User login
List of medications linked to drug-induced lupus expands
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
FROM ANNALS OF THE RHEUMATIC DISEASES
Ehlers-Danlos syndrome: Increased IUGR risk reported
LAS VEGAS – Women with Ehlers-Danlos syndrome who became pregnant were more likely to experience antepartum hemorrhage, placenta previa, cervical incompetence, and preterm birth, according to a retrospective cohort study of national birth data. Long hospital stays also were more likely among these women.

Infants born to women with Ehlers-Danlos syndrome (EDS) were significantly more likely to have intrauterine growth retardation (IUGR) as well, an unexpected and as-yet unexplained finding, said the study’s first author, Laura Nicholls-Dempsey, MD, speaking at a poster session at the meeting sponsored by the Society for Maternal-Fetal Medicine.
Complications were infrequent overall, with a very low rate of intrauterine demise and no maternal mortality seen in the 910 women with EDS who were studied, said Dr. Nicholls-Dempsey, an ob.gyn. resident at McGill University, Montreal.
In counseling women with EDS, Dr. Nicholls-Dempsey said that she would advise them that “these are the types of things we’re going to watch out for, and we’ll see how the pregnancy goes. But we have to be careful about these: preterm birth, antepartum bleeding, placenta previa. We’ll watch the growth of the baby; we just have to be more careful about these specific things.”
Compared with women without EDS, those with the inherited connective tissue disorder had adjusted odds ratios (AORs) of 3.2 for cervical incompetence (95% confidence interval, 2.0-5.1) and 2.2 for placenta previa (95% CI, 1.3-3.9; P less than .01 for both). Absolute rates for these complications were 0.8% and 0.7% for women without EDS and 2.1% and 1.4% for women with EDS, respectively.
Women with EDS also had AORs of 1.8 for antepartum hemorrhage (2.8% versus 1.6%; 95% CI, 1.2-2.7; P less than .01). Cesarean delivery was more likely in women with EDS, with an AOR of 1.6 (37.4% versus 26.9%; 95% CI, 2.0-5.1); conversely, instrumental vaginal delivery was less likely in women with EDS (AOR = 0.5; 95% CI, 0.4-0.7; P less than .01 for both), meaning that spontaneous vaginal delivery was less likely in the EDS cohort.
The higher frequency of Cesarean deliveries may be attributable to anticipatory management by physicians seeking to avoid such complications as antepartum hemorrhage, as well as to the increased rate of placenta previa seen among the EDS cohort, Dr. Nicholls-Dempsey said.
After statistical adjustment, women in the EDS cohort were more than three times as likely to have hospital stays of both more than 7 days and 14 days (5.7% versus 2.1%, AOR = 3.1 for 7 days; 2.3% versus 0.7%, AOR = 3.8 for 14 days; P less than .01 for both).
Rates of some other maternal complications, such as pre-eclampsia, eclampsia, and gestational hypertension, were not elevated in the EDS cohort. Rates of premature rupture of membranes, chorioamnionitis, uterine rupture, postpartum hemorrhage, perineal laceration, and venous thromboembolism were also similar between groups.
However, not only was the AOR for preterm birth 1.5 for infants of women with EDS, but IUGR was more common in these neonates as well (AOR = 1.7, P less than .01 for both). The latter finding was unexpected, and Dr. Nicholls-Dempsey and her colleagues currently don’t have a mechanistic explanation for the higher IUGR rate.
Dr. Nicholls-Dempsey explained that she and her colleagues used data from the United States’ Health Care Cost and Utilization Project’s Nationwide Inpatient Sample (HCUP-NIS) to compare outcomes of women with EDS with the national sample as a whole.
Between 1999 and 2013, 13,881,592 births occurred in the HCUP-NIS cohort, with 910 deliveries to women who had EDS. These women were identified by ICD-9 codes, she said.
Comparing women with EDS to the non-EDS cohort, women with EDS were more likely to be Caucasian, have a higher income, and to be smokers; the cohorts were otherwise similar.
Ehlers-Danlos syndrome is a heterogeneous disorder involving abnormalities of collagen synthesis, with 13 known subtypes not captured in the HCUP-NIS data, Dr. Nicholls-Dempsey acknowledged. She characterized this as both a limitation but also a potential strength of the study.
“I really like this study because ... we know there’s 13 types of EDS that are genetically different ... They have their overlapping symptoms, but each one is different,” she said. “In an ideal world, we would have each subtype, and we would run this type of analysis on each subtype, to really be able to say to a patient, ‘You have this mutation, and this complication is going to be a big problem for you.’” The numbers of each subtype are so small that this is infeasible, she noted.
Still, the national sample acquired over many years offers real-world outcomes that clinicians can use in shared decision-making with EDS patients who are contemplating pregnancy or are already pregnant. Also, knowing which complications are more likely in patients with EDS can help plan optimal management of labor and delivery, Dr. Nicholls-Dempsey said.
Over the study’s 14-year span, the overall arc of EDS pregnancy outcomes is well captured regardless of mutation type. “It’s very applicable to the general population” of individuals with EDS, she noted. “Because it’s not type-specific, it’s really a good overview of what you can expect in EDS patients, regardless of the type.”
Dr. Nicholls-Dempsey reported no conflicts of interest and no outside sources of funding.
SOURCE: Nicholls-Dempsey L et al. Am J Obstet Gynecol. 2019 Jan;220(1):S381-382. Abstract 574
LAS VEGAS – Women with Ehlers-Danlos syndrome who became pregnant were more likely to experience antepartum hemorrhage, placenta previa, cervical incompetence, and preterm birth, according to a retrospective cohort study of national birth data. Long hospital stays also were more likely among these women.
Infants born to women with Ehlers-Danlos syndrome (EDS) were significantly more likely to have intrauterine growth retardation (IUGR) as well, an unexpected and as-yet unexplained finding, said the study’s first author, Laura Nicholls-Dempsey, MD, speaking at a poster session at the meeting sponsored by the Society for Maternal-Fetal Medicine.
Complications were infrequent overall, with a very low rate of intrauterine demise and no maternal mortality seen in the 910 women with EDS who were studied, said Dr. Nicholls-Dempsey, an ob.gyn. resident at McGill University, Montreal.
In counseling women with EDS, Dr. Nicholls-Dempsey said that she would advise them that “these are the types of things we’re going to watch out for, and we’ll see how the pregnancy goes. But we have to be careful about these: preterm birth, antepartum bleeding, placenta previa. We’ll watch the growth of the baby; we just have to be more careful about these specific things.”
Compared with women without EDS, those with the inherited connective tissue disorder had adjusted odds ratios (AORs) of 3.2 for cervical incompetence (95% confidence interval, 2.0-5.1) and 2.2 for placenta previa (95% CI, 1.3-3.9; P less than .01 for both). Absolute rates for these complications were 0.8% and 0.7% for women without EDS and 2.1% and 1.4% for women with EDS, respectively.
Women with EDS also had AORs of 1.8 for antepartum hemorrhage (2.8% versus 1.6%; 95% CI, 1.2-2.7; P less than .01). Cesarean delivery was more likely in women with EDS, with an AOR of 1.6 (37.4% versus 26.9%; 95% CI, 2.0-5.1); conversely, instrumental vaginal delivery was less likely in women with EDS (AOR = 0.5; 95% CI, 0.4-0.7; P less than .01 for both), meaning that spontaneous vaginal delivery was less likely in the EDS cohort.
The higher frequency of Cesarean deliveries may be attributable to anticipatory management by physicians seeking to avoid such complications as antepartum hemorrhage, as well as to the increased rate of placenta previa seen among the EDS cohort, Dr. Nicholls-Dempsey said.
After statistical adjustment, women in the EDS cohort were more than three times as likely to have hospital stays of both more than 7 days and 14 days (5.7% versus 2.1%, AOR = 3.1 for 7 days; 2.3% versus 0.7%, AOR = 3.8 for 14 days; P less than .01 for both).
Rates of some other maternal complications, such as pre-eclampsia, eclampsia, and gestational hypertension, were not elevated in the EDS cohort. Rates of premature rupture of membranes, chorioamnionitis, uterine rupture, postpartum hemorrhage, perineal laceration, and venous thromboembolism were also similar between groups.
However, not only was the AOR for preterm birth 1.5 for infants of women with EDS, but IUGR was more common in these neonates as well (AOR = 1.7, P less than .01 for both). The latter finding was unexpected, and Dr. Nicholls-Dempsey and her colleagues currently don’t have a mechanistic explanation for the higher IUGR rate.
Dr. Nicholls-Dempsey explained that she and her colleagues used data from the United States’ Health Care Cost and Utilization Project’s Nationwide Inpatient Sample (HCUP-NIS) to compare outcomes of women with EDS with the national sample as a whole.
Between 1999 and 2013, 13,881,592 births occurred in the HCUP-NIS cohort, with 910 deliveries to women who had EDS. These women were identified by ICD-9 codes, she said.
Comparing women with EDS to the non-EDS cohort, women with EDS were more likely to be Caucasian, have a higher income, and to be smokers; the cohorts were otherwise similar.
Ehlers-Danlos syndrome is a heterogeneous disorder involving abnormalities of collagen synthesis, with 13 known subtypes not captured in the HCUP-NIS data, Dr. Nicholls-Dempsey acknowledged. She characterized this as both a limitation but also a potential strength of the study.
“I really like this study because ... we know there’s 13 types of EDS that are genetically different ... They have their overlapping symptoms, but each one is different,” she said. “In an ideal world, we would have each subtype, and we would run this type of analysis on each subtype, to really be able to say to a patient, ‘You have this mutation, and this complication is going to be a big problem for you.’” The numbers of each subtype are so small that this is infeasible, she noted.
Still, the national sample acquired over many years offers real-world outcomes that clinicians can use in shared decision-making with EDS patients who are contemplating pregnancy or are already pregnant. Also, knowing which complications are more likely in patients with EDS can help plan optimal management of labor and delivery, Dr. Nicholls-Dempsey said.
Over the study’s 14-year span, the overall arc of EDS pregnancy outcomes is well captured regardless of mutation type. “It’s very applicable to the general population” of individuals with EDS, she noted. “Because it’s not type-specific, it’s really a good overview of what you can expect in EDS patients, regardless of the type.”
Dr. Nicholls-Dempsey reported no conflicts of interest and no outside sources of funding.
SOURCE: Nicholls-Dempsey L et al. Am J Obstet Gynecol. 2019 Jan;220(1):S381-382. Abstract 574
LAS VEGAS – Women with Ehlers-Danlos syndrome who became pregnant were more likely to experience antepartum hemorrhage, placenta previa, cervical incompetence, and preterm birth, according to a retrospective cohort study of national birth data. Long hospital stays also were more likely among these women.
Infants born to women with Ehlers-Danlos syndrome (EDS) were significantly more likely to have intrauterine growth retardation (IUGR) as well, an unexpected and as-yet unexplained finding, said the study’s first author, Laura Nicholls-Dempsey, MD, speaking at a poster session at the meeting sponsored by the Society for Maternal-Fetal Medicine.
Complications were infrequent overall, with a very low rate of intrauterine demise and no maternal mortality seen in the 910 women with EDS who were studied, said Dr. Nicholls-Dempsey, an ob.gyn. resident at McGill University, Montreal.
In counseling women with EDS, Dr. Nicholls-Dempsey said that she would advise them that “these are the types of things we’re going to watch out for, and we’ll see how the pregnancy goes. But we have to be careful about these: preterm birth, antepartum bleeding, placenta previa. We’ll watch the growth of the baby; we just have to be more careful about these specific things.”
Compared with women without EDS, those with the inherited connective tissue disorder had adjusted odds ratios (AORs) of 3.2 for cervical incompetence (95% confidence interval, 2.0-5.1) and 2.2 for placenta previa (95% CI, 1.3-3.9; P less than .01 for both). Absolute rates for these complications were 0.8% and 0.7% for women without EDS and 2.1% and 1.4% for women with EDS, respectively.
Women with EDS also had AORs of 1.8 for antepartum hemorrhage (2.8% versus 1.6%; 95% CI, 1.2-2.7; P less than .01). Cesarean delivery was more likely in women with EDS, with an AOR of 1.6 (37.4% versus 26.9%; 95% CI, 2.0-5.1); conversely, instrumental vaginal delivery was less likely in women with EDS (AOR = 0.5; 95% CI, 0.4-0.7; P less than .01 for both), meaning that spontaneous vaginal delivery was less likely in the EDS cohort.
The higher frequency of Cesarean deliveries may be attributable to anticipatory management by physicians seeking to avoid such complications as antepartum hemorrhage, as well as to the increased rate of placenta previa seen among the EDS cohort, Dr. Nicholls-Dempsey said.
After statistical adjustment, women in the EDS cohort were more than three times as likely to have hospital stays of both more than 7 days and 14 days (5.7% versus 2.1%, AOR = 3.1 for 7 days; 2.3% versus 0.7%, AOR = 3.8 for 14 days; P less than .01 for both).
Rates of some other maternal complications, such as pre-eclampsia, eclampsia, and gestational hypertension, were not elevated in the EDS cohort. Rates of premature rupture of membranes, chorioamnionitis, uterine rupture, postpartum hemorrhage, perineal laceration, and venous thromboembolism were also similar between groups.
However, not only was the AOR for preterm birth 1.5 for infants of women with EDS, but IUGR was more common in these neonates as well (AOR = 1.7, P less than .01 for both). The latter finding was unexpected, and Dr. Nicholls-Dempsey and her colleagues currently don’t have a mechanistic explanation for the higher IUGR rate.
Dr. Nicholls-Dempsey explained that she and her colleagues used data from the United States’ Health Care Cost and Utilization Project’s Nationwide Inpatient Sample (HCUP-NIS) to compare outcomes of women with EDS with the national sample as a whole.
Between 1999 and 2013, 13,881,592 births occurred in the HCUP-NIS cohort, with 910 deliveries to women who had EDS. These women were identified by ICD-9 codes, she said.
Comparing women with EDS to the non-EDS cohort, women with EDS were more likely to be Caucasian, have a higher income, and to be smokers; the cohorts were otherwise similar.
Ehlers-Danlos syndrome is a heterogeneous disorder involving abnormalities of collagen synthesis, with 13 known subtypes not captured in the HCUP-NIS data, Dr. Nicholls-Dempsey acknowledged. She characterized this as both a limitation but also a potential strength of the study.
“I really like this study because ... we know there’s 13 types of EDS that are genetically different ... They have their overlapping symptoms, but each one is different,” she said. “In an ideal world, we would have each subtype, and we would run this type of analysis on each subtype, to really be able to say to a patient, ‘You have this mutation, and this complication is going to be a big problem for you.’” The numbers of each subtype are so small that this is infeasible, she noted.
Still, the national sample acquired over many years offers real-world outcomes that clinicians can use in shared decision-making with EDS patients who are contemplating pregnancy or are already pregnant. Also, knowing which complications are more likely in patients with EDS can help plan optimal management of labor and delivery, Dr. Nicholls-Dempsey said.
Over the study’s 14-year span, the overall arc of EDS pregnancy outcomes is well captured regardless of mutation type. “It’s very applicable to the general population” of individuals with EDS, she noted. “Because it’s not type-specific, it’s really a good overview of what you can expect in EDS patients, regardless of the type.”
Dr. Nicholls-Dempsey reported no conflicts of interest and no outside sources of funding.
SOURCE: Nicholls-Dempsey L et al. Am J Obstet Gynecol. 2019 Jan;220(1):S381-382. Abstract 574
REPORTING FROM THE PREGNANCY MEETING
Cutaneous Collagenous Vasculopathy
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
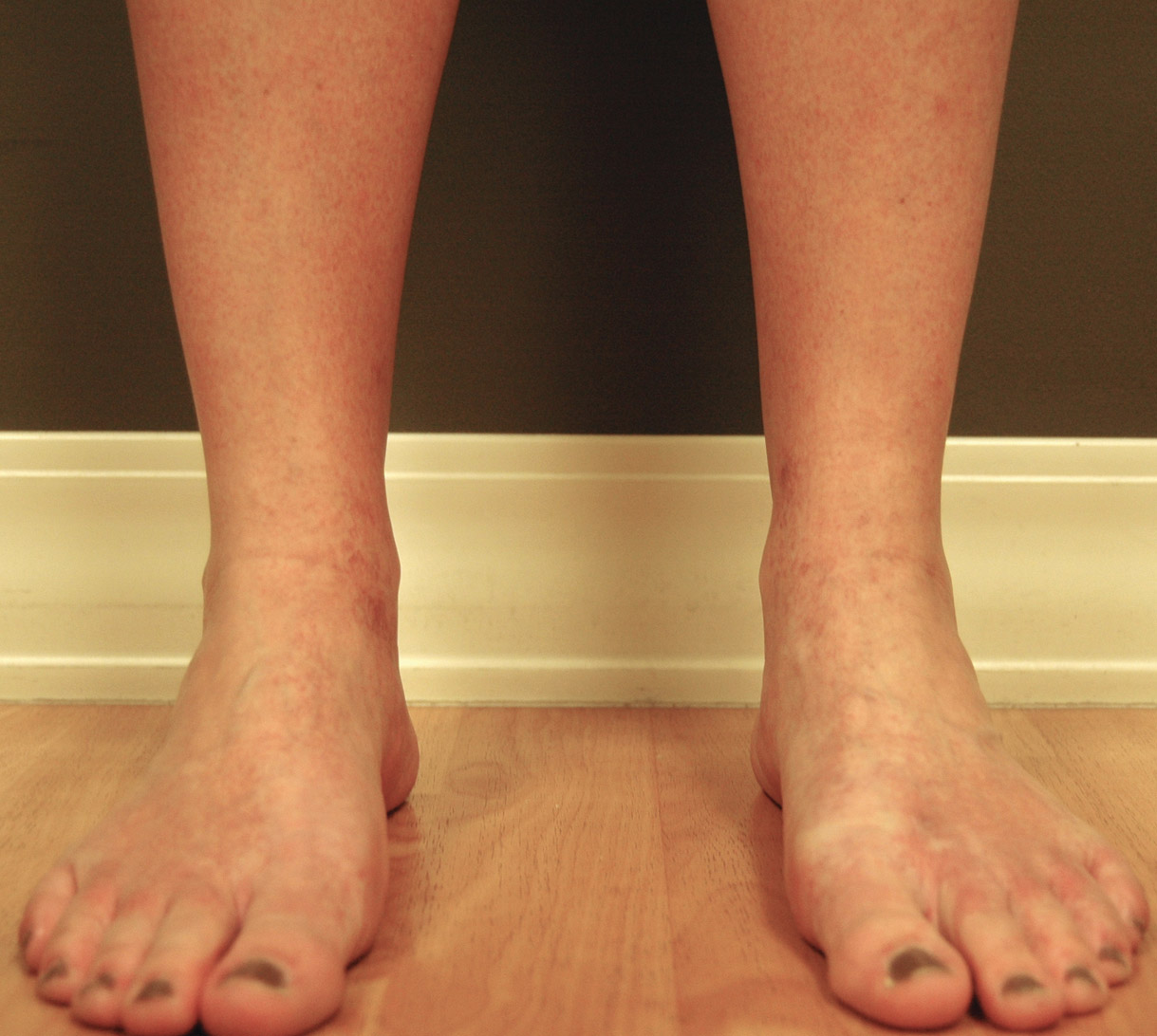
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
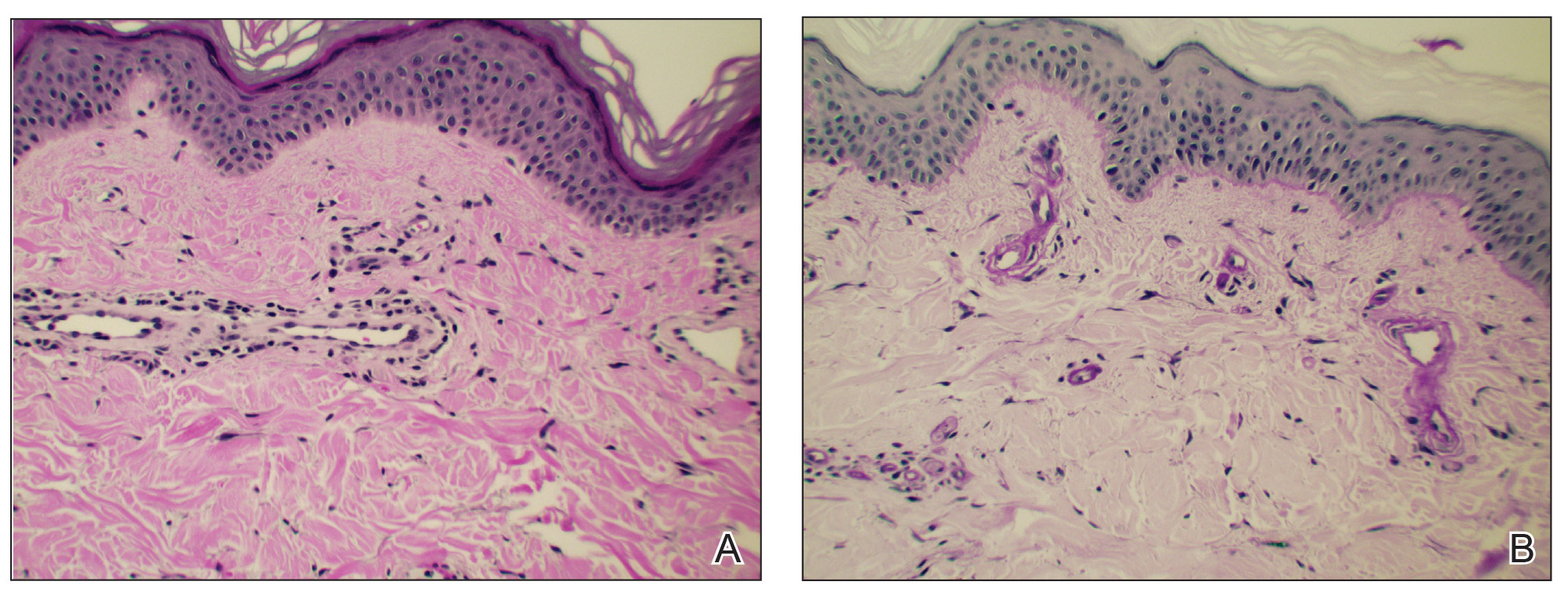
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
Practice Points
- In cutaneous collagenous vasculopathy (CCV), skin biopsy may demonstrate eosinophilic hyaline thickening of superficial dermal blood vessels with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes.
- Lack of mucous membrane and nail involvement differentiates CCV from hereditary hemorrhagic telangiectasia.
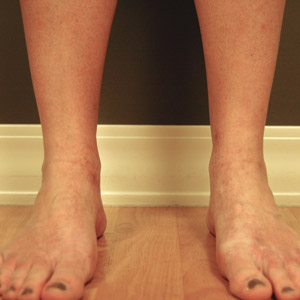
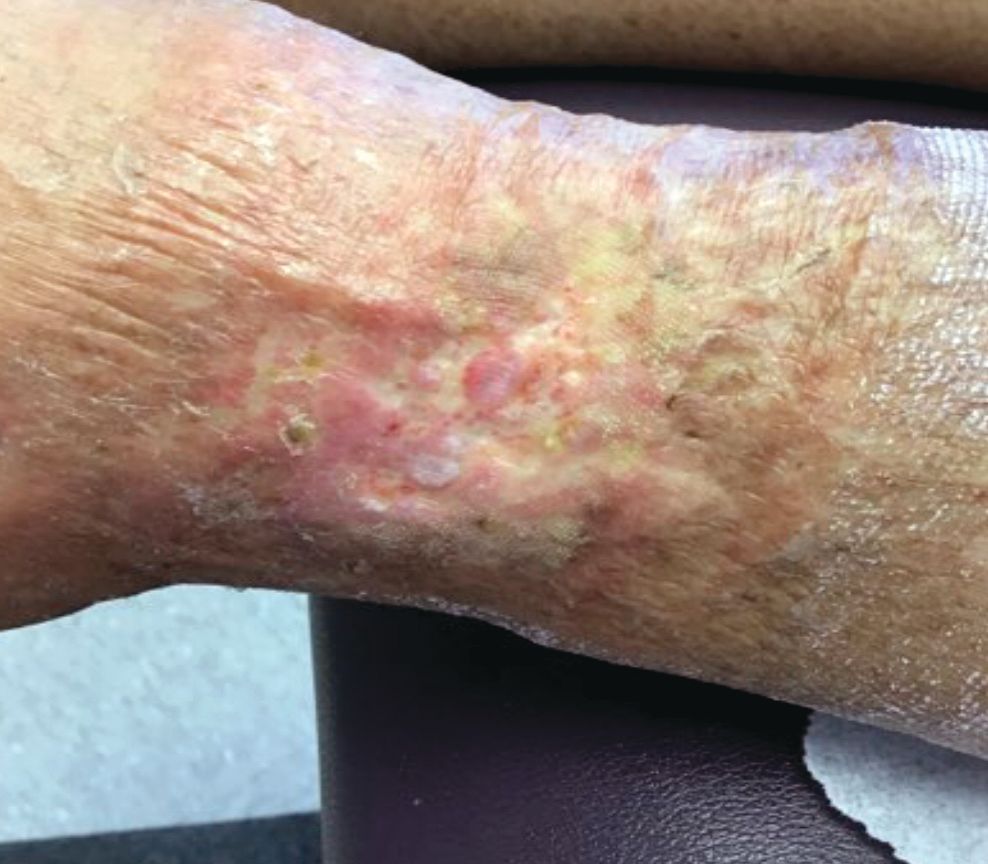
A 60-year-old white woman presented with a 3-month history of a painful, nonhealing ulceration on her left lateral lower leg
It is a vasculopathy rather than a vasculitis as the former is caused by occlusion of blood vessels and the latter results from inflammation of the vessels. Middle-aged women tend to be affected more frequently. Although the exact cause is unclear, systemic diseases, such as hypercoagulable states, may predispose vessels to develop occlusion. Associated disorders include antiphospholipid syndrome, protein C deficiency, factor V mutation, arteriosclerosis, hyperhomocysteinemia, and hepatitis C.

Typically, lesions begin as painful purpura or reticulated macules on the lower extremities that ulcerate and heal very slowly. Ankles, particularly malleoli, are more frequently affected. When they heal, they form painless white stellate scars typical of atrophie blanche. Surrounding erythema, telangiectasias, and sclerosis may be present; livedo reticularis may be seen as well.
Histologically, the epidermis may be atrophic or necrotic. Hyaline thickening of the blood vessel walls is seen. Thrombi may be present. Direct immunofluorescence of perilesional skin may be positive for complement C3 and immunoglobulin (IgM) in dermal blood vessels.
Livedoid vasculopathy can be difficult to treat. Treatment is aimed at reducing clotting and improving blood flow and includes antiplatelet drugs (low-dose aspirin, dipyridamole), anticoagulants, and vasodilating agents (nifedipine). Pentoxifylline two or three times daily may help by altering blood viscosity. A recent literature search reports success in topical dapsone applied to lesions twice daily under occlusion. Leg elevation and compression stockings help healing. Livedoid vasculopathy may have periods of activity and remission.
The case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected]
It is a vasculopathy rather than a vasculitis as the former is caused by occlusion of blood vessels and the latter results from inflammation of the vessels. Middle-aged women tend to be affected more frequently. Although the exact cause is unclear, systemic diseases, such as hypercoagulable states, may predispose vessels to develop occlusion. Associated disorders include antiphospholipid syndrome, protein C deficiency, factor V mutation, arteriosclerosis, hyperhomocysteinemia, and hepatitis C.
Typically, lesions begin as painful purpura or reticulated macules on the lower extremities that ulcerate and heal very slowly. Ankles, particularly malleoli, are more frequently affected. When they heal, they form painless white stellate scars typical of atrophie blanche. Surrounding erythema, telangiectasias, and sclerosis may be present; livedo reticularis may be seen as well.
Histologically, the epidermis may be atrophic or necrotic. Hyaline thickening of the blood vessel walls is seen. Thrombi may be present. Direct immunofluorescence of perilesional skin may be positive for complement C3 and immunoglobulin (IgM) in dermal blood vessels.
Livedoid vasculopathy can be difficult to treat. Treatment is aimed at reducing clotting and improving blood flow and includes antiplatelet drugs (low-dose aspirin, dipyridamole), anticoagulants, and vasodilating agents (nifedipine). Pentoxifylline two or three times daily may help by altering blood viscosity. A recent literature search reports success in topical dapsone applied to lesions twice daily under occlusion. Leg elevation and compression stockings help healing. Livedoid vasculopathy may have periods of activity and remission.
The case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected]
It is a vasculopathy rather than a vasculitis as the former is caused by occlusion of blood vessels and the latter results from inflammation of the vessels. Middle-aged women tend to be affected more frequently. Although the exact cause is unclear, systemic diseases, such as hypercoagulable states, may predispose vessels to develop occlusion. Associated disorders include antiphospholipid syndrome, protein C deficiency, factor V mutation, arteriosclerosis, hyperhomocysteinemia, and hepatitis C.
Typically, lesions begin as painful purpura or reticulated macules on the lower extremities that ulcerate and heal very slowly. Ankles, particularly malleoli, are more frequently affected. When they heal, they form painless white stellate scars typical of atrophie blanche. Surrounding erythema, telangiectasias, and sclerosis may be present; livedo reticularis may be seen as well.
Histologically, the epidermis may be atrophic or necrotic. Hyaline thickening of the blood vessel walls is seen. Thrombi may be present. Direct immunofluorescence of perilesional skin may be positive for complement C3 and immunoglobulin (IgM) in dermal blood vessels.
Livedoid vasculopathy can be difficult to treat. Treatment is aimed at reducing clotting and improving blood flow and includes antiplatelet drugs (low-dose aspirin, dipyridamole), anticoagulants, and vasodilating agents (nifedipine). Pentoxifylline two or three times daily may help by altering blood viscosity. A recent literature search reports success in topical dapsone applied to lesions twice daily under occlusion. Leg elevation and compression stockings help healing. Livedoid vasculopathy may have periods of activity and remission.
The case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected]
Trial supports less aggressive myeloma treatment
For patients with multiple myeloma that remains symptomatic within a year of starting therapy, neither a second autologous stem cell transplant nor more intensive consolidation therapy offered survival benefits superior to those seen with a single first autologous transplant and lenalidomide maintenance, reported investigators in a multicenter U.S. trial.
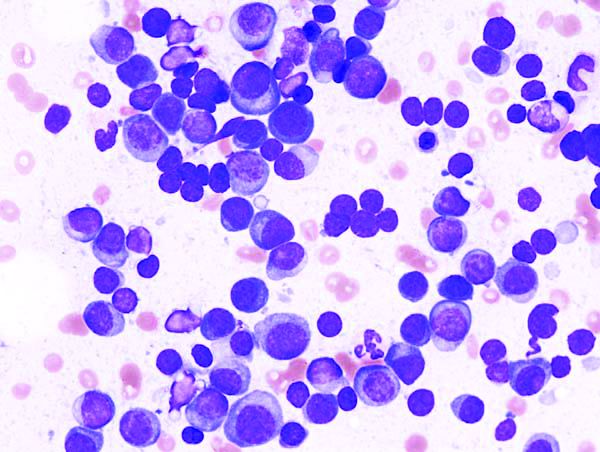
Among 758 patients with multiple myeloma (MM) who underwent standard induction therapy, followed by melphalan conditioning and autologous hematopoietic cell transplant (AHCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) between the three treatment arms, reported Edward A. Stadtmauer, MD, from the University of Pennsylvania, Philadelphia, and his colleagues.
Patients were randomized to either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide, bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant followed by lenalidomide maintenance.
“Single AHCT followed by len[alidomide] remains the standard of care. Greater than 80% of patients were alive at 38 months, which highlights excellent contemporary outcomes of patients with MM when treated with a standard approach of a multidrug induction followed by AHCT consolidation and maintenance,” they wrote in the Journal of Clinical Oncology.
The investigators hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and AHCT would improve survival, compared with a second AHCT.
To test this idea, they enrolled 758 patients from 54 U.S. centers and randomized them to one of three post-transplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and AHCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta-2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
The patients, who were a median age of 56 years old, had symptomatic multiple myeloma 12 months from the start of therapy without disease progression. They were randomly assigned to either AHCT followed by a second transplant and lenalidomide maintenance (247 patients), single transplant followed by RVD and lenalidomide maintenance (254), or single AHCT plus lenalidomide maintenance (257).
There were no significant differences between the groups in the primary endpoint of PFS at 38 months, with rates of 58.5% for the dual AHCT plus lenalidomide group, 57.8% for AHCT/RVD/lenalidomide, and 53.9% for AHCT/lenalidomide. Respective OS rates also did not differ significantly, at 81.8%, 85.4%, and 83.7%.
Complete response rates at 1 year were 50.5%, 58.4%, and 47.1%, respectively.
The three regimens also were similar in their toxicity profiles and in the risk of second malignancies.
The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
SOURCE: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
For patients with multiple myeloma that remains symptomatic within a year of starting therapy, neither a second autologous stem cell transplant nor more intensive consolidation therapy offered survival benefits superior to those seen with a single first autologous transplant and lenalidomide maintenance, reported investigators in a multicenter U.S. trial.
Among 758 patients with multiple myeloma (MM) who underwent standard induction therapy, followed by melphalan conditioning and autologous hematopoietic cell transplant (AHCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) between the three treatment arms, reported Edward A. Stadtmauer, MD, from the University of Pennsylvania, Philadelphia, and his colleagues.
Patients were randomized to either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide, bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant followed by lenalidomide maintenance.
“Single AHCT followed by len[alidomide] remains the standard of care. Greater than 80% of patients were alive at 38 months, which highlights excellent contemporary outcomes of patients with MM when treated with a standard approach of a multidrug induction followed by AHCT consolidation and maintenance,” they wrote in the Journal of Clinical Oncology.
The investigators hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and AHCT would improve survival, compared with a second AHCT.
To test this idea, they enrolled 758 patients from 54 U.S. centers and randomized them to one of three post-transplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and AHCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta-2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
The patients, who were a median age of 56 years old, had symptomatic multiple myeloma 12 months from the start of therapy without disease progression. They were randomly assigned to either AHCT followed by a second transplant and lenalidomide maintenance (247 patients), single transplant followed by RVD and lenalidomide maintenance (254), or single AHCT plus lenalidomide maintenance (257).
There were no significant differences between the groups in the primary endpoint of PFS at 38 months, with rates of 58.5% for the dual AHCT plus lenalidomide group, 57.8% for AHCT/RVD/lenalidomide, and 53.9% for AHCT/lenalidomide. Respective OS rates also did not differ significantly, at 81.8%, 85.4%, and 83.7%.
Complete response rates at 1 year were 50.5%, 58.4%, and 47.1%, respectively.
The three regimens also were similar in their toxicity profiles and in the risk of second malignancies.
The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
SOURCE: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
For patients with multiple myeloma that remains symptomatic within a year of starting therapy, neither a second autologous stem cell transplant nor more intensive consolidation therapy offered survival benefits superior to those seen with a single first autologous transplant and lenalidomide maintenance, reported investigators in a multicenter U.S. trial.
Among 758 patients with multiple myeloma (MM) who underwent standard induction therapy, followed by melphalan conditioning and autologous hematopoietic cell transplant (AHCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) between the three treatment arms, reported Edward A. Stadtmauer, MD, from the University of Pennsylvania, Philadelphia, and his colleagues.
Patients were randomized to either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide, bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant followed by lenalidomide maintenance.
“Single AHCT followed by len[alidomide] remains the standard of care. Greater than 80% of patients were alive at 38 months, which highlights excellent contemporary outcomes of patients with MM when treated with a standard approach of a multidrug induction followed by AHCT consolidation and maintenance,” they wrote in the Journal of Clinical Oncology.
The investigators hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and AHCT would improve survival, compared with a second AHCT.
To test this idea, they enrolled 758 patients from 54 U.S. centers and randomized them to one of three post-transplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and AHCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta-2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
The patients, who were a median age of 56 years old, had symptomatic multiple myeloma 12 months from the start of therapy without disease progression. They were randomly assigned to either AHCT followed by a second transplant and lenalidomide maintenance (247 patients), single transplant followed by RVD and lenalidomide maintenance (254), or single AHCT plus lenalidomide maintenance (257).
There were no significant differences between the groups in the primary endpoint of PFS at 38 months, with rates of 58.5% for the dual AHCT plus lenalidomide group, 57.8% for AHCT/RVD/lenalidomide, and 53.9% for AHCT/lenalidomide. Respective OS rates also did not differ significantly, at 81.8%, 85.4%, and 83.7%.
Complete response rates at 1 year were 50.5%, 58.4%, and 47.1%, respectively.
The three regimens also were similar in their toxicity profiles and in the risk of second malignancies.
The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
SOURCE: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: There were no differences in progression-free survival or overall survival among the three trial arms.
Study details: Randomized clinical trial with 758 patients with multiple myeloma.
Disclosures: The trial was supported by grants from the National Institutes of Health, research groups, Celgene, and Millennium (Takeda) Pharmaceuticals. Dr. Stadtmauer reported ties to Celgene, Takeda, and other companies. Multiple coauthors reported relationships with industry.
Source: Stadtmauer E et al. J Clin Oncol. 2019 Jan 17. doi: 10.1200/JCO.18.00685.
FDA approves caplacizumab for aTTP
The Food and Drug Administration has approved caplacizumab (Cablivi) in combination with plasma exchange and immunosuppressive therapy for the treatment of adults with acquired thrombotic thrombocytopenic purpura (aTTP).

Caplacizumab is an anti–von Willebrand factor nanobody designed to inhibit the interaction between von Willebrand factor and platelets. The injection previously received orphan drug designation from the FDA and was approved under priority review.
The FDA’s approval of caplacizumab was based on results from the phase 3 HERCULES trial (N Engl J Med 2019 Jan 24;380:335-46).
The trial (NCT02553317) included 145 adults with aTTP. They were randomized to receive caplacizumab (n = 72) or placebo (n = 73), in addition to plasma exchange and immunosuppression.
The study’s primary endpoint was the time to platelet count response (normalization), which was defined as a platelet count of at least 150 x 109/L with subsequent stop of daily plasma exchange within 5 days.
There was a significant reduction in time to platelet count response in the caplacizumab arm, compared with the placebo arm – 2.69 days and 2.88 days, respectively. The platelet normalization rate ratio was 1.55 (P less than .01).
A secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least one major thromboembolic event during study treatment. The incidence of this combined endpoint was 12% in the caplacizumab arm and 49% in the placebo arm (P less than .001).
The most common treatment-emergent adverse events (occurring in at least 15% of patients in the caplacizumab and placebo arms, respectively) were epistaxis (32% and 3%), headache (23% and 8%), urticaria (17% and 7%), and hypokalemia (9% and 19%).
During the treatment period, there were no deaths in the caplacizumab arm and three deaths in the placebo arm. There was one death (from cerebral ischemia) in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab.
For more details on caplacizumab, see the full prescribing information.
The Food and Drug Administration has approved caplacizumab (Cablivi) in combination with plasma exchange and immunosuppressive therapy for the treatment of adults with acquired thrombotic thrombocytopenic purpura (aTTP).
Caplacizumab is an anti–von Willebrand factor nanobody designed to inhibit the interaction between von Willebrand factor and platelets. The injection previously received orphan drug designation from the FDA and was approved under priority review.
The FDA’s approval of caplacizumab was based on results from the phase 3 HERCULES trial (N Engl J Med 2019 Jan 24;380:335-46).
The trial (NCT02553317) included 145 adults with aTTP. They were randomized to receive caplacizumab (n = 72) or placebo (n = 73), in addition to plasma exchange and immunosuppression.
The study’s primary endpoint was the time to platelet count response (normalization), which was defined as a platelet count of at least 150 x 109/L with subsequent stop of daily plasma exchange within 5 days.
There was a significant reduction in time to platelet count response in the caplacizumab arm, compared with the placebo arm – 2.69 days and 2.88 days, respectively. The platelet normalization rate ratio was 1.55 (P less than .01).
A secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least one major thromboembolic event during study treatment. The incidence of this combined endpoint was 12% in the caplacizumab arm and 49% in the placebo arm (P less than .001).
The most common treatment-emergent adverse events (occurring in at least 15% of patients in the caplacizumab and placebo arms, respectively) were epistaxis (32% and 3%), headache (23% and 8%), urticaria (17% and 7%), and hypokalemia (9% and 19%).
During the treatment period, there were no deaths in the caplacizumab arm and three deaths in the placebo arm. There was one death (from cerebral ischemia) in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab.
For more details on caplacizumab, see the full prescribing information.
The Food and Drug Administration has approved caplacizumab (Cablivi) in combination with plasma exchange and immunosuppressive therapy for the treatment of adults with acquired thrombotic thrombocytopenic purpura (aTTP).
Caplacizumab is an anti–von Willebrand factor nanobody designed to inhibit the interaction between von Willebrand factor and platelets. The injection previously received orphan drug designation from the FDA and was approved under priority review.
The FDA’s approval of caplacizumab was based on results from the phase 3 HERCULES trial (N Engl J Med 2019 Jan 24;380:335-46).
The trial (NCT02553317) included 145 adults with aTTP. They were randomized to receive caplacizumab (n = 72) or placebo (n = 73), in addition to plasma exchange and immunosuppression.
The study’s primary endpoint was the time to platelet count response (normalization), which was defined as a platelet count of at least 150 x 109/L with subsequent stop of daily plasma exchange within 5 days.
There was a significant reduction in time to platelet count response in the caplacizumab arm, compared with the placebo arm – 2.69 days and 2.88 days, respectively. The platelet normalization rate ratio was 1.55 (P less than .01).
A secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least one major thromboembolic event during study treatment. The incidence of this combined endpoint was 12% in the caplacizumab arm and 49% in the placebo arm (P less than .001).
The most common treatment-emergent adverse events (occurring in at least 15% of patients in the caplacizumab and placebo arms, respectively) were epistaxis (32% and 3%), headache (23% and 8%), urticaria (17% and 7%), and hypokalemia (9% and 19%).
During the treatment period, there were no deaths in the caplacizumab arm and three deaths in the placebo arm. There was one death (from cerebral ischemia) in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab.
For more details on caplacizumab, see the full prescribing information.
Meta-analysis: IVIG bests anti-D on platelet count in pediatric ITP
For patients with pediatric immune thrombocytopenia (ITP), treatment with intravenous immunoglobulins (IVIG) is more likely to raise platelet count in the short-term, compared with anti-D immunoglobulins (anti-D), according the authors of a recent systematic review and meta-analysis.
Although findings from the meta-analysis support recommendations for first-line IVIG, not all studies reported bleeding symptoms, so the clinical effects of differing platelet responses remain unknown, reported lead author Bertrand Lioger, MD, of François-Rabelais University in Tours, France, and his colleagues.
“To date, no meta-analysis has compared the efficacy and safety of IVIG vs. anti-D,” the investigators wrote in The Journal of Pediatrics.
Each treatment approach has strengths and weaknesses, the investigators noted. Namely, IVIG is more expensive, while anti-D is more likely to cause adverse drugs reactions (ADRs), such as disseminated intravascular coagulation and hemolysis.
The present review evaluated 11 studies comparing the efficacy of IVIG with that of anti-D in 704 children with ITP. Platelet response and bleeding were the main efficacy outcomes. The investigators used response thresholds defined by each study because several did not use standardized levels. Other outcomes considered were mortality, disease course, splenectomy, and ADRs. The ADRs included serious adverse reactions, infusion reactions, transfusions, hemoglobin loss, and hemolysis.
In alignment with previous guidelines, anti-D therapy was most often given to RhD positive, nonsplenectomized children at a dose of 50-75 mcg/kg, whereas IVIG was dosed at 0.8-1 g/kg for 1 or 2 consecutive days.
Results showed that patients treated with IVIG were 15% more likely to have platelet counts greater than 20 × 109/L within 24-72 hours, compared with those given anti-D. This disparity rose to 25% in favor of IVIG when using a threshold of 50 × 109/L.
Treatment risk was lower and general symptoms were less common after treatment with anti-D infusion, compared with IVIG (24.6% vs. 31.4%), but this was only true for trials foregoing premedication. Anti-D was more often associated with hemolysis, making transfusion necessary for some patients.
Although platelet count is often used as a surrogate measure of bleeding risk, the investigators decided that a lack of bleeding data among the studies precluded an accurate determination of clinical superiority between the treatments.
“Severe hemolysis remains an important issue when using anti-D immunoglobulins and premedication reduces the incidence of general symptoms observed with IVIG,” the investigators wrote. “Our conclusions should, however, be cautiously considered due to the poor overall quality of included studies and to limited data about clinically relevant outcomes.”
The study was not supported by outside funding. The investigators reported financial relationships with Amgen, Novartis, Roche Pharma, Sanofi, and others.
SOURCE: Lioger B et al. J Pediatr. 2019;204:225-33.
For patients with pediatric immune thrombocytopenia (ITP), treatment with intravenous immunoglobulins (IVIG) is more likely to raise platelet count in the short-term, compared with anti-D immunoglobulins (anti-D), according the authors of a recent systematic review and meta-analysis.
Although findings from the meta-analysis support recommendations for first-line IVIG, not all studies reported bleeding symptoms, so the clinical effects of differing platelet responses remain unknown, reported lead author Bertrand Lioger, MD, of François-Rabelais University in Tours, France, and his colleagues.
“To date, no meta-analysis has compared the efficacy and safety of IVIG vs. anti-D,” the investigators wrote in The Journal of Pediatrics.
Each treatment approach has strengths and weaknesses, the investigators noted. Namely, IVIG is more expensive, while anti-D is more likely to cause adverse drugs reactions (ADRs), such as disseminated intravascular coagulation and hemolysis.
The present review evaluated 11 studies comparing the efficacy of IVIG with that of anti-D in 704 children with ITP. Platelet response and bleeding were the main efficacy outcomes. The investigators used response thresholds defined by each study because several did not use standardized levels. Other outcomes considered were mortality, disease course, splenectomy, and ADRs. The ADRs included serious adverse reactions, infusion reactions, transfusions, hemoglobin loss, and hemolysis.
In alignment with previous guidelines, anti-D therapy was most often given to RhD positive, nonsplenectomized children at a dose of 50-75 mcg/kg, whereas IVIG was dosed at 0.8-1 g/kg for 1 or 2 consecutive days.
Results showed that patients treated with IVIG were 15% more likely to have platelet counts greater than 20 × 109/L within 24-72 hours, compared with those given anti-D. This disparity rose to 25% in favor of IVIG when using a threshold of 50 × 109/L.
Treatment risk was lower and general symptoms were less common after treatment with anti-D infusion, compared with IVIG (24.6% vs. 31.4%), but this was only true for trials foregoing premedication. Anti-D was more often associated with hemolysis, making transfusion necessary for some patients.
Although platelet count is often used as a surrogate measure of bleeding risk, the investigators decided that a lack of bleeding data among the studies precluded an accurate determination of clinical superiority between the treatments.
“Severe hemolysis remains an important issue when using anti-D immunoglobulins and premedication reduces the incidence of general symptoms observed with IVIG,” the investigators wrote. “Our conclusions should, however, be cautiously considered due to the poor overall quality of included studies and to limited data about clinically relevant outcomes.”
The study was not supported by outside funding. The investigators reported financial relationships with Amgen, Novartis, Roche Pharma, Sanofi, and others.
SOURCE: Lioger B et al. J Pediatr. 2019;204:225-33.
For patients with pediatric immune thrombocytopenia (ITP), treatment with intravenous immunoglobulins (IVIG) is more likely to raise platelet count in the short-term, compared with anti-D immunoglobulins (anti-D), according the authors of a recent systematic review and meta-analysis.
Although findings from the meta-analysis support recommendations for first-line IVIG, not all studies reported bleeding symptoms, so the clinical effects of differing platelet responses remain unknown, reported lead author Bertrand Lioger, MD, of François-Rabelais University in Tours, France, and his colleagues.
“To date, no meta-analysis has compared the efficacy and safety of IVIG vs. anti-D,” the investigators wrote in The Journal of Pediatrics.
Each treatment approach has strengths and weaknesses, the investigators noted. Namely, IVIG is more expensive, while anti-D is more likely to cause adverse drugs reactions (ADRs), such as disseminated intravascular coagulation and hemolysis.
The present review evaluated 11 studies comparing the efficacy of IVIG with that of anti-D in 704 children with ITP. Platelet response and bleeding were the main efficacy outcomes. The investigators used response thresholds defined by each study because several did not use standardized levels. Other outcomes considered were mortality, disease course, splenectomy, and ADRs. The ADRs included serious adverse reactions, infusion reactions, transfusions, hemoglobin loss, and hemolysis.
In alignment with previous guidelines, anti-D therapy was most often given to RhD positive, nonsplenectomized children at a dose of 50-75 mcg/kg, whereas IVIG was dosed at 0.8-1 g/kg for 1 or 2 consecutive days.
Results showed that patients treated with IVIG were 15% more likely to have platelet counts greater than 20 × 109/L within 24-72 hours, compared with those given anti-D. This disparity rose to 25% in favor of IVIG when using a threshold of 50 × 109/L.
Treatment risk was lower and general symptoms were less common after treatment with anti-D infusion, compared with IVIG (24.6% vs. 31.4%), but this was only true for trials foregoing premedication. Anti-D was more often associated with hemolysis, making transfusion necessary for some patients.
Although platelet count is often used as a surrogate measure of bleeding risk, the investigators decided that a lack of bleeding data among the studies precluded an accurate determination of clinical superiority between the treatments.
“Severe hemolysis remains an important issue when using anti-D immunoglobulins and premedication reduces the incidence of general symptoms observed with IVIG,” the investigators wrote. “Our conclusions should, however, be cautiously considered due to the poor overall quality of included studies and to limited data about clinically relevant outcomes.”
The study was not supported by outside funding. The investigators reported financial relationships with Amgen, Novartis, Roche Pharma, Sanofi, and others.
SOURCE: Lioger B et al. J Pediatr. 2019;204:225-33.
FROM THE JOURNAL OF PEDIATRICS
Key clinical point:
Major finding: Treatment with IVIG was 15% more likely than anti-D immunoglobulin to raise platelet counts higher than 20 × 109/L within 24-72 hours.
Study details: A systematic review and meta-analysis of 11 studies comparing the efficacy of IVIG with that of anti-D in 704 children with ITP.
Disclosures: The meta-analysis did not have outside funding. The investigators reported financial relationships with Amgen, Novartis, Roche Pharma, Sanofi, and others.
Source: Lioger B et al. J Pediatr. 2019; 204:225-33.
Top cancer advance: Treatment of rare diseases
The American Society of Clinical Oncology (ASCO) named “Progress in Treating Rare Cancers” as the Advance of the Year for 2018, citing five major studies as examples of significant breakthroughs.
In an ASCO Special Article published in the Journal of Clinical Oncology, Sumanta K. Pal, MD, of City of Hope Comprehensive Cancer Center, Duarte, Calif., and colleagues, identified five studies that notably advanced cancer research.
Each study “reflects the impressive gains we have made in understanding these so-called orphan diseases and in tailoring treatments to target their unique characteristics,” wrote ASCO president Monica M. Bertagnolli, MD, in an introduction to the report.
One of the significant advances included use of a new combination of targeted therapies for a rare thyroid cancer that elicited responses in more than two-thirds of patients. A second study showed sorafenib improving progression-free survival for patients with desmoid tumors. In addition, patients with advanced midgut neuroendocrine tumors had a 79% lower risk of disease progression or death when treated with a new therapy of targeted radiation to tumor cells, lutetium-177 (177Lu)–Dotatate, compared with standard therapy; and trastuzumab, a standard treatment for human epidermal growth factor receptor 2 (HER2)–positive breast cancer, expanded its reach and significantly slowed progression of HER2-positive uterine serous carcinoma, the authors wrote. Finally, the “first promising therapy – the colony-stimulating factor-1 (CSF-1) inhibitor pexidartinib – for a rare cancer of the joints known as tenosynovial giant cell tumor, showed an overall response rate of 39.3% in patients taking pexidartinib versus 0% in patients taking a placebo,” they said.
For the first time, the ASCO progress report included a list of priorities to guide future research efforts, stated as follows:
- Identify strategies that better predict response to immunotherapies.
- Better define the patient populations that benefit from postoperative (adjuvant) therapy.
- Translate innovations in cellular therapies to solid tumors.
- Increase precision medicine research and treatment approaches in pediatric cancers.
- Optimize care for older adults with cancer.
- Increase equitable access to cancer clinical trials.
- Reduce the long-term consequences of cancer treatment.
- Reduce obesity and its impact on cancer incidence and outcomes.
- Identify strategies to detect and treat premalignant lesions.
“These priority areas, listed in no particular order, address an unmet need or help fill a knowledge gap in areas critical to improving patient care and outcomes,” the authors wrote.
The report acknowledged the value of federally funded research and the importance of ongoing federal investment in cancer research.
Dr. Pal disclosed relationships with Pfizer, Novartis, Aveo, Myriad Pharmaceuticals, Genentech, Exelixis, Bristol-Myers Squibb, Astellas Pharma, Ipsen, Eisai, and Medivation. Coauthors disclosed relationships with these and other companies.
SOURCE: Pal SK et al. J Clin Oncol. 2019 Jan 31. doi: 10.1200/JCO.18.02037.
The American Society of Clinical Oncology (ASCO) named “Progress in Treating Rare Cancers” as the Advance of the Year for 2018, citing five major studies as examples of significant breakthroughs.
In an ASCO Special Article published in the Journal of Clinical Oncology, Sumanta K. Pal, MD, of City of Hope Comprehensive Cancer Center, Duarte, Calif., and colleagues, identified five studies that notably advanced cancer research.
Each study “reflects the impressive gains we have made in understanding these so-called orphan diseases and in tailoring treatments to target their unique characteristics,” wrote ASCO president Monica M. Bertagnolli, MD, in an introduction to the report.
One of the significant advances included use of a new combination of targeted therapies for a rare thyroid cancer that elicited responses in more than two-thirds of patients. A second study showed sorafenib improving progression-free survival for patients with desmoid tumors. In addition, patients with advanced midgut neuroendocrine tumors had a 79% lower risk of disease progression or death when treated with a new therapy of targeted radiation to tumor cells, lutetium-177 (177Lu)–Dotatate, compared with standard therapy; and trastuzumab, a standard treatment for human epidermal growth factor receptor 2 (HER2)–positive breast cancer, expanded its reach and significantly slowed progression of HER2-positive uterine serous carcinoma, the authors wrote. Finally, the “first promising therapy – the colony-stimulating factor-1 (CSF-1) inhibitor pexidartinib – for a rare cancer of the joints known as tenosynovial giant cell tumor, showed an overall response rate of 39.3% in patients taking pexidartinib versus 0% in patients taking a placebo,” they said.
For the first time, the ASCO progress report included a list of priorities to guide future research efforts, stated as follows:
- Identify strategies that better predict response to immunotherapies.
- Better define the patient populations that benefit from postoperative (adjuvant) therapy.
- Translate innovations in cellular therapies to solid tumors.
- Increase precision medicine research and treatment approaches in pediatric cancers.
- Optimize care for older adults with cancer.
- Increase equitable access to cancer clinical trials.
- Reduce the long-term consequences of cancer treatment.
- Reduce obesity and its impact on cancer incidence and outcomes.
- Identify strategies to detect and treat premalignant lesions.
“These priority areas, listed in no particular order, address an unmet need or help fill a knowledge gap in areas critical to improving patient care and outcomes,” the authors wrote.
The report acknowledged the value of federally funded research and the importance of ongoing federal investment in cancer research.
Dr. Pal disclosed relationships with Pfizer, Novartis, Aveo, Myriad Pharmaceuticals, Genentech, Exelixis, Bristol-Myers Squibb, Astellas Pharma, Ipsen, Eisai, and Medivation. Coauthors disclosed relationships with these and other companies.
SOURCE: Pal SK et al. J Clin Oncol. 2019 Jan 31. doi: 10.1200/JCO.18.02037.
The American Society of Clinical Oncology (ASCO) named “Progress in Treating Rare Cancers” as the Advance of the Year for 2018, citing five major studies as examples of significant breakthroughs.
In an ASCO Special Article published in the Journal of Clinical Oncology, Sumanta K. Pal, MD, of City of Hope Comprehensive Cancer Center, Duarte, Calif., and colleagues, identified five studies that notably advanced cancer research.
Each study “reflects the impressive gains we have made in understanding these so-called orphan diseases and in tailoring treatments to target their unique characteristics,” wrote ASCO president Monica M. Bertagnolli, MD, in an introduction to the report.
One of the significant advances included use of a new combination of targeted therapies for a rare thyroid cancer that elicited responses in more than two-thirds of patients. A second study showed sorafenib improving progression-free survival for patients with desmoid tumors. In addition, patients with advanced midgut neuroendocrine tumors had a 79% lower risk of disease progression or death when treated with a new therapy of targeted radiation to tumor cells, lutetium-177 (177Lu)–Dotatate, compared with standard therapy; and trastuzumab, a standard treatment for human epidermal growth factor receptor 2 (HER2)–positive breast cancer, expanded its reach and significantly slowed progression of HER2-positive uterine serous carcinoma, the authors wrote. Finally, the “first promising therapy – the colony-stimulating factor-1 (CSF-1) inhibitor pexidartinib – for a rare cancer of the joints known as tenosynovial giant cell tumor, showed an overall response rate of 39.3% in patients taking pexidartinib versus 0% in patients taking a placebo,” they said.
For the first time, the ASCO progress report included a list of priorities to guide future research efforts, stated as follows:
- Identify strategies that better predict response to immunotherapies.
- Better define the patient populations that benefit from postoperative (adjuvant) therapy.
- Translate innovations in cellular therapies to solid tumors.
- Increase precision medicine research and treatment approaches in pediatric cancers.
- Optimize care for older adults with cancer.
- Increase equitable access to cancer clinical trials.
- Reduce the long-term consequences of cancer treatment.
- Reduce obesity and its impact on cancer incidence and outcomes.
- Identify strategies to detect and treat premalignant lesions.
“These priority areas, listed in no particular order, address an unmet need or help fill a knowledge gap in areas critical to improving patient care and outcomes,” the authors wrote.
The report acknowledged the value of federally funded research and the importance of ongoing federal investment in cancer research.
Dr. Pal disclosed relationships with Pfizer, Novartis, Aveo, Myriad Pharmaceuticals, Genentech, Exelixis, Bristol-Myers Squibb, Astellas Pharma, Ipsen, Eisai, and Medivation. Coauthors disclosed relationships with these and other companies.
SOURCE: Pal SK et al. J Clin Oncol. 2019 Jan 31. doi: 10.1200/JCO.18.02037.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Clinical benefits persist 5 years after thymectomy for myasthenia gravis
Thymectomy may continue to benefit patients with myasthenia gravis 5 years after the procedure, according to an extension study published in Lancet Neurology.

The study evaluated the clinical status, medication requirements, and adverse events of patients with myasthenia gravis who completed a randomized controlled trial of thymectomy plus prednisone versus prednisone alone and agreed to participate in a rater-blinded 2-year extension.
“Thymectomy within the first few years of the disease course in addition to prednisone therapy confers benefits that persist for 5 years ... in patients with generalized nonthymomatous myasthenia gravis,” said lead study author Gil I. Wolfe, MD, chair of the department of neurology at the University at Buffalo in New York, and his research colleagues. “Results from the extension study provide further support for the use of thymectomy in management of myasthenia gravis and should encourage serious consideration of this treatment option in discussions between clinicians and their patients,” they wrote. “Our results should lead to revision of clinical guidelines in favor of thymectomy and could potentially reverse downward trends in the use of thymectomy in overall management of myasthenia gravis.”
The main 3-year results of the Thymectomy Trial in Nonthymomatous Myasthenia Gravis Patients Receiving Prednisone (MGTX) were reported in 2016; the international trial found that thymectomy plus prednisone was superior to prednisone alone at 3 years (N Engl J Med. 2016 Aug 11;375[6]:511-22). The extension study aimed to assess the durability of the treatment response.
MGTX enrolled patients aged 18-65 years who had generalized nonthymomatous myasthenia gravis of less than 5 years’ duration and Myasthenia Gravis Foundation of America Clinical Classification Class II-IV disease. Of 111 patients who completed MGTX, 68 entered the extension study, and 50 completed the 60-month assessment (24 patients in the prednisone alone group and 26 patients in the prednisone plus thymectomy group).
At 5 years, patients in the thymectomy plus prednisone group had significantly lower time-weighted average Quantitative Myasthenia Gravis (QMG) scores (5.47 vs. 9.34) and mean alternate-day prednisone doses (24 mg vs. 48 mg), compared with patients who received prednisone alone. Twelve of 35 patients in the thymectomy group and 14 of 33 patients in the prednisone group had at least one adverse event by month 60. No treatment-related deaths occurred in the extension phase.
At 5 years, significantly more patients who underwent thymectomy had minimal manifestation status (i.e., no functional limitations from the disease other than some muscle weakness) – 88% versus 58%. The corresponding figures at 3 years were 67% and 47%.
In addition, 3-year and 5-year data indicate that the need for hospitalization is reduced after surgery, compared with medical therapy alone, Dr. Wolfe said.
Two patients in each treatment arm had an increase of 2 points or more in the QMG score, indicating clinical worsening.
“Our current findings reinforce the benefit of thymectomy seen in [MGTX], dispelling doubts about the procedure’s benefits and how long those benefits last,” said Dr. Wolfe. “We do hope that the new findings help reverse the apparent reluctance to do thymectomy and that the proportion of patients with myasthenia gravis who undergo thymectomy will increase.”
The authors noted that the small sample size of the extension study may limit its generalizability.
The study received funding from the National Institutes of Health. Dr. Wolfe reported grants from the NIH, the Muscular Dystrophy Association, the Myasthenia Gravis Foundation of America, CSL-Behring, and ArgenX, as well as personal fees from Grifols, Shire, and Alexion Pharmaceuticals. Coauthors reported working with and receiving funds from agencies, foundations, and pharmaceutical companies.
SOURCE: Wolfe GI et al. Lancet Neurol. 2019 Jan 25. doi: 10.1016/S1474-4422(18)30392-2.
Thymectomy may continue to benefit patients with myasthenia gravis 5 years after the procedure, according to an extension study published in Lancet Neurology.
The study evaluated the clinical status, medication requirements, and adverse events of patients with myasthenia gravis who completed a randomized controlled trial of thymectomy plus prednisone versus prednisone alone and agreed to participate in a rater-blinded 2-year extension.
“Thymectomy within the first few years of the disease course in addition to prednisone therapy confers benefits that persist for 5 years ... in patients with generalized nonthymomatous myasthenia gravis,” said lead study author Gil I. Wolfe, MD, chair of the department of neurology at the University at Buffalo in New York, and his research colleagues. “Results from the extension study provide further support for the use of thymectomy in management of myasthenia gravis and should encourage serious consideration of this treatment option in discussions between clinicians and their patients,” they wrote. “Our results should lead to revision of clinical guidelines in favor of thymectomy and could potentially reverse downward trends in the use of thymectomy in overall management of myasthenia gravis.”
The main 3-year results of the Thymectomy Trial in Nonthymomatous Myasthenia Gravis Patients Receiving Prednisone (MGTX) were reported in 2016; the international trial found that thymectomy plus prednisone was superior to prednisone alone at 3 years (N Engl J Med. 2016 Aug 11;375[6]:511-22). The extension study aimed to assess the durability of the treatment response.
MGTX enrolled patients aged 18-65 years who had generalized nonthymomatous myasthenia gravis of less than 5 years’ duration and Myasthenia Gravis Foundation of America Clinical Classification Class II-IV disease. Of 111 patients who completed MGTX, 68 entered the extension study, and 50 completed the 60-month assessment (24 patients in the prednisone alone group and 26 patients in the prednisone plus thymectomy group).
At 5 years, patients in the thymectomy plus prednisone group had significantly lower time-weighted average Quantitative Myasthenia Gravis (QMG) scores (5.47 vs. 9.34) and mean alternate-day prednisone doses (24 mg vs. 48 mg), compared with patients who received prednisone alone. Twelve of 35 patients in the thymectomy group and 14 of 33 patients in the prednisone group had at least one adverse event by month 60. No treatment-related deaths occurred in the extension phase.
At 5 years, significantly more patients who underwent thymectomy had minimal manifestation status (i.e., no functional limitations from the disease other than some muscle weakness) – 88% versus 58%. The corresponding figures at 3 years were 67% and 47%.
In addition, 3-year and 5-year data indicate that the need for hospitalization is reduced after surgery, compared with medical therapy alone, Dr. Wolfe said.
Two patients in each treatment arm had an increase of 2 points or more in the QMG score, indicating clinical worsening.
“Our current findings reinforce the benefit of thymectomy seen in [MGTX], dispelling doubts about the procedure’s benefits and how long those benefits last,” said Dr. Wolfe. “We do hope that the new findings help reverse the apparent reluctance to do thymectomy and that the proportion of patients with myasthenia gravis who undergo thymectomy will increase.”
The authors noted that the small sample size of the extension study may limit its generalizability.
The study received funding from the National Institutes of Health. Dr. Wolfe reported grants from the NIH, the Muscular Dystrophy Association, the Myasthenia Gravis Foundation of America, CSL-Behring, and ArgenX, as well as personal fees from Grifols, Shire, and Alexion Pharmaceuticals. Coauthors reported working with and receiving funds from agencies, foundations, and pharmaceutical companies.
SOURCE: Wolfe GI et al. Lancet Neurol. 2019 Jan 25. doi: 10.1016/S1474-4422(18)30392-2.
Thymectomy may continue to benefit patients with myasthenia gravis 5 years after the procedure, according to an extension study published in Lancet Neurology.
The study evaluated the clinical status, medication requirements, and adverse events of patients with myasthenia gravis who completed a randomized controlled trial of thymectomy plus prednisone versus prednisone alone and agreed to participate in a rater-blinded 2-year extension.
“Thymectomy within the first few years of the disease course in addition to prednisone therapy confers benefits that persist for 5 years ... in patients with generalized nonthymomatous myasthenia gravis,” said lead study author Gil I. Wolfe, MD, chair of the department of neurology at the University at Buffalo in New York, and his research colleagues. “Results from the extension study provide further support for the use of thymectomy in management of myasthenia gravis and should encourage serious consideration of this treatment option in discussions between clinicians and their patients,” they wrote. “Our results should lead to revision of clinical guidelines in favor of thymectomy and could potentially reverse downward trends in the use of thymectomy in overall management of myasthenia gravis.”
The main 3-year results of the Thymectomy Trial in Nonthymomatous Myasthenia Gravis Patients Receiving Prednisone (MGTX) were reported in 2016; the international trial found that thymectomy plus prednisone was superior to prednisone alone at 3 years (N Engl J Med. 2016 Aug 11;375[6]:511-22). The extension study aimed to assess the durability of the treatment response.
MGTX enrolled patients aged 18-65 years who had generalized nonthymomatous myasthenia gravis of less than 5 years’ duration and Myasthenia Gravis Foundation of America Clinical Classification Class II-IV disease. Of 111 patients who completed MGTX, 68 entered the extension study, and 50 completed the 60-month assessment (24 patients in the prednisone alone group and 26 patients in the prednisone plus thymectomy group).
At 5 years, patients in the thymectomy plus prednisone group had significantly lower time-weighted average Quantitative Myasthenia Gravis (QMG) scores (5.47 vs. 9.34) and mean alternate-day prednisone doses (24 mg vs. 48 mg), compared with patients who received prednisone alone. Twelve of 35 patients in the thymectomy group and 14 of 33 patients in the prednisone group had at least one adverse event by month 60. No treatment-related deaths occurred in the extension phase.
At 5 years, significantly more patients who underwent thymectomy had minimal manifestation status (i.e., no functional limitations from the disease other than some muscle weakness) – 88% versus 58%. The corresponding figures at 3 years were 67% and 47%.
In addition, 3-year and 5-year data indicate that the need for hospitalization is reduced after surgery, compared with medical therapy alone, Dr. Wolfe said.
Two patients in each treatment arm had an increase of 2 points or more in the QMG score, indicating clinical worsening.
“Our current findings reinforce the benefit of thymectomy seen in [MGTX], dispelling doubts about the procedure’s benefits and how long those benefits last,” said Dr. Wolfe. “We do hope that the new findings help reverse the apparent reluctance to do thymectomy and that the proportion of patients with myasthenia gravis who undergo thymectomy will increase.”
The authors noted that the small sample size of the extension study may limit its generalizability.
The study received funding from the National Institutes of Health. Dr. Wolfe reported grants from the NIH, the Muscular Dystrophy Association, the Myasthenia Gravis Foundation of America, CSL-Behring, and ArgenX, as well as personal fees from Grifols, Shire, and Alexion Pharmaceuticals. Coauthors reported working with and receiving funds from agencies, foundations, and pharmaceutical companies.
SOURCE: Wolfe GI et al. Lancet Neurol. 2019 Jan 25. doi: 10.1016/S1474-4422(18)30392-2.
FROM LANCET NEUROLOGY
Key clinical point: The benefits of thymectomy for myasthenia gravis persist 5 years after the procedure.
Major finding: Patients who undergo thymectomy and receive prednisone have lower time-weighted average Quantitative Myasthenia Gravis scores (5.47 vs. 9.34) and mean alternate-day prednisone doses (24 mg vs. 48 mg), compared with patients who receive prednisone alone.
Study details: A rater-blinded 2-year extension study that enrolled 68 patients who had completed a 3-year randomized controlled trial.
Disclosures: The study received funding from the National Institutes of Health. Dr. Wolfe reported grants from the NIH, the Muscular Dystrophy Association, the Myasthenia Gravis Foundation of America, CSL-Behring, and ArgenX, as well as personal fees from Grifols, Shire, and Alexion Pharmaceuticals. Other authors reported working with and receiving funds from various agencies, foundations, and pharmaceutical companies.
Source: Wolfe GI et al. Lancet Neurol. 2019 Jan 25. doi: 10.1016/S1474-4422(18)30392-2.
Science Has Spoken: Undetectable Equals Untransmittable
The consensus is in: Undetectable is Untransmittable (U = U). That is, scientific experts are finally willing to say that the concept of “Undetectable is Untransmittable” for HIV treatment is now “firmly established.” Anthony Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID), writing in JAMA, says an “overwhelming” body of clinical evidence provides a firm basis for accepting the concept as scientifically sound.
In the JAMA commentary, Fauci and colleagues review the results of clinical trials validating U = U. One landmark study, for instance, showed that no linked HIV transmissions occurred among HIV serodifferent heterosexual couples when the partner living with HIV had a durably suppressed viral load. Subsequent studies confirmed the findings and extended them to male-male couples.
The key, the researchers all agree, is to be absolutely adherent to antiretroviral therapy (ART). Viral suppression measured at 6 months after starting therapy is required for U = U. Stopping ART represents a “significant challenge” to successful implementation of U = U. According to the clinical trials, when ART is stopped, viral rebound usually occurs within 2 to 3 weeks. In 2 studies, stopping ART caused viral rebound to levels that would have been associated with increased risk of HIV transmission.
The NIH experts say this consensus has a variety of implications. It gives incentive to people living with HIV to start and adhere to treatment, removes the sense of fear and guilt they may have about harming others, and reduces the risk of legal penalties arising from putting virus-free partners at risk. And because “prevention as control” is a critical tool, the U = U concept can support worldwide efforts to control—or even eliminate—the pandemic.
Source:
Eisinger RW, Dieffenbach CW, Fauci AS. JAMA. 2019.
doi: 10.1001/jama.2018.21167. [Epub ahead of print.]
The consensus is in: Undetectable is Untransmittable (U = U). That is, scientific experts are finally willing to say that the concept of “Undetectable is Untransmittable” for HIV treatment is now “firmly established.” Anthony Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID), writing in JAMA, says an “overwhelming” body of clinical evidence provides a firm basis for accepting the concept as scientifically sound.
In the JAMA commentary, Fauci and colleagues review the results of clinical trials validating U = U. One landmark study, for instance, showed that no linked HIV transmissions occurred among HIV serodifferent heterosexual couples when the partner living with HIV had a durably suppressed viral load. Subsequent studies confirmed the findings and extended them to male-male couples.
The key, the researchers all agree, is to be absolutely adherent to antiretroviral therapy (ART). Viral suppression measured at 6 months after starting therapy is required for U = U. Stopping ART represents a “significant challenge” to successful implementation of U = U. According to the clinical trials, when ART is stopped, viral rebound usually occurs within 2 to 3 weeks. In 2 studies, stopping ART caused viral rebound to levels that would have been associated with increased risk of HIV transmission.
The NIH experts say this consensus has a variety of implications. It gives incentive to people living with HIV to start and adhere to treatment, removes the sense of fear and guilt they may have about harming others, and reduces the risk of legal penalties arising from putting virus-free partners at risk. And because “prevention as control” is a critical tool, the U = U concept can support worldwide efforts to control—or even eliminate—the pandemic.
Source:
Eisinger RW, Dieffenbach CW, Fauci AS. JAMA. 2019.
doi: 10.1001/jama.2018.21167. [Epub ahead of print.]
The consensus is in: Undetectable is Untransmittable (U = U). That is, scientific experts are finally willing to say that the concept of “Undetectable is Untransmittable” for HIV treatment is now “firmly established.” Anthony Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID), writing in JAMA, says an “overwhelming” body of clinical evidence provides a firm basis for accepting the concept as scientifically sound.
In the JAMA commentary, Fauci and colleagues review the results of clinical trials validating U = U. One landmark study, for instance, showed that no linked HIV transmissions occurred among HIV serodifferent heterosexual couples when the partner living with HIV had a durably suppressed viral load. Subsequent studies confirmed the findings and extended them to male-male couples.
The key, the researchers all agree, is to be absolutely adherent to antiretroviral therapy (ART). Viral suppression measured at 6 months after starting therapy is required for U = U. Stopping ART represents a “significant challenge” to successful implementation of U = U. According to the clinical trials, when ART is stopped, viral rebound usually occurs within 2 to 3 weeks. In 2 studies, stopping ART caused viral rebound to levels that would have been associated with increased risk of HIV transmission.
The NIH experts say this consensus has a variety of implications. It gives incentive to people living with HIV to start and adhere to treatment, removes the sense of fear and guilt they may have about harming others, and reduces the risk of legal penalties arising from putting virus-free partners at risk. And because “prevention as control” is a critical tool, the U = U concept can support worldwide efforts to control—or even eliminate—the pandemic.
Source:
Eisinger RW, Dieffenbach CW, Fauci AS. JAMA. 2019.
doi: 10.1001/jama.2018.21167. [Epub ahead of print.]