User login
FDA approves risankizumab (Skyrizi) for psoriatic arthritis
The Food and Drug Administration on Jan. 21 approved risankizumab-rzaa (Skyrizi) for a second indication – treating adults with active psoriatic arthritis (PsA) – making it the second anti–interleukin-23 monoclonal antibody available to treat PsA, according to an announcement from manufacturer AbbVie.
The agency previously approved risankizumab in April 2019 for adults with moderate to severe plaque psoriasis.
The dosing regimen for PsA is the same as it is for patients with moderate to severe plaque psoriasis: a single 150-mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4), and it can be administered alone or in combination with disease-modifying antirheumatic drugs (DMARDs).
Two phase 3 trials, KEEPsAKE 1 and KEEPsAKE 2, were the basis for the approval. These two trials tested the biologic agent in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or nonbiologic DMARDs. Fulfillment of the trials’ primary endpoint of at least a 20% improvement in American College of Rheumatology response criteria at 24 weeks occurred in 51.3%-57.3% of patients, compared with 26.5%-33.5% of placebo-treated patients.
Those on risankizumab also achieved significantly higher rates of ACR50 and ACR70 responses than those on placebo. In addition, patients with preexisting dactylitis and enthesitis experienced improvements in these PsA manifestations. Risankizumab was also associated with an improvement in physical function at 24 weeks on the Health Assessment Questionnaire–Disability Index, bettering placebo by a mean difference of 0.16-0.20 points in the two trials. A significantly higher percentage of patients who had psoriatic skin lesions experienced at least 90% improvement with risankizumab on the Psoriasis Area and Severity Index, compared with placebo.
AbbVie said that the safety profile of risankizumab in patients with PsA has been generally consistent with its effects in patients with plaque psoriasis.
The KEEPsAKE 1 and KEEPsAKE 2 studies are ongoing, and patients in the long-term extensions of the trials remain blinded to the original randomized allocation for the duration of the studies.
Phase 3 trials of risankizumab are also ongoing in patients with Crohn’s disease and ulcerative colitis.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on Jan. 21 approved risankizumab-rzaa (Skyrizi) for a second indication – treating adults with active psoriatic arthritis (PsA) – making it the second anti–interleukin-23 monoclonal antibody available to treat PsA, according to an announcement from manufacturer AbbVie.
The agency previously approved risankizumab in April 2019 for adults with moderate to severe plaque psoriasis.
The dosing regimen for PsA is the same as it is for patients with moderate to severe plaque psoriasis: a single 150-mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4), and it can be administered alone or in combination with disease-modifying antirheumatic drugs (DMARDs).
Two phase 3 trials, KEEPsAKE 1 and KEEPsAKE 2, were the basis for the approval. These two trials tested the biologic agent in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or nonbiologic DMARDs. Fulfillment of the trials’ primary endpoint of at least a 20% improvement in American College of Rheumatology response criteria at 24 weeks occurred in 51.3%-57.3% of patients, compared with 26.5%-33.5% of placebo-treated patients.
Those on risankizumab also achieved significantly higher rates of ACR50 and ACR70 responses than those on placebo. In addition, patients with preexisting dactylitis and enthesitis experienced improvements in these PsA manifestations. Risankizumab was also associated with an improvement in physical function at 24 weeks on the Health Assessment Questionnaire–Disability Index, bettering placebo by a mean difference of 0.16-0.20 points in the two trials. A significantly higher percentage of patients who had psoriatic skin lesions experienced at least 90% improvement with risankizumab on the Psoriasis Area and Severity Index, compared with placebo.
AbbVie said that the safety profile of risankizumab in patients with PsA has been generally consistent with its effects in patients with plaque psoriasis.
The KEEPsAKE 1 and KEEPsAKE 2 studies are ongoing, and patients in the long-term extensions of the trials remain blinded to the original randomized allocation for the duration of the studies.
Phase 3 trials of risankizumab are also ongoing in patients with Crohn’s disease and ulcerative colitis.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on Jan. 21 approved risankizumab-rzaa (Skyrizi) for a second indication – treating adults with active psoriatic arthritis (PsA) – making it the second anti–interleukin-23 monoclonal antibody available to treat PsA, according to an announcement from manufacturer AbbVie.
The agency previously approved risankizumab in April 2019 for adults with moderate to severe plaque psoriasis.
The dosing regimen for PsA is the same as it is for patients with moderate to severe plaque psoriasis: a single 150-mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4), and it can be administered alone or in combination with disease-modifying antirheumatic drugs (DMARDs).
Two phase 3 trials, KEEPsAKE 1 and KEEPsAKE 2, were the basis for the approval. These two trials tested the biologic agent in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or nonbiologic DMARDs. Fulfillment of the trials’ primary endpoint of at least a 20% improvement in American College of Rheumatology response criteria at 24 weeks occurred in 51.3%-57.3% of patients, compared with 26.5%-33.5% of placebo-treated patients.
Those on risankizumab also achieved significantly higher rates of ACR50 and ACR70 responses than those on placebo. In addition, patients with preexisting dactylitis and enthesitis experienced improvements in these PsA manifestations. Risankizumab was also associated with an improvement in physical function at 24 weeks on the Health Assessment Questionnaire–Disability Index, bettering placebo by a mean difference of 0.16-0.20 points in the two trials. A significantly higher percentage of patients who had psoriatic skin lesions experienced at least 90% improvement with risankizumab on the Psoriasis Area and Severity Index, compared with placebo.
AbbVie said that the safety profile of risankizumab in patients with PsA has been generally consistent with its effects in patients with plaque psoriasis.
The KEEPsAKE 1 and KEEPsAKE 2 studies are ongoing, and patients in the long-term extensions of the trials remain blinded to the original randomized allocation for the duration of the studies.
Phase 3 trials of risankizumab are also ongoing in patients with Crohn’s disease and ulcerative colitis.
A version of this article first appeared on Medscape.com.
Allopurinol found safe in patients with concomitant gout, CKD
Allopurinol treatment is not associated with increased mortality in patients with gout and chronic kidney disease even at 5 years after starting treatment, a study has found.
Around one in five patients with gout also have chronic kidney disease, and previous research suggests that hyperuricemia is itself a contributor to renal disease, which is why there has been interest in the use of serum urate–lowering medication in patients with both conditions.
Since the publication of two earlier randomized controlled trials suggested a twofold increase in mortality among patients with renal disease who were treated with allopurinol in an attempt to slow progression, there has been wariness about the drug in patients with compromised renal function.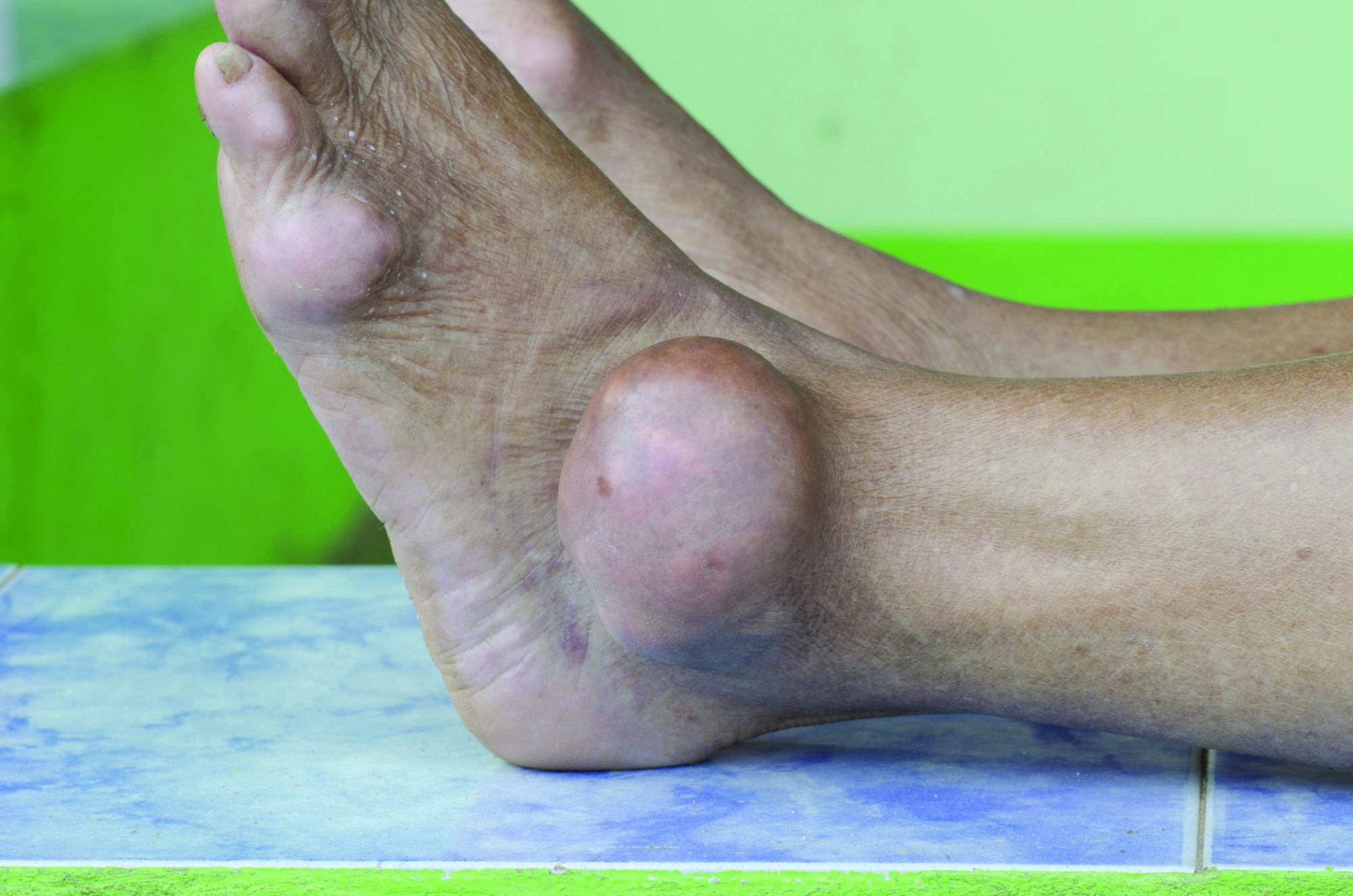
In a study published in Annals of Internal Medicine, Jie Wei, PhD, of Xiangya Hospital at Central South University in Changsha, China, and coauthors report the results of their retrospective, population-based study of 5,277 adults aged 40 and older with gout and moderate to severe chronic kidney disease who were initiated on allopurinol and 5,277 matched individuals not on allopurinol.
At 5 years after the patients started allopurinol, the study found that mortality was a statistically significant 15% lower (hazard ratio, 0.85; 95% confidence interval, 0.77-0.93) among those on allopurinol, compared with those not taking the drug. The rate was 4.9 deaths per 100 person-years among those on allopurinol, compared with 5.8 among those not taking it.
The researchers also created two simulated randomized clinical trials from the data for initiators of allopurinol, replicating each initiator twice. The first trial assigned patient replicates either to achieving a target serum urate level of less than 0.36 mmol/L within a year or not achieving it. The second assigned patient replicates to either an allopurinol dose-escalation group or no dose escalation.
For the target serum urate level study, 1,484 achieved the target, and this was associated with a 13% lower hazard ratio for mortality that just missed statistical significance (HR, 0.87; 95% confidence interval, 0.75-1.01).
In the dose-escalation study, there were 773 participants who increased their dose of allopurinol in the first year after initiation – from a median of 100 mg/day to a median final dose of 300 mg/day – and 2,923 who didn’t. Those who escalated their dose had a nonsignificant 12% lower risk of mortality (HR, 0.88; 95% CI, 0.73-1.07), compared with those who didn’t.
The authors suggest that this could be the result of confounding, as patients who achieved target serum urate levels may have been of better health generally than those who didn’t, which could also have contributed to lower mortality.
Coauthor of the study Yuqing Zhang, DSc, of Massachusetts General Hospital and Harvard Medical School, Boston, said there had previously been a theory that allopurinol could protect against progression of renal disease. However, the two randomized, controlled trials in patients with chronic kidney disease but not gout published in 2020 suggested that allopurinol was instead associated with a doubling of mortality in this group.
“This study really shows convincing evidence that among gout patients with renal disease, allopurinol does not increase mortality,” Dr. Zhang told this news organization. He suggested the reason that the earlier studies had found higher mortality among patients on allopurinol was because those patients did not have gout. Given that gout can increase mortality, treating it effectively with allopurinol may therefore reduce mortality even in patients with concurrent chronic kidney disease.
Commenting on the study, Angelo Gaffo, MD, from the Birmingham VA Medical Center and the division of rheumatology at the University of Alabama at Birmingham, said that, while there had been data suggesting increased mortality, the findings from this “very well-done” study were reassuring and even suggested a possible decrease in mortality associated with allopurinol.

“I wouldn’t scream it out loud because it needs confirmation, but it’s something also that we have a sense that could be true,” he said.
Dr. Gaffo noted that patients treated with allopurinol tended to be those with fewer comorbidities. “Patients who have a lot of comorbidities probably are less likely to have their dose of allopurinol started or increased because of some concerns that practitioners may have about putting them on another medicine or increasing the dose of that medicine,” he said.
He also stressed that the findings still need replication in other large database studies, given that a prospective, randomized clinical trial addressing such a question would be difficult to conduct.
The study was supported by the Project Program of National Clinical Research Center for Geriatric Disorders, the National Natural Science Foundation of China, and the U.S. National Institutes of Health. Two authors reported consulting fees from the pharmaceutical sector unrelated to the study. No other conflicts of interest were declared.
A version of this article first appeared on Medscape.com.
Allopurinol treatment is not associated with increased mortality in patients with gout and chronic kidney disease even at 5 years after starting treatment, a study has found.
Around one in five patients with gout also have chronic kidney disease, and previous research suggests that hyperuricemia is itself a contributor to renal disease, which is why there has been interest in the use of serum urate–lowering medication in patients with both conditions.
Since the publication of two earlier randomized controlled trials suggested a twofold increase in mortality among patients with renal disease who were treated with allopurinol in an attempt to slow progression, there has been wariness about the drug in patients with compromised renal function.
In a study published in Annals of Internal Medicine, Jie Wei, PhD, of Xiangya Hospital at Central South University in Changsha, China, and coauthors report the results of their retrospective, population-based study of 5,277 adults aged 40 and older with gout and moderate to severe chronic kidney disease who were initiated on allopurinol and 5,277 matched individuals not on allopurinol.
At 5 years after the patients started allopurinol, the study found that mortality was a statistically significant 15% lower (hazard ratio, 0.85; 95% confidence interval, 0.77-0.93) among those on allopurinol, compared with those not taking the drug. The rate was 4.9 deaths per 100 person-years among those on allopurinol, compared with 5.8 among those not taking it.
The researchers also created two simulated randomized clinical trials from the data for initiators of allopurinol, replicating each initiator twice. The first trial assigned patient replicates either to achieving a target serum urate level of less than 0.36 mmol/L within a year or not achieving it. The second assigned patient replicates to either an allopurinol dose-escalation group or no dose escalation.
For the target serum urate level study, 1,484 achieved the target, and this was associated with a 13% lower hazard ratio for mortality that just missed statistical significance (HR, 0.87; 95% confidence interval, 0.75-1.01).
In the dose-escalation study, there were 773 participants who increased their dose of allopurinol in the first year after initiation – from a median of 100 mg/day to a median final dose of 300 mg/day – and 2,923 who didn’t. Those who escalated their dose had a nonsignificant 12% lower risk of mortality (HR, 0.88; 95% CI, 0.73-1.07), compared with those who didn’t.
The authors suggest that this could be the result of confounding, as patients who achieved target serum urate levels may have been of better health generally than those who didn’t, which could also have contributed to lower mortality.
Coauthor of the study Yuqing Zhang, DSc, of Massachusetts General Hospital and Harvard Medical School, Boston, said there had previously been a theory that allopurinol could protect against progression of renal disease. However, the two randomized, controlled trials in patients with chronic kidney disease but not gout published in 2020 suggested that allopurinol was instead associated with a doubling of mortality in this group.
“This study really shows convincing evidence that among gout patients with renal disease, allopurinol does not increase mortality,” Dr. Zhang told this news organization. He suggested the reason that the earlier studies had found higher mortality among patients on allopurinol was because those patients did not have gout. Given that gout can increase mortality, treating it effectively with allopurinol may therefore reduce mortality even in patients with concurrent chronic kidney disease.
Commenting on the study, Angelo Gaffo, MD, from the Birmingham VA Medical Center and the division of rheumatology at the University of Alabama at Birmingham, said that, while there had been data suggesting increased mortality, the findings from this “very well-done” study were reassuring and even suggested a possible decrease in mortality associated with allopurinol.
“I wouldn’t scream it out loud because it needs confirmation, but it’s something also that we have a sense that could be true,” he said.
Dr. Gaffo noted that patients treated with allopurinol tended to be those with fewer comorbidities. “Patients who have a lot of comorbidities probably are less likely to have their dose of allopurinol started or increased because of some concerns that practitioners may have about putting them on another medicine or increasing the dose of that medicine,” he said.
He also stressed that the findings still need replication in other large database studies, given that a prospective, randomized clinical trial addressing such a question would be difficult to conduct.
The study was supported by the Project Program of National Clinical Research Center for Geriatric Disorders, the National Natural Science Foundation of China, and the U.S. National Institutes of Health. Two authors reported consulting fees from the pharmaceutical sector unrelated to the study. No other conflicts of interest were declared.
A version of this article first appeared on Medscape.com.
Allopurinol treatment is not associated with increased mortality in patients with gout and chronic kidney disease even at 5 years after starting treatment, a study has found.
Around one in five patients with gout also have chronic kidney disease, and previous research suggests that hyperuricemia is itself a contributor to renal disease, which is why there has been interest in the use of serum urate–lowering medication in patients with both conditions.
Since the publication of two earlier randomized controlled trials suggested a twofold increase in mortality among patients with renal disease who were treated with allopurinol in an attempt to slow progression, there has been wariness about the drug in patients with compromised renal function.
In a study published in Annals of Internal Medicine, Jie Wei, PhD, of Xiangya Hospital at Central South University in Changsha, China, and coauthors report the results of their retrospective, population-based study of 5,277 adults aged 40 and older with gout and moderate to severe chronic kidney disease who were initiated on allopurinol and 5,277 matched individuals not on allopurinol.
At 5 years after the patients started allopurinol, the study found that mortality was a statistically significant 15% lower (hazard ratio, 0.85; 95% confidence interval, 0.77-0.93) among those on allopurinol, compared with those not taking the drug. The rate was 4.9 deaths per 100 person-years among those on allopurinol, compared with 5.8 among those not taking it.
The researchers also created two simulated randomized clinical trials from the data for initiators of allopurinol, replicating each initiator twice. The first trial assigned patient replicates either to achieving a target serum urate level of less than 0.36 mmol/L within a year or not achieving it. The second assigned patient replicates to either an allopurinol dose-escalation group or no dose escalation.
For the target serum urate level study, 1,484 achieved the target, and this was associated with a 13% lower hazard ratio for mortality that just missed statistical significance (HR, 0.87; 95% confidence interval, 0.75-1.01).
In the dose-escalation study, there were 773 participants who increased their dose of allopurinol in the first year after initiation – from a median of 100 mg/day to a median final dose of 300 mg/day – and 2,923 who didn’t. Those who escalated their dose had a nonsignificant 12% lower risk of mortality (HR, 0.88; 95% CI, 0.73-1.07), compared with those who didn’t.
The authors suggest that this could be the result of confounding, as patients who achieved target serum urate levels may have been of better health generally than those who didn’t, which could also have contributed to lower mortality.
Coauthor of the study Yuqing Zhang, DSc, of Massachusetts General Hospital and Harvard Medical School, Boston, said there had previously been a theory that allopurinol could protect against progression of renal disease. However, the two randomized, controlled trials in patients with chronic kidney disease but not gout published in 2020 suggested that allopurinol was instead associated with a doubling of mortality in this group.
“This study really shows convincing evidence that among gout patients with renal disease, allopurinol does not increase mortality,” Dr. Zhang told this news organization. He suggested the reason that the earlier studies had found higher mortality among patients on allopurinol was because those patients did not have gout. Given that gout can increase mortality, treating it effectively with allopurinol may therefore reduce mortality even in patients with concurrent chronic kidney disease.
Commenting on the study, Angelo Gaffo, MD, from the Birmingham VA Medical Center and the division of rheumatology at the University of Alabama at Birmingham, said that, while there had been data suggesting increased mortality, the findings from this “very well-done” study were reassuring and even suggested a possible decrease in mortality associated with allopurinol.
“I wouldn’t scream it out loud because it needs confirmation, but it’s something also that we have a sense that could be true,” he said.
Dr. Gaffo noted that patients treated with allopurinol tended to be those with fewer comorbidities. “Patients who have a lot of comorbidities probably are less likely to have their dose of allopurinol started or increased because of some concerns that practitioners may have about putting them on another medicine or increasing the dose of that medicine,” he said.
He also stressed that the findings still need replication in other large database studies, given that a prospective, randomized clinical trial addressing such a question would be difficult to conduct.
The study was supported by the Project Program of National Clinical Research Center for Geriatric Disorders, the National Natural Science Foundation of China, and the U.S. National Institutes of Health. Two authors reported consulting fees from the pharmaceutical sector unrelated to the study. No other conflicts of interest were declared.
A version of this article first appeared on Medscape.com.
FROM ANNALS OF INTERNAL MEDICINE
Rituximab and COVID-19 vaccines: Studies begin to answer key questions
Rituximab has presented something of a conundrum for patients taking the monoclonal antibody during the COVID-19 pandemic.
Used to manage a variety of autoimmune diseases and cancers, rituximab acts against CD20 proteins expressed on the surface of B cells, causing B-cell depletion. However, it is this B-cell depletion that may put these patients at greater risk of COVID-19 development, progression to more severe disease, and in-hospital mortality. Evidence for this appears to be mixed, with studies showing both that patients using rituximab to manage various diseases are and are not at increased risk for SARS-CoV-2 infection, COVID-19 progression, and mortality.
As COVID-19 vaccine rollouts take place across the world, more questions have been raised about the relationship between B-cell depletion from anti-CD20 therapies and COVID-19 vaccines. Do rituximab and other anti-CD20 therapies affect a patient’s response to COVID-19 vaccines? If this is the case, does the timing of anti-CD20 treatment matter to maximize B-cell levels and improve the vaccine’s effectiveness? And how do COVID-19 vaccine booster doses factor into the equation?
Humoral and cell-mediated responses following COVID-19 vaccination
First, the bad news: The vaccine is unquestionably safe to administer in patients taking rituximab, but one thing that has been well established is that antibody response to COVID-19 vaccination in these individuals does is reduced. This isn’t entirely unprecedented, as previous studies have shown a weakened immune response to pneumococcal polysaccharide and keyhole limpet hemocyanin vaccines among patients taking rituximab.

“Compromised immunogenicity to the SARS-CoV-2 vaccines has been demonstrated in rituximab-treated patients, which is of particular concern given the observation that B-cell–depleting therapies may be associated with worse COVID outcomes,” Robert F. Spiera, MD, director of the Scleroderma, Vasculitis, and Myositis Center at the Hospital for Special Surgery in New York, said in an interview.
For example, in a recent study from the Medical University of Vienna, 29 (39%) of 74 patients receiving rituximab (43% as monotherapy, 57% with conventional-synthetic disease-modifying antirheumatic drugs) who were vaccinated with either the Comirnaty (Pfizer-BioNTech) or Spikevax (Moderna) COVID-19 vaccine achieved seroconversion, compared with 100% of patients in a healthy control group, and all but 1 patient without detectable CD19+ peripheral B cells did not develop anti–SARS-CoV-2 receptor-binding domain antibodies.
“There is an increasing number of studies in this field, and they confirm that patients treated with rituximab and other anti-CD20 agents have severely reduced serological responses to COVID-19 vaccines,” Ingrid Jyssum, MD, of the division of rheumatology and research at Diakonhjemmet Hospital in Oslo, said in an interview.

One silver lining is that patients treated with anti-CD20 therapies appear to have a cell-mediated response following vaccination even if they don’t develop SARS-CoV-2 antibodies. “Studies that also investigate T-cell responses are starting to emerge, and so far, they show that, even if the patients do not have antibodies, they may have T-cell responses,” Dr. Jyssum said.
One study of 24 patients with autoimmune diseases taking rituximab that evaluated humoral and T-cell responses following vaccination with the Comirnaty vaccine found that none had a humoral response to the vaccine, but the T-cell response from that group did not significantly differ from 35 patients receiving other immunosuppressants and 26 patients in a healthy control group. In another study of rituximab- or ocrelizumab-treated patients who received mRNA-based COVID-19 vaccines, 69.4% developed SARS-CoV-2–specific antibodies, compared with a control group, but 96.2% of patients taking ocrelizumab and 81.8% of patients taking rituximab mounted a spike-specific CD8+ T-cell response, compared with 66.7% in the control group, and there were comparable rates (85%-90%) of spike-specific CD4+ T cells in all groups. In the study from the Medical University of Vienna, T-cell response was detected in rituximab-treated patients who both did and did not mount an antibody response.
The clinical relevance of how a blunted humoral immune response but a respectable T-cell response to COVID-19 vaccines affects patients treated with anti-CD20 therapies isn’t currently known, Dr. Jyssum said.
While these data are reassuring, they’re also incomplete, Dr. Spiera noted. “The ultimate outcome of relevance to assess vaccine efficacy is protection from COVID and from severe outcomes of COVID infection (i.e., hospitalization, mechanical ventilation, death). That data will require assessment of very large numbers of rituximab-treated vaccinated patients to be compared with rituximab-treated unvaccinated patients, and is unlikely to be forthcoming in the very near future.
“In the meantime, however, achieving serologic positivity, meaning having evidence of serologic as well as cellular immunity following vaccination, is a desired outcome, and likely implies more robust immunity.”
Does treatment timing impact COVID-19 vaccine response?
Given enough time, B-cell reconstitution will occur in patients taking rituximab. With that in mind, is it beneficial to wait a certain amount of time after a patient has stopped rituximab therapy or time since their last dose before giving them a COVID-19 vaccine? In their guidance on COVID-19 vaccines for patients with rheumatic and musculoskeletal diseases, the American College of Rheumatology said there is moderate evidence to consider “optimal timing of dosing and vaccination with the rheumatology provider before proceeding.”
“Guidelines and preliminary studies of serologic response to COVID vaccine in rituximab-treated patients have suggested that longer time from last rituximab exposure is associated with a greater likelihood of a serologic response,” Dr. Spiera said.
In a brief report published in Arthritis & Rheumatology, Dr. Spiera and colleagues performed a retrospective chart review of 56 patients with varying levels of last exposure to rituximab who received a COVID-19 vaccine. Their results showed that, when patients were vaccinated 6-12 months after the last rituximab dose, 55% were seronegative, and when this was more than 12 months, only 13% were seronegative, compared with seronegativity in 86% who were vaccinated less than 6 months after their last rituximab dose.
The RituxiVac trial, conducted by researchers in Switzerland, also examined vaccine responses of 96 rituximab-treated patients who received Comirnaty or Spikevax; results recently published in The Lancet Rheumatology showed findings similar to other studies, with reduced humoral and cell-mediated responses. In the RituxiVac trial, the median time to last anti-CD20 treatment was 1.07 years.
“The typical interval between rituximab doses [for treatment of rheumatoid arthritis, as well as for remission maintenance in antineutrophil cytoplasmic antibody–associated vasculitis] is typically 6 months, and this has become widely used as the interval from last rituximab to time of COVID vaccination, with a recommendation to wait 4 weeks (if possible) from time of vaccination until the next rituximab administration,” Dr. Spiera explained. However, this window seems to vary depending on the study.
Recent research published in Arthritis & Rheumatology indicates B-cell levels could be a relevant indicator for humoral and cell-mediated response in patients with rheumatic diseases treated with rituximab, with a level of 10 B cells/mcL (0.4% of lymphocytes) identified as one potential marker for likely seroconversion following COVID-19 vaccination.
“In some smaller case series, it has been further recognized that rituximab-treated patients who were beginning to reconstitute peripheral B cells were most likely to respond serologically. Our present study confirmed those findings, demonstrating that the presence of detectable B cells was strongly associated with vaccine responsiveness, and affords complementary information to time from last [rituximab dose] in informing the likelihood of a vaccine response,” Dr. Spiera said.
However, the literature is limited in this area, and an exact cutoff for B-cell counts in these patients isn’t currently known, Dr. Jyssum said. A better metric is time away from anti-CD20 therapies, with CD19 cell count being highly correlated with last infusion.
Dr. Spiera agreed that there is no consistent B-cell percentage that works as a cutoff. “In our study, we looked at it as a binary variable, although we did find that a higher percentage of B cells in the peripheral lymphocyte population was associated with a higher likelihood of seroconversion. We did not, however, identify a ‘threshold’ for vaccine serologic responsiveness.”
Should clinicians measure antibodies?
The Food and Drug Administration and the Centers for Disease Control and Prevention have recommended that health care providers and the public not use COVID-19 antibody tests as a way to gauge immunity after exposure to SARS-CoV-2 and after receiving a COVID-19 vaccination. The ACR’s guidance on COVID-19 vaccination for patients with rheumatic and musculoskeletal diseases strongly recommends against ordering antibody tests for patients with autoimmune inflammatory rheumatic diseases as a way to measure immunity.
“Generally, such measurements are not recommended as the clinical correlate of various antibody levels are not known,” Dr. Jyssum said. “With regular infusions of rituximab or other anti-CD20 agents, one cannot expect that these patients will develop significant levels of antibodies.”
However, she said there might be situations where it’s useful to know whether a patient has developed antibodies at all. “Assessing the significance of specific antibody levels is difficult, and the subject of scientific studies. Patients lacking a humoral vaccine response are left to rely on their T-cell responses and on infectious control measures to prevent disease.”
Dr. Spiera said he disagreed with guidelines recommending against checking antibody levels after vaccination, “particularly in patients treated with immunosuppressive medications that might be expected to blunt their serologic response to the vaccines.
“Although we cannot be sure what level of measurable antibodies offer what level of protection, most clinicians would agree that patients who demonstrate no detectable antibodies (which is a common finding in rituximab-treated patients) should be considered at higher risk,” he said. “Indeed, recommendations regarding booster vaccine administration in general was initially based on the observation of declining antibody levels with longer time from vaccination.”
Do COVID-19 vaccine boosters help patients on anti-CD20 therapy?
As of January 2022, the FDA and CDC have recommended a third primary series shot of COVID-19 vaccines for some moderately to severely immunocompromised patients as young as 5 years old (for Comirnaty vaccine) or a booster shot of either Comirnaty or Spikevax for everyone aged 12 years and older, including immunocompromised people, while the ACR goes into more detail and recommends clinicians time a patient’s booster shot with temporary treatment interruption.
In The Lancet Rheumatology, Dr. Jyssum and colleagues recently published results from the prospective Nor-vaC study examining the humoral and cell-mediated immune responses of 87 patients with RA being treated with rituximab who received the Comirnaty, Spikevax, or Vaxzevria (AstraZeneca) COVID-19 vaccines; of these, 49 patients received a booster dose at a median of 70 days after completing their primary series. The results showed 19 patients (28.1%) had a serologic response after their primary series, while 8 of 49 patients (16.3%) who received their booster dose had a serologic response.
All patients who received a third dose in the study had a T-cell response, Dr. Jyssum said. “This is reassuring for patients and clinicians. T cells have been found to be important in countering COVID-19 disease, but whether we can rely on the T-cell response alone in the absence of antibodies to protect patients from infection or from serious COVID disease is still not determined,” she said.
When asked if she would recommend COVID-19 vaccine booster doses for patients on rituximab, Dr. Jyssum replied: “Absolutely.”
Another study, recently published in Annals of the Rheumatic Diseases, examined heterologous and homologous booster doses for 60 patients receiving rituximab without seroconversion after their COVID-19 vaccine primary series. The results showed no significant difference in new seroconversion at 4 weeks based on whether the patient received a vector or mRNA vaccine (22% vs. 32%), but all patients who received a booster dose with a vector vaccine had specific T-cell responses, compared with 81% of patients who received an mRNA vaccine booster. There was a new humoral and/or cellular response in 9 of 11 patients (82%), and most patients with peripheral B cells (12 of 18 patients; 67%) achieved seroconversion.
“Our data show that a cellular and/or humoral immune response can be achieved on a third COVID-19 vaccination in most of the patients who initially developed neither a humoral nor a cellular immune response,” the researchers concluded. “The efficacy data together with the safety data seen in our trial provide a favorable risk/benefit ratio and support the implementation of a third vaccination for nonseroconverted high-risk autoimmune disease patients treated with B-cell–depleting agents.”
Dr. Spiera said booster doses are an important part of the equation, and “it is important to consider factors that would be associated with a greater likelihood of achieving a serologic response, particularly in those patients who did not demonstrate a serologic response to the initial vaccines series.
“Preliminary data shows that the beginnings of B-cell reconstitution is also associated with a positive serologic response following a booster of the COVID-19 vaccine,” he said.
The authors of the cited studies reported numerous relevant financial disclosures. Dr. Spiera and Dr. Jyssum reported no relevant financial disclosures.
Rituximab has presented something of a conundrum for patients taking the monoclonal antibody during the COVID-19 pandemic.
Used to manage a variety of autoimmune diseases and cancers, rituximab acts against CD20 proteins expressed on the surface of B cells, causing B-cell depletion. However, it is this B-cell depletion that may put these patients at greater risk of COVID-19 development, progression to more severe disease, and in-hospital mortality. Evidence for this appears to be mixed, with studies showing both that patients using rituximab to manage various diseases are and are not at increased risk for SARS-CoV-2 infection, COVID-19 progression, and mortality.
As COVID-19 vaccine rollouts take place across the world, more questions have been raised about the relationship between B-cell depletion from anti-CD20 therapies and COVID-19 vaccines. Do rituximab and other anti-CD20 therapies affect a patient’s response to COVID-19 vaccines? If this is the case, does the timing of anti-CD20 treatment matter to maximize B-cell levels and improve the vaccine’s effectiveness? And how do COVID-19 vaccine booster doses factor into the equation?
Humoral and cell-mediated responses following COVID-19 vaccination
First, the bad news: The vaccine is unquestionably safe to administer in patients taking rituximab, but one thing that has been well established is that antibody response to COVID-19 vaccination in these individuals does is reduced. This isn’t entirely unprecedented, as previous studies have shown a weakened immune response to pneumococcal polysaccharide and keyhole limpet hemocyanin vaccines among patients taking rituximab.
“Compromised immunogenicity to the SARS-CoV-2 vaccines has been demonstrated in rituximab-treated patients, which is of particular concern given the observation that B-cell–depleting therapies may be associated with worse COVID outcomes,” Robert F. Spiera, MD, director of the Scleroderma, Vasculitis, and Myositis Center at the Hospital for Special Surgery in New York, said in an interview.
For example, in a recent study from the Medical University of Vienna, 29 (39%) of 74 patients receiving rituximab (43% as monotherapy, 57% with conventional-synthetic disease-modifying antirheumatic drugs) who were vaccinated with either the Comirnaty (Pfizer-BioNTech) or Spikevax (Moderna) COVID-19 vaccine achieved seroconversion, compared with 100% of patients in a healthy control group, and all but 1 patient without detectable CD19+ peripheral B cells did not develop anti–SARS-CoV-2 receptor-binding domain antibodies.
“There is an increasing number of studies in this field, and they confirm that patients treated with rituximab and other anti-CD20 agents have severely reduced serological responses to COVID-19 vaccines,” Ingrid Jyssum, MD, of the division of rheumatology and research at Diakonhjemmet Hospital in Oslo, said in an interview.
One silver lining is that patients treated with anti-CD20 therapies appear to have a cell-mediated response following vaccination even if they don’t develop SARS-CoV-2 antibodies. “Studies that also investigate T-cell responses are starting to emerge, and so far, they show that, even if the patients do not have antibodies, they may have T-cell responses,” Dr. Jyssum said.
One study of 24 patients with autoimmune diseases taking rituximab that evaluated humoral and T-cell responses following vaccination with the Comirnaty vaccine found that none had a humoral response to the vaccine, but the T-cell response from that group did not significantly differ from 35 patients receiving other immunosuppressants and 26 patients in a healthy control group. In another study of rituximab- or ocrelizumab-treated patients who received mRNA-based COVID-19 vaccines, 69.4% developed SARS-CoV-2–specific antibodies, compared with a control group, but 96.2% of patients taking ocrelizumab and 81.8% of patients taking rituximab mounted a spike-specific CD8+ T-cell response, compared with 66.7% in the control group, and there were comparable rates (85%-90%) of spike-specific CD4+ T cells in all groups. In the study from the Medical University of Vienna, T-cell response was detected in rituximab-treated patients who both did and did not mount an antibody response.
The clinical relevance of how a blunted humoral immune response but a respectable T-cell response to COVID-19 vaccines affects patients treated with anti-CD20 therapies isn’t currently known, Dr. Jyssum said.
While these data are reassuring, they’re also incomplete, Dr. Spiera noted. “The ultimate outcome of relevance to assess vaccine efficacy is protection from COVID and from severe outcomes of COVID infection (i.e., hospitalization, mechanical ventilation, death). That data will require assessment of very large numbers of rituximab-treated vaccinated patients to be compared with rituximab-treated unvaccinated patients, and is unlikely to be forthcoming in the very near future.
“In the meantime, however, achieving serologic positivity, meaning having evidence of serologic as well as cellular immunity following vaccination, is a desired outcome, and likely implies more robust immunity.”
Does treatment timing impact COVID-19 vaccine response?
Given enough time, B-cell reconstitution will occur in patients taking rituximab. With that in mind, is it beneficial to wait a certain amount of time after a patient has stopped rituximab therapy or time since their last dose before giving them a COVID-19 vaccine? In their guidance on COVID-19 vaccines for patients with rheumatic and musculoskeletal diseases, the American College of Rheumatology said there is moderate evidence to consider “optimal timing of dosing and vaccination with the rheumatology provider before proceeding.”
“Guidelines and preliminary studies of serologic response to COVID vaccine in rituximab-treated patients have suggested that longer time from last rituximab exposure is associated with a greater likelihood of a serologic response,” Dr. Spiera said.
In a brief report published in Arthritis & Rheumatology, Dr. Spiera and colleagues performed a retrospective chart review of 56 patients with varying levels of last exposure to rituximab who received a COVID-19 vaccine. Their results showed that, when patients were vaccinated 6-12 months after the last rituximab dose, 55% were seronegative, and when this was more than 12 months, only 13% were seronegative, compared with seronegativity in 86% who were vaccinated less than 6 months after their last rituximab dose.
The RituxiVac trial, conducted by researchers in Switzerland, also examined vaccine responses of 96 rituximab-treated patients who received Comirnaty or Spikevax; results recently published in The Lancet Rheumatology showed findings similar to other studies, with reduced humoral and cell-mediated responses. In the RituxiVac trial, the median time to last anti-CD20 treatment was 1.07 years.
“The typical interval between rituximab doses [for treatment of rheumatoid arthritis, as well as for remission maintenance in antineutrophil cytoplasmic antibody–associated vasculitis] is typically 6 months, and this has become widely used as the interval from last rituximab to time of COVID vaccination, with a recommendation to wait 4 weeks (if possible) from time of vaccination until the next rituximab administration,” Dr. Spiera explained. However, this window seems to vary depending on the study.
Recent research published in Arthritis & Rheumatology indicates B-cell levels could be a relevant indicator for humoral and cell-mediated response in patients with rheumatic diseases treated with rituximab, with a level of 10 B cells/mcL (0.4% of lymphocytes) identified as one potential marker for likely seroconversion following COVID-19 vaccination.
“In some smaller case series, it has been further recognized that rituximab-treated patients who were beginning to reconstitute peripheral B cells were most likely to respond serologically. Our present study confirmed those findings, demonstrating that the presence of detectable B cells was strongly associated with vaccine responsiveness, and affords complementary information to time from last [rituximab dose] in informing the likelihood of a vaccine response,” Dr. Spiera said.
However, the literature is limited in this area, and an exact cutoff for B-cell counts in these patients isn’t currently known, Dr. Jyssum said. A better metric is time away from anti-CD20 therapies, with CD19 cell count being highly correlated with last infusion.
Dr. Spiera agreed that there is no consistent B-cell percentage that works as a cutoff. “In our study, we looked at it as a binary variable, although we did find that a higher percentage of B cells in the peripheral lymphocyte population was associated with a higher likelihood of seroconversion. We did not, however, identify a ‘threshold’ for vaccine serologic responsiveness.”
Should clinicians measure antibodies?
The Food and Drug Administration and the Centers for Disease Control and Prevention have recommended that health care providers and the public not use COVID-19 antibody tests as a way to gauge immunity after exposure to SARS-CoV-2 and after receiving a COVID-19 vaccination. The ACR’s guidance on COVID-19 vaccination for patients with rheumatic and musculoskeletal diseases strongly recommends against ordering antibody tests for patients with autoimmune inflammatory rheumatic diseases as a way to measure immunity.
“Generally, such measurements are not recommended as the clinical correlate of various antibody levels are not known,” Dr. Jyssum said. “With regular infusions of rituximab or other anti-CD20 agents, one cannot expect that these patients will develop significant levels of antibodies.”
However, she said there might be situations where it’s useful to know whether a patient has developed antibodies at all. “Assessing the significance of specific antibody levels is difficult, and the subject of scientific studies. Patients lacking a humoral vaccine response are left to rely on their T-cell responses and on infectious control measures to prevent disease.”
Dr. Spiera said he disagreed with guidelines recommending against checking antibody levels after vaccination, “particularly in patients treated with immunosuppressive medications that might be expected to blunt their serologic response to the vaccines.
“Although we cannot be sure what level of measurable antibodies offer what level of protection, most clinicians would agree that patients who demonstrate no detectable antibodies (which is a common finding in rituximab-treated patients) should be considered at higher risk,” he said. “Indeed, recommendations regarding booster vaccine administration in general was initially based on the observation of declining antibody levels with longer time from vaccination.”
Do COVID-19 vaccine boosters help patients on anti-CD20 therapy?
As of January 2022, the FDA and CDC have recommended a third primary series shot of COVID-19 vaccines for some moderately to severely immunocompromised patients as young as 5 years old (for Comirnaty vaccine) or a booster shot of either Comirnaty or Spikevax for everyone aged 12 years and older, including immunocompromised people, while the ACR goes into more detail and recommends clinicians time a patient’s booster shot with temporary treatment interruption.
In The Lancet Rheumatology, Dr. Jyssum and colleagues recently published results from the prospective Nor-vaC study examining the humoral and cell-mediated immune responses of 87 patients with RA being treated with rituximab who received the Comirnaty, Spikevax, or Vaxzevria (AstraZeneca) COVID-19 vaccines; of these, 49 patients received a booster dose at a median of 70 days after completing their primary series. The results showed 19 patients (28.1%) had a serologic response after their primary series, while 8 of 49 patients (16.3%) who received their booster dose had a serologic response.
All patients who received a third dose in the study had a T-cell response, Dr. Jyssum said. “This is reassuring for patients and clinicians. T cells have been found to be important in countering COVID-19 disease, but whether we can rely on the T-cell response alone in the absence of antibodies to protect patients from infection or from serious COVID disease is still not determined,” she said.
When asked if she would recommend COVID-19 vaccine booster doses for patients on rituximab, Dr. Jyssum replied: “Absolutely.”
Another study, recently published in Annals of the Rheumatic Diseases, examined heterologous and homologous booster doses for 60 patients receiving rituximab without seroconversion after their COVID-19 vaccine primary series. The results showed no significant difference in new seroconversion at 4 weeks based on whether the patient received a vector or mRNA vaccine (22% vs. 32%), but all patients who received a booster dose with a vector vaccine had specific T-cell responses, compared with 81% of patients who received an mRNA vaccine booster. There was a new humoral and/or cellular response in 9 of 11 patients (82%), and most patients with peripheral B cells (12 of 18 patients; 67%) achieved seroconversion.
“Our data show that a cellular and/or humoral immune response can be achieved on a third COVID-19 vaccination in most of the patients who initially developed neither a humoral nor a cellular immune response,” the researchers concluded. “The efficacy data together with the safety data seen in our trial provide a favorable risk/benefit ratio and support the implementation of a third vaccination for nonseroconverted high-risk autoimmune disease patients treated with B-cell–depleting agents.”
Dr. Spiera said booster doses are an important part of the equation, and “it is important to consider factors that would be associated with a greater likelihood of achieving a serologic response, particularly in those patients who did not demonstrate a serologic response to the initial vaccines series.
“Preliminary data shows that the beginnings of B-cell reconstitution is also associated with a positive serologic response following a booster of the COVID-19 vaccine,” he said.
The authors of the cited studies reported numerous relevant financial disclosures. Dr. Spiera and Dr. Jyssum reported no relevant financial disclosures.
Rituximab has presented something of a conundrum for patients taking the monoclonal antibody during the COVID-19 pandemic.
Used to manage a variety of autoimmune diseases and cancers, rituximab acts against CD20 proteins expressed on the surface of B cells, causing B-cell depletion. However, it is this B-cell depletion that may put these patients at greater risk of COVID-19 development, progression to more severe disease, and in-hospital mortality. Evidence for this appears to be mixed, with studies showing both that patients using rituximab to manage various diseases are and are not at increased risk for SARS-CoV-2 infection, COVID-19 progression, and mortality.
As COVID-19 vaccine rollouts take place across the world, more questions have been raised about the relationship between B-cell depletion from anti-CD20 therapies and COVID-19 vaccines. Do rituximab and other anti-CD20 therapies affect a patient’s response to COVID-19 vaccines? If this is the case, does the timing of anti-CD20 treatment matter to maximize B-cell levels and improve the vaccine’s effectiveness? And how do COVID-19 vaccine booster doses factor into the equation?
Humoral and cell-mediated responses following COVID-19 vaccination
First, the bad news: The vaccine is unquestionably safe to administer in patients taking rituximab, but one thing that has been well established is that antibody response to COVID-19 vaccination in these individuals does is reduced. This isn’t entirely unprecedented, as previous studies have shown a weakened immune response to pneumococcal polysaccharide and keyhole limpet hemocyanin vaccines among patients taking rituximab.
“Compromised immunogenicity to the SARS-CoV-2 vaccines has been demonstrated in rituximab-treated patients, which is of particular concern given the observation that B-cell–depleting therapies may be associated with worse COVID outcomes,” Robert F. Spiera, MD, director of the Scleroderma, Vasculitis, and Myositis Center at the Hospital for Special Surgery in New York, said in an interview.
For example, in a recent study from the Medical University of Vienna, 29 (39%) of 74 patients receiving rituximab (43% as monotherapy, 57% with conventional-synthetic disease-modifying antirheumatic drugs) who were vaccinated with either the Comirnaty (Pfizer-BioNTech) or Spikevax (Moderna) COVID-19 vaccine achieved seroconversion, compared with 100% of patients in a healthy control group, and all but 1 patient without detectable CD19+ peripheral B cells did not develop anti–SARS-CoV-2 receptor-binding domain antibodies.
“There is an increasing number of studies in this field, and they confirm that patients treated with rituximab and other anti-CD20 agents have severely reduced serological responses to COVID-19 vaccines,” Ingrid Jyssum, MD, of the division of rheumatology and research at Diakonhjemmet Hospital in Oslo, said in an interview.
One silver lining is that patients treated with anti-CD20 therapies appear to have a cell-mediated response following vaccination even if they don’t develop SARS-CoV-2 antibodies. “Studies that also investigate T-cell responses are starting to emerge, and so far, they show that, even if the patients do not have antibodies, they may have T-cell responses,” Dr. Jyssum said.
One study of 24 patients with autoimmune diseases taking rituximab that evaluated humoral and T-cell responses following vaccination with the Comirnaty vaccine found that none had a humoral response to the vaccine, but the T-cell response from that group did not significantly differ from 35 patients receiving other immunosuppressants and 26 patients in a healthy control group. In another study of rituximab- or ocrelizumab-treated patients who received mRNA-based COVID-19 vaccines, 69.4% developed SARS-CoV-2–specific antibodies, compared with a control group, but 96.2% of patients taking ocrelizumab and 81.8% of patients taking rituximab mounted a spike-specific CD8+ T-cell response, compared with 66.7% in the control group, and there were comparable rates (85%-90%) of spike-specific CD4+ T cells in all groups. In the study from the Medical University of Vienna, T-cell response was detected in rituximab-treated patients who both did and did not mount an antibody response.
The clinical relevance of how a blunted humoral immune response but a respectable T-cell response to COVID-19 vaccines affects patients treated with anti-CD20 therapies isn’t currently known, Dr. Jyssum said.
While these data are reassuring, they’re also incomplete, Dr. Spiera noted. “The ultimate outcome of relevance to assess vaccine efficacy is protection from COVID and from severe outcomes of COVID infection (i.e., hospitalization, mechanical ventilation, death). That data will require assessment of very large numbers of rituximab-treated vaccinated patients to be compared with rituximab-treated unvaccinated patients, and is unlikely to be forthcoming in the very near future.
“In the meantime, however, achieving serologic positivity, meaning having evidence of serologic as well as cellular immunity following vaccination, is a desired outcome, and likely implies more robust immunity.”
Does treatment timing impact COVID-19 vaccine response?
Given enough time, B-cell reconstitution will occur in patients taking rituximab. With that in mind, is it beneficial to wait a certain amount of time after a patient has stopped rituximab therapy or time since their last dose before giving them a COVID-19 vaccine? In their guidance on COVID-19 vaccines for patients with rheumatic and musculoskeletal diseases, the American College of Rheumatology said there is moderate evidence to consider “optimal timing of dosing and vaccination with the rheumatology provider before proceeding.”
“Guidelines and preliminary studies of serologic response to COVID vaccine in rituximab-treated patients have suggested that longer time from last rituximab exposure is associated with a greater likelihood of a serologic response,” Dr. Spiera said.
In a brief report published in Arthritis & Rheumatology, Dr. Spiera and colleagues performed a retrospective chart review of 56 patients with varying levels of last exposure to rituximab who received a COVID-19 vaccine. Their results showed that, when patients were vaccinated 6-12 months after the last rituximab dose, 55% were seronegative, and when this was more than 12 months, only 13% were seronegative, compared with seronegativity in 86% who were vaccinated less than 6 months after their last rituximab dose.
The RituxiVac trial, conducted by researchers in Switzerland, also examined vaccine responses of 96 rituximab-treated patients who received Comirnaty or Spikevax; results recently published in The Lancet Rheumatology showed findings similar to other studies, with reduced humoral and cell-mediated responses. In the RituxiVac trial, the median time to last anti-CD20 treatment was 1.07 years.
“The typical interval between rituximab doses [for treatment of rheumatoid arthritis, as well as for remission maintenance in antineutrophil cytoplasmic antibody–associated vasculitis] is typically 6 months, and this has become widely used as the interval from last rituximab to time of COVID vaccination, with a recommendation to wait 4 weeks (if possible) from time of vaccination until the next rituximab administration,” Dr. Spiera explained. However, this window seems to vary depending on the study.
Recent research published in Arthritis & Rheumatology indicates B-cell levels could be a relevant indicator for humoral and cell-mediated response in patients with rheumatic diseases treated with rituximab, with a level of 10 B cells/mcL (0.4% of lymphocytes) identified as one potential marker for likely seroconversion following COVID-19 vaccination.
“In some smaller case series, it has been further recognized that rituximab-treated patients who were beginning to reconstitute peripheral B cells were most likely to respond serologically. Our present study confirmed those findings, demonstrating that the presence of detectable B cells was strongly associated with vaccine responsiveness, and affords complementary information to time from last [rituximab dose] in informing the likelihood of a vaccine response,” Dr. Spiera said.
However, the literature is limited in this area, and an exact cutoff for B-cell counts in these patients isn’t currently known, Dr. Jyssum said. A better metric is time away from anti-CD20 therapies, with CD19 cell count being highly correlated with last infusion.
Dr. Spiera agreed that there is no consistent B-cell percentage that works as a cutoff. “In our study, we looked at it as a binary variable, although we did find that a higher percentage of B cells in the peripheral lymphocyte population was associated with a higher likelihood of seroconversion. We did not, however, identify a ‘threshold’ for vaccine serologic responsiveness.”
Should clinicians measure antibodies?
The Food and Drug Administration and the Centers for Disease Control and Prevention have recommended that health care providers and the public not use COVID-19 antibody tests as a way to gauge immunity after exposure to SARS-CoV-2 and after receiving a COVID-19 vaccination. The ACR’s guidance on COVID-19 vaccination for patients with rheumatic and musculoskeletal diseases strongly recommends against ordering antibody tests for patients with autoimmune inflammatory rheumatic diseases as a way to measure immunity.
“Generally, such measurements are not recommended as the clinical correlate of various antibody levels are not known,” Dr. Jyssum said. “With regular infusions of rituximab or other anti-CD20 agents, one cannot expect that these patients will develop significant levels of antibodies.”
However, she said there might be situations where it’s useful to know whether a patient has developed antibodies at all. “Assessing the significance of specific antibody levels is difficult, and the subject of scientific studies. Patients lacking a humoral vaccine response are left to rely on their T-cell responses and on infectious control measures to prevent disease.”
Dr. Spiera said he disagreed with guidelines recommending against checking antibody levels after vaccination, “particularly in patients treated with immunosuppressive medications that might be expected to blunt their serologic response to the vaccines.
“Although we cannot be sure what level of measurable antibodies offer what level of protection, most clinicians would agree that patients who demonstrate no detectable antibodies (which is a common finding in rituximab-treated patients) should be considered at higher risk,” he said. “Indeed, recommendations regarding booster vaccine administration in general was initially based on the observation of declining antibody levels with longer time from vaccination.”
Do COVID-19 vaccine boosters help patients on anti-CD20 therapy?
As of January 2022, the FDA and CDC have recommended a third primary series shot of COVID-19 vaccines for some moderately to severely immunocompromised patients as young as 5 years old (for Comirnaty vaccine) or a booster shot of either Comirnaty or Spikevax for everyone aged 12 years and older, including immunocompromised people, while the ACR goes into more detail and recommends clinicians time a patient’s booster shot with temporary treatment interruption.
In The Lancet Rheumatology, Dr. Jyssum and colleagues recently published results from the prospective Nor-vaC study examining the humoral and cell-mediated immune responses of 87 patients with RA being treated with rituximab who received the Comirnaty, Spikevax, or Vaxzevria (AstraZeneca) COVID-19 vaccines; of these, 49 patients received a booster dose at a median of 70 days after completing their primary series. The results showed 19 patients (28.1%) had a serologic response after their primary series, while 8 of 49 patients (16.3%) who received their booster dose had a serologic response.
All patients who received a third dose in the study had a T-cell response, Dr. Jyssum said. “This is reassuring for patients and clinicians. T cells have been found to be important in countering COVID-19 disease, but whether we can rely on the T-cell response alone in the absence of antibodies to protect patients from infection or from serious COVID disease is still not determined,” she said.
When asked if she would recommend COVID-19 vaccine booster doses for patients on rituximab, Dr. Jyssum replied: “Absolutely.”
Another study, recently published in Annals of the Rheumatic Diseases, examined heterologous and homologous booster doses for 60 patients receiving rituximab without seroconversion after their COVID-19 vaccine primary series. The results showed no significant difference in new seroconversion at 4 weeks based on whether the patient received a vector or mRNA vaccine (22% vs. 32%), but all patients who received a booster dose with a vector vaccine had specific T-cell responses, compared with 81% of patients who received an mRNA vaccine booster. There was a new humoral and/or cellular response in 9 of 11 patients (82%), and most patients with peripheral B cells (12 of 18 patients; 67%) achieved seroconversion.
“Our data show that a cellular and/or humoral immune response can be achieved on a third COVID-19 vaccination in most of the patients who initially developed neither a humoral nor a cellular immune response,” the researchers concluded. “The efficacy data together with the safety data seen in our trial provide a favorable risk/benefit ratio and support the implementation of a third vaccination for nonseroconverted high-risk autoimmune disease patients treated with B-cell–depleting agents.”
Dr. Spiera said booster doses are an important part of the equation, and “it is important to consider factors that would be associated with a greater likelihood of achieving a serologic response, particularly in those patients who did not demonstrate a serologic response to the initial vaccines series.
“Preliminary data shows that the beginnings of B-cell reconstitution is also associated with a positive serologic response following a booster of the COVID-19 vaccine,” he said.
The authors of the cited studies reported numerous relevant financial disclosures. Dr. Spiera and Dr. Jyssum reported no relevant financial disclosures.
How safe is a drug holiday from bisphosphonates for osteoporosis?
Researchers found a small but greater risk of a hip fracture after 2 years of taking a “drug holiday” – stopping therapy – after long-term (≥3-year) use of one bisphosphonate, risedronate, versus another, alendronate.
The risk of a hip fracture after stopping either of these oral bisphosphonate osteoporosis drugs was similar until 2 years, suggesting that patients who take a drug holiday from risedronate should be revaluated before 2 years.
These top-line findings from a propensity-matched cohort study of older patients in Ontario, Canada, were reported at the annual American Society of Bone and Mineral Research (ASBMR) last fall.
The full study, led by Kaleen N. Hayes, PharmD, PhD, Brown University School of Public Health, Providence, R.I., was published online in the Annals of Internal Medicine.
“We emphasize that our results do not indicate that alendronate therapy should be preferred over risedronate therapy,” the researchers stress, as several real-world studies found a similar risk of fractures while patients were receiving either drug.
“The decision to initiate alendronate or risedronate therapy [the two most commonly prescribed bisphosphonates] is driven by the prescriber,” they note, adding that some patients may prefer risedronate because it is available as a monthly dose or a weekly delayed-release formula that does not require fasting.
“We found little difference in the association between risedronate versus alendronate drug holidays and hip fractures until approximately 2 years of not receiving therapy,” Dr. Hayes and colleagues summarize.
Over 3 years, risedronate drug holidays were associated with an 18% relative and 0.6% absolute increased risk for hip fracture compared with alendronate drug holidays.
“To further inform clinical decision-making on drug holidays,” they conclude, “future research should examine when to start and restart osteoporosis therapy on the basis of initial length and type of treatment, patient characteristics, and relative risk for hip fractures versus [atypical femoral fracture].”
Hip fracture risk with risedronate vs. alendronate drug holiday
Long-term bisphosphonate use is associated with a rare risk of osteonecrosis of the jaw or atypical femoral fractures. At the same time, bisphosphonates continue to have a therapeutic effect after therapy is discontinued.
Guidelines recommend that patients at low risk of fracture should therefore have a “drug holiday” after 3 to 5 years of bisphosphonate use and be reassessed 2 to 3 years later, largely based on the Fracture Intervention Trial Long-Term Extension (FLEX) study of alendronate. But risedronate has a shorter half-life, so it may provide shorter residual fracture protection.
Using Ontario administrative data, Dr. Hayes and associates identified more than 60,000 patients who were over aged 65, had received at least 3 years of continuous alendronate or risedronate, and had a subsequent 3-year drug holiday between 2000 and 2020.
They excluded patients who had a fracture or entered a nursing home within 120 days of starting a drug holiday who may have stopped the bisphosphonate due to declining health rather than a drug holiday.
Roughly half (55%) had been taking risedronate and 45% had been taking alendronate.
Using propensity scores, the researchers matched 25,077 patients who had been taking risedronate with an equal number who had been taking alendronate.
Most of the patients were women (82%) and were White.
They started the drug holiday when they were on average 81 years old, after taking the bisphosphonate for 5.9 years on average.
During the 3-year drug holiday, 915 of the 50,154 patients had hip fractures.
This was equivalent to 12.4 hip fractures per 1,000 patients per year during a risedronate holiday and 10.6 hip fractures per 1,000 patients per year during an alendronate holiday (hazard ratio, 1.18).
The risk of hip fracture was not significantly higher at 1 year (HR, 1.03) or at 2 years of a risedronate holiday versus an alendronate holiday (HR, 1.14).
However, the risk of a hip fracture was significantly higher at 2 to 3 years of a risedronate holiday than after an alendronate holiday (HR, 1.34).
There was no significant difference in the risk of any osteoporotic fracture overall (including hip, vertebrae, pelvis, ribs, forearm), however, during a 3-year risedronate versus alendronate drug holiday (HR, 1.07).
The research was supported by the Canadian Institutes of Health Research and Institute for Clinical Evaluative Sciences. Dr. Hayes was supported by a CIHR doctoral research award. The authors have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers found a small but greater risk of a hip fracture after 2 years of taking a “drug holiday” – stopping therapy – after long-term (≥3-year) use of one bisphosphonate, risedronate, versus another, alendronate.
The risk of a hip fracture after stopping either of these oral bisphosphonate osteoporosis drugs was similar until 2 years, suggesting that patients who take a drug holiday from risedronate should be revaluated before 2 years.
These top-line findings from a propensity-matched cohort study of older patients in Ontario, Canada, were reported at the annual American Society of Bone and Mineral Research (ASBMR) last fall.
The full study, led by Kaleen N. Hayes, PharmD, PhD, Brown University School of Public Health, Providence, R.I., was published online in the Annals of Internal Medicine.
“We emphasize that our results do not indicate that alendronate therapy should be preferred over risedronate therapy,” the researchers stress, as several real-world studies found a similar risk of fractures while patients were receiving either drug.
“The decision to initiate alendronate or risedronate therapy [the two most commonly prescribed bisphosphonates] is driven by the prescriber,” they note, adding that some patients may prefer risedronate because it is available as a monthly dose or a weekly delayed-release formula that does not require fasting.
“We found little difference in the association between risedronate versus alendronate drug holidays and hip fractures until approximately 2 years of not receiving therapy,” Dr. Hayes and colleagues summarize.
Over 3 years, risedronate drug holidays were associated with an 18% relative and 0.6% absolute increased risk for hip fracture compared with alendronate drug holidays.
“To further inform clinical decision-making on drug holidays,” they conclude, “future research should examine when to start and restart osteoporosis therapy on the basis of initial length and type of treatment, patient characteristics, and relative risk for hip fractures versus [atypical femoral fracture].”
Hip fracture risk with risedronate vs. alendronate drug holiday
Long-term bisphosphonate use is associated with a rare risk of osteonecrosis of the jaw or atypical femoral fractures. At the same time, bisphosphonates continue to have a therapeutic effect after therapy is discontinued.
Guidelines recommend that patients at low risk of fracture should therefore have a “drug holiday” after 3 to 5 years of bisphosphonate use and be reassessed 2 to 3 years later, largely based on the Fracture Intervention Trial Long-Term Extension (FLEX) study of alendronate. But risedronate has a shorter half-life, so it may provide shorter residual fracture protection.
Using Ontario administrative data, Dr. Hayes and associates identified more than 60,000 patients who were over aged 65, had received at least 3 years of continuous alendronate or risedronate, and had a subsequent 3-year drug holiday between 2000 and 2020.
They excluded patients who had a fracture or entered a nursing home within 120 days of starting a drug holiday who may have stopped the bisphosphonate due to declining health rather than a drug holiday.
Roughly half (55%) had been taking risedronate and 45% had been taking alendronate.
Using propensity scores, the researchers matched 25,077 patients who had been taking risedronate with an equal number who had been taking alendronate.
Most of the patients were women (82%) and were White.
They started the drug holiday when they were on average 81 years old, after taking the bisphosphonate for 5.9 years on average.
During the 3-year drug holiday, 915 of the 50,154 patients had hip fractures.
This was equivalent to 12.4 hip fractures per 1,000 patients per year during a risedronate holiday and 10.6 hip fractures per 1,000 patients per year during an alendronate holiday (hazard ratio, 1.18).
The risk of hip fracture was not significantly higher at 1 year (HR, 1.03) or at 2 years of a risedronate holiday versus an alendronate holiday (HR, 1.14).
However, the risk of a hip fracture was significantly higher at 2 to 3 years of a risedronate holiday than after an alendronate holiday (HR, 1.34).
There was no significant difference in the risk of any osteoporotic fracture overall (including hip, vertebrae, pelvis, ribs, forearm), however, during a 3-year risedronate versus alendronate drug holiday (HR, 1.07).
The research was supported by the Canadian Institutes of Health Research and Institute for Clinical Evaluative Sciences. Dr. Hayes was supported by a CIHR doctoral research award. The authors have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers found a small but greater risk of a hip fracture after 2 years of taking a “drug holiday” – stopping therapy – after long-term (≥3-year) use of one bisphosphonate, risedronate, versus another, alendronate.
The risk of a hip fracture after stopping either of these oral bisphosphonate osteoporosis drugs was similar until 2 years, suggesting that patients who take a drug holiday from risedronate should be revaluated before 2 years.
These top-line findings from a propensity-matched cohort study of older patients in Ontario, Canada, were reported at the annual American Society of Bone and Mineral Research (ASBMR) last fall.
The full study, led by Kaleen N. Hayes, PharmD, PhD, Brown University School of Public Health, Providence, R.I., was published online in the Annals of Internal Medicine.
“We emphasize that our results do not indicate that alendronate therapy should be preferred over risedronate therapy,” the researchers stress, as several real-world studies found a similar risk of fractures while patients were receiving either drug.
“The decision to initiate alendronate or risedronate therapy [the two most commonly prescribed bisphosphonates] is driven by the prescriber,” they note, adding that some patients may prefer risedronate because it is available as a monthly dose or a weekly delayed-release formula that does not require fasting.
“We found little difference in the association between risedronate versus alendronate drug holidays and hip fractures until approximately 2 years of not receiving therapy,” Dr. Hayes and colleagues summarize.
Over 3 years, risedronate drug holidays were associated with an 18% relative and 0.6% absolute increased risk for hip fracture compared with alendronate drug holidays.
“To further inform clinical decision-making on drug holidays,” they conclude, “future research should examine when to start and restart osteoporosis therapy on the basis of initial length and type of treatment, patient characteristics, and relative risk for hip fractures versus [atypical femoral fracture].”
Hip fracture risk with risedronate vs. alendronate drug holiday
Long-term bisphosphonate use is associated with a rare risk of osteonecrosis of the jaw or atypical femoral fractures. At the same time, bisphosphonates continue to have a therapeutic effect after therapy is discontinued.
Guidelines recommend that patients at low risk of fracture should therefore have a “drug holiday” after 3 to 5 years of bisphosphonate use and be reassessed 2 to 3 years later, largely based on the Fracture Intervention Trial Long-Term Extension (FLEX) study of alendronate. But risedronate has a shorter half-life, so it may provide shorter residual fracture protection.
Using Ontario administrative data, Dr. Hayes and associates identified more than 60,000 patients who were over aged 65, had received at least 3 years of continuous alendronate or risedronate, and had a subsequent 3-year drug holiday between 2000 and 2020.
They excluded patients who had a fracture or entered a nursing home within 120 days of starting a drug holiday who may have stopped the bisphosphonate due to declining health rather than a drug holiday.
Roughly half (55%) had been taking risedronate and 45% had been taking alendronate.
Using propensity scores, the researchers matched 25,077 patients who had been taking risedronate with an equal number who had been taking alendronate.
Most of the patients were women (82%) and were White.
They started the drug holiday when they were on average 81 years old, after taking the bisphosphonate for 5.9 years on average.
During the 3-year drug holiday, 915 of the 50,154 patients had hip fractures.
This was equivalent to 12.4 hip fractures per 1,000 patients per year during a risedronate holiday and 10.6 hip fractures per 1,000 patients per year during an alendronate holiday (hazard ratio, 1.18).
The risk of hip fracture was not significantly higher at 1 year (HR, 1.03) or at 2 years of a risedronate holiday versus an alendronate holiday (HR, 1.14).
However, the risk of a hip fracture was significantly higher at 2 to 3 years of a risedronate holiday than after an alendronate holiday (HR, 1.34).
There was no significant difference in the risk of any osteoporotic fracture overall (including hip, vertebrae, pelvis, ribs, forearm), however, during a 3-year risedronate versus alendronate drug holiday (HR, 1.07).
The research was supported by the Canadian Institutes of Health Research and Institute for Clinical Evaluative Sciences. Dr. Hayes was supported by a CIHR doctoral research award. The authors have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Super-low uric acid may not be best for erosive gout
Lowering the serum urate target to less than 0.20 mmol/L (<3.6 mg/dL) for patients with erosive gout does not achieve better gout outcomes and leads to more medication use and subsequent side effects, according to findings from a 2-year, double-blind, randomized, controlled trial.
Nicola Dalbeth, MD, of the bone and joint research group, department of medicine, faculty of medical and health sciences at University of Auckland (New Zealand), and coauthors noted that intensive serum urate lowering is difficult to achieve with oral urate-lowering therapy (ULT) and their findings suggest lower is not always better.
Their data, published in Arthritis & Rheumatology, suggest the less-intensive standard target of less than 0.30 mmol/L (<5.4 mg/dL), currently recommended by rheumatology guidelines, is sufficient.
The more intensive target leads to a high medication burden and does not improve bone erosion score in erosive gout, the authors found.
Rheumatologist Angelo Gaffo, MD, associate professor of medicine at the University of Alabama at Birmingham, who was not part of the study, said erosion scores are the best way to test outcomes and this study provides support for current gout treatment approaches.
“It is reassuring that the approach of treating to target is a good approach,” Dr. Gaffo said. “The very, very low targets were not better than the [standard target].”
The trial included 104 participants with erosive gout on oral ULT who were randomized either to a serum urate target of less than 0.20 mmol/L or less than 0.30 mmol/L.
Ninety participants completed the study: 44 (85%) in the intensive target group and 46 (88%) in the standard target group. All were included in the primary intention-to-treat analysis. Participants were mostly men with an average age of 61. Average period of disease was 19 years and about half had a gout flare in the 3 months before enrollment in the study.
Fewer in intensive group hit target
The researchers found that serum urate at year 2 was significantly lower in the intensive target group, compared with the level in the standard target group (P = .002), but fewer participants in the intensive group hit their target, compared with those in the standard group (62% vs. 83%; P < .05).
The intensive group also required more medication. Participants in that group needed higher doses of the first-line treatment allopurinol (mean, 746 mg/day vs. 496 mg/day; P < .001). They also used more combination therapy (P = .0004).
Bone erosion scores were slightly better in both groups over 2 years, but there was no between-group difference (P = .20).
Rates of adverse and serious adverse events were similar between the groups.
The authors noted that a previous study has shown that escalating doses of allopurinol to achieve a target lower than 0.36 mmol/L (6.48 mg/dL) can reduce progression of bone erosion in gout.
“However, improved erosion scores were not observed in this study,” the authors noted.
The authors said that emerging data on intensive serum urate lowering “may lead to erosion healing in gout,” particularly with pegloticase (Krystexxa), a treatment that leads to profound reductions in serum urate.
They highlighted a small longitudinal study of patients treated with pegloticase in whom researchers observed the filling in of bone erosions over a year.
Pegloticase not available outside United States
However, the authors explained, use of pegloticase is unlikely to be widespread for erosive gout because of its lack of availability outside the United States and the need for infusions every 2 weeks. Therefore, more feasible strategies are needed.
Guidelines suggest the serum urate target of less than 0.30 mmol/L (5.4 mg/dL) for people with severe gout, including those with chronic arthropathy.
Managing gout is a long-term process
Herbert S.B. Baraf, MD, a rheumatologist in a large group practice in the Washington, D.C., area and clinical professor of medicine at George Washington University, Washington, who was not part of this study, said he would not come to the conclusion that some cynics might draw that there’s no point in trying to continually lower uric acid.

“Managing gout is a long-term proposition, and the long-term benefit of continuous uric acid lowering continue to accumulate over a period of time,” Dr. Baraf said.
He agreed with Dr. Dalbeth and colleagues that trying to get serum uric acid to less than 0.20 mmol/L is very difficult to achieve with oral drugs.
He said: “The study was not able to show a change in erosions because the amount of uric acid lowering wasn’t profound enough over a short enough period of time to show that, but over a longer period of time it might well show that.”
He said oral therapies work more slowly than enzyme-based therapies, such as pegloticase, but agreed there are barriers to using pegloticase.
“A drug like pegloticase costs about $26,000 per infusion every 2 weeks for a 6-month period. It’s not practical, and we tend to use it for people who are severely functionally impaired,” said Dr. Baraf.
It would still be a goal to keep the arthritis from progressing by using oral therapies, he said.
“I wouldn’t denigrate the fact that oral therapies are effective in decreasing flares over time, decreasing tophaceous deposits and probably – over a longer period of time allowing bone to heal. But 2 years is not enough time to show that.” He said showing benefit on erosions may take 5-10 years instead.
The study authors noted that the trial’s results “are not relevant to those without erosive disease, and to health care systems without access to a broad range of urate-lowering agents.”
Dr. Dalbeth reports personal fees (all less than $10,000) from AstraZeneca, Dyve BioSciences, Selecta, Arthrosi, Horizon, AbbVie, JW Pharmaceuticals, and PK Med outside the submitted work. The other authors have no disclosures. Dr. Gaffo reported no relevant financial relationships. Dr. Baraf has been an investigator/consultant and speaker for Horizon Therapeutics, maker of pegloticase; is an investigator and a consultant to Selecta Biosciences; and has been an investigator, speaker, and consultant for Takeda.
Lowering the serum urate target to less than 0.20 mmol/L (<3.6 mg/dL) for patients with erosive gout does not achieve better gout outcomes and leads to more medication use and subsequent side effects, according to findings from a 2-year, double-blind, randomized, controlled trial.
Nicola Dalbeth, MD, of the bone and joint research group, department of medicine, faculty of medical and health sciences at University of Auckland (New Zealand), and coauthors noted that intensive serum urate lowering is difficult to achieve with oral urate-lowering therapy (ULT) and their findings suggest lower is not always better.
Their data, published in Arthritis & Rheumatology, suggest the less-intensive standard target of less than 0.30 mmol/L (<5.4 mg/dL), currently recommended by rheumatology guidelines, is sufficient.
The more intensive target leads to a high medication burden and does not improve bone erosion score in erosive gout, the authors found.
Rheumatologist Angelo Gaffo, MD, associate professor of medicine at the University of Alabama at Birmingham, who was not part of the study, said erosion scores are the best way to test outcomes and this study provides support for current gout treatment approaches.
“It is reassuring that the approach of treating to target is a good approach,” Dr. Gaffo said. “The very, very low targets were not better than the [standard target].”
The trial included 104 participants with erosive gout on oral ULT who were randomized either to a serum urate target of less than 0.20 mmol/L or less than 0.30 mmol/L.
Ninety participants completed the study: 44 (85%) in the intensive target group and 46 (88%) in the standard target group. All were included in the primary intention-to-treat analysis. Participants were mostly men with an average age of 61. Average period of disease was 19 years and about half had a gout flare in the 3 months before enrollment in the study.
Fewer in intensive group hit target
The researchers found that serum urate at year 2 was significantly lower in the intensive target group, compared with the level in the standard target group (P = .002), but fewer participants in the intensive group hit their target, compared with those in the standard group (62% vs. 83%; P < .05).
The intensive group also required more medication. Participants in that group needed higher doses of the first-line treatment allopurinol (mean, 746 mg/day vs. 496 mg/day; P < .001). They also used more combination therapy (P = .0004).
Bone erosion scores were slightly better in both groups over 2 years, but there was no between-group difference (P = .20).
Rates of adverse and serious adverse events were similar between the groups.
The authors noted that a previous study has shown that escalating doses of allopurinol to achieve a target lower than 0.36 mmol/L (6.48 mg/dL) can reduce progression of bone erosion in gout.
“However, improved erosion scores were not observed in this study,” the authors noted.
The authors said that emerging data on intensive serum urate lowering “may lead to erosion healing in gout,” particularly with pegloticase (Krystexxa), a treatment that leads to profound reductions in serum urate.
They highlighted a small longitudinal study of patients treated with pegloticase in whom researchers observed the filling in of bone erosions over a year.
Pegloticase not available outside United States
However, the authors explained, use of pegloticase is unlikely to be widespread for erosive gout because of its lack of availability outside the United States and the need for infusions every 2 weeks. Therefore, more feasible strategies are needed.
Guidelines suggest the serum urate target of less than 0.30 mmol/L (5.4 mg/dL) for people with severe gout, including those with chronic arthropathy.
Managing gout is a long-term process
Herbert S.B. Baraf, MD, a rheumatologist in a large group practice in the Washington, D.C., area and clinical professor of medicine at George Washington University, Washington, who was not part of this study, said he would not come to the conclusion that some cynics might draw that there’s no point in trying to continually lower uric acid.
“Managing gout is a long-term proposition, and the long-term benefit of continuous uric acid lowering continue to accumulate over a period of time,” Dr. Baraf said.
He agreed with Dr. Dalbeth and colleagues that trying to get serum uric acid to less than 0.20 mmol/L is very difficult to achieve with oral drugs.
He said: “The study was not able to show a change in erosions because the amount of uric acid lowering wasn’t profound enough over a short enough period of time to show that, but over a longer period of time it might well show that.”
He said oral therapies work more slowly than enzyme-based therapies, such as pegloticase, but agreed there are barriers to using pegloticase.
“A drug like pegloticase costs about $26,000 per infusion every 2 weeks for a 6-month period. It’s not practical, and we tend to use it for people who are severely functionally impaired,” said Dr. Baraf.
It would still be a goal to keep the arthritis from progressing by using oral therapies, he said.
“I wouldn’t denigrate the fact that oral therapies are effective in decreasing flares over time, decreasing tophaceous deposits and probably – over a longer period of time allowing bone to heal. But 2 years is not enough time to show that.” He said showing benefit on erosions may take 5-10 years instead.
The study authors noted that the trial’s results “are not relevant to those without erosive disease, and to health care systems without access to a broad range of urate-lowering agents.”
Dr. Dalbeth reports personal fees (all less than $10,000) from AstraZeneca, Dyve BioSciences, Selecta, Arthrosi, Horizon, AbbVie, JW Pharmaceuticals, and PK Med outside the submitted work. The other authors have no disclosures. Dr. Gaffo reported no relevant financial relationships. Dr. Baraf has been an investigator/consultant and speaker for Horizon Therapeutics, maker of pegloticase; is an investigator and a consultant to Selecta Biosciences; and has been an investigator, speaker, and consultant for Takeda.
Lowering the serum urate target to less than 0.20 mmol/L (<3.6 mg/dL) for patients with erosive gout does not achieve better gout outcomes and leads to more medication use and subsequent side effects, according to findings from a 2-year, double-blind, randomized, controlled trial.
Nicola Dalbeth, MD, of the bone and joint research group, department of medicine, faculty of medical and health sciences at University of Auckland (New Zealand), and coauthors noted that intensive serum urate lowering is difficult to achieve with oral urate-lowering therapy (ULT) and their findings suggest lower is not always better.
Their data, published in Arthritis & Rheumatology, suggest the less-intensive standard target of less than 0.30 mmol/L (<5.4 mg/dL), currently recommended by rheumatology guidelines, is sufficient.
The more intensive target leads to a high medication burden and does not improve bone erosion score in erosive gout, the authors found.
Rheumatologist Angelo Gaffo, MD, associate professor of medicine at the University of Alabama at Birmingham, who was not part of the study, said erosion scores are the best way to test outcomes and this study provides support for current gout treatment approaches.
“It is reassuring that the approach of treating to target is a good approach,” Dr. Gaffo said. “The very, very low targets were not better than the [standard target].”
The trial included 104 participants with erosive gout on oral ULT who were randomized either to a serum urate target of less than 0.20 mmol/L or less than 0.30 mmol/L.
Ninety participants completed the study: 44 (85%) in the intensive target group and 46 (88%) in the standard target group. All were included in the primary intention-to-treat analysis. Participants were mostly men with an average age of 61. Average period of disease was 19 years and about half had a gout flare in the 3 months before enrollment in the study.
Fewer in intensive group hit target
The researchers found that serum urate at year 2 was significantly lower in the intensive target group, compared with the level in the standard target group (P = .002), but fewer participants in the intensive group hit their target, compared with those in the standard group (62% vs. 83%; P < .05).
The intensive group also required more medication. Participants in that group needed higher doses of the first-line treatment allopurinol (mean, 746 mg/day vs. 496 mg/day; P < .001). They also used more combination therapy (P = .0004).
Bone erosion scores were slightly better in both groups over 2 years, but there was no between-group difference (P = .20).
Rates of adverse and serious adverse events were similar between the groups.
The authors noted that a previous study has shown that escalating doses of allopurinol to achieve a target lower than 0.36 mmol/L (6.48 mg/dL) can reduce progression of bone erosion in gout.
“However, improved erosion scores were not observed in this study,” the authors noted.
The authors said that emerging data on intensive serum urate lowering “may lead to erosion healing in gout,” particularly with pegloticase (Krystexxa), a treatment that leads to profound reductions in serum urate.
They highlighted a small longitudinal study of patients treated with pegloticase in whom researchers observed the filling in of bone erosions over a year.
Pegloticase not available outside United States
However, the authors explained, use of pegloticase is unlikely to be widespread for erosive gout because of its lack of availability outside the United States and the need for infusions every 2 weeks. Therefore, more feasible strategies are needed.
Guidelines suggest the serum urate target of less than 0.30 mmol/L (5.4 mg/dL) for people with severe gout, including those with chronic arthropathy.
Managing gout is a long-term process
Herbert S.B. Baraf, MD, a rheumatologist in a large group practice in the Washington, D.C., area and clinical professor of medicine at George Washington University, Washington, who was not part of this study, said he would not come to the conclusion that some cynics might draw that there’s no point in trying to continually lower uric acid.
“Managing gout is a long-term proposition, and the long-term benefit of continuous uric acid lowering continue to accumulate over a period of time,” Dr. Baraf said.
He agreed with Dr. Dalbeth and colleagues that trying to get serum uric acid to less than 0.20 mmol/L is very difficult to achieve with oral drugs.
He said: “The study was not able to show a change in erosions because the amount of uric acid lowering wasn’t profound enough over a short enough period of time to show that, but over a longer period of time it might well show that.”
He said oral therapies work more slowly than enzyme-based therapies, such as pegloticase, but agreed there are barriers to using pegloticase.
“A drug like pegloticase costs about $26,000 per infusion every 2 weeks for a 6-month period. It’s not practical, and we tend to use it for people who are severely functionally impaired,” said Dr. Baraf.
It would still be a goal to keep the arthritis from progressing by using oral therapies, he said.
“I wouldn’t denigrate the fact that oral therapies are effective in decreasing flares over time, decreasing tophaceous deposits and probably – over a longer period of time allowing bone to heal. But 2 years is not enough time to show that.” He said showing benefit on erosions may take 5-10 years instead.
The study authors noted that the trial’s results “are not relevant to those without erosive disease, and to health care systems without access to a broad range of urate-lowering agents.”
Dr. Dalbeth reports personal fees (all less than $10,000) from AstraZeneca, Dyve BioSciences, Selecta, Arthrosi, Horizon, AbbVie, JW Pharmaceuticals, and PK Med outside the submitted work. The other authors have no disclosures. Dr. Gaffo reported no relevant financial relationships. Dr. Baraf has been an investigator/consultant and speaker for Horizon Therapeutics, maker of pegloticase; is an investigator and a consultant to Selecta Biosciences; and has been an investigator, speaker, and consultant for Takeda.
FROM ARTHRITIS & RHEUMATOLOGY
Proactive infliximab monitoring found best for sustaining control of inflammatory diseases
A new study has found that proactive therapeutic drug monitoring (TDM) with maintenance infliximab is more effective than standard therapy in sustaining control of immune-mediated inflammatory diseases.
The findings from the Norwegian Drug Monitoring B (NOR-DRUM B) trial, published Dec. 21, 2021, in JAMA, provide greater support to the usefulness of TDM in proactively monitoring serum drug levels and antidrug antibodies to infliximab, which has been previously shown to have benefit in patients with inflammatory bowel disease, but leave the benefits of proactive versus reactive monitoring and the cost-effectiveness of the approach in individual immune-mediated inflammatory diseases still open to questioning.
TDM is ‘not the holy grail,’ and that’s OK
“This is an important milestone in the field of TDM with biologics for immunoinflammatory diseases,” Niels Vande Casteele, PharmD, PhD, of the University of California, San Diego, told this news organization. He was not involved in the study.
“When you read through the study, you can see the authors used the TAXIT trial results to inform their study design and the sample size,” he added, referencing his 2015 study on infliximab guide dosing for patients with inflammatory bowel disease, “the first-ever randomized, controlled trial of proactive TDM with any biologic.”
For the TAXIT study’s primary outcome of clinical and biochemical remission at 1 year, “continued concentration-based dosing was not superior to clinically based dosing for achieving remission.” But in regard to their secondary outcome of sustained remission, their results were quite similar to the results of NOR-DRUM B.

“If anything, we already showed a benefit of proactive TDM in 2015,” he said, “but I’m very glad that the authors looked at the trial design and teased out where TDM could be the most important and have the biggest impact, which is to maintain that sustained disease remission over a prolonged period.”
As for next steps, Dr. Vande Casteele noted that TDM isn’t a one-size-fits-all upgrade for drug treatments. But that doesn’t mean it won’t be very useful in many patients.
“What the paper is saying, and what we’ve been finding all along, is that TDM is not the holy grail,” he said. “But it is a tool in the physicians’ toolbox to optimize treatments and maximize efficacy, and there are some patients who truly benefit from it.”
Study details
To determine if proactive TDM with infliximab led to more sustained disease control than standard therapy, first author Silje Watterdal Syversen, MD, PhD, of Diakonhjemmet Hospital in Oslo, and coauthors conducted a 52-week, randomized, parallel-group, open-label trial. From 20 Norwegian hospitals, they recruited 458 patients with rheumatoid arthritis (n = 80), spondyloarthritis (n = 138), psoriatic arthritis (n = 54), ulcerative colitis (n = 81), Crohn’s disease (n = 68), or psoriasis (n = 37) who were undergoing maintenance therapy with the biologic.

The 454 patients who received at least one randomly allocated dose of infliximab were treated with one of two strategies: TDM (n = 227) or standard therapy (n = 227). The TDM group received dose and interval adjustments based on an algorithm that factored in serum drug levels and antidrug antibodies. The standard therapy group was treated on the basis of clinical judgment and physician discretion. The average age across groups was roughly 45 years, and just under 50% were women.
Overall, sustained disease control without worsening was achieved in 167 patients (73.6%) in the TDM group and 127 patients (55.9%) in the standard therapy group, with an estimated adjusted difference of 17.6% (95% confidence interval, 9.0%-26.2%; P < .001). The estimated hazard ratio of disease worsening was 2.1 (95% CI, 1.5-2.9) for standard therapy, compared with TDM. A total of 27 patients (15%) in the standard therapy group and 21 patients (9.2%) in the TDM group developed significant levels of antidrug antibodies, defined here as 50 mcg/L or more.
A total of 34 patients discontinued infliximab in each group; in the TDM group, most discontinued because of antidrug antibody formation, while the main reason for discontinuing in the standard therapy group was disease worsening. Adverse events were reported in 137 patients (60%) in the TDM group and 142 patients (63%) in the standard therapy group.
Removing barriers to TDM
It’s not clear that proactive TDM will benefit treatment with all biologic disease-modifying antirheumatic drugs (bDMARDs), but the findings from Dr. Syversen and colleagues state the clear value of using drug monitoring to guide maintenance therapy with infliximab, Zachary S. Wallace, MD, and Jeffrey A. Sparks, MD, wrote in an accompanying editorial.

“The relatively large sample size and rigorous study design ... helped to overcome some limitations of previous observational studies and small clinical trials that yielded conflicting results regarding TDM,” they added, noting that these findings contrasted somewhat with the NOR-DRUM A trial in which TDM did not improve remission induction in patients initiating infliximab therapy.
Along those lines, they recognized that TDM appears to have a greater effect in patients on maintenance infliximab, compared with those just starting the drug, surmising – among several explanations – that achieving remission in someone beginning treatment is a more difficult outcome to achieve than controlling disease in a patient already in remission.

For now, more clinical trials assessing specific diseases and involving other bDMARDs are needed; Dr. Wallace and Dr. Sparks stated that it’s time to remove barriers to implementing TDM – including the need for medical insurance preauthorization before increasing drug doses – and potentially “introduce a new era in treatment approach to maintenance therapy for patients with immune-mediated inflammatory diseases.”
The authors acknowledged their study’s limitations, including disease worsening being measured in part by patient-physician consensus and thus potentially subject to bias. In addition, they did not have the statistical ability to test TDM effectiveness in each of the six disease groups, noting that “these diseases have inherent differences, and findings may not be completely generalizable across groups.”
The study was funded by grants from the Norwegian Regional Health Authorities and the South-Eastern Norway Regional Health Authorities. The authors reported numerous potential conflicts of interest, including receiving personal fees and grants from various pharmaceutical companies. Dr. Wallace and Dr. Sparks also reported receiving research support and fees from pharmaceutical companies. Dr. Vande Casteele reported receiving research grants and personal fees from multiple pharmaceutical companies, all outside of the reviewed work.
A version of this article first appeared on Medscape.com.
A new study has found that proactive therapeutic drug monitoring (TDM) with maintenance infliximab is more effective than standard therapy in sustaining control of immune-mediated inflammatory diseases.
The findings from the Norwegian Drug Monitoring B (NOR-DRUM B) trial, published Dec. 21, 2021, in JAMA, provide greater support to the usefulness of TDM in proactively monitoring serum drug levels and antidrug antibodies to infliximab, which has been previously shown to have benefit in patients with inflammatory bowel disease, but leave the benefits of proactive versus reactive monitoring and the cost-effectiveness of the approach in individual immune-mediated inflammatory diseases still open to questioning.
TDM is ‘not the holy grail,’ and that’s OK
“This is an important milestone in the field of TDM with biologics for immunoinflammatory diseases,” Niels Vande Casteele, PharmD, PhD, of the University of California, San Diego, told this news organization. He was not involved in the study.
“When you read through the study, you can see the authors used the TAXIT trial results to inform their study design and the sample size,” he added, referencing his 2015 study on infliximab guide dosing for patients with inflammatory bowel disease, “the first-ever randomized, controlled trial of proactive TDM with any biologic.”
For the TAXIT study’s primary outcome of clinical and biochemical remission at 1 year, “continued concentration-based dosing was not superior to clinically based dosing for achieving remission.” But in regard to their secondary outcome of sustained remission, their results were quite similar to the results of NOR-DRUM B.
“If anything, we already showed a benefit of proactive TDM in 2015,” he said, “but I’m very glad that the authors looked at the trial design and teased out where TDM could be the most important and have the biggest impact, which is to maintain that sustained disease remission over a prolonged period.”
As for next steps, Dr. Vande Casteele noted that TDM isn’t a one-size-fits-all upgrade for drug treatments. But that doesn’t mean it won’t be very useful in many patients.
“What the paper is saying, and what we’ve been finding all along, is that TDM is not the holy grail,” he said. “But it is a tool in the physicians’ toolbox to optimize treatments and maximize efficacy, and there are some patients who truly benefit from it.”
Study details
To determine if proactive TDM with infliximab led to more sustained disease control than standard therapy, first author Silje Watterdal Syversen, MD, PhD, of Diakonhjemmet Hospital in Oslo, and coauthors conducted a 52-week, randomized, parallel-group, open-label trial. From 20 Norwegian hospitals, they recruited 458 patients with rheumatoid arthritis (n = 80), spondyloarthritis (n = 138), psoriatic arthritis (n = 54), ulcerative colitis (n = 81), Crohn’s disease (n = 68), or psoriasis (n = 37) who were undergoing maintenance therapy with the biologic.
The 454 patients who received at least one randomly allocated dose of infliximab were treated with one of two strategies: TDM (n = 227) or standard therapy (n = 227). The TDM group received dose and interval adjustments based on an algorithm that factored in serum drug levels and antidrug antibodies. The standard therapy group was treated on the basis of clinical judgment and physician discretion. The average age across groups was roughly 45 years, and just under 50% were women.
Overall, sustained disease control without worsening was achieved in 167 patients (73.6%) in the TDM group and 127 patients (55.9%) in the standard therapy group, with an estimated adjusted difference of 17.6% (95% confidence interval, 9.0%-26.2%; P < .001). The estimated hazard ratio of disease worsening was 2.1 (95% CI, 1.5-2.9) for standard therapy, compared with TDM. A total of 27 patients (15%) in the standard therapy group and 21 patients (9.2%) in the TDM group developed significant levels of antidrug antibodies, defined here as 50 mcg/L or more.
A total of 34 patients discontinued infliximab in each group; in the TDM group, most discontinued because of antidrug antibody formation, while the main reason for discontinuing in the standard therapy group was disease worsening. Adverse events were reported in 137 patients (60%) in the TDM group and 142 patients (63%) in the standard therapy group.
Removing barriers to TDM
It’s not clear that proactive TDM will benefit treatment with all biologic disease-modifying antirheumatic drugs (bDMARDs), but the findings from Dr. Syversen and colleagues state the clear value of using drug monitoring to guide maintenance therapy with infliximab, Zachary S. Wallace, MD, and Jeffrey A. Sparks, MD, wrote in an accompanying editorial.
“The relatively large sample size and rigorous study design ... helped to overcome some limitations of previous observational studies and small clinical trials that yielded conflicting results regarding TDM,” they added, noting that these findings contrasted somewhat with the NOR-DRUM A trial in which TDM did not improve remission induction in patients initiating infliximab therapy.
Along those lines, they recognized that TDM appears to have a greater effect in patients on maintenance infliximab, compared with those just starting the drug, surmising – among several explanations – that achieving remission in someone beginning treatment is a more difficult outcome to achieve than controlling disease in a patient already in remission.
For now, more clinical trials assessing specific diseases and involving other bDMARDs are needed; Dr. Wallace and Dr. Sparks stated that it’s time to remove barriers to implementing TDM – including the need for medical insurance preauthorization before increasing drug doses – and potentially “introduce a new era in treatment approach to maintenance therapy for patients with immune-mediated inflammatory diseases.”
The authors acknowledged their study’s limitations, including disease worsening being measured in part by patient-physician consensus and thus potentially subject to bias. In addition, they did not have the statistical ability to test TDM effectiveness in each of the six disease groups, noting that “these diseases have inherent differences, and findings may not be completely generalizable across groups.”
The study was funded by grants from the Norwegian Regional Health Authorities and the South-Eastern Norway Regional Health Authorities. The authors reported numerous potential conflicts of interest, including receiving personal fees and grants from various pharmaceutical companies. Dr. Wallace and Dr. Sparks also reported receiving research support and fees from pharmaceutical companies. Dr. Vande Casteele reported receiving research grants and personal fees from multiple pharmaceutical companies, all outside of the reviewed work.
A version of this article first appeared on Medscape.com.
A new study has found that proactive therapeutic drug monitoring (TDM) with maintenance infliximab is more effective than standard therapy in sustaining control of immune-mediated inflammatory diseases.
The findings from the Norwegian Drug Monitoring B (NOR-DRUM B) trial, published Dec. 21, 2021, in JAMA, provide greater support to the usefulness of TDM in proactively monitoring serum drug levels and antidrug antibodies to infliximab, which has been previously shown to have benefit in patients with inflammatory bowel disease, but leave the benefits of proactive versus reactive monitoring and the cost-effectiveness of the approach in individual immune-mediated inflammatory diseases still open to questioning.
TDM is ‘not the holy grail,’ and that’s OK
“This is an important milestone in the field of TDM with biologics for immunoinflammatory diseases,” Niels Vande Casteele, PharmD, PhD, of the University of California, San Diego, told this news organization. He was not involved in the study.
“When you read through the study, you can see the authors used the TAXIT trial results to inform their study design and the sample size,” he added, referencing his 2015 study on infliximab guide dosing for patients with inflammatory bowel disease, “the first-ever randomized, controlled trial of proactive TDM with any biologic.”
For the TAXIT study’s primary outcome of clinical and biochemical remission at 1 year, “continued concentration-based dosing was not superior to clinically based dosing for achieving remission.” But in regard to their secondary outcome of sustained remission, their results were quite similar to the results of NOR-DRUM B.
“If anything, we already showed a benefit of proactive TDM in 2015,” he said, “but I’m very glad that the authors looked at the trial design and teased out where TDM could be the most important and have the biggest impact, which is to maintain that sustained disease remission over a prolonged period.”
As for next steps, Dr. Vande Casteele noted that TDM isn’t a one-size-fits-all upgrade for drug treatments. But that doesn’t mean it won’t be very useful in many patients.
“What the paper is saying, and what we’ve been finding all along, is that TDM is not the holy grail,” he said. “But it is a tool in the physicians’ toolbox to optimize treatments and maximize efficacy, and there are some patients who truly benefit from it.”
Study details
To determine if proactive TDM with infliximab led to more sustained disease control than standard therapy, first author Silje Watterdal Syversen, MD, PhD, of Diakonhjemmet Hospital in Oslo, and coauthors conducted a 52-week, randomized, parallel-group, open-label trial. From 20 Norwegian hospitals, they recruited 458 patients with rheumatoid arthritis (n = 80), spondyloarthritis (n = 138), psoriatic arthritis (n = 54), ulcerative colitis (n = 81), Crohn’s disease (n = 68), or psoriasis (n = 37) who were undergoing maintenance therapy with the biologic.
The 454 patients who received at least one randomly allocated dose of infliximab were treated with one of two strategies: TDM (n = 227) or standard therapy (n = 227). The TDM group received dose and interval adjustments based on an algorithm that factored in serum drug levels and antidrug antibodies. The standard therapy group was treated on the basis of clinical judgment and physician discretion. The average age across groups was roughly 45 years, and just under 50% were women.
Overall, sustained disease control without worsening was achieved in 167 patients (73.6%) in the TDM group and 127 patients (55.9%) in the standard therapy group, with an estimated adjusted difference of 17.6% (95% confidence interval, 9.0%-26.2%; P < .001). The estimated hazard ratio of disease worsening was 2.1 (95% CI, 1.5-2.9) for standard therapy, compared with TDM. A total of 27 patients (15%) in the standard therapy group and 21 patients (9.2%) in the TDM group developed significant levels of antidrug antibodies, defined here as 50 mcg/L or more.
A total of 34 patients discontinued infliximab in each group; in the TDM group, most discontinued because of antidrug antibody formation, while the main reason for discontinuing in the standard therapy group was disease worsening. Adverse events were reported in 137 patients (60%) in the TDM group and 142 patients (63%) in the standard therapy group.
Removing barriers to TDM
It’s not clear that proactive TDM will benefit treatment with all biologic disease-modifying antirheumatic drugs (bDMARDs), but the findings from Dr. Syversen and colleagues state the clear value of using drug monitoring to guide maintenance therapy with infliximab, Zachary S. Wallace, MD, and Jeffrey A. Sparks, MD, wrote in an accompanying editorial.
“The relatively large sample size and rigorous study design ... helped to overcome some limitations of previous observational studies and small clinical trials that yielded conflicting results regarding TDM,” they added, noting that these findings contrasted somewhat with the NOR-DRUM A trial in which TDM did not improve remission induction in patients initiating infliximab therapy.
Along those lines, they recognized that TDM appears to have a greater effect in patients on maintenance infliximab, compared with those just starting the drug, surmising – among several explanations – that achieving remission in someone beginning treatment is a more difficult outcome to achieve than controlling disease in a patient already in remission.
For now, more clinical trials assessing specific diseases and involving other bDMARDs are needed; Dr. Wallace and Dr. Sparks stated that it’s time to remove barriers to implementing TDM – including the need for medical insurance preauthorization before increasing drug doses – and potentially “introduce a new era in treatment approach to maintenance therapy for patients with immune-mediated inflammatory diseases.”
The authors acknowledged their study’s limitations, including disease worsening being measured in part by patient-physician consensus and thus potentially subject to bias. In addition, they did not have the statistical ability to test TDM effectiveness in each of the six disease groups, noting that “these diseases have inherent differences, and findings may not be completely generalizable across groups.”
The study was funded by grants from the Norwegian Regional Health Authorities and the South-Eastern Norway Regional Health Authorities. The authors reported numerous potential conflicts of interest, including receiving personal fees and grants from various pharmaceutical companies. Dr. Wallace and Dr. Sparks also reported receiving research support and fees from pharmaceutical companies. Dr. Vande Casteele reported receiving research grants and personal fees from multiple pharmaceutical companies, all outside of the reviewed work.
A version of this article first appeared on Medscape.com.
FROM JAMA
Therapeutic aquatic exercise superior to physical therapy for back pain in study
“This is the first study to compare the efficacy of therapeutic aquatic exercise and physical therapy modalities in the treatment of chronic low back pain,” senior coauthors Pei-Jie Chen, PhD and Xue-Qiang Wang, PhD, both of the department of sport rehabilitation, Shanghai (China) University of Sport, wrote in JAMA Network Open. “Therapeutic aquatic exercise is a safe treatment for chronic low back pain and most participants who received it were willing to recommend it to other patients with chronic low back pain.”
As compared with individuals in the physical therapy modalities arm, the therapeutic aquatic exercise experienced greater relief of disability at all time points assessed: after the 3-month intervention, at the 6-month follow-up, and at the 12-month follow-up.
Commenting on the study, Linda Girgis, MD, FAAFP, a family physician in private practice in South River, N.J., agreed that aquatic therapy is a great tool for many chronic low back patients. “It helps them get active for one and do things that may exacerbate their symptoms doing the same exercises on land,” noted Dr. Girgis, who also is a clinical assistant professor at Robert Wood Johnson Medical School, New Brunswick.
She pointed out that access to a pool can be a problem. “But I have found a few physical therapy places in my area that do have access to a pool, and I refer appropriate patients there,” added Dr. Girgis, who was not involved with the study. “I have also found it works well for other types of pain, such as knee and hip pain. It is not for everyone but I have seen some patients get great benefit from it when they didn’t get any with traditional physical therapy.”
Aquatic therapy was more beneficial
Low back pain is a common condition, and clinical practice guidelines currently recommend therapeutic exercise and physical therapy modalities. Among the modalities that are available, therapeutic aquatic exercise is often prescribed for chronic low back pain, and it is becoming increasingly popular for treatment of chronic low back pain, the authors stated in their paper. The authors noted that water is an ideal environment for conducting an exercise program given its various properties, including buoyancy pressure, density, thermal capacity, and conductivity.
Two previously published systematic reviews have suggested that therapeutic aquatic exercise may be able to reduce the intensity of back pain and improve function in this population. But to date, evidence regarding long-term benefits in patients with chronic low back pain is very limited and there haven’t been any studies comparing the efficacy of therapeutic aquatic exercise and physical therapy modalities for chronic low back pain, according to the authors.
In this study, 113 individuals with chronic low back pain were randomized to either therapeutic aquatic exercise or to physical therapy, with an endpoint of efficacy regarding disability. This was measured using the Roland-Morris Disability Questionnaire.
Scores ranged from 0 to 24, with higher scores indicating more severe disability. Secondary endpoints included pain intensity, quality of life, sleep quality, and recommendation of intervention, and these were rated using various standardized tools.
Those randomized to the therapeutic aquatic exercise group had about an hour of therapy, beginning with a 10-minute active warm-up session to enhance neuromuscular activation, then an exercise session for 40 minutes followed by a 10-minute cooldown.
The physical therapy group received transcutaneous electrical nerve stimulation and infrared ray thermal therapy, also for 60 minutes. Both groups received these interventions twice a week for 3 months.
The overall mean age of the cohort was 31.0 years, and they were almost evenly divided by gender; 54 were men (47.8%), and 59 were women (52.2%).
As compared with the physical therapy group, individuals participating in therapeutic aquatic exercise group showed improvement in disability by an additional −1.77 points (95% confidence interval, −3.02 to −0.51) at the end of the 3-month intervention; at 6 months it was −2.42 points (95% CI, −4.13 to −0.70) and −3.61 points (95% CI, −5.63 to −1.58) at the 12-month follow-up (P < .001 for overall group x time interaction).
Functional improvement did not appear to be significantly affected by confounders that included age, sex, body mass index, low back pain duration, educational level, or pain level.
For secondary outcomes, those in the therapeutic aquatic exercise group demonstrated improvement in the most severe pain by an additional −0.79 points (95% CI, −1.31 to −0.27) after the 3-month intervention, −1.34 points (95% CI, −2.06 to −0.62) at 6 months, and −2.04 points (95% CI, −2.75 to −1.34) at the 12-month follow-up (P < .001 for overall group x time interaction), as compared with the physical therapy group. All pain scores differed significantly between the two groups at every time point.
In addition, individuals in the therapeutic aquatic exercise group showed more improvements on the 36-item Short-form Health Survey (overall group x time interaction, P = .003), Pittsburgh Sleep Quality Index (overall group x time interaction, P = .02), Tampa Scale for Kinesiophobia (overall group x time interaction, P < .001), and Fear-Avoidance Beliefs Questionnaire (physical activity subscale overall group x time interaction, P = .04), as compared with the physical therapy group. These improvements were also not influenced by confounders.
Finally, at the 12-month follow-up point, those in the aquatic therapy group had significantly greater improvements in the number of participants who met the minimal clinically important difference in pain (at least a 2-point improvement on the numeric rating scale).
More outside experts’ takes
“The current research evidence does suggest indeed that aquatic exercise therapy is suitable and often better than land exercise, passive relaxation, or other treatments for many people with low back pain,” commented Stelios Psycharakis PhD, senior lecturer in biomechanics, Institute for Sport, Physical Education and Health Sciences, University of Edinburgh.
He also noted that since low back pain is an issue affecting about 80% of all people at some stage of their life, it is “improbable that one could identify a single type of treatment or exercise therapy that would be suitable for every person with this problem.”
Dr. Psycharakis pointed out that there are also some contraindications for aquatic therapy, such as incontinence and skin conditions. “Other than that though, clinicians should definitely consider aquatic exercise therapy when advising people with chronic low back pain,” he said.
Justin M. Lantz, DPT, agreed that the study showed therapeutic aquatic exercise appears to be safe and beneficial in some patients with chronic low back pain, but he also shared limitations of the new research.
“The study has notable limitations as it did not include patients above 65 years old, pain levels were generally low for the subjects involved, and it did not include a treatment group with land therapeutic exercise – so it is difficult to determine if the beneficial effects reported were due to active exercise or because the exercises were performed in water,” said Dr. Lantz, director of the spine physical therapy fellowship program at the University of Southern California, Los Angeles, and an assistant professor of clinical physical therapy.
He also pointed out that, since active exercise has been shown to be beneficial and is advocated in multiple clinical practice guidelines for chronic low back pain, “it would be helpful to determine if the true effects on pain and disability were due to the water environment or the effect of active exercise itself.”
“Due to the significant positive long-term effects and limited adverse events reported, I believe this study supports the use of therapeutic aquatic exercise in select patient populations with chronic low back pain and should be considered as a part of a rehabilitation treatment plan if accessibility is feasible,” Dr. Lantz said.
The authors of the paper, Dr. Girgis, and Dr. Psycharakis had no conflicts of interest. Justin Lantz is a physical therapy consultant to SI-Bone.
“This is the first study to compare the efficacy of therapeutic aquatic exercise and physical therapy modalities in the treatment of chronic low back pain,” senior coauthors Pei-Jie Chen, PhD and Xue-Qiang Wang, PhD, both of the department of sport rehabilitation, Shanghai (China) University of Sport, wrote in JAMA Network Open. “Therapeutic aquatic exercise is a safe treatment for chronic low back pain and most participants who received it were willing to recommend it to other patients with chronic low back pain.”
As compared with individuals in the physical therapy modalities arm, the therapeutic aquatic exercise experienced greater relief of disability at all time points assessed: after the 3-month intervention, at the 6-month follow-up, and at the 12-month follow-up.
Commenting on the study, Linda Girgis, MD, FAAFP, a family physician in private practice in South River, N.J., agreed that aquatic therapy is a great tool for many chronic low back patients. “It helps them get active for one and do things that may exacerbate their symptoms doing the same exercises on land,” noted Dr. Girgis, who also is a clinical assistant professor at Robert Wood Johnson Medical School, New Brunswick.
She pointed out that access to a pool can be a problem. “But I have found a few physical therapy places in my area that do have access to a pool, and I refer appropriate patients there,” added Dr. Girgis, who was not involved with the study. “I have also found it works well for other types of pain, such as knee and hip pain. It is not for everyone but I have seen some patients get great benefit from it when they didn’t get any with traditional physical therapy.”
Aquatic therapy was more beneficial
Low back pain is a common condition, and clinical practice guidelines currently recommend therapeutic exercise and physical therapy modalities. Among the modalities that are available, therapeutic aquatic exercise is often prescribed for chronic low back pain, and it is becoming increasingly popular for treatment of chronic low back pain, the authors stated in their paper. The authors noted that water is an ideal environment for conducting an exercise program given its various properties, including buoyancy pressure, density, thermal capacity, and conductivity.
Two previously published systematic reviews have suggested that therapeutic aquatic exercise may be able to reduce the intensity of back pain and improve function in this population. But to date, evidence regarding long-term benefits in patients with chronic low back pain is very limited and there haven’t been any studies comparing the efficacy of therapeutic aquatic exercise and physical therapy modalities for chronic low back pain, according to the authors.
In this study, 113 individuals with chronic low back pain were randomized to either therapeutic aquatic exercise or to physical therapy, with an endpoint of efficacy regarding disability. This was measured using the Roland-Morris Disability Questionnaire.
Scores ranged from 0 to 24, with higher scores indicating more severe disability. Secondary endpoints included pain intensity, quality of life, sleep quality, and recommendation of intervention, and these were rated using various standardized tools.
Those randomized to the therapeutic aquatic exercise group had about an hour of therapy, beginning with a 10-minute active warm-up session to enhance neuromuscular activation, then an exercise session for 40 minutes followed by a 10-minute cooldown.
The physical therapy group received transcutaneous electrical nerve stimulation and infrared ray thermal therapy, also for 60 minutes. Both groups received these interventions twice a week for 3 months.
The overall mean age of the cohort was 31.0 years, and they were almost evenly divided by gender; 54 were men (47.8%), and 59 were women (52.2%).
As compared with the physical therapy group, individuals participating in therapeutic aquatic exercise group showed improvement in disability by an additional −1.77 points (95% confidence interval, −3.02 to −0.51) at the end of the 3-month intervention; at 6 months it was −2.42 points (95% CI, −4.13 to −0.70) and −3.61 points (95% CI, −5.63 to −1.58) at the 12-month follow-up (P < .001 for overall group x time interaction).
Functional improvement did not appear to be significantly affected by confounders that included age, sex, body mass index, low back pain duration, educational level, or pain level.
For secondary outcomes, those in the therapeutic aquatic exercise group demonstrated improvement in the most severe pain by an additional −0.79 points (95% CI, −1.31 to −0.27) after the 3-month intervention, −1.34 points (95% CI, −2.06 to −0.62) at 6 months, and −2.04 points (95% CI, −2.75 to −1.34) at the 12-month follow-up (P < .001 for overall group x time interaction), as compared with the physical therapy group. All pain scores differed significantly between the two groups at every time point.
In addition, individuals in the therapeutic aquatic exercise group showed more improvements on the 36-item Short-form Health Survey (overall group x time interaction, P = .003), Pittsburgh Sleep Quality Index (overall group x time interaction, P = .02), Tampa Scale for Kinesiophobia (overall group x time interaction, P < .001), and Fear-Avoidance Beliefs Questionnaire (physical activity subscale overall group x time interaction, P = .04), as compared with the physical therapy group. These improvements were also not influenced by confounders.
Finally, at the 12-month follow-up point, those in the aquatic therapy group had significantly greater improvements in the number of participants who met the minimal clinically important difference in pain (at least a 2-point improvement on the numeric rating scale).
More outside experts’ takes
“The current research evidence does suggest indeed that aquatic exercise therapy is suitable and often better than land exercise, passive relaxation, or other treatments for many people with low back pain,” commented Stelios Psycharakis PhD, senior lecturer in biomechanics, Institute for Sport, Physical Education and Health Sciences, University of Edinburgh.
He also noted that since low back pain is an issue affecting about 80% of all people at some stage of their life, it is “improbable that one could identify a single type of treatment or exercise therapy that would be suitable for every person with this problem.”
Dr. Psycharakis pointed out that there are also some contraindications for aquatic therapy, such as incontinence and skin conditions. “Other than that though, clinicians should definitely consider aquatic exercise therapy when advising people with chronic low back pain,” he said.
Justin M. Lantz, DPT, agreed that the study showed therapeutic aquatic exercise appears to be safe and beneficial in some patients with chronic low back pain, but he also shared limitations of the new research.
“The study has notable limitations as it did not include patients above 65 years old, pain levels were generally low for the subjects involved, and it did not include a treatment group with land therapeutic exercise – so it is difficult to determine if the beneficial effects reported were due to active exercise or because the exercises were performed in water,” said Dr. Lantz, director of the spine physical therapy fellowship program at the University of Southern California, Los Angeles, and an assistant professor of clinical physical therapy.
He also pointed out that, since active exercise has been shown to be beneficial and is advocated in multiple clinical practice guidelines for chronic low back pain, “it would be helpful to determine if the true effects on pain and disability were due to the water environment or the effect of active exercise itself.”
“Due to the significant positive long-term effects and limited adverse events reported, I believe this study supports the use of therapeutic aquatic exercise in select patient populations with chronic low back pain and should be considered as a part of a rehabilitation treatment plan if accessibility is feasible,” Dr. Lantz said.
The authors of the paper, Dr. Girgis, and Dr. Psycharakis had no conflicts of interest. Justin Lantz is a physical therapy consultant to SI-Bone.
“This is the first study to compare the efficacy of therapeutic aquatic exercise and physical therapy modalities in the treatment of chronic low back pain,” senior coauthors Pei-Jie Chen, PhD and Xue-Qiang Wang, PhD, both of the department of sport rehabilitation, Shanghai (China) University of Sport, wrote in JAMA Network Open. “Therapeutic aquatic exercise is a safe treatment for chronic low back pain and most participants who received it were willing to recommend it to other patients with chronic low back pain.”
As compared with individuals in the physical therapy modalities arm, the therapeutic aquatic exercise experienced greater relief of disability at all time points assessed: after the 3-month intervention, at the 6-month follow-up, and at the 12-month follow-up.
Commenting on the study, Linda Girgis, MD, FAAFP, a family physician in private practice in South River, N.J., agreed that aquatic therapy is a great tool for many chronic low back patients. “It helps them get active for one and do things that may exacerbate their symptoms doing the same exercises on land,” noted Dr. Girgis, who also is a clinical assistant professor at Robert Wood Johnson Medical School, New Brunswick.
She pointed out that access to a pool can be a problem. “But I have found a few physical therapy places in my area that do have access to a pool, and I refer appropriate patients there,” added Dr. Girgis, who was not involved with the study. “I have also found it works well for other types of pain, such as knee and hip pain. It is not for everyone but I have seen some patients get great benefit from it when they didn’t get any with traditional physical therapy.”
Aquatic therapy was more beneficial
Low back pain is a common condition, and clinical practice guidelines currently recommend therapeutic exercise and physical therapy modalities. Among the modalities that are available, therapeutic aquatic exercise is often prescribed for chronic low back pain, and it is becoming increasingly popular for treatment of chronic low back pain, the authors stated in their paper. The authors noted that water is an ideal environment for conducting an exercise program given its various properties, including buoyancy pressure, density, thermal capacity, and conductivity.
Two previously published systematic reviews have suggested that therapeutic aquatic exercise may be able to reduce the intensity of back pain and improve function in this population. But to date, evidence regarding long-term benefits in patients with chronic low back pain is very limited and there haven’t been any studies comparing the efficacy of therapeutic aquatic exercise and physical therapy modalities for chronic low back pain, according to the authors.
In this study, 113 individuals with chronic low back pain were randomized to either therapeutic aquatic exercise or to physical therapy, with an endpoint of efficacy regarding disability. This was measured using the Roland-Morris Disability Questionnaire.
Scores ranged from 0 to 24, with higher scores indicating more severe disability. Secondary endpoints included pain intensity, quality of life, sleep quality, and recommendation of intervention, and these were rated using various standardized tools.
Those randomized to the therapeutic aquatic exercise group had about an hour of therapy, beginning with a 10-minute active warm-up session to enhance neuromuscular activation, then an exercise session for 40 minutes followed by a 10-minute cooldown.
The physical therapy group received transcutaneous electrical nerve stimulation and infrared ray thermal therapy, also for 60 minutes. Both groups received these interventions twice a week for 3 months.
The overall mean age of the cohort was 31.0 years, and they were almost evenly divided by gender; 54 were men (47.8%), and 59 were women (52.2%).
As compared with the physical therapy group, individuals participating in therapeutic aquatic exercise group showed improvement in disability by an additional −1.77 points (95% confidence interval, −3.02 to −0.51) at the end of the 3-month intervention; at 6 months it was −2.42 points (95% CI, −4.13 to −0.70) and −3.61 points (95% CI, −5.63 to −1.58) at the 12-month follow-up (P < .001 for overall group x time interaction).
Functional improvement did not appear to be significantly affected by confounders that included age, sex, body mass index, low back pain duration, educational level, or pain level.
For secondary outcomes, those in the therapeutic aquatic exercise group demonstrated improvement in the most severe pain by an additional −0.79 points (95% CI, −1.31 to −0.27) after the 3-month intervention, −1.34 points (95% CI, −2.06 to −0.62) at 6 months, and −2.04 points (95% CI, −2.75 to −1.34) at the 12-month follow-up (P < .001 for overall group x time interaction), as compared with the physical therapy group. All pain scores differed significantly between the two groups at every time point.
In addition, individuals in the therapeutic aquatic exercise group showed more improvements on the 36-item Short-form Health Survey (overall group x time interaction, P = .003), Pittsburgh Sleep Quality Index (overall group x time interaction, P = .02), Tampa Scale for Kinesiophobia (overall group x time interaction, P < .001), and Fear-Avoidance Beliefs Questionnaire (physical activity subscale overall group x time interaction, P = .04), as compared with the physical therapy group. These improvements were also not influenced by confounders.
Finally, at the 12-month follow-up point, those in the aquatic therapy group had significantly greater improvements in the number of participants who met the minimal clinically important difference in pain (at least a 2-point improvement on the numeric rating scale).
More outside experts’ takes
“The current research evidence does suggest indeed that aquatic exercise therapy is suitable and often better than land exercise, passive relaxation, or other treatments for many people with low back pain,” commented Stelios Psycharakis PhD, senior lecturer in biomechanics, Institute for Sport, Physical Education and Health Sciences, University of Edinburgh.
He also noted that since low back pain is an issue affecting about 80% of all people at some stage of their life, it is “improbable that one could identify a single type of treatment or exercise therapy that would be suitable for every person with this problem.”
Dr. Psycharakis pointed out that there are also some contraindications for aquatic therapy, such as incontinence and skin conditions. “Other than that though, clinicians should definitely consider aquatic exercise therapy when advising people with chronic low back pain,” he said.
Justin M. Lantz, DPT, agreed that the study showed therapeutic aquatic exercise appears to be safe and beneficial in some patients with chronic low back pain, but he also shared limitations of the new research.
“The study has notable limitations as it did not include patients above 65 years old, pain levels were generally low for the subjects involved, and it did not include a treatment group with land therapeutic exercise – so it is difficult to determine if the beneficial effects reported were due to active exercise or because the exercises were performed in water,” said Dr. Lantz, director of the spine physical therapy fellowship program at the University of Southern California, Los Angeles, and an assistant professor of clinical physical therapy.
He also pointed out that, since active exercise has been shown to be beneficial and is advocated in multiple clinical practice guidelines for chronic low back pain, “it would be helpful to determine if the true effects on pain and disability were due to the water environment or the effect of active exercise itself.”
“Due to the significant positive long-term effects and limited adverse events reported, I believe this study supports the use of therapeutic aquatic exercise in select patient populations with chronic low back pain and should be considered as a part of a rehabilitation treatment plan if accessibility is feasible,” Dr. Lantz said.
The authors of the paper, Dr. Girgis, and Dr. Psycharakis had no conflicts of interest. Justin Lantz is a physical therapy consultant to SI-Bone.
FROM JAMA NETWORK OPEN
Duloxetine added to usual care doesn’t improve hip, knee OA pain
A small, open-label, randomized trial of patients with chronic pain from hip and knee osteoarthritis in the Netherlands shows that adding duloxetine to usual care doesn’t significantly improve clinical outcomes.
The results, published on Jan. 6 in Arthritis & Rheumatology, also showed duloxetine did not affect outcomes for a subgroup of patients who had symptoms of centrally sensitized pain, according to Jacoline J. van den Driest, MD, of the department of general practice at Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues.
The researchers acknowledged their findings contrast with other studies that showed a “small to moderate effect of duloxetine” for patients with chronic pain from hip and knee OA. There was also a higher rate of discontinuation of duloxetine around 3 months in the current trial, compared with previous studies, the authors said, which they attributed to the fact that clinicians were asked to discontinue treatment at 3 months if patients saw no effect or increased side effects.
“This difference in outcome can be due to the fact that we studied the effectiveness of duloxetine in primary care, while the other studies examined the efficacy in placebo-controlled trials in secondary care,” the researchers wrote. Patients in the current trial were also older, had more comorbidities, and had been living with OA symptoms “for a longer time” than patients in other trials, they explained.
“It is known that, in these more ‘real-life’ primary care populations and in effectiveness studies, smaller effects are found than in highly controlled efficacy trials,” they noted.
Dr. van den Driest and colleagues evaluated 132 patients with hip or knee OA between January 2016 and February 2019 who were cluster randomized at 66 general practitioner practice sites to receive duloxetine (30 mg/day in the first week, 60 mg/day in the second week and beyond) in addition to usual care that consisted of analgesics, physiotherapy, patient education, diet, and lifestyle advice. Patients were included in the study if they were at least 18 years old, met the American College of Rheumatology criteria for hip or knee OA, and experienced chronic pain for “most days” over 3 months that was not improved through use of NSAIDs or acetaminophen or were unable to use NSAIDs because of contraindications or adverse effects. They were excluded if taking duloxetine was contraindicated for them, if they were taking an antidepressant or neuropathic pain medication, and if they had rheumatoid arthritis or were scheduled for total hip or total knee replacement.
The researchers assessed patients’ Western Ontario McMaster Universities (WOMAC) Osteoarthritis Index pain scores at 3 months, compared with baseline, as a primary outcome, with secondary outcomes of WOMAC pain and function at 1 year, and cost-effectiveness as measured by the EQ-5D-5L. A modified painDETECT questionnaire was also used at baseline to identify a subset of patients with presence of centralized pain, which was defined as a score >12.
At 12 months, 80.3% of patients in both groups completed follow-up. Patient characteristics differed in duloxetine and usual-care groups, with the duloxetine group being younger (63.2 years vs. 65.4 years) and having fewer women (59.1% vs. 75.8%). The duloxetine group also had a lower percentage of patients with knee OA (77.3% vs. 86.4%) and a lower percentage of patients with two or more comorbidities (15.2% vs. 33.2%).
Duloxetine led to a nonsignificant improvement in WOMAC-measured pain at 3 months, compared with usual care (adjusted difference, –0.58; 95% confidence interval, –1.80 to 0.63), and at 12 months (adjusted difference, –0.26; 95% CI, –1.86 to 1.34). Among a subgroup of patients with central sensitization symptoms, there was a nonsignificant improvement in WOMAC-measured pain at 3 months (adjusted difference, –0.32; 95% CI, –2.32 to 1.67) and 12 months (adjusted difference, 1.02; 95% CI, –1.22 to 3.27).
Duloxetine also did not significantly improve WOMAC-measured function at 3 months (adjusted difference, –2.10; 95% CI, –6.39 to 2.20) or 12 months (adjusted difference, –1.79; 95% CI, –7.22 to 3.64).
For other secondary outcomes of quality of life, patient satisfaction, and Outcome Measures in Rheumatology (OMERACT)-Osteoarthritis Research Society International (OARSI) responder criteria, Dr. van den Driest and colleagues noted that “none of the differences between the two groups were clinically relevant or statistically significant.”
Some patients may likely still benefit from duloxetine
Commenting on the results, Joshua F. Baker, MD, MSCE, associate professor of rheumatology and epidemiology at the University of Pennsylvania and Philadelphia VA Medical Center, said the study by van den Driest and colleagues is pragmatic and demonstrates the “ ‘real-world’ benefits of trying duloxetine” – one of the study’s strengths.
“As we would probably expect, the benefits are small, and somewhat smaller in this setting than what was observed in more standard clinical trials evaluating this question,” he said, noting that the study is limited by a small sample size and loss to follow-up, as well as its open-label design and the fact that most patients stopped treatment during follow-up.
Dr. Baker also explained that while patients on average did not have a meaningful effect after taking duloxetine, “that doesn’t mean that the therapy didn’t have a meaningful effect for some people.”
“In fact, though most people didn’t receive a meaningful benefit in this study, some did,” he said. “[A]ccording to these data, treating 8 people would be expected to result in 1 person achieving an [OMERACT-OARSI] response. That’s pretty good for a disease with few things that work.”
Future study of duloxetine should focus on who is most likely to benefit from treatment “since while most probably don’t benefit a lot, some probably do,” he said.
Dr. Baker also called attention to the questions surrounding use of antidepressants. “Use of antidepressants has been questioned by some, since the average clinical benefit is low, even for conditions like depression,” he explained. “However, some would argue that even small benefits may be important since there are few things that do work very well, and because a multimodal approach that provides multiple small benefits to patients can add up to a meaningful benefit.”
This study was funded by The Netherlands Organization for Health Research and Development. One author reported receiving grants from The Netherlands Organization for Health Research and Development, the European Union, FOREUM, and the Dutch Arthritis Association, as well as personal fees from OARSI and Pfizer. The other authors reported no relevant financial disclosures.
* This story was updated 1/6/22.
A small, open-label, randomized trial of patients with chronic pain from hip and knee osteoarthritis in the Netherlands shows that adding duloxetine to usual care doesn’t significantly improve clinical outcomes.
The results, published on Jan. 6 in Arthritis & Rheumatology, also showed duloxetine did not affect outcomes for a subgroup of patients who had symptoms of centrally sensitized pain, according to Jacoline J. van den Driest, MD, of the department of general practice at Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues.
The researchers acknowledged their findings contrast with other studies that showed a “small to moderate effect of duloxetine” for patients with chronic pain from hip and knee OA. There was also a higher rate of discontinuation of duloxetine around 3 months in the current trial, compared with previous studies, the authors said, which they attributed to the fact that clinicians were asked to discontinue treatment at 3 months if patients saw no effect or increased side effects.
“This difference in outcome can be due to the fact that we studied the effectiveness of duloxetine in primary care, while the other studies examined the efficacy in placebo-controlled trials in secondary care,” the researchers wrote. Patients in the current trial were also older, had more comorbidities, and had been living with OA symptoms “for a longer time” than patients in other trials, they explained.
“It is known that, in these more ‘real-life’ primary care populations and in effectiveness studies, smaller effects are found than in highly controlled efficacy trials,” they noted.
Dr. van den Driest and colleagues evaluated 132 patients with hip or knee OA between January 2016 and February 2019 who were cluster randomized at 66 general practitioner practice sites to receive duloxetine (30 mg/day in the first week, 60 mg/day in the second week and beyond) in addition to usual care that consisted of analgesics, physiotherapy, patient education, diet, and lifestyle advice. Patients were included in the study if they were at least 18 years old, met the American College of Rheumatology criteria for hip or knee OA, and experienced chronic pain for “most days” over 3 months that was not improved through use of NSAIDs or acetaminophen or were unable to use NSAIDs because of contraindications or adverse effects. They were excluded if taking duloxetine was contraindicated for them, if they were taking an antidepressant or neuropathic pain medication, and if they had rheumatoid arthritis or were scheduled for total hip or total knee replacement.
The researchers assessed patients’ Western Ontario McMaster Universities (WOMAC) Osteoarthritis Index pain scores at 3 months, compared with baseline, as a primary outcome, with secondary outcomes of WOMAC pain and function at 1 year, and cost-effectiveness as measured by the EQ-5D-5L. A modified painDETECT questionnaire was also used at baseline to identify a subset of patients with presence of centralized pain, which was defined as a score >12.
At 12 months, 80.3% of patients in both groups completed follow-up. Patient characteristics differed in duloxetine and usual-care groups, with the duloxetine group being younger (63.2 years vs. 65.4 years) and having fewer women (59.1% vs. 75.8%). The duloxetine group also had a lower percentage of patients with knee OA (77.3% vs. 86.4%) and a lower percentage of patients with two or more comorbidities (15.2% vs. 33.2%).
Duloxetine led to a nonsignificant improvement in WOMAC-measured pain at 3 months, compared with usual care (adjusted difference, –0.58; 95% confidence interval, –1.80 to 0.63), and at 12 months (adjusted difference, –0.26; 95% CI, –1.86 to 1.34). Among a subgroup of patients with central sensitization symptoms, there was a nonsignificant improvement in WOMAC-measured pain at 3 months (adjusted difference, –0.32; 95% CI, –2.32 to 1.67) and 12 months (adjusted difference, 1.02; 95% CI, –1.22 to 3.27).
Duloxetine also did not significantly improve WOMAC-measured function at 3 months (adjusted difference, –2.10; 95% CI, –6.39 to 2.20) or 12 months (adjusted difference, –1.79; 95% CI, –7.22 to 3.64).
For other secondary outcomes of quality of life, patient satisfaction, and Outcome Measures in Rheumatology (OMERACT)-Osteoarthritis Research Society International (OARSI) responder criteria, Dr. van den Driest and colleagues noted that “none of the differences between the two groups were clinically relevant or statistically significant.”
Some patients may likely still benefit from duloxetine
Commenting on the results, Joshua F. Baker, MD, MSCE, associate professor of rheumatology and epidemiology at the University of Pennsylvania and Philadelphia VA Medical Center, said the study by van den Driest and colleagues is pragmatic and demonstrates the “ ‘real-world’ benefits of trying duloxetine” – one of the study’s strengths.
“As we would probably expect, the benefits are small, and somewhat smaller in this setting than what was observed in more standard clinical trials evaluating this question,” he said, noting that the study is limited by a small sample size and loss to follow-up, as well as its open-label design and the fact that most patients stopped treatment during follow-up.
Dr. Baker also explained that while patients on average did not have a meaningful effect after taking duloxetine, “that doesn’t mean that the therapy didn’t have a meaningful effect for some people.”
“In fact, though most people didn’t receive a meaningful benefit in this study, some did,” he said. “[A]ccording to these data, treating 8 people would be expected to result in 1 person achieving an [OMERACT-OARSI] response. That’s pretty good for a disease with few things that work.”
Future study of duloxetine should focus on who is most likely to benefit from treatment “since while most probably don’t benefit a lot, some probably do,” he said.
Dr. Baker also called attention to the questions surrounding use of antidepressants. “Use of antidepressants has been questioned by some, since the average clinical benefit is low, even for conditions like depression,” he explained. “However, some would argue that even small benefits may be important since there are few things that do work very well, and because a multimodal approach that provides multiple small benefits to patients can add up to a meaningful benefit.”
This study was funded by The Netherlands Organization for Health Research and Development. One author reported receiving grants from The Netherlands Organization for Health Research and Development, the European Union, FOREUM, and the Dutch Arthritis Association, as well as personal fees from OARSI and Pfizer. The other authors reported no relevant financial disclosures.
* This story was updated 1/6/22.
A small, open-label, randomized trial of patients with chronic pain from hip and knee osteoarthritis in the Netherlands shows that adding duloxetine to usual care doesn’t significantly improve clinical outcomes.
The results, published on Jan. 6 in Arthritis & Rheumatology, also showed duloxetine did not affect outcomes for a subgroup of patients who had symptoms of centrally sensitized pain, according to Jacoline J. van den Driest, MD, of the department of general practice at Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues.
The researchers acknowledged their findings contrast with other studies that showed a “small to moderate effect of duloxetine” for patients with chronic pain from hip and knee OA. There was also a higher rate of discontinuation of duloxetine around 3 months in the current trial, compared with previous studies, the authors said, which they attributed to the fact that clinicians were asked to discontinue treatment at 3 months if patients saw no effect or increased side effects.
“This difference in outcome can be due to the fact that we studied the effectiveness of duloxetine in primary care, while the other studies examined the efficacy in placebo-controlled trials in secondary care,” the researchers wrote. Patients in the current trial were also older, had more comorbidities, and had been living with OA symptoms “for a longer time” than patients in other trials, they explained.
“It is known that, in these more ‘real-life’ primary care populations and in effectiveness studies, smaller effects are found than in highly controlled efficacy trials,” they noted.
Dr. van den Driest and colleagues evaluated 132 patients with hip or knee OA between January 2016 and February 2019 who were cluster randomized at 66 general practitioner practice sites to receive duloxetine (30 mg/day in the first week, 60 mg/day in the second week and beyond) in addition to usual care that consisted of analgesics, physiotherapy, patient education, diet, and lifestyle advice. Patients were included in the study if they were at least 18 years old, met the American College of Rheumatology criteria for hip or knee OA, and experienced chronic pain for “most days” over 3 months that was not improved through use of NSAIDs or acetaminophen or were unable to use NSAIDs because of contraindications or adverse effects. They were excluded if taking duloxetine was contraindicated for them, if they were taking an antidepressant or neuropathic pain medication, and if they had rheumatoid arthritis or were scheduled for total hip or total knee replacement.
The researchers assessed patients’ Western Ontario McMaster Universities (WOMAC) Osteoarthritis Index pain scores at 3 months, compared with baseline, as a primary outcome, with secondary outcomes of WOMAC pain and function at 1 year, and cost-effectiveness as measured by the EQ-5D-5L. A modified painDETECT questionnaire was also used at baseline to identify a subset of patients with presence of centralized pain, which was defined as a score >12.
At 12 months, 80.3% of patients in both groups completed follow-up. Patient characteristics differed in duloxetine and usual-care groups, with the duloxetine group being younger (63.2 years vs. 65.4 years) and having fewer women (59.1% vs. 75.8%). The duloxetine group also had a lower percentage of patients with knee OA (77.3% vs. 86.4%) and a lower percentage of patients with two or more comorbidities (15.2% vs. 33.2%).
Duloxetine led to a nonsignificant improvement in WOMAC-measured pain at 3 months, compared with usual care (adjusted difference, –0.58; 95% confidence interval, –1.80 to 0.63), and at 12 months (adjusted difference, –0.26; 95% CI, –1.86 to 1.34). Among a subgroup of patients with central sensitization symptoms, there was a nonsignificant improvement in WOMAC-measured pain at 3 months (adjusted difference, –0.32; 95% CI, –2.32 to 1.67) and 12 months (adjusted difference, 1.02; 95% CI, –1.22 to 3.27).
Duloxetine also did not significantly improve WOMAC-measured function at 3 months (adjusted difference, –2.10; 95% CI, –6.39 to 2.20) or 12 months (adjusted difference, –1.79; 95% CI, –7.22 to 3.64).
For other secondary outcomes of quality of life, patient satisfaction, and Outcome Measures in Rheumatology (OMERACT)-Osteoarthritis Research Society International (OARSI) responder criteria, Dr. van den Driest and colleagues noted that “none of the differences between the two groups were clinically relevant or statistically significant.”
Some patients may likely still benefit from duloxetine
Commenting on the results, Joshua F. Baker, MD, MSCE, associate professor of rheumatology and epidemiology at the University of Pennsylvania and Philadelphia VA Medical Center, said the study by van den Driest and colleagues is pragmatic and demonstrates the “ ‘real-world’ benefits of trying duloxetine” – one of the study’s strengths.
“As we would probably expect, the benefits are small, and somewhat smaller in this setting than what was observed in more standard clinical trials evaluating this question,” he said, noting that the study is limited by a small sample size and loss to follow-up, as well as its open-label design and the fact that most patients stopped treatment during follow-up.
Dr. Baker also explained that while patients on average did not have a meaningful effect after taking duloxetine, “that doesn’t mean that the therapy didn’t have a meaningful effect for some people.”
“In fact, though most people didn’t receive a meaningful benefit in this study, some did,” he said. “[A]ccording to these data, treating 8 people would be expected to result in 1 person achieving an [OMERACT-OARSI] response. That’s pretty good for a disease with few things that work.”
Future study of duloxetine should focus on who is most likely to benefit from treatment “since while most probably don’t benefit a lot, some probably do,” he said.
Dr. Baker also called attention to the questions surrounding use of antidepressants. “Use of antidepressants has been questioned by some, since the average clinical benefit is low, even for conditions like depression,” he explained. “However, some would argue that even small benefits may be important since there are few things that do work very well, and because a multimodal approach that provides multiple small benefits to patients can add up to a meaningful benefit.”
This study was funded by The Netherlands Organization for Health Research and Development. One author reported receiving grants from The Netherlands Organization for Health Research and Development, the European Union, FOREUM, and the Dutch Arthritis Association, as well as personal fees from OARSI and Pfizer. The other authors reported no relevant financial disclosures.
* This story was updated 1/6/22.
FROM ARTHRITIS & RHEUMATOLOGY
Rheumatology achieves 95% fill rate in 2021 MSMP Match; pediatric subspecialty lags
Rheumatologists seeking fellowships continue to show a preference for adult programs. Adult rheumatology programs filled nearly 100% of positions this year, but pediatric rheumatology programs filled only 69% of available slots, echoing trends of previous years.
The National Resident Matching Program (NRMP) issued results for 2021’s Medical Specialties Matching Program (MSMP) and Pediatric Specialties Match (PSM) in December.
“In pediatric rheumatology, like many other pediatric specialties, the limiting factor is the number of interested candidates. The number of available positions has not really changed over the last several years, but multiple positions again remained unfilled this year,” said Beth Marston, MD, chair of the American College of Rheumatology’s Committee on Rheumatology Training and Workforce Issues, in a statement.
Rheumatology was one of seven medical specialties that filled at least 95% of fellowship positions this year.
The specialty filled 120 of 125 enrolled programs (96%) and 266 of 272 certified positions (97.8%) in 2021. A total of 42.1% of the matched applicants comprised MD graduates, followed by foreign (27.1%), U.S. foreign (16.5%), and DO graduates (14.3%).
Among 357 applicants preferring this specialty in 2021, 73.9% (n = 264) matched to rheumatology. This meant that 22.1% did not match to any program. This scenario has played out over the last several years, Dr. Marston noted. Additional support for funding and the creation of more fellowship positions would translate to an increase in rheumatology graduates entering the workforce.
This could help mitigate workforce shortages that the ACR projected in 2015, she added.
Pediatric program applicants remain stagnant
For pediatric fellowships, the numbers weren’t as robust. Just 60% of 32 enrolled programs and 69.2% positions filled in 2021. MD graduates comprised most of the matched applicants (77.8%), followed by U.S. foreign (14.8%) and foreign and DO graduates (3.7% each). Overall, 27 out of 28 applicants or 96.4% matched to this specialty, a metric that’s remained steady but has not grown in recent years, Dr. Marston said.
This “suggests the need for additional efforts to understand and address barriers to choosing rheumatology fellowship training as a career path for pediatricians,” she said. The ACR’s Committee on Training and Workforce recently initiated a survey of combined medicine-pediatrics graduates in rheumatology to gain insights on why these graduates chose this career path.
The ACR is also looking into increasing access to rheumatology specialty care in underserved areas and finding creative solutions for increasing and filling rheumatology fellowship positions.
Largest match on record
Overall, 7,435 applicants participated in the 2021 MSMP, the largest on record. NRMP reported that 2,277 programs submitted rank order lists and offered 6,368 positions, an increase of more than 11% from 2020, respectively. A total of 90.4% positions (n = 5,759) were filled.
“The 2021 MSMP matched a record number of applicants to subspecialty training programs for positions set to begin July 2022,” NRMP President and CEO Donna L. Lamb, DHSc, MBA, said in a statement. “It’s rewarding to watch the MSMP grow, not only in terms of applicant interest and available training positions, but also from its launch 20 years ago with only three internal medicine subspecialties.”
NRMP largely attributed the increase in positions to the addition of critical care medicine, the latest subspecialty to join the MSMP. All fellows begin their training in July 2022.
The PSM also saw notable increases this year in several metrics. It offered 1,735 positions this year, a 5.9% increase from 2020. Overall, 1,507 positions (86.9%) were filled, a 6.6% increase from last year. 854 programs participated.
Rheumatologists seeking fellowships continue to show a preference for adult programs. Adult rheumatology programs filled nearly 100% of positions this year, but pediatric rheumatology programs filled only 69% of available slots, echoing trends of previous years.
The National Resident Matching Program (NRMP) issued results for 2021’s Medical Specialties Matching Program (MSMP) and Pediatric Specialties Match (PSM) in December.
“In pediatric rheumatology, like many other pediatric specialties, the limiting factor is the number of interested candidates. The number of available positions has not really changed over the last several years, but multiple positions again remained unfilled this year,” said Beth Marston, MD, chair of the American College of Rheumatology’s Committee on Rheumatology Training and Workforce Issues, in a statement.
Rheumatology was one of seven medical specialties that filled at least 95% of fellowship positions this year.
The specialty filled 120 of 125 enrolled programs (96%) and 266 of 272 certified positions (97.8%) in 2021. A total of 42.1% of the matched applicants comprised MD graduates, followed by foreign (27.1%), U.S. foreign (16.5%), and DO graduates (14.3%).
Among 357 applicants preferring this specialty in 2021, 73.9% (n = 264) matched to rheumatology. This meant that 22.1% did not match to any program. This scenario has played out over the last several years, Dr. Marston noted. Additional support for funding and the creation of more fellowship positions would translate to an increase in rheumatology graduates entering the workforce.
This could help mitigate workforce shortages that the ACR projected in 2015, she added.
Pediatric program applicants remain stagnant
For pediatric fellowships, the numbers weren’t as robust. Just 60% of 32 enrolled programs and 69.2% positions filled in 2021. MD graduates comprised most of the matched applicants (77.8%), followed by U.S. foreign (14.8%) and foreign and DO graduates (3.7% each). Overall, 27 out of 28 applicants or 96.4% matched to this specialty, a metric that’s remained steady but has not grown in recent years, Dr. Marston said.
This “suggests the need for additional efforts to understand and address barriers to choosing rheumatology fellowship training as a career path for pediatricians,” she said. The ACR’s Committee on Training and Workforce recently initiated a survey of combined medicine-pediatrics graduates in rheumatology to gain insights on why these graduates chose this career path.
The ACR is also looking into increasing access to rheumatology specialty care in underserved areas and finding creative solutions for increasing and filling rheumatology fellowship positions.
Largest match on record
Overall, 7,435 applicants participated in the 2021 MSMP, the largest on record. NRMP reported that 2,277 programs submitted rank order lists and offered 6,368 positions, an increase of more than 11% from 2020, respectively. A total of 90.4% positions (n = 5,759) were filled.
“The 2021 MSMP matched a record number of applicants to subspecialty training programs for positions set to begin July 2022,” NRMP President and CEO Donna L. Lamb, DHSc, MBA, said in a statement. “It’s rewarding to watch the MSMP grow, not only in terms of applicant interest and available training positions, but also from its launch 20 years ago with only three internal medicine subspecialties.”
NRMP largely attributed the increase in positions to the addition of critical care medicine, the latest subspecialty to join the MSMP. All fellows begin their training in July 2022.
The PSM also saw notable increases this year in several metrics. It offered 1,735 positions this year, a 5.9% increase from 2020. Overall, 1,507 positions (86.9%) were filled, a 6.6% increase from last year. 854 programs participated.
Rheumatologists seeking fellowships continue to show a preference for adult programs. Adult rheumatology programs filled nearly 100% of positions this year, but pediatric rheumatology programs filled only 69% of available slots, echoing trends of previous years.
The National Resident Matching Program (NRMP) issued results for 2021’s Medical Specialties Matching Program (MSMP) and Pediatric Specialties Match (PSM) in December.
“In pediatric rheumatology, like many other pediatric specialties, the limiting factor is the number of interested candidates. The number of available positions has not really changed over the last several years, but multiple positions again remained unfilled this year,” said Beth Marston, MD, chair of the American College of Rheumatology’s Committee on Rheumatology Training and Workforce Issues, in a statement.
Rheumatology was one of seven medical specialties that filled at least 95% of fellowship positions this year.
The specialty filled 120 of 125 enrolled programs (96%) and 266 of 272 certified positions (97.8%) in 2021. A total of 42.1% of the matched applicants comprised MD graduates, followed by foreign (27.1%), U.S. foreign (16.5%), and DO graduates (14.3%).
Among 357 applicants preferring this specialty in 2021, 73.9% (n = 264) matched to rheumatology. This meant that 22.1% did not match to any program. This scenario has played out over the last several years, Dr. Marston noted. Additional support for funding and the creation of more fellowship positions would translate to an increase in rheumatology graduates entering the workforce.
This could help mitigate workforce shortages that the ACR projected in 2015, she added.
Pediatric program applicants remain stagnant
For pediatric fellowships, the numbers weren’t as robust. Just 60% of 32 enrolled programs and 69.2% positions filled in 2021. MD graduates comprised most of the matched applicants (77.8%), followed by U.S. foreign (14.8%) and foreign and DO graduates (3.7% each). Overall, 27 out of 28 applicants or 96.4% matched to this specialty, a metric that’s remained steady but has not grown in recent years, Dr. Marston said.
This “suggests the need for additional efforts to understand and address barriers to choosing rheumatology fellowship training as a career path for pediatricians,” she said. The ACR’s Committee on Training and Workforce recently initiated a survey of combined medicine-pediatrics graduates in rheumatology to gain insights on why these graduates chose this career path.
The ACR is also looking into increasing access to rheumatology specialty care in underserved areas and finding creative solutions for increasing and filling rheumatology fellowship positions.
Largest match on record
Overall, 7,435 applicants participated in the 2021 MSMP, the largest on record. NRMP reported that 2,277 programs submitted rank order lists and offered 6,368 positions, an increase of more than 11% from 2020, respectively. A total of 90.4% positions (n = 5,759) were filled.
“The 2021 MSMP matched a record number of applicants to subspecialty training programs for positions set to begin July 2022,” NRMP President and CEO Donna L. Lamb, DHSc, MBA, said in a statement. “It’s rewarding to watch the MSMP grow, not only in terms of applicant interest and available training positions, but also from its launch 20 years ago with only three internal medicine subspecialties.”
NRMP largely attributed the increase in positions to the addition of critical care medicine, the latest subspecialty to join the MSMP. All fellows begin their training in July 2022.
The PSM also saw notable increases this year in several metrics. It offered 1,735 positions this year, a 5.9% increase from 2020. Overall, 1,507 positions (86.9%) were filled, a 6.6% increase from last year. 854 programs participated.
Tofacitinib approved for new ankylosing spondylitis indication
The Food and Drug Administration approved a supplemental new drug application for tofacitinib (Xeljanz, Xeljanz XR) that adds active ankylosing spondylitis in adults to its list of indications, according to a Dec. 14 announcement from manufacturer Pfizer.

The approval makes the drug the first Janus kinase (JAK) inhibitor to be approved for ankylosing spondylitis, joining tofacitinib’s other indications of rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, and polyarticular-course juvenile idiopathic arthritis.
Like other JAK inhibitors that are indicated for immune-mediated inflammatory diseases, tofacitinib’s use for all indications is limited to patients who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers.
The agency based its decision on the results of a phase 3, multicenter, randomized, double-blind, placebo-controlled trial in 269 adults with active ankylosing spondylitis that tested tofacitinib 5 mg twice daily.
The study met its primary endpoint showing that at week 16 the percentage of tofacitinib-treated patients who achieved 20% improvement in Assessment in SpondyloArthritis International Society response criteria (ASAS20) was significantly greater than with placebo (56.4% vs. 29.4%; P < .0001). The percentage of responders for ASAS40 criteria was likewise significantly greater with tofacitinib vs. placebo (40.6% vs. 12.5%; P < .0001). Pfizer said that the safety profile of tofacitinib observed in patients with ankylosing spondylitis was consistent with the safety profile observed in patients with either rheumatoid arthritis or psoriatic arthritis.
Pfizer noted in its announcement that the FDA updated the prescribing information this month for tofacitinib (and other JAK inhibitors approved for immune-mediated inflammatory conditions, upadacitinib [Rinvoq] and baricitinib [Olumiant]). This update included a new boxed warning for major adverse cardiovascular events and updated boxed warnings regarding mortality, malignancies, and thrombosis. These changes were made in light of results from the ORAL Surveillance postmarketing study of patients with rheumatoid arthritis aged 50 years and older with at least one cardiovascular risk factor. That study found an association between tofacitinib and increased risk of heart attack or stroke, cancer, blood clots, and death in comparison with patients who took the TNF blockers adalimumab or etanercept.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration approved a supplemental new drug application for tofacitinib (Xeljanz, Xeljanz XR) that adds active ankylosing spondylitis in adults to its list of indications, according to a Dec. 14 announcement from manufacturer Pfizer.
The approval makes the drug the first Janus kinase (JAK) inhibitor to be approved for ankylosing spondylitis, joining tofacitinib’s other indications of rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, and polyarticular-course juvenile idiopathic arthritis.
Like other JAK inhibitors that are indicated for immune-mediated inflammatory diseases, tofacitinib’s use for all indications is limited to patients who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers.
The agency based its decision on the results of a phase 3, multicenter, randomized, double-blind, placebo-controlled trial in 269 adults with active ankylosing spondylitis that tested tofacitinib 5 mg twice daily.
The study met its primary endpoint showing that at week 16 the percentage of tofacitinib-treated patients who achieved 20% improvement in Assessment in SpondyloArthritis International Society response criteria (ASAS20) was significantly greater than with placebo (56.4% vs. 29.4%; P < .0001). The percentage of responders for ASAS40 criteria was likewise significantly greater with tofacitinib vs. placebo (40.6% vs. 12.5%; P < .0001). Pfizer said that the safety profile of tofacitinib observed in patients with ankylosing spondylitis was consistent with the safety profile observed in patients with either rheumatoid arthritis or psoriatic arthritis.
Pfizer noted in its announcement that the FDA updated the prescribing information this month for tofacitinib (and other JAK inhibitors approved for immune-mediated inflammatory conditions, upadacitinib [Rinvoq] and baricitinib [Olumiant]). This update included a new boxed warning for major adverse cardiovascular events and updated boxed warnings regarding mortality, malignancies, and thrombosis. These changes were made in light of results from the ORAL Surveillance postmarketing study of patients with rheumatoid arthritis aged 50 years and older with at least one cardiovascular risk factor. That study found an association between tofacitinib and increased risk of heart attack or stroke, cancer, blood clots, and death in comparison with patients who took the TNF blockers adalimumab or etanercept.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration approved a supplemental new drug application for tofacitinib (Xeljanz, Xeljanz XR) that adds active ankylosing spondylitis in adults to its list of indications, according to a Dec. 14 announcement from manufacturer Pfizer.
The approval makes the drug the first Janus kinase (JAK) inhibitor to be approved for ankylosing spondylitis, joining tofacitinib’s other indications of rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, and polyarticular-course juvenile idiopathic arthritis.
Like other JAK inhibitors that are indicated for immune-mediated inflammatory diseases, tofacitinib’s use for all indications is limited to patients who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers.
The agency based its decision on the results of a phase 3, multicenter, randomized, double-blind, placebo-controlled trial in 269 adults with active ankylosing spondylitis that tested tofacitinib 5 mg twice daily.
The study met its primary endpoint showing that at week 16 the percentage of tofacitinib-treated patients who achieved 20% improvement in Assessment in SpondyloArthritis International Society response criteria (ASAS20) was significantly greater than with placebo (56.4% vs. 29.4%; P < .0001). The percentage of responders for ASAS40 criteria was likewise significantly greater with tofacitinib vs. placebo (40.6% vs. 12.5%; P < .0001). Pfizer said that the safety profile of tofacitinib observed in patients with ankylosing spondylitis was consistent with the safety profile observed in patients with either rheumatoid arthritis or psoriatic arthritis.
Pfizer noted in its announcement that the FDA updated the prescribing information this month for tofacitinib (and other JAK inhibitors approved for immune-mediated inflammatory conditions, upadacitinib [Rinvoq] and baricitinib [Olumiant]). This update included a new boxed warning for major adverse cardiovascular events and updated boxed warnings regarding mortality, malignancies, and thrombosis. These changes were made in light of results from the ORAL Surveillance postmarketing study of patients with rheumatoid arthritis aged 50 years and older with at least one cardiovascular risk factor. That study found an association between tofacitinib and increased risk of heart attack or stroke, cancer, blood clots, and death in comparison with patients who took the TNF blockers adalimumab or etanercept.
A version of this article first appeared on Medscape.com.