User login
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
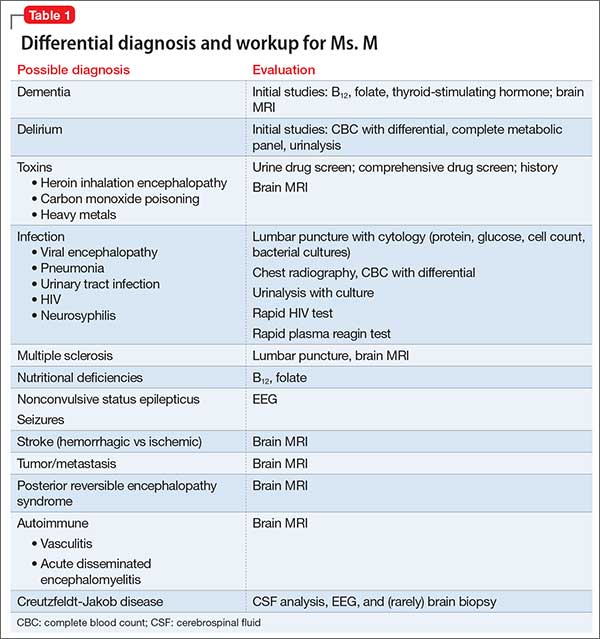
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.

Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
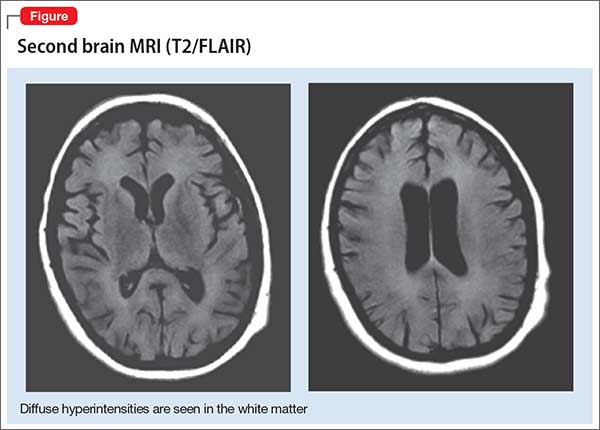
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.