User login
Cardiac pleomorphic sarcoma after placement of a Dacron graft
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-month mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis. TSJ
Correspondence
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-month mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis. TSJ
Correspondence
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-month mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis. TSJ
Correspondence
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Cardiac pleomorphic sarcoma after placement of Dacron graft
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.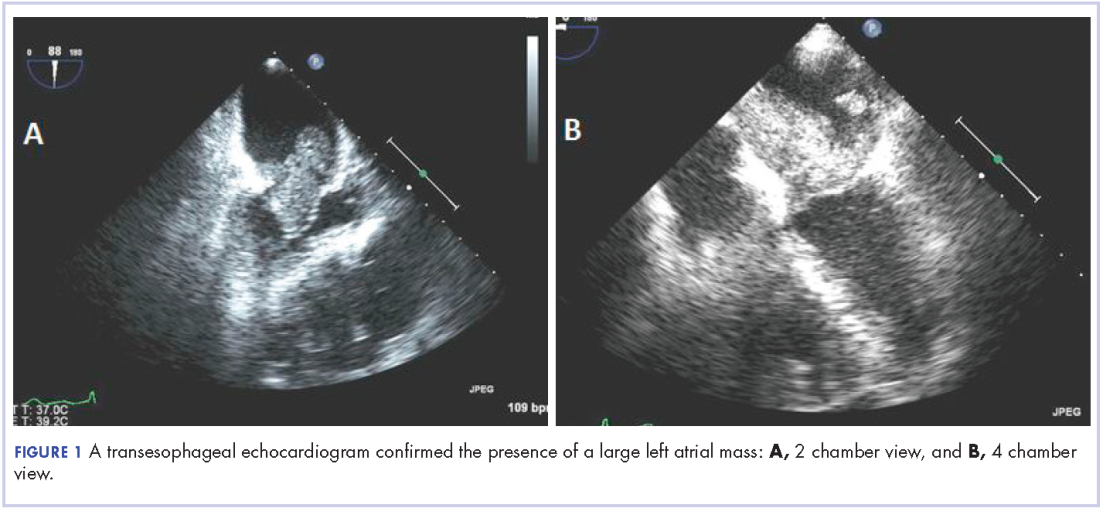
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).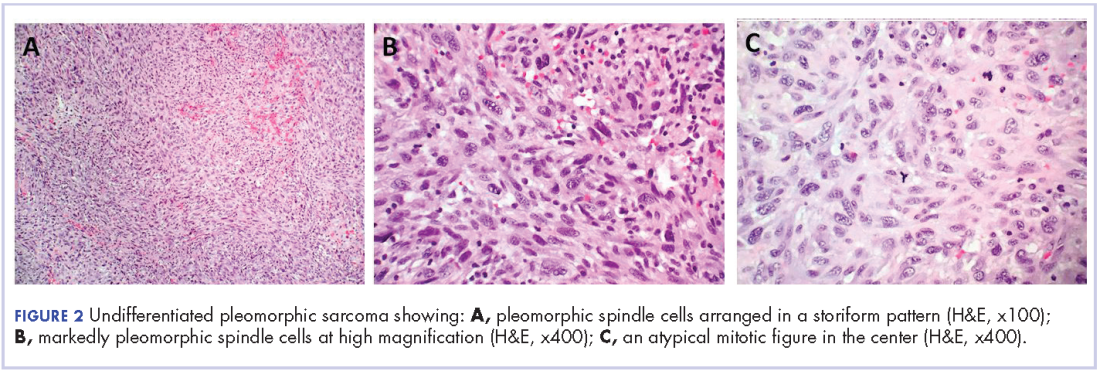
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-months mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis.
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-months mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-months mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis.
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Introducing The Sarcoma Journal—The Official Journal of the Sarcoma Foundation of America ™ : An Exciting Initiative in Peer-Reviewed Professional Education and Patient Advocacy
The Sarcoma Journal — Official Journal of the Sarcoma Foundation of America™ represents a new and exciting initiative in professional education. We invite you to share in the excitement surrounding the launch of a medical journal designed to be your most authoritative and comprehensive source of scientific information on the diagnosis and treatment of sarcomas and sarcoma sub-types.
On behalf of myself, our editorial board, and editorial staff, I welcome you to this journal as we explore new treatment paradigms for this disease, translational research that bridges the bench and the clinic, and a broad range of science to encompass the many facets of sarcoma. In my opinion, the startup of this publication could not come at a better time.
As cancer specialists and allied health care professionals who attend regular meetings of your peers, including ASCO and CTOS, we have seen a dramatic shift in management within the last few years. In many ways we are at a threshold of a new era in sarcoma management, and the spectrum of treatment is expanding across subspecialties, promising more effective strategies for our patients that are based on an improved understanding of disease biology. We need a resource to maintain and clarify our focus on this disease as research opens new avenues for us to consider in the management of patients with sarcoma.
When I was approached to serve as Editor-in-Chief of The Sarcoma Journal by the Sarcoma Foundation of America, I began to recruit an esteemed group of colleagues whose knowledge, worldwide reputation as thought leaders, and dedicated work as researchers would reflect our commitment toward finding a cure for sarcoma. Many of the colleagues who will join me on the Editorial Advisory Board have long-standing affiliations with the Sarcoma Foundation of America and its comprehensive program of sarcoma research, patient support and education and advocacy. As you explore the first issue of the journal, you will discover how our editorial content is an extension of this three-tiered approach. The SFA program is characterized by a multi-dimensional and uniquely coordinated outreach program of videos and webinars, websites (a new journal website is launching as well) a sarcoma-specific clinical trials database, newsletters and related materials— all aimed ultimately at finding a cure for this disease. This professional journal complements and extends the SFA’s mission.
Although The Sarcoma Journal has a position within the SFA umbrella, my focus is foremost on ensuring that The Sarcoma Journal contains the most accurate, relevant and up to date information available. I urge you to explore our highly informative and relevant sarcoma-specific content—including original reports, review articles, a Journal Club, expert opinion, meeting reports, and patient advocacy that encapsulates the latest findings from the bench with implications for the bedside.
Whether it is discussing the latest findings in advanced sarcoma sub-types or implications of genetics as a prognostic factor, you will find the information in this journal, reliably analyzed by our team of experts who are leading sarcoma clinicians and investigators. All of the content we provide is presented in a thought-provoking, lively and peer-reviewed format; we welcome your comments and suggestions to keep us on the forefront of patient care as we cover a rapidly evolving landscape of new information in the treatment of sarcomas and frame it within a context directly applicable to enhancing the quality of patient care.
The Sarcoma Journal — Official Journal of the Sarcoma Foundation of America™ represents a new and exciting initiative in professional education. We invite you to share in the excitement surrounding the launch of a medical journal designed to be your most authoritative and comprehensive source of scientific information on the diagnosis and treatment of sarcomas and sarcoma sub-types.
On behalf of myself, our editorial board, and editorial staff, I welcome you to this journal as we explore new treatment paradigms for this disease, translational research that bridges the bench and the clinic, and a broad range of science to encompass the many facets of sarcoma. In my opinion, the startup of this publication could not come at a better time.
As cancer specialists and allied health care professionals who attend regular meetings of your peers, including ASCO and CTOS, we have seen a dramatic shift in management within the last few years. In many ways we are at a threshold of a new era in sarcoma management, and the spectrum of treatment is expanding across subspecialties, promising more effective strategies for our patients that are based on an improved understanding of disease biology. We need a resource to maintain and clarify our focus on this disease as research opens new avenues for us to consider in the management of patients with sarcoma.
When I was approached to serve as Editor-in-Chief of The Sarcoma Journal by the Sarcoma Foundation of America, I began to recruit an esteemed group of colleagues whose knowledge, worldwide reputation as thought leaders, and dedicated work as researchers would reflect our commitment toward finding a cure for sarcoma. Many of the colleagues who will join me on the Editorial Advisory Board have long-standing affiliations with the Sarcoma Foundation of America and its comprehensive program of sarcoma research, patient support and education and advocacy. As you explore the first issue of the journal, you will discover how our editorial content is an extension of this three-tiered approach. The SFA program is characterized by a multi-dimensional and uniquely coordinated outreach program of videos and webinars, websites (a new journal website is launching as well) a sarcoma-specific clinical trials database, newsletters and related materials— all aimed ultimately at finding a cure for this disease. This professional journal complements and extends the SFA’s mission.
Although The Sarcoma Journal has a position within the SFA umbrella, my focus is foremost on ensuring that The Sarcoma Journal contains the most accurate, relevant and up to date information available. I urge you to explore our highly informative and relevant sarcoma-specific content—including original reports, review articles, a Journal Club, expert opinion, meeting reports, and patient advocacy that encapsulates the latest findings from the bench with implications for the bedside.
Whether it is discussing the latest findings in advanced sarcoma sub-types or implications of genetics as a prognostic factor, you will find the information in this journal, reliably analyzed by our team of experts who are leading sarcoma clinicians and investigators. All of the content we provide is presented in a thought-provoking, lively and peer-reviewed format; we welcome your comments and suggestions to keep us on the forefront of patient care as we cover a rapidly evolving landscape of new information in the treatment of sarcomas and frame it within a context directly applicable to enhancing the quality of patient care.
The Sarcoma Journal — Official Journal of the Sarcoma Foundation of America™ represents a new and exciting initiative in professional education. We invite you to share in the excitement surrounding the launch of a medical journal designed to be your most authoritative and comprehensive source of scientific information on the diagnosis and treatment of sarcomas and sarcoma sub-types.
On behalf of myself, our editorial board, and editorial staff, I welcome you to this journal as we explore new treatment paradigms for this disease, translational research that bridges the bench and the clinic, and a broad range of science to encompass the many facets of sarcoma. In my opinion, the startup of this publication could not come at a better time.
As cancer specialists and allied health care professionals who attend regular meetings of your peers, including ASCO and CTOS, we have seen a dramatic shift in management within the last few years. In many ways we are at a threshold of a new era in sarcoma management, and the spectrum of treatment is expanding across subspecialties, promising more effective strategies for our patients that are based on an improved understanding of disease biology. We need a resource to maintain and clarify our focus on this disease as research opens new avenues for us to consider in the management of patients with sarcoma.
When I was approached to serve as Editor-in-Chief of The Sarcoma Journal by the Sarcoma Foundation of America, I began to recruit an esteemed group of colleagues whose knowledge, worldwide reputation as thought leaders, and dedicated work as researchers would reflect our commitment toward finding a cure for sarcoma. Many of the colleagues who will join me on the Editorial Advisory Board have long-standing affiliations with the Sarcoma Foundation of America and its comprehensive program of sarcoma research, patient support and education and advocacy. As you explore the first issue of the journal, you will discover how our editorial content is an extension of this three-tiered approach. The SFA program is characterized by a multi-dimensional and uniquely coordinated outreach program of videos and webinars, websites (a new journal website is launching as well) a sarcoma-specific clinical trials database, newsletters and related materials— all aimed ultimately at finding a cure for this disease. This professional journal complements and extends the SFA’s mission.
Although The Sarcoma Journal has a position within the SFA umbrella, my focus is foremost on ensuring that The Sarcoma Journal contains the most accurate, relevant and up to date information available. I urge you to explore our highly informative and relevant sarcoma-specific content—including original reports, review articles, a Journal Club, expert opinion, meeting reports, and patient advocacy that encapsulates the latest findings from the bench with implications for the bedside.
Whether it is discussing the latest findings in advanced sarcoma sub-types or implications of genetics as a prognostic factor, you will find the information in this journal, reliably analyzed by our team of experts who are leading sarcoma clinicians and investigators. All of the content we provide is presented in a thought-provoking, lively and peer-reviewed format; we welcome your comments and suggestions to keep us on the forefront of patient care as we cover a rapidly evolving landscape of new information in the treatment of sarcomas and frame it within a context directly applicable to enhancing the quality of patient care.
Pulmonary sarcomatoid carcinoma presenting as a necrotizing cavitary lung lesion: diagnostic dilemma
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear. The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsy of the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung, likely due to the underlying cavitary lesion (Figure 2B). Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic mass within the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2A) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3). Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine. Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was
recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade andnwith equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment. Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis and timely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high plateletderived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies. TSJ
Correspondence
Gaurang Nandkishor Vaidya, MD
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso. biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear. The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsy of the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung, likely due to the underlying cavitary lesion (Figure 2B). Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic mass within the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2A) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3). Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine. Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was
recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade andnwith equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment. Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis and timely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high plateletderived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies. TSJ
Correspondence
Gaurang Nandkishor Vaidya, MD
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso. biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear. The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsy of the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung, likely due to the underlying cavitary lesion (Figure 2B). Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic mass within the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2A) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3). Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine. Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was
recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade andnwith equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment. Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis and timely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high plateletderived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies. TSJ
Correspondence
Gaurang Nandkishor Vaidya, MD
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso. biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
This article was originally published in the Journal of Community and Supportive Oncology (JCSO 2017;15(2):103-105). doi: https://doi.org/10.12788/jcso.0259. It is reproduced here with permission of the copyright owner. Further reproduction is prohibited without permission.
Bilateral chylothorax in an AIDS patient with newly diagnosed Kaposi sarcoma
Kaposi sarcoma is an angioproliferative tumor that is associated with human herpes virus-B (HIV-B). Mucocutaneous disease is the most common site for manifestation of AIDS-related Kaposi sarcoma, commonly affecting the lower extremeties, oral mucosa, face, and genitalia. Pleural effusions can occur in 36%-60% of patients with Kaposi sarcoma, and it has been documented that chylothorax is a rare, but plausible presentation in patients with Kaposi sarcoma.1 We present here a case of bilateral chylothorax in a patient with AIDS-related Kaposi sarcoma.
Case presentation and summary
A 52-year-old MSM male with AIDS (CD4, <20 mm3 ; viral load, 58 copies/ml) presented to the emergency department with complaints of shortness of breath, productive cough, and diarrhea for 2 days prior to presentation. His medical history also included chronic obstructive pulmonary disease, coronary artery disease, and hyperlipidemia.The patient was not on HAART because of his history of noncompliance. The results of a chest X-ray and computed-tomography (CT) scan showed that the patient had bilateral pleural effusion and a spiculated 14-mm nodule in the left upper lobe.
The patient underwent ultrasound-guided placement of a 12-French left-sided chest catheter, and a milky white fluid was aspirated from the left pleural space. Laboratory analysis of the pleural fluid confirmed an exudate with an elevated triglyceride level of 120 mg/dL (chylous, >110 mg/dL) indicating chylothorax.
On close physical examination, the patient was found to have multiple irregular plaques on the back and lower extremities. As described by dermatology, there was a violaceous indurated plaque on the left axillae, violaceous indurated plaques with superficial scale grouped on the left midlateral back, and hyperpigmented lichenified plaques and papules on bilateral shins, with some with plate-like scale. Two punch biopsies were taken of the skin lesions, which confirmed Kaposi sarcoma, plaque stage from the lesion biopsied on the back, and patch stage from the lesion biopsied in the left axilla. Cytology of the pleural fluid was negative for malignant cells. On review by the radiologist of the CT scan of the chest, there was no indication of gross distention of the thoracic duct. Treatment options were offered to the patient, and the patient was considering options for chemotherapy and home hospice given his advanced disease state at the time of discharge.
Discussion
Chylothorax occurs with a thoracic duct obstruction, which results in leakage of lymphatic fluid into the pleural cavity. The two leading causes of chylothorax are trauma and malignancy, with lymphoma being the most common cause of chylothorax among those with malignancy.2 Chylothorax, however, is a rare but documented complication of Kaposi sarcoma. Marais and colleagues reported the case of a 3-year-old HIV-positive patient with newly diagnosed Kaposi sarcoma who was found to have tumor infiltration in the thoracic duct leading to bilateral chylothorax.3 Maradona and colleagues described a 40-year-old man with AIDS-related Kaposi sarcoma who was found to have pleural and pericardial Kaposi sarcoma with chylothorax.4 Priest and colleagues wrote about a 32-year-old patient with AIDS with biopsy-proven Kaposi sarcoma who required multiple therapeutic thoracenteses for rapidly recurrent left chylothorax effusions.5
There are two leading discussions as to the pathophysiology of chylothorax that is related to Kaposi sarcoma: chylothorax developing secondary to metastatic disease or the development of chylothorax secondary to primary Kaposi sarcoma arising from the pleural region.6 One case report examined pleural and lung biopsies in a 34-year-old patient with AIDS-related Kaposi sarcoma that showed immunohistochemical staining that was suggestive of early-stage Kaposi sarcoma of lymphatic endothelial origin. The authors were attempting to illustrate that Kaposi sarcoma may have a stem-cell origin which can differentiate into lymph cells. Kontantinopoulos and colleagues postulated that in situ Kaposi sarcoma can arise from the lymphatic system with a resultant clinical presentation of chylothorax.7 The more mainstream thought however, is that chylothorax has been found to develop secondary to metastatic disease. The present case, therefore, illustrates an unusual presentation of cytology negative chylothorax in a patient with AIDS-related Kaposi sarcoma. TSJ
Rebecca E Neril, MD; Department of Internal Medicine, SBH Health System, Bronx, New York.
References
1. Sridar S, Garza EG, Cox J, Rumbak MJ. Serosanguineous pleural effusions in a patient with HIV and Kaposi sarcoma: pleuroscopic findings. J Bronchology Interv Pulmonol. 2011;18(4):337-339.
2. Light RW. Chylothorax and pseudochylothorax. In: Light RW, ed. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013:412-426.
3. Marais BJ, Pienaar J, Gie RP. Kaposi sarcoma with upper airway obstruction and bilateral chylothoraces. Pediatr Infect Dis J. 2003;22:926-928.
4. Maradona JA, Carton JA, Asensi V, Rodriguez-Guardado A. AIDSrelated Kaposi sarcoma with chylothorax and pericardial involvement satisfactorily treated with liposomal doxorubicin. AIDS. 2002;16(5):806.
5. Priest ER, Weiss R. Chylothorax with Kaposi sarcoma. South Med J. 1991;84:806-807.
6. Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190.
7. Konstantinopoulos PA, Dezube BJ, Pantanowitz L. Morphologic and immunophenotypic evidence of in situ Kaposi sarcoma. BMC Clin Pathol. 2006;30:6:7.
Kaposi sarcoma is an angioproliferative tumor that is associated with human herpes virus-B (HIV-B). Mucocutaneous disease is the most common site for manifestation of AIDS-related Kaposi sarcoma, commonly affecting the lower extremeties, oral mucosa, face, and genitalia. Pleural effusions can occur in 36%-60% of patients with Kaposi sarcoma, and it has been documented that chylothorax is a rare, but plausible presentation in patients with Kaposi sarcoma.1 We present here a case of bilateral chylothorax in a patient with AIDS-related Kaposi sarcoma.
Case presentation and summary
A 52-year-old MSM male with AIDS (CD4, <20 mm3 ; viral load, 58 copies/ml) presented to the emergency department with complaints of shortness of breath, productive cough, and diarrhea for 2 days prior to presentation. His medical history also included chronic obstructive pulmonary disease, coronary artery disease, and hyperlipidemia.The patient was not on HAART because of his history of noncompliance. The results of a chest X-ray and computed-tomography (CT) scan showed that the patient had bilateral pleural effusion and a spiculated 14-mm nodule in the left upper lobe.
The patient underwent ultrasound-guided placement of a 12-French left-sided chest catheter, and a milky white fluid was aspirated from the left pleural space. Laboratory analysis of the pleural fluid confirmed an exudate with an elevated triglyceride level of 120 mg/dL (chylous, >110 mg/dL) indicating chylothorax.
On close physical examination, the patient was found to have multiple irregular plaques on the back and lower extremities. As described by dermatology, there was a violaceous indurated plaque on the left axillae, violaceous indurated plaques with superficial scale grouped on the left midlateral back, and hyperpigmented lichenified plaques and papules on bilateral shins, with some with plate-like scale. Two punch biopsies were taken of the skin lesions, which confirmed Kaposi sarcoma, plaque stage from the lesion biopsied on the back, and patch stage from the lesion biopsied in the left axilla. Cytology of the pleural fluid was negative for malignant cells. On review by the radiologist of the CT scan of the chest, there was no indication of gross distention of the thoracic duct. Treatment options were offered to the patient, and the patient was considering options for chemotherapy and home hospice given his advanced disease state at the time of discharge.
Discussion
Chylothorax occurs with a thoracic duct obstruction, which results in leakage of lymphatic fluid into the pleural cavity. The two leading causes of chylothorax are trauma and malignancy, with lymphoma being the most common cause of chylothorax among those with malignancy.2 Chylothorax, however, is a rare but documented complication of Kaposi sarcoma. Marais and colleagues reported the case of a 3-year-old HIV-positive patient with newly diagnosed Kaposi sarcoma who was found to have tumor infiltration in the thoracic duct leading to bilateral chylothorax.3 Maradona and colleagues described a 40-year-old man with AIDS-related Kaposi sarcoma who was found to have pleural and pericardial Kaposi sarcoma with chylothorax.4 Priest and colleagues wrote about a 32-year-old patient with AIDS with biopsy-proven Kaposi sarcoma who required multiple therapeutic thoracenteses for rapidly recurrent left chylothorax effusions.5
There are two leading discussions as to the pathophysiology of chylothorax that is related to Kaposi sarcoma: chylothorax developing secondary to metastatic disease or the development of chylothorax secondary to primary Kaposi sarcoma arising from the pleural region.6 One case report examined pleural and lung biopsies in a 34-year-old patient with AIDS-related Kaposi sarcoma that showed immunohistochemical staining that was suggestive of early-stage Kaposi sarcoma of lymphatic endothelial origin. The authors were attempting to illustrate that Kaposi sarcoma may have a stem-cell origin which can differentiate into lymph cells. Kontantinopoulos and colleagues postulated that in situ Kaposi sarcoma can arise from the lymphatic system with a resultant clinical presentation of chylothorax.7 The more mainstream thought however, is that chylothorax has been found to develop secondary to metastatic disease. The present case, therefore, illustrates an unusual presentation of cytology negative chylothorax in a patient with AIDS-related Kaposi sarcoma. TSJ
Rebecca E Neril, MD; Department of Internal Medicine, SBH Health System, Bronx, New York.
Kaposi sarcoma is an angioproliferative tumor that is associated with human herpes virus-B (HIV-B). Mucocutaneous disease is the most common site for manifestation of AIDS-related Kaposi sarcoma, commonly affecting the lower extremeties, oral mucosa, face, and genitalia. Pleural effusions can occur in 36%-60% of patients with Kaposi sarcoma, and it has been documented that chylothorax is a rare, but plausible presentation in patients with Kaposi sarcoma.1 We present here a case of bilateral chylothorax in a patient with AIDS-related Kaposi sarcoma.
Case presentation and summary
A 52-year-old MSM male with AIDS (CD4, <20 mm3 ; viral load, 58 copies/ml) presented to the emergency department with complaints of shortness of breath, productive cough, and diarrhea for 2 days prior to presentation. His medical history also included chronic obstructive pulmonary disease, coronary artery disease, and hyperlipidemia.The patient was not on HAART because of his history of noncompliance. The results of a chest X-ray and computed-tomography (CT) scan showed that the patient had bilateral pleural effusion and a spiculated 14-mm nodule in the left upper lobe.
The patient underwent ultrasound-guided placement of a 12-French left-sided chest catheter, and a milky white fluid was aspirated from the left pleural space. Laboratory analysis of the pleural fluid confirmed an exudate with an elevated triglyceride level of 120 mg/dL (chylous, >110 mg/dL) indicating chylothorax.
On close physical examination, the patient was found to have multiple irregular plaques on the back and lower extremities. As described by dermatology, there was a violaceous indurated plaque on the left axillae, violaceous indurated plaques with superficial scale grouped on the left midlateral back, and hyperpigmented lichenified plaques and papules on bilateral shins, with some with plate-like scale. Two punch biopsies were taken of the skin lesions, which confirmed Kaposi sarcoma, plaque stage from the lesion biopsied on the back, and patch stage from the lesion biopsied in the left axilla. Cytology of the pleural fluid was negative for malignant cells. On review by the radiologist of the CT scan of the chest, there was no indication of gross distention of the thoracic duct. Treatment options were offered to the patient, and the patient was considering options for chemotherapy and home hospice given his advanced disease state at the time of discharge.
Discussion
Chylothorax occurs with a thoracic duct obstruction, which results in leakage of lymphatic fluid into the pleural cavity. The two leading causes of chylothorax are trauma and malignancy, with lymphoma being the most common cause of chylothorax among those with malignancy.2 Chylothorax, however, is a rare but documented complication of Kaposi sarcoma. Marais and colleagues reported the case of a 3-year-old HIV-positive patient with newly diagnosed Kaposi sarcoma who was found to have tumor infiltration in the thoracic duct leading to bilateral chylothorax.3 Maradona and colleagues described a 40-year-old man with AIDS-related Kaposi sarcoma who was found to have pleural and pericardial Kaposi sarcoma with chylothorax.4 Priest and colleagues wrote about a 32-year-old patient with AIDS with biopsy-proven Kaposi sarcoma who required multiple therapeutic thoracenteses for rapidly recurrent left chylothorax effusions.5
There are two leading discussions as to the pathophysiology of chylothorax that is related to Kaposi sarcoma: chylothorax developing secondary to metastatic disease or the development of chylothorax secondary to primary Kaposi sarcoma arising from the pleural region.6 One case report examined pleural and lung biopsies in a 34-year-old patient with AIDS-related Kaposi sarcoma that showed immunohistochemical staining that was suggestive of early-stage Kaposi sarcoma of lymphatic endothelial origin. The authors were attempting to illustrate that Kaposi sarcoma may have a stem-cell origin which can differentiate into lymph cells. Kontantinopoulos and colleagues postulated that in situ Kaposi sarcoma can arise from the lymphatic system with a resultant clinical presentation of chylothorax.7 The more mainstream thought however, is that chylothorax has been found to develop secondary to metastatic disease. The present case, therefore, illustrates an unusual presentation of cytology negative chylothorax in a patient with AIDS-related Kaposi sarcoma. TSJ
Rebecca E Neril, MD; Department of Internal Medicine, SBH Health System, Bronx, New York.
References
1. Sridar S, Garza EG, Cox J, Rumbak MJ. Serosanguineous pleural effusions in a patient with HIV and Kaposi sarcoma: pleuroscopic findings. J Bronchology Interv Pulmonol. 2011;18(4):337-339.
2. Light RW. Chylothorax and pseudochylothorax. In: Light RW, ed. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013:412-426.
3. Marais BJ, Pienaar J, Gie RP. Kaposi sarcoma with upper airway obstruction and bilateral chylothoraces. Pediatr Infect Dis J. 2003;22:926-928.
4. Maradona JA, Carton JA, Asensi V, Rodriguez-Guardado A. AIDSrelated Kaposi sarcoma with chylothorax and pericardial involvement satisfactorily treated with liposomal doxorubicin. AIDS. 2002;16(5):806.
5. Priest ER, Weiss R. Chylothorax with Kaposi sarcoma. South Med J. 1991;84:806-807.
6. Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190.
7. Konstantinopoulos PA, Dezube BJ, Pantanowitz L. Morphologic and immunophenotypic evidence of in situ Kaposi sarcoma. BMC Clin Pathol. 2006;30:6:7.
References
1. Sridar S, Garza EG, Cox J, Rumbak MJ. Serosanguineous pleural effusions in a patient with HIV and Kaposi sarcoma: pleuroscopic findings. J Bronchology Interv Pulmonol. 2011;18(4):337-339.
2. Light RW. Chylothorax and pseudochylothorax. In: Light RW, ed. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013:412-426.
3. Marais BJ, Pienaar J, Gie RP. Kaposi sarcoma with upper airway obstruction and bilateral chylothoraces. Pediatr Infect Dis J. 2003;22:926-928.
4. Maradona JA, Carton JA, Asensi V, Rodriguez-Guardado A. AIDSrelated Kaposi sarcoma with chylothorax and pericardial involvement satisfactorily treated with liposomal doxorubicin. AIDS. 2002;16(5):806.
5. Priest ER, Weiss R. Chylothorax with Kaposi sarcoma. South Med J. 1991;84:806-807.
6. Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190.
7. Konstantinopoulos PA, Dezube BJ, Pantanowitz L. Morphologic and immunophenotypic evidence of in situ Kaposi sarcoma. BMC Clin Pathol. 2006;30:6:7.
This article was originally published in the Journal of Community and Supportive Oncology (JCSO 2017;15(3):e174-e175). doi: https://doi.org/10.12788/jcso.0261. It is reproduced here with permission of the copyright owner. Further reproduction is prohibited without permission.
Soft Tissue Sarcoma: Diagnosis and Treatment
Introduction
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
Epidemiology and Classification
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
Clinical Evaluation
› Case Presentation
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (
• What is the typical presentation for sarcomas?
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
Management
› Case Continued
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
Surgery
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups.The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
› Case Continued
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
Radiation THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
› Case Continued
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
Chemotherapy
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study.35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
› Case Continued
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dysp-nea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
› Case Continued
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51 A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
› Case Continued
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
Conclusion
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma. TSJ
CORRESPONDENCE
Ashley Pariser, MD, Resident, Department of Medicine, Northwestern University Feinberg School of Medicine Chicago, IL. Accepted for publication Jan/Feb 2017; Hosp Phys; Vol. 12, Part1
References
1. American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
2. National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
3. Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
4. Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4: 252–66.
5. Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
6. Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
7. Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
8. Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
9. Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.

10. Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
12. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
13. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
14. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
15. Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
16. Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
17. Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
18. Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
19. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
20. Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
21. Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
22. Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
23. Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
24. Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
25. Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
26. Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
27. Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
28. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
29. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
30. Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
31. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
32. Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
33. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
34. Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
35. Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
36. Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
37. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
38. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
39. Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
40. Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
41. Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
42. Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
43. Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
44. Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
45. Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
46. Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
47. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
48. Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
49. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
50. Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
51. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
52. Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
53. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
54. Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
55. Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
56. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
57. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
58. Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
59. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
Introduction
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
Epidemiology and Classification
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
Clinical Evaluation
› Case Presentation
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (
• What is the typical presentation for sarcomas?
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
Management
› Case Continued
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
Surgery
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups.The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
› Case Continued
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
Radiation THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
› Case Continued
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
Chemotherapy
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study.35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
› Case Continued
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dysp-nea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
› Case Continued
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51 A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
› Case Continued
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
Conclusion
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma. TSJ
CORRESPONDENCE
Ashley Pariser, MD, Resident, Department of Medicine, Northwestern University Feinberg School of Medicine Chicago, IL. Accepted for publication Jan/Feb 2017; Hosp Phys; Vol. 12, Part1
Introduction
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
Epidemiology and Classification
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
Clinical Evaluation
› Case Presentation
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (
• What is the typical presentation for sarcomas?
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
Management
› Case Continued
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
Surgery
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups.The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
› Case Continued
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
Radiation THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
› Case Continued
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
Chemotherapy
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study.35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
› Case Continued
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dysp-nea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
› Case Continued
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51 A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
› Case Continued
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
Conclusion
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma. TSJ
CORRESPONDENCE
Ashley Pariser, MD, Resident, Department of Medicine, Northwestern University Feinberg School of Medicine Chicago, IL. Accepted for publication Jan/Feb 2017; Hosp Phys; Vol. 12, Part1
References
1. American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
2. National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
3. Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
4. Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4: 252–66.
5. Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
6. Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
7. Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
8. Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
9. Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
10. Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
12. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
13. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
14. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
15. Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
16. Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
17. Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
18. Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
19. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
20. Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
21. Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
22. Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
23. Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
24. Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
25. Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
26. Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
27. Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
28. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
29. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
30. Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
31. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
32. Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
33. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
34. Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
35. Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
36. Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
37. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
38. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
39. Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
40. Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
41. Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
42. Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
43. Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
44. Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
45. Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
46. Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
47. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
48. Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
49. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
50. Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
51. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
52. Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
53. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
54. Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
55. Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
56. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
57. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
58. Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
59. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
References
1. American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
2. National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
3. Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
4. Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4: 252–66.
5. Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
6. Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
7. Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
8. Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
9. Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
10. Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
12. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
13. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
14. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
15. Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
16. Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
17. Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
18. Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
19. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
20. Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
21. Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
22. Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
23. Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
24. Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
25. Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
26. Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
27. Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
28. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
29. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
30. Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
31. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
32. Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
33. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
34. Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
35. Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
36. Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
37. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
38. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
39. Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
40. Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
41. Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
42. Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
43. Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
44. Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
45. Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
46. Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
47. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
48. Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
49. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
50. Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
51. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
52. Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
53. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
54. Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
55. Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
56. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
57. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
58. Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
59. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
Onodera’s Prognostic Nutritional Index in soft tissue sarcoma patients as a predictor of wound complications
Wound complications after pre- or post-operative radiation for soft tissue sarcomas are well established.1 The ability to predict who will have a wound complication remains difficult. Some studies have looked at risk factors such as smoking, and the preoperative nutritional status of patients has been identified as a risk factor for wound complication in patients with elective orthopedic surgical procedures.2 One validated method of measuring preoperative nutritional status in patients with gastrointestinal malignant tumors has been with Onodera’s Prognostic Nutritional Index (OPNI). It uses the patient’s preoperative albumin (g/dL) and absolute lymphocyte values (per mm3). The prognostic value of the OPNI has been demonstrated in patients with colorectal, esophageal, and gastric cancers, and has been shown to be prognostic for postoperative wound healing and overall prognosis.3-5 In this study, we investigate the significance of preoperative nutritional status, measured by OPNI, as a predictor of wound complications in patients treated with pre- or postoperative radiation for soft tissue sarcoma.
Methods
After receiving Institutional Review Board approval for the study, we conducted a retrospective review of consecutive patients treated during July 2012-April 2016 for a soft tissue sarcoma by the orthopedic oncology division at Cooper University Hospital in Camden, New Jersey. Inclusion criteria were patients with biopsy-proven soft tissue sarcoma, who were older than 18 years, had received pre- or postoperative radiation, and who had a recorded preoperative albumin and total lymphocyte count. A minimum follow-up of 3 months was required to assess for postoperative wound complications. Exclusion criteria included patients who had a bone sarcoma, had not received radiation therapy, or had a missing preoperative albumin or total lymphocyte count.
All of the surgeries were performed by 2 fellowshiptrained orthopedic oncologists. Patients received either pre- or postoperative radiation therapy by multiple radiation oncologists.
The OPNI was calculated based on the published formula OPNI = (10*albumin level [g/dL]) + (0.005*total lymphocyte count [per mm3]). The albumin level and total lymphocyte counts closest to the index operation were chosen.
Demographic information including gender, age at diagnosis, height, and weight were recorded. Data related to the patients’ pathologic diagnosis, stage at presentation, radiation therapy, and surgical resection were collected. A minor wound complication was defined as a wound problem that did not require operative intervention. Major wound complication was defined as a complication requiring operative intervention with or without flap reconstruction. Wound complications occurring within the 3-month postoperative period were considered.
Univariate and multiple variable analysis was performed. A P value <.05 was considered significant. A receiver operating curve as well as recursive partitioning was performed for OPNI and age to determine the best cut-off point to use in the analysis. The Sobel test was used to evaluate mediation. All statistical analysis was performed using SAS v9.4 and JMP10. (SAS Institute, Cary, NC).
Results
In all, 44 patients (28 men, 16 women) were included in the study. Their mean age was 61.2 years (range, 19-94). The average size of the tumors was 8.5 cm in greatest dimension (range, 1.2-27.4 cm), and all of the patients had nonmetastatic disease at the time of surgical resection; 37 patients had R0 resections, and 7 patients had a positive margin from an outside hospital, but obtained R0 resections on a subsequent resection (Table 1 and Table 2).
In all, 30 patients received preoperative radiation, 14 patients received postoperative radiation, 32 patients received external beam radiation, 8 received Cyberknife treatment, and information for 4 patients was not unavailable. Mean preoperative external beam radiation and Cyberknife dose was 4,931 Gy and 3,750 Gy, respectively. Mean postoperative external beam and Cyberknife radiation dose was 6,077 Gy and 4,000 Gy, respectively. When evaluating radiation dose delivered between those who had wound complications and those who did not, there was no significant difference (Table 3).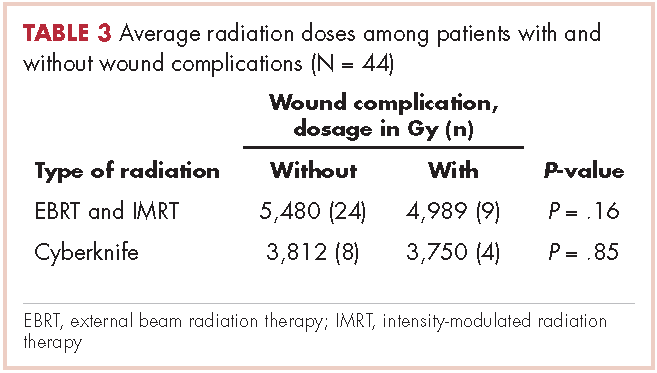
Of the total, 13 patients had a wound complication (30%). Ten patients had preoperative radiation, and 3 had postoperative radiation. Ten patients had major wound complications requiring a combined 27 surgeries. Three patients had minor wound complications, which resolved with conservative management. One patient had a major wound complication in the group that had an initial R1 resection.
The OPNI was calculated based on the aforementioned formula. When the univariate analysis was performed, only age and OPNI were statistically significant. Patients older than 72.6 years had a 6.8 times higher risk of a wound complication (P = .01; 95% confidence interval [CI], 1.6-28.7). When the OPNI value of 45.4 was used as the threshold, a patient with a preoperative OPNI value of <45.4 had a 7.5 times increased risk of developing a wound complication (P = .005; 95% CI, 1.8-31.0).
When the receiver operating curve and recursive partitioning was performed, an OPNI value of 45.4 showed a sensitivity of 62% and specificity of 82% in predicting wound complications (Figure 1).
When a multiple variable analysis was performed, OPNI and age were not statistically significant (P = .06 and P = .11, respectively). A test for mediation was performed, and the OPNI seemed to mediate the effect age has on wound complications, accounting for 36% of the total effect (Sobel test statistic, 1.79; P = .07).
Discussion
Wound complications after pre- and postoperative radiation for soft tissue sarcomas are well known. The best study to date to demonstrate that relationship was a randomized controlled trial performed in Canada, which showed that preoperative radiation resulted in 37% wound complications, compared with 17% for postoperative radiation.6 In that study, of the wound complications in both radiation types, more than 50%-60% required a secondary surgical procedure, designating it as a major wound complication. Other variables that have been shown to contribute to wound complications include being older than 40 years and/or having large tumors, diabetes, peripheral vascular disease, and begin a smoker.7-10
In our study, we applied OPNI to orthopedic oncology and showed that the patient’s age and preoperative nutritional status were significant predictors of developing a wound complication. An OPNI of <45.4 increased the chance of a wound complication by 7.5 times. Being older than 73 years increased the risk of a wound complication by 6.8 times. Most of these wound complications were major and required surgical intervention.
In general surgical oncology, the evaluation of nutritional status has had a significant impact on the care of patients, especially for those patients undergoing gastrointestinal surgery. The OPNI was initially designed to assess the nutritional and immunological statuses of patients undergoing gastrointestinal surgery.11 Preoperative OPNI has been shown to be a good predictor of postoperative complications and survival in patients with colorectal cancer, malignant mesothelioma, hepatocellular carcinoma and in patients who undergo total gastrectomy.12-15 Chen and colleagues evaluated the significance of OPNI in patients with colorectal cancer. They found an optimal cut-off value of 45. An OPNI value <45 has a sensitivity and specificity of 85% and 69%, respectively, in predicting 5-year overall survival.16 Hong and colleagues noted that an OPNI cut-off value of 52.6 as a predictor of overall survival.17
Poor preoperative nutritional status has been shown to have a negative impact on wound healing. In patients who underwent emergency laparotomy, a low OPNI had significantly higher rates of wound dehiscence and infection.18 This happens because protein deficiency leads to decreased wound tensile strength, decreased T-cell function, decreased phagocytic activity, which ultimately diminish the patient’s ability to heal and defend against wound infections.19-21
In soft tissue sarcoma patients, poor preoperative nutritional status is further compromised by radiation therapy to the wound. Gu and colleagues showed that radiation to wounds in mice showed early inhibition of the inflammatory phase, injury and inhibition of fibroblasts, and collagen formation, and then prolonged re-epithelialization.22 This “double hit” with radiation onto host tissue that is already nutritionally compromised could be an important cause of why wound complications occur at such high rates in our soft tissue sarcoma patients.
There are several limitations to this study. First, the study has a small sample size, which was a direct result of the number of patients who were excluded because an OPNI value could not be calculated for them. Second, we could not determine if the OPNI was more valuable in patients who underwent pre- or postoperative radiation. This study did not look at other nutritional indices such as prealbumin and vitamin levels. Third, the radiation was provided by different providers, so technique was variable, but the patients received nearly equivalent doses and variability in technique is likely limited. Fourth, we were not able to meaningfully analyze the role of chemotherapy in this patient population because there was a significant heterogeneity of patients receiving pre- and postoperative chemotherapy.
Our findings strongly suggest that a preoperative OPNI of <45.4 and being older than 73 years are strong predictors of patients who will experience a wound complication after radiation therapy for soft tissue sarcomas. This study has led us to start measuring preoperative albumin levels and assess complete metabolic panels. Our goal is to identify patients who are at high risk of wound complication and perform interventions to improve nutrition, then to study whether the interventions help lower the rates of wound complications.
1. Ormsby MV, Hilaris BS, Nori D, Brennan MF. Wound complications of adjuvant radiation therapy in patients with soft-tissue sarcomas. Ann Surg. 1989;210(1):93-99.
2. Greene KA, Wilde AH, Stulberg BN. Preoperative nutritional status of total joint patients: relationship to postoperative wound complications. J Arthroplasty. 1991;6(4):321-325.
3. Nozoe T, Kimura Y, Ishida M, Saeki H, Korenaga D, Sugimachi K. Correlation of pre-operative nutritional condition with post-operative complications in surgical treatment for oesophageal carcinoma. Eur J Surg Oncol. 2002;28(4):396-400.
4. Nozoe T, Kohno M, Iguchi T, et al. The prognostic nutritional index can be a prognostic indicator in colorectal carcinoma. Surg Today. 2012;42(6):532-535.
5. Nozoe T, Ninomiya M, Maeda T, Matsukuma A, Nakashima H, Ezaki T. Prognostic nutritional index: a tool to predict the biological aggressiveness of gastric carcinoma. Surg Today. 2010;40(5):440-443.
6. O’Sullivan B, Davis AM, Turcotte R, Bell R, Catton C, Chabot P, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002;359(9325):2235-2241.
7. Peat BG, Bell RS, Davis A, et al. Wound-healing complications after soft-tissue sarcoma surgery. Plast Reconstr Surg. 1994;93(5):980-987.
8. Kunisada T, Ngan SY, Powell G, Choong PF. Wound complications following pre-operative radiotherapy for soft tissue sarcoma. Eur J Surg Oncol. 2002;28(1):75-79.
9. Saddegh MK, Bauer HC. Wound complication in surgery of soft tissue sarcoma: analysis of 103 consecutive patients managed without adjuvant therapy. Clin Orthop Relat Res. 1993;289:247-253.
10. Tseng JF, Ballo MT, Langstein HN, et al. The effect of preoperative radiotherapy and reconstructive surgery on wound complications after resection of extremity soft-tissue sarcomas. Ann Surg Oncol. 2006;13(9):1209-1215.
11. Smale BF, Mullen JL, Buzby GP, Rosato EF. The efficacy of nutritional assessment and support in cancer surgery. Cancer. 1981;47(10):2375-2381.
12. Mohri Y, Inoue Y, Tanaka K, Hiro J, Uchida K, Kusunoki M. Prognostic nutritional index predicts postoperative outcome in colorectal cancer. World J Surg. 2013;37(11):2688-2692.
13. Jiang N, Deng JY, Ding XW, et al. Prognostic nutritional index predicts postoperative complications and long-term outcomes of gastric cancer. World J Gastroenterol. 2014;20(30):10537-10544.
14. Pinato DJ, North BV, Sharma R. A novel, externally validated inflammation-based prognostic algorithm in hepatocellular carcinoma: the prognostic nutritional index (PNI). Brit J Cancer. 2012;106(8):1439-1445.
15. Yao ZH, Tian GY, Wan YY, et al. Prognostic nutritional index predicts outcomes of malignant pleural mesothelioma. J Cancer Res Clin Oncol. 2013;139(12):2117-2123.
16. Jian-Hui C, Iskandar EA, Cai Sh I, et al. Significance of Onodera’s prognostic nutritional index in patients with colorectal cancer: a large cohort study in a single Chinese institution. Tumour Biol. 2016;37(3):3277-3283.
17. Hong S, Zhou T, Fang W, et al. The prognostic nutritional index (PNI) predicts overall survival of small-cell lung cancer patients. Tumour Biol. 2015;36(5):3389-9337.
18. Mohil RS, Agarwal A, Singh N, Arora J, Bhatnagar D. Does nutritional status play a role in patients undergoing emergency laparotomy? E Spen Eur E J Clin Nutr Metab. 2008;3(5):e226-e231.
19. Kay SP, Moreland JR, Schmitter E. Nutritional status and wound healing in lower extremity amputations. Clin Orthop Relat Res. 1987;(217):253-256.
20. Dickhaut SC, DeLee JC, Page CP. Nutritional status: importance in predicting wound-healing after amputation. J Bone Joint Surg Am. 1984;66(1):71-75.
21. Casey J, Flinn WR, Yao JS, Fahey V, Pawlowski J, Bergan JJ. Correlation of immune and nutritional status with wound complications in patients undergoing vascular operations. Surgery. 1983;93(6):822-827.
22. Gu Q, Wang D, Cui C, Gao Y, Xia G, Cui X. Effects of radiation on wound healing. J Environ Pathol Toxicol Oncol. 1998;17(2):117-123.
Wound complications after pre- or post-operative radiation for soft tissue sarcomas are well established.1 The ability to predict who will have a wound complication remains difficult. Some studies have looked at risk factors such as smoking, and the preoperative nutritional status of patients has been identified as a risk factor for wound complication in patients with elective orthopedic surgical procedures.2 One validated method of measuring preoperative nutritional status in patients with gastrointestinal malignant tumors has been with Onodera’s Prognostic Nutritional Index (OPNI). It uses the patient’s preoperative albumin (g/dL) and absolute lymphocyte values (per mm3). The prognostic value of the OPNI has been demonstrated in patients with colorectal, esophageal, and gastric cancers, and has been shown to be prognostic for postoperative wound healing and overall prognosis.3-5 In this study, we investigate the significance of preoperative nutritional status, measured by OPNI, as a predictor of wound complications in patients treated with pre- or postoperative radiation for soft tissue sarcoma.
Methods
After receiving Institutional Review Board approval for the study, we conducted a retrospective review of consecutive patients treated during July 2012-April 2016 for a soft tissue sarcoma by the orthopedic oncology division at Cooper University Hospital in Camden, New Jersey. Inclusion criteria were patients with biopsy-proven soft tissue sarcoma, who were older than 18 years, had received pre- or postoperative radiation, and who had a recorded preoperative albumin and total lymphocyte count. A minimum follow-up of 3 months was required to assess for postoperative wound complications. Exclusion criteria included patients who had a bone sarcoma, had not received radiation therapy, or had a missing preoperative albumin or total lymphocyte count.
All of the surgeries were performed by 2 fellowshiptrained orthopedic oncologists. Patients received either pre- or postoperative radiation therapy by multiple radiation oncologists.
The OPNI was calculated based on the published formula OPNI = (10*albumin level [g/dL]) + (0.005*total lymphocyte count [per mm3]). The albumin level and total lymphocyte counts closest to the index operation were chosen.
Demographic information including gender, age at diagnosis, height, and weight were recorded. Data related to the patients’ pathologic diagnosis, stage at presentation, radiation therapy, and surgical resection were collected. A minor wound complication was defined as a wound problem that did not require operative intervention. Major wound complication was defined as a complication requiring operative intervention with or without flap reconstruction. Wound complications occurring within the 3-month postoperative period were considered.
Univariate and multiple variable analysis was performed. A P value <.05 was considered significant. A receiver operating curve as well as recursive partitioning was performed for OPNI and age to determine the best cut-off point to use in the analysis. The Sobel test was used to evaluate mediation. All statistical analysis was performed using SAS v9.4 and JMP10. (SAS Institute, Cary, NC).
Results
In all, 44 patients (28 men, 16 women) were included in the study. Their mean age was 61.2 years (range, 19-94). The average size of the tumors was 8.5 cm in greatest dimension (range, 1.2-27.4 cm), and all of the patients had nonmetastatic disease at the time of surgical resection; 37 patients had R0 resections, and 7 patients had a positive margin from an outside hospital, but obtained R0 resections on a subsequent resection (Table 1 and Table 2).
In all, 30 patients received preoperative radiation, 14 patients received postoperative radiation, 32 patients received external beam radiation, 8 received Cyberknife treatment, and information for 4 patients was not unavailable. Mean preoperative external beam radiation and Cyberknife dose was 4,931 Gy and 3,750 Gy, respectively. Mean postoperative external beam and Cyberknife radiation dose was 6,077 Gy and 4,000 Gy, respectively. When evaluating radiation dose delivered between those who had wound complications and those who did not, there was no significant difference (Table 3).
Of the total, 13 patients had a wound complication (30%). Ten patients had preoperative radiation, and 3 had postoperative radiation. Ten patients had major wound complications requiring a combined 27 surgeries. Three patients had minor wound complications, which resolved with conservative management. One patient had a major wound complication in the group that had an initial R1 resection.
The OPNI was calculated based on the aforementioned formula. When the univariate analysis was performed, only age and OPNI were statistically significant. Patients older than 72.6 years had a 6.8 times higher risk of a wound complication (P = .01; 95% confidence interval [CI], 1.6-28.7). When the OPNI value of 45.4 was used as the threshold, a patient with a preoperative OPNI value of <45.4 had a 7.5 times increased risk of developing a wound complication (P = .005; 95% CI, 1.8-31.0).
When the receiver operating curve and recursive partitioning was performed, an OPNI value of 45.4 showed a sensitivity of 62% and specificity of 82% in predicting wound complications (Figure 1).
When a multiple variable analysis was performed, OPNI and age were not statistically significant (P = .06 and P = .11, respectively). A test for mediation was performed, and the OPNI seemed to mediate the effect age has on wound complications, accounting for 36% of the total effect (Sobel test statistic, 1.79; P = .07).
Discussion
Wound complications after pre- and postoperative radiation for soft tissue sarcomas are well known. The best study to date to demonstrate that relationship was a randomized controlled trial performed in Canada, which showed that preoperative radiation resulted in 37% wound complications, compared with 17% for postoperative radiation.6 In that study, of the wound complications in both radiation types, more than 50%-60% required a secondary surgical procedure, designating it as a major wound complication. Other variables that have been shown to contribute to wound complications include being older than 40 years and/or having large tumors, diabetes, peripheral vascular disease, and begin a smoker.7-10
In our study, we applied OPNI to orthopedic oncology and showed that the patient’s age and preoperative nutritional status were significant predictors of developing a wound complication. An OPNI of <45.4 increased the chance of a wound complication by 7.5 times. Being older than 73 years increased the risk of a wound complication by 6.8 times. Most of these wound complications were major and required surgical intervention.
In general surgical oncology, the evaluation of nutritional status has had a significant impact on the care of patients, especially for those patients undergoing gastrointestinal surgery. The OPNI was initially designed to assess the nutritional and immunological statuses of patients undergoing gastrointestinal surgery.11 Preoperative OPNI has been shown to be a good predictor of postoperative complications and survival in patients with colorectal cancer, malignant mesothelioma, hepatocellular carcinoma and in patients who undergo total gastrectomy.12-15 Chen and colleagues evaluated the significance of OPNI in patients with colorectal cancer. They found an optimal cut-off value of 45. An OPNI value <45 has a sensitivity and specificity of 85% and 69%, respectively, in predicting 5-year overall survival.16 Hong and colleagues noted that an OPNI cut-off value of 52.6 as a predictor of overall survival.17
Poor preoperative nutritional status has been shown to have a negative impact on wound healing. In patients who underwent emergency laparotomy, a low OPNI had significantly higher rates of wound dehiscence and infection.18 This happens because protein deficiency leads to decreased wound tensile strength, decreased T-cell function, decreased phagocytic activity, which ultimately diminish the patient’s ability to heal and defend against wound infections.19-21
In soft tissue sarcoma patients, poor preoperative nutritional status is further compromised by radiation therapy to the wound. Gu and colleagues showed that radiation to wounds in mice showed early inhibition of the inflammatory phase, injury and inhibition of fibroblasts, and collagen formation, and then prolonged re-epithelialization.22 This “double hit” with radiation onto host tissue that is already nutritionally compromised could be an important cause of why wound complications occur at such high rates in our soft tissue sarcoma patients.
There are several limitations to this study. First, the study has a small sample size, which was a direct result of the number of patients who were excluded because an OPNI value could not be calculated for them. Second, we could not determine if the OPNI was more valuable in patients who underwent pre- or postoperative radiation. This study did not look at other nutritional indices such as prealbumin and vitamin levels. Third, the radiation was provided by different providers, so technique was variable, but the patients received nearly equivalent doses and variability in technique is likely limited. Fourth, we were not able to meaningfully analyze the role of chemotherapy in this patient population because there was a significant heterogeneity of patients receiving pre- and postoperative chemotherapy.
Our findings strongly suggest that a preoperative OPNI of <45.4 and being older than 73 years are strong predictors of patients who will experience a wound complication after radiation therapy for soft tissue sarcomas. This study has led us to start measuring preoperative albumin levels and assess complete metabolic panels. Our goal is to identify patients who are at high risk of wound complication and perform interventions to improve nutrition, then to study whether the interventions help lower the rates of wound complications.
Wound complications after pre- or post-operative radiation for soft tissue sarcomas are well established.1 The ability to predict who will have a wound complication remains difficult. Some studies have looked at risk factors such as smoking, and the preoperative nutritional status of patients has been identified as a risk factor for wound complication in patients with elective orthopedic surgical procedures.2 One validated method of measuring preoperative nutritional status in patients with gastrointestinal malignant tumors has been with Onodera’s Prognostic Nutritional Index (OPNI). It uses the patient’s preoperative albumin (g/dL) and absolute lymphocyte values (per mm3). The prognostic value of the OPNI has been demonstrated in patients with colorectal, esophageal, and gastric cancers, and has been shown to be prognostic for postoperative wound healing and overall prognosis.3-5 In this study, we investigate the significance of preoperative nutritional status, measured by OPNI, as a predictor of wound complications in patients treated with pre- or postoperative radiation for soft tissue sarcoma.
Methods
After receiving Institutional Review Board approval for the study, we conducted a retrospective review of consecutive patients treated during July 2012-April 2016 for a soft tissue sarcoma by the orthopedic oncology division at Cooper University Hospital in Camden, New Jersey. Inclusion criteria were patients with biopsy-proven soft tissue sarcoma, who were older than 18 years, had received pre- or postoperative radiation, and who had a recorded preoperative albumin and total lymphocyte count. A minimum follow-up of 3 months was required to assess for postoperative wound complications. Exclusion criteria included patients who had a bone sarcoma, had not received radiation therapy, or had a missing preoperative albumin or total lymphocyte count.
All of the surgeries were performed by 2 fellowshiptrained orthopedic oncologists. Patients received either pre- or postoperative radiation therapy by multiple radiation oncologists.
The OPNI was calculated based on the published formula OPNI = (10*albumin level [g/dL]) + (0.005*total lymphocyte count [per mm3]). The albumin level and total lymphocyte counts closest to the index operation were chosen.
Demographic information including gender, age at diagnosis, height, and weight were recorded. Data related to the patients’ pathologic diagnosis, stage at presentation, radiation therapy, and surgical resection were collected. A minor wound complication was defined as a wound problem that did not require operative intervention. Major wound complication was defined as a complication requiring operative intervention with or without flap reconstruction. Wound complications occurring within the 3-month postoperative period were considered.
Univariate and multiple variable analysis was performed. A P value <.05 was considered significant. A receiver operating curve as well as recursive partitioning was performed for OPNI and age to determine the best cut-off point to use in the analysis. The Sobel test was used to evaluate mediation. All statistical analysis was performed using SAS v9.4 and JMP10. (SAS Institute, Cary, NC).
Results
In all, 44 patients (28 men, 16 women) were included in the study. Their mean age was 61.2 years (range, 19-94). The average size of the tumors was 8.5 cm in greatest dimension (range, 1.2-27.4 cm), and all of the patients had nonmetastatic disease at the time of surgical resection; 37 patients had R0 resections, and 7 patients had a positive margin from an outside hospital, but obtained R0 resections on a subsequent resection (Table 1 and Table 2).
In all, 30 patients received preoperative radiation, 14 patients received postoperative radiation, 32 patients received external beam radiation, 8 received Cyberknife treatment, and information for 4 patients was not unavailable. Mean preoperative external beam radiation and Cyberknife dose was 4,931 Gy and 3,750 Gy, respectively. Mean postoperative external beam and Cyberknife radiation dose was 6,077 Gy and 4,000 Gy, respectively. When evaluating radiation dose delivered between those who had wound complications and those who did not, there was no significant difference (Table 3).
Of the total, 13 patients had a wound complication (30%). Ten patients had preoperative radiation, and 3 had postoperative radiation. Ten patients had major wound complications requiring a combined 27 surgeries. Three patients had minor wound complications, which resolved with conservative management. One patient had a major wound complication in the group that had an initial R1 resection.
The OPNI was calculated based on the aforementioned formula. When the univariate analysis was performed, only age and OPNI were statistically significant. Patients older than 72.6 years had a 6.8 times higher risk of a wound complication (P = .01; 95% confidence interval [CI], 1.6-28.7). When the OPNI value of 45.4 was used as the threshold, a patient with a preoperative OPNI value of <45.4 had a 7.5 times increased risk of developing a wound complication (P = .005; 95% CI, 1.8-31.0).
When the receiver operating curve and recursive partitioning was performed, an OPNI value of 45.4 showed a sensitivity of 62% and specificity of 82% in predicting wound complications (Figure 1).
When a multiple variable analysis was performed, OPNI and age were not statistically significant (P = .06 and P = .11, respectively). A test for mediation was performed, and the OPNI seemed to mediate the effect age has on wound complications, accounting for 36% of the total effect (Sobel test statistic, 1.79; P = .07).
Discussion
Wound complications after pre- and postoperative radiation for soft tissue sarcomas are well known. The best study to date to demonstrate that relationship was a randomized controlled trial performed in Canada, which showed that preoperative radiation resulted in 37% wound complications, compared with 17% for postoperative radiation.6 In that study, of the wound complications in both radiation types, more than 50%-60% required a secondary surgical procedure, designating it as a major wound complication. Other variables that have been shown to contribute to wound complications include being older than 40 years and/or having large tumors, diabetes, peripheral vascular disease, and begin a smoker.7-10
In our study, we applied OPNI to orthopedic oncology and showed that the patient’s age and preoperative nutritional status were significant predictors of developing a wound complication. An OPNI of <45.4 increased the chance of a wound complication by 7.5 times. Being older than 73 years increased the risk of a wound complication by 6.8 times. Most of these wound complications were major and required surgical intervention.
In general surgical oncology, the evaluation of nutritional status has had a significant impact on the care of patients, especially for those patients undergoing gastrointestinal surgery. The OPNI was initially designed to assess the nutritional and immunological statuses of patients undergoing gastrointestinal surgery.11 Preoperative OPNI has been shown to be a good predictor of postoperative complications and survival in patients with colorectal cancer, malignant mesothelioma, hepatocellular carcinoma and in patients who undergo total gastrectomy.12-15 Chen and colleagues evaluated the significance of OPNI in patients with colorectal cancer. They found an optimal cut-off value of 45. An OPNI value <45 has a sensitivity and specificity of 85% and 69%, respectively, in predicting 5-year overall survival.16 Hong and colleagues noted that an OPNI cut-off value of 52.6 as a predictor of overall survival.17
Poor preoperative nutritional status has been shown to have a negative impact on wound healing. In patients who underwent emergency laparotomy, a low OPNI had significantly higher rates of wound dehiscence and infection.18 This happens because protein deficiency leads to decreased wound tensile strength, decreased T-cell function, decreased phagocytic activity, which ultimately diminish the patient’s ability to heal and defend against wound infections.19-21
In soft tissue sarcoma patients, poor preoperative nutritional status is further compromised by radiation therapy to the wound. Gu and colleagues showed that radiation to wounds in mice showed early inhibition of the inflammatory phase, injury and inhibition of fibroblasts, and collagen formation, and then prolonged re-epithelialization.22 This “double hit” with radiation onto host tissue that is already nutritionally compromised could be an important cause of why wound complications occur at such high rates in our soft tissue sarcoma patients.
There are several limitations to this study. First, the study has a small sample size, which was a direct result of the number of patients who were excluded because an OPNI value could not be calculated for them. Second, we could not determine if the OPNI was more valuable in patients who underwent pre- or postoperative radiation. This study did not look at other nutritional indices such as prealbumin and vitamin levels. Third, the radiation was provided by different providers, so technique was variable, but the patients received nearly equivalent doses and variability in technique is likely limited. Fourth, we were not able to meaningfully analyze the role of chemotherapy in this patient population because there was a significant heterogeneity of patients receiving pre- and postoperative chemotherapy.
Our findings strongly suggest that a preoperative OPNI of <45.4 and being older than 73 years are strong predictors of patients who will experience a wound complication after radiation therapy for soft tissue sarcomas. This study has led us to start measuring preoperative albumin levels and assess complete metabolic panels. Our goal is to identify patients who are at high risk of wound complication and perform interventions to improve nutrition, then to study whether the interventions help lower the rates of wound complications.
1. Ormsby MV, Hilaris BS, Nori D, Brennan MF. Wound complications of adjuvant radiation therapy in patients with soft-tissue sarcomas. Ann Surg. 1989;210(1):93-99.
2. Greene KA, Wilde AH, Stulberg BN. Preoperative nutritional status of total joint patients: relationship to postoperative wound complications. J Arthroplasty. 1991;6(4):321-325.
3. Nozoe T, Kimura Y, Ishida M, Saeki H, Korenaga D, Sugimachi K. Correlation of pre-operative nutritional condition with post-operative complications in surgical treatment for oesophageal carcinoma. Eur J Surg Oncol. 2002;28(4):396-400.
4. Nozoe T, Kohno M, Iguchi T, et al. The prognostic nutritional index can be a prognostic indicator in colorectal carcinoma. Surg Today. 2012;42(6):532-535.
5. Nozoe T, Ninomiya M, Maeda T, Matsukuma A, Nakashima H, Ezaki T. Prognostic nutritional index: a tool to predict the biological aggressiveness of gastric carcinoma. Surg Today. 2010;40(5):440-443.
6. O’Sullivan B, Davis AM, Turcotte R, Bell R, Catton C, Chabot P, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002;359(9325):2235-2241.
7. Peat BG, Bell RS, Davis A, et al. Wound-healing complications after soft-tissue sarcoma surgery. Plast Reconstr Surg. 1994;93(5):980-987.
8. Kunisada T, Ngan SY, Powell G, Choong PF. Wound complications following pre-operative radiotherapy for soft tissue sarcoma. Eur J Surg Oncol. 2002;28(1):75-79.
9. Saddegh MK, Bauer HC. Wound complication in surgery of soft tissue sarcoma: analysis of 103 consecutive patients managed without adjuvant therapy. Clin Orthop Relat Res. 1993;289:247-253.
10. Tseng JF, Ballo MT, Langstein HN, et al. The effect of preoperative radiotherapy and reconstructive surgery on wound complications after resection of extremity soft-tissue sarcomas. Ann Surg Oncol. 2006;13(9):1209-1215.
11. Smale BF, Mullen JL, Buzby GP, Rosato EF. The efficacy of nutritional assessment and support in cancer surgery. Cancer. 1981;47(10):2375-2381.
12. Mohri Y, Inoue Y, Tanaka K, Hiro J, Uchida K, Kusunoki M. Prognostic nutritional index predicts postoperative outcome in colorectal cancer. World J Surg. 2013;37(11):2688-2692.
13. Jiang N, Deng JY, Ding XW, et al. Prognostic nutritional index predicts postoperative complications and long-term outcomes of gastric cancer. World J Gastroenterol. 2014;20(30):10537-10544.
14. Pinato DJ, North BV, Sharma R. A novel, externally validated inflammation-based prognostic algorithm in hepatocellular carcinoma: the prognostic nutritional index (PNI). Brit J Cancer. 2012;106(8):1439-1445.
15. Yao ZH, Tian GY, Wan YY, et al. Prognostic nutritional index predicts outcomes of malignant pleural mesothelioma. J Cancer Res Clin Oncol. 2013;139(12):2117-2123.
16. Jian-Hui C, Iskandar EA, Cai Sh I, et al. Significance of Onodera’s prognostic nutritional index in patients with colorectal cancer: a large cohort study in a single Chinese institution. Tumour Biol. 2016;37(3):3277-3283.
17. Hong S, Zhou T, Fang W, et al. The prognostic nutritional index (PNI) predicts overall survival of small-cell lung cancer patients. Tumour Biol. 2015;36(5):3389-9337.
18. Mohil RS, Agarwal A, Singh N, Arora J, Bhatnagar D. Does nutritional status play a role in patients undergoing emergency laparotomy? E Spen Eur E J Clin Nutr Metab. 2008;3(5):e226-e231.
19. Kay SP, Moreland JR, Schmitter E. Nutritional status and wound healing in lower extremity amputations. Clin Orthop Relat Res. 1987;(217):253-256.
20. Dickhaut SC, DeLee JC, Page CP. Nutritional status: importance in predicting wound-healing after amputation. J Bone Joint Surg Am. 1984;66(1):71-75.
21. Casey J, Flinn WR, Yao JS, Fahey V, Pawlowski J, Bergan JJ. Correlation of immune and nutritional status with wound complications in patients undergoing vascular operations. Surgery. 1983;93(6):822-827.
22. Gu Q, Wang D, Cui C, Gao Y, Xia G, Cui X. Effects of radiation on wound healing. J Environ Pathol Toxicol Oncol. 1998;17(2):117-123.
1. Ormsby MV, Hilaris BS, Nori D, Brennan MF. Wound complications of adjuvant radiation therapy in patients with soft-tissue sarcomas. Ann Surg. 1989;210(1):93-99.
2. Greene KA, Wilde AH, Stulberg BN. Preoperative nutritional status of total joint patients: relationship to postoperative wound complications. J Arthroplasty. 1991;6(4):321-325.
3. Nozoe T, Kimura Y, Ishida M, Saeki H, Korenaga D, Sugimachi K. Correlation of pre-operative nutritional condition with post-operative complications in surgical treatment for oesophageal carcinoma. Eur J Surg Oncol. 2002;28(4):396-400.
4. Nozoe T, Kohno M, Iguchi T, et al. The prognostic nutritional index can be a prognostic indicator in colorectal carcinoma. Surg Today. 2012;42(6):532-535.
5. Nozoe T, Ninomiya M, Maeda T, Matsukuma A, Nakashima H, Ezaki T. Prognostic nutritional index: a tool to predict the biological aggressiveness of gastric carcinoma. Surg Today. 2010;40(5):440-443.
6. O’Sullivan B, Davis AM, Turcotte R, Bell R, Catton C, Chabot P, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002;359(9325):2235-2241.
7. Peat BG, Bell RS, Davis A, et al. Wound-healing complications after soft-tissue sarcoma surgery. Plast Reconstr Surg. 1994;93(5):980-987.
8. Kunisada T, Ngan SY, Powell G, Choong PF. Wound complications following pre-operative radiotherapy for soft tissue sarcoma. Eur J Surg Oncol. 2002;28(1):75-79.
9. Saddegh MK, Bauer HC. Wound complication in surgery of soft tissue sarcoma: analysis of 103 consecutive patients managed without adjuvant therapy. Clin Orthop Relat Res. 1993;289:247-253.
10. Tseng JF, Ballo MT, Langstein HN, et al. The effect of preoperative radiotherapy and reconstructive surgery on wound complications after resection of extremity soft-tissue sarcomas. Ann Surg Oncol. 2006;13(9):1209-1215.
11. Smale BF, Mullen JL, Buzby GP, Rosato EF. The efficacy of nutritional assessment and support in cancer surgery. Cancer. 1981;47(10):2375-2381.
12. Mohri Y, Inoue Y, Tanaka K, Hiro J, Uchida K, Kusunoki M. Prognostic nutritional index predicts postoperative outcome in colorectal cancer. World J Surg. 2013;37(11):2688-2692.
13. Jiang N, Deng JY, Ding XW, et al. Prognostic nutritional index predicts postoperative complications and long-term outcomes of gastric cancer. World J Gastroenterol. 2014;20(30):10537-10544.
14. Pinato DJ, North BV, Sharma R. A novel, externally validated inflammation-based prognostic algorithm in hepatocellular carcinoma: the prognostic nutritional index (PNI). Brit J Cancer. 2012;106(8):1439-1445.
15. Yao ZH, Tian GY, Wan YY, et al. Prognostic nutritional index predicts outcomes of malignant pleural mesothelioma. J Cancer Res Clin Oncol. 2013;139(12):2117-2123.
16. Jian-Hui C, Iskandar EA, Cai Sh I, et al. Significance of Onodera’s prognostic nutritional index in patients with colorectal cancer: a large cohort study in a single Chinese institution. Tumour Biol. 2016;37(3):3277-3283.
17. Hong S, Zhou T, Fang W, et al. The prognostic nutritional index (PNI) predicts overall survival of small-cell lung cancer patients. Tumour Biol. 2015;36(5):3389-9337.
18. Mohil RS, Agarwal A, Singh N, Arora J, Bhatnagar D. Does nutritional status play a role in patients undergoing emergency laparotomy? E Spen Eur E J Clin Nutr Metab. 2008;3(5):e226-e231.
19. Kay SP, Moreland JR, Schmitter E. Nutritional status and wound healing in lower extremity amputations. Clin Orthop Relat Res. 1987;(217):253-256.
20. Dickhaut SC, DeLee JC, Page CP. Nutritional status: importance in predicting wound-healing after amputation. J Bone Joint Surg Am. 1984;66(1):71-75.
21. Casey J, Flinn WR, Yao JS, Fahey V, Pawlowski J, Bergan JJ. Correlation of immune and nutritional status with wound complications in patients undergoing vascular operations. Surgery. 1983;93(6):822-827.
22. Gu Q, Wang D, Cui C, Gao Y, Xia G, Cui X. Effects of radiation on wound healing. J Environ Pathol Toxicol Oncol. 1998;17(2):117-123.
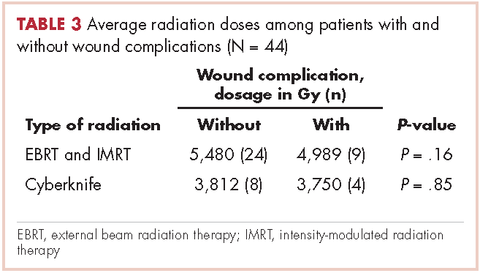
Eribulin superior to dacarbazine in advanced liposarcoma
In patients with advanced liposarcoma, eribulin was superior to dacarbazine in improving both overall and progression-free survival, investigators report.
In a phase 3 trial that included 452 patients with advanced liposarcoma (LPS), those treated with eribulin had an overall survival of 15.6 months, compared with 8.4 months with dacarbazine (hazard ratio, 0.51; 95% CI, 0.35 to 0.75; P less than .001).
Longer overall survival was also seen in all histologic subtypes of LPS and in all geographic regions included in the study.
Median progression-free survival was significantly longer among patients in the eribulin arm, compared with those who received dacarbazine, at 2.9 versus 1.7 months (HR, 0.521; 95% CI, 0.35 to 0.78; nominal P = .0015).
“Our findings demonstrate that eribulin is an important treatment option for patients with previously treated LPS, a sarcoma subtype for which limited treatment options are available,” wrote lead author George D. Demetri, MD, of the Dana-Farber Cancer Institute and Brigham and Women’s Cancer Center, both in Boston, and his colleagues (J Clin Oncol. 2017 Aug 30. doi: 10.1200/JCO.2016.71.6605).
A larger randomized, phase 3 prospective study had found that eribulin significantly improved overall survival versus dacarbazine (13.5 versus 11.5 months, respectively; HR, 0.77; 95% CI, 0.62-0.95; P = .0169) in patients with LPS and leiomyosarcoma. Toxicity was also manageable and similar to that observed when used to treat breast cancer.
However, the survival benefit with eribulin was greater in patients with LPS than those with leiomyosarcoma; on the basis of this trial, eribulin was approved in both the United States and European Union for use in advanced LPS in patients who have received a prior anthracycline-containing regimen.
The current study is a subgroup analysis of 452 patients with advanced or metastatic LPS that is dedifferentiated, myxoid/round cell, or pleomorphic and has been deemed incurable by surgery or radiotherapy; patients were randomized to receive either eribulin mesylate (1.4 mg/m2 intravenously on days 1 and 8) or dacarbazine (850, 1,000, or 1,200 mg/m2 intravenously on day 1) every 21 days.
The largest difference in overall survival for eribulin versus dacarbazine was observed in patients with pleomorphic LPS (22.2 versus 6.7 months), but there were only 23 patients (16.2%) with this disease subtype included in the study. Thus, the authors caution that this particular finding should be interpreted carefully.
All patients who received eribulin and 95.8% treated with dacarbazine experienced adverse events, with the majority being treatment related (95.7% versus 84.7% in the eribulin and dacarbazine arms, respectively).
“On the basis of its activity in patients with advanced LPS, eribulin merits further exploration in earlier lines of treatment, either alone or in combination with other therapies,” the authors concluded.
In patients with advanced liposarcoma, eribulin was superior to dacarbazine in improving both overall and progression-free survival, investigators report.
In a phase 3 trial that included 452 patients with advanced liposarcoma (LPS), those treated with eribulin had an overall survival of 15.6 months, compared with 8.4 months with dacarbazine (hazard ratio, 0.51; 95% CI, 0.35 to 0.75; P less than .001).
Longer overall survival was also seen in all histologic subtypes of LPS and in all geographic regions included in the study.
Median progression-free survival was significantly longer among patients in the eribulin arm, compared with those who received dacarbazine, at 2.9 versus 1.7 months (HR, 0.521; 95% CI, 0.35 to 0.78; nominal P = .0015).
“Our findings demonstrate that eribulin is an important treatment option for patients with previously treated LPS, a sarcoma subtype for which limited treatment options are available,” wrote lead author George D. Demetri, MD, of the Dana-Farber Cancer Institute and Brigham and Women’s Cancer Center, both in Boston, and his colleagues (J Clin Oncol. 2017 Aug 30. doi: 10.1200/JCO.2016.71.6605).
A larger randomized, phase 3 prospective study had found that eribulin significantly improved overall survival versus dacarbazine (13.5 versus 11.5 months, respectively; HR, 0.77; 95% CI, 0.62-0.95; P = .0169) in patients with LPS and leiomyosarcoma. Toxicity was also manageable and similar to that observed when used to treat breast cancer.
However, the survival benefit with eribulin was greater in patients with LPS than those with leiomyosarcoma; on the basis of this trial, eribulin was approved in both the United States and European Union for use in advanced LPS in patients who have received a prior anthracycline-containing regimen.
The current study is a subgroup analysis of 452 patients with advanced or metastatic LPS that is dedifferentiated, myxoid/round cell, or pleomorphic and has been deemed incurable by surgery or radiotherapy; patients were randomized to receive either eribulin mesylate (1.4 mg/m2 intravenously on days 1 and 8) or dacarbazine (850, 1,000, or 1,200 mg/m2 intravenously on day 1) every 21 days.
The largest difference in overall survival for eribulin versus dacarbazine was observed in patients with pleomorphic LPS (22.2 versus 6.7 months), but there were only 23 patients (16.2%) with this disease subtype included in the study. Thus, the authors caution that this particular finding should be interpreted carefully.
All patients who received eribulin and 95.8% treated with dacarbazine experienced adverse events, with the majority being treatment related (95.7% versus 84.7% in the eribulin and dacarbazine arms, respectively).
“On the basis of its activity in patients with advanced LPS, eribulin merits further exploration in earlier lines of treatment, either alone or in combination with other therapies,” the authors concluded.
In patients with advanced liposarcoma, eribulin was superior to dacarbazine in improving both overall and progression-free survival, investigators report.
In a phase 3 trial that included 452 patients with advanced liposarcoma (LPS), those treated with eribulin had an overall survival of 15.6 months, compared with 8.4 months with dacarbazine (hazard ratio, 0.51; 95% CI, 0.35 to 0.75; P less than .001).
Longer overall survival was also seen in all histologic subtypes of LPS and in all geographic regions included in the study.
Median progression-free survival was significantly longer among patients in the eribulin arm, compared with those who received dacarbazine, at 2.9 versus 1.7 months (HR, 0.521; 95% CI, 0.35 to 0.78; nominal P = .0015).
“Our findings demonstrate that eribulin is an important treatment option for patients with previously treated LPS, a sarcoma subtype for which limited treatment options are available,” wrote lead author George D. Demetri, MD, of the Dana-Farber Cancer Institute and Brigham and Women’s Cancer Center, both in Boston, and his colleagues (J Clin Oncol. 2017 Aug 30. doi: 10.1200/JCO.2016.71.6605).
A larger randomized, phase 3 prospective study had found that eribulin significantly improved overall survival versus dacarbazine (13.5 versus 11.5 months, respectively; HR, 0.77; 95% CI, 0.62-0.95; P = .0169) in patients with LPS and leiomyosarcoma. Toxicity was also manageable and similar to that observed when used to treat breast cancer.
However, the survival benefit with eribulin was greater in patients with LPS than those with leiomyosarcoma; on the basis of this trial, eribulin was approved in both the United States and European Union for use in advanced LPS in patients who have received a prior anthracycline-containing regimen.
The current study is a subgroup analysis of 452 patients with advanced or metastatic LPS that is dedifferentiated, myxoid/round cell, or pleomorphic and has been deemed incurable by surgery or radiotherapy; patients were randomized to receive either eribulin mesylate (1.4 mg/m2 intravenously on days 1 and 8) or dacarbazine (850, 1,000, or 1,200 mg/m2 intravenously on day 1) every 21 days.
The largest difference in overall survival for eribulin versus dacarbazine was observed in patients with pleomorphic LPS (22.2 versus 6.7 months), but there were only 23 patients (16.2%) with this disease subtype included in the study. Thus, the authors caution that this particular finding should be interpreted carefully.
All patients who received eribulin and 95.8% treated with dacarbazine experienced adverse events, with the majority being treatment related (95.7% versus 84.7% in the eribulin and dacarbazine arms, respectively).
“On the basis of its activity in patients with advanced LPS, eribulin merits further exploration in earlier lines of treatment, either alone or in combination with other therapies,” the authors concluded.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Eribulin is superior to dacarbazine in improving outcomes in advanced liposarcoma.
Major finding: Overall survival with eribulin was 15.6 months versus 8.4 months with dacarbazine (hazard ratio, 0.51; 95% CI, 0.35 to 0.75; P less than .001).
Data source: Phase 3 randomized, prospective clinical trial that included 452 patients with advanced liposarcoma.
Disclosures: The study was supported by Eisai. Dr. Demetri and several of the coauthors report relationships with industry including Eisai.
Management of high-grade pleomorphic sarcoma with colon metastasis
Soft tissue sarcomas (STS) are a heterogeneous group of tumors of mesenchymal origin that represent a rare form of adult malignancy. According to the World Health Organization classification system, there are more than 100 histologic subtypes of sarcoma based on tissue of origin. Staging criteria most commonly use the American Joint Committee on Cancer’s TNM Classification of Malignant Tumours. About 50% of soft tissue sarcomas originate in the lower extremity.1 Advancements in the use of multimodal therapy have reduced the need for amputation and allowed for equally effective treatment strategies that use limb-sparing surgical resections.
Although most sarcoma metastases spread in a hematogenous fashion, nodal spread is underestimated. Certain histologic subtypes carry a higher predilection for nodal involvement: these include rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, vascular sarcoma, and clear-cell sarcoma.2-4 Fong and colleagues have reported that 2.6% of sarcomas have lymphatic spread.3 In the current report, we describe a rare observation of locoregional pelvic nodal metastases from a large undifferentiated pleomorphic sarcoma of the right thigh.
Case presentation and summary
A 63-year-old white woman had a 1-year history of a right thigh mass and an unintentional weight loss of 40 lb. After a year of chiropractic care, she was referred to a physician because of palpable inguinal adenopathy and a 20-cm mass in the medial compartment of the right thigh, with heterogeneous appearance on a magnetic-resonance imaging scan (Figure 1). The patient was referred to the sarcoma transdisciplinary team for evaluation. She was diagnosed by core needle biopsy with a high-grade malignant epithelioid and spindle-cell neoplasm, favoring pleomorphic sarcoma. The metastatic work-up confirmed locoregional right inguinal and retroperitoneal lymph node disease, with 2 lung nodules that were too small to characterize. She also had a paraneoplastic leukocytosis with a white blood cell count of 45,500 cells/ml (normal 10,500 cells/ml).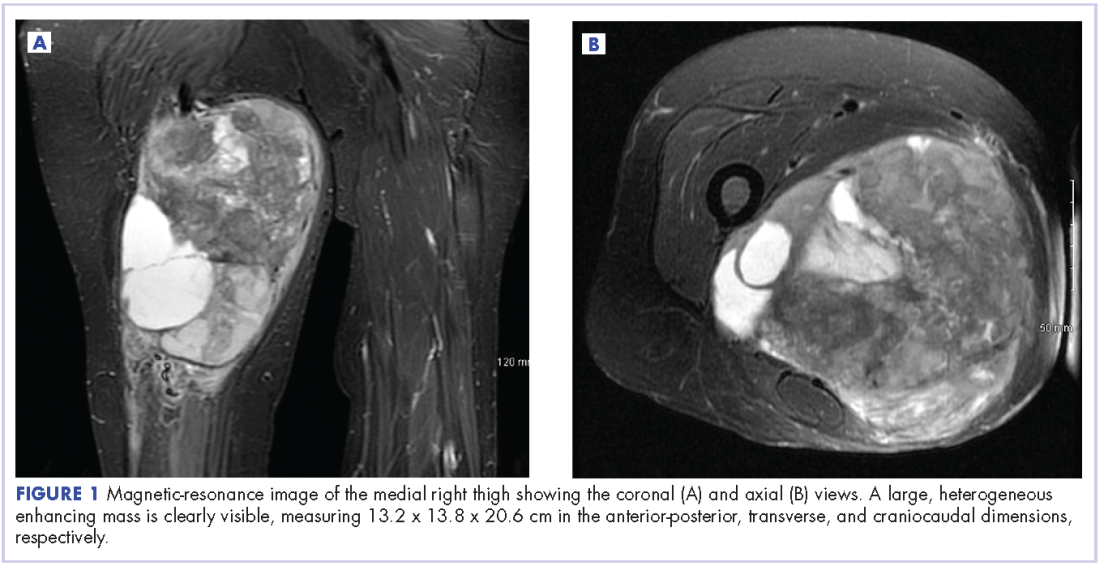
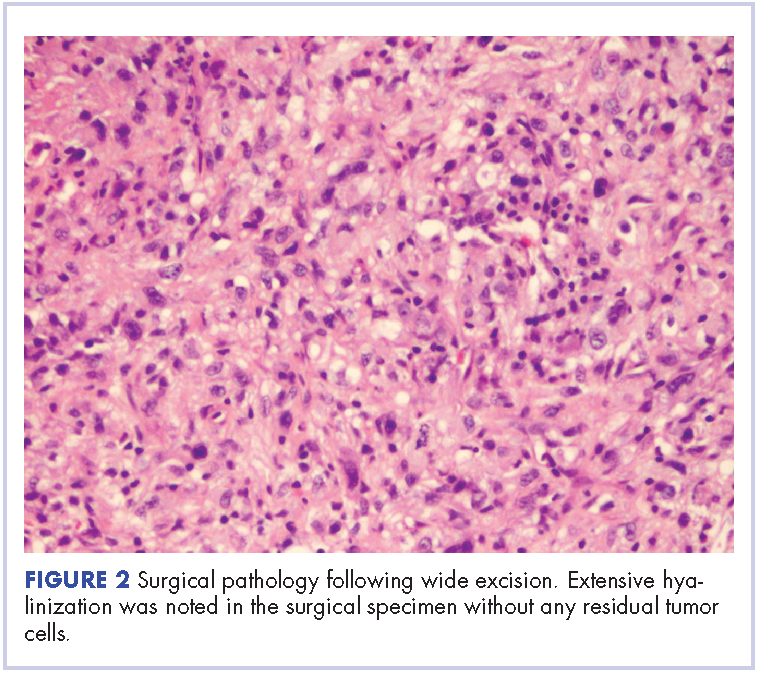
About 8 months after surgery, she presented to the emergency room with a 3-day history of blood per rectum, anemia, and fatigue. She also reported a weight loss of 10 lb in the previous month. She was admitted for hydration and monitoring. Although computed-tomography (CT) scans of the abdomen and chest done during the previous month and had been interpreted as having no evidence of recurrence or any lymph node disease, the results of an inpatient colonoscopy revealed 2 colonic masses, 1 in the ascending colon and another in the transverse colon. The biopsy findings were consistent with undifferentiated pleomorphic sarcoma, favoring epithelioid histology. The CT scans were re-evaluated in light of these colonoscopic findings. These masses were visible retrospectively on imaging but had been interpreted as stool given the lack of abnormality on imaging 3 months before.
Adequate re-staging was complete, and without other evidence of disease, an extended right hemicolectomy was performed. The postoperative pathology report described geographically 2 distinct masses: a 7-cm mass in the ascending colon, about 3 cm from the ileocecal valve; and a 4-cm mass in the transverse colon, about 7.5 cm from the distal margin of resection. Both masses were identified as high-grade pleomorphic sarcoma. Again, all nodes recovered were negative for malignancy (0/5). Of note is that the background colonic mucosa showed active multifocal colitis with deep inflammatory activity likely consistent with a paraneoplastic syndrome.
Discussion
Surgery remains the primary treatment modality for localized soft tissue sarcoma. Obtaining a margin-free resection, while maintaining optimal function, is the objective with extremity sarcoma. In addition to surgical resection, doxorubicin-based adjuvant chemotherapy remains the standard of care with modest improvement in overall survival and disease-free survival, especially in sarcomas of the extremities.5,6 Gemcitabine also has activity in soft tissue sarcomas7 and might synergize with docetaxel. High response rates of 53% with fixed dose infusion rates of these agents in uterine leiomyosarcoma led the Sarcoma Alliance for Research through Collaboration investigators to consider this regimen for other STS.8 An overall response rate of 16% was noted across all sarcoma subtypes, and undifferentiated pleomorphic sarcoma had a response rate of 32%.
In our experience, a modified schedule of gemcitabine and docetaxel is better tolerated than the standard every 3 weeks regimen or doxorubicin-based chemotherapies. As previously described, gemcitabine and docetaxel were administered to this patient every 2 weeks.9 This regimen, in our experience, is less toxic and preserves the dose intensity of the regimen. A complete pathologic necrosis of the primary tumor and regional nodal basin was observed, as well as pulmonary nodule regression, enabling an R0-wide excision and regional lymphadenectomy. The colonic recurrence was surprising, but easily managed with surgical resection.
Pathologic complete response after neoadjuvant chemotherapy is a rare event, observed in about 10% of patients. However, when observed, complete pathologic necrosis (>95%) provided a distant recurrence-free survival of 100% at 3 years.10-12
Our patient, despite achieving a complete pathologic response and excellent initial local control, ultimately experienced an isolated metastatic recurrence in the colon within 1 year of therapy. Data supports performing metastatectomy for stage IV extremity sarcoma for isolated pulmonary or hepatic burden in selected patients with improved survival rates.13,14 As far as we know, there is no published literature describing isolated colon metastases in the absence of liver burden from lower extremity soft tissue sarcomas, or the outcome of surgical resection and adjuvant therapy in these cases.
In conclusion, high-grade pleomorphic sarcoma often follows an aggressive clinical course with ultimately local and distant recurrence. Use of multimodal therapy may have a role in improving local control. Complete pathologic necrosis is a rare event that is predictive of improved outcome. Our case represents an unusual pattern of recurrence among patients with a complete pathologic response to neoadjuvant therapy with isolated colon metastases. Timely, comprehensive management together with vigilant surveillance remain key priorities in the long-term management of high-risk sarcoma.
1. Lawrence W Jr, Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. A pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349-359.
2. Riad S, Griffin AM, Liberman B, et al. Lymph node metastasis in soft tissue sarcoma in an extremity. Clin Orthop Relat Res. 2004:129-134.
3. Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217:72-77.
4. Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800-1808.
5. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647-1654.
6. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft tissue sarcoma. Cancer. 2008;113:573-581.
7. Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483-3489.
8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755-2763.
9. Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23:785-790.
10. MacDermed DM, Miller LL, Peabody TD, et al. Primary tumor necrosis predicts distant control in locally advanced soft tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2010;76:1147-1153.
11. Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol. 2001;19:3203-3209.
12. Shah D, Borys D, Martinez SR, et al. Complete pathologic response to neoadjuvant radiotherapy is predictive of oncological outcome in patients with soft tissue sarcoma. Anticancer Res. 2012;32:3911-3915.
13. Rehders A, Peiper M, Stoecklein NH, et al. Hepatic metastasectomy for soft tissue sarcomas: is it justified? World J Surg. 2009;33:111-117.
14. Pfannschmidt J, Hoffmann H, Schneider T, Dienemann H. Pulmonary metastasectomy for soft tissue sarcomas: is it justified? Recent Results Cancer Res. 2009;179:321-336.
Soft tissue sarcomas (STS) are a heterogeneous group of tumors of mesenchymal origin that represent a rare form of adult malignancy. According to the World Health Organization classification system, there are more than 100 histologic subtypes of sarcoma based on tissue of origin. Staging criteria most commonly use the American Joint Committee on Cancer’s TNM Classification of Malignant Tumours. About 50% of soft tissue sarcomas originate in the lower extremity.1 Advancements in the use of multimodal therapy have reduced the need for amputation and allowed for equally effective treatment strategies that use limb-sparing surgical resections.
Although most sarcoma metastases spread in a hematogenous fashion, nodal spread is underestimated. Certain histologic subtypes carry a higher predilection for nodal involvement: these include rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, vascular sarcoma, and clear-cell sarcoma.2-4 Fong and colleagues have reported that 2.6% of sarcomas have lymphatic spread.3 In the current report, we describe a rare observation of locoregional pelvic nodal metastases from a large undifferentiated pleomorphic sarcoma of the right thigh.
Case presentation and summary
A 63-year-old white woman had a 1-year history of a right thigh mass and an unintentional weight loss of 40 lb. After a year of chiropractic care, she was referred to a physician because of palpable inguinal adenopathy and a 20-cm mass in the medial compartment of the right thigh, with heterogeneous appearance on a magnetic-resonance imaging scan (Figure 1). The patient was referred to the sarcoma transdisciplinary team for evaluation. She was diagnosed by core needle biopsy with a high-grade malignant epithelioid and spindle-cell neoplasm, favoring pleomorphic sarcoma. The metastatic work-up confirmed locoregional right inguinal and retroperitoneal lymph node disease, with 2 lung nodules that were too small to characterize. She also had a paraneoplastic leukocytosis with a white blood cell count of 45,500 cells/ml (normal 10,500 cells/ml).
About 8 months after surgery, she presented to the emergency room with a 3-day history of blood per rectum, anemia, and fatigue. She also reported a weight loss of 10 lb in the previous month. She was admitted for hydration and monitoring. Although computed-tomography (CT) scans of the abdomen and chest done during the previous month and had been interpreted as having no evidence of recurrence or any lymph node disease, the results of an inpatient colonoscopy revealed 2 colonic masses, 1 in the ascending colon and another in the transverse colon. The biopsy findings were consistent with undifferentiated pleomorphic sarcoma, favoring epithelioid histology. The CT scans were re-evaluated in light of these colonoscopic findings. These masses were visible retrospectively on imaging but had been interpreted as stool given the lack of abnormality on imaging 3 months before.
Adequate re-staging was complete, and without other evidence of disease, an extended right hemicolectomy was performed. The postoperative pathology report described geographically 2 distinct masses: a 7-cm mass in the ascending colon, about 3 cm from the ileocecal valve; and a 4-cm mass in the transverse colon, about 7.5 cm from the distal margin of resection. Both masses were identified as high-grade pleomorphic sarcoma. Again, all nodes recovered were negative for malignancy (0/5). Of note is that the background colonic mucosa showed active multifocal colitis with deep inflammatory activity likely consistent with a paraneoplastic syndrome.
Discussion
Surgery remains the primary treatment modality for localized soft tissue sarcoma. Obtaining a margin-free resection, while maintaining optimal function, is the objective with extremity sarcoma. In addition to surgical resection, doxorubicin-based adjuvant chemotherapy remains the standard of care with modest improvement in overall survival and disease-free survival, especially in sarcomas of the extremities.5,6 Gemcitabine also has activity in soft tissue sarcomas7 and might synergize with docetaxel. High response rates of 53% with fixed dose infusion rates of these agents in uterine leiomyosarcoma led the Sarcoma Alliance for Research through Collaboration investigators to consider this regimen for other STS.8 An overall response rate of 16% was noted across all sarcoma subtypes, and undifferentiated pleomorphic sarcoma had a response rate of 32%.
In our experience, a modified schedule of gemcitabine and docetaxel is better tolerated than the standard every 3 weeks regimen or doxorubicin-based chemotherapies. As previously described, gemcitabine and docetaxel were administered to this patient every 2 weeks.9 This regimen, in our experience, is less toxic and preserves the dose intensity of the regimen. A complete pathologic necrosis of the primary tumor and regional nodal basin was observed, as well as pulmonary nodule regression, enabling an R0-wide excision and regional lymphadenectomy. The colonic recurrence was surprising, but easily managed with surgical resection.
Pathologic complete response after neoadjuvant chemotherapy is a rare event, observed in about 10% of patients. However, when observed, complete pathologic necrosis (>95%) provided a distant recurrence-free survival of 100% at 3 years.10-12
Our patient, despite achieving a complete pathologic response and excellent initial local control, ultimately experienced an isolated metastatic recurrence in the colon within 1 year of therapy. Data supports performing metastatectomy for stage IV extremity sarcoma for isolated pulmonary or hepatic burden in selected patients with improved survival rates.13,14 As far as we know, there is no published literature describing isolated colon metastases in the absence of liver burden from lower extremity soft tissue sarcomas, or the outcome of surgical resection and adjuvant therapy in these cases.
In conclusion, high-grade pleomorphic sarcoma often follows an aggressive clinical course with ultimately local and distant recurrence. Use of multimodal therapy may have a role in improving local control. Complete pathologic necrosis is a rare event that is predictive of improved outcome. Our case represents an unusual pattern of recurrence among patients with a complete pathologic response to neoadjuvant therapy with isolated colon metastases. Timely, comprehensive management together with vigilant surveillance remain key priorities in the long-term management of high-risk sarcoma.
Soft tissue sarcomas (STS) are a heterogeneous group of tumors of mesenchymal origin that represent a rare form of adult malignancy. According to the World Health Organization classification system, there are more than 100 histologic subtypes of sarcoma based on tissue of origin. Staging criteria most commonly use the American Joint Committee on Cancer’s TNM Classification of Malignant Tumours. About 50% of soft tissue sarcomas originate in the lower extremity.1 Advancements in the use of multimodal therapy have reduced the need for amputation and allowed for equally effective treatment strategies that use limb-sparing surgical resections.
Although most sarcoma metastases spread in a hematogenous fashion, nodal spread is underestimated. Certain histologic subtypes carry a higher predilection for nodal involvement: these include rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, vascular sarcoma, and clear-cell sarcoma.2-4 Fong and colleagues have reported that 2.6% of sarcomas have lymphatic spread.3 In the current report, we describe a rare observation of locoregional pelvic nodal metastases from a large undifferentiated pleomorphic sarcoma of the right thigh.
Case presentation and summary
A 63-year-old white woman had a 1-year history of a right thigh mass and an unintentional weight loss of 40 lb. After a year of chiropractic care, she was referred to a physician because of palpable inguinal adenopathy and a 20-cm mass in the medial compartment of the right thigh, with heterogeneous appearance on a magnetic-resonance imaging scan (Figure 1). The patient was referred to the sarcoma transdisciplinary team for evaluation. She was diagnosed by core needle biopsy with a high-grade malignant epithelioid and spindle-cell neoplasm, favoring pleomorphic sarcoma. The metastatic work-up confirmed locoregional right inguinal and retroperitoneal lymph node disease, with 2 lung nodules that were too small to characterize. She also had a paraneoplastic leukocytosis with a white blood cell count of 45,500 cells/ml (normal 10,500 cells/ml).
About 8 months after surgery, she presented to the emergency room with a 3-day history of blood per rectum, anemia, and fatigue. She also reported a weight loss of 10 lb in the previous month. She was admitted for hydration and monitoring. Although computed-tomography (CT) scans of the abdomen and chest done during the previous month and had been interpreted as having no evidence of recurrence or any lymph node disease, the results of an inpatient colonoscopy revealed 2 colonic masses, 1 in the ascending colon and another in the transverse colon. The biopsy findings were consistent with undifferentiated pleomorphic sarcoma, favoring epithelioid histology. The CT scans were re-evaluated in light of these colonoscopic findings. These masses were visible retrospectively on imaging but had been interpreted as stool given the lack of abnormality on imaging 3 months before.
Adequate re-staging was complete, and without other evidence of disease, an extended right hemicolectomy was performed. The postoperative pathology report described geographically 2 distinct masses: a 7-cm mass in the ascending colon, about 3 cm from the ileocecal valve; and a 4-cm mass in the transverse colon, about 7.5 cm from the distal margin of resection. Both masses were identified as high-grade pleomorphic sarcoma. Again, all nodes recovered were negative for malignancy (0/5). Of note is that the background colonic mucosa showed active multifocal colitis with deep inflammatory activity likely consistent with a paraneoplastic syndrome.
Discussion
Surgery remains the primary treatment modality for localized soft tissue sarcoma. Obtaining a margin-free resection, while maintaining optimal function, is the objective with extremity sarcoma. In addition to surgical resection, doxorubicin-based adjuvant chemotherapy remains the standard of care with modest improvement in overall survival and disease-free survival, especially in sarcomas of the extremities.5,6 Gemcitabine also has activity in soft tissue sarcomas7 and might synergize with docetaxel. High response rates of 53% with fixed dose infusion rates of these agents in uterine leiomyosarcoma led the Sarcoma Alliance for Research through Collaboration investigators to consider this regimen for other STS.8 An overall response rate of 16% was noted across all sarcoma subtypes, and undifferentiated pleomorphic sarcoma had a response rate of 32%.
In our experience, a modified schedule of gemcitabine and docetaxel is better tolerated than the standard every 3 weeks regimen or doxorubicin-based chemotherapies. As previously described, gemcitabine and docetaxel were administered to this patient every 2 weeks.9 This regimen, in our experience, is less toxic and preserves the dose intensity of the regimen. A complete pathologic necrosis of the primary tumor and regional nodal basin was observed, as well as pulmonary nodule regression, enabling an R0-wide excision and regional lymphadenectomy. The colonic recurrence was surprising, but easily managed with surgical resection.
Pathologic complete response after neoadjuvant chemotherapy is a rare event, observed in about 10% of patients. However, when observed, complete pathologic necrosis (>95%) provided a distant recurrence-free survival of 100% at 3 years.10-12
Our patient, despite achieving a complete pathologic response and excellent initial local control, ultimately experienced an isolated metastatic recurrence in the colon within 1 year of therapy. Data supports performing metastatectomy for stage IV extremity sarcoma for isolated pulmonary or hepatic burden in selected patients with improved survival rates.13,14 As far as we know, there is no published literature describing isolated colon metastases in the absence of liver burden from lower extremity soft tissue sarcomas, or the outcome of surgical resection and adjuvant therapy in these cases.
In conclusion, high-grade pleomorphic sarcoma often follows an aggressive clinical course with ultimately local and distant recurrence. Use of multimodal therapy may have a role in improving local control. Complete pathologic necrosis is a rare event that is predictive of improved outcome. Our case represents an unusual pattern of recurrence among patients with a complete pathologic response to neoadjuvant therapy with isolated colon metastases. Timely, comprehensive management together with vigilant surveillance remain key priorities in the long-term management of high-risk sarcoma.
1. Lawrence W Jr, Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. A pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349-359.
2. Riad S, Griffin AM, Liberman B, et al. Lymph node metastasis in soft tissue sarcoma in an extremity. Clin Orthop Relat Res. 2004:129-134.
3. Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217:72-77.
4. Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800-1808.
5. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647-1654.
6. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft tissue sarcoma. Cancer. 2008;113:573-581.
7. Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483-3489.
8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755-2763.
9. Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23:785-790.
10. MacDermed DM, Miller LL, Peabody TD, et al. Primary tumor necrosis predicts distant control in locally advanced soft tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2010;76:1147-1153.
11. Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol. 2001;19:3203-3209.
12. Shah D, Borys D, Martinez SR, et al. Complete pathologic response to neoadjuvant radiotherapy is predictive of oncological outcome in patients with soft tissue sarcoma. Anticancer Res. 2012;32:3911-3915.
13. Rehders A, Peiper M, Stoecklein NH, et al. Hepatic metastasectomy for soft tissue sarcomas: is it justified? World J Surg. 2009;33:111-117.
14. Pfannschmidt J, Hoffmann H, Schneider T, Dienemann H. Pulmonary metastasectomy for soft tissue sarcomas: is it justified? Recent Results Cancer Res. 2009;179:321-336.
1. Lawrence W Jr, Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. A pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349-359.
2. Riad S, Griffin AM, Liberman B, et al. Lymph node metastasis in soft tissue sarcoma in an extremity. Clin Orthop Relat Res. 2004:129-134.
3. Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217:72-77.
4. Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800-1808.
5. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647-1654.
6. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft tissue sarcoma. Cancer. 2008;113:573-581.
7. Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483-3489.
8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755-2763.
9. Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23:785-790.
10. MacDermed DM, Miller LL, Peabody TD, et al. Primary tumor necrosis predicts distant control in locally advanced soft tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2010;76:1147-1153.
11. Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol. 2001;19:3203-3209.
12. Shah D, Borys D, Martinez SR, et al. Complete pathologic response to neoadjuvant radiotherapy is predictive of oncological outcome in patients with soft tissue sarcoma. Anticancer Res. 2012;32:3911-3915.
13. Rehders A, Peiper M, Stoecklein NH, et al. Hepatic metastasectomy for soft tissue sarcomas: is it justified? World J Surg. 2009;33:111-117.
14. Pfannschmidt J, Hoffmann H, Schneider T, Dienemann H. Pulmonary metastasectomy for soft tissue sarcomas: is it justified? Recent Results Cancer Res. 2009;179:321-336.
Approval makes olaratumab the first first-line treatment option for soft tissue sarcoma in more than 40 years
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.