next best step in management?
a) discontinue quetiapine
b) replace quetiapine with clozapine
c) increase quetiapine to target manic symptoms and reassess in a few weeks
d) continue quetiapine and treat abnormal movements with benztropine
TREATMENT Increase dosage
We increase quetiapine to 150 mg/d to target Ms. X’s manic symptoms. She is scheduled for a follow-up visit in 4 weeks but is instructed to return to the clinic earlier if her manic symptoms do not improve. At the 4-week follow-up visit, Ms. X does not have any abnormal movements and her manic symptoms have resolved. Her AIMS score is 4. Her husband reports that her abnormal movements resolved 4 days after increasing quetiapine to 150 mg/d.
The authors’ observations
Second-generation antipsychotics are known to have a lower risk of extrapyramidal adverse reactions compared with older first-generation antipsychotics.2,3 TD differs from other extrapyramidal symptoms (EPS) because of its delayed onset. Risk factors for TD include:
• female sex
• age >50
• history of brain damage
• long-term antipsychotic use
• diagnosis of a mood disorder.
Gardos et al4 described 2 other forms of delayed dyskinesias related to antipsychotic use but resulting from antipsychotic discontinuation: withdrawal dyskinesia and covert dyskinesia. Evidence for these types of antipsychotic discontinuation syndromes mostly is anecdotal.5,6Table 1 highlights 3 different types of dyskinesias and their management.
Withdrawal dyskinesia has been described as a syndrome resembling TD that appears after discontinuation or dosage reduction of an antipsychotic in a patient who does not have an earlier TD diagnosis.7 The prevalence of withdrawal dyskinesia among patients undergoing antipsychotic discontinuation is approximately 30%.8 Cases of withdrawal dyskinesia are self-limited and resolve in 1 to 3 months.9,10 We believe that Ms. X’s movement disorder was withdrawal dyskinesia from aripiprazole because her symptoms started 10 days after the drug was discontinued, and was self-limited and reversible.
Similar to TD, withdrawal dyskinesia can present in different forms:
• tongue protrusion movements
• facial grimacing
• ticks
• chorea
• tremors
• athetosis
• involuntary vocalizations
• abnormal movements of hands and legs
• “dyspnea” due to involvement of respiratory musculature.5,11
There may be a sex difference in duration of withdrawal dyskinesias, because symptoms persist longer in females.9
Although covert dyskinesia also develops after discontinuation or dosage reduction of a dopamine-blocking agent, the symptoms usually are permanent, and could require reintroducing the antipsychotic or management with evidence-based treatments for TD, such as tetrabenazine or amantadine.6,12
What is the cause of Ms. X’s abnormal involuntary movements?
a) quetiapine-induced D2 receptor hypersensitivity
b) aripiprazole-induced cholinergic overactivity
c) quetiapine-induced cholinergic overactivity
d) aripiprazole-induced D2 receptor hypersensitivity
The authors’ observations
Pathophysiology of this condition is unknown but different theories have been proposed. D2 receptor up-regulation and hypersensitivity to compensate for chronic D2 receptor blockade by antipsychotics is a commonly cited theory.7,13 Discontinuation of an antipsychotic can make this D2 receptor up-regulation and hypersensitivity manifest as withdrawal dyskinesia by creating a temporary hyperdopaminergic state in basal ganglia. Other theories implicate decrease of γ-aminobutyric acid (GABA) in the globus pallidus (GP) and substantia nigra (SN) regions of the brain, and oxidative damage to GABAergic interneurons in GP and SN from excess production of catecholamines in response to chronic dopamine blockade.14
It has been proposed that patients with withdrawal dyskinesia might be in an early phase of D2 receptor modulation that, if continued because of use of the antipsychotic implicated in withdrawal dyskinesia, can lead to development of TD.4,7,8 A feature of withdrawal dyskinesia that differentiates it from TD is that it usually remits spontaneously within several weeks to a few months.4,7 Because of this characteristic, Schultz et al8 propose that, if withdrawal dyskinesia is identified early in treatment, it may be possible to prevent development of persistent TD.
Look carefully for dyskinetic movements in patients who have recently discontinued or decreased the dosage of their antipsychotic. Non-compliance and partial compliance are common problems among patients taking an antipsychotic.15 Therefore, careful watchfulness for withdrawal dyskinesias at all times can be beneficial. Inquiring about recent history of these dyskinesias in such patients is probably more useful than an exam because the dyskinesias may not be evident on exam when these patients show up for their follow-up visit, because of their self-limited nature.8
Treatment options
If a patient is noted to have a withdrawal-emergent dyskinesia, a clinician has options to prevent TD, including:
• decreasing the dosage of the antipsychotic
• switching from a typical antipsychotic to an atypical antipsychotic
• switching from one atypical to another with lesser affinity for striatal D2 receptor, such as clozapine or quetiapine.16,17
In addition, researchers are investigating the use of vitamin B6, Ginkgo biloba, amantadine, levetiracetam, melatonin, tetrabenazine, zonisamide, branched chain amino acids, clonazepam, and vitamin E as treatment alternatives for TD.
Tetrabenazine acts by blocking vesicular monoamine transporter type 2, thereby inhibiting release of monoamines, including dopamine into synaptic cleft area in basal ganglia.18 Clonazepam’s benefit for TD relates to its facilitation of GABAergic neurotransmission, because reduced GABAergic transmission in GP and SN has been associ ated with hyperkinetic movements, including TD.14Ginkgo biloba and melatonin exert their beneficial effects in TD through their antioxidant function.14
The agents listed in Table 219 could be used on a short-term basis for symptomatic treatment of withdrawal dyskinesias.1,18,20
Withdrawal dyskinesia has been reported with aripiprazole discontinuation and is thought to be related to aripiprazole’s strong affinity for D2 receptors.21 Aripiprazole at dosages of 15 to 30 mg/d can occupy more than 80% of the striatal D2 dopamine receptors. The dosage of ≥30 mg/d can lead to receptor occupancy of >90%.22 Studies have shown that EPS correlate with D2 receptor occupancy in steady-state conditions, and occupancy exceeding 80% results in these symptoms.22
Compared with aripiprazole, quetiapine has weak affinity for D2 receptors (Table 3), making it an unlikely culprit if dyskinesia emerges within 2 weeks of initation.22 We believe that, in Ms. X’s case, quetiapine might have masked the severity of aripiprazole withdrawal dyskinesia by causing some degree of D2 receptor blockade. It may have decreased the duration of withdrawal dyskinesia by the same effect on D2 receptors. It may have lasted longer if aripiprazole was not replaced by another antipsychotic. This is particularly evident because dyskinesia improved quickly when quetiapine was titrated to 150 mg/d. The higher quetiapine dosage of 150 mg/d is closer to 5 mg/d of aripiprazole in terms of D2 receptor occupancy and affinity. However, quetiapine is weaker than aripiprazole in terms of D2 receptor occupancy at all dosages, and therefore less likely to cause EPS.16
Summing up
Withdrawal dyskinesia in the absence of a history of TD is a common symptom of antipsychotic discontinuation or dosage reduction after long-term use of an antipsychotic. It is more commonly seen with antipsychotics with high D2 receptor occupancy, and has been hypothesized to be related to D2 receptor supersensitivity to ambient dopamine, resulting as a compensatory response to chronic D2 blockade by this class of medication.
Evidence suggests that reversible withdrawal dyskinesia could represent a prodrome to irreversible TD. Therefore, keeping a watchful eye for these movements during the exam, along with specific inquiry about withdrawal dyskinesias while taking a history at every follow-up visit, is important because doing so can:
• inform the clinician about partial compliance or noncompliance to these medications, which could lead to treatment failure
• help prevent development of irreversible TD syndrome.
Ms. X’s case reminds clinicians (1) to be aware of this unexpected side effect occurring even with second-generation antipsychotics and (2) that they should consider EPS in patients while they are discontinuing their drugs. Furthermore, it is important for clinical and medicolegal reasons to inform our patients that different forms of dyskinesias can be potential side effects of antipsychotics.
Bottom Line
Dyskinesias can result from withdrawal of both typical and atypical antipsychotics, and usually are self-limited. Withdrawal dyskinesia may represent a prodrome to tardive dyskinesia; early recognition may aid in preventing development of persistent tardive dyskinesia.
Related Resources
• Abnormal Involuntary Movement Scale. http://www.cqaimh.org/pdf/toolaims.pdf.
• Goldberg JF, Ernst CL. Managing the side effects of psychotropic medications. Arlington, VA: American Psychiatric Publishing, Inc; 2012.
• Tarsay D. Tardive dyskinesia: prevention and treatment. http:// www.uptodate.com/contents/tardive-dyskinesia-prevention-and-treatment?topicKey=NEURO%2F4908&elapsedTimeMs=3 &view=print&displayedView=full#.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Benztropine • Cogentin
Carvedilol • Coreg
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Divalproex sodium • Depakote
Donepezil • Aricept
Enalapril • Vasotec
Haloperidol • Haldol
Lamotrigine • Lamictal
Levetiracetam • Keppra
Levothyroxine • Levoxyl, Synthroid
Metformin • Glucophage
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Omeprazole • Prilosec
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trazodone • Desyrel, Oleptro
Ziprasidone • Geodon
Zonisamide • Zonegran
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
1. Bhidayasiri R1, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
2. Dolder CR, Jeste DV. Incidence of tardive dyskinesia with typical versus atypical antipsychotics in very high risk patients. Biol Psychiatry. 2003;53(12):1142-1145.
3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
4. Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135(11):1321-1324.
5. Salomon C, Hamilton B. Antipsychotic discontinuation syndromes: a narrative review of the evidence and its integration into Australian mental health nursing textbooks. Int J Ment Health Nurs. 2014;23(1):69-78.
6. Moseley CN, Simpson-Khanna HA, Catalano G, et al. Covert dyskinesia associated with aripiprazole: a case report and review of the literature. Clin Neuropharmacol. 2013;36(4):128-130.
7. Anand VS, Dewan MJ. Withdrawal-emergent dyskinesia in a patient on risperidone undergoing dosage reduction. Ann Clin Psychiatry. 1996;8(3):179-182.
8. Schultz SK, Miller DD, Arndt S, et al. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry. 1995;38(11):713-719.
9. Degkwitz R, Bauer MP, Gruber M, et al. Time relationship between the appearance of persisting extrapyramidal hyperkineses and psychotic recurrences following sudden interruption of prolonged neuroleptic therapy of chronic schizophrenic patients [in German]. Arzneimittelforschung. 1970;20(7):890-893.
10. Sethi KD. Tardive dyskinesias. In: Adler CH, Ahlskog JE, eds. Parkinson’s disease and movement disorders: diagnosis and treatment guidelines for the practicing physician. New York, NY: Humana Press; 2000:331-338.
11. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
12. Horváth K, Aschermann Z, Komoly S, et al. Treatment of tardive syndromes [in Hungarian]. Psychiatr Hung. 2014;29(2):214-224.
13. Samaha AN, Seeman P, Stewart J, et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci. 2007;27(11):2979-2986.
14. Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9(9):1285-1306.
15. Keith SJ, Kane JM. Partial compliance and patient consequences in schizophrenia: our patients can do better. J Clin Psychiatry. 2003;64(11):1308-1315.
16. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
17. Farah A. Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry. 2005;7(6):268-274.
18. Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther. 2013;7:1329-1340.
19. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
20. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176.
21. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
22. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2-receptor occupancy. Eur Psychiatry. 2007;22(5):267-275.
Head pain and psychiatric illness: Applying the biopsychosocial model to care
More than 45% of people worldwide suffer from headache at some point in their life.1 Head pain can lead to disability and functional decline, yet headache disorders often are underdiagnosed and poorly assessed. For example, 60% of migraine and tension-type headaches go undiagnosed and 50% of persons suffering from migraine have severe functional disability or require bed rest.2-4
Because head pain can be associated with secondary medical and psychiatric conditions, diagnosis can be challenging. This article reviews the medical and psychological aspects of major headaches and assists with clinical assessment. We present clinical interviewing tools and a diagram to enhance focused, efficient assessment and inform treatment plans.
Classification of headache
Headache is a common complaint, yet it is often underdiagnosed and ineffectively treated. The World Health Organization estimates that, globally, 50% of people with headache self-treat their pain.5 The International Headache Society classifies headache as primary or secondary; approximately 90% of complaints are from primary headache.6
Assessment and diagnosis of headache can be complex because of overlapping, subjective symptoms. It is important to have a general understanding of primary and secondary causes of headache so that interrelated symptoms do not obscure the most accurate diagnosis and effective treatment course. Although most headache complaints are benign, ruling out secondary causes helps gauge the likelihood of developing severe sequelae from underlying pathology.
By definition, primary headaches are idiopathic and commonly include migraine, tension-type, cluster, and hemicrania continua headache. Secondary headaches have an underlying pathology, which could improve by targeting the disorder. Common secondary causes of headache include:
• trauma
• vascular abnormalities
• structural abnormalities
• chemical (including medications)
• inflammation or infection
• metabolic conditions
• diseases of the neck and pericranial and intracranial structures
• psychiatric conditions.
Table 1 illustrates common causes of head pain. More definitive criteria for symptoms and diagnosis can be found in the International Classification of Headache Disorders.7

Primary headache
Tension-type is the most common primary headache, accounting for more than one-half of all headaches.7 Patients usually describe a tight pain in a bilateral band-like distribution, which could be caused by sustained neck muscle contraction. Pain usually builds in intensity and can last 30 minutes to several days. There is a well-established association between emotional stress or depression and the development of tension-type headaches.8
Migraine typically causes pulsating pain in a localized area of the head that lasts as long as 72 hours and can be associated with nausea, vomiting, photophobia, phono-phobia, and aura. Patients report varying precipitating factors but commonly cite certain foods, menstruation, and sleep deprivation. Although rare, migraine with aura has been linked to ischemic stroke; most cases have been reported in female smokers and oral contraceptive users age <45.9
Because migraines can be debilitating, some patients—typically those with ≥4 attacks a month—opt for prophylactic medication. Effective prophylactics include amitriptyline, propranolol, divalproex sodium, and topiramate, which should be monitored closely and given a trial for several months before switching to another drug. Commonly used abortive treatments include triptans and anti-emetics such as metoclopramide.
Meperidine and ketorolac are popular second-line agents for migraine. Botulinum toxin A also has been used in severe cases to reduce the number of headache days in chronic migraine patients.6
Cluster headache is rare, but typically exhibits repeated burning and intense unilateral periorbital or retro-orbital pain that lasts 15 minutes to 3 hours over several weeks. Men are predominantly affected. Cluster headaches typically improve with oxygen treatment.
Biopsychosocial model of head pain
The biomedical model has helped iden tify pathophysiological pain mechanisms and pharmacotherapeutic agents for headache. However, during assessment, limiting one’s attention to the linear relationship between pathology, mechanism of action, and pain oversimplifies common questions clinicians face when assessing chronic head pain.
Advancements in the last 3 decades have expanded the conceptualization of head pain to integrate sociocultural, environmental, behavioral, affective, cognitive, and biological variables—otherwise known as the biopsychosocial model.10,11 The biopsychosocial model is a multidimensional theory that helps answer difficult clinical assessment questions and complex patient presentations (Table 2).10-13 Many unusual responses to pain treatment, questionable validity of pain behavior, and disproportionate pain perception and functional decline are explained by non-pathophysiological and non-biomechanical models.

Psychiatric comorbidity and head pain
Psychiatric conditions are highly prevalent among persons with primary headache. Verri et al14 found that 90% of chronic daily headache patients had ≥1 psychiatric condition; depression and anxiety were most common. Of concern, 1 study found that headache is associated with increased frequency of suicidal ideation among patients with chronic pain.15 It is critical for clinicians to screen for psychiatric comorbidities in patients with chronic headache. Conversely, clinicians might want to screen for headache in their patients with psychiatric illness.
Migraine. Mood disorders are common among patients who suffer from migraine. The rate of depression is 2 to 4 times higher in those with migraine compared with healthy controls.16,17 In a large-scale study, patients with migraine had a 1.9-fold higher risk (compared with controls) of having a comorbid depressive episode; a 2-fold higher risk of manic episodes; and a 3-fold higher risk of both mania and depression.18 In a study of 62 inpatients, Fasmer19 reported that 46% of patients with unipolar depression and 44% of patients with bipolar disorder experienced migraine (77% of the bipolar disorder patients with migraine had bipolar II disorder). Patients with migraine are at increased risk of suicide attempts (odds ratio 4.3; 95% CI, 1.2-15.7).20
Tension-type headache. The relationship between psychiatric comorbidity in tension-type headache is well established. In contrast to what is seen with migraines, Puca et al21 found a higher prevalence of anxiety disorders (52.5%) than depressive disorders (36.4%) in patients with tension-type headache. Generalized anxiety disorder was one of the most prevalent anxiety conditions (83.3%), and dysthymia was the most prevalent mood disorder (45.6%). In the same study, 21.7% of patients were found to have a comorbid somatoform disorder.21
Emotional and cognitive factors can co-occur in patients with tension-type headache and a comorbid psychiatric condition. For example, difficulty identifying or recognizing emotions—commonly referred to as alexithymia—has been linked to tension-type headache.22 Additionally, maladaptive cognitive appraisal of stress is more common among patients with tension-type headache when compared with those without headaches.23 Being mindful of and recognizing these co-occurring emotional and cognitive factors will help clinicians construct a more accurate assessment and effective behavioral treatment plan.
Clinical assessment with a useful mnemonic
Clinical assessment of psychiatric illness is essential when evaluating chronic pain patients. Using the acronym AMPS (Anxiety, Mood, Psychosis, and Substance use disorders) (Table 3) is an efficient way for the clinician to ask pertinent questions regarding common psychiatric conditions that could have a direct effect on chronic pain.24 Head pain can be more intense when combined with untreated anxiety, depression, psychosis, or a substance use disorder. Untreated anxiety, for example, can amplify sympathetic response to pain and complicate treatment.

Investigating head pain patients for an underlying mood disorder is essential to providing successful treatment. Consider:
• starting psychotherapy modalities that address both pain and psychiatric illness, such as cognitive-behavioral therapy (CBT)
• reframing unhelpful pain beliefs
• managing activity-rest levels
• biofeedback
• supportive group therapy
• reducing family members’ reinforcement of the patient’s pain behavior or sick role.25
Assessing for somatic symptom disorders
In addition to using the AMPS approach for psychiatric assessment, clinicians should evaluate for somatization, which can present as head pain. Somatic symptom disorders (SSD) are a class of conditions that are impacted by affective, cognitive, and reinforcing factors that might or might not be consciously or intentionally produced. Patients with an SSD have somatic symptoms that are distressing or cause significant disruption of daily life because of excessive thoughts, feelings, or behaviors related to the somatic symptoms, for ≥6 months. The Figure outlines SSD, related conditions, and their respective prominent symptoms to assist in the differential diagnosis.26
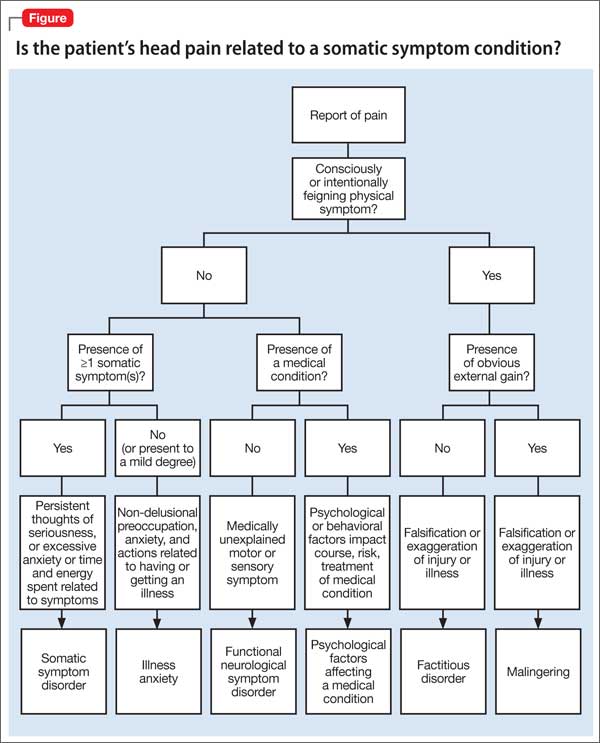
Note that some headache conditions present with severe distress because of their abrupt onset and severity of symptoms (eg, cluster headaches). Therefore, the expectation and likelihood of psychological disturbance should be factored into a diagnosis of SSD and related conditions as seen in the Figure.
Secondary factors of unusual pain behavior or treatment response. The role of thoughts, affect, and behaviors is clinically meaningful in understanding SSD and similar conditions. Specific questions about cultural beliefs and rituals as they relate to exacerbations of head pain are of value. Table 413,27 lists behavioral, cognitive, and affective dimensions of head pain using the biopsychosocial model, and further clarifies common questions that arise with unusual pain response and complex patient presentations, which were outlined in the beginning of the article.
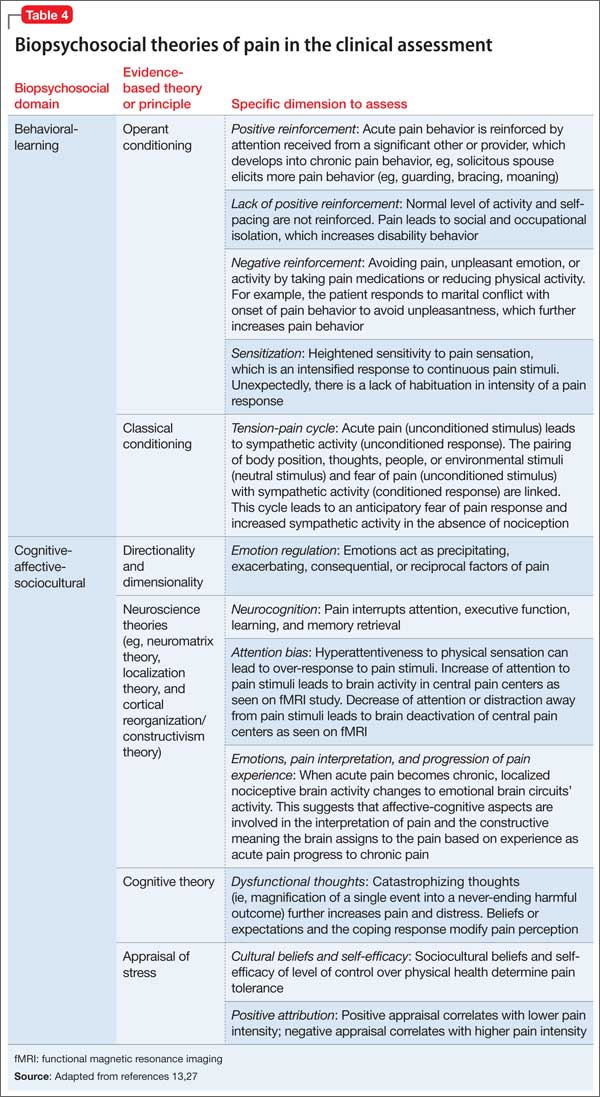
Because depression and anxiety can be comorbid with head pain, it is important to recognize psychological factors that contribute to pain perception. Indifference or denial of emotional stress as a result of severe pain and disability can imply a somatization process, which could suggest emotional disconnection or dissociation from somatic functioning.28 This finding can be a component of alexithymia, in which a person is disconnected from emotions and how emotions impact the body. Therefore, recognizing alexithymia assists in identifying psychological factors when patients deny mood symptoms, particularly in tension-type headache.
Functional assessment to rule out the disproportional impact of pain on daily activities is helpful in understanding the somatization process. Neurocognitive functioning should be assessed, particularly because frontal and subcortical dysregulation has been observed in head pain sufferers.29,30 Patients with cognitive changes as a result of a medical illness (eg, stroke, head concussion, brain tumor, or seizures) are especially at risk for neurocognitive dysfunction.
Neuropsychological assessment can be useful, not only to assess neurocognitive functioning (eg, Repeated Battery for the Assessment of Neuropsychological Status) but to identify objective test profiles associated with altered motivation (eg, Rey 15-Item Test, Minnesota Multiphasic Personality Inventory-2-Restructured Form F Scale, Personality Assessment Inventory [PAI] Negative Impression Management) and somatization processes (eg, PAI Somatization Scale). These instruments help to identify the severity of psychiatric and neurocognitive symptoms by comparing scores to normative (eg, healthy control group), clinical (eg, somatization, traumatic brain injury, mild cognitive impairment), and altered motivation (eg, persons instructed to exaggerate symptoms) databases.
If the clinician pursues neurocognitive assessment, direct referral to a neuropsychologist, referral to neurologist, or administration of a cognitive screening tool such as the Montreal Cognitive Assessment, Saint Louis University Mental Status, or Cognitive Log is recommended. If the cognitive screening is positive, next steps include: referring for full neuropsychological assessment, which includes complete cognitive and motor testing, personality testing, and integration of neuroimaging data (eg, MRI, CT scans, and/or EEG).
Assessing the patients’ self-talk or thought patterns as they describe their head pain will help clinicians understand belief systems that may be distorting the reality of the medical condition. For example, a patient might report that “my pain feels like someone is hitting me with an axe”; this is a catastrophic thought that can distort the clarity and perceptibility of pain. Encouraging patients to monitor and analyze their anxiety and associated negative thoughts is an important strategy for improving mood and decreasing somatization. Recording daily thoughts and CBT can help the patient identify and appropriately address his (her) cognitive distortions and futile thinking.
When implementing a treatment plan for somatization disorder, we propose the mnemonic device CARE MD:
• CBT
• Assess (by ruling out a medical cause for somatic complaints)
• Regular visits
• Empathy
• Med-psych interface (help the patient connect physical complaints and emotional stressors)
• Do no harm.
Clinical recommendations
Chronic head pain can be debilitating; psychodiagnostic assessment should therefore be considered an important part of the diagnosis and treatment plan. After ruling out common and emergent primary or secondary causes of head pain, consider psychiatric comorbidities. Depression and anxiety have a strong bidirectional relationship with chronic headache; therefore, we suggest evaluating patients with the intention of alleviating both psychiatric symptoms and head pain.
It is important to diligently assess for common psychiatric comorbidities; using the AMPS and CARE MD mnemonics, along with screening for somatization disorders, is an easy and effective way to evaluate for relevant psychiatric conditions associated with chronic head pain. Because many patients have unusual and complicated responses to head pain that can be explained by non-pathophysiological and non-biomechanical models, using the biopsychosocial model is essential for effective diagnosis, assessment, and treatment. Abortive and prophylactic medical interventions, as well as behavioral, sociocultural, and cognitive assessment, are vital to a comprehensive treatment approach.
Bottom Line
The psychodiagnostic assessment can help the astute clinician identify comorbid psychiatric conditions, psychological factors, and somatic symptoms to develop a comprehensive biopsychosocial treatment plan for patients with chronic head pain. Rule out primary and secondary causes of pain and screen for somatization disorders. Consider medication and psychotherapeutic treatment options.
Related Resources
• Pompili M, Di Cosimo D, Innamorati M, et al. Psychiatric comorbidity in patients with chronic daily headache and migraine: a selective overview including personality traits and suicide risk. J Headache Pain. 2009;10(4):283-290.
• Sinclair AJ, Sturrock A, Davies B, et al. Headache management: pharmacological approaches [published online July 3, 2015]. Pract Neurol. doi: 10.1136/practneurol-2015-001167.
Drug Brand Names
Amitriptyline • Elavil Meperidine • Demerol
Botulinum toxin A • Botox Metoclopramide • Reglan
Divalproex sodium • Depakote Propranolol • Inderide
Ketorolac • Toradol Topiramate • Topamax
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stovner LJ, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646-657.
3. The World Health Report 2001: Mental health: new understanding new hope. Geneva, Switzerland: World Health Organization; 2001.
4. World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_ disease/GBD_report_2004update_full.pdf. Published 2004. Accessed July 31, 2015.
5. World Health Organization. Headache disorders. http:// www.who.int/mediacentre/factsheets/fs277/en/. Published October 2012. Accessed July 31, 2015.
6. Clinch C. Evaluation & management of headache. In: South- Paul JE, Matheny SC, Lewis EL. eds. Current diagnosis & treatment in family medicine, 4th ed. New York, NY: McGraw-Hill; 2015:293-297.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
8. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia. 2015;35(2):165-181.
9. Janke EA, Holroyd KA, Romanek K. Depression increases onset of tension-type headache following laboratory stress. Pain. 2004;111(3):230-238.
10. Engel GL. The need for a new medical model: a challenge for biomedicine. Science. 1977;196(4286):129-136.
11. Engel GL. The clinical application of the biopsychosocial model. Am J Psychiatry. 1980;137(5):535-544.
12. Turk DC, Flor H. Chronic pain: a biobehavioral perspective. In: Gatchel RJ, Turk DC, eds. Psychosocial factors in pain: critical perspectives. New York, NY: Guilford Press; 1999:18-34.
13. Andrasik F, Flor H, Turk DC. An expanded view of psychological aspects in head pain: the biopsychosocial model. Neurol Sci. 2005;26(suppl 2):s87-s91.
14. Verri AP, Proietti Cecchini A, Galli C, et al. Psychiatric comorbidity in chronic daily headache. Cephalalgia. 1998; 18(suppl 21):45-49.
15. Ilgen MA, Zivin K, McCammon RJ, et al. Pain and suicidal thoughts, plans and attempts in the United States. Gen Hosp Psychiatry. 2008;30(6):521-527.
16. Kowacs F, Socal MP, Ziomkowski SC, et al. Symptoms of depression and anxiety, and screening for mental disorders in migrainous patients. Cephalalgia. 2003;23(2):79-89.
17. Hamelsky SW, Lipton RB. Psychiatric comorbidity of migraine. Headache. 2006;46(9):1327-1333.
18. Nguyen TV, Low NC. Comorbidity of migraine and mood episodes in a nationally representative population-based sample. Headache. 2013;53(3):498-506.
19. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001; 21(9):894-899.
20. Breslau N. Migraine, suicidal ideation, and suicide attempts. Neurology. 1992;42(2):392-395.
21. Puca F, Genco S, Prudenzano MP, et al. Psychiatric comorbidity and psychosocial stress in patients with tension-type headache from headache centers in Italy. Cephalalgia. 1999;19(3):159-164.
22. Yücel B, Kora K, Ozyalçín S, et al. Depression, automatic thoughts, alexithymia, and assertiveness in patients with tension-type headache. Headache. 2002;42(3):194-199.
23. Wittrock DA, Myers TC. The comparison of individuals with recurrent tension-type headache and headache-free controls in physiological response, appraisal, and coping with stressors: a review of the literature. Ann Behav Med. 1998;20(2):118-134.
24. Onate J, Xiong G, McCarron R. The primary care psychiatric interview. In: McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott, Williams and Wilkins; 2009:3-4.
25. Songer D. Psychotherapeutic approaches in the treatment of pain. Psychiatry (Edgmont). 2005;2(5):19-24.
26. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
27. Hashmi JA, Baliki MN, Huang L, et al. Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain. 2013;136(pt 9):2751-2768.
28. Packard RC. Conversion headache. Headache. 1980;20(5):266-268.
29. Mongini F, Keller R, Deregibus A, et al. Frontal lobe dysfunction in patients with chronic migraine: a clinical-neuropsychological study. Psychiatry Res. 2005;133(1):101-106.
30. Martelli MF, Grayson RL, Zasler ND. Posttraumatic headache: neuropsychological and psychological effects and treatment implications. J Head Trauma Rehabil. 1999;14(1):49-69.
More than 45% of people worldwide suffer from headache at some point in their life.1 Head pain can lead to disability and functional decline, yet headache disorders often are underdiagnosed and poorly assessed. For example, 60% of migraine and tension-type headaches go undiagnosed and 50% of persons suffering from migraine have severe functional disability or require bed rest.2-4
Because head pain can be associated with secondary medical and psychiatric conditions, diagnosis can be challenging. This article reviews the medical and psychological aspects of major headaches and assists with clinical assessment. We present clinical interviewing tools and a diagram to enhance focused, efficient assessment and inform treatment plans.
Classification of headache
Headache is a common complaint, yet it is often underdiagnosed and ineffectively treated. The World Health Organization estimates that, globally, 50% of people with headache self-treat their pain.5 The International Headache Society classifies headache as primary or secondary; approximately 90% of complaints are from primary headache.6
Assessment and diagnosis of headache can be complex because of overlapping, subjective symptoms. It is important to have a general understanding of primary and secondary causes of headache so that interrelated symptoms do not obscure the most accurate diagnosis and effective treatment course. Although most headache complaints are benign, ruling out secondary causes helps gauge the likelihood of developing severe sequelae from underlying pathology.
By definition, primary headaches are idiopathic and commonly include migraine, tension-type, cluster, and hemicrania continua headache. Secondary headaches have an underlying pathology, which could improve by targeting the disorder. Common secondary causes of headache include:
• trauma
• vascular abnormalities
• structural abnormalities
• chemical (including medications)
• inflammation or infection
• metabolic conditions
• diseases of the neck and pericranial and intracranial structures
• psychiatric conditions.
Table 1 illustrates common causes of head pain. More definitive criteria for symptoms and diagnosis can be found in the International Classification of Headache Disorders.7
Primary headache
Tension-type is the most common primary headache, accounting for more than one-half of all headaches.7 Patients usually describe a tight pain in a bilateral band-like distribution, which could be caused by sustained neck muscle contraction. Pain usually builds in intensity and can last 30 minutes to several days. There is a well-established association between emotional stress or depression and the development of tension-type headaches.8
Migraine typically causes pulsating pain in a localized area of the head that lasts as long as 72 hours and can be associated with nausea, vomiting, photophobia, phono-phobia, and aura. Patients report varying precipitating factors but commonly cite certain foods, menstruation, and sleep deprivation. Although rare, migraine with aura has been linked to ischemic stroke; most cases have been reported in female smokers and oral contraceptive users age <45.9
Because migraines can be debilitating, some patients—typically those with ≥4 attacks a month—opt for prophylactic medication. Effective prophylactics include amitriptyline, propranolol, divalproex sodium, and topiramate, which should be monitored closely and given a trial for several months before switching to another drug. Commonly used abortive treatments include triptans and anti-emetics such as metoclopramide.
Meperidine and ketorolac are popular second-line agents for migraine. Botulinum toxin A also has been used in severe cases to reduce the number of headache days in chronic migraine patients.6
Cluster headache is rare, but typically exhibits repeated burning and intense unilateral periorbital or retro-orbital pain that lasts 15 minutes to 3 hours over several weeks. Men are predominantly affected. Cluster headaches typically improve with oxygen treatment.
Biopsychosocial model of head pain
The biomedical model has helped iden tify pathophysiological pain mechanisms and pharmacotherapeutic agents for headache. However, during assessment, limiting one’s attention to the linear relationship between pathology, mechanism of action, and pain oversimplifies common questions clinicians face when assessing chronic head pain.
Advancements in the last 3 decades have expanded the conceptualization of head pain to integrate sociocultural, environmental, behavioral, affective, cognitive, and biological variables—otherwise known as the biopsychosocial model.10,11 The biopsychosocial model is a multidimensional theory that helps answer difficult clinical assessment questions and complex patient presentations (Table 2).10-13 Many unusual responses to pain treatment, questionable validity of pain behavior, and disproportionate pain perception and functional decline are explained by non-pathophysiological and non-biomechanical models.
Psychiatric comorbidity and head pain
Psychiatric conditions are highly prevalent among persons with primary headache. Verri et al14 found that 90% of chronic daily headache patients had ≥1 psychiatric condition; depression and anxiety were most common. Of concern, 1 study found that headache is associated with increased frequency of suicidal ideation among patients with chronic pain.15 It is critical for clinicians to screen for psychiatric comorbidities in patients with chronic headache. Conversely, clinicians might want to screen for headache in their patients with psychiatric illness.
Migraine. Mood disorders are common among patients who suffer from migraine. The rate of depression is 2 to 4 times higher in those with migraine compared with healthy controls.16,17 In a large-scale study, patients with migraine had a 1.9-fold higher risk (compared with controls) of having a comorbid depressive episode; a 2-fold higher risk of manic episodes; and a 3-fold higher risk of both mania and depression.18 In a study of 62 inpatients, Fasmer19 reported that 46% of patients with unipolar depression and 44% of patients with bipolar disorder experienced migraine (77% of the bipolar disorder patients with migraine had bipolar II disorder). Patients with migraine are at increased risk of suicide attempts (odds ratio 4.3; 95% CI, 1.2-15.7).20
Tension-type headache. The relationship between psychiatric comorbidity in tension-type headache is well established. In contrast to what is seen with migraines, Puca et al21 found a higher prevalence of anxiety disorders (52.5%) than depressive disorders (36.4%) in patients with tension-type headache. Generalized anxiety disorder was one of the most prevalent anxiety conditions (83.3%), and dysthymia was the most prevalent mood disorder (45.6%). In the same study, 21.7% of patients were found to have a comorbid somatoform disorder.21
Emotional and cognitive factors can co-occur in patients with tension-type headache and a comorbid psychiatric condition. For example, difficulty identifying or recognizing emotions—commonly referred to as alexithymia—has been linked to tension-type headache.22 Additionally, maladaptive cognitive appraisal of stress is more common among patients with tension-type headache when compared with those without headaches.23 Being mindful of and recognizing these co-occurring emotional and cognitive factors will help clinicians construct a more accurate assessment and effective behavioral treatment plan.
Clinical assessment with a useful mnemonic
Clinical assessment of psychiatric illness is essential when evaluating chronic pain patients. Using the acronym AMPS (Anxiety, Mood, Psychosis, and Substance use disorders) (Table 3) is an efficient way for the clinician to ask pertinent questions regarding common psychiatric conditions that could have a direct effect on chronic pain.24 Head pain can be more intense when combined with untreated anxiety, depression, psychosis, or a substance use disorder. Untreated anxiety, for example, can amplify sympathetic response to pain and complicate treatment.
Investigating head pain patients for an underlying mood disorder is essential to providing successful treatment. Consider:
• starting psychotherapy modalities that address both pain and psychiatric illness, such as cognitive-behavioral therapy (CBT)
• reframing unhelpful pain beliefs
• managing activity-rest levels
• biofeedback
• supportive group therapy
• reducing family members’ reinforcement of the patient’s pain behavior or sick role.25
Assessing for somatic symptom disorders
In addition to using the AMPS approach for psychiatric assessment, clinicians should evaluate for somatization, which can present as head pain. Somatic symptom disorders (SSD) are a class of conditions that are impacted by affective, cognitive, and reinforcing factors that might or might not be consciously or intentionally produced. Patients with an SSD have somatic symptoms that are distressing or cause significant disruption of daily life because of excessive thoughts, feelings, or behaviors related to the somatic symptoms, for ≥6 months. The Figure outlines SSD, related conditions, and their respective prominent symptoms to assist in the differential diagnosis.26
Note that some headache conditions present with severe distress because of their abrupt onset and severity of symptoms (eg, cluster headaches). Therefore, the expectation and likelihood of psychological disturbance should be factored into a diagnosis of SSD and related conditions as seen in the Figure.
Secondary factors of unusual pain behavior or treatment response. The role of thoughts, affect, and behaviors is clinically meaningful in understanding SSD and similar conditions. Specific questions about cultural beliefs and rituals as they relate to exacerbations of head pain are of value. Table 413,27 lists behavioral, cognitive, and affective dimensions of head pain using the biopsychosocial model, and further clarifies common questions that arise with unusual pain response and complex patient presentations, which were outlined in the beginning of the article.
Because depression and anxiety can be comorbid with head pain, it is important to recognize psychological factors that contribute to pain perception. Indifference or denial of emotional stress as a result of severe pain and disability can imply a somatization process, which could suggest emotional disconnection or dissociation from somatic functioning.28 This finding can be a component of alexithymia, in which a person is disconnected from emotions and how emotions impact the body. Therefore, recognizing alexithymia assists in identifying psychological factors when patients deny mood symptoms, particularly in tension-type headache.
Functional assessment to rule out the disproportional impact of pain on daily activities is helpful in understanding the somatization process. Neurocognitive functioning should be assessed, particularly because frontal and subcortical dysregulation has been observed in head pain sufferers.29,30 Patients with cognitive changes as a result of a medical illness (eg, stroke, head concussion, brain tumor, or seizures) are especially at risk for neurocognitive dysfunction.
Neuropsychological assessment can be useful, not only to assess neurocognitive functioning (eg, Repeated Battery for the Assessment of Neuropsychological Status) but to identify objective test profiles associated with altered motivation (eg, Rey 15-Item Test, Minnesota Multiphasic Personality Inventory-2-Restructured Form F Scale, Personality Assessment Inventory [PAI] Negative Impression Management) and somatization processes (eg, PAI Somatization Scale). These instruments help to identify the severity of psychiatric and neurocognitive symptoms by comparing scores to normative (eg, healthy control group), clinical (eg, somatization, traumatic brain injury, mild cognitive impairment), and altered motivation (eg, persons instructed to exaggerate symptoms) databases.
If the clinician pursues neurocognitive assessment, direct referral to a neuropsychologist, referral to neurologist, or administration of a cognitive screening tool such as the Montreal Cognitive Assessment, Saint Louis University Mental Status, or Cognitive Log is recommended. If the cognitive screening is positive, next steps include: referring for full neuropsychological assessment, which includes complete cognitive and motor testing, personality testing, and integration of neuroimaging data (eg, MRI, CT scans, and/or EEG).
Assessing the patients’ self-talk or thought patterns as they describe their head pain will help clinicians understand belief systems that may be distorting the reality of the medical condition. For example, a patient might report that “my pain feels like someone is hitting me with an axe”; this is a catastrophic thought that can distort the clarity and perceptibility of pain. Encouraging patients to monitor and analyze their anxiety and associated negative thoughts is an important strategy for improving mood and decreasing somatization. Recording daily thoughts and CBT can help the patient identify and appropriately address his (her) cognitive distortions and futile thinking.
When implementing a treatment plan for somatization disorder, we propose the mnemonic device CARE MD:
• CBT
• Assess (by ruling out a medical cause for somatic complaints)
• Regular visits
• Empathy
• Med-psych interface (help the patient connect physical complaints and emotional stressors)
• Do no harm.
Clinical recommendations
Chronic head pain can be debilitating; psychodiagnostic assessment should therefore be considered an important part of the diagnosis and treatment plan. After ruling out common and emergent primary or secondary causes of head pain, consider psychiatric comorbidities. Depression and anxiety have a strong bidirectional relationship with chronic headache; therefore, we suggest evaluating patients with the intention of alleviating both psychiatric symptoms and head pain.
It is important to diligently assess for common psychiatric comorbidities; using the AMPS and CARE MD mnemonics, along with screening for somatization disorders, is an easy and effective way to evaluate for relevant psychiatric conditions associated with chronic head pain. Because many patients have unusual and complicated responses to head pain that can be explained by non-pathophysiological and non-biomechanical models, using the biopsychosocial model is essential for effective diagnosis, assessment, and treatment. Abortive and prophylactic medical interventions, as well as behavioral, sociocultural, and cognitive assessment, are vital to a comprehensive treatment approach.
Bottom Line
The psychodiagnostic assessment can help the astute clinician identify comorbid psychiatric conditions, psychological factors, and somatic symptoms to develop a comprehensive biopsychosocial treatment plan for patients with chronic head pain. Rule out primary and secondary causes of pain and screen for somatization disorders. Consider medication and psychotherapeutic treatment options.
Related Resources
• Pompili M, Di Cosimo D, Innamorati M, et al. Psychiatric comorbidity in patients with chronic daily headache and migraine: a selective overview including personality traits and suicide risk. J Headache Pain. 2009;10(4):283-290.
• Sinclair AJ, Sturrock A, Davies B, et al. Headache management: pharmacological approaches [published online July 3, 2015]. Pract Neurol. doi: 10.1136/practneurol-2015-001167.
Drug Brand Names
Amitriptyline • Elavil Meperidine • Demerol
Botulinum toxin A • Botox Metoclopramide • Reglan
Divalproex sodium • Depakote Propranolol • Inderide
Ketorolac • Toradol Topiramate • Topamax
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
More than 45% of people worldwide suffer from headache at some point in their life.1 Head pain can lead to disability and functional decline, yet headache disorders often are underdiagnosed and poorly assessed. For example, 60% of migraine and tension-type headaches go undiagnosed and 50% of persons suffering from migraine have severe functional disability or require bed rest.2-4
Because head pain can be associated with secondary medical and psychiatric conditions, diagnosis can be challenging. This article reviews the medical and psychological aspects of major headaches and assists with clinical assessment. We present clinical interviewing tools and a diagram to enhance focused, efficient assessment and inform treatment plans.
Classification of headache
Headache is a common complaint, yet it is often underdiagnosed and ineffectively treated. The World Health Organization estimates that, globally, 50% of people with headache self-treat their pain.5 The International Headache Society classifies headache as primary or secondary; approximately 90% of complaints are from primary headache.6
Assessment and diagnosis of headache can be complex because of overlapping, subjective symptoms. It is important to have a general understanding of primary and secondary causes of headache so that interrelated symptoms do not obscure the most accurate diagnosis and effective treatment course. Although most headache complaints are benign, ruling out secondary causes helps gauge the likelihood of developing severe sequelae from underlying pathology.
By definition, primary headaches are idiopathic and commonly include migraine, tension-type, cluster, and hemicrania continua headache. Secondary headaches have an underlying pathology, which could improve by targeting the disorder. Common secondary causes of headache include:
• trauma
• vascular abnormalities
• structural abnormalities
• chemical (including medications)
• inflammation or infection
• metabolic conditions
• diseases of the neck and pericranial and intracranial structures
• psychiatric conditions.
Table 1 illustrates common causes of head pain. More definitive criteria for symptoms and diagnosis can be found in the International Classification of Headache Disorders.7
Primary headache
Tension-type is the most common primary headache, accounting for more than one-half of all headaches.7 Patients usually describe a tight pain in a bilateral band-like distribution, which could be caused by sustained neck muscle contraction. Pain usually builds in intensity and can last 30 minutes to several days. There is a well-established association between emotional stress or depression and the development of tension-type headaches.8
Migraine typically causes pulsating pain in a localized area of the head that lasts as long as 72 hours and can be associated with nausea, vomiting, photophobia, phono-phobia, and aura. Patients report varying precipitating factors but commonly cite certain foods, menstruation, and sleep deprivation. Although rare, migraine with aura has been linked to ischemic stroke; most cases have been reported in female smokers and oral contraceptive users age <45.9
Because migraines can be debilitating, some patients—typically those with ≥4 attacks a month—opt for prophylactic medication. Effective prophylactics include amitriptyline, propranolol, divalproex sodium, and topiramate, which should be monitored closely and given a trial for several months before switching to another drug. Commonly used abortive treatments include triptans and anti-emetics such as metoclopramide.
Meperidine and ketorolac are popular second-line agents for migraine. Botulinum toxin A also has been used in severe cases to reduce the number of headache days in chronic migraine patients.6
Cluster headache is rare, but typically exhibits repeated burning and intense unilateral periorbital or retro-orbital pain that lasts 15 minutes to 3 hours over several weeks. Men are predominantly affected. Cluster headaches typically improve with oxygen treatment.
Biopsychosocial model of head pain
The biomedical model has helped iden tify pathophysiological pain mechanisms and pharmacotherapeutic agents for headache. However, during assessment, limiting one’s attention to the linear relationship between pathology, mechanism of action, and pain oversimplifies common questions clinicians face when assessing chronic head pain.
Advancements in the last 3 decades have expanded the conceptualization of head pain to integrate sociocultural, environmental, behavioral, affective, cognitive, and biological variables—otherwise known as the biopsychosocial model.10,11 The biopsychosocial model is a multidimensional theory that helps answer difficult clinical assessment questions and complex patient presentations (Table 2).10-13 Many unusual responses to pain treatment, questionable validity of pain behavior, and disproportionate pain perception and functional decline are explained by non-pathophysiological and non-biomechanical models.
Psychiatric comorbidity and head pain
Psychiatric conditions are highly prevalent among persons with primary headache. Verri et al14 found that 90% of chronic daily headache patients had ≥1 psychiatric condition; depression and anxiety were most common. Of concern, 1 study found that headache is associated with increased frequency of suicidal ideation among patients with chronic pain.15 It is critical for clinicians to screen for psychiatric comorbidities in patients with chronic headache. Conversely, clinicians might want to screen for headache in their patients with psychiatric illness.
Migraine. Mood disorders are common among patients who suffer from migraine. The rate of depression is 2 to 4 times higher in those with migraine compared with healthy controls.16,17 In a large-scale study, patients with migraine had a 1.9-fold higher risk (compared with controls) of having a comorbid depressive episode; a 2-fold higher risk of manic episodes; and a 3-fold higher risk of both mania and depression.18 In a study of 62 inpatients, Fasmer19 reported that 46% of patients with unipolar depression and 44% of patients with bipolar disorder experienced migraine (77% of the bipolar disorder patients with migraine had bipolar II disorder). Patients with migraine are at increased risk of suicide attempts (odds ratio 4.3; 95% CI, 1.2-15.7).20
Tension-type headache. The relationship between psychiatric comorbidity in tension-type headache is well established. In contrast to what is seen with migraines, Puca et al21 found a higher prevalence of anxiety disorders (52.5%) than depressive disorders (36.4%) in patients with tension-type headache. Generalized anxiety disorder was one of the most prevalent anxiety conditions (83.3%), and dysthymia was the most prevalent mood disorder (45.6%). In the same study, 21.7% of patients were found to have a comorbid somatoform disorder.21
Emotional and cognitive factors can co-occur in patients with tension-type headache and a comorbid psychiatric condition. For example, difficulty identifying or recognizing emotions—commonly referred to as alexithymia—has been linked to tension-type headache.22 Additionally, maladaptive cognitive appraisal of stress is more common among patients with tension-type headache when compared with those without headaches.23 Being mindful of and recognizing these co-occurring emotional and cognitive factors will help clinicians construct a more accurate assessment and effective behavioral treatment plan.
Clinical assessment with a useful mnemonic
Clinical assessment of psychiatric illness is essential when evaluating chronic pain patients. Using the acronym AMPS (Anxiety, Mood, Psychosis, and Substance use disorders) (Table 3) is an efficient way for the clinician to ask pertinent questions regarding common psychiatric conditions that could have a direct effect on chronic pain.24 Head pain can be more intense when combined with untreated anxiety, depression, psychosis, or a substance use disorder. Untreated anxiety, for example, can amplify sympathetic response to pain and complicate treatment.
Investigating head pain patients for an underlying mood disorder is essential to providing successful treatment. Consider:
• starting psychotherapy modalities that address both pain and psychiatric illness, such as cognitive-behavioral therapy (CBT)
• reframing unhelpful pain beliefs
• managing activity-rest levels
• biofeedback
• supportive group therapy
• reducing family members’ reinforcement of the patient’s pain behavior or sick role.25
Assessing for somatic symptom disorders
In addition to using the AMPS approach for psychiatric assessment, clinicians should evaluate for somatization, which can present as head pain. Somatic symptom disorders (SSD) are a class of conditions that are impacted by affective, cognitive, and reinforcing factors that might or might not be consciously or intentionally produced. Patients with an SSD have somatic symptoms that are distressing or cause significant disruption of daily life because of excessive thoughts, feelings, or behaviors related to the somatic symptoms, for ≥6 months. The Figure outlines SSD, related conditions, and their respective prominent symptoms to assist in the differential diagnosis.26
Note that some headache conditions present with severe distress because of their abrupt onset and severity of symptoms (eg, cluster headaches). Therefore, the expectation and likelihood of psychological disturbance should be factored into a diagnosis of SSD and related conditions as seen in the Figure.
Secondary factors of unusual pain behavior or treatment response. The role of thoughts, affect, and behaviors is clinically meaningful in understanding SSD and similar conditions. Specific questions about cultural beliefs and rituals as they relate to exacerbations of head pain are of value. Table 413,27 lists behavioral, cognitive, and affective dimensions of head pain using the biopsychosocial model, and further clarifies common questions that arise with unusual pain response and complex patient presentations, which were outlined in the beginning of the article.
Because depression and anxiety can be comorbid with head pain, it is important to recognize psychological factors that contribute to pain perception. Indifference or denial of emotional stress as a result of severe pain and disability can imply a somatization process, which could suggest emotional disconnection or dissociation from somatic functioning.28 This finding can be a component of alexithymia, in which a person is disconnected from emotions and how emotions impact the body. Therefore, recognizing alexithymia assists in identifying psychological factors when patients deny mood symptoms, particularly in tension-type headache.
Functional assessment to rule out the disproportional impact of pain on daily activities is helpful in understanding the somatization process. Neurocognitive functioning should be assessed, particularly because frontal and subcortical dysregulation has been observed in head pain sufferers.29,30 Patients with cognitive changes as a result of a medical illness (eg, stroke, head concussion, brain tumor, or seizures) are especially at risk for neurocognitive dysfunction.
Neuropsychological assessment can be useful, not only to assess neurocognitive functioning (eg, Repeated Battery for the Assessment of Neuropsychological Status) but to identify objective test profiles associated with altered motivation (eg, Rey 15-Item Test, Minnesota Multiphasic Personality Inventory-2-Restructured Form F Scale, Personality Assessment Inventory [PAI] Negative Impression Management) and somatization processes (eg, PAI Somatization Scale). These instruments help to identify the severity of psychiatric and neurocognitive symptoms by comparing scores to normative (eg, healthy control group), clinical (eg, somatization, traumatic brain injury, mild cognitive impairment), and altered motivation (eg, persons instructed to exaggerate symptoms) databases.
If the clinician pursues neurocognitive assessment, direct referral to a neuropsychologist, referral to neurologist, or administration of a cognitive screening tool such as the Montreal Cognitive Assessment, Saint Louis University Mental Status, or Cognitive Log is recommended. If the cognitive screening is positive, next steps include: referring for full neuropsychological assessment, which includes complete cognitive and motor testing, personality testing, and integration of neuroimaging data (eg, MRI, CT scans, and/or EEG).
Assessing the patients’ self-talk or thought patterns as they describe their head pain will help clinicians understand belief systems that may be distorting the reality of the medical condition. For example, a patient might report that “my pain feels like someone is hitting me with an axe”; this is a catastrophic thought that can distort the clarity and perceptibility of pain. Encouraging patients to monitor and analyze their anxiety and associated negative thoughts is an important strategy for improving mood and decreasing somatization. Recording daily thoughts and CBT can help the patient identify and appropriately address his (her) cognitive distortions and futile thinking.
When implementing a treatment plan for somatization disorder, we propose the mnemonic device CARE MD:
• CBT
• Assess (by ruling out a medical cause for somatic complaints)
• Regular visits
• Empathy
• Med-psych interface (help the patient connect physical complaints and emotional stressors)
• Do no harm.
Clinical recommendations
Chronic head pain can be debilitating; psychodiagnostic assessment should therefore be considered an important part of the diagnosis and treatment plan. After ruling out common and emergent primary or secondary causes of head pain, consider psychiatric comorbidities. Depression and anxiety have a strong bidirectional relationship with chronic headache; therefore, we suggest evaluating patients with the intention of alleviating both psychiatric symptoms and head pain.
It is important to diligently assess for common psychiatric comorbidities; using the AMPS and CARE MD mnemonics, along with screening for somatization disorders, is an easy and effective way to evaluate for relevant psychiatric conditions associated with chronic head pain. Because many patients have unusual and complicated responses to head pain that can be explained by non-pathophysiological and non-biomechanical models, using the biopsychosocial model is essential for effective diagnosis, assessment, and treatment. Abortive and prophylactic medical interventions, as well as behavioral, sociocultural, and cognitive assessment, are vital to a comprehensive treatment approach.
Bottom Line
The psychodiagnostic assessment can help the astute clinician identify comorbid psychiatric conditions, psychological factors, and somatic symptoms to develop a comprehensive biopsychosocial treatment plan for patients with chronic head pain. Rule out primary and secondary causes of pain and screen for somatization disorders. Consider medication and psychotherapeutic treatment options.
Related Resources
• Pompili M, Di Cosimo D, Innamorati M, et al. Psychiatric comorbidity in patients with chronic daily headache and migraine: a selective overview including personality traits and suicide risk. J Headache Pain. 2009;10(4):283-290.
• Sinclair AJ, Sturrock A, Davies B, et al. Headache management: pharmacological approaches [published online July 3, 2015]. Pract Neurol. doi: 10.1136/practneurol-2015-001167.
Drug Brand Names
Amitriptyline • Elavil Meperidine • Demerol
Botulinum toxin A • Botox Metoclopramide • Reglan
Divalproex sodium • Depakote Propranolol • Inderide
Ketorolac • Toradol Topiramate • Topamax
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stovner LJ, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646-657.
3. The World Health Report 2001: Mental health: new understanding new hope. Geneva, Switzerland: World Health Organization; 2001.
4. World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_ disease/GBD_report_2004update_full.pdf. Published 2004. Accessed July 31, 2015.
5. World Health Organization. Headache disorders. http:// www.who.int/mediacentre/factsheets/fs277/en/. Published October 2012. Accessed July 31, 2015.
6. Clinch C. Evaluation & management of headache. In: South- Paul JE, Matheny SC, Lewis EL. eds. Current diagnosis & treatment in family medicine, 4th ed. New York, NY: McGraw-Hill; 2015:293-297.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
8. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia. 2015;35(2):165-181.
9. Janke EA, Holroyd KA, Romanek K. Depression increases onset of tension-type headache following laboratory stress. Pain. 2004;111(3):230-238.
10. Engel GL. The need for a new medical model: a challenge for biomedicine. Science. 1977;196(4286):129-136.
11. Engel GL. The clinical application of the biopsychosocial model. Am J Psychiatry. 1980;137(5):535-544.
12. Turk DC, Flor H. Chronic pain: a biobehavioral perspective. In: Gatchel RJ, Turk DC, eds. Psychosocial factors in pain: critical perspectives. New York, NY: Guilford Press; 1999:18-34.
13. Andrasik F, Flor H, Turk DC. An expanded view of psychological aspects in head pain: the biopsychosocial model. Neurol Sci. 2005;26(suppl 2):s87-s91.
14. Verri AP, Proietti Cecchini A, Galli C, et al. Psychiatric comorbidity in chronic daily headache. Cephalalgia. 1998; 18(suppl 21):45-49.
15. Ilgen MA, Zivin K, McCammon RJ, et al. Pain and suicidal thoughts, plans and attempts in the United States. Gen Hosp Psychiatry. 2008;30(6):521-527.
16. Kowacs F, Socal MP, Ziomkowski SC, et al. Symptoms of depression and anxiety, and screening for mental disorders in migrainous patients. Cephalalgia. 2003;23(2):79-89.
17. Hamelsky SW, Lipton RB. Psychiatric comorbidity of migraine. Headache. 2006;46(9):1327-1333.
18. Nguyen TV, Low NC. Comorbidity of migraine and mood episodes in a nationally representative population-based sample. Headache. 2013;53(3):498-506.
19. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001; 21(9):894-899.
20. Breslau N. Migraine, suicidal ideation, and suicide attempts. Neurology. 1992;42(2):392-395.
21. Puca F, Genco S, Prudenzano MP, et al. Psychiatric comorbidity and psychosocial stress in patients with tension-type headache from headache centers in Italy. Cephalalgia. 1999;19(3):159-164.
22. Yücel B, Kora K, Ozyalçín S, et al. Depression, automatic thoughts, alexithymia, and assertiveness in patients with tension-type headache. Headache. 2002;42(3):194-199.
23. Wittrock DA, Myers TC. The comparison of individuals with recurrent tension-type headache and headache-free controls in physiological response, appraisal, and coping with stressors: a review of the literature. Ann Behav Med. 1998;20(2):118-134.
24. Onate J, Xiong G, McCarron R. The primary care psychiatric interview. In: McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott, Williams and Wilkins; 2009:3-4.
25. Songer D. Psychotherapeutic approaches in the treatment of pain. Psychiatry (Edgmont). 2005;2(5):19-24.
26. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
27. Hashmi JA, Baliki MN, Huang L, et al. Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain. 2013;136(pt 9):2751-2768.
28. Packard RC. Conversion headache. Headache. 1980;20(5):266-268.
29. Mongini F, Keller R, Deregibus A, et al. Frontal lobe dysfunction in patients with chronic migraine: a clinical-neuropsychological study. Psychiatry Res. 2005;133(1):101-106.
30. Martelli MF, Grayson RL, Zasler ND. Posttraumatic headache: neuropsychological and psychological effects and treatment implications. J Head Trauma Rehabil. 1999;14(1):49-69.
1. Stovner LJ, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41(7):646-657.
3. The World Health Report 2001: Mental health: new understanding new hope. Geneva, Switzerland: World Health Organization; 2001.
4. World Health Organization. The global burden of disease: 2004 update. http://www.who.int/healthinfo/global_burden_ disease/GBD_report_2004update_full.pdf. Published 2004. Accessed July 31, 2015.
5. World Health Organization. Headache disorders. http:// www.who.int/mediacentre/factsheets/fs277/en/. Published October 2012. Accessed July 31, 2015.
6. Clinch C. Evaluation & management of headache. In: South- Paul JE, Matheny SC, Lewis EL. eds. Current diagnosis & treatment in family medicine, 4th ed. New York, NY: McGraw-Hill; 2015:293-297.
7. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
8. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia. 2015;35(2):165-181.
9. Janke EA, Holroyd KA, Romanek K. Depression increases onset of tension-type headache following laboratory stress. Pain. 2004;111(3):230-238.
10. Engel GL. The need for a new medical model: a challenge for biomedicine. Science. 1977;196(4286):129-136.
11. Engel GL. The clinical application of the biopsychosocial model. Am J Psychiatry. 1980;137(5):535-544.
12. Turk DC, Flor H. Chronic pain: a biobehavioral perspective. In: Gatchel RJ, Turk DC, eds. Psychosocial factors in pain: critical perspectives. New York, NY: Guilford Press; 1999:18-34.
13. Andrasik F, Flor H, Turk DC. An expanded view of psychological aspects in head pain: the biopsychosocial model. Neurol Sci. 2005;26(suppl 2):s87-s91.
14. Verri AP, Proietti Cecchini A, Galli C, et al. Psychiatric comorbidity in chronic daily headache. Cephalalgia. 1998; 18(suppl 21):45-49.
15. Ilgen MA, Zivin K, McCammon RJ, et al. Pain and suicidal thoughts, plans and attempts in the United States. Gen Hosp Psychiatry. 2008;30(6):521-527.
16. Kowacs F, Socal MP, Ziomkowski SC, et al. Symptoms of depression and anxiety, and screening for mental disorders in migrainous patients. Cephalalgia. 2003;23(2):79-89.
17. Hamelsky SW, Lipton RB. Psychiatric comorbidity of migraine. Headache. 2006;46(9):1327-1333.
18. Nguyen TV, Low NC. Comorbidity of migraine and mood episodes in a nationally representative population-based sample. Headache. 2013;53(3):498-506.
19. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001; 21(9):894-899.
20. Breslau N. Migraine, suicidal ideation, and suicide attempts. Neurology. 1992;42(2):392-395.
21. Puca F, Genco S, Prudenzano MP, et al. Psychiatric comorbidity and psychosocial stress in patients with tension-type headache from headache centers in Italy. Cephalalgia. 1999;19(3):159-164.
22. Yücel B, Kora K, Ozyalçín S, et al. Depression, automatic thoughts, alexithymia, and assertiveness in patients with tension-type headache. Headache. 2002;42(3):194-199.
23. Wittrock DA, Myers TC. The comparison of individuals with recurrent tension-type headache and headache-free controls in physiological response, appraisal, and coping with stressors: a review of the literature. Ann Behav Med. 1998;20(2):118-134.
24. Onate J, Xiong G, McCarron R. The primary care psychiatric interview. In: McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott, Williams and Wilkins; 2009:3-4.
25. Songer D. Psychotherapeutic approaches in the treatment of pain. Psychiatry (Edgmont). 2005;2(5):19-24.
26. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
27. Hashmi JA, Baliki MN, Huang L, et al. Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain. 2013;136(pt 9):2751-2768.
28. Packard RC. Conversion headache. Headache. 1980;20(5):266-268.
29. Mongini F, Keller R, Deregibus A, et al. Frontal lobe dysfunction in patients with chronic migraine: a clinical-neuropsychological study. Psychiatry Res. 2005;133(1):101-106.
30. Martelli MF, Grayson RL, Zasler ND. Posttraumatic headache: neuropsychological and psychological effects and treatment implications. J Head Trauma Rehabil. 1999;14(1):49-69.
